User login
Outpatient Management and Follow-up Recommendations for Adverse Drug Reactions: Guidelines for Posthospitalization Care
It has been estimated that 2 million serious adverse drug reactions (ADRs) occur annually in the United States, resulting in 100,000 deaths.1 Although the acute morbidity and mortality of these ADRs are readily apparent, postdischarge sequalae are critical aspects of a patient’s care. Herein, we present an approach to outpatient dermatologic follow-up of 3 ADRs: acute generalized exanthematous pustulosis (AGEP), drug rash with eosinophilia and systemic symptoms (DRESS) syndrome, and Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN). For these ADRs, the first step is prompt diagnosis and discontinuation of any potentially causative medications.
ACUTE GENERALIZED EXANTHEMATOUS PUSTULOSIS
Ninety percent of the time, AGEP is caused by medications, most commonly antibiotics, and less often it is caused by viruses.2-4 It presents as a cutaneous eruption with nonfollicular sterile pustules, fever, and leukocytosis, usually within 5 days after starting a causative medication.5 After stopping the medication, cutaneous findings generally improve within 1 week, and leukocytosis often resolves within 1 week.3
Notable Sequelae
Although AGEP typically is considered benign,2 there have been reports of severe sequelae including death from a systemic inflammatory response and complications such as bacterial superinfection and sepsis.6,7 Visceral involvement can be seen in up to 20% of AGEP patients, with systemic symptoms similar to those seen in DRESS syndrome. Mortality has been reported in up to 5% of cases, mainly in patients with comorbidities and notable mucosal involvement.8 More severe disease can be seen in patients with known dermatologic disease, as AGEP can provoke an isomorphic phenomenon.9 Laboratory alterations typically seen in AGEP include neutrophilia, eosinophilia, and elevated liver enzymes.2
Follow-up Recommendations
Patients should be informed of the expected timeline for resolution and should be counseled on the possibility of rare systemic symptoms. Laboratory abnormalities should be monitored every 2 to 4 weeks until normalized.
DRESS SYNDROME
DRESS syndrome is characterized by a morbilliform eruption that can be accompanied by fever; eosinophilia; purpura; facial edema; lymphadenopathy; and liver, renal, or other organ dysfunction. DRESS syndrome most often presents within 8 weeks of exposure to a causative drug.10,11 The most common causative agents are anticonvulsants, antimicrobials, and allopurinol.12 Treatment includes topical corticosteroids and systemic corticosteroids for internal organ involvement.10
Short-term Sequelae
Several potential sequelae may occur within 6 months of resolution of DRESS syndrome, resulting from both the ADR itself and/or systemic corticosteroids that often are required for treatment.13 Complications secondary to herpesviruses have been reported.14 Cases of cytomegalovirus-induced gastric ulcers can lead to gastrointestinal tract bleeds.15
Infections including Cryptococcus species and herpes zoster also have been reported.16 Patients, particularly those treated with systemic corticosteroids, should be monitored with close follow-up for infectious complications and treatment-related adverse effects.13
Long-term Sequelae
Endocrine
Thyroid gland abnormalities secondary to DRESS syndrome include Graves disease and Hashimoto disease as well as variations in biomarkers including elevated free thyroxine and low and elevated thyroid-stimulating hormone levels.16,17 Type 1 diabetes mellitus also has been seen after DRESS syndrome, developing within the first 10 months after onset with unknown pathogenesis.18
Autoimmune
Other reported sequelae of DRESS syndrome include elevated antinuclear antibodies with possible development into systemic lupus erythematosus, autoimmune hemolytic anemia, vitiligo, and rheumatoid arthritis.11,16 Symptoms may be exacerbated in patients with preexisting autoimmune diseases such as systemic lupus erythematosus and rheumatoid arthritis, and patients with preexisting renal disease are at an increased risk for requiring lifelong hemodialysis after DRESS syndrome.16
Other
Studies have demonstrated that pneumonia, thrombosis, and alopecia can be complications of DRESS syndrome.11,16 Psychiatric disturbances including fear of taking new medications, anxiety, and depression also have been reported.19 Children with DRESS syndrome may develop vitiligo, alopecia, sclerodermatous lesions, photophobia, uveitis, and Vogt-Koyanagi-Harada disease.17
Follow-up Recommendations
It is important to inform patients of both the potential short-term and long-term sequelae of DRESS syndrome, including those associated with treatment. A thorough review of systems should be performed at each visit, along with laboratory evaluation including a complete blood cell count with differential and liver function testing every 1 to 2 weeks after discharge until normalized, with monthly monitoring of glucose, thyroid-stimulating hormone, and free thyroxine levels for 3 months after discharge.
STEVENS-JOHNSON SYNDROME/TOXIC EPIDERMAL NECROLYSIS
Stevens-Johnson syndrome/toxic epidermal necrolysis are severe ADRs that present with dusky violaceous macules. Inciting medications include nonsteroidal anti-inflammatory drugs, allopurinol, antibiotics, and anticonvulsants, and symptoms begin 1 to 3 weeks after medication exposure.12 Initially, the lesions often begin on the trunk and can progress to full-body erythema and exfoliation with a necrotic epidermis and mucosal involvement.12,20
Notable Sequelae
Cutaneous
Chronic eczema can present at any time and can vary in severity in SJS/TEN patients.21 Xerosis and pruritus can be treated with emollients.11 Dyschromia is common. Hypertrophic and keloidal scarring can result from surgical debridement and are best prevented with the use of nonadherent dressings.22 Nail changes such as anonychia, dystrophy, longitudinal ridges, and pterygium also are seen, and topical steroids can be helpful. Other reported dermatologic sequelae include dyschromia and eruption of ectopic sebaceous glands.21,22
Ocular
Ocular sequelae include dry eyes, photophobia, symblepharon, corneal scarring, corneal neovascularization, corneal xerosis, trichiasis, reduced visual acuity, blindness, and subconjunctival fibrosis. The most common sequelae are bilateral conjunctivitis and corneal ulcerations.22,23 Early and regular ophthalmologic follow-up is recommended, as SJS/TEN-induced blindness can result from delayed therapy, destroying corneal stem cells.21 Amniotic membrane transplantation replaces the damaged corneal membrane, which may reduce corneal inflammation.24
Chronic dry eye syndrome can recur for years after SJS/TEN resolves and progresses over time.22 Frequent use of nonpreserved artificial tears and salivary gland transplantation can be helpful.24 Unfortunately, ocular disease may develop months after discharge; therefore, it is recommended that dermatologists ask all SJS/TEN patients about ocular symptoms in follow-up visits. If ocular involvement was present initially, patients should be followed by ophthalmology for at least 1 year after discharge.23
Genitourinary
Genitourinary sequelae in SJS/TEN include adhesions, particularly in the female urethra and vaginal opening; vaginal adenosis; vulvovaginal endometriosis; and persistent genital ulcerations most commonly reported in females.22 Prompt inpatient gynecologic or urologic consultation is critical to reduce these potentially permanent outcomes. Topical corticosteroid therapy is recommended in the acute phase.22
Psychologic
Posttraumatic stress disorder may occur in patients with SJS/TEN. One study showed that 23% (7/30) of patients had posttraumatic stress disorder 6 months after hospitalization for SJS/TEN. The investigators recommended routine psychiatric assessment in the acute disease period and for at least 1 year after discharge.25
Pulmonary, Gastrointestinal, and Renal
Interstitial pneumonia and obliterative bronchitis/bronchiolitis can be caused by SJS/TEN. Interstitial pneumonia tends to occur during the acute course, while obliterative airway disease manifests after resolution of SJS/TEN.21,22 Abnormal pulmonary function testing can be seen in more than half of SJS/TEN patients 2 months after the ADR.22 Gastrointestinal sequelae include esophageal strictures, intestinal ulceration, and cholestasis.22 Renal sequelae include acute kidney injury and glomerulonephritis, which may be secondary to the volume loss seen in SJS/TEN but may be irreversible.21
Special Populations
A correlation with infertility in women has been documented in patients with SJS/TEN; thus, follow-up with obstetrics and gynecology is recommended in women of child-bearing potential. The most considerable risk in pregnant women with SJS/TEN is premature birth, and mucosal necrosis of SJS/TEN can impair vaginal delivery.26 Antiretrovirals can be a cause of SJS/TEN in the human immunodeficiency virus–positive population.27 In those cases, it is best to discontinue the medication and find an alternative.
Risk factors for children can be different and can include viral and febrile illnesses as well as mycoplasma infection.28 Children also can be at an increased risk for poor ocular outcomes, such as permanent deficiency in visual acuity and blindness.29
Follow-up Recommendations
Patients should be counseled regarding sequelae and the multisystem nature of SJS/TEN. Inpatient referrals should be given as needed. It is important to watch for ocular symptoms for 1 year after SJS/TEN resolution. When ocular involvement is present, follow-up with ophthalmology is recommended within 1 month of discharge and then at the discretion of the ophthalmologist. Pulmonary function should be monitored for 1 year after SJS/TEN, starting 1 month after discharge and then at the discretion of the pulmonologist. Patients also should be screened for psychologic sequelae for at least 1 year after discharge.
FINAL THOUGHTS
Adverse drug reactions are notable causes of inpatient hospitalization and may lead to considerable sequelae. These ADRs range in severity from more common and benign maculopapular exanthems to severe multiorgan ADRs such as DRESS syndrome and SJS/TEN.
In AGEP, it is important to monitor patients with preexisting dermatologic diseases and to screen for visceral involvement. DRESS syndrome has the potential to cause immune dysregulation and variable long-term adverse sequelae, both from the disease itself and from corticosteroid therapy. Mucocutaneous sequelae of SJS/TEN can potentially affect a patient’s cutaneous, ocular, genitourinary, mental, pulmonary, gastrointestinal, and renal health.
The baseline recommendations provided here warrant more frequent monitoring if the findings and symptoms are severe. In all of these cases, if a causative medication is identified, it should be added to the patient’s allergy list and the patient should be counseled extensively to avoid this medication and other medications in the same class. If a single agent cannot be identified, referrals for patch testing may be of some utility, particularly in AGEP and DRESS syndrome.30,31
- Preventable adverse drug reactions: a focus on drug interactions. US Food and Drug Administration website. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm110632.htm. Updated March 6, 2018. Accessed April 12, 2019.
- Thienvibul C, Vachiramon V, Chanprapaph K. Five-year retrospective review of acute generalized exanthematous pustulosis. Dermatol Res Pract. 2015;3:1-8.
- Lee HY, Chou D, Pang SM, et al. Acute generalized exanthematous pustulosis: analysis of cases managed in a tertiary hospital in Singapore. Int J Dermatol. 2010
;49:507-512. - Ropars N, Darrieux L, Tisseau L, et al. Acute generalized exanthematous pustulosis associated with primary Epstein-Barr virus infection. JAAD Case Rep. 2014;1:9-11.
- Hattem S, Beerthuizen G, Kardaun S. Severe flucloxacillin‐induced acute generalized exanthematous pustulosis (AGEP), with toxic epidermal necrolysis (TEN)‐like features: does overlap between AGEP and TEN exist? clinical report and review of the literature. Br J Dermatol. 2014;171:1539-1545.
- Tajmir-Riahi A, Wörl P, Harrer T, et al. Life-threatening atypical case of acute generalized exanthematous pustulosis. Int Arch Allergy Immunol. 2017;174:108-111.
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:E1214.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP). a review and update. J Am Acad Dermatol. 2015;73:843-848.
- Totonchy MB, McNiff JM, Bunick CG. Koebnerization of Hailey-Hailey disease into a cutaneous drug eruption of acute generalized exanthematous pustulosis associated with systemic symptoms. J Cutan Pathol. 2016;43:1031-1035.
- Husain Z, Reddy BY, Schwartz RA. DRESS syndrome: part II. management and therapeutics. J Am Acad Dermatol. 2013;68:709.e1-e9; quiz 718-720.
- Kano Y, Shiohara T. Long-term outcome of patients with severe cutaneous adverse reactions. Dermatologica Sinica. 2013;31:211-216.
- Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 1. Philadelphia, PA: Elsevier Saunders; 2012.
- Ushigome Y, Kano Y, Ishida T, et al. Short- and long-term outcomes of 34 patients with drug-induced hypersensitivity syndrome in a single institution. J Am Acad Dermatol. 2013;68:721-728.
- Ljungman P, Wang FZ, Clark DA, et al. High levels of human herpesvirus 6 DNA in peripheral blood leucocytes are correlated to platelet engraftment and disease in allogeneic stem cell transplant patients. Br J Haematol. 2000;111:774-781.
- Asano Y, Kagawa H, Kano Y, et al. Cytomegalovirus disease during severe drug eruptions: report of 2 cases and retrospective study of 18 patients with drug-induced hypersensitivity syndrome. Arch Dermatol. 2009;145:1030-1036.
- Kano Y , Tohyama M, Aihara M, et al. Sequelae in 145 patients with drug‐induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms: survey conducted by the Asian Research Committee on Severe Cutaneous Adverse Reactions (ASCAR). J Dermatol. 2015;42:276-282.
- Morita C, Yanase T, Shiohara T, et al. Aggressive treatment in paediatric or young patients with drug-induced hypersensitivity syndrome (DiHS)/ drug reaction with eosinophilia and systemic symptoms (DRESS) is associated with future development of type III polyglandular autoimmune syndrome [published online October 27, 2018]. BMJ Case Rep. doi:10.1136/bcr-2018-225528.
- Chiang A, Shiu J, Elsensohn AN, et al. Classic autoimmune type 1 diabetes mellitus after a case of drug reaction with eosinophilia and systemic symptoms (DRESS). JAAD Case Rep. 2018;4:295-297.
- Lew TT, Creamer D, Mackenzie J, et al. Post-traumatic stress disorder following drug reaction with eosinophilia and systemic symptoms. Br J Dermatol. 2015;172:836-837.
- Kumar R, Das A, Das S. Management of Stevens-Johnson syndrome-toxic epidermal necrolysis: looking beyond guidelines! Indian J Dermatol. 2018;63:117-124.
- Yang CW, Cho YT, Chen KL, et al. Long-term sequelae of Stevens-Johnson syndrome/toxic epidermal necrolysis. Acta Derm Venereol. 2016;96:525-529.
- Lee HY, Walsh SA, Creamer D. Long‐term complications of Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN): the spectrum of chronic problems in patients who survive an episode of SJS/TEN necessitates multidisciplinary follow‐up. Br J Dermatol. 2017;177:924-935.
- Hsu M, Jayaram A, Verner R, et al. Indications and outcomes of amniotic membrane transplantation in the management of acute Stevens-Johnson syndrome and toxic epidermal necrolysis: a case-control study. Cornea. 2012;31:1394-1402.
- Sant’ Anna AE, Hazarbassanov RM, de Freitas D, et al. Minor salivary glands and labial mucous membrane graft in the treatment of severe symblepharon and dry eye in patients with Stevens-Johnson syndrome. Br J Ophthalmol. 2012;96:234-239.
- Hefez L, Zaghbib K, Sbidian E, et al. Post-traumatic stress disorder in Stevens-Johnson syndrome and toxic epidermal necrolysis: prevalence and risk factors. a prospective study of 31 patients [published online October 3, 2018]. Br J Dermatol. doi:10.1111/bjd.17267.
- Knight L, Todd G, Muloiwa R, et al. Stevens Johnson syndrome and toxic epidermal necrolysis: maternal and foetal outcomes intwenty-two consecutive pregnant HIV infected women [published online August 12, 2015]. PLoS One. doi:10.1371/journal.pone.0135501.
- Tchetnya X, Ngwasiri CA, Munge T, et al. Severe eye complications from toxic epidermal necrolysis following initiation of nevirapine based HAART regimen in a child with HIV infection: a case from Cameroon. BMC Pediatr. 2018;18:108.
- Antoon JW, Goldman JL, Lee B, et al. Incidence, outcomes, and resource use in children with Stevens-Johnson syndrome and toxic epidermal necrolysis. Pediatr Dermatol. 2018;35:182-187.
- Basu S, Shanbhag SS, Gokani A, et al. Chronic ocular sequelae of Stevens-Johnson syndrome in children: long-term impact of appropriate therapy on natural history of disease. Am J Ophthalmol. 2018;189:17-28.
- Pinho A, Marta A, Coutinho I, et al. Long‐term reproducibility of positive patch test reactions in patients with non‐immediate cutaneous adverse drug reactions to antibiotics. Contact Dermatitis. 2017;76:204-209.
- Barbaud A, Collet E, Milpied B, et al. A multicentre study to determine the value and safety of drug patch tests for the three main classes of severe cutaneous adverse drug reactions. Br J Dermatol. 2013;168:555-562.
It has been estimated that 2 million serious adverse drug reactions (ADRs) occur annually in the United States, resulting in 100,000 deaths.1 Although the acute morbidity and mortality of these ADRs are readily apparent, postdischarge sequalae are critical aspects of a patient’s care. Herein, we present an approach to outpatient dermatologic follow-up of 3 ADRs: acute generalized exanthematous pustulosis (AGEP), drug rash with eosinophilia and systemic symptoms (DRESS) syndrome, and Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN). For these ADRs, the first step is prompt diagnosis and discontinuation of any potentially causative medications.
ACUTE GENERALIZED EXANTHEMATOUS PUSTULOSIS
Ninety percent of the time, AGEP is caused by medications, most commonly antibiotics, and less often it is caused by viruses.2-4 It presents as a cutaneous eruption with nonfollicular sterile pustules, fever, and leukocytosis, usually within 5 days after starting a causative medication.5 After stopping the medication, cutaneous findings generally improve within 1 week, and leukocytosis often resolves within 1 week.3
Notable Sequelae
Although AGEP typically is considered benign,2 there have been reports of severe sequelae including death from a systemic inflammatory response and complications such as bacterial superinfection and sepsis.6,7 Visceral involvement can be seen in up to 20% of AGEP patients, with systemic symptoms similar to those seen in DRESS syndrome. Mortality has been reported in up to 5% of cases, mainly in patients with comorbidities and notable mucosal involvement.8 More severe disease can be seen in patients with known dermatologic disease, as AGEP can provoke an isomorphic phenomenon.9 Laboratory alterations typically seen in AGEP include neutrophilia, eosinophilia, and elevated liver enzymes.2
Follow-up Recommendations
Patients should be informed of the expected timeline for resolution and should be counseled on the possibility of rare systemic symptoms. Laboratory abnormalities should be monitored every 2 to 4 weeks until normalized.
DRESS SYNDROME
DRESS syndrome is characterized by a morbilliform eruption that can be accompanied by fever; eosinophilia; purpura; facial edema; lymphadenopathy; and liver, renal, or other organ dysfunction. DRESS syndrome most often presents within 8 weeks of exposure to a causative drug.10,11 The most common causative agents are anticonvulsants, antimicrobials, and allopurinol.12 Treatment includes topical corticosteroids and systemic corticosteroids for internal organ involvement.10
Short-term Sequelae
Several potential sequelae may occur within 6 months of resolution of DRESS syndrome, resulting from both the ADR itself and/or systemic corticosteroids that often are required for treatment.13 Complications secondary to herpesviruses have been reported.14 Cases of cytomegalovirus-induced gastric ulcers can lead to gastrointestinal tract bleeds.15
Infections including Cryptococcus species and herpes zoster also have been reported.16 Patients, particularly those treated with systemic corticosteroids, should be monitored with close follow-up for infectious complications and treatment-related adverse effects.13
Long-term Sequelae
Endocrine
Thyroid gland abnormalities secondary to DRESS syndrome include Graves disease and Hashimoto disease as well as variations in biomarkers including elevated free thyroxine and low and elevated thyroid-stimulating hormone levels.16,17 Type 1 diabetes mellitus also has been seen after DRESS syndrome, developing within the first 10 months after onset with unknown pathogenesis.18
Autoimmune
Other reported sequelae of DRESS syndrome include elevated antinuclear antibodies with possible development into systemic lupus erythematosus, autoimmune hemolytic anemia, vitiligo, and rheumatoid arthritis.11,16 Symptoms may be exacerbated in patients with preexisting autoimmune diseases such as systemic lupus erythematosus and rheumatoid arthritis, and patients with preexisting renal disease are at an increased risk for requiring lifelong hemodialysis after DRESS syndrome.16
Other
Studies have demonstrated that pneumonia, thrombosis, and alopecia can be complications of DRESS syndrome.11,16 Psychiatric disturbances including fear of taking new medications, anxiety, and depression also have been reported.19 Children with DRESS syndrome may develop vitiligo, alopecia, sclerodermatous lesions, photophobia, uveitis, and Vogt-Koyanagi-Harada disease.17
Follow-up Recommendations
It is important to inform patients of both the potential short-term and long-term sequelae of DRESS syndrome, including those associated with treatment. A thorough review of systems should be performed at each visit, along with laboratory evaluation including a complete blood cell count with differential and liver function testing every 1 to 2 weeks after discharge until normalized, with monthly monitoring of glucose, thyroid-stimulating hormone, and free thyroxine levels for 3 months after discharge.
STEVENS-JOHNSON SYNDROME/TOXIC EPIDERMAL NECROLYSIS
Stevens-Johnson syndrome/toxic epidermal necrolysis are severe ADRs that present with dusky violaceous macules. Inciting medications include nonsteroidal anti-inflammatory drugs, allopurinol, antibiotics, and anticonvulsants, and symptoms begin 1 to 3 weeks after medication exposure.12 Initially, the lesions often begin on the trunk and can progress to full-body erythema and exfoliation with a necrotic epidermis and mucosal involvement.12,20
Notable Sequelae
Cutaneous
Chronic eczema can present at any time and can vary in severity in SJS/TEN patients.21 Xerosis and pruritus can be treated with emollients.11 Dyschromia is common. Hypertrophic and keloidal scarring can result from surgical debridement and are best prevented with the use of nonadherent dressings.22 Nail changes such as anonychia, dystrophy, longitudinal ridges, and pterygium also are seen, and topical steroids can be helpful. Other reported dermatologic sequelae include dyschromia and eruption of ectopic sebaceous glands.21,22
Ocular
Ocular sequelae include dry eyes, photophobia, symblepharon, corneal scarring, corneal neovascularization, corneal xerosis, trichiasis, reduced visual acuity, blindness, and subconjunctival fibrosis. The most common sequelae are bilateral conjunctivitis and corneal ulcerations.22,23 Early and regular ophthalmologic follow-up is recommended, as SJS/TEN-induced blindness can result from delayed therapy, destroying corneal stem cells.21 Amniotic membrane transplantation replaces the damaged corneal membrane, which may reduce corneal inflammation.24
Chronic dry eye syndrome can recur for years after SJS/TEN resolves and progresses over time.22 Frequent use of nonpreserved artificial tears and salivary gland transplantation can be helpful.24 Unfortunately, ocular disease may develop months after discharge; therefore, it is recommended that dermatologists ask all SJS/TEN patients about ocular symptoms in follow-up visits. If ocular involvement was present initially, patients should be followed by ophthalmology for at least 1 year after discharge.23
Genitourinary
Genitourinary sequelae in SJS/TEN include adhesions, particularly in the female urethra and vaginal opening; vaginal adenosis; vulvovaginal endometriosis; and persistent genital ulcerations most commonly reported in females.22 Prompt inpatient gynecologic or urologic consultation is critical to reduce these potentially permanent outcomes. Topical corticosteroid therapy is recommended in the acute phase.22
Psychologic
Posttraumatic stress disorder may occur in patients with SJS/TEN. One study showed that 23% (7/30) of patients had posttraumatic stress disorder 6 months after hospitalization for SJS/TEN. The investigators recommended routine psychiatric assessment in the acute disease period and for at least 1 year after discharge.25
Pulmonary, Gastrointestinal, and Renal
Interstitial pneumonia and obliterative bronchitis/bronchiolitis can be caused by SJS/TEN. Interstitial pneumonia tends to occur during the acute course, while obliterative airway disease manifests after resolution of SJS/TEN.21,22 Abnormal pulmonary function testing can be seen in more than half of SJS/TEN patients 2 months after the ADR.22 Gastrointestinal sequelae include esophageal strictures, intestinal ulceration, and cholestasis.22 Renal sequelae include acute kidney injury and glomerulonephritis, which may be secondary to the volume loss seen in SJS/TEN but may be irreversible.21
Special Populations
A correlation with infertility in women has been documented in patients with SJS/TEN; thus, follow-up with obstetrics and gynecology is recommended in women of child-bearing potential. The most considerable risk in pregnant women with SJS/TEN is premature birth, and mucosal necrosis of SJS/TEN can impair vaginal delivery.26 Antiretrovirals can be a cause of SJS/TEN in the human immunodeficiency virus–positive population.27 In those cases, it is best to discontinue the medication and find an alternative.
