User login
Asymptomatic Pink Plaque on the Scapula
The Diagnosis: Primary Cutaneous Follicle Center Lymphoma
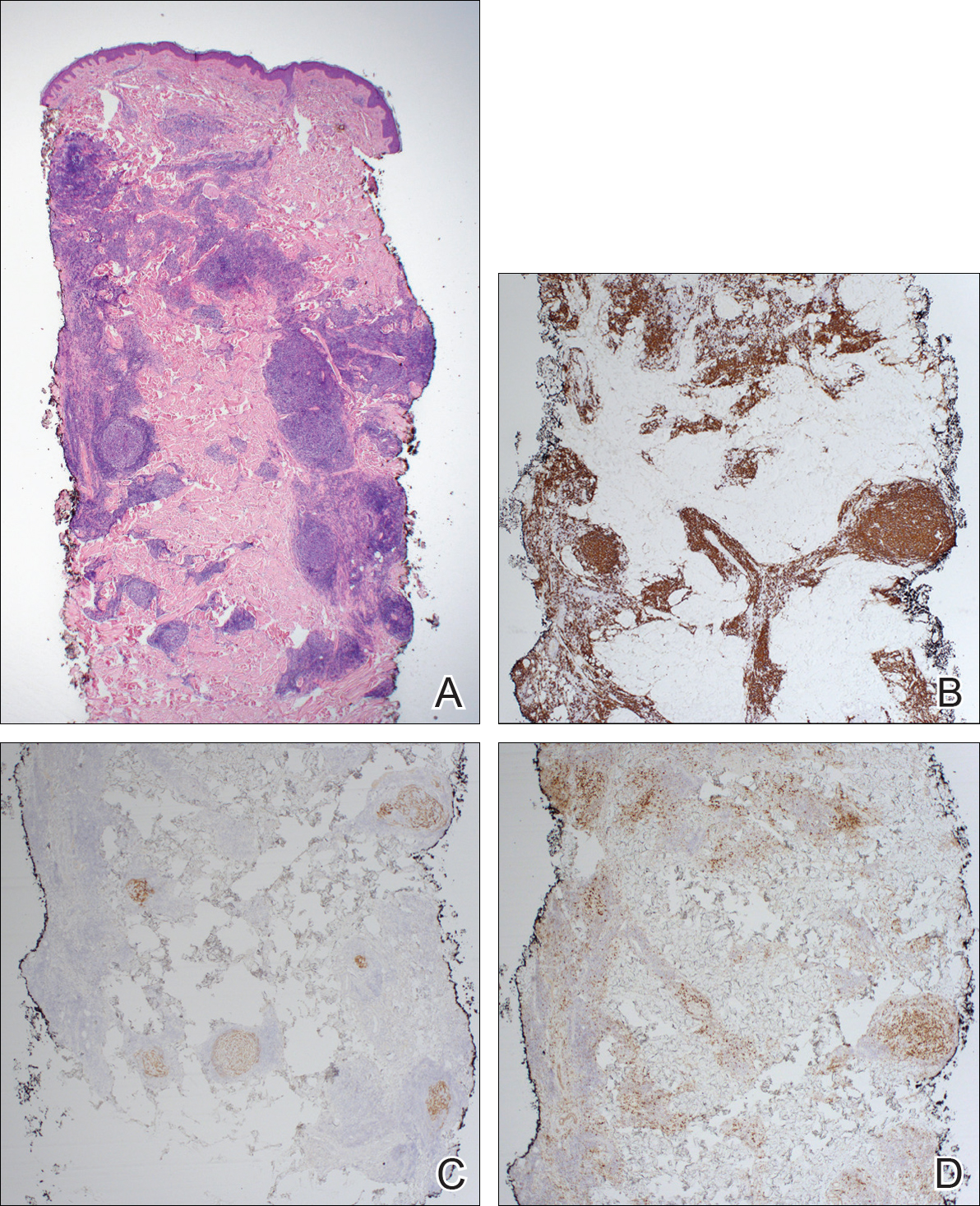
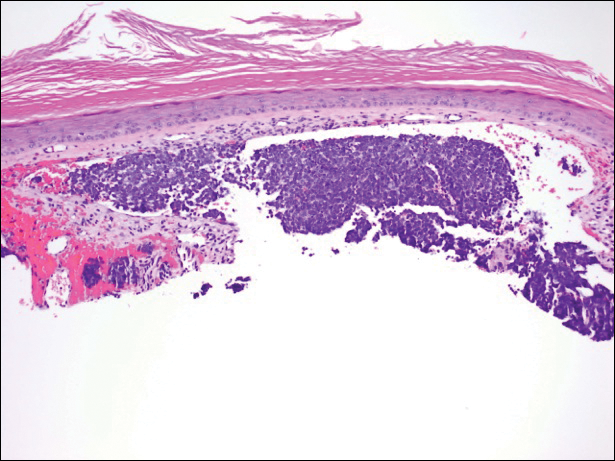
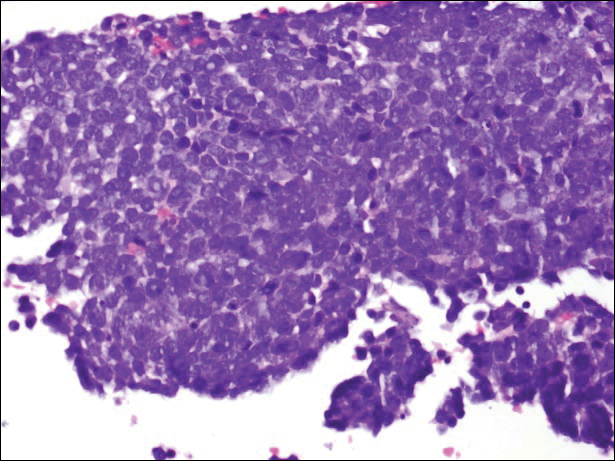
Immunohistochemistry revealed a nodular infiltrate consisting of small to large atypical lymphocytes forming an irregular germinal center with notably thinned mantle zones and lack of polarization (Figure, A). Atypical cells stained positively with Bcl-6, and CD20 was diffusely positive (Figure, B-D). Bcl-2 and CD3 colocalized to the reactive T-cell infiltrate, and CD10 was largely negative. Further workup with bone marrow biopsy and full-body positron emission tomography-computed tomography was unremarkable. Given these findings, a diagnosis of primary cutaneous follicle center lymphoma (FCL) was made. At 1 month following radiation therapy, complete clinical clearance of the lymphoma was achieved.
Follicle center lymphoma, also known as cutaneous follicular lymphoma, is the most common subtype of primary cutaneous B-cell lymphomas, representing approximately 57% of cases.1 Follicle center lymphoma typically affects older, non-Hispanic white adults with a median age of onset of 60 years. It has a predilection for the head, neck, and trunk.2 Lesions present as solitary erythematous to violaceous papules, plaques, or nodules, but they can more rarely be multifocal.3 Clinical diagnosis of FCL can be difficult, with papular lesions resembling acne, rosacea, folliculitis, or arthropod assault.4,5 As such, diagnosis of FCL typically relies on histopathologic analysis.
Histologically, FCL can present in several different patterns including follicular, nodular, diffuse, or a pleomorphic mix of these.2,6 The cells are comprised of germinal center B cells, staining positively for Bcl-6, CD20, and CD79a.7 Tumor cells do not exhibit the t(14;18) translocation seen in nodal follicular lymphomas.2,8 Unlike marginal zone lymphoma, FCL stains negatively for Bcl-2 and multiple myeloma 1/interferon regulatory factor 4 (MUM1/IRF-4).2,9 Forkhead box P1 (FOXP1) also is usually negative, but its presence can indicate a poorer prognosis.2 It is important to distinguish primary cutaneous B-cell lymphomas from systemic B-cell lymphoma with secondary cutaneous involvement, as they have a different clinical prognosis and management course. Further workup includes bone marrow biopsy, serum analysis for clonal involvement, and positron emission tomography-computed tomography imaging. Follicle center lymphoma generally has an indolent disease course with a favorable 5-year survival rate of approximately 95%.6,8
Untreated lesions may enlarge slowly or even spontaneously involute.10 The histologic growth pattern and number of lesions do not affect prognosis, but presence on the legs has a 5-year survival rate of 41%.2 Extracutaneous dissemination can occur in 5% to 10% of cases.2 Given the slow progression of FCL, conservative management with observation is an option. However, curative treatment can be reasonably attempted for solitary lesions by excision or radiation. Treatment of FCL often can be complicated by its predilection for the head and neck. Other treatment modalities include topical steroids, imiquimod, nitrogen mustard, and bexarotene.10 More generalized involvement may require systemic therapy with rituximab or chemotherapy. Recurrence after therapy is common, reported in 46.5% of patients, but does not affect prognosis.2
- Zinzani PL, Quaglino P, Pimpinelli N, et al. Prognostic factors in primary cutaneous B-cell lymphoma: The Italian Study Group for Cutaneous Lymphomas. J Clin Oncol. 2006;24:1376-1382.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:1-13.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in primary cutaneous large B-cell lymphomas: a European multicenter study. J Clin Oncol. 2001;19:3602-3610.
- Soon CW, Pincus LB, Ai WZ, et al. Acneiform presentation of primary cutaneous follicle center lymphoma. J Am Acad Dermatol. 2011;65:887-889.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: a report of 18 cases. J Am Acad Dermatol. 2011;65:749-755.
- Wilcox RA. CME information: cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Franco R, Fernandez-Vazquez A, Rodriguez-Peralto JL, et al. Cutaneous follicular B-cell lymphoma: description of a series of 18 cases. Am J Surg Pathol. 2001;25:875-883.
- Kempf W, Denisjuk N, Kerl K, et al. Primary cutaneous B-cell lymphomas. J Dtsch Dermatol Ges. 2012;10:12-22; quiz 23.
- de Leval L HN, Longtine J, Ferry JA, et al. Cutaneous B-cell lymphomas of follicular and marginal zone types: use of Bcl-6, CD10, Bcl-2, and CD21 in differential diagnosis and classification. Am J Surg Pathol. 2001;25:732-741.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:1-11.
The Diagnosis: Primary Cutaneous Follicle Center Lymphoma
Immunohistochemistry revealed a nodular infiltrate consisting of small to large atypical lymphocytes forming an irregular germinal center with notably thinned mantle zones and lack of polarization (Figure, A). Atypical cells stained positively with Bcl-6, and CD20 was diffusely positive (Figure, B-D). Bcl-2 and CD3 colocalized to the reactive T-cell infiltrate, and CD10 was largely negative. Further workup with bone marrow biopsy and full-body positron emission tomography-computed tomography was unremarkable. Given these findings, a diagnosis of primary cutaneous follicle center lymphoma (FCL) was made. At 1 month following radiation therapy, complete clinical clearance of the lymphoma was achieved.
Follicle center lymphoma, also known as cutaneous follicular lymphoma, is the most common subtype of primary cutaneous B-cell lymphomas, representing approximately 57% of cases.1 Follicle center lymphoma typically affects older, non-Hispanic white adults with a median age of onset of 60 years. It has a predilection for the head, neck, and trunk.2 Lesions present as solitary erythematous to violaceous papules, plaques, or nodules, but they can more rarely be multifocal.3 Clinical diagnosis of FCL can be difficult, with papular lesions resembling acne, rosacea, folliculitis, or arthropod assault.4,5 As such, diagnosis of FCL typically relies on histopathologic analysis.
Histologically, FCL can present in several different patterns including follicular, nodular, diffuse, or a pleomorphic mix of these.2,6 The cells are comprised of germinal center B cells, staining positively for Bcl-6, CD20, and CD79a.7 Tumor cells do not exhibit the t(14;18) translocation seen in nodal follicular lymphomas.2,8 Unlike marginal zone lymphoma, FCL stains negatively for Bcl-2 and multiple myeloma 1/interferon regulatory factor 4 (MUM1/IRF-4).2,9 Forkhead box P1 (FOXP1) also is usually negative, but its presence can indicate a poorer prognosis.2 It is important to distinguish primary cutaneous B-cell lymphomas from systemic B-cell lymphoma with secondary cutaneous involvement, as they have a different clinical prognosis and management course. Further workup includes bone marrow biopsy, serum analysis for clonal involvement, and positron emission tomography-computed tomography imaging. Follicle center lymphoma generally has an indolent disease course with a favorable 5-year survival rate of approximately 95%.6,8
Untreated lesions may enlarge slowly or even spontaneously involute.10 The histologic growth pattern and number of lesions do not affect prognosis, but presence on the legs has a 5-year survival rate of 41%.2 Extracutaneous dissemination can occur in 5% to 10% of cases.2 Given the slow progression of FCL, conservative management with observation is an option. However, curative treatment can be reasonably attempted for solitary lesions by excision or radiation. Treatment of FCL often can be complicated by its predilection for the head and neck. Other treatment modalities include topical steroids, imiquimod, nitrogen mustard, and bexarotene.10 More generalized involvement may require systemic therapy with rituximab or chemotherapy. Recurrence after therapy is common, reported in 46.5% of patients, but does not affect prognosis.2
The Diagnosis: Primary Cutaneous Follicle Center Lymphoma
Immunohistochemistry revealed a nodular infiltrate consisting of small to large atypical lymphocytes forming an irregular germinal center with notably thinned mantle zones and lack of polarization (Figure, A). Atypical cells stained positively with Bcl-6, and CD20 was diffusely positive (Figure, B-D). Bcl-2 and CD3 colocalized to the reactive T-cell infiltrate, and CD10 was largely negative. Further workup with bone marrow biopsy and full-body positron emission tomography-computed tomography was unremarkable. Given these findings, a diagnosis of primary cutaneous follicle center lymphoma (FCL) was made. At 1 month following radiation therapy, complete clinical clearance of the lymphoma was achieved.
Follicle center lymphoma, also known as cutaneous follicular lymphoma, is the most common subtype of primary cutaneous B-cell lymphomas, representing approximately 57% of cases.1 Follicle center lymphoma typically affects older, non-Hispanic white adults with a median age of onset of 60 years. It has a predilection for the head, neck, and trunk.2 Lesions present as solitary erythematous to violaceous papules, plaques, or nodules, but they can more rarely be multifocal.3 Clinical diagnosis of FCL can be difficult, with papular lesions resembling acne, rosacea, folliculitis, or arthropod assault.4,5 As such, diagnosis of FCL typically relies on histopathologic analysis.
Histologically, FCL can present in several different patterns including follicular, nodular, diffuse, or a pleomorphic mix of these.2,6 The cells are comprised of germinal center B cells, staining positively for Bcl-6, CD20, and CD79a.7 Tumor cells do not exhibit the t(14;18) translocation seen in nodal follicular lymphomas.2,8 Unlike marginal zone lymphoma, FCL stains negatively for Bcl-2 and multiple myeloma 1/interferon regulatory factor 4 (MUM1/IRF-4).2,9 Forkhead box P1 (FOXP1) also is usually negative, but its presence can indicate a poorer prognosis.2 It is important to distinguish primary cutaneous B-cell lymphomas from systemic B-cell lymphoma with secondary cutaneous involvement, as they have a different clinical prognosis and management course. Further workup includes bone marrow biopsy, serum analysis for clonal involvement, and positron emission tomography-computed tomography imaging. Follicle center lymphoma generally has an indolent disease course with a favorable 5-year survival rate of approximately 95%.6,8
Untreated lesions may enlarge slowly or even spontaneously involute.10 The histologic growth pattern and number of lesions do not affect prognosis, but presence on the legs has a 5-year survival rate of 41%.2 Extracutaneous dissemination can occur in 5% to 10% of cases.2 Given the slow progression of FCL, conservative management with observation is an option. However, curative treatment can be reasonably attempted for solitary lesions by excision or radiation. Treatment of FCL often can be complicated by its predilection for the head and neck. Other treatment modalities include topical steroids, imiquimod, nitrogen mustard, and bexarotene.10 More generalized involvement may require systemic therapy with rituximab or chemotherapy. Recurrence after therapy is common, reported in 46.5% of patients, but does not affect prognosis.2
- Zinzani PL, Quaglino P, Pimpinelli N, et al. Prognostic factors in primary cutaneous B-cell lymphoma: The Italian Study Group for Cutaneous Lymphomas. J Clin Oncol. 2006;24:1376-1382.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:1-13.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in primary cutaneous large B-cell lymphomas: a European multicenter study. J Clin Oncol. 2001;19:3602-3610.
- Soon CW, Pincus LB, Ai WZ, et al. Acneiform presentation of primary cutaneous follicle center lymphoma. J Am Acad Dermatol. 2011;65:887-889.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: a report of 18 cases. J Am Acad Dermatol. 2011;65:749-755.
- Wilcox RA. CME information: cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Franco R, Fernandez-Vazquez A, Rodriguez-Peralto JL, et al. Cutaneous follicular B-cell lymphoma: description of a series of 18 cases. Am J Surg Pathol. 2001;25:875-883.
- Kempf W, Denisjuk N, Kerl K, et al. Primary cutaneous B-cell lymphomas. J Dtsch Dermatol Ges. 2012;10:12-22; quiz 23.
- de Leval L HN, Longtine J, Ferry JA, et al. Cutaneous B-cell lymphomas of follicular and marginal zone types: use of Bcl-6, CD10, Bcl-2, and CD21 in differential diagnosis and classification. Am J Surg Pathol. 2001;25:732-741.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:1-11.
- Zinzani PL, Quaglino P, Pimpinelli N, et al. Prognostic factors in primary cutaneous B-cell lymphoma: The Italian Study Group for Cutaneous Lymphomas. J Clin Oncol. 2006;24:1376-1382.
- Suárez AL, Pulitzer M, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part I. clinical features, diagnosis, and classification. J Am Acad Dermatol. 2013;69:1-13.
- Grange F, Bekkenk MW, Wechsler J, et al. Prognostic factors in primary cutaneous large B-cell lymphomas: a European multicenter study. J Clin Oncol. 2001;19:3602-3610.
- Soon CW, Pincus LB, Ai WZ, et al. Acneiform presentation of primary cutaneous follicle center lymphoma. J Am Acad Dermatol. 2011;65:887-889.
- Massone C, Fink-Puches R, Laimer M, et al. Miliary and agminated-type primary cutaneous follicle center lymphoma: a report of 18 cases. J Am Acad Dermatol. 2011;65:749-755.
- Wilcox RA. CME information: cutaneous B-cell lymphomas: 2015 update on diagnosis, risk-stratification, and management. Am J Hematol. 2015;90:73-76.
- Franco R, Fernandez-Vazquez A, Rodriguez-Peralto JL, et al. Cutaneous follicular B-cell lymphoma: description of a series of 18 cases. Am J Surg Pathol. 2001;25:875-883.
- Kempf W, Denisjuk N, Kerl K, et al. Primary cutaneous B-cell lymphomas. J Dtsch Dermatol Ges. 2012;10:12-22; quiz 23.
- de Leval L HN, Longtine J, Ferry JA, et al. Cutaneous B-cell lymphomas of follicular and marginal zone types: use of Bcl-6, CD10, Bcl-2, and CD21 in differential diagnosis and classification. Am J Surg Pathol. 2001;25:732-741.
- Suárez AL, Querfeld C, Horwitz S, et al. Primary cutaneous B-cell lymphomas: part II. therapy and future directions. J Am Acad Dermatol. 2013;69:1-11.
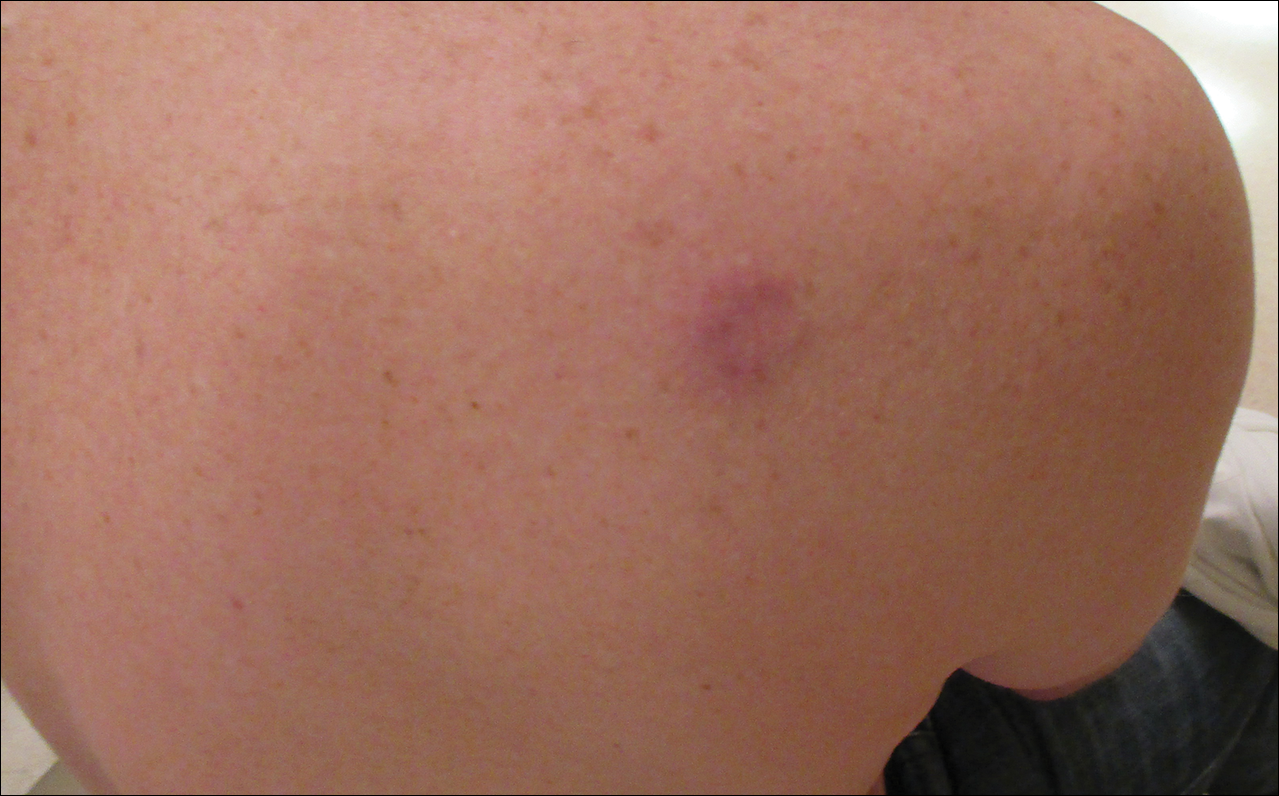
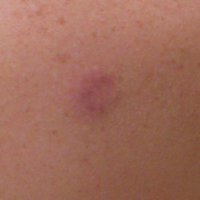
A 36-year-old man presented with a pink plaque on the right side of the scapula of 1 year's duration. The plaque had not grown and was completely asymptomatic. Physical examination revealed a violaceous, pink, 2-cm nodule with overlying telangiectasia. No other concerning lesions were identified on total-body skin examination. A punch biopsy was obtained.
Red Scaly Rash Following Tattoo Application
The Diagnosis: Isomorphic Psoriasis
Tattooing has become an increasingly popular trend among young people. Currently, there are no guidelines in the United States regulating the production of tattoo ink and pigments.1 Henna tattooing, a form of temporary skin painting, also has risks of allergic contact dermatitis from paraphenylenediamine dye.2 Complications following tattoo application include an allergic contact dermatitis to tattoo pigments, infection, granulomatous and lichenoid reactions, and skin disease localized to the tattooed area.3
Localized dermatosis arising in a traumatized area, or the Koebner phenomenon, was first described by Heinrich Koebner in 1877.4 He described the formation of psoriasiform lesions at the site of cutaneous trauma.5 These isomorphic lesions can occur in 25% of patients with psoriasis after trauma to the skin such as tattooing.6 Other dermatologic diseases that can present as an isomorphic response to tattooing include lichen planus, Darier disease, vitiligo, and autoimmune bullous disease.3,5,6
Various causes of trauma such as burns, insect bites, physical trauma, and needle trauma have been shown to produce new psoriatic lesions.6 The time period from trauma to formation of psoriasiform lesions usually ranges from 10 to 20 days; however, an initial reaction can occur as early as 3 days or as long as 2 years after trauma.4 Although the pathophysiology of the isomorphic response is not well known, it has been shown that nerve growth factor has a role. Raychaudhuri et al7 demonstrated the upregulation of nerve growth factor in the development of a psoriatic lesion, influencing keratinocyte proliferation, angiogenesis, and T-cell activation.
