User login
Risk Factors Predicting Cellulitis Diagnosis in a Prospective Cohort Undergoing Dermatology Consultation in the Emergency Department
Cellulitis is an infection of the skin and skin-associated structures characterized by redness, warmth, swelling, and pain of the affected area. Cellulitis most commonly occurs in middle-aged and older adults and frequently affects the lower extremities.1 Serious complications of cellulitis such as bacteremia, metastatic infection, and sepsis are rare, and most cases of cellulitis in patients with normal vital signs and mental status can be managed with outpatient treatment.2
Diagnosis of cellulitis can be confounded by a number of similarly presenting conditions collectively known as pseudocellulitis, such as venous stasis dermatitis and deep vein thrombosis.1 Misdiagnosis of cellulitis is common, with rates exceeding 30% among hospitalized patients initially diagnosed with cellulitis.3,4 Dermatology or infectious disease assessment is considered the diagnostic gold standard for cellulitis4,5 but is not always readily available, especially in resource-constrained settings.
Most cases of uncomplicated cellulitis can be managed with outpatient treatment, especially because serious complications are rare. Frequent misdiagnosis leads to repeat or unnecessary hospitalization and antibiosis. Exceptions necessitating hospitalization usually are predicated on signs of systemic infection, severe immunocompromised states, or failure of prior outpatient therapy.6 Such presentations can be distinguished by corresponding notable historical or examination factors, such as vital sign abnormalities suggesting systemic infection or history of malignancy leading to an immunocompromised state.
We sought to evaluate factors leading to the diagnosis of cellulitis in a cohort of patients with uncomplicated presentations receiving dermatology consultation to emphasize findings indicative of cellulitis in the absence of clinical or historical factors suggestive of other conditions necessitating hospitalization, such as systemic infection.
Methods
Study Participants—A prospective cohort study of patients presenting to an emergency department (ED) between October 2012 and January 2017 at an urban academic medical center in Boston, Massachusetts, was conducted with approval of study design and procedures by the relevant institutional review board. Patients older than 18 years were eligible for inclusion if given an initial diagnosis of cellulitis by an ED physician. Patients were excluded if incarcerated, pregnant, or unable to provide informed consent. Other exclusion criteria includedinfections overlying temporary or permanent indwelling hardware, animal or human bites, or sites of recent surgery (within the prior 4 weeks); preceding antibiotic treatment for more than 24 hours; or clinical or radiographic evidence of complications requiring alternative management such as osteomyelitis or abscess. Patients presenting with an elevated heart rate (>100 beats per minute) or body temperature (>100.5 °F [38.1 °C]) also were excluded. Eligible patients were enrolled upon providing written informed consent, and no remuneration was offered for participation.
Dermatology Consultation Intervention—A random subset of enrolled patients received dermatology consultation within 24 hours of presentation. Consultation consisted of a patient interview and physical examination with care recommendations to relevant ED and inpatient teams. Consultations confirmed the presence or absence of cellulitis as the primary outcome and also noted the presence of any pseudocellulitis diagnoses either occurring concomitantly with or mimicking cellulitis as a secondary outcome.
Statistical Analysis—Patient characteristics were analyzed to identify factors independently associated with the diagnosis of cellulitis in cases affecting the lower extremities. Factors were recorded with categorical variables reported as counts and percentages and continuous variables as means and standard deviations. Univariate analyses between categorical variables or discretized continuous variables and cellulitis diagnosis were conducted via Fisher exact test to identify a preliminary set of potential risk factors. Continuous variables were discretized at multiple incremental values with the discretization most significantly associated with cellulitis diagnosis selected as a preliminary risk factor. Multivariate analyses involved using any objective preliminary factor meeting a significance threshold of P<.1 in univariate comparisons in a multivariate logistic regression model for prediction of cellulitis diagnosis with corresponding calculation of odds ratios with confidence intervals and receiver operating characteristic. Factors with confidence intervals that excluded 1 were considered significant independent predictors of cellulitis. Analyses were performed using Python version 3.8 (Python Software Foundation).
Results
Of 1359 patients screened for eligibility, 104 patients with presumed lower extremity cellulitis undergoing dermatology consultation were included in this study (Figure). The mean patient age (SD) was 60.4 (19.2) years, and 63.5% of patients were male. In the study population, 63 (60.6%) patients received a final diagnosis of cellulitis. The most common pseudocellulitis diagnosis identified was venous stasis dermatitis, which occurred in 12 (11.5%) patients with concomitant cellulitis and in 12 (11.5%) patients mimicking cellulitis (Table).
.")
Univariate comparisons revealed a diverse set of historical, examination, and laboratory factors associated with cellulitis diagnosis. Diagnosis of cellulitis was associated with unilateral presentation, recent trauma to the affected site, and history of cellulitis or onychomycosis. Diagnosis of cellulitis also was associated with elevated white blood cell count, absolute neutrophil count, C-reactive protein, body mass index, hematocrit, and platelet count; age less than 75 years; and lower serum sodium and serum chloride levels. These were the independent factors included in the multivariate analysis, which consisted of a logistic regression model for prediction of cellulitis (eTable).

Multivariate logistic regression on all preliminary factors significantly associated with cellulitis diagnosis in univariate comparisons demonstrated leukocytosis, which was defined as having a white blood cell count exceeding 11,000/μL, unilateral presentation, history of onychomycosis, and trauma to the affected site as significant independent predictors of cellulitis diagnosis; history of cellulitis approached significance (eTable). Unilateral presentation and leukocytosis were the strongest predictors; having either of these factors had a sensitivity of 93.7% and a negative predictive value of 76.5%.

Comment
Importance of Identifying Pseudocellulitis—Successful diagnosis of cellulitis can be confounded by pseudocellulitis that can present concomitantly with or in lieu of cellulitis itself. Although cellulitis mostly affects the lower extremities in adults, pseudocellulitis also was common in this study population of patients with suspected lower extremity cellulitis, occurring both as a mimicker and concomitantly with cellulitis with substantial frequency. Notably, among patients with both venous stasis dermatitis and cellulitis diagnosed, most patients (n=10/12; 83.3%) had unilateral presentations of cellulitis as evidenced by signs and symptoms more notably affecting one lower extremity than the other. These findings suggest that certain pseudocellulitis diagnoses may predispose patients to cellulitis by disrupting the skin barrier, leading to bacterial infiltration; however, these pseudocellulitis diagnoses typically affect both lower extremities equally,1 and asymmetric involvement suggests the presence of overlying cellulitis. Furthermore, the most common pseudocellulitis entities found, such as venous stasis dermatitis, hematoma, and eczema, do not benefit from antibiotic treatment and require alternative therapy.1 Successful discrimination of these pseudocellulitis entities is critical to bolster proper antibiotic stewardship and discourage unnecessary hospitalization.
Independent Predictors of Cellulitis—Unilateral presentation and leukocytosis each emerged as strong independent predictors of cellulitis diagnosis in this study. Having either of these factors furthermore demonstrated high sensitivity and negative predictive value for cellulitis diagnosis. Other notable risk factors were history of onychomycosis, cellulitis, and trauma to the affected site. Prior studies have identified similar historical factors as predisposing patients to cellulitis.7-9 Interestingly, warmth of the affected area on physical examination emerged as strongly associated with cellulitis but was not included in the final predictive model because of its subjective determination. These factors may be especially important in diagnosing cellulitis in patients without concerning vital signs and with concomitant or prior pseudocellulitis.
Study Limitations—This study was limited to patients with uncomplicated presentations to emphasize discrimination of factors associated with cellulitis in the absence of suggestive signs of infection, such as vital sign abnormalities. Signs such as fever and tachypnea have been previously correlated to outpatient treatment failure and necessity for hospitalization.10-12 This study instead focused on patients without concerning vital signs to reduce confounding by such factors in more severe presentations that heighten suspicion for infection and increase likelihood of additional treatment measures. For such patients, suggestive historical factors, such as those discovered in this study, should be considered instead. Interestingly, increased age did not emerge as a significant predictor in this population in contrast to other predictive models that included patients with vital sign abnormalities. Notably, older patients tend to have more variable vital signs, especially in response to physiologic stressors such as infection.13 As such, age may serve as a proxy for vital sign abnormalities to some degree in such predictive models, leading to heightened suspicion for infection in older patients. This study demonstrated that in the absence of concerning vital signs, historical rather than demographic factors are more predictive of cellulitis.
Conclusion
Unilateral presentation and leukocytosis emerged as strong independent predictors of lower extremity cellulitis in patients with uncomplicated presentations. Having either of these factors had a sensitivity of 93.7% and a negative predictive value of 76.5%. Other factors such as history of cellulitis, onychomycosis, and recent trauma to the affected site emerged as additional predictors. These historical, examination, and laboratory characteristics may be especially useful for successful diagnosis of cellulitis in varied practice settings, including outpatient clinics and EDs.
- Raff AB, Kroshinsky D. Cellulitis: a review. JAMA. 2016;316:325-337.
- Gunderson CG, Cherry BM, Fisher A. Do patients with cellulitis need to be hospitalized? a systematic review and meta-analysis of mortality rates of inpatients with cellulitis. J Gen Intern Med. 2018;33:1553-1560.
- Ko LN, Garza-Mayers AC, St. John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis: a randomized clinical trial. JAMA Dermatol. 2018;154:529-536.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J. 2011;17:1.
- Hughey LC. The impact dermatologists can have on misdiagnosis of cellulitis and overuse of antibiotics: closing the gap. JAMA Dermatol. 2014;150:1061-1062.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59:147-159.
- Björnsdóttir S, Gottfredsson M, Thórisdóttir AS, et al. Risk factors for acute cellulitis of the lower limb: a prospective case-control study. Clin Infect Dis. 2005;41:1416-1422.
- Roujeau JC, Sigurgeirsson B, Korting HC, et al. Chronic dermatomycoses of the foot as risk factors for acute bacterial cellulitis of the leg: a case-control study. Dermatology. 2004;209:301-307.
- McNamara DR, Tleyjeh IM, Berbari EF, et al. A predictive model of recurrent lower extremity cellulitis in a population-based cohort. Arch Intern Med. 2007;167:709-715.
- Yadav K, Suh KN, Eagles D, et al. Predictors of oral antibiotic treatment failure for nonpurulent skin and soft tissue infections in the emergency department. Acad Emerg Med. 2019;26:51-59.
- Peterson D, McLeod S, Woolfrey K, et al. Predictors of failure of empiric outpatient antibiotic therapy in emergency department patients with uncomplicated cellulitis. Acad Emerg Med. 2014;21:526-531.
- Volz KA, Canham L, Kaplan E, et al. Identifying patients with cellulitis who are likely to require inpatient admission after a stay in an ED observation unit. Am J Emerg Med. 2013;31:360-364.
- Chester JG, Rudolph JL. Vital signs in older patients: age-related changes. J Am Med Dir Assoc. 2011;12:337-343.
Cellulitis is an infection of the skin and skin-associated structures characterized by redness, warmth, swelling, and pain of the affected area. Cellulitis most commonly occurs in middle-aged and older adults and frequently affects the lower extremities.1 Serious complications of cellulitis such as bacteremia, metastatic infection, and sepsis are rare, and most cases of cellulitis in patients with normal vital signs and mental status can be managed with outpatient treatment.2
Diagnosis of cellulitis can be confounded by a number of similarly presenting conditions collectively known as pseudocellulitis, such as venous stasis dermatitis and deep vein thrombosis.1 Misdiagnosis of cellulitis is common, with rates exceeding 30% among hospitalized patients initially diagnosed with cellulitis.3,4 Dermatology or infectious disease assessment is considered the diagnostic gold standard for cellulitis4,5 but is not always readily available, especially in resource-constrained settings.
Most cases of uncomplicated cellulitis can be managed with outpatient treatment, especially because serious complications are rare. Frequent misdiagnosis leads to repeat or unnecessary hospitalization and antibiosis. Exceptions necessitating hospitalization usually are predicated on signs of systemic infection, severe immunocompromised states, or failure of prior outpatient therapy.6 Such presentations can be distinguished by corresponding notable historical or examination factors, such as vital sign abnormalities suggesting systemic infection or history of malignancy leading to an immunocompromised state.
We sought to evaluate factors leading to the diagnosis of cellulitis in a cohort of patients with uncomplicated presentations receiving dermatology consultation to emphasize findings indicative of cellulitis in the absence of clinical or historical factors suggestive of other conditions necessitating hospitalization, such as systemic infection.
Methods
Study Participants—A prospective cohort study of patients presenting to an emergency department (ED) between October 2012 and January 2017 at an urban academic medical center in Boston, Massachusetts, was conducted with approval of study design and procedures by the relevant institutional review board. Patients older than 18 years were eligible for inclusion if given an initial diagnosis of cellulitis by an ED physician. Patients were excluded if incarcerated, pregnant, or unable to provide informed consent. Other exclusion criteria includedinfections overlying temporary or permanent indwelling hardware, animal or human bites, or sites of recent surgery (within the prior 4 weeks); preceding antibiotic treatment for more than 24 hours; or clinical or radiographic evidence of complications requiring alternative management such as osteomyelitis or abscess. Patients presenting with an elevated heart rate (>100 beats per minute) or body temperature (>100.5 °F [38.1 °C]) also were excluded. Eligible patients were enrolled upon providing written informed consent, and no remuneration was offered for participation.
Dermatology Consultation Intervention—A random subset of enrolled patients received dermatology consultation within 24 hours of presentation. Consultation consisted of a patient interview and physical examination with care recommendations to relevant ED and inpatient teams. Consultations confirmed the presence or absence of cellulitis as the primary outcome and also noted the presence of any pseudocellulitis diagnoses either occurring concomitantly with or mimicking cellulitis as a secondary outcome.
Statistical Analysis—Patient characteristics were analyzed to identify factors independently associated with the diagnosis of cellulitis in cases affecting the lower extremities. Factors were recorded with categorical variables reported as counts and percentages and continuous variables as means and standard deviations. Univariate analyses between categorical variables or discretized continuous variables and cellulitis diagnosis were conducted via Fisher exact test to identify a preliminary set of potential risk factors. Continuous variables were discretized at multiple incremental values with the discretization most significantly associated with cellulitis diagnosis selected as a preliminary risk factor. Multivariate analyses involved using any objective preliminary factor meeting a significance threshold of P<.1 in univariate comparisons in a multivariate logistic regression model for prediction of cellulitis diagnosis with corresponding calculation of odds ratios with confidence intervals and receiver operating characteristic. Factors with confidence intervals that excluded 1 were considered significant independent predictors of cellulitis. Analyses were performed using Python version 3.8 (Python Software Foundation).
Results
Of 1359 patients screened for eligibility, 104 patients with presumed lower extremity cellulitis undergoing dermatology consultation were included in this study (Figure). The mean patient age (SD) was 60.4 (19.2) years, and 63.5% of patients were male. In the study population, 63 (60.6%) patients received a final diagnosis of cellulitis. The most common pseudocellulitis diagnosis identified was venous stasis dermatitis, which occurred in 12 (11.5%) patients with concomitant cellulitis and in 12 (11.5%) patients mimicking cellulitis (Table).
Univariate comparisons revealed a diverse set of historical, examination, and laboratory factors associated with cellulitis diagnosis. Diagnosis of cellulitis was associated with unilateral presentation, recent trauma to the affected site, and history of cellulitis or onychomycosis. Diagnosis of cellulitis also was associated with elevated white blood cell count, absolute neutrophil count, C-reactive protein, body mass index, hematocrit, and platelet count; age less than 75 years; and lower serum sodium and serum chloride levels. These were the independent factors included in the multivariate analysis, which consisted of a logistic regression model for prediction of cellulitis (eTable).
Multivariate logistic regression on all preliminary factors significantly associated with cellulitis diagnosis in univariate comparisons demonstrated leukocytosis, which was defined as having a white blood cell count exceeding 11,000/μL, unilateral presentation, history of onychomycosis, and trauma to the affected site as significant independent predictors of cellulitis diagnosis; history of cellulitis approached significance (eTable). Unilateral presentation and leukocytosis were the strongest predictors; having either of these factors had a sensitivity of 93.7% and a negative predictive value of 76.5%.
Comment
Importance of Identifying Pseudocellulitis—Successful diagnosis of cellulitis can be confounded by pseudocellulitis that can present concomitantly with or in lieu of cellulitis itself. Although cellulitis mostly affects the lower extremities in adults, pseudocellulitis also was common in this study population of patients with suspected lower extremity cellulitis, occurring both as a mimicker and concomitantly with cellulitis with substantial frequency. Notably, among patients with both venous stasis dermatitis and cellulitis diagnosed, most patients (n=10/12; 83.3%) had unilateral presentations of cellulitis as evidenced by signs and symptoms more notably affecting one lower extremity than the other. These findings suggest that certain pseudocellulitis diagnoses may predispose patients to cellulitis by disrupting the skin barrier, leading to bacterial infiltration; however, these pseudocellulitis diagnoses typically affect both lower extremities equally,1 and asymmetric involvement suggests the presence of overlying cellulitis. Furthermore, the most common pseudocellulitis entities found, such as venous stasis dermatitis, hematoma, and eczema, do not benefit from antibiotic treatment and require alternative therapy.1 Successful discrimination of these pseudocellulitis entities is critical to bolster proper antibiotic stewardship and discourage unnecessary hospitalization.
Independent Predictors of Cellulitis—Unilateral presentation and leukocytosis each emerged as strong independent predictors of cellulitis diagnosis in this study. Having either of these factors furthermore demonstrated high sensitivity and negative predictive value for cellulitis diagnosis. Other notable risk factors were history of onychomycosis, cellulitis, and trauma to the affected site. Prior studies have identified similar historical factors as predisposing patients to cellulitis.7-9 Interestingly, warmth of the affected area on physical examination emerged as strongly associated with cellulitis but was not included in the final predictive model because of its subjective determination. These factors may be especially important in diagnosing cellulitis in patients without concerning vital signs and with concomitant or prior pseudocellulitis.
Study Limitations—This study was limited to patients with uncomplicated presentations to emphasize discrimination of factors associated with cellulitis in the absence of suggestive signs of infection, such as vital sign abnormalities. Signs such as fever and tachypnea have been previously correlated to outpatient treatment failure and necessity for hospitalization.10-12 This study instead focused on patients without concerning vital signs to reduce confounding by such factors in more severe presentations that heighten suspicion for infection and increase likelihood of additional treatment measures. For such patients, suggestive historical factors, such as those discovered in this study, should be considered instead. Interestingly, increased age did not emerge as a significant predictor in this population in contrast to other predictive models that included patients with vital sign abnormalities. Notably, older patients tend to have more variable vital signs, especially in response to physiologic stressors such as infection.13 As such, age may serve as a proxy for vital sign abnormalities to some degree in such predictive models, leading to heightened suspicion for infection in older patients. This study demonstrated that in the absence of concerning vital signs, historical rather than demographic factors are more predictive of cellulitis.
Conclusion
Unilateral presentation and leukocytosis emerged as strong independent predictors of lower extremity cellulitis in patients with uncomplicated presentations. Having either of these factors had a sensitivity of 93.7% and a negative predictive value of 76.5%. Other factors such as history of cellulitis, onychomycosis, and recent trauma to the affected site emerged as additional predictors. These historical, examination, and laboratory characteristics may be especially useful for successful diagnosis of cellulitis in varied practice settings, including outpatient clinics and EDs.
Cellulitis is an infection of the skin and skin-associated structures characterized by redness, warmth, swelling, and pain of the affected area. Cellulitis most commonly occurs in middle-aged and older adults and frequently affects the lower extremities.1 Serious complications of cellulitis such as bacteremia, metastatic infection, and sepsis are rare, and most cases of cellulitis in patients with normal vital signs and mental status can be managed with outpatient treatment.2
Diagnosis of cellulitis can be confounded by a number of similarly presenting conditions collectively known as pseudocellulitis, such as venous stasis dermatitis and deep vein thrombosis.1 Misdiagnosis of cellulitis is common, with rates exceeding 30% among hospitalized patients initially diagnosed with cellulitis.3,4 Dermatology or infectious disease assessment is considered the diagnostic gold standard for cellulitis4,5 but is not always readily available, especially in resource-constrained settings.
Most cases of uncomplicated cellulitis can be managed with outpatient treatment, especially because serious complications are rare. Frequent misdiagnosis leads to repeat or unnecessary hospitalization and antibiosis. Exceptions necessitating hospitalization usually are predicated on signs of systemic infection, severe immunocompromised states, or failure of prior outpatient therapy.6 Such presentations can be distinguished by corresponding notable historical or examination factors, such as vital sign abnormalities suggesting systemic infection or history of malignancy leading to an immunocompromised state.
We sought to evaluate factors leading to the diagnosis of cellulitis in a cohort of patients with uncomplicated presentations receiving dermatology consultation to emphasize findings indicative of cellulitis in the absence of clinical or historical factors suggestive of other conditions necessitating hospitalization, such as systemic infection.
Methods
Study Participants—A prospective cohort study of patients presenting to an emergency department (ED) between October 2012 and January 2017 at an urban academic medical center in Boston, Massachusetts, was conducted with approval of study design and procedures by the relevant institutional review board. Patients older than 18 years were eligible for inclusion if given an initial diagnosis of cellulitis by an ED physician. Patients were excluded if incarcerated, pregnant, or unable to provide informed consent. Other exclusion criteria includedinfections overlying temporary or permanent indwelling hardware, animal or human bites, or sites of recent surgery (within the prior 4 weeks); preceding antibiotic treatment for more than 24 hours; or clinical or radiographic evidence of complications requiring alternative management such as osteomyelitis or abscess. Patients presenting with an elevated heart rate (>100 beats per minute) or body temperature (>100.5 °F [38.1 °C]) also were excluded. Eligible patients were enrolled upon providing written informed consent, and no remuneration was offered for participation.
Dermatology Consultation Intervention—A random subset of enrolled patients received dermatology consultation within 24 hours of presentation. Consultation consisted of a patient interview and physical examination with care recommendations to relevant ED and inpatient teams. Consultations confirmed the presence or absence of cellulitis as the primary outcome and also noted the presence of any pseudocellulitis diagnoses either occurring concomitantly with or mimicking cellulitis as a secondary outcome.
Statistical Analysis—Patient characteristics were analyzed to identify factors independently associated with the diagnosis of cellulitis in cases affecting the lower extremities. Factors were recorded with categorical variables reported as counts and percentages and continuous variables as means and standard deviations. Univariate analyses between categorical variables or discretized continuous variables and cellulitis diagnosis were conducted via Fisher exact test to identify a preliminary set of potential risk factors. Continuous variables were discretized at multiple incremental values with the discretization most significantly associated with cellulitis diagnosis selected as a preliminary risk factor. Multivariate analyses involved using any objective preliminary factor meeting a significance threshold of P<.1 in univariate comparisons in a multivariate logistic regression model for prediction of cellulitis diagnosis with corresponding calculation of odds ratios with confidence intervals and receiver operating characteristic. Factors with confidence intervals that excluded 1 were considered significant independent predictors of cellulitis. Analyses were performed using Python version 3.8 (Python Software Foundation).
Results
Of 1359 patients screened for eligibility, 104 patients with presumed lower extremity cellulitis undergoing dermatology consultation were included in this study (Figure). The mean patient age (SD) was 60.4 (19.2) years, and 63.5% of patients were male. In the study population, 63 (60.6%) patients received a final diagnosis of cellulitis. The most common pseudocellulitis diagnosis identified was venous stasis dermatitis, which occurred in 12 (11.5%) patients with concomitant cellulitis and in 12 (11.5%) patients mimicking cellulitis (Table).
Univariate comparisons revealed a diverse set of historical, examination, and laboratory factors associated with cellulitis diagnosis. Diagnosis of cellulitis was associated with unilateral presentation, recent trauma to the affected site, and history of cellulitis or onychomycosis. Diagnosis of cellulitis also was associated with elevated white blood cell count, absolute neutrophil count, C-reactive protein, body mass index, hematocrit, and platelet count; age less than 75 years; and lower serum sodium and serum chloride levels. These were the independent factors included in the multivariate analysis, which consisted of a logistic regression model for prediction of cellulitis (eTable).
Multivariate logistic regression on all preliminary factors significantly associated with cellulitis diagnosis in univariate comparisons demonstrated leukocytosis, which was defined as having a white blood cell count exceeding 11,000/μL, unilateral presentation, history of onychomycosis, and trauma to the affected site as significant independent predictors of cellulitis diagnosis; history of cellulitis approached significance (eTable). Unilateral presentation and leukocytosis were the strongest predictors; having either of these factors had a sensitivity of 93.7% and a negative predictive value of 76.5%.
Comment
Importance of Identifying Pseudocellulitis—Successful diagnosis of cellulitis can be confounded by pseudocellulitis that can present concomitantly with or in lieu of cellulitis itself. Although cellulitis mostly affects the lower extremities in adults, pseudocellulitis also was common in this study population of patients with suspected lower extremity cellulitis, occurring both as a mimicker and concomitantly with cellulitis with substantial frequency. Notably, among patients with both venous stasis dermatitis and cellulitis diagnosed, most patients (n=10/12; 83.3%) had unilateral presentations of cellulitis as evidenced by signs and symptoms more notably affecting one lower extremity than the other. These findings suggest that certain pseudocellulitis diagnoses may predispose patients to cellulitis by disrupting the skin barrier, leading to bacterial infiltration; however, these pseudocellulitis diagnoses typically affect both lower extremities equally,1 and asymmetric involvement suggests the presence of overlying cellulitis. Furthermore, the most common pseudocellulitis entities found, such as venous stasis dermatitis, hematoma, and eczema, do not benefit from antibiotic treatment and require alternative therapy.1 Successful discrimination of these pseudocellulitis entities is critical to bolster proper antibiotic stewardship and discourage unnecessary hospitalization.
Independent Predictors of Cellulitis—Unilateral presentation and leukocytosis each emerged as strong independent predictors of cellulitis diagnosis in this study. Having either of these factors furthermore demonstrated high sensitivity and negative predictive value for cellulitis diagnosis. Other notable risk factors were history of onychomycosis, cellulitis, and trauma to the affected site. Prior studies have identified similar historical factors as predisposing patients to cellulitis.7-9 Interestingly, warmth of the affected area on physical examination emerged as strongly associated with cellulitis but was not included in the final predictive model because of its subjective determination. These factors may be especially important in diagnosing cellulitis in patients without concerning vital signs and with concomitant or prior pseudocellulitis.
Study Limitations—This study was limited to patients with uncomplicated presentations to emphasize discrimination of factors associated with cellulitis in the absence of suggestive signs of infection, such as vital sign abnormalities. Signs such as fever and tachypnea have been previously correlated to outpatient treatment failure and necessity for hospitalization.10-12 This study instead focused on patients without concerning vital signs to reduce confounding by such factors in more severe presentations that heighten suspicion for infection and increase likelihood of additional treatment measures. For such patients, suggestive historical factors, such as those discovered in this study, should be considered instead. Interestingly, increased age did not emerge as a significant predictor in this population in contrast to other predictive models that included patients with vital sign abnormalities. Notably, older patients tend to have more variable vital signs, especially in response to physiologic stressors such as infection.13 As such, age may serve as a proxy for vital sign abnormalities to some degree in such predictive models, leading to heightened suspicion for infection in older patients. This study demonstrated that in the absence of concerning vital signs, historical rather than demographic factors are more predictive of cellulitis.
Conclusion
Unilateral presentation and leukocytosis emerged as strong independent predictors of lower extremity cellulitis in patients with uncomplicated presentations. Having either of these factors had a sensitivity of 93.7% and a negative predictive value of 76.5%. Other factors such as history of cellulitis, onychomycosis, and recent trauma to the affected site emerged as additional predictors. These historical, examination, and laboratory characteristics may be especially useful for successful diagnosis of cellulitis in varied practice settings, including outpatient clinics and EDs.
- Raff AB, Kroshinsky D. Cellulitis: a review. JAMA. 2016;316:325-337.
- Gunderson CG, Cherry BM, Fisher A. Do patients with cellulitis need to be hospitalized? a systematic review and meta-analysis of mortality rates of inpatients with cellulitis. J Gen Intern Med. 2018;33:1553-1560.
- Ko LN, Garza-Mayers AC, St. John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis: a randomized clinical trial. JAMA Dermatol. 2018;154:529-536.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J. 2011;17:1.
- Hughey LC. The impact dermatologists can have on misdiagnosis of cellulitis and overuse of antibiotics: closing the gap. JAMA Dermatol. 2014;150:1061-1062.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59:147-159.
- Björnsdóttir S, Gottfredsson M, Thórisdóttir AS, et al. Risk factors for acute cellulitis of the lower limb: a prospective case-control study. Clin Infect Dis. 2005;41:1416-1422.
- Roujeau JC, Sigurgeirsson B, Korting HC, et al. Chronic dermatomycoses of the foot as risk factors for acute bacterial cellulitis of the leg: a case-control study. Dermatology. 2004;209:301-307.
- McNamara DR, Tleyjeh IM, Berbari EF, et al. A predictive model of recurrent lower extremity cellulitis in a population-based cohort. Arch Intern Med. 2007;167:709-715.
- Yadav K, Suh KN, Eagles D, et al. Predictors of oral antibiotic treatment failure for nonpurulent skin and soft tissue infections in the emergency department. Acad Emerg Med. 2019;26:51-59.
- Peterson D, McLeod S, Woolfrey K, et al. Predictors of failure of empiric outpatient antibiotic therapy in emergency department patients with uncomplicated cellulitis. Acad Emerg Med. 2014;21:526-531.
- Volz KA, Canham L, Kaplan E, et al. Identifying patients with cellulitis who are likely to require inpatient admission after a stay in an ED observation unit. Am J Emerg Med. 2013;31:360-364.
- Chester JG, Rudolph JL. Vital signs in older patients: age-related changes. J Am Med Dir Assoc. 2011;12:337-343.
- Raff AB, Kroshinsky D. Cellulitis: a review. JAMA. 2016;316:325-337.
- Gunderson CG, Cherry BM, Fisher A. Do patients with cellulitis need to be hospitalized? a systematic review and meta-analysis of mortality rates of inpatients with cellulitis. J Gen Intern Med. 2018;33:1553-1560.
- Ko LN, Garza-Mayers AC, St. John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis: a randomized clinical trial. JAMA Dermatol. 2018;154:529-536.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J. 2011;17:1.
- Hughey LC. The impact dermatologists can have on misdiagnosis of cellulitis and overuse of antibiotics: closing the gap. JAMA Dermatol. 2014;150:1061-1062.
- Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59:147-159.
- Björnsdóttir S, Gottfredsson M, Thórisdóttir AS, et al. Risk factors for acute cellulitis of the lower limb: a prospective case-control study. Clin Infect Dis. 2005;41:1416-1422.
- Roujeau JC, Sigurgeirsson B, Korting HC, et al. Chronic dermatomycoses of the foot as risk factors for acute bacterial cellulitis of the leg: a case-control study. Dermatology. 2004;209:301-307.
- McNamara DR, Tleyjeh IM, Berbari EF, et al. A predictive model of recurrent lower extremity cellulitis in a population-based cohort. Arch Intern Med. 2007;167:709-715.
- Yadav K, Suh KN, Eagles D, et al. Predictors of oral antibiotic treatment failure for nonpurulent skin and soft tissue infections in the emergency department. Acad Emerg Med. 2019;26:51-59.
- Peterson D, McLeod S, Woolfrey K, et al. Predictors of failure of empiric outpatient antibiotic therapy in emergency department patients with uncomplicated cellulitis. Acad Emerg Med. 2014;21:526-531.
- Volz KA, Canham L, Kaplan E, et al. Identifying patients with cellulitis who are likely to require inpatient admission after a stay in an ED observation unit. Am J Emerg Med. 2013;31:360-364.
- Chester JG, Rudolph JL. Vital signs in older patients: age-related changes. J Am Med Dir Assoc. 2011;12:337-343.
Practice Points
- Unilateral involvement and leukocytosis are both highly predictive of lower extremity cellulitis in uncomplicated presentations.
- Historical factors such as history of onychomycosis and trauma to the affected site are more predictive of lower extremity cellulitis than demographic factors such as age in uncomplicated presentations of cellulitis.
Current Recommendations for the Systemic Treatment of Cutaneous Lupus Erythematosus During Pregnancy
Cutaneous lupus erythematosus (CLE) is a heterogeneous autoimmune disease that involves the skin. Cutaneous lupus erythematosus can be classified into various subtypes.1 These include, but are not limited to, acute CLE, subacute CLE, chronic CLE, intermittent CLE, lupus tumidus, and lupus profundus.1,2 The CLE subtypes have variable associations with systemic lupus erythematosus. For instance, some subtypes, such as acute CLE, are more strongly associated with systemic lupus erythematosus.
Treatment of CLE is similar to other autoimmune disorders. Although the US Food and Drug Administration (FDA) has not approved any treatments for CLE,3,4 the most common therapeutic options are disease-modifying antirheumatic drugs. Unfortunately, many of these treatments carry teratogenic effects. Because CLE predominantly affects women, particularly those of childbearing age, it is imperative to understand the available treatment options for those who are pregnant or considering pregnancy for an informed discussion with patients.5
For years, the gold standard when considering a medication during pregnancy was the FDA’s classification system. According to this system, medications were classified into 5 letter categories based on their potential teratogenicity, including A (no fetal risk), B (potential animal risk but inconclusive human studies), C (risk cannot be ruled out), D (evidence of fetal risk), and X (contraindicated in pregnancy). In 2014, the FDA decided to no longer use this classification system for medications approved after 2000.6 However, because many proposed treatment options for CLE were approved prior to 2001, we have summarized the commonly prescribed medications for CLE according to their prior FDA letter categories.
Treatment Options for CLE During Pregnancy
Prior to initiating systemic medications for the treatment of CLE, topical medications should be considered. Recommended treatment options include corticosteroids and calcineurin inhibitors.7 Compared with systemic medications, topical treatments carry minimal side effects, such as skin atrophy, that typically remain localized to areas of application.8 Moreover, even with extensive application, no correlation has been found between topical corticosteroid use and fetal growth,9 which suggests that topical steroids are safe in pregnancy and should be considered as a first-line treatment option for CLE. Calcineurin inhibitors also are considered safe based on their low level of absorption through the skin and are considered second-line topical treatment options in pregnancy.10

Although topical medications are effective for the treatment of CLE, many patients require the administration of systemic therapeutics for severe or refractory disease. Based on previously published reports, Figure 1 describes the current recommended systemic treatment options for CLE.11 Unfortunately, many of these medications carry teratogenic risks during pregnancy. The risks and side effects of the medications are described in detail in the following sections and summarized in the eTable.

Category B
Systemic Steroids—Systemic steroids are one of the most prescribed medications during pregnancy.12 Oral steroids have been associated with fast symptom relief, making this class of medications particularly effective during CLE flares; however, long-term management is not recommended because of the side effects, which include osteoporosis and impaired glucose metabolism.13
With low transmission across the placenta, there are 3 glucocorticoids that carry the safest profile in pregnancy: prednisone, cortisone, and hydrocortisone.14 Dexamethasone and betamethasone should be avoided, as both readily cross the placenta and increase fetal exposure.15 Although teratogenic effects have been associated with steroid use, most studies involving pregnant patients have inconclusive results. For instance, one study described an association between cleft lip/palate with in utero glucocorticoid exposure.16 However, multiple follow-up studies found no association between the two.17,18 Studies investigating the relationship between steroids and miscarriages or steroids and low birth weight also are inconclusive. Of note, if used throughout pregnancy, administration of a loading dose of glucocorticoids prior to delivery is recommended because of the increased stress brought on during labor.19
Sulfasalazine—Sulfasalazine is an immunomodulator commonly used for the treatment of inflammatory bowel disease and rheumatoid arthritis. However, studies also have shown that sulfasalazine is an effective treatment of CLE if standard treatments have failed.20,21
During pregnancy, patients exposed to sulfasalazine experienced minimal side effects despite transportation across the placenta.22 In comparison with control, pregnant women taking sulfasalazine experienced no increased risk for low fetal weight,23 congenital abnormalities,24 or spontaneous abortions.25 Of note, sulfasalazine can affect sperm, so male patients also should be counselled.
Category C
Hydroxychloroquine—Hydroxychloroquine is considered a first-line medication for those with CLE based on a symptomatic relief rate of 50% to 70%.26 For those taking hydroxychloroquine during pregnancy, the majority of studies have shown no association between the medication and adverse fetal events, including congenital abnormalities, prematurity, or spontaneous abortions.27-29 Therefore, hydroxychloroquine is considered safe in pregnancy, and those on the medication should continue standard monitoring, including retinopathy screening.30
Of note, hydroxychloroquine can be stored in tissue for weeks to months after discontinuation.5 Therefore, if patients wish to avoid hydroxychloroquine in pregnancy, one should stop taking the medication several months prior to conception.
Dapsone—Dapsone, a medication with both antimicrobial and immunomodulatory properties, is an effective second-line therapy for CLE.31 Although large-scale human trials have not been performed, multiple case reports and observational studies have supported the safe use of dapsone in pregnancy.32-34 However, there are notable side effects, including dose-dependent hemolysis, methemoglobinemia, and hypersensitivity reactions.13 Therefore, once treatment is initiated or continued, folic acid supplementation (5 mg daily) and regular serum analysis, including complete blood cell counts, are recommended in pregnant patients.19
Rituximab—Recent studies have demonstrated that rituximab can be an effective treatment of subacute and chronic CLE.35,36 Through inhibition of CD20, rituximab causes a decrease in circulating B cells and a reduced immune response. Therefore, experts recommend discontinuation of rituximab for 12 months prior to conception to reduce potential side effects to the fetus, which may include a transient reduction of circulating fetal B cells.37
If continued during pregnancy, most studies suggest discontinuation of rituximab during the third trimester, as it has been associated with neonatal infections and congenital abnormalities.19,37 However, these results are based on limited case reports, and thus robust research is needed to better understand the effect of rituximab in utero.
Intravenous Immunoglobulin Infusion—Intravenous immunoglobulin (IVIG) infusion is a well-tolerated treatment for many autoimmune disorders.38 Although not first line, limited case studies have demonstrated remission of refractory CLE following IVIG.39,40 Although no studies have directly investigated the effect of IVIG on fetal development, it has been frequently administered and well tolerated during pregnancy, especially in those with multiple sclerosis or antiphospholipid syndrome.41 Commonly reported side effects include headache and fatigue, and a rare associated side effect to be aware of is embolic events.42,43
Cyclosporine—Cyclosporine rarely is used in the treatment of localized CLE due to its extensive side-effect profile, most notably nephrotoxicity.44 However, studies have shown that cyclosporine may be efficacious if symptoms extend beyond the skin, involve multiple organs, and/or other treatments have failed.39 For those who are pregnant and wish to continue cyclosporine use, studies have associated low birth weight and premature delivery with its exposure in utero.44
Category D
Mycophenolate Mofetil—In conjunction with standard therapy, mycophenolate mofetil (MMF) is an adequate treatment of refractory CLE.45 Unfortunately, case reports have demonstrated an increased risk for fetal congenital abnormalities and first-trimester spontaneous abortion with use of MMF during pregnancy.46,47 Therefore, it is recommended that patients on MMF discontinue the medication at least 6 weeks prior to conception.46
Azathioprine—Although azathioprine has been shown to provide relief of discoid lupus erythematosus symptoms,48 it currently is only utilized for refractory disease, largely due to notable side effects that particularly affect the gastrointestinal tract and liver.4 Moreover, azathioprine use during pregnancy has been associated with prematurity, congenital anomalies, fetal cytopenia, and low birth weight.49 With that said, and although not recommended, if patients decide to continue treatment, experts recommend limiting the dose to 2 mg/kg daily to reduce potential adverse events.
Category X
Oral Retinoids—According to the American Academy of Dermatology, retinoids such as isotretinoin and acitretin are considered second-line therapy for CLE.50 With that being said, there are well-documented effects on fetal development associated with oral retinoid use, including central nervous system, cardiovascular system, and craniofacial abnormalities.51 Therefore, its use is contraindicated during pregnancy. To prevent pregnancy while taking isotretinoin, patients must enroll in an online monitoring program called iPLEDGE. This program requires monthly updates by both the physician and the patient, including a negative pregnancy test every month for female patients actively taking the medication.52
The half-lives of the oral retinoids isotretinoin and acitretin are 10 to 20 hours and 50 to 60 hours, respectively.53,54 However, alcohol consumption converts acitretin into the metabolite etretinate, which can remain in tissue for up to 120 days.54,55 Therefore, women are advised to avoid alcohol while taking acitretin and avoid conception for 2 to 3 years after cessation of the medication.55 For those wishing to restart retinoids after pregnancy, studies show the medication can be safely reinstated 35 days after delivery for those interested in continued treatment.56
Thalidomide—Although low-dose thalidomide can treat refractory CLE, its use is restricted because of its known teratogenicity, most notably limb deformities.57 If prescribed thalidomide, women will need to enroll in the System for Thalidomide Education and Prescribing Safety program, similar to the iPLEDGE program, and use 2 forms of contraception when sexually active.58 Contraception should be continued for 4 weeks following the last dose of thalidomide. After this point, conception is considered safe.59
Methotrexate—For nonpregnant patients, low-dose methotrexate (MTX) with folate supplementation is a treatment option for CLE.60 However, for those who are pregnant, low-dose MTX is an abortive agent and has been associated with aminopterin syndrome, which includes skull deficits, craniofacial abnormalities, and limb deformities in live births.19,61 Therefore, MTX is not recommended in pregnancy. Of note, MTX can affect sperm; male patients also should be counselled.
Final Thoughts
Overall, it is recommended to limit medication use as much as possible in pregnancy. To reduce these exposures, it is imperative to reduce triggers that may lead to symptomatic flares of CLE. Because CLE can be triggered by sun exposure, we advise topical sunscreen to prevent CLE flares that may require additional oral medication.62,63
Various medications are considered safe for the treatment of CLE in pregnant patients (Figure 2). Based on studies in animal and clinical trials, hydroxychloroquine is considered a safe and effective medication for CLE in pregnancy and is a first-line therapy in nonpregnant patients.26,27 If flares occur, IVIG or a short course of oral steroids should be considered to manage symptoms.13,39 For those with severe flares, treatment is difficult, and personalized approaches may be necessary.

Part of the question for the childbearing population is when a patient would like to conceive. For severe cases when hydroxychloroquine is not effective as monotherapy, using a treatment that can encourage remission prior to conception attempts can be a beneficial strategy. Rituximab is an excellent example of such a therapy, as the therapeutic effect outlasts the immunosuppressive effect and therefore is unlikely to affect a future fetus.64 Thalidomide also is a potential option prior to conception, based on its short washout period and its ability to achieve notable remission rates in patients with CLE.57,59 Regardless, patients with CLE should still consult their dermatologist and rheumatologist (if applicable) prior to conception.
Patients of childbearing potential represent a population in which discussion about life goals greatly affects medication options. Having these discussions early and often allows for an open, more successful approach so that treatment regimens are not derailed at the time of conception.
- Renner R, Sticherling M. The different faces of cutaneous lupus erythematosus. G Ital Dermatol Venereol. 2009;144:135-147.
- Kuhn A, Landmann A. The classification and diagnosis of cutaneous lupus erythematosus. J Autoimmun. 2014;48:14-19.
- Shi H, Gudjonsson J, Kahlenberg J. Treatment of cutaneous lupus erythematosus: current approaches and future strategies. Curr Opin Rheumatol. 2020;32:208-214.
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Jacobson DL, Gange SJ, Rose NR, et al. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997;84:223-243.
- Pernia S, DeMaagd G. The new pregnancy and lactation labeling rule. P T. 2016;41:713-715.
- Fanouriakis A, Kostopoulou M, Alunno A, et al. 2019 Update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. 2019;78:736-745.
- Kuhn A, Aberer E, Bata‐Csörgö Z, et al. S2k guideline for treatment of cutaneous lupus erythematosus—guided by the European Dermatology Forum (EDF) in cooperation with the European Academyof Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2017;31:389-404.
- Andersson NW, Skov L, Andersen JT. Evaluation of topical corticosteroid use in pregnancy and risk of newborns being small for gestational age and having low birth weight. JAMA Dermatol. 2021;157:788-795.
- Undre NA, Moloney FJ, Ahmadi S, et al. Skin and systemic pharmacokinetics of tacrolimus following topical application of tacrolimus ointment in adults with moderate to severe atopic dermatitis. Br J Dermatol. 2009;160:665-669.
- Xiong W, Lahita RG. Pragmatic approaches to therapy for systemic lupus erythematosus. Nat Rev Rheumatol. 2014;10:97-107.
- Kuriya B, Hernández‐Díaz S, Liu J, et al. Patterns of medication use during pregnancy in rheumatoid arthritis. Arthritis Care Res. 2011;63:721-728.
- Chang A, Werth V. Treatment of cutaneous lupus. Curr Rheumatol Rep. 2011;13:300-307.
- Beitins IZ, Bayard F, Ances IG, et al. The transplacental passage of prednisone and prednisolone in pregnancy near term. J Pediatr. 1972;81:936-945.
- Ogueh O, Johnson MR. The metabolic effect of antenatal corticosteroid therapy. Hum Reprod Update. 2000;6:169-176.
- Park-Wyllie L, Mazzotta P, Pastuszak A, et al. Birth defects after maternal exposure to corticosteroids: prospective cohort study and meta-analysis of epidemiological studies. Teratology. 2000;62:385-392.
- Bay Bjørn A, Ehrenstein V, Hundborg HH, et al. Use of corticosteroids in early pregnancy is not associated with risk of oral clefts and other congenital malformations in offspring. Am J Ther. 2014;21:73-80.
- Hviid A, Mølgaard-Nielsen D. Corticosteroid use during pregnancy and risk of orofacial clefts. CMAJ. 2011;183:796-804.
- Krause ML, Amin S, Makol A. Use of DMARDs and biologics during pregnancy and lactation in rheumatoid arthritis: what the rheumatologist needs to know. Ther Adv Musculoskelet Dis. 2014;6:169-184.
- Artuz F, Lenk N, Deniz N, et al. Efficacy of sulfasalazine in discoid lupus erythematosus. Int J Dermatol. 1996;35:746-748.
- Delaporte E, Catteau B, Sabbagh N, et al. Treatment of discoid lupus erythematosus with sulfasalazine: 11 cases [in French]. Ann Dermatol Venereol. 1997;124:151-156.
- Järnerot G, Into-Malmberg MB, Esbjörner E. Placental transfer of sulphasalazine and sulphapyridine and some of its metabolites. Scand J Gastroenterol. 1981;16:693-697.
- Norgard B, Pedersen L, Christensen LA, et al. Therapeutic drug use in women with Crohn’s disease and birth outcomes: a Danish nationwide cohort study. Am J Gastroenterol. 2007;102:1406-1413.
- Nørgård B, Czeizel AE, Rockenbauer M, et al. Population-based case control study of the safety of sulfasalazine use during pregnancy. Aliment Pharmacol Ther. 2001;15:483-486.
- Rahimi R, Nikfar S, Rezaie A, et al. Pregnancy outcome in women with inflammatory bowel disease following exposure to 5-aminosalicylic acid drugs: a meta-analysis. Reprod Toxicol. 2008;25:271-275.
- Callen JP. Chronic cutaneous lupus erythematosus: clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol. 1982;118:412-416.
- Buchanan NM, Toubi E, Khamashta MA, et al. Hydroxychloroquine and lupus pregnancy: review of a series of 36 cases. Ann Rheum Dis. 1996;55:486-488.
- Costedoat‐Chalumeau N, Amoura Z, Duhaut P, et al. Safety of hydroxychloroquine in pregnant patients with connective tissue diseases: a study of one hundred thirty‐three cases compared with a control group. Arthritis Rheum. 2003;48:3207-3211.
- Sperber K, Hom C, Chao CP, et al. Systematic review of hydroxychloroquine use in pregnant patients with autoimmune diseases. Pediatr Rheumatol Online J. 2009;7:9.
- Marmor MF, Carr RE, Easterbrook M, et al. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy: a report by the American Academy of Ophthalmology. Ophthalmology. 2002;109:1377-1382.
- Klebes M, Wutte N, Aberer E. Dapsone as second-line treatment for cutaneous lupus erythematosus? a retrospective analysis of 34 patients and a review of the literature. Dermatology. 2016;232:91-96.
- Tuffanelli DL. Successful pregnancy in a patient with dermatitis herpetiformis treated with low-dose dapsone. Arch Dermatol. 1982;118:876.
- Varghese L, Viswabandya A, Mathew AJ. Dapsone, danazol, and intrapartum splenectomy in refractory ITP complicating pregnancy. Indian J Med Sci. 2008;62:452-455.
- Kahn G. Dapsone is safe during pregnancy. J Am Acad Dermatol. 1985;13:838-839.
- Quelhas da Costa R, Aguirre-Alastuey ME, Isenberg DA, et al. Assessment of response to B-cell depletion using rituximab in cutaneous lupus erythematosus. JAMA Dermatol. 2018;154:1432-1440.
- Alsanafi S, Kovarik C, Mermelstein A, et al. Rituximab in thetreatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506.
- Fernandez AP, Kerdel FA. The use of i.v. IG therapy in dermatology. Dermatol Ther. 2007;20:288-305.
- Kuhn A, Ruland V, Bonsmann G. Cutaneous lupus erythematosus: update of therapeutic options part II. J Am Acad Dermatol. 2011;65:E195-E213.
- Singh H, Naidu G, Sharma A. Intravenous immunoglobulin for the rescue in refractory cutaneous lupus. Indian Dermatol Online J. 2020;11:1003-1004.
- Clark AL. Clinical uses of intravenous immunoglobulin in pregnancy. Clin Obstet Gynecol. 1999;42:368-380.
- Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med. 2001;345:747-755.
- Woodruff RK, Grigg AP, Firkin FC, et al. Fatal thrombotic events during treatment of autoimmune thrombocytopenia with intravenous immunoglobulin in elderly patients. Lancet. 1986;2:217-218.
- Paziana K, Del Monaco M, Cardonick E, et al. Ciclosporin use during pregnancy. Drug Saf. 2013;36:279-294.
- Gammon B, Hansen C, Costner MI. Efficacy of mycophenolate mofetil in antimalarial-resistant cutaneous lupus erythematosus. J Am Acad Dermatol. 2010;65:717-721.e2.
- Abdulaziz HM, Shemies RS, Taman M, et al. Fetal proximal and distal limb anomalies following exposure to mycophenolate mofetil during pregnancy: a case report and review of the literature. Lupus. 2021;30:1522-1525.
- Pisoni CN, D’Cruz DP. The safety of mycophenolate mofetil in pregnancy. Exp Opin Drug Saf. 2008;7:219-222.
- Ashinoff R, Werth VP, Franks AG. Resistant discoid lupus erythematosus of palms and soles: successful treatment with azathioprine. J Am Acad Dermatol. 1988;19:961-965. doi:10.1016/S0190-9622(88)70259-5
- Goldstein LH, Dolinsky G, Greenberg R, et al. Pregnancy outcome of women exposed to azathioprine during pregnancy. Birth Defects Res A Clin Mol Teratol. 2007;79:696-701.
- Drake LA, Dinehart SM, Farmer ER, et al. Guidelines of care for cutaneous lupus erythematosus. American Academy of Dermatology. J Am Acad Dermatol. 1996;34:830-836.
- Sladden MJ, Harman KE. What is the chance of a normal pregnancy in a woman whose fetus has been exposed to isotretinoin? Arch Dermatol. 2007;143:1187-1188.
- Shin J, Cheetham TC, Wong L, et al. The impact of the iPLEDGE program on isotretinoin fetal exposure in an integrated health care system. J Am Acad Dermatol. 2011;65:1117-1125.
- Brazzell RK, Colburn WA. Pharmacokinetics of the retinoids isotretinoin and etretinate. J Am Acad Dermatol. 1982;6:643-651.
- Pilkington T, Brogden RN. Acitretin: a review of its pharmacology and therapeutic use. Drugs. 1992;43:597-627.
- Gronhoj Larsen F, Steinkjer B, Jakobsen P, et al. Acitretin is converted to etretinate only during concomitant alcohol intake. Br J Dermatol. 2000;143:1164-1169.
- Jajoria H, Mysore V. Washout period for pregnancy post isotretinoin therapy. Indian Dermatol Online J. 2020;11:239-242.
- Cortés-Hernández J, Torres-Salido M, Castro-Marrero J, et al. Thalidomide in the treatment of refractory cutaneous lupus erythematosus: prognostic factors of clinical outcome. Br J Dermatol. 2012;166:616-623.
- Zeldis JB, Williams BA, Thomas SD, et al. S.T.E.P.S.™: a comprehensive program for controlling and monitoring access to thalidomide. Clin Ther. 1999;21:319-330.
- C.S. Mott Children’s Hospital. University of Michigan Health. Thalidomide. Updated March 26, 2020. Accessed January 14, 2022. https://www.mottchildren.org/health-library/d04331a1
- Boehm IB, Boehm GA, Bauer R. Management of cutaneous lupus erythematosus with low-dose methotrexate: indication for modulation of inflammatory mechanisms. Rheumatol Int. 1998;18:59-62.
- Buckley LM, Bullaboy CA, Leichtman L, et al. Multiple congenital anomalies associated with weekly low‐dose methotrexate treatment of the mother. Arthritis Rheum. 1997;40:971-973.
- Foering K, Okawa J, Rose M, et al. Characterization of photosensitivity and poor quality of life in lupus. J Invest Dermatol. 2010;130(suppl):S10.
- Kuhn A, Herrmann M, Kleber S, et al. Accumulation of apoptotic cells in the epidermis of patients with cutaneous lupus erythematosus after ultraviolet irradiation. Arthritis Rheum. 2006;54:939-950.
- Lake EP, Huang Y, Aronson IK. Rituximab treatment of pemphigus in women of childbearing age: experience with two patients. J Dermatol Treat. 2017;28:751-752.
Cutaneous lupus erythematosus (CLE) is a heterogeneous autoimmune disease that involves the skin. Cutaneous lupus erythematosus can be classified into various subtypes.1 These include, but are not limited to, acute CLE, subacute CLE, chronic CLE, intermittent CLE, lupus tumidus, and lupus profundus.1,2 The CLE subtypes have variable associations with systemic lupus erythematosus. For instance, some subtypes, such as acute CLE, are more strongly associated with systemic lupus erythematosus.
Treatment of CLE is similar to other autoimmune disorders. Although the US Food and Drug Administration (FDA) has not approved any treatments for CLE,3,4 the most common therapeutic options are disease-modifying antirheumatic drugs. Unfortunately, many of these treatments carry teratogenic effects. Because CLE predominantly affects women, particularly those of childbearing age, it is imperative to understand the available treatment options for those who are pregnant or considering pregnancy for an informed discussion with patients.5
For years, the gold standard when considering a medication during pregnancy was the FDA’s classification system. According to this system, medications were classified into 5 letter categories based on their potential teratogenicity, including A (no fetal risk), B (potential animal risk but inconclusive human studies), C (risk cannot be ruled out), D (evidence of fetal risk), and X (contraindicated in pregnancy). In 2014, the FDA decided to no longer use this classification system for medications approved after 2000.6 However, because many proposed treatment options for CLE were approved prior to 2001, we have summarized the commonly prescribed medications for CLE according to their prior FDA letter categories.
Treatment Options for CLE During Pregnancy
Prior to initiating systemic medications for the treatment of CLE, topical medications should be considered. Recommended treatment options include corticosteroids and calcineurin inhibitors.7 Compared with systemic medications, topical treatments carry minimal side effects, such as skin atrophy, that typically remain localized to areas of application.8 Moreover, even with extensive application, no correlation has been found between topical corticosteroid use and fetal growth,9 which suggests that topical steroids are safe in pregnancy and should be considered as a first-line treatment option for CLE. Calcineurin inhibitors also are considered safe based on their low level of absorption through the skin and are considered second-line topical treatment options in pregnancy.10
Although topical medications are effective for the treatment of CLE, many patients require the administration of systemic therapeutics for severe or refractory disease. Based on previously published reports, Figure 1 describes the current recommended systemic treatment options for CLE.11 Unfortunately, many of these medications carry teratogenic risks during pregnancy. The risks and side effects of the medications are described in detail in the following sections and summarized in the eTable.
Category B
Systemic Steroids—Systemic steroids are one of the most prescribed medications during pregnancy.12 Oral steroids have been associated with fast symptom relief, making this class of medications particularly effective during CLE flares; however, long-term management is not recommended because of the side effects, which include osteoporosis and impaired glucose metabolism.13
With low transmission across the placenta, there are 3 glucocorticoids that carry the safest profile in pregnancy: prednisone, cortisone, and hydrocortisone.14 Dexamethasone and betamethasone should be avoided, as both readily cross the placenta and increase fetal exposure.15 Although teratogenic effects have been associated with steroid use, most studies involving pregnant patients have inconclusive results. For instance, one study described an association between cleft lip/palate with in utero glucocorticoid exposure.16 However, multiple follow-up studies found no association between the two.17,18 Studies investigating the relationship between steroids and miscarriages or steroids and low birth weight also are inconclusive. Of note, if used throughout pregnancy, administration of a loading dose of glucocorticoids prior to delivery is recommended because of the increased stress brought on during labor.19
Sulfasalazine—Sulfasalazine is an immunomodulator commonly used for the treatment of inflammatory bowel disease and rheumatoid arthritis. However, studies also have shown that sulfasalazine is an effective treatment of CLE if standard treatments have failed.20,21
During pregnancy, patients exposed to sulfasalazine experienced minimal side effects despite transportation across the placenta.22 In comparison with control, pregnant women taking sulfasalazine experienced no increased risk for low fetal weight,23 congenital abnormalities,24 or spontaneous abortions.25 Of note, sulfasalazine can affect sperm, so male patients also should be counselled.
Category C
Hydroxychloroquine—Hydroxychloroquine is considered a first-line medication for those with CLE based on a symptomatic relief rate of 50% to 70%.26 For those taking hydroxychloroquine during pregnancy, the majority of studies have shown no association between the medication and adverse fetal events, including congenital abnormalities, prematurity, or spontaneous abortions.27-29 Therefore, hydroxychloroquine is considered safe in pregnancy, and those on the medication should continue standard monitoring, including retinopathy screening.30
Of note, hydroxychloroquine can be stored in tissue for weeks to months after discontinuation.5 Therefore, if patients wish to avoid hydroxychloroquine in pregnancy, one should stop taking the medication several months prior to conception.
Dapsone—Dapsone, a medication with both antimicrobial and immunomodulatory properties, is an effective second-line therapy for CLE.31 Although large-scale human trials have not been performed, multiple case reports and observational studies have supported the safe use of dapsone in pregnancy.32-34 However, there are notable side effects, including dose-dependent hemolysis, methemoglobinemia, and hypersensitivity reactions.13 Therefore, once treatment is initiated or continued, folic acid supplementation (5 mg daily) and regular serum analysis, including complete blood cell counts, are recommended in pregnant patients.19
Rituximab—Recent studies have demonstrated that rituximab can be an effective treatment of subacute and chronic CLE.35,36 Through inhibition of CD20, rituximab causes a decrease in circulating B cells and a reduced immune response. Therefore, experts recommend discontinuation of rituximab for 12 months prior to conception to reduce potential side effects to the fetus, which may include a transient reduction of circulating fetal B cells.37
If continued during pregnancy, most studies suggest discontinuation of rituximab during the third trimester, as it has been associated with neonatal infections and congenital abnormalities.19,37 However, these results are based on limited case reports, and thus robust research is needed to better understand the effect of rituximab in utero.
Intravenous Immunoglobulin Infusion—Intravenous immunoglobulin (IVIG) infusion is a well-tolerated treatment for many autoimmune disorders.38 Although not first line, limited case studies have demonstrated remission of refractory CLE following IVIG.39,40 Although no studies have directly investigated the effect of IVIG on fetal development, it has been frequently administered and well tolerated during pregnancy, especially in those with multiple sclerosis or antiphospholipid syndrome.41 Commonly reported side effects include headache and fatigue, and a rare associated side effect to be aware of is embolic events.42,43
Cyclosporine—Cyclosporine rarely is used in the treatment of localized CLE due to its extensive side-effect profile, most notably nephrotoxicity.44 However, studies have shown that cyclosporine may be efficacious if symptoms extend beyond the skin, involve multiple organs, and/or other treatments have failed.39 For those who are pregnant and wish to continue cyclosporine use, studies have associated low birth weight and premature delivery with its exposure in utero.44
Category D
Mycophenolate Mofetil—In conjunction with standard therapy, mycophenolate mofetil (MMF) is an adequate treatment of refractory CLE.45 Unfortunately, case reports have demonstrated an increased risk for fetal congenital abnormalities and first-trimester spontaneous abortion with use of MMF during pregnancy.46,47 Therefore, it is recommended that patients on MMF discontinue the medication at least 6 weeks prior to conception.46
Azathioprine—Although azathioprine has been shown to provide relief of discoid lupus erythematosus symptoms,48 it currently is only utilized for refractory disease, largely due to notable side effects that particularly affect the gastrointestinal tract and liver.4 Moreover, azathioprine use during pregnancy has been associated with prematurity, congenital anomalies, fetal cytopenia, and low birth weight.49 With that said, and although not recommended, if patients decide to continue treatment, experts recommend limiting the dose to 2 mg/kg daily to reduce potential adverse events.
Category X
Oral Retinoids—According to the American Academy of Dermatology, retinoids such as isotretinoin and acitretin are considered second-line therapy for CLE.50 With that being said, there are well-documented effects on fetal development associated with oral retinoid use, including central nervous system, cardiovascular system, and craniofacial abnormalities.51 Therefore, its use is contraindicated during pregnancy. To prevent pregnancy while taking isotretinoin, patients must enroll in an online monitoring program called iPLEDGE. This program requires monthly updates by both the physician and the patient, including a negative pregnancy test every month for female patients actively taking the medication.52
The half-lives of the oral retinoids isotretinoin and acitretin are 10 to 20 hours and 50 to 60 hours, respectively.53,54 However, alcohol consumption converts acitretin into the metabolite etretinate, which can remain in tissue for up to 120 days.54,55 Therefore, women are advised to avoid alcohol while taking acitretin and avoid conception for 2 to 3 years after cessation of the medication.55 For those wishing to restart retinoids after pregnancy, studies show the medication can be safely reinstated 35 days after delivery for those interested in continued treatment.56
Thalidomide—Although low-dose thalidomide can treat refractory CLE, its use is restricted because of its known teratogenicity, most notably limb deformities.57 If prescribed thalidomide, women will need to enroll in the System for Thalidomide Education and Prescribing Safety program, similar to the iPLEDGE program, and use 2 forms of contraception when sexually active.58 Contraception should be continued for 4 weeks following the last dose of thalidomide. After this point, conception is considered safe.59
Methotrexate—For nonpregnant patients, low-dose methotrexate (MTX) with folate supplementation is a treatment option for CLE.60 However, for those who are pregnant, low-dose MTX is an abortive agent and has been associated with aminopterin syndrome, which includes skull deficits, craniofacial abnormalities, and limb deformities in live births.19,61 Therefore, MTX is not recommended in pregnancy. Of note, MTX can affect sperm; male patients also should be counselled.
Final Thoughts
Overall, it is recommended to limit medication use as much as possible in pregnancy. To reduce these exposures, it is imperative to reduce triggers that may lead to symptomatic flares of CLE. Because CLE can be triggered by sun exposure, we advise topical sunscreen to prevent CLE flares that may require additional oral medication.62,63
Various medications are considered safe for the treatment of CLE in pregnant patients (Figure 2). Based on studies in animal and clinical trials, hydroxychloroquine is considered a safe and effective medication for CLE in pregnancy and is a first-line therapy in nonpregnant patients.26,27 If flares occur, IVIG or a short course of oral steroids should be considered to manage symptoms.13,39 For those with severe flares, treatment is difficult, and personalized approaches may be necessary.
Part of the question for the childbearing population is when a patient would like to conceive. For severe cases when hydroxychloroquine is not effective as monotherapy, using a treatment that can encourage remission prior to conception attempts can be a beneficial strategy. Rituximab is an excellent example of such a therapy, as the therapeutic effect outlasts the immunosuppressive effect and therefore is unlikely to affect a future fetus.64 Thalidomide also is a potential option prior to conception, based on its short washout period and its ability to achieve notable remission rates in patients with CLE.57,59 Regardless, patients with CLE should still consult their dermatologist and rheumatologist (if applicable) prior to conception.
Patients of childbearing potential represent a population in which discussion about life goals greatly affects medication options. Having these discussions early and often allows for an open, more successful approach so that treatment regimens are not derailed at the time of conception.
Cutaneous lupus erythematosus (CLE) is a heterogeneous autoimmune disease that involves the skin. Cutaneous lupus erythematosus can be classified into various subtypes.1 These include, but are not limited to, acute CLE, subacute CLE, chronic CLE, intermittent CLE, lupus tumidus, and lupus profundus.1,2 The CLE subtypes have variable associations with systemic lupus erythematosus. For instance, some subtypes, such as acute CLE, are more strongly associated with systemic lupus erythematosus.
Treatment of CLE is similar to other autoimmune disorders. Although the US Food and Drug Administration (FDA) has not approved any treatments for CLE,3,4 the most common therapeutic options are disease-modifying antirheumatic drugs. Unfortunately, many of these treatments carry teratogenic effects. Because CLE predominantly affects women, particularly those of childbearing age, it is imperative to understand the available treatment options for those who are pregnant or considering pregnancy for an informed discussion with patients.5
For years, the gold standard when considering a medication during pregnancy was the FDA’s classification system. According to this system, medications were classified into 5 letter categories based on their potential teratogenicity, including A (no fetal risk), B (potential animal risk but inconclusive human studies), C (risk cannot be ruled out), D (evidence of fetal risk), and X (contraindicated in pregnancy). In 2014, the FDA decided to no longer use this classification system for medications approved after 2000.6 However, because many proposed treatment options for CLE were approved prior to 2001, we have summarized the commonly prescribed medications for CLE according to their prior FDA letter categories.
Treatment Options for CLE During Pregnancy
Prior to initiating systemic medications for the treatment of CLE, topical medications should be considered. Recommended treatment options include corticosteroids and calcineurin inhibitors.7 Compared with systemic medications, topical treatments carry minimal side effects, such as skin atrophy, that typically remain localized to areas of application.8 Moreover, even with extensive application, no correlation has been found between topical corticosteroid use and fetal growth,9 which suggests that topical steroids are safe in pregnancy and should be considered as a first-line treatment option for CLE. Calcineurin inhibitors also are considered safe based on their low level of absorption through the skin and are considered second-line topical treatment options in pregnancy.10
Although topical medications are effective for the treatment of CLE, many patients require the administration of systemic therapeutics for severe or refractory disease. Based on previously published reports, Figure 1 describes the current recommended systemic treatment options for CLE.11 Unfortunately, many of these medications carry teratogenic risks during pregnancy. The risks and side effects of the medications are described in detail in the following sections and summarized in the eTable.
Category B
Systemic Steroids—Systemic steroids are one of the most prescribed medications during pregnancy.12 Oral steroids have been associated with fast symptom relief, making this class of medications particularly effective during CLE flares; however, long-term management is not recommended because of the side effects, which include osteoporosis and impaired glucose metabolism.13
With low transmission across the placenta, there are 3 glucocorticoids that carry the safest profile in pregnancy: prednisone, cortisone, and hydrocortisone.14 Dexamethasone and betamethasone should be avoided, as both readily cross the placenta and increase fetal exposure.15 Although teratogenic effects have been associated with steroid use, most studies involving pregnant patients have inconclusive results. For instance, one study described an association between cleft lip/palate with in utero glucocorticoid exposure.16 However, multiple follow-up studies found no association between the two.17,18 Studies investigating the relationship between steroids and miscarriages or steroids and low birth weight also are inconclusive. Of note, if used throughout pregnancy, administration of a loading dose of glucocorticoids prior to delivery is recommended because of the increased stress brought on during labor.19
Sulfasalazine—Sulfasalazine is an immunomodulator commonly used for the treatment of inflammatory bowel disease and rheumatoid arthritis. However, studies also have shown that sulfasalazine is an effective treatment of CLE if standard treatments have failed.20,21
During pregnancy, patients exposed to sulfasalazine experienced minimal side effects despite transportation across the placenta.22 In comparison with control, pregnant women taking sulfasalazine experienced no increased risk for low fetal weight,23 congenital abnormalities,24 or spontaneous abortions.25 Of note, sulfasalazine can affect sperm, so male patients also should be counselled.
Category C
Hydroxychloroquine—Hydroxychloroquine is considered a first-line medication for those with CLE based on a symptomatic relief rate of 50% to 70%.26 For those taking hydroxychloroquine during pregnancy, the majority of studies have shown no association between the medication and adverse fetal events, including congenital abnormalities, prematurity, or spontaneous abortions.27-29 Therefore, hydroxychloroquine is considered safe in pregnancy, and those on the medication should continue standard monitoring, including retinopathy screening.30
Of note, hydroxychloroquine can be stored in tissue for weeks to months after discontinuation.5 Therefore, if patients wish to avoid hydroxychloroquine in pregnancy, one should stop taking the medication several months prior to conception.
Dapsone—Dapsone, a medication with both antimicrobial and immunomodulatory properties, is an effective second-line therapy for CLE.31 Although large-scale human trials have not been performed, multiple case reports and observational studies have supported the safe use of dapsone in pregnancy.32-34 However, there are notable side effects, including dose-dependent hemolysis, methemoglobinemia, and hypersensitivity reactions.13 Therefore, once treatment is initiated or continued, folic acid supplementation (5 mg daily) and regular serum analysis, including complete blood cell counts, are recommended in pregnant patients.19
Rituximab—Recent studies have demonstrated that rituximab can be an effective treatment of subacute and chronic CLE.35,36 Through inhibition of CD20, rituximab causes a decrease in circulating B cells and a reduced immune response. Therefore, experts recommend discontinuation of rituximab for 12 months prior to conception to reduce potential side effects to the fetus, which may include a transient reduction of circulating fetal B cells.37
If continued during pregnancy, most studies suggest discontinuation of rituximab during the third trimester, as it has been associated with neonatal infections and congenital abnormalities.19,37 However, these results are based on limited case reports, and thus robust research is needed to better understand the effect of rituximab in utero.
Intravenous Immunoglobulin Infusion—Intravenous immunoglobulin (IVIG) infusion is a well-tolerated treatment for many autoimmune disorders.38 Although not first line, limited case studies have demonstrated remission of refractory CLE following IVIG.39,40 Although no studies have directly investigated the effect of IVIG on fetal development, it has been frequently administered and well tolerated during pregnancy, especially in those with multiple sclerosis or antiphospholipid syndrome.41 Commonly reported side effects include headache and fatigue, and a rare associated side effect to be aware of is embolic events.42,43
Cyclosporine—Cyclosporine rarely is used in the treatment of localized CLE due to its extensive side-effect profile, most notably nephrotoxicity.44 However, studies have shown that cyclosporine may be efficacious if symptoms extend beyond the skin, involve multiple organs, and/or other treatments have failed.39 For those who are pregnant and wish to continue cyclosporine use, studies have associated low birth weight and premature delivery with its exposure in utero.44
Category D
Mycophenolate Mofetil—In conjunction with standard therapy, mycophenolate mofetil (MMF) is an adequate treatment of refractory CLE.45 Unfortunately, case reports have demonstrated an increased risk for fetal congenital abnormalities and first-trimester spontaneous abortion with use of MMF during pregnancy.46,47 Therefore, it is recommended that patients on MMF discontinue the medication at least 6 weeks prior to conception.46
Azathioprine—Although azathioprine has been shown to provide relief of discoid lupus erythematosus symptoms,48 it currently is only utilized for refractory disease, largely due to notable side effects that particularly affect the gastrointestinal tract and liver.4 Moreover, azathioprine use during pregnancy has been associated with prematurity, congenital anomalies, fetal cytopenia, and low birth weight.49 With that said, and although not recommended, if patients decide to continue treatment, experts recommend limiting the dose to 2 mg/kg daily to reduce potential adverse events.
Category X
Oral Retinoids—According to the American Academy of Dermatology, retinoids such as isotretinoin and acitretin are considered second-line therapy for CLE.50 With that being said, there are well-documented effects on fetal development associated with oral retinoid use, including central nervous system, cardiovascular system, and craniofacial abnormalities.51 Therefore, its use is contraindicated during pregnancy. To prevent pregnancy while taking isotretinoin, patients must enroll in an online monitoring program called iPLEDGE. This program requires monthly updates by both the physician and the patient, including a negative pregnancy test every month for female patients actively taking the medication.52
The half-lives of the oral retinoids isotretinoin and acitretin are 10 to 20 hours and 50 to 60 hours, respectively.53,54 However, alcohol consumption converts acitretin into the metabolite etretinate, which can remain in tissue for up to 120 days.54,55 Therefore, women are advised to avoid alcohol while taking acitretin and avoid conception for 2 to 3 years after cessation of the medication.55 For those wishing to restart retinoids after pregnancy, studies show the medication can be safely reinstated 35 days after delivery for those interested in continued treatment.56
Thalidomide—Although low-dose thalidomide can treat refractory CLE, its use is restricted because of its known teratogenicity, most notably limb deformities.57 If prescribed thalidomide, women will need to enroll in the System for Thalidomide Education and Prescribing Safety program, similar to the iPLEDGE program, and use 2 forms of contraception when sexually active.58 Contraception should be continued for 4 weeks following the last dose of thalidomide. After this point, conception is considered safe.59
Methotrexate—For nonpregnant patients, low-dose methotrexate (MTX) with folate supplementation is a treatment option for CLE.60 However, for those who are pregnant, low-dose MTX is an abortive agent and has been associated with aminopterin syndrome, which includes skull deficits, craniofacial abnormalities, and limb deformities in live births.19,61 Therefore, MTX is not recommended in pregnancy. Of note, MTX can affect sperm; male patients also should be counselled.
Final Thoughts
Overall, it is recommended to limit medication use as much as possible in pregnancy. To reduce these exposures, it is imperative to reduce triggers that may lead to symptomatic flares of CLE. Because CLE can be triggered by sun exposure, we advise topical sunscreen to prevent CLE flares that may require additional oral medication.62,63
Various medications are considered safe for the treatment of CLE in pregnant patients (Figure 2). Based on studies in animal and clinical trials, hydroxychloroquine is considered a safe and effective medication for CLE in pregnancy and is a first-line therapy in nonpregnant patients.26,27 If flares occur, IVIG or a short course of oral steroids should be considered to manage symptoms.13,39 For those with severe flares, treatment is difficult, and personalized approaches may be necessary.
Part of the question for the childbearing population is when a patient would like to conceive. For severe cases when hydroxychloroquine is not effective as monotherapy, using a treatment that can encourage remission prior to conception attempts can be a beneficial strategy. Rituximab is an excellent example of such a therapy, as the therapeutic effect outlasts the immunosuppressive effect and therefore is unlikely to affect a future fetus.64 Thalidomide also is a potential option prior to conception, based on its short washout period and its ability to achieve notable remission rates in patients with CLE.57,59 Regardless, patients with CLE should still consult their dermatologist and rheumatologist (if applicable) prior to conception.
Patients of childbearing potential represent a population in which discussion about life goals greatly affects medication options. Having these discussions early and often allows for an open, more successful approach so that treatment regimens are not derailed at the time of conception.
- Renner R, Sticherling M. The different faces of cutaneous lupus erythematosus. G Ital Dermatol Venereol. 2009;144:135-147.
- Kuhn A, Landmann A. The classification and diagnosis of cutaneous lupus erythematosus. J Autoimmun. 2014;48:14-19.
- Shi H, Gudjonsson J, Kahlenberg J. Treatment of cutaneous lupus erythematosus: current approaches and future strategies. Curr Opin Rheumatol. 2020;32:208-214.
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Jacobson DL, Gange SJ, Rose NR, et al. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997;84:223-243.
- Pernia S, DeMaagd G. The new pregnancy and lactation labeling rule. P T. 2016;41:713-715.
- Fanouriakis A, Kostopoulou M, Alunno A, et al. 2019 Update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. 2019;78:736-745.
- Kuhn A, Aberer E, Bata‐Csörgö Z, et al. S2k guideline for treatment of cutaneous lupus erythematosus—guided by the European Dermatology Forum (EDF) in cooperation with the European Academyof Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2017;31:389-404.
- Andersson NW, Skov L, Andersen JT. Evaluation of topical corticosteroid use in pregnancy and risk of newborns being small for gestational age and having low birth weight. JAMA Dermatol. 2021;157:788-795.
- Undre NA, Moloney FJ, Ahmadi S, et al. Skin and systemic pharmacokinetics of tacrolimus following topical application of tacrolimus ointment in adults with moderate to severe atopic dermatitis. Br J Dermatol. 2009;160:665-669.
- Xiong W, Lahita RG. Pragmatic approaches to therapy for systemic lupus erythematosus. Nat Rev Rheumatol. 2014;10:97-107.
- Kuriya B, Hernández‐Díaz S, Liu J, et al. Patterns of medication use during pregnancy in rheumatoid arthritis. Arthritis Care Res. 2011;63:721-728.
- Chang A, Werth V. Treatment of cutaneous lupus. Curr Rheumatol Rep. 2011;13:300-307.
- Beitins IZ, Bayard F, Ances IG, et al. The transplacental passage of prednisone and prednisolone in pregnancy near term. J Pediatr. 1972;81:936-945.
- Ogueh O, Johnson MR. The metabolic effect of antenatal corticosteroid therapy. Hum Reprod Update. 2000;6:169-176.
- Park-Wyllie L, Mazzotta P, Pastuszak A, et al. Birth defects after maternal exposure to corticosteroids: prospective cohort study and meta-analysis of epidemiological studies. Teratology. 2000;62:385-392.
- Bay Bjørn A, Ehrenstein V, Hundborg HH, et al. Use of corticosteroids in early pregnancy is not associated with risk of oral clefts and other congenital malformations in offspring. Am J Ther. 2014;21:73-80.
- Hviid A, Mølgaard-Nielsen D. Corticosteroid use during pregnancy and risk of orofacial clefts. CMAJ. 2011;183:796-804.
- Krause ML, Amin S, Makol A. Use of DMARDs and biologics during pregnancy and lactation in rheumatoid arthritis: what the rheumatologist needs to know. Ther Adv Musculoskelet Dis. 2014;6:169-184.
- Artuz F, Lenk N, Deniz N, et al. Efficacy of sulfasalazine in discoid lupus erythematosus. Int J Dermatol. 1996;35:746-748.
- Delaporte E, Catteau B, Sabbagh N, et al. Treatment of discoid lupus erythematosus with sulfasalazine: 11 cases [in French]. Ann Dermatol Venereol. 1997;124:151-156.
- Järnerot G, Into-Malmberg MB, Esbjörner E. Placental transfer of sulphasalazine and sulphapyridine and some of its metabolites. Scand J Gastroenterol. 1981;16:693-697.
- Norgard B, Pedersen L, Christensen LA, et al. Therapeutic drug use in women with Crohn’s disease and birth outcomes: a Danish nationwide cohort study. Am J Gastroenterol. 2007;102:1406-1413.
