User login
FDA extends Liletta IUD duration of use to 4 years
The Food and Drug Administration has approved a supplemental New Drug Application to extend the duration of use for Liletta (levonorgestrel-releasing intrauterine system) 52 mg, for up to 4 years.
The approval, issued Aug. 3, adds 1 year to the duration of use on the drug label. It is based on additional efficacy and safety data from ACCESS IUS (A Comprehensive Contraceptive Efficacy & Safety Study of an Intrauterine System), an ongoing phase 3 trial with 1,751 U.S. women.

There are three other levonorgestrel-releasing IUDs currently on the market: Mirena and Kyleena, which are both approved for up to 5 years of use; and Skyla, which is approved for up to 3 years of use.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
The Food and Drug Administration has approved a supplemental New Drug Application to extend the duration of use for Liletta (levonorgestrel-releasing intrauterine system) 52 mg, for up to 4 years.
The approval, issued Aug. 3, adds 1 year to the duration of use on the drug label. It is based on additional efficacy and safety data from ACCESS IUS (A Comprehensive Contraceptive Efficacy & Safety Study of an Intrauterine System), an ongoing phase 3 trial with 1,751 U.S. women.
There are three other levonorgestrel-releasing IUDs currently on the market: Mirena and Kyleena, which are both approved for up to 5 years of use; and Skyla, which is approved for up to 3 years of use.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
The Food and Drug Administration has approved a supplemental New Drug Application to extend the duration of use for Liletta (levonorgestrel-releasing intrauterine system) 52 mg, for up to 4 years.
The approval, issued Aug. 3, adds 1 year to the duration of use on the drug label. It is based on additional efficacy and safety data from ACCESS IUS (A Comprehensive Contraceptive Efficacy & Safety Study of an Intrauterine System), an ongoing phase 3 trial with 1,751 U.S. women.
There are three other levonorgestrel-releasing IUDs currently on the market: Mirena and Kyleena, which are both approved for up to 5 years of use; and Skyla, which is approved for up to 3 years of use.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
Vemurafenib granted sNDA, priority review for Erdheim-Chester disease
Vemurafenib (Zelboraf) has been granted a supplemental new drug application and priority review by the Food and Drug Administration for the treatment of Erdheim-Chester disease with BRAF V600 mutation, according to a press release issued by Genentech.
The FDA is expected to make a decision on the indication by Dec. 7, 2017. Vemurafenib is approved for the treatment of unresectable or metastatic melanoma with BRAF V600E mutation.
The supportive data for the application came from VE-BASKET, a phase 2, nonrandomized study investigating the use of vemurafenib for people with various BRAF V600 mutation–positive cancers and other diseases. Final results for the 22 people with Erdheim-Chester disease showed a best overall response rate of 54.5% by RECIST v1.1 criteria.
The median duration of response, progression-free survival, and overall survival were not reached at a median follow-up time of 26.6 months. The most common grade 3 or higher adverse events were new skin cancers, high blood pressure, rash, and joint pain. Initial study results were published in the New England Journal of Medicine in August 2015.
Based on available published data, there are fewer than 500 cases of Erdheim-Chester disease in the United States. More than half of affected people have BRAF V600 mutation–positive disease, and there are no approved treatments, according to the release.
Vemurafenib (Zelboraf) has been granted a supplemental new drug application and priority review by the Food and Drug Administration for the treatment of Erdheim-Chester disease with BRAF V600 mutation, according to a press release issued by Genentech.
The FDA is expected to make a decision on the indication by Dec. 7, 2017. Vemurafenib is approved for the treatment of unresectable or metastatic melanoma with BRAF V600E mutation.
The supportive data for the application came from VE-BASKET, a phase 2, nonrandomized study investigating the use of vemurafenib for people with various BRAF V600 mutation–positive cancers and other diseases. Final results for the 22 people with Erdheim-Chester disease showed a best overall response rate of 54.5% by RECIST v1.1 criteria.
The median duration of response, progression-free survival, and overall survival were not reached at a median follow-up time of 26.6 months. The most common grade 3 or higher adverse events were new skin cancers, high blood pressure, rash, and joint pain. Initial study results were published in the New England Journal of Medicine in August 2015.
Based on available published data, there are fewer than 500 cases of Erdheim-Chester disease in the United States. More than half of affected people have BRAF V600 mutation–positive disease, and there are no approved treatments, according to the release.
Vemurafenib (Zelboraf) has been granted a supplemental new drug application and priority review by the Food and Drug Administration for the treatment of Erdheim-Chester disease with BRAF V600 mutation, according to a press release issued by Genentech.
The FDA is expected to make a decision on the indication by Dec. 7, 2017. Vemurafenib is approved for the treatment of unresectable or metastatic melanoma with BRAF V600E mutation.
The supportive data for the application came from VE-BASKET, a phase 2, nonrandomized study investigating the use of vemurafenib for people with various BRAF V600 mutation–positive cancers and other diseases. Final results for the 22 people with Erdheim-Chester disease showed a best overall response rate of 54.5% by RECIST v1.1 criteria.
The median duration of response, progression-free survival, and overall survival were not reached at a median follow-up time of 26.6 months. The most common grade 3 or higher adverse events were new skin cancers, high blood pressure, rash, and joint pain. Initial study results were published in the New England Journal of Medicine in August 2015.
Based on available published data, there are fewer than 500 cases of Erdheim-Chester disease in the United States. More than half of affected people have BRAF V600 mutation–positive disease, and there are no approved treatments, according to the release.
FDA committee recommends approval of tofacitinib for PsA
Convinced largely by encouraging efficacy data, the Food and Drug Administration’s Arthritis Advisory Committee voted overwhelmingly in favor of approval of tofacitinib for the treatment of adult patients with active psoriatic arthritis.
If approved by the FDA, which usually adheres to advisory committee recommendations, the oral inhibitor of Janus-associated kinases (JAK) would be the first JAK inhibitor approved for the treatment of psoriatic arthritis (PsA). Pfizer submitted supplemental new drug applications (sNDAs) for both tofacitinib tablets (Xeljanz) and tofacitinib extended-release tablets (Xeljanz XR) at a dose of 5 mg twice daily and 11 mg once daily, respectively and, despite some reservations with respect to adverse events and lack of evidence regarding inhibition of radiographic progression, the committee voted 10-1 in favor of approval at an Aug. 3 meeting.
