User login
FDA approves pembrolizumab for BCG-unresponsive NMIBC with CIS
The Food and Drug Administration has approved pembrolizumab (Keytruda) for the treatment of patients with Bacillus Calmette-Guérin (BCG)–unresponsive, high-risk, non–muscle invasive bladder cancer (NMIBC) with carcinoma in situ (CIS), with or without papillary tumors, who are ineligible for or have elected not to undergo cystectomy.

The approval was based on response in a single-arm trial of 148 patients with high-risk NMIBC, 96 of whom had BCG-unresponsive CIS with or without papillary tumors, the FDA said in a statement.
The complete response rate in the 96 patients with high-risk BCG-unresponsive NMIBC with CIS was 41% (95% confidence interval, 31-51) and median response duration was 16.2 months.
The most common adverse reactions in patients who received pembrolizumab in the trial were fatigue, diarrhea, rash, pruritus, musculoskeletal pain, hematuria, cough, arthralgia, nausea, constipation, urinary tract infection, peripheral edema, hypothyroidism, and nasopharyngitis.
The recommended dose is 200 mg every 3 weeks, the FDA said.
The Food and Drug Administration has approved pembrolizumab (Keytruda) for the treatment of patients with Bacillus Calmette-Guérin (BCG)–unresponsive, high-risk, non–muscle invasive bladder cancer (NMIBC) with carcinoma in situ (CIS), with or without papillary tumors, who are ineligible for or have elected not to undergo cystectomy.
The approval was based on response in a single-arm trial of 148 patients with high-risk NMIBC, 96 of whom had BCG-unresponsive CIS with or without papillary tumors, the FDA said in a statement.
The complete response rate in the 96 patients with high-risk BCG-unresponsive NMIBC with CIS was 41% (95% confidence interval, 31-51) and median response duration was 16.2 months.
The most common adverse reactions in patients who received pembrolizumab in the trial were fatigue, diarrhea, rash, pruritus, musculoskeletal pain, hematuria, cough, arthralgia, nausea, constipation, urinary tract infection, peripheral edema, hypothyroidism, and nasopharyngitis.
The recommended dose is 200 mg every 3 weeks, the FDA said.
The Food and Drug Administration has approved pembrolizumab (Keytruda) for the treatment of patients with Bacillus Calmette-Guérin (BCG)–unresponsive, high-risk, non–muscle invasive bladder cancer (NMIBC) with carcinoma in situ (CIS), with or without papillary tumors, who are ineligible for or have elected not to undergo cystectomy.
The approval was based on response in a single-arm trial of 148 patients with high-risk NMIBC, 96 of whom had BCG-unresponsive CIS with or without papillary tumors, the FDA said in a statement.
The complete response rate in the 96 patients with high-risk BCG-unresponsive NMIBC with CIS was 41% (95% confidence interval, 31-51) and median response duration was 16.2 months.
The most common adverse reactions in patients who received pembrolizumab in the trial were fatigue, diarrhea, rash, pruritus, musculoskeletal pain, hematuria, cough, arthralgia, nausea, constipation, urinary tract infection, peripheral edema, hypothyroidism, and nasopharyngitis.
The recommended dose is 200 mg every 3 weeks, the FDA said.
Mystery pneumonia in China has health officials on alert
An according to a statement from the Centers for Disease Control and Prevention.![]()
As of Jan. 5, 2020, 59 cases of the disease have been reported by the Wuhan Municipal Health Commission. The cluster of cases is linked to the Wuhan South China Seafood City market where – in addition to seafood – chickens, bats, marmots, and other animals were sold. That market has been closed since Jan. 1, 2020, for cleaning and disinfection.
Wuhan health authorities are closely monitoring over 150 contacts for symptoms. Laboratory results have been negative for influenza, avian influenza, adenovirus, and the viruses that caused SARS (severe acute respiratory syndrome) and MERS (Middle East respiratory syndrome). So far, there are no reports of person-to-person transmission or health care worker infection of this pneumonia.
The World Health Organization reported that, as of Dec. 31, 2019, about one-quarter of patients were severely ill with the pneumonia and the rest were stable. Symptoms reported include fever, difficulty breathing, and chest radiographs showing invasive lesions in both lungs. All patients are being treated in isolation and efforts to identify the pathogen are ongoing.

The WHO is monitoring the situation closely and is in close contact with Chinese health authorities.
The CDC has recommended that travelers to Wuhan, a city of over 19 million people, avoid animal and meat markets, avoid contact with sick people, and wash hands often with soap and water. Travelers who have been in Wuhan recently and who experience respiratory symptoms should notify the local health department immediately. In addition, the CDC has issued a Level 1 travel alert, which recommends travelers observe usual precautions against infectious disease.
In addition, the CDC recommends that, for symptomatic patients with a history of travel to Wuhan, caution should be exercised in the health care setting. “Ask such patients to don a surgical mask as soon as they are identified. Conduct their evaluation in a private room with the door closed. Personnel entering the room to evaluate the patient should use contact precautions and wear an N95 disposable facepiece respirator. For patients admitted for inpatient care, implement contact and airborne isolation precautions, in addition to standard precautions, until further information becomes available. For additional infection control guidance see: www.cdc.gov/infectioncontrol/guidelines/isolation/index.html.”
An according to a statement from the Centers for Disease Control and Prevention.![]()
As of Jan. 5, 2020, 59 cases of the disease have been reported by the Wuhan Municipal Health Commission. The cluster of cases is linked to the Wuhan South China Seafood City market where – in addition to seafood – chickens, bats, marmots, and other animals were sold. That market has been closed since Jan. 1, 2020, for cleaning and disinfection.
Wuhan health authorities are closely monitoring over 150 contacts for symptoms. Laboratory results have been negative for influenza, avian influenza, adenovirus, and the viruses that caused SARS (severe acute respiratory syndrome) and MERS (Middle East respiratory syndrome). So far, there are no reports of person-to-person transmission or health care worker infection of this pneumonia.
The World Health Organization reported that, as of Dec. 31, 2019, about one-quarter of patients were severely ill with the pneumonia and the rest were stable. Symptoms reported include fever, difficulty breathing, and chest radiographs showing invasive lesions in both lungs. All patients are being treated in isolation and efforts to identify the pathogen are ongoing.
The WHO is monitoring the situation closely and is in close contact with Chinese health authorities.
The CDC has recommended that travelers to Wuhan, a city of over 19 million people, avoid animal and meat markets, avoid contact with sick people, and wash hands often with soap and water. Travelers who have been in Wuhan recently and who experience respiratory symptoms should notify the local health department immediately. In addition, the CDC has issued a Level 1 travel alert, which recommends travelers observe usual precautions against infectious disease.
In addition, the CDC recommends that, for symptomatic patients with a history of travel to Wuhan, caution should be exercised in the health care setting. “Ask such patients to don a surgical mask as soon as they are identified. Conduct their evaluation in a private room with the door closed. Personnel entering the room to evaluate the patient should use contact precautions and wear an N95 disposable facepiece respirator. For patients admitted for inpatient care, implement contact and airborne isolation precautions, in addition to standard precautions, until further information becomes available. For additional infection control guidance see: www.cdc.gov/infectioncontrol/guidelines/isolation/index.html.”
An according to a statement from the Centers for Disease Control and Prevention.![]()
As of Jan. 5, 2020, 59 cases of the disease have been reported by the Wuhan Municipal Health Commission. The cluster of cases is linked to the Wuhan South China Seafood City market where – in addition to seafood – chickens, bats, marmots, and other animals were sold. That market has been closed since Jan. 1, 2020, for cleaning and disinfection.
Wuhan health authorities are closely monitoring over 150 contacts for symptoms. Laboratory results have been negative for influenza, avian influenza, adenovirus, and the viruses that caused SARS (severe acute respiratory syndrome) and MERS (Middle East respiratory syndrome). So far, there are no reports of person-to-person transmission or health care worker infection of this pneumonia.
The World Health Organization reported that, as of Dec. 31, 2019, about one-quarter of patients were severely ill with the pneumonia and the rest were stable. Symptoms reported include fever, difficulty breathing, and chest radiographs showing invasive lesions in both lungs. All patients are being treated in isolation and efforts to identify the pathogen are ongoing.
The WHO is monitoring the situation closely and is in close contact with Chinese health authorities.
The CDC has recommended that travelers to Wuhan, a city of over 19 million people, avoid animal and meat markets, avoid contact with sick people, and wash hands often with soap and water. Travelers who have been in Wuhan recently and who experience respiratory symptoms should notify the local health department immediately. In addition, the CDC has issued a Level 1 travel alert, which recommends travelers observe usual precautions against infectious disease.
In addition, the CDC recommends that, for symptomatic patients with a history of travel to Wuhan, caution should be exercised in the health care setting. “Ask such patients to don a surgical mask as soon as they are identified. Conduct their evaluation in a private room with the door closed. Personnel entering the room to evaluate the patient should use contact precautions and wear an N95 disposable facepiece respirator. For patients admitted for inpatient care, implement contact and airborne isolation precautions, in addition to standard precautions, until further information becomes available. For additional infection control guidance see: www.cdc.gov/infectioncontrol/guidelines/isolation/index.html.”
Flu records most active December since 2003
The 2019-2020 flu season took a big jump in severity during the last full week of 2019, according to the Centers for Disease Control and Prevention.
