User login
ID experts urge widespread flu vaccination for 2018-2019 season
WASHINGTON – The flu vaccine may not be perfect, but it can reduce the severity of illness and curb the risk of spreading the disease to others, William Schaffner, MD, emphasized at a press conference held by the National Foundation for Infectious Diseases.
“Give the vaccine credit for softening the blow,” said Dr. Schaffner, medical director of NFID and a professor at Vanderbilt University in Nashville.
Dr. Schaffner and a panel of experts including U.S. Surgeon General Jerome M. Adams, MD, encouraged the public and the health care community to follow recommendation from the Centers for Disease Control & Prevention that everyone aged 6 months and older receive an influenza vaccine.
Dr. Schaffner shared recent data showing that complications from the flu don’t stop when the acute illness resolves. Acute influenza causes a whole-body inflammatory reaction, and consequently “there is an increased risk of heart attack and stroke during the 2-4 weeks of recovery from acute influenza,” he said. In addition, older adults who experience acute flu and are already frail may never regain their pre-flu level of function, as the flu can start a “domino effect of decline and disability.”
Despite last year’s severe flu season that included 180 deaths in children, vaccination remains the most effective protection against the flu, Dr. Adams said.
This year, between 163 million and 168 million doses of vaccine will be available in the United States. The vaccine is available in a range of settings including doctors’ offices, pharmacies, grocery stores, and workplaces, said Dr. Adams.
Flu vaccine choices this year include a return of the live-attenuated influenza vaccine (LAIV) given via nasal spray, along with the standard influenza vaccine that includes either three influenza viruses (trivalent, with two influenza A and one influenza B) or four influenza viruses (quadrivalent, with two influenza A and two influenza B). Other options are adjuvanted vaccine and high-dose vaccine for adults aged 65 years and older, and a cell-based and recombinant vaccine as alternatives to egg-based vaccines.
Dr. Adams emphasized the importance of healthy people getting vaccinated to protect the community. “All the people who died from the flu caught it from someone else,” he said.

The message to health care providers remains the same: Recommend the flu vaccine to patients at every opportunity, and lead by example and get vaccinated yourself, Dr. Adams said. He noted this year’s strategies to promote flu vaccination on social media, and encouraged clinicians to recommend the flu shot to their patients and to showcase their own shots via the #FightFlu hashtag.
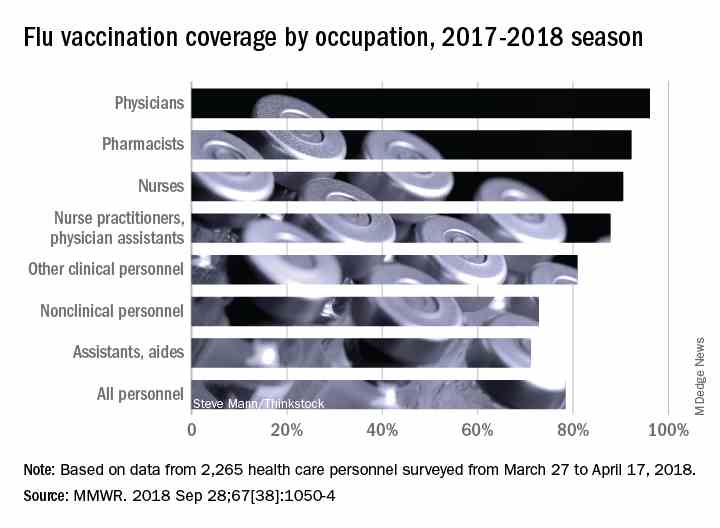
Vaccination among health care personnel last year was approximately 78%, which is a plateau over the past several years (MMWR 2018; 67:1050-54).
Be prepared to offer antivirals to patients as appropriate, and to promote the pneumococcal vaccine to eligible older adults as well, to protect not only themselves, but their contacts and the community, Dr. Adams emphasized. Currently approved antiviral drugs recommended for the 2018-2019 flu season: oseltamivir, zanamivir, and peramivir.
