User login
Liraglutide approved for cardiovascular event reduction
The type 2 diabetes treatment Victoza (liraglutide) has been approved by the Food and Drug Administration for a new indication – to reduce the risk of several adverse cardiovascular events, according to a press release from Novo Nordisk.
FDA approval was based on results from the LEADER (NCT01179048) trial, where people with type 2 diabetes who received liraglutide were 13% less likely to experience cardiovascular death, nonfatal heart attack, or nonfatal stroke then type 2 diabetes patients who received a placebo, with an absolute risk reduction of 1.9%. Notably, risk of cardiovascular death was reduced by 22% in the liraglutide group, compared with the control group, and all-cause death was reduced by 15%.
“Today’s news is significant for millions of Americans living with type 2 diabetes because, even when controlled, diabetes puts patients at a greater risk for cardiovascular events. More treatment options like Victoza that address critical aspects of diabetes care beyond glucose lowering are essential to confront this pervasive issue,” Steve Marso, MD, medical director at the Cardiovascular Services HCA Midwest Health Heart and Vascular Institute and one of the primary investigators in LEADER said in the press release.
Find the full press release on the Novo Nordisk website.
The type 2 diabetes treatment Victoza (liraglutide) has been approved by the Food and Drug Administration for a new indication – to reduce the risk of several adverse cardiovascular events, according to a press release from Novo Nordisk.
FDA approval was based on results from the LEADER (NCT01179048) trial, where people with type 2 diabetes who received liraglutide were 13% less likely to experience cardiovascular death, nonfatal heart attack, or nonfatal stroke then type 2 diabetes patients who received a placebo, with an absolute risk reduction of 1.9%. Notably, risk of cardiovascular death was reduced by 22% in the liraglutide group, compared with the control group, and all-cause death was reduced by 15%.
“Today’s news is significant for millions of Americans living with type 2 diabetes because, even when controlled, diabetes puts patients at a greater risk for cardiovascular events. More treatment options like Victoza that address critical aspects of diabetes care beyond glucose lowering are essential to confront this pervasive issue,” Steve Marso, MD, medical director at the Cardiovascular Services HCA Midwest Health Heart and Vascular Institute and one of the primary investigators in LEADER said in the press release.
Find the full press release on the Novo Nordisk website.
The type 2 diabetes treatment Victoza (liraglutide) has been approved by the Food and Drug Administration for a new indication – to reduce the risk of several adverse cardiovascular events, according to a press release from Novo Nordisk.
FDA approval was based on results from the LEADER (NCT01179048) trial, where people with type 2 diabetes who received liraglutide were 13% less likely to experience cardiovascular death, nonfatal heart attack, or nonfatal stroke then type 2 diabetes patients who received a placebo, with an absolute risk reduction of 1.9%. Notably, risk of cardiovascular death was reduced by 22% in the liraglutide group, compared with the control group, and all-cause death was reduced by 15%.
“Today’s news is significant for millions of Americans living with type 2 diabetes because, even when controlled, diabetes puts patients at a greater risk for cardiovascular events. More treatment options like Victoza that address critical aspects of diabetes care beyond glucose lowering are essential to confront this pervasive issue,” Steve Marso, MD, medical director at the Cardiovascular Services HCA Midwest Health Heart and Vascular Institute and one of the primary investigators in LEADER said in the press release.
Find the full press release on the Novo Nordisk website.
FDA approves once-daily treatment for hyperuricemia in gout
The Food and Drug Administration announced Aug. 21 the approval of Duzallo, a once-daily oral treatment for hyperuricemia associated with gout in patients who have not achieved target serum uric acid (sUA) levels with a medically appropriate daily dose of allopurinol alone.
Duzallo is a fixed-dose combination of lesinurad 200 mg and allopurinol 300 mg that will be marketed by Ironwood Pharmaceuticals. It will also be available in a lesinurad 200 mg plus allopurinol 200 mg dosage.
“With Duzallo, nearly twice as many patients with uncontrolled gout may be able to achieve target serum uric acid levels compared to those patients taking allopurinol alone, which is important, considering the significant unmet need among uncontrolled gout patients to get to goal of under 6 mg/dL,” said Tom McCourt, senior vice president of marketing and sales and chief commercial officer at Ironwood in an announcement from the company.
Duzallo is the first drug that combines the current standard of care for the treatment of hyperuricemia associated with gout, allopurinol, with the most recent FDA-approved treatment for gout, lesinurad (Zurampic). Allopurinol is an xanthine oxidase inhibitor whose action differs from that of uricosuric agents such as lesinurad. Allopurinol reduces the production of uric acid, whereas lesinurad increases renal excretion of uric acid by selectively inhibiting the action of URAT1, the uric acid transporter responsible for the majority of renal uric acid reabsorption.
Duzallo is not recommended for the treatment of asymptomatic hyperuricemia. It has a boxed warning regarding the risk of acute renal failure.