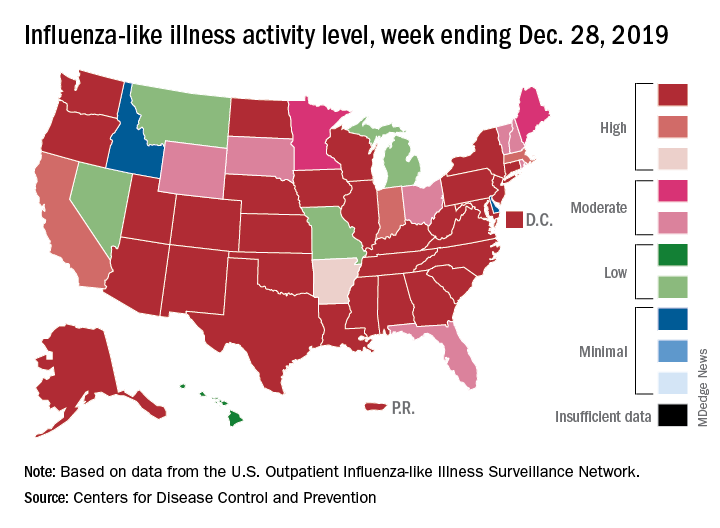
For the week ending Dec. 28, 6.9% of all outpatient visits to health care providers were for influenza-like illness (ILI), the CDC’s influenza division reported Jan. 3. That is up from 5.1% the previous week and is the highest rate recorded in December since 2003. During the flu pandemic season of 2009-2010, the rate peaked in October and dropped to relatively normal levels by the end of November, CDC data show.
This marks the eighth consecutive week that the outpatient visit rate has been at or above the nation’s baseline level of 2.4%, but the data for this week “may in part be influenced by changes in healthcare-seeking behavior that can occur during the holidays,” the CDC suggested.
All those outpatient visits mean that the ILI activity map is getting quite red. Thirty states, as well as the District of Columbia and Puerto Rico, were at the highest level on the CDC’s 1-10 activity scale during the week ending Dec. 28, compared with 20 the week before. Four states were categorized in the “high” range with activity levels of 8 and 9.
There have been approximately 6.4 million flu illnesses so far this season, the CDC estimated, along with 55,000 hospitalizations, although the ILI admission rate of 9.2 per 100,000 population is fairly typical for this time of year.
The week of Dec. 28 also brought reports of five more ILI-related pediatric deaths, which all occurred in the two previous weeks. A total of 27 children have died from the flu so far during the 2019-2020 season, the CDC said.
The 2019-2020 flu season took a big jump in severity during the last full week of 2019, according to the Centers for Disease Control and Prevention.
For the week ending Dec. 28, 6.9% of all outpatient visits to health care providers were for influenza-like illness (ILI), the CDC’s influenza division reported Jan. 3. That is up from 5.1% the previous week and is the highest rate recorded in December since 2003. During the flu pandemic season of 2009-2010, the rate peaked in October and dropped to relatively normal levels by the end of November, CDC data show.
This marks the eighth consecutive week that the outpatient visit rate has been at or above the nation’s baseline level of 2.4%, but the data for this week “may in part be influenced by changes in healthcare-seeking behavior that can occur during the holidays,” the CDC suggested.
All those outpatient visits mean that the ILI activity map is getting quite red. Thirty states, as well as the District of Columbia and Puerto Rico, were at the highest level on the CDC’s 1-10 activity scale during the week ending Dec. 28, compared with 20 the week before. Four states were categorized in the “high” range with activity levels of 8 and 9.
There have been approximately 6.4 million flu illnesses so far this season, the CDC estimated, along with 55,000 hospitalizations, although the ILI admission rate of 9.2 per 100,000 population is fairly typical for this time of year.
The week of Dec. 28 also brought reports of five more ILI-related pediatric deaths, which all occurred in the two previous weeks. A total of 27 children have died from the flu so far during the 2019-2020 season, the CDC said.
The 2019-2020 flu season took a big jump in severity during the last full week of 2019, according to the Centers for Disease Control and Prevention.
For the week ending Dec. 28, 6.9% of all outpatient visits to health care providers were for influenza-like illness (ILI), the CDC’s influenza division reported Jan. 3. That is up from 5.1% the previous week and is the highest rate recorded in December since 2003. During the flu pandemic season of 2009-2010, the rate peaked in October and dropped to relatively normal levels by the end of November, CDC data show.
This marks the eighth consecutive week that the outpatient visit rate has been at or above the nation’s baseline level of 2.4%, but the data for this week “may in part be influenced by changes in healthcare-seeking behavior that can occur during the holidays,” the CDC suggested.
All those outpatient visits mean that the ILI activity map is getting quite red. Thirty states, as well as the District of Columbia and Puerto Rico, were at the highest level on the CDC’s 1-10 activity scale during the week ending Dec. 28, compared with 20 the week before. Four states were categorized in the “high” range with activity levels of 8 and 9.
There have been approximately 6.4 million flu illnesses so far this season, the CDC estimated, along with 55,000 hospitalizations, although the ILI admission rate of 9.2 per 100,000 population is fairly typical for this time of year.
The week of Dec. 28 also brought reports of five more ILI-related pediatric deaths, which all occurred in the two previous weeks. A total of 27 children have died from the flu so far during the 2019-2020 season, the CDC said.
FDA targets flavored cartridge-based e-cigarettes, but says it is not a ‘ban’
but states it is not a “ban.”

On Jan. 2, the agency issued enforcement guidance alerting companies that manufacture, distribute, and sell unauthorized flavored cartridge-based e-cigarettes within the next 30 days will risk FDA enforcement action.
FDA has had the authority to require premarket authorization of all e-cigarettes and other electronic nicotine delivery systems (ENDS) since August 2016, but thus far has exercised enforcement discretion regarding the need for premarket authorization for these types of products.
“By prioritizing enforcement against the products that are most widely used by children, our action today seeks to strike the right public health balance by maintaining e-cigarettes as a potential off-ramp for adults using combustible tobacco while ensuring these products don’t provide an on-ramp to nicotine addiction for our youth,” Department of Health & Human Services Secretary Alex Azar said in a statement.
The action comes in the wake of more than 2,500 vaping-related injuries being reported, including more than 50 deaths associated with vaping reported by the Centers for Disease Control and Prevention (although many are related to the use of tetrahydrocannabinol [THC] within vaping products) and a continued rise in youth use of e-cigarettes noted in government surveys.
The agency noted in a Jan. 2 statement announcing the enforcement action that, to date, no ENDS products have received a premarket authorization, “meaning that all ENDS products currently on the market are considered illegally marketed and are subject to enforcement, at any time, in the FDA’s discretion.”
FDA said it is prioritizing enforcement in 30 days against:
- Any flavored, cartridge-based ENDS product, other than those with a tobacco or menthol flavoring.
- All other ENDS products for which manufacturers are failing to take adequate measures to prevent access by minors.
- Any ENDS product that is targeted to minors or is likely to promote use by minors.
In the last category, this might include labeling or advertising resembling “kid-friendly food and drinks such as juice boxes or kid-friendly cereal; products marketed directly to minors by promoting ease of concealing the product or disguising it as another product; and products marketed with characters designed to appeal to youth,” according to the FDA statement.
As of May 12, FDA also will prioritize enforcement against any ENDS product for which the manufacturer has not submitted a premarket application. The agency will continue to exercise enforcement discretion for up to 1 year on these products if an application has been submitted, pending the review of that application.
“By not prioritizing enforcement against other flavored ENDS products in the same way as flavored cartridge-based ENDS products, the FDA has attempted to balance the public health concerns related to youth use of ENDS products with consideration regarding addicted adult cigarette smokers who may try to use ENDS products to transition away from combustible tobacco products,” the agency stated, adding that cartridge-based ENDS products are most commonly used among youth.
The FDA statement noted that the enforcement priorities outlined in the guidance document were not a “ban” on flavored or cartridge-based ENDS, noting the agency “has already accepted and begun review of several premarket applications for flavored ENDS products through the pathway that Congress established in the Tobacco Control Act. ... If a company can demonstrate to the FDA that a specific product meets the applicable standard set forth by Congress, including considering how the marketing of the product may affect youth initiation and use, then the FDA could authorize that product for sale.”
“Coupled with the recently signed legislation increasing the minimum age of sale of tobacco to 21, we believe this policy balances the urgency with which we must address the public health threat of youth use of e-cigarette products with the potential role that e-cigarettes may play in helping adult smokers transition completely away from combustible tobacco to a potentially less risky form of nicotine delivery,” FDA Commissioner Stephen Hahn, MD, said in a statement. “While we expect that responsible members of industry will comply with premarket requirements, we’re ready to take action against any unauthorized e-cigarette products as outlined in our priorities. We’ll also closely monitor the use rates of all e-cigarette products and take additional steps to address youth use as necessary.”
The American Medical Association criticized the action as not going far enough, even though it was a step in the right direction.
“The AMA is disappointed that menthol flavors, one of the most popular, will still be allowed, and that flavored e-liquids will remain on the market, leaving young people with easy access to alternative flavored e-cigarette products,” AMA President Patrice A. Harris, MD, said in a statement. “If we are serious about tackling this epidemic and keeping these harmful products out of the hands of young people, a total ban on all flavored e-cigarettes, in all forms and at all locations, is prudent and urgently needed. We are pleased the administration committed today to closely monitoring the situation and trends in e-cigarette use among young people, and to taking further action if needed.”
but states it is not a “ban.”