Wendy Sue Swanson, MD, of Seattle Children’s Hospital, stressed the importance of flu vaccination for all children, given their ability to spread viral infections. She noted a concerning 2% drop in vaccinations for children aged 6 months to 4 years, although vaccination coverage in this group was highest among children overall, at approximately 68% last season.
Last year, approximately 80% of the child deaths from flu occurred in unvaccinated children, but the vaccine has been shown to reduce the likelihood of hospitalization or death even if a child does become ill, Dr. Swanson said.
Laura E. Riley, MD, of Weill Cornell Medical Center, noted that vaccination of pregnant women has plateaued in recent years, and was 49% last year. “Our goal is 80% plus,” she said. Data show that pregnant women who received flu vaccination were 40% less likely to be hospitalized for the flu, she noted. The American College of Obstetricians and Gynecologists recommends flu vaccination as safe during any trimester, and valuable to both mothers and newborns because it provides protective antibodies during the first 6 months of life before babies can receive their own vaccinations, Dr. Riley said.
More information about this year’s flu season is available from the CDC and NFID.
WASHINGTON – The flu vaccine may not be perfect, but it can reduce the severity of illness and curb the risk of spreading the disease to others, William Schaffner, MD, emphasized at a press conference held by the National Foundation for Infectious Diseases.
“Give the vaccine credit for softening the blow,” said Dr. Schaffner, medical director of NFID and a professor at Vanderbilt University in Nashville.
Dr. Schaffner and a panel of experts including U.S. Surgeon General Jerome M. Adams, MD, encouraged the public and the health care community to follow recommendation from the Centers for Disease Control & Prevention that everyone aged 6 months and older receive an influenza vaccine.
Dr. Schaffner shared recent data showing that complications from the flu don’t stop when the acute illness resolves. Acute influenza causes a whole-body inflammatory reaction, and consequently “there is an increased risk of heart attack and stroke during the 2-4 weeks of recovery from acute influenza,” he said. In addition, older adults who experience acute flu and are already frail may never regain their pre-flu level of function, as the flu can start a “domino effect of decline and disability.”
Despite last year’s severe flu season that included 180 deaths in children, vaccination remains the most effective protection against the flu, Dr. Adams said.
This year, between 163 million and 168 million doses of vaccine will be available in the United States. The vaccine is available in a range of settings including doctors’ offices, pharmacies, grocery stores, and workplaces, said Dr. Adams.
Flu vaccine choices this year include a return of the live-attenuated influenza vaccine (LAIV) given via nasal spray, along with the standard influenza vaccine that includes either three influenza viruses (trivalent, with two influenza A and one influenza B) or four influenza viruses (quadrivalent, with two influenza A and two influenza B). Other options are adjuvanted vaccine and high-dose vaccine for adults aged 65 years and older, and a cell-based and recombinant vaccine as alternatives to egg-based vaccines.
Dr. Adams emphasized the importance of healthy people getting vaccinated to protect the community. “All the people who died from the flu caught it from someone else,” he said.
The message to health care providers remains the same: Recommend the flu vaccine to patients at every opportunity, and lead by example and get vaccinated yourself, Dr. Adams said. He noted this year’s strategies to promote flu vaccination on social media, and encouraged clinicians to recommend the flu shot to their patients and to showcase their own shots via the #FightFlu hashtag.
Vaccination among health care personnel last year was approximately 78%, which is a plateau over the past several years (MMWR 2018; 67:1050-54).
Be prepared to offer antivirals to patients as appropriate, and to promote the pneumococcal vaccine to eligible older adults as well, to protect not only themselves, but their contacts and the community, Dr. Adams emphasized. Currently approved antiviral drugs recommended for the 2018-2019 flu season: oseltamivir, zanamivir, and peramivir.
Wendy Sue Swanson, MD, of Seattle Children’s Hospital, stressed the importance of flu vaccination for all children, given their ability to spread viral infections. She noted a concerning 2% drop in vaccinations for children aged 6 months to 4 years, although vaccination coverage in this group was highest among children overall, at approximately 68% last season.
Last year, approximately 80% of the child deaths from flu occurred in unvaccinated children, but the vaccine has been shown to reduce the likelihood of hospitalization or death even if a child does become ill, Dr. Swanson said.
