User login
How a vaccine may reduce the risk of ALL

Photo by Petr Kratochvil
Researchers believe they have discovered how a commonly administered vaccine protects children from developing acute lymphoblastic leukemia (ALL).
The Haemophilus influenzae Type b (Hib) vaccine is part of the standard vaccination schedule recommended for children by the US Centers for Disease Control and Prevention. The vaccine is routinely given in 4 doses before children reach 15 months of age.
The Hib vaccine prevents ear infections and meningitis caused by the Hib bacterium, but it may also protect against ALL.
This protection has been suggested in previous studies, but it is not well-known among the public at large, and the mechanism underlying this effect has been poorly understood.
Now, researchers have shown that recurrent Hib infections can shift certain genes into overdrive, converting pre-leukemic cells into full-blown cancer. The team described this work in Nature Immunology.
“These experiments help explain why the incidence of leukemia has been dramatically reduced since the advent of regular vaccinations during infancy,” said study author Markus Müschen, MD, PhD, of the University of California San Francisco.
“Hib and other childhood infections can cause recurrent and vehement immune responses, which we have found could lead to leukemia, but infants that have received vaccines are largely protected and acquire long-term immunity through very mild immune reactions.”
For this study, Dr Müschen and his colleagues tested the idea that chronic inflammation caused by recurrent infections might cause additional genetic lesions in blood cells already carrying an oncogene, promoting their transformation to overt disease.
The team conducted experiments in mice and discovered that the enzymes AID and RAG1-RAG2 drive this process. AID and RAG1-RAG2 introduce mutations in DNA that allow immune cells to adapt to infectious challenges, and these enzymes are necessary for a normal and efficient immune response.
The researchers found that, in the presence of chronic infection, AID and RAG1-RAG2 were hyperactivated, randomly cutting and mutating genes.
Additional experiments revealed that AID and RAG1-RAG2 working together is critical to introduce the additional lesions that result in ALL.
Though the researchers focused on a bacterial infection in this study, they believe the same mechanisms may be at work in viral infections.
The team is currently conducting experiments to determine if protection against leukemia is provided by vaccines against viral infections, such as the measles-mumps-rubella vaccine. ![]()
Photo by Petr Kratochvil
Researchers believe they have discovered how a commonly administered vaccine protects children from developing acute lymphoblastic leukemia (ALL).
The Haemophilus influenzae Type b (Hib) vaccine is part of the standard vaccination schedule recommended for children by the US Centers for Disease Control and Prevention. The vaccine is routinely given in 4 doses before children reach 15 months of age.
The Hib vaccine prevents ear infections and meningitis caused by the Hib bacterium, but it may also protect against ALL.
This protection has been suggested in previous studies, but it is not well-known among the public at large, and the mechanism underlying this effect has been poorly understood.
Now, researchers have shown that recurrent Hib infections can shift certain genes into overdrive, converting pre-leukemic cells into full-blown cancer. The team described this work in Nature Immunology.
“These experiments help explain why the incidence of leukemia has been dramatically reduced since the advent of regular vaccinations during infancy,” said study author Markus Müschen, MD, PhD, of the University of California San Francisco.
“Hib and other childhood infections can cause recurrent and vehement immune responses, which we have found could lead to leukemia, but infants that have received vaccines are largely protected and acquire long-term immunity through very mild immune reactions.”
For this study, Dr Müschen and his colleagues tested the idea that chronic inflammation caused by recurrent infections might cause additional genetic lesions in blood cells already carrying an oncogene, promoting their transformation to overt disease.
The team conducted experiments in mice and discovered that the enzymes AID and RAG1-RAG2 drive this process. AID and RAG1-RAG2 introduce mutations in DNA that allow immune cells to adapt to infectious challenges, and these enzymes are necessary for a normal and efficient immune response.
The researchers found that, in the presence of chronic infection, AID and RAG1-RAG2 were hyperactivated, randomly cutting and mutating genes.
Additional experiments revealed that AID and RAG1-RAG2 working together is critical to introduce the additional lesions that result in ALL.
Though the researchers focused on a bacterial infection in this study, they believe the same mechanisms may be at work in viral infections.
The team is currently conducting experiments to determine if protection against leukemia is provided by vaccines against viral infections, such as the measles-mumps-rubella vaccine. ![]()
Photo by Petr Kratochvil
Researchers believe they have discovered how a commonly administered vaccine protects children from developing acute lymphoblastic leukemia (ALL).
The Haemophilus influenzae Type b (Hib) vaccine is part of the standard vaccination schedule recommended for children by the US Centers for Disease Control and Prevention. The vaccine is routinely given in 4 doses before children reach 15 months of age.
The Hib vaccine prevents ear infections and meningitis caused by the Hib bacterium, but it may also protect against ALL.
This protection has been suggested in previous studies, but it is not well-known among the public at large, and the mechanism underlying this effect has been poorly understood.
Now, researchers have shown that recurrent Hib infections can shift certain genes into overdrive, converting pre-leukemic cells into full-blown cancer. The team described this work in Nature Immunology.
“These experiments help explain why the incidence of leukemia has been dramatically reduced since the advent of regular vaccinations during infancy,” said study author Markus Müschen, MD, PhD, of the University of California San Francisco.
“Hib and other childhood infections can cause recurrent and vehement immune responses, which we have found could lead to leukemia, but infants that have received vaccines are largely protected and acquire long-term immunity through very mild immune reactions.”
For this study, Dr Müschen and his colleagues tested the idea that chronic inflammation caused by recurrent infections might cause additional genetic lesions in blood cells already carrying an oncogene, promoting their transformation to overt disease.
The team conducted experiments in mice and discovered that the enzymes AID and RAG1-RAG2 drive this process. AID and RAG1-RAG2 introduce mutations in DNA that allow immune cells to adapt to infectious challenges, and these enzymes are necessary for a normal and efficient immune response.
The researchers found that, in the presence of chronic infection, AID and RAG1-RAG2 were hyperactivated, randomly cutting and mutating genes.
Additional experiments revealed that AID and RAG1-RAG2 working together is critical to introduce the additional lesions that result in ALL.
Though the researchers focused on a bacterial infection in this study, they believe the same mechanisms may be at work in viral infections.
The team is currently conducting experiments to determine if protection against leukemia is provided by vaccines against viral infections, such as the measles-mumps-rubella vaccine. ![]()
MRD doesn’t suggest need for more treatment
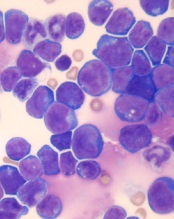
© Hind Medyouf, German
Cancer Research Center
A new study suggests that minimal residual disease (MRD) alone is not predictive of outcomes in children with T-cell acute lymphoblastic leukemia (T-ALL).
Study investigators analyzed a small group of T-ALL patients who received similar treatment regimens, comparing those with and without MRD after induction.
None of the MRD-positive patients relapsed within the follow-up period, despite not receiving intensified treatment to fully eradicate their disease.
These results, published in Pediatric Blood & Cancer, suggest T-ALL patients with MRD may not need intensified treatment and can therefore avoid additional toxicities.
“Until now, the dogma has been that, for patients with leukemia who have minimal residual disease at the end of induction, we need to intensify their treatment, which also increases side effects,” said study author Hisham Abdel-Azim, MD, of Children’s Hospital Los Angeles in California.
“We have found, for T-ALL, patients have excellent outcomes without therapy intensification and its associated toxicities.”
Dr Abdel-Azim and his colleagues studied 33 children (ages 1 to 21) with newly diagnosed T-ALL. Their treatment included induction, augmented consolidation, interim maintenance (high-dose [5 g/m2] or escalating-dose intravenous methotrexate), 1 delayed intensification, and maintenance. Twenty-one patients underwent cranial irradiation, and 1 received a transplant.
After induction, 19 of the 32 patients analyzed had MRD, which was defined as ≥ 0.01% residual leukemia cells. At the end of consolidation, 6 of the 11 patients tested were MRD-positive. And at the end of interim maintenance, 2 of the 4 patients tested were MRD-positive.
The 19 patients who were MRD-positive after induction were in continuous complete remission at a median follow-up of 4 years (range, 1.3-7.1 years). The same was true for 13 of the 14 MRD-negative patients. One of the MRD-negative patients relapsed 18 months after diagnosis but was still alive with refractory disease at last follow-up.
Dr Abdel-Azim and his colleagues noted that there were no significant differences in treatment variables between MRD-positive patients and MRD-negative patients. However, 1 patient did receive a transplant for rising MRD at 5.4 months after diagnosis.
The investigators also said they did not find any associations between MRD positivity after induction and patients’ age, sex, ethnicity, weight, white blood cell count at diagnosis, cytogenetics, immunophenotype, or the type of steroid they received during induction therapy.
The team said these results may be explained by the fact that clearance of leukemia cells from the blood is slower in patients with T-ALL than in those with B-cell ALL. However, the leukemia cells ultimately clear in T-ALL without changes in therapy. ![]()
© Hind Medyouf, German
Cancer Research Center
A new study suggests that minimal residual disease (MRD) alone is not predictive of outcomes in children with T-cell acute lymphoblastic leukemia (T-ALL).
Study investigators analyzed a small group of T-ALL patients who received similar treatment regimens, comparing those with and without MRD after induction.
None of the MRD-positive patients relapsed within the follow-up period, despite not receiving intensified treatment to fully eradicate their disease.
These results, published in Pediatric Blood & Cancer, suggest T-ALL patients with MRD may not need intensified treatment and can therefore avoid additional toxicities.
“Until now, the dogma has been that, for patients with leukemia who have minimal residual disease at the end of induction, we need to intensify their treatment, which also increases side effects,” said study author Hisham Abdel-Azim, MD, of Children’s Hospital Los Angeles in California.
“We have found, for T-ALL, patients have excellent outcomes without therapy intensification and its associated toxicities.”
Dr Abdel-Azim and his colleagues studied 33 children (ages 1 to 21) with newly diagnosed T-ALL. Their treatment included induction, augmented consolidation, interim maintenance (high-dose [5 g/m2] or escalating-dose intravenous methotrexate), 1 delayed intensification, and maintenance. Twenty-one patients underwent cranial irradiation, and 1 received a transplant.