Physical trauma such as tattooing has been shown to cause an isomorphic response in psoriasis. We describe a case of isomorphic psoriasis in a patient after tattoo application. Our patient had a several-month history of well-controlled psoriasis prior to obtaining the new tattoo. Several days after receiving the tattoo, the patient reported an increase in psoriatic lesions, including at the site of the tattoo. The trauma causing the isomorphic response could have been either a response to the tattoo pigment or needle injury to the skin.6
Psoriasis and isomorphic lesions can be treated with topical corticosteroids as well as systemic and biologic agents. Our patient was treated with triamcinolone cream with good response.8
- Haugh IM, Laumann SL, Laumann AE. Regulation of tattoo ink production and the tattoo business in the US. Curr Probl Dermatol. 2015;48:248-252.
- Marcoux D, Couture-Trudel PM, Rboulet-Delmas G, et al. Sensitization to paraphenylenediame from a streetside temporary tattoo. Pediatr Dermatol. 2002;19:498-502.
- Bassi A, Campolmi P, Cannarozzo G, et al. Tattoo-associated skin reaction: the importance of an early diagnosis and proper treatment [published online July 23, 2014]. Biomed Res Int. 2014;2014:354608.
- Weiss G, Shemer A, Trau H. The Koebner phenomenon: review of the literature. J Eur Acad Dermatol Venereol. 2002;16:241-248.
- Sagi L, Trau H. The Koebner phenomenon. Clin Dermatol. 2011;29:231-236.
- Orzan OA, Popa LG, Vexler ES, et al. Tattoo-induced psoriasis. J Med Life. 2014;7:65-68.
- Raychaudhuri SP, Jiang WY, Raychaudhuri SK. Revisiting the Koebner phenomenon: role of NGF and its receptor system in the pathogenesis of psoriasis. Am J Pathol. 2008;172:961-971.
- Gottlieb AB. Therapeutic options in the treatment of psoriasis and atopic dermatitis. J Am Acad Dermatol. 2005;53(1 suppl 1):S3-S16.
The Diagnosis: Isomorphic Psoriasis
Tattooing has become an increasingly popular trend among young people. Currently, there are no guidelines in the United States regulating the production of tattoo ink and pigments.1 Henna tattooing, a form of temporary skin painting, also has risks of allergic contact dermatitis from paraphenylenediamine dye.2 Complications following tattoo application include an allergic contact dermatitis to tattoo pigments, infection, granulomatous and lichenoid reactions, and skin disease localized to the tattooed area.3
Localized dermatosis arising in a traumatized area, or the Koebner phenomenon, was first described by Heinrich Koebner in 1877.4 He described the formation of psoriasiform lesions at the site of cutaneous trauma.5 These isomorphic lesions can occur in 25% of patients with psoriasis after trauma to the skin such as tattooing.6 Other dermatologic diseases that can present as an isomorphic response to tattooing include lichen planus, Darier disease, vitiligo, and autoimmune bullous disease.3,5,6
Various causes of trauma such as burns, insect bites, physical trauma, and needle trauma have been shown to produce new psoriatic lesions.6 The time period from trauma to formation of psoriasiform lesions usually ranges from 10 to 20 days; however, an initial reaction can occur as early as 3 days or as long as 2 years after trauma.4 Although the pathophysiology of the isomorphic response is not well known, it has been shown that nerve growth factor has a role. Raychaudhuri et al7 demonstrated the upregulation of nerve growth factor in the development of a psoriatic lesion, influencing keratinocyte proliferation, angiogenesis, and T-cell activation.
Physical trauma such as tattooing has been shown to cause an isomorphic response in psoriasis. We describe a case of isomorphic psoriasis in a patient after tattoo application. Our patient had a several-month history of well-controlled psoriasis prior to obtaining the new tattoo. Several days after receiving the tattoo, the patient reported an increase in psoriatic lesions, including at the site of the tattoo. The trauma causing the isomorphic response could have been either a response to the tattoo pigment or needle injury to the skin.6
Psoriasis and isomorphic lesions can be treated with topical corticosteroids as well as systemic and biologic agents. Our patient was treated with triamcinolone cream with good response.8
The Diagnosis: Isomorphic Psoriasis
Tattooing has become an increasingly popular trend among young people. Currently, there are no guidelines in the United States regulating the production of tattoo ink and pigments.1 Henna tattooing, a form of temporary skin painting, also has risks of allergic contact dermatitis from paraphenylenediamine dye.2 Complications following tattoo application include an allergic contact dermatitis to tattoo pigments, infection, granulomatous and lichenoid reactions, and skin disease localized to the tattooed area.3
Localized dermatosis arising in a traumatized area, or the Koebner phenomenon, was first described by Heinrich Koebner in 1877.4 He described the formation of psoriasiform lesions at the site of cutaneous trauma.5 These isomorphic lesions can occur in 25% of patients with psoriasis after trauma to the skin such as tattooing.6 Other dermatologic diseases that can present as an isomorphic response to tattooing include lichen planus, Darier disease, vitiligo, and autoimmune bullous disease.3,5,6
Various causes of trauma such as burns, insect bites, physical trauma, and needle trauma have been shown to produce new psoriatic lesions.6 The time period from trauma to formation of psoriasiform lesions usually ranges from 10 to 20 days; however, an initial reaction can occur as early as 3 days or as long as 2 years after trauma.4 Although the pathophysiology of the isomorphic response is not well known, it has been shown that nerve growth factor has a role. Raychaudhuri et al7 demonstrated the upregulation of nerve growth factor in the development of a psoriatic lesion, influencing keratinocyte proliferation, angiogenesis, and T-cell activation.
Physical trauma such as tattooing has been shown to cause an isomorphic response in psoriasis. We describe a case of isomorphic psoriasis in a patient after tattoo application. Our patient had a several-month history of well-controlled psoriasis prior to obtaining the new tattoo. Several days after receiving the tattoo, the patient reported an increase in psoriatic lesions, including at the site of the tattoo. The trauma causing the isomorphic response could have been either a response to the tattoo pigment or needle injury to the skin.6
Psoriasis and isomorphic lesions can be treated with topical corticosteroids as well as systemic and biologic agents. Our patient was treated with triamcinolone cream with good response.8
- Haugh IM, Laumann SL, Laumann AE. Regulation of tattoo ink production and the tattoo business in the US. Curr Probl Dermatol. 2015;48:248-252.
- Marcoux D, Couture-Trudel PM, Rboulet-Delmas G, et al. Sensitization to paraphenylenediame from a streetside temporary tattoo. Pediatr Dermatol. 2002;19:498-502.
- Bassi A, Campolmi P, Cannarozzo G, et al. Tattoo-associated skin reaction: the importance of an early diagnosis and proper treatment [published online July 23, 2014]. Biomed Res Int. 2014;2014:354608.
- Weiss G, Shemer A, Trau H. The Koebner phenomenon: review of the literature. J Eur Acad Dermatol Venereol. 2002;16:241-248.
- Sagi L, Trau H. The Koebner phenomenon. Clin Dermatol. 2011;29:231-236.
- Orzan OA, Popa LG, Vexler ES, et al. Tattoo-induced psoriasis. J Med Life. 2014;7:65-68.
- Raychaudhuri SP, Jiang WY, Raychaudhuri SK. Revisiting the Koebner phenomenon: role of NGF and its receptor system in the pathogenesis of psoriasis. Am J Pathol. 2008;172:961-971.
- Gottlieb AB. Therapeutic options in the treatment of psoriasis and atopic dermatitis. J Am Acad Dermatol. 2005;53(1 suppl 1):S3-S16.
- Haugh IM, Laumann SL, Laumann AE. Regulation of tattoo ink production and the tattoo business in the US. Curr Probl Dermatol. 2015;48:248-252.
- Marcoux D, Couture-Trudel PM, Rboulet-Delmas G, et al. Sensitization to paraphenylenediame from a streetside temporary tattoo. Pediatr Dermatol. 2002;19:498-502.
- Bassi A, Campolmi P, Cannarozzo G, et al. Tattoo-associated skin reaction: the importance of an early diagnosis and proper treatment [published online July 23, 2014]. Biomed Res Int. 2014;2014:354608.
- Weiss G, Shemer A, Trau H. The Koebner phenomenon: review of the literature. J Eur Acad Dermatol Venereol. 2002;16:241-248.
- Sagi L, Trau H. The Koebner phenomenon. Clin Dermatol. 2011;29:231-236.
- Orzan OA, Popa LG, Vexler ES, et al. Tattoo-induced psoriasis. J Med Life. 2014;7:65-68.
- Raychaudhuri SP, Jiang WY, Raychaudhuri SK. Revisiting the Koebner phenomenon: role of NGF and its receptor system in the pathogenesis of psoriasis. Am J Pathol. 2008;172:961-971.
- Gottlieb AB. Therapeutic options in the treatment of psoriasis and atopic dermatitis. J Am Acad Dermatol. 2005;53(1 suppl 1):S3-S16.

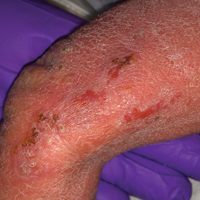
A 26-year-old man presented with a mildly pruritic red scaly rash on the right arm of 3 weeks' duration. He reported having a tattoo placed on previously normal skin on the right lateral arm prior to the development of the rash. Two weeks after receiving the tattoo, he developed scaling and redness of the skin involved in the tattoo. He also had similar papules and plaques over the rest of his body. Physical examination showed well-demarcated, erythematous, scaly papules and plaques following the design of a black-pigmented tattoo on the lateral aspect of the right arm. There also were similar erythematous scaly plaques scattered over both arms and the trunk. He denied any pain or blister formation of the involved areas.

Black Eschars on the Face and Body
The Diagnosis: Lymphomatoid Papulosis
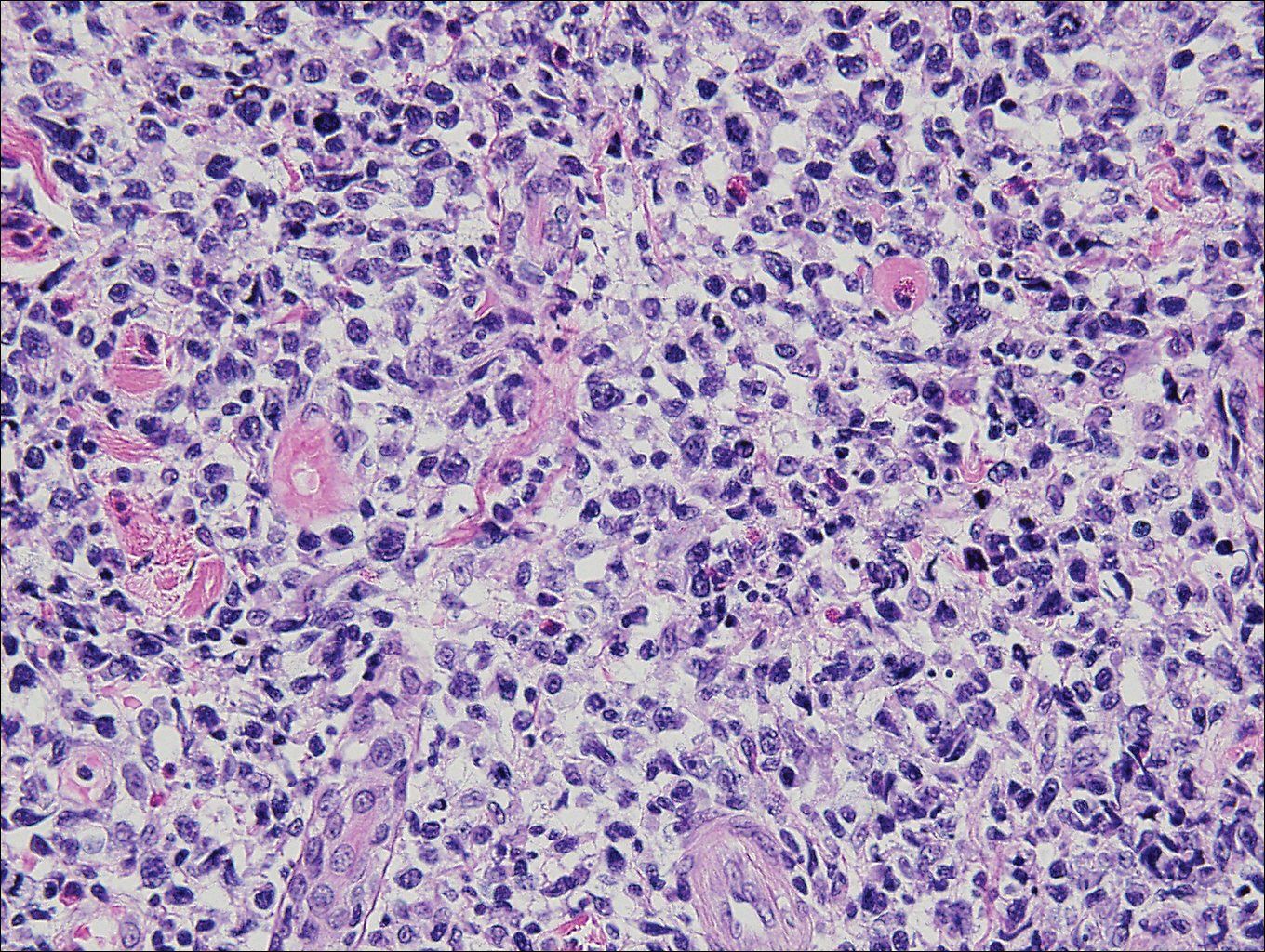
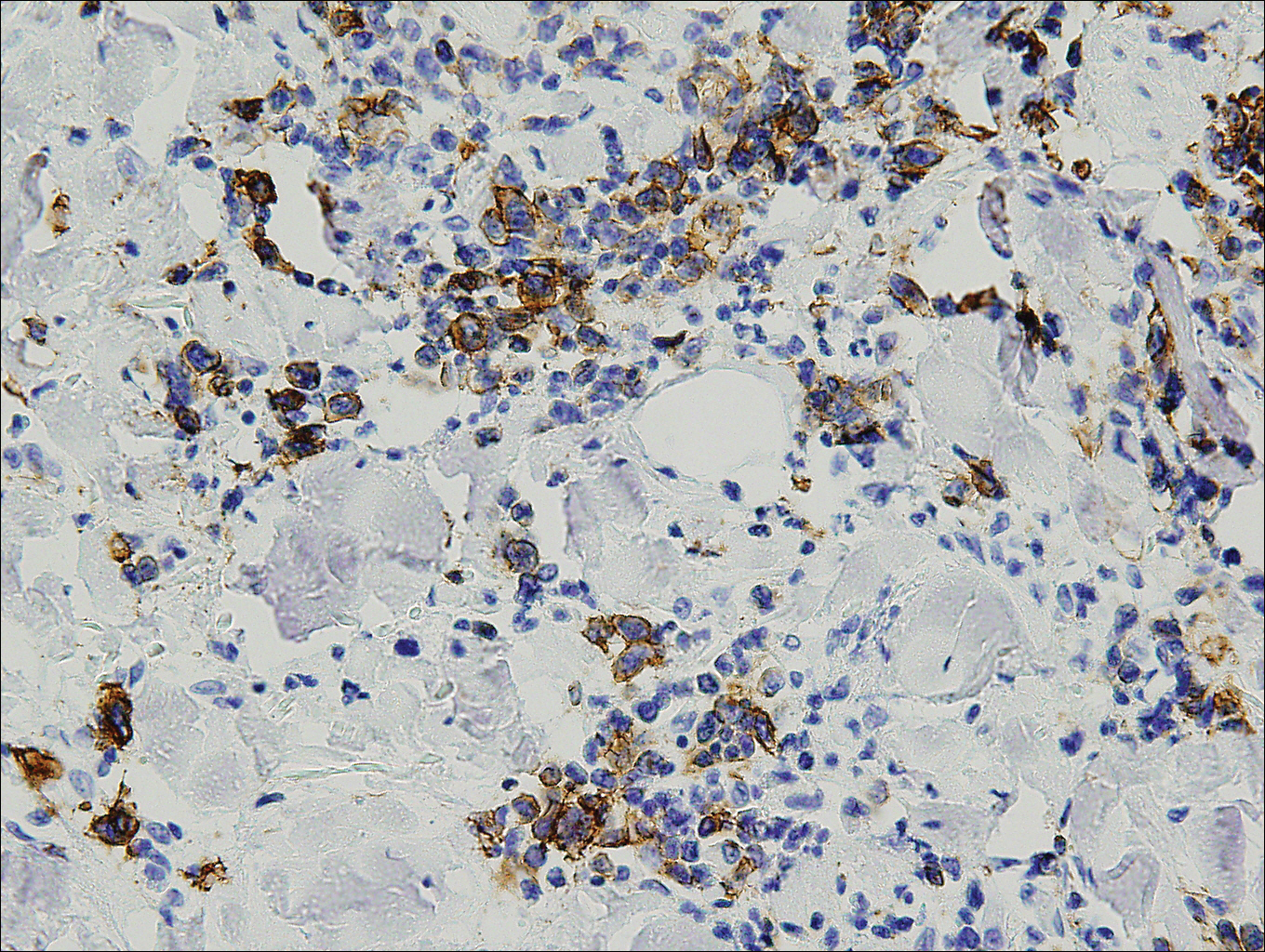
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
The Diagnosis: Lymphomatoid Papulosis
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
The Diagnosis: Lymphomatoid Papulosis
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
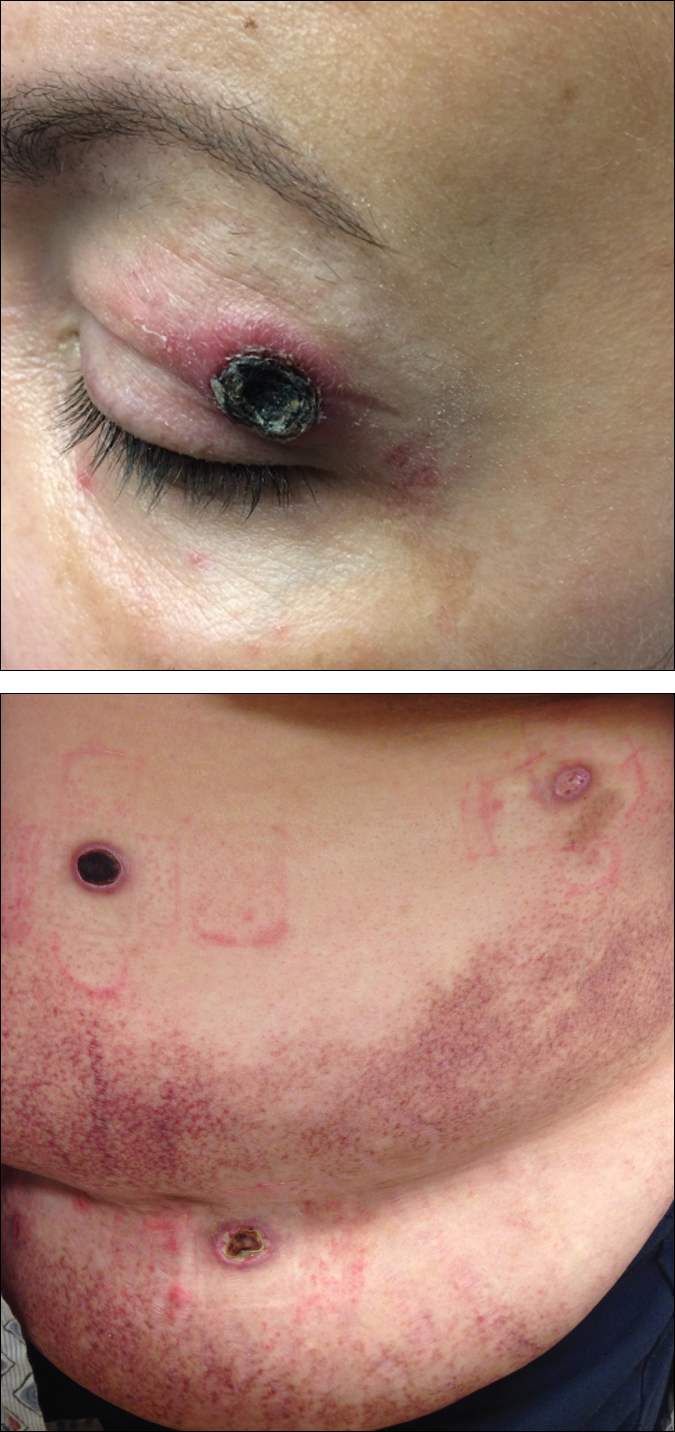
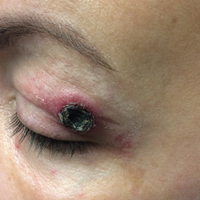
A 50-year-old woman presented for evaluation of black eschars on the face and body. Over the preceding 8 weeks she had developed several asymptomatic papules that gradually enlarged, ulcerated, and formed a black eschar, prior to gradually self-resolving over the course of several weeks. During this time, new lesions were forming. The resulting skin revealed dyspigmentation and scar formation. Prior to presentation, antimicrobial therapy had been initiated for a presumed infectious etiology; however, the eruption continued to progress. The patient denied sick contacts, livestock exposure, or recent travel. A complete review of systems, including fever, chills, or lymphadenopathy, was negative. Physical examination revealed 6 circular necrotic ulcers with an overlying black eschar on the face (top), trunk (bottom), hands, and thighs, all in various stages of healing. In addition, large, reticulated, poikilodermatous patches were incidentally noted in areas free of ulcers and eschars on the trunk (bottom) and bilateral arms and legs. Upon questioning, the patient said these patches had been present for more than 30 years. A punch biopsy from an ulcer on the chest was obtained and sent for histopathologic and immunohistochemical examination.
Painless Telangiectatic Lesion on the Wrist
The Diagnosis: Merkel Cell Carcinoma
A partial biopsy was performed during the dermatology examination. Histopathology demonstrated a dense dermal infiltrate of small, dark blue, pleomorphic cells (Figure 1). On high power, the individual cells were noted to have vesicular nuclei with finely granular and dusty chromatin (Figure 2). Numerous mitotic figures were present. Immunohistochemical stains were performed and revealed positive staining for cytokeratin 20 (with a perinuclear dot pattern), synaptophysin, and chromogranin.
Merkel cell carcinoma (MCC) is an uncommon carcinoma of the epidermal neuroendocrine cells with approximately 1500 cases a year in the United States.1 Merkel cell carcinoma has a poor prognosis with approximately one-third of cases resulting in death within 5 years and with a survival rate strongly dependent on the stage of disease at presentation.2 A complete surgical excision with histologically verified clear margins is the main form of treatment of the primary cancer.3 Although the effectiveness of adjuvant therapy for MCC has been debated,4 retrospective analysis has shown that the high local recurrence rate of the primary tumor can be reduced by combining surgical excision with a form of radiation therapy.5
A systematic cohort study of 195 patients diagnosed with MCC summarized its most clinical factors with the acronym AEIOU: asymptomatic, expanding rapidly, immunosuppression, older than 50 years of age, and UV-exposed site on a fair-skinned individual.6 The role of immune function in MCC was highlighted by a 16-fold overrepresentation of immunosuppressed patients in the studied cohort as compared to the general US population. The immunosuppressed patients included individuals with human immunodeficiency virus, chronic lymphocytic leukemia, and iatrogenic suppression secondary to solid organ transplantation.6
In 2008, Merkel cell polyomavirus (MCPyV) was found in 80% (8/10) of MCC tumors tested.7 Since then, many different studies have suggested that MCPyV is an etiologic agent of MCC.8-10 A natural component of skin flora, MCPyV only becomes tumorigenic after integration into the host DNA and with mutations to the viral genome.11 Although there currently is no difference in treatment of MCPyV-positive and MCPyV-negative MCC,12 research is being done to determine how the discovery of the MCPyV could impact the treatment of MCC.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2009;37:20-27.
- Allen PJ, Bowne WB, Jaques DP, et al. Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol. 2005;23:2300-2309.
- Eng TY, Boersma MG, Fuller CD, et al. A comprehensive review of the treatment of Merkel cell carcinoma. Am J Clin Oncol. 2007;30:624-636.
- Beenken SW, Urist MM. Treatment options for Merkel cell carcinoma. J Natl Compr Canc Netw. 2004;2:89-92.
- Decker RH, Wilson LD. Role of radiotherapy in the management of Merkel cell carcinoma of the skin. J Natl Compr Canc Netw. 2006;4:713-718.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the "AEIOU" features. J Am Acad Dermatol. 2008;58:375-381.
- Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
- Duncavage EJ, Zehnbauer BA, Pfeifer JD. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol. 2009;22:516-521.
- Sastre-Garau X, Peter M, Avril MF, et al. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. J Pathol. 2009;218:48-56.
- Varga E, Kiss M, Szabó K, et al. Detection of Merkel cell polyomavirus DNA in Merkel cell carcinomas. Br J Dermatol. 2009;161:930-932.
- Wendzicki JA, Moore PS, Chang Y. Large T and small T antigens of Merkel cell carcinoma. Curr Opin Virol. 2015;11:38-43.