Laura E. Riley, MD, of Weill Cornell Medical Center, noted that vaccination of pregnant women has plateaued in recent years, and was 49% last year. “Our goal is 80% plus,” she said. Data show that pregnant women who received flu vaccination were 40% less likely to be hospitalized for the flu, she noted. The American College of Obstetricians and Gynecologists recommends flu vaccination as safe during any trimester, and valuable to both mothers and newborns because it provides protective antibodies during the first 6 months of life before babies can receive their own vaccinations, Dr. Riley said.
More information about this year’s flu season is available from the CDC and NFID.
WASHINGTON – The flu vaccine may not be perfect, but it can reduce the severity of illness and curb the risk of spreading the disease to others, William Schaffner, MD, emphasized at a press conference held by the National Foundation for Infectious Diseases.
“Give the vaccine credit for softening the blow,” said Dr. Schaffner, medical director of NFID and a professor at Vanderbilt University in Nashville.
Dr. Schaffner and a panel of experts including U.S. Surgeon General Jerome M. Adams, MD, encouraged the public and the health care community to follow recommendation from the Centers for Disease Control & Prevention that everyone aged 6 months and older receive an influenza vaccine.
Dr. Schaffner shared recent data showing that complications from the flu don’t stop when the acute illness resolves. Acute influenza causes a whole-body inflammatory reaction, and consequently “there is an increased risk of heart attack and stroke during the 2-4 weeks of recovery from acute influenza,” he said. In addition, older adults who experience acute flu and are already frail may never regain their pre-flu level of function, as the flu can start a “domino effect of decline and disability.”
Despite last year’s severe flu season that included 180 deaths in children, vaccination remains the most effective protection against the flu, Dr. Adams said.
This year, between 163 million and 168 million doses of vaccine will be available in the United States. The vaccine is available in a range of settings including doctors’ offices, pharmacies, grocery stores, and workplaces, said Dr. Adams.
Flu vaccine choices this year include a return of the live-attenuated influenza vaccine (LAIV) given via nasal spray, along with the standard influenza vaccine that includes either three influenza viruses (trivalent, with two influenza A and one influenza B) or four influenza viruses (quadrivalent, with two influenza A and two influenza B). Other options are adjuvanted vaccine and high-dose vaccine for adults aged 65 years and older, and a cell-based and recombinant vaccine as alternatives to egg-based vaccines.
Dr. Adams emphasized the importance of healthy people getting vaccinated to protect the community. “All the people who died from the flu caught it from someone else,” he said.
The message to health care providers remains the same: Recommend the flu vaccine to patients at every opportunity, and lead by example and get vaccinated yourself, Dr. Adams said. He noted this year’s strategies to promote flu vaccination on social media, and encouraged clinicians to recommend the flu shot to their patients and to showcase their own shots via the #FightFlu hashtag.
Vaccination among health care personnel last year was approximately 78%, which is a plateau over the past several years (MMWR 2018; 67:1050-54).
Be prepared to offer antivirals to patients as appropriate, and to promote the pneumococcal vaccine to eligible older adults as well, to protect not only themselves, but their contacts and the community, Dr. Adams emphasized. Currently approved antiviral drugs recommended for the 2018-2019 flu season: oseltamivir, zanamivir, and peramivir.
Wendy Sue Swanson, MD, of Seattle Children’s Hospital, stressed the importance of flu vaccination for all children, given their ability to spread viral infections. She noted a concerning 2% drop in vaccinations for children aged 6 months to 4 years, although vaccination coverage in this group was highest among children overall, at approximately 68% last season.
Last year, approximately 80% of the child deaths from flu occurred in unvaccinated children, but the vaccine has been shown to reduce the likelihood of hospitalization or death even if a child does become ill, Dr. Swanson said.
Laura E. Riley, MD, of Weill Cornell Medical Center, noted that vaccination of pregnant women has plateaued in recent years, and was 49% last year. “Our goal is 80% plus,” she said. Data show that pregnant women who received flu vaccination were 40% less likely to be hospitalized for the flu, she noted. The American College of Obstetricians and Gynecologists recommends flu vaccination as safe during any trimester, and valuable to both mothers and newborns because it provides protective antibodies during the first 6 months of life before babies can receive their own vaccinations, Dr. Riley said.
