User login
Strategies for managing medication-induced hyperprolactinemia
Ms. E, age 23, presents to your office for a routine visit for management of bipolar I disorder and posttraumatic stress disorder with comorbid type 2 diabetes mellitus. She currently is taking
Ms. E has a history of self-discontinuing medication when adverse events occur. She has been hospitalized twice for psychosis and suicide attempts. Past psychotropic medications that have been discontinued due to adverse effects include ziprasidone (mild abnormal lip movement), olanzapine (ineffective and drowsy), valproic acid (tremor and abdominal discomfort), lithium (rash), and aripiprazole (increased fasting blood sugar and labile mood).
At her appointment today, Ms. E says she is concerned because she has been experiencing galactorrhea for the past 4 weeks. Her prolactin level is 14.4 ng/mL; a normal level for a woman who is not pregnant is <25 ng/mL. However, a repeat prolactin level is obtained, and is found to be elevated at 38 ng/mL.
Prolactin, a polypeptide hormone that is secreted from the pituitary gland, has many functions, including involvement in the synthesis and maintenance of breast milk production, in reproductive behavior, and in luteal function.1,2 Hyperprolactinemia—an elevated prolactin level—is a common endocrinologic disorder of the hypothalamic–pituitary–axis.3 Children, adolescents, premenopausal women, and women in the perinatal period are more vulnerable to medication-induced hyperprolactinemia.4 If not asymptomatic, patients with hyperprolactinemia may experience amenorrhea, galactorrhea, hypogonadism, sexual dysfunction, or infertility.1,4 Chronic hyperprolactinemia may increase the risk for long-term complications, such as decreased bone mineral density and osteoporosis, although available evidence has conflicting findings.1

Hyperprolactinemia is diagnosed by a prolactin concentration above the upper reference range.3 Various hormones and neurotransmitters can impact inhibition or stimulation of prolactin release.5 For example, dopamine tonically inhibits prolactin release and synthesis, whereas estrogen stimulates prolactin secretion.1,5 Prolactin also can be elevated under several physiologic and pathologic conditions, such as during stressful situations, meals, or sexual activity.1,5 A prolactin level >250 ng/mL is usually indicative of a prolactinoma; however, some medications, such as strong D2 receptor antagonists (eg, risperidone, haloperidol), can cause significant elevation without evidence of prolactinoma.3 In the absence of a tumor, medications are often identified as the cause of hyperprolactinemia.3 According to the Endocrinology Society clinical practice guideline, medication-induced elevated prolactin levels are typically between 25 to 100 ng/mL.3
Medication-induced hyperprolactinemia
Antipsychotics, antidepressants, hormonal preparations, antihypertensives, and gastrointestinal agents have been associated with hyperprolactinemia (Table 11,3,5-11). These medication classes increase prolactin by decreasing dopamine, which facilitates disinhibition of prolactin synthesis and release, or increasing prolactin stimulating hormones, such as serotonin or estrogen.5
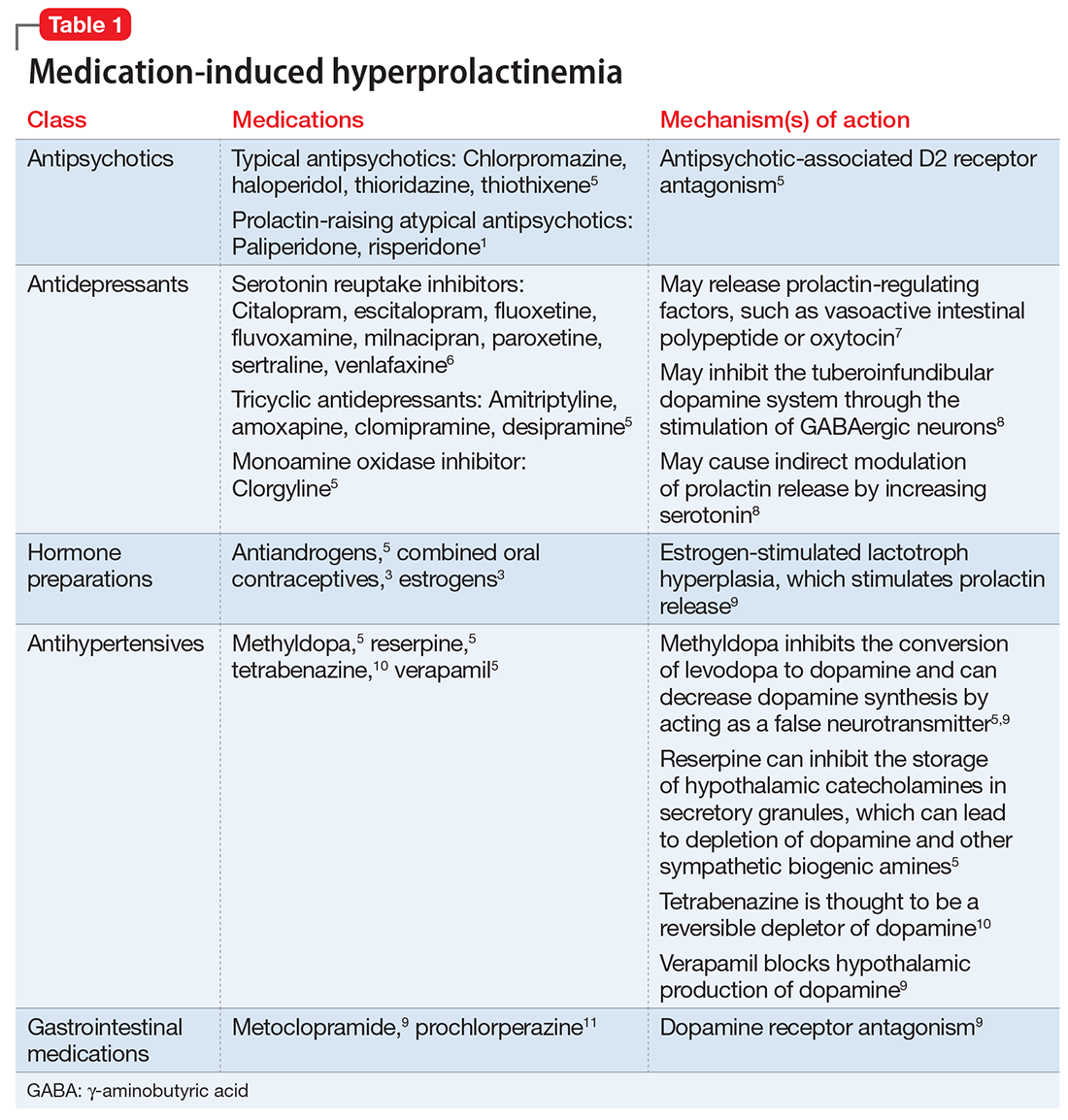
Antipsychotics are the most common medication-related cause of hyperprolactinemia.3 Typical antipsychotics are more likely to cause hyperprolactinemia than atypical antipsychotics; the incidence among patients taking typical antipsychotics is 40% to 90%.3 Atypical antipsychotics, except risperidone and paliperidone, are considered to cause less endocrinologic effects than typical antipsychotics through various mechanisms: serotonergic receptor antagonism, fast dissociation from D2 receptors, D2 receptor partial agonism, and preferential binding of D3 vs D2 receptors.1,5 By having transient D2 receptor association, clozapine and quetiapine are considered to have less risk of hyperprolactinemia compared with other atypical antipsychotics.1,5 Aripiprazole, brexpiprazole, and cariprazine are partial D2 receptor agonists, and cariprazine is the only agent that exhibits preferential binding to D3 receptors.12,13 Based on limited data, brexpiprazole and cariprazine may have prolactin-sparing properties given their partial D2 receptor agonism.12,13 However, one study found increased prolactin levels in some patients after treatment with brexpiprazole, 4 mg/d.14 Similarly, another study found that cariprazine could increase prolactin levels as much as 4.1 ng/mL, depending on the dose.15 Except for aripiprazole, brexpiprazole, cariprazine, and clozapine, all other atypical antipsychotics marketed in the United States have a standard warning in the package insert regarding prolactin elevations.1,16,17
Because antidepressants are less well-studied as a cause of medication-induced hyperprolactinemia, drawing definitive conclusions regarding incidence rates is limited, but the incidence seems to be fairly low.6,18 A French pharmacovigilance study found that of 182,836 spontaneous adverse drug events reported between 1985 and 2009, there were 159 reports of selective serotonin reuptake inhibitors (SSRIs) inducing hyperprolactinemia.6 F
Mirtazapine and bupropion have been found to be prolactin-neutral.5 Bupropion also has been reported to decrease prolactin levels, potentially via its ability to block dopamine reuptake.19
Managing medication-induced hyperprolactinemia
Screening for and identifying clinically significant hyperprolactinemia is critical, because adverse effects of medications can lead to nonadherence and clinical decompensation.20 Patients must be informed of potential symptoms of hyperprolactinemia, and clinicians should inquire about such symptoms at each visit. Routine monitoring of prolactin levels in asymptomatic patients is not necessary, because the Endocrine Society Clinical Practice Guideline does not recommend treating patients with asymptomatic medication-induced hyperprolactinemia.3
In patients who report hyperprolactinemia symptoms, clinicians should review the patient’s prescribed medications and past medical history (eg, chronic renal failure, hypothyroidism) for potential causes or exacerbations, and address these factors accordingly.3 Order a measurement of prolactin level. A patient with a prolactin level >100 ng/mL should be referred to Endocrinology to rule out prolactinoma.1
If a patient’s prolactin level is between 25 and 100 ng/mL, review the patient’s medications (Table 11,3,5-11), because prolactin levels within this range usually signal a medication-induced cause.3 For patients with antipsychotic-induced hyperprolactinemia, there are several management strategies (Table 21,3,4,9,16,17,21-27):
- Watch and wait may be warranted when the patient is experiencing mild hyperprolactinemia symptoms.
- Discontinue. If the patient can be maintained without an antipsychotic, discontinuing the antipsychotic would be a first-line option.3
- Reduce the dose. Reducing the antipsychotic dose may be the preferred strategy for patients with moderate to severe hyperprolactinemia symptoms who responded to the antipsychotic and do not wish to start adjunctive therapy.4
- Switching to a prolactin-sparing antipsychotic may help normalize prolactin levels and may be preferred when the risk of relapse is low.3 Dopamine agonists can treat medication-induced hyperprolactinemia, but may worsen psychiatric symptoms.28,29 Therefore, this may be the preferred strategy if the offending medication cannot be discontinued or switched, or if the patient has a comorbid prolactinoma.

Less data exist on managing hyperprolactinemia that is induced by a medication other than an antipsychotic; however, it seems reasonable that the same strategies could be implemented. Specifically, for SSRI–induced hyperprolactinemia, if clinically appropriate, switching to or adding an alternative antidepressant that may be prolactin-sparing, such as mirtazapine or bupropion, could be attempted.8 One study found that fluoxetine-induced galactorrhea ceased within 10 days of discontinuing the medication.30
CASE CONTINUED
Because Ms. E has been on the same medication regimen for 3 years and recently developed galactorrhea, it seems unlikely that her hyperprolactinemia is medication-induced. However, a tumor-related cause is less likely because the prolactin level is <100 ng/mL. Based on the literature, the only possible medication-induced cause of her galactorrhea is risperidone. Ms. E agrees to a trial of adjunctive oral aripiprazole, 5 mg/d, with close monitoring of her type 2 diabetes mellitus. Because of the long elimination half-life of aripiprazole, 1 month is required to monitor for improvement in galactorrhea. Ms. E is advised to use breast pads as a nonpharmacologic strategy in the interim. After 1 month of treatment, Ms. E denies galactorrhea symptoms and no longer requires the use of breast pads.
1. Peuskens J, Pani L, Detraux J, et al. The effects of novel and newly approved antipsychotics on serum prolactin levels: a comprehensive review. CNS Drugs.2014;28(5):421-453.
2. Freeman ME, Kanyicska B, Lerant A, et al. Prolactin: structure, function, and regulation of secretion. Physiol Rev. 2000;80(4):1523-1631.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical practice guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
4. Bostwick JR, Guthrie SK, Ellingrod VL. Antipsychotic-induced hyperprolactinemia. Pharmacotherapy. 2009;29(1):64-73.
5. La Torre D, Falorni A. Pharmacological causes of hyperprolactinemia. Ther Clin Risk Manag. 2007;3(5):929-951.
6. Petit A, Piednoir D, Germain ML, et al. Drug-induced hyperprolactinemia: a case-non-case study from the national pharmacovigilance database [in French]. Therapie. 2003;58(2):159-163.
7. Emiliano AB, Fudge JL. From galactorrhea to osteopenia: rethinking serotonin-prolactin interactions. Neuropsychopharmacology. 2004;29(5):833-846.
8. Coker F, Taylor D. Antidepressant-induced hyperprolactinaemia: incidence, mechanisms and management. CNS Drugs. 2010;24(7):563-574.
9. Molitch ME. Medication induced hyperprolactinemia. Mayo Clin Proc. 2005;80(8):1050-1057.
10. Xenazine (tetrabenazine) [package insert]. Washington, DC: Prestwick Pharmaceuticals, Inc.; 2008.
11. Peña KS, Rosenfeld JA. Evaluation and treatment of galactorrhea. Am Fam Physician 2001;63(9):1763-1770.
12. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
13. Das S, Barnwal P, Winston AB, et al. Brexpiprazole: so far so good. Ther Adv Psychopharmacol. 2016;6(1):39-54.
14. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
15. Durgam S, Earley W, Guo H, et al. Efficacy and safety of adjunctive cariprazine in inadequate responders to antidepressants: a randomized, double-blind, placebo-controlled study in adult patients with major depressive disorder. J Clin Pscyhiatry. 2016;77(3):371-378.
16. Rexulti (brexpiprazole) [package insert]. Tokyo, Japan: Otsuka Pharmaceuticals Inc.; 2015.
17. Cariprazine (Vraylar) [package insert]. Parsippany, New Jersey: Actavis Pharmacueitcals Inc.; 2015.
18. Marken PA, Haykal RF, Fisher JN. Management of psychotropic-induced hyperprolactinemia. Clin Pharm. 1992;11(10):851-856.
19. Meltzer HY, Fang VS, Tricou BJ, et al. Effect of antidepressants on neuroendocrine axis in humans. Adv Biochem Psychopharmacol. 1982;32:303-316.
20. Tsuboi T, Bies RR, Suzuki T, et al. Hyperprolactinemia and estimated dopamine D2 receptor occupancy in patients with schizophrenia: analysis of the CATIE data. Prog Neuropsychopharmacol Biol Psychiatry. 2013;45:178-182.
21. Lee BH, Kim YK, Park SH. Using aripiprazole to resolve antipsychotic-induced symptomatic hyperprolactinemia: a pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(4):714-717.
22. Lu ML, Shen WW, Chen CH. Time course of the changes in antipsychotic-induced hyperprolactinemia following the switch to aripiprazole. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1978-1981.
23. Mendhekar DN, Andrade C. Galactorrhea with aripiprazole. Can J Psychiatry. 2005;50(4):243.
24. Joseph SP. Aripiprazole induced hyperprolactinemia in a young female with delusional disorder. Indian J Psychol Med. 2016;38(3):260-262.
25. Meng M, Li W, Zhang S, et al. Using aripiprazole to reduce antipsychotic-induced hyperprolactinemia: meta-analysis of currently available randomized controlled trials. Shaghai Arch Psychiatry. 2015;27(1):4-17.
26. Tollin SR. Use of the dopamine agonists bromocriptine and cabergoline in the management of risperidone induced hyperprolactinemia in patients with psychotic disorders. J Endocrinol Invest. 2000;23(11):765-70.
27. Yuan HN, Wang CY, Sze CW, et al. A randomized, crossover comparison of herbal medicine and bromocriptine against risperidone-induced hyperprolactinemia in patients with schizophrenia. J Clin Psychopharmacol. 2008;28(3):264-370.
28. Chang SC, Chen CH, Lu ML. Cabergoline-induced psychotic exacerbation in schizophrenic patients. General Hospital Psychiatry. 2008;30(4):378-380.
29. Ishitobi M, Kosaka H, Shukunami K, et al. Adjunctive treatment with low-dosage pramipexole for risperidone-associated hyperprolactinemia and sexual dysfunction in a male patient with schizophrenia. J Clin Psychopharmacol 2011;31(2):243-245.
30. Peterson MC. Reversible galactorrhea and prolactin elevation related to fluoxetine use. Mayo Clin Proc. 2001;76(2):215-216.
Ms. E, age 23, presents to your office for a routine visit for management of bipolar I disorder and posttraumatic stress disorder with comorbid type 2 diabetes mellitus. She currently is taking
Ms. E has a history of self-discontinuing medication when adverse events occur. She has been hospitalized twice for psychosis and suicide attempts. Past psychotropic medications that have been discontinued due to adverse effects include ziprasidone (mild abnormal lip movement), olanzapine (ineffective and drowsy), valproic acid (tremor and abdominal discomfort), lithium (rash), and aripiprazole (increased fasting blood sugar and labile mood).
At her appointment today, Ms. E says she is concerned because she has been experiencing galactorrhea for the past 4 weeks. Her prolactin level is 14.4 ng/mL; a normal level for a woman who is not pregnant is <25 ng/mL. However, a repeat prolactin level is obtained, and is found to be elevated at 38 ng/mL.