Risk factors for children can be different and can include viral and febrile illnesses as well as mycoplasma infection.28 Children also can be at an increased risk for poor ocular outcomes, such as permanent deficiency in visual acuity and blindness.29
Follow-up Recommendations
Patients should be counseled regarding sequelae and the multisystem nature of SJS/TEN. Inpatient referrals should be given as needed. It is important to watch for ocular symptoms for 1 year after SJS/TEN resolution. When ocular involvement is present, follow-up with ophthalmology is recommended within 1 month of discharge and then at the discretion of the ophthalmologist. Pulmonary function should be monitored for 1 year after SJS/TEN, starting 1 month after discharge and then at the discretion of the pulmonologist. Patients also should be screened for psychologic sequelae for at least 1 year after discharge.
FINAL THOUGHTS
Adverse drug reactions are notable causes of inpatient hospitalization and may lead to considerable sequelae. These ADRs range in severity from more common and benign maculopapular exanthems to severe multiorgan ADRs such as DRESS syndrome and SJS/TEN.
In AGEP, it is important to monitor patients with preexisting dermatologic diseases and to screen for visceral involvement. DRESS syndrome has the potential to cause immune dysregulation and variable long-term adverse sequelae, both from the disease itself and from corticosteroid therapy. Mucocutaneous sequelae of SJS/TEN can potentially affect a patient’s cutaneous, ocular, genitourinary, mental, pulmonary, gastrointestinal, and renal health.
The baseline recommendations provided here warrant more frequent monitoring if the findings and symptoms are severe. In all of these cases, if a causative medication is identified, it should be added to the patient’s allergy list and the patient should be counseled extensively to avoid this medication and other medications in the same class. If a single agent cannot be identified, referrals for patch testing may be of some utility, particularly in AGEP and DRESS syndrome.30,31
It has been estimated that 2 million serious adverse drug reactions (ADRs) occur annually in the United States, resulting in 100,000 deaths.1 Although the acute morbidity and mortality of these ADRs are readily apparent, postdischarge sequalae are critical aspects of a patient’s care. Herein, we present an approach to outpatient dermatologic follow-up of 3 ADRs: acute generalized exanthematous pustulosis (AGEP), drug rash with eosinophilia and systemic symptoms (DRESS) syndrome, and Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN). For these ADRs, the first step is prompt diagnosis and discontinuation of any potentially causative medications.
ACUTE GENERALIZED EXANTHEMATOUS PUSTULOSIS
Ninety percent of the time, AGEP is caused by medications, most commonly antibiotics, and less often it is caused by viruses.2-4 It presents as a cutaneous eruption with nonfollicular sterile pustules, fever, and leukocytosis, usually within 5 days after starting a causative medication.5 After stopping the medication, cutaneous findings generally improve within 1 week, and leukocytosis often resolves within 1 week.3
Notable Sequelae
Although AGEP typically is considered benign,2 there have been reports of severe sequelae including death from a systemic inflammatory response and complications such as bacterial superinfection and sepsis.6,7 Visceral involvement can be seen in up to 20% of AGEP patients, with systemic symptoms similar to those seen in DRESS syndrome. Mortality has been reported in up to 5% of cases, mainly in patients with comorbidities and notable mucosal involvement.8 More severe disease can be seen in patients with known dermatologic disease, as AGEP can provoke an isomorphic phenomenon.9 Laboratory alterations typically seen in AGEP include neutrophilia, eosinophilia, and elevated liver enzymes.2
Follow-up Recommendations
Patients should be informed of the expected timeline for resolution and should be counseled on the possibility of rare systemic symptoms. Laboratory abnormalities should be monitored every 2 to 4 weeks until normalized.
DRESS SYNDROME
DRESS syndrome is characterized by a morbilliform eruption that can be accompanied by fever; eosinophilia; purpura; facial edema; lymphadenopathy; and liver, renal, or other organ dysfunction. DRESS syndrome most often presents within 8 weeks of exposure to a causative drug.10,11 The most common causative agents are anticonvulsants, antimicrobials, and allopurinol.12 Treatment includes topical corticosteroids and systemic corticosteroids for internal organ involvement.10
Short-term Sequelae
Several potential sequelae may occur within 6 months of resolution of DRESS syndrome, resulting from both the ADR itself and/or systemic corticosteroids that often are required for treatment.13 Complications secondary to herpesviruses have been reported.14 Cases of cytomegalovirus-induced gastric ulcers can lead to gastrointestinal tract bleeds.15
Infections including Cryptococcus species and herpes zoster also have been reported.16 Patients, particularly those treated with systemic corticosteroids, should be monitored with close follow-up for infectious complications and treatment-related adverse effects.13
Long-term Sequelae
Endocrine
Thyroid gland abnormalities secondary to DRESS syndrome include Graves disease and Hashimoto disease as well as variations in biomarkers including elevated free thyroxine and low and elevated thyroid-stimulating hormone levels.16,17 Type 1 diabetes mellitus also has been seen after DRESS syndrome, developing within the first 10 months after onset with unknown pathogenesis.18
Autoimmune
Other reported sequelae of DRESS syndrome include elevated antinuclear antibodies with possible development into systemic lupus erythematosus, autoimmune hemolytic anemia, vitiligo, and rheumatoid arthritis.11,16 Symptoms may be exacerbated in patients with preexisting autoimmune diseases such as systemic lupus erythematosus and rheumatoid arthritis, and patients with preexisting renal disease are at an increased risk for requiring lifelong hemodialysis after DRESS syndrome.16
Other
Studies have demonstrated that pneumonia, thrombosis, and alopecia can be complications of DRESS syndrome.11,16 Psychiatric disturbances including fear of taking new medications, anxiety, and depression also have been reported.19 Children with DRESS syndrome may develop vitiligo, alopecia, sclerodermatous lesions, photophobia, uveitis, and Vogt-Koyanagi-Harada disease.17
Follow-up Recommendations
It is important to inform patients of both the potential short-term and long-term sequelae of DRESS syndrome, including those associated with treatment. A thorough review of systems should be performed at each visit, along with laboratory evaluation including a complete blood cell count with differential and liver function testing every 1 to 2 weeks after discharge until normalized, with monthly monitoring of glucose, thyroid-stimulating hormone, and free thyroxine levels for 3 months after discharge.
STEVENS-JOHNSON SYNDROME/TOXIC EPIDERMAL NECROLYSIS
Stevens-Johnson syndrome/toxic epidermal necrolysis are severe ADRs that present with dusky violaceous macules. Inciting medications include nonsteroidal anti-inflammatory drugs, allopurinol, antibiotics, and anticonvulsants, and symptoms begin 1 to 3 weeks after medication exposure.12 Initially, the lesions often begin on the trunk and can progress to full-body erythema and exfoliation with a necrotic epidermis and mucosal involvement.12,20
Notable Sequelae
Cutaneous
Chronic eczema can present at any time and can vary in severity in SJS/TEN patients.21 Xerosis and pruritus can be treated with emollients.11 Dyschromia is common. Hypertrophic and keloidal scarring can result from surgical debridement and are best prevented with the use of nonadherent dressings.22 Nail changes such as anonychia, dystrophy, longitudinal ridges, and pterygium also are seen, and topical steroids can be helpful. Other reported dermatologic sequelae include dyschromia and eruption of ectopic sebaceous glands.21,22
Ocular
Ocular sequelae include dry eyes, photophobia, symblepharon, corneal scarring, corneal neovascularization, corneal xerosis, trichiasis, reduced visual acuity, blindness, and subconjunctival fibrosis. The most common sequelae are bilateral conjunctivitis and corneal ulcerations.22,23 Early and regular ophthalmologic follow-up is recommended, as SJS/TEN-induced blindness can result from delayed therapy, destroying corneal stem cells.21 Amniotic membrane transplantation replaces the damaged corneal membrane, which may reduce corneal inflammation.24
Chronic dry eye syndrome can recur for years after SJS/TEN resolves and progresses over time.22 Frequent use of nonpreserved artificial tears and salivary gland transplantation can be helpful.24 Unfortunately, ocular disease may develop months after discharge; therefore, it is recommended that dermatologists ask all SJS/TEN patients about ocular symptoms in follow-up visits. If ocular involvement was present initially, patients should be followed by ophthalmology for at least 1 year after discharge.23
Genitourinary
Genitourinary sequelae in SJS/TEN include adhesions, particularly in the female urethra and vaginal opening; vaginal adenosis; vulvovaginal endometriosis; and persistent genital ulcerations most commonly reported in females.22 Prompt inpatient gynecologic or urologic consultation is critical to reduce these potentially permanent outcomes. Topical corticosteroid therapy is recommended in the acute phase.22
Psychologic
Posttraumatic stress disorder may occur in patients with SJS/TEN. One study showed that 23% (7/30) of patients had posttraumatic stress disorder 6 months after hospitalization for SJS/TEN. The investigators recommended routine psychiatric assessment in the acute disease period and for at least 1 year after discharge.25
Pulmonary, Gastrointestinal, and Renal
Interstitial pneumonia and obliterative bronchitis/bronchiolitis can be caused by SJS/TEN. Interstitial pneumonia tends to occur during the acute course, while obliterative airway disease manifests after resolution of SJS/TEN.21,22 Abnormal pulmonary function testing can be seen in more than half of SJS/TEN patients 2 months after the ADR.22 Gastrointestinal sequelae include esophageal strictures, intestinal ulceration, and cholestasis.22 Renal sequelae include acute kidney injury and glomerulonephritis, which may be secondary to the volume loss seen in SJS/TEN but may be irreversible.21
Special Populations
A correlation with infertility in women has been documented in patients with SJS/TEN; thus, follow-up with obstetrics and gynecology is recommended in women of child-bearing potential. The most considerable risk in pregnant women with SJS/TEN is premature birth, and mucosal necrosis of SJS/TEN can impair vaginal delivery.26 Antiretrovirals can be a cause of SJS/TEN in the human immunodeficiency virus–positive population.27 In those cases, it is best to discontinue the medication and find an alternative.
Risk factors for children can be different and can include viral and febrile illnesses as well as mycoplasma infection.28 Children also can be at an increased risk for poor ocular outcomes, such as permanent deficiency in visual acuity and blindness.29
Follow-up Recommendations
Patients should be counseled regarding sequelae and the multisystem nature of SJS/TEN. Inpatient referrals should be given as needed. It is important to watch for ocular symptoms for 1 year after SJS/TEN resolution. When ocular involvement is present, follow-up with ophthalmology is recommended within 1 month of discharge and then at the discretion of the ophthalmologist. Pulmonary function should be monitored for 1 year after SJS/TEN, starting 1 month after discharge and then at the discretion of the pulmonologist. Patients also should be screened for psychologic sequelae for at least 1 year after discharge.
FINAL THOUGHTS
Adverse drug reactions are notable causes of inpatient hospitalization and may lead to considerable sequelae. These ADRs range in severity from more common and benign maculopapular exanthems to severe multiorgan ADRs such as DRESS syndrome and SJS/TEN.
In AGEP, it is important to monitor patients with preexisting dermatologic diseases and to screen for visceral involvement. DRESS syndrome has the potential to cause immune dysregulation and variable long-term adverse sequelae, both from the disease itself and from corticosteroid therapy. Mucocutaneous sequelae of SJS/TEN can potentially affect a patient’s cutaneous, ocular, genitourinary, mental, pulmonary, gastrointestinal, and renal health.
The baseline recommendations provided here warrant more frequent monitoring if the findings and symptoms are severe. In all of these cases, if a causative medication is identified, it should be added to the patient’s allergy list and the patient should be counseled extensively to avoid this medication and other medications in the same class. If a single agent cannot be identified, referrals for patch testing may be of some utility, particularly in AGEP and DRESS syndrome.30,31
- Preventable adverse drug reactions: a focus on drug interactions. US Food and Drug Administration website. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm110632.htm. Updated March 6, 2018. Accessed April 12, 2019.
- Thienvibul C, Vachiramon V, Chanprapaph K. Five-year retrospective review of acute generalized exanthematous pustulosis. Dermatol Res Pract. 2015;3:1-8.
- Lee HY, Chou D, Pang SM, et al. Acute generalized exanthematous pustulosis: analysis of cases managed in a tertiary hospital in Singapore. Int J Dermatol. 2010
;49:507-512. - Ropars N, Darrieux L, Tisseau L, et al. Acute generalized exanthematous pustulosis associated with primary Epstein-Barr virus infection. JAAD Case Rep. 2014;1:9-11.
- Hattem S, Beerthuizen G, Kardaun S. Severe flucloxacillin‐induced acute generalized exanthematous pustulosis (AGEP), with toxic epidermal necrolysis (TEN)‐like features: does overlap between AGEP and TEN exist? clinical report and review of the literature. Br J Dermatol. 2014;171:1539-1545.
- Tajmir-Riahi A, Wörl P, Harrer T, et al. Life-threatening atypical case of acute generalized exanthematous pustulosis. Int Arch Allergy Immunol. 2017;174:108-111.
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:E1214.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP). a review and update. J Am Acad Dermatol. 2015;73:843-848.
- Totonchy MB, McNiff JM, Bunick CG. Koebnerization of Hailey-Hailey disease into a cutaneous drug eruption of acute generalized exanthematous pustulosis associated with systemic symptoms. J Cutan Pathol. 2016;43:1031-1035.
- Husain Z, Reddy BY, Schwartz RA. DRESS syndrome: part II. management and therapeutics. J Am Acad Dermatol. 2013;68:709.e1-e9; quiz 718-720.
- Kano Y, Shiohara T. Long-term outcome of patients with severe cutaneous adverse reactions. Dermatologica Sinica. 2013;31:211-216.
- Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 1. Philadelphia, PA: Elsevier Saunders; 2012.
- Ushigome Y, Kano Y, Ishida T, et al. Short- and long-term outcomes of 34 patients with drug-induced hypersensitivity syndrome in a single institution. J Am Acad Dermatol. 2013;68:721-728.
- Ljungman P, Wang FZ, Clark DA, et al. High levels of human herpesvirus 6 DNA in peripheral blood leucocytes are correlated to platelet engraftment and disease in allogeneic stem cell transplant patients. Br J Haematol. 2000;111:774-781.
- Asano Y, Kagawa H, Kano Y, et al. Cytomegalovirus disease during severe drug eruptions: report of 2 cases and retrospective study of 18 patients with drug-induced hypersensitivity syndrome. Arch Dermatol. 2009;145:1030-1036.
- Kano Y , Tohyama M, Aihara M, et al. Sequelae in 145 patients with drug‐induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms: survey conducted by the Asian Research Committee on Severe Cutaneous Adverse Reactions (ASCAR). J Dermatol. 2015;42:276-282.
- Morita C, Yanase T, Shiohara T, et al. Aggressive treatment in paediatric or young patients with drug-induced hypersensitivity syndrome (DiHS)/ drug reaction with eosinophilia and systemic symptoms (DRESS) is associated with future development of type III polyglandular autoimmune syndrome [published online October 27, 2018]. BMJ Case Rep. doi:10.1136/bcr-2018-225528.
- Chiang A, Shiu J, Elsensohn AN, et al. Classic autoimmune type 1 diabetes mellitus after a case of drug reaction with eosinophilia and systemic symptoms (DRESS). JAAD Case Rep. 2018;4:295-297.
- Lew TT, Creamer D, Mackenzie J, et al. Post-traumatic stress disorder following drug reaction with eosinophilia and systemic symptoms. Br J Dermatol. 2015;172:836-837.
- Kumar R, Das A, Das S. Management of Stevens-Johnson syndrome-toxic epidermal necrolysis: looking beyond guidelines! Indian J Dermatol. 2018;63:117-124.
- Yang CW, Cho YT, Chen KL, et al. Long-term sequelae of Stevens-Johnson syndrome/toxic epidermal necrolysis. Acta Derm Venereol. 2016;96:525-529.
- Lee HY, Walsh SA, Creamer D. Long‐term complications of Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN): the spectrum of chronic problems in patients who survive an episode of SJS/TEN necessitates multidisciplinary follow‐up. Br J Dermatol. 2017;177:924-935.
- Hsu M, Jayaram A, Verner R, et al. Indications and outcomes of amniotic membrane transplantation in the management of acute Stevens-Johnson syndrome and toxic epidermal necrolysis: a case-control study. Cornea. 2012;31:1394-1402.
- Sant’ Anna AE, Hazarbassanov RM, de Freitas D, et al. Minor salivary glands and labial mucous membrane graft in the treatment of severe symblepharon and dry eye in patients with Stevens-Johnson syndrome. Br J Ophthalmol. 2012;96:234-239.
- Hefez L, Zaghbib K, Sbidian E, et al. Post-traumatic stress disorder in Stevens-Johnson syndrome and toxic epidermal necrolysis: prevalence and risk factors. a prospective study of 31 patients [published online October 3, 2018]. Br J Dermatol. doi:10.1111/bjd.17267.
- Knight L, Todd G, Muloiwa R, et al. Stevens Johnson syndrome and toxic epidermal necrolysis: maternal and foetal outcomes intwenty-two consecutive pregnant HIV infected women [published online August 12, 2015]. PLoS One. doi:10.1371/journal.pone.0135501.
- Tchetnya X, Ngwasiri CA, Munge T, et al. Severe eye complications from toxic epidermal necrolysis following initiation of nevirapine based HAART regimen in a child with HIV infection: a case from Cameroon. BMC Pediatr. 2018;18:108.
- Antoon JW, Goldman JL, Lee B, et al. Incidence, outcomes, and resource use in children with Stevens-Johnson syndrome and toxic epidermal necrolysis. Pediatr Dermatol. 2018;35:182-187.
- Basu S, Shanbhag SS, Gokani A, et al. Chronic ocular sequelae of Stevens-Johnson syndrome in children: long-term impact of appropriate therapy on natural history of disease. Am J Ophthalmol. 2018;189:17-28.
- Pinho A, Marta A, Coutinho I, et al. Long‐term reproducibility of positive patch test reactions in patients with non‐immediate cutaneous adverse drug reactions to antibiotics. Contact Dermatitis. 2017;76:204-209.
- Barbaud A, Collet E, Milpied B, et al. A multicentre study to determine the value and safety of drug patch tests for the three main classes of severe cutaneous adverse drug reactions. Br J Dermatol. 2013;168:555-562.
- Preventable adverse drug reactions: a focus on drug interactions. US Food and Drug Administration website. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm110632.htm. Updated March 6, 2018. Accessed April 12, 2019.
- Thienvibul C, Vachiramon V, Chanprapaph K. Five-year retrospective review of acute generalized exanthematous pustulosis. Dermatol Res Pract. 2015;3:1-8.
- Lee HY, Chou D, Pang SM, et al. Acute generalized exanthematous pustulosis: analysis of cases managed in a tertiary hospital in Singapore. Int J Dermatol. 2010
;49:507-512. - Ropars N, Darrieux L, Tisseau L, et al. Acute generalized exanthematous pustulosis associated with primary Epstein-Barr virus infection. JAAD Case Rep. 2014;1:9-11.
- Hattem S, Beerthuizen G, Kardaun S. Severe flucloxacillin‐induced acute generalized exanthematous pustulosis (AGEP), with toxic epidermal necrolysis (TEN)‐like features: does overlap between AGEP and TEN exist? clinical report and review of the literature. Br J Dermatol. 2014;171:1539-1545.
- Tajmir-Riahi A, Wörl P, Harrer T, et al. Life-threatening atypical case of acute generalized exanthematous pustulosis. Int Arch Allergy Immunol. 2017;174:108-111.
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:E1214.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP). a review and update. J Am Acad Dermatol. 2015;73:843-848.
- Totonchy MB, McNiff JM, Bunick CG. Koebnerization of Hailey-Hailey disease into a cutaneous drug eruption of acute generalized exanthematous pustulosis associated with systemic symptoms. J Cutan Pathol. 2016;43:1031-1035.
- Husain Z, Reddy BY, Schwartz RA. DRESS syndrome: part II. management and therapeutics. J Am Acad Dermatol. 2013;68:709.e1-e9; quiz 718-720.
- Kano Y, Shiohara T. Long-term outcome of patients with severe cutaneous adverse reactions. Dermatologica Sinica. 2013;31:211-216.
- Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 1. Philadelphia, PA: Elsevier Saunders; 2012.
- Ushigome Y, Kano Y, Ishida T, et al. Short- and long-term outcomes of 34 patients with drug-induced hypersensitivity syndrome in a single institution. J Am Acad Dermatol. 2013;68:721-728.
- Ljungman P, Wang FZ, Clark DA, et al. High levels of human herpesvirus 6 DNA in peripheral blood leucocytes are correlated to platelet engraftment and disease in allogeneic stem cell transplant patients. Br J Haematol. 2000;111:774-781.
- Asano Y, Kagawa H, Kano Y, et al. Cytomegalovirus disease during severe drug eruptions: report of 2 cases and retrospective study of 18 patients with drug-induced hypersensitivity syndrome. Arch Dermatol. 2009;145:1030-1036.
- Kano Y , Tohyama M, Aihara M, et al. Sequelae in 145 patients with drug‐induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms: survey conducted by the Asian Research Committee on Severe Cutaneous Adverse Reactions (ASCAR). J Dermatol. 2015;42:276-282.
- Morita C, Yanase T, Shiohara T, et al. Aggressive treatment in paediatric or young patients with drug-induced hypersensitivity syndrome (DiHS)/ drug reaction with eosinophilia and systemic symptoms (DRESS) is associated with future development of type III polyglandular autoimmune syndrome [published online October 27, 2018]. BMJ Case Rep. doi:10.1136/bcr-2018-225528.
- Chiang A, Shiu J, Elsensohn AN, et al. Classic autoimmune type 1 diabetes mellitus after a case of drug reaction with eosinophilia and systemic symptoms (DRESS). JAAD Case Rep. 2018;4:295-297.
- Lew TT, Creamer D, Mackenzie J, et al. Post-traumatic stress disorder following drug reaction with eosinophilia and systemic symptoms. Br J Dermatol. 2015;172:836-837.