Duzallo is expected to be commercially available early in the fourth quarter of 2017.
The Food and Drug Administration announced Aug. 21 the approval of Duzallo, a once-daily oral treatment for hyperuricemia associated with gout in patients who have not achieved target serum uric acid (sUA) levels with a medically appropriate daily dose of allopurinol alone.
Duzallo is a fixed-dose combination of lesinurad 200 mg and allopurinol 300 mg that will be marketed by Ironwood Pharmaceuticals. It will also be available in a lesinurad 200 mg plus allopurinol 200 mg dosage.
“With Duzallo, nearly twice as many patients with uncontrolled gout may be able to achieve target serum uric acid levels compared to those patients taking allopurinol alone, which is important, considering the significant unmet need among uncontrolled gout patients to get to goal of under 6 mg/dL,” said Tom McCourt, senior vice president of marketing and sales and chief commercial officer at Ironwood in an announcement from the company.
Duzallo is the first drug that combines the current standard of care for the treatment of hyperuricemia associated with gout, allopurinol, with the most recent FDA-approved treatment for gout, lesinurad (Zurampic). Allopurinol is an xanthine oxidase inhibitor whose action differs from that of uricosuric agents such as lesinurad. Allopurinol reduces the production of uric acid, whereas lesinurad increases renal excretion of uric acid by selectively inhibiting the action of URAT1, the uric acid transporter responsible for the majority of renal uric acid reabsorption.
Duzallo is not recommended for the treatment of asymptomatic hyperuricemia. It has a boxed warning regarding the risk of acute renal failure.
Duzallo is expected to be commercially available early in the fourth quarter of 2017.
The Food and Drug Administration announced Aug. 21 the approval of Duzallo, a once-daily oral treatment for hyperuricemia associated with gout in patients who have not achieved target serum uric acid (sUA) levels with a medically appropriate daily dose of allopurinol alone.
Duzallo is a fixed-dose combination of lesinurad 200 mg and allopurinol 300 mg that will be marketed by Ironwood Pharmaceuticals. It will also be available in a lesinurad 200 mg plus allopurinol 200 mg dosage.
“With Duzallo, nearly twice as many patients with uncontrolled gout may be able to achieve target serum uric acid levels compared to those patients taking allopurinol alone, which is important, considering the significant unmet need among uncontrolled gout patients to get to goal of under 6 mg/dL,” said Tom McCourt, senior vice president of marketing and sales and chief commercial officer at Ironwood in an announcement from the company.
Duzallo is the first drug that combines the current standard of care for the treatment of hyperuricemia associated with gout, allopurinol, with the most recent FDA-approved treatment for gout, lesinurad (Zurampic). Allopurinol is an xanthine oxidase inhibitor whose action differs from that of uricosuric agents such as lesinurad. Allopurinol reduces the production of uric acid, whereas lesinurad increases renal excretion of uric acid by selectively inhibiting the action of URAT1, the uric acid transporter responsible for the majority of renal uric acid reabsorption.
Duzallo is not recommended for the treatment of asymptomatic hyperuricemia. It has a boxed warning regarding the risk of acute renal failure.
Duzallo is expected to be commercially available early in the fourth quarter of 2017.
Extended-release amantadine approved for treatment of dyskinesia in Parkinson’s
An extended-release formulation of amantadine received approval from the Food and Drug Administration on Aug. 24 for the treatment of dyskinesia in patients with Parkinson’s disease. The significant increase in functional time for Parkinson’s disease patients with dyskinesia who took extended-release amantadine was attributable both to a reduction in off-time and to a decrease in troublesome dyskinesia during on-time.
This is the first FDA approval for a drug to treat levodopa therapy-related dyskinesia in patients with Parkinson’s disease, according to an announcement from its manufacturer, Adamas Pharmaceuticals.
The second study also showed clinically relevant and statistically significant results, with a 46% reduction on the UDysRS for those taking ER amantadine, compared with a 16% reduction for those taking placebo.
The oral ER amantadine formulation delivers 274 mg of amantadine once daily at bedtime, allowing sustained high levels of the drug during waking hours, with peak levels delivered during the morning and throughout the day and a trough near bedtime.
When investigators of the two studies analyzed diaries that had been kept by Parkinson’s disease patients, they found that patients in the two studies who were taking ER amantadine experienced a placebo-adjusted reduction in off-time of about 1 hour per day.
Patients in the ER amantadine arm of the first study also had an increase of 3.6 hours per day of functional time, compared with a 0.8-hour increase for patients taking placebo. In the second study, functional time went up by 4.0 hours per day for patients in the ER amantadine arm, compared with an increase of 2.1 hours per day for those on placebo. Functional time was defined as on-time without troublesome dyskinesia.
