User login
Study tests a simpler low disease activity measure for lupus
An alternative disease activity index for patients with systemic lupus erythematosus called the SLE-DAS (Disease Activity Score) has shown similar results to the Lupus Low Disease Activity State (LLDAS) in classifying low disease activity but may be easier to potentially apply in daily clinical practice in treat-to-target strategies, according to research presented at the annual European Congress of Rheumatology, held online this year because of COVID-19.

A treat-to-target approach, in which therapies are adjusted and the patient monitored to achieve the desired endpoint, has been proposed for patients with SLE. Clinical remission is the ideal goal, followed by achieving low disease activity (LDA) when clinical remission is unattainable, the first author of the SLE-DAS study, Helena Assunção, MD, of the department of rheumatology at Centro Hospitalar e Universitário de Coimbra (Portugal), said in an interview prior to the presentation of the study at the e-congress.
But to conduct a treat-to-target approach in the clinical setting, clinicians must have reliable, user-friendly targets to assess a patient’s progress, she said. But that’s not available right now. Proposed definitions of LDA, such as the LLDAS, are based on the Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K). This index doesn’t address some important manifestations of SLE and it is scored dichotomously – for example, giving a similar score for thrombocytopenia when platelet count is reduced to 100,000 or to 10,000.
To compensate for these limitations, the current LLDAS definition also requires the Physician Global Assessment and other steps, including a review of medication and changes to treatment or clinical status since the previous visit.
“It is not easy to apply,” Dr. Assunção said.
The SLE-DAS is a continuous index involving 17 parameters (4 continuous: arthritis, proteinuria, thrombocytopenia, and leukopenia), assigning higher scores when a manifestation is more severe, and has manifestation information that SLEDAI-2K lacks (cardiopulmonary involvement, lupus enteritis, and hemolytic anemia).
In contrast, the LLDAS is defined as:
- A SLEDAI-2k score of 4 or less with no major organ involvement
- No new disease activity
- A physician global assessment of the patient of 1 or less on a 0-3 scale
- Maintenance on a prednisolone dosage of 7.5 mg/day or less
- Maintenance on a standard immunosuppressive regimen
A previous study validated the SLE-DAS (Ann Rheum Dis. 2019 Mar;78[3]:365-71), and another exploratory study identified a cutoff SLE-DAS value of 3.77 or lower for LDA with SLE-DAS (Ann Rheum Dis. 2019;78:411-2).
Her group compared LDA status as measured with LLDAS versus the SLE-DAS in a cross-sectional study of 292 consecutive patients at their hospital. LDA on the SLE-DAS was defined as a score 3.77 or lower and a prednisolone dose of 7.5 mg/day or less. A total of 85% of patients were in LDA with SLE-DAS and 83.9% with LLDAS, and the agreement between LLDAS and SLE-DAS LDA was very high (Cohen’s kappa coefficient test; kappa = 0.831; P < .01). Out of 292 patients, only 13 were classified differently by the two definitions, 8 of which were classified as LDA by SLE-DAS, and 5 by LLDAS. Overall, 87% of patients were women and had a mean age of nearly 49 years, with a mean disease duration of about 14 years.
Dr. Assunção feels that the SLE-DAS LDA should be sufficient to monitor disease activity without adding the Physician Global Assessment and other steps, which would make it easier to apply than LLDAS. The fact that it is based on a continuous index is also an important difference. “Especially for low disease activity, it’s very good to be able to define it with a continuous index, because you are not that bad, but not that good, you’re in the middle,” she said.
The study should be regarded as exploratory, she said, but the results were encouraging. “We got similar results, and it’s definitely easier to apply.” She can also personally attest that the new model is easier to use, since she personally collected data for LLDAS assignment. “I had to check this, and this, and this … [SLE-DAS] is easier.”
Future work from her group will aim at deriving and validating a more robust definition of LDA, which will again be compared with the current LLDAS definition.
Her colleagues have already developed and validated a definition for clinical remission using SLE-DAS, although those results have not yet been published. They hope to define activity states using SLE-DAS, including mild, moderate, and high disease activity.
The team has produced an online SLE-DAS calculator (http://sle-das.eu/) where clinicians can score the 17 parameters. “You just input the values and it gives a number reflecting disease activity. Using this definition of SLE-DAS LDA you only need that number and to verify that the prednisolone dose is equal to or inferior to 7.5 mg/day,” said Dr. Assunção.
The study received no funding. Dr. Assunção has no financial disclosures, but one coauthor reported receiving grant/research support from Pfizer and AbbVie and serving as a consultant to Pfizer, AbbVie, Roche, Lilly, and Novartis.
SOURCE: Assunção H et al. Ann Rheum Dis 2020;79[suppl 1]:60, Abstract OP0092.
An alternative disease activity index for patients with systemic lupus erythematosus called the SLE-DAS (Disease Activity Score) has shown similar results to the Lupus Low Disease Activity State (LLDAS) in classifying low disease activity but may be easier to potentially apply in daily clinical practice in treat-to-target strategies, according to research presented at the annual European Congress of Rheumatology, held online this year because of COVID-19.
A treat-to-target approach, in which therapies are adjusted and the patient monitored to achieve the desired endpoint, has been proposed for patients with SLE. Clinical remission is the ideal goal, followed by achieving low disease activity (LDA) when clinical remission is unattainable, the first author of the SLE-DAS study, Helena Assunção, MD, of the department of rheumatology at Centro Hospitalar e Universitário de Coimbra (Portugal), said in an interview prior to the presentation of the study at the e-congress.
But to conduct a treat-to-target approach in the clinical setting, clinicians must have reliable, user-friendly targets to assess a patient’s progress, she said. But that’s not available right now. Proposed definitions of LDA, such as the LLDAS, are based on the Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K). This index doesn’t address some important manifestations of SLE and it is scored dichotomously – for example, giving a similar score for thrombocytopenia when platelet count is reduced to 100,000 or to 10,000.
To compensate for these limitations, the current LLDAS definition also requires the Physician Global Assessment and other steps, including a review of medication and changes to treatment or clinical status since the previous visit.
“It is not easy to apply,” Dr. Assunção said.
The SLE-DAS is a continuous index involving 17 parameters (4 continuous: arthritis, proteinuria, thrombocytopenia, and leukopenia), assigning higher scores when a manifestation is more severe, and has manifestation information that SLEDAI-2K lacks (cardiopulmonary involvement, lupus enteritis, and hemolytic anemia).
In contrast, the LLDAS is defined as:
- A SLEDAI-2k score of 4 or less with no major organ involvement
- No new disease activity
- A physician global assessment of the patient of 1 or less on a 0-3 scale
- Maintenance on a prednisolone dosage of 7.5 mg/day or less
- Maintenance on a standard immunosuppressive regimen
A previous study validated the SLE-DAS (Ann Rheum Dis. 2019 Mar;78[3]:365-71), and another exploratory study identified a cutoff SLE-DAS value of 3.77 or lower for LDA with SLE-DAS (Ann Rheum Dis. 2019;78:411-2).
Her group compared LDA status as measured with LLDAS versus the SLE-DAS in a cross-sectional study of 292 consecutive patients at their hospital. LDA on the SLE-DAS was defined as a score 3.77 or lower and a prednisolone dose of 7.5 mg/day or less. A total of 85% of patients were in LDA with SLE-DAS and 83.9% with LLDAS, and the agreement between LLDAS and SLE-DAS LDA was very high (Cohen’s kappa coefficient test; kappa = 0.831; P < .01). Out of 292 patients, only 13 were classified differently by the two definitions, 8 of which were classified as LDA by SLE-DAS, and 5 by LLDAS. Overall, 87% of patients were women and had a mean age of nearly 49 years, with a mean disease duration of about 14 years.
Dr. Assunção feels that the SLE-DAS LDA should be sufficient to monitor disease activity without adding the Physician Global Assessment and other steps, which would make it easier to apply than LLDAS. The fact that it is based on a continuous index is also an important difference. “Especially for low disease activity, it’s very good to be able to define it with a continuous index, because you are not that bad, but not that good, you’re in the middle,” she said.
The study should be regarded as exploratory, she said, but the results were encouraging. “We got similar results, and it’s definitely easier to apply.” She can also personally attest that the new model is easier to use, since she personally collected data for LLDAS assignment. “I had to check this, and this, and this … [SLE-DAS] is easier.”
Future work from her group will aim at deriving and validating a more robust definition of LDA, which will again be compared with the current LLDAS definition.
Her colleagues have already developed and validated a definition for clinical remission using SLE-DAS, although those results have not yet been published. They hope to define activity states using SLE-DAS, including mild, moderate, and high disease activity.
The team has produced an online SLE-DAS calculator (http://sle-das.eu/) where clinicians can score the 17 parameters. “You just input the values and it gives a number reflecting disease activity. Using this definition of SLE-DAS LDA you only need that number and to verify that the prednisolone dose is equal to or inferior to 7.5 mg/day,” said Dr. Assunção.
The study received no funding. Dr. Assunção has no financial disclosures, but one coauthor reported receiving grant/research support from Pfizer and AbbVie and serving as a consultant to Pfizer, AbbVie, Roche, Lilly, and Novartis.
SOURCE: Assunção H et al. Ann Rheum Dis 2020;79[suppl 1]:60, Abstract OP0092.
An alternative disease activity index for patients with systemic lupus erythematosus called the SLE-DAS (Disease Activity Score) has shown similar results to the Lupus Low Disease Activity State (LLDAS) in classifying low disease activity but may be easier to potentially apply in daily clinical practice in treat-to-target strategies, according to research presented at the annual European Congress of Rheumatology, held online this year because of COVID-19.
A treat-to-target approach, in which therapies are adjusted and the patient monitored to achieve the desired endpoint, has been proposed for patients with SLE. Clinical remission is the ideal goal, followed by achieving low disease activity (LDA) when clinical remission is unattainable, the first author of the SLE-DAS study, Helena Assunção, MD, of the department of rheumatology at Centro Hospitalar e Universitário de Coimbra (Portugal), said in an interview prior to the presentation of the study at the e-congress.
But to conduct a treat-to-target approach in the clinical setting, clinicians must have reliable, user-friendly targets to assess a patient’s progress, she said. But that’s not available right now. Proposed definitions of LDA, such as the LLDAS, are based on the Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K). This index doesn’t address some important manifestations of SLE and it is scored dichotomously – for example, giving a similar score for thrombocytopenia when platelet count is reduced to 100,000 or to 10,000.
To compensate for these limitations, the current LLDAS definition also requires the Physician Global Assessment and other steps, including a review of medication and changes to treatment or clinical status since the previous visit.
“It is not easy to apply,” Dr. Assunção said.
The SLE-DAS is a continuous index involving 17 parameters (4 continuous: arthritis, proteinuria, thrombocytopenia, and leukopenia), assigning higher scores when a manifestation is more severe, and has manifestation information that SLEDAI-2K lacks (cardiopulmonary involvement, lupus enteritis, and hemolytic anemia).
In contrast, the LLDAS is defined as:
- A SLEDAI-2k score of 4 or less with no major organ involvement
- No new disease activity
- A physician global assessment of the patient of 1 or less on a 0-3 scale
- Maintenance on a prednisolone dosage of 7.5 mg/day or less
- Maintenance on a standard immunosuppressive regimen
A previous study validated the SLE-DAS (Ann Rheum Dis. 2019 Mar;78[3]:365-71), and another exploratory study identified a cutoff SLE-DAS value of 3.77 or lower for LDA with SLE-DAS (Ann Rheum Dis. 2019;78:411-2).
Her group compared LDA status as measured with LLDAS versus the SLE-DAS in a cross-sectional study of 292 consecutive patients at their hospital. LDA on the SLE-DAS was defined as a score 3.77 or lower and a prednisolone dose of 7.5 mg/day or less. A total of 85% of patients were in LDA with SLE-DAS and 83.9% with LLDAS, and the agreement between LLDAS and SLE-DAS LDA was very high (Cohen’s kappa coefficient test; kappa = 0.831; P < .01). Out of 292 patients, only 13 were classified differently by the two definitions, 8 of which were classified as LDA by SLE-DAS, and 5 by LLDAS. Overall, 87% of patients were women and had a mean age of nearly 49 years, with a mean disease duration of about 14 years.
Dr. Assunção feels that the SLE-DAS LDA should be sufficient to monitor disease activity without adding the Physician Global Assessment and other steps, which would make it easier to apply than LLDAS. The fact that it is based on a continuous index is also an important difference. “Especially for low disease activity, it’s very good to be able to define it with a continuous index, because you are not that bad, but not that good, you’re in the middle,” she said.
The study should be regarded as exploratory, she said, but the results were encouraging. “We got similar results, and it’s definitely easier to apply.” She can also personally attest that the new model is easier to use, since she personally collected data for LLDAS assignment. “I had to check this, and this, and this … [SLE-DAS] is easier.”
Future work from her group will aim at deriving and validating a more robust definition of LDA, which will again be compared with the current LLDAS definition.
Her colleagues have already developed and validated a definition for clinical remission using SLE-DAS, although those results have not yet been published. They hope to define activity states using SLE-DAS, including mild, moderate, and high disease activity.
The team has produced an online SLE-DAS calculator (http://sle-das.eu/) where clinicians can score the 17 parameters. “You just input the values and it gives a number reflecting disease activity. Using this definition of SLE-DAS LDA you only need that number and to verify that the prednisolone dose is equal to or inferior to 7.5 mg/day,” said Dr. Assunção.
The study received no funding. Dr. Assunção has no financial disclosures, but one coauthor reported receiving grant/research support from Pfizer and AbbVie and serving as a consultant to Pfizer, AbbVie, Roche, Lilly, and Novartis.
SOURCE: Assunção H et al. Ann Rheum Dis 2020;79[suppl 1]:60, Abstract OP0092.
FROM EULAR 2020 E-CONGRESS
Emerging Therapies for Cutaneous Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a chronic autoimmune inflammatory disease that can have devastating effects on many organs. Despite the considerable morbidity and mortality associated with SLE, treatment options have been largely unchanged since the 1950s.1 It was not until the last decade that a new biologic medication was approved, and several other promising treatments currently are being evaluated in clinical trials. Dermatologists are most likely to encounter cutaneous lupus erythematosus (CLE) with or without SLE, which can present with a variety of skin manifestations. Cutaneous lupus erythematosus can have devastating effects on quality of life and can be a visible sign of the internal activity and damage of SLE.2,3 Although many trials have been completed evaluating SLE treatments, few medications have been evaluated specifically for CLE despite the availability of validated measures of CLE skin activity.4 There is a recent shortage of antimalarial medications, the current first-line therapy for CLE, due to both an import alert in the United States on quinacrine placed in 2019 as well as the use of hydroxychloroquine and chloroquine in treating coronavirus disease 2019.5,6 Due to this shortage, the need for new and effective treatments is more critical than ever, as alternatives to first-line therapy frequently require immunosuppression. We review recent drug approvals for SLE and their efficacy in CLE. We also provide an update on new agents currently being studied to treat this disease.
Belimumab
Belimumab is a B-lymphocyte stimulator–specific inhibitor that was first approved for treatment of SLE in 2011. It was the first monoclonal antibody approved to treat SLE.7 B-lymphocyte stimulator plays a critical role in B-cell survival; thus, its inhibition increases apoptosis of autoreactive B cells involved in the pathogenesis of SLE. More recently, belimumab was approved for pediatric SLE in April 2019 based on the PLUTO study, a phase 2 randomized, double-blind study of 93 patients.8 Although patients with cutaneous manifestations of lupus were included in trials for belimumab, they lacked CLE-specific outcome measurements to truly evaluate the efficacy in treating skin disease.9 This medication currently is not approved by the US Food and Drug Administration (FDA) for CLE; however, it is used off label in some cases for recalcitrant disease.10
Baricitinib
Baricitinib is a selective and reversible inhibitor of JAK1 and JAK2 that was granted fast-track status by the FDA in December 2018. In a phase 2 trial, baricitinib was superior to placebo plus standard of care, primarily for arthritis and lupus nephritis.11 Although improvement of cutaneous disease was measured as an end point, it did not show significant improvement in disease. The presence of skin disease was high, but the activity of disease was low, which can make it difficult to show meaningful improvement, as there is not much room for patients to objectively improve.12 Showing meaningful improvement in skin disease often is difficult in phase 2 trials, especially when the trial design is focused on SLE rather than CLE activity. Further studies of baricitinib that include more severe patients with CLE disease are needed to truly understand its effects on the skin.
Lenalidomide
There have been several CLE studies in the last several years surrounding lenalidomide, an analog of thalidomide.13-15 This molecule has a number of immunomodulatory effects including antiangiogenic effects, increased natural killer cell–dependent cytotoxicity, and cytokine and interleukin inhibition. Lenalidomide is of particular interest in treating CLE, as it was shown to be more potent than thalidomide at low doses and with a better side-effect profile. Multiple small, open-label trials have shown lenalidomide to be both safe and efficacious in the treatment of CLE.13,14 In addition, iberdomide, a derivative of lenalidomide, recently completed a phase 2 dose-escalation study showing improvement in both SLE and CLE end points.16 A phase 2b proof-of-concept study currently is underway (ClinicalTrials.gov Identifier NCT03161483).
Monoclonal Antibodies
Many developing therapies target specific components of the type I interferon pathway, which is a primary driver of CLE lesions. Innate immune system pathways involving type I interferon were shown to be active in the pathogenesis of CLE, and levels of interferon correlate with skin disease activity.17 One molecule in development that targets this pathway is BIIB059, a humanized IgG1 monoclonal antibody that binds to blood dendritic cell antigen 2. This cell surface protein is uniquely expressed on plasmacytoid dendritic cells, which are the main source of type I interferon overproduction in SLE. The binding of this antibody to the blood dendritic cell antigen 2 receptor both blocks type I interferon production and decreases the overall number of active plasmacytoid dendritic cells present.18 In the completed phase 1b study, a response in cutaneous disease was shown through a reduction in the CLE disease area and severity index score following single-dose administration.19 More recently, a phase 2 study met primary end points in both SLE and CLE compared to placebo.20
Anifrolumab is a human IgG1k monoclonal antibody that binds to type I interferon receptor, blocking all type I interferon signaling. Following a successful phase 2 trial, it failed to meet its primary end point in its first phase 3 trial.21 Several secondary end points suggested a clinical benefit. A second phase 3 trial of 362 patients randomized to treatment with anifrolumab or placebo over 48 weeks showed anifrolumab to be superior to placebo for multiple end points, including the overall disease primary end point as well as a notable reduction in skin activity.22
Final Thoughts
Outside of the approval of belimumab, there have been no new FDA-approved treatments for SLE since the approval of antimalarial agents nearly 50 years ago. For CLE specifically, there is an even greater scarcity of evidence-based treatments. Recently studied medications, such as belimumab and lenalidomide, are available off label for CLE patients when other options have failed. Recent studies have evaluated the efficacy of these agents in the treatment of CLE using the CLE disease area and severity index.10,13,14 Enrollment in CLE trials is difficult due to the rarity of the disease, and careful attention must be paid to evaluating skin end points. As experts in CLE and the nuances of these assessments, it is critical that dermatologists be involved in clinical trials. Future SLE trials must consider CLE as an important end point for CLE patients to get access to much-needed novel therapies.