“I voted yes and, although there are safety concerns, I feel like it’s nothing different than what we see with other biologics, and I want to make sure that patients have options,” said Jennifer Horonjeff, PhD, a research fellow and patient advocate with the Center for Immune Disease with Onset in Childhood at Columbia University Medical Center, New York, and a consumer representative on the committee.
Dr. Horonjeff added that she hopes there is continued conversation between the sponsor and the FDA on “what we can do to make patients aware of these risks.”
Similarly, committee member Daniel H. Solomon, MD, a professor of medicine at Harvard Medical School and chief of the section of clinical sciences in the divisions of rheumatology and pharmacoepidemiology at Brigham and Women’s Hospital, both in Boston, said he sees a “great opportunity for risk mitigation that the sponsor and the [FDA] can take together.”
“We have a clear risk, we have a clear strategy for mitigating the risk, and there are going to be a lot more people exposed to this drug with a known risk, so let’s do something about it,” Dr. Solomon said about a plan put forward by Pfizer, and discussed at some length, to mitigate risks through measures such as vaccination against herpes zoster and additional study.
Temporary voting member Steven Meisel, PharmD, system director of patient safety at Fairview Health Services in Minneapolis added: “These are nasty drugs, but I think those who use them understand that, and this is no different than any of the other nasty drugs in these categories.”
Diane Aronson, a patient representative and temporary voting member on the committee, cast the only vote against approval, citing concerns about the lack of inhibition of radiographic progression of the disease and about the infection risks in a vulnerable population.
Tofacitinib was initially approved in 2012 at a dose of 5 mg, twice daily, for the treatment of adults with moderately to severely active rheumatoid arthritis who had an inadequate response or intolerance to methotrexate. The extended-release formulation was approved in 2016 at a dose of 11 mg daily.
With respect to the current sNDAs, Pfizer presented data from two placebo-controlled phase 3 trials in patients with psoriatic arthritis. The FDA deemed these trials to be adequate and well-controlled, providing “corroborating evidence of the efficacy of tofacitinib for reducing signs and symptoms of PsA, based on the proportion of patients experiencing the American College of Rheumatology (ACR) 20% response criteria,” according to a report presented to the committee. The report also noted that both phase 3 trials provided evidence of improvement in physical function, but did not provide sufficient evidence that tofacitinib inhibits radiographic progression in PsA.
The report also stated that the safety profile of tofacitinib in PsA was consistent with that established in RA; risks include serious infections, opportunistic infections, malignancy, gastrointestinal perforation, and laboratory abnormalities, including elevations in low-density lipoprotein and triglycerides.
“No new safety signals were identified in PsA,” the report states.
Of note, the sNDAs do not include an indication for generalized psoriasis; an application for that indication was withdrawn in 2016, and Dr. Meisel cautioned against any “unintentional leakage of the use of this drug for generalized psoriasis.”
He and others also cautioned against any implied endorsement in labeling that the drug inhibits radiographic progression of PsA.
Two individuals who participated in the open public hearing portion of the meeting each urged the committee to recommend approval of the sNDAs, with one, Stephen Marmaras, manager of state and national advocacy for the Global Healthy Living Foundation, noting that the joint pain and stiffness associated with PsA are a primary concern of patients.
“Our members with psoriatic arthritis overwhelmingly prioritize joint pain and stiffness as the most bothersome symptoms they experience,” Mr. Marmaras said. “With that in mind, we were encouraged to read that tofacitinib has particularly notable efficacy in treating the joint symptoms of the disease in clinical trials.”
All voting advisory committee members were screened and cleared with respect to potential conflicts of interest.
Convinced largely by encouraging efficacy data, the Food and Drug Administration’s Arthritis Advisory Committee voted overwhelmingly in favor of approval of tofacitinib for the treatment of adult patients with active psoriatic arthritis.
If approved by the FDA, which usually adheres to advisory committee recommendations, the oral inhibitor of Janus-associated kinases (JAK) would be the first JAK inhibitor approved for the treatment of psoriatic arthritis (PsA). Pfizer submitted supplemental new drug applications (sNDAs) for both tofacitinib tablets (Xeljanz) and tofacitinib extended-release tablets (Xeljanz XR) at a dose of 5 mg twice daily and 11 mg once daily, respectively and, despite some reservations with respect to adverse events and lack of evidence regarding inhibition of radiographic progression, the committee voted 10-1 in favor of approval at an Aug. 3 meeting.
“I voted yes and, although there are safety concerns, I feel like it’s nothing different than what we see with other biologics, and I want to make sure that patients have options,” said Jennifer Horonjeff, PhD, a research fellow and patient advocate with the Center for Immune Disease with Onset in Childhood at Columbia University Medical Center, New York, and a consumer representative on the committee.
Dr. Horonjeff added that she hopes there is continued conversation between the sponsor and the FDA on “what we can do to make patients aware of these risks.”
Similarly, committee member Daniel H. Solomon, MD, a professor of medicine at Harvard Medical School and chief of the section of clinical sciences in the divisions of rheumatology and pharmacoepidemiology at Brigham and Women’s Hospital, both in Boston, said he sees a “great opportunity for risk mitigation that the sponsor and the [FDA] can take together.”
“We have a clear risk, we have a clear strategy for mitigating the risk, and there are going to be a lot more people exposed to this drug with a known risk, so let’s do something about it,” Dr. Solomon said about a plan put forward by Pfizer, and discussed at some length, to mitigate risks through measures such as vaccination against herpes zoster and additional study.
Temporary voting member Steven Meisel, PharmD, system director of patient safety at Fairview Health Services in Minneapolis added: “These are nasty drugs, but I think those who use them understand that, and this is no different than any of the other nasty drugs in these categories.”
Diane Aronson, a patient representative and temporary voting member on the committee, cast the only vote against approval, citing concerns about the lack of inhibition of radiographic progression of the disease and about the infection risks in a vulnerable population.
Tofacitinib was initially approved in 2012 at a dose of 5 mg, twice daily, for the treatment of adults with moderately to severely active rheumatoid arthritis who had an inadequate response or intolerance to methotrexate. The extended-release formulation was approved in 2016 at a dose of 11 mg daily.
With respect to the current sNDAs, Pfizer presented data from two placebo-controlled phase 3 trials in patients with psoriatic arthritis. The FDA deemed these trials to be adequate and well-controlled, providing “corroborating evidence of the efficacy of tofacitinib for reducing signs and symptoms of PsA, based on the proportion of patients experiencing the American College of Rheumatology (ACR) 20% response criteria,” according to a report presented to the committee. The report also noted that both phase 3 trials provided evidence of improvement in physical function, but did not provide sufficient evidence that tofacitinib inhibits radiographic progression in PsA.