- Kumar R, Das A, Das S. Management of Stevens-Johnson syndrome-toxic epidermal necrolysis: looking beyond guidelines! Indian J Dermatol. 2018;63:117-124.
- Yang CW, Cho YT, Chen KL, et al. Long-term sequelae of Stevens-Johnson syndrome/toxic epidermal necrolysis. Acta Derm Venereol. 2016;96:525-529.
- Lee HY, Walsh SA, Creamer D. Long‐term complications of Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN): the spectrum of chronic problems in patients who survive an episode of SJS/TEN necessitates multidisciplinary follow‐up. Br J Dermatol. 2017;177:924-935.
- Hsu M, Jayaram A, Verner R, et al. Indications and outcomes of amniotic membrane transplantation in the management of acute Stevens-Johnson syndrome and toxic epidermal necrolysis: a case-control study. Cornea. 2012;31:1394-1402.
- Sant’ Anna AE, Hazarbassanov RM, de Freitas D, et al. Minor salivary glands and labial mucous membrane graft in the treatment of severe symblepharon and dry eye in patients with Stevens-Johnson syndrome. Br J Ophthalmol. 2012;96:234-239.
- Hefez L, Zaghbib K, Sbidian E, et al. Post-traumatic stress disorder in Stevens-Johnson syndrome and toxic epidermal necrolysis: prevalence and risk factors. a prospective study of 31 patients [published online October 3, 2018]. Br J Dermatol. doi:10.1111/bjd.17267.
- Knight L, Todd G, Muloiwa R, et al. Stevens Johnson syndrome and toxic epidermal necrolysis: maternal and foetal outcomes intwenty-two consecutive pregnant HIV infected women [published online August 12, 2015]. PLoS One. doi:10.1371/journal.pone.0135501.
- Tchetnya X, Ngwasiri CA, Munge T, et al. Severe eye complications from toxic epidermal necrolysis following initiation of nevirapine based HAART regimen in a child with HIV infection: a case from Cameroon. BMC Pediatr. 2018;18:108.
- Antoon JW, Goldman JL, Lee B, et al. Incidence, outcomes, and resource use in children with Stevens-Johnson syndrome and toxic epidermal necrolysis. Pediatr Dermatol. 2018;35:182-187.
- Basu S, Shanbhag SS, Gokani A, et al. Chronic ocular sequelae of Stevens-Johnson syndrome in children: long-term impact of appropriate therapy on natural history of disease. Am J Ophthalmol. 2018;189:17-28.
- Pinho A, Marta A, Coutinho I, et al. Long‐term reproducibility of positive patch test reactions in patients with non‐immediate cutaneous adverse drug reactions to antibiotics. Contact Dermatitis. 2017;76:204-209.
- Barbaud A, Collet E, Milpied B, et al. A multicentre study to determine the value and safety of drug patch tests for the three main classes of severe cutaneous adverse drug reactions. Br J Dermatol. 2013;168:555-562.
Practice Points
- In the setting of an adverse drug reaction (ADR), discontinuing the concerning medication is the first and most important step.
- Acute generalized exanthematous pustulosis, drug rash with eosinophilia and systemic symptoms (DRESS) syndrome, and Stevens-Johnson syndrome/toxic epidermal necrolysis all require specific outpatient follow-up after discharge.

Update on Calciphylaxis Etiopathogenesis, Diagnosis, and Management
Calciphylaxis, also known as calcific uremic arteriolopathy, is a painful skin condition classically seen in patients with end-stage renal disease (ESRD), particularly those on chronic dialysis.1,2 It also has increasingly been reported in patients with normal renal function and calcium and phosphate homeostasis.3,4 Effective diagnosis and management of calciphylaxis remains challenging for physicians.2,5 The condition is characterized by tissue ischemia caused by calcification of cutaneous arteriolar vessels. As a result, calciphylaxis is associated with high mortality rates, ranging from 60% to 80%.5,6 Excruciating pain and nonhealing ulcers often lead to recurrent hospitalizations and infectious complications,7 and poor nutritional status, chronic pain, depression, and insomnia can further complicate recovery and lead to poor quality of life.8
We provide an update on calciphylaxis etiopathogenesis, diagnosis, and management. We also highlight some challenges faced in managing this potentially fatal condition.
Epidemiology
Calciphylaxis is considered a rare dermatosis with an estimated annual incidence of 1% to 4% in ESRD patients on dialysis. Recent data suggest that incidence of calciphylaxis is rising,5,7,9 which may stem from an increased use of calcium-based phosphate binders, an actual rise in disease incidence, and/or increased recognition of the disease.5 It is difficult to estimate the exact disease burden of calciphylaxis because the diagnostic criteria are not well defined, often leading to missed or delayed diagnosis.3,10 Furthermore, there is no centralized registry for calciphylaxis cases.3
Etiology and Pathogenesis
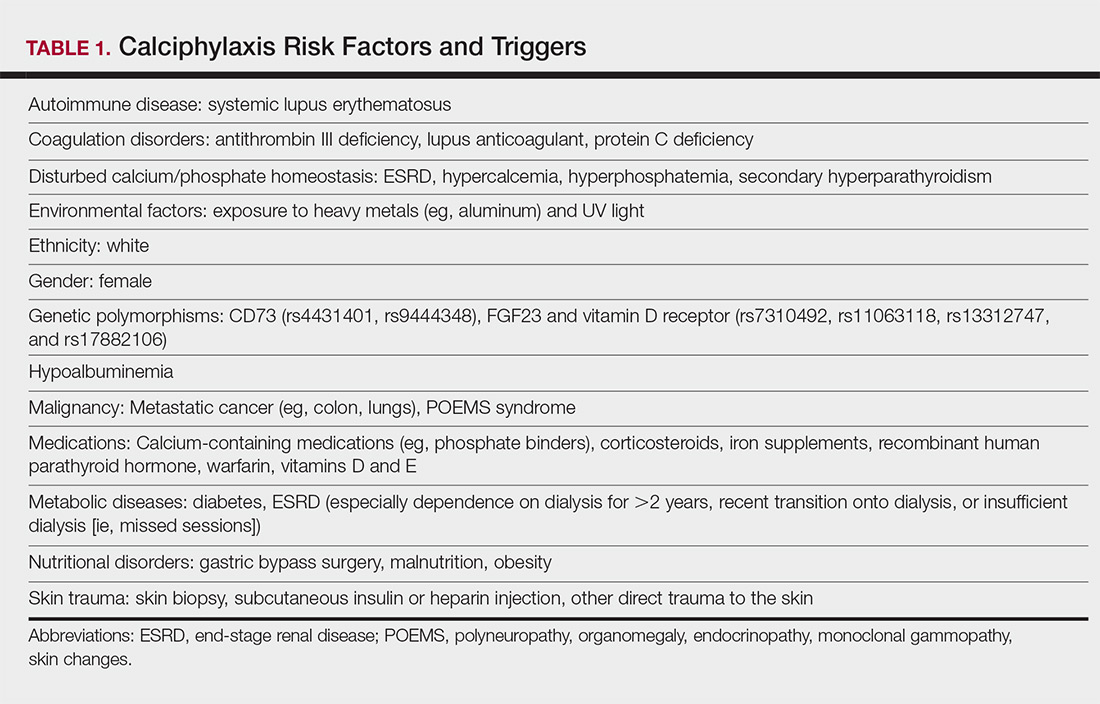
Calciphylaxis is thought to have a multifactorial etiology with the exact cause or trigger unknown.7 A long list of risk factors and triggers is associated with the condition (Table 1). Calciphylaxis primarily affects small arteries (40–600 μm in diameter) that become calcified due to an imbalance between inhibitors and promoters of calcification.2,11 Fetuin-A and matrix Gla protein inhibit vascular calcification and are downregulated in calciphylaxis.12,13 Dysfunctional calcium, phosphate, and parathyroid hormone regulatory pathways provide an increased substrate for the process of calcification, which causes endothelial damage and microthrombosis, resulting in tissue ischemia and infarction.14,15 Notably, there is growing interest in the role of vitamin K in the pathogenesis of calciphylaxis. Vitamin K inhibits vascular calcification, possibly by increasing the circulating levels of carboxylated matrix Gla protein.16
Clinical Features
Calciphylaxis is most commonly seen on the legs, abdomen, and buttocks.2 Patients with ESRD commonly develop proximal lesions affecting adipose-rich sites and have a poor prognosis. Distal lesions are more common in patients with nonuremic calciphylaxis, and mortality rates are lower in this population.2
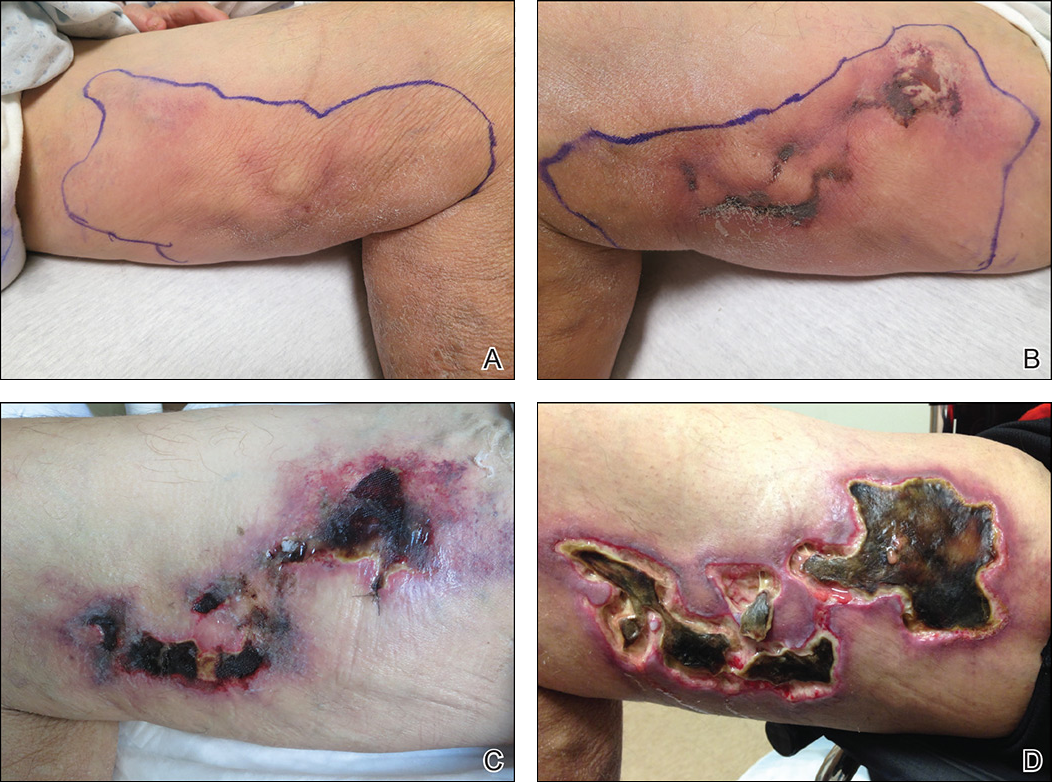
Early lesions present as painful skin nodules or indurated plaques that often are rock-hard or firm to palpation with overlying mottling or a livedoid pattern (Figure, A). Early lesions progress from livedo reticularis to livedo racemosa and then to retiform purpura (Figure, B). Purpuric lesions later evolve into black eschars (Figure, C), then to necrotic, ulcerated, malodorous plaques or nodules in later stages of the disease (Figure, D). Lesions also may develop a gangrenous sclerotic appearance.2,5
Although most patients with calciphylaxis have ESRD, nonuremic patients also can develop the disease. Those with calciphylaxis who do not have renal dysfunction frequently have other risk factors for the disease and often report another notable health problem in the weeks or months prior to presentation.4 More than half of patients with calciphylaxis become bedridden or require use of a wheelchair.17 Pain is characteristically severe throughout the course of the disease; it may even precede the appearance of the skin lesions.18 Because the pain is associated with ischemia, it tends to be relatively refractory to treatment with opioids. Rare extracutaneous vascular calcifications may lead to visual impairment, gastrointestinal tract bleeding, and myopathy.5,9,19,20
Diagnosis
Considering the high morbidity and mortality associated with calciphylaxis, it is important to provide accurate and timely diagnosis; however, there currently are no validated diagnostic criteria for calciphylaxis. Careful correlation of clinical and histologic findings is required. Calciphylaxis biopsies have demonstrated medial calcification and proliferation of the intima of small- to medium-sized arteries.21 Lobular and septal panniculitis and extravascular soft-tissue calcification, particularly stippled calcification of the eccrine sweat glands, also has been seen.2,22 Special calcium stains (eg, von Kossa, Alizarin red) increase the sensitivity of biopsy by highlighting subtle areas of intravascular and extravascular calcification.5,23 Sufficient sampling of subcutaneous tissue and specimen evaluation by an experienced dermatopathologist are necessary to ensure proper interpretation of the histologic findings.
Despite these measures, skin biopsies may be nondiagnostic or falsely negative; therefore, when there is high clinical suspicion, it may be appropriate to move forward with a presumptive diagnosis of calciphylaxis even if the histologic findings are nondiagnostic.1,9,24 It also is worth noting that localized progression and ulceration may occur following skin biopsy, such that biopsy may even be contraindicated in certain cases (eg, penile calciphylaxis).
Standard laboratory workup for calciphylaxis includes evaluation for associated risk factors as well as exclusion of other conditions in the differential diagnosis (Table 2). Blood tests to evaluate for risk factors include liver and renal function tests, a complete metabolic panel, parathyroid hormone level, and serum albumin level.5 Elevated calcium and phosphate levels may signal disturbed calcium and phosphate homeostasis but are neither sensitive nor specific for the diagnosis.25 Complete blood cell count, blood cultures, thorough hypercoagulability workup (including but not limited to antiphospholipid antibodies, proteins C and S, factor V Leiden, antithrombin III, homocysteine, methylenetetrahydrofolate reductase mutation, and cryoglobulins), rheumatoid factor, antineutrophil cytoplasmic antibodies, and antinuclear antibody testing may be relevant to help identify contributing factors or mimickers of calciphylaxis.5 Various imaging modalities also have been used to evaluate for the presence of soft-tissue calcification in areas of suspected calciphylaxis, including radiography, mammography, computed tomography, ultrasonography, nuclear bone scintigraphy, and spectroscopy.2,26,27 Unfortunately, there currently is no standardized reproducible imaging modality for reliable diagnosis of calciphylaxis. Ultimately, histologic and radiographic findings should always be interpreted in the context of relevant clinical findings.2,9

Prevention
Reduction of the net calcium phosphorus product may help reduce the risk of calciphylaxis in ESRD patients, which can be accomplished by using non–calcium-phosphate binders, adequate dialysis, and restricting use of vitamin D and vitamin K antagonists.2,5 There are limited data regarding the benefits of using bisphosphonates and cinacalcet in ESRD patients on dialysis to prevent calciphylaxis.28,29
Management
Management of calciphylaxis is multifactorial. Besides dermatology and nephrology, specialists in pain management, wound care, plastic surgery, and nutrition are critical partners in management.1,5,9,30 Nephrologists can help optimize calcium and phosphate balance and ensure adequate dialysis. Pain specialists can aid in creating aggressive multiagent pain regimens that target the neuropathic/ischemic and physical aspects of calciphylaxis pain. When appropriate, nutrition specialists can help establish high-protein, low-phosphorus diets, and wound specialists can provide access to advanced wound dressings and adjunctive hyperbaric oxygen therapy. Plastic surgeons can provide conservative debridement procedures in a subset of patients, usually those with distal stable disease.
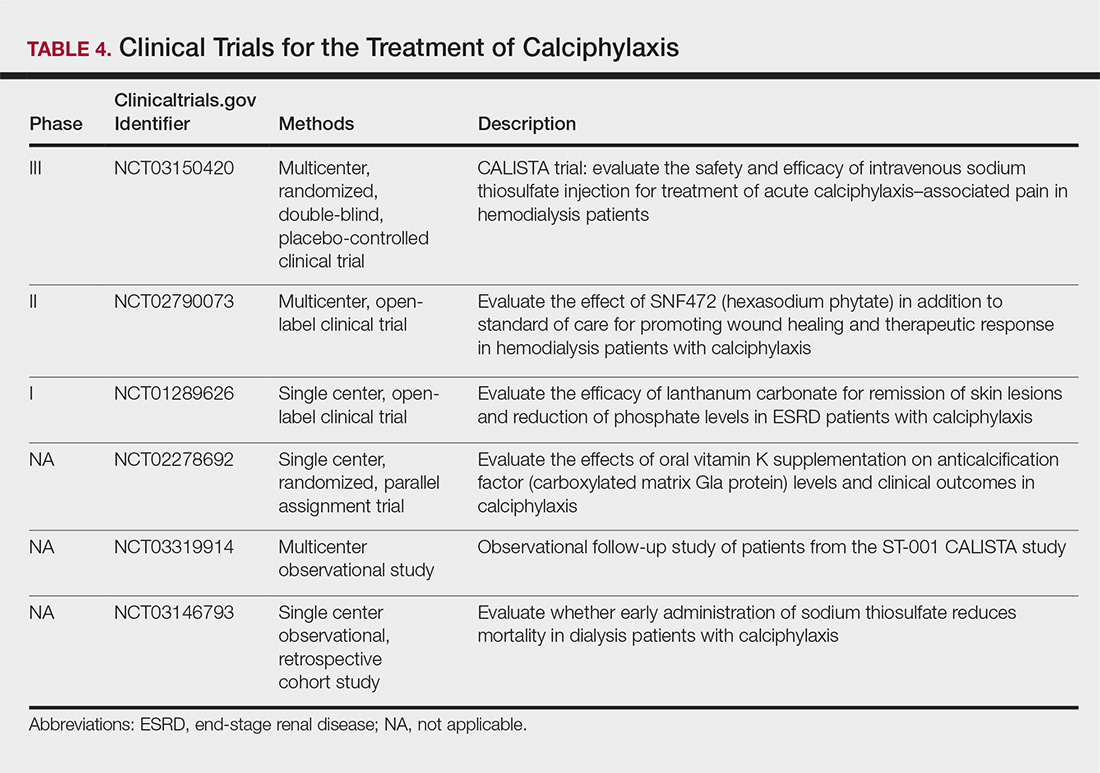
The limited understanding of the etiopathogenesis of calciphylaxis and the lack of data on its management are reflected in the limited treatment options for the disease (Table 3).2,5,9 There are no formal algorithms for the treatment of calciphylaxis. Therapeutic trials are scarce, and most of the current treatment recommendations are based on small retrospective reports or case series. Sodium thiosulfate has been the most widely used treatment option since 2004, when its use in calciphylaxis was first reported.31 Sodium thiosulfate chelates calcium and is thought to have antioxidant and vasodilatory properties.32 There are a few promising clinical trials and large-scale studies (Table 4) that aim to evaluate the efficacy of existing treatments (eg, sodium thiosulfate) as well as novel treatment options such as lanthanum carbonate, SNF472 (hexasodium phytate), and vitamin K.33-36

Prognosis
Calciphylaxis is a potentially fatal condition with a poor prognosis and a median survival rate of approximately 1 year following the appearance of skin lesions.37-39 Patients with proximal lesions and those on peritoneal dialysis (as opposed to hemodialysis) have a worse prognosis.40 Mortality rates are estimated to be 30% at 6 months, 50% at 12 months, and 80% at 2 years, with sepsis secondary to infection of cutaneous ulcers being the leading cause of death.37-39 The impact of calciphylaxis on patient quality of life and activities of daily living is severe.8,17
Future Directions
Multi-institution cohort studies and collaborative registries are needed to provide updated information related to the epidemiology, diagnosis, treatment, morbidity, and mortality associated with calciphylaxis and to help formulate evidence-based diagnostic criteria. Radiographic and histologic studies, as well as other tools for early and accurate diagnosis of calciphylaxis, should be studied for feasibility, accuracy, and reproducibility. The incidence of nonuremic calciphylaxis points toward pathogenic pathways besides those based on the bone-mineral axis. Basic science research directed at improving understanding of the pathophysiology of calciphylaxis would be helpful in devising new treatment strategies targeting these pathways. Establishment of a collaborative, multi-institutional calciphylaxis working group would enable experts to formulate therapeutic guidelines based on current evidence. Such a group could facilitate initiation of large prospective studies to establish the efficacy of existing and new treatment modalities for calciphylaxis. A working group within the Society for Dermatology Hospitalists has been tasked with addressing these issues and is currently establishing a multicenter calciphylaxis database.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
- Nigwekar SU, Thadhani RI, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714.
- Davis JM. The relationship between obesity and calciphylaxis: a review of the literature. Ostomy Wound Manage. 2016;62:12-18.
- Bajaj R, Courbebaisse M, Kroshinsky D, et al. Calciphylaxis in patients with normal renal function: a case series and systematic review. Mayo Clin Proc. 2018;93:1202-1212.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Westphal SG, Plumb T. Calciphylaxis. In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2018. https://www.ncbi.nlm.nih.gov/books/NBK519020. Accessed November 12, 2018.
- Riemer CA, El-Azhary RA, Wu KL, et al. Underreported use of palliative care and patient-reported outcome measures to address reduced quality of life in patients with calciphylaxis: a systematic review. Br J Dermatol. 2017;177:1510-1518.
- Nigwekar SU. Calciphylaxis. Curr Opin Nephrol Hypertens. 2017;26:276-281.
- Fine A, Fontaine B. Calciphylaxis: the beginning of the end? Perit Dial Int. 2008;28:268-270.
- Lin WT, Chao CM. Tumoral calcinosis in renal failure. QJM. 2014;107:387.
- Schafer C, Heiss A, Schwarz A, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Bleyer AJ, Choi M, Igwemezie B, et al. A case control study of proximal calciphylaxis. Am J Kidney Dis. 1998;32:376-383.
- Ahmed S, O’Neill KD, Hood AF, et al. Calciphylaxis is associated with hyperphosphatemia and increased osteopontin expression by vascular smooth muscle cells. Am J Kidney Dis. 2001;37:267-276.
- Nigwekar SU, Bloch DB, Nazarian RM, et al. Vitamin K-dependent carboxylation of matrix gla protein influences the risk of calciphylaxis. J Am Soc Nephrol. 2017;28:1717-1722.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Polizzotto MN, Bryan T, Ashby MA, et al. Symptomatic management of calciphylaxis: a case series and review of the literature. J Pain Symptom Manage. 2006;32:186-190.
- Gupta N, Haq KF, Mahajan S, et al. Gastrointestinal bleeding secondary to calciphylaxis. Am J Case Rep. 2015;16:818-822.
- Edelstein CL, Wickham MK, Kirby PA. Systemic calciphylaxis presenting as a painful, proximal myopathy. Postgrad Med J. 1992;68:209-211.
- Mochel MC, Arakari RY, Wang G, et al. Cutaneous calciphylaxis: a retrospective histopathologic evaluation. Am J Dermatopathol. 2013;35:582-586.
- Chen TY, Lehman JS, Gibson LE, et al. Histopathology of calciphylaxis: cohort study with clinical correlations. Am J Dermatopathol. 2017;39:795-802.
- Cassius C, Moguelet P, Monfort JB, et al. Calciphylaxis in haemodialysed patients: diagnostic value of calcifications in cutaneous biopsy. Br J Dermatol. 2018;178:292-293.