Prolactin, a polypeptide hormone that is secreted from the pituitary gland, has many functions, including involvement in the synthesis and maintenance of breast milk production, in reproductive behavior, and in luteal function.1,2 Hyperprolactinemia—an elevated prolactin level—is a common endocrinologic disorder of the hypothalamic–pituitary–axis.3 Children, adolescents, premenopausal women, and women in the perinatal period are more vulnerable to medication-induced hyperprolactinemia.4 If not asymptomatic, patients with hyperprolactinemia may experience amenorrhea, galactorrhea, hypogonadism, sexual dysfunction, or infertility.1,4 Chronic hyperprolactinemia may increase the risk for long-term complications, such as decreased bone mineral density and osteoporosis, although available evidence has conflicting findings.1
Hyperprolactinemia is diagnosed by a prolactin concentration above the upper reference range.3 Various hormones and neurotransmitters can impact inhibition or stimulation of prolactin release.5 For example, dopamine tonically inhibits prolactin release and synthesis, whereas estrogen stimulates prolactin secretion.1,5 Prolactin also can be elevated under several physiologic and pathologic conditions, such as during stressful situations, meals, or sexual activity.1,5 A prolactin level >250 ng/mL is usually indicative of a prolactinoma; however, some medications, such as strong D2 receptor antagonists (eg, risperidone, haloperidol), can cause significant elevation without evidence of prolactinoma.3 In the absence of a tumor, medications are often identified as the cause of hyperprolactinemia.3 According to the Endocrinology Society clinical practice guideline, medication-induced elevated prolactin levels are typically between 25 to 100 ng/mL.3
Medication-induced hyperprolactinemia
Antipsychotics, antidepressants, hormonal preparations, antihypertensives, and gastrointestinal agents have been associated with hyperprolactinemia (Table 11,3,5-11). These medication classes increase prolactin by decreasing dopamine, which facilitates disinhibition of prolactin synthesis and release, or increasing prolactin stimulating hormones, such as serotonin or estrogen.5
Antipsychotics are the most common medication-related cause of hyperprolactinemia.3 Typical antipsychotics are more likely to cause hyperprolactinemia than atypical antipsychotics; the incidence among patients taking typical antipsychotics is 40% to 90%.3 Atypical antipsychotics, except risperidone and paliperidone, are considered to cause less endocrinologic effects than typical antipsychotics through various mechanisms: serotonergic receptor antagonism, fast dissociation from D2 receptors, D2 receptor partial agonism, and preferential binding of D3 vs D2 receptors.1,5 By having transient D2 receptor association, clozapine and quetiapine are considered to have less risk of hyperprolactinemia compared with other atypical antipsychotics.1,5 Aripiprazole, brexpiprazole, and cariprazine are partial D2 receptor agonists, and cariprazine is the only agent that exhibits preferential binding to D3 receptors.12,13 Based on limited data, brexpiprazole and cariprazine may have prolactin-sparing properties given their partial D2 receptor agonism.12,13 However, one study found increased prolactin levels in some patients after treatment with brexpiprazole, 4 mg/d.14 Similarly, another study found that cariprazine could increase prolactin levels as much as 4.1 ng/mL, depending on the dose.15 Except for aripiprazole, brexpiprazole, cariprazine, and clozapine, all other atypical antipsychotics marketed in the United States have a standard warning in the package insert regarding prolactin elevations.1,16,17
Because antidepressants are less well-studied as a cause of medication-induced hyperprolactinemia, drawing definitive conclusions regarding incidence rates is limited, but the incidence seems to be fairly low.6,18 A French pharmacovigilance study found that of 182,836 spontaneous adverse drug events reported between 1985 and 2009, there were 159 reports of selective serotonin reuptake inhibitors (SSRIs) inducing hyperprolactinemia.6 F
Mirtazapine and bupropion have been found to be prolactin-neutral.5 Bupropion also has been reported to decrease prolactin levels, potentially via its ability to block dopamine reuptake.19
Managing medication-induced hyperprolactinemia
Screening for and identifying clinically significant hyperprolactinemia is critical, because adverse effects of medications can lead to nonadherence and clinical decompensation.20 Patients must be informed of potential symptoms of hyperprolactinemia, and clinicians should inquire about such symptoms at each visit. Routine monitoring of prolactin levels in asymptomatic patients is not necessary, because the Endocrine Society Clinical Practice Guideline does not recommend treating patients with asymptomatic medication-induced hyperprolactinemia.3
In patients who report hyperprolactinemia symptoms, clinicians should review the patient’s prescribed medications and past medical history (eg, chronic renal failure, hypothyroidism) for potential causes or exacerbations, and address these factors accordingly.3 Order a measurement of prolactin level. A patient with a prolactin level >100 ng/mL should be referred to Endocrinology to rule out prolactinoma.1
If a patient’s prolactin level is between 25 and 100 ng/mL, review the patient’s medications (Table 11,3,5-11), because prolactin levels within this range usually signal a medication-induced cause.3 For patients with antipsychotic-induced hyperprolactinemia, there are several management strategies (Table 21,3,4,9,16,17,21-27):
- Watch and wait may be warranted when the patient is experiencing mild hyperprolactinemia symptoms.
- Discontinue. If the patient can be maintained without an antipsychotic, discontinuing the antipsychotic would be a first-line option.3
- Reduce the dose. Reducing the antipsychotic dose may be the preferred strategy for patients with moderate to severe hyperprolactinemia symptoms who responded to the antipsychotic and do not wish to start adjunctive therapy.4
- Switching to a prolactin-sparing antipsychotic may help normalize prolactin levels and may be preferred when the risk of relapse is low.3 Dopamine agonists can treat medication-induced hyperprolactinemia, but may worsen psychiatric symptoms.28,29 Therefore, this may be the preferred strategy if the offending medication cannot be discontinued or switched, or if the patient has a comorbid prolactinoma.
Less data exist on managing hyperprolactinemia that is induced by a medication other than an antipsychotic; however, it seems reasonable that the same strategies could be implemented. Specifically, for SSRI–induced hyperprolactinemia, if clinically appropriate, switching to or adding an alternative antidepressant that may be prolactin-sparing, such as mirtazapine or bupropion, could be attempted.8 One study found that fluoxetine-induced galactorrhea ceased within 10 days of discontinuing the medication.30
CASE CONTINUED
Because Ms. E has been on the same medication regimen for 3 years and recently developed galactorrhea, it seems unlikely that her hyperprolactinemia is medication-induced. However, a tumor-related cause is less likely because the prolactin level is <100 ng/mL. Based on the literature, the only possible medication-induced cause of her galactorrhea is risperidone. Ms. E agrees to a trial of adjunctive oral aripiprazole, 5 mg/d, with close monitoring of her type 2 diabetes mellitus. Because of the long elimination half-life of aripiprazole, 1 month is required to monitor for improvement in galactorrhea. Ms. E is advised to use breast pads as a nonpharmacologic strategy in the interim. After 1 month of treatment, Ms. E denies galactorrhea symptoms and no longer requires the use of breast pads.
Ms. E, age 23, presents to your office for a routine visit for management of bipolar I disorder and posttraumatic stress disorder with comorbid type 2 diabetes mellitus. She currently is taking
Ms. E has a history of self-discontinuing medication when adverse events occur. She has been hospitalized twice for psychosis and suicide attempts. Past psychotropic medications that have been discontinued due to adverse effects include ziprasidone (mild abnormal lip movement), olanzapine (ineffective and drowsy), valproic acid (tremor and abdominal discomfort), lithium (rash), and aripiprazole (increased fasting blood sugar and labile mood).
At her appointment today, Ms. E says she is concerned because she has been experiencing galactorrhea for the past 4 weeks. Her prolactin level is 14.4 ng/mL; a normal level for a woman who is not pregnant is <25 ng/mL. However, a repeat prolactin level is obtained, and is found to be elevated at 38 ng/mL.
Prolactin, a polypeptide hormone that is secreted from the pituitary gland, has many functions, including involvement in the synthesis and maintenance of breast milk production, in reproductive behavior, and in luteal function.1,2 Hyperprolactinemia—an elevated prolactin level—is a common endocrinologic disorder of the hypothalamic–pituitary–axis.3 Children, adolescents, premenopausal women, and women in the perinatal period are more vulnerable to medication-induced hyperprolactinemia.4 If not asymptomatic, patients with hyperprolactinemia may experience amenorrhea, galactorrhea, hypogonadism, sexual dysfunction, or infertility.1,4 Chronic hyperprolactinemia may increase the risk for long-term complications, such as decreased bone mineral density and osteoporosis, although available evidence has conflicting findings.1
Hyperprolactinemia is diagnosed by a prolactin concentration above the upper reference range.3 Various hormones and neurotransmitters can impact inhibition or stimulation of prolactin release.5 For example, dopamine tonically inhibits prolactin release and synthesis, whereas estrogen stimulates prolactin secretion.1,5 Prolactin also can be elevated under several physiologic and pathologic conditions, such as during stressful situations, meals, or sexual activity.1,5 A prolactin level >250 ng/mL is usually indicative of a prolactinoma; however, some medications, such as strong D2 receptor antagonists (eg, risperidone, haloperidol), can cause significant elevation without evidence of prolactinoma.3 In the absence of a tumor, medications are often identified as the cause of hyperprolactinemia.3 According to the Endocrinology Society clinical practice guideline, medication-induced elevated prolactin levels are typically between 25 to 100 ng/mL.3
Medication-induced hyperprolactinemia
Antipsychotics, antidepressants, hormonal preparations, antihypertensives, and gastrointestinal agents have been associated with hyperprolactinemia (Table 11,3,5-11). These medication classes increase prolactin by decreasing dopamine, which facilitates disinhibition of prolactin synthesis and release, or increasing prolactin stimulating hormones, such as serotonin or estrogen.5
Antipsychotics are the most common medication-related cause of hyperprolactinemia.3 Typical antipsychotics are more likely to cause hyperprolactinemia than atypical antipsychotics; the incidence among patients taking typical antipsychotics is 40% to 90%.3 Atypical antipsychotics, except risperidone and paliperidone, are considered to cause less endocrinologic effects than typical antipsychotics through various mechanisms: serotonergic receptor antagonism, fast dissociation from D2 receptors, D2 receptor partial agonism, and preferential binding of D3 vs D2 receptors.1,5 By having transient D2 receptor association, clozapine and quetiapine are considered to have less risk of hyperprolactinemia compared with other atypical antipsychotics.1,5 Aripiprazole, brexpiprazole, and cariprazine are partial D2 receptor agonists, and cariprazine is the only agent that exhibits preferential binding to D3 receptors.12,13 Based on limited data, brexpiprazole and cariprazine may have prolactin-sparing properties given their partial D2 receptor agonism.12,13 However, one study found increased prolactin levels in some patients after treatment with brexpiprazole, 4 mg/d.14 Similarly, another study found that cariprazine could increase prolactin levels as much as 4.1 ng/mL, depending on the dose.15 Except for aripiprazole, brexpiprazole, cariprazine, and clozapine, all other atypical antipsychotics marketed in the United States have a standard warning in the package insert regarding prolactin elevations.1,16,17
Because antidepressants are less well-studied as a cause of medication-induced hyperprolactinemia, drawing definitive conclusions regarding incidence rates is limited, but the incidence seems to be fairly low.6,18 A French pharmacovigilance study found that of 182,836 spontaneous adverse drug events reported between 1985 and 2009, there were 159 reports of selective serotonin reuptake inhibitors (SSRIs) inducing hyperprolactinemia.6 F
Mirtazapine and bupropion have been found to be prolactin-neutral.5 Bupropion also has been reported to decrease prolactin levels, potentially via its ability to block dopamine reuptake.19
Managing medication-induced hyperprolactinemia
Screening for and identifying clinically significant hyperprolactinemia is critical, because adverse effects of medications can lead to nonadherence and clinical decompensation.20 Patients must be informed of potential symptoms of hyperprolactinemia, and clinicians should inquire about such symptoms at each visit. Routine monitoring of prolactin levels in asymptomatic patients is not necessary, because the Endocrine Society Clinical Practice Guideline does not recommend treating patients with asymptomatic medication-induced hyperprolactinemia.3
In patients who report hyperprolactinemia symptoms, clinicians should review the patient’s prescribed medications and past medical history (eg, chronic renal failure, hypothyroidism) for potential causes or exacerbations, and address these factors accordingly.3 Order a measurement of prolactin level. A patient with a prolactin level >100 ng/mL should be referred to Endocrinology to rule out prolactinoma.1
If a patient’s prolactin level is between 25 and 100 ng/mL, review the patient’s medications (Table 11,3,5-11), because prolactin levels within this range usually signal a medication-induced cause.3 For patients with antipsychotic-induced hyperprolactinemia, there are several management strategies (Table 21,3,4,9,16,17,21-27):
- Watch and wait may be warranted when the patient is experiencing mild hyperprolactinemia symptoms.
- Discontinue. If the patient can be maintained without an antipsychotic, discontinuing the antipsychotic would be a first-line option.3
- Reduce the dose. Reducing the antipsychotic dose may be the preferred strategy for patients with moderate to severe hyperprolactinemia symptoms who responded to the antipsychotic and do not wish to start adjunctive therapy.4
- Switching to a prolactin-sparing antipsychotic may help normalize prolactin levels and may be preferred when the risk of relapse is low.3 Dopamine agonists can treat medication-induced hyperprolactinemia, but may worsen psychiatric symptoms.28,29 Therefore, this may be the preferred strategy if the offending medication cannot be discontinued or switched, or if the patient has a comorbid prolactinoma.
Less data exist on managing hyperprolactinemia that is induced by a medication other than an antipsychotic; however, it seems reasonable that the same strategies could be implemented. Specifically, for SSRI–induced hyperprolactinemia, if clinically appropriate, switching to or adding an alternative antidepressant that may be prolactin-sparing, such as mirtazapine or bupropion, could be attempted.8 One study found that fluoxetine-induced galactorrhea ceased within 10 days of discontinuing the medication.30
CASE CONTINUED
Because Ms. E has been on the same medication regimen for 3 years and recently developed galactorrhea, it seems unlikely that her hyperprolactinemia is medication-induced. However, a tumor-related cause is less likely because the prolactin level is <100 ng/mL. Based on the literature, the only possible medication-induced cause of her galactorrhea is risperidone. Ms. E agrees to a trial of adjunctive oral aripiprazole, 5 mg/d, with close monitoring of her type 2 diabetes mellitus. Because of the long elimination half-life of aripiprazole, 1 month is required to monitor for improvement in galactorrhea. Ms. E is advised to use breast pads as a nonpharmacologic strategy in the interim. After 1 month of treatment, Ms. E denies galactorrhea symptoms and no longer requires the use of breast pads.
1. Peuskens J, Pani L, Detraux J, et al. The effects of novel and newly approved antipsychotics on serum prolactin levels: a comprehensive review. CNS Drugs.2014;28(5):421-453.
2. Freeman ME, Kanyicska B, Lerant A, et al. Prolactin: structure, function, and regulation of secretion. Physiol Rev. 2000;80(4):1523-1631.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical practice guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
4. Bostwick JR, Guthrie SK, Ellingrod VL. Antipsychotic-induced hyperprolactinemia. Pharmacotherapy. 2009;29(1):64-73.
5. La Torre D, Falorni A. Pharmacological causes of hyperprolactinemia. Ther Clin Risk Manag. 2007;3(5):929-951.
6. Petit A, Piednoir D, Germain ML, et al. Drug-induced hyperprolactinemia: a case-non-case study from the national pharmacovigilance database [in French]. Therapie. 2003;58(2):159-163.
7. Emiliano AB, Fudge JL. From galactorrhea to osteopenia: rethinking serotonin-prolactin interactions. Neuropsychopharmacology. 2004;29(5):833-846.
8. Coker F, Taylor D. Antidepressant-induced hyperprolactinaemia: incidence, mechanisms and management. CNS Drugs. 2010;24(7):563-574.
9. Molitch ME. Medication induced hyperprolactinemia. Mayo Clin Proc. 2005;80(8):1050-1057.
10. Xenazine (tetrabenazine) [package insert]. Washington, DC: Prestwick Pharmaceuticals, Inc.; 2008.
11. Peña KS, Rosenfeld JA. Evaluation and treatment of galactorrhea. Am Fam Physician 2001;63(9):1763-1770.
12. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
13. Das S, Barnwal P, Winston AB, et al. Brexpiprazole: so far so good. Ther Adv Psychopharmacol. 2016;6(1):39-54.
14. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
15. Durgam S, Earley W, Guo H, et al. Efficacy and safety of adjunctive cariprazine in inadequate responders to antidepressants: a randomized, double-blind, placebo-controlled study in adult patients with major depressive disorder. J Clin Pscyhiatry. 2016;77(3):371-378.
16. Rexulti (brexpiprazole) [package insert]. Tokyo, Japan: Otsuka Pharmaceuticals Inc.; 2015.
17. Cariprazine (Vraylar) [package insert]. Parsippany, New Jersey: Actavis Pharmacueitcals Inc.; 2015.
18. Marken PA, Haykal RF, Fisher JN. Management of psychotropic-induced hyperprolactinemia. Clin Pharm. 1992;11(10):851-856.
19. Meltzer HY, Fang VS, Tricou BJ, et al. Effect of antidepressants on neuroendocrine axis in humans. Adv Biochem Psychopharmacol. 1982;32:303-316.
20. Tsuboi T, Bies RR, Suzuki T, et al. Hyperprolactinemia and estimated dopamine D2 receptor occupancy in patients with schizophrenia: analysis of the CATIE data. Prog Neuropsychopharmacol Biol Psychiatry. 2013;45:178-182.
21. Lee BH, Kim YK, Park SH. Using aripiprazole to resolve antipsychotic-induced symptomatic hyperprolactinemia: a pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(4):714-717.
22. Lu ML, Shen WW, Chen CH. Time course of the changes in antipsychotic-induced hyperprolactinemia following the switch to aripiprazole. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1978-1981.