The report also stated that the safety profile of tofacitinib in PsA was consistent with that established in RA; risks include serious infections, opportunistic infections, malignancy, gastrointestinal perforation, and laboratory abnormalities, including elevations in low-density lipoprotein and triglycerides.
“No new safety signals were identified in PsA,” the report states.
Of note, the sNDAs do not include an indication for generalized psoriasis; an application for that indication was withdrawn in 2016, and Dr. Meisel cautioned against any “unintentional leakage of the use of this drug for generalized psoriasis.”
He and others also cautioned against any implied endorsement in labeling that the drug inhibits radiographic progression of PsA.
Two individuals who participated in the open public hearing portion of the meeting each urged the committee to recommend approval of the sNDAs, with one, Stephen Marmaras, manager of state and national advocacy for the Global Healthy Living Foundation, noting that the joint pain and stiffness associated with PsA are a primary concern of patients.
“Our members with psoriatic arthritis overwhelmingly prioritize joint pain and stiffness as the most bothersome symptoms they experience,” Mr. Marmaras said. “With that in mind, we were encouraged to read that tofacitinib has particularly notable efficacy in treating the joint symptoms of the disease in clinical trials.”
All voting advisory committee members were screened and cleared with respect to potential conflicts of interest.
Convinced largely by encouraging efficacy data, the Food and Drug Administration’s Arthritis Advisory Committee voted overwhelmingly in favor of approval of tofacitinib for the treatment of adult patients with active psoriatic arthritis.
If approved by the FDA, which usually adheres to advisory committee recommendations, the oral inhibitor of Janus-associated kinases (JAK) would be the first JAK inhibitor approved for the treatment of psoriatic arthritis (PsA). Pfizer submitted supplemental new drug applications (sNDAs) for both tofacitinib tablets (Xeljanz) and tofacitinib extended-release tablets (Xeljanz XR) at a dose of 5 mg twice daily and 11 mg once daily, respectively and, despite some reservations with respect to adverse events and lack of evidence regarding inhibition of radiographic progression, the committee voted 10-1 in favor of approval at an Aug. 3 meeting.
“I voted yes and, although there are safety concerns, I feel like it’s nothing different than what we see with other biologics, and I want to make sure that patients have options,” said Jennifer Horonjeff, PhD, a research fellow and patient advocate with the Center for Immune Disease with Onset in Childhood at Columbia University Medical Center, New York, and a consumer representative on the committee.
Dr. Horonjeff added that she hopes there is continued conversation between the sponsor and the FDA on “what we can do to make patients aware of these risks.”
Similarly, committee member Daniel H. Solomon, MD, a professor of medicine at Harvard Medical School and chief of the section of clinical sciences in the divisions of rheumatology and pharmacoepidemiology at Brigham and Women’s Hospital, both in Boston, said he sees a “great opportunity for risk mitigation that the sponsor and the [FDA] can take together.”
“We have a clear risk, we have a clear strategy for mitigating the risk, and there are going to be a lot more people exposed to this drug with a known risk, so let’s do something about it,” Dr. Solomon said about a plan put forward by Pfizer, and discussed at some length, to mitigate risks through measures such as vaccination against herpes zoster and additional study.
Temporary voting member Steven Meisel, PharmD, system director of patient safety at Fairview Health Services in Minneapolis added: “These are nasty drugs, but I think those who use them understand that, and this is no different than any of the other nasty drugs in these categories.”
Diane Aronson, a patient representative and temporary voting member on the committee, cast the only vote against approval, citing concerns about the lack of inhibition of radiographic progression of the disease and about the infection risks in a vulnerable population.
Tofacitinib was initially approved in 2012 at a dose of 5 mg, twice daily, for the treatment of adults with moderately to severely active rheumatoid arthritis who had an inadequate response or intolerance to methotrexate. The extended-release formulation was approved in 2016 at a dose of 11 mg daily.
With respect to the current sNDAs, Pfizer presented data from two placebo-controlled phase 3 trials in patients with psoriatic arthritis. The FDA deemed these trials to be adequate and well-controlled, providing “corroborating evidence of the efficacy of tofacitinib for reducing signs and symptoms of PsA, based on the proportion of patients experiencing the American College of Rheumatology (ACR) 20% response criteria,” according to a report presented to the committee. The report also noted that both phase 3 trials provided evidence of improvement in physical function, but did not provide sufficient evidence that tofacitinib inhibits radiographic progression in PsA.
The report also stated that the safety profile of tofacitinib in PsA was consistent with that established in RA; risks include serious infections, opportunistic infections, malignancy, gastrointestinal perforation, and laboratory abnormalities, including elevations in low-density lipoprotein and triglycerides.
“No new safety signals were identified in PsA,” the report states.
Of note, the sNDAs do not include an indication for generalized psoriasis; an application for that indication was withdrawn in 2016, and Dr. Meisel cautioned against any “unintentional leakage of the use of this drug for generalized psoriasis.”
He and others also cautioned against any implied endorsement in labeling that the drug inhibits radiographic progression of PsA.
Two individuals who participated in the open public hearing portion of the meeting each urged the committee to recommend approval of the sNDAs, with one, Stephen Marmaras, manager of state and national advocacy for the Global Healthy Living Foundation, noting that the joint pain and stiffness associated with PsA are a primary concern of patients.
“Our members with psoriatic arthritis overwhelmingly prioritize joint pain and stiffness as the most bothersome symptoms they experience,” Mr. Marmaras said. “With that in mind, we were encouraged to read that tofacitinib has particularly notable efficacy in treating the joint symptoms of the disease in clinical trials.”
All voting advisory committee members were screened and cleared with respect to potential conflicts of interest.
Liposomal daunorubicin and cytarabine approved for t-AML, AML-MRC
, the Food and Drug Administration announced on Aug. 3.
Vyxeos is the first FDA-approved treatment specifically for patients with t-AML or AML-MRC, the FDA said in its press release announcing the approval.
“Vyxeos is the first chemotherapy to demonstrate an overall survival advantage over the standard of care in a phase 3 randomized study of older adults with newly-diagnosed therapy-related AML or AML with myelodysplasia-related changes,” Jeffrey E. Lancet, MD, an investigator in the clinical trials of Vyxeos and chair of the department of malignant hematology at the H. Lee Moffitt Cancer Center in Tampa, Fla., said in a press release.