- Sreedhar A, Sheikh HA, Scagliotti CJ, et al. Advanced-stage calciphylaxis: think before you punch. Cleve Clin J Med. 2016;83:562-564.
- Brandenburg VM, Kramann R, Rothe H, et al. Calcific uraemic arteriolopathy (calciphylaxis): data from a large nation-wide registry. Nephrol Dial Transplant. 2017;32:126-132.
- Paul S, Rabito CA, Vedak P, et al. The role of bone scintigraphy in the diagnosis of calciphylaxis. JAMA Dermatol. 2017;153:101-103.
- Shmidt E, Murthy NS, Knudsen JM, et al. Net-like pattern of calcification on plain soft-tissue radiographs in patients with calciphylaxis. J Am Acad Dermatol. 2012;67:1296-1301.
- EVOLVE Trial Investigators; Chertow GM, Block GA, Correa-Rotter R, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367:2482-2494.
- Rogers NM, Teubner DJO, Coates PT. Calcific uremic arteriolopathy: advances in pathogenesis and treatment. Semin Dial. 2007;20:150-157.
- Nigwekar SU. Multidisciplinary approach to calcific uremic arteriolopathy. Curr Opin Nephrol Hypertens. 2015;24:531-537.
- Cicone JS, Petronis JB, Embert CD, et al. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis. 2004;43:1104-1108.
- Chen NX, O’Neill K, Akl NK, et al. Adipocyte induced arterial calcification is prevented with sodium thiosulfate. Biochem Biophys Res Commun. 2014;449:151-156.
- Chan MR, Ghandour F, Murali NS, et al. Pilot study of the effect of lanthanum carbonate in patients with calciphylaxis: a Wisconsin Network for Health Research (WiNHR) study. J Nephrol Ther. 2014;4:1000162.
- Perelló J, Gómez M, Ferrer MD, et al. SNF472, a novel inhibitor of vascular calcification, could be administered during hemodialysis to attain potentially therapeutic phytate levels. J Nephrol. 2018;31:287-296.
- Christiadi D, Singer RF. Calciphylaxis in a dialysis patient successfully treated with high-dose vitamin K supplementation. Clin Kidney J. 2018;11:528-529.
- Caluwe R, Vandecasteele S, Van Vlem B, et al. Vitamin K2 supplementation in haemodialysis patients: a randomized dose-finding study. Nephrol Dial Transplant. 2014;29:1385-1390.
- McCarthy JT, El-Azhary RA, Patzelt MT, et al. Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin Proc. 2016;91:1384-1394.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Nigwekar SU, Zhao S, Wenger J, et al. A nationally representative study of calcific uremic arteriolopathy risk factors. J Am Soc Nephrol. 2016;27:3421-3429.
- Zhang Y, Corapi KM, Luongo M, et al. Calciphylaxis in peritoneal dialysis patients: a single center cohort study. Int J Nephrol Renovasc Dis. 2016;9:235-241.
Calciphylaxis, also known as calcific uremic arteriolopathy, is a painful skin condition classically seen in patients with end-stage renal disease (ESRD), particularly those on chronic dialysis.1,2 It also has increasingly been reported in patients with normal renal function and calcium and phosphate homeostasis.3,4 Effective diagnosis and management of calciphylaxis remains challenging for physicians.2,5 The condition is characterized by tissue ischemia caused by calcification of cutaneous arteriolar vessels. As a result, calciphylaxis is associated with high mortality rates, ranging from 60% to 80%.5,6 Excruciating pain and nonhealing ulcers often lead to recurrent hospitalizations and infectious complications,7 and poor nutritional status, chronic pain, depression, and insomnia can further complicate recovery and lead to poor quality of life.8
We provide an update on calciphylaxis etiopathogenesis, diagnosis, and management. We also highlight some challenges faced in managing this potentially fatal condition.
Epidemiology
Calciphylaxis is considered a rare dermatosis with an estimated annual incidence of 1% to 4% in ESRD patients on dialysis. Recent data suggest that incidence of calciphylaxis is rising,5,7,9 which may stem from an increased use of calcium-based phosphate binders, an actual rise in disease incidence, and/or increased recognition of the disease.5 It is difficult to estimate the exact disease burden of calciphylaxis because the diagnostic criteria are not well defined, often leading to missed or delayed diagnosis.3,10 Furthermore, there is no centralized registry for calciphylaxis cases.3
Etiology and Pathogenesis
Calciphylaxis is thought to have a multifactorial etiology with the exact cause or trigger unknown.7 A long list of risk factors and triggers is associated with the condition (Table 1). Calciphylaxis primarily affects small arteries (40–600 μm in diameter) that become calcified due to an imbalance between inhibitors and promoters of calcification.2,11 Fetuin-A and matrix Gla protein inhibit vascular calcification and are downregulated in calciphylaxis.12,13 Dysfunctional calcium, phosphate, and parathyroid hormone regulatory pathways provide an increased substrate for the process of calcification, which causes endothelial damage and microthrombosis, resulting in tissue ischemia and infarction.14,15 Notably, there is growing interest in the role of vitamin K in the pathogenesis of calciphylaxis. Vitamin K inhibits vascular calcification, possibly by increasing the circulating levels of carboxylated matrix Gla protein.16
Clinical Features
Calciphylaxis is most commonly seen on the legs, abdomen, and buttocks.2 Patients with ESRD commonly develop proximal lesions affecting adipose-rich sites and have a poor prognosis. Distal lesions are more common in patients with nonuremic calciphylaxis, and mortality rates are lower in this population.2
Early lesions present as painful skin nodules or indurated plaques that often are rock-hard or firm to palpation with overlying mottling or a livedoid pattern (Figure, A). Early lesions progress from livedo reticularis to livedo racemosa and then to retiform purpura (Figure, B). Purpuric lesions later evolve into black eschars (Figure, C), then to necrotic, ulcerated, malodorous plaques or nodules in later stages of the disease (Figure, D). Lesions also may develop a gangrenous sclerotic appearance.2,5
Although most patients with calciphylaxis have ESRD, nonuremic patients also can develop the disease. Those with calciphylaxis who do not have renal dysfunction frequently have other risk factors for the disease and often report another notable health problem in the weeks or months prior to presentation.4 More than half of patients with calciphylaxis become bedridden or require use of a wheelchair.17 Pain is characteristically severe throughout the course of the disease; it may even precede the appearance of the skin lesions.18 Because the pain is associated with ischemia, it tends to be relatively refractory to treatment with opioids. Rare extracutaneous vascular calcifications may lead to visual impairment, gastrointestinal tract bleeding, and myopathy.5,9,19,20
Diagnosis
Considering the high morbidity and mortality associated with calciphylaxis, it is important to provide accurate and timely diagnosis; however, there currently are no validated diagnostic criteria for calciphylaxis. Careful correlation of clinical and histologic findings is required. Calciphylaxis biopsies have demonstrated medial calcification and proliferation of the intima of small- to medium-sized arteries.21 Lobular and septal panniculitis and extravascular soft-tissue calcification, particularly stippled calcification of the eccrine sweat glands, also has been seen.2,22 Special calcium stains (eg, von Kossa, Alizarin red) increase the sensitivity of biopsy by highlighting subtle areas of intravascular and extravascular calcification.5,23 Sufficient sampling of subcutaneous tissue and specimen evaluation by an experienced dermatopathologist are necessary to ensure proper interpretation of the histologic findings.
Despite these measures, skin biopsies may be nondiagnostic or falsely negative; therefore, when there is high clinical suspicion, it may be appropriate to move forward with a presumptive diagnosis of calciphylaxis even if the histologic findings are nondiagnostic.1,9,24 It also is worth noting that localized progression and ulceration may occur following skin biopsy, such that biopsy may even be contraindicated in certain cases (eg, penile calciphylaxis).
Standard laboratory workup for calciphylaxis includes evaluation for associated risk factors as well as exclusion of other conditions in the differential diagnosis (Table 2). Blood tests to evaluate for risk factors include liver and renal function tests, a complete metabolic panel, parathyroid hormone level, and serum albumin level.5 Elevated calcium and phosphate levels may signal disturbed calcium and phosphate homeostasis but are neither sensitive nor specific for the diagnosis.25 Complete blood cell count, blood cultures, thorough hypercoagulability workup (including but not limited to antiphospholipid antibodies, proteins C and S, factor V Leiden, antithrombin III, homocysteine, methylenetetrahydrofolate reductase mutation, and cryoglobulins), rheumatoid factor, antineutrophil cytoplasmic antibodies, and antinuclear antibody testing may be relevant to help identify contributing factors or mimickers of calciphylaxis.5 Various imaging modalities also have been used to evaluate for the presence of soft-tissue calcification in areas of suspected calciphylaxis, including radiography, mammography, computed tomography, ultrasonography, nuclear bone scintigraphy, and spectroscopy.2,26,27 Unfortunately, there currently is no standardized reproducible imaging modality for reliable diagnosis of calciphylaxis. Ultimately, histologic and radiographic findings should always be interpreted in the context of relevant clinical findings.2,9
Prevention
Reduction of the net calcium phosphorus product may help reduce the risk of calciphylaxis in ESRD patients, which can be accomplished by using non–calcium-phosphate binders, adequate dialysis, and restricting use of vitamin D and vitamin K antagonists.2,5 There are limited data regarding the benefits of using bisphosphonates and cinacalcet in ESRD patients on dialysis to prevent calciphylaxis.28,29
Management
Management of calciphylaxis is multifactorial. Besides dermatology and nephrology, specialists in pain management, wound care, plastic surgery, and nutrition are critical partners in management.1,5,9,30 Nephrologists can help optimize calcium and phosphate balance and ensure adequate dialysis. Pain specialists can aid in creating aggressive multiagent pain regimens that target the neuropathic/ischemic and physical aspects of calciphylaxis pain. When appropriate, nutrition specialists can help establish high-protein, low-phosphorus diets, and wound specialists can provide access to advanced wound dressings and adjunctive hyperbaric oxygen therapy. Plastic surgeons can provide conservative debridement procedures in a subset of patients, usually those with distal stable disease.
The limited understanding of the etiopathogenesis of calciphylaxis and the lack of data on its management are reflected in the limited treatment options for the disease (Table 3).2,5,9 There are no formal algorithms for the treatment of calciphylaxis. Therapeutic trials are scarce, and most of the current treatment recommendations are based on small retrospective reports or case series. Sodium thiosulfate has been the most widely used treatment option since 2004, when its use in calciphylaxis was first reported.31 Sodium thiosulfate chelates calcium and is thought to have antioxidant and vasodilatory properties.32 There are a few promising clinical trials and large-scale studies (Table 4) that aim to evaluate the efficacy of existing treatments (eg, sodium thiosulfate) as well as novel treatment options such as lanthanum carbonate, SNF472 (hexasodium phytate), and vitamin K.33-36
Prognosis
Calciphylaxis is a potentially fatal condition with a poor prognosis and a median survival rate of approximately 1 year following the appearance of skin lesions.37-39 Patients with proximal lesions and those on peritoneal dialysis (as opposed to hemodialysis) have a worse prognosis.40 Mortality rates are estimated to be 30% at 6 months, 50% at 12 months, and 80% at 2 years, with sepsis secondary to infection of cutaneous ulcers being the leading cause of death.37-39 The impact of calciphylaxis on patient quality of life and activities of daily living is severe.8,17
Future Directions
Multi-institution cohort studies and collaborative registries are needed to provide updated information related to the epidemiology, diagnosis, treatment, morbidity, and mortality associated with calciphylaxis and to help formulate evidence-based diagnostic criteria. Radiographic and histologic studies, as well as other tools for early and accurate diagnosis of calciphylaxis, should be studied for feasibility, accuracy, and reproducibility. The incidence of nonuremic calciphylaxis points toward pathogenic pathways besides those based on the bone-mineral axis. Basic science research directed at improving understanding of the pathophysiology of calciphylaxis would be helpful in devising new treatment strategies targeting these pathways. Establishment of a collaborative, multi-institutional calciphylaxis working group would enable experts to formulate therapeutic guidelines based on current evidence. Such a group could facilitate initiation of large prospective studies to establish the efficacy of existing and new treatment modalities for calciphylaxis. A working group within the Society for Dermatology Hospitalists has been tasked with addressing these issues and is currently establishing a multicenter calciphylaxis database.
Calciphylaxis, also known as calcific uremic arteriolopathy, is a painful skin condition classically seen in patients with end-stage renal disease (ESRD), particularly those on chronic dialysis.1,2 It also has increasingly been reported in patients with normal renal function and calcium and phosphate homeostasis.3,4 Effective diagnosis and management of calciphylaxis remains challenging for physicians.2,5 The condition is characterized by tissue ischemia caused by calcification of cutaneous arteriolar vessels. As a result, calciphylaxis is associated with high mortality rates, ranging from 60% to 80%.5,6 Excruciating pain and nonhealing ulcers often lead to recurrent hospitalizations and infectious complications,7 and poor nutritional status, chronic pain, depression, and insomnia can further complicate recovery and lead to poor quality of life.8
We provide an update on calciphylaxis etiopathogenesis, diagnosis, and management. We also highlight some challenges faced in managing this potentially fatal condition.
Epidemiology
Calciphylaxis is considered a rare dermatosis with an estimated annual incidence of 1% to 4% in ESRD patients on dialysis. Recent data suggest that incidence of calciphylaxis is rising,5,7,9 which may stem from an increased use of calcium-based phosphate binders, an actual rise in disease incidence, and/or increased recognition of the disease.5 It is difficult to estimate the exact disease burden of calciphylaxis because the diagnostic criteria are not well defined, often leading to missed or delayed diagnosis.3,10 Furthermore, there is no centralized registry for calciphylaxis cases.3
Etiology and Pathogenesis
Calciphylaxis is thought to have a multifactorial etiology with the exact cause or trigger unknown.7 A long list of risk factors and triggers is associated with the condition (Table 1). Calciphylaxis primarily affects small arteries (40–600 μm in diameter) that become calcified due to an imbalance between inhibitors and promoters of calcification.2,11 Fetuin-A and matrix Gla protein inhibit vascular calcification and are downregulated in calciphylaxis.12,13 Dysfunctional calcium, phosphate, and parathyroid hormone regulatory pathways provide an increased substrate for the process of calcification, which causes endothelial damage and microthrombosis, resulting in tissue ischemia and infarction.14,15 Notably, there is growing interest in the role of vitamin K in the pathogenesis of calciphylaxis. Vitamin K inhibits vascular calcification, possibly by increasing the circulating levels of carboxylated matrix Gla protein.16
Clinical Features
Calciphylaxis is most commonly seen on the legs, abdomen, and buttocks.2 Patients with ESRD commonly develop proximal lesions affecting adipose-rich sites and have a poor prognosis. Distal lesions are more common in patients with nonuremic calciphylaxis, and mortality rates are lower in this population.2
Early lesions present as painful skin nodules or indurated plaques that often are rock-hard or firm to palpation with overlying mottling or a livedoid pattern (Figure, A). Early lesions progress from livedo reticularis to livedo racemosa and then to retiform purpura (Figure, B). Purpuric lesions later evolve into black eschars (Figure, C), then to necrotic, ulcerated, malodorous plaques or nodules in later stages of the disease (Figure, D). Lesions also may develop a gangrenous sclerotic appearance.2,5
Although most patients with calciphylaxis have ESRD, nonuremic patients also can develop the disease. Those with calciphylaxis who do not have renal dysfunction frequently have other risk factors for the disease and often report another notable health problem in the weeks or months prior to presentation.4 More than half of patients with calciphylaxis become bedridden or require use of a wheelchair.17 Pain is characteristically severe throughout the course of the disease; it may even precede the appearance of the skin lesions.18 Because the pain is associated with ischemia, it tends to be relatively refractory to treatment with opioids. Rare extracutaneous vascular calcifications may lead to visual impairment, gastrointestinal tract bleeding, and myopathy.5,9,19,20
Diagnosis
Considering the high morbidity and mortality associated with calciphylaxis, it is important to provide accurate and timely diagnosis; however, there currently are no validated diagnostic criteria for calciphylaxis. Careful correlation of clinical and histologic findings is required. Calciphylaxis biopsies have demonstrated medial calcification and proliferation of the intima of small- to medium-sized arteries.21 Lobular and septal panniculitis and extravascular soft-tissue calcification, particularly stippled calcification of the eccrine sweat glands, also has been seen.2,22 Special calcium stains (eg, von Kossa, Alizarin red) increase the sensitivity of biopsy by highlighting subtle areas of intravascular and extravascular calcification.5,23 Sufficient sampling of subcutaneous tissue and specimen evaluation by an experienced dermatopathologist are necessary to ensure proper interpretation of the histologic findings.
Despite these measures, skin biopsies may be nondiagnostic or falsely negative; therefore, when there is high clinical suspicion, it may be appropriate to move forward with a presumptive diagnosis of calciphylaxis even if the histologic findings are nondiagnostic.1,9,24 It also is worth noting that localized progression and ulceration may occur following skin biopsy, such that biopsy may even be contraindicated in certain cases (eg, penile calciphylaxis).
Standard laboratory workup for calciphylaxis includes evaluation for associated risk factors as well as exclusion of other conditions in the differential diagnosis (Table 2). Blood tests to evaluate for risk factors include liver and renal function tests, a complete metabolic panel, parathyroid hormone level, and serum albumin level.5 Elevated calcium and phosphate levels may signal disturbed calcium and phosphate homeostasis but are neither sensitive nor specific for the diagnosis.25 Complete blood cell count, blood cultures, thorough hypercoagulability workup (including but not limited to antiphospholipid antibodies, proteins C and S, factor V Leiden, antithrombin III, homocysteine, methylenetetrahydrofolate reductase mutation, and cryoglobulins), rheumatoid factor, antineutrophil cytoplasmic antibodies, and antinuclear antibody testing may be relevant to help identify contributing factors or mimickers of calciphylaxis.5 Various imaging modalities also have been used to evaluate for the presence of soft-tissue calcification in areas of suspected calciphylaxis, including radiography, mammography, computed tomography, ultrasonography, nuclear bone scintigraphy, and spectroscopy.2,26,27 Unfortunately, there currently is no standardized reproducible imaging modality for reliable diagnosis of calciphylaxis. Ultimately, histologic and radiographic findings should always be interpreted in the context of relevant clinical findings.2,9
Prevention
Reduction of the net calcium phosphorus product may help reduce the risk of calciphylaxis in ESRD patients, which can be accomplished by using non–calcium-phosphate binders, adequate dialysis, and restricting use of vitamin D and vitamin K antagonists.2,5 There are limited data regarding the benefits of using bisphosphonates and cinacalcet in ESRD patients on dialysis to prevent calciphylaxis.28,29
Management
Management of calciphylaxis is multifactorial. Besides dermatology and nephrology, specialists in pain management, wound care, plastic surgery, and nutrition are critical partners in management.1,5,9,30 Nephrologists can help optimize calcium and phosphate balance and ensure adequate dialysis. Pain specialists can aid in creating aggressive multiagent pain regimens that target the neuropathic/ischemic and physical aspects of calciphylaxis pain. When appropriate, nutrition specialists can help establish high-protein, low-phosphorus diets, and wound specialists can provide access to advanced wound dressings and adjunctive hyperbaric oxygen therapy. Plastic surgeons can provide conservative debridement procedures in a subset of patients, usually those with distal stable disease.
The limited understanding of the etiopathogenesis of calciphylaxis and the lack of data on its management are reflected in the limited treatment options for the disease (Table 3).2,5,9 There are no formal algorithms for the treatment of calciphylaxis. Therapeutic trials are scarce, and most of the current treatment recommendations are based on small retrospective reports or case series. Sodium thiosulfate has been the most widely used treatment option since 2004, when its use in calciphylaxis was first reported.31 Sodium thiosulfate chelates calcium and is thought to have antioxidant and vasodilatory properties.32 There are a few promising clinical trials and large-scale studies (Table 4) that aim to evaluate the efficacy of existing treatments (eg, sodium thiosulfate) as well as novel treatment options such as lanthanum carbonate, SNF472 (hexasodium phytate), and vitamin K.33-36
Prognosis
Calciphylaxis is a potentially fatal condition with a poor prognosis and a median survival rate of approximately 1 year following the appearance of skin lesions.37-39 Patients with proximal lesions and those on peritoneal dialysis (as opposed to hemodialysis) have a worse prognosis.40 Mortality rates are estimated to be 30% at 6 months, 50% at 12 months, and 80% at 2 years, with sepsis secondary to infection of cutaneous ulcers being the leading cause of death.37-39 The impact of calciphylaxis on patient quality of life and activities of daily living is severe.8,17
Future Directions
Multi-institution cohort studies and collaborative registries are needed to provide updated information related to the epidemiology, diagnosis, treatment, morbidity, and mortality associated with calciphylaxis and to help formulate evidence-based diagnostic criteria. Radiographic and histologic studies, as well as other tools for early and accurate diagnosis of calciphylaxis, should be studied for feasibility, accuracy, and reproducibility. The incidence of nonuremic calciphylaxis points toward pathogenic pathways besides those based on the bone-mineral axis. Basic science research directed at improving understanding of the pathophysiology of calciphylaxis would be helpful in devising new treatment strategies targeting these pathways. Establishment of a collaborative, multi-institutional calciphylaxis working group would enable experts to formulate therapeutic guidelines based on current evidence. Such a group could facilitate initiation of large prospective studies to establish the efficacy of existing and new treatment modalities for calciphylaxis. A working group within the Society for Dermatology Hospitalists has been tasked with addressing these issues and is currently establishing a multicenter calciphylaxis database.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
- Nigwekar SU, Thadhani RI, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714.
- Davis JM. The relationship between obesity and calciphylaxis: a review of the literature. Ostomy Wound Manage. 2016;62:12-18.
- Bajaj R, Courbebaisse M, Kroshinsky D, et al. Calciphylaxis in patients with normal renal function: a case series and systematic review. Mayo Clin Proc. 2018;93:1202-1212.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Westphal SG, Plumb T. Calciphylaxis. In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2018. https://www.ncbi.nlm.nih.gov/books/NBK519020. Accessed November 12, 2018.
- Riemer CA, El-Azhary RA, Wu KL, et al. Underreported use of palliative care and patient-reported outcome measures to address reduced quality of life in patients with calciphylaxis: a systematic review. Br J Dermatol. 2017;177:1510-1518.
- Nigwekar SU. Calciphylaxis. Curr Opin Nephrol Hypertens. 2017;26:276-281.
- Fine A, Fontaine B. Calciphylaxis: the beginning of the end? Perit Dial Int. 2008;28:268-270.
- Lin WT, Chao CM. Tumoral calcinosis in renal failure. QJM. 2014;107:387.
- Schafer C, Heiss A, Schwarz A, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Bleyer AJ, Choi M, Igwemezie B, et al. A case control study of proximal calciphylaxis. Am J Kidney Dis. 1998;32:376-383.
- Ahmed S, O’Neill KD, Hood AF, et al. Calciphylaxis is associated with hyperphosphatemia and increased osteopontin expression by vascular smooth muscle cells. Am J Kidney Dis. 2001;37:267-276.
- Nigwekar SU, Bloch DB, Nazarian RM, et al. Vitamin K-dependent carboxylation of matrix gla protein influences the risk of calciphylaxis. J Am Soc Nephrol. 2017;28:1717-1722.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Polizzotto MN, Bryan T, Ashby MA, et al. Symptomatic management of calciphylaxis: a case series and review of the literature. J Pain Symptom Manage. 2006;32:186-190.