23. Mendhekar DN, Andrade C. Galactorrhea with aripiprazole. Can J Psychiatry. 2005;50(4):243.
24. Joseph SP. Aripiprazole induced hyperprolactinemia in a young female with delusional disorder. Indian J Psychol Med. 2016;38(3):260-262.
25. Meng M, Li W, Zhang S, et al. Using aripiprazole to reduce antipsychotic-induced hyperprolactinemia: meta-analysis of currently available randomized controlled trials. Shaghai Arch Psychiatry. 2015;27(1):4-17.
26. Tollin SR. Use of the dopamine agonists bromocriptine and cabergoline in the management of risperidone induced hyperprolactinemia in patients with psychotic disorders. J Endocrinol Invest. 2000;23(11):765-70.
27. Yuan HN, Wang CY, Sze CW, et al. A randomized, crossover comparison of herbal medicine and bromocriptine against risperidone-induced hyperprolactinemia in patients with schizophrenia. J Clin Psychopharmacol. 2008;28(3):264-370.
28. Chang SC, Chen CH, Lu ML. Cabergoline-induced psychotic exacerbation in schizophrenic patients. General Hospital Psychiatry. 2008;30(4):378-380.
29. Ishitobi M, Kosaka H, Shukunami K, et al. Adjunctive treatment with low-dosage pramipexole for risperidone-associated hyperprolactinemia and sexual dysfunction in a male patient with schizophrenia. J Clin Psychopharmacol 2011;31(2):243-245.
30. Peterson MC. Reversible galactorrhea and prolactin elevation related to fluoxetine use. Mayo Clin Proc. 2001;76(2):215-216.
1. Peuskens J, Pani L, Detraux J, et al. The effects of novel and newly approved antipsychotics on serum prolactin levels: a comprehensive review. CNS Drugs.2014;28(5):421-453.
2. Freeman ME, Kanyicska B, Lerant A, et al. Prolactin: structure, function, and regulation of secretion. Physiol Rev. 2000;80(4):1523-1631.
3. Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society Clinical practice guideline. J Clin Endocrinol Metab. 2011;96(2):273-288.
4. Bostwick JR, Guthrie SK, Ellingrod VL. Antipsychotic-induced hyperprolactinemia. Pharmacotherapy. 2009;29(1):64-73.
5. La Torre D, Falorni A. Pharmacological causes of hyperprolactinemia. Ther Clin Risk Manag. 2007;3(5):929-951.
6. Petit A, Piednoir D, Germain ML, et al. Drug-induced hyperprolactinemia: a case-non-case study from the national pharmacovigilance database [in French]. Therapie. 2003;58(2):159-163.
7. Emiliano AB, Fudge JL. From galactorrhea to osteopenia: rethinking serotonin-prolactin interactions. Neuropsychopharmacology. 2004;29(5):833-846.
8. Coker F, Taylor D. Antidepressant-induced hyperprolactinaemia: incidence, mechanisms and management. CNS Drugs. 2010;24(7):563-574.
9. Molitch ME. Medication induced hyperprolactinemia. Mayo Clin Proc. 2005;80(8):1050-1057.
10. Xenazine (tetrabenazine) [package insert]. Washington, DC: Prestwick Pharmaceuticals, Inc.; 2008.
11. Peña KS, Rosenfeld JA. Evaluation and treatment of galactorrhea. Am Fam Physician 2001;63(9):1763-1770.
12. Durgam S, Starace A, Li D, et al. The efficacy and tolerability of cariprazine in acute mania associated with bipolar I disorder: a phase II trial. Bipolar Disord. 2015;17(1):63-75.
13. Das S, Barnwal P, Winston AB, et al. Brexpiprazole: so far so good. Ther Adv Psychopharmacol. 2016;6(1):39-54.
14. Correll CU, Skuban A, Ouyang J, et al. Efficacy and safety of brexpiprazole for the treatment of acute schizophrenia: a 6-week randomized, double-blind, placebo-controlled trial. Am J Psychiatry. 2015;172(9):870-880.
15. Durgam S, Earley W, Guo H, et al. Efficacy and safety of adjunctive cariprazine in inadequate responders to antidepressants: a randomized, double-blind, placebo-controlled study in adult patients with major depressive disorder. J Clin Pscyhiatry. 2016;77(3):371-378.
16. Rexulti (brexpiprazole) [package insert]. Tokyo, Japan: Otsuka Pharmaceuticals Inc.; 2015.
17. Cariprazine (Vraylar) [package insert]. Parsippany, New Jersey: Actavis Pharmacueitcals Inc.; 2015.
18. Marken PA, Haykal RF, Fisher JN. Management of psychotropic-induced hyperprolactinemia. Clin Pharm. 1992;11(10):851-856.
19. Meltzer HY, Fang VS, Tricou BJ, et al. Effect of antidepressants on neuroendocrine axis in humans. Adv Biochem Psychopharmacol. 1982;32:303-316.
20. Tsuboi T, Bies RR, Suzuki T, et al. Hyperprolactinemia and estimated dopamine D2 receptor occupancy in patients with schizophrenia: analysis of the CATIE data. Prog Neuropsychopharmacol Biol Psychiatry. 2013;45:178-182.
21. Lee BH, Kim YK, Park SH. Using aripiprazole to resolve antipsychotic-induced symptomatic hyperprolactinemia: a pilot study. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(4):714-717.
22. Lu ML, Shen WW, Chen CH. Time course of the changes in antipsychotic-induced hyperprolactinemia following the switch to aripiprazole. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(8):1978-1981.
23. Mendhekar DN, Andrade C. Galactorrhea with aripiprazole. Can J Psychiatry. 2005;50(4):243.
24. Joseph SP. Aripiprazole induced hyperprolactinemia in a young female with delusional disorder. Indian J Psychol Med. 2016;38(3):260-262.
25. Meng M, Li W, Zhang S, et al. Using aripiprazole to reduce antipsychotic-induced hyperprolactinemia: meta-analysis of currently available randomized controlled trials. Shaghai Arch Psychiatry. 2015;27(1):4-17.
26. Tollin SR. Use of the dopamine agonists bromocriptine and cabergoline in the management of risperidone induced hyperprolactinemia in patients with psychotic disorders. J Endocrinol Invest. 2000;23(11):765-70.
27. Yuan HN, Wang CY, Sze CW, et al. A randomized, crossover comparison of herbal medicine and bromocriptine against risperidone-induced hyperprolactinemia in patients with schizophrenia. J Clin Psychopharmacol. 2008;28(3):264-370.
28. Chang SC, Chen CH, Lu ML. Cabergoline-induced psychotic exacerbation in schizophrenic patients. General Hospital Psychiatry. 2008;30(4):378-380.
29. Ishitobi M, Kosaka H, Shukunami K, et al. Adjunctive treatment with low-dosage pramipexole for risperidone-associated hyperprolactinemia and sexual dysfunction in a male patient with schizophrenia. J Clin Psychopharmacol 2011;31(2):243-245.
30. Peterson MC. Reversible galactorrhea and prolactin elevation related to fluoxetine use. Mayo Clin Proc. 2001;76(2):215-216.
Antipsychotics for obsessive-compulsive disorder: Weighing risks vs benefits
Mr. E, age 37, has a 20-year history of obsessive-compulsive disorder (OCD), with comorbid generalized anxiety disorder and hypertension. His medication regimen consists of

Box
Antipsychotics for OCD: What the guidelines recommend
The 2013 American Psychiatric Association (APA) obsessive-compulsive disorder (OCD) treatment guidelines include recommendations regarding the use of antipsychotics in patients who do not respond to first-line treatment with selective serotonin reuptake inhibitors (SSRIs) and/or cognitive-behavioral therapy (CBT). The APA recommends evaluating contributing factors, including comorbidities, family support, and ability to tolerate psychotherapy or maximum recommended drug doses, before augmenting or switching therapies.1
In patients with a partial response to SSRIs and/or CBT, the APA suggests that augmentation may be preferable to switching treatments. Augmentation strategies for SSRIs include antipsychotics or CBT with Exposure Response Prevention (ERP); augmentation strategies for CBT include SSRIs. Combining SSRIs and CBT may decrease the chance of relapse when medication is discontinued. If the patient has a partial response to ERP, intensification of therapy also can be considered based on patient-specific factors. In non-responders, switching therapies may be necessary. Alternative treatments including a different SSRI; an antidepressant from a difference class, such as clomipramine or mirtazapine; an antipsychotic; or CBT.
The 2006 National Institute for Health and Clinical Excellence guidelines for OCD recommend additional high-intensity CBT, adding an antipsychotic to an SSRI or clomipramine, or combining clomipramine with citalopram in non-responders. There is no guidance regarding the order in which these treatments should be trialed. Antipsychotics are recommended as an entire class, and there are no recommendations regarding dosing or long-term risks. These guidelines are based on limited evidence, including only 1 trial of quetiapine and 1 trial of olanzapine.2,3
Efficacy
The 2013 National Institute for Health Care and Excellence Evidence Update included a 2010 Cochrane Review of 11 randomized controlled trials (RCTs) of antipsychotics as adjunctive treatment to SSRIs.5 All trials were <6 months, and most were limited regarding quality aspects. Two trials found no statistically significant difference with olanzapine in efficacy measures (Y-BOCS mean difference [MD] −2.96; 95% confidence interval [CI] −7.41 to 1.22; effect size d = −2.96 [−7.14, 1.22]). Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was no significant difference between groups (n = 44, 1 RCT, odds ratio [OR] 0.76; 95% CI 0.17 to 3.29; effect size d = 0.76 [0.17, 3.29]). Studies found increased weight gain with olanzapine compared with antidepressant monotherapy.
Statistically significant differences were demonstrated with the addition of quetiapine to antidepressant monotherapy as shown in Y-BOCS score at endpoint (Y-BOCS MD −2.28; 95% CI −4.05 to −0.52; effect size d −2.28 [−4.05, −0.52]). Quetiapine also demonstrated benefit for depressive and anxiety symptoms. Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was a significant difference between groups (n = 80, 2 RCTs, OR 0.27; 95% CI 0.09 to 0.87; effect size d = 0.27 [0.09, 0.87]).
Adjunctive treatment with risperidone was superior to antidepressant monotherapy for participants without a significant response in OCD symptom severity of at least 25% with validated measures (OR 0.17; 95% CI 0.04 to 0.66; effect size d = 0.17 [0.04, 0.66]), and in depressive and anxiety symptoms. Mean reduction in Y-BOCS scores was not statistically significant with risperidone (MD −3.35; 95% CI −8.25 to 1.55; effect size d = −3.35 [−8.25, 1.55]).5
A 2014 meta-analysis by Veale et al3 included double-blind, randomized trials that examined atypical antipsychotics compared with placebo for adults with OCD that used an intention-to-treat analysis. Unlike the Cochrane Review, these studies used the Y-BOCS as a primary outcome measure. Participants had a Y-BOCS score of ≥16; had at least 1 appropriate trial of an SSRI or clomipramine (defined as the maximum dose tolerated for at least 8 weeks); and had to continue taking the SSRI or
Fourteen articles were included in the meta-analysis, but all had small sample sizes and no long-term follow-up data.3 Antipsychotics in the meta-analysis included risperidone (4 studies), quetiapine (5 studies), olanzapine (2 studies),
The overall difference in Y-BOCS score change between drug and placebo groups was 2.34 points, which had an overall effect size of d = 0.40. Those taking antipsychotics had approximately a 10% reduction in Y-BOCS score over time. The overall difference was statistically significant with risperidone (overall mean reduction of 3.89 points on the Y-BOCS; 95% CI 1.43 to 5.48; effect size of d = 0.53) and aripiprazole (difference in Y-BOCS outcome 0.1 scores of 6.29 points; effect size of d = 1.11). One trial of risperidone used a low dose (0.5 mg) and had a larger effect size than the studies that used moderate doses. The overall difference was not statistically significant for quetiapine (difference of Y-BOCS outcome scores of 0.81 points) or olanzapine (difference in Y-BOCS outcome scores of −0.19; indicating <1 point difference on the Y-BOCS).3
Studies included in the meta-analysis ranged in durations from 6 to 16 weeks; duration of ≥4 weeks did not make a difference in response. One study demonstrated a worsening of symptoms in the quetiapine group between weeks 4 and 12. Only 4 studies included most patients that had a previous trial of CBT. One study with an additional treatment arm evaluating CBT found that adding CBT was superior to adjunctive risperidone or placebo. Another study found that adding clomipramine or placebo to
Two studies included in the meta-analysis classified OCD symptoms by subtype, such as by dimensions of checking; symmetry, ordering, counting, and repeating; contamination and cleaning; and hoarding. Currently, no clinically significant predictor of outcome of antipsychotic therapy has been identified. Two studies included in the meta-analysis assessed patients with comorbid tic disorders and found no difference by treatment. One study demonstrated benefit of haloperidol in patients with comorbid tic disorders compared with those without comorbid tic disorders. Of note, none of the studies included in the meta-analysis excluded patients with hoarding characteristics, which generally indicate a worse prognosis with treatment.3
In 2015, Dold et al6 provided an update to a 2013 meta-analysis7 assessing antipsychotic augmentation of SSRIs in treatment-resistant OCD. This update included 2 new RCTs. The 2013 analysis7 concluded that risperidone should be considered first-line and is preferred over olanzapine and quetiapine. However, the update found the highest effect size for aripiprazole (d = −1.35), followed by haloperidol (d = −0.82), risperidone (d = −0.59), quetiapine (d = −0.50), olanzapine (d = −0.49), and paliperidone (d = −0.21).6,7
The 2015 update6 concluded that the antipsychotic doses used in trials were moderate and that there was no association between dose and treatment response, indicating that high doses of antipsychotics may not be more effective. Dold et al6 postulated that the antipsychotic doses required for treating OCD are similar to those used in treating major depressive disorder and lower than doses used in treating schizophrenia. The 2013 meta-analysis demonstrated that moderate doses of antipsychotics resulted in statistically significant efficacy (relative risk [RR] = 3.99, 95% CI 1.92 to 8.27), while low doses did not demonstrate statistical significance (RR = 1.06, 95% CI 0.45 to 2.53).6,7
The 2015 subgroup analysis update evaluated the duration of SSRI treatment prior to the antipsychotic augmentation phase, but did not demonstrate statistically significant efficacy for studies with <8 weeks’ duration of SSRI treatment, further highlighting the need for extended duration of treatment with an SSRI prior to augmentation.6
The 2013 meta-analysis discussed populations with comorbid tic disorders, including a study that found that patients with OCD and comorbid tic disorders benefit more from adjunctive antipsychotic therapy than those without the comorbidity. The 2015 update excluded trials that included patients with comorbid tic disorders to reduce bias, which did not affect the overall effect sizes of the data.6,7
In summary, efficacy has been demonstrated for risperidone and aripiprazole. There has been no benefit demonstrated with olanzapine and limited benefit with quetiapine. One study suggested worsening of symptoms with quetiapine the longer that treatment persisted.3,5-7
Safety
Assessing potential harms related to the use of antipsychotics in treating OCD is complicated, because this information is not always assessed in trials. Instead, researchers often focus on exploring potential benefits because long-term effects of antipsychotics, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects, are well documented.3
Trials included in the meta-analysis by Veale et al3 had a maximum duration of 16 weeks, so it is likely that many of the potential harms of antipsychotic use would not yet have been measurable. The authors cautioned that, although aripiprazole and risperidone demonstrated benefit, their benefit must be weighed against the potential physical risks of long-term antipsychotic use.3One study that was not included in the meta-analysis by Veale et al3 evaluated individuals who did not respond to a SSRI, and randomly assigned them to quetiapine, olanzapine, or risperidone plus CBT. At 1-year follow-up, 50% of participants receiving an antipsychotic had an increase of >10% in body mass index (BMI) and had higher fasting blood sugars compared with only 15.2% of participants with increased BMI in the comparison group (SSRI responders).3
Foa et al8 investigated long-term outcomes (ie, 6 months) of SSRI augmentation with ERP or risperidone in patients with OCD. Forty patients were randomized to receive risperidone, and 9 were considered responders. Only 8 chose to enter the maintenance phase, and of those participants, 5 did not complete the study. Two withdrew due to worsening depression, 2 withdrew due to intolerable adverse effects, and 1 was lost to follow-up. Unfortunately, there was no further discussion of what the intolerable adverse effects were.8
Patients with comorbid schizophrenia and OCD face additional risks. Lifetime prevalence rates of OCD are greater in persons with schizophrenia compared with the general population (26% vs 8%, respectively). Most studies have demonstrated poor prognosis and medication adherence among patients with comorbid schizophrenia and OCD. Fonseka et al9 assessed the risk of antipsychotic induction and exacerbation of OCD symptoms in patients with schizophrenia. Induction and exacerbation of OCD symptoms
Evidence of olanzapine induction and exacerbation of OCD symptoms is also limited to case reports and retrospective studies. However, some studies have estimated induction of OCD symptoms with olanzapine in 11% to 20% of patients.9 There is insufficient evidence to form conclusions regarding other antipsychotics. Fonseka et al9 recommends switching to an antipsychotic with lower 5HT-2 binding affinity or adding an SSRI, such as fluvoxamine, if induction or exacerbation of OCD symptoms occurs.
Consider long-term risks
The evidence for benefits with antipsychotics in treatment-resistant OCD is limited by different populations recruited, small sample sizes, and lack of long-term follow-up. Most evidence supports using ERP over antipsychotics for treating OCD symptoms that have not responded to SSRIs. However, ERP poses its own challenges that may limit clinical utility, such as economic and time restraints. Therefore, benefits with antipsychotics, such as risperidone and aripiprazole, must be weighed against potential long-term risks of treatment, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects.