More information about this year’s flu season is available from the CDC and NFID.
FROM AN NFID PRESS CONFERENCE
FDA approves new drug for CLL/SLL and follicular lymphoma
The Food and Drug Administration has approved duvelisib (Copiktra), a dual PI3K delta/gamma inhibitor, for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and follicular lymphoma.

who have received at least two prior therapies. Duvelisib also has accelerated approval to treat adults with relapsed or refractory follicular lymphoma (FL) who have received at least two prior systemic therapies.
Accelerated approval is based on a surrogate or intermediate endpoint – in this case, overall response rate – that is reasonably likely to predict clinical benefit. Continued approval of duvelisib in FL may be contingent upon results of confirmatory trials verifying that the drug provides a clinical benefit.
Duvelisib will be available in the U.S. immediately, according to Verastem, the company marketing the drug. The prescribing information for duvelisib includes a boxed warning detailing four fatal and/or serious toxicities associated with the drug – infections, diarrhea or colitis, cutaneous reactions, and pneumonitis. Verastem said it is implementing an informational risk evaluation and mitigation strategy to provide appropriate dosing and safety information for duvelisib.
The recommended dose of duvelisib is 25 mg orally twice daily, taken continuously in 28-day treatment cycles.
The FDA’s approval of duvelisib is supported by data from the phase 3 DUO trial and the phase 2 DYNAMO trial. The DUO trial included 319 patients with CLL (n=312) or SLL (n=7) who had received at least one prior therapy. They were randomized to receive either duvelisib (25 mg orally twice daily) or ofatumumab (initial infusion of 300 mg followed by 7 weekly infusions and 4 monthly infusions of 2,000 mg).
Efficacy results are based on patients who had received at least two prior therapies, including 95 patients in the duvelisib arm and 101 in the ofatumumab arm. The overall response rate was 78% in the duvelisib arm and 39% in the ofatumumab arm. All responses in both arms were partial responses.
The median progression-free survival was 16.4 months with duvelisib and 9.1 months with ofatumumab.
The safety results include all patients treated with duvelisib or ofatumumab in this trial. In the duvelisib arm, 12% of patients had fatal adverse events (AEs) within 30 days of the last dose. The same was true of 4% of patients treated with ofatumumab. Serious AEs occurred in 73% of patients treated with duvelisib. The most common were infection and diarrhea/colitis. The DYNAMO trial enrolled patients with indolent non-Hodgkin lymphoma whose disease was refractory to both rituximab and chemotherapy or radioimmunotherapy. There were 83 patients with FL.
Patients received duvelisib at 25 mg orally twice daily until disease progression or unacceptable toxicity.
The overall response rate was 42%. One patient achieved a complete response, and 34 had a partial response.
Forty-three percent of responders maintained their response at 6 months, and 17% maintained their response at 12 months.
Serious AEs occurred in 58% of FL patients. The most common were diarrhea/colitis, pneumonia, renal insufficiency, rash, and sepsis.
The Food and Drug Administration has approved duvelisib (Copiktra), a dual PI3K delta/gamma inhibitor, for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and follicular lymphoma.
who have received at least two prior therapies. Duvelisib also has accelerated approval to treat adults with relapsed or refractory follicular lymphoma (FL) who have received at least two prior systemic therapies.
Accelerated approval is based on a surrogate or intermediate endpoint – in this case, overall response rate – that is reasonably likely to predict clinical benefit. Continued approval of duvelisib in FL may be contingent upon results of confirmatory trials verifying that the drug provides a clinical benefit.
Duvelisib will be available in the U.S. immediately, according to Verastem, the company marketing the drug. The prescribing information for duvelisib includes a boxed warning detailing four fatal and/or serious toxicities associated with the drug – infections, diarrhea or colitis, cutaneous reactions, and pneumonitis. Verastem said it is implementing an informational risk evaluation and mitigation strategy to provide appropriate dosing and safety information for duvelisib.
