User login
Relapse rate drives stem cell transplant failure in pediatric ALL patients
Relapse was the main impediment to successful hematopoietic stem cell transplant (HSCT) in high-risk pediatric acute lymphocytic leukemia (ALL), according to one of the largest single-center experiences reported to date.
The effects of relapse were especially evident for patients with haploidentical donors; these patients had a 3-year cumulative relapse incidence of 47% and event-free survival rate of 35%, both significantly higher than what was seen in other transplant recipients treated at the same center.
The findings, recently published in the journal Biology of Blood and Marrow Transplantation, suggest a substantial unmet need in the treatment of high-risk patients in first remission.
“Newer methods to improve graft-versus-leukemia effect are being tested and will need to be incorporated into the management of high-risk patients,” Asaf D. Yanir, MD, and coauthors at Baylor College of Medicine in Houston said in the report.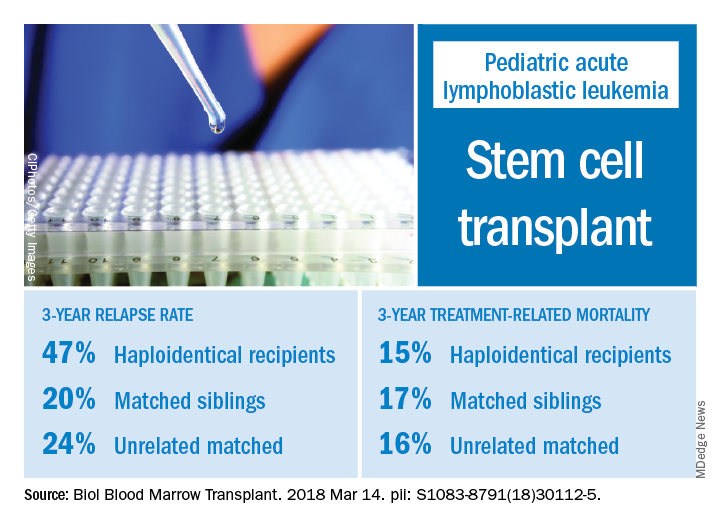
Dr. Yanir and colleagues reported recent outcomes for 124 patients who had undergone HSCT for ALL at their center during 2008-2016. That group included 20 haploidentical transplant recipients, 48 patients with matched sibling donors, and 56 with unrelated matched donors.
The 3-year cumulative incidence of relapse was 47% for haploidentical recipients, compared with 20% for matched sibling donors recipients and 24% for unrelated matched donors recipients (P = .02), according to their findings.
The main cause of HSCT failure was relapse, occurring in 47% of haploidentical transplant recipients, compared with 20% for those with matched sibling donors and 24% for those with unrelated matched donors (P = .02).
Those findings are in line with other studies showing inferior outcomes following haploidentical donor HSCT. However, in contrast to those studies, Dr. Yanir and colleagues did find a rate of treatment-related mortality comparable with other transplant approaches. The 3-year incidence of treatment-related mortality was 15% for the haploidentical group and, similarly, 17% in the matched sibling donor group and 16% in the unrelated matched donor group.
That lower rate of treatment-related mortality in the haploidentical group may be caused by improvements in procedures and supportive care. “Unfortunately, the benefits gained by reducing treatment-related mortality were offset by the high rate of relapse, which remains the main obstacle to successful haploidentical donor HSCT,” Dr. Yanir and coauthors wrote in their report.
New strategies are being studied to retain the graft-versus-leukemia effect or enhance it in patients who’ve undergone haploidentical HSCT, such as selectively depleting alloreactive T cells while sparing other immune effectors, investigators wrote.
“Given evolving practices, it is important to continually evaluate the projected event-free survival for pediatric ALL following HSCT, based on the donor type used,” they wrote.
Dr. Yanir and coauthors had no financial disclosures or conflicts of interest to report.
SOURCE: Yanir AD et al. Biol Blood Marrow Transplant. 2018 Mar 14. doi: 10.1016/j.bbmt.2018.03.001.
Relapse was the main impediment to successful hematopoietic stem cell transplant (HSCT) in high-risk pediatric acute lymphocytic leukemia (ALL), according to one of the largest single-center experiences reported to date.
The effects of relapse were especially evident for patients with haploidentical donors; these patients had a 3-year cumulative relapse incidence of 47% and event-free survival rate of 35%, both significantly higher than what was seen in other transplant recipients treated at the same center.
The findings, recently published in the journal Biology of Blood and Marrow Transplantation, suggest a substantial unmet need in the treatment of high-risk patients in first remission.
“Newer methods to improve graft-versus-leukemia effect are being tested and will need to be incorporated into the management of high-risk patients,” Asaf D. Yanir, MD, and coauthors at Baylor College of Medicine in Houston said in the report.
Dr. Yanir and colleagues reported recent outcomes for 124 patients who had undergone HSCT for ALL at their center during 2008-2016. That group included 20 haploidentical transplant recipients, 48 patients with matched sibling donors, and 56 with unrelated matched donors.
The 3-year cumulative incidence of relapse was 47% for haploidentical recipients, compared with 20% for matched sibling donors recipients and 24% for unrelated matched donors recipients (P = .02), according to their findings.
The main cause of HSCT failure was relapse, occurring in 47% of haploidentical transplant recipients, compared with 20% for those with matched sibling donors and 24% for those with unrelated matched donors (P = .02).
Those findings are in line with other studies showing inferior outcomes following haploidentical donor HSCT. However, in contrast to those studies, Dr. Yanir and colleagues did find a rate of treatment-related mortality comparable with other transplant approaches. The 3-year incidence of treatment-related mortality was 15% for the haploidentical group and, similarly, 17% in the matched sibling donor group and 16% in the unrelated matched donor group.
That lower rate of treatment-related mortality in the haploidentical group may be caused by improvements in procedures and supportive care. “Unfortunately, the benefits gained by reducing treatment-related mortality were offset by the high rate of relapse, which remains the main obstacle to successful haploidentical donor HSCT,” Dr. Yanir and coauthors wrote in their report.
New strategies are being studied to retain the graft-versus-leukemia effect or enhance it in patients who’ve undergone haploidentical HSCT, such as selectively depleting alloreactive T cells while sparing other immune effectors, investigators wrote.
“Given evolving practices, it is important to continually evaluate the projected event-free survival for pediatric ALL following HSCT, based on the donor type used,” they wrote.
Dr. Yanir and coauthors had no financial disclosures or conflicts of interest to report.
SOURCE: Yanir AD et al. Biol Blood Marrow Transplant. 2018 Mar 14. doi: 10.1016/j.bbmt.2018.03.001.
Relapse was the main impediment to successful hematopoietic stem cell transplant (HSCT) in high-risk pediatric acute lymphocytic leukemia (ALL), according to one of the largest single-center experiences reported to date.
The effects of relapse were especially evident for patients with haploidentical donors; these patients had a 3-year cumulative relapse incidence of 47% and event-free survival rate of 35%, both significantly higher than what was seen in other transplant recipients treated at the same center.
The findings, recently published in the journal Biology of Blood and Marrow Transplantation, suggest a substantial unmet need in the treatment of high-risk patients in first remission.
“Newer methods to improve graft-versus-leukemia effect are being tested and will need to be incorporated into the management of high-risk patients,” Asaf D. Yanir, MD, and coauthors at Baylor College of Medicine in Houston said in the report.
Dr. Yanir and colleagues reported recent outcomes for 124 patients who had undergone HSCT for ALL at their center during 2008-2016. That group included 20 haploidentical transplant recipients, 48 patients with matched sibling donors, and 56 with unrelated matched donors.
The 3-year cumulative incidence of relapse was 47% for haploidentical recipients, compared with 20% for matched sibling donors recipients and 24% for unrelated matched donors recipients (P = .02), according to their findings.
The main cause of HSCT failure was relapse, occurring in 47% of haploidentical transplant recipients, compared with 20% for those with matched sibling donors and 24% for those with unrelated matched donors (P = .02).
Those findings are in line with other studies showing inferior outcomes following haploidentical donor HSCT. However, in contrast to those studies, Dr. Yanir and colleagues did find a rate of treatment-related mortality comparable with other transplant approaches. The 3-year incidence of treatment-related mortality was 15% for the haploidentical group and, similarly, 17% in the matched sibling donor group and 16% in the unrelated matched donor group.
That lower rate of treatment-related mortality in the haploidentical group may be caused by improvements in procedures and supportive care. “Unfortunately, the benefits gained by reducing treatment-related mortality were offset by the high rate of relapse, which remains the main obstacle to successful haploidentical donor HSCT,” Dr. Yanir and coauthors wrote in their report.
New strategies are being studied to retain the graft-versus-leukemia effect or enhance it in patients who’ve undergone haploidentical HSCT, such as selectively depleting alloreactive T cells while sparing other immune effectors, investigators wrote.
“Given evolving practices, it is important to continually evaluate the projected event-free survival for pediatric ALL following HSCT, based on the donor type used,” they wrote.
Dr. Yanir and coauthors had no financial disclosures or conflicts of interest to report.
SOURCE: Yanir AD et al. Biol Blood Marrow Transplant. 2018 Mar 14. doi: 10.1016/j.bbmt.2018.03.001.
FROM BIOLOGY OF BLOOD AND MARROW TRANSPLANTATION
Key clinical point: In pediatric acute lymphocytic leukemia (ALL) patients, relapse is the main barrier to successful hematopoietic stem cell transplant (HSCT), especially for those who receive haploidentical donor grafts.
Major finding: The main cause of HSCT failure was relapse, occurring in 47% of haploidentical transplant recipients, compared with 20% for those with matched sibling donors and 24% for those with unrelated matched donors (P = .02).
Study details: A retrospective analysis of 124 transplants performed at a single center during 2008-2016.
Disclosures: Authors had no financial disclosures or conflicts of interest to report.
Source: Yanir AD et al. Biol Blood Marrow Transplant. doi: 10.1016/j.bbmt.2018.03.001.
Drug receives orphan designation for ALL

The US Food and Drug Administration (FDA) has granted orphan drug designation to LBS-007 as a treatment for acute lymphoblastic leukemia (ALL).
LBS-007 is a non-ATP cell-cycle inhibitor targeting a range of cancers.
LBS-007 functions by blocking the kinase activity of CDC7, a key regulator of the cancer cell cycle.
Inhibiting CDC7 stops the proliferation of tumor cells and results in cell death.