- Gupta N, Haq KF, Mahajan S, et al. Gastrointestinal bleeding secondary to calciphylaxis. Am J Case Rep. 2015;16:818-822.
- Edelstein CL, Wickham MK, Kirby PA. Systemic calciphylaxis presenting as a painful, proximal myopathy. Postgrad Med J. 1992;68:209-211.
- Mochel MC, Arakari RY, Wang G, et al. Cutaneous calciphylaxis: a retrospective histopathologic evaluation. Am J Dermatopathol. 2013;35:582-586.
- Chen TY, Lehman JS, Gibson LE, et al. Histopathology of calciphylaxis: cohort study with clinical correlations. Am J Dermatopathol. 2017;39:795-802.
- Cassius C, Moguelet P, Monfort JB, et al. Calciphylaxis in haemodialysed patients: diagnostic value of calcifications in cutaneous biopsy. Br J Dermatol. 2018;178:292-293.
- Sreedhar A, Sheikh HA, Scagliotti CJ, et al. Advanced-stage calciphylaxis: think before you punch. Cleve Clin J Med. 2016;83:562-564.
- Brandenburg VM, Kramann R, Rothe H, et al. Calcific uraemic arteriolopathy (calciphylaxis): data from a large nation-wide registry. Nephrol Dial Transplant. 2017;32:126-132.
- Paul S, Rabito CA, Vedak P, et al. The role of bone scintigraphy in the diagnosis of calciphylaxis. JAMA Dermatol. 2017;153:101-103.
- Shmidt E, Murthy NS, Knudsen JM, et al. Net-like pattern of calcification on plain soft-tissue radiographs in patients with calciphylaxis. J Am Acad Dermatol. 2012;67:1296-1301.
- EVOLVE Trial Investigators; Chertow GM, Block GA, Correa-Rotter R, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367:2482-2494.
- Rogers NM, Teubner DJO, Coates PT. Calcific uremic arteriolopathy: advances in pathogenesis and treatment. Semin Dial. 2007;20:150-157.
- Nigwekar SU. Multidisciplinary approach to calcific uremic arteriolopathy. Curr Opin Nephrol Hypertens. 2015;24:531-537.
- Cicone JS, Petronis JB, Embert CD, et al. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis. 2004;43:1104-1108.
- Chen NX, O’Neill K, Akl NK, et al. Adipocyte induced arterial calcification is prevented with sodium thiosulfate. Biochem Biophys Res Commun. 2014;449:151-156.
- Chan MR, Ghandour F, Murali NS, et al. Pilot study of the effect of lanthanum carbonate in patients with calciphylaxis: a Wisconsin Network for Health Research (WiNHR) study. J Nephrol Ther. 2014;4:1000162.
- Perelló J, Gómez M, Ferrer MD, et al. SNF472, a novel inhibitor of vascular calcification, could be administered during hemodialysis to attain potentially therapeutic phytate levels. J Nephrol. 2018;31:287-296.
- Christiadi D, Singer RF. Calciphylaxis in a dialysis patient successfully treated with high-dose vitamin K supplementation. Clin Kidney J. 2018;11:528-529.
- Caluwe R, Vandecasteele S, Van Vlem B, et al. Vitamin K2 supplementation in haemodialysis patients: a randomized dose-finding study. Nephrol Dial Transplant. 2014;29:1385-1390.
- McCarthy JT, El-Azhary RA, Patzelt MT, et al. Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin Proc. 2016;91:1384-1394.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Nigwekar SU, Zhao S, Wenger J, et al. A nationally representative study of calcific uremic arteriolopathy risk factors. J Am Soc Nephrol. 2016;27:3421-3429.
- Zhang Y, Corapi KM, Luongo M, et al. Calciphylaxis in peritoneal dialysis patients: a single center cohort study. Int J Nephrol Renovasc Dis. 2016;9:235-241.
- Nigwekar SU, Kroshinsky D, Nazarian RM, et al. Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis. 2015;66:133-146.
- Nigwekar SU, Thadhani RI, Brandenburg VM. Calciphylaxis. N Engl J Med. 2018;378:1704-1714.
- Davis JM. The relationship between obesity and calciphylaxis: a review of the literature. Ostomy Wound Manage. 2016;62:12-18.
- Bajaj R, Courbebaisse M, Kroshinsky D, et al. Calciphylaxis in patients with normal renal function: a case series and systematic review. Mayo Clin Proc. 2018;93:1202-1212.
- Hafner J, Keusch G, Wahl C, et al. Uremic small-artery disease with medial calcification and intimal hyperplasia (so-called calciphylaxis): a complication of chronic renal failure and benefit from parathyroidectomy. J Am Acad Dermatol. 1995;33:954-962.
- Jeong HS, Dominguez AR. Calciphylaxis: controversies in pathogenesis, diagnosis and treatment. Am J Med Sci. 2016;351:217-227.
- Westphal SG, Plumb T. Calciphylaxis. In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2018. https://www.ncbi.nlm.nih.gov/books/NBK519020. Accessed November 12, 2018.
- Riemer CA, El-Azhary RA, Wu KL, et al. Underreported use of palliative care and patient-reported outcome measures to address reduced quality of life in patients with calciphylaxis: a systematic review. Br J Dermatol. 2017;177:1510-1518.
- Nigwekar SU. Calciphylaxis. Curr Opin Nephrol Hypertens. 2017;26:276-281.
- Fine A, Fontaine B. Calciphylaxis: the beginning of the end? Perit Dial Int. 2008;28:268-270.
- Lin WT, Chao CM. Tumoral calcinosis in renal failure. QJM. 2014;107:387.
- Schafer C, Heiss A, Schwarz A, et al. The serum protein alpha 2-Heremans-Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. 2003;112:357-366.
- Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78-81.
- Bleyer AJ, Choi M, Igwemezie B, et al. A case control study of proximal calciphylaxis. Am J Kidney Dis. 1998;32:376-383.
- Ahmed S, O’Neill KD, Hood AF, et al. Calciphylaxis is associated with hyperphosphatemia and increased osteopontin expression by vascular smooth muscle cells. Am J Kidney Dis. 2001;37:267-276.
- Nigwekar SU, Bloch DB, Nazarian RM, et al. Vitamin K-dependent carboxylation of matrix gla protein influences the risk of calciphylaxis. J Am Soc Nephrol. 2017;28:1717-1722.
- Weenig RH, Sewell LD, Davis MD, et al. Calciphylaxis: natural history, risk factor analysis, and outcome. J Am Acad Dermatol. 2007;56:569-579.
- Polizzotto MN, Bryan T, Ashby MA, et al. Symptomatic management of calciphylaxis: a case series and review of the literature. J Pain Symptom Manage. 2006;32:186-190.
- Gupta N, Haq KF, Mahajan S, et al. Gastrointestinal bleeding secondary to calciphylaxis. Am J Case Rep. 2015;16:818-822.
- Edelstein CL, Wickham MK, Kirby PA. Systemic calciphylaxis presenting as a painful, proximal myopathy. Postgrad Med J. 1992;68:209-211.
- Mochel MC, Arakari RY, Wang G, et al. Cutaneous calciphylaxis: a retrospective histopathologic evaluation. Am J Dermatopathol. 2013;35:582-586.
- Chen TY, Lehman JS, Gibson LE, et al. Histopathology of calciphylaxis: cohort study with clinical correlations. Am J Dermatopathol. 2017;39:795-802.
- Cassius C, Moguelet P, Monfort JB, et al. Calciphylaxis in haemodialysed patients: diagnostic value of calcifications in cutaneous biopsy. Br J Dermatol. 2018;178:292-293.
- Sreedhar A, Sheikh HA, Scagliotti CJ, et al. Advanced-stage calciphylaxis: think before you punch. Cleve Clin J Med. 2016;83:562-564.
- Brandenburg VM, Kramann R, Rothe H, et al. Calcific uraemic arteriolopathy (calciphylaxis): data from a large nation-wide registry. Nephrol Dial Transplant. 2017;32:126-132.
- Paul S, Rabito CA, Vedak P, et al. The role of bone scintigraphy in the diagnosis of calciphylaxis. JAMA Dermatol. 2017;153:101-103.
- Shmidt E, Murthy NS, Knudsen JM, et al. Net-like pattern of calcification on plain soft-tissue radiographs in patients with calciphylaxis. J Am Acad Dermatol. 2012;67:1296-1301.
- EVOLVE Trial Investigators; Chertow GM, Block GA, Correa-Rotter R, et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367:2482-2494.
- Rogers NM, Teubner DJO, Coates PT. Calcific uremic arteriolopathy: advances in pathogenesis and treatment. Semin Dial. 2007;20:150-157.
- Nigwekar SU. Multidisciplinary approach to calcific uremic arteriolopathy. Curr Opin Nephrol Hypertens. 2015;24:531-537.
- Cicone JS, Petronis JB, Embert CD, et al. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am J Kidney Dis. 2004;43:1104-1108.
- Chen NX, O’Neill K, Akl NK, et al. Adipocyte induced arterial calcification is prevented with sodium thiosulfate. Biochem Biophys Res Commun. 2014;449:151-156.
- Chan MR, Ghandour F, Murali NS, et al. Pilot study of the effect of lanthanum carbonate in patients with calciphylaxis: a Wisconsin Network for Health Research (WiNHR) study. J Nephrol Ther. 2014;4:1000162.
- Perelló J, Gómez M, Ferrer MD, et al. SNF472, a novel inhibitor of vascular calcification, could be administered during hemodialysis to attain potentially therapeutic phytate levels. J Nephrol. 2018;31:287-296.
- Christiadi D, Singer RF. Calciphylaxis in a dialysis patient successfully treated with high-dose vitamin K supplementation. Clin Kidney J. 2018;11:528-529.
- Caluwe R, Vandecasteele S, Van Vlem B, et al. Vitamin K2 supplementation in haemodialysis patients: a randomized dose-finding study. Nephrol Dial Transplant. 2014;29:1385-1390.
- McCarthy JT, El-Azhary RA, Patzelt MT, et al. Survival, risk factors, and effect of treatment in 101 patients with calciphylaxis. Mayo Clin Proc. 2016;91:1384-1394.
- Fine A, Zacharias J. Calciphylaxis is usually non-ulcerating: risk factors, outcome and therapy. Kidney Int. 2002;61:2210-2217.
- Nigwekar SU, Zhao S, Wenger J, et al. A nationally representative study of calcific uremic arteriolopathy risk factors. J Am Soc Nephrol. 2016;27:3421-3429.
- Zhang Y, Corapi KM, Luongo M, et al. Calciphylaxis in peritoneal dialysis patients: a single center cohort study. Int J Nephrol Renovasc Dis. 2016;9:235-241.
Practice Points
- Maintain a high index of suspicion for calciphylaxis in patients with end-stage renal disease on chronic dialysis presenting with severely painful livedoid plaques or retiform purpura, particularly in fat-rich body sites.
- Skin biopsies may be limited by biopsy site, inadequate biopsy depth, missed areas of microcalcification, and absence of definitive histologic criteria. Special calcium stains and review by an experienced dermatopathologist may lower the rate of false-negative biopsies.
- In cases where the most likely clinical diagnosis is calciphylaxis, treatment should be initiated even if definitive histopathology findings are lacking.
- Treatment should be multimodal, including elimination of risk factors, intravenous sodium thiosulfate, agents addressing calcium-phosphate metabolism, and surgical debridement, if indicated.
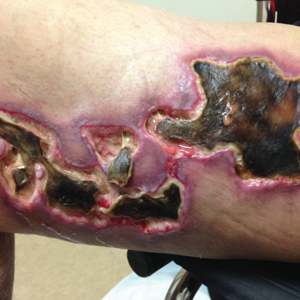
DRESS Syndrome: Clinical Myths and Pearls
Drug rash with eosinophilia and systemic symptoms (DRESS syndrome), also known as drug-induced hypersensitivity syndrome, is an uncommon severe systemic hypersensitivity drug reaction. It is estimated to occur in 1 in every 1000 to 10,000 drug exposures.1 It can affect patients of all ages and typically presents 2 to 6 weeks after exposure to a culprit medication. Classically, DRESS syndrome presents with often widespread rash, facial edema, systemic symptoms such as fever, lymphadenopathy, and evidence of visceral organ involvement. Peripheral blood eosinophilia is frequently but not universally observed.1,2
Even with proper management, reported DRESS syndrome mortality rates worldwide are approximately 10%2 or higher depending on the degree and type of other organ involvement (eg, cardiac).3 Beyond the acute manifestations of DRESS syndrome, this condition is unique in that some patients develop late-onset sequelae such as myocarditis or autoimmune conditions even years after the initial cutaneous eruption.4 Therefore, longitudinal evaluation is a key component of management.
The clinical myths and pearls presented here highlight some of the commonly held assumptions regarding DRESS syndrome in an effort to illuminate subtleties of managing patients with this condition (Table).
Myth: DRESS syndrome may only be diagnosed when the clinical criteria satisfy one of the established scoring systems.
Patients with DRESS syndrome can have heterogeneous manifestations. As a result, patients may develop a drug hypersensitivity with biological behavior and a natural history compatible with DRESS syndrome that does not fulfill published diagnostic criteria.5 The syndrome also may reveal its component manifestations gradually, thus delaying the diagnosis. The terms mini-DRESS and skirt syndrome have been employed to describe drug eruptions that clearly have systemic symptoms and more complex and pernicious biologic behavior than a simple drug exanthema but do not meet DRESS syndrome criteria. Ultimately, it is important to note that in clinical practice, DRESS syndrome exists on a spectrum of severity and the diagnosis remains a clinical one.
Pearl: The most commonly involved organ in DRESS syndrome is the liver.
Liver involvement is the most common visceral organ involved in DRESS syndrome and is estimated to occur in approximately 45.0% to 86.1% of cases.6,7 If a patient develops the characteristic rash, peripheral blood eosinophilia, and evidence of liver injury, DRESS syndrome must be included in the differential diagnosis.
Hepatitis presenting in DRESS syndrome can be hepatocellular, cholestatic, or mixed.6,7 Case series are varied in whether the transaminitis of DRESS syndrome tends to be more hepatocellular8 or cholestatic.7 Liver dysfunction in DRESS syndrome often lasts longer than in other severe cutaneous adverse drug reactions, and patients may improve anywhere from a few days in milder cases to months to achieve resolution of abnormalities.6,7 Severe hepatic involvement is thought to be the most notable cause of mortality.9
Pearl: New-onset proteinuria, hematuria, and sterile pyuria indicate acute interstitial nephritis that may be associated with DRESS syndrome.
Acute interstitial nephritis (AIN) is a drug-induced form of acute kidney injury that can co-occur with DRESS syndrome. Acute interstitial nephritis can present with some combination of acute kidney injury, morbilliform eruption, eosinophilia, fever, and sometimes eosinophiluria. Although AIN can be distinct from DRESS syndrome, there are cases of DRESS syndrome associated with AIN.10 In the correct clinical context, urinalysis may help by showing new-onset proteinuria, new-onset hematuria, and sterile pyuria. More common causes of acute kidney injury such as prerenal etiologies and acute tubular necrosis have a bland urinary sediment.
Myth: If the eruption is not morbilliform, then it is not DRESS syndrome.
The most common morphology of DRESS syndrome is a morbilliform eruption (Figure 1), but urticarial and atypical targetoid (erythema multiforme–like) eruptions also have been described.9 Rarely, DRESS syndrome secondary to use of allopurinol or anticonvulsants may have a pustular morphology (Figure 2), which is distinguished from acute generalized exanthematous pustulosis by its delayed onset, more severe visceral involvement, and prolonged course.11
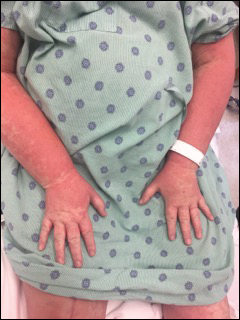
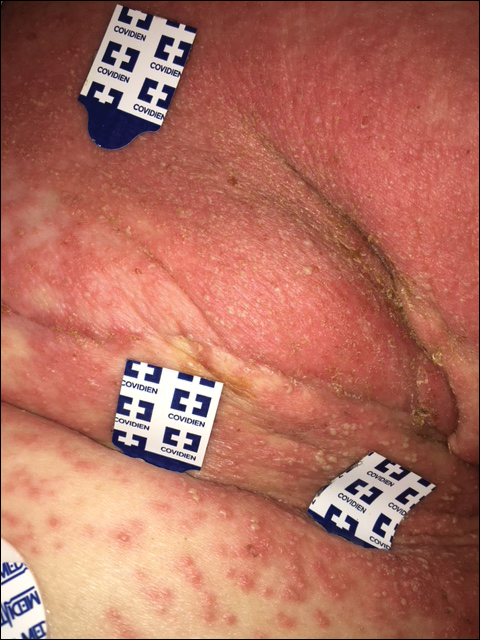
Another reported variant demonstrates overlapping features between Stevens-Johnson syndrome/toxic epidermal necrolysis and DRESS syndrome. It may present with mucositis, atypical targetoid lesions, and vesiculobullous lesions.12 It is unclear whether this reported variant is indeed a true subtype of DRESS syndrome, as Stevens-Johnson syndrome/toxic epidermal necrolysis may present with systemic symptoms, lymphadenopathy, hepatic, renal, and pulmonary complications, among other systemic disturbances.12
Pearl: Facial edema noted during physical examination is an important clue of DRESS syndrome.
Perhaps the most helpful findings in the diagnosis of DRESS syndrome are facial edema and anasarca (Figure 3), as facial edema is not a usual finding in sepsis. Facial edema can be severe enough that the patient’s features are dramatically altered. It may be useful to ask family members if the patient’s face appears swollen or to compare the current appearance to the patient’s driver’s license photograph. An important complication to note is laryngeal edema, which may complicate airway management and may manifest as respiratory distress, stridor, and the need for emergent intubation.13
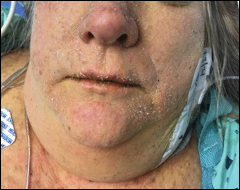
Myth: Patients who have had an allergic reaction to sulfonamide antibiotics will have a cross-reaction to nonantibiotic sulfonamides.
A common question is, if a patient has had a prior allergy to sulfonamide antibiotics, then are nonantibiotic sulfones such as a sulfonylurea, thiazide diuretic, or furosemide likely to cause a a cross-reaction? In one study (N=969), only 9.9% of patients with a prior sulfone antibiotic allergy developed hypersensitivity when exposed to a nonantibiotic sulfone, which is thought to be due to an overall increased propensity for hypersensitivity rather than a true cross-reaction. In fact, the risk for developing a hypersensitivity reaction to penicillin (14.0% [717/5115]) was higher than the risk for developing a reaction from a nonantibiotic sulfone among these patients.14 This study bolsters the argument that if there are other potential culprit medications and the time course for a patient’s nonantibiotic sulfone is not consistent with the timeline for DRESS syndrome, it may be beneficial to look for a different causative agent.
Pearl: Vancomycin is an important cause of DRESS syndrome.
Guidelines for treating endocarditis and osteomyelitis caused by methicillin-resistant Staphylococcus aureus infection recommend intravenous vancomycin for 4 to 6 weeks.15 This duration is within the relevant time frame of exposure for the development of DRESS syndrome de novo.
One case series noted that 37.5% (12/32) of DRESS syndrome cases in a 3-year period were caused by vancomycin, which notably was the most common medication associated with DRESS syndrome.16 There were caveats to this case series in that no standardized drug causality score was used and the sample size over the 3-year period was small; however, the increased use (and misuse) of antibiotics and perhaps increased recognition of rash in outpatient parenteral antibiotic therapy clinics may play a role if vancomycin-induced DRESS syndrome is indeed becoming more common.
Myth: Myocarditis secondary to DRESS syndrome will present with chest pain at the time of the cutaneous eruption.
Few patients with DRESS syndrome–associated myocarditis actually are symptomatic during their hospitalization.4 In asymptomatic patients, the primary team and consultants should be vigilant for the potential of subclinical myocarditis or the possibility of developing cardiac involvement after discharge, as myocarditis secondary to DRESS syndrome may present any time from rash onset up to 4 months later.4 Therefore, DRESS patients should be especially attentive to any new cardiac symptoms and notify their provider if any develop.
Although no standard cardiac screening guidelines exist for DRESS syndrome, some have recommended that baseline cardiac screening tests including electrocardiogram, troponin levels, and echocardiogram be considered at the time of diagnosis.5 If any testing is abnormal, DRESS syndrome–associated myocarditis should be suspected and an endomyocardial biopsy, which is the diagnostic gold standard, may be necessary.4 If the cardiac screening tests are normal, some investigators recommend serial outpatient echocardiograms for all DRESS patients, even those who remain asymptomatic.17 An alternative is an empiric approach in which a thorough review of systems is performed and testing is done if patients develop symptoms that are concerning for myocarditis.
Pearl: Steroids are not the only treatment used to control DRESS syndrome.
A prolonged taper of systemic steroids is the first-line treatment of DRESS syndrome. Steroids at the equivalent of 1 to 2 mg/kg daily (once or divided into 2 doses) of prednisone typically are used. For severe and/or recalcitrant DRESS syndrome, 2 mg/kg daily (once or divided into 2 doses) typically is used, and less than 1 mg/kg daily may be used for mini-DRESS syndrome.
Clinical improvement of DRESS syndrome has been demonstrated in several case reports with intravenous immunoglobulin, cyclosporine, cyclophosphamide, mycophenolate mofetil, and plasmapheresis.18-21 Each of these therapies typically were initiated as second-line therapeutic agents when initial treatment with steroids failed. It is important to note that large prospective studies regarding these treatments are lacking; however, there have been case reports of acute necrotizing eosinophilic myocarditis that did not respond to the combination of steroids and cyclosporine.4,22
Although there have been successful case reports using intravenous immunoglobulin, a 2012 prospective open-label clinical trial reported notable side effects in 5 of 6 (83.3%) patients with only 1 of 6 (16.6%) achieving the primary end point of control of fever/symptoms at day 7 and clinical remission without steroids on day 30.23
Pearl: DRESS patients need to be monitored for long-term sequelae such as autoimmune disease.
Several autoimmune conditions may develop as a delayed complication of DRESS syndrome, including autoimmune thyroiditis, systemic lupus erythematosus, type 1 diabetes mellitus, and autoimmune hemolytic anemia.24-26 Incidence rates of autoimmunity following DRESS syndrome range from 3% to 5% among small case series.24,25
Autoimmune thyroiditis, which may present as Graves disease, Hashimoto thyroiditis, or painless thyroiditis, is the most common autoimmune disorder to develop in DRESS patients and appears from several weeks to up to 3 years after DRESS.24 Therefore, all DRESS patients should be monitored longitudinally for several years for signs or symptoms suggestive of an autoimmune condition.5,24,26
Because no guidelines exist regarding serial monitoring for autoimmune sequelae, it may be reasonable to check thyroid function tests at the time of diagnosis and regularly for at least 2 years after diagnosis.5 Alternatively, clinicians may consider an empiric approach to laboratory testing that is guided by the development of clinical symptoms.
Pearl: Small cases series suggest differences between adult and pediatric DRESS syndrome, but there are no large studies in children.