Regarding Mr. E’s case, because he had been maximized on SSRI therapy for an adequate duration (escitalopram, 40 mg/d, for 12 weeks) and completed CBT with ERP with a partial response, adding risperidone, 0.5 mg at bedtime, was an appropriate treatment option that is supported by the available guidelines and evidence. The risperidone dose is reflective of the initial dosing strategies used in clinical trials. It is recommended to assess efficacy of treatment at 8 weeks with a validated measure, such as the Y-BOCS. A dose increase may be needed to achieve clinically significant symptom improvement, because moderate doses of risperidone have demonstrated efficacy in trials; however, high doses of risperidone are unlikely to provide additional benefit and increase the risk of adverse effects. If risperidone does not provide a clinically favorable risk–benefit ratio for Mr. E, aripiprazole is a potential alternative.
1. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/ocd.pdf. Published July 2007. Accessed December 11, 2017.
2. National Institute for Health and Care Excellence (NICE). Obsessive compulsive disorder. http://arms.evidence.nhs.uk/resources/hub/1028833/attachment. Updated September 18, 2013. Accessed December 11, 2017.
3. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and
4. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
5. Komossa K, Depping AM, Meyer M, et al. Second-generation antipsychotics for obsessive compulsive disorder. Cochrane Database Syst Rev. 2010;12:1-44.
6. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: an update meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2015;18(9). doi: 10.1093/ijnp/pyv047.
7. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2013;16(3):557-574.
8. Foa EB, Simpson HB, Rosenfield D, et al. Six-month outcomes from a randomized trial augmenting serotonin reuptake inhibitors with exposure and response prevention or risperidone in adults with obsessive-compulsive disorder. J Clin Psychiatry. 2015;76(4):440-446.
9. Fonseka TM, Richter MA, Muller DJ. Second generation antipsychotic-induced obsessive-compulsive symptoms in schizophrenia: a review of the experimental literature. Curr Psychiatry Rep. 2014;16(11):510.
Mr. E, age 37, has a 20-year history of obsessive-compulsive disorder (OCD), with comorbid generalized anxiety disorder and hypertension. His medication regimen consists of
Box
Antipsychotics for OCD: What the guidelines recommend
The 2013 American Psychiatric Association (APA) obsessive-compulsive disorder (OCD) treatment guidelines include recommendations regarding the use of antipsychotics in patients who do not respond to first-line treatment with selective serotonin reuptake inhibitors (SSRIs) and/or cognitive-behavioral therapy (CBT). The APA recommends evaluating contributing factors, including comorbidities, family support, and ability to tolerate psychotherapy or maximum recommended drug doses, before augmenting or switching therapies.1
In patients with a partial response to SSRIs and/or CBT, the APA suggests that augmentation may be preferable to switching treatments. Augmentation strategies for SSRIs include antipsychotics or CBT with Exposure Response Prevention (ERP); augmentation strategies for CBT include SSRIs. Combining SSRIs and CBT may decrease the chance of relapse when medication is discontinued. If the patient has a partial response to ERP, intensification of therapy also can be considered based on patient-specific factors. In non-responders, switching therapies may be necessary. Alternative treatments including a different SSRI; an antidepressant from a difference class, such as clomipramine or mirtazapine; an antipsychotic; or CBT.
The 2006 National Institute for Health and Clinical Excellence guidelines for OCD recommend additional high-intensity CBT, adding an antipsychotic to an SSRI or clomipramine, or combining clomipramine with citalopram in non-responders. There is no guidance regarding the order in which these treatments should be trialed. Antipsychotics are recommended as an entire class, and there are no recommendations regarding dosing or long-term risks. These guidelines are based on limited evidence, including only 1 trial of quetiapine and 1 trial of olanzapine.2,3
Efficacy
The 2013 National Institute for Health Care and Excellence Evidence Update included a 2010 Cochrane Review of 11 randomized controlled trials (RCTs) of antipsychotics as adjunctive treatment to SSRIs.5 All trials were <6 months, and most were limited regarding quality aspects. Two trials found no statistically significant difference with olanzapine in efficacy measures (Y-BOCS mean difference [MD] −2.96; 95% confidence interval [CI] −7.41 to 1.22; effect size d = −2.96 [−7.14, 1.22]). Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was no significant difference between groups (n = 44, 1 RCT, odds ratio [OR] 0.76; 95% CI 0.17 to 3.29; effect size d = 0.76 [0.17, 3.29]). Studies found increased weight gain with olanzapine compared with antidepressant monotherapy.
Statistically significant differences were demonstrated with the addition of quetiapine to antidepressant monotherapy as shown in Y-BOCS score at endpoint (Y-BOCS MD −2.28; 95% CI −4.05 to −0.52; effect size d −2.28 [−4.05, −0.52]). Quetiapine also demonstrated benefit for depressive and anxiety symptoms. Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was a significant difference between groups (n = 80, 2 RCTs, OR 0.27; 95% CI 0.09 to 0.87; effect size d = 0.27 [0.09, 0.87]).
Adjunctive treatment with risperidone was superior to antidepressant monotherapy for participants without a significant response in OCD symptom severity of at least 25% with validated measures (OR 0.17; 95% CI 0.04 to 0.66; effect size d = 0.17 [0.04, 0.66]), and in depressive and anxiety symptoms. Mean reduction in Y-BOCS scores was not statistically significant with risperidone (MD −3.35; 95% CI −8.25 to 1.55; effect size d = −3.35 [−8.25, 1.55]).5
A 2014 meta-analysis by Veale et al3 included double-blind, randomized trials that examined atypical antipsychotics compared with placebo for adults with OCD that used an intention-to-treat analysis. Unlike the Cochrane Review, these studies used the Y-BOCS as a primary outcome measure. Participants had a Y-BOCS score of ≥16; had at least 1 appropriate trial of an SSRI or clomipramine (defined as the maximum dose tolerated for at least 8 weeks); and had to continue taking the SSRI or
Fourteen articles were included in the meta-analysis, but all had small sample sizes and no long-term follow-up data.3 Antipsychotics in the meta-analysis included risperidone (4 studies), quetiapine (5 studies), olanzapine (2 studies),
The overall difference in Y-BOCS score change between drug and placebo groups was 2.34 points, which had an overall effect size of d = 0.40. Those taking antipsychotics had approximately a 10% reduction in Y-BOCS score over time. The overall difference was statistically significant with risperidone (overall mean reduction of 3.89 points on the Y-BOCS; 95% CI 1.43 to 5.48; effect size of d = 0.53) and aripiprazole (difference in Y-BOCS outcome 0.1 scores of 6.29 points; effect size of d = 1.11). One trial of risperidone used a low dose (0.5 mg) and had a larger effect size than the studies that used moderate doses. The overall difference was not statistically significant for quetiapine (difference of Y-BOCS outcome scores of 0.81 points) or olanzapine (difference in Y-BOCS outcome scores of −0.19; indicating <1 point difference on the Y-BOCS).3
Studies included in the meta-analysis ranged in durations from 6 to 16 weeks; duration of ≥4 weeks did not make a difference in response. One study demonstrated a worsening of symptoms in the quetiapine group between weeks 4 and 12. Only 4 studies included most patients that had a previous trial of CBT. One study with an additional treatment arm evaluating CBT found that adding CBT was superior to adjunctive risperidone or placebo. Another study found that adding clomipramine or placebo to
Two studies included in the meta-analysis classified OCD symptoms by subtype, such as by dimensions of checking; symmetry, ordering, counting, and repeating; contamination and cleaning; and hoarding. Currently, no clinically significant predictor of outcome of antipsychotic therapy has been identified. Two studies included in the meta-analysis assessed patients with comorbid tic disorders and found no difference by treatment. One study demonstrated benefit of haloperidol in patients with comorbid tic disorders compared with those without comorbid tic disorders. Of note, none of the studies included in the meta-analysis excluded patients with hoarding characteristics, which generally indicate a worse prognosis with treatment.3
In 2015, Dold et al6 provided an update to a 2013 meta-analysis7 assessing antipsychotic augmentation of SSRIs in treatment-resistant OCD. This update included 2 new RCTs. The 2013 analysis7 concluded that risperidone should be considered first-line and is preferred over olanzapine and quetiapine. However, the update found the highest effect size for aripiprazole (d = −1.35), followed by haloperidol (d = −0.82), risperidone (d = −0.59), quetiapine (d = −0.50), olanzapine (d = −0.49), and paliperidone (d = −0.21).6,7
The 2015 update6 concluded that the antipsychotic doses used in trials were moderate and that there was no association between dose and treatment response, indicating that high doses of antipsychotics may not be more effective. Dold et al6 postulated that the antipsychotic doses required for treating OCD are similar to those used in treating major depressive disorder and lower than doses used in treating schizophrenia. The 2013 meta-analysis demonstrated that moderate doses of antipsychotics resulted in statistically significant efficacy (relative risk [RR] = 3.99, 95% CI 1.92 to 8.27), while low doses did not demonstrate statistical significance (RR = 1.06, 95% CI 0.45 to 2.53).6,7
The 2015 subgroup analysis update evaluated the duration of SSRI treatment prior to the antipsychotic augmentation phase, but did not demonstrate statistically significant efficacy for studies with <8 weeks’ duration of SSRI treatment, further highlighting the need for extended duration of treatment with an SSRI prior to augmentation.6
The 2013 meta-analysis discussed populations with comorbid tic disorders, including a study that found that patients with OCD and comorbid tic disorders benefit more from adjunctive antipsychotic therapy than those without the comorbidity. The 2015 update excluded trials that included patients with comorbid tic disorders to reduce bias, which did not affect the overall effect sizes of the data.6,7
In summary, efficacy has been demonstrated for risperidone and aripiprazole. There has been no benefit demonstrated with olanzapine and limited benefit with quetiapine. One study suggested worsening of symptoms with quetiapine the longer that treatment persisted.3,5-7
Safety
Assessing potential harms related to the use of antipsychotics in treating OCD is complicated, because this information is not always assessed in trials. Instead, researchers often focus on exploring potential benefits because long-term effects of antipsychotics, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects, are well documented.3
Trials included in the meta-analysis by Veale et al3 had a maximum duration of 16 weeks, so it is likely that many of the potential harms of antipsychotic use would not yet have been measurable. The authors cautioned that, although aripiprazole and risperidone demonstrated benefit, their benefit must be weighed against the potential physical risks of long-term antipsychotic use.3One study that was not included in the meta-analysis by Veale et al3 evaluated individuals who did not respond to a SSRI, and randomly assigned them to quetiapine, olanzapine, or risperidone plus CBT. At 1-year follow-up, 50% of participants receiving an antipsychotic had an increase of >10% in body mass index (BMI) and had higher fasting blood sugars compared with only 15.2% of participants with increased BMI in the comparison group (SSRI responders).3
Foa et al8 investigated long-term outcomes (ie, 6 months) of SSRI augmentation with ERP or risperidone in patients with OCD. Forty patients were randomized to receive risperidone, and 9 were considered responders. Only 8 chose to enter the maintenance phase, and of those participants, 5 did not complete the study. Two withdrew due to worsening depression, 2 withdrew due to intolerable adverse effects, and 1 was lost to follow-up. Unfortunately, there was no further discussion of what the intolerable adverse effects were.8
Patients with comorbid schizophrenia and OCD face additional risks. Lifetime prevalence rates of OCD are greater in persons with schizophrenia compared with the general population (26% vs 8%, respectively). Most studies have demonstrated poor prognosis and medication adherence among patients with comorbid schizophrenia and OCD. Fonseka et al9 assessed the risk of antipsychotic induction and exacerbation of OCD symptoms in patients with schizophrenia. Induction and exacerbation of OCD symptoms
Evidence of olanzapine induction and exacerbation of OCD symptoms is also limited to case reports and retrospective studies. However, some studies have estimated induction of OCD symptoms with olanzapine in 11% to 20% of patients.9 There is insufficient evidence to form conclusions regarding other antipsychotics. Fonseka et al9 recommends switching to an antipsychotic with lower 5HT-2 binding affinity or adding an SSRI, such as fluvoxamine, if induction or exacerbation of OCD symptoms occurs.
Consider long-term risks
The evidence for benefits with antipsychotics in treatment-resistant OCD is limited by different populations recruited, small sample sizes, and lack of long-term follow-up. Most evidence supports using ERP over antipsychotics for treating OCD symptoms that have not responded to SSRIs. However, ERP poses its own challenges that may limit clinical utility, such as economic and time restraints. Therefore, benefits with antipsychotics, such as risperidone and aripiprazole, must be weighed against potential long-term risks of treatment, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects.
Regarding Mr. E’s case, because he had been maximized on SSRI therapy for an adequate duration (escitalopram, 40 mg/d, for 12 weeks) and completed CBT with ERP with a partial response, adding risperidone, 0.5 mg at bedtime, was an appropriate treatment option that is supported by the available guidelines and evidence. The risperidone dose is reflective of the initial dosing strategies used in clinical trials. It is recommended to assess efficacy of treatment at 8 weeks with a validated measure, such as the Y-BOCS. A dose increase may be needed to achieve clinically significant symptom improvement, because moderate doses of risperidone have demonstrated efficacy in trials; however, high doses of risperidone are unlikely to provide additional benefit and increase the risk of adverse effects. If risperidone does not provide a clinically favorable risk–benefit ratio for Mr. E, aripiprazole is a potential alternative.
Mr. E, age 37, has a 20-year history of obsessive-compulsive disorder (OCD), with comorbid generalized anxiety disorder and hypertension. His medication regimen consists of
Box
Antipsychotics for OCD: What the guidelines recommend
The 2013 American Psychiatric Association (APA) obsessive-compulsive disorder (OCD) treatment guidelines include recommendations regarding the use of antipsychotics in patients who do not respond to first-line treatment with selective serotonin reuptake inhibitors (SSRIs) and/or cognitive-behavioral therapy (CBT). The APA recommends evaluating contributing factors, including comorbidities, family support, and ability to tolerate psychotherapy or maximum recommended drug doses, before augmenting or switching therapies.1
In patients with a partial response to SSRIs and/or CBT, the APA suggests that augmentation may be preferable to switching treatments. Augmentation strategies for SSRIs include antipsychotics or CBT with Exposure Response Prevention (ERP); augmentation strategies for CBT include SSRIs. Combining SSRIs and CBT may decrease the chance of relapse when medication is discontinued. If the patient has a partial response to ERP, intensification of therapy also can be considered based on patient-specific factors. In non-responders, switching therapies may be necessary. Alternative treatments including a different SSRI; an antidepressant from a difference class, such as clomipramine or mirtazapine; an antipsychotic; or CBT.
The 2006 National Institute for Health and Clinical Excellence guidelines for OCD recommend additional high-intensity CBT, adding an antipsychotic to an SSRI or clomipramine, or combining clomipramine with citalopram in non-responders. There is no guidance regarding the order in which these treatments should be trialed. Antipsychotics are recommended as an entire class, and there are no recommendations regarding dosing or long-term risks. These guidelines are based on limited evidence, including only 1 trial of quetiapine and 1 trial of olanzapine.2,3
Efficacy
The 2013 National Institute for Health Care and Excellence Evidence Update included a 2010 Cochrane Review of 11 randomized controlled trials (RCTs) of antipsychotics as adjunctive treatment to SSRIs.5 All trials were <6 months, and most were limited regarding quality aspects. Two trials found no statistically significant difference with olanzapine in efficacy measures (Y-BOCS mean difference [MD] −2.96; 95% confidence interval [CI] −7.41 to 1.22; effect size d = −2.96 [−7.14, 1.22]). Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was no significant difference between groups (n = 44, 1 RCT, odds ratio [OR] 0.76; 95% CI 0.17 to 3.29; effect size d = 0.76 [0.17, 3.29]). Studies found increased weight gain with olanzapine compared with antidepressant monotherapy.
Statistically significant differences were demonstrated with the addition of quetiapine to antidepressant monotherapy as shown in Y-BOCS score at endpoint (Y-BOCS MD −2.28; 95% CI −4.05 to −0.52; effect size d −2.28 [−4.05, −0.52]). Quetiapine also demonstrated benefit for depressive and anxiety symptoms. Among patients with no clinically significant change (defined as ≤35% reduction in Y-BOCS), there was a significant difference between groups (n = 80, 2 RCTs, OR 0.27; 95% CI 0.09 to 0.87; effect size d = 0.27 [0.09, 0.87]).
