User login
Study supports sequential MRD monitoring in certain patients

Photo by Chad McNeeley
Based on results of a prospective study, investigators are advocating sequential minimal residual disease (MRD) monitoring in pediatric patients with acute lymphoblastic leukemia (ALL) who have detectable MRD after remission induction therapy.
The researchers said their findings show that MRD levels during remission induction treatment have important therapeutic indications, even in the context of MRD-guided therapy, as patients with higher MRD levels have worse outcomes.
Ching-Hon Pui, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues described the findings in The Lancet Oncology.
The team analyzed 498 pediatric patients newly diagnosed with ALL, 492 of whom (99%) attained a complete remission following induction therapy and 491 of whom were monitored for MRD.
The researchers first estimated patients’ risk of relapse according to baseline clinical and laboratory features, provisionally classifying them as having a low, standard, or high risk of relapse.
But the investigators also took MRD levels into consideration. They measured MRD on days 19 and 46 of remission induction, on week 7 of maintenance treatment, and on weeks 17, 48, and 120 (the end of treatment).
The team found that 10-year event-free survival (EFS) was significantly worse for patients with 1% or greater MRD levels on day 19, regardless of their initial risk assessment. Ten-year EFS was 64.1% in these patients, compared to 90.7% in patients with lower or no detectable MRD (P<0.001).
Thirteen percent of patients who were deemed low-risk initially and 28% of patients deemed standard-risk initially had 1% or higher MRD levels on day 19. And these levels were associated with worse 10-year EFS.
In the provisional low-risk group, EFS was 69.2% in the high-MRD patients and 95.5% in the low-MRD patients (P<0.001). And in the provisional standard-risk group, 10-year EFS was 65.1% and 82.9%, respectively (P=0.01).
MRD levels at day 46 also appeared to have a bearing on EFS. For patients in the provisional low-risk group who had 1% or higher MRD on day 19 but became MRD-negative on day 46, 10-year EFS was 88.9%, compared to 59.2% for other provisionally low-risk patients who had detectable MRD on day 46 (P=0.02).
MRD levels on days 19 and 46 led to the reclassification of 50 patients from low-risk to a higher risk group that warranted more intensive therapy. The researchers credited the change with boosting survival.
“This analysis shows that MRD-directed therapy clearly contributed to the unprecedented high rates of long-term survival that patients in this study achieved,” Dr Pui said. “MRD proved to be a powerful way to identify high-risk patients who needed more intensive therapy and helped us avoid over-treatment of low-risk patients by reducing their exposure to chemotherapy.”
Still, MRD assessments at days 19 and 46 were not perfect predictors of patient outcomes. Of the patients who were MRD negative after remission induction, MRD re-emerged in 6 patients—4 of the 382 patients studied on week 7, 1 of the 448 studied at week 17, and 1 of the 437 studied at week 48. All but 1 of these patients died despite additional treatment.
On the other hand, relapse occurred in 2 of the 11 patients who had decreasing MRD levels between the end of induction and week 7 of maintenance therapy and were treated with chemotherapy alone.
Taking these results together, the investigators concluded that measuring MRD at days 19 and 46 was sufficient to guide the treatment of most pediatric ALL patients. However, MRD measurements should continue to guide treatment for patients with detectable MRD on day 46. ![]()
Photo by Chad McNeeley
Based on results of a prospective study, investigators are advocating sequential minimal residual disease (MRD) monitoring in pediatric patients with acute lymphoblastic leukemia (ALL) who have detectable MRD after remission induction therapy.
The researchers said their findings show that MRD levels during remission induction treatment have important therapeutic indications, even in the context of MRD-guided therapy, as patients with higher MRD levels have worse outcomes.
Ching-Hon Pui, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues described the findings in The Lancet Oncology.
The team analyzed 498 pediatric patients newly diagnosed with ALL, 492 of whom (99%) attained a complete remission following induction therapy and 491 of whom were monitored for MRD.
The researchers first estimated patients’ risk of relapse according to baseline clinical and laboratory features, provisionally classifying them as having a low, standard, or high risk of relapse.
But the investigators also took MRD levels into consideration. They measured MRD on days 19 and 46 of remission induction, on week 7 of maintenance treatment, and on weeks 17, 48, and 120 (the end of treatment).
The team found that 10-year event-free survival (EFS) was significantly worse for patients with 1% or greater MRD levels on day 19, regardless of their initial risk assessment. Ten-year EFS was 64.1% in these patients, compared to 90.7% in patients with lower or no detectable MRD (P<0.001).
Thirteen percent of patients who were deemed low-risk initially and 28% of patients deemed standard-risk initially had 1% or higher MRD levels on day 19. And these levels were associated with worse 10-year EFS.
In the provisional low-risk group, EFS was 69.2% in the high-MRD patients and 95.5% in the low-MRD patients (P<0.001). And in the provisional standard-risk group, 10-year EFS was 65.1% and 82.9%, respectively (P=0.01).
MRD levels at day 46 also appeared to have a bearing on EFS. For patients in the provisional low-risk group who had 1% or higher MRD on day 19 but became MRD-negative on day 46, 10-year EFS was 88.9%, compared to 59.2% for other provisionally low-risk patients who had detectable MRD on day 46 (P=0.02).
MRD levels on days 19 and 46 led to the reclassification of 50 patients from low-risk to a higher risk group that warranted more intensive therapy. The researchers credited the change with boosting survival.
“This analysis shows that MRD-directed therapy clearly contributed to the unprecedented high rates of long-term survival that patients in this study achieved,” Dr Pui said. “MRD proved to be a powerful way to identify high-risk patients who needed more intensive therapy and helped us avoid over-treatment of low-risk patients by reducing their exposure to chemotherapy.”
Still, MRD assessments at days 19 and 46 were not perfect predictors of patient outcomes. Of the patients who were MRD negative after remission induction, MRD re-emerged in 6 patients—4 of the 382 patients studied on week 7, 1 of the 448 studied at week 17, and 1 of the 437 studied at week 48. All but 1 of these patients died despite additional treatment.
On the other hand, relapse occurred in 2 of the 11 patients who had decreasing MRD levels between the end of induction and week 7 of maintenance therapy and were treated with chemotherapy alone.
Taking these results together, the investigators concluded that measuring MRD at days 19 and 46 was sufficient to guide the treatment of most pediatric ALL patients. However, MRD measurements should continue to guide treatment for patients with detectable MRD on day 46. ![]()
Photo by Chad McNeeley
Based on results of a prospective study, investigators are advocating sequential minimal residual disease (MRD) monitoring in pediatric patients with acute lymphoblastic leukemia (ALL) who have detectable MRD after remission induction therapy.
The researchers said their findings show that MRD levels during remission induction treatment have important therapeutic indications, even in the context of MRD-guided therapy, as patients with higher MRD levels have worse outcomes.
Ching-Hon Pui, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues described the findings in The Lancet Oncology.
The team analyzed 498 pediatric patients newly diagnosed with ALL, 492 of whom (99%) attained a complete remission following induction therapy and 491 of whom were monitored for MRD.
The researchers first estimated patients’ risk of relapse according to baseline clinical and laboratory features, provisionally classifying them as having a low, standard, or high risk of relapse.
But the investigators also took MRD levels into consideration. They measured MRD on days 19 and 46 of remission induction, on week 7 of maintenance treatment, and on weeks 17, 48, and 120 (the end of treatment).
The team found that 10-year event-free survival (EFS) was significantly worse for patients with 1% or greater MRD levels on day 19, regardless of their initial risk assessment. Ten-year EFS was 64.1% in these patients, compared to 90.7% in patients with lower or no detectable MRD (P<0.001).
Thirteen percent of patients who were deemed low-risk initially and 28% of patients deemed standard-risk initially had 1% or higher MRD levels on day 19. And these levels were associated with worse 10-year EFS.
In the provisional low-risk group, EFS was 69.2% in the high-MRD patients and 95.5% in the low-MRD patients (P<0.001). And in the provisional standard-risk group, 10-year EFS was 65.1% and 82.9%, respectively (P=0.01).
MRD levels at day 46 also appeared to have a bearing on EFS. For patients in the provisional low-risk group who had 1% or higher MRD on day 19 but became MRD-negative on day 46, 10-year EFS was 88.9%, compared to 59.2% for other provisionally low-risk patients who had detectable MRD on day 46 (P=0.02).
MRD levels on days 19 and 46 led to the reclassification of 50 patients from low-risk to a higher risk group that warranted more intensive therapy. The researchers credited the change with boosting survival.
“This analysis shows that MRD-directed therapy clearly contributed to the unprecedented high rates of long-term survival that patients in this study achieved,” Dr Pui said. “MRD proved to be a powerful way to identify high-risk patients who needed more intensive therapy and helped us avoid over-treatment of low-risk patients by reducing their exposure to chemotherapy.”
Still, MRD assessments at days 19 and 46 were not perfect predictors of patient outcomes. Of the patients who were MRD negative after remission induction, MRD re-emerged in 6 patients—4 of the 382 patients studied on week 7, 1 of the 448 studied at week 17, and 1 of the 437 studied at week 48. All but 1 of these patients died despite additional treatment.
On the other hand, relapse occurred in 2 of the 11 patients who had decreasing MRD levels between the end of induction and week 7 of maintenance therapy and were treated with chemotherapy alone.
Taking these results together, the investigators concluded that measuring MRD at days 19 and 46 was sufficient to guide the treatment of most pediatric ALL patients. However, MRD measurements should continue to guide treatment for patients with detectable MRD on day 46. ![]()
Group traces clonal evolution of B-ALL

In tracing the clonal evolution of B-cell acute lymphoblastic leukemia (B-ALL) from diagnosis to relapse, researchers discovered that clonal diversity is comparable in both states.