After induction, 19 of the 32 patients analyzed had MRD, which was defined as ≥ 0.01% residual leukemia cells. At the end of consolidation, 6 of the 11 patients tested were MRD-positive. And at the end of interim maintenance, 2 of the 4 patients tested were MRD-positive.
The 19 patients who were MRD-positive after induction were in continuous complete remission at a median follow-up of 4 years (range, 1.3-7.1 years). The same was true for 13 of the 14 MRD-negative patients. One of the MRD-negative patients relapsed 18 months after diagnosis but was still alive with refractory disease at last follow-up.
Dr Abdel-Azim and his colleagues noted that there were no significant differences in treatment variables between MRD-positive patients and MRD-negative patients. However, 1 patient did receive a transplant for rising MRD at 5.4 months after diagnosis.
The investigators also said they did not find any associations between MRD positivity after induction and patients’ age, sex, ethnicity, weight, white blood cell count at diagnosis, cytogenetics, immunophenotype, or the type of steroid they received during induction therapy.
The team said these results may be explained by the fact that clearance of leukemia cells from the blood is slower in patients with T-ALL than in those with B-cell ALL. However, the leukemia cells ultimately clear in T-ALL without changes in therapy. ![]()
© Hind Medyouf, German
Cancer Research Center
A new study suggests that minimal residual disease (MRD) alone is not predictive of outcomes in children with T-cell acute lymphoblastic leukemia (T-ALL).
Study investigators analyzed a small group of T-ALL patients who received similar treatment regimens, comparing those with and without MRD after induction.
None of the MRD-positive patients relapsed within the follow-up period, despite not receiving intensified treatment to fully eradicate their disease.
These results, published in Pediatric Blood & Cancer, suggest T-ALL patients with MRD may not need intensified treatment and can therefore avoid additional toxicities.
“Until now, the dogma has been that, for patients with leukemia who have minimal residual disease at the end of induction, we need to intensify their treatment, which also increases side effects,” said study author Hisham Abdel-Azim, MD, of Children’s Hospital Los Angeles in California.
“We have found, for T-ALL, patients have excellent outcomes without therapy intensification and its associated toxicities.”
Dr Abdel-Azim and his colleagues studied 33 children (ages 1 to 21) with newly diagnosed T-ALL. Their treatment included induction, augmented consolidation, interim maintenance (high-dose [5 g/m2] or escalating-dose intravenous methotrexate), 1 delayed intensification, and maintenance. Twenty-one patients underwent cranial irradiation, and 1 received a transplant.
After induction, 19 of the 32 patients analyzed had MRD, which was defined as ≥ 0.01% residual leukemia cells. At the end of consolidation, 6 of the 11 patients tested were MRD-positive. And at the end of interim maintenance, 2 of the 4 patients tested were MRD-positive.
The 19 patients who were MRD-positive after induction were in continuous complete remission at a median follow-up of 4 years (range, 1.3-7.1 years). The same was true for 13 of the 14 MRD-negative patients. One of the MRD-negative patients relapsed 18 months after diagnosis but was still alive with refractory disease at last follow-up.
Dr Abdel-Azim and his colleagues noted that there were no significant differences in treatment variables between MRD-positive patients and MRD-negative patients. However, 1 patient did receive a transplant for rising MRD at 5.4 months after diagnosis.
The investigators also said they did not find any associations between MRD positivity after induction and patients’ age, sex, ethnicity, weight, white blood cell count at diagnosis, cytogenetics, immunophenotype, or the type of steroid they received during induction therapy.
The team said these results may be explained by the fact that clearance of leukemia cells from the blood is slower in patients with T-ALL than in those with B-cell ALL. However, the leukemia cells ultimately clear in T-ALL without changes in therapy. ![]()
Inhibitor may benefit certain ALL patients
PHILADELPHIA—Results of preclinical research suggest the BCL-2 inhibitor ABT-199 (venetoclax) may be effective in certain pediatric patients with acute lymphoblastic leukemia (ALL).
In xenograft models of various ALL subtypes, ABT-199 produced an objective response rate below 30%.
However, additional analyses unearthed information that could potentially help us identify which ALL patients might respond to the drug.
Santi Suryani, PhD, of the Children’s Cancer Institute in Sydney, New South Wales, Australia, and her colleagues presented this research at the AACR Annual Meeting 2015 (abstract 3276*). The work was supported by AbbVie, one of the companies developing ABT-199.
Dr Suryani and her colleagues decided to investigate ABT-199 in pediatric ALL after observing mixed results with the BCL-2/BCL-W/BCL-XL inhibitor ABT-263 (navitoclax).
ABT-263 delayed ALL progression in nearly all of the xenograft models the team tested and produced a 61% response rate. However, the drug also induced BCL-XL-mediated thrombocytopenia.
As ABT-199 doesn’t target BCL-XL, the researchers thought the drug might produce similar responses as ABT-263 without inducing thrombocytopenia.
“When ABT-199 came into the picture, we were very excited,” Dr Suryani said. “We thought, ‘This is a wonder drug. This will cure pediatric ALL.’”
To test this hypothesis, the team compared ABT-199 (100 mg/kg x 21 days) and vehicle control in 19 pediatric ALL patient-derived xenografts, including infant mixed-lineage leukemia (MLL) ALL (n=4), B-cell precursor (BCP) ALL (n=5), BCP-ALL categorized as Ph-like (n=4), T-cell ALL (n=4), and early T-cell precursor (ETP) ALL (n=2).
ABT-199 significantly delayed progression in 12 xenografts (63%) for periods ranging from 0.4 days to 28 days. And the drug produced objective responses in 5 xenografts (26%).
Responses occurred in MLL-ALL, BCP-ALL, and Ph-like BCP ALL, but not T-cell ALL or ETP-ALL. Complete responses were seen in MLL-ALL (n=1) and BCP-ALL (n=2), and partial responses occurred in MLL-ALL (n=1) and Ph-like BCP-ALL (n=1).
As the response rate with ABT-263 was more than double that of ABT-199 (61% vs 26%), the researchers found the results with ABT-199 “a little bit disappointing,” according to Dr Suryani.
“But we thought, ‘That’s okay. That already tells us the science behind it—that pediatric ALL is probably more BCL-XL-dependent, rather than BCL-2-dependent,’” she said. “We wondered if there was any way we could come up with a predictive biomarker so we could select patients who will benefit from this treatment.”
With that in mind, the researchers evaluated the link between protein expression and response. They looked at BCL-2 and BCL-XL, as well as a range of other proteins, including BCL-W, MCL1, BAK1, and BAX, among others.
And they found that high BCL-XL and low BCL-2 expression were significantly associated with ABT-199 resistance.
The researchers are still investigating ways to guide treatment with ABT-199 in ALL. They are also hoping to improve responses by administering the drug in combination with other agents. ![]()
*Information in the abstract differs from that presented at the meeting.
PHILADELPHIA—Results of preclinical research suggest the BCL-2 inhibitor ABT-199 (venetoclax) may be effective in certain pediatric patients with acute lymphoblastic leukemia (ALL).
In xenograft models of various ALL subtypes, ABT-199 produced an objective response rate below 30%.
However, additional analyses unearthed information that could potentially help us identify which ALL patients might respond to the drug.
Santi Suryani, PhD, of the Children’s Cancer Institute in Sydney, New South Wales, Australia, and her colleagues presented this research at the AACR Annual Meeting 2015 (abstract 3276*). The work was supported by AbbVie, one of the companies developing ABT-199.
Dr Suryani and her colleagues decided to investigate ABT-199 in pediatric ALL after observing mixed results with the BCL-2/BCL-W/BCL-XL inhibitor ABT-263 (navitoclax).
ABT-263 delayed ALL progression in nearly all of the xenograft models the team tested and produced a 61% response rate. However, the drug also induced BCL-XL-mediated thrombocytopenia.
As ABT-199 doesn’t target BCL-XL, the researchers thought the drug might produce similar responses as ABT-263 without inducing thrombocytopenia.
“When ABT-199 came into the picture, we were very excited,” Dr Suryani said. “We thought, ‘This is a wonder drug. This will cure pediatric ALL.’”
To test this hypothesis, the team compared ABT-199 (100 mg/kg x 21 days) and vehicle control in 19 pediatric ALL patient-derived xenografts, including infant mixed-lineage leukemia (MLL) ALL (n=4), B-cell precursor (BCP) ALL (n=5), BCP-ALL categorized as Ph-like (n=4), T-cell ALL (n=4), and early T-cell precursor (ETP) ALL (n=2).
ABT-199 significantly delayed progression in 12 xenografts (63%) for periods ranging from 0.4 days to 28 days. And the drug produced objective responses in 5 xenografts (26%).
Responses occurred in MLL-ALL, BCP-ALL, and Ph-like BCP ALL, but not T-cell ALL or ETP-ALL. Complete responses were seen in MLL-ALL (n=1) and BCP-ALL (n=2), and partial responses occurred in MLL-ALL (n=1) and Ph-like BCP-ALL (n=1).
As the response rate with ABT-263 was more than double that of ABT-199 (61% vs 26%), the researchers found the results with ABT-199 “a little bit disappointing,” according to Dr Suryani.
“But we thought, ‘That’s okay. That already tells us the science behind it—that pediatric ALL is probably more BCL-XL-dependent, rather than BCL-2-dependent,’” she said. “We wondered if there was any way we could come up with a predictive biomarker so we could select patients who will benefit from this treatment.”
With that in mind, the researchers evaluated the link between protein expression and response. They looked at BCL-2 and BCL-XL, as well as a range of other proteins, including BCL-W, MCL1, BAK1, and BAX, among others.
And they found that high BCL-XL and low BCL-2 expression were significantly associated with ABT-199 resistance.
The researchers are still investigating ways to guide treatment with ABT-199 in ALL. They are also hoping to improve responses by administering the drug in combination with other agents. ![]()
*Information in the abstract differs from that presented at the meeting.