- Bernatsky S, Boivin JF, Joseph L, et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006;54:2550-2557.
- Vasquez R, Wang D, Tran QP, et al. A multicentre, cross-sectional study on quality of life in patients with cutaneous lupus erythematosus. Br J Dermatol. 2013;168:145-153.
- Klein R, Moghadam-Kia S, Taylor L, et al. Quality of life in cutaneous lupus erythematosus. J Am Acad Dermatol. 2011;64:849-858.
- Klein R, Moghadam-Kia S, LoMonico J, et al. Development of the CLASI as a tool to measure disease severity and responsiveness to therapy in cutaneous lupus erythematosus. Arch Dermatol. 2011;147:203-208.
- Jakhar D, Kaur I. Potential of chloroquine and hydroxychloroquine to treat COVID-19 causes fears of shortages among people with systemic lupus erythematosus. Nat Med. 2020;26:632.
- American College of Rheumatology. Quinacrine shortage & what the ACR is doing about it. https://www.the-rheumatologist.org/article/quinacrine-shortage-what-the-acr-is-doing-about-it/. Published February 8, 2019. Accessed May 15, 2020.
- Dubey AK, Handu SS, Dubey S, et al. Belimumab: first targeted biological treatment for systemic lupus erythematosus. J Pharmacol Pharmacother. 2011;2:317-319.
- Brunner H, Abud-Mendoza C, Viola D, et al. Efficacy and safety of intravenous belimumab in children with systemic lupus erythematosus [abstract]. Arthritis Rheumatol. 2018;70(suppl 10). https://acrabstracts.org/abstract/efficacy-and-safety-of-intravenous-belimumab-in-children-with-systemic-lupus-erythematosus/. Accessed May 7, 2020.
- Hui-Yuen JS, Reddy A, Taylor J, et al. Safety and efficacy of belimumab to treat systemic lupus erythematosus in academic clinical practices. J Rheumatol. 2015;42:2288-2295.
- Vashisht P, Borghoff K, O’Dell JR, et al. Belimumab for the treatment of recalcitrant cutaneous lupus. Lupus. 2017;26:857-864.
- Wallace DJ, Furie RA, Tanaka Y, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2018;392:222-231.
- Werth VP, Merrill JT. A double-blind, randomized, placebo-controlled, phase II trial of baricitinib for systemic lupus erythematosus: how to optimize lupus trials to examine effects on cutaneous lupus erythematosus. Br J Dermatol. 2019;180:964-965.
- Cortés-Hernández J, Ávila G, Vilardell-Tarrés M, et al. Efficacy and safety of lenalidomide for refractory cutaneous lupus erythematosus. Arthritis Res Ther. 2012;14:R265.
- Okon L, Rosenbach M, Krathen M, et al. Lenalidomide in treatment-refractory cutaneous lupus erythematosus: efficacy and safety in a 52-week trial. J Am Acad Dermatol. 2014;70:583-584.
- Fennira F, Chasset F, Soubrier M, et al. Lenalidomide for refractory chronic and subacute cutaneous lupus erythematosus: 16 patients. J Am Acad Dermatol. 2016;74:1248-1251.
- Furie R, Werth V, Gaudy A, et al. A randomized, placebo-controlled, double-blind, ascending-dose, safety, and pharmacokinetics study of CC-220 in subjects with systemic LUPUS erythematosus [abstract]. Arthritis Rheumatol. 2017;69(suppl 10). https://acrabstracts.org/abstract/a-randomized-placebo-controlled-double-blind-ascending-dose-safety-and-pharmacokinetics-study-of-cc-220-in-subjects-with-systemic-lupus-erythematosus/. Accessed May 7, 2020.
- Braunstein I, Klein R, Okawa J, et al. The interferon-regulated gene signature is elevated in subacute cutaneous lupus erythematosus and discoid lupus erythematosus and correlates with the cutaneous lupus area and severity index score. Br J Dermatol. 2012;166:971-975.
- Kim JM, Park SH, Kim HY, et al. A plasmacytoid dendritic cells-type I interferon axis is critically implicated in the pathogenesis of systemic lupus erythematosus. Int J Mol Sci. 2015;16:14158-14170.
- Furie R, Werth VP, Merola JF, et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J Clin Invest. 2019;129:1359-1371.
- Werth V, Musselli C, Furie R, et al. BIIB059, a humanized monoclonal antibody targeting BDCA2 on plasmacytoid dendritic cells (pDC), shows dose-related efficacy in the phase 2 LILAC study in patients (pts) with active cutaneous lupus erythematosus (CLE). Ann Rheum Dis. In press.
- Furie R, Morand EF, Bruce I, et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): a randomised, controlled, phase 3 trial. Lancet Rheumatol. 2019;1:E208-E219.
- Morand EF, Furie R, Tanaka Y, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. 2020;382:211-221.
Systemic lupus erythematosus (SLE) is a chronic autoimmune inflammatory disease that can have devastating effects on many organs. Despite the considerable morbidity and mortality associated with SLE, treatment options have been largely unchanged since the 1950s.1 It was not until the last decade that a new biologic medication was approved, and several other promising treatments currently are being evaluated in clinical trials. Dermatologists are most likely to encounter cutaneous lupus erythematosus (CLE) with or without SLE, which can present with a variety of skin manifestations. Cutaneous lupus erythematosus can have devastating effects on quality of life and can be a visible sign of the internal activity and damage of SLE.2,3 Although many trials have been completed evaluating SLE treatments, few medications have been evaluated specifically for CLE despite the availability of validated measures of CLE skin activity.4 There is a recent shortage of antimalarial medications, the current first-line therapy for CLE, due to both an import alert in the United States on quinacrine placed in 2019 as well as the use of hydroxychloroquine and chloroquine in treating coronavirus disease 2019.5,6 Due to this shortage, the need for new and effective treatments is more critical than ever, as alternatives to first-line therapy frequently require immunosuppression. We review recent drug approvals for SLE and their efficacy in CLE. We also provide an update on new agents currently being studied to treat this disease.
Belimumab
Belimumab is a B-lymphocyte stimulator–specific inhibitor that was first approved for treatment of SLE in 2011. It was the first monoclonal antibody approved to treat SLE.7 B-lymphocyte stimulator plays a critical role in B-cell survival; thus, its inhibition increases apoptosis of autoreactive B cells involved in the pathogenesis of SLE. More recently, belimumab was approved for pediatric SLE in April 2019 based on the PLUTO study, a phase 2 randomized, double-blind study of 93 patients.8 Although patients with cutaneous manifestations of lupus were included in trials for belimumab, they lacked CLE-specific outcome measurements to truly evaluate the efficacy in treating skin disease.9 This medication currently is not approved by the US Food and Drug Administration (FDA) for CLE; however, it is used off label in some cases for recalcitrant disease.10
Baricitinib
Baricitinib is a selective and reversible inhibitor of JAK1 and JAK2 that was granted fast-track status by the FDA in December 2018. In a phase 2 trial, baricitinib was superior to placebo plus standard of care, primarily for arthritis and lupus nephritis.11 Although improvement of cutaneous disease was measured as an end point, it did not show significant improvement in disease. The presence of skin disease was high, but the activity of disease was low, which can make it difficult to show meaningful improvement, as there is not much room for patients to objectively improve.12 Showing meaningful improvement in skin disease often is difficult in phase 2 trials, especially when the trial design is focused on SLE rather than CLE activity. Further studies of baricitinib that include more severe patients with CLE disease are needed to truly understand its effects on the skin.
Lenalidomide
There have been several CLE studies in the last several years surrounding lenalidomide, an analog of thalidomide.13-15 This molecule has a number of immunomodulatory effects including antiangiogenic effects, increased natural killer cell–dependent cytotoxicity, and cytokine and interleukin inhibition. Lenalidomide is of particular interest in treating CLE, as it was shown to be more potent than thalidomide at low doses and with a better side-effect profile. Multiple small, open-label trials have shown lenalidomide to be both safe and efficacious in the treatment of CLE.13,14 In addition, iberdomide, a derivative of lenalidomide, recently completed a phase 2 dose-escalation study showing improvement in both SLE and CLE end points.16 A phase 2b proof-of-concept study currently is underway (ClinicalTrials.gov Identifier NCT03161483).
Monoclonal Antibodies
Many developing therapies target specific components of the type I interferon pathway, which is a primary driver of CLE lesions. Innate immune system pathways involving type I interferon were shown to be active in the pathogenesis of CLE, and levels of interferon correlate with skin disease activity.17 One molecule in development that targets this pathway is BIIB059, a humanized IgG1 monoclonal antibody that binds to blood dendritic cell antigen 2. This cell surface protein is uniquely expressed on plasmacytoid dendritic cells, which are the main source of type I interferon overproduction in SLE. The binding of this antibody to the blood dendritic cell antigen 2 receptor both blocks type I interferon production and decreases the overall number of active plasmacytoid dendritic cells present.18 In the completed phase 1b study, a response in cutaneous disease was shown through a reduction in the CLE disease area and severity index score following single-dose administration.19 More recently, a phase 2 study met primary end points in both SLE and CLE compared to placebo.20
Anifrolumab is a human IgG1k monoclonal antibody that binds to type I interferon receptor, blocking all type I interferon signaling. Following a successful phase 2 trial, it failed to meet its primary end point in its first phase 3 trial.21 Several secondary end points suggested a clinical benefit. A second phase 3 trial of 362 patients randomized to treatment with anifrolumab or placebo over 48 weeks showed anifrolumab to be superior to placebo for multiple end points, including the overall disease primary end point as well as a notable reduction in skin activity.22
Final Thoughts
Outside of the approval of belimumab, there have been no new FDA-approved treatments for SLE since the approval of antimalarial agents nearly 50 years ago. For CLE specifically, there is an even greater scarcity of evidence-based treatments. Recently studied medications, such as belimumab and lenalidomide, are available off label for CLE patients when other options have failed. Recent studies have evaluated the efficacy of these agents in the treatment of CLE using the CLE disease area and severity index.10,13,14 Enrollment in CLE trials is difficult due to the rarity of the disease, and careful attention must be paid to evaluating skin end points. As experts in CLE and the nuances of these assessments, it is critical that dermatologists be involved in clinical trials. Future SLE trials must consider CLE as an important end point for CLE patients to get access to much-needed novel therapies.
Systemic lupus erythematosus (SLE) is a chronic autoimmune inflammatory disease that can have devastating effects on many organs. Despite the considerable morbidity and mortality associated with SLE, treatment options have been largely unchanged since the 1950s.1 It was not until the last decade that a new biologic medication was approved, and several other promising treatments currently are being evaluated in clinical trials. Dermatologists are most likely to encounter cutaneous lupus erythematosus (CLE) with or without SLE, which can present with a variety of skin manifestations. Cutaneous lupus erythematosus can have devastating effects on quality of life and can be a visible sign of the internal activity and damage of SLE.2,3 Although many trials have been completed evaluating SLE treatments, few medications have been evaluated specifically for CLE despite the availability of validated measures of CLE skin activity.4 There is a recent shortage of antimalarial medications, the current first-line therapy for CLE, due to both an import alert in the United States on quinacrine placed in 2019 as well as the use of hydroxychloroquine and chloroquine in treating coronavirus disease 2019.5,6 Due to this shortage, the need for new and effective treatments is more critical than ever, as alternatives to first-line therapy frequently require immunosuppression. We review recent drug approvals for SLE and their efficacy in CLE. We also provide an update on new agents currently being studied to treat this disease.
Belimumab
Belimumab is a B-lymphocyte stimulator–specific inhibitor that was first approved for treatment of SLE in 2011. It was the first monoclonal antibody approved to treat SLE.7 B-lymphocyte stimulator plays a critical role in B-cell survival; thus, its inhibition increases apoptosis of autoreactive B cells involved in the pathogenesis of SLE. More recently, belimumab was approved for pediatric SLE in April 2019 based on the PLUTO study, a phase 2 randomized, double-blind study of 93 patients.8 Although patients with cutaneous manifestations of lupus were included in trials for belimumab, they lacked CLE-specific outcome measurements to truly evaluate the efficacy in treating skin disease.9 This medication currently is not approved by the US Food and Drug Administration (FDA) for CLE; however, it is used off label in some cases for recalcitrant disease.10
Baricitinib
Baricitinib is a selective and reversible inhibitor of JAK1 and JAK2 that was granted fast-track status by the FDA in December 2018. In a phase 2 trial, baricitinib was superior to placebo plus standard of care, primarily for arthritis and lupus nephritis.11 Although improvement of cutaneous disease was measured as an end point, it did not show significant improvement in disease. The presence of skin disease was high, but the activity of disease was low, which can make it difficult to show meaningful improvement, as there is not much room for patients to objectively improve.12 Showing meaningful improvement in skin disease often is difficult in phase 2 trials, especially when the trial design is focused on SLE rather than CLE activity. Further studies of baricitinib that include more severe patients with CLE disease are needed to truly understand its effects on the skin.
Lenalidomide
There have been several CLE studies in the last several years surrounding lenalidomide, an analog of thalidomide.13-15 This molecule has a number of immunomodulatory effects including antiangiogenic effects, increased natural killer cell–dependent cytotoxicity, and cytokine and interleukin inhibition. Lenalidomide is of particular interest in treating CLE, as it was shown to be more potent than thalidomide at low doses and with a better side-effect profile. Multiple small, open-label trials have shown lenalidomide to be both safe and efficacious in the treatment of CLE.13,14 In addition, iberdomide, a derivative of lenalidomide, recently completed a phase 2 dose-escalation study showing improvement in both SLE and CLE end points.16 A phase 2b proof-of-concept study currently is underway (ClinicalTrials.gov Identifier NCT03161483).
Monoclonal Antibodies
Many developing therapies target specific components of the type I interferon pathway, which is a primary driver of CLE lesions. Innate immune system pathways involving type I interferon were shown to be active in the pathogenesis of CLE, and levels of interferon correlate with skin disease activity.17 One molecule in development that targets this pathway is BIIB059, a humanized IgG1 monoclonal antibody that binds to blood dendritic cell antigen 2. This cell surface protein is uniquely expressed on plasmacytoid dendritic cells, which are the main source of type I interferon overproduction in SLE. The binding of this antibody to the blood dendritic cell antigen 2 receptor both blocks type I interferon production and decreases the overall number of active plasmacytoid dendritic cells present.18 In the completed phase 1b study, a response in cutaneous disease was shown through a reduction in the CLE disease area and severity index score following single-dose administration.19 More recently, a phase 2 study met primary end points in both SLE and CLE compared to placebo.20
Anifrolumab is a human IgG1k monoclonal antibody that binds to type I interferon receptor, blocking all type I interferon signaling. Following a successful phase 2 trial, it failed to meet its primary end point in its first phase 3 trial.21 Several secondary end points suggested a clinical benefit. A second phase 3 trial of 362 patients randomized to treatment with anifrolumab or placebo over 48 weeks showed anifrolumab to be superior to placebo for multiple end points, including the overall disease primary end point as well as a notable reduction in skin activity.22
Final Thoughts
Outside of the approval of belimumab, there have been no new FDA-approved treatments for SLE since the approval of antimalarial agents nearly 50 years ago. For CLE specifically, there is an even greater scarcity of evidence-based treatments. Recently studied medications, such as belimumab and lenalidomide, are available off label for CLE patients when other options have failed. Recent studies have evaluated the efficacy of these agents in the treatment of CLE using the CLE disease area and severity index.10,13,14 Enrollment in CLE trials is difficult due to the rarity of the disease, and careful attention must be paid to evaluating skin end points. As experts in CLE and the nuances of these assessments, it is critical that dermatologists be involved in clinical trials. Future SLE trials must consider CLE as an important end point for CLE patients to get access to much-needed novel therapies.
- Bernatsky S, Boivin JF, Joseph L, et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006;54:2550-2557.
- Vasquez R, Wang D, Tran QP, et al. A multicentre, cross-sectional study on quality of life in patients with cutaneous lupus erythematosus. Br J Dermatol. 2013;168:145-153.
- Klein R, Moghadam-Kia S, Taylor L, et al. Quality of life in cutaneous lupus erythematosus. J Am Acad Dermatol. 2011;64:849-858.
- Klein R, Moghadam-Kia S, LoMonico J, et al. Development of the CLASI as a tool to measure disease severity and responsiveness to therapy in cutaneous lupus erythematosus. Arch Dermatol. 2011;147:203-208.
- Jakhar D, Kaur I. Potential of chloroquine and hydroxychloroquine to treat COVID-19 causes fears of shortages among people with systemic lupus erythematosus. Nat Med. 2020;26:632.
- American College of Rheumatology. Quinacrine shortage & what the ACR is doing about it. https://www.the-rheumatologist.org/article/quinacrine-shortage-what-the-acr-is-doing-about-it/. Published February 8, 2019. Accessed May 15, 2020.
- Dubey AK, Handu SS, Dubey S, et al. Belimumab: first targeted biological treatment for systemic lupus erythematosus. J Pharmacol Pharmacother. 2011;2:317-319.
- Brunner H, Abud-Mendoza C, Viola D, et al. Efficacy and safety of intravenous belimumab in children with systemic lupus erythematosus [abstract]. Arthritis Rheumatol. 2018;70(suppl 10). https://acrabstracts.org/abstract/efficacy-and-safety-of-intravenous-belimumab-in-children-with-systemic-lupus-erythematosus/. Accessed May 7, 2020.
