User login
Team creates public repository of xenografts

Photo by Rhoda Baer
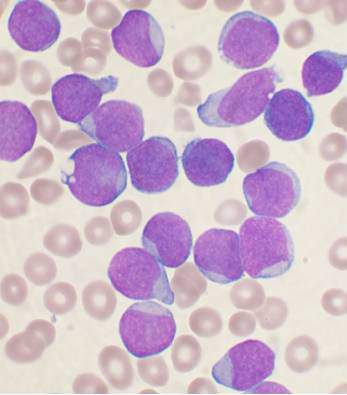
Researchers have established a public repository of leukemia and lymphoma xenografts, according to a report in Cancer Cell.
The repository, known as the Public Repository of Xenografts (PRoXe), contains material from bone marrow, peripheral blood, and lymph nodes of mice.
It also contains information on the patients from whom the cancer tissues were derived and details on the characteristics of the tumors themselves.
PRoXe is based at Dana-Farber Cancer Institute in Boston, Massachusetts, but it has a web portal that can be accessed by researchers around the world.
Those who register with PRoxE can access the repository and search for cells from patients with specific hematologic malignancies.
Researchers can then have frozen cells shipped to them and transplant the cells into mice to create patient-derived xenograft (PDX) models for testing drugs.
“About 90% of compounds that show anticancer activity in preclinical tests don’t work when given to patients,” said David Weinstock, MD, of the Dana-Farber Cancer Institute.
“By trying drugs in PDX models, we can ‘mimic’ large and expensive human clinical trials and get answers about efficacy more quickly, less expensively, and without the need for patients to get investigational drugs that won’t work.”
To demonstrate how PRoXe can be used, Dr Weinstock and his colleagues tested the MDM2 inhibitor CGM097 against B-cell acute lymphoblastic leukemia (B-ALL) in 2 groups of mice. One group had a mutation in the TP53 tumor suppressor gene, and the other did not.
The researchers observed superior survival in the mice with wild-type TP53. This result corresponds with the results of previous research, which showed that inhibitors that disrupt the MDM2-p53 interaction can be effective in tumor models and patient tumors with wild-type TP53.
Dr Weinstock and his colleagues said their results provide strong preclinical evidence for testing CGM097 in patients who have been extensively treated for B-ALL and have wild-type TP53.
Dr Weinstock also noted that the leaders of ProXe are negotiating with a number of academic centers in an attempt to incorporate their PDX models into the repository.
The Cancer Cell manuscript had 95 authors from 14 different centers that contributed samples, models, and/or effort to the project. ![]()
Photo by Rhoda Baer
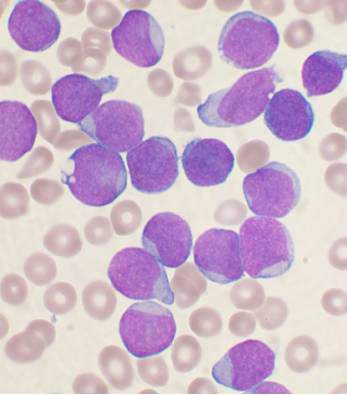
Researchers have established a public repository of leukemia and lymphoma xenografts, according to a report in Cancer Cell.
The repository, known as the Public Repository of Xenografts (PRoXe), contains material from bone marrow, peripheral blood, and lymph nodes of mice.
It also contains information on the patients from whom the cancer tissues were derived and details on the characteristics of the tumors themselves.
PRoXe is based at Dana-Farber Cancer Institute in Boston, Massachusetts, but it has a web portal that can be accessed by researchers around the world.
Those who register with PRoxE can access the repository and search for cells from patients with specific hematologic malignancies.
Researchers can then have frozen cells shipped to them and transplant the cells into mice to create patient-derived xenograft (PDX) models for testing drugs.
“About 90% of compounds that show anticancer activity in preclinical tests don’t work when given to patients,” said David Weinstock, MD, of the Dana-Farber Cancer Institute.
“By trying drugs in PDX models, we can ‘mimic’ large and expensive human clinical trials and get answers about efficacy more quickly, less expensively, and without the need for patients to get investigational drugs that won’t work.”
To demonstrate how PRoXe can be used, Dr Weinstock and his colleagues tested the MDM2 inhibitor CGM097 against B-cell acute lymphoblastic leukemia (B-ALL) in 2 groups of mice. One group had a mutation in the TP53 tumor suppressor gene, and the other did not.
The researchers observed superior survival in the mice with wild-type TP53. This result corresponds with the results of previous research, which showed that inhibitors that disrupt the MDM2-p53 interaction can be effective in tumor models and patient tumors with wild-type TP53.
Dr Weinstock and his colleagues said their results provide strong preclinical evidence for testing CGM097 in patients who have been extensively treated for B-ALL and have wild-type TP53.
Dr Weinstock also noted that the leaders of ProXe are negotiating with a number of academic centers in an attempt to incorporate their PDX models into the repository.
The Cancer Cell manuscript had 95 authors from 14 different centers that contributed samples, models, and/or effort to the project. ![]()
Photo by Rhoda Baer
Researchers have established a public repository of leukemia and lymphoma xenografts, according to a report in Cancer Cell.
The repository, known as the Public Repository of Xenografts (PRoXe), contains material from bone marrow, peripheral blood, and lymph nodes of mice.
It also contains information on the patients from whom the cancer tissues were derived and details on the characteristics of the tumors themselves.
PRoXe is based at Dana-Farber Cancer Institute in Boston, Massachusetts, but it has a web portal that can be accessed by researchers around the world.
Those who register with PRoxE can access the repository and search for cells from patients with specific hematologic malignancies.
Researchers can then have frozen cells shipped to them and transplant the cells into mice to create patient-derived xenograft (PDX) models for testing drugs.
“About 90% of compounds that show anticancer activity in preclinical tests don’t work when given to patients,” said David Weinstock, MD, of the Dana-Farber Cancer Institute.
“By trying drugs in PDX models, we can ‘mimic’ large and expensive human clinical trials and get answers about efficacy more quickly, less expensively, and without the need for patients to get investigational drugs that won’t work.”
To demonstrate how PRoXe can be used, Dr Weinstock and his colleagues tested the MDM2 inhibitor CGM097 against B-cell acute lymphoblastic leukemia (B-ALL) in 2 groups of mice. One group had a mutation in the TP53 tumor suppressor gene, and the other did not.
The researchers observed superior survival in the mice with wild-type TP53. This result corresponds with the results of previous research, which showed that inhibitors that disrupt the MDM2-p53 interaction can be effective in tumor models and patient tumors with wild-type TP53.
Dr Weinstock and his colleagues said their results provide strong preclinical evidence for testing CGM097 in patients who have been extensively treated for B-ALL and have wild-type TP53.
Dr Weinstock also noted that the leaders of ProXe are negotiating with a number of academic centers in an attempt to incorporate their PDX models into the repository.
The Cancer Cell manuscript had 95 authors from 14 different centers that contributed samples, models, and/or effort to the project. ![]()
Combo appears effective against B-ALL

Combining a MEK inhibitor and a BCL-2/BCL-XL inhibitor may be a feasible treatment option for B-cell acute lymphoblastic leukemia (B-ALL), according to preclinical research published in Cell Death and Disease.
Researchers found that, when given alone, the MEK inhibitor trametinib did not block B-ALL cell growth.
And the BCL-2/BCL-XL inhibitors navitoclax (ABT-263) and venetoclax (ABT-199) did not prove particularly effective either.
However, combining trametinib with navitoclax or venetoclax successfully induced apoptosis in B-ALL cells.
“Cancer cells often outwit us by rewiring themselves, but this early research offers a promising idea to get ahead of them,” said study author Richard Marais, PhD, of the Cancer Research UK Manchester Institute.
“We’ll still need to do further research to prove that this is the case beyond cancer cells in the laboratory, and it may take many years before we see it in the clinic, but it’s the first step to finding a new, effective drug combination for B-cell acute lymphoblastic leukemia.”
Dr Marais and his colleagues found that, although the MEK/ERK pathway is activated in B-ALL cells driven by different oncogenes, MEK inhibition alone did not suppress B-ALL cell growth.
And although B-ALL cells were sensitive to treatment with navitoclax or venetoclax alone, the researchers did not see complete loss of cell viability at clinically achievable doses.
However, trametinib did synergize with either navitoclax or venetoclax to suppress proliferation and induce apoptosis in B-ALL cells.
Further investigation revealed that the resistance of B-ALL cells to BCL-2/BCL-XL inhibition is mediated by MCL-1. And the synergism between trametinib and navitoclax/venetoclax is mediated by the pro-apoptotic factor BIM.
BIM is dephosphorylated as a result of MEK inhibition, which allows it to bind to and neutralize MCL-1, thereby enhancing BCL-2/BCL-XL inhibitor-induced cell death.
The researchers said they observed this effect in B-ALL cells driven by a range of genetic abnormalities, so the combination of a MEK inhibitor and a BCL-2/BCL-XL inhibitor could have therapeutic potential in a range of B-ALL subtypes. ![]()
Combining a MEK inhibitor and a BCL-2/BCL-XL inhibitor may be a feasible treatment option for B-cell acute lymphoblastic leukemia (B-ALL), according to preclinical research published in Cell Death and Disease.
Researchers found that, when given alone, the MEK inhibitor trametinib did not block B-ALL cell growth.
And the BCL-2/BCL-XL inhibitors navitoclax (ABT-263) and venetoclax (ABT-199) did not prove particularly effective either.