PHILADELPHIA—Results of preclinical research suggest the BCL-2 inhibitor ABT-199 (venetoclax) may be effective in certain pediatric patients with acute lymphoblastic leukemia (ALL).
In xenograft models of various ALL subtypes, ABT-199 produced an objective response rate below 30%.
However, additional analyses unearthed information that could potentially help us identify which ALL patients might respond to the drug.
Santi Suryani, PhD, of the Children’s Cancer Institute in Sydney, New South Wales, Australia, and her colleagues presented this research at the AACR Annual Meeting 2015 (abstract 3276*). The work was supported by AbbVie, one of the companies developing ABT-199.
Dr Suryani and her colleagues decided to investigate ABT-199 in pediatric ALL after observing mixed results with the BCL-2/BCL-W/BCL-XL inhibitor ABT-263 (navitoclax).
ABT-263 delayed ALL progression in nearly all of the xenograft models the team tested and produced a 61% response rate. However, the drug also induced BCL-XL-mediated thrombocytopenia.
As ABT-199 doesn’t target BCL-XL, the researchers thought the drug might produce similar responses as ABT-263 without inducing thrombocytopenia.
“When ABT-199 came into the picture, we were very excited,” Dr Suryani said. “We thought, ‘This is a wonder drug. This will cure pediatric ALL.’”
To test this hypothesis, the team compared ABT-199 (100 mg/kg x 21 days) and vehicle control in 19 pediatric ALL patient-derived xenografts, including infant mixed-lineage leukemia (MLL) ALL (n=4), B-cell precursor (BCP) ALL (n=5), BCP-ALL categorized as Ph-like (n=4), T-cell ALL (n=4), and early T-cell precursor (ETP) ALL (n=2).
ABT-199 significantly delayed progression in 12 xenografts (63%) for periods ranging from 0.4 days to 28 days. And the drug produced objective responses in 5 xenografts (26%).
Responses occurred in MLL-ALL, BCP-ALL, and Ph-like BCP ALL, but not T-cell ALL or ETP-ALL. Complete responses were seen in MLL-ALL (n=1) and BCP-ALL (n=2), and partial responses occurred in MLL-ALL (n=1) and Ph-like BCP-ALL (n=1).
As the response rate with ABT-263 was more than double that of ABT-199 (61% vs 26%), the researchers found the results with ABT-199 “a little bit disappointing,” according to Dr Suryani.
“But we thought, ‘That’s okay. That already tells us the science behind it—that pediatric ALL is probably more BCL-XL-dependent, rather than BCL-2-dependent,’” she said. “We wondered if there was any way we could come up with a predictive biomarker so we could select patients who will benefit from this treatment.”
With that in mind, the researchers evaluated the link between protein expression and response. They looked at BCL-2 and BCL-XL, as well as a range of other proteins, including BCL-W, MCL1, BAK1, and BAX, among others.
And they found that high BCL-XL and low BCL-2 expression were significantly associated with ABT-199 resistance.
The researchers are still investigating ways to guide treatment with ABT-199 in ALL. They are also hoping to improve responses by administering the drug in combination with other agents. ![]()
*Information in the abstract differs from that presented at the meeting.
Reversing glucocorticoid resistance in ALL

Photo courtesy of St. Jude
Researchers say they have identified a mechanism that helps leukemia cells resist glucocorticoids.
They believe the mechanism is responsible for about a third of steroid resistance in children and adolescents with acute lymphoblastic leukemia (ALL).
However, additional research is needed to determine if the process is at work in adults with ALL, where steroid resistance is more common and long-term survival lags.
The researchers described the mechanism in Nature Genetics.
“Based on these findings, research has already begun to identify small molecules with the potential to reverse glucocorticoid resistance, leading to more effective treatment and increased survival,” said study author William Evans, PharmD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Dr Evans and his colleagues analyzed samples from 444 newly diagnosed ALL patients being treated at St. Jude or in clinical trials sponsored by the Dutch Childhood Oncology Group and the German Cooperative Study Group for Childhood ALL.
The team also analyzed samples collected at diagnosis and relapse from 49 pediatric ALL patients enrolled in clinical trials organized by the Children’s Oncology Group.
The researchers found differences in gene expression that correlated with sensitivity to steroids. CASP1 and NLRP3 were among the genes with increased activity in steroid-resistant leukemia cells.
The team also identified a reason for the increased gene activity. Leukemia cells overexpressing CASP1 and NLRP3 had lower levels of methylation compared to cells with normal expression.
Previous research showed that steroid resistance was more common in young ALL patients who relapsed than in newly diagnosed patients. Dr Evans and his colleagues found that expression of CASP1 and NLRP3 was significantly higher in ALL patients who relapsed.
The researchers also discovered that CASP1 blocks glucocorticoids by splitting the receptor where the drug binds and therefore blocks its access to the nucleus.
“Cells that overexpress CASP1 are chewing up their glucocorticoid receptor,” Dr Evans said. “That means when steroids enter the cell, there is no receptor for the drugs to bind to or fulfill its therapeutic function.”
To confirm that CASP1 cleavage of the steroid receptor is pivotal to ALL steroid resistance, the researchers engineered a receptor that lacked the CASP1 cleavage site. When they introduced the genetically engineered receptors into ALL cells that expressed high levels of CASP1, the cells remained sensitive to steroids.
Using a variety of techniques, the researchers showed that steroid resistance rose or fell in leukemia cells based on CASP1 levels. Overexpression of CASP1 rendered ALL cells 5 to 15 times more resistant to the glucocorticoids dexamethasone and prednisolone.
However, reducing CASP1 using genetic, pharmacologic, and other methods restored steroid sensitivity in leukemia cells. ![]()
Photo courtesy of St. Jude
Researchers say they have identified a mechanism that helps leukemia cells resist glucocorticoids.
They believe the mechanism is responsible for about a third of steroid resistance in children and adolescents with acute lymphoblastic leukemia (ALL).
However, additional research is needed to determine if the process is at work in adults with ALL, where steroid resistance is more common and long-term survival lags.
The researchers described the mechanism in Nature Genetics.
“Based on these findings, research has already begun to identify small molecules with the potential to reverse glucocorticoid resistance, leading to more effective treatment and increased survival,” said study author William Evans, PharmD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Dr Evans and his colleagues analyzed samples from 444 newly diagnosed ALL patients being treated at St. Jude or in clinical trials sponsored by the Dutch Childhood Oncology Group and the German Cooperative Study Group for Childhood ALL.
The team also analyzed samples collected at diagnosis and relapse from 49 pediatric ALL patients enrolled in clinical trials organized by the Children’s Oncology Group.
The researchers found differences in gene expression that correlated with sensitivity to steroids. CASP1 and NLRP3 were among the genes with increased activity in steroid-resistant leukemia cells.
The team also identified a reason for the increased gene activity. Leukemia cells overexpressing CASP1 and NLRP3 had lower levels of methylation compared to cells with normal expression.
Previous research showed that steroid resistance was more common in young ALL patients who relapsed than in newly diagnosed patients. Dr Evans and his colleagues found that expression of CASP1 and NLRP3 was significantly higher in ALL patients who relapsed.
The researchers also discovered that CASP1 blocks glucocorticoids by splitting the receptor where the drug binds and therefore blocks its access to the nucleus.
“Cells that overexpress CASP1 are chewing up their glucocorticoid receptor,” Dr Evans said. “That means when steroids enter the cell, there is no receptor for the drugs to bind to or fulfill its therapeutic function.”
To confirm that CASP1 cleavage of the steroid receptor is pivotal to ALL steroid resistance, the researchers engineered a receptor that lacked the CASP1 cleavage site. When they introduced the genetically engineered receptors into ALL cells that expressed high levels of CASP1, the cells remained sensitive to steroids.
Using a variety of techniques, the researchers showed that steroid resistance rose or fell in leukemia cells based on CASP1 levels. Overexpression of CASP1 rendered ALL cells 5 to 15 times more resistant to the glucocorticoids dexamethasone and prednisolone.
However, reducing CASP1 using genetic, pharmacologic, and other methods restored steroid sensitivity in leukemia cells. ![]()
Photo courtesy of St. Jude
Researchers say they have identified a mechanism that helps leukemia cells resist glucocorticoids.
They believe the mechanism is responsible for about a third of steroid resistance in children and adolescents with acute lymphoblastic leukemia (ALL).
However, additional research is needed to determine if the process is at work in adults with ALL, where steroid resistance is more common and long-term survival lags.
The researchers described the mechanism in Nature Genetics.
“Based on these findings, research has already begun to identify small molecules with the potential to reverse glucocorticoid resistance, leading to more effective treatment and increased survival,” said study author William Evans, PharmD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
Dr Evans and his colleagues analyzed samples from 444 newly diagnosed ALL patients being treated at St. Jude or in clinical trials sponsored by the Dutch Childhood Oncology Group and the German Cooperative Study Group for Childhood ALL.
The team also analyzed samples collected at diagnosis and relapse from 49 pediatric ALL patients enrolled in clinical trials organized by the Children’s Oncology Group.
The researchers found differences in gene expression that correlated with sensitivity to steroids. CASP1 and NLRP3 were among the genes with increased activity in steroid-resistant leukemia cells.
The team also identified a reason for the increased gene activity. Leukemia cells overexpressing CASP1 and NLRP3 had lower levels of methylation compared to cells with normal expression.
Previous research showed that steroid resistance was more common in young ALL patients who relapsed than in newly diagnosed patients. Dr Evans and his colleagues found that expression of CASP1 and NLRP3 was significantly higher in ALL patients who relapsed.
The researchers also discovered that CASP1 blocks glucocorticoids by splitting the receptor where the drug binds and therefore blocks its access to the nucleus.
“Cells that overexpress CASP1 are chewing up their glucocorticoid receptor,” Dr Evans said. “That means when steroids enter the cell, there is no receptor for the drugs to bind to or fulfill its therapeutic function.”
To confirm that CASP1 cleavage of the steroid receptor is pivotal to ALL steroid resistance, the researchers engineered a receptor that lacked the CASP1 cleavage site. When they introduced the genetically engineered receptors into ALL cells that expressed high levels of CASP1, the cells remained sensitive to steroids.