- Hui-Yuen JS, Reddy A, Taylor J, et al. Safety and efficacy of belimumab to treat systemic lupus erythematosus in academic clinical practices. J Rheumatol. 2015;42:2288-2295.
- Vashisht P, Borghoff K, O’Dell JR, et al. Belimumab for the treatment of recalcitrant cutaneous lupus. Lupus. 2017;26:857-864.
- Wallace DJ, Furie RA, Tanaka Y, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2018;392:222-231.
- Werth VP, Merrill JT. A double-blind, randomized, placebo-controlled, phase II trial of baricitinib for systemic lupus erythematosus: how to optimize lupus trials to examine effects on cutaneous lupus erythematosus. Br J Dermatol. 2019;180:964-965.
- Cortés-Hernández J, Ávila G, Vilardell-Tarrés M, et al. Efficacy and safety of lenalidomide for refractory cutaneous lupus erythematosus. Arthritis Res Ther. 2012;14:R265.
- Okon L, Rosenbach M, Krathen M, et al. Lenalidomide in treatment-refractory cutaneous lupus erythematosus: efficacy and safety in a 52-week trial. J Am Acad Dermatol. 2014;70:583-584.
- Fennira F, Chasset F, Soubrier M, et al. Lenalidomide for refractory chronic and subacute cutaneous lupus erythematosus: 16 patients. J Am Acad Dermatol. 2016;74:1248-1251.
- Furie R, Werth V, Gaudy A, et al. A randomized, placebo-controlled, double-blind, ascending-dose, safety, and pharmacokinetics study of CC-220 in subjects with systemic LUPUS erythematosus [abstract]. Arthritis Rheumatol. 2017;69(suppl 10). https://acrabstracts.org/abstract/a-randomized-placebo-controlled-double-blind-ascending-dose-safety-and-pharmacokinetics-study-of-cc-220-in-subjects-with-systemic-lupus-erythematosus/. Accessed May 7, 2020.
- Braunstein I, Klein R, Okawa J, et al. The interferon-regulated gene signature is elevated in subacute cutaneous lupus erythematosus and discoid lupus erythematosus and correlates with the cutaneous lupus area and severity index score. Br J Dermatol. 2012;166:971-975.
- Kim JM, Park SH, Kim HY, et al. A plasmacytoid dendritic cells-type I interferon axis is critically implicated in the pathogenesis of systemic lupus erythematosus. Int J Mol Sci. 2015;16:14158-14170.
- Furie R, Werth VP, Merola JF, et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J Clin Invest. 2019;129:1359-1371.
- Werth V, Musselli C, Furie R, et al. BIIB059, a humanized monoclonal antibody targeting BDCA2 on plasmacytoid dendritic cells (pDC), shows dose-related efficacy in the phase 2 LILAC study in patients (pts) with active cutaneous lupus erythematosus (CLE). Ann Rheum Dis. In press.
- Furie R, Morand EF, Bruce I, et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): a randomised, controlled, phase 3 trial. Lancet Rheumatol. 2019;1:E208-E219.
- Morand EF, Furie R, Tanaka Y, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. 2020;382:211-221.
- Bernatsky S, Boivin JF, Joseph L, et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006;54:2550-2557.
- Vasquez R, Wang D, Tran QP, et al. A multicentre, cross-sectional study on quality of life in patients with cutaneous lupus erythematosus. Br J Dermatol. 2013;168:145-153.
- Klein R, Moghadam-Kia S, Taylor L, et al. Quality of life in cutaneous lupus erythematosus. J Am Acad Dermatol. 2011;64:849-858.
- Klein R, Moghadam-Kia S, LoMonico J, et al. Development of the CLASI as a tool to measure disease severity and responsiveness to therapy in cutaneous lupus erythematosus. Arch Dermatol. 2011;147:203-208.
- Jakhar D, Kaur I. Potential of chloroquine and hydroxychloroquine to treat COVID-19 causes fears of shortages among people with systemic lupus erythematosus. Nat Med. 2020;26:632.
- American College of Rheumatology. Quinacrine shortage & what the ACR is doing about it. https://www.the-rheumatologist.org/article/quinacrine-shortage-what-the-acr-is-doing-about-it/. Published February 8, 2019. Accessed May 15, 2020.
- Dubey AK, Handu SS, Dubey S, et al. Belimumab: first targeted biological treatment for systemic lupus erythematosus. J Pharmacol Pharmacother. 2011;2:317-319.
- Brunner H, Abud-Mendoza C, Viola D, et al. Efficacy and safety of intravenous belimumab in children with systemic lupus erythematosus [abstract]. Arthritis Rheumatol. 2018;70(suppl 10). https://acrabstracts.org/abstract/efficacy-and-safety-of-intravenous-belimumab-in-children-with-systemic-lupus-erythematosus/. Accessed May 7, 2020.
- Hui-Yuen JS, Reddy A, Taylor J, et al. Safety and efficacy of belimumab to treat systemic lupus erythematosus in academic clinical practices. J Rheumatol. 2015;42:2288-2295.
- Vashisht P, Borghoff K, O’Dell JR, et al. Belimumab for the treatment of recalcitrant cutaneous lupus. Lupus. 2017;26:857-864.
- Wallace DJ, Furie RA, Tanaka Y, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2018;392:222-231.
- Werth VP, Merrill JT. A double-blind, randomized, placebo-controlled, phase II trial of baricitinib for systemic lupus erythematosus: how to optimize lupus trials to examine effects on cutaneous lupus erythematosus. Br J Dermatol. 2019;180:964-965.
- Cortés-Hernández J, Ávila G, Vilardell-Tarrés M, et al. Efficacy and safety of lenalidomide for refractory cutaneous lupus erythematosus. Arthritis Res Ther. 2012;14:R265.
- Okon L, Rosenbach M, Krathen M, et al. Lenalidomide in treatment-refractory cutaneous lupus erythematosus: efficacy and safety in a 52-week trial. J Am Acad Dermatol. 2014;70:583-584.
- Fennira F, Chasset F, Soubrier M, et al. Lenalidomide for refractory chronic and subacute cutaneous lupus erythematosus: 16 patients. J Am Acad Dermatol. 2016;74:1248-1251.
- Furie R, Werth V, Gaudy A, et al. A randomized, placebo-controlled, double-blind, ascending-dose, safety, and pharmacokinetics study of CC-220 in subjects with systemic LUPUS erythematosus [abstract]. Arthritis Rheumatol. 2017;69(suppl 10). https://acrabstracts.org/abstract/a-randomized-placebo-controlled-double-blind-ascending-dose-safety-and-pharmacokinetics-study-of-cc-220-in-subjects-with-systemic-lupus-erythematosus/. Accessed May 7, 2020.
- Braunstein I, Klein R, Okawa J, et al. The interferon-regulated gene signature is elevated in subacute cutaneous lupus erythematosus and discoid lupus erythematosus and correlates with the cutaneous lupus area and severity index score. Br J Dermatol. 2012;166:971-975.
- Kim JM, Park SH, Kim HY, et al. A plasmacytoid dendritic cells-type I interferon axis is critically implicated in the pathogenesis of systemic lupus erythematosus. Int J Mol Sci. 2015;16:14158-14170.
- Furie R, Werth VP, Merola JF, et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J Clin Invest. 2019;129:1359-1371.
- Werth V, Musselli C, Furie R, et al. BIIB059, a humanized monoclonal antibody targeting BDCA2 on plasmacytoid dendritic cells (pDC), shows dose-related efficacy in the phase 2 LILAC study in patients (pts) with active cutaneous lupus erythematosus (CLE). Ann Rheum Dis. In press.
- Furie R, Morand EF, Bruce I, et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): a randomised, controlled, phase 3 trial. Lancet Rheumatol. 2019;1:E208-E219.
- Morand EF, Furie R, Tanaka Y, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. 2020;382:211-221.
A toddler with a fever and desquamating perineal rash
Kawasaki disease
Given (KD). An echocardiogram revealed diffuse dilation of the left anterior descending artery without evidence of an aneurysm. The patient was promptly started on 2 g/kg IVIG and high-dose aspirin. She was later transitioned to low-dose aspirin. Long-term follow-up thus far has revealed no cardiac sequelae.
KD, or mucocutaneous lymph node syndrome, is a multisystem vasculitis with predilection for the coronary arteries that most commonly affects children between 6 months and 5 years of age.1 While the etiology remains unclear, the pathogenesis is thought to be the result of an immune response to an infection in the setting of genetic susceptibility.1 Approximately 90% of patients have mucocutaneous manifestations, highlighting the important role dermatologists play in the diagnosis and early intervention to prevent cardiovascular morbidity.
The diagnostic criteria include fever for at least 5 days accompanied by at least four of the following:
- Bilateral bulbar conjunctival injection without exudate that is classically limbal sparing.
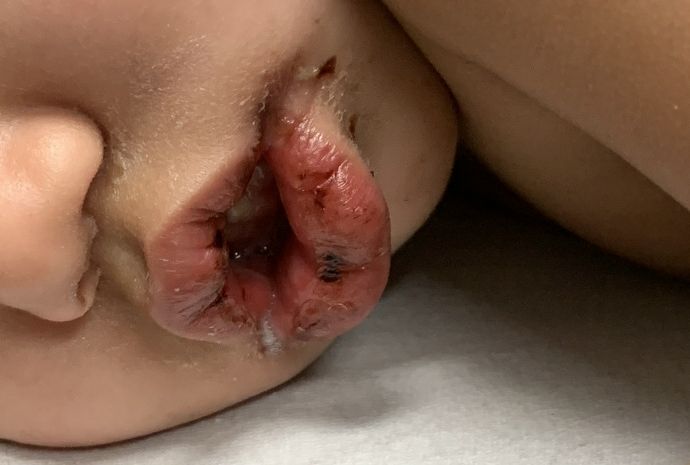
- Oral mucosal changes with cracked fissured lips, “strawberry tongue,” or erythema of the lips and mucosa.
- Changes in the extremities: erythema, swelling, or periungual peeling.
- Polymorphous exanthem.
- Cervical lymphadenopathy, often unilateral (greater than 1.5 cm).
Although nonspecific for diagnosis, laboratory abnormalities are common, including anemia, thrombocytosis, leukocytosis, elevated inflammatory markers, elevated alanine aminotransferase (ALT), hypoalbuminemia, and sterile pyuria on urine analysis.1
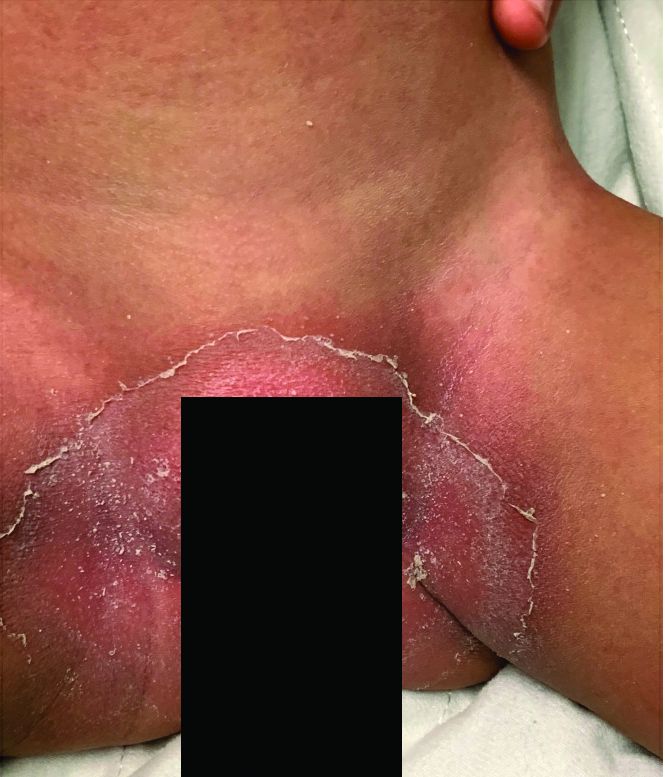
Notably, a classic finding of KD is perineal dermatitis with desquamation occurring in the acute phase of disease in 80%-90% of patients.2-5 In a retrospective review, up to 67% of patients with KD developed a perineal rash in the first week, most often beginning in the diaper area.2 The perineal rash classically desquamates early during the acute phase of the disease.1
While most individuals with KD follow a benign disease course, it is the most common cause of acquired heart disease in the United States.1 Treatment is aimed at decreasing the risk of developing coronary abnormalities through the prompt administration of IVIG and high-dose aspirin initiated early in the acute phase.6 A second dose of IVIG may be given to patients who remain febrile within 24-48 hours after treatment.6 Infliximab has been used safely and effectively in patients with refractory KD.7 Long-term cardiac follow-up of KD patients is recommended.
Recently, there has been an emerging association between COVID-19 and pediatric multi-system inflammatory syndrome, which shares features with KD. Patients with pediatric multi-system inflammatory syndrome who meet clinical criteria for KD should be promptly treated with IVIG and aspirin to avoid long-term cardiac sequelae.
This case and the photos were submitted by Dr. Elizabeth H. Cusick and Dr. Molly E. Plovanich, both with the department of dermatology at the University of Rochester (N.Y.). Dr. Donna Bilu Martin edited the case.

Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. Bayers S et al. (2013). J Am Acad Dermatol. 2013 Oct;69(4):501.e1-11.
2. Friter BS and Lucky AW. Arch Dermatol. 1988 Dec;124(12):1805-10.
3. Urbach AH et al. Am J Dis Child. 1988 Nov;142(11):1174-6.
4. Fink CW. Pediatr Infect Dis. 1983 Mar-Apr; 2(2):140-1.
5. Aballi A J and Bisken LC. Pediatr Infect Dis. 1984 Mar-Apr;3(2):187.
6. McCrindle BW et al. Circulation. 2017 Apr 25;135(17):e927-e99.
7.Sauvaget E et al. J Pediatr. 2012 May; 160(5),875-6.
Kawasaki disease
Given (KD). An echocardiogram revealed diffuse dilation of the left anterior descending artery without evidence of an aneurysm. The patient was promptly started on 2 g/kg IVIG and high-dose aspirin. She was later transitioned to low-dose aspirin. Long-term follow-up thus far has revealed no cardiac sequelae.
KD, or mucocutaneous lymph node syndrome, is a multisystem vasculitis with predilection for the coronary arteries that most commonly affects children between 6 months and 5 years of age.1 While the etiology remains unclear, the pathogenesis is thought to be the result of an immune response to an infection in the setting of genetic susceptibility.1 Approximately 90% of patients have mucocutaneous manifestations, highlighting the important role dermatologists play in the diagnosis and early intervention to prevent cardiovascular morbidity.
The diagnostic criteria include fever for at least 5 days accompanied by at least four of the following:
- Bilateral bulbar conjunctival injection without exudate that is classically limbal sparing.
- Oral mucosal changes with cracked fissured lips, “strawberry tongue,” or erythema of the lips and mucosa.
- Changes in the extremities: erythema, swelling, or periungual peeling.
- Polymorphous exanthem.
- Cervical lymphadenopathy, often unilateral (greater than 1.5 cm).
Although nonspecific for diagnosis, laboratory abnormalities are common, including anemia, thrombocytosis, leukocytosis, elevated inflammatory markers, elevated alanine aminotransferase (ALT), hypoalbuminemia, and sterile pyuria on urine analysis.1
Notably, a classic finding of KD is perineal dermatitis with desquamation occurring in the acute phase of disease in 80%-90% of patients.2-5 In a retrospective review, up to 67% of patients with KD developed a perineal rash in the first week, most often beginning in the diaper area.2 The perineal rash classically desquamates early during the acute phase of the disease.1
While most individuals with KD follow a benign disease course, it is the most common cause of acquired heart disease in the United States.1 Treatment is aimed at decreasing the risk of developing coronary abnormalities through the prompt administration of IVIG and high-dose aspirin initiated early in the acute phase.6 A second dose of IVIG may be given to patients who remain febrile within 24-48 hours after treatment.6 Infliximab has been used safely and effectively in patients with refractory KD.7 Long-term cardiac follow-up of KD patients is recommended.
Recently, there has been an emerging association between COVID-19 and pediatric multi-system inflammatory syndrome, which shares features with KD. Patients with pediatric multi-system inflammatory syndrome who meet clinical criteria for KD should be promptly treated with IVIG and aspirin to avoid long-term cardiac sequelae.
This case and the photos were submitted by Dr. Elizabeth H. Cusick and Dr. Molly E. Plovanich, both with the department of dermatology at the University of Rochester (N.Y.). Dr. Donna Bilu Martin edited the case.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. Bayers S et al. (2013). J Am Acad Dermatol. 2013 Oct;69(4):501.e1-11.
2. Friter BS and Lucky AW. Arch Dermatol. 1988 Dec;124(12):1805-10.
3. Urbach AH et al. Am J Dis Child. 1988 Nov;142(11):1174-6.
4. Fink CW. Pediatr Infect Dis. 1983 Mar-Apr; 2(2):140-1.
5. Aballi A J and Bisken LC. Pediatr Infect Dis. 1984 Mar-Apr;3(2):187.
6. McCrindle BW et al. Circulation. 2017 Apr 25;135(17):e927-e99.
7.Sauvaget E et al. J Pediatr. 2012 May; 160(5),875-6.
Kawasaki disease
Given (KD). An echocardiogram revealed diffuse dilation of the left anterior descending artery without evidence of an aneurysm. The patient was promptly started on 2 g/kg IVIG and high-dose aspirin. She was later transitioned to low-dose aspirin. Long-term follow-up thus far has revealed no cardiac sequelae.
KD, or mucocutaneous lymph node syndrome, is a multisystem vasculitis with predilection for the coronary arteries that most commonly affects children between 6 months and 5 years of age.1 While the etiology remains unclear, the pathogenesis is thought to be the result of an immune response to an infection in the setting of genetic susceptibility.1 Approximately 90% of patients have mucocutaneous manifestations, highlighting the important role dermatologists play in the diagnosis and early intervention to prevent cardiovascular morbidity.
The diagnostic criteria include fever for at least 5 days accompanied by at least four of the following:
- Bilateral bulbar conjunctival injection without exudate that is classically limbal sparing.
- Oral mucosal changes with cracked fissured lips, “strawberry tongue,” or erythema of the lips and mucosa.
- Changes in the extremities: erythema, swelling, or periungual peeling.
- Polymorphous exanthem.