However, combining trametinib with navitoclax or venetoclax successfully induced apoptosis in B-ALL cells.
“Cancer cells often outwit us by rewiring themselves, but this early research offers a promising idea to get ahead of them,” said study author Richard Marais, PhD, of the Cancer Research UK Manchester Institute.
“We’ll still need to do further research to prove that this is the case beyond cancer cells in the laboratory, and it may take many years before we see it in the clinic, but it’s the first step to finding a new, effective drug combination for B-cell acute lymphoblastic leukemia.”
Dr Marais and his colleagues found that, although the MEK/ERK pathway is activated in B-ALL cells driven by different oncogenes, MEK inhibition alone did not suppress B-ALL cell growth.
And although B-ALL cells were sensitive to treatment with navitoclax or venetoclax alone, the researchers did not see complete loss of cell viability at clinically achievable doses.
However, trametinib did synergize with either navitoclax or venetoclax to suppress proliferation and induce apoptosis in B-ALL cells.
Further investigation revealed that the resistance of B-ALL cells to BCL-2/BCL-XL inhibition is mediated by MCL-1. And the synergism between trametinib and navitoclax/venetoclax is mediated by the pro-apoptotic factor BIM.
BIM is dephosphorylated as a result of MEK inhibition, which allows it to bind to and neutralize MCL-1, thereby enhancing BCL-2/BCL-XL inhibitor-induced cell death.
The researchers said they observed this effect in B-ALL cells driven by a range of genetic abnormalities, so the combination of a MEK inhibitor and a BCL-2/BCL-XL inhibitor could have therapeutic potential in a range of B-ALL subtypes. ![]()
Combining a MEK inhibitor and a BCL-2/BCL-XL inhibitor may be a feasible treatment option for B-cell acute lymphoblastic leukemia (B-ALL), according to preclinical research published in Cell Death and Disease.
Researchers found that, when given alone, the MEK inhibitor trametinib did not block B-ALL cell growth.
And the BCL-2/BCL-XL inhibitors navitoclax (ABT-263) and venetoclax (ABT-199) did not prove particularly effective either.
However, combining trametinib with navitoclax or venetoclax successfully induced apoptosis in B-ALL cells.
“Cancer cells often outwit us by rewiring themselves, but this early research offers a promising idea to get ahead of them,” said study author Richard Marais, PhD, of the Cancer Research UK Manchester Institute.
“We’ll still need to do further research to prove that this is the case beyond cancer cells in the laboratory, and it may take many years before we see it in the clinic, but it’s the first step to finding a new, effective drug combination for B-cell acute lymphoblastic leukemia.”
Dr Marais and his colleagues found that, although the MEK/ERK pathway is activated in B-ALL cells driven by different oncogenes, MEK inhibition alone did not suppress B-ALL cell growth.
And although B-ALL cells were sensitive to treatment with navitoclax or venetoclax alone, the researchers did not see complete loss of cell viability at clinically achievable doses.
However, trametinib did synergize with either navitoclax or venetoclax to suppress proliferation and induce apoptosis in B-ALL cells.
Further investigation revealed that the resistance of B-ALL cells to BCL-2/BCL-XL inhibition is mediated by MCL-1. And the synergism between trametinib and navitoclax/venetoclax is mediated by the pro-apoptotic factor BIM.
BIM is dephosphorylated as a result of MEK inhibition, which allows it to bind to and neutralize MCL-1, thereby enhancing BCL-2/BCL-XL inhibitor-induced cell death.
The researchers said they observed this effect in B-ALL cells driven by a range of genetic abnormalities, so the combination of a MEK inhibitor and a BCL-2/BCL-XL inhibitor could have therapeutic potential in a range of B-ALL subtypes. ![]()
Inhibitor could overcome TKI resistance in Ph+ B-ALL

Results of preclinical research indicate that combining 2 kinase inhibitors may be a promising treatment strategy for Philadelphia chromosome-positive B-cell acute lymphoblastic leukemia (Ph+ B-ALL).
Researchers found that combining a tyrosine kinase inhibitor (TKI) and an inhibitor of focal adhesion kinase (FAK) was “remarkably effective” against Ph+ B-ALL in vitro and in vivo.
The TKI dasatinib and the FAK inhibitor VS-4718 decreased leukemic cell survival and adhesion, inhibited tumor growth, and prolonged survival in mouse models of Ph+ B-ALL.
Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and reported their results in JCI Insight.
The researchers noted that patients with Ph+ ALL have shown resistance to TKI therapy, and this resistance has been tied to alterations in IKZF1.
As FAK expression is elevated in IKZF1-mutated leukemias, the team speculated that adding a FAK inhibitor to TKI therapy might lead to better results.
First, the researchers set out to confirm that FAK is overexpressed in Ph+ B-ALL. Their experiments revealed upregulation of the FAK pathway in Ph+ B-ALL cells, with further overexpression of FAK in IKZF1-mutated Ph+ B-ALL cells.
When they inhibited FAK with VS-4718, the team observed decreases in the survival, clonogenicity, and adhesion of IKZF1-mutated Ph+ B-ALL cells from both mice and humans.
Next, the researchers found that VS-4718 synergizes with the TKI dasatinib in vitro and in vivo.
In in vitro experiments with both mouse and human Ph+ B-ALL cells, the combination decreased cell survival and adhesion and inhibited downstream targets of FAK.
In mouse models of Ph+ B-ALL, VS-4718 proved ineffective when given alone.
However, the researchers said the combination of VS-4718 and dasatinib “dramatically” decreased leukemic burden and extended the lives of mice.
In fact, 1 long-term survivor achieved a complete remission that endured after treatment was stopped.
The researchers said these results suggest that targeting FAK with VS-4718 can overcome the deleterious effects of FAK overexpression in Ph+ B-ALL, potentiating responsiveness to TKIs. ![]()
Results of preclinical research indicate that combining 2 kinase inhibitors may be a promising treatment strategy for Philadelphia chromosome-positive B-cell acute lymphoblastic leukemia (Ph+ B-ALL).
Researchers found that combining a tyrosine kinase inhibitor (TKI) and an inhibitor of focal adhesion kinase (FAK) was “remarkably effective” against Ph+ B-ALL in vitro and in vivo.
The TKI dasatinib and the FAK inhibitor VS-4718 decreased leukemic cell survival and adhesion, inhibited tumor growth, and prolonged survival in mouse models of Ph+ B-ALL.
Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and reported their results in JCI Insight.
The researchers noted that patients with Ph+ ALL have shown resistance to TKI therapy, and this resistance has been tied to alterations in IKZF1.
As FAK expression is elevated in IKZF1-mutated leukemias, the team speculated that adding a FAK inhibitor to TKI therapy might lead to better results.
First, the researchers set out to confirm that FAK is overexpressed in Ph+ B-ALL. Their experiments revealed upregulation of the FAK pathway in Ph+ B-ALL cells, with further overexpression of FAK in IKZF1-mutated Ph+ B-ALL cells.
When they inhibited FAK with VS-4718, the team observed decreases in the survival, clonogenicity, and adhesion of IKZF1-mutated Ph+ B-ALL cells from both mice and humans.
Next, the researchers found that VS-4718 synergizes with the TKI dasatinib in vitro and in vivo.
In in vitro experiments with both mouse and human Ph+ B-ALL cells, the combination decreased cell survival and adhesion and inhibited downstream targets of FAK.
In mouse models of Ph+ B-ALL, VS-4718 proved ineffective when given alone.
However, the researchers said the combination of VS-4718 and dasatinib “dramatically” decreased leukemic burden and extended the lives of mice.
In fact, 1 long-term survivor achieved a complete remission that endured after treatment was stopped.
The researchers said these results suggest that targeting FAK with VS-4718 can overcome the deleterious effects of FAK overexpression in Ph+ B-ALL, potentiating responsiveness to TKIs. ![]()
Results of preclinical research indicate that combining 2 kinase inhibitors may be a promising treatment strategy for Philadelphia chromosome-positive B-cell acute lymphoblastic leukemia (Ph+ B-ALL).
Researchers found that combining a tyrosine kinase inhibitor (TKI) and an inhibitor of focal adhesion kinase (FAK) was “remarkably effective” against Ph+ B-ALL in vitro and in vivo.
The TKI dasatinib and the FAK inhibitor VS-4718 decreased leukemic cell survival and adhesion, inhibited tumor growth, and prolonged survival in mouse models of Ph+ B-ALL.
Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee, and his colleagues conducted this research and reported their results in JCI Insight.
The researchers noted that patients with Ph+ ALL have shown resistance to TKI therapy, and this resistance has been tied to alterations in IKZF1.
As FAK expression is elevated in IKZF1-mutated leukemias, the team speculated that adding a FAK inhibitor to TKI therapy might lead to better results.
First, the researchers set out to confirm that FAK is overexpressed in Ph+ B-ALL. Their experiments revealed upregulation of the FAK pathway in Ph+ B-ALL cells, with further overexpression of FAK in IKZF1-mutated Ph+ B-ALL cells.
When they inhibited FAK with VS-4718, the team observed decreases in the survival, clonogenicity, and adhesion of IKZF1-mutated Ph+ B-ALL cells from both mice and humans.
Next, the researchers found that VS-4718 synergizes with the TKI dasatinib in vitro and in vivo.
In in vitro experiments with both mouse and human Ph+ B-ALL cells, the combination decreased cell survival and adhesion and inhibited downstream targets of FAK.
In mouse models of Ph+ B-ALL, VS-4718 proved ineffective when given alone.