Vyxeos was associated with a median overall survival of 9.6 months and a standard combination of cytarabine and daunorubicin (7+3) was associated with a median survival of 5.9 months in a randomized, multicenter, open-label trial of 309 patients aged 60-75 years with newly-diagnosed t-AML or AML-MRC. Data from the study, which is NCT01696084, was the basis for the drug’s approval.
Vyxeos is a fixed-dose combination with each Vyxeos vial containing 44 mg daunorubicin and 100 mg cytarabine encapsulated together in liposomes. As dosing is based on the daunorubicin component, the corresponding cytarabine dose does not need to be calculated. Daunorubicin dosing is calculated on the basis of body surface area (mg/m2).
For the first induction cycle, the recommended Vyxeos dose is daunorubicin 44 mg/m2 (cytarabine 100 mg/m2) infused intravenously over 90 minutes on days 1, 3, and 5. If a second induction cycle is needed, the same dose is administered on days 1 and 3. The recommended dose of Vyxeos for each cycle of consolidation therapy is daunorubicin 29 mg/m2 (cytarabine 65 mg/m2) liposome via intravenous infusion over 90 minutes on days 1 and 3.
Adverse reactions occurring in at least 25% of treated patients in the clinical trial included hemorrhage, febrile neutropenia, rash, edema, nausea, mucositis, diarrhea, constipation, musculoskeletal pain, fatigue, abdominal pain, dyspnea, headache, cough, decreased appetite, arrhythmia, pneumonia, bacteremia, chills, sleep disorders, and vomiting.
The prescribing information includes a boxed warning not to substitute Vyxeos with other daunorubicin- or cytarabine-containing products. Full prescribing information is available at: www.accessdata.fda.gov/drugsatfda_docs/label/2017/209401s000lbl.pdf
The maker of Vyxeos is Jazz Pharmaceuticals.
mdales@frontlinemedcom.com
On Twitter @maryjodales
, the Food and Drug Administration announced on Aug. 3.
Vyxeos is the first FDA-approved treatment specifically for patients with t-AML or AML-MRC, the FDA said in its press release announcing the approval.
“Vyxeos is the first chemotherapy to demonstrate an overall survival advantage over the standard of care in a phase 3 randomized study of older adults with newly-diagnosed therapy-related AML or AML with myelodysplasia-related changes,” Jeffrey E. Lancet, MD, an investigator in the clinical trials of Vyxeos and chair of the department of malignant hematology at the H. Lee Moffitt Cancer Center in Tampa, Fla., said in a press release.
Vyxeos was associated with a median overall survival of 9.6 months and a standard combination of cytarabine and daunorubicin (7+3) was associated with a median survival of 5.9 months in a randomized, multicenter, open-label trial of 309 patients aged 60-75 years with newly-diagnosed t-AML or AML-MRC. Data from the study, which is NCT01696084, was the basis for the drug’s approval.
Vyxeos is a fixed-dose combination with each Vyxeos vial containing 44 mg daunorubicin and 100 mg cytarabine encapsulated together in liposomes. As dosing is based on the daunorubicin component, the corresponding cytarabine dose does not need to be calculated. Daunorubicin dosing is calculated on the basis of body surface area (mg/m2).
For the first induction cycle, the recommended Vyxeos dose is daunorubicin 44 mg/m2 (cytarabine 100 mg/m2) infused intravenously over 90 minutes on days 1, 3, and 5. If a second induction cycle is needed, the same dose is administered on days 1 and 3. The recommended dose of Vyxeos for each cycle of consolidation therapy is daunorubicin 29 mg/m2 (cytarabine 65 mg/m2) liposome via intravenous infusion over 90 minutes on days 1 and 3.
Adverse reactions occurring in at least 25% of treated patients in the clinical trial included hemorrhage, febrile neutropenia, rash, edema, nausea, mucositis, diarrhea, constipation, musculoskeletal pain, fatigue, abdominal pain, dyspnea, headache, cough, decreased appetite, arrhythmia, pneumonia, bacteremia, chills, sleep disorders, and vomiting.
The prescribing information includes a boxed warning not to substitute Vyxeos with other daunorubicin- or cytarabine-containing products. Full prescribing information is available at: www.accessdata.fda.gov/drugsatfda_docs/label/2017/209401s000lbl.pdf
The maker of Vyxeos is Jazz Pharmaceuticals.
mdales@frontlinemedcom.com
On Twitter @maryjodales
, the Food and Drug Administration announced on Aug. 3.
Vyxeos is the first FDA-approved treatment specifically for patients with t-AML or AML-MRC, the FDA said in its press release announcing the approval.
“Vyxeos is the first chemotherapy to demonstrate an overall survival advantage over the standard of care in a phase 3 randomized study of older adults with newly-diagnosed therapy-related AML or AML with myelodysplasia-related changes,” Jeffrey E. Lancet, MD, an investigator in the clinical trials of Vyxeos and chair of the department of malignant hematology at the H. Lee Moffitt Cancer Center in Tampa, Fla., said in a press release.
Vyxeos was associated with a median overall survival of 9.6 months and a standard combination of cytarabine and daunorubicin (7+3) was associated with a median survival of 5.9 months in a randomized, multicenter, open-label trial of 309 patients aged 60-75 years with newly-diagnosed t-AML or AML-MRC. Data from the study, which is NCT01696084, was the basis for the drug’s approval.
Vyxeos is a fixed-dose combination with each Vyxeos vial containing 44 mg daunorubicin and 100 mg cytarabine encapsulated together in liposomes. As dosing is based on the daunorubicin component, the corresponding cytarabine dose does not need to be calculated. Daunorubicin dosing is calculated on the basis of body surface area (mg/m2).
For the first induction cycle, the recommended Vyxeos dose is daunorubicin 44 mg/m2 (cytarabine 100 mg/m2) infused intravenously over 90 minutes on days 1, 3, and 5. If a second induction cycle is needed, the same dose is administered on days 1 and 3. The recommended dose of Vyxeos for each cycle of consolidation therapy is daunorubicin 29 mg/m2 (cytarabine 65 mg/m2) liposome via intravenous infusion over 90 minutes on days 1 and 3.
Adverse reactions occurring in at least 25% of treated patients in the clinical trial included hemorrhage, febrile neutropenia, rash, edema, nausea, mucositis, diarrhea, constipation, musculoskeletal pain, fatigue, abdominal pain, dyspnea, headache, cough, decreased appetite, arrhythmia, pneumonia, bacteremia, chills, sleep disorders, and vomiting.
