User login
Novel study offers clues to sex bias in lupus
Systemic lupus erythematosus (SLE), or lupus, shows a marked sex bias, affecting about nine females for every one male, according to Susan Kovats, PhD, who studies sex differences in immunity at the Oklahoma Medical Research Foundation in Oklahoma City. This characteristic of lupus suggests that hormones are involved in the pathogenesis of the disease. It also suggests, Dr. Kovats said, that the X chromosome might play a role.
Though studies since the 1970s have indicated a significant role for hormones, the issue is still complex and not well understood, and relatively little research has been done on the molecular mechanisms that might be responsible. This may be because of difficulties with influencing the immune system in vitro, said George A. Robinson, PhD, of University College London’s Centre for Rheumatology.

But Dr. Robinson and his team found a unique way of investigating the role of sex chromosomes and hormones in the inflammatory profiles across subjects of different sex, gender, age, and disease status. In research published online in The Lancet Rheumatology, Dr. Robinson and his team looked at immune cells taken from both cisgender men and women and transgender men and women, and thus were able to “get a more physiological view of what sex hormones are doing to the immune system,” he said.
Dr. Kovats agreed that it was a useful approach. “The transgender people provided an opportunity to effectively separate sex hormone levels from chromosome content,” she said in an interview.
Methods and findings
Peripheral blood mononuclear cell (PBMC) samples were taken from cisgender individuals with and without juvenile-onset lupus and assessed for 28 immune-cell subsets, including different T-cell, B-cell, and monotype subsets. Subjects included 39 postpubertal cisgender men and women (17 men and 22 women) who did not have juvenile-onset lupus, and 35 postpubertal cisgender men and women (12 men and 23 women) who did have juvenile onset lupus. All were aged 16-25 years. The transgender group included five transgender men and five transgender women (aged 18-19) who were undergoing gender-affirming sex hormone treatment.
The analysis found that one of the key differences between young postpubertal cisgender men and age-matched cisgender women was that the men had significantly elevated frequencies of regulatory T cells (T-reg cells), and the T-reg cells from young cisgender men had greater suppressive capacity in vitro than did those from cisgender women. In addition, RNA sequencing data from isolated T-reg cells showed the transcriptomic signature of the cisgender men’s T-regs were significantly enriched for genes in the P13K-AKT signaling pathway. The frequency of T-reg cells was not influenced by sex hormones, but their transcriptomic profile was affected.
“These results are beginning to give us an indication of which genes might be differentially regulated by sex hormones and how these are associated with autoimmunity,” Dr. Robinson said. “We’ve also found that, depending on whether you’re a cisgender man or woman, you may have a different pathogenic process to developing lupus. It’s not necessarily that one mechanism drives the disease across both sexes.”
New approaches, better insights
Dr. Kovats was particularly impressed by the methods of this study. “It was a natural study, the kind of thing we can usually do only in mice,” she said.

“One problem with studies on the effects of hormones in disease is that historically researchers have not paid that much attention to the actual hormone levels in the humans they studied. They might look at 100 women and 100 men, roughly between the ages of 20 and 50. We’re starting to see more, but there aren’t a lot of studies correlating numbers of cells in blood with actual hormone levels in the person. And as we know, just because someone’s a certain age doesn’t mean that they have a textbook hormone level. Early menopause, birth-control pills, many things can affect those levels.”
The researchers hope that these findings will shed light on the mechanisms that create sexual bias in autoimmune diseases, particularly lupus, as well as help researchers to better understand the innate and adaptive immunological differences between men and women. It could also be useful in the clinical setting, Dr. Robinson said. Because of the extreme sex bias in lupus, doctors see far more women with the illness than men. When they do see men with lupus, they need to be able to consider how the patient’s sex affects the development and course of the disease. “I think that people need to start looking at patients as clinically different, depending on their sex and gender,” he said. Information like that analyzed in this study could help with that. This could be especially important because as Dr. Kovats pointed out, although men get lupus far less often than women, when they do have it, they tend to have more severe disease.
Help from machines
This study was groundbreaking in another area as well. The researchers used machine learning to analyze the data. “We’ve started working a lot more with these analysis methods to try to answer as much as we can with these smaller data sets,” Dr. Robinson said. “Rather than the conventional analysis that we would typically perform, we’re able to use machine learning and artificial intelligence to try and learn from the data and increase the numbers that we’re working with by using a training data set. This allows us to interrogate the data with a lot more precision.”
The authors declared no competing interests.
Systemic lupus erythematosus (SLE), or lupus, shows a marked sex bias, affecting about nine females for every one male, according to Susan Kovats, PhD, who studies sex differences in immunity at the Oklahoma Medical Research Foundation in Oklahoma City. This characteristic of lupus suggests that hormones are involved in the pathogenesis of the disease. It also suggests, Dr. Kovats said, that the X chromosome might play a role.
Though studies since the 1970s have indicated a significant role for hormones, the issue is still complex and not well understood, and relatively little research has been done on the molecular mechanisms that might be responsible. This may be because of difficulties with influencing the immune system in vitro, said George A. Robinson, PhD, of University College London’s Centre for Rheumatology.
But Dr. Robinson and his team found a unique way of investigating the role of sex chromosomes and hormones in the inflammatory profiles across subjects of different sex, gender, age, and disease status. In research published online in The Lancet Rheumatology, Dr. Robinson and his team looked at immune cells taken from both cisgender men and women and transgender men and women, and thus were able to “get a more physiological view of what sex hormones are doing to the immune system,” he said.
Dr. Kovats agreed that it was a useful approach. “The transgender people provided an opportunity to effectively separate sex hormone levels from chromosome content,” she said in an interview.
Methods and findings
Peripheral blood mononuclear cell (PBMC) samples were taken from cisgender individuals with and without juvenile-onset lupus and assessed for 28 immune-cell subsets, including different T-cell, B-cell, and monotype subsets. Subjects included 39 postpubertal cisgender men and women (17 men and 22 women) who did not have juvenile-onset lupus, and 35 postpubertal cisgender men and women (12 men and 23 women) who did have juvenile onset lupus. All were aged 16-25 years. The transgender group included five transgender men and five transgender women (aged 18-19) who were undergoing gender-affirming sex hormone treatment.
The analysis found that one of the key differences between young postpubertal cisgender men and age-matched cisgender women was that the men had significantly elevated frequencies of regulatory T cells (T-reg cells), and the T-reg cells from young cisgender men had greater suppressive capacity in vitro than did those from cisgender women. In addition, RNA sequencing data from isolated T-reg cells showed the transcriptomic signature of the cisgender men’s T-regs were significantly enriched for genes in the P13K-AKT signaling pathway. The frequency of T-reg cells was not influenced by sex hormones, but their transcriptomic profile was affected.
“These results are beginning to give us an indication of which genes might be differentially regulated by sex hormones and how these are associated with autoimmunity,” Dr. Robinson said. “We’ve also found that, depending on whether you’re a cisgender man or woman, you may have a different pathogenic process to developing lupus. It’s not necessarily that one mechanism drives the disease across both sexes.”
New approaches, better insights
Dr. Kovats was particularly impressed by the methods of this study. “It was a natural study, the kind of thing we can usually do only in mice,” she said.
“One problem with studies on the effects of hormones in disease is that historically researchers have not paid that much attention to the actual hormone levels in the humans they studied. They might look at 100 women and 100 men, roughly between the ages of 20 and 50. We’re starting to see more, but there aren’t a lot of studies correlating numbers of cells in blood with actual hormone levels in the person. And as we know, just because someone’s a certain age doesn’t mean that they have a textbook hormone level. Early menopause, birth-control pills, many things can affect those levels.”
The researchers hope that these findings will shed light on the mechanisms that create sexual bias in autoimmune diseases, particularly lupus, as well as help researchers to better understand the innate and adaptive immunological differences between men and women. It could also be useful in the clinical setting, Dr. Robinson said. Because of the extreme sex bias in lupus, doctors see far more women with the illness than men. When they do see men with lupus, they need to be able to consider how the patient’s sex affects the development and course of the disease. “I think that people need to start looking at patients as clinically different, depending on their sex and gender,” he said. Information like that analyzed in this study could help with that. This could be especially important because as Dr. Kovats pointed out, although men get lupus far less often than women, when they do have it, they tend to have more severe disease.
Help from machines
This study was groundbreaking in another area as well. The researchers used machine learning to analyze the data. “We’ve started working a lot more with these analysis methods to try to answer as much as we can with these smaller data sets,” Dr. Robinson said. “Rather than the conventional analysis that we would typically perform, we’re able to use machine learning and artificial intelligence to try and learn from the data and increase the numbers that we’re working with by using a training data set. This allows us to interrogate the data with a lot more precision.”
The authors declared no competing interests.
Systemic lupus erythematosus (SLE), or lupus, shows a marked sex bias, affecting about nine females for every one male, according to Susan Kovats, PhD, who studies sex differences in immunity at the Oklahoma Medical Research Foundation in Oklahoma City. This characteristic of lupus suggests that hormones are involved in the pathogenesis of the disease. It also suggests, Dr. Kovats said, that the X chromosome might play a role.
Though studies since the 1970s have indicated a significant role for hormones, the issue is still complex and not well understood, and relatively little research has been done on the molecular mechanisms that might be responsible. This may be because of difficulties with influencing the immune system in vitro, said George A. Robinson, PhD, of University College London’s Centre for Rheumatology.
But Dr. Robinson and his team found a unique way of investigating the role of sex chromosomes and hormones in the inflammatory profiles across subjects of different sex, gender, age, and disease status. In research published online in The Lancet Rheumatology, Dr. Robinson and his team looked at immune cells taken from both cisgender men and women and transgender men and women, and thus were able to “get a more physiological view of what sex hormones are doing to the immune system,” he said.
Dr. Kovats agreed that it was a useful approach. “The transgender people provided an opportunity to effectively separate sex hormone levels from chromosome content,” she said in an interview.
Methods and findings
Peripheral blood mononuclear cell (PBMC) samples were taken from cisgender individuals with and without juvenile-onset lupus and assessed for 28 immune-cell subsets, including different T-cell, B-cell, and monotype subsets. Subjects included 39 postpubertal cisgender men and women (17 men and 22 women) who did not have juvenile-onset lupus, and 35 postpubertal cisgender men and women (12 men and 23 women) who did have juvenile onset lupus. All were aged 16-25 years. The transgender group included five transgender men and five transgender women (aged 18-19) who were undergoing gender-affirming sex hormone treatment.
The analysis found that one of the key differences between young postpubertal cisgender men and age-matched cisgender women was that the men had significantly elevated frequencies of regulatory T cells (T-reg cells), and the T-reg cells from young cisgender men had greater suppressive capacity in vitro than did those from cisgender women. In addition, RNA sequencing data from isolated T-reg cells showed the transcriptomic signature of the cisgender men’s T-regs were significantly enriched for genes in the P13K-AKT signaling pathway. The frequency of T-reg cells was not influenced by sex hormones, but their transcriptomic profile was affected.
“These results are beginning to give us an indication of which genes might be differentially regulated by sex hormones and how these are associated with autoimmunity,” Dr. Robinson said. “We’ve also found that, depending on whether you’re a cisgender man or woman, you may have a different pathogenic process to developing lupus. It’s not necessarily that one mechanism drives the disease across both sexes.”
New approaches, better insights
Dr. Kovats was particularly impressed by the methods of this study. “It was a natural study, the kind of thing we can usually do only in mice,” she said.
“One problem with studies on the effects of hormones in disease is that historically researchers have not paid that much attention to the actual hormone levels in the humans they studied. They might look at 100 women and 100 men, roughly between the ages of 20 and 50. We’re starting to see more, but there aren’t a lot of studies correlating numbers of cells in blood with actual hormone levels in the person. And as we know, just because someone’s a certain age doesn’t mean that they have a textbook hormone level. Early menopause, birth-control pills, many things can affect those levels.”
The researchers hope that these findings will shed light on the mechanisms that create sexual bias in autoimmune diseases, particularly lupus, as well as help researchers to better understand the innate and adaptive immunological differences between men and women. It could also be useful in the clinical setting, Dr. Robinson said. Because of the extreme sex bias in lupus, doctors see far more women with the illness than men. When they do see men with lupus, they need to be able to consider how the patient’s sex affects the development and course of the disease. “I think that people need to start looking at patients as clinically different, depending on their sex and gender,” he said. Information like that analyzed in this study could help with that. This could be especially important because as Dr. Kovats pointed out, although men get lupus far less often than women, when they do have it, they tend to have more severe disease.
Help from machines
This study was groundbreaking in another area as well. The researchers used machine learning to analyze the data. “We’ve started working a lot more with these analysis methods to try to answer as much as we can with these smaller data sets,” Dr. Robinson said. “Rather than the conventional analysis that we would typically perform, we’re able to use machine learning and artificial intelligence to try and learn from the data and increase the numbers that we’re working with by using a training data set. This allows us to interrogate the data with a lot more precision.”
The authors declared no competing interests.
FROM THE LANCET RHEUMATOLOGY
Surgical Deroofing for Hidradenitis Suppurativa
Practice Gap
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterized by inflammatory nodules, abscesses, sinus tracts, fistulae, and scarring, mainly in intertriginous areas. The extent of disease—classified using the Hurley staging system (stages I–III)—helps guide treatment, which includes medical management and surgical intervention in later stages.
First-line treatment of HS includes topical or systemic medications, or both. Surgical therapy typically is reserved for refractory HS in moderate to severe disease (Hurley stages II and III) and is combined with pharmacotherapy. Specifically, clinical management guidelines issued by an expert committee of the United States and Canadian Hidradenitis Suppurativa Foundations recommend excision or deroofing for recurrent nodules and tunnels.1
Surgical options for HS that are available to the outpatient dermatologist include incision and drainage, electrosurgery, CO2 laser evaporation, excision, and deroofing (also known as unroofing).2 Deroofing is a fairly novel therapy; many dermatologists are unfamiliar with the procedure. A PubMed search of articles indexed for MEDLINE related to HS prior to 2010 revealed only 1 article containing the word deroofing and only 4 articles containing unroofing.
The pathophysiology of HS has important implications for successful treatment. Inflammation of the follicular pilosebaceous unit along with follicular occlusion create challenges with treatment.3 It is postulated that a defect in the glassy membrane of the infra-infundibular wall predisposes the pilosebaceous follicle to lose its structural integrality as pressure builds from plugging of the duct,4 which can result in the clinical hallmarks of HS including tunneling tracts, bridging nodules, abscesses, and fistulae that form with lateral expansion of the plugged follicle.
Leaking of the contents of these plugged follicles into surrounding tissue produces an inflammatory response in characteristic HS lesions. Because debris within the lesions moves laterally instead of being able to burst to the surface, the lesions have difficulty fully healing. Unroofing the lesions and removing built-up debris allows them to heal more expediently and quiets the underlying immune response by removing the stimulus.4
Herein, we describe the benefits, risks, and surgical process of deroofing for HS.
Technique and Tools
Deroofing is performed under local anesthesia, stepwise as follows:
1. Identify sinus tracts and infiltrate the area with lidocaine (Figure, A).
2. Use a blunt probe to define the borders of the area to be unroofed and to evaluate for any communicating sinus tracts (Figure, B).
3. Remove the roof of underlying abscesses and tracts, using a probe as a guide (Figure, C).
4. Enter through the skin or sinus opening using electrocautery or with a scalpel or scissors; perform blunt dissection.
5. Reflect back the entirety of skin overlying the probed areas and remove the skin to expose the base of the lesion (Figure, D).
6. Explore the exposed base and walls of the lesion with the probe again to assess for hidden tracts; take care not to create false tracts.
7. Debride the surgical wound using curettage or rough gauze grattage to remove remaining inflammatory debris or biofilm. To achieve hemostasis, apply aluminum chloride or ferric chloride. Coat the wound with petroleum jelly and gauze and allow it to heal by secondary intention.
8. Educate the patient on wound care—once-daily gentle cleansing with soap and water, followed by application of a moist dressing—which is similar to wound healing by secondary intention from other causes.2,4

Practice Implications
A deroofing procedure has many benefits compared to other surgical modalities for the treatment of HS. Deroofing requires only a probe, curette, and electrocautery device, making the procedure more cost-effective than excision, which requires a full tray of equipment and sutures. Furthermore, margins do not need to be taken with deroofing, and no undermining or closure is needed, which saves time during the operation and minimizes the risk for complications, including dehiscence and formation of new sinus tracts.4 No specialized equipment, such as a CO2 laser, is required, which makes deroofing accessible to every clinical dermatologist in any demographic or geographic setting.
Evidence of Benefit—Saylor and colleagues5 found that deroofing carries a 12.5% complication rate, which includes postoperative bleeding, hypergranulation tissue, and rarely wound infection. This rate is significantly lower than the 26% complication rate associated with local excision, which includes wound dehiscence, infection, and contracture (P<.001). Deroofing also was found to have an HS recurrence rate of 14.5%, which is significantly less than the 30% recurrence rate seen with local excision (P=.015). Saylor et al5 also concluded that incision and drainage was recommended only for immediate relief of HS because of its 100% recurrence rate.
van der Zee2 reported on 88 lesions from 44 patients that were treated by surgical deroofing, resulting in an average defect of 3.0 cm in length and a mean healing time of 14 days. The typical outcome was cosmetically acceptable scarring; this finding was supported by a postoperative survey (>1 year), to which 37 of 44 patients responded and assigned an average satisfaction score of 8 (of a possible 10) and a recommendation rate of 90%.2
Procedural Coding—Specific Current Procedural Terminology codes (11450-11471) from the International Classification of Diseases, Tenth Revision, exist for HS deroofing procedures; the applicable code for a given case depends on the final length of the surgical defect. Documentation to support these codes is similar to the note for an excision procedure, taking care to include location, depth, and length of the excision; healing by secondary intention; and the diagnosis of HS.
Final Thoughts
Deroofing is a surgical option that can be beneficial to patients with HS. It is a relatively simple procedure available to any dermatologist, regardless of setting. We encourage dermatologists to consider deroofing, even in patients with Hurley stage II lesions, because it can yield cosmetically acceptable and definitive results, given the variety of therapies available for HS. Deroofing also can be superior to standard excision, especially because of the potential complications with standard excision and quicker operative time with deroofing. As more providers become familiar with the deroofing procedure for HS, further studies can be undertaken to add to the paucity of data about deroofing and how it compares to other surgical treatments.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j.jaad.2019.02.067
- van der Zee HH, Prens EP, Boer J. Deroofing: a tissue-saving surgical technique for the treatment of mild to moderate hidradenitis suppurativa lesions. J Am Acad Dermatol. 2010;63:475-480. doi:10.1016/j.jaad.2009.12.018
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115. doi:10.2147/CCID.S111019
- Danby FW. Commentary: unroofing for hidradenitis suppurativa, why and how. J Am Acad Dermatol. 2010;63:481.e1-481.e3. doi:10.1016/j.jaad.2010.01.033
- Saylor DK, Brownstone ND, Naik HB. Office-based surgical intervention for hidradenitis suppurativa (HS): a focused review for dermatologists. Dermatol Ther (Heidelb). 2020;10:529-549. doi:10.1007/s13555-020-00391-x
Practice Gap
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterized by inflammatory nodules, abscesses, sinus tracts, fistulae, and scarring, mainly in intertriginous areas. The extent of disease—classified using the Hurley staging system (stages I–III)—helps guide treatment, which includes medical management and surgical intervention in later stages.
First-line treatment of HS includes topical or systemic medications, or both. Surgical therapy typically is reserved for refractory HS in moderate to severe disease (Hurley stages II and III) and is combined with pharmacotherapy. Specifically, clinical management guidelines issued by an expert committee of the United States and Canadian Hidradenitis Suppurativa Foundations recommend excision or deroofing for recurrent nodules and tunnels.1
Surgical options for HS that are available to the outpatient dermatologist include incision and drainage, electrosurgery, CO2 laser evaporation, excision, and deroofing (also known as unroofing).2 Deroofing is a fairly novel therapy; many dermatologists are unfamiliar with the procedure. A PubMed search of articles indexed for MEDLINE related to HS prior to 2010 revealed only 1 article containing the word deroofing and only 4 articles containing unroofing.
The pathophysiology of HS has important implications for successful treatment. Inflammation of the follicular pilosebaceous unit along with follicular occlusion create challenges with treatment.3 It is postulated that a defect in the glassy membrane of the infra-infundibular wall predisposes the pilosebaceous follicle to lose its structural integrality as pressure builds from plugging of the duct,4 which can result in the clinical hallmarks of HS including tunneling tracts, bridging nodules, abscesses, and fistulae that form with lateral expansion of the plugged follicle.
Leaking of the contents of these plugged follicles into surrounding tissue produces an inflammatory response in characteristic HS lesions. Because debris within the lesions moves laterally instead of being able to burst to the surface, the lesions have difficulty fully healing. Unroofing the lesions and removing built-up debris allows them to heal more expediently and quiets the underlying immune response by removing the stimulus.4
Herein, we describe the benefits, risks, and surgical process of deroofing for HS.
Technique and Tools
Deroofing is performed under local anesthesia, stepwise as follows:
1. Identify sinus tracts and infiltrate the area with lidocaine (Figure, A).
2. Use a blunt probe to define the borders of the area to be unroofed and to evaluate for any communicating sinus tracts (Figure, B).
3. Remove the roof of underlying abscesses and tracts, using a probe as a guide (Figure, C).
4. Enter through the skin or sinus opening using electrocautery or with a scalpel or scissors; perform blunt dissection.
5. Reflect back the entirety of skin overlying the probed areas and remove the skin to expose the base of the lesion (Figure, D).
6. Explore the exposed base and walls of the lesion with the probe again to assess for hidden tracts; take care not to create false tracts.
7. Debride the surgical wound using curettage or rough gauze grattage to remove remaining inflammatory debris or biofilm. To achieve hemostasis, apply aluminum chloride or ferric chloride. Coat the wound with petroleum jelly and gauze and allow it to heal by secondary intention.
8. Educate the patient on wound care—once-daily gentle cleansing with soap and water, followed by application of a moist dressing—which is similar to wound healing by secondary intention from other causes.2,4
Practice Implications
A deroofing procedure has many benefits compared to other surgical modalities for the treatment of HS. Deroofing requires only a probe, curette, and electrocautery device, making the procedure more cost-effective than excision, which requires a full tray of equipment and sutures. Furthermore, margins do not need to be taken with deroofing, and no undermining or closure is needed, which saves time during the operation and minimizes the risk for complications, including dehiscence and formation of new sinus tracts.4 No specialized equipment, such as a CO2 laser, is required, which makes deroofing accessible to every clinical dermatologist in any demographic or geographic setting.
Evidence of Benefit—Saylor and colleagues5 found that deroofing carries a 12.5% complication rate, which includes postoperative bleeding, hypergranulation tissue, and rarely wound infection. This rate is significantly lower than the 26% complication rate associated with local excision, which includes wound dehiscence, infection, and contracture (P<.001). Deroofing also was found to have an HS recurrence rate of 14.5%, which is significantly less than the 30% recurrence rate seen with local excision (P=.015). Saylor et al5 also concluded that incision and drainage was recommended only for immediate relief of HS because of its 100% recurrence rate.
van der Zee2 reported on 88 lesions from 44 patients that were treated by surgical deroofing, resulting in an average defect of 3.0 cm in length and a mean healing time of 14 days. The typical outcome was cosmetically acceptable scarring; this finding was supported by a postoperative survey (>1 year), to which 37 of 44 patients responded and assigned an average satisfaction score of 8 (of a possible 10) and a recommendation rate of 90%.2
Procedural Coding—Specific Current Procedural Terminology codes (11450-11471) from the International Classification of Diseases, Tenth Revision, exist for HS deroofing procedures; the applicable code for a given case depends on the final length of the surgical defect. Documentation to support these codes is similar to the note for an excision procedure, taking care to include location, depth, and length of the excision; healing by secondary intention; and the diagnosis of HS.
Final Thoughts
Deroofing is a surgical option that can be beneficial to patients with HS. It is a relatively simple procedure available to any dermatologist, regardless of setting. We encourage dermatologists to consider deroofing, even in patients with Hurley stage II lesions, because it can yield cosmetically acceptable and definitive results, given the variety of therapies available for HS. Deroofing also can be superior to standard excision, especially because of the potential complications with standard excision and quicker operative time with deroofing. As more providers become familiar with the deroofing procedure for HS, further studies can be undertaken to add to the paucity of data about deroofing and how it compares to other surgical treatments.
Practice Gap
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterized by inflammatory nodules, abscesses, sinus tracts, fistulae, and scarring, mainly in intertriginous areas. The extent of disease—classified using the Hurley staging system (stages I–III)—helps guide treatment, which includes medical management and surgical intervention in later stages.
First-line treatment of HS includes topical or systemic medications, or both. Surgical therapy typically is reserved for refractory HS in moderate to severe disease (Hurley stages II and III) and is combined with pharmacotherapy. Specifically, clinical management guidelines issued by an expert committee of the United States and Canadian Hidradenitis Suppurativa Foundations recommend excision or deroofing for recurrent nodules and tunnels.1
Surgical options for HS that are available to the outpatient dermatologist include incision and drainage, electrosurgery, CO2 laser evaporation, excision, and deroofing (also known as unroofing).2 Deroofing is a fairly novel therapy; many dermatologists are unfamiliar with the procedure. A PubMed search of articles indexed for MEDLINE related to HS prior to 2010 revealed only 1 article containing the word deroofing and only 4 articles containing unroofing.
The pathophysiology of HS has important implications for successful treatment. Inflammation of the follicular pilosebaceous unit along with follicular occlusion create challenges with treatment.3 It is postulated that a defect in the glassy membrane of the infra-infundibular wall predisposes the pilosebaceous follicle to lose its structural integrality as pressure builds from plugging of the duct,4 which can result in the clinical hallmarks of HS including tunneling tracts, bridging nodules, abscesses, and fistulae that form with lateral expansion of the plugged follicle.
Leaking of the contents of these plugged follicles into surrounding tissue produces an inflammatory response in characteristic HS lesions. Because debris within the lesions moves laterally instead of being able to burst to the surface, the lesions have difficulty fully healing. Unroofing the lesions and removing built-up debris allows them to heal more expediently and quiets the underlying immune response by removing the stimulus.4
Herein, we describe the benefits, risks, and surgical process of deroofing for HS.
Technique and Tools
Deroofing is performed under local anesthesia, stepwise as follows:
1. Identify sinus tracts and infiltrate the area with lidocaine (Figure, A).
2. Use a blunt probe to define the borders of the area to be unroofed and to evaluate for any communicating sinus tracts (Figure, B).
3. Remove the roof of underlying abscesses and tracts, using a probe as a guide (Figure, C).
4. Enter through the skin or sinus opening using electrocautery or with a scalpel or scissors; perform blunt dissection.
5. Reflect back the entirety of skin overlying the probed areas and remove the skin to expose the base of the lesion (Figure, D).
6. Explore the exposed base and walls of the lesion with the probe again to assess for hidden tracts; take care not to create false tracts.
7. Debride the surgical wound using curettage or rough gauze grattage to remove remaining inflammatory debris or biofilm. To achieve hemostasis, apply aluminum chloride or ferric chloride. Coat the wound with petroleum jelly and gauze and allow it to heal by secondary intention.
8. Educate the patient on wound care—once-daily gentle cleansing with soap and water, followed by application of a moist dressing—which is similar to wound healing by secondary intention from other causes.2,4
Practice Implications
A deroofing procedure has many benefits compared to other surgical modalities for the treatment of HS. Deroofing requires only a probe, curette, and electrocautery device, making the procedure more cost-effective than excision, which requires a full tray of equipment and sutures. Furthermore, margins do not need to be taken with deroofing, and no undermining or closure is needed, which saves time during the operation and minimizes the risk for complications, including dehiscence and formation of new sinus tracts.4 No specialized equipment, such as a CO2 laser, is required, which makes deroofing accessible to every clinical dermatologist in any demographic or geographic setting.
Evidence of Benefit—Saylor and colleagues5 found that deroofing carries a 12.5% complication rate, which includes postoperative bleeding, hypergranulation tissue, and rarely wound infection. This rate is significantly lower than the 26% complication rate associated with local excision, which includes wound dehiscence, infection, and contracture (P<.001). Deroofing also was found to have an HS recurrence rate of 14.5%, which is significantly less than the 30% recurrence rate seen with local excision (P=.015). Saylor et al5 also concluded that incision and drainage was recommended only for immediate relief of HS because of its 100% recurrence rate.
van der Zee2 reported on 88 lesions from 44 patients that were treated by surgical deroofing, resulting in an average defect of 3.0 cm in length and a mean healing time of 14 days. The typical outcome was cosmetically acceptable scarring; this finding was supported by a postoperative survey (>1 year), to which 37 of 44 patients responded and assigned an average satisfaction score of 8 (of a possible 10) and a recommendation rate of 90%.2
Procedural Coding—Specific Current Procedural Terminology codes (11450-11471) from the International Classification of Diseases, Tenth Revision, exist for HS deroofing procedures; the applicable code for a given case depends on the final length of the surgical defect. Documentation to support these codes is similar to the note for an excision procedure, taking care to include location, depth, and length of the excision; healing by secondary intention; and the diagnosis of HS.
Final Thoughts
Deroofing is a surgical option that can be beneficial to patients with HS. It is a relatively simple procedure available to any dermatologist, regardless of setting. We encourage dermatologists to consider deroofing, even in patients with Hurley stage II lesions, because it can yield cosmetically acceptable and definitive results, given the variety of therapies available for HS. Deroofing also can be superior to standard excision, especially because of the potential complications with standard excision and quicker operative time with deroofing. As more providers become familiar with the deroofing procedure for HS, further studies can be undertaken to add to the paucity of data about deroofing and how it compares to other surgical treatments.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j.jaad.2019.02.067
- van der Zee HH, Prens EP, Boer J. Deroofing: a tissue-saving surgical technique for the treatment of mild to moderate hidradenitis suppurativa lesions. J Am Acad Dermatol. 2010;63:475-480. doi:10.1016/j.jaad.2009.12.018
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115. doi:10.2147/CCID.S111019
- Danby FW. Commentary: unroofing for hidradenitis suppurativa, why and how. J Am Acad Dermatol. 2010;63:481.e1-481.e3. doi:10.1016/j.jaad.2010.01.033
- Saylor DK, Brownstone ND, Naik HB. Office-based surgical intervention for hidradenitis suppurativa (HS): a focused review for dermatologists. Dermatol Ther (Heidelb). 2020;10:529-549. doi:10.1007/s13555-020-00391-x
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90. doi:10.1016/j.jaad.2019.02.067
- van der Zee HH, Prens EP, Boer J. Deroofing: a tissue-saving surgical technique for the treatment of mild to moderate hidradenitis suppurativa lesions. J Am Acad Dermatol. 2010;63:475-480. doi:10.1016/j.jaad.2009.12.018
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115. doi:10.2147/CCID.S111019
- Danby FW. Commentary: unroofing for hidradenitis suppurativa, why and how. J Am Acad Dermatol. 2010;63:481.e1-481.e3. doi:10.1016/j.jaad.2010.01.033
- Saylor DK, Brownstone ND, Naik HB. Office-based surgical intervention for hidradenitis suppurativa (HS): a focused review for dermatologists. Dermatol Ther (Heidelb). 2020;10:529-549. doi:10.1007/s13555-020-00391-x