- Duprat JP, Landman G, Salvajoli JV, et al. A review of the epidemiology and treatment of Merkel cell carcinoma. Clinics (Sao Paulo). 2011;66:1817-1823.
The Diagnosis: Merkel Cell Carcinoma
A partial biopsy was performed during the dermatology examination. Histopathology demonstrated a dense dermal infiltrate of small, dark blue, pleomorphic cells (Figure 1). On high power, the individual cells were noted to have vesicular nuclei with finely granular and dusty chromatin (Figure 2). Numerous mitotic figures were present. Immunohistochemical stains were performed and revealed positive staining for cytokeratin 20 (with a perinuclear dot pattern), synaptophysin, and chromogranin.
Merkel cell carcinoma (MCC) is an uncommon carcinoma of the epidermal neuroendocrine cells with approximately 1500 cases a year in the United States.1 Merkel cell carcinoma has a poor prognosis with approximately one-third of cases resulting in death within 5 years and with a survival rate strongly dependent on the stage of disease at presentation.2 A complete surgical excision with histologically verified clear margins is the main form of treatment of the primary cancer.3 Although the effectiveness of adjuvant therapy for MCC has been debated,4 retrospective analysis has shown that the high local recurrence rate of the primary tumor can be reduced by combining surgical excision with a form of radiation therapy.5
A systematic cohort study of 195 patients diagnosed with MCC summarized its most clinical factors with the acronym AEIOU: asymptomatic, expanding rapidly, immunosuppression, older than 50 years of age, and UV-exposed site on a fair-skinned individual.6 The role of immune function in MCC was highlighted by a 16-fold overrepresentation of immunosuppressed patients in the studied cohort as compared to the general US population. The immunosuppressed patients included individuals with human immunodeficiency virus, chronic lymphocytic leukemia, and iatrogenic suppression secondary to solid organ transplantation.6
In 2008, Merkel cell polyomavirus (MCPyV) was found in 80% (8/10) of MCC tumors tested.7 Since then, many different studies have suggested that MCPyV is an etiologic agent of MCC.8-10 A natural component of skin flora, MCPyV only becomes tumorigenic after integration into the host DNA and with mutations to the viral genome.11 Although there currently is no difference in treatment of MCPyV-positive and MCPyV-negative MCC,12 research is being done to determine how the discovery of the MCPyV could impact the treatment of MCC.
The Diagnosis: Merkel Cell Carcinoma
A partial biopsy was performed during the dermatology examination. Histopathology demonstrated a dense dermal infiltrate of small, dark blue, pleomorphic cells (Figure 1). On high power, the individual cells were noted to have vesicular nuclei with finely granular and dusty chromatin (Figure 2). Numerous mitotic figures were present. Immunohistochemical stains were performed and revealed positive staining for cytokeratin 20 (with a perinuclear dot pattern), synaptophysin, and chromogranin.
Merkel cell carcinoma (MCC) is an uncommon carcinoma of the epidermal neuroendocrine cells with approximately 1500 cases a year in the United States.1 Merkel cell carcinoma has a poor prognosis with approximately one-third of cases resulting in death within 5 years and with a survival rate strongly dependent on the stage of disease at presentation.2 A complete surgical excision with histologically verified clear margins is the main form of treatment of the primary cancer.3 Although the effectiveness of adjuvant therapy for MCC has been debated,4 retrospective analysis has shown that the high local recurrence rate of the primary tumor can be reduced by combining surgical excision with a form of radiation therapy.5
A systematic cohort study of 195 patients diagnosed with MCC summarized its most clinical factors with the acronym AEIOU: asymptomatic, expanding rapidly, immunosuppression, older than 50 years of age, and UV-exposed site on a fair-skinned individual.6 The role of immune function in MCC was highlighted by a 16-fold overrepresentation of immunosuppressed patients in the studied cohort as compared to the general US population. The immunosuppressed patients included individuals with human immunodeficiency virus, chronic lymphocytic leukemia, and iatrogenic suppression secondary to solid organ transplantation.6
In 2008, Merkel cell polyomavirus (MCPyV) was found in 80% (8/10) of MCC tumors tested.7 Since then, many different studies have suggested that MCPyV is an etiologic agent of MCC.8-10 A natural component of skin flora, MCPyV only becomes tumorigenic after integration into the host DNA and with mutations to the viral genome.11 Although there currently is no difference in treatment of MCPyV-positive and MCPyV-negative MCC,12 research is being done to determine how the discovery of the MCPyV could impact the treatment of MCC.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2009;37:20-27.
- Allen PJ, Bowne WB, Jaques DP, et al. Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol. 2005;23:2300-2309.
- Eng TY, Boersma MG, Fuller CD, et al. A comprehensive review of the treatment of Merkel cell carcinoma. Am J Clin Oncol. 2007;30:624-636.
- Beenken SW, Urist MM. Treatment options for Merkel cell carcinoma. J Natl Compr Canc Netw. 2004;2:89-92.
- Decker RH, Wilson LD. Role of radiotherapy in the management of Merkel cell carcinoma of the skin. J Natl Compr Canc Netw. 2006;4:713-718.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the "AEIOU" features. J Am Acad Dermatol. 2008;58:375-381.
- Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
- Duncavage EJ, Zehnbauer BA, Pfeifer JD. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol. 2009;22:516-521.
- Sastre-Garau X, Peter M, Avril MF, et al. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. J Pathol. 2009;218:48-56.
- Varga E, Kiss M, Szabó K, et al. Detection of Merkel cell polyomavirus DNA in Merkel cell carcinomas. Br J Dermatol. 2009;161:930-932.
- Wendzicki JA, Moore PS, Chang Y. Large T and small T antigens of Merkel cell carcinoma. Curr Opin Virol. 2015;11:38-43.
- Duprat JP, Landman G, Salvajoli JV, et al. A review of the epidemiology and treatment of Merkel cell carcinoma. Clinics (Sao Paulo). 2011;66:1817-1823.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2009;37:20-27.
- Allen PJ, Bowne WB, Jaques DP, et al. Merkel cell carcinoma: prognosis and treatment of patients from a single institution. J Clin Oncol. 2005;23:2300-2309.
- Eng TY, Boersma MG, Fuller CD, et al. A comprehensive review of the treatment of Merkel cell carcinoma. Am J Clin Oncol. 2007;30:624-636.
- Beenken SW, Urist MM. Treatment options for Merkel cell carcinoma. J Natl Compr Canc Netw. 2004;2:89-92.
- Decker RH, Wilson LD. Role of radiotherapy in the management of Merkel cell carcinoma of the skin. J Natl Compr Canc Netw. 2006;4:713-718.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the "AEIOU" features. J Am Acad Dermatol. 2008;58:375-381.
- Feng H, Shuda M, Chang Y, et al. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096-1100.
- Duncavage EJ, Zehnbauer BA, Pfeifer JD. Prevalence of Merkel cell polyomavirus in Merkel cell carcinoma. Mod Pathol. 2009;22:516-521.
- Sastre-Garau X, Peter M, Avril MF, et al. Merkel cell carcinoma of the skin: pathological and molecular evidence for a causative role of MCV in oncogenesis. J Pathol. 2009;218:48-56.
- Varga E, Kiss M, Szabó K, et al. Detection of Merkel cell polyomavirus DNA in Merkel cell carcinomas. Br J Dermatol. 2009;161:930-932.
- Wendzicki JA, Moore PS, Chang Y. Large T and small T antigens of Merkel cell carcinoma. Curr Opin Virol. 2015;11:38-43.
- Duprat JP, Landman G, Salvajoli JV, et al. A review of the epidemiology and treatment of Merkel cell carcinoma. Clinics (Sao Paulo). 2011;66:1817-1823.
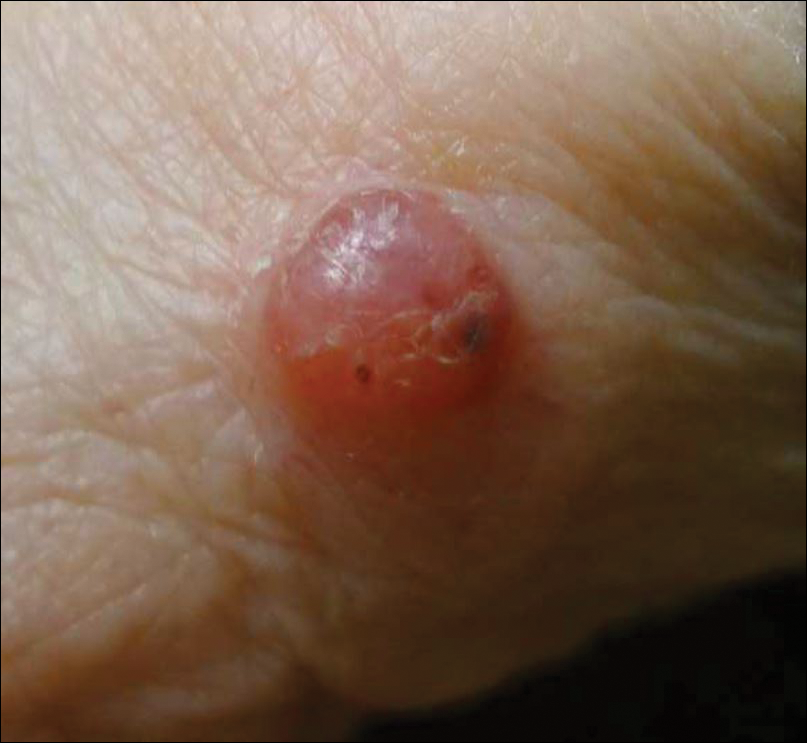
A 91-year-old white man with a history of atrial fibrillation, benign prostatic hyperplasia, dysphagia, gastroesophageal reflux disease, hypertension, hypothyroidism, osteoarthritis, and laryngeal cancer presented with an 8-mm firm, painless, pink lesion with telangiectasia on the left wrist. The lesion had been present for an unknown period of time and was asymptomatic at presentation.

Irregular Erythematous Patch on the Face of an Infant
The Diagnosis: Phakomatosis Pigmentovascularis With Sturge-Weber Syndrome
The erythematous patches were identified as capillary malformations (port-wine stains) and the slate gray pigmentary changes as dermal melanocytosis (Mongolian spots)(Figure). In fact, the diagnosis of phakomatosis pigmentovascularis (PPV) type II requires dermal melanocytosis and capillary malformation with and without nevus anemicus.1 In one case series, 46% (7/15) of patients with PPV had nevus anemicus2 but our patient did not.

Phakomatosis pigmentovascularis was divided into 4 types in 1985,3 then later 5 types.4 Subcategories of the 5 types include type A, which denotes a lack of extracutaneous involvement, and type B, which is used when internal manifestations have been exhibited. Since 1947, approximately 222 cases of PPV have been described in the literature.2
A case of PPV associated with Sturge-Weber syndrome (SWS) was reported in 1997.5 Since then, PPV occasionally has been linked with SWS,5-9 though there have been other syndromic associations including Klippel-Trenaunay-Weber syndrome and melanosis oculi.2 The incidence and prevalence of overlap of PPV and SWS is unknown but is likely to be rare. In our case, magnetic resonance imaging of the patient's brain did not reveal the characteristic tram-track appearance of SWS; however, the diagnosis of SWS type II only requires facial angioma with or without glaucoma.9,10 Most cases of PPV originate from Japan, Argentina, and Mexico.2 Interestingly, our patient's parents were both of Mexican ancestry. Phakomatosis pigmentovascularis type IIb is the most common, followed by type IIa.2 Most cases have been described as sporadic, though our patient's mother also exhibited a port-wine stain on the right neck, suggesting a possible genetic association.
The etiology of PPV has been postulated as twin spotting or didymosis (Greek for twin), most commonly seen in plants and animals. A previous review defined twin spotting as 2 mutant tissues situated adjacent to one another and unique from the normal tissue surrounding both of them.2 When the cell loses its heterozygosity, this phenomenon appears. An alternative etiology supplants that a drug or virus toxic to the nervous system causes aberrant angioblasts and melanoblasts.11,12 The etiology of SWS also is unknown, though vasomotor instability has been postulated as a cause.6,13
It is important to exclude associated internal organ involvement with both of these syndromes because approximately 50% of PPV cases have extracutaneous organ involvement.2,14 In fact, PPV is known to involve the brain, skeletal system, and eye, potentially manifesting as deafness, hydrocephalus, extremity overgrowth, scoliosis, cataracts, and more.2 Patients with SWS often exhibit brain and eye symptoms including seizures.1 To screen for extracutaneous involvement, multiple imaging studies should be performed. In our patient, an echocardiogram revealed a patent foramen ovale and normal cardiac anatomy for his age. Brain imaging revealed a hypoplastic left sigmoid and transverse sinus without venous thrombosis and unremarkable appearance of the brain. An ultrasound of the liver, spleen, kidneys, and pancreas revealed no evidence of solid, cystic, or vascular lesions, though the gallbladder exhibited hyperechoic areas.
To manage the skin lesions, some authors recommend Q-switched lasers for pigmented lesions and pulsed dye lasers for capillary malformations.15 Paller and Mancini1 cited evidence that pulsed dye laser treatment before the age of 1 year may offer a psychological advantage, while other views have been offered.16 Some physicians believe that no urgent treatment of capillary malformations is needed unless internal organs are involved.2,15
- Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 4th ed. New York, NY: Elsevier/Saunders; 2011.
- Fernández-Guarino M, Boixeda P, de Las Heras E, et al. Phakomatosis pigmentovascularis: clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88-93.
- Hasegawa Y, Yasuhara M. Phakomatosis pigmentovascularis type VIa. Arch Dermatol. 1985;121:651-655.
- Torrelo A, Zambrano A, Happle R. Cutis marmorata telangiectatica congenita and extensive Mongolian spots: type V phacomatosis pigmentovascularis. Br J Dermatol. 2003;148:342-345.
- Teekhasaenee C, Ritch R. Glaucoma in phakomatosis pigmentovascularis. Ophthalmology. 1997;104:150-157.
- Patil B, Sinha G, Nayak B, et al. Bilateral Sturge-Weber and phakomatosis pigmentovascularis with glaucoma, an overlap syndrome [published online May 6, 2015]. Case Rep Ophthalmol Med. 2015;2015:106932.
- Hagiwara K, Uezato H, Nonaka S. Phacomatosis pigmentovascularis type IIb associated with Sturge-Weber syndrome and pyogenic granuloma. J Dermatol. 1998;25:721-729.
- Al Robaee A, Banka N, Alfadley A. Phakomatosis pigmentovascularis type IIb associated with Sturge-Weber syndrome. Pediatr Dermatol. 2004;21:642-645.
- Yang Y, Guo X, Xu J, et al. Phakomatosis pigmentovascularis associated with Sturge-Weber syndrome, ota nevus, and congenital glaucoma. Medicine (Baltimore). 2015;94:E1025.
- Roach ES. Neurocutaneous syndromes. Pediatr Clin North Am. 1992;39:591-620.
- Happle R. Mosaicism in human skin, understanding the patterns and mechanisms. Arch Dermatol. 1993;129:1460-1470.
- Happle R. Loss of heterozygosity in human skin. J Am Acad Dermatol. 1999;85:355-358.
- Comi AM. Pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509-516.
- Kim YC, Park HJ, Cinn YW. Phakomatosis pigmentovascularis type IIa with generalized vitiligo. Br J Dermatol. 2002;147:1028-1029.
- Brittain P, Walsh EJ, Smidt AC. Blotchy baby: a case of phakomatosis pigmentovascularis [published online February 1, 2013]. J Pediatr. 2013;162:1293.
- Van der Horst CM, Koster PH, de Borgie CA, et al. Effect of the timing of treatment of port-wine stains with the flash-lamp-pumped pulsed-dye laser. N Engl J Med. 1998;338:1028-1033.
The Diagnosis: Phakomatosis Pigmentovascularis With Sturge-Weber Syndrome
The erythematous patches were identified as capillary malformations (port-wine stains) and the slate gray pigmentary changes as dermal melanocytosis (Mongolian spots)(Figure). In fact, the diagnosis of phakomatosis pigmentovascularis (PPV) type II requires dermal melanocytosis and capillary malformation with and without nevus anemicus.1 In one case series, 46% (7/15) of patients with PPV had nevus anemicus2 but our patient did not.
Phakomatosis pigmentovascularis was divided into 4 types in 1985,3 then later 5 types.4 Subcategories of the 5 types include type A, which denotes a lack of extracutaneous involvement, and type B, which is used when internal manifestations have been exhibited. Since 1947, approximately 222 cases of PPV have been described in the literature.2
A case of PPV associated with Sturge-Weber syndrome (SWS) was reported in 1997.5 Since then, PPV occasionally has been linked with SWS,5-9 though there have been other syndromic associations including Klippel-Trenaunay-Weber syndrome and melanosis oculi.2 The incidence and prevalence of overlap of PPV and SWS is unknown but is likely to be rare. In our case, magnetic resonance imaging of the patient's brain did not reveal the characteristic tram-track appearance of SWS; however, the diagnosis of SWS type II only requires facial angioma with or without glaucoma.9,10 Most cases of PPV originate from Japan, Argentina, and Mexico.2 Interestingly, our patient's parents were both of Mexican ancestry. Phakomatosis pigmentovascularis type IIb is the most common, followed by type IIa.2 Most cases have been described as sporadic, though our patient's mother also exhibited a port-wine stain on the right neck, suggesting a possible genetic association.
The etiology of PPV has been postulated as twin spotting or didymosis (Greek for twin), most commonly seen in plants and animals. A previous review defined twin spotting as 2 mutant tissues situated adjacent to one another and unique from the normal tissue surrounding both of them.2 When the cell loses its heterozygosity, this phenomenon appears. An alternative etiology supplants that a drug or virus toxic to the nervous system causes aberrant angioblasts and melanoblasts.11,12 The etiology of SWS also is unknown, though vasomotor instability has been postulated as a cause.6,13
It is important to exclude associated internal organ involvement with both of these syndromes because approximately 50% of PPV cases have extracutaneous organ involvement.2,14 In fact, PPV is known to involve the brain, skeletal system, and eye, potentially manifesting as deafness, hydrocephalus, extremity overgrowth, scoliosis, cataracts, and more.2 Patients with SWS often exhibit brain and eye symptoms including seizures.1 To screen for extracutaneous involvement, multiple imaging studies should be performed. In our patient, an echocardiogram revealed a patent foramen ovale and normal cardiac anatomy for his age. Brain imaging revealed a hypoplastic left sigmoid and transverse sinus without venous thrombosis and unremarkable appearance of the brain. An ultrasound of the liver, spleen, kidneys, and pancreas revealed no evidence of solid, cystic, or vascular lesions, though the gallbladder exhibited hyperechoic areas.
To manage the skin lesions, some authors recommend Q-switched lasers for pigmented lesions and pulsed dye lasers for capillary malformations.15 Paller and Mancini1 cited evidence that pulsed dye laser treatment before the age of 1 year may offer a psychological advantage, while other views have been offered.16 Some physicians believe that no urgent treatment of capillary malformations is needed unless internal organs are involved.2,15
The Diagnosis: Phakomatosis Pigmentovascularis With Sturge-Weber Syndrome
The erythematous patches were identified as capillary malformations (port-wine stains) and the slate gray pigmentary changes as dermal melanocytosis (Mongolian spots)(Figure). In fact, the diagnosis of phakomatosis pigmentovascularis (PPV) type II requires dermal melanocytosis and capillary malformation with and without nevus anemicus.1 In one case series, 46% (7/15) of patients with PPV had nevus anemicus2 but our patient did not.
Phakomatosis pigmentovascularis was divided into 4 types in 1985,3 then later 5 types.4 Subcategories of the 5 types include type A, which denotes a lack of extracutaneous involvement, and type B, which is used when internal manifestations have been exhibited. Since 1947, approximately 222 cases of PPV have been described in the literature.2
A case of PPV associated with Sturge-Weber syndrome (SWS) was reported in 1997.5 Since then, PPV occasionally has been linked with SWS,5-9 though there have been other syndromic associations including Klippel-Trenaunay-Weber syndrome and melanosis oculi.2 The incidence and prevalence of overlap of PPV and SWS is unknown but is likely to be rare. In our case, magnetic resonance imaging of the patient's brain did not reveal the characteristic tram-track appearance of SWS; however, the diagnosis of SWS type II only requires facial angioma with or without glaucoma.9,10 Most cases of PPV originate from Japan, Argentina, and Mexico.2 Interestingly, our patient's parents were both of Mexican ancestry. Phakomatosis pigmentovascularis type IIb is the most common, followed by type IIa.2 Most cases have been described as sporadic, though our patient's mother also exhibited a port-wine stain on the right neck, suggesting a possible genetic association.
The etiology of PPV has been postulated as twin spotting or didymosis (Greek for twin), most commonly seen in plants and animals. A previous review defined twin spotting as 2 mutant tissues situated adjacent to one another and unique from the normal tissue surrounding both of them.2 When the cell loses its heterozygosity, this phenomenon appears. An alternative etiology supplants that a drug or virus toxic to the nervous system causes aberrant angioblasts and melanoblasts.11,12 The etiology of SWS also is unknown, though vasomotor instability has been postulated as a cause.6,13
It is important to exclude associated internal organ involvement with both of these syndromes because approximately 50% of PPV cases have extracutaneous organ involvement.2,14 In fact, PPV is known to involve the brain, skeletal system, and eye, potentially manifesting as deafness, hydrocephalus, extremity overgrowth, scoliosis, cataracts, and more.2 Patients with SWS often exhibit brain and eye symptoms including seizures.1 To screen for extracutaneous involvement, multiple imaging studies should be performed. In our patient, an echocardiogram revealed a patent foramen ovale and normal cardiac anatomy for his age. Brain imaging revealed a hypoplastic left sigmoid and transverse sinus without venous thrombosis and unremarkable appearance of the brain. An ultrasound of the liver, spleen, kidneys, and pancreas revealed no evidence of solid, cystic, or vascular lesions, though the gallbladder exhibited hyperechoic areas.
To manage the skin lesions, some authors recommend Q-switched lasers for pigmented lesions and pulsed dye lasers for capillary malformations.15 Paller and Mancini1 cited evidence that pulsed dye laser treatment before the age of 1 year may offer a psychological advantage, while other views have been offered.16 Some physicians believe that no urgent treatment of capillary malformations is needed unless internal organs are involved.2,15
- Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 4th ed. New York, NY: Elsevier/Saunders; 2011.
- Fernández-Guarino M, Boixeda P, de Las Heras E, et al. Phakomatosis pigmentovascularis: clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88-93.
- Hasegawa Y, Yasuhara M. Phakomatosis pigmentovascularis type VIa. Arch Dermatol. 1985;121:651-655.
- Torrelo A, Zambrano A, Happle R. Cutis marmorata telangiectatica congenita and extensive Mongolian spots: type V phacomatosis pigmentovascularis. Br J Dermatol. 2003;148:342-345.
- Teekhasaenee C, Ritch R. Glaucoma in phakomatosis pigmentovascularis. Ophthalmology. 1997;104:150-157.
- Patil B, Sinha G, Nayak B, et al. Bilateral Sturge-Weber and phakomatosis pigmentovascularis with glaucoma, an overlap syndrome [published online May 6, 2015]. Case Rep Ophthalmol Med. 2015;2015:106932.
- Hagiwara K, Uezato H, Nonaka S. Phacomatosis pigmentovascularis type IIb associated with Sturge-Weber syndrome and pyogenic granuloma. J Dermatol. 1998;25:721-729.
- Al Robaee A, Banka N, Alfadley A. Phakomatosis pigmentovascularis type IIb associated with Sturge-Weber syndrome. Pediatr Dermatol. 2004;21:642-645.
- Yang Y, Guo X, Xu J, et al. Phakomatosis pigmentovascularis associated with Sturge-Weber syndrome, ota nevus, and congenital glaucoma. Medicine (Baltimore). 2015;94:E1025.
- Roach ES. Neurocutaneous syndromes. Pediatr Clin North Am. 1992;39:591-620.
- Happle R. Mosaicism in human skin, understanding the patterns and mechanisms. Arch Dermatol. 1993;129:1460-1470.
- Happle R. Loss of heterozygosity in human skin. J Am Acad Dermatol. 1999;85:355-358.
- Comi AM. Pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509-516.
- Kim YC, Park HJ, Cinn YW. Phakomatosis pigmentovascularis type IIa with generalized vitiligo. Br J Dermatol. 2002;147:1028-1029.
- Brittain P, Walsh EJ, Smidt AC. Blotchy baby: a case of phakomatosis pigmentovascularis [published online February 1, 2013]. J Pediatr. 2013;162:1293.
- Van der Horst CM, Koster PH, de Borgie CA, et al. Effect of the timing of treatment of port-wine stains with the flash-lamp-pumped pulsed-dye laser. N Engl J Med. 1998;338:1028-1033.
- Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. 4th ed. New York, NY: Elsevier/Saunders; 2011.