- Cervical lymphadenopathy, often unilateral (greater than 1.5 cm).
Although nonspecific for diagnosis, laboratory abnormalities are common, including anemia, thrombocytosis, leukocytosis, elevated inflammatory markers, elevated alanine aminotransferase (ALT), hypoalbuminemia, and sterile pyuria on urine analysis.1
Notably, a classic finding of KD is perineal dermatitis with desquamation occurring in the acute phase of disease in 80%-90% of patients.2-5 In a retrospective review, up to 67% of patients with KD developed a perineal rash in the first week, most often beginning in the diaper area.2 The perineal rash classically desquamates early during the acute phase of the disease.1
While most individuals with KD follow a benign disease course, it is the most common cause of acquired heart disease in the United States.1 Treatment is aimed at decreasing the risk of developing coronary abnormalities through the prompt administration of IVIG and high-dose aspirin initiated early in the acute phase.6 A second dose of IVIG may be given to patients who remain febrile within 24-48 hours after treatment.6 Infliximab has been used safely and effectively in patients with refractory KD.7 Long-term cardiac follow-up of KD patients is recommended.
Recently, there has been an emerging association between COVID-19 and pediatric multi-system inflammatory syndrome, which shares features with KD. Patients with pediatric multi-system inflammatory syndrome who meet clinical criteria for KD should be promptly treated with IVIG and aspirin to avoid long-term cardiac sequelae.
This case and the photos were submitted by Dr. Elizabeth H. Cusick and Dr. Molly E. Plovanich, both with the department of dermatology at the University of Rochester (N.Y.). Dr. Donna Bilu Martin edited the case.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
1. Bayers S et al. (2013). J Am Acad Dermatol. 2013 Oct;69(4):501.e1-11.
2. Friter BS and Lucky AW. Arch Dermatol. 1988 Dec;124(12):1805-10.
3. Urbach AH et al. Am J Dis Child. 1988 Nov;142(11):1174-6.
4. Fink CW. Pediatr Infect Dis. 1983 Mar-Apr; 2(2):140-1.
5. Aballi A J and Bisken LC. Pediatr Infect Dis. 1984 Mar-Apr;3(2):187.
6. McCrindle BW et al. Circulation. 2017 Apr 25;135(17):e927-e99.
7.Sauvaget E et al. J Pediatr. 2012 May; 160(5),875-6.
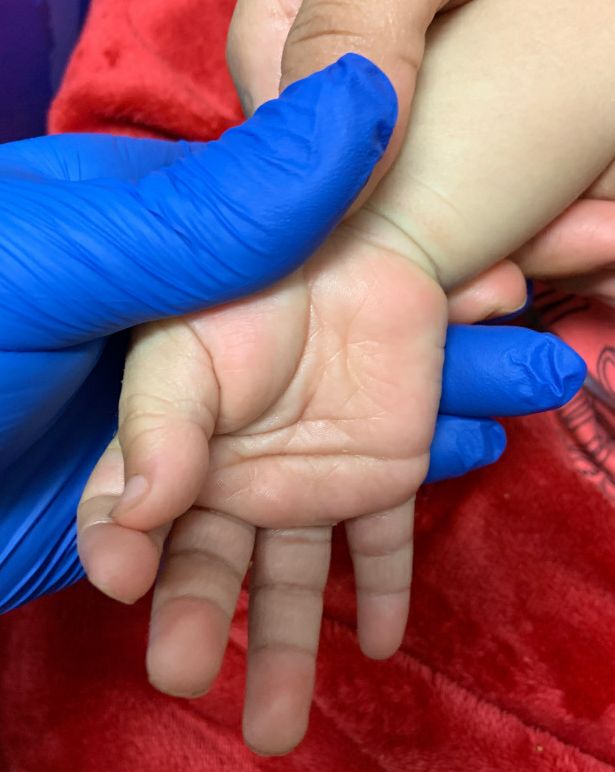
An otherwise healthy 18-month-old female presented to the emergency department with 5 days of fever, erythema, fissuring of the lips, conjunctival injection, and a desquamating perineal rash. In addition, she had nasal congestion and cough for which she was started on amoxicillin 2 days prior to presentation given concern for pneumonia.
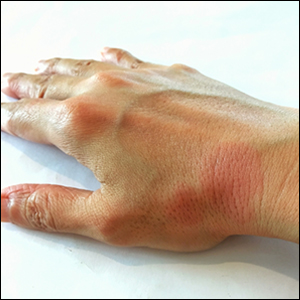
On exam, she was also noted to have several palpable cervical lymph nodes and edematous hands with overlying erythema. Laboratory evaluation was notable for respiratory syncytial virus positivity by polymerase chain reaction assay, leukocytosis, and elevated inflammatory markers (erythrocyte sedimentation rate and C-reactive protein).
Anti-Ro52 autoantibodies signal interstitial lung disease in juvenile dermatomyositis teaser
MAUI, HAWAII – , Anne M. Stevens, MD, PhD, said at the 2020 Rheumatology Winter Clinical Symposium.
And in a recent potential treatment advance, Janus kinase inhibition shows promise as a novel therapy for ILD in patients with juvenile dermatomyositis (JDM), added Dr. Stevens, a pediatric rheumatologist at the University of Washington, Seattle, and senior director for the adaptive immunity research program at Janssen Pharmaceuticals.
Autoantibodies predict ILD in JDM
Dr. Stevens highlighted recent work by Sara Sabbagh, DO, of the National Institute of Arthritis and Musculoskeletal and Skin Diseases and coinvestigators in the Childhood Myositis Heterogeneity Collaborative Study Group. They reported the presence of anti-Ro52 autoantibodies in 14% of a cohort of 302 patients with JDM as well as in 3 (12%) of 25 patients with juvenile polymyositis and in 8 (18%) of 44 youths with juvenile connective tissue disease–myositis overlap. In addition, 13% of patients were positive for autoantibodies previously identified as being associated with ILD in these forms of juvenile myositis: namely, 9% of the cohort were positive for anti–melanoma differentiation–associated protein 5 (anti-MDA5) autoantibodies, and antiaminoacyl-tRNA synthetase (anti-Jo-1) autoantibodies were present in 4%.
Thirty-three of the 371 juvenile myositis patients had ILD based upon CT imaging, chest x-ray, dyspnea on exertion, and/or biopsy. Most patients with anti-Ro52 also had other autoantibodies associated with ILD. Indeed, 31% of patients with anti-MDA5 autoantibodies also had anti-Ro52, as did 64% of those with anti-Jo1. After controlling for the presence of these other myositis-specific autoantibodies, anti-Ro52 autoantibodies were independently associated with ILD, which was present in 36% of those with and just 4% of those without anti-Ro52 autoantibodies.
Importantly, if a patient with JDM or another form of juvenile myositis had both anti-Ro52 and another myositis-specific autoantibody, the risk for ILD rose dramatically, climbing to 70% in patients with anti-Ro52 and anti-MDA5 autoantibodies, and to 100% in those who were both anti-Ro52 and anti-Jo1 positive (Ann Rheum Dis. 2019 Jul;78[7]:988-95).
Patients with anti-Ro autoantibodies had a worse prognosis, with more severe and chronic disease, Dr. Stevens noted.
Potential treatment for ILD in JDM: JAK inhibitors
Standard treatment of ILD in JDM in all cases includes high-dose pulsed corticosteroids, IVIG, and either methotrexate or mycophenolate mofetil. Consideration should be given to adding cyclosporine, particularly when a macrophage activation–syndrome component is present. In addition, several exciting recent lines of evidence suggest a potential role for Janus kinase (JAK) inhibitors in the subset of JDM patients with anti-MDA5 autoantibody–positive disease, according to Dr. Stevens.
For one, Dr. Sabbagh and colleagues have reported impressive success with the use of the JAK 1/3 inhibitor tofacitinib (Xeljanz) in two patients with anti-MDA5 autoantibody–positive refractory JDM with ILD. Both patients experienced moderate clinical improvement in disease activity in their skin, muscles, and other target organs. But particularly striking was what the investigators termed the “remarkable” improvement in ILD, including near resolution of abnormal findings on high-resolution CT imaging and a more robust performance on pulmonary function testing.
Both of these hitherto treatment-refractory patients were able to wean or discontinue their immunosuppressive medications. The patients’ elevated blood interferon-response gene signature improved significantly in response to tofacitinib, and their problematic upregulation of STAT1 phosphorylation of CD4+ T cells and monocytes stimulated with interferon-gamma was tamed, dropping to levels typically seen in healthy individuals (Brain. 2019 Nov 1;142[11]:e59).
Also, French pediatric rheumatologists have identified key phenotypic and cytokine differences between 13 patients with JDM or juvenile overlap myositis who were anti-MDA5 autoantibody–positive at presentation and 51 others who were not. The anti-MDA5 autoantibody–positive group had a higher frequency of ILD, arthritis, skin ulcerations, and lupus features, but milder muscle involvement than the anti-MDA5 autoantibody–negative group. The anti-MDA5 autoantibody–positive patients demonstrated enhanced interferon-alpha signaling based upon their significantly higher serum interferon-alpha levels, compared with the anti-MDA5-negative group, and those levels decreased following treatment with improvement in symptoms (Rheumatology [Oxford]. 2019 Nov 22. doi: 10.1093/rheumatology/kez525. [Epub ahead of print]).
The French investigators proposed that interferon-alpha may constitute a novel therapeutic target in the subgroup of patients with severe, refractory juvenile myositis and anti-MDA5 autoantibodies – and, as it happens, it’s known that JAK inhibitors modulate the interferon pathway.
Dr. Stevens reported research collaborations with Kineta and Seattle Genetics in addition to her employment at Janssen Pharmaceuticals.
MAUI, HAWAII – , Anne M. Stevens, MD, PhD, said at the 2020 Rheumatology Winter Clinical Symposium.
And in a recent potential treatment advance, Janus kinase inhibition shows promise as a novel therapy for ILD in patients with juvenile dermatomyositis (JDM), added Dr. Stevens, a pediatric rheumatologist at the University of Washington, Seattle, and senior director for the adaptive immunity research program at Janssen Pharmaceuticals.
Autoantibodies predict ILD in JDM
Dr. Stevens highlighted recent work by Sara Sabbagh, DO, of the National Institute of Arthritis and Musculoskeletal and Skin Diseases and coinvestigators in the Childhood Myositis Heterogeneity Collaborative Study Group. They reported the presence of anti-Ro52 autoantibodies in 14% of a cohort of 302 patients with JDM as well as in 3 (12%) of 25 patients with juvenile polymyositis and in 8 (18%) of 44 youths with juvenile connective tissue disease–myositis overlap. In addition, 13% of patients were positive for autoantibodies previously identified as being associated with ILD in these forms of juvenile myositis: namely, 9% of the cohort were positive for anti–melanoma differentiation–associated protein 5 (anti-MDA5) autoantibodies, and antiaminoacyl-tRNA synthetase (anti-Jo-1) autoantibodies were present in 4%.
Thirty-three of the 371 juvenile myositis patients had ILD based upon CT imaging, chest x-ray, dyspnea on exertion, and/or biopsy. Most patients with anti-Ro52 also had other autoantibodies associated with ILD. Indeed, 31% of patients with anti-MDA5 autoantibodies also had anti-Ro52, as did 64% of those with anti-Jo1. After controlling for the presence of these other myositis-specific autoantibodies, anti-Ro52 autoantibodies were independently associated with ILD, which was present in 36% of those with and just 4% of those without anti-Ro52 autoantibodies.
Importantly, if a patient with JDM or another form of juvenile myositis had both anti-Ro52 and another myositis-specific autoantibody, the risk for ILD rose dramatically, climbing to 70% in patients with anti-Ro52 and anti-MDA5 autoantibodies, and to 100% in those who were both anti-Ro52 and anti-Jo1 positive (Ann Rheum Dis. 2019 Jul;78[7]:988-95).
Patients with anti-Ro autoantibodies had a worse prognosis, with more severe and chronic disease, Dr. Stevens noted.
Potential treatment for ILD in JDM: JAK inhibitors
Standard treatment of ILD in JDM in all cases includes high-dose pulsed corticosteroids, IVIG, and either methotrexate or mycophenolate mofetil. Consideration should be given to adding cyclosporine, particularly when a macrophage activation–syndrome component is present. In addition, several exciting recent lines of evidence suggest a potential role for Janus kinase (JAK) inhibitors in the subset of JDM patients with anti-MDA5 autoantibody–positive disease, according to Dr. Stevens.
For one, Dr. Sabbagh and colleagues have reported impressive success with the use of the JAK 1/3 inhibitor tofacitinib (Xeljanz) in two patients with anti-MDA5 autoantibody–positive refractory JDM with ILD. Both patients experienced moderate clinical improvement in disease activity in their skin, muscles, and other target organs. But particularly striking was what the investigators termed the “remarkable” improvement in ILD, including near resolution of abnormal findings on high-resolution CT imaging and a more robust performance on pulmonary function testing.
Both of these hitherto treatment-refractory patients were able to wean or discontinue their immunosuppressive medications. The patients’ elevated blood interferon-response gene signature improved significantly in response to tofacitinib, and their problematic upregulation of STAT1 phosphorylation of CD4+ T cells and monocytes stimulated with interferon-gamma was tamed, dropping to levels typically seen in healthy individuals (Brain. 2019 Nov 1;142[11]:e59).
Also, French pediatric rheumatologists have identified key phenotypic and cytokine differences between 13 patients with JDM or juvenile overlap myositis who were anti-MDA5 autoantibody–positive at presentation and 51 others who were not. The anti-MDA5 autoantibody–positive group had a higher frequency of ILD, arthritis, skin ulcerations, and lupus features, but milder muscle involvement than the anti-MDA5 autoantibody–negative group. The anti-MDA5 autoantibody–positive patients demonstrated enhanced interferon-alpha signaling based upon their significantly higher serum interferon-alpha levels, compared with the anti-MDA5-negative group, and those levels decreased following treatment with improvement in symptoms (Rheumatology [Oxford]. 2019 Nov 22. doi: 10.1093/rheumatology/kez525. [Epub ahead of print]).
The French investigators proposed that interferon-alpha may constitute a novel therapeutic target in the subgroup of patients with severe, refractory juvenile myositis and anti-MDA5 autoantibodies – and, as it happens, it’s known that JAK inhibitors modulate the interferon pathway.
Dr. Stevens reported research collaborations with Kineta and Seattle Genetics in addition to her employment at Janssen Pharmaceuticals.
MAUI, HAWAII – , Anne M. Stevens, MD, PhD, said at the 2020 Rheumatology Winter Clinical Symposium.
And in a recent potential treatment advance, Janus kinase inhibition shows promise as a novel therapy for ILD in patients with juvenile dermatomyositis (JDM), added Dr. Stevens, a pediatric rheumatologist at the University of Washington, Seattle, and senior director for the adaptive immunity research program at Janssen Pharmaceuticals.
Autoantibodies predict ILD in JDM
Dr. Stevens highlighted recent work by Sara Sabbagh, DO, of the National Institute of Arthritis and Musculoskeletal and Skin Diseases and coinvestigators in the Childhood Myositis Heterogeneity Collaborative Study Group. They reported the presence of anti-Ro52 autoantibodies in 14% of a cohort of 302 patients with JDM as well as in 3 (12%) of 25 patients with juvenile polymyositis and in 8 (18%) of 44 youths with juvenile connective tissue disease–myositis overlap. In addition, 13% of patients were positive for autoantibodies previously identified as being associated with ILD in these forms of juvenile myositis: namely, 9% of the cohort were positive for anti–melanoma differentiation–associated protein 5 (anti-MDA5) autoantibodies, and antiaminoacyl-tRNA synthetase (anti-Jo-1) autoantibodies were present in 4%.
Thirty-three of the 371 juvenile myositis patients had ILD based upon CT imaging, chest x-ray, dyspnea on exertion, and/or biopsy. Most patients with anti-Ro52 also had other autoantibodies associated with ILD. Indeed, 31% of patients with anti-MDA5 autoantibodies also had anti-Ro52, as did 64% of those with anti-Jo1. After controlling for the presence of these other myositis-specific autoantibodies, anti-Ro52 autoantibodies were independently associated with ILD, which was present in 36% of those with and just 4% of those without anti-Ro52 autoantibodies.
Importantly, if a patient with JDM or another form of juvenile myositis had both anti-Ro52 and another myositis-specific autoantibody, the risk for ILD rose dramatically, climbing to 70% in patients with anti-Ro52 and anti-MDA5 autoantibodies, and to 100% in those who were both anti-Ro52 and anti-Jo1 positive (Ann Rheum Dis. 2019 Jul;78[7]:988-95).
Patients with anti-Ro autoantibodies had a worse prognosis, with more severe and chronic disease, Dr. Stevens noted.
Potential treatment for ILD in JDM: JAK inhibitors
Standard treatment of ILD in JDM in all cases includes high-dose pulsed corticosteroids, IVIG, and either methotrexate or mycophenolate mofetil. Consideration should be given to adding cyclosporine, particularly when a macrophage activation–syndrome component is present. In addition, several exciting recent lines of evidence suggest a potential role for Janus kinase (JAK) inhibitors in the subset of JDM patients with anti-MDA5 autoantibody–positive disease, according to Dr. Stevens.
For one, Dr. Sabbagh and colleagues have reported impressive success with the use of the JAK 1/3 inhibitor tofacitinib (Xeljanz) in two patients with anti-MDA5 autoantibody–positive refractory JDM with ILD. Both patients experienced moderate clinical improvement in disease activity in their skin, muscles, and other target organs. But particularly striking was what the investigators termed the “remarkable” improvement in ILD, including near resolution of abnormal findings on high-resolution CT imaging and a more robust performance on pulmonary function testing.
Both of these hitherto treatment-refractory patients were able to wean or discontinue their immunosuppressive medications. The patients’ elevated blood interferon-response gene signature improved significantly in response to tofacitinib, and their problematic upregulation of STAT1 phosphorylation of CD4+ T cells and monocytes stimulated with interferon-gamma was tamed, dropping to levels typically seen in healthy individuals (Brain. 2019 Nov 1;142[11]:e59).