Inhaled, systemic steroids linked to changes in brain structure
New research links the use of glucocorticoids with changes in white matter microstructure – which may explain the development of anxiety, depression, and other neuropsychiatric side effects related to these drugs, investigators say.
Results from a cross-sectional study showed that use of both systemic and inhaled glucocorticoids was associated with widespread reductions in fractional anisotropy (FA) and increases in mean diffusivity.
Glucocorticoids have “a whole catalogue” of adverse events, and effects on brain structure “adds to the list,” co-investigator Onno C. Meijer, PhD, professor of molecular neuroendocrinology of corticosteroids, department of medicine, Leiden University Medical Center, the Netherlands, told this news organization.

The findings should encourage clinicians to consider whether doses they are prescribing are too high, said Dr. Meijer. He added that the negative effect of glucocorticoids on the brain was also found in those using inhalers, such as patients with asthma.
The findings were published online in the BMJ Open.
Serious side effects
Glucocorticoids, a class of synthetic steroids with immunosuppressive properties, are prescribed for a wide range of conditions, including rheumatoid arthritis and asthma.
However, they are also associated with potentially serious metabolic, cardiovascular, and musculoskeletal side effects as well as neuropsychiatric side effects such as depression, mania, and cognitive impairment.
About 1 in 3 patients exposed to “quite a lot of these drugs” will experience neuropsychiatric symptoms, Dr. Meijer said.
Most previous studies that investigated effects from high levels of glucocorticoids on brain structure have been small and involved selected populations, such as those with Cushing disease.
The new study included participants from the UK Biobank, a large population-based cohort. Participants had undergone imaging and did not have a history of psychiatric disease – although they could have conditions associated with glucocorticoid use, including anxiety, depression, mania, or delirium.
The analysis included 222 patients using oral or parenteral glucocorticoids at the time of imaging (systemic group), 557 using inhaled glucocorticoids, and 24,106 not using glucocorticoids (the control group).
Inhaled steroids target the lungs, whereas a steroid in pill form “travels in the blood and reaches each and every organ and cell in the body and typically requires higher doses,” Dr. Meijer noted.
The groups were similar with respect to sex, education, and smoking status. However, the systemic glucocorticoid group was slightly older (mean age, 66.1 years vs. 63.3 years for inhaled glucocorticoid users and 63.5 years for the control group).
In addition to age, researchers adjusted for sex, education level, head position in the scanner, head size, assessment center, and year of imaging.
Imaging analyses
Imaging analyses showed systemic glucocorticoid use was associated with reduced global FA (adjusted mean difference, -3.7e-3; 95% confidence interval, -6.4e-3 to 1.0e-3), and reductions in regional FA in the body and genu of the corpus callosum versus the control group.
Inhaled glucocorticoid use was associated with reduced global FA (AMD, -2.3e-3; 95% CI, -4.0e-3 to -5.7e-4), and lower FA in the splenium of the corpus callosum and the cingulum of the hippocampus.
Global mean diffusivity was higher in systemic glucocorticoid users (AMD, 7.2e-6; 95% CI, 3.2e-6 to 1.1e-5) and inhaled glucocorticoid users (AMD, 2.7e-6; 95% CI, 1.7e-7 to 5.2e-6), compared with the control group.
The effects of glucocorticoids on white matter were “pervasive,” and the “most important finding” of the study, Dr. Meijer said. “We were impressed by the fact white matter is so sensitive to these drugs.”
He noted that it is likely that functional connectivity between brain regions is affected by use of glucocorticoids. “You could say communication between brain regions is probably somewhat impaired or challenged,” he said.
Subgroup analyses among participants using glucocorticoids chronically, defined as reported at two consecutive visits, suggested a potential dose-dependent or duration-dependent effect of glucocorticoids on white matter microstructure.
Systemic glucocorticoid use was also associated with an increase in total and grey matter volume of the caudate nucleus.
In addition, there was a significant association between inhaled glucocorticoid use and decreased grey matter volume of the amygdala, which Dr. Meijer said was surprising because studies have shown that glucocorticoids “can drive amygdala big time.”
Move away from ‘one dose for all’?
Another surprise was that the results showed no hippocampal volume differences with steroid use, Dr. Meijer noted.
The modest association between glucocorticoid use and brain volumes could indicate that white matter integrity is more sensitive to glucocorticoids than is grey matter volume, “at least at the structural level,” he said.
He added that longer use or higher doses may be necessary to also induce volumetric changes.
Participants also completed a questionnaire to assess mood over the previous 2 weeks. Systemic glucocorticoid users had more depressive symptoms, disinterest, tenseness/restlessness, and tiredness/lethargy, compared with the control group. Inhaled glucocorticoid users only reported more tiredness/lethargy.
The investigators note that mood-related effects could be linked to the condition for which glucocorticoids were prescribed: for example, rheumatoid arthritis or chronic obstructive pulmonary disease.
In terms of cognition, systemic glucocorticoid users performed significantly worse on the symbol digit substitution task, compared with participants in the control group.
In light of these findings, pharmaceutical companies that make inhaled corticosteroids “should perhaps find out if glucocorticoids can be dosed by kilogram body weight rather than simply one dose fits all,” which is currently the case, Dr. Meijer said.
Impressive, but several limitations
Commenting on the findings, E. Sherwood Brown, MD, PhD, Distinguished Chair in Psychiatric Research and professor and vice chair for clinical research, department of psychiatry, The University of Texas Southwestern Medical Center, Dallas, called the study sample size “impressive.”
In addition, the study is the first to look at systemic as well as inhaled corticosteroids, said Dr. Brown, who was not involved with the research. He noted that previously, there had been only case reports of psychiatric symptoms with inhaled corticosteroids.
That results are in the same direction but greater with systemic, compared with inhaled corticosteroids, is “particularly interesting” because this might suggest dose-dependent effects, Dr. Brown said.
He noted that cognitive differences were also only observed with systemic corticosteroids.
Some study observations, such as smaller amygdala volume with inhaled but not systemic corticosteroids, “are harder to understand,” said Dr. Brown.
However, he pointed out some study limitations. For example, data were apparently unavailable for verbal and declarative memory test data, despite corticosteroids probably affecting the hippocampus and causing memory changes.
Other drawbacks were that the dose and duration of corticosteroid use, as well as the medical histories of study participants, were not available, Dr. Brown said.
No study funding was reported. Dr. Meijer has received research grants and honorariums from Corcept Therapeutics and a speakers’ fee from Ipsen. Dr. Brown is on an advisory board for Sage Pharmaceuticals, which is developing neurosteroids (not corticosteroids) for mood disorders. He is also on a Medscape advisory board related to bipolar disorder.
A version of this article first appeared on Medscape.com.
New research links the use of glucocorticoids with changes in white matter microstructure – which may explain the development of anxiety, depression, and other neuropsychiatric side effects related to these drugs, investigators say.
Results from a cross-sectional study showed that use of both systemic and inhaled glucocorticoids was associated with widespread reductions in fractional anisotropy (FA) and increases in mean diffusivity.
Glucocorticoids have “a whole catalogue” of adverse events, and effects on brain structure “adds to the list,” co-investigator Onno C. Meijer, PhD, professor of molecular neuroendocrinology of corticosteroids, department of medicine, Leiden University Medical Center, the Netherlands, told this news organization.
The findings should encourage clinicians to consider whether doses they are prescribing are too high, said Dr. Meijer. He added that the negative effect of glucocorticoids on the brain was also found in those using inhalers, such as patients with asthma.
The findings were published online in the BMJ Open.
Serious side effects
Glucocorticoids, a class of synthetic steroids with immunosuppressive properties, are prescribed for a wide range of conditions, including rheumatoid arthritis and asthma.
However, they are also associated with potentially serious metabolic, cardiovascular, and musculoskeletal side effects as well as neuropsychiatric side effects such as depression, mania, and cognitive impairment.
About 1 in 3 patients exposed to “quite a lot of these drugs” will experience neuropsychiatric symptoms, Dr. Meijer said.
Most previous studies that investigated effects from high levels of glucocorticoids on brain structure have been small and involved selected populations, such as those with Cushing disease.
The new study included participants from the UK Biobank, a large population-based cohort. Participants had undergone imaging and did not have a history of psychiatric disease – although they could have conditions associated with glucocorticoid use, including anxiety, depression, mania, or delirium.
The analysis included 222 patients using oral or parenteral glucocorticoids at the time of imaging (systemic group), 557 using inhaled glucocorticoids, and 24,106 not using glucocorticoids (the control group).
Inhaled steroids target the lungs, whereas a steroid in pill form “travels in the blood and reaches each and every organ and cell in the body and typically requires higher doses,” Dr. Meijer noted.
The groups were similar with respect to sex, education, and smoking status. However, the systemic glucocorticoid group was slightly older (mean age, 66.1 years vs. 63.3 years for inhaled glucocorticoid users and 63.5 years for the control group).
In addition to age, researchers adjusted for sex, education level, head position in the scanner, head size, assessment center, and year of imaging.
Imaging analyses
Imaging analyses showed systemic glucocorticoid use was associated with reduced global FA (adjusted mean difference, -3.7e-3; 95% confidence interval, -6.4e-3 to 1.0e-3), and reductions in regional FA in the body and genu of the corpus callosum versus the control group.
Inhaled glucocorticoid use was associated with reduced global FA (AMD, -2.3e-3; 95% CI, -4.0e-3 to -5.7e-4), and lower FA in the splenium of the corpus callosum and the cingulum of the hippocampus.
Global mean diffusivity was higher in systemic glucocorticoid users (AMD, 7.2e-6; 95% CI, 3.2e-6 to 1.1e-5) and inhaled glucocorticoid users (AMD, 2.7e-6; 95% CI, 1.7e-7 to 5.2e-6), compared with the control group.
The effects of glucocorticoids on white matter were “pervasive,” and the “most important finding” of the study, Dr. Meijer said. “We were impressed by the fact white matter is so sensitive to these drugs.”
He noted that it is likely that functional connectivity between brain regions is affected by use of glucocorticoids. “You could say communication between brain regions is probably somewhat impaired or challenged,” he said.
Subgroup analyses among participants using glucocorticoids chronically, defined as reported at two consecutive visits, suggested a potential dose-dependent or duration-dependent effect of glucocorticoids on white matter microstructure.
Systemic glucocorticoid use was also associated with an increase in total and grey matter volume of the caudate nucleus.
In addition, there was a significant association between inhaled glucocorticoid use and decreased grey matter volume of the amygdala, which Dr. Meijer said was surprising because studies have shown that glucocorticoids “can drive amygdala big time.”
Move away from ‘one dose for all’?
Another surprise was that the results showed no hippocampal volume differences with steroid use, Dr. Meijer noted.
The modest association between glucocorticoid use and brain volumes could indicate that white matter integrity is more sensitive to glucocorticoids than is grey matter volume, “at least at the structural level,” he said.
He added that longer use or higher doses may be necessary to also induce volumetric changes.
Participants also completed a questionnaire to assess mood over the previous 2 weeks. Systemic glucocorticoid users had more depressive symptoms, disinterest, tenseness/restlessness, and tiredness/lethargy, compared with the control group. Inhaled glucocorticoid users only reported more tiredness/lethargy.
The investigators note that mood-related effects could be linked to the condition for which glucocorticoids were prescribed: for example, rheumatoid arthritis or chronic obstructive pulmonary disease.
In terms of cognition, systemic glucocorticoid users performed significantly worse on the symbol digit substitution task, compared with participants in the control group.
In light of these findings, pharmaceutical companies that make inhaled corticosteroids “should perhaps find out if glucocorticoids can be dosed by kilogram body weight rather than simply one dose fits all,” which is currently the case, Dr. Meijer said.
Impressive, but several limitations
Commenting on the findings, E. Sherwood Brown, MD, PhD, Distinguished Chair in Psychiatric Research and professor and vice chair for clinical research, department of psychiatry, The University of Texas Southwestern Medical Center, Dallas, called the study sample size “impressive.”
In addition, the study is the first to look at systemic as well as inhaled corticosteroids, said Dr. Brown, who was not involved with the research. He noted that previously, there had been only case reports of psychiatric symptoms with inhaled corticosteroids.
That results are in the same direction but greater with systemic, compared with inhaled corticosteroids, is “particularly interesting” because this might suggest dose-dependent effects, Dr. Brown said.
He noted that cognitive differences were also only observed with systemic corticosteroids.
Some study observations, such as smaller amygdala volume with inhaled but not systemic corticosteroids, “are harder to understand,” said Dr. Brown.
However, he pointed out some study limitations. For example, data were apparently unavailable for verbal and declarative memory test data, despite corticosteroids probably affecting the hippocampus and causing memory changes.
Other drawbacks were that the dose and duration of corticosteroid use, as well as the medical histories of study participants, were not available, Dr. Brown said.
No study funding was reported. Dr. Meijer has received research grants and honorariums from Corcept Therapeutics and a speakers’ fee from Ipsen. Dr. Brown is on an advisory board for Sage Pharmaceuticals, which is developing neurosteroids (not corticosteroids) for mood disorders. He is also on a Medscape advisory board related to bipolar disorder.
A version of this article first appeared on Medscape.com.
New research links the use of glucocorticoids with changes in white matter microstructure – which may explain the development of anxiety, depression, and other neuropsychiatric side effects related to these drugs, investigators say.
Results from a cross-sectional study showed that use of both systemic and inhaled glucocorticoids was associated with widespread reductions in fractional anisotropy (FA) and increases in mean diffusivity.
Glucocorticoids have “a whole catalogue” of adverse events, and effects on brain structure “adds to the list,” co-investigator Onno C. Meijer, PhD, professor of molecular neuroendocrinology of corticosteroids, department of medicine, Leiden University Medical Center, the Netherlands, told this news organization.
The findings should encourage clinicians to consider whether doses they are prescribing are too high, said Dr. Meijer. He added that the negative effect of glucocorticoids on the brain was also found in those using inhalers, such as patients with asthma.
The findings were published online in the BMJ Open.
Serious side effects
Glucocorticoids, a class of synthetic steroids with immunosuppressive properties, are prescribed for a wide range of conditions, including rheumatoid arthritis and asthma.
However, they are also associated with potentially serious metabolic, cardiovascular, and musculoskeletal side effects as well as neuropsychiatric side effects such as depression, mania, and cognitive impairment.
About 1 in 3 patients exposed to “quite a lot of these drugs” will experience neuropsychiatric symptoms, Dr. Meijer said.
Most previous studies that investigated effects from high levels of glucocorticoids on brain structure have been small and involved selected populations, such as those with Cushing disease.
The new study included participants from the UK Biobank, a large population-based cohort. Participants had undergone imaging and did not have a history of psychiatric disease – although they could have conditions associated with glucocorticoid use, including anxiety, depression, mania, or delirium.
The analysis included 222 patients using oral or parenteral glucocorticoids at the time of imaging (systemic group), 557 using inhaled glucocorticoids, and 24,106 not using glucocorticoids (the control group).
Inhaled steroids target the lungs, whereas a steroid in pill form “travels in the blood and reaches each and every organ and cell in the body and typically requires higher doses,” Dr. Meijer noted.
The groups were similar with respect to sex, education, and smoking status. However, the systemic glucocorticoid group was slightly older (mean age, 66.1 years vs. 63.3 years for inhaled glucocorticoid users and 63.5 years for the control group).
In addition to age, researchers adjusted for sex, education level, head position in the scanner, head size, assessment center, and year of imaging.
Imaging analyses
Imaging analyses showed systemic glucocorticoid use was associated with reduced global FA (adjusted mean difference, -3.7e-3; 95% confidence interval, -6.4e-3 to 1.0e-3), and reductions in regional FA in the body and genu of the corpus callosum versus the control group.
Inhaled glucocorticoid use was associated with reduced global FA (AMD, -2.3e-3; 95% CI, -4.0e-3 to -5.7e-4), and lower FA in the splenium of the corpus callosum and the cingulum of the hippocampus.
Global mean diffusivity was higher in systemic glucocorticoid users (AMD, 7.2e-6; 95% CI, 3.2e-6 to 1.1e-5) and inhaled glucocorticoid users (AMD, 2.7e-6; 95% CI, 1.7e-7 to 5.2e-6), compared with the control group.
The effects of glucocorticoids on white matter were “pervasive,” and the “most important finding” of the study, Dr. Meijer said. “We were impressed by the fact white matter is so sensitive to these drugs.”
He noted that it is likely that functional connectivity between brain regions is affected by use of glucocorticoids. “You could say communication between brain regions is probably somewhat impaired or challenged,” he said.
Subgroup analyses among participants using glucocorticoids chronically, defined as reported at two consecutive visits, suggested a potential dose-dependent or duration-dependent effect of glucocorticoids on white matter microstructure.
Systemic glucocorticoid use was also associated with an increase in total and grey matter volume of the caudate nucleus.
In addition, there was a significant association between inhaled glucocorticoid use and decreased grey matter volume of the amygdala, which Dr. Meijer said was surprising because studies have shown that glucocorticoids “can drive amygdala big time.”
Move away from ‘one dose for all’?
Another surprise was that the results showed no hippocampal volume differences with steroid use, Dr. Meijer noted.
The modest association between glucocorticoid use and brain volumes could indicate that white matter integrity is more sensitive to glucocorticoids than is grey matter volume, “at least at the structural level,” he said.
He added that longer use or higher doses may be necessary to also induce volumetric changes.
Participants also completed a questionnaire to assess mood over the previous 2 weeks. Systemic glucocorticoid users had more depressive symptoms, disinterest, tenseness/restlessness, and tiredness/lethargy, compared with the control group. Inhaled glucocorticoid users only reported more tiredness/lethargy.
The investigators note that mood-related effects could be linked to the condition for which glucocorticoids were prescribed: for example, rheumatoid arthritis or chronic obstructive pulmonary disease.
In terms of cognition, systemic glucocorticoid users performed significantly worse on the symbol digit substitution task, compared with participants in the control group.
In light of these findings, pharmaceutical companies that make inhaled corticosteroids “should perhaps find out if glucocorticoids can be dosed by kilogram body weight rather than simply one dose fits all,” which is currently the case, Dr. Meijer said.
Impressive, but several limitations
Commenting on the findings, E. Sherwood Brown, MD, PhD, Distinguished Chair in Psychiatric Research and professor and vice chair for clinical research, department of psychiatry, The University of Texas Southwestern Medical Center, Dallas, called the study sample size “impressive.”
In addition, the study is the first to look at systemic as well as inhaled corticosteroids, said Dr. Brown, who was not involved with the research. He noted that previously, there had been only case reports of psychiatric symptoms with inhaled corticosteroids.
That results are in the same direction but greater with systemic, compared with inhaled corticosteroids, is “particularly interesting” because this might suggest dose-dependent effects, Dr. Brown said.
He noted that cognitive differences were also only observed with systemic corticosteroids.
Some study observations, such as smaller amygdala volume with inhaled but not systemic corticosteroids, “are harder to understand,” said Dr. Brown.
However, he pointed out some study limitations. For example, data were apparently unavailable for verbal and declarative memory test data, despite corticosteroids probably affecting the hippocampus and causing memory changes.
Other drawbacks were that the dose and duration of corticosteroid use, as well as the medical histories of study participants, were not available, Dr. Brown said.
No study funding was reported. Dr. Meijer has received research grants and honorariums from Corcept Therapeutics and a speakers’ fee from Ipsen. Dr. Brown is on an advisory board for Sage Pharmaceuticals, which is developing neurosteroids (not corticosteroids) for mood disorders. He is also on a Medscape advisory board related to bipolar disorder.
A version of this article first appeared on Medscape.com.
FROM BMJ OPEN
Anti-BDCA2 antibody meets primary endpoint in phase 2 cutaneous lupus trial
Treatment with the humanized monoclonal antibody litifilimab improved scores on a validated measure of skin disease activity in an international phase 2 trial of patients with cutaneous lupus erythematosus (CLE).
Improvements in Cutaneous Lupus Erythematosus Disease Area and Severity Index–Activity (CLASI-A) scores in patients randomly assigned to receive subcutaneous litifilimab were superior to changes in patients randomly assigned to placebo over the trial period of 16 weeks. The double-blind study was published in the New England Journal of Medicine.