- Fernández-Guarino M, Boixeda P, de Las Heras E, et al. Phakomatosis pigmentovascularis: clinical findings in 15 patients and review of the literature. J Am Acad Dermatol. 2008;58:88-93.
- Hasegawa Y, Yasuhara M. Phakomatosis pigmentovascularis type VIa. Arch Dermatol. 1985;121:651-655.
- Torrelo A, Zambrano A, Happle R. Cutis marmorata telangiectatica congenita and extensive Mongolian spots: type V phacomatosis pigmentovascularis. Br J Dermatol. 2003;148:342-345.
- Teekhasaenee C, Ritch R. Glaucoma in phakomatosis pigmentovascularis. Ophthalmology. 1997;104:150-157.
- Patil B, Sinha G, Nayak B, et al. Bilateral Sturge-Weber and phakomatosis pigmentovascularis with glaucoma, an overlap syndrome [published online May 6, 2015]. Case Rep Ophthalmol Med. 2015;2015:106932.
- Hagiwara K, Uezato H, Nonaka S. Phacomatosis pigmentovascularis type IIb associated with Sturge-Weber syndrome and pyogenic granuloma. J Dermatol. 1998;25:721-729.
- Al Robaee A, Banka N, Alfadley A. Phakomatosis pigmentovascularis type IIb associated with Sturge-Weber syndrome. Pediatr Dermatol. 2004;21:642-645.
- Yang Y, Guo X, Xu J, et al. Phakomatosis pigmentovascularis associated with Sturge-Weber syndrome, ota nevus, and congenital glaucoma. Medicine (Baltimore). 2015;94:E1025.
- Roach ES. Neurocutaneous syndromes. Pediatr Clin North Am. 1992;39:591-620.
- Happle R. Mosaicism in human skin, understanding the patterns and mechanisms. Arch Dermatol. 1993;129:1460-1470.
- Happle R. Loss of heterozygosity in human skin. J Am Acad Dermatol. 1999;85:355-358.
- Comi AM. Pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509-516.
- Kim YC, Park HJ, Cinn YW. Phakomatosis pigmentovascularis type IIa with generalized vitiligo. Br J Dermatol. 2002;147:1028-1029.
- Brittain P, Walsh EJ, Smidt AC. Blotchy baby: a case of phakomatosis pigmentovascularis [published online February 1, 2013]. J Pediatr. 2013;162:1293.
- Van der Horst CM, Koster PH, de Borgie CA, et al. Effect of the timing of treatment of port-wine stains with the flash-lamp-pumped pulsed-dye laser. N Engl J Med. 1998;338:1028-1033.
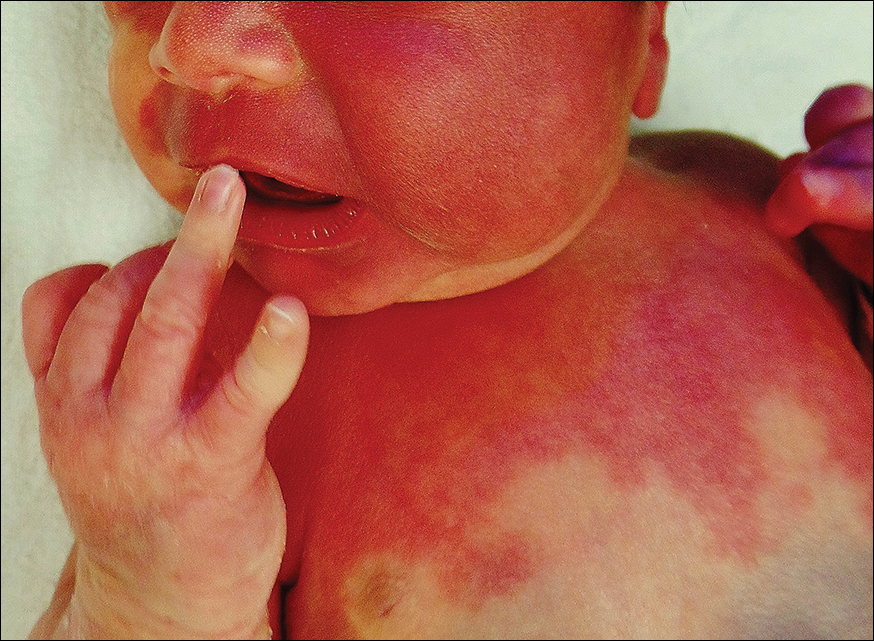
A newborn presented with an irregular and well-demarcated erythematous patch on the face, trunk, buttocks, and toes on the left foot. Another red patch was present on the right side of the face, while a slate gray patch covered the flanks and back. The limbs appeared symmetric and he exhibited no gross deformities. On close physical examination, he was noted to have a cloudy left eye. An ophthalmology consultation revealed a choroidal hemangioma and congenital glaucoma in the left eye.
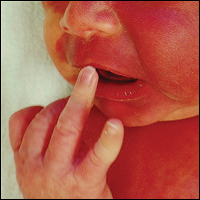
Cyanosis of the Foot
The Diagnosis: Antiphospholipid Antibody Syndrome
A biopsy demonstrated scattered intravascular thrombi in the dermis and subcutis, intact vascular walls, and scant lymphocytic inflammation in a background of stasis (Figure 1). A periodic acid-Schiff stain was negative for fungal elements and highlighted the intravascular thrombi. Histologic findings were consistent with thrombotic vasculopathy. On further laboratory workup, lupus anticoagulant studies, including a mixing study, diluted Russell viper venom test, and hexagonal phase phospholipid neutralization test, were abnormal. Titers of anticardiolipin and β2-glycoprotein I antibodies were elevated (anticardiolipin IgG, 137.7 calculated units [normal, <15 calculated units]; β2-glycoprotein I IgG, 256.4 calculated units [normal, <20 calculated units]). Tissue cultures showed no growth of microorganisms and studies for cryoglobulinemia were negative.

The patient was diagnosed with primary antiphospholipid syndrome (APS). He remained on anticoagulation therapy with fondaparinux as an inpatient and was treated with pulse-dose intravenous (IV) corticosteroids followed by a slow oral taper, daily plasmapheresis for 1 week, IV immunoglobulin (0.5 g/kg) for 3 doses, and 4 weekly doses of rituximab (375 mg/m2). His cutaneous findings slowly improved over the next several weeks (Figure 2).
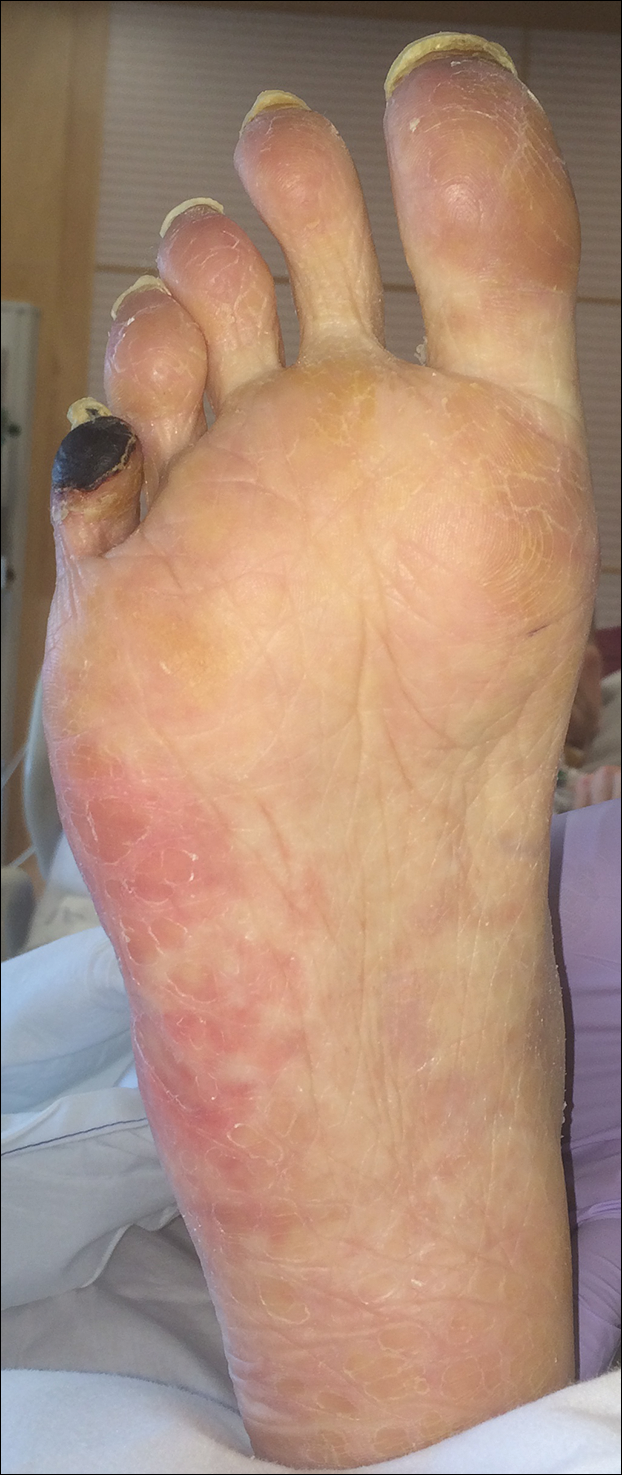
Antiphospholipid syndrome is an autoimmune disorder characterized by thrombotic events and the presence of autoantibodies. The syndrome is defined by 2 major criteria: (1) the occurrence of at least 1 clinical feature of either an episode of vascular thrombosis or pregnancy morbidity such as unexplained fetal death beyond 10 weeks of gestation or recurrent unexplained pregnancy losses; and (2) the presence of at least 1 type of autoantibody, including lupus anticoagulant, anticardiolipin, or β2-glycoprotein antibodies, on 2 separate occasions at least 12 weeks apart.1 Antiphospholipid syndrome can either be primary with no identifiable associated rheumatologic disease or secondary to another autoimmune disease such as systemic lupus erythematosus. Cutaneous manifestations are common and frequently are the first sign of disease in 30% to 40% of patients.2 The most common skin finding is persistent livedo reticularis, which can be seen in 20% to 25% of patients. Patients also may develop skin necrosis, ulcerations, digital gangrene, splinter hemorrhages, and livedoid vasculopathy.2 Systemic manifestations of APS include thrombocytopenia, nephropathy, cognitive dysfunction, and cardiac valve abnormalities.
The exact pathogenesis of APS remains unknown. It is thought to be due to the combination of an inflammatory stimulus that has yet to be characterized in conjunction with autoantibodies that affect multiple target cells including monocytes, platelets, and endothelial cells, which results in activation of the complement system and clotting cascade.3 In rare cases, the disorder can progress to catastrophic antiphospholipid syndrome (CAPS), which requires fulfillment of 4 criteria: (1) evidence of involvement of 3 organs, tissues, or systems; (2) development of manifestations simultaneously or in less than 1 week; (3) laboratory confirmation of the presence of antiphospholipid antibodies; and (4) confirmation by histopathology of small vessel occlusion.4 Probable CAPS is diagnosed when 3 of 4 criteria are present. Our patient met criteria for probable CAPS, as his antibody titers remained elevated 15 weeks after initial presentation. Precipitating factors that can lead to CAPS are thought to include infection, surgical procedures, medications, or discontinuation of anticoagulation drugs.2 Although the mainstay of management of APS is anticoagulation therapy with warfarin and antiplatelet agents such as aspirin, first-line treatment of CAPS involves high-dose systemic glucocorticoids and plasma exchange. Intravenous immunoglobulin also may be employed in treatment. Data from the CAPS registry demonstrate a role for rituximab, an anti-CD20 antibody, at 375 mg/m2 weekly for 4 weeks (the regimen described in our case) or 1 g every 14 days for 2 sessions.5 A majority of the registry patients treated with rituximab recovered (75% [15/20]) and had no recurrent thrombosis (87% [13/15]) at follow-up.5 Data also are emerging on the role of eculizumab, an anti-C5 antibody that inhibits the terminal complement cascade, as a therapy in difficult-to-treat or refractory CAPS.6-8 The prognosis for CAPS patients without treatment is poor, and mortality has been reported in up to 44% of patients. However, with intervention mortality is reduced by more than 2-fold.9,10
It is important to recognize that acral cyanosis with persistent livedo reticularis and digital gangrene can be a presenting manifestation of APS. These cutaneous manifestations should prompt histologic evaluation for thrombotic vasculopathy in addition to serologic tests for APS autoantibodies. Although APS may be treated with anticoagulants and antiplatelet agents, CAPS may require more aggressive therapy with systemic steroids, plasma exchange, IV immunoglobulin, rituximab, and/or eculizumab.
- Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309-1311.
- Pinto-Almeida T, Caetano M, Sanches M, et al. Cutaneous manifestations of antiphospholipid syndrome: a review of the clinical features, diagnosis and management. Acta Reumatol Port. 2013;38:10-18.
- Meroni PL, Chighizola CB, Rovelli F, et al. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16:209.
- Asherson RA, Cervera R, de Grott PG, et al; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12:530-534.
- Berman H, Rodríguez-Pintó I, Cervera R, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab [published online June 15, 2013]. Autoimmun Rev. 2013;12:1085-1090.
- Shapira I, Andrade D, Allen SL, et al. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64:2719-2723.
- Strakhan M, Hurtado-Sbordoni M, Galeas N, et al. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. 2014;2014:704371.
- Lonze BE, Zachary AA, Magro CM, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459-465.
- Bucciarelli S, Espinosa G, Cervera R, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54:2568-2576.
- Asherson RA, Cervera R, Piette JC, et al. Catastrophic antiphospholipid syndrome. clinical and laboratory features of 50 patients. Medicine (Baltimore). 1998;77:195-207.
The Diagnosis: Antiphospholipid Antibody Syndrome
A biopsy demonstrated scattered intravascular thrombi in the dermis and subcutis, intact vascular walls, and scant lymphocytic inflammation in a background of stasis (Figure 1). A periodic acid-Schiff stain was negative for fungal elements and highlighted the intravascular thrombi. Histologic findings were consistent with thrombotic vasculopathy. On further laboratory workup, lupus anticoagulant studies, including a mixing study, diluted Russell viper venom test, and hexagonal phase phospholipid neutralization test, were abnormal. Titers of anticardiolipin and β2-glycoprotein I antibodies were elevated (anticardiolipin IgG, 137.7 calculated units [normal, <15 calculated units]; β2-glycoprotein I IgG, 256.4 calculated units [normal, <20 calculated units]). Tissue cultures showed no growth of microorganisms and studies for cryoglobulinemia were negative.
The patient was diagnosed with primary antiphospholipid syndrome (APS). He remained on anticoagulation therapy with fondaparinux as an inpatient and was treated with pulse-dose intravenous (IV) corticosteroids followed by a slow oral taper, daily plasmapheresis for 1 week, IV immunoglobulin (0.5 g/kg) for 3 doses, and 4 weekly doses of rituximab (375 mg/m2). His cutaneous findings slowly improved over the next several weeks (Figure 2).
Antiphospholipid syndrome is an autoimmune disorder characterized by thrombotic events and the presence of autoantibodies. The syndrome is defined by 2 major criteria: (1) the occurrence of at least 1 clinical feature of either an episode of vascular thrombosis or pregnancy morbidity such as unexplained fetal death beyond 10 weeks of gestation or recurrent unexplained pregnancy losses; and (2) the presence of at least 1 type of autoantibody, including lupus anticoagulant, anticardiolipin, or β2-glycoprotein antibodies, on 2 separate occasions at least 12 weeks apart.1 Antiphospholipid syndrome can either be primary with no identifiable associated rheumatologic disease or secondary to another autoimmune disease such as systemic lupus erythematosus. Cutaneous manifestations are common and frequently are the first sign of disease in 30% to 40% of patients.2 The most common skin finding is persistent livedo reticularis, which can be seen in 20% to 25% of patients. Patients also may develop skin necrosis, ulcerations, digital gangrene, splinter hemorrhages, and livedoid vasculopathy.2 Systemic manifestations of APS include thrombocytopenia, nephropathy, cognitive dysfunction, and cardiac valve abnormalities.
The exact pathogenesis of APS remains unknown. It is thought to be due to the combination of an inflammatory stimulus that has yet to be characterized in conjunction with autoantibodies that affect multiple target cells including monocytes, platelets, and endothelial cells, which results in activation of the complement system and clotting cascade.3 In rare cases, the disorder can progress to catastrophic antiphospholipid syndrome (CAPS), which requires fulfillment of 4 criteria: (1) evidence of involvement of 3 organs, tissues, or systems; (2) development of manifestations simultaneously or in less than 1 week; (3) laboratory confirmation of the presence of antiphospholipid antibodies; and (4) confirmation by histopathology of small vessel occlusion.4 Probable CAPS is diagnosed when 3 of 4 criteria are present. Our patient met criteria for probable CAPS, as his antibody titers remained elevated 15 weeks after initial presentation. Precipitating factors that can lead to CAPS are thought to include infection, surgical procedures, medications, or discontinuation of anticoagulation drugs.2 Although the mainstay of management of APS is anticoagulation therapy with warfarin and antiplatelet agents such as aspirin, first-line treatment of CAPS involves high-dose systemic glucocorticoids and plasma exchange. Intravenous immunoglobulin also may be employed in treatment. Data from the CAPS registry demonstrate a role for rituximab, an anti-CD20 antibody, at 375 mg/m2 weekly for 4 weeks (the regimen described in our case) or 1 g every 14 days for 2 sessions.5 A majority of the registry patients treated with rituximab recovered (75% [15/20]) and had no recurrent thrombosis (87% [13/15]) at follow-up.5 Data also are emerging on the role of eculizumab, an anti-C5 antibody that inhibits the terminal complement cascade, as a therapy in difficult-to-treat or refractory CAPS.6-8 The prognosis for CAPS patients without treatment is poor, and mortality has been reported in up to 44% of patients. However, with intervention mortality is reduced by more than 2-fold.9,10
It is important to recognize that acral cyanosis with persistent livedo reticularis and digital gangrene can be a presenting manifestation of APS. These cutaneous manifestations should prompt histologic evaluation for thrombotic vasculopathy in addition to serologic tests for APS autoantibodies. Although APS may be treated with anticoagulants and antiplatelet agents, CAPS may require more aggressive therapy with systemic steroids, plasma exchange, IV immunoglobulin, rituximab, and/or eculizumab.
The Diagnosis: Antiphospholipid Antibody Syndrome
A biopsy demonstrated scattered intravascular thrombi in the dermis and subcutis, intact vascular walls, and scant lymphocytic inflammation in a background of stasis (Figure 1). A periodic acid-Schiff stain was negative for fungal elements and highlighted the intravascular thrombi. Histologic findings were consistent with thrombotic vasculopathy. On further laboratory workup, lupus anticoagulant studies, including a mixing study, diluted Russell viper venom test, and hexagonal phase phospholipid neutralization test, were abnormal. Titers of anticardiolipin and β2-glycoprotein I antibodies were elevated (anticardiolipin IgG, 137.7 calculated units [normal, <15 calculated units]; β2-glycoprotein I IgG, 256.4 calculated units [normal, <20 calculated units]). Tissue cultures showed no growth of microorganisms and studies for cryoglobulinemia were negative.
The patient was diagnosed with primary antiphospholipid syndrome (APS). He remained on anticoagulation therapy with fondaparinux as an inpatient and was treated with pulse-dose intravenous (IV) corticosteroids followed by a slow oral taper, daily plasmapheresis for 1 week, IV immunoglobulin (0.5 g/kg) for 3 doses, and 4 weekly doses of rituximab (375 mg/m2). His cutaneous findings slowly improved over the next several weeks (Figure 2).
Antiphospholipid syndrome is an autoimmune disorder characterized by thrombotic events and the presence of autoantibodies. The syndrome is defined by 2 major criteria: (1) the occurrence of at least 1 clinical feature of either an episode of vascular thrombosis or pregnancy morbidity such as unexplained fetal death beyond 10 weeks of gestation or recurrent unexplained pregnancy losses; and (2) the presence of at least 1 type of autoantibody, including lupus anticoagulant, anticardiolipin, or β2-glycoprotein antibodies, on 2 separate occasions at least 12 weeks apart.1 Antiphospholipid syndrome can either be primary with no identifiable associated rheumatologic disease or secondary to another autoimmune disease such as systemic lupus erythematosus. Cutaneous manifestations are common and frequently are the first sign of disease in 30% to 40% of patients.2 The most common skin finding is persistent livedo reticularis, which can be seen in 20% to 25% of patients. Patients also may develop skin necrosis, ulcerations, digital gangrene, splinter hemorrhages, and livedoid vasculopathy.2 Systemic manifestations of APS include thrombocytopenia, nephropathy, cognitive dysfunction, and cardiac valve abnormalities.
The exact pathogenesis of APS remains unknown. It is thought to be due to the combination of an inflammatory stimulus that has yet to be characterized in conjunction with autoantibodies that affect multiple target cells including monocytes, platelets, and endothelial cells, which results in activation of the complement system and clotting cascade.3 In rare cases, the disorder can progress to catastrophic antiphospholipid syndrome (CAPS), which requires fulfillment of 4 criteria: (1) evidence of involvement of 3 organs, tissues, or systems; (2) development of manifestations simultaneously or in less than 1 week; (3) laboratory confirmation of the presence of antiphospholipid antibodies; and (4) confirmation by histopathology of small vessel occlusion.4 Probable CAPS is diagnosed when 3 of 4 criteria are present. Our patient met criteria for probable CAPS, as his antibody titers remained elevated 15 weeks after initial presentation. Precipitating factors that can lead to CAPS are thought to include infection, surgical procedures, medications, or discontinuation of anticoagulation drugs.2 Although the mainstay of management of APS is anticoagulation therapy with warfarin and antiplatelet agents such as aspirin, first-line treatment of CAPS involves high-dose systemic glucocorticoids and plasma exchange. Intravenous immunoglobulin also may be employed in treatment. Data from the CAPS registry demonstrate a role for rituximab, an anti-CD20 antibody, at 375 mg/m2 weekly for 4 weeks (the regimen described in our case) or 1 g every 14 days for 2 sessions.5 A majority of the registry patients treated with rituximab recovered (75% [15/20]) and had no recurrent thrombosis (87% [13/15]) at follow-up.5 Data also are emerging on the role of eculizumab, an anti-C5 antibody that inhibits the terminal complement cascade, as a therapy in difficult-to-treat or refractory CAPS.6-8 The prognosis for CAPS patients without treatment is poor, and mortality has been reported in up to 44% of patients. However, with intervention mortality is reduced by more than 2-fold.9,10
It is important to recognize that acral cyanosis with persistent livedo reticularis and digital gangrene can be a presenting manifestation of APS. These cutaneous manifestations should prompt histologic evaluation for thrombotic vasculopathy in addition to serologic tests for APS autoantibodies. Although APS may be treated with anticoagulants and antiplatelet agents, CAPS may require more aggressive therapy with systemic steroids, plasma exchange, IV immunoglobulin, rituximab, and/or eculizumab.
- Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309-1311.
- Pinto-Almeida T, Caetano M, Sanches M, et al. Cutaneous manifestations of antiphospholipid syndrome: a review of the clinical features, diagnosis and management. Acta Reumatol Port. 2013;38:10-18.
- Meroni PL, Chighizola CB, Rovelli F, et al. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16:209.
- Asherson RA, Cervera R, de Grott PG, et al; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12:530-534.
- Berman H, Rodríguez-Pintó I, Cervera R, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab [published online June 15, 2013]. Autoimmun Rev. 2013;12:1085-1090.
- Shapira I, Andrade D, Allen SL, et al. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64:2719-2723.
- Strakhan M, Hurtado-Sbordoni M, Galeas N, et al. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. 2014;2014:704371.
- Lonze BE, Zachary AA, Magro CM, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459-465.
- Bucciarelli S, Espinosa G, Cervera R, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54:2568-2576.
- Asherson RA, Cervera R, Piette JC, et al. Catastrophic antiphospholipid syndrome. clinical and laboratory features of 50 patients. Medicine (Baltimore). 1998;77:195-207.
- Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309-1311.
- Pinto-Almeida T, Caetano M, Sanches M, et al. Cutaneous manifestations of antiphospholipid syndrome: a review of the clinical features, diagnosis and management. Acta Reumatol Port. 2013;38:10-18.
- Meroni PL, Chighizola CB, Rovelli F, et al. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16:209.
- Asherson RA, Cervera R, de Grott PG, et al; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12:530-534.
- Berman H, Rodríguez-Pintó I, Cervera R, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab [published online June 15, 2013]. Autoimmun Rev. 2013;12:1085-1090.
- Shapira I, Andrade D, Allen SL, et al. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64:2719-2723.
- Strakhan M, Hurtado-Sbordoni M, Galeas N, et al. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. 2014;2014:704371.