Lin BioScience, Inc., the company developing LBS-007, said the drug has demonstrated “very potent activity” against leukemia and solid tumors in preclinical studies.
The company is expected to launch a phase 1 trial of LBS-007 in drug-resistant and refractory acute leukemia in the fourth quarter of 2018.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The US Food and Drug Administration (FDA) has granted orphan drug designation to LBS-007 as a treatment for acute lymphoblastic leukemia (ALL).
LBS-007 is a non-ATP cell-cycle inhibitor targeting a range of cancers.
LBS-007 functions by blocking the kinase activity of CDC7, a key regulator of the cancer cell cycle.
Inhibiting CDC7 stops the proliferation of tumor cells and results in cell death.
Lin BioScience, Inc., the company developing LBS-007, said the drug has demonstrated “very potent activity” against leukemia and solid tumors in preclinical studies.
The company is expected to launch a phase 1 trial of LBS-007 in drug-resistant and refractory acute leukemia in the fourth quarter of 2018.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The US Food and Drug Administration (FDA) has granted orphan drug designation to LBS-007 as a treatment for acute lymphoblastic leukemia (ALL).
LBS-007 is a non-ATP cell-cycle inhibitor targeting a range of cancers.
LBS-007 functions by blocking the kinase activity of CDC7, a key regulator of the cancer cell cycle.
Inhibiting CDC7 stops the proliferation of tumor cells and results in cell death.
Lin BioScience, Inc., the company developing LBS-007, said the drug has demonstrated “very potent activity” against leukemia and solid tumors in preclinical studies.
The company is expected to launch a phase 1 trial of LBS-007 in drug-resistant and refractory acute leukemia in the fourth quarter of 2018.
About orphan designation
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
Art education benefits blood cancer patients

New research suggests a bedside visual art intervention (BVAI) can reduce pain and anxiety in inpatients with hematologic malignancies, including those undergoing transplant.
The BVAI involved an educator teaching patients art technique one-on-one for approximately 30 minutes.
After a single session, patients had significant improvements in positive mood and pain scores, as well as decreases in negative mood and anxiety.
Alexandra P. Wolanskyj, MD, of Mayo Clinic in Rochester, Minnesota, and her colleagues reported these results in the European Journal of Cancer Care.
The study included 21 patients, 19 of them female. Their median age was 53.5 (range, 19-75). Six patients were undergoing hematopoietic stem cell transplant.
The patients had multiple myeloma (n=5), acute myeloid leukemia (n=5), non-Hodgkin lymphoma (n=3), Hodgkin lymphoma (n=2), acute lymphoblastic leukemia (n=1), chronic lymphocytic leukemia (n=1), amyloidosis (n=1), Gardner-Diamond syndrome (n=1), myelodysplastic syndrome (n=1), and Waldenstrom’s macroglobulinemia (n=1).
Nearly half of patients had relapsed disease (47.6%), 23.8% had active and new disease, 19.0% had active disease with primary resistance on chemotherapy, and 9.5% of patients were in remission.
Intervention
The researchers recruited an educator from a community art center to teach art at the patients’ bedsides. Sessions were intended to be about 30 minutes. However, patients could stop at any time or continue beyond 30 minutes.
Patients and their families could make art or just observe. Materials used included watercolors, oil pastels, colored pencils, and clay (all non-toxic and odorless). The materials were left with patients so they could continue to use them after the sessions.
Results
The researchers assessed patients’ pain, anxiety, and mood at baseline and after the patients had a session with the art educator.
After the BVAI, patients had a significant decrease in pain, according to the Visual Analog Scale (VAS). The 14 patients who reported any pain at baseline had a mean reduction in VAS score of 1.5, or a 35.1% reduction in pain (P=0.017).
Patients had a 21.6% reduction in anxiety after the BVAI. Among the 20 patients who completed this assessment, there was a mean 9.2-point decrease in State-Trait Anxiety Inventory (STAI) score (P=0.001).
In addition, patients had a significant increase in positive mood and a significant decrease in negative mood after the BVAI. Mood was assessed in 20 patients using the Positive and Negative Affect Schedule (PANAS) scale.
Positive mood increased 14.6% (P=0.003), and negative mood decreased 18.0% (P=0.015) after the BVAI. Patients’ mean PANAS scores increased 4.6 points for positive mood and decreased 3.3 points for negative mood.
All 21 patients completed a questionnaire on the BVAI. All but 1 patient (95%) said the intervention was positive overall, and 85% of patients (n=18) said they would be interested in participating in future art-based interventions.
The researchers said these results suggest experiences provided by artists in the community may be an adjunct to conventional treatments in patients with cancer-related mood symptoms and pain.
New research suggests a bedside visual art intervention (BVAI) can reduce pain and anxiety in inpatients with hematologic malignancies, including those undergoing transplant.
The BVAI involved an educator teaching patients art technique one-on-one for approximately 30 minutes.
After a single session, patients had significant improvements in positive mood and pain scores, as well as decreases in negative mood and anxiety.
Alexandra P. Wolanskyj, MD, of Mayo Clinic in Rochester, Minnesota, and her colleagues reported these results in the European Journal of Cancer Care.
The study included 21 patients, 19 of them female. Their median age was 53.5 (range, 19-75). Six patients were undergoing hematopoietic stem cell transplant.
The patients had multiple myeloma (n=5), acute myeloid leukemia (n=5), non-Hodgkin lymphoma (n=3), Hodgkin lymphoma (n=2), acute lymphoblastic leukemia (n=1), chronic lymphocytic leukemia (n=1), amyloidosis (n=1), Gardner-Diamond syndrome (n=1), myelodysplastic syndrome (n=1), and Waldenstrom’s macroglobulinemia (n=1).
Nearly half of patients had relapsed disease (47.6%), 23.8% had active and new disease, 19.0% had active disease with primary resistance on chemotherapy, and 9.5% of patients were in remission.
Intervention
The researchers recruited an educator from a community art center to teach art at the patients’ bedsides. Sessions were intended to be about 30 minutes. However, patients could stop at any time or continue beyond 30 minutes.
Patients and their families could make art or just observe. Materials used included watercolors, oil pastels, colored pencils, and clay (all non-toxic and odorless). The materials were left with patients so they could continue to use them after the sessions.
Results
The researchers assessed patients’ pain, anxiety, and mood at baseline and after the patients had a session with the art educator.
After the BVAI, patients had a significant decrease in pain, according to the Visual Analog Scale (VAS). The 14 patients who reported any pain at baseline had a mean reduction in VAS score of 1.5, or a 35.1% reduction in pain (P=0.017).
Patients had a 21.6% reduction in anxiety after the BVAI. Among the 20 patients who completed this assessment, there was a mean 9.2-point decrease in State-Trait Anxiety Inventory (STAI) score (P=0.001).
In addition, patients had a significant increase in positive mood and a significant decrease in negative mood after the BVAI. Mood was assessed in 20 patients using the Positive and Negative Affect Schedule (PANAS) scale.
Positive mood increased 14.6% (P=0.003), and negative mood decreased 18.0% (P=0.015) after the BVAI. Patients’ mean PANAS scores increased 4.6 points for positive mood and decreased 3.3 points for negative mood.
All 21 patients completed a questionnaire on the BVAI. All but 1 patient (95%) said the intervention was positive overall, and 85% of patients (n=18) said they would be interested in participating in future art-based interventions.
The researchers said these results suggest experiences provided by artists in the community may be an adjunct to conventional treatments in patients with cancer-related mood symptoms and pain.
New research suggests a bedside visual art intervention (BVAI) can reduce pain and anxiety in inpatients with hematologic malignancies, including those undergoing transplant.
The BVAI involved an educator teaching patients art technique one-on-one for approximately 30 minutes.
After a single session, patients had significant improvements in positive mood and pain scores, as well as decreases in negative mood and anxiety.
Alexandra P. Wolanskyj, MD, of Mayo Clinic in Rochester, Minnesota, and her colleagues reported these results in the European Journal of Cancer Care.
The study included 21 patients, 19 of them female. Their median age was 53.5 (range, 19-75). Six patients were undergoing hematopoietic stem cell transplant.
The patients had multiple myeloma (n=5), acute myeloid leukemia (n=5), non-Hodgkin lymphoma (n=3), Hodgkin lymphoma (n=2), acute lymphoblastic leukemia (n=1), chronic lymphocytic leukemia (n=1), amyloidosis (n=1), Gardner-Diamond syndrome (n=1), myelodysplastic syndrome (n=1), and Waldenstrom’s macroglobulinemia (n=1).
Nearly half of patients had relapsed disease (47.6%), 23.8% had active and new disease, 19.0% had active disease with primary resistance on chemotherapy, and 9.5% of patients were in remission.
Intervention
The researchers recruited an educator from a community art center to teach art at the patients’ bedsides. Sessions were intended to be about 30 minutes. However, patients could stop at any time or continue beyond 30 minutes.
Patients and their families could make art or just observe. Materials used included watercolors, oil pastels, colored pencils, and clay (all non-toxic and odorless). The materials were left with patients so they could continue to use them after the sessions.
Results
The researchers assessed patients’ pain, anxiety, and mood at baseline and after the patients had a session with the art educator.
After the BVAI, patients had a significant decrease in pain, according to the Visual Analog Scale (VAS). The 14 patients who reported any pain at baseline had a mean reduction in VAS score of 1.5, or a 35.1% reduction in pain (P=0.017).
Patients had a 21.6% reduction in anxiety after the BVAI. Among the 20 patients who completed this assessment, there was a mean 9.2-point decrease in State-Trait Anxiety Inventory (STAI) score (P=0.001).
In addition, patients had a significant increase in positive mood and a significant decrease in negative mood after the BVAI. Mood was assessed in 20 patients using the Positive and Negative Affect Schedule (PANAS) scale.
Positive mood increased 14.6% (P=0.003), and negative mood decreased 18.0% (P=0.015) after the BVAI. Patients’ mean PANAS scores increased 4.6 points for positive mood and decreased 3.3 points for negative mood.
All 21 patients completed a questionnaire on the BVAI. All but 1 patient (95%) said the intervention was positive overall, and 85% of patients (n=18) said they would be interested in participating in future art-based interventions.
The researchers said these results suggest experiences provided by artists in the community may be an adjunct to conventional treatments in patients with cancer-related mood symptoms and pain.