- Nørgård B, Czeizel AE, Rockenbauer M, et al. Population-based case control study of the safety of sulfasalazine use during pregnancy. Aliment Pharmacol Ther. 2001;15:483-486.
- Rahimi R, Nikfar S, Rezaie A, et al. Pregnancy outcome in women with inflammatory bowel disease following exposure to 5-aminosalicylic acid drugs: a meta-analysis. Reprod Toxicol. 2008;25:271-275.
- Callen JP. Chronic cutaneous lupus erythematosus: clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol. 1982;118:412-416.
- Buchanan NM, Toubi E, Khamashta MA, et al. Hydroxychloroquine and lupus pregnancy: review of a series of 36 cases. Ann Rheum Dis. 1996;55:486-488.
- Costedoat‐Chalumeau N, Amoura Z, Duhaut P, et al. Safety of hydroxychloroquine in pregnant patients with connective tissue diseases: a study of one hundred thirty‐three cases compared with a control group. Arthritis Rheum. 2003;48:3207-3211.
- Sperber K, Hom C, Chao CP, et al. Systematic review of hydroxychloroquine use in pregnant patients with autoimmune diseases. Pediatr Rheumatol Online J. 2009;7:9.
- Marmor MF, Carr RE, Easterbrook M, et al. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy: a report by the American Academy of Ophthalmology. Ophthalmology. 2002;109:1377-1382.
- Klebes M, Wutte N, Aberer E. Dapsone as second-line treatment for cutaneous lupus erythematosus? a retrospective analysis of 34 patients and a review of the literature. Dermatology. 2016;232:91-96.
- Tuffanelli DL. Successful pregnancy in a patient with dermatitis herpetiformis treated with low-dose dapsone. Arch Dermatol. 1982;118:876.
- Varghese L, Viswabandya A, Mathew AJ. Dapsone, danazol, and intrapartum splenectomy in refractory ITP complicating pregnancy. Indian J Med Sci. 2008;62:452-455.
- Kahn G. Dapsone is safe during pregnancy. J Am Acad Dermatol. 1985;13:838-839.
- Quelhas da Costa R, Aguirre-Alastuey ME, Isenberg DA, et al. Assessment of response to B-cell depletion using rituximab in cutaneous lupus erythematosus. JAMA Dermatol. 2018;154:1432-1440.
- Alsanafi S, Kovarik C, Mermelstein A, et al. Rituximab in thetreatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506.
- Fernandez AP, Kerdel FA. The use of i.v. IG therapy in dermatology. Dermatol Ther. 2007;20:288-305.
- Kuhn A, Ruland V, Bonsmann G. Cutaneous lupus erythematosus: update of therapeutic options part II. J Am Acad Dermatol. 2011;65:E195-E213.
- Singh H, Naidu G, Sharma A. Intravenous immunoglobulin for the rescue in refractory cutaneous lupus. Indian Dermatol Online J. 2020;11:1003-1004.
- Clark AL. Clinical uses of intravenous immunoglobulin in pregnancy. Clin Obstet Gynecol. 1999;42:368-380.
- Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med. 2001;345:747-755.
- Woodruff RK, Grigg AP, Firkin FC, et al. Fatal thrombotic events during treatment of autoimmune thrombocytopenia with intravenous immunoglobulin in elderly patients. Lancet. 1986;2:217-218.
- Paziana K, Del Monaco M, Cardonick E, et al. Ciclosporin use during pregnancy. Drug Saf. 2013;36:279-294.
- Gammon B, Hansen C, Costner MI. Efficacy of mycophenolate mofetil in antimalarial-resistant cutaneous lupus erythematosus. J Am Acad Dermatol. 2010;65:717-721.e2.
- Abdulaziz HM, Shemies RS, Taman M, et al. Fetal proximal and distal limb anomalies following exposure to mycophenolate mofetil during pregnancy: a case report and review of the literature. Lupus. 2021;30:1522-1525.
- Pisoni CN, D’Cruz DP. The safety of mycophenolate mofetil in pregnancy. Exp Opin Drug Saf. 2008;7:219-222.
- Ashinoff R, Werth VP, Franks AG. Resistant discoid lupus erythematosus of palms and soles: successful treatment with azathioprine. J Am Acad Dermatol. 1988;19:961-965. doi:10.1016/S0190-9622(88)70259-5
- Goldstein LH, Dolinsky G, Greenberg R, et al. Pregnancy outcome of women exposed to azathioprine during pregnancy. Birth Defects Res A Clin Mol Teratol. 2007;79:696-701.
- Drake LA, Dinehart SM, Farmer ER, et al. Guidelines of care for cutaneous lupus erythematosus. American Academy of Dermatology. J Am Acad Dermatol. 1996;34:830-836.
- Sladden MJ, Harman KE. What is the chance of a normal pregnancy in a woman whose fetus has been exposed to isotretinoin? Arch Dermatol. 2007;143:1187-1188.
- Shin J, Cheetham TC, Wong L, et al. The impact of the iPLEDGE program on isotretinoin fetal exposure in an integrated health care system. J Am Acad Dermatol. 2011;65:1117-1125.
- Brazzell RK, Colburn WA. Pharmacokinetics of the retinoids isotretinoin and etretinate. J Am Acad Dermatol. 1982;6:643-651.
- Pilkington T, Brogden RN. Acitretin: a review of its pharmacology and therapeutic use. Drugs. 1992;43:597-627.
- Gronhoj Larsen F, Steinkjer B, Jakobsen P, et al. Acitretin is converted to etretinate only during concomitant alcohol intake. Br J Dermatol. 2000;143:1164-1169.
- Jajoria H, Mysore V. Washout period for pregnancy post isotretinoin therapy. Indian Dermatol Online J. 2020;11:239-242.
- Cortés-Hernández J, Torres-Salido M, Castro-Marrero J, et al. Thalidomide in the treatment of refractory cutaneous lupus erythematosus: prognostic factors of clinical outcome. Br J Dermatol. 2012;166:616-623.
- Zeldis JB, Williams BA, Thomas SD, et al. S.T.E.P.S.™: a comprehensive program for controlling and monitoring access to thalidomide. Clin Ther. 1999;21:319-330.
- C.S. Mott Children’s Hospital. University of Michigan Health. Thalidomide. Updated March 26, 2020. Accessed January 14, 2022. https://www.mottchildren.org/health-library/d04331a1
- Boehm IB, Boehm GA, Bauer R. Management of cutaneous lupus erythematosus with low-dose methotrexate: indication for modulation of inflammatory mechanisms. Rheumatol Int. 1998;18:59-62.
- Buckley LM, Bullaboy CA, Leichtman L, et al. Multiple congenital anomalies associated with weekly low‐dose methotrexate treatment of the mother. Arthritis Rheum. 1997;40:971-973.
- Foering K, Okawa J, Rose M, et al. Characterization of photosensitivity and poor quality of life in lupus. J Invest Dermatol. 2010;130(suppl):S10.
- Kuhn A, Herrmann M, Kleber S, et al. Accumulation of apoptotic cells in the epidermis of patients with cutaneous lupus erythematosus after ultraviolet irradiation. Arthritis Rheum. 2006;54:939-950.
- Lake EP, Huang Y, Aronson IK. Rituximab treatment of pemphigus in women of childbearing age: experience with two patients. J Dermatol Treat. 2017;28:751-752.
- Renner R, Sticherling M. The different faces of cutaneous lupus erythematosus. G Ital Dermatol Venereol. 2009;144:135-147.
- Kuhn A, Landmann A. The classification and diagnosis of cutaneous lupus erythematosus. J Autoimmun. 2014;48:14-19.
- Shi H, Gudjonsson J, Kahlenberg J. Treatment of cutaneous lupus erythematosus: current approaches and future strategies. Curr Opin Rheumatol. 2020;32:208-214.
- Winkelmann RR, Kim GK, Del Rosso JQ. Treatment of cutaneous lupus erythematosus: review and assessment of treatment benefits based on Oxford Centre for Evidence-based Medicine criteria. J Clin Aesthet Dermatol. 2013;6:27-38.
- Jacobson DL, Gange SJ, Rose NR, et al. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997;84:223-243.
- Pernia S, DeMaagd G. The new pregnancy and lactation labeling rule. P T. 2016;41:713-715.
- Fanouriakis A, Kostopoulou M, Alunno A, et al. 2019 Update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann Rheum Dis. 2019;78:736-745.
- Kuhn A, Aberer E, Bata‐Csörgö Z, et al. S2k guideline for treatment of cutaneous lupus erythematosus—guided by the European Dermatology Forum (EDF) in cooperation with the European Academyof Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2017;31:389-404.
- Andersson NW, Skov L, Andersen JT. Evaluation of topical corticosteroid use in pregnancy and risk of newborns being small for gestational age and having low birth weight. JAMA Dermatol. 2021;157:788-795.
- Undre NA, Moloney FJ, Ahmadi S, et al. Skin and systemic pharmacokinetics of tacrolimus following topical application of tacrolimus ointment in adults with moderate to severe atopic dermatitis. Br J Dermatol. 2009;160:665-669.
- Xiong W, Lahita RG. Pragmatic approaches to therapy for systemic lupus erythematosus. Nat Rev Rheumatol. 2014;10:97-107.
- Kuriya B, Hernández‐Díaz S, Liu J, et al. Patterns of medication use during pregnancy in rheumatoid arthritis. Arthritis Care Res. 2011;63:721-728.
- Chang A, Werth V. Treatment of cutaneous lupus. Curr Rheumatol Rep. 2011;13:300-307.
- Beitins IZ, Bayard F, Ances IG, et al. The transplacental passage of prednisone and prednisolone in pregnancy near term. J Pediatr. 1972;81:936-945.
- Ogueh O, Johnson MR. The metabolic effect of antenatal corticosteroid therapy. Hum Reprod Update. 2000;6:169-176.
- Park-Wyllie L, Mazzotta P, Pastuszak A, et al. Birth defects after maternal exposure to corticosteroids: prospective cohort study and meta-analysis of epidemiological studies. Teratology. 2000;62:385-392.
- Bay Bjørn A, Ehrenstein V, Hundborg HH, et al. Use of corticosteroids in early pregnancy is not associated with risk of oral clefts and other congenital malformations in offspring. Am J Ther. 2014;21:73-80.
- Hviid A, Mølgaard-Nielsen D. Corticosteroid use during pregnancy and risk of orofacial clefts. CMAJ. 2011;183:796-804.
- Krause ML, Amin S, Makol A. Use of DMARDs and biologics during pregnancy and lactation in rheumatoid arthritis: what the rheumatologist needs to know. Ther Adv Musculoskelet Dis. 2014;6:169-184.
- Artuz F, Lenk N, Deniz N, et al. Efficacy of sulfasalazine in discoid lupus erythematosus. Int J Dermatol. 1996;35:746-748.
- Delaporte E, Catteau B, Sabbagh N, et al. Treatment of discoid lupus erythematosus with sulfasalazine: 11 cases [in French]. Ann Dermatol Venereol. 1997;124:151-156.
- Järnerot G, Into-Malmberg MB, Esbjörner E. Placental transfer of sulphasalazine and sulphapyridine and some of its metabolites. Scand J Gastroenterol. 1981;16:693-697.
- Norgard B, Pedersen L, Christensen LA, et al. Therapeutic drug use in women with Crohn’s disease and birth outcomes: a Danish nationwide cohort study. Am J Gastroenterol. 2007;102:1406-1413.
- Nørgård B, Czeizel AE, Rockenbauer M, et al. Population-based case control study of the safety of sulfasalazine use during pregnancy. Aliment Pharmacol Ther. 2001;15:483-486.
- Rahimi R, Nikfar S, Rezaie A, et al. Pregnancy outcome in women with inflammatory bowel disease following exposure to 5-aminosalicylic acid drugs: a meta-analysis. Reprod Toxicol. 2008;25:271-275.
- Callen JP. Chronic cutaneous lupus erythematosus: clinical, laboratory, therapeutic, and prognostic examination of 62 patients. Arch Dermatol. 1982;118:412-416.
- Buchanan NM, Toubi E, Khamashta MA, et al. Hydroxychloroquine and lupus pregnancy: review of a series of 36 cases. Ann Rheum Dis. 1996;55:486-488.
- Costedoat‐Chalumeau N, Amoura Z, Duhaut P, et al. Safety of hydroxychloroquine in pregnant patients with connective tissue diseases: a study of one hundred thirty‐three cases compared with a control group. Arthritis Rheum. 2003;48:3207-3211.
- Sperber K, Hom C, Chao CP, et al. Systematic review of hydroxychloroquine use in pregnant patients with autoimmune diseases. Pediatr Rheumatol Online J. 2009;7:9.
- Marmor MF, Carr RE, Easterbrook M, et al. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy: a report by the American Academy of Ophthalmology. Ophthalmology. 2002;109:1377-1382.
- Klebes M, Wutte N, Aberer E. Dapsone as second-line treatment for cutaneous lupus erythematosus? a retrospective analysis of 34 patients and a review of the literature. Dermatology. 2016;232:91-96.
- Tuffanelli DL. Successful pregnancy in a patient with dermatitis herpetiformis treated with low-dose dapsone. Arch Dermatol. 1982;118:876.
- Varghese L, Viswabandya A, Mathew AJ. Dapsone, danazol, and intrapartum splenectomy in refractory ITP complicating pregnancy. Indian J Med Sci. 2008;62:452-455.
- Kahn G. Dapsone is safe during pregnancy. J Am Acad Dermatol. 1985;13:838-839.
- Quelhas da Costa R, Aguirre-Alastuey ME, Isenberg DA, et al. Assessment of response to B-cell depletion using rituximab in cutaneous lupus erythematosus. JAMA Dermatol. 2018;154:1432-1440.
- Alsanafi S, Kovarik C, Mermelstein A, et al. Rituximab in thetreatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117:1499-1506.
- Fernandez AP, Kerdel FA. The use of i.v. IG therapy in dermatology. Dermatol Ther. 2007;20:288-305.
- Kuhn A, Ruland V, Bonsmann G. Cutaneous lupus erythematosus: update of therapeutic options part II. J Am Acad Dermatol. 2011;65:E195-E213.
- Singh H, Naidu G, Sharma A. Intravenous immunoglobulin for the rescue in refractory cutaneous lupus. Indian Dermatol Online J. 2020;11:1003-1004.
- Clark AL. Clinical uses of intravenous immunoglobulin in pregnancy. Clin Obstet Gynecol. 1999;42:368-380.
- Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med. 2001;345:747-755.
- Woodruff RK, Grigg AP, Firkin FC, et al. Fatal thrombotic events during treatment of autoimmune thrombocytopenia with intravenous immunoglobulin in elderly patients. Lancet. 1986;2:217-218.
- Paziana K, Del Monaco M, Cardonick E, et al. Ciclosporin use during pregnancy. Drug Saf. 2013;36:279-294.
- Gammon B, Hansen C, Costner MI. Efficacy of mycophenolate mofetil in antimalarial-resistant cutaneous lupus erythematosus. J Am Acad Dermatol. 2010;65:717-721.e2.
- Abdulaziz HM, Shemies RS, Taman M, et al. Fetal proximal and distal limb anomalies following exposure to mycophenolate mofetil during pregnancy: a case report and review of the literature. Lupus. 2021;30:1522-1525.
- Pisoni CN, D’Cruz DP. The safety of mycophenolate mofetil in pregnancy. Exp Opin Drug Saf. 2008;7:219-222.
- Ashinoff R, Werth VP, Franks AG. Resistant discoid lupus erythematosus of palms and soles: successful treatment with azathioprine. J Am Acad Dermatol. 1988;19:961-965. doi:10.1016/S0190-9622(88)70259-5
- Goldstein LH, Dolinsky G, Greenberg R, et al. Pregnancy outcome of women exposed to azathioprine during pregnancy. Birth Defects Res A Clin Mol Teratol. 2007;79:696-701.
- Drake LA, Dinehart SM, Farmer ER, et al. Guidelines of care for cutaneous lupus erythematosus. American Academy of Dermatology. J Am Acad Dermatol. 1996;34:830-836.
- Sladden MJ, Harman KE. What is the chance of a normal pregnancy in a woman whose fetus has been exposed to isotretinoin? Arch Dermatol. 2007;143:1187-1188.
- Shin J, Cheetham TC, Wong L, et al. The impact of the iPLEDGE program on isotretinoin fetal exposure in an integrated health care system. J Am Acad Dermatol. 2011;65:1117-1125.
- Brazzell RK, Colburn WA. Pharmacokinetics of the retinoids isotretinoin and etretinate. J Am Acad Dermatol. 1982;6:643-651.
- Pilkington T, Brogden RN. Acitretin: a review of its pharmacology and therapeutic use. Drugs. 1992;43:597-627.
- Gronhoj Larsen F, Steinkjer B, Jakobsen P, et al. Acitretin is converted to etretinate only during concomitant alcohol intake. Br J Dermatol. 2000;143:1164-1169.
- Jajoria H, Mysore V. Washout period for pregnancy post isotretinoin therapy. Indian Dermatol Online J. 2020;11:239-242.
- Cortés-Hernández J, Torres-Salido M, Castro-Marrero J, et al. Thalidomide in the treatment of refractory cutaneous lupus erythematosus: prognostic factors of clinical outcome. Br J Dermatol. 2012;166:616-623.
- Zeldis JB, Williams BA, Thomas SD, et al. S.T.E.P.S.™: a comprehensive program for controlling and monitoring access to thalidomide. Clin Ther. 1999;21:319-330.
- C.S. Mott Children’s Hospital. University of Michigan Health. Thalidomide. Updated March 26, 2020. Accessed January 14, 2022. https://www.mottchildren.org/health-library/d04331a1
- Boehm IB, Boehm GA, Bauer R. Management of cutaneous lupus erythematosus with low-dose methotrexate: indication for modulation of inflammatory mechanisms. Rheumatol Int. 1998;18:59-62.
- Buckley LM, Bullaboy CA, Leichtman L, et al. Multiple congenital anomalies associated with weekly low‐dose methotrexate treatment of the mother. Arthritis Rheum. 1997;40:971-973.
- Foering K, Okawa J, Rose M, et al. Characterization of photosensitivity and poor quality of life in lupus. J Invest Dermatol. 2010;130(suppl):S10.
- Kuhn A, Herrmann M, Kleber S, et al. Accumulation of apoptotic cells in the epidermis of patients with cutaneous lupus erythematosus after ultraviolet irradiation. Arthritis Rheum. 2006;54:939-950.
- Lake EP, Huang Y, Aronson IK. Rituximab treatment of pemphigus in women of childbearing age: experience with two patients. J Dermatol Treat. 2017;28:751-752.
Practice Points
- Patients should consult their primary dermatologist when discussing medication options for cutaneous lupus erythematosus (CLE) prior to pregnancy.
- Hydroxychloroquine is a first-line medication for maintenance treatment of CLE, while oral steroids are effective for CLE flares in pregnancy. Second-line medications include dapsone and intravenous immunoglobulin. These classes of medications are considered safe in pregnancy.
- Cutaneous lupus erythematosus medications contraindicated in pregnancy include oral retinoids, mycophenolate mofetil, thalidomide, and methotrexate.
The Role of Inpatient Dermatology Consultations
Dermatology is an often-underutilized resource in the hospital setting. As the health care landscape has evolved, so has the role of the inpatient dermatologist.1-3 Structural changes in the health system and advances in therapies have shifted dermatology from an admitting service to an almost exclusively outpatient practice. Improved treatment modalities led to decreases in the number of patients requiring admission for chronic dermatoses, and outpatient clinics began offering therapies once limited to hospitals.1,4 Inpatient dermatology consultations emerged and continue to have profound effects on hospitalized patients regardless of their reason for admission.1-11
Inpatient dermatologists supply knowledge in areas primary medical teams lack, and there is evidence that dermatology consultations improve the quality of care while decreasing cost.2,5-7 Establishing correct diagnoses, preventing exposure to unnecessary medications, and reducing hospitalization duration and readmission rates are a few ways dermatology consultations positively impact hospitalized patients.2,5-7,9,10 This study highlights the role of the dermatologist in the care of hospitalized patients at a large academic medical center in an urban setting and reveals how consultation supports the efficiency and efficacy of other services.
Materials and Methods
Study Design—This single-institution, cross-sectional retrospective study included all hospitalized patients at the Thomas Jefferson University Hospital (Philadelphia, Pennsylvania), who received an inpatient dermatology consultation completed by physicians of Jefferson Dermatology Associates between January 1, 2019, and December 31, 2019. The institutional review board at Thomas Jefferson University approved this study.
Data Collection—A list of all inpatient dermatology consultations in 2019 was provided by Jefferson Dermatology Associates. Through a retrospective chart review, data regarding the consultations were collected from the electronic medical record (Epic Systems) and recorded into the Research Electronic Data Capture system. Data on patient demographics, the primary medical team, the dermatology evaluation, and the hospital course of the patient were collected.
Results
Patient Characteristics—Dermatology received 253 inpatient consultation requests during this time period; 53% of patients were female and 47% were male, with a mean age of 55 years. Most patients were White (57%), while 34% were Black. Five percent and 4% of patients were Asian and Hispanic or Latino, respectively (Table 1). The mean duration of hospitalization for all patients was 15 days, and the average number of days to discharge following the first encounter with dermatology was 10 days.
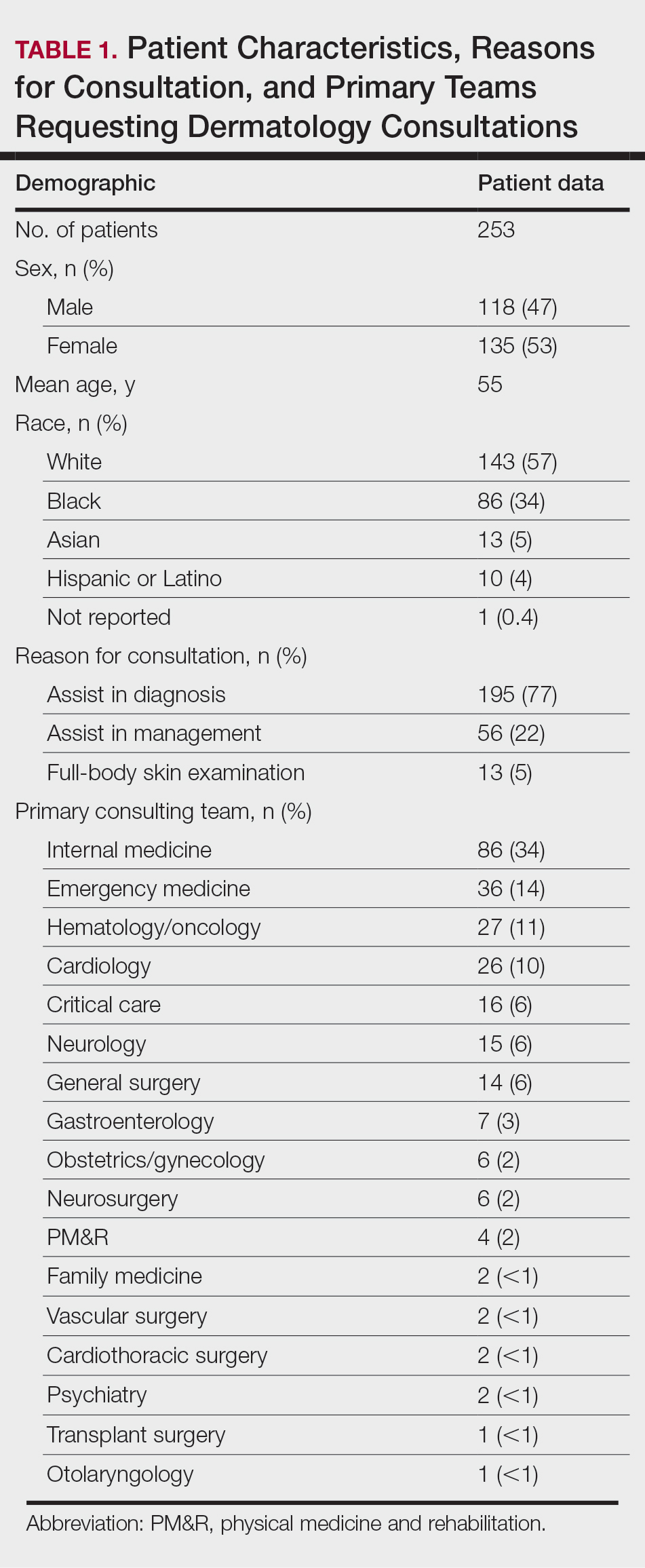
Requesting Team and Reason for Consultation—Internal medicine consulted dermatology most frequently (34% of all consultations), followed by emergency medicine (14%) and a variety of other services (Table 1). Most dermatology consultations were placed to assist in achieving a diagnosis of a cutaneous condition (77%), while a minority were to assist in the management of a previously diagnosed disease (22%). A small fraction of consultations (5%) were to complete full-body skin examinations (FBSEs) to rule out infection or malignancy in candidates for organ transplantation, left ventricular assist devices, or certain chemotherapies. One FBSE was conducted to search for a primary tumor in a patient diagnosed with metastatic melanoma.
Most Common Final Diagnoses and Consultation Impact—Table 2 lists the most common final diagnosis categories, as well as the effects of the consultation on diagnosis, management, biopsies, hospitalization, and clinical improvement as documented by the primary medical provider. The most common final diagnoses were inflammatory and autoimmune (39%), such as contact dermatitis and seborrheic dermatitis; infectious (23%), such as varicella (primary or zoster) and bacterial furunculosis; drug reactions (20%), such as morbilliform drug eruptions; vascular (8%), such as vasculitis and calciphylaxis; neoplastic (7%), such as keratinocyte carcinomas and leukemia cutis; and other (15%), such as xerosis, keratosis pilaris, and miliaria rubra.
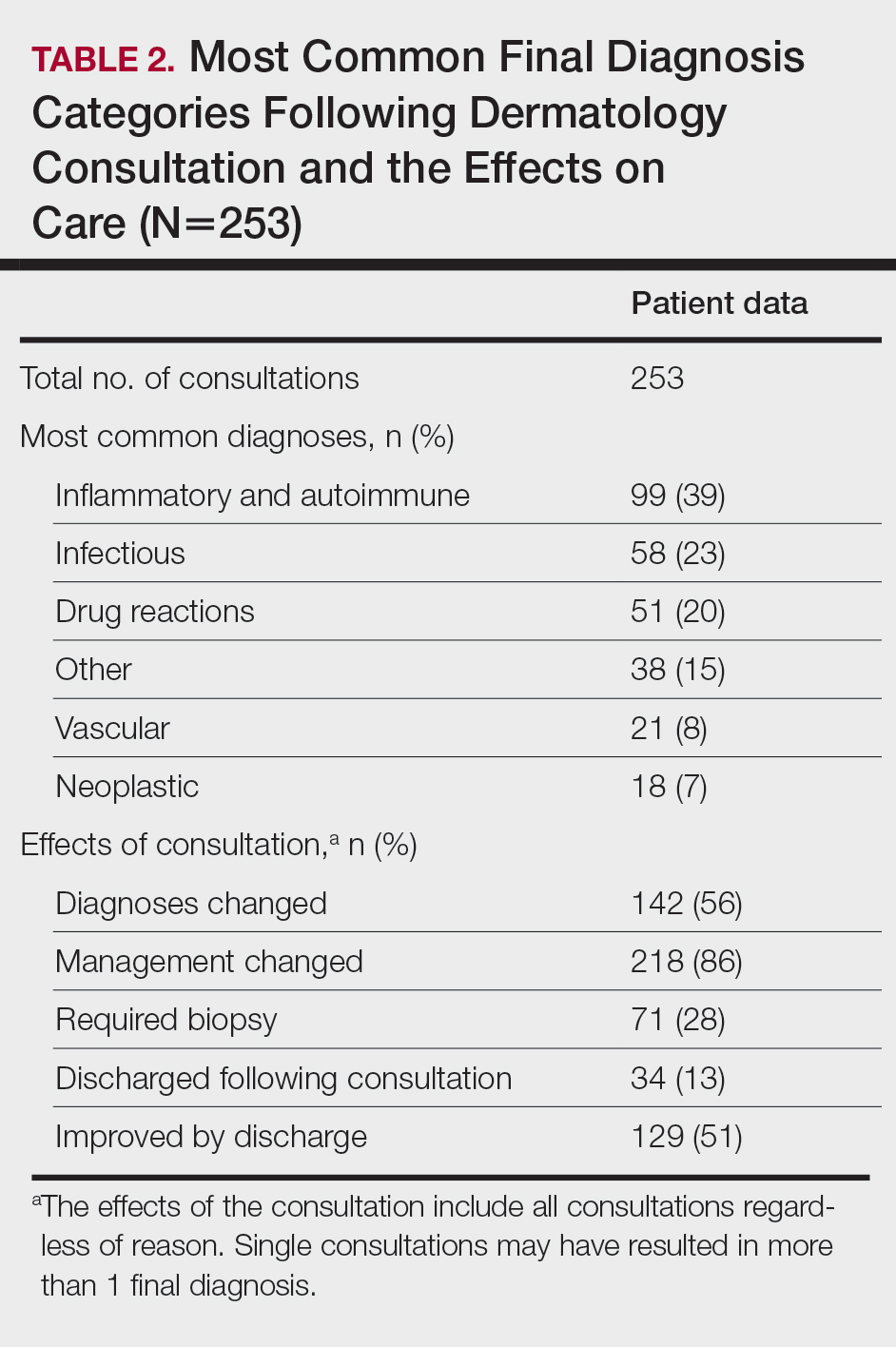
Impact on Diagnosis—Fifty-six percent of all consultations resulted in a change in diagnosis. When dermatology was consulted specifically to assist in the diagnosis of a patient (195 consultations), the working diagnosis of the primary team was changed 69% of the time. Thirty-five of these consultation requests had no preliminary diagnosis, and the primary team listed the working diagnosis as either rash or a morphologic description of the lesion(s). Sixty-three percent of suspected drug eruptions ended with a diagnosis of a form of drug eruption, while 20% of consultations for suspected cellulitis or bacterial infections were confirmed to be cellulitis or soft tissue infections.
Impact on Management—Regardless of the reason for the consultation, most consultations (86%) resulted in a change in management. The remaining 14% consisted of FBSEs with benign findings; cases of cutaneous metastases and leukemia cutis managed by oncology; as well as select cases of purpura fulminans, postfebrile desquamation, and postinflammatory hyperpigmentation.
Changes in management included alterations in medications, requests for additional laboratory work or imaging, additional consultation requests, biopsies, or specific wound care instructions. Seventy-five percent of all consultations were given specific medication recommendations by dermatology. Most (61%) were recommended to be given a topical steroid, antibiotic, or both. However, 45% of all consultations were recommended to initiate a systemic medication, most commonly antihistamines, antibiotics, steroids, antivirals, or immunomodulators. Dermatology recommended discontinuing specific medications in 16% of all consultations, with antibiotics being the most frequent culprit (17 antibiotics discontinued), owing to drug eruptions or misdiagnosed infections. Vancomycin, piperacillin-tazobactam, and trimethoprim-sulfamethoxazole were the most frequently discontinued antibiotics.
Dermatology was consulted for assistance in management of previously diagnosed cutaneous conditions 56 times (22% of all consultations), often regarding complicated cases of hidradenitis suppurativa (9 cases), pyoderma gangrenosum (5 cases), bullous pemphigoid (4 cases), or erythroderma (4 cases). Most of these cases required a single dermatology encounter to provide recommendations (71%), and 21% required 1 additional follow-up. Sixty-three percent of patients consulted for management assistance were noted to have improvement in their cutaneous condition by time of discharge, as documented by the primary provider in the medical record.
Twenty-eight percent of all consultations required at least 1 biopsy. Seventy-two percent of all biopsies were consistent with the dermatologist’s working diagnosis or highest-ranked differential diagnosis, and 16% of biopsy results were consistent with the second- or third-ranked diagnosis. The primary teams requested a biopsy 38 times to assist in diagnosis, as documented in the progress note or consultation request. Only 21 of these consultations (55% of requests) received at least 1 biopsy, as the remaining consultations did not require a biopsy to establish a diagnosis. The most common final diagnoses of consultations receiving biopsies included drug eruptions (5), leukemia cutis (4), vasculopathies (4), vasculitis (4), and calciphylaxis (3).
Impact on Hospitalization and Efficacy—Dermatology performed 217 consultations regarding patients already admitted to the hospital, and 92% remained hospitalized either due to comorbidities or complicated cutaneous conditions following the consultation. The remaining 8% were cleared for discharge. Dermatology received 36 consultation requests from emergency medicine physicians. Fifty-three percent of these patients were admitted, while the remaining 47% were discharged from the emergency department or its observation unit following evaluation.
Fifty-one percent of all consultations were noted to have improvement in their cutaneous condition by the time of discharge, as noted in the physical examination, progress note, or discharge summary of the primary team. Thirty percent of cases remained stable, where improvement was not noted in in the medical record. Most of these cases involved keratinocyte carcinomas scheduled for outpatient excision, benign melanocytic nevi found on FBSE, and benign etiologies that led to immediate discharge following consultation. Three percent of all consultations were noted to have worsened following consultation, including cases of calciphylaxis, vasculopathies, and purpura fulminans, as well as patients who elected for palliative care and hospice. The cutaneous condition by the time of discharge could not be determined from the medical record in 16% of all consultations.
Eighty-five percent of all consultations required a single encounter with dermatology. An additional 10% required a single follow-up with dermatology, while only 5% of patients required 3 or more encounters. Notably, these cases included patients with 1 or more severe cutaneous diseases, such as Sweet syndrome, calciphylaxis, Stevens-Johnson syndrome/toxic epidermal necrolysis, and hidradenitis suppurativa.
Comment
Although dermatology often is viewed as an outpatient specialty, this study provides a glimpse into the ways inpatient dermatology consultations optimize the care of hospitalized patients. Most consultations involved assistance in diagnosing an unknown condition, but several regarded pre-existing skin disorders requiring management aid. As a variety of medical specialties requested consultations, dermatology was able to provide care to a diverse group of patients with conditions varying in complexity and severity. Several specialties benefited from niche dermatologic expertise: hematology and oncology frequently requested dermatology to assist in diagnosis and management of the toxic effects of chemotherapy, cutaneous metastasis, or suspected cutaneous infections in immunocompromised patients. Cardiology patients were frequently evaluated for potential malignancy or infection prior to heart transplantation and initiation of antirejection immunosuppressants. Dermatology was consulted to differentiate cutaneous manifestations of critical illness from underlying systemic disease in the intensive care unit, and patients presenting to the emergency department often were examined to determine if hospital admission was necessary, with 47% of these consultations resulting in a discharge following evaluation by a dermatologist.
Our results were consistent with prior studies1,5,6 that have reported frequent changes in final diagnosis following dermatology consultation, with 69% of working diagnoses changed in this study when consultation was requested for diagnostic assistance. When dermatology was consulted for diagnostic assistance, several of these cases lacked a preliminary differential diagnosis. Although the absence of a documented differential diagnosis may not necessarily reflect a lack of suspicion for a particular etiology, 86% of all consultations included a ranked differential or working diagnosis either in the consultation request or progress note prior to consultation. The final diagnoses of consultations without a preliminary diagnosis varied from the mild and localized to systemic and severe, further suggesting these cases reflected knowledge gaps of the primary medical team.
Integration of dermatology into the care of hospitalized patients could provide an opportunity for education of primary medical teams. With frequent consultation, primary medical teams may become more comfortable diagnosing and managing common cutaneous conditions specific to their specialty or extended hospitalizations.
Several consultations were requested to aid in management of cases of hidradenitis suppurativa, pyoderma gangrenosum, or bullous pemphigoid that either failed outpatient therapy or were complicated by superinfections. Despite the ranges in complexity, the majority of all consultations required a single encounter and led to improvement by the time of discharge, demonstrating the efficacy and efficiency of inpatient dermatologists.
Dermatology consultations often led to changes in management involving medications and additional workup. Changes in management also extended to specific wound care instructions provided by dermatology, as expected for cases of Stevens-Johnson syndrome/toxic epidermal necrolysis, Sweet syndrome, hidradenitis suppurativa, and pyoderma gangrenosum. However, patients with the sequelae of extended hospitalizations, such as chronic wounds, pressure ulcers, and edema bullae, also benefited from this expertise.
When patients required a biopsy, the final diagnoses were consistent with the dermatologist’s number one differential diagnosis or top 3 differential diagnoses 72% and 88% of the time, respectively. Only 55% of cases where the primary team requested a biopsy ultimately required a biopsy, as many involved clinical diagnoses such as urticaria. Not only was dermatology accurate in their preliminary diagnoses, but they decreased cost and morbidity by avoiding unnecessary procedures.
This study provided additional evidence to support the integration of dermatology into the hospital setting for the benefit of patients, primary medical teams, and hospital systems. Dermatology offers high-value care through the efficient diagnosis and management of hospitalized patients, which contributes to decreased cost and improved outcomes.2,5-7,9,10 This study highlighted lesser-known areas of impact, such as the various specialty-specific services dermatology provides as well as the high rates of reported improvement following consultation. Future studies should continue to explore the field’s unique impact on hospitalized medicine as well as other avenues of care delivery, such as telemedicine, that may encourage dermatologists to participate in consultations and increase the volume of patients who may benefit from their care.
- Madigan LM, Fox LP. Where are we now with inpatient consultative dermatology?: assessing the value and evolution of this subspecialty over the past decade. J Am Acad Dermatol. 2019;80:1804-1808. doi:10.1016/j.jaad.2019.01.031
- Noe MH, Rosenbach M. Inpatient dermatologists—crucial for the management of skin diseases in hospitalized patients [editorial]. JAMA Dermatol. 2018;154:524-525. doi:10.1001/jamadermatol.2017.6195
- Strowd LC. Inpatient dermatology: a paradigm shift in the management of skin disease in the hospital. Br J Dermatol. 2019;180:966-967. doi:10.1111/bjd.17778
- Kirsner RS, Yang DG, Kerdel FA. The changing status of inpatient dermatology at American academic dermatology programs. J Am Acad Dermatol. 1999;40:755-757. doi:10.1016/s0190-9622(99)70158-1
- Kroshinsky D, Cotliar J, Hughey LC, et al. Association of dermatology consultation with accuracy of cutaneous disorder diagnoses in hospitalized patients: a multicenter analysis. JAMA Dermatol. 2016;152:477-480. doi:10.1001/jamadermatol.2015.5098
- Ko LN, Garza-Mayers AC, St John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis. JAMA Dermatol. 2018;154:529-533. doi:10.1001/jamadermatol.2017.6196
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543. doi:10.1001/jamadermatol.2017.6197
- Milani-Nejad N, Zhang M, Kaffenberger BH. Association of dermatology consultations with patient care outcomes in hospitalized patients with inflammatory skin diseases. JAMA Dermatol. 2017;153:523-528. doi:10.1001/jamadermatol.2016.6130
- Imadojemu S, Rosenbach M. Dermatologists must take an active role in the diagnosis of cellulitis. JAMA Dermatol. 2017;153:134-135. doi:10.1001/jamadermatol.2016.4230
- Hughey LC. The impact dermatologists can have on misdiagnosis of cellulitis and overuse of antibiotics: closing the gap. JAMA Dermatol. 2014;150:1061-1062. doi:10.1001/jamadermatol.2014.1164
- Ko LN, Kroshinsky D. Dermatology hospitalists: a multicenter survey study characterizing the infrastructure of consultative dermatology in select American hospitals. Int J Dermatol. 2018;57:553-558. doi:10.1111/ijd.13939
Dermatology is an often-underutilized resource in the hospital setting. As the health care landscape has evolved, so has the role of the inpatient dermatologist.1-3 Structural changes in the health system and advances in therapies have shifted dermatology from an admitting service to an almost exclusively outpatient practice. Improved treatment modalities led to decreases in the number of patients requiring admission for chronic dermatoses, and outpatient clinics began offering therapies once limited to hospitals.1,4 Inpatient dermatology consultations emerged and continue to have profound effects on hospitalized patients regardless of their reason for admission.1-11
Inpatient dermatologists supply knowledge in areas primary medical teams lack, and there is evidence that dermatology consultations improve the quality of care while decreasing cost.2,5-7 Establishing correct diagnoses, preventing exposure to unnecessary medications, and reducing hospitalization duration and readmission rates are a few ways dermatology consultations positively impact hospitalized patients.2,5-7,9,10 This study highlights the role of the dermatologist in the care of hospitalized patients at a large academic medical center in an urban setting and reveals how consultation supports the efficiency and efficacy of other services.
Materials and Methods
Study Design—This single-institution, cross-sectional retrospective study included all hospitalized patients at the Thomas Jefferson University Hospital (Philadelphia, Pennsylvania), who received an inpatient dermatology consultation completed by physicians of Jefferson Dermatology Associates between January 1, 2019, and December 31, 2019. The institutional review board at Thomas Jefferson University approved this study.
Data Collection—A list of all inpatient dermatology consultations in 2019 was provided by Jefferson Dermatology Associates. Through a retrospective chart review, data regarding the consultations were collected from the electronic medical record (Epic Systems) and recorded into the Research Electronic Data Capture system. Data on patient demographics, the primary medical team, the dermatology evaluation, and the hospital course of the patient were collected.
Results
Patient Characteristics—Dermatology received 253 inpatient consultation requests during this time period; 53% of patients were female and 47% were male, with a mean age of 55 years. Most patients were White (57%), while 34% were Black. Five percent and 4% of patients were Asian and Hispanic or Latino, respectively (Table 1). The mean duration of hospitalization for all patients was 15 days, and the average number of days to discharge following the first encounter with dermatology was 10 days.
Requesting Team and Reason for Consultation—Internal medicine consulted dermatology most frequently (34% of all consultations), followed by emergency medicine (14%) and a variety of other services (Table 1). Most dermatology consultations were placed to assist in achieving a diagnosis of a cutaneous condition (77%), while a minority were to assist in the management of a previously diagnosed disease (22%). A small fraction of consultations (5%) were to complete full-body skin examinations (FBSEs) to rule out infection or malignancy in candidates for organ transplantation, left ventricular assist devices, or certain chemotherapies. One FBSE was conducted to search for a primary tumor in a patient diagnosed with metastatic melanoma.
Most Common Final Diagnoses and Consultation Impact—Table 2 lists the most common final diagnosis categories, as well as the effects of the consultation on diagnosis, management, biopsies, hospitalization, and clinical improvement as documented by the primary medical provider. The most common final diagnoses were inflammatory and autoimmune (39%), such as contact dermatitis and seborrheic dermatitis; infectious (23%), such as varicella (primary or zoster) and bacterial furunculosis; drug reactions (20%), such as morbilliform drug eruptions; vascular (8%), such as vasculitis and calciphylaxis; neoplastic (7%), such as keratinocyte carcinomas and leukemia cutis; and other (15%), such as xerosis, keratosis pilaris, and miliaria rubra.
Impact on Diagnosis—Fifty-six percent of all consultations resulted in a change in diagnosis. When dermatology was consulted specifically to assist in the diagnosis of a patient (195 consultations), the working diagnosis of the primary team was changed 69% of the time. Thirty-five of these consultation requests had no preliminary diagnosis, and the primary team listed the working diagnosis as either rash or a morphologic description of the lesion(s). Sixty-three percent of suspected drug eruptions ended with a diagnosis of a form of drug eruption, while 20% of consultations for suspected cellulitis or bacterial infections were confirmed to be cellulitis or soft tissue infections.
Impact on Management—Regardless of the reason for the consultation, most consultations (86%) resulted in a change in management. The remaining 14% consisted of FBSEs with benign findings; cases of cutaneous metastases and leukemia cutis managed by oncology; as well as select cases of purpura fulminans, postfebrile desquamation, and postinflammatory hyperpigmentation.
Changes in management included alterations in medications, requests for additional laboratory work or imaging, additional consultation requests, biopsies, or specific wound care instructions. Seventy-five percent of all consultations were given specific medication recommendations by dermatology. Most (61%) were recommended to be given a topical steroid, antibiotic, or both. However, 45% of all consultations were recommended to initiate a systemic medication, most commonly antihistamines, antibiotics, steroids, antivirals, or immunomodulators. Dermatology recommended discontinuing specific medications in 16% of all consultations, with antibiotics being the most frequent culprit (17 antibiotics discontinued), owing to drug eruptions or misdiagnosed infections. Vancomycin, piperacillin-tazobactam, and trimethoprim-sulfamethoxazole were the most frequently discontinued antibiotics.
Dermatology was consulted for assistance in management of previously diagnosed cutaneous conditions 56 times (22% of all consultations), often regarding complicated cases of hidradenitis suppurativa (9 cases), pyoderma gangrenosum (5 cases), bullous pemphigoid (4 cases), or erythroderma (4 cases). Most of these cases required a single dermatology encounter to provide recommendations (71%), and 21% required 1 additional follow-up. Sixty-three percent of patients consulted for management assistance were noted to have improvement in their cutaneous condition by time of discharge, as documented by the primary provider in the medical record.
Twenty-eight percent of all consultations required at least 1 biopsy. Seventy-two percent of all biopsies were consistent with the dermatologist’s working diagnosis or highest-ranked differential diagnosis, and 16% of biopsy results were consistent with the second- or third-ranked diagnosis. The primary teams requested a biopsy 38 times to assist in diagnosis, as documented in the progress note or consultation request. Only 21 of these consultations (55% of requests) received at least 1 biopsy, as the remaining consultations did not require a biopsy to establish a diagnosis. The most common final diagnoses of consultations receiving biopsies included drug eruptions (5), leukemia cutis (4), vasculopathies (4), vasculitis (4), and calciphylaxis (3).
Impact on Hospitalization and Efficacy—Dermatology performed 217 consultations regarding patients already admitted to the hospital, and 92% remained hospitalized either due to comorbidities or complicated cutaneous conditions following the consultation. The remaining 8% were cleared for discharge. Dermatology received 36 consultation requests from emergency medicine physicians. Fifty-three percent of these patients were admitted, while the remaining 47% were discharged from the emergency department or its observation unit following evaluation.
Fifty-one percent of all consultations were noted to have improvement in their cutaneous condition by the time of discharge, as noted in the physical examination, progress note, or discharge summary of the primary team. Thirty percent of cases remained stable, where improvement was not noted in in the medical record. Most of these cases involved keratinocyte carcinomas scheduled for outpatient excision, benign melanocytic nevi found on FBSE, and benign etiologies that led to immediate discharge following consultation. Three percent of all consultations were noted to have worsened following consultation, including cases of calciphylaxis, vasculopathies, and purpura fulminans, as well as patients who elected for palliative care and hospice. The cutaneous condition by the time of discharge could not be determined from the medical record in 16% of all consultations.
Eighty-five percent of all consultations required a single encounter with dermatology. An additional 10% required a single follow-up with dermatology, while only 5% of patients required 3 or more encounters. Notably, these cases included patients with 1 or more severe cutaneous diseases, such as Sweet syndrome, calciphylaxis, Stevens-Johnson syndrome/toxic epidermal necrolysis, and hidradenitis suppurativa.
Comment
Although dermatology often is viewed as an outpatient specialty, this study provides a glimpse into the ways inpatient dermatology consultations optimize the care of hospitalized patients. Most consultations involved assistance in diagnosing an unknown condition, but several regarded pre-existing skin disorders requiring management aid. As a variety of medical specialties requested consultations, dermatology was able to provide care to a diverse group of patients with conditions varying in complexity and severity. Several specialties benefited from niche dermatologic expertise: hematology and oncology frequently requested dermatology to assist in diagnosis and management of the toxic effects of chemotherapy, cutaneous metastasis, or suspected cutaneous infections in immunocompromised patients. Cardiology patients were frequently evaluated for potential malignancy or infection prior to heart transplantation and initiation of antirejection immunosuppressants. Dermatology was consulted to differentiate cutaneous manifestations of critical illness from underlying systemic disease in the intensive care unit, and patients presenting to the emergency department often were examined to determine if hospital admission was necessary, with 47% of these consultations resulting in a discharge following evaluation by a dermatologist.
Our results were consistent with prior studies1,5,6 that have reported frequent changes in final diagnosis following dermatology consultation, with 69% of working diagnoses changed in this study when consultation was requested for diagnostic assistance. When dermatology was consulted for diagnostic assistance, several of these cases lacked a preliminary differential diagnosis. Although the absence of a documented differential diagnosis may not necessarily reflect a lack of suspicion for a particular etiology, 86% of all consultations included a ranked differential or working diagnosis either in the consultation request or progress note prior to consultation. The final diagnoses of consultations without a preliminary diagnosis varied from the mild and localized to systemic and severe, further suggesting these cases reflected knowledge gaps of the primary medical team.
Integration of dermatology into the care of hospitalized patients could provide an opportunity for education of primary medical teams. With frequent consultation, primary medical teams may become more comfortable diagnosing and managing common cutaneous conditions specific to their specialty or extended hospitalizations.
Several consultations were requested to aid in management of cases of hidradenitis suppurativa, pyoderma gangrenosum, or bullous pemphigoid that either failed outpatient therapy or were complicated by superinfections. Despite the ranges in complexity, the majority of all consultations required a single encounter and led to improvement by the time of discharge, demonstrating the efficacy and efficiency of inpatient dermatologists.
Dermatology consultations often led to changes in management involving medications and additional workup. Changes in management also extended to specific wound care instructions provided by dermatology, as expected for cases of Stevens-Johnson syndrome/toxic epidermal necrolysis, Sweet syndrome, hidradenitis suppurativa, and pyoderma gangrenosum. However, patients with the sequelae of extended hospitalizations, such as chronic wounds, pressure ulcers, and edema bullae, also benefited from this expertise.
When patients required a biopsy, the final diagnoses were consistent with the dermatologist’s number one differential diagnosis or top 3 differential diagnoses 72% and 88% of the time, respectively. Only 55% of cases where the primary team requested a biopsy ultimately required a biopsy, as many involved clinical diagnoses such as urticaria. Not only was dermatology accurate in their preliminary diagnoses, but they decreased cost and morbidity by avoiding unnecessary procedures.
This study provided additional evidence to support the integration of dermatology into the hospital setting for the benefit of patients, primary medical teams, and hospital systems. Dermatology offers high-value care through the efficient diagnosis and management of hospitalized patients, which contributes to decreased cost and improved outcomes.2,5-7,9,10 This study highlighted lesser-known areas of impact, such as the various specialty-specific services dermatology provides as well as the high rates of reported improvement following consultation. Future studies should continue to explore the field’s unique impact on hospitalized medicine as well as other avenues of care delivery, such as telemedicine, that may encourage dermatologists to participate in consultations and increase the volume of patients who may benefit from their care.
Dermatology is an often-underutilized resource in the hospital setting. As the health care landscape has evolved, so has the role of the inpatient dermatologist.1-3 Structural changes in the health system and advances in therapies have shifted dermatology from an admitting service to an almost exclusively outpatient practice. Improved treatment modalities led to decreases in the number of patients requiring admission for chronic dermatoses, and outpatient clinics began offering therapies once limited to hospitals.1,4 Inpatient dermatology consultations emerged and continue to have profound effects on hospitalized patients regardless of their reason for admission.1-11
Inpatient dermatologists supply knowledge in areas primary medical teams lack, and there is evidence that dermatology consultations improve the quality of care while decreasing cost.2,5-7 Establishing correct diagnoses, preventing exposure to unnecessary medications, and reducing hospitalization duration and readmission rates are a few ways dermatology consultations positively impact hospitalized patients.2,5-7,9,10 This study highlights the role of the dermatologist in the care of hospitalized patients at a large academic medical center in an urban setting and reveals how consultation supports the efficiency and efficacy of other services.
Materials and Methods
Study Design—This single-institution, cross-sectional retrospective study included all hospitalized patients at the Thomas Jefferson University Hospital (Philadelphia, Pennsylvania), who received an inpatient dermatology consultation completed by physicians of Jefferson Dermatology Associates between January 1, 2019, and December 31, 2019. The institutional review board at Thomas Jefferson University approved this study.
Data Collection—A list of all inpatient dermatology consultations in 2019 was provided by Jefferson Dermatology Associates. Through a retrospective chart review, data regarding the consultations were collected from the electronic medical record (Epic Systems) and recorded into the Research Electronic Data Capture system. Data on patient demographics, the primary medical team, the dermatology evaluation, and the hospital course of the patient were collected.
Results
Patient Characteristics—Dermatology received 253 inpatient consultation requests during this time period; 53% of patients were female and 47% were male, with a mean age of 55 years. Most patients were White (57%), while 34% were Black. Five percent and 4% of patients were Asian and Hispanic or Latino, respectively (Table 1). The mean duration of hospitalization for all patients was 15 days, and the average number of days to discharge following the first encounter with dermatology was 10 days.
Requesting Team and Reason for Consultation—Internal medicine consulted dermatology most frequently (34% of all consultations), followed by emergency medicine (14%) and a variety of other services (Table 1). Most dermatology consultations were placed to assist in achieving a diagnosis of a cutaneous condition (77%), while a minority were to assist in the management of a previously diagnosed disease (22%). A small fraction of consultations (5%) were to complete full-body skin examinations (FBSEs) to rule out infection or malignancy in candidates for organ transplantation, left ventricular assist devices, or certain chemotherapies. One FBSE was conducted to search for a primary tumor in a patient diagnosed with metastatic melanoma.
Most Common Final Diagnoses and Consultation Impact—Table 2 lists the most common final diagnosis categories, as well as the effects of the consultation on diagnosis, management, biopsies, hospitalization, and clinical improvement as documented by the primary medical provider. The most common final diagnoses were inflammatory and autoimmune (39%), such as contact dermatitis and seborrheic dermatitis; infectious (23%), such as varicella (primary or zoster) and bacterial furunculosis; drug reactions (20%), such as morbilliform drug eruptions; vascular (8%), such as vasculitis and calciphylaxis; neoplastic (7%), such as keratinocyte carcinomas and leukemia cutis; and other (15%), such as xerosis, keratosis pilaris, and miliaria rubra.
Impact on Diagnosis—Fifty-six percent of all consultations resulted in a change in diagnosis. When dermatology was consulted specifically to assist in the diagnosis of a patient (195 consultations), the working diagnosis of the primary team was changed 69% of the time. Thirty-five of these consultation requests had no preliminary diagnosis, and the primary team listed the working diagnosis as either rash or a morphologic description of the lesion(s). Sixty-three percent of suspected drug eruptions ended with a diagnosis of a form of drug eruption, while 20% of consultations for suspected cellulitis or bacterial infections were confirmed to be cellulitis or soft tissue infections.
Impact on Management—Regardless of the reason for the consultation, most consultations (86%) resulted in a change in management. The remaining 14% consisted of FBSEs with benign findings; cases of cutaneous metastases and leukemia cutis managed by oncology; as well as select cases of purpura fulminans, postfebrile desquamation, and postinflammatory hyperpigmentation.
Changes in management included alterations in medications, requests for additional laboratory work or imaging, additional consultation requests, biopsies, or specific wound care instructions. Seventy-five percent of all consultations were given specific medication recommendations by dermatology. Most (61%) were recommended to be given a topical steroid, antibiotic, or both. However, 45% of all consultations were recommended to initiate a systemic medication, most commonly antihistamines, antibiotics, steroids, antivirals, or immunomodulators. Dermatology recommended discontinuing specific medications in 16% of all consultations, with antibiotics being the most frequent culprit (17 antibiotics discontinued), owing to drug eruptions or misdiagnosed infections. Vancomycin, piperacillin-tazobactam, and trimethoprim-sulfamethoxazole were the most frequently discontinued antibiotics.
Dermatology was consulted for assistance in management of previously diagnosed cutaneous conditions 56 times (22% of all consultations), often regarding complicated cases of hidradenitis suppurativa (9 cases), pyoderma gangrenosum (5 cases), bullous pemphigoid (4 cases), or erythroderma (4 cases). Most of these cases required a single dermatology encounter to provide recommendations (71%), and 21% required 1 additional follow-up. Sixty-three percent of patients consulted for management assistance were noted to have improvement in their cutaneous condition by time of discharge, as documented by the primary provider in the medical record.
Twenty-eight percent of all consultations required at least 1 biopsy. Seventy-two percent of all biopsies were consistent with the dermatologist’s working diagnosis or highest-ranked differential diagnosis, and 16% of biopsy results were consistent with the second- or third-ranked diagnosis. The primary teams requested a biopsy 38 times to assist in diagnosis, as documented in the progress note or consultation request. Only 21 of these consultations (55% of requests) received at least 1 biopsy, as the remaining consultations did not require a biopsy to establish a diagnosis. The most common final diagnoses of consultations receiving biopsies included drug eruptions (5), leukemia cutis (4), vasculopathies (4), vasculitis (4), and calciphylaxis (3).
Impact on Hospitalization and Efficacy—Dermatology performed 217 consultations regarding patients already admitted to the hospital, and 92% remained hospitalized either due to comorbidities or complicated cutaneous conditions following the consultation. The remaining 8% were cleared for discharge. Dermatology received 36 consultation requests from emergency medicine physicians. Fifty-three percent of these patients were admitted, while the remaining 47% were discharged from the emergency department or its observation unit following evaluation.
Fifty-one percent of all consultations were noted to have improvement in their cutaneous condition by the time of discharge, as noted in the physical examination, progress note, or discharge summary of the primary team. Thirty percent of cases remained stable, where improvement was not noted in in the medical record. Most of these cases involved keratinocyte carcinomas scheduled for outpatient excision, benign melanocytic nevi found on FBSE, and benign etiologies that led to immediate discharge following consultation. Three percent of all consultations were noted to have worsened following consultation, including cases of calciphylaxis, vasculopathies, and purpura fulminans, as well as patients who elected for palliative care and hospice. The cutaneous condition by the time of discharge could not be determined from the medical record in 16% of all consultations.
Eighty-five percent of all consultations required a single encounter with dermatology. An additional 10% required a single follow-up with dermatology, while only 5% of patients required 3 or more encounters. Notably, these cases included patients with 1 or more severe cutaneous diseases, such as Sweet syndrome, calciphylaxis, Stevens-Johnson syndrome/toxic epidermal necrolysis, and hidradenitis suppurativa.
Comment
Although dermatology often is viewed as an outpatient specialty, this study provides a glimpse into the ways inpatient dermatology consultations optimize the care of hospitalized patients. Most consultations involved assistance in diagnosing an unknown condition, but several regarded pre-existing skin disorders requiring management aid. As a variety of medical specialties requested consultations, dermatology was able to provide care to a diverse group of patients with conditions varying in complexity and severity. Several specialties benefited from niche dermatologic expertise: hematology and oncology frequently requested dermatology to assist in diagnosis and management of the toxic effects of chemotherapy, cutaneous metastasis, or suspected cutaneous infections in immunocompromised patients. Cardiology patients were frequently evaluated for potential malignancy or infection prior to heart transplantation and initiation of antirejection immunosuppressants. Dermatology was consulted to differentiate cutaneous manifestations of critical illness from underlying systemic disease in the intensive care unit, and patients presenting to the emergency department often were examined to determine if hospital admission was necessary, with 47% of these consultations resulting in a discharge following evaluation by a dermatologist.
Our results were consistent with prior studies1,5,6 that have reported frequent changes in final diagnosis following dermatology consultation, with 69% of working diagnoses changed in this study when consultation was requested for diagnostic assistance. When dermatology was consulted for diagnostic assistance, several of these cases lacked a preliminary differential diagnosis. Although the absence of a documented differential diagnosis may not necessarily reflect a lack of suspicion for a particular etiology, 86% of all consultations included a ranked differential or working diagnosis either in the consultation request or progress note prior to consultation. The final diagnoses of consultations without a preliminary diagnosis varied from the mild and localized to systemic and severe, further suggesting these cases reflected knowledge gaps of the primary medical team.
Integration of dermatology into the care of hospitalized patients could provide an opportunity for education of primary medical teams. With frequent consultation, primary medical teams may become more comfortable diagnosing and managing common cutaneous conditions specific to their specialty or extended hospitalizations.
Several consultations were requested to aid in management of cases of hidradenitis suppurativa, pyoderma gangrenosum, or bullous pemphigoid that either failed outpatient therapy or were complicated by superinfections. Despite the ranges in complexity, the majority of all consultations required a single encounter and led to improvement by the time of discharge, demonstrating the efficacy and efficiency of inpatient dermatologists.
Dermatology consultations often led to changes in management involving medications and additional workup. Changes in management also extended to specific wound care instructions provided by dermatology, as expected for cases of Stevens-Johnson syndrome/toxic epidermal necrolysis, Sweet syndrome, hidradenitis suppurativa, and pyoderma gangrenosum. However, patients with the sequelae of extended hospitalizations, such as chronic wounds, pressure ulcers, and edema bullae, also benefited from this expertise.
When patients required a biopsy, the final diagnoses were consistent with the dermatologist’s number one differential diagnosis or top 3 differential diagnoses 72% and 88% of the time, respectively. Only 55% of cases where the primary team requested a biopsy ultimately required a biopsy, as many involved clinical diagnoses such as urticaria. Not only was dermatology accurate in their preliminary diagnoses, but they decreased cost and morbidity by avoiding unnecessary procedures.
This study provided additional evidence to support the integration of dermatology into the hospital setting for the benefit of patients, primary medical teams, and hospital systems. Dermatology offers high-value care through the efficient diagnosis and management of hospitalized patients, which contributes to decreased cost and improved outcomes.2,5-7,9,10 This study highlighted lesser-known areas of impact, such as the various specialty-specific services dermatology provides as well as the high rates of reported improvement following consultation. Future studies should continue to explore the field’s unique impact on hospitalized medicine as well as other avenues of care delivery, such as telemedicine, that may encourage dermatologists to participate in consultations and increase the volume of patients who may benefit from their care.
- Madigan LM, Fox LP. Where are we now with inpatient consultative dermatology?: assessing the value and evolution of this subspecialty over the past decade. J Am Acad Dermatol. 2019;80:1804-1808. doi:10.1016/j.jaad.2019.01.031
- Noe MH, Rosenbach M. Inpatient dermatologists—crucial for the management of skin diseases in hospitalized patients [editorial]. JAMA Dermatol. 2018;154:524-525. doi:10.1001/jamadermatol.2017.6195
- Strowd LC. Inpatient dermatology: a paradigm shift in the management of skin disease in the hospital. Br J Dermatol. 2019;180:966-967. doi:10.1111/bjd.17778
- Kirsner RS, Yang DG, Kerdel FA. The changing status of inpatient dermatology at American academic dermatology programs. J Am Acad Dermatol. 1999;40:755-757. doi:10.1016/s0190-9622(99)70158-1
- Kroshinsky D, Cotliar J, Hughey LC, et al. Association of dermatology consultation with accuracy of cutaneous disorder diagnoses in hospitalized patients: a multicenter analysis. JAMA Dermatol. 2016;152:477-480. doi:10.1001/jamadermatol.2015.5098
- Ko LN, Garza-Mayers AC, St John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis. JAMA Dermatol. 2018;154:529-533. doi:10.1001/jamadermatol.2017.6196
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543. doi:10.1001/jamadermatol.2017.6197
- Milani-Nejad N, Zhang M, Kaffenberger BH. Association of dermatology consultations with patient care outcomes in hospitalized patients with inflammatory skin diseases. JAMA Dermatol. 2017;153:523-528. doi:10.1001/jamadermatol.2016.6130
- Imadojemu S, Rosenbach M. Dermatologists must take an active role in the diagnosis of cellulitis. JAMA Dermatol. 2017;153:134-135. doi:10.1001/jamadermatol.2016.4230
- Hughey LC. The impact dermatologists can have on misdiagnosis of cellulitis and overuse of antibiotics: closing the gap. JAMA Dermatol. 2014;150:1061-1062. doi:10.1001/jamadermatol.2014.1164
- Ko LN, Kroshinsky D. Dermatology hospitalists: a multicenter survey study characterizing the infrastructure of consultative dermatology in select American hospitals. Int J Dermatol. 2018;57:553-558. doi:10.1111/ijd.13939
- Madigan LM, Fox LP. Where are we now with inpatient consultative dermatology?: assessing the value and evolution of this subspecialty over the past decade. J Am Acad Dermatol. 2019;80:1804-1808. doi:10.1016/j.jaad.2019.01.031
- Noe MH, Rosenbach M. Inpatient dermatologists—crucial for the management of skin diseases in hospitalized patients [editorial]. JAMA Dermatol. 2018;154:524-525. doi:10.1001/jamadermatol.2017.6195
- Strowd LC. Inpatient dermatology: a paradigm shift in the management of skin disease in the hospital. Br J Dermatol. 2019;180:966-967. doi:10.1111/bjd.17778
- Kirsner RS, Yang DG, Kerdel FA. The changing status of inpatient dermatology at American academic dermatology programs. J Am Acad Dermatol. 1999;40:755-757. doi:10.1016/s0190-9622(99)70158-1
- Kroshinsky D, Cotliar J, Hughey LC, et al. Association of dermatology consultation with accuracy of cutaneous disorder diagnoses in hospitalized patients: a multicenter analysis. JAMA Dermatol. 2016;152:477-480. doi:10.1001/jamadermatol.2015.5098
- Ko LN, Garza-Mayers AC, St John J, et al. Effect of dermatology consultation on outcomes for patients with presumed cellulitis. JAMA Dermatol. 2018;154:529-533. doi:10.1001/jamadermatol.2017.6196
- Li DG, Xia FD, Khosravi H, et al. Outcomes of early dermatology consultation for inpatients diagnosed with cellulitis. JAMA Dermatol. 2018;154:537-543. doi:10.1001/jamadermatol.2017.6197
- Milani-Nejad N, Zhang M, Kaffenberger BH. Association of dermatology consultations with patient care outcomes in hospitalized patients with inflammatory skin diseases. JAMA Dermatol. 2017;153:523-528. doi:10.1001/jamadermatol.2016.6130
- Imadojemu S, Rosenbach M. Dermatologists must take an active role in the diagnosis of cellulitis. JAMA Dermatol. 2017;153:134-135. doi:10.1001/jamadermatol.2016.4230
- Hughey LC. The impact dermatologists can have on misdiagnosis of cellulitis and overuse of antibiotics: closing the gap. JAMA Dermatol. 2014;150:1061-1062. doi:10.1001/jamadermatol.2014.1164
- Ko LN, Kroshinsky D. Dermatology hospitalists: a multicenter survey study characterizing the infrastructure of consultative dermatology in select American hospitals. Int J Dermatol. 2018;57:553-558. doi:10.1111/ijd.13939
Practice Points
- Inpatient dermatologists fill knowledge gaps that often alter the diagnosis, management, and hospital course of hospitalized patients.
- Several medical specialties benefit from niche expertise of inpatient dermatologists specific to their patient population.
- Integration of inpatient dermatology consultations can prevent unnecessary hospital admissions and medication administration.
Cutaneous Manifestations of COVID-19: Characteristics, Pathogenesis, and the Role of Dermatology in the Pandemic
The virus that causes COVID-19—SARS-CoV-2—has infected more than 128 million individuals, resulting in more than 2.8 million deaths worldwide between December 2019 and April 2021. Disease mortality primarily is driven by hypoxemic respiratory failure and systemic hypercoagulability, resulting in multisystem organ failure.1 With more than 17 million Americans infected, the virus is estimated to have impacted someone within the social circle of nearly every American.2
The COVID-19 pandemic has highlighted resource limitations, delayed elective and preventive care, and rapidly increased the adoption of telemedicine, presenting a host of new challenges to providers in every medical specialty, including dermatology. Although COVID-19 primarily is a respiratory disease, clinical manifestations have been observed in nearly every organ, including the skin. The cutaneous manifestations of COVID-19 provide insight into disease diagnosis, prognosis, and pathophysiology. In this article, we review the cutaneous manifestations of COVID-19 and explore the state of knowledge regarding their pathophysiology and clinical significance. Finally, we discuss the role of dermatology consultants in the care of patients with COVID-19, and the impact of the pandemic on the field of dermatology.
Prevalence of Cutaneous Findings in COVID-19
Early reports characterizing the clinical presentation of patients hospitalized with COVID-19 suggested skin findings associated with the disease were rare. Cohort studies from Europe, China, and New York City in January through March 2020 reported a low prevalence or made no mention of rash.3-7 However, reports from dermatologists in Italy that emerged in May 2020 indicated a substantially higher proportion of cutaneous disease: 18 of 88 (20.4%) hospitalized patients were found to have cutaneous involvement, primarily consisting of erythematous rash, along with some cases of urticarial and vesicular lesions.8 In October 2020, a retrospective cohort study from Spain examining 2761 patients presenting to the emergency department or admitted to the hospital for COVID-19 found that 58 (2.1%) patients had skin lesions attributed to COVID-19.9
The wide range in reported prevalence of skin lesions may be due to variable involvement of dermatologic specialists in patient care, particularly in China.10 Some variation also may be due to variability in the timing of clinical examination, as well as demographic and clinical differences in patient populations. Of note, a multisystem inflammatory disease seen in US children subsequent to infection with COVID-19 has been associated with rash in as many as 74% of cases.11 Although COVID-19 disproportionately impacts people with skin of color, there are few reports of cutaneous manifestations in that population,12 highlighting the challenges of the dermatologic examination in individuals with darker skin and suggesting the prevalence of dermatologic disease in COVID-19 may be greater than reported.
Morphologic Patterns of Cutaneous Involvement in COVID-19
Researchers in Europe and the United States have attempted to classify the cutaneous manifestations of COVID-19. A registry established through the American Academy of Dermatology published a compilation of reports from 31 countries, totaling 716 patient profiles.13 A prospective Spanish study detailed the cutaneous involvement of 375 patients with suspected or confirmed COVID-19.14 Together, these efforts have revealed several distinct patterns of cutaneous involvement associated with COVID-19 (Table).9,15-18
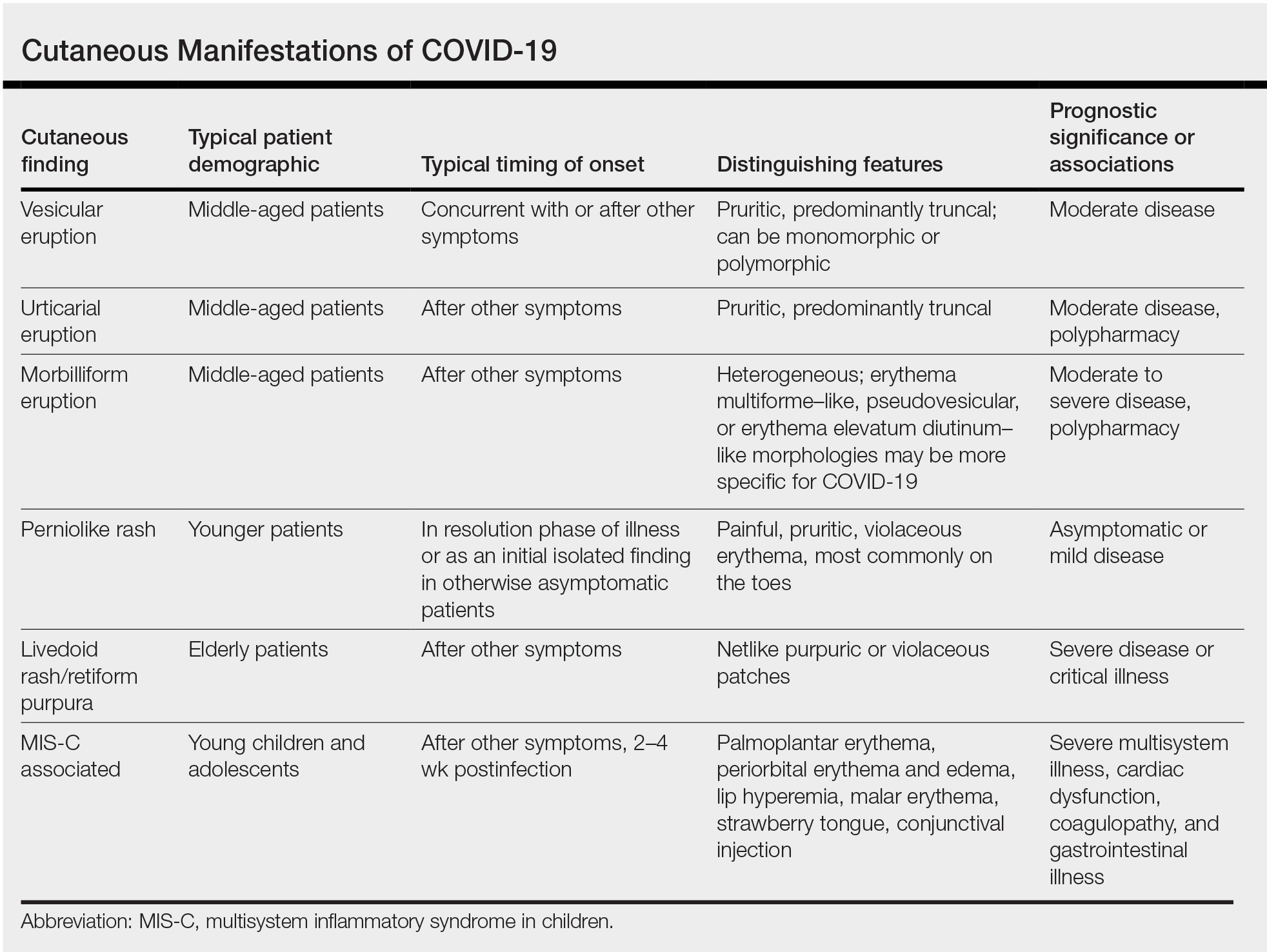
Vesicular Rash
Vesicular rash associated with COVID-19 has been described in several studies and case series8,13,14 and is considered, along with the pseudopernio (or pseudochilblains) morphology, to be one of the more disease-specific patterns in COVID-19.14,18 Vesicular rash appears to comprise roughly one-tenth of all COVID-19–associated rashes.13,14 It usually is described as pruritic, with 72% to 83% of patients reporting itch.13,16
Small monomorphic or polymorphic vesicles predominantly on the trunk and to a lesser extent the extremities and head have been described by multiple authors.14,16 Vesicular rash is most common among middle-aged individuals, with studies reporting median and mean ages ranging from 40.5 to 55 years.9,13,14,16
Vesicular rash develops concurrent with or after other presenting symptoms of COVID-19; in 2 studies, vesicular rash preceded development of other symptoms in only 15% and 5.6% of cases, respectively.13,14 Prognostically, vesicular rash is associated with moderate disease severity.14,16 It may persist for an average of 8 to 10 days.14,16,18
Histopathologic examination reveals basal layer vacuolar degeneration, hyperchromatic keratinocytes, acantholysis, and dyskeratosis.9,16,18
Urticarial Rash
Urticarial lesions represent approximately 7% to 19% of reported COVID-19–associated rashes.9,13,14 Urticarial rashes in patients testing positive for SARS-CoV-2 primarily occur on the trunk.14 The urticaria, which typically last about 1 week,14 are seen most frequently in middle-aged patients (mean/median age, 42–48 years)13,14 and are associated with pruritus, which has been reported in 74% to 92% of patients.13,14 Urticarial lesions typically do not precede other symptoms of COVID-19 and are nonspecific, making them less useful diagnostically.14
Urticaria appears to be associated with more severe COVID-19 illness in several studies, but this finding may be confounded by several factors, including older age, increased tobacco use, and polypharmacy. Of 104 patients with reported urticarial rash and suspected or confirmed COVID-19 across 3 studies, only 1 death was reported.9,13,14
The histopathologic appearance is that of typical hives, demonstrating a perivascular infiltrate of lymphocytes and eosinophils with edema of the upper dermis.9,19
Morbilliform Eruption
Morbilliform eruption is a commonly reported morphology associated with COVID-19, accounting for 20% to 47% of rashes.9,13,14 This categorization may have limited utility from a diagnostic and prognostic perspective, given that morbilliform eruptions are common, nonspecific, and heterogenous and can arise from many causes.9,13,14 Onset of morbilliform eruption appears to coincide with14 or follow13,20,21 the development of other COVID-19–related symptoms, with 5% of patients reporting morbilliform rash as the initial manifestation of infection.13,14 Morbilliform eruptions have been observed to occur in patients with more severe disease.9,13,14
Certain morphologic subtypes, such as erythema multiforme–like, erythema elevatum diutinum–like, or pseudovesicular, may be more specific to COVID-19 infection.14 A small case series highlighted 4 patients with erythema multiforme–like eruptions, 3 of whom also were found to have petechial enanthem occurring after COVID-19 diagnosis; however, the investigators were unable to exclude drug reaction as a potential cause of rash in these patients.22 Another case series of 21 patients with COVID-19 and skin rash described a (primarily) petechial enanthem on the palate in 6 (28.5%) patients.23 It is unclear to what extent oral enanthem may be underrecognized given that some physicians may be disinclined to remove the masks of known COVID-19–positive patients to examine the oral cavity.