The prescribing information includes a boxed warning not to substitute Vyxeos with other daunorubicin- or cytarabine-containing products. Full prescribing information is available at: www.accessdata.fda.gov/drugsatfda_docs/label/2017/209401s000lbl.pdf
The maker of Vyxeos is Jazz Pharmaceuticals.
mdales@frontlinemedcom.com
On Twitter @maryjodales
FDA committee rejects sirukumab approval on safety concerns
Citing safety concerns, the Food and Drug Administration’s Arthritis Advisory Committee voted overwhelmingly against recommending FDA approval of the interleukin-6 inhibitor sirukumab for refractory rheumatoid arthritis.
Janssen Biotech submitted a biologics license application (BLA) for the monoclonal antibody, seeking an indication for adults with moderately to severely active rheumatoid arthritis (RA) who had an inadequate response or intolerance to one or more prior disease-modifying antirheumatic drugs, but despite agreeing unanimously that the data presented by the applicant provided substantial evidence of efficacy for this indication, the committee voted 12-1 against approval at an Aug. 2 meeting.
“I’m not sure whether the safety signal is of concern or not. I don’t think there’s enough data here to know that. It’s concerning, and it may be just noise, but it may also be real and I’m not willing to ... be supportive of the notion that it’s safe enough to take its place along with other biologicals,” said temporary voting member David T. Felson, MD, professor of medicine and public health at Boston University.
Similarly, temporary voting member Erica Brittain, PhD, said the mortality concerns swayed her vote.
“It was a very close call for me. I do think there’s a real argument to be made about bias in the analysis that shows some possibility of a difference. On the other hand, I just couldn’t get past the uncertainty, and when we’re talking about mortality it’s hard to dismiss that,” said Dr. Brittain, a mathematical statistician and deputy chief of the Biostatistics Research Branch at the National Institute of Allergy and Infectious Diseases.
Michael H. Weisman, MD, also a temporary voting member and chair of rheumatology at Cedars-Sinai Medical Center in Los Angeles, said that if the indication had been narrowed – perhaps to those who showed a biologic response – he would have voted “yes.”
Temporary voting member James Katz, MD, was the only committee member to vote in favor of approval.
“I actually voted yes because this drug doesn’t scare me any more than all the other drugs I use. I’m very scared by all the biological agents, and this is no different,” said Dr. Katz, director of the Rheumatology Fellowship and Training Branch at the National Institute of Arthritis and Musculoskeletal Diseases.
Sirukumab differs from two other approved monoclonal antibodies that target the IL-6 pathway for the treatment of patients with RA – tocilizumab (Actemra) and sarilumab (Kevzara) – in that it targets IL-6, while the others target the IL-6 receptor. This slight difference could make a difference for some highly refractory patients who had failed to respond to prior treatments, according to applicant presentations at the meeting.
The efficacy and safety of the agent were assessed in a phase 2 dose-ranging study, as well in three pivotal phase 3 studies. Two of the phase 3 studies compared 100 mg twice weekly and 50 mg four times weekly doses of subcutaneous sirukumab to placebo – which were shown to have similar efficacy for reducing the signs and symptoms of RA – and a third compared those two doses against adalimumab (Humira) and showed that it was not superior to adalimumab for efficacy.
One phase 3 placebo-controlled study involving 878 refractory patients was published in February in The Lancet and showed that sirukumab was associated with rapid and sustained improvements in RA signs and symptoms, physical function, and health status, as well as improvement in physical and mental well-being.
However, a safety signal – a trend of increased overall mortality with sirukumab vs. placebo – emerged from the studies. The mortality was mainly associated with major adverse cardiovascular events, infection, and malignancy.
Three speakers, including RA patients or patient representatives, participated in the open public hearing portion of the committee meeting, and all spoke in favor of approval of sirukumab, but ultimately the committee agreed that the limited benefits of the agent – given that it does not involve an entirely new mechanism of action – did not outweigh the unknowns regarding safety.
Committee chairperson Daniel H. Solomon, MD, professor of medicine at Harvard Medical School and chief of the section of clinical sciences at Brigham and Women’s Hospital, both in Boston, said longer-term outcomes data with a clear comparator are needed.
The FDA will now consider the committee’s recommendations in making its final determination regarding the BLA.
All voting advisory committee members were screened and cleared with respect to potential conflicts of interest.
Citing safety concerns, the Food and Drug Administration’s Arthritis Advisory Committee voted overwhelmingly against recommending FDA approval of the interleukin-6 inhibitor sirukumab for refractory rheumatoid arthritis.
Janssen Biotech submitted a biologics license application (BLA) for the monoclonal antibody, seeking an indication for adults with moderately to severely active rheumatoid arthritis (RA) who had an inadequate response or intolerance to one or more prior disease-modifying antirheumatic drugs, but despite agreeing unanimously that the data presented by the applicant provided substantial evidence of efficacy for this indication, the committee voted 12-1 against approval at an Aug. 2 meeting.
“I’m not sure whether the safety signal is of concern or not. I don’t think there’s enough data here to know that. It’s concerning, and it may be just noise, but it may also be real and I’m not willing to ... be supportive of the notion that it’s safe enough to take its place along with other biologicals,” said temporary voting member David T. Felson, MD, professor of medicine and public health at Boston University.
Similarly, temporary voting member Erica Brittain, PhD, said the mortality concerns swayed her vote.
“It was a very close call for me. I do think there’s a real argument to be made about bias in the analysis that shows some possibility of a difference. On the other hand, I just couldn’t get past the uncertainty, and when we’re talking about mortality it’s hard to dismiss that,” said Dr. Brittain, a mathematical statistician and deputy chief of the Biostatistics Research Branch at the National Institute of Allergy and Infectious Diseases.
Michael H. Weisman, MD, also a temporary voting member and chair of rheumatology at Cedars-Sinai Medical Center in Los Angeles, said that if the indication had been narrowed – perhaps to those who showed a biologic response – he would have voted “yes.”
Temporary voting member James Katz, MD, was the only committee member to vote in favor of approval.
“I actually voted yes because this drug doesn’t scare me any more than all the other drugs I use. I’m very scared by all the biological agents, and this is no different,” said Dr. Katz, director of the Rheumatology Fellowship and Training Branch at the National Institute of Arthritis and Musculoskeletal Diseases.