However, the researchers said the combination of VS-4718 and dasatinib “dramatically” decreased leukemic burden and extended the lives of mice.
In fact, 1 long-term survivor achieved a complete remission that endured after treatment was stopped.
The researchers said these results suggest that targeting FAK with VS-4718 can overcome the deleterious effects of FAK overexpression in Ph+ B-ALL, potentiating responsiveness to TKIs. ![]()
Protein plays key role in B-ALL subtype
An RNA binding protein promotes the development of MLL-rearranged B-cell acute lymphoblastic leukemia (B-ALL), according to research published in The Journal of Clinical Investigation.
Researchers found an overabundance of the protein, IGF2BP3, in MLL-rearranged B-ALL.
They also identified genes that are directly regulated by IFG2BP3, and many of them turned out to be oncogenes that have already been implicated in cancers.
The overall effect of IFG2BP3 in MLL-rearranged B-ALL is to promote the proliferation of B cells by shifting the expression of a large number of genes, explained study author Jeremy Sanford, PhD, of the University of California Santa Cruz.
“This protein, IFG2BP3, has been correlated with many types of malignancies and with the worst prognoses,” he noted. “What is exciting about this study is that it goes beyond correlation and shows causation, because we demonstrated, for the first time, that aberrant expression of this protein is sufficient to induce pathology.”
This research began in the lab of Dinesh Rao, PhD, an assistant professor at the University of California Los Angeles who was studying MLL-rearranged B-ALL.
After researchers in Dr Rao’s lab identified IGF2BP3 as one of the top dysregulated genes in this malignancy, they began working with Dr Sanford’s lab to figure out which genes were being directly regulated by IGF2BP3.
Dr Sanford’s lab was among the few using individual nucleotide resolution crosslinking immunoprecipitation (iCLIP), a technique that can capture RNA molecules bound to a particular protein.
iCLIP enabled the researchers to identify IGF2BP3 binding sites in several hundred RNA transcripts in 2 B-ALL cell lines.
The work also revealed that IGF2BP3 enhanced the expression of MYC and other oncogenes in hematopoietic stem cells.
In experiments with mice, the researchers found that overexpression of IGF2BP3 in the bone marrow leads to proliferation of hematopoietic stem cells and B-cell progenitors, reproducing some features of MLL-rearranged B-ALL.
“Understanding its mechanism of action is important for thinking about therapeutics that might interfere with the action of this protein in disease,” Dr Sanford said.
“One possibility is an RNA-based therapeutic that could sequester the protein and keep it from binding to RNA transcripts. That would be a way to influence the expression of many genes involved in the proliferation of cancer cells.” ![]()
An RNA binding protein promotes the development of MLL-rearranged B-cell acute lymphoblastic leukemia (B-ALL), according to research published in The Journal of Clinical Investigation.
Researchers found an overabundance of the protein, IGF2BP3, in MLL-rearranged B-ALL.
They also identified genes that are directly regulated by IFG2BP3, and many of them turned out to be oncogenes that have already been implicated in cancers.
The overall effect of IFG2BP3 in MLL-rearranged B-ALL is to promote the proliferation of B cells by shifting the expression of a large number of genes, explained study author Jeremy Sanford, PhD, of the University of California Santa Cruz.
“This protein, IFG2BP3, has been correlated with many types of malignancies and with the worst prognoses,” he noted. “What is exciting about this study is that it goes beyond correlation and shows causation, because we demonstrated, for the first time, that aberrant expression of this protein is sufficient to induce pathology.”
This research began in the lab of Dinesh Rao, PhD, an assistant professor at the University of California Los Angeles who was studying MLL-rearranged B-ALL.
After researchers in Dr Rao’s lab identified IGF2BP3 as one of the top dysregulated genes in this malignancy, they began working with Dr Sanford’s lab to figure out which genes were being directly regulated by IGF2BP3.
Dr Sanford’s lab was among the few using individual nucleotide resolution crosslinking immunoprecipitation (iCLIP), a technique that can capture RNA molecules bound to a particular protein.
iCLIP enabled the researchers to identify IGF2BP3 binding sites in several hundred RNA transcripts in 2 B-ALL cell lines.
The work also revealed that IGF2BP3 enhanced the expression of MYC and other oncogenes in hematopoietic stem cells.
In experiments with mice, the researchers found that overexpression of IGF2BP3 in the bone marrow leads to proliferation of hematopoietic stem cells and B-cell progenitors, reproducing some features of MLL-rearranged B-ALL.
“Understanding its mechanism of action is important for thinking about therapeutics that might interfere with the action of this protein in disease,” Dr Sanford said.
“One possibility is an RNA-based therapeutic that could sequester the protein and keep it from binding to RNA transcripts. That would be a way to influence the expression of many genes involved in the proliferation of cancer cells.” ![]()
An RNA binding protein promotes the development of MLL-rearranged B-cell acute lymphoblastic leukemia (B-ALL), according to research published in The Journal of Clinical Investigation.
Researchers found an overabundance of the protein, IGF2BP3, in MLL-rearranged B-ALL.
They also identified genes that are directly regulated by IFG2BP3, and many of them turned out to be oncogenes that have already been implicated in cancers.
The overall effect of IFG2BP3 in MLL-rearranged B-ALL is to promote the proliferation of B cells by shifting the expression of a large number of genes, explained study author Jeremy Sanford, PhD, of the University of California Santa Cruz.
“This protein, IFG2BP3, has been correlated with many types of malignancies and with the worst prognoses,” he noted. “What is exciting about this study is that it goes beyond correlation and shows causation, because we demonstrated, for the first time, that aberrant expression of this protein is sufficient to induce pathology.”
This research began in the lab of Dinesh Rao, PhD, an assistant professor at the University of California Los Angeles who was studying MLL-rearranged B-ALL.
After researchers in Dr Rao’s lab identified IGF2BP3 as one of the top dysregulated genes in this malignancy, they began working with Dr Sanford’s lab to figure out which genes were being directly regulated by IGF2BP3.
Dr Sanford’s lab was among the few using individual nucleotide resolution crosslinking immunoprecipitation (iCLIP), a technique that can capture RNA molecules bound to a particular protein.
iCLIP enabled the researchers to identify IGF2BP3 binding sites in several hundred RNA transcripts in 2 B-ALL cell lines.
The work also revealed that IGF2BP3 enhanced the expression of MYC and other oncogenes in hematopoietic stem cells.
In experiments with mice, the researchers found that overexpression of IGF2BP3 in the bone marrow leads to proliferation of hematopoietic stem cells and B-cell progenitors, reproducing some features of MLL-rearranged B-ALL.
“Understanding its mechanism of action is important for thinking about therapeutics that might interfere with the action of this protein in disease,” Dr Sanford said.
“One possibility is an RNA-based therapeutic that could sequester the protein and keep it from binding to RNA transcripts. That would be a way to influence the expression of many genes involved in the proliferation of cancer cells.” ![]()
Guidelines on asparaginase hypersensitivity and silent inactivation in ALL
Recommendations for identification and management of asparaginase hypersensitivity and silent inactivation have been issued in consensus expert guidelines written by Dr. Inge M. van der Sluis of Sophia Children’s Hospital, Rotterdam, The Netherlands, and her colleagues.
The guidelines were published in Haematologica, the Journal of the European Hematology Association (Haematologica 2016 March;101:279-85).
Hypersensitivity reactions to asparaginase therapy can occur in 30% of children with acute lymphoblastic leukemia (ALL), and “silent inactivation” with the formation of neutralizing antibodies and reduced asparaginase activity can occur in the absence of a clinically evident allergic reaction. The guidelines seek to reduce the risk that patients will receive an inadequate course of asparaginase therapy due to either intolerable side effects or silent inactivation. The recommendations address therapeutic drug monitoring through serum asparaginase level assessment, indications for switching asparaginase preparations, and recommendations for monitoring after changes in asparaginase preparation.
The guidelines conclude that measures of serum asparaginase activity levels are the best and most reliable indicators of asparaginase efficacy. Trough asparaginase activity levels of at least 0.1 IU/mL appear to be a safe target level to ensure therapeutic benefit. Anti-asparaginase antibodies and asparagine measurements are not indicated for clinical decision making outside the context of a clinical trial.
Should clinical hypersensitivity occur, the patient has likely developed anti-asparaginase antibodies. Continued use of asparaginase of the same formulation will be ineffective in the treatment of leukemia and should be discouraged, even when it is clinically possible to administer the same preparation by using premedication such as steroids and antihistamines or by decreasing the infusion rate. Such measures reduce the symptoms of the allergy, but do not prevent asparaginase inactivation.
In patients with grade 1 allergic reactions (transient flushing or rash without need for intervention), the guideline writers recommend monitoring serum asparaginase level in real-time within 7 days to identify inactivation. In those with grade 2-4 reactions, the guideline writers recommend switching the asparaginase preparation, and there is no definite need to check asparaginase levels.
Screening for silent inactivation should be considered in all patients undergoing therapy for ALL with asparaginase, especially when there have been gaps in asparaginase therapy or in the setting of the treatment of relapsed leukemia.
“We recommend the testing of serum asparaginase activity level after the first dose of E. coli–derived asparaginase. With the use of pegaspargase, this should be done within 7 days of the dose. If the level is detectable but less than 0.1 IU/mL, activity should be rechecked at day 14,” according to the authors.