Adverse reactions to ER amantadine that occurred in more than 10% of patients in the active arms of the study, and which occurred more frequently than in those taking placebo, included hallucinations, falls, orthostatic hypotension, dizziness, peripheral edema, dry mouth, and constipation.
The medication is contraindicated in those with creatinine clearance below 15 mL/min/1.73 m2. Prescribing information advises that amantadine ER be avoided or used with caution in patients with a history of suicidality and depression, hallucinations or psychotic behavior, and orthostatic hypotension or dizziness.
Patients taking ER amantadine may also experience impulsivity and sexual, spending, or gambling urges. Abrupt withdrawal or rapid dose reduction may result in withdrawal-emergent hyperpyrexia or confusion, including delirium, hallucinations, stupor, and slurred speech.
The clinical trials upon which the approval is based were funded by Adamas Pharmaceuticals.
koakes@frontlinemedcom.com
On Twitter @karioakes
An extended-release formulation of amantadine received approval from the Food and Drug Administration on Aug. 24 for the treatment of dyskinesia in patients with Parkinson’s disease. The significant increase in functional time for Parkinson’s disease patients with dyskinesia who took extended-release amantadine was attributable both to a reduction in off-time and to a decrease in troublesome dyskinesia during on-time.
This is the first FDA approval for a drug to treat levodopa therapy-related dyskinesia in patients with Parkinson’s disease, according to an announcement from its manufacturer, Adamas Pharmaceuticals.
The second study also showed clinically relevant and statistically significant results, with a 46% reduction on the UDysRS for those taking ER amantadine, compared with a 16% reduction for those taking placebo.
The oral ER amantadine formulation delivers 274 mg of amantadine once daily at bedtime, allowing sustained high levels of the drug during waking hours, with peak levels delivered during the morning and throughout the day and a trough near bedtime.
When investigators of the two studies analyzed diaries that had been kept by Parkinson’s disease patients, they found that patients in the two studies who were taking ER amantadine experienced a placebo-adjusted reduction in off-time of about 1 hour per day.
Patients in the ER amantadine arm of the first study also had an increase of 3.6 hours per day of functional time, compared with a 0.8-hour increase for patients taking placebo. In the second study, functional time went up by 4.0 hours per day for patients in the ER amantadine arm, compared with an increase of 2.1 hours per day for those on placebo. Functional time was defined as on-time without troublesome dyskinesia.
Adverse reactions to ER amantadine that occurred in more than 10% of patients in the active arms of the study, and which occurred more frequently than in those taking placebo, included hallucinations, falls, orthostatic hypotension, dizziness, peripheral edema, dry mouth, and constipation.
The medication is contraindicated in those with creatinine clearance below 15 mL/min/1.73 m2. Prescribing information advises that amantadine ER be avoided or used with caution in patients with a history of suicidality and depression, hallucinations or psychotic behavior, and orthostatic hypotension or dizziness.
Patients taking ER amantadine may also experience impulsivity and sexual, spending, or gambling urges. Abrupt withdrawal or rapid dose reduction may result in withdrawal-emergent hyperpyrexia or confusion, including delirium, hallucinations, stupor, and slurred speech.
The clinical trials upon which the approval is based were funded by Adamas Pharmaceuticals.
koakes@frontlinemedcom.com
On Twitter @karioakes
An extended-release formulation of amantadine received approval from the Food and Drug Administration on Aug. 24 for the treatment of dyskinesia in patients with Parkinson’s disease. The significant increase in functional time for Parkinson’s disease patients with dyskinesia who took extended-release amantadine was attributable both to a reduction in off-time and to a decrease in troublesome dyskinesia during on-time.
This is the first FDA approval for a drug to treat levodopa therapy-related dyskinesia in patients with Parkinson’s disease, according to an announcement from its manufacturer, Adamas Pharmaceuticals.
The second study also showed clinically relevant and statistically significant results, with a 46% reduction on the UDysRS for those taking ER amantadine, compared with a 16% reduction for those taking placebo.
The oral ER amantadine formulation delivers 274 mg of amantadine once daily at bedtime, allowing sustained high levels of the drug during waking hours, with peak levels delivered during the morning and throughout the day and a trough near bedtime.
When investigators of the two studies analyzed diaries that had been kept by Parkinson’s disease patients, they found that patients in the two studies who were taking ER amantadine experienced a placebo-adjusted reduction in off-time of about 1 hour per day.
Patients in the ER amantadine arm of the first study also had an increase of 3.6 hours per day of functional time, compared with a 0.8-hour increase for patients taking placebo. In the second study, functional time went up by 4.0 hours per day for patients in the ER amantadine arm, compared with an increase of 2.1 hours per day for those on placebo. Functional time was defined as on-time without troublesome dyskinesia.
Adverse reactions to ER amantadine that occurred in more than 10% of patients in the active arms of the study, and which occurred more frequently than in those taking placebo, included hallucinations, falls, orthostatic hypotension, dizziness, peripheral edema, dry mouth, and constipation.