“This validated measure is working, and it’s very important to now go into phase 3 using the instrument that worked in phase 2 to measure improvement in the skin,” Victoria P. Werth, MD, professor of dermatology at the University of Pennsylvania, Philadelphia, and lead author of the study, said in an interview.
Research on lupus erythematosus has focused on systemic lupus erythematosus (SLE), with few randomized controlled trials addressing CLE, she said, and no Food and Drug Administration–approved treatments for CLE in the last 50 years.
Asked to comment on the results, Alisa Femia, MD, associate professor and director of autoimmune connective tissue disease in the department of dermatology at New York University, who was not involved in the research, said it is “exciting to have a trial that specifically investigates the effect of a drug on cutaneous lupus, as well-designed investigations into this potentially disfiguring disease are relatively sparse and novel treatment pathways are needed.”
The investigational drug targets blood dendritic cell antigen 2 (BDCA2) – a receptor expressed solely on the surface of plasmacytoid dendritic cells (pDCs) – and inhibits the production of type 1 interferon and other inflammatory cytokines and chemokines believed to play a major role in the pathogenesis of cutaneous and systemic lupus, the investigators said.

Rheumatologist Edward Vital, MD, who leads a lupus research group at the University of Leeds (England), said he’s most interested in how the therapy works. The “idea [has been] that pDCs are the main source of type 1 interferon. But there’s a lot of data emerging at present that suggests there are many other sources of interferons, and the drug may work in other ways,” Dr. Vital, an associate professor at the university, said in an interview. He was not involved with the study.
“Maybe pDCs have other important roles. Or maybe other cells are targeted by the therapy, too,” he said. “Understanding this will help us understand the pathogenesis of lupus and which patients will benefit the most.”
Improvements in CLASI-A scores
Across 54 centers, the study enrolled 132 patients with primarily moderate to severe active subacute CLE or chronic CLE (including discoid lupus erythematosus), or both subacute and chronic CLE with or without systemic manifestations. Active CLE was defined as a score of at least 8 on CLASI-A, which measures erythema and scaling or hypertrophy in 13 skin regions.
Patients were randomly assigned to receive placebo or litifilimab at doses of 50 mg, 150 mg, or 450 mg subcutaneously at weeks 0, 2, 4, 8, and 12. Mean CLASI-A scores at baseline for placebo and each of the dosage groups were 16.5, 15.2, 18.4, and 16.5, respectively.
The investigators used a test of dose-response to assess response across the four groups on the basis of the percent change in CLASI-A scores from baseline to 16 weeks, the primary endpoint. The percent changes in CLASI-A score were –38.8 ± 7.5 in the 50-mg group; –47.9 ± 7.5 in the 150-mg group; –42.5 ± 5.5 in the 450-mg group; and –14.5 ± 6.4 in the placebo group. (Negative value indicates improvement from baseline.)
When compared with placebo, the change in CLASI-A scores in each of the litifilimab groups was –24.3 percentage points for the 50-mg dose (95% confidence interval, –43.7 to –4.9); –33.4 percentage points for the 150-mg dose (95% CI, –52.7 to –14.1); and –28.0 percentage points for the 450-mg dose (95% CI, –44.6 to –11.4).
“All three dosages caused a similar skin response,” said Dr. Werth. “And importantly, the placebo response is fairly low, much lower than in SLE trials, possibly because the background therapies tend to be less overall [including with slightly lower doses of prednisone]. So we can really see the broad effect of the drug.”
Just under half of participants – 42%-48% of patients receiving litifilimab and 42% of those in the placebo group – had concomitant SLE with low to moderate disease activity as measured by the Systemic Lupus Erythematosus Disease Activity Index 2000. Patients could meet SLE criteria based on previous findings, and “didn’t have to have active SLE,” Dr. Werth noted.
The trial allowed background therapy as long as treatment had begun at least 12 weeks before randomization, with a stable dose starting at least 4 weeks before randomization and maintained throughout the trial period.
Most patients had moderate to severe CLE at baseline “despite approximately 90% having received concomitant background therapy and 80% of those participants having received antimalarial drugs, either alone or with other agents,” Dr. Werth and coinvestigators wrote.
CLASI-A has been shown to correlate to patients’ quality of life, Dr. Werth emphasized in the interview.
Most of the reported side effects in the phase 2 CLE trial were mild or moderate. The treatment was associated with three cases of hypersensitivity, three cases of oral herpes infection, and one case of herpes zoster infection. One case of herpes zoster meningitis occurred 4 months after the last dose of litifilimab.
Approximately 10% of study participants who reported race and ethnicity were Black or African American.
Phase 3 trials
The trial was one part of a two-part phase 2 study of litifilimab, named the LILAC trial, sponsored by Biogen. The other part, which will be published separately, involved patients who had SLE with active joint and skin manifestations.
Biogen is currently enrolling patients in phase 3 studies – the TOPAZ-1 and TOPAZ-2 studies – to evaluate the efficacy and safety of the drug in patients with active SLE. As secondary endpoints, both trials will measure the percentage of participants with a CLASI-A score of at least 10 at baseline who achieve improvement in the score, including a 50% improvement from baseline to week 16, Nathalie Franchimont, MD, PhD, of Biogen, a coauthor of the NEJM study, said in an email.
Biogen also has “plans to initiate a pivotal study in CLE this year,” she said.

With respect to the newly published phase 2 study, Dr. Femia said that, while “conclusions about the magnitude of efficacy are difficult to extrapolate in this trial design, there’s reason for cautious optimism.” There is “good theoretical basis to be optimistic about a drug such as litifilimab, that ultimately reduces type 1 interferon response,” she added.
Anifrolumab, a type 1 interferon receptor monoclonal antibody marketed as Saphnelo, was approved by the FDA for SLE in July 2021, but CLE subtypes were not characterized in trials and CLE was not studied independently of SLE, the authors pointed out in their NEJM article.
The study was supported by Biogen. In addition to working with Biogen, Dr. Werth serves as a consultant to Gilead Sciences and other pharmaceutical companies. Dr. Vital has research grants and has received honoraria from AstraZeneca. Dr. Femia disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Treatment with the humanized monoclonal antibody litifilimab improved scores on a validated measure of skin disease activity in an international phase 2 trial of patients with cutaneous lupus erythematosus (CLE).
Improvements in Cutaneous Lupus Erythematosus Disease Area and Severity Index–Activity (CLASI-A) scores in patients randomly assigned to receive subcutaneous litifilimab were superior to changes in patients randomly assigned to placebo over the trial period of 16 weeks. The double-blind study was published in the New England Journal of Medicine.
“This validated measure is working, and it’s very important to now go into phase 3 using the instrument that worked in phase 2 to measure improvement in the skin,” Victoria P. Werth, MD, professor of dermatology at the University of Pennsylvania, Philadelphia, and lead author of the study, said in an interview.
Research on lupus erythematosus has focused on systemic lupus erythematosus (SLE), with few randomized controlled trials addressing CLE, she said, and no Food and Drug Administration–approved treatments for CLE in the last 50 years.
Asked to comment on the results, Alisa Femia, MD, associate professor and director of autoimmune connective tissue disease in the department of dermatology at New York University, who was not involved in the research, said it is “exciting to have a trial that specifically investigates the effect of a drug on cutaneous lupus, as well-designed investigations into this potentially disfiguring disease are relatively sparse and novel treatment pathways are needed.”
The investigational drug targets blood dendritic cell antigen 2 (BDCA2) – a receptor expressed solely on the surface of plasmacytoid dendritic cells (pDCs) – and inhibits the production of type 1 interferon and other inflammatory cytokines and chemokines believed to play a major role in the pathogenesis of cutaneous and systemic lupus, the investigators said.
Rheumatologist Edward Vital, MD, who leads a lupus research group at the University of Leeds (England), said he’s most interested in how the therapy works. The “idea [has been] that pDCs are the main source of type 1 interferon. But there’s a lot of data emerging at present that suggests there are many other sources of interferons, and the drug may work in other ways,” Dr. Vital, an associate professor at the university, said in an interview. He was not involved with the study.
“Maybe pDCs have other important roles. Or maybe other cells are targeted by the therapy, too,” he said. “Understanding this will help us understand the pathogenesis of lupus and which patients will benefit the most.”
Improvements in CLASI-A scores
Across 54 centers, the study enrolled 132 patients with primarily moderate to severe active subacute CLE or chronic CLE (including discoid lupus erythematosus), or both subacute and chronic CLE with or without systemic manifestations. Active CLE was defined as a score of at least 8 on CLASI-A, which measures erythema and scaling or hypertrophy in 13 skin regions.
Patients were randomly assigned to receive placebo or litifilimab at doses of 50 mg, 150 mg, or 450 mg subcutaneously at weeks 0, 2, 4, 8, and 12. Mean CLASI-A scores at baseline for placebo and each of the dosage groups were 16.5, 15.2, 18.4, and 16.5, respectively.
The investigators used a test of dose-response to assess response across the four groups on the basis of the percent change in CLASI-A scores from baseline to 16 weeks, the primary endpoint. The percent changes in CLASI-A score were –38.8 ± 7.5 in the 50-mg group; –47.9 ± 7.5 in the 150-mg group; –42.5 ± 5.5 in the 450-mg group; and –14.5 ± 6.4 in the placebo group. (Negative value indicates improvement from baseline.)
When compared with placebo, the change in CLASI-A scores in each of the litifilimab groups was –24.3 percentage points for the 50-mg dose (95% confidence interval, –43.7 to –4.9); –33.4 percentage points for the 150-mg dose (95% CI, –52.7 to –14.1); and –28.0 percentage points for the 450-mg dose (95% CI, –44.6 to –11.4).
“All three dosages caused a similar skin response,” said Dr. Werth. “And importantly, the placebo response is fairly low, much lower than in SLE trials, possibly because the background therapies tend to be less overall [including with slightly lower doses of prednisone]. So we can really see the broad effect of the drug.”
Just under half of participants – 42%-48% of patients receiving litifilimab and 42% of those in the placebo group – had concomitant SLE with low to moderate disease activity as measured by the Systemic Lupus Erythematosus Disease Activity Index 2000. Patients could meet SLE criteria based on previous findings, and “didn’t have to have active SLE,” Dr. Werth noted.
The trial allowed background therapy as long as treatment had begun at least 12 weeks before randomization, with a stable dose starting at least 4 weeks before randomization and maintained throughout the trial period.
Most patients had moderate to severe CLE at baseline “despite approximately 90% having received concomitant background therapy and 80% of those participants having received antimalarial drugs, either alone or with other agents,” Dr. Werth and coinvestigators wrote.
CLASI-A has been shown to correlate to patients’ quality of life, Dr. Werth emphasized in the interview.
Most of the reported side effects in the phase 2 CLE trial were mild or moderate. The treatment was associated with three cases of hypersensitivity, three cases of oral herpes infection, and one case of herpes zoster infection. One case of herpes zoster meningitis occurred 4 months after the last dose of litifilimab.
Approximately 10% of study participants who reported race and ethnicity were Black or African American.
Phase 3 trials
The trial was one part of a two-part phase 2 study of litifilimab, named the LILAC trial, sponsored by Biogen. The other part, which will be published separately, involved patients who had SLE with active joint and skin manifestations.
Biogen is currently enrolling patients in phase 3 studies – the TOPAZ-1 and TOPAZ-2 studies – to evaluate the efficacy and safety of the drug in patients with active SLE. As secondary endpoints, both trials will measure the percentage of participants with a CLASI-A score of at least 10 at baseline who achieve improvement in the score, including a 50% improvement from baseline to week 16, Nathalie Franchimont, MD, PhD, of Biogen, a coauthor of the NEJM study, said in an email.
Biogen also has “plans to initiate a pivotal study in CLE this year,” she said.
With respect to the newly published phase 2 study, Dr. Femia said that, while “conclusions about the magnitude of efficacy are difficult to extrapolate in this trial design, there’s reason for cautious optimism.” There is “good theoretical basis to be optimistic about a drug such as litifilimab, that ultimately reduces type 1 interferon response,” she added.
Anifrolumab, a type 1 interferon receptor monoclonal antibody marketed as Saphnelo, was approved by the FDA for SLE in July 2021, but CLE subtypes were not characterized in trials and CLE was not studied independently of SLE, the authors pointed out in their NEJM article.
The study was supported by Biogen. In addition to working with Biogen, Dr. Werth serves as a consultant to Gilead Sciences and other pharmaceutical companies. Dr. Vital has research grants and has received honoraria from AstraZeneca. Dr. Femia disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Treatment with the humanized monoclonal antibody litifilimab improved scores on a validated measure of skin disease activity in an international phase 2 trial of patients with cutaneous lupus erythematosus (CLE).
Improvements in Cutaneous Lupus Erythematosus Disease Area and Severity Index–Activity (CLASI-A) scores in patients randomly assigned to receive subcutaneous litifilimab were superior to changes in patients randomly assigned to placebo over the trial period of 16 weeks. The double-blind study was published in the New England Journal of Medicine.
“This validated measure is working, and it’s very important to now go into phase 3 using the instrument that worked in phase 2 to measure improvement in the skin,” Victoria P. Werth, MD, professor of dermatology at the University of Pennsylvania, Philadelphia, and lead author of the study, said in an interview.
Research on lupus erythematosus has focused on systemic lupus erythematosus (SLE), with few randomized controlled trials addressing CLE, she said, and no Food and Drug Administration–approved treatments for CLE in the last 50 years.
Asked to comment on the results, Alisa Femia, MD, associate professor and director of autoimmune connective tissue disease in the department of dermatology at New York University, who was not involved in the research, said it is “exciting to have a trial that specifically investigates the effect of a drug on cutaneous lupus, as well-designed investigations into this potentially disfiguring disease are relatively sparse and novel treatment pathways are needed.”
The investigational drug targets blood dendritic cell antigen 2 (BDCA2) – a receptor expressed solely on the surface of plasmacytoid dendritic cells (pDCs) – and inhibits the production of type 1 interferon and other inflammatory cytokines and chemokines believed to play a major role in the pathogenesis of cutaneous and systemic lupus, the investigators said.
Rheumatologist Edward Vital, MD, who leads a lupus research group at the University of Leeds (England), said he’s most interested in how the therapy works. The “idea [has been] that pDCs are the main source of type 1 interferon. But there’s a lot of data emerging at present that suggests there are many other sources of interferons, and the drug may work in other ways,” Dr. Vital, an associate professor at the university, said in an interview. He was not involved with the study.
“Maybe pDCs have other important roles. Or maybe other cells are targeted by the therapy, too,” he said. “Understanding this will help us understand the pathogenesis of lupus and which patients will benefit the most.”
Improvements in CLASI-A scores
Across 54 centers, the study enrolled 132 patients with primarily moderate to severe active subacute CLE or chronic CLE (including discoid lupus erythematosus), or both subacute and chronic CLE with or without systemic manifestations. Active CLE was defined as a score of at least 8 on CLASI-A, which measures erythema and scaling or hypertrophy in 13 skin regions.
Patients were randomly assigned to receive placebo or litifilimab at doses of 50 mg, 150 mg, or 450 mg subcutaneously at weeks 0, 2, 4, 8, and 12. Mean CLASI-A scores at baseline for placebo and each of the dosage groups were 16.5, 15.2, 18.4, and 16.5, respectively.
The investigators used a test of dose-response to assess response across the four groups on the basis of the percent change in CLASI-A scores from baseline to 16 weeks, the primary endpoint. The percent changes in CLASI-A score were –38.8 ± 7.5 in the 50-mg group; –47.9 ± 7.5 in the 150-mg group; –42.5 ± 5.5 in the 450-mg group; and –14.5 ± 6.4 in the placebo group. (Negative value indicates improvement from baseline.)
When compared with placebo, the change in CLASI-A scores in each of the litifilimab groups was –24.3 percentage points for the 50-mg dose (95% confidence interval, –43.7 to –4.9); –33.4 percentage points for the 150-mg dose (95% CI, –52.7 to –14.1); and –28.0 percentage points for the 450-mg dose (95% CI, –44.6 to –11.4).
“All three dosages caused a similar skin response,” said Dr. Werth. “And importantly, the placebo response is fairly low, much lower than in SLE trials, possibly because the background therapies tend to be less overall [including with slightly lower doses of prednisone]. So we can really see the broad effect of the drug.”
Just under half of participants – 42%-48% of patients receiving litifilimab and 42% of those in the placebo group – had concomitant SLE with low to moderate disease activity as measured by the Systemic Lupus Erythematosus Disease Activity Index 2000. Patients could meet SLE criteria based on previous findings, and “didn’t have to have active SLE,” Dr. Werth noted.
The trial allowed background therapy as long as treatment had begun at least 12 weeks before randomization, with a stable dose starting at least 4 weeks before randomization and maintained throughout the trial period.
Most patients had moderate to severe CLE at baseline “despite approximately 90% having received concomitant background therapy and 80% of those participants having received antimalarial drugs, either alone or with other agents,” Dr. Werth and coinvestigators wrote.
CLASI-A has been shown to correlate to patients’ quality of life, Dr. Werth emphasized in the interview.
Most of the reported side effects in the phase 2 CLE trial were mild or moderate. The treatment was associated with three cases of hypersensitivity, three cases of oral herpes infection, and one case of herpes zoster infection. One case of herpes zoster meningitis occurred 4 months after the last dose of litifilimab.
Approximately 10% of study participants who reported race and ethnicity were Black or African American.
Phase 3 trials
The trial was one part of a two-part phase 2 study of litifilimab, named the LILAC trial, sponsored by Biogen. The other part, which will be published separately, involved patients who had SLE with active joint and skin manifestations.
Biogen is currently enrolling patients in phase 3 studies – the TOPAZ-1 and TOPAZ-2 studies – to evaluate the efficacy and safety of the drug in patients with active SLE. As secondary endpoints, both trials will measure the percentage of participants with a CLASI-A score of at least 10 at baseline who achieve improvement in the score, including a 50% improvement from baseline to week 16, Nathalie Franchimont, MD, PhD, of Biogen, a coauthor of the NEJM study, said in an email.
Biogen also has “plans to initiate a pivotal study in CLE this year,” she said.
With respect to the newly published phase 2 study, Dr. Femia said that, while “conclusions about the magnitude of efficacy are difficult to extrapolate in this trial design, there’s reason for cautious optimism.” There is “good theoretical basis to be optimistic about a drug such as litifilimab, that ultimately reduces type 1 interferon response,” she added.
Anifrolumab, a type 1 interferon receptor monoclonal antibody marketed as Saphnelo, was approved by the FDA for SLE in July 2021, but CLE subtypes were not characterized in trials and CLE was not studied independently of SLE, the authors pointed out in their NEJM article.
The study was supported by Biogen. In addition to working with Biogen, Dr. Werth serves as a consultant to Gilead Sciences and other pharmaceutical companies. Dr. Vital has research grants and has received honoraria from AstraZeneca. Dr. Femia disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
No more ‘escape hatch’: Post Roe, new worries about meds linked to birth defects
As states ban or limit abortion in the wake of the demise of Roe v. Wade, physicians are turning their attention to widely-used drugs that can cause birth defects. At issue: Should these drugs still be prescribed to women of childbearing age if they don’t have the option of terminating their pregnancies?

