User login
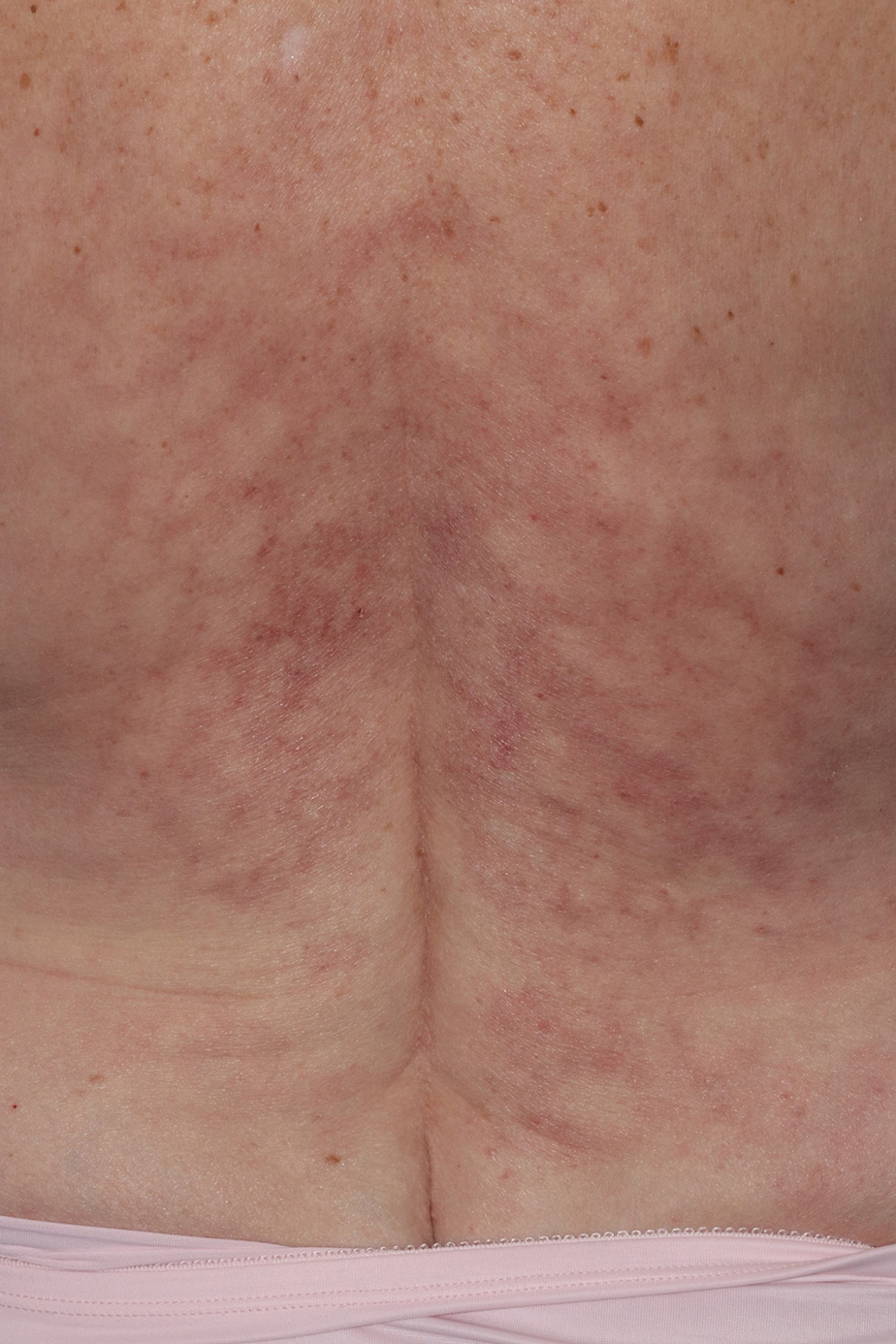
Reticulated Brownish Erythema on the Lower Back
The Diagnosis: Erythema Ab Igne
Based on the patient's long-standing history of back pain treated with heating pads as well as the normal laboratory findings and skin examination, a diagnosis of erythema ab igne (EAI) was made.
Erythema ab igne presents as reticulated brownish erythema or hyperpigmentation on sites exposed to prolonged use of heat sources such as heating pads, laptops, and space heaters. Erythema ab igne most commonly affects the lower back, thighs, or legs1-6; however, EAI can appear on atypical sites such as the forehead and eyebrows due to newer technology (eg, virtual reality headsets).7 The level of heat required for EAI to occur is below the threshold for thermal burns (<45 °C [113 °F]).1 Erythema ab igne can occur at any age, and woman are more commonly affected than men.8 The pathophysiology currently is unknown; however, recurrent and prolonged heat exposure may damage superficial vessels. As a result, hemosiderin accumulates in the skin, and hyperpigmentation subsequently occurs.9
The diagnosis of EAI is clinical, and early stages of the rash present as blanching reticulated erythema in areas associated with heat exposure. If the offending source of heat is not removed, EAI can progress to nonblanching, fixed, hyperpigmented plaques with skin atrophy, bullae, or hyperkeratosis. Patients often are asymptomatic; however, mild burning may occur.2 Histopathology reveals cellular atypia, epidermal atrophy, dilation of dermal blood vessels, a minute inflammatory infiltrate, and keratinocyte apoptosis.10 Skin biopsy may be necessary in cases of suspected malignancy due to chronic heat exposure. Lesions that ulcerate or evolve should raise suspicion for malignancy.11 Squamous cell carcinoma is the most common malignancy associated with EAI; other malignancies that may manifest include basal cell carcinoma, Merkel cell carcinoma, or cutaneous marginal zone lymphoma.2,12-14
Erythema ab igne often is mistaken for livedo reticularis, which appears more erythematous without hyperpigmentation or epidermal changes and may be associated with a pathologic state.15 The differential diagnosis in our patient, who was in her 40s with a history of fatigue and joint pain, included livedo reticularis associated with lupus; however, the history of heating pad use, normal laboratory findings, and presence of epidermal changes suggested EAI. Lupus typically affects the hand and knee joints.16 Additionally, livedo reticularis more commonly appears on the legs.15
Other differentials for EAI include livedo racemosa, cutaneous T-cell lymphoma, and cutis marmorata. Livedo racemosa presents with broken rings of erythema in young to middle-aged women and primarily affects the trunk and proximal limbs. It is associated with an underlying condition such as polyarteritis nodosa and less commonly with lupus erythematosus with antiphospholipid or Sneddon syndrome.15,17 Cutaneous T-cell lymphoma typically manifests with poikilodermatous patches larger than the palm, especially in covered areas of skin.18 Cutis marmorata is transient and temperature dependent.9
The key intervention for EAI is removal of the offending heat source.2 Patients should be counseled that the erythema and hyperpigmentation may take months to years to resolve. Topical hydroquinone or tretinoin may be used in cases of persistent hyperpigmentation.19 Patients who continue to use heating pads for long-standing pain should be advised to limit their use to short intervals without occlusion. If malignancy is a concern, a biopsy should be performed.20
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Patel DP. The evolving nomenclature of erythema ab igne-redness from fire. JAMA Dermatol. 2017;153:685. doi:10.1001/jamadermatol.2017.2021
- Arnold AW, Itin PH. Laptop computer-induced erythema ab igne in a child and review of the literature. Pediatrics. 2010;126:E1227-E1230. doi:10.1542/peds.2010-1390
- Riahi RR, Cohen PR. Laptop-induced erythema ab igne: report and review of literature. Dermatol Online J. 2012;18:5.
- Haleem Z, Philip J, Muhammad S. Erythema ab igne: a rare presentation of toasted skin syndrome with the use of a space heater. Cureus. 2021;13:e13401. doi:10.7759/cureus.13401
- Moreau T, Benzaquen M, Gueissaz F. Erythema ab igne after using a virtual reality headset: a new phenomenon to know. J Eur Acad Dermatol Venereol. 2022;36:E932-E933. doi:10.1111/jdv.18371
- Ozturk M, An I. Clinical features and etiology of patients with erythema ab igne: a retrospective multicenter study. J Cosmet Dermatol. 2020;19:1774-1779. doi:10.1111/jocd.13210
- Gmuca S, Yu J, Weiss PF, et al. Erythema ab igne in an adolescent with chronic pain: an alarming cutaneous eruption from heat exposure. Pediatr Emerg Care. 2020;36:E236-E238. doi:10.1097 /PEC.0000000000001460
- Wells A, Desai A, Rudnick EW, et al. Erythema ab igne with features resembling keratosis lichenoides chronica. J Cutan Pathol. 2021;48:151-153. doi:10.1111/cup.13885
- Milchak M, Smucker J, Chung CG, et al. Erythema ab igne due to heating pad use: a case report and review of clinical presentation, prevention, and complications. Case Rep Med. 2016;2016:1862480. doi:10.1155/2016/1862480
- Daneshvar E, Seraji S, Kamyab-Hesari K, et al. Basal cell carcinoma associated with erythema ab igne. Dermatol Online J. 2020;26:13030 /qt3kz985b4.
- Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124:110-113.
- Wharton J, Roffwarg D, Miller J, et al. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62:1080-1081. doi:10.1016/j.jaad.2009.08.005
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103 /2229-5178.164493
- Grossman JM. Lupus arthritis. Best Pract Res Clin Rheumatol. 2009;23:495-506. doi:10.1016/j.berh.2009.04.003
- Aria AB, Chen L, Silapunt S. Erythema ab igne from heating pad use: a report of three clinical cases and a differential diagnosis. Cureus. 2018;10:E2635. doi:10.7759/cureus.2635
- Wilcox RA. Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92:1085-1102. doi:10.1002/ajh.24876
- Pennitz A, Kinberger M, Avila Valle G, et al. Self-applied topical interventions for melasma: a systematic review and meta-analysis of data from randomized, investigator-blinded clinical trials. Br J Dermatol. 2022;187:309-317.
- Sahl WJ, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol. 1992;27:109-110.
The Diagnosis: Erythema Ab Igne
Based on the patient's long-standing history of back pain treated with heating pads as well as the normal laboratory findings and skin examination, a diagnosis of erythema ab igne (EAI) was made.
Erythema ab igne presents as reticulated brownish erythema or hyperpigmentation on sites exposed to prolonged use of heat sources such as heating pads, laptops, and space heaters. Erythema ab igne most commonly affects the lower back, thighs, or legs1-6; however, EAI can appear on atypical sites such as the forehead and eyebrows due to newer technology (eg, virtual reality headsets).7 The level of heat required for EAI to occur is below the threshold for thermal burns (<45 °C [113 °F]).1 Erythema ab igne can occur at any age, and woman are more commonly affected than men.8 The pathophysiology currently is unknown; however, recurrent and prolonged heat exposure may damage superficial vessels. As a result, hemosiderin accumulates in the skin, and hyperpigmentation subsequently occurs.9
The diagnosis of EAI is clinical, and early stages of the rash present as blanching reticulated erythema in areas associated with heat exposure. If the offending source of heat is not removed, EAI can progress to nonblanching, fixed, hyperpigmented plaques with skin atrophy, bullae, or hyperkeratosis. Patients often are asymptomatic; however, mild burning may occur.2 Histopathology reveals cellular atypia, epidermal atrophy, dilation of dermal blood vessels, a minute inflammatory infiltrate, and keratinocyte apoptosis.10 Skin biopsy may be necessary in cases of suspected malignancy due to chronic heat exposure. Lesions that ulcerate or evolve should raise suspicion for malignancy.11 Squamous cell carcinoma is the most common malignancy associated with EAI; other malignancies that may manifest include basal cell carcinoma, Merkel cell carcinoma, or cutaneous marginal zone lymphoma.2,12-14
Erythema ab igne often is mistaken for livedo reticularis, which appears more erythematous without hyperpigmentation or epidermal changes and may be associated with a pathologic state.15 The differential diagnosis in our patient, who was in her 40s with a history of fatigue and joint pain, included livedo reticularis associated with lupus; however, the history of heating pad use, normal laboratory findings, and presence of epidermal changes suggested EAI. Lupus typically affects the hand and knee joints.16 Additionally, livedo reticularis more commonly appears on the legs.15
Other differentials for EAI include livedo racemosa, cutaneous T-cell lymphoma, and cutis marmorata. Livedo racemosa presents with broken rings of erythema in young to middle-aged women and primarily affects the trunk and proximal limbs. It is associated with an underlying condition such as polyarteritis nodosa and less commonly with lupus erythematosus with antiphospholipid or Sneddon syndrome.15,17 Cutaneous T-cell lymphoma typically manifests with poikilodermatous patches larger than the palm, especially in covered areas of skin.18 Cutis marmorata is transient and temperature dependent.9
The key intervention for EAI is removal of the offending heat source.2 Patients should be counseled that the erythema and hyperpigmentation may take months to years to resolve. Topical hydroquinone or tretinoin may be used in cases of persistent hyperpigmentation.19 Patients who continue to use heating pads for long-standing pain should be advised to limit their use to short intervals without occlusion. If malignancy is a concern, a biopsy should be performed.20
The Diagnosis: Erythema Ab Igne
Based on the patient's long-standing history of back pain treated with heating pads as well as the normal laboratory findings and skin examination, a diagnosis of erythema ab igne (EAI) was made.
Erythema ab igne presents as reticulated brownish erythema or hyperpigmentation on sites exposed to prolonged use of heat sources such as heating pads, laptops, and space heaters. Erythema ab igne most commonly affects the lower back, thighs, or legs1-6; however, EAI can appear on atypical sites such as the forehead and eyebrows due to newer technology (eg, virtual reality headsets).7 The level of heat required for EAI to occur is below the threshold for thermal burns (<45 °C [113 °F]).1 Erythema ab igne can occur at any age, and woman are more commonly affected than men.8 The pathophysiology currently is unknown; however, recurrent and prolonged heat exposure may damage superficial vessels. As a result, hemosiderin accumulates in the skin, and hyperpigmentation subsequently occurs.9
The diagnosis of EAI is clinical, and early stages of the rash present as blanching reticulated erythema in areas associated with heat exposure. If the offending source of heat is not removed, EAI can progress to nonblanching, fixed, hyperpigmented plaques with skin atrophy, bullae, or hyperkeratosis. Patients often are asymptomatic; however, mild burning may occur.2 Histopathology reveals cellular atypia, epidermal atrophy, dilation of dermal blood vessels, a minute inflammatory infiltrate, and keratinocyte apoptosis.10 Skin biopsy may be necessary in cases of suspected malignancy due to chronic heat exposure. Lesions that ulcerate or evolve should raise suspicion for malignancy.11 Squamous cell carcinoma is the most common malignancy associated with EAI; other malignancies that may manifest include basal cell carcinoma, Merkel cell carcinoma, or cutaneous marginal zone lymphoma.2,12-14
Erythema ab igne often is mistaken for livedo reticularis, which appears more erythematous without hyperpigmentation or epidermal changes and may be associated with a pathologic state.15 The differential diagnosis in our patient, who was in her 40s with a history of fatigue and joint pain, included livedo reticularis associated with lupus; however, the history of heating pad use, normal laboratory findings, and presence of epidermal changes suggested EAI. Lupus typically affects the hand and knee joints.16 Additionally, livedo reticularis more commonly appears on the legs.15
Other differentials for EAI include livedo racemosa, cutaneous T-cell lymphoma, and cutis marmorata. Livedo racemosa presents with broken rings of erythema in young to middle-aged women and primarily affects the trunk and proximal limbs. It is associated with an underlying condition such as polyarteritis nodosa and less commonly with lupus erythematosus with antiphospholipid or Sneddon syndrome.15,17 Cutaneous T-cell lymphoma typically manifests with poikilodermatous patches larger than the palm, especially in covered areas of skin.18 Cutis marmorata is transient and temperature dependent.9
The key intervention for EAI is removal of the offending heat source.2 Patients should be counseled that the erythema and hyperpigmentation may take months to years to resolve. Topical hydroquinone or tretinoin may be used in cases of persistent hyperpigmentation.19 Patients who continue to use heating pads for long-standing pain should be advised to limit their use to short intervals without occlusion. If malignancy is a concern, a biopsy should be performed.20
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Patel DP. The evolving nomenclature of erythema ab igne-redness from fire. JAMA Dermatol. 2017;153:685. doi:10.1001/jamadermatol.2017.2021
- Arnold AW, Itin PH. Laptop computer-induced erythema ab igne in a child and review of the literature. Pediatrics. 2010;126:E1227-E1230. doi:10.1542/peds.2010-1390
- Riahi RR, Cohen PR. Laptop-induced erythema ab igne: report and review of literature. Dermatol Online J. 2012;18:5.
- Haleem Z, Philip J, Muhammad S. Erythema ab igne: a rare presentation of toasted skin syndrome with the use of a space heater. Cureus. 2021;13:e13401. doi:10.7759/cureus.13401
- Moreau T, Benzaquen M, Gueissaz F. Erythema ab igne after using a virtual reality headset: a new phenomenon to know. J Eur Acad Dermatol Venereol. 2022;36:E932-E933. doi:10.1111/jdv.18371
- Ozturk M, An I. Clinical features and etiology of patients with erythema ab igne: a retrospective multicenter study. J Cosmet Dermatol. 2020;19:1774-1779. doi:10.1111/jocd.13210
- Gmuca S, Yu J, Weiss PF, et al. Erythema ab igne in an adolescent with chronic pain: an alarming cutaneous eruption from heat exposure. Pediatr Emerg Care. 2020;36:E236-E238. doi:10.1097 /PEC.0000000000001460
- Wells A, Desai A, Rudnick EW, et al. Erythema ab igne with features resembling keratosis lichenoides chronica. J Cutan Pathol. 2021;48:151-153. doi:10.1111/cup.13885
- Milchak M, Smucker J, Chung CG, et al. Erythema ab igne due to heating pad use: a case report and review of clinical presentation, prevention, and complications. Case Rep Med. 2016;2016:1862480. doi:10.1155/2016/1862480
- Daneshvar E, Seraji S, Kamyab-Hesari K, et al. Basal cell carcinoma associated with erythema ab igne. Dermatol Online J. 2020;26:13030 /qt3kz985b4.
- Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124:110-113.
- Wharton J, Roffwarg D, Miller J, et al. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62:1080-1081. doi:10.1016/j.jaad.2009.08.005
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103 /2229-5178.164493
- Grossman JM. Lupus arthritis. Best Pract Res Clin Rheumatol. 2009;23:495-506. doi:10.1016/j.berh.2009.04.003
- Aria AB, Chen L, Silapunt S. Erythema ab igne from heating pad use: a report of three clinical cases and a differential diagnosis. Cureus. 2018;10:E2635. doi:10.7759/cureus.2635
- Wilcox RA. Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92:1085-1102. doi:10.1002/ajh.24876
- Pennitz A, Kinberger M, Avila Valle G, et al. Self-applied topical interventions for melasma: a systematic review and meta-analysis of data from randomized, investigator-blinded clinical trials. Br J Dermatol. 2022;187:309-317.
- Sahl WJ, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol. 1992;27:109-110.
- Wipf AJ, Brown MR. Malignant transformation of erythema ab igne. JAAD Case Rep. 2022;26:85-87. doi:10.1016/j.jdcr.2022.06.018
- Sigmon JR, Cantrell J, Teague D, et al. Poorly differentiated carcinoma arising in the setting of erythema ab igne. Am J Dermatopathol. 2013;35:676-678. doi:10.1097/DAD.0b013e3182871648
- Patel DP. The evolving nomenclature of erythema ab igne-redness from fire. JAMA Dermatol. 2017;153:685. doi:10.1001/jamadermatol.2017.2021
- Arnold AW, Itin PH. Laptop computer-induced erythema ab igne in a child and review of the literature. Pediatrics. 2010;126:E1227-E1230. doi:10.1542/peds.2010-1390
- Riahi RR, Cohen PR. Laptop-induced erythema ab igne: report and review of literature. Dermatol Online J. 2012;18:5.
- Haleem Z, Philip J, Muhammad S. Erythema ab igne: a rare presentation of toasted skin syndrome with the use of a space heater. Cureus. 2021;13:e13401. doi:10.7759/cureus.13401
- Moreau T, Benzaquen M, Gueissaz F. Erythema ab igne after using a virtual reality headset: a new phenomenon to know. J Eur Acad Dermatol Venereol. 2022;36:E932-E933. doi:10.1111/jdv.18371
- Ozturk M, An I. Clinical features and etiology of patients with erythema ab igne: a retrospective multicenter study. J Cosmet Dermatol. 2020;19:1774-1779. doi:10.1111/jocd.13210
- Gmuca S, Yu J, Weiss PF, et al. Erythema ab igne in an adolescent with chronic pain: an alarming cutaneous eruption from heat exposure. Pediatr Emerg Care. 2020;36:E236-E238. doi:10.1097 /PEC.0000000000001460
- Wells A, Desai A, Rudnick EW, et al. Erythema ab igne with features resembling keratosis lichenoides chronica. J Cutan Pathol. 2021;48:151-153. doi:10.1111/cup.13885
- Milchak M, Smucker J, Chung CG, et al. Erythema ab igne due to heating pad use: a case report and review of clinical presentation, prevention, and complications. Case Rep Med. 2016;2016:1862480. doi:10.1155/2016/1862480
- Daneshvar E, Seraji S, Kamyab-Hesari K, et al. Basal cell carcinoma associated with erythema ab igne. Dermatol Online J. 2020;26:13030 /qt3kz985b4.
- Jones CS, Tyring SK, Lee PC, et al. Development of neuroendocrine (Merkel cell) carcinoma mixed with squamous cell carcinoma in erythema ab igne. Arch Dermatol. 1988;124:110-113.
- Wharton J, Roffwarg D, Miller J, et al. Cutaneous marginal zone lymphoma arising in the setting of erythema ab igne. J Am Acad Dermatol. 2010;62:1080-1081. doi:10.1016/j.jaad.2009.08.005
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103 /2229-5178.164493
- Grossman JM. Lupus arthritis. Best Pract Res Clin Rheumatol. 2009;23:495-506. doi:10.1016/j.berh.2009.04.003
- Aria AB, Chen L, Silapunt S. Erythema ab igne from heating pad use: a report of three clinical cases and a differential diagnosis. Cureus. 2018;10:E2635. doi:10.7759/cureus.2635
- Wilcox RA. Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92:1085-1102. doi:10.1002/ajh.24876
- Pennitz A, Kinberger M, Avila Valle G, et al. Self-applied topical interventions for melasma: a systematic review and meta-analysis of data from randomized, investigator-blinded clinical trials. Br J Dermatol. 2022;187:309-317.
- Sahl WJ, Taira JW. Erythema ab igne: treatment with 5-fluorouracil cream. J Am Acad Dermatol. 1992;27:109-110.
A 42-year-old woman presented with an asymptomatic, erythematous, lacelike rash on the lower back of 8 months’ duration that was first noticed by her husband. The patient had a long-standing history of chronic fatigue and lower back pain treated with acetaminophen, diclofenac gel, and heating pads. Physical examination revealed reticulated brownish erythema confined to the lower back. Laboratory findings were unremarkable.