Adjunctive treatment with risperidone was superior to antidepressant monotherapy for participants without a significant response in OCD symptom severity of at least 25% with validated measures (OR 0.17; 95% CI 0.04 to 0.66; effect size d = 0.17 [0.04, 0.66]), and in depressive and anxiety symptoms. Mean reduction in Y-BOCS scores was not statistically significant with risperidone (MD −3.35; 95% CI −8.25 to 1.55; effect size d = −3.35 [−8.25, 1.55]).5
A 2014 meta-analysis by Veale et al3 included double-blind, randomized trials that examined atypical antipsychotics compared with placebo for adults with OCD that used an intention-to-treat analysis. Unlike the Cochrane Review, these studies used the Y-BOCS as a primary outcome measure. Participants had a Y-BOCS score of ≥16; had at least 1 appropriate trial of an SSRI or clomipramine (defined as the maximum dose tolerated for at least 8 weeks); and had to continue taking the SSRI or
Fourteen articles were included in the meta-analysis, but all had small sample sizes and no long-term follow-up data.3 Antipsychotics in the meta-analysis included risperidone (4 studies), quetiapine (5 studies), olanzapine (2 studies),
The overall difference in Y-BOCS score change between drug and placebo groups was 2.34 points, which had an overall effect size of d = 0.40. Those taking antipsychotics had approximately a 10% reduction in Y-BOCS score over time. The overall difference was statistically significant with risperidone (overall mean reduction of 3.89 points on the Y-BOCS; 95% CI 1.43 to 5.48; effect size of d = 0.53) and aripiprazole (difference in Y-BOCS outcome 0.1 scores of 6.29 points; effect size of d = 1.11). One trial of risperidone used a low dose (0.5 mg) and had a larger effect size than the studies that used moderate doses. The overall difference was not statistically significant for quetiapine (difference of Y-BOCS outcome scores of 0.81 points) or olanzapine (difference in Y-BOCS outcome scores of −0.19; indicating <1 point difference on the Y-BOCS).3
Studies included in the meta-analysis ranged in durations from 6 to 16 weeks; duration of ≥4 weeks did not make a difference in response. One study demonstrated a worsening of symptoms in the quetiapine group between weeks 4 and 12. Only 4 studies included most patients that had a previous trial of CBT. One study with an additional treatment arm evaluating CBT found that adding CBT was superior to adjunctive risperidone or placebo. Another study found that adding clomipramine or placebo to
Two studies included in the meta-analysis classified OCD symptoms by subtype, such as by dimensions of checking; symmetry, ordering, counting, and repeating; contamination and cleaning; and hoarding. Currently, no clinically significant predictor of outcome of antipsychotic therapy has been identified. Two studies included in the meta-analysis assessed patients with comorbid tic disorders and found no difference by treatment. One study demonstrated benefit of haloperidol in patients with comorbid tic disorders compared with those without comorbid tic disorders. Of note, none of the studies included in the meta-analysis excluded patients with hoarding characteristics, which generally indicate a worse prognosis with treatment.3
In 2015, Dold et al6 provided an update to a 2013 meta-analysis7 assessing antipsychotic augmentation of SSRIs in treatment-resistant OCD. This update included 2 new RCTs. The 2013 analysis7 concluded that risperidone should be considered first-line and is preferred over olanzapine and quetiapine. However, the update found the highest effect size for aripiprazole (d = −1.35), followed by haloperidol (d = −0.82), risperidone (d = −0.59), quetiapine (d = −0.50), olanzapine (d = −0.49), and paliperidone (d = −0.21).6,7
The 2015 update6 concluded that the antipsychotic doses used in trials were moderate and that there was no association between dose and treatment response, indicating that high doses of antipsychotics may not be more effective. Dold et al6 postulated that the antipsychotic doses required for treating OCD are similar to those used in treating major depressive disorder and lower than doses used in treating schizophrenia. The 2013 meta-analysis demonstrated that moderate doses of antipsychotics resulted in statistically significant efficacy (relative risk [RR] = 3.99, 95% CI 1.92 to 8.27), while low doses did not demonstrate statistical significance (RR = 1.06, 95% CI 0.45 to 2.53).6,7
The 2015 subgroup analysis update evaluated the duration of SSRI treatment prior to the antipsychotic augmentation phase, but did not demonstrate statistically significant efficacy for studies with <8 weeks’ duration of SSRI treatment, further highlighting the need for extended duration of treatment with an SSRI prior to augmentation.6
The 2013 meta-analysis discussed populations with comorbid tic disorders, including a study that found that patients with OCD and comorbid tic disorders benefit more from adjunctive antipsychotic therapy than those without the comorbidity. The 2015 update excluded trials that included patients with comorbid tic disorders to reduce bias, which did not affect the overall effect sizes of the data.6,7
In summary, efficacy has been demonstrated for risperidone and aripiprazole. There has been no benefit demonstrated with olanzapine and limited benefit with quetiapine. One study suggested worsening of symptoms with quetiapine the longer that treatment persisted.3,5-7
Safety
Assessing potential harms related to the use of antipsychotics in treating OCD is complicated, because this information is not always assessed in trials. Instead, researchers often focus on exploring potential benefits because long-term effects of antipsychotics, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects, are well documented.3
Trials included in the meta-analysis by Veale et al3 had a maximum duration of 16 weeks, so it is likely that many of the potential harms of antipsychotic use would not yet have been measurable. The authors cautioned that, although aripiprazole and risperidone demonstrated benefit, their benefit must be weighed against the potential physical risks of long-term antipsychotic use.3One study that was not included in the meta-analysis by Veale et al3 evaluated individuals who did not respond to a SSRI, and randomly assigned them to quetiapine, olanzapine, or risperidone plus CBT. At 1-year follow-up, 50% of participants receiving an antipsychotic had an increase of >10% in body mass index (BMI) and had higher fasting blood sugars compared with only 15.2% of participants with increased BMI in the comparison group (SSRI responders).3
Foa et al8 investigated long-term outcomes (ie, 6 months) of SSRI augmentation with ERP or risperidone in patients with OCD. Forty patients were randomized to receive risperidone, and 9 were considered responders. Only 8 chose to enter the maintenance phase, and of those participants, 5 did not complete the study. Two withdrew due to worsening depression, 2 withdrew due to intolerable adverse effects, and 1 was lost to follow-up. Unfortunately, there was no further discussion of what the intolerable adverse effects were.8
Patients with comorbid schizophrenia and OCD face additional risks. Lifetime prevalence rates of OCD are greater in persons with schizophrenia compared with the general population (26% vs 8%, respectively). Most studies have demonstrated poor prognosis and medication adherence among patients with comorbid schizophrenia and OCD. Fonseka et al9 assessed the risk of antipsychotic induction and exacerbation of OCD symptoms in patients with schizophrenia. Induction and exacerbation of OCD symptoms
Evidence of olanzapine induction and exacerbation of OCD symptoms is also limited to case reports and retrospective studies. However, some studies have estimated induction of OCD symptoms with olanzapine in 11% to 20% of patients.9 There is insufficient evidence to form conclusions regarding other antipsychotics. Fonseka et al9 recommends switching to an antipsychotic with lower 5HT-2 binding affinity or adding an SSRI, such as fluvoxamine, if induction or exacerbation of OCD symptoms occurs.
Consider long-term risks
The evidence for benefits with antipsychotics in treatment-resistant OCD is limited by different populations recruited, small sample sizes, and lack of long-term follow-up. Most evidence supports using ERP over antipsychotics for treating OCD symptoms that have not responded to SSRIs. However, ERP poses its own challenges that may limit clinical utility, such as economic and time restraints. Therefore, benefits with antipsychotics, such as risperidone and aripiprazole, must be weighed against potential long-term risks of treatment, including sedation, weight gain, metabolic syndrome, and extrapyramidal side effects.
Regarding Mr. E’s case, because he had been maximized on SSRI therapy for an adequate duration (escitalopram, 40 mg/d, for 12 weeks) and completed CBT with ERP with a partial response, adding risperidone, 0.5 mg at bedtime, was an appropriate treatment option that is supported by the available guidelines and evidence. The risperidone dose is reflective of the initial dosing strategies used in clinical trials. It is recommended to assess efficacy of treatment at 8 weeks with a validated measure, such as the Y-BOCS. A dose increase may be needed to achieve clinically significant symptom improvement, because moderate doses of risperidone have demonstrated efficacy in trials; however, high doses of risperidone are unlikely to provide additional benefit and increase the risk of adverse effects. If risperidone does not provide a clinically favorable risk–benefit ratio for Mr. E, aripiprazole is a potential alternative.
1. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/ocd.pdf. Published July 2007. Accessed December 11, 2017.
2. National Institute for Health and Care Excellence (NICE). Obsessive compulsive disorder. http://arms.evidence.nhs.uk/resources/hub/1028833/attachment. Updated September 18, 2013. Accessed December 11, 2017.
3. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and
4. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
5. Komossa K, Depping AM, Meyer M, et al. Second-generation antipsychotics for obsessive compulsive disorder. Cochrane Database Syst Rev. 2010;12:1-44.
6. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: an update meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2015;18(9). doi: 10.1093/ijnp/pyv047.
7. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2013;16(3):557-574.
8. Foa EB, Simpson HB, Rosenfield D, et al. Six-month outcomes from a randomized trial augmenting serotonin reuptake inhibitors with exposure and response prevention or risperidone in adults with obsessive-compulsive disorder. J Clin Psychiatry. 2015;76(4):440-446.
9. Fonseka TM, Richter MA, Muller DJ. Second generation antipsychotic-induced obsessive-compulsive symptoms in schizophrenia: a review of the experimental literature. Curr Psychiatry Rep. 2014;16(11):510.
1. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/ocd.pdf. Published July 2007. Accessed December 11, 2017.
2. National Institute for Health and Care Excellence (NICE). Obsessive compulsive disorder. http://arms.evidence.nhs.uk/resources/hub/1028833/attachment. Updated September 18, 2013. Accessed December 11, 2017.
3. Veale D, Miles S, Smallcombe N, et al. Atypical antipsychotic augmentation in SSRI treatment refractory obsessive-compulsive disorder: a systematic review and
4. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Publishing; 2013.
5. Komossa K, Depping AM, Meyer M, et al. Second-generation antipsychotics for obsessive compulsive disorder. Cochrane Database Syst Rev. 2010;12:1-44.
6. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: an update meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2015;18(9). doi: 10.1093/ijnp/pyv047.
7. Dold M, Aigner M, Lanzenberger R, et al. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials. Int J Neuropsychopharmacol. 2013;16(3):557-574.
8. Foa EB, Simpson HB, Rosenfield D, et al. Six-month outcomes from a randomized trial augmenting serotonin reuptake inhibitors with exposure and response prevention or risperidone in adults with obsessive-compulsive disorder. J Clin Psychiatry. 2015;76(4):440-446.
9. Fonseka TM, Richter MA, Muller DJ. Second generation antipsychotic-induced obsessive-compulsive symptoms in schizophrenia: a review of the experimental literature. Curr Psychiatry Rep. 2014;16(11):510.
Using pharmacogenetics guidelines when prescribing: What’s available
Ms. C, age 45, has a history of generalized anxiety disorder, which has been controlled for the past 6 weeks with extended-release (ER) venlafaxine, 37.5 mg/d. Previous medication trials included fluvoxamine, 300 mg/d, for 2 weeks; paroxetine, 20 mg/d, for 1 week; sertraline, 100 mg/d, for 1 week; and citalopram, 20 mg/d, for 2 weeks. For each trial, Ms. C was unable to tolerate standard doses because of substantial adverse effects; she complained that her anxiety would significantly worsen with each course of treatment. Although the adverse effects would eventually subside with continued treatment, they appeared to be the dose-limiting factor for treatment, even when much lower doses were started.
Ms. C’s son recently suggested that she undergo pharmacogenomics testing, and she brings in the results of this testing. The report states that Ms. C has cytochrome P450 (CYP) pharmacogenotypes CYP2D6 *5/*9, CYP2C19 *2/*3, CYP2C9 *2/*2, and CYP1A2 *1A/*1F. Ms. C wants to know if these results explain some of the issues she has had with previous medication trials, and if these results mean that she should be taking a different medication.
The human genome project was a vast, international effort to sequence the entire human genome1 and identify individual differences in drug response, which serves as the basis for pharmacogenomics. Since completion of the human genome project in the early 2000s, the field of pharmacogenomics has advanced, and using pharmacogenomic testing to make therapeutic decisions for medication management is becoming commonplace.2 Although this critical change to how medicine is practiced is exciting, implementation of pharmacogenomics into practice has been varied.2 Therefore, having an understanding of the resources available to guide pharmacogenomics into practice is critical, because the FDA now lists >160 medications that include specific pharmacogenomics information within their package insert.3

CPIC provides guidance for implementing pharmacogenomics

In 2000, the National Institutes of Health established the Pharmacogenomics Knowledge Base (PharmGKB) and the Pharmacogenomics Research Network (PGRN). These 2 resources provide information from cutting-edge research on genomic variation and therapeutic and adverse events, as well as practical implementation of this research.4 As part of their partnership, PharmGKB and PGRN established the Clinical Pharmacogenomics Implementation Consortium (CPIC), which has begun to provide clinical practice guidelines for implementing pharmacogenomic results. Although CPIC does not advocate for pharmacogenomics testing as a standard, it recognizes that this testing is becoming more commonplace, and therefore its guidelines can help clinicians make rational prescribing decisions.4
In a recent partnership among several PGRN members, investigators found that 1 out of 4 pharmacogenomic test results had a potential clinically actionable outcome.2 There are currently >43 gene/drug pairs for which CPIC has provided guidelines; however, >200 other gene/drug pairs are being evaluated.5
Table 15 lists the current CPIC gene/drug combinations with accompanying published guidelines that are pertinent to psychiatry. For each of these guidelines, experts reviewed the available literature to provide graded therapeutic recommendations: A (“preponderance of evidence is high or moderate in favor of changing prescribing”), B (“preponderance of evidence is weak with little conflicting data”), and C and D (“evidence levels can vary”).4 Looking at the specific genotypes for Ms. C, we can use the information within the CPIC to assign a drug metabolism phenotype for her genotype combinations (Table 2).6

Consider additional resources
In addition to those from the CPIC, guidelines have been developed by other scientific groups, such as the Dutch Pharmacogenetics Working Group and the European Pharmacogenomics Implementation Consortium. Although most of these guidelines are concordant with CPIC, differences exist, which makes it important to be aware of all available resources.
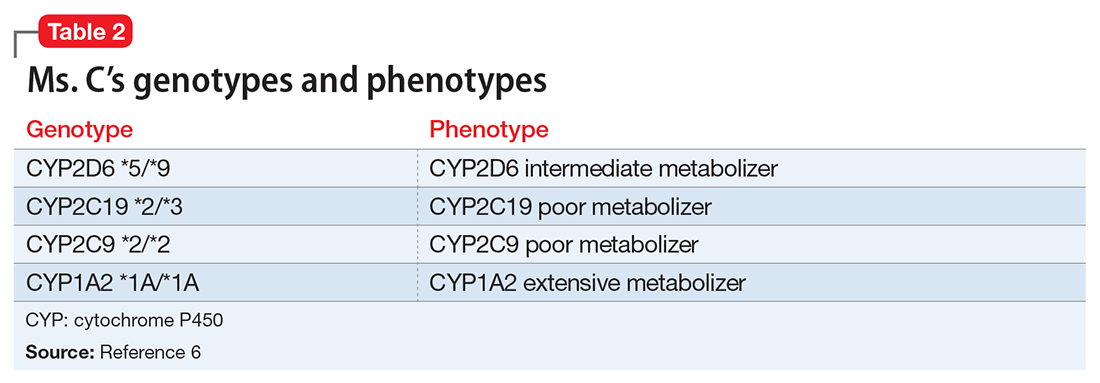
As well as working on the CPIC guidelines, PGRN investigators also provide numerous free online educational resources related to the principles behind pharmacogenomics, including additional resources necessary for systematic implementation. Examples include tables that outline all possible diplotypes (genotypes) for genes in the guidelines and how these are related to the metabolic phenotypes.2,4 Drug metabolizing phenotypes, for example, often are described as poor, intermediate, extensive, and ultra-rapid; in this system, metabolizing ability labeled as poor is less-than-average, and ultra-rapid describes greater-than-average ability. The extensive phenotype is considered average. The data files on the CPIC Web site also can be used as resources to “double check” interpretation results for the diplotype phenotype combinations currently available from various pharmacogenomics companies.7
Based on Ms. C’s presentation, as well as information from the CPIC guidelines, we expect that she might experience substantial adverse effects from most selective serotonin reuptake inhibitors and tricycle antidepressants because of her intermediate metabolizer status for CYP2D6 and poor metabolizer status for CYP2C19. The CPIC’s recommendation for using paroxetine and fluvoxamine in patients with a CYP2D6 intermediate metabolism phenotype is to initiate the recommend starting dose, but acknowledge that reduced metabolic capacity through CYP2D6 may result in higher blood levels and greater probability of adverse drug reactions. For a patient with the CYP2C19 poor metabolizer phenotype, the recommendation is to reduce the starting dose of citalopram or sertraline by 50%, or to prescribe a drug that is not metabolized by CYP2C19.8 Therefore, this pharmacogenomic information may help us understand why Ms. C is unable to tolerate these medications.
Although the CPIC guidelines do not address venlafaxine, the PharmGKB Web site contains literature supporting CYP2D6 as important in venlafaxine metabolism. Current recommendations from the Dutch Pharmacogenetics Working Group Guidelines9 are to either use a non–CYP2D6 metabolized medication or to adjust the dose to clinical response. Because Ms. C has been taking venlafaxine ER for the last 6 weeks and is taking a relatively low but effective dose, our recommendation is to continue current therapy.
It is also important to consider drug interactions when interpreting pharmacogenomic test results. In Ms. C’s case, the impact of a CYP2D6 intermediate metabolism phenotype would be increased if she also was taking a strong CYP2D6 inhibitor such as bupropion. Pharmacogenomics is another clinical tool and discontinuation of an effective treatment that is adequately tolerated should not be done based on pharmacogenomics recommendations alone.
1. Collins FS, Patrinos A, Jordan E, et al. New goals for the U.S. Human Genome Project: 1998-2003. Science. 1998;282(5389):682-689.
2. Luzum JA, Pakyz RE, Elsey AR, et al; Pharmacogenomics Research Network Translational Pharmacogenetics Program. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: outcomes and metrics of pharmacogenetic implementations across diverse healthcare systems. Clin Pharmacol Ther. 2017;102(3):502-510.
3. U.S. Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Updated October 3, 2017. Accessed October 23, 2017.
4. Caudle KE, Gammal RS, Whirl-Carrillo M, et al. Evidence and resources to implement pharmacogenetic knowledge for precision medicine. Am J Health Syst Pharm. 2016;73(23):1977-1985.
5. Clinical Pharmacogenomics Implementation Consortium. Genes-drugs. https://cpicpgx.org/genes-drugs. Updated October 2, 2017. Accessed October 23, 2017.