- Lonze BE, Zachary AA, Magro CM, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459-465.
- Bucciarelli S, Espinosa G, Cervera R, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54:2568-2576.
- Asherson RA, Cervera R, Piette JC, et al. Catastrophic antiphospholipid syndrome. clinical and laboratory features of 50 patients. Medicine (Baltimore). 1998;77:195-207.
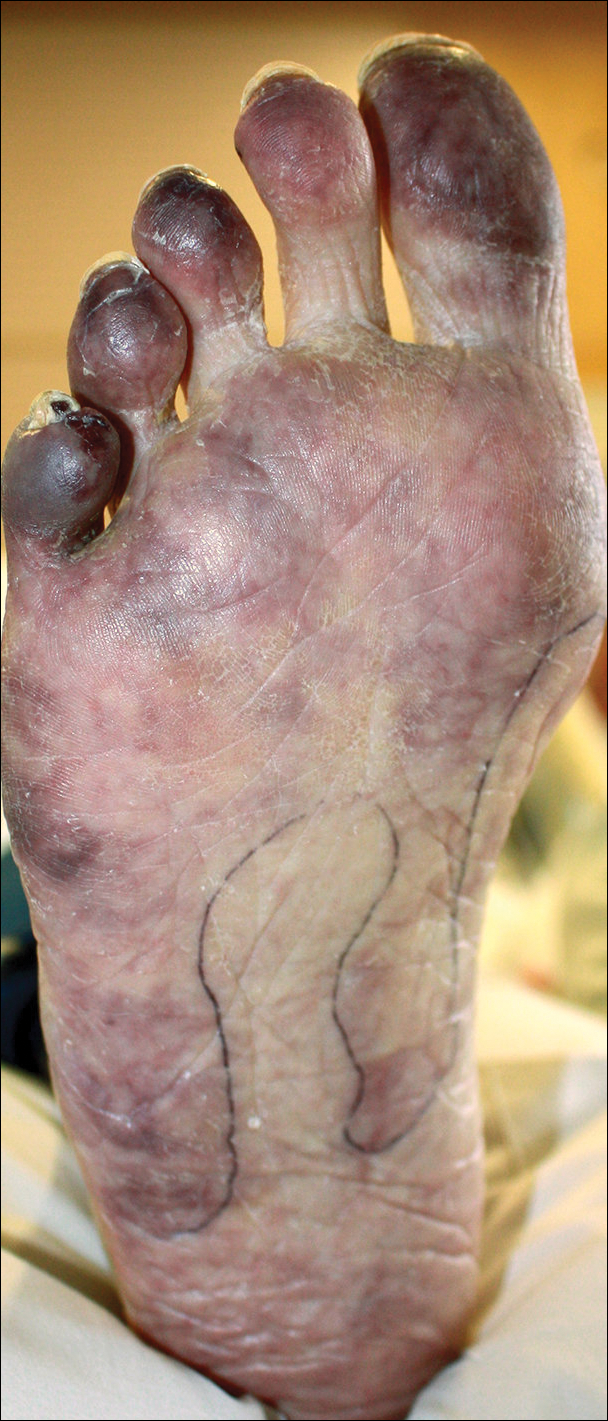
A man in his 50s with a medical history of arterial thrombosis of the right arm, multiple deep vein thromboses (DVTs) of the legs on long-term warfarin, ischemic stroke, atrial fibrillation, and peripheral arterial disease presented with discoloration of the right foot and increasing tenderness of 1 month's duration. There was no history of trauma or recent change in outpatient medications. A family history was notable for an aunt and 2 cousins with DVTs and protein S deficiency. Physical examination revealed livedo reticularis on the sole and lateral aspect of the right foot. There was violaceous discoloration of the volar aspects of all 5 toes and a focal area of ulceration on the fifth toe. Pulses were palpable bilaterally. Initial laboratory evaluation was notable for thrombocytopenia, and preliminary blood cultures revealed no growth of bacterial or fungal organisms. Imaging studies revealed increased arterial stenosis of the right leg as well as DVT of the right great saphenous vein. A punch biopsy of the right medial foot was performed for hematoxylin and eosin stain as well as tissue culture.
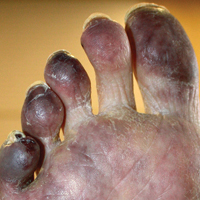
Recalcitrant Ulcer on the Lower Leg
The Diagnosis: Nonuremic Calciphylaxis
Histopathologic findings revealed ischemic necrosis and a subepidermal blister (Figure 1) with arteriosclerotic changes and fat necrosis. Foci of calcification were noted within the fat lobules. Arterioles within the deeper dermis and subcutis showed thickened hyalinized walls, narrowed lumina, and medial calcification (Figure 2). Multiple sections did not reveal any granulomatous inflammation. Periodic acid-Schiff and Gram stains were negative for fungal and bacterial elements, respectively. No dense neutrophilic infiltrate was seen. Multifocal calcific deposits within fat lobules and vessel walls (endothelium highlighted by the CD31 stain) suggested calciphylaxis.
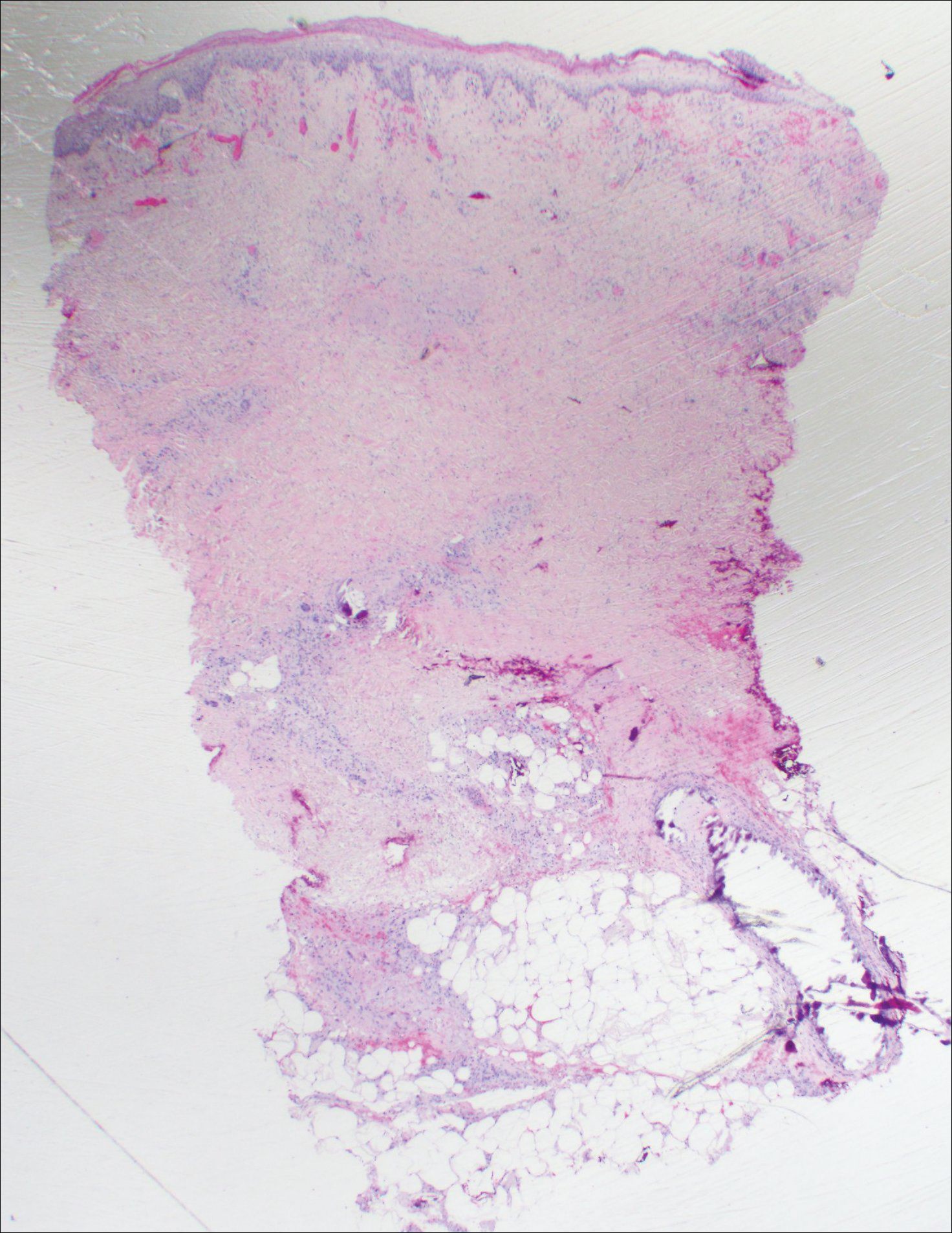
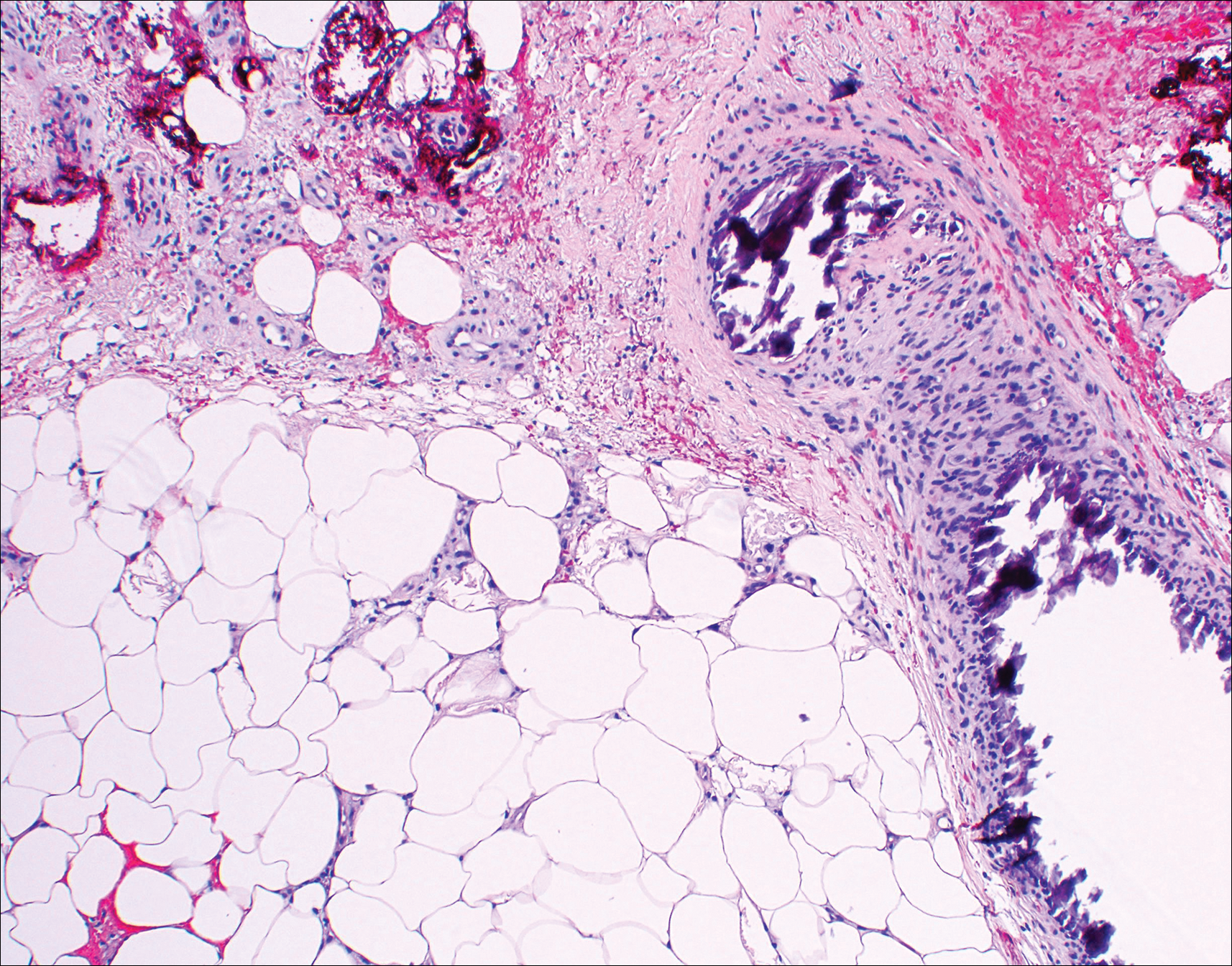
Laboratory test results revealed a normal white blood cell count, international normalized ratio level of 4 (on warfarin), and an elevated sedimentation rate at 72 mm/h (reference range, 0-20 mm/h). Serum creatinine was 1.1 mg/dL (reference range, 0.6-1.2 mg/dL) and the calcium-phosphorous product was 40.8 mg2/dL (reference range, <55 mg2/dL). Hemoglobin A1C (glycated hemoglobin) was 8.2% (reference range, 4%-7%). Wound cultures grew Proteus mirabilis sensitive to cefazolin. Acid-fast bacilli and fungal cultures were negative. Computed tomography of the left lower leg without contrast showed no evidence of osteomyelitis. Of note, the popliteal arteries and distal vessels showed moderate vascular calcification.
Histopathology findings as well as a clinical picture of painful ulceration on the distal extremities and uncontrolled diabetes with normal renal function favored a diagnosis of nonuremic calciphylaxis (NUC). The patient was treated with intravenous infusions of sodium thiosulfate 25 mg 3 times weekly and oral cefazolin for superadded bacterial infection. Local wound care included collagenase dressings with light compression. Warfarin was discontinued, as it can worsen calciphylaxis. Complete reepithelialization of the ulcer along with substantial reduction in pain was noted within 4 weeks.
Ulceration of the lower legs is a relatively common condition in the Western world, the prevalence of which increases up to 5% in patients older than 65 years.1 Of the myriad of causes that lead to ulceration of the distal aspect of the leg, NUC is a rare but known phenomenon. The pathogenesis of NUC is complicated based on theories of derangement of receptor activator of nuclear factor κβ, receptor activator of nuclear factor κβ ligand, and osteoprotegerin, leading to calcium deposits in the media of the arteries.2 This deposition precipitates vascular occlusion coupled with ischemic necrosis of the subcutaneous tissue and skin.3 Some of the more common causes of NUC are primary hyperparathyroidism, malignancy, and rheumatoid arthritis. Type 2 diabetes mellitus is a less common cause but often is found in association with NUC, as noted by Nigwekar et al.2 According to their study, the laboratory parameters commonly found in NUC included a calcium-phosphorous product greater than 50 mg2/dL and serum creatinine of 1.2 mg/dL or less.2
Our patient displayed these laboratory findings. However, distinguishing NUC from other atypical lower extremity ulcers such as Martorell hypertensive ischemic ulcer, pyoderma gangrenosum, and warfarin necrosis can pose a challenge to the dermatologist. Martorell hypertensive ischemic ulcer is excruciatingly painful and occurs more frequently near the Achilles tendon, responding well to surgical debridement. Histopathologically, medial calcinosis and arteriosclerosis are seen.4
Pyoderma gangrenosum is a neutrophilic dermatosis wherein the classical ulcerative variant is painful. It occurs mostly on the pretibial area and worsens after debridement.5 Clinically and histopathologically, it is a diagnosis of exclusion in which a dense neutrophilic to mixed lymphocytic infiltrate is seen with necrosis of dermal vessels.6
Warfarin necrosis is extremely rare, affecting 0.01% to 0.1% of patients on warfarin-derived anticoagulant therapy.7 Necrosis occurs mostly on fat-bearing areas such as the breasts, abdomen, and thighs 3 to 5 days after initiating treatment. Histologically, fibrin deposits occlude dermal vessels without perivascular inflammation.8
Necrobiosis lipoidica is a rare cutaneous entity seen in 0.3% of diabetic patients.9 The exact pathogenesis is unknown; however, microangiopathy in collaboration with cross-linking of abnormal collagen fibers play a role. These lesions appear as erythematous plaques with a slightly depressed to atrophic center, ultimately taking on a waxy porcelain appearance. Although most of these lesions either resolve or become chronically persistent, approximately 15% undergo ulceration, which can be painful. Histologically, with hematoxylin and eosin staining, areas of necrobiosis are seen surrounded by an inflammatory infiltrate comprised mainly of histiocytes along with lymphocytes and plasma cells.9
Nonuremic calciphylaxis can mimic the aforementioned conditions to a greater extent in female patients with obesity, diabetes mellitus, and hypertension. However, microscopic calcium deposition in the media of dermal arterioles, extravascular calcification within fat lobules, and cutaneous necrosis, along with remarkable response to intravenous sodium thiosulfate, confirmed a diagnosis of NUC in our patient. Sodium thiosulfate scavenges reactive oxygen species and promotes nitric oxygen generation, thereby reducing endothelial damage.10 Although there are no randomized controlled trials to support its use, sodium thiosulfate has been successfully used to treat established cases of NUC.11
- Spentzouris G, Labropoulos N. The evaluation of lower-extremity ulcers. Semin Intervent Radiol. 2009;26:286-295.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Bardin T. Musculoskeletal manifestations of chronic renal failure. Curr Opin Rheumatol. 2003;15:48-54.
- Hafner J, Nobbe S, Partsch H, et al. Martorell hypertensive ischemic leg ulcer: a model of ischemic subcutaneous arteriolosclerosis. Arch Dermatol. 2010;146:961-968.
- Sedda S, Caruso R, Marafini I, et al. Pyoderma gangrenosum in refractory celiac disease: a case report. BMC Gastroenterol. 2013;13:162.
- Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Breakey W, Hall C, Vann Jones S, et al. Warfarin-induced skin necrosis progressing to calciphylaxis. J Plast Reconstr Aesthet Surg. 2014;67:244-246.
- Kakagia DD, Papanas N, Karadimas E, et al. Warfarin-induced skin necrosis. Ann Dermatol. 2014;26:96-98.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23:258-262.
- Ning MS, Dahir KM, Castellanos EH, et al. Sodium thiosulfate in the treatment of non-uremic calciphylaxis. J Dermatol. 2013;40:649-652.
The Diagnosis: Nonuremic Calciphylaxis
Histopathologic findings revealed ischemic necrosis and a subepidermal blister (Figure 1) with arteriosclerotic changes and fat necrosis. Foci of calcification were noted within the fat lobules. Arterioles within the deeper dermis and subcutis showed thickened hyalinized walls, narrowed lumina, and medial calcification (Figure 2). Multiple sections did not reveal any granulomatous inflammation. Periodic acid-Schiff and Gram stains were negative for fungal and bacterial elements, respectively. No dense neutrophilic infiltrate was seen. Multifocal calcific deposits within fat lobules and vessel walls (endothelium highlighted by the CD31 stain) suggested calciphylaxis.
Laboratory test results revealed a normal white blood cell count, international normalized ratio level of 4 (on warfarin), and an elevated sedimentation rate at 72 mm/h (reference range, 0-20 mm/h). Serum creatinine was 1.1 mg/dL (reference range, 0.6-1.2 mg/dL) and the calcium-phosphorous product was 40.8 mg2/dL (reference range, <55 mg2/dL). Hemoglobin A1C (glycated hemoglobin) was 8.2% (reference range, 4%-7%). Wound cultures grew Proteus mirabilis sensitive to cefazolin. Acid-fast bacilli and fungal cultures were negative. Computed tomography of the left lower leg without contrast showed no evidence of osteomyelitis. Of note, the popliteal arteries and distal vessels showed moderate vascular calcification.
Histopathology findings as well as a clinical picture of painful ulceration on the distal extremities and uncontrolled diabetes with normal renal function favored a diagnosis of nonuremic calciphylaxis (NUC). The patient was treated with intravenous infusions of sodium thiosulfate 25 mg 3 times weekly and oral cefazolin for superadded bacterial infection. Local wound care included collagenase dressings with light compression. Warfarin was discontinued, as it can worsen calciphylaxis. Complete reepithelialization of the ulcer along with substantial reduction in pain was noted within 4 weeks.
Ulceration of the lower legs is a relatively common condition in the Western world, the prevalence of which increases up to 5% in patients older than 65 years.1 Of the myriad of causes that lead to ulceration of the distal aspect of the leg, NUC is a rare but known phenomenon. The pathogenesis of NUC is complicated based on theories of derangement of receptor activator of nuclear factor κβ, receptor activator of nuclear factor κβ ligand, and osteoprotegerin, leading to calcium deposits in the media of the arteries.2 This deposition precipitates vascular occlusion coupled with ischemic necrosis of the subcutaneous tissue and skin.3 Some of the more common causes of NUC are primary hyperparathyroidism, malignancy, and rheumatoid arthritis. Type 2 diabetes mellitus is a less common cause but often is found in association with NUC, as noted by Nigwekar et al.2 According to their study, the laboratory parameters commonly found in NUC included a calcium-phosphorous product greater than 50 mg2/dL and serum creatinine of 1.2 mg/dL or less.2
Our patient displayed these laboratory findings. However, distinguishing NUC from other atypical lower extremity ulcers such as Martorell hypertensive ischemic ulcer, pyoderma gangrenosum, and warfarin necrosis can pose a challenge to the dermatologist. Martorell hypertensive ischemic ulcer is excruciatingly painful and occurs more frequently near the Achilles tendon, responding well to surgical debridement. Histopathologically, medial calcinosis and arteriosclerosis are seen.4
Pyoderma gangrenosum is a neutrophilic dermatosis wherein the classical ulcerative variant is painful. It occurs mostly on the pretibial area and worsens after debridement.5 Clinically and histopathologically, it is a diagnosis of exclusion in which a dense neutrophilic to mixed lymphocytic infiltrate is seen with necrosis of dermal vessels.6
Warfarin necrosis is extremely rare, affecting 0.01% to 0.1% of patients on warfarin-derived anticoagulant therapy.7 Necrosis occurs mostly on fat-bearing areas such as the breasts, abdomen, and thighs 3 to 5 days after initiating treatment. Histologically, fibrin deposits occlude dermal vessels without perivascular inflammation.8
Necrobiosis lipoidica is a rare cutaneous entity seen in 0.3% of diabetic patients.9 The exact pathogenesis is unknown; however, microangiopathy in collaboration with cross-linking of abnormal collagen fibers play a role. These lesions appear as erythematous plaques with a slightly depressed to atrophic center, ultimately taking on a waxy porcelain appearance. Although most of these lesions either resolve or become chronically persistent, approximately 15% undergo ulceration, which can be painful. Histologically, with hematoxylin and eosin staining, areas of necrobiosis are seen surrounded by an inflammatory infiltrate comprised mainly of histiocytes along with lymphocytes and plasma cells.9
Nonuremic calciphylaxis can mimic the aforementioned conditions to a greater extent in female patients with obesity, diabetes mellitus, and hypertension. However, microscopic calcium deposition in the media of dermal arterioles, extravascular calcification within fat lobules, and cutaneous necrosis, along with remarkable response to intravenous sodium thiosulfate, confirmed a diagnosis of NUC in our patient. Sodium thiosulfate scavenges reactive oxygen species and promotes nitric oxygen generation, thereby reducing endothelial damage.10 Although there are no randomized controlled trials to support its use, sodium thiosulfate has been successfully used to treat established cases of NUC.11
The Diagnosis: Nonuremic Calciphylaxis
Histopathologic findings revealed ischemic necrosis and a subepidermal blister (Figure 1) with arteriosclerotic changes and fat necrosis. Foci of calcification were noted within the fat lobules. Arterioles within the deeper dermis and subcutis showed thickened hyalinized walls, narrowed lumina, and medial calcification (Figure 2). Multiple sections did not reveal any granulomatous inflammation. Periodic acid-Schiff and Gram stains were negative for fungal and bacterial elements, respectively. No dense neutrophilic infiltrate was seen. Multifocal calcific deposits within fat lobules and vessel walls (endothelium highlighted by the CD31 stain) suggested calciphylaxis.
Laboratory test results revealed a normal white blood cell count, international normalized ratio level of 4 (on warfarin), and an elevated sedimentation rate at 72 mm/h (reference range, 0-20 mm/h). Serum creatinine was 1.1 mg/dL (reference range, 0.6-1.2 mg/dL) and the calcium-phosphorous product was 40.8 mg2/dL (reference range, <55 mg2/dL). Hemoglobin A1C (glycated hemoglobin) was 8.2% (reference range, 4%-7%). Wound cultures grew Proteus mirabilis sensitive to cefazolin. Acid-fast bacilli and fungal cultures were negative. Computed tomography of the left lower leg without contrast showed no evidence of osteomyelitis. Of note, the popliteal arteries and distal vessels showed moderate vascular calcification.