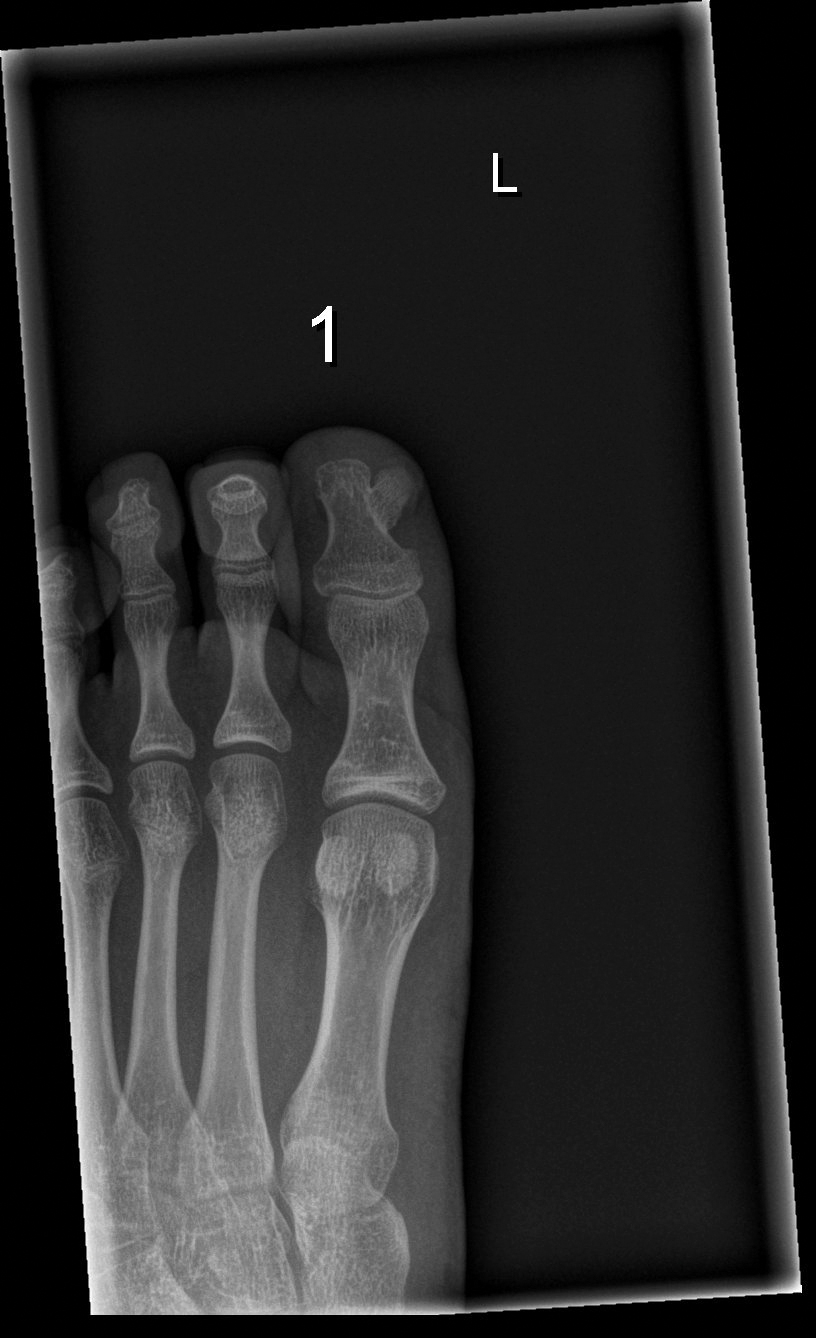
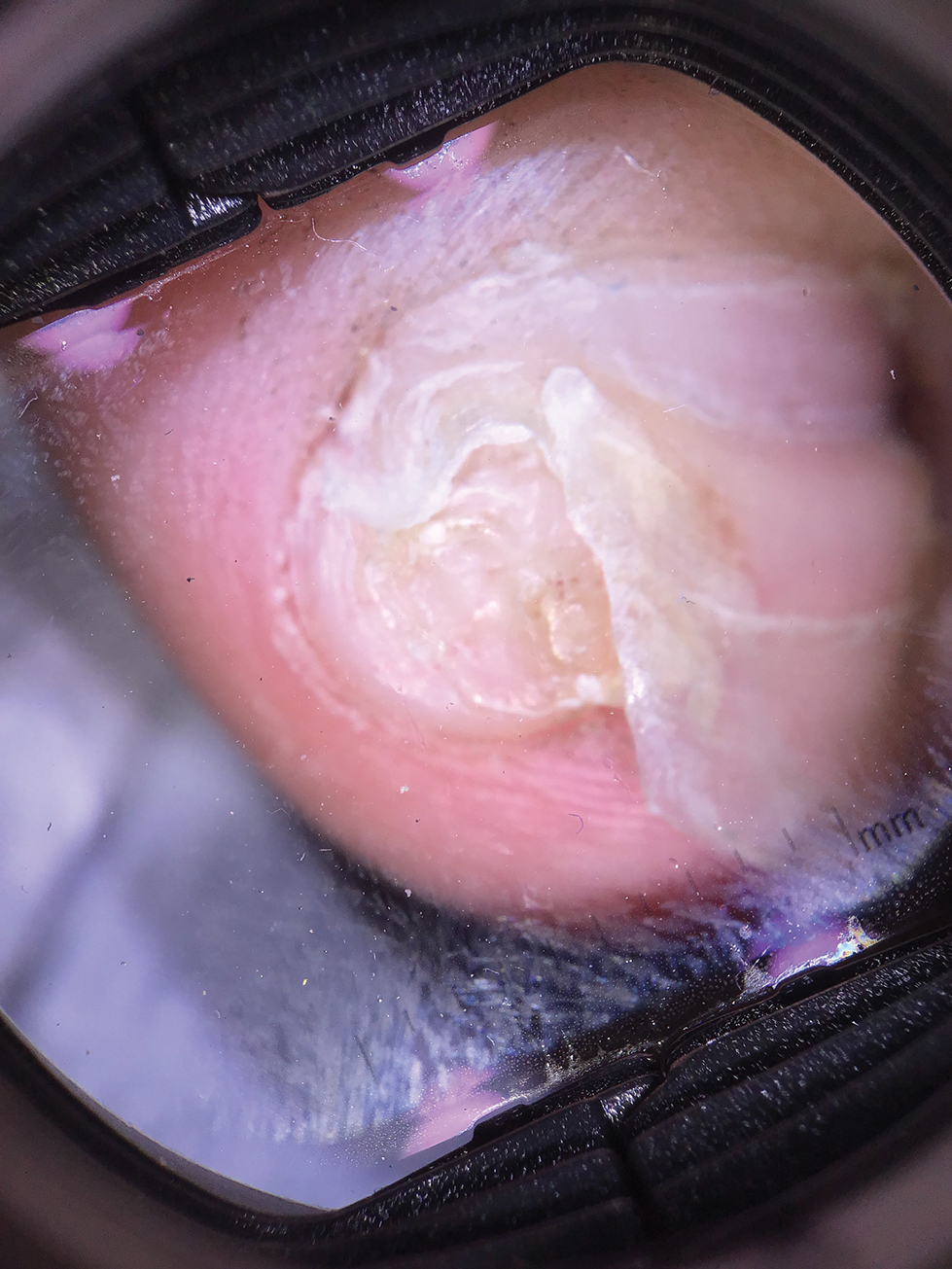
Subungual Nodule in a Pediatric Patient
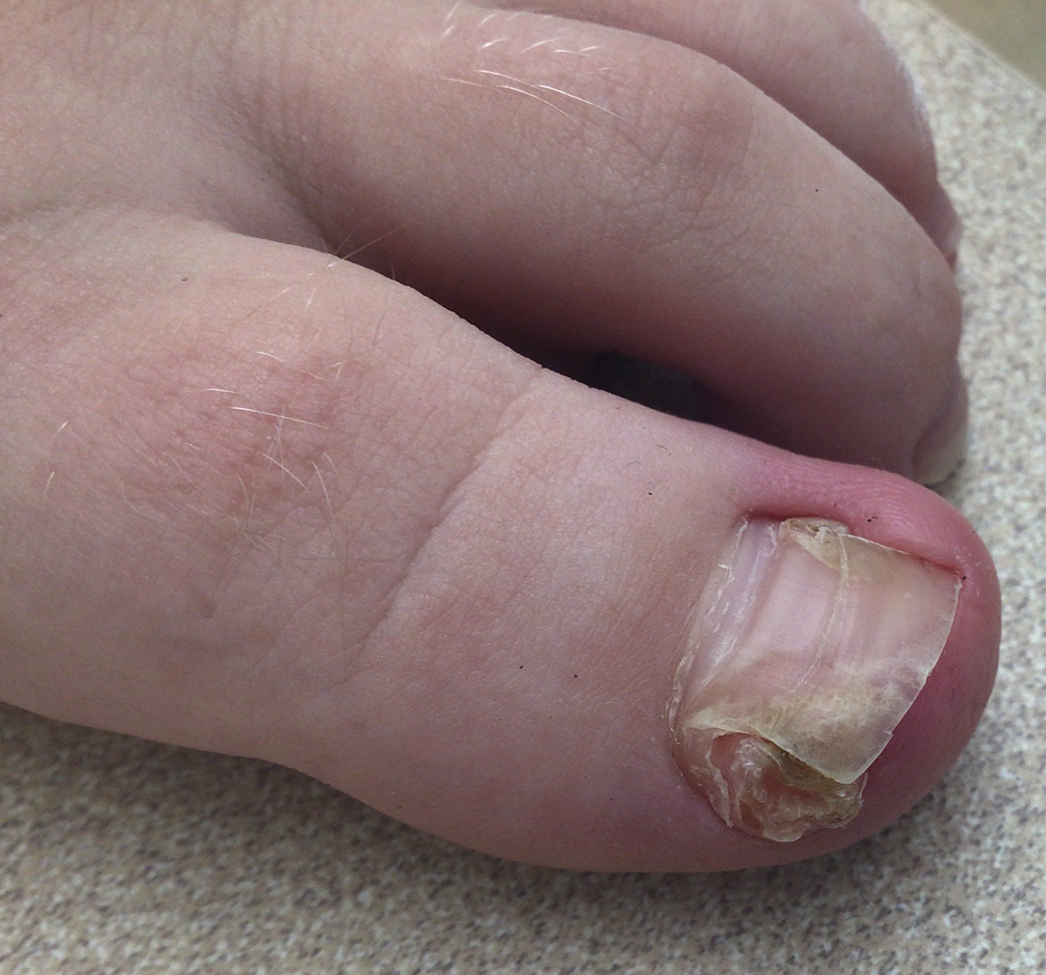
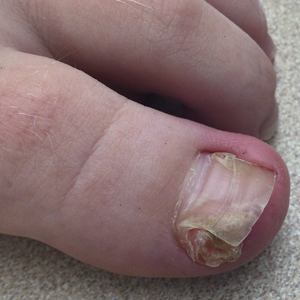
The Diagnosis: Subungual Exostosis
Subungual exostosis should be considered as a possible cause of an exophytic subungual nodule in a young active female. In our patient, the involvement of the great toe was a clue, as the hallux is the most common location of subungual exostosis. The patient’s age and sex also were supportive, as subungual exostosis is most common in female children and adolescents— particularly those who are active, as trauma is thought to play a possible role in development of this benign tumor.1-3 Radiography is the preferred modality for diagnosis; in our case, it showed a trabecular bony overgrowth (Figure 1), which confirmed the diagnosis. Subungual exostosis is a rare, benign, osteocartilaginous tumor of trabecular bone. The etiology is unknown but is hypothesized to be related to trauma, infection, or activation of a cartilaginous cyst.1,3 The subungual nodule may be asymptomatic or painful. Disruption and elevation of the nail plate is common.4 The differential diagnosis includes amelanotic melanoma, fibroma, fibrokeratoma, osteochondroma, pyogenic granuloma, squamous cell carcinoma, glomus tumor, and verruca vulgaris, among others.5
Physical examination demonstrates a firm, fixed, subungual nodule, often with an accompanying nail deformity. Further workup is required to confirm the benign nature of the lesion and exclude nail tumors such as melanoma or squamous cell carcinoma. Radiography is the gold standard for diagnosis, demonstrating a trabecular bony overgrowth.6 Performing a radiograph as the initial diagnostic test spares the patient from unnecessary procedures such as biopsy or expensive imaging techniques such as magnetic resonance imaging. Early lesions may not demonstrate sufficient bone formation shown on radiography. In these situations, a combination of dermoscopy and histopathologic examination may aid in diagnosis (Figure 2).4 Vascular ectasia, hyperkeratosis, onycholysis, and ulceration are the most common findings on dermoscopy (in ascending order).7 Histopathology typically demonstrates a base or stalk of normal-appearing trabecular bone with a fibrocartilage cap.8 However, initial clinical workup via radiography allows for the least-invasive and highest-yield intervention. Clinical suspicion for this condition is important, as it can be diagnosed with noninvasive inexpensive imaging rather than biopsy or more specialized imaging modalities. Appropriate recognition can save young patients from unnecessary and expensive procedures. Treatment typically involves surgical excision; to prevent regrowth, removal of the lesion at the base of the bone is recommended.2
Although amelanotic melanoma also can manifest as a subungual nail tumor, it would be unusual in a young child and would not be expected to show characteristic changes on radiography. A glomus tumor would be painful, is more common on the fingers than on the toes, and typically has a bluish hue.9 Verruca vulgaris can occur subungually but is more common around the nailfold and often has the characteristic dermoscopic finding of thrombosed capillaries. It also would not be expected to show characteristic radiographic findings. Osteochondroma can occur in young patients and can appear clinically similar to subungual exostosis; however, it typically is painful.10
- Pascoal D, Balaco I, Alves C, et al. Subungual exostosis—treatment results with preservation of the nail bed. J Pediatr Orthop B. 2020;29:382-386.
- Yousefian F, Davis B, Browning JC. Pediatric subungual exostosis. Cutis. 2021;108:256-257.
- Chiheb S, Slimani Y, Karam R, et al. Subungual exostosis: a case series of 48 patients. Skin Appendage Disord. 2021;7:475-479.
- Zhang W, Gu L, Fan H, et al. Subungual exostosis with an unusual dermoscopic feature. JAAD Case Rep. 2020;6:725-726.
- Demirdag HG, Tugrul Ayanoglu B, Akay BN. Dermoscopic features of subungual exostosis. Australas J Dermatol. 2019;60:E138-E141.
- Tritto M, Mirkin G, Hao X. Subungual exostosis on the right hallux. J Am Podiatr Med Assoc. 2021;111.
- Piccolo V, Argenziano G, Alessandrini AM, et al. Dermoscopy of subungual exostosis: a retrospective study of 10 patients. Dermatology. 2017;233:80-85.
- Lee SK, Jung MS, Lee YH, et al. Two distinctive subungual pathologies: subungual exostosis and subungual osteochondroma. Foot Ankle Int. 2007;28:595-601. doi:10.3113/FAI.2007.0595
- Samaniego E, Crespo A, Sanz A. Key diagnostic features and treatment of subungual glomus tumor. Actas Dermosifiliogr. 2009;100:875-882.
- Glick S. Subungual osteochondroma of the third toe. Consult.360. 2013;12.
The Diagnosis: Subungual Exostosis
Subungual exostosis should be considered as a possible cause of an exophytic subungual nodule in a young active female. In our patient, the involvement of the great toe was a clue, as the hallux is the most common location of subungual exostosis. The patient’s age and sex also were supportive, as subungual exostosis is most common in female children and adolescents— particularly those who are active, as trauma is thought to play a possible role in development of this benign tumor.1-3 Radiography is the preferred modality for diagnosis; in our case, it showed a trabecular bony overgrowth (Figure 1), which confirmed the diagnosis. Subungual exostosis is a rare, benign, osteocartilaginous tumor of trabecular bone. The etiology is unknown but is hypothesized to be related to trauma, infection, or activation of a cartilaginous cyst.1,3 The subungual nodule may be asymptomatic or painful. Disruption and elevation of the nail plate is common.4 The differential diagnosis includes amelanotic melanoma, fibroma, fibrokeratoma, osteochondroma, pyogenic granuloma, squamous cell carcinoma, glomus tumor, and verruca vulgaris, among others.5
Physical examination demonstrates a firm, fixed, subungual nodule, often with an accompanying nail deformity. Further workup is required to confirm the benign nature of the lesion and exclude nail tumors such as melanoma or squamous cell carcinoma. Radiography is the gold standard for diagnosis, demonstrating a trabecular bony overgrowth.6 Performing a radiograph as the initial diagnostic test spares the patient from unnecessary procedures such as biopsy or expensive imaging techniques such as magnetic resonance imaging. Early lesions may not demonstrate sufficient bone formation shown on radiography. In these situations, a combination of dermoscopy and histopathologic examination may aid in diagnosis (Figure 2).4 Vascular ectasia, hyperkeratosis, onycholysis, and ulceration are the most common findings on dermoscopy (in ascending order).7 Histopathology typically demonstrates a base or stalk of normal-appearing trabecular bone with a fibrocartilage cap.8 However, initial clinical workup via radiography allows for the least-invasive and highest-yield intervention. Clinical suspicion for this condition is important, as it can be diagnosed with noninvasive inexpensive imaging rather than biopsy or more specialized imaging modalities. Appropriate recognition can save young patients from unnecessary and expensive procedures. Treatment typically involves surgical excision; to prevent regrowth, removal of the lesion at the base of the bone is recommended.2
Although amelanotic melanoma also can manifest as a subungual nail tumor, it would be unusual in a young child and would not be expected to show characteristic changes on radiography. A glomus tumor would be painful, is more common on the fingers than on the toes, and typically has a bluish hue.9 Verruca vulgaris can occur subungually but is more common around the nailfold and often has the characteristic dermoscopic finding of thrombosed capillaries. It also would not be expected to show characteristic radiographic findings. Osteochondroma can occur in young patients and can appear clinically similar to subungual exostosis; however, it typically is painful.10
The Diagnosis: Subungual Exostosis
Subungual exostosis should be considered as a possible cause of an exophytic subungual nodule in a young active female. In our patient, the involvement of the great toe was a clue, as the hallux is the most common location of subungual exostosis. The patient’s age and sex also were supportive, as subungual exostosis is most common in female children and adolescents— particularly those who are active, as trauma is thought to play a possible role in development of this benign tumor.1-3 Radiography is the preferred modality for diagnosis; in our case, it showed a trabecular bony overgrowth (Figure 1), which confirmed the diagnosis. Subungual exostosis is a rare, benign, osteocartilaginous tumor of trabecular bone. The etiology is unknown but is hypothesized to be related to trauma, infection, or activation of a cartilaginous cyst.1,3 The subungual nodule may be asymptomatic or painful. Disruption and elevation of the nail plate is common.4 The differential diagnosis includes amelanotic melanoma, fibroma, fibrokeratoma, osteochondroma, pyogenic granuloma, squamous cell carcinoma, glomus tumor, and verruca vulgaris, among others.5
Physical examination demonstrates a firm, fixed, subungual nodule, often with an accompanying nail deformity. Further workup is required to confirm the benign nature of the lesion and exclude nail tumors such as melanoma or squamous cell carcinoma. Radiography is the gold standard for diagnosis, demonstrating a trabecular bony overgrowth.6 Performing a radiograph as the initial diagnostic test spares the patient from unnecessary procedures such as biopsy or expensive imaging techniques such as magnetic resonance imaging. Early lesions may not demonstrate sufficient bone formation shown on radiography. In these situations, a combination of dermoscopy and histopathologic examination may aid in diagnosis (Figure 2).4 Vascular ectasia, hyperkeratosis, onycholysis, and ulceration are the most common findings on dermoscopy (in ascending order).7 Histopathology typically demonstrates a base or stalk of normal-appearing trabecular bone with a fibrocartilage cap.8 However, initial clinical workup via radiography allows for the least-invasive and highest-yield intervention. Clinical suspicion for this condition is important, as it can be diagnosed with noninvasive inexpensive imaging rather than biopsy or more specialized imaging modalities. Appropriate recognition can save young patients from unnecessary and expensive procedures. Treatment typically involves surgical excision; to prevent regrowth, removal of the lesion at the base of the bone is recommended.2
Although amelanotic melanoma also can manifest as a subungual nail tumor, it would be unusual in a young child and would not be expected to show characteristic changes on radiography. A glomus tumor would be painful, is more common on the fingers than on the toes, and typically has a bluish hue.9 Verruca vulgaris can occur subungually but is more common around the nailfold and often has the characteristic dermoscopic finding of thrombosed capillaries. It also would not be expected to show characteristic radiographic findings. Osteochondroma can occur in young patients and can appear clinically similar to subungual exostosis; however, it typically is painful.10
- Pascoal D, Balaco I, Alves C, et al. Subungual exostosis—treatment results with preservation of the nail bed. J Pediatr Orthop B. 2020;29:382-386.
- Yousefian F, Davis B, Browning JC. Pediatric subungual exostosis. Cutis. 2021;108:256-257.
- Chiheb S, Slimani Y, Karam R, et al. Subungual exostosis: a case series of 48 patients. Skin Appendage Disord. 2021;7:475-479.
- Zhang W, Gu L, Fan H, et al. Subungual exostosis with an unusual dermoscopic feature. JAAD Case Rep. 2020;6:725-726.
- Demirdag HG, Tugrul Ayanoglu B, Akay BN. Dermoscopic features of subungual exostosis. Australas J Dermatol. 2019;60:E138-E141.
- Tritto M, Mirkin G, Hao X. Subungual exostosis on the right hallux. J Am Podiatr Med Assoc. 2021;111.
- Piccolo V, Argenziano G, Alessandrini AM, et al. Dermoscopy of subungual exostosis: a retrospective study of 10 patients. Dermatology. 2017;233:80-85.
- Lee SK, Jung MS, Lee YH, et al. Two distinctive subungual pathologies: subungual exostosis and subungual osteochondroma. Foot Ankle Int. 2007;28:595-601. doi:10.3113/FAI.2007.0595
- Samaniego E, Crespo A, Sanz A. Key diagnostic features and treatment of subungual glomus tumor. Actas Dermosifiliogr. 2009;100:875-882.
- Glick S. Subungual osteochondroma of the third toe. Consult.360. 2013;12.
- Pascoal D, Balaco I, Alves C, et al. Subungual exostosis—treatment results with preservation of the nail bed. J Pediatr Orthop B. 2020;29:382-386.
- Yousefian F, Davis B, Browning JC. Pediatric subungual exostosis. Cutis. 2021;108:256-257.
- Chiheb S, Slimani Y, Karam R, et al. Subungual exostosis: a case series of 48 patients. Skin Appendage Disord. 2021;7:475-479.
- Zhang W, Gu L, Fan H, et al. Subungual exostosis with an unusual dermoscopic feature. JAAD Case Rep. 2020;6:725-726.
- Demirdag HG, Tugrul Ayanoglu B, Akay BN. Dermoscopic features of subungual exostosis. Australas J Dermatol. 2019;60:E138-E141.
- Tritto M, Mirkin G, Hao X. Subungual exostosis on the right hallux. J Am Podiatr Med Assoc. 2021;111.
- Piccolo V, Argenziano G, Alessandrini AM, et al. Dermoscopy of subungual exostosis: a retrospective study of 10 patients. Dermatology. 2017;233:80-85.
- Lee SK, Jung MS, Lee YH, et al. Two distinctive subungual pathologies: subungual exostosis and subungual osteochondroma. Foot Ankle Int. 2007;28:595-601. doi:10.3113/FAI.2007.0595
- Samaniego E, Crespo A, Sanz A. Key diagnostic features and treatment of subungual glomus tumor. Actas Dermosifiliogr. 2009;100:875-882.
- Glick S. Subungual osteochondroma of the third toe. Consult.360. 2013;12.
A 13-year-old girl presented to her pediatrician with a small pink bump under the left great toenail of 8 months’ duration that was slowly growing. Months later, she developed an ingrown nail on the same toe, which was treated with partial nail avulsion by the pediatrician. Given continued nail dystrophy and a visible bump under the nail, the patient was referred to dermatology. Physical examination revealed a subungual, flesh-colored, sessile nodule causing distortion of the nail plate on the left great toe with associated intermittent redness and swelling. She denied wearing new shoes or experiencing any pain, pruritus, or purulent drainage or bleeding from the lesion. She reported being physically active and playing tennis.
Transient Symmetric Blanching Macules on a Background of Reticulate Erythema
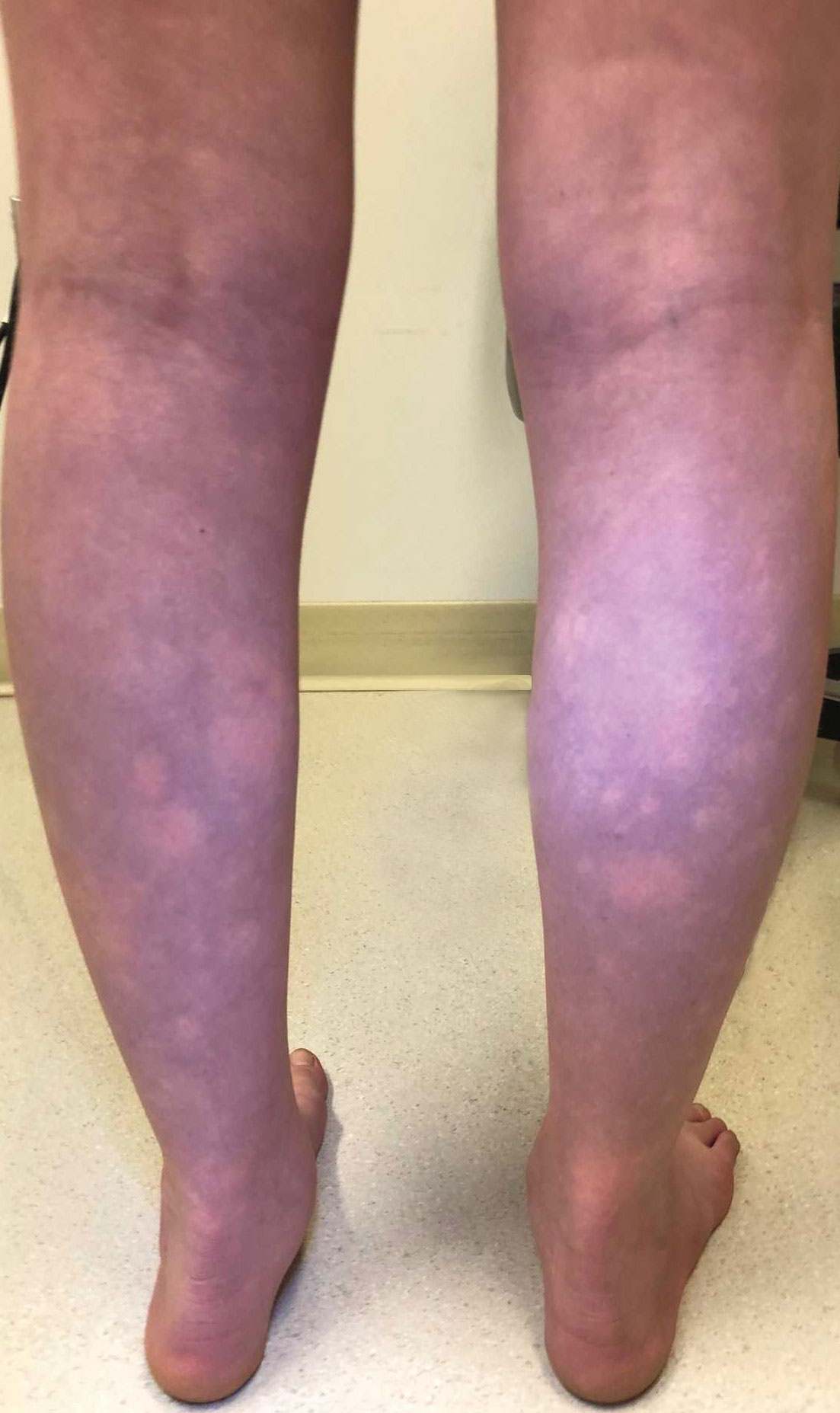
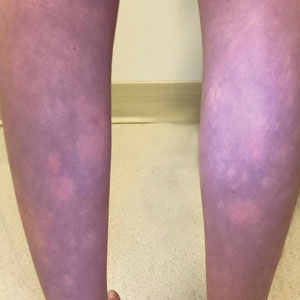
The Diagnosis: BASCULE Syndrome
The patient had previously been thought to have livedo reticularis by primary care. Repeat antinuclear antibody (ANA) testing was positive (1:1280 homogeneous [reflexive titers all negative]). However, upon dermatologic evaluation, the manifestation of the rash in addition to onset occurring with postural changes challenged the livedo reticularis diagnosis. Extensive research and consultation with dermatologic colleagues led to the diagnosis of the rare entity BASCULE syndrome. BASCULE (Bier anemic spots, cyanosis, and urticarialike eruption) syndrome was described by Bessis et al1 in 2016. It is a rare condition but may be underreported.2 It is a benign pediatric disorder in the vascular acrosyndrome family that is characterized by underlying vasomotor dysfunction in distal regions of the body. Raynaud phenomenon is a widely known member of this family. As seen in our patient, it typically presents on the distal legs and feet with numerous irregular hypopigmented macules on a cyanotic background. Red-orange papules may appear on the hypopigmented macules and often are pruritic. Lesions on the distal upper extremities are less common, and a case involving the trunk has been reported.3 Onset generally begins within a couple of minutes of standing or mechanical compression of the lower legs, with full reversal of symptoms occurring within minutes of laying down or walking. Commonly reported associated symptoms include tenderness, pruritus, edema, and pain; however, the cutaneous lesions may be asymptomatic. The condition tends to affect adolescents, as seen in our patient; however, there have been reports in infants as young as 3 months to adults aged 19 years.2
The pathophysiology behind BASCULE syndrome remains unclear but is believed to be centered around the role of physiologic venous stasis that occurs when standing. The hypoxia secondary to stasis is thought to induce amplified vasoconstriction of arterioles. These responses are further exaggerated due to absence of venoarteriolar reflexes in dermal ascending arterioles, leading to Bier spots.2 The role of mast cells and eosinophils remains unclear. It is a clinical diagnosis without clear histologic findings; therefore, biopsy was not pursued in our patient.