Study reveals gene variants that predispose kids to ALL

Germline variants in IKZF1 can predispose carriers to acute lymphoblastic leukemia (ALL), according to a study published in Cancer Cell.
The research began with the discovery of an IKZF1 variant in 3 generations of a German family affected by pediatric ALL.
Researchers then analyzed data from nearly 5000 children with ALL and identified 27 additional germline variants in IKZF1.
These variants were present in 0.9% of the patients analyzed, and most of the patients with the variants had B-cell ALL.
“This finding adds to the growing body of evidence that, while germline variations still account for a small percentage of pediatric ALL cases overall, more children than previously recognized inherit a predisposition to develop ALL,” said Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
In the Cancer Cell paper, Dr Mullighan and his colleagues report the discovery of a germline deletion variant in IKZF1 (c.del556 or D186fs), which was present in 3 generations of a family.
Two of the 6 family members with this variant had developed B-ALL as children and died. The remaining 4 subjects are apparently healthy, despite having reduced numbers of B cells.
To build upon this discovery, the researchers performed targeted sequencing of IKZF1 in 4963 children with ALL.
This revealed 27 additional IKZF1 variants in 43 patients, most of whom had B-ALL. (One patient had T-cell ALL, and, for 8 patients, their subtype was unknown.)
The researchers noted that the variants were distributed across the gene.
“The pattern of IKZF1 variants was surprising because many of the variants were in regions of the gene that are rarely mutated in leukemic cells,” said study author Jun J. Yang, PhD, of St. Jude. “These regions of the gene have not been well characterized.”
The researchers also found that 22 of the 28 IKZF1 variants adversely affect gene function, while the remaining 6 variants appear to be benign.
The team said the deleterious variants impair DNA binding and regulation of transcriptional targets, induce aberrant leukemic cell adhesion, and reduce ALL cells’ sensitivity to treatment with dasatinib and dexamethasone.
The researchers identified the most deleterious variants as 5 that are located outside of the zinc-finger domains (M31V, M347V, R423C, A434G, and L449F), 2 variants affecting the N-terminal DNA-binding domain (R162P and H163Y), 2 truncating nonsense variants (M306* and C394*), and the frameshift variant discovered in the German family (D186fs).
“This [research] will expand the number of genes to consider when screening for predisposition to leukemia, particularly B-ALL,” said study author Kim Nichols, of St. Jude.
“And while not everyone carrying a germline IKZF1 variant will develop leukemia, these results will help us educate families about the potential risk of leukemia.”
Germline variants in IKZF1 can predispose carriers to acute lymphoblastic leukemia (ALL), according to a study published in Cancer Cell.
The research began with the discovery of an IKZF1 variant in 3 generations of a German family affected by pediatric ALL.
Researchers then analyzed data from nearly 5000 children with ALL and identified 27 additional germline variants in IKZF1.
These variants were present in 0.9% of the patients analyzed, and most of the patients with the variants had B-cell ALL.
“This finding adds to the growing body of evidence that, while germline variations still account for a small percentage of pediatric ALL cases overall, more children than previously recognized inherit a predisposition to develop ALL,” said Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
In the Cancer Cell paper, Dr Mullighan and his colleagues report the discovery of a germline deletion variant in IKZF1 (c.del556 or D186fs), which was present in 3 generations of a family.
Two of the 6 family members with this variant had developed B-ALL as children and died. The remaining 4 subjects are apparently healthy, despite having reduced numbers of B cells.
To build upon this discovery, the researchers performed targeted sequencing of IKZF1 in 4963 children with ALL.
This revealed 27 additional IKZF1 variants in 43 patients, most of whom had B-ALL. (One patient had T-cell ALL, and, for 8 patients, their subtype was unknown.)
The researchers noted that the variants were distributed across the gene.
“The pattern of IKZF1 variants was surprising because many of the variants were in regions of the gene that are rarely mutated in leukemic cells,” said study author Jun J. Yang, PhD, of St. Jude. “These regions of the gene have not been well characterized.”
The researchers also found that 22 of the 28 IKZF1 variants adversely affect gene function, while the remaining 6 variants appear to be benign.
The team said the deleterious variants impair DNA binding and regulation of transcriptional targets, induce aberrant leukemic cell adhesion, and reduce ALL cells’ sensitivity to treatment with dasatinib and dexamethasone.
The researchers identified the most deleterious variants as 5 that are located outside of the zinc-finger domains (M31V, M347V, R423C, A434G, and L449F), 2 variants affecting the N-terminal DNA-binding domain (R162P and H163Y), 2 truncating nonsense variants (M306* and C394*), and the frameshift variant discovered in the German family (D186fs).
“This [research] will expand the number of genes to consider when screening for predisposition to leukemia, particularly B-ALL,” said study author Kim Nichols, of St. Jude.
“And while not everyone carrying a germline IKZF1 variant will develop leukemia, these results will help us educate families about the potential risk of leukemia.”
Germline variants in IKZF1 can predispose carriers to acute lymphoblastic leukemia (ALL), according to a study published in Cancer Cell.
The research began with the discovery of an IKZF1 variant in 3 generations of a German family affected by pediatric ALL.
Researchers then analyzed data from nearly 5000 children with ALL and identified 27 additional germline variants in IKZF1.
These variants were present in 0.9% of the patients analyzed, and most of the patients with the variants had B-cell ALL.
“This finding adds to the growing body of evidence that, while germline variations still account for a small percentage of pediatric ALL cases overall, more children than previously recognized inherit a predisposition to develop ALL,” said Charles Mullighan, MBBS, MD, of St. Jude Children’s Research Hospital in Memphis, Tennessee.
In the Cancer Cell paper, Dr Mullighan and his colleagues report the discovery of a germline deletion variant in IKZF1 (c.del556 or D186fs), which was present in 3 generations of a family.
Two of the 6 family members with this variant had developed B-ALL as children and died. The remaining 4 subjects are apparently healthy, despite having reduced numbers of B cells.
To build upon this discovery, the researchers performed targeted sequencing of IKZF1 in 4963 children with ALL.
This revealed 27 additional IKZF1 variants in 43 patients, most of whom had B-ALL. (One patient had T-cell ALL, and, for 8 patients, their subtype was unknown.)
The researchers noted that the variants were distributed across the gene.
“The pattern of IKZF1 variants was surprising because many of the variants were in regions of the gene that are rarely mutated in leukemic cells,” said study author Jun J. Yang, PhD, of St. Jude. “These regions of the gene have not been well characterized.”
The researchers also found that 22 of the 28 IKZF1 variants adversely affect gene function, while the remaining 6 variants appear to be benign.
The team said the deleterious variants impair DNA binding and regulation of transcriptional targets, induce aberrant leukemic cell adhesion, and reduce ALL cells’ sensitivity to treatment with dasatinib and dexamethasone.
The researchers identified the most deleterious variants as 5 that are located outside of the zinc-finger domains (M31V, M347V, R423C, A434G, and L449F), 2 variants affecting the N-terminal DNA-binding domain (R162P and H163Y), 2 truncating nonsense variants (M306* and C394*), and the frameshift variant discovered in the German family (D186fs).
“This [research] will expand the number of genes to consider when screening for predisposition to leukemia, particularly B-ALL,” said study author Kim Nichols, of St. Jude.
“And while not everyone carrying a germline IKZF1 variant will develop leukemia, these results will help us educate families about the potential risk of leukemia.”
Team uses iPSCs to create ‘universal’ CAR T cells
CHICAGO—Researchers have used induced pluripotent stem cells (iPSCs) to create a “universal” chimeric antigen receptor (CAR) T-cell therapy known as FT819.
The team says FT819 has the potential to be mass-produced, stored, and made readily available for cancer patients.
In in vitro experiments, FT819 demonstrated activity against leukemia and lymphoma.
These results were presented at the AACR Annual Meeting 2018 (abstract LB-108).
The research was conducted by employees of Fate Therapeutics, Inc., the company developing FT819, as well as Memorial Sloan-Kettering Cancer Center.
About FT819
FT819 is produced from a master iPSC line generated using T cells from healthy donors.
“A master iPSC line has unlimited capacity to self-renew and can be banked and renewably used,” said Bob Valamehr, PhD, vice-president of cancer immunotherapy at Fate Therapeutics, Inc.
“We started with cells from a healthy donor rather than the patient, created a master cell line, and used the master cell line to produce large quantities of ‘universal’ CAR19 T cells that are not patient-restricted. These first-of-kind CAR19 T cells, called FT819, can be packaged, stored, and made readily available for treatment of a large number of patients.”
FT819 has 2 targeting receptors—a CAR targeting CD19-positive tumor cells and a CD16 Fc receptor that can engage other therapies (such as tumor antigen-targeting monoclonal antibodies) to overcome antigen escape.
The master iPSC line used for the production of FT819 is engineered in a one-time event to insert a CD19 CAR into the T-cell receptor α constant (TRAC) locus. This is done to eliminate T-cell receptor expression and reduce the likelihood of graft-versus-host disease.
Previous research showed that targeting a CAR to the TRAC locus results in uniform CAR expression and enhances T-cell potency. In fact, TRAC-CAR T cells outperformed conventionally generated CAR T cells by preventing T-cell exhaustion in a mouse model of acute lymphoblastic leukemia.
In vitro experiments
With the current work, the researchers found that FT819 displayed an efficient cytotoxic T-cell response when challenged with CD19-positive tumor cells. FT819 produced cytokines (IFN-gamma, TNF-alpha, and IL-2) and mediators of cell death (CD107a/b, perforin, and granzyme B).
FT819 was also target-specific, attacking only CD19-positive tumor cells and sparing CD19-negative tumor cells in experiments with Raji (Burkitt lymphoma) and Nalm-6 (B-cell acute lymphoblastic leukemia) cell lines.
The researchers said they observed consistent antigen-specific cytotoxicity against Nalm-6 cells with FT819 but variability in antigen-specific cytotoxicity with conventional CAR T cells.
In addition, when combined with rituximab, FT819 elicited antibody-dependent cell-mediated cytotoxicity against CD19-negative, CD20-positive tumor cells.
“Through the development of FT819, we believe there is significant opportunity to lower the cost of CAR T-cell manufacture, enhance the quality of the product, and create a readily available supply of a more efficacious product to reach more patients in need,” Dr Valamehr said.
CHICAGO—Researchers have used induced pluripotent stem cells (iPSCs) to create a “universal” chimeric antigen receptor (CAR) T-cell therapy known as FT819.