The recommended dose of duvelisib is 25 mg orally twice daily, taken continuously in 28-day treatment cycles.
The FDA’s approval of duvelisib is supported by data from the phase 3 DUO trial and the phase 2 DYNAMO trial. The DUO trial included 319 patients with CLL (n=312) or SLL (n=7) who had received at least one prior therapy. They were randomized to receive either duvelisib (25 mg orally twice daily) or ofatumumab (initial infusion of 300 mg followed by 7 weekly infusions and 4 monthly infusions of 2,000 mg).
Efficacy results are based on patients who had received at least two prior therapies, including 95 patients in the duvelisib arm and 101 in the ofatumumab arm. The overall response rate was 78% in the duvelisib arm and 39% in the ofatumumab arm. All responses in both arms were partial responses.
The median progression-free survival was 16.4 months with duvelisib and 9.1 months with ofatumumab.
The safety results include all patients treated with duvelisib or ofatumumab in this trial. In the duvelisib arm, 12% of patients had fatal adverse events (AEs) within 30 days of the last dose. The same was true of 4% of patients treated with ofatumumab. Serious AEs occurred in 73% of patients treated with duvelisib. The most common were infection and diarrhea/colitis. The DYNAMO trial enrolled patients with indolent non-Hodgkin lymphoma whose disease was refractory to both rituximab and chemotherapy or radioimmunotherapy. There were 83 patients with FL.
Patients received duvelisib at 25 mg orally twice daily until disease progression or unacceptable toxicity.
The overall response rate was 42%. One patient achieved a complete response, and 34 had a partial response.
Forty-three percent of responders maintained their response at 6 months, and 17% maintained their response at 12 months.
Serious AEs occurred in 58% of FL patients. The most common were diarrhea/colitis, pneumonia, renal insufficiency, rash, and sepsis.
The Food and Drug Administration has approved duvelisib (Copiktra), a dual PI3K delta/gamma inhibitor, for the treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) and follicular lymphoma.
who have received at least two prior therapies. Duvelisib also has accelerated approval to treat adults with relapsed or refractory follicular lymphoma (FL) who have received at least two prior systemic therapies.
Accelerated approval is based on a surrogate or intermediate endpoint – in this case, overall response rate – that is reasonably likely to predict clinical benefit. Continued approval of duvelisib in FL may be contingent upon results of confirmatory trials verifying that the drug provides a clinical benefit.
Duvelisib will be available in the U.S. immediately, according to Verastem, the company marketing the drug. The prescribing information for duvelisib includes a boxed warning detailing four fatal and/or serious toxicities associated with the drug – infections, diarrhea or colitis, cutaneous reactions, and pneumonitis. Verastem said it is implementing an informational risk evaluation and mitigation strategy to provide appropriate dosing and safety information for duvelisib.
The recommended dose of duvelisib is 25 mg orally twice daily, taken continuously in 28-day treatment cycles.
The FDA’s approval of duvelisib is supported by data from the phase 3 DUO trial and the phase 2 DYNAMO trial. The DUO trial included 319 patients with CLL (n=312) or SLL (n=7) who had received at least one prior therapy. They were randomized to receive either duvelisib (25 mg orally twice daily) or ofatumumab (initial infusion of 300 mg followed by 7 weekly infusions and 4 monthly infusions of 2,000 mg).
Efficacy results are based on patients who had received at least two prior therapies, including 95 patients in the duvelisib arm and 101 in the ofatumumab arm. The overall response rate was 78% in the duvelisib arm and 39% in the ofatumumab arm. All responses in both arms were partial responses.
The median progression-free survival was 16.4 months with duvelisib and 9.1 months with ofatumumab.
The safety results include all patients treated with duvelisib or ofatumumab in this trial. In the duvelisib arm, 12% of patients had fatal adverse events (AEs) within 30 days of the last dose. The same was true of 4% of patients treated with ofatumumab. Serious AEs occurred in 73% of patients treated with duvelisib. The most common were infection and diarrhea/colitis. The DYNAMO trial enrolled patients with indolent non-Hodgkin lymphoma whose disease was refractory to both rituximab and chemotherapy or radioimmunotherapy. There were 83 patients with FL.