Although BASCULE syndrome is a benign entity, it is imperative that it be recognized to avoid a time consuming, expensive, and anxiety-producing diagnostic workup, as occurred in our patient. Although not a manifestation of systemic disease, BASCULE syndrome may be associated with orthostatic hypotension in up to 20% of cases.2,4 Therefore, these patients should undergo orthostatic testing, including the tilt table test. In our patient, these manifestations were not appreciated.
There are no current guidelines for effective treatment of BASCULE syndrome. Given the possible role of mast cells in the condition, H1 antihistamines are proposed as first-line treatment. Desloratadine (10 mg/d for 7 days) has been found to be associated with improvement of pruritus. However, a recent literature review found little evidence to support the use of H1 antihistamines for resolution of other symptoms.2
The differential diagnosis includes livedo reticularis, Bier spots, Sneddon syndrome, and urticarial vasculitis. Livedo reticularis presents as distinct, netlike, blue-erythematousviolaceous discoloration, which differs from the distinct orange-red macules in BASCULE syndrome.5 In addition to distinct variances in dermatologic presentation, livedo reticularis typically is associated with cold exposure as a causative agent, with cold avoidance as the treatment for this benign and often transient condition.6 This phenomenon was not appreciated in our patient. Livedo reticularis commonly occurs with antiphospholipid syndrome.5 This association in combination with our patient's positive ANA findings and her mother's history of miscarriages resulted in the misdiagnosis as livedo reticularis.
Bier spots manifest as white macules with surrounding erythema and typically present in young adults. When first described in the literature, it was debated if BASCULE syndrome was simply another manifestation of Bier spots or postural orthostatic intolerance,4 as there was a large consensus that postural orthostatic intolerance was associated with BASCULE syndrome, with the majority of patients not meeting criteria for the condition. Heymann4 addressed the differences in BASCULE manifestations vs typical Bier spots. The author extended the syndrome to include cyanosis, an urticarialike eruption of red-orange macules with central papules located centrally, pruritus, tenderness, and partial or diffuse edema, in addition to Bier spots.4
Sneddon syndrome is a rare progressive disorder that affects small- to medium-sized blood vessels resulting in multiple episodes of ischemia in the brain. Skin manifestations of these repeated strokes are similar to livedo reticularis, typically manifesting as livedo racemosa—irregular reticular patterns of skin mottling with reddish-blue hues.6 However, Sneddon syndrome is more generalized and widespread and differs from BASCULE syndrome in shape and histologic findings. Our patient presented with findings on the legs, which is more characteristic of livedo reticularis vs livedo racemosa. Our patient experienced resolution upon laying down and sitting, and Sneddon syndrome persists beyond postural changes. Furthermore, patients with Sneddon syndrome present with neurologic symptoms such as prodromal headaches.6
Urticarial vasculitis was ruled out in our patient because of the duration of symptoms as well as the spatial changes. Urticarial vasculitis is a rare skin condition characterized by chronic recurring urticarial lesions that may persist for more than a day. This condition typically presents in middle-aged women and rarely in children. Urticarial vasculitis is thought to be immune-complex mediated, but its cause is largely unknown. It is a common manifestation of underlying conditions such as systemic lupus erythematosus.6 Our patient had a positive ANA and possible autoimmune history from her mother; however, urticarial vasculitis does not present transiently on the legs or in the rash pattern appreciated in our patient.
- Bessis D, Jeziorski E, Rigau V, et al. Bier anaemic spots, cyanosis with urticaria-like eruption (BASCULE) syndrome: a new entity? Br J Dermatol. 2016;175:218-220. doi:10.1111/bjd.14589
- Baurens N, Briand C, Giovannini-Chami L, et al. Case report, practices survey and literature review of an under-recognized pediatric vascular disorder: the BASCULE syndrome. Front Pediatr. 2022;10:849914. doi:10.3389/fped.2022.849914
- Jiménez-Gallo D, Collantes-Rodríguez C, Ossorio-García L, et al. Bier anaemic spots, cyanosis with urticaria-like eruption (BASCULE) syndrome on trunk and upper limbs. Pediatr Dermatol. 2018;35:E313-E315. doi:10.1111/pde.13558
- Heymann WR. BASCULE syndrome: is something brewing with Bier spots? Dermatology World Insights and Inquiries. September 7, 2022. https://www.aad.org/dw/dw-insights-and-inquiries/archive/2022/bascule-syndrome
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103/2229-5178.164493
- Gu SL, Jorizzo JL. Urticarial vasculitis. Int J Womens Dermatol. 2021;7:290-297. doi:10.1016/j.ijwd.2021.01.021
The Diagnosis: BASCULE Syndrome
The patient had previously been thought to have livedo reticularis by primary care. Repeat antinuclear antibody (ANA) testing was positive (1:1280 homogeneous [reflexive titers all negative]). However, upon dermatologic evaluation, the manifestation of the rash in addition to onset occurring with postural changes challenged the livedo reticularis diagnosis. Extensive research and consultation with dermatologic colleagues led to the diagnosis of the rare entity BASCULE syndrome. BASCULE (Bier anemic spots, cyanosis, and urticarialike eruption) syndrome was described by Bessis et al1 in 2016. It is a rare condition but may be underreported.2 It is a benign pediatric disorder in the vascular acrosyndrome family that is characterized by underlying vasomotor dysfunction in distal regions of the body. Raynaud phenomenon is a widely known member of this family. As seen in our patient, it typically presents on the distal legs and feet with numerous irregular hypopigmented macules on a cyanotic background. Red-orange papules may appear on the hypopigmented macules and often are pruritic. Lesions on the distal upper extremities are less common, and a case involving the trunk has been reported.3 Onset generally begins within a couple of minutes of standing or mechanical compression of the lower legs, with full reversal of symptoms occurring within minutes of laying down or walking. Commonly reported associated symptoms include tenderness, pruritus, edema, and pain; however, the cutaneous lesions may be asymptomatic. The condition tends to affect adolescents, as seen in our patient; however, there have been reports in infants as young as 3 months to adults aged 19 years.2
The pathophysiology behind BASCULE syndrome remains unclear but is believed to be centered around the role of physiologic venous stasis that occurs when standing. The hypoxia secondary to stasis is thought to induce amplified vasoconstriction of arterioles. These responses are further exaggerated due to absence of venoarteriolar reflexes in dermal ascending arterioles, leading to Bier spots.2 The role of mast cells and eosinophils remains unclear. It is a clinical diagnosis without clear histologic findings; therefore, biopsy was not pursued in our patient.
Although BASCULE syndrome is a benign entity, it is imperative that it be recognized to avoid a time consuming, expensive, and anxiety-producing diagnostic workup, as occurred in our patient. Although not a manifestation of systemic disease, BASCULE syndrome may be associated with orthostatic hypotension in up to 20% of cases.2,4 Therefore, these patients should undergo orthostatic testing, including the tilt table test. In our patient, these manifestations were not appreciated.
There are no current guidelines for effective treatment of BASCULE syndrome. Given the possible role of mast cells in the condition, H1 antihistamines are proposed as first-line treatment. Desloratadine (10 mg/d for 7 days) has been found to be associated with improvement of pruritus. However, a recent literature review found little evidence to support the use of H1 antihistamines for resolution of other symptoms.2
The differential diagnosis includes livedo reticularis, Bier spots, Sneddon syndrome, and urticarial vasculitis. Livedo reticularis presents as distinct, netlike, blue-erythematousviolaceous discoloration, which differs from the distinct orange-red macules in BASCULE syndrome.5 In addition to distinct variances in dermatologic presentation, livedo reticularis typically is associated with cold exposure as a causative agent, with cold avoidance as the treatment for this benign and often transient condition.6 This phenomenon was not appreciated in our patient. Livedo reticularis commonly occurs with antiphospholipid syndrome.5 This association in combination with our patient's positive ANA findings and her mother's history of miscarriages resulted in the misdiagnosis as livedo reticularis.
Bier spots manifest as white macules with surrounding erythema and typically present in young adults. When first described in the literature, it was debated if BASCULE syndrome was simply another manifestation of Bier spots or postural orthostatic intolerance,4 as there was a large consensus that postural orthostatic intolerance was associated with BASCULE syndrome, with the majority of patients not meeting criteria for the condition. Heymann4 addressed the differences in BASCULE manifestations vs typical Bier spots. The author extended the syndrome to include cyanosis, an urticarialike eruption of red-orange macules with central papules located centrally, pruritus, tenderness, and partial or diffuse edema, in addition to Bier spots.4
Sneddon syndrome is a rare progressive disorder that affects small- to medium-sized blood vessels resulting in multiple episodes of ischemia in the brain. Skin manifestations of these repeated strokes are similar to livedo reticularis, typically manifesting as livedo racemosa—irregular reticular patterns of skin mottling with reddish-blue hues.6 However, Sneddon syndrome is more generalized and widespread and differs from BASCULE syndrome in shape and histologic findings. Our patient presented with findings on the legs, which is more characteristic of livedo reticularis vs livedo racemosa. Our patient experienced resolution upon laying down and sitting, and Sneddon syndrome persists beyond postural changes. Furthermore, patients with Sneddon syndrome present with neurologic symptoms such as prodromal headaches.6
Urticarial vasculitis was ruled out in our patient because of the duration of symptoms as well as the spatial changes. Urticarial vasculitis is a rare skin condition characterized by chronic recurring urticarial lesions that may persist for more than a day. This condition typically presents in middle-aged women and rarely in children. Urticarial vasculitis is thought to be immune-complex mediated, but its cause is largely unknown. It is a common manifestation of underlying conditions such as systemic lupus erythematosus.6 Our patient had a positive ANA and possible autoimmune history from her mother; however, urticarial vasculitis does not present transiently on the legs or in the rash pattern appreciated in our patient.
The Diagnosis: BASCULE Syndrome
The patient had previously been thought to have livedo reticularis by primary care. Repeat antinuclear antibody (ANA) testing was positive (1:1280 homogeneous [reflexive titers all negative]). However, upon dermatologic evaluation, the manifestation of the rash in addition to onset occurring with postural changes challenged the livedo reticularis diagnosis. Extensive research and consultation with dermatologic colleagues led to the diagnosis of the rare entity BASCULE syndrome. BASCULE (Bier anemic spots, cyanosis, and urticarialike eruption) syndrome was described by Bessis et al1 in 2016. It is a rare condition but may be underreported.2 It is a benign pediatric disorder in the vascular acrosyndrome family that is characterized by underlying vasomotor dysfunction in distal regions of the body. Raynaud phenomenon is a widely known member of this family. As seen in our patient, it typically presents on the distal legs and feet with numerous irregular hypopigmented macules on a cyanotic background. Red-orange papules may appear on the hypopigmented macules and often are pruritic. Lesions on the distal upper extremities are less common, and a case involving the trunk has been reported.3 Onset generally begins within a couple of minutes of standing or mechanical compression of the lower legs, with full reversal of symptoms occurring within minutes of laying down or walking. Commonly reported associated symptoms include tenderness, pruritus, edema, and pain; however, the cutaneous lesions may be asymptomatic. The condition tends to affect adolescents, as seen in our patient; however, there have been reports in infants as young as 3 months to adults aged 19 years.2
The pathophysiology behind BASCULE syndrome remains unclear but is believed to be centered around the role of physiologic venous stasis that occurs when standing. The hypoxia secondary to stasis is thought to induce amplified vasoconstriction of arterioles. These responses are further exaggerated due to absence of venoarteriolar reflexes in dermal ascending arterioles, leading to Bier spots.2 The role of mast cells and eosinophils remains unclear. It is a clinical diagnosis without clear histologic findings; therefore, biopsy was not pursued in our patient.
Although BASCULE syndrome is a benign entity, it is imperative that it be recognized to avoid a time consuming, expensive, and anxiety-producing diagnostic workup, as occurred in our patient. Although not a manifestation of systemic disease, BASCULE syndrome may be associated with orthostatic hypotension in up to 20% of cases.2,4 Therefore, these patients should undergo orthostatic testing, including the tilt table test. In our patient, these manifestations were not appreciated.
There are no current guidelines for effective treatment of BASCULE syndrome. Given the possible role of mast cells in the condition, H1 antihistamines are proposed as first-line treatment. Desloratadine (10 mg/d for 7 days) has been found to be associated with improvement of pruritus. However, a recent literature review found little evidence to support the use of H1 antihistamines for resolution of other symptoms.2
The differential diagnosis includes livedo reticularis, Bier spots, Sneddon syndrome, and urticarial vasculitis. Livedo reticularis presents as distinct, netlike, blue-erythematousviolaceous discoloration, which differs from the distinct orange-red macules in BASCULE syndrome.5 In addition to distinct variances in dermatologic presentation, livedo reticularis typically is associated with cold exposure as a causative agent, with cold avoidance as the treatment for this benign and often transient condition.6 This phenomenon was not appreciated in our patient. Livedo reticularis commonly occurs with antiphospholipid syndrome.5 This association in combination with our patient's positive ANA findings and her mother's history of miscarriages resulted in the misdiagnosis as livedo reticularis.
Bier spots manifest as white macules with surrounding erythema and typically present in young adults. When first described in the literature, it was debated if BASCULE syndrome was simply another manifestation of Bier spots or postural orthostatic intolerance,4 as there was a large consensus that postural orthostatic intolerance was associated with BASCULE syndrome, with the majority of patients not meeting criteria for the condition. Heymann4 addressed the differences in BASCULE manifestations vs typical Bier spots. The author extended the syndrome to include cyanosis, an urticarialike eruption of red-orange macules with central papules located centrally, pruritus, tenderness, and partial or diffuse edema, in addition to Bier spots.4
Sneddon syndrome is a rare progressive disorder that affects small- to medium-sized blood vessels resulting in multiple episodes of ischemia in the brain. Skin manifestations of these repeated strokes are similar to livedo reticularis, typically manifesting as livedo racemosa—irregular reticular patterns of skin mottling with reddish-blue hues.6 However, Sneddon syndrome is more generalized and widespread and differs from BASCULE syndrome in shape and histologic findings. Our patient presented with findings on the legs, which is more characteristic of livedo reticularis vs livedo racemosa. Our patient experienced resolution upon laying down and sitting, and Sneddon syndrome persists beyond postural changes. Furthermore, patients with Sneddon syndrome present with neurologic symptoms such as prodromal headaches.6
Urticarial vasculitis was ruled out in our patient because of the duration of symptoms as well as the spatial changes. Urticarial vasculitis is a rare skin condition characterized by chronic recurring urticarial lesions that may persist for more than a day. This condition typically presents in middle-aged women and rarely in children. Urticarial vasculitis is thought to be immune-complex mediated, but its cause is largely unknown. It is a common manifestation of underlying conditions such as systemic lupus erythematosus.6 Our patient had a positive ANA and possible autoimmune history from her mother; however, urticarial vasculitis does not present transiently on the legs or in the rash pattern appreciated in our patient.
- Bessis D, Jeziorski E, Rigau V, et al. Bier anaemic spots, cyanosis with urticaria-like eruption (BASCULE) syndrome: a new entity? Br J Dermatol. 2016;175:218-220. doi:10.1111/bjd.14589
- Baurens N, Briand C, Giovannini-Chami L, et al. Case report, practices survey and literature review of an under-recognized pediatric vascular disorder: the BASCULE syndrome. Front Pediatr. 2022;10:849914. doi:10.3389/fped.2022.849914
- Jiménez-Gallo D, Collantes-Rodríguez C, Ossorio-García L, et al. Bier anaemic spots, cyanosis with urticaria-like eruption (BASCULE) syndrome on trunk and upper limbs. Pediatr Dermatol. 2018;35:E313-E315. doi:10.1111/pde.13558
- Heymann WR. BASCULE syndrome: is something brewing with Bier spots? Dermatology World Insights and Inquiries. September 7, 2022. https://www.aad.org/dw/dw-insights-and-inquiries/archive/2022/bascule-syndrome
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103/2229-5178.164493
- Gu SL, Jorizzo JL. Urticarial vasculitis. Int J Womens Dermatol. 2021;7:290-297. doi:10.1016/j.ijwd.2021.01.021
- Bessis D, Jeziorski E, Rigau V, et al. Bier anaemic spots, cyanosis with urticaria-like eruption (BASCULE) syndrome: a new entity? Br J Dermatol. 2016;175:218-220. doi:10.1111/bjd.14589
- Baurens N, Briand C, Giovannini-Chami L, et al. Case report, practices survey and literature review of an under-recognized pediatric vascular disorder: the BASCULE syndrome. Front Pediatr. 2022;10:849914. doi:10.3389/fped.2022.849914
- Jiménez-Gallo D, Collantes-Rodríguez C, Ossorio-García L, et al. Bier anaemic spots, cyanosis with urticaria-like eruption (BASCULE) syndrome on trunk and upper limbs. Pediatr Dermatol. 2018;35:E313-E315. doi:10.1111/pde.13558
- Heymann WR. BASCULE syndrome: is something brewing with Bier spots? Dermatology World Insights and Inquiries. September 7, 2022. https://www.aad.org/dw/dw-insights-and-inquiries/archive/2022/bascule-syndrome
- Sajjan VV, Lunge S, Swamy MB, et al. Livedo reticularis: a review of the literature. Indian Dermatol Online J. 2015;6:315-321. doi:10.4103/2229-5178.164493
- Gu SL, Jorizzo JL. Urticarial vasculitis. Int J Womens Dermatol. 2021;7:290-297. doi:10.1016/j.ijwd.2021.01.021
An 11-year-old girl was referred to the dermatology clinic for evaluation of a rash on the legs and feet of 1 year’s duration. The rash appeared every time she was standing for longer than 10 to 15 minutes and resolved when sitting or laying down. After the initial onset, the rash did not spread to other body areas but became more prominent in appearance. The patient endorsed intense pruritus associated with the rash. A review of systems was negative for fever, headaches, history of blood clots, and joint pain. She did not have any known medical conditions or take any medications. The patient’s mother reported that the patient experienced episodes of leg numbness while sitting in vehicles from 6 to 10 years of age. There was no family history of rheumatologic, hematologic, or cardiac conditions. The patient’s mother had experienced 2 miscarriages but denied any other obstetric complications. The patient had 1 sibling who was unaffected. Physical examination revealed reticulate erythema on the calves with scattered regions of blanching and evanescent pink macules as well as dermatographism.
One month prior to presenting to dermatology, the patient was evaluated by rheumatology, endocrinology, and hematology. Laboratory workup completed at age 3 years included antinuclear antibody, anticardiolipin antibody, and antithrombin III activity; factor V Leiden; cryoglobulins; quantitation (human chorionic gonadotropin); proteins S and C activity; antineutrophil cytoplasmic antibody screen; thyroid studies; prothrombin time; and partial thromboplastin time. All laboratory results were within reference range.
Purpuric Eruption in a Patient With Hairy Cell Leukemia
The Diagnosis: Purpuric Drug Eruption
Histopathology revealed interface dermatitis, spongiosis, and a perivascular lymphocytic infiltrate with extravasated red blood cells consistent with a purpuric drug eruption. Our patient achieved remission of hairy cell leukemia after receiving only 2 of 5 expected doses of cladribine. The rash resolved completely in 3 weeks following a prednisone taper (Figure).
Hairy cell leukemia is a rare indolent lymphoproliferative disorder of B cells that accounts for approximately 2% of adult leukemias in the United States. Cladribine, a purine nucleoside analog that impairs DNA synthesis and repair, has become the mainstay of therapy, demonstrating a 95% complete response rate.1 Although few reports have addressed the cutaneous reactions seen with cladribine therapy, they can occur in more than 50% of patients.1,2 The most common skin manifestation associated with cladribine therapy is a morbilliform rash, but Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN) have been reported.1
Few cases of purpuric eruption secondary to cladribine treatment have been described, and nearly all reports involve concomitant medications such as allopurinol, which our patient was taking, and antibiotics including trimethoprim-sulfamethoxazole and penicillins.1,3,4 In a cohort of 35 patients receiving cladribine,1 only concomitant treatment with cladribine and allopurinol caused cutaneous reactions, further supporting the hypothesis of cladribine-induced drug sensitivity. Allopurinol often is prescribed during induction therapy for prophylaxis against tumor lysis syndrome; similarly, antibiotics frequently are given prophylactically and therapeutically for neutropenic fever. It is believed that T-cell imbalance and profound lymphopenia induced by cladribine increase susceptibility to drug hypersensitivity reactions.1,3
The typical purpuric eruption develops within 2 days of starting cladribine therapy. Diascopy will reveal petechiae, and biopsy should be performed to rule out other serious drug-induced reactions, such as erythema multiforme, Stevens-Johnson syndrome, and TEN. A cladribine-induced purpuric eruption typically is self-resolving and carries a favorable prognosis, though high-dose corticosteroids often are prescribed to hasten recovery. The rare reports of serious cutaneous reactions secondary to cladribine therapy have been with maculopapular, not purpuric eruptions.2 Based on limited available data, cladribine-induced purpura should not be a limitation to continued treatment in patients who need it.1 Careful consideration of concomitant drug use is necessary, as the current literature demonstrates resolution of rash with withdrawal of other therapies, namely allopurinol.2-4 Future studies are needed to examine the safety of withholding offending medications and to further elucidate the mechanisms contributing to drug hypersensitivity due to cladribine.
Widespread purpura and petechiae can pose a wide differential; the patient’s recent history of cladribine administration pointed to a classic purpuric eruption. Other diagnoses such as toxic erythema of chemotherapy (TEC) and TEN are not purpuric, though plaques can be violaceous. Lack of bullae, blisters, and facial or mucosal surface involvement suggest TEN.5 Thrombotic thrombocytopenic purpura and disseminated intravascular coagulation do manifest with petechiae and purpura, though such a robust eruption in the context of recent cladribine therapy is less likely. The classic retiform purpura and necrosis were not present to suggest purpura fulminans from disseminated intravascular coagulation.
Several of the proposed diagnoses as well as a purpuric drug eruption would demonstrate extravasated red blood cells on histopathology, but the presence of interface dermatitis narrows the differential to a purpuric drug eruption. Necrotic keratinocytes and full-thickness necrosis were not present on biopsy to support a diagnosis of TEN in our patient. Characteristic features of TEC—including eccrine squamous syringometaplasia, dermal edema, and keratinocyte atypia—were not present on biopsy.6 Finally, although TEN should resolve with steroid treatment, TEC is self-limited and thrombotic thrombocytopenic purpura and disseminated intravascular coagulation would not resolve with use of steroids alone.
- Ganzel C, Gatt ME, Maly A, et al. High incidence of skin rash in patients with hairy cell leukemia treated with cladribine. Leuk Lymphoma. 2012;53:1169-1173. doi:10.3109/10428194.2011.635864
- Chubar Y, Bennett M. Cutaneous reactions in hairy cell leukaemia treated with 2-chlorodeoxyadenosine and allopurinol. Br J Haematol. 2003;122:768-770. doi:10.1046/j.1365-2141.2003.04506.x
- Espinosa Lara P, Quirós Redondo V, Aguado Lobo M, et al. Purpuric exanthema in a patient with hairy cell leukemia treated with cladribine and allopurinol. Ann Hematol. 2017;96:1209-1210. doi:10.1007 /s00277-017-2992-z
- Hendrick A. Purpuric rash following treatment with 2-chlorodeoxyadenosine. Clin Lab Haematol. 2001;23:67-68. doi:10.1046 /j.1365-2257.2001.0346b.x
- Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw-Hill Education; 2019.
- Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59:524-529.
The Diagnosis: Purpuric Drug Eruption
Histopathology revealed interface dermatitis, spongiosis, and a perivascular lymphocytic infiltrate with extravasated red blood cells consistent with a purpuric drug eruption. Our patient achieved remission of hairy cell leukemia after receiving only 2 of 5 expected doses of cladribine. The rash resolved completely in 3 weeks following a prednisone taper (Figure).
Hairy cell leukemia is a rare indolent lymphoproliferative disorder of B cells that accounts for approximately 2% of adult leukemias in the United States. Cladribine, a purine nucleoside analog that impairs DNA synthesis and repair, has become the mainstay of therapy, demonstrating a 95% complete response rate.1 Although few reports have addressed the cutaneous reactions seen with cladribine therapy, they can occur in more than 50% of patients.1,2 The most common skin manifestation associated with cladribine therapy is a morbilliform rash, but Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN) have been reported.1
Few cases of purpuric eruption secondary to cladribine treatment have been described, and nearly all reports involve concomitant medications such as allopurinol, which our patient was taking, and antibiotics including trimethoprim-sulfamethoxazole and penicillins.1,3,4 In a cohort of 35 patients receiving cladribine,1 only concomitant treatment with cladribine and allopurinol caused cutaneous reactions, further supporting the hypothesis of cladribine-induced drug sensitivity. Allopurinol often is prescribed during induction therapy for prophylaxis against tumor lysis syndrome; similarly, antibiotics frequently are given prophylactically and therapeutically for neutropenic fever. It is believed that T-cell imbalance and profound lymphopenia induced by cladribine increase susceptibility to drug hypersensitivity reactions.1,3
The typical purpuric eruption develops within 2 days of starting cladribine therapy. Diascopy will reveal petechiae, and biopsy should be performed to rule out other serious drug-induced reactions, such as erythema multiforme, Stevens-Johnson syndrome, and TEN. A cladribine-induced purpuric eruption typically is self-resolving and carries a favorable prognosis, though high-dose corticosteroids often are prescribed to hasten recovery. The rare reports of serious cutaneous reactions secondary to cladribine therapy have been with maculopapular, not purpuric eruptions.2 Based on limited available data, cladribine-induced purpura should not be a limitation to continued treatment in patients who need it.1 Careful consideration of concomitant drug use is necessary, as the current literature demonstrates resolution of rash with withdrawal of other therapies, namely allopurinol.2-4 Future studies are needed to examine the safety of withholding offending medications and to further elucidate the mechanisms contributing to drug hypersensitivity due to cladribine.