The histologic appearance of morbilliform rash seen in association with COVID-19 has been described as spongiotic with interface dermatitis with perivascular lymphocytic inflammation.9,21
COVID Toes, Pseudochilblains Rash, Perniolike Rash, and Acral Erythema/Edema
Of all the rashes associated with COVID-19, COVID toes, or pseudochilblains rash, has perhaps attracted the most attention. The characteristic violaceous erythema on the fingers and/or toes may be itchy or painful, presenting similar to idiopathic cases of pernio (Figure 1).14 The entity has been controversial because of an absence of a clear correlation with a positive SARS-CoV-2 polymerase chain reaction test or antibodies to the virus in a subset of reported cases.24,25 Onset of the rash late in the disease course, generally after symptom resolution in mild or asymptomatic cases, may explain the absence of viral DNA in the nasopharynx by the time of lesion appearance.14,26 Seronegative patients may have cleared SARS-CoV-2 infection before humoral immunity could occur via a strong type 1 interferon response.25
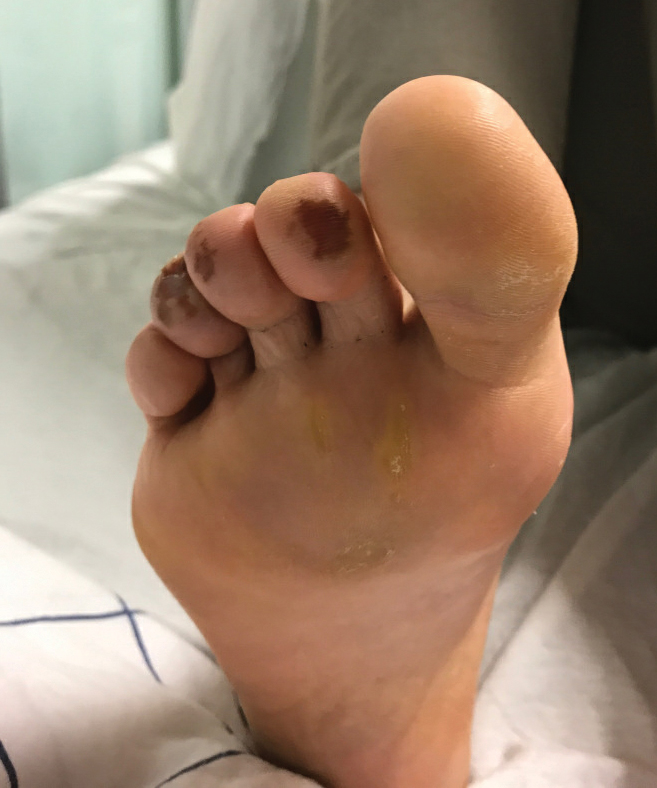
Across 3 studies, perniolike skin lesions constituted 18% to 29% of COVID-19–associated skin findings9,13,14 and persisted for an average of 12 to 14 days.13,14 Perniolike lesions portend a favorable outcome; patients with COVID toes rarely present with systemic symptoms or laboratory or imaging abnormalities9 and less commonly require hospitalization for severe illness. Perniolike lesions have been reported most frequently in younger patients, with a median or mean age of 32 to 35 years.13,14
Histology demonstrates lichenoid dermatitis with perivascular and periadnexal lymphocytic infiltrates.9 Notably, one study observed interface dermatitis of the intraepidermal portion of the acrosyringium, a rare finding in chilblain lupus, in 83% of patients (N=40).25 Direct immunofluorescence demonstrates a vasculopathic pattern, with some patients showing deposition of IgM or IgG, C3, and fibrinogen in dermal blood vessels. Vascular C9 deposits also have been demonstrated on immunohistochemistry.9 Biopsies of perniolike lesions in COVID-19 patients have demonstrated the presence of SARS-CoV-2 RNA,27 have identified SARS-CoV-2 spike protein in endothelial cells on immunohistochemistry, and have visualized intracytoplasmic viral particles in vascular endothelium on electron microscopy.28
Livedoid Rash/Retiform Purpura
Netlike purpuric or violaceous patches signifying vessel damage or occlusion have been seen in association with COVID-19, constituting approximately 6% of COVID-19–associated skin findings in 2 studies.13,14 Livedoid rash (Figure 2) and retiform purpura (Figure 3) are associated with older age and occur primarily in severely ill patients, including those requiring intensive care. In a registry of 716 patients with COVID-19, 100% of patients with retiform purpura were hospitalized, and 82% had acute respiratory distress syndrome.13 In another study, 33% (7/21) of patients with livedoid and necrotic lesions required intensive care, and 10% (2/21) died.14
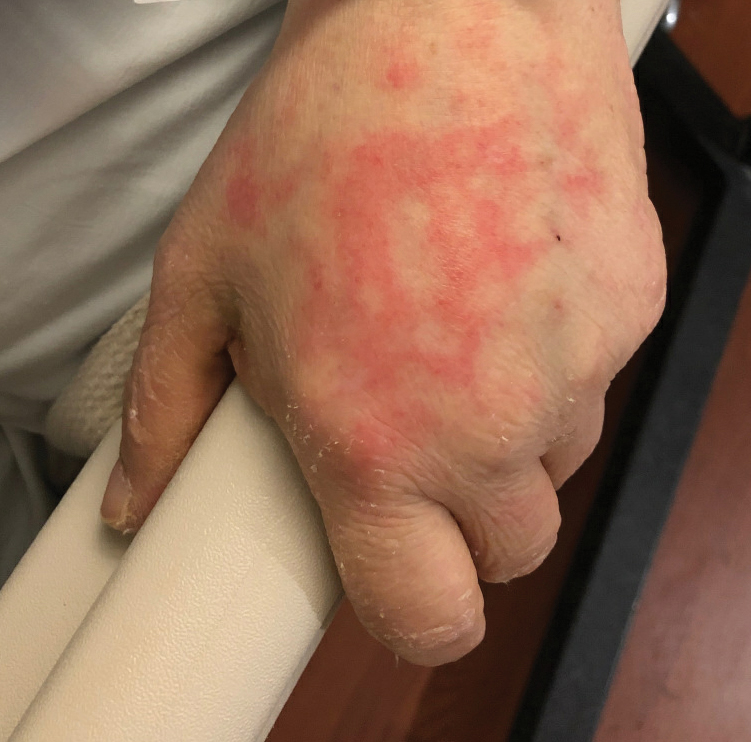
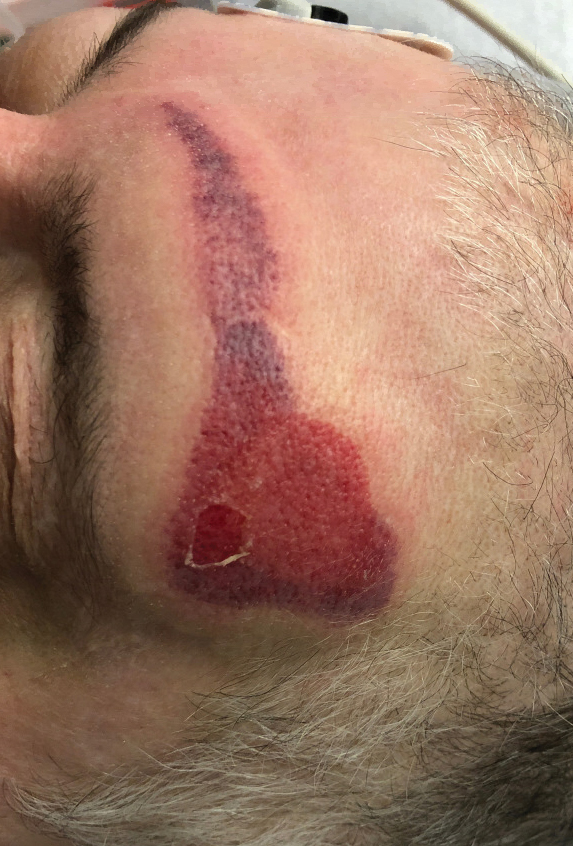
Livedoid lesions and retiform purpura represent thrombotic disease in the skin due to vasculopathy/coagulopathy. Dermatopathology available through the American Academy of Dermatology registry revealed thrombotic vasculopathy.13 A case series of 4 patients with livedo racemosa and retiform purpura demonstrated pauci-inflammatory thrombogenic vasculopathy involving capillaries, venules, and arterioles with complement deposition.29 Livedoid and retiform lesions in the skin may be associated with a COVID-19–induced coagulopathy, a propensity for systemic clotting including pulmonary embolism, which mostly occurs in hospitalized patients with severe illness.30
Multisystem Inflammatory Disease in Children
A hyperinflammatory syndrome similar to Kawasaki disease and toxic shock syndrome associated with mucocutaneous, cardiac, and gastrointestinal manifestations has been reported following COVID-19 infection.31 This syndrome, known as multisystem inflammatory syndrome in children (MIS-C), predominantly affects adolescents and children older than 5 years,11 typically occurs 2 to 4 weeks after infection, and appears to be at least 100-times less common than COVID-19 infection among the same age group.31 Sixty percent31 to 74%11 of affected patients have mucocutaneous involvement, with the most common clinical findings being conjunctival injection, palmoplantar erythema, lip hyperemia, periorbital erythema and edema, strawberry tongue, and malar erythema, respectively.32
Because this condition appears to reflect an immune response to the virus, the majority of cases demonstrate negative SARS-CoV-2 polymerase chain reaction and positive antibody testing.33 Although cutaneous findings are similar to those seen in Kawasaki disease, certain findings have been noted in MIS-C that are not typical of Kawasaki disease, including heliotrope rash–like periorbital edema and erythema as well as erythema infectiosum–like malar erythema and reticulated erythematous eruptions.32
The course of MIS-C can be severe; in one case series of patients presenting with MIS-C, 80% (79/99) required intensive care unit admission, with 10% requiring mechanical ventilation and 2% of patients dying during admission.31 Cardiac dysfunction, coagulopathy, and gastrointestinal symptoms are common.11,31 It has been postulated that a superantigenlike region of the SARS-CoV-2 spike protein, similar to that of staphylococcal enterotoxin B, may underlie MIS-C and account for its similarities to toxic shock syndrome.34 Of note, a similar multisystem inflammatory syndrome associated with COVID-19 also has been described in adults, and it too may present with rash as a cardinal feature.35
Pathophysiology of COVID-19: What the Skin May Reveal About the Disease
The diverse range of cutaneous manifestations in COVID-19 reflects a spectrum of host immunologicresponses to SARS-CoV-2 and may inform the pathophysiology of the disease as well as potential treatment modalities.
Host Response to SARS-CoV-2
The body’s response to viral infection is 2-pronged, involving activation of cellular antiviral defenses mediated by type I and III interferons, as well as recruitment of leukocytes, mobilized by cytokines and chemokines.36,37 Infection with SARS-CoV-2 results in a unique inflammatory response characterized by suppression of interferons, juxtaposed with a rampant proinflammatory cytokine and chemokine response, reminiscent of a cytokine storm. Reflective of this imbalance, a study of 50 COVID-19 patients and 20 healthy controls found decreased natural killer cells and CD3+ T cells in COVID-19 patients, particularly severely or critically ill patients, with an increase in B cells and monocytes.38 This distinctive immune imbalance positions SARS-CoV-2 to thrive in the absence of inhibitory interferon activity while submitting the host to the deleterious effects of a cytokine surge.36
Type I Interferons
The perniolike lesions associated with mild COVID-19 disease14 may represent a robust immune response via effective stimulation of type I interferons (IFN-1). Similar perniolike lesions are observed in Aicardi-Goutières syndrome37 and familial chilblain lupus, hereditary interferonopathies associated with mutations in the TREX1 (three prime repair exonuclease 1) gene and characterized by inappropriate upregulation of IFN-1,39 resulting in chilblains. It has been suggested that perniolike lesions in COVID-19 result from IFN-1 activation—a robust effective immunologic response to the virus.14,26,40
On the other end of the spectrum, patients with severe COVID-19 may have a blunted IFN-1 response and reduced IFN-1–stimulated gene expression.36,38 Notably, low IFN-1 response preceded clinical deterioration and was associated with increased risk for evolution to critical illness.38 Severe disease from COVID-19 also is more commonly observed in older patients and those with comorbidities,1 both of which are known factors associated with depressed IFN-1 function.38,41 Reflective of this disparate IFN-1 response, biopsies of COVID-19 perniosis have demonstrated striking expression of myxovirus resistance protein A (MXA), a marker for IFN-1 signaling in tissue, whereas its expression is absent in COVID-19 livedo/retiform purpura.27
Familial chilblain lupus may be effectively treated by the Janus kinase inhibitor baricitinib,39 which inhibits IFN-1 signaling. Baricitinib recently received emergency use authorization by the US Food and Drug Administration for treatment of severe COVID-19 pneumonia,42,43 hinting to disordered IFN-1 signaling in the COVID-19 pathophysiology.
The impaired IFN-1 response in COVID-19 patients may be due to a unique characteristic of SARS-CoV-2: its ORF3b gene is a potent IFN-1 antagonist. In a series of experiments comparing SARS-CoV-2 to the related virus severe acute respiratory disease coronavirus (which was responsible for an epidemic in 2002), Konno et al44 found that SARS-CoV-2 is more effectively able to downregulate host IFN-1, likely due to premature stop codons on ORF3b that produce a truncated version of the gene with amplified anti–IFN-1 activity.
Cytokine Storm and Coagulation Cascade
This dulled interferon response is juxtaposed with a surge of inflammatory chemokines and cytokines, including IL-6, IL-8, IL-10, and tumor necrosis factor α, impairing innate immunity and leading to end-organ damage. This inflammatory response is associated with the influx of innate immune cells, specifically neutrophils and monocytes, which likely contribute to lung injury in COVID-19 acute respiratory distress syndrome.38 It also is thought to lead to downstream activation of coagulation, with a high incidence of thrombotic events observed in patients with severe COVID-19.1 In a retrospective study of 184 intensive care patients with COVID-19 receiving at least standard doses of thromboprophylaxis, venous thromboembolism occurred in 27% and arterial thrombotic events occurred in 3.7%.45
Livedo racemosa and retiform purpura are cutaneous markers of hypercoagulability, which indicate an increased risk for systemic clotting in COVID-19. A positive feedback loop between the complement and coagulation cascades appears to be important.13,14,29,46-48 In addition, a few studies have reported antiphospholipid antibody positivity in hospitalized COVID-19 patients.49,50
The high incidence of coagulopathy in severe COVID-19 has prompted many institutions to develop aggressive prophylactic anticoagulation protocols. Elevation of proinflammatory cytokines and observation of terminal complement activation in the skin and other organs has led to therapeutic trials of IL-6 inhibitors such as tocilizumab,51 complement inhibitors such as eculizumab, and Janus kinase inhibitors such as ruxolitinib and baricitinib.42,48
COVID Long-Haulers
The long-term effects of immune dysregulation in COVID-19 patients remain to be seen. Viral triggering of autoimmune disease is a well-established phenomenon, seen in DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome and other dermatologic diseases, raising the possibility that dermatologists will see a rising incidence of cutaneous autoimmune disease in the aftermath of the pandemic. Disordered interferon stimulation could lead to increased incidence of interferon-mediated disorders, such as sarcoidosis and other granulomatous diseases. Vasculitislike skin lesions could persist beyond the acute infectious period. Recent data from a registry of 990 COVID-19 cases from 39 countries suggest that COVID-19 perniolike lesions may persist as long as 150 days.52 In a time of many unknowns, these questions serve as a call to action for rigorous data collection, contribution to existing registries for dermatologic manifestations of COVID-19, and long-term follow-up of COVID-19 patients by the dermatology community.
Pandemic Dermatology
The pandemic has posed unprecedented challenges for patient care. The use of hydroxychloroquine as a popular but unproven treatment for COVID-19, 53 particularly early in the pandemic, has resulted in drug shortages for patients with lupus and other autoimmune skin diseases. Meanwhile, the need for patients with complex dermatologic conditions to receive systemic immunosuppression has had to be balanced against the associated risks during a global pandemic. To help dermatologists navigate this dilemma, various subspecialty groups have issued guidelines, including the COVID-19 Task Force of the Medical Dermatology Society and Society of Dermatology Hospitalists, which recommends a stepwise approach to shared decision-making with the goal of minimizing both the risk for disease flare and that of infection. The use of systemic steroids and rituximab, as well as the dose of immunosuppression—particularly broad-acting immunosuppression—should be limited where permitted. 54
Rapid adoption of telemedicine and remote monitoring strategies has enabled dermatologists to provide safe and timely care when in-person visits have not been possible, including for patients with confirmed or suspected COVID-19, as well as for hospitalized patients. 55-57 Use of telemedicine has facilitated preservation of personal protective equipment at a time when these important resources have been scarce. For patients with transportation or scheduling barriers, telemedicine has even expanded access to care.
However, this strategy cannot completely replace comprehensive in-person evaluation. Variability in video and photographic quality limits evaluation, while in-person physical examination can reveal subtle morphologic clues necessary for diagnosis. 5 8 Additionally, unequal access to technology may disadvantage some patients. For dermatologists to provide optimal care and continue to contribute accurate and insightful observations into COVID-19, it is essential to be physically present in the clinic and in the hospital when necessary, caring for patients in need of dermatologic expertise. Creative management strategies developed during this time will benefit patients and expand the reach of the specialty . 5 8
Final Thoughts
The COVID-19 pandemic has profoundly challenged the medical community and dermatology is no exception. By documenting and characterizing the diverse cutaneous manifestations of this novel disease, dermatologists have furthered understanding of its pathophysiology and management. By adapting quickly and developing creative ways to deliver care, dermatologists have found ways to contribute, both large and small. As we take stock at this juncture of the pandemic, it is clear there remains much to learn. We hope dermatologists will continue to take an active role in meeting the challenges of this time.
- Wiersinga WJ, Rhodes A, Cheng AC, et al. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA . 2020;324:782-793. doi:10.1001/jama.2020.12839
- New York Times . Updated December 23, 2020. Accessed March 22, 2021. https://www.nytimes.com/2020/11/15/us/coronavirus-us-cases-deaths.html
- Guan W, Ni Z, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med . 2020;382:1708-1720. doi:10.1056/NEJMoa2002032
- Lechien JR, Chiesa-Estomba CM, Place S, et al. Clinical and epidemiological characteristics of 1420 European patients with mild-to-moderate coronavirus disease 2019. J Intern Med . 2020;288:335-344. doi:https://doi.org/10.1111/joim.13089
- Wu J, Liu J, Zhao X, et al. Clinical characteristics of imported cases of coronavirus disease 2019 (COVID-19) in Jiangsu province: a multicenter descriptive study. Clin Infect Dis . 2020;71:706-712. doi:10.1093/cid/ciaa199
- Goyal P, Choi JJ, Pinheiro LC, et al. Clinical characteristics of COVID-19 in New York City. N Engl J Med . 2020;382:2372-2374. doi:10.1056/NEJMc2010419
- Sun L, Shen L, Fan J, et al. Clinical features of patients with coronavirus disease 2019 from a designated hospital in Beijing, China. J Med Virol . 2020;92:2055-2066. https://doi.org/10.1002/jmv.25966
- Recalcati S. Cutaneous manifestations in COVID-19: a first perspective. J Eur Acad Dermatology Venereol . 2020;34:E212-E213. https://doi.org/10.1111/jdv.16387
- Giavedoni P, Podlipnik S, Pericàs JM, et al. Skin manifestations in COVID-19: prevalence and relationship with disease severity. J Clin Med . 2020;9:3261. doi:10.3390/jcm9103261
- Jimenez-Cauhe J, Ortega-Quijano D, Prieto-Barrios M, et al. Reply to “COVID-19 can present with a rash and be mistaken for dengue”: petechial rash in a patient with COVID-19 infection. J Am Acad Dermatol . 2020;83:E141-E142. doi:10.1016/j.jaad.2020.04.016
- Feldstein LR, Rose EB, Horwitz SM, et al. Multisystem inflammatory syndrome in U.S. children and adolescents. N Engl J Med . 2020;383:334-346. doi:10.1056/NEJMoa2021680
- Shinkai K, Bruckner AL. Dermatology and COVID-19. JAMA . 2020;324:1133-1134. doi:10.1001/jama.2020.15276
- Freeman EE, McMahon DE, Lipoff JB, et al. The spectrum of COVID-19-associated dermatologic manifestations: an international registry of 716 patients from 31 countries. J Am Acad Dermatol . 2020;83:1118-1129. doi:10.1016/j.jaad.2020.06.1016
- Galván Casas C, Català A, Carretero Hernández G, et al. Classification of the cutaneous manifestations of COVID-19: a rapid prospective nationwide consensus study in Spain with 375 cases. Br J Dermatol . 2020;183:71-77. https://doi.org/10.1111/bjd.19163
- Bouaziz JD, Duong TA, Jachiet M, et al. Vascular skin symptoms in COVID-19: a French observational study. J Eur Acad Dermatology Venereol . 2020;34:E451-E452. https://doi.org/10.1111/jdv.16544
- Fernandez-Nieto D, Ortega-Quijano D, Jimenez-Cauhe J, et al. Clinical and histological characterization of vesicular COVID-19 rashes: a prospective study in a tertiary care hospital. Clin Exp Dermatol . 2020;45:872-875. https://doi.org/10.1111/ced.14277
- Fernandez-Nieto D, Jimenez-Cauhe J, Suarez-Valle A, et al. Characterization of acute acral skin lesions in nonhospitalized patients: a case series of 132 patients during the COVID-19 outbreak. J Am Acad Dermatol . 2020;83:E61-E63. doi:10.1016/j.jaad.2020.04.093
- Marzano AV, Genovese G, Fabbrocini G, et al. Varicella-like exanthem as a specific COVID-19-associated skin manifestation: Multicenter case series of 22 patients. J Am Acad Dermatol . 2020;83:280-285. doi:10.1016/j.jaad.2020.04.044
- Fernandez-Nieto D, Ortega-Quijano D, Segurado-Miravalles G, et al. Comment on: cutaneous manifestations in COVID-19: a first perspective. safety concerns of clinical images and skin biopsies. J Eur Acad Dermatol Venereol . 2020;34:E252-E254. https://doi.org/10.1111/jdv.16470
- Herrero-Moyano M, Capusan TM, Andreu-Barasoain M, et al. A clinicopathological study of eight patients with COVID-19 pneumonia and a late-onset exanthema. J Eur Acad Dermatol Venereol . 2020;34:E460-E464. https://doi.org/10.1111/jdv.16631
- Rubio-Muniz CA, Puerta-Peñ a M, Falkenhain-L ópez D, et al. The broad spectrum of dermatological manifestations in COVID-19: clinical and histopathological features learned from a series of 34 cases. J Eur Acad Dermatol Venereol . 2020;34:E574-E576. https://doi.org/10.1111/jdv.16734
- Jimenez-Cauhe J, Ortega-Quijano D, Carretero-Barrio I, et al. Erythema multiforme-like eruption in patients with COVID-19 infection: clinical and histological findings. Clin Exp Dermatol . 2020;45:892-895. https://doi.org/10.1111/ced.14281
- Jimenez-Cauhe J, Ortega-Quijano D, de Perosanz-Lobo D, et al. Enanthem in patients with COVID-19 and skin rash. JAMA Dermatol . 2020;156:1134-1136. doi:10.1001/jamadermatol.2020.2550
- Le Cleach L, Dousset L, Assier H, et al. Most chilblains observed during the COVID-19 outbreak occur in patients who are negative for COVID-19 on polymerase chain reaction and serology testing. Br J Dermatol . 2020;183:866-874. https://doi.org/10.1111/bjd.19377
- Hubiche T, Cardot-Leccia N, Le Duff F, et al. Clinical, laboratory, and interferon-alpha response characteristics of patients with chilblain-like lesions during the COVID-19 pandemic [published online November 25, 2020]. JAMA Dermatol . doi:10.1001/jamadermatol.2020.4324
- Freeman EE, McMahon DE, Lipoff JB, et al. Pernio-like skin lesions associated with COVID-19: a case series of 318 patients from 8 countries. J Am Acad Dermatol . 2020;83:486-492. doi:10.1016/j.jaad.2020.05.109
- Magro CM, Mulvey JJ, Laurence J, et al. The differing pathophysiologies that underlie COVID-19-associated perniosis and thrombotic retiform purpura: a case series. Br J Dermatol . 2021;184:141-150. https://doi.org/10.1111/bjd.19415
- Colmenero I, Santonja C, Alonso-Riaño M, et al. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: histopathological, immunohistochemical and ultrastructural study of seven paediatric cases. Br J Dermatol . 2020;183:729-737. doi:10.1111/bjd.19327
- Droesch C, Do MH, DeSancho M, et al. Livedoid and purpuric skin eruptions associated with coagulopathy in severe COVID-19. JAMA Dermatol . 2020;156:1-3. doi:10.1001/jamadermatol.2020.2800
- Asakura H, Ogawa H. COVID-19-associated coagulopathy and disseminated intravascular coagulation. Int J Hematol . 2021;113:45-57. doi:10.1007/s12185-020-03029-y
- Dufort EM, Koumans EH, Chow EJ, et al. Multisystem inflammatory syndrome in children in New York State. N Engl J Med . 2020;383:347-358. doi:10.1056/NEJMoa2021756
- Young TK, Shaw KS, Shah JK, et al. Mucocutaneous manifestations of multisystem inflammatory syndrome in children during the COVID-19 pandemic. JAMA Dermatol . 2021;157:207-212. doi:10.1001/jamadermatol.2020.4779
- Whittaker E, Bamford A, Kenny J, et al. Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. JAMA. 2020;324:259-269. doi:10.1001/jama.2020.10369
- Cheng MH, Zhang S, Porritt RA, et al. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation.
- Morris SB, Schwartz NG, Patel P, et al. Case series of multisystem inflammatory syndrome in adults associated with SARS-CoV-2 Infection—United Kingdom and United States, March–August 2020. MMWR Morb Mortal Wkly Rep. 2020;69:1450-1456. doi:10.15585/mmwr.mm6940e1
- Blanco-Melo D, Nilsson-Payant BE, Liu W-C, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036.e9-1045.e9. doi:10.1016/j.cell.2020.04.026
- Crow YJ, Manel N. Aicardi–Goutières syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15:429-440. doi:10.1038/nri3850
- Hadjadj J, Yatim N, Barnabei L, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718-724. doi:10.1126/science.abc6027
- Zimmermann N, Wolf C, Schwenke R, et al. Assessment of clinical response to janus kinase inhibition in patients with familial chilblain lupus and TREX1 mutation. JAMA Dermatol. 2019;155:342-346. doi:10.1001/jamadermatol.2018.5077
- Hubiche T, Le Duff F, Chiaverini C, et al. Negative SARS-CoV-2 PCR in patients with chilblain-like lesions. Lancet Infect Dis. 2021;21:315-316. doi:10.1016/S1473-3099(20)30518-1
- Agrawal A. Mechanisms and implications of age-associated impaired innate interferon secretion by dendritic cells: a mini-review. Gerontology. 2013;59:421-426. doi:10.1159/000350536
- Kalil AC, Patterson TF, Mehta AK, et al. Baricitinib plus remdesivir for hospitalized adults with COVID-19. N Engl J Med. 2021;384:795-807. doi:10.1056/NEJMoa2031994
- US Food and Drug Administration. Fact sheet for healthcare providers: emergency use authorization (EUA) of baricitinib. Accessed March 29, 2021. https://www.fda.gov/media/143823/download
- Konno Y, Kimura I, Uriu K, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 2020;32:108185. doi:10.1016/j.celrep.2020.108185
- Sacks D, Baxter B, Campbell BCV, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke: from the American Association of Neurological Surgeons (AANS), American Society of Neuroradiology (ASNR), Cardiovascular and Interventional Radiology Society of Europe (CIRSE), Canadian Interventional Radiology Association (CIRA), Congress of Neurological Surgeons (CNS), European Society of Minimally Invasive Neurological Therapy (ESMINT), European Society of Neuroradiology (ESNR), European Stroke Organization (ESO), Society for Cardiovascular Angiography and Interventions (SCAI), Society of Interventional Radiology (SIR), Society of NeuroInterventional Surgery (SNIS), and World Stroke Organization (WSO). J Vasc Interv Radiol. 2018;29:441-453. doi:10.1016/j.jvir.2017.11.026
- Lo MW, Kemper C, Woodruff TM. COVID-19: complement, coagulation, and collateral damage. J Immunol. 2020;205:1488-1495. doi:10.4049/jimmunol.2000644
- Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1-13. doi:10.1016/j.trsl.2020.04.007
- Yan B, Freiwald T, Chauss D, et al. SARS-CoV2 drives JAK1/2-dependent local and systemic complement hyper-activation [published online June 9, 2020]. Res Sq. doi:10.21203/rs.3.rs-33390/v1
- Marietta M, Coluccio V, Luppi M. COVID-19, coagulopathy and venous thromboembolism: more questions than answers. Intern Emerg Med. 2020;15:1375-1387. doi:10.1007/s11739-020-02432-x
- Zuo Y, Estes SK, Ali RA, et al. Prothrombotic antiphospholipid antibodies in COVID-19 [published online June 17, 2020]. medRxiv. doi:10.1101/2020.06.15.20131607
- Lan S-H, Lai C-C, Huang H-T, et al. Tocilizumab for severe COVID-19: a systematic review and meta-analysis. Int J Antimicrob Agents. 2020;56:106103. doi:10.1016/j.ijantimicag.2020.106103
- McMahon D, Gallman A, Hruza G, et al. COVID-19 “long-haulers” in dermatology? duration of dermatologic symptoms in an international registry from 39 countries. Abstract presented at: 29th EADV Congress; October 29, 2020. Accessed March 29, 2020. https://eadvdistribute.m-anage.com/from.storage?image=PXQEdDtICIihN3sM_8nAmh7p_y9AFijhQlf2-_KjrtYgOsOXNVwGxDdti95GZ2Yh0
- Saag MS. Misguided use of hydroxychloroquine for COVID-19: the infusion of politics into science. JAMA. 2020;324:2161-2162. doi:10.1001/jama.2020.22389
- Zahedi Niaki O, Anadkat MJ, Chen ST, et al. Navigating immunosuppression in a pandemic: a guide for the dermatologist from the COVID Task Force of the Medical Dermatology Society and Society of Dermatology Hospitalists. J Am Acad Dermatol. 2020;83:1150-1159. doi:10.1016/j.jaad.2020.06.051
- Hammond MI, Sharma TR, Cooper KD, et al. Conducting inpatient dermatology consultations and maintaining resident education in the COVID-19 telemedicine era. J Am Acad Dermatol. 2020;83:E317-E318. doi:10.1016/j.jaad.2020.07.008
- Brunasso AMG, Massone C. Teledermatologic monitoring for chronic cutaneous autoimmune diseases with smartworking during COVID-19 emergency in a tertiary center in Italy. Dermatol Ther. 2020;33:E13495-E13495. doi:10.1111/dth.13695
- Trinidad J, Kroshinsky D, Kaffenberger BH, et al. Telemedicine for inpatient dermatology consultations in response to the COVID-19 pandemic. J Am Acad Dermatol. 2020;83:E69-E71. doi:10.1016/j.jaad.2020.04.096
- Madigan LM, Micheletti RG, Shinkai K. How dermatologists can learn and contribute at the leading edge of the COVID-19 global pandemic. JAMA Dermatology. 2020;156:733-734. doi:10.1001/jamadermatol.2020.1438
The virus that causes COVID-19—SARS-CoV-2—has infected more than 128 million individuals, resulting in more than 2.8 million deaths worldwide between December 2019 and April 2021. Disease mortality primarily is driven by hypoxemic respiratory failure and systemic hypercoagulability, resulting in multisystem organ failure.1 With more than 17 million Americans infected, the virus is estimated to have impacted someone within the social circle of nearly every American.2
The COVID-19 pandemic has highlighted resource limitations, delayed elective and preventive care, and rapidly increased the adoption of telemedicine, presenting a host of new challenges to providers in every medical specialty, including dermatology. Although COVID-19 primarily is a respiratory disease, clinical manifestations have been observed in nearly every organ, including the skin. The cutaneous manifestations of COVID-19 provide insight into disease diagnosis, prognosis, and pathophysiology. In this article, we review the cutaneous manifestations of COVID-19 and explore the state of knowledge regarding their pathophysiology and clinical significance. Finally, we discuss the role of dermatology consultants in the care of patients with COVID-19, and the impact of the pandemic on the field of dermatology.
Prevalence of Cutaneous Findings in COVID-19
Early reports characterizing the clinical presentation of patients hospitalized with COVID-19 suggested skin findings associated with the disease were rare. Cohort studies from Europe, China, and New York City in January through March 2020 reported a low prevalence or made no mention of rash.3-7 However, reports from dermatologists in Italy that emerged in May 2020 indicated a substantially higher proportion of cutaneous disease: 18 of 88 (20.4%) hospitalized patients were found to have cutaneous involvement, primarily consisting of erythematous rash, along with some cases of urticarial and vesicular lesions.8 In October 2020, a retrospective cohort study from Spain examining 2761 patients presenting to the emergency department or admitted to the hospital for COVID-19 found that 58 (2.1%) patients had skin lesions attributed to COVID-19.9
The wide range in reported prevalence of skin lesions may be due to variable involvement of dermatologic specialists in patient care, particularly in China.10 Some variation also may be due to variability in the timing of clinical examination, as well as demographic and clinical differences in patient populations. Of note, a multisystem inflammatory disease seen in US children subsequent to infection with COVID-19 has been associated with rash in as many as 74% of cases.11 Although COVID-19 disproportionately impacts people with skin of color, there are few reports of cutaneous manifestations in that population,12 highlighting the challenges of the dermatologic examination in individuals with darker skin and suggesting the prevalence of dermatologic disease in COVID-19 may be greater than reported.
Morphologic Patterns of Cutaneous Involvement in COVID-19
Researchers in Europe and the United States have attempted to classify the cutaneous manifestations of COVID-19. A registry established through the American Academy of Dermatology published a compilation of reports from 31 countries, totaling 716 patient profiles.13 A prospective Spanish study detailed the cutaneous involvement of 375 patients with suspected or confirmed COVID-19.14 Together, these efforts have revealed several distinct patterns of cutaneous involvement associated with COVID-19 (Table).9,15-18
Vesicular Rash
Vesicular rash associated with COVID-19 has been described in several studies and case series8,13,14 and is considered, along with the pseudopernio (or pseudochilblains) morphology, to be one of the more disease-specific patterns in COVID-19.14,18 Vesicular rash appears to comprise roughly one-tenth of all COVID-19–associated rashes.13,14 It usually is described as pruritic, with 72% to 83% of patients reporting itch.13,16
Small monomorphic or polymorphic vesicles predominantly on the trunk and to a lesser extent the extremities and head have been described by multiple authors.14,16 Vesicular rash is most common among middle-aged individuals, with studies reporting median and mean ages ranging from 40.5 to 55 years.9,13,14,16
Vesicular rash develops concurrent with or after other presenting symptoms of COVID-19; in 2 studies, vesicular rash preceded development of other symptoms in only 15% and 5.6% of cases, respectively.13,14 Prognostically, vesicular rash is associated with moderate disease severity.14,16 It may persist for an average of 8 to 10 days.14,16,18
Histopathologic examination reveals basal layer vacuolar degeneration, hyperchromatic keratinocytes, acantholysis, and dyskeratosis.9,16,18
Urticarial Rash
Urticarial lesions represent approximately 7% to 19% of reported COVID-19–associated rashes.9,13,14 Urticarial rashes in patients testing positive for SARS-CoV-2 primarily occur on the trunk.14 The urticaria, which typically last about 1 week,14 are seen most frequently in middle-aged patients (mean/median age, 42–48 years)13,14 and are associated with pruritus, which has been reported in 74% to 92% of patients.13,14 Urticarial lesions typically do not precede other symptoms of COVID-19 and are nonspecific, making them less useful diagnostically.14
Urticaria appears to be associated with more severe COVID-19 illness in several studies, but this finding may be confounded by several factors, including older age, increased tobacco use, and polypharmacy. Of 104 patients with reported urticarial rash and suspected or confirmed COVID-19 across 3 studies, only 1 death was reported.9,13,14
The histopathologic appearance is that of typical hives, demonstrating a perivascular infiltrate of lymphocytes and eosinophils with edema of the upper dermis.9,19
Morbilliform Eruption
Morbilliform eruption is a commonly reported morphology associated with COVID-19, accounting for 20% to 47% of rashes.9,13,14 This categorization may have limited utility from a diagnostic and prognostic perspective, given that morbilliform eruptions are common, nonspecific, and heterogenous and can arise from many causes.9,13,14 Onset of morbilliform eruption appears to coincide with14 or follow13,20,21 the development of other COVID-19–related symptoms, with 5% of patients reporting morbilliform rash as the initial manifestation of infection.13,14 Morbilliform eruptions have been observed to occur in patients with more severe disease.9,13,14
Certain morphologic subtypes, such as erythema multiforme–like, erythema elevatum diutinum–like, or pseudovesicular, may be more specific to COVID-19 infection.14 A small case series highlighted 4 patients with erythema multiforme–like eruptions, 3 of whom also were found to have petechial enanthem occurring after COVID-19 diagnosis; however, the investigators were unable to exclude drug reaction as a potential cause of rash in these patients.22 Another case series of 21 patients with COVID-19 and skin rash described a (primarily) petechial enanthem on the palate in 6 (28.5%) patients.23 It is unclear to what extent oral enanthem may be underrecognized given that some physicians may be disinclined to remove the masks of known COVID-19–positive patients to examine the oral cavity.
The histologic appearance of morbilliform rash seen in association with COVID-19 has been described as spongiotic with interface dermatitis with perivascular lymphocytic inflammation.9,21
COVID Toes, Pseudochilblains Rash, Perniolike Rash, and Acral Erythema/Edema
Of all the rashes associated with COVID-19, COVID toes, or pseudochilblains rash, has perhaps attracted the most attention. The characteristic violaceous erythema on the fingers and/or toes may be itchy or painful, presenting similar to idiopathic cases of pernio (Figure 1).14 The entity has been controversial because of an absence of a clear correlation with a positive SARS-CoV-2 polymerase chain reaction test or antibodies to the virus in a subset of reported cases.24,25 Onset of the rash late in the disease course, generally after symptom resolution in mild or asymptomatic cases, may explain the absence of viral DNA in the nasopharynx by the time of lesion appearance.14,26 Seronegative patients may have cleared SARS-CoV-2 infection before humoral immunity could occur via a strong type 1 interferon response.25
Across 3 studies, perniolike skin lesions constituted 18% to 29% of COVID-19–associated skin findings9,13,14 and persisted for an average of 12 to 14 days.13,14 Perniolike lesions portend a favorable outcome; patients with COVID toes rarely present with systemic symptoms or laboratory or imaging abnormalities9 and less commonly require hospitalization for severe illness. Perniolike lesions have been reported most frequently in younger patients, with a median or mean age of 32 to 35 years.13,14
Histology demonstrates lichenoid dermatitis with perivascular and periadnexal lymphocytic infiltrates.9 Notably, one study observed interface dermatitis of the intraepidermal portion of the acrosyringium, a rare finding in chilblain lupus, in 83% of patients (N=40).25 Direct immunofluorescence demonstrates a vasculopathic pattern, with some patients showing deposition of IgM or IgG, C3, and fibrinogen in dermal blood vessels. Vascular C9 deposits also have been demonstrated on immunohistochemistry.9 Biopsies of perniolike lesions in COVID-19 patients have demonstrated the presence of SARS-CoV-2 RNA,27 have identified SARS-CoV-2 spike protein in endothelial cells on immunohistochemistry, and have visualized intracytoplasmic viral particles in vascular endothelium on electron microscopy.28
Livedoid Rash/Retiform Purpura
Netlike purpuric or violaceous patches signifying vessel damage or occlusion have been seen in association with COVID-19, constituting approximately 6% of COVID-19–associated skin findings in 2 studies.13,14 Livedoid rash (Figure 2) and retiform purpura (Figure 3) are associated with older age and occur primarily in severely ill patients, including those requiring intensive care. In a registry of 716 patients with COVID-19, 100% of patients with retiform purpura were hospitalized, and 82% had acute respiratory distress syndrome.13 In another study, 33% (7/21) of patients with livedoid and necrotic lesions required intensive care, and 10% (2/21) died.14
Livedoid lesions and retiform purpura represent thrombotic disease in the skin due to vasculopathy/coagulopathy. Dermatopathology available through the American Academy of Dermatology registry revealed thrombotic vasculopathy.13 A case series of 4 patients with livedo racemosa and retiform purpura demonstrated pauci-inflammatory thrombogenic vasculopathy involving capillaries, venules, and arterioles with complement deposition.29 Livedoid and retiform lesions in the skin may be associated with a COVID-19–induced coagulopathy, a propensity for systemic clotting including pulmonary embolism, which mostly occurs in hospitalized patients with severe illness.30
Multisystem Inflammatory Disease in Children
A hyperinflammatory syndrome similar to Kawasaki disease and toxic shock syndrome associated with mucocutaneous, cardiac, and gastrointestinal manifestations has been reported following COVID-19 infection.31 This syndrome, known as multisystem inflammatory syndrome in children (MIS-C), predominantly affects adolescents and children older than 5 years,11 typically occurs 2 to 4 weeks after infection, and appears to be at least 100-times less common than COVID-19 infection among the same age group.31 Sixty percent31 to 74%11 of affected patients have mucocutaneous involvement, with the most common clinical findings being conjunctival injection, palmoplantar erythema, lip hyperemia, periorbital erythema and edema, strawberry tongue, and malar erythema, respectively.32
Because this condition appears to reflect an immune response to the virus, the majority of cases demonstrate negative SARS-CoV-2 polymerase chain reaction and positive antibody testing.33 Although cutaneous findings are similar to those seen in Kawasaki disease, certain findings have been noted in MIS-C that are not typical of Kawasaki disease, including heliotrope rash–like periorbital edema and erythema as well as erythema infectiosum–like malar erythema and reticulated erythematous eruptions.32
The course of MIS-C can be severe; in one case series of patients presenting with MIS-C, 80% (79/99) required intensive care unit admission, with 10% requiring mechanical ventilation and 2% of patients dying during admission.31 Cardiac dysfunction, coagulopathy, and gastrointestinal symptoms are common.11,31 It has been postulated that a superantigenlike region of the SARS-CoV-2 spike protein, similar to that of staphylococcal enterotoxin B, may underlie MIS-C and account for its similarities to toxic shock syndrome.34 Of note, a similar multisystem inflammatory syndrome associated with COVID-19 also has been described in adults, and it too may present with rash as a cardinal feature.35
Pathophysiology of COVID-19: What the Skin May Reveal About the Disease
The diverse range of cutaneous manifestations in COVID-19 reflects a spectrum of host immunologicresponses to SARS-CoV-2 and may inform the pathophysiology of the disease as well as potential treatment modalities.
Host Response to SARS-CoV-2
The body’s response to viral infection is 2-pronged, involving activation of cellular antiviral defenses mediated by type I and III interferons, as well as recruitment of leukocytes, mobilized by cytokines and chemokines.36,37 Infection with SARS-CoV-2 results in a unique inflammatory response characterized by suppression of interferons, juxtaposed with a rampant proinflammatory cytokine and chemokine response, reminiscent of a cytokine storm. Reflective of this imbalance, a study of 50 COVID-19 patients and 20 healthy controls found decreased natural killer cells and CD3+ T cells in COVID-19 patients, particularly severely or critically ill patients, with an increase in B cells and monocytes.38 This distinctive immune imbalance positions SARS-CoV-2 to thrive in the absence of inhibitory interferon activity while submitting the host to the deleterious effects of a cytokine surge.36
Type I Interferons
The perniolike lesions associated with mild COVID-19 disease14 may represent a robust immune response via effective stimulation of type I interferons (IFN-1). Similar perniolike lesions are observed in Aicardi-Goutières syndrome37 and familial chilblain lupus, hereditary interferonopathies associated with mutations in the TREX1 (three prime repair exonuclease 1) gene and characterized by inappropriate upregulation of IFN-1,39 resulting in chilblains. It has been suggested that perniolike lesions in COVID-19 result from IFN-1 activation—a robust effective immunologic response to the virus.14,26,40
On the other end of the spectrum, patients with severe COVID-19 may have a blunted IFN-1 response and reduced IFN-1–stimulated gene expression.36,38 Notably, low IFN-1 response preceded clinical deterioration and was associated with increased risk for evolution to critical illness.38 Severe disease from COVID-19 also is more commonly observed in older patients and those with comorbidities,1 both of which are known factors associated with depressed IFN-1 function.38,41 Reflective of this disparate IFN-1 response, biopsies of COVID-19 perniosis have demonstrated striking expression of myxovirus resistance protein A (MXA), a marker for IFN-1 signaling in tissue, whereas its expression is absent in COVID-19 livedo/retiform purpura.27
Familial chilblain lupus may be effectively treated by the Janus kinase inhibitor baricitinib,39 which inhibits IFN-1 signaling. Baricitinib recently received emergency use authorization by the US Food and Drug Administration for treatment of severe COVID-19 pneumonia,42,43 hinting to disordered IFN-1 signaling in the COVID-19 pathophysiology.
The impaired IFN-1 response in COVID-19 patients may be due to a unique characteristic of SARS-CoV-2: its ORF3b gene is a potent IFN-1 antagonist. In a series of experiments comparing SARS-CoV-2 to the related virus severe acute respiratory disease coronavirus (which was responsible for an epidemic in 2002), Konno et al44 found that SARS-CoV-2 is more effectively able to downregulate host IFN-1, likely due to premature stop codons on ORF3b that produce a truncated version of the gene with amplified anti–IFN-1 activity.
Cytokine Storm and Coagulation Cascade
This dulled interferon response is juxtaposed with a surge of inflammatory chemokines and cytokines, including IL-6, IL-8, IL-10, and tumor necrosis factor α, impairing innate immunity and leading to end-organ damage. This inflammatory response is associated with the influx of innate immune cells, specifically neutrophils and monocytes, which likely contribute to lung injury in COVID-19 acute respiratory distress syndrome.38 It also is thought to lead to downstream activation of coagulation, with a high incidence of thrombotic events observed in patients with severe COVID-19.1 In a retrospective study of 184 intensive care patients with COVID-19 receiving at least standard doses of thromboprophylaxis, venous thromboembolism occurred in 27% and arterial thrombotic events occurred in 3.7%.45
Livedo racemosa and retiform purpura are cutaneous markers of hypercoagulability, which indicate an increased risk for systemic clotting in COVID-19. A positive feedback loop between the complement and coagulation cascades appears to be important.13,14,29,46-48 In addition, a few studies have reported antiphospholipid antibody positivity in hospitalized COVID-19 patients.49,50
The high incidence of coagulopathy in severe COVID-19 has prompted many institutions to develop aggressive prophylactic anticoagulation protocols. Elevation of proinflammatory cytokines and observation of terminal complement activation in the skin and other organs has led to therapeutic trials of IL-6 inhibitors such as tocilizumab,51 complement inhibitors such as eculizumab, and Janus kinase inhibitors such as ruxolitinib and baricitinib.42,48
COVID Long-Haulers
The long-term effects of immune dysregulation in COVID-19 patients remain to be seen. Viral triggering of autoimmune disease is a well-established phenomenon, seen in DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome and other dermatologic diseases, raising the possibility that dermatologists will see a rising incidence of cutaneous autoimmune disease in the aftermath of the pandemic. Disordered interferon stimulation could lead to increased incidence of interferon-mediated disorders, such as sarcoidosis and other granulomatous diseases. Vasculitislike skin lesions could persist beyond the acute infectious period. Recent data from a registry of 990 COVID-19 cases from 39 countries suggest that COVID-19 perniolike lesions may persist as long as 150 days.52 In a time of many unknowns, these questions serve as a call to action for rigorous data collection, contribution to existing registries for dermatologic manifestations of COVID-19, and long-term follow-up of COVID-19 patients by the dermatology community.
Pandemic Dermatology
The pandemic has posed unprecedented challenges for patient care. The use of hydroxychloroquine as a popular but unproven treatment for COVID-19, 53 particularly early in the pandemic, has resulted in drug shortages for patients with lupus and other autoimmune skin diseases. Meanwhile, the need for patients with complex dermatologic conditions to receive systemic immunosuppression has had to be balanced against the associated risks during a global pandemic. To help dermatologists navigate this dilemma, various subspecialty groups have issued guidelines, including the COVID-19 Task Force of the Medical Dermatology Society and Society of Dermatology Hospitalists, which recommends a stepwise approach to shared decision-making with the goal of minimizing both the risk for disease flare and that of infection. The use of systemic steroids and rituximab, as well as the dose of immunosuppression—particularly broad-acting immunosuppression—should be limited where permitted. 54
Rapid adoption of telemedicine and remote monitoring strategies has enabled dermatologists to provide safe and timely care when in-person visits have not been possible, including for patients with confirmed or suspected COVID-19, as well as for hospitalized patients. 55-57 Use of telemedicine has facilitated preservation of personal protective equipment at a time when these important resources have been scarce. For patients with transportation or scheduling barriers, telemedicine has even expanded access to care.
However, this strategy cannot completely replace comprehensive in-person evaluation. Variability in video and photographic quality limits evaluation, while in-person physical examination can reveal subtle morphologic clues necessary for diagnosis. 5 8 Additionally, unequal access to technology may disadvantage some patients. For dermatologists to provide optimal care and continue to contribute accurate and insightful observations into COVID-19, it is essential to be physically present in the clinic and in the hospital when necessary, caring for patients in need of dermatologic expertise. Creative management strategies developed during this time will benefit patients and expand the reach of the specialty . 5 8
Final Thoughts
The COVID-19 pandemic has profoundly challenged the medical community and dermatology is no exception. By documenting and characterizing the diverse cutaneous manifestations of this novel disease, dermatologists have furthered understanding of its pathophysiology and management. By adapting quickly and developing creative ways to deliver care, dermatologists have found ways to contribute, both large and small. As we take stock at this juncture of the pandemic, it is clear there remains much to learn. We hope dermatologists will continue to take an active role in meeting the challenges of this time.
The virus that causes COVID-19—SARS-CoV-2—has infected more than 128 million individuals, resulting in more than 2.8 million deaths worldwide between December 2019 and April 2021. Disease mortality primarily is driven by hypoxemic respiratory failure and systemic hypercoagulability, resulting in multisystem organ failure.1 With more than 17 million Americans infected, the virus is estimated to have impacted someone within the social circle of nearly every American.2
The COVID-19 pandemic has highlighted resource limitations, delayed elective and preventive care, and rapidly increased the adoption of telemedicine, presenting a host of new challenges to providers in every medical specialty, including dermatology. Although COVID-19 primarily is a respiratory disease, clinical manifestations have been observed in nearly every organ, including the skin. The cutaneous manifestations of COVID-19 provide insight into disease diagnosis, prognosis, and pathophysiology. In this article, we review the cutaneous manifestations of COVID-19 and explore the state of knowledge regarding their pathophysiology and clinical significance. Finally, we discuss the role of dermatology consultants in the care of patients with COVID-19, and the impact of the pandemic on the field of dermatology.
Prevalence of Cutaneous Findings in COVID-19
Early reports characterizing the clinical presentation of patients hospitalized with COVID-19 suggested skin findings associated with the disease were rare. Cohort studies from Europe, China, and New York City in January through March 2020 reported a low prevalence or made no mention of rash.3-7 However, reports from dermatologists in Italy that emerged in May 2020 indicated a substantially higher proportion of cutaneous disease: 18 of 88 (20.4%) hospitalized patients were found to have cutaneous involvement, primarily consisting of erythematous rash, along with some cases of urticarial and vesicular lesions.8 In October 2020, a retrospective cohort study from Spain examining 2761 patients presenting to the emergency department or admitted to the hospital for COVID-19 found that 58 (2.1%) patients had skin lesions attributed to COVID-19.9
The wide range in reported prevalence of skin lesions may be due to variable involvement of dermatologic specialists in patient care, particularly in China.10 Some variation also may be due to variability in the timing of clinical examination, as well as demographic and clinical differences in patient populations. Of note, a multisystem inflammatory disease seen in US children subsequent to infection with COVID-19 has been associated with rash in as many as 74% of cases.11 Although COVID-19 disproportionately impacts people with skin of color, there are few reports of cutaneous manifestations in that population,12 highlighting the challenges of the dermatologic examination in individuals with darker skin and suggesting the prevalence of dermatologic disease in COVID-19 may be greater than reported.
Morphologic Patterns of Cutaneous Involvement in COVID-19
Researchers in Europe and the United States have attempted to classify the cutaneous manifestations of COVID-19. A registry established through the American Academy of Dermatology published a compilation of reports from 31 countries, totaling 716 patient profiles.13 A prospective Spanish study detailed the cutaneous involvement of 375 patients with suspected or confirmed COVID-19.14 Together, these efforts have revealed several distinct patterns of cutaneous involvement associated with COVID-19 (Table).9,15-18
Vesicular Rash
Vesicular rash associated with COVID-19 has been described in several studies and case series8,13,14 and is considered, along with the pseudopernio (or pseudochilblains) morphology, to be one of the more disease-specific patterns in COVID-19.14,18 Vesicular rash appears to comprise roughly one-tenth of all COVID-19–associated rashes.13,14 It usually is described as pruritic, with 72% to 83% of patients reporting itch.13,16
Small monomorphic or polymorphic vesicles predominantly on the trunk and to a lesser extent the extremities and head have been described by multiple authors.14,16 Vesicular rash is most common among middle-aged individuals, with studies reporting median and mean ages ranging from 40.5 to 55 years.9,13,14,16
Vesicular rash develops concurrent with or after other presenting symptoms of COVID-19; in 2 studies, vesicular rash preceded development of other symptoms in only 15% and 5.6% of cases, respectively.13,14 Prognostically, vesicular rash is associated with moderate disease severity.14,16 It may persist for an average of 8 to 10 days.14,16,18
Histopathologic examination reveals basal layer vacuolar degeneration, hyperchromatic keratinocytes, acantholysis, and dyskeratosis.9,16,18
Urticarial Rash
Urticarial lesions represent approximately 7% to 19% of reported COVID-19–associated rashes.9,13,14 Urticarial rashes in patients testing positive for SARS-CoV-2 primarily occur on the trunk.14 The urticaria, which typically last about 1 week,14 are seen most frequently in middle-aged patients (mean/median age, 42–48 years)13,14 and are associated with pruritus, which has been reported in 74% to 92% of patients.13,14 Urticarial lesions typically do not precede other symptoms of COVID-19 and are nonspecific, making them less useful diagnostically.14
Urticaria appears to be associated with more severe COVID-19 illness in several studies, but this finding may be confounded by several factors, including older age, increased tobacco use, and polypharmacy. Of 104 patients with reported urticarial rash and suspected or confirmed COVID-19 across 3 studies, only 1 death was reported.9,13,14
The histopathologic appearance is that of typical hives, demonstrating a perivascular infiltrate of lymphocytes and eosinophils with edema of the upper dermis.9,19
Morbilliform Eruption
Morbilliform eruption is a commonly reported morphology associated with COVID-19, accounting for 20% to 47% of rashes.9,13,14 This categorization may have limited utility from a diagnostic and prognostic perspective, given that morbilliform eruptions are common, nonspecific, and heterogenous and can arise from many causes.9,13,14 Onset of morbilliform eruption appears to coincide with14 or follow13,20,21 the development of other COVID-19–related symptoms, with 5% of patients reporting morbilliform rash as the initial manifestation of infection.13,14 Morbilliform eruptions have been observed to occur in patients with more severe disease.9,13,14
Certain morphologic subtypes, such as erythema multiforme–like, erythema elevatum diutinum–like, or pseudovesicular, may be more specific to COVID-19 infection.14 A small case series highlighted 4 patients with erythema multiforme–like eruptions, 3 of whom also were found to have petechial enanthem occurring after COVID-19 diagnosis; however, the investigators were unable to exclude drug reaction as a potential cause of rash in these patients.22 Another case series of 21 patients with COVID-19 and skin rash described a (primarily) petechial enanthem on the palate in 6 (28.5%) patients.23 It is unclear to what extent oral enanthem may be underrecognized given that some physicians may be disinclined to remove the masks of known COVID-19–positive patients to examine the oral cavity.
The histologic appearance of morbilliform rash seen in association with COVID-19 has been described as spongiotic with interface dermatitis with perivascular lymphocytic inflammation.9,21
COVID Toes, Pseudochilblains Rash, Perniolike Rash, and Acral Erythema/Edema
Of all the rashes associated with COVID-19, COVID toes, or pseudochilblains rash, has perhaps attracted the most attention. The characteristic violaceous erythema on the fingers and/or toes may be itchy or painful, presenting similar to idiopathic cases of pernio (Figure 1).14 The entity has been controversial because of an absence of a clear correlation with a positive SARS-CoV-2 polymerase chain reaction test or antibodies to the virus in a subset of reported cases.24,25 Onset of the rash late in the disease course, generally after symptom resolution in mild or asymptomatic cases, may explain the absence of viral DNA in the nasopharynx by the time of lesion appearance.14,26 Seronegative patients may have cleared SARS-CoV-2 infection before humoral immunity could occur via a strong type 1 interferon response.25
Across 3 studies, perniolike skin lesions constituted 18% to 29% of COVID-19–associated skin findings9,13,14 and persisted for an average of 12 to 14 days.13,14 Perniolike lesions portend a favorable outcome; patients with COVID toes rarely present with systemic symptoms or laboratory or imaging abnormalities9 and less commonly require hospitalization for severe illness. Perniolike lesions have been reported most frequently in younger patients, with a median or mean age of 32 to 35 years.13,14
Histology demonstrates lichenoid dermatitis with perivascular and periadnexal lymphocytic infiltrates.9 Notably, one study observed interface dermatitis of the intraepidermal portion of the acrosyringium, a rare finding in chilblain lupus, in 83% of patients (N=40).25 Direct immunofluorescence demonstrates a vasculopathic pattern, with some patients showing deposition of IgM or IgG, C3, and fibrinogen in dermal blood vessels. Vascular C9 deposits also have been demonstrated on immunohistochemistry.9 Biopsies of perniolike lesions in COVID-19 patients have demonstrated the presence of SARS-CoV-2 RNA,27 have identified SARS-CoV-2 spike protein in endothelial cells on immunohistochemistry, and have visualized intracytoplasmic viral particles in vascular endothelium on electron microscopy.28
Livedoid Rash/Retiform Purpura
Netlike purpuric or violaceous patches signifying vessel damage or occlusion have been seen in association with COVID-19, constituting approximately 6% of COVID-19–associated skin findings in 2 studies.13,14 Livedoid rash (Figure 2) and retiform purpura (Figure 3) are associated with older age and occur primarily in severely ill patients, including those requiring intensive care. In a registry of 716 patients with COVID-19, 100% of patients with retiform purpura were hospitalized, and 82% had acute respiratory distress syndrome.13 In another study, 33% (7/21) of patients with livedoid and necrotic lesions required intensive care, and 10% (2/21) died.14
Livedoid lesions and retiform purpura represent thrombotic disease in the skin due to vasculopathy/coagulopathy. Dermatopathology available through the American Academy of Dermatology registry revealed thrombotic vasculopathy.13 A case series of 4 patients with livedo racemosa and retiform purpura demonstrated pauci-inflammatory thrombogenic vasculopathy involving capillaries, venules, and arterioles with complement deposition.29 Livedoid and retiform lesions in the skin may be associated with a COVID-19–induced coagulopathy, a propensity for systemic clotting including pulmonary embolism, which mostly occurs in hospitalized patients with severe illness.30
Multisystem Inflammatory Disease in Children
A hyperinflammatory syndrome similar to Kawasaki disease and toxic shock syndrome associated with mucocutaneous, cardiac, and gastrointestinal manifestations has been reported following COVID-19 infection.31 This syndrome, known as multisystem inflammatory syndrome in children (MIS-C), predominantly affects adolescents and children older than 5 years,11 typically occurs 2 to 4 weeks after infection, and appears to be at least 100-times less common than COVID-19 infection among the same age group.31 Sixty percent31 to 74%11 of affected patients have mucocutaneous involvement, with the most common clinical findings being conjunctival injection, palmoplantar erythema, lip hyperemia, periorbital erythema and edema, strawberry tongue, and malar erythema, respectively.32
Because this condition appears to reflect an immune response to the virus, the majority of cases demonstrate negative SARS-CoV-2 polymerase chain reaction and positive antibody testing.33 Although cutaneous findings are similar to those seen in Kawasaki disease, certain findings have been noted in MIS-C that are not typical of Kawasaki disease, including heliotrope rash–like periorbital edema and erythema as well as erythema infectiosum–like malar erythema and reticulated erythematous eruptions.32
The course of MIS-C can be severe; in one case series of patients presenting with MIS-C, 80% (79/99) required intensive care unit admission, with 10% requiring mechanical ventilation and 2% of patients dying during admission.31 Cardiac dysfunction, coagulopathy, and gastrointestinal symptoms are common.11,31 It has been postulated that a superantigenlike region of the SARS-CoV-2 spike protein, similar to that of staphylococcal enterotoxin B, may underlie MIS-C and account for its similarities to toxic shock syndrome.34 Of note, a similar multisystem inflammatory syndrome associated with COVID-19 also has been described in adults, and it too may present with rash as a cardinal feature.35
Pathophysiology of COVID-19: What the Skin May Reveal About the Disease
The diverse range of cutaneous manifestations in COVID-19 reflects a spectrum of host immunologicresponses to SARS-CoV-2 and may inform the pathophysiology of the disease as well as potential treatment modalities.
Host Response to SARS-CoV-2
The body’s response to viral infection is 2-pronged, involving activation of cellular antiviral defenses mediated by type I and III interferons, as well as recruitment of leukocytes, mobilized by cytokines and chemokines.36,37 Infection with SARS-CoV-2 results in a unique inflammatory response characterized by suppression of interferons, juxtaposed with a rampant proinflammatory cytokine and chemokine response, reminiscent of a cytokine storm. Reflective of this imbalance, a study of 50 COVID-19 patients and 20 healthy controls found decreased natural killer cells and CD3+ T cells in COVID-19 patients, particularly severely or critically ill patients, with an increase in B cells and monocytes.38 This distinctive immune imbalance positions SARS-CoV-2 to thrive in the absence of inhibitory interferon activity while submitting the host to the deleterious effects of a cytokine surge.36
Type I Interferons
The perniolike lesions associated with mild COVID-19 disease14 may represent a robust immune response via effective stimulation of type I interferons (IFN-1). Similar perniolike lesions are observed in Aicardi-Goutières syndrome37 and familial chilblain lupus, hereditary interferonopathies associated with mutations in the TREX1 (three prime repair exonuclease 1) gene and characterized by inappropriate upregulation of IFN-1,39 resulting in chilblains. It has been suggested that perniolike lesions in COVID-19 result from IFN-1 activation—a robust effective immunologic response to the virus.14,26,40
On the other end of the spectrum, patients with severe COVID-19 may have a blunted IFN-1 response and reduced IFN-1–stimulated gene expression.36,38 Notably, low IFN-1 response preceded clinical deterioration and was associated with increased risk for evolution to critical illness.38 Severe disease from COVID-19 also is more commonly observed in older patients and those with comorbidities,1 both of which are known factors associated with depressed IFN-1 function.38,41 Reflective of this disparate IFN-1 response, biopsies of COVID-19 perniosis have demonstrated striking expression of myxovirus resistance protein A (MXA), a marker for IFN-1 signaling in tissue, whereas its expression is absent in COVID-19 livedo/retiform purpura.27
Familial chilblain lupus may be effectively treated by the Janus kinase inhibitor baricitinib,39 which inhibits IFN-1 signaling. Baricitinib recently received emergency use authorization by the US Food and Drug Administration for treatment of severe COVID-19 pneumonia,42,43 hinting to disordered IFN-1 signaling in the COVID-19 pathophysiology.
The impaired IFN-1 response in COVID-19 patients may be due to a unique characteristic of SARS-CoV-2: its ORF3b gene is a potent IFN-1 antagonist. In a series of experiments comparing SARS-CoV-2 to the related virus severe acute respiratory disease coronavirus (which was responsible for an epidemic in 2002), Konno et al44 found that SARS-CoV-2 is more effectively able to downregulate host IFN-1, likely due to premature stop codons on ORF3b that produce a truncated version of the gene with amplified anti–IFN-1 activity.
Cytokine Storm and Coagulation Cascade
This dulled interferon response is juxtaposed with a surge of inflammatory chemokines and cytokines, including IL-6, IL-8, IL-10, and tumor necrosis factor α, impairing innate immunity and leading to end-organ damage. This inflammatory response is associated with the influx of innate immune cells, specifically neutrophils and monocytes, which likely contribute to lung injury in COVID-19 acute respiratory distress syndrome.38 It also is thought to lead to downstream activation of coagulation, with a high incidence of thrombotic events observed in patients with severe COVID-19.1 In a retrospective study of 184 intensive care patients with COVID-19 receiving at least standard doses of thromboprophylaxis, venous thromboembolism occurred in 27% and arterial thrombotic events occurred in 3.7%.45
Livedo racemosa and retiform purpura are cutaneous markers of hypercoagulability, which indicate an increased risk for systemic clotting in COVID-19. A positive feedback loop between the complement and coagulation cascades appears to be important.13,14,29,46-48 In addition, a few studies have reported antiphospholipid antibody positivity in hospitalized COVID-19 patients.49,50
The high incidence of coagulopathy in severe COVID-19 has prompted many institutions to develop aggressive prophylactic anticoagulation protocols. Elevation of proinflammatory cytokines and observation of terminal complement activation in the skin and other organs has led to therapeutic trials of IL-6 inhibitors such as tocilizumab,51 complement inhibitors such as eculizumab, and Janus kinase inhibitors such as ruxolitinib and baricitinib.42,48
COVID Long-Haulers
The long-term effects of immune dysregulation in COVID-19 patients remain to be seen. Viral triggering of autoimmune disease is a well-established phenomenon, seen in DRESS (drug reaction with eosinophilia and systemic symptoms) syndrome and other dermatologic diseases, raising the possibility that dermatologists will see a rising incidence of cutaneous autoimmune disease in the aftermath of the pandemic. Disordered interferon stimulation could lead to increased incidence of interferon-mediated disorders, such as sarcoidosis and other granulomatous diseases. Vasculitislike skin lesions could persist beyond the acute infectious period. Recent data from a registry of 990 COVID-19 cases from 39 countries suggest that COVID-19 perniolike lesions may persist as long as 150 days.52 In a time of many unknowns, these questions serve as a call to action for rigorous data collection, contribution to existing registries for dermatologic manifestations of COVID-19, and long-term follow-up of COVID-19 patients by the dermatology community.
Pandemic Dermatology
The pandemic has posed unprecedented challenges for patient care. The use of hydroxychloroquine as a popular but unproven treatment for COVID-19, 53 particularly early in the pandemic, has resulted in drug shortages for patients with lupus and other autoimmune skin diseases. Meanwhile, the need for patients with complex dermatologic conditions to receive systemic immunosuppression has had to be balanced against the associated risks during a global pandemic. To help dermatologists navigate this dilemma, various subspecialty groups have issued guidelines, including the COVID-19 Task Force of the Medical Dermatology Society and Society of Dermatology Hospitalists, which recommends a stepwise approach to shared decision-making with the goal of minimizing both the risk for disease flare and that of infection. The use of systemic steroids and rituximab, as well as the dose of immunosuppression—particularly broad-acting immunosuppression—should be limited where permitted. 54
Rapid adoption of telemedicine and remote monitoring strategies has enabled dermatologists to provide safe and timely care when in-person visits have not been possible, including for patients with confirmed or suspected COVID-19, as well as for hospitalized patients. 55-57 Use of telemedicine has facilitated preservation of personal protective equipment at a time when these important resources have been scarce. For patients with transportation or scheduling barriers, telemedicine has even expanded access to care.
However, this strategy cannot completely replace comprehensive in-person evaluation. Variability in video and photographic quality limits evaluation, while in-person physical examination can reveal subtle morphologic clues necessary for diagnosis. 5 8 Additionally, unequal access to technology may disadvantage some patients. For dermatologists to provide optimal care and continue to contribute accurate and insightful observations into COVID-19, it is essential to be physically present in the clinic and in the hospital when necessary, caring for patients in need of dermatologic expertise. Creative management strategies developed during this time will benefit patients and expand the reach of the specialty . 5 8
Final Thoughts
The COVID-19 pandemic has profoundly challenged the medical community and dermatology is no exception. By documenting and characterizing the diverse cutaneous manifestations of this novel disease, dermatologists have furthered understanding of its pathophysiology and management. By adapting quickly and developing creative ways to deliver care, dermatologists have found ways to contribute, both large and small. As we take stock at this juncture of the pandemic, it is clear there remains much to learn. We hope dermatologists will continue to take an active role in meeting the challenges of this time.
- Wiersinga WJ, Rhodes A, Cheng AC, et al. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA . 2020;324:782-793. doi:10.1001/jama.2020.12839
- New York Times . Updated December 23, 2020. Accessed March 22, 2021. https://www.nytimes.com/2020/11/15/us/coronavirus-us-cases-deaths.html
- Guan W, Ni Z, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med . 2020;382:1708-1720. doi:10.1056/NEJMoa2002032
- Lechien JR, Chiesa-Estomba CM, Place S, et al. Clinical and epidemiological characteristics of 1420 European patients with mild-to-moderate coronavirus disease 2019. J Intern Med . 2020;288:335-344. doi:https://doi.org/10.1111/joim.13089
- Wu J, Liu J, Zhao X, et al. Clinical characteristics of imported cases of coronavirus disease 2019 (COVID-19) in Jiangsu province: a multicenter descriptive study. Clin Infect Dis . 2020;71:706-712. doi:10.1093/cid/ciaa199
- Goyal P, Choi JJ, Pinheiro LC, et al. Clinical characteristics of COVID-19 in New York City. N Engl J Med . 2020;382:2372-2374. doi:10.1056/NEJMc2010419
- Sun L, Shen L, Fan J, et al. Clinical features of patients with coronavirus disease 2019 from a designated hospital in Beijing, China. J Med Virol . 2020;92:2055-2066. https://doi.org/10.1002/jmv.25966
- Recalcati S. Cutaneous manifestations in COVID-19: a first perspective. J Eur Acad Dermatology Venereol . 2020;34:E212-E213. https://doi.org/10.1111/jdv.16387
- Giavedoni P, Podlipnik S, Pericàs JM, et al. Skin manifestations in COVID-19: prevalence and relationship with disease severity. J Clin Med . 2020;9:3261. doi:10.3390/jcm9103261
- Jimenez-Cauhe J, Ortega-Quijano D, Prieto-Barrios M, et al. Reply to “COVID-19 can present with a rash and be mistaken for dengue”: petechial rash in a patient with COVID-19 infection. J Am Acad Dermatol . 2020;83:E141-E142. doi:10.1016/j.jaad.2020.04.016
- Feldstein LR, Rose EB, Horwitz SM, et al. Multisystem inflammatory syndrome in U.S. children and adolescents. N Engl J Med . 2020;383:334-346. doi:10.1056/NEJMoa2021680
- Shinkai K, Bruckner AL. Dermatology and COVID-19. JAMA . 2020;324:1133-1134. doi:10.1001/jama.2020.15276
- Freeman EE, McMahon DE, Lipoff JB, et al. The spectrum of COVID-19-associated dermatologic manifestations: an international registry of 716 patients from 31 countries. J Am Acad Dermatol . 2020;83:1118-1129. doi:10.1016/j.jaad.2020.06.1016
- Galván Casas C, Català A, Carretero Hernández G, et al. Classification of the cutaneous manifestations of COVID-19: a rapid prospective nationwide consensus study in Spain with 375 cases. Br J Dermatol . 2020;183:71-77. https://doi.org/10.1111/bjd.19163
- Bouaziz JD, Duong TA, Jachiet M, et al. Vascular skin symptoms in COVID-19: a French observational study. J Eur Acad Dermatology Venereol . 2020;34:E451-E452. https://doi.org/10.1111/jdv.16544
- Fernandez-Nieto D, Ortega-Quijano D, Jimenez-Cauhe J, et al. Clinical and histological characterization of vesicular COVID-19 rashes: a prospective study in a tertiary care hospital. Clin Exp Dermatol . 2020;45:872-875. https://doi.org/10.1111/ced.14277
- Fernandez-Nieto D, Jimenez-Cauhe J, Suarez-Valle A, et al. Characterization of acute acral skin lesions in nonhospitalized patients: a case series of 132 patients during the COVID-19 outbreak. J Am Acad Dermatol . 2020;83:E61-E63. doi:10.1016/j.jaad.2020.04.093
- Marzano AV, Genovese G, Fabbrocini G, et al. Varicella-like exanthem as a specific COVID-19-associated skin manifestation: Multicenter case series of 22 patients. J Am Acad Dermatol . 2020;83:280-285. doi:10.1016/j.jaad.2020.04.044
- Fernandez-Nieto D, Ortega-Quijano D, Segurado-Miravalles G, et al. Comment on: cutaneous manifestations in COVID-19: a first perspective. safety concerns of clinical images and skin biopsies. J Eur Acad Dermatol Venereol . 2020;34:E252-E254. https://doi.org/10.1111/jdv.16470
- Herrero-Moyano M, Capusan TM, Andreu-Barasoain M, et al. A clinicopathological study of eight patients with COVID-19 pneumonia and a late-onset exanthema. J Eur Acad Dermatol Venereol . 2020;34:E460-E464. https://doi.org/10.1111/jdv.16631
- Rubio-Muniz CA, Puerta-Peñ a M, Falkenhain-L ópez D, et al. The broad spectrum of dermatological manifestations in COVID-19: clinical and histopathological features learned from a series of 34 cases. J Eur Acad Dermatol Venereol . 2020;34:E574-E576. https://doi.org/10.1111/jdv.16734
- Jimenez-Cauhe J, Ortega-Quijano D, Carretero-Barrio I, et al. Erythema multiforme-like eruption in patients with COVID-19 infection: clinical and histological findings. Clin Exp Dermatol . 2020;45:892-895. https://doi.org/10.1111/ced.14281
- Jimenez-Cauhe J, Ortega-Quijano D, de Perosanz-Lobo D, et al. Enanthem in patients with COVID-19 and skin rash. JAMA Dermatol . 2020;156:1134-1136. doi:10.1001/jamadermatol.2020.2550
- Le Cleach L, Dousset L, Assier H, et al. Most chilblains observed during the COVID-19 outbreak occur in patients who are negative for COVID-19 on polymerase chain reaction and serology testing. Br J Dermatol . 2020;183:866-874. https://doi.org/10.1111/bjd.19377
- Hubiche T, Cardot-Leccia N, Le Duff F, et al. Clinical, laboratory, and interferon-alpha response characteristics of patients with chilblain-like lesions during the COVID-19 pandemic [published online November 25, 2020]. JAMA Dermatol . doi:10.1001/jamadermatol.2020.4324
- Freeman EE, McMahon DE, Lipoff JB, et al. Pernio-like skin lesions associated with COVID-19: a case series of 318 patients from 8 countries. J Am Acad Dermatol . 2020;83:486-492. doi:10.1016/j.jaad.2020.05.109
- Magro CM, Mulvey JJ, Laurence J, et al. The differing pathophysiologies that underlie COVID-19-associated perniosis and thrombotic retiform purpura: a case series. Br J Dermatol . 2021;184:141-150. https://doi.org/10.1111/bjd.19415
- Colmenero I, Santonja C, Alonso-Riaño M, et al. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: histopathological, immunohistochemical and ultrastructural study of seven paediatric cases. Br J Dermatol . 2020;183:729-737. doi:10.1111/bjd.19327
- Droesch C, Do MH, DeSancho M, et al. Livedoid and purpuric skin eruptions associated with coagulopathy in severe COVID-19. JAMA Dermatol . 2020;156:1-3. doi:10.1001/jamadermatol.2020.2800
- Asakura H, Ogawa H. COVID-19-associated coagulopathy and disseminated intravascular coagulation. Int J Hematol . 2021;113:45-57. doi:10.1007/s12185-020-03029-y
- Dufort EM, Koumans EH, Chow EJ, et al. Multisystem inflammatory syndrome in children in New York State. N Engl J Med . 2020;383:347-358. doi:10.1056/NEJMoa2021756
- Young TK, Shaw KS, Shah JK, et al. Mucocutaneous manifestations of multisystem inflammatory syndrome in children during the COVID-19 pandemic. JAMA Dermatol . 2021;157:207-212. doi:10.1001/jamadermatol.2020.4779
- Whittaker E, Bamford A, Kenny J, et al. Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. JAMA. 2020;324:259-269. doi:10.1001/jama.2020.10369
- Cheng MH, Zhang S, Porritt RA, et al. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation.