“Doctors are going to understandably be terrified that a patient may become pregnant using a teratogen that they have prescribed,” said University of Pittsburgh rheumatologist Mehret Birru Talabi, MD, PhD, who works in a state where the future of abortion rights is uncertain. “While this was a feared outcome before Roe v. Wade was overturned, abortion provided an escape hatch by which women could avoid having to continue a pregnancy and potentially raise a child with congenital anomalies. I believe that prescribing is going to become much more defensive and conservative. Some clinicians may choose not to prescribe these medications to patients who have childbearing potential, even if they don’t have much risk for pregnancy.”
Other physicians expressed similar concerns in interviews. Duke University, Durham, N.C., rheumatologist Megan E. B. Clowse, MD, MPH, fears that physicians will be wary of prescribing a variety of medications – including new ones for which there are few pregnancy data – if abortion is unavailable. “Women who receive these new or teratogenic medications will likely lose their reproductive autonomy and be forced to choose between having sexual relationships with men, obtaining procedures that make them permanently sterile, or using contraception that may cause intolerable side effects,” she said. “I am very concerned that young women with rheumatic disease will now be left with active disease resulting in joint damage and renal failure.”
Abortion is now banned in at least six states, according to The New York Times. That number may rise to 16 as more restrictions become law. Another five states aren’t expected to ban abortion soon but have implemented gestational age limits on abortion or are expected to adopt them. In another nine states, courts or lawmakers will decide whether abortion remains legal.
Only 20 states and the District of Columbia have firm abortion protections in place.
Numerous drugs are considered teratogens, which means they may cause birth defects. Thalidomide is the most infamous, but there are many more, including several used in rheumatology, dermatology, and gastroenterology. Among the most widely used teratogenic medications are the acne drugs isotretinoin and methotrexate, which are used to treat a variety of conditions, such as cancer, rheumatoid arthritis, and psoriasis.
Dr. Clowse, who helps manage an industry-supported website devoted to reproductive care for women with lupus (www.LupusPregnancy.org), noted that several drugs linked to birth defects and pregnancy loss are commonly prescribed in rheumatology.
“Methotrexate is the most common medication and has been the cornerstone of rheumatoid arthritis [treatment] for at least two decades,” she said. “Mycophenolate is our best medication to treat lupus nephritis, which is inflammation in the kidneys caused by lupus. This is a common complication for young women with lupus, and all of our guideline-recommended treatment regimens include a medication that causes pregnancy loss and birth defects, either mycophenolate or cyclophosphamide.”
Rheumatologists also prescribe a large number of new drugs for which there are few data about pregnancy risks. “It typically takes about two decades to have sufficient data about the safety of our medications,” she said.
Reflecting the sensitivity of the topic, Dr. Clowse made clear that her opinions don’t represent the views of her institution. She works in North Carolina, where the fate of abortion rights is uncertain, according to The New York Times.
What about alternatives? “The short answer is that some of these medications work really well and sometimes much better than the nonteratogenic alternatives,” said Dr. Birru Talabi. “I’m worried about methotrexate. It has been used to induce abortions but is primarily used in the United States as a highly effective treatment for cancer as well as a myriad of rheumatic diseases. If legislators try to restrict access to methotrexate, we may see increasing disability and even death among people who need this medication but cannot access it.”
Rheumatologists aren’t the only physicians who are worrying about the fates of their patients in a new era of abortion restrictions. Gastroenterologist Sunanda Kane, MD, MSPH, of the Mayo Clinic, Rochester, Minn., said several teratogenic medications are used in her field to treat constipation, viral hepatitis, and inflammatory bowel disease.
“When treating women of childbearing age, there are usually alternatives. If we do prescribe a medication with a high teratogenic potential, we counsel and document that we have discussed two forms of birth control to avoid pregnancy. We usually do not prescribe a drug with teratogenic potential with the ‘out’ being an abortion if a pregnancy does occur,” she said. However, “if abortion is not even on the table as an option, we may be much less likely to prescribe these medications. This will be particularly true in patients who clearly do not have the means to travel to have an abortion in any situation.”
Abortion is expected to remain legal in Minnesota, where Dr. Kane practices, but it may be restricted or banned in nearby Wisconsin, depending on the state legislature. None of her patients have had abortions after becoming pregnant while taking the medications, she said, although she “did have a patient who because of her religious faith did not have an abortion after exposure and ended up with a stillbirth.”
The crackdown on abortion won’t just pose risks to patients who take potentially dangerous medications, physicians said. Dr. Kane said pregnancy itself is a significant risk for patients with “very active, uncontrolled gastrointestinal conditions where a pregnancy could be harmful to the mother’s health or result in offspring that are very unhealthy.” These include decompensated cirrhosis, uncontrolled Crohn’s disease or ulcerative colitis, refractory gastroparesis, uncontrolled celiac sprue, and chronic pancreatitis, she said.
“There have been times when after shared decisionmaking, a patient with very active inflammatory bowel disease has decided to terminate the pregnancy because of her own ongoing health issues,” she said. “Not having this option will potentially lead to disastrous results.”
Dr. Clowse, the Duke University rheumatologist, echoed Dr. Kane’s concerns about women who are too sick to bear children. “The removal of abortion rights puts the lives and quality of life for women with rheumatic disease at risk. For patients with lupus and other systemic rheumatic disease, pregnancy can be medically catastrophic, leading to permanent harm and even death to the woman and her offspring. I am worried that women in these conditions will die without lifesaving pregnancy terminations, due to worries about the legal consequences for their physicians.”
The U.S. Supreme Court’s ruling that overturned Roe v. Wade has also raised the prospect that the court could ultimately allow birth control to be restricted or outlawed.
While the ruling states that “nothing in this opinion should be understood to cast doubt on precedents that do not concern abortion,” Justice Clarence Thomas wrote a concurrence in which he said that the court should reconsider a 1960s ruling that forbids the banning of contraceptives. Republicans have dismissed concerns about bans being allowed, although Democrats, including the president and vice president, starkly warn that they could happen.
“If we as providers have to be concerned that there will be an unplanned pregnancy because of the lack of access to contraception,” Dr. Kane said, “this will have significant downstream consequences to the kind of care we can provide and might just drive some providers to not give care to female patients at all given this concern.”
The physicians quoted in this article report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
As states ban or limit abortion in the wake of the demise of Roe v. Wade, physicians are turning their attention to widely-used drugs that can cause birth defects. At issue: Should these drugs still be prescribed to women of childbearing age if they don’t have the option of terminating their pregnancies?
“Doctors are going to understandably be terrified that a patient may become pregnant using a teratogen that they have prescribed,” said University of Pittsburgh rheumatologist Mehret Birru Talabi, MD, PhD, who works in a state where the future of abortion rights is uncertain. “While this was a feared outcome before Roe v. Wade was overturned, abortion provided an escape hatch by which women could avoid having to continue a pregnancy and potentially raise a child with congenital anomalies. I believe that prescribing is going to become much more defensive and conservative. Some clinicians may choose not to prescribe these medications to patients who have childbearing potential, even if they don’t have much risk for pregnancy.”
Other physicians expressed similar concerns in interviews. Duke University, Durham, N.C., rheumatologist Megan E. B. Clowse, MD, MPH, fears that physicians will be wary of prescribing a variety of medications – including new ones for which there are few pregnancy data – if abortion is unavailable. “Women who receive these new or teratogenic medications will likely lose their reproductive autonomy and be forced to choose between having sexual relationships with men, obtaining procedures that make them permanently sterile, or using contraception that may cause intolerable side effects,” she said. “I am very concerned that young women with rheumatic disease will now be left with active disease resulting in joint damage and renal failure.”
Abortion is now banned in at least six states, according to The New York Times. That number may rise to 16 as more restrictions become law. Another five states aren’t expected to ban abortion soon but have implemented gestational age limits on abortion or are expected to adopt them. In another nine states, courts or lawmakers will decide whether abortion remains legal.
Only 20 states and the District of Columbia have firm abortion protections in place.
Numerous drugs are considered teratogens, which means they may cause birth defects. Thalidomide is the most infamous, but there are many more, including several used in rheumatology, dermatology, and gastroenterology. Among the most widely used teratogenic medications are the acne drugs isotretinoin and methotrexate, which are used to treat a variety of conditions, such as cancer, rheumatoid arthritis, and psoriasis.
Dr. Clowse, who helps manage an industry-supported website devoted to reproductive care for women with lupus (www.LupusPregnancy.org), noted that several drugs linked to birth defects and pregnancy loss are commonly prescribed in rheumatology.
“Methotrexate is the most common medication and has been the cornerstone of rheumatoid arthritis [treatment] for at least two decades,” she said. “Mycophenolate is our best medication to treat lupus nephritis, which is inflammation in the kidneys caused by lupus. This is a common complication for young women with lupus, and all of our guideline-recommended treatment regimens include a medication that causes pregnancy loss and birth defects, either mycophenolate or cyclophosphamide.”
Rheumatologists also prescribe a large number of new drugs for which there are few data about pregnancy risks. “It typically takes about two decades to have sufficient data about the safety of our medications,” she said.
Reflecting the sensitivity of the topic, Dr. Clowse made clear that her opinions don’t represent the views of her institution. She works in North Carolina, where the fate of abortion rights is uncertain, according to The New York Times.
What about alternatives? “The short answer is that some of these medications work really well and sometimes much better than the nonteratogenic alternatives,” said Dr. Birru Talabi. “I’m worried about methotrexate. It has been used to induce abortions but is primarily used in the United States as a highly effective treatment for cancer as well as a myriad of rheumatic diseases. If legislators try to restrict access to methotrexate, we may see increasing disability and even death among people who need this medication but cannot access it.”
Rheumatologists aren’t the only physicians who are worrying about the fates of their patients in a new era of abortion restrictions. Gastroenterologist Sunanda Kane, MD, MSPH, of the Mayo Clinic, Rochester, Minn., said several teratogenic medications are used in her field to treat constipation, viral hepatitis, and inflammatory bowel disease.
“When treating women of childbearing age, there are usually alternatives. If we do prescribe a medication with a high teratogenic potential, we counsel and document that we have discussed two forms of birth control to avoid pregnancy. We usually do not prescribe a drug with teratogenic potential with the ‘out’ being an abortion if a pregnancy does occur,” she said. However, “if abortion is not even on the table as an option, we may be much less likely to prescribe these medications. This will be particularly true in patients who clearly do not have the means to travel to have an abortion in any situation.”
Abortion is expected to remain legal in Minnesota, where Dr. Kane practices, but it may be restricted or banned in nearby Wisconsin, depending on the state legislature. None of her patients have had abortions after becoming pregnant while taking the medications, she said, although she “did have a patient who because of her religious faith did not have an abortion after exposure and ended up with a stillbirth.”
The crackdown on abortion won’t just pose risks to patients who take potentially dangerous medications, physicians said. Dr. Kane said pregnancy itself is a significant risk for patients with “very active, uncontrolled gastrointestinal conditions where a pregnancy could be harmful to the mother’s health or result in offspring that are very unhealthy.” These include decompensated cirrhosis, uncontrolled Crohn’s disease or ulcerative colitis, refractory gastroparesis, uncontrolled celiac sprue, and chronic pancreatitis, she said.
“There have been times when after shared decisionmaking, a patient with very active inflammatory bowel disease has decided to terminate the pregnancy because of her own ongoing health issues,” she said. “Not having this option will potentially lead to disastrous results.”
Dr. Clowse, the Duke University rheumatologist, echoed Dr. Kane’s concerns about women who are too sick to bear children. “The removal of abortion rights puts the lives and quality of life for women with rheumatic disease at risk. For patients with lupus and other systemic rheumatic disease, pregnancy can be medically catastrophic, leading to permanent harm and even death to the woman and her offspring. I am worried that women in these conditions will die without lifesaving pregnancy terminations, due to worries about the legal consequences for their physicians.”
The U.S. Supreme Court’s ruling that overturned Roe v. Wade has also raised the prospect that the court could ultimately allow birth control to be restricted or outlawed.
While the ruling states that “nothing in this opinion should be understood to cast doubt on precedents that do not concern abortion,” Justice Clarence Thomas wrote a concurrence in which he said that the court should reconsider a 1960s ruling that forbids the banning of contraceptives. Republicans have dismissed concerns about bans being allowed, although Democrats, including the president and vice president, starkly warn that they could happen.
“If we as providers have to be concerned that there will be an unplanned pregnancy because of the lack of access to contraception,” Dr. Kane said, “this will have significant downstream consequences to the kind of care we can provide and might just drive some providers to not give care to female patients at all given this concern.”
The physicians quoted in this article report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
As states ban or limit abortion in the wake of the demise of Roe v. Wade, physicians are turning their attention to widely-used drugs that can cause birth defects. At issue: Should these drugs still be prescribed to women of childbearing age if they don’t have the option of terminating their pregnancies?
“Doctors are going to understandably be terrified that a patient may become pregnant using a teratogen that they have prescribed,” said University of Pittsburgh rheumatologist Mehret Birru Talabi, MD, PhD, who works in a state where the future of abortion rights is uncertain. “While this was a feared outcome before Roe v. Wade was overturned, abortion provided an escape hatch by which women could avoid having to continue a pregnancy and potentially raise a child with congenital anomalies. I believe that prescribing is going to become much more defensive and conservative. Some clinicians may choose not to prescribe these medications to patients who have childbearing potential, even if they don’t have much risk for pregnancy.”
Other physicians expressed similar concerns in interviews. Duke University, Durham, N.C., rheumatologist Megan E. B. Clowse, MD, MPH, fears that physicians will be wary of prescribing a variety of medications – including new ones for which there are few pregnancy data – if abortion is unavailable. “Women who receive these new or teratogenic medications will likely lose their reproductive autonomy and be forced to choose between having sexual relationships with men, obtaining procedures that make them permanently sterile, or using contraception that may cause intolerable side effects,” she said. “I am very concerned that young women with rheumatic disease will now be left with active disease resulting in joint damage and renal failure.”
Abortion is now banned in at least six states, according to The New York Times. That number may rise to 16 as more restrictions become law. Another five states aren’t expected to ban abortion soon but have implemented gestational age limits on abortion or are expected to adopt them. In another nine states, courts or lawmakers will decide whether abortion remains legal.
Only 20 states and the District of Columbia have firm abortion protections in place.
Numerous drugs are considered teratogens, which means they may cause birth defects. Thalidomide is the most infamous, but there are many more, including several used in rheumatology, dermatology, and gastroenterology. Among the most widely used teratogenic medications are the acne drugs isotretinoin and methotrexate, which are used to treat a variety of conditions, such as cancer, rheumatoid arthritis, and psoriasis.
Dr. Clowse, who helps manage an industry-supported website devoted to reproductive care for women with lupus (www.LupusPregnancy.org), noted that several drugs linked to birth defects and pregnancy loss are commonly prescribed in rheumatology.
“Methotrexate is the most common medication and has been the cornerstone of rheumatoid arthritis [treatment] for at least two decades,” she said. “Mycophenolate is our best medication to treat lupus nephritis, which is inflammation in the kidneys caused by lupus. This is a common complication for young women with lupus, and all of our guideline-recommended treatment regimens include a medication that causes pregnancy loss and birth defects, either mycophenolate or cyclophosphamide.”
Rheumatologists also prescribe a large number of new drugs for which there are few data about pregnancy risks. “It typically takes about two decades to have sufficient data about the safety of our medications,” she said.
Reflecting the sensitivity of the topic, Dr. Clowse made clear that her opinions don’t represent the views of her institution. She works in North Carolina, where the fate of abortion rights is uncertain, according to The New York Times.
What about alternatives? “The short answer is that some of these medications work really well and sometimes much better than the nonteratogenic alternatives,” said Dr. Birru Talabi. “I’m worried about methotrexate. It has been used to induce abortions but is primarily used in the United States as a highly effective treatment for cancer as well as a myriad of rheumatic diseases. If legislators try to restrict access to methotrexate, we may see increasing disability and even death among people who need this medication but cannot access it.”
Rheumatologists aren’t the only physicians who are worrying about the fates of their patients in a new era of abortion restrictions. Gastroenterologist Sunanda Kane, MD, MSPH, of the Mayo Clinic, Rochester, Minn., said several teratogenic medications are used in her field to treat constipation, viral hepatitis, and inflammatory bowel disease.
“When treating women of childbearing age, there are usually alternatives. If we do prescribe a medication with a high teratogenic potential, we counsel and document that we have discussed two forms of birth control to avoid pregnancy. We usually do not prescribe a drug with teratogenic potential with the ‘out’ being an abortion if a pregnancy does occur,” she said. However, “if abortion is not even on the table as an option, we may be much less likely to prescribe these medications. This will be particularly true in patients who clearly do not have the means to travel to have an abortion in any situation.”
Abortion is expected to remain legal in Minnesota, where Dr. Kane practices, but it may be restricted or banned in nearby Wisconsin, depending on the state legislature. None of her patients have had abortions after becoming pregnant while taking the medications, she said, although she “did have a patient who because of her religious faith did not have an abortion after exposure and ended up with a stillbirth.”
The crackdown on abortion won’t just pose risks to patients who take potentially dangerous medications, physicians said. Dr. Kane said pregnancy itself is a significant risk for patients with “very active, uncontrolled gastrointestinal conditions where a pregnancy could be harmful to the mother’s health or result in offspring that are very unhealthy.” These include decompensated cirrhosis, uncontrolled Crohn’s disease or ulcerative colitis, refractory gastroparesis, uncontrolled celiac sprue, and chronic pancreatitis, she said.
“There have been times when after shared decisionmaking, a patient with very active inflammatory bowel disease has decided to terminate the pregnancy because of her own ongoing health issues,” she said. “Not having this option will potentially lead to disastrous results.”
Dr. Clowse, the Duke University rheumatologist, echoed Dr. Kane’s concerns about women who are too sick to bear children. “The removal of abortion rights puts the lives and quality of life for women with rheumatic disease at risk. For patients with lupus and other systemic rheumatic disease, pregnancy can be medically catastrophic, leading to permanent harm and even death to the woman and her offspring. I am worried that women in these conditions will die without lifesaving pregnancy terminations, due to worries about the legal consequences for their physicians.”
The U.S. Supreme Court’s ruling that overturned Roe v. Wade has also raised the prospect that the court could ultimately allow birth control to be restricted or outlawed.
While the ruling states that “nothing in this opinion should be understood to cast doubt on precedents that do not concern abortion,” Justice Clarence Thomas wrote a concurrence in which he said that the court should reconsider a 1960s ruling that forbids the banning of contraceptives. Republicans have dismissed concerns about bans being allowed, although Democrats, including the president and vice president, starkly warn that they could happen.
“If we as providers have to be concerned that there will be an unplanned pregnancy because of the lack of access to contraception,” Dr. Kane said, “this will have significant downstream consequences to the kind of care we can provide and might just drive some providers to not give care to female patients at all given this concern.”
The physicians quoted in this article report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Autoimmune disease linked to better late-stage breast cancer survival
CHICAGO – Comorbid autoimmune disease is associated with a greater chance of survival among women with stage IV breast cancer, according to a retrospective study presented at the annual meeting of the American Society of Clinical Oncology.
“It’s counterintuitive that, if you have two diseases instead of one, that you live longer, so then we had to scratch our heads a little bit and think about why these people are living longer,” said lead author Demitrios Dedousis, MD, University Hospitals, Case Medical Center, Cleveland.
Dr. Dedousis and colleagues conducted a retrospective analysis of patients from Surveillance, Epidemiology, and End Results–Medicare databases between 2007 and 2014 with breast cancer. The study included data from 137,324 patients diagnosed between 2007 and 2012, before the widespread use of immunotherapy. 27% of patients had an autoimmune disease, most commonly rheumatoid arthritis (23%), psoriasis (2.4%), and systemic lupus erythematosus (1.1%).
When all patients were included in the analysis, those with autoimmune disorders had slightly longer survival times, but these weren’t clinically significant. A subanalysis found a greater difference in survival.
The association appears more pronounced in metastatic cancer. Patients with stage 4 breast cancer and autoimmune disease had a longer mean overall survival (36 months vs. 30 months; hazard ratio, 1.46; P < .0001. Cancer-specific survival: HR, 1.39; P < .0001). Patients with autoimmune disease and stage 1-3 breast cancer had lower overall survival (P < .0001, P < 0.0001, and P = 0.026 respectively), compared with patients without autoimmune disease.
“What we thought was happening is that the lack of increased survival in stages 1 through 3 was hiding the increase in survival among the stage IV patients when looking at the overall cohort,” Dr. Dedousis said.
The retrospective nature of the study makes it impossible to draw any firm conclusions about causation. It could be that patients who have already been diagnosed with an autoimmune disease are more vigilant about going to health checkups. “There are other possible explanations, but the one that’s most interesting to us is that their immune system is involved in fighting the cancer. Our study certainly didn’t prove that, but it’s suggesting that’s a possibility,” Dr. Dedousis said.
He and his coauthors anticipate conducting similar studies in other cancers to see if there are similar relationships. Some preliminary work has already suggested something similar in lung cancer. “I think demonstrating this in a few kinds of cancer goes part of the way towards showing that this is a real biological phenomenon,” he said.
Another research avenue is to examine the immune systems and pathology specimens in patients with both an autoimmune disease and cancer to see if there is a greater immune response within the tumor. If so, that could suggest new immunotherapy strategies.
Another possibility is to look at the specific immune pathways within “protective” autoimmune conditions. “For the sake of argument, if we find a particular autoimmune condition is improving survival across multiple kinds of cancers, we could look at those pathways that are specifically involved in that autoimmune condition. It might help us identify a target for drug development,” Dr. Dedousis said.
Asked why a potential benefit might be more apparent in late-stage disease, he suggested that, in early-stage breast cancer, surgery and other treatments may be so effective that the immune system’s role only rarely makes a difference. It could play a larger role in late-stage disease when there are less effective therapies. It could also be that the immune system doesn’t recognize the cancer until it has spread beyond the regional lymph nodes on its way to metastasizing.
According to the National Cancer Institute, 10%-30% of people with cancer also have an autoimmune disease.
Dr. Dedousis has no relevant financial disclosures.
CHICAGO – Comorbid autoimmune disease is associated with a greater chance of survival among women with stage IV breast cancer, according to a retrospective study presented at the annual meeting of the American Society of Clinical Oncology.
“It’s counterintuitive that, if you have two diseases instead of one, that you live longer, so then we had to scratch our heads a little bit and think about why these people are living longer,” said lead author Demitrios Dedousis, MD, University Hospitals, Case Medical Center, Cleveland.
Dr. Dedousis and colleagues conducted a retrospective analysis of patients from Surveillance, Epidemiology, and End Results–Medicare databases between 2007 and 2014 with breast cancer. The study included data from 137,324 patients diagnosed between 2007 and 2012, before the widespread use of immunotherapy. 27% of patients had an autoimmune disease, most commonly rheumatoid arthritis (23%), psoriasis (2.4%), and systemic lupus erythematosus (1.1%).
When all patients were included in the analysis, those with autoimmune disorders had slightly longer survival times, but these weren’t clinically significant. A subanalysis found a greater difference in survival.
The association appears more pronounced in metastatic cancer. Patients with stage 4 breast cancer and autoimmune disease had a longer mean overall survival (36 months vs. 30 months; hazard ratio, 1.46; P < .0001. Cancer-specific survival: HR, 1.39; P < .0001). Patients with autoimmune disease and stage 1-3 breast cancer had lower overall survival (P < .0001, P < 0.0001, and P = 0.026 respectively), compared with patients without autoimmune disease.
“What we thought was happening is that the lack of increased survival in stages 1 through 3 was hiding the increase in survival among the stage IV patients when looking at the overall cohort,” Dr. Dedousis said.
The retrospective nature of the study makes it impossible to draw any firm conclusions about causation. It could be that patients who have already been diagnosed with an autoimmune disease are more vigilant about going to health checkups. “There are other possible explanations, but the one that’s most interesting to us is that their immune system is involved in fighting the cancer. Our study certainly didn’t prove that, but it’s suggesting that’s a possibility,” Dr. Dedousis said.
He and his coauthors anticipate conducting similar studies in other cancers to see if there are similar relationships. Some preliminary work has already suggested something similar in lung cancer. “I think demonstrating this in a few kinds of cancer goes part of the way towards showing that this is a real biological phenomenon,” he said.
Another research avenue is to examine the immune systems and pathology specimens in patients with both an autoimmune disease and cancer to see if there is a greater immune response within the tumor. If so, that could suggest new immunotherapy strategies.
Another possibility is to look at the specific immune pathways within “protective” autoimmune conditions. “For the sake of argument, if we find a particular autoimmune condition is improving survival across multiple kinds of cancers, we could look at those pathways that are specifically involved in that autoimmune condition. It might help us identify a target for drug development,” Dr. Dedousis said.
Asked why a potential benefit might be more apparent in late-stage disease, he suggested that, in early-stage breast cancer, surgery and other treatments may be so effective that the immune system’s role only rarely makes a difference. It could play a larger role in late-stage disease when there are less effective therapies. It could also be that the immune system doesn’t recognize the cancer until it has spread beyond the regional lymph nodes on its way to metastasizing.
According to the National Cancer Institute, 10%-30% of people with cancer also have an autoimmune disease.
Dr. Dedousis has no relevant financial disclosures.
CHICAGO – Comorbid autoimmune disease is associated with a greater chance of survival among women with stage IV breast cancer, according to a retrospective study presented at the annual meeting of the American Society of Clinical Oncology.
“It’s counterintuitive that, if you have two diseases instead of one, that you live longer, so then we had to scratch our heads a little bit and think about why these people are living longer,” said lead author Demitrios Dedousis, MD, University Hospitals, Case Medical Center, Cleveland.
Dr. Dedousis and colleagues conducted a retrospective analysis of patients from Surveillance, Epidemiology, and End Results–Medicare databases between 2007 and 2014 with breast cancer. The study included data from 137,324 patients diagnosed between 2007 and 2012, before the widespread use of immunotherapy. 27% of patients had an autoimmune disease, most commonly rheumatoid arthritis (23%), psoriasis (2.4%), and systemic lupus erythematosus (1.1%).
When all patients were included in the analysis, those with autoimmune disorders had slightly longer survival times, but these weren’t clinically significant. A subanalysis found a greater difference in survival.
The association appears more pronounced in metastatic cancer. Patients with stage 4 breast cancer and autoimmune disease had a longer mean overall survival (36 months vs. 30 months; hazard ratio, 1.46; P < .0001. Cancer-specific survival: HR, 1.39; P < .0001). Patients with autoimmune disease and stage 1-3 breast cancer had lower overall survival (P < .0001, P < 0.0001, and P = 0.026 respectively), compared with patients without autoimmune disease.
“What we thought was happening is that the lack of increased survival in stages 1 through 3 was hiding the increase in survival among the stage IV patients when looking at the overall cohort,” Dr. Dedousis said.
The retrospective nature of the study makes it impossible to draw any firm conclusions about causation. It could be that patients who have already been diagnosed with an autoimmune disease are more vigilant about going to health checkups. “There are other possible explanations, but the one that’s most interesting to us is that their immune system is involved in fighting the cancer. Our study certainly didn’t prove that, but it’s suggesting that’s a possibility,” Dr. Dedousis said.
He and his coauthors anticipate conducting similar studies in other cancers to see if there are similar relationships. Some preliminary work has already suggested something similar in lung cancer. “I think demonstrating this in a few kinds of cancer goes part of the way towards showing that this is a real biological phenomenon,” he said.
Another research avenue is to examine the immune systems and pathology specimens in patients with both an autoimmune disease and cancer to see if there is a greater immune response within the tumor. If so, that could suggest new immunotherapy strategies.
Another possibility is to look at the specific immune pathways within “protective” autoimmune conditions. “For the sake of argument, if we find a particular autoimmune condition is improving survival across multiple kinds of cancers, we could look at those pathways that are specifically involved in that autoimmune condition. It might help us identify a target for drug development,” Dr. Dedousis said.
Asked why a potential benefit might be more apparent in late-stage disease, he suggested that, in early-stage breast cancer, surgery and other treatments may be so effective that the immune system’s role only rarely makes a difference. It could play a larger role in late-stage disease when there are less effective therapies. It could also be that the immune system doesn’t recognize the cancer until it has spread beyond the regional lymph nodes on its way to metastasizing.
According to the National Cancer Institute, 10%-30% of people with cancer also have an autoimmune disease.
Dr. Dedousis has no relevant financial disclosures.
AT ASCO 2022
Deucravacitinib and orelabrutinib perform well in early lupus trials
Deucravacitinib and orelabrutinib – two novel oral drugs under investigation for the treatment of systemic lupus erythematosus (SLE) – have performed well in early clinical trials reported as late-breaking abstracts at the annual European Congress of Rheumatology.
In the phase 2 PAISLEY study, up to 58% of patients treated with deucravacitinib versus 34% of placebo-treated patients met the primary study endpoint of an SLE Responder Index-4 (SRI-4) after 38 weeks of treatment. Deucravacitinib also “achieved or meaningfully improved” all of the secondary endpoints set out in the 363-patient trial and was reported to have a safety and tolerability profile that was generally similar to placebo.