Using a variety of techniques, the researchers showed that steroid resistance rose or fell in leukemia cells based on CASP1 levels. Overexpression of CASP1 rendered ALL cells 5 to 15 times more resistant to the glucocorticoids dexamethasone and prednisolone.
However, reducing CASP1 using genetic, pharmacologic, and other methods restored steroid sensitivity in leukemia cells. ![]()
CDK inhibitor proves active against AML, ALL
PHILADELPHIA—Preclinical research suggests a cyclin-dependent kinase (CDK) inhibitor is active against acute leukemias, particularly those with mixed-lineage leukemia rearrangements (MLL-r).
CYC065 selectively inhibits CDK2, which drives cell-cycle transition and activates major DNA double-strand break repair pathways; CDK5, which drives metastatic spread; and CDK9, which regulates the transcription of genes important for the proliferation and survival of malignant cells.
Experiments have shown that CYC065 exhibits activity against acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL), with and without MLL-r.
Daniella Zheleva, PhD, and her colleagues described these experiments in a poster at the AACR Annual Meeting 2015 (abstract 1650). All of the researchers are employees of Cyclacel Ltd., the company developing CYC065.
The researchers tested CYC065 in a panel of AML cell lines with wild-type MLL (HEL, HL60, Kasumi-1, KG-1, OCI-AML5, and PL21) and MLL-r (EOL-1, ML-2, MOLM-13, MV4-11, Nomo-1, OCI-AML2, and THP-1).
They found that MLL-r cell lines were “highly sensitive” to CYC065, but the sensitivity of cell lines with wild-type MLL correlated with the level of Bcl-2 family proteins. In the wild-type cell lines, IC50/70/90 values were correlated with BclXL and inversely correlated with Bak.
Six-hour pulse treatment of CYC065 at 0.5 µM to 1 µM was sufficient to cause 90% or greater cell death in sensitive cell lines. And cell lines with reduced sensitivity to the drug could be targeted by exposure to 10-hour pulse treatments of CYC065, or to CYC065 in combination with short pulses of Bcl-2 inhibitors.
The researchers observed “potent antitumor activity” when they administered CYC065 in AML xenograft models.
In an EOL-1 model, the median tumor growth inhibition on day 19 was 97% for mice that received CYC065 at 40 mg/kg (every day on days 1-5 and 8-12), 95% for mice that received CYC065 at 20 mg/kg every day on days 1-5 and 8-12), and 41% for mice that received cytarabine at 100 mg/kg (every day on days 1-5).
In the HL60 model, the median tumor growth inhibition on day 11 was 90% for mice that received CYC065 at 70 mg/kg (every day on days 1-5 and 8-12). And 2 mice achieved a complete response to treatment.
The researchers also found that CYC065 synergizes with cytarabine, particularly when CYC065 is given first. In fact, the combination could overcome the cytarabine resistance observed in the MV4-11 cell line.
CYC065 was “strongly synergistic” with Bcl2/BclXL inhibitors as well, the researchers said. CYC065 synergized with ABT-199, ABT-263, and ABT-737 in both AML cell lines (THP-1 and HEL) and ALL cell lines (Jurkat and SEM).
The researchers said the potent in vitro and in vivo activity of CYC065 and the ability to combine the drug with other antileukemic agents suggest that it may have therapeutic potential in AML and ALL. ![]()
PHILADELPHIA—Preclinical research suggests a cyclin-dependent kinase (CDK) inhibitor is active against acute leukemias, particularly those with mixed-lineage leukemia rearrangements (MLL-r).
CYC065 selectively inhibits CDK2, which drives cell-cycle transition and activates major DNA double-strand break repair pathways; CDK5, which drives metastatic spread; and CDK9, which regulates the transcription of genes important for the proliferation and survival of malignant cells.
Experiments have shown that CYC065 exhibits activity against acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL), with and without MLL-r.
Daniella Zheleva, PhD, and her colleagues described these experiments in a poster at the AACR Annual Meeting 2015 (abstract 1650). All of the researchers are employees of Cyclacel Ltd., the company developing CYC065.
The researchers tested CYC065 in a panel of AML cell lines with wild-type MLL (HEL, HL60, Kasumi-1, KG-1, OCI-AML5, and PL21) and MLL-r (EOL-1, ML-2, MOLM-13, MV4-11, Nomo-1, OCI-AML2, and THP-1).
They found that MLL-r cell lines were “highly sensitive” to CYC065, but the sensitivity of cell lines with wild-type MLL correlated with the level of Bcl-2 family proteins. In the wild-type cell lines, IC50/70/90 values were correlated with BclXL and inversely correlated with Bak.
Six-hour pulse treatment of CYC065 at 0.5 µM to 1 µM was sufficient to cause 90% or greater cell death in sensitive cell lines. And cell lines with reduced sensitivity to the drug could be targeted by exposure to 10-hour pulse treatments of CYC065, or to CYC065 in combination with short pulses of Bcl-2 inhibitors.
The researchers observed “potent antitumor activity” when they administered CYC065 in AML xenograft models.
In an EOL-1 model, the median tumor growth inhibition on day 19 was 97% for mice that received CYC065 at 40 mg/kg (every day on days 1-5 and 8-12), 95% for mice that received CYC065 at 20 mg/kg every day on days 1-5 and 8-12), and 41% for mice that received cytarabine at 100 mg/kg (every day on days 1-5).
In the HL60 model, the median tumor growth inhibition on day 11 was 90% for mice that received CYC065 at 70 mg/kg (every day on days 1-5 and 8-12). And 2 mice achieved a complete response to treatment.
The researchers also found that CYC065 synergizes with cytarabine, particularly when CYC065 is given first. In fact, the combination could overcome the cytarabine resistance observed in the MV4-11 cell line.
CYC065 was “strongly synergistic” with Bcl2/BclXL inhibitors as well, the researchers said. CYC065 synergized with ABT-199, ABT-263, and ABT-737 in both AML cell lines (THP-1 and HEL) and ALL cell lines (Jurkat and SEM).
The researchers said the potent in vitro and in vivo activity of CYC065 and the ability to combine the drug with other antileukemic agents suggest that it may have therapeutic potential in AML and ALL. ![]()
PHILADELPHIA—Preclinical research suggests a cyclin-dependent kinase (CDK) inhibitor is active against acute leukemias, particularly those with mixed-lineage leukemia rearrangements (MLL-r).
CYC065 selectively inhibits CDK2, which drives cell-cycle transition and activates major DNA double-strand break repair pathways; CDK5, which drives metastatic spread; and CDK9, which regulates the transcription of genes important for the proliferation and survival of malignant cells.
Experiments have shown that CYC065 exhibits activity against acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL), with and without MLL-r.
Daniella Zheleva, PhD, and her colleagues described these experiments in a poster at the AACR Annual Meeting 2015 (abstract 1650). All of the researchers are employees of Cyclacel Ltd., the company developing CYC065.
The researchers tested CYC065 in a panel of AML cell lines with wild-type MLL (HEL, HL60, Kasumi-1, KG-1, OCI-AML5, and PL21) and MLL-r (EOL-1, ML-2, MOLM-13, MV4-11, Nomo-1, OCI-AML2, and THP-1).
They found that MLL-r cell lines were “highly sensitive” to CYC065, but the sensitivity of cell lines with wild-type MLL correlated with the level of Bcl-2 family proteins. In the wild-type cell lines, IC50/70/90 values were correlated with BclXL and inversely correlated with Bak.
Six-hour pulse treatment of CYC065 at 0.5 µM to 1 µM was sufficient to cause 90% or greater cell death in sensitive cell lines. And cell lines with reduced sensitivity to the drug could be targeted by exposure to 10-hour pulse treatments of CYC065, or to CYC065 in combination with short pulses of Bcl-2 inhibitors.
The researchers observed “potent antitumor activity” when they administered CYC065 in AML xenograft models.
In an EOL-1 model, the median tumor growth inhibition on day 19 was 97% for mice that received CYC065 at 40 mg/kg (every day on days 1-5 and 8-12), 95% for mice that received CYC065 at 20 mg/kg every day on days 1-5 and 8-12), and 41% for mice that received cytarabine at 100 mg/kg (every day on days 1-5).
In the HL60 model, the median tumor growth inhibition on day 11 was 90% for mice that received CYC065 at 70 mg/kg (every day on days 1-5 and 8-12). And 2 mice achieved a complete response to treatment.
The researchers also found that CYC065 synergizes with cytarabine, particularly when CYC065 is given first. In fact, the combination could overcome the cytarabine resistance observed in the MV4-11 cell line.
CYC065 was “strongly synergistic” with Bcl2/BclXL inhibitors as well, the researchers said. CYC065 synergized with ABT-199, ABT-263, and ABT-737 in both AML cell lines (THP-1 and HEL) and ALL cell lines (Jurkat and SEM).
The researchers said the potent in vitro and in vivo activity of CYC065 and the ability to combine the drug with other antileukemic agents suggest that it may have therapeutic potential in AML and ALL. ![]()
Drug approved to treat CML, ALL in Canada

Photo courtesy of the FDA
Health Canada has approved ponatinib hydrochloride (Iclusig) to treat adults with any phase of chronic myeloid leukemia (CML) or Philadelphia
chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) for whom other tyrosine kinase inhibitor (TKI) therapy is not appropriate, including CML or Ph+ ALL patients with the T315I mutation and those who have exhibited prior TKI resistance or intolerance.
Ponatinib is approved under the Notice of Compliance with Conditions policy based on promising evidence of clinical effectiveness.
Products approved under this policy are intended for the treatment, prevention, or diagnosis of a serious, life-threatening, or severely debilitating illness. The products must have demonstrated promising benefit, be of high quality, and possess an acceptable safety profile based on a benefit/risk assessment.
These products either respond to a serious unmet medical need in Canada or have demonstrated a significant improvement in the benefit/risk profile over existing therapies.
Ponatinib will be made available in Canada through a controlled distribution program. Prescribers who have completed the certification procedure will be able to prescribe the drug. Trained pharmacies will verify the prescriber’s certified status prior to dispensing ponatinib to the patient.