The team says FT819 has the potential to be mass-produced, stored, and made readily available for cancer patients.
In in vitro experiments, FT819 demonstrated activity against leukemia and lymphoma.
These results were presented at the AACR Annual Meeting 2018 (abstract LB-108).
The research was conducted by employees of Fate Therapeutics, Inc., the company developing FT819, as well as Memorial Sloan-Kettering Cancer Center.
About FT819
FT819 is produced from a master iPSC line generated using T cells from healthy donors.
“A master iPSC line has unlimited capacity to self-renew and can be banked and renewably used,” said Bob Valamehr, PhD, vice-president of cancer immunotherapy at Fate Therapeutics, Inc.
“We started with cells from a healthy donor rather than the patient, created a master cell line, and used the master cell line to produce large quantities of ‘universal’ CAR19 T cells that are not patient-restricted. These first-of-kind CAR19 T cells, called FT819, can be packaged, stored, and made readily available for treatment of a large number of patients.”
FT819 has 2 targeting receptors—a CAR targeting CD19-positive tumor cells and a CD16 Fc receptor that can engage other therapies (such as tumor antigen-targeting monoclonal antibodies) to overcome antigen escape.
The master iPSC line used for the production of FT819 is engineered in a one-time event to insert a CD19 CAR into the T-cell receptor α constant (TRAC) locus. This is done to eliminate T-cell receptor expression and reduce the likelihood of graft-versus-host disease.
Previous research showed that targeting a CAR to the TRAC locus results in uniform CAR expression and enhances T-cell potency. In fact, TRAC-CAR T cells outperformed conventionally generated CAR T cells by preventing T-cell exhaustion in a mouse model of acute lymphoblastic leukemia.
In vitro experiments
With the current work, the researchers found that FT819 displayed an efficient cytotoxic T-cell response when challenged with CD19-positive tumor cells. FT819 produced cytokines (IFN-gamma, TNF-alpha, and IL-2) and mediators of cell death (CD107a/b, perforin, and granzyme B).
FT819 was also target-specific, attacking only CD19-positive tumor cells and sparing CD19-negative tumor cells in experiments with Raji (Burkitt lymphoma) and Nalm-6 (B-cell acute lymphoblastic leukemia) cell lines.
The researchers said they observed consistent antigen-specific cytotoxicity against Nalm-6 cells with FT819 but variability in antigen-specific cytotoxicity with conventional CAR T cells.
In addition, when combined with rituximab, FT819 elicited antibody-dependent cell-mediated cytotoxicity against CD19-negative, CD20-positive tumor cells.
“Through the development of FT819, we believe there is significant opportunity to lower the cost of CAR T-cell manufacture, enhance the quality of the product, and create a readily available supply of a more efficacious product to reach more patients in need,” Dr Valamehr said.
CHICAGO—Researchers have used induced pluripotent stem cells (iPSCs) to create a “universal” chimeric antigen receptor (CAR) T-cell therapy known as FT819.
The team says FT819 has the potential to be mass-produced, stored, and made readily available for cancer patients.
In in vitro experiments, FT819 demonstrated activity against leukemia and lymphoma.
These results were presented at the AACR Annual Meeting 2018 (abstract LB-108).
The research was conducted by employees of Fate Therapeutics, Inc., the company developing FT819, as well as Memorial Sloan-Kettering Cancer Center.
About FT819
FT819 is produced from a master iPSC line generated using T cells from healthy donors.
“A master iPSC line has unlimited capacity to self-renew and can be banked and renewably used,” said Bob Valamehr, PhD, vice-president of cancer immunotherapy at Fate Therapeutics, Inc.
“We started with cells from a healthy donor rather than the patient, created a master cell line, and used the master cell line to produce large quantities of ‘universal’ CAR19 T cells that are not patient-restricted. These first-of-kind CAR19 T cells, called FT819, can be packaged, stored, and made readily available for treatment of a large number of patients.”
FT819 has 2 targeting receptors—a CAR targeting CD19-positive tumor cells and a CD16 Fc receptor that can engage other therapies (such as tumor antigen-targeting monoclonal antibodies) to overcome antigen escape.
The master iPSC line used for the production of FT819 is engineered in a one-time event to insert a CD19 CAR into the T-cell receptor α constant (TRAC) locus. This is done to eliminate T-cell receptor expression and reduce the likelihood of graft-versus-host disease.
Previous research showed that targeting a CAR to the TRAC locus results in uniform CAR expression and enhances T-cell potency. In fact, TRAC-CAR T cells outperformed conventionally generated CAR T cells by preventing T-cell exhaustion in a mouse model of acute lymphoblastic leukemia.
In vitro experiments
With the current work, the researchers found that FT819 displayed an efficient cytotoxic T-cell response when challenged with CD19-positive tumor cells. FT819 produced cytokines (IFN-gamma, TNF-alpha, and IL-2) and mediators of cell death (CD107a/b, perforin, and granzyme B).
FT819 was also target-specific, attacking only CD19-positive tumor cells and sparing CD19-negative tumor cells in experiments with Raji (Burkitt lymphoma) and Nalm-6 (B-cell acute lymphoblastic leukemia) cell lines.
The researchers said they observed consistent antigen-specific cytotoxicity against Nalm-6 cells with FT819 but variability in antigen-specific cytotoxicity with conventional CAR T cells.
In addition, when combined with rituximab, FT819 elicited antibody-dependent cell-mediated cytotoxicity against CD19-negative, CD20-positive tumor cells.
“Through the development of FT819, we believe there is significant opportunity to lower the cost of CAR T-cell manufacture, enhance the quality of the product, and create a readily available supply of a more efficacious product to reach more patients in need,” Dr Valamehr said.
Blinatumomab triggers complete MRD response in ALL
After treatment with blinatumomab, most patients with minimal residual disease–positive acute lymphoblastic leukemia (ALL) achieved complete MRD response, according to results of a single-arm phase 2 study.
Achieving complete MRD response was associated with significantly longer relapse-free and overall survival in the patients, who were already in hematologic complete remission, researchers reported in the journal Blood.
“Our results suggest that targeted treatment in early stages of MRD is a viable therapeutic strategy for patients with B-cell precursor ALL and that it should also be evaluated in other hematologic malignancies,” Nicola Gökbuget, MD, University Hospital, Frankfurt, Germany, and her coauthors wrote.
This is the first international multicenter study to specifically enroll MRD-positive ALL patients and evaluate them for an MRD-based primary outcome in a cohort of MRD-positive ALL patients, according to the authors.
Preemptively treating low but measurable disease in ALL in remission, instead of waiting for overt relapse, is a strategy that may prolong overall survival, Dr. Gökbuget and her colleagues said in describing the rationale for their study. While there is no standard therapy yet for ALL patients with detectable MRD after intensive chemotherapy, hematopoietic stem cell transplantation (HSCT) is recommended, based on data that it may improve outcomes in patients with persistent MRD. However, other studies suggest detectable MRD before HSCT is associated with higher relapse rates, and many patients relapse while waiting for HSCT, the researchers noted.
To test an MRD-directed treatment strategy, Dr. Gökbuget and colleagues at 46 centers in Europe and Russia conducted an open-label, single-arm, phase 2 study including 116 patients with B-cell precursor ALL in hematologic complete remission. Patients in the study received up to four cycles of blinatumomab, a bispecific, T cell–engager antibody construct that enables T cells to recognize and eliminate CD19-positive cells.
Of 113 evaluable patients, 88 (78%) achieved complete MRD response after one cycle, the primary end point of the study. Relapse-free survival at 18 months was estimated at 54% and median overall survival was 36.5 months in the subset of 110 patients with Philadelphia chromosome–negative ALL in hematologic remission.
Complete MRD responders had improved relapse-free survival versus MRD nonresponders (23.6 vs. 5.7 months; P = .002), they reported. Likewise, overall survival was improved for MRD responders (38.9 vs. 12.5 months; P = .002).
Adverse events were consistent with what was previously reported for blinatumomab and included grade 3 and 4 neurologic events in 12 patients (10%) and 3 patients (3%), respectively. Cytokine-release syndrome was seen in four patients, with grade 1 and grade 3 cases.
The study was not designed to assess the impact of HSCT, which most patients (n = 76) underwent. However, a number of patients with complete MRD response but no HSCT remained in long-term remission, confirming results of an earlier blinatumomab pilot study, according to the researchers.
“This observation might be of relevance for the development of future treatment strategies, particularly for less fit and elderly patients,” Dr. Gökbuget and her coauthors wrote.
Additional studies are needed to clarify the role and indications for HSCT in this setting, they added.
The study was designed by Amgen Research in collaboration with the researchers. Dr. Gökbuget reported financial relationships with Amgen and Pfizer. Other authors reported ties to various pharmaceutical companies.
SOURCE: Gökbuget N et al. Blood. 2018 Apr 5;131(14):1522-31.
The study by Dr. Gökbuget and her colleagues provides “strong evidence” that blinatumomab immunotherapy eliminates residual B-cell acute lymphoblastic leukemia (ALL) cells, thereby preventing relapse and improving survival, according to Patrick Brown, MD.
“This addresses the most important unsolved clinical problem in adults with B-ALL: the development of chemotherapy-resistant relapsed disease,” Dr. Brown wrote in an editorial.
Persistence of minimal residual disease (MRD) is the strongest independent predictor of outcomes in B-cell ALL, and is seen in up to 50% of adult patients after chemotherapy, according to Dr. Brown.
The “well-designed and well-executed” multicenter phase 2 study demonstrated an MRD clearance rate of 78% after one cycle of blinatumomab with modest adverse effects, according to Dr. Brown. Moreover, the results show a doubling of overall survival and tripling of relapse-free survival in MRD responders versus nonresponders, he said.
“An important caveat, however, is that, although the MRD clearance rate was no lower in the 35% of patients who had already relapsed once before enrolling, these patients had a substantially inferior RFS [relapse-free survival] and OS [overall survival], compared with those treated in first remission,” he added. “The clear lesson is that the impact of immunotherapeutic clearance of MRD on survival is greatest when applied early in the disease course.
The “most pressing question” not answered by this study is the impact of hematopoietic stem cell transplantation after complete MRD response, since the study allowed optional HSCT.
Patrick A. Brown, MD, is with Johns Hopkins University, Baltimore. These comments are adapted from his editorial in Blood (2018;131:1497-8). Dr. Brown reported having no competing financial interests related to his editorial.