In patients who receive pegylated asparaginase every 14 days without a gap in dosing, it would be reasonable to confirm a low or undetectable level after a subsequent dose, and to change to an Erwinia preparation, for example, if two consecutive levels are undetectable.
“When there is a gap between asparaginase doses, we recommend checking a level after the first dose of asparaginase administered after the gap, with a gap defined as a period in which asparaginase activity level will have decreased to less than the [lower limit of quantification] between doses. In practice, this is usually the case when there is an interval of at least 4 weeks between pegylated asparaginase doses. With native E. coli asparaginase, we recommend measuring a trough level after the first dose and after every reintroduction of asparaginase,” the authors wrote.
When a limited number of asparaginase doses is planned or there are prolonged gaps between doses, the guidelines advise screening for silent inactivation after every asparaginase dose, and considering a switching in preparation based upon a single undetectable or low level.
The guidelines also recommend more insight into the costs of treatment with the various asparaginase preparations. In addition to the detection of silent inactivation, therapeutic drug monitoring can be used to individualize dosing based on activity levels and has the potential to reduce medication costs.
Several of the consensus writers have participated in advisory boards of companies involved in asparaginase, including Sigma-Tau, Medac, and Jazz Pharmaceuticals. Jazz Pharmaceuticals provided financial support for the investigator-initiated consensus meeting.
On Twitter @maryjodales
Recommendations for identification and management of asparaginase hypersensitivity and silent inactivation have been issued in consensus expert guidelines written by Dr. Inge M. van der Sluis of Sophia Children’s Hospital, Rotterdam, The Netherlands, and her colleagues.
The guidelines were published in Haematologica, the Journal of the European Hematology Association (Haematologica 2016 March;101:279-85).
Hypersensitivity reactions to asparaginase therapy can occur in 30% of children with acute lymphoblastic leukemia (ALL), and “silent inactivation” with the formation of neutralizing antibodies and reduced asparaginase activity can occur in the absence of a clinically evident allergic reaction. The guidelines seek to reduce the risk that patients will receive an inadequate course of asparaginase therapy due to either intolerable side effects or silent inactivation. The recommendations address therapeutic drug monitoring through serum asparaginase level assessment, indications for switching asparaginase preparations, and recommendations for monitoring after changes in asparaginase preparation.
The guidelines conclude that measures of serum asparaginase activity levels are the best and most reliable indicators of asparaginase efficacy. Trough asparaginase activity levels of at least 0.1 IU/mL appear to be a safe target level to ensure therapeutic benefit. Anti-asparaginase antibodies and asparagine measurements are not indicated for clinical decision making outside the context of a clinical trial.
Should clinical hypersensitivity occur, the patient has likely developed anti-asparaginase antibodies. Continued use of asparaginase of the same formulation will be ineffective in the treatment of leukemia and should be discouraged, even when it is clinically possible to administer the same preparation by using premedication such as steroids and antihistamines or by decreasing the infusion rate. Such measures reduce the symptoms of the allergy, but do not prevent asparaginase inactivation.
In patients with grade 1 allergic reactions (transient flushing or rash without need for intervention), the guideline writers recommend monitoring serum asparaginase level in real-time within 7 days to identify inactivation. In those with grade 2-4 reactions, the guideline writers recommend switching the asparaginase preparation, and there is no definite need to check asparaginase levels.
Screening for silent inactivation should be considered in all patients undergoing therapy for ALL with asparaginase, especially when there have been gaps in asparaginase therapy or in the setting of the treatment of relapsed leukemia.
“We recommend the testing of serum asparaginase activity level after the first dose of E. coli–derived asparaginase. With the use of pegaspargase, this should be done within 7 days of the dose. If the level is detectable but less than 0.1 IU/mL, activity should be rechecked at day 14,” according to the authors.
In patients who receive pegylated asparaginase every 14 days without a gap in dosing, it would be reasonable to confirm a low or undetectable level after a subsequent dose, and to change to an Erwinia preparation, for example, if two consecutive levels are undetectable.
“When there is a gap between asparaginase doses, we recommend checking a level after the first dose of asparaginase administered after the gap, with a gap defined as a period in which asparaginase activity level will have decreased to less than the [lower limit of quantification] between doses. In practice, this is usually the case when there is an interval of at least 4 weeks between pegylated asparaginase doses. With native E. coli asparaginase, we recommend measuring a trough level after the first dose and after every reintroduction of asparaginase,” the authors wrote.
When a limited number of asparaginase doses is planned or there are prolonged gaps between doses, the guidelines advise screening for silent inactivation after every asparaginase dose, and considering a switching in preparation based upon a single undetectable or low level.
The guidelines also recommend more insight into the costs of treatment with the various asparaginase preparations. In addition to the detection of silent inactivation, therapeutic drug monitoring can be used to individualize dosing based on activity levels and has the potential to reduce medication costs.
Several of the consensus writers have participated in advisory boards of companies involved in asparaginase, including Sigma-Tau, Medac, and Jazz Pharmaceuticals. Jazz Pharmaceuticals provided financial support for the investigator-initiated consensus meeting.
On Twitter @maryjodales
Recommendations for identification and management of asparaginase hypersensitivity and silent inactivation have been issued in consensus expert guidelines written by Dr. Inge M. van der Sluis of Sophia Children’s Hospital, Rotterdam, The Netherlands, and her colleagues.
The guidelines were published in Haematologica, the Journal of the European Hematology Association (Haematologica 2016 March;101:279-85).
Hypersensitivity reactions to asparaginase therapy can occur in 30% of children with acute lymphoblastic leukemia (ALL), and “silent inactivation” with the formation of neutralizing antibodies and reduced asparaginase activity can occur in the absence of a clinically evident allergic reaction. The guidelines seek to reduce the risk that patients will receive an inadequate course of asparaginase therapy due to either intolerable side effects or silent inactivation. The recommendations address therapeutic drug monitoring through serum asparaginase level assessment, indications for switching asparaginase preparations, and recommendations for monitoring after changes in asparaginase preparation.
The guidelines conclude that measures of serum asparaginase activity levels are the best and most reliable indicators of asparaginase efficacy. Trough asparaginase activity levels of at least 0.1 IU/mL appear to be a safe target level to ensure therapeutic benefit. Anti-asparaginase antibodies and asparagine measurements are not indicated for clinical decision making outside the context of a clinical trial.
Should clinical hypersensitivity occur, the patient has likely developed anti-asparaginase antibodies. Continued use of asparaginase of the same formulation will be ineffective in the treatment of leukemia and should be discouraged, even when it is clinically possible to administer the same preparation by using premedication such as steroids and antihistamines or by decreasing the infusion rate. Such measures reduce the symptoms of the allergy, but do not prevent asparaginase inactivation.
In patients with grade 1 allergic reactions (transient flushing or rash without need for intervention), the guideline writers recommend monitoring serum asparaginase level in real-time within 7 days to identify inactivation. In those with grade 2-4 reactions, the guideline writers recommend switching the asparaginase preparation, and there is no definite need to check asparaginase levels.
Screening for silent inactivation should be considered in all patients undergoing therapy for ALL with asparaginase, especially when there have been gaps in asparaginase therapy or in the setting of the treatment of relapsed leukemia.
“We recommend the testing of serum asparaginase activity level after the first dose of E. coli–derived asparaginase. With the use of pegaspargase, this should be done within 7 days of the dose. If the level is detectable but less than 0.1 IU/mL, activity should be rechecked at day 14,” according to the authors.
In patients who receive pegylated asparaginase every 14 days without a gap in dosing, it would be reasonable to confirm a low or undetectable level after a subsequent dose, and to change to an Erwinia preparation, for example, if two consecutive levels are undetectable.
“When there is a gap between asparaginase doses, we recommend checking a level after the first dose of asparaginase administered after the gap, with a gap defined as a period in which asparaginase activity level will have decreased to less than the [lower limit of quantification] between doses. In practice, this is usually the case when there is an interval of at least 4 weeks between pegylated asparaginase doses. With native E. coli asparaginase, we recommend measuring a trough level after the first dose and after every reintroduction of asparaginase,” the authors wrote.
When a limited number of asparaginase doses is planned or there are prolonged gaps between doses, the guidelines advise screening for silent inactivation after every asparaginase dose, and considering a switching in preparation based upon a single undetectable or low level.
The guidelines also recommend more insight into the costs of treatment with the various asparaginase preparations. In addition to the detection of silent inactivation, therapeutic drug monitoring can be used to individualize dosing based on activity levels and has the potential to reduce medication costs.
Several of the consensus writers have participated in advisory boards of companies involved in asparaginase, including Sigma-Tau, Medac, and Jazz Pharmaceuticals. Jazz Pharmaceuticals provided financial support for the investigator-initiated consensus meeting.
On Twitter @maryjodales
FROM HAEMATOLOGICA
Study elucidates MYC’s role in T-ALL

Image by Juha Klefstrom
Research has revealed a relationship between the oncogene MYC and 2 cell-surface proteins that protect cancer cells from the immune system—CD47 and PD-L1.
Researchers discovered that MYC regulates the expression of CD47 and PD-L1 in T-cell acute lymphoblastic leukemia (T-ALL) and several solid tumor malignancies.
The team said this study is the first to link 2 critical steps in cancer development—uncontrolled cell growth (courtesy of mutated or misregulated MYC) and an ability to “outsmart” the immune molecules meant to stop it (via CD47 and PD-L1).
The study was published in Science.
“Our findings describe an intimate, causal connection between how oncogenes like MYC cause cancer and how those cancer cells manage to evade the immune system,” said study author Dean Felsher, MD, PhD, of the Stanford University School of Medicine in California.