“Deucravacitinib shows promise as a novel therapy for SLE and warrants further investigation in phase 3 trials,” said Eric F. Morand, MD, PhD, a clinical rheumatologist and head of the School of Clinical Sciences at Monash University in Melbourne.
In a separate, ongoing phase 1b/2a study designed to evaluate orelabrutinib as a potential treatment for SLE, no safety concerns were seen with the investigational drug, along with “trending efficacy,” that supports “further studies in larger and longer-term trials,” according to the study’s investigators.
“What sets these two new drugs apart from currently available targeted therapies are their mode of action,” said Md Yuzaiful Md Yusof, MBChB, PhD, who was not involved in either study.

“The results from the PAISLEY study are promising, and it’s good to see the patients recruited were of diverse ethnicity [50%–60% were White],” added Dr. Md Yusof, a senior research fellow within the Leeds (England) Institute of Rheumatic and Musculoskeletal Medicine and a consultant rheumatologist at Leeds Teaching Hospitals NHS Trust.
He noted that the placebo rate was also low: “This could be contributed to by keeping the background prednisolone dose low, which is often a challenge in designing SLE trials.”
Deucravacitinib – the distant cousin of the JAK family?
“Deucravacitinib is a compound you might not have heard of before,” Dr. Morand said.
“It’s an inhibitor of a kinase called TYK2, which, broadly speaking, is a member of JAK [Janus kinase] family,” he explained in an interview. TYK2 regulates signal transduction downstream of receptors for interleukin (IL)-23 and IL-12 pathways and the type I interferon family.
“It’s a very finite set of cytokine signals” that are being blocked with deucravacitinib, he said, adding that this means it’s more directly targeting SLE pathogenic mechanisms than perhaps other JAK inhibitor compounds.
“It also means that it shouldn’t have some of the downsides of the other JAK inhibitors,” Dr. Morand said, “such as hematopoietic side effects, including cytopenias.”
The phase 2 PAISLEY study
This study involved 363 patients with moderate to severe, active SLE were recruited and randomized to receive placebo (n = 90) or one of three doses of deucravacitinib: 3 mg twice daily (n = 91), 6 mg twice daily (n = 93), or 12 mg once daily (n = 89). Most patients were also taking multiple background therapies, but this was similar across the four treatment arms.
The SRI-4 primary endpoint after 38 weeks of treatment was met by 34.4% of patients who received placebo, but 58.2% of those treated with deucravacitinib 3 mg twice daily (P = .0006 versus placebo), 49.5% (P = .021) of those treated with 6 mg twice daily, and 44.9% (P = .078) treated with 12 mg once daily.
“All secondary outcome measures were achieved or meaningfully improved at week 48, including SRI-4, BICLA [British Isles Lupus Assessment Group-based Composite Lupus Assessment], low-level disease activity state [LLDAS], reduction in skin disease and reduction in arthritis,” Dr. Morand said.
In addition, early biomarker results showed reductions in double-stranded DNA titers and increases in serum C4 complement with deucravacitinib across the duration of the study.
In discussion, Dr. Morand was asked about the seemingly negative or inverse dose response seen in the trial, with the best results seen with the 3-mg twice daily dose, then lower effects seen with two higher doses.
“Our analysis is that it’s not an inverse dose response, but rather a flat dose response above the 3-mg [twice daily] dose,” he said, noting that there was a higher dropout rate because of adverse effects in the 12-mg once daily group and those participants were recorded as nonresponders.
“We think what we’ve seen here is that 3 mg twice daily is a sufficient dose and there was no additional therapeutic gain above that.”
Rates of adverse events (AEs), serious AEs, and AEs of interest were overall fairly similar between deucravacitinib and placebo groups. The most common side effects seen with deucravacitinib were upper respiratory tract infection, nasopharyngitis, headache, and urinary tract infection. Skin reactions, such as acne, rash, and pruritis, among others, were more common in deucravacitinib- than in placebo-treated patients.
Importantly, Dr. Morand noted that there were no major cardiac events or thrombotic events and no deaths seen in the study. There was no signal for an increase in serious or opportunistic infections, including herpes zoster. There was no effect on common laboratory parameters.
“These are very encouraging results for patients with SLE,” Albert Roy, executive director of Lupus Therapeutics, said in a press release issued by the Lupus Research Alliance.
“We are honored to have played a role in this exciting work by helping to conduct this clinical trial through our Lupus Clinical Investigators Network of renowned North American academic centers.”
In an interview, he added: “We’re cautiously optimistic. Hopefully, if it continues to progress through phase 3, it’ll be the first oral agent that would be approved for lupus, notwithstanding prednisone and Plaquenil [hydroxychloroquine], back in the 50s.”
Orelabrutinib phase 2 study in SLE
Another approach to oral route of administration under investigation in SLE is the use of orelabrutinib, an irreversible inhibitor of Bruton’s tyrosine kinase (BTK) that was approved in China in December 2020 for the treatment of certain lymphomas and leukemias.
The rationale for testing it in SLE comes from two preclinical studies that had suggested a possible benefit in reducing disease activity, explained Zhanguo Li, MD, PhD, professor at Peking University People’s Hospital in Beijing. He presented the results of an ongoing randomized, double-blind, placebo-controlled, phase Ib/IIa dose-finding study comparing three different doses of orelabrutinib (50, 80, and 100 mg, once daily) to placebo.
As in the deucravacitinib trial, the SRI-4 was used to assess the potential efficacy of orelabrutinib, although in a much smaller patient population (n = 92) and at a shorter time point (12 weeks). Results showed an 11%-20% difference between the percentage of patients who met SRI-4 response criteria with orelabrutinib and those on placebo, at a respective 46.5%, 53.3%, 56.3% and 35.7%.
SLE Disease Activity Index (SLEDAI) scores showed a similar benefit of orelabrutinib over placebo, with 54%-63% and 30% of patients, respectively, achieving a score of 8 or more.
Adverse event rates were similar to those of placebo with most events being of mild or moderate nature. Three patients treated with orelabrutinib experienced serious adverse events, of which one was grade 3, but there were no reported deaths.
Pharmacokinetic and pharmacodynamic data showed a dose effect, and nearly complete occupancy of BTK was achieved at all dose levels for 24 hours, consistent with once-daily dosing.
“BTK plays an important role in B-cell regulation, thus B-cell and myeloid-cell blockade through BTK inhibition is an interesting potential new target for SLE,” Dr. Md Yusof said.
“Data from this early dose-ranging trial is encouraging. No major safety signal apart from mild reduction in lymphocyte and white cell counts,” he added.
“There are still plenty of challenges ahead for this drug’s development, particularly as none of the BTK inhibitors have yet to succeed in phase 3 trials in rheumatic and musculoskeletal diseases,” Dr. Md Yusof said.
Early days for both agents
While both seem currently promising, it’s very early days for deucravacitinib and orelabrutinib as possible new agents for SLE.
Aside from SLE, deucravacitinib is being tested across multiple immune-mediated diseases. This includes psoriasis, where two phase 3 trials – POETYK PSO-1 and POETYK PSO-2 – have already been completed, and psoriatic arthritis, where a phase 2 trial has been reported; all with positive results.
Phase 3 testing of deucravacitinib will go ahead and recruitment may start toward the end of this year, but it’ll take years to complete the studies, Dr. Morand said. Even if the trials prove positive, neither agent is going to be available for clinical use for several years.
A case in point is anifrolumab (Saphnelo), which Dr. Morand was involved in assessing. Despite gaining approval in the United States and across much of the world, the drug still going through reimbursement processes.
“The trial data, and lots of post hoc analysis, show clearly that it’s a major step forward in treating lupus,” he said in an interview, but “access is limited in most places, so hands-on experience with that new treatment is still limited for most clinicians.”
As for all the other new targeted approaches under investigation, “although there’s a lot of trial activity, there’s still a couple of years away before any of the current trials deliver new treatment. That’s if they provide positive findings. Indeed, there have been numerous agents that have shown promise at phase 2 but then fall at the final phase 3 hurdle, including baricitinib, which Dr. Morand reported on in a separate poster presentation.
Phase 3 data proved disappointing: “Results are not sufficiently positive for that to go forward,” he said, adding that “transitioning from a successful phase 2 to a successful phase 3 is challenging, and many products have failed.”
Dr. Morand added: “It’s a very exciting time to be in lupus research, and there’s a lot of optimism about the future. But when I go back to my clinic tomorrow, I treat my patients exactly the same as I did last week and last year.”
It’s yet to be seen if deucravacitinib will fulfill its early promise, but it’s off to an impressive start. A positive for patients is that it’s an oral drug, with the potential to improve access to treatment across the world where getting infusions may be an issue.
“These are some of the most exciting data that I’ve seen at the phase 2 level in terms of effect size across all the readouts that are used,” Dr. Morand said. “There’s no guesswork here; it worked across all the measures. That’s very reassuring.”
The PAISLEY study was sponsored by Bristol-Myers Squibb. Dr. Morand has acted as a consultant to the company and received research support for the conduct of the trial. He disclosed acting as a consultant or receiving research funding from AbbVie, Amgen, AstraZeneca, Biogen, Eli Lilly, EMD Serono, Janssen, Genentech, Servier, Novartis, and UCB. Mr. Roy is the executive director of Lupus Therapeutics, which manages the Lupus Clinical Investigators Network based in North America. Lupus Therapeutics is the clinical trials arm of the Lupus Research Alliance, a nongovernmental, nonprofit funder of lupus research worldwide. The orelabrutinib study was sponsored by InnoCare Pharma. Dr. Li is the principal investigator for the trial but had no conflicts of interest to declare. Dr. Md Yusof disclosed receiving consultancy fees from Aurinia Pharmaceuticals.
Deucravacitinib and orelabrutinib – two novel oral drugs under investigation for the treatment of systemic lupus erythematosus (SLE) – have performed well in early clinical trials reported as late-breaking abstracts at the annual European Congress of Rheumatology.
In the phase 2 PAISLEY study, up to 58% of patients treated with deucravacitinib versus 34% of placebo-treated patients met the primary study endpoint of an SLE Responder Index-4 (SRI-4) after 38 weeks of treatment. Deucravacitinib also “achieved or meaningfully improved” all of the secondary endpoints set out in the 363-patient trial and was reported to have a safety and tolerability profile that was generally similar to placebo.
“Deucravacitinib shows promise as a novel therapy for SLE and warrants further investigation in phase 3 trials,” said Eric F. Morand, MD, PhD, a clinical rheumatologist and head of the School of Clinical Sciences at Monash University in Melbourne.
In a separate, ongoing phase 1b/2a study designed to evaluate orelabrutinib as a potential treatment for SLE, no safety concerns were seen with the investigational drug, along with “trending efficacy,” that supports “further studies in larger and longer-term trials,” according to the study’s investigators.
“What sets these two new drugs apart from currently available targeted therapies are their mode of action,” said Md Yuzaiful Md Yusof, MBChB, PhD, who was not involved in either study.
“The results from the PAISLEY study are promising, and it’s good to see the patients recruited were of diverse ethnicity [50%–60% were White],” added Dr. Md Yusof, a senior research fellow within the Leeds (England) Institute of Rheumatic and Musculoskeletal Medicine and a consultant rheumatologist at Leeds Teaching Hospitals NHS Trust.
He noted that the placebo rate was also low: “This could be contributed to by keeping the background prednisolone dose low, which is often a challenge in designing SLE trials.”
Deucravacitinib – the distant cousin of the JAK family?
“Deucravacitinib is a compound you might not have heard of before,” Dr. Morand said.
“It’s an inhibitor of a kinase called TYK2, which, broadly speaking, is a member of JAK [Janus kinase] family,” he explained in an interview. TYK2 regulates signal transduction downstream of receptors for interleukin (IL)-23 and IL-12 pathways and the type I interferon family.
“It’s a very finite set of cytokine signals” that are being blocked with deucravacitinib, he said, adding that this means it’s more directly targeting SLE pathogenic mechanisms than perhaps other JAK inhibitor compounds.
“It also means that it shouldn’t have some of the downsides of the other JAK inhibitors,” Dr. Morand said, “such as hematopoietic side effects, including cytopenias.”
The phase 2 PAISLEY study
This study involved 363 patients with moderate to severe, active SLE were recruited and randomized to receive placebo (n = 90) or one of three doses of deucravacitinib: 3 mg twice daily (n = 91), 6 mg twice daily (n = 93), or 12 mg once daily (n = 89). Most patients were also taking multiple background therapies, but this was similar across the four treatment arms.
The SRI-4 primary endpoint after 38 weeks of treatment was met by 34.4% of patients who received placebo, but 58.2% of those treated with deucravacitinib 3 mg twice daily (P = .0006 versus placebo), 49.5% (P = .021) of those treated with 6 mg twice daily, and 44.9% (P = .078) treated with 12 mg once daily.
“All secondary outcome measures were achieved or meaningfully improved at week 48, including SRI-4, BICLA [British Isles Lupus Assessment Group-based Composite Lupus Assessment], low-level disease activity state [LLDAS], reduction in skin disease and reduction in arthritis,” Dr. Morand said.
In addition, early biomarker results showed reductions in double-stranded DNA titers and increases in serum C4 complement with deucravacitinib across the duration of the study.
In discussion, Dr. Morand was asked about the seemingly negative or inverse dose response seen in the trial, with the best results seen with the 3-mg twice daily dose, then lower effects seen with two higher doses.
“Our analysis is that it’s not an inverse dose response, but rather a flat dose response above the 3-mg [twice daily] dose,” he said, noting that there was a higher dropout rate because of adverse effects in the 12-mg once daily group and those participants were recorded as nonresponders.
“We think what we’ve seen here is that 3 mg twice daily is a sufficient dose and there was no additional therapeutic gain above that.”
Rates of adverse events (AEs), serious AEs, and AEs of interest were overall fairly similar between deucravacitinib and placebo groups. The most common side effects seen with deucravacitinib were upper respiratory tract infection, nasopharyngitis, headache, and urinary tract infection. Skin reactions, such as acne, rash, and pruritis, among others, were more common in deucravacitinib- than in placebo-treated patients.
Importantly, Dr. Morand noted that there were no major cardiac events or thrombotic events and no deaths seen in the study. There was no signal for an increase in serious or opportunistic infections, including herpes zoster. There was no effect on common laboratory parameters.
“These are very encouraging results for patients with SLE,” Albert Roy, executive director of Lupus Therapeutics, said in a press release issued by the Lupus Research Alliance.
“We are honored to have played a role in this exciting work by helping to conduct this clinical trial through our Lupus Clinical Investigators Network of renowned North American academic centers.”
In an interview, he added: “We’re cautiously optimistic. Hopefully, if it continues to progress through phase 3, it’ll be the first oral agent that would be approved for lupus, notwithstanding prednisone and Plaquenil [hydroxychloroquine], back in the 50s.”
Orelabrutinib phase 2 study in SLE
Another approach to oral route of administration under investigation in SLE is the use of orelabrutinib, an irreversible inhibitor of Bruton’s tyrosine kinase (BTK) that was approved in China in December 2020 for the treatment of certain lymphomas and leukemias.
The rationale for testing it in SLE comes from two preclinical studies that had suggested a possible benefit in reducing disease activity, explained Zhanguo Li, MD, PhD, professor at Peking University People’s Hospital in Beijing. He presented the results of an ongoing randomized, double-blind, placebo-controlled, phase Ib/IIa dose-finding study comparing three different doses of orelabrutinib (50, 80, and 100 mg, once daily) to placebo.
As in the deucravacitinib trial, the SRI-4 was used to assess the potential efficacy of orelabrutinib, although in a much smaller patient population (n = 92) and at a shorter time point (12 weeks). Results showed an 11%-20% difference between the percentage of patients who met SRI-4 response criteria with orelabrutinib and those on placebo, at a respective 46.5%, 53.3%, 56.3% and 35.7%.
SLE Disease Activity Index (SLEDAI) scores showed a similar benefit of orelabrutinib over placebo, with 54%-63% and 30% of patients, respectively, achieving a score of 8 or more.
Adverse event rates were similar to those of placebo with most events being of mild or moderate nature. Three patients treated with orelabrutinib experienced serious adverse events, of which one was grade 3, but there were no reported deaths.
Pharmacokinetic and pharmacodynamic data showed a dose effect, and nearly complete occupancy of BTK was achieved at all dose levels for 24 hours, consistent with once-daily dosing.
“BTK plays an important role in B-cell regulation, thus B-cell and myeloid-cell blockade through BTK inhibition is an interesting potential new target for SLE,” Dr. Md Yusof said.
“Data from this early dose-ranging trial is encouraging. No major safety signal apart from mild reduction in lymphocyte and white cell counts,” he added.
“There are still plenty of challenges ahead for this drug’s development, particularly as none of the BTK inhibitors have yet to succeed in phase 3 trials in rheumatic and musculoskeletal diseases,” Dr. Md Yusof said.
Early days for both agents
While both seem currently promising, it’s very early days for deucravacitinib and orelabrutinib as possible new agents for SLE.
Aside from SLE, deucravacitinib is being tested across multiple immune-mediated diseases. This includes psoriasis, where two phase 3 trials – POETYK PSO-1 and POETYK PSO-2 – have already been completed, and psoriatic arthritis, where a phase 2 trial has been reported; all with positive results.
Phase 3 testing of deucravacitinib will go ahead and recruitment may start toward the end of this year, but it’ll take years to complete the studies, Dr. Morand said. Even if the trials prove positive, neither agent is going to be available for clinical use for several years.
A case in point is anifrolumab (Saphnelo), which Dr. Morand was involved in assessing. Despite gaining approval in the United States and across much of the world, the drug still going through reimbursement processes.
“The trial data, and lots of post hoc analysis, show clearly that it’s a major step forward in treating lupus,” he said in an interview, but “access is limited in most places, so hands-on experience with that new treatment is still limited for most clinicians.”
As for all the other new targeted approaches under investigation, “although there’s a lot of trial activity, there’s still a couple of years away before any of the current trials deliver new treatment. That’s if they provide positive findings. Indeed, there have been numerous agents that have shown promise at phase 2 but then fall at the final phase 3 hurdle, including baricitinib, which Dr. Morand reported on in a separate poster presentation.
Phase 3 data proved disappointing: “Results are not sufficiently positive for that to go forward,” he said, adding that “transitioning from a successful phase 2 to a successful phase 3 is challenging, and many products have failed.”
Dr. Morand added: “It’s a very exciting time to be in lupus research, and there’s a lot of optimism about the future. But when I go back to my clinic tomorrow, I treat my patients exactly the same as I did last week and last year.”
It’s yet to be seen if deucravacitinib will fulfill its early promise, but it’s off to an impressive start. A positive for patients is that it’s an oral drug, with the potential to improve access to treatment across the world where getting infusions may be an issue.
“These are some of the most exciting data that I’ve seen at the phase 2 level in terms of effect size across all the readouts that are used,” Dr. Morand said. “There’s no guesswork here; it worked across all the measures. That’s very reassuring.”
The PAISLEY study was sponsored by Bristol-Myers Squibb. Dr. Morand has acted as a consultant to the company and received research support for the conduct of the trial. He disclosed acting as a consultant or receiving research funding from AbbVie, Amgen, AstraZeneca, Biogen, Eli Lilly, EMD Serono, Janssen, Genentech, Servier, Novartis, and UCB. Mr. Roy is the executive director of Lupus Therapeutics, which manages the Lupus Clinical Investigators Network based in North America. Lupus Therapeutics is the clinical trials arm of the Lupus Research Alliance, a nongovernmental, nonprofit funder of lupus research worldwide. The orelabrutinib study was sponsored by InnoCare Pharma. Dr. Li is the principal investigator for the trial but had no conflicts of interest to declare. Dr. Md Yusof disclosed receiving consultancy fees from Aurinia Pharmaceuticals.
Deucravacitinib and orelabrutinib – two novel oral drugs under investigation for the treatment of systemic lupus erythematosus (SLE) – have performed well in early clinical trials reported as late-breaking abstracts at the annual European Congress of Rheumatology.
In the phase 2 PAISLEY study, up to 58% of patients treated with deucravacitinib versus 34% of placebo-treated patients met the primary study endpoint of an SLE Responder Index-4 (SRI-4) after 38 weeks of treatment. Deucravacitinib also “achieved or meaningfully improved” all of the secondary endpoints set out in the 363-patient trial and was reported to have a safety and tolerability profile that was generally similar to placebo.
“Deucravacitinib shows promise as a novel therapy for SLE and warrants further investigation in phase 3 trials,” said Eric F. Morand, MD, PhD, a clinical rheumatologist and head of the School of Clinical Sciences at Monash University in Melbourne.
In a separate, ongoing phase 1b/2a study designed to evaluate orelabrutinib as a potential treatment for SLE, no safety concerns were seen with the investigational drug, along with “trending efficacy,” that supports “further studies in larger and longer-term trials,” according to the study’s investigators.
“What sets these two new drugs apart from currently available targeted therapies are their mode of action,” said Md Yuzaiful Md Yusof, MBChB, PhD, who was not involved in either study.
“The results from the PAISLEY study are promising, and it’s good to see the patients recruited were of diverse ethnicity [50%–60% were White],” added Dr. Md Yusof, a senior research fellow within the Leeds (England) Institute of Rheumatic and Musculoskeletal Medicine and a consultant rheumatologist at Leeds Teaching Hospitals NHS Trust.
He noted that the placebo rate was also low: “This could be contributed to by keeping the background prednisolone dose low, which is often a challenge in designing SLE trials.”
Deucravacitinib – the distant cousin of the JAK family?
“Deucravacitinib is a compound you might not have heard of before,” Dr. Morand said.
“It’s an inhibitor of a kinase called TYK2, which, broadly speaking, is a member of JAK [Janus kinase] family,” he explained in an interview. TYK2 regulates signal transduction downstream of receptors for interleukin (IL)-23 and IL-12 pathways and the type I interferon family.
“It’s a very finite set of cytokine signals” that are being blocked with deucravacitinib, he said, adding that this means it’s more directly targeting SLE pathogenic mechanisms than perhaps other JAK inhibitor compounds.
“It also means that it shouldn’t have some of the downsides of the other JAK inhibitors,” Dr. Morand said, “such as hematopoietic side effects, including cytopenias.”
The phase 2 PAISLEY study
This study involved 363 patients with moderate to severe, active SLE were recruited and randomized to receive placebo (n = 90) or one of three doses of deucravacitinib: 3 mg twice daily (n = 91), 6 mg twice daily (n = 93), or 12 mg once daily (n = 89). Most patients were also taking multiple background therapies, but this was similar across the four treatment arms.
The SRI-4 primary endpoint after 38 weeks of treatment was met by 34.4% of patients who received placebo, but 58.2% of those treated with deucravacitinib 3 mg twice daily (P = .0006 versus placebo), 49.5% (P = .021) of those treated with 6 mg twice daily, and 44.9% (P = .078) treated with 12 mg once daily.
“All secondary outcome measures were achieved or meaningfully improved at week 48, including SRI-4, BICLA [British Isles Lupus Assessment Group-based Composite Lupus Assessment], low-level disease activity state [LLDAS], reduction in skin disease and reduction in arthritis,” Dr. Morand said.
In addition, early biomarker results showed reductions in double-stranded DNA titers and increases in serum C4 complement with deucravacitinib across the duration of the study.
In discussion, Dr. Morand was asked about the seemingly negative or inverse dose response seen in the trial, with the best results seen with the 3-mg twice daily dose, then lower effects seen with two higher doses.
“Our analysis is that it’s not an inverse dose response, but rather a flat dose response above the 3-mg [twice daily] dose,” he said, noting that there was a higher dropout rate because of adverse effects in the 12-mg once daily group and those participants were recorded as nonresponders.
“We think what we’ve seen here is that 3 mg twice daily is a sufficient dose and there was no additional therapeutic gain above that.”
Rates of adverse events (AEs), serious AEs, and AEs of interest were overall fairly similar between deucravacitinib and placebo groups. The most common side effects seen with deucravacitinib were upper respiratory tract infection, nasopharyngitis, headache, and urinary tract infection. Skin reactions, such as acne, rash, and pruritis, among others, were more common in deucravacitinib- than in placebo-treated patients.
Importantly, Dr. Morand noted that there were no major cardiac events or thrombotic events and no deaths seen in the study. There was no signal for an increase in serious or opportunistic infections, including herpes zoster. There was no effect on common laboratory parameters.
“These are very encouraging results for patients with SLE,” Albert Roy, executive director of Lupus Therapeutics, said in a press release issued by the Lupus Research Alliance.
“We are honored to have played a role in this exciting work by helping to conduct this clinical trial through our Lupus Clinical Investigators Network of renowned North American academic centers.”
In an interview, he added: “We’re cautiously optimistic. Hopefully, if it continues to progress through phase 3, it’ll be the first oral agent that would be approved for lupus, notwithstanding prednisone and Plaquenil [hydroxychloroquine], back in the 50s.”
Orelabrutinib phase 2 study in SLE
Another approach to oral route of administration under investigation in SLE is the use of orelabrutinib, an irreversible inhibitor of Bruton’s tyrosine kinase (BTK) that was approved in China in December 2020 for the treatment of certain lymphomas and leukemias.
The rationale for testing it in SLE comes from two preclinical studies that had suggested a possible benefit in reducing disease activity, explained Zhanguo Li, MD, PhD, professor at Peking University People’s Hospital in Beijing. He presented the results of an ongoing randomized, double-blind, placebo-controlled, phase Ib/IIa dose-finding study comparing three different doses of orelabrutinib (50, 80, and 100 mg, once daily) to placebo.
As in the deucravacitinib trial, the SRI-4 was used to assess the potential efficacy of orelabrutinib, although in a much smaller patient population (n = 92) and at a shorter time point (12 weeks). Results showed an 11%-20% difference between the percentage of patients who met SRI-4 response criteria with orelabrutinib and those on placebo, at a respective 46.5%, 53.3%, 56.3% and 35.7%.
SLE Disease Activity Index (SLEDAI) scores showed a similar benefit of orelabrutinib over placebo, with 54%-63% and 30% of patients, respectively, achieving a score of 8 or more.
Adverse event rates were similar to those of placebo with most events being of mild or moderate nature. Three patients treated with orelabrutinib experienced serious adverse events, of which one was grade 3, but there were no reported deaths.
Pharmacokinetic and pharmacodynamic data showed a dose effect, and nearly complete occupancy of BTK was achieved at all dose levels for 24 hours, consistent with once-daily dosing.
“BTK plays an important role in B-cell regulation, thus B-cell and myeloid-cell blockade through BTK inhibition is an interesting potential new target for SLE,” Dr. Md Yusof said.
“Data from this early dose-ranging trial is encouraging. No major safety signal apart from mild reduction in lymphocyte and white cell counts,” he added.
“There are still plenty of challenges ahead for this drug’s development, particularly as none of the BTK inhibitors have yet to succeed in phase 3 trials in rheumatic and musculoskeletal diseases,” Dr. Md Yusof said.
Early days for both agents
While both seem currently promising, it’s very early days for deucravacitinib and orelabrutinib as possible new agents for SLE.
Aside from SLE, deucravacitinib is being tested across multiple immune-mediated diseases. This includes psoriasis, where two phase 3 trials – POETYK PSO-1 and POETYK PSO-2 – have already been completed, and psoriatic arthritis, where a phase 2 trial has been reported; all with positive results.
Phase 3 testing of deucravacitinib will go ahead and recruitment may start toward the end of this year, but it’ll take years to complete the studies, Dr. Morand said. Even if the trials prove positive, neither agent is going to be available for clinical use for several years.
A case in point is anifrolumab (Saphnelo), which Dr. Morand was involved in assessing. Despite gaining approval in the United States and across much of the world, the drug still going through reimbursement processes.
“The trial data, and lots of post hoc analysis, show clearly that it’s a major step forward in treating lupus,” he said in an interview, but “access is limited in most places, so hands-on experience with that new treatment is still limited for most clinicians.”
As for all the other new targeted approaches under investigation, “although there’s a lot of trial activity, there’s still a couple of years away before any of the current trials deliver new treatment. That’s if they provide positive findings. Indeed, there have been numerous agents that have shown promise at phase 2 but then fall at the final phase 3 hurdle, including baricitinib, which Dr. Morand reported on in a separate poster presentation.
Phase 3 data proved disappointing: “Results are not sufficiently positive for that to go forward,” he said, adding that “transitioning from a successful phase 2 to a successful phase 3 is challenging, and many products have failed.”
Dr. Morand added: “It’s a very exciting time to be in lupus research, and there’s a lot of optimism about the future. But when I go back to my clinic tomorrow, I treat my patients exactly the same as I did last week and last year.”
It’s yet to be seen if deucravacitinib will fulfill its early promise, but it’s off to an impressive start. A positive for patients is that it’s an oral drug, with the potential to improve access to treatment across the world where getting infusions may be an issue.
“These are some of the most exciting data that I’ve seen at the phase 2 level in terms of effect size across all the readouts that are used,” Dr. Morand said. “There’s no guesswork here; it worked across all the measures. That’s very reassuring.”
The PAISLEY study was sponsored by Bristol-Myers Squibb. Dr. Morand has acted as a consultant to the company and received research support for the conduct of the trial. He disclosed acting as a consultant or receiving research funding from AbbVie, Amgen, AstraZeneca, Biogen, Eli Lilly, EMD Serono, Janssen, Genentech, Servier, Novartis, and UCB. Mr. Roy is the executive director of Lupus Therapeutics, which manages the Lupus Clinical Investigators Network based in North America. Lupus Therapeutics is the clinical trials arm of the Lupus Research Alliance, a nongovernmental, nonprofit funder of lupus research worldwide. The orelabrutinib study was sponsored by InnoCare Pharma. Dr. Li is the principal investigator for the trial but had no conflicts of interest to declare. Dr. Md Yusof disclosed receiving consultancy fees from Aurinia Pharmaceuticals.
FROM THE EULAR 2022 CONGRESS
Lupus mutation may unlock targeted drugs for patient subset
Scientists have confirmed that a receptor long suspected to be linked to lupus is, in fact, a major driver of the autoimmune disease for at least some subset of patients, according to a study recently published in Nature. Researchers discovered the crucial role of toll-like receptor 7 (TLR7) because of a rare mutation in a pediatric patient with systemic lupus erythematosus (SLE) who had a particularly severe presentation.
“Sometimes it’s valuable to find these very severe cases where there is one mutation that has a strong effect because if we understand how those mutations work, the lessons we learn can generally tell us about disease mechanisms,” explained senior author Carola G. Vinuesa, MD, PhD, of the Centre for Personalised Immunology at Australian National University in Canberra and The Francis Crick Institute in London.