Widespread purpura and petechiae can pose a wide differential; the patient’s recent history of cladribine administration pointed to a classic purpuric eruption. Other diagnoses such as toxic erythema of chemotherapy (TEC) and TEN are not purpuric, though plaques can be violaceous. Lack of bullae, blisters, and facial or mucosal surface involvement suggest TEN.5 Thrombotic thrombocytopenic purpura and disseminated intravascular coagulation do manifest with petechiae and purpura, though such a robust eruption in the context of recent cladribine therapy is less likely. The classic retiform purpura and necrosis were not present to suggest purpura fulminans from disseminated intravascular coagulation.
Several of the proposed diagnoses as well as a purpuric drug eruption would demonstrate extravasated red blood cells on histopathology, but the presence of interface dermatitis narrows the differential to a purpuric drug eruption. Necrotic keratinocytes and full-thickness necrosis were not present on biopsy to support a diagnosis of TEN in our patient. Characteristic features of TEC—including eccrine squamous syringometaplasia, dermal edema, and keratinocyte atypia—were not present on biopsy.6 Finally, although TEN should resolve with steroid treatment, TEC is self-limited and thrombotic thrombocytopenic purpura and disseminated intravascular coagulation would not resolve with use of steroids alone.
The Diagnosis: Purpuric Drug Eruption
Histopathology revealed interface dermatitis, spongiosis, and a perivascular lymphocytic infiltrate with extravasated red blood cells consistent with a purpuric drug eruption. Our patient achieved remission of hairy cell leukemia after receiving only 2 of 5 expected doses of cladribine. The rash resolved completely in 3 weeks following a prednisone taper (Figure).
Hairy cell leukemia is a rare indolent lymphoproliferative disorder of B cells that accounts for approximately 2% of adult leukemias in the United States. Cladribine, a purine nucleoside analog that impairs DNA synthesis and repair, has become the mainstay of therapy, demonstrating a 95% complete response rate.1 Although few reports have addressed the cutaneous reactions seen with cladribine therapy, they can occur in more than 50% of patients.1,2 The most common skin manifestation associated with cladribine therapy is a morbilliform rash, but Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN) have been reported.1
Few cases of purpuric eruption secondary to cladribine treatment have been described, and nearly all reports involve concomitant medications such as allopurinol, which our patient was taking, and antibiotics including trimethoprim-sulfamethoxazole and penicillins.1,3,4 In a cohort of 35 patients receiving cladribine,1 only concomitant treatment with cladribine and allopurinol caused cutaneous reactions, further supporting the hypothesis of cladribine-induced drug sensitivity. Allopurinol often is prescribed during induction therapy for prophylaxis against tumor lysis syndrome; similarly, antibiotics frequently are given prophylactically and therapeutically for neutropenic fever. It is believed that T-cell imbalance and profound lymphopenia induced by cladribine increase susceptibility to drug hypersensitivity reactions.1,3
The typical purpuric eruption develops within 2 days of starting cladribine therapy. Diascopy will reveal petechiae, and biopsy should be performed to rule out other serious drug-induced reactions, such as erythema multiforme, Stevens-Johnson syndrome, and TEN. A cladribine-induced purpuric eruption typically is self-resolving and carries a favorable prognosis, though high-dose corticosteroids often are prescribed to hasten recovery. The rare reports of serious cutaneous reactions secondary to cladribine therapy have been with maculopapular, not purpuric eruptions.2 Based on limited available data, cladribine-induced purpura should not be a limitation to continued treatment in patients who need it.1 Careful consideration of concomitant drug use is necessary, as the current literature demonstrates resolution of rash with withdrawal of other therapies, namely allopurinol.2-4 Future studies are needed to examine the safety of withholding offending medications and to further elucidate the mechanisms contributing to drug hypersensitivity due to cladribine.
Widespread purpura and petechiae can pose a wide differential; the patient’s recent history of cladribine administration pointed to a classic purpuric eruption. Other diagnoses such as toxic erythema of chemotherapy (TEC) and TEN are not purpuric, though plaques can be violaceous. Lack of bullae, blisters, and facial or mucosal surface involvement suggest TEN.5 Thrombotic thrombocytopenic purpura and disseminated intravascular coagulation do manifest with petechiae and purpura, though such a robust eruption in the context of recent cladribine therapy is less likely. The classic retiform purpura and necrosis were not present to suggest purpura fulminans from disseminated intravascular coagulation.
Several of the proposed diagnoses as well as a purpuric drug eruption would demonstrate extravasated red blood cells on histopathology, but the presence of interface dermatitis narrows the differential to a purpuric drug eruption. Necrotic keratinocytes and full-thickness necrosis were not present on biopsy to support a diagnosis of TEN in our patient. Characteristic features of TEC—including eccrine squamous syringometaplasia, dermal edema, and keratinocyte atypia—were not present on biopsy.6 Finally, although TEN should resolve with steroid treatment, TEC is self-limited and thrombotic thrombocytopenic purpura and disseminated intravascular coagulation would not resolve with use of steroids alone.
- Ganzel C, Gatt ME, Maly A, et al. High incidence of skin rash in patients with hairy cell leukemia treated with cladribine. Leuk Lymphoma. 2012;53:1169-1173. doi:10.3109/10428194.2011.635864
- Chubar Y, Bennett M. Cutaneous reactions in hairy cell leukaemia treated with 2-chlorodeoxyadenosine and allopurinol. Br J Haematol. 2003;122:768-770. doi:10.1046/j.1365-2141.2003.04506.x
- Espinosa Lara P, Quirós Redondo V, Aguado Lobo M, et al. Purpuric exanthema in a patient with hairy cell leukemia treated with cladribine and allopurinol. Ann Hematol. 2017;96:1209-1210. doi:10.1007 /s00277-017-2992-z
- Hendrick A. Purpuric rash following treatment with 2-chlorodeoxyadenosine. Clin Lab Haematol. 2001;23:67-68. doi:10.1046 /j.1365-2257.2001.0346b.x
- Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw-Hill Education; 2019.
- Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59:524-529.
- Ganzel C, Gatt ME, Maly A, et al. High incidence of skin rash in patients with hairy cell leukemia treated with cladribine. Leuk Lymphoma. 2012;53:1169-1173. doi:10.3109/10428194.2011.635864
- Chubar Y, Bennett M. Cutaneous reactions in hairy cell leukaemia treated with 2-chlorodeoxyadenosine and allopurinol. Br J Haematol. 2003;122:768-770. doi:10.1046/j.1365-2141.2003.04506.x
- Espinosa Lara P, Quirós Redondo V, Aguado Lobo M, et al. Purpuric exanthema in a patient with hairy cell leukemia treated with cladribine and allopurinol. Ann Hematol. 2017;96:1209-1210. doi:10.1007 /s00277-017-2992-z
- Hendrick A. Purpuric rash following treatment with 2-chlorodeoxyadenosine. Clin Lab Haematol. 2001;23:67-68. doi:10.1046 /j.1365-2257.2001.0346b.x
- Kang S, Amagai M, Bruckner AL, et al, eds. Fitzpatrick’s Dermatology. 9th ed. McGraw-Hill Education; 2019.
- Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59:524-529.
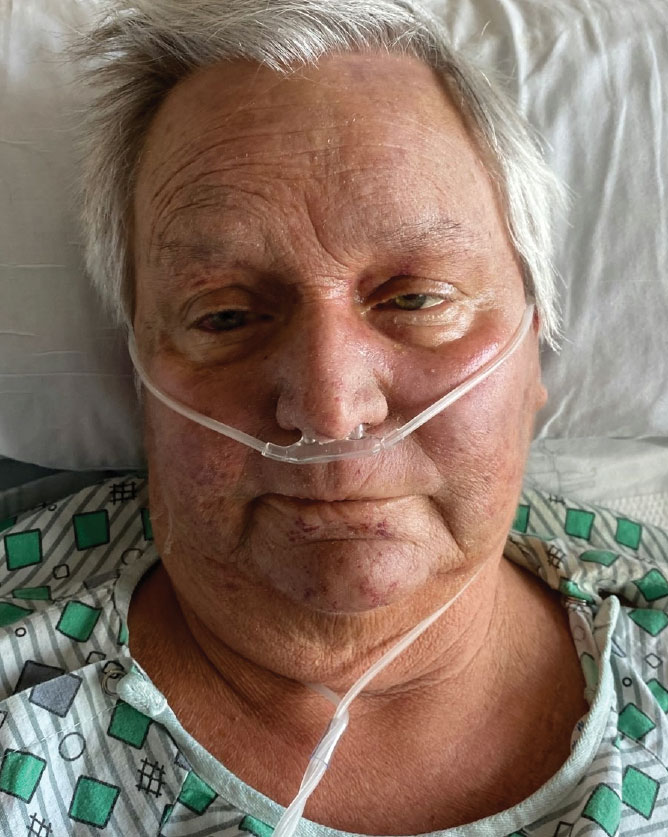
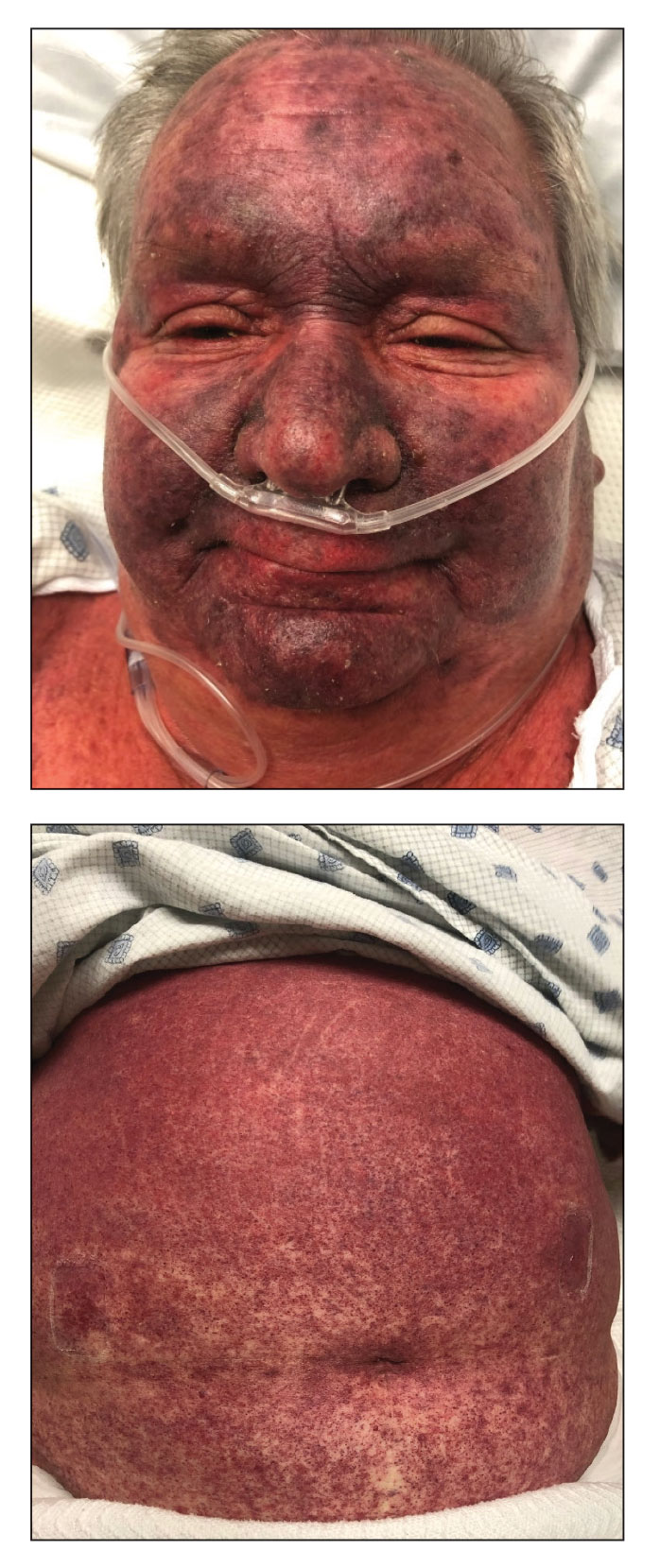
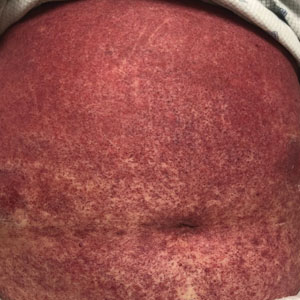
A 68-year-old woman presented to the emergency department with neutropenic fever and a rash over the body after receiving 2 doses of cladribine therapy for hairy cell leukemia. Physical examination demonstrated marked facial (top), lip, and tongue swelling, as well as a diffuse dusky nonpalpable purpuric rash on the abdomen (bottom) and back involving 90% of the body surface area. Bilateral ear edema was appreciated with accentuation of the earlobe crease. The patient exhibited subconjunctival hemorrhage, ectropion, and scleral injection. A punch biopsy of the thigh was performed.
Chronic Cribriform Ulcerated Plaque on the Left Calf
The Diagnosis: Nodular Basal Cell Carcinoma
Histopathology of the lesion showed a large basaloid lobule with focal epidermal attachment, peripheral nuclear palisading with cleft formation between the tumor and surrounding stroma, fibromyxoid stroma and mild pleomorphism, and variable mitotic activity and apoptosis (Figure). Based on the clinical presentation and histopathology, the patient was diagnosed with nodular basal cell carcinoma (BCC). He underwent a wide local excision of the affected area that was repaired with a splitthickness skin graft.
")
Basal cell carcinoma is the most common skin cancer worldwide and typically occurs due to years of UV radiation damage on sun-exposed skin, which accounts for a higher frequency of BCC occurring in patients residing in geographic locations with greater UV exposure (eg, higher and lower latitudes). In addition to cumulative UV dose, the duration of the exposure as well as its intensity also play a role in the development of BCC, particularly in early childhood and adolescence. Nevertheless, UV exposure is not the only risk factor, as 20% of BCCs arise in skin that is not exposed to the sun. Other risk factors include exposure to ionizing radiation and arsenic, immunosuppression, and genetic predisposition.1 Although these malignancies typically do not metastasize, growth can lead to local tissue destruction and major disfigurement if not treated in a timely fashion.2
In our patient, the differential diagnosis included pyoderma gangrenosum (PG) given the clinical appearance of the cribriform base and violaceous undermined rim of the ulcer. Pyoderma gangrenosum is a rare neutrophilic disorder that often results in ulcers that have been associated with various systemic autoimmune and inflammatory conditions, such as inflammatory bowel disease. There are 4 main subtypes of PG: the classic ulcerative type (our patient); the pustular type, which most often is seen in patients with inflammatory bowel disease; the bullous type, which can be seen in patients with an associated lymphoproliferative disorder; and the vegetative type. It frequently is thought of as both a clinical and histologic diagnosis of exclusion due to the nonspecific histopathologic features; most lesions demonstrate an infiltrate of neutrophils in the dermis. A biopsy was crucial in our patient, considering that diagnosis and treatment would have been further delayed had the patient been empirically treated with oral and topical steroids for presumed PG, which is precisely why PG is a diagnosis of exclusion. It is imperative for clinicians to rule out other pathologies, such as infection or malignancy, as demonstrated in our patient. The progressive onset and slow evolution of the lesion over years along with a lack of pain were more suggestive of BCC rather than PG. However, there is a report in the literature of PG mimicking BCC with both clinical and dermoscopic findings.3
Venous or stasis ulcers are painless, and although they rarely occur on the calf, they typically are seen lower on the leg such as on the medial ankles. Our patient endorsed occasional swelling of the affected leg and presented with edema, but overlying stasis change and other signs of venous insufficiency were absent.
Buruli ulcer is a painless chronic debilitating cutaneous disease resulting in indolent necrotizing skin as well as subcutaneous and bone lesions. It is caused by the environmental organism Mycobacterium ulcerans and typically is reported in Africa, Central/South America, the Western Pacific Region, and Australia.4 Histopathology usually demonstrates necrosis of subcutaneous tissue and dermal collagen accompanied by inflammation and acidfast bacilli highlighted by Ziehl-Neelsen stain.5 Smears of the lesions as well as culture and polymerase chain reaction for acid-fast bacilli also can be performed. Our patient reported no recent travel to any endemic areas and had no other risk factors or exposures to the pathogen responsible for this condition.
Traumatic ulcer also was included in the differential diagnosis, but the patient denied preceding trauma to the area, and the contralateral foot prosthesis did not rub on or impact the affected leg.
Basal cell carcinoma typically is treated surgically, but choice of treatment can depend on the subtype, size, tumor site, and/or patient preference.1 Other treatment modalities include electrodesiccation and curettage, cryosurgical destruction, photodynamic therapy, radiation, topical therapies, and systemic medications. Radiotherapy can be considered as a primary treatment option for BCC if surgery is contraindicated or declined by the patient, but it also is useful as an adjuvant therapy when there is perineural invasion of the tumor or positive margins. Hedgehog pathway inhibitors such as vismodegib currently are indicated for patients who are not candidates for surgery or radiation as well as for those with metastatic or locally advanced, recurrent BCC. There is no single treatment method ideal for every lesion or patient. Specific populations such as the elderly, the immunosuppressed, or those with poor baseline functional status may warrant a nonsurgical approach. The clinician must take into consideration all factors while at the same time thinking about how to best accomplish the goals of recurrencefree tumor removal, correction of any underlying functional impairment from the tumor, and maintenance of cosmesis.1
- McDaniel B, Badri T, Steele RB. Basal cell carcinoma. In: StatPearls. StatPearls; 2022.
- Marzuka AG, Book SE. Basal cell carcinoma: pathogenesis, epidemiology, clinical features, diagnosis, histopathology, and management. Yale J Biol Med. 2015;88:167-179.
- Rosina P, Papagrigoraki A, Colato C. A case of superficial granulomatous pyoderma mimicking a basal cell carcinoma. Acta Dermatovenerol Croat. 2014;22:48-51.
- Yotsu RR, Suzuki K, Simmonds RE, et al. Buruli ulcer: a review of the current knowledge. Curr Trop Med Rep. 2018;5:247-256.
- Guarner J, Bartlett J, Whitney EA, et al. Histopathologic features of Mycobacterium ulcerans infection. Emerg Infect Dis. 2003;9:651-656.
The Diagnosis: Nodular Basal Cell Carcinoma
Histopathology of the lesion showed a large basaloid lobule with focal epidermal attachment, peripheral nuclear palisading with cleft formation between the tumor and surrounding stroma, fibromyxoid stroma and mild pleomorphism, and variable mitotic activity and apoptosis (Figure). Based on the clinical presentation and histopathology, the patient was diagnosed with nodular basal cell carcinoma (BCC). He underwent a wide local excision of the affected area that was repaired with a splitthickness skin graft.
Basal cell carcinoma is the most common skin cancer worldwide and typically occurs due to years of UV radiation damage on sun-exposed skin, which accounts for a higher frequency of BCC occurring in patients residing in geographic locations with greater UV exposure (eg, higher and lower latitudes). In addition to cumulative UV dose, the duration of the exposure as well as its intensity also play a role in the development of BCC, particularly in early childhood and adolescence. Nevertheless, UV exposure is not the only risk factor, as 20% of BCCs arise in skin that is not exposed to the sun. Other risk factors include exposure to ionizing radiation and arsenic, immunosuppression, and genetic predisposition.1 Although these malignancies typically do not metastasize, growth can lead to local tissue destruction and major disfigurement if not treated in a timely fashion.2
In our patient, the differential diagnosis included pyoderma gangrenosum (PG) given the clinical appearance of the cribriform base and violaceous undermined rim of the ulcer. Pyoderma gangrenosum is a rare neutrophilic disorder that often results in ulcers that have been associated with various systemic autoimmune and inflammatory conditions, such as inflammatory bowel disease. There are 4 main subtypes of PG: the classic ulcerative type (our patient); the pustular type, which most often is seen in patients with inflammatory bowel disease; the bullous type, which can be seen in patients with an associated lymphoproliferative disorder; and the vegetative type. It frequently is thought of as both a clinical and histologic diagnosis of exclusion due to the nonspecific histopathologic features; most lesions demonstrate an infiltrate of neutrophils in the dermis. A biopsy was crucial in our patient, considering that diagnosis and treatment would have been further delayed had the patient been empirically treated with oral and topical steroids for presumed PG, which is precisely why PG is a diagnosis of exclusion. It is imperative for clinicians to rule out other pathologies, such as infection or malignancy, as demonstrated in our patient. The progressive onset and slow evolution of the lesion over years along with a lack of pain were more suggestive of BCC rather than PG. However, there is a report in the literature of PG mimicking BCC with both clinical and dermoscopic findings.3
Venous or stasis ulcers are painless, and although they rarely occur on the calf, they typically are seen lower on the leg such as on the medial ankles. Our patient endorsed occasional swelling of the affected leg and presented with edema, but overlying stasis change and other signs of venous insufficiency were absent.
Buruli ulcer is a painless chronic debilitating cutaneous disease resulting in indolent necrotizing skin as well as subcutaneous and bone lesions. It is caused by the environmental organism Mycobacterium ulcerans and typically is reported in Africa, Central/South America, the Western Pacific Region, and Australia.4 Histopathology usually demonstrates necrosis of subcutaneous tissue and dermal collagen accompanied by inflammation and acidfast bacilli highlighted by Ziehl-Neelsen stain.5 Smears of the lesions as well as culture and polymerase chain reaction for acid-fast bacilli also can be performed. Our patient reported no recent travel to any endemic areas and had no other risk factors or exposures to the pathogen responsible for this condition.
Traumatic ulcer also was included in the differential diagnosis, but the patient denied preceding trauma to the area, and the contralateral foot prosthesis did not rub on or impact the affected leg.
Basal cell carcinoma typically is treated surgically, but choice of treatment can depend on the subtype, size, tumor site, and/or patient preference.1 Other treatment modalities include electrodesiccation and curettage, cryosurgical destruction, photodynamic therapy, radiation, topical therapies, and systemic medications. Radiotherapy can be considered as a primary treatment option for BCC if surgery is contraindicated or declined by the patient, but it also is useful as an adjuvant therapy when there is perineural invasion of the tumor or positive margins. Hedgehog pathway inhibitors such as vismodegib currently are indicated for patients who are not candidates for surgery or radiation as well as for those with metastatic or locally advanced, recurrent BCC. There is no single treatment method ideal for every lesion or patient. Specific populations such as the elderly, the immunosuppressed, or those with poor baseline functional status may warrant a nonsurgical approach. The clinician must take into consideration all factors while at the same time thinking about how to best accomplish the goals of recurrencefree tumor removal, correction of any underlying functional impairment from the tumor, and maintenance of cosmesis.1
The Diagnosis: Nodular Basal Cell Carcinoma
Histopathology of the lesion showed a large basaloid lobule with focal epidermal attachment, peripheral nuclear palisading with cleft formation between the tumor and surrounding stroma, fibromyxoid stroma and mild pleomorphism, and variable mitotic activity and apoptosis (Figure). Based on the clinical presentation and histopathology, the patient was diagnosed with nodular basal cell carcinoma (BCC). He underwent a wide local excision of the affected area that was repaired with a splitthickness skin graft.
Basal cell carcinoma is the most common skin cancer worldwide and typically occurs due to years of UV radiation damage on sun-exposed skin, which accounts for a higher frequency of BCC occurring in patients residing in geographic locations with greater UV exposure (eg, higher and lower latitudes). In addition to cumulative UV dose, the duration of the exposure as well as its intensity also play a role in the development of BCC, particularly in early childhood and adolescence. Nevertheless, UV exposure is not the only risk factor, as 20% of BCCs arise in skin that is not exposed to the sun. Other risk factors include exposure to ionizing radiation and arsenic, immunosuppression, and genetic predisposition.1 Although these malignancies typically do not metastasize, growth can lead to local tissue destruction and major disfigurement if not treated in a timely fashion.2
In our patient, the differential diagnosis included pyoderma gangrenosum (PG) given the clinical appearance of the cribriform base and violaceous undermined rim of the ulcer. Pyoderma gangrenosum is a rare neutrophilic disorder that often results in ulcers that have been associated with various systemic autoimmune and inflammatory conditions, such as inflammatory bowel disease. There are 4 main subtypes of PG: the classic ulcerative type (our patient); the pustular type, which most often is seen in patients with inflammatory bowel disease; the bullous type, which can be seen in patients with an associated lymphoproliferative disorder; and the vegetative type. It frequently is thought of as both a clinical and histologic diagnosis of exclusion due to the nonspecific histopathologic features; most lesions demonstrate an infiltrate of neutrophils in the dermis. A biopsy was crucial in our patient, considering that diagnosis and treatment would have been further delayed had the patient been empirically treated with oral and topical steroids for presumed PG, which is precisely why PG is a diagnosis of exclusion. It is imperative for clinicians to rule out other pathologies, such as infection or malignancy, as demonstrated in our patient. The progressive onset and slow evolution of the lesion over years along with a lack of pain were more suggestive of BCC rather than PG. However, there is a report in the literature of PG mimicking BCC with both clinical and dermoscopic findings.3
Venous or stasis ulcers are painless, and although they rarely occur on the calf, they typically are seen lower on the leg such as on the medial ankles. Our patient endorsed occasional swelling of the affected leg and presented with edema, but overlying stasis change and other signs of venous insufficiency were absent.
Buruli ulcer is a painless chronic debilitating cutaneous disease resulting in indolent necrotizing skin as well as subcutaneous and bone lesions. It is caused by the environmental organism Mycobacterium ulcerans and typically is reported in Africa, Central/South America, the Western Pacific Region, and Australia.4 Histopathology usually demonstrates necrosis of subcutaneous tissue and dermal collagen accompanied by inflammation and acidfast bacilli highlighted by Ziehl-Neelsen stain.5 Smears of the lesions as well as culture and polymerase chain reaction for acid-fast bacilli also can be performed. Our patient reported no recent travel to any endemic areas and had no other risk factors or exposures to the pathogen responsible for this condition.
Traumatic ulcer also was included in the differential diagnosis, but the patient denied preceding trauma to the area, and the contralateral foot prosthesis did not rub on or impact the affected leg.