- Morris SB, Schwartz NG, Patel P, et al. Case series of multisystem inflammatory syndrome in adults associated with SARS-CoV-2 Infection—United Kingdom and United States, March–August 2020. MMWR Morb Mortal Wkly Rep. 2020;69:1450-1456. doi:10.15585/mmwr.mm6940e1
- Blanco-Melo D, Nilsson-Payant BE, Liu W-C, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036.e9-1045.e9. doi:10.1016/j.cell.2020.04.026
- Crow YJ, Manel N. Aicardi–Goutières syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15:429-440. doi:10.1038/nri3850
- Hadjadj J, Yatim N, Barnabei L, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718-724. doi:10.1126/science.abc6027
- Zimmermann N, Wolf C, Schwenke R, et al. Assessment of clinical response to janus kinase inhibition in patients with familial chilblain lupus and TREX1 mutation. JAMA Dermatol. 2019;155:342-346. doi:10.1001/jamadermatol.2018.5077
- Hubiche T, Le Duff F, Chiaverini C, et al. Negative SARS-CoV-2 PCR in patients with chilblain-like lesions. Lancet Infect Dis. 2021;21:315-316. doi:10.1016/S1473-3099(20)30518-1
- Agrawal A. Mechanisms and implications of age-associated impaired innate interferon secretion by dendritic cells: a mini-review. Gerontology. 2013;59:421-426. doi:10.1159/000350536
- Kalil AC, Patterson TF, Mehta AK, et al. Baricitinib plus remdesivir for hospitalized adults with COVID-19. N Engl J Med. 2021;384:795-807. doi:10.1056/NEJMoa2031994
- US Food and Drug Administration. Fact sheet for healthcare providers: emergency use authorization (EUA) of baricitinib. Accessed March 29, 2021. https://www.fda.gov/media/143823/download
- Konno Y, Kimura I, Uriu K, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 2020;32:108185. doi:10.1016/j.celrep.2020.108185
- Sacks D, Baxter B, Campbell BCV, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke: from the American Association of Neurological Surgeons (AANS), American Society of Neuroradiology (ASNR), Cardiovascular and Interventional Radiology Society of Europe (CIRSE), Canadian Interventional Radiology Association (CIRA), Congress of Neurological Surgeons (CNS), European Society of Minimally Invasive Neurological Therapy (ESMINT), European Society of Neuroradiology (ESNR), European Stroke Organization (ESO), Society for Cardiovascular Angiography and Interventions (SCAI), Society of Interventional Radiology (SIR), Society of NeuroInterventional Surgery (SNIS), and World Stroke Organization (WSO). J Vasc Interv Radiol. 2018;29:441-453. doi:10.1016/j.jvir.2017.11.026
- Lo MW, Kemper C, Woodruff TM. COVID-19: complement, coagulation, and collateral damage. J Immunol. 2020;205:1488-1495. doi:10.4049/jimmunol.2000644
- Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1-13. doi:10.1016/j.trsl.2020.04.007
- Yan B, Freiwald T, Chauss D, et al. SARS-CoV2 drives JAK1/2-dependent local and systemic complement hyper-activation [published online June 9, 2020]. Res Sq. doi:10.21203/rs.3.rs-33390/v1
- Marietta M, Coluccio V, Luppi M. COVID-19, coagulopathy and venous thromboembolism: more questions than answers. Intern Emerg Med. 2020;15:1375-1387. doi:10.1007/s11739-020-02432-x
- Zuo Y, Estes SK, Ali RA, et al. Prothrombotic antiphospholipid antibodies in COVID-19 [published online June 17, 2020]. medRxiv. doi:10.1101/2020.06.15.20131607
- Lan S-H, Lai C-C, Huang H-T, et al. Tocilizumab for severe COVID-19: a systematic review and meta-analysis. Int J Antimicrob Agents. 2020;56:106103. doi:10.1016/j.ijantimicag.2020.106103
- McMahon D, Gallman A, Hruza G, et al. COVID-19 “long-haulers” in dermatology? duration of dermatologic symptoms in an international registry from 39 countries. Abstract presented at: 29th EADV Congress; October 29, 2020. Accessed March 29, 2020. https://eadvdistribute.m-anage.com/from.storage?image=PXQEdDtICIihN3sM_8nAmh7p_y9AFijhQlf2-_KjrtYgOsOXNVwGxDdti95GZ2Yh0
- Saag MS. Misguided use of hydroxychloroquine for COVID-19: the infusion of politics into science. JAMA. 2020;324:2161-2162. doi:10.1001/jama.2020.22389
- Zahedi Niaki O, Anadkat MJ, Chen ST, et al. Navigating immunosuppression in a pandemic: a guide for the dermatologist from the COVID Task Force of the Medical Dermatology Society and Society of Dermatology Hospitalists. J Am Acad Dermatol. 2020;83:1150-1159. doi:10.1016/j.jaad.2020.06.051
- Hammond MI, Sharma TR, Cooper KD, et al. Conducting inpatient dermatology consultations and maintaining resident education in the COVID-19 telemedicine era. J Am Acad Dermatol. 2020;83:E317-E318. doi:10.1016/j.jaad.2020.07.008
- Brunasso AMG, Massone C. Teledermatologic monitoring for chronic cutaneous autoimmune diseases with smartworking during COVID-19 emergency in a tertiary center in Italy. Dermatol Ther. 2020;33:E13495-E13495. doi:10.1111/dth.13695
- Trinidad J, Kroshinsky D, Kaffenberger BH, et al. Telemedicine for inpatient dermatology consultations in response to the COVID-19 pandemic. J Am Acad Dermatol. 2020;83:E69-E71. doi:10.1016/j.jaad.2020.04.096
- Madigan LM, Micheletti RG, Shinkai K. How dermatologists can learn and contribute at the leading edge of the COVID-19 global pandemic. JAMA Dermatology. 2020;156:733-734. doi:10.1001/jamadermatol.2020.1438
- Wiersinga WJ, Rhodes A, Cheng AC, et al. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): a review. JAMA . 2020;324:782-793. doi:10.1001/jama.2020.12839
- New York Times . Updated December 23, 2020. Accessed March 22, 2021. https://www.nytimes.com/2020/11/15/us/coronavirus-us-cases-deaths.html
- Guan W, Ni Z, Hu Y, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med . 2020;382:1708-1720. doi:10.1056/NEJMoa2002032
- Lechien JR, Chiesa-Estomba CM, Place S, et al. Clinical and epidemiological characteristics of 1420 European patients with mild-to-moderate coronavirus disease 2019. J Intern Med . 2020;288:335-344. doi:https://doi.org/10.1111/joim.13089
- Wu J, Liu J, Zhao X, et al. Clinical characteristics of imported cases of coronavirus disease 2019 (COVID-19) in Jiangsu province: a multicenter descriptive study. Clin Infect Dis . 2020;71:706-712. doi:10.1093/cid/ciaa199
- Goyal P, Choi JJ, Pinheiro LC, et al. Clinical characteristics of COVID-19 in New York City. N Engl J Med . 2020;382:2372-2374. doi:10.1056/NEJMc2010419
- Sun L, Shen L, Fan J, et al. Clinical features of patients with coronavirus disease 2019 from a designated hospital in Beijing, China. J Med Virol . 2020;92:2055-2066. https://doi.org/10.1002/jmv.25966
- Recalcati S. Cutaneous manifestations in COVID-19: a first perspective. J Eur Acad Dermatology Venereol . 2020;34:E212-E213. https://doi.org/10.1111/jdv.16387
- Giavedoni P, Podlipnik S, Pericàs JM, et al. Skin manifestations in COVID-19: prevalence and relationship with disease severity. J Clin Med . 2020;9:3261. doi:10.3390/jcm9103261
- Jimenez-Cauhe J, Ortega-Quijano D, Prieto-Barrios M, et al. Reply to “COVID-19 can present with a rash and be mistaken for dengue”: petechial rash in a patient with COVID-19 infection. J Am Acad Dermatol . 2020;83:E141-E142. doi:10.1016/j.jaad.2020.04.016
- Feldstein LR, Rose EB, Horwitz SM, et al. Multisystem inflammatory syndrome in U.S. children and adolescents. N Engl J Med . 2020;383:334-346. doi:10.1056/NEJMoa2021680
- Shinkai K, Bruckner AL. Dermatology and COVID-19. JAMA . 2020;324:1133-1134. doi:10.1001/jama.2020.15276
- Freeman EE, McMahon DE, Lipoff JB, et al. The spectrum of COVID-19-associated dermatologic manifestations: an international registry of 716 patients from 31 countries. J Am Acad Dermatol . 2020;83:1118-1129. doi:10.1016/j.jaad.2020.06.1016
- Galván Casas C, Català A, Carretero Hernández G, et al. Classification of the cutaneous manifestations of COVID-19: a rapid prospective nationwide consensus study in Spain with 375 cases. Br J Dermatol . 2020;183:71-77. https://doi.org/10.1111/bjd.19163
- Bouaziz JD, Duong TA, Jachiet M, et al. Vascular skin symptoms in COVID-19: a French observational study. J Eur Acad Dermatology Venereol . 2020;34:E451-E452. https://doi.org/10.1111/jdv.16544
- Fernandez-Nieto D, Ortega-Quijano D, Jimenez-Cauhe J, et al. Clinical and histological characterization of vesicular COVID-19 rashes: a prospective study in a tertiary care hospital. Clin Exp Dermatol . 2020;45:872-875. https://doi.org/10.1111/ced.14277
- Fernandez-Nieto D, Jimenez-Cauhe J, Suarez-Valle A, et al. Characterization of acute acral skin lesions in nonhospitalized patients: a case series of 132 patients during the COVID-19 outbreak. J Am Acad Dermatol . 2020;83:E61-E63. doi:10.1016/j.jaad.2020.04.093
- Marzano AV, Genovese G, Fabbrocini G, et al. Varicella-like exanthem as a specific COVID-19-associated skin manifestation: Multicenter case series of 22 patients. J Am Acad Dermatol . 2020;83:280-285. doi:10.1016/j.jaad.2020.04.044
- Fernandez-Nieto D, Ortega-Quijano D, Segurado-Miravalles G, et al. Comment on: cutaneous manifestations in COVID-19: a first perspective. safety concerns of clinical images and skin biopsies. J Eur Acad Dermatol Venereol . 2020;34:E252-E254. https://doi.org/10.1111/jdv.16470
- Herrero-Moyano M, Capusan TM, Andreu-Barasoain M, et al. A clinicopathological study of eight patients with COVID-19 pneumonia and a late-onset exanthema. J Eur Acad Dermatol Venereol . 2020;34:E460-E464. https://doi.org/10.1111/jdv.16631
- Rubio-Muniz CA, Puerta-Peñ a M, Falkenhain-L ópez D, et al. The broad spectrum of dermatological manifestations in COVID-19: clinical and histopathological features learned from a series of 34 cases. J Eur Acad Dermatol Venereol . 2020;34:E574-E576. https://doi.org/10.1111/jdv.16734
- Jimenez-Cauhe J, Ortega-Quijano D, Carretero-Barrio I, et al. Erythema multiforme-like eruption in patients with COVID-19 infection: clinical and histological findings. Clin Exp Dermatol . 2020;45:892-895. https://doi.org/10.1111/ced.14281
- Jimenez-Cauhe J, Ortega-Quijano D, de Perosanz-Lobo D, et al. Enanthem in patients with COVID-19 and skin rash. JAMA Dermatol . 2020;156:1134-1136. doi:10.1001/jamadermatol.2020.2550
- Le Cleach L, Dousset L, Assier H, et al. Most chilblains observed during the COVID-19 outbreak occur in patients who are negative for COVID-19 on polymerase chain reaction and serology testing. Br J Dermatol . 2020;183:866-874. https://doi.org/10.1111/bjd.19377
- Hubiche T, Cardot-Leccia N, Le Duff F, et al. Clinical, laboratory, and interferon-alpha response characteristics of patients with chilblain-like lesions during the COVID-19 pandemic [published online November 25, 2020]. JAMA Dermatol . doi:10.1001/jamadermatol.2020.4324
- Freeman EE, McMahon DE, Lipoff JB, et al. Pernio-like skin lesions associated with COVID-19: a case series of 318 patients from 8 countries. J Am Acad Dermatol . 2020;83:486-492. doi:10.1016/j.jaad.2020.05.109
- Magro CM, Mulvey JJ, Laurence J, et al. The differing pathophysiologies that underlie COVID-19-associated perniosis and thrombotic retiform purpura: a case series. Br J Dermatol . 2021;184:141-150. https://doi.org/10.1111/bjd.19415
- Colmenero I, Santonja C, Alonso-Riaño M, et al. SARS-CoV-2 endothelial infection causes COVID-19 chilblains: histopathological, immunohistochemical and ultrastructural study of seven paediatric cases. Br J Dermatol . 2020;183:729-737. doi:10.1111/bjd.19327
- Droesch C, Do MH, DeSancho M, et al. Livedoid and purpuric skin eruptions associated with coagulopathy in severe COVID-19. JAMA Dermatol . 2020;156:1-3. doi:10.1001/jamadermatol.2020.2800
- Asakura H, Ogawa H. COVID-19-associated coagulopathy and disseminated intravascular coagulation. Int J Hematol . 2021;113:45-57. doi:10.1007/s12185-020-03029-y
- Dufort EM, Koumans EH, Chow EJ, et al. Multisystem inflammatory syndrome in children in New York State. N Engl J Med . 2020;383:347-358. doi:10.1056/NEJMoa2021756
- Young TK, Shaw KS, Shah JK, et al. Mucocutaneous manifestations of multisystem inflammatory syndrome in children during the COVID-19 pandemic. JAMA Dermatol . 2021;157:207-212. doi:10.1001/jamadermatol.2020.4779
- Whittaker E, Bamford A, Kenny J, et al. Clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. JAMA. 2020;324:259-269. doi:10.1001/jama.2020.10369
- Cheng MH, Zhang S, Porritt RA, et al. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation.
- Morris SB, Schwartz NG, Patel P, et al. Case series of multisystem inflammatory syndrome in adults associated with SARS-CoV-2 Infection—United Kingdom and United States, March–August 2020. MMWR Morb Mortal Wkly Rep. 2020;69:1450-1456. doi:10.15585/mmwr.mm6940e1
- Blanco-Melo D, Nilsson-Payant BE, Liu W-C, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036.e9-1045.e9. doi:10.1016/j.cell.2020.04.026
- Crow YJ, Manel N. Aicardi–Goutières syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15:429-440. doi:10.1038/nri3850
- Hadjadj J, Yatim N, Barnabei L, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718-724. doi:10.1126/science.abc6027
- Zimmermann N, Wolf C, Schwenke R, et al. Assessment of clinical response to janus kinase inhibition in patients with familial chilblain lupus and TREX1 mutation. JAMA Dermatol. 2019;155:342-346. doi:10.1001/jamadermatol.2018.5077
- Hubiche T, Le Duff F, Chiaverini C, et al. Negative SARS-CoV-2 PCR in patients with chilblain-like lesions. Lancet Infect Dis. 2021;21:315-316. doi:10.1016/S1473-3099(20)30518-1
- Agrawal A. Mechanisms and implications of age-associated impaired innate interferon secretion by dendritic cells: a mini-review. Gerontology. 2013;59:421-426. doi:10.1159/000350536
- Kalil AC, Patterson TF, Mehta AK, et al. Baricitinib plus remdesivir for hospitalized adults with COVID-19. N Engl J Med. 2021;384:795-807. doi:10.1056/NEJMoa2031994
- US Food and Drug Administration. Fact sheet for healthcare providers: emergency use authorization (EUA) of baricitinib. Accessed March 29, 2021. https://www.fda.gov/media/143823/download
- Konno Y, Kimura I, Uriu K, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep. 2020;32:108185. doi:10.1016/j.celrep.2020.108185
- Sacks D, Baxter B, Campbell BCV, et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke: from the American Association of Neurological Surgeons (AANS), American Society of Neuroradiology (ASNR), Cardiovascular and Interventional Radiology Society of Europe (CIRSE), Canadian Interventional Radiology Association (CIRA), Congress of Neurological Surgeons (CNS), European Society of Minimally Invasive Neurological Therapy (ESMINT), European Society of Neuroradiology (ESNR), European Stroke Organization (ESO), Society for Cardiovascular Angiography and Interventions (SCAI), Society of Interventional Radiology (SIR), Society of NeuroInterventional Surgery (SNIS), and World Stroke Organization (WSO). J Vasc Interv Radiol. 2018;29:441-453. doi:10.1016/j.jvir.2017.11.026
- Lo MW, Kemper C, Woodruff TM. COVID-19: complement, coagulation, and collateral damage. J Immunol. 2020;205:1488-1495. doi:10.4049/jimmunol.2000644
- Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1-13. doi:10.1016/j.trsl.2020.04.007
- Yan B, Freiwald T, Chauss D, et al. SARS-CoV2 drives JAK1/2-dependent local and systemic complement hyper-activation [published online June 9, 2020]. Res Sq. doi:10.21203/rs.3.rs-33390/v1
- Marietta M, Coluccio V, Luppi M. COVID-19, coagulopathy and venous thromboembolism: more questions than answers. Intern Emerg Med. 2020;15:1375-1387. doi:10.1007/s11739-020-02432-x
- Zuo Y, Estes SK, Ali RA, et al. Prothrombotic antiphospholipid antibodies in COVID-19 [published online June 17, 2020]. medRxiv. doi:10.1101/2020.06.15.20131607
- Lan S-H, Lai C-C, Huang H-T, et al. Tocilizumab for severe COVID-19: a systematic review and meta-analysis. Int J Antimicrob Agents. 2020;56:106103. doi:10.1016/j.ijantimicag.2020.106103
- McMahon D, Gallman A, Hruza G, et al. COVID-19 “long-haulers” in dermatology? duration of dermatologic symptoms in an international registry from 39 countries. Abstract presented at: 29th EADV Congress; October 29, 2020. Accessed March 29, 2020. https://eadvdistribute.m-anage.com/from.storage?image=PXQEdDtICIihN3sM_8nAmh7p_y9AFijhQlf2-_KjrtYgOsOXNVwGxDdti95GZ2Yh0
- Saag MS. Misguided use of hydroxychloroquine for COVID-19: the infusion of politics into science. JAMA. 2020;324:2161-2162. doi:10.1001/jama.2020.22389
- Zahedi Niaki O, Anadkat MJ, Chen ST, et al. Navigating immunosuppression in a pandemic: a guide for the dermatologist from the COVID Task Force of the Medical Dermatology Society and Society of Dermatology Hospitalists. J Am Acad Dermatol. 2020;83:1150-1159. doi:10.1016/j.jaad.2020.06.051
- Hammond MI, Sharma TR, Cooper KD, et al. Conducting inpatient dermatology consultations and maintaining resident education in the COVID-19 telemedicine era. J Am Acad Dermatol. 2020;83:E317-E318. doi:10.1016/j.jaad.2020.07.008
- Brunasso AMG, Massone C. Teledermatologic monitoring for chronic cutaneous autoimmune diseases with smartworking during COVID-19 emergency in a tertiary center in Italy. Dermatol Ther. 2020;33:E13495-E13495. doi:10.1111/dth.13695
- Trinidad J, Kroshinsky D, Kaffenberger BH, et al. Telemedicine for inpatient dermatology consultations in response to the COVID-19 pandemic. J Am Acad Dermatol. 2020;83:E69-E71. doi:10.1016/j.jaad.2020.04.096
- Madigan LM, Micheletti RG, Shinkai K. How dermatologists can learn and contribute at the leading edge of the COVID-19 global pandemic. JAMA Dermatology. 2020;156:733-734. doi:10.1001/jamadermatol.2020.1438
Practice Points
- Cutaneous manifestations of COVID-19 may reflect the range of host immunologic responses to SARS-CoV-2.
- Perniosis appears to be a late manifestation of COVID-19 associated with a comparatively benign disease course, whereas livedoid or other vasculopathic lesions portend poorer outcomes and may warrant further workup for occult thrombotic disease.
- Maculopapular, vesicular, and urticarial eruptions may be seen in association with COVID-19 but are nonspecific and necessitate a broad differential and workup.
- Challenges posed by the COVID-19 pandemic necessitate creative management strategies for immunosuppression and clinical assessment.
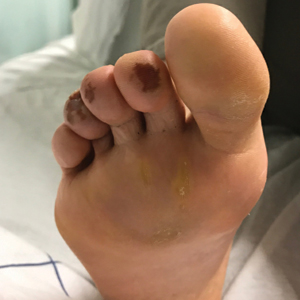
Management of Classic Ulcerative Pyoderma Gangrenosum
Pyoderma gangrenosum (PG) is a rare, chronic, ulcerative, neutrophilic dermatosis of unclear etiology. Large, multicentered, randomized controlled trials (RCTs) are challenging due to the rarity of PG and the lack of a diagnostic confirmatory test; therefore, evidence-based guidelines for diagnosis and treatment are not well established. Current management of PG primarily is guided by case series, small clinical trials, and expert opinion.1-4 We conducted a survey of expert medical dermatologists to highlight best practices in diagnostic and therapeutic approaches to PG.
Methods
The Society of Dermatology Hospitalists (SDH) Scientific Task Force gathered expert opinions from members of the SDH and Rheumatologic Dermatology Society (RDS) regarding PG workup and treatment through an online survey of 15 items (eTable 1). Subscribers of the SDH and RDS LISTSERVs were invited via email to participate in the survey from January 2016 to February 2016. Anonymous survey responses were collected and collated using SurveyMonkey. The survey results identified expert recommendations for evaluation, diagnosis, and treatment of PG and are reported as the sum of the percentage of respondents who answered always (almost 100% of the time) or often (more than half the time) following a particular course of action. A subanalysis was performed defining 2 groups of respondents based on the number of cases of PG treated per year (≥10 vs <10). Survey responses between each group were compared using χ2 analysis with statistical significance set at P=.05.

Results
Fifty-one respondents completed the survey out of 140 surveyed (36% response rate). All respondents were dermatologists, and 96% (49/51) were affiliated with an academic institution. Among the respondents, the number of PG cases managed per year ranged from 2 to 35.
Respondents consistently ordered skin biopsies (92% [47/51]) and tissue cultures (90% [46/51]), as well as certain ancillary tests, including complete blood cell count (96% [49/51]), complete metabolic panel (86% [44/51]), serum protein electrophoresis (76% [39/51]), and hepatitis panel (71% [36/51]). Other frequently ordered studies were rheumatoid factor (69% [35/51]), antinuclear antibodies (67% [34/51]), and antineutrophilic antibodies (65% [33/51]). Respondents frequently ordered erythrocyte sedimentation rate (59% [30/51]), C-reactive protein (55% [28/51]), cryoglobulins (53% [27/51]), urine protein electrophoresis (53% [27/51]), hypercoagulability workup (49% [25/51]), and serum immunofixation test (49% [25/51]). Human immunodeficiency virus testing (43% [22/51]), chest radiograph (41% [21/51]), colonoscopy (41% [21/51]) and referral to other specialties for workup—gastroenterology (38% [19/51]), hematology/oncology (14% [7/51]), and rheumatology (10% [5/51])—were less frequently ordered (eTable 2).
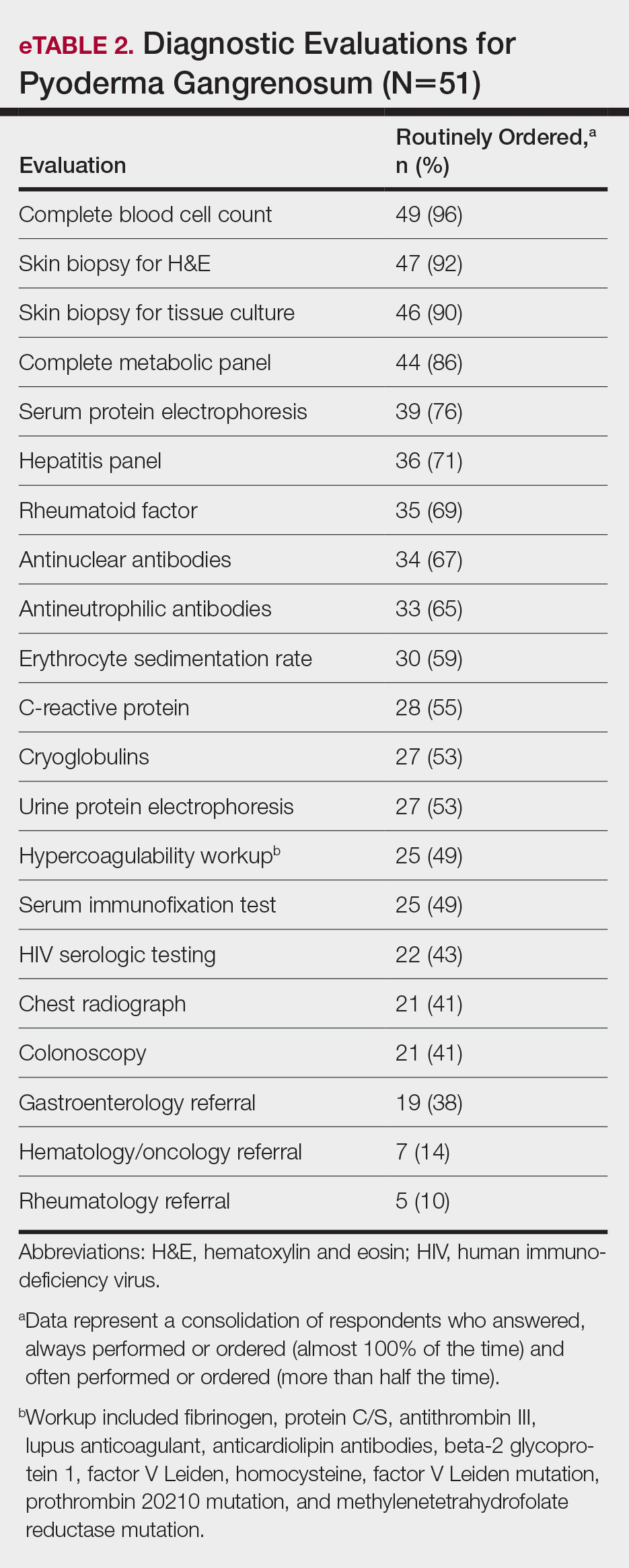
Systemic corticosteroids were reported as first-line therapy by most respondents (94% [48/51]), followed by topical immunomodulatory therapies (63% [32/51]). Topical corticosteroids (75% [38/51]) were the most common first-line topical agents. Thirty-nine percent of respondents (20/51) prescribed topical calcineurin inhibitors as first-line topical therapy. Additional therapies frequently used included systemic cyclosporine (47% [24/51]), antineutrophilic agents (41% [21/51]), and biologic agents (37% [19/51]). Fifty-seven percent of respondents (29/51) supported using combination topical and systemic therapy (Table).
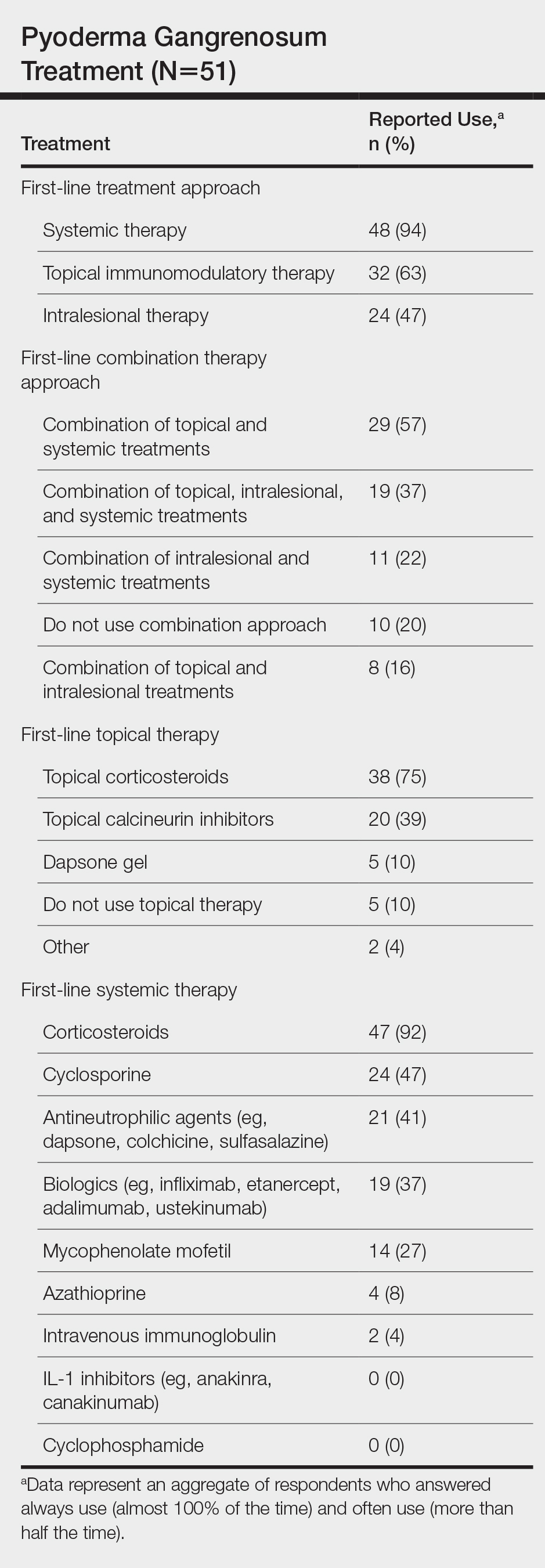
A wide variety of wound care practices were reported in the management of PG. Seventy-six percent of respondents (39/51) favored petroleum-impregnated gauze, 69% (35/51) used nonadhesive dressings, and 43% (22/51) added antimicrobial therapy for PG wound care (eTable 3). In the subanalysis, there were no significant differences in the majority of answer responses in patients treating 10 or more PG cases per year vs fewer than 10 PG cases, except with regard to the practice of combination therapy. Those treating more than 10 cases of PG per year more frequently reported use of combination therapies compared to respondents treating fewer than 10 cases (P=.04).
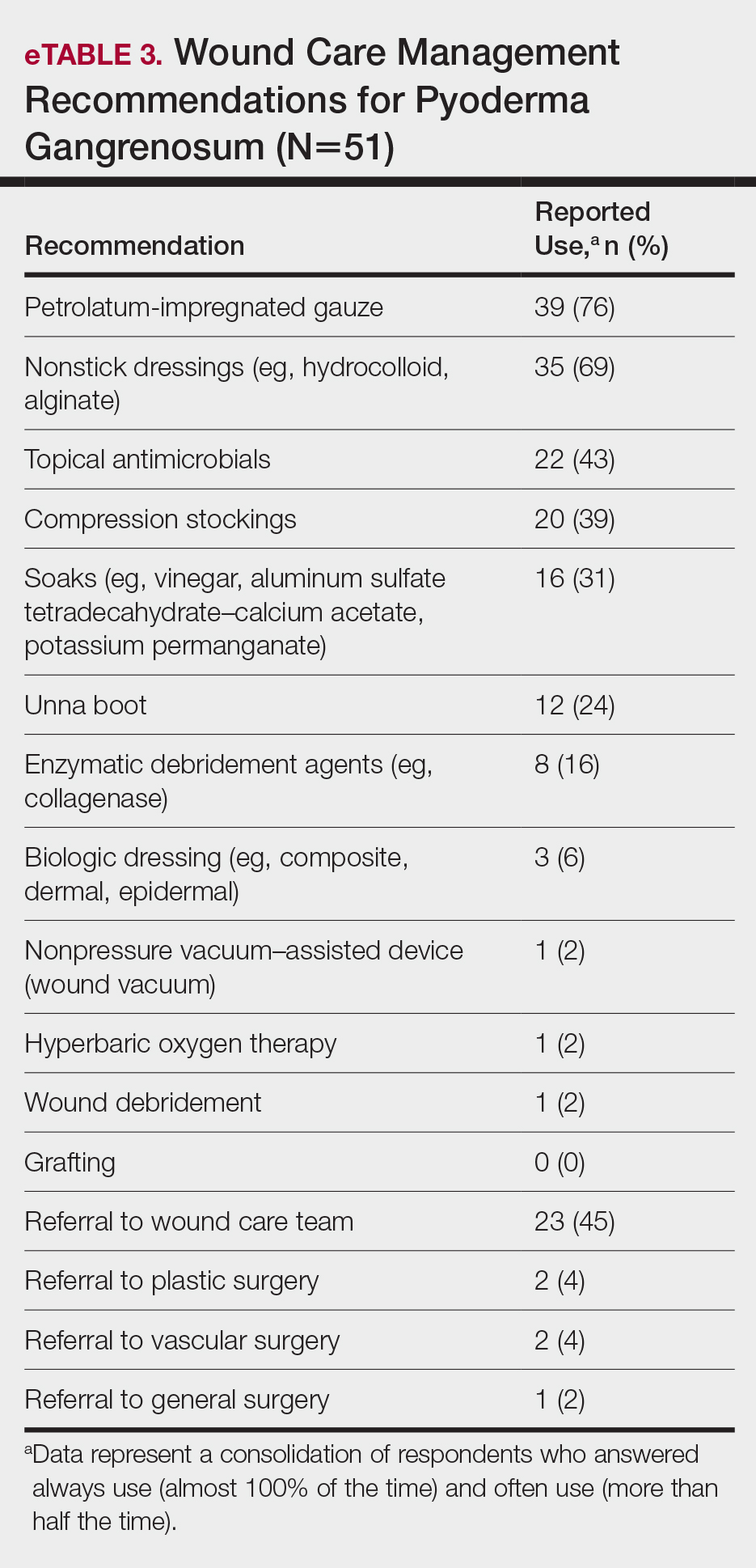
Comment
Skin biopsies and tissue cultures were strongly recommended (>90% survey respondents) for the initial evaluation of lesions suspected to be PG to evaluate for typical histopathologic changes that appear early in the disease, to rule out PG mimickers such as infectious or vascular causes, and to prevent the detrimental effects of inappropriate treatment and delayed diagnosis.5
Suspected PG warrants a reasonable search for related conditions because more than 50% of PG cases are associated with comorbidities such as rheumatoid arthritis, inflammatory bowel disease, and hematologic disease/malignancy.6,7 A complete blood cell count and comprehensive metabolic panel were recommended by most respondents, aiding in the preliminary screening for hematologic and infectious causes as well as detecting liver and kidney dysfunction associated with systemic conditions. Additionally, exclusion of infection or malignancy may be particularly important if the patient will undergo systemic immunosuppression. In challenging PG cases when initial findings are inconclusive and the clinical presentation does not direct workup (eg, colonoscopy to evaluate gastrointestinal tract symptoms), serum protein electrophoresis, hepatitis panel, rheumatoid factor, antinuclear antibodies, and antineutrophilic antibody tests also were frequently ordered by respondents to further evaluate for underlying or associated conditions.
This consensus regarding skin biopsies and certain ancillary tests is consistent with the proposed diagnostic criteria for classic ulcerative PG in which the absence or exclusion of other relevant causes of cutaneous ulcers is required based on the criteria.8 The importance of ensuring an accurate diagnosis is paramount, as a 10% misdiagnosis rate has been documented in the literature.5
Importantly, a stepwise diagnostic workup for PG is proposed based on survey results, which may limit unnecessary testing and the associated costs to the health care system (Figure 1). Selection of additional testing is guided by initial test results and features of the patient’s clinical presentation, including age, review of systems, and associated comorbidities. Available data suggest that underlying inflammatory bowel disease is more frequent in PG patients who are younger than 65 years, whereas those who are 65 years and older are more likely to have inflammatory arthritis, cancer, or an underlying hematologic disorder.9
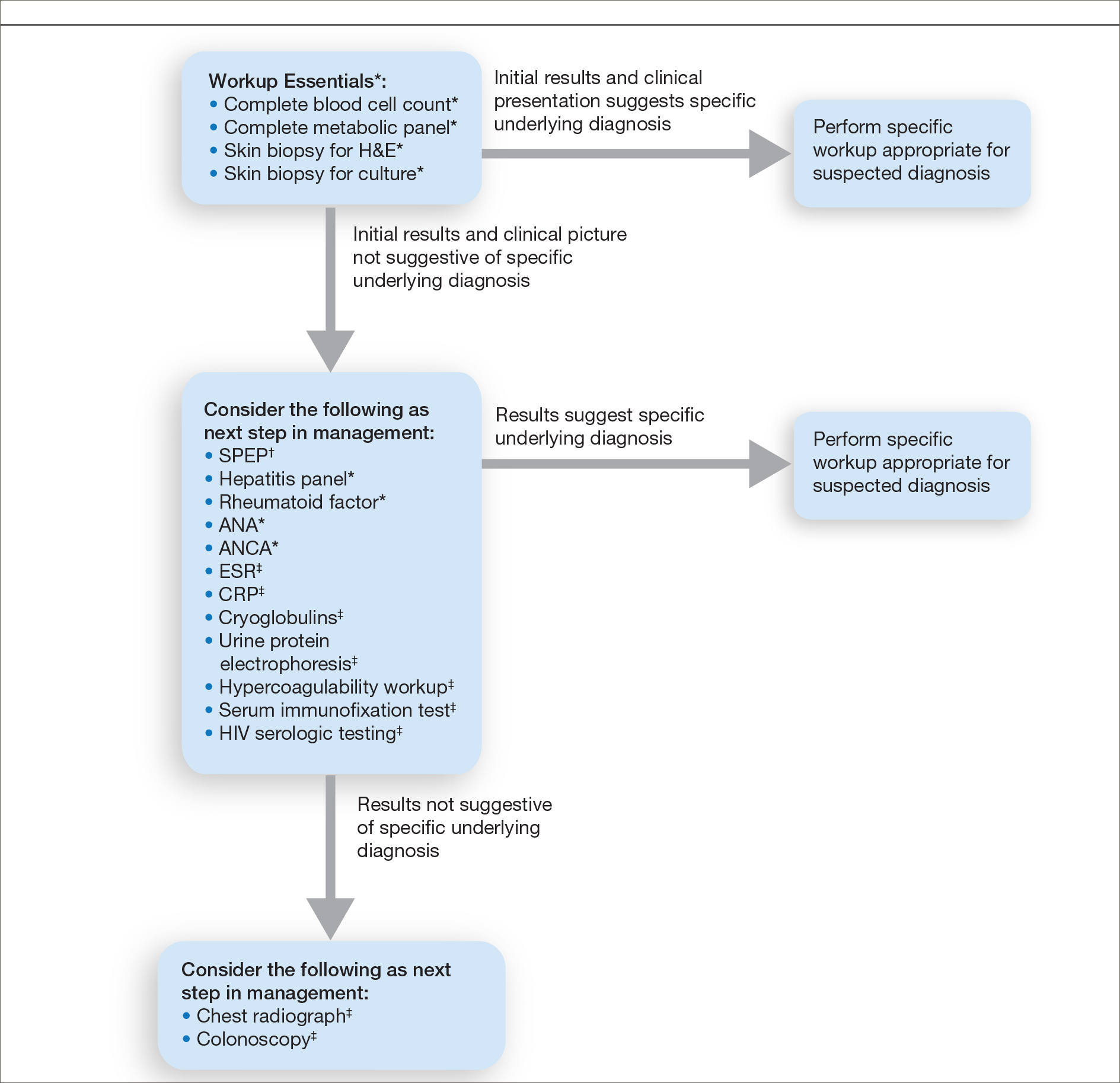
Treatment of PG should address both the inflammatory and wound components of the disease (Figure 2).7 In our survey results, systemic corticosteroids were identified as an important first-line therapy supported by reasonable evidence and were favored for their rapid response and minimal cost.1,10,11 Many respondents endorsed the use of systemic therapy in combination with topical steroids or calcineurin inhibitors. Combination therapy may provide more immediate control of rapidly progressing disease while minimizing adverse effects of long-term systemic corticosteroid use. A survey of German wound experts similarly endorsed frequent use of topical calcineurin inhibitors and combination systemic and topical glucocorticoid therapy as common therapeutic approaches.1
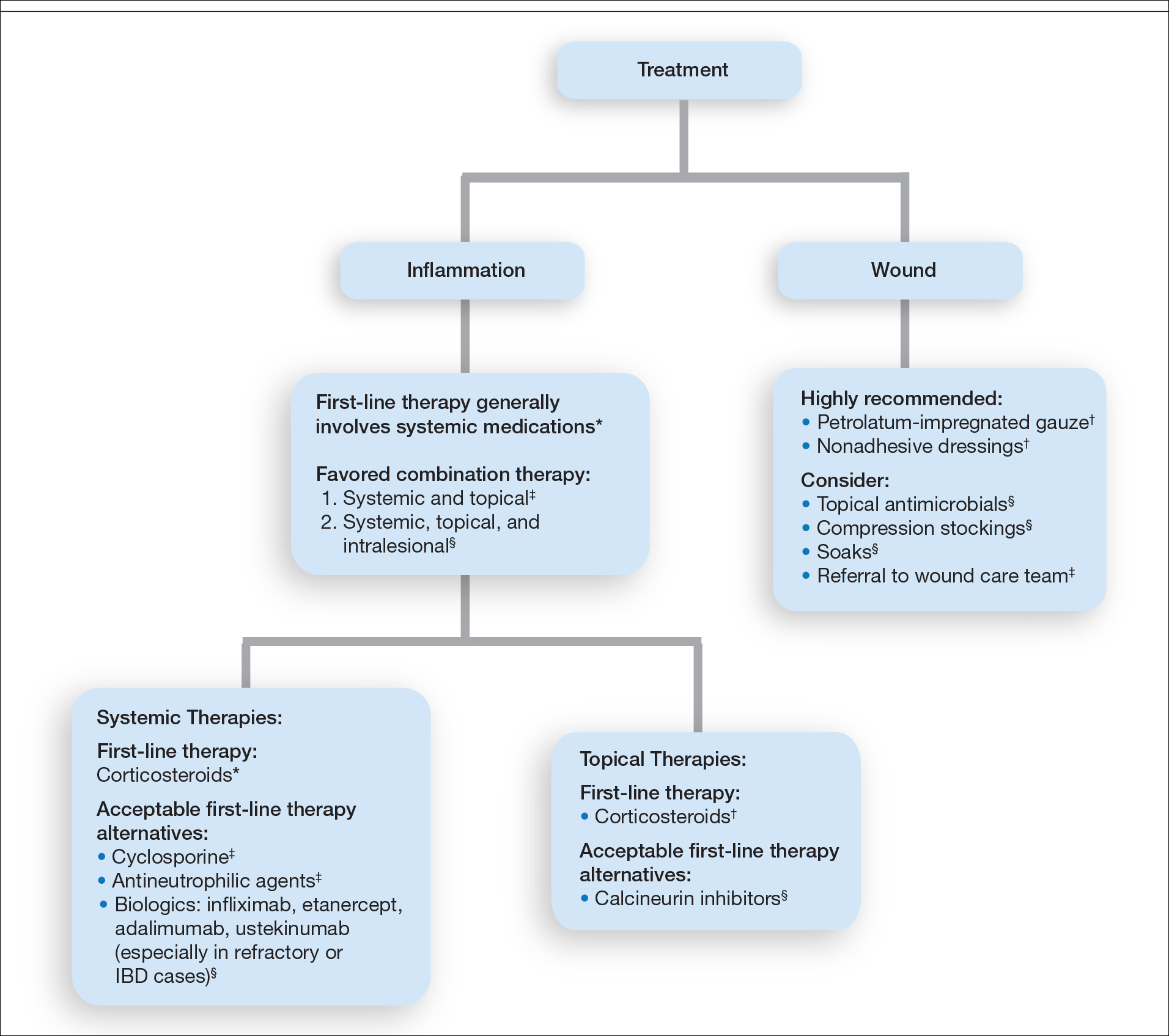
Importantly, treatments may vary depending on patient characteristics, comorbidities, and underlying disease, which underscores the need for individualized treatment approaches. Alternative first-line systemic treatments favored by respondents were cyclosporine, biologic medications, and antineutrophilic agents such as dapsone. Cyclosporine has demonstrated comparable efficacy to systemic glucocorticoids in one RCT and is considered an important steroid-sparing alternative for PG treatment.2 Biologic agents, especially tumor necrosis factor inhibitors, may be effective in treating cases of refractory PG or for concomitant inflammatory bowel disease management, as demonstrated by a small RCT documenting improvement of PG following infliximab infusion.3
Respondents strongly recommended petrolatum-impregnated gauze and other nonadhesive dressings, including alginate and hydrocolloid dressings, as part of PG wound care. Topical antimicrobials and compression stockings also were recommended by respondents. These practices aim to promote moist environments for healing, avoid maceration, prevent superinfection, optimize wound healing, and minimize damage from adhesive injury.12 Wound debridement and grafting generally were not recommended. However, pathergy is not a universal phenomenon in PG, and wounds that are no longer in the inflammatory phase may benefit from gentle debridement of necrotic tissue and/or grafting in select cases.10
Conclusion
An approach to modifying PG management based on clinical presentation and the practice of combination therapy with multiple systemic agents in refractory PG cases was not addressed in our survey. The low response rate is a limitation; however, the opinions of 51 medical dermatologist experts who regularly manage PG (in contrast to papers based on individualized clinical experience) can provide important clinical guidance until more scientific evidence is established.
Acknowledgments
We would like to thank the SDH and RDS membership for their participation in this survey. We especially acknowledge the other members of the SDH Scientific Task Force for their feedback: Misha Rosenbach, MD (Philadelphia, Pennsylvania); Robert G. Micheletti, MD (Philadelphia, Pennsylvania); Karolyn Wanat, MD (Milwaukee, Wisconsin); Amy Chen, MD (Cromwell, Connecticut); and A. Rambi Cardones, MD (Durham, North Carolina).
- Al Ghazal P, Dissemond J. Therapy of pyoderma gangrenosum in Germany: results of a survey among wound experts. J Dtsch Dermatol Ges . 2015;13:317-324.
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958.
- Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Al Ghazal P, Klode J, Dissemond J. Diagnostic criteria for pyoderma gangrenosum: results of a survey among dermatologic wound experts in Germany. J Dtsch Dermatol Ges. 2014;12:1129-1131.
- Weenig RH, Davis MD, Dahl PR, et al. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002;347:1412-1418.
- Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34:395-409.
- Bennett ML, Jackson JM, Jorizzo JL, et al. Pyoderma gangrenosum: a comparison of typical and atypical forms with an emphasis on time to remission. case review of 86 patients from 2 institutions. Medicine. 2000;79:37-46.
- Su WP, Davis MD, Weening RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Aschyan H, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Binus AM, Qureshi AA, Li VW, et al. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol. 2011;165:1244-1250.
- Reichrath J, Bens G, Bonowitz A, et al. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005;53:273-283.
- Miller J, Yentzer BA, Clark A, et al. Pyoderma gangrenosum: a review and update on new therapies. J Am Acad Dermatol. 2010;62:646-654.
Pyoderma gangrenosum (PG) is a rare, chronic, ulcerative, neutrophilic dermatosis of unclear etiology. Large, multicentered, randomized controlled trials (RCTs) are challenging due to the rarity of PG and the lack of a diagnostic confirmatory test; therefore, evidence-based guidelines for diagnosis and treatment are not well established. Current management of PG primarily is guided by case series, small clinical trials, and expert opinion.1-4 We conducted a survey of expert medical dermatologists to highlight best practices in diagnostic and therapeutic approaches to PG.
Methods
The Society of Dermatology Hospitalists (SDH) Scientific Task Force gathered expert opinions from members of the SDH and Rheumatologic Dermatology Society (RDS) regarding PG workup and treatment through an online survey of 15 items (eTable 1). Subscribers of the SDH and RDS LISTSERVs were invited via email to participate in the survey from January 2016 to February 2016. Anonymous survey responses were collected and collated using SurveyMonkey. The survey results identified expert recommendations for evaluation, diagnosis, and treatment of PG and are reported as the sum of the percentage of respondents who answered always (almost 100% of the time) or often (more than half the time) following a particular course of action. A subanalysis was performed defining 2 groups of respondents based on the number of cases of PG treated per year (≥10 vs <10). Survey responses between each group were compared using χ2 analysis with statistical significance set at P=.05.
Results
Fifty-one respondents completed the survey out of 140 surveyed (36% response rate). All respondents were dermatologists, and 96% (49/51) were affiliated with an academic institution. Among the respondents, the number of PG cases managed per year ranged from 2 to 35.
Respondents consistently ordered skin biopsies (92% [47/51]) and tissue cultures (90% [46/51]), as well as certain ancillary tests, including complete blood cell count (96% [49/51]), complete metabolic panel (86% [44/51]), serum protein electrophoresis (76% [39/51]), and hepatitis panel (71% [36/51]). Other frequently ordered studies were rheumatoid factor (69% [35/51]), antinuclear antibodies (67% [34/51]), and antineutrophilic antibodies (65% [33/51]). Respondents frequently ordered erythrocyte sedimentation rate (59% [30/51]), C-reactive protein (55% [28/51]), cryoglobulins (53% [27/51]), urine protein electrophoresis (53% [27/51]), hypercoagulability workup (49% [25/51]), and serum immunofixation test (49% [25/51]). Human immunodeficiency virus testing (43% [22/51]), chest radiograph (41% [21/51]), colonoscopy (41% [21/51]) and referral to other specialties for workup—gastroenterology (38% [19/51]), hematology/oncology (14% [7/51]), and rheumatology (10% [5/51])—were less frequently ordered (eTable 2).
Systemic corticosteroids were reported as first-line therapy by most respondents (94% [48/51]), followed by topical immunomodulatory therapies (63% [32/51]). Topical corticosteroids (75% [38/51]) were the most common first-line topical agents. Thirty-nine percent of respondents (20/51) prescribed topical calcineurin inhibitors as first-line topical therapy. Additional therapies frequently used included systemic cyclosporine (47% [24/51]), antineutrophilic agents (41% [21/51]), and biologic agents (37% [19/51]). Fifty-seven percent of respondents (29/51) supported using combination topical and systemic therapy (Table).
A wide variety of wound care practices were reported in the management of PG. Seventy-six percent of respondents (39/51) favored petroleum-impregnated gauze, 69% (35/51) used nonadhesive dressings, and 43% (22/51) added antimicrobial therapy for PG wound care (eTable 3). In the subanalysis, there were no significant differences in the majority of answer responses in patients treating 10 or more PG cases per year vs fewer than 10 PG cases, except with regard to the practice of combination therapy. Those treating more than 10 cases of PG per year more frequently reported use of combination therapies compared to respondents treating fewer than 10 cases (P=.04).
Comment
Skin biopsies and tissue cultures were strongly recommended (>90% survey respondents) for the initial evaluation of lesions suspected to be PG to evaluate for typical histopathologic changes that appear early in the disease, to rule out PG mimickers such as infectious or vascular causes, and to prevent the detrimental effects of inappropriate treatment and delayed diagnosis.5
Suspected PG warrants a reasonable search for related conditions because more than 50% of PG cases are associated with comorbidities such as rheumatoid arthritis, inflammatory bowel disease, and hematologic disease/malignancy.6,7 A complete blood cell count and comprehensive metabolic panel were recommended by most respondents, aiding in the preliminary screening for hematologic and infectious causes as well as detecting liver and kidney dysfunction associated with systemic conditions. Additionally, exclusion of infection or malignancy may be particularly important if the patient will undergo systemic immunosuppression. In challenging PG cases when initial findings are inconclusive and the clinical presentation does not direct workup (eg, colonoscopy to evaluate gastrointestinal tract symptoms), serum protein electrophoresis, hepatitis panel, rheumatoid factor, antinuclear antibodies, and antineutrophilic antibody tests also were frequently ordered by respondents to further evaluate for underlying or associated conditions.
This consensus regarding skin biopsies and certain ancillary tests is consistent with the proposed diagnostic criteria for classic ulcerative PG in which the absence or exclusion of other relevant causes of cutaneous ulcers is required based on the criteria.8 The importance of ensuring an accurate diagnosis is paramount, as a 10% misdiagnosis rate has been documented in the literature.5
Importantly, a stepwise diagnostic workup for PG is proposed based on survey results, which may limit unnecessary testing and the associated costs to the health care system (Figure 1). Selection of additional testing is guided by initial test results and features of the patient’s clinical presentation, including age, review of systems, and associated comorbidities. Available data suggest that underlying inflammatory bowel disease is more frequent in PG patients who are younger than 65 years, whereas those who are 65 years and older are more likely to have inflammatory arthritis, cancer, or an underlying hematologic disorder.9
Treatment of PG should address both the inflammatory and wound components of the disease (Figure 2).7 In our survey results, systemic corticosteroids were identified as an important first-line therapy supported by reasonable evidence and were favored for their rapid response and minimal cost.1,10,11 Many respondents endorsed the use of systemic therapy in combination with topical steroids or calcineurin inhibitors. Combination therapy may provide more immediate control of rapidly progressing disease while minimizing adverse effects of long-term systemic corticosteroid use. A survey of German wound experts similarly endorsed frequent use of topical calcineurin inhibitors and combination systemic and topical glucocorticoid therapy as common therapeutic approaches.1
Importantly, treatments may vary depending on patient characteristics, comorbidities, and underlying disease, which underscores the need for individualized treatment approaches. Alternative first-line systemic treatments favored by respondents were cyclosporine, biologic medications, and antineutrophilic agents such as dapsone. Cyclosporine has demonstrated comparable efficacy to systemic glucocorticoids in one RCT and is considered an important steroid-sparing alternative for PG treatment.2 Biologic agents, especially tumor necrosis factor inhibitors, may be effective in treating cases of refractory PG or for concomitant inflammatory bowel disease management, as demonstrated by a small RCT documenting improvement of PG following infliximab infusion.3
Respondents strongly recommended petrolatum-impregnated gauze and other nonadhesive dressings, including alginate and hydrocolloid dressings, as part of PG wound care. Topical antimicrobials and compression stockings also were recommended by respondents. These practices aim to promote moist environments for healing, avoid maceration, prevent superinfection, optimize wound healing, and minimize damage from adhesive injury.12 Wound debridement and grafting generally were not recommended. However, pathergy is not a universal phenomenon in PG, and wounds that are no longer in the inflammatory phase may benefit from gentle debridement of necrotic tissue and/or grafting in select cases.10
Conclusion
An approach to modifying PG management based on clinical presentation and the practice of combination therapy with multiple systemic agents in refractory PG cases was not addressed in our survey. The low response rate is a limitation; however, the opinions of 51 medical dermatologist experts who regularly manage PG (in contrast to papers based on individualized clinical experience) can provide important clinical guidance until more scientific evidence is established.
Acknowledgments
We would like to thank the SDH and RDS membership for their participation in this survey. We especially acknowledge the other members of the SDH Scientific Task Force for their feedback: Misha Rosenbach, MD (Philadelphia, Pennsylvania); Robert G. Micheletti, MD (Philadelphia, Pennsylvania); Karolyn Wanat, MD (Milwaukee, Wisconsin); Amy Chen, MD (Cromwell, Connecticut); and A. Rambi Cardones, MD (Durham, North Carolina).
Pyoderma gangrenosum (PG) is a rare, chronic, ulcerative, neutrophilic dermatosis of unclear etiology. Large, multicentered, randomized controlled trials (RCTs) are challenging due to the rarity of PG and the lack of a diagnostic confirmatory test; therefore, evidence-based guidelines for diagnosis and treatment are not well established. Current management of PG primarily is guided by case series, small clinical trials, and expert opinion.1-4 We conducted a survey of expert medical dermatologists to highlight best practices in diagnostic and therapeutic approaches to PG.
Methods
The Society of Dermatology Hospitalists (SDH) Scientific Task Force gathered expert opinions from members of the SDH and Rheumatologic Dermatology Society (RDS) regarding PG workup and treatment through an online survey of 15 items (eTable 1). Subscribers of the SDH and RDS LISTSERVs were invited via email to participate in the survey from January 2016 to February 2016. Anonymous survey responses were collected and collated using SurveyMonkey. The survey results identified expert recommendations for evaluation, diagnosis, and treatment of PG and are reported as the sum of the percentage of respondents who answered always (almost 100% of the time) or often (more than half the time) following a particular course of action. A subanalysis was performed defining 2 groups of respondents based on the number of cases of PG treated per year (≥10 vs <10). Survey responses between each group were compared using χ2 analysis with statistical significance set at P=.05.
Results
Fifty-one respondents completed the survey out of 140 surveyed (36% response rate). All respondents were dermatologists, and 96% (49/51) were affiliated with an academic institution. Among the respondents, the number of PG cases managed per year ranged from 2 to 35.
Respondents consistently ordered skin biopsies (92% [47/51]) and tissue cultures (90% [46/51]), as well as certain ancillary tests, including complete blood cell count (96% [49/51]), complete metabolic panel (86% [44/51]), serum protein electrophoresis (76% [39/51]), and hepatitis panel (71% [36/51]). Other frequently ordered studies were rheumatoid factor (69% [35/51]), antinuclear antibodies (67% [34/51]), and antineutrophilic antibodies (65% [33/51]). Respondents frequently ordered erythrocyte sedimentation rate (59% [30/51]), C-reactive protein (55% [28/51]), cryoglobulins (53% [27/51]), urine protein electrophoresis (53% [27/51]), hypercoagulability workup (49% [25/51]), and serum immunofixation test (49% [25/51]). Human immunodeficiency virus testing (43% [22/51]), chest radiograph (41% [21/51]), colonoscopy (41% [21/51]) and referral to other specialties for workup—gastroenterology (38% [19/51]), hematology/oncology (14% [7/51]), and rheumatology (10% [5/51])—were less frequently ordered (eTable 2).
Systemic corticosteroids were reported as first-line therapy by most respondents (94% [48/51]), followed by topical immunomodulatory therapies (63% [32/51]). Topical corticosteroids (75% [38/51]) were the most common first-line topical agents. Thirty-nine percent of respondents (20/51) prescribed topical calcineurin inhibitors as first-line topical therapy. Additional therapies frequently used included systemic cyclosporine (47% [24/51]), antineutrophilic agents (41% [21/51]), and biologic agents (37% [19/51]). Fifty-seven percent of respondents (29/51) supported using combination topical and systemic therapy (Table).
A wide variety of wound care practices were reported in the management of PG. Seventy-six percent of respondents (39/51) favored petroleum-impregnated gauze, 69% (35/51) used nonadhesive dressings, and 43% (22/51) added antimicrobial therapy for PG wound care (eTable 3). In the subanalysis, there were no significant differences in the majority of answer responses in patients treating 10 or more PG cases per year vs fewer than 10 PG cases, except with regard to the practice of combination therapy. Those treating more than 10 cases of PG per year more frequently reported use of combination therapies compared to respondents treating fewer than 10 cases (P=.04).
Comment
Skin biopsies and tissue cultures were strongly recommended (>90% survey respondents) for the initial evaluation of lesions suspected to be PG to evaluate for typical histopathologic changes that appear early in the disease, to rule out PG mimickers such as infectious or vascular causes, and to prevent the detrimental effects of inappropriate treatment and delayed diagnosis.5
Suspected PG warrants a reasonable search for related conditions because more than 50% of PG cases are associated with comorbidities such as rheumatoid arthritis, inflammatory bowel disease, and hematologic disease/malignancy.6,7 A complete blood cell count and comprehensive metabolic panel were recommended by most respondents, aiding in the preliminary screening for hematologic and infectious causes as well as detecting liver and kidney dysfunction associated with systemic conditions. Additionally, exclusion of infection or malignancy may be particularly important if the patient will undergo systemic immunosuppression. In challenging PG cases when initial findings are inconclusive and the clinical presentation does not direct workup (eg, colonoscopy to evaluate gastrointestinal tract symptoms), serum protein electrophoresis, hepatitis panel, rheumatoid factor, antinuclear antibodies, and antineutrophilic antibody tests also were frequently ordered by respondents to further evaluate for underlying or associated conditions.
This consensus regarding skin biopsies and certain ancillary tests is consistent with the proposed diagnostic criteria for classic ulcerative PG in which the absence or exclusion of other relevant causes of cutaneous ulcers is required based on the criteria.8 The importance of ensuring an accurate diagnosis is paramount, as a 10% misdiagnosis rate has been documented in the literature.5
Importantly, a stepwise diagnostic workup for PG is proposed based on survey results, which may limit unnecessary testing and the associated costs to the health care system (Figure 1). Selection of additional testing is guided by initial test results and features of the patient’s clinical presentation, including age, review of systems, and associated comorbidities. Available data suggest that underlying inflammatory bowel disease is more frequent in PG patients who are younger than 65 years, whereas those who are 65 years and older are more likely to have inflammatory arthritis, cancer, or an underlying hematologic disorder.9
Treatment of PG should address both the inflammatory and wound components of the disease (Figure 2).7 In our survey results, systemic corticosteroids were identified as an important first-line therapy supported by reasonable evidence and were favored for their rapid response and minimal cost.1,10,11 Many respondents endorsed the use of systemic therapy in combination with topical steroids or calcineurin inhibitors. Combination therapy may provide more immediate control of rapidly progressing disease while minimizing adverse effects of long-term systemic corticosteroid use. A survey of German wound experts similarly endorsed frequent use of topical calcineurin inhibitors and combination systemic and topical glucocorticoid therapy as common therapeutic approaches.1
Importantly, treatments may vary depending on patient characteristics, comorbidities, and underlying disease, which underscores the need for individualized treatment approaches. Alternative first-line systemic treatments favored by respondents were cyclosporine, biologic medications, and antineutrophilic agents such as dapsone. Cyclosporine has demonstrated comparable efficacy to systemic glucocorticoids in one RCT and is considered an important steroid-sparing alternative for PG treatment.2 Biologic agents, especially tumor necrosis factor inhibitors, may be effective in treating cases of refractory PG or for concomitant inflammatory bowel disease management, as demonstrated by a small RCT documenting improvement of PG following infliximab infusion.3
Respondents strongly recommended petrolatum-impregnated gauze and other nonadhesive dressings, including alginate and hydrocolloid dressings, as part of PG wound care. Topical antimicrobials and compression stockings also were recommended by respondents. These practices aim to promote moist environments for healing, avoid maceration, prevent superinfection, optimize wound healing, and minimize damage from adhesive injury.12 Wound debridement and grafting generally were not recommended. However, pathergy is not a universal phenomenon in PG, and wounds that are no longer in the inflammatory phase may benefit from gentle debridement of necrotic tissue and/or grafting in select cases.10
Conclusion
An approach to modifying PG management based on clinical presentation and the practice of combination therapy with multiple systemic agents in refractory PG cases was not addressed in our survey. The low response rate is a limitation; however, the opinions of 51 medical dermatologist experts who regularly manage PG (in contrast to papers based on individualized clinical experience) can provide important clinical guidance until more scientific evidence is established.
Acknowledgments
We would like to thank the SDH and RDS membership for their participation in this survey. We especially acknowledge the other members of the SDH Scientific Task Force for their feedback: Misha Rosenbach, MD (Philadelphia, Pennsylvania); Robert G. Micheletti, MD (Philadelphia, Pennsylvania); Karolyn Wanat, MD (Milwaukee, Wisconsin); Amy Chen, MD (Cromwell, Connecticut); and A. Rambi Cardones, MD (Durham, North Carolina).
- Al Ghazal P, Dissemond J. Therapy of pyoderma gangrenosum in Germany: results of a survey among wound experts. J Dtsch Dermatol Ges . 2015;13:317-324.
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958.
- Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Al Ghazal P, Klode J, Dissemond J. Diagnostic criteria for pyoderma gangrenosum: results of a survey among dermatologic wound experts in Germany. J Dtsch Dermatol Ges. 2014;12:1129-1131.
- Weenig RH, Davis MD, Dahl PR, et al. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002;347:1412-1418.
- Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34:395-409.
- Bennett ML, Jackson JM, Jorizzo JL, et al. Pyoderma gangrenosum: a comparison of typical and atypical forms with an emphasis on time to remission. case review of 86 patients from 2 institutions. Medicine. 2000;79:37-46.
- Su WP, Davis MD, Weening RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Aschyan H, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Binus AM, Qureshi AA, Li VW, et al. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol. 2011;165:1244-1250.
- Reichrath J, Bens G, Bonowitz A, et al. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005;53:273-283.
- Miller J, Yentzer BA, Clark A, et al. Pyoderma gangrenosum: a review and update on new therapies. J Am Acad Dermatol. 2010;62:646-654.
- Al Ghazal P, Dissemond J. Therapy of pyoderma gangrenosum in Germany: results of a survey among wound experts. J Dtsch Dermatol Ges . 2015;13:317-324.
- Ormerod AD, Thomas KS, Craig FE, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350:h2958.
- Brooklyn TN, Dunnill MG, Shetty A, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Al Ghazal P, Klode J, Dissemond J. Diagnostic criteria for pyoderma gangrenosum: results of a survey among dermatologic wound experts in Germany. J Dtsch Dermatol Ges. 2014;12:1129-1131.
- Weenig RH, Davis MD, Dahl PR, et al. Skin ulcers misdiagnosed as pyoderma gangrenosum. N Engl J Med. 2002;347:1412-1418.
- Powell FC, Su WP, Perry HO. Pyoderma gangrenosum: classification and management. J Am Acad Dermatol. 1996;34:395-409.
- Bennett ML, Jackson JM, Jorizzo JL, et al. Pyoderma gangrenosum: a comparison of typical and atypical forms with an emphasis on time to remission. case review of 86 patients from 2 institutions. Medicine. 2000;79:37-46.
- Su WP, Davis MD, Weening RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Aschyan H, Butler DC, Nelson CA, et al. The association of age with clinical presentation and comorbidities of pyoderma gangrenosum. JAMA Dermatol. 2018;154:409-413.
- Binus AM, Qureshi AA, Li VW, et al. Pyoderma gangrenosum: a retrospective review of patient characteristics, comorbidities and therapy in 103 patients. Br J Dermatol. 2011;165:1244-1250.
- Reichrath J, Bens G, Bonowitz A, et al. Treatment recommendations for pyoderma gangrenosum: an evidence-based review of the literature based on more than 350 patients. J Am Acad Dermatol. 2005;53:273-283.
- Miller J, Yentzer BA, Clark A, et al. Pyoderma gangrenosum: a review and update on new therapies. J Am Acad Dermatol. 2010;62:646-654.
Practice Points
- The diagnosis of pyoderma gangrenosum (PG) poses a challenge in clinical practice that could be minimized by following a stepwise algorithm based on initial test results (including skin biopsies) and features of the patient’s clinical presentation.
- As there is no US Food and Drug Administration–approved treatment for PG, a stepwise algorithm approach in combination with the clinical experience addressing inflammation and wound care is essential to reach control and remission of PG.
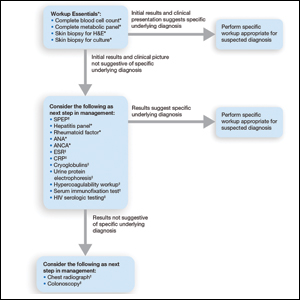
Nutritional Dermatoses in the Hospitalized Patient
The World Health Organization defines malnutrition as deficiencies, excesses, or imbalances in an individual’s intake of energy and/or nutrients.1 This review will focus on undernutrition, which may result from macronutrient or micronutrient deficiencies. Undernutrition in the hospitalized patient is a common yet underrecognized phenomenon, with an estimated prevalence of 20% to 50% worldwide.2 Malnutrition is an independent risk factor for patient morbidity and mortality and has been associated with increased health care costs.3 Nutritional deficiencies may arise from inadequate nutrient intake, abnormal nutrient absorption, or improper nutrient utilization.4 Unfortunately, no standardized algorithm for screening and diagnosing patients with malnutrition exists, making early physical examination findings of utmost importance. Herein, we present a review of acquired nutritional deficiency dermatoses in the inpatient setting.
Protein-Energy Malnutrition
Protein-energy malnutrition (PEM) refers to a set of related disorders that include marasmus, kwashiorkor (KW), and marasmic KW. These conditions frequently are seen in developing countries but also have been reported in developed nations.5 Marasmus occurs from a chronic deficiency of protein and calories. Decreased insulin production and unopposed catabolism result in sarcopenia and loss of bone and subcutaneous fat.6 Affected patients include children who are less than 60% ideal body weight (IBW) without edema or hypoproteinemia.7 Kwashiorkor is the edematous form of PEM that develops from isolated protein deficiency, resulting in edema, diarrhea, and immunosuppression.6 Micronutrient deficiencies, oxidative stress, slow protein catabolism, and excess antidiuretic hormone have been proposed as potential drivers of KW.8 Kwashiorkor affects children between 60% and 80% IBW. Marasmic KW has features of both diseases, including children who are less than 60% IBW but with associated edema and/or hypoproteinemia.9
Although PEM is uncommon in adults, hospitalized patients carry many predisposing risk factors, including infections, malabsorptive conditions, psychiatric disease, and chronic illness (eTable). Patients with chronic infections present with findings consistent with marasmic KW due to lean body mass loss.
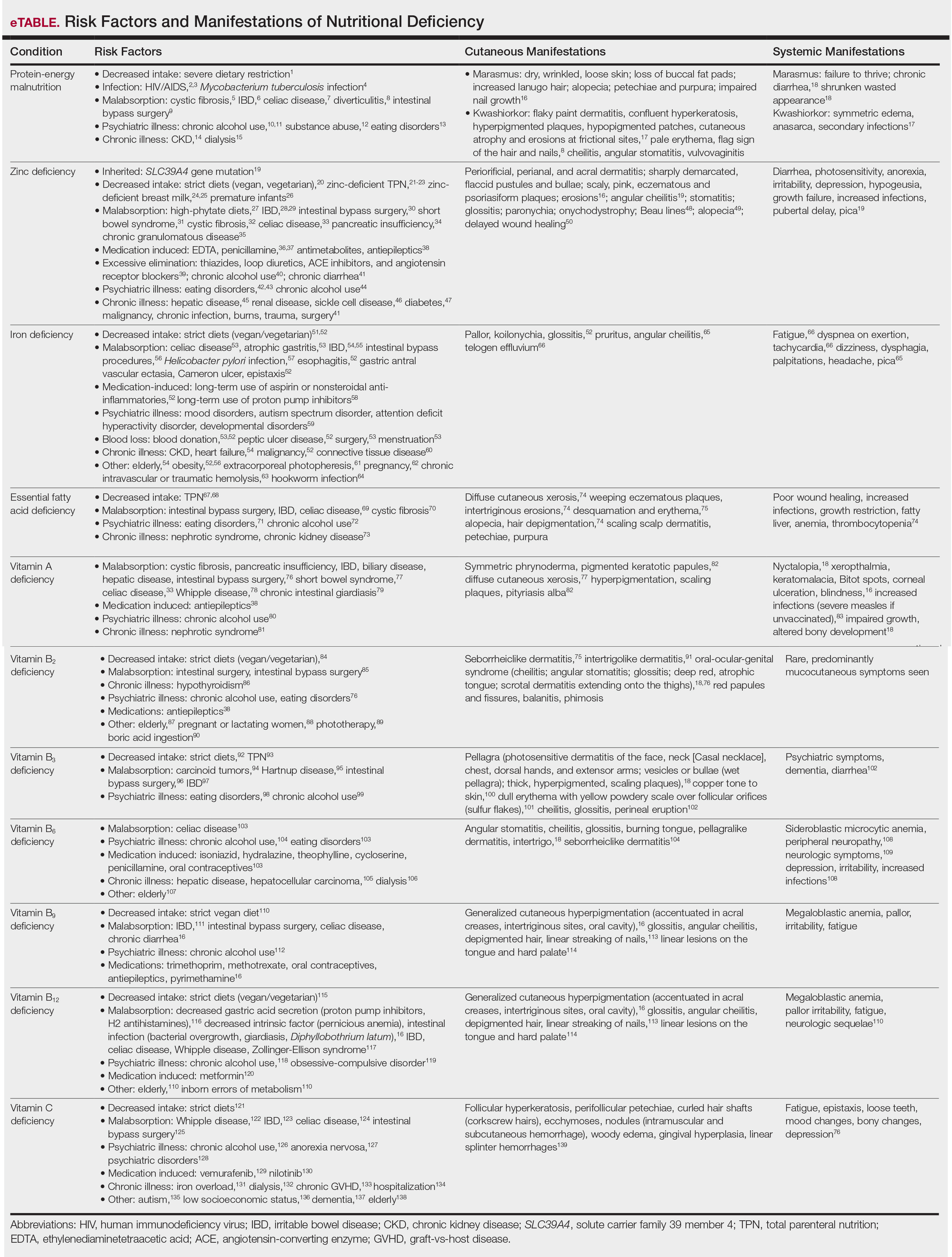
The cutaneous findings in PEM are related to dysmaturation of epidermal keratinocytes and resultant epidermal atrophy.10 Patients with marasmus exhibit dry, wrinkled, loose skin due to subcutaneous fat loss. Emaciated children often lose their buccal fat pads, and reduced perianal adipose may lead to rectal prolapse. Increased lanugo hair may be present on the face, and alopecia of the scalp may occur.6 In KW, cutaneous disease progresses from confluent hyperkeratosis to a dry atrophic epidermis that erodes easily, leaving underlying pale erythema. The resultant pattern is one of hyperpigmented plaques with slightly raised borders, and hypopigmented patches and erosions described as flaky paint dermatitis (Figure 1).5 Lesions appear first in areas of friction. The hair often is dry and brittle; curly hair may straighten and scale.11 Red-yellow to gray-white hypopigmentation may develop, denoting periods of inadequate nutrition. The flag sign describes alternating horizontal bands of hypopigmentation interspersed with bands of pigmented hair. The nails usually are thin and soft and may exhibit the nail flag sign, characterized by horizontal bands of white and red.12 Cheilitis, angular stomatitis, and vulvovaginitis may be present.6
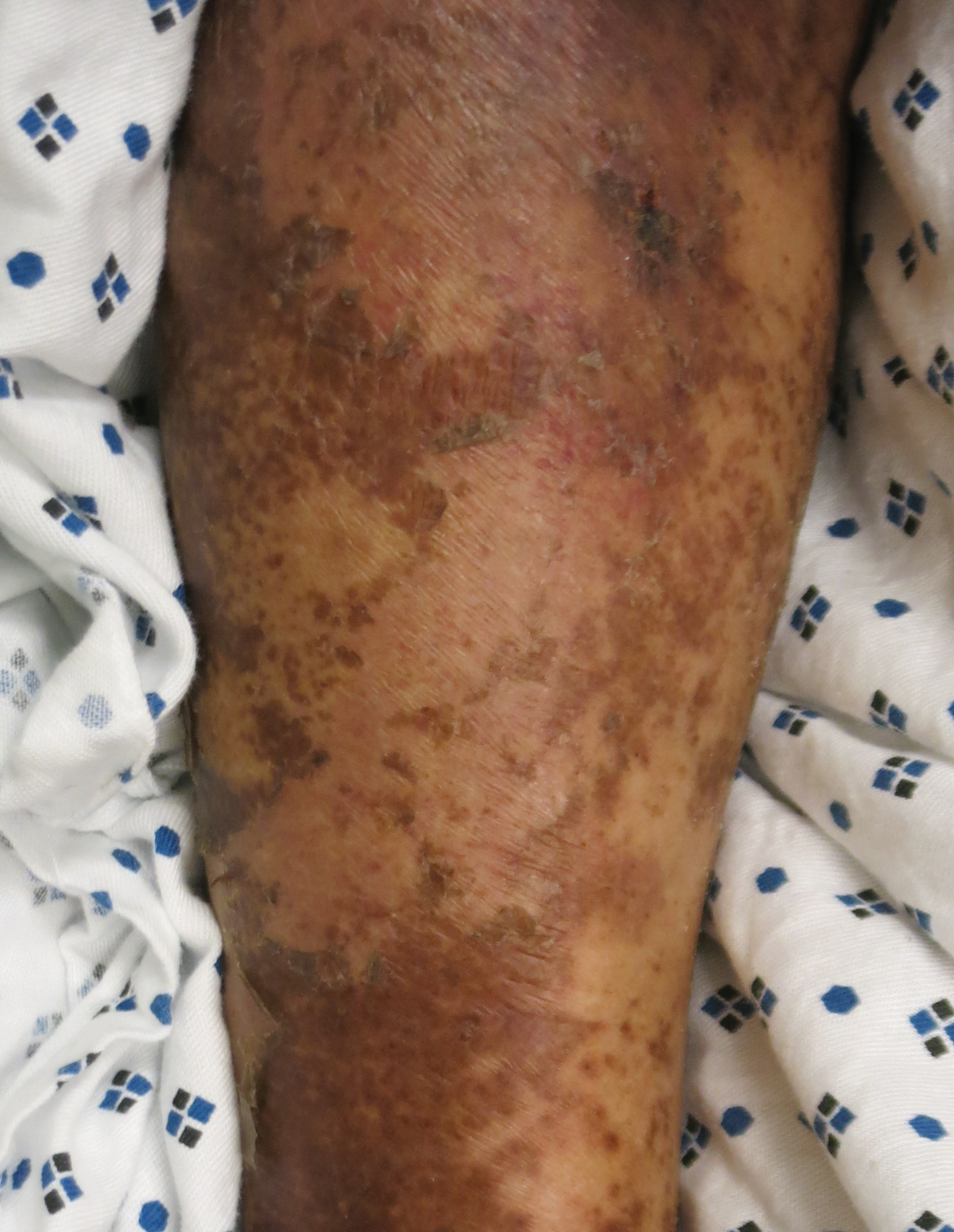
In adults, weight loss and body mass index can be used to assess nutritional status, along with a focused history and physical examination. Complete blood cell count, electrolyte levels, and blood urea nitrogen should be assessed, as hypoglycemia and anemia often accompany PEM.13 In KW, hypoalbuminemia and hypoproteinemia are invariably present. Although prealbumin may be a valid prognostic indicator of disease outcomes and mortality in patients at risk for malnutrition, checking other serum biomarkers remains controversial.14 Focused testing may be warranted in patients with risk factors for chronic infectious processes, such as human immunodeficiency virus or tuberculosis.6 Skin biopsy may solidify the diagnosis of PEM. Hypertrophy of the stratum corneum, atrophy of the stratum spinosum and stratum granulosum, and increased basal layer melanin have been reported.15
Treatment involves initial fluid resuscitation and correction of electrolyte imbalances, followed by nutritional replacement.13 Oral or enteral tube feedings are preferred over total parenteral nutrition (TPN), as they enhance recovery of the gastrointestinal tract.16 Refeeding should occur in small amounts and frequent intervals.5 Skin-directed therapy is aimed at restoring epidermal function and hydration, with regular moisturization and application of barrier creams, such as zinc oxide ointment or petrolatum.10
Zinc Deficiency
Zinc is an essential trace element that provides regulatory, structural, and catalytic functions across multiple biochemical pathways6 and serves as an enzymatic cofactor and key component for numerous transcription factors.17 Zinc is derived from food sources, and its concentration correlates with protein content.18 Zinc is found in both animal and plant-based proteins, albeit with a lower oral bioavailability in the latter. Zinc deficiency may be inherited or acquired. Primary acrodermatitis enteropathica is an autosomal-recessive disorder of the solute carrier family 39 member 4 gene, SLC39A4 (encodes zinc transporter ZIP4 on enterocytes); the result is abnormal zinc absorption from the small intestine.18
Acquired zinc deficiency occurs from decreased dietary zinc intake, impaired intestinal zinc absorption, excessive zinc elimination, or systemic states of high catabolism or low albumin (eTable). Total parenteral nutrition–associated deficiency has arisen when nutritional formulations did not contain trace elements during national shortages or when prolonged TPN was not anticipated and trace elements were removed.19 Zinc levels may already be low in patients with chronic illness or inflammation, so even a short period on TPN can precipitate deficiency.18,19 Diets high in phytate may result in zinc deficiency, as phytate impairs intestinal zinc absorption.20 Approximately 15% of patients with inflammatory bowel disease experienced zinc deficiency worldwide.21 In Crohn disease, zinc deficiency has been associated with active intestinal inflammation, increased risk for hospitalization, surgeries, and disease-related complications.22,23
Medications such as antiepileptics, antimetabolites, or penicillamine may induce zinc deficiency, highlighting the importance of medication review for hospitalized patients (eTable). Catabolic states, frequently encountered in hospitalized patients, increase the risk for zinc deficiency.24 Patients with necrolytic migratory erythema (associated with pancreatic glucagonomas) often experience low serum zinc levels.25
The skin is the third most zinc-abundant tissue in the human body. Within keratinocytes, zinc is critical to normal proliferation and suppression of inflammation.17 Zinc also plays an important role in cutaneous immune function.26 Zinc deficiency presents with sharply demarcated, flaccid pustules and bullae that erode into scaly, pink, eczematous or psoriasiform plaques. Lesions are found preferentially in acral and periorificial sites, often with crusting and exudate. The groin and flexural surfaces may be affected. Erosions often become secondarily impetiginized. Other cutaneous findings include angular cheilitis, stomatitis, glossitis, paronychia, onychodystrophy, generalized alopecia, and delayed wound healing.26 Histopathology of skin lesions is characterized by granular layer loss, epidermal pallor, confluent parakeratosis, spongiosis, dyskeratosis, and psoriasiform hyperplasia.27 Acquired bullous acrodermatitis enteropathica has been reported as a histologic mimicker of pemphigus foliaceous in patients on TPN.28
Diagnosis of zinc deficiency is made by measuring plasma zinc levels. Fasting levels should be drawn in the morning, as they can fluctuate based on the time of day, stress levels, or inflammation.6 Sample hemolysis and anticoagulants high in zinc may falsely elevate plasma zinc. A normal zinc level is greater than 70 µg/dL; however, normal levels do not rule out deficiency.18 Measurement of zinc-dependent enzymes, such as alkaline phosphatase, can be a quick way to assess zinc status. Serum albumin also should be measured; because zinc is carried by albumin in the blood, hypoalbuminemia may result in secondary zinc deficiency.18
Zinc replacement therapy is largely through oral supplementation and should start at 0.5 to 2.0 mg/kg/d in adults with acquired disease.29,30 Zinc sulfate is the most affordable and is the supplement of choice, with 50 mg of elemental zinc per 220 mg of zinc sulfate (~23% elemental zinc).31 Alternative zinc salts, such as zinc gluconate (13% elemental zinc), may be used. Patients with malabsorptive disorders often require parenteral supplementation.32 Clinical symptoms often will resolve within 1 to 2 weeks of supplementation.29 In patients with primary acrodermatitis enteropathica, lifelong supplementation with 3 mg/kg/d elemental zinc should occur.6 Calcium and folate may reduce zinc absorption, while zinc supplementation can interfere with copper and iron absorption.33
Iron Deficiency
Iron is an essential component of the hemoglobin molecule. Iron homeostasis and metabolism are tightly regulated processes that drive erythropoiesis. Only 5% to 10% of dietary iron is absorbed through nutrition, while the remainder is recycled from red cell breakdown. Both normal iron levels and iron deficiency (ID) are defined by age and gender.34 Iron-deficiency anemia (IDA) is one of the most common cause-specific anemias worldwide.35
Fatigue is the most common and earliest symptom of ID. In a single study, pallor was predictive of anemia in hospitalized patients; however, absence of pallor did not rule out anemia.34 Dyspnea on exertion, tachycardia, dysphagia, and pica also may be reported. Cutaneous manifestations include koilonychia (Figure 2), glossitis, pruritus, angular cheilitis, and telogen effluvium. Plummer-Vinson syndrome is characterized by microcytic anemia, glossitis, and dysphagia.