The study by Dr. Gökbuget and her colleagues provides “strong evidence” that blinatumomab immunotherapy eliminates residual B-cell acute lymphoblastic leukemia (ALL) cells, thereby preventing relapse and improving survival, according to Patrick Brown, MD.
“This addresses the most important unsolved clinical problem in adults with B-ALL: the development of chemotherapy-resistant relapsed disease,” Dr. Brown wrote in an editorial.
Persistence of minimal residual disease (MRD) is the strongest independent predictor of outcomes in B-cell ALL, and is seen in up to 50% of adult patients after chemotherapy, according to Dr. Brown.
The “well-designed and well-executed” multicenter phase 2 study demonstrated an MRD clearance rate of 78% after one cycle of blinatumomab with modest adverse effects, according to Dr. Brown. Moreover, the results show a doubling of overall survival and tripling of relapse-free survival in MRD responders versus nonresponders, he said.
“An important caveat, however, is that, although the MRD clearance rate was no lower in the 35% of patients who had already relapsed once before enrolling, these patients had a substantially inferior RFS [relapse-free survival] and OS [overall survival], compared with those treated in first remission,” he added. “The clear lesson is that the impact of immunotherapeutic clearance of MRD on survival is greatest when applied early in the disease course.
The “most pressing question” not answered by this study is the impact of hematopoietic stem cell transplantation after complete MRD response, since the study allowed optional HSCT.
Patrick A. Brown, MD, is with Johns Hopkins University, Baltimore. These comments are adapted from his editorial in Blood (2018;131:1497-8). Dr. Brown reported having no competing financial interests related to his editorial.
The study by Dr. Gökbuget and her colleagues provides “strong evidence” that blinatumomab immunotherapy eliminates residual B-cell acute lymphoblastic leukemia (ALL) cells, thereby preventing relapse and improving survival, according to Patrick Brown, MD.
“This addresses the most important unsolved clinical problem in adults with B-ALL: the development of chemotherapy-resistant relapsed disease,” Dr. Brown wrote in an editorial.
Persistence of minimal residual disease (MRD) is the strongest independent predictor of outcomes in B-cell ALL, and is seen in up to 50% of adult patients after chemotherapy, according to Dr. Brown.
The “well-designed and well-executed” multicenter phase 2 study demonstrated an MRD clearance rate of 78% after one cycle of blinatumomab with modest adverse effects, according to Dr. Brown. Moreover, the results show a doubling of overall survival and tripling of relapse-free survival in MRD responders versus nonresponders, he said.
“An important caveat, however, is that, although the MRD clearance rate was no lower in the 35% of patients who had already relapsed once before enrolling, these patients had a substantially inferior RFS [relapse-free survival] and OS [overall survival], compared with those treated in first remission,” he added. “The clear lesson is that the impact of immunotherapeutic clearance of MRD on survival is greatest when applied early in the disease course.
The “most pressing question” not answered by this study is the impact of hematopoietic stem cell transplantation after complete MRD response, since the study allowed optional HSCT.
Patrick A. Brown, MD, is with Johns Hopkins University, Baltimore. These comments are adapted from his editorial in Blood (2018;131:1497-8). Dr. Brown reported having no competing financial interests related to his editorial.
After treatment with blinatumomab, most patients with minimal residual disease–positive acute lymphoblastic leukemia (ALL) achieved complete MRD response, according to results of a single-arm phase 2 study.
Achieving complete MRD response was associated with significantly longer relapse-free and overall survival in the patients, who were already in hematologic complete remission, researchers reported in the journal Blood.
“Our results suggest that targeted treatment in early stages of MRD is a viable therapeutic strategy for patients with B-cell precursor ALL and that it should also be evaluated in other hematologic malignancies,” Nicola Gökbuget, MD, University Hospital, Frankfurt, Germany, and her coauthors wrote.
This is the first international multicenter study to specifically enroll MRD-positive ALL patients and evaluate them for an MRD-based primary outcome in a cohort of MRD-positive ALL patients, according to the authors.
Preemptively treating low but measurable disease in ALL in remission, instead of waiting for overt relapse, is a strategy that may prolong overall survival, Dr. Gökbuget and her colleagues said in describing the rationale for their study. While there is no standard therapy yet for ALL patients with detectable MRD after intensive chemotherapy, hematopoietic stem cell transplantation (HSCT) is recommended, based on data that it may improve outcomes in patients with persistent MRD. However, other studies suggest detectable MRD before HSCT is associated with higher relapse rates, and many patients relapse while waiting for HSCT, the researchers noted.
To test an MRD-directed treatment strategy, Dr. Gökbuget and colleagues at 46 centers in Europe and Russia conducted an open-label, single-arm, phase 2 study including 116 patients with B-cell precursor ALL in hematologic complete remission. Patients in the study received up to four cycles of blinatumomab, a bispecific, T cell–engager antibody construct that enables T cells to recognize and eliminate CD19-positive cells.
Of 113 evaluable patients, 88 (78%) achieved complete MRD response after one cycle, the primary end point of the study. Relapse-free survival at 18 months was estimated at 54% and median overall survival was 36.5 months in the subset of 110 patients with Philadelphia chromosome–negative ALL in hematologic remission.
Complete MRD responders had improved relapse-free survival versus MRD nonresponders (23.6 vs. 5.7 months; P = .002), they reported. Likewise, overall survival was improved for MRD responders (38.9 vs. 12.5 months; P = .002).
Adverse events were consistent with what was previously reported for blinatumomab and included grade 3 and 4 neurologic events in 12 patients (10%) and 3 patients (3%), respectively. Cytokine-release syndrome was seen in four patients, with grade 1 and grade 3 cases.
The study was not designed to assess the impact of HSCT, which most patients (n = 76) underwent. However, a number of patients with complete MRD response but no HSCT remained in long-term remission, confirming results of an earlier blinatumomab pilot study, according to the researchers.
“This observation might be of relevance for the development of future treatment strategies, particularly for less fit and elderly patients,” Dr. Gökbuget and her coauthors wrote.
Additional studies are needed to clarify the role and indications for HSCT in this setting, they added.
The study was designed by Amgen Research in collaboration with the researchers. Dr. Gökbuget reported financial relationships with Amgen and Pfizer. Other authors reported ties to various pharmaceutical companies.
SOURCE: Gökbuget N et al. Blood. 2018 Apr 5;131(14):1522-31.
After treatment with blinatumomab, most patients with minimal residual disease–positive acute lymphoblastic leukemia (ALL) achieved complete MRD response, according to results of a single-arm phase 2 study.
Achieving complete MRD response was associated with significantly longer relapse-free and overall survival in the patients, who were already in hematologic complete remission, researchers reported in the journal Blood.
“Our results suggest that targeted treatment in early stages of MRD is a viable therapeutic strategy for patients with B-cell precursor ALL and that it should also be evaluated in other hematologic malignancies,” Nicola Gökbuget, MD, University Hospital, Frankfurt, Germany, and her coauthors wrote.
This is the first international multicenter study to specifically enroll MRD-positive ALL patients and evaluate them for an MRD-based primary outcome in a cohort of MRD-positive ALL patients, according to the authors.
Preemptively treating low but measurable disease in ALL in remission, instead of waiting for overt relapse, is a strategy that may prolong overall survival, Dr. Gökbuget and her colleagues said in describing the rationale for their study. While there is no standard therapy yet for ALL patients with detectable MRD after intensive chemotherapy, hematopoietic stem cell transplantation (HSCT) is recommended, based on data that it may improve outcomes in patients with persistent MRD. However, other studies suggest detectable MRD before HSCT is associated with higher relapse rates, and many patients relapse while waiting for HSCT, the researchers noted.
To test an MRD-directed treatment strategy, Dr. Gökbuget and colleagues at 46 centers in Europe and Russia conducted an open-label, single-arm, phase 2 study including 116 patients with B-cell precursor ALL in hematologic complete remission. Patients in the study received up to four cycles of blinatumomab, a bispecific, T cell–engager antibody construct that enables T cells to recognize and eliminate CD19-positive cells.
Of 113 evaluable patients, 88 (78%) achieved complete MRD response after one cycle, the primary end point of the study. Relapse-free survival at 18 months was estimated at 54% and median overall survival was 36.5 months in the subset of 110 patients with Philadelphia chromosome–negative ALL in hematologic remission.
Complete MRD responders had improved relapse-free survival versus MRD nonresponders (23.6 vs. 5.7 months; P = .002), they reported. Likewise, overall survival was improved for MRD responders (38.9 vs. 12.5 months; P = .002).
Adverse events were consistent with what was previously reported for blinatumomab and included grade 3 and 4 neurologic events in 12 patients (10%) and 3 patients (3%), respectively. Cytokine-release syndrome was seen in four patients, with grade 1 and grade 3 cases.
The study was not designed to assess the impact of HSCT, which most patients (n = 76) underwent. However, a number of patients with complete MRD response but no HSCT remained in long-term remission, confirming results of an earlier blinatumomab pilot study, according to the researchers.
“This observation might be of relevance for the development of future treatment strategies, particularly for less fit and elderly patients,” Dr. Gökbuget and her coauthors wrote.
Additional studies are needed to clarify the role and indications for HSCT in this setting, they added.
The study was designed by Amgen Research in collaboration with the researchers. Dr. Gökbuget reported financial relationships with Amgen and Pfizer. Other authors reported ties to various pharmaceutical companies.
SOURCE: Gökbuget N et al. Blood. 2018 Apr 5;131(14):1522-31.
FROM BLOOD
Key clinical point:
Major finding: Complete MRD response, seen in 78% of blinatumomab-treated patients, was associated with improved relapse-free and overall survival.
Study details: An open-label, single-arm, phase 2 study including 116 patients with B-cell precursor ALL in hematologic complete remission, conducted at 46 centers in Europe and Russia.
Disclosures: The study was designed by Amgen Research in collaboration with the researchers. Dr. Gökbuget reported financial relationships with Amgen and Pfizer. Other authors reported ties to various pharmaceutical companies.
Source: Gökbuget N et al. Blood. 2018 Apr 5;131(14):1522-31.
Gene variants linked to survival after HSCT

New research has revealed a link between rare gene variants and survival after hematopoietic stem cell transplant (HSCT).
Researchers performed exome sequencing in nearly 2500 HSCT recipients and their matched, unrelated donors.