6. PharmGKB. PGx gene-specific information tables. https://www.pharmgkb.org/page/pgxGeneRef. Accessed October 27, 2017.
7. Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
8. Hicks JK, Bishop JR, Sangkuhl K, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther. 2015;98(2):127-134.
9
Ms. C, age 45, has a history of generalized anxiety disorder, which has been controlled for the past 6 weeks with extended-release (ER) venlafaxine, 37.5 mg/d. Previous medication trials included fluvoxamine, 300 mg/d, for 2 weeks; paroxetine, 20 mg/d, for 1 week; sertraline, 100 mg/d, for 1 week; and citalopram, 20 mg/d, for 2 weeks. For each trial, Ms. C was unable to tolerate standard doses because of substantial adverse effects; she complained that her anxiety would significantly worsen with each course of treatment. Although the adverse effects would eventually subside with continued treatment, they appeared to be the dose-limiting factor for treatment, even when much lower doses were started.
Ms. C’s son recently suggested that she undergo pharmacogenomics testing, and she brings in the results of this testing. The report states that Ms. C has cytochrome P450 (CYP) pharmacogenotypes CYP2D6 *5/*9, CYP2C19 *2/*3, CYP2C9 *2/*2, and CYP1A2 *1A/*1F. Ms. C wants to know if these results explain some of the issues she has had with previous medication trials, and if these results mean that she should be taking a different medication.
The human genome project was a vast, international effort to sequence the entire human genome1 and identify individual differences in drug response, which serves as the basis for pharmacogenomics. Since completion of the human genome project in the early 2000s, the field of pharmacogenomics has advanced, and using pharmacogenomic testing to make therapeutic decisions for medication management is becoming commonplace.2 Although this critical change to how medicine is practiced is exciting, implementation of pharmacogenomics into practice has been varied.2 Therefore, having an understanding of the resources available to guide pharmacogenomics into practice is critical, because the FDA now lists >160 medications that include specific pharmacogenomics information within their package insert.3
CPIC provides guidance for implementing pharmacogenomics
In 2000, the National Institutes of Health established the Pharmacogenomics Knowledge Base (PharmGKB) and the Pharmacogenomics Research Network (PGRN). These 2 resources provide information from cutting-edge research on genomic variation and therapeutic and adverse events, as well as practical implementation of this research.4 As part of their partnership, PharmGKB and PGRN established the Clinical Pharmacogenomics Implementation Consortium (CPIC), which has begun to provide clinical practice guidelines for implementing pharmacogenomic results. Although CPIC does not advocate for pharmacogenomics testing as a standard, it recognizes that this testing is becoming more commonplace, and therefore its guidelines can help clinicians make rational prescribing decisions.4
In a recent partnership among several PGRN members, investigators found that 1 out of 4 pharmacogenomic test results had a potential clinically actionable outcome.2 There are currently >43 gene/drug pairs for which CPIC has provided guidelines; however, >200 other gene/drug pairs are being evaluated.5
Table 15 lists the current CPIC gene/drug combinations with accompanying published guidelines that are pertinent to psychiatry. For each of these guidelines, experts reviewed the available literature to provide graded therapeutic recommendations: A (“preponderance of evidence is high or moderate in favor of changing prescribing”), B (“preponderance of evidence is weak with little conflicting data”), and C and D (“evidence levels can vary”).4 Looking at the specific genotypes for Ms. C, we can use the information within the CPIC to assign a drug metabolism phenotype for her genotype combinations (Table 2).6
Consider additional resources
In addition to those from the CPIC, guidelines have been developed by other scientific groups, such as the Dutch Pharmacogenetics Working Group and the European Pharmacogenomics Implementation Consortium. Although most of these guidelines are concordant with CPIC, differences exist, which makes it important to be aware of all available resources.
As well as working on the CPIC guidelines, PGRN investigators also provide numerous free online educational resources related to the principles behind pharmacogenomics, including additional resources necessary for systematic implementation. Examples include tables that outline all possible diplotypes (genotypes) for genes in the guidelines and how these are related to the metabolic phenotypes.2,4 Drug metabolizing phenotypes, for example, often are described as poor, intermediate, extensive, and ultra-rapid; in this system, metabolizing ability labeled as poor is less-than-average, and ultra-rapid describes greater-than-average ability. The extensive phenotype is considered average. The data files on the CPIC Web site also can be used as resources to “double check” interpretation results for the diplotype phenotype combinations currently available from various pharmacogenomics companies.7
Based on Ms. C’s presentation, as well as information from the CPIC guidelines, we expect that she might experience substantial adverse effects from most selective serotonin reuptake inhibitors and tricycle antidepressants because of her intermediate metabolizer status for CYP2D6 and poor metabolizer status for CYP2C19. The CPIC’s recommendation for using paroxetine and fluvoxamine in patients with a CYP2D6 intermediate metabolism phenotype is to initiate the recommend starting dose, but acknowledge that reduced metabolic capacity through CYP2D6 may result in higher blood levels and greater probability of adverse drug reactions. For a patient with the CYP2C19 poor metabolizer phenotype, the recommendation is to reduce the starting dose of citalopram or sertraline by 50%, or to prescribe a drug that is not metabolized by CYP2C19.8 Therefore, this pharmacogenomic information may help us understand why Ms. C is unable to tolerate these medications.
Although the CPIC guidelines do not address venlafaxine, the PharmGKB Web site contains literature supporting CYP2D6 as important in venlafaxine metabolism. Current recommendations from the Dutch Pharmacogenetics Working Group Guidelines9 are to either use a non–CYP2D6 metabolized medication or to adjust the dose to clinical response. Because Ms. C has been taking venlafaxine ER for the last 6 weeks and is taking a relatively low but effective dose, our recommendation is to continue current therapy.
It is also important to consider drug interactions when interpreting pharmacogenomic test results. In Ms. C’s case, the impact of a CYP2D6 intermediate metabolism phenotype would be increased if she also was taking a strong CYP2D6 inhibitor such as bupropion. Pharmacogenomics is another clinical tool and discontinuation of an effective treatment that is adequately tolerated should not be done based on pharmacogenomics recommendations alone.
Ms. C, age 45, has a history of generalized anxiety disorder, which has been controlled for the past 6 weeks with extended-release (ER) venlafaxine, 37.5 mg/d. Previous medication trials included fluvoxamine, 300 mg/d, for 2 weeks; paroxetine, 20 mg/d, for 1 week; sertraline, 100 mg/d, for 1 week; and citalopram, 20 mg/d, for 2 weeks. For each trial, Ms. C was unable to tolerate standard doses because of substantial adverse effects; she complained that her anxiety would significantly worsen with each course of treatment. Although the adverse effects would eventually subside with continued treatment, they appeared to be the dose-limiting factor for treatment, even when much lower doses were started.
Ms. C’s son recently suggested that she undergo pharmacogenomics testing, and she brings in the results of this testing. The report states that Ms. C has cytochrome P450 (CYP) pharmacogenotypes CYP2D6 *5/*9, CYP2C19 *2/*3, CYP2C9 *2/*2, and CYP1A2 *1A/*1F. Ms. C wants to know if these results explain some of the issues she has had with previous medication trials, and if these results mean that she should be taking a different medication.
The human genome project was a vast, international effort to sequence the entire human genome1 and identify individual differences in drug response, which serves as the basis for pharmacogenomics. Since completion of the human genome project in the early 2000s, the field of pharmacogenomics has advanced, and using pharmacogenomic testing to make therapeutic decisions for medication management is becoming commonplace.2 Although this critical change to how medicine is practiced is exciting, implementation of pharmacogenomics into practice has been varied.2 Therefore, having an understanding of the resources available to guide pharmacogenomics into practice is critical, because the FDA now lists >160 medications that include specific pharmacogenomics information within their package insert.3
CPIC provides guidance for implementing pharmacogenomics
In 2000, the National Institutes of Health established the Pharmacogenomics Knowledge Base (PharmGKB) and the Pharmacogenomics Research Network (PGRN). These 2 resources provide information from cutting-edge research on genomic variation and therapeutic and adverse events, as well as practical implementation of this research.4 As part of their partnership, PharmGKB and PGRN established the Clinical Pharmacogenomics Implementation Consortium (CPIC), which has begun to provide clinical practice guidelines for implementing pharmacogenomic results. Although CPIC does not advocate for pharmacogenomics testing as a standard, it recognizes that this testing is becoming more commonplace, and therefore its guidelines can help clinicians make rational prescribing decisions.4
In a recent partnership among several PGRN members, investigators found that 1 out of 4 pharmacogenomic test results had a potential clinically actionable outcome.2 There are currently >43 gene/drug pairs for which CPIC has provided guidelines; however, >200 other gene/drug pairs are being evaluated.5
Table 15 lists the current CPIC gene/drug combinations with accompanying published guidelines that are pertinent to psychiatry. For each of these guidelines, experts reviewed the available literature to provide graded therapeutic recommendations: A (“preponderance of evidence is high or moderate in favor of changing prescribing”), B (“preponderance of evidence is weak with little conflicting data”), and C and D (“evidence levels can vary”).4 Looking at the specific genotypes for Ms. C, we can use the information within the CPIC to assign a drug metabolism phenotype for her genotype combinations (Table 2).6
Consider additional resources
In addition to those from the CPIC, guidelines have been developed by other scientific groups, such as the Dutch Pharmacogenetics Working Group and the European Pharmacogenomics Implementation Consortium. Although most of these guidelines are concordant with CPIC, differences exist, which makes it important to be aware of all available resources.
As well as working on the CPIC guidelines, PGRN investigators also provide numerous free online educational resources related to the principles behind pharmacogenomics, including additional resources necessary for systematic implementation. Examples include tables that outline all possible diplotypes (genotypes) for genes in the guidelines and how these are related to the metabolic phenotypes.2,4 Drug metabolizing phenotypes, for example, often are described as poor, intermediate, extensive, and ultra-rapid; in this system, metabolizing ability labeled as poor is less-than-average, and ultra-rapid describes greater-than-average ability. The extensive phenotype is considered average. The data files on the CPIC Web site also can be used as resources to “double check” interpretation results for the diplotype phenotype combinations currently available from various pharmacogenomics companies.7
Based on Ms. C’s presentation, as well as information from the CPIC guidelines, we expect that she might experience substantial adverse effects from most selective serotonin reuptake inhibitors and tricycle antidepressants because of her intermediate metabolizer status for CYP2D6 and poor metabolizer status for CYP2C19. The CPIC’s recommendation for using paroxetine and fluvoxamine in patients with a CYP2D6 intermediate metabolism phenotype is to initiate the recommend starting dose, but acknowledge that reduced metabolic capacity through CYP2D6 may result in higher blood levels and greater probability of adverse drug reactions. For a patient with the CYP2C19 poor metabolizer phenotype, the recommendation is to reduce the starting dose of citalopram or sertraline by 50%, or to prescribe a drug that is not metabolized by CYP2C19.8 Therefore, this pharmacogenomic information may help us understand why Ms. C is unable to tolerate these medications.
Although the CPIC guidelines do not address venlafaxine, the PharmGKB Web site contains literature supporting CYP2D6 as important in venlafaxine metabolism. Current recommendations from the Dutch Pharmacogenetics Working Group Guidelines9 are to either use a non–CYP2D6 metabolized medication or to adjust the dose to clinical response. Because Ms. C has been taking venlafaxine ER for the last 6 weeks and is taking a relatively low but effective dose, our recommendation is to continue current therapy.
It is also important to consider drug interactions when interpreting pharmacogenomic test results. In Ms. C’s case, the impact of a CYP2D6 intermediate metabolism phenotype would be increased if she also was taking a strong CYP2D6 inhibitor such as bupropion. Pharmacogenomics is another clinical tool and discontinuation of an effective treatment that is adequately tolerated should not be done based on pharmacogenomics recommendations alone.
1. Collins FS, Patrinos A, Jordan E, et al. New goals for the U.S. Human Genome Project: 1998-2003. Science. 1998;282(5389):682-689.
2. Luzum JA, Pakyz RE, Elsey AR, et al; Pharmacogenomics Research Network Translational Pharmacogenetics Program. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: outcomes and metrics of pharmacogenetic implementations across diverse healthcare systems. Clin Pharmacol Ther. 2017;102(3):502-510.
3. U.S. Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Updated October 3, 2017. Accessed October 23, 2017.
4. Caudle KE, Gammal RS, Whirl-Carrillo M, et al. Evidence and resources to implement pharmacogenetic knowledge for precision medicine. Am J Health Syst Pharm. 2016;73(23):1977-1985.
5. Clinical Pharmacogenomics Implementation Consortium. Genes-drugs. https://cpicpgx.org/genes-drugs. Updated October 2, 2017. Accessed October 23, 2017.
6. PharmGKB. PGx gene-specific information tables. https://www.pharmgkb.org/page/pgxGeneRef. Accessed October 27, 2017.
7. Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
8. Hicks JK, Bishop JR, Sangkuhl K, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther. 2015;98(2):127-134.
9
1. Collins FS, Patrinos A, Jordan E, et al. New goals for the U.S. Human Genome Project: 1998-2003. Science. 1998;282(5389):682-689.
2. Luzum JA, Pakyz RE, Elsey AR, et al; Pharmacogenomics Research Network Translational Pharmacogenetics Program. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: outcomes and metrics of pharmacogenetic implementations across diverse healthcare systems. Clin Pharmacol Ther. 2017;102(3):502-510.
3. U.S. Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labeling. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Updated October 3, 2017. Accessed October 23, 2017.
4. Caudle KE, Gammal RS, Whirl-Carrillo M, et al. Evidence and resources to implement pharmacogenetic knowledge for precision medicine. Am J Health Syst Pharm. 2016;73(23):1977-1985.
5. Clinical Pharmacogenomics Implementation Consortium. Genes-drugs. https://cpicpgx.org/genes-drugs. Updated October 2, 2017. Accessed October 23, 2017.
6. PharmGKB. PGx gene-specific information tables. https://www.pharmgkb.org/page/pgxGeneRef. Accessed October 27, 2017.
7. Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414-417.
8. Hicks JK, Bishop JR, Sangkuhl K, et al; Clinical Pharmacogenetics Implementation Consortium. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 genotypes and dosing of selective serotonin reuptake inhibitors. Clin Pharmacol Ther. 2015;98(2):127-134.
9

Lithium-induced bradycardia: A rare but serious adverse effect
Mr. C, age 30, with schizoaffective disorder, bipolar type, Cannabis abuse, and nicotine dependence, has been enrolled in a Program of Assertive Community Treatment (PACT) for approximately 5 years. He presents to the PACT clinic for follow-up with his psychiatrist. Mr. C reports dizziness, lightheadedness, blurred vision, and nausea worsening over the last few days, and he appears drowsy and hypoactive. He does not report any chest pain, abdominal pain, swelling, cold extremities, shortness of breath, vomiting, diarrhea, or blood loss. Mr. C admits he has eaten only once daily for several weeks because of delusional ideation that he is responsible for others suffering from anorexia nervosa.
His medical history includes gastroesophageal reflux disease. Mr. C’s medication regimen for the past year included total daily oral doses of
Because of Mr. C’s complaints, appearance, and low HR, the psychiatrist calls emergency medical services (EMS). Although the paramedics recommend emergency transport to the hospital, Mr. C refuses. The psychiatrist instructs Mr. C to stop taking lithium because of suspected lithium-induced bradycardia and a concern that he may be more susceptible to lithium toxicity with prolonged anorexia nervosa. When nursing staff evaluate Mr. C the next day, his vitals are HR 60 bpm, respirations 20 breaths per minute, and blood pressure 124/81 mm Hg; his dizziness, blurred vision, lightheadedness, and nausea are resolved.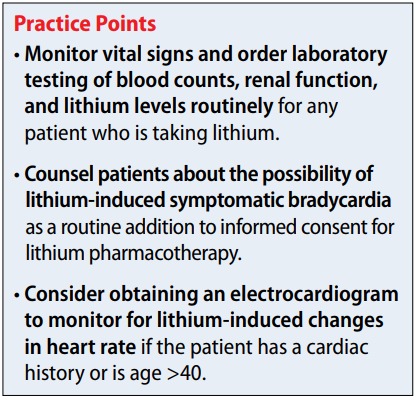
Bradycardia is defined as a HR <60 bpm; however, symptoms may not occur until the HR is <50 bpm. Symptoms include fatigue, dizziness, lightheadedness, chest pain, shortness of breath, and syncope. The incidence of bradycardia during lithium treatment is unknown; it is considered a rare but serious adverse effect. A literature review reveals several case reports of bradycardia with lithium treatment,2-4 including symptomatic bradycardia after a single dose of lithium.5 Other possible causes of bradycardia include anorexia nervosa, hypothermia, hypothyroidism, hypoxia, infection, stroke, acute myocardial infarction, sedative or opiate use, increased vagal tone with exercise conditioning, and other medications including fluphenazine.6
Mr. C’s symptoms may have been assumed to be secondary to several possible causes, including bradycardia, dehydration from poor oral intake, lithium toxicity, or an undiagnosed medical condition. The combination of nausea, dizziness, anorexia nervosa, blurred vision, and lightheadedness in a patient receiving lithium would certainly trigger a clinician’s concern for lithium toxicity, but he (she) may not be aware of the risk of bradycardia as an adverse effect of lithium. Because Mr. C refused hospital transportation by EMS, discontinuing lithium appears to have been the safest option. Laboratory studies from the day after Mr. C presented to the clinic appeared to lessen the probability that lithium toxicity, hypothyroidism,
Although psychiatrists may be vigilant about monitoring for signs and symptoms of toxicity with lithium use by utilizing regular laboratory studies, they may not be as vigilant with monitoring vital signs at every patient visit (Table). This case demonstrates the importance of regular vital sign measurements to be able to detect this rare but serious adverse effect.