Histopathology findings as well as a clinical picture of painful ulceration on the distal extremities and uncontrolled diabetes with normal renal function favored a diagnosis of nonuremic calciphylaxis (NUC). The patient was treated with intravenous infusions of sodium thiosulfate 25 mg 3 times weekly and oral cefazolin for superadded bacterial infection. Local wound care included collagenase dressings with light compression. Warfarin was discontinued, as it can worsen calciphylaxis. Complete reepithelialization of the ulcer along with substantial reduction in pain was noted within 4 weeks.
Ulceration of the lower legs is a relatively common condition in the Western world, the prevalence of which increases up to 5% in patients older than 65 years.1 Of the myriad of causes that lead to ulceration of the distal aspect of the leg, NUC is a rare but known phenomenon. The pathogenesis of NUC is complicated based on theories of derangement of receptor activator of nuclear factor κβ, receptor activator of nuclear factor κβ ligand, and osteoprotegerin, leading to calcium deposits in the media of the arteries.2 This deposition precipitates vascular occlusion coupled with ischemic necrosis of the subcutaneous tissue and skin.3 Some of the more common causes of NUC are primary hyperparathyroidism, malignancy, and rheumatoid arthritis. Type 2 diabetes mellitus is a less common cause but often is found in association with NUC, as noted by Nigwekar et al.2 According to their study, the laboratory parameters commonly found in NUC included a calcium-phosphorous product greater than 50 mg2/dL and serum creatinine of 1.2 mg/dL or less.2
Our patient displayed these laboratory findings. However, distinguishing NUC from other atypical lower extremity ulcers such as Martorell hypertensive ischemic ulcer, pyoderma gangrenosum, and warfarin necrosis can pose a challenge to the dermatologist. Martorell hypertensive ischemic ulcer is excruciatingly painful and occurs more frequently near the Achilles tendon, responding well to surgical debridement. Histopathologically, medial calcinosis and arteriosclerosis are seen.4
Pyoderma gangrenosum is a neutrophilic dermatosis wherein the classical ulcerative variant is painful. It occurs mostly on the pretibial area and worsens after debridement.5 Clinically and histopathologically, it is a diagnosis of exclusion in which a dense neutrophilic to mixed lymphocytic infiltrate is seen with necrosis of dermal vessels.6
Warfarin necrosis is extremely rare, affecting 0.01% to 0.1% of patients on warfarin-derived anticoagulant therapy.7 Necrosis occurs mostly on fat-bearing areas such as the breasts, abdomen, and thighs 3 to 5 days after initiating treatment. Histologically, fibrin deposits occlude dermal vessels without perivascular inflammation.8
Necrobiosis lipoidica is a rare cutaneous entity seen in 0.3% of diabetic patients.9 The exact pathogenesis is unknown; however, microangiopathy in collaboration with cross-linking of abnormal collagen fibers play a role. These lesions appear as erythematous plaques with a slightly depressed to atrophic center, ultimately taking on a waxy porcelain appearance. Although most of these lesions either resolve or become chronically persistent, approximately 15% undergo ulceration, which can be painful. Histologically, with hematoxylin and eosin staining, areas of necrobiosis are seen surrounded by an inflammatory infiltrate comprised mainly of histiocytes along with lymphocytes and plasma cells.9
Nonuremic calciphylaxis can mimic the aforementioned conditions to a greater extent in female patients with obesity, diabetes mellitus, and hypertension. However, microscopic calcium deposition in the media of dermal arterioles, extravascular calcification within fat lobules, and cutaneous necrosis, along with remarkable response to intravenous sodium thiosulfate, confirmed a diagnosis of NUC in our patient. Sodium thiosulfate scavenges reactive oxygen species and promotes nitric oxygen generation, thereby reducing endothelial damage.10 Although there are no randomized controlled trials to support its use, sodium thiosulfate has been successfully used to treat established cases of NUC.11
- Spentzouris G, Labropoulos N. The evaluation of lower-extremity ulcers. Semin Intervent Radiol. 2009;26:286-295.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Bardin T. Musculoskeletal manifestations of chronic renal failure. Curr Opin Rheumatol. 2003;15:48-54.
- Hafner J, Nobbe S, Partsch H, et al. Martorell hypertensive ischemic leg ulcer: a model of ischemic subcutaneous arteriolosclerosis. Arch Dermatol. 2010;146:961-968.
- Sedda S, Caruso R, Marafini I, et al. Pyoderma gangrenosum in refractory celiac disease: a case report. BMC Gastroenterol. 2013;13:162.
- Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Breakey W, Hall C, Vann Jones S, et al. Warfarin-induced skin necrosis progressing to calciphylaxis. J Plast Reconstr Aesthet Surg. 2014;67:244-246.
- Kakagia DD, Papanas N, Karadimas E, et al. Warfarin-induced skin necrosis. Ann Dermatol. 2014;26:96-98.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23:258-262.
- Ning MS, Dahir KM, Castellanos EH, et al. Sodium thiosulfate in the treatment of non-uremic calciphylaxis. J Dermatol. 2013;40:649-652.
- Spentzouris G, Labropoulos N. The evaluation of lower-extremity ulcers. Semin Intervent Radiol. 2009;26:286-295.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Bardin T. Musculoskeletal manifestations of chronic renal failure. Curr Opin Rheumatol. 2003;15:48-54.
- Hafner J, Nobbe S, Partsch H, et al. Martorell hypertensive ischemic leg ulcer: a model of ischemic subcutaneous arteriolosclerosis. Arch Dermatol. 2010;146:961-968.
- Sedda S, Caruso R, Marafini I, et al. Pyoderma gangrenosum in refractory celiac disease: a case report. BMC Gastroenterol. 2013;13:162.
- Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Breakey W, Hall C, Vann Jones S, et al. Warfarin-induced skin necrosis progressing to calciphylaxis. J Plast Reconstr Aesthet Surg. 2014;67:244-246.
- Kakagia DD, Papanas N, Karadimas E, et al. Warfarin-induced skin necrosis. Ann Dermatol. 2014;26:96-98.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23:258-262.
- Ning MS, Dahir KM, Castellanos EH, et al. Sodium thiosulfate in the treatment of non-uremic calciphylaxis. J Dermatol. 2013;40:649-652.
An 80-year-old woman with a medical history notable for obesity (body mass index, 31.2), type 2 diabetes mellitus, hypertension, and chronic atrial fibrillation treated with warfarin presented with a chronic painful wound on the left lower calf of 1 month's duration. A 7×7-cm ulcer on the posterior aspect of the left calf with necrotic debris was seen surrounded by skin of mottled purple discoloration. The edge of the ulcer was not undermined. There were tense nonhemorrhagic bullae on the medial aspect of the left leg and on bilateral anterior tibial areas. Two punch biopsy specimens were obtained from the anterior tibial bulla and the edge of the ulcer.
Tender Edematous Nodules on the Hand
The Diagnosis: Ecthyma Contagiosum (Orf)
Orf, or ecthyma contagiosum, is a zoonotic cutaneous infection caused by the orf DNA virus of the genus Parapoxvirus of the family Poxviridae. It is transmitted to humans through direct contact with infected animals, namely sheep and goats, and as such is most commonly seen in patients with occupational exposure to these animals such as butchers, farmers, veterinarians, and shepherds.1,2 Human-to-human transmission is exceedingly rare in immunocompetent patients.2,3 In affected animals, lesions usually are found around the mouth, muzzle, and eyes. In humans, hands are the most commonly affected site, and lesions occur 3 to 10 days after contact. Clinically, the lesions are nonspecific, and our patient presented with tender, erythematous, edematous nodules on the left hand. The differential diagnosis is broad and includes a milker's nodule, pyogenic granuloma, tularemia, anthrax, atypical mycobacterial infection, and sporotrichosis.1,4,5
The diagnosis usually is made with a thorough history and examination, but in cases of uncertainty, routine pathology with hematoxylin and eosin staining, electron microscopy, or real-time polymerase chain reaction may be used.2-4 Histopathologically, lesions demonstrate intraepidermal vesicles, vacuolization of keratinocytes of the upper epidermis with characteristic cytoplasmic inclusion bodies, rete ridge elongation, and dilated vessels in the intervening dermal papillae. Central necrosis may occur in well-developed lesions.2,6 Interestingly, our patient's biopsy exhibited all of these findings (Figure). Immunostains for cytomegalovirus and herpes simplex virus were negative, and Grocott-Gomori methenamine-silver and acid-fast bacillus stains also were negative.

Our patient also developed lymphangitic streaking suggestive of a bacterial superinfection and was treated with a course of intravenous antibiotics. She eventually was discharged with reassurance, wound care instructions, and outpatient antibiotics. She returned to an outside institution's emergency department for further evaluation, and she was admitted for workup. A lesional swab was sent for real-time polymerase chain reaction, which confirmed the diagnosis as orf. When the patient was contacted for follow-up 1 week after biopsy, the hand lesions had notably improved.
Orf is self-limited and typically resolves within 4 to 8 weeks after undergoing evolution through 5 described stages. The maculopapular stage is denoted by enlarging erythematous macule. The targetoid stage is described by a red center within a white halo surrounded by a broader red halo. The nodular stage is self-descriptive. The regenerative and regression stages describe the progressively improving, drier, and crusted nodules.3
Because orf is self-limited, no treatment is required, and patients should be counseled that their lesions should resolve within weeks. Complications include lymphangitis, secondary bacterial infection, and erythema multiforme.1,2,4,5 Immunocompromised patients may develop recalcitrant, giant, or multiple lesions that may be treated with topical imiquimod, topical cidofovir, intralesional interferon alfa, or surgical excision.1,2,4,7
We present a case of orf to remind practitioners of this rare entity. Although the disease is endemic worldwide, it likely is underreported due to its self-limited nature.2,4 A careful history may reveal the diagnosis, and overtreatment with antibiotics, many of which have their own significant side-effect profile, can then be avoided.
Acknowledgment
We thank Eric Behling, MD (Camden, New Jersey), for his contributions in obtaining the histologic images.
- Veraldi S, Nazzaro G, Vaira F, et al. Presentation of orf (ecthyma contagiosum) after sheep slaughtering for religious feasts. Infection. 2014;42:767-769.
- Al-Salam S, Nowotny N, Sohail MR, et al. Ecthyma contagiosum (orf)--report of a human case from the United Arab Emirates and review of the literature. J Cutan Pathol. 2008;35:603-607.
- Thurman RJ, Fitch RW. Images in clinical medicine. contagious ecthyma. N Engl J Med. 2015;372:E12.
- Meier R, Sommacal A, Stahel A, et al. Orf--an orphan disease? JRSM Open. 2015;6:2054270415593718.
- Joseph RH, Haddad FA, Matthews AL, et al. Erythema multiforme after orf virus infection: a report of two cases and literature review. Epidemiol Infect. 2015;143:385-390.
- Xu X, Yun SJ, Erikson L, et al. Diseases caused by viruses. In: Elder DE, Elenitsas R, Rosenbach M, eds. Lever's Histopathology of the Skin. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:781-815.
- Koufakis T, Katsaitis P, Gabranis I. Orf disease: a report of a case. Braz J Infect Dis. 2014;18:568-569.
The Diagnosis: Ecthyma Contagiosum (Orf)
Orf, or ecthyma contagiosum, is a zoonotic cutaneous infection caused by the orf DNA virus of the genus Parapoxvirus of the family Poxviridae. It is transmitted to humans through direct contact with infected animals, namely sheep and goats, and as such is most commonly seen in patients with occupational exposure to these animals such as butchers, farmers, veterinarians, and shepherds.1,2 Human-to-human transmission is exceedingly rare in immunocompetent patients.2,3 In affected animals, lesions usually are found around the mouth, muzzle, and eyes. In humans, hands are the most commonly affected site, and lesions occur 3 to 10 days after contact. Clinically, the lesions are nonspecific, and our patient presented with tender, erythematous, edematous nodules on the left hand. The differential diagnosis is broad and includes a milker's nodule, pyogenic granuloma, tularemia, anthrax, atypical mycobacterial infection, and sporotrichosis.1,4,5
The diagnosis usually is made with a thorough history and examination, but in cases of uncertainty, routine pathology with hematoxylin and eosin staining, electron microscopy, or real-time polymerase chain reaction may be used.2-4 Histopathologically, lesions demonstrate intraepidermal vesicles, vacuolization of keratinocytes of the upper epidermis with characteristic cytoplasmic inclusion bodies, rete ridge elongation, and dilated vessels in the intervening dermal papillae. Central necrosis may occur in well-developed lesions.2,6 Interestingly, our patient's biopsy exhibited all of these findings (Figure). Immunostains for cytomegalovirus and herpes simplex virus were negative, and Grocott-Gomori methenamine-silver and acid-fast bacillus stains also were negative.
Our patient also developed lymphangitic streaking suggestive of a bacterial superinfection and was treated with a course of intravenous antibiotics. She eventually was discharged with reassurance, wound care instructions, and outpatient antibiotics. She returned to an outside institution's emergency department for further evaluation, and she was admitted for workup. A lesional swab was sent for real-time polymerase chain reaction, which confirmed the diagnosis as orf. When the patient was contacted for follow-up 1 week after biopsy, the hand lesions had notably improved.
Orf is self-limited and typically resolves within 4 to 8 weeks after undergoing evolution through 5 described stages. The maculopapular stage is denoted by enlarging erythematous macule. The targetoid stage is described by a red center within a white halo surrounded by a broader red halo. The nodular stage is self-descriptive. The regenerative and regression stages describe the progressively improving, drier, and crusted nodules.3
Because orf is self-limited, no treatment is required, and patients should be counseled that their lesions should resolve within weeks. Complications include lymphangitis, secondary bacterial infection, and erythema multiforme.1,2,4,5 Immunocompromised patients may develop recalcitrant, giant, or multiple lesions that may be treated with topical imiquimod, topical cidofovir, intralesional interferon alfa, or surgical excision.1,2,4,7
We present a case of orf to remind practitioners of this rare entity. Although the disease is endemic worldwide, it likely is underreported due to its self-limited nature.2,4 A careful history may reveal the diagnosis, and overtreatment with antibiotics, many of which have their own significant side-effect profile, can then be avoided.
Acknowledgment
We thank Eric Behling, MD (Camden, New Jersey), for his contributions in obtaining the histologic images.
The Diagnosis: Ecthyma Contagiosum (Orf)
Orf, or ecthyma contagiosum, is a zoonotic cutaneous infection caused by the orf DNA virus of the genus Parapoxvirus of the family Poxviridae. It is transmitted to humans through direct contact with infected animals, namely sheep and goats, and as such is most commonly seen in patients with occupational exposure to these animals such as butchers, farmers, veterinarians, and shepherds.1,2 Human-to-human transmission is exceedingly rare in immunocompetent patients.2,3 In affected animals, lesions usually are found around the mouth, muzzle, and eyes. In humans, hands are the most commonly affected site, and lesions occur 3 to 10 days after contact. Clinically, the lesions are nonspecific, and our patient presented with tender, erythematous, edematous nodules on the left hand. The differential diagnosis is broad and includes a milker's nodule, pyogenic granuloma, tularemia, anthrax, atypical mycobacterial infection, and sporotrichosis.1,4,5
The diagnosis usually is made with a thorough history and examination, but in cases of uncertainty, routine pathology with hematoxylin and eosin staining, electron microscopy, or real-time polymerase chain reaction may be used.2-4 Histopathologically, lesions demonstrate intraepidermal vesicles, vacuolization of keratinocytes of the upper epidermis with characteristic cytoplasmic inclusion bodies, rete ridge elongation, and dilated vessels in the intervening dermal papillae. Central necrosis may occur in well-developed lesions.2,6 Interestingly, our patient's biopsy exhibited all of these findings (Figure). Immunostains for cytomegalovirus and herpes simplex virus were negative, and Grocott-Gomori methenamine-silver and acid-fast bacillus stains also were negative.
Our patient also developed lymphangitic streaking suggestive of a bacterial superinfection and was treated with a course of intravenous antibiotics. She eventually was discharged with reassurance, wound care instructions, and outpatient antibiotics. She returned to an outside institution's emergency department for further evaluation, and she was admitted for workup. A lesional swab was sent for real-time polymerase chain reaction, which confirmed the diagnosis as orf. When the patient was contacted for follow-up 1 week after biopsy, the hand lesions had notably improved.
Orf is self-limited and typically resolves within 4 to 8 weeks after undergoing evolution through 5 described stages. The maculopapular stage is denoted by enlarging erythematous macule. The targetoid stage is described by a red center within a white halo surrounded by a broader red halo. The nodular stage is self-descriptive. The regenerative and regression stages describe the progressively improving, drier, and crusted nodules.3
Because orf is self-limited, no treatment is required, and patients should be counseled that their lesions should resolve within weeks. Complications include lymphangitis, secondary bacterial infection, and erythema multiforme.1,2,4,5 Immunocompromised patients may develop recalcitrant, giant, or multiple lesions that may be treated with topical imiquimod, topical cidofovir, intralesional interferon alfa, or surgical excision.1,2,4,7
We present a case of orf to remind practitioners of this rare entity. Although the disease is endemic worldwide, it likely is underreported due to its self-limited nature.2,4 A careful history may reveal the diagnosis, and overtreatment with antibiotics, many of which have their own significant side-effect profile, can then be avoided.
Acknowledgment
We thank Eric Behling, MD (Camden, New Jersey), for his contributions in obtaining the histologic images.
- Veraldi S, Nazzaro G, Vaira F, et al. Presentation of orf (ecthyma contagiosum) after sheep slaughtering for religious feasts. Infection. 2014;42:767-769.
- Al-Salam S, Nowotny N, Sohail MR, et al. Ecthyma contagiosum (orf)--report of a human case from the United Arab Emirates and review of the literature. J Cutan Pathol. 2008;35:603-607.
- Thurman RJ, Fitch RW. Images in clinical medicine. contagious ecthyma. N Engl J Med. 2015;372:E12.
- Meier R, Sommacal A, Stahel A, et al. Orf--an orphan disease? JRSM Open. 2015;6:2054270415593718.
- Joseph RH, Haddad FA, Matthews AL, et al. Erythema multiforme after orf virus infection: a report of two cases and literature review. Epidemiol Infect. 2015;143:385-390.
- Xu X, Yun SJ, Erikson L, et al. Diseases caused by viruses. In: Elder DE, Elenitsas R, Rosenbach M, eds. Lever's Histopathology of the Skin. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:781-815.
- Koufakis T, Katsaitis P, Gabranis I. Orf disease: a report of a case. Braz J Infect Dis. 2014;18:568-569.
- Veraldi S, Nazzaro G, Vaira F, et al. Presentation of orf (ecthyma contagiosum) after sheep slaughtering for religious feasts. Infection. 2014;42:767-769.
- Al-Salam S, Nowotny N, Sohail MR, et al. Ecthyma contagiosum (orf)--report of a human case from the United Arab Emirates and review of the literature. J Cutan Pathol. 2008;35:603-607.
- Thurman RJ, Fitch RW. Images in clinical medicine. contagious ecthyma. N Engl J Med. 2015;372:E12.
- Meier R, Sommacal A, Stahel A, et al. Orf--an orphan disease? JRSM Open. 2015;6:2054270415593718.
- Joseph RH, Haddad FA, Matthews AL, et al. Erythema multiforme after orf virus infection: a report of two cases and literature review. Epidemiol Infect. 2015;143:385-390.
- Xu X, Yun SJ, Erikson L, et al. Diseases caused by viruses. In: Elder DE, Elenitsas R, Rosenbach M, eds. Lever's Histopathology of the Skin. 11th ed. Philadelphia, PA: Wolters Kluwer; 2015:781-815.
- Koufakis T, Katsaitis P, Gabranis I. Orf disease: a report of a case. Braz J Infect Dis. 2014;18:568-569.
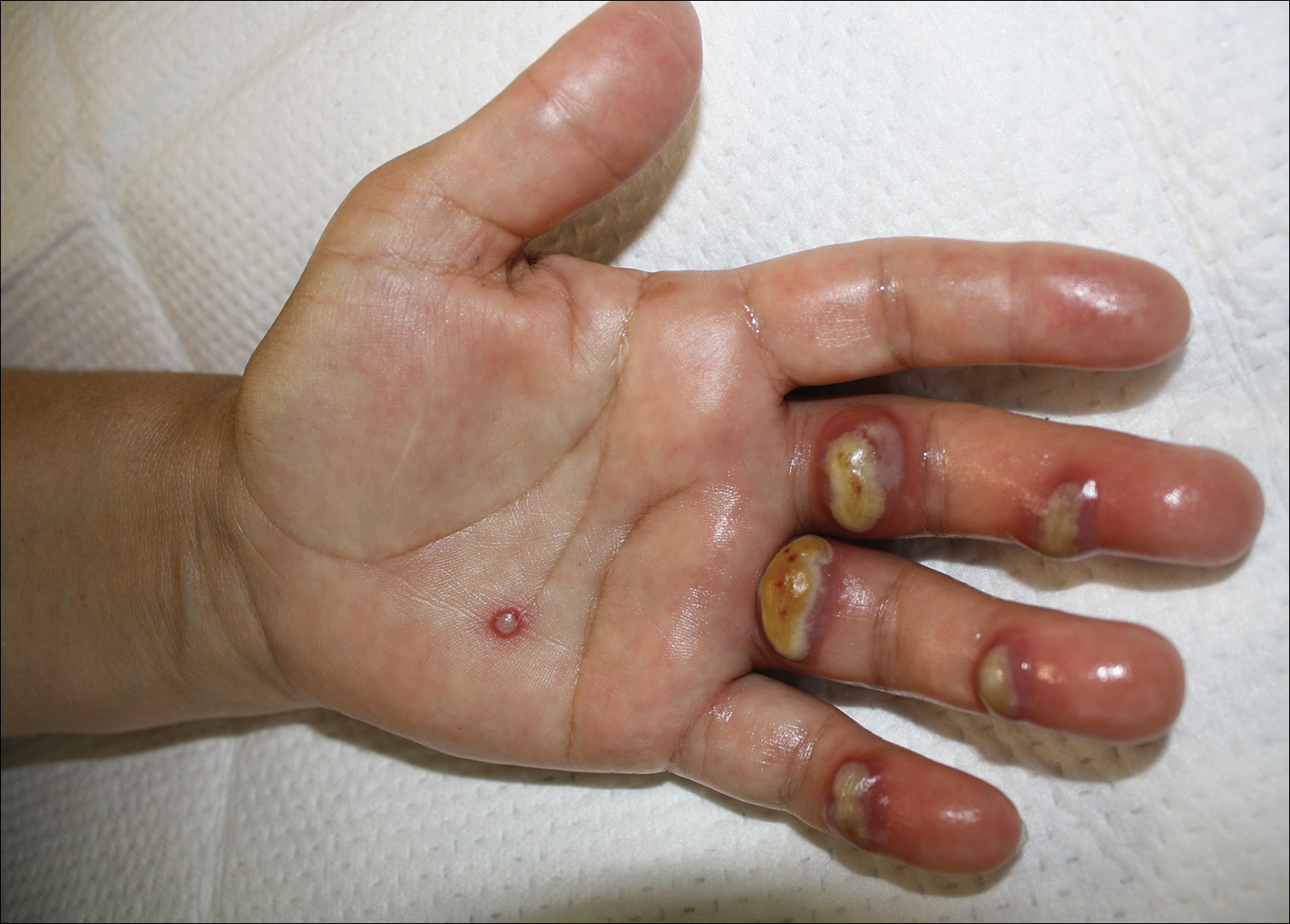
A 57-year-old woman presented to the emergency department (ED) for evaluation of a rash on the left hand of 2 weeks' duration. She described pinpoint red lesions on the left palm, as well as the third, fourth, and fifth fingers, which gradually enlarged and became painful. She denied any specific trauma but recalled cutting her hand on a piece of metal in the ground prior to the onset of the rash. She worked on a farm and bottle-fed sheep and chickens. Physical examination revealed tender edematous nodules with central gray pustules, and the left axillary lymph node was enlarged and tender. Ulceration was not appreciated. Various antibiotics including cephalexin, trimethoprim-sulfamethoxazole, and clindamycin were prescribed during prior ED visits, but she reported no improvement with these medications. She remained afebrile throughout the course of the hand rash, and laboratory workup was consistently unremarkable. Two sets of herpes simplex virus cultures from the ED visits showed no growth, and a hand radiograph also was normal. Medical history included coronary artery disease, myocardial infarction, mitral regurgitation, and hyperlipidemia.
Hyperpigmented Patch on the Leg
The Diagnosis: Lichen Aureus
The clinicopathological findings were diagnostic of lichen aureus (LA). Microscopic examination revealed a relatively sparse, superficial, perivascular and interstitial lymphohistiocytic infiltrate with scattered siderophages in the upper dermis. Extravasation of red blood cells also was noted (Figure 1). An immunohistochemical stain for Melan-A highlighted a normal number and distribution of single melanocytes at the dermoepidermal junction with no evidence of pagetoid scatter. A Perls Prussian blue stain for iron demonstrated abundant hemosiderin in the dermis (Figure 2).