The medication is contraindicated in those with creatinine clearance below 15 mL/min/1.73 m2. Prescribing information advises that amantadine ER be avoided or used with caution in patients with a history of suicidality and depression, hallucinations or psychotic behavior, and orthostatic hypotension or dizziness.
Patients taking ER amantadine may also experience impulsivity and sexual, spending, or gambling urges. Abrupt withdrawal or rapid dose reduction may result in withdrawal-emergent hyperpyrexia or confusion, including delirium, hallucinations, stupor, and slurred speech.
The clinical trials upon which the approval is based were funded by Adamas Pharmaceuticals.
koakes@frontlinemedcom.com
On Twitter @karioakes
L-glutamine to prevent sickle cell complications featured in FDA podcast
The recent approval of L-glutamine, marketed as Endari, to reduce the acute complications of sickle cell disease in adult and pediatric patients 5 years of age and older is discussed in the Drug Information Soundcast in Clinical Oncology (DISCO) from a Food and Drug Adminstration podcast series that provides information about new product approvals, emerging safety information for cancer treatments, and other current topics in cancer drug development.
The basis for the approval was discussed in our coverage of the FDA’s Oncologic Drugs Advisory Committee meeting.
This episode of DISCO is hosted by Sanjeeve Bala, MD, and was developed by Abhilasha Nair, MD; Dr. Bala; Kathy M. Robie Suh, MD; Ann T. Farrell, MD; Kirsten B. Goldberg, and Richard Pazdur, MD. All are with the FDA’s Oncology Center of Excellence and the Office of Hematology and Oncology Products. Steven Jackson of the FDA’s Division of Drug Information was the sound producer.
The recent approval of L-glutamine, marketed as Endari, to reduce the acute complications of sickle cell disease in adult and pediatric patients 5 years of age and older is discussed in the Drug Information Soundcast in Clinical Oncology (DISCO) from a Food and Drug Adminstration podcast series that provides information about new product approvals, emerging safety information for cancer treatments, and other current topics in cancer drug development.
The basis for the approval was discussed in our coverage of the FDA’s Oncologic Drugs Advisory Committee meeting.
This episode of DISCO is hosted by Sanjeeve Bala, MD, and was developed by Abhilasha Nair, MD; Dr. Bala; Kathy M. Robie Suh, MD; Ann T. Farrell, MD; Kirsten B. Goldberg, and Richard Pazdur, MD. All are with the FDA’s Oncology Center of Excellence and the Office of Hematology and Oncology Products. Steven Jackson of the FDA’s Division of Drug Information was the sound producer.
The recent approval of L-glutamine, marketed as Endari, to reduce the acute complications of sickle cell disease in adult and pediatric patients 5 years of age and older is discussed in the Drug Information Soundcast in Clinical Oncology (DISCO) from a Food and Drug Adminstration podcast series that provides information about new product approvals, emerging safety information for cancer treatments, and other current topics in cancer drug development.
The basis for the approval was discussed in our coverage of the FDA’s Oncologic Drugs Advisory Committee meeting.
This episode of DISCO is hosted by Sanjeeve Bala, MD, and was developed by Abhilasha Nair, MD; Dr. Bala; Kathy M. Robie Suh, MD; Ann T. Farrell, MD; Kirsten B. Goldberg, and Richard Pazdur, MD. All are with the FDA’s Oncology Center of Excellence and the Office of Hematology and Oncology Products. Steven Jackson of the FDA’s Division of Drug Information was the sound producer.
Standardized infection ratio for CLABSI almost halved since 2009
The standardized infection ratio (SIR) for central line–associated bloodstream infections dropped 42% from 2009 to 2014, according to the Agency for Healthcare Research and Quality.
For acute care hospitalizations, the SIR for central line–associated bloodstream infections (CLABSIs) fell from 0.854 in 2009 to 0.495 in 2014. Over that same time period, the SIR for surgical site infections involving Surgical Care Improvement Project procedures decreased from 0.981 to 0.827 – almost 16%, the AHRQ said in its annual National Healthcare Quality and Disparities Report.