They also identified mutations associated with B-ALL relapse and found that clonal survival is not dependent upon mutation burden.
In most of the cases the researchers analyzed, a single, minor clone survived therapy, acquired additional mutations, and drove disease relapse.
Jinghui Zhang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and her colleagues recounted these findings in Nature Communications.
The researchers performed deep, whole-exome sequencing on cell samples from 20 young patients (ages 2 to 19) with relapsed B-ALL. The samples were collected at diagnosis, remission, and relapse.
“[W]e wanted to find out the underlying mechanism leading to cancer relapse,” Dr Zhang said. “When the cancer recurs, is it a completely different cancer, or is it an extension, or change, arising from pre-existing cancer?”
The researchers were able to detect the mutations in both the “rising” and “falling” clones—those that survive therapy and those that don’t—at the different disease stages and pinpoint the mutations that drove the leukemia.
Seven genes were highly likely to be mutated in relapsed disease—NT5C2, CREBBP, WHSC1, TP53, USH2A, NRAS, and IKZF1.
The researchers also characterized how diverse those mutations were at diagnosis and relapse. They found that B-ALL cells were mutating just as wildly and diversely in one phase of disease as in the other.
“This finding was interesting, because most people think that the clone that has the most mutations is more likely to survive therapy and evolve, but that doesn’t seem to be the case,” Dr Zhang said.
In most cases, relapse was driven by a minor subclone that had survived therapy and was present at an extremely low level. The researchers said this finding suggests a need to change the way we assess patients after treatment to determine the likelihood of relapse.
“When we are analyzing for the level of minimum residual disease in monitoring remission in patients, we should not only pay attention to the mutations in the predominant clone,” Dr Zhang said. “We should also be tracking what kinds of mutations exist in the minor subclones.” ![]()
In tracing the clonal evolution of B-cell acute lymphoblastic leukemia (B-ALL) from diagnosis to relapse, researchers discovered that clonal diversity is comparable in both states.
They also identified mutations associated with B-ALL relapse and found that clonal survival is not dependent upon mutation burden.
In most of the cases the researchers analyzed, a single, minor clone survived therapy, acquired additional mutations, and drove disease relapse.
Jinghui Zhang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and her colleagues recounted these findings in Nature Communications.
The researchers performed deep, whole-exome sequencing on cell samples from 20 young patients (ages 2 to 19) with relapsed B-ALL. The samples were collected at diagnosis, remission, and relapse.
“[W]e wanted to find out the underlying mechanism leading to cancer relapse,” Dr Zhang said. “When the cancer recurs, is it a completely different cancer, or is it an extension, or change, arising from pre-existing cancer?”
The researchers were able to detect the mutations in both the “rising” and “falling” clones—those that survive therapy and those that don’t—at the different disease stages and pinpoint the mutations that drove the leukemia.
Seven genes were highly likely to be mutated in relapsed disease—NT5C2, CREBBP, WHSC1, TP53, USH2A, NRAS, and IKZF1.
The researchers also characterized how diverse those mutations were at diagnosis and relapse. They found that B-ALL cells were mutating just as wildly and diversely in one phase of disease as in the other.
“This finding was interesting, because most people think that the clone that has the most mutations is more likely to survive therapy and evolve, but that doesn’t seem to be the case,” Dr Zhang said.
In most cases, relapse was driven by a minor subclone that had survived therapy and was present at an extremely low level. The researchers said this finding suggests a need to change the way we assess patients after treatment to determine the likelihood of relapse.
“When we are analyzing for the level of minimum residual disease in monitoring remission in patients, we should not only pay attention to the mutations in the predominant clone,” Dr Zhang said. “We should also be tracking what kinds of mutations exist in the minor subclones.” ![]()
In tracing the clonal evolution of B-cell acute lymphoblastic leukemia (B-ALL) from diagnosis to relapse, researchers discovered that clonal diversity is comparable in both states.
They also identified mutations associated with B-ALL relapse and found that clonal survival is not dependent upon mutation burden.
In most of the cases the researchers analyzed, a single, minor clone survived therapy, acquired additional mutations, and drove disease relapse.
Jinghui Zhang, PhD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and her colleagues recounted these findings in Nature Communications.
The researchers performed deep, whole-exome sequencing on cell samples from 20 young patients (ages 2 to 19) with relapsed B-ALL. The samples were collected at diagnosis, remission, and relapse.
“[W]e wanted to find out the underlying mechanism leading to cancer relapse,” Dr Zhang said. “When the cancer recurs, is it a completely different cancer, or is it an extension, or change, arising from pre-existing cancer?”
The researchers were able to detect the mutations in both the “rising” and “falling” clones—those that survive therapy and those that don’t—at the different disease stages and pinpoint the mutations that drove the leukemia.
Seven genes were highly likely to be mutated in relapsed disease—NT5C2, CREBBP, WHSC1, TP53, USH2A, NRAS, and IKZF1.
The researchers also characterized how diverse those mutations were at diagnosis and relapse. They found that B-ALL cells were mutating just as wildly and diversely in one phase of disease as in the other.
“This finding was interesting, because most people think that the clone that has the most mutations is more likely to survive therapy and evolve, but that doesn’t seem to be the case,” Dr Zhang said.
In most cases, relapse was driven by a minor subclone that had survived therapy and was present at an extremely low level. The researchers said this finding suggests a need to change the way we assess patients after treatment to determine the likelihood of relapse.
“When we are analyzing for the level of minimum residual disease in monitoring remission in patients, we should not only pay attention to the mutations in the predominant clone,” Dr Zhang said. “We should also be tracking what kinds of mutations exist in the minor subclones.” ![]()
Group reprograms B-ALL cells into macrophage-like cells
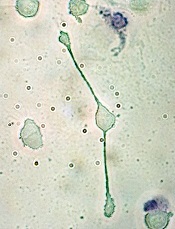
pseudopodia to engulf particles
Investigators have reported methods for reprogramming leukemia cells into non-leukemic, macrophage-like cells.
The team reprogrammed cells derived from patients with BCR-ABL1+ precursor B-cell acute lymphoblastic leukemia (B-ALL) by exposing the cells to myeloid differentiation-promoting cytokines or by transient expression of certain transcription factors.
The group described this work in Proceedings of the National Academy of Sciences.
The research began when the investigators were trying to keep patient-derived leukemia cells alive in culture.
“We were throwing everything at them to help them survive,” said Ravindra Majeti, MD, PhD, of Stanford University School of Medicine in California.
Then, James Scott McClellan, MD, PhD, also of Stanford University School of Medicine, mentioned that some of the cells were changing shape and size, morphing into what looked like macrophages.
Dr Majeti concurred with that observation, but the reasons for the changed cells were a mystery. That is, until he remembered an old research paper, which showed that early B-cell mouse progenitor cells could be forced to become macrophages when exposed to certain transcription factors.
So he and his colleagues set out to confirm that they could transform leukemic cells into macrophage-like cells.
The team isolated CD19+CD34+ blasts from 12 adults with BCR-ABL1+ B-ALL and cultured the blasts in the presence of myeloid-differentiation-promoting cytokines. This resulted in CD14high/CD19low cells that expressed the surface markers and had the functional properties typical of normal macrophages.
The investigators also cultured B-ALL cells with the myeloid transcription factor C/EBPα or the myeloid/lymphoid transcription factor PU.1. Both factors were able to reprogram blasts into macrophage-like cells.
Experiments in mice revealed that reprogramming the blasts into macrophage-like cells eliminated their leukemogenicity.
And the investigators’ final experiments suggested that myeloid reprogramming occurs, to some degree, in humans. In samples from patients with BCR-ABL1+ B-ALL, the team found primary CD14+ monocytes/macrophages that had the BCR-ABL1+ translocation and clonally recombined VDJ regions.
Dr Majeti and his colleagues said they have reason to hope that, when the leukemic cells become macrophage-like cells, they are not only neutralized but may actually assist in fighting the leukemia.
“Because the macrophage cells came from the cancer cells, they will already carry with them the chemical signals that will identify the cancer cells, making an immune attack against the cancer more likely,” Dr Majeti said. ![]()
pseudopodia to engulf particles
Investigators have reported methods for reprogramming leukemia cells into non-leukemic, macrophage-like cells.
The team reprogrammed cells derived from patients with BCR-ABL1+ precursor B-cell acute lymphoblastic leukemia (B-ALL) by exposing the cells to myeloid differentiation-promoting cytokines or by transient expression of certain transcription factors.
The group described this work in Proceedings of the National Academy of Sciences.
The research began when the investigators were trying to keep patient-derived leukemia cells alive in culture.
“We were throwing everything at them to help them survive,” said Ravindra Majeti, MD, PhD, of Stanford University School of Medicine in California.
Then, James Scott McClellan, MD, PhD, also of Stanford University School of Medicine, mentioned that some of the cells were changing shape and size, morphing into what looked like macrophages.
Dr Majeti concurred with that observation, but the reasons for the changed cells were a mystery. That is, until he remembered an old research paper, which showed that early B-cell mouse progenitor cells could be forced to become macrophages when exposed to certain transcription factors.
So he and his colleagues set out to confirm that they could transform leukemic cells into macrophage-like cells.
The team isolated CD19+CD34+ blasts from 12 adults with BCR-ABL1+ B-ALL and cultured the blasts in the presence of myeloid-differentiation-promoting cytokines. This resulted in CD14high/CD19low cells that expressed the surface markers and had the functional properties typical of normal macrophages.
The investigators also cultured B-ALL cells with the myeloid transcription factor C/EBPα or the myeloid/lymphoid transcription factor PU.1. Both factors were able to reprogram blasts into macrophage-like cells.