“It’s quite difficult to find one mutation that can alone cause the entire disease,” Dr. Vinuesa added, but what it reveals about how the disease develops may lead to more effective targeted therapies than the immune suppressants most often used to treat lupus currently.
The mutation they found was in the TLR7 gene that encodes the TLR7 protein. TLR7 is a receptor used by immune cells to identify viral RNA so they can fight off viral infections, including COVID-19. But if the body’s own genetic material binds to TLR7 in susceptible individuals, it can lead to an overproduction of type 1 interferons, which are cytokines that trigger or exacerbate the immune reactions that lead to lupus symptoms. The TLR7 gene occurs on the X chromosome, which may explain men’s greater susceptibility to COVID-19 and the greater incidence of lupus in women, who have two X chromosomes instead of the one that men have, Dr. Vinuesa said.
Previous research had shown an association between TLR7 and lupus, but this new study is the first to provide definitive proof that a TLR7 mutation by itself can directly cause human lupus. After discovering the variant in the patient, Dr. Vinuesa’s team used CRISPR to edit the genome of a mouse model and introduce the same mutation the patient had. “And they developed full-blown disease, just with this one single base-pair substitution – 1 letter in the 3 billion letters of the genome,” Dr. Vinuesa said. “It tells us that these receptors are not just there to recognize viral RNA, that in some circumstances, they could be triggered by our own nucleic acids.”
One pathway among many?
The finding does not mean that every lupus patient has this mutation, which remains rare, but suggests that overactivity in this receptor already reported in many lupus patients may be causally related to disease, Dr. Vinuesa said.

Noa Schwartz, MD, an assistant professor of medicine at Albert Einstein College of Medicine, New York, and director of the Montefiore-Einstein Institute for Lupus Care and Research, said in an interview that lupus is thought of as a syndrome, a collection of different but similar diseases that don’t necessarily have a single cause. But finding a single gene mutation that could potentially lead to lupus is an important piece of the puzzle, said Dr. Schwartz, who was not involved in the study. Based on past research in mice models, “we’ve hypothesized that TLR7 is important in humans as well, but this is the last nail in the coffin.”
One of the key questions this finding has prompted is how many patients’ disease results from TLR7 activity. “Because of the evidence from Ignacio Sanz’s group demonstrating TLR7 overactivity in a significant fraction of SLE patients, we believe that it is probably going to be pretty important,” Dr. Vinuesa said. “My feeling is that it is going to be quite a central pathway in lupus pathogenesis, if not the central pathway.”
Dr. Schwartz was more cautious, noting that it is probably important for a subset of patients but may “have a limited effect on the general lupus population.” While it’s not yet clear how large that subset is, it is possible it will include people with cutaneous lupus, those with primarily dermatologic symptoms.
“Hydroxychloroquine works particularly well for cutaneous manifestations of lupus, and one of the ways that works is by inhibiting TLR7 and TLR9, so this [finding] potentially matters for skin disease and lupus, but it’s very early,” Dr. Schwartz said. If it does turn out that TLR7 activity is particularly associated with cutaneous lupus, it may mean therapies with fewer side effects, she said. “Specifically for cutaneous lupus, the concept of suppressing the entire immune system for skin illness sometimes feels, especially to patients, very extreme, so they are [patients] who directed therapy could be so especially relevant for.”

Laura Lewandowski, MD, an assistant clinical investigator and head of the lupus genomics and global health disparities unit at the National Institute of Arthritis and Musculoskeletal and Skin Disease, described this study as particularly remarkable in the way it revealed the mechanism leading to lupus symptoms.
“As whole genome sequencing becomes faster and less expensive, more and more people are employing them in their studies,” most of which report changes in certain genes, Dr. Lewandowski said. “One of the most striking findings about this paper was that they took it to the next step and did a really elegant study on the exact way this gain-of-function TLR7 mutation leads to the autoimmunity that we see in lupus. The detail of mechanism in this paper is really unique.”
A step toward personalized medicine
Dr. Lewandowski is part of a team that recently presented a poster related to genomic sequencing in lupus patients at the annual meeting of the Childhood Arthritis and Rheumatology Research Alliance. Her study reported on the whole genome sequencing of patients with childhood-onset SLE who were already enrolled in the CARRA Lupus Registry. Children with lupus may be more likely than adults to have rare genetic variants, so a registry of childhood-onset SLE patients with fully sequenced genomes provides an opportunity to look for single-gene mutations specifically linked to lupus, said Dr. Lewandowski, who has recently begun a research collaboration with Dr. Vinuesa.
“As we move forward and more and more patients are included in these studies, we will understand a little bit more about the genetic architecture of patients who have rare variations leading to disease, or even common variations,” Dr. Lewandowski said about the intersection between her research and Dr. Vinuesa’s study. The more data they gather, the more they can explore the possible interactions of rare and common variants that play a role in SLE as well as what environmental triggers, such as viral infection or pollution exposure, might tip someone into having an autoimmune disease. “We’re just starting to peek under the hood,” Dr. Lewandowski said.
If further research can reveal the relative contribution of genetics to the disease and what those genetic drivers are, it may allow for greater precision in therapies and “ultimately improve the quality of life for our patients, the ultimate goal of all of these studies,” Dr. Lewandowski said.
Drugs that target TLR7 already exist for other indications, and clinical trials have already begun to see if these TLR7 inhibitors benefit lupus patients.
“If the clinical trials work, this will be quite a nice, targeted therapy with potentially much less side effects than other therapies on the market at the moment,” Dr. Vinuesa said. She is cautiously hopeful, saying it’s likely to make an impact on lupus treatment, but it’s too early to say precisely how much.
“It allows us to understand the disease mechanisms a little bit better and to try and assess what percentage of patients’ disease can be explained by overactivity in this receptor,” Dr. Vinuesa said. She thinks it’s possible that TLR7 over activation may be relevant to other systemic autoimmune diseases as well, such as Sjögren’s syndrome, rheumatoid arthritis, or juvenile dermatomyositis, but it will take more studies to find out.
“Right now, we have medicines that broadly inhibit the immune system and aren’t as targeted, but we have a lot more clinical and scientific work to do before we move this field forward for lupus patients,” Dr. Lewandowski said. “This is one case where they were able to find the exact molecular defect, and it’s not the end of the path of precision medicine — it’s the beginning.”
Dr. Vinuesa, Dr. Schwartz, and Dr. Lewandowski reported no disclosures.
A version of this article first appeared on Medscape.com.
Scientists have confirmed that a receptor long suspected to be linked to lupus is, in fact, a major driver of the autoimmune disease for at least some subset of patients, according to a study recently published in Nature. Researchers discovered the crucial role of toll-like receptor 7 (TLR7) because of a rare mutation in a pediatric patient with systemic lupus erythematosus (SLE) who had a particularly severe presentation.
“Sometimes it’s valuable to find these very severe cases where there is one mutation that has a strong effect because if we understand how those mutations work, the lessons we learn can generally tell us about disease mechanisms,” explained senior author Carola G. Vinuesa, MD, PhD, of the Centre for Personalised Immunology at Australian National University in Canberra and The Francis Crick Institute in London.
“It’s quite difficult to find one mutation that can alone cause the entire disease,” Dr. Vinuesa added, but what it reveals about how the disease develops may lead to more effective targeted therapies than the immune suppressants most often used to treat lupus currently.
The mutation they found was in the TLR7 gene that encodes the TLR7 protein. TLR7 is a receptor used by immune cells to identify viral RNA so they can fight off viral infections, including COVID-19. But if the body’s own genetic material binds to TLR7 in susceptible individuals, it can lead to an overproduction of type 1 interferons, which are cytokines that trigger or exacerbate the immune reactions that lead to lupus symptoms. The TLR7 gene occurs on the X chromosome, which may explain men’s greater susceptibility to COVID-19 and the greater incidence of lupus in women, who have two X chromosomes instead of the one that men have, Dr. Vinuesa said.
Previous research had shown an association between TLR7 and lupus, but this new study is the first to provide definitive proof that a TLR7 mutation by itself can directly cause human lupus. After discovering the variant in the patient, Dr. Vinuesa’s team used CRISPR to edit the genome of a mouse model and introduce the same mutation the patient had. “And they developed full-blown disease, just with this one single base-pair substitution – 1 letter in the 3 billion letters of the genome,” Dr. Vinuesa said. “It tells us that these receptors are not just there to recognize viral RNA, that in some circumstances, they could be triggered by our own nucleic acids.”
One pathway among many?
The finding does not mean that every lupus patient has this mutation, which remains rare, but suggests that overactivity in this receptor already reported in many lupus patients may be causally related to disease, Dr. Vinuesa said.
Noa Schwartz, MD, an assistant professor of medicine at Albert Einstein College of Medicine, New York, and director of the Montefiore-Einstein Institute for Lupus Care and Research, said in an interview that lupus is thought of as a syndrome, a collection of different but similar diseases that don’t necessarily have a single cause. But finding a single gene mutation that could potentially lead to lupus is an important piece of the puzzle, said Dr. Schwartz, who was not involved in the study. Based on past research in mice models, “we’ve hypothesized that TLR7 is important in humans as well, but this is the last nail in the coffin.”
One of the key questions this finding has prompted is how many patients’ disease results from TLR7 activity. “Because of the evidence from Ignacio Sanz’s group demonstrating TLR7 overactivity in a significant fraction of SLE patients, we believe that it is probably going to be pretty important,” Dr. Vinuesa said. “My feeling is that it is going to be quite a central pathway in lupus pathogenesis, if not the central pathway.”
Dr. Schwartz was more cautious, noting that it is probably important for a subset of patients but may “have a limited effect on the general lupus population.” While it’s not yet clear how large that subset is, it is possible it will include people with cutaneous lupus, those with primarily dermatologic symptoms.
“Hydroxychloroquine works particularly well for cutaneous manifestations of lupus, and one of the ways that works is by inhibiting TLR7 and TLR9, so this [finding] potentially matters for skin disease and lupus, but it’s very early,” Dr. Schwartz said. If it does turn out that TLR7 activity is particularly associated with cutaneous lupus, it may mean therapies with fewer side effects, she said. “Specifically for cutaneous lupus, the concept of suppressing the entire immune system for skin illness sometimes feels, especially to patients, very extreme, so they are [patients] who directed therapy could be so especially relevant for.”
Laura Lewandowski, MD, an assistant clinical investigator and head of the lupus genomics and global health disparities unit at the National Institute of Arthritis and Musculoskeletal and Skin Disease, described this study as particularly remarkable in the way it revealed the mechanism leading to lupus symptoms.
“As whole genome sequencing becomes faster and less expensive, more and more people are employing them in their studies,” most of which report changes in certain genes, Dr. Lewandowski said. “One of the most striking findings about this paper was that they took it to the next step and did a really elegant study on the exact way this gain-of-function TLR7 mutation leads to the autoimmunity that we see in lupus. The detail of mechanism in this paper is really unique.”
A step toward personalized medicine
Dr. Lewandowski is part of a team that recently presented a poster related to genomic sequencing in lupus patients at the annual meeting of the Childhood Arthritis and Rheumatology Research Alliance. Her study reported on the whole genome sequencing of patients with childhood-onset SLE who were already enrolled in the CARRA Lupus Registry. Children with lupus may be more likely than adults to have rare genetic variants, so a registry of childhood-onset SLE patients with fully sequenced genomes provides an opportunity to look for single-gene mutations specifically linked to lupus, said Dr. Lewandowski, who has recently begun a research collaboration with Dr. Vinuesa.
“As we move forward and more and more patients are included in these studies, we will understand a little bit more about the genetic architecture of patients who have rare variations leading to disease, or even common variations,” Dr. Lewandowski said about the intersection between her research and Dr. Vinuesa’s study. The more data they gather, the more they can explore the possible interactions of rare and common variants that play a role in SLE as well as what environmental triggers, such as viral infection or pollution exposure, might tip someone into having an autoimmune disease. “We’re just starting to peek under the hood,” Dr. Lewandowski said.
If further research can reveal the relative contribution of genetics to the disease and what those genetic drivers are, it may allow for greater precision in therapies and “ultimately improve the quality of life for our patients, the ultimate goal of all of these studies,” Dr. Lewandowski said.
Drugs that target TLR7 already exist for other indications, and clinical trials have already begun to see if these TLR7 inhibitors benefit lupus patients.
“If the clinical trials work, this will be quite a nice, targeted therapy with potentially much less side effects than other therapies on the market at the moment,” Dr. Vinuesa said. She is cautiously hopeful, saying it’s likely to make an impact on lupus treatment, but it’s too early to say precisely how much.
“It allows us to understand the disease mechanisms a little bit better and to try and assess what percentage of patients’ disease can be explained by overactivity in this receptor,” Dr. Vinuesa said. She thinks it’s possible that TLR7 over activation may be relevant to other systemic autoimmune diseases as well, such as Sjögren’s syndrome, rheumatoid arthritis, or juvenile dermatomyositis, but it will take more studies to find out.
“Right now, we have medicines that broadly inhibit the immune system and aren’t as targeted, but we have a lot more clinical and scientific work to do before we move this field forward for lupus patients,” Dr. Lewandowski said. “This is one case where they were able to find the exact molecular defect, and it’s not the end of the path of precision medicine — it’s the beginning.”
Dr. Vinuesa, Dr. Schwartz, and Dr. Lewandowski reported no disclosures.
A version of this article first appeared on Medscape.com.
Scientists have confirmed that a receptor long suspected to be linked to lupus is, in fact, a major driver of the autoimmune disease for at least some subset of patients, according to a study recently published in Nature. Researchers discovered the crucial role of toll-like receptor 7 (TLR7) because of a rare mutation in a pediatric patient with systemic lupus erythematosus (SLE) who had a particularly severe presentation.
“Sometimes it’s valuable to find these very severe cases where there is one mutation that has a strong effect because if we understand how those mutations work, the lessons we learn can generally tell us about disease mechanisms,” explained senior author Carola G. Vinuesa, MD, PhD, of the Centre for Personalised Immunology at Australian National University in Canberra and The Francis Crick Institute in London.
“It’s quite difficult to find one mutation that can alone cause the entire disease,” Dr. Vinuesa added, but what it reveals about how the disease develops may lead to more effective targeted therapies than the immune suppressants most often used to treat lupus currently.
The mutation they found was in the TLR7 gene that encodes the TLR7 protein. TLR7 is a receptor used by immune cells to identify viral RNA so they can fight off viral infections, including COVID-19. But if the body’s own genetic material binds to TLR7 in susceptible individuals, it can lead to an overproduction of type 1 interferons, which are cytokines that trigger or exacerbate the immune reactions that lead to lupus symptoms. The TLR7 gene occurs on the X chromosome, which may explain men’s greater susceptibility to COVID-19 and the greater incidence of lupus in women, who have two X chromosomes instead of the one that men have, Dr. Vinuesa said.
Previous research had shown an association between TLR7 and lupus, but this new study is the first to provide definitive proof that a TLR7 mutation by itself can directly cause human lupus. After discovering the variant in the patient, Dr. Vinuesa’s team used CRISPR to edit the genome of a mouse model and introduce the same mutation the patient had. “And they developed full-blown disease, just with this one single base-pair substitution – 1 letter in the 3 billion letters of the genome,” Dr. Vinuesa said. “It tells us that these receptors are not just there to recognize viral RNA, that in some circumstances, they could be triggered by our own nucleic acids.”
One pathway among many?
The finding does not mean that every lupus patient has this mutation, which remains rare, but suggests that overactivity in this receptor already reported in many lupus patients may be causally related to disease, Dr. Vinuesa said.
Noa Schwartz, MD, an assistant professor of medicine at Albert Einstein College of Medicine, New York, and director of the Montefiore-Einstein Institute for Lupus Care and Research, said in an interview that lupus is thought of as a syndrome, a collection of different but similar diseases that don’t necessarily have a single cause. But finding a single gene mutation that could potentially lead to lupus is an important piece of the puzzle, said Dr. Schwartz, who was not involved in the study. Based on past research in mice models, “we’ve hypothesized that TLR7 is important in humans as well, but this is the last nail in the coffin.”
One of the key questions this finding has prompted is how many patients’ disease results from TLR7 activity. “Because of the evidence from Ignacio Sanz’s group demonstrating TLR7 overactivity in a significant fraction of SLE patients, we believe that it is probably going to be pretty important,” Dr. Vinuesa said. “My feeling is that it is going to be quite a central pathway in lupus pathogenesis, if not the central pathway.”
Dr. Schwartz was more cautious, noting that it is probably important for a subset of patients but may “have a limited effect on the general lupus population.” While it’s not yet clear how large that subset is, it is possible it will include people with cutaneous lupus, those with primarily dermatologic symptoms.
“Hydroxychloroquine works particularly well for cutaneous manifestations of lupus, and one of the ways that works is by inhibiting TLR7 and TLR9, so this [finding] potentially matters for skin disease and lupus, but it’s very early,” Dr. Schwartz said. If it does turn out that TLR7 activity is particularly associated with cutaneous lupus, it may mean therapies with fewer side effects, she said. “Specifically for cutaneous lupus, the concept of suppressing the entire immune system for skin illness sometimes feels, especially to patients, very extreme, so they are [patients] who directed therapy could be so especially relevant for.”
Laura Lewandowski, MD, an assistant clinical investigator and head of the lupus genomics and global health disparities unit at the National Institute of Arthritis and Musculoskeletal and Skin Disease, described this study as particularly remarkable in the way it revealed the mechanism leading to lupus symptoms.
“As whole genome sequencing becomes faster and less expensive, more and more people are employing them in their studies,” most of which report changes in certain genes, Dr. Lewandowski said. “One of the most striking findings about this paper was that they took it to the next step and did a really elegant study on the exact way this gain-of-function TLR7 mutation leads to the autoimmunity that we see in lupus. The detail of mechanism in this paper is really unique.”
A step toward personalized medicine
Dr. Lewandowski is part of a team that recently presented a poster related to genomic sequencing in lupus patients at the annual meeting of the Childhood Arthritis and Rheumatology Research Alliance. Her study reported on the whole genome sequencing of patients with childhood-onset SLE who were already enrolled in the CARRA Lupus Registry. Children with lupus may be more likely than adults to have rare genetic variants, so a registry of childhood-onset SLE patients with fully sequenced genomes provides an opportunity to look for single-gene mutations specifically linked to lupus, said Dr. Lewandowski, who has recently begun a research collaboration with Dr. Vinuesa.
“As we move forward and more and more patients are included in these studies, we will understand a little bit more about the genetic architecture of patients who have rare variations leading to disease, or even common variations,” Dr. Lewandowski said about the intersection between her research and Dr. Vinuesa’s study. The more data they gather, the more they can explore the possible interactions of rare and common variants that play a role in SLE as well as what environmental triggers, such as viral infection or pollution exposure, might tip someone into having an autoimmune disease. “We’re just starting to peek under the hood,” Dr. Lewandowski said.
If further research can reveal the relative contribution of genetics to the disease and what those genetic drivers are, it may allow for greater precision in therapies and “ultimately improve the quality of life for our patients, the ultimate goal of all of these studies,” Dr. Lewandowski said.
Drugs that target TLR7 already exist for other indications, and clinical trials have already begun to see if these TLR7 inhibitors benefit lupus patients.
“If the clinical trials work, this will be quite a nice, targeted therapy with potentially much less side effects than other therapies on the market at the moment,” Dr. Vinuesa said. She is cautiously hopeful, saying it’s likely to make an impact on lupus treatment, but it’s too early to say precisely how much.
“It allows us to understand the disease mechanisms a little bit better and to try and assess what percentage of patients’ disease can be explained by overactivity in this receptor,” Dr. Vinuesa said. She thinks it’s possible that TLR7 over activation may be relevant to other systemic autoimmune diseases as well, such as Sjögren’s syndrome, rheumatoid arthritis, or juvenile dermatomyositis, but it will take more studies to find out.
“Right now, we have medicines that broadly inhibit the immune system and aren’t as targeted, but we have a lot more clinical and scientific work to do before we move this field forward for lupus patients,” Dr. Lewandowski said. “This is one case where they were able to find the exact molecular defect, and it’s not the end of the path of precision medicine — it’s the beginning.”
Dr. Vinuesa, Dr. Schwartz, and Dr. Lewandowski reported no disclosures.
A version of this article first appeared on Medscape.com.
FROM NATURE
Pemphigus Vulgaris Aggravated: Rifampicin Found at the Scene of the Crime
Case Report
A 60-year-old man presented with eroded areas in the mouth and blistering eruptions on the scalp, face, trunk, arms, and legs. He initially presented to an outside hospital 4 years prior and was treated with oral prednisone 50 mg daily, to which the eruptions responded rapidly; however, following a nearly 5-mg reduction of the dose per week by the patient and irregular oral administration, he experienced several episodes of recurrence, but he could not remember the exact dosage of prednisone he had taken during that period. Subsequently, he was admitted to our hospital because of large areas of erythema and erosions on the scalp, trunk, arms, and legs.
Since starting the prednisone regimen 4 years prior, the patient had experienced onset of hypertension, diabetes, glaucoma, cataracts, optic nerve atrophy, aseptic necrosis of the femoral head, and osteoporosis. Biopsy of a new skin lesion