From 2010 to 2014, the SIR for catheter-associated urinary tract infections increased 6.7% from 0.937 to 1.000, but that change was not significant. For laboratory-identified hospital-onset Clostridium difficile infection, the SIR dropped from 0.963 to 0.924 – about 4% – from 2012 to 2014, the AHRQ reported using data from the National Center for Emerging and Zoonotic Infectious Diseases and the National Healthcare Safety Network.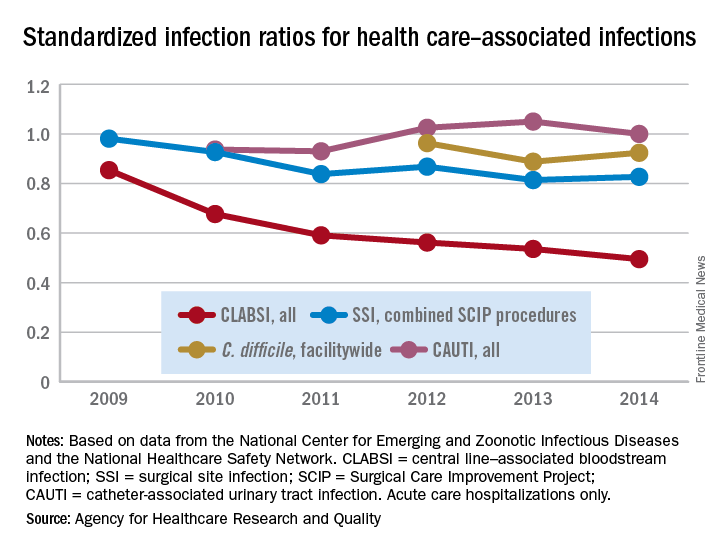
The standardized infection ratio (SIR) for central line–associated bloodstream infections dropped 42% from 2009 to 2014, according to the Agency for Healthcare Research and Quality.
For acute care hospitalizations, the SIR for central line–associated bloodstream infections (CLABSIs) fell from 0.854 in 2009 to 0.495 in 2014. Over that same time period, the SIR for surgical site infections involving Surgical Care Improvement Project procedures decreased from 0.981 to 0.827 – almost 16%, the AHRQ said in its annual National Healthcare Quality and Disparities Report.
From 2010 to 2014, the SIR for catheter-associated urinary tract infections increased 6.7% from 0.937 to 1.000, but that change was not significant. For laboratory-identified hospital-onset Clostridium difficile infection, the SIR dropped from 0.963 to 0.924 – about 4% – from 2012 to 2014, the AHRQ reported using data from the National Center for Emerging and Zoonotic Infectious Diseases and the National Healthcare Safety Network.
The standardized infection ratio (SIR) for central line–associated bloodstream infections dropped 42% from 2009 to 2014, according to the Agency for Healthcare Research and Quality.
For acute care hospitalizations, the SIR for central line–associated bloodstream infections (CLABSIs) fell from 0.854 in 2009 to 0.495 in 2014. Over that same time period, the SIR for surgical site infections involving Surgical Care Improvement Project procedures decreased from 0.981 to 0.827 – almost 16%, the AHRQ said in its annual National Healthcare Quality and Disparities Report.
From 2010 to 2014, the SIR for catheter-associated urinary tract infections increased 6.7% from 0.937 to 1.000, but that change was not significant. For laboratory-identified hospital-onset Clostridium difficile infection, the SIR dropped from 0.963 to 0.924 – about 4% – from 2012 to 2014, the AHRQ reported using data from the National Center for Emerging and Zoonotic Infectious Diseases and the National Healthcare Safety Network.
Exelixis seeks expanded indication for cabozantinib in RCC
Exelixis has submitted a supplemental New Drug Application to the Food and Drug Administration for cabozantinib (Cabometyx) for the treatment of previously untreated advanced renal cell carcinoma (RCC).
The application, announced on Aug. 16, seeks to allow the manufacturer to modify the label. Cabozantinib was approved in April 2016 for treatment of patients with advanced RCC who had previously received antiangiogenic therapy.![]()
The results of the trial were published in the Journal of Clinical Oncology (2017 Feb 20;35[6]:591-7). An independent review committee confirmed the primary efficacy endpoint results in June 2017.
Exelixis has submitted a supplemental New Drug Application to the Food and Drug Administration for cabozantinib (Cabometyx) for the treatment of previously untreated advanced renal cell carcinoma (RCC).
The application, announced on Aug. 16, seeks to allow the manufacturer to modify the label. Cabozantinib was approved in April 2016 for treatment of patients with advanced RCC who had previously received antiangiogenic therapy.![]()
The results of the trial were published in the Journal of Clinical Oncology (2017 Feb 20;35[6]:591-7). An independent review committee confirmed the primary efficacy endpoint results in June 2017.
Exelixis has submitted a supplemental New Drug Application to the Food and Drug Administration for cabozantinib (Cabometyx) for the treatment of previously untreated advanced renal cell carcinoma (RCC).
The application, announced on Aug. 16, seeks to allow the manufacturer to modify the label. Cabozantinib was approved in April 2016 for treatment of patients with advanced RCC who had previously received antiangiogenic therapy.![]()
The results of the trial were published in the Journal of Clinical Oncology (2017 Feb 20;35[6]:591-7). An independent review committee confirmed the primary efficacy endpoint results in June 2017.