On Jan. 2, the agency issued enforcement guidance alerting companies that manufacture, distribute, and sell unauthorized flavored cartridge-based e-cigarettes within the next 30 days will risk FDA enforcement action.
FDA has had the authority to require premarket authorization of all e-cigarettes and other electronic nicotine delivery systems (ENDS) since August 2016, but thus far has exercised enforcement discretion regarding the need for premarket authorization for these types of products.
“By prioritizing enforcement against the products that are most widely used by children, our action today seeks to strike the right public health balance by maintaining e-cigarettes as a potential off-ramp for adults using combustible tobacco while ensuring these products don’t provide an on-ramp to nicotine addiction for our youth,” Department of Health & Human Services Secretary Alex Azar said in a statement.
The action comes in the wake of more than 2,500 vaping-related injuries being reported, including more than 50 deaths associated with vaping reported by the Centers for Disease Control and Prevention (although many are related to the use of tetrahydrocannabinol [THC] within vaping products) and a continued rise in youth use of e-cigarettes noted in government surveys.
The agency noted in a Jan. 2 statement announcing the enforcement action that, to date, no ENDS products have received a premarket authorization, “meaning that all ENDS products currently on the market are considered illegally marketed and are subject to enforcement, at any time, in the FDA’s discretion.”
FDA said it is prioritizing enforcement in 30 days against:
- Any flavored, cartridge-based ENDS product, other than those with a tobacco or menthol flavoring.
- All other ENDS products for which manufacturers are failing to take adequate measures to prevent access by minors.
- Any ENDS product that is targeted to minors or is likely to promote use by minors.
In the last category, this might include labeling or advertising resembling “kid-friendly food and drinks such as juice boxes or kid-friendly cereal; products marketed directly to minors by promoting ease of concealing the product or disguising it as another product; and products marketed with characters designed to appeal to youth,” according to the FDA statement.
As of May 12, FDA also will prioritize enforcement against any ENDS product for which the manufacturer has not submitted a premarket application. The agency will continue to exercise enforcement discretion for up to 1 year on these products if an application has been submitted, pending the review of that application.
“By not prioritizing enforcement against other flavored ENDS products in the same way as flavored cartridge-based ENDS products, the FDA has attempted to balance the public health concerns related to youth use of ENDS products with consideration regarding addicted adult cigarette smokers who may try to use ENDS products to transition away from combustible tobacco products,” the agency stated, adding that cartridge-based ENDS products are most commonly used among youth.
The FDA statement noted that the enforcement priorities outlined in the guidance document were not a “ban” on flavored or cartridge-based ENDS, noting the agency “has already accepted and begun review of several premarket applications for flavored ENDS products through the pathway that Congress established in the Tobacco Control Act. ... If a company can demonstrate to the FDA that a specific product meets the applicable standard set forth by Congress, including considering how the marketing of the product may affect youth initiation and use, then the FDA could authorize that product for sale.”
“Coupled with the recently signed legislation increasing the minimum age of sale of tobacco to 21, we believe this policy balances the urgency with which we must address the public health threat of youth use of e-cigarette products with the potential role that e-cigarettes may play in helping adult smokers transition completely away from combustible tobacco to a potentially less risky form of nicotine delivery,” FDA Commissioner Stephen Hahn, MD, said in a statement. “While we expect that responsible members of industry will comply with premarket requirements, we’re ready to take action against any unauthorized e-cigarette products as outlined in our priorities. We’ll also closely monitor the use rates of all e-cigarette products and take additional steps to address youth use as necessary.”
The American Medical Association criticized the action as not going far enough, even though it was a step in the right direction.
“The AMA is disappointed that menthol flavors, one of the most popular, will still be allowed, and that flavored e-liquids will remain on the market, leaving young people with easy access to alternative flavored e-cigarette products,” AMA President Patrice A. Harris, MD, said in a statement. “If we are serious about tackling this epidemic and keeping these harmful products out of the hands of young people, a total ban on all flavored e-cigarettes, in all forms and at all locations, is prudent and urgently needed. We are pleased the administration committed today to closely monitoring the situation and trends in e-cigarette use among young people, and to taking further action if needed.”
but states it is not a “ban.”
On Jan. 2, the agency issued enforcement guidance alerting companies that manufacture, distribute, and sell unauthorized flavored cartridge-based e-cigarettes within the next 30 days will risk FDA enforcement action.
FDA has had the authority to require premarket authorization of all e-cigarettes and other electronic nicotine delivery systems (ENDS) since August 2016, but thus far has exercised enforcement discretion regarding the need for premarket authorization for these types of products.
“By prioritizing enforcement against the products that are most widely used by children, our action today seeks to strike the right public health balance by maintaining e-cigarettes as a potential off-ramp for adults using combustible tobacco while ensuring these products don’t provide an on-ramp to nicotine addiction for our youth,” Department of Health & Human Services Secretary Alex Azar said in a statement.
The action comes in the wake of more than 2,500 vaping-related injuries being reported, including more than 50 deaths associated with vaping reported by the Centers for Disease Control and Prevention (although many are related to the use of tetrahydrocannabinol [THC] within vaping products) and a continued rise in youth use of e-cigarettes noted in government surveys.
The agency noted in a Jan. 2 statement announcing the enforcement action that, to date, no ENDS products have received a premarket authorization, “meaning that all ENDS products currently on the market are considered illegally marketed and are subject to enforcement, at any time, in the FDA’s discretion.”
FDA said it is prioritizing enforcement in 30 days against:
- Any flavored, cartridge-based ENDS product, other than those with a tobacco or menthol flavoring.
- All other ENDS products for which manufacturers are failing to take adequate measures to prevent access by minors.
- Any ENDS product that is targeted to minors or is likely to promote use by minors.
In the last category, this might include labeling or advertising resembling “kid-friendly food and drinks such as juice boxes or kid-friendly cereal; products marketed directly to minors by promoting ease of concealing the product or disguising it as another product; and products marketed with characters designed to appeal to youth,” according to the FDA statement.
As of May 12, FDA also will prioritize enforcement against any ENDS product for which the manufacturer has not submitted a premarket application. The agency will continue to exercise enforcement discretion for up to 1 year on these products if an application has been submitted, pending the review of that application.
“By not prioritizing enforcement against other flavored ENDS products in the same way as flavored cartridge-based ENDS products, the FDA has attempted to balance the public health concerns related to youth use of ENDS products with consideration regarding addicted adult cigarette smokers who may try to use ENDS products to transition away from combustible tobacco products,” the agency stated, adding that cartridge-based ENDS products are most commonly used among youth.
The FDA statement noted that the enforcement priorities outlined in the guidance document were not a “ban” on flavored or cartridge-based ENDS, noting the agency “has already accepted and begun review of several premarket applications for flavored ENDS products through the pathway that Congress established in the Tobacco Control Act. ... If a company can demonstrate to the FDA that a specific product meets the applicable standard set forth by Congress, including considering how the marketing of the product may affect youth initiation and use, then the FDA could authorize that product for sale.”
“Coupled with the recently signed legislation increasing the minimum age of sale of tobacco to 21, we believe this policy balances the urgency with which we must address the public health threat of youth use of e-cigarette products with the potential role that e-cigarettes may play in helping adult smokers transition completely away from combustible tobacco to a potentially less risky form of nicotine delivery,” FDA Commissioner Stephen Hahn, MD, said in a statement. “While we expect that responsible members of industry will comply with premarket requirements, we’re ready to take action against any unauthorized e-cigarette products as outlined in our priorities. We’ll also closely monitor the use rates of all e-cigarette products and take additional steps to address youth use as necessary.”
The American Medical Association criticized the action as not going far enough, even though it was a step in the right direction.
“The AMA is disappointed that menthol flavors, one of the most popular, will still be allowed, and that flavored e-liquids will remain on the market, leaving young people with easy access to alternative flavored e-cigarette products,” AMA President Patrice A. Harris, MD, said in a statement. “If we are serious about tackling this epidemic and keeping these harmful products out of the hands of young people, a total ban on all flavored e-cigarettes, in all forms and at all locations, is prudent and urgently needed. We are pleased the administration committed today to closely monitoring the situation and trends in e-cigarette use among young people, and to taking further action if needed.”
Early increase in flu activity shows no signs of slowing
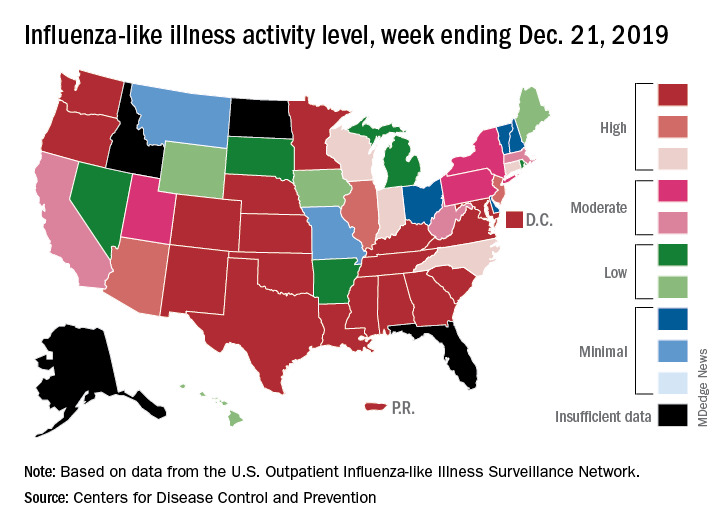
An important measure of U.S. flu activity for the 2019-2020 season has already surpassed last season’s high, and more than half the states are experiencing high levels of activity, according to the Centers for Disease Control and Prevention.
reported Dec. 27.