Sirukumab differs from two other approved monoclonal antibodies that target the IL-6 pathway for the treatment of patients with RA – tocilizumab (Actemra) and sarilumab (Kevzara) – in that it targets IL-6, while the others target the IL-6 receptor. This slight difference could make a difference for some highly refractory patients who had failed to respond to prior treatments, according to applicant presentations at the meeting.
The efficacy and safety of the agent were assessed in a phase 2 dose-ranging study, as well in three pivotal phase 3 studies. Two of the phase 3 studies compared 100 mg twice weekly and 50 mg four times weekly doses of subcutaneous sirukumab to placebo – which were shown to have similar efficacy for reducing the signs and symptoms of RA – and a third compared those two doses against adalimumab (Humira) and showed that it was not superior to adalimumab for efficacy.
One phase 3 placebo-controlled study involving 878 refractory patients was published in February in The Lancet and showed that sirukumab was associated with rapid and sustained improvements in RA signs and symptoms, physical function, and health status, as well as improvement in physical and mental well-being.
However, a safety signal – a trend of increased overall mortality with sirukumab vs. placebo – emerged from the studies. The mortality was mainly associated with major adverse cardiovascular events, infection, and malignancy.
Three speakers, including RA patients or patient representatives, participated in the open public hearing portion of the committee meeting, and all spoke in favor of approval of sirukumab, but ultimately the committee agreed that the limited benefits of the agent – given that it does not involve an entirely new mechanism of action – did not outweigh the unknowns regarding safety.
Committee chairperson Daniel H. Solomon, MD, professor of medicine at Harvard Medical School and chief of the section of clinical sciences at Brigham and Women’s Hospital, both in Boston, said longer-term outcomes data with a clear comparator are needed.
The FDA will now consider the committee’s recommendations in making its final determination regarding the BLA.
All voting advisory committee members were screened and cleared with respect to potential conflicts of interest.
Citing safety concerns, the Food and Drug Administration’s Arthritis Advisory Committee voted overwhelmingly against recommending FDA approval of the interleukin-6 inhibitor sirukumab for refractory rheumatoid arthritis.
Janssen Biotech submitted a biologics license application (BLA) for the monoclonal antibody, seeking an indication for adults with moderately to severely active rheumatoid arthritis (RA) who had an inadequate response or intolerance to one or more prior disease-modifying antirheumatic drugs, but despite agreeing unanimously that the data presented by the applicant provided substantial evidence of efficacy for this indication, the committee voted 12-1 against approval at an Aug. 2 meeting.
“I’m not sure whether the safety signal is of concern or not. I don’t think there’s enough data here to know that. It’s concerning, and it may be just noise, but it may also be real and I’m not willing to ... be supportive of the notion that it’s safe enough to take its place along with other biologicals,” said temporary voting member David T. Felson, MD, professor of medicine and public health at Boston University.
Similarly, temporary voting member Erica Brittain, PhD, said the mortality concerns swayed her vote.
“It was a very close call for me. I do think there’s a real argument to be made about bias in the analysis that shows some possibility of a difference. On the other hand, I just couldn’t get past the uncertainty, and when we’re talking about mortality it’s hard to dismiss that,” said Dr. Brittain, a mathematical statistician and deputy chief of the Biostatistics Research Branch at the National Institute of Allergy and Infectious Diseases.
Michael H. Weisman, MD, also a temporary voting member and chair of rheumatology at Cedars-Sinai Medical Center in Los Angeles, said that if the indication had been narrowed – perhaps to those who showed a biologic response – he would have voted “yes.”
Temporary voting member James Katz, MD, was the only committee member to vote in favor of approval.
“I actually voted yes because this drug doesn’t scare me any more than all the other drugs I use. I’m very scared by all the biological agents, and this is no different,” said Dr. Katz, director of the Rheumatology Fellowship and Training Branch at the National Institute of Arthritis and Musculoskeletal Diseases.
Sirukumab differs from two other approved monoclonal antibodies that target the IL-6 pathway for the treatment of patients with RA – tocilizumab (Actemra) and sarilumab (Kevzara) – in that it targets IL-6, while the others target the IL-6 receptor. This slight difference could make a difference for some highly refractory patients who had failed to respond to prior treatments, according to applicant presentations at the meeting.
The efficacy and safety of the agent were assessed in a phase 2 dose-ranging study, as well in three pivotal phase 3 studies. Two of the phase 3 studies compared 100 mg twice weekly and 50 mg four times weekly doses of subcutaneous sirukumab to placebo – which were shown to have similar efficacy for reducing the signs and symptoms of RA – and a third compared those two doses against adalimumab (Humira) and showed that it was not superior to adalimumab for efficacy.
One phase 3 placebo-controlled study involving 878 refractory patients was published in February in The Lancet and showed that sirukumab was associated with rapid and sustained improvements in RA signs and symptoms, physical function, and health status, as well as improvement in physical and mental well-being.
However, a safety signal – a trend of increased overall mortality with sirukumab vs. placebo – emerged from the studies. The mortality was mainly associated with major adverse cardiovascular events, infection, and malignancy.
Three speakers, including RA patients or patient representatives, participated in the open public hearing portion of the committee meeting, and all spoke in favor of approval of sirukumab, but ultimately the committee agreed that the limited benefits of the agent – given that it does not involve an entirely new mechanism of action – did not outweigh the unknowns regarding safety.
Committee chairperson Daniel H. Solomon, MD, professor of medicine at Harvard Medical School and chief of the section of clinical sciences at Brigham and Women’s Hospital, both in Boston, said longer-term outcomes data with a clear comparator are needed.
The FDA will now consider the committee’s recommendations in making its final determination regarding the BLA.
All voting advisory committee members were screened and cleared with respect to potential conflicts of interest.
Abilify Maintena OK’d by FDA for adults with bipolar I disorder
The Food and Drug Administration has approved a monthly injectable formulation of aripiprazole, (Abilify Maintena) , for a maintenance monotherapy treatment of bipolar I disorder for adults, Otsuka and Lundbeck have announced.
Patients treated with the injectable formulation of the atypical antipsychotic must continue to take a daily oral antipsychotic for the first 14 days. After that, however, the long-acting injectable (LAI) – which must be administered by a health care professional – can replace the daily medication.
Joseph R. Calabrese, MD, director of the mood disorders program at University Hospitals Cleveland Medical Center, said in the July 28 announcement that the LAI is a new treatment option for bipolar I patients “who have established tolerability with oral aripiprazole.”