Also, French pediatric rheumatologists have identified key phenotypic and cytokine differences between 13 patients with JDM or juvenile overlap myositis who were anti-MDA5 autoantibody–positive at presentation and 51 others who were not. The anti-MDA5 autoantibody–positive group had a higher frequency of ILD, arthritis, skin ulcerations, and lupus features, but milder muscle involvement than the anti-MDA5 autoantibody–negative group. The anti-MDA5 autoantibody–positive patients demonstrated enhanced interferon-alpha signaling based upon their significantly higher serum interferon-alpha levels, compared with the anti-MDA5-negative group, and those levels decreased following treatment with improvement in symptoms (Rheumatology [Oxford]. 2019 Nov 22. doi: 10.1093/rheumatology/kez525. [Epub ahead of print]).
The French investigators proposed that interferon-alpha may constitute a novel therapeutic target in the subgroup of patients with severe, refractory juvenile myositis and anti-MDA5 autoantibodies – and, as it happens, it’s known that JAK inhibitors modulate the interferon pathway.
Dr. Stevens reported research collaborations with Kineta and Seattle Genetics in addition to her employment at Janssen Pharmaceuticals.
REPORTING FROM RWCS 2020
Dermatomyositis without dermatitis correlates with autoantibodies
The prevalence of dermatomyositis without dermatitis among patients with biopsy-confirmed dermatomyositis was approximately 8% in a Japanese cohort study. “Dermatomyositis sine dermatitis does exist and is significantly associated with anti–nuclear matrix protein 2 [anti-NXP-2] autoantibodies,” the researchers reported in JAMA Neurology.
Few case reports of dermatomyositis sine dermatitis have been documented. To confirm the existence of the condition, study its prevalence, and characterize its serologic features, Michio Inoue, MD, PhD, of the National Center of Neurology and Psychiatry in Tokyo, and colleagues conducted a cohort study of patients seen at the center between January 2009 and August 2019.
Of more than 8,800 patients whose muscle biopsies were examined for diagnostic purposes, 199 were tested for dermatomyositis-specific autoantibodies. The investigators excluded patients who did not have myxovirus resistance protein A expression in myofibers on muscle biopsy. In all, 182 patients with dermatomyositis were enrolled in the study (51% women; median age at biopsy, 56 years). Fourteen patients without a skin rash at the time of muscle biopsy received a diagnosis of dermatomyositis sine dermatitis. Before the muscle biopsy, most patients without a rash had a diagnosis of polymyositis.
Association with anti-NXP-2 autoantibodies
Anti-NXP-2 autoantibodies were detected in 86% of the patients without a rash at the time of biopsy, compared with 28% of the patients with rashes. “No other clinical or pathological characteristics were associated with [dermatomyositis sine dermatitis] except increased probability of developing perifascicular atrophy (71% vs. 43%),” Dr. Inoue and colleagues said.
During a median follow-up of 34 months, patients with dermatomyositis sine dermatitis received oral prednisolone with or without additional immunotherapy, and two patients had subcutaneous edema. Calcification was not seen during follow-up. “One patient with ... anti-NXP-2 autoantibodies had severe interstitial lung disease and needed noninvasive positive-pressure ventilation support,” the researchers said.
Four of the 14 patients with dermatomyositis sine dermatitis “developed skin rashes after muscle biopsy,” the researchers noted. “Similarly, a patient with [dermatomyositis sine dermatitis] was reported to have developed a skin rash 2 years after muscle biopsy.”
Potential therapies for refractory dermatomyositis, such as Janus kinase inhibitors, may not be effective for other types of myositis, so identifying patients with dermatomyositis may be “more essential than ever,” the authors said.
Effects on organ systems vary
The study is the first to systematically examine dermatomyositis sine dermatitis, said David Fiorentino, MD, PhD, professor of dermatology and director of the multidisciplinary rheumatic skin disease clinic at the Stanford (Calif.) University.

On the one hand, the results are not surprising because dermatomyositis is a systemic autoimmune disease. “There are no rules about which organs it will or won’t affect in a given individual,” Dr. Fiorentino said in an interview.
At the same time, dermatomyositis’s historical association with rash persists even though there is “no biological reason why that would have to be the case.”
Some patients with dermatomyositis have skin-predominant disease without clinically significant muscle involvement. Lung-predominant disease also may exist, although it has not been carefully studied, he said.
The findings remind clinicians that they need to consider the diagnosis of dermatomyositis “even if they do not have the skin findings,” he said. Dr. Fiorentino cautioned against interpreting the results to mean that certain patients never have signs of cutaneous inflammation. In the study, about a one-third of patients without dermatitis at the time of biopsy developed a rash. In addition, clinicians often miss subtle disease under the fingernails or on the scalp, or mild rash on the elbows.
The cohort of patients who underwent muscle biopsy may not be representative of the spectrum of patients with dermatomyositis, and the findings need to be verified in other populations, Dr. Fiorentino said.
The study was supported by an intramural research grant of the National Center of Neurology and Psychiatry and a grant from the Japan Society for the Promotion of Science. Authors disclosed personal fees from pharmaceutical companies and government and corporate grants outside the submitted work. Dr. Fiorentino had no relevant disclosures.
SOURCE: Inoue M et al. JAMA Neurol. 2020 Apr 20. doi: 10.1001/jamaneurol.2020.0673.
The prevalence of dermatomyositis without dermatitis among patients with biopsy-confirmed dermatomyositis was approximately 8% in a Japanese cohort study. “Dermatomyositis sine dermatitis does exist and is significantly associated with anti–nuclear matrix protein 2 [anti-NXP-2] autoantibodies,” the researchers reported in JAMA Neurology.
Few case reports of dermatomyositis sine dermatitis have been documented. To confirm the existence of the condition, study its prevalence, and characterize its serologic features, Michio Inoue, MD, PhD, of the National Center of Neurology and Psychiatry in Tokyo, and colleagues conducted a cohort study of patients seen at the center between January 2009 and August 2019.
Of more than 8,800 patients whose muscle biopsies were examined for diagnostic purposes, 199 were tested for dermatomyositis-specific autoantibodies. The investigators excluded patients who did not have myxovirus resistance protein A expression in myofibers on muscle biopsy. In all, 182 patients with dermatomyositis were enrolled in the study (51% women; median age at biopsy, 56 years). Fourteen patients without a skin rash at the time of muscle biopsy received a diagnosis of dermatomyositis sine dermatitis. Before the muscle biopsy, most patients without a rash had a diagnosis of polymyositis.
Association with anti-NXP-2 autoantibodies
Anti-NXP-2 autoantibodies were detected in 86% of the patients without a rash at the time of biopsy, compared with 28% of the patients with rashes. “No other clinical or pathological characteristics were associated with [dermatomyositis sine dermatitis] except increased probability of developing perifascicular atrophy (71% vs. 43%),” Dr. Inoue and colleagues said.
During a median follow-up of 34 months, patients with dermatomyositis sine dermatitis received oral prednisolone with or without additional immunotherapy, and two patients had subcutaneous edema. Calcification was not seen during follow-up. “One patient with ... anti-NXP-2 autoantibodies had severe interstitial lung disease and needed noninvasive positive-pressure ventilation support,” the researchers said.
Four of the 14 patients with dermatomyositis sine dermatitis “developed skin rashes after muscle biopsy,” the researchers noted. “Similarly, a patient with [dermatomyositis sine dermatitis] was reported to have developed a skin rash 2 years after muscle biopsy.”
Potential therapies for refractory dermatomyositis, such as Janus kinase inhibitors, may not be effective for other types of myositis, so identifying patients with dermatomyositis may be “more essential than ever,” the authors said.
Effects on organ systems vary
The study is the first to systematically examine dermatomyositis sine dermatitis, said David Fiorentino, MD, PhD, professor of dermatology and director of the multidisciplinary rheumatic skin disease clinic at the Stanford (Calif.) University.
On the one hand, the results are not surprising because dermatomyositis is a systemic autoimmune disease. “There are no rules about which organs it will or won’t affect in a given individual,” Dr. Fiorentino said in an interview.
At the same time, dermatomyositis’s historical association with rash persists even though there is “no biological reason why that would have to be the case.”
Some patients with dermatomyositis have skin-predominant disease without clinically significant muscle involvement. Lung-predominant disease also may exist, although it has not been carefully studied, he said.
The findings remind clinicians that they need to consider the diagnosis of dermatomyositis “even if they do not have the skin findings,” he said. Dr. Fiorentino cautioned against interpreting the results to mean that certain patients never have signs of cutaneous inflammation. In the study, about a one-third of patients without dermatitis at the time of biopsy developed a rash. In addition, clinicians often miss subtle disease under the fingernails or on the scalp, or mild rash on the elbows.
The cohort of patients who underwent muscle biopsy may not be representative of the spectrum of patients with dermatomyositis, and the findings need to be verified in other populations, Dr. Fiorentino said.
The study was supported by an intramural research grant of the National Center of Neurology and Psychiatry and a grant from the Japan Society for the Promotion of Science. Authors disclosed personal fees from pharmaceutical companies and government and corporate grants outside the submitted work. Dr. Fiorentino had no relevant disclosures.
SOURCE: Inoue M et al. JAMA Neurol. 2020 Apr 20. doi: 10.1001/jamaneurol.2020.0673.
The prevalence of dermatomyositis without dermatitis among patients with biopsy-confirmed dermatomyositis was approximately 8% in a Japanese cohort study. “Dermatomyositis sine dermatitis does exist and is significantly associated with anti–nuclear matrix protein 2 [anti-NXP-2] autoantibodies,” the researchers reported in JAMA Neurology.
Few case reports of dermatomyositis sine dermatitis have been documented. To confirm the existence of the condition, study its prevalence, and characterize its serologic features, Michio Inoue, MD, PhD, of the National Center of Neurology and Psychiatry in Tokyo, and colleagues conducted a cohort study of patients seen at the center between January 2009 and August 2019.
Of more than 8,800 patients whose muscle biopsies were examined for diagnostic purposes, 199 were tested for dermatomyositis-specific autoantibodies. The investigators excluded patients who did not have myxovirus resistance protein A expression in myofibers on muscle biopsy. In all, 182 patients with dermatomyositis were enrolled in the study (51% women; median age at biopsy, 56 years). Fourteen patients without a skin rash at the time of muscle biopsy received a diagnosis of dermatomyositis sine dermatitis. Before the muscle biopsy, most patients without a rash had a diagnosis of polymyositis.
Association with anti-NXP-2 autoantibodies
Anti-NXP-2 autoantibodies were detected in 86% of the patients without a rash at the time of biopsy, compared with 28% of the patients with rashes. “No other clinical or pathological characteristics were associated with [dermatomyositis sine dermatitis] except increased probability of developing perifascicular atrophy (71% vs. 43%),” Dr. Inoue and colleagues said.
During a median follow-up of 34 months, patients with dermatomyositis sine dermatitis received oral prednisolone with or without additional immunotherapy, and two patients had subcutaneous edema. Calcification was not seen during follow-up. “One patient with ... anti-NXP-2 autoantibodies had severe interstitial lung disease and needed noninvasive positive-pressure ventilation support,” the researchers said.
Four of the 14 patients with dermatomyositis sine dermatitis “developed skin rashes after muscle biopsy,” the researchers noted. “Similarly, a patient with [dermatomyositis sine dermatitis] was reported to have developed a skin rash 2 years after muscle biopsy.”
Potential therapies for refractory dermatomyositis, such as Janus kinase inhibitors, may not be effective for other types of myositis, so identifying patients with dermatomyositis may be “more essential than ever,” the authors said.
Effects on organ systems vary
The study is the first to systematically examine dermatomyositis sine dermatitis, said David Fiorentino, MD, PhD, professor of dermatology and director of the multidisciplinary rheumatic skin disease clinic at the Stanford (Calif.) University.
On the one hand, the results are not surprising because dermatomyositis is a systemic autoimmune disease. “There are no rules about which organs it will or won’t affect in a given individual,” Dr. Fiorentino said in an interview.
At the same time, dermatomyositis’s historical association with rash persists even though there is “no biological reason why that would have to be the case.”
Some patients with dermatomyositis have skin-predominant disease without clinically significant muscle involvement. Lung-predominant disease also may exist, although it has not been carefully studied, he said.
The findings remind clinicians that they need to consider the diagnosis of dermatomyositis “even if they do not have the skin findings,” he said. Dr. Fiorentino cautioned against interpreting the results to mean that certain patients never have signs of cutaneous inflammation. In the study, about a one-third of patients without dermatitis at the time of biopsy developed a rash. In addition, clinicians often miss subtle disease under the fingernails or on the scalp, or mild rash on the elbows.
The cohort of patients who underwent muscle biopsy may not be representative of the spectrum of patients with dermatomyositis, and the findings need to be verified in other populations, Dr. Fiorentino said.
The study was supported by an intramural research grant of the National Center of Neurology and Psychiatry and a grant from the Japan Society for the Promotion of Science. Authors disclosed personal fees from pharmaceutical companies and government and corporate grants outside the submitted work. Dr. Fiorentino had no relevant disclosures.
SOURCE: Inoue M et al. JAMA Neurol. 2020 Apr 20. doi: 10.1001/jamaneurol.2020.0673.
FROM JAMA NEUROLOGY
Itchy, vesicular rash
Pemphigoid gestationis
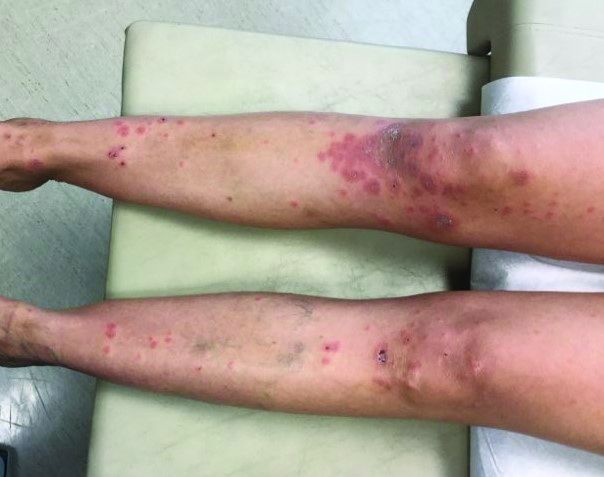
It typically presents with the abrupt onset of very pruritic urticarial plaques and papules, which start around the umbilicus and then spread to involve the trunk and extremities. The papules and plaques evolve to generalized tense blisters, which typically spare the face, palms, soles, and mucous membranes. Half of affected patients may present in an atypical distribution involving the extremities, palms, or soles. Patients may be at an increased risk for the development of Graves disease.
The cause of pemphigoid gestationis is a factor known as “herpes gestationis factor” that induces C3 deposition along the dermal-epidermal junction. As in bullous pemphigoid, patients with pemphigoid gestationis have antibodies to a transmembrane hemidesmosomal protein called BPAG2/BP180/collagen XVII.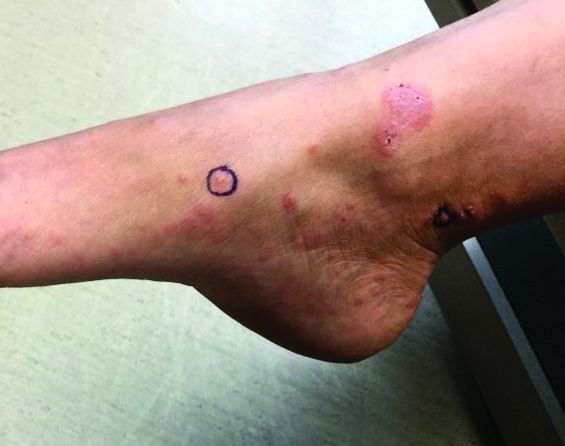
Three-quarters of patients worsen at the time of delivery and up to 10% of newborns will have bullous lesions secondary to placental transfer of antibodies. In most cases, lesions will spontaneously resolve over a few weeks following delivery. Recurrence with future pregnancies is common, with severity increasing with each pregnancy. Recurrence with menstruation and with the use of oral contraceptives can also occur. Although there is no increase in maternal mortality, onset in the first or second trimester and presence of blisters is associated with decreased gestational age of baby at delivery and lower-birth-weight infants. There is no increase in fetal mortality.
Histopathology reveals a subepidermal vesicle and perivascular infiltrate consisting of lymphocytes and eosinophils. Diagnosis can be confirmed with direct immunofluorescence showing C3 in a linear band along the basement membrane zone. IgG may be present as well. Complement added indirect immunofluorescence reveals circulating anti–basement zone IgG, which allows differentiation from pruritic urticarial papules and plaques of pregnancy.
Treatment for localized disease includes class I topical steroids and oral antihistamines. More severe cases require systemic corticosteroid treatment. Systemic steroids may cause lower-birth-weight infants.
This case and the photos were submitted by Dr. Hanson of Associated Skin Care Specialists in Eden Prairie, Minn. The case was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Pemphigoid gestationis
It typically presents with the abrupt onset of very pruritic urticarial plaques and papules, which start around the umbilicus and then spread to involve the trunk and extremities. The papules and plaques evolve to generalized tense blisters, which typically spare the face, palms, soles, and mucous membranes. Half of affected patients may present in an atypical distribution involving the extremities, palms, or soles. Patients may be at an increased risk for the development of Graves disease.
The cause of pemphigoid gestationis is a factor known as “herpes gestationis factor” that induces C3 deposition along the dermal-epidermal junction. As in bullous pemphigoid, patients with pemphigoid gestationis have antibodies to a transmembrane hemidesmosomal protein called BPAG2/BP180/collagen XVII.
Three-quarters of patients worsen at the time of delivery and up to 10% of newborns will have bullous lesions secondary to placental transfer of antibodies. In most cases, lesions will spontaneously resolve over a few weeks following delivery. Recurrence with future pregnancies is common, with severity increasing with each pregnancy. Recurrence with menstruation and with the use of oral contraceptives can also occur. Although there is no increase in maternal mortality, onset in the first or second trimester and presence of blisters is associated with decreased gestational age of baby at delivery and lower-birth-weight infants. There is no increase in fetal mortality.
Histopathology reveals a subepidermal vesicle and perivascular infiltrate consisting of lymphocytes and eosinophils. Diagnosis can be confirmed with direct immunofluorescence showing C3 in a linear band along the basement membrane zone. IgG may be present as well. Complement added indirect immunofluorescence reveals circulating anti–basement zone IgG, which allows differentiation from pruritic urticarial papules and plaques of pregnancy.
Treatment for localized disease includes class I topical steroids and oral antihistamines. More severe cases require systemic corticosteroid treatment. Systemic steroids may cause lower-birth-weight infants.