The last time the outpatient visit rate was higher than that was in February of the 2017-2018 season, when it peaked at 7.5%. The peak month of flu activity occurs most often – about once every 3 years – in February, and the odds of a December peak are about one in five, the CDC has said.
Outpatient illness activity also increased at the state level during the week ending Dec. 21. There were 20 jurisdictions – 18 states, the District of Columbia, and Puerto Rico – at level 10 on the CDC’s 1-10 scale of activity, compared with 13 the previous week, and the number of jurisdictions in the “high” range (levels 8-10) jumped from 21 to 28, the CDC data show.
The influenza division estimated that there have been 4.6 million flu illnesses so far this season, nearly a million more than the total after last week, along with 39,000 hospitalizations. The overall hospitalization rate for the season is up to 6.6 per 100,000 population, which is about average at this point. The proportion of deaths attributed to pneumonia and influenza increased to 5.7%, which is below the epidemic threshold, the CDC said.
Three pediatric deaths related to influenza-like illness were reported during the week ending Dec. 21, two of which occurred in an earlier week. For the 2019-2020 season so far, a total of 22 pediatric deaths have been reported to the CDC.
An important measure of U.S. flu activity for the 2019-2020 season has already surpassed last season’s high, and more than half the states are experiencing high levels of activity, according to the Centers for Disease Control and Prevention.
reported Dec. 27.
The last time the outpatient visit rate was higher than that was in February of the 2017-2018 season, when it peaked at 7.5%. The peak month of flu activity occurs most often – about once every 3 years – in February, and the odds of a December peak are about one in five, the CDC has said.
Outpatient illness activity also increased at the state level during the week ending Dec. 21. There were 20 jurisdictions – 18 states, the District of Columbia, and Puerto Rico – at level 10 on the CDC’s 1-10 scale of activity, compared with 13 the previous week, and the number of jurisdictions in the “high” range (levels 8-10) jumped from 21 to 28, the CDC data show.
The influenza division estimated that there have been 4.6 million flu illnesses so far this season, nearly a million more than the total after last week, along with 39,000 hospitalizations. The overall hospitalization rate for the season is up to 6.6 per 100,000 population, which is about average at this point. The proportion of deaths attributed to pneumonia and influenza increased to 5.7%, which is below the epidemic threshold, the CDC said.
Three pediatric deaths related to influenza-like illness were reported during the week ending Dec. 21, two of which occurred in an earlier week. For the 2019-2020 season so far, a total of 22 pediatric deaths have been reported to the CDC.
An important measure of U.S. flu activity for the 2019-2020 season has already surpassed last season’s high, and more than half the states are experiencing high levels of activity, according to the Centers for Disease Control and Prevention.
reported Dec. 27.
The last time the outpatient visit rate was higher than that was in February of the 2017-2018 season, when it peaked at 7.5%. The peak month of flu activity occurs most often – about once every 3 years – in February, and the odds of a December peak are about one in five, the CDC has said.
Outpatient illness activity also increased at the state level during the week ending Dec. 21. There were 20 jurisdictions – 18 states, the District of Columbia, and Puerto Rico – at level 10 on the CDC’s 1-10 scale of activity, compared with 13 the previous week, and the number of jurisdictions in the “high” range (levels 8-10) jumped from 21 to 28, the CDC data show.
The influenza division estimated that there have been 4.6 million flu illnesses so far this season, nearly a million more than the total after last week, along with 39,000 hospitalizations. The overall hospitalization rate for the season is up to 6.6 per 100,000 population, which is about average at this point. The proportion of deaths attributed to pneumonia and influenza increased to 5.7%, which is below the epidemic threshold, the CDC said.
Three pediatric deaths related to influenza-like illness were reported during the week ending Dec. 21, two of which occurred in an earlier week. For the 2019-2020 season so far, a total of 22 pediatric deaths have been reported to the CDC.
The measles comeback of 2019
Measles made a comeback in 2019.
The Centers for Disease Control and Prevention reported that, as of Dec. 5, 2019, 1,276 individual cases of measles of measles were confirmed in 31 states, the largest number since 1992. This number is a major uptick in cases, compared with previous years since 2000 when the CDC declared measles eliminated from the United States. No deaths have been reported for 2019.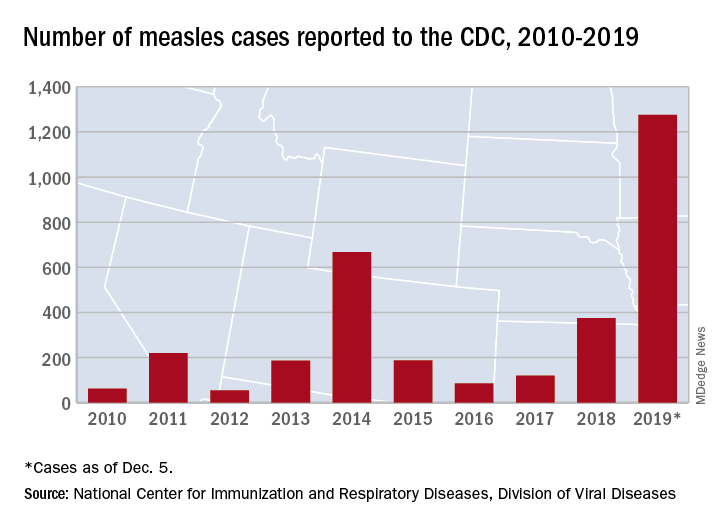
Three-quarters of these cases in 2019 were linked to recent outbreaks in New York and occurred in primarily in underimmunized, close-knit communities and in patients with links to international travel. A total of 124 of the people who got measles this year were hospitalized, and 61 reported having complications, including pneumonia and encephalitis. The overall median patient age was 6 years (31% aged 1-4 years, 27% aged 5-17 years, and 29% aged at least 18 years).
The good news is that most of these cases occurred in unvaccinated patients. The national vaccination rate for the almost 4 million kindergartners reported as enrolled in 2018-2019 was 94.7% for two doses of the MMR vaccine, falling just short of the CDC recommended 95% vaccination rate threshold. The CDC reported an approximate 2.5% rate of vaccination exemptions among school-age children.
The bad news is that, despite the high rate of MMR vaccination rates among U.S. children, there are gaps in measles protection in the U.S. population because of factors leaving patients immunocompromised and antivaccination sentiment that has led some parents to defer or refuse the MMR.
In addition, adults who were vaccinated prior to 1968 with either inactivated measles vaccine or measles vaccine of unknown type may have limited immunity. The inactivated measles vaccine, which was available in 1963-1967, did not achieve effective measles protection.
A global measles surge
While antivaccination sentiment contributed to the 2019 measles cases, a more significant factor may be the global surge of measles. More than 140,000 people worldwide died from measles in 2018, according to the World Health Organization and the CDC.
“[Recent data on measles] indicates that during the first 6 months of the year there have been more measles cases reported worldwide than in any year since 2006. From Jan. 1 to July 31, 2019, 182 countries reported 364,808 measles cases to the WHO. This surpasses the 129,239 reported during the same time period in 2018. WHO regions with the biggest increases in cases include the African region (900%), the Western Pacific region (230%), and the European region (150%),” according to a CDC report.
Studies on hospitalization and complications linked to measles in the United States are scarce, but two outbreaks in Minnesota (2011 and 2017) provided some data on what to expect if the measles surge continues into 2020. The investigators found that poor feeding was a primary reason for admission (97%); additional complications included otitis media (42%), pneumonia (30%), and tracheitis (6%). Three-quarters received antibiotics, 30% required oxygen, and 21% received vitamin A. Median length of stay was 3.7 days (range, 1.1-26.2 days) (Pediatr Infect Dis J. 2019 Jun;38[6]:547-52. doi: 10.1097/INF.0000000000002221).
‘Immunological amnesia’
Infection with the measles virus appears to reduce immunity to other pathogens, according to a paper published in Science (2019 Nov 1;366[6465]599-606).
The hypothesis that the measles virus could cause “immunological amnesia” by impairing immune memory is supported by early research showing children with measles had negative cutaneous tuberculin reactions after having previously tested positive.
“Subsequent studies have shown decreased interferon signaling, skewed cytokine responses, lymphopenia, and suppression of lymphocyte proliferation shortly after infection,” wrote Michael Mina, MD, from Brigham and Women’s Hospital in Boston, and coauthors.
“Given the variation in the degree of immune repertoire modulation we observed, we anticipate that future risk of morbidity and mortality after measles would not be homogeneous but would be skewed toward individuals with the most severe elimination of immunological memory,” they wrote. “These findings underscore the crucial need for continued widespread vaccination.”
In this study, researchers compared the levels of around 400 pathogen-specific antibodies in blood samples from 77 unvaccinated children, taken before and 2 months after natural measles infection, with 5 unvaccinated children who did not contract measles. A total of 34 children experienced mild measles, and 43 had severe measles.
They found that the samples taken after measles infection showed “substantial” reductions in the number of pathogen epitopes, compared with the samples from children who did not get infected with measles.
This amounted to approximately a 20% mean reduction in overall diversity or size of the antibody repertoire. However, in children who experienced severe measles, there was a median loss of 40% (range, 11%-62%) of antibody repertoire, compared with a median of 33% (range, 12%-73%) range in children who experienced mild infection. Meanwhile, the control subjects retained approximately 90% of their antibody repertoire over a similar or longer time period. Some children lost up to 70% of antibodies for specific pathogens.