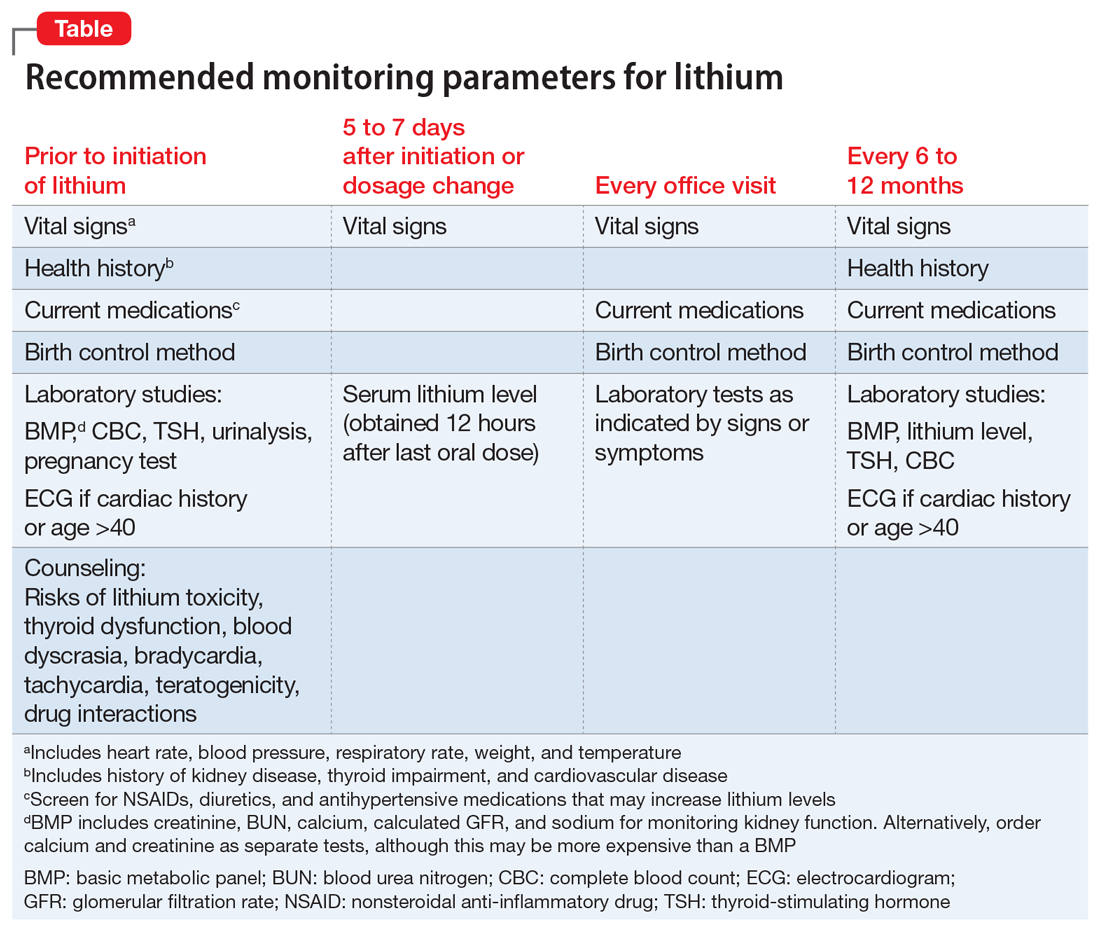
1. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
2. White B, Larry J, Kantharia BK. Protracted presyncope and profound bradycardia due to lithium toxicity. Int J Cardiol. 2008;125(3):e48-e50.
3. Palatnik A, Kates R. Bradycardia and medications: identify the dangerous pace. Nurs Manage. 2003;34(6):56A-56F.
4. La Rocca R, Foschi A, Preston NM, et al. QT interval prolongation and bradycardia in lithium-induced nephrogenic diabetes insipidus. Int J Cardiol. 2012;162(1):e1-e2.
5. Sabharwal MS, Annapureddy N, Agarwal SK, et al. Severe bradycardia caused by a single dose of lithium. Intern Med. 2013;52(7):767-769.
6. Homoud MK. Sinus bradycardia. UpToDate. www.uptodate.com/contents/sinus-bradycardia. Updated June 7, 2017. Accessed August 28, 2017.
Mr. C, age 30, with schizoaffective disorder, bipolar type, Cannabis abuse, and nicotine dependence, has been enrolled in a Program of Assertive Community Treatment (PACT) for approximately 5 years. He presents to the PACT clinic for follow-up with his psychiatrist. Mr. C reports dizziness, lightheadedness, blurred vision, and nausea worsening over the last few days, and he appears drowsy and hypoactive. He does not report any chest pain, abdominal pain, swelling, cold extremities, shortness of breath, vomiting, diarrhea, or blood loss. Mr. C admits he has eaten only once daily for several weeks because of delusional ideation that he is responsible for others suffering from anorexia nervosa.
His medical history includes gastroesophageal reflux disease. Mr. C’s medication regimen for the past year included total daily oral doses of
Because of Mr. C’s complaints, appearance, and low HR, the psychiatrist calls emergency medical services (EMS). Although the paramedics recommend emergency transport to the hospital, Mr. C refuses. The psychiatrist instructs Mr. C to stop taking lithium because of suspected lithium-induced bradycardia and a concern that he may be more susceptible to lithium toxicity with prolonged anorexia nervosa. When nursing staff evaluate Mr. C the next day, his vitals are HR 60 bpm, respirations 20 breaths per minute, and blood pressure 124/81 mm Hg; his dizziness, blurred vision, lightheadedness, and nausea are resolved.
Bradycardia is defined as a HR <60 bpm; however, symptoms may not occur until the HR is <50 bpm. Symptoms include fatigue, dizziness, lightheadedness, chest pain, shortness of breath, and syncope. The incidence of bradycardia during lithium treatment is unknown; it is considered a rare but serious adverse effect. A literature review reveals several case reports of bradycardia with lithium treatment,2-4 including symptomatic bradycardia after a single dose of lithium.5 Other possible causes of bradycardia include anorexia nervosa, hypothermia, hypothyroidism, hypoxia, infection, stroke, acute myocardial infarction, sedative or opiate use, increased vagal tone with exercise conditioning, and other medications including fluphenazine.6
Mr. C’s symptoms may have been assumed to be secondary to several possible causes, including bradycardia, dehydration from poor oral intake, lithium toxicity, or an undiagnosed medical condition. The combination of nausea, dizziness, anorexia nervosa, blurred vision, and lightheadedness in a patient receiving lithium would certainly trigger a clinician’s concern for lithium toxicity, but he (she) may not be aware of the risk of bradycardia as an adverse effect of lithium. Because Mr. C refused hospital transportation by EMS, discontinuing lithium appears to have been the safest option. Laboratory studies from the day after Mr. C presented to the clinic appeared to lessen the probability that lithium toxicity, hypothyroidism,
Although psychiatrists may be vigilant about monitoring for signs and symptoms of toxicity with lithium use by utilizing regular laboratory studies, they may not be as vigilant with monitoring vital signs at every patient visit (Table). This case demonstrates the importance of regular vital sign measurements to be able to detect this rare but serious adverse effect.
Mr. C, age 30, with schizoaffective disorder, bipolar type, Cannabis abuse, and nicotine dependence, has been enrolled in a Program of Assertive Community Treatment (PACT) for approximately 5 years. He presents to the PACT clinic for follow-up with his psychiatrist. Mr. C reports dizziness, lightheadedness, blurred vision, and nausea worsening over the last few days, and he appears drowsy and hypoactive. He does not report any chest pain, abdominal pain, swelling, cold extremities, shortness of breath, vomiting, diarrhea, or blood loss. Mr. C admits he has eaten only once daily for several weeks because of delusional ideation that he is responsible for others suffering from anorexia nervosa.
His medical history includes gastroesophageal reflux disease. Mr. C’s medication regimen for the past year included total daily oral doses of
Because of Mr. C’s complaints, appearance, and low HR, the psychiatrist calls emergency medical services (EMS). Although the paramedics recommend emergency transport to the hospital, Mr. C refuses. The psychiatrist instructs Mr. C to stop taking lithium because of suspected lithium-induced bradycardia and a concern that he may be more susceptible to lithium toxicity with prolonged anorexia nervosa. When nursing staff evaluate Mr. C the next day, his vitals are HR 60 bpm, respirations 20 breaths per minute, and blood pressure 124/81 mm Hg; his dizziness, blurred vision, lightheadedness, and nausea are resolved.
Bradycardia is defined as a HR <60 bpm; however, symptoms may not occur until the HR is <50 bpm. Symptoms include fatigue, dizziness, lightheadedness, chest pain, shortness of breath, and syncope. The incidence of bradycardia during lithium treatment is unknown; it is considered a rare but serious adverse effect. A literature review reveals several case reports of bradycardia with lithium treatment,2-4 including symptomatic bradycardia after a single dose of lithium.5 Other possible causes of bradycardia include anorexia nervosa, hypothermia, hypothyroidism, hypoxia, infection, stroke, acute myocardial infarction, sedative or opiate use, increased vagal tone with exercise conditioning, and other medications including fluphenazine.6
Mr. C’s symptoms may have been assumed to be secondary to several possible causes, including bradycardia, dehydration from poor oral intake, lithium toxicity, or an undiagnosed medical condition. The combination of nausea, dizziness, anorexia nervosa, blurred vision, and lightheadedness in a patient receiving lithium would certainly trigger a clinician’s concern for lithium toxicity, but he (she) may not be aware of the risk of bradycardia as an adverse effect of lithium. Because Mr. C refused hospital transportation by EMS, discontinuing lithium appears to have been the safest option. Laboratory studies from the day after Mr. C presented to the clinic appeared to lessen the probability that lithium toxicity, hypothyroidism,
Although psychiatrists may be vigilant about monitoring for signs and symptoms of toxicity with lithium use by utilizing regular laboratory studies, they may not be as vigilant with monitoring vital signs at every patient visit (Table). This case demonstrates the importance of regular vital sign measurements to be able to detect this rare but serious adverse effect.
1. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
2. White B, Larry J, Kantharia BK. Protracted presyncope and profound bradycardia due to lithium toxicity. Int J Cardiol. 2008;125(3):e48-e50.
3. Palatnik A, Kates R. Bradycardia and medications: identify the dangerous pace. Nurs Manage. 2003;34(6):56A-56F.
4. La Rocca R, Foschi A, Preston NM, et al. QT interval prolongation and bradycardia in lithium-induced nephrogenic diabetes insipidus. Int J Cardiol. 2012;162(1):e1-e2.
5. Sabharwal MS, Annapureddy N, Agarwal SK, et al. Severe bradycardia caused by a single dose of lithium. Intern Med. 2013;52(7):767-769.
6. Homoud MK. Sinus bradycardia. UpToDate. www.uptodate.com/contents/sinus-bradycardia. Updated June 7, 2017. Accessed August 28, 2017.
1. Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30(2):239-245.
2. White B, Larry J, Kantharia BK. Protracted presyncope and profound bradycardia due to lithium toxicity. Int J Cardiol. 2008;125(3):e48-e50.
3. Palatnik A, Kates R. Bradycardia and medications: identify the dangerous pace. Nurs Manage. 2003;34(6):56A-56F.
4. La Rocca R, Foschi A, Preston NM, et al. QT interval prolongation and bradycardia in lithium-induced nephrogenic diabetes insipidus. Int J Cardiol. 2012;162(1):e1-e2.
5. Sabharwal MS, Annapureddy N, Agarwal SK, et al. Severe bradycardia caused by a single dose of lithium. Intern Med. 2013;52(7):767-769.
6. Homoud MK. Sinus bradycardia. UpToDate. www.uptodate.com/contents/sinus-bradycardia. Updated June 7, 2017. Accessed August 28, 2017.
When should you consider combining 2 long-acting injectable antipsychotics?
Ms. S, age 39, with a 15-year history of schizophrenia and severe paranoid delusions, is admitted after physically assaulting a staff member at a group home. She is receiving paliperidone palmitate, 234 mg every 4 weeks. This has reduced the severity of her symptoms, but she continues to have persistent delusions that affect her ability to accept redirection from staff. Ms. S frequently accuses staff and peers of sexual assault, says that she is pregnant, and does not adhere to treatment recommendations for laboratory monitoring because the “staff uses her blood for experiments.”
Ms. S frequently requires administration of oral and IM haloperidol, as needed, when she becomes aggressive with the staff. She has poor insight into her mental illness and does not believe that she needs medication. Ms. S has a long history of stopping her oral antipsychotic after a few days, reporting that it is “harming her baby.” Monotherapy has been tried with various long-acting injectable antipsychotics (LAIAs), but she still exhibits persistent delusions. The treatment team decides to add a second LAIA, haloperidol decanoate, 200 mg every 4 weeks, to her regimen.

Ladds et al.7 A 49-year-old woman with schizophrenia who was hospitalized for aggressive and bizarre behavior and had been institutionalized for 20 years stopped taking her medication regimen.7 She started taking 8-hour showers with bleach, talking incoherently, and believing that someone was poisoning her. She had poor response to oral risperidone monotherapy; however, 2 months after adding oral fluphenazine and benztropine to her regimen, her symptoms substantially improved (doses not reported). Because she had impaired insight into the need for daily medication, she was started on depot fluphenazine decanoate and risperidone microspheres (doses not reported) before discharge. No substantial adverse effects were noted with this regimen.
Wartelsteiner and Hofer.8 A man who had been diagnosed with paranoid schizophrenia at age 20 presented with thought blocking, incoherence, persecutory delusions, and uncontrolled self-damaging behavior.8 He had been admitted 27 times over 7 years; during this time he received many antipsychotic monotherapies and combination regimens. A total of 8 oral antipsychotics (including clozapine) and 5 LAIAs had been administered during these trials. He significantly improved with the combination of olanzapine and risperidone. Both medications were switched to LAIA formulations to address medication nonadherence. His symptoms remained stable with risperidone microspheres, 100 mg, and olanzapine pamoate, 300 mg, each administered every 2 weeks. He did not experience any adverse effects with this combination therapy.
Scangos et al.9 A 26-year-old Vietnamese man with schizophrenia and an extensive history of unprovoked, psychotically driven assaults was given multiple antipsychotics (including clozapine) during hospitalizations, and his medication regimen consistently included 2 antipsychotics. After contracting viral gastroenteritis, he refused oral medications and required short-acting IM administration of both haloperidol, 5 mg, twice a day, and olanzapine, 10 mg, twice a day. Because of concerns about continuing this regimen, he was switched to haloperidol decanoate (dose not reported) and olanzapine pamoate, 405 mg, administered once per month. The injections were scheduled to alternate so that the patient would receive 1 injection every 2 weeks. The patient’s assaultive behavior was significantly reduced, and no adverse effects were reported.
Ross and Fabian.11 An African American man, age 44, was receiving haloperidol decanoate, 400 mg every 2 weeks, and oral haloperidol, 20 mg/d.11 Because of residual symptoms, a history of nonadherence, and concerns about increasing the haloperidol decanoate dose or frequency, oral haloperidol was discontinued and paliperidone palmitate, 156 mg every 4 weeks, was started. The patient was able to transition into a step-down unit, and no adverse effects were reported.
What to consider before initiating dual LAIA treatment
Evaluate the frequency of administration, flexibility of dosing, administration site, adverse effects, and monitoring requirements of each LAIA (Table 212-19) to ensure the patient’s optimal tolerability of the regimen. Previous tolerability of each medication must be confirmed by evaluating the patient’s medication history or oral or IM administration of each agent prior to initiating the LAIA.
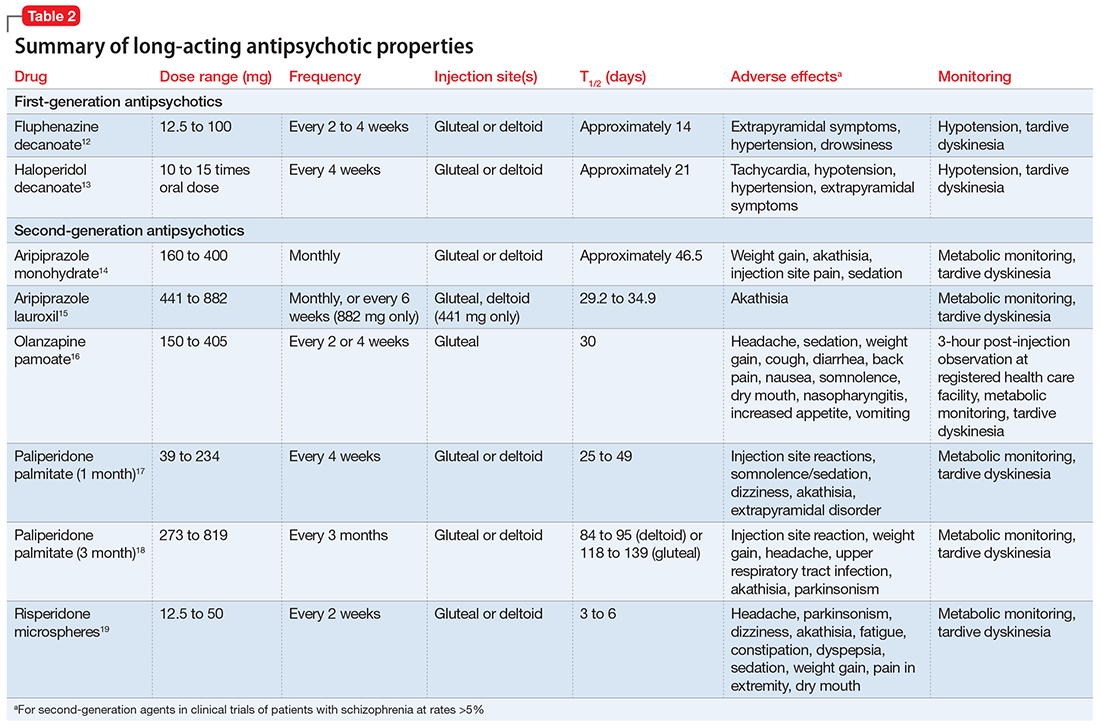
When choosing 2 agents that are each administered once every 4 weeks, consider administering the medications together every 4 weeks or alternating administration so that the patient receives an injection every 2 weeks. Receiving an injection once every 2 weeks might be beneficial for patients who need close follow-up or are more sensitive to injection site reactions, whereas a regimen of once every 4 weeks might be beneficial for patients who are more resistant to receiving the injections, so there is potentially less time spent agitated or anxious leading up to the date of the injection.