Basal cell carcinoma typically is treated surgically, but choice of treatment can depend on the subtype, size, tumor site, and/or patient preference.1 Other treatment modalities include electrodesiccation and curettage, cryosurgical destruction, photodynamic therapy, radiation, topical therapies, and systemic medications. Radiotherapy can be considered as a primary treatment option for BCC if surgery is contraindicated or declined by the patient, but it also is useful as an adjuvant therapy when there is perineural invasion of the tumor or positive margins. Hedgehog pathway inhibitors such as vismodegib currently are indicated for patients who are not candidates for surgery or radiation as well as for those with metastatic or locally advanced, recurrent BCC. There is no single treatment method ideal for every lesion or patient. Specific populations such as the elderly, the immunosuppressed, or those with poor baseline functional status may warrant a nonsurgical approach. The clinician must take into consideration all factors while at the same time thinking about how to best accomplish the goals of recurrencefree tumor removal, correction of any underlying functional impairment from the tumor, and maintenance of cosmesis.1
- McDaniel B, Badri T, Steele RB. Basal cell carcinoma. In: StatPearls. StatPearls; 2022.
- Marzuka AG, Book SE. Basal cell carcinoma: pathogenesis, epidemiology, clinical features, diagnosis, histopathology, and management. Yale J Biol Med. 2015;88:167-179.
- Rosina P, Papagrigoraki A, Colato C. A case of superficial granulomatous pyoderma mimicking a basal cell carcinoma. Acta Dermatovenerol Croat. 2014;22:48-51.
- Yotsu RR, Suzuki K, Simmonds RE, et al. Buruli ulcer: a review of the current knowledge. Curr Trop Med Rep. 2018;5:247-256.
- Guarner J, Bartlett J, Whitney EA, et al. Histopathologic features of Mycobacterium ulcerans infection. Emerg Infect Dis. 2003;9:651-656.
- McDaniel B, Badri T, Steele RB. Basal cell carcinoma. In: StatPearls. StatPearls; 2022.
- Marzuka AG, Book SE. Basal cell carcinoma: pathogenesis, epidemiology, clinical features, diagnosis, histopathology, and management. Yale J Biol Med. 2015;88:167-179.
- Rosina P, Papagrigoraki A, Colato C. A case of superficial granulomatous pyoderma mimicking a basal cell carcinoma. Acta Dermatovenerol Croat. 2014;22:48-51.
- Yotsu RR, Suzuki K, Simmonds RE, et al. Buruli ulcer: a review of the current knowledge. Curr Trop Med Rep. 2018;5:247-256.
- Guarner J, Bartlett J, Whitney EA, et al. Histopathologic features of Mycobacterium ulcerans infection. Emerg Infect Dis. 2003;9:651-656.
A 61-year-old man presented to the dermatology clinic for evaluation of a painless nonhealing wound on the left calf of 4 years’ duration. The patient had a history of amputation of the right foot as an infant, for which he wore an orthopedic prosthesis. He also had chronic lymphedema of the left leg, hyperlipidemia, and osteoarthritis of the right hip. There was no history of gastrointestinal tract issues. The lesion initially was small, then grew and began to ulcerate and bleed. His presentation to dermatology was delayed due to office closures during the COVID-19 pandemic. Physical examination revealed a 5-cm, erythematous, cribriform ulcer with a violaceous undermined rim. A punch biopsy was performed on the edge of the ulcer.


Occipital Scalp Nodule in a Newborn
The Diagnosis: Subcutaneous Fat Necrosis
Histopathology revealed lobular panniculitis with lymphohistiocytic inflammation, lipid crystals, and calcifications in our patient (Figure). Subcutaneous fat necrosis (SCFN) was diagnosed based on these characteristic histopathologic findings. No further treatment was pursued.

Subcutaneous fat necrosis is a rare, self-limiting panniculitis that typically resolves within several weeks to months without scarring. It manifests as red or violaceous subcutaneous nodules or plaques most commonly on the buttocks, trunk, proximal arms and legs, and cheeks.1 Histopathology reveals lobular panniculitis with dense granulomatous infiltrates of histiocytes, eosinophils, and multinucleated giant cells with needle-shaped crystals. Focal areas of fat necrosis with calcification also can be seen.2
The epidemiology of SCFN is unknown. Most cases occur in healthy full-term to postterm neonates who experience hypoxia, other prenatal stressors, or therapeutic hypothermia for the treatment of hypoxic-ischemic encephalopathy.3 Although the etiology is unclear, certain inciting factors such as local tissue hypoxia, cold exposure, meconium aspiration, maternal diabetes, preeclampsia, and mechanical pressure have been proposed. Our patient underwent hypothermic cooling protocol, and it has been suggested that the increased saturated to unsaturated fat concentration in the skin of newborns increases the melting point, thus predisposing them to fat crystalization.4 Cases of SCFN involving the scalp are rare; therefore, any newborns receiving hypothermic therapy for hypoxic-ischemic encephalopathy should have a thorough skin examination with possible biopsy of lesions that are characteristic of SCFN, such as red or violaceous subcutaneous nodules or plaques, for specific disease identification.
The main complication of SCFN is hypercalcemia, which occurs in approximately 50% of cases. Other serum abnormalities include hyperglycemia, hypertriglyceridemia, and thrombocytopenia, though these findings are not as well associated.4 Patients with associated hypercalcemia may be asymptomatic, as in our patient, but other presentations include irritability, weakness, anorexia, vomiting, renal failure, failure to thrive, and encephalopathy. Nephrocalcinosis is a common complication of severe hypercalcemia; however, there is little evidence of associated major renal dysfunction.5 The exact mechanism of hypercalcemia is poorly understood. A widely accepted theory postulates that a granulomatous inflammatory infiltrate upregulates 1-α-hydroxylase activity, which enzymatically converts 25-hydroxyvitamin D to its active form, 1,25-dihydroxycholecalciferol, which increases bone resorption and calcium absorption through the gastrointestinal tract and renal systems. Treatments for hypercalcemia include hyperhydration, calcium-wasting diuretics, and low calcium intake.6 Furthermore, calcium levels should be obtained at the time of diagnosis and 30, 45, and 60 days after the lesions resolve.4
Subcutaneous fat necrosis needs to be differentiated from the more severe panniculitis, sclerema neonatorum (SN), which typically affects critically ill, preterm, and small-for-gestational-age newborns. It is associated with a high mortality rate and is characterized by skin and subadjacent tissue structures. The process typically begins in the thighs, buttocks, or trunk and spreads diffusely, sparing the fat-free palms, soles, and genitalia.7 Although our patient was born preterm, the physical characteristics of the nodule and the lack of severe illness placed SN lower on our differential. Histopathologic differences between SCFN and SN involve the extent of tissue fibrosis and presence of inflammatory cells. Sclerema neonatorum typically manifests with thickened connective tissue with a sparse inflammatory infiltrate, including lymphocytes, histiocytes, and multinucleated giant cells.7 Conversely, SCFN manifests with fat necrosis with an extensive inflammatory infiltrate. It is important to be able to distinguish between these 2 conditions, as both have vastly different prognoses.
Cold panniculitis, sometimes called “popsicle panniculitis,” is a phenomenon in which cold contact with the skin causes eruption of firm, erythematous, indurated plaques at the site of exposure. This self-limiting condition typically appears hours to days after cold exposure and spontaneously resolves in a few weeks.8 Therapeutic hypothermic protocol treatment involves using cooling devices to lower the body temperature for a short duration. The temperature typically is lowered to approximately 32 °C to 36 °C. These temperatures are not low enough to induce cold panniculitis, which is more commonly seen in facial ice applications when managing supraventricular tachycardia in neonates.
Cephalohematoma is a birthing injury that causes blood accumulation within the subperiosteal space. During parturition, the compressive and sheering forces on the calvarium rupture the vessels passing through the periosteum, causing blood to pool slowly into the subperiostium; thus, a cephalohematoma usually manifests later at 1 to 3 days of life as localized head swelling.9 The bleeding typically does not cross suture lines and is primarily found in the occipital or parietal regions. The incidence has been reported to be 0.4% to 2.5% of all live births.10 Although the location of the nodule in our patient was in the occipital region, imaging and biopsy results did not show hemorrhagic findings consistent with cephalohematoma. Management of cephalohematoma mainly is observational, as the mass slowly regresses and the accumulated blood gradually is reabsorbed.
Fungal scalp infections (tinea capitis) are common in the pediatric population. The peak incidence of this infection has been reported in children aged 3 to 7 years, with Trichophyton tonsurans and Microsporum canis as the usual causative organisms.11 Clinical features of tinea capitis include scaly patches with hair loss, hair loss with black pigmented dots at the follicular openings, diffuse scalp scaling with subtle hair loss, and cervical lymphadenopathy.12 Although less common, tinea capitis can progress to a more severe form known as a kerion, which is characterized by a tender plaque with pustules and crusting. A kerion can result in permanent scarring and alopecia if left untreated.12 In our patient, a nodule with scaling and faint erythema was observed, but no black pigmented dots at the follicular orifices were present. Therefore, a potassium hydroxide wet mount preparation used to diagnose tinea capitis was unnecessary. Systemic oral antifungal therapy such as fluconazole or terbinafine is the standard treatment for tinea capitis.
- Coondoo A, Lahiry R, Choudhury A, et al. Tender skin nodules in a newborn. Indian J Dermatol. 2013;58:328. doi:10.4103/0019-5154.113983
- Mitra S, Dove J, Somisetty SK. Subcutaneous fat necrosis in newbornan unusual case and review of literature. Eur J Pediatr. 2011;170:1107- 1110. doi:10.1007/s00431-011-1405-x
- Velasquez JH, Mendez MD. Newborn subcutaneous fat necrosis. In: StatPearls. StatPearls Publishing; 2022.
- Stefanko NS, Drolet BA. Subcutaneous fat necrosis of the newborn and associated hypercalcemia: a systematic review of the literature. Pediatr Dermatol. 2019;36:24-30. doi:10.1111/pde.13640
- Shumer DE, Thaker V, Taylor GA, et al. Severe hypercalcaemia due to subcutaneous fat necrosis: presentation, management and complications. Arch Dis Child Fetal Neonatal Ed. 2014;99:F419-F421. doi:10.1136/ archdischild-2014-306069
- Farooque A, Moss C, Zehnder D, et al. Expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in subcutaneous fat necrosis. Br J Dermatol. 2009;160:423-425. doi:10.1111/j.1365-2133.2008.08844.x
- Zeb A, Darmstadt GL. Sclerema neonatorum: a review of nomenclature, clinical presentation, histological features, differential diagnoses and management. J Perinatol. 2008;28:453-460. doi:10.1038/jp.2008.33
- Quesada-Cortés A, Campos-Muñoz L, Díaz-Díaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:485-489, vii. doi:10.1016 /j.det.2008.05.015
- Raines DA, Krawiec C, Jain S. Cephalohematoma. In: StatPearls. StatPearls Publishing; 2023.
- Chung HY, Chung JY, Lee DG, et al. Surgical treatment of ossified cephalhematoma. J Craniofac Surg. 2004;15:774-779. doi:10.1097/00001665- 200409000-00015
- Leung AKC, Hon KL, Leong KF, et al. Tinea capitis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:58-68. doi:10.2174/1872 213x14666200106145624
- Kovitwanichkanont T, Chong A. Superficial fungal infections. Aust J Gen Pract. 2019;48:706-711. doi:10.31128/ajgp-05-19-4930
The Diagnosis: Subcutaneous Fat Necrosis
Histopathology revealed lobular panniculitis with lymphohistiocytic inflammation, lipid crystals, and calcifications in our patient (Figure). Subcutaneous fat necrosis (SCFN) was diagnosed based on these characteristic histopathologic findings. No further treatment was pursued.
Subcutaneous fat necrosis is a rare, self-limiting panniculitis that typically resolves within several weeks to months without scarring. It manifests as red or violaceous subcutaneous nodules or plaques most commonly on the buttocks, trunk, proximal arms and legs, and cheeks.1 Histopathology reveals lobular panniculitis with dense granulomatous infiltrates of histiocytes, eosinophils, and multinucleated giant cells with needle-shaped crystals. Focal areas of fat necrosis with calcification also can be seen.2
The epidemiology of SCFN is unknown. Most cases occur in healthy full-term to postterm neonates who experience hypoxia, other prenatal stressors, or therapeutic hypothermia for the treatment of hypoxic-ischemic encephalopathy.3 Although the etiology is unclear, certain inciting factors such as local tissue hypoxia, cold exposure, meconium aspiration, maternal diabetes, preeclampsia, and mechanical pressure have been proposed. Our patient underwent hypothermic cooling protocol, and it has been suggested that the increased saturated to unsaturated fat concentration in the skin of newborns increases the melting point, thus predisposing them to fat crystalization.4 Cases of SCFN involving the scalp are rare; therefore, any newborns receiving hypothermic therapy for hypoxic-ischemic encephalopathy should have a thorough skin examination with possible biopsy of lesions that are characteristic of SCFN, such as red or violaceous subcutaneous nodules or plaques, for specific disease identification.
The main complication of SCFN is hypercalcemia, which occurs in approximately 50% of cases. Other serum abnormalities include hyperglycemia, hypertriglyceridemia, and thrombocytopenia, though these findings are not as well associated.4 Patients with associated hypercalcemia may be asymptomatic, as in our patient, but other presentations include irritability, weakness, anorexia, vomiting, renal failure, failure to thrive, and encephalopathy. Nephrocalcinosis is a common complication of severe hypercalcemia; however, there is little evidence of associated major renal dysfunction.5 The exact mechanism of hypercalcemia is poorly understood. A widely accepted theory postulates that a granulomatous inflammatory infiltrate upregulates 1-α-hydroxylase activity, which enzymatically converts 25-hydroxyvitamin D to its active form, 1,25-dihydroxycholecalciferol, which increases bone resorption and calcium absorption through the gastrointestinal tract and renal systems. Treatments for hypercalcemia include hyperhydration, calcium-wasting diuretics, and low calcium intake.6 Furthermore, calcium levels should be obtained at the time of diagnosis and 30, 45, and 60 days after the lesions resolve.4
Subcutaneous fat necrosis needs to be differentiated from the more severe panniculitis, sclerema neonatorum (SN), which typically affects critically ill, preterm, and small-for-gestational-age newborns. It is associated with a high mortality rate and is characterized by skin and subadjacent tissue structures. The process typically begins in the thighs, buttocks, or trunk and spreads diffusely, sparing the fat-free palms, soles, and genitalia.7 Although our patient was born preterm, the physical characteristics of the nodule and the lack of severe illness placed SN lower on our differential. Histopathologic differences between SCFN and SN involve the extent of tissue fibrosis and presence of inflammatory cells. Sclerema neonatorum typically manifests with thickened connective tissue with a sparse inflammatory infiltrate, including lymphocytes, histiocytes, and multinucleated giant cells.7 Conversely, SCFN manifests with fat necrosis with an extensive inflammatory infiltrate. It is important to be able to distinguish between these 2 conditions, as both have vastly different prognoses.
Cold panniculitis, sometimes called “popsicle panniculitis,” is a phenomenon in which cold contact with the skin causes eruption of firm, erythematous, indurated plaques at the site of exposure. This self-limiting condition typically appears hours to days after cold exposure and spontaneously resolves in a few weeks.8 Therapeutic hypothermic protocol treatment involves using cooling devices to lower the body temperature for a short duration. The temperature typically is lowered to approximately 32 °C to 36 °C. These temperatures are not low enough to induce cold panniculitis, which is more commonly seen in facial ice applications when managing supraventricular tachycardia in neonates.
Cephalohematoma is a birthing injury that causes blood accumulation within the subperiosteal space. During parturition, the compressive and sheering forces on the calvarium rupture the vessels passing through the periosteum, causing blood to pool slowly into the subperiostium; thus, a cephalohematoma usually manifests later at 1 to 3 days of life as localized head swelling.9 The bleeding typically does not cross suture lines and is primarily found in the occipital or parietal regions. The incidence has been reported to be 0.4% to 2.5% of all live births.10 Although the location of the nodule in our patient was in the occipital region, imaging and biopsy results did not show hemorrhagic findings consistent with cephalohematoma. Management of cephalohematoma mainly is observational, as the mass slowly regresses and the accumulated blood gradually is reabsorbed.
Fungal scalp infections (tinea capitis) are common in the pediatric population. The peak incidence of this infection has been reported in children aged 3 to 7 years, with Trichophyton tonsurans and Microsporum canis as the usual causative organisms.11 Clinical features of tinea capitis include scaly patches with hair loss, hair loss with black pigmented dots at the follicular openings, diffuse scalp scaling with subtle hair loss, and cervical lymphadenopathy.12 Although less common, tinea capitis can progress to a more severe form known as a kerion, which is characterized by a tender plaque with pustules and crusting. A kerion can result in permanent scarring and alopecia if left untreated.12 In our patient, a nodule with scaling and faint erythema was observed, but no black pigmented dots at the follicular orifices were present. Therefore, a potassium hydroxide wet mount preparation used to diagnose tinea capitis was unnecessary. Systemic oral antifungal therapy such as fluconazole or terbinafine is the standard treatment for tinea capitis.
The Diagnosis: Subcutaneous Fat Necrosis
Histopathology revealed lobular panniculitis with lymphohistiocytic inflammation, lipid crystals, and calcifications in our patient (Figure). Subcutaneous fat necrosis (SCFN) was diagnosed based on these characteristic histopathologic findings. No further treatment was pursued.
Subcutaneous fat necrosis is a rare, self-limiting panniculitis that typically resolves within several weeks to months without scarring. It manifests as red or violaceous subcutaneous nodules or plaques most commonly on the buttocks, trunk, proximal arms and legs, and cheeks.1 Histopathology reveals lobular panniculitis with dense granulomatous infiltrates of histiocytes, eosinophils, and multinucleated giant cells with needle-shaped crystals. Focal areas of fat necrosis with calcification also can be seen.2
The epidemiology of SCFN is unknown. Most cases occur in healthy full-term to postterm neonates who experience hypoxia, other prenatal stressors, or therapeutic hypothermia for the treatment of hypoxic-ischemic encephalopathy.3 Although the etiology is unclear, certain inciting factors such as local tissue hypoxia, cold exposure, meconium aspiration, maternal diabetes, preeclampsia, and mechanical pressure have been proposed. Our patient underwent hypothermic cooling protocol, and it has been suggested that the increased saturated to unsaturated fat concentration in the skin of newborns increases the melting point, thus predisposing them to fat crystalization.4 Cases of SCFN involving the scalp are rare; therefore, any newborns receiving hypothermic therapy for hypoxic-ischemic encephalopathy should have a thorough skin examination with possible biopsy of lesions that are characteristic of SCFN, such as red or violaceous subcutaneous nodules or plaques, for specific disease identification.
The main complication of SCFN is hypercalcemia, which occurs in approximately 50% of cases. Other serum abnormalities include hyperglycemia, hypertriglyceridemia, and thrombocytopenia, though these findings are not as well associated.4 Patients with associated hypercalcemia may be asymptomatic, as in our patient, but other presentations include irritability, weakness, anorexia, vomiting, renal failure, failure to thrive, and encephalopathy. Nephrocalcinosis is a common complication of severe hypercalcemia; however, there is little evidence of associated major renal dysfunction.5 The exact mechanism of hypercalcemia is poorly understood. A widely accepted theory postulates that a granulomatous inflammatory infiltrate upregulates 1-α-hydroxylase activity, which enzymatically converts 25-hydroxyvitamin D to its active form, 1,25-dihydroxycholecalciferol, which increases bone resorption and calcium absorption through the gastrointestinal tract and renal systems. Treatments for hypercalcemia include hyperhydration, calcium-wasting diuretics, and low calcium intake.6 Furthermore, calcium levels should be obtained at the time of diagnosis and 30, 45, and 60 days after the lesions resolve.4
Subcutaneous fat necrosis needs to be differentiated from the more severe panniculitis, sclerema neonatorum (SN), which typically affects critically ill, preterm, and small-for-gestational-age newborns. It is associated with a high mortality rate and is characterized by skin and subadjacent tissue structures. The process typically begins in the thighs, buttocks, or trunk and spreads diffusely, sparing the fat-free palms, soles, and genitalia.7 Although our patient was born preterm, the physical characteristics of the nodule and the lack of severe illness placed SN lower on our differential. Histopathologic differences between SCFN and SN involve the extent of tissue fibrosis and presence of inflammatory cells. Sclerema neonatorum typically manifests with thickened connective tissue with a sparse inflammatory infiltrate, including lymphocytes, histiocytes, and multinucleated giant cells.7 Conversely, SCFN manifests with fat necrosis with an extensive inflammatory infiltrate. It is important to be able to distinguish between these 2 conditions, as both have vastly different prognoses.
Cold panniculitis, sometimes called “popsicle panniculitis,” is a phenomenon in which cold contact with the skin causes eruption of firm, erythematous, indurated plaques at the site of exposure. This self-limiting condition typically appears hours to days after cold exposure and spontaneously resolves in a few weeks.8 Therapeutic hypothermic protocol treatment involves using cooling devices to lower the body temperature for a short duration. The temperature typically is lowered to approximately 32 °C to 36 °C. These temperatures are not low enough to induce cold panniculitis, which is more commonly seen in facial ice applications when managing supraventricular tachycardia in neonates.
Cephalohematoma is a birthing injury that causes blood accumulation within the subperiosteal space. During parturition, the compressive and sheering forces on the calvarium rupture the vessels passing through the periosteum, causing blood to pool slowly into the subperiostium; thus, a cephalohematoma usually manifests later at 1 to 3 days of life as localized head swelling.9 The bleeding typically does not cross suture lines and is primarily found in the occipital or parietal regions. The incidence has been reported to be 0.4% to 2.5% of all live births.10 Although the location of the nodule in our patient was in the occipital region, imaging and biopsy results did not show hemorrhagic findings consistent with cephalohematoma. Management of cephalohematoma mainly is observational, as the mass slowly regresses and the accumulated blood gradually is reabsorbed.
Fungal scalp infections (tinea capitis) are common in the pediatric population. The peak incidence of this infection has been reported in children aged 3 to 7 years, with Trichophyton tonsurans and Microsporum canis as the usual causative organisms.11 Clinical features of tinea capitis include scaly patches with hair loss, hair loss with black pigmented dots at the follicular openings, diffuse scalp scaling with subtle hair loss, and cervical lymphadenopathy.12 Although less common, tinea capitis can progress to a more severe form known as a kerion, which is characterized by a tender plaque with pustules and crusting. A kerion can result in permanent scarring and alopecia if left untreated.12 In our patient, a nodule with scaling and faint erythema was observed, but no black pigmented dots at the follicular orifices were present. Therefore, a potassium hydroxide wet mount preparation used to diagnose tinea capitis was unnecessary. Systemic oral antifungal therapy such as fluconazole or terbinafine is the standard treatment for tinea capitis.
- Coondoo A, Lahiry R, Choudhury A, et al. Tender skin nodules in a newborn. Indian J Dermatol. 2013;58:328. doi:10.4103/0019-5154.113983
- Mitra S, Dove J, Somisetty SK. Subcutaneous fat necrosis in newbornan unusual case and review of literature. Eur J Pediatr. 2011;170:1107- 1110. doi:10.1007/s00431-011-1405-x
- Velasquez JH, Mendez MD. Newborn subcutaneous fat necrosis. In: StatPearls. StatPearls Publishing; 2022.
- Stefanko NS, Drolet BA. Subcutaneous fat necrosis of the newborn and associated hypercalcemia: a systematic review of the literature. Pediatr Dermatol. 2019;36:24-30. doi:10.1111/pde.13640
- Shumer DE, Thaker V, Taylor GA, et al. Severe hypercalcaemia due to subcutaneous fat necrosis: presentation, management and complications. Arch Dis Child Fetal Neonatal Ed. 2014;99:F419-F421. doi:10.1136/ archdischild-2014-306069
- Farooque A, Moss C, Zehnder D, et al. Expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in subcutaneous fat necrosis. Br J Dermatol. 2009;160:423-425. doi:10.1111/j.1365-2133.2008.08844.x
- Zeb A, Darmstadt GL. Sclerema neonatorum: a review of nomenclature, clinical presentation, histological features, differential diagnoses and management. J Perinatol. 2008;28:453-460. doi:10.1038/jp.2008.33
- Quesada-Cortés A, Campos-Muñoz L, Díaz-Díaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:485-489, vii. doi:10.1016 /j.det.2008.05.015
- Raines DA, Krawiec C, Jain S. Cephalohematoma. In: StatPearls. StatPearls Publishing; 2023.
- Chung HY, Chung JY, Lee DG, et al. Surgical treatment of ossified cephalhematoma. J Craniofac Surg. 2004;15:774-779. doi:10.1097/00001665- 200409000-00015
- Leung AKC, Hon KL, Leong KF, et al. Tinea capitis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:58-68. doi:10.2174/1872 213x14666200106145624
- Kovitwanichkanont T, Chong A. Superficial fungal infections. Aust J Gen Pract. 2019;48:706-711. doi:10.31128/ajgp-05-19-4930
- Coondoo A, Lahiry R, Choudhury A, et al. Tender skin nodules in a newborn. Indian J Dermatol. 2013;58:328. doi:10.4103/0019-5154.113983
- Mitra S, Dove J, Somisetty SK. Subcutaneous fat necrosis in newbornan unusual case and review of literature. Eur J Pediatr. 2011;170:1107- 1110. doi:10.1007/s00431-011-1405-x
- Velasquez JH, Mendez MD. Newborn subcutaneous fat necrosis. In: StatPearls. StatPearls Publishing; 2022.
- Stefanko NS, Drolet BA. Subcutaneous fat necrosis of the newborn and associated hypercalcemia: a systematic review of the literature. Pediatr Dermatol. 2019;36:24-30. doi:10.1111/pde.13640
- Shumer DE, Thaker V, Taylor GA, et al. Severe hypercalcaemia due to subcutaneous fat necrosis: presentation, management and complications. Arch Dis Child Fetal Neonatal Ed. 2014;99:F419-F421. doi:10.1136/ archdischild-2014-306069
- Farooque A, Moss C, Zehnder D, et al. Expression of 25-hydroxyvitamin D3-1alpha-hydroxylase in subcutaneous fat necrosis. Br J Dermatol. 2009;160:423-425. doi:10.1111/j.1365-2133.2008.08844.x
- Zeb A, Darmstadt GL. Sclerema neonatorum: a review of nomenclature, clinical presentation, histological features, differential diagnoses and management. J Perinatol. 2008;28:453-460. doi:10.1038/jp.2008.33
- Quesada-Cortés A, Campos-Muñoz L, Díaz-Díaz RM, et al. Cold panniculitis. Dermatol Clin. 2008;26:485-489, vii. doi:10.1016 /j.det.2008.05.015
- Raines DA, Krawiec C, Jain S. Cephalohematoma. In: StatPearls. StatPearls Publishing; 2023.