Health Canada’s decision to approve ponatinib was based on 2-year data from the phase 2 PACE trial.
A trial set to begin in mid-2015 will serve as the confirmatory trial for the Health Canada approval. Investigators will evaluate 3 starting doses of ponatinib in patients with refractory, chronic-phase CML who are resistant to at least 2 approved TKIs.
PACE trial
Researchers conducted this trial in patients with CML or Ph+ ALL who were resistant or intolerant to prior TKI therapy, or who had the T315I mutation.
Ponatinib demonstrated anti-leukemic activity in these patients, prompting a major cytogenetic response (MCyR) in 56% of chronic-phase CML patients and in 70% of patients with the T315I mutation. MCyR within the first 12 months of treatment was the primary endpoint for chronic-phase patients.
In patients with advanced disease, 57% of accelerated-phase CML patients and 31% of blast-phase CML patients achieved a major hematologic response (MaHR). MaHR within the first 6 months was the primary endpoint for patients with advanced disease. In patients with Ph+ ALL, 41% achieved MaHR.
Common non-hematologic adverse events included rash (38%), abdominal pain (38%), headache (35%), dry skin (35%), constipation (34%), fatigue (27%), pyrexia (27%), nausea (26%), arthralgia (25%), hypertension (21%), increased lipase (19%), and increased amylase (7%).
Hematologic events of any grade included thrombocytopenia (42%), neutropenia (24%), and anemia (20%). Serious adverse events of arterial thromboembolism, including arterial stenosis, occurred in patients with cardiovascular risk factors.
Extended follow-up data from the PACE trial, collected in 2013, suggested ponatinib can increase the risk of thrombotic events. When these data came to light, officials in the European Union and the US, where ponatinib had already been approved, began to investigate the drug.
Ponatinib was pulled from the US market for a little over 2 months, and trials of the drug were placed on partial hold while the Food and Drug Administration evaluated the drug’s safety. Ponatinib went back on the market in January 2014, with new safety measures in place.
The drug was not pulled from the market in the European Union, but the European Medicine’s Agency released recommendations for safer use of ponatinib. The Committee for Medicinal Products for Human Use reviewed data on ponatinib and decided the drug’s benefits outweigh its risks. ![]()
Photo courtesy of the FDA
Health Canada has approved ponatinib hydrochloride (Iclusig) to treat adults with any phase of chronic myeloid leukemia (CML) or Philadelphia
chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) for whom other tyrosine kinase inhibitor (TKI) therapy is not appropriate, including CML or Ph+ ALL patients with the T315I mutation and those who have exhibited prior TKI resistance or intolerance.
Ponatinib is approved under the Notice of Compliance with Conditions policy based on promising evidence of clinical effectiveness.
Products approved under this policy are intended for the treatment, prevention, or diagnosis of a serious, life-threatening, or severely debilitating illness. The products must have demonstrated promising benefit, be of high quality, and possess an acceptable safety profile based on a benefit/risk assessment.
These products either respond to a serious unmet medical need in Canada or have demonstrated a significant improvement in the benefit/risk profile over existing therapies.
Ponatinib will be made available in Canada through a controlled distribution program. Prescribers who have completed the certification procedure will be able to prescribe the drug. Trained pharmacies will verify the prescriber’s certified status prior to dispensing ponatinib to the patient.
Health Canada’s decision to approve ponatinib was based on 2-year data from the phase 2 PACE trial.
A trial set to begin in mid-2015 will serve as the confirmatory trial for the Health Canada approval. Investigators will evaluate 3 starting doses of ponatinib in patients with refractory, chronic-phase CML who are resistant to at least 2 approved TKIs.
PACE trial
Researchers conducted this trial in patients with CML or Ph+ ALL who were resistant or intolerant to prior TKI therapy, or who had the T315I mutation.
Ponatinib demonstrated anti-leukemic activity in these patients, prompting a major cytogenetic response (MCyR) in 56% of chronic-phase CML patients and in 70% of patients with the T315I mutation. MCyR within the first 12 months of treatment was the primary endpoint for chronic-phase patients.
In patients with advanced disease, 57% of accelerated-phase CML patients and 31% of blast-phase CML patients achieved a major hematologic response (MaHR). MaHR within the first 6 months was the primary endpoint for patients with advanced disease. In patients with Ph+ ALL, 41% achieved MaHR.
Common non-hematologic adverse events included rash (38%), abdominal pain (38%), headache (35%), dry skin (35%), constipation (34%), fatigue (27%), pyrexia (27%), nausea (26%), arthralgia (25%), hypertension (21%), increased lipase (19%), and increased amylase (7%).
Hematologic events of any grade included thrombocytopenia (42%), neutropenia (24%), and anemia (20%). Serious adverse events of arterial thromboembolism, including arterial stenosis, occurred in patients with cardiovascular risk factors.
Extended follow-up data from the PACE trial, collected in 2013, suggested ponatinib can increase the risk of thrombotic events. When these data came to light, officials in the European Union and the US, where ponatinib had already been approved, began to investigate the drug.
Ponatinib was pulled from the US market for a little over 2 months, and trials of the drug were placed on partial hold while the Food and Drug Administration evaluated the drug’s safety. Ponatinib went back on the market in January 2014, with new safety measures in place.
The drug was not pulled from the market in the European Union, but the European Medicine’s Agency released recommendations for safer use of ponatinib. The Committee for Medicinal Products for Human Use reviewed data on ponatinib and decided the drug’s benefits outweigh its risks. ![]()
Photo courtesy of the FDA
Health Canada has approved ponatinib hydrochloride (Iclusig) to treat adults with any phase of chronic myeloid leukemia (CML) or Philadelphia
chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) for whom other tyrosine kinase inhibitor (TKI) therapy is not appropriate, including CML or Ph+ ALL patients with the T315I mutation and those who have exhibited prior TKI resistance or intolerance.
Ponatinib is approved under the Notice of Compliance with Conditions policy based on promising evidence of clinical effectiveness.
Products approved under this policy are intended for the treatment, prevention, or diagnosis of a serious, life-threatening, or severely debilitating illness. The products must have demonstrated promising benefit, be of high quality, and possess an acceptable safety profile based on a benefit/risk assessment.
These products either respond to a serious unmet medical need in Canada or have demonstrated a significant improvement in the benefit/risk profile over existing therapies.
Ponatinib will be made available in Canada through a controlled distribution program. Prescribers who have completed the certification procedure will be able to prescribe the drug. Trained pharmacies will verify the prescriber’s certified status prior to dispensing ponatinib to the patient.
Health Canada’s decision to approve ponatinib was based on 2-year data from the phase 2 PACE trial.
A trial set to begin in mid-2015 will serve as the confirmatory trial for the Health Canada approval. Investigators will evaluate 3 starting doses of ponatinib in patients with refractory, chronic-phase CML who are resistant to at least 2 approved TKIs.
PACE trial
Researchers conducted this trial in patients with CML or Ph+ ALL who were resistant or intolerant to prior TKI therapy, or who had the T315I mutation.
Ponatinib demonstrated anti-leukemic activity in these patients, prompting a major cytogenetic response (MCyR) in 56% of chronic-phase CML patients and in 70% of patients with the T315I mutation. MCyR within the first 12 months of treatment was the primary endpoint for chronic-phase patients.
In patients with advanced disease, 57% of accelerated-phase CML patients and 31% of blast-phase CML patients achieved a major hematologic response (MaHR). MaHR within the first 6 months was the primary endpoint for patients with advanced disease. In patients with Ph+ ALL, 41% achieved MaHR.
Common non-hematologic adverse events included rash (38%), abdominal pain (38%), headache (35%), dry skin (35%), constipation (34%), fatigue (27%), pyrexia (27%), nausea (26%), arthralgia (25%), hypertension (21%), increased lipase (19%), and increased amylase (7%).
Hematologic events of any grade included thrombocytopenia (42%), neutropenia (24%), and anemia (20%). Serious adverse events of arterial thromboembolism, including arterial stenosis, occurred in patients with cardiovascular risk factors.
Extended follow-up data from the PACE trial, collected in 2013, suggested ponatinib can increase the risk of thrombotic events. When these data came to light, officials in the European Union and the US, where ponatinib had already been approved, began to investigate the drug.
Ponatinib was pulled from the US market for a little over 2 months, and trials of the drug were placed on partial hold while the Food and Drug Administration evaluated the drug’s safety. Ponatinib went back on the market in January 2014, with new safety measures in place.
The drug was not pulled from the market in the European Union, but the European Medicine’s Agency released recommendations for safer use of ponatinib. The Committee for Medicinal Products for Human Use reviewed data on ponatinib and decided the drug’s benefits outweigh its risks. ![]()
Engineered protein overcomes radiation resistance in ALL

Photo courtesy of Dr Uckun
An engineered protein can enhance the effects of radiation and even overcome radiation resistance to treat B-precursor acute lymphoblastic leukemia (ALL), according to research published in EBioMedicine.
The protein, CD19L-sTRAIL, is a fusion of the CD19 ligand protein, which seeks out and binds to leukemia cells, with soluble TRAIL, a protein that can amplify the potency of radiation if it can be anchored on the membrane of leukemia cells.
Researchers found that CD19L-sTRAIL augmented the potency of radiation therapy even against the most aggressive and radiation-resistant forms of leukemia.
“Even very low doses of radiation were highly effective in mice challenged with aggressive human leukemia cells, when it was combined with [CD19L-sTRAIL],” said Fatih M. Uckun, MD, PhD, of The Saban Research Institute of Children’s Hospital Los Angeles in California.
“Due to its ability to selectively anchor to the surface of leukemia cells via its CD19L portion, CD19L-sTRAIL was 100,000-fold more potent than sTRAIL and consistently killed aggressive leukemia cells taken directly from children with ALL—not only in the test tube but also in mice.”
The researchers found that a combination of low-dose total body irradiation (TBI) and CD19L-sTRAIL greatly improved event-free survival (EFS) in mice that had received a typically fatal dose of cells from a patient with relapsed B-precursor ALL.