The patient initially was started again prednisone 50 mg daily, to which the skin eruptions responded, and 2 weeks later, the disease was considered controlled. The prednisone dosage was tapered to 20 mg daily 3 months later with no new blister formation. However, 2 weeks later, the patient was diagnosed by a tuberculosis specialist with pulmonary tuberculosis, and a daily regimen of isoniazid, rifampicin, ethambutol, and levofloxacin was instituted.
Ten days after starting antituberculosis therapy, the patient developed new erythematous blisters that could not be controlled and self-adjusted the prednisone dose to 50 mg daily. Two months later, blister formation continued.
Six months after the initial presentation, the patient returned to our hospital because of uncontrollable rashes (Figure 2). On admission, he had a Pemphigus Disease Area Index (PDAI) score of 32 with disease involving 30% of the body surface area. Laboratory testing showed a desmoglein 1 level of 233 U/mL and desmoglein 3 level of 228 U/mL. A tuberculosis specialist from an outside hospital was consulted to evaluate the patient’s condition and assist in treatment. Based on findings from a pulmonary computed tomography scan, which showed the inflammation was considerably absorbed, treatment was adjusted to stop using ethambutol and levofloxacin and continue rifampicin and isoniazid. For the PV, prednisone was titrated upward to 75 mg daily, mycophenolate mofetil (MMF) 1 g twice daily was added, and IVIG 400 mg/kg daily was administered for 7 days. After 3 weeks, the rash still expanded.

In considering possible interactions between the drugs, we consulted the literature and found reports1-3 that rifampicin accelerated glucocorticoid metabolism, of which the tuberculosis specialist that we consulted was not aware. Therefore, rifampicin was stopped, and the antituberculosis therapy was adjusted to levofloxacin and isoniazid. Meanwhile, the steroid was changed to methylprednisolone 120 mg daily for 3 days, then to 80 mg daily for 2 days.
After 5 days, the rash was controlled with no new development and the patient was discharged. He continued on prednisone 80 mg daily and MMF 1 g twice daily.
At 2-month follow-up, no new rash had developed. The patient had already self-discontinued the MMF for 1 month because it was difficult to obtain at local hospitals. The prednisone was reduced to 40 mg daily. Pulmonary computed tomography showed no signs of reactivation of tuberculosis.
Comment
Drugs that depend on these enzymes for their metabolism are prone to
Rifampicin causes a marked reduction in dose-corrected mycophenolic acid exposure when administered simultaneously with MMF through induction of glucuronidation activity and inhibition of enterohepatic recirculation.5,10In in vitro studies, rifampin and other cytochrome P450 inducers have been identified as potentially useful for increasing the rate of cyclophosphamide and ifosfamide (an isomeric analogue of cyclophosphamide) 4-hydroxylation in the human liver in a manner that could have a favorable impact on the clinical pharmacokinetics of these anticancer prodrugs.11 However, clinical analysis of 16 patients indicated that co-administration of ifosfamide with rifampin did not result in changes in the pharmacokinetics of the parent drug or its metabolites.12
The steroids and
Conclusion
In our patient, the use of rifapentine resulted in a recurrence of previously controlled PV and resistance to treatment. The patient’s disease was quickly controlled after discontinuation of rifampicin and with a short-term course of high-dose methylprednisolone and remained stable when the dosages of MMF and prednisone were reduced.
- Miyagawa S, Yamashina Y, Okuchi T, et al. Exacerbation of pemphigus by rifampicin. Br J Dermatol. 1986;114:729-732. doi:10.1111/j.1365-2133.1986.tb04882.x
- Gange RW, Rhodes EL, Edwards CO, et al. Pemphigus induced by rifampicin. Br J Dermatol. 1976;95:445-448. doi:10.1111/j.1365-2133.1976.tb00849.x
- Bergrem H, Refvem OK. Altered prednisolone pharmacokinetics in patients treated with rifampicin. Acta Med Scand. 1983;213:339-343. doi:10.1111/j.0954-6820.1983.tb03748.x
- McAllister WA, Thompson PJ, Al-Habet SM, et al. Rifampicin reduces effectiveness and bioavailability of prednisolone. Br Med J (Clin Res Ed). 1983;286:923-925. doi:10.1136/bmj.286.6369.923
- Tavakolpour S. Pemphigus trigger factors: special focus on pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol Res. 2018;310:95-106. doi:10.1007/s00403-017-1790-8
- Barman H, Dass R, Duwarah SG. Use of high-dose prednisolone to overcome rifampicin-induced corticosteroid non-responsiveness in childhood nephrotic syndrome. Saudi J Kidney Dis Transpl. 2016;27:157-160. doi:10.4103/1319-2442.174198
- Okey AB, Roberts EA, Harper PA, et al. Induction of drug-metabolizing enzymes: mechanisms and consequences. Clin Biochem. 1986;19:132-141. doi:10.1016/s0009-9120(86)80060-1
- Venkatesan K. Pharmacokinetic interactions with rifampicin. Clin Pharmacokinet. 1992;22:47-65. doi:10.2165/00003088-199222010-00005
- Naesens M, Kuypers DRJ, Streit F, et al. Rifampin induces alterations in mycophenolic acid glucuronidation and elimination: implications for drug exposure in renal allograft recipients. Clin Pharmacol Ther. 2006;80:509-521. doi:10.1016/j.clpt.2006.08.002
- Kuypers DRJ, Verleden G, Naesens M, et al. Drug interaction between mycophenolate mofetil and rifampin: possible induction of uridine diphosphate–glucuronosyltransferase. Clin Pharmacol Ther. 2005;78:81-88. doi:10.1016/j.clpt.2005.03.004
- Chenhsu RY, Loong CC, Chou MH, et al. Renal allograft dysfunction associated with rifampin–tacrolimus interaction. Ann Pharmacother. 2000;34:27-31. doi:10.1345/aph.19069
- Douglas JG, McLeod MJ. Pharmacokinetic factors in the modern drug treatment of tuberculosis. Clin Pharmacokinet. 1999;37:127-146. doi:10.2165/00003088-199937020-00003
Case Report
A 60-year-old man presented with eroded areas in the mouth and blistering eruptions on the scalp, face, trunk, arms, and legs. He initially presented to an outside hospital 4 years prior and was treated with oral prednisone 50 mg daily, to which the eruptions responded rapidly; however, following a nearly 5-mg reduction of the dose per week by the patient and irregular oral administration, he experienced several episodes of recurrence, but he could not remember the exact dosage of prednisone he had taken during that period. Subsequently, he was admitted to our hospital because of large areas of erythema and erosions on the scalp, trunk, arms, and legs.
Since starting the prednisone regimen 4 years prior, the patient had experienced onset of hypertension, diabetes, glaucoma, cataracts, optic nerve atrophy, aseptic necrosis of the femoral head, and osteoporosis. Biopsy of a new skin lesion
The patient initially was started again prednisone 50 mg daily, to which the skin eruptions responded, and 2 weeks later, the disease was considered controlled. The prednisone dosage was tapered to 20 mg daily 3 months later with no new blister formation. However, 2 weeks later, the patient was diagnosed by a tuberculosis specialist with pulmonary tuberculosis, and a daily regimen of isoniazid, rifampicin, ethambutol, and levofloxacin was instituted.
Ten days after starting antituberculosis therapy, the patient developed new erythematous blisters that could not be controlled and self-adjusted the prednisone dose to 50 mg daily. Two months later, blister formation continued.
Six months after the initial presentation, the patient returned to our hospital because of uncontrollable rashes (Figure 2). On admission, he had a Pemphigus Disease Area Index (PDAI) score of 32 with disease involving 30% of the body surface area. Laboratory testing showed a desmoglein 1 level of 233 U/mL and desmoglein 3 level of 228 U/mL. A tuberculosis specialist from an outside hospital was consulted to evaluate the patient’s condition and assist in treatment. Based on findings from a pulmonary computed tomography scan, which showed the inflammation was considerably absorbed, treatment was adjusted to stop using ethambutol and levofloxacin and continue rifampicin and isoniazid. For the PV, prednisone was titrated upward to 75 mg daily, mycophenolate mofetil (MMF) 1 g twice daily was added, and IVIG 400 mg/kg daily was administered for 7 days. After 3 weeks, the rash still expanded.
In considering possible interactions between the drugs, we consulted the literature and found reports1-3 that rifampicin accelerated glucocorticoid metabolism, of which the tuberculosis specialist that we consulted was not aware. Therefore, rifampicin was stopped, and the antituberculosis therapy was adjusted to levofloxacin and isoniazid. Meanwhile, the steroid was changed to methylprednisolone 120 mg daily for 3 days, then to 80 mg daily for 2 days.
After 5 days, the rash was controlled with no new development and the patient was discharged. He continued on prednisone 80 mg daily and MMF 1 g twice daily.
At 2-month follow-up, no new rash had developed. The patient had already self-discontinued the MMF for 1 month because it was difficult to obtain at local hospitals. The prednisone was reduced to 40 mg daily. Pulmonary computed tomography showed no signs of reactivation of tuberculosis.
Comment
Drugs that depend on these enzymes for their metabolism are prone to
Rifampicin causes a marked reduction in dose-corrected mycophenolic acid exposure when administered simultaneously with MMF through induction of glucuronidation activity and inhibition of enterohepatic recirculation.5,10In in vitro studies, rifampin and other cytochrome P450 inducers have been identified as potentially useful for increasing the rate of cyclophosphamide and ifosfamide (an isomeric analogue of cyclophosphamide) 4-hydroxylation in the human liver in a manner that could have a favorable impact on the clinical pharmacokinetics of these anticancer prodrugs.11 However, clinical analysis of 16 patients indicated that co-administration of ifosfamide with rifampin did not result in changes in the pharmacokinetics of the parent drug or its metabolites.12
The steroids and
Conclusion
In our patient, the use of rifapentine resulted in a recurrence of previously controlled PV and resistance to treatment. The patient’s disease was quickly controlled after discontinuation of rifampicin and with a short-term course of high-dose methylprednisolone and remained stable when the dosages of MMF and prednisone were reduced.
Case Report
A 60-year-old man presented with eroded areas in the mouth and blistering eruptions on the scalp, face, trunk, arms, and legs. He initially presented to an outside hospital 4 years prior and was treated with oral prednisone 50 mg daily, to which the eruptions responded rapidly; however, following a nearly 5-mg reduction of the dose per week by the patient and irregular oral administration, he experienced several episodes of recurrence, but he could not remember the exact dosage of prednisone he had taken during that period. Subsequently, he was admitted to our hospital because of large areas of erythema and erosions on the scalp, trunk, arms, and legs.
Since starting the prednisone regimen 4 years prior, the patient had experienced onset of hypertension, diabetes, glaucoma, cataracts, optic nerve atrophy, aseptic necrosis of the femoral head, and osteoporosis. Biopsy of a new skin lesion
The patient initially was started again prednisone 50 mg daily, to which the skin eruptions responded, and 2 weeks later, the disease was considered controlled. The prednisone dosage was tapered to 20 mg daily 3 months later with no new blister formation. However, 2 weeks later, the patient was diagnosed by a tuberculosis specialist with pulmonary tuberculosis, and a daily regimen of isoniazid, rifampicin, ethambutol, and levofloxacin was instituted.
Ten days after starting antituberculosis therapy, the patient developed new erythematous blisters that could not be controlled and self-adjusted the prednisone dose to 50 mg daily. Two months later, blister formation continued.
Six months after the initial presentation, the patient returned to our hospital because of uncontrollable rashes (Figure 2). On admission, he had a Pemphigus Disease Area Index (PDAI) score of 32 with disease involving 30% of the body surface area. Laboratory testing showed a desmoglein 1 level of 233 U/mL and desmoglein 3 level of 228 U/mL. A tuberculosis specialist from an outside hospital was consulted to evaluate the patient’s condition and assist in treatment. Based on findings from a pulmonary computed tomography scan, which showed the inflammation was considerably absorbed, treatment was adjusted to stop using ethambutol and levofloxacin and continue rifampicin and isoniazid. For the PV, prednisone was titrated upward to 75 mg daily, mycophenolate mofetil (MMF) 1 g twice daily was added, and IVIG 400 mg/kg daily was administered for 7 days. After 3 weeks, the rash still expanded.
In considering possible interactions between the drugs, we consulted the literature and found reports1-3 that rifampicin accelerated glucocorticoid metabolism, of which the tuberculosis specialist that we consulted was not aware. Therefore, rifampicin was stopped, and the antituberculosis therapy was adjusted to levofloxacin and isoniazid. Meanwhile, the steroid was changed to methylprednisolone 120 mg daily for 3 days, then to 80 mg daily for 2 days.
After 5 days, the rash was controlled with no new development and the patient was discharged. He continued on prednisone 80 mg daily and MMF 1 g twice daily.
At 2-month follow-up, no new rash had developed. The patient had already self-discontinued the MMF for 1 month because it was difficult to obtain at local hospitals. The prednisone was reduced to 40 mg daily. Pulmonary computed tomography showed no signs of reactivation of tuberculosis.
Comment
Drugs that depend on these enzymes for their metabolism are prone to
Rifampicin causes a marked reduction in dose-corrected mycophenolic acid exposure when administered simultaneously with MMF through induction of glucuronidation activity and inhibition of enterohepatic recirculation.5,10In in vitro studies, rifampin and other cytochrome P450 inducers have been identified as potentially useful for increasing the rate of cyclophosphamide and ifosfamide (an isomeric analogue of cyclophosphamide) 4-hydroxylation in the human liver in a manner that could have a favorable impact on the clinical pharmacokinetics of these anticancer prodrugs.11 However, clinical analysis of 16 patients indicated that co-administration of ifosfamide with rifampin did not result in changes in the pharmacokinetics of the parent drug or its metabolites.12
The steroids and
Conclusion
In our patient, the use of rifapentine resulted in a recurrence of previously controlled PV and resistance to treatment. The patient’s disease was quickly controlled after discontinuation of rifampicin and with a short-term course of high-dose methylprednisolone and remained stable when the dosages of MMF and prednisone were reduced.
- Miyagawa S, Yamashina Y, Okuchi T, et al. Exacerbation of pemphigus by rifampicin. Br J Dermatol. 1986;114:729-732. doi:10.1111/j.1365-2133.1986.tb04882.x
- Gange RW, Rhodes EL, Edwards CO, et al. Pemphigus induced by rifampicin. Br J Dermatol. 1976;95:445-448. doi:10.1111/j.1365-2133.1976.tb00849.x
- Bergrem H, Refvem OK. Altered prednisolone pharmacokinetics in patients treated with rifampicin. Acta Med Scand. 1983;213:339-343. doi:10.1111/j.0954-6820.1983.tb03748.x
- McAllister WA, Thompson PJ, Al-Habet SM, et al. Rifampicin reduces effectiveness and bioavailability of prednisolone. Br Med J (Clin Res Ed). 1983;286:923-925. doi:10.1136/bmj.286.6369.923
- Tavakolpour S. Pemphigus trigger factors: special focus on pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol Res. 2018;310:95-106. doi:10.1007/s00403-017-1790-8
- Barman H, Dass R, Duwarah SG. Use of high-dose prednisolone to overcome rifampicin-induced corticosteroid non-responsiveness in childhood nephrotic syndrome. Saudi J Kidney Dis Transpl. 2016;27:157-160. doi:10.4103/1319-2442.174198
- Okey AB, Roberts EA, Harper PA, et al. Induction of drug-metabolizing enzymes: mechanisms and consequences. Clin Biochem. 1986;19:132-141. doi:10.1016/s0009-9120(86)80060-1
- Venkatesan K. Pharmacokinetic interactions with rifampicin. Clin Pharmacokinet. 1992;22:47-65. doi:10.2165/00003088-199222010-00005
- Naesens M, Kuypers DRJ, Streit F, et al. Rifampin induces alterations in mycophenolic acid glucuronidation and elimination: implications for drug exposure in renal allograft recipients. Clin Pharmacol Ther. 2006;80:509-521. doi:10.1016/j.clpt.2006.08.002
- Kuypers DRJ, Verleden G, Naesens M, et al. Drug interaction between mycophenolate mofetil and rifampin: possible induction of uridine diphosphate–glucuronosyltransferase. Clin Pharmacol Ther. 2005;78:81-88. doi:10.1016/j.clpt.2005.03.004
- Chenhsu RY, Loong CC, Chou MH, et al. Renal allograft dysfunction associated with rifampin–tacrolimus interaction. Ann Pharmacother. 2000;34:27-31. doi:10.1345/aph.19069
- Douglas JG, McLeod MJ. Pharmacokinetic factors in the modern drug treatment of tuberculosis. Clin Pharmacokinet. 1999;37:127-146. doi:10.2165/00003088-199937020-00003
- Miyagawa S, Yamashina Y, Okuchi T, et al. Exacerbation of pemphigus by rifampicin. Br J Dermatol. 1986;114:729-732. doi:10.1111/j.1365-2133.1986.tb04882.x
- Gange RW, Rhodes EL, Edwards CO, et al. Pemphigus induced by rifampicin. Br J Dermatol. 1976;95:445-448. doi:10.1111/j.1365-2133.1976.tb00849.x
- Bergrem H, Refvem OK. Altered prednisolone pharmacokinetics in patients treated with rifampicin. Acta Med Scand. 1983;213:339-343. doi:10.1111/j.0954-6820.1983.tb03748.x
- McAllister WA, Thompson PJ, Al-Habet SM, et al. Rifampicin reduces effectiveness and bioavailability of prednisolone. Br Med J (Clin Res Ed). 1983;286:923-925. doi:10.1136/bmj.286.6369.923
- Tavakolpour S. Pemphigus trigger factors: special focus on pemphigus vulgaris and pemphigus foliaceus. Arch Dermatol Res. 2018;310:95-106. doi:10.1007/s00403-017-1790-8
- Barman H, Dass R, Duwarah SG. Use of high-dose prednisolone to overcome rifampicin-induced corticosteroid non-responsiveness in childhood nephrotic syndrome. Saudi J Kidney Dis Transpl. 2016;27:157-160. doi:10.4103/1319-2442.174198
- Okey AB, Roberts EA, Harper PA, et al. Induction of drug-metabolizing enzymes: mechanisms and consequences. Clin Biochem. 1986;19:132-141. doi:10.1016/s0009-9120(86)80060-1
- Venkatesan K. Pharmacokinetic interactions with rifampicin. Clin Pharmacokinet. 1992;22:47-65. doi:10.2165/00003088-199222010-00005
- Naesens M, Kuypers DRJ, Streit F, et al. Rifampin induces alterations in mycophenolic acid glucuronidation and elimination: implications for drug exposure in renal allograft recipients. Clin Pharmacol Ther. 2006;80:509-521. doi:10.1016/j.clpt.2006.08.002
- Kuypers DRJ, Verleden G, Naesens M, et al. Drug interaction between mycophenolate mofetil and rifampin: possible induction of uridine diphosphate–glucuronosyltransferase. Clin Pharmacol Ther. 2005;78:81-88. doi:10.1016/j.clpt.2005.03.004
- Chenhsu RY, Loong CC, Chou MH, et al. Renal allograft dysfunction associated with rifampin–tacrolimus interaction. Ann Pharmacother. 2000;34:27-31. doi:10.1345/aph.19069
- Douglas JG, McLeod MJ. Pharmacokinetic factors in the modern drug treatment of tuberculosis. Clin Pharmacokinet. 1999;37:127-146. doi:10.2165/00003088-199937020-00003
Practice Points
- Long-term use of immunosuppressants requires constant attention for infections, especially latent infections in the body.
- Clinicians should carefully inquire with patients about concomitant diseases and medications used, and be vigilant about drug interactions.