Experiments in mice revealed that reprogramming the blasts into macrophage-like cells eliminated their leukemogenicity.
And the investigators’ final experiments suggested that myeloid reprogramming occurs, to some degree, in humans. In samples from patients with BCR-ABL1+ B-ALL, the team found primary CD14+ monocytes/macrophages that had the BCR-ABL1+ translocation and clonally recombined VDJ regions.
Dr Majeti and his colleagues said they have reason to hope that, when the leukemic cells become macrophage-like cells, they are not only neutralized but may actually assist in fighting the leukemia.
“Because the macrophage cells came from the cancer cells, they will already carry with them the chemical signals that will identify the cancer cells, making an immune attack against the cancer more likely,” Dr Majeti said. ![]()
pseudopodia to engulf particles
Investigators have reported methods for reprogramming leukemia cells into non-leukemic, macrophage-like cells.
The team reprogrammed cells derived from patients with BCR-ABL1+ precursor B-cell acute lymphoblastic leukemia (B-ALL) by exposing the cells to myeloid differentiation-promoting cytokines or by transient expression of certain transcription factors.
The group described this work in Proceedings of the National Academy of Sciences.
The research began when the investigators were trying to keep patient-derived leukemia cells alive in culture.
“We were throwing everything at them to help them survive,” said Ravindra Majeti, MD, PhD, of Stanford University School of Medicine in California.
Then, James Scott McClellan, MD, PhD, also of Stanford University School of Medicine, mentioned that some of the cells were changing shape and size, morphing into what looked like macrophages.
Dr Majeti concurred with that observation, but the reasons for the changed cells were a mystery. That is, until he remembered an old research paper, which showed that early B-cell mouse progenitor cells could be forced to become macrophages when exposed to certain transcription factors.
So he and his colleagues set out to confirm that they could transform leukemic cells into macrophage-like cells.
The team isolated CD19+CD34+ blasts from 12 adults with BCR-ABL1+ B-ALL and cultured the blasts in the presence of myeloid-differentiation-promoting cytokines. This resulted in CD14high/CD19low cells that expressed the surface markers and had the functional properties typical of normal macrophages.
The investigators also cultured B-ALL cells with the myeloid transcription factor C/EBPα or the myeloid/lymphoid transcription factor PU.1. Both factors were able to reprogram blasts into macrophage-like cells.
Experiments in mice revealed that reprogramming the blasts into macrophage-like cells eliminated their leukemogenicity.
And the investigators’ final experiments suggested that myeloid reprogramming occurs, to some degree, in humans. In samples from patients with BCR-ABL1+ B-ALL, the team found primary CD14+ monocytes/macrophages that had the BCR-ABL1+ translocation and clonally recombined VDJ regions.
Dr Majeti and his colleagues said they have reason to hope that, when the leukemic cells become macrophage-like cells, they are not only neutralized but may actually assist in fighting the leukemia.
“Because the macrophage cells came from the cancer cells, they will already carry with them the chemical signals that will identify the cancer cells, making an immune attack against the cancer more likely,” Dr Majeti said. ![]()
Immunotoxin could treat B-cell malignancies, team says

Photo courtesy of the
University of Minnesota
A bispecific ligand-directed diphtheria toxin known as DT2219 shows promise for treating patients with relapsed/refractory B-cell malignancies, according to researchers.
DT2219 produced responses in 2 of 25 patients analyzed in a phase 1 study. The maximum tolerated dose of DT2219 was not reached, although 2 patients experienced dose-limiting toxicities.
“In this phase 1 trial, we found a safe dose of the drug that has biological activity,” said Daniel Vallera, PhD, of the University of Minnesota in Minneapolis.
“We are planning a phase 2 trial with this drug. It will focus on giving more cycles of treatment, which we believe will dramatically enhance the response rates.”
Dr Vallera and his colleagues detailed the phase 1 results in Clinical Cancer Research.
To develop DT2219, the researchers chose 2 antibody fragments that each selectively bind to CD19 and CD22. They used genetic engineering to attach these two antibodies to the bacterial diphtheria toxin.
When the antibody fragments bind to the two targets on the cancer cell, the entire drug enters the cell, and the toxin kills the cell.
To test DT2219, the researchers enrolled 25 patients with chemo-refractory pre-B acute lymphoblastic leukemia (n=10), chronic lymphocytic leukemia (n=5), or non-Hodgkin lymphoma (n=10). All tumors had CD19 and/or CD22 proteins.
Patients had received a median of 3 prior therapies (range, 2 to 5), and 8 patients had undergone an unsuccessful stem cell transplant (5 autologous and 3 allogeneic).
Patients received DT2219 intravenously over 2 hours every other day for 4 total doses. The dose was escalated from 0.5 μg/kg/day to 80 μg/kg/day in 9 dose cohorts until a dose-limiting toxicity occurred.
All but 1 patient received a single course of DT2219. That patient received a second, 4-dose course after attaining a partial response.
Outcomes
The 12 patients who received doses ranging from 0.5 mg/kg/day to 20 mg/kg/day had minimal or no adverse events (AEs). But all 13 patients who received 4 doses of DT2219 at 40 mg/kg or greater every other day experienced treatment-related AEs.
Grade 1-2 treatment-related AEs included capillary leak syndrome (n=7), ALT/AST elevation (n=4), fatigue (n=3), fever (n=3), hypokalemia (n=2), hypoalbuminemia (n=1), hearing loss (n=1), hypocalcemia (n=1), anemia (n=1), and vomiting (n=1).
Grade 3-4 treatment-related AEs were thrombocytopenia (n=2), neutropenia (n=1), neutropenic fever (n=1), capillary leak syndrome (n=1), hypokalemia (n=1), and leg weakness (n=1). The grade 3 leg weakness and grade 3 capillary leak syndrome were dose-limiting toxicities.
The maximum tolerated dose was not reached, but clinical responses occurred between doses of 40 to 80 µg/kg.
All 25 patients were evaluable for response, but only 9 patients in the highest dose cohorts had measurable drug levels.
Two patients had durable, objective responses. One patient had chronic lymphocytic leukemia, and the other had diffuse large B-cell lymphoma. The latter patient’s response was a complete remission that occurred after 2 treatment cycles.
“We were surprised that the drug was effective enough to entirely eliminate the cancer in one of our patients,” Dr Vallera said. “Further, we expected the patients to make antibodies against the bacterial toxin and, thus, reject our drug. Surprisingly, this did not occur in the majority of our patients [70%].”
“We need to study more patients to understand why they did not produce neutralizing antibodies. However, we also have been working to create a less immunogenic form of the toxin for the next-generation drug.”
“Another important fact about our drug is that it was home-grown, meaning there was no commercial partner, which is rare. The drug was funded mostly with private donations, including individuals that have lost loved ones to cancer.” ![]()
Photo courtesy of the
University of Minnesota
A bispecific ligand-directed diphtheria toxin known as DT2219 shows promise for treating patients with relapsed/refractory B-cell malignancies, according to researchers.
DT2219 produced responses in 2 of 25 patients analyzed in a phase 1 study. The maximum tolerated dose of DT2219 was not reached, although 2 patients experienced dose-limiting toxicities.
“In this phase 1 trial, we found a safe dose of the drug that has biological activity,” said Daniel Vallera, PhD, of the University of Minnesota in Minneapolis.
“We are planning a phase 2 trial with this drug. It will focus on giving more cycles of treatment, which we believe will dramatically enhance the response rates.”
Dr Vallera and his colleagues detailed the phase 1 results in Clinical Cancer Research.
To develop DT2219, the researchers chose 2 antibody fragments that each selectively bind to CD19 and CD22. They used genetic engineering to attach these two antibodies to the bacterial diphtheria toxin.
When the antibody fragments bind to the two targets on the cancer cell, the entire drug enters the cell, and the toxin kills the cell.
To test DT2219, the researchers enrolled 25 patients with chemo-refractory pre-B acute lymphoblastic leukemia (n=10), chronic lymphocytic leukemia (n=5), or non-Hodgkin lymphoma (n=10). All tumors had CD19 and/or CD22 proteins.
Patients had received a median of 3 prior therapies (range, 2 to 5), and 8 patients had undergone an unsuccessful stem cell transplant (5 autologous and 3 allogeneic).
Patients received DT2219 intravenously over 2 hours every other day for 4 total doses. The dose was escalated from 0.5 μg/kg/day to 80 μg/kg/day in 9 dose cohorts until a dose-limiting toxicity occurred.
All but 1 patient received a single course of DT2219. That patient received a second, 4-dose course after attaining a partial response.
Outcomes
The 12 patients who received doses ranging from 0.5 mg/kg/day to 20 mg/kg/day had minimal or no adverse events (AEs). But all 13 patients who received 4 doses of DT2219 at 40 mg/kg or greater every other day experienced treatment-related AEs.
Grade 1-2 treatment-related AEs included capillary leak syndrome (n=7), ALT/AST elevation (n=4), fatigue (n=3), fever (n=3), hypokalemia (n=2), hypoalbuminemia (n=1), hearing loss (n=1), hypocalcemia (n=1), anemia (n=1), and vomiting (n=1).
Grade 3-4 treatment-related AEs were thrombocytopenia (n=2), neutropenia (n=1), neutropenic fever (n=1), capillary leak syndrome (n=1), hypokalemia (n=1), and leg weakness (n=1). The grade 3 leg weakness and grade 3 capillary leak syndrome were dose-limiting toxicities.
The maximum tolerated dose was not reached, but clinical responses occurred between doses of 40 to 80 µg/kg.
All 25 patients were evaluable for response, but only 9 patients in the highest dose cohorts had measurable drug levels.