The median EFS for mice treated with CD19L-sTRAIL plus low-dose TBI was 72 days, which was significantly longer than the EFS for untreated control mice (17 days, P<0.0001), mice that received TBI alone (64 days, P=0.0014), mice that received CD19L-sTRAIL alone (20 days, P=0.0022), and mice that received a combination of vincristine, dexamethasone, and PEG-asparaginase (17 days, P=0.0033).
Dr Uckun and his colleagues noted that none of the mice that received CD19L-sTRAIL and TBI experienced a toxic death or signs of treatment-related toxicity.
The team also found that TBI plus CD19L-sTRAIL improved progression-free survival (PFS) in CD22ΔE12xBCR-ABL double transgenic mice with radiation-resistant, advanced stage, CD19+ murine B-precursor ALL with lymphomatous features.
The mean PFS was 24.0 ± 4.0 days for mice that received CD19L-sTRAIL plus TBI, which was significantly longer than the PFS for control mice (0 ± 0 days, P<0.0001), mice that received CD19L-sTRAIL alone (3.4 ± 0.9 days, P=0.0003), and mice that received TBI alone (9.0 ± 4.6 days, P=0.020).
Based on these results, Dr Uckun and his colleagues believe that incorporating CD19L-sTRAIL into the pre-transplant TBI regimens of patients with very high-risk B-precursor ALL could improve survival after hematopoietic stem cell transplant.
“We are hopeful that the knowledge gained from this study will open a new range of effective treatment opportunities for children with recurrent leukemia,” Dr Uckun said. ![]()
Photo courtesy of Dr Uckun
An engineered protein can enhance the effects of radiation and even overcome radiation resistance to treat B-precursor acute lymphoblastic leukemia (ALL), according to research published in EBioMedicine.
The protein, CD19L-sTRAIL, is a fusion of the CD19 ligand protein, which seeks out and binds to leukemia cells, with soluble TRAIL, a protein that can amplify the potency of radiation if it can be anchored on the membrane of leukemia cells.
Researchers found that CD19L-sTRAIL augmented the potency of radiation therapy even against the most aggressive and radiation-resistant forms of leukemia.
“Even very low doses of radiation were highly effective in mice challenged with aggressive human leukemia cells, when it was combined with [CD19L-sTRAIL],” said Fatih M. Uckun, MD, PhD, of The Saban Research Institute of Children’s Hospital Los Angeles in California.
“Due to its ability to selectively anchor to the surface of leukemia cells via its CD19L portion, CD19L-sTRAIL was 100,000-fold more potent than sTRAIL and consistently killed aggressive leukemia cells taken directly from children with ALL—not only in the test tube but also in mice.”
The researchers found that a combination of low-dose total body irradiation (TBI) and CD19L-sTRAIL greatly improved event-free survival (EFS) in mice that had received a typically fatal dose of cells from a patient with relapsed B-precursor ALL.
The median EFS for mice treated with CD19L-sTRAIL plus low-dose TBI was 72 days, which was significantly longer than the EFS for untreated control mice (17 days, P<0.0001), mice that received TBI alone (64 days, P=0.0014), mice that received CD19L-sTRAIL alone (20 days, P=0.0022), and mice that received a combination of vincristine, dexamethasone, and PEG-asparaginase (17 days, P=0.0033).
Dr Uckun and his colleagues noted that none of the mice that received CD19L-sTRAIL and TBI experienced a toxic death or signs of treatment-related toxicity.
The team also found that TBI plus CD19L-sTRAIL improved progression-free survival (PFS) in CD22ΔE12xBCR-ABL double transgenic mice with radiation-resistant, advanced stage, CD19+ murine B-precursor ALL with lymphomatous features.
The mean PFS was 24.0 ± 4.0 days for mice that received CD19L-sTRAIL plus TBI, which was significantly longer than the PFS for control mice (0 ± 0 days, P<0.0001), mice that received CD19L-sTRAIL alone (3.4 ± 0.9 days, P=0.0003), and mice that received TBI alone (9.0 ± 4.6 days, P=0.020).
Based on these results, Dr Uckun and his colleagues believe that incorporating CD19L-sTRAIL into the pre-transplant TBI regimens of patients with very high-risk B-precursor ALL could improve survival after hematopoietic stem cell transplant.
“We are hopeful that the knowledge gained from this study will open a new range of effective treatment opportunities for children with recurrent leukemia,” Dr Uckun said. ![]()
Photo courtesy of Dr Uckun
An engineered protein can enhance the effects of radiation and even overcome radiation resistance to treat B-precursor acute lymphoblastic leukemia (ALL), according to research published in EBioMedicine.
The protein, CD19L-sTRAIL, is a fusion of the CD19 ligand protein, which seeks out and binds to leukemia cells, with soluble TRAIL, a protein that can amplify the potency of radiation if it can be anchored on the membrane of leukemia cells.
Researchers found that CD19L-sTRAIL augmented the potency of radiation therapy even against the most aggressive and radiation-resistant forms of leukemia.
“Even very low doses of radiation were highly effective in mice challenged with aggressive human leukemia cells, when it was combined with [CD19L-sTRAIL],” said Fatih M. Uckun, MD, PhD, of The Saban Research Institute of Children’s Hospital Los Angeles in California.
“Due to its ability to selectively anchor to the surface of leukemia cells via its CD19L portion, CD19L-sTRAIL was 100,000-fold more potent than sTRAIL and consistently killed aggressive leukemia cells taken directly from children with ALL—not only in the test tube but also in mice.”
The researchers found that a combination of low-dose total body irradiation (TBI) and CD19L-sTRAIL greatly improved event-free survival (EFS) in mice that had received a typically fatal dose of cells from a patient with relapsed B-precursor ALL.
The median EFS for mice treated with CD19L-sTRAIL plus low-dose TBI was 72 days, which was significantly longer than the EFS for untreated control mice (17 days, P<0.0001), mice that received TBI alone (64 days, P=0.0014), mice that received CD19L-sTRAIL alone (20 days, P=0.0022), and mice that received a combination of vincristine, dexamethasone, and PEG-asparaginase (17 days, P=0.0033).
Dr Uckun and his colleagues noted that none of the mice that received CD19L-sTRAIL and TBI experienced a toxic death or signs of treatment-related toxicity.
The team also found that TBI plus CD19L-sTRAIL improved progression-free survival (PFS) in CD22ΔE12xBCR-ABL double transgenic mice with radiation-resistant, advanced stage, CD19+ murine B-precursor ALL with lymphomatous features.
The mean PFS was 24.0 ± 4.0 days for mice that received CD19L-sTRAIL plus TBI, which was significantly longer than the PFS for control mice (0 ± 0 days, P<0.0001), mice that received CD19L-sTRAIL alone (3.4 ± 0.9 days, P=0.0003), and mice that received TBI alone (9.0 ± 4.6 days, P=0.020).
Based on these results, Dr Uckun and his colleagues believe that incorporating CD19L-sTRAIL into the pre-transplant TBI regimens of patients with very high-risk B-precursor ALL could improve survival after hematopoietic stem cell transplant.
“We are hopeful that the knowledge gained from this study will open a new range of effective treatment opportunities for children with recurrent leukemia,” Dr Uckun said.
Increasing cell signaling to eradicate Ph+ ALL

Photo by Aaron Logan
Increasing B-cell antigen receptor (BCR) signaling beyond “a point of no return” can lead to the selective elimination of leukemic cells, according to a group of researchers.
The team knew that proximal pre-BCR signaling is toxic to Philadelphia-chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) cells, and their experiments revealed that SYK tyrosine kinase activity mimicked constitutively active pre-BCR signaling.
So it was no surprise that pharmacologic hyperactivation of SYK prompted the removal of self-reactive B cells and selective killing of Ph+ ALL cells in vivo.
Markus Müschen, MD, PhD, of the University of California, San Francisco, and his colleagues described these findings in Nature.
When the researchers tested proximal pre-BCR signaling in mouse BCR-ABL1 cells, they found that an incremental increase of SYK activity could induce cell death.
Additional experiments showed that patient-derived Ph+ ALL cells have high levels of the inhibitory receptors PECAM1, CD300A, and LAIR1. And these receptors are needed to calibrate oncogenic signaling strength via recruitment of the inhibitory phosphatases PTPN6 and INPP5D.
So the researchers wondered if a small-molecule inhibitor of INPP5D, known as 3-a-aminocholestane (3AC), could induce SYK hyperactivation and target Ph+ ALL cells in mice.
They found that 3AC eliminated imatinib-resistant Ph+ ALL cells via rapid and massive cell death, and this significantly prolonged survival in the mice.
Dr Müschen and his colleagues noted that only Ph+ ALL cells were marked for destruction, which suggests a BCR-targeted drug could overcome imatinib resistance without affecting normal B cells.
The team also pointed out that a short exposure to 3AC was sufficient to commit the leukemia cells to death and to clear most of the disease burden from mice.
Dr Müschen said 3AC’s fast action was encouraging, because it remains unknown whether prolonged BCR hyperactivation is safe. That is why he and his colleagues are now focusing on formulating hyperactivating drugs that could be administered for only a few hours.
“These experiments show that we can engage signaling checkpoints in a very short period of time and that, once these checkpoints are engaged, the cell is irreversibly slated for death; it’s a point of no return,” Dr Müschen said.
“The next step is to work with medicinal chemists to make better ALL drugs that will overstimulate the B-cell receptor pathway and . . . could be used on a treatment schedule to elicit a very strong, but time-limited, spike in signaling to engage negative B-cell selection.”
Photo by Aaron Logan
Increasing B-cell antigen receptor (BCR) signaling beyond “a point of no return” can lead to the selective elimination of leukemic cells, according to a group of researchers.
The team knew that proximal pre-BCR signaling is toxic to Philadelphia-chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) cells, and their experiments revealed that SYK tyrosine kinase activity mimicked constitutively active pre-BCR signaling.
So it was no surprise that pharmacologic hyperactivation of SYK prompted the removal of self-reactive B cells and selective killing of Ph+ ALL cells in vivo.
Markus Müschen, MD, PhD, of the University of California, San Francisco, and his colleagues described these findings in Nature.