The sequencing revealed several gene variants—in both donors and recipients—that were significantly associated with overall survival (OS), transplant-related mortality (TRM), and disease-related mortality (DRM) after HSCT.
Qianqian Zhu, PhD, of Roswell Park Comprehensive Cancer Center in Buffalo, New York, and her colleagues described these findings in Blood.
The team performed exome sequencing—using the Illumina HumanExome BeadChip—in patients who participated in the DISCOVeRY-BMT study.
This included 2473 HSCT recipients who had acute myeloid leukemia, acute lymphoblastic leukemia, or myelodysplastic syndromes. It also included 2221 donors who were a 10/10 human leukocyte antigen match for each recipient.
The researchers looked at genetic variants in donors and recipients and assessed the variants’ associations with OS, TRM, and DRM.
Variants in recipients
Analyses revealed an increased risk of TRM when there was a mismatch between donors and recipients for a variant in TEX38—rs200092801. The increased risk was even more pronounced when either the recipient or the donor was female.
Among the recipients mismatched with their donors at rs200092801, every female recipient and every recipient with a female donor died from TRM. In comparison, 44% of the male recipients with male donors died from TRM.
The researchers said the rs200092801 variant may prompt the production of a mutant peptide that can be presented by MHC-I molecules to immune cells to trigger downstream immune response and TRM.
Dr Zhu and her colleagues also identified variants that appeared to have a positive impact on TRM and OS.
Recipients who had any of 6 variants in the gene OR51D1 had a decreased risk of TRM and improved OS.
The variants (rs138224979, rs148606808, rs141786655, rs61745314, rs200394876, and rs149135276) were not associated with DRM, so the researchers concluded that the improvement in OS was driven by protection against TRM.
Donor variants linked to OS
Donors had variants in 4 genes—ALPP, EMID1, SLC44A5, and LRP1—that were associated with OS but not TRM or DRM.
The 3 variants identified in ALPP (rs144454460, rs140078460, and rs142493383) were associated with improved OS.
And the 2 variants in SLC44A5 (rs143004355 and rs149696907) were associated with worse OS.
There were 2 variants in EMID1. One was associated with improved OS (rs34772704), and the other was associated with decreased OS (rs139996840).
And there were 27 variants in LRP1. Some had a positive association with OS, and others had a negative association.
Donor variants linked to TRM and DRM
Six variants in the HHAT gene were associated with TRM. Five of the variants appeared to have a protective effect against TRM (rs145455128, rs146916002, rs61744143, rs149597734, and rs145943928). For the other variant (rs141591165), the apparent effect was inconsistent between patient cohorts.
There were 3 variants in LYZL4 associated with DRM. Two were associated with an increased risk of DRM (rs147770623 and rs76947105), and 1 appeared to have a protective effect (rs181886204).
Six variants in NT5E appeared to have a protective effect against DRM (rs200250022, rs200369370, rs41271617, rs200648774, rs144719925, and rs145505137).
The researchers said the variants in NT5E probably reduce the enzyme activity of the gene. This supports preclinical findings showing that targeted blockade of NT5E can slow tumor growth.
“We have just started to uncover the biological relevance of these new and unexpected genes to a patient’s survival after [HSCT],” Dr Zhu said.
“Our findings shed light on new areas that were not considered before, but we need to further replicate and test our findings. We’re hoping that additional studies of this type will continue to discover novel genes leading to improved outcomes for patients.”
New research has revealed a link between rare gene variants and survival after hematopoietic stem cell transplant (HSCT).
Researchers performed exome sequencing in nearly 2500 HSCT recipients and their matched, unrelated donors.
The sequencing revealed several gene variants—in both donors and recipients—that were significantly associated with overall survival (OS), transplant-related mortality (TRM), and disease-related mortality (DRM) after HSCT.
Qianqian Zhu, PhD, of Roswell Park Comprehensive Cancer Center in Buffalo, New York, and her colleagues described these findings in Blood.
The team performed exome sequencing—using the Illumina HumanExome BeadChip—in patients who participated in the DISCOVeRY-BMT study.
This included 2473 HSCT recipients who had acute myeloid leukemia, acute lymphoblastic leukemia, or myelodysplastic syndromes. It also included 2221 donors who were a 10/10 human leukocyte antigen match for each recipient.
The researchers looked at genetic variants in donors and recipients and assessed the variants’ associations with OS, TRM, and DRM.
Variants in recipients
Analyses revealed an increased risk of TRM when there was a mismatch between donors and recipients for a variant in TEX38—rs200092801. The increased risk was even more pronounced when either the recipient or the donor was female.
Among the recipients mismatched with their donors at rs200092801, every female recipient and every recipient with a female donor died from TRM. In comparison, 44% of the male recipients with male donors died from TRM.
The researchers said the rs200092801 variant may prompt the production of a mutant peptide that can be presented by MHC-I molecules to immune cells to trigger downstream immune response and TRM.
Dr Zhu and her colleagues also identified variants that appeared to have a positive impact on TRM and OS.
Recipients who had any of 6 variants in the gene OR51D1 had a decreased risk of TRM and improved OS.
The variants (rs138224979, rs148606808, rs141786655, rs61745314, rs200394876, and rs149135276) were not associated with DRM, so the researchers concluded that the improvement in OS was driven by protection against TRM.
Donor variants linked to OS
Donors had variants in 4 genes—ALPP, EMID1, SLC44A5, and LRP1—that were associated with OS but not TRM or DRM.
The 3 variants identified in ALPP (rs144454460, rs140078460, and rs142493383) were associated with improved OS.
And the 2 variants in SLC44A5 (rs143004355 and rs149696907) were associated with worse OS.
There were 2 variants in EMID1. One was associated with improved OS (rs34772704), and the other was associated with decreased OS (rs139996840).
And there were 27 variants in LRP1. Some had a positive association with OS, and others had a negative association.
Donor variants linked to TRM and DRM
Six variants in the HHAT gene were associated with TRM. Five of the variants appeared to have a protective effect against TRM (rs145455128, rs146916002, rs61744143, rs149597734, and rs145943928). For the other variant (rs141591165), the apparent effect was inconsistent between patient cohorts.
There were 3 variants in LYZL4 associated with DRM. Two were associated with an increased risk of DRM (rs147770623 and rs76947105), and 1 appeared to have a protective effect (rs181886204).
Six variants in NT5E appeared to have a protective effect against DRM (rs200250022, rs200369370, rs41271617, rs200648774, rs144719925, and rs145505137).
The researchers said the variants in NT5E probably reduce the enzyme activity of the gene. This supports preclinical findings showing that targeted blockade of NT5E can slow tumor growth.
“We have just started to uncover the biological relevance of these new and unexpected genes to a patient’s survival after [HSCT],” Dr Zhu said.
“Our findings shed light on new areas that were not considered before, but we need to further replicate and test our findings. We’re hoping that additional studies of this type will continue to discover novel genes leading to improved outcomes for patients.”
New research has revealed a link between rare gene variants and survival after hematopoietic stem cell transplant (HSCT).
Researchers performed exome sequencing in nearly 2500 HSCT recipients and their matched, unrelated donors.
The sequencing revealed several gene variants—in both donors and recipients—that were significantly associated with overall survival (OS), transplant-related mortality (TRM), and disease-related mortality (DRM) after HSCT.
Qianqian Zhu, PhD, of Roswell Park Comprehensive Cancer Center in Buffalo, New York, and her colleagues described these findings in Blood.
The team performed exome sequencing—using the Illumina HumanExome BeadChip—in patients who participated in the DISCOVeRY-BMT study.
This included 2473 HSCT recipients who had acute myeloid leukemia, acute lymphoblastic leukemia, or myelodysplastic syndromes. It also included 2221 donors who were a 10/10 human leukocyte antigen match for each recipient.
The researchers looked at genetic variants in donors and recipients and assessed the variants’ associations with OS, TRM, and DRM.
Variants in recipients
Analyses revealed an increased risk of TRM when there was a mismatch between donors and recipients for a variant in TEX38—rs200092801. The increased risk was even more pronounced when either the recipient or the donor was female.
Among the recipients mismatched with their donors at rs200092801, every female recipient and every recipient with a female donor died from TRM. In comparison, 44% of the male recipients with male donors died from TRM.
The researchers said the rs200092801 variant may prompt the production of a mutant peptide that can be presented by MHC-I molecules to immune cells to trigger downstream immune response and TRM.
Dr Zhu and her colleagues also identified variants that appeared to have a positive impact on TRM and OS.
Recipients who had any of 6 variants in the gene OR51D1 had a decreased risk of TRM and improved OS.
The variants (rs138224979, rs148606808, rs141786655, rs61745314, rs200394876, and rs149135276) were not associated with DRM, so the researchers concluded that the improvement in OS was driven by protection against TRM.
Donor variants linked to OS
Donors had variants in 4 genes—ALPP, EMID1, SLC44A5, and LRP1—that were associated with OS but not TRM or DRM.
The 3 variants identified in ALPP (rs144454460, rs140078460, and rs142493383) were associated with improved OS.
And the 2 variants in SLC44A5 (rs143004355 and rs149696907) were associated with worse OS.
There were 2 variants in EMID1. One was associated with improved OS (rs34772704), and the other was associated with decreased OS (rs139996840).
And there were 27 variants in LRP1. Some had a positive association with OS, and others had a negative association.
Donor variants linked to TRM and DRM
Six variants in the HHAT gene were associated with TRM. Five of the variants appeared to have a protective effect against TRM (rs145455128, rs146916002, rs61744143, rs149597734, and rs145943928). For the other variant (rs141591165), the apparent effect was inconsistent between patient cohorts.
There were 3 variants in LYZL4 associated with DRM. Two were associated with an increased risk of DRM (rs147770623 and rs76947105), and 1 appeared to have a protective effect (rs181886204).
Six variants in NT5E appeared to have a protective effect against DRM (rs200250022, rs200369370, rs41271617, rs200648774, rs144719925, and rs145505137).
The researchers said the variants in NT5E probably reduce the enzyme activity of the gene. This supports preclinical findings showing that targeted blockade of NT5E can slow tumor growth.
“We have just started to uncover the biological relevance of these new and unexpected genes to a patient’s survival after [HSCT],” Dr Zhu said.
“Our findings shed light on new areas that were not considered before, but we need to further replicate and test our findings. We’re hoping that additional studies of this type will continue to discover novel genes leading to improved outcomes for patients.”