Lenabasum improved skin symptoms in dermatomyositis, but future is uncertain
An – some of it statistically significant – in a phase 2, double-blind, randomized, controlled study.
Patients taking lenabasum experienced greater reductions in the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI) – a validated outcome designed to assess inflammatory skin involvement in the rare autoimmune disease – and improvements in patient-reported and biomarker outcomes, compared with those on placebo, dermatologist Victoria P. Werth, MD, and coinvestigators reported.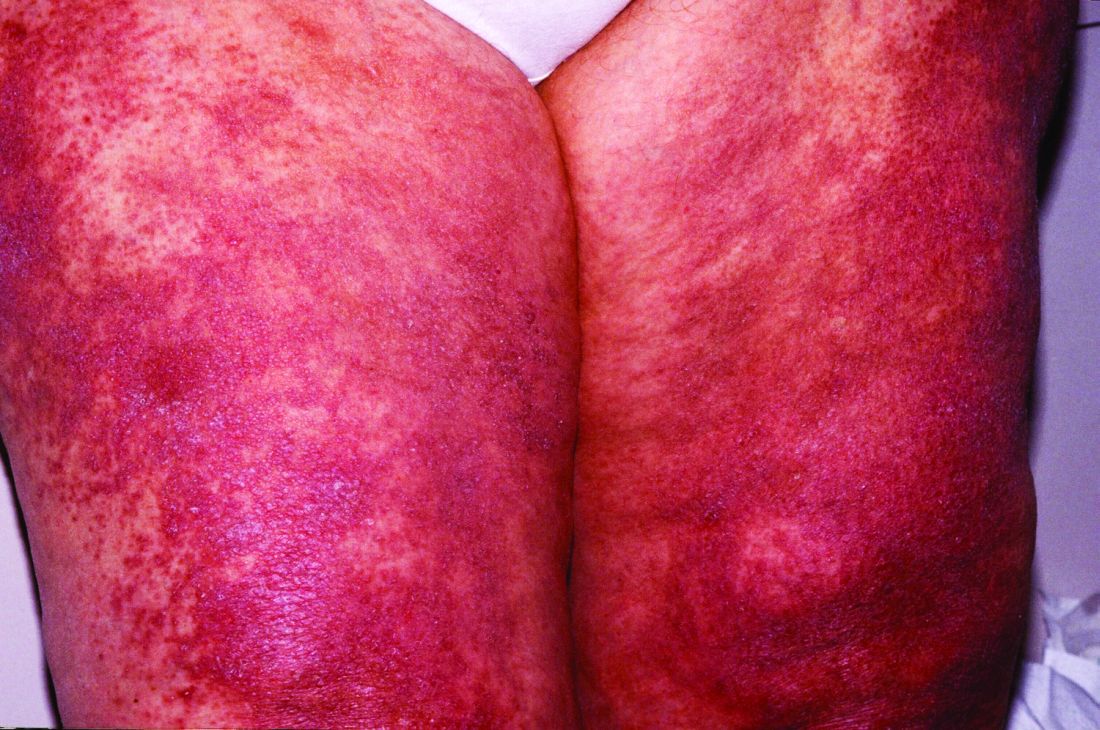
And in a recently completed phase 3 trial, reported by the manufacturer, a subpopulation of patients with active skin disease and no active muscle disease again showed greater reductions in CDASI activity scores – a secondary outcome in the trial.
However, the phase 3 DETERMINE trial produced negative findings overall. It enrolled a more heterogeneous group of patients – including those with both muscle weakness and skin involvement – and its primary outcome measure was a broader composite measure, the Total Improvement Score. The trial failed to meet this primary endpoint, Corbus Pharmaceuticals, the developer of lenabasum, announced in a press release in June 2021.
The phase 3 results are “frustrating” for patients with symptomatic and refractory skin manifestations of dermatomyositis (DM), given the promising findings from the phase 2 trial and from an open-label extension study, said Dr. Werth, professor of dermatology and medicine, University of Pennsylvania, Philadelphia, and principal investigator and coprincipal investigator of the phase 2 and phase 3 studies, respectively.
Dr. Werth is scheduled to present the results from the phase 3 trial at the annual European Alliance of Associations for Rheumatology meeting in June.
“With lenabasum, we have a therapy that doesn’t work for every patient, but does work for quite a number of them,” Dr. Werth said in an interview. “It’s oral, it’s not really that immunosuppressing, and there aren’t many side effects. Right now, patients are often being managed with steroids ... we really need treatments that are not as toxic.”
Robert Spiera, MD, a rheumatologist who led trials of lenabasum for treatment of diffuse cutaneous systemic sclerosis (dcSSc), agreed. “The CB2 agonist strategy is appealing because it’s nonimmunosuppressing and has both anti-inflammatory and antifibrotic properties,” he said in an interview. “I wouldn’t want to give up on it ... especially [for patients] with scleroderma and dermatomyositis who are treated with substantial drugs that are associated with morbidity.”
Lenabasum, he said, has proven to be “incredibly safe, and incredibly safe in the long term.”
While the phase 2 trial of the drug for dcSSc showed clear benefit over placebo, the phase 3 trial did not meet its primary endpoint using the American College of Rheumatology Combined Response Index in Diffuse Cutaneous Systemic Sclerosis.

It allowed background immunosuppressant therapy to reflect real-world clinical practice, and “there was such a high response rate to [that therapy, largely mycophenolate] that there was little room to show benefit beyond that,” said Dr. Spiera, director of the vasculitis and scleroderma program, Hospital for Special Surgery, New York.
The drug led to more improvement in the small subset of participants who were not receiving background immunotherapy during the trial, he noted.
Corbus is currently “seeking a partnership to further explore the drug” for treatment in different subpopulations, according to a company spokesperson. Results of a phase 2 trial of lenabasum for the treatment of systemic lupus erythematosus – with a pain rating as the primary outcome measure – are expected soon.
Phase 2 findings
The single-center phase 2 trial of lenabasum for DM enrolled 22 adults with minimal muscle involvement as evidenced by normal maximal resistance on muscle testing at entry and throughout the study. Most were taking immunosuppressant medication, and all had CDASI scores of at least 20, with mean scores in the severe range (> 26). Symptoms registered on patient-reported outcome measures were moderate to severe.
Patients received a half-dose of lenabasum (20 mg daily) for 1 month and a full dose (20 mg twice daily) for 2 months, or placebo, and were followed for an additional month without dosing.
Starting at day 43 – approximately 2 weeks after the dose was increased – there was “a trend for the change from baseline CDASI to be greater” in the lenabasum group, compared with those on placebo, Dr. Werth and colleagues reported. The differences reached statistical significance on day 113 (P = .038), a month after patients discontinued lenabasum, “suggesting that the modulation of the inflammatory response by lenabasum continued beyond its last dose.”
Five of the 11 patients treated with lenabasum (45%), and none of those on placebo, achieved at least a 40% reduction in the CDASI activity score by the end of the study.
Patients in the lenabasum group also had greater improvement in the Skindex-29 Symptoms scores – an objective measure of itch – and improvements in other secondary efficacy outcomes, including pain, though these did not reach statistical significance.
Skin biopsies before and after treatment showed significant reductions in inflammatory cytokines relevant to DM pathogenesis. Patients treated with the CB2 agonist had a downward trend in the CD4+ T cell population, which correlated with decreased CDASI activity scores, for instance, and a decrease in IL-31 protein expression, which correlated with decreased Skindex-29 Symptoms scores, the investigators reported.
There were no serious adverse events related to the CB2 agonist, and no treatment discontinuations.
The main part of the phase 2 trial, conducted from 2015 to 2017, was followed by a 3-year, open-label extension, in which 20 of the 22 patients took lenabasum 20 mg twice a day. The drug continued to be safe and well tolerated, and the CDASI activity score and other outcomes improved through year 1 and remained stable thereafter, according to a poster presented by Dr. Werth at the 2021 EULAR meeting.
After 1 year in the open-label extension, 60%-70% of patients had achieved mild skin disease, and 75% had achieved at least a 40% reduction in CDASI activity.
“A lot of patients, even if they weren’t completely cleared, were much happier in terms of their itch,” said Dr. Werth, also chief of dermatology, Corporal Michael J. Crescenz Veterans Affairs Medical Center, Philadelphia. “It’s been difficult for a lot of them now that they’re off the long-term extension ... a lot of them are flaring.”
The future
In the lab, with funding from the National Institutes of Health, Dr. Werth is continuing to investigate how lenabasum may be working in DM. A paper just published in the open access journal Arthritis Research & Therapy describes CB2 receptor distribution and up-regulation on key immune cells in the skin and blood, and how, in DM skin, its highest expression is on dendritic cells.
Through both mechanistic and more clinical research, “it’s important to understand the characteristics of the people [lenabasum] worked in or didn’t work in,” she said.
And in clinical trials, it’s important to capture meaningful improvement from the patient perspective. “It may be,” she noted, “that more global, systemic assessments are not the way to go for autoimmune skin disease.”
For dcSSc, Dr. Spiera said, it’s possible that a CB2 agonist may be helpful for patients who have been on immunosuppressants, particularly mycophenolate, for more than 6 months “and are still struggling.”
The phase 2 trial in DM was funded by the National Institutes of Health, the Department of Veterans Affairs, and Corbus Pharmaceuticals. The phase 3 trials in DM and in dcSSc were funded by Corbus. Dr. Werth disclosed grant support from Corbus and several other pharmaceutical companies. Dr. Spiera disclosed that he has received grant support or consulting fees from Roche-Genentech, GlaxoSmithKline, and several other pharmaceutical companies.
A version of this article first appeared on Medscape.com.
An – some of it statistically significant – in a phase 2, double-blind, randomized, controlled study.
Patients taking lenabasum experienced greater reductions in the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI) – a validated outcome designed to assess inflammatory skin involvement in the rare autoimmune disease – and improvements in patient-reported and biomarker outcomes, compared with those on placebo, dermatologist Victoria P. Werth, MD, and coinvestigators reported.
And in a recently completed phase 3 trial, reported by the manufacturer, a subpopulation of patients with active skin disease and no active muscle disease again showed greater reductions in CDASI activity scores – a secondary outcome in the trial.
However, the phase 3 DETERMINE trial produced negative findings overall. It enrolled a more heterogeneous group of patients – including those with both muscle weakness and skin involvement – and its primary outcome measure was a broader composite measure, the Total Improvement Score. The trial failed to meet this primary endpoint, Corbus Pharmaceuticals, the developer of lenabasum, announced in a press release in June 2021.
The phase 3 results are “frustrating” for patients with symptomatic and refractory skin manifestations of dermatomyositis (DM), given the promising findings from the phase 2 trial and from an open-label extension study, said Dr. Werth, professor of dermatology and medicine, University of Pennsylvania, Philadelphia, and principal investigator and coprincipal investigator of the phase 2 and phase 3 studies, respectively.
Dr. Werth is scheduled to present the results from the phase 3 trial at the annual European Alliance of Associations for Rheumatology meeting in June.
“With lenabasum, we have a therapy that doesn’t work for every patient, but does work for quite a number of them,” Dr. Werth said in an interview. “It’s oral, it’s not really that immunosuppressing, and there aren’t many side effects. Right now, patients are often being managed with steroids ... we really need treatments that are not as toxic.”
Robert Spiera, MD, a rheumatologist who led trials of lenabasum for treatment of diffuse cutaneous systemic sclerosis (dcSSc), agreed. “The CB2 agonist strategy is appealing because it’s nonimmunosuppressing and has both anti-inflammatory and antifibrotic properties,” he said in an interview. “I wouldn’t want to give up on it ... especially [for patients] with scleroderma and dermatomyositis who are treated with substantial drugs that are associated with morbidity.”
Lenabasum, he said, has proven to be “incredibly safe, and incredibly safe in the long term.”
While the phase 2 trial of the drug for dcSSc showed clear benefit over placebo, the phase 3 trial did not meet its primary endpoint using the American College of Rheumatology Combined Response Index in Diffuse Cutaneous Systemic Sclerosis.
It allowed background immunosuppressant therapy to reflect real-world clinical practice, and “there was such a high response rate to [that therapy, largely mycophenolate] that there was little room to show benefit beyond that,” said Dr. Spiera, director of the vasculitis and scleroderma program, Hospital for Special Surgery, New York.
The drug led to more improvement in the small subset of participants who were not receiving background immunotherapy during the trial, he noted.
Corbus is currently “seeking a partnership to further explore the drug” for treatment in different subpopulations, according to a company spokesperson. Results of a phase 2 trial of lenabasum for the treatment of systemic lupus erythematosus – with a pain rating as the primary outcome measure – are expected soon.
Phase 2 findings
The single-center phase 2 trial of lenabasum for DM enrolled 22 adults with minimal muscle involvement as evidenced by normal maximal resistance on muscle testing at entry and throughout the study. Most were taking immunosuppressant medication, and all had CDASI scores of at least 20, with mean scores in the severe range (> 26). Symptoms registered on patient-reported outcome measures were moderate to severe.
Patients received a half-dose of lenabasum (20 mg daily) for 1 month and a full dose (20 mg twice daily) for 2 months, or placebo, and were followed for an additional month without dosing.
Starting at day 43 – approximately 2 weeks after the dose was increased – there was “a trend for the change from baseline CDASI to be greater” in the lenabasum group, compared with those on placebo, Dr. Werth and colleagues reported. The differences reached statistical significance on day 113 (P = .038), a month after patients discontinued lenabasum, “suggesting that the modulation of the inflammatory response by lenabasum continued beyond its last dose.”
Five of the 11 patients treated with lenabasum (45%), and none of those on placebo, achieved at least a 40% reduction in the CDASI activity score by the end of the study.
Patients in the lenabasum group also had greater improvement in the Skindex-29 Symptoms scores – an objective measure of itch – and improvements in other secondary efficacy outcomes, including pain, though these did not reach statistical significance.
Skin biopsies before and after treatment showed significant reductions in inflammatory cytokines relevant to DM pathogenesis. Patients treated with the CB2 agonist had a downward trend in the CD4+ T cell population, which correlated with decreased CDASI activity scores, for instance, and a decrease in IL-31 protein expression, which correlated with decreased Skindex-29 Symptoms scores, the investigators reported.
There were no serious adverse events related to the CB2 agonist, and no treatment discontinuations.
The main part of the phase 2 trial, conducted from 2015 to 2017, was followed by a 3-year, open-label extension, in which 20 of the 22 patients took lenabasum 20 mg twice a day. The drug continued to be safe and well tolerated, and the CDASI activity score and other outcomes improved through year 1 and remained stable thereafter, according to a poster presented by Dr. Werth at the 2021 EULAR meeting.
After 1 year in the open-label extension, 60%-70% of patients had achieved mild skin disease, and 75% had achieved at least a 40% reduction in CDASI activity.
“A lot of patients, even if they weren’t completely cleared, were much happier in terms of their itch,” said Dr. Werth, also chief of dermatology, Corporal Michael J. Crescenz Veterans Affairs Medical Center, Philadelphia. “It’s been difficult for a lot of them now that they’re off the long-term extension ... a lot of them are flaring.”
The future
In the lab, with funding from the National Institutes of Health, Dr. Werth is continuing to investigate how lenabasum may be working in DM. A paper just published in the open access journal Arthritis Research & Therapy describes CB2 receptor distribution and up-regulation on key immune cells in the skin and blood, and how, in DM skin, its highest expression is on dendritic cells.
Through both mechanistic and more clinical research, “it’s important to understand the characteristics of the people [lenabasum] worked in or didn’t work in,” she said.
And in clinical trials, it’s important to capture meaningful improvement from the patient perspective. “It may be,” she noted, “that more global, systemic assessments are not the way to go for autoimmune skin disease.”
For dcSSc, Dr. Spiera said, it’s possible that a CB2 agonist may be helpful for patients who have been on immunosuppressants, particularly mycophenolate, for more than 6 months “and are still struggling.”
The phase 2 trial in DM was funded by the National Institutes of Health, the Department of Veterans Affairs, and Corbus Pharmaceuticals. The phase 3 trials in DM and in dcSSc were funded by Corbus. Dr. Werth disclosed grant support from Corbus and several other pharmaceutical companies. Dr. Spiera disclosed that he has received grant support or consulting fees from Roche-Genentech, GlaxoSmithKline, and several other pharmaceutical companies.
A version of this article first appeared on Medscape.com.
An – some of it statistically significant – in a phase 2, double-blind, randomized, controlled study.
Patients taking lenabasum experienced greater reductions in the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI) – a validated outcome designed to assess inflammatory skin involvement in the rare autoimmune disease – and improvements in patient-reported and biomarker outcomes, compared with those on placebo, dermatologist Victoria P. Werth, MD, and coinvestigators reported.
And in a recently completed phase 3 trial, reported by the manufacturer, a subpopulation of patients with active skin disease and no active muscle disease again showed greater reductions in CDASI activity scores – a secondary outcome in the trial.
However, the phase 3 DETERMINE trial produced negative findings overall. It enrolled a more heterogeneous group of patients – including those with both muscle weakness and skin involvement – and its primary outcome measure was a broader composite measure, the Total Improvement Score. The trial failed to meet this primary endpoint, Corbus Pharmaceuticals, the developer of lenabasum, announced in a press release in June 2021.
The phase 3 results are “frustrating” for patients with symptomatic and refractory skin manifestations of dermatomyositis (DM), given the promising findings from the phase 2 trial and from an open-label extension study, said Dr. Werth, professor of dermatology and medicine, University of Pennsylvania, Philadelphia, and principal investigator and coprincipal investigator of the phase 2 and phase 3 studies, respectively.
Dr. Werth is scheduled to present the results from the phase 3 trial at the annual European Alliance of Associations for Rheumatology meeting in June.
“With lenabasum, we have a therapy that doesn’t work for every patient, but does work for quite a number of them,” Dr. Werth said in an interview. “It’s oral, it’s not really that immunosuppressing, and there aren’t many side effects. Right now, patients are often being managed with steroids ... we really need treatments that are not as toxic.”
Robert Spiera, MD, a rheumatologist who led trials of lenabasum for treatment of diffuse cutaneous systemic sclerosis (dcSSc), agreed. “The CB2 agonist strategy is appealing because it’s nonimmunosuppressing and has both anti-inflammatory and antifibrotic properties,” he said in an interview. “I wouldn’t want to give up on it ... especially [for patients] with scleroderma and dermatomyositis who are treated with substantial drugs that are associated with morbidity.”
Lenabasum, he said, has proven to be “incredibly safe, and incredibly safe in the long term.”
While the phase 2 trial of the drug for dcSSc showed clear benefit over placebo, the phase 3 trial did not meet its primary endpoint using the American College of Rheumatology Combined Response Index in Diffuse Cutaneous Systemic Sclerosis.
It allowed background immunosuppressant therapy to reflect real-world clinical practice, and “there was such a high response rate to [that therapy, largely mycophenolate] that there was little room to show benefit beyond that,” said Dr. Spiera, director of the vasculitis and scleroderma program, Hospital for Special Surgery, New York.
The drug led to more improvement in the small subset of participants who were not receiving background immunotherapy during the trial, he noted.
Corbus is currently “seeking a partnership to further explore the drug” for treatment in different subpopulations, according to a company spokesperson. Results of a phase 2 trial of lenabasum for the treatment of systemic lupus erythematosus – with a pain rating as the primary outcome measure – are expected soon.
Phase 2 findings
The single-center phase 2 trial of lenabasum for DM enrolled 22 adults with minimal muscle involvement as evidenced by normal maximal resistance on muscle testing at entry and throughout the study. Most were taking immunosuppressant medication, and all had CDASI scores of at least 20, with mean scores in the severe range (> 26). Symptoms registered on patient-reported outcome measures were moderate to severe.
Patients received a half-dose of lenabasum (20 mg daily) for 1 month and a full dose (20 mg twice daily) for 2 months, or placebo, and were followed for an additional month without dosing.
Starting at day 43 – approximately 2 weeks after the dose was increased – there was “a trend for the change from baseline CDASI to be greater” in the lenabasum group, compared with those on placebo, Dr. Werth and colleagues reported. The differences reached statistical significance on day 113 (P = .038), a month after patients discontinued lenabasum, “suggesting that the modulation of the inflammatory response by lenabasum continued beyond its last dose.”
Five of the 11 patients treated with lenabasum (45%), and none of those on placebo, achieved at least a 40% reduction in the CDASI activity score by the end of the study.
Patients in the lenabasum group also had greater improvement in the Skindex-29 Symptoms scores – an objective measure of itch – and improvements in other secondary efficacy outcomes, including pain, though these did not reach statistical significance.
Skin biopsies before and after treatment showed significant reductions in inflammatory cytokines relevant to DM pathogenesis. Patients treated with the CB2 agonist had a downward trend in the CD4+ T cell population, which correlated with decreased CDASI activity scores, for instance, and a decrease in IL-31 protein expression, which correlated with decreased Skindex-29 Symptoms scores, the investigators reported.
There were no serious adverse events related to the CB2 agonist, and no treatment discontinuations.
The main part of the phase 2 trial, conducted from 2015 to 2017, was followed by a 3-year, open-label extension, in which 20 of the 22 patients took lenabasum 20 mg twice a day. The drug continued to be safe and well tolerated, and the CDASI activity score and other outcomes improved through year 1 and remained stable thereafter, according to a poster presented by Dr. Werth at the 2021 EULAR meeting.
After 1 year in the open-label extension, 60%-70% of patients had achieved mild skin disease, and 75% had achieved at least a 40% reduction in CDASI activity.
“A lot of patients, even if they weren’t completely cleared, were much happier in terms of their itch,” said Dr. Werth, also chief of dermatology, Corporal Michael J. Crescenz Veterans Affairs Medical Center, Philadelphia. “It’s been difficult for a lot of them now that they’re off the long-term extension ... a lot of them are flaring.”
The future
In the lab, with funding from the National Institutes of Health, Dr. Werth is continuing to investigate how lenabasum may be working in DM. A paper just published in the open access journal Arthritis Research & Therapy describes CB2 receptor distribution and up-regulation on key immune cells in the skin and blood, and how, in DM skin, its highest expression is on dendritic cells.
Through both mechanistic and more clinical research, “it’s important to understand the characteristics of the people [lenabasum] worked in or didn’t work in,” she said.
And in clinical trials, it’s important to capture meaningful improvement from the patient perspective. “It may be,” she noted, “that more global, systemic assessments are not the way to go for autoimmune skin disease.”
For dcSSc, Dr. Spiera said, it’s possible that a CB2 agonist may be helpful for patients who have been on immunosuppressants, particularly mycophenolate, for more than 6 months “and are still struggling.”
The phase 2 trial in DM was funded by the National Institutes of Health, the Department of Veterans Affairs, and Corbus Pharmaceuticals. The phase 3 trials in DM and in dcSSc were funded by Corbus. Dr. Werth disclosed grant support from Corbus and several other pharmaceutical companies. Dr. Spiera disclosed that he has received grant support or consulting fees from Roche-Genentech, GlaxoSmithKline, and several other pharmaceutical companies.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF INVESTIGATIVE DERMATOLOGY