When the researchers tested proximal pre-BCR signaling in mouse BCR-ABL1 cells, they found that an incremental increase of SYK activity could induce cell death.
Additional experiments showed that patient-derived Ph+ ALL cells have high levels of the inhibitory receptors PECAM1, CD300A, and LAIR1. And these receptors are needed to calibrate oncogenic signaling strength via recruitment of the inhibitory phosphatases PTPN6 and INPP5D.
So the researchers wondered if a small-molecule inhibitor of INPP5D, known as 3-a-aminocholestane (3AC), could induce SYK hyperactivation and target Ph+ ALL cells in mice.
They found that 3AC eliminated imatinib-resistant Ph+ ALL cells via rapid and massive cell death, and this significantly prolonged survival in the mice.
Dr Müschen and his colleagues noted that only Ph+ ALL cells were marked for destruction, which suggests a BCR-targeted drug could overcome imatinib resistance without affecting normal B cells.
The team also pointed out that a short exposure to 3AC was sufficient to commit the leukemia cells to death and to clear most of the disease burden from mice.
Dr Müschen said 3AC’s fast action was encouraging, because it remains unknown whether prolonged BCR hyperactivation is safe. That is why he and his colleagues are now focusing on formulating hyperactivating drugs that could be administered for only a few hours.
“These experiments show that we can engage signaling checkpoints in a very short period of time and that, once these checkpoints are engaged, the cell is irreversibly slated for death; it’s a point of no return,” Dr Müschen said.
“The next step is to work with medicinal chemists to make better ALL drugs that will overstimulate the B-cell receptor pathway and . . . could be used on a treatment schedule to elicit a very strong, but time-limited, spike in signaling to engage negative B-cell selection.”
Photo by Aaron Logan
Increasing B-cell antigen receptor (BCR) signaling beyond “a point of no return” can lead to the selective elimination of leukemic cells, according to a group of researchers.
The team knew that proximal pre-BCR signaling is toxic to Philadelphia-chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) cells, and their experiments revealed that SYK tyrosine kinase activity mimicked constitutively active pre-BCR signaling.
So it was no surprise that pharmacologic hyperactivation of SYK prompted the removal of self-reactive B cells and selective killing of Ph+ ALL cells in vivo.
Markus Müschen, MD, PhD, of the University of California, San Francisco, and his colleagues described these findings in Nature.
When the researchers tested proximal pre-BCR signaling in mouse BCR-ABL1 cells, they found that an incremental increase of SYK activity could induce cell death.
Additional experiments showed that patient-derived Ph+ ALL cells have high levels of the inhibitory receptors PECAM1, CD300A, and LAIR1. And these receptors are needed to calibrate oncogenic signaling strength via recruitment of the inhibitory phosphatases PTPN6 and INPP5D.
So the researchers wondered if a small-molecule inhibitor of INPP5D, known as 3-a-aminocholestane (3AC), could induce SYK hyperactivation and target Ph+ ALL cells in mice.
They found that 3AC eliminated imatinib-resistant Ph+ ALL cells via rapid and massive cell death, and this significantly prolonged survival in the mice.
Dr Müschen and his colleagues noted that only Ph+ ALL cells were marked for destruction, which suggests a BCR-targeted drug could overcome imatinib resistance without affecting normal B cells.
The team also pointed out that a short exposure to 3AC was sufficient to commit the leukemia cells to death and to clear most of the disease burden from mice.
Dr Müschen said 3AC’s fast action was encouraging, because it remains unknown whether prolonged BCR hyperactivation is safe. That is why he and his colleagues are now focusing on formulating hyperactivating drugs that could be administered for only a few hours.
“These experiments show that we can engage signaling checkpoints in a very short period of time and that, once these checkpoints are engaged, the cell is irreversibly slated for death; it’s a point of no return,” Dr Müschen said.
“The next step is to work with medicinal chemists to make better ALL drugs that will overstimulate the B-cell receptor pathway and . . . could be used on a treatment schedule to elicit a very strong, but time-limited, spike in signaling to engage negative B-cell selection.”
Israel approves ponatinib for CML, Ph+ ALL
Photo courtesy of the US FDA
The Israeli Ministry of Health has granted regulatory approval for the kinase inhibitor ponatinib (Iclusig) to treat certain adults with chronic myeloid leukemia (CML) or Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL).
The drug can now be used to treat adults with any phase of CML who have the T315I mutation or are resistant to/cannot tolerate dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate.
Ponatinib is also approved to treat patients with Ph+ ALL who have the T315I mutation or are resistant to/cannot tolerate dasatinib and for whom subsequent treatment with imatinib is not clinically appropriate.
Ariad Pharmaceuticals, Inc., the company developing ponatinib, said the drug should be available in Israel in the second quarter of 2015.
Trial results
The Ministry of Health’s decision to approve ponatinib was based on results from the phase 2 PACE trial, which included patients with CML or Ph+ ALL who were resistant to or intolerant of prior tyrosine kinase inhibitor therapy, or who had the T315I mutation.
The median follow-up times were 15.3 months in chronic-phase CML patients, 15.8 months in accelerated-phase CML patients, and 6.2 months in patients with blast-phase CML or Ph+ ALL.
In chronic-phase CML, the primary endpoint was major cytogenetic response, and it occurred in 56% of patients. Among chronic-phase patients with the T315I mutation, 70% achieved a major cytogenetic response. Among patients who had failed treatment with dasatinib or nilotinib, 51% achieved a major cytogenetic response.
In accelerated-phase CML, the primary endpoint was major hematologic response. This occurred in 57% of all patients in this group, 50% of patients with the T315I mutation, and 58% of patients who had failed treatment with dasatinib or nilotinib.
The primary endpoint was major hematologic response in blast-phase CML/Ph+ ALL as well. Thirty-four percent of all patients in this group met this endpoint, as did 33% of patients with the T315I mutation and 35% of patients who had failed treatment with dasatinib or nilotinib.
Common non-hematologic adverse events included rash (38%), abdominal pain (38%), headache (35%), dry skin (35%), constipation (34%), fatigue (27%), pyrexia (27%), nausea (26%), arthralgia (25%), hypertension (21%), increased lipase (19%), and increased amylase (7%).
Hematologic events of any grade included thrombocytopenia (42%), neutropenia (24%), and anemia (20%). Serious adverse events of arterial thromboembolism, including arterial stenosis, occurred in patients with cardiovascular risk factors.
Safety issues
Extended follow-up data from the PACE trial, collected in 2013, suggested ponatinib can increase the risk of thrombotic events. When these data came to light, officials in the European Union and the US, where ponatinib had already been approved, began to investigate the drug.
Ponatinib was pulled from the US market for a little over 2 months, and trials of the drug were placed on partial hold while the Food and Drug Administration evaluated the drug’s safety. Ponatinib went back on the market in January 2014, with new safety measures in place.
The drug was not pulled from the market in the European Union, but the European Medicine’s Agency released recommendations for safer use of ponatinib. The Committee for Medicinal Products for Human Use reviewed data on ponatinib and decided the drug’s benefits outweigh its risks.
Photo courtesy of the US FDA
The Israeli Ministry of Health has granted regulatory approval for the kinase inhibitor ponatinib (Iclusig) to treat certain adults with chronic myeloid leukemia (CML) or Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL).
The drug can now be used to treat adults with any phase of CML who have the T315I mutation or are resistant to/cannot tolerate dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate.
Ponatinib is also approved to treat patients with Ph+ ALL who have the T315I mutation or are resistant to/cannot tolerate dasatinib and for whom subsequent treatment with imatinib is not clinically appropriate.
Ariad Pharmaceuticals, Inc., the company developing ponatinib, said the drug should be available in Israel in the second quarter of 2015.
Trial results
The Ministry of Health’s decision to approve ponatinib was based on results from the phase 2 PACE trial, which included patients with CML or Ph+ ALL who were resistant to or intolerant of prior tyrosine kinase inhibitor therapy, or who had the T315I mutation.
The median follow-up times were 15.3 months in chronic-phase CML patients, 15.8 months in accelerated-phase CML patients, and 6.2 months in patients with blast-phase CML or Ph+ ALL.
In chronic-phase CML, the primary endpoint was major cytogenetic response, and it occurred in 56% of patients. Among chronic-phase patients with the T315I mutation, 70% achieved a major cytogenetic response. Among patients who had failed treatment with dasatinib or nilotinib, 51% achieved a major cytogenetic response.
In accelerated-phase CML, the primary endpoint was major hematologic response. This occurred in 57% of all patients in this group, 50% of patients with the T315I mutation, and 58% of patients who had failed treatment with dasatinib or nilotinib.
The primary endpoint was major hematologic response in blast-phase CML/Ph+ ALL as well. Thirty-four percent of all patients in this group met this endpoint, as did 33% of patients with the T315I mutation and 35% of patients who had failed treatment with dasatinib or nilotinib.
Common non-hematologic adverse events included rash (38%), abdominal pain (38%), headache (35%), dry skin (35%), constipation (34%), fatigue (27%), pyrexia (27%), nausea (26%), arthralgia (25%), hypertension (21%), increased lipase (19%), and increased amylase (7%).
Hematologic events of any grade included thrombocytopenia (42%), neutropenia (24%), and anemia (20%). Serious adverse events of arterial thromboembolism, including arterial stenosis, occurred in patients with cardiovascular risk factors.
Safety issues
Extended follow-up data from the PACE trial, collected in 2013, suggested ponatinib can increase the risk of thrombotic events. When these data came to light, officials in the European Union and the US, where ponatinib had already been approved, began to investigate the drug.
Ponatinib was pulled from the US market for a little over 2 months, and trials of the drug were placed on partial hold while the Food and Drug Administration evaluated the drug’s safety. Ponatinib went back on the market in January 2014, with new safety measures in place.
The drug was not pulled from the market in the European Union, but the European Medicine’s Agency released recommendations for safer use of ponatinib. The Committee for Medicinal Products for Human Use reviewed data on ponatinib and decided the drug’s benefits outweigh its risks.