Inotuzumab ozogamicin approved for relapsed/refractory ALL
The Food and Drug Administration has approved the antibody drug conjugate inotuzumab ozogamicin for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
The treatment, to be marketed by Pfizer as Besponsa, won approval based on the results of the INO-VATE ALL trial, which randomized 326 patients to receive either inotuzumab ozogamicin (164 patients) or a chemotherapy regimen of the investigator’s choice (162 patients). To be considered for inclusion in the trial, patients with Philadelphia chromosome–negative or –positive relapsed or refractory B-cell precursor ALL were required to have at least 5% bone marrow blasts and have received one or two induction chemotherapy regimens.
Adverse events that occurred in more than 20% of patients included thrombocytopenia, neutropenia, anemia, leukopenia, fatigue, hemorrhage, pyrexia, nausea, headache, febrile neutropenia, abdominal pain, and hyperbilirubinemia, as well as increases in gamma-glutamyltransferase and transaminases. Adverse events that led to discontinuation of treatment were infection, thrombocytopenia, hyperbilirubinemia, hemorrhage, and increases in transaminases.
Preliminary results were published in August 2016 (N Engl J Med. 2016;375:740-53).
Inotuzumab ozogamicin was granted orphan drug and breakthrough status, as well as priority review, by the FDA in February 2017.
dfulton@frontlinemedcom.com
On Twitter @denisefulton
The Food and Drug Administration has approved the antibody drug conjugate inotuzumab ozogamicin for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
The treatment, to be marketed by Pfizer as Besponsa, won approval based on the results of the INO-VATE ALL trial, which randomized 326 patients to receive either inotuzumab ozogamicin (164 patients) or a chemotherapy regimen of the investigator’s choice (162 patients). To be considered for inclusion in the trial, patients with Philadelphia chromosome–negative or –positive relapsed or refractory B-cell precursor ALL were required to have at least 5% bone marrow blasts and have received one or two induction chemotherapy regimens.
Adverse events that occurred in more than 20% of patients included thrombocytopenia, neutropenia, anemia, leukopenia, fatigue, hemorrhage, pyrexia, nausea, headache, febrile neutropenia, abdominal pain, and hyperbilirubinemia, as well as increases in gamma-glutamyltransferase and transaminases. Adverse events that led to discontinuation of treatment were infection, thrombocytopenia, hyperbilirubinemia, hemorrhage, and increases in transaminases.
Preliminary results were published in August 2016 (N Engl J Med. 2016;375:740-53).
Inotuzumab ozogamicin was granted orphan drug and breakthrough status, as well as priority review, by the FDA in February 2017.
dfulton@frontlinemedcom.com
On Twitter @denisefulton
The Food and Drug Administration has approved the antibody drug conjugate inotuzumab ozogamicin for the treatment of adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL).
The treatment, to be marketed by Pfizer as Besponsa, won approval based on the results of the INO-VATE ALL trial, which randomized 326 patients to receive either inotuzumab ozogamicin (164 patients) or a chemotherapy regimen of the investigator’s choice (162 patients). To be considered for inclusion in the trial, patients with Philadelphia chromosome–negative or –positive relapsed or refractory B-cell precursor ALL were required to have at least 5% bone marrow blasts and have received one or two induction chemotherapy regimens.
Adverse events that occurred in more than 20% of patients included thrombocytopenia, neutropenia, anemia, leukopenia, fatigue, hemorrhage, pyrexia, nausea, headache, febrile neutropenia, abdominal pain, and hyperbilirubinemia, as well as increases in gamma-glutamyltransferase and transaminases. Adverse events that led to discontinuation of treatment were infection, thrombocytopenia, hyperbilirubinemia, hemorrhage, and increases in transaminases.
Preliminary results were published in August 2016 (N Engl J Med. 2016;375:740-53).
Inotuzumab ozogamicin was granted orphan drug and breakthrough status, as well as priority review, by the FDA in February 2017.
dfulton@frontlinemedcom.com
On Twitter @denisefulton
FDA advisory committee to consider adjuvant sunitinib for RCC
The Oncologic Drugs Advisory Committee to the Food and Drug Administration will meet on Sept. 19 to discuss a supplemental new drug application for sunitinib (Sutent), for the adjuvant treatment of adult patients at high risk of recurrent renal cell carcinoma (RCC) following nephrectomy.
Sunitinib is an oral antiangiogenic agent that has been approved for the treatment of advanced RCC since 2006.
The FDA accepted the new drug application in May and is expected to issue a decision by January 2018.
Results for sunitinib as adjuvant treatment have been mixed. No significant differences in disease-free survival or overall survival were found in the phase 3 ASSURE between patients receiving adjuvant sunitinib and those receiving placebo, according to results published in The Lancet. However, adjuvant sunitinib prolonged disease-free survival by 1.2 years, compared with placebo, in the phase 3 S-TRAC trial, presented at the 2016 ESMO Congress and published in the New England Journal of Medicine. S-TRAC results are the basis for the new drug application submitted by Pfizer, Inc., according to a press release.