Small case series have suggested there may be noteworthy differences between DRESS syndrome in adults and children. Although human herpesvirus 6 (HHV-6) positivity in DRESS syndrome in adults may be as high as 80%, 13% of pediatric patients in one cohort tested positive for HHV-6, though the study size was limited at 29 total patients.27 In children, DRESS syndrome secondary to antibiotics was associated with a shorter latency time as compared to cases secondary to nonantibiotics. In contrast to the typical 2- to 6-week timeline, Sasidharanpillai et al28 reported an average onset 5.8 days after drug administration in antibiotic-associated DRESS syndrome compared to 23.9 days for anticonvulsants, though this study only included 11 total patients. Other reports have suggested a similar trend.27
The role of HHV-6 positivity in pediatric DRESS syndrome and its influence on prognosis remains unclear. One study showed a worse prognosis for pediatric patients with positive HHV-6 antibodies.27 However, with such a small sample size—only 4 HHV-6–positive patients of 29 pediatric DRESS cases—larger studies are needed to better characterize the relationship between HHV-6 positivity and prognosis.
- Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med, 2011;124:588-597.
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
- Intarasupht J, Kanchanomai A, Leelasattakul W, et al. Prevalence, risk factors, and mortality outcome in the drug reaction with eosinophilia and systemic symptoms patients with cardiac involvement. Int J Dermatol. 2018;57:1187-1191.
- Bourgeois GP, Cafardi JA, Groysman V, et al. A review of DRESS-associated myocarditis. J Am Acad Dermatol. 2012;66:E229-E236.
- Husain Z, Reddy BY, Schwartz RA. DRESS syndrome: part I. clinical perspectives. J Am Acad Dermatol. 2013;68:693.e1-693.e14; quiz 706-708.
- Lee T, Lee YS, Yoon SY, et al. Characteristics of liver injury in drug-induced systemic hypersensitivity reactions. J Am Acad Dermatol. 2013;69:407-415.
- Lin IC, Yang HC, Strong C, et al. Liver injury in patients with DRESS: a clinical study of 72 cases. J Am Acad Dermatol. 2015;72:984-991.
- Peyrière H, Dereure O, Breton H, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2006;155:422-428.
- Walsh S, Diaz-Cano S, Higgins E, et al. Drug reaction with eosinophilia and systemic symptoms: is cutaneous phenotype a prognostic marker for outcome? a review of clinicopathological features of 27 cases. Br J Dermatol. 2013;168:391-401.
- Raghavan R, Eknoyan G. Acute interstitial nephritis—a reappraisal and update. Clin Nephrol. 2014;82:149-162.
- Matsuda H, Saito K, Takayanagi Y, et al. Pustular-type drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms due to carbamazepine with systemic muscle involvement. J Dermatol. 2013;40:118-122.
- Wolf R, Davidovici B, Matz H, et al. Drug rash with eosinophilia and systemic symptoms versus Stevens-Johnson Syndrome—a case that indicates a stumbling block in the current classification. Int Arch Allergy Immunol. 2006;141:308-310.
- Kumar A, Goldfarb JW, Bittner EA. A case of drug rash with eosinophilia and systemic symptoms (DRESS) syndrome complicating airway management. Can J Anaesth. 2012;59:295-298.
- Strom BL, Schinnar R, Apter AJ, et al. Absence of cross-reactivity between sulfonamide antibiotics and sulfonamide nonantibiotics. N Engl J Med. 2003;349:1628-1635.
- Berbari EF, Kanj SS, Kowalski TJ, et al; Infectious Diseases Society of America. 2015 Infectious Diseases Society of America (IDSA) clinical practice guidelines for the diagnosis and treatment of native vertebral osteomyelitis in adults. Clin Infect Dis. 2015;61:E26-E46.
- Lam BD, Miller MM, Sutton AV, et al. Vancomycin and DRESS: a retrospective chart review of 32 cases in Los Angeles, California. J Am Acad Dermatol. 2017;77:973-975.
- Eppenberger M, Hack D, Ammann P, et al. Acute eosinophilic myocarditis with dramatic response to steroid therapy: the central role of echocardiography in diagnosis and follow-up. Tex Heart Inst J. 2013;40:326-330.
- Kirchhof MG, Wong A, Dutz JP. Cyclosporine treatment of drug-induced hypersensitivity syndrome. JAMA Dermatol. 2016;152:1254-1257.
- Singer EM, Wanat KA, Rosenbach MA. A case of recalcitrant DRESS syndrome with multiple autoimmune sequelae treated with intravenous immunoglobulins. JAMA Dermatol. 2013;149:494-495.
- Bommersbach TJ, Lapid MI, Leung JG, et al. Management of psychotropic drug-induced DRESS syndrome: a systematic review. Mayo Clin Proc. 2016;91:787-801.
- Alexander T, Iglesia E, Park Y, et al. Severe DRESS syndrome managed with therapeutic plasma exchange. Pediatrics. 2013;131:E945-E949.
- Daoulah A, Alqahtani AA, Ocheltree SR, et al. Acute myocardial infarction in a 56-year-old female patient treated with sulfasalazine. Am J Emerg Med. 2012;30:638.e1-638.e3.
- Joly P, Janela B, Tetart F, et al. Poor benefit/risk balance of intravenous immunoglobulins in DRESS. Arch Dermatol. 2012;148:543-544.
- Kano Y, Tohyama M, Aihara M, et al. Sequelae in 145 patients with drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms: survey conducted by the Asian Research Committee on Severe Cutaneous Adverse Reactions (ASCAR). J Dermatol. 2015;42:276-282.
- Ushigome Y, Kano Y, Ishida T, et al. Short- and long-term outcomes of 34 patients with drug-induced hypersensitivity syndrome in a single institution. J Am Acad Dermatol. 2013;68:721-728.
- Matta JM, Flores SM, Cherit JD. Drug reaction with eosinophilia and systemic symptoms (DRESS) and its relation with autoimmunity in a reference center in Mexico. An Bras Dermatol. 2017;92:30-33.
- Ahluwalia J, Abuabara K, Perman MJ, et al. Human herpesvirus 6 involvement in paediatric drug hypersensitivity syndrome. Br J Dermatol. 2015;172:1090-1095.
- Sasidharanpillai S, Sabitha S, Riyaz N, et al. Drug reaction with eosinophilia and systemic symptoms in children: a prospective study. Pediatr Dermatol. 2016;33:E162-E165.
Drug rash with eosinophilia and systemic symptoms (DRESS syndrome), also known as drug-induced hypersensitivity syndrome, is an uncommon severe systemic hypersensitivity drug reaction. It is estimated to occur in 1 in every 1000 to 10,000 drug exposures.1 It can affect patients of all ages and typically presents 2 to 6 weeks after exposure to a culprit medication. Classically, DRESS syndrome presents with often widespread rash, facial edema, systemic symptoms such as fever, lymphadenopathy, and evidence of visceral organ involvement. Peripheral blood eosinophilia is frequently but not universally observed.1,2
Even with proper management, reported DRESS syndrome mortality rates worldwide are approximately 10%2 or higher depending on the degree and type of other organ involvement (eg, cardiac).3 Beyond the acute manifestations of DRESS syndrome, this condition is unique in that some patients develop late-onset sequelae such as myocarditis or autoimmune conditions even years after the initial cutaneous eruption.4 Therefore, longitudinal evaluation is a key component of management.
The clinical myths and pearls presented here highlight some of the commonly held assumptions regarding DRESS syndrome in an effort to illuminate subtleties of managing patients with this condition (Table).
Myth: DRESS syndrome may only be diagnosed when the clinical criteria satisfy one of the established scoring systems.
Patients with DRESS syndrome can have heterogeneous manifestations. As a result, patients may develop a drug hypersensitivity with biological behavior and a natural history compatible with DRESS syndrome that does not fulfill published diagnostic criteria.5 The syndrome also may reveal its component manifestations gradually, thus delaying the diagnosis. The terms mini-DRESS and skirt syndrome have been employed to describe drug eruptions that clearly have systemic symptoms and more complex and pernicious biologic behavior than a simple drug exanthema but do not meet DRESS syndrome criteria. Ultimately, it is important to note that in clinical practice, DRESS syndrome exists on a spectrum of severity and the diagnosis remains a clinical one.
Pearl: The most commonly involved organ in DRESS syndrome is the liver.
Liver involvement is the most common visceral organ involved in DRESS syndrome and is estimated to occur in approximately 45.0% to 86.1% of cases.6,7 If a patient develops the characteristic rash, peripheral blood eosinophilia, and evidence of liver injury, DRESS syndrome must be included in the differential diagnosis.
Hepatitis presenting in DRESS syndrome can be hepatocellular, cholestatic, or mixed.6,7 Case series are varied in whether the transaminitis of DRESS syndrome tends to be more hepatocellular8 or cholestatic.7 Liver dysfunction in DRESS syndrome often lasts longer than in other severe cutaneous adverse drug reactions, and patients may improve anywhere from a few days in milder cases to months to achieve resolution of abnormalities.6,7 Severe hepatic involvement is thought to be the most notable cause of mortality.9
Pearl: New-onset proteinuria, hematuria, and sterile pyuria indicate acute interstitial nephritis that may be associated with DRESS syndrome.
Acute interstitial nephritis (AIN) is a drug-induced form of acute kidney injury that can co-occur with DRESS syndrome. Acute interstitial nephritis can present with some combination of acute kidney injury, morbilliform eruption, eosinophilia, fever, and sometimes eosinophiluria. Although AIN can be distinct from DRESS syndrome, there are cases of DRESS syndrome associated with AIN.10 In the correct clinical context, urinalysis may help by showing new-onset proteinuria, new-onset hematuria, and sterile pyuria. More common causes of acute kidney injury such as prerenal etiologies and acute tubular necrosis have a bland urinary sediment.
Myth: If the eruption is not morbilliform, then it is not DRESS syndrome.
The most common morphology of DRESS syndrome is a morbilliform eruption (Figure 1), but urticarial and atypical targetoid (erythema multiforme–like) eruptions also have been described.9 Rarely, DRESS syndrome secondary to use of allopurinol or anticonvulsants may have a pustular morphology (Figure 2), which is distinguished from acute generalized exanthematous pustulosis by its delayed onset, more severe visceral involvement, and prolonged course.11
Another reported variant demonstrates overlapping features between Stevens-Johnson syndrome/toxic epidermal necrolysis and DRESS syndrome. It may present with mucositis, atypical targetoid lesions, and vesiculobullous lesions.12 It is unclear whether this reported variant is indeed a true subtype of DRESS syndrome, as Stevens-Johnson syndrome/toxic epidermal necrolysis may present with systemic symptoms, lymphadenopathy, hepatic, renal, and pulmonary complications, among other systemic disturbances.12
Pearl: Facial edema noted during physical examination is an important clue of DRESS syndrome.
Perhaps the most helpful findings in the diagnosis of DRESS syndrome are facial edema and anasarca (Figure 3), as facial edema is not a usual finding in sepsis. Facial edema can be severe enough that the patient’s features are dramatically altered. It may be useful to ask family members if the patient’s face appears swollen or to compare the current appearance to the patient’s driver’s license photograph. An important complication to note is laryngeal edema, which may complicate airway management and may manifest as respiratory distress, stridor, and the need for emergent intubation.13
Myth: Patients who have had an allergic reaction to sulfonamide antibiotics will have a cross-reaction to nonantibiotic sulfonamides.
A common question is, if a patient has had a prior allergy to sulfonamide antibiotics, then are nonantibiotic sulfones such as a sulfonylurea, thiazide diuretic, or furosemide likely to cause a a cross-reaction? In one study (N=969), only 9.9% of patients with a prior sulfone antibiotic allergy developed hypersensitivity when exposed to a nonantibiotic sulfone, which is thought to be due to an overall increased propensity for hypersensitivity rather than a true cross-reaction. In fact, the risk for developing a hypersensitivity reaction to penicillin (14.0% [717/5115]) was higher than the risk for developing a reaction from a nonantibiotic sulfone among these patients.14 This study bolsters the argument that if there are other potential culprit medications and the time course for a patient’s nonantibiotic sulfone is not consistent with the timeline for DRESS syndrome, it may be beneficial to look for a different causative agent.
Pearl: Vancomycin is an important cause of DRESS syndrome.
Guidelines for treating endocarditis and osteomyelitis caused by methicillin-resistant Staphylococcus aureus infection recommend intravenous vancomycin for 4 to 6 weeks.15 This duration is within the relevant time frame of exposure for the development of DRESS syndrome de novo.
One case series noted that 37.5% (12/32) of DRESS syndrome cases in a 3-year period were caused by vancomycin, which notably was the most common medication associated with DRESS syndrome.16 There were caveats to this case series in that no standardized drug causality score was used and the sample size over the 3-year period was small; however, the increased use (and misuse) of antibiotics and perhaps increased recognition of rash in outpatient parenteral antibiotic therapy clinics may play a role if vancomycin-induced DRESS syndrome is indeed becoming more common.
Myth: Myocarditis secondary to DRESS syndrome will present with chest pain at the time of the cutaneous eruption.
Few patients with DRESS syndrome–associated myocarditis actually are symptomatic during their hospitalization.4 In asymptomatic patients, the primary team and consultants should be vigilant for the potential of subclinical myocarditis or the possibility of developing cardiac involvement after discharge, as myocarditis secondary to DRESS syndrome may present any time from rash onset up to 4 months later.4 Therefore, DRESS patients should be especially attentive to any new cardiac symptoms and notify their provider if any develop.
Although no standard cardiac screening guidelines exist for DRESS syndrome, some have recommended that baseline cardiac screening tests including electrocardiogram, troponin levels, and echocardiogram be considered at the time of diagnosis.5 If any testing is abnormal, DRESS syndrome–associated myocarditis should be suspected and an endomyocardial biopsy, which is the diagnostic gold standard, may be necessary.4 If the cardiac screening tests are normal, some investigators recommend serial outpatient echocardiograms for all DRESS patients, even those who remain asymptomatic.17 An alternative is an empiric approach in which a thorough review of systems is performed and testing is done if patients develop symptoms that are concerning for myocarditis.
Pearl: Steroids are not the only treatment used to control DRESS syndrome.
A prolonged taper of systemic steroids is the first-line treatment of DRESS syndrome. Steroids at the equivalent of 1 to 2 mg/kg daily (once or divided into 2 doses) of prednisone typically are used. For severe and/or recalcitrant DRESS syndrome, 2 mg/kg daily (once or divided into 2 doses) typically is used, and less than 1 mg/kg daily may be used for mini-DRESS syndrome.
Clinical improvement of DRESS syndrome has been demonstrated in several case reports with intravenous immunoglobulin, cyclosporine, cyclophosphamide, mycophenolate mofetil, and plasmapheresis.18-21 Each of these therapies typically were initiated as second-line therapeutic agents when initial treatment with steroids failed. It is important to note that large prospective studies regarding these treatments are lacking; however, there have been case reports of acute necrotizing eosinophilic myocarditis that did not respond to the combination of steroids and cyclosporine.4,22
Although there have been successful case reports using intravenous immunoglobulin, a 2012 prospective open-label clinical trial reported notable side effects in 5 of 6 (83.3%) patients with only 1 of 6 (16.6%) achieving the primary end point of control of fever/symptoms at day 7 and clinical remission without steroids on day 30.23
Pearl: DRESS patients need to be monitored for long-term sequelae such as autoimmune disease.
Several autoimmune conditions may develop as a delayed complication of DRESS syndrome, including autoimmune thyroiditis, systemic lupus erythematosus, type 1 diabetes mellitus, and autoimmune hemolytic anemia.24-26 Incidence rates of autoimmunity following DRESS syndrome range from 3% to 5% among small case series.24,25
Autoimmune thyroiditis, which may present as Graves disease, Hashimoto thyroiditis, or painless thyroiditis, is the most common autoimmune disorder to develop in DRESS patients and appears from several weeks to up to 3 years after DRESS.24 Therefore, all DRESS patients should be monitored longitudinally for several years for signs or symptoms suggestive of an autoimmune condition.5,24,26
Because no guidelines exist regarding serial monitoring for autoimmune sequelae, it may be reasonable to check thyroid function tests at the time of diagnosis and regularly for at least 2 years after diagnosis.5 Alternatively, clinicians may consider an empiric approach to laboratory testing that is guided by the development of clinical symptoms.
Pearl: Small cases series suggest differences between adult and pediatric DRESS syndrome, but there are no large studies in children.
Small case series have suggested there may be noteworthy differences between DRESS syndrome in adults and children. Although human herpesvirus 6 (HHV-6) positivity in DRESS syndrome in adults may be as high as 80%, 13% of pediatric patients in one cohort tested positive for HHV-6, though the study size was limited at 29 total patients.27 In children, DRESS syndrome secondary to antibiotics was associated with a shorter latency time as compared to cases secondary to nonantibiotics. In contrast to the typical 2- to 6-week timeline, Sasidharanpillai et al28 reported an average onset 5.8 days after drug administration in antibiotic-associated DRESS syndrome compared to 23.9 days for anticonvulsants, though this study only included 11 total patients. Other reports have suggested a similar trend.27
The role of HHV-6 positivity in pediatric DRESS syndrome and its influence on prognosis remains unclear. One study showed a worse prognosis for pediatric patients with positive HHV-6 antibodies.27 However, with such a small sample size—only 4 HHV-6–positive patients of 29 pediatric DRESS cases—larger studies are needed to better characterize the relationship between HHV-6 positivity and prognosis.
Drug rash with eosinophilia and systemic symptoms (DRESS syndrome), also known as drug-induced hypersensitivity syndrome, is an uncommon severe systemic hypersensitivity drug reaction. It is estimated to occur in 1 in every 1000 to 10,000 drug exposures.1 It can affect patients of all ages and typically presents 2 to 6 weeks after exposure to a culprit medication. Classically, DRESS syndrome presents with often widespread rash, facial edema, systemic symptoms such as fever, lymphadenopathy, and evidence of visceral organ involvement. Peripheral blood eosinophilia is frequently but not universally observed.1,2
Even with proper management, reported DRESS syndrome mortality rates worldwide are approximately 10%2 or higher depending on the degree and type of other organ involvement (eg, cardiac).3 Beyond the acute manifestations of DRESS syndrome, this condition is unique in that some patients develop late-onset sequelae such as myocarditis or autoimmune conditions even years after the initial cutaneous eruption.4 Therefore, longitudinal evaluation is a key component of management.
The clinical myths and pearls presented here highlight some of the commonly held assumptions regarding DRESS syndrome in an effort to illuminate subtleties of managing patients with this condition (Table).
Myth: DRESS syndrome may only be diagnosed when the clinical criteria satisfy one of the established scoring systems.
Patients with DRESS syndrome can have heterogeneous manifestations. As a result, patients may develop a drug hypersensitivity with biological behavior and a natural history compatible with DRESS syndrome that does not fulfill published diagnostic criteria.5 The syndrome also may reveal its component manifestations gradually, thus delaying the diagnosis. The terms mini-DRESS and skirt syndrome have been employed to describe drug eruptions that clearly have systemic symptoms and more complex and pernicious biologic behavior than a simple drug exanthema but do not meet DRESS syndrome criteria. Ultimately, it is important to note that in clinical practice, DRESS syndrome exists on a spectrum of severity and the diagnosis remains a clinical one.
Pearl: The most commonly involved organ in DRESS syndrome is the liver.
Liver involvement is the most common visceral organ involved in DRESS syndrome and is estimated to occur in approximately 45.0% to 86.1% of cases.6,7 If a patient develops the characteristic rash, peripheral blood eosinophilia, and evidence of liver injury, DRESS syndrome must be included in the differential diagnosis.
Hepatitis presenting in DRESS syndrome can be hepatocellular, cholestatic, or mixed.6,7 Case series are varied in whether the transaminitis of DRESS syndrome tends to be more hepatocellular8 or cholestatic.7 Liver dysfunction in DRESS syndrome often lasts longer than in other severe cutaneous adverse drug reactions, and patients may improve anywhere from a few days in milder cases to months to achieve resolution of abnormalities.6,7 Severe hepatic involvement is thought to be the most notable cause of mortality.9
Pearl: New-onset proteinuria, hematuria, and sterile pyuria indicate acute interstitial nephritis that may be associated with DRESS syndrome.
Acute interstitial nephritis (AIN) is a drug-induced form of acute kidney injury that can co-occur with DRESS syndrome. Acute interstitial nephritis can present with some combination of acute kidney injury, morbilliform eruption, eosinophilia, fever, and sometimes eosinophiluria. Although AIN can be distinct from DRESS syndrome, there are cases of DRESS syndrome associated with AIN.10 In the correct clinical context, urinalysis may help by showing new-onset proteinuria, new-onset hematuria, and sterile pyuria. More common causes of acute kidney injury such as prerenal etiologies and acute tubular necrosis have a bland urinary sediment.
Myth: If the eruption is not morbilliform, then it is not DRESS syndrome.
The most common morphology of DRESS syndrome is a morbilliform eruption (Figure 1), but urticarial and atypical targetoid (erythema multiforme–like) eruptions also have been described.9 Rarely, DRESS syndrome secondary to use of allopurinol or anticonvulsants may have a pustular morphology (Figure 2), which is distinguished from acute generalized exanthematous pustulosis by its delayed onset, more severe visceral involvement, and prolonged course.11
Another reported variant demonstrates overlapping features between Stevens-Johnson syndrome/toxic epidermal necrolysis and DRESS syndrome. It may present with mucositis, atypical targetoid lesions, and vesiculobullous lesions.12 It is unclear whether this reported variant is indeed a true subtype of DRESS syndrome, as Stevens-Johnson syndrome/toxic epidermal necrolysis may present with systemic symptoms, lymphadenopathy, hepatic, renal, and pulmonary complications, among other systemic disturbances.12
Pearl: Facial edema noted during physical examination is an important clue of DRESS syndrome.
Perhaps the most helpful findings in the diagnosis of DRESS syndrome are facial edema and anasarca (Figure 3), as facial edema is not a usual finding in sepsis. Facial edema can be severe enough that the patient’s features are dramatically altered. It may be useful to ask family members if the patient’s face appears swollen or to compare the current appearance to the patient’s driver’s license photograph. An important complication to note is laryngeal edema, which may complicate airway management and may manifest as respiratory distress, stridor, and the need for emergent intubation.13
Myth: Patients who have had an allergic reaction to sulfonamide antibiotics will have a cross-reaction to nonantibiotic sulfonamides.
A common question is, if a patient has had a prior allergy to sulfonamide antibiotics, then are nonantibiotic sulfones such as a sulfonylurea, thiazide diuretic, or furosemide likely to cause a a cross-reaction? In one study (N=969), only 9.9% of patients with a prior sulfone antibiotic allergy developed hypersensitivity when exposed to a nonantibiotic sulfone, which is thought to be due to an overall increased propensity for hypersensitivity rather than a true cross-reaction. In fact, the risk for developing a hypersensitivity reaction to penicillin (14.0% [717/5115]) was higher than the risk for developing a reaction from a nonantibiotic sulfone among these patients.14 This study bolsters the argument that if there are other potential culprit medications and the time course for a patient’s nonantibiotic sulfone is not consistent with the timeline for DRESS syndrome, it may be beneficial to look for a different causative agent.
Pearl: Vancomycin is an important cause of DRESS syndrome.