This case and the photos were submitted by Dr. Hanson of Associated Skin Care Specialists in Eden Prairie, Minn. The case was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Pemphigoid gestationis
It typically presents with the abrupt onset of very pruritic urticarial plaques and papules, which start around the umbilicus and then spread to involve the trunk and extremities. The papules and plaques evolve to generalized tense blisters, which typically spare the face, palms, soles, and mucous membranes. Half of affected patients may present in an atypical distribution involving the extremities, palms, or soles. Patients may be at an increased risk for the development of Graves disease.
The cause of pemphigoid gestationis is a factor known as “herpes gestationis factor” that induces C3 deposition along the dermal-epidermal junction. As in bullous pemphigoid, patients with pemphigoid gestationis have antibodies to a transmembrane hemidesmosomal protein called BPAG2/BP180/collagen XVII.
Three-quarters of patients worsen at the time of delivery and up to 10% of newborns will have bullous lesions secondary to placental transfer of antibodies. In most cases, lesions will spontaneously resolve over a few weeks following delivery. Recurrence with future pregnancies is common, with severity increasing with each pregnancy. Recurrence with menstruation and with the use of oral contraceptives can also occur. Although there is no increase in maternal mortality, onset in the first or second trimester and presence of blisters is associated with decreased gestational age of baby at delivery and lower-birth-weight infants. There is no increase in fetal mortality.
Histopathology reveals a subepidermal vesicle and perivascular infiltrate consisting of lymphocytes and eosinophils. Diagnosis can be confirmed with direct immunofluorescence showing C3 in a linear band along the basement membrane zone. IgG may be present as well. Complement added indirect immunofluorescence reveals circulating anti–basement zone IgG, which allows differentiation from pruritic urticarial papules and plaques of pregnancy.
Treatment for localized disease includes class I topical steroids and oral antihistamines. More severe cases require systemic corticosteroid treatment. Systemic steroids may cause lower-birth-weight infants.
This case and the photos were submitted by Dr. Hanson of Associated Skin Care Specialists in Eden Prairie, Minn. The case was edited by Donna Bilu Martin, MD.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
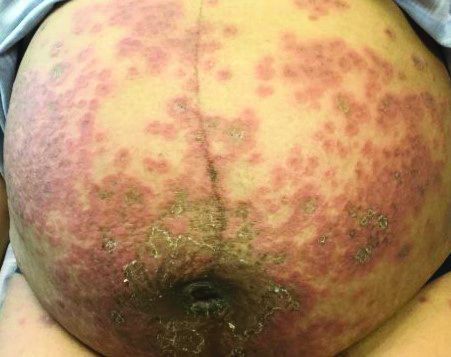
U.S. prevalence of antinuclear antibodies has steadily risen, study finds
Between 1988 and 2012, the prevalence of antinuclear antibodies in the United States increased from 11% to 15.9%, especially among adolescents, males, and non-Hispanic whites.
The finding comes from a retrospective, cross-sectional analysis of serum samples from individuals who participated in the U.S. National Health and Nutrition Examination Survey over three time periods: 1988-1991, 1999-2004, and 2011-2012.
“Autoimmune diseases are a diverse group of disorders characterized by damaging immune responses to self-antigens and, for the most part, are of unknown etiology,” authors led by Gregg E. Dinse, ScD, wrote in a study published in Arthritis & Rheumatology. “They are thought to impact 3%-5% of the population, with rising rates noted several decades ago. Recent studies suggest continued increases for certain autoimmune diseases, but it is unclear whether these trends are due to changes in recognition and diagnosis, or are true temporal changes in incidence.”
Dr. Dinse, of the National Institute of Environmental Health Sciences in Research Triangle Park, N.C., and his colleagues evaluated sera samples of 14,211 survey participants aged 12 years and older at 1:80 dilution for antinuclear antibodies (ANA) using a standard indirect immunofluorescence assay (HEp-2 assay). The samples that received a grade of 3 or 4 on a 0-4 scale (compared with standard references, with values of 1-4 indicating positivity) underwent additional assessment by sequential ANA titers up to 1:1,280 dilution. To estimate changes in ANA prevalence over the time periods, they used logistic regression adjusted for age, sex, race/ethnicity, and survey design variables.
The researchers observed an ANA prevalence of 11% in 1988-1991, 11.5% in 1999-2004, and 15.9% in 2011-2012. This corresponds to 22, 27, and 41 million affected individuals, respectively. Females were more likely than males to have ANA (odds ratios of 2.53, 2.97, and 1.94 in 1988-1991, 1999-2004, and 2011-2012, respectively; P less than .0001), as were older adults relative to adolescents (ORs of 3.63, 1.80, and 1.71; P less than .002). Among adolescents, the prevalence of ANA rose steeply, with odds ratios of 2.02 in 1999-2004 and 2.88 in 2011-2012 in the second and third time periods relative to the first (trend P less than .0001). The researchers also found that, compared with non-Hispanic whites, the odds of having ANA were higher for non-Hispanic blacks (OR, 1.75) and Mexican-Americans (OR, 1.87) in 1988-1991, but racial/ethnic differences diminished in 1999-2004 and 2011-2012.
After adjustment for covariates, the researchers found that the estimated odds ratios for the second and third time periods relative to the first were 1.02 and 1.47, respectively, reflecting an overall ANA time trend (P less than .0001). Increases in ANA prevalence among cohorts did not correlate with contemporaneous trends in body mass index, smoking, or alcohol consumption.
Dr. Dinse and his colleagues acknowledged certain limitations of the study, including the fact that associations were based on cross-sectional data rather than repeated measures, and that some variables were self-reported, including the limited questionnaire data on autoimmune diseases.

In an interview, David S. Pisetsky, MD, professor of medicine/rheumatology and immunology at Duke University, Durham, N.C., characterized the study findings as “hypothesis generating” and said that he would like to know if the researchers would find the same results if they used a different ANA assay. “There’s a lot of variability from ANA kit to ANA kit – much greater than what was thought,” said Dr. Pisetsky, who is an authority on the topic. “One thing that needs to be done is to find out what the frequency is with other tests. One should recognize that the actual frequency is going to vary by the assay used. In another test format, the frequency may have been lower; it could have been higher.”
He added that the precise reasons why the prevalence of ANAs are rising in the general population remains elusive. “We know the target antigens in people with autoantibody-associated rheumatic disease,” Dr. Pisetsky said. “But what we don’t know a lot of times is, what are the target antigens in the otherwise healthy population? There has only been one antibody system that people have felt is associated with the otherwise healthy population. Those are called anti-DFS-70 antibodies, but there is even uncertainty about those. If you know what the antigens recognized were, then I think you could begin to speculate more about what’s going on in the population that’s increasing the frequency [of ANAs].”
In an accompanying editorial, Richard J. Bucala, MD, chief of rheumatology, allergy, and immunology at Yale University, New Haven, Conn., noted that the origins of autoantibodies in different rheumatic diseases and the steps that lead to disease progression remain elusive. “Modern societies experience an ever increasing variety of exposures due to travel and population migration, an increase in both the internationalization of agriculture and the industrialization of food production, a higher environmental burden of synthetic chemicals, emerging pathogens, and the inexorable effects of climate change,” Dr. Bucala wrote. “The speed and intensity of these influences is arguably unprecedented in human history and clearly outpace the possibility of protective genetic mechanisms to evolve and adapt.” He went on to note that the study’s findings “give impetus to multidisciplinary efforts aimed at preventative strategies, identifying environmental hazards, defining high-risk individuals, and preventing disease development in susceptible populations.”
The study was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences. The authors reported having no disclosures.
SOURCE: Dinse G et al. Arthritis Rheumatol. 2020 April 7. doi: 10.1002/ART.41214.
Between 1988 and 2012, the prevalence of antinuclear antibodies in the United States increased from 11% to 15.9%, especially among adolescents, males, and non-Hispanic whites.
The finding comes from a retrospective, cross-sectional analysis of serum samples from individuals who participated in the U.S. National Health and Nutrition Examination Survey over three time periods: 1988-1991, 1999-2004, and 2011-2012.
“Autoimmune diseases are a diverse group of disorders characterized by damaging immune responses to self-antigens and, for the most part, are of unknown etiology,” authors led by Gregg E. Dinse, ScD, wrote in a study published in Arthritis & Rheumatology. “They are thought to impact 3%-5% of the population, with rising rates noted several decades ago. Recent studies suggest continued increases for certain autoimmune diseases, but it is unclear whether these trends are due to changes in recognition and diagnosis, or are true temporal changes in incidence.”
Dr. Dinse, of the National Institute of Environmental Health Sciences in Research Triangle Park, N.C., and his colleagues evaluated sera samples of 14,211 survey participants aged 12 years and older at 1:80 dilution for antinuclear antibodies (ANA) using a standard indirect immunofluorescence assay (HEp-2 assay). The samples that received a grade of 3 or 4 on a 0-4 scale (compared with standard references, with values of 1-4 indicating positivity) underwent additional assessment by sequential ANA titers up to 1:1,280 dilution. To estimate changes in ANA prevalence over the time periods, they used logistic regression adjusted for age, sex, race/ethnicity, and survey design variables.
The researchers observed an ANA prevalence of 11% in 1988-1991, 11.5% in 1999-2004, and 15.9% in 2011-2012. This corresponds to 22, 27, and 41 million affected individuals, respectively. Females were more likely than males to have ANA (odds ratios of 2.53, 2.97, and 1.94 in 1988-1991, 1999-2004, and 2011-2012, respectively; P less than .0001), as were older adults relative to adolescents (ORs of 3.63, 1.80, and 1.71; P less than .002). Among adolescents, the prevalence of ANA rose steeply, with odds ratios of 2.02 in 1999-2004 and 2.88 in 2011-2012 in the second and third time periods relative to the first (trend P less than .0001). The researchers also found that, compared with non-Hispanic whites, the odds of having ANA were higher for non-Hispanic blacks (OR, 1.75) and Mexican-Americans (OR, 1.87) in 1988-1991, but racial/ethnic differences diminished in 1999-2004 and 2011-2012.
After adjustment for covariates, the researchers found that the estimated odds ratios for the second and third time periods relative to the first were 1.02 and 1.47, respectively, reflecting an overall ANA time trend (P less than .0001). Increases in ANA prevalence among cohorts did not correlate with contemporaneous trends in body mass index, smoking, or alcohol consumption.
Dr. Dinse and his colleagues acknowledged certain limitations of the study, including the fact that associations were based on cross-sectional data rather than repeated measures, and that some variables were self-reported, including the limited questionnaire data on autoimmune diseases.
In an interview, David S. Pisetsky, MD, professor of medicine/rheumatology and immunology at Duke University, Durham, N.C., characterized the study findings as “hypothesis generating” and said that he would like to know if the researchers would find the same results if they used a different ANA assay. “There’s a lot of variability from ANA kit to ANA kit – much greater than what was thought,” said Dr. Pisetsky, who is an authority on the topic. “One thing that needs to be done is to find out what the frequency is with other tests. One should recognize that the actual frequency is going to vary by the assay used. In another test format, the frequency may have been lower; it could have been higher.”
He added that the precise reasons why the prevalence of ANAs are rising in the general population remains elusive. “We know the target antigens in people with autoantibody-associated rheumatic disease,” Dr. Pisetsky said. “But what we don’t know a lot of times is, what are the target antigens in the otherwise healthy population? There has only been one antibody system that people have felt is associated with the otherwise healthy population. Those are called anti-DFS-70 antibodies, but there is even uncertainty about those. If you know what the antigens recognized were, then I think you could begin to speculate more about what’s going on in the population that’s increasing the frequency [of ANAs].”
In an accompanying editorial, Richard J. Bucala, MD, chief of rheumatology, allergy, and immunology at Yale University, New Haven, Conn., noted that the origins of autoantibodies in different rheumatic diseases and the steps that lead to disease progression remain elusive. “Modern societies experience an ever increasing variety of exposures due to travel and population migration, an increase in both the internationalization of agriculture and the industrialization of food production, a higher environmental burden of synthetic chemicals, emerging pathogens, and the inexorable effects of climate change,” Dr. Bucala wrote. “The speed and intensity of these influences is arguably unprecedented in human history and clearly outpace the possibility of protective genetic mechanisms to evolve and adapt.” He went on to note that the study’s findings “give impetus to multidisciplinary efforts aimed at preventative strategies, identifying environmental hazards, defining high-risk individuals, and preventing disease development in susceptible populations.”
The study was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences. The authors reported having no disclosures.
SOURCE: Dinse G et al. Arthritis Rheumatol. 2020 April 7. doi: 10.1002/ART.41214.
Between 1988 and 2012, the prevalence of antinuclear antibodies in the United States increased from 11% to 15.9%, especially among adolescents, males, and non-Hispanic whites.
The finding comes from a retrospective, cross-sectional analysis of serum samples from individuals who participated in the U.S. National Health and Nutrition Examination Survey over three time periods: 1988-1991, 1999-2004, and 2011-2012.
“Autoimmune diseases are a diverse group of disorders characterized by damaging immune responses to self-antigens and, for the most part, are of unknown etiology,” authors led by Gregg E. Dinse, ScD, wrote in a study published in Arthritis & Rheumatology. “They are thought to impact 3%-5% of the population, with rising rates noted several decades ago. Recent studies suggest continued increases for certain autoimmune diseases, but it is unclear whether these trends are due to changes in recognition and diagnosis, or are true temporal changes in incidence.”
Dr. Dinse, of the National Institute of Environmental Health Sciences in Research Triangle Park, N.C., and his colleagues evaluated sera samples of 14,211 survey participants aged 12 years and older at 1:80 dilution for antinuclear antibodies (ANA) using a standard indirect immunofluorescence assay (HEp-2 assay). The samples that received a grade of 3 or 4 on a 0-4 scale (compared with standard references, with values of 1-4 indicating positivity) underwent additional assessment by sequential ANA titers up to 1:1,280 dilution. To estimate changes in ANA prevalence over the time periods, they used logistic regression adjusted for age, sex, race/ethnicity, and survey design variables.
The researchers observed an ANA prevalence of 11% in 1988-1991, 11.5% in 1999-2004, and 15.9% in 2011-2012. This corresponds to 22, 27, and 41 million affected individuals, respectively. Females were more likely than males to have ANA (odds ratios of 2.53, 2.97, and 1.94 in 1988-1991, 1999-2004, and 2011-2012, respectively; P less than .0001), as were older adults relative to adolescents (ORs of 3.63, 1.80, and 1.71; P less than .002). Among adolescents, the prevalence of ANA rose steeply, with odds ratios of 2.02 in 1999-2004 and 2.88 in 2011-2012 in the second and third time periods relative to the first (trend P less than .0001). The researchers also found that, compared with non-Hispanic whites, the odds of having ANA were higher for non-Hispanic blacks (OR, 1.75) and Mexican-Americans (OR, 1.87) in 1988-1991, but racial/ethnic differences diminished in 1999-2004 and 2011-2012.
After adjustment for covariates, the researchers found that the estimated odds ratios for the second and third time periods relative to the first were 1.02 and 1.47, respectively, reflecting an overall ANA time trend (P less than .0001). Increases in ANA prevalence among cohorts did not correlate with contemporaneous trends in body mass index, smoking, or alcohol consumption.
Dr. Dinse and his colleagues acknowledged certain limitations of the study, including the fact that associations were based on cross-sectional data rather than repeated measures, and that some variables were self-reported, including the limited questionnaire data on autoimmune diseases.
In an interview, David S. Pisetsky, MD, professor of medicine/rheumatology and immunology at Duke University, Durham, N.C., characterized the study findings as “hypothesis generating” and said that he would like to know if the researchers would find the same results if they used a different ANA assay. “There’s a lot of variability from ANA kit to ANA kit – much greater than what was thought,” said Dr. Pisetsky, who is an authority on the topic. “One thing that needs to be done is to find out what the frequency is with other tests. One should recognize that the actual frequency is going to vary by the assay used. In another test format, the frequency may have been lower; it could have been higher.”
He added that the precise reasons why the prevalence of ANAs are rising in the general population remains elusive. “We know the target antigens in people with autoantibody-associated rheumatic disease,” Dr. Pisetsky said. “But what we don’t know a lot of times is, what are the target antigens in the otherwise healthy population? There has only been one antibody system that people have felt is associated with the otherwise healthy population. Those are called anti-DFS-70 antibodies, but there is even uncertainty about those. If you know what the antigens recognized were, then I think you could begin to speculate more about what’s going on in the population that’s increasing the frequency [of ANAs].”
In an accompanying editorial, Richard J. Bucala, MD, chief of rheumatology, allergy, and immunology at Yale University, New Haven, Conn., noted that the origins of autoantibodies in different rheumatic diseases and the steps that lead to disease progression remain elusive. “Modern societies experience an ever increasing variety of exposures due to travel and population migration, an increase in both the internationalization of agriculture and the industrialization of food production, a higher environmental burden of synthetic chemicals, emerging pathogens, and the inexorable effects of climate change,” Dr. Bucala wrote. “The speed and intensity of these influences is arguably unprecedented in human history and clearly outpace the possibility of protective genetic mechanisms to evolve and adapt.” He went on to note that the study’s findings “give impetus to multidisciplinary efforts aimed at preventative strategies, identifying environmental hazards, defining high-risk individuals, and preventing disease development in susceptible populations.”
The study was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences. The authors reported having no disclosures.
SOURCE: Dinse G et al. Arthritis Rheumatol. 2020 April 7. doi: 10.1002/ART.41214.
FROM ARTHRITIS & RHEUMATOLOGY
Is There an Association Between Hidradenitis Suppurativa and Fibromyalgia?
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory condition that affects approximately 1% to 4% of the worldwide population and is 3 times more common in females than in males.1 The condition is characterized by painful inflamed nodules in apocrine gland–bearing regions that can progress to abscesses, sinus tracts, and/or scarring. Hidradenitis suppurativa is associated with intense pain, work disability, and poor quality of life.1
Recent evidence has suggested that HS is an autoimmune disease resulting from dysregulation of the γ-secretase/Notch pathway, leading to stimulation of the toll-like receptor–mediated innate immunity that contributes to occlusion and inflammation of the hair follicle. Additionally, elevated levels of proinflammatory cytokines such as tumor necrosis factor α and IL-17 are seen in HS lesions.2 The autoimmune nature of HS may account for its increased association with other autoimmune disorders such as thyroid disease and potentially with other unexplored conditions such as fibromyalgia.3
Fibromyalgia is a chronic pain condition that primarily affects females and is commonly associated with other autoimmune conditions.4 The primary objective of this retrospective study was to determine the prevalence of fibromyalgia in HS patients and assess if there is an association between HS disease severity and development of fibromyalgia.