Two patients had durable, objective responses. One patient had chronic lymphocytic leukemia, and the other had diffuse large B-cell lymphoma. The latter patient’s response was a complete remission that occurred after 2 treatment cycles.
“We were surprised that the drug was effective enough to entirely eliminate the cancer in one of our patients,” Dr Vallera said. “Further, we expected the patients to make antibodies against the bacterial toxin and, thus, reject our drug. Surprisingly, this did not occur in the majority of our patients [70%].”
“We need to study more patients to understand why they did not produce neutralizing antibodies. However, we also have been working to create a less immunogenic form of the toxin for the next-generation drug.”
“Another important fact about our drug is that it was home-grown, meaning there was no commercial partner, which is rare. The drug was funded mostly with private donations, including individuals that have lost loved ones to cancer.” ![]()
Photo courtesy of the
University of Minnesota
A bispecific ligand-directed diphtheria toxin known as DT2219 shows promise for treating patients with relapsed/refractory B-cell malignancies, according to researchers.
DT2219 produced responses in 2 of 25 patients analyzed in a phase 1 study. The maximum tolerated dose of DT2219 was not reached, although 2 patients experienced dose-limiting toxicities.
“In this phase 1 trial, we found a safe dose of the drug that has biological activity,” said Daniel Vallera, PhD, of the University of Minnesota in Minneapolis.
“We are planning a phase 2 trial with this drug. It will focus on giving more cycles of treatment, which we believe will dramatically enhance the response rates.”
Dr Vallera and his colleagues detailed the phase 1 results in Clinical Cancer Research.
To develop DT2219, the researchers chose 2 antibody fragments that each selectively bind to CD19 and CD22. They used genetic engineering to attach these two antibodies to the bacterial diphtheria toxin.
When the antibody fragments bind to the two targets on the cancer cell, the entire drug enters the cell, and the toxin kills the cell.
To test DT2219, the researchers enrolled 25 patients with chemo-refractory pre-B acute lymphoblastic leukemia (n=10), chronic lymphocytic leukemia (n=5), or non-Hodgkin lymphoma (n=10). All tumors had CD19 and/or CD22 proteins.
Patients had received a median of 3 prior therapies (range, 2 to 5), and 8 patients had undergone an unsuccessful stem cell transplant (5 autologous and 3 allogeneic).
Patients received DT2219 intravenously over 2 hours every other day for 4 total doses. The dose was escalated from 0.5 μg/kg/day to 80 μg/kg/day in 9 dose cohorts until a dose-limiting toxicity occurred.
All but 1 patient received a single course of DT2219. That patient received a second, 4-dose course after attaining a partial response.
Outcomes
The 12 patients who received doses ranging from 0.5 mg/kg/day to 20 mg/kg/day had minimal or no adverse events (AEs). But all 13 patients who received 4 doses of DT2219 at 40 mg/kg or greater every other day experienced treatment-related AEs.
Grade 1-2 treatment-related AEs included capillary leak syndrome (n=7), ALT/AST elevation (n=4), fatigue (n=3), fever (n=3), hypokalemia (n=2), hypoalbuminemia (n=1), hearing loss (n=1), hypocalcemia (n=1), anemia (n=1), and vomiting (n=1).
Grade 3-4 treatment-related AEs were thrombocytopenia (n=2), neutropenia (n=1), neutropenic fever (n=1), capillary leak syndrome (n=1), hypokalemia (n=1), and leg weakness (n=1). The grade 3 leg weakness and grade 3 capillary leak syndrome were dose-limiting toxicities.
The maximum tolerated dose was not reached, but clinical responses occurred between doses of 40 to 80 µg/kg.
All 25 patients were evaluable for response, but only 9 patients in the highest dose cohorts had measurable drug levels.
Two patients had durable, objective responses. One patient had chronic lymphocytic leukemia, and the other had diffuse large B-cell lymphoma. The latter patient’s response was a complete remission that occurred after 2 treatment cycles.
“We were surprised that the drug was effective enough to entirely eliminate the cancer in one of our patients,” Dr Vallera said. “Further, we expected the patients to make antibodies against the bacterial toxin and, thus, reject our drug. Surprisingly, this did not occur in the majority of our patients [70%].”
“We need to study more patients to understand why they did not produce neutralizing antibodies. However, we also have been working to create a less immunogenic form of the toxin for the next-generation drug.”
“Another important fact about our drug is that it was home-grown, meaning there was no commercial partner, which is rare. The drug was funded mostly with private donations, including individuals that have lost loved ones to cancer.” ![]()
Group identifies new subtype of ALL

Photo by Steven Harbour
Researchers say they have discovered a new subtype of acute lymphoblastic leukemia (ALL) that is sensitive to drugs already approved to treat other hematologic malignancies.
The team uncovered cases of ALL that were dependent upon tonic pre-B-cell-receptor (BCR) signaling and therefore sensitive to drugs that inhibit tyrosine kinases downstream of the pre-BCR.
The group also developed a test that can identify patients with this subtype of ALL.
“We hope patients in this newly identified subset can be treated with these targeted drugs, . . . which are powerfully effective in the mouse experiments we have conducted on ALL,” said Markus Müschen, MD, PhD, of the University of California, San Francisco.
Dr Müschen and his colleagues described this work in Cancer Cell.
The researchers studied samples from 830 patients enrolled in 4 ongoing ALL trials and found tonic pre-BCR signaling in 112 patients (13.5%). Virtually all of the bone marrow slices from these patients showed “beautiful staining” of BCL6 expression, Dr Müschen said. (Two of the patients had weak staining.)
On the other hand, no BCL6 staining was observed in patients lacking pre-BCR signaling. These results suggest that BCL6 is a biomarker for pre-BCR signaling. And by testing patients for BCL6, we may be able to identify those who will respond to treatment with pre-BCR signaling inhibitors, the researchers said.
The team tested a range of pre-BCR signaling inhibitors in vitro. And they found a few compounds that were effective against pre-BCR+ ALL—the SYK inhibitor PRT062607, the BTK inhibitor ibrutinib, the SRC inhibitor dasatinib, and the PIK3δ inhibitor idelalisib.
Subsequent experiments revealed that dasatinib had the strongest antileukemic effect, so the researchers tested the drug in mouse models of pre-BCR+ ALL. Dasatinib significantly delayed leukemic expansion and prolonged overall survival in some mice, while completely eradicating the disease in other mice.
Dr Müschen said that dasatinib and other pre-BCR signaling inhibitors may be able to reduce the amount of conventional chemotherapy given to patients with pre-BCR+ ALL, or even replace chemotherapy altogether.
“In our experiments with mice, both combination therapy with low-dose chemotherapy and single-agent targeted therapy each worked very well,” he said. ![]()
Photo by Steven Harbour
Researchers say they have discovered a new subtype of acute lymphoblastic leukemia (ALL) that is sensitive to drugs already approved to treat other hematologic malignancies.
The team uncovered cases of ALL that were dependent upon tonic pre-B-cell-receptor (BCR) signaling and therefore sensitive to drugs that inhibit tyrosine kinases downstream of the pre-BCR.
The group also developed a test that can identify patients with this subtype of ALL.
“We hope patients in this newly identified subset can be treated with these targeted drugs, . . . which are powerfully effective in the mouse experiments we have conducted on ALL,” said Markus Müschen, MD, PhD, of the University of California, San Francisco.
Dr Müschen and his colleagues described this work in Cancer Cell.
The researchers studied samples from 830 patients enrolled in 4 ongoing ALL trials and found tonic pre-BCR signaling in 112 patients (13.5%). Virtually all of the bone marrow slices from these patients showed “beautiful staining” of BCL6 expression, Dr Müschen said. (Two of the patients had weak staining.)
On the other hand, no BCL6 staining was observed in patients lacking pre-BCR signaling. These results suggest that BCL6 is a biomarker for pre-BCR signaling. And by testing patients for BCL6, we may be able to identify those who will respond to treatment with pre-BCR signaling inhibitors, the researchers said.
The team tested a range of pre-BCR signaling inhibitors in vitro. And they found a few compounds that were effective against pre-BCR+ ALL—the SYK inhibitor PRT062607, the BTK inhibitor ibrutinib, the SRC inhibitor dasatinib, and the PIK3δ inhibitor idelalisib.
Subsequent experiments revealed that dasatinib had the strongest antileukemic effect, so the researchers tested the drug in mouse models of pre-BCR+ ALL. Dasatinib significantly delayed leukemic expansion and prolonged overall survival in some mice, while completely eradicating the disease in other mice.
Dr Müschen said that dasatinib and other pre-BCR signaling inhibitors may be able to reduce the amount of conventional chemotherapy given to patients with pre-BCR+ ALL, or even replace chemotherapy altogether.
“In our experiments with mice, both combination therapy with low-dose chemotherapy and single-agent targeted therapy each worked very well,” he said. ![]()
Photo by Steven Harbour
Researchers say they have discovered a new subtype of acute lymphoblastic leukemia (ALL) that is sensitive to drugs already approved to treat other hematologic malignancies.
The team uncovered cases of ALL that were dependent upon tonic pre-B-cell-receptor (BCR) signaling and therefore sensitive to drugs that inhibit tyrosine kinases downstream of the pre-BCR.
The group also developed a test that can identify patients with this subtype of ALL.
“We hope patients in this newly identified subset can be treated with these targeted drugs, . . . which are powerfully effective in the mouse experiments we have conducted on ALL,” said Markus Müschen, MD, PhD, of the University of California, San Francisco.
Dr Müschen and his colleagues described this work in Cancer Cell.
The researchers studied samples from 830 patients enrolled in 4 ongoing ALL trials and found tonic pre-BCR signaling in 112 patients (13.5%). Virtually all of the bone marrow slices from these patients showed “beautiful staining” of BCL6 expression, Dr Müschen said. (Two of the patients had weak staining.)