Guidelines for treating endocarditis and osteomyelitis caused by methicillin-resistant Staphylococcus aureus infection recommend intravenous vancomycin for 4 to 6 weeks.15 This duration is within the relevant time frame of exposure for the development of DRESS syndrome de novo.
One case series noted that 37.5% (12/32) of DRESS syndrome cases in a 3-year period were caused by vancomycin, which notably was the most common medication associated with DRESS syndrome.16 There were caveats to this case series in that no standardized drug causality score was used and the sample size over the 3-year period was small; however, the increased use (and misuse) of antibiotics and perhaps increased recognition of rash in outpatient parenteral antibiotic therapy clinics may play a role if vancomycin-induced DRESS syndrome is indeed becoming more common.
Myth: Myocarditis secondary to DRESS syndrome will present with chest pain at the time of the cutaneous eruption.
Few patients with DRESS syndrome–associated myocarditis actually are symptomatic during their hospitalization.4 In asymptomatic patients, the primary team and consultants should be vigilant for the potential of subclinical myocarditis or the possibility of developing cardiac involvement after discharge, as myocarditis secondary to DRESS syndrome may present any time from rash onset up to 4 months later.4 Therefore, DRESS patients should be especially attentive to any new cardiac symptoms and notify their provider if any develop.
Although no standard cardiac screening guidelines exist for DRESS syndrome, some have recommended that baseline cardiac screening tests including electrocardiogram, troponin levels, and echocardiogram be considered at the time of diagnosis.5 If any testing is abnormal, DRESS syndrome–associated myocarditis should be suspected and an endomyocardial biopsy, which is the diagnostic gold standard, may be necessary.4 If the cardiac screening tests are normal, some investigators recommend serial outpatient echocardiograms for all DRESS patients, even those who remain asymptomatic.17 An alternative is an empiric approach in which a thorough review of systems is performed and testing is done if patients develop symptoms that are concerning for myocarditis.
Pearl: Steroids are not the only treatment used to control DRESS syndrome.
A prolonged taper of systemic steroids is the first-line treatment of DRESS syndrome. Steroids at the equivalent of 1 to 2 mg/kg daily (once or divided into 2 doses) of prednisone typically are used. For severe and/or recalcitrant DRESS syndrome, 2 mg/kg daily (once or divided into 2 doses) typically is used, and less than 1 mg/kg daily may be used for mini-DRESS syndrome.
Clinical improvement of DRESS syndrome has been demonstrated in several case reports with intravenous immunoglobulin, cyclosporine, cyclophosphamide, mycophenolate mofetil, and plasmapheresis.18-21 Each of these therapies typically were initiated as second-line therapeutic agents when initial treatment with steroids failed. It is important to note that large prospective studies regarding these treatments are lacking; however, there have been case reports of acute necrotizing eosinophilic myocarditis that did not respond to the combination of steroids and cyclosporine.4,22
Although there have been successful case reports using intravenous immunoglobulin, a 2012 prospective open-label clinical trial reported notable side effects in 5 of 6 (83.3%) patients with only 1 of 6 (16.6%) achieving the primary end point of control of fever/symptoms at day 7 and clinical remission without steroids on day 30.23
Pearl: DRESS patients need to be monitored for long-term sequelae such as autoimmune disease.
Several autoimmune conditions may develop as a delayed complication of DRESS syndrome, including autoimmune thyroiditis, systemic lupus erythematosus, type 1 diabetes mellitus, and autoimmune hemolytic anemia.24-26 Incidence rates of autoimmunity following DRESS syndrome range from 3% to 5% among small case series.24,25
Autoimmune thyroiditis, which may present as Graves disease, Hashimoto thyroiditis, or painless thyroiditis, is the most common autoimmune disorder to develop in DRESS patients and appears from several weeks to up to 3 years after DRESS.24 Therefore, all DRESS patients should be monitored longitudinally for several years for signs or symptoms suggestive of an autoimmune condition.5,24,26
Because no guidelines exist regarding serial monitoring for autoimmune sequelae, it may be reasonable to check thyroid function tests at the time of diagnosis and regularly for at least 2 years after diagnosis.5 Alternatively, clinicians may consider an empiric approach to laboratory testing that is guided by the development of clinical symptoms.
Pearl: Small cases series suggest differences between adult and pediatric DRESS syndrome, but there are no large studies in children.
Small case series have suggested there may be noteworthy differences between DRESS syndrome in adults and children. Although human herpesvirus 6 (HHV-6) positivity in DRESS syndrome in adults may be as high as 80%, 13% of pediatric patients in one cohort tested positive for HHV-6, though the study size was limited at 29 total patients.27 In children, DRESS syndrome secondary to antibiotics was associated with a shorter latency time as compared to cases secondary to nonantibiotics. In contrast to the typical 2- to 6-week timeline, Sasidharanpillai et al28 reported an average onset 5.8 days after drug administration in antibiotic-associated DRESS syndrome compared to 23.9 days for anticonvulsants, though this study only included 11 total patients. Other reports have suggested a similar trend.27
The role of HHV-6 positivity in pediatric DRESS syndrome and its influence on prognosis remains unclear. One study showed a worse prognosis for pediatric patients with positive HHV-6 antibodies.27 However, with such a small sample size—only 4 HHV-6–positive patients of 29 pediatric DRESS cases—larger studies are needed to better characterize the relationship between HHV-6 positivity and prognosis.
- Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med, 2011;124:588-597.
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
- Intarasupht J, Kanchanomai A, Leelasattakul W, et al. Prevalence, risk factors, and mortality outcome in the drug reaction with eosinophilia and systemic symptoms patients with cardiac involvement. Int J Dermatol. 2018;57:1187-1191.
- Bourgeois GP, Cafardi JA, Groysman V, et al. A review of DRESS-associated myocarditis. J Am Acad Dermatol. 2012;66:E229-E236.
- Husain Z, Reddy BY, Schwartz RA. DRESS syndrome: part I. clinical perspectives. J Am Acad Dermatol. 2013;68:693.e1-693.e14; quiz 706-708.
- Lee T, Lee YS, Yoon SY, et al. Characteristics of liver injury in drug-induced systemic hypersensitivity reactions. J Am Acad Dermatol. 2013;69:407-415.
- Lin IC, Yang HC, Strong C, et al. Liver injury in patients with DRESS: a clinical study of 72 cases. J Am Acad Dermatol. 2015;72:984-991.
- Peyrière H, Dereure O, Breton H, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2006;155:422-428.
- Walsh S, Diaz-Cano S, Higgins E, et al. Drug reaction with eosinophilia and systemic symptoms: is cutaneous phenotype a prognostic marker for outcome? a review of clinicopathological features of 27 cases. Br J Dermatol. 2013;168:391-401.
- Raghavan R, Eknoyan G. Acute interstitial nephritis—a reappraisal and update. Clin Nephrol. 2014;82:149-162.
- Matsuda H, Saito K, Takayanagi Y, et al. Pustular-type drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms due to carbamazepine with systemic muscle involvement. J Dermatol. 2013;40:118-122.
- Wolf R, Davidovici B, Matz H, et al. Drug rash with eosinophilia and systemic symptoms versus Stevens-Johnson Syndrome—a case that indicates a stumbling block in the current classification. Int Arch Allergy Immunol. 2006;141:308-310.
- Kumar A, Goldfarb JW, Bittner EA. A case of drug rash with eosinophilia and systemic symptoms (DRESS) syndrome complicating airway management. Can J Anaesth. 2012;59:295-298.
- Strom BL, Schinnar R, Apter AJ, et al. Absence of cross-reactivity between sulfonamide antibiotics and sulfonamide nonantibiotics. N Engl J Med. 2003;349:1628-1635.
- Berbari EF, Kanj SS, Kowalski TJ, et al; Infectious Diseases Society of America. 2015 Infectious Diseases Society of America (IDSA) clinical practice guidelines for the diagnosis and treatment of native vertebral osteomyelitis in adults. Clin Infect Dis. 2015;61:E26-E46.
- Lam BD, Miller MM, Sutton AV, et al. Vancomycin and DRESS: a retrospective chart review of 32 cases in Los Angeles, California. J Am Acad Dermatol. 2017;77:973-975.
- Eppenberger M, Hack D, Ammann P, et al. Acute eosinophilic myocarditis with dramatic response to steroid therapy: the central role of echocardiography in diagnosis and follow-up. Tex Heart Inst J. 2013;40:326-330.
- Kirchhof MG, Wong A, Dutz JP. Cyclosporine treatment of drug-induced hypersensitivity syndrome. JAMA Dermatol. 2016;152:1254-1257.
- Singer EM, Wanat KA, Rosenbach MA. A case of recalcitrant DRESS syndrome with multiple autoimmune sequelae treated with intravenous immunoglobulins. JAMA Dermatol. 2013;149:494-495.
- Bommersbach TJ, Lapid MI, Leung JG, et al. Management of psychotropic drug-induced DRESS syndrome: a systematic review. Mayo Clin Proc. 2016;91:787-801.
- Alexander T, Iglesia E, Park Y, et al. Severe DRESS syndrome managed with therapeutic plasma exchange. Pediatrics. 2013;131:E945-E949.
- Daoulah A, Alqahtani AA, Ocheltree SR, et al. Acute myocardial infarction in a 56-year-old female patient treated with sulfasalazine. Am J Emerg Med. 2012;30:638.e1-638.e3.
- Joly P, Janela B, Tetart F, et al. Poor benefit/risk balance of intravenous immunoglobulins in DRESS. Arch Dermatol. 2012;148:543-544.
- Kano Y, Tohyama M, Aihara M, et al. Sequelae in 145 patients with drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms: survey conducted by the Asian Research Committee on Severe Cutaneous Adverse Reactions (ASCAR). J Dermatol. 2015;42:276-282.
- Ushigome Y, Kano Y, Ishida T, et al. Short- and long-term outcomes of 34 patients with drug-induced hypersensitivity syndrome in a single institution. J Am Acad Dermatol. 2013;68:721-728.
- Matta JM, Flores SM, Cherit JD. Drug reaction with eosinophilia and systemic symptoms (DRESS) and its relation with autoimmunity in a reference center in Mexico. An Bras Dermatol. 2017;92:30-33.
- Ahluwalia J, Abuabara K, Perman MJ, et al. Human herpesvirus 6 involvement in paediatric drug hypersensitivity syndrome. Br J Dermatol. 2015;172:1090-1095.
- Sasidharanpillai S, Sabitha S, Riyaz N, et al. Drug reaction with eosinophilia and systemic symptoms in children: a prospective study. Pediatr Dermatol. 2016;33:E162-E165.
- Cacoub P, Musette P, Descamps V, et al. The DRESS syndrome: a literature review. Am J Med, 2011;124:588-597.
- Kardaun SH, Sekula P, Valeyrie-Allanore L, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. results from the prospective RegiSCAR study. Br J Dermatol. 2013;169:1071-1080.
- Intarasupht J, Kanchanomai A, Leelasattakul W, et al. Prevalence, risk factors, and mortality outcome in the drug reaction with eosinophilia and systemic symptoms patients with cardiac involvement. Int J Dermatol. 2018;57:1187-1191.
- Bourgeois GP, Cafardi JA, Groysman V, et al. A review of DRESS-associated myocarditis. J Am Acad Dermatol. 2012;66:E229-E236.
- Husain Z, Reddy BY, Schwartz RA. DRESS syndrome: part I. clinical perspectives. J Am Acad Dermatol. 2013;68:693.e1-693.e14; quiz 706-708.
- Lee T, Lee YS, Yoon SY, et al. Characteristics of liver injury in drug-induced systemic hypersensitivity reactions. J Am Acad Dermatol. 2013;69:407-415.
- Lin IC, Yang HC, Strong C, et al. Liver injury in patients with DRESS: a clinical study of 72 cases. J Am Acad Dermatol. 2015;72:984-991.
- Peyrière H, Dereure O, Breton H, et al. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2006;155:422-428.
- Walsh S, Diaz-Cano S, Higgins E, et al. Drug reaction with eosinophilia and systemic symptoms: is cutaneous phenotype a prognostic marker for outcome? a review of clinicopathological features of 27 cases. Br J Dermatol. 2013;168:391-401.
- Raghavan R, Eknoyan G. Acute interstitial nephritis—a reappraisal and update. Clin Nephrol. 2014;82:149-162.
- Matsuda H, Saito K, Takayanagi Y, et al. Pustular-type drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms due to carbamazepine with systemic muscle involvement. J Dermatol. 2013;40:118-122.
- Wolf R, Davidovici B, Matz H, et al. Drug rash with eosinophilia and systemic symptoms versus Stevens-Johnson Syndrome—a case that indicates a stumbling block in the current classification. Int Arch Allergy Immunol. 2006;141:308-310.
- Kumar A, Goldfarb JW, Bittner EA. A case of drug rash with eosinophilia and systemic symptoms (DRESS) syndrome complicating airway management. Can J Anaesth. 2012;59:295-298.
- Strom BL, Schinnar R, Apter AJ, et al. Absence of cross-reactivity between sulfonamide antibiotics and sulfonamide nonantibiotics. N Engl J Med. 2003;349:1628-1635.
- Berbari EF, Kanj SS, Kowalski TJ, et al; Infectious Diseases Society of America. 2015 Infectious Diseases Society of America (IDSA) clinical practice guidelines for the diagnosis and treatment of native vertebral osteomyelitis in adults. Clin Infect Dis. 2015;61:E26-E46.
- Lam BD, Miller MM, Sutton AV, et al. Vancomycin and DRESS: a retrospective chart review of 32 cases in Los Angeles, California. J Am Acad Dermatol. 2017;77:973-975.
- Eppenberger M, Hack D, Ammann P, et al. Acute eosinophilic myocarditis with dramatic response to steroid therapy: the central role of echocardiography in diagnosis and follow-up. Tex Heart Inst J. 2013;40:326-330.
- Kirchhof MG, Wong A, Dutz JP. Cyclosporine treatment of drug-induced hypersensitivity syndrome. JAMA Dermatol. 2016;152:1254-1257.
- Singer EM, Wanat KA, Rosenbach MA. A case of recalcitrant DRESS syndrome with multiple autoimmune sequelae treated with intravenous immunoglobulins. JAMA Dermatol. 2013;149:494-495.
- Bommersbach TJ, Lapid MI, Leung JG, et al. Management of psychotropic drug-induced DRESS syndrome: a systematic review. Mayo Clin Proc. 2016;91:787-801.
- Alexander T, Iglesia E, Park Y, et al. Severe DRESS syndrome managed with therapeutic plasma exchange. Pediatrics. 2013;131:E945-E949.
- Daoulah A, Alqahtani AA, Ocheltree SR, et al. Acute myocardial infarction in a 56-year-old female patient treated with sulfasalazine. Am J Emerg Med. 2012;30:638.e1-638.e3.
- Joly P, Janela B, Tetart F, et al. Poor benefit/risk balance of intravenous immunoglobulins in DRESS. Arch Dermatol. 2012;148:543-544.
- Kano Y, Tohyama M, Aihara M, et al. Sequelae in 145 patients with drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms: survey conducted by the Asian Research Committee on Severe Cutaneous Adverse Reactions (ASCAR). J Dermatol. 2015;42:276-282.
- Ushigome Y, Kano Y, Ishida T, et al. Short- and long-term outcomes of 34 patients with drug-induced hypersensitivity syndrome in a single institution. J Am Acad Dermatol. 2013;68:721-728.
- Matta JM, Flores SM, Cherit JD. Drug reaction with eosinophilia and systemic symptoms (DRESS) and its relation with autoimmunity in a reference center in Mexico. An Bras Dermatol. 2017;92:30-33.
- Ahluwalia J, Abuabara K, Perman MJ, et al. Human herpesvirus 6 involvement in paediatric drug hypersensitivity syndrome. Br J Dermatol. 2015;172:1090-1095.
- Sasidharanpillai S, Sabitha S, Riyaz N, et al. Drug reaction with eosinophilia and systemic symptoms in children: a prospective study. Pediatr Dermatol. 2016;33:E162-E165.
Practice Points
- Drug rash with eosinophilia and systemic symptoms (DRESS syndrome) is a clinical diagnosis, and incomplete forms may not meet formal criteria-based diagnosis.
- Although DRESS syndrome typically has a morbilliform eruption, different rash morphologies may be observed.
- The myocarditis of DRESS syndrome may not present with chest pain; a high index of suspicion is warranted.
- Autoimmune sequelae are more frequent in patients who have had an episode of DRESS syndrome.
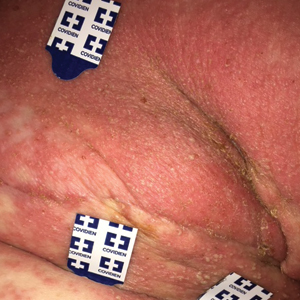
Investing in the Future of Inpatient Dermatology: The Evolution and Impact of Specialized Dermatologic Consultation in Hospitalized Patients
The practice of inpatient dermatology has a rich history rooted in specialized hospital wards that housed patients with chronic dermatoses. Because systemic agents were limited, the care of these patients required skilled nursing and a distinctive knowledge of the application of numerous topical agents, including washes, baths, powders, lotions, and pastes1; however, with the evolving nature of health care in the last half a century, such dermatologic inpatient units are now rare, with only 2 units remaining in the United States, specifically at the Mayo Clinic in Minnesota and at the University of Miami.2
Although the shift away from a primary dermatologic admitting service is likely multifactorial, what is more sobering is that the majority of inpatients with dermatologic disorders are cared for by nondermatologists.2 Although the dynamics for such a diminished presence are due to various personal and professional concerns, the essential outcome for patients hospitalized with a cutaneous concern—whether directly related to their hospitalization or iatrogenic in nature—is the potential for suboptimal care.3
Fortunately, the practice of inpatient dermatology currently is undergoing a renaissance. With this renewed interest in hospital-based dermatology, there is a growing body of evidence that demonstrates how the dermatology hospitalist has become a vital member of the inpatient team, adding value to the care of patients across all specialties.
To explore the impact of consultative dermatology services, there has been a push by members of the Society for Dermatology Hospitalists to elucidate the contributions of dermatologists in the inpatient setting, which has been accomplished primarily by defining and characterizing the types of patients that dermatology hospitalists care for and, more recently, by demonstrating the improved outcomes that result from expert consultation.
Breadth of Inpatient Dermatologic Consultations
With the adaptation of dermatology consultation services, the scope of practice has shifted from the skilled management of chronic dermatoses to one with an emphasis on the identification of various acute dermatologic diseases. Although the extent of such acute disease states in the inpatient setting is vast, it is interesting to note that the majority of consultations are for common conditions, namely cutaneous infections, venous stasis dermatitis, contact dermatitis, atopic dermatitis, and cutaneous drug eruptions (Table).4,5

Moreover, for the services that obtain dermatologic consultation, the majority of requests originate from internal medicine and hematology/oncology.4,5 Although internal medicine often is the largest-represented specialty in the hospital and provides a proportional amount of dermatology consultations, hematology/oncology patients represent a distinct cohort who are prone to unique mucocutaneous dermatoses related to underlying malignancies, immunosuppression, and cancer-specific therapies (eg, chemotherapy, immunotherapy, stem cell transplantation). Within this subset of patients, cutaneous infections and drug eruptions constitute the majority of cases, while graft-versus-host disease and neutrophilic dermatoses account for a smaller percentage of dermatologic disease in this population. Given the complex and uncommon nature of these dermatoses, timely intervention by a dermatologist can have a considerable impact on morbidity and mortality associated with such disease states.6,7
Among pediatric patients, dermatology consultation patterns mimic those seen among adult patients, with common conditions such as atopic dermatitis and contact dermatitis representing the majority of consultations.8-11 Vascular lesions further represent a unique source of consultation among pediatric patients. Although they often are considered an outpatient concern, one group found that the majority of inpatient consultations for vascular lesions led to early identification of a syndromic association and/or complication (eg, ulceration).10 Identifying these cases in the hospital provides early opportunities for intervention and multidisciplinary care.
Adding Value to the Care of Hospitalized Patients
Following other inpatient models, hospitalist dermatology has begun to demonstrate feasibility, advances in quality improvement, and most importantly improved health care outcomes. In an effort to better characterize the enhancement of such health care delivery, recent literature around the impact of inpatient dermatology consultation has centered on improving key objective hospital-based quality measures, namely diagnosis and management as well as hospital length of stay (LOS) and readmission rates.5,12-18
When identifying cutaneous disease, recent evidence points to the increased diagnostic accuracy by way of dermatology consultation. Specifically, diagnoses were changed 30% to 70% of the time when consultations were provided.6,12-15 Interestingly, misdiagnosis regularly centered on common diagnoses, specifically cellulitis, stasis dermatitis, and hypersensitivity reactions.6,12-16 In a multi-institutional retrospective study that examined the national incidence of cellulitis misdiagnosis, the authors found that when a dermatology consultation for presumed cellulitis was called, approximately 75% (N=55) of cases represented mimickers of cellulitis, such as stasis dermatitis, contact dermatitis, and cutaneous fungal infections. Moreover, in more than 38% (N=21) of such cellulitis consultations, patients often had more than one ongoing disease process, further speaking to the diagnostic accuracy obtained from expert consultation.16 The result of such misdiagnosis is not trivial, as unnecessary hospital admission or inappropriate treatment due to misdiagnosis of cutaneous disease often leads to avoidable complications and preventable health care spending. In a cross-sectional analysis of patients diagnosed with presumed lower extremity cellulitis (N=259), approximately 30% were misdiagnosed. In these cases, more than 90% of patients received unnecessary antibiotics, with approximately 30% of them experiencing a complication or avoidable utilization of health care related to their misdiagnosis.17
Along with the profound impact on diagnostic accuracy, management and treatment are almost universally affected after dermatology consultation.5,12-14 Such findings bear importance on optimizing hospital LOS as well as readmission rates. For hospital LOS, a recent study demonstrated reductions in LOS by 2.64 days as well as 1-year cutaneous disease-specific readmissions for patients who received dermatologic consultation for their inflammatory skin disease.18 Similarly, in a recent prospective cohort study of patients diagnosed with presumed lower extremity cellulitis, hospital LOS decreased by 2 days following a diagnosis of pseudocellulitis via timely dermatologic consultation. Across the United States, such reductions in LOS associated with unnecessary hospitalization due to pseudocellulitis can result in annual health care savings of $100 to $200 million.13 As such, early dermatologic intervention plays a vital role in diagnostic accuracy, appropriate treatment implementation, expedited discharge, and the overall economics of health care delivery and utilization, thereby supporting the utility of clinical decision support through expert consultation.
Conclusion
There is a clear and distinct value that results in having specialized inpatient dermatology services. Such expert consultation enhances quality of care and reduces health care costs. Although the implementation and success of inpatient dermatology services has primarily been observed at large hospitals/tertiary care centers, there is incredible potential to further our impact through engagement in our community hospitals. With that said, all practicing dermatologists should feel empowered to employ their expert skillset in their own communities, as such access to care and specialty support is desperately needed and can remarkably impact health care outcomes. Moreover, in addition to the direct impact on health care delivery and economics, the intangible benefits of an inpatient dermatology presence are innumerable, as opportunities to promote quality research and improve trainee education also demonstrate our value. These facets together provide a positive perspective on the potential contribution that our field can have on shaping the outlook of hospital medicine. As such, in addition to enjoying the current renaissance of inpatient dermatology, it is imperative that dermatologists build on this momentum and invest in the future of consultative dermatology.