We conducted a retrospective chart review of patients at Wake Forest Baptist Medical Center (Winston-Salem, North Carolina) who were 18 years and older and had a diagnosis of both HS and fibromyalgia from January 2008 to November 2018. The primary end point was the prevalence of fibromyalgia in the HS population. The secondary end point was the association of HS disease severity with the development of fibromyalgia. Hidradenitis disease severity was defined according to the number of body areas affected by HS: mild disease involved 1 body area, moderate disease involved 2 body areas, and severe disease involved 3 or more body areas. Patient age, sex, and race also were recorded.
A total of 1356 patients were seen during this time period for HS. The prevalence of fibromyalgia in the HS population was 3.2% (n=44). Ninety-five percent (42/44) of patients with HS and fibromyalgia were women; 22 (50%) patients had severe disease, 12 (27%) had moderate disease, 7 (16%) had mild disease, and 3 (7%) had an unknown number of affected body areas. Fifty-seven percent (25/44) of patients were diagnosed with HS prior to the diagnosis of fibromyalgia (Table).

In our study, the prevalence of fibromyalgia in HS patients was lower than the overall prevalence estimates of up to 6% in the United States.5 Although fibromyalgia is associated with other autoimmune conditions, it does not appear that fibromyalgia occurs more frequently in the HS population than the general population. A limitation of this study was that we only included academic outpatient clinic visits at one institution, which may not be representative of the entire HS population. Fibromyalgia was one of the many pain disorders in this population of patients. In this population of HS patients, many had pain issues with diagnose
- Smith MK, Nichlson CL, Parks-Miller A, et al. Hidradenitis suppurativa: an update on connecting the tracts. F1000Res. 2017;6:1272.
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115.
- Miller IM, Vinding G, Sorensen HA, et al. Thyroid function in hidradenitis suppurativa: a population-based cross-sectional study from Denmark. Clin Exp Dermatol. 2018;43:899-905.
- Giacomelli C, Talarico R, Bombardieri S, et al. The interaction between autoimmune diseases and fibromyalgia: risk, disease course and management. Expert Rev Clin Immunol. 2013;9:1069-1076.
- Queiroz LP. Worldwide epidemiology of fibromyalgia. Curr Pain Headache Rep. 2013;17:356.
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory condition that affects approximately 1% to 4% of the worldwide population and is 3 times more common in females than in males.1 The condition is characterized by painful inflamed nodules in apocrine gland–bearing regions that can progress to abscesses, sinus tracts, and/or scarring. Hidradenitis suppurativa is associated with intense pain, work disability, and poor quality of life.1
Recent evidence has suggested that HS is an autoimmune disease resulting from dysregulation of the γ-secretase/Notch pathway, leading to stimulation of the toll-like receptor–mediated innate immunity that contributes to occlusion and inflammation of the hair follicle. Additionally, elevated levels of proinflammatory cytokines such as tumor necrosis factor α and IL-17 are seen in HS lesions.2 The autoimmune nature of HS may account for its increased association with other autoimmune disorders such as thyroid disease and potentially with other unexplored conditions such as fibromyalgia.3
Fibromyalgia is a chronic pain condition that primarily affects females and is commonly associated with other autoimmune conditions.4 The primary objective of this retrospective study was to determine the prevalence of fibromyalgia in HS patients and assess if there is an association between HS disease severity and development of fibromyalgia.
We conducted a retrospective chart review of patients at Wake Forest Baptist Medical Center (Winston-Salem, North Carolina) who were 18 years and older and had a diagnosis of both HS and fibromyalgia from January 2008 to November 2018. The primary end point was the prevalence of fibromyalgia in the HS population. The secondary end point was the association of HS disease severity with the development of fibromyalgia. Hidradenitis disease severity was defined according to the number of body areas affected by HS: mild disease involved 1 body area, moderate disease involved 2 body areas, and severe disease involved 3 or more body areas. Patient age, sex, and race also were recorded.
A total of 1356 patients were seen during this time period for HS. The prevalence of fibromyalgia in the HS population was 3.2% (n=44). Ninety-five percent (42/44) of patients with HS and fibromyalgia were women; 22 (50%) patients had severe disease, 12 (27%) had moderate disease, 7 (16%) had mild disease, and 3 (7%) had an unknown number of affected body areas. Fifty-seven percent (25/44) of patients were diagnosed with HS prior to the diagnosis of fibromyalgia (Table).
In our study, the prevalence of fibromyalgia in HS patients was lower than the overall prevalence estimates of up to 6% in the United States.5 Although fibromyalgia is associated with other autoimmune conditions, it does not appear that fibromyalgia occurs more frequently in the HS population than the general population. A limitation of this study was that we only included academic outpatient clinic visits at one institution, which may not be representative of the entire HS population. Fibromyalgia was one of the many pain disorders in this population of patients. In this population of HS patients, many had pain issues with diagnose
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory condition that affects approximately 1% to 4% of the worldwide population and is 3 times more common in females than in males.1 The condition is characterized by painful inflamed nodules in apocrine gland–bearing regions that can progress to abscesses, sinus tracts, and/or scarring. Hidradenitis suppurativa is associated with intense pain, work disability, and poor quality of life.1
Recent evidence has suggested that HS is an autoimmune disease resulting from dysregulation of the γ-secretase/Notch pathway, leading to stimulation of the toll-like receptor–mediated innate immunity that contributes to occlusion and inflammation of the hair follicle. Additionally, elevated levels of proinflammatory cytokines such as tumor necrosis factor α and IL-17 are seen in HS lesions.2 The autoimmune nature of HS may account for its increased association with other autoimmune disorders such as thyroid disease and potentially with other unexplored conditions such as fibromyalgia.3
Fibromyalgia is a chronic pain condition that primarily affects females and is commonly associated with other autoimmune conditions.4 The primary objective of this retrospective study was to determine the prevalence of fibromyalgia in HS patients and assess if there is an association between HS disease severity and development of fibromyalgia.
We conducted a retrospective chart review of patients at Wake Forest Baptist Medical Center (Winston-Salem, North Carolina) who were 18 years and older and had a diagnosis of both HS and fibromyalgia from January 2008 to November 2018. The primary end point was the prevalence of fibromyalgia in the HS population. The secondary end point was the association of HS disease severity with the development of fibromyalgia. Hidradenitis disease severity was defined according to the number of body areas affected by HS: mild disease involved 1 body area, moderate disease involved 2 body areas, and severe disease involved 3 or more body areas. Patient age, sex, and race also were recorded.
A total of 1356 patients were seen during this time period for HS. The prevalence of fibromyalgia in the HS population was 3.2% (n=44). Ninety-five percent (42/44) of patients with HS and fibromyalgia were women; 22 (50%) patients had severe disease, 12 (27%) had moderate disease, 7 (16%) had mild disease, and 3 (7%) had an unknown number of affected body areas. Fifty-seven percent (25/44) of patients were diagnosed with HS prior to the diagnosis of fibromyalgia (Table).
In our study, the prevalence of fibromyalgia in HS patients was lower than the overall prevalence estimates of up to 6% in the United States.5 Although fibromyalgia is associated with other autoimmune conditions, it does not appear that fibromyalgia occurs more frequently in the HS population than the general population. A limitation of this study was that we only included academic outpatient clinic visits at one institution, which may not be representative of the entire HS population. Fibromyalgia was one of the many pain disorders in this population of patients. In this population of HS patients, many had pain issues with diagnose
- Smith MK, Nichlson CL, Parks-Miller A, et al. Hidradenitis suppurativa: an update on connecting the tracts. F1000Res. 2017;6:1272.
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115.
- Miller IM, Vinding G, Sorensen HA, et al. Thyroid function in hidradenitis suppurativa: a population-based cross-sectional study from Denmark. Clin Exp Dermatol. 2018;43:899-905.
- Giacomelli C, Talarico R, Bombardieri S, et al. The interaction between autoimmune diseases and fibromyalgia: risk, disease course and management. Expert Rev Clin Immunol. 2013;9:1069-1076.
- Queiroz LP. Worldwide epidemiology of fibromyalgia. Curr Pain Headache Rep. 2013;17:356.
- Smith MK, Nichlson CL, Parks-Miller A, et al. Hidradenitis suppurativa: an update on connecting the tracts. F1000Res. 2017;6:1272.
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115.
- Miller IM, Vinding G, Sorensen HA, et al. Thyroid function in hidradenitis suppurativa: a population-based cross-sectional study from Denmark. Clin Exp Dermatol. 2018;43:899-905.
- Giacomelli C, Talarico R, Bombardieri S, et al. The interaction between autoimmune diseases and fibromyalgia: risk, disease course and management. Expert Rev Clin Immunol. 2013;9:1069-1076.
- Queiroz LP. Worldwide epidemiology of fibromyalgia. Curr Pain Headache Rep. 2013;17:356.
Practice Point
- Although fibromyalgia does not occur more frequently in hidradenitis suppurativa (HS) patients, it is important to recognize that HS patients can have comorbidities that should be addressed when possible to improve overall quality of life.
Belimumab may improve skin in scleroderma
MAUI, HAWAII – Belimumab shows promise as a novel biologic treatment for skin involvement in early diffuse cutaneous systemic sclerosis, Janet E. Pope, MD, said at the 2020 Rheumatology Winter Clinical Symposium.

She highlighted a single-center, double-blind, placebo-controlled, New York pilot study including 20 patients with early diffuse cutaneous systemic sclerosis and moderate skin involvement. Participants had recently started on background mycophenolate mofetil (MMF) at 1,000 mg twice daily and were then randomized to add-on belimumab (Benlysta) at the dosing approved for systemic lupus erythematosus or to infusions of normal saline.
At 52 weeks, the modified Rodnan skin thickness score (mRSS) decreased by a median of 10 points from a baseline of 27 in the belimumab group, compared with just a 3-point reduction in controls on MMF plus placebo.
This small study raises several key points. It definitely warrants confirmation in a large phase 3 trial, according to Dr. Pope, professor of medicine at the University of Western Ontario and head of the division of rheumatology at St. Joseph’s Health Care, both in London.
For one thing, the pilot study makes a good case for multidrug therapy in scleroderma. “In rheumatoid arthritis, if in general one drug is not as good as two, why would we ever think, in our most difficult-to-treat disease, one drug would be okay?” the rheumatologist observed.
The belimumab study also highlights the role of abnormalities in B-cell function in the pathogenesis of skin involvement in early diffuse cutaneous systemic sclerosis. Belimumab is a fully human monoclonal antibody which binds to soluble B-lymphocyte stimulator and inhibits autoantibody production.
Belimumab’s mechanism of benefit was as expected: The improvement in skin scores in the belimumab group was accompanied by decreased expression of profibrotic genes and B-cell signaling, changes that didn’t occur in the controls on MMF alone.
The belimumab study makes another important point: MMF, despite its growing popularity for treatment of skin manifestations of scleroderma, is actually a wimpy drug for that purpose, achieving a mere 3-point reduction in mRSS.
“To be quite honest, mycophenolate mofetil is not all that great on skin,” Dr. Pope said.
Nonetheless, when she and her coworkers recently polled 170 scleroderma experts as to their favored treatments directed at various target organs impaired by the disease, as she had previously done in 2012, a clear trend was evident. “There’s a shift in that mycophenolate mofetil is moving to first-line treatment across the board for skin,” Dr. Pope observed.
Indeed, in the more recent survey, 71% of the experts agreed upon a scleroderma skin involvement treatment algorithm in which the first-line treatment for severe skin disease as defined by an mRSS of 32 was MMF, with methotrexate as second line, intravenous cyclophosphamide third, and autologous stem cell transplantation as fourth line for the small number of patients who qualify for it.
For moderate skin involvement, with an mRSS of 24, methotrexate was endorsed as first line, although by the narrowest of margins, over MMF, with intravenous cyclophosphamide as third line. For mild disease, with an mRSS of 10, methotrexate again narrowly beat out MMF by expert consensus as the preferred first-line therapy.
When asked about concomitant use of corticosteroids for treatment of skin involvement, 35% of experts said they never prescribe them for that indication, 33% do so occasionally, 19% sometimes, and 13% routinely. There was an even split on dosing among those who prescribe steroids: 49% suggested using prednisone at less than 7.5 mg/day, and 51% recommended 7.5-20 mg/day.
The purpose in polling the experts, who were drawn from the Scleroderma Clinical Trials Consortium and the Canadian Scleroderma Research Group, was to provide treatment guidance to general rheumatologists and dermatologists who may not see many patients with scleroderma. In contrast, the great majority of the polled experts see more than 50 scleroderma patients per year. And they had a high level of total agreement for treatment algorithms addressing not only skin disease, but also pulmonary arterial hypertension, interstitial lung disease, Raynaud’s phenomenon, renal crisis, digital ulcers, inflammatory arthritis, cardiac involvement, and gastrointestinal disease, Dr. Pope noted.
She attributed the experts’ rising enthusiasm for MMF for scleroderma skin involvement to the results of the Scleroderma Lung Study II, the first randomized, controlled trial to compare MMF and cyclophosphamide for the treatment of symptomatic scleroderma interstitial lung disease. Two years of MMF improved forced vital capacity as much as 1 year of oral cyclophosphamide. At 2 years of follow-up, the mRSS dropped modestly from baseline by an average of 6.1 points in the cyclophosphamide group and 2.9 points with MMF, a nonsignificant difference. But the incidence of serious adverse events was roughly three times higher and deaths were twice as frequent in the cyclophosphamide group.
“I think mycophenolate mofetil is surging for treatment of skin because of the lung protection and it was safer, but it’s hard for me to know if the deaths were more common in the cyclophosphamide group because of the cyclophosphamide or because of no treatment in year 2,” Dr. Pope commented.
She reported receiving research grants from Bristol-Myers Squibb, Merck, Roche, Seattle Genetics, and UCB, and serving as a consultant to more than a dozen pharmaceutical companies.
MAUI, HAWAII – Belimumab shows promise as a novel biologic treatment for skin involvement in early diffuse cutaneous systemic sclerosis, Janet E. Pope, MD, said at the 2020 Rheumatology Winter Clinical Symposium.
She highlighted a single-center, double-blind, placebo-controlled, New York pilot study including 20 patients with early diffuse cutaneous systemic sclerosis and moderate skin involvement. Participants had recently started on background mycophenolate mofetil (MMF) at 1,000 mg twice daily and were then randomized to add-on belimumab (Benlysta) at the dosing approved for systemic lupus erythematosus or to infusions of normal saline.
At 52 weeks, the modified Rodnan skin thickness score (mRSS) decreased by a median of 10 points from a baseline of 27 in the belimumab group, compared with just a 3-point reduction in controls on MMF plus placebo.
This small study raises several key points. It definitely warrants confirmation in a large phase 3 trial, according to Dr. Pope, professor of medicine at the University of Western Ontario and head of the division of rheumatology at St. Joseph’s Health Care, both in London.
For one thing, the pilot study makes a good case for multidrug therapy in scleroderma. “In rheumatoid arthritis, if in general one drug is not as good as two, why would we ever think, in our most difficult-to-treat disease, one drug would be okay?” the rheumatologist observed.
The belimumab study also highlights the role of abnormalities in B-cell function in the pathogenesis of skin involvement in early diffuse cutaneous systemic sclerosis. Belimumab is a fully human monoclonal antibody which binds to soluble B-lymphocyte stimulator and inhibits autoantibody production.
Belimumab’s mechanism of benefit was as expected: The improvement in skin scores in the belimumab group was accompanied by decreased expression of profibrotic genes and B-cell signaling, changes that didn’t occur in the controls on MMF alone.
The belimumab study makes another important point: MMF, despite its growing popularity for treatment of skin manifestations of scleroderma, is actually a wimpy drug for that purpose, achieving a mere 3-point reduction in mRSS.
“To be quite honest, mycophenolate mofetil is not all that great on skin,” Dr. Pope said.
Nonetheless, when she and her coworkers recently polled 170 scleroderma experts as to their favored treatments directed at various target organs impaired by the disease, as she had previously done in 2012, a clear trend was evident. “There’s a shift in that mycophenolate mofetil is moving to first-line treatment across the board for skin,” Dr. Pope observed.
Indeed, in the more recent survey, 71% of the experts agreed upon a scleroderma skin involvement treatment algorithm in which the first-line treatment for severe skin disease as defined by an mRSS of 32 was MMF, with methotrexate as second line, intravenous cyclophosphamide third, and autologous stem cell transplantation as fourth line for the small number of patients who qualify for it.
For moderate skin involvement, with an mRSS of 24, methotrexate was endorsed as first line, although by the narrowest of margins, over MMF, with intravenous cyclophosphamide as third line. For mild disease, with an mRSS of 10, methotrexate again narrowly beat out MMF by expert consensus as the preferred first-line therapy.
When asked about concomitant use of corticosteroids for treatment of skin involvement, 35% of experts said they never prescribe them for that indication, 33% do so occasionally, 19% sometimes, and 13% routinely. There was an even split on dosing among those who prescribe steroids: 49% suggested using prednisone at less than 7.5 mg/day, and 51% recommended 7.5-20 mg/day.
The purpose in polling the experts, who were drawn from the Scleroderma Clinical Trials Consortium and the Canadian Scleroderma Research Group, was to provide treatment guidance to general rheumatologists and dermatologists who may not see many patients with scleroderma. In contrast, the great majority of the polled experts see more than 50 scleroderma patients per year. And they had a high level of total agreement for treatment algorithms addressing not only skin disease, but also pulmonary arterial hypertension, interstitial lung disease, Raynaud’s phenomenon, renal crisis, digital ulcers, inflammatory arthritis, cardiac involvement, and gastrointestinal disease, Dr. Pope noted.