Maternal-acquired immunity fades
In another study of measles immunity, maternal antibodies were found to be insufficient to provide immunity to infants after 6 months.
The study of 196 infants showed that maternal measles antibodies had dropped below the protective threshold by 3 months of age – well before the recommended age of 12-15 months for the first dose of MMR vaccine.
The odds of inadequate protection doubled for each additional month of age, Michelle Science, MD, of the University of Toronto and associates reported in Pediatrics (2019 Dec 1. doi 10.1542/peds.2019-0630).
“The widening gap between loss of maternal antibodies and measles vaccination described in our study leaves infants vulnerable to measles for much of their infancy and highlights the need for further research to support public health policy,” Dr. Science and colleagues wrote.
The researchers randomly selected 25 samples for each of eight different age groups: up to 30 days old; 1 month (31-60 days), 2 months (61-89 days), 3 months (90-119 days), 4 months, 5 months, 6-9 months, and 9-11 months.
Just over half the babies (56%) were male, and 35% had an underlying condition, but none had conditions that might affect antibody levels. The conditions were primarily a developmental delay or otherwise affecting the central nervous system, liver, or gastrointestinal function. Mean maternal age was 32 years.
To ensure high test sensitivity, the researchers used the plaque-reduction neutralization test to test for measles-neutralizing antibodies instead of using enzyme-linked immunosorbent assay, because “ELISA sensitivity decreases as antibody titers decrease,” Dr. Science and colleagues wrote. They used a neutralization titer of less than 192 mIU/mL as the threshold for protection against measles.
When the researchers calculated the predicted standardized mean antibody titer for infants with a mother aged 32 years, they determined their mean to be 541 mIU/mL at 1 month, 142 mIU/mL at 3 months (below the measles threshold of susceptibility of 192 mIU/mL), and 64 mIU/mL at 6 months. None of the infants had measles antibodies above the protective threshold at 6 months old, the authors noted.
Children’s odds of susceptibility to measles doubled for each additional month of age, after adjustment for infant sex and maternal age (odds ratio, 2.13). Children’s likelihood of susceptibility to measles modestly increased as maternal age increased in 5-year increments from 25 to 40 years.
Children with an underlying conditions had greater susceptibility to measles (83%), compared with those without a comorbidity (68%, P = .03). No difference in susceptibility existed between males and females or based on gestational age at birth (ranging from 37 to 41 weeks).
The Advisory Committee on Immunization Practices permits measles vaccination “as early as 6 months for infants who plan to travel internationally, infants with ongoing risk for exposure during measles outbreaks and as postexposure prophylaxis,” Huong Q. McLean, PhD, of Marshfield (Wisc.) Clinic Research Institute, and Walter A. Orenstein, MD, of Emory University, Atlanta, noted in an editorial.
The research was funded by the Public Health Ontario Project Initiation Fund. The authors had no relevant financial disclosures.
Bianca Nogrady and Tara Haelle contributed to this story.
Measles made a comeback in 2019.
The Centers for Disease Control and Prevention reported that, as of Dec. 5, 2019, 1,276 individual cases of measles of measles were confirmed in 31 states, the largest number since 1992. This number is a major uptick in cases, compared with previous years since 2000 when the CDC declared measles eliminated from the United States. No deaths have been reported for 2019.
Three-quarters of these cases in 2019 were linked to recent outbreaks in New York and occurred in primarily in underimmunized, close-knit communities and in patients with links to international travel. A total of 124 of the people who got measles this year were hospitalized, and 61 reported having complications, including pneumonia and encephalitis. The overall median patient age was 6 years (31% aged 1-4 years, 27% aged 5-17 years, and 29% aged at least 18 years).
The good news is that most of these cases occurred in unvaccinated patients. The national vaccination rate for the almost 4 million kindergartners reported as enrolled in 2018-2019 was 94.7% for two doses of the MMR vaccine, falling just short of the CDC recommended 95% vaccination rate threshold. The CDC reported an approximate 2.5% rate of vaccination exemptions among school-age children.
The bad news is that, despite the high rate of MMR vaccination rates among U.S. children, there are gaps in measles protection in the U.S. population because of factors leaving patients immunocompromised and antivaccination sentiment that has led some parents to defer or refuse the MMR.
In addition, adults who were vaccinated prior to 1968 with either inactivated measles vaccine or measles vaccine of unknown type may have limited immunity. The inactivated measles vaccine, which was available in 1963-1967, did not achieve effective measles protection.
A global measles surge
While antivaccination sentiment contributed to the 2019 measles cases, a more significant factor may be the global surge of measles. More than 140,000 people worldwide died from measles in 2018, according to the World Health Organization and the CDC.
“[Recent data on measles] indicates that during the first 6 months of the year there have been more measles cases reported worldwide than in any year since 2006. From Jan. 1 to July 31, 2019, 182 countries reported 364,808 measles cases to the WHO. This surpasses the 129,239 reported during the same time period in 2018. WHO regions with the biggest increases in cases include the African region (900%), the Western Pacific region (230%), and the European region (150%),” according to a CDC report.
Studies on hospitalization and complications linked to measles in the United States are scarce, but two outbreaks in Minnesota (2011 and 2017) provided some data on what to expect if the measles surge continues into 2020. The investigators found that poor feeding was a primary reason for admission (97%); additional complications included otitis media (42%), pneumonia (30%), and tracheitis (6%). Three-quarters received antibiotics, 30% required oxygen, and 21% received vitamin A. Median length of stay was 3.7 days (range, 1.1-26.2 days) (Pediatr Infect Dis J. 2019 Jun;38[6]:547-52. doi: 10.1097/INF.0000000000002221).
‘Immunological amnesia’
Infection with the measles virus appears to reduce immunity to other pathogens, according to a paper published in Science (2019 Nov 1;366[6465]599-606).
The hypothesis that the measles virus could cause “immunological amnesia” by impairing immune memory is supported by early research showing children with measles had negative cutaneous tuberculin reactions after having previously tested positive.
“Subsequent studies have shown decreased interferon signaling, skewed cytokine responses, lymphopenia, and suppression of lymphocyte proliferation shortly after infection,” wrote Michael Mina, MD, from Brigham and Women’s Hospital in Boston, and coauthors.
“Given the variation in the degree of immune repertoire modulation we observed, we anticipate that future risk of morbidity and mortality after measles would not be homogeneous but would be skewed toward individuals with the most severe elimination of immunological memory,” they wrote. “These findings underscore the crucial need for continued widespread vaccination.”
In this study, researchers compared the levels of around 400 pathogen-specific antibodies in blood samples from 77 unvaccinated children, taken before and 2 months after natural measles infection, with 5 unvaccinated children who did not contract measles. A total of 34 children experienced mild measles, and 43 had severe measles.
They found that the samples taken after measles infection showed “substantial” reductions in the number of pathogen epitopes, compared with the samples from children who did not get infected with measles.
This amounted to approximately a 20% mean reduction in overall diversity or size of the antibody repertoire. However, in children who experienced severe measles, there was a median loss of 40% (range, 11%-62%) of antibody repertoire, compared with a median of 33% (range, 12%-73%) range in children who experienced mild infection. Meanwhile, the control subjects retained approximately 90% of their antibody repertoire over a similar or longer time period. Some children lost up to 70% of antibodies for specific pathogens.
Maternal-acquired immunity fades
In another study of measles immunity, maternal antibodies were found to be insufficient to provide immunity to infants after 6 months.
The study of 196 infants showed that maternal measles antibodies had dropped below the protective threshold by 3 months of age – well before the recommended age of 12-15 months for the first dose of MMR vaccine.
The odds of inadequate protection doubled for each additional month of age, Michelle Science, MD, of the University of Toronto and associates reported in Pediatrics (2019 Dec 1. doi 10.1542/peds.2019-0630).
“The widening gap between loss of maternal antibodies and measles vaccination described in our study leaves infants vulnerable to measles for much of their infancy and highlights the need for further research to support public health policy,” Dr. Science and colleagues wrote.
The researchers randomly selected 25 samples for each of eight different age groups: up to 30 days old; 1 month (31-60 days), 2 months (61-89 days), 3 months (90-119 days), 4 months, 5 months, 6-9 months, and 9-11 months.
Just over half the babies (56%) were male, and 35% had an underlying condition, but none had conditions that might affect antibody levels. The conditions were primarily a developmental delay or otherwise affecting the central nervous system, liver, or gastrointestinal function. Mean maternal age was 32 years.
To ensure high test sensitivity, the researchers used the plaque-reduction neutralization test to test for measles-neutralizing antibodies instead of using enzyme-linked immunosorbent assay, because “ELISA sensitivity decreases as antibody titers decrease,” Dr. Science and colleagues wrote. They used a neutralization titer of less than 192 mIU/mL as the threshold for protection against measles.
When the researchers calculated the predicted standardized mean antibody titer for infants with a mother aged 32 years, they determined their mean to be 541 mIU/mL at 1 month, 142 mIU/mL at 3 months (below the measles threshold of susceptibility of 192 mIU/mL), and 64 mIU/mL at 6 months. None of the infants had measles antibodies above the protective threshold at 6 months old, the authors noted.
Children’s odds of susceptibility to measles doubled for each additional month of age, after adjustment for infant sex and maternal age (odds ratio, 2.13). Children’s likelihood of susceptibility to measles modestly increased as maternal age increased in 5-year increments from 25 to 40 years.