- Albert MR, Mackool BT. A dermatology ward at the beginning of the 20th century. J Am Acad Dermatol. 2000;42(1, pt 1):113-123.
- Ko LN, Kroshinsky D. Dermatology hospitalists: a multicenter survey study characterizing the infrastructure of consultative dermatology in select American hospitals. Int J Dermatol. 2018;57:553-558.
- Helms AE, Helms SE, Brodell RT. Hospital consultations: time to address an unmet need? J Am Acad Dermatol. 2009;60:308-311.
- Storan ER, McEvoy MT, Wetter DA, et al. Experience of a year of adult hospital dermatology consultations. Int J Dermatol. 2015;54:1150-1156.
- Galimberti F, Guren L, Fernandez AP, et al. Dermatology consultations significantly contribute quality to care of hospitalized patients: a prospective study of dermatology inpatient consults at a tertiary care center. Int J Dermatol. 2016;55:E547-E551.
- Tracey EH, Forrestel A, Rosenbach M, et al. Inpatient dermatology consultation in patients with hematologic malignancies. J Am Acad Dermatol. 2016;75:835-836.
- Phillips GS, Freites-Martinez A, Hsu M, et al. Inflammatory dermatoses, infections, and drug eruptions are the most common skin conditions in hospitalized cancer patients. J Am Acad Dermatol. 2018;78:1102-1109.
- Storan ER, McEvoy MT, Wetter DA, et al. Pediatric hospital dermatology: experience with inpatient and consult services at the Mayo Clinic. Pediatr Dermatol. 2013;30:433-437.
- Afsar FS. Analysis of pediatric dermatology inpatient consultations in a pediatric teaching hospital. Arch Argent Pediatr. 2017;115:E377-E384.
- McMahon P, Goddard D, Frieden IJ. Pediatric dermatology inpatient consultations: a retrospective study of 427 cases. J Am Acad Dermatol. 2013;68:926-931.
- Peñate Y, Borrego L, Hernández N, et al. Pediatric dermatology consultations: a retrospective analysis of inpatient consultations referred to the dermatology service. Pediatr Dermatol. 2012;29:115-118.
- Hu L, Haynes H, Ferrazza D, et al. Impact of specialist consultations on inpatient admissions for dermatology-specific and related DRGs. J Gen Intern Med. 2013;28:1477-1482.
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543.
- Falanga V, Schachner LA, Rae V, et al. Dermatologic consultations in the hospital setting. Arch Dermatol. 1994;130:1022-1025.
- Ko LN, Garza-Mayers AC, St John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis: a randomized clinical trial. JAMA Dermatol. 2018;154:529-536.
- Strazzula L, Cotliar J, Fox LP, et al. Inpatient dermatology consultation aids diagnosis of cellulitis among hospitalized patients: a multi-institutional analysis. J Am Acad Dermatol. 2015;73:70-75.
- Weng QY, Raff AB, Cohen JM, et al. Costs and consequences associated with misdiagnosed lower extremity cellulitis [published online November 2, 2016]. JAMA Dermatol. doi:10.1001/jamadermatol.2016.3816.
- Milani-Nejad N, Zhang M, Kaffenberger BH. Association of dermatology consultations with patient care outcomes in hospitalized patients with inflammatory skin diseases. JAMA Dermatol. 2017;153:523-528.
The practice of inpatient dermatology has a rich history rooted in specialized hospital wards that housed patients with chronic dermatoses. Because systemic agents were limited, the care of these patients required skilled nursing and a distinctive knowledge of the application of numerous topical agents, including washes, baths, powders, lotions, and pastes1; however, with the evolving nature of health care in the last half a century, such dermatologic inpatient units are now rare, with only 2 units remaining in the United States, specifically at the Mayo Clinic in Minnesota and at the University of Miami.2
Although the shift away from a primary dermatologic admitting service is likely multifactorial, what is more sobering is that the majority of inpatients with dermatologic disorders are cared for by nondermatologists.2 Although the dynamics for such a diminished presence are due to various personal and professional concerns, the essential outcome for patients hospitalized with a cutaneous concern—whether directly related to their hospitalization or iatrogenic in nature—is the potential for suboptimal care.3
Fortunately, the practice of inpatient dermatology currently is undergoing a renaissance. With this renewed interest in hospital-based dermatology, there is a growing body of evidence that demonstrates how the dermatology hospitalist has become a vital member of the inpatient team, adding value to the care of patients across all specialties.
To explore the impact of consultative dermatology services, there has been a push by members of the Society for Dermatology Hospitalists to elucidate the contributions of dermatologists in the inpatient setting, which has been accomplished primarily by defining and characterizing the types of patients that dermatology hospitalists care for and, more recently, by demonstrating the improved outcomes that result from expert consultation.
Breadth of Inpatient Dermatologic Consultations
With the adaptation of dermatology consultation services, the scope of practice has shifted from the skilled management of chronic dermatoses to one with an emphasis on the identification of various acute dermatologic diseases. Although the extent of such acute disease states in the inpatient setting is vast, it is interesting to note that the majority of consultations are for common conditions, namely cutaneous infections, venous stasis dermatitis, contact dermatitis, atopic dermatitis, and cutaneous drug eruptions (Table).4,5
Moreover, for the services that obtain dermatologic consultation, the majority of requests originate from internal medicine and hematology/oncology.4,5 Although internal medicine often is the largest-represented specialty in the hospital and provides a proportional amount of dermatology consultations, hematology/oncology patients represent a distinct cohort who are prone to unique mucocutaneous dermatoses related to underlying malignancies, immunosuppression, and cancer-specific therapies (eg, chemotherapy, immunotherapy, stem cell transplantation). Within this subset of patients, cutaneous infections and drug eruptions constitute the majority of cases, while graft-versus-host disease and neutrophilic dermatoses account for a smaller percentage of dermatologic disease in this population. Given the complex and uncommon nature of these dermatoses, timely intervention by a dermatologist can have a considerable impact on morbidity and mortality associated with such disease states.6,7
Among pediatric patients, dermatology consultation patterns mimic those seen among adult patients, with common conditions such as atopic dermatitis and contact dermatitis representing the majority of consultations.8-11 Vascular lesions further represent a unique source of consultation among pediatric patients. Although they often are considered an outpatient concern, one group found that the majority of inpatient consultations for vascular lesions led to early identification of a syndromic association and/or complication (eg, ulceration).10 Identifying these cases in the hospital provides early opportunities for intervention and multidisciplinary care.
Adding Value to the Care of Hospitalized Patients
Following other inpatient models, hospitalist dermatology has begun to demonstrate feasibility, advances in quality improvement, and most importantly improved health care outcomes. In an effort to better characterize the enhancement of such health care delivery, recent literature around the impact of inpatient dermatology consultation has centered on improving key objective hospital-based quality measures, namely diagnosis and management as well as hospital length of stay (LOS) and readmission rates.5,12-18
When identifying cutaneous disease, recent evidence points to the increased diagnostic accuracy by way of dermatology consultation. Specifically, diagnoses were changed 30% to 70% of the time when consultations were provided.6,12-15 Interestingly, misdiagnosis regularly centered on common diagnoses, specifically cellulitis, stasis dermatitis, and hypersensitivity reactions.6,12-16 In a multi-institutional retrospective study that examined the national incidence of cellulitis misdiagnosis, the authors found that when a dermatology consultation for presumed cellulitis was called, approximately 75% (N=55) of cases represented mimickers of cellulitis, such as stasis dermatitis, contact dermatitis, and cutaneous fungal infections. Moreover, in more than 38% (N=21) of such cellulitis consultations, patients often had more than one ongoing disease process, further speaking to the diagnostic accuracy obtained from expert consultation.16 The result of such misdiagnosis is not trivial, as unnecessary hospital admission or inappropriate treatment due to misdiagnosis of cutaneous disease often leads to avoidable complications and preventable health care spending. In a cross-sectional analysis of patients diagnosed with presumed lower extremity cellulitis (N=259), approximately 30% were misdiagnosed. In these cases, more than 90% of patients received unnecessary antibiotics, with approximately 30% of them experiencing a complication or avoidable utilization of health care related to their misdiagnosis.17
Along with the profound impact on diagnostic accuracy, management and treatment are almost universally affected after dermatology consultation.5,12-14 Such findings bear importance on optimizing hospital LOS as well as readmission rates. For hospital LOS, a recent study demonstrated reductions in LOS by 2.64 days as well as 1-year cutaneous disease-specific readmissions for patients who received dermatologic consultation for their inflammatory skin disease.18 Similarly, in a recent prospective cohort study of patients diagnosed with presumed lower extremity cellulitis, hospital LOS decreased by 2 days following a diagnosis of pseudocellulitis via timely dermatologic consultation. Across the United States, such reductions in LOS associated with unnecessary hospitalization due to pseudocellulitis can result in annual health care savings of $100 to $200 million.13 As such, early dermatologic intervention plays a vital role in diagnostic accuracy, appropriate treatment implementation, expedited discharge, and the overall economics of health care delivery and utilization, thereby supporting the utility of clinical decision support through expert consultation.
Conclusion
There is a clear and distinct value that results in having specialized inpatient dermatology services. Such expert consultation enhances quality of care and reduces health care costs. Although the implementation and success of inpatient dermatology services has primarily been observed at large hospitals/tertiary care centers, there is incredible potential to further our impact through engagement in our community hospitals. With that said, all practicing dermatologists should feel empowered to employ their expert skillset in their own communities, as such access to care and specialty support is desperately needed and can remarkably impact health care outcomes. Moreover, in addition to the direct impact on health care delivery and economics, the intangible benefits of an inpatient dermatology presence are innumerable, as opportunities to promote quality research and improve trainee education also demonstrate our value. These facets together provide a positive perspective on the potential contribution that our field can have on shaping the outlook of hospital medicine. As such, in addition to enjoying the current renaissance of inpatient dermatology, it is imperative that dermatologists build on this momentum and invest in the future of consultative dermatology.
The practice of inpatient dermatology has a rich history rooted in specialized hospital wards that housed patients with chronic dermatoses. Because systemic agents were limited, the care of these patients required skilled nursing and a distinctive knowledge of the application of numerous topical agents, including washes, baths, powders, lotions, and pastes1; however, with the evolving nature of health care in the last half a century, such dermatologic inpatient units are now rare, with only 2 units remaining in the United States, specifically at the Mayo Clinic in Minnesota and at the University of Miami.2
Although the shift away from a primary dermatologic admitting service is likely multifactorial, what is more sobering is that the majority of inpatients with dermatologic disorders are cared for by nondermatologists.2 Although the dynamics for such a diminished presence are due to various personal and professional concerns, the essential outcome for patients hospitalized with a cutaneous concern—whether directly related to their hospitalization or iatrogenic in nature—is the potential for suboptimal care.3
Fortunately, the practice of inpatient dermatology currently is undergoing a renaissance. With this renewed interest in hospital-based dermatology, there is a growing body of evidence that demonstrates how the dermatology hospitalist has become a vital member of the inpatient team, adding value to the care of patients across all specialties.
To explore the impact of consultative dermatology services, there has been a push by members of the Society for Dermatology Hospitalists to elucidate the contributions of dermatologists in the inpatient setting, which has been accomplished primarily by defining and characterizing the types of patients that dermatology hospitalists care for and, more recently, by demonstrating the improved outcomes that result from expert consultation.
Breadth of Inpatient Dermatologic Consultations
With the adaptation of dermatology consultation services, the scope of practice has shifted from the skilled management of chronic dermatoses to one with an emphasis on the identification of various acute dermatologic diseases. Although the extent of such acute disease states in the inpatient setting is vast, it is interesting to note that the majority of consultations are for common conditions, namely cutaneous infections, venous stasis dermatitis, contact dermatitis, atopic dermatitis, and cutaneous drug eruptions (Table).4,5
Moreover, for the services that obtain dermatologic consultation, the majority of requests originate from internal medicine and hematology/oncology.4,5 Although internal medicine often is the largest-represented specialty in the hospital and provides a proportional amount of dermatology consultations, hematology/oncology patients represent a distinct cohort who are prone to unique mucocutaneous dermatoses related to underlying malignancies, immunosuppression, and cancer-specific therapies (eg, chemotherapy, immunotherapy, stem cell transplantation). Within this subset of patients, cutaneous infections and drug eruptions constitute the majority of cases, while graft-versus-host disease and neutrophilic dermatoses account for a smaller percentage of dermatologic disease in this population. Given the complex and uncommon nature of these dermatoses, timely intervention by a dermatologist can have a considerable impact on morbidity and mortality associated with such disease states.6,7
Among pediatric patients, dermatology consultation patterns mimic those seen among adult patients, with common conditions such as atopic dermatitis and contact dermatitis representing the majority of consultations.8-11 Vascular lesions further represent a unique source of consultation among pediatric patients. Although they often are considered an outpatient concern, one group found that the majority of inpatient consultations for vascular lesions led to early identification of a syndromic association and/or complication (eg, ulceration).10 Identifying these cases in the hospital provides early opportunities for intervention and multidisciplinary care.
Adding Value to the Care of Hospitalized Patients
Following other inpatient models, hospitalist dermatology has begun to demonstrate feasibility, advances in quality improvement, and most importantly improved health care outcomes. In an effort to better characterize the enhancement of such health care delivery, recent literature around the impact of inpatient dermatology consultation has centered on improving key objective hospital-based quality measures, namely diagnosis and management as well as hospital length of stay (LOS) and readmission rates.5,12-18
When identifying cutaneous disease, recent evidence points to the increased diagnostic accuracy by way of dermatology consultation. Specifically, diagnoses were changed 30% to 70% of the time when consultations were provided.6,12-15 Interestingly, misdiagnosis regularly centered on common diagnoses, specifically cellulitis, stasis dermatitis, and hypersensitivity reactions.6,12-16 In a multi-institutional retrospective study that examined the national incidence of cellulitis misdiagnosis, the authors found that when a dermatology consultation for presumed cellulitis was called, approximately 75% (N=55) of cases represented mimickers of cellulitis, such as stasis dermatitis, contact dermatitis, and cutaneous fungal infections. Moreover, in more than 38% (N=21) of such cellulitis consultations, patients often had more than one ongoing disease process, further speaking to the diagnostic accuracy obtained from expert consultation.16 The result of such misdiagnosis is not trivial, as unnecessary hospital admission or inappropriate treatment due to misdiagnosis of cutaneous disease often leads to avoidable complications and preventable health care spending. In a cross-sectional analysis of patients diagnosed with presumed lower extremity cellulitis (N=259), approximately 30% were misdiagnosed. In these cases, more than 90% of patients received unnecessary antibiotics, with approximately 30% of them experiencing a complication or avoidable utilization of health care related to their misdiagnosis.17
Along with the profound impact on diagnostic accuracy, management and treatment are almost universally affected after dermatology consultation.5,12-14 Such findings bear importance on optimizing hospital LOS as well as readmission rates. For hospital LOS, a recent study demonstrated reductions in LOS by 2.64 days as well as 1-year cutaneous disease-specific readmissions for patients who received dermatologic consultation for their inflammatory skin disease.18 Similarly, in a recent prospective cohort study of patients diagnosed with presumed lower extremity cellulitis, hospital LOS decreased by 2 days following a diagnosis of pseudocellulitis via timely dermatologic consultation. Across the United States, such reductions in LOS associated with unnecessary hospitalization due to pseudocellulitis can result in annual health care savings of $100 to $200 million.13 As such, early dermatologic intervention plays a vital role in diagnostic accuracy, appropriate treatment implementation, expedited discharge, and the overall economics of health care delivery and utilization, thereby supporting the utility of clinical decision support through expert consultation.
Conclusion
There is a clear and distinct value that results in having specialized inpatient dermatology services. Such expert consultation enhances quality of care and reduces health care costs. Although the implementation and success of inpatient dermatology services has primarily been observed at large hospitals/tertiary care centers, there is incredible potential to further our impact through engagement in our community hospitals. With that said, all practicing dermatologists should feel empowered to employ their expert skillset in their own communities, as such access to care and specialty support is desperately needed and can remarkably impact health care outcomes. Moreover, in addition to the direct impact on health care delivery and economics, the intangible benefits of an inpatient dermatology presence are innumerable, as opportunities to promote quality research and improve trainee education also demonstrate our value. These facets together provide a positive perspective on the potential contribution that our field can have on shaping the outlook of hospital medicine. As such, in addition to enjoying the current renaissance of inpatient dermatology, it is imperative that dermatologists build on this momentum and invest in the future of consultative dermatology.
- Albert MR, Mackool BT. A dermatology ward at the beginning of the 20th century. J Am Acad Dermatol. 2000;42(1, pt 1):113-123.
- Ko LN, Kroshinsky D. Dermatology hospitalists: a multicenter survey study characterizing the infrastructure of consultative dermatology in select American hospitals. Int J Dermatol. 2018;57:553-558.
- Helms AE, Helms SE, Brodell RT. Hospital consultations: time to address an unmet need? J Am Acad Dermatol. 2009;60:308-311.
- Storan ER, McEvoy MT, Wetter DA, et al. Experience of a year of adult hospital dermatology consultations. Int J Dermatol. 2015;54:1150-1156.
- Galimberti F, Guren L, Fernandez AP, et al. Dermatology consultations significantly contribute quality to care of hospitalized patients: a prospective study of dermatology inpatient consults at a tertiary care center. Int J Dermatol. 2016;55:E547-E551.
- Tracey EH, Forrestel A, Rosenbach M, et al. Inpatient dermatology consultation in patients with hematologic malignancies. J Am Acad Dermatol. 2016;75:835-836.
- Phillips GS, Freites-Martinez A, Hsu M, et al. Inflammatory dermatoses, infections, and drug eruptions are the most common skin conditions in hospitalized cancer patients. J Am Acad Dermatol. 2018;78:1102-1109.
- Storan ER, McEvoy MT, Wetter DA, et al. Pediatric hospital dermatology: experience with inpatient and consult services at the Mayo Clinic. Pediatr Dermatol. 2013;30:433-437.
- Afsar FS. Analysis of pediatric dermatology inpatient consultations in a pediatric teaching hospital. Arch Argent Pediatr. 2017;115:E377-E384.
- McMahon P, Goddard D, Frieden IJ. Pediatric dermatology inpatient consultations: a retrospective study of 427 cases. J Am Acad Dermatol. 2013;68:926-931.
- Peñate Y, Borrego L, Hernández N, et al. Pediatric dermatology consultations: a retrospective analysis of inpatient consultations referred to the dermatology service. Pediatr Dermatol. 2012;29:115-118.
- Hu L, Haynes H, Ferrazza D, et al. Impact of specialist consultations on inpatient admissions for dermatology-specific and related DRGs. J Gen Intern Med. 2013;28:1477-1482.
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543.
- Falanga V, Schachner LA, Rae V, et al. Dermatologic consultations in the hospital setting. Arch Dermatol. 1994;130:1022-1025.
- Ko LN, Garza-Mayers AC, St John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis: a randomized clinical trial. JAMA Dermatol. 2018;154:529-536.
- Strazzula L, Cotliar J, Fox LP, et al. Inpatient dermatology consultation aids diagnosis of cellulitis among hospitalized patients: a multi-institutional analysis. J Am Acad Dermatol. 2015;73:70-75.
- Weng QY, Raff AB, Cohen JM, et al. Costs and consequences associated with misdiagnosed lower extremity cellulitis [published online November 2, 2016]. JAMA Dermatol. doi:10.1001/jamadermatol.2016.3816.
- Milani-Nejad N, Zhang M, Kaffenberger BH. Association of dermatology consultations with patient care outcomes in hospitalized patients with inflammatory skin diseases. JAMA Dermatol. 2017;153:523-528.
- Albert MR, Mackool BT. A dermatology ward at the beginning of the 20th century. J Am Acad Dermatol. 2000;42(1, pt 1):113-123.
- Ko LN, Kroshinsky D. Dermatology hospitalists: a multicenter survey study characterizing the infrastructure of consultative dermatology in select American hospitals. Int J Dermatol. 2018;57:553-558.
- Helms AE, Helms SE, Brodell RT. Hospital consultations: time to address an unmet need? J Am Acad Dermatol. 2009;60:308-311.
- Storan ER, McEvoy MT, Wetter DA, et al. Experience of a year of adult hospital dermatology consultations. Int J Dermatol. 2015;54:1150-1156.
- Galimberti F, Guren L, Fernandez AP, et al. Dermatology consultations significantly contribute quality to care of hospitalized patients: a prospective study of dermatology inpatient consults at a tertiary care center. Int J Dermatol. 2016;55:E547-E551.
- Tracey EH, Forrestel A, Rosenbach M, et al. Inpatient dermatology consultation in patients with hematologic malignancies. J Am Acad Dermatol. 2016;75:835-836.
- Phillips GS, Freites-Martinez A, Hsu M, et al. Inflammatory dermatoses, infections, and drug eruptions are the most common skin conditions in hospitalized cancer patients. J Am Acad Dermatol. 2018;78:1102-1109.
- Storan ER, McEvoy MT, Wetter DA, et al. Pediatric hospital dermatology: experience with inpatient and consult services at the Mayo Clinic. Pediatr Dermatol. 2013;30:433-437.
- Afsar FS. Analysis of pediatric dermatology inpatient consultations in a pediatric teaching hospital. Arch Argent Pediatr. 2017;115:E377-E384.
- McMahon P, Goddard D, Frieden IJ. Pediatric dermatology inpatient consultations: a retrospective study of 427 cases. J Am Acad Dermatol. 2013;68:926-931.
- Peñate Y, Borrego L, Hernández N, et al. Pediatric dermatology consultations: a retrospective analysis of inpatient consultations referred to the dermatology service. Pediatr Dermatol. 2012;29:115-118.
- Hu L, Haynes H, Ferrazza D, et al. Impact of specialist consultations on inpatient admissions for dermatology-specific and related DRGs. J Gen Intern Med. 2013;28:1477-1482.
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543.
- Falanga V, Schachner LA, Rae V, et al. Dermatologic consultations in the hospital setting. Arch Dermatol. 1994;130:1022-1025.
- Ko LN, Garza-Mayers AC, St John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis: a randomized clinical trial. JAMA Dermatol. 2018;154:529-536.
- Strazzula L, Cotliar J, Fox LP, et al. Inpatient dermatology consultation aids diagnosis of cellulitis among hospitalized patients: a multi-institutional analysis. J Am Acad Dermatol. 2015;73:70-75.
- Weng QY, Raff AB, Cohen JM, et al. Costs and consequences associated with misdiagnosed lower extremity cellulitis [published online November 2, 2016]. JAMA Dermatol. doi:10.1001/jamadermatol.2016.3816.
- Milani-Nejad N, Zhang M, Kaffenberger BH. Association of dermatology consultations with patient care outcomes in hospitalized patients with inflammatory skin diseases. JAMA Dermatol. 2017;153:523-528.
Practice Points
- Dermatology inpatient consultation enhances quality of care and reduces health care costs.
- Dermatology input in the inpatient setting leads to a diagnosis change in up to 70% of consultations.
- The majority of dermatologic misdiagnoses by nondermatologists involves common dermatoses such as cellulitis, stasis dermatitis, and hypersensitivity reactions.