Patients received duvelisib at 25 mg orally twice daily until disease progression or unacceptable toxicity.
The overall response rate was 42%. One patient achieved a complete response, and 34 had a partial response.
Forty-three percent of responders maintained their response at 6 months, and 17% maintained their response at 12 months.
Serious AEs occurred in 58% of FL patients. The most common were diarrhea/colitis, pneumonia, renal insufficiency, rash, and sepsis.
Congenital syphilis rates continue skyrocketing alongside other STDs
Rapidly increasing cases of newborn syphilis have reached their highest prevalence in 2 decades, according to a new report by the Centers for Disease Control and Prevention on sexually transmitted disease surveillance in 2017.
Newborn syphilis incidence has more than doubled, from 362 cases in 2013 to 918 cases in 2017, resulting in 64 syphilitic stillbirths and 13 infant deaths that year, according to data published in Sexually Transmitted Disease Surveillance 2017.
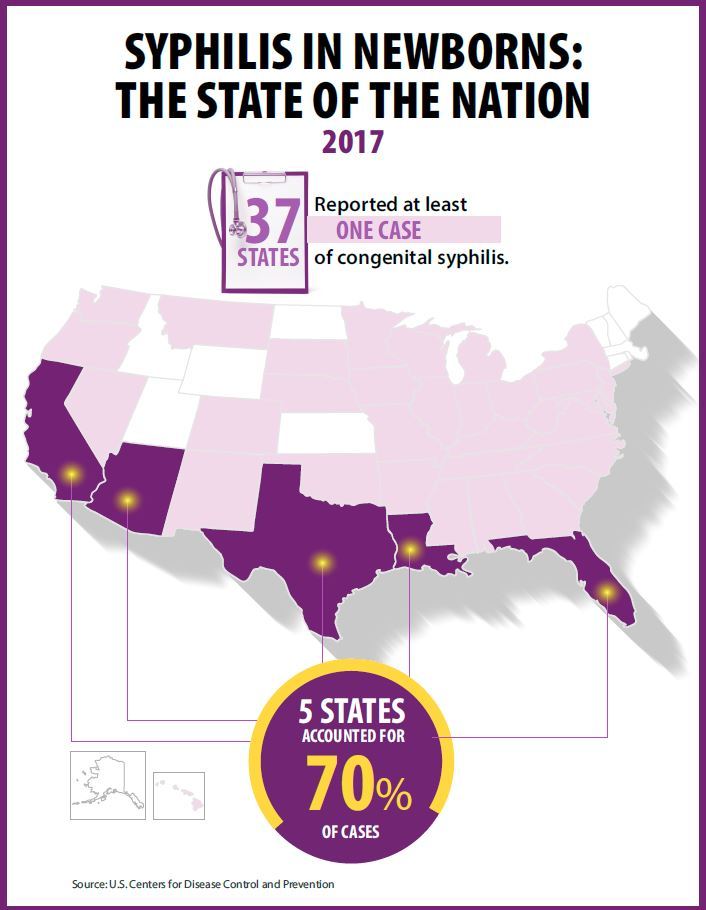
At least one case was reported in 37 states last year, and the greatest burden of cases occurred in California, Arizona, Texas, Louisiana, and Florida, together accounting for 70% of all 2017 cases.
“The resurgence of syphilis, and particularly congenital syphilis, is not an arbitrary event, but rather a symptom of a deteriorating public health infrastructure and lack of access to health care,” wrote Gail Bolan, MD, director of the Division of STD Prevention at the CDC’s National Center for HIV/AIDS, Viral Hepatitis, STD and TB Prevention. “It is exposing hidden, fragile populations in need that are not getting the health care and preventive services they deserve.”
Dr. Bolan recommends modernizing surveillance to capture more of the cases in populations without ready access to diagnosis and treatment and in those choosing not to access care.