Children with an underlying conditions had greater susceptibility to measles (83%), compared with those without a comorbidity (68%, P = .03). No difference in susceptibility existed between males and females or based on gestational age at birth (ranging from 37 to 41 weeks).
The Advisory Committee on Immunization Practices permits measles vaccination “as early as 6 months for infants who plan to travel internationally, infants with ongoing risk for exposure during measles outbreaks and as postexposure prophylaxis,” Huong Q. McLean, PhD, of Marshfield (Wisc.) Clinic Research Institute, and Walter A. Orenstein, MD, of Emory University, Atlanta, noted in an editorial.
The research was funded by the Public Health Ontario Project Initiation Fund. The authors had no relevant financial disclosures.
Bianca Nogrady and Tara Haelle contributed to this story.
Measles made a comeback in 2019.
The Centers for Disease Control and Prevention reported that, as of Dec. 5, 2019, 1,276 individual cases of measles of measles were confirmed in 31 states, the largest number since 1992. This number is a major uptick in cases, compared with previous years since 2000 when the CDC declared measles eliminated from the United States. No deaths have been reported for 2019.
Three-quarters of these cases in 2019 were linked to recent outbreaks in New York and occurred in primarily in underimmunized, close-knit communities and in patients with links to international travel. A total of 124 of the people who got measles this year were hospitalized, and 61 reported having complications, including pneumonia and encephalitis. The overall median patient age was 6 years (31% aged 1-4 years, 27% aged 5-17 years, and 29% aged at least 18 years).
The good news is that most of these cases occurred in unvaccinated patients. The national vaccination rate for the almost 4 million kindergartners reported as enrolled in 2018-2019 was 94.7% for two doses of the MMR vaccine, falling just short of the CDC recommended 95% vaccination rate threshold. The CDC reported an approximate 2.5% rate of vaccination exemptions among school-age children.
The bad news is that, despite the high rate of MMR vaccination rates among U.S. children, there are gaps in measles protection in the U.S. population because of factors leaving patients immunocompromised and antivaccination sentiment that has led some parents to defer or refuse the MMR.
In addition, adults who were vaccinated prior to 1968 with either inactivated measles vaccine or measles vaccine of unknown type may have limited immunity. The inactivated measles vaccine, which was available in 1963-1967, did not achieve effective measles protection.
A global measles surge
While antivaccination sentiment contributed to the 2019 measles cases, a more significant factor may be the global surge of measles. More than 140,000 people worldwide died from measles in 2018, according to the World Health Organization and the CDC.
“[Recent data on measles] indicates that during the first 6 months of the year there have been more measles cases reported worldwide than in any year since 2006. From Jan. 1 to July 31, 2019, 182 countries reported 364,808 measles cases to the WHO. This surpasses the 129,239 reported during the same time period in 2018. WHO regions with the biggest increases in cases include the African region (900%), the Western Pacific region (230%), and the European region (150%),” according to a CDC report.
Studies on hospitalization and complications linked to measles in the United States are scarce, but two outbreaks in Minnesota (2011 and 2017) provided some data on what to expect if the measles surge continues into 2020. The investigators found that poor feeding was a primary reason for admission (97%); additional complications included otitis media (42%), pneumonia (30%), and tracheitis (6%). Three-quarters received antibiotics, 30% required oxygen, and 21% received vitamin A. Median length of stay was 3.7 days (range, 1.1-26.2 days) (Pediatr Infect Dis J. 2019 Jun;38[6]:547-52. doi: 10.1097/INF.0000000000002221).
‘Immunological amnesia’
Infection with the measles virus appears to reduce immunity to other pathogens, according to a paper published in Science (2019 Nov 1;366[6465]599-606).
The hypothesis that the measles virus could cause “immunological amnesia” by impairing immune memory is supported by early research showing children with measles had negative cutaneous tuberculin reactions after having previously tested positive.
“Subsequent studies have shown decreased interferon signaling, skewed cytokine responses, lymphopenia, and suppression of lymphocyte proliferation shortly after infection,” wrote Michael Mina, MD, from Brigham and Women’s Hospital in Boston, and coauthors.
“Given the variation in the degree of immune repertoire modulation we observed, we anticipate that future risk of morbidity and mortality after measles would not be homogeneous but would be skewed toward individuals with the most severe elimination of immunological memory,” they wrote. “These findings underscore the crucial need for continued widespread vaccination.”
In this study, researchers compared the levels of around 400 pathogen-specific antibodies in blood samples from 77 unvaccinated children, taken before and 2 months after natural measles infection, with 5 unvaccinated children who did not contract measles. A total of 34 children experienced mild measles, and 43 had severe measles.
They found that the samples taken after measles infection showed “substantial” reductions in the number of pathogen epitopes, compared with the samples from children who did not get infected with measles.
This amounted to approximately a 20% mean reduction in overall diversity or size of the antibody repertoire. However, in children who experienced severe measles, there was a median loss of 40% (range, 11%-62%) of antibody repertoire, compared with a median of 33% (range, 12%-73%) range in children who experienced mild infection. Meanwhile, the control subjects retained approximately 90% of their antibody repertoire over a similar or longer time period. Some children lost up to 70% of antibodies for specific pathogens.
Maternal-acquired immunity fades
In another study of measles immunity, maternal antibodies were found to be insufficient to provide immunity to infants after 6 months.
The study of 196 infants showed that maternal measles antibodies had dropped below the protective threshold by 3 months of age – well before the recommended age of 12-15 months for the first dose of MMR vaccine.
The odds of inadequate protection doubled for each additional month of age, Michelle Science, MD, of the University of Toronto and associates reported in Pediatrics (2019 Dec 1. doi 10.1542/peds.2019-0630).
“The widening gap between loss of maternal antibodies and measles vaccination described in our study leaves infants vulnerable to measles for much of their infancy and highlights the need for further research to support public health policy,” Dr. Science and colleagues wrote.
The researchers randomly selected 25 samples for each of eight different age groups: up to 30 days old; 1 month (31-60 days), 2 months (61-89 days), 3 months (90-119 days), 4 months, 5 months, 6-9 months, and 9-11 months.
Just over half the babies (56%) were male, and 35% had an underlying condition, but none had conditions that might affect antibody levels. The conditions were primarily a developmental delay or otherwise affecting the central nervous system, liver, or gastrointestinal function. Mean maternal age was 32 years.
To ensure high test sensitivity, the researchers used the plaque-reduction neutralization test to test for measles-neutralizing antibodies instead of using enzyme-linked immunosorbent assay, because “ELISA sensitivity decreases as antibody titers decrease,” Dr. Science and colleagues wrote. They used a neutralization titer of less than 192 mIU/mL as the threshold for protection against measles.
When the researchers calculated the predicted standardized mean antibody titer for infants with a mother aged 32 years, they determined their mean to be 541 mIU/mL at 1 month, 142 mIU/mL at 3 months (below the measles threshold of susceptibility of 192 mIU/mL), and 64 mIU/mL at 6 months. None of the infants had measles antibodies above the protective threshold at 6 months old, the authors noted.
Children’s odds of susceptibility to measles doubled for each additional month of age, after adjustment for infant sex and maternal age (odds ratio, 2.13). Children’s likelihood of susceptibility to measles modestly increased as maternal age increased in 5-year increments from 25 to 40 years.
Children with an underlying conditions had greater susceptibility to measles (83%), compared with those without a comorbidity (68%, P = .03). No difference in susceptibility existed between males and females or based on gestational age at birth (ranging from 37 to 41 weeks).
The Advisory Committee on Immunization Practices permits measles vaccination “as early as 6 months for infants who plan to travel internationally, infants with ongoing risk for exposure during measles outbreaks and as postexposure prophylaxis,” Huong Q. McLean, PhD, of Marshfield (Wisc.) Clinic Research Institute, and Walter A. Orenstein, MD, of Emory University, Atlanta, noted in an editorial.
The research was funded by the Public Health Ontario Project Initiation Fund. The authors had no relevant financial disclosures.
Bianca Nogrady and Tara Haelle contributed to this story.
FDA okays first generics for Eliquis
The Food and Drug Administration has approved two applications for first generic versions of apixaban (Eliquis, Bristol-Myers Squibb/Pfizer) tablets to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation.

The FDA gave the go-ahead to market generic versions of apixaban to Micro Labs Limited and Mylan Pharmaceuticals.
“Today’s approvals of the first generics of apixaban are an example of how the FDA’s generic drug program improves access to lower-cost, safe, and high-quality medicines,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement today. “These approvals mark the first generic approvals of a direct oral anticoagulant.”
It is estimated that between 2.7 and 6.1 million people in the United States have atrial fibrillation. Many of these individuals use anticoagulants or anticlotting drugs to reduce that risk. Direct oral anticoagulants, however, do not require repeated blood testing.
Apixaban was approved by the FDA in December 2012 for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Additional indications in the United States are to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE) and as DVT/PE prophylaxis in adults who have undergone hip or knee replacement surgery.
The FDA reminds providers that, as with brand name apixaban, generic versions must be dispensed with a medication guide that provides important instructions on the drug’s uses and risks. Healthcare professionals should counsel patients on signs and symptoms of possible bleeding.
As with other FDA-approved anticlotting drugs, bleeding, including life-threatening and fatal bleeding, is the most serious risk with apixaban.