The drug label includes a warning that elderly patients with dementia-related psychosis who are treated with antipsychotics are at a higher mortality risk. Adverse reactions that have been associated with treatment with aripiprazole include weight gain, akathisia, injection site pain, sedation, and certain compulsive behaviors.
Created by Otsuka, and marketed by Otsuka and Lundbeck, the LAI was approved in the United States for treating adults with schizophrenia in 2013.
The Food and Drug Administration has approved a monthly injectable formulation of aripiprazole, (Abilify Maintena) , for a maintenance monotherapy treatment of bipolar I disorder for adults, Otsuka and Lundbeck have announced.
Patients treated with the injectable formulation of the atypical antipsychotic must continue to take a daily oral antipsychotic for the first 14 days. After that, however, the long-acting injectable (LAI) – which must be administered by a health care professional – can replace the daily medication.
Joseph R. Calabrese, MD, director of the mood disorders program at University Hospitals Cleveland Medical Center, said in the July 28 announcement that the LAI is a new treatment option for bipolar I patients “who have established tolerability with oral aripiprazole.”
The drug label includes a warning that elderly patients with dementia-related psychosis who are treated with antipsychotics are at a higher mortality risk. Adverse reactions that have been associated with treatment with aripiprazole include weight gain, akathisia, injection site pain, sedation, and certain compulsive behaviors.
Created by Otsuka, and marketed by Otsuka and Lundbeck, the LAI was approved in the United States for treating adults with schizophrenia in 2013.
The Food and Drug Administration has approved a monthly injectable formulation of aripiprazole, (Abilify Maintena) , for a maintenance monotherapy treatment of bipolar I disorder for adults, Otsuka and Lundbeck have announced.
Patients treated with the injectable formulation of the atypical antipsychotic must continue to take a daily oral antipsychotic for the first 14 days. After that, however, the long-acting injectable (LAI) – which must be administered by a health care professional – can replace the daily medication.
Joseph R. Calabrese, MD, director of the mood disorders program at University Hospitals Cleveland Medical Center, said in the July 28 announcement that the LAI is a new treatment option for bipolar I patients “who have established tolerability with oral aripiprazole.”
The drug label includes a warning that elderly patients with dementia-related psychosis who are treated with antipsychotics are at a higher mortality risk. Adverse reactions that have been associated with treatment with aripiprazole include weight gain, akathisia, injection site pain, sedation, and certain compulsive behaviors.
Created by Otsuka, and marketed by Otsuka and Lundbeck, the LAI was approved in the United States for treating adults with schizophrenia in 2013.
FDA approves nivolumab for metastatic CRC
The Food and Drug Administration has granted accelerated approval to checkpoint inhibitor nivolumab for the treatment of patients with mismatch repair deficient (dMMR) and microsatellite instability high (MSI-H) metastatic colorectal cancer (CRC) that has progressed following treatment with fluoropyrimidine, oxaliplatin, and irinotecan.
The indication covers patients aged 12 years and older. Efficacy for adolescent patients with MSI-H or dMMR metastatic CRC is extrapolated from the results in the respective adult population, the FDA said in a statement.
Approval of nivolumab in the adult population was based on an objective response rate of 28% in CHECKMATE 142, an open-label, single-arm study of 53 patients with locally determined dMMR or MSI-H metastatic CRC who had disease progression during, after, or were intolerant to prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy.
The most common adverse reactions to nivolumab, marketed as Opdivo by Bristol-Myers Squibb, include fatigue, rash, musculoskeletal pain, pruritus, diarrhea, nausea, asthenia, cough, dyspnea, constipation, decreased appetite, back pain, arthralgia, upper respiratory tract infection, and pyrexia, the FDA said.
The recommended nivolumab dose is 240 mg every 2 weeks.
The Food and Drug Administration has granted accelerated approval to checkpoint inhibitor nivolumab for the treatment of patients with mismatch repair deficient (dMMR) and microsatellite instability high (MSI-H) metastatic colorectal cancer (CRC) that has progressed following treatment with fluoropyrimidine, oxaliplatin, and irinotecan.
The indication covers patients aged 12 years and older. Efficacy for adolescent patients with MSI-H or dMMR metastatic CRC is extrapolated from the results in the respective adult population, the FDA said in a statement.
Approval of nivolumab in the adult population was based on an objective response rate of 28% in CHECKMATE 142, an open-label, single-arm study of 53 patients with locally determined dMMR or MSI-H metastatic CRC who had disease progression during, after, or were intolerant to prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy.
The most common adverse reactions to nivolumab, marketed as Opdivo by Bristol-Myers Squibb, include fatigue, rash, musculoskeletal pain, pruritus, diarrhea, nausea, asthenia, cough, dyspnea, constipation, decreased appetite, back pain, arthralgia, upper respiratory tract infection, and pyrexia, the FDA said.
The recommended nivolumab dose is 240 mg every 2 weeks.
The Food and Drug Administration has granted accelerated approval to checkpoint inhibitor nivolumab for the treatment of patients with mismatch repair deficient (dMMR) and microsatellite instability high (MSI-H) metastatic colorectal cancer (CRC) that has progressed following treatment with fluoropyrimidine, oxaliplatin, and irinotecan.
The indication covers patients aged 12 years and older. Efficacy for adolescent patients with MSI-H or dMMR metastatic CRC is extrapolated from the results in the respective adult population, the FDA said in a statement.
Approval of nivolumab in the adult population was based on an objective response rate of 28% in CHECKMATE 142, an open-label, single-arm study of 53 patients with locally determined dMMR or MSI-H metastatic CRC who had disease progression during, after, or were intolerant to prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy.
The most common adverse reactions to nivolumab, marketed as Opdivo by Bristol-Myers Squibb, include fatigue, rash, musculoskeletal pain, pruritus, diarrhea, nausea, asthenia, cough, dyspnea, constipation, decreased appetite, back pain, arthralgia, upper respiratory tract infection, and pyrexia, the FDA said.
The recommended nivolumab dose is 240 mg every 2 weeks.
FDA grants priority review of acalabrutinib for second-line treatment of MCL
The Food and Drug Administration has granted a priority review for acalabrutinib, a Bruton tyrosine kinase inhibitor, for the treatment of patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.
The new drug application is based on results from the phase 2 ACE-LY-004 trial, which evaluated the safety and efficacy of acalabrutinib in patients with relapsed/refractory MCL who had received at least one prior therapy.