On the other hand, no BCL6 staining was observed in patients lacking pre-BCR signaling. These results suggest that BCL6 is a biomarker for pre-BCR signaling. And by testing patients for BCL6, we may be able to identify those who will respond to treatment with pre-BCR signaling inhibitors, the researchers said.
The team tested a range of pre-BCR signaling inhibitors in vitro. And they found a few compounds that were effective against pre-BCR+ ALL—the SYK inhibitor PRT062607, the BTK inhibitor ibrutinib, the SRC inhibitor dasatinib, and the PIK3δ inhibitor idelalisib.
Subsequent experiments revealed that dasatinib had the strongest antileukemic effect, so the researchers tested the drug in mouse models of pre-BCR+ ALL. Dasatinib significantly delayed leukemic expansion and prolonged overall survival in some mice, while completely eradicating the disease in other mice.
Dr Müschen said that dasatinib and other pre-BCR signaling inhibitors may be able to reduce the amount of conventional chemotherapy given to patients with pre-BCR+ ALL, or even replace chemotherapy altogether.
“In our experiments with mice, both combination therapy with low-dose chemotherapy and single-agent targeted therapy each worked very well,” he said. ![]()
Aggressive infant leukemia has few mutations

Photo by Vera Kratochvil
Infants who have acute lymphoblastic leukemia (ALL) with MLL rearrangements have few other mutations, according to new research.
The findings suggest that targeting MLL rearrangements in these patients is likely the key to improving their survival.
“We frequently associate a cancer’s aggressiveness with its mutation rate, but this work indicates that the two don’t always go hand-in-hand,” said Richard K. Wilson, PhD, of the Washington University School of Medicine in St Louis, Missouri.
“Still, our findings provide a new direction for developing more effective treatments for these very young patients.”
Dr Wilson and his colleagues reported their findings in Nature Genetics.
The researchers performed whole-genome, exome, RNA, and targeted DNA sequencing to identify genetic alterations in 65 infants with ALL, including 47 with the MLL rearrangement.
The team was surprised to find that, despite being an aggressive leukemia, the MLL-rearranged subtype had among the lowest mutation rates reported for any cancer. The predominant leukemic clone carried a mean of 1.3 non-silent mutations.
“These results show that, to improve survival for patients with this aggressive leukemia, we need to develop drugs that target the abnormal proteins produced by the MLL fusion gene or that interact with the abnormal MLL fusion protein to shut down the cellular machinery that drives their tumors,” said James R. Downing, MD, of St Jude Research Hospital in Memphis, Tennessee. “That will not be easy, but this study found no obvious cooperating mutations to target.”
Almost half of infants with MLL-rearranged ALL (47%) had activating mutations in the kinase-PI3K-RAS signaling pathway. But the mutations were often present in only some of the leukemic cells.
Furthermore, the researchers analyzed leukemia cells in infants whose cancer returned after treatment and found that, at the time of relapse, the cells lacked these mutations.
“The fact that the mutations were often lost at relapse suggests that patients are unlikely to benefit from therapeutically targeting these mutations at diagnosis,” Dr Downing said.
The researchers also found that older children with MLL-rearranged leukemia had significantly more mutations than infants—a mean of 6.5 mutations per case (P=7.15 × 10−5).
Furthermore, 45% of the older children had mutations in genes that encode epigenetic regulatory proteins. And, aside from MLL, epigenetic regulators were rarely mutated in infants with MLL-rearranged ALL.
“While MLL belongs to a family of genes that encode epigenetic regulatory proteins, there was a striking difference between infants and older children regarding the frequency of mutations in other epigenetic regulatory genes,” said Anna Andersson, PhD, of Lund University in Sweden.
“This observation raises the possibility of a fundamental difference in the cell targeted for transformation in infants versus older patients,” said Tanja Gruber, MD, PhD, of St Jude.
“Our working hypothesis is that, in infants, the MLL rearrangement occurs in a developing blood cell, a prenatal progenitor cell, which requires fewer additional mutations to fully transform into leukemia. In contrast, in older patients, the MLL rearrangement isn’t enough on its own.” ![]()
Photo by Vera Kratochvil
Infants who have acute lymphoblastic leukemia (ALL) with MLL rearrangements have few other mutations, according to new research.
The findings suggest that targeting MLL rearrangements in these patients is likely the key to improving their survival.
“We frequently associate a cancer’s aggressiveness with its mutation rate, but this work indicates that the two don’t always go hand-in-hand,” said Richard K. Wilson, PhD, of the Washington University School of Medicine in St Louis, Missouri.
“Still, our findings provide a new direction for developing more effective treatments for these very young patients.”
Dr Wilson and his colleagues reported their findings in Nature Genetics.
The researchers performed whole-genome, exome, RNA, and targeted DNA sequencing to identify genetic alterations in 65 infants with ALL, including 47 with the MLL rearrangement.
The team was surprised to find that, despite being an aggressive leukemia, the MLL-rearranged subtype had among the lowest mutation rates reported for any cancer. The predominant leukemic clone carried a mean of 1.3 non-silent mutations.
“These results show that, to improve survival for patients with this aggressive leukemia, we need to develop drugs that target the abnormal proteins produced by the MLL fusion gene or that interact with the abnormal MLL fusion protein to shut down the cellular machinery that drives their tumors,” said James R. Downing, MD, of St Jude Research Hospital in Memphis, Tennessee. “That will not be easy, but this study found no obvious cooperating mutations to target.”
Almost half of infants with MLL-rearranged ALL (47%) had activating mutations in the kinase-PI3K-RAS signaling pathway. But the mutations were often present in only some of the leukemic cells.
Furthermore, the researchers analyzed leukemia cells in infants whose cancer returned after treatment and found that, at the time of relapse, the cells lacked these mutations.
“The fact that the mutations were often lost at relapse suggests that patients are unlikely to benefit from therapeutically targeting these mutations at diagnosis,” Dr Downing said.
The researchers also found that older children with MLL-rearranged leukemia had significantly more mutations than infants—a mean of 6.5 mutations per case (P=7.15 × 10−5).
Furthermore, 45% of the older children had mutations in genes that encode epigenetic regulatory proteins. And, aside from MLL, epigenetic regulators were rarely mutated in infants with MLL-rearranged ALL.
“While MLL belongs to a family of genes that encode epigenetic regulatory proteins, there was a striking difference between infants and older children regarding the frequency of mutations in other epigenetic regulatory genes,” said Anna Andersson, PhD, of Lund University in Sweden.
“This observation raises the possibility of a fundamental difference in the cell targeted for transformation in infants versus older patients,” said Tanja Gruber, MD, PhD, of St Jude.
“Our working hypothesis is that, in infants, the MLL rearrangement occurs in a developing blood cell, a prenatal progenitor cell, which requires fewer additional mutations to fully transform into leukemia. In contrast, in older patients, the MLL rearrangement isn’t enough on its own.” ![]()
Photo by Vera Kratochvil
Infants who have acute lymphoblastic leukemia (ALL) with MLL rearrangements have few other mutations, according to new research.
The findings suggest that targeting MLL rearrangements in these patients is likely the key to improving their survival.
“We frequently associate a cancer’s aggressiveness with its mutation rate, but this work indicates that the two don’t always go hand-in-hand,” said Richard K. Wilson, PhD, of the Washington University School of Medicine in St Louis, Missouri.
“Still, our findings provide a new direction for developing more effective treatments for these very young patients.”
Dr Wilson and his colleagues reported their findings in Nature Genetics.
The researchers performed whole-genome, exome, RNA, and targeted DNA sequencing to identify genetic alterations in 65 infants with ALL, including 47 with the MLL rearrangement.
The team was surprised to find that, despite being an aggressive leukemia, the MLL-rearranged subtype had among the lowest mutation rates reported for any cancer. The predominant leukemic clone carried a mean of 1.3 non-silent mutations.
“These results show that, to improve survival for patients with this aggressive leukemia, we need to develop drugs that target the abnormal proteins produced by the MLL fusion gene or that interact with the abnormal MLL fusion protein to shut down the cellular machinery that drives their tumors,” said James R. Downing, MD, of St Jude Research Hospital in Memphis, Tennessee. “That will not be easy, but this study found no obvious cooperating mutations to target.”
Almost half of infants with MLL-rearranged ALL (47%) had activating mutations in the kinase-PI3K-RAS signaling pathway. But the mutations were often present in only some of the leukemic cells.
Furthermore, the researchers analyzed leukemia cells in infants whose cancer returned after treatment and found that, at the time of relapse, the cells lacked these mutations.
“The fact that the mutations were often lost at relapse suggests that patients are unlikely to benefit from therapeutically targeting these mutations at diagnosis,” Dr Downing said.
The researchers also found that older children with MLL-rearranged leukemia had significantly more mutations than infants—a mean of 6.5 mutations per case (P=7.15 × 10−5).
Furthermore, 45% of the older children had mutations in genes that encode epigenetic regulatory proteins. And, aside from MLL, epigenetic regulators were rarely mutated in infants with MLL-rearranged ALL.
“While MLL belongs to a family of genes that encode epigenetic regulatory proteins, there was a striking difference between infants and older children regarding the frequency of mutations in other epigenetic regulatory genes,” said Anna Andersson, PhD, of Lund University in Sweden.
“This observation raises the possibility of a fundamental difference in the cell targeted for transformation in infants versus older patients,” said Tanja Gruber, MD, PhD, of St Jude.