She attributed the experts’ rising enthusiasm for MMF for scleroderma skin involvement to the results of the Scleroderma Lung Study II, the first randomized, controlled trial to compare MMF and cyclophosphamide for the treatment of symptomatic scleroderma interstitial lung disease. Two years of MMF improved forced vital capacity as much as 1 year of oral cyclophosphamide. At 2 years of follow-up, the mRSS dropped modestly from baseline by an average of 6.1 points in the cyclophosphamide group and 2.9 points with MMF, a nonsignificant difference. But the incidence of serious adverse events was roughly three times higher and deaths were twice as frequent in the cyclophosphamide group.
“I think mycophenolate mofetil is surging for treatment of skin because of the lung protection and it was safer, but it’s hard for me to know if the deaths were more common in the cyclophosphamide group because of the cyclophosphamide or because of no treatment in year 2,” Dr. Pope commented.
She reported receiving research grants from Bristol-Myers Squibb, Merck, Roche, Seattle Genetics, and UCB, and serving as a consultant to more than a dozen pharmaceutical companies.
MAUI, HAWAII – Belimumab shows promise as a novel biologic treatment for skin involvement in early diffuse cutaneous systemic sclerosis, Janet E. Pope, MD, said at the 2020 Rheumatology Winter Clinical Symposium.
She highlighted a single-center, double-blind, placebo-controlled, New York pilot study including 20 patients with early diffuse cutaneous systemic sclerosis and moderate skin involvement. Participants had recently started on background mycophenolate mofetil (MMF) at 1,000 mg twice daily and were then randomized to add-on belimumab (Benlysta) at the dosing approved for systemic lupus erythematosus or to infusions of normal saline.
At 52 weeks, the modified Rodnan skin thickness score (mRSS) decreased by a median of 10 points from a baseline of 27 in the belimumab group, compared with just a 3-point reduction in controls on MMF plus placebo.
This small study raises several key points. It definitely warrants confirmation in a large phase 3 trial, according to Dr. Pope, professor of medicine at the University of Western Ontario and head of the division of rheumatology at St. Joseph’s Health Care, both in London.
For one thing, the pilot study makes a good case for multidrug therapy in scleroderma. “In rheumatoid arthritis, if in general one drug is not as good as two, why would we ever think, in our most difficult-to-treat disease, one drug would be okay?” the rheumatologist observed.
The belimumab study also highlights the role of abnormalities in B-cell function in the pathogenesis of skin involvement in early diffuse cutaneous systemic sclerosis. Belimumab is a fully human monoclonal antibody which binds to soluble B-lymphocyte stimulator and inhibits autoantibody production.
Belimumab’s mechanism of benefit was as expected: The improvement in skin scores in the belimumab group was accompanied by decreased expression of profibrotic genes and B-cell signaling, changes that didn’t occur in the controls on MMF alone.
The belimumab study makes another important point: MMF, despite its growing popularity for treatment of skin manifestations of scleroderma, is actually a wimpy drug for that purpose, achieving a mere 3-point reduction in mRSS.
“To be quite honest, mycophenolate mofetil is not all that great on skin,” Dr. Pope said.
Nonetheless, when she and her coworkers recently polled 170 scleroderma experts as to their favored treatments directed at various target organs impaired by the disease, as she had previously done in 2012, a clear trend was evident. “There’s a shift in that mycophenolate mofetil is moving to first-line treatment across the board for skin,” Dr. Pope observed.
Indeed, in the more recent survey, 71% of the experts agreed upon a scleroderma skin involvement treatment algorithm in which the first-line treatment for severe skin disease as defined by an mRSS of 32 was MMF, with methotrexate as second line, intravenous cyclophosphamide third, and autologous stem cell transplantation as fourth line for the small number of patients who qualify for it.
For moderate skin involvement, with an mRSS of 24, methotrexate was endorsed as first line, although by the narrowest of margins, over MMF, with intravenous cyclophosphamide as third line. For mild disease, with an mRSS of 10, methotrexate again narrowly beat out MMF by expert consensus as the preferred first-line therapy.
When asked about concomitant use of corticosteroids for treatment of skin involvement, 35% of experts said they never prescribe them for that indication, 33% do so occasionally, 19% sometimes, and 13% routinely. There was an even split on dosing among those who prescribe steroids: 49% suggested using prednisone at less than 7.5 mg/day, and 51% recommended 7.5-20 mg/day.
The purpose in polling the experts, who were drawn from the Scleroderma Clinical Trials Consortium and the Canadian Scleroderma Research Group, was to provide treatment guidance to general rheumatologists and dermatologists who may not see many patients with scleroderma. In contrast, the great majority of the polled experts see more than 50 scleroderma patients per year. And they had a high level of total agreement for treatment algorithms addressing not only skin disease, but also pulmonary arterial hypertension, interstitial lung disease, Raynaud’s phenomenon, renal crisis, digital ulcers, inflammatory arthritis, cardiac involvement, and gastrointestinal disease, Dr. Pope noted.
She attributed the experts’ rising enthusiasm for MMF for scleroderma skin involvement to the results of the Scleroderma Lung Study II, the first randomized, controlled trial to compare MMF and cyclophosphamide for the treatment of symptomatic scleroderma interstitial lung disease. Two years of MMF improved forced vital capacity as much as 1 year of oral cyclophosphamide. At 2 years of follow-up, the mRSS dropped modestly from baseline by an average of 6.1 points in the cyclophosphamide group and 2.9 points with MMF, a nonsignificant difference. But the incidence of serious adverse events was roughly three times higher and deaths were twice as frequent in the cyclophosphamide group.
“I think mycophenolate mofetil is surging for treatment of skin because of the lung protection and it was safer, but it’s hard for me to know if the deaths were more common in the cyclophosphamide group because of the cyclophosphamide or because of no treatment in year 2,” Dr. Pope commented.
She reported receiving research grants from Bristol-Myers Squibb, Merck, Roche, Seattle Genetics, and UCB, and serving as a consultant to more than a dozen pharmaceutical companies.
REPORTING FROM RWCS 2020
Use of mHealth technology lags in lupus care, research
Most mobile apps are poor in quality, review finds.
and have limited “The use of mobile technologies to support health (mHealth technologies), specifically mHealth applications (apps), has the potential to improve outcomes in SLE by empowering patients through education, symptom tracking, and peer support,” first author Lucas Ogura Dantas, of Tufts Medical Center, Boston, and coauthors wrote in Lupus. “These may be particularly powerful tools in SLE which commonly affects young adults, who are typically avid smartphone users familiar with the use of mobile apps.”

The authors’ review of 19 mHealth apps on Google Play and the Apple App Store (and 1 not on either platform) gave an overall average score of 2.3 out of a possible 5.0 from individual mean scores for engagement, functionality, aesthetics, and information quality on the 23-item Mobile App Rating Scale, each item of which is rated on a 0-5 point scale. Overall, 10 apps offered educational content, 7 offered tools for tracking patient-reported symptoms, 5 offered interactive online communities, 1 offered emojis to share through text messages or email for the purpose of entertainment, and 1 could not be fully evaluated.
The researchers noted that “most apps scored poorly based on design, user interface, functionality, and credibility,” with mean scores of 2.5 for engagement, 2.9 for functionality, 2.2 for aesthetics, and 1.6 for information. “The majority of the apps provided low-quality information from questionable sources (i.e., sources were not cited or their legitimacy was unknown or unverifiable),” they wrote.
The three highest-rated apps – LupusMinder (overall mean score, 3.3), Lupus Corner Health Manager (3.2), and PatientsLikeMe (3.1) – all focused on tracking patient-reported outcomes, but none used validated outcome measures; offered connectivity with wearable devices; or passively collected data such as step count, walking distances, flights climbed, calories expenditure, or sleep monitoring. However, two did have social network components and interactive support groups. Despite these apps’ interactivity and customizability, none offered “features for patients to create and track goals, directly connect with a physician or expert in the field, or synchronize data with electronic health records. None of the apps provided patients with feedback.”
The authors wrote that the Lupus Corner Health Manager was “the only one that addresses the majority of the preferences of SLE patients identified in our literature review. This app provides educational material, a symptom and medication tracker, and a discussion group for communicating with others living with lupus – all key features of mHealth technologies in the management of chronic diseases.”
However, they noted that the “ideal mHealth app for patients with SLE would incorporate evidence-based educational material, customizable symptom and medication trackers, logs for personalized health goals, and connectivity with external hardware devices to enrich data collection. Additional useful features would include gamification components to engage users, the provision of tailored feedback based on collected data, and secure mechanisms of communication and data access between users and health care providers to facilitate treatment planning and coordination of care.”
In the systematic literature review, the researchers identified a total of 21 original research studies “related to the development or use of mHealth technologies targeting people of all ages with an SLE diagnosis,” including 2 randomized trials, 10 observational studies, 4 qualitative studies, 3 review articles, and 2 study protocols for future randomized trials.
These papers most often focused on developing and using mHealth for providing patient information (11 papers), followed by mHealth interventions (5); study protocols (2); and developing mHealth apps, websites, or mHealth interventions (3).
Seven studies implemented mHealth technologies, including two with wearable devices, two with text-messaging interventions, and three that used web-based systems. These had mixed results and small samples sizes ranging from 9 to 41 patients, making their interpretation difficult, the authors wrote. A total of 11 studies examined the development of mHealth technologies, including 7 that recognized “the need for more interactive educational platforms with high-quality information,” 2 that described a need for “novel methods of disease monitoring,” and 2 that revealed “a need for sources of support such as virtual communities.”
“Though our systematic literature review found that patients seek to use mHealth technologies to aid with disease management, we identified few studies exploring mHealth-based interventions to improve health outcomes, with the limited published literature devoted to the use of mHealth platforms to provide educational information,” Mr. Dantas and associates wrote.
The lack of evidence for mHealth technologies in SLE patients “contrast with the robust evidence supporting the effectiveness of mHealth interventions in improving outcomes in several chronic conditions,” such as chronic low back and musculoskeletal pain and blood pressure control, they noted.
The authors reported no potential conflicts of interest. The study authors received funding from the Saõ Paulo Research Foundation, the National Center for Complementary and Integrative Health, and the National Center for Advancing Translational Sciences.
SOURCE: Dantas LO et al. Lupus. 2020;29:144-56.
Most mobile apps are poor in quality, review finds.
Most mobile apps are poor in quality, review finds.
and have limited “The use of mobile technologies to support health (mHealth technologies), specifically mHealth applications (apps), has the potential to improve outcomes in SLE by empowering patients through education, symptom tracking, and peer support,” first author Lucas Ogura Dantas, of Tufts Medical Center, Boston, and coauthors wrote in Lupus. “These may be particularly powerful tools in SLE which commonly affects young adults, who are typically avid smartphone users familiar with the use of mobile apps.”
The authors’ review of 19 mHealth apps on Google Play and the Apple App Store (and 1 not on either platform) gave an overall average score of 2.3 out of a possible 5.0 from individual mean scores for engagement, functionality, aesthetics, and information quality on the 23-item Mobile App Rating Scale, each item of which is rated on a 0-5 point scale. Overall, 10 apps offered educational content, 7 offered tools for tracking patient-reported symptoms, 5 offered interactive online communities, 1 offered emojis to share through text messages or email for the purpose of entertainment, and 1 could not be fully evaluated.
The researchers noted that “most apps scored poorly based on design, user interface, functionality, and credibility,” with mean scores of 2.5 for engagement, 2.9 for functionality, 2.2 for aesthetics, and 1.6 for information. “The majority of the apps provided low-quality information from questionable sources (i.e., sources were not cited or their legitimacy was unknown or unverifiable),” they wrote.
The three highest-rated apps – LupusMinder (overall mean score, 3.3), Lupus Corner Health Manager (3.2), and PatientsLikeMe (3.1) – all focused on tracking patient-reported outcomes, but none used validated outcome measures; offered connectivity with wearable devices; or passively collected data such as step count, walking distances, flights climbed, calories expenditure, or sleep monitoring. However, two did have social network components and interactive support groups. Despite these apps’ interactivity and customizability, none offered “features for patients to create and track goals, directly connect with a physician or expert in the field, or synchronize data with electronic health records. None of the apps provided patients with feedback.”
The authors wrote that the Lupus Corner Health Manager was “the only one that addresses the majority of the preferences of SLE patients identified in our literature review. This app provides educational material, a symptom and medication tracker, and a discussion group for communicating with others living with lupus – all key features of mHealth technologies in the management of chronic diseases.”
However, they noted that the “ideal mHealth app for patients with SLE would incorporate evidence-based educational material, customizable symptom and medication trackers, logs for personalized health goals, and connectivity with external hardware devices to enrich data collection. Additional useful features would include gamification components to engage users, the provision of tailored feedback based on collected data, and secure mechanisms of communication and data access between users and health care providers to facilitate treatment planning and coordination of care.”
In the systematic literature review, the researchers identified a total of 21 original research studies “related to the development or use of mHealth technologies targeting people of all ages with an SLE diagnosis,” including 2 randomized trials, 10 observational studies, 4 qualitative studies, 3 review articles, and 2 study protocols for future randomized trials.
These papers most often focused on developing and using mHealth for providing patient information (11 papers), followed by mHealth interventions (5); study protocols (2); and developing mHealth apps, websites, or mHealth interventions (3).
Seven studies implemented mHealth technologies, including two with wearable devices, two with text-messaging interventions, and three that used web-based systems. These had mixed results and small samples sizes ranging from 9 to 41 patients, making their interpretation difficult, the authors wrote. A total of 11 studies examined the development of mHealth technologies, including 7 that recognized “the need for more interactive educational platforms with high-quality information,” 2 that described a need for “novel methods of disease monitoring,” and 2 that revealed “a need for sources of support such as virtual communities.”
“Though our systematic literature review found that patients seek to use mHealth technologies to aid with disease management, we identified few studies exploring mHealth-based interventions to improve health outcomes, with the limited published literature devoted to the use of mHealth platforms to provide educational information,” Mr. Dantas and associates wrote.
The lack of evidence for mHealth technologies in SLE patients “contrast with the robust evidence supporting the effectiveness of mHealth interventions in improving outcomes in several chronic conditions,” such as chronic low back and musculoskeletal pain and blood pressure control, they noted.
The authors reported no potential conflicts of interest. The study authors received funding from the Saõ Paulo Research Foundation, the National Center for Complementary and Integrative Health, and the National Center for Advancing Translational Sciences.
SOURCE: Dantas LO et al. Lupus. 2020;29:144-56.
and have limited “The use of mobile technologies to support health (mHealth technologies), specifically mHealth applications (apps), has the potential to improve outcomes in SLE by empowering patients through education, symptom tracking, and peer support,” first author Lucas Ogura Dantas, of Tufts Medical Center, Boston, and coauthors wrote in Lupus. “These may be particularly powerful tools in SLE which commonly affects young adults, who are typically avid smartphone users familiar with the use of mobile apps.”
The authors’ review of 19 mHealth apps on Google Play and the Apple App Store (and 1 not on either platform) gave an overall average score of 2.3 out of a possible 5.0 from individual mean scores for engagement, functionality, aesthetics, and information quality on the 23-item Mobile App Rating Scale, each item of which is rated on a 0-5 point scale. Overall, 10 apps offered educational content, 7 offered tools for tracking patient-reported symptoms, 5 offered interactive online communities, 1 offered emojis to share through text messages or email for the purpose of entertainment, and 1 could not be fully evaluated.
The researchers noted that “most apps scored poorly based on design, user interface, functionality, and credibility,” with mean scores of 2.5 for engagement, 2.9 for functionality, 2.2 for aesthetics, and 1.6 for information. “The majority of the apps provided low-quality information from questionable sources (i.e., sources were not cited or their legitimacy was unknown or unverifiable),” they wrote.
The three highest-rated apps – LupusMinder (overall mean score, 3.3), Lupus Corner Health Manager (3.2), and PatientsLikeMe (3.1) – all focused on tracking patient-reported outcomes, but none used validated outcome measures; offered connectivity with wearable devices; or passively collected data such as step count, walking distances, flights climbed, calories expenditure, or sleep monitoring. However, two did have social network components and interactive support groups. Despite these apps’ interactivity and customizability, none offered “features for patients to create and track goals, directly connect with a physician or expert in the field, or synchronize data with electronic health records. None of the apps provided patients with feedback.”
The authors wrote that the Lupus Corner Health Manager was “the only one that addresses the majority of the preferences of SLE patients identified in our literature review. This app provides educational material, a symptom and medication tracker, and a discussion group for communicating with others living with lupus – all key features of mHealth technologies in the management of chronic diseases.”
However, they noted that the “ideal mHealth app for patients with SLE would incorporate evidence-based educational material, customizable symptom and medication trackers, logs for personalized health goals, and connectivity with external hardware devices to enrich data collection. Additional useful features would include gamification components to engage users, the provision of tailored feedback based on collected data, and secure mechanisms of communication and data access between users and health care providers to facilitate treatment planning and coordination of care.”
In the systematic literature review, the researchers identified a total of 21 original research studies “related to the development or use of mHealth technologies targeting people of all ages with an SLE diagnosis,” including 2 randomized trials, 10 observational studies, 4 qualitative studies, 3 review articles, and 2 study protocols for future randomized trials.
These papers most often focused on developing and using mHealth for providing patient information (11 papers), followed by mHealth interventions (5); study protocols (2); and developing mHealth apps, websites, or mHealth interventions (3).
Seven studies implemented mHealth technologies, including two with wearable devices, two with text-messaging interventions, and three that used web-based systems. These had mixed results and small samples sizes ranging from 9 to 41 patients, making their interpretation difficult, the authors wrote. A total of 11 studies examined the development of mHealth technologies, including 7 that recognized “the need for more interactive educational platforms with high-quality information,” 2 that described a need for “novel methods of disease monitoring,” and 2 that revealed “a need for sources of support such as virtual communities.”
“Though our systematic literature review found that patients seek to use mHealth technologies to aid with disease management, we identified few studies exploring mHealth-based interventions to improve health outcomes, with the limited published literature devoted to the use of mHealth platforms to provide educational information,” Mr. Dantas and associates wrote.
The lack of evidence for mHealth technologies in SLE patients “contrast with the robust evidence supporting the effectiveness of mHealth interventions in improving outcomes in several chronic conditions,” such as chronic low back and musculoskeletal pain and blood pressure control, they noted.
The authors reported no potential conflicts of interest. The study authors received funding from the Saõ Paulo Research Foundation, the National Center for Complementary and Integrative Health, and the National Center for Advancing Translational Sciences.
SOURCE: Dantas LO et al. Lupus. 2020;29:144-56.
FROM LUPUS