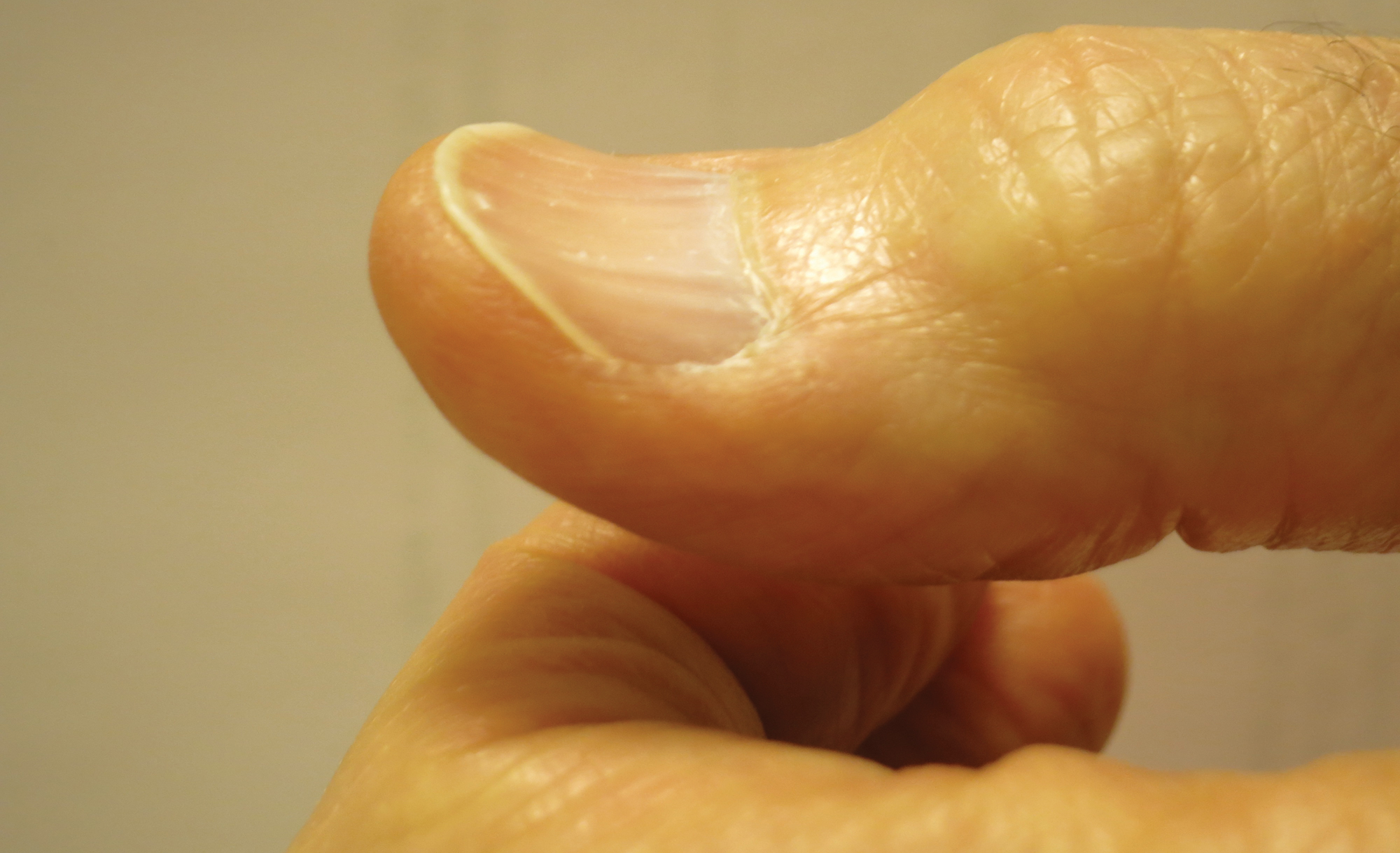
Risk factors for ID include insufficient dietary consumption,36 blood loss, malabsorptive states,37,38 and increased iron requirements (eTable). Patient fragility (eg, elderly, chronic disease) is a newly described risk factor where correction of ID may impact morbidity, mortality, and quality of life.35
Iron deficiency can be present despite a normal hemoglobin level. Serum ferritin and percentage transferrin saturation are key to early identification of IDA.35 Ferritin levels lower than 30 µg/L confirm the diagnosis. Decreased transferrin saturation and increased total iron binding capacity aid in the diagnosis of IDA. Serum ferritin is an acute-phase reactant, and levels may be falsely elevated in the setting of inflammation or infection.
Treatment includes reversing the cause of deficiency and supplementing iron. Calculation of the total iron deficit can help inform iron supplementation. First-line therapy for IDA is oral ferrous sulfate 325 mg (65 mg elemental iron) 3 times daily. Newer studies suggest 40 to 80 mg oral iron should be taken every other day to increase absorption.39 Other iron salts, such as ferrous gluconate (325 mg is equivalent to 38 mg elemental iron), have been used. Iron absorption is enhanced by an acidic environment. Parenteral iron is utilized in patients with uncorrectable blood loss, malabsorption, renal failure, intolerance to oral iron, and nonadherence in those who are unable to receive transfusions. Iron infusions are favored in frail patients, such as the elderly and those with chronic kidney disease or heart failure.35 Multiple parenteral iron formulations exist, and their use should be driven by underlying patient comorbidities and potential risks. Packed red blood cell transfusions should be considered in acute blood loss, hypoxia, or cardiac insufficiency.
Essential Fatty Acid Deficiency
Essential fatty acids (EFAs) including linoleic and α-linolenic acid cannot be synthesized by the human body and must be obtained through diet (mostly plant oils). Essential fatty acids have various functions, including maintaining phospholipid membrane integrity, forming prostaglandins and leukotrienes, and storing energy.40 Essential fatty acids are important in the structure and function of the stratum corneum and are crucial in maintaining epidermal barrier function.41 Increased epidermal permeability and transepidermal water loss may be the first signs of EFA deficiency (EFAD).42
The cutaneous manifestations of EFAD include xerosis, weeping eczematous plaques, and erosions in intertriginous sites. The lesions may progress to widespread desquamation and erythema. With time, the skin can become thick and leathery. Alopecia may occur, and hair may depigment.7 Additional findings include poor wound healing and increased susceptibility to infections.43,44
Essential fatty acid deficiency may occur when dietary fat intake is severely restricted or in malabsorptive states.45,46 It develops in patients on prolonged TPN, typically when receiving fat-restricted nutrition,47,48 as occurs in hypertriglyceridemia.47 Essential fatty acid deficiency has developed in patients on TPN containing EFAs,47 as the introduction of novel intravenous lipid emulsions has resulted in varying proportions of EFA.40 Premature neonates are particularly at risk for EFAD.49
The diagnosis of EFAD involves the measurement of the triene to tetraene ratio. A ratio of more than 0.2 suggests EFAD, but the clinical signs are not seen until the ratio is over 0.4.40 Low plasma levels of linoleic, linolenic, and arachidonic acids also are seen. Elevated liver function tests are supportive of the diagnosis. Biochemical findings typically are seen before cutaneous manifestations.40
Treatment of EFAD includes topical, oral, or intravenous replacement of EFAs. Improvement of EFAD with the application of topical linoleic acid to the skin has been reported.50 Patients receiving TPN should undergo assessment of parenteral lipid emulsion to ensure adequate fatty acid composition.
Vitamin A Deficiency
Vitamin A (retinol) is a fat-soluble vitamin that plays a critical role in keratinization, epithelial proliferation, and cellular differentiation.6 Vitamin A is found in animal products as retinyl esters and in plants as beta-carotene. Vitamin A has 2 clinically important forms: all-trans retinoic acid and 11-cis-retinal. All-trans retinoic acid is involved in cellular differentiation and regulating gene transcription, while 11-cis-retinal is key to rhodopsin generation required for vision. Vitamin A deficiency presents with early ophthalmologic findings, specifically nyctalopia, or delayed adaptation to the dark.51 Xerophthalmia, abnormal conjunctival keratinization, and Bitot spots subsequently develop and may progress to corneal ulceration and blindness.6
Vitamin A deficiency manifests in the skin as follicular hyperkeratosis, or phrynoderma. Notably, numerous other micronutrient deficiencies may result in phrynoderma. Clinically, multiple pigmented keratotic papules of various sizes, many with a central keratinous plug, are distributed symmetrically on the extensor elbows, knees, shoulders, buttocks, and extremities. The skin surrounding these lesions may be scaly and hyperpigmented.52 Generalized xerosis without preceding nyctalopia has been reported.53 Accompanying pityriasis alba may develop.52 Lesions on the face may mimic acne, while lesions on the extremities may simulate a perforating disorder. Histopathology of phrynoderma reveals epidermal hyperkeratosis, follicular hyperkeratosis, and follicular plugging.52
Patients at risk for vitamin A deficiency include those with conditions that affect intestinal fat absorption, underlying psychiatric illness, or chronic disease (eTable). Chronic alcohol use predisposes patients to a multitude of micronutrient deficiencies, including vitamin A deficiency.54 In chronic alcohol use, even mild cutaneous changes may be the first clue to low serum retinol.55
Vitamin A deficiency can be diagnosed by measuring serum retinol levels, with levels lower than 20 µg/dL being diagnostic of deficiency.56 Decreased serum retinol in patients hospitalized with flaring irritable bowel disorder has been repeatedly reported.57-59 Notably, serum retinol concentration does not decline until liver reserves of vitamin A are nearing exhaustion.33
The US Food and Drug Administration requires manufacturers to list retinol activity equivalents on labels. One international unit of retinol is equivalent to 0.3 µg of retinol activity equivalents.60 The treatment of vitamin A deficiency involves high-dose oral supplementation when possible.61 Although dependent on age, the treatment dose for most adults with vitamin A deficiency is 3000 µg (10,000 IU) once daily.
Phrynoderma has been specifically treated with salicylic acid ointment 3% and intramuscular vitamin A.62 Topical urea cream also may treat phrynoderma.63
Vitamin B2
Vitamin B2 (riboflavin) is absorbed in the small intestine and converted into 2 biologically active forms—flavin adenine dinucleotide and flavin mononucleotide—which serve as cofactors in metabolic and oxidation-reduction reactions. Malabsorptive disorders and bowel resection can lead to riboflavin deficiency.64 Other at-risk populations include those with restrictive diets,65 psychiatric illness, or systemic illness (eTable). Riboflavin can be degraded by light (deficiency has been reported after phototherapy for neonatal jaundice66) and following boric acid ingestion.67 Medications, including long-term treatment with antiepileptics, may lead to riboflavin deficiency.68
Riboflavin is critical to maintaining collagen production. Riboflavin deficiency may manifest clinically with extensive seborrheiclike dermatitis,44 intertrigolike dermatitis,69 or oral-ocular-genital syndrome.70 Angular cheilitis may accompany an atrophic tongue that is deep red in color. The scrotum is characteristically involved in men, with confluent dermatitis extending onto the thighs and sparing the midline. Red papules and painful fissures may develop. Balanitis and phimosis have been reported. Testing for riboflavin deficiency should be considered in patients with refractory seborrheic dermatitis.
Riboflavin stores are assessed by the erythrocyte glutathione reductase activity coefficient.44 A level of 1.4 or higher is consistent with deficiency. Serum riboflavin levels, performed after a 12-hour fast, may support the diagnosis but are less sensitive. Patients with glucose-6-phosphate deficiency cannot be assessed via the erythrocyte glutathione reductase activity coefficient and may instead require evaluation of 24-hour urine riboflavin level.44
Vitamin B3
Vitamin B3 (niacin, nicotinamide, nicotinic acid) is found in plant and animal products or can be derived from its amino acid precursor tryptophan. Niacin deficiency results in pellagra, characterized by dermatitis, dementia, and diarrhea.71 The most prominent feature is a symmetrically distributed photosensitive dermatitis of the face, neck (called Casal necklace)(Figure 3), chest, dorsal hands, and extensor arms. The eruption may begin with erythema, vesicles, or bullae (wet pellagra) and evolve into thick, hyperpigmented, scaling plaques.71 The skin may take on a copper tone and become atrophic.72 Dull erythema with overlying yellow powdery scale (called sulfur flakes) at follicular orifices has been described on the nasal bridge.73
Causes of niacin deficiency include malabsorptive conditions, malignancy (including carcinoid tumors), parenteral nutrition, psychiatric disease,74,75 and restrictive diets (eTable).76 Carcinoid tumors divert tryptophan to serotonin resulting in niacin deficiency.77
The diagnosis of niacin deficiency is based on clinical findings and response to supplementation.75 Low niacin urinary metabolites (N-methylnicotinamide and 2-pyridone) may aid in diagnosis.6 Treatment generally includes oral nicotinamide 100 mg every 6 hours; the dose can then be tapered to 50 mg every 8 to 12 hours until symptoms resolve. Severe deficiency may require parenteral nicotinamide 1 g 3 to 4 times daily.75
Vitamin B6
Vitamin B6 (pyridoxine, pyridoxamine, pyridoxal) is found in whole grains and plant and animal products. Vitamin B6 functions as a coenzyme in many metabolic pathways and is involved in the conversion of tryptophan to niacin.44 Absorption requires hydrolysis by intestinal phosphates and transport to the liver for rephosphorylation prior to release in active form.6
Cutaneous findings associated with vitamin B6 deficiency include periorificial and perineal seborrheic dermatitis,78 angular stomatitis, and cheilitis, with associated burning, redness, and tongue edema.6 Vitamin B6 deficiency is a rarely reported cause of burning mouth syndrome.79 Because vitamin B6 is involved in the conversion of tryptophan to niacin, deficiency also may present with pellagralike findings.70 Other clinical symptoms are outlined in the eTable.80,81
Conditions that increase risk for vitamin B6 deficiency are highlighted in the eTable and include malabsorptive disorders; psychiatric illness82; and chronic disease, especially end-stage renal disease.83 Vitamin B6 deficiency associated with chronic alcohol use is due to both inadequate vitamin B6 intake as well as reduced hepatic storage.78 Medications such as isoniazid, hydralazine, and oral contraceptives may decrease vitamin B6 levels (eTable).82
Vitamin B6 can be measured in the plasma as pyridoxal 5′-phosphate. Plasma concentrations of less than 20 nmol/L are suggestive of deficiency.82 Indirect tests include tryptophan and methionine loading.6 The treatment of vitamin B6 deficiency is determined by symptom severity. Recommendations for oral supplementation range from 25 to 600 mg daily.82 Symptoms typically improve on 100 mg daily.6
Vitamins B9 and B12
Deficiencies of vitamins B9 (folic acid, folate) and B12 (cobalamin) have similar clinical presentations. Folate is essential in the metabolism of amino acids, purines, and pyrimidines.6 Cobalamin, found in animal products, is a cofactor for methionine synthase and methylmalonyl-CoA mutase.84 Megaloblastic anemia is the main finding in folate or cobalamin deficiency. Neurologic findings only accompany cobalamin deficiency. Risk factors for folate deficiency include malabsorptive conditions,6 chronic alcohol use,85 and antifolate medication use (eTable).6
Cobalamin absorption requires gastric acid and intrinsic factor binding in the duodenum. Deficiency may occur from strict diets, psychiatric illness, old age,86 decreased gastric acid secretion,87 abnormal intrinsic factor function, or intestinal infections.6
Generalized cutaneous hyperpigmentation may be the first manifestation of vitamins B9 and B12 deficiency.88 Typically accentuated in acral creases and the oral cavity, pigmentation may mimic Addison disease. Hair depigmentation and linear streaking of the nails are reported.84 The tongue becomes painful and red with atrophy of the filiform papillae (Hunter glossitis).78 Linear lesions on the tongue and hard palate may serve as an early sign of cobalamin deficiency.89
Folate deficiency is diagnosed by measuring the plasma folate level; coincidental cobalamin deficiency should be excluded. Deficiency is managed with oral supplementation (when possible) with 1 to 5 mg of folate daily.6 Cobalamin deficiency is based on low serum levels (<150 pg/mL is diagnostic).86 Cobalamin deficiency may take years to develop, as vitamin B12 exists in large body stores.6 Serum methylmalonic acid may be elevated in patients with clinical features but normal-low serum vitamin B12 level.86 Treatment of vitamin B12 deficiency is with oral (2 mg once daily) or parenteral (1 mg every 4 weeks then maintained at once monthly) cyanocobalamin. For patients with neurologic symptoms, intramuscular injection should be given.86 The underlying cause of deficiency must be elucidated and treated.
Vitamin C Deficiency
Vitamin C (ascorbic acid) is an essential cofactor for the hydroxylation of proline and lysine residues in collagen synthesis. Plant-based foods are the main dietary source of vitamin C, and deficiency presents clinically as scurvy. Cutaneous findings include follicular hyperkeratosis, perifollicular petechiae, and curled hair shafts (corkscrew hairs)(Figure 4). Ecchymoses of the lower extremities, forearms, and abdomen may be seen. Nodules representing intramuscular and subcutaneous hemorrhage can be present.90 Woody edema may mimic cellulitis, while lower extremity hemorrhage may mimic vasculitis. Gingival hyperplasia, hemorrhage, and edema may occur,90 along with linear splinter hemorrhages.91
Hypovitaminosis C has been routinely demonstrated in hospitalized patients.92 Scurvy may occur in patients on strict diets,93 chronic alcohol use,94 psychiatric illness,95 or gastrointestinal tract disease (eTable).96-99 Those with low socioeconomic status70 or dementia100 as well as the elderly also are at risk.101 Scurvy has developed in patients with iron overload and those who are on hemodialysis44 as well as in association with nilotinib use.102 Patients with chronic mucous membrane graft-vs-host disease may exhibit vitamin C deficiency.103
Scurvy is a clinical diagnosis. Vitamin C levels normalize quickly with supplementation. Cutaneous biopsy will exhibit follicular hyperkeratosis, perifollicular hemorrhage, and fibrosis.91
Oral ascorbic acid supplementation should be initiated at 500 to 1000 mg daily in adults.104 The cause of deficiency should be identified, and further supplementation should be decided based on patient risk factors. Lifestyle modifications, such as cessation of smoking and chronic alcohol use, is recommended. The diagnosis of scurvy should prompt workup for additional nutrient deficiencies.
Final Thoughts
Dermatologists play an important role in the early recognition of nutritional deficiencies, as cutaneous manifestations often are the first clue to diagnosis. Nutritional deficiencies are common yet underrecognized in the hospitalized patient and serve as an independent risk factor for patient morbidity and mortality.3 Awareness of the cutaneous manifestations of undernutrition as well as the risk factors for nutritional deficiency may expedite diagnosis and supplementation, thereby improving outcomes for hospitalized patients.
- Mehta NM, Corkins MR, Lyman B, et al. Defining pediatric malnutrition: a paradigm shift toward etiology-related definitions. JPEN J Parenter Enteral Nutr. 2013;37:460-481.
- Barker LA, Gout BS, Crowe TC. Hospital malnutrition: prevalence, identification and impact on patients and the healthcare system. Int J Environ Res Public Health. 2011;8:514-527.
- Bharadwaj S, Ginoya S, Tandon P, et al. Malnutrition: laboratory markers vs nutritional assessment. Gastroenterol Rep (Oxf). 2016;4:272-280.
- Basavaraj KH, Seemanthini C, Rashmi R. Diet in dermatology: present perspectives. Indian J Dermatol. 2010;55:205-210.
- Grover Z, Ee LC. Protein energy malnutrition. Pediatr Clin North Am. 2009;56:1055-1068.
- Jen M, Yan AC. Syndromes associated with nutritional deficiency and excess. Clin Dermatol. 2010;28:669-685.
- Lekwuttikarn R, Teng JMC. Cutaneous manifestations of nutritional deficiency. Curr Opin Pediatr. 2018;30:505-513.
- Jaffe AT, Heymann WR. Kwashiorkor/zinc deficiency overlap following partial gastrectomy. Int J Dermatol. 1998;37:134-137.
- Listernick R, Christoffel K, Pace J, et al. Severe primary malnutrition in US children. Am J Dis Child. 1985;139:1157-1160.
- Heilskov S, Rytter MJ, Vestergaard C, et al. Dermatosis in children with oedematous malnutrition (Kwashiorkor): a review of the literature. J Eur Acad Dermatol Venereol. 2014;28:995-1001.
- Bradfield RB. Hair tissue as a medium for the differential diagnosis of protein-calorie malnutrition: a commentary. J Pediatr. 1974;84:294-296.
- Cohen PR. The nail flag sign: case report in a man with diverticulitis and review of dermatology flag sign of the hair, skin, and nails. Cureus. 2018;10:e2929.
- Management of Severe Malnutrition: A Manual for Physicians and Other Senior Health Workers. Geneva, Switzerland: World Health Organization; 1999. https://www.who.int/nutrition/publications/en/manage_severe_malnutrition_eng.pdf. Accessed May 19, 2020.
- Keller U. Nutritional laboratory markers in malnutrition. J Clin Med. 2019;8:775.
- Thavaraj V, Sesikeran B. Histopathological changes in skin of children with clinical protein energy malnutrition before and after recovery. J Trop Pediatr. 1989;35:105-108.
- McClave SA, Heyland DK. The physiologic response and associated clinical benefits from provision of early enteral nutrition. Nutr Clin Pract. 2009;24:305-315.
- Ogawa Y, Kinoshita M, Shimada S, et al. Zinc and skin disorders. Nutrients. 2018;10:199.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Wiznia LE, Bhansali S, Brinster N, et al. Acquired acrodermatitis enteropathica due to zinc-depleted parenteral nutrition. Pediatr Dermatol. 2019;36:520-523.
- Sandstead HH, Freeland-Graves JH. Dietary phytate, zinc and hidden zinc deficiency. J Trace Elem Med Biol. 2014;28:414-417.
- Vagianos K, Bector S, McConnell J, et al. Nutrition assessment of patients with inflammatory bowel disease. JPEN J Parenter Enteral Nutr. 2007;31:311-319.
- Schoelmerich J, Becher MS, Hoppe-Seyler P, et al. Zinc and vitamin A deficiency in patients with Crohn’s disease is correlated with activity but not with localization or extent of the disease. Hepatogastroenterology. 1985;32:34-38.
- Siva S, Rubin DT, Gulotta G, et al. Zinc deficiency is associated with poor clinical outcomes in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2017;23:152-157.
- Semrad CE. Zinc and intestinal function. Curr Gastroenterol Rep. 1999;1:398-403.
- Sinclair SA, Reynolds NJ. Necrolytic migratory erythema and zinc deficiency. Br J Dermatol. 1997;136:783-785.
- Gammoh NZ, Rink L. Zinc in infection and inflammation. Nutrients. 2017;9:624.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Wu D, Fung MA, Kiuru M, et al. Acquired bullous acrodermatitis enteropathica as a histologic mimic of pemphigus foliaceus in a patient on parenteral nutrition. Dermatol Online J. 2018;24:20.
- Maxfield L, Crane J. Zinc Deficiency. Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK493231/Updated November 14, 2019. Accessed May 19, 2020.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Wegmüller R, Tay F, Zeder C, et al. Zinc absorption by young adults from supplemental zinc citrate is comparable with that from zinc gluconate and higher than from zinc oxide. J Nutr. 2014;144:132-136.
- Vick G, Mahmoudizad R, Fiala K. Intravenous zinc therapy for acquired zinc deficiency secondary to gastric bypass surgery: a case report. Dermatol Ther. 2015;28:222-225.
- Ghishan FK, Kiela PR. Vitamins and minerals in inflammatory bowel disease. Gastroenterol Clin North Am. 2017;46:797-808.
- Killip S, Bennett JM, Chambers MD. Iron deficiency anemia. Am Fam Physician. 2007;75:671-678.
- De Franceschi L, Iolascon A, Taher A, et al. Clinical management of iron deficiency anemia in adults: systemic review on advances in diagnosis and treatment. Eur J Intern Med. 2017;42:16-23.
- Haider LM, Schwingshackl L, Hoffmann G, et al. The effect of vegetarian diets on iron status in adults: a systematic review and meta-analysis. Crit Rev Food Sci Nutr. 2018;58:1359-1374.
- Enani G, Bilgic E, Lebedeva E, et al. The incidence of iron deficiency anemia post-Roux-en-Y gastric bypass and sleeve gastrectomy: a systematic review [published online September 4, 2019]. Surg Endosc. doi:10.1007/s00464-019-07092-3.
- Kaitha S, Bashir M, Ali T. Iron deficiency anemia in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2015;6:62-72.
- Moretti D, Goede JS, Zeder C, et al. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood. 2015;126:1981-1989.
- Gramlich L, Meddings L, Alberda C, et al. Essential fatty acid deficiency in 2015: the impact of novel intravenous lipid emulsions. JPEN J Parenter Enteral Nutr. 2015;39(1 suppl):61S-66S.
- Khnykin D, Miner JH, Jahnsen F. Role of fatty acid transporters in epidermis: implications for health and disease. Dermatoendocrinol. 2011;3:53-61.
- Wright S. Essential fatty acids and the skin. Br J Dermatol. 1991;125:503-515.
- Lakdawala N, Grant-Kels JM. Acrodermatitis caused by nutritional deficiency and metabolic disorders. Clin Dermatol. 2017;35:64-67.
- DiBaise M, Tarleton SM. Hair, nails, and skin: differentiating cutaneous manifestations of micronutrient deficiency. Nutr Clin Pract. 2019;34:490-503.
- Aldámiz-Echevarría L, Bilbao A, Andrade F, et al. Fatty acid deficiency profile in children with food allergy managed with elimination diets. Acta Paediatr. 2008;97:1572-1576.
- Jeppesen PB, Christensen MS, Høy CE, et al. Essential fatty acid deficiency in patients with severe fat malabsorption. Am J Clin Nutr. 1997;65:837-843.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published online June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
- Fleming CR, Smith LM, Hodges RE. Essential fatty acid deficiency in adults receiving total parenteral nutrition. Am J Clin Nutr. 1976;29:976-983.
- Cooke RJ, Zee P, Yeh YY. Essential fatty acid status of the premature infant during short-term fat-free parenteral nutrition. J Pediatr Gastroenterol Nutr. 1984;3:446-449.
- Skolnik P, Eaglstein WH, Ziboh VA. Human essential fatty acid deficiency: treatment by topical application of linoleic acid. Arch Dermatol. 1977;113:939-941.
- Vahlquist A. Clinical use of vitamin A and its derivatives—physiological and pharmacological aspects. Clin Exp Dermatol. 1985;10:133-143.
- Ragunatha S, Kumar VJ, Murugesh SB. A clinical study of 125 patients with phrynoderma. Indian J Dermatol. 2011;56:389-392.
- Phanachet P, Shantavasinkul PC, Chantrathammachart P, et al. Unusual manifestation of vitamin A deficiency presenting with generalized xerosis without night blindness. Clin Case Rep. 2018;6:878-882.
- Fuchs J. Alcoholism, malnutrition, vitamin deficiencies, and the skin. Clin Dermatol. 1999;17:457-461.
- Uhoda E, Petit L, Piérard-Franchimont C, et al. Ultraviolet light-enhanced visualization of cutaneous signs of carotene and vitamin A dietary deficiency. Acta Clin Belg. 2004;59:97-101.
- de Pee S, Dary O. Biochemical indicators of vitamin A deficiency: serum retinol and serum retinol binding protein. J Nutr. 2002;132(9 suppl):2895S-2901S.
- Fernandez-Banares F, Abad-Lacruz A, Xiol X, et al. Vitamin status in patients with inflammatory bowel disease. Am J Gastroenterol. 1989;84:744-748.
- Main AN, Mills PR, Russell RI, et al. Vitamin A deficiency in Crohn’s disease. Gut. 1983;24:1169-1175.
- Cobos G, Cornejo C, McMahon P. A case of phrynoderma in a patient with Crohn’s disease. Pediatr Dermatol. 2015;32:234-236.
- Trumbo P, Yates AA, Schlicker S, et al. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. 2001;101:294-301.
- Ross DA. Recommendations for vitamin A supplementation. J Nutr. 2002;132(9 suppl):2902S-2906S.
- Ragunatha S, Jagannath Kumar V, Murugesh SB, et al. Therapeutic response of vitamin A, vitamin B complex, essential fatty acids (EFA) and vitamin E in the treatment of phrynoderma: a randomized controlled study. J Clin Diagn Res. 2014;8:116-118.
- Nakjang Y, Yuttanavivat T. Phrynoderma: a review of 105 cases. J Dermatol. 1988;15:531-534.
- Pinto JT, Zempleni J. Riboflavin. Adv Nutr. 2016;7:973-975.
- Larsson CL, Johansson GK. Dietary intake and nutritional status of young vegans and omnivores in Sweden. Am J Clin Nutr. 2002;76:100-106.
- Gromisch DS, Lopez R, Cole HS, et al. Light (phototherapy)—induced riboflavin deficiency in the neonate. J Pediatr. 1977;90:118-122.
- Pinto J, Huang YP, McConnell RJ, et al. Increased urinary riboflavin excretion resulting from boric acid ingestion. J Lab Clin Med. 1978;92:126-134.
- Soltani D, Ghaffar Pour M, et al. Nutritional aspects of treatment in epileptic patients. Iran J Child Neurol. 2016;10:1-12.
- Roe DA. Riboflavin deficiency: mucocutaneous signs of acute and chronic deficiency. Semin Dermatol. 1991;10:293-295.
- Galimberti F, Mesinkovska NA. Skin findings associated with nutritional deficiencies. Cleve Clin J Med. 2016;83:731-739.
- Karthikeyan K, Thappa DM. Pellagra and skin. Int J Dermatol. 2002;41:476-481.
- Nogueira A, Duarte AF, Magina S, et al. Pellagra associated with esophageal carcinoma and alcoholism. Dermatol Online J. 2009;15:8.
- Wan P, Moat S, Anstey A. Pellagra: a review with emphasis on photosensitivity. Br J Dermatol. 2011;164:1188-1200.
- Jagielska G, Tomaszewicz-Libudzic EC, Brzozowska A. Pellagra: a rare complication of anorexia nervosa. Eur Child Adolesc Psychiatry. 2007;16:417-420.
- Li R, Yu K, Wang Q, et al. Pellagra secondary to medication and alcoholism: a case report and review of the literature. Nutr Clin Pract. 2016;31:785-789.
- Ladoyanni E, Cheung ST, North J, et al. Pellagra occurring in a patient with atopic dermatitis and food allergy. J Eur Acad Dermatol Venereol. 2007;21:394-396.
- Bell HK, Poston GJ, Vora J, et al. Cutaneous manifestations of the malignant carcinoid syndrome. Br J Dermatol. 2005;152:71-75.
- Barthelemy H, Chouvet B, Cambazard F. Skin and mucosal manifestations in vitamin deficiency. J Am Acad Dermatol. 1986;15:1263-1274.
- Lamey PJ, Hammond A, Allam BF, et al. Vitamin status of patients with burning mouth syndrome and the response to replacement therapy. Br Dent J. 1986;160:81-84.
- Stover PJ, Field MS. Vitamin B-6. Adv Nutr. 2015;6:132-133.
- Gerlach AT, Thomas S, Stawicki SP, et al. Vitamin B6 deficiency: a potential cause of refractory seizures in adults. JPEN J Parenter Enteral Nutr. 2011;35:272-275.
- Spinneker A, Sola R, Lemmen V, et al. Vitamin B6 status, deficiency and its consequences—an overview. Nutr Hosp. 2007;22:7-24.
- Ross EA, Shah GM, Reynolds RD, et al. Vitamin B6 requirements of patients on chronic peritoneal dialysis. Kidney Int. 1989;36:702-706.
- Brescoll J, Daveluy S. A review of vitamin B12 in dermatology. Am J Clin Dermatol. 2015;16:27-33.
- Sanvisens A, Zuluaga P, Pineda M, et al. Folate deficiency in patients seeking treatment of alcohol use disorder. Drug Alcohol Depend. 2017;180:417-422.
- Langan RC, Goodbred AJ. Vitamin B12 deficiency: recognition and management. Am Fam Physician. 2017;96:384-389.
- Bradford GS, Taylor CT. Omeprazole and vitamin B12 deficiency. Ann Pharmacother. 1999;33:641-643.
- Srivastava N, Chand S, Bansal M, et al. Reversible hyperpigmentation as the first manifestation of dietary vitamin B12 deficiency. Indian J Dermatol Venereol Leprol. 2006;72:389-390.
- Graells J, Ojeda RM, Muniesa C, et al. Glossitis with linear lesions: an early sign of vitamin B12 deficiency. J Am Acad Dermatol. 2009;60:498-500.
- Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999;41:895-906; quiz 907-810.
- Shaath T, Fischer R, Goeser M, et al. Scurvy in the present times: vitamin C allergy leading to strict fast food diet. Dermatol Online J. 2016;22:13030/qt50b8w28b.
- Fain O, Pariés J, Jacquart B, et al. Hypovitaminosis C in hospitalized patients. Eur J Intern Med. 2003;14:419-425.
- Ahmad SA, Al Thobiti TA, El Toum M, et al. Florid scurvy in an autistic child on a ketogenic diet [published online November 19, 2018]. Pediatr Emerg Care. doi:10.1097/PEC.0000000000001695.
- Lux-Battistelli C, Battistelli D. Latent scurvy with tiredness and leg pain in alcoholics: an underestimated disease three case reports. Medicine (Baltimore). 2017;96:e8861.
- Christopher K, Tammaro D, Wing EJ. Early scurvy complicating anorexia nervosa. South Med J. 2002;95:1065-1066.
- Berger ML, Siegel DM, Lee EL. Scurvy as an initial manifestation of Whipple’s disease. Ann Intern Med. 1984;101:58-59.
- Imes S, Dinwoodie A, Walker K, et al. Vitamin C status in 137 outpatients with Crohn’s disease. effect of diet counseling. J Clin Gastroenterol. 1986;8:443-446.
- Echeverría Zudaire L, García Cuartero B, Campelo Moreno O, et al. Scurvy associated with celiac disease [in Spanish]. An Esp Pediatr. 2002;57:587.
- Hansen EP, Metzsche C, Henningsen E, et al. Severe scurvy after gastric bypass surgery and a poor postoperative diet. J Clin Med Res. 2012;4:135-137.
- Rivière S, Birlouez-Aragon I, Nourhashémi F, et al. Low plasma vitamin C in Alzheimer patients despite an adequate diet. Int J Geriatr Psychiatry. 1998;13:749-754.
- Bhattacharyya P, Giannoutsos J, Eslick GD, et al. Scurvy: an unrecognized and emerging public health issue in developed economies. Mayo Clin Proc. 2019;94:2594-2597.
- Oak AS, Jaleel T, Fening K, et al. A case of scurvy associated with nilotinib. J Cutan Pathol. 2016;43:725-726.
- Kletzel M, Powers K, Hayes M. Scurvy: a new problem for patients with chronic GVHD involving mucous membranes; an easy problem to resolve. Pediatr Transplant. 2014;18:524-526.
- Maxfield L, Crane JS. Vitamin C Deficiency (Scurvy). Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK493187/. Updated November 19, 2019. Accessed May 19, 2020.
The World Health Organization defines malnutrition as deficiencies, excesses, or imbalances in an individual’s intake of energy and/or nutrients.1 This review will focus on undernutrition, which may result from macronutrient or micronutrient deficiencies. Undernutrition in the hospitalized patient is a common yet underrecognized phenomenon, with an estimated prevalence of 20% to 50% worldwide.2 Malnutrition is an independent risk factor for patient morbidity and mortality and has been associated with increased health care costs.3 Nutritional deficiencies may arise from inadequate nutrient intake, abnormal nutrient absorption, or improper nutrient utilization.4 Unfortunately, no standardized algorithm for screening and diagnosing patients with malnutrition exists, making early physical examination findings of utmost importance. Herein, we present a review of acquired nutritional deficiency dermatoses in the inpatient setting.
Protein-Energy Malnutrition
Protein-energy malnutrition (PEM) refers to a set of related disorders that include marasmus, kwashiorkor (KW), and marasmic KW. These conditions frequently are seen in developing countries but also have been reported in developed nations.5 Marasmus occurs from a chronic deficiency of protein and calories. Decreased insulin production and unopposed catabolism result in sarcopenia and loss of bone and subcutaneous fat.6 Affected patients include children who are less than 60% ideal body weight (IBW) without edema or hypoproteinemia.7 Kwashiorkor is the edematous form of PEM that develops from isolated protein deficiency, resulting in edema, diarrhea, and immunosuppression.6 Micronutrient deficiencies, oxidative stress, slow protein catabolism, and excess antidiuretic hormone have been proposed as potential drivers of KW.8 Kwashiorkor affects children between 60% and 80% IBW. Marasmic KW has features of both diseases, including children who are less than 60% IBW but with associated edema and/or hypoproteinemia.9
Although PEM is uncommon in adults, hospitalized patients carry many predisposing risk factors, including infections, malabsorptive conditions, psychiatric disease, and chronic illness (eTable). Patients with chronic infections present with findings consistent with marasmic KW due to lean body mass loss.
The cutaneous findings in PEM are related to dysmaturation of epidermal keratinocytes and resultant epidermal atrophy.10 Patients with marasmus exhibit dry, wrinkled, loose skin due to subcutaneous fat loss. Emaciated children often lose their buccal fat pads, and reduced perianal adipose may lead to rectal prolapse. Increased lanugo hair may be present on the face, and alopecia of the scalp may occur.6 In KW, cutaneous disease progresses from confluent hyperkeratosis to a dry atrophic epidermis that erodes easily, leaving underlying pale erythema. The resultant pattern is one of hyperpigmented plaques with slightly raised borders, and hypopigmented patches and erosions described as flaky paint dermatitis (Figure 1).5 Lesions appear first in areas of friction. The hair often is dry and brittle; curly hair may straighten and scale.11 Red-yellow to gray-white hypopigmentation may develop, denoting periods of inadequate nutrition. The flag sign describes alternating horizontal bands of hypopigmentation interspersed with bands of pigmented hair. The nails usually are thin and soft and may exhibit the nail flag sign, characterized by horizontal bands of white and red.12 Cheilitis, angular stomatitis, and vulvovaginitis may be present.6
In adults, weight loss and body mass index can be used to assess nutritional status, along with a focused history and physical examination. Complete blood cell count, electrolyte levels, and blood urea nitrogen should be assessed, as hypoglycemia and anemia often accompany PEM.13 In KW, hypoalbuminemia and hypoproteinemia are invariably present. Although prealbumin may be a valid prognostic indicator of disease outcomes and mortality in patients at risk for malnutrition, checking other serum biomarkers remains controversial.14 Focused testing may be warranted in patients with risk factors for chronic infectious processes, such as human immunodeficiency virus or tuberculosis.6 Skin biopsy may solidify the diagnosis of PEM. Hypertrophy of the stratum corneum, atrophy of the stratum spinosum and stratum granulosum, and increased basal layer melanin have been reported.15
Treatment involves initial fluid resuscitation and correction of electrolyte imbalances, followed by nutritional replacement.13 Oral or enteral tube feedings are preferred over total parenteral nutrition (TPN), as they enhance recovery of the gastrointestinal tract.16 Refeeding should occur in small amounts and frequent intervals.5 Skin-directed therapy is aimed at restoring epidermal function and hydration, with regular moisturization and application of barrier creams, such as zinc oxide ointment or petrolatum.10
Zinc Deficiency
Zinc is an essential trace element that provides regulatory, structural, and catalytic functions across multiple biochemical pathways6 and serves as an enzymatic cofactor and key component for numerous transcription factors.17 Zinc is derived from food sources, and its concentration correlates with protein content.18 Zinc is found in both animal and plant-based proteins, albeit with a lower oral bioavailability in the latter. Zinc deficiency may be inherited or acquired. Primary acrodermatitis enteropathica is an autosomal-recessive disorder of the solute carrier family 39 member 4 gene, SLC39A4 (encodes zinc transporter ZIP4 on enterocytes); the result is abnormal zinc absorption from the small intestine.18
Acquired zinc deficiency occurs from decreased dietary zinc intake, impaired intestinal zinc absorption, excessive zinc elimination, or systemic states of high catabolism or low albumin (eTable). Total parenteral nutrition–associated deficiency has arisen when nutritional formulations did not contain trace elements during national shortages or when prolonged TPN was not anticipated and trace elements were removed.19 Zinc levels may already be low in patients with chronic illness or inflammation, so even a short period on TPN can precipitate deficiency.18,19 Diets high in phytate may result in zinc deficiency, as phytate impairs intestinal zinc absorption.20 Approximately 15% of patients with inflammatory bowel disease experienced zinc deficiency worldwide.21 In Crohn disease, zinc deficiency has been associated with active intestinal inflammation, increased risk for hospitalization, surgeries, and disease-related complications.22,23
Medications such as antiepileptics, antimetabolites, or penicillamine may induce zinc deficiency, highlighting the importance of medication review for hospitalized patients (eTable). Catabolic states, frequently encountered in hospitalized patients, increase the risk for zinc deficiency.24 Patients with necrolytic migratory erythema (associated with pancreatic glucagonomas) often experience low serum zinc levels.25
The skin is the third most zinc-abundant tissue in the human body. Within keratinocytes, zinc is critical to normal proliferation and suppression of inflammation.17 Zinc also plays an important role in cutaneous immune function.26 Zinc deficiency presents with sharply demarcated, flaccid pustules and bullae that erode into scaly, pink, eczematous or psoriasiform plaques. Lesions are found preferentially in acral and periorificial sites, often with crusting and exudate. The groin and flexural surfaces may be affected. Erosions often become secondarily impetiginized. Other cutaneous findings include angular cheilitis, stomatitis, glossitis, paronychia, onychodystrophy, generalized alopecia, and delayed wound healing.26 Histopathology of skin lesions is characterized by granular layer loss, epidermal pallor, confluent parakeratosis, spongiosis, dyskeratosis, and psoriasiform hyperplasia.27 Acquired bullous acrodermatitis enteropathica has been reported as a histologic mimicker of pemphigus foliaceous in patients on TPN.28
Diagnosis of zinc deficiency is made by measuring plasma zinc levels. Fasting levels should be drawn in the morning, as they can fluctuate based on the time of day, stress levels, or inflammation.6 Sample hemolysis and anticoagulants high in zinc may falsely elevate plasma zinc. A normal zinc level is greater than 70 µg/dL; however, normal levels do not rule out deficiency.18 Measurement of zinc-dependent enzymes, such as alkaline phosphatase, can be a quick way to assess zinc status. Serum albumin also should be measured; because zinc is carried by albumin in the blood, hypoalbuminemia may result in secondary zinc deficiency.18
Zinc replacement therapy is largely through oral supplementation and should start at 0.5 to 2.0 mg/kg/d in adults with acquired disease.29,30 Zinc sulfate is the most affordable and is the supplement of choice, with 50 mg of elemental zinc per 220 mg of zinc sulfate (~23% elemental zinc).31 Alternative zinc salts, such as zinc gluconate (13% elemental zinc), may be used. Patients with malabsorptive disorders often require parenteral supplementation.32 Clinical symptoms often will resolve within 1 to 2 weeks of supplementation.29 In patients with primary acrodermatitis enteropathica, lifelong supplementation with 3 mg/kg/d elemental zinc should occur.6 Calcium and folate may reduce zinc absorption, while zinc supplementation can interfere with copper and iron absorption.33
Iron Deficiency
Iron is an essential component of the hemoglobin molecule. Iron homeostasis and metabolism are tightly regulated processes that drive erythropoiesis. Only 5% to 10% of dietary iron is absorbed through nutrition, while the remainder is recycled from red cell breakdown. Both normal iron levels and iron deficiency (ID) are defined by age and gender.34 Iron-deficiency anemia (IDA) is one of the most common cause-specific anemias worldwide.35
Fatigue is the most common and earliest symptom of ID. In a single study, pallor was predictive of anemia in hospitalized patients; however, absence of pallor did not rule out anemia.34 Dyspnea on exertion, tachycardia, dysphagia, and pica also may be reported. Cutaneous manifestations include koilonychia (Figure 2), glossitis, pruritus, angular cheilitis, and telogen effluvium. Plummer-Vinson syndrome is characterized by microcytic anemia, glossitis, and dysphagia.
Risk factors for ID include insufficient dietary consumption,36 blood loss, malabsorptive states,37,38 and increased iron requirements (eTable). Patient fragility (eg, elderly, chronic disease) is a newly described risk factor where correction of ID may impact morbidity, mortality, and quality of life.35
Iron deficiency can be present despite a normal hemoglobin level. Serum ferritin and percentage transferrin saturation are key to early identification of IDA.35 Ferritin levels lower than 30 µg/L confirm the diagnosis. Decreased transferrin saturation and increased total iron binding capacity aid in the diagnosis of IDA. Serum ferritin is an acute-phase reactant, and levels may be falsely elevated in the setting of inflammation or infection.
Treatment includes reversing the cause of deficiency and supplementing iron. Calculation of the total iron deficit can help inform iron supplementation. First-line therapy for IDA is oral ferrous sulfate 325 mg (65 mg elemental iron) 3 times daily. Newer studies suggest 40 to 80 mg oral iron should be taken every other day to increase absorption.39 Other iron salts, such as ferrous gluconate (325 mg is equivalent to 38 mg elemental iron), have been used. Iron absorption is enhanced by an acidic environment. Parenteral iron is utilized in patients with uncorrectable blood loss, malabsorption, renal failure, intolerance to oral iron, and nonadherence in those who are unable to receive transfusions. Iron infusions are favored in frail patients, such as the elderly and those with chronic kidney disease or heart failure.35 Multiple parenteral iron formulations exist, and their use should be driven by underlying patient comorbidities and potential risks. Packed red blood cell transfusions should be considered in acute blood loss, hypoxia, or cardiac insufficiency.
Essential Fatty Acid Deficiency
Essential fatty acids (EFAs) including linoleic and α-linolenic acid cannot be synthesized by the human body and must be obtained through diet (mostly plant oils). Essential fatty acids have various functions, including maintaining phospholipid membrane integrity, forming prostaglandins and leukotrienes, and storing energy.40 Essential fatty acids are important in the structure and function of the stratum corneum and are crucial in maintaining epidermal barrier function.41 Increased epidermal permeability and transepidermal water loss may be the first signs of EFA deficiency (EFAD).42
The cutaneous manifestations of EFAD include xerosis, weeping eczematous plaques, and erosions in intertriginous sites. The lesions may progress to widespread desquamation and erythema. With time, the skin can become thick and leathery. Alopecia may occur, and hair may depigment.7 Additional findings include poor wound healing and increased susceptibility to infections.43,44
Essential fatty acid deficiency may occur when dietary fat intake is severely restricted or in malabsorptive states.45,46 It develops in patients on prolonged TPN, typically when receiving fat-restricted nutrition,47,48 as occurs in hypertriglyceridemia.47 Essential fatty acid deficiency has developed in patients on TPN containing EFAs,47 as the introduction of novel intravenous lipid emulsions has resulted in varying proportions of EFA.40 Premature neonates are particularly at risk for EFAD.49
The diagnosis of EFAD involves the measurement of the triene to tetraene ratio. A ratio of more than 0.2 suggests EFAD, but the clinical signs are not seen until the ratio is over 0.4.40 Low plasma levels of linoleic, linolenic, and arachidonic acids also are seen. Elevated liver function tests are supportive of the diagnosis. Biochemical findings typically are seen before cutaneous manifestations.40
Treatment of EFAD includes topical, oral, or intravenous replacement of EFAs. Improvement of EFAD with the application of topical linoleic acid to the skin has been reported.50 Patients receiving TPN should undergo assessment of parenteral lipid emulsion to ensure adequate fatty acid composition.
Vitamin A Deficiency
Vitamin A (retinol) is a fat-soluble vitamin that plays a critical role in keratinization, epithelial proliferation, and cellular differentiation.6 Vitamin A is found in animal products as retinyl esters and in plants as beta-carotene. Vitamin A has 2 clinically important forms: all-trans retinoic acid and 11-cis-retinal. All-trans retinoic acid is involved in cellular differentiation and regulating gene transcription, while 11-cis-retinal is key to rhodopsin generation required for vision. Vitamin A deficiency presents with early ophthalmologic findings, specifically nyctalopia, or delayed adaptation to the dark.51 Xerophthalmia, abnormal conjunctival keratinization, and Bitot spots subsequently develop and may progress to corneal ulceration and blindness.6
Vitamin A deficiency manifests in the skin as follicular hyperkeratosis, or phrynoderma. Notably, numerous other micronutrient deficiencies may result in phrynoderma. Clinically, multiple pigmented keratotic papules of various sizes, many with a central keratinous plug, are distributed symmetrically on the extensor elbows, knees, shoulders, buttocks, and extremities. The skin surrounding these lesions may be scaly and hyperpigmented.52 Generalized xerosis without preceding nyctalopia has been reported.53 Accompanying pityriasis alba may develop.52 Lesions on the face may mimic acne, while lesions on the extremities may simulate a perforating disorder. Histopathology of phrynoderma reveals epidermal hyperkeratosis, follicular hyperkeratosis, and follicular plugging.52
Patients at risk for vitamin A deficiency include those with conditions that affect intestinal fat absorption, underlying psychiatric illness, or chronic disease (eTable). Chronic alcohol use predisposes patients to a multitude of micronutrient deficiencies, including vitamin A deficiency.54 In chronic alcohol use, even mild cutaneous changes may be the first clue to low serum retinol.55
Vitamin A deficiency can be diagnosed by measuring serum retinol levels, with levels lower than 20 µg/dL being diagnostic of deficiency.56 Decreased serum retinol in patients hospitalized with flaring irritable bowel disorder has been repeatedly reported.57-59 Notably, serum retinol concentration does not decline until liver reserves of vitamin A are nearing exhaustion.33
The US Food and Drug Administration requires manufacturers to list retinol activity equivalents on labels. One international unit of retinol is equivalent to 0.3 µg of retinol activity equivalents.60 The treatment of vitamin A deficiency involves high-dose oral supplementation when possible.61 Although dependent on age, the treatment dose for most adults with vitamin A deficiency is 3000 µg (10,000 IU) once daily.
Phrynoderma has been specifically treated with salicylic acid ointment 3% and intramuscular vitamin A.62 Topical urea cream also may treat phrynoderma.63
Vitamin B2
Vitamin B2 (riboflavin) is absorbed in the small intestine and converted into 2 biologically active forms—flavin adenine dinucleotide and flavin mononucleotide—which serve as cofactors in metabolic and oxidation-reduction reactions. Malabsorptive disorders and bowel resection can lead to riboflavin deficiency.64 Other at-risk populations include those with restrictive diets,65 psychiatric illness, or systemic illness (eTable). Riboflavin can be degraded by light (deficiency has been reported after phototherapy for neonatal jaundice66) and following boric acid ingestion.67 Medications, including long-term treatment with antiepileptics, may lead to riboflavin deficiency.68
Riboflavin is critical to maintaining collagen production. Riboflavin deficiency may manifest clinically with extensive seborrheiclike dermatitis,44 intertrigolike dermatitis,69 or oral-ocular-genital syndrome.70 Angular cheilitis may accompany an atrophic tongue that is deep red in color. The scrotum is characteristically involved in men, with confluent dermatitis extending onto the thighs and sparing the midline. Red papules and painful fissures may develop. Balanitis and phimosis have been reported. Testing for riboflavin deficiency should be considered in patients with refractory seborrheic dermatitis.
Riboflavin stores are assessed by the erythrocyte glutathione reductase activity coefficient.44 A level of 1.4 or higher is consistent with deficiency. Serum riboflavin levels, performed after a 12-hour fast, may support the diagnosis but are less sensitive. Patients with glucose-6-phosphate deficiency cannot be assessed via the erythrocyte glutathione reductase activity coefficient and may instead require evaluation of 24-hour urine riboflavin level.44
Vitamin B3
Vitamin B3 (niacin, nicotinamide, nicotinic acid) is found in plant and animal products or can be derived from its amino acid precursor tryptophan. Niacin deficiency results in pellagra, characterized by dermatitis, dementia, and diarrhea.71 The most prominent feature is a symmetrically distributed photosensitive dermatitis of the face, neck (called Casal necklace)(Figure 3), chest, dorsal hands, and extensor arms. The eruption may begin with erythema, vesicles, or bullae (wet pellagra) and evolve into thick, hyperpigmented, scaling plaques.71 The skin may take on a copper tone and become atrophic.72 Dull erythema with overlying yellow powdery scale (called sulfur flakes) at follicular orifices has been described on the nasal bridge.73
Causes of niacin deficiency include malabsorptive conditions, malignancy (including carcinoid tumors), parenteral nutrition, psychiatric disease,74,75 and restrictive diets (eTable).76 Carcinoid tumors divert tryptophan to serotonin resulting in niacin deficiency.77
The diagnosis of niacin deficiency is based on clinical findings and response to supplementation.75 Low niacin urinary metabolites (N-methylnicotinamide and 2-pyridone) may aid in diagnosis.6 Treatment generally includes oral nicotinamide 100 mg every 6 hours; the dose can then be tapered to 50 mg every 8 to 12 hours until symptoms resolve. Severe deficiency may require parenteral nicotinamide 1 g 3 to 4 times daily.75
Vitamin B6
Vitamin B6 (pyridoxine, pyridoxamine, pyridoxal) is found in whole grains and plant and animal products. Vitamin B6 functions as a coenzyme in many metabolic pathways and is involved in the conversion of tryptophan to niacin.44 Absorption requires hydrolysis by intestinal phosphates and transport to the liver for rephosphorylation prior to release in active form.6
Cutaneous findings associated with vitamin B6 deficiency include periorificial and perineal seborrheic dermatitis,78 angular stomatitis, and cheilitis, with associated burning, redness, and tongue edema.6 Vitamin B6 deficiency is a rarely reported cause of burning mouth syndrome.79 Because vitamin B6 is involved in the conversion of tryptophan to niacin, deficiency also may present with pellagralike findings.70 Other clinical symptoms are outlined in the eTable.80,81
Conditions that increase risk for vitamin B6 deficiency are highlighted in the eTable and include malabsorptive disorders; psychiatric illness82; and chronic disease, especially end-stage renal disease.83 Vitamin B6 deficiency associated with chronic alcohol use is due to both inadequate vitamin B6 intake as well as reduced hepatic storage.78 Medications such as isoniazid, hydralazine, and oral contraceptives may decrease vitamin B6 levels (eTable).82
Vitamin B6 can be measured in the plasma as pyridoxal 5′-phosphate. Plasma concentrations of less than 20 nmol/L are suggestive of deficiency.82 Indirect tests include tryptophan and methionine loading.6 The treatment of vitamin B6 deficiency is determined by symptom severity. Recommendations for oral supplementation range from 25 to 600 mg daily.82 Symptoms typically improve on 100 mg daily.6
Vitamins B9 and B12
Deficiencies of vitamins B9 (folic acid, folate) and B12 (cobalamin) have similar clinical presentations. Folate is essential in the metabolism of amino acids, purines, and pyrimidines.6 Cobalamin, found in animal products, is a cofactor for methionine synthase and methylmalonyl-CoA mutase.84 Megaloblastic anemia is the main finding in folate or cobalamin deficiency. Neurologic findings only accompany cobalamin deficiency. Risk factors for folate deficiency include malabsorptive conditions,6 chronic alcohol use,85 and antifolate medication use (eTable).6
Cobalamin absorption requires gastric acid and intrinsic factor binding in the duodenum. Deficiency may occur from strict diets, psychiatric illness, old age,86 decreased gastric acid secretion,87 abnormal intrinsic factor function, or intestinal infections.6
Generalized cutaneous hyperpigmentation may be the first manifestation of vitamins B9 and B12 deficiency.88 Typically accentuated in acral creases and the oral cavity, pigmentation may mimic Addison disease. Hair depigmentation and linear streaking of the nails are reported.84 The tongue becomes painful and red with atrophy of the filiform papillae (Hunter glossitis).78 Linear lesions on the tongue and hard palate may serve as an early sign of cobalamin deficiency.89
Folate deficiency is diagnosed by measuring the plasma folate level; coincidental cobalamin deficiency should be excluded. Deficiency is managed with oral supplementation (when possible) with 1 to 5 mg of folate daily.6 Cobalamin deficiency is based on low serum levels (<150 pg/mL is diagnostic).86 Cobalamin deficiency may take years to develop, as vitamin B12 exists in large body stores.6 Serum methylmalonic acid may be elevated in patients with clinical features but normal-low serum vitamin B12 level.86 Treatment of vitamin B12 deficiency is with oral (2 mg once daily) or parenteral (1 mg every 4 weeks then maintained at once monthly) cyanocobalamin. For patients with neurologic symptoms, intramuscular injection should be given.86 The underlying cause of deficiency must be elucidated and treated.
Vitamin C Deficiency
Vitamin C (ascorbic acid) is an essential cofactor for the hydroxylation of proline and lysine residues in collagen synthesis. Plant-based foods are the main dietary source of vitamin C, and deficiency presents clinically as scurvy. Cutaneous findings include follicular hyperkeratosis, perifollicular petechiae, and curled hair shafts (corkscrew hairs)(Figure 4). Ecchymoses of the lower extremities, forearms, and abdomen may be seen. Nodules representing intramuscular and subcutaneous hemorrhage can be present.90 Woody edema may mimic cellulitis, while lower extremity hemorrhage may mimic vasculitis. Gingival hyperplasia, hemorrhage, and edema may occur,90 along with linear splinter hemorrhages.91
Hypovitaminosis C has been routinely demonstrated in hospitalized patients.92 Scurvy may occur in patients on strict diets,93 chronic alcohol use,94 psychiatric illness,95 or gastrointestinal tract disease (eTable).96-99 Those with low socioeconomic status70 or dementia100 as well as the elderly also are at risk.101 Scurvy has developed in patients with iron overload and those who are on hemodialysis44 as well as in association with nilotinib use.102 Patients with chronic mucous membrane graft-vs-host disease may exhibit vitamin C deficiency.103
Scurvy is a clinical diagnosis. Vitamin C levels normalize quickly with supplementation. Cutaneous biopsy will exhibit follicular hyperkeratosis, perifollicular hemorrhage, and fibrosis.91
Oral ascorbic acid supplementation should be initiated at 500 to 1000 mg daily in adults.104 The cause of deficiency should be identified, and further supplementation should be decided based on patient risk factors. Lifestyle modifications, such as cessation of smoking and chronic alcohol use, is recommended. The diagnosis of scurvy should prompt workup for additional nutrient deficiencies.
Final Thoughts
Dermatologists play an important role in the early recognition of nutritional deficiencies, as cutaneous manifestations often are the first clue to diagnosis. Nutritional deficiencies are common yet underrecognized in the hospitalized patient and serve as an independent risk factor for patient morbidity and mortality.3 Awareness of the cutaneous manifestations of undernutrition as well as the risk factors for nutritional deficiency may expedite diagnosis and supplementation, thereby improving outcomes for hospitalized patients.
The World Health Organization defines malnutrition as deficiencies, excesses, or imbalances in an individual’s intake of energy and/or nutrients.1 This review will focus on undernutrition, which may result from macronutrient or micronutrient deficiencies. Undernutrition in the hospitalized patient is a common yet underrecognized phenomenon, with an estimated prevalence of 20% to 50% worldwide.2 Malnutrition is an independent risk factor for patient morbidity and mortality and has been associated with increased health care costs.3 Nutritional deficiencies may arise from inadequate nutrient intake, abnormal nutrient absorption, or improper nutrient utilization.4 Unfortunately, no standardized algorithm for screening and diagnosing patients with malnutrition exists, making early physical examination findings of utmost importance. Herein, we present a review of acquired nutritional deficiency dermatoses in the inpatient setting.
Protein-Energy Malnutrition
Protein-energy malnutrition (PEM) refers to a set of related disorders that include marasmus, kwashiorkor (KW), and marasmic KW. These conditions frequently are seen in developing countries but also have been reported in developed nations.5 Marasmus occurs from a chronic deficiency of protein and calories. Decreased insulin production and unopposed catabolism result in sarcopenia and loss of bone and subcutaneous fat.6 Affected patients include children who are less than 60% ideal body weight (IBW) without edema or hypoproteinemia.7 Kwashiorkor is the edematous form of PEM that develops from isolated protein deficiency, resulting in edema, diarrhea, and immunosuppression.6 Micronutrient deficiencies, oxidative stress, slow protein catabolism, and excess antidiuretic hormone have been proposed as potential drivers of KW.8 Kwashiorkor affects children between 60% and 80% IBW. Marasmic KW has features of both diseases, including children who are less than 60% IBW but with associated edema and/or hypoproteinemia.9
Although PEM is uncommon in adults, hospitalized patients carry many predisposing risk factors, including infections, malabsorptive conditions, psychiatric disease, and chronic illness (eTable). Patients with chronic infections present with findings consistent with marasmic KW due to lean body mass loss.
The cutaneous findings in PEM are related to dysmaturation of epidermal keratinocytes and resultant epidermal atrophy.10 Patients with marasmus exhibit dry, wrinkled, loose skin due to subcutaneous fat loss. Emaciated children often lose their buccal fat pads, and reduced perianal adipose may lead to rectal prolapse. Increased lanugo hair may be present on the face, and alopecia of the scalp may occur.6 In KW, cutaneous disease progresses from confluent hyperkeratosis to a dry atrophic epidermis that erodes easily, leaving underlying pale erythema. The resultant pattern is one of hyperpigmented plaques with slightly raised borders, and hypopigmented patches and erosions described as flaky paint dermatitis (Figure 1).5 Lesions appear first in areas of friction. The hair often is dry and brittle; curly hair may straighten and scale.11 Red-yellow to gray-white hypopigmentation may develop, denoting periods of inadequate nutrition. The flag sign describes alternating horizontal bands of hypopigmentation interspersed with bands of pigmented hair. The nails usually are thin and soft and may exhibit the nail flag sign, characterized by horizontal bands of white and red.12 Cheilitis, angular stomatitis, and vulvovaginitis may be present.6
In adults, weight loss and body mass index can be used to assess nutritional status, along with a focused history and physical examination. Complete blood cell count, electrolyte levels, and blood urea nitrogen should be assessed, as hypoglycemia and anemia often accompany PEM.13 In KW, hypoalbuminemia and hypoproteinemia are invariably present. Although prealbumin may be a valid prognostic indicator of disease outcomes and mortality in patients at risk for malnutrition, checking other serum biomarkers remains controversial.14 Focused testing may be warranted in patients with risk factors for chronic infectious processes, such as human immunodeficiency virus or tuberculosis.6 Skin biopsy may solidify the diagnosis of PEM. Hypertrophy of the stratum corneum, atrophy of the stratum spinosum and stratum granulosum, and increased basal layer melanin have been reported.15
Treatment involves initial fluid resuscitation and correction of electrolyte imbalances, followed by nutritional replacement.13 Oral or enteral tube feedings are preferred over total parenteral nutrition (TPN), as they enhance recovery of the gastrointestinal tract.16 Refeeding should occur in small amounts and frequent intervals.5 Skin-directed therapy is aimed at restoring epidermal function and hydration, with regular moisturization and application of barrier creams, such as zinc oxide ointment or petrolatum.10
Zinc Deficiency
Zinc is an essential trace element that provides regulatory, structural, and catalytic functions across multiple biochemical pathways6 and serves as an enzymatic cofactor and key component for numerous transcription factors.17 Zinc is derived from food sources, and its concentration correlates with protein content.18 Zinc is found in both animal and plant-based proteins, albeit with a lower oral bioavailability in the latter. Zinc deficiency may be inherited or acquired. Primary acrodermatitis enteropathica is an autosomal-recessive disorder of the solute carrier family 39 member 4 gene, SLC39A4 (encodes zinc transporter ZIP4 on enterocytes); the result is abnormal zinc absorption from the small intestine.18
Acquired zinc deficiency occurs from decreased dietary zinc intake, impaired intestinal zinc absorption, excessive zinc elimination, or systemic states of high catabolism or low albumin (eTable). Total parenteral nutrition–associated deficiency has arisen when nutritional formulations did not contain trace elements during national shortages or when prolonged TPN was not anticipated and trace elements were removed.19 Zinc levels may already be low in patients with chronic illness or inflammation, so even a short period on TPN can precipitate deficiency.18,19 Diets high in phytate may result in zinc deficiency, as phytate impairs intestinal zinc absorption.20 Approximately 15% of patients with inflammatory bowel disease experienced zinc deficiency worldwide.21 In Crohn disease, zinc deficiency has been associated with active intestinal inflammation, increased risk for hospitalization, surgeries, and disease-related complications.22,23
Medications such as antiepileptics, antimetabolites, or penicillamine may induce zinc deficiency, highlighting the importance of medication review for hospitalized patients (eTable). Catabolic states, frequently encountered in hospitalized patients, increase the risk for zinc deficiency.24 Patients with necrolytic migratory erythema (associated with pancreatic glucagonomas) often experience low serum zinc levels.25
The skin is the third most zinc-abundant tissue in the human body. Within keratinocytes, zinc is critical to normal proliferation and suppression of inflammation.17 Zinc also plays an important role in cutaneous immune function.26 Zinc deficiency presents with sharply demarcated, flaccid pustules and bullae that erode into scaly, pink, eczematous or psoriasiform plaques. Lesions are found preferentially in acral and periorificial sites, often with crusting and exudate. The groin and flexural surfaces may be affected. Erosions often become secondarily impetiginized. Other cutaneous findings include angular cheilitis, stomatitis, glossitis, paronychia, onychodystrophy, generalized alopecia, and delayed wound healing.26 Histopathology of skin lesions is characterized by granular layer loss, epidermal pallor, confluent parakeratosis, spongiosis, dyskeratosis, and psoriasiform hyperplasia.27 Acquired bullous acrodermatitis enteropathica has been reported as a histologic mimicker of pemphigus foliaceous in patients on TPN.28
Diagnosis of zinc deficiency is made by measuring plasma zinc levels. Fasting levels should be drawn in the morning, as they can fluctuate based on the time of day, stress levels, or inflammation.6 Sample hemolysis and anticoagulants high in zinc may falsely elevate plasma zinc. A normal zinc level is greater than 70 µg/dL; however, normal levels do not rule out deficiency.18 Measurement of zinc-dependent enzymes, such as alkaline phosphatase, can be a quick way to assess zinc status. Serum albumin also should be measured; because zinc is carried by albumin in the blood, hypoalbuminemia may result in secondary zinc deficiency.18
Zinc replacement therapy is largely through oral supplementation and should start at 0.5 to 2.0 mg/kg/d in adults with acquired disease.29,30 Zinc sulfate is the most affordable and is the supplement of choice, with 50 mg of elemental zinc per 220 mg of zinc sulfate (~23% elemental zinc).31 Alternative zinc salts, such as zinc gluconate (13% elemental zinc), may be used. Patients with malabsorptive disorders often require parenteral supplementation.32 Clinical symptoms often will resolve within 1 to 2 weeks of supplementation.29 In patients with primary acrodermatitis enteropathica, lifelong supplementation with 3 mg/kg/d elemental zinc should occur.6 Calcium and folate may reduce zinc absorption, while zinc supplementation can interfere with copper and iron absorption.33
Iron Deficiency
Iron is an essential component of the hemoglobin molecule. Iron homeostasis and metabolism are tightly regulated processes that drive erythropoiesis. Only 5% to 10% of dietary iron is absorbed through nutrition, while the remainder is recycled from red cell breakdown. Both normal iron levels and iron deficiency (ID) are defined by age and gender.34 Iron-deficiency anemia (IDA) is one of the most common cause-specific anemias worldwide.35
Fatigue is the most common and earliest symptom of ID. In a single study, pallor was predictive of anemia in hospitalized patients; however, absence of pallor did not rule out anemia.34 Dyspnea on exertion, tachycardia, dysphagia, and pica also may be reported. Cutaneous manifestations include koilonychia (Figure 2), glossitis, pruritus, angular cheilitis, and telogen effluvium. Plummer-Vinson syndrome is characterized by microcytic anemia, glossitis, and dysphagia.
Risk factors for ID include insufficient dietary consumption,36 blood loss, malabsorptive states,37,38 and increased iron requirements (eTable). Patient fragility (eg, elderly, chronic disease) is a newly described risk factor where correction of ID may impact morbidity, mortality, and quality of life.35
Iron deficiency can be present despite a normal hemoglobin level. Serum ferritin and percentage transferrin saturation are key to early identification of IDA.35 Ferritin levels lower than 30 µg/L confirm the diagnosis. Decreased transferrin saturation and increased total iron binding capacity aid in the diagnosis of IDA. Serum ferritin is an acute-phase reactant, and levels may be falsely elevated in the setting of inflammation or infection.
Treatment includes reversing the cause of deficiency and supplementing iron. Calculation of the total iron deficit can help inform iron supplementation. First-line therapy for IDA is oral ferrous sulfate 325 mg (65 mg elemental iron) 3 times daily. Newer studies suggest 40 to 80 mg oral iron should be taken every other day to increase absorption.39 Other iron salts, such as ferrous gluconate (325 mg is equivalent to 38 mg elemental iron), have been used. Iron absorption is enhanced by an acidic environment. Parenteral iron is utilized in patients with uncorrectable blood loss, malabsorption, renal failure, intolerance to oral iron, and nonadherence in those who are unable to receive transfusions. Iron infusions are favored in frail patients, such as the elderly and those with chronic kidney disease or heart failure.35 Multiple parenteral iron formulations exist, and their use should be driven by underlying patient comorbidities and potential risks. Packed red blood cell transfusions should be considered in acute blood loss, hypoxia, or cardiac insufficiency.
Essential Fatty Acid Deficiency
Essential fatty acids (EFAs) including linoleic and α-linolenic acid cannot be synthesized by the human body and must be obtained through diet (mostly plant oils). Essential fatty acids have various functions, including maintaining phospholipid membrane integrity, forming prostaglandins and leukotrienes, and storing energy.40 Essential fatty acids are important in the structure and function of the stratum corneum and are crucial in maintaining epidermal barrier function.41 Increased epidermal permeability and transepidermal water loss may be the first signs of EFA deficiency (EFAD).42
The cutaneous manifestations of EFAD include xerosis, weeping eczematous plaques, and erosions in intertriginous sites. The lesions may progress to widespread desquamation and erythema. With time, the skin can become thick and leathery. Alopecia may occur, and hair may depigment.7 Additional findings include poor wound healing and increased susceptibility to infections.43,44
Essential fatty acid deficiency may occur when dietary fat intake is severely restricted or in malabsorptive states.45,46 It develops in patients on prolonged TPN, typically when receiving fat-restricted nutrition,47,48 as occurs in hypertriglyceridemia.47 Essential fatty acid deficiency has developed in patients on TPN containing EFAs,47 as the introduction of novel intravenous lipid emulsions has resulted in varying proportions of EFA.40 Premature neonates are particularly at risk for EFAD.49
The diagnosis of EFAD involves the measurement of the triene to tetraene ratio. A ratio of more than 0.2 suggests EFAD, but the clinical signs are not seen until the ratio is over 0.4.40 Low plasma levels of linoleic, linolenic, and arachidonic acids also are seen. Elevated liver function tests are supportive of the diagnosis. Biochemical findings typically are seen before cutaneous manifestations.40
Treatment of EFAD includes topical, oral, or intravenous replacement of EFAs. Improvement of EFAD with the application of topical linoleic acid to the skin has been reported.50 Patients receiving TPN should undergo assessment of parenteral lipid emulsion to ensure adequate fatty acid composition.
Vitamin A Deficiency
Vitamin A (retinol) is a fat-soluble vitamin that plays a critical role in keratinization, epithelial proliferation, and cellular differentiation.6 Vitamin A is found in animal products as retinyl esters and in plants as beta-carotene. Vitamin A has 2 clinically important forms: all-trans retinoic acid and 11-cis-retinal. All-trans retinoic acid is involved in cellular differentiation and regulating gene transcription, while 11-cis-retinal is key to rhodopsin generation required for vision. Vitamin A deficiency presents with early ophthalmologic findings, specifically nyctalopia, or delayed adaptation to the dark.51 Xerophthalmia, abnormal conjunctival keratinization, and Bitot spots subsequently develop and may progress to corneal ulceration and blindness.6
Vitamin A deficiency manifests in the skin as follicular hyperkeratosis, or phrynoderma. Notably, numerous other micronutrient deficiencies may result in phrynoderma. Clinically, multiple pigmented keratotic papules of various sizes, many with a central keratinous plug, are distributed symmetrically on the extensor elbows, knees, shoulders, buttocks, and extremities. The skin surrounding these lesions may be scaly and hyperpigmented.52 Generalized xerosis without preceding nyctalopia has been reported.53 Accompanying pityriasis alba may develop.52 Lesions on the face may mimic acne, while lesions on the extremities may simulate a perforating disorder. Histopathology of phrynoderma reveals epidermal hyperkeratosis, follicular hyperkeratosis, and follicular plugging.52
Patients at risk for vitamin A deficiency include those with conditions that affect intestinal fat absorption, underlying psychiatric illness, or chronic disease (eTable). Chronic alcohol use predisposes patients to a multitude of micronutrient deficiencies, including vitamin A deficiency.54 In chronic alcohol use, even mild cutaneous changes may be the first clue to low serum retinol.55
Vitamin A deficiency can be diagnosed by measuring serum retinol levels, with levels lower than 20 µg/dL being diagnostic of deficiency.56 Decreased serum retinol in patients hospitalized with flaring irritable bowel disorder has been repeatedly reported.57-59 Notably, serum retinol concentration does not decline until liver reserves of vitamin A are nearing exhaustion.33
The US Food and Drug Administration requires manufacturers to list retinol activity equivalents on labels. One international unit of retinol is equivalent to 0.3 µg of retinol activity equivalents.60 The treatment of vitamin A deficiency involves high-dose oral supplementation when possible.61 Although dependent on age, the treatment dose for most adults with vitamin A deficiency is 3000 µg (10,000 IU) once daily.
Phrynoderma has been specifically treated with salicylic acid ointment 3% and intramuscular vitamin A.62 Topical urea cream also may treat phrynoderma.63
Vitamin B2
Vitamin B2 (riboflavin) is absorbed in the small intestine and converted into 2 biologically active forms—flavin adenine dinucleotide and flavin mononucleotide—which serve as cofactors in metabolic and oxidation-reduction reactions. Malabsorptive disorders and bowel resection can lead to riboflavin deficiency.64 Other at-risk populations include those with restrictive diets,65 psychiatric illness, or systemic illness (eTable). Riboflavin can be degraded by light (deficiency has been reported after phototherapy for neonatal jaundice66) and following boric acid ingestion.67 Medications, including long-term treatment with antiepileptics, may lead to riboflavin deficiency.68
Riboflavin is critical to maintaining collagen production. Riboflavin deficiency may manifest clinically with extensive seborrheiclike dermatitis,44 intertrigolike dermatitis,69 or oral-ocular-genital syndrome.70 Angular cheilitis may accompany an atrophic tongue that is deep red in color. The scrotum is characteristically involved in men, with confluent dermatitis extending onto the thighs and sparing the midline. Red papules and painful fissures may develop. Balanitis and phimosis have been reported. Testing for riboflavin deficiency should be considered in patients with refractory seborrheic dermatitis.
Riboflavin stores are assessed by the erythrocyte glutathione reductase activity coefficient.44 A level of 1.4 or higher is consistent with deficiency. Serum riboflavin levels, performed after a 12-hour fast, may support the diagnosis but are less sensitive. Patients with glucose-6-phosphate deficiency cannot be assessed via the erythrocyte glutathione reductase activity coefficient and may instead require evaluation of 24-hour urine riboflavin level.44
Vitamin B3
Vitamin B3 (niacin, nicotinamide, nicotinic acid) is found in plant and animal products or can be derived from its amino acid precursor tryptophan. Niacin deficiency results in pellagra, characterized by dermatitis, dementia, and diarrhea.71 The most prominent feature is a symmetrically distributed photosensitive dermatitis of the face, neck (called Casal necklace)(Figure 3), chest, dorsal hands, and extensor arms. The eruption may begin with erythema, vesicles, or bullae (wet pellagra) and evolve into thick, hyperpigmented, scaling plaques.71 The skin may take on a copper tone and become atrophic.72 Dull erythema with overlying yellow powdery scale (called sulfur flakes) at follicular orifices has been described on the nasal bridge.73
Causes of niacin deficiency include malabsorptive conditions, malignancy (including carcinoid tumors), parenteral nutrition, psychiatric disease,74,75 and restrictive diets (eTable).76 Carcinoid tumors divert tryptophan to serotonin resulting in niacin deficiency.77
The diagnosis of niacin deficiency is based on clinical findings and response to supplementation.75 Low niacin urinary metabolites (N-methylnicotinamide and 2-pyridone) may aid in diagnosis.6 Treatment generally includes oral nicotinamide 100 mg every 6 hours; the dose can then be tapered to 50 mg every 8 to 12 hours until symptoms resolve. Severe deficiency may require parenteral nicotinamide 1 g 3 to 4 times daily.75
Vitamin B6
Vitamin B6 (pyridoxine, pyridoxamine, pyridoxal) is found in whole grains and plant and animal products. Vitamin B6 functions as a coenzyme in many metabolic pathways and is involved in the conversion of tryptophan to niacin.44 Absorption requires hydrolysis by intestinal phosphates and transport to the liver for rephosphorylation prior to release in active form.6
Cutaneous findings associated with vitamin B6 deficiency include periorificial and perineal seborrheic dermatitis,78 angular stomatitis, and cheilitis, with associated burning, redness, and tongue edema.6 Vitamin B6 deficiency is a rarely reported cause of burning mouth syndrome.79 Because vitamin B6 is involved in the conversion of tryptophan to niacin, deficiency also may present with pellagralike findings.70 Other clinical symptoms are outlined in the eTable.80,81
Conditions that increase risk for vitamin B6 deficiency are highlighted in the eTable and include malabsorptive disorders; psychiatric illness82; and chronic disease, especially end-stage renal disease.83 Vitamin B6 deficiency associated with chronic alcohol use is due to both inadequate vitamin B6 intake as well as reduced hepatic storage.78 Medications such as isoniazid, hydralazine, and oral contraceptives may decrease vitamin B6 levels (eTable).82
Vitamin B6 can be measured in the plasma as pyridoxal 5′-phosphate. Plasma concentrations of less than 20 nmol/L are suggestive of deficiency.82 Indirect tests include tryptophan and methionine loading.6 The treatment of vitamin B6 deficiency is determined by symptom severity. Recommendations for oral supplementation range from 25 to 600 mg daily.82 Symptoms typically improve on 100 mg daily.6
Vitamins B9 and B12
Deficiencies of vitamins B9 (folic acid, folate) and B12 (cobalamin) have similar clinical presentations. Folate is essential in the metabolism of amino acids, purines, and pyrimidines.6 Cobalamin, found in animal products, is a cofactor for methionine synthase and methylmalonyl-CoA mutase.84 Megaloblastic anemia is the main finding in folate or cobalamin deficiency. Neurologic findings only accompany cobalamin deficiency. Risk factors for folate deficiency include malabsorptive conditions,6 chronic alcohol use,85 and antifolate medication use (eTable).6
Cobalamin absorption requires gastric acid and intrinsic factor binding in the duodenum. Deficiency may occur from strict diets, psychiatric illness, old age,86 decreased gastric acid secretion,87 abnormal intrinsic factor function, or intestinal infections.6
Generalized cutaneous hyperpigmentation may be the first manifestation of vitamins B9 and B12 deficiency.88 Typically accentuated in acral creases and the oral cavity, pigmentation may mimic Addison disease. Hair depigmentation and linear streaking of the nails are reported.84 The tongue becomes painful and red with atrophy of the filiform papillae (Hunter glossitis).78 Linear lesions on the tongue and hard palate may serve as an early sign of cobalamin deficiency.89
Folate deficiency is diagnosed by measuring the plasma folate level; coincidental cobalamin deficiency should be excluded. Deficiency is managed with oral supplementation (when possible) with 1 to 5 mg of folate daily.6 Cobalamin deficiency is based on low serum levels (<150 pg/mL is diagnostic).86 Cobalamin deficiency may take years to develop, as vitamin B12 exists in large body stores.6 Serum methylmalonic acid may be elevated in patients with clinical features but normal-low serum vitamin B12 level.86 Treatment of vitamin B12 deficiency is with oral (2 mg once daily) or parenteral (1 mg every 4 weeks then maintained at once monthly) cyanocobalamin. For patients with neurologic symptoms, intramuscular injection should be given.86 The underlying cause of deficiency must be elucidated and treated.