Photo courtesy of the US FDA
The Israeli Ministry of Health has granted regulatory approval for the kinase inhibitor ponatinib (Iclusig) to treat certain adults with chronic myeloid leukemia (CML) or Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL).
The drug can now be used to treat adults with any phase of CML who have the T315I mutation or are resistant to/cannot tolerate dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate.
Ponatinib is also approved to treat patients with Ph+ ALL who have the T315I mutation or are resistant to/cannot tolerate dasatinib and for whom subsequent treatment with imatinib is not clinically appropriate.
Ariad Pharmaceuticals, Inc., the company developing ponatinib, said the drug should be available in Israel in the second quarter of 2015.
Trial results
The Ministry of Health’s decision to approve ponatinib was based on results from the phase 2 PACE trial, which included patients with CML or Ph+ ALL who were resistant to or intolerant of prior tyrosine kinase inhibitor therapy, or who had the T315I mutation.
The median follow-up times were 15.3 months in chronic-phase CML patients, 15.8 months in accelerated-phase CML patients, and 6.2 months in patients with blast-phase CML or Ph+ ALL.
In chronic-phase CML, the primary endpoint was major cytogenetic response, and it occurred in 56% of patients. Among chronic-phase patients with the T315I mutation, 70% achieved a major cytogenetic response. Among patients who had failed treatment with dasatinib or nilotinib, 51% achieved a major cytogenetic response.
In accelerated-phase CML, the primary endpoint was major hematologic response. This occurred in 57% of all patients in this group, 50% of patients with the T315I mutation, and 58% of patients who had failed treatment with dasatinib or nilotinib.
The primary endpoint was major hematologic response in blast-phase CML/Ph+ ALL as well. Thirty-four percent of all patients in this group met this endpoint, as did 33% of patients with the T315I mutation and 35% of patients who had failed treatment with dasatinib or nilotinib.
Common non-hematologic adverse events included rash (38%), abdominal pain (38%), headache (35%), dry skin (35%), constipation (34%), fatigue (27%), pyrexia (27%), nausea (26%), arthralgia (25%), hypertension (21%), increased lipase (19%), and increased amylase (7%).
Hematologic events of any grade included thrombocytopenia (42%), neutropenia (24%), and anemia (20%). Serious adverse events of arterial thromboembolism, including arterial stenosis, occurred in patients with cardiovascular risk factors.
Safety issues
Extended follow-up data from the PACE trial, collected in 2013, suggested ponatinib can increase the risk of thrombotic events. When these data came to light, officials in the European Union and the US, where ponatinib had already been approved, began to investigate the drug.
Ponatinib was pulled from the US market for a little over 2 months, and trials of the drug were placed on partial hold while the Food and Drug Administration evaluated the drug’s safety. Ponatinib went back on the market in January 2014, with new safety measures in place.
The drug was not pulled from the market in the European Union, but the European Medicine’s Agency released recommendations for safer use of ponatinib. The Committee for Medicinal Products for Human Use reviewed data on ponatinib and decided the drug’s benefits outweigh its risks.
Germline mutations linked to ALL

Photo by Rhoda Baer
New research suggests that heritable mutations in the gene ETV6 can predispose people to acute lymphoblastic leukemia (ALL).
Somatic mutations in ETV6 have previously been implicated in the development of ALL and other hematologic malignancies.
The new study, published in Nature Genetics, has shown that germline mutations in ETV6 are associated with thrombocytopenia, red blood cell (RBC) macrocytosis, and predisposition to ALL.
The research began with a family (family 1) that had autosomal dominant thrombocytopenia (67,000-132,000 platelets/μL), high RBC mean corpuscular volume (MCV, 92.5-101.5 fl), and 2 cases of B-cell-precursor ALL.
“All of them had big red blood cells, low platelet counts, and propensity to bleed,” said study author Christopher Porter, MD, of the University of Colorado Denver.
This familial link to abnormal blood dynamics and predisposition to ALL implied a common genetic denominator. To find it, the researchers performed whole-exome sequencing and found a heterozygous single-nucleotide change in ETV6, c.641C>T, encoding a p.Pro214Leu substitution in the central domain.
The researchers then screened 23 other families with similar phenotypes as the first and identified 2 families with ETV6 mutations.
One family (family 2) had members with platelet counts ranging from 44,000 to 115,000 platelets/μL, RBC MCVs ranging from 88 to 97 fl, and a member with ALL. All affected family members had a c.641C>T mutation identical to the one identified in family 1.
Another family (family 3) had members with platelet counts ranging from 99,000 to 101,000 platelets/μL and RBC MCVs ranging from 93 to 98 fl but no malignancies. Members of this family had a c.1252A>G transition producing a p.Arg418Gly substitution in the DNA-binding domain, with alternative splicing and exon skipping.
The researchers said these mutations partially disrupt ETV6 transcriptional repression in vitro and cause aberrant cytoplasmic localization of both mutant and endogenous ETV6, suggesting a dominant-negative effect. The mutations also impair megakaryocyte development and proplatelet formation in culture.
Dr Porter and his colleagues noted that another team of researchers recently discovered germline missense mutations in ETV6 in 3 unrelated families. Dr Porter’s group hopes future work will show the prevalence of germline mutations in ETV6.
“It’s not common in a general population, but we think it might be much more common in people who develop ALL,” Dr Porter said. “[Our] paper highlights this gene in the development of leukemia. By studying this mutation, we should be able to gather a better understanding of how leukemia develops.”
Photo by Rhoda Baer
New research suggests that heritable mutations in the gene ETV6 can predispose people to acute lymphoblastic leukemia (ALL).
Somatic mutations in ETV6 have previously been implicated in the development of ALL and other hematologic malignancies.
The new study, published in Nature Genetics, has shown that germline mutations in ETV6 are associated with thrombocytopenia, red blood cell (RBC) macrocytosis, and predisposition to ALL.
The research began with a family (family 1) that had autosomal dominant thrombocytopenia (67,000-132,000 platelets/μL), high RBC mean corpuscular volume (MCV, 92.5-101.5 fl), and 2 cases of B-cell-precursor ALL.
“All of them had big red blood cells, low platelet counts, and propensity to bleed,” said study author Christopher Porter, MD, of the University of Colorado Denver.
This familial link to abnormal blood dynamics and predisposition to ALL implied a common genetic denominator. To find it, the researchers performed whole-exome sequencing and found a heterozygous single-nucleotide change in ETV6, c.641C>T, encoding a p.Pro214Leu substitution in the central domain.
The researchers then screened 23 other families with similar phenotypes as the first and identified 2 families with ETV6 mutations.
One family (family 2) had members with platelet counts ranging from 44,000 to 115,000 platelets/μL, RBC MCVs ranging from 88 to 97 fl, and a member with ALL. All affected family members had a c.641C>T mutation identical to the one identified in family 1.
Another family (family 3) had members with platelet counts ranging from 99,000 to 101,000 platelets/μL and RBC MCVs ranging from 93 to 98 fl but no malignancies. Members of this family had a c.1252A>G transition producing a p.Arg418Gly substitution in the DNA-binding domain, with alternative splicing and exon skipping.
The researchers said these mutations partially disrupt ETV6 transcriptional repression in vitro and cause aberrant cytoplasmic localization of both mutant and endogenous ETV6, suggesting a dominant-negative effect. The mutations also impair megakaryocyte development and proplatelet formation in culture.
Dr Porter and his colleagues noted that another team of researchers recently discovered germline missense mutations in ETV6 in 3 unrelated families. Dr Porter’s group hopes future work will show the prevalence of germline mutations in ETV6.
“It’s not common in a general population, but we think it might be much more common in people who develop ALL,” Dr Porter said. “[Our] paper highlights this gene in the development of leukemia. By studying this mutation, we should be able to gather a better understanding of how leukemia develops.”
Photo by Rhoda Baer
New research suggests that heritable mutations in the gene ETV6 can predispose people to acute lymphoblastic leukemia (ALL).
Somatic mutations in ETV6 have previously been implicated in the development of ALL and other hematologic malignancies.
The new study, published in Nature Genetics, has shown that germline mutations in ETV6 are associated with thrombocytopenia, red blood cell (RBC) macrocytosis, and predisposition to ALL.
The research began with a family (family 1) that had autosomal dominant thrombocytopenia (67,000-132,000 platelets/μL), high RBC mean corpuscular volume (MCV, 92.5-101.5 fl), and 2 cases of B-cell-precursor ALL.
“All of them had big red blood cells, low platelet counts, and propensity to bleed,” said study author Christopher Porter, MD, of the University of Colorado Denver.
This familial link to abnormal blood dynamics and predisposition to ALL implied a common genetic denominator. To find it, the researchers performed whole-exome sequencing and found a heterozygous single-nucleotide change in ETV6, c.641C>T, encoding a p.Pro214Leu substitution in the central domain.
The researchers then screened 23 other families with similar phenotypes as the first and identified 2 families with ETV6 mutations.
One family (family 2) had members with platelet counts ranging from 44,000 to 115,000 platelets/μL, RBC MCVs ranging from 88 to 97 fl, and a member with ALL. All affected family members had a c.641C>T mutation identical to the one identified in family 1.
Another family (family 3) had members with platelet counts ranging from 99,000 to 101,000 platelets/μL and RBC MCVs ranging from 93 to 98 fl but no malignancies. Members of this family had a c.1252A>G transition producing a p.Arg418Gly substitution in the DNA-binding domain, with alternative splicing and exon skipping.
The researchers said these mutations partially disrupt ETV6 transcriptional repression in vitro and cause aberrant cytoplasmic localization of both mutant and endogenous ETV6, suggesting a dominant-negative effect. The mutations also impair megakaryocyte development and proplatelet formation in culture.
Dr Porter and his colleagues noted that another team of researchers recently discovered germline missense mutations in ETV6 in 3 unrelated families. Dr Porter’s group hopes future work will show the prevalence of germline mutations in ETV6.
“It’s not common in a general population, but we think it might be much more common in people who develop ALL,” Dr Porter said. “[Our] paper highlights this gene in the development of leukemia. By studying this mutation, we should be able to gather a better understanding of how leukemia develops.”