Full prescribing information for the drug also warns about the increased risk for stroke in patients who discontinue use of the drug without taking some other form of anticoagulation. Epidural or spinal hematoma, which may cause long-term or permanent paralysis, may occur in patients treated with apixaban who are undergoing spinal epidural anesthesia or spinal puncture.
This story first appeared on Medscape.com.
The Food and Drug Administration has approved two applications for first generic versions of apixaban (Eliquis, Bristol-Myers Squibb/Pfizer) tablets to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation.
The FDA gave the go-ahead to market generic versions of apixaban to Micro Labs Limited and Mylan Pharmaceuticals.
“Today’s approvals of the first generics of apixaban are an example of how the FDA’s generic drug program improves access to lower-cost, safe, and high-quality medicines,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement today. “These approvals mark the first generic approvals of a direct oral anticoagulant.”
It is estimated that between 2.7 and 6.1 million people in the United States have atrial fibrillation. Many of these individuals use anticoagulants or anticlotting drugs to reduce that risk. Direct oral anticoagulants, however, do not require repeated blood testing.
Apixaban was approved by the FDA in December 2012 for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Additional indications in the United States are to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE) and as DVT/PE prophylaxis in adults who have undergone hip or knee replacement surgery.
The FDA reminds providers that, as with brand name apixaban, generic versions must be dispensed with a medication guide that provides important instructions on the drug’s uses and risks. Healthcare professionals should counsel patients on signs and symptoms of possible bleeding.
As with other FDA-approved anticlotting drugs, bleeding, including life-threatening and fatal bleeding, is the most serious risk with apixaban.
Full prescribing information for the drug also warns about the increased risk for stroke in patients who discontinue use of the drug without taking some other form of anticoagulation. Epidural or spinal hematoma, which may cause long-term or permanent paralysis, may occur in patients treated with apixaban who are undergoing spinal epidural anesthesia or spinal puncture.
This story first appeared on Medscape.com.
The Food and Drug Administration has approved two applications for first generic versions of apixaban (Eliquis, Bristol-Myers Squibb/Pfizer) tablets to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation.
The FDA gave the go-ahead to market generic versions of apixaban to Micro Labs Limited and Mylan Pharmaceuticals.
“Today’s approvals of the first generics of apixaban are an example of how the FDA’s generic drug program improves access to lower-cost, safe, and high-quality medicines,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement today. “These approvals mark the first generic approvals of a direct oral anticoagulant.”
It is estimated that between 2.7 and 6.1 million people in the United States have atrial fibrillation. Many of these individuals use anticoagulants or anticlotting drugs to reduce that risk. Direct oral anticoagulants, however, do not require repeated blood testing.
Apixaban was approved by the FDA in December 2012 for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Additional indications in the United States are to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE) and as DVT/PE prophylaxis in adults who have undergone hip or knee replacement surgery.
The FDA reminds providers that, as with brand name apixaban, generic versions must be dispensed with a medication guide that provides important instructions on the drug’s uses and risks. Healthcare professionals should counsel patients on signs and symptoms of possible bleeding.
As with other FDA-approved anticlotting drugs, bleeding, including life-threatening and fatal bleeding, is the most serious risk with apixaban.
Full prescribing information for the drug also warns about the increased risk for stroke in patients who discontinue use of the drug without taking some other form of anticoagulation. Epidural or spinal hematoma, which may cause long-term or permanent paralysis, may occur in patients treated with apixaban who are undergoing spinal epidural anesthesia or spinal puncture.
This story first appeared on Medscape.com.
FDA okays ubrogepant for acute migraine treatment
The Food and Drug Administration has approved ubrogepant (Ubrelvy, Allergan) for the acute treatment of migraine with or without aura in adults.
Ubrogepant is the first drug in the class of oral calcitonin gene–related peptide receptor antagonists approved for the acute treatment of migraine. It is approved in two dose strengths (50 mg and 100 mg).
The drug is not indicated, however, for the preventive treatment of migraine.
“Migraine is an often disabling condition that affects an estimated 37 million people in the U.S.,” Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research, said in an FDA news release.
Ubrogepant represents “an important new option for the acute treatment of migraine in adults, as it is the first drug in its class approved for this indication. The FDA is pleased to approve a novel treatment for patients suffering from migraine and will continue to work with stakeholders to promote the development of new safe and effective migraine therapies,” added Dr. Dunn.
The safety and efficacy of ubrogepant for the acute treatment of migraine was demonstrated in two randomized, double-blind, placebo-controlled trials (ACHIEVE I and ACHIEVE II). In total, 1,439 adults with a history of migraine, with and without aura, received ubrogepant to treat an ongoing migraine.
“Both 50-mg and 100-mg dose strengths demonstrated significantly greater rates of pain freedom and freedom from the most bothersome migraine-associated symptom at 2 hours, compared with placebo,” Allergan said in a news release announcing approval.
The most common side effects reported by patients in the clinical trials were nausea, tiredness, and dry mouth. Ubrogepant is contraindicated for coadministration with strong CYP3A4 inhibitors.
The company expects to have ubrogepant available in the first quarter of 2020.
A version of this story originally appeared on Medscape.com.
The Food and Drug Administration has approved ubrogepant (Ubrelvy, Allergan) for the acute treatment of migraine with or without aura in adults.
Ubrogepant is the first drug in the class of oral calcitonin gene–related peptide receptor antagonists approved for the acute treatment of migraine. It is approved in two dose strengths (50 mg and 100 mg).
The drug is not indicated, however, for the preventive treatment of migraine.
“Migraine is an often disabling condition that affects an estimated 37 million people in the U.S.,” Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research, said in an FDA news release.
Ubrogepant represents “an important new option for the acute treatment of migraine in adults, as it is the first drug in its class approved for this indication. The FDA is pleased to approve a novel treatment for patients suffering from migraine and will continue to work with stakeholders to promote the development of new safe and effective migraine therapies,” added Dr. Dunn.
The safety and efficacy of ubrogepant for the acute treatment of migraine was demonstrated in two randomized, double-blind, placebo-controlled trials (ACHIEVE I and ACHIEVE II). In total, 1,439 adults with a history of migraine, with and without aura, received ubrogepant to treat an ongoing migraine.
“Both 50-mg and 100-mg dose strengths demonstrated significantly greater rates of pain freedom and freedom from the most bothersome migraine-associated symptom at 2 hours, compared with placebo,” Allergan said in a news release announcing approval.
The most common side effects reported by patients in the clinical trials were nausea, tiredness, and dry mouth. Ubrogepant is contraindicated for coadministration with strong CYP3A4 inhibitors.
The company expects to have ubrogepant available in the first quarter of 2020.
A version of this story originally appeared on Medscape.com.
The Food and Drug Administration has approved ubrogepant (Ubrelvy, Allergan) for the acute treatment of migraine with or without aura in adults.
Ubrogepant is the first drug in the class of oral calcitonin gene–related peptide receptor antagonists approved for the acute treatment of migraine. It is approved in two dose strengths (50 mg and 100 mg).
The drug is not indicated, however, for the preventive treatment of migraine.
“Migraine is an often disabling condition that affects an estimated 37 million people in the U.S.,” Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research, said in an FDA news release.
Ubrogepant represents “an important new option for the acute treatment of migraine in adults, as it is the first drug in its class approved for this indication. The FDA is pleased to approve a novel treatment for patients suffering from migraine and will continue to work with stakeholders to promote the development of new safe and effective migraine therapies,” added Dr. Dunn.
The safety and efficacy of ubrogepant for the acute treatment of migraine was demonstrated in two randomized, double-blind, placebo-controlled trials (ACHIEVE I and ACHIEVE II). In total, 1,439 adults with a history of migraine, with and without aura, received ubrogepant to treat an ongoing migraine.
“Both 50-mg and 100-mg dose strengths demonstrated significantly greater rates of pain freedom and freedom from the most bothersome migraine-associated symptom at 2 hours, compared with placebo,” Allergan said in a news release announcing approval.
The most common side effects reported by patients in the clinical trials were nausea, tiredness, and dry mouth. Ubrogepant is contraindicated for coadministration with strong CYP3A4 inhibitors.
The company expects to have ubrogepant available in the first quarter of 2020.
A version of this story originally appeared on Medscape.com.
EVALI readmissions and deaths prompt guideline change
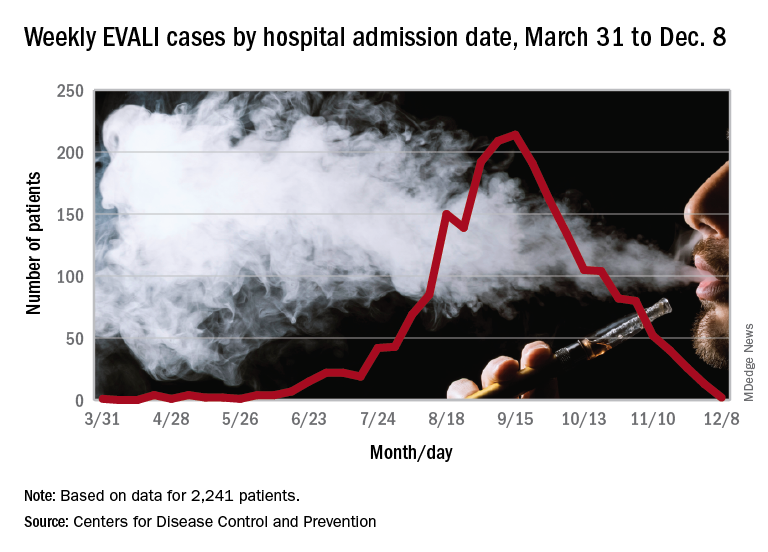
Those who required rehospitalization for e-cigarette or vaping product use–associated lung injury (EVALI) and those who died after discharge were more likely to have one or more chronic conditions than were other EVALI patients, and those “who died also were more likely to have been admitted to an intensive care unit, experienced respiratory failure necessitating intubation and mechanical ventilation, and were significantly older,” Christina A. Mikosz, MD, and associates wrote in the Morbidity and Mortality Weekly Report.