“It is imperative that federal, state, and local programs employ strategies that maximize long-term population impact by reducing STD incidence and promoting sexual, reproductive, maternal, and infant health,” she wrote. “Further, it will be important for us to measure and monitor the adverse health consequences of STDs, such as ocular and neurosyphilis, pelvic inflammatory disease, ectopic pregnancy, infertility, HIV, congenital syphilis, and neonatal herpes.”
Multiple sources contributed data to the report: state and local STD programs’ notifiable disease reporting, private and federal national surveys, and specific projects that collect STD prevalence data, including the National Job Training Program, the STD Surveillance Network and the Gonococcal Isolate Surveillance Project.
The four nationally notifiable STDs are chlamydia, gonorrhea, syphilis, and chancroid.
The rise in newborn syphilis cases, currently at 23.3 cases per 100,000 live births, mirrors the increased U.S. prevalence of both primary and secondary syphilis in 2017, with 9.5 cases per 100,000 people. Syphilis has increased every year since 2000-2001, when prevalence was at a record low.
Chlamydia and gonorrhea rates climb too
The report also noted increases in the prevalence of other STDs. Chlamydia, the most common STD, increased 6.9% as compared to 2016, with 528.8 cases per 100,000 people. This increase occurred in all U.S. regions and independently of sex, race, or ethnicity, though rates were highest in teens and young adults. Nearly two-thirds of chlamydia cases in 2017 occurred in people ages 15-24 years old.
Reported rates were higher in women than in men, likely due to women’s increased likelihood of undergoing screening, the report suggested. Better surveillance may also partly explain the climb in men’s cases.
“Increases in rates among men may reflect an increased number of men, including gay, bisexual and other men who have sex with men (collectively referred to as MSM) being tested and diagnosed with a chlamydial infection due to increased availability of urine testing and extragenital screening,” according to the report.
The CDC received reports of more than a half million gonorrhea infections in 2017 (555,608 cases), an increase of 18.6% since the previous year, including a 19.3% increase among men and a 17.8% increase among women.
“The magnitude of the increase among men suggests either increased transmission, increased case ascertainment (e.g., through increased extra-genital screening among MSM), or both,” the authors wrote. “The concurrent increase in cases reported among women suggests parallel increases in heterosexual transmission, increased screening among women, or both.”
Overall, gonorrhea cases have skyrocketed 75.2% since their historic low in 2009, compounding the problem of antibiotic resistance that has limited CDC-recommended treatment to just ceftriaxone and azithromycin.
The report was supported by the Centers for Disease Control and Prevention. The authors did not report having any disclosures.
SOURCE: Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance 2017; https://www.cdc.gov/std/stats
Rapidly increasing cases of newborn syphilis have reached their highest prevalence in 2 decades, according to a new report by the Centers for Disease Control and Prevention on sexually transmitted disease surveillance in 2017.
Newborn syphilis incidence has more than doubled, from 362 cases in 2013 to 918 cases in 2017, resulting in 64 syphilitic stillbirths and 13 infant deaths that year, according to data published in Sexually Transmitted Disease Surveillance 2017.
At least one case was reported in 37 states last year, and the greatest burden of cases occurred in California, Arizona, Texas, Louisiana, and Florida, together accounting for 70% of all 2017 cases.
“The resurgence of syphilis, and particularly congenital syphilis, is not an arbitrary event, but rather a symptom of a deteriorating public health infrastructure and lack of access to health care,” wrote Gail Bolan, MD, director of the Division of STD Prevention at the CDC’s National Center for HIV/AIDS, Viral Hepatitis, STD and TB Prevention. “It is exposing hidden, fragile populations in need that are not getting the health care and preventive services they deserve.”
Dr. Bolan recommends modernizing surveillance to capture more of the cases in populations without ready access to diagnosis and treatment and in those choosing not to access care.
“It is imperative that federal, state, and local programs employ strategies that maximize long-term population impact by reducing STD incidence and promoting sexual, reproductive, maternal, and infant health,” she wrote. “Further, it will be important for us to measure and monitor the adverse health consequences of STDs, such as ocular and neurosyphilis, pelvic inflammatory disease, ectopic pregnancy, infertility, HIV, congenital syphilis, and neonatal herpes.”