“Our working hypothesis is that, in infants, the MLL rearrangement occurs in a developing blood cell, a prenatal progenitor cell, which requires fewer additional mutations to fully transform into leukemia. In contrast, in older patients, the MLL rearrangement isn’t enough on its own.” ![]()
Variant may predict risk of toxicity in ALL

Kristine Crews, Barthelemy
Diouf, and William Evans
Photo by Seth Dixon
A gene variant may help predict vincristine-related toxicity in children with acute lymphoblastic leukemia (ALL).
A study of more than 300 children with ALL showed that patients with an inherited variant in the gene CEP72 were more likely to develop peripheral neuropathy after receiving the chemotherapy drug vincristine.
If these results can be replicated in additional populations, they may provide a basis for safer dosing of the drug, according to investigators.
William Evans, PharmD, of St Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and shared the results in JAMA.
The investigators analyzed patients in 2 prospective clinical trials for childhood ALL that included treatment with 36 to 39 doses of vincristine. The team performed genetic analyses and assessed vincristine-induced peripheral neuropathy in 321 patients in whom DNA data were available.
This included 222 patients with a median age of 6 years who were enrolled in a St Jude Children’s Research Hospital study from 1994 to 1998, as well as 99 patients with a median age of 11.4 years who were enrolled in a Children’s Oncology Group (COG) study from 2007 to 2010.
Grade 2 to 4 vincristine-induced peripheral neuropathy occurred in 28.8% of patients (64/222) in the St Jude cohort and in 22.2% (22/99) in the COG cohort.
The investigators found that a single nucleotide polymorphism (SNP) in the promoter region of the CEP72 gene, which encodes a centrosomal protein involved in microtubule formation, was significantly associated with vincristine-induced neuropathy (P=6.3×10-9).
This SNP had a minor allele frequency of 37%, and 16% of patients (50/321) were homozygous for the risk allele (TT at rs924607).
Among patients with the high-risk CEP72 genotype, 56% (28/50) had at least 1 episode of grade 2 to 4 neuropathy. This was significantly higher than in patients with the CEP72 CC or CT genotypes. About 21% of those patients (58/271) had at least 1 episode of grade 2 to 4 neuropathy (P=2.4×10-6).
In addition, the severity of neuropathy was greater in patients who were homozygous for the TT genotype than in patients with the CC or CT genotype—2.4-fold greater by Poisson regression (P<0.0001) and 2.7-fold greater based on the mean grade of neuropathy (P=0.004).
Finally, in lab experiments, the investigators found that reducing CEP72 expression in human neurons and leukemia cells increased their sensitivity to vincristine.
In a related editorial, Howard L. McLeod, PharmD, of the Moffitt Cancer Center in Tampa, Florida, noted that this study has several strengths: genome-wide discovery in patients from well-conducted clinical trials, replication in a multicenter cohort, statistical robustness, and laboratory correlative findings that contribute to biologic plausibility.
However, he also pointed out that vincristine remains a component of the most widely accepted treatment regimens for childhood ALL.
“It is not clear that vincristine can be removed from the treatment options for a child with CEP72 variants, although this study suggests that the resulting increase in leukemia cellular sensitivity makes vincristine dose reductions possible without compromising antileukemic effect,” he wrote.
“However, there is value in the association of CEP72 with vincristine-induced peripheral neuropathy (VIPN). The ability to objectively ascribe a degree of heightened VIPN risk will allow for greater transparency in discussions of risk and benefits of therapy with patients and their family members.”
“This also may lead to developmental therapeutic approaches to modulate CEP72 function as either primary prevention or treatment of chronic VIPN. This study also represents an initial robust effort to generate predictors for adverse drug reactions in cancer care.” ![]()
Kristine Crews, Barthelemy
Diouf, and William Evans
Photo by Seth Dixon
A gene variant may help predict vincristine-related toxicity in children with acute lymphoblastic leukemia (ALL).
A study of more than 300 children with ALL showed that patients with an inherited variant in the gene CEP72 were more likely to develop peripheral neuropathy after receiving the chemotherapy drug vincristine.
If these results can be replicated in additional populations, they may provide a basis for safer dosing of the drug, according to investigators.
William Evans, PharmD, of St Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and shared the results in JAMA.
The investigators analyzed patients in 2 prospective clinical trials for childhood ALL that included treatment with 36 to 39 doses of vincristine. The team performed genetic analyses and assessed vincristine-induced peripheral neuropathy in 321 patients in whom DNA data were available.
This included 222 patients with a median age of 6 years who were enrolled in a St Jude Children’s Research Hospital study from 1994 to 1998, as well as 99 patients with a median age of 11.4 years who were enrolled in a Children’s Oncology Group (COG) study from 2007 to 2010.
Grade 2 to 4 vincristine-induced peripheral neuropathy occurred in 28.8% of patients (64/222) in the St Jude cohort and in 22.2% (22/99) in the COG cohort.
The investigators found that a single nucleotide polymorphism (SNP) in the promoter region of the CEP72 gene, which encodes a centrosomal protein involved in microtubule formation, was significantly associated with vincristine-induced neuropathy (P=6.3×10-9).
This SNP had a minor allele frequency of 37%, and 16% of patients (50/321) were homozygous for the risk allele (TT at rs924607).
Among patients with the high-risk CEP72 genotype, 56% (28/50) had at least 1 episode of grade 2 to 4 neuropathy. This was significantly higher than in patients with the CEP72 CC or CT genotypes. About 21% of those patients (58/271) had at least 1 episode of grade 2 to 4 neuropathy (P=2.4×10-6).
In addition, the severity of neuropathy was greater in patients who were homozygous for the TT genotype than in patients with the CC or CT genotype—2.4-fold greater by Poisson regression (P<0.0001) and 2.7-fold greater based on the mean grade of neuropathy (P=0.004).
Finally, in lab experiments, the investigators found that reducing CEP72 expression in human neurons and leukemia cells increased their sensitivity to vincristine.
In a related editorial, Howard L. McLeod, PharmD, of the Moffitt Cancer Center in Tampa, Florida, noted that this study has several strengths: genome-wide discovery in patients from well-conducted clinical trials, replication in a multicenter cohort, statistical robustness, and laboratory correlative findings that contribute to biologic plausibility.
However, he also pointed out that vincristine remains a component of the most widely accepted treatment regimens for childhood ALL.
“It is not clear that vincristine can be removed from the treatment options for a child with CEP72 variants, although this study suggests that the resulting increase in leukemia cellular sensitivity makes vincristine dose reductions possible without compromising antileukemic effect,” he wrote.
“However, there is value in the association of CEP72 with vincristine-induced peripheral neuropathy (VIPN). The ability to objectively ascribe a degree of heightened VIPN risk will allow for greater transparency in discussions of risk and benefits of therapy with patients and their family members.”
“This also may lead to developmental therapeutic approaches to modulate CEP72 function as either primary prevention or treatment of chronic VIPN. This study also represents an initial robust effort to generate predictors for adverse drug reactions in cancer care.” ![]()
Kristine Crews, Barthelemy
Diouf, and William Evans
Photo by Seth Dixon
A gene variant may help predict vincristine-related toxicity in children with acute lymphoblastic leukemia (ALL).
A study of more than 300 children with ALL showed that patients with an inherited variant in the gene CEP72 were more likely to develop peripheral neuropathy after receiving the chemotherapy drug vincristine.
If these results can be replicated in additional populations, they may provide a basis for safer dosing of the drug, according to investigators.
William Evans, PharmD, of St Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and shared the results in JAMA.
The investigators analyzed patients in 2 prospective clinical trials for childhood ALL that included treatment with 36 to 39 doses of vincristine. The team performed genetic analyses and assessed vincristine-induced peripheral neuropathy in 321 patients in whom DNA data were available.
This included 222 patients with a median age of 6 years who were enrolled in a St Jude Children’s Research Hospital study from 1994 to 1998, as well as 99 patients with a median age of 11.4 years who were enrolled in a Children’s Oncology Group (COG) study from 2007 to 2010.
Grade 2 to 4 vincristine-induced peripheral neuropathy occurred in 28.8% of patients (64/222) in the St Jude cohort and in 22.2% (22/99) in the COG cohort.
The investigators found that a single nucleotide polymorphism (SNP) in the promoter region of the CEP72 gene, which encodes a centrosomal protein involved in microtubule formation, was significantly associated with vincristine-induced neuropathy (P=6.3×10-9).
This SNP had a minor allele frequency of 37%, and 16% of patients (50/321) were homozygous for the risk allele (TT at rs924607).
Among patients with the high-risk CEP72 genotype, 56% (28/50) had at least 1 episode of grade 2 to 4 neuropathy. This was significantly higher than in patients with the CEP72 CC or CT genotypes. About 21% of those patients (58/271) had at least 1 episode of grade 2 to 4 neuropathy (P=2.4×10-6).
In addition, the severity of neuropathy was greater in patients who were homozygous for the TT genotype than in patients with the CC or CT genotype—2.4-fold greater by Poisson regression (P<0.0001) and 2.7-fold greater based on the mean grade of neuropathy (P=0.004).
Finally, in lab experiments, the investigators found that reducing CEP72 expression in human neurons and leukemia cells increased their sensitivity to vincristine.
In a related editorial, Howard L. McLeod, PharmD, of the Moffitt Cancer Center in Tampa, Florida, noted that this study has several strengths: genome-wide discovery in patients from well-conducted clinical trials, replication in a multicenter cohort, statistical robustness, and laboratory correlative findings that contribute to biologic plausibility.
However, he also pointed out that vincristine remains a component of the most widely accepted treatment regimens for childhood ALL.
“It is not clear that vincristine can be removed from the treatment options for a child with CEP72 variants, although this study suggests that the resulting increase in leukemia cellular sensitivity makes vincristine dose reductions possible without compromising antileukemic effect,” he wrote.