Researchers in Dr Felsher’s lab have been studying MYC for more than a decade, focusing on oncogene addiction, in which tumor cells are completely dependent on the expression of the oncogene. Blocking the expression of MYC in these cases causes the complete regression of tumors in animals.
In 2010, Dr Felsher and his colleagues showed this regression could only occur in animals with an intact immune system, but it wasn’t clear why.
“Since then, I’ve had it in the back of my mind that there must be a relationship between MYC and the immune system,” Dr Felsher said.
So he and his colleagues decided to see if there was a link between MYC expression and the levels of CD47 and PD-L1 proteins on the surface of cancer cells. They investigated what would happen if they actively turned off MYC expression in tumor cells from mice or humans.
The researchers found that a reduction in MYC caused a similar reduction in the levels of CD47 and PD-L1 proteins on the surface of mouse and human T-ALL cells, mouse and human liver cancer cells, human skin cancer cells, and human non-small-cell lung cancer cells.
In contrast, levels of other immune regulatory molecules found on the surface of the cells were unaffected.
In gene expression data on tumor samples from hundreds of patients, the researchers found that levels of MYC expression correlated strongly with expression levels of CD47 and PD-L1 genes in liver, kidney, and colorectal tumors.
The team then looked directly at the regulatory regions in the CD47 and PD-L1 genes. They found high levels of the MYC protein bound directly to the promoter regions of CD47 and PD-L1 in mouse T-ALL cells and in a human osteosarcoma cell line.
The researchers were also able to verify that this binding increased the expression of CD47 in a human B cell line.
Finally, the team engineered mouse T-ALL cells to constantly express CD47 or PD-L1 regardless of MYC expression status.
These cells were better able than control cells to evade the detection of immune cells like macrophages and T cells. And, unlike in previous experiments, tumors arising from these cells did not regress when MYC expression was deactivated.
“What we’re learning is that if CD47 and PD-L1 are present on the surfaces of cancer cells, even if you shut down a cancer gene, the animal doesn’t mount an adequate immune response, and the tumors don’t regress,” Dr Felsher said.
Therefore, this work suggests a combination of therapies targeting the expression of both MYC and CD47 or PD-L1 could possibly have a synergistic effect by slowing or stopping tumor growth and waving a red flag at the immune system.
“There is a growing sense of tremendous excitement in the field of cancer immunotherapy,” Dr Felsher said. “In many cases, it’s working, but it’s not been clear why some cancers are more sensitive than others. Our work highlights a direct link between oncogene expression and immune regulation that could be exploited to help patients.” ![]()
Image by Juha Klefstrom
Research has revealed a relationship between the oncogene MYC and 2 cell-surface proteins that protect cancer cells from the immune system—CD47 and PD-L1.
Researchers discovered that MYC regulates the expression of CD47 and PD-L1 in T-cell acute lymphoblastic leukemia (T-ALL) and several solid tumor malignancies.
The team said this study is the first to link 2 critical steps in cancer development—uncontrolled cell growth (courtesy of mutated or misregulated MYC) and an ability to “outsmart” the immune molecules meant to stop it (via CD47 and PD-L1).
The study was published in Science.
“Our findings describe an intimate, causal connection between how oncogenes like MYC cause cancer and how those cancer cells manage to evade the immune system,” said study author Dean Felsher, MD, PhD, of the Stanford University School of Medicine in California.
Researchers in Dr Felsher’s lab have been studying MYC for more than a decade, focusing on oncogene addiction, in which tumor cells are completely dependent on the expression of the oncogene. Blocking the expression of MYC in these cases causes the complete regression of tumors in animals.
In 2010, Dr Felsher and his colleagues showed this regression could only occur in animals with an intact immune system, but it wasn’t clear why.
“Since then, I’ve had it in the back of my mind that there must be a relationship between MYC and the immune system,” Dr Felsher said.
So he and his colleagues decided to see if there was a link between MYC expression and the levels of CD47 and PD-L1 proteins on the surface of cancer cells. They investigated what would happen if they actively turned off MYC expression in tumor cells from mice or humans.
The researchers found that a reduction in MYC caused a similar reduction in the levels of CD47 and PD-L1 proteins on the surface of mouse and human T-ALL cells, mouse and human liver cancer cells, human skin cancer cells, and human non-small-cell lung cancer cells.
In contrast, levels of other immune regulatory molecules found on the surface of the cells were unaffected.
In gene expression data on tumor samples from hundreds of patients, the researchers found that levels of MYC expression correlated strongly with expression levels of CD47 and PD-L1 genes in liver, kidney, and colorectal tumors.
The team then looked directly at the regulatory regions in the CD47 and PD-L1 genes. They found high levels of the MYC protein bound directly to the promoter regions of CD47 and PD-L1 in mouse T-ALL cells and in a human osteosarcoma cell line.
The researchers were also able to verify that this binding increased the expression of CD47 in a human B cell line.
Finally, the team engineered mouse T-ALL cells to constantly express CD47 or PD-L1 regardless of MYC expression status.
These cells were better able than control cells to evade the detection of immune cells like macrophages and T cells. And, unlike in previous experiments, tumors arising from these cells did not regress when MYC expression was deactivated.
“What we’re learning is that if CD47 and PD-L1 are present on the surfaces of cancer cells, even if you shut down a cancer gene, the animal doesn’t mount an adequate immune response, and the tumors don’t regress,” Dr Felsher said.
Therefore, this work suggests a combination of therapies targeting the expression of both MYC and CD47 or PD-L1 could possibly have a synergistic effect by slowing or stopping tumor growth and waving a red flag at the immune system.
“There is a growing sense of tremendous excitement in the field of cancer immunotherapy,” Dr Felsher said. “In many cases, it’s working, but it’s not been clear why some cancers are more sensitive than others. Our work highlights a direct link between oncogene expression and immune regulation that could be exploited to help patients.” ![]()
Image by Juha Klefstrom
Research has revealed a relationship between the oncogene MYC and 2 cell-surface proteins that protect cancer cells from the immune system—CD47 and PD-L1.
Researchers discovered that MYC regulates the expression of CD47 and PD-L1 in T-cell acute lymphoblastic leukemia (T-ALL) and several solid tumor malignancies.
The team said this study is the first to link 2 critical steps in cancer development—uncontrolled cell growth (courtesy of mutated or misregulated MYC) and an ability to “outsmart” the immune molecules meant to stop it (via CD47 and PD-L1).
The study was published in Science.
“Our findings describe an intimate, causal connection between how oncogenes like MYC cause cancer and how those cancer cells manage to evade the immune system,” said study author Dean Felsher, MD, PhD, of the Stanford University School of Medicine in California.
Researchers in Dr Felsher’s lab have been studying MYC for more than a decade, focusing on oncogene addiction, in which tumor cells are completely dependent on the expression of the oncogene. Blocking the expression of MYC in these cases causes the complete regression of tumors in animals.
In 2010, Dr Felsher and his colleagues showed this regression could only occur in animals with an intact immune system, but it wasn’t clear why.
“Since then, I’ve had it in the back of my mind that there must be a relationship between MYC and the immune system,” Dr Felsher said.
So he and his colleagues decided to see if there was a link between MYC expression and the levels of CD47 and PD-L1 proteins on the surface of cancer cells. They investigated what would happen if they actively turned off MYC expression in tumor cells from mice or humans.
The researchers found that a reduction in MYC caused a similar reduction in the levels of CD47 and PD-L1 proteins on the surface of mouse and human T-ALL cells, mouse and human liver cancer cells, human skin cancer cells, and human non-small-cell lung cancer cells.
In contrast, levels of other immune regulatory molecules found on the surface of the cells were unaffected.
In gene expression data on tumor samples from hundreds of patients, the researchers found that levels of MYC expression correlated strongly with expression levels of CD47 and PD-L1 genes in liver, kidney, and colorectal tumors.
The team then looked directly at the regulatory regions in the CD47 and PD-L1 genes. They found high levels of the MYC protein bound directly to the promoter regions of CD47 and PD-L1 in mouse T-ALL cells and in a human osteosarcoma cell line.
The researchers were also able to verify that this binding increased the expression of CD47 in a human B cell line.
Finally, the team engineered mouse T-ALL cells to constantly express CD47 or PD-L1 regardless of MYC expression status.
These cells were better able than control cells to evade the detection of immune cells like macrophages and T cells. And, unlike in previous experiments, tumors arising from these cells did not regress when MYC expression was deactivated.
“What we’re learning is that if CD47 and PD-L1 are present on the surfaces of cancer cells, even if you shut down a cancer gene, the animal doesn’t mount an adequate immune response, and the tumors don’t regress,” Dr Felsher said.
Therefore, this work suggests a combination of therapies targeting the expression of both MYC and CD47 or PD-L1 could possibly have a synergistic effect by slowing or stopping tumor growth and waving a red flag at the immune system.
“There is a growing sense of tremendous excitement in the field of cancer immunotherapy,” Dr Felsher said. “In many cases, it’s working, but it’s not been clear why some cancers are more sensitive than others. Our work highlights a direct link between oncogene expression and immune regulation that could be exploited to help patients.” ![]()
Compound could treat T-ALL subtype

Photo courtesy of
The Ottawa Hospital
An experimental compound is “highly promising” as a potential treatment for a subtype of T-cell acute lymphoblastic leukemia (T-ALL), according to researchers.
The team found the histone demethylase UTX is important for the maintenance of TAL1-positive T-ALL.