Their analysis included the 1,139 EVALI patients who were discharged on or after Oct. 31, 2019. Of that group, 31 (2.7%) patients were rehospitalized and subsequently discharged and another 7 died after the initial discharge. The median age was 54 years for those who died, 27 years for those who were rehospitalized, and 23 for those who survived without rehospitalization, said Dr. Mikosz of the CDC National Center for Injury Prevention and Control, Atlanta, and associates.
Those findings, along with the rates of one or more comorbidities – 83% for those who died, 71% for those who were rehospitalized, and 26% for those who did not die or get readmitted – prompted the CDC to update its guidance for postdischarge follow-up of EVALI patients.
That update involves six specific recommendations to determine readiness for discharge, which include “confirming no clinically significant fluctuations in vital signs for at least 24-48 hours before discharge [and] preparation for hospital discharge and postdischarge care coordination to reduce risk of rehospitalization and death,” Mary E. Evans, MD, and associates said in a separate CDC communication (MMWR. 2019 Dec. 20. 68[early release]:1-6).
As of Dec. 17, the CDC reports that 2,506 patients have been hospitalized with EVALI since March 31, 2019, and 54 deaths have been confirmed in 27 states and the District of Columbia. The outbreak appears to have peaked in September, but cases are still being reported: 13 during the week of Dec. 1-7 and one case for the week of Dec. 8-14.
SOURCE: Mikosz CA et al. MMWR. 2019 Dec. 20. 68[early release]:1-7.
Those who required rehospitalization for e-cigarette or vaping product use–associated lung injury (EVALI) and those who died after discharge were more likely to have one or more chronic conditions than were other EVALI patients, and those “who died also were more likely to have been admitted to an intensive care unit, experienced respiratory failure necessitating intubation and mechanical ventilation, and were significantly older,” Christina A. Mikosz, MD, and associates wrote in the Morbidity and Mortality Weekly Report.
Their analysis included the 1,139 EVALI patients who were discharged on or after Oct. 31, 2019. Of that group, 31 (2.7%) patients were rehospitalized and subsequently discharged and another 7 died after the initial discharge. The median age was 54 years for those who died, 27 years for those who were rehospitalized, and 23 for those who survived without rehospitalization, said Dr. Mikosz of the CDC National Center for Injury Prevention and Control, Atlanta, and associates.
Those findings, along with the rates of one or more comorbidities – 83% for those who died, 71% for those who were rehospitalized, and 26% for those who did not die or get readmitted – prompted the CDC to update its guidance for postdischarge follow-up of EVALI patients.
That update involves six specific recommendations to determine readiness for discharge, which include “confirming no clinically significant fluctuations in vital signs for at least 24-48 hours before discharge [and] preparation for hospital discharge and postdischarge care coordination to reduce risk of rehospitalization and death,” Mary E. Evans, MD, and associates said in a separate CDC communication (MMWR. 2019 Dec. 20. 68[early release]:1-6).
As of Dec. 17, the CDC reports that 2,506 patients have been hospitalized with EVALI since March 31, 2019, and 54 deaths have been confirmed in 27 states and the District of Columbia. The outbreak appears to have peaked in September, but cases are still being reported: 13 during the week of Dec. 1-7 and one case for the week of Dec. 8-14.
SOURCE: Mikosz CA et al. MMWR. 2019 Dec. 20. 68[early release]:1-7.
Those who required rehospitalization for e-cigarette or vaping product use–associated lung injury (EVALI) and those who died after discharge were more likely to have one or more chronic conditions than were other EVALI patients, and those “who died also were more likely to have been admitted to an intensive care unit, experienced respiratory failure necessitating intubation and mechanical ventilation, and were significantly older,” Christina A. Mikosz, MD, and associates wrote in the Morbidity and Mortality Weekly Report.
Their analysis included the 1,139 EVALI patients who were discharged on or after Oct. 31, 2019. Of that group, 31 (2.7%) patients were rehospitalized and subsequently discharged and another 7 died after the initial discharge. The median age was 54 years for those who died, 27 years for those who were rehospitalized, and 23 for those who survived without rehospitalization, said Dr. Mikosz of the CDC National Center for Injury Prevention and Control, Atlanta, and associates.
Those findings, along with the rates of one or more comorbidities – 83% for those who died, 71% for those who were rehospitalized, and 26% for those who did not die or get readmitted – prompted the CDC to update its guidance for postdischarge follow-up of EVALI patients.
That update involves six specific recommendations to determine readiness for discharge, which include “confirming no clinically significant fluctuations in vital signs for at least 24-48 hours before discharge [and] preparation for hospital discharge and postdischarge care coordination to reduce risk of rehospitalization and death,” Mary E. Evans, MD, and associates said in a separate CDC communication (MMWR. 2019 Dec. 20. 68[early release]:1-6).
As of Dec. 17, the CDC reports that 2,506 patients have been hospitalized with EVALI since March 31, 2019, and 54 deaths have been confirmed in 27 states and the District of Columbia. The outbreak appears to have peaked in September, but cases are still being reported: 13 during the week of Dec. 1-7 and one case for the week of Dec. 8-14.
SOURCE: Mikosz CA et al. MMWR. 2019 Dec. 20. 68[early release]:1-7.
FROM MMWR
FDA approves Dayvigo for insomnia
Lemborexant will be available in 5-mg and 10-mg tablets after the Drug Enforcement Administration schedules the drug, which is expected to occur within 90 days, according to a statement from Eisai.
Lemborexant is an orexin receptor antagonist. Its approval is based on two phase 3 studies, SUNRISE 1 and SUNRISE 2, that included approximately 2,000 adults with insomnia. Investigators assessed lemborexant versus active comparators for as long as 1 month and versus placebo for 6 months.
In these studies, lemborexant significantly improved objective and subjective measures of sleep onset and sleep maintenance, compared with placebo. The medication was not associated with rebound insomnia or withdrawal effects after treatment discontinuation.
In the phase 3 trials, somnolence was the most common adverse reaction that occurred in at least 5% of patients who received lemborexant and at twice the rate in patients who received placebo (lemborexant 10 mg, 10%; lemborexant 5 mg, 7%; placebo, 1%).
In a middle-of-the-night safety study, lemborexant was associated with dose-dependent worsening on measures of attention and memory, compared with placebo. Treatment did not meaningfully affect ability to awaken to sound, however.
Previously reported data showed that lemborexant was effective in male and female patients and was well tolerated by both sexes and that the medication did not impair postural stability and driving performance.
Lemborexant will be available in 5-mg and 10-mg tablets after the Drug Enforcement Administration schedules the drug, which is expected to occur within 90 days, according to a statement from Eisai.
Lemborexant is an orexin receptor antagonist. Its approval is based on two phase 3 studies, SUNRISE 1 and SUNRISE 2, that included approximately 2,000 adults with insomnia. Investigators assessed lemborexant versus active comparators for as long as 1 month and versus placebo for 6 months.
In these studies, lemborexant significantly improved objective and subjective measures of sleep onset and sleep maintenance, compared with placebo. The medication was not associated with rebound insomnia or withdrawal effects after treatment discontinuation.
In the phase 3 trials, somnolence was the most common adverse reaction that occurred in at least 5% of patients who received lemborexant and at twice the rate in patients who received placebo (lemborexant 10 mg, 10%; lemborexant 5 mg, 7%; placebo, 1%).
In a middle-of-the-night safety study, lemborexant was associated with dose-dependent worsening on measures of attention and memory, compared with placebo. Treatment did not meaningfully affect ability to awaken to sound, however.
Previously reported data showed that lemborexant was effective in male and female patients and was well tolerated by both sexes and that the medication did not impair postural stability and driving performance.
Lemborexant will be available in 5-mg and 10-mg tablets after the Drug Enforcement Administration schedules the drug, which is expected to occur within 90 days, according to a statement from Eisai.
Lemborexant is an orexin receptor antagonist. Its approval is based on two phase 3 studies, SUNRISE 1 and SUNRISE 2, that included approximately 2,000 adults with insomnia. Investigators assessed lemborexant versus active comparators for as long as 1 month and versus placebo for 6 months.
In these studies, lemborexant significantly improved objective and subjective measures of sleep onset and sleep maintenance, compared with placebo. The medication was not associated with rebound insomnia or withdrawal effects after treatment discontinuation.
In the phase 3 trials, somnolence was the most common adverse reaction that occurred in at least 5% of patients who received lemborexant and at twice the rate in patients who received placebo (lemborexant 10 mg, 10%; lemborexant 5 mg, 7%; placebo, 1%).
In a middle-of-the-night safety study, lemborexant was associated with dose-dependent worsening on measures of attention and memory, compared with placebo. Treatment did not meaningfully affect ability to awaken to sound, however.
Previously reported data showed that lemborexant was effective in male and female patients and was well tolerated by both sexes and that the medication did not impair postural stability and driving performance.