- Chung HY, Chung JY, Lee DG, et al. Surgical treatment of ossified cephalhematoma. J Craniofac Surg. 2004;15:774-779. doi:10.1097/00001665- 200409000-00015
- Leung AKC, Hon KL, Leong KF, et al. Tinea capitis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:58-68. doi:10.2174/1872 213x14666200106145624
- Kovitwanichkanont T, Chong A. Superficial fungal infections. Aust J Gen Pract. 2019;48:706-711. doi:10.31128/ajgp-05-19-4930
A 4-week-old male infant was referred to dermatology for evaluation of a nodule on the occipital protuberance of 2 weeks’ duration. The patient was born at 36 weeks and 6 days’ gestation via an emergency cesarean delivery due to fetal distress. He later was found to have hypoxic-ischemic encephalopathy, pulmonary hypertension, and hypertrophic cardiomyopathy. He underwent therapeutic hypothermia protocol treatment starting at less than 6 hours after birth. At the current presentation, physical examination showed a 2.5-cm, erythematous, firm, mobile nodule on the occipital scalp with some overlying crusting and minimal surrounding erythema. No other cutaneous features or lesions were present. Initial laboratory findings were remarkable for hypercalcemia at 11 mg/dL (reference range, 8.5-10.5 mg/dL). Magnetic resonance imaging showed a faint abnormality in the subcutaneous tissue in this region without a noted connection to the underlying brain/meningeal matter. A punch biopsy was performed.


Progressively Worsening Scaly Patches and Plaques in an Infant
The Diagnosis: Erythrodermic Allergic Contact Dermatitis
The worsening symptoms in our patient prompted intervention rather than observation and reassurance. Contact allergy to lanolin was suspected given the worsening presentation after the addition of Minerin, which was immediately discontinued. The patient’s family applied betamethasone cream 0.1% twice daily to severe plaques, pimecrolimus cream 1% to the face, and triamcinolone cream 0.1% to the rest of the body. At follow-up 1 week later, he experienced complete resolution of symptoms, which supported the diagnosis of erythrodermic allergic contact dermatitis (ACD).
The prevalence of ACD caused by lanolin varies among the general population from 1.2% to 6.9%.1 Lanolin recently was named Allergen of the Year in 2023 by the American Contact Dermatitis Society.2 It can be found in various commercial products, including creams, soaps, and ointments. Atopic dermatitis (AD) is a common pediatric inflammatory skin disorder that typically is treated with these products.3 In a study analyzing 533 products, up to 6% of skin care products for babies and children contained lanolin.4 Therefore, exposure to lanolin-containing products may be fairly common in the pediatric population.
Lanolin is a fatlike substance derived from sheep sebaceous gland secretions and extracted from sheep’s wool. Its composition varies by sheep breed, location, and extraction and purification methods. The most common allergens involve the alcoholic fraction produced by hydrolysis of lanolin.4 In 1996, Wolf5 described the “lanolin paradox,” which argued the difficulty with identifying lanolin as an allergen (similar to Fisher’s “paraben paradox”) based on 4 principles: (1) lanolin-containing topical medicaments tend to be more sensitizing than lanolin-containing cosmetics; (2) patients with ACD after applying lanolin-containing topical medicaments to damaged or ulcerated skin often can apply lanolin-containing cosmetics to normal or unaffected skin without a reaction; (3) false-negative patch test results often occur in lanolin-sensitive patients; and (4) patch testing with a single lanolin-containing agent (lanolin alcohol [30% in petrolatum]) is an unreliable and inadequate method of detecting lanolin allergy.6,7 This theory elucidates the challenge of diagnosing contact allergies, particularly lanolin contact allergies.
Clinical features of acute ACD vary by skin type. Lighter skin types may have well-demarcated, pruritic, eczematous patches and plaques affecting the flexor surfaces. Asian patients may present with psoriasiform plaques with more well-demarcated borders and increased scaling and lichenification. In patients with darker skin types, dermatitis may manifest as papulation, lichenification, and color changes (violet, gray, or darker brown) along extensor surfaces.8 Chronic dermatitis manifests as lichenified scaly plaques. Given the diversity in dermatitis manifestation and the challenges of identifying erythema, especially in skin of color, clinicians may misidentify disease severity. These features aid in diagnosing and treating patients presenting with diffuse erythroderma and worsening eczematous patches and plaques despite use of typical topical treatments.
The differential diagnosis includes irritant contact dermatitis, AD, seborrheic dermatitis, and chronic plaque psoriasis. Negative patch testing suggests contact dermatitis based on exposure to a product. A thorough medication and personal history helps distinguish ACD from AD. Atopic dermatitis classically appears on the flexural areas, face, eyelids, and hands of patients with a personal or family history of atopy. Greasy scaly plaques on the central part of the face, eyelids, and scalp commonly are found in seborrheic dermatitis. In chronic plaque psoriasis, lesions typically are described as welldemarcated, inflamed plaques with notable scale located primarily in the scalp and diaper area in newborns and children until the age of 2 years. Our patient presented with scaly plaques throughout most of the body. The history of Minerin use over the course of 3 to 5 months and worsening skin eruptions involving a majority of the skin surface suggested continued exposure.
Patch testing assists in the diagnosis of ACD, with varying results due to manufacturing and processing inconsistencies in the composition of various substances used in the standard test sets, often making it difficult to diagnose lanolin as an allergen. According to Lee and Warshaw,6 the lack of uniformity within testing of lanolin-containing products may cause false-positive results, poor patch-test reproducibility, and loss of allergic contact response. A 2019 study utilized a combination of Amerchol L101 and lanolin alcohol to improve the diagnosis of lanolin allergy, as standard testing may not identify patients with lanolin sensitivities.1 A study with the North American Contact Dermatitis Group from 2005 to 2012 demonstrated that positive patch testing among children was the most consistent method for diagnosing ACD, and results were clinically relevant.9 However, the different lanolin-containing products are not standardized in patch testing, which often causes mixed reactions and does not definitely demonstrate classic positive results, even with the use of repeated open application tests.2 Although there has been an emphasis on refining the standardization of the lanolin used for patch testing, lanolin contact allergy remains a predominantly clinical diagnosis.
Both AD and ACD are common pediatric skin findings, and mixed positive and neutral associations between AD and allergy to lanolin have been described in a few studies.1,3,9,10 A history of atopy is more notable in a pediatric patient vs an adult, as sensitivities tend to subside into adulthood.9 Further studies and more precise testing are needed to investigate the relationship between AD and ACD.
- Knijp J, Bruynzeel DP, Rustemeyer T. Diagnosing lanolin contact allergy with lanolin alcohol and Amerchol L101. Contact Dermatitis. 2019;80:298-303. doi:10.1111/cod.13210
- Jenkins BA, Belsito DV. Lanolin. Dermatitis. 2023;34:4-12. doi:10.1089 /derm.2022.0002
- Jacob SE, McGowan M, Silverberg NB, et al. Pediatric Contact Dermatitis Registry data on contact allergy in children with atopic dermatitis. JAMA Dermatol. 2017;153:765-770. doi:10.1001/jamadermatol .2016.6136
- Bonchak JG, Prouty ME, de la Feld SF. Prevalence of contact allergens in personal care products for babies and children. Dermatitis. 2018; 29:81-84. doi:10.1097/DER.0000000000000348
- Wolf R. The lanolin paradox. Dermatology. 1996;192:198-202. doi:10.1159/000246365
- Lee B, Warshaw E. Lanolin allergy: history, epidemiology, responsible allergens, and management. Dermatitis. 2008;19:63-72.
- Miest RY, Yiannias JA, Chang YH, et al. Diagnosis and prevalence of lanolin allergy. Dermatitis. 2013;24:119-123. doi:10.1097 /DER.0b013e3182937aa4
- Sangha AM. Dermatological conditions in SKIN OF COLOR-: managing atopic dermatitis. J Clin Aesthet Dermatol. 2021;14(3 Suppl 1):S20-S22.
- Zug KA, Pham AK, Belsito DV, et al. Patch testing in children from 2005 to 2012: results from the North American contact dermatitis group. Dermatitis. 2014;25:345-355. doi:10.1097/DER.0000000000000083
- Wakelin SH, Smith H, White IR, et al. A retrospective analysis of contact allergy to lanolin. Br J Dermatol. 2001;145:28-31. doi:10.1046 /j.1365-2133.2001.04277.x
The Diagnosis: Erythrodermic Allergic Contact Dermatitis
The worsening symptoms in our patient prompted intervention rather than observation and reassurance. Contact allergy to lanolin was suspected given the worsening presentation after the addition of Minerin, which was immediately discontinued. The patient’s family applied betamethasone cream 0.1% twice daily to severe plaques, pimecrolimus cream 1% to the face, and triamcinolone cream 0.1% to the rest of the body. At follow-up 1 week later, he experienced complete resolution of symptoms, which supported the diagnosis of erythrodermic allergic contact dermatitis (ACD).
The prevalence of ACD caused by lanolin varies among the general population from 1.2% to 6.9%.1 Lanolin recently was named Allergen of the Year in 2023 by the American Contact Dermatitis Society.2 It can be found in various commercial products, including creams, soaps, and ointments. Atopic dermatitis (AD) is a common pediatric inflammatory skin disorder that typically is treated with these products.3 In a study analyzing 533 products, up to 6% of skin care products for babies and children contained lanolin.4 Therefore, exposure to lanolin-containing products may be fairly common in the pediatric population.
Lanolin is a fatlike substance derived from sheep sebaceous gland secretions and extracted from sheep’s wool. Its composition varies by sheep breed, location, and extraction and purification methods. The most common allergens involve the alcoholic fraction produced by hydrolysis of lanolin.4 In 1996, Wolf5 described the “lanolin paradox,” which argued the difficulty with identifying lanolin as an allergen (similar to Fisher’s “paraben paradox”) based on 4 principles: (1) lanolin-containing topical medicaments tend to be more sensitizing than lanolin-containing cosmetics; (2) patients with ACD after applying lanolin-containing topical medicaments to damaged or ulcerated skin often can apply lanolin-containing cosmetics to normal or unaffected skin without a reaction; (3) false-negative patch test results often occur in lanolin-sensitive patients; and (4) patch testing with a single lanolin-containing agent (lanolin alcohol [30% in petrolatum]) is an unreliable and inadequate method of detecting lanolin allergy.6,7 This theory elucidates the challenge of diagnosing contact allergies, particularly lanolin contact allergies.
Clinical features of acute ACD vary by skin type. Lighter skin types may have well-demarcated, pruritic, eczematous patches and plaques affecting the flexor surfaces. Asian patients may present with psoriasiform plaques with more well-demarcated borders and increased scaling and lichenification. In patients with darker skin types, dermatitis may manifest as papulation, lichenification, and color changes (violet, gray, or darker brown) along extensor surfaces.8 Chronic dermatitis manifests as lichenified scaly plaques. Given the diversity in dermatitis manifestation and the challenges of identifying erythema, especially in skin of color, clinicians may misidentify disease severity. These features aid in diagnosing and treating patients presenting with diffuse erythroderma and worsening eczematous patches and plaques despite use of typical topical treatments.
The differential diagnosis includes irritant contact dermatitis, AD, seborrheic dermatitis, and chronic plaque psoriasis. Negative patch testing suggests contact dermatitis based on exposure to a product. A thorough medication and personal history helps distinguish ACD from AD. Atopic dermatitis classically appears on the flexural areas, face, eyelids, and hands of patients with a personal or family history of atopy. Greasy scaly plaques on the central part of the face, eyelids, and scalp commonly are found in seborrheic dermatitis. In chronic plaque psoriasis, lesions typically are described as welldemarcated, inflamed plaques with notable scale located primarily in the scalp and diaper area in newborns and children until the age of 2 years. Our patient presented with scaly plaques throughout most of the body. The history of Minerin use over the course of 3 to 5 months and worsening skin eruptions involving a majority of the skin surface suggested continued exposure.
Patch testing assists in the diagnosis of ACD, with varying results due to manufacturing and processing inconsistencies in the composition of various substances used in the standard test sets, often making it difficult to diagnose lanolin as an allergen. According to Lee and Warshaw,6 the lack of uniformity within testing of lanolin-containing products may cause false-positive results, poor patch-test reproducibility, and loss of allergic contact response. A 2019 study utilized a combination of Amerchol L101 and lanolin alcohol to improve the diagnosis of lanolin allergy, as standard testing may not identify patients with lanolin sensitivities.1 A study with the North American Contact Dermatitis Group from 2005 to 2012 demonstrated that positive patch testing among children was the most consistent method for diagnosing ACD, and results were clinically relevant.9 However, the different lanolin-containing products are not standardized in patch testing, which often causes mixed reactions and does not definitely demonstrate classic positive results, even with the use of repeated open application tests.2 Although there has been an emphasis on refining the standardization of the lanolin used for patch testing, lanolin contact allergy remains a predominantly clinical diagnosis.
Both AD and ACD are common pediatric skin findings, and mixed positive and neutral associations between AD and allergy to lanolin have been described in a few studies.1,3,9,10 A history of atopy is more notable in a pediatric patient vs an adult, as sensitivities tend to subside into adulthood.9 Further studies and more precise testing are needed to investigate the relationship between AD and ACD.
The Diagnosis: Erythrodermic Allergic Contact Dermatitis
The worsening symptoms in our patient prompted intervention rather than observation and reassurance. Contact allergy to lanolin was suspected given the worsening presentation after the addition of Minerin, which was immediately discontinued. The patient’s family applied betamethasone cream 0.1% twice daily to severe plaques, pimecrolimus cream 1% to the face, and triamcinolone cream 0.1% to the rest of the body. At follow-up 1 week later, he experienced complete resolution of symptoms, which supported the diagnosis of erythrodermic allergic contact dermatitis (ACD).
The prevalence of ACD caused by lanolin varies among the general population from 1.2% to 6.9%.1 Lanolin recently was named Allergen of the Year in 2023 by the American Contact Dermatitis Society.2 It can be found in various commercial products, including creams, soaps, and ointments. Atopic dermatitis (AD) is a common pediatric inflammatory skin disorder that typically is treated with these products.3 In a study analyzing 533 products, up to 6% of skin care products for babies and children contained lanolin.4 Therefore, exposure to lanolin-containing products may be fairly common in the pediatric population.
Lanolin is a fatlike substance derived from sheep sebaceous gland secretions and extracted from sheep’s wool. Its composition varies by sheep breed, location, and extraction and purification methods. The most common allergens involve the alcoholic fraction produced by hydrolysis of lanolin.4 In 1996, Wolf5 described the “lanolin paradox,” which argued the difficulty with identifying lanolin as an allergen (similar to Fisher’s “paraben paradox”) based on 4 principles: (1) lanolin-containing topical medicaments tend to be more sensitizing than lanolin-containing cosmetics; (2) patients with ACD after applying lanolin-containing topical medicaments to damaged or ulcerated skin often can apply lanolin-containing cosmetics to normal or unaffected skin without a reaction; (3) false-negative patch test results often occur in lanolin-sensitive patients; and (4) patch testing with a single lanolin-containing agent (lanolin alcohol [30% in petrolatum]) is an unreliable and inadequate method of detecting lanolin allergy.6,7 This theory elucidates the challenge of diagnosing contact allergies, particularly lanolin contact allergies.
Clinical features of acute ACD vary by skin type. Lighter skin types may have well-demarcated, pruritic, eczematous patches and plaques affecting the flexor surfaces. Asian patients may present with psoriasiform plaques with more well-demarcated borders and increased scaling and lichenification. In patients with darker skin types, dermatitis may manifest as papulation, lichenification, and color changes (violet, gray, or darker brown) along extensor surfaces.8 Chronic dermatitis manifests as lichenified scaly plaques. Given the diversity in dermatitis manifestation and the challenges of identifying erythema, especially in skin of color, clinicians may misidentify disease severity. These features aid in diagnosing and treating patients presenting with diffuse erythroderma and worsening eczematous patches and plaques despite use of typical topical treatments.
The differential diagnosis includes irritant contact dermatitis, AD, seborrheic dermatitis, and chronic plaque psoriasis. Negative patch testing suggests contact dermatitis based on exposure to a product. A thorough medication and personal history helps distinguish ACD from AD. Atopic dermatitis classically appears on the flexural areas, face, eyelids, and hands of patients with a personal or family history of atopy. Greasy scaly plaques on the central part of the face, eyelids, and scalp commonly are found in seborrheic dermatitis. In chronic plaque psoriasis, lesions typically are described as welldemarcated, inflamed plaques with notable scale located primarily in the scalp and diaper area in newborns and children until the age of 2 years. Our patient presented with scaly plaques throughout most of the body. The history of Minerin use over the course of 3 to 5 months and worsening skin eruptions involving a majority of the skin surface suggested continued exposure.
Patch testing assists in the diagnosis of ACD, with varying results due to manufacturing and processing inconsistencies in the composition of various substances used in the standard test sets, often making it difficult to diagnose lanolin as an allergen. According to Lee and Warshaw,6 the lack of uniformity within testing of lanolin-containing products may cause false-positive results, poor patch-test reproducibility, and loss of allergic contact response. A 2019 study utilized a combination of Amerchol L101 and lanolin alcohol to improve the diagnosis of lanolin allergy, as standard testing may not identify patients with lanolin sensitivities.1 A study with the North American Contact Dermatitis Group from 2005 to 2012 demonstrated that positive patch testing among children was the most consistent method for diagnosing ACD, and results were clinically relevant.9 However, the different lanolin-containing products are not standardized in patch testing, which often causes mixed reactions and does not definitely demonstrate classic positive results, even with the use of repeated open application tests.2 Although there has been an emphasis on refining the standardization of the lanolin used for patch testing, lanolin contact allergy remains a predominantly clinical diagnosis.
Both AD and ACD are common pediatric skin findings, and mixed positive and neutral associations between AD and allergy to lanolin have been described in a few studies.1,3,9,10 A history of atopy is more notable in a pediatric patient vs an adult, as sensitivities tend to subside into adulthood.9 Further studies and more precise testing are needed to investigate the relationship between AD and ACD.
- Knijp J, Bruynzeel DP, Rustemeyer T. Diagnosing lanolin contact allergy with lanolin alcohol and Amerchol L101. Contact Dermatitis. 2019;80:298-303. doi:10.1111/cod.13210
- Jenkins BA, Belsito DV. Lanolin. Dermatitis. 2023;34:4-12. doi:10.1089 /derm.2022.0002
- Jacob SE, McGowan M, Silverberg NB, et al. Pediatric Contact Dermatitis Registry data on contact allergy in children with atopic dermatitis. JAMA Dermatol. 2017;153:765-770. doi:10.1001/jamadermatol .2016.6136
- Bonchak JG, Prouty ME, de la Feld SF. Prevalence of contact allergens in personal care products for babies and children. Dermatitis. 2018; 29:81-84. doi:10.1097/DER.0000000000000348
- Wolf R. The lanolin paradox. Dermatology. 1996;192:198-202. doi:10.1159/000246365
- Lee B, Warshaw E. Lanolin allergy: history, epidemiology, responsible allergens, and management. Dermatitis. 2008;19:63-72.
- Miest RY, Yiannias JA, Chang YH, et al. Diagnosis and prevalence of lanolin allergy. Dermatitis. 2013;24:119-123. doi:10.1097 /DER.0b013e3182937aa4
- Sangha AM. Dermatological conditions in SKIN OF COLOR-: managing atopic dermatitis. J Clin Aesthet Dermatol. 2021;14(3 Suppl 1):S20-S22.
- Zug KA, Pham AK, Belsito DV, et al. Patch testing in children from 2005 to 2012: results from the North American contact dermatitis group. Dermatitis. 2014;25:345-355. doi:10.1097/DER.0000000000000083
- Wakelin SH, Smith H, White IR, et al. A retrospective analysis of contact allergy to lanolin. Br J Dermatol. 2001;145:28-31. doi:10.1046 /j.1365-2133.2001.04277.x
- Knijp J, Bruynzeel DP, Rustemeyer T. Diagnosing lanolin contact allergy with lanolin alcohol and Amerchol L101. Contact Dermatitis. 2019;80:298-303. doi:10.1111/cod.13210
- Jenkins BA, Belsito DV. Lanolin. Dermatitis. 2023;34:4-12. doi:10.1089 /derm.2022.0002
- Jacob SE, McGowan M, Silverberg NB, et al. Pediatric Contact Dermatitis Registry data on contact allergy in children with atopic dermatitis. JAMA Dermatol. 2017;153:765-770. doi:10.1001/jamadermatol .2016.6136
- Bonchak JG, Prouty ME, de la Feld SF. Prevalence of contact allergens in personal care products for babies and children. Dermatitis. 2018; 29:81-84. doi:10.1097/DER.0000000000000348
- Wolf R. The lanolin paradox. Dermatology. 1996;192:198-202. doi:10.1159/000246365
- Lee B, Warshaw E. Lanolin allergy: history, epidemiology, responsible allergens, and management. Dermatitis. 2008;19:63-72.
- Miest RY, Yiannias JA, Chang YH, et al. Diagnosis and prevalence of lanolin allergy. Dermatitis. 2013;24:119-123. doi:10.1097 /DER.0b013e3182937aa4
- Sangha AM. Dermatological conditions in SKIN OF COLOR-: managing atopic dermatitis. J Clin Aesthet Dermatol. 2021;14(3 Suppl 1):S20-S22.
- Zug KA, Pham AK, Belsito DV, et al. Patch testing in children from 2005 to 2012: results from the North American contact dermatitis group. Dermatitis. 2014;25:345-355. doi:10.1097/DER.0000000000000083
- Wakelin SH, Smith H, White IR, et al. A retrospective analysis of contact allergy to lanolin. Br J Dermatol. 2001;145:28-31. doi:10.1046 /j.1365-2133.2001.04277.x
A 5-month-old male with moderately brown skin that rarely burns and tans profusely presented to the emergency department with a worsening red rash of more than 4 months’ duration. The patient had diffuse erythroderma and eczematous patches and plaques covering 95% of the total body surface area, including lichenified plaques on the arms and elbows, with no signs of infection. He initially presented for his 1-month appointment at the pediatric clinic with scaly patches and plaques on the face and trunk as well as diffuse xerosis. He was prescribed daily oatmeal baths and topical Minerin (Major Pharmaceuticals)—containing water, petrolatum, mineral oil, mineral wax, lanolin alcohol, methylchloroisothiazolinone, and methylisothiazolinone—to be applied to the whole body twice daily. At the patient’s 2-month well visit, symptoms persisted. The patient’s pediatrician increased application of Minerin to 2 to 3 times daily, and hydrocortisone cream 2.5% application 2 to 3 times daily was added.


Lichenoid Dermatosis on the Feet
The Diagnosis: Hypertrophic Lichen Planus
Two biopsies from the left lateral foot revealed hyperkeratosis, wedge-shaped hypergranulosis, irregular acanthosis, and a bandlike lymphocytic infiltrate in the superficial dermis with a classic sawtooth pattern of the rete ridges (Figure 1). Based on the clinical findings and histopathology, the patient was diagnosed with hypertrophic lichen planus (LP) and was treated with clobetasol ointment 0.05%, which resulted in progression of the symptoms. She experienced notable improvement 3 months after adding methotrexate 12.5 mg weekly (Figure 2).

Lichen planus is an idiopathic chronic inflammatory condition of the skin and mucous membranes that classically manifests as pruritic violaceous papules and plaques, which commonly are found on the wrists, lower back, and ankles.1 The most common variants of LP are hypertrophic, linear, mucosal, actinic, follicular, pigmented, annular, atrophic, and guttate.2 The clinical presentation and biopsy results in our patient were consistent with the hypertrophic variant of LP, which is a chronic condition that most often manifests on the lower legs, especially around the ankles, as hyperkeratotic papules, plaques, and nodules.2,3 The exact pathophysiology of hypertrophic LP is unknown, but there is evidence that the immune system plays a role in its development and that the Koebner phenomenon may contribute to its exacerbation.4 There is a well-known association between LP and hepatitis. Patients with chronic LP may develop squamous cell carcinoma.4 The variants of LP can overlap and do not exist independent of one another. Recognizing the overlap in these variants allows for earlier diagnosis and therapeutic intervention of the disease process to limit disease progression and patient clinic visits and to improve patient quality of life.

The differential diagnosis for hyperkeratotic plaques of the feet and ankles can be broad and may include keratosis lichenoides chronica, palmoplantar keratoderma, palmoplantar psoriasis, or lichen amyloidosis. These conditions are classified based on various criteria that include extent of disease manifestations, morphology of palmoplantar skin involvement, inheritance patterns, and molecular pathogenesis.5 Keratosis lichenoides chronica is a rare dermatosis that presents as a distinctive seborrheic dermatitis–like facial eruption. The facial eruption is accompanied by violaceous papular and nodular lesions that appear on the extremities and trunk, typically arranged in a linear or reticular pattern.6 Palmoplantar keratoderma represents a group of acquired and hereditary conditions that are characterized by excessive thickening of the palms and soles.5 Palmoplantar psoriasis is a variant of psoriasis that affects the palms and soles and can manifest as hyperkeratosis, pustular, or mixed morphology.7 Lichen amyloidosis is a subtype of primary localized cutaneous amyloidosis that manifests as multiple pruritic, firm, hyperpigmented, hyperkeratotic papules on the shins that later coalesce in a rippled pattern.8,9
The first-line treatment for hypertrophic LP is topical corticosteroids. Alternative therapies include mycophenolate mofetil, acitretin, and intralesional corticosteroid injections.4 Treatment is similar for all of the LP variants.
- Arnold DL, Krishnamurthy K. Lichen planus. In: StatPearls. StatPearls Publishing; 2022.