The advisory committee will consider comments from the public if submitted by Sept. 5, as electronic comments through the electronic filing system or by mail/hand delivery/courier at Division of Dockets Management (HFA-305), Food and Drug Administration, 5630 Fishers Lane, Rm. 1061, Rockville, MD 20852.
The docket number is FDA-2017-N-1063.
The Oncologic Drugs Advisory Committee to the Food and Drug Administration will meet on Sept. 19 to discuss a supplemental new drug application for sunitinib (Sutent), for the adjuvant treatment of adult patients at high risk of recurrent renal cell carcinoma (RCC) following nephrectomy.
Sunitinib is an oral antiangiogenic agent that has been approved for the treatment of advanced RCC since 2006.
The FDA accepted the new drug application in May and is expected to issue a decision by January 2018.
Results for sunitinib as adjuvant treatment have been mixed. No significant differences in disease-free survival or overall survival were found in the phase 3 ASSURE between patients receiving adjuvant sunitinib and those receiving placebo, according to results published in The Lancet. However, adjuvant sunitinib prolonged disease-free survival by 1.2 years, compared with placebo, in the phase 3 S-TRAC trial, presented at the 2016 ESMO Congress and published in the New England Journal of Medicine. S-TRAC results are the basis for the new drug application submitted by Pfizer, Inc., according to a press release.
The advisory committee will consider comments from the public if submitted by Sept. 5, as electronic comments through the electronic filing system or by mail/hand delivery/courier at Division of Dockets Management (HFA-305), Food and Drug Administration, 5630 Fishers Lane, Rm. 1061, Rockville, MD 20852.
The docket number is FDA-2017-N-1063.
The Oncologic Drugs Advisory Committee to the Food and Drug Administration will meet on Sept. 19 to discuss a supplemental new drug application for sunitinib (Sutent), for the adjuvant treatment of adult patients at high risk of recurrent renal cell carcinoma (RCC) following nephrectomy.
Sunitinib is an oral antiangiogenic agent that has been approved for the treatment of advanced RCC since 2006.
The FDA accepted the new drug application in May and is expected to issue a decision by January 2018.
Results for sunitinib as adjuvant treatment have been mixed. No significant differences in disease-free survival or overall survival were found in the phase 3 ASSURE between patients receiving adjuvant sunitinib and those receiving placebo, according to results published in The Lancet. However, adjuvant sunitinib prolonged disease-free survival by 1.2 years, compared with placebo, in the phase 3 S-TRAC trial, presented at the 2016 ESMO Congress and published in the New England Journal of Medicine. S-TRAC results are the basis for the new drug application submitted by Pfizer, Inc., according to a press release.
The advisory committee will consider comments from the public if submitted by Sept. 5, as electronic comments through the electronic filing system or by mail/hand delivery/courier at Division of Dockets Management (HFA-305), Food and Drug Administration, 5630 Fishers Lane, Rm. 1061, Rockville, MD 20852.
The docket number is FDA-2017-N-1063.
CDC: 3.4 million Americans have epilepsy
Epilepsy estimates available for the first time for every state show that the disorder is widespread, with at least 3.4 million people affected, according to the Centers for Disease Control and Prevention.
The CDC data also show that the number of people with epilepsy is increasing, probably as a result of population growth. The number of affected adults went from 2.3 million in 2010 to 3 million in 2015, and the number of children with epilepsy rose from 450,000 in 2007 to 470,000 in 2015, CDC investigators reported (MMWR. 2017 Aug 11;66[31]:821-5).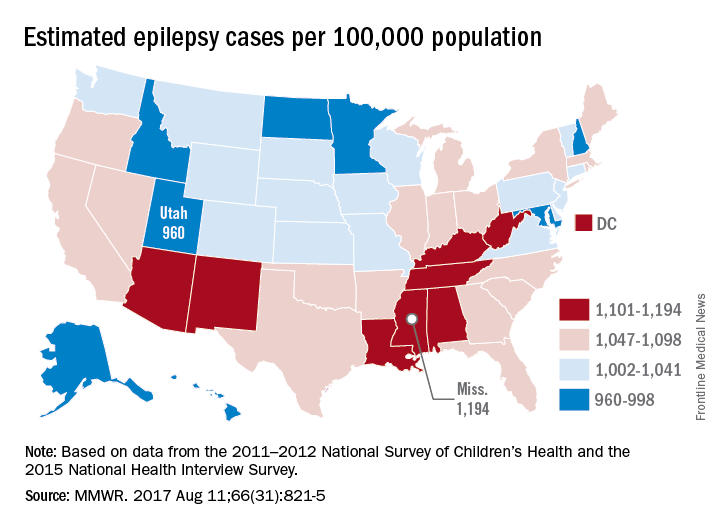
“Millions of Americans are impacted by epilepsy, and unfortunately, this study shows cases are on the rise,” CDC Director Brenda Fitzgerald said in a separate statement. “Proper diagnosis is key to finding an effective treatment – and at CDC we are committed to researching, testing, and sharing strategies that will improve the lives of people with epilepsy.”