Vitamin C Deficiency
Vitamin C (ascorbic acid) is an essential cofactor for the hydroxylation of proline and lysine residues in collagen synthesis. Plant-based foods are the main dietary source of vitamin C, and deficiency presents clinically as scurvy. Cutaneous findings include follicular hyperkeratosis, perifollicular petechiae, and curled hair shafts (corkscrew hairs)(Figure 4). Ecchymoses of the lower extremities, forearms, and abdomen may be seen. Nodules representing intramuscular and subcutaneous hemorrhage can be present.90 Woody edema may mimic cellulitis, while lower extremity hemorrhage may mimic vasculitis. Gingival hyperplasia, hemorrhage, and edema may occur,90 along with linear splinter hemorrhages.91
Hypovitaminosis C has been routinely demonstrated in hospitalized patients.92 Scurvy may occur in patients on strict diets,93 chronic alcohol use,94 psychiatric illness,95 or gastrointestinal tract disease (eTable).96-99 Those with low socioeconomic status70 or dementia100 as well as the elderly also are at risk.101 Scurvy has developed in patients with iron overload and those who are on hemodialysis44 as well as in association with nilotinib use.102 Patients with chronic mucous membrane graft-vs-host disease may exhibit vitamin C deficiency.103
Scurvy is a clinical diagnosis. Vitamin C levels normalize quickly with supplementation. Cutaneous biopsy will exhibit follicular hyperkeratosis, perifollicular hemorrhage, and fibrosis.91
Oral ascorbic acid supplementation should be initiated at 500 to 1000 mg daily in adults.104 The cause of deficiency should be identified, and further supplementation should be decided based on patient risk factors. Lifestyle modifications, such as cessation of smoking and chronic alcohol use, is recommended. The diagnosis of scurvy should prompt workup for additional nutrient deficiencies.
Final Thoughts
Dermatologists play an important role in the early recognition of nutritional deficiencies, as cutaneous manifestations often are the first clue to diagnosis. Nutritional deficiencies are common yet underrecognized in the hospitalized patient and serve as an independent risk factor for patient morbidity and mortality.3 Awareness of the cutaneous manifestations of undernutrition as well as the risk factors for nutritional deficiency may expedite diagnosis and supplementation, thereby improving outcomes for hospitalized patients.
- Mehta NM, Corkins MR, Lyman B, et al. Defining pediatric malnutrition: a paradigm shift toward etiology-related definitions. JPEN J Parenter Enteral Nutr. 2013;37:460-481.
- Barker LA, Gout BS, Crowe TC. Hospital malnutrition: prevalence, identification and impact on patients and the healthcare system. Int J Environ Res Public Health. 2011;8:514-527.
- Bharadwaj S, Ginoya S, Tandon P, et al. Malnutrition: laboratory markers vs nutritional assessment. Gastroenterol Rep (Oxf). 2016;4:272-280.
- Basavaraj KH, Seemanthini C, Rashmi R. Diet in dermatology: present perspectives. Indian J Dermatol. 2010;55:205-210.
- Grover Z, Ee LC. Protein energy malnutrition. Pediatr Clin North Am. 2009;56:1055-1068.
- Jen M, Yan AC. Syndromes associated with nutritional deficiency and excess. Clin Dermatol. 2010;28:669-685.
- Lekwuttikarn R, Teng JMC. Cutaneous manifestations of nutritional deficiency. Curr Opin Pediatr. 2018;30:505-513.
- Jaffe AT, Heymann WR. Kwashiorkor/zinc deficiency overlap following partial gastrectomy. Int J Dermatol. 1998;37:134-137.
- Listernick R, Christoffel K, Pace J, et al. Severe primary malnutrition in US children. Am J Dis Child. 1985;139:1157-1160.
- Heilskov S, Rytter MJ, Vestergaard C, et al. Dermatosis in children with oedematous malnutrition (Kwashiorkor): a review of the literature. J Eur Acad Dermatol Venereol. 2014;28:995-1001.
- Bradfield RB. Hair tissue as a medium for the differential diagnosis of protein-calorie malnutrition: a commentary. J Pediatr. 1974;84:294-296.
- Cohen PR. The nail flag sign: case report in a man with diverticulitis and review of dermatology flag sign of the hair, skin, and nails. Cureus. 2018;10:e2929.
- Management of Severe Malnutrition: A Manual for Physicians and Other Senior Health Workers. Geneva, Switzerland: World Health Organization; 1999. https://www.who.int/nutrition/publications/en/manage_severe_malnutrition_eng.pdf. Accessed May 19, 2020.
- Keller U. Nutritional laboratory markers in malnutrition. J Clin Med. 2019;8:775.
- Thavaraj V, Sesikeran B. Histopathological changes in skin of children with clinical protein energy malnutrition before and after recovery. J Trop Pediatr. 1989;35:105-108.
- McClave SA, Heyland DK. The physiologic response and associated clinical benefits from provision of early enteral nutrition. Nutr Clin Pract. 2009;24:305-315.
- Ogawa Y, Kinoshita M, Shimada S, et al. Zinc and skin disorders. Nutrients. 2018;10:199.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Wiznia LE, Bhansali S, Brinster N, et al. Acquired acrodermatitis enteropathica due to zinc-depleted parenteral nutrition. Pediatr Dermatol. 2019;36:520-523.
- Sandstead HH, Freeland-Graves JH. Dietary phytate, zinc and hidden zinc deficiency. J Trace Elem Med Biol. 2014;28:414-417.
- Vagianos K, Bector S, McConnell J, et al. Nutrition assessment of patients with inflammatory bowel disease. JPEN J Parenter Enteral Nutr. 2007;31:311-319.
- Schoelmerich J, Becher MS, Hoppe-Seyler P, et al. Zinc and vitamin A deficiency in patients with Crohn’s disease is correlated with activity but not with localization or extent of the disease. Hepatogastroenterology. 1985;32:34-38.
- Siva S, Rubin DT, Gulotta G, et al. Zinc deficiency is associated with poor clinical outcomes in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2017;23:152-157.
- Semrad CE. Zinc and intestinal function. Curr Gastroenterol Rep. 1999;1:398-403.
- Sinclair SA, Reynolds NJ. Necrolytic migratory erythema and zinc deficiency. Br J Dermatol. 1997;136:783-785.
- Gammoh NZ, Rink L. Zinc in infection and inflammation. Nutrients. 2017;9:624.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Wu D, Fung MA, Kiuru M, et al. Acquired bullous acrodermatitis enteropathica as a histologic mimic of pemphigus foliaceus in a patient on parenteral nutrition. Dermatol Online J. 2018;24:20.
- Maxfield L, Crane J. Zinc Deficiency. Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK493231/Updated November 14, 2019. Accessed May 19, 2020.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Wegmüller R, Tay F, Zeder C, et al. Zinc absorption by young adults from supplemental zinc citrate is comparable with that from zinc gluconate and higher than from zinc oxide. J Nutr. 2014;144:132-136.
- Vick G, Mahmoudizad R, Fiala K. Intravenous zinc therapy for acquired zinc deficiency secondary to gastric bypass surgery: a case report. Dermatol Ther. 2015;28:222-225.
- Ghishan FK, Kiela PR. Vitamins and minerals in inflammatory bowel disease. Gastroenterol Clin North Am. 2017;46:797-808.
- Killip S, Bennett JM, Chambers MD. Iron deficiency anemia. Am Fam Physician. 2007;75:671-678.
- De Franceschi L, Iolascon A, Taher A, et al. Clinical management of iron deficiency anemia in adults: systemic review on advances in diagnosis and treatment. Eur J Intern Med. 2017;42:16-23.
- Haider LM, Schwingshackl L, Hoffmann G, et al. The effect of vegetarian diets on iron status in adults: a systematic review and meta-analysis. Crit Rev Food Sci Nutr. 2018;58:1359-1374.
- Enani G, Bilgic E, Lebedeva E, et al. The incidence of iron deficiency anemia post-Roux-en-Y gastric bypass and sleeve gastrectomy: a systematic review [published online September 4, 2019]. Surg Endosc. doi:10.1007/s00464-019-07092-3.
- Kaitha S, Bashir M, Ali T. Iron deficiency anemia in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2015;6:62-72.
- Moretti D, Goede JS, Zeder C, et al. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood. 2015;126:1981-1989.
- Gramlich L, Meddings L, Alberda C, et al. Essential fatty acid deficiency in 2015: the impact of novel intravenous lipid emulsions. JPEN J Parenter Enteral Nutr. 2015;39(1 suppl):61S-66S.
- Khnykin D, Miner JH, Jahnsen F. Role of fatty acid transporters in epidermis: implications for health and disease. Dermatoendocrinol. 2011;3:53-61.
- Wright S. Essential fatty acids and the skin. Br J Dermatol. 1991;125:503-515.
- Lakdawala N, Grant-Kels JM. Acrodermatitis caused by nutritional deficiency and metabolic disorders. Clin Dermatol. 2017;35:64-67.
- DiBaise M, Tarleton SM. Hair, nails, and skin: differentiating cutaneous manifestations of micronutrient deficiency. Nutr Clin Pract. 2019;34:490-503.
- Aldámiz-Echevarría L, Bilbao A, Andrade F, et al. Fatty acid deficiency profile in children with food allergy managed with elimination diets. Acta Paediatr. 2008;97:1572-1576.
- Jeppesen PB, Christensen MS, Høy CE, et al. Essential fatty acid deficiency in patients with severe fat malabsorption. Am J Clin Nutr. 1997;65:837-843.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published online June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
- Fleming CR, Smith LM, Hodges RE. Essential fatty acid deficiency in adults receiving total parenteral nutrition. Am J Clin Nutr. 1976;29:976-983.
- Cooke RJ, Zee P, Yeh YY. Essential fatty acid status of the premature infant during short-term fat-free parenteral nutrition. J Pediatr Gastroenterol Nutr. 1984;3:446-449.
- Skolnik P, Eaglstein WH, Ziboh VA. Human essential fatty acid deficiency: treatment by topical application of linoleic acid. Arch Dermatol. 1977;113:939-941.
- Vahlquist A. Clinical use of vitamin A and its derivatives—physiological and pharmacological aspects. Clin Exp Dermatol. 1985;10:133-143.
- Ragunatha S, Kumar VJ, Murugesh SB. A clinical study of 125 patients with phrynoderma. Indian J Dermatol. 2011;56:389-392.
- Phanachet P, Shantavasinkul PC, Chantrathammachart P, et al. Unusual manifestation of vitamin A deficiency presenting with generalized xerosis without night blindness. Clin Case Rep. 2018;6:878-882.
- Fuchs J. Alcoholism, malnutrition, vitamin deficiencies, and the skin. Clin Dermatol. 1999;17:457-461.
- Uhoda E, Petit L, Piérard-Franchimont C, et al. Ultraviolet light-enhanced visualization of cutaneous signs of carotene and vitamin A dietary deficiency. Acta Clin Belg. 2004;59:97-101.
- de Pee S, Dary O. Biochemical indicators of vitamin A deficiency: serum retinol and serum retinol binding protein. J Nutr. 2002;132(9 suppl):2895S-2901S.
- Fernandez-Banares F, Abad-Lacruz A, Xiol X, et al. Vitamin status in patients with inflammatory bowel disease. Am J Gastroenterol. 1989;84:744-748.
- Main AN, Mills PR, Russell RI, et al. Vitamin A deficiency in Crohn’s disease. Gut. 1983;24:1169-1175.
- Cobos G, Cornejo C, McMahon P. A case of phrynoderma in a patient with Crohn’s disease. Pediatr Dermatol. 2015;32:234-236.
- Trumbo P, Yates AA, Schlicker S, et al. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. 2001;101:294-301.
- Ross DA. Recommendations for vitamin A supplementation. J Nutr. 2002;132(9 suppl):2902S-2906S.
- Ragunatha S, Jagannath Kumar V, Murugesh SB, et al. Therapeutic response of vitamin A, vitamin B complex, essential fatty acids (EFA) and vitamin E in the treatment of phrynoderma: a randomized controlled study. J Clin Diagn Res. 2014;8:116-118.
- Nakjang Y, Yuttanavivat T. Phrynoderma: a review of 105 cases. J Dermatol. 1988;15:531-534.
- Pinto JT, Zempleni J. Riboflavin. Adv Nutr. 2016;7:973-975.
- Larsson CL, Johansson GK. Dietary intake and nutritional status of young vegans and omnivores in Sweden. Am J Clin Nutr. 2002;76:100-106.
- Gromisch DS, Lopez R, Cole HS, et al. Light (phototherapy)—induced riboflavin deficiency in the neonate. J Pediatr. 1977;90:118-122.
- Pinto J, Huang YP, McConnell RJ, et al. Increased urinary riboflavin excretion resulting from boric acid ingestion. J Lab Clin Med. 1978;92:126-134.
- Soltani D, Ghaffar Pour M, et al. Nutritional aspects of treatment in epileptic patients. Iran J Child Neurol. 2016;10:1-12.
- Roe DA. Riboflavin deficiency: mucocutaneous signs of acute and chronic deficiency. Semin Dermatol. 1991;10:293-295.
- Galimberti F, Mesinkovska NA. Skin findings associated with nutritional deficiencies. Cleve Clin J Med. 2016;83:731-739.
- Karthikeyan K, Thappa DM. Pellagra and skin. Int J Dermatol. 2002;41:476-481.
- Nogueira A, Duarte AF, Magina S, et al. Pellagra associated with esophageal carcinoma and alcoholism. Dermatol Online J. 2009;15:8.
- Wan P, Moat S, Anstey A. Pellagra: a review with emphasis on photosensitivity. Br J Dermatol. 2011;164:1188-1200.
- Jagielska G, Tomaszewicz-Libudzic EC, Brzozowska A. Pellagra: a rare complication of anorexia nervosa. Eur Child Adolesc Psychiatry. 2007;16:417-420.
- Li R, Yu K, Wang Q, et al. Pellagra secondary to medication and alcoholism: a case report and review of the literature. Nutr Clin Pract. 2016;31:785-789.
- Ladoyanni E, Cheung ST, North J, et al. Pellagra occurring in a patient with atopic dermatitis and food allergy. J Eur Acad Dermatol Venereol. 2007;21:394-396.
- Bell HK, Poston GJ, Vora J, et al. Cutaneous manifestations of the malignant carcinoid syndrome. Br J Dermatol. 2005;152:71-75.
- Barthelemy H, Chouvet B, Cambazard F. Skin and mucosal manifestations in vitamin deficiency. J Am Acad Dermatol. 1986;15:1263-1274.
- Lamey PJ, Hammond A, Allam BF, et al. Vitamin status of patients with burning mouth syndrome and the response to replacement therapy. Br Dent J. 1986;160:81-84.
- Stover PJ, Field MS. Vitamin B-6. Adv Nutr. 2015;6:132-133.
- Gerlach AT, Thomas S, Stawicki SP, et al. Vitamin B6 deficiency: a potential cause of refractory seizures in adults. JPEN J Parenter Enteral Nutr. 2011;35:272-275.
- Spinneker A, Sola R, Lemmen V, et al. Vitamin B6 status, deficiency and its consequences—an overview. Nutr Hosp. 2007;22:7-24.
- Ross EA, Shah GM, Reynolds RD, et al. Vitamin B6 requirements of patients on chronic peritoneal dialysis. Kidney Int. 1989;36:702-706.
- Brescoll J, Daveluy S. A review of vitamin B12 in dermatology. Am J Clin Dermatol. 2015;16:27-33.
- Sanvisens A, Zuluaga P, Pineda M, et al. Folate deficiency in patients seeking treatment of alcohol use disorder. Drug Alcohol Depend. 2017;180:417-422.
- Langan RC, Goodbred AJ. Vitamin B12 deficiency: recognition and management. Am Fam Physician. 2017;96:384-389.
- Bradford GS, Taylor CT. Omeprazole and vitamin B12 deficiency. Ann Pharmacother. 1999;33:641-643.
- Srivastava N, Chand S, Bansal M, et al. Reversible hyperpigmentation as the first manifestation of dietary vitamin B12 deficiency. Indian J Dermatol Venereol Leprol. 2006;72:389-390.
- Graells J, Ojeda RM, Muniesa C, et al. Glossitis with linear lesions: an early sign of vitamin B12 deficiency. J Am Acad Dermatol. 2009;60:498-500.
- Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999;41:895-906; quiz 907-810.
- Shaath T, Fischer R, Goeser M, et al. Scurvy in the present times: vitamin C allergy leading to strict fast food diet. Dermatol Online J. 2016;22:13030/qt50b8w28b.
- Fain O, Pariés J, Jacquart B, et al. Hypovitaminosis C in hospitalized patients. Eur J Intern Med. 2003;14:419-425.
- Ahmad SA, Al Thobiti TA, El Toum M, et al. Florid scurvy in an autistic child on a ketogenic diet [published online November 19, 2018]. Pediatr Emerg Care. doi:10.1097/PEC.0000000000001695.
- Lux-Battistelli C, Battistelli D. Latent scurvy with tiredness and leg pain in alcoholics: an underestimated disease three case reports. Medicine (Baltimore). 2017;96:e8861.
- Christopher K, Tammaro D, Wing EJ. Early scurvy complicating anorexia nervosa. South Med J. 2002;95:1065-1066.
- Berger ML, Siegel DM, Lee EL. Scurvy as an initial manifestation of Whipple’s disease. Ann Intern Med. 1984;101:58-59.
- Imes S, Dinwoodie A, Walker K, et al. Vitamin C status in 137 outpatients with Crohn’s disease. effect of diet counseling. J Clin Gastroenterol. 1986;8:443-446.
- Echeverría Zudaire L, García Cuartero B, Campelo Moreno O, et al. Scurvy associated with celiac disease [in Spanish]. An Esp Pediatr. 2002;57:587.
- Hansen EP, Metzsche C, Henningsen E, et al. Severe scurvy after gastric bypass surgery and a poor postoperative diet. J Clin Med Res. 2012;4:135-137.
- Rivière S, Birlouez-Aragon I, Nourhashémi F, et al. Low plasma vitamin C in Alzheimer patients despite an adequate diet. Int J Geriatr Psychiatry. 1998;13:749-754.
- Bhattacharyya P, Giannoutsos J, Eslick GD, et al. Scurvy: an unrecognized and emerging public health issue in developed economies. Mayo Clin Proc. 2019;94:2594-2597.
- Oak AS, Jaleel T, Fening K, et al. A case of scurvy associated with nilotinib. J Cutan Pathol. 2016;43:725-726.
- Kletzel M, Powers K, Hayes M. Scurvy: a new problem for patients with chronic GVHD involving mucous membranes; an easy problem to resolve. Pediatr Transplant. 2014;18:524-526.
- Maxfield L, Crane JS. Vitamin C Deficiency (Scurvy). Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK493187/. Updated November 19, 2019. Accessed May 19, 2020.
- Mehta NM, Corkins MR, Lyman B, et al. Defining pediatric malnutrition: a paradigm shift toward etiology-related definitions. JPEN J Parenter Enteral Nutr. 2013;37:460-481.
- Barker LA, Gout BS, Crowe TC. Hospital malnutrition: prevalence, identification and impact on patients and the healthcare system. Int J Environ Res Public Health. 2011;8:514-527.
- Bharadwaj S, Ginoya S, Tandon P, et al. Malnutrition: laboratory markers vs nutritional assessment. Gastroenterol Rep (Oxf). 2016;4:272-280.
- Basavaraj KH, Seemanthini C, Rashmi R. Diet in dermatology: present perspectives. Indian J Dermatol. 2010;55:205-210.
- Grover Z, Ee LC. Protein energy malnutrition. Pediatr Clin North Am. 2009;56:1055-1068.
- Jen M, Yan AC. Syndromes associated with nutritional deficiency and excess. Clin Dermatol. 2010;28:669-685.
- Lekwuttikarn R, Teng JMC. Cutaneous manifestations of nutritional deficiency. Curr Opin Pediatr. 2018;30:505-513.
- Jaffe AT, Heymann WR. Kwashiorkor/zinc deficiency overlap following partial gastrectomy. Int J Dermatol. 1998;37:134-137.
- Listernick R, Christoffel K, Pace J, et al. Severe primary malnutrition in US children. Am J Dis Child. 1985;139:1157-1160.
- Heilskov S, Rytter MJ, Vestergaard C, et al. Dermatosis in children with oedematous malnutrition (Kwashiorkor): a review of the literature. J Eur Acad Dermatol Venereol. 2014;28:995-1001.
- Bradfield RB. Hair tissue as a medium for the differential diagnosis of protein-calorie malnutrition: a commentary. J Pediatr. 1974;84:294-296.
- Cohen PR. The nail flag sign: case report in a man with diverticulitis and review of dermatology flag sign of the hair, skin, and nails. Cureus. 2018;10:e2929.
- Management of Severe Malnutrition: A Manual for Physicians and Other Senior Health Workers. Geneva, Switzerland: World Health Organization; 1999. https://www.who.int/nutrition/publications/en/manage_severe_malnutrition_eng.pdf. Accessed May 19, 2020.
- Keller U. Nutritional laboratory markers in malnutrition. J Clin Med. 2019;8:775.
- Thavaraj V, Sesikeran B. Histopathological changes in skin of children with clinical protein energy malnutrition before and after recovery. J Trop Pediatr. 1989;35:105-108.
- McClave SA, Heyland DK. The physiologic response and associated clinical benefits from provision of early enteral nutrition. Nutr Clin Pract. 2009;24:305-315.
- Ogawa Y, Kinoshita M, Shimada S, et al. Zinc and skin disorders. Nutrients. 2018;10:199.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Wiznia LE, Bhansali S, Brinster N, et al. Acquired acrodermatitis enteropathica due to zinc-depleted parenteral nutrition. Pediatr Dermatol. 2019;36:520-523.
- Sandstead HH, Freeland-Graves JH. Dietary phytate, zinc and hidden zinc deficiency. J Trace Elem Med Biol. 2014;28:414-417.
- Vagianos K, Bector S, McConnell J, et al. Nutrition assessment of patients with inflammatory bowel disease. JPEN J Parenter Enteral Nutr. 2007;31:311-319.
- Schoelmerich J, Becher MS, Hoppe-Seyler P, et al. Zinc and vitamin A deficiency in patients with Crohn’s disease is correlated with activity but not with localization or extent of the disease. Hepatogastroenterology. 1985;32:34-38.
- Siva S, Rubin DT, Gulotta G, et al. Zinc deficiency is associated with poor clinical outcomes in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2017;23:152-157.
- Semrad CE. Zinc and intestinal function. Curr Gastroenterol Rep. 1999;1:398-403.
- Sinclair SA, Reynolds NJ. Necrolytic migratory erythema and zinc deficiency. Br J Dermatol. 1997;136:783-785.
- Gammoh NZ, Rink L. Zinc in infection and inflammation. Nutrients. 2017;9:624.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Wu D, Fung MA, Kiuru M, et al. Acquired bullous acrodermatitis enteropathica as a histologic mimic of pemphigus foliaceus in a patient on parenteral nutrition. Dermatol Online J. 2018;24:20.
- Maxfield L, Crane J. Zinc Deficiency. Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK493231/Updated November 14, 2019. Accessed May 19, 2020.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Wegmüller R, Tay F, Zeder C, et al. Zinc absorption by young adults from supplemental zinc citrate is comparable with that from zinc gluconate and higher than from zinc oxide. J Nutr. 2014;144:132-136.
- Vick G, Mahmoudizad R, Fiala K. Intravenous zinc therapy for acquired zinc deficiency secondary to gastric bypass surgery: a case report. Dermatol Ther. 2015;28:222-225.
- Ghishan FK, Kiela PR. Vitamins and minerals in inflammatory bowel disease. Gastroenterol Clin North Am. 2017;46:797-808.
- Killip S, Bennett JM, Chambers MD. Iron deficiency anemia. Am Fam Physician. 2007;75:671-678.
- De Franceschi L, Iolascon A, Taher A, et al. Clinical management of iron deficiency anemia in adults: systemic review on advances in diagnosis and treatment. Eur J Intern Med. 2017;42:16-23.
- Haider LM, Schwingshackl L, Hoffmann G, et al. The effect of vegetarian diets on iron status in adults: a systematic review and meta-analysis. Crit Rev Food Sci Nutr. 2018;58:1359-1374.
- Enani G, Bilgic E, Lebedeva E, et al. The incidence of iron deficiency anemia post-Roux-en-Y gastric bypass and sleeve gastrectomy: a systematic review [published online September 4, 2019]. Surg Endosc. doi:10.1007/s00464-019-07092-3.
- Kaitha S, Bashir M, Ali T. Iron deficiency anemia in inflammatory bowel disease. World J Gastrointest Pathophysiol. 2015;6:62-72.
- Moretti D, Goede JS, Zeder C, et al. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood. 2015;126:1981-1989.
- Gramlich L, Meddings L, Alberda C, et al. Essential fatty acid deficiency in 2015: the impact of novel intravenous lipid emulsions. JPEN J Parenter Enteral Nutr. 2015;39(1 suppl):61S-66S.
- Khnykin D, Miner JH, Jahnsen F. Role of fatty acid transporters in epidermis: implications for health and disease. Dermatoendocrinol. 2011;3:53-61.
- Wright S. Essential fatty acids and the skin. Br J Dermatol. 1991;125:503-515.
- Lakdawala N, Grant-Kels JM. Acrodermatitis caused by nutritional deficiency and metabolic disorders. Clin Dermatol. 2017;35:64-67.
- DiBaise M, Tarleton SM. Hair, nails, and skin: differentiating cutaneous manifestations of micronutrient deficiency. Nutr Clin Pract. 2019;34:490-503.
- Aldámiz-Echevarría L, Bilbao A, Andrade F, et al. Fatty acid deficiency profile in children with food allergy managed with elimination diets. Acta Paediatr. 2008;97:1572-1576.
- Jeppesen PB, Christensen MS, Høy CE, et al. Essential fatty acid deficiency in patients with severe fat malabsorption. Am J Clin Nutr. 1997;65:837-843.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published online June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
- Fleming CR, Smith LM, Hodges RE. Essential fatty acid deficiency in adults receiving total parenteral nutrition. Am J Clin Nutr. 1976;29:976-983.
- Cooke RJ, Zee P, Yeh YY. Essential fatty acid status of the premature infant during short-term fat-free parenteral nutrition. J Pediatr Gastroenterol Nutr. 1984;3:446-449.
- Skolnik P, Eaglstein WH, Ziboh VA. Human essential fatty acid deficiency: treatment by topical application of linoleic acid. Arch Dermatol. 1977;113:939-941.
- Vahlquist A. Clinical use of vitamin A and its derivatives—physiological and pharmacological aspects. Clin Exp Dermatol. 1985;10:133-143.
- Ragunatha S, Kumar VJ, Murugesh SB. A clinical study of 125 patients with phrynoderma. Indian J Dermatol. 2011;56:389-392.
- Phanachet P, Shantavasinkul PC, Chantrathammachart P, et al. Unusual manifestation of vitamin A deficiency presenting with generalized xerosis without night blindness. Clin Case Rep. 2018;6:878-882.
- Fuchs J. Alcoholism, malnutrition, vitamin deficiencies, and the skin. Clin Dermatol. 1999;17:457-461.
- Uhoda E, Petit L, Piérard-Franchimont C, et al. Ultraviolet light-enhanced visualization of cutaneous signs of carotene and vitamin A dietary deficiency. Acta Clin Belg. 2004;59:97-101.
- de Pee S, Dary O. Biochemical indicators of vitamin A deficiency: serum retinol and serum retinol binding protein. J Nutr. 2002;132(9 suppl):2895S-2901S.
- Fernandez-Banares F, Abad-Lacruz A, Xiol X, et al. Vitamin status in patients with inflammatory bowel disease. Am J Gastroenterol. 1989;84:744-748.
- Main AN, Mills PR, Russell RI, et al. Vitamin A deficiency in Crohn’s disease. Gut. 1983;24:1169-1175.
- Cobos G, Cornejo C, McMahon P. A case of phrynoderma in a patient with Crohn’s disease. Pediatr Dermatol. 2015;32:234-236.
- Trumbo P, Yates AA, Schlicker S, et al. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J Am Diet Assoc. 2001;101:294-301.
- Ross DA. Recommendations for vitamin A supplementation. J Nutr. 2002;132(9 suppl):2902S-2906S.
- Ragunatha S, Jagannath Kumar V, Murugesh SB, et al. Therapeutic response of vitamin A, vitamin B complex, essential fatty acids (EFA) and vitamin E in the treatment of phrynoderma: a randomized controlled study. J Clin Diagn Res. 2014;8:116-118.
- Nakjang Y, Yuttanavivat T. Phrynoderma: a review of 105 cases. J Dermatol. 1988;15:531-534.
- Pinto JT, Zempleni J. Riboflavin. Adv Nutr. 2016;7:973-975.
- Larsson CL, Johansson GK. Dietary intake and nutritional status of young vegans and omnivores in Sweden. Am J Clin Nutr. 2002;76:100-106.
- Gromisch DS, Lopez R, Cole HS, et al. Light (phototherapy)—induced riboflavin deficiency in the neonate. J Pediatr. 1977;90:118-122.
- Pinto J, Huang YP, McConnell RJ, et al. Increased urinary riboflavin excretion resulting from boric acid ingestion. J Lab Clin Med. 1978;92:126-134.
- Soltani D, Ghaffar Pour M, et al. Nutritional aspects of treatment in epileptic patients. Iran J Child Neurol. 2016;10:1-12.
- Roe DA. Riboflavin deficiency: mucocutaneous signs of acute and chronic deficiency. Semin Dermatol. 1991;10:293-295.
- Galimberti F, Mesinkovska NA. Skin findings associated with nutritional deficiencies. Cleve Clin J Med. 2016;83:731-739.
- Karthikeyan K, Thappa DM. Pellagra and skin. Int J Dermatol. 2002;41:476-481.
- Nogueira A, Duarte AF, Magina S, et al. Pellagra associated with esophageal carcinoma and alcoholism. Dermatol Online J. 2009;15:8.
- Wan P, Moat S, Anstey A. Pellagra: a review with emphasis on photosensitivity. Br J Dermatol. 2011;164:1188-1200.
- Jagielska G, Tomaszewicz-Libudzic EC, Brzozowska A. Pellagra: a rare complication of anorexia nervosa. Eur Child Adolesc Psychiatry. 2007;16:417-420.
- Li R, Yu K, Wang Q, et al. Pellagra secondary to medication and alcoholism: a case report and review of the literature. Nutr Clin Pract. 2016;31:785-789.
- Ladoyanni E, Cheung ST, North J, et al. Pellagra occurring in a patient with atopic dermatitis and food allergy. J Eur Acad Dermatol Venereol. 2007;21:394-396.
- Bell HK, Poston GJ, Vora J, et al. Cutaneous manifestations of the malignant carcinoid syndrome. Br J Dermatol. 2005;152:71-75.
- Barthelemy H, Chouvet B, Cambazard F. Skin and mucosal manifestations in vitamin deficiency. J Am Acad Dermatol. 1986;15:1263-1274.
- Lamey PJ, Hammond A, Allam BF, et al. Vitamin status of patients with burning mouth syndrome and the response to replacement therapy. Br Dent J. 1986;160:81-84.
- Stover PJ, Field MS. Vitamin B-6. Adv Nutr. 2015;6:132-133.
- Gerlach AT, Thomas S, Stawicki SP, et al. Vitamin B6 deficiency: a potential cause of refractory seizures in adults. JPEN J Parenter Enteral Nutr. 2011;35:272-275.
- Spinneker A, Sola R, Lemmen V, et al. Vitamin B6 status, deficiency and its consequences—an overview. Nutr Hosp. 2007;22:7-24.
- Ross EA, Shah GM, Reynolds RD, et al. Vitamin B6 requirements of patients on chronic peritoneal dialysis. Kidney Int. 1989;36:702-706.
- Brescoll J, Daveluy S. A review of vitamin B12 in dermatology. Am J Clin Dermatol. 2015;16:27-33.
- Sanvisens A, Zuluaga P, Pineda M, et al. Folate deficiency in patients seeking treatment of alcohol use disorder. Drug Alcohol Depend. 2017;180:417-422.
- Langan RC, Goodbred AJ. Vitamin B12 deficiency: recognition and management. Am Fam Physician. 2017;96:384-389.
- Bradford GS, Taylor CT. Omeprazole and vitamin B12 deficiency. Ann Pharmacother. 1999;33:641-643.
- Srivastava N, Chand S, Bansal M, et al. Reversible hyperpigmentation as the first manifestation of dietary vitamin B12 deficiency. Indian J Dermatol Venereol Leprol. 2006;72:389-390.
- Graells J, Ojeda RM, Muniesa C, et al. Glossitis with linear lesions: an early sign of vitamin B12 deficiency. J Am Acad Dermatol. 2009;60:498-500.
- Hirschmann JV, Raugi GJ. Adult scurvy. J Am Acad Dermatol. 1999;41:895-906; quiz 907-810.
- Shaath T, Fischer R, Goeser M, et al. Scurvy in the present times: vitamin C allergy leading to strict fast food diet. Dermatol Online J. 2016;22:13030/qt50b8w28b.
- Fain O, Pariés J, Jacquart B, et al. Hypovitaminosis C in hospitalized patients. Eur J Intern Med. 2003;14:419-425.
- Ahmad SA, Al Thobiti TA, El Toum M, et al. Florid scurvy in an autistic child on a ketogenic diet [published online November 19, 2018]. Pediatr Emerg Care. doi:10.1097/PEC.0000000000001695.
- Lux-Battistelli C, Battistelli D. Latent scurvy with tiredness and leg pain in alcoholics: an underestimated disease three case reports. Medicine (Baltimore). 2017;96:e8861.
- Christopher K, Tammaro D, Wing EJ. Early scurvy complicating anorexia nervosa. South Med J. 2002;95:1065-1066.
- Berger ML, Siegel DM, Lee EL. Scurvy as an initial manifestation of Whipple’s disease. Ann Intern Med. 1984;101:58-59.
- Imes S, Dinwoodie A, Walker K, et al. Vitamin C status in 137 outpatients with Crohn’s disease. effect of diet counseling. J Clin Gastroenterol. 1986;8:443-446.
- Echeverría Zudaire L, García Cuartero B, Campelo Moreno O, et al. Scurvy associated with celiac disease [in Spanish]. An Esp Pediatr. 2002;57:587.
- Hansen EP, Metzsche C, Henningsen E, et al. Severe scurvy after gastric bypass surgery and a poor postoperative diet. J Clin Med Res. 2012;4:135-137.
- Rivière S, Birlouez-Aragon I, Nourhashémi F, et al. Low plasma vitamin C in Alzheimer patients despite an adequate diet. Int J Geriatr Psychiatry. 1998;13:749-754.
- Bhattacharyya P, Giannoutsos J, Eslick GD, et al. Scurvy: an unrecognized and emerging public health issue in developed economies. Mayo Clin Proc. 2019;94:2594-2597.
- Oak AS, Jaleel T, Fening K, et al. A case of scurvy associated with nilotinib. J Cutan Pathol. 2016;43:725-726.
- Kletzel M, Powers K, Hayes M. Scurvy: a new problem for patients with chronic GVHD involving mucous membranes; an easy problem to resolve. Pediatr Transplant. 2014;18:524-526.
- Maxfield L, Crane JS. Vitamin C Deficiency (Scurvy). Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK493187/. Updated November 19, 2019. Accessed May 19, 2020.
Practice Points
- Nutritional deficiencies are common in hospitalized patients and often go unrecognized.
- Awareness of the risk factors predisposing patients to nutritional deficiencies and the cutaneous manifestations associated with undernutrition can promote early diagnosis.
- When investigating cutaneous findings, undernutrition should be considered in patients with chronic infections, malabsorptive states, psychiatric illness, and strict dietary practices, as well as in those using certain medications.
- Prompt nutritional supplementation can prevent patient morbidity and mortality and reverse skin disease.
Pediatric Dermatology Emergencies
Many pediatric skin conditions can be safely monitored with minimal intervention, but certain skin conditions are emergent and require immediate attention and proper assessment of the neonate, infant, or child. The skin may provide the first presentation of a potentially fatal disease with serious sequelae. Cutaneous findings may indicate the need for further evaluation. Therefore, it is important to differentiate skin conditions with benign etiologies from those that require immediate diagnosis and treatment, as early intervention of some of these conditions can be lifesaving. Herein, we discuss pertinent pediatric dermatology emergencies that dermatologists should keep in mind so that these diagnoses are never missed.
Staphylococcal Scalded Skin Syndrome
Presentation
Staphylococcal scalded skin syndrome (SSSS), or Ritter disease, is a potentially fatal pediatric emergency, especially in newborns.1 The mortality rate for SSSS in the United States is 3.6% to 11% in children.2 It typically presents with a prodrome of tenderness, fever, and confluent erythematous patches on the folds of the skin such as the groin, axillae, nose, and ears, with eventual spread to the legs and trunk.1,2 Within 24 to 48 hours of symptom onset, blistering and fluid accumulation will appear diffusely. Bullae are flaccid, and tangential and gentle pressure on involved unblistered skin may lead to shearing of the epithelium, which is a positive Nikolsky sign.1,2
Causes
Staphylococcal scalded skin syndrome is caused by exfoliative toxins A and B, toxigenic strains of Staphylococcus aureus. Exfoliative toxins A and B are serine proteases that target and cleave desmoglein 1, which binds keratinocytes in the stratum granulosum.1,3 Exfoliative toxins disrupt the adhesion of keratinocytes, resulting in bullae formation and subsequently diffuse sheetlike desquamation.1,4,5 Although up to 30% of the human population are asymptomatically and permanently colonized with nasal S aureus,6 the exfoliative toxins are produced by only 5% of species.1
In neonates, the immune and renal systems are underdeveloped; therefore, patients are susceptible to SSSS due to lack of neutralizing antibodies and decreased renal toxin excretion.4 Potential complications of SSSS are deeper soft-tissue infection, septicemia (blood-borne infection), and fluid and electrolyte imbalance.1,4
Diagnosis and Treatment
The condition is diagnosed clinically based on the findings of tender erythroderma, bullae, and desquamation with a scalded appearance, especially in friction zones; periorificial crusting; positive Nikolsky sign; and lack of mucosal involvement (Figure 1).1 Histopathology can aid in complicated clinical scenarios as well as culture from affected areas, including the upper respiratory tract, diaper region, and umbilicus.1,4 Hospitalization is required for SSSS for intravenous antibiotics, fluids, and electrolyte repletion.
Differential Diagnosis
There are multiple diagnoses to consider in the setting of flaccid bullae in the pediatric population. Stevens-Johnson syndrome or toxic epidermal necrolysis also can present with fever and superficial desquamation or bullae; however, exposure to medications and mucosal involvement often are absent in SSSS (Figure 2).2 Pemphigus, particularly paraneoplastic pemphigus, also often includes mucosal involvement and scalding thermal burns that are often geometric or focal. Epidermolysis bullosa and toxic shock syndrome also should be considered.1
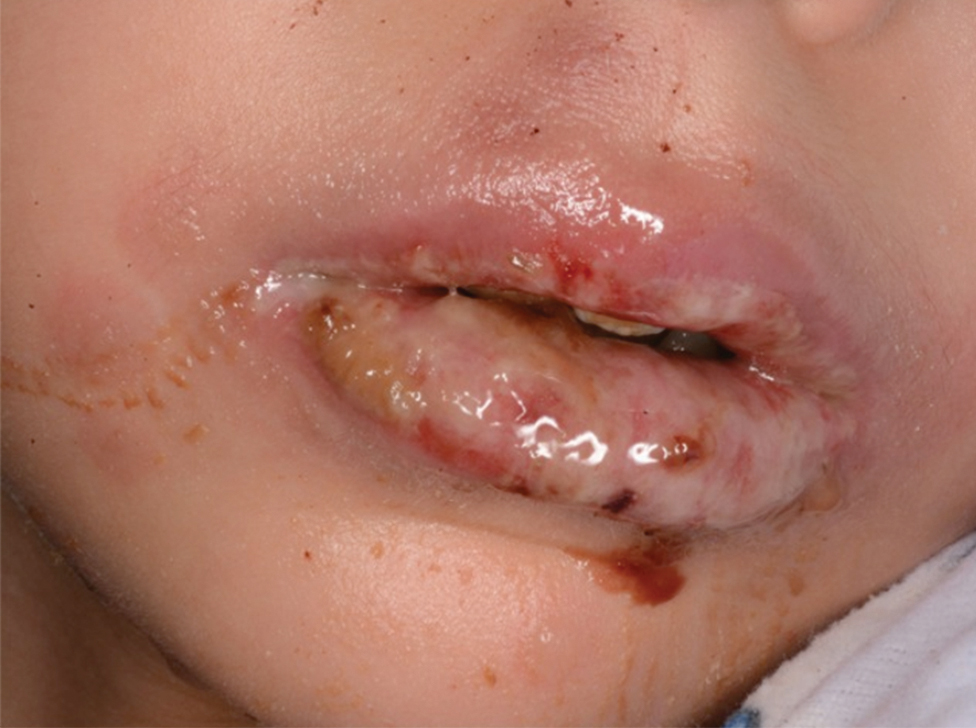
Impetigo
Presentation
Impetigo is the most common bacterial skin infection in children caused by S aureus or Streptococcus pyogenes.7-9 It begins as erythematous papules transitioning to thin-walled vesicles that rapidly rupture and result in honey-crusted papules.7,9,10 Individuals of any age can be affected by nonbullous impetigo, but it is the most common skin infection in children aged 2 to 5 years.7
Bullous impetigo primarily is seen in children, especially infants, and rarely can occur in teenagers or adults.7 It most commonly is caused by the exfoliative toxins of S aureus. Bullous impetigo presents as small vesicles that may converge into larger flaccid bullae or pustules.7-10 Once the bullae rupture, an erythematous base with a collarette of scale remains without the formation of a honey-colored crust.8 Bullous impetigo usually affects moist intertriginous areas such as the axillae, neck, and diaper area8,10 (Figure 3). Complications may result in cellulitis, septicemia, osteomyelitis, poststreptococcal glomerulonephritis associated with S pyogenes, and S aureus–induced SSSS.7-9
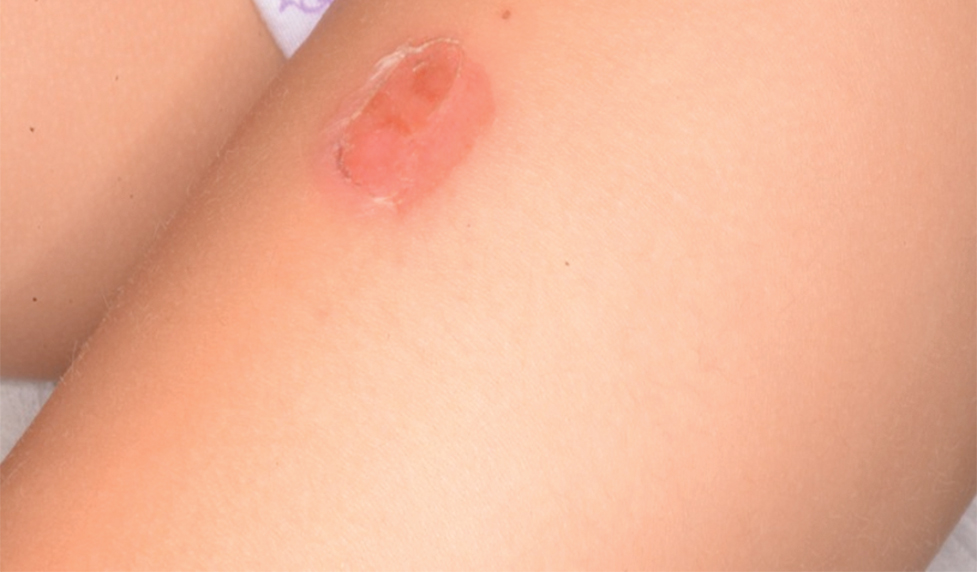
Diagnosis
Nonbullous and bullous impetigo are largely clinical diagnoses that can be confirmed by culture of a vesicle or pustular fluid.10 Treatment of impetigo includes topical or systemic antibiotics.7,10 Patients should be advised to keep lesions covered and avoid contact with others until all lesions resolve, as lesions are contagious.9
Eczema Herpeticum
Presentation
Eczema herpeticum (EH), also known as Kaposi varicelliform eruption, is a disseminated herpes simplex virus infection of impaired skin, most commonly in patients with atopic dermatitis (AD).11 Eczema herpeticum presents as a widespread eruption of erythematous monomorphic vesicles that progress to punched-out erosions with hemorrhagic crusting (Figure 4). Patients may have associated fever or lymphadenopathy.12,13
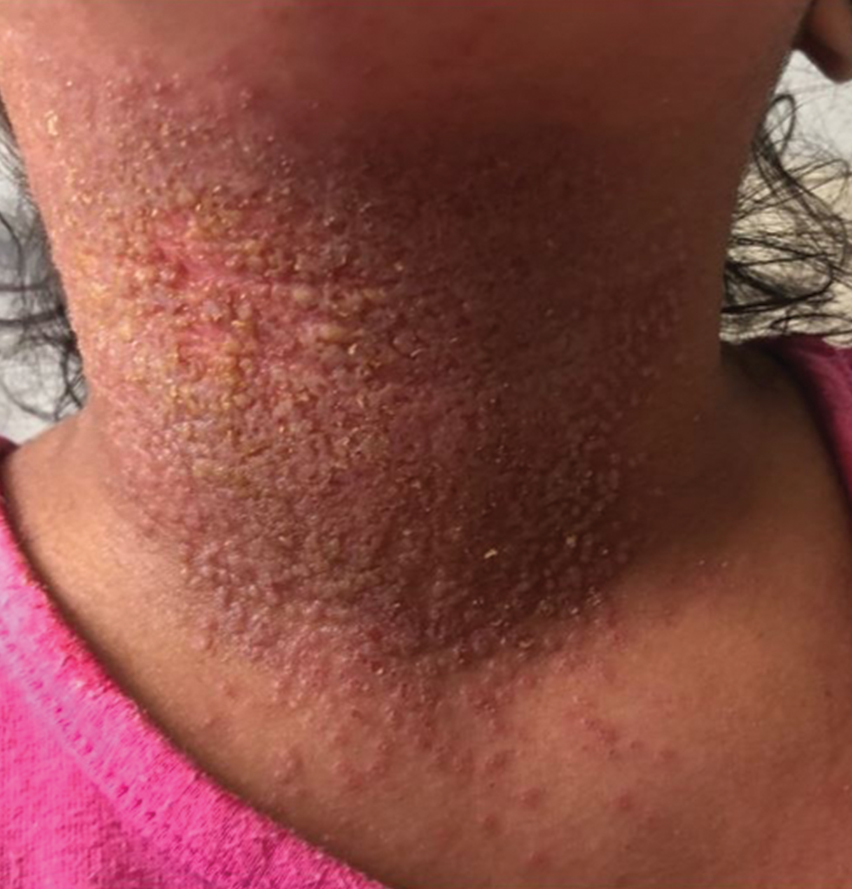
Causes
The number of children hospitalized annually for EH in the United States is approximately 4 to 7 cases per million children. Less than 3% of pediatric AD patients are affected, with a particularly increased risk in patients with severe and earlier-onset AD.12-15 Patients with AD have skin barrier defects, and decreased IFN-γ expression and cathelicidins predispose patients with AD to developing EH.12,16,17
Diagnosis
Viral polymerase chain reaction for herpes simplex virus types 1 and 2 is the standard for confirmatory diagnosis. Herpes simplex virus cultures from cutaneous scrapings, direct fluorescent antibody testing, or Tzanck test revealing multinucleated giant cells also may help establish the diagnosis.11,12,17
Management
Individuals with severe AD and other dermatologic conditions with cutaneous barrier compromise are at risk for developing EH, which is a medical emergency requiring hospitalization and prompt treatment with antiviral therapy such as acyclovir, often intravenously, as death can result if left untreated.11,17 Topical or systemic antibiotic therapy should be initiated if there is suspicion for secondary bacterial superinfection. Patients should be evaluated for multiorgan involvement such as keratoconjunctivitis, meningitis, encephalitis, and systemic viremia due to increased mortality, especially in infants.12,15,16
Langerhans Cell Histiocytosis
Presentation
Langerhans cell histiocytosis (LCH) has a variable clinical presentation and can involve a single or multiple organ systems, including the bones and skin. Cutaneous LCH can present as violaceous papules, nodules, or ulcerations and crusted erosions (Figure 5). The lymph nodes, liver, spleen, oral mucosa, and respiratory and central nervous systems also may be involved.
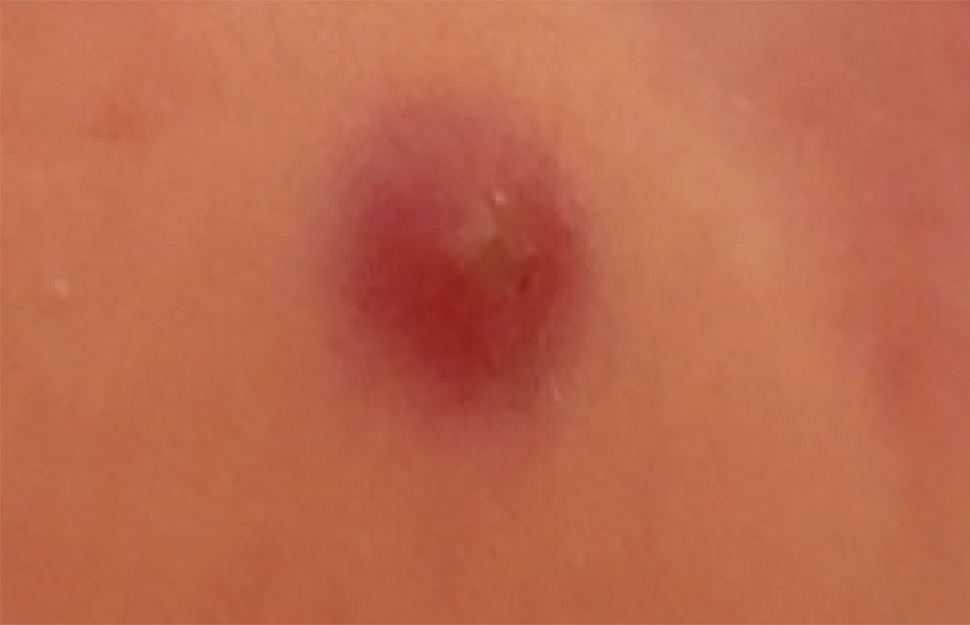
Langerhans cell histiocytosis affects individuals of any age group but more often is seen in pediatric patients. The incidence of LCH is approximately 4.6 cases per million children.18 The pathogenesis is secondary to pathologic Langerhans cells, characterized as a clonal myeloid malignancy and dysregulation of the immune system.18,19
Diagnosis
A thorough physical examination is essential in patients with suspected LCH. Additionally, diagnosis of LCH is heavily based on histopathology of tissue from the involved organ system(s) with features of positive S-100 protein, CD1a, and CD207, and identification of Birbeck granules.20 Imaging and laboratory studies also are indicated and can include a skeletal survey (to assess osteolytic and organ involvement), a complete hematologic panel, coagulation studies, and liver function tests.18,21
Management
Management of LCH varies based on the organ system(s) involved along with the extent of the disease. Dermatology referral may be indicated in patients presenting with nonresolving cutaneous lesions as well as in severe cases. Single-organ and multisystem disease may require one treatment modality or a combination of chemotherapy, surgery, radiation, and/or immunotherapy.21
Infantile Hemangioma
Presentation
Infantile hemangioma (IH) is the most common benign tumor of infancy and usually is apparent a few weeks after birth. Lesions appear as bright red papules, nodules, or plaques. Deep or subcutaneous lesions present as raised, flesh-colored nodules with a blue hue and bruiselike appearance with or without a central patch of telangiectasia22-24 (Figure 6). Although all IHs eventually resolve, residual skin changes such as scarring, atrophy, and fibrosis can persist.24

The incidence of IH has been reported to occur in up to 4% to 5% of infants in the United States.23,25 Infantile hemangiomas also have been found to be more common among white, preterm, and multiple-gestation infants.25 The proposed pathogenesis of IHs includes angiogenic and vasogenic factors that cause rapid proliferation of blood vessels, likely driven by tissue hypoxia.23,26,27
Diagnosis
Infantile hemangioma is diagnosed clinically; however, immunohistochemical staining showing positivity for glucose transporter 1 also is helpful.26,27 Imaging modalities such as ultrasonography and magnetic resonance imaging also can be utilized to visualize the extent of lesions if necessary.25
Management
Around 15% to 25% of IHs are considered complicated and require intervention.25,27 Infantile hemangiomas can interfere with function depending on location or have potentially fatal complications. Based on the location and extent of involvement, these findings can include ulceration; hemorrhage; impairment of feeding, hearing, and/or vision; facial deformities; airway obstruction; hypothyroidism; and congestive heart failure.25,28 Early treatment with topical or oral beta-blockers is imperative for potentially life-threatening IHs, which can be seen due to large size or dangerous location.28,29 Because the rapid proliferative phase of IHs is thought to begin around 6 weeks of life, treatment should be initiated as early as possible. Initiation of beta-blocker therapy in the first few months of life can prevent functional impairment, ulceration, and permanent cosmetic changes. Additionally, surgery or pulsed dye laser treatment have been found to be effective for skin changes found after involution of IH.25,29
Differential Diagnosis
The differential diagnosis for IH includes vascular malformations, which are present at birth and do not undergo rapid proliferation; sarcoma; and kaposiform hemangioendothelioma, which causes the Kasabach-Merritt phenomenon secondary to platelet trapping. Careful attention to the history of the skin lesion provides good support for diagnosis of IH in most cases.
IgA Vasculitis
Presentation
IgA vasculitis, or Henoch-Schönlein purpura, classically presents as a tetrad of palpable purpura, acute-onset arthritis or arthralgia, abdominal pain, and renal disease with proteinuria or hematuria.30 Skin involvement is seen in almost all cases and is essential for diagnosis of IgA vasculitis. The initial dermatosis may be pruritic and present as an erythematous macular or urticarial wheal that evolves into petechiae, along with palpable purpura that is most frequently located on the legs or buttocks (Figure 7).30-34
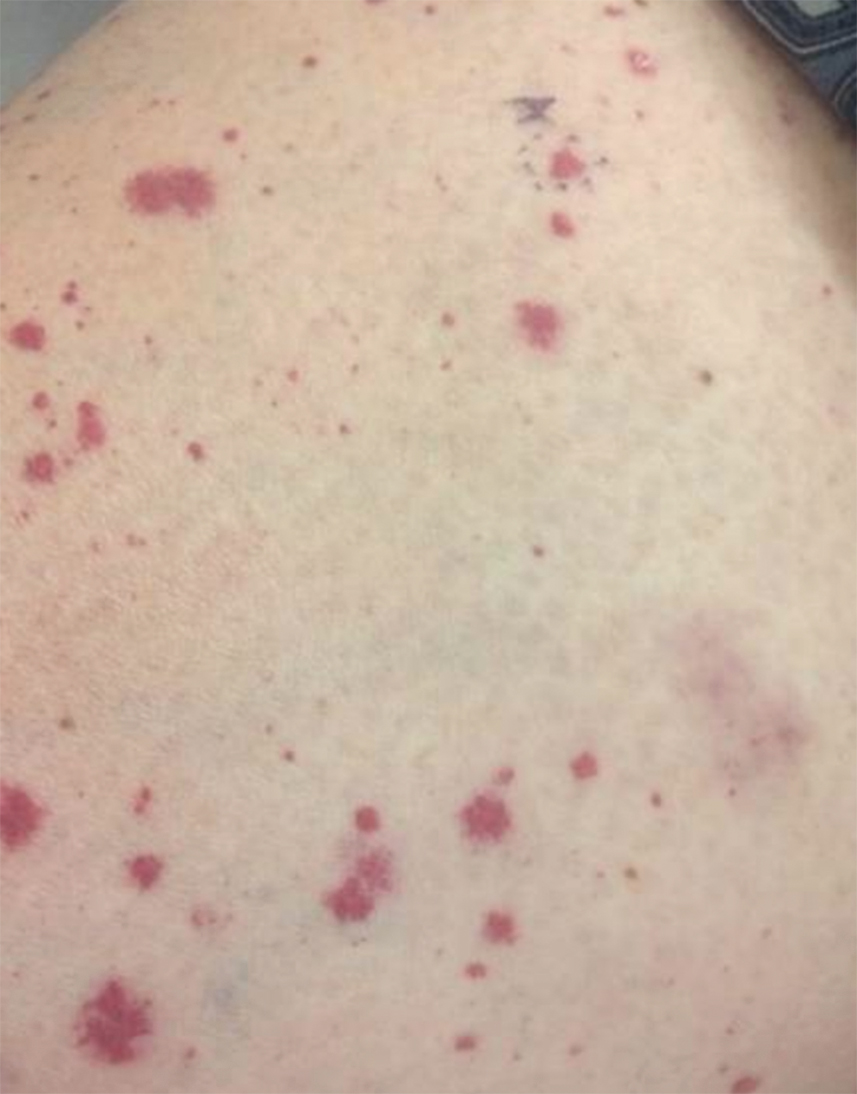
IgA vasculitis is an immune-mediated small vessel vasculitis with deposition of IgA in the small vessels. The underlying cause remains unknown, though infection, dietary allergens, drugs, vaccinations, and chemical triggers have been recognized in literature.32,35,36 IgA vasculitis is largely a pediatric diagnosis, with 90% of affected individuals younger than 10 years worldwide.37 In the pediatric population, the incidence has been reported to be 3 to 26.7 cases per 100,000 children.32
Diagnosis
Diagnosis is based on the clinical presentation and histopathology.30 On direct immunofluorescence, IgA deposition is seen in the vessel walls.35 Laboratory testing is not diagnostic, but urinalysis is mandatory to identify involvement of renal vasculature. Imaging studies may be used in patients with abdominal symptoms, as an ultrasound can be used to visualize bowel structure and abnormalities such as intussusception.33
Management
The majority of cases of IgA vasculitis recover spontaneously, with patients requiring hospital admission based on severity of symptoms.30 The primary approach to management involves providing supportive care including hydration, adequate rest, and symptomatic pain relief of the joints and abdomen with oral analgesics. Systemic corticosteroids or steroid-sparing agents such as dapsone or colchicine can be used to treat cutaneous manifestations in addition to severe pain symptoms.30,31 Patients with IgA vasculitis must be monitored for proteinuria or hematuria to assess the extent of renal involvement. Although much more common in adults, long-term renal impairment can result from childhood cases of IgA vasculitis.34
Final Thoughts
Pediatric dermatology emergencies can be difficult to detect and accurately diagnose. Many of these diseases are potential emergencies that that may result in delayed treatment and considerable morbidity and mortality if missed. Clinicians should be aware that timely recognition and diagnosis, along with possible referral to pediatric dermatology, are essential to avoid complications.
- Leung AKC, Barankin B, Leong KF. Staphylococcal-scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr. 2018;14:116-120.
- Handler MZ, Schwartz RA. Staphylococcal scalded skin syndrome: diagnosis and management in children and adults. J Eur Acad Dermatol Venereol. 2014;28:1418-1423.
- Davidson J, Polly S, Hayes P, et al. Recurrent staphylococcal scalded skin syndrome in an extremely low-birth-weight neonate. AJP Rep. 2017;7:E134-E137.
- Mishra AK, Yadav P, Mishra A. A systemic review on staphylococcal scalded skin syndrome (SSSS): a rare and critical disease of neonates. Open Microbiol J. 2016;10:150-159.
- Berk D. Staphylococcal scalded skin syndrome. Cancer Therapy Advisor website. https://www.cancertherapyadvisor.com/home/decision-support-in-medicine/pediatrics/staphylococcal-scalded-skin-syndrome/. Published 2017. Accessed February 19, 2020.
- Sakr A, Brégeon F, Mège JL, et al. Staphylococcus aureus nasal colonization: an update on mechanisms, epidemiology, risk factors, and subsequent infections [published online October 8, 2018]. Front Microbiol. 2018;9:2419.
- Pereira LB. Impetigo review. An Bras Dermatol. 2014;89:293-299.
- Nardi NM, Schaefer TJ. Impetigo. In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK430974/. Accessed February 21, 2020.
- Koning S, van der Sande R, Verhagen AP, et al. Interventions for impetigo. Cochrane Database Syst Rev. 2012;1:CD003261.
- Sommer LL, Reboli AC, Heymann WR. Bacterial diseases. In: Bolognia, JL Schaffer, JV Cerroni L, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier; 2018:1259-1295.
- Micali G, Lacarrubba F. Eczema herpeticum. N Engl J Med. 2017;377:e9.
- Leung DY. Why is eczema herpeticum unexpectedly rare? Antiviral Res. 2013;98:153-157.
- Seegräber M, Worm M, Werfel T, et al. Recurrent eczema herpeticum—a retrospective European multicenter study evaluating the clinical characteristics of eczema herpeticum cases in atopic dermatitis patients [published online November 16, 2019]. J Eur Acad Dermatology Venereol. doi:10.1111/jdv.16090.
- Sun D, Ong PY. Infectious complications in atopic dermatitis. Immunol Allergy Clin North Am. 2017;37:75-93.
- Hsu DY, Shinkai K, Silverberg JI. Epidemiology of eczema herpeticum in hospitalized U.S. children: analysis of a nationwide cohort [published online September 17, 2018]. J Invest Dermatol. 2018;138:265-272.
- Leung DY, Gao PS, Grigoryev DN, et al. Human atopic dermatitis complicated by eczema herpeticum is associated with abnormalities in IFN-γ response. J Allergy Clin Immunol. 2011;127:965-73.e1-5.
- Darji K, Frisch S, Adjei Boakye E, et al. Characterization of children with recurrent eczema herpeticum and response to treatment with interferon-gamma. Pediatr Dermatol. 2017;34:686-689.
- Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. 2018;379:856-868.
- Abla O, Weitzman S. Treatment of Langerhans cell histiocytosis: role of BRAF/MAPK inhibition. Hematology Am Soc Hematol Educ Program. 2015;2015:565-570.
- Allen CE, Li L, Peters TL, et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol. 2010;184:4557-4567.
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184.
- Holland KE, Drolet BA. Infantile hemangioma [published online August 21, 2010]. Pediatr Clin North Am. 2010;57:1069-1083.
- Chen TS, Eichenfield LF, Friedlander SF. Infantile hemangiomas: an update on pathogenesis and therapy. Pediatrics. 2013;131:99-108.
- George A, Mani V, Noufal A. Update on the classification of hemangioma. J Oral Maxillofac Pathol. 2014;18(suppl 1):S117-S120.
- Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136:786-791.
- Munden A, Butschek R, Tom WL, et al. Prospective study of infantile haemangiomas: incidence, clinical characteristics and association with placental anomalies. Br J Dermatol. 2014;170:907-913.
- de Jong S, Itinteang T, Withers AH, et al. Does hypoxia play a role in infantile hemangioma? Arch Dermatol Res. 2016;308:219-227.
- Hogeling M, Adams S, Wargon O. A randomized controlled trial of propranolol for infantile hemangiomas. Pediatrics. 2011;128:E259-E266.
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas [published online January 2019]. Pediatrics. doi:10.1542/peds.2018-3475.
- Sohagia AB, Gunturu SG, Tong TR, et al. Henoch-Schönlein purpura—a case report and review of the literature [published online May 23, 2010]. Gastroenterol Res Pract. doi:10.1155/2010/597648.
- Rigante D, Castellazzi L, Bosco A, et al. Is there a crossroad between infections, genetics, and Henoch-Schönlein purpura? Autoimmun Rev. 2013;12:1016-1021.
- Piram M, Mahr A. Epidemiology of immunoglobulin A vasculitis (Henoch–Schönlein): current state of knowledge. Curr Opin Rheumatol. 2013;25:171-178.
- Carlson JA. The histological assessment of cutaneous vasculitis. Histopathology. 2010;56:3-23.
- Eleftheriou D, Batu ED, Ozen S, et al. Vasculitis in children. Nephrol Dial Transplant. 2014;30:I94-I103.
- van Timmeren MM, Heeringa P, Kallenberg CG. Infectious triggers for vasculitis. Curr Opin Rheumatol. 2014;26:416-423.
- Scott DGI, Watts RA. Epidemiology and clinical features of systemic vasculitis [published online July 11, 2013]. Clin Exp Nephrol. 2013;17:607-610.
- He X, Yu C, Zhao P, et al. The genetics of Henoch-Schönlein purpura: a systematic review and meta-analysis. Rheumatol Int. 2013;33:1387-1395.
Many pediatric skin conditions can be safely monitored with minimal intervention, but certain skin conditions are emergent and require immediate attention and proper assessment of the neonate, infant, or child. The skin may provide the first presentation of a potentially fatal disease with serious sequelae. Cutaneous findings may indicate the need for further evaluation. Therefore, it is important to differentiate skin conditions with benign etiologies from those that require immediate diagnosis and treatment, as early intervention of some of these conditions can be lifesaving. Herein, we discuss pertinent pediatric dermatology emergencies that dermatologists should keep in mind so that these diagnoses are never missed.
Staphylococcal Scalded Skin Syndrome
Presentation
Staphylococcal scalded skin syndrome (SSSS), or Ritter disease, is a potentially fatal pediatric emergency, especially in newborns.1 The mortality rate for SSSS in the United States is 3.6% to 11% in children.2 It typically presents with a prodrome of tenderness, fever, and confluent erythematous patches on the folds of the skin such as the groin, axillae, nose, and ears, with eventual spread to the legs and trunk.1,2 Within 24 to 48 hours of symptom onset, blistering and fluid accumulation will appear diffusely. Bullae are flaccid, and tangential and gentle pressure on involved unblistered skin may lead to shearing of the epithelium, which is a positive Nikolsky sign.1,2
Causes
Staphylococcal scalded skin syndrome is caused by exfoliative toxins A and B, toxigenic strains of Staphylococcus aureus. Exfoliative toxins A and B are serine proteases that target and cleave desmoglein 1, which binds keratinocytes in the stratum granulosum.1,3 Exfoliative toxins disrupt the adhesion of keratinocytes, resulting in bullae formation and subsequently diffuse sheetlike desquamation.1,4,5 Although up to 30% of the human population are asymptomatically and permanently colonized with nasal S aureus,6 the exfoliative toxins are produced by only 5% of species.1
In neonates, the immune and renal systems are underdeveloped; therefore, patients are susceptible to SSSS due to lack of neutralizing antibodies and decreased renal toxin excretion.4 Potential complications of SSSS are deeper soft-tissue infection, septicemia (blood-borne infection), and fluid and electrolyte imbalance.1,4
Diagnosis and Treatment
The condition is diagnosed clinically based on the findings of tender erythroderma, bullae, and desquamation with a scalded appearance, especially in friction zones; periorificial crusting; positive Nikolsky sign; and lack of mucosal involvement (Figure 1).1 Histopathology can aid in complicated clinical scenarios as well as culture from affected areas, including the upper respiratory tract, diaper region, and umbilicus.1,4 Hospitalization is required for SSSS for intravenous antibiotics, fluids, and electrolyte repletion.
Differential Diagnosis
There are multiple diagnoses to consider in the setting of flaccid bullae in the pediatric population. Stevens-Johnson syndrome or toxic epidermal necrolysis also can present with fever and superficial desquamation or bullae; however, exposure to medications and mucosal involvement often are absent in SSSS (Figure 2).2 Pemphigus, particularly paraneoplastic pemphigus, also often includes mucosal involvement and scalding thermal burns that are often geometric or focal. Epidermolysis bullosa and toxic shock syndrome also should be considered.1
Impetigo
Presentation
Impetigo is the most common bacterial skin infection in children caused by S aureus or Streptococcus pyogenes.7-9 It begins as erythematous papules transitioning to thin-walled vesicles that rapidly rupture and result in honey-crusted papules.7,9,10 Individuals of any age can be affected by nonbullous impetigo, but it is the most common skin infection in children aged 2 to 5 years.7
Bullous impetigo primarily is seen in children, especially infants, and rarely can occur in teenagers or adults.7 It most commonly is caused by the exfoliative toxins of S aureus. Bullous impetigo presents as small vesicles that may converge into larger flaccid bullae or pustules.7-10 Once the bullae rupture, an erythematous base with a collarette of scale remains without the formation of a honey-colored crust.8 Bullous impetigo usually affects moist intertriginous areas such as the axillae, neck, and diaper area8,10 (Figure 3). Complications may result in cellulitis, septicemia, osteomyelitis, poststreptococcal glomerulonephritis associated with S pyogenes, and S aureus–induced SSSS.7-9
Diagnosis
Nonbullous and bullous impetigo are largely clinical diagnoses that can be confirmed by culture of a vesicle or pustular fluid.10 Treatment of impetigo includes topical or systemic antibiotics.7,10 Patients should be advised to keep lesions covered and avoid contact with others until all lesions resolve, as lesions are contagious.9
Eczema Herpeticum
Presentation
Eczema herpeticum (EH), also known as Kaposi varicelliform eruption, is a disseminated herpes simplex virus infection of impaired skin, most commonly in patients with atopic dermatitis (AD).11 Eczema herpeticum presents as a widespread eruption of erythematous monomorphic vesicles that progress to punched-out erosions with hemorrhagic crusting (Figure 4). Patients may have associated fever or lymphadenopathy.12,13
Causes
The number of children hospitalized annually for EH in the United States is approximately 4 to 7 cases per million children. Less than 3% of pediatric AD patients are affected, with a particularly increased risk in patients with severe and earlier-onset AD.12-15 Patients with AD have skin barrier defects, and decreased IFN-γ expression and cathelicidins predispose patients with AD to developing EH.12,16,17
Diagnosis
Viral polymerase chain reaction for herpes simplex virus types 1 and 2 is the standard for confirmatory diagnosis. Herpes simplex virus cultures from cutaneous scrapings, direct fluorescent antibody testing, or Tzanck test revealing multinucleated giant cells also may help establish the diagnosis.11,12,17
Management
Individuals with severe AD and other dermatologic conditions with cutaneous barrier compromise are at risk for developing EH, which is a medical emergency requiring hospitalization and prompt treatment with antiviral therapy such as acyclovir, often intravenously, as death can result if left untreated.11,17 Topical or systemic antibiotic therapy should be initiated if there is suspicion for secondary bacterial superinfection. Patients should be evaluated for multiorgan involvement such as keratoconjunctivitis, meningitis, encephalitis, and systemic viremia due to increased mortality, especially in infants.12,15,16
Langerhans Cell Histiocytosis
Presentation
Langerhans cell histiocytosis (LCH) has a variable clinical presentation and can involve a single or multiple organ systems, including the bones and skin. Cutaneous LCH can present as violaceous papules, nodules, or ulcerations and crusted erosions (Figure 5). The lymph nodes, liver, spleen, oral mucosa, and respiratory and central nervous systems also may be involved.
Langerhans cell histiocytosis affects individuals of any age group but more often is seen in pediatric patients. The incidence of LCH is approximately 4.6 cases per million children.18 The pathogenesis is secondary to pathologic Langerhans cells, characterized as a clonal myeloid malignancy and dysregulation of the immune system.18,19
Diagnosis
A thorough physical examination is essential in patients with suspected LCH. Additionally, diagnosis of LCH is heavily based on histopathology of tissue from the involved organ system(s) with features of positive S-100 protein, CD1a, and CD207, and identification of Birbeck granules.20 Imaging and laboratory studies also are indicated and can include a skeletal survey (to assess osteolytic and organ involvement), a complete hematologic panel, coagulation studies, and liver function tests.18,21
Management
Management of LCH varies based on the organ system(s) involved along with the extent of the disease. Dermatology referral may be indicated in patients presenting with nonresolving cutaneous lesions as well as in severe cases. Single-organ and multisystem disease may require one treatment modality or a combination of chemotherapy, surgery, radiation, and/or immunotherapy.21
Infantile Hemangioma
Presentation
Infantile hemangioma (IH) is the most common benign tumor of infancy and usually is apparent a few weeks after birth. Lesions appear as bright red papules, nodules, or plaques. Deep or subcutaneous lesions present as raised, flesh-colored nodules with a blue hue and bruiselike appearance with or without a central patch of telangiectasia22-24 (Figure 6). Although all IHs eventually resolve, residual skin changes such as scarring, atrophy, and fibrosis can persist.24
The incidence of IH has been reported to occur in up to 4% to 5% of infants in the United States.23,25 Infantile hemangiomas also have been found to be more common among white, preterm, and multiple-gestation infants.25 The proposed pathogenesis of IHs includes angiogenic and vasogenic factors that cause rapid proliferation of blood vessels, likely driven by tissue hypoxia.23,26,27
Diagnosis
Infantile hemangioma is diagnosed clinically; however, immunohistochemical staining showing positivity for glucose transporter 1 also is helpful.26,27 Imaging modalities such as ultrasonography and magnetic resonance imaging also can be utilized to visualize the extent of lesions if necessary.25
Management
Around 15% to 25% of IHs are considered complicated and require intervention.25,27 Infantile hemangiomas can interfere with function depending on location or have potentially fatal complications. Based on the location and extent of involvement, these findings can include ulceration; hemorrhage; impairment of feeding, hearing, and/or vision; facial deformities; airway obstruction; hypothyroidism; and congestive heart failure.25,28 Early treatment with topical or oral beta-blockers is imperative for potentially life-threatening IHs, which can be seen due to large size or dangerous location.28,29 Because the rapid proliferative phase of IHs is thought to begin around 6 weeks of life, treatment should be initiated as early as possible. Initiation of beta-blocker therapy in the first few months of life can prevent functional impairment, ulceration, and permanent cosmetic changes. Additionally, surgery or pulsed dye laser treatment have been found to be effective for skin changes found after involution of IH.25,29
Differential Diagnosis
The differential diagnosis for IH includes vascular malformations, which are present at birth and do not undergo rapid proliferation; sarcoma; and kaposiform hemangioendothelioma, which causes the Kasabach-Merritt phenomenon secondary to platelet trapping. Careful attention to the history of the skin lesion provides good support for diagnosis of IH in most cases.
IgA Vasculitis
Presentation
IgA vasculitis, or Henoch-Schönlein purpura, classically presents as a tetrad of palpable purpura, acute-onset arthritis or arthralgia, abdominal pain, and renal disease with proteinuria or hematuria.30 Skin involvement is seen in almost all cases and is essential for diagnosis of IgA vasculitis. The initial dermatosis may be pruritic and present as an erythematous macular or urticarial wheal that evolves into petechiae, along with palpable purpura that is most frequently located on the legs or buttocks (Figure 7).30-34
IgA vasculitis is an immune-mediated small vessel vasculitis with deposition of IgA in the small vessels. The underlying cause remains unknown, though infection, dietary allergens, drugs, vaccinations, and chemical triggers have been recognized in literature.32,35,36 IgA vasculitis is largely a pediatric diagnosis, with 90% of affected individuals younger than 10 years worldwide.37 In the pediatric population, the incidence has been reported to be 3 to 26.7 cases per 100,000 children.32
Diagnosis
Diagnosis is based on the clinical presentation and histopathology.30 On direct immunofluorescence, IgA deposition is seen in the vessel walls.35 Laboratory testing is not diagnostic, but urinalysis is mandatory to identify involvement of renal vasculature. Imaging studies may be used in patients with abdominal symptoms, as an ultrasound can be used to visualize bowel structure and abnormalities such as intussusception.33
Management
The majority of cases of IgA vasculitis recover spontaneously, with patients requiring hospital admission based on severity of symptoms.30 The primary approach to management involves providing supportive care including hydration, adequate rest, and symptomatic pain relief of the joints and abdomen with oral analgesics. Systemic corticosteroids or steroid-sparing agents such as dapsone or colchicine can be used to treat cutaneous manifestations in addition to severe pain symptoms.30,31 Patients with IgA vasculitis must be monitored for proteinuria or hematuria to assess the extent of renal involvement. Although much more common in adults, long-term renal impairment can result from childhood cases of IgA vasculitis.34
Final Thoughts
Pediatric dermatology emergencies can be difficult to detect and accurately diagnose. Many of these diseases are potential emergencies that that may result in delayed treatment and considerable morbidity and mortality if missed. Clinicians should be aware that timely recognition and diagnosis, along with possible referral to pediatric dermatology, are essential to avoid complications.
Many pediatric skin conditions can be safely monitored with minimal intervention, but certain skin conditions are emergent and require immediate attention and proper assessment of the neonate, infant, or child. The skin may provide the first presentation of a potentially fatal disease with serious sequelae. Cutaneous findings may indicate the need for further evaluation. Therefore, it is important to differentiate skin conditions with benign etiologies from those that require immediate diagnosis and treatment, as early intervention of some of these conditions can be lifesaving. Herein, we discuss pertinent pediatric dermatology emergencies that dermatologists should keep in mind so that these diagnoses are never missed.
Staphylococcal Scalded Skin Syndrome
Presentation
Staphylococcal scalded skin syndrome (SSSS), or Ritter disease, is a potentially fatal pediatric emergency, especially in newborns.1 The mortality rate for SSSS in the United States is 3.6% to 11% in children.2 It typically presents with a prodrome of tenderness, fever, and confluent erythematous patches on the folds of the skin such as the groin, axillae, nose, and ears, with eventual spread to the legs and trunk.1,2 Within 24 to 48 hours of symptom onset, blistering and fluid accumulation will appear diffusely. Bullae are flaccid, and tangential and gentle pressure on involved unblistered skin may lead to shearing of the epithelium, which is a positive Nikolsky sign.1,2
Causes
Staphylococcal scalded skin syndrome is caused by exfoliative toxins A and B, toxigenic strains of Staphylococcus aureus. Exfoliative toxins A and B are serine proteases that target and cleave desmoglein 1, which binds keratinocytes in the stratum granulosum.1,3 Exfoliative toxins disrupt the adhesion of keratinocytes, resulting in bullae formation and subsequently diffuse sheetlike desquamation.1,4,5 Although up to 30% of the human population are asymptomatically and permanently colonized with nasal S aureus,6 the exfoliative toxins are produced by only 5% of species.1
In neonates, the immune and renal systems are underdeveloped; therefore, patients are susceptible to SSSS due to lack of neutralizing antibodies and decreased renal toxin excretion.4 Potential complications of SSSS are deeper soft-tissue infection, septicemia (blood-borne infection), and fluid and electrolyte imbalance.1,4
Diagnosis and Treatment
The condition is diagnosed clinically based on the findings of tender erythroderma, bullae, and desquamation with a scalded appearance, especially in friction zones; periorificial crusting; positive Nikolsky sign; and lack of mucosal involvement (Figure 1).1 Histopathology can aid in complicated clinical scenarios as well as culture from affected areas, including the upper respiratory tract, diaper region, and umbilicus.1,4 Hospitalization is required for SSSS for intravenous antibiotics, fluids, and electrolyte repletion.