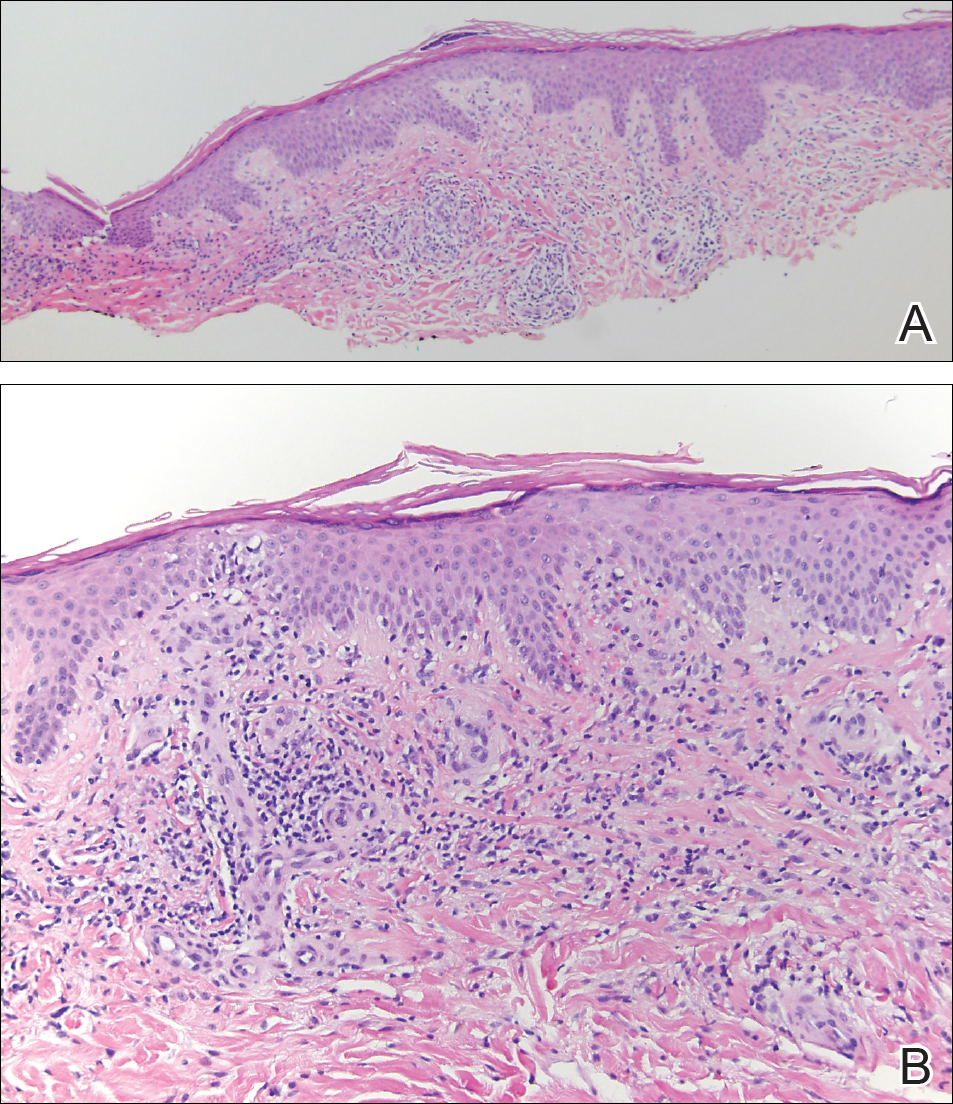
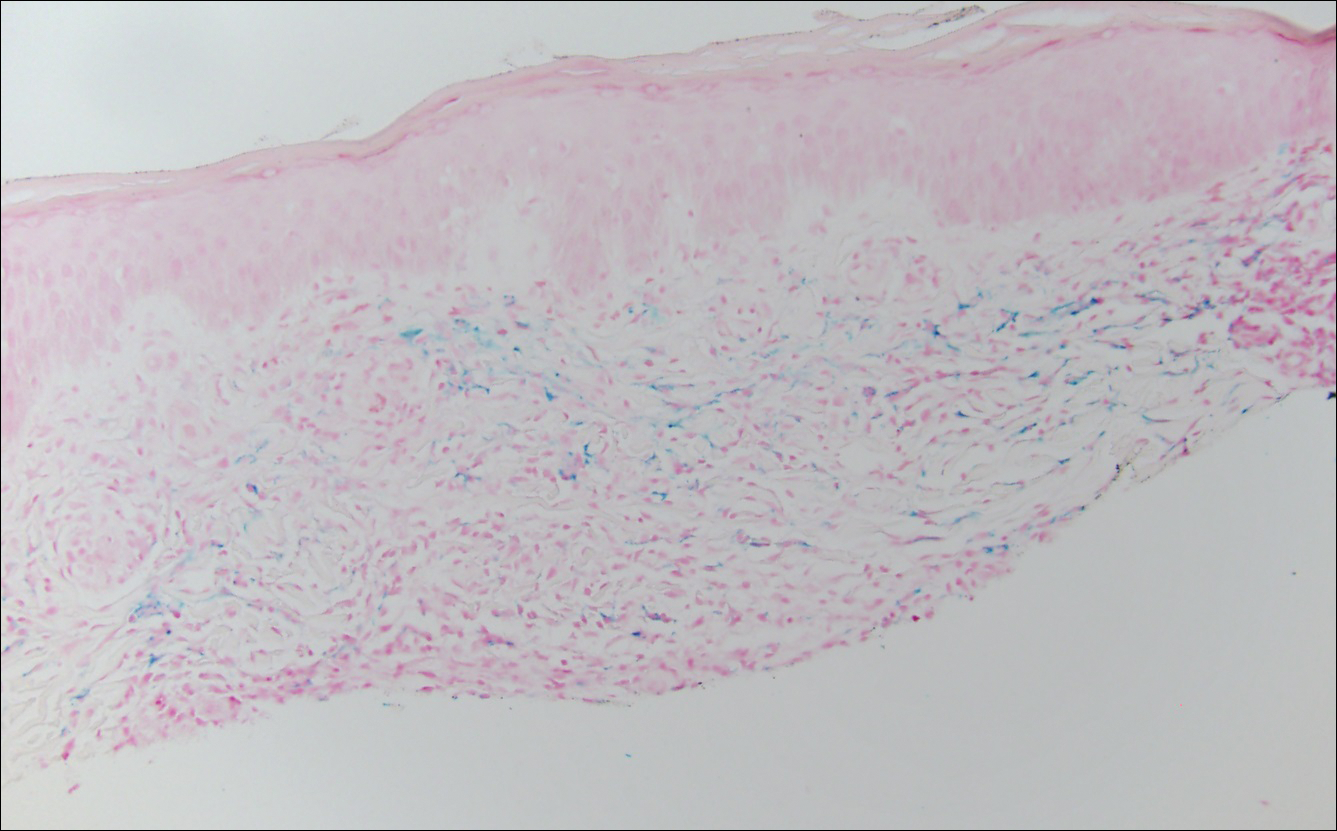
Pigmented purpuric dermatosis (PPD) describes a group of cutaneous lesions that are characterized by petechiae and pigmentary changes. These lesions most commonly present on the lower limbs; however, other sites have been reported.1 This group includes several major clinical forms such as Schamberg disease, LA, purpura annularis telangiectodes of Majocchi, eczematidlike purpura of Doucas and Kapetanakis, and lichenoid PPD of Gougerot and Blum. Lesions typically demonstrate a striking golden brown color clinically and by definition occur in the absence of platelet defects or vasculitis.1
Factors implicated in the pathogenesis of pigmented purpura include gravitational dependency, venous stasis, infection, and drugs.2 It is suggested that cellular immunity may play a role in the development of the disease based on the presence of CD4+ T lymphocytes in the infiltrate and the expression of HLA-DR by these lymphocytes and the keratinocytes.3 Lichen aureus differs in that it relates to increased intravascular pressure from an incompetent valve in an underlying perforating vein.4
Lichen aureus, also referred to as lichen purpuricus, is one major variant of PPD. The name reflects both the characteristic golden brown color and the histopathologic pattern of inflammation.1 Lichen aureus usually presents as a unilateral, asymptomatic, confined single lesion located mainly on the leg,1 though it can develop at other sites or as a localized group of lesions. Extensive lesions have been reported5 and cases with a segmental distribution have been described.6 In contrast, Schamberg disease demonstrates pinhead-sized reddish lesions giving the characteristic cayenne pepper pigmentation. These lesions coalesce to form thumbprint patches that progress proximally.1 Majocchi purpura is annular and telangiectatic, while lichenoid purpura of Gougerot and Blum presents with flat-topped, polygonal, violaceous papules that turn brown over time.
Some authors have championed a role for dermoscopy in diagnosis of LA.7 By dermoscopy, LA demonstrates a diffuse copper background reflecting the lymphohistiocytic dermal infiltrate, red dots and globules representing the extravasated red blood cells and the dilated swollen vessels, and grey dots that reflect the hemosiderin present in the dermis.8
Histologically, LA demonstrates a superficial perivascular infiltrate composed mainly of CD4+ lymphocytes surrounding the superficial capillaries. Over time, red cell extravasation leads to the formation of hemosiderin-laden macrophages, which can be highlighted with Perls Prussian blue stain. A bandlike infiltrate with thin strands of collagen separating it from the epidermis also may be noted.9
An important consideration in the differential diagnosis of PPD is mycosis fungoides (MF). Mycosis fungoides is a cutaneous T-cell lymphoma that clinically presents as a single or multiple hypopigmented or hyperpigmented patches or as erythematous scaly lesions in the patch or plaque stage. These lesions eventually may evolve into tumor stage.10 Mycosis fungoides may mimic PPD clinically and/or histopathologically, and rarely PPD also may precede MF.11 Involvement of the trunk, especially the lower abdomen and buttock region, favors a diagnosis of MF. Typically, histopathologic examination of MF demonstrates an epidermotropic lymphocytic infiltrate composed of atypical cerebriform lymphocytes overlying papillary dermal fibrosis. Although classic MF would be difficult to confuse with PPD, the atrophic lichenoid pattern of MF may show remarkable overlap with PPD.12 Such cases require clinicopathologic correlation, immunophenotyping of the epidermotropic lymphocytes, and occasionally T-cell clonality studies.
Lichen aureus is a chronic persistent disease unless the underlying incompetent perforator vessel is ligated. Various treatments have been used for other forms of pigmented purpura including topical corticosteroids, topical tacrolimus, systemic vasodilators such as prostacyclin and pentoxifylline, and phototherapy.1 Clinical follow-up is recommended for lesions that show some clinical or histopathological overlap with MF. Additional biopsies also may prove useful in establishing a definitive diagnosis in ambiguous cases.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Aiba S, Tagami H. Immunohistologic studies in Schamberg's disease. evidence for cellular immune reaction in lesional skin. Arch Dermatol. 1988;124:1058-1062.
- English J. Lichen aureus. J Am Acad Dermatol. 1985;12(2, pt 1):377-379.
- Duhra P, Tan CY. Lichen aureus. Br J Dermatol. 1986;114:395.
- Moche J, Glassman S, Modi D, et al. Segmental lichen aureus: a report of two cases treated with methylprednisolone aceponate. Australas J Dermatol. 2011;52:E15-E18.
- Zaballos P, Puig S, Malvehy J. Dermoscopy of pigmented purpuric dermatoses (lichen aureus): a useful tool for clinical diagnosis. Arch Dermatol. 2004;140:1290-1291.
- Portela PS, Melo DF, Ormiga P, et al. Dermoscopy of lichen aureus. An Bras Dermatol. 2013;88:253-255.
- Smoller BR, Kamel OW. Pigmented purpuric eruptions: immunopathologic studies supportive of a common immunophenotype. J Cutan Pathol. 1991;18:423-427.
- Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. a progress report. Am J Clin Pathol. 1999;111(1 suppl 1):S8-S12.
- Hanna S, Walsh N, D'Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
- Toro JR, Sander CA, LeBoit PE. Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
The Diagnosis: Lichen Aureus
The clinicopathological findings were diagnostic of lichen aureus (LA). Microscopic examination revealed a relatively sparse, superficial, perivascular and interstitial lymphohistiocytic infiltrate with scattered siderophages in the upper dermis. Extravasation of red blood cells also was noted (Figure 1). An immunohistochemical stain for Melan-A highlighted a normal number and distribution of single melanocytes at the dermoepidermal junction with no evidence of pagetoid scatter. A Perls Prussian blue stain for iron demonstrated abundant hemosiderin in the dermis (Figure 2).
Pigmented purpuric dermatosis (PPD) describes a group of cutaneous lesions that are characterized by petechiae and pigmentary changes. These lesions most commonly present on the lower limbs; however, other sites have been reported.1 This group includes several major clinical forms such as Schamberg disease, LA, purpura annularis telangiectodes of Majocchi, eczematidlike purpura of Doucas and Kapetanakis, and lichenoid PPD of Gougerot and Blum. Lesions typically demonstrate a striking golden brown color clinically and by definition occur in the absence of platelet defects or vasculitis.1
Factors implicated in the pathogenesis of pigmented purpura include gravitational dependency, venous stasis, infection, and drugs.2 It is suggested that cellular immunity may play a role in the development of the disease based on the presence of CD4+ T lymphocytes in the infiltrate and the expression of HLA-DR by these lymphocytes and the keratinocytes.3 Lichen aureus differs in that it relates to increased intravascular pressure from an incompetent valve in an underlying perforating vein.4
Lichen aureus, also referred to as lichen purpuricus, is one major variant of PPD. The name reflects both the characteristic golden brown color and the histopathologic pattern of inflammation.1 Lichen aureus usually presents as a unilateral, asymptomatic, confined single lesion located mainly on the leg,1 though it can develop at other sites or as a localized group of lesions. Extensive lesions have been reported5 and cases with a segmental distribution have been described.6 In contrast, Schamberg disease demonstrates pinhead-sized reddish lesions giving the characteristic cayenne pepper pigmentation. These lesions coalesce to form thumbprint patches that progress proximally.1 Majocchi purpura is annular and telangiectatic, while lichenoid purpura of Gougerot and Blum presents with flat-topped, polygonal, violaceous papules that turn brown over time.
Some authors have championed a role for dermoscopy in diagnosis of LA.7 By dermoscopy, LA demonstrates a diffuse copper background reflecting the lymphohistiocytic dermal infiltrate, red dots and globules representing the extravasated red blood cells and the dilated swollen vessels, and grey dots that reflect the hemosiderin present in the dermis.8
Histologically, LA demonstrates a superficial perivascular infiltrate composed mainly of CD4+ lymphocytes surrounding the superficial capillaries. Over time, red cell extravasation leads to the formation of hemosiderin-laden macrophages, which can be highlighted with Perls Prussian blue stain. A bandlike infiltrate with thin strands of collagen separating it from the epidermis also may be noted.9
An important consideration in the differential diagnosis of PPD is mycosis fungoides (MF). Mycosis fungoides is a cutaneous T-cell lymphoma that clinically presents as a single or multiple hypopigmented or hyperpigmented patches or as erythematous scaly lesions in the patch or plaque stage. These lesions eventually may evolve into tumor stage.10 Mycosis fungoides may mimic PPD clinically and/or histopathologically, and rarely PPD also may precede MF.11 Involvement of the trunk, especially the lower abdomen and buttock region, favors a diagnosis of MF. Typically, histopathologic examination of MF demonstrates an epidermotropic lymphocytic infiltrate composed of atypical cerebriform lymphocytes overlying papillary dermal fibrosis. Although classic MF would be difficult to confuse with PPD, the atrophic lichenoid pattern of MF may show remarkable overlap with PPD.12 Such cases require clinicopathologic correlation, immunophenotyping of the epidermotropic lymphocytes, and occasionally T-cell clonality studies.
Lichen aureus is a chronic persistent disease unless the underlying incompetent perforator vessel is ligated. Various treatments have been used for other forms of pigmented purpura including topical corticosteroids, topical tacrolimus, systemic vasodilators such as prostacyclin and pentoxifylline, and phototherapy.1 Clinical follow-up is recommended for lesions that show some clinical or histopathological overlap with MF. Additional biopsies also may prove useful in establishing a definitive diagnosis in ambiguous cases.
The Diagnosis: Lichen Aureus
The clinicopathological findings were diagnostic of lichen aureus (LA). Microscopic examination revealed a relatively sparse, superficial, perivascular and interstitial lymphohistiocytic infiltrate with scattered siderophages in the upper dermis. Extravasation of red blood cells also was noted (Figure 1). An immunohistochemical stain for Melan-A highlighted a normal number and distribution of single melanocytes at the dermoepidermal junction with no evidence of pagetoid scatter. A Perls Prussian blue stain for iron demonstrated abundant hemosiderin in the dermis (Figure 2).
Pigmented purpuric dermatosis (PPD) describes a group of cutaneous lesions that are characterized by petechiae and pigmentary changes. These lesions most commonly present on the lower limbs; however, other sites have been reported.1 This group includes several major clinical forms such as Schamberg disease, LA, purpura annularis telangiectodes of Majocchi, eczematidlike purpura of Doucas and Kapetanakis, and lichenoid PPD of Gougerot and Blum. Lesions typically demonstrate a striking golden brown color clinically and by definition occur in the absence of platelet defects or vasculitis.1
Factors implicated in the pathogenesis of pigmented purpura include gravitational dependency, venous stasis, infection, and drugs.2 It is suggested that cellular immunity may play a role in the development of the disease based on the presence of CD4+ T lymphocytes in the infiltrate and the expression of HLA-DR by these lymphocytes and the keratinocytes.3 Lichen aureus differs in that it relates to increased intravascular pressure from an incompetent valve in an underlying perforating vein.4
Lichen aureus, also referred to as lichen purpuricus, is one major variant of PPD. The name reflects both the characteristic golden brown color and the histopathologic pattern of inflammation.1 Lichen aureus usually presents as a unilateral, asymptomatic, confined single lesion located mainly on the leg,1 though it can develop at other sites or as a localized group of lesions. Extensive lesions have been reported5 and cases with a segmental distribution have been described.6 In contrast, Schamberg disease demonstrates pinhead-sized reddish lesions giving the characteristic cayenne pepper pigmentation. These lesions coalesce to form thumbprint patches that progress proximally.1 Majocchi purpura is annular and telangiectatic, while lichenoid purpura of Gougerot and Blum presents with flat-topped, polygonal, violaceous papules that turn brown over time.
Some authors have championed a role for dermoscopy in diagnosis of LA.7 By dermoscopy, LA demonstrates a diffuse copper background reflecting the lymphohistiocytic dermal infiltrate, red dots and globules representing the extravasated red blood cells and the dilated swollen vessels, and grey dots that reflect the hemosiderin present in the dermis.8
Histologically, LA demonstrates a superficial perivascular infiltrate composed mainly of CD4+ lymphocytes surrounding the superficial capillaries. Over time, red cell extravasation leads to the formation of hemosiderin-laden macrophages, which can be highlighted with Perls Prussian blue stain. A bandlike infiltrate with thin strands of collagen separating it from the epidermis also may be noted.9
An important consideration in the differential diagnosis of PPD is mycosis fungoides (MF). Mycosis fungoides is a cutaneous T-cell lymphoma that clinically presents as a single or multiple hypopigmented or hyperpigmented patches or as erythematous scaly lesions in the patch or plaque stage. These lesions eventually may evolve into tumor stage.10 Mycosis fungoides may mimic PPD clinically and/or histopathologically, and rarely PPD also may precede MF.11 Involvement of the trunk, especially the lower abdomen and buttock region, favors a diagnosis of MF. Typically, histopathologic examination of MF demonstrates an epidermotropic lymphocytic infiltrate composed of atypical cerebriform lymphocytes overlying papillary dermal fibrosis. Although classic MF would be difficult to confuse with PPD, the atrophic lichenoid pattern of MF may show remarkable overlap with PPD.12 Such cases require clinicopathologic correlation, immunophenotyping of the epidermotropic lymphocytes, and occasionally T-cell clonality studies.
Lichen aureus is a chronic persistent disease unless the underlying incompetent perforator vessel is ligated. Various treatments have been used for other forms of pigmented purpura including topical corticosteroids, topical tacrolimus, systemic vasodilators such as prostacyclin and pentoxifylline, and phototherapy.1 Clinical follow-up is recommended for lesions that show some clinical or histopathological overlap with MF. Additional biopsies also may prove useful in establishing a definitive diagnosis in ambiguous cases.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Aiba S, Tagami H. Immunohistologic studies in Schamberg's disease. evidence for cellular immune reaction in lesional skin. Arch Dermatol. 1988;124:1058-1062.
- English J. Lichen aureus. J Am Acad Dermatol. 1985;12(2, pt 1):377-379.
- Duhra P, Tan CY. Lichen aureus. Br J Dermatol. 1986;114:395.
- Moche J, Glassman S, Modi D, et al. Segmental lichen aureus: a report of two cases treated with methylprednisolone aceponate. Australas J Dermatol. 2011;52:E15-E18.
- Zaballos P, Puig S, Malvehy J. Dermoscopy of pigmented purpuric dermatoses (lichen aureus): a useful tool for clinical diagnosis. Arch Dermatol. 2004;140:1290-1291.
- Portela PS, Melo DF, Ormiga P, et al. Dermoscopy of lichen aureus. An Bras Dermatol. 2013;88:253-255.
- Smoller BR, Kamel OW. Pigmented purpuric eruptions: immunopathologic studies supportive of a common immunophenotype. J Cutan Pathol. 1991;18:423-427.
- Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. a progress report. Am J Clin Pathol. 1999;111(1 suppl 1):S8-S12.
- Hanna S, Walsh N, D'Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
- Toro JR, Sander CA, LeBoit PE. Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Aiba S, Tagami H. Immunohistologic studies in Schamberg's disease. evidence for cellular immune reaction in lesional skin. Arch Dermatol. 1988;124:1058-1062.
- English J. Lichen aureus. J Am Acad Dermatol. 1985;12(2, pt 1):377-379.
- Duhra P, Tan CY. Lichen aureus. Br J Dermatol. 1986;114:395.
- Moche J, Glassman S, Modi D, et al. Segmental lichen aureus: a report of two cases treated with methylprednisolone aceponate. Australas J Dermatol. 2011;52:E15-E18.
- Zaballos P, Puig S, Malvehy J. Dermoscopy of pigmented purpuric dermatoses (lichen aureus): a useful tool for clinical diagnosis. Arch Dermatol. 2004;140:1290-1291.
- Portela PS, Melo DF, Ormiga P, et al. Dermoscopy of lichen aureus. An Bras Dermatol. 2013;88:253-255.
- Smoller BR, Kamel OW. Pigmented purpuric eruptions: immunopathologic studies supportive of a common immunophenotype. J Cutan Pathol. 1991;18:423-427.
- Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. a progress report. Am J Clin Pathol. 1999;111(1 suppl 1):S8-S12.