“However, there is value in the association of CEP72 with vincristine-induced peripheral neuropathy (VIPN). The ability to objectively ascribe a degree of heightened VIPN risk will allow for greater transparency in discussions of risk and benefits of therapy with patients and their family members.”
“This also may lead to developmental therapeutic approaches to modulate CEP72 function as either primary prevention or treatment of chronic VIPN. This study also represents an initial robust effort to generate predictors for adverse drug reactions in cancer care.”
VIDEO: Search for genetic risk factors may improve vincristine therapy
Discovery of a genetic variation that may boost the risk of peripheral neuropathy for some cancer patients on vincristine therapy could lead to better treatment regimens for those patients.
Investigators uncovered the genetic variation in a study that analyzed DNA samples from 321 children with acute lymphoblastic leukemia who received standard vincristine therapy while participating in two prospective clinical trials (JAMA 2015 Feb. 24 [doi:10.1001/jama.2015.0894]).
William E. Evans, Pharm.D., and Kristine R. Crews, Pharm.D., both of St. Jude Children’s Research Hospital, Memphis, Tenn., discussed what spurred the search for genetic risk factors and what’s next for researchers looking to cut the chance of side effects associated with vincristine treatment in a video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Discovery of a genetic variation that may boost the risk of peripheral neuropathy for some cancer patients on vincristine therapy could lead to better treatment regimens for those patients.
Investigators uncovered the genetic variation in a study that analyzed DNA samples from 321 children with acute lymphoblastic leukemia who received standard vincristine therapy while participating in two prospective clinical trials (JAMA 2015 Feb. 24 [doi:10.1001/jama.2015.0894]).
William E. Evans, Pharm.D., and Kristine R. Crews, Pharm.D., both of St. Jude Children’s Research Hospital, Memphis, Tenn., discussed what spurred the search for genetic risk factors and what’s next for researchers looking to cut the chance of side effects associated with vincristine treatment in a video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Discovery of a genetic variation that may boost the risk of peripheral neuropathy for some cancer patients on vincristine therapy could lead to better treatment regimens for those patients.
Investigators uncovered the genetic variation in a study that analyzed DNA samples from 321 children with acute lymphoblastic leukemia who received standard vincristine therapy while participating in two prospective clinical trials (JAMA 2015 Feb. 24 [doi:10.1001/jama.2015.0894]).
William E. Evans, Pharm.D., and Kristine R. Crews, Pharm.D., both of St. Jude Children’s Research Hospital, Memphis, Tenn., discussed what spurred the search for genetic risk factors and what’s next for researchers looking to cut the chance of side effects associated with vincristine treatment in a video interview.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
FROM JAMA

Predicting outcomes of allo-HSCT in ALL
![]()
Photo by Chad McNeeley
SAN DIEGO—A retrospective study has revealed a few factors that may predict outcomes of allogeneic hematopoietic stem cell transplant (allo-HSCT) in adults with acute lymphoblastic leukemia (ALL).
The study showed that cytogenetics at diagnosis did not impact survival rates, although having high-risk cytogenetics was associated with an increased incidence of relapse in patients who were transplanted in their first complete remission (CR1).
Patients who were not in CR1 at transplant tended to have worse survival and higher relapse rates.
And patients who received a tacrolimus/sirolimus-based regimen as graft-vs-host disease (GVHD) prophylaxis had better survival rates than their peers, but their relapse rates did not differ.
Ibrahim Aldoss, MD, of City of Hope Medical Center in Duarte, California, presented these findings at the 2015 BMT Tandem Meetings as abstract 69.*
Dr Aldoss said there is a lack of data addressing individual ALL-related prognostic factors for transplant outcomes. So he and his colleagues decided to analyze 358 adult ALL patients who received allo-HSCT at the City of Hope from January 2004 through March 2014.
The patients’ median age was 38 (range, 18 to 72), and most patients (91%) had B-cell disease. At diagnosis, 2% of patients had good-risk cytogenetics, 43% had intermediate-risk, and 46% had poor-risk. For 9% of patients, the cytogenetic risk group was unknown.
At transplant, 60% of patients were in CR1, 17% were in CR2, and 23% were in a subsequent CR or had refractory disease.
Most patients received peripheral blood stem cell transplant (86%), 7% of patients received bone marrow, and the same percentage received cord blood. Fifty-four percent of patients had a matched sibling donor, 45% had an unrelated donor, and 1% had a related donor.
Eighty-one percent of patients received myeloablative conditioning, and the same percentage received a tacrolimus/sirolimus-based regimen for GVHD prophylaxis.
The 3-year estimated overall survival (OS) rate was 54%, leukemia-free survival (LFS) was 47%, and the cumulative incidence of relapse (CIR) was 27%. The 1-year non-relapse mortality (NRM) rate was 19%.
In multivariable analyses, disease status at allo-HSCT was an independent predictor of OS, LFS, and CIR. For OS, when the researchers compared patients in CR1 to those in CR2, the hazard ratio (HR) was 1.87 (P<0.01). When patients in CR1 were compared to other patients, the HR was 2.79 (P<0.01).
For LFS, the HRs were 1.69 (P=0.02) for CR1 vs CR2 and 2.94 (P<0.01) for CR1 vs others. And for CIR, the HRs were 2.21 (P<0.01) and 3.55 (P<0.01), respectively.
The analyses also revealed that tacrolimus/sirolimus-based GVHD prophylaxis was an independent predictor of OS, LFS, and NRM. The HRs were 1.58 (P=0.03), 1.5 (P=0.03), and 1.75 (P=0.03), respectively.
“So cytogenetics at diagnosis did not impact overall survival or leukemia-free survival among adult ALL patients who underwent allogeneic stem cell transplantation,” Dr Aldoss said in closing. “However, high-risk cytogenetics was associated with an increased cumulative incidence of relapse in patients transplanted in CR1.”
“Non-CR1 status at the time of transplant adversely affected overall survival, leukemia-free survival, and cumulative incidence of relapse. And a tacrolimus/sirolimus-based GVHD prophylaxis regimen was associated with improved overall survival, leukemia-free survival, and non-relapse mortality but did not influence the cumulative incidence of relapse.”
*Information in the abstract differs from that presented at the meeting.
![]()
Photo by Chad McNeeley
SAN DIEGO—A retrospective study has revealed a few factors that may predict outcomes of allogeneic hematopoietic stem cell transplant (allo-HSCT) in adults with acute lymphoblastic leukemia (ALL).
The study showed that cytogenetics at diagnosis did not impact survival rates, although having high-risk cytogenetics was associated with an increased incidence of relapse in patients who were transplanted in their first complete remission (CR1).
Patients who were not in CR1 at transplant tended to have worse survival and higher relapse rates.
And patients who received a tacrolimus/sirolimus-based regimen as graft-vs-host disease (GVHD) prophylaxis had better survival rates than their peers, but their relapse rates did not differ.
Ibrahim Aldoss, MD, of City of Hope Medical Center in Duarte, California, presented these findings at the 2015 BMT Tandem Meetings as abstract 69.*
Dr Aldoss said there is a lack of data addressing individual ALL-related prognostic factors for transplant outcomes. So he and his colleagues decided to analyze 358 adult ALL patients who received allo-HSCT at the City of Hope from January 2004 through March 2014.
The patients’ median age was 38 (range, 18 to 72), and most patients (91%) had B-cell disease. At diagnosis, 2% of patients had good-risk cytogenetics, 43% had intermediate-risk, and 46% had poor-risk. For 9% of patients, the cytogenetic risk group was unknown.
At transplant, 60% of patients were in CR1, 17% were in CR2, and 23% were in a subsequent CR or had refractory disease.
Most patients received peripheral blood stem cell transplant (86%), 7% of patients received bone marrow, and the same percentage received cord blood. Fifty-four percent of patients had a matched sibling donor, 45% had an unrelated donor, and 1% had a related donor.
Eighty-one percent of patients received myeloablative conditioning, and the same percentage received a tacrolimus/sirolimus-based regimen for GVHD prophylaxis.
The 3-year estimated overall survival (OS) rate was 54%, leukemia-free survival (LFS) was 47%, and the cumulative incidence of relapse (CIR) was 27%. The 1-year non-relapse mortality (NRM) rate was 19%.
In multivariable analyses, disease status at allo-HSCT was an independent predictor of OS, LFS, and CIR. For OS, when the researchers compared patients in CR1 to those in CR2, the hazard ratio (HR) was 1.87 (P<0.01). When patients in CR1 were compared to other patients, the HR was 2.79 (P<0.01).
For LFS, the HRs were 1.69 (P=0.02) for CR1 vs CR2 and 2.94 (P<0.01) for CR1 vs others. And for CIR, the HRs were 2.21 (P<0.01) and 3.55 (P<0.01), respectively.
The analyses also revealed that tacrolimus/sirolimus-based GVHD prophylaxis was an independent predictor of OS, LFS, and NRM. The HRs were 1.58 (P=0.03), 1.5 (P=0.03), and 1.75 (P=0.03), respectively.
“So cytogenetics at diagnosis did not impact overall survival or leukemia-free survival among adult ALL patients who underwent allogeneic stem cell transplantation,” Dr Aldoss said in closing. “However, high-risk cytogenetics was associated with an increased cumulative incidence of relapse in patients transplanted in CR1.”
“Non-CR1 status at the time of transplant adversely affected overall survival, leukemia-free survival, and cumulative incidence of relapse. And a tacrolimus/sirolimus-based GVHD prophylaxis regimen was associated with improved overall survival, leukemia-free survival, and non-relapse mortality but did not influence the cumulative incidence of relapse.”
*Information in the abstract differs from that presented at the meeting.