Inhibiting UTX with a compound known as GSK-J4 proved toxic to TAL1-positive T-ALL cells in vitro and in vivo, but normal cells were spared.
The researchers reported these findings in Genes & Development.
“It’s very exciting because this is the first time anyone has found a potential personalized treatment for this aggressive disease,” said study author Marjorie Brand, PhD, of The Ottawa Hospital Research Institute in Ontario, Canada.
“Unlike current therapies, ours targets the offending gene without harming the rest of the body.”
Dr Brand and her colleagues decided the best way to find a better treatment for TAL1-positive T-ALL was to investigate exactly how it works at a molecular level. So they analyzed the TAL1 gene, which, in certain circumstances, can transform T-cell precursors into leukemic cells.
They found that TAL1 needs UTX to trigger leukemia production. This was surprising because UTX was previously described as a tumor suppressor in T-ALL.
The team said their work suggests UTX is actually a pro-oncogenic cofactor that is essential for leukemia maintenance in TAL1-positive—but not TAL1-negative—T-ALL.
The researchers found that targeting UTX with the H3K27 demethylase inhibitor GSK-J4 could completely halt the growth of TAL1-positive leukemia cells and induce apoptosis in these cells in vitro. But the compound did not have the same effect in TAL1-negative T-ALL.
The team also tested GSK-J4 in mouse models of T-ALL. After 3 weeks of treatment, there was a “dramatic decrease” in the percentage of leukemic blasts in the bone marrow of mice with TAL1-positive T-ALL. These mice also had a reduction in the infiltration of leukemic cells in the spleen.
However, mice with TAL1-negative T-ALL did not experience the same benefits.
The researchers said GSK-J4 appeared to be well-tolerated. None of the treated mice experienced weight loss or adverse effects in the intestine, spleen, liver, kidney, or hematopoietic system.
“While our study is a proof-of-concept, these promising results might one day lead to a similar targeted treatment for humans,” said study author Carmen Palii, PhD, also of The Ottawa Hospital Research Institute.
In the meantime, the researchers are conducting additional studies in mice, testing higher doses of GSK-J4 and evaluating long-term side effects of the compound. ![]()
Photo courtesy of
The Ottawa Hospital
An experimental compound is “highly promising” as a potential treatment for a subtype of T-cell acute lymphoblastic leukemia (T-ALL), according to researchers.
The team found the histone demethylase UTX is important for the maintenance of TAL1-positive T-ALL.
Inhibiting UTX with a compound known as GSK-J4 proved toxic to TAL1-positive T-ALL cells in vitro and in vivo, but normal cells were spared.
The researchers reported these findings in Genes & Development.
“It’s very exciting because this is the first time anyone has found a potential personalized treatment for this aggressive disease,” said study author Marjorie Brand, PhD, of The Ottawa Hospital Research Institute in Ontario, Canada.
“Unlike current therapies, ours targets the offending gene without harming the rest of the body.”
Dr Brand and her colleagues decided the best way to find a better treatment for TAL1-positive T-ALL was to investigate exactly how it works at a molecular level. So they analyzed the TAL1 gene, which, in certain circumstances, can transform T-cell precursors into leukemic cells.
They found that TAL1 needs UTX to trigger leukemia production. This was surprising because UTX was previously described as a tumor suppressor in T-ALL.
The team said their work suggests UTX is actually a pro-oncogenic cofactor that is essential for leukemia maintenance in TAL1-positive—but not TAL1-negative—T-ALL.
The researchers found that targeting UTX with the H3K27 demethylase inhibitor GSK-J4 could completely halt the growth of TAL1-positive leukemia cells and induce apoptosis in these cells in vitro. But the compound did not have the same effect in TAL1-negative T-ALL.
The team also tested GSK-J4 in mouse models of T-ALL. After 3 weeks of treatment, there was a “dramatic decrease” in the percentage of leukemic blasts in the bone marrow of mice with TAL1-positive T-ALL. These mice also had a reduction in the infiltration of leukemic cells in the spleen.
However, mice with TAL1-negative T-ALL did not experience the same benefits.
The researchers said GSK-J4 appeared to be well-tolerated. None of the treated mice experienced weight loss or adverse effects in the intestine, spleen, liver, kidney, or hematopoietic system.
“While our study is a proof-of-concept, these promising results might one day lead to a similar targeted treatment for humans,” said study author Carmen Palii, PhD, also of The Ottawa Hospital Research Institute.
In the meantime, the researchers are conducting additional studies in mice, testing higher doses of GSK-J4 and evaluating long-term side effects of the compound. ![]()
Photo courtesy of
The Ottawa Hospital
An experimental compound is “highly promising” as a potential treatment for a subtype of T-cell acute lymphoblastic leukemia (T-ALL), according to researchers.
The team found the histone demethylase UTX is important for the maintenance of TAL1-positive T-ALL.
Inhibiting UTX with a compound known as GSK-J4 proved toxic to TAL1-positive T-ALL cells in vitro and in vivo, but normal cells were spared.
The researchers reported these findings in Genes & Development.
“It’s very exciting because this is the first time anyone has found a potential personalized treatment for this aggressive disease,” said study author Marjorie Brand, PhD, of The Ottawa Hospital Research Institute in Ontario, Canada.
“Unlike current therapies, ours targets the offending gene without harming the rest of the body.”
Dr Brand and her colleagues decided the best way to find a better treatment for TAL1-positive T-ALL was to investigate exactly how it works at a molecular level. So they analyzed the TAL1 gene, which, in certain circumstances, can transform T-cell precursors into leukemic cells.
They found that TAL1 needs UTX to trigger leukemia production. This was surprising because UTX was previously described as a tumor suppressor in T-ALL.
The team said their work suggests UTX is actually a pro-oncogenic cofactor that is essential for leukemia maintenance in TAL1-positive—but not TAL1-negative—T-ALL.
The researchers found that targeting UTX with the H3K27 demethylase inhibitor GSK-J4 could completely halt the growth of TAL1-positive leukemia cells and induce apoptosis in these cells in vitro. But the compound did not have the same effect in TAL1-negative T-ALL.
The team also tested GSK-J4 in mouse models of T-ALL. After 3 weeks of treatment, there was a “dramatic decrease” in the percentage of leukemic blasts in the bone marrow of mice with TAL1-positive T-ALL. These mice also had a reduction in the infiltration of leukemic cells in the spleen.
However, mice with TAL1-negative T-ALL did not experience the same benefits.
The researchers said GSK-J4 appeared to be well-tolerated. None of the treated mice experienced weight loss or adverse effects in the intestine, spleen, liver, kidney, or hematopoietic system.
“While our study is a proof-of-concept, these promising results might one day lead to a similar targeted treatment for humans,” said study author Carmen Palii, PhD, also of The Ottawa Hospital Research Institute.
In the meantime, the researchers are conducting additional studies in mice, testing higher doses of GSK-J4 and evaluating long-term side effects of the compound. ![]()
Changes in chromosome structure contribute to T-ALL, other cancers
acute lymphoblastic leukemia
Image by Hind Medyouf
Breaches in looping chromosomal structures known as insulated neighborhoods can activate oncogenes capable of fueling aggressive tumor growth, according to research published in Science.
These neighborhood breaches were particularly frequent in T-cell acute lymphoblastic leukemia (T-ALL) and esophageal and liver carcinoma.
In some cases, the breaches allowed enhancer elements to activate previously silent oncogenes.
“This new understanding of the role of chromosome structure in cancer gene misregulation reveals the powerful influence of the genome’s structure in human health and disease,” said study author Richard Young, PhD, of the Whitehead Institute for Biomedical Research in Cambridge, Massachusetts.
These findings build on previous work in which Dr Young and his colleagues charted human genome structure and described its influence on gene control in healthy cells.
By mapping the genome’s 3-dimensional conformation, the researchers found that key genes controlling cell identity are found in insulated neighborhoods, whose loops are maintained through anchor sites bound by the protein CTCF.
All essential gene regulation, including the control of proper activation and repression, takes place within these enclosed neighborhoods.
The researchers also found these CTCF loop anchor sites are maintained across various cell types in the human body and are highly conserved in primate genomes. Such widespread structural conservation led the team to hypothesize that disruptions in genome conformation might be associated with disease, including cancers.
Sure enough, subsequent systematic genomic analysis of more than 50 cancer cell types revealed mutations affecting CTCF anchor sites, which led to the loss of insulated neighborhood boundaries.
By mapping insulated neighborhoods in T-ALL, the researchers found that tumor cell genomes contain recurrent microdeletions that eliminate the boundary sites of insulated neighborhoods containing prominent T-ALL proto-oncogenes.
The team also found the genomes of esophageal and liver carcinoma samples were enriched for boundary CTCF site mutations. The genes located in the most frequently mutated neighborhoods included known proto-oncogenes and genes not previously associated with these malignancies.
“We hadn’t known if these types of mutations contributed to cancer,” Dr Young said. “Now, we have multiple examples where these disruptions activate oncogenes that play major roles in tumorigenesis.”
The researchers noted that this oncogenic mechanism may be valuable for identifying genes that drive poorly understood cancers.
“In some cancers, such as esophageal carcinoma, the most frequent genetic mutation occurs at the CTCF sites, which is quite striking,” said Denes Hnisz, PhD, a researcher in the Young lab.
“In addition, there are still many cancers whose driver mutations and oncogenes are not known, and mapping altered structures may reveal the key oncogenes in these cancers.”