- Namazi MR, Bahmani M. Diagnosis: hypertrophic lichen planus. Ann Saudi Med. 2008;28:1-2. doi:10.5144/0256-4947.2008.222
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:e3555. doi:10.7759 /cureus.3555
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149. doi:10.1016/j .ijwd.2015.04.001
- Has C, Technau-Hafsi K. Palmoplantar keratodermas: clinical and genetic aspects. J Dtsch Dermatol Ges. 2016;14:123-139; quiz 140. doi:10.1111/ddg.12930
- Konstantinov KN, Søndergaard J, Izuno G, et al. Keratosis lichenoides chronica. J Am Acad Dermatol. 1998;38(2 Pt 2):306-309. doi:10.1016 /s0190-9622(98)70570-5
- Miceli A, Schmieder GJ. Palmoplantar psoriasis. In: StatPearls. StatPearls Publishing; 2023.
- Tay CH, Dacosta JL. Lichen amyloidosis—clinical study of 40 cases. Br J Dermatol. 1970;82:129-136.
- Salim T, Shenoi SD, Balachandran C, et al. Lichen amyloidosis: a study of clinical, histopathologic and immunofluorescence findings in 30 cases. Indian J Dermatol Venereol Leprol. 2005;71:166-169.
The Diagnosis: Hypertrophic Lichen Planus
Two biopsies from the left lateral foot revealed hyperkeratosis, wedge-shaped hypergranulosis, irregular acanthosis, and a bandlike lymphocytic infiltrate in the superficial dermis with a classic sawtooth pattern of the rete ridges (Figure 1). Based on the clinical findings and histopathology, the patient was diagnosed with hypertrophic lichen planus (LP) and was treated with clobetasol ointment 0.05%, which resulted in progression of the symptoms. She experienced notable improvement 3 months after adding methotrexate 12.5 mg weekly (Figure 2).
Lichen planus is an idiopathic chronic inflammatory condition of the skin and mucous membranes that classically manifests as pruritic violaceous papules and plaques, which commonly are found on the wrists, lower back, and ankles.1 The most common variants of LP are hypertrophic, linear, mucosal, actinic, follicular, pigmented, annular, atrophic, and guttate.2 The clinical presentation and biopsy results in our patient were consistent with the hypertrophic variant of LP, which is a chronic condition that most often manifests on the lower legs, especially around the ankles, as hyperkeratotic papules, plaques, and nodules.2,3 The exact pathophysiology of hypertrophic LP is unknown, but there is evidence that the immune system plays a role in its development and that the Koebner phenomenon may contribute to its exacerbation.4 There is a well-known association between LP and hepatitis. Patients with chronic LP may develop squamous cell carcinoma.4 The variants of LP can overlap and do not exist independent of one another. Recognizing the overlap in these variants allows for earlier diagnosis and therapeutic intervention of the disease process to limit disease progression and patient clinic visits and to improve patient quality of life.
The differential diagnosis for hyperkeratotic plaques of the feet and ankles can be broad and may include keratosis lichenoides chronica, palmoplantar keratoderma, palmoplantar psoriasis, or lichen amyloidosis. These conditions are classified based on various criteria that include extent of disease manifestations, morphology of palmoplantar skin involvement, inheritance patterns, and molecular pathogenesis.5 Keratosis lichenoides chronica is a rare dermatosis that presents as a distinctive seborrheic dermatitis–like facial eruption. The facial eruption is accompanied by violaceous papular and nodular lesions that appear on the extremities and trunk, typically arranged in a linear or reticular pattern.6 Palmoplantar keratoderma represents a group of acquired and hereditary conditions that are characterized by excessive thickening of the palms and soles.5 Palmoplantar psoriasis is a variant of psoriasis that affects the palms and soles and can manifest as hyperkeratosis, pustular, or mixed morphology.7 Lichen amyloidosis is a subtype of primary localized cutaneous amyloidosis that manifests as multiple pruritic, firm, hyperpigmented, hyperkeratotic papules on the shins that later coalesce in a rippled pattern.8,9
The first-line treatment for hypertrophic LP is topical corticosteroids. Alternative therapies include mycophenolate mofetil, acitretin, and intralesional corticosteroid injections.4 Treatment is similar for all of the LP variants.
The Diagnosis: Hypertrophic Lichen Planus
Two biopsies from the left lateral foot revealed hyperkeratosis, wedge-shaped hypergranulosis, irregular acanthosis, and a bandlike lymphocytic infiltrate in the superficial dermis with a classic sawtooth pattern of the rete ridges (Figure 1). Based on the clinical findings and histopathology, the patient was diagnosed with hypertrophic lichen planus (LP) and was treated with clobetasol ointment 0.05%, which resulted in progression of the symptoms. She experienced notable improvement 3 months after adding methotrexate 12.5 mg weekly (Figure 2).
Lichen planus is an idiopathic chronic inflammatory condition of the skin and mucous membranes that classically manifests as pruritic violaceous papules and plaques, which commonly are found on the wrists, lower back, and ankles.1 The most common variants of LP are hypertrophic, linear, mucosal, actinic, follicular, pigmented, annular, atrophic, and guttate.2 The clinical presentation and biopsy results in our patient were consistent with the hypertrophic variant of LP, which is a chronic condition that most often manifests on the lower legs, especially around the ankles, as hyperkeratotic papules, plaques, and nodules.2,3 The exact pathophysiology of hypertrophic LP is unknown, but there is evidence that the immune system plays a role in its development and that the Koebner phenomenon may contribute to its exacerbation.4 There is a well-known association between LP and hepatitis. Patients with chronic LP may develop squamous cell carcinoma.4 The variants of LP can overlap and do not exist independent of one another. Recognizing the overlap in these variants allows for earlier diagnosis and therapeutic intervention of the disease process to limit disease progression and patient clinic visits and to improve patient quality of life.
The differential diagnosis for hyperkeratotic plaques of the feet and ankles can be broad and may include keratosis lichenoides chronica, palmoplantar keratoderma, palmoplantar psoriasis, or lichen amyloidosis. These conditions are classified based on various criteria that include extent of disease manifestations, morphology of palmoplantar skin involvement, inheritance patterns, and molecular pathogenesis.5 Keratosis lichenoides chronica is a rare dermatosis that presents as a distinctive seborrheic dermatitis–like facial eruption. The facial eruption is accompanied by violaceous papular and nodular lesions that appear on the extremities and trunk, typically arranged in a linear or reticular pattern.6 Palmoplantar keratoderma represents a group of acquired and hereditary conditions that are characterized by excessive thickening of the palms and soles.5 Palmoplantar psoriasis is a variant of psoriasis that affects the palms and soles and can manifest as hyperkeratosis, pustular, or mixed morphology.7 Lichen amyloidosis is a subtype of primary localized cutaneous amyloidosis that manifests as multiple pruritic, firm, hyperpigmented, hyperkeratotic papules on the shins that later coalesce in a rippled pattern.8,9
The first-line treatment for hypertrophic LP is topical corticosteroids. Alternative therapies include mycophenolate mofetil, acitretin, and intralesional corticosteroid injections.4 Treatment is similar for all of the LP variants.
- Arnold DL, Krishnamurthy K. Lichen planus. In: StatPearls. StatPearls Publishing; 2022.
- Namazi MR, Bahmani M. Diagnosis: hypertrophic lichen planus. Ann Saudi Med. 2008;28:1-2. doi:10.5144/0256-4947.2008.222
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:e3555. doi:10.7759 /cureus.3555
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149. doi:10.1016/j .ijwd.2015.04.001
- Has C, Technau-Hafsi K. Palmoplantar keratodermas: clinical and genetic aspects. J Dtsch Dermatol Ges. 2016;14:123-139; quiz 140. doi:10.1111/ddg.12930
- Konstantinov KN, Søndergaard J, Izuno G, et al. Keratosis lichenoides chronica. J Am Acad Dermatol. 1998;38(2 Pt 2):306-309. doi:10.1016 /s0190-9622(98)70570-5
- Miceli A, Schmieder GJ. Palmoplantar psoriasis. In: StatPearls. StatPearls Publishing; 2023.
- Tay CH, Dacosta JL. Lichen amyloidosis—clinical study of 40 cases. Br J Dermatol. 1970;82:129-136.
- Salim T, Shenoi SD, Balachandran C, et al. Lichen amyloidosis: a study of clinical, histopathologic and immunofluorescence findings in 30 cases. Indian J Dermatol Venereol Leprol. 2005;71:166-169.
- Arnold DL, Krishnamurthy K. Lichen planus. In: StatPearls. StatPearls Publishing; 2022.
- Namazi MR, Bahmani M. Diagnosis: hypertrophic lichen planus. Ann Saudi Med. 2008;28:1-2. doi:10.5144/0256-4947.2008.222
- Riahi RR, Cohen PR. Hypertrophic lichen planus mimicking verrucous lupus erythematosus. Cureus. 2018;10:e3555. doi:10.7759 /cureus.3555
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149. doi:10.1016/j .ijwd.2015.04.001
- Has C, Technau-Hafsi K. Palmoplantar keratodermas: clinical and genetic aspects. J Dtsch Dermatol Ges. 2016;14:123-139; quiz 140. doi:10.1111/ddg.12930
- Konstantinov KN, Søndergaard J, Izuno G, et al. Keratosis lichenoides chronica. J Am Acad Dermatol. 1998;38(2 Pt 2):306-309. doi:10.1016 /s0190-9622(98)70570-5
- Miceli A, Schmieder GJ. Palmoplantar psoriasis. In: StatPearls. StatPearls Publishing; 2023.
- Tay CH, Dacosta JL. Lichen amyloidosis—clinical study of 40 cases. Br J Dermatol. 1970;82:129-136.
- Salim T, Shenoi SD, Balachandran C, et al. Lichen amyloidosis: a study of clinical, histopathologic and immunofluorescence findings in 30 cases. Indian J Dermatol Venereol Leprol. 2005;71:166-169.
An 83-year-old woman presented for evaluation of hyperkeratotic plaques on the medial and lateral aspects of the left heel (top). Physical examination also revealed onychodystrophy of the toenails on the halluces (bottom). A crusted friable plaque on the lower lip and white plaques with peripheral reticulation and erosions on the buccal mucosa also were present. The patient had a history of nummular eczema, stasis dermatitis, and hand dermatitis. She denied a history of cold sores.


Asymptomatic Erythematous Plaque in an Outdoorsman
The Diagnosis: Erythema Migrans
The patient was clinically diagnosed with erythema migrans. He did not recall a tick bite but spent a lot of time outdoors. He was treated with 10 days of doxycycline 100 mg twice daily with complete resolution of the rash.
Lyme disease is a spirochete infection caused by the Borrelia burgdorferi sensu lato species complex and transmitted by the Ixodidae tick family. It is the most common tick-borne disease in the United States and mostly is reported in the northeastern and upper midwestern states during the warmer seasons, but it is prevalent worldwide. In geographic areas where Lyme disease is common, the incidence is approximately 40 cases per 100,000 individuals.1 Our patient resided in coastal South Carolina. Lyme disease is more commonly reported in White individuals. The skin lesions may be more difficult to discern and diagnose in patients with darker skin types, leading to delayed diagnosis and treatment.2,3
Patients may be diagnosed with early localized, early disseminated, or late Lyme disease. Erythema migrans is the early localized form of the disease and is classically described as an erythematous targetlike plaque with raised borders arising at the site of the tick bite 1 to 2 weeks later.4 However, many patients simply have a homogeneous erythematous plaque with raised advancing borders ranging in size from 5 to 68 cm.5 In a 2022 study of 69 patients with suspected Lyme disease, only 35 (50.7%) were determined to truly have acute Lyme disease.6 Of them, only 2 (5.7%) had the classic ringwithin- a-ring pattern. Most plaques were uniform, pink, oval-shaped lesions with well-demarcated borders.6
The rash may present with a burning sensation, or patients may experience no symptoms at all, which can lead to delayed diagnosis and progression to late disease. Patients may develop malaise, fever, headache, body aches, or joint pain. Early disseminated disease manifests similarly. Patients with disseminated disease also may develop more serious complications, including lymphadenopathy; cranial nerve palsies; ocular involvement; meningitis; or cardiac abnormalities such as myocarditis, pericarditis, or arrhythmia. Late disease most often causes arthritis of the large joints, though it also can have cardiac or neurologic manifestations. Some patients with chronic disease—the majority of whom were diagnosed in Europe—may develop acrodermatitis chronica atrophicans with edematous blue-red plaques that become atrophic and hyperpigmented fibrotic plaques over the course of years.
Allergic contact dermatitis to a plant more likely would cause itchy or painful, oozy, weepy, vesicular lesions arranged in a linear pattern. A dermatophyte infection likely would cause a scaly eruption. Although our patient presented with a sharply demarcated, raised, erythematous lesion, the distribution did not follow normal clothing lines and would be unusual for a photosensitive drug eruption. Cellulitis likely would be associated with tenderness or warmth to the touch. Finally, southern tick-associated rash illness, which is associated with Amblyomma americanum (lone star tick) bites, may appear with a similar rash but few systemic symptoms. It also can be treated with tetracycline antibiotics.7
Our case in South Carolina demonstrates the importance of keeping Lyme disease in the differential. Clinicians should remember to ask patients about their travel history. In endemic areas, patients with erythema migrans can be started on treatment without waiting for serology. Patients with early Lyme disease may or may not have positive serologies at the time of presentation.6 Guidelines for the treatment of Lyme disease have been revised in recent years to decrease patient antibiotic exposure by reducing the number of days of antibiotic therapy.8 A recent randomized controlled trial found no significant difference in recurrence for patients treated with 7 days of doxycycline compared with 14 days.9 We typically prescribe a 10-day course of doxycycline, which also is adequate for concurrent rickettsial disease. Patients who develop malarialike symptoms should be evaluated for babesiosis, which is treated with clindamycin.
- Skar GL, Simonsen KA. Lyme disease. StatPearls [Internet]. Updated February 4, 2024. Accessed March 20, 2024. https://www.ncbi.nlm.nih.gov/books/NBK431066/
- Dennison R, Novak C, Rebman A, et al. Lyme disease with erythema migrans and seventh nerve palsy in an African-American man. Cureus. 2019;11:E6509.
- Bax CE, Clark AK, Oboite M, et al. A case of disseminated Lyme disease in a child with skin of color. Pediatr Dermatol. 2021;38 (suppl 2):140-141.
- Shah AS, Varatharaj Palraj BR. Multiple erythema migrans rashes characteristic of early disseminated lyme disease, before and after therapy. Mayo Clin Proc. 2019;94:172-173.
- Feder HM Jr, Abeles M, Bernstein M, et al. Diagnosis, treatment, and prognosis of erythema migrans and Lyme arthritis. Clin Dermatol. 2006;24:509-520.
- Schotthoefer AM, Green CB, Dempsey G, et al. The spectrum of erythema migrans in early Lyme disease: can we improve its recognition? Cureus. 2022;14:E30673.
- Strle F, Wormser GP. Early Lyme disease (erythema migrans) and its mimics (southern tick-associated rash illness and tick-associated rash illness). Infect Dis Clin North Am. 2022;36:523-539.
- Torbahn G, Hofmann H, Rücker G, et al. Efficacy and safety of antibiotic therapy in early cutaneous Lyme borreliosis: a network meta-analysis. JAMA Dermatol. 2018;154:1292-1303.
- Stupica D, Collinet-Adler S, Blagus R, et al. Treatment of erythema migrans with doxycycline for 7 days versus 14 days in Slovenia: a randomised open-label non-inferiority trial. Lancet Infect Dis. 2023;23:371-379.
The Diagnosis: Erythema Migrans
The patient was clinically diagnosed with erythema migrans. He did not recall a tick bite but spent a lot of time outdoors. He was treated with 10 days of doxycycline 100 mg twice daily with complete resolution of the rash.
Lyme disease is a spirochete infection caused by the Borrelia burgdorferi sensu lato species complex and transmitted by the Ixodidae tick family. It is the most common tick-borne disease in the United States and mostly is reported in the northeastern and upper midwestern states during the warmer seasons, but it is prevalent worldwide. In geographic areas where Lyme disease is common, the incidence is approximately 40 cases per 100,000 individuals.1 Our patient resided in coastal South Carolina. Lyme disease is more commonly reported in White individuals. The skin lesions may be more difficult to discern and diagnose in patients with darker skin types, leading to delayed diagnosis and treatment.2,3
Patients may be diagnosed with early localized, early disseminated, or late Lyme disease. Erythema migrans is the early localized form of the disease and is classically described as an erythematous targetlike plaque with raised borders arising at the site of the tick bite 1 to 2 weeks later.4 However, many patients simply have a homogeneous erythematous plaque with raised advancing borders ranging in size from 5 to 68 cm.5 In a 2022 study of 69 patients with suspected Lyme disease, only 35 (50.7%) were determined to truly have acute Lyme disease.6 Of them, only 2 (5.7%) had the classic ringwithin- a-ring pattern. Most plaques were uniform, pink, oval-shaped lesions with well-demarcated borders.6
The rash may present with a burning sensation, or patients may experience no symptoms at all, which can lead to delayed diagnosis and progression to late disease. Patients may develop malaise, fever, headache, body aches, or joint pain. Early disseminated disease manifests similarly. Patients with disseminated disease also may develop more serious complications, including lymphadenopathy; cranial nerve palsies; ocular involvement; meningitis; or cardiac abnormalities such as myocarditis, pericarditis, or arrhythmia. Late disease most often causes arthritis of the large joints, though it also can have cardiac or neurologic manifestations. Some patients with chronic disease—the majority of whom were diagnosed in Europe—may develop acrodermatitis chronica atrophicans with edematous blue-red plaques that become atrophic and hyperpigmented fibrotic plaques over the course of years.
Allergic contact dermatitis to a plant more likely would cause itchy or painful, oozy, weepy, vesicular lesions arranged in a linear pattern. A dermatophyte infection likely would cause a scaly eruption. Although our patient presented with a sharply demarcated, raised, erythematous lesion, the distribution did not follow normal clothing lines and would be unusual for a photosensitive drug eruption. Cellulitis likely would be associated with tenderness or warmth to the touch. Finally, southern tick-associated rash illness, which is associated with Amblyomma americanum (lone star tick) bites, may appear with a similar rash but few systemic symptoms. It also can be treated with tetracycline antibiotics.7
Our case in South Carolina demonstrates the importance of keeping Lyme disease in the differential. Clinicians should remember to ask patients about their travel history. In endemic areas, patients with erythema migrans can be started on treatment without waiting for serology. Patients with early Lyme disease may or may not have positive serologies at the time of presentation.6 Guidelines for the treatment of Lyme disease have been revised in recent years to decrease patient antibiotic exposure by reducing the number of days of antibiotic therapy.8 A recent randomized controlled trial found no significant difference in recurrence for patients treated with 7 days of doxycycline compared with 14 days.9 We typically prescribe a 10-day course of doxycycline, which also is adequate for concurrent rickettsial disease. Patients who develop malarialike symptoms should be evaluated for babesiosis, which is treated with clindamycin.
The Diagnosis: Erythema Migrans
The patient was clinically diagnosed with erythema migrans. He did not recall a tick bite but spent a lot of time outdoors. He was treated with 10 days of doxycycline 100 mg twice daily with complete resolution of the rash.
Lyme disease is a spirochete infection caused by the Borrelia burgdorferi sensu lato species complex and transmitted by the Ixodidae tick family. It is the most common tick-borne disease in the United States and mostly is reported in the northeastern and upper midwestern states during the warmer seasons, but it is prevalent worldwide. In geographic areas where Lyme disease is common, the incidence is approximately 40 cases per 100,000 individuals.1 Our patient resided in coastal South Carolina. Lyme disease is more commonly reported in White individuals. The skin lesions may be more difficult to discern and diagnose in patients with darker skin types, leading to delayed diagnosis and treatment.2,3
Patients may be diagnosed with early localized, early disseminated, or late Lyme disease. Erythema migrans is the early localized form of the disease and is classically described as an erythematous targetlike plaque with raised borders arising at the site of the tick bite 1 to 2 weeks later.4 However, many patients simply have a homogeneous erythematous plaque with raised advancing borders ranging in size from 5 to 68 cm.5 In a 2022 study of 69 patients with suspected Lyme disease, only 35 (50.7%) were determined to truly have acute Lyme disease.6 Of them, only 2 (5.7%) had the classic ringwithin- a-ring pattern. Most plaques were uniform, pink, oval-shaped lesions with well-demarcated borders.6
The rash may present with a burning sensation, or patients may experience no symptoms at all, which can lead to delayed diagnosis and progression to late disease. Patients may develop malaise, fever, headache, body aches, or joint pain. Early disseminated disease manifests similarly. Patients with disseminated disease also may develop more serious complications, including lymphadenopathy; cranial nerve palsies; ocular involvement; meningitis; or cardiac abnormalities such as myocarditis, pericarditis, or arrhythmia. Late disease most often causes arthritis of the large joints, though it also can have cardiac or neurologic manifestations. Some patients with chronic disease—the majority of whom were diagnosed in Europe—may develop acrodermatitis chronica atrophicans with edematous blue-red plaques that become atrophic and hyperpigmented fibrotic plaques over the course of years.
Allergic contact dermatitis to a plant more likely would cause itchy or painful, oozy, weepy, vesicular lesions arranged in a linear pattern. A dermatophyte infection likely would cause a scaly eruption. Although our patient presented with a sharply demarcated, raised, erythematous lesion, the distribution did not follow normal clothing lines and would be unusual for a photosensitive drug eruption. Cellulitis likely would be associated with tenderness or warmth to the touch. Finally, southern tick-associated rash illness, which is associated with Amblyomma americanum (lone star tick) bites, may appear with a similar rash but few systemic symptoms. It also can be treated with tetracycline antibiotics.7
Our case in South Carolina demonstrates the importance of keeping Lyme disease in the differential. Clinicians should remember to ask patients about their travel history. In endemic areas, patients with erythema migrans can be started on treatment without waiting for serology. Patients with early Lyme disease may or may not have positive serologies at the time of presentation.6 Guidelines for the treatment of Lyme disease have been revised in recent years to decrease patient antibiotic exposure by reducing the number of days of antibiotic therapy.8 A recent randomized controlled trial found no significant difference in recurrence for patients treated with 7 days of doxycycline compared with 14 days.9 We typically prescribe a 10-day course of doxycycline, which also is adequate for concurrent rickettsial disease. Patients who develop malarialike symptoms should be evaluated for babesiosis, which is treated with clindamycin.
- Skar GL, Simonsen KA. Lyme disease. StatPearls [Internet]. Updated February 4, 2024. Accessed March 20, 2024. https://www.ncbi.nlm.nih.gov/books/NBK431066/
- Dennison R, Novak C, Rebman A, et al. Lyme disease with erythema migrans and seventh nerve palsy in an African-American man. Cureus. 2019;11:E6509.
- Bax CE, Clark AK, Oboite M, et al. A case of disseminated Lyme disease in a child with skin of color. Pediatr Dermatol. 2021;38 (suppl 2):140-141.
- Shah AS, Varatharaj Palraj BR. Multiple erythema migrans rashes characteristic of early disseminated lyme disease, before and after therapy. Mayo Clin Proc. 2019;94:172-173.
- Feder HM Jr, Abeles M, Bernstein M, et al. Diagnosis, treatment, and prognosis of erythema migrans and Lyme arthritis. Clin Dermatol. 2006;24:509-520.
- Schotthoefer AM, Green CB, Dempsey G, et al. The spectrum of erythema migrans in early Lyme disease: can we improve its recognition? Cureus. 2022;14:E30673.
- Strle F, Wormser GP. Early Lyme disease (erythema migrans) and its mimics (southern tick-associated rash illness and tick-associated rash illness). Infect Dis Clin North Am. 2022;36:523-539.
- Torbahn G, Hofmann H, Rücker G, et al. Efficacy and safety of antibiotic therapy in early cutaneous Lyme borreliosis: a network meta-analysis. JAMA Dermatol. 2018;154:1292-1303.
- Stupica D, Collinet-Adler S, Blagus R, et al. Treatment of erythema migrans with doxycycline for 7 days versus 14 days in Slovenia: a randomised open-label non-inferiority trial. Lancet Infect Dis. 2023;23:371-379.
- Skar GL, Simonsen KA. Lyme disease. StatPearls [Internet]. Updated February 4, 2024. Accessed March 20, 2024. https://www.ncbi.nlm.nih.gov/books/NBK431066/
- Dennison R, Novak C, Rebman A, et al. Lyme disease with erythema migrans and seventh nerve palsy in an African-American man. Cureus. 2019;11:E6509.
- Bax CE, Clark AK, Oboite M, et al. A case of disseminated Lyme disease in a child with skin of color. Pediatr Dermatol. 2021;38 (suppl 2):140-141.
- Shah AS, Varatharaj Palraj BR. Multiple erythema migrans rashes characteristic of early disseminated lyme disease, before and after therapy. Mayo Clin Proc. 2019;94:172-173.
- Feder HM Jr, Abeles M, Bernstein M, et al. Diagnosis, treatment, and prognosis of erythema migrans and Lyme arthritis. Clin Dermatol. 2006;24:509-520.
- Schotthoefer AM, Green CB, Dempsey G, et al. The spectrum of erythema migrans in early Lyme disease: can we improve its recognition? Cureus. 2022;14:E30673.
- Strle F, Wormser GP. Early Lyme disease (erythema migrans) and its mimics (southern tick-associated rash illness and tick-associated rash illness). Infect Dis Clin North Am. 2022;36:523-539.
- Torbahn G, Hofmann H, Rücker G, et al. Efficacy and safety of antibiotic therapy in early cutaneous Lyme borreliosis: a network meta-analysis. JAMA Dermatol. 2018;154:1292-1303.
- Stupica D, Collinet-Adler S, Blagus R, et al. Treatment of erythema migrans with doxycycline for 7 days versus 14 days in Slovenia: a randomised open-label non-inferiority trial. Lancet Infect Dis. 2023;23:371-379.
A middle-aged man presented with a well-demarcated, hyperpigmented, erythematous patch with an annular erythematous border that extended from the mid-back to the lower back. The patient was otherwise asymptomatic. He was an avid gardener who resided in South Carolina and had recently adopted 2 puppies.