![]()
Photo by Chad McNeeley
SAN DIEGO—A retrospective study has revealed a few factors that may predict outcomes of allogeneic hematopoietic stem cell transplant (allo-HSCT) in adults with acute lymphoblastic leukemia (ALL).
The study showed that cytogenetics at diagnosis did not impact survival rates, although having high-risk cytogenetics was associated with an increased incidence of relapse in patients who were transplanted in their first complete remission (CR1).
Patients who were not in CR1 at transplant tended to have worse survival and higher relapse rates.
And patients who received a tacrolimus/sirolimus-based regimen as graft-vs-host disease (GVHD) prophylaxis had better survival rates than their peers, but their relapse rates did not differ.
Ibrahim Aldoss, MD, of City of Hope Medical Center in Duarte, California, presented these findings at the 2015 BMT Tandem Meetings as abstract 69.*
Dr Aldoss said there is a lack of data addressing individual ALL-related prognostic factors for transplant outcomes. So he and his colleagues decided to analyze 358 adult ALL patients who received allo-HSCT at the City of Hope from January 2004 through March 2014.
The patients’ median age was 38 (range, 18 to 72), and most patients (91%) had B-cell disease. At diagnosis, 2% of patients had good-risk cytogenetics, 43% had intermediate-risk, and 46% had poor-risk. For 9% of patients, the cytogenetic risk group was unknown.
At transplant, 60% of patients were in CR1, 17% were in CR2, and 23% were in a subsequent CR or had refractory disease.
Most patients received peripheral blood stem cell transplant (86%), 7% of patients received bone marrow, and the same percentage received cord blood. Fifty-four percent of patients had a matched sibling donor, 45% had an unrelated donor, and 1% had a related donor.
Eighty-one percent of patients received myeloablative conditioning, and the same percentage received a tacrolimus/sirolimus-based regimen for GVHD prophylaxis.
The 3-year estimated overall survival (OS) rate was 54%, leukemia-free survival (LFS) was 47%, and the cumulative incidence of relapse (CIR) was 27%. The 1-year non-relapse mortality (NRM) rate was 19%.
In multivariable analyses, disease status at allo-HSCT was an independent predictor of OS, LFS, and CIR. For OS, when the researchers compared patients in CR1 to those in CR2, the hazard ratio (HR) was 1.87 (P<0.01). When patients in CR1 were compared to other patients, the HR was 2.79 (P<0.01).
For LFS, the HRs were 1.69 (P=0.02) for CR1 vs CR2 and 2.94 (P<0.01) for CR1 vs others. And for CIR, the HRs were 2.21 (P<0.01) and 3.55 (P<0.01), respectively.
The analyses also revealed that tacrolimus/sirolimus-based GVHD prophylaxis was an independent predictor of OS, LFS, and NRM. The HRs were 1.58 (P=0.03), 1.5 (P=0.03), and 1.75 (P=0.03), respectively.
“So cytogenetics at diagnosis did not impact overall survival or leukemia-free survival among adult ALL patients who underwent allogeneic stem cell transplantation,” Dr Aldoss said in closing. “However, high-risk cytogenetics was associated with an increased cumulative incidence of relapse in patients transplanted in CR1.”
“Non-CR1 status at the time of transplant adversely affected overall survival, leukemia-free survival, and cumulative incidence of relapse. And a tacrolimus/sirolimus-based GVHD prophylaxis regimen was associated with improved overall survival, leukemia-free survival, and non-relapse mortality but did not influence the cumulative incidence of relapse.”
*Information in the abstract differs from that presented at the meeting.
Approved TKI could treat drug-resistant CML, ALL

Photo courtesy of CDC
New research indicates that a tyrosine kinase inhibitor (TKI) approved to treat advanced renal cell carcinoma could prove useful in treating patients with drug-resistant chronic myeloid leukemia (CML) or acute lymphoblastic leukemia (ALL).
The study showed that the TKI, axitinib, can inhibit BCR-ABL1 (T315I), a mutation known to confer drug resistance in CML and ALL.
“Since axitinib is already used to treat cancer, its safety is known,” said Kimmo Porkka, MD, PhD, of the University of Helsinki in Finland.
“[A] formal exploration of its clinical utility in drug-resistant leukemia can now be done in a fast-track mode. Thus, the normally very long path from lab bench to bedside is now significantly shortened.”
Dr Porkka and his colleagues described the newfound activity of axitinib in Nature.
The researchers used a drug sensitivity and resistance testing method developed at the University of Helsinki’s Institute for Molecular Medicine Finland (FIMM) to examine how patient-derived leukemia cells responded to a large panel of drugs.
In this way, the group identified axitinib as a promising drug candidate for CML and ALL. Axitinib effectively eliminated drug-resistant leukemia cells.
The TKI inhibited BCR-ABL1 (T315I) at biochemical and cellular levels by binding to the active form of ABL1 (T315I) in a mutation-selective binding mode.
The researchers said this suggests the T315I mutation shifts the conformational equilibrium of the kinase in favor of an active (DFG-in) A-loop conformation, which has more optimal binding interactions with axitinib.
“If you think of the targeted protein as a lock into which the cancer drug fits in as a key, the resistant protein changes in such a way that we need a different key,” said study author Brion Murray, PhD, of Pfizer Worldwide Research & Development in San Diego, California.
“In the case of axitinib, it acts as two distinct keys—one for renal cell carcinoma and one for leukemia.”
The researchers also treated a CML patient with axitinib and observed a “rapid reduction” of T315I-positive cells in the patient’s bone marrow.
“Further research will determine whether these findings have the potential to significantly improve the standard of care for this select group of CML patients and patients with other related leukemias,” Dr Murray concluded.
Photo courtesy of CDC
New research indicates that a tyrosine kinase inhibitor (TKI) approved to treat advanced renal cell carcinoma could prove useful in treating patients with drug-resistant chronic myeloid leukemia (CML) or acute lymphoblastic leukemia (ALL).
The study showed that the TKI, axitinib, can inhibit BCR-ABL1 (T315I), a mutation known to confer drug resistance in CML and ALL.
“Since axitinib is already used to treat cancer, its safety is known,” said Kimmo Porkka, MD, PhD, of the University of Helsinki in Finland.
“[A] formal exploration of its clinical utility in drug-resistant leukemia can now be done in a fast-track mode. Thus, the normally very long path from lab bench to bedside is now significantly shortened.”
Dr Porkka and his colleagues described the newfound activity of axitinib in Nature.
The researchers used a drug sensitivity and resistance testing method developed at the University of Helsinki’s Institute for Molecular Medicine Finland (FIMM) to examine how patient-derived leukemia cells responded to a large panel of drugs.
In this way, the group identified axitinib as a promising drug candidate for CML and ALL. Axitinib effectively eliminated drug-resistant leukemia cells.
The TKI inhibited BCR-ABL1 (T315I) at biochemical and cellular levels by binding to the active form of ABL1 (T315I) in a mutation-selective binding mode.
The researchers said this suggests the T315I mutation shifts the conformational equilibrium of the kinase in favor of an active (DFG-in) A-loop conformation, which has more optimal binding interactions with axitinib.
“If you think of the targeted protein as a lock into which the cancer drug fits in as a key, the resistant protein changes in such a way that we need a different key,” said study author Brion Murray, PhD, of Pfizer Worldwide Research & Development in San Diego, California.
“In the case of axitinib, it acts as two distinct keys—one for renal cell carcinoma and one for leukemia.”
The researchers also treated a CML patient with axitinib and observed a “rapid reduction” of T315I-positive cells in the patient’s bone marrow.
“Further research will determine whether these findings have the potential to significantly improve the standard of care for this select group of CML patients and patients with other related leukemias,” Dr Murray concluded.
Photo courtesy of CDC
New research indicates that a tyrosine kinase inhibitor (TKI) approved to treat advanced renal cell carcinoma could prove useful in treating patients with drug-resistant chronic myeloid leukemia (CML) or acute lymphoblastic leukemia (ALL).
The study showed that the TKI, axitinib, can inhibit BCR-ABL1 (T315I), a mutation known to confer drug resistance in CML and ALL.
“Since axitinib is already used to treat cancer, its safety is known,” said Kimmo Porkka, MD, PhD, of the University of Helsinki in Finland.
“[A] formal exploration of its clinical utility in drug-resistant leukemia can now be done in a fast-track mode. Thus, the normally very long path from lab bench to bedside is now significantly shortened.”
Dr Porkka and his colleagues described the newfound activity of axitinib in Nature.
The researchers used a drug sensitivity and resistance testing method developed at the University of Helsinki’s Institute for Molecular Medicine Finland (FIMM) to examine how patient-derived leukemia cells responded to a large panel of drugs.
In this way, the group identified axitinib as a promising drug candidate for CML and ALL. Axitinib effectively eliminated drug-resistant leukemia cells.
The TKI inhibited BCR-ABL1 (T315I) at biochemical and cellular levels by binding to the active form of ABL1 (T315I) in a mutation-selective binding mode.
The researchers said this suggests the T315I mutation shifts the conformational equilibrium of the kinase in favor of an active (DFG-in) A-loop conformation, which has more optimal binding interactions with axitinib.
“If you think of the targeted protein as a lock into which the cancer drug fits in as a key, the resistant protein changes in such a way that we need a different key,” said study author Brion Murray, PhD, of Pfizer Worldwide Research & Development in San Diego, California.
“In the case of axitinib, it acts as two distinct keys—one for renal cell carcinoma and one for leukemia.”
The researchers also treated a CML patient with axitinib and observed a “rapid reduction” of T315I-positive cells in the patient’s bone marrow.
“Further research will determine whether these findings have the potential to significantly improve the standard of care for this select group of CML patients and patients with other related leukemias,” Dr Murray concluded.