Health Canada approves product for adult ALL
Health Canada has approved inotuzumab ozogamicin (Besponsa™) as monotherapy for adults with relapsed or refractory, CD22-positive, B-cell precursor acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin is the first and only CD22-directed antibody-drug conjugate approved for this indication.
The product consists of a monoclonal antibody targeting CD22 and a cytotoxic agent known as calicheamicin.
Health Canada’s approval of inotuzumab ozogamicin is based on results from the phase 3 INO-VATE trial, which were published in NEJM in June 2016.
The trial enrolled 326 adults with relapsed or refractory B-cell precursor ALL.
Patients received inotuzumab ozogamicin or 1 of 3 chemotherapy regimens—high-dose cytarabine; cytarabine plus mitoxantrone; or fludarabine, cytarabine, and granulocyte colony-stimulating factor.
The rate of complete remission, including incomplete hematologic recovery, was 80.7% in the inotuzumab arm and 29.4% in the chemotherapy arm (P<0.001). The median duration of remission was 4.6 months and 3.1 months, respectively (P=0.03).
Forty-one percent of patients treated with inotuzumab and 11% of those who received chemotherapy proceeded to stem cell transplant directly after treatment (P<0.001).
The median progression-free survival was 5.0 months in the inotuzumab arm and 1.8 months in the chemotherapy arm (P<0.001).
The median overall survival was 7.7 months and 6.7 months, respectively (P=0.04). This did not meet the prespecified boundary of significance (P=0.0208).
Liver-related adverse events were more common in the inotuzumab arm than the chemotherapy arm. The most frequent of these were increased aspartate aminotransferase level (20% vs 10%), hyperbilirubinemia (15% vs 10%), and increased alanine aminotransferase level (14% vs 11%).
Veno-occlusive liver disease occurred in 11% of patients in the inotuzumab arm and 1% in the chemotherapy arm.
There were 17 deaths during treatment in the inotuzumab arm and 11 in the chemotherapy arm. Four deaths were considered related to inotuzumab, and 2 were deemed related to chemotherapy.
Health Canada has approved inotuzumab ozogamicin (Besponsa™) as monotherapy for adults with relapsed or refractory, CD22-positive, B-cell precursor acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin is the first and only CD22-directed antibody-drug conjugate approved for this indication.
The product consists of a monoclonal antibody targeting CD22 and a cytotoxic agent known as calicheamicin.
Health Canada’s approval of inotuzumab ozogamicin is based on results from the phase 3 INO-VATE trial, which were published in NEJM in June 2016.
The trial enrolled 326 adults with relapsed or refractory B-cell precursor ALL.
Patients received inotuzumab ozogamicin or 1 of 3 chemotherapy regimens—high-dose cytarabine; cytarabine plus mitoxantrone; or fludarabine, cytarabine, and granulocyte colony-stimulating factor.
The rate of complete remission, including incomplete hematologic recovery, was 80.7% in the inotuzumab arm and 29.4% in the chemotherapy arm (P<0.001). The median duration of remission was 4.6 months and 3.1 months, respectively (P=0.03).
Forty-one percent of patients treated with inotuzumab and 11% of those who received chemotherapy proceeded to stem cell transplant directly after treatment (P<0.001).
The median progression-free survival was 5.0 months in the inotuzumab arm and 1.8 months in the chemotherapy arm (P<0.001).
The median overall survival was 7.7 months and 6.7 months, respectively (P=0.04). This did not meet the prespecified boundary of significance (P=0.0208).
Liver-related adverse events were more common in the inotuzumab arm than the chemotherapy arm. The most frequent of these were increased aspartate aminotransferase level (20% vs 10%), hyperbilirubinemia (15% vs 10%), and increased alanine aminotransferase level (14% vs 11%).
Veno-occlusive liver disease occurred in 11% of patients in the inotuzumab arm and 1% in the chemotherapy arm.
There were 17 deaths during treatment in the inotuzumab arm and 11 in the chemotherapy arm. Four deaths were considered related to inotuzumab, and 2 were deemed related to chemotherapy.
Health Canada has approved inotuzumab ozogamicin (Besponsa™) as monotherapy for adults with relapsed or refractory, CD22-positive, B-cell precursor acute lymphoblastic leukemia (ALL).
Inotuzumab ozogamicin is the first and only CD22-directed antibody-drug conjugate approved for this indication.
The product consists of a monoclonal antibody targeting CD22 and a cytotoxic agent known as calicheamicin.
Health Canada’s approval of inotuzumab ozogamicin is based on results from the phase 3 INO-VATE trial, which were published in NEJM in June 2016.
The trial enrolled 326 adults with relapsed or refractory B-cell precursor ALL.
Patients received inotuzumab ozogamicin or 1 of 3 chemotherapy regimens—high-dose cytarabine; cytarabine plus mitoxantrone; or fludarabine, cytarabine, and granulocyte colony-stimulating factor.
The rate of complete remission, including incomplete hematologic recovery, was 80.7% in the inotuzumab arm and 29.4% in the chemotherapy arm (P<0.001). The median duration of remission was 4.6 months and 3.1 months, respectively (P=0.03).
Forty-one percent of patients treated with inotuzumab and 11% of those who received chemotherapy proceeded to stem cell transplant directly after treatment (P<0.001).
The median progression-free survival was 5.0 months in the inotuzumab arm and 1.8 months in the chemotherapy arm (P<0.001).
The median overall survival was 7.7 months and 6.7 months, respectively (P=0.04). This did not meet the prespecified boundary of significance (P=0.0208).
Liver-related adverse events were more common in the inotuzumab arm than the chemotherapy arm. The most frequent of these were increased aspartate aminotransferase level (20% vs 10%), hyperbilirubinemia (15% vs 10%), and increased alanine aminotransferase level (14% vs 11%).
Veno-occlusive liver disease occurred in 11% of patients in the inotuzumab arm and 1% in the chemotherapy arm.
There were 17 deaths during treatment in the inotuzumab arm and 11 in the chemotherapy arm. Four deaths were considered related to inotuzumab, and 2 were deemed related to chemotherapy.
Team maps genetic evolution of T-ALL subtype
Single-cell analysis has revealed key genetic events in a type of T-cell acute lymphoblastic leukemia (T-ALL), according to researchers.
The team tracked the branching pattern of evolution in STIL-TAL1-positive T-ALL and identified mutations that may trigger development of the disease.
The researchers believe their findings could be used in minimal residual disease assessments as well as for the development of new targeted drugs.
Caroline Furness, MD, of Institute of Cancer Research in London, UK, and her colleagues described this research in Leukemia.
To determine the sequence of mutational events in STIL-TAL1-positive T-ALL, the researchers examined individual leukemia cells from 19 children and young adults with the disease, as well as 1 cell line with the STIL-TAL1 rearrangement (RPMI 8402).
The team found the STIL-TAL1 gene fusion and inactivation of the CDKN2A gene occurred very early in leukemia development.
The researchers said it was difficult to tell whether STIL-TAL1 fusion or CDKN2A loss are initiating events in this disease, and it isn’t clear which mutational event occurs first.
However, the team believes the STIL-TAL1 fusion is likely a founder or truncal event for this type of leukemia, so targeting the TAL1 regulatory complex could be an effective way to treat the disease.
The researchers also identified mutations in NOTCH1 and PTEN as secondary subclonal events.
Half of the samples examined had errors affecting PTEN, which suggests these events are key to maintaining leukemia growth and survival in STIL-TAL1-positive T-ALL, according to the researchers.
“We need to understand how cancers evolve and unpick which mutations are key to triggering cancer development and which are important for driving its growth and spread,” Dr Furness said.
“Our study uncovered these crucial mutations in a type of leukemia that accounts for around a quarter of cases of T-cell leukemia in children and young adults. This will help us to develop more effective treatments, especially in those children who relapse, and kinder treatments that won’t cause life-long side effects.”
Single-cell analysis has revealed key genetic events in a type of T-cell acute lymphoblastic leukemia (T-ALL), according to researchers.
The team tracked the branching pattern of evolution in STIL-TAL1-positive T-ALL and identified mutations that may trigger development of the disease.
The researchers believe their findings could be used in minimal residual disease assessments as well as for the development of new targeted drugs.
Caroline Furness, MD, of Institute of Cancer Research in London, UK, and her colleagues described this research in Leukemia.
To determine the sequence of mutational events in STIL-TAL1-positive T-ALL, the researchers examined individual leukemia cells from 19 children and young adults with the disease, as well as 1 cell line with the STIL-TAL1 rearrangement (RPMI 8402).
The team found the STIL-TAL1 gene fusion and inactivation of the CDKN2A gene occurred very early in leukemia development.
The researchers said it was difficult to tell whether STIL-TAL1 fusion or CDKN2A loss are initiating events in this disease, and it isn’t clear which mutational event occurs first.
However, the team believes the STIL-TAL1 fusion is likely a founder or truncal event for this type of leukemia, so targeting the TAL1 regulatory complex could be an effective way to treat the disease.
The researchers also identified mutations in NOTCH1 and PTEN as secondary subclonal events.
Half of the samples examined had errors affecting PTEN, which suggests these events are key to maintaining leukemia growth and survival in STIL-TAL1-positive T-ALL, according to the researchers.
“We need to understand how cancers evolve and unpick which mutations are key to triggering cancer development and which are important for driving its growth and spread,” Dr Furness said.
“Our study uncovered these crucial mutations in a type of leukemia that accounts for around a quarter of cases of T-cell leukemia in children and young adults. This will help us to develop more effective treatments, especially in those children who relapse, and kinder treatments that won’t cause life-long side effects.”
Single-cell analysis has revealed key genetic events in a type of T-cell acute lymphoblastic leukemia (T-ALL), according to researchers.
The team tracked the branching pattern of evolution in STIL-TAL1-positive T-ALL and identified mutations that may trigger development of the disease.
The researchers believe their findings could be used in minimal residual disease assessments as well as for the development of new targeted drugs.
Caroline Furness, MD, of Institute of Cancer Research in London, UK, and her colleagues described this research in Leukemia.
To determine the sequence of mutational events in STIL-TAL1-positive T-ALL, the researchers examined individual leukemia cells from 19 children and young adults with the disease, as well as 1 cell line with the STIL-TAL1 rearrangement (RPMI 8402).
The team found the STIL-TAL1 gene fusion and inactivation of the CDKN2A gene occurred very early in leukemia development.