Skin Lesions on the Face and Chest
The Diagnosis: Blastic Plasmacytoid Dendritic Cell Neoplasm
Cutaneous plasmacytoma initially was suspected because of the patient’s history of monoclonal gammopathy as well as angiosarcoma due to the purpuric vascular appearance of the lesions. However, histopathology revealed a pleomorphic cellular dermal infiltrate characterized by atypical cells with mediumlarge nuclei, fine chromatin, and small nucleoli; the cells also had little cytoplasm (Figure). The infiltrate did not involve the epidermis but extended into the subcutaneous tissue. Immunohistochemistry revealed that the cells were positive for CD45, CD43, CD4, CD7, CD56, CD123, CD33, T-cell leukemia/lymphoma protein 1, and CD68. The cells were negative for CD2, CD3, CD5, CD8, T-cell intracellular antigen 1, CD13, CD15, CD19, CD20, CD21, CD23, cyclin D1, Bcl-2, Bcl-6, CD10, PAX5, MUM1, lysozyme, myeloperoxidase, perforin, granzyme B, CD57, CD34, CD117, terminal deoxynucleotidyl transferase, activin receptorlike kinase 1 βF1, Epstein-Barr virus– encoded small RNA, CD30, CD163, and pancytokeratin. Thus, the clinical and histopathologic findings led to a diagnosis of blastic plasmacytoid dendritic cell neoplasm (BPDCN), a rare and aggressive hematologic malignancy.

Blastic plasmacytoid dendritic cell neoplasm affects males older than 60 years.1 It is characterized by the clonal proliferation of precursor plasmacytoid dendritic cells—otherwise known as professional type I interferonproducing cells or plasmacytoid monocytes—of myeloid origin. Plasmacytoid dendritic cells have been renamed on several occasions, reflecting uncertainties of their histogenesis. The diagnosis of BPDCN requires a biopsy showing the morphology of plasmacytoid dendritic blast cells and immunophenotypic criteria established by either immunohistochemistry or flow cytometry.2,3 Tumor cells morphologically show an immature blastic appearance, and the diagnosis rests upon the demonstration of CD4 and CD56, together with markers more restricted to plasmacytoid dendritic cells (eg, BDCA-2, CD123, T-cell leukemia/lymphoma protein 1, CD2AP, BCL11A) and negativity for lymphoid and myeloid lineage–associated antigens.1,4
Blastic plasmacytoid dendritic cell neoplasms account for less than 1% of all hematopoietic neoplasms. Cutaneous lesions occur in 64% of patients with the disease and often are the reason patients seek medical care.5 Clinical findings include numerous erythematous and violaceous papules, nodules, and plaques that resemble purpura or vasculitis. Cutaneous lesions can vary in size from a few millimeters to 10 cm and vary in color. Moreover, patients often present with bruiselike patches, disseminated lesions, or mucosal lesions.1 Extracutaneous involvement includes lymphadenopathy, splenomegaly, and cytopenia caused by bone marrow infiltration, which may be present at diagnosis or during disease progression. Bone marrow involvement often is present with thrombocytopenia, anemia, and neutropenia. One-third of patients with BPDCN have central nervous system involvement and no disease relapse.6 Other affected sites include the liver, lungs, tonsils, soft tissues, and eyes. Patients with BPDCN may present with a history of myeloid neoplasms, such as acute/chronic myeloid leukemia, chronic myelomonocytic leukemia, or myelodysplastic syndrome.4 Our case highlights the importance of skin biopsy for making the correct diagnosis, as BPDCN manifests with cutaneous lesions that are nonspecific for neoplastic or nonneoplastic etiologies.
Given the aggressive nature of BPDCN, along with its potential for acute leukemic transformation, treatment has been challenging due to both poor response rates and lack of consensus and treatment strategies. Historically, patients who have received high-dose acute leukemia–based chemotherapy followed by an allogeneic stem cell transplant during the first remission appeared to have the best outcomes.7 Conventional treatments have included surgical excision with radiation and various leukemia-based chemotherapy regimens, with hyper- CVAD (fractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone-methotrexate, and cytarabine) being the most commonly used regimen.7,8 Venetoclax, a B-cell lymphoma 2 protein inhibitor, has shown promise when used in combination with hyper-CVAD. For older patients who may not tolerate aggressive chemotherapy, hypomethylating agents are preferred for their tolerability. Although tagraxofusp, a CD123-directed cytotoxin, has been utilized, Sapienza et al9 demonstrated an association with capillary leak syndrome.
Leukemia cutis is characterized by infiltration of the skin by malignant leukocytes, often associated with a prior diagnosis of systemic leukemia or myelodysplasia. Extramedullary accumulation of leukemic cells typically is referred to as myeloid sarcoma, while leukemia cutis serves as a general term for specific skin involvement.10 In rare instances, cutaneous lesions may manifest as the initial sign of systemic disease.
Cutaneous T-cell lymphomas comprise a diverse group of non-Hodgkin lymphomas that manifest as malignant monoclonal T-lymphocyte infiltration in the skin. Mycosis fungoides, Sézary syndrome, and primary cutaneous peripheral T-cell lymphomas are among the key subtypes. Histologically, differentiating these conditions from benign inflammatory disorders can be challenging due to subtle features such as haloed lymphocytes, epidermotropism, and Pautrier microabscesses seen in mycosis fungoides.11
Multiple myeloma involves monoclonal plasma cell proliferation, primarily affecting bone and bone marrow. Extramedullary plasmacytomas can occur outside these sites through hematogenous spread or adjacent infiltration, while metastatic plasmacytomas result from metastasis. Cutaneous plasmacytomas may arise from hematogenous dissemination or infiltration from neighboring structures.12
Extranodal natural killer/T-cell lymphoma, nasal type, manifests as aggressive mid-facial necrotizing lesions with extranodal involvement, notably in the nasal/paranasal area. These lesions can cause local destruction of cartilage, bone, and soft tissues and may progress through stages or arise de novo. Diagnostic challenges arise from the historical variety of terms used to describe extranodal natural killer/T-cell lymphoma, including midline lethal granuloma and lymphomatoid granulomatosis.13
- Cheng W, Yu TT, Tang AP, et al. Blastic plasmacytoid dendritic cell neoplasm: progress in cell origin, molecular biology, diagnostic criteria and therapeutic approaches. Curr Med Sci. 2021;41:405-419. doi:10.1007/s11596-021-2393-3
- Chang HJ, Lee MD, Yi HG, et al. A case of blastic plasmacytoid dendritic cell neoplasm initially mimicking cutaneous lupus erythematosus. Cancer Res Treat. 2010;42:239-243. doi:10.4143/crt.2010.42.4.239
- Garnache-Ottou F, Vidal C, Biichlé S, et al. How should we diagnose and treat blastic plasmacytoid dendritic cell neoplasm patients? Blood Adv. 2019;3:4238-4251. doi:10.1182/bloodadvances.2019000647
- Sweet K. Blastic plasmacytoid dendritic cell neoplasm. Curr Opin Hematol. 2020;27:103-107. doi:10.1097/moh.0000000000000569
- Julia F, Petrella T, Beylot-Barry M, et al. Blastic plasmacytoid dendritic cell neoplasm: clinical features in 90 patients. Br J Dermatol. 2013;169:579-586. doi:10.1111/bjd.12412
- Molina Castro D, Perilla Suárez O, Cuervo-Sierra J, et al. Blastic plasmacytoid dendritic cell neoplasm with central nervous system involvement: a case report. Cureus. 2022;14:e23888. doi:10.7759 /cureus.23888
- Grushchak S, Joy C, Gray A, et al. Novel treatment of blastic plasmacytoid dendritic cell neoplasm: a case report. Medicine (Baltimore). 2017;96:E9452.
- Lim MS, Lemmert K, Enjeti A. Blastic plasmacytoid dendritic cell neoplasm (BPDCN): a rare entity. BMJ Case Rep. 2016;2016:bcr2015214093. doi:10.1136/bcr-2015-214093
- Sapienza MR, Pileri A, Derenzini E, et al. Blastic plasmacytoid dendritic cell neoplasm: state of the art and prospects. Cancers (Basel). 2019;11:595. doi:10.3390/cancers11050595
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826. doi:10.1001/jamadermatol.2019.0052
- Ralfkiaer U, Hagedorn PH, Bangsgaard N, et al. Diagnostic micro RNA profiling in cutaneous T-cell lymphoma (CTCL). Blood. 2011;118: 5891-5900. doi:10.1182/blood-2011-06-358382
- Tsang DS, Le LW, Kukreti V. Treatment and outcomes for primary cutaneous extramedullary plasmacytoma: a case series. Curr Oncol. 2016;23:630-646. doi:10.3747/co.23.3288
- Lee J, Kim W, Park Y, et al. Nasal-type NK/T cell lymphoma: clinical features and treatment outcome. Br J Cancer. 2005;92:1226-1230. doi:10.1038/sj.bjc.6602502
The Diagnosis: Blastic Plasmacytoid Dendritic Cell Neoplasm
Cutaneous plasmacytoma initially was suspected because of the patient’s history of monoclonal gammopathy as well as angiosarcoma due to the purpuric vascular appearance of the lesions. However, histopathology revealed a pleomorphic cellular dermal infiltrate characterized by atypical cells with mediumlarge nuclei, fine chromatin, and small nucleoli; the cells also had little cytoplasm (Figure). The infiltrate did not involve the epidermis but extended into the subcutaneous tissue. Immunohistochemistry revealed that the cells were positive for CD45, CD43, CD4, CD7, CD56, CD123, CD33, T-cell leukemia/lymphoma protein 1, and CD68. The cells were negative for CD2, CD3, CD5, CD8, T-cell intracellular antigen 1, CD13, CD15, CD19, CD20, CD21, CD23, cyclin D1, Bcl-2, Bcl-6, CD10, PAX5, MUM1, lysozyme, myeloperoxidase, perforin, granzyme B, CD57, CD34, CD117, terminal deoxynucleotidyl transferase, activin receptorlike kinase 1 βF1, Epstein-Barr virus– encoded small RNA, CD30, CD163, and pancytokeratin. Thus, the clinical and histopathologic findings led to a diagnosis of blastic plasmacytoid dendritic cell neoplasm (BPDCN), a rare and aggressive hematologic malignancy.
Blastic plasmacytoid dendritic cell neoplasm affects males older than 60 years.1 It is characterized by the clonal proliferation of precursor plasmacytoid dendritic cells—otherwise known as professional type I interferonproducing cells or plasmacytoid monocytes—of myeloid origin. Plasmacytoid dendritic cells have been renamed on several occasions, reflecting uncertainties of their histogenesis. The diagnosis of BPDCN requires a biopsy showing the morphology of plasmacytoid dendritic blast cells and immunophenotypic criteria established by either immunohistochemistry or flow cytometry.2,3 Tumor cells morphologically show an immature blastic appearance, and the diagnosis rests upon the demonstration of CD4 and CD56, together with markers more restricted to plasmacytoid dendritic cells (eg, BDCA-2, CD123, T-cell leukemia/lymphoma protein 1, CD2AP, BCL11A) and negativity for lymphoid and myeloid lineage–associated antigens.1,4
Blastic plasmacytoid dendritic cell neoplasms account for less than 1% of all hematopoietic neoplasms. Cutaneous lesions occur in 64% of patients with the disease and often are the reason patients seek medical care.5 Clinical findings include numerous erythematous and violaceous papules, nodules, and plaques that resemble purpura or vasculitis. Cutaneous lesions can vary in size from a few millimeters to 10 cm and vary in color. Moreover, patients often present with bruiselike patches, disseminated lesions, or mucosal lesions.1 Extracutaneous involvement includes lymphadenopathy, splenomegaly, and cytopenia caused by bone marrow infiltration, which may be present at diagnosis or during disease progression. Bone marrow involvement often is present with thrombocytopenia, anemia, and neutropenia. One-third of patients with BPDCN have central nervous system involvement and no disease relapse.6 Other affected sites include the liver, lungs, tonsils, soft tissues, and eyes. Patients with BPDCN may present with a history of myeloid neoplasms, such as acute/chronic myeloid leukemia, chronic myelomonocytic leukemia, or myelodysplastic syndrome.4 Our case highlights the importance of skin biopsy for making the correct diagnosis, as BPDCN manifests with cutaneous lesions that are nonspecific for neoplastic or nonneoplastic etiologies.
Given the aggressive nature of BPDCN, along with its potential for acute leukemic transformation, treatment has been challenging due to both poor response rates and lack of consensus and treatment strategies. Historically, patients who have received high-dose acute leukemia–based chemotherapy followed by an allogeneic stem cell transplant during the first remission appeared to have the best outcomes.7 Conventional treatments have included surgical excision with radiation and various leukemia-based chemotherapy regimens, with hyper- CVAD (fractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone-methotrexate, and cytarabine) being the most commonly used regimen.7,8 Venetoclax, a B-cell lymphoma 2 protein inhibitor, has shown promise when used in combination with hyper-CVAD. For older patients who may not tolerate aggressive chemotherapy, hypomethylating agents are preferred for their tolerability. Although tagraxofusp, a CD123-directed cytotoxin, has been utilized, Sapienza et al9 demonstrated an association with capillary leak syndrome.
Leukemia cutis is characterized by infiltration of the skin by malignant leukocytes, often associated with a prior diagnosis of systemic leukemia or myelodysplasia. Extramedullary accumulation of leukemic cells typically is referred to as myeloid sarcoma, while leukemia cutis serves as a general term for specific skin involvement.10 In rare instances, cutaneous lesions may manifest as the initial sign of systemic disease.
Cutaneous T-cell lymphomas comprise a diverse group of non-Hodgkin lymphomas that manifest as malignant monoclonal T-lymphocyte infiltration in the skin. Mycosis fungoides, Sézary syndrome, and primary cutaneous peripheral T-cell lymphomas are among the key subtypes. Histologically, differentiating these conditions from benign inflammatory disorders can be challenging due to subtle features such as haloed lymphocytes, epidermotropism, and Pautrier microabscesses seen in mycosis fungoides.11
Multiple myeloma involves monoclonal plasma cell proliferation, primarily affecting bone and bone marrow. Extramedullary plasmacytomas can occur outside these sites through hematogenous spread or adjacent infiltration, while metastatic plasmacytomas result from metastasis. Cutaneous plasmacytomas may arise from hematogenous dissemination or infiltration from neighboring structures.12
Extranodal natural killer/T-cell lymphoma, nasal type, manifests as aggressive mid-facial necrotizing lesions with extranodal involvement, notably in the nasal/paranasal area. These lesions can cause local destruction of cartilage, bone, and soft tissues and may progress through stages or arise de novo. Diagnostic challenges arise from the historical variety of terms used to describe extranodal natural killer/T-cell lymphoma, including midline lethal granuloma and lymphomatoid granulomatosis.13
The Diagnosis: Blastic Plasmacytoid Dendritic Cell Neoplasm
Cutaneous plasmacytoma initially was suspected because of the patient’s history of monoclonal gammopathy as well as angiosarcoma due to the purpuric vascular appearance of the lesions. However, histopathology revealed a pleomorphic cellular dermal infiltrate characterized by atypical cells with mediumlarge nuclei, fine chromatin, and small nucleoli; the cells also had little cytoplasm (Figure). The infiltrate did not involve the epidermis but extended into the subcutaneous tissue. Immunohistochemistry revealed that the cells were positive for CD45, CD43, CD4, CD7, CD56, CD123, CD33, T-cell leukemia/lymphoma protein 1, and CD68. The cells were negative for CD2, CD3, CD5, CD8, T-cell intracellular antigen 1, CD13, CD15, CD19, CD20, CD21, CD23, cyclin D1, Bcl-2, Bcl-6, CD10, PAX5, MUM1, lysozyme, myeloperoxidase, perforin, granzyme B, CD57, CD34, CD117, terminal deoxynucleotidyl transferase, activin receptorlike kinase 1 βF1, Epstein-Barr virus– encoded small RNA, CD30, CD163, and pancytokeratin. Thus, the clinical and histopathologic findings led to a diagnosis of blastic plasmacytoid dendritic cell neoplasm (BPDCN), a rare and aggressive hematologic malignancy.
Blastic plasmacytoid dendritic cell neoplasm affects males older than 60 years.1 It is characterized by the clonal proliferation of precursor plasmacytoid dendritic cells—otherwise known as professional type I interferonproducing cells or plasmacytoid monocytes—of myeloid origin. Plasmacytoid dendritic cells have been renamed on several occasions, reflecting uncertainties of their histogenesis. The diagnosis of BPDCN requires a biopsy showing the morphology of plasmacytoid dendritic blast cells and immunophenotypic criteria established by either immunohistochemistry or flow cytometry.2,3 Tumor cells morphologically show an immature blastic appearance, and the diagnosis rests upon the demonstration of CD4 and CD56, together with markers more restricted to plasmacytoid dendritic cells (eg, BDCA-2, CD123, T-cell leukemia/lymphoma protein 1, CD2AP, BCL11A) and negativity for lymphoid and myeloid lineage–associated antigens.1,4
Blastic plasmacytoid dendritic cell neoplasms account for less than 1% of all hematopoietic neoplasms. Cutaneous lesions occur in 64% of patients with the disease and often are the reason patients seek medical care.5 Clinical findings include numerous erythematous and violaceous papules, nodules, and plaques that resemble purpura or vasculitis. Cutaneous lesions can vary in size from a few millimeters to 10 cm and vary in color. Moreover, patients often present with bruiselike patches, disseminated lesions, or mucosal lesions.1 Extracutaneous involvement includes lymphadenopathy, splenomegaly, and cytopenia caused by bone marrow infiltration, which may be present at diagnosis or during disease progression. Bone marrow involvement often is present with thrombocytopenia, anemia, and neutropenia. One-third of patients with BPDCN have central nervous system involvement and no disease relapse.6 Other affected sites include the liver, lungs, tonsils, soft tissues, and eyes. Patients with BPDCN may present with a history of myeloid neoplasms, such as acute/chronic myeloid leukemia, chronic myelomonocytic leukemia, or myelodysplastic syndrome.4 Our case highlights the importance of skin biopsy for making the correct diagnosis, as BPDCN manifests with cutaneous lesions that are nonspecific for neoplastic or nonneoplastic etiologies.
Given the aggressive nature of BPDCN, along with its potential for acute leukemic transformation, treatment has been challenging due to both poor response rates and lack of consensus and treatment strategies. Historically, patients who have received high-dose acute leukemia–based chemotherapy followed by an allogeneic stem cell transplant during the first remission appeared to have the best outcomes.7 Conventional treatments have included surgical excision with radiation and various leukemia-based chemotherapy regimens, with hyper- CVAD (fractionated cyclophosphamide, vincristine, doxorubicin, dexamethasone-methotrexate, and cytarabine) being the most commonly used regimen.7,8 Venetoclax, a B-cell lymphoma 2 protein inhibitor, has shown promise when used in combination with hyper-CVAD. For older patients who may not tolerate aggressive chemotherapy, hypomethylating agents are preferred for their tolerability. Although tagraxofusp, a CD123-directed cytotoxin, has been utilized, Sapienza et al9 demonstrated an association with capillary leak syndrome.
Leukemia cutis is characterized by infiltration of the skin by malignant leukocytes, often associated with a prior diagnosis of systemic leukemia or myelodysplasia. Extramedullary accumulation of leukemic cells typically is referred to as myeloid sarcoma, while leukemia cutis serves as a general term for specific skin involvement.10 In rare instances, cutaneous lesions may manifest as the initial sign of systemic disease.
Cutaneous T-cell lymphomas comprise a diverse group of non-Hodgkin lymphomas that manifest as malignant monoclonal T-lymphocyte infiltration in the skin. Mycosis fungoides, Sézary syndrome, and primary cutaneous peripheral T-cell lymphomas are among the key subtypes. Histologically, differentiating these conditions from benign inflammatory disorders can be challenging due to subtle features such as haloed lymphocytes, epidermotropism, and Pautrier microabscesses seen in mycosis fungoides.11
Multiple myeloma involves monoclonal plasma cell proliferation, primarily affecting bone and bone marrow. Extramedullary plasmacytomas can occur outside these sites through hematogenous spread or adjacent infiltration, while metastatic plasmacytomas result from metastasis. Cutaneous plasmacytomas may arise from hematogenous dissemination or infiltration from neighboring structures.12
Extranodal natural killer/T-cell lymphoma, nasal type, manifests as aggressive mid-facial necrotizing lesions with extranodal involvement, notably in the nasal/paranasal area. These lesions can cause local destruction of cartilage, bone, and soft tissues and may progress through stages or arise de novo. Diagnostic challenges arise from the historical variety of terms used to describe extranodal natural killer/T-cell lymphoma, including midline lethal granuloma and lymphomatoid granulomatosis.13
- Cheng W, Yu TT, Tang AP, et al. Blastic plasmacytoid dendritic cell neoplasm: progress in cell origin, molecular biology, diagnostic criteria and therapeutic approaches. Curr Med Sci. 2021;41:405-419. doi:10.1007/s11596-021-2393-3
- Chang HJ, Lee MD, Yi HG, et al. A case of blastic plasmacytoid dendritic cell neoplasm initially mimicking cutaneous lupus erythematosus. Cancer Res Treat. 2010;42:239-243. doi:10.4143/crt.2010.42.4.239
- Garnache-Ottou F, Vidal C, Biichlé S, et al. How should we diagnose and treat blastic plasmacytoid dendritic cell neoplasm patients? Blood Adv. 2019;3:4238-4251. doi:10.1182/bloodadvances.2019000647
- Sweet K. Blastic plasmacytoid dendritic cell neoplasm. Curr Opin Hematol. 2020;27:103-107. doi:10.1097/moh.0000000000000569
- Julia F, Petrella T, Beylot-Barry M, et al. Blastic plasmacytoid dendritic cell neoplasm: clinical features in 90 patients. Br J Dermatol. 2013;169:579-586. doi:10.1111/bjd.12412
- Molina Castro D, Perilla Suárez O, Cuervo-Sierra J, et al. Blastic plasmacytoid dendritic cell neoplasm with central nervous system involvement: a case report. Cureus. 2022;14:e23888. doi:10.7759 /cureus.23888
- Grushchak S, Joy C, Gray A, et al. Novel treatment of blastic plasmacytoid dendritic cell neoplasm: a case report. Medicine (Baltimore). 2017;96:E9452.
- Lim MS, Lemmert K, Enjeti A. Blastic plasmacytoid dendritic cell neoplasm (BPDCN): a rare entity. BMJ Case Rep. 2016;2016:bcr2015214093. doi:10.1136/bcr-2015-214093
- Sapienza MR, Pileri A, Derenzini E, et al. Blastic plasmacytoid dendritic cell neoplasm: state of the art and prospects. Cancers (Basel). 2019;11:595. doi:10.3390/cancers11050595
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826. doi:10.1001/jamadermatol.2019.0052
- Ralfkiaer U, Hagedorn PH, Bangsgaard N, et al. Diagnostic micro RNA profiling in cutaneous T-cell lymphoma (CTCL). Blood. 2011;118: 5891-5900. doi:10.1182/blood-2011-06-358382
- Tsang DS, Le LW, Kukreti V. Treatment and outcomes for primary cutaneous extramedullary plasmacytoma: a case series. Curr Oncol. 2016;23:630-646. doi:10.3747/co.23.3288
- Lee J, Kim W, Park Y, et al. Nasal-type NK/T cell lymphoma: clinical features and treatment outcome. Br J Cancer. 2005;92:1226-1230. doi:10.1038/sj.bjc.6602502
- Cheng W, Yu TT, Tang AP, et al. Blastic plasmacytoid dendritic cell neoplasm: progress in cell origin, molecular biology, diagnostic criteria and therapeutic approaches. Curr Med Sci. 2021;41:405-419. doi:10.1007/s11596-021-2393-3
- Chang HJ, Lee MD, Yi HG, et al. A case of blastic plasmacytoid dendritic cell neoplasm initially mimicking cutaneous lupus erythematosus. Cancer Res Treat. 2010;42:239-243. doi:10.4143/crt.2010.42.4.239
- Garnache-Ottou F, Vidal C, Biichlé S, et al. How should we diagnose and treat blastic plasmacytoid dendritic cell neoplasm patients? Blood Adv. 2019;3:4238-4251. doi:10.1182/bloodadvances.2019000647
- Sweet K. Blastic plasmacytoid dendritic cell neoplasm. Curr Opin Hematol. 2020;27:103-107. doi:10.1097/moh.0000000000000569
- Julia F, Petrella T, Beylot-Barry M, et al. Blastic plasmacytoid dendritic cell neoplasm: clinical features in 90 patients. Br J Dermatol. 2013;169:579-586. doi:10.1111/bjd.12412
- Molina Castro D, Perilla Suárez O, Cuervo-Sierra J, et al. Blastic plasmacytoid dendritic cell neoplasm with central nervous system involvement: a case report. Cureus. 2022;14:e23888. doi:10.7759 /cureus.23888
- Grushchak S, Joy C, Gray A, et al. Novel treatment of blastic plasmacytoid dendritic cell neoplasm: a case report. Medicine (Baltimore). 2017;96:E9452.
- Lim MS, Lemmert K, Enjeti A. Blastic plasmacytoid dendritic cell neoplasm (BPDCN): a rare entity. BMJ Case Rep. 2016;2016:bcr2015214093. doi:10.1136/bcr-2015-214093
- Sapienza MR, Pileri A, Derenzini E, et al. Blastic plasmacytoid dendritic cell neoplasm: state of the art and prospects. Cancers (Basel). 2019;11:595. doi:10.3390/cancers11050595
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826. doi:10.1001/jamadermatol.2019.0052
- Ralfkiaer U, Hagedorn PH, Bangsgaard N, et al. Diagnostic micro RNA profiling in cutaneous T-cell lymphoma (CTCL). Blood. 2011;118: 5891-5900. doi:10.1182/blood-2011-06-358382
- Tsang DS, Le LW, Kukreti V. Treatment and outcomes for primary cutaneous extramedullary plasmacytoma: a case series. Curr Oncol. 2016;23:630-646. doi:10.3747/co.23.3288
- Lee J, Kim W, Park Y, et al. Nasal-type NK/T cell lymphoma: clinical features and treatment outcome. Br J Cancer. 2005;92:1226-1230. doi:10.1038/sj.bjc.6602502
A 79-year-old man presented to the dermatology clinic with multiple skin lesions of 4 months’ duration. The patient had a history of monoclonal gammopathy and reported no changes in medication, travel, or trauma. He reported tenderness only when trying to comb hair over the left occipital nodule. He denied fevers, night sweats, weight loss, or poor appetite. Physical examination revealed 4 concerning skin lesions: a 3×3-cm violaceous nodule with underlying ecchymosis on the right medial jaw (top), a 3×2.5-cm violaceous nodule on the posterior occiput, a pink plaque with 1-mm vascular papules on the right mid-chest (bottom), and a 4×2.5-cm oval pink patch on the left side of the lower back. Punch biopsies were performed on the right medial jaw nodule and right mid-chest plaque.