In an attempt to confirm the relationship between structural disruption and oncogenesis, the researchers used genome editing techniques to introduce CTCF anchor site deletions in non-malignant cells. They found these mutations were sufficient to activate oncogenes that are silent in normal cells.
The researchers said these findings suggest future mapping of genome structure in individual cancer patients might improve diagnosis and help guide treatment protocols.
“Now that we understand how perturbations in the genome’s structure can contribute to oncogenesis, we’re developing strategies to efficiently diagnose and potentially fix these faulty neighborhoods,” said Abe Weintraub, a graduate student in the Young lab. ![]()
acute lymphoblastic leukemia
Image by Hind Medyouf
Breaches in looping chromosomal structures known as insulated neighborhoods can activate oncogenes capable of fueling aggressive tumor growth, according to research published in Science.
These neighborhood breaches were particularly frequent in T-cell acute lymphoblastic leukemia (T-ALL) and esophageal and liver carcinoma.
In some cases, the breaches allowed enhancer elements to activate previously silent oncogenes.
“This new understanding of the role of chromosome structure in cancer gene misregulation reveals the powerful influence of the genome’s structure in human health and disease,” said study author Richard Young, PhD, of the Whitehead Institute for Biomedical Research in Cambridge, Massachusetts.
These findings build on previous work in which Dr Young and his colleagues charted human genome structure and described its influence on gene control in healthy cells.
By mapping the genome’s 3-dimensional conformation, the researchers found that key genes controlling cell identity are found in insulated neighborhoods, whose loops are maintained through anchor sites bound by the protein CTCF.
All essential gene regulation, including the control of proper activation and repression, takes place within these enclosed neighborhoods.
The researchers also found these CTCF loop anchor sites are maintained across various cell types in the human body and are highly conserved in primate genomes. Such widespread structural conservation led the team to hypothesize that disruptions in genome conformation might be associated with disease, including cancers.
Sure enough, subsequent systematic genomic analysis of more than 50 cancer cell types revealed mutations affecting CTCF anchor sites, which led to the loss of insulated neighborhood boundaries.
By mapping insulated neighborhoods in T-ALL, the researchers found that tumor cell genomes contain recurrent microdeletions that eliminate the boundary sites of insulated neighborhoods containing prominent T-ALL proto-oncogenes.
The team also found the genomes of esophageal and liver carcinoma samples were enriched for boundary CTCF site mutations. The genes located in the most frequently mutated neighborhoods included known proto-oncogenes and genes not previously associated with these malignancies.
“We hadn’t known if these types of mutations contributed to cancer,” Dr Young said. “Now, we have multiple examples where these disruptions activate oncogenes that play major roles in tumorigenesis.”
The researchers noted that this oncogenic mechanism may be valuable for identifying genes that drive poorly understood cancers.
“In some cancers, such as esophageal carcinoma, the most frequent genetic mutation occurs at the CTCF sites, which is quite striking,” said Denes Hnisz, PhD, a researcher in the Young lab.
“In addition, there are still many cancers whose driver mutations and oncogenes are not known, and mapping altered structures may reveal the key oncogenes in these cancers.”
In an attempt to confirm the relationship between structural disruption and oncogenesis, the researchers used genome editing techniques to introduce CTCF anchor site deletions in non-malignant cells. They found these mutations were sufficient to activate oncogenes that are silent in normal cells.
The researchers said these findings suggest future mapping of genome structure in individual cancer patients might improve diagnosis and help guide treatment protocols.
“Now that we understand how perturbations in the genome’s structure can contribute to oncogenesis, we’re developing strategies to efficiently diagnose and potentially fix these faulty neighborhoods,” said Abe Weintraub, a graduate student in the Young lab. ![]()
acute lymphoblastic leukemia
Image by Hind Medyouf
Breaches in looping chromosomal structures known as insulated neighborhoods can activate oncogenes capable of fueling aggressive tumor growth, according to research published in Science.
These neighborhood breaches were particularly frequent in T-cell acute lymphoblastic leukemia (T-ALL) and esophageal and liver carcinoma.
In some cases, the breaches allowed enhancer elements to activate previously silent oncogenes.
“This new understanding of the role of chromosome structure in cancer gene misregulation reveals the powerful influence of the genome’s structure in human health and disease,” said study author Richard Young, PhD, of the Whitehead Institute for Biomedical Research in Cambridge, Massachusetts.
These findings build on previous work in which Dr Young and his colleagues charted human genome structure and described its influence on gene control in healthy cells.
By mapping the genome’s 3-dimensional conformation, the researchers found that key genes controlling cell identity are found in insulated neighborhoods, whose loops are maintained through anchor sites bound by the protein CTCF.
All essential gene regulation, including the control of proper activation and repression, takes place within these enclosed neighborhoods.
The researchers also found these CTCF loop anchor sites are maintained across various cell types in the human body and are highly conserved in primate genomes. Such widespread structural conservation led the team to hypothesize that disruptions in genome conformation might be associated with disease, including cancers.
Sure enough, subsequent systematic genomic analysis of more than 50 cancer cell types revealed mutations affecting CTCF anchor sites, which led to the loss of insulated neighborhood boundaries.
By mapping insulated neighborhoods in T-ALL, the researchers found that tumor cell genomes contain recurrent microdeletions that eliminate the boundary sites of insulated neighborhoods containing prominent T-ALL proto-oncogenes.
The team also found the genomes of esophageal and liver carcinoma samples were enriched for boundary CTCF site mutations. The genes located in the most frequently mutated neighborhoods included known proto-oncogenes and genes not previously associated with these malignancies.
“We hadn’t known if these types of mutations contributed to cancer,” Dr Young said. “Now, we have multiple examples where these disruptions activate oncogenes that play major roles in tumorigenesis.”
The researchers noted that this oncogenic mechanism may be valuable for identifying genes that drive poorly understood cancers.
“In some cancers, such as esophageal carcinoma, the most frequent genetic mutation occurs at the CTCF sites, which is quite striking,” said Denes Hnisz, PhD, a researcher in the Young lab.
“In addition, there are still many cancers whose driver mutations and oncogenes are not known, and mapping altered structures may reveal the key oncogenes in these cancers.”
In an attempt to confirm the relationship between structural disruption and oncogenesis, the researchers used genome editing techniques to introduce CTCF anchor site deletions in non-malignant cells. They found these mutations were sufficient to activate oncogenes that are silent in normal cells.
The researchers said these findings suggest future mapping of genome structure in individual cancer patients might improve diagnosis and help guide treatment protocols.
“Now that we understand how perturbations in the genome’s structure can contribute to oncogenesis, we’re developing strategies to efficiently diagnose and potentially fix these faulty neighborhoods,” said Abe Weintraub, a graduate student in the Young lab.
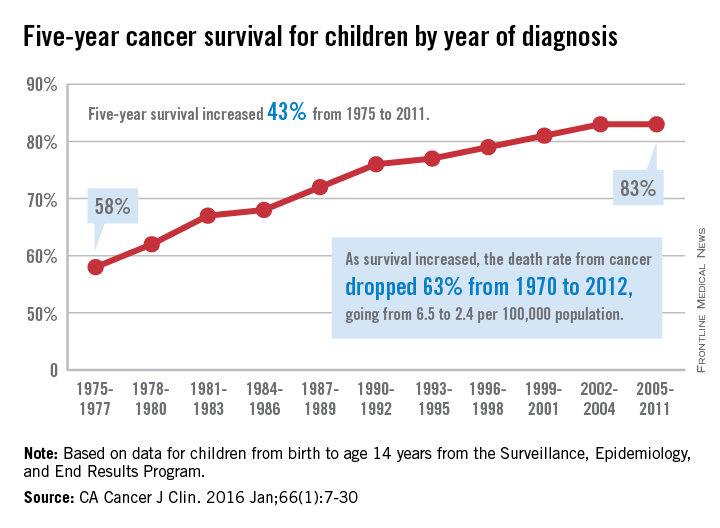
Children’s cancer survival steadily increasing
The 5-year cancer survival rate for children younger than 15 years old is up by 43% since 1975, according to investigators from the American Cancer Society.
The 5-year survival rate for all cancers showed a statistically significant rise from 58% in 1975 to 83% in 2011, said Rebecca L. Siegel and her associates at the ACS (CA Cancer J Clin. 2016 Jan;66[1]:7-30).
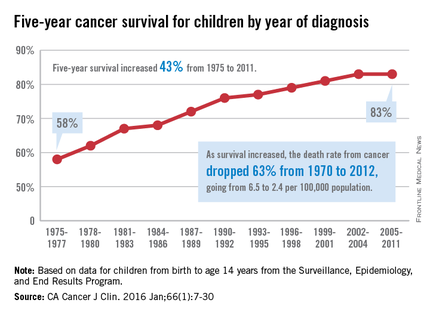
“The substantial progress for all of the major childhood cancers reflects both improvements in treatment and high levels of participation in clinical trials,” they wrote.
Survival for cancers of the brain and nervous system – now the leading cause of cancer death for those younger than 20 years old – increased from 57% in 1975 to 74% in 2011. The next-most-common cause of cancer death in children and adolescents is leukemia, and 5-year survival for acute myeloid leukemia went from 19% in 1975 to 67% in 2011, while 5-year survival for acute lymphocytic leukemia rose from 57% to 91% over that time period, the investigators reported.
The authors reported no conflicts of interest.
The 5-year cancer survival rate for children younger than 15 years old is up by 43% since 1975, according to investigators from the American Cancer Society.