- Hanna S, Walsh N, D'Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
- Toro JR, Sander CA, LeBoit PE. Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
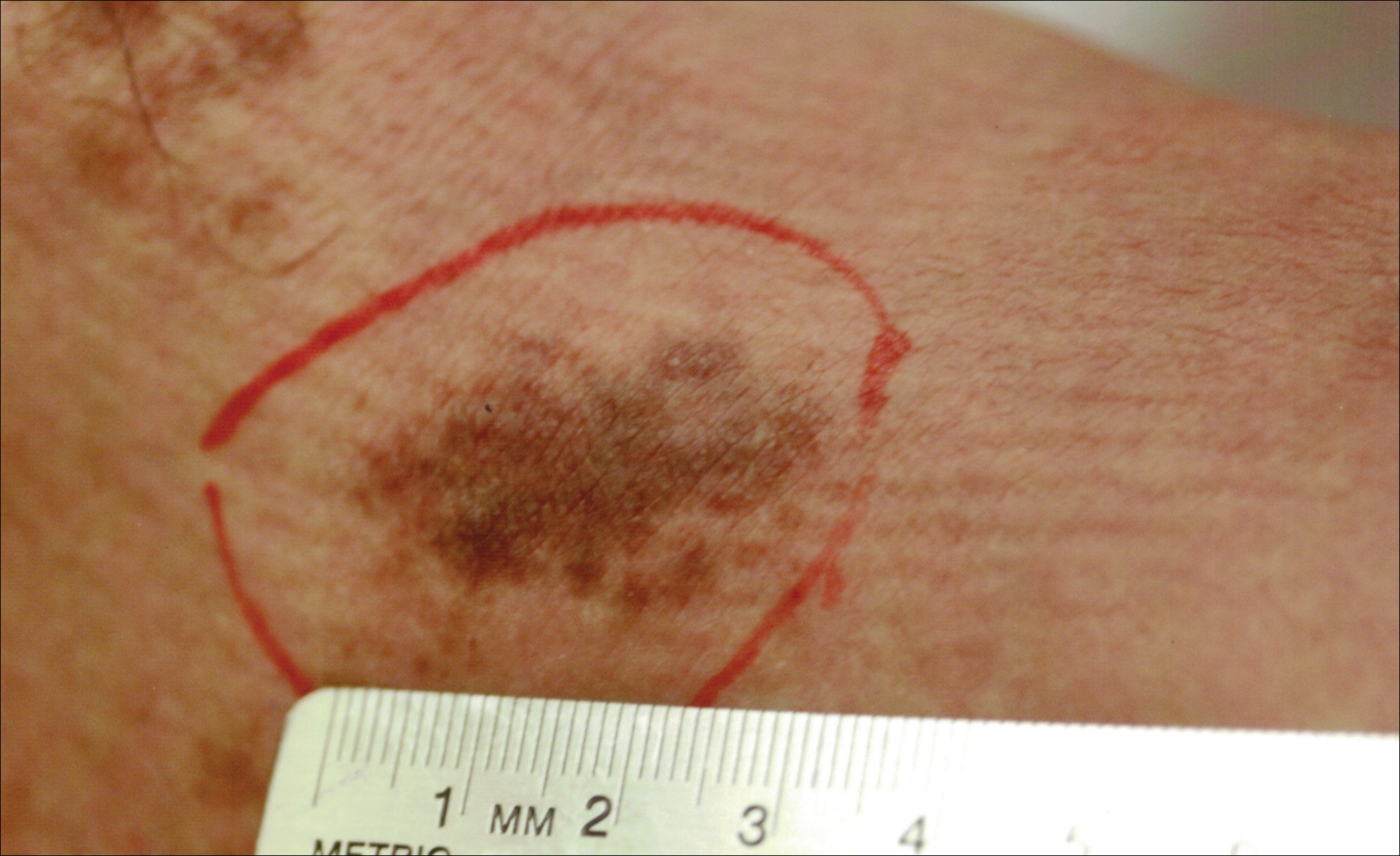
A 32-year-old man presented with an asymptomatic pigmented lesion on the left foot that developed over the course of 4 months. Physical examination revealed a 4-cm asymmetrical, deeply pigmented macule on the left foot. A shave biopsy of the lesion was performed.
Bullous Lesions in a Neonate
The Diagnosis: Incontinentia Pigmenti
The infant's mother was noted to have diffuse hypopigmented patches over the trunk, arms, and legs (present since adolescence) with whorled cicatricial alopecia of the vertex scalp and peg-shaped teeth (Figure). Together, these findings suggested incontinentia pigmenti (IP), which the mother revealed she had been diagnosed with in childhood. The infant's characteristic lesions in the setting of her mother's diagnosed genodermatosis confirmed the diagnosis of IP.
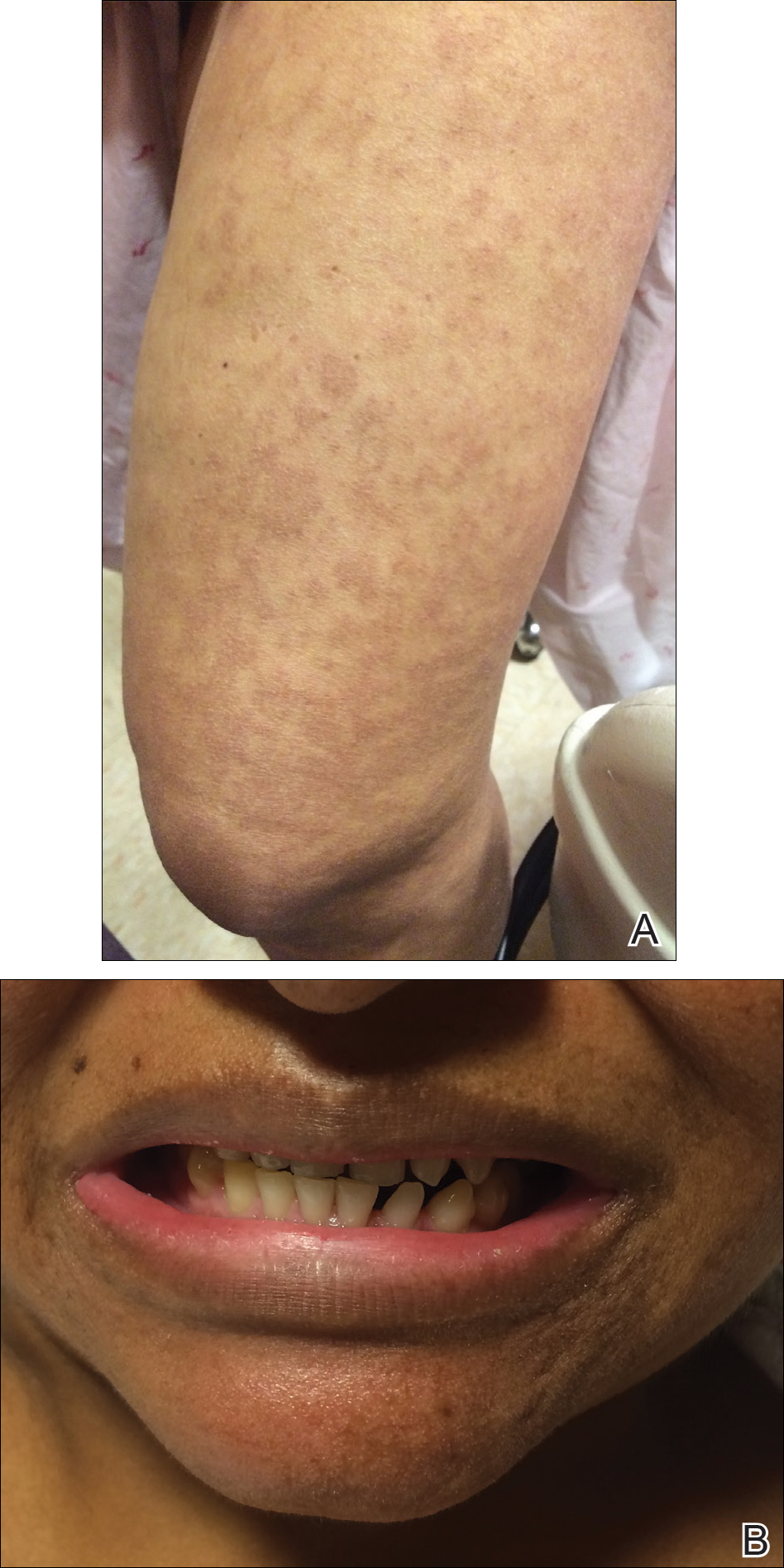
Incontinentia pigmenti is an X-linked dominant disorder that presents with many classic dermatologic, dental, neurologic, and ophthalmologic findings. The causative mutation occurs in IKBKG/NEMO (inhibitor of κ polypeptide gene enhancer in B-cells, kinase γ/nuclear factor-κB essential modulator) gene on Xq28, disabling the resultant protein that normally protects cells from tumor necrosis factor family-induced apoptosis.1 Incontinentia pigmenti usually is lethal in males and causes an unbalanced X-inactivation in surviving female IP patients. Occurring at a rate of 1.2 per 100,000 births,2 IP typically presents in female infants with skin lesions patterned along Blaschko lines that evolve in 4 stages over a lifetime.3 Stage I, presenting in the neonatal period, manifests as vesiculobullous eruptions on the limbs and scalp. Stages II to IV vary in duration from months to years and are comprised of a verrucous stage, a hyperpigmented stage, and a hypopigmented stage, respectively.3 All stages of IP can overlap and coexist.
The vesiculobullous findings in infants with IP may be mistakenly attributed to other diseases with prominent vesicular or bullous components including herpes simplex virus, epidermolysis bullosa, and infantile acropustulosis. With neonatal herpes simplex virus infection, vesicular skin or mucocutaneous lesions occur 9 to 11 days after birth and can be confirmed by specimen culture or qualitative polymerase chain reaction, while stage I of IP appears within the first 6 to 8 weeks of life and can be present at birth.4 The hallmark of epidermolysis bullosa, caused by mutations in keratins 5 and 14, is blistering erosions of the skin in response to frictional stress,1 thus these lesions do not follow Blaschko lines. Infantile acropustulosis, a nonheritable vesiculopustular eruption of the hands and feet, rarely occurs in the immediate newborn period; it most often appears in the 3- to 6-month age range with recurrent eruptions at 3- to 4-week intervals.5 Focal dermal hypoplasia is another X-linked dominant disorder with blaschkolinear findings at birth that presents with pink or red, angular, atrophic macules, in contrast to the bullous lesions of IP.6
Incontinentia pigmenti may encompass a wide range of systemic symptoms in addition to the classic dermatologic findings. Notably, central nervous system defects are concurrent in up to 40% of IP cases, with seizures, mental retardation, and spastic paresis being the most common sequelae.7 Teeth defects, seen in 35% of patients, include delayed primary dentition and peg-shaped teeth. Many patients will experience ophthalmologic defects including vision problems (16%) and retinopathy (15%).7
The cutaneous eruptions of IP may be treated with topical corticosteroids or topical tacrolimus, and vesicles should be left intact and monitored for signs of infection.8,9 Seizures, if present, should be treated with anticonvulsants, and regular neuropsychiatric monitoring and physical rehabilitation may be warranted. Patients should be regularly monitored for retinopathy beginning at the time of diagnosis. Retinal fibrovascular proliferation is treated with xenon laser photocoagulation to reduce the high risk for retinal detachment in this population.10,11 Older and younger at-risk relatives must be evaluated by genetic testing or thorough physical examination to clarify their disease status and determine the need for additional genetic counseling.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012.
- Prevalence and incidence of rare diseases: bibliographic data. Orphanet Report Series, Rare Diseases collection. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Published June 2017. Accessed July 13, 2017.
- Scheuerle AE, Ursini MV. Incontinentia pigmenti. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. Seattle, WA: University of Washington; 2015. http://www.ncbi.nlm.nih.gov/books/NBK1472/. Accessed July 25, 2017.
- James SH, Kimberlin DW. Neonatal herpes simplex virus infection. Infect Dis Clin North Am. 2015;29:391-400.
- Eichenfield LF, Frieden IJ, Mathes E, et al, eds. Neonatal and Infant Dermatology. Philadelphia, PA: Saunders; 2015.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Fusco F, Paciolla M, Conte MI, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014;9:93.
- Jessup CJ, Morgan SC, Cohen LM, et al. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8:944-946.
- Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti [published online June 1, 2009]. Clin Exp Dermatol. 2009;34:E611-E613.
- Nguyen JK, Brady-Mccreery KM. Laser photocoagulation in preproliferative retinopathy of incontinentia pigmenti. J AAPOS. 2001;5:258-259.
- Chen CJ, Han IC, Tian J, et al. Extended follow-up of treated and untreated retinopathy in incontinentia pigmenti: analysis of peripheral vascular changes and incidence of retinal detachment. JAMA Ophthalmol. 2015;133:542-548.
The Diagnosis: Incontinentia Pigmenti
The infant's mother was noted to have diffuse hypopigmented patches over the trunk, arms, and legs (present since adolescence) with whorled cicatricial alopecia of the vertex scalp and peg-shaped teeth (Figure). Together, these findings suggested incontinentia pigmenti (IP), which the mother revealed she had been diagnosed with in childhood. The infant's characteristic lesions in the setting of her mother's diagnosed genodermatosis confirmed the diagnosis of IP.
Incontinentia pigmenti is an X-linked dominant disorder that presents with many classic dermatologic, dental, neurologic, and ophthalmologic findings. The causative mutation occurs in IKBKG/NEMO (inhibitor of κ polypeptide gene enhancer in B-cells, kinase γ/nuclear factor-κB essential modulator) gene on Xq28, disabling the resultant protein that normally protects cells from tumor necrosis factor family-induced apoptosis.1 Incontinentia pigmenti usually is lethal in males and causes an unbalanced X-inactivation in surviving female IP patients. Occurring at a rate of 1.2 per 100,000 births,2 IP typically presents in female infants with skin lesions patterned along Blaschko lines that evolve in 4 stages over a lifetime.3 Stage I, presenting in the neonatal period, manifests as vesiculobullous eruptions on the limbs and scalp. Stages II to IV vary in duration from months to years and are comprised of a verrucous stage, a hyperpigmented stage, and a hypopigmented stage, respectively.3 All stages of IP can overlap and coexist.
The vesiculobullous findings in infants with IP may be mistakenly attributed to other diseases with prominent vesicular or bullous components including herpes simplex virus, epidermolysis bullosa, and infantile acropustulosis. With neonatal herpes simplex virus infection, vesicular skin or mucocutaneous lesions occur 9 to 11 days after birth and can be confirmed by specimen culture or qualitative polymerase chain reaction, while stage I of IP appears within the first 6 to 8 weeks of life and can be present at birth.4 The hallmark of epidermolysis bullosa, caused by mutations in keratins 5 and 14, is blistering erosions of the skin in response to frictional stress,1 thus these lesions do not follow Blaschko lines. Infantile acropustulosis, a nonheritable vesiculopustular eruption of the hands and feet, rarely occurs in the immediate newborn period; it most often appears in the 3- to 6-month age range with recurrent eruptions at 3- to 4-week intervals.5 Focal dermal hypoplasia is another X-linked dominant disorder with blaschkolinear findings at birth that presents with pink or red, angular, atrophic macules, in contrast to the bullous lesions of IP.6
Incontinentia pigmenti may encompass a wide range of systemic symptoms in addition to the classic dermatologic findings. Notably, central nervous system defects are concurrent in up to 40% of IP cases, with seizures, mental retardation, and spastic paresis being the most common sequelae.7 Teeth defects, seen in 35% of patients, include delayed primary dentition and peg-shaped teeth. Many patients will experience ophthalmologic defects including vision problems (16%) and retinopathy (15%).7
The cutaneous eruptions of IP may be treated with topical corticosteroids or topical tacrolimus, and vesicles should be left intact and monitored for signs of infection.8,9 Seizures, if present, should be treated with anticonvulsants, and regular neuropsychiatric monitoring and physical rehabilitation may be warranted. Patients should be regularly monitored for retinopathy beginning at the time of diagnosis. Retinal fibrovascular proliferation is treated with xenon laser photocoagulation to reduce the high risk for retinal detachment in this population.10,11 Older and younger at-risk relatives must be evaluated by genetic testing or thorough physical examination to clarify their disease status and determine the need for additional genetic counseling.
The Diagnosis: Incontinentia Pigmenti
The infant's mother was noted to have diffuse hypopigmented patches over the trunk, arms, and legs (present since adolescence) with whorled cicatricial alopecia of the vertex scalp and peg-shaped teeth (Figure). Together, these findings suggested incontinentia pigmenti (IP), which the mother revealed she had been diagnosed with in childhood. The infant's characteristic lesions in the setting of her mother's diagnosed genodermatosis confirmed the diagnosis of IP.
Incontinentia pigmenti is an X-linked dominant disorder that presents with many classic dermatologic, dental, neurologic, and ophthalmologic findings. The causative mutation occurs in IKBKG/NEMO (inhibitor of κ polypeptide gene enhancer in B-cells, kinase γ/nuclear factor-κB essential modulator) gene on Xq28, disabling the resultant protein that normally protects cells from tumor necrosis factor family-induced apoptosis.1 Incontinentia pigmenti usually is lethal in males and causes an unbalanced X-inactivation in surviving female IP patients. Occurring at a rate of 1.2 per 100,000 births,2 IP typically presents in female infants with skin lesions patterned along Blaschko lines that evolve in 4 stages over a lifetime.3 Stage I, presenting in the neonatal period, manifests as vesiculobullous eruptions on the limbs and scalp. Stages II to IV vary in duration from months to years and are comprised of a verrucous stage, a hyperpigmented stage, and a hypopigmented stage, respectively.3 All stages of IP can overlap and coexist.
The vesiculobullous findings in infants with IP may be mistakenly attributed to other diseases with prominent vesicular or bullous components including herpes simplex virus, epidermolysis bullosa, and infantile acropustulosis. With neonatal herpes simplex virus infection, vesicular skin or mucocutaneous lesions occur 9 to 11 days after birth and can be confirmed by specimen culture or qualitative polymerase chain reaction, while stage I of IP appears within the first 6 to 8 weeks of life and can be present at birth.4 The hallmark of epidermolysis bullosa, caused by mutations in keratins 5 and 14, is blistering erosions of the skin in response to frictional stress,1 thus these lesions do not follow Blaschko lines. Infantile acropustulosis, a nonheritable vesiculopustular eruption of the hands and feet, rarely occurs in the immediate newborn period; it most often appears in the 3- to 6-month age range with recurrent eruptions at 3- to 4-week intervals.5 Focal dermal hypoplasia is another X-linked dominant disorder with blaschkolinear findings at birth that presents with pink or red, angular, atrophic macules, in contrast to the bullous lesions of IP.6
Incontinentia pigmenti may encompass a wide range of systemic symptoms in addition to the classic dermatologic findings. Notably, central nervous system defects are concurrent in up to 40% of IP cases, with seizures, mental retardation, and spastic paresis being the most common sequelae.7 Teeth defects, seen in 35% of patients, include delayed primary dentition and peg-shaped teeth. Many patients will experience ophthalmologic defects including vision problems (16%) and retinopathy (15%).7
The cutaneous eruptions of IP may be treated with topical corticosteroids or topical tacrolimus, and vesicles should be left intact and monitored for signs of infection.8,9 Seizures, if present, should be treated with anticonvulsants, and regular neuropsychiatric monitoring and physical rehabilitation may be warranted. Patients should be regularly monitored for retinopathy beginning at the time of diagnosis. Retinal fibrovascular proliferation is treated with xenon laser photocoagulation to reduce the high risk for retinal detachment in this population.10,11 Older and younger at-risk relatives must be evaluated by genetic testing or thorough physical examination to clarify their disease status and determine the need for additional genetic counseling.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012.
- Prevalence and incidence of rare diseases: bibliographic data. Orphanet Report Series, Rare Diseases collection. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Published June 2017. Accessed July 13, 2017.
- Scheuerle AE, Ursini MV. Incontinentia pigmenti. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. Seattle, WA: University of Washington; 2015. http://www.ncbi.nlm.nih.gov/books/NBK1472/. Accessed July 25, 2017.
- James SH, Kimberlin DW. Neonatal herpes simplex virus infection. Infect Dis Clin North Am. 2015;29:391-400.
- Eichenfield LF, Frieden IJ, Mathes E, et al, eds. Neonatal and Infant Dermatology. Philadelphia, PA: Saunders; 2015.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Fusco F, Paciolla M, Conte MI, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014;9:93.
- Jessup CJ, Morgan SC, Cohen LM, et al. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8:944-946.
- Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti [published online June 1, 2009]. Clin Exp Dermatol. 2009;34:E611-E613.
- Nguyen JK, Brady-Mccreery KM. Laser photocoagulation in preproliferative retinopathy of incontinentia pigmenti. J AAPOS. 2001;5:258-259.
- Chen CJ, Han IC, Tian J, et al. Extended follow-up of treated and untreated retinopathy in incontinentia pigmenti: analysis of peripheral vascular changes and incidence of retinal detachment. JAMA Ophthalmol. 2015;133:542-548.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012.
- Prevalence and incidence of rare diseases: bibliographic data. Orphanet Report Series, Rare Diseases collection. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Published June 2017. Accessed July 13, 2017.
- Scheuerle AE, Ursini MV. Incontinentia pigmenti. In: Pagon RA, Adam MP, Ardinger HH, et al, eds. GeneReviews. Seattle, WA: University of Washington; 2015. http://www.ncbi.nlm.nih.gov/books/NBK1472/. Accessed July 25, 2017.
- James SH, Kimberlin DW. Neonatal herpes simplex virus infection. Infect Dis Clin North Am. 2015;29:391-400.
- Eichenfield LF, Frieden IJ, Mathes E, et al, eds. Neonatal and Infant Dermatology. Philadelphia, PA: Saunders; 2015.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Fusco F, Paciolla M, Conte MI, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014;9:93.
- Jessup CJ, Morgan SC, Cohen LM, et al. Incontinentia pigmenti: treatment of IP with topical tacrolimus. J Drugs Dermatol. 2009;8:944-946.
- Kaya TI, Tursen U, Ikizoglu G. Therapeutic use of topical corticosteroids in the vesiculobullous lesions of incontinentia pigmenti [published online June 1, 2009]. Clin Exp Dermatol. 2009;34:E611-E613.
- Nguyen JK, Brady-Mccreery KM. Laser photocoagulation in preproliferative retinopathy of incontinentia pigmenti. J AAPOS. 2001;5:258-259.
- Chen CJ, Han IC, Tian J, et al. Extended follow-up of treated and untreated retinopathy in incontinentia pigmenti: analysis of peripheral vascular changes and incidence of retinal detachment. JAMA Ophthalmol. 2015;133:542-548.
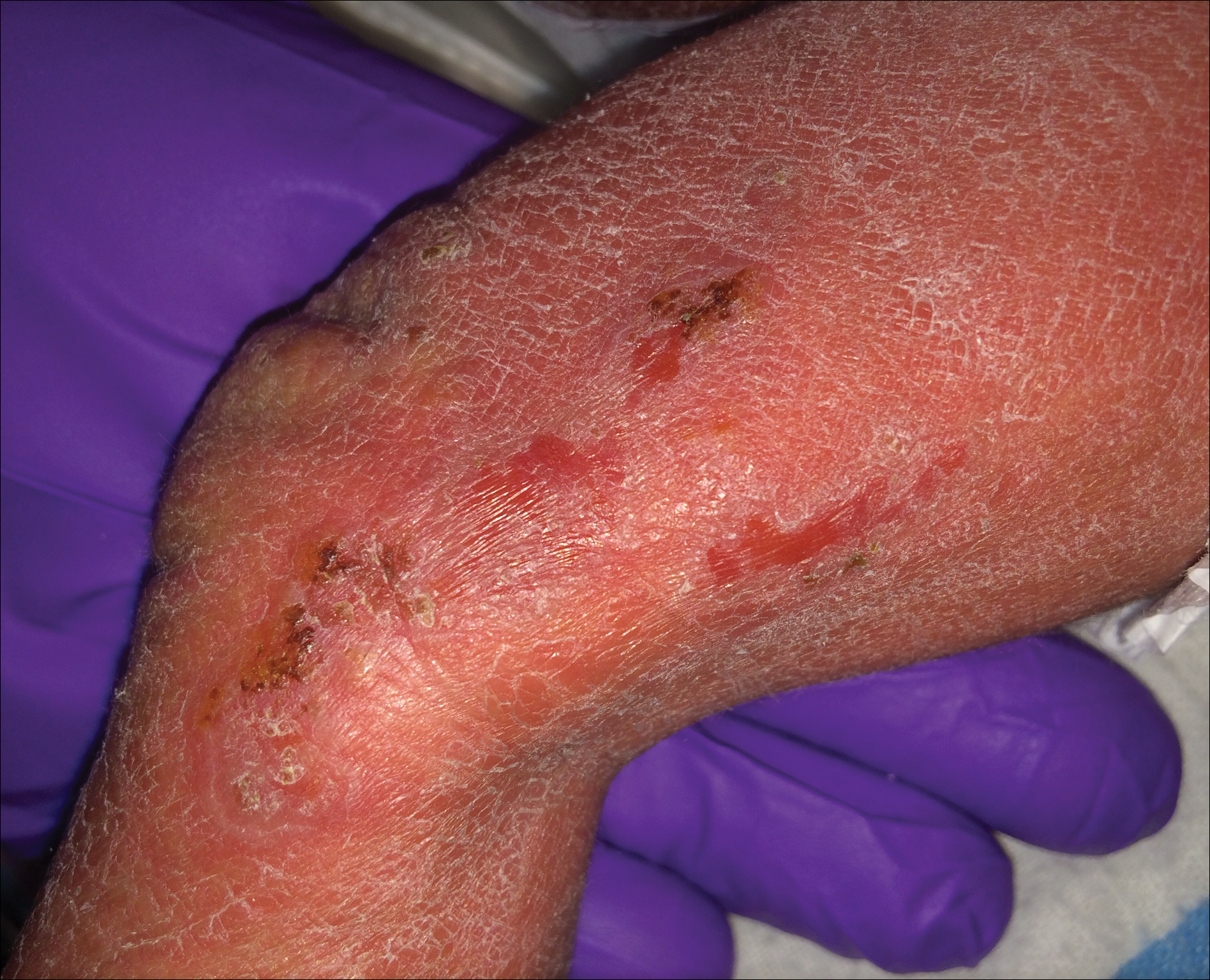
A 1-day-old Hispanic female infant was born via uncomplicated vaginal delivery at 41 weeks' gestation after a normal pregnancy. Linear plaques containing multiple ruptured vesicles and bullae following Blaschko lines were noted on the right medial thigh and anterior arm. The infant was afebrile and generally well-appearing.