The Food and Drug Administration has granted a priority review for acalabrutinib, a Bruton tyrosine kinase inhibitor, for the treatment of patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.
The new drug application is based on results from the phase 2 ACE-LY-004 trial, which evaluated the safety and efficacy of acalabrutinib in patients with relapsed/refractory MCL who had received at least one prior therapy.
The Food and Drug Administration has granted a priority review for acalabrutinib, a Bruton tyrosine kinase inhibitor, for the treatment of patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.
The new drug application is based on results from the phase 2 ACE-LY-004 trial, which evaluated the safety and efficacy of acalabrutinib in patients with relapsed/refractory MCL who had received at least one prior therapy.
Ibrutinib becomes first FDA-approved treatment for chronic GVHD
Ibrutinib (Imbruvica) added another notch on its indications belt with its Aug. 2 approval by the U.S. Food and Drug Administration for the treatment of adult patients with chronic graft versus host disease (cGVHD) after failure of one or more lines of systemic therapy.
The new indication makes ibrutinib the first FDA-approved therapy for the treatment of cGVHD, according to an FDA press release.
Ibrutinib’s other approved indications include chronic lymphocytic leukemia/small lymphocytic lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma with 17p deletion, Waldenström’s macroglobulinemia, marginal zone lymphoma, and mantle cell lymphoma, according to a press release from the FDA.
The recommended dose of ibrutinib for cGVHD is 420 mg (three 140 mg capsules once daily). Prescribing information is available on the FDA website.
Imbruvica is manufactured by Pharmacyclics.
mdales@frontlinemedcom.com
On Twitter @maryjodales
Ibrutinib (Imbruvica) added another notch on its indications belt with its Aug. 2 approval by the U.S. Food and Drug Administration for the treatment of adult patients with chronic graft versus host disease (cGVHD) after failure of one or more lines of systemic therapy.
The new indication makes ibrutinib the first FDA-approved therapy for the treatment of cGVHD, according to an FDA press release.
Ibrutinib’s other approved indications include chronic lymphocytic leukemia/small lymphocytic lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma with 17p deletion, Waldenström’s macroglobulinemia, marginal zone lymphoma, and mantle cell lymphoma, according to a press release from the FDA.
The recommended dose of ibrutinib for cGVHD is 420 mg (three 140 mg capsules once daily). Prescribing information is available on the FDA website.
Imbruvica is manufactured by Pharmacyclics.
mdales@frontlinemedcom.com
On Twitter @maryjodales
Ibrutinib (Imbruvica) added another notch on its indications belt with its Aug. 2 approval by the U.S. Food and Drug Administration for the treatment of adult patients with chronic graft versus host disease (cGVHD) after failure of one or more lines of systemic therapy.
The new indication makes ibrutinib the first FDA-approved therapy for the treatment of cGVHD, according to an FDA press release.
The approval was based on an open-label, multicenter, single-arm clinical trial that found a 67% response rate in 42 patients with cGVHD who did not respond to first-line corticosteroid therapy. The median time to response was 12.3 weeks, and responses persisted for at least 5 months in half of the patients. Treatment was discontinued because of adverse events in 24% of patients; 26% of the patients needed dose reductions. All of the specifics of that trial were covered in an article by our reporter at the annual congress of the European Hematology Association in Madrid. (Ibrutinib dons new anti-GVHD hat.)
Ibrutinib’s other approved indications include chronic lymphocytic leukemia/small lymphocytic lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma with 17p deletion, Waldenström’s macroglobulinemia, marginal zone lymphoma, and mantle cell lymphoma, according to a press release from the FDA.
The recommended dose of ibrutinib for cGVHD is 420 mg (three 140 mg capsules once daily). Prescribing information is available on the FDA website.
Imbruvica is manufactured by Pharmacyclics.
mdales@frontlinemedcom.com
On Twitter @maryjodales
Pain frequency varies by employment status
Adults who were previously employed are twice as likely to report daily or almost-daily pain than are those who are currently employed, according to the Centers for Disease Control and Prevention.
In an ongoing survey, just over 30% of adults aged 18 years and older who were previously employed reported that they experienced pain on “most days or every day” in the past 6 months, compared with 15% of those who were currently employed and 19% of those classified as never employed, investigators from the CDC estimated (MMWR. 2017 Jul 28;66[29]:796).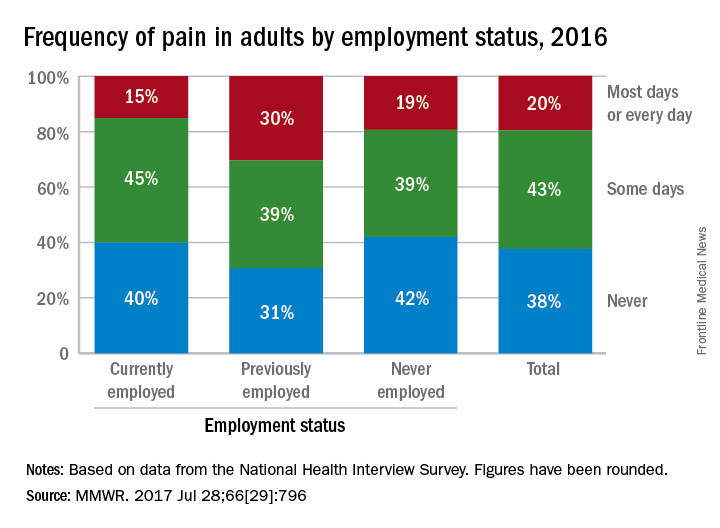
Adults who were previously employed are twice as likely to report daily or almost-daily pain than are those who are currently employed, according to the Centers for Disease Control and Prevention.
In an ongoing survey, just over 30% of adults aged 18 years and older who were previously employed reported that they experienced pain on “most days or every day” in the past 6 months, compared with 15% of those who were currently employed and 19% of those classified as never employed, investigators from the CDC estimated (MMWR. 2017 Jul 28;66[29]:796).
Adults who were previously employed are twice as likely to report daily or almost-daily pain than are those who are currently employed, according to the Centers for Disease Control and Prevention.
In an ongoing survey, just over 30% of adults aged 18 years and older who were previously employed reported that they experienced pain on “most days or every day” in the past 6 months, compared with 15% of those who were currently employed and 19% of those classified as never employed, investigators from the CDC estimated (MMWR. 2017 Jul 28;66[29]:796).
FROM MMWR