The CDC investigators based their estimates for children under age 18 years on data from the 2011-2012 National Survey of Children’s Health; estimates for those age 18 and over are based on data from the 2015 National Health Interview Survey.
Epilepsy estimates available for the first time for every state show that the disorder is widespread, with at least 3.4 million people affected, according to the Centers for Disease Control and Prevention.
The CDC data also show that the number of people with epilepsy is increasing, probably as a result of population growth. The number of affected adults went from 2.3 million in 2010 to 3 million in 2015, and the number of children with epilepsy rose from 450,000 in 2007 to 470,000 in 2015, CDC investigators reported (MMWR. 2017 Aug 11;66[31]:821-5).
“Millions of Americans are impacted by epilepsy, and unfortunately, this study shows cases are on the rise,” CDC Director Brenda Fitzgerald said in a separate statement. “Proper diagnosis is key to finding an effective treatment – and at CDC we are committed to researching, testing, and sharing strategies that will improve the lives of people with epilepsy.”
The CDC investigators based their estimates for children under age 18 years on data from the 2011-2012 National Survey of Children’s Health; estimates for those age 18 and over are based on data from the 2015 National Health Interview Survey.
Epilepsy estimates available for the first time for every state show that the disorder is widespread, with at least 3.4 million people affected, according to the Centers for Disease Control and Prevention.
The CDC data also show that the number of people with epilepsy is increasing, probably as a result of population growth. The number of affected adults went from 2.3 million in 2010 to 3 million in 2015, and the number of children with epilepsy rose from 450,000 in 2007 to 470,000 in 2015, CDC investigators reported (MMWR. 2017 Aug 11;66[31]:821-5).
“Millions of Americans are impacted by epilepsy, and unfortunately, this study shows cases are on the rise,” CDC Director Brenda Fitzgerald said in a separate statement. “Proper diagnosis is key to finding an effective treatment – and at CDC we are committed to researching, testing, and sharing strategies that will improve the lives of people with epilepsy.”
The CDC investigators based their estimates for children under age 18 years on data from the 2011-2012 National Survey of Children’s Health; estimates for those age 18 and over are based on data from the 2015 National Health Interview Survey.
FROM MMWR
FDA approves first spironolactone oral suspension
The Food and Drug Administration has approved CaroSpir, the first oral suspension form of spironolactone, the aldosterone antagonist that was first approved in 1960, according to an announcement from CMP Pharma.
CaroSpir is intended for the treatment of New York Heart Association class III-IV heart failure and reduced ejection fraction, usually in combination with other treatments. CaroSpir is also indicated as an add-on medication for the treatment of hypertension, and for the treatment of edema in cirrhotic patients who have not adequately responded to fluid and sodium restriction.
“CaroSpir provides a stable, ready to use, and consistent liquid treatment option for adult patients. Up until now, these patients have been prescribed a pharmacy-compounded liquid form of spironolactone. The dosing inconsistencies of compounded liquids have long been a persistent challenge for physicians,” Gerald Sakowski, CEO at CMP Pharma, said in the press release.
Find the full press release on the CMP Pharma website.
The Food and Drug Administration has approved CaroSpir, the first oral suspension form of spironolactone, the aldosterone antagonist that was first approved in 1960, according to an announcement from CMP Pharma.
CaroSpir is intended for the treatment of New York Heart Association class III-IV heart failure and reduced ejection fraction, usually in combination with other treatments. CaroSpir is also indicated as an add-on medication for the treatment of hypertension, and for the treatment of edema in cirrhotic patients who have not adequately responded to fluid and sodium restriction.
“CaroSpir provides a stable, ready to use, and consistent liquid treatment option for adult patients. Up until now, these patients have been prescribed a pharmacy-compounded liquid form of spironolactone. The dosing inconsistencies of compounded liquids have long been a persistent challenge for physicians,” Gerald Sakowski, CEO at CMP Pharma, said in the press release.
Find the full press release on the CMP Pharma website.
The Food and Drug Administration has approved CaroSpir, the first oral suspension form of spironolactone, the aldosterone antagonist that was first approved in 1960, according to an announcement from CMP Pharma.
CaroSpir is intended for the treatment of New York Heart Association class III-IV heart failure and reduced ejection fraction, usually in combination with other treatments. CaroSpir is also indicated as an add-on medication for the treatment of hypertension, and for the treatment of edema in cirrhotic patients who have not adequately responded to fluid and sodium restriction.
“CaroSpir provides a stable, ready to use, and consistent liquid treatment option for adult patients. Up until now, these patients have been prescribed a pharmacy-compounded liquid form of spironolactone. The dosing inconsistencies of compounded liquids have long been a persistent challenge for physicians,” Gerald Sakowski, CEO at CMP Pharma, said in the press release.
Find the full press release on the CMP Pharma website.