Differential Diagnosis
There are multiple diagnoses to consider in the setting of flaccid bullae in the pediatric population. Stevens-Johnson syndrome or toxic epidermal necrolysis also can present with fever and superficial desquamation or bullae; however, exposure to medications and mucosal involvement often are absent in SSSS (Figure 2).2 Pemphigus, particularly paraneoplastic pemphigus, also often includes mucosal involvement and scalding thermal burns that are often geometric or focal. Epidermolysis bullosa and toxic shock syndrome also should be considered.1
Impetigo
Presentation
Impetigo is the most common bacterial skin infection in children caused by S aureus or Streptococcus pyogenes.7-9 It begins as erythematous papules transitioning to thin-walled vesicles that rapidly rupture and result in honey-crusted papules.7,9,10 Individuals of any age can be affected by nonbullous impetigo, but it is the most common skin infection in children aged 2 to 5 years.7
Bullous impetigo primarily is seen in children, especially infants, and rarely can occur in teenagers or adults.7 It most commonly is caused by the exfoliative toxins of S aureus. Bullous impetigo presents as small vesicles that may converge into larger flaccid bullae or pustules.7-10 Once the bullae rupture, an erythematous base with a collarette of scale remains without the formation of a honey-colored crust.8 Bullous impetigo usually affects moist intertriginous areas such as the axillae, neck, and diaper area8,10 (Figure 3). Complications may result in cellulitis, septicemia, osteomyelitis, poststreptococcal glomerulonephritis associated with S pyogenes, and S aureus–induced SSSS.7-9
Diagnosis
Nonbullous and bullous impetigo are largely clinical diagnoses that can be confirmed by culture of a vesicle or pustular fluid.10 Treatment of impetigo includes topical or systemic antibiotics.7,10 Patients should be advised to keep lesions covered and avoid contact with others until all lesions resolve, as lesions are contagious.9
Eczema Herpeticum
Presentation
Eczema herpeticum (EH), also known as Kaposi varicelliform eruption, is a disseminated herpes simplex virus infection of impaired skin, most commonly in patients with atopic dermatitis (AD).11 Eczema herpeticum presents as a widespread eruption of erythematous monomorphic vesicles that progress to punched-out erosions with hemorrhagic crusting (Figure 4). Patients may have associated fever or lymphadenopathy.12,13
Causes
The number of children hospitalized annually for EH in the United States is approximately 4 to 7 cases per million children. Less than 3% of pediatric AD patients are affected, with a particularly increased risk in patients with severe and earlier-onset AD.12-15 Patients with AD have skin barrier defects, and decreased IFN-γ expression and cathelicidins predispose patients with AD to developing EH.12,16,17
Diagnosis
Viral polymerase chain reaction for herpes simplex virus types 1 and 2 is the standard for confirmatory diagnosis. Herpes simplex virus cultures from cutaneous scrapings, direct fluorescent antibody testing, or Tzanck test revealing multinucleated giant cells also may help establish the diagnosis.11,12,17
Management
Individuals with severe AD and other dermatologic conditions with cutaneous barrier compromise are at risk for developing EH, which is a medical emergency requiring hospitalization and prompt treatment with antiviral therapy such as acyclovir, often intravenously, as death can result if left untreated.11,17 Topical or systemic antibiotic therapy should be initiated if there is suspicion for secondary bacterial superinfection. Patients should be evaluated for multiorgan involvement such as keratoconjunctivitis, meningitis, encephalitis, and systemic viremia due to increased mortality, especially in infants.12,15,16
Langerhans Cell Histiocytosis
Presentation
Langerhans cell histiocytosis (LCH) has a variable clinical presentation and can involve a single or multiple organ systems, including the bones and skin. Cutaneous LCH can present as violaceous papules, nodules, or ulcerations and crusted erosions (Figure 5). The lymph nodes, liver, spleen, oral mucosa, and respiratory and central nervous systems also may be involved.
Langerhans cell histiocytosis affects individuals of any age group but more often is seen in pediatric patients. The incidence of LCH is approximately 4.6 cases per million children.18 The pathogenesis is secondary to pathologic Langerhans cells, characterized as a clonal myeloid malignancy and dysregulation of the immune system.18,19
Diagnosis
A thorough physical examination is essential in patients with suspected LCH. Additionally, diagnosis of LCH is heavily based on histopathology of tissue from the involved organ system(s) with features of positive S-100 protein, CD1a, and CD207, and identification of Birbeck granules.20 Imaging and laboratory studies also are indicated and can include a skeletal survey (to assess osteolytic and organ involvement), a complete hematologic panel, coagulation studies, and liver function tests.18,21
Management
Management of LCH varies based on the organ system(s) involved along with the extent of the disease. Dermatology referral may be indicated in patients presenting with nonresolving cutaneous lesions as well as in severe cases. Single-organ and multisystem disease may require one treatment modality or a combination of chemotherapy, surgery, radiation, and/or immunotherapy.21
Infantile Hemangioma
Presentation
Infantile hemangioma (IH) is the most common benign tumor of infancy and usually is apparent a few weeks after birth. Lesions appear as bright red papules, nodules, or plaques. Deep or subcutaneous lesions present as raised, flesh-colored nodules with a blue hue and bruiselike appearance with or without a central patch of telangiectasia22-24 (Figure 6). Although all IHs eventually resolve, residual skin changes such as scarring, atrophy, and fibrosis can persist.24
The incidence of IH has been reported to occur in up to 4% to 5% of infants in the United States.23,25 Infantile hemangiomas also have been found to be more common among white, preterm, and multiple-gestation infants.25 The proposed pathogenesis of IHs includes angiogenic and vasogenic factors that cause rapid proliferation of blood vessels, likely driven by tissue hypoxia.23,26,27
Diagnosis
Infantile hemangioma is diagnosed clinically; however, immunohistochemical staining showing positivity for glucose transporter 1 also is helpful.26,27 Imaging modalities such as ultrasonography and magnetic resonance imaging also can be utilized to visualize the extent of lesions if necessary.25
Management
Around 15% to 25% of IHs are considered complicated and require intervention.25,27 Infantile hemangiomas can interfere with function depending on location or have potentially fatal complications. Based on the location and extent of involvement, these findings can include ulceration; hemorrhage; impairment of feeding, hearing, and/or vision; facial deformities; airway obstruction; hypothyroidism; and congestive heart failure.25,28 Early treatment with topical or oral beta-blockers is imperative for potentially life-threatening IHs, which can be seen due to large size or dangerous location.28,29 Because the rapid proliferative phase of IHs is thought to begin around 6 weeks of life, treatment should be initiated as early as possible. Initiation of beta-blocker therapy in the first few months of life can prevent functional impairment, ulceration, and permanent cosmetic changes. Additionally, surgery or pulsed dye laser treatment have been found to be effective for skin changes found after involution of IH.25,29
Differential Diagnosis
The differential diagnosis for IH includes vascular malformations, which are present at birth and do not undergo rapid proliferation; sarcoma; and kaposiform hemangioendothelioma, which causes the Kasabach-Merritt phenomenon secondary to platelet trapping. Careful attention to the history of the skin lesion provides good support for diagnosis of IH in most cases.
IgA Vasculitis
Presentation
IgA vasculitis, or Henoch-Schönlein purpura, classically presents as a tetrad of palpable purpura, acute-onset arthritis or arthralgia, abdominal pain, and renal disease with proteinuria or hematuria.30 Skin involvement is seen in almost all cases and is essential for diagnosis of IgA vasculitis. The initial dermatosis may be pruritic and present as an erythematous macular or urticarial wheal that evolves into petechiae, along with palpable purpura that is most frequently located on the legs or buttocks (Figure 7).30-34
IgA vasculitis is an immune-mediated small vessel vasculitis with deposition of IgA in the small vessels. The underlying cause remains unknown, though infection, dietary allergens, drugs, vaccinations, and chemical triggers have been recognized in literature.32,35,36 IgA vasculitis is largely a pediatric diagnosis, with 90% of affected individuals younger than 10 years worldwide.37 In the pediatric population, the incidence has been reported to be 3 to 26.7 cases per 100,000 children.32
Diagnosis
Diagnosis is based on the clinical presentation and histopathology.30 On direct immunofluorescence, IgA deposition is seen in the vessel walls.35 Laboratory testing is not diagnostic, but urinalysis is mandatory to identify involvement of renal vasculature. Imaging studies may be used in patients with abdominal symptoms, as an ultrasound can be used to visualize bowel structure and abnormalities such as intussusception.33
Management
The majority of cases of IgA vasculitis recover spontaneously, with patients requiring hospital admission based on severity of symptoms.30 The primary approach to management involves providing supportive care including hydration, adequate rest, and symptomatic pain relief of the joints and abdomen with oral analgesics. Systemic corticosteroids or steroid-sparing agents such as dapsone or colchicine can be used to treat cutaneous manifestations in addition to severe pain symptoms.30,31 Patients with IgA vasculitis must be monitored for proteinuria or hematuria to assess the extent of renal involvement. Although much more common in adults, long-term renal impairment can result from childhood cases of IgA vasculitis.34
Final Thoughts
Pediatric dermatology emergencies can be difficult to detect and accurately diagnose. Many of these diseases are potential emergencies that that may result in delayed treatment and considerable morbidity and mortality if missed. Clinicians should be aware that timely recognition and diagnosis, along with possible referral to pediatric dermatology, are essential to avoid complications.
- Leung AKC, Barankin B, Leong KF. Staphylococcal-scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr. 2018;14:116-120.
- Handler MZ, Schwartz RA. Staphylococcal scalded skin syndrome: diagnosis and management in children and adults. J Eur Acad Dermatol Venereol. 2014;28:1418-1423.
- Davidson J, Polly S, Hayes P, et al. Recurrent staphylococcal scalded skin syndrome in an extremely low-birth-weight neonate. AJP Rep. 2017;7:E134-E137.
- Mishra AK, Yadav P, Mishra A. A systemic review on staphylococcal scalded skin syndrome (SSSS): a rare and critical disease of neonates. Open Microbiol J. 2016;10:150-159.
- Berk D. Staphylococcal scalded skin syndrome. Cancer Therapy Advisor website. https://www.cancertherapyadvisor.com/home/decision-support-in-medicine/pediatrics/staphylococcal-scalded-skin-syndrome/. Published 2017. Accessed February 19, 2020.
- Sakr A, Brégeon F, Mège JL, et al. Staphylococcus aureus nasal colonization: an update on mechanisms, epidemiology, risk factors, and subsequent infections [published online October 8, 2018]. Front Microbiol. 2018;9:2419.
- Pereira LB. Impetigo review. An Bras Dermatol. 2014;89:293-299.
- Nardi NM, Schaefer TJ. Impetigo. In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK430974/. Accessed February 21, 2020.
- Koning S, van der Sande R, Verhagen AP, et al. Interventions for impetigo. Cochrane Database Syst Rev. 2012;1:CD003261.
- Sommer LL, Reboli AC, Heymann WR. Bacterial diseases. In: Bolognia, JL Schaffer, JV Cerroni L, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier; 2018:1259-1295.
- Micali G, Lacarrubba F. Eczema herpeticum. N Engl J Med. 2017;377:e9.
- Leung DY. Why is eczema herpeticum unexpectedly rare? Antiviral Res. 2013;98:153-157.
- Seegräber M, Worm M, Werfel T, et al. Recurrent eczema herpeticum—a retrospective European multicenter study evaluating the clinical characteristics of eczema herpeticum cases in atopic dermatitis patients [published online November 16, 2019]. J Eur Acad Dermatology Venereol. doi:10.1111/jdv.16090.
- Sun D, Ong PY. Infectious complications in atopic dermatitis. Immunol Allergy Clin North Am. 2017;37:75-93.
- Hsu DY, Shinkai K, Silverberg JI. Epidemiology of eczema herpeticum in hospitalized U.S. children: analysis of a nationwide cohort [published online September 17, 2018]. J Invest Dermatol. 2018;138:265-272.
- Leung DY, Gao PS, Grigoryev DN, et al. Human atopic dermatitis complicated by eczema herpeticum is associated with abnormalities in IFN-γ response. J Allergy Clin Immunol. 2011;127:965-73.e1-5.
- Darji K, Frisch S, Adjei Boakye E, et al. Characterization of children with recurrent eczema herpeticum and response to treatment with interferon-gamma. Pediatr Dermatol. 2017;34:686-689.
- Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. 2018;379:856-868.
- Abla O, Weitzman S. Treatment of Langerhans cell histiocytosis: role of BRAF/MAPK inhibition. Hematology Am Soc Hematol Educ Program. 2015;2015:565-570.
- Allen CE, Li L, Peters TL, et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol. 2010;184:4557-4567.
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184.
- Holland KE, Drolet BA. Infantile hemangioma [published online August 21, 2010]. Pediatr Clin North Am. 2010;57:1069-1083.
- Chen TS, Eichenfield LF, Friedlander SF. Infantile hemangiomas: an update on pathogenesis and therapy. Pediatrics. 2013;131:99-108.
- George A, Mani V, Noufal A. Update on the classification of hemangioma. J Oral Maxillofac Pathol. 2014;18(suppl 1):S117-S120.
- Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136:786-791.
- Munden A, Butschek R, Tom WL, et al. Prospective study of infantile haemangiomas: incidence, clinical characteristics and association with placental anomalies. Br J Dermatol. 2014;170:907-913.
- de Jong S, Itinteang T, Withers AH, et al. Does hypoxia play a role in infantile hemangioma? Arch Dermatol Res. 2016;308:219-227.
- Hogeling M, Adams S, Wargon O. A randomized controlled trial of propranolol for infantile hemangiomas. Pediatrics. 2011;128:E259-E266.
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas [published online January 2019]. Pediatrics. doi:10.1542/peds.2018-3475.
- Sohagia AB, Gunturu SG, Tong TR, et al. Henoch-Schönlein purpura—a case report and review of the literature [published online May 23, 2010]. Gastroenterol Res Pract. doi:10.1155/2010/597648.
- Rigante D, Castellazzi L, Bosco A, et al. Is there a crossroad between infections, genetics, and Henoch-Schönlein purpura? Autoimmun Rev. 2013;12:1016-1021.
- Piram M, Mahr A. Epidemiology of immunoglobulin A vasculitis (Henoch–Schönlein): current state of knowledge. Curr Opin Rheumatol. 2013;25:171-178.
- Carlson JA. The histological assessment of cutaneous vasculitis. Histopathology. 2010;56:3-23.
- Eleftheriou D, Batu ED, Ozen S, et al. Vasculitis in children. Nephrol Dial Transplant. 2014;30:I94-I103.
- van Timmeren MM, Heeringa P, Kallenberg CG. Infectious triggers for vasculitis. Curr Opin Rheumatol. 2014;26:416-423.
- Scott DGI, Watts RA. Epidemiology and clinical features of systemic vasculitis [published online July 11, 2013]. Clin Exp Nephrol. 2013;17:607-610.
- He X, Yu C, Zhao P, et al. The genetics of Henoch-Schönlein purpura: a systematic review and meta-analysis. Rheumatol Int. 2013;33:1387-1395.
- Leung AKC, Barankin B, Leong KF. Staphylococcal-scalded skin syndrome: evaluation, diagnosis, and management. World J Pediatr. 2018;14:116-120.
- Handler MZ, Schwartz RA. Staphylococcal scalded skin syndrome: diagnosis and management in children and adults. J Eur Acad Dermatol Venereol. 2014;28:1418-1423.
- Davidson J, Polly S, Hayes P, et al. Recurrent staphylococcal scalded skin syndrome in an extremely low-birth-weight neonate. AJP Rep. 2017;7:E134-E137.
- Mishra AK, Yadav P, Mishra A. A systemic review on staphylococcal scalded skin syndrome (SSSS): a rare and critical disease of neonates. Open Microbiol J. 2016;10:150-159.
- Berk D. Staphylococcal scalded skin syndrome. Cancer Therapy Advisor website. https://www.cancertherapyadvisor.com/home/decision-support-in-medicine/pediatrics/staphylococcal-scalded-skin-syndrome/. Published 2017. Accessed February 19, 2020.
- Sakr A, Brégeon F, Mège JL, et al. Staphylococcus aureus nasal colonization: an update on mechanisms, epidemiology, risk factors, and subsequent infections [published online October 8, 2018]. Front Microbiol. 2018;9:2419.
- Pereira LB. Impetigo review. An Bras Dermatol. 2014;89:293-299.
- Nardi NM, Schaefer TJ. Impetigo. In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2019. https://www.ncbi.nlm.nih.gov/books/NBK430974/. Accessed February 21, 2020.
- Koning S, van der Sande R, Verhagen AP, et al. Interventions for impetigo. Cochrane Database Syst Rev. 2012;1:CD003261.
- Sommer LL, Reboli AC, Heymann WR. Bacterial diseases. In: Bolognia, JL Schaffer, JV Cerroni L, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier; 2018:1259-1295.
- Micali G, Lacarrubba F. Eczema herpeticum. N Engl J Med. 2017;377:e9.
- Leung DY. Why is eczema herpeticum unexpectedly rare? Antiviral Res. 2013;98:153-157.
- Seegräber M, Worm M, Werfel T, et al. Recurrent eczema herpeticum—a retrospective European multicenter study evaluating the clinical characteristics of eczema herpeticum cases in atopic dermatitis patients [published online November 16, 2019]. J Eur Acad Dermatology Venereol. doi:10.1111/jdv.16090.
- Sun D, Ong PY. Infectious complications in atopic dermatitis. Immunol Allergy Clin North Am. 2017;37:75-93.
- Hsu DY, Shinkai K, Silverberg JI. Epidemiology of eczema herpeticum in hospitalized U.S. children: analysis of a nationwide cohort [published online September 17, 2018]. J Invest Dermatol. 2018;138:265-272.
- Leung DY, Gao PS, Grigoryev DN, et al. Human atopic dermatitis complicated by eczema herpeticum is associated with abnormalities in IFN-γ response. J Allergy Clin Immunol. 2011;127:965-73.e1-5.
- Darji K, Frisch S, Adjei Boakye E, et al. Characterization of children with recurrent eczema herpeticum and response to treatment with interferon-gamma. Pediatr Dermatol. 2017;34:686-689.
- Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. 2018;379:856-868.
- Abla O, Weitzman S. Treatment of Langerhans cell histiocytosis: role of BRAF/MAPK inhibition. Hematology Am Soc Hematol Educ Program. 2015;2015:565-570.
- Allen CE, Li L, Peters TL, et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J Immunol. 2010;184:4557-4567.
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60:175-184.
- Holland KE, Drolet BA. Infantile hemangioma [published online August 21, 2010]. Pediatr Clin North Am. 2010;57:1069-1083.
- Chen TS, Eichenfield LF, Friedlander SF. Infantile hemangiomas: an update on pathogenesis and therapy. Pediatrics. 2013;131:99-108.
- George A, Mani V, Noufal A. Update on the classification of hemangioma. J Oral Maxillofac Pathol. 2014;18(suppl 1):S117-S120.
- Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136:786-791.
- Munden A, Butschek R, Tom WL, et al. Prospective study of infantile haemangiomas: incidence, clinical characteristics and association with placental anomalies. Br J Dermatol. 2014;170:907-913.
- de Jong S, Itinteang T, Withers AH, et al. Does hypoxia play a role in infantile hemangioma? Arch Dermatol Res. 2016;308:219-227.
- Hogeling M, Adams S, Wargon O. A randomized controlled trial of propranolol for infantile hemangiomas. Pediatrics. 2011;128:E259-E266.
- Krowchuk DP, Frieden IJ, Mancini AJ, et al. Clinical practice guideline for the management of infantile hemangiomas [published online January 2019]. Pediatrics. doi:10.1542/peds.2018-3475.
- Sohagia AB, Gunturu SG, Tong TR, et al. Henoch-Schönlein purpura—a case report and review of the literature [published online May 23, 2010]. Gastroenterol Res Pract. doi:10.1155/2010/597648.
- Rigante D, Castellazzi L, Bosco A, et al. Is there a crossroad between infections, genetics, and Henoch-Schönlein purpura? Autoimmun Rev. 2013;12:1016-1021.
- Piram M, Mahr A. Epidemiology of immunoglobulin A vasculitis (Henoch–Schönlein): current state of knowledge. Curr Opin Rheumatol. 2013;25:171-178.
- Carlson JA. The histological assessment of cutaneous vasculitis. Histopathology. 2010;56:3-23.
- Eleftheriou D, Batu ED, Ozen S, et al. Vasculitis in children. Nephrol Dial Transplant. 2014;30:I94-I103.
- van Timmeren MM, Heeringa P, Kallenberg CG. Infectious triggers for vasculitis. Curr Opin Rheumatol. 2014;26:416-423.
- Scott DGI, Watts RA. Epidemiology and clinical features of systemic vasculitis [published online July 11, 2013]. Clin Exp Nephrol. 2013;17:607-610.
- He X, Yu C, Zhao P, et al. The genetics of Henoch-Schönlein purpura: a systematic review and meta-analysis. Rheumatol Int. 2013;33:1387-1395.
Practice Points
- Staphylococcal scalded skin syndrome, impetigo, eczema herpeticum, Langerhans cell histiocytosis, infantile hemangiomas, and IgA vasculitis all present potential emergencies in pediatric patients in dermatologic settings.
- Early and accurate identification and management of these entities is critical to avoid short-term and long-term negative sequalae.
Clinical Characterization of Leukemia Cutis Presentation
Leukemia is a malignant, life-threatening neoplasm affecting the hematopoietic system. Extramedullary manifestations can occur in various organs, including skin.1 Skin findings in leukemia patients are common and varied, including pallor secondary to anemia, petechiae or ecchymoses due to thrombocytopenia, and skin manifestations of neutropenia and chemotherapy.2 When patients with leukemia develop skin lesions without leukemic infiltration, the resulting nonspecific cutaneous manifestations are known as leukemids.3 Specific cutaneous manifestations of leukemia resulting from direct invasion of leukemic cells into the epidermis, dermis, or subcutis are referred to as leukemia cutis (LC).2,3
Acute myeloid leukemia (AML) is the most common type of leukemia associated with LC, but LC also is seen in other leukemias with various frequencies.1 The lesions of LC can present anywhere on skin, though it has been reported that LC has a tendency to occur at sites of prior ongoing inflammation,2,4 most commonly the extremities, trunk, and face.2,5,6 LC lesions have a range of morphological findings and most commonly present as nodules, papules, and plaques.1,7
Most reports of LC in the literature are case reports or case series with small numbers of subjects.3,6,8 A study of LC patients (N=75) in Korea by Kang et al7 has been the only one to analyze clinical characteristics of LC since 2000.
The aim of this study was to further contribute to the knowledge of clinical characteristics of LC. Clinical patterns of 46 patients were analyzed to further characterize the presentation of LC and to compare our results with those in the literature.
Methods
We conducted a single-institution retrospective review of medical records of patients with LC diagnosed in the Department of Dermatology at Wake Forest School of Medicine (Winston-Salem, North Carolina) over a 17-year period (2001-2017). The study protocol was approved by the institutional review board of Wake Forest University School of Medicine (IRB No. 00054474). Patients had a leukemia diagnosis established by bone marrow biopsy. Patients were included in this analysis if they had ongoing active leukemia and a skin biopsy consistent with LC. Patients of all sexes and ages were included in the cohort. Patients were excluded if they presented only with nonspecific cutaneous lesions associated with leukemia (leukemids). After removing duplicate records from a total of 60 patients initially identified, 46 unique patients were included in this study.
Results
Demographics
Fifty-six percent (26/46) of patients were male. The average age at diagnosis of leukemia was 58 years (range, 8.5 months–84 years). Eighty-five percent of patients were white (39/46), 11% were black (5/46), 2% were Hispanic (1/46), and 2% were of unknown ethnicity (1/46).
Eighty percent (37/46) of patients with LC had AML; 3 of these patients had a prior diagnosis of chronic myeloid leukemia (CML) and 2 had myelodysplastic syndrome (MDS) that did not develop LC until after they had transitioned to AML. Other subtypes of leukemia in this patient population included acute lymphoblastic leukemia (ALL)(n=2), plasma cell leukemia (PCL)(n=2), undifferentiated leukemia (n=2), chronic lymphocytic leukemia (CLL)(n=1), myelodysplastic syndrome (n=1), and Burkitt-type leukemia (n=1).
Distribution and Morphology of LC Lesions
The clinical appearance of LC was widely variable in morphology and anatomic location (Table 1 and Figure). Eighty-four percent of LC occurrences involved more than one lesion (n=32); 14% were a solitary lesion (n=6). For the 2 patients who had 2 separate episodes of LC, the initial presentation of LC was multiple lesions; recurrent LC at relapse presented as a solitary lesion in both cases. Most LC lesions (77% [67/87]) occurred on the trunk or extremities; 23% (20/87) of LC lesions occurred on less common sites, such as the groin, face, hands, feet, and mucosa. Papules (38% [22/58]) and nodules (31% [18/58]) were the most common morphology; macules, plaques, and ulcers were observed less frequently. Clinical descriptions of LC lesions varied widely, with the most common descriptive characteristics being erythematous (57% [20/35]), violaceous (31% [11/35]), and asymptomatic (84% [32/38]). Rare descriptors included flesh colored, hyperpigmented, tender, pruritic, edema, crusting, and confluent erythematous.
Interval Between Leukemia Diagnosis and LC Diagnosis
Approximately 59% (n=27) of patients had LC as a presenting finding of their leukemia (Table 2). Twenty-two percent (n=10) developed LC at the time of leukemia relapse; 20% (n=9) developed LC during consolidation or salvage chemotherapy. Two AML patients had recurrent episodes of LC both at initial presentation of leukemia and when AML relapsed. Two other AML patients received a diagnosis of LC at the same time as a negative concurrent bone marrow biopsy (ie, aleukemic LC). Mean duration between diagnosis of leukemia and diagnosis of LC was 0.4 months (CLL), 1.0 month (ALL), 4.7 months (AML), and 7.15 months (PCL). In cases of MDS and CML transformation to AML, the interval was 6.5 and 4.9 months, respectively.
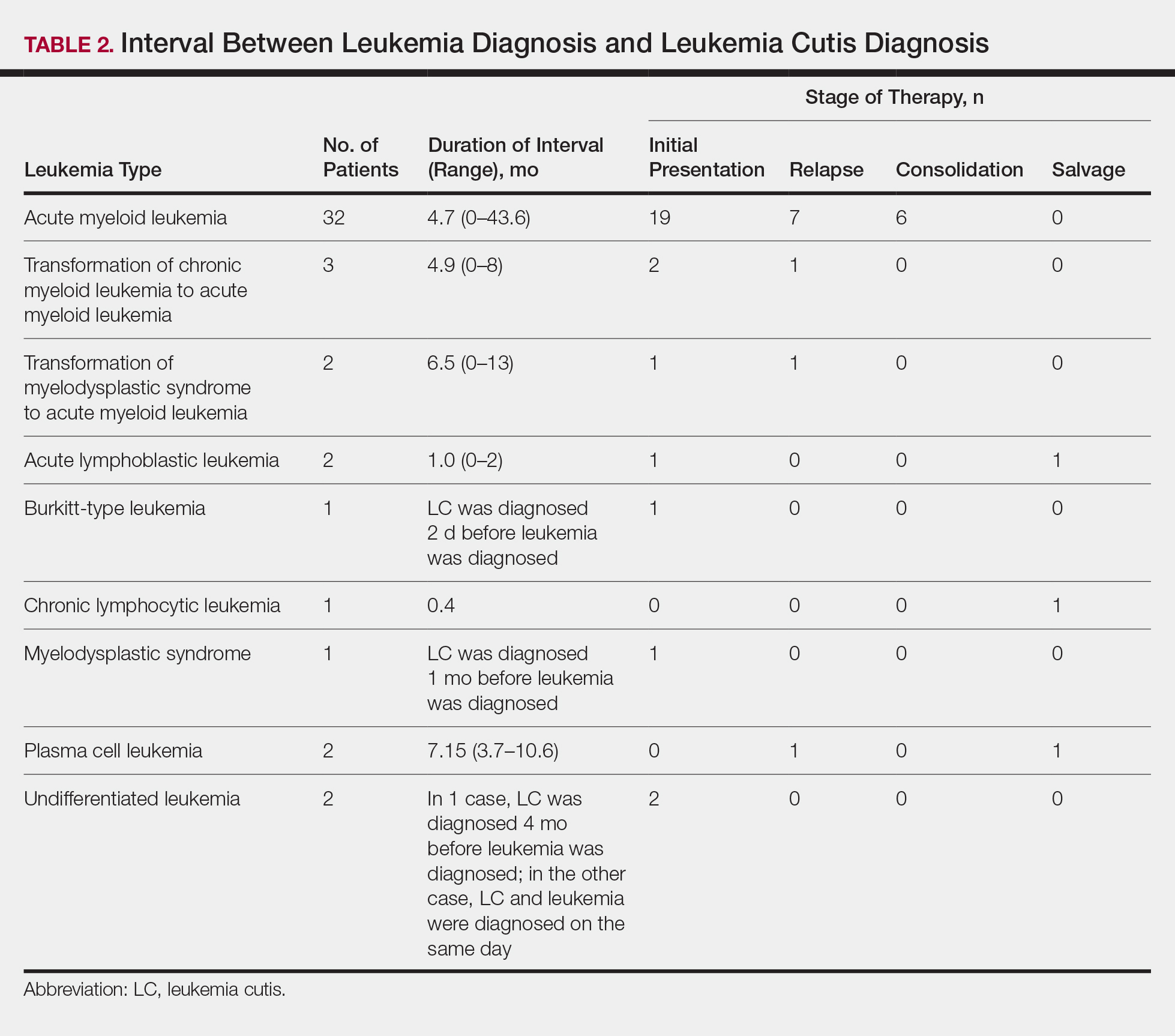
Interval Between LC Diagnosis and Death
As a whole, 17% (n=8) of patients were living at the time this article was written (eTable). Of patients who are still living, 10.9% (n=5) have AML. Looking at the cohort of patients with AML and LC, average age at AML diagnosis was 59.8 years. Average time from diagnosis of leukemia to death was 17.3 months (range, 0.6–49.6 months) for AML; 17.0 months (range, 10.0–24.0 months) for CML transformation to AML; 15.0 months (range, 12.0–18.0 months) for PCL; 14.75 months (range, 11.0–18.5 months) for undifferentiated leukemia; and 8.95 months (range, 4.2–13.7 months) for MDS transformation to AML. The interval between leukemia diagnosis and death was notably shorter for the CLL patient (4.0 months) and the deceased ALL patient (2.4 months). Mean duration between LC diagnosis and death was 11.7 months (AML), 11.2 months (undifferentiated leukemia), 9.9 months (CML transformation to AML), 2.75 months (PCL), and 2.4 months (MDS transformation to AML). The shortest intervals between LC diagnosis and death were seen in CLL (0.5 months) and ALL (0.4 months).
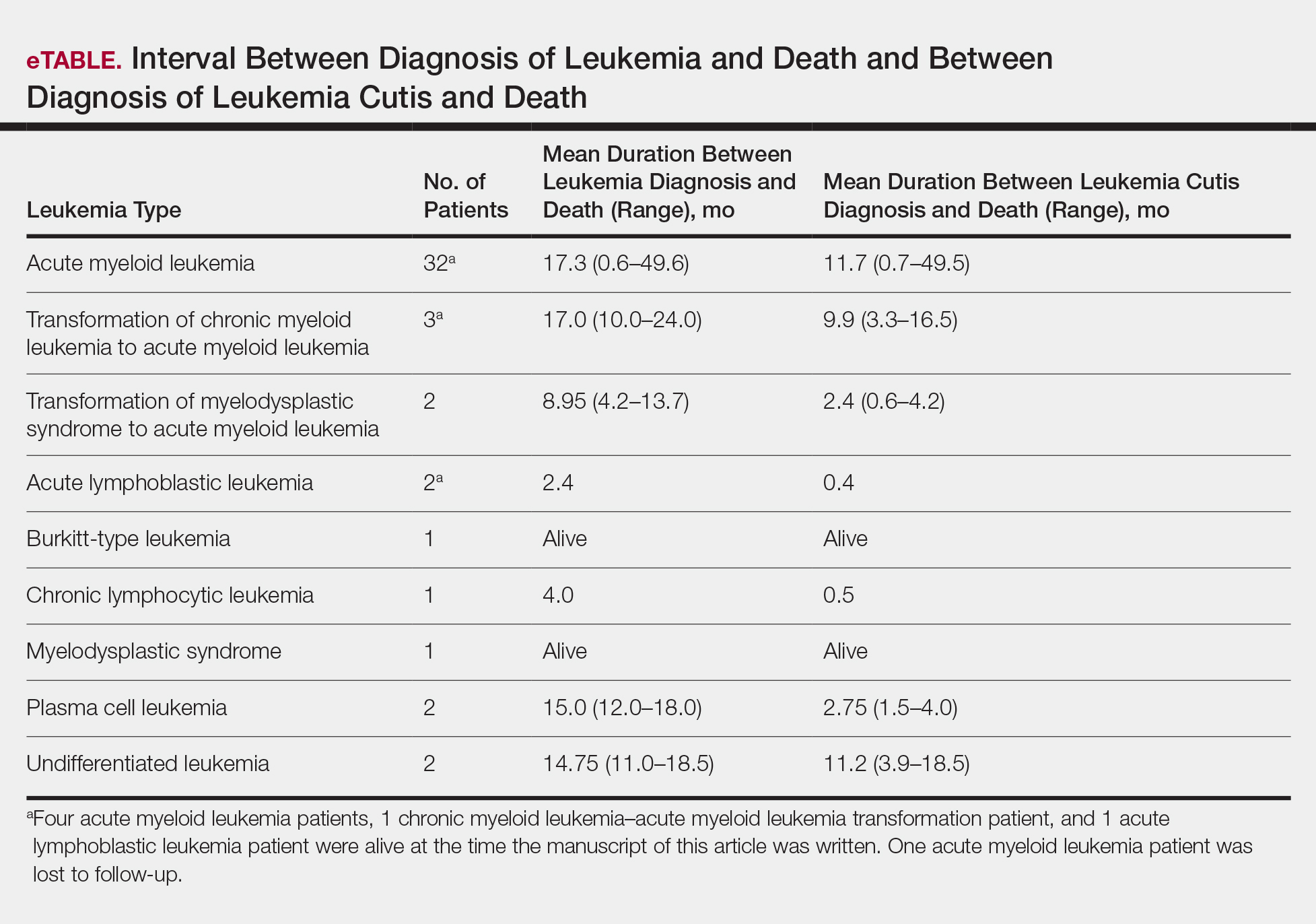
Discussion
Cutaneous manifestations are not uncommon in leukemia patients and can have a number of causes, including paraneoplastic cutaneous manifestations, such as pyoderma gangrenosum and Sweet syndrome; infection; cutaneous toxicities from antineoplastic agents; and LC.2 Leukemia cutis can be confused with other skin lesions in leukemia patients; diagnosis requires biopsy.2,9
We analyzed clinical characteristics and prognosis of 46 patients with LC over a 17-year period. To the best of our knowledge, this is the largest study of LC patients published in the United States. A similar study by Kang et al7 analyzed 75 patients in Korea; however, the incidence of LC among different types of leukemia in the Korean population cannot be applied to Western countries. We did compare the clinical characteristics of our cohort of patients to those reported by Kang et al7 and other studies including a smaller number of patients.
In this study, the male to female ratio was 1.3 to 1 compared to the 2:1 ratio reported by Kang et al.7 The mean age of leukemia diagnosis among our patients was 58 years, which is notably older than the mean age previously reported.7 In this cohort, 4 patients (8.7%) were 34 years or younger, including 1 infant aged 8.5 months; 24 (52.2%) were aged 35 to 64 years; and 18 (39.1%) were 65 years and older.
Consistent with other studies,2,5,7 the most common type of leukemia in patients who developed LC was AML (80%). Among AML patients, the mean age at AML diagnosis (59.8 years) was notably younger than the reported US average age of patients who had a diagnosis of AML (68 years).10 Gender breakdown was slightly different than US statistics: 63% of AML patients in our group were male, whereas AML is only slightly more common among men in the United States.10
Clinically, skin lesions observed most commonly were (in decreasing order) papules, nodules, macules, plaques, and ulcers. Papules (38%) were the most common lesion overall in our study, which differed from the Kang et al7 report in which nodules were the most common. Nodules (31%) were the second most common LC morphology among our patients. Among AML patients, papules were seen in 56% of patients (18/32); nodules were seen in 44% (14/32). The extremities (when combined together) were the most common location of LC lesions (46% [arms, 24%; legs, 22%]); the trunk was the second most common body region (31%). Our study did not find a difference among most common LC anatomic sites compared to other studies.5,7 Less common sites in our cohort included the head, scalp/ears, neck, hands, mucosa, and feet. All body sites were represented, including ocular and oral mucosa and groin, a finding that underscores the importance of complete and comprehensive skin examinations in this patient population. The terms erythematous and violaceous were used to describe the color of most lesions (88%), which commonly presented as multiple lesions (84%) and often were asymptomatic (84%).
It has been reported that, first, in most cases of LC, the condition develops in patients who have already been given a diagnosis of leukemia and, second, simultaneous manifestation of systemic leukemia and LC is less common.11,12 Leukemia cutis also can precede peripheral or bone marrow leukemia (known as aleukemic LC).1,13 Two AML patients (4.3% [2/46]) in this study met criteria for aleukemic LC because they had LC at the same time as negative bone marrow biopsy, which is consistent with a prior report that aleukemic LC can affect as many as 7% of patients.1 Our results differed slightly from prior studies in that most of our patients had LC as one of the presenting manifestations of their leukemia.3,7
Regardless of leukemia type, patients were likely to die within 1 year of LC diagnosis, on average, which is consistent with prior reports.7,11,12 However, the time between diagnosis of LC and death varied greatly among our patients (range, 12 days to 4.1 years). From 2007 to 2013, the 5-year relative survival rate overall for leukemia patients in the US population (by type) was 86.2% (CLL), 71.0% (ALL), 68.0% (CML), and 27.4% (AML).14 Compared to these national statistics, the relative survival rate in LC is poor, with patients who have AML surviving, on average, less than 8 months from time of leukemia diagnosis, whereas ALL and CLL patients survive less than 6 months.
When LC is a late presentation of B-cell CLL or when it presents as myeloid leukemia, blastic transformation (Richter syndrome), or T-cell CLL, it is occasionally associated with poor prognosis, though LC does not affect survival.15-17 In a study of the association of LC with survival in AML, 5-year survival among 62 AML patients with LC was 8.6%, shorter than 28.3% among the 186 matched patients with AML without LC.18 Similarly, the estimated 5-year survival for all patients with AML, according to Surveillance, Epidemiology, and End Results Program data (2007-2013), was 27.4%.14 Based on those results, LC might be a good prognostic indicator in patients with AML.
Conclusion
This study characterized the clinical presentation of LC, which is highly variable in appearance, symptoms, distribution, and stage of leukemia at presentation. In our study cohort, LC most commonly presented as asymptomatic erythematous or violaceous papules or nodules in older male AML patients at leukemia diagnosis. Given such wide variability, dermatologists and oncologists need to keep LC in the differential diagnosis for any new skin lesion and to have a low threshold for performing skin biopsy. Complete and thorough skin examinations should be performed on leukemia patients throughout the course of their disease to identify LC early so that treatment can be implemented in a timely fashion at initial diagnosis, first sign of relapse, or change in disease state.
- Wagner G, Fenchel K, Back W, et al. Leukemia cutis—epidemiology, clinical presentation, and differential diagnoses. J Dtsch Dermatol Ges. 2012;10:27-36.
- Grunwald MR, McDonnell MH, Induru R, et al. Cutaneous manifestations in leukemia patients. Semin Oncol. 2016;43:359-365.
- Martínez-Leboráns L, Victoria-Martínez A, Torregrosa-Calatayu JL, et al. Leukemia cutis: a report of 17 cases and a review of literature. Actas Dermosifiliogr. 2016;107:e65-e69.
- Li L, Wang Y, Lian CG, et al. Clinical and pathological features of myeloid leukemia cutis. An Bras Dermatol. 2018;93:216-221.
- Paydas¸ S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Lee JI, Park HJ, Oh ST, et al. A case of leukemia cutis at the site of a prior catheter insertion. Ann Dermatol. 2009;21:193-196.
- Kang YS, Kim HS, Park HJ, et al. Clinical characteristics of 75 patients with leukemia cutis. J Korean Med Sci. 2013;28:614-619.
- Stern M, Halter J, Buser A, et al. Leukemia cutis preceding systemic relapse of acute myeloid leukemia. Int J Hematol. 2008;87:108-109.
- Patel LM, Maghari A, Schwartz RA, et al. Myeloid leukemia cutis in the setting of myelodysplastic syndrome: a crucial dermatological diagnosis. Int J Dermatol. 2012;51:383-388.
- American Cancer Society. Cancer Facts & Figures 2019. Atlanta, GA: American Cancer Society; 2019. http://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf. Accessed November 21, 2019.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
- Barzilai A, Lyakhovitsky A, Goldberg I, et al. Aleukemic monocytic leukemia cutis. Cutis. 2002;69:301-304
- Howlader N, Noone AM, Krapcho M, et al, eds. SEER cancer statistics review (CSR) 1975-2014. Bethesda, MD: National Cancer Institute; April 2017. https://seer.cancer.gov/archive/csr/1975_2014/. Accessed November 21, 2019.
- Cerroni L, Zenahlik P, Höfler G, et al. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol. 1996;20:1000-1010.
- Colburn DE, Welch MA, Giles FJ. Skin infiltration with chronic lymphocytic leukemia is consistent with a good prognosis. Hematology. 2002;7:187-188.
- Ratnam KV, Khor CJ, Su WP. Leukemia cutis. Dermatol Clin. 1994;12:419-431.
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826-832.
Leukemia is a malignant, life-threatening neoplasm affecting the hematopoietic system. Extramedullary manifestations can occur in various organs, including skin.1 Skin findings in leukemia patients are common and varied, including pallor secondary to anemia, petechiae or ecchymoses due to thrombocytopenia, and skin manifestations of neutropenia and chemotherapy.2 When patients with leukemia develop skin lesions without leukemic infiltration, the resulting nonspecific cutaneous manifestations are known as leukemids.3 Specific cutaneous manifestations of leukemia resulting from direct invasion of leukemic cells into the epidermis, dermis, or subcutis are referred to as leukemia cutis (LC).2,3
Acute myeloid leukemia (AML) is the most common type of leukemia associated with LC, but LC also is seen in other leukemias with various frequencies.1 The lesions of LC can present anywhere on skin, though it has been reported that LC has a tendency to occur at sites of prior ongoing inflammation,2,4 most commonly the extremities, trunk, and face.2,5,6 LC lesions have a range of morphological findings and most commonly present as nodules, papules, and plaques.1,7
Most reports of LC in the literature are case reports or case series with small numbers of subjects.3,6,8 A study of LC patients (N=75) in Korea by Kang et al7 has been the only one to analyze clinical characteristics of LC since 2000.
The aim of this study was to further contribute to the knowledge of clinical characteristics of LC. Clinical patterns of 46 patients were analyzed to further characterize the presentation of LC and to compare our results with those in the literature.
Methods
We conducted a single-institution retrospective review of medical records of patients with LC diagnosed in the Department of Dermatology at Wake Forest School of Medicine (Winston-Salem, North Carolina) over a 17-year period (2001-2017). The study protocol was approved by the institutional review board of Wake Forest University School of Medicine (IRB No. 00054474). Patients had a leukemia diagnosis established by bone marrow biopsy. Patients were included in this analysis if they had ongoing active leukemia and a skin biopsy consistent with LC. Patients of all sexes and ages were included in the cohort. Patients were excluded if they presented only with nonspecific cutaneous lesions associated with leukemia (leukemids). After removing duplicate records from a total of 60 patients initially identified, 46 unique patients were included in this study.
Results
Demographics
Fifty-six percent (26/46) of patients were male. The average age at diagnosis of leukemia was 58 years (range, 8.5 months–84 years). Eighty-five percent of patients were white (39/46), 11% were black (5/46), 2% were Hispanic (1/46), and 2% were of unknown ethnicity (1/46).
Eighty percent (37/46) of patients with LC had AML; 3 of these patients had a prior diagnosis of chronic myeloid leukemia (CML) and 2 had myelodysplastic syndrome (MDS) that did not develop LC until after they had transitioned to AML. Other subtypes of leukemia in this patient population included acute lymphoblastic leukemia (ALL)(n=2), plasma cell leukemia (PCL)(n=2), undifferentiated leukemia (n=2), chronic lymphocytic leukemia (CLL)(n=1), myelodysplastic syndrome (n=1), and Burkitt-type leukemia (n=1).
Distribution and Morphology of LC Lesions
The clinical appearance of LC was widely variable in morphology and anatomic location (Table 1 and Figure). Eighty-four percent of LC occurrences involved more than one lesion (n=32); 14% were a solitary lesion (n=6). For the 2 patients who had 2 separate episodes of LC, the initial presentation of LC was multiple lesions; recurrent LC at relapse presented as a solitary lesion in both cases. Most LC lesions (77% [67/87]) occurred on the trunk or extremities; 23% (20/87) of LC lesions occurred on less common sites, such as the groin, face, hands, feet, and mucosa. Papules (38% [22/58]) and nodules (31% [18/58]) were the most common morphology; macules, plaques, and ulcers were observed less frequently. Clinical descriptions of LC lesions varied widely, with the most common descriptive characteristics being erythematous (57% [20/35]), violaceous (31% [11/35]), and asymptomatic (84% [32/38]). Rare descriptors included flesh colored, hyperpigmented, tender, pruritic, edema, crusting, and confluent erythematous.
Interval Between Leukemia Diagnosis and LC Diagnosis
Approximately 59% (n=27) of patients had LC as a presenting finding of their leukemia (Table 2). Twenty-two percent (n=10) developed LC at the time of leukemia relapse; 20% (n=9) developed LC during consolidation or salvage chemotherapy. Two AML patients had recurrent episodes of LC both at initial presentation of leukemia and when AML relapsed. Two other AML patients received a diagnosis of LC at the same time as a negative concurrent bone marrow biopsy (ie, aleukemic LC). Mean duration between diagnosis of leukemia and diagnosis of LC was 0.4 months (CLL), 1.0 month (ALL), 4.7 months (AML), and 7.15 months (PCL). In cases of MDS and CML transformation to AML, the interval was 6.5 and 4.9 months, respectively.
Interval Between LC Diagnosis and Death
As a whole, 17% (n=8) of patients were living at the time this article was written (eTable). Of patients who are still living, 10.9% (n=5) have AML. Looking at the cohort of patients with AML and LC, average age at AML diagnosis was 59.8 years. Average time from diagnosis of leukemia to death was 17.3 months (range, 0.6–49.6 months) for AML; 17.0 months (range, 10.0–24.0 months) for CML transformation to AML; 15.0 months (range, 12.0–18.0 months) for PCL; 14.75 months (range, 11.0–18.5 months) for undifferentiated leukemia; and 8.95 months (range, 4.2–13.7 months) for MDS transformation to AML. The interval between leukemia diagnosis and death was notably shorter for the CLL patient (4.0 months) and the deceased ALL patient (2.4 months). Mean duration between LC diagnosis and death was 11.7 months (AML), 11.2 months (undifferentiated leukemia), 9.9 months (CML transformation to AML), 2.75 months (PCL), and 2.4 months (MDS transformation to AML). The shortest intervals between LC diagnosis and death were seen in CLL (0.5 months) and ALL (0.4 months).
Discussion
Cutaneous manifestations are not uncommon in leukemia patients and can have a number of causes, including paraneoplastic cutaneous manifestations, such as pyoderma gangrenosum and Sweet syndrome; infection; cutaneous toxicities from antineoplastic agents; and LC.2 Leukemia cutis can be confused with other skin lesions in leukemia patients; diagnosis requires biopsy.2,9
We analyzed clinical characteristics and prognosis of 46 patients with LC over a 17-year period. To the best of our knowledge, this is the largest study of LC patients published in the United States. A similar study by Kang et al7 analyzed 75 patients in Korea; however, the incidence of LC among different types of leukemia in the Korean population cannot be applied to Western countries. We did compare the clinical characteristics of our cohort of patients to those reported by Kang et al7 and other studies including a smaller number of patients.
In this study, the male to female ratio was 1.3 to 1 compared to the 2:1 ratio reported by Kang et al.7 The mean age of leukemia diagnosis among our patients was 58 years, which is notably older than the mean age previously reported.7 In this cohort, 4 patients (8.7%) were 34 years or younger, including 1 infant aged 8.5 months; 24 (52.2%) were aged 35 to 64 years; and 18 (39.1%) were 65 years and older.
Consistent with other studies,2,5,7 the most common type of leukemia in patients who developed LC was AML (80%). Among AML patients, the mean age at AML diagnosis (59.8 years) was notably younger than the reported US average age of patients who had a diagnosis of AML (68 years).10 Gender breakdown was slightly different than US statistics: 63% of AML patients in our group were male, whereas AML is only slightly more common among men in the United States.10
Clinically, skin lesions observed most commonly were (in decreasing order) papules, nodules, macules, plaques, and ulcers. Papules (38%) were the most common lesion overall in our study, which differed from the Kang et al7 report in which nodules were the most common. Nodules (31%) were the second most common LC morphology among our patients. Among AML patients, papules were seen in 56% of patients (18/32); nodules were seen in 44% (14/32). The extremities (when combined together) were the most common location of LC lesions (46% [arms, 24%; legs, 22%]); the trunk was the second most common body region (31%). Our study did not find a difference among most common LC anatomic sites compared to other studies.5,7 Less common sites in our cohort included the head, scalp/ears, neck, hands, mucosa, and feet. All body sites were represented, including ocular and oral mucosa and groin, a finding that underscores the importance of complete and comprehensive skin examinations in this patient population. The terms erythematous and violaceous were used to describe the color of most lesions (88%), which commonly presented as multiple lesions (84%) and often were asymptomatic (84%).
It has been reported that, first, in most cases of LC, the condition develops in patients who have already been given a diagnosis of leukemia and, second, simultaneous manifestation of systemic leukemia and LC is less common.11,12 Leukemia cutis also can precede peripheral or bone marrow leukemia (known as aleukemic LC).1,13 Two AML patients (4.3% [2/46]) in this study met criteria for aleukemic LC because they had LC at the same time as negative bone marrow biopsy, which is consistent with a prior report that aleukemic LC can affect as many as 7% of patients.1 Our results differed slightly from prior studies in that most of our patients had LC as one of the presenting manifestations of their leukemia.3,7
Regardless of leukemia type, patients were likely to die within 1 year of LC diagnosis, on average, which is consistent with prior reports.7,11,12 However, the time between diagnosis of LC and death varied greatly among our patients (range, 12 days to 4.1 years). From 2007 to 2013, the 5-year relative survival rate overall for leukemia patients in the US population (by type) was 86.2% (CLL), 71.0% (ALL), 68.0% (CML), and 27.4% (AML).14 Compared to these national statistics, the relative survival rate in LC is poor, with patients who have AML surviving, on average, less than 8 months from time of leukemia diagnosis, whereas ALL and CLL patients survive less than 6 months.
When LC is a late presentation of B-cell CLL or when it presents as myeloid leukemia, blastic transformation (Richter syndrome), or T-cell CLL, it is occasionally associated with poor prognosis, though LC does not affect survival.15-17 In a study of the association of LC with survival in AML, 5-year survival among 62 AML patients with LC was 8.6%, shorter than 28.3% among the 186 matched patients with AML without LC.18 Similarly, the estimated 5-year survival for all patients with AML, according to Surveillance, Epidemiology, and End Results Program data (2007-2013), was 27.4%.14 Based on those results, LC might be a good prognostic indicator in patients with AML.
Conclusion
This study characterized the clinical presentation of LC, which is highly variable in appearance, symptoms, distribution, and stage of leukemia at presentation. In our study cohort, LC most commonly presented as asymptomatic erythematous or violaceous papules or nodules in older male AML patients at leukemia diagnosis. Given such wide variability, dermatologists and oncologists need to keep LC in the differential diagnosis for any new skin lesion and to have a low threshold for performing skin biopsy. Complete and thorough skin examinations should be performed on leukemia patients throughout the course of their disease to identify LC early so that treatment can be implemented in a timely fashion at initial diagnosis, first sign of relapse, or change in disease state.
Leukemia is a malignant, life-threatening neoplasm affecting the hematopoietic system. Extramedullary manifestations can occur in various organs, including skin.1 Skin findings in leukemia patients are common and varied, including pallor secondary to anemia, petechiae or ecchymoses due to thrombocytopenia, and skin manifestations of neutropenia and chemotherapy.2 When patients with leukemia develop skin lesions without leukemic infiltration, the resulting nonspecific cutaneous manifestations are known as leukemids.3 Specific cutaneous manifestations of leukemia resulting from direct invasion of leukemic cells into the epidermis, dermis, or subcutis are referred to as leukemia cutis (LC).2,3
Acute myeloid leukemia (AML) is the most common type of leukemia associated with LC, but LC also is seen in other leukemias with various frequencies.1 The lesions of LC can present anywhere on skin, though it has been reported that LC has a tendency to occur at sites of prior ongoing inflammation,2,4 most commonly the extremities, trunk, and face.2,5,6 LC lesions have a range of morphological findings and most commonly present as nodules, papules, and plaques.1,7
Most reports of LC in the literature are case reports or case series with small numbers of subjects.3,6,8 A study of LC patients (N=75) in Korea by Kang et al7 has been the only one to analyze clinical characteristics of LC since 2000.
The aim of this study was to further contribute to the knowledge of clinical characteristics of LC. Clinical patterns of 46 patients were analyzed to further characterize the presentation of LC and to compare our results with those in the literature.
Methods
We conducted a single-institution retrospective review of medical records of patients with LC diagnosed in the Department of Dermatology at Wake Forest School of Medicine (Winston-Salem, North Carolina) over a 17-year period (2001-2017). The study protocol was approved by the institutional review board of Wake Forest University School of Medicine (IRB No. 00054474). Patients had a leukemia diagnosis established by bone marrow biopsy. Patients were included in this analysis if they had ongoing active leukemia and a skin biopsy consistent with LC. Patients of all sexes and ages were included in the cohort. Patients were excluded if they presented only with nonspecific cutaneous lesions associated with leukemia (leukemids). After removing duplicate records from a total of 60 patients initially identified, 46 unique patients were included in this study.
Results
Demographics
Fifty-six percent (26/46) of patients were male. The average age at diagnosis of leukemia was 58 years (range, 8.5 months–84 years). Eighty-five percent of patients were white (39/46), 11% were black (5/46), 2% were Hispanic (1/46), and 2% were of unknown ethnicity (1/46).
Eighty percent (37/46) of patients with LC had AML; 3 of these patients had a prior diagnosis of chronic myeloid leukemia (CML) and 2 had myelodysplastic syndrome (MDS) that did not develop LC until after they had transitioned to AML. Other subtypes of leukemia in this patient population included acute lymphoblastic leukemia (ALL)(n=2), plasma cell leukemia (PCL)(n=2), undifferentiated leukemia (n=2), chronic lymphocytic leukemia (CLL)(n=1), myelodysplastic syndrome (n=1), and Burkitt-type leukemia (n=1).
Distribution and Morphology of LC Lesions
The clinical appearance of LC was widely variable in morphology and anatomic location (Table 1 and Figure). Eighty-four percent of LC occurrences involved more than one lesion (n=32); 14% were a solitary lesion (n=6). For the 2 patients who had 2 separate episodes of LC, the initial presentation of LC was multiple lesions; recurrent LC at relapse presented as a solitary lesion in both cases. Most LC lesions (77% [67/87]) occurred on the trunk or extremities; 23% (20/87) of LC lesions occurred on less common sites, such as the groin, face, hands, feet, and mucosa. Papules (38% [22/58]) and nodules (31% [18/58]) were the most common morphology; macules, plaques, and ulcers were observed less frequently. Clinical descriptions of LC lesions varied widely, with the most common descriptive characteristics being erythematous (57% [20/35]), violaceous (31% [11/35]), and asymptomatic (84% [32/38]). Rare descriptors included flesh colored, hyperpigmented, tender, pruritic, edema, crusting, and confluent erythematous.
Interval Between Leukemia Diagnosis and LC Diagnosis
Approximately 59% (n=27) of patients had LC as a presenting finding of their leukemia (Table 2). Twenty-two percent (n=10) developed LC at the time of leukemia relapse; 20% (n=9) developed LC during consolidation or salvage chemotherapy. Two AML patients had recurrent episodes of LC both at initial presentation of leukemia and when AML relapsed. Two other AML patients received a diagnosis of LC at the same time as a negative concurrent bone marrow biopsy (ie, aleukemic LC). Mean duration between diagnosis of leukemia and diagnosis of LC was 0.4 months (CLL), 1.0 month (ALL), 4.7 months (AML), and 7.15 months (PCL). In cases of MDS and CML transformation to AML, the interval was 6.5 and 4.9 months, respectively.
Interval Between LC Diagnosis and Death
As a whole, 17% (n=8) of patients were living at the time this article was written (eTable). Of patients who are still living, 10.9% (n=5) have AML. Looking at the cohort of patients with AML and LC, average age at AML diagnosis was 59.8 years. Average time from diagnosis of leukemia to death was 17.3 months (range, 0.6–49.6 months) for AML; 17.0 months (range, 10.0–24.0 months) for CML transformation to AML; 15.0 months (range, 12.0–18.0 months) for PCL; 14.75 months (range, 11.0–18.5 months) for undifferentiated leukemia; and 8.95 months (range, 4.2–13.7 months) for MDS transformation to AML. The interval between leukemia diagnosis and death was notably shorter for the CLL patient (4.0 months) and the deceased ALL patient (2.4 months). Mean duration between LC diagnosis and death was 11.7 months (AML), 11.2 months (undifferentiated leukemia), 9.9 months (CML transformation to AML), 2.75 months (PCL), and 2.4 months (MDS transformation to AML). The shortest intervals between LC diagnosis and death were seen in CLL (0.5 months) and ALL (0.4 months).
Discussion
Cutaneous manifestations are not uncommon in leukemia patients and can have a number of causes, including paraneoplastic cutaneous manifestations, such as pyoderma gangrenosum and Sweet syndrome; infection; cutaneous toxicities from antineoplastic agents; and LC.2 Leukemia cutis can be confused with other skin lesions in leukemia patients; diagnosis requires biopsy.2,9
We analyzed clinical characteristics and prognosis of 46 patients with LC over a 17-year period. To the best of our knowledge, this is the largest study of LC patients published in the United States. A similar study by Kang et al7 analyzed 75 patients in Korea; however, the incidence of LC among different types of leukemia in the Korean population cannot be applied to Western countries. We did compare the clinical characteristics of our cohort of patients to those reported by Kang et al7 and other studies including a smaller number of patients.
In this study, the male to female ratio was 1.3 to 1 compared to the 2:1 ratio reported by Kang et al.7 The mean age of leukemia diagnosis among our patients was 58 years, which is notably older than the mean age previously reported.7 In this cohort, 4 patients (8.7%) were 34 years or younger, including 1 infant aged 8.5 months; 24 (52.2%) were aged 35 to 64 years; and 18 (39.1%) were 65 years and older.
Consistent with other studies,2,5,7 the most common type of leukemia in patients who developed LC was AML (80%). Among AML patients, the mean age at AML diagnosis (59.8 years) was notably younger than the reported US average age of patients who had a diagnosis of AML (68 years).10 Gender breakdown was slightly different than US statistics: 63% of AML patients in our group were male, whereas AML is only slightly more common among men in the United States.10
Clinically, skin lesions observed most commonly were (in decreasing order) papules, nodules, macules, plaques, and ulcers. Papules (38%) were the most common lesion overall in our study, which differed from the Kang et al7 report in which nodules were the most common. Nodules (31%) were the second most common LC morphology among our patients. Among AML patients, papules were seen in 56% of patients (18/32); nodules were seen in 44% (14/32). The extremities (when combined together) were the most common location of LC lesions (46% [arms, 24%; legs, 22%]); the trunk was the second most common body region (31%). Our study did not find a difference among most common LC anatomic sites compared to other studies.5,7 Less common sites in our cohort included the head, scalp/ears, neck, hands, mucosa, and feet. All body sites were represented, including ocular and oral mucosa and groin, a finding that underscores the importance of complete and comprehensive skin examinations in this patient population. The terms erythematous and violaceous were used to describe the color of most lesions (88%), which commonly presented as multiple lesions (84%) and often were asymptomatic (84%).
It has been reported that, first, in most cases of LC, the condition develops in patients who have already been given a diagnosis of leukemia and, second, simultaneous manifestation of systemic leukemia and LC is less common.11,12 Leukemia cutis also can precede peripheral or bone marrow leukemia (known as aleukemic LC).1,13 Two AML patients (4.3% [2/46]) in this study met criteria for aleukemic LC because they had LC at the same time as negative bone marrow biopsy, which is consistent with a prior report that aleukemic LC can affect as many as 7% of patients.1 Our results differed slightly from prior studies in that most of our patients had LC as one of the presenting manifestations of their leukemia.3,7
Regardless of leukemia type, patients were likely to die within 1 year of LC diagnosis, on average, which is consistent with prior reports.7,11,12 However, the time between diagnosis of LC and death varied greatly among our patients (range, 12 days to 4.1 years). From 2007 to 2013, the 5-year relative survival rate overall for leukemia patients in the US population (by type) was 86.2% (CLL), 71.0% (ALL), 68.0% (CML), and 27.4% (AML).14 Compared to these national statistics, the relative survival rate in LC is poor, with patients who have AML surviving, on average, less than 8 months from time of leukemia diagnosis, whereas ALL and CLL patients survive less than 6 months.
When LC is a late presentation of B-cell CLL or when it presents as myeloid leukemia, blastic transformation (Richter syndrome), or T-cell CLL, it is occasionally associated with poor prognosis, though LC does not affect survival.15-17 In a study of the association of LC with survival in AML, 5-year survival among 62 AML patients with LC was 8.6%, shorter than 28.3% among the 186 matched patients with AML without LC.18 Similarly, the estimated 5-year survival for all patients with AML, according to Surveillance, Epidemiology, and End Results Program data (2007-2013), was 27.4%.14 Based on those results, LC might be a good prognostic indicator in patients with AML.
Conclusion
This study characterized the clinical presentation of LC, which is highly variable in appearance, symptoms, distribution, and stage of leukemia at presentation. In our study cohort, LC most commonly presented as asymptomatic erythematous or violaceous papules or nodules in older male AML patients at leukemia diagnosis. Given such wide variability, dermatologists and oncologists need to keep LC in the differential diagnosis for any new skin lesion and to have a low threshold for performing skin biopsy. Complete and thorough skin examinations should be performed on leukemia patients throughout the course of their disease to identify LC early so that treatment can be implemented in a timely fashion at initial diagnosis, first sign of relapse, or change in disease state.
- Wagner G, Fenchel K, Back W, et al. Leukemia cutis—epidemiology, clinical presentation, and differential diagnoses. J Dtsch Dermatol Ges. 2012;10:27-36.
- Grunwald MR, McDonnell MH, Induru R, et al. Cutaneous manifestations in leukemia patients. Semin Oncol. 2016;43:359-365.
- Martínez-Leboráns L, Victoria-Martínez A, Torregrosa-Calatayu JL, et al. Leukemia cutis: a report of 17 cases and a review of literature. Actas Dermosifiliogr. 2016;107:e65-e69.
- Li L, Wang Y, Lian CG, et al. Clinical and pathological features of myeloid leukemia cutis. An Bras Dermatol. 2018;93:216-221.
- Paydas¸ S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Lee JI, Park HJ, Oh ST, et al. A case of leukemia cutis at the site of a prior catheter insertion. Ann Dermatol. 2009;21:193-196.
- Kang YS, Kim HS, Park HJ, et al. Clinical characteristics of 75 patients with leukemia cutis. J Korean Med Sci. 2013;28:614-619.
- Stern M, Halter J, Buser A, et al. Leukemia cutis preceding systemic relapse of acute myeloid leukemia. Int J Hematol. 2008;87:108-109.
- Patel LM, Maghari A, Schwartz RA, et al. Myeloid leukemia cutis in the setting of myelodysplastic syndrome: a crucial dermatological diagnosis. Int J Dermatol. 2012;51:383-388.
- American Cancer Society. Cancer Facts & Figures 2019. Atlanta, GA: American Cancer Society; 2019. http://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf. Accessed November 21, 2019.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
- Barzilai A, Lyakhovitsky A, Goldberg I, et al. Aleukemic monocytic leukemia cutis. Cutis. 2002;69:301-304
- Howlader N, Noone AM, Krapcho M, et al, eds. SEER cancer statistics review (CSR) 1975-2014. Bethesda, MD: National Cancer Institute; April 2017. https://seer.cancer.gov/archive/csr/1975_2014/. Accessed November 21, 2019.
- Cerroni L, Zenahlik P, Höfler G, et al. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol. 1996;20:1000-1010.
- Colburn DE, Welch MA, Giles FJ. Skin infiltration with chronic lymphocytic leukemia is consistent with a good prognosis. Hematology. 2002;7:187-188.
- Ratnam KV, Khor CJ, Su WP. Leukemia cutis. Dermatol Clin. 1994;12:419-431.
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826-832.
- Wagner G, Fenchel K, Back W, et al. Leukemia cutis—epidemiology, clinical presentation, and differential diagnoses. J Dtsch Dermatol Ges. 2012;10:27-36.
- Grunwald MR, McDonnell MH, Induru R, et al. Cutaneous manifestations in leukemia patients. Semin Oncol. 2016;43:359-365.
- Martínez-Leboráns L, Victoria-Martínez A, Torregrosa-Calatayu JL, et al. Leukemia cutis: a report of 17 cases and a review of literature. Actas Dermosifiliogr. 2016;107:e65-e69.
- Li L, Wang Y, Lian CG, et al. Clinical and pathological features of myeloid leukemia cutis. An Bras Dermatol. 2018;93:216-221.
- Paydas¸ S, Zorludemir S. Leukaemia cutis and leukaemic vasculitis. Br J Dermatol. 2000;143:773-779.
- Lee JI, Park HJ, Oh ST, et al. A case of leukemia cutis at the site of a prior catheter insertion. Ann Dermatol. 2009;21:193-196.
- Kang YS, Kim HS, Park HJ, et al. Clinical characteristics of 75 patients with leukemia cutis. J Korean Med Sci. 2013;28:614-619.
- Stern M, Halter J, Buser A, et al. Leukemia cutis preceding systemic relapse of acute myeloid leukemia. Int J Hematol. 2008;87:108-109.
- Patel LM, Maghari A, Schwartz RA, et al. Myeloid leukemia cutis in the setting of myelodysplastic syndrome: a crucial dermatological diagnosis. Int J Dermatol. 2012;51:383-388.
- American Cancer Society. Cancer Facts & Figures 2019. Atlanta, GA: American Cancer Society; 2019. http://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf. Accessed November 21, 2019.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Su WP. Clinical, histopathologic, and immunohistochemical correlations in leukemia cutis. Semin Dermatol. 1994;13:223-230.
- Barzilai A, Lyakhovitsky A, Goldberg I, et al. Aleukemic monocytic leukemia cutis. Cutis. 2002;69:301-304
- Howlader N, Noone AM, Krapcho M, et al, eds. SEER cancer statistics review (CSR) 1975-2014. Bethesda, MD: National Cancer Institute; April 2017. https://seer.cancer.gov/archive/csr/1975_2014/. Accessed November 21, 2019.
- Cerroni L, Zenahlik P, Höfler G, et al. Specific cutaneous infiltrates of B-cell chronic lymphocytic leukemia: a clinicopathologic and prognostic study of 42 patients. Am J Surg Pathol. 1996;20:1000-1010.
- Colburn DE, Welch MA, Giles FJ. Skin infiltration with chronic lymphocytic leukemia is consistent with a good prognosis. Hematology. 2002;7:187-188.
- Ratnam KV, Khor CJ, Su WP. Leukemia cutis. Dermatol Clin. 1994;12:419-431.
- Wang CX, Pusic I, Anadkat MJ. Association of leukemia cutis with survival in acute myeloid leukemia. JAMA Dermatol. 2019;155:826-832.
Practice Points
- Complete and comprehensive skin examination is important in leukemia patients, as leukemia cutis (LC) lesions can present in all body sites including ocular and oral mucosa as well as the groin.
- Given the wide variability in appearance, symptoms, distribution, and stage of leukemia at presentation, dermatologists and oncologists need to keep LC in the differential diagnosis for any new skin lesion and to have a low threshold for performing skin biopsy.
- Performing thorough skin examination on leukemia patients throughout the course of their disease may help identify LC early so that treatment can be implemented in a timely fashion at initial diagnosis, first sign of relapse, or change in disease state.
Hidradenitis Suppurativa for the Dermatologic Hospitalist
Hidradenitis suppurativa (HS) is a common chronic inflammatory skin disease characterized by purulent subcutaneous nodules, papules, abscesses, and fistula tracts that lead to scarring and fibrosis. Lesions develop primarily in the axilla, groin, and other intertriginous and hair-bearing areas.
The natural history of the disease is characterized by periods of disease flare, followed by periods of disease quiescence. Patients might have weeks or months of low disease activity but frequently develop multiple exacerbating episodes over the course of weeks or months. The condition primarily presents in adolescent and peripubescent years, continuing throughout adulthood. Some evidence suggests a bimodal disease distribution, with a second peak of incidence in middle-aged adults. Women and men are affected equally; however, the disease can be phenotypically different in men and women.
Patients frequently present in emergency and inpatient settings for evaluation because of the pain and severity of HS flares as well as associated systemic symptoms. Inpatient and emergency department (ED) care are unique opportunities for dermatologic hospitalist and dermatologic consultative services to educate other physicians about the condition and initiate aggressive treatments that are frequently necessary to control HS flares. This article aims to address best methods for treating HS in these settings.
Pathophysiology
Although the exact pathophysiology of the condition is unknown, HS is thought to begin with follicular occlusion with downstream inflammation mediating neutrophilic activity and scarring. Hyperplasia of the infundibular epithelium is observed on histology, and the resulting occlusion, contained keratin, and follicular rupture initiate robust downstream inflammation.1,2 Follicular occlusion might be initially androgen mediated3 or might occur in combination with friction4 and genetic or acquired factors involving Notch signaling. Although HS characteristically presents in areas of high apocrine density, apocrine glands are not thought to be the primary mediator of disease activity.5 IL-17, IL-23, tumor necrosis factor α, and IL-1β are implicated in the pathogenesis of HS, but it is unknown if these cytokines are the driving pathologic factor in HS or if they are merely secondary sequelae.6
Demographics and Prevalence in Hospitalized Patients
Although increasing treatment availability has brought more attention to HS, true prevalence is unknown. A prevalence of 1% has been reported in many European countries.7 Global prevalence has been more difficult to determine, with variable data suggesting a prevalence of 0.03% to 8%, depending on the population included.8 Most patients studied in a US-based claims database were aged 30 to 64 years, and the overall prevalence was 0.05%.9 Despite prevalence similar to psoriasis, utilization of high-cost emergency and inpatient admissions is notably higher among patients with HS. Recent claims data suggest that HS patients utilize the ED at a rate 3 times higher than psoriasis patients and are admitted as inpatients at a rate 5 times higher.10 Similar data suggest an associated increased cost of care for patients with HS vs other conditions, such as psoriasis, due to frequent ED and inpatient stays.11 Although HS frequently presents in the inpatient and emergency settings, there is little literature on best methods for managing patients in these settings.
Pearls for Inpatient and Emergency Evaluation and Management
Initial Evaluation
When dermatologic consultative services are asked to evaluate patients with HS, preliminary evaluation should reflect the acuity of the patient. Vital signs and toxicity should be reviewed to ensure that there is no evidence of severe infection necessitating critical or acute care.
History
History-taking should reflect assessment of the patient’s baseline disease, including date of initial onset; exacerbating factors, such as friction, smoking, pregnancy, and menses; and the current history of the patient’s flare. A history of antibiotics, immunosuppression, topical therapy, antiandrogen therapy, and vitamin A analog therapy also should be reviewed. If an initial diagnosis is made in the ED or inpatient setting, a family and personal history should focus on specific risk factors and disease associations, including inflammatory bowel disease,12 pilonidal cysts,13 polycystic ovary syndrome,14 and metabolic syndrome.15
Physical Examination
As with all dermatologic consultations, a full-body skin examination, with special attention to the axilla, inframammary skin, groin, buttocks, and perineum, should be undertaken. In addition to these common areas of disease progression, examination should focus on atypical sites for disease manifestation, including the posterior auricular scalp, skin folds in the pannus and back, and the beard area in men. Evaluation of axillary and gluteal hair should note features of folliculitis and hair removal, which can exacerbate HS. Examination also should include an investigation of cutaneous manifestations of comorbid conditions, including acanthosis nigricans, contiguous or metastatic cutaneous Crohn disease, erythema nodosum, and pilonidal cysts. Caution should be exercised when diagnosing pilonidal cysts, as isolated or evolving HS in the gluteal cleft often is misdiagnosed as a pilonidal cyst.
Laboratory Evaluation
Testing often is misleading in patients with HS, especially in the acute setting, because the condition is a chronic inflammatory process. The C-reactive protein level as well as the absolute white blood cell and neutrophil counts often are elevated, even in the absence of acute infection.16 In fact, although patients often are treated with intravenous antibiotics by inpatient and emergency teams in the setting of these 3 laboratory abnormalities, these findings often reflect disease activity, not frank infection. Fever, especially low-grade fever, also can reflect ongoing disease activity. Thrombocytosis and anemia also are anecdotally common, though these findings have not been reported specifically in the literature.
Bacterial Cultures
The role of lesional and perilesional bacterial cultures is controversial in HS. Prior studies have demonstrated that biofilm formation may be associated with the chronic inflammation seen in HS.17 However, most data to date suggest that infection is not the primary driver of HS disease flares, as demonstrated by the frequency of sterile cultures and the variable response of the disease to penicillin and related antibiotics.18
Imaging
Ultrasonography and magnetic resonance imaging can be conducted if there is concern about deeper abscesses that are not apparent on examination. When interpreted by nondermatologic practitioners, however, the findings of these modalities can result in unnecessary surgical intervention, given the concern for development of infectious abscess.19
Diagnosis
Many patients with HS experience a notable delay in time to diagnosis, living with symptoms for 7 years on average prior to being given a name for their condition.20 Often, patients seek ED care at initial presentation because lesions can present quickly and are associated with remarkable pain. Inpatient dermatologic evaluation can provide patients with definitive diagnosis, appropriate counseling that provides an overview of the natural history of the disease, lifestyle recommendations, and expedited connection to outpatient longitudinal care.
Diagnosis is made clinically by assessment for typical lesions, such as painful or tender papules, nodules, or abscesses in the axillae, inframammary region, groin, thighs, and perineal and perianal regions. Cordlike scarring often is seen in the absence of active inflammatory lesions.21 Double-headed open comedones and prominent follicular occlusion are seen in some phenotypes but are not required for diagnosis.22
Multiple scoring modalities are in use23; the Hurley staging system, initially developed for surgical staging, has become a commonly used method in the clinical setting24:
• Hurley stage I: isolated nodules or abscesses;
• Hurley stage II: widely separate lesions and sinus tracts or scarring are suggestive; and
• Hurley stage III: multiple lesions with near-diffuse involvement and formation of sinus tracts and scarring.
Other scoring modalities, such as the Hidradenitis Suppurativa Clinical Response (HiSCR), are more commonly used in the clinical trial setting and quantitatively capture lesion count improvement while the patient is being treated.25
Treatment
Evaluation in the ED might necessitate recommendations for inpatient admission. Dermatologic consultation can be helpful in providing ED physicians with context for interpretation of laboratory results and clinical findings. Specifically, dermatologic evaluation can help differentiate presentations consistent with a primary infection from a more common presentation of HS flaring and associated bacterial colonization. Indications for inpatient admission are severe pain; concern for systemic infection, including high fever or sepsis; and need for surgical intervention. Patients with severe disease who do not have a longitudinal care plan or who lack the ability to care for lesions at home also are candidates for inpatient admission, where they can receive more intensive nursing and wound care as well as outpatient logistical management.
Acute care should be aimed at treatments that work quickly and aggressively and have both anti-inflammatory and antimicrobial effects. Severe flares require aggressive initial treatment to ensure more long-term remission. Adalimumab, maintained at 40 mg/wk after a loading dose, is the mainstay of evidence-based treatment for moderate to severe HS in patients 12 years or older; however, this treatment might not be easy to initiate in the inpatient setting because of its cost and availability and the fact that it is not as fast acting as other therapies.26 For patients with severe disease flares, prednisone,27 infliximab,28 or cyclosporine29 can be used in combination with antimicrobial therapy in the inpatient setting to quickly control active flaring. Intravenous antimicrobial therapy might be necessary in severe disease and should include coverage of gram-positive30 and anaerobic organisms.31
Although management of acute flares is critical, especially for hospitalized patients, initiating longitudinal treatment modalities while the patient is an inpatient will help prevent future readmissions, facilitate better outcomes, and enable longer periods of disease-free progression. Specific treatments, stratified by disease severity, are listed in the Table.