The researchers said it was difficult to tell whether STIL-TAL1 fusion or CDKN2A loss are initiating events in this disease, and it isn’t clear which mutational event occurs first.
However, the team believes the STIL-TAL1 fusion is likely a founder or truncal event for this type of leukemia, so targeting the TAL1 regulatory complex could be an effective way to treat the disease.
The researchers also identified mutations in NOTCH1 and PTEN as secondary subclonal events.
Half of the samples examined had errors affecting PTEN, which suggests these events are key to maintaining leukemia growth and survival in STIL-TAL1-positive T-ALL, according to the researchers.
“We need to understand how cancers evolve and unpick which mutations are key to triggering cancer development and which are important for driving its growth and spread,” Dr Furness said.
“Our study uncovered these crucial mutations in a type of leukemia that accounts for around a quarter of cases of T-cell leukemia in children and young adults. This will help us to develop more effective treatments, especially in those children who relapse, and kinder treatments that won’t cause life-long side effects.”
Ponatinib bests older TKIs against Ph+ALL
Every generation aspires to be better than its predecessors, and for the third-generation tyrosine kinase inhibitor ponatinib (Iclusig), that might just be true, investigators claim.
A retrospective analysis comparing clinical trial outcomes for patients with newly diagnosed acute lymphoblastic leukemia positive for the Philadelphia chromosome (Ph+ALL) suggests that first-line ponatinib offers modestly better complete molecular response (CMR) rates and 3-year overall survival (OS) than either first-generation TKIs such as imatinib (Gleevec) or second-generation agents such as dasatinib (Sprycel) and nilotinib (Tasigna).
“Although only 1 relevant study of ponatinib combined with chemotherapy in Ph+ALL has been reported and our ability to adjust for baseline patient characteristics was limited, the results suggest that ponatinib combined with chemotherapy might represent a more effective front-line treatment option than chemotherapy combined with an earlier generation TKI for patients with newly diagnosed Ph+ALL, including those who cannot or choose not to undergo [stem cell transplant],” wrote Elias Jabbour, MD, of the University of Texas MD Anderson Cancer Center in Houston, and his colleagues.
They based their conclusions on a meta-regression analysis of 25 studies looking at first- or second-generation TKIs and one study of ponatinib as frontline therapy for patients with Ph+ALL. They described their results in Clinical Lymphoma, Myeloma & Leukemia.
The investigators created pooled estimates of outcomes from studies of earlier-generation TKIs plus combination chemotherapy using a random-effects meta-analysis method. For the sole ponatinib study – a single-arm trial of combination chemotherapy plus ponatinib – they used a binomial distribution method to calculate 95% confidence intervals (CI). The method essentially estimates the probability of success or failure of a repeated experiment.
They found that 79% of patients in the ponatinib trial achieved a CMR, compared with 34% of patients treated with earlier generation TKIs plus chemotherapy. This translates into an odds ratio (OR) for CMR with ponatinib of 6.09 (P = .034).
Two-year OS rates were 83% with ponatinib versus 58% for all patients treated with other TKIs. Although the OR (3.70) seemed to run in favor of ponatinib, the difference was not statistically significant (P = .062).
For 3-year OS, however, ponatinib was superior to the pooled data for the other TKIs, at 79% versus 50%, translating into an OR of 4.49 (P = .050).
The authors acknowledged that the study was limited by differences in treatment regimens and centers, and a limited ability to adjust for patient characteristics, due to the study’s reliance on only those covariates published across different studies. Head-to-head comparison trials are needed to confirm their results, they noted.
Nonetheless, “[w]e believe that the improved efficacy of ponatinib combined with chemotherapy for newly diagnosed Ph+ALL might prevent the need for allogeneic SCT,” they wrote.
Ariad Pharmaceuticals funded the study. Dr. Jabbour and three coauthors reported research funding from the company; other study authors reported employment or other financial relationships with Ariad or its parent company Takeda.
SOURCE: Jabbour E et al. Clin Lymphoma Myeloma Leuk. 2018 18(4):257-65.
Every generation aspires to be better than its predecessors, and for the third-generation tyrosine kinase inhibitor ponatinib (Iclusig), that might just be true, investigators claim.
A retrospective analysis comparing clinical trial outcomes for patients with newly diagnosed acute lymphoblastic leukemia positive for the Philadelphia chromosome (Ph+ALL) suggests that first-line ponatinib offers modestly better complete molecular response (CMR) rates and 3-year overall survival (OS) than either first-generation TKIs such as imatinib (Gleevec) or second-generation agents such as dasatinib (Sprycel) and nilotinib (Tasigna).
“Although only 1 relevant study of ponatinib combined with chemotherapy in Ph+ALL has been reported and our ability to adjust for baseline patient characteristics was limited, the results suggest that ponatinib combined with chemotherapy might represent a more effective front-line treatment option than chemotherapy combined with an earlier generation TKI for patients with newly diagnosed Ph+ALL, including those who cannot or choose not to undergo [stem cell transplant],” wrote Elias Jabbour, MD, of the University of Texas MD Anderson Cancer Center in Houston, and his colleagues.
They based their conclusions on a meta-regression analysis of 25 studies looking at first- or second-generation TKIs and one study of ponatinib as frontline therapy for patients with Ph+ALL. They described their results in Clinical Lymphoma, Myeloma & Leukemia.
The investigators created pooled estimates of outcomes from studies of earlier-generation TKIs plus combination chemotherapy using a random-effects meta-analysis method. For the sole ponatinib study – a single-arm trial of combination chemotherapy plus ponatinib – they used a binomial distribution method to calculate 95% confidence intervals (CI). The method essentially estimates the probability of success or failure of a repeated experiment.
They found that 79% of patients in the ponatinib trial achieved a CMR, compared with 34% of patients treated with earlier generation TKIs plus chemotherapy. This translates into an odds ratio (OR) for CMR with ponatinib of 6.09 (P = .034).
Two-year OS rates were 83% with ponatinib versus 58% for all patients treated with other TKIs. Although the OR (3.70) seemed to run in favor of ponatinib, the difference was not statistically significant (P = .062).
For 3-year OS, however, ponatinib was superior to the pooled data for the other TKIs, at 79% versus 50%, translating into an OR of 4.49 (P = .050).
The authors acknowledged that the study was limited by differences in treatment regimens and centers, and a limited ability to adjust for patient characteristics, due to the study’s reliance on only those covariates published across different studies. Head-to-head comparison trials are needed to confirm their results, they noted.
Nonetheless, “[w]e believe that the improved efficacy of ponatinib combined with chemotherapy for newly diagnosed Ph+ALL might prevent the need for allogeneic SCT,” they wrote.
Ariad Pharmaceuticals funded the study. Dr. Jabbour and three coauthors reported research funding from the company; other study authors reported employment or other financial relationships with Ariad or its parent company Takeda.
SOURCE: Jabbour E et al. Clin Lymphoma Myeloma Leuk. 2018 18(4):257-65.
Every generation aspires to be better than its predecessors, and for the third-generation tyrosine kinase inhibitor ponatinib (Iclusig), that might just be true, investigators claim.
A retrospective analysis comparing clinical trial outcomes for patients with newly diagnosed acute lymphoblastic leukemia positive for the Philadelphia chromosome (Ph+ALL) suggests that first-line ponatinib offers modestly better complete molecular response (CMR) rates and 3-year overall survival (OS) than either first-generation TKIs such as imatinib (Gleevec) or second-generation agents such as dasatinib (Sprycel) and nilotinib (Tasigna).
“Although only 1 relevant study of ponatinib combined with chemotherapy in Ph+ALL has been reported and our ability to adjust for baseline patient characteristics was limited, the results suggest that ponatinib combined with chemotherapy might represent a more effective front-line treatment option than chemotherapy combined with an earlier generation TKI for patients with newly diagnosed Ph+ALL, including those who cannot or choose not to undergo [stem cell transplant],” wrote Elias Jabbour, MD, of the University of Texas MD Anderson Cancer Center in Houston, and his colleagues.
They based their conclusions on a meta-regression analysis of 25 studies looking at first- or second-generation TKIs and one study of ponatinib as frontline therapy for patients with Ph+ALL. They described their results in Clinical Lymphoma, Myeloma & Leukemia.
The investigators created pooled estimates of outcomes from studies of earlier-generation TKIs plus combination chemotherapy using a random-effects meta-analysis method. For the sole ponatinib study – a single-arm trial of combination chemotherapy plus ponatinib – they used a binomial distribution method to calculate 95% confidence intervals (CI). The method essentially estimates the probability of success or failure of a repeated experiment.
They found that 79% of patients in the ponatinib trial achieved a CMR, compared with 34% of patients treated with earlier generation TKIs plus chemotherapy. This translates into an odds ratio (OR) for CMR with ponatinib of 6.09 (P = .034).
Two-year OS rates were 83% with ponatinib versus 58% for all patients treated with other TKIs. Although the OR (3.70) seemed to run in favor of ponatinib, the difference was not statistically significant (P = .062).
For 3-year OS, however, ponatinib was superior to the pooled data for the other TKIs, at 79% versus 50%, translating into an OR of 4.49 (P = .050).
The authors acknowledged that the study was limited by differences in treatment regimens and centers, and a limited ability to adjust for patient characteristics, due to the study’s reliance on only those covariates published across different studies. Head-to-head comparison trials are needed to confirm their results, they noted.
Nonetheless, “[w]e believe that the improved efficacy of ponatinib combined with chemotherapy for newly diagnosed Ph+ALL might prevent the need for allogeneic SCT,” they wrote.
Ariad Pharmaceuticals funded the study. Dr. Jabbour and three coauthors reported research funding from the company; other study authors reported employment or other financial relationships with Ariad or its parent company Takeda.
SOURCE: Jabbour E et al. Clin Lymphoma Myeloma Leuk. 2018 18(4):257-65.
FROM CLINICAL LYMPHOMA, MYELOMA & LEUKEMIA
Key clinical point:
Major finding: Complete molecular response rates and 3-year overall survival were better with ponatinib in combination with chemotherapy.
Study details: Meta-regression analysis of 26 studies of TKIs in combination with chemotherapy as first-line therapy for patients with Ph+ALL.
Disclosures: Ariad Pharmaceuticals funded the study. Dr. Jabbour and three coauthors reported research funding from the company; other study authors reported employment or other financial relationships with Ariad or its parent company, Takeda.
Source: Jabbour E et al. Clin Lymphoma Myeloma Leuk. 2018 18(4):257-65.