The 5-year survival rate for all cancers showed a statistically significant rise from 58% in 1975 to 83% in 2011, said Rebecca L. Siegel and her associates at the ACS (CA Cancer J Clin. 2016 Jan;66[1]:7-30).
“The substantial progress for all of the major childhood cancers reflects both improvements in treatment and high levels of participation in clinical trials,” they wrote.
Survival for cancers of the brain and nervous system – now the leading cause of cancer death for those younger than 20 years old – increased from 57% in 1975 to 74% in 2011. The next-most-common cause of cancer death in children and adolescents is leukemia, and 5-year survival for acute myeloid leukemia went from 19% in 1975 to 67% in 2011, while 5-year survival for acute lymphocytic leukemia rose from 57% to 91% over that time period, the investigators reported.
The authors reported no conflicts of interest.
The 5-year cancer survival rate for children younger than 15 years old is up by 43% since 1975, according to investigators from the American Cancer Society.
The 5-year survival rate for all cancers showed a statistically significant rise from 58% in 1975 to 83% in 2011, said Rebecca L. Siegel and her associates at the ACS (CA Cancer J Clin. 2016 Jan;66[1]:7-30).
“The substantial progress for all of the major childhood cancers reflects both improvements in treatment and high levels of participation in clinical trials,” they wrote.
Survival for cancers of the brain and nervous system – now the leading cause of cancer death for those younger than 20 years old – increased from 57% in 1975 to 74% in 2011. The next-most-common cause of cancer death in children and adolescents is leukemia, and 5-year survival for acute myeloid leukemia went from 19% in 1975 to 67% in 2011, while 5-year survival for acute lymphocytic leukemia rose from 57% to 91% over that time period, the investigators reported.
The authors reported no conflicts of interest.
FROM CA: A CANCER JOURNAL FOR CLINICIANS
Pre-labor cesarean delivery linked to ALL

Photo by Nina Matthews
Researchers have found a potential correlation between pre-labor cesarean delivery (PLCD) and acute lymphoblastic leukemia (ALL).
The team analyzed data from 13 studies and found a 23% increase in the risk of ALL in children born by PLCD.
However, there was no link between PLCD and acute myeloid leukemia (AML), and there was no correlation between emergency cesareans and ALL or AML.
Erin Marcotte, PhD, of the University of Minnesota, and her colleagues reported these results in The Lancet Haematology.
The researchers analyzed data from the Childhood Leukemia International Consortium. They looked at 33,571 subjects overall, including 23,351 control subjects, 8780 cases of ALL, and 1332 cases of AML.
The analyses were controlled for a number of outside factors, including breastfeeding, parental education levels, and ethnicity.
“Our goal was to determine if there was an association between cesarean deliveries and ALL [and] to identify potential new targets for research into cancer prevention if there is a correlation,” Dr Marcotte said.
“While the link between overall cesarean delivery and childhood leukemia was not statistically significant, it was notable to find an association between pre-labor cesarean delivery and ALL.”
The data suggested AML was not associated with cesarean delivery. The odds ratios (ORs) were 0.99 for all cesarean deliveries, 0.83 for PLCD, and 1.05 for emergency cesarean delivery.
The ORs for ALL were 1.06 overall, 1.02 for emergency cesarean delivery, and 1.23 (P=0.018) for PLCD.
The reason for the increased risk of ALL with PLCD is not known. The researchers said several mechanisms may be at play, including the stress response in the fetus caused by labor and the colonization of microbiota a newborn experiences during a vaginal delivery that is missed during a cesarean birth.
“The most plausible explanation for the association between ALL and pre-labor cesarean delivery is in the cortisol, or stress-related, mechanism,” Dr Marcotte said. “Because ALL is not associated with all cesarean deliveries, it seems less likely the microbiota colonization is a significant factor in this phenomenon. We believe further investigation into this cortisol-mechanism link is warranted due to these findings.”
The researchers said the strength of association in these findings is comparable to other studies looking at cesarean delivery rates and other childhood outcomes, including Type I diabetes and asthma. They believe further investigation into this study’s findings is needed, utilizing more detailed and reliable delivery information.
“This association deserves a closer look to better determine what’s behind the link,” said Logan Spector, PhD, of the University of Minnesota.
“Cortisol exposure is plausible since similar compounds are used to treat ALL. We also know that some are born with cells that are on the path to becoming leukemia. Thus, our working hypothesis is that cortisol exposure at birth may eliminate these pre-leukemic cells.”
Photo by Nina Matthews
Researchers have found a potential correlation between pre-labor cesarean delivery (PLCD) and acute lymphoblastic leukemia (ALL).
The team analyzed data from 13 studies and found a 23% increase in the risk of ALL in children born by PLCD.
However, there was no link between PLCD and acute myeloid leukemia (AML), and there was no correlation between emergency cesareans and ALL or AML.
Erin Marcotte, PhD, of the University of Minnesota, and her colleagues reported these results in The Lancet Haematology.
The researchers analyzed data from the Childhood Leukemia International Consortium. They looked at 33,571 subjects overall, including 23,351 control subjects, 8780 cases of ALL, and 1332 cases of AML.
The analyses were controlled for a number of outside factors, including breastfeeding, parental education levels, and ethnicity.
“Our goal was to determine if there was an association between cesarean deliveries and ALL [and] to identify potential new targets for research into cancer prevention if there is a correlation,” Dr Marcotte said.
“While the link between overall cesarean delivery and childhood leukemia was not statistically significant, it was notable to find an association between pre-labor cesarean delivery and ALL.”
The data suggested AML was not associated with cesarean delivery. The odds ratios (ORs) were 0.99 for all cesarean deliveries, 0.83 for PLCD, and 1.05 for emergency cesarean delivery.
The ORs for ALL were 1.06 overall, 1.02 for emergency cesarean delivery, and 1.23 (P=0.018) for PLCD.
The reason for the increased risk of ALL with PLCD is not known. The researchers said several mechanisms may be at play, including the stress response in the fetus caused by labor and the colonization of microbiota a newborn experiences during a vaginal delivery that is missed during a cesarean birth.
“The most plausible explanation for the association between ALL and pre-labor cesarean delivery is in the cortisol, or stress-related, mechanism,” Dr Marcotte said. “Because ALL is not associated with all cesarean deliveries, it seems less likely the microbiota colonization is a significant factor in this phenomenon. We believe further investigation into this cortisol-mechanism link is warranted due to these findings.”
The researchers said the strength of association in these findings is comparable to other studies looking at cesarean delivery rates and other childhood outcomes, including Type I diabetes and asthma. They believe further investigation into this study’s findings is needed, utilizing more detailed and reliable delivery information.
“This association deserves a closer look to better determine what’s behind the link,” said Logan Spector, PhD, of the University of Minnesota.
“Cortisol exposure is plausible since similar compounds are used to treat ALL. We also know that some are born with cells that are on the path to becoming leukemia. Thus, our working hypothesis is that cortisol exposure at birth may eliminate these pre-leukemic cells.”
Photo by Nina Matthews
Researchers have found a potential correlation between pre-labor cesarean delivery (PLCD) and acute lymphoblastic leukemia (ALL).
The team analyzed data from 13 studies and found a 23% increase in the risk of ALL in children born by PLCD.
However, there was no link between PLCD and acute myeloid leukemia (AML), and there was no correlation between emergency cesareans and ALL or AML.
Erin Marcotte, PhD, of the University of Minnesota, and her colleagues reported these results in The Lancet Haematology.
The researchers analyzed data from the Childhood Leukemia International Consortium. They looked at 33,571 subjects overall, including 23,351 control subjects, 8780 cases of ALL, and 1332 cases of AML.
The analyses were controlled for a number of outside factors, including breastfeeding, parental education levels, and ethnicity.
“Our goal was to determine if there was an association between cesarean deliveries and ALL [and] to identify potential new targets for research into cancer prevention if there is a correlation,” Dr Marcotte said.
“While the link between overall cesarean delivery and childhood leukemia was not statistically significant, it was notable to find an association between pre-labor cesarean delivery and ALL.”
The data suggested AML was not associated with cesarean delivery. The odds ratios (ORs) were 0.99 for all cesarean deliveries, 0.83 for PLCD, and 1.05 for emergency cesarean delivery.
The ORs for ALL were 1.06 overall, 1.02 for emergency cesarean delivery, and 1.23 (P=0.018) for PLCD.
The reason for the increased risk of ALL with PLCD is not known. The researchers said several mechanisms may be at play, including the stress response in the fetus caused by labor and the colonization of microbiota a newborn experiences during a vaginal delivery that is missed during a cesarean birth.
“The most plausible explanation for the association between ALL and pre-labor cesarean delivery is in the cortisol, or stress-related, mechanism,” Dr Marcotte said. “Because ALL is not associated with all cesarean deliveries, it seems less likely the microbiota colonization is a significant factor in this phenomenon. We believe further investigation into this cortisol-mechanism link is warranted due to these findings.”
The researchers said the strength of association in these findings is comparable to other studies looking at cesarean delivery rates and other childhood outcomes, including Type I diabetes and asthma. They believe further investigation into this study’s findings is needed, utilizing more detailed and reliable delivery information.
“This association deserves a closer look to better determine what’s behind the link,” said Logan Spector, PhD, of the University of Minnesota.
“Cortisol exposure is plausible since similar compounds are used to treat ALL. We also know that some are born with cells that are on the path to becoming leukemia. Thus, our working hypothesis is that cortisol exposure at birth may eliminate these pre-leukemic cells.”