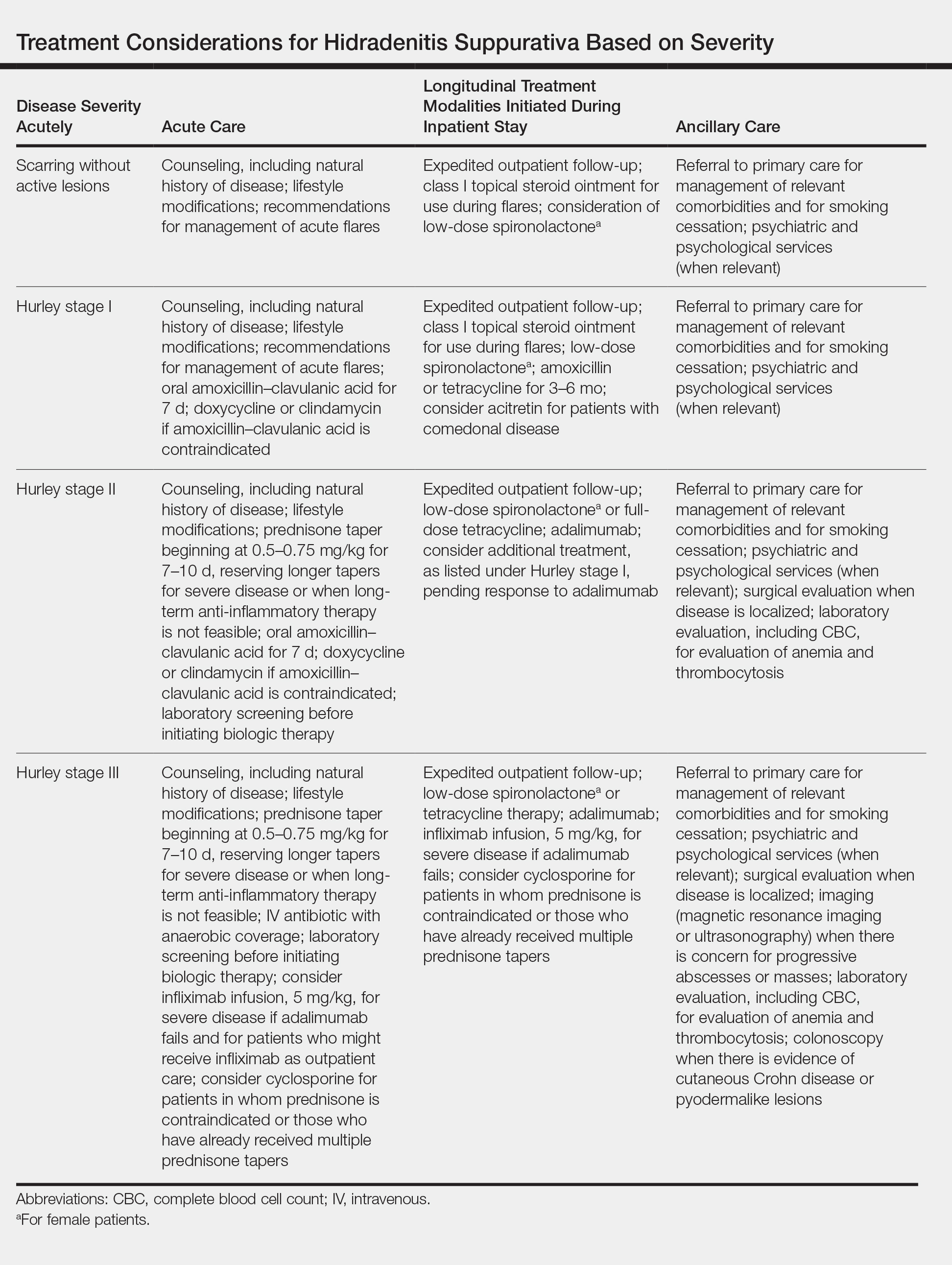
Postdischarge Lifestyle Modification
All disease management should include recommendations for lifestyle modification, including counseling on terminal hair removal (ie, avoid shaving, plucking, and waxing) and recommendations for daily and weekly decolonization with chlorhexidine or other antimicrobial soap, a weekly vinegar bath, and antiperspirant use in the groin and axilla. Avoiding tight clothes and humidity might also be helpful.
Other beneficial postdischarge strategies include smoking cessation and weight loss, which often are beneficial but difficult for many patients to achieve on their own; connecting patients with a primary care provider, which can facilitate better long-term outcomes; informing patients of the natural history of the disease and providing strategies for them to implement for acute flares to help avoid readmission and ED visits; and writing a “pill-in-pocket” prescription for a course of an antibiotic that provides good staphylococcal and anaerobic coverage, which can be helpful for patients who are prone to infrequent flares.
Lastly, appropriate postdischarge maintenance therapy also can be initiated during the inpatient stay, including maintenance antibiotic therapy, spironolactone32 for female patients, and acitretin33 for comedonal-predominant patients.
Final Thoughts
Hidradenitis suppurativa is a common dermatologic condition that frequently presents in emergency and inpatient settings, given its association with painful and acutely indurated lesions that often appear concerning for infection. Elevated inflammatory markers and fever are common in HS and are not necessarily suggestive of infection. As such, while antibiotics may be part of acute management of HS, care also should address the inflammatory component of the disease. Longitudinal outpatient care coordination with a dermatologist and primary care physician is imperative for limiting ED and inpatient care utilization.
- Jemec GB, Hansen U. Histology of hidradenitis suppurativa. J Am Acad Dermatol. 1996;34:994-999.
- Prens E, Deckers I. Pathophysiology of hidradenitis suppurativa: an update. J Am Acad Dermatol. 2015;73(suppl 1):S8-S11.
- Barth JH, Kealey T. Androgen metabolism by isolated human axillary apocrine glands in hidradenitis suppurativa. Br J Dermatol. 1991;125:304-308.
- de Winter K, van der Zee HH, Prens EP. Is mechanical stress an important pathogenic factor in hidradenitis suppurativa? Exp Dermatol. 2012;21:176-177.
- Yu CC, Cook MG. Hidradenitis suppurativa: a disease of follicular epithelium, rather than apocrine glands. Br J Dermatol. 1990;122:763-769.
- Deckers IE, van der Zee HH, Prens EP. Epidemiology of hidradenitis suppurativa: prevalence, pathogenesis, and factors associated with the development of HS. Curr Dermatol Rep. 2014;3:54-60.
- Revuz JE, Canoui-Poitrine F, Wolkenstein P, et al. Prevalence and factors associated with hidradenitis suppurativa: Results from two case-control studies. J Am Acad Dermatol. 2008;59:596-601.
- Jemec GE, Kimball AB. Hidradenitis suppurativa: epidemiology and scope of the problem. J Am Acad Dermatol. 2015;73(suppl 1):S4-S7.
- Cosmatos I, Matcho A, Weinstein R, et al. Analysis of patient claims data to determine the prevalence of hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2013;68:412-419.
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944.
- Deckers IE, Benhadou F, Koldijk MJ, et al. Inflammatory bowel disease is associated with hidradenitis suppurativa: results from a multicenter cross-sectional study. J Am Acad Dermatol. 2017;76:49-53.
- Benhadou F, Van der Zee HH, Pascual JC, et al. Pilonidal sinus disease: an intergluteal localization of hidradenitis suppurativa/acne inversa: a cross-sectional study among 2465 patients [published online March 27, 2019]. Br J Dermatol. doi:10.1111/bjd.17927.
- Garg A, Neuren E, Strunk A. Hidradenitis suppurativa is associated with polycystic ovary syndrome: a population-based analysis in the United States. J Invest Dermatol. 2018;138:1288-1292.
- Porter ML, Kimball AB. Comorbidities of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:55-57.
- Hessam S, Sand M, Gambichler T, et al. Correlation of inflammatory serum markers with disease severity in patients with hidradenitis suppurativa (HS). J Am Acad Dermatol. 2015;73:998-1005.
- Ring HC, Bay L, Nilsson M, et al. Bacterial biofilm in chronic lesions of hidradenitis suppurativa. Br J Dermatol. 2017;176:993-1000.
- Yazdanyar S, Jemec GB. Hidradenitis suppurativa: a review of cause and treatment. Curr Opin Infect Dis. 2011;24:118-123.
- Wortsman X. Imaging of hidradenitis suppurativa. Dermatol Clin. 2016;34:59-68.
- Saunte DM, Boer J, Stratigos A, et al. Diagnostic delay in hidradenitis suppurativa is a global problem. Br J Dermatol. 2015;173:1546-1549.
- Revuz JE, Jemec GB. Diagnosing hidradenitis suppurativa. Dermatol Clin. 2016;34:1-5.
- Canoui-Poitrine F, Le Thuaut A, Revuz JE, et al. Identification of three hidradenitis suppurativa phenotypes: latent class analysis of a cross-sectional study. J Invest Dermatol. 2013;133:1506-1511.
- Porter ML, Kimball AB. Hidradenitis suppurativa scoring systems: can we choose just one? Cutis. 2017;99:156-157.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa, and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH, Jr, eds. Dermatologic Surgery: Principles and Practice. New York, NY: Marcel Dekker, Inc; 1989:732-738.
- Kimball AB, Sobell JM, Zouboulis CC, et al. HiSCR (Hidradenitis Suppurativa Clinical Response): a novel clinical endpoint to evaluate therapeutic outcomes in patients with hidradenitis suppurativa from the placebo-controlled portion of a phase 2 adalimumab study. J Eur Acad Dermatol Venereol. 2016;30:989-994.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Wong D, Walsh S, Alhusayen R. Low-dose systemic corticosteroid treatment for recalcitrant hidradenitis suppurativa. J Am Acad Dermatol. 2016;75:1059-1062.
- Sullivan TP, Welsh E, Kerdel FA. Infliximab for hidradenitis suppurativa. Br J Dermatol. 2003;149:1046-1049.
- Anderson MD, Zauli S, Bettoli V, et al. Cyclosporine treatment of severe hidradenitis suppurativa—a case series. J Dermatolog Treat. 2016;27:247-250.
- Ring HC, Riis Mikkelsen P, Miller IM, et al. The bacteriology of hidradenitis suppurativa: a systematic review. Exp Dermatol. 2015;24:727-731.
- Guet-Revillet H, Coignard-Biehler H, Jais JP, et al. Bacterial pathogens associated with hidradenitis suppurativa, France. Emerg Infect Dis. 2014;20:1990-1998.
- Golbari NM, Porter ML, Kimball AB. Antiandrogen therapy with spironolactone for the treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;80:114-119.
- Matusiak L, Bieniek A, Szepietowski JC. Acitretin treatment for hidradenitis suppurativa: a prospective series of 17 patients. Br J Dermatol. 2014;171:170-174.
Hidradenitis suppurativa (HS) is a common chronic inflammatory skin disease characterized by purulent subcutaneous nodules, papules, abscesses, and fistula tracts that lead to scarring and fibrosis. Lesions develop primarily in the axilla, groin, and other intertriginous and hair-bearing areas.
The natural history of the disease is characterized by periods of disease flare, followed by periods of disease quiescence. Patients might have weeks or months of low disease activity but frequently develop multiple exacerbating episodes over the course of weeks or months. The condition primarily presents in adolescent and peripubescent years, continuing throughout adulthood. Some evidence suggests a bimodal disease distribution, with a second peak of incidence in middle-aged adults. Women and men are affected equally; however, the disease can be phenotypically different in men and women.
Patients frequently present in emergency and inpatient settings for evaluation because of the pain and severity of HS flares as well as associated systemic symptoms. Inpatient and emergency department (ED) care are unique opportunities for dermatologic hospitalist and dermatologic consultative services to educate other physicians about the condition and initiate aggressive treatments that are frequently necessary to control HS flares. This article aims to address best methods for treating HS in these settings.
Pathophysiology
Although the exact pathophysiology of the condition is unknown, HS is thought to begin with follicular occlusion with downstream inflammation mediating neutrophilic activity and scarring. Hyperplasia of the infundibular epithelium is observed on histology, and the resulting occlusion, contained keratin, and follicular rupture initiate robust downstream inflammation.1,2 Follicular occlusion might be initially androgen mediated3 or might occur in combination with friction4 and genetic or acquired factors involving Notch signaling. Although HS characteristically presents in areas of high apocrine density, apocrine glands are not thought to be the primary mediator of disease activity.5 IL-17, IL-23, tumor necrosis factor α, and IL-1β are implicated in the pathogenesis of HS, but it is unknown if these cytokines are the driving pathologic factor in HS or if they are merely secondary sequelae.6
Demographics and Prevalence in Hospitalized Patients
Although increasing treatment availability has brought more attention to HS, true prevalence is unknown. A prevalence of 1% has been reported in many European countries.7 Global prevalence has been more difficult to determine, with variable data suggesting a prevalence of 0.03% to 8%, depending on the population included.8 Most patients studied in a US-based claims database were aged 30 to 64 years, and the overall prevalence was 0.05%.9 Despite prevalence similar to psoriasis, utilization of high-cost emergency and inpatient admissions is notably higher among patients with HS. Recent claims data suggest that HS patients utilize the ED at a rate 3 times higher than psoriasis patients and are admitted as inpatients at a rate 5 times higher.10 Similar data suggest an associated increased cost of care for patients with HS vs other conditions, such as psoriasis, due to frequent ED and inpatient stays.11 Although HS frequently presents in the inpatient and emergency settings, there is little literature on best methods for managing patients in these settings.
Pearls for Inpatient and Emergency Evaluation and Management
Initial Evaluation
When dermatologic consultative services are asked to evaluate patients with HS, preliminary evaluation should reflect the acuity of the patient. Vital signs and toxicity should be reviewed to ensure that there is no evidence of severe infection necessitating critical or acute care.
History
History-taking should reflect assessment of the patient’s baseline disease, including date of initial onset; exacerbating factors, such as friction, smoking, pregnancy, and menses; and the current history of the patient’s flare. A history of antibiotics, immunosuppression, topical therapy, antiandrogen therapy, and vitamin A analog therapy also should be reviewed. If an initial diagnosis is made in the ED or inpatient setting, a family and personal history should focus on specific risk factors and disease associations, including inflammatory bowel disease,12 pilonidal cysts,13 polycystic ovary syndrome,14 and metabolic syndrome.15
Physical Examination
As with all dermatologic consultations, a full-body skin examination, with special attention to the axilla, inframammary skin, groin, buttocks, and perineum, should be undertaken. In addition to these common areas of disease progression, examination should focus on atypical sites for disease manifestation, including the posterior auricular scalp, skin folds in the pannus and back, and the beard area in men. Evaluation of axillary and gluteal hair should note features of folliculitis and hair removal, which can exacerbate HS. Examination also should include an investigation of cutaneous manifestations of comorbid conditions, including acanthosis nigricans, contiguous or metastatic cutaneous Crohn disease, erythema nodosum, and pilonidal cysts. Caution should be exercised when diagnosing pilonidal cysts, as isolated or evolving HS in the gluteal cleft often is misdiagnosed as a pilonidal cyst.
Laboratory Evaluation
Testing often is misleading in patients with HS, especially in the acute setting, because the condition is a chronic inflammatory process. The C-reactive protein level as well as the absolute white blood cell and neutrophil counts often are elevated, even in the absence of acute infection.16 In fact, although patients often are treated with intravenous antibiotics by inpatient and emergency teams in the setting of these 3 laboratory abnormalities, these findings often reflect disease activity, not frank infection. Fever, especially low-grade fever, also can reflect ongoing disease activity. Thrombocytosis and anemia also are anecdotally common, though these findings have not been reported specifically in the literature.
Bacterial Cultures
The role of lesional and perilesional bacterial cultures is controversial in HS. Prior studies have demonstrated that biofilm formation may be associated with the chronic inflammation seen in HS.17 However, most data to date suggest that infection is not the primary driver of HS disease flares, as demonstrated by the frequency of sterile cultures and the variable response of the disease to penicillin and related antibiotics.18
Imaging
Ultrasonography and magnetic resonance imaging can be conducted if there is concern about deeper abscesses that are not apparent on examination. When interpreted by nondermatologic practitioners, however, the findings of these modalities can result in unnecessary surgical intervention, given the concern for development of infectious abscess.19
Diagnosis
Many patients with HS experience a notable delay in time to diagnosis, living with symptoms for 7 years on average prior to being given a name for their condition.20 Often, patients seek ED care at initial presentation because lesions can present quickly and are associated with remarkable pain. Inpatient dermatologic evaluation can provide patients with definitive diagnosis, appropriate counseling that provides an overview of the natural history of the disease, lifestyle recommendations, and expedited connection to outpatient longitudinal care.
Diagnosis is made clinically by assessment for typical lesions, such as painful or tender papules, nodules, or abscesses in the axillae, inframammary region, groin, thighs, and perineal and perianal regions. Cordlike scarring often is seen in the absence of active inflammatory lesions.21 Double-headed open comedones and prominent follicular occlusion are seen in some phenotypes but are not required for diagnosis.22
Multiple scoring modalities are in use23; the Hurley staging system, initially developed for surgical staging, has become a commonly used method in the clinical setting24:
• Hurley stage I: isolated nodules or abscesses;
• Hurley stage II: widely separate lesions and sinus tracts or scarring are suggestive; and
• Hurley stage III: multiple lesions with near-diffuse involvement and formation of sinus tracts and scarring.
Other scoring modalities, such as the Hidradenitis Suppurativa Clinical Response (HiSCR), are more commonly used in the clinical trial setting and quantitatively capture lesion count improvement while the patient is being treated.25
Treatment
Evaluation in the ED might necessitate recommendations for inpatient admission. Dermatologic consultation can be helpful in providing ED physicians with context for interpretation of laboratory results and clinical findings. Specifically, dermatologic evaluation can help differentiate presentations consistent with a primary infection from a more common presentation of HS flaring and associated bacterial colonization. Indications for inpatient admission are severe pain; concern for systemic infection, including high fever or sepsis; and need for surgical intervention. Patients with severe disease who do not have a longitudinal care plan or who lack the ability to care for lesions at home also are candidates for inpatient admission, where they can receive more intensive nursing and wound care as well as outpatient logistical management.
Acute care should be aimed at treatments that work quickly and aggressively and have both anti-inflammatory and antimicrobial effects. Severe flares require aggressive initial treatment to ensure more long-term remission. Adalimumab, maintained at 40 mg/wk after a loading dose, is the mainstay of evidence-based treatment for moderate to severe HS in patients 12 years or older; however, this treatment might not be easy to initiate in the inpatient setting because of its cost and availability and the fact that it is not as fast acting as other therapies.26 For patients with severe disease flares, prednisone,27 infliximab,28 or cyclosporine29 can be used in combination with antimicrobial therapy in the inpatient setting to quickly control active flaring. Intravenous antimicrobial therapy might be necessary in severe disease and should include coverage of gram-positive30 and anaerobic organisms.31
Although management of acute flares is critical, especially for hospitalized patients, initiating longitudinal treatment modalities while the patient is an inpatient will help prevent future readmissions, facilitate better outcomes, and enable longer periods of disease-free progression. Specific treatments, stratified by disease severity, are listed in the Table.
Postdischarge Lifestyle Modification
All disease management should include recommendations for lifestyle modification, including counseling on terminal hair removal (ie, avoid shaving, plucking, and waxing) and recommendations for daily and weekly decolonization with chlorhexidine or other antimicrobial soap, a weekly vinegar bath, and antiperspirant use in the groin and axilla. Avoiding tight clothes and humidity might also be helpful.
Other beneficial postdischarge strategies include smoking cessation and weight loss, which often are beneficial but difficult for many patients to achieve on their own; connecting patients with a primary care provider, which can facilitate better long-term outcomes; informing patients of the natural history of the disease and providing strategies for them to implement for acute flares to help avoid readmission and ED visits; and writing a “pill-in-pocket” prescription for a course of an antibiotic that provides good staphylococcal and anaerobic coverage, which can be helpful for patients who are prone to infrequent flares.
Lastly, appropriate postdischarge maintenance therapy also can be initiated during the inpatient stay, including maintenance antibiotic therapy, spironolactone32 for female patients, and acitretin33 for comedonal-predominant patients.
Final Thoughts
Hidradenitis suppurativa is a common dermatologic condition that frequently presents in emergency and inpatient settings, given its association with painful and acutely indurated lesions that often appear concerning for infection. Elevated inflammatory markers and fever are common in HS and are not necessarily suggestive of infection. As such, while antibiotics may be part of acute management of HS, care also should address the inflammatory component of the disease. Longitudinal outpatient care coordination with a dermatologist and primary care physician is imperative for limiting ED and inpatient care utilization.
Hidradenitis suppurativa (HS) is a common chronic inflammatory skin disease characterized by purulent subcutaneous nodules, papules, abscesses, and fistula tracts that lead to scarring and fibrosis. Lesions develop primarily in the axilla, groin, and other intertriginous and hair-bearing areas.
The natural history of the disease is characterized by periods of disease flare, followed by periods of disease quiescence. Patients might have weeks or months of low disease activity but frequently develop multiple exacerbating episodes over the course of weeks or months. The condition primarily presents in adolescent and peripubescent years, continuing throughout adulthood. Some evidence suggests a bimodal disease distribution, with a second peak of incidence in middle-aged adults. Women and men are affected equally; however, the disease can be phenotypically different in men and women.
Patients frequently present in emergency and inpatient settings for evaluation because of the pain and severity of HS flares as well as associated systemic symptoms. Inpatient and emergency department (ED) care are unique opportunities for dermatologic hospitalist and dermatologic consultative services to educate other physicians about the condition and initiate aggressive treatments that are frequently necessary to control HS flares. This article aims to address best methods for treating HS in these settings.
Pathophysiology
Although the exact pathophysiology of the condition is unknown, HS is thought to begin with follicular occlusion with downstream inflammation mediating neutrophilic activity and scarring. Hyperplasia of the infundibular epithelium is observed on histology, and the resulting occlusion, contained keratin, and follicular rupture initiate robust downstream inflammation.1,2 Follicular occlusion might be initially androgen mediated3 or might occur in combination with friction4 and genetic or acquired factors involving Notch signaling. Although HS characteristically presents in areas of high apocrine density, apocrine glands are not thought to be the primary mediator of disease activity.5 IL-17, IL-23, tumor necrosis factor α, and IL-1β are implicated in the pathogenesis of HS, but it is unknown if these cytokines are the driving pathologic factor in HS or if they are merely secondary sequelae.6
Demographics and Prevalence in Hospitalized Patients
Although increasing treatment availability has brought more attention to HS, true prevalence is unknown. A prevalence of 1% has been reported in many European countries.7 Global prevalence has been more difficult to determine, with variable data suggesting a prevalence of 0.03% to 8%, depending on the population included.8 Most patients studied in a US-based claims database were aged 30 to 64 years, and the overall prevalence was 0.05%.9 Despite prevalence similar to psoriasis, utilization of high-cost emergency and inpatient admissions is notably higher among patients with HS. Recent claims data suggest that HS patients utilize the ED at a rate 3 times higher than psoriasis patients and are admitted as inpatients at a rate 5 times higher.10 Similar data suggest an associated increased cost of care for patients with HS vs other conditions, such as psoriasis, due to frequent ED and inpatient stays.11 Although HS frequently presents in the inpatient and emergency settings, there is little literature on best methods for managing patients in these settings.
Pearls for Inpatient and Emergency Evaluation and Management
Initial Evaluation
When dermatologic consultative services are asked to evaluate patients with HS, preliminary evaluation should reflect the acuity of the patient. Vital signs and toxicity should be reviewed to ensure that there is no evidence of severe infection necessitating critical or acute care.
History
History-taking should reflect assessment of the patient’s baseline disease, including date of initial onset; exacerbating factors, such as friction, smoking, pregnancy, and menses; and the current history of the patient’s flare. A history of antibiotics, immunosuppression, topical therapy, antiandrogen therapy, and vitamin A analog therapy also should be reviewed. If an initial diagnosis is made in the ED or inpatient setting, a family and personal history should focus on specific risk factors and disease associations, including inflammatory bowel disease,12 pilonidal cysts,13 polycystic ovary syndrome,14 and metabolic syndrome.15
Physical Examination
As with all dermatologic consultations, a full-body skin examination, with special attention to the axilla, inframammary skin, groin, buttocks, and perineum, should be undertaken. In addition to these common areas of disease progression, examination should focus on atypical sites for disease manifestation, including the posterior auricular scalp, skin folds in the pannus and back, and the beard area in men. Evaluation of axillary and gluteal hair should note features of folliculitis and hair removal, which can exacerbate HS. Examination also should include an investigation of cutaneous manifestations of comorbid conditions, including acanthosis nigricans, contiguous or metastatic cutaneous Crohn disease, erythema nodosum, and pilonidal cysts. Caution should be exercised when diagnosing pilonidal cysts, as isolated or evolving HS in the gluteal cleft often is misdiagnosed as a pilonidal cyst.
Laboratory Evaluation
Testing often is misleading in patients with HS, especially in the acute setting, because the condition is a chronic inflammatory process. The C-reactive protein level as well as the absolute white blood cell and neutrophil counts often are elevated, even in the absence of acute infection.16 In fact, although patients often are treated with intravenous antibiotics by inpatient and emergency teams in the setting of these 3 laboratory abnormalities, these findings often reflect disease activity, not frank infection. Fever, especially low-grade fever, also can reflect ongoing disease activity. Thrombocytosis and anemia also are anecdotally common, though these findings have not been reported specifically in the literature.
Bacterial Cultures
The role of lesional and perilesional bacterial cultures is controversial in HS. Prior studies have demonstrated that biofilm formation may be associated with the chronic inflammation seen in HS.17 However, most data to date suggest that infection is not the primary driver of HS disease flares, as demonstrated by the frequency of sterile cultures and the variable response of the disease to penicillin and related antibiotics.18
Imaging
Ultrasonography and magnetic resonance imaging can be conducted if there is concern about deeper abscesses that are not apparent on examination. When interpreted by nondermatologic practitioners, however, the findings of these modalities can result in unnecessary surgical intervention, given the concern for development of infectious abscess.19
Diagnosis
Many patients with HS experience a notable delay in time to diagnosis, living with symptoms for 7 years on average prior to being given a name for their condition.20 Often, patients seek ED care at initial presentation because lesions can present quickly and are associated with remarkable pain. Inpatient dermatologic evaluation can provide patients with definitive diagnosis, appropriate counseling that provides an overview of the natural history of the disease, lifestyle recommendations, and expedited connection to outpatient longitudinal care.
Diagnosis is made clinically by assessment for typical lesions, such as painful or tender papules, nodules, or abscesses in the axillae, inframammary region, groin, thighs, and perineal and perianal regions. Cordlike scarring often is seen in the absence of active inflammatory lesions.21 Double-headed open comedones and prominent follicular occlusion are seen in some phenotypes but are not required for diagnosis.22
Multiple scoring modalities are in use23; the Hurley staging system, initially developed for surgical staging, has become a commonly used method in the clinical setting24:
• Hurley stage I: isolated nodules or abscesses;
• Hurley stage II: widely separate lesions and sinus tracts or scarring are suggestive; and
• Hurley stage III: multiple lesions with near-diffuse involvement and formation of sinus tracts and scarring.
Other scoring modalities, such as the Hidradenitis Suppurativa Clinical Response (HiSCR), are more commonly used in the clinical trial setting and quantitatively capture lesion count improvement while the patient is being treated.25
Treatment
Evaluation in the ED might necessitate recommendations for inpatient admission. Dermatologic consultation can be helpful in providing ED physicians with context for interpretation of laboratory results and clinical findings. Specifically, dermatologic evaluation can help differentiate presentations consistent with a primary infection from a more common presentation of HS flaring and associated bacterial colonization. Indications for inpatient admission are severe pain; concern for systemic infection, including high fever or sepsis; and need for surgical intervention. Patients with severe disease who do not have a longitudinal care plan or who lack the ability to care for lesions at home also are candidates for inpatient admission, where they can receive more intensive nursing and wound care as well as outpatient logistical management.
Acute care should be aimed at treatments that work quickly and aggressively and have both anti-inflammatory and antimicrobial effects. Severe flares require aggressive initial treatment to ensure more long-term remission. Adalimumab, maintained at 40 mg/wk after a loading dose, is the mainstay of evidence-based treatment for moderate to severe HS in patients 12 years or older; however, this treatment might not be easy to initiate in the inpatient setting because of its cost and availability and the fact that it is not as fast acting as other therapies.26 For patients with severe disease flares, prednisone,27 infliximab,28 or cyclosporine29 can be used in combination with antimicrobial therapy in the inpatient setting to quickly control active flaring. Intravenous antimicrobial therapy might be necessary in severe disease and should include coverage of gram-positive30 and anaerobic organisms.31
Although management of acute flares is critical, especially for hospitalized patients, initiating longitudinal treatment modalities while the patient is an inpatient will help prevent future readmissions, facilitate better outcomes, and enable longer periods of disease-free progression. Specific treatments, stratified by disease severity, are listed in the Table.
Postdischarge Lifestyle Modification
All disease management should include recommendations for lifestyle modification, including counseling on terminal hair removal (ie, avoid shaving, plucking, and waxing) and recommendations for daily and weekly decolonization with chlorhexidine or other antimicrobial soap, a weekly vinegar bath, and antiperspirant use in the groin and axilla. Avoiding tight clothes and humidity might also be helpful.
Other beneficial postdischarge strategies include smoking cessation and weight loss, which often are beneficial but difficult for many patients to achieve on their own; connecting patients with a primary care provider, which can facilitate better long-term outcomes; informing patients of the natural history of the disease and providing strategies for them to implement for acute flares to help avoid readmission and ED visits; and writing a “pill-in-pocket” prescription for a course of an antibiotic that provides good staphylococcal and anaerobic coverage, which can be helpful for patients who are prone to infrequent flares.
Lastly, appropriate postdischarge maintenance therapy also can be initiated during the inpatient stay, including maintenance antibiotic therapy, spironolactone32 for female patients, and acitretin33 for comedonal-predominant patients.
Final Thoughts
Hidradenitis suppurativa is a common dermatologic condition that frequently presents in emergency and inpatient settings, given its association with painful and acutely indurated lesions that often appear concerning for infection. Elevated inflammatory markers and fever are common in HS and are not necessarily suggestive of infection. As such, while antibiotics may be part of acute management of HS, care also should address the inflammatory component of the disease. Longitudinal outpatient care coordination with a dermatologist and primary care physician is imperative for limiting ED and inpatient care utilization.
- Jemec GB, Hansen U. Histology of hidradenitis suppurativa. J Am Acad Dermatol. 1996;34:994-999.
- Prens E, Deckers I. Pathophysiology of hidradenitis suppurativa: an update. J Am Acad Dermatol. 2015;73(suppl 1):S8-S11.
- Barth JH, Kealey T. Androgen metabolism by isolated human axillary apocrine glands in hidradenitis suppurativa. Br J Dermatol. 1991;125:304-308.
- de Winter K, van der Zee HH, Prens EP. Is mechanical stress an important pathogenic factor in hidradenitis suppurativa? Exp Dermatol. 2012;21:176-177.
- Yu CC, Cook MG. Hidradenitis suppurativa: a disease of follicular epithelium, rather than apocrine glands. Br J Dermatol. 1990;122:763-769.
- Deckers IE, van der Zee HH, Prens EP. Epidemiology of hidradenitis suppurativa: prevalence, pathogenesis, and factors associated with the development of HS. Curr Dermatol Rep. 2014;3:54-60.
- Revuz JE, Canoui-Poitrine F, Wolkenstein P, et al. Prevalence and factors associated with hidradenitis suppurativa: Results from two case-control studies. J Am Acad Dermatol. 2008;59:596-601.
- Jemec GE, Kimball AB. Hidradenitis suppurativa: epidemiology and scope of the problem. J Am Acad Dermatol. 2015;73(suppl 1):S4-S7.
- Cosmatos I, Matcho A, Weinstein R, et al. Analysis of patient claims data to determine the prevalence of hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2013;68:412-419.
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944.
- Deckers IE, Benhadou F, Koldijk MJ, et al. Inflammatory bowel disease is associated with hidradenitis suppurativa: results from a multicenter cross-sectional study. J Am Acad Dermatol. 2017;76:49-53.
- Benhadou F, Van der Zee HH, Pascual JC, et al. Pilonidal sinus disease: an intergluteal localization of hidradenitis suppurativa/acne inversa: a cross-sectional study among 2465 patients [published online March 27, 2019]. Br J Dermatol. doi:10.1111/bjd.17927.
- Garg A, Neuren E, Strunk A. Hidradenitis suppurativa is associated with polycystic ovary syndrome: a population-based analysis in the United States. J Invest Dermatol. 2018;138:1288-1292.
- Porter ML, Kimball AB. Comorbidities of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:55-57.
- Hessam S, Sand M, Gambichler T, et al. Correlation of inflammatory serum markers with disease severity in patients with hidradenitis suppurativa (HS). J Am Acad Dermatol. 2015;73:998-1005.
- Ring HC, Bay L, Nilsson M, et al. Bacterial biofilm in chronic lesions of hidradenitis suppurativa. Br J Dermatol. 2017;176:993-1000.
- Yazdanyar S, Jemec GB. Hidradenitis suppurativa: a review of cause and treatment. Curr Opin Infect Dis. 2011;24:118-123.
- Wortsman X. Imaging of hidradenitis suppurativa. Dermatol Clin. 2016;34:59-68.
- Saunte DM, Boer J, Stratigos A, et al. Diagnostic delay in hidradenitis suppurativa is a global problem. Br J Dermatol. 2015;173:1546-1549.
- Revuz JE, Jemec GB. Diagnosing hidradenitis suppurativa. Dermatol Clin. 2016;34:1-5.
- Canoui-Poitrine F, Le Thuaut A, Revuz JE, et al. Identification of three hidradenitis suppurativa phenotypes: latent class analysis of a cross-sectional study. J Invest Dermatol. 2013;133:1506-1511.
- Porter ML, Kimball AB. Hidradenitis suppurativa scoring systems: can we choose just one? Cutis. 2017;99:156-157.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa, and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH, Jr, eds. Dermatologic Surgery: Principles and Practice. New York, NY: Marcel Dekker, Inc; 1989:732-738.
- Kimball AB, Sobell JM, Zouboulis CC, et al. HiSCR (Hidradenitis Suppurativa Clinical Response): a novel clinical endpoint to evaluate therapeutic outcomes in patients with hidradenitis suppurativa from the placebo-controlled portion of a phase 2 adalimumab study. J Eur Acad Dermatol Venereol. 2016;30:989-994.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Wong D, Walsh S, Alhusayen R. Low-dose systemic corticosteroid treatment for recalcitrant hidradenitis suppurativa. J Am Acad Dermatol. 2016;75:1059-1062.
- Sullivan TP, Welsh E, Kerdel FA. Infliximab for hidradenitis suppurativa. Br J Dermatol. 2003;149:1046-1049.
- Anderson MD, Zauli S, Bettoli V, et al. Cyclosporine treatment of severe hidradenitis suppurativa—a case series. J Dermatolog Treat. 2016;27:247-250.
- Ring HC, Riis Mikkelsen P, Miller IM, et al. The bacteriology of hidradenitis suppurativa: a systematic review. Exp Dermatol. 2015;24:727-731.
- Guet-Revillet H, Coignard-Biehler H, Jais JP, et al. Bacterial pathogens associated with hidradenitis suppurativa, France. Emerg Infect Dis. 2014;20:1990-1998.
- Golbari NM, Porter ML, Kimball AB. Antiandrogen therapy with spironolactone for the treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;80:114-119.
- Matusiak L, Bieniek A, Szepietowski JC. Acitretin treatment for hidradenitis suppurativa: a prospective series of 17 patients. Br J Dermatol. 2014;171:170-174.
- Jemec GB, Hansen U. Histology of hidradenitis suppurativa. J Am Acad Dermatol. 1996;34:994-999.
- Prens E, Deckers I. Pathophysiology of hidradenitis suppurativa: an update. J Am Acad Dermatol. 2015;73(suppl 1):S8-S11.
- Barth JH, Kealey T. Androgen metabolism by isolated human axillary apocrine glands in hidradenitis suppurativa. Br J Dermatol. 1991;125:304-308.
- de Winter K, van der Zee HH, Prens EP. Is mechanical stress an important pathogenic factor in hidradenitis suppurativa? Exp Dermatol. 2012;21:176-177.
- Yu CC, Cook MG. Hidradenitis suppurativa: a disease of follicular epithelium, rather than apocrine glands. Br J Dermatol. 1990;122:763-769.
- Deckers IE, van der Zee HH, Prens EP. Epidemiology of hidradenitis suppurativa: prevalence, pathogenesis, and factors associated with the development of HS. Curr Dermatol Rep. 2014;3:54-60.
- Revuz JE, Canoui-Poitrine F, Wolkenstein P, et al. Prevalence and factors associated with hidradenitis suppurativa: Results from two case-control studies. J Am Acad Dermatol. 2008;59:596-601.
- Jemec GE, Kimball AB. Hidradenitis suppurativa: epidemiology and scope of the problem. J Am Acad Dermatol. 2015;73(suppl 1):S4-S7.
- Cosmatos I, Matcho A, Weinstein R, et al. Analysis of patient claims data to determine the prevalence of hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2013;68:412-419.
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944.
- Deckers IE, Benhadou F, Koldijk MJ, et al. Inflammatory bowel disease is associated with hidradenitis suppurativa: results from a multicenter cross-sectional study. J Am Acad Dermatol. 2017;76:49-53.
- Benhadou F, Van der Zee HH, Pascual JC, et al. Pilonidal sinus disease: an intergluteal localization of hidradenitis suppurativa/acne inversa: a cross-sectional study among 2465 patients [published online March 27, 2019]. Br J Dermatol. doi:10.1111/bjd.17927.
- Garg A, Neuren E, Strunk A. Hidradenitis suppurativa is associated with polycystic ovary syndrome: a population-based analysis in the United States. J Invest Dermatol. 2018;138:1288-1292.
- Porter ML, Kimball AB. Comorbidities of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:55-57.
- Hessam S, Sand M, Gambichler T, et al. Correlation of inflammatory serum markers with disease severity in patients with hidradenitis suppurativa (HS). J Am Acad Dermatol. 2015;73:998-1005.
- Ring HC, Bay L, Nilsson M, et al. Bacterial biofilm in chronic lesions of hidradenitis suppurativa. Br J Dermatol. 2017;176:993-1000.
- Yazdanyar S, Jemec GB. Hidradenitis suppurativa: a review of cause and treatment. Curr Opin Infect Dis. 2011;24:118-123.
- Wortsman X. Imaging of hidradenitis suppurativa. Dermatol Clin. 2016;34:59-68.
- Saunte DM, Boer J, Stratigos A, et al. Diagnostic delay in hidradenitis suppurativa is a global problem. Br J Dermatol. 2015;173:1546-1549.
- Revuz JE, Jemec GB. Diagnosing hidradenitis suppurativa. Dermatol Clin. 2016;34:1-5.
- Canoui-Poitrine F, Le Thuaut A, Revuz JE, et al. Identification of three hidradenitis suppurativa phenotypes: latent class analysis of a cross-sectional study. J Invest Dermatol. 2013;133:1506-1511.
- Porter ML, Kimball AB. Hidradenitis suppurativa scoring systems: can we choose just one? Cutis. 2017;99:156-157.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa, and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH, Jr, eds. Dermatologic Surgery: Principles and Practice. New York, NY: Marcel Dekker, Inc; 1989:732-738.
- Kimball AB, Sobell JM, Zouboulis CC, et al. HiSCR (Hidradenitis Suppurativa Clinical Response): a novel clinical endpoint to evaluate therapeutic outcomes in patients with hidradenitis suppurativa from the placebo-controlled portion of a phase 2 adalimumab study. J Eur Acad Dermatol Venereol. 2016;30:989-994.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Wong D, Walsh S, Alhusayen R. Low-dose systemic corticosteroid treatment for recalcitrant hidradenitis suppurativa. J Am Acad Dermatol. 2016;75:1059-1062.
- Sullivan TP, Welsh E, Kerdel FA. Infliximab for hidradenitis suppurativa. Br J Dermatol. 2003;149:1046-1049.
- Anderson MD, Zauli S, Bettoli V, et al. Cyclosporine treatment of severe hidradenitis suppurativa—a case series. J Dermatolog Treat. 2016;27:247-250.
- Ring HC, Riis Mikkelsen P, Miller IM, et al. The bacteriology of hidradenitis suppurativa: a systematic review. Exp Dermatol. 2015;24:727-731.
- Guet-Revillet H, Coignard-Biehler H, Jais JP, et al. Bacterial pathogens associated with hidradenitis suppurativa, France. Emerg Infect Dis. 2014;20:1990-1998.
- Golbari NM, Porter ML, Kimball AB. Antiandrogen therapy with spironolactone for the treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;80:114-119.
- Matusiak L, Bieniek A, Szepietowski JC. Acitretin treatment for hidradenitis suppurativa: a prospective series of 17 patients. Br J Dermatol. 2014;171:170-174.
Practice Points
- Hidradenitis suppurativa (HS) is a common dermatologic condition that frequently presents in emergency and inpatient settings.
- Anemia, leukocytosis, neutrophilia, an elevated erythrocyte sedimentation rate, and an elevated C-reactive protein level are common markers of chronic inflammation in HS patients and might not signify infection.
- Acute management of HS should focus on anti-inflammatory and antibiotic regimens, with increasing severity dictating the need for more aggressive therapy.
- Longitudinal outpatient care coordination with a dermatologist and primary care physician is imperative for limiting emergency department and inpatient care utilization.

Barriers and Job Satisfaction Among Dermatology Hospitalists
Consultative dermatologists, or dermatology hospitalists (DHs), perform a critical role in the care of inpatients with skin disease, providing efficient diagnosis and management of patients with complex skin conditions as well as education of patients and trainees in the hospital setting.1 In 2013, 27% of the US population was seen by a physician for a skin disease.2 In 2014, there were nearly 650,000 hospital admissions principally for skin disease.3 Input by dermatologists facilitates accurate diagnosis and management of inpatients with skin disease,4 including a substantial number of cutaneous malignancies diagnosed in the inpatient setting.5 Several studies have highlighted the generally low level of diagnostic concordance between referring services and dermatology consultants,4,6 with dermatology consultants frequently noting diagnoses not considered by referring services,7 reinforcing the importance of having access to dermatologists in the hospital setting.
The care of skin disease in the inpatient setting has become increasingly complex. The Society for Dermatology Hospitalists (SDH) was created in 2009 to address this complexity, with the goal to “strive to develop the highest standards of clinical care of hospitalized patients with skin disease.”8 A recent survey found that 50% of DHs spend between 41 to 52 weeks per year on service.9 Despite this degree of commitment, there are considerable barriers that prevent the majority of dermatologists from efficiently providing inpatient consultative care. The inpatient and outpatient provision of dermatology care varies greatly, including the variety of ethical situations encountered and the diversity of skin conditions treated.10-12 Additionally, the transition between inpatient and outpatient care can be challenging for providers.13
The goal of this study was to evaluate the overall job satisfaction of DHs and further describe potential barriers to inpatient dermatology consultations.
Methods
An anonymous 31-question electronic survey was sent via email to all current members of the SDH from November 20 to December 10, 2018. The study was reviewed and determined to be exempt from federal human subjects regulations by the University of Washington Human Subjects Division (Seattle, Washington)(STUDY00005832).
Results
At the time of survey distribution, the SDH had 145 members, including attending-level dermatologists and resident members. Thirty-seven self-identified DHs (46% [17/37] women; 54% [20/37] men) completed the survey. The majority of respondents were junior faculty, with 46% (17/37) assistant professors, 5% (2/37) acting instructors, 32% (12/37) associate professors, and 16% (6/37) professors. All regions of the United States were represented.
Time Dedicated to Providing Inpatient Dermatology Consultations
The majority of those surveyed were satisfied or very satisfied (68% [25/37]) with the amount of time allotted for inpatient dermatology consultations, while 14% (5/37) were unsatisfied or very unsatisfied. Of those surveyed, 46% (17/37) reported that 21% to 50% of their time is dedicated to inpatient dermatology consultations. The majority (57% [21/37]) reported that their outpatient clinic efforts are reduced when providing dermatology inpatient consultations.
Regarding travel to the inpatient practice site, 60% (22/37) rated their travel time/effort as very easy, with 38% (14/37) reporting that the sites at which they provide inpatient dermatology consultations and their main outpatient clinics are the same physical location; 38% (14/37) reported travel times of less than 15 minutes between clinical practice sites.
Eighty-nine percent (33/37) of respondents said they are able to spend more time teaching trainees when providing inpatient dermatology consultations compared to their time spent in clinic. Similarly, 70% (26/37) said they are able to spend more time learning about patients and their conditions when providing inpatient dermatology consultations. Respondents also reported additional time expenditures because of inpatient dermatology consultations, primarily additional teaching requirements (49% [18/37]), additional electronic medical record training (35% [13/37]), and credentialing requirements (24% [9/37]).
Infrastructure for Providing Inpatient Dermatology Consultations
For many respondents (30% [11/37]), only 2 faculty dermatologists regularly provide inpatient dermatology consultations at their institutions. Four respondents reported having at least 5 faculty dermatologists who regularly provide inpatient dermatology consultations; excluding these, the average number of DHs was 2.42 faculty per institution.
Most respondents (57% [21/37]) reported their institutions support inpatient dermatology services by providing salary support for residents to cover services. Other methods of support included dedicated office spaces (30% [11/37]), free hospital parking while providing inpatient consultations (24% [9/37]), and administrative support (11% [4/37]).
Consultation Composition
Respondents indicated that requests for DH consultations most often come from medical services, including medical intensive care, internal medicine, and family medicine (95% [35/37]); the emergency department (95% [35/37]); surgical services (92% [34/37]); and hematology/oncology (89% [33/37]). Fewer DHs reported receiving consultation requests from pediatrics (70% [26/37]).
Many respondents (49% [18/37]) reported consulting for patients with skin disorders that they considered to be life-threatening or potentially life-threatening either very frequently (daily) or frequently (several times weekly), with only 16% (6/37) responding that they see such patients about once per month.
Compensation for Inpatient Dermatology Consultation
The most commonly reported compensation models for DHs were fixed salary plus productivity or performance incentives and fixed salary only models (49% [18/37] and 32% [12/37], respectively), with relative value unit (RVU) models and other models less frequently reported (16% [6/37] and 3% [1/37], respectively). Only 46% (17/37) of respondents were satisfied or very satisfied with their institutions’ compensation models; the remainder (54% [20/37]) were either neutral, unsatisfied, or very unsatisfied regarding their institutions’ compensation models. Overall compensation satisfaction was higher, with 60% (22/37) of DHs reporting they were satisfied or very satisfied with their salaries and 41% (15/37) reporting they were either neutral or not satisfied. The majority (60% [2/37]) of respondents felt that fixed salary plus productivity or performance incentives models would be the ideal compensation model for DHs.
Of the DHs whose compensations models were RVU based (6/37 [16%]), 67% (4/6) said they receive incentive pay upon meeting their RVU targets. No respondents reported that they were expected to generate an equivalent number of RVUs when performing inpatient consultations as compared to an outpatient session. Only 32% (12/37) of respondents reported keeping the revenue/RVUs generated by inpatient dermatology consultations; most (57% [21/37]) noted that their dermatology divisions/departments keep the revenue/RVUs, followed by university hospitals (27% [10/37]), schools of medicine (11% [4/37]), and departments of medicine (3% [1/37]). The remainder of respondents (22% [8/37]) were unsure who keeps the revenue/RVUs generated by inpatient dermatology consultations.
Most respondents (70% [26/37]) reported that the revenue (or RVU equivalent) generated by inpatient dermatology consultations does not fully support their salary for the time spent as consultants. Rather, these DHs noted sources of additional financial support, primarily the DHs themselves (69% [18/26]), followed by dermatology divisions/departments (50% [13/26]), departments of medicine (23% [6/26]), university hospitals (23% [6/26]), and schools of medicine (12% [3/26]).
Job Fulfillment Among DHs
Most respondents said they choose to provide inpatient dermatology consultations due to their interest in complex medical dermatology and their desire to work with other medical teams and specialties (92% [34/37] and 76% [28/37], respectively). Seventy percent (26/37) said they choose to provide inpatient consultations to be able to teach medical students and residents as well as to take advantage of the added opportunities to practice in a variety of settings beyond their outpatient clinics (57% [21/37]). Only 3% (1/37) of respondents reported that they provide inpatient dermatology consultations because they are “required to do so.”
Most DHs (84% [31/37]) said they feel their institutions as well as their dermatology divisions/departments value having access to inpatient dermatology services, though some did not feel this way (16% [6/37] neutral or strongly disagree). Nearly all respondents (97% [36/37]) felt they provide a critical service when performing inpatient dermatology consultations. All respondents (100%) said they found providing inpatient dermatology consultations fulfilling, and 65% (24/37) said they prefer providing inpatient dermatology consultations to spending time in clinic. Of the DHs who were surveyed, 68% (25/37) said they were satisfied with the balance of outpatient and inpatient services in their clinical practice and 30% (11/37) said they were not.
Comment
Factors such as patient care, hospital infrastructure, and procedural support have all been cited by DHs as crucial aspects of their contributions to the care of hospitalized pa
Dermatology is primarily an outpatient specialty, and our study highlighted several important challenges for providers performing inpatient dermatology consultations. A major issue is time expenditures, including additional teaching requirements, additional electronic medical record training, and credentialing requirements. Travel time to inpatient hospital sites does not appear to be one of these hindering factors; nearly 60% of respondents rated their travel time/effort as very easy, with approximately 75% of respondents’ consultation locations being either at the same physical location as their main outpatient clinic or less than 15 minutes away. Maintaining easy travel between outpatient and inpatient settings is important to the success of the DH.
Our data suggest that compensation of DHs is a potential limitation to providing inpatient dermatology care. Our survey reinforced that providers who do inpatient dermatology consultations generally do not generate the revenue necessary to cover these efforts. More than 40% of DH respondents said they either feel neutral about or unsatisfied with their overall salary, and more than half said they feel similarly regarding their institutions’ compensation models. Most respondents said that a fixed salary model plus productivity or performance incentives is the ideal compensation model for those providing inpatient dermatology consultations, though only half said they actually are compensated according to this model. This discrepancy highlights the disconnect between the current accepted compensation models and the DH’s ideal model and provides direction for dermatology chairs and division heads as to what compensation model is preferable to support the success of DHs at their institutions.
Despite the barriers and compensation constraints we identified, DHs report high job satisfaction, which we hypothesize could combat burnout. In our study, all DHs surveyed say they find providing inpatient dermatology consultations fulfilling, and most were satisfied with the amount of time allotted for consultations. Some of the possible reasons why DHs may find their work fulfilling include increased time for teaching trainees and learning about patients and their conditions while consulting, as well as a preference for providing inpatient dermatology consultations to spending time in clinic. Most DHs said they choose to provide inpatient dermatology consultation rather than do so as a requirement, primarily due to their interest in complex medical dermatology and their desire to work with other medical teams/specialties; thankfully, only a small percentage said they provide these consultations because they are required to do so.
This study was conducted to analyze job satisfaction among DHs who provided inpatient dermatology consultations and determine common barriers and obstacles to their job satisfaction. Limitations to our study included the small sample size and the possibly limited representation of the intended population, as only the members of the SDH were surveyed, potentially excluding providers who regularly perform inpatient dermatology consultations but are not members of the SDH. Further limitations included recall bias and the qualitative nature of the survey instrument.
Final Thoughts
There was near-unanimous agreement among the DHs we surveyed regarding the importance of the role they play in their divisions/departments, but there are clear barriers to provision of inpatient dermatology consultation, specifically relating to extraneous time expenditures and compensation. Despite these barriers, the majority of respondents said they are very satisfied with the role they play in the inpatient setting and feel that their contributions are valued by the institutions where they work. Protecting these benefits of providing dermatology hospital consultations will be critical for maintaining this high job satisfaction and balancing out the barriers to providing these consultations. Protecting the time required to provide consultations is paramount so DHs continue to gain fulfillment from teaching trainees, caring for complex patients, and maintaining their place as valuable colleagues in the hospital setting.
Acknowledgment
The authors thank the members of the SDH for their participation in this survey.
- Biesbroeck LK, Shinohara MM. Inpatient consultative dermatology. Med Clin North Am. 2015;99:1349-1364.
- Lim HW, Collins SAB, Resneck JS Jr, et al. The burden of skin disease in the United States. J Am Acad Dermatol. 2017;76:958-972.e2.
- Arnold JD, Yoon SJ, Kirkorian AY. The national burden of inpatient dermatology in adults. J Am Acad Dermatol. 2018;80:425-432.
- Mancusi S, Festa Neto C. Inpatient dermatological consultations in a university hospital. Clinics (Sao Paulo). 2010;65:851-855.
- Tsai S, Scott JF, Keller JJ, et al. Cutaneous malignancies identified in an inpatient dermatology consultation service. Br J Dermatol. 2017;177:e116-e118.
- Pereira AR, Porro AM, Seque CA, et al. Inpatient dermatology consultations in renal transplant recipients. Actas Dermosifiliogr. 2018;109:900-907.
- Tracey EH, Forrestel A, Rosenbach M, et al. Inpatient dermatology consultation in patients with hematologic malignancies. J Am Acad Dermatol. 2016;75:835-836.
- Fox LP, Cotliar J, Hughey L, et al. Hospitalist dermatology. J Am Acad Dermatol. 2009;61:153-154.
- Ko LN, Kroshinsky D. Dermatology hospitalists: a multicenter survey study characterizing the infrastructure of consultative dermatology in select American hospitals. Int J Dermatol. 2018;57:553-558.
- Hansra NK, Shinkai K, Fox LP. Ethical issues in inpatient consultative dermatology. Clin Dermatol. 2012;30:496-500.
- El-Azhary R, Weenig RH, Gibson LE. The dermatology hospitalist: creating value by rapid clinical pathologic correlation in a patient-centered care model. Int J Dermatol. 2012;51:1461-1466.
- Ahronowitz I, Fox LP. Herpes zoster in hospitalized adults: practice gaps, new evidence, and remaining questions. J Am Acad Dermatol. 2018;78:223-230.e3.
- Rosenbach M. The logistics of an inpatient dermatology service. Semin Cutan Med Surg. 2017;36:3-8.
- Ackerman L, Kessler M. The efficient, effective community hospital inpatient dermatology consult. Semin Cutan Med Surg. 2017;36:9-11.
Consultative dermatologists, or dermatology hospitalists (DHs), perform a critical role in the care of inpatients with skin disease, providing efficient diagnosis and management of patients with complex skin conditions as well as education of patients and trainees in the hospital setting.1 In 2013, 27% of the US population was seen by a physician for a skin disease.2 In 2014, there were nearly 650,000 hospital admissions principally for skin disease.3 Input by dermatologists facilitates accurate diagnosis and management of inpatients with skin disease,4 including a substantial number of cutaneous malignancies diagnosed in the inpatient setting.5 Several studies have highlighted the generally low level of diagnostic concordance between referring services and dermatology consultants,4,6 with dermatology consultants frequently noting diagnoses not considered by referring services,7 reinforcing the importance of having access to dermatologists in the hospital setting.
The care of skin disease in the inpatient setting has become increasingly complex. The Society for Dermatology Hospitalists (SDH) was created in 2009 to address this complexity, with the goal to “strive to develop the highest standards of clinical care of hospitalized patients with skin disease.”8 A recent survey found that 50% of DHs spend between 41 to 52 weeks per year on service.9 Despite this degree of commitment, there are considerable barriers that prevent the majority of dermatologists from efficiently providing inpatient consultative care. The inpatient and outpatient provision of dermatology care varies greatly, including the variety of ethical situations encountered and the diversity of skin conditions treated.10-12 Additionally, the transition between inpatient and outpatient care can be challenging for providers.13
The goal of this study was to evaluate the overall job satisfaction of DHs and further describe potential barriers to inpatient dermatology consultations.
Methods
An anonymous 31-question electronic survey was sent via email to all current members of the SDH from November 20 to December 10, 2018. The study was reviewed and determined to be exempt from federal human subjects regulations by the University of Washington Human Subjects Division (Seattle, Washington)(STUDY00005832).
Results
At the time of survey distribution, the SDH had 145 members, including attending-level dermatologists and resident members. Thirty-seven self-identified DHs (46% [17/37] women; 54% [20/37] men) completed the survey. The majority of respondents were junior faculty, with 46% (17/37) assistant professors, 5% (2/37) acting instructors, 32% (12/37) associate professors, and 16% (6/37) professors. All regions of the United States were represented.
Time Dedicated to Providing Inpatient Dermatology Consultations
The majority of those surveyed were satisfied or very satisfied (68% [25/37]) with the amount of time allotted for inpatient dermatology consultations, while 14% (5/37) were unsatisfied or very unsatisfied. Of those surveyed, 46% (17/37) reported that 21% to 50% of their time is dedicated to inpatient dermatology consultations. The majority (57% [21/37]) reported that their outpatient clinic efforts are reduced when providing dermatology inpatient consultations.
Regarding travel to the inpatient practice site, 60% (22/37) rated their travel time/effort as very easy, with 38% (14/37) reporting that the sites at which they provide inpatient dermatology consultations and their main outpatient clinics are the same physical location; 38% (14/37) reported travel times of less than 15 minutes between clinical practice sites.
Eighty-nine percent (33/37) of respondents said they are able to spend more time teaching trainees when providing inpatient dermatology consultations compared to their time spent in clinic. Similarly, 70% (26/37) said they are able to spend more time learning about patients and their conditions when providing inpatient dermatology consultations. Respondents also reported additional time expenditures because of inpatient dermatology consultations, primarily additional teaching requirements (49% [18/37]), additional electronic medical record training (35% [13/37]), and credentialing requirements (24% [9/37]).
Infrastructure for Providing Inpatient Dermatology Consultations
For many respondents (30% [11/37]), only 2 faculty dermatologists regularly provide inpatient dermatology consultations at their institutions. Four respondents reported having at least 5 faculty dermatologists who regularly provide inpatient dermatology consultations; excluding these, the average number of DHs was 2.42 faculty per institution.
Most respondents (57% [21/37]) reported their institutions support inpatient dermatology services by providing salary support for residents to cover services. Other methods of support included dedicated office spaces (30% [11/37]), free hospital parking while providing inpatient consultations (24% [9/37]), and administrative support (11% [4/37]).
Consultation Composition
Respondents indicated that requests for DH consultations most often come from medical services, including medical intensive care, internal medicine, and family medicine (95% [35/37]); the emergency department (95% [35/37]); surgical services (92% [34/37]); and hematology/oncology (89% [33/37]). Fewer DHs reported receiving consultation requests from pediatrics (70% [26/37]).
Many respondents (49% [18/37]) reported consulting for patients with skin disorders that they considered to be life-threatening or potentially life-threatening either very frequently (daily) or frequently (several times weekly), with only 16% (6/37) responding that they see such patients about once per month.
Compensation for Inpatient Dermatology Consultation
The most commonly reported compensation models for DHs were fixed salary plus productivity or performance incentives and fixed salary only models (49% [18/37] and 32% [12/37], respectively), with relative value unit (RVU) models and other models less frequently reported (16% [6/37] and 3% [1/37], respectively). Only 46% (17/37) of respondents were satisfied or very satisfied with their institutions’ compensation models; the remainder (54% [20/37]) were either neutral, unsatisfied, or very unsatisfied regarding their institutions’ compensation models. Overall compensation satisfaction was higher, with 60% (22/37) of DHs reporting they were satisfied or very satisfied with their salaries and 41% (15/37) reporting they were either neutral or not satisfied. The majority (60% [2/37]) of respondents felt that fixed salary plus productivity or performance incentives models would be the ideal compensation model for DHs.
Of the DHs whose compensations models were RVU based (6/37 [16%]), 67% (4/6) said they receive incentive pay upon meeting their RVU targets. No respondents reported that they were expected to generate an equivalent number of RVUs when performing inpatient consultations as compared to an outpatient session. Only 32% (12/37) of respondents reported keeping the revenue/RVUs generated by inpatient dermatology consultations; most (57% [21/37]) noted that their dermatology divisions/departments keep the revenue/RVUs, followed by university hospitals (27% [10/37]), schools of medicine (11% [4/37]), and departments of medicine (3% [1/37]). The remainder of respondents (22% [8/37]) were unsure who keeps the revenue/RVUs generated by inpatient dermatology consultations.
Most respondents (70% [26/37]) reported that the revenue (or RVU equivalent) generated by inpatient dermatology consultations does not fully support their salary for the time spent as consultants. Rather, these DHs noted sources of additional financial support, primarily the DHs themselves (69% [18/26]), followed by dermatology divisions/departments (50% [13/26]), departments of medicine (23% [6/26]), university hospitals (23% [6/26]), and schools of medicine (12% [3/26]).
Job Fulfillment Among DHs
Most respondents said they choose to provide inpatient dermatology consultations due to their interest in complex medical dermatology and their desire to work with other medical teams and specialties (92% [34/37] and 76% [28/37], respectively). Seventy percent (26/37) said they choose to provide inpatient consultations to be able to teach medical students and residents as well as to take advantage of the added opportunities to practice in a variety of settings beyond their outpatient clinics (57% [21/37]). Only 3% (1/37) of respondents reported that they provide inpatient dermatology consultations because they are “required to do so.”
Most DHs (84% [31/37]) said they feel their institutions as well as their dermatology divisions/departments value having access to inpatient dermatology services, though some did not feel this way (16% [6/37] neutral or strongly disagree). Nearly all respondents (97% [36/37]) felt they provide a critical service when performing inpatient dermatology consultations. All respondents (100%) said they found providing inpatient dermatology consultations fulfilling, and 65% (24/37) said they prefer providing inpatient dermatology consultations to spending time in clinic. Of the DHs who were surveyed, 68% (25/37) said they were satisfied with the balance of outpatient and inpatient services in their clinical practice and 30% (11/37) said they were not.
Comment
Factors such as patient care, hospital infrastructure, and procedural support have all been cited by DHs as crucial aspects of their contributions to the care of hospitalized pa
Dermatology is primarily an outpatient specialty, and our study highlighted several important challenges for providers performing inpatient dermatology consultations. A major issue is time expenditures, including additional teaching requirements, additional electronic medical record training, and credentialing requirements. Travel time to inpatient hospital sites does not appear to be one of these hindering factors; nearly 60% of respondents rated their travel time/effort as very easy, with approximately 75% of respondents’ consultation locations being either at the same physical location as their main outpatient clinic or less than 15 minutes away. Maintaining easy travel between outpatient and inpatient settings is important to the success of the DH.
Our data suggest that compensation of DHs is a potential limitation to providing inpatient dermatology care. Our survey reinforced that providers who do inpatient dermatology consultations generally do not generate the revenue necessary to cover these efforts. More than 40% of DH respondents said they either feel neutral about or unsatisfied with their overall salary, and more than half said they feel similarly regarding their institutions’ compensation models. Most respondents said that a fixed salary model plus productivity or performance incentives is the ideal compensation model for those providing inpatient dermatology consultations, though only half said they actually are compensated according to this model. This discrepancy highlights the disconnect between the current accepted compensation models and the DH’s ideal model and provides direction for dermatology chairs and division heads as to what compensation model is preferable to support the success of DHs at their institutions.
Despite the barriers and compensation constraints we identified, DHs report high job satisfaction, which we hypothesize could combat burnout. In our study, all DHs surveyed say they find providing inpatient dermatology consultations fulfilling, and most were satisfied with the amount of time allotted for consultations. Some of the possible reasons why DHs may find their work fulfilling include increased time for teaching trainees and learning about patients and their conditions while consulting, as well as a preference for providing inpatient dermatology consultations to spending time in clinic. Most DHs said they choose to provide inpatient dermatology consultation rather than do so as a requirement, primarily due to their interest in complex medical dermatology and their desire to work with other medical teams/specialties; thankfully, only a small percentage said they provide these consultations because they are required to do so.
This study was conducted to analyze job satisfaction among DHs who provided inpatient dermatology consultations and determine common barriers and obstacles to their job satisfaction. Limitations to our study included the small sample size and the possibly limited representation of the intended population, as only the members of the SDH were surveyed, potentially excluding providers who regularly perform inpatient dermatology consultations but are not members of the SDH. Further limitations included recall bias and the qualitative nature of the survey instrument.
Final Thoughts
There was near-unanimous agreement among the DHs we surveyed regarding the importance of the role they play in their divisions/departments, but there are clear barriers to provision of inpatient dermatology consultation, specifically relating to extraneous time expenditures and compensation. Despite these barriers, the majority of respondents said they are very satisfied with the role they play in the inpatient setting and feel that their contributions are valued by the institutions where they work. Protecting these benefits of providing dermatology hospital consultations will be critical for maintaining this high job satisfaction and balancing out the barriers to providing these consultations. Protecting the time required to provide consultations is paramount so DHs continue to gain fulfillment from teaching trainees, caring for complex patients, and maintaining their place as valuable colleagues in the hospital setting.
Acknowledgment
The authors thank the members of the SDH for their participation in this survey.
Consultative dermatologists, or dermatology hospitalists (DHs), perform a critical role in the care of inpatients with skin disease, providing efficient diagnosis and management of patients with complex skin conditions as well as education of patients and trainees in the hospital setting.1 In 2013, 27% of the US population was seen by a physician for a skin disease.2 In 2014, there were nearly 650,000 hospital admissions principally for skin disease.3 Input by dermatologists facilitates accurate diagnosis and management of inpatients with skin disease,4 including a substantial number of cutaneous malignancies diagnosed in the inpatient setting.5 Several studies have highlighted the generally low level of diagnostic concordance between referring services and dermatology consultants,4,6 with dermatology consultants frequently noting diagnoses not considered by referring services,7 reinforcing the importance of having access to dermatologists in the hospital setting.
The care of skin disease in the inpatient setting has become increasingly complex. The Society for Dermatology Hospitalists (SDH) was created in 2009 to address this complexity, with the goal to “strive to develop the highest standards of clinical care of hospitalized patients with skin disease.”8 A recent survey found that 50% of DHs spend between 41 to 52 weeks per year on service.9 Despite this degree of commitment, there are considerable barriers that prevent the majority of dermatologists from efficiently providing inpatient consultative care. The inpatient and outpatient provision of dermatology care varies greatly, including the variety of ethical situations encountered and the diversity of skin conditions treated.10-12 Additionally, the transition between inpatient and outpatient care can be challenging for providers.13
The goal of this study was to evaluate the overall job satisfaction of DHs and further describe potential barriers to inpatient dermatology consultations.
Methods
An anonymous 31-question electronic survey was sent via email to all current members of the SDH from November 20 to December 10, 2018. The study was reviewed and determined to be exempt from federal human subjects regulations by the University of Washington Human Subjects Division (Seattle, Washington)(STUDY00005832).
Results
At the time of survey distribution, the SDH had 145 members, including attending-level dermatologists and resident members. Thirty-seven self-identified DHs (46% [17/37] women; 54% [20/37] men) completed the survey. The majority of respondents were junior faculty, with 46% (17/37) assistant professors, 5% (2/37) acting instructors, 32% (12/37) associate professors, and 16% (6/37) professors. All regions of the United States were represented.
Time Dedicated to Providing Inpatient Dermatology Consultations
The majority of those surveyed were satisfied or very satisfied (68% [25/37]) with the amount of time allotted for inpatient dermatology consultations, while 14% (5/37) were unsatisfied or very unsatisfied. Of those surveyed, 46% (17/37) reported that 21% to 50% of their time is dedicated to inpatient dermatology consultations. The majority (57% [21/37]) reported that their outpatient clinic efforts are reduced when providing dermatology inpatient consultations.
Regarding travel to the inpatient practice site, 60% (22/37) rated their travel time/effort as very easy, with 38% (14/37) reporting that the sites at which they provide inpatient dermatology consultations and their main outpatient clinics are the same physical location; 38% (14/37) reported travel times of less than 15 minutes between clinical practice sites.
Eighty-nine percent (33/37) of respondents said they are able to spend more time teaching trainees when providing inpatient dermatology consultations compared to their time spent in clinic. Similarly, 70% (26/37) said they are able to spend more time learning about patients and their conditions when providing inpatient dermatology consultations. Respondents also reported additional time expenditures because of inpatient dermatology consultations, primarily additional teaching requirements (49% [18/37]), additional electronic medical record training (35% [13/37]), and credentialing requirements (24% [9/37]).
Infrastructure for Providing Inpatient Dermatology Consultations
For many respondents (30% [11/37]), only 2 faculty dermatologists regularly provide inpatient dermatology consultations at their institutions. Four respondents reported having at least 5 faculty dermatologists who regularly provide inpatient dermatology consultations; excluding these, the average number of DHs was 2.42 faculty per institution.
Most respondents (57% [21/37]) reported their institutions support inpatient dermatology services by providing salary support for residents to cover services. Other methods of support included dedicated office spaces (30% [11/37]), free hospital parking while providing inpatient consultations (24% [9/37]), and administrative support (11% [4/37]).
Consultation Composition
Respondents indicated that requests for DH consultations most often come from medical services, including medical intensive care, internal medicine, and family medicine (95% [35/37]); the emergency department (95% [35/37]); surgical services (92% [34/37]); and hematology/oncology (89% [33/37]). Fewer DHs reported receiving consultation requests from pediatrics (70% [26/37]).
Many respondents (49% [18/37]) reported consulting for patients with skin disorders that they considered to be life-threatening or potentially life-threatening either very frequently (daily) or frequently (several times weekly), with only 16% (6/37) responding that they see such patients about once per month.
Compensation for Inpatient Dermatology Consultation
The most commonly reported compensation models for DHs were fixed salary plus productivity or performance incentives and fixed salary only models (49% [18/37] and 32% [12/37], respectively), with relative value unit (RVU) models and other models less frequently reported (16% [6/37] and 3% [1/37], respectively). Only 46% (17/37) of respondents were satisfied or very satisfied with their institutions’ compensation models; the remainder (54% [20/37]) were either neutral, unsatisfied, or very unsatisfied regarding their institutions’ compensation models. Overall compensation satisfaction was higher, with 60% (22/37) of DHs reporting they were satisfied or very satisfied with their salaries and 41% (15/37) reporting they were either neutral or not satisfied. The majority (60% [2/37]) of respondents felt that fixed salary plus productivity or performance incentives models would be the ideal compensation model for DHs.
Of the DHs whose compensations models were RVU based (6/37 [16%]), 67% (4/6) said they receive incentive pay upon meeting their RVU targets. No respondents reported that they were expected to generate an equivalent number of RVUs when performing inpatient consultations as compared to an outpatient session. Only 32% (12/37) of respondents reported keeping the revenue/RVUs generated by inpatient dermatology consultations; most (57% [21/37]) noted that their dermatology divisions/departments keep the revenue/RVUs, followed by university hospitals (27% [10/37]), schools of medicine (11% [4/37]), and departments of medicine (3% [1/37]). The remainder of respondents (22% [8/37]) were unsure who keeps the revenue/RVUs generated by inpatient dermatology consultations.
Most respondents (70% [26/37]) reported that the revenue (or RVU equivalent) generated by inpatient dermatology consultations does not fully support their salary for the time spent as consultants. Rather, these DHs noted sources of additional financial support, primarily the DHs themselves (69% [18/26]), followed by dermatology divisions/departments (50% [13/26]), departments of medicine (23% [6/26]), university hospitals (23% [6/26]), and schools of medicine (12% [3/26]).
Job Fulfillment Among DHs
Most respondents said they choose to provide inpatient dermatology consultations due to their interest in complex medical dermatology and their desire to work with other medical teams and specialties (92% [34/37] and 76% [28/37], respectively). Seventy percent (26/37) said they choose to provide inpatient consultations to be able to teach medical students and residents as well as to take advantage of the added opportunities to practice in a variety of settings beyond their outpatient clinics (57% [21/37]). Only 3% (1/37) of respondents reported that they provide inpatient dermatology consultations because they are “required to do so.”
Most DHs (84% [31/37]) said they feel their institutions as well as their dermatology divisions/departments value having access to inpatient dermatology services, though some did not feel this way (16% [6/37] neutral or strongly disagree). Nearly all respondents (97% [36/37]) felt they provide a critical service when performing inpatient dermatology consultations. All respondents (100%) said they found providing inpatient dermatology consultations fulfilling, and 65% (24/37) said they prefer providing inpatient dermatology consultations to spending time in clinic. Of the DHs who were surveyed, 68% (25/37) said they were satisfied with the balance of outpatient and inpatient services in their clinical practice and 30% (11/37) said they were not.
Comment
Factors such as patient care, hospital infrastructure, and procedural support have all been cited by DHs as crucial aspects of their contributions to the care of hospitalized pa
Dermatology is primarily an outpatient specialty, and our study highlighted several important challenges for providers performing inpatient dermatology consultations. A major issue is time expenditures, including additional teaching requirements, additional electronic medical record training, and credentialing requirements. Travel time to inpatient hospital sites does not appear to be one of these hindering factors; nearly 60% of respondents rated their travel time/effort as very easy, with approximately 75% of respondents’ consultation locations being either at the same physical location as their main outpatient clinic or less than 15 minutes away. Maintaining easy travel between outpatient and inpatient settings is important to the success of the DH.
Our data suggest that compensation of DHs is a potential limitation to providing inpatient dermatology care. Our survey reinforced that providers who do inpatient dermatology consultations generally do not generate the revenue necessary to cover these efforts. More than 40% of DH respondents said they either feel neutral about or unsatisfied with their overall salary, and more than half said they feel similarly regarding their institutions’ compensation models. Most respondents said that a fixed salary model plus productivity or performance incentives is the ideal compensation model for those providing inpatient dermatology consultations, though only half said they actually are compensated according to this model. This discrepancy highlights the disconnect between the current accepted compensation models and the DH’s ideal model and provides direction for dermatology chairs and division heads as to what compensation model is preferable to support the success of DHs at their institutions.
Despite the barriers and compensation constraints we identified, DHs report high job satisfaction, which we hypothesize could combat burnout. In our study, all DHs surveyed say they find providing inpatient dermatology consultations fulfilling, and most were satisfied with the amount of time allotted for consultations. Some of the possible reasons why DHs may find their work fulfilling include increased time for teaching trainees and learning about patients and their conditions while consulting, as well as a preference for providing inpatient dermatology consultations to spending time in clinic. Most DHs said they choose to provide inpatient dermatology consultation rather than do so as a requirement, primarily due to their interest in complex medical dermatology and their desire to work with other medical teams/specialties; thankfully, only a small percentage said they provide these consultations because they are required to do so.
This study was conducted to analyze job satisfaction among DHs who provided inpatient dermatology consultations and determine common barriers and obstacles to their job satisfaction. Limitations to our study included the small sample size and the possibly limited representation of the intended population, as only the members of the SDH were surveyed, potentially excluding providers who regularly perform inpatient dermatology consultations but are not members of the SDH. Further limitations included recall bias and the qualitative nature of the survey instrument.
Final Thoughts
There was near-unanimous agreement among the DHs we surveyed regarding the importance of the role they play in their divisions/departments, but there are clear barriers to provision of inpatient dermatology consultation, specifically relating to extraneous time expenditures and compensation. Despite these barriers, the majority of respondents said they are very satisfied with the role they play in the inpatient setting and feel that their contributions are valued by the institutions where they work. Protecting these benefits of providing dermatology hospital consultations will be critical for maintaining this high job satisfaction and balancing out the barriers to providing these consultations. Protecting the time required to provide consultations is paramount so DHs continue to gain fulfillment from teaching trainees, caring for complex patients, and maintaining their place as valuable colleagues in the hospital setting.
Acknowledgment
The authors thank the members of the SDH for their participation in this survey.
- Biesbroeck LK, Shinohara MM. Inpatient consultative dermatology. Med Clin North Am. 2015;99:1349-1364.
- Lim HW, Collins SAB, Resneck JS Jr, et al. The burden of skin disease in the United States. J Am Acad Dermatol. 2017;76:958-972.e2.
- Arnold JD, Yoon SJ, Kirkorian AY. The national burden of inpatient dermatology in adults. J Am Acad Dermatol. 2018;80:425-432.
- Mancusi S, Festa Neto C. Inpatient dermatological consultations in a university hospital. Clinics (Sao Paulo). 2010;65:851-855.
- Tsai S, Scott JF, Keller JJ, et al. Cutaneous malignancies identified in an inpatient dermatology consultation service. Br J Dermatol. 2017;177:e116-e118.
- Pereira AR, Porro AM, Seque CA, et al. Inpatient dermatology consultations in renal transplant recipients. Actas Dermosifiliogr. 2018;109:900-907.
- Tracey EH, Forrestel A, Rosenbach M, et al. Inpatient dermatology consultation in patients with hematologic malignancies. J Am Acad Dermatol. 2016;75:835-836.
- Fox LP, Cotliar J, Hughey L, et al. Hospitalist dermatology. J Am Acad Dermatol. 2009;61:153-154.
- Ko LN, Kroshinsky D. Dermatology hospitalists: a multicenter survey study characterizing the infrastructure of consultative dermatology in select American hospitals. Int J Dermatol. 2018;57:553-558.
- Hansra NK, Shinkai K, Fox LP. Ethical issues in inpatient consultative dermatology. Clin Dermatol. 2012;30:496-500.
- El-Azhary R, Weenig RH, Gibson LE. The dermatology hospitalist: creating value by rapid clinical pathologic correlation in a patient-centered care model. Int J Dermatol. 2012;51:1461-1466.
- Ahronowitz I, Fox LP. Herpes zoster in hospitalized adults: practice gaps, new evidence, and remaining questions. J Am Acad Dermatol. 2018;78:223-230.e3.
- Rosenbach M. The logistics of an inpatient dermatology service. Semin Cutan Med Surg. 2017;36:3-8.
- Ackerman L, Kessler M. The efficient, effective community hospital inpatient dermatology consult. Semin Cutan Med Surg. 2017;36:9-11.
- Biesbroeck LK, Shinohara MM. Inpatient consultative dermatology. Med Clin North Am. 2015;99:1349-1364.
- Lim HW, Collins SAB, Resneck JS Jr, et al. The burden of skin disease in the United States. J Am Acad Dermatol. 2017;76:958-972.e2.
- Arnold JD, Yoon SJ, Kirkorian AY. The national burden of inpatient dermatology in adults. J Am Acad Dermatol. 2018;80:425-432.
- Mancusi S, Festa Neto C. Inpatient dermatological consultations in a university hospital. Clinics (Sao Paulo). 2010;65:851-855.
- Tsai S, Scott JF, Keller JJ, et al. Cutaneous malignancies identified in an inpatient dermatology consultation service. Br J Dermatol. 2017;177:e116-e118.
- Pereira AR, Porro AM, Seque CA, et al. Inpatient dermatology consultations in renal transplant recipients. Actas Dermosifiliogr. 2018;109:900-907.
- Tracey EH, Forrestel A, Rosenbach M, et al. Inpatient dermatology consultation in patients with hematologic malignancies. J Am Acad Dermatol. 2016;75:835-836.
- Fox LP, Cotliar J, Hughey L, et al. Hospitalist dermatology. J Am Acad Dermatol. 2009;61:153-154.
- Ko LN, Kroshinsky D. Dermatology hospitalists: a multicenter survey study characterizing the infrastructure of consultative dermatology in select American hospitals. Int J Dermatol. 2018;57:553-558.
- Hansra NK, Shinkai K, Fox LP. Ethical issues in inpatient consultative dermatology. Clin Dermatol. 2012;30:496-500.
- El-Azhary R, Weenig RH, Gibson LE. The dermatology hospitalist: creating value by rapid clinical pathologic correlation in a patient-centered care model. Int J Dermatol. 2012;51:1461-1466.
- Ahronowitz I, Fox LP. Herpes zoster in hospitalized adults: practice gaps, new evidence, and remaining questions. J Am Acad Dermatol. 2018;78:223-230.e3.
- Rosenbach M. The logistics of an inpatient dermatology service. Semin Cutan Med Surg. 2017;36:3-8.
- Ackerman L, Kessler M. The efficient, effective community hospital inpatient dermatology consult. Semin Cutan Med Surg. 2017;36:9-11.
Practice Points
• Dermatology hospitalists play a critical role in the specialized care of hospitalized patients with
skin conditions.
• Dermatology hospitalists have high job satisfaction, with opportunities to teach trainees and practice complex medical dermatology.
• Most dermatology hospitalists do not generate sufficient revenue providing inpatient dermatology consultations to fully support their salary for the time spent as consultants; alternate payment models are needed to maintain dermatology’s presence in the hospital.