Use the lowest effective dose of each LAIA to limit adverse effects and improve tolerability of the regimen. Monitor patients closely for adverse reactions and discontinue the regimen as soon as possible if a severe adverse reaction occurs.
Cost may influence the decision to use 2 LAIAs. The majority of LAIAs in the United States are available only as branded formulations. Insurance companies may require prior authorization for the use of 2 LAIAs.
Although there are no treatment guidelines for combining 2 LAIAs, this practice has been used. A few case reports have described successful use of dual LAIA treatment, but one should consider the risk of the publication’s bias. Overall, the decision to use 2 LAIAs is difficult because there is lack of a large evidence base supporting the practice or direction from treatment guidelines. Because of this, dual LAIA treatment should not be used for most patients. In cases of treatment-resistant schizophrenia where clozapine is not an option and adherence is a concern, it is reasonable to consider this strategy on a case-by-case basis.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Lehman A, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
3. Hasan A, Falkai P, Wobrock T, et al; the WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-78.
4. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620. 5. Hasan A, Falkai P, Wobrock T, et al; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44.
6. Kreyenbuhl J, Buchanan RW, Dickerson FB, et al; Schizophrenia Patient Outcomes Research Team (PORT). The Schizophrenia Patient Outcomes Research Team (PORT): updated treatment recommendations 2009. Schizophr Bull. 2010;36(1):94-103.
7. Ladds B, Cosme R, Rivera F. Concurrent use of two depot antipsychotic medications in schizophrenia. The Internet Journal of Psychiatry. 2009;1(1):1-3.
8. Wartelsteiner F, Hofer A. Treating schizophrenia with 2 long-acting injectable antipsychotic drugs: a case report. J Clin Psychopharmacol. 2015;35(4):474-475.
9. Scangos KW, Caton M, Newman WJ. Multiple long-acting injectable antipsychotics for treatment-resistant schizophrenia: case report. J Clin Psychopharmacol. 2016;36(3):283-285.
10. Yazdi K, Rosenleitner J, Pischinger B. Combination of two depot antipsychotic drugs [in German]. Nervenarzt. 2014;85(7):870-871.
11. Ross C, Fabian T. High dose haloperidol decanoate augmentation with paliperidone palmitate. Presented at: College of Psychiatric and Neurologic Pharmacists 16th Annual Meeting; April 21-24, 2013; Colorado Springs, CO.
12. Fluphenazine decanoate [package insert]. Schaumburg, IL: APP Pharmaceuticals, LLC; 2010.
13. Haloperidol decanoate [package insert]. Rockford, IL: Mylan; 2014.
14. Abilify Maintena [package insert]. Rockville, MD: Otsuka America Pharmaceutical, Inc.; 2016.
15. Aristada [package insert]. Waltham, MA: Alkermes; 2016.
16. Zyprexa Relprevv [package insert]. Indianapolis, IN: Lilly USA, LLC; 2016.
17. Invega Sustenna [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2009.
18. Invega Trinza [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2015.
19. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2007.
Ms. S, age 39, with a 15-year history of schizophrenia and severe paranoid delusions, is admitted after physically assaulting a staff member at a group home. She is receiving paliperidone palmitate, 234 mg every 4 weeks. This has reduced the severity of her symptoms, but she continues to have persistent delusions that affect her ability to accept redirection from staff. Ms. S frequently accuses staff and peers of sexual assault, says that she is pregnant, and does not adhere to treatment recommendations for laboratory monitoring because the “staff uses her blood for experiments.”
Ms. S frequently requires administration of oral and IM haloperidol, as needed, when she becomes aggressive with the staff. She has poor insight into her mental illness and does not believe that she needs medication. Ms. S has a long history of stopping her oral antipsychotic after a few days, reporting that it is “harming her baby.” Monotherapy has been tried with various long-acting injectable antipsychotics (LAIAs), but she still exhibits persistent delusions. The treatment team decides to add a second LAIA, haloperidol decanoate, 200 mg every 4 weeks, to her regimen.
Ladds et al.7 A 49-year-old woman with schizophrenia who was hospitalized for aggressive and bizarre behavior and had been institutionalized for 20 years stopped taking her medication regimen.7 She started taking 8-hour showers with bleach, talking incoherently, and believing that someone was poisoning her. She had poor response to oral risperidone monotherapy; however, 2 months after adding oral fluphenazine and benztropine to her regimen, her symptoms substantially improved (doses not reported). Because she had impaired insight into the need for daily medication, she was started on depot fluphenazine decanoate and risperidone microspheres (doses not reported) before discharge. No substantial adverse effects were noted with this regimen.
Wartelsteiner and Hofer.8 A man who had been diagnosed with paranoid schizophrenia at age 20 presented with thought blocking, incoherence, persecutory delusions, and uncontrolled self-damaging behavior.8 He had been admitted 27 times over 7 years; during this time he received many antipsychotic monotherapies and combination regimens. A total of 8 oral antipsychotics (including clozapine) and 5 LAIAs had been administered during these trials. He significantly improved with the combination of olanzapine and risperidone. Both medications were switched to LAIA formulations to address medication nonadherence. His symptoms remained stable with risperidone microspheres, 100 mg, and olanzapine pamoate, 300 mg, each administered every 2 weeks. He did not experience any adverse effects with this combination therapy.
Scangos et al.9 A 26-year-old Vietnamese man with schizophrenia and an extensive history of unprovoked, psychotically driven assaults was given multiple antipsychotics (including clozapine) during hospitalizations, and his medication regimen consistently included 2 antipsychotics. After contracting viral gastroenteritis, he refused oral medications and required short-acting IM administration of both haloperidol, 5 mg, twice a day, and olanzapine, 10 mg, twice a day. Because of concerns about continuing this regimen, he was switched to haloperidol decanoate (dose not reported) and olanzapine pamoate, 405 mg, administered once per month. The injections were scheduled to alternate so that the patient would receive 1 injection every 2 weeks. The patient’s assaultive behavior was significantly reduced, and no adverse effects were reported.
Ross and Fabian.11 An African American man, age 44, was receiving haloperidol decanoate, 400 mg every 2 weeks, and oral haloperidol, 20 mg/d.11 Because of residual symptoms, a history of nonadherence, and concerns about increasing the haloperidol decanoate dose or frequency, oral haloperidol was discontinued and paliperidone palmitate, 156 mg every 4 weeks, was started. The patient was able to transition into a step-down unit, and no adverse effects were reported.
What to consider before initiating dual LAIA treatment
Evaluate the frequency of administration, flexibility of dosing, administration site, adverse effects, and monitoring requirements of each LAIA (Table 212-19) to ensure the patient’s optimal tolerability of the regimen. Previous tolerability of each medication must be confirmed by evaluating the patient’s medication history or oral or IM administration of each agent prior to initiating the LAIA.
When choosing 2 agents that are each administered once every 4 weeks, consider administering the medications together every 4 weeks or alternating administration so that the patient receives an injection every 2 weeks. Receiving an injection once every 2 weeks might be beneficial for patients who need close follow-up or are more sensitive to injection site reactions, whereas a regimen of once every 4 weeks might be beneficial for patients who are more resistant to receiving the injections, so there is potentially less time spent agitated or anxious leading up to the date of the injection.
Use the lowest effective dose of each LAIA to limit adverse effects and improve tolerability of the regimen. Monitor patients closely for adverse reactions and discontinue the regimen as soon as possible if a severe adverse reaction occurs.
Cost may influence the decision to use 2 LAIAs. The majority of LAIAs in the United States are available only as branded formulations. Insurance companies may require prior authorization for the use of 2 LAIAs.
Although there are no treatment guidelines for combining 2 LAIAs, this practice has been used. A few case reports have described successful use of dual LAIA treatment, but one should consider the risk of the publication’s bias. Overall, the decision to use 2 LAIAs is difficult because there is lack of a large evidence base supporting the practice or direction from treatment guidelines. Because of this, dual LAIA treatment should not be used for most patients. In cases of treatment-resistant schizophrenia where clozapine is not an option and adherence is a concern, it is reasonable to consider this strategy on a case-by-case basis.
Ms. S, age 39, with a 15-year history of schizophrenia and severe paranoid delusions, is admitted after physically assaulting a staff member at a group home. She is receiving paliperidone palmitate, 234 mg every 4 weeks. This has reduced the severity of her symptoms, but she continues to have persistent delusions that affect her ability to accept redirection from staff. Ms. S frequently accuses staff and peers of sexual assault, says that she is pregnant, and does not adhere to treatment recommendations for laboratory monitoring because the “staff uses her blood for experiments.”
Ms. S frequently requires administration of oral and IM haloperidol, as needed, when she becomes aggressive with the staff. She has poor insight into her mental illness and does not believe that she needs medication. Ms. S has a long history of stopping her oral antipsychotic after a few days, reporting that it is “harming her baby.” Monotherapy has been tried with various long-acting injectable antipsychotics (LAIAs), but she still exhibits persistent delusions. The treatment team decides to add a second LAIA, haloperidol decanoate, 200 mg every 4 weeks, to her regimen.
Ladds et al.7 A 49-year-old woman with schizophrenia who was hospitalized for aggressive and bizarre behavior and had been institutionalized for 20 years stopped taking her medication regimen.7 She started taking 8-hour showers with bleach, talking incoherently, and believing that someone was poisoning her. She had poor response to oral risperidone monotherapy; however, 2 months after adding oral fluphenazine and benztropine to her regimen, her symptoms substantially improved (doses not reported). Because she had impaired insight into the need for daily medication, she was started on depot fluphenazine decanoate and risperidone microspheres (doses not reported) before discharge. No substantial adverse effects were noted with this regimen.
Wartelsteiner and Hofer.8 A man who had been diagnosed with paranoid schizophrenia at age 20 presented with thought blocking, incoherence, persecutory delusions, and uncontrolled self-damaging behavior.8 He had been admitted 27 times over 7 years; during this time he received many antipsychotic monotherapies and combination regimens. A total of 8 oral antipsychotics (including clozapine) and 5 LAIAs had been administered during these trials. He significantly improved with the combination of olanzapine and risperidone. Both medications were switched to LAIA formulations to address medication nonadherence. His symptoms remained stable with risperidone microspheres, 100 mg, and olanzapine pamoate, 300 mg, each administered every 2 weeks. He did not experience any adverse effects with this combination therapy.
Scangos et al.9 A 26-year-old Vietnamese man with schizophrenia and an extensive history of unprovoked, psychotically driven assaults was given multiple antipsychotics (including clozapine) during hospitalizations, and his medication regimen consistently included 2 antipsychotics. After contracting viral gastroenteritis, he refused oral medications and required short-acting IM administration of both haloperidol, 5 mg, twice a day, and olanzapine, 10 mg, twice a day. Because of concerns about continuing this regimen, he was switched to haloperidol decanoate (dose not reported) and olanzapine pamoate, 405 mg, administered once per month. The injections were scheduled to alternate so that the patient would receive 1 injection every 2 weeks. The patient’s assaultive behavior was significantly reduced, and no adverse effects were reported.
Ross and Fabian.11 An African American man, age 44, was receiving haloperidol decanoate, 400 mg every 2 weeks, and oral haloperidol, 20 mg/d.11 Because of residual symptoms, a history of nonadherence, and concerns about increasing the haloperidol decanoate dose or frequency, oral haloperidol was discontinued and paliperidone palmitate, 156 mg every 4 weeks, was started. The patient was able to transition into a step-down unit, and no adverse effects were reported.
What to consider before initiating dual LAIA treatment
Evaluate the frequency of administration, flexibility of dosing, administration site, adverse effects, and monitoring requirements of each LAIA (Table 212-19) to ensure the patient’s optimal tolerability of the regimen. Previous tolerability of each medication must be confirmed by evaluating the patient’s medication history or oral or IM administration of each agent prior to initiating the LAIA.
When choosing 2 agents that are each administered once every 4 weeks, consider administering the medications together every 4 weeks or alternating administration so that the patient receives an injection every 2 weeks. Receiving an injection once every 2 weeks might be beneficial for patients who need close follow-up or are more sensitive to injection site reactions, whereas a regimen of once every 4 weeks might be beneficial for patients who are more resistant to receiving the injections, so there is potentially less time spent agitated or anxious leading up to the date of the injection.
Use the lowest effective dose of each LAIA to limit adverse effects and improve tolerability of the regimen. Monitor patients closely for adverse reactions and discontinue the regimen as soon as possible if a severe adverse reaction occurs.
Cost may influence the decision to use 2 LAIAs. The majority of LAIAs in the United States are available only as branded formulations. Insurance companies may require prior authorization for the use of 2 LAIAs.
Although there are no treatment guidelines for combining 2 LAIAs, this practice has been used. A few case reports have described successful use of dual LAIA treatment, but one should consider the risk of the publication’s bias. Overall, the decision to use 2 LAIAs is difficult because there is lack of a large evidence base supporting the practice or direction from treatment guidelines. Because of this, dual LAIA treatment should not be used for most patients. In cases of treatment-resistant schizophrenia where clozapine is not an option and adherence is a concern, it is reasonable to consider this strategy on a case-by-case basis.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Lehman A, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
3. Hasan A, Falkai P, Wobrock T, et al; the WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-78.
4. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620. 5. Hasan A, Falkai P, Wobrock T, et al; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44.
6. Kreyenbuhl J, Buchanan RW, Dickerson FB, et al; Schizophrenia Patient Outcomes Research Team (PORT). The Schizophrenia Patient Outcomes Research Team (PORT): updated treatment recommendations 2009. Schizophr Bull. 2010;36(1):94-103.
7. Ladds B, Cosme R, Rivera F. Concurrent use of two depot antipsychotic medications in schizophrenia. The Internet Journal of Psychiatry. 2009;1(1):1-3.
8. Wartelsteiner F, Hofer A. Treating schizophrenia with 2 long-acting injectable antipsychotic drugs: a case report. J Clin Psychopharmacol. 2015;35(4):474-475.
9. Scangos KW, Caton M, Newman WJ. Multiple long-acting injectable antipsychotics for treatment-resistant schizophrenia: case report. J Clin Psychopharmacol. 2016;36(3):283-285.
10. Yazdi K, Rosenleitner J, Pischinger B. Combination of two depot antipsychotic drugs [in German]. Nervenarzt. 2014;85(7):870-871.
11. Ross C, Fabian T. High dose haloperidol decanoate augmentation with paliperidone palmitate. Presented at: College of Psychiatric and Neurologic Pharmacists 16th Annual Meeting; April 21-24, 2013; Colorado Springs, CO.
12. Fluphenazine decanoate [package insert]. Schaumburg, IL: APP Pharmaceuticals, LLC; 2010.
13. Haloperidol decanoate [package insert]. Rockford, IL: Mylan; 2014.
14. Abilify Maintena [package insert]. Rockville, MD: Otsuka America Pharmaceutical, Inc.; 2016.
15. Aristada [package insert]. Waltham, MA: Alkermes; 2016.
16. Zyprexa Relprevv [package insert]. Indianapolis, IN: Lilly USA, LLC; 2016.
17. Invega Sustenna [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2009.
18. Invega Trinza [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2015.
19. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2007.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Lehman A, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
3. Hasan A, Falkai P, Wobrock T, et al; the WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-78.
4. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620. 5. Hasan A, Falkai P, Wobrock T, et al; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44.
6. Kreyenbuhl J, Buchanan RW, Dickerson FB, et al; Schizophrenia Patient Outcomes Research Team (PORT). The Schizophrenia Patient Outcomes Research Team (PORT): updated treatment recommendations 2009. Schizophr Bull. 2010;36(1):94-103.
7. Ladds B, Cosme R, Rivera F. Concurrent use of two depot antipsychotic medications in schizophrenia. The Internet Journal of Psychiatry. 2009;1(1):1-3.
8. Wartelsteiner F, Hofer A. Treating schizophrenia with 2 long-acting injectable antipsychotic drugs: a case report. J Clin Psychopharmacol. 2015;35(4):474-475.
9. Scangos KW, Caton M, Newman WJ. Multiple long-acting injectable antipsychotics for treatment-resistant schizophrenia: case report. J Clin Psychopharmacol. 2016;36(3):283-285.
10. Yazdi K, Rosenleitner J, Pischinger B. Combination of two depot antipsychotic drugs [in German]. Nervenarzt. 2014;85(7):870-871.
11. Ross C, Fabian T. High dose haloperidol decanoate augmentation with paliperidone palmitate. Presented at: College of Psychiatric and Neurologic Pharmacists 16th Annual Meeting; April 21-24, 2013; Colorado Springs, CO.
12. Fluphenazine decanoate [package insert]. Schaumburg, IL: APP Pharmaceuticals, LLC; 2010.
13. Haloperidol decanoate [package insert]. Rockford, IL: Mylan; 2014.
14. Abilify Maintena [package insert]. Rockville, MD: Otsuka America Pharmaceutical, Inc.; 2016.
15. Aristada [package insert]. Waltham, MA: Alkermes; 2016.
16. Zyprexa Relprevv [package insert]. Indianapolis, IN: Lilly USA, LLC; 2016.
17. Invega Sustenna [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2009.
18. Invega Trinza [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2015.
19. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2007.