Multiple sources contributed data to the report: state and local STD programs’ notifiable disease reporting, private and federal national surveys, and specific projects that collect STD prevalence data, including the National Job Training Program, the STD Surveillance Network and the Gonococcal Isolate Surveillance Project.
The four nationally notifiable STDs are chlamydia, gonorrhea, syphilis, and chancroid.
The rise in newborn syphilis cases, currently at 23.3 cases per 100,000 live births, mirrors the increased U.S. prevalence of both primary and secondary syphilis in 2017, with 9.5 cases per 100,000 people. Syphilis has increased every year since 2000-2001, when prevalence was at a record low.
Chlamydia and gonorrhea rates climb too
The report also noted increases in the prevalence of other STDs. Chlamydia, the most common STD, increased 6.9% as compared to 2016, with 528.8 cases per 100,000 people. This increase occurred in all U.S. regions and independently of sex, race, or ethnicity, though rates were highest in teens and young adults. Nearly two-thirds of chlamydia cases in 2017 occurred in people ages 15-24 years old.
Reported rates were higher in women than in men, likely due to women’s increased likelihood of undergoing screening, the report suggested. Better surveillance may also partly explain the climb in men’s cases.
“Increases in rates among men may reflect an increased number of men, including gay, bisexual and other men who have sex with men (collectively referred to as MSM) being tested and diagnosed with a chlamydial infection due to increased availability of urine testing and extragenital screening,” according to the report.
The CDC received reports of more than a half million gonorrhea infections in 2017 (555,608 cases), an increase of 18.6% since the previous year, including a 19.3% increase among men and a 17.8% increase among women.
“The magnitude of the increase among men suggests either increased transmission, increased case ascertainment (e.g., through increased extra-genital screening among MSM), or both,” the authors wrote. “The concurrent increase in cases reported among women suggests parallel increases in heterosexual transmission, increased screening among women, or both.”
Overall, gonorrhea cases have skyrocketed 75.2% since their historic low in 2009, compounding the problem of antibiotic resistance that has limited CDC-recommended treatment to just ceftriaxone and azithromycin.
The report was supported by the Centers for Disease Control and Prevention. The authors did not report having any disclosures.
SOURCE: Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance 2017; https://www.cdc.gov/std/stats
Rapidly increasing cases of newborn syphilis have reached their highest prevalence in 2 decades, according to a new report by the Centers for Disease Control and Prevention on sexually transmitted disease surveillance in 2017.
Newborn syphilis incidence has more than doubled, from 362 cases in 2013 to 918 cases in 2017, resulting in 64 syphilitic stillbirths and 13 infant deaths that year, according to data published in Sexually Transmitted Disease Surveillance 2017.
At least one case was reported in 37 states last year, and the greatest burden of cases occurred in California, Arizona, Texas, Louisiana, and Florida, together accounting for 70% of all 2017 cases.
“The resurgence of syphilis, and particularly congenital syphilis, is not an arbitrary event, but rather a symptom of a deteriorating public health infrastructure and lack of access to health care,” wrote Gail Bolan, MD, director of the Division of STD Prevention at the CDC’s National Center for HIV/AIDS, Viral Hepatitis, STD and TB Prevention. “It is exposing hidden, fragile populations in need that are not getting the health care and preventive services they deserve.”
Dr. Bolan recommends modernizing surveillance to capture more of the cases in populations without ready access to diagnosis and treatment and in those choosing not to access care.
“It is imperative that federal, state, and local programs employ strategies that maximize long-term population impact by reducing STD incidence and promoting sexual, reproductive, maternal, and infant health,” she wrote. “Further, it will be important for us to measure and monitor the adverse health consequences of STDs, such as ocular and neurosyphilis, pelvic inflammatory disease, ectopic pregnancy, infertility, HIV, congenital syphilis, and neonatal herpes.”
Multiple sources contributed data to the report: state and local STD programs’ notifiable disease reporting, private and federal national surveys, and specific projects that collect STD prevalence data, including the National Job Training Program, the STD Surveillance Network and the Gonococcal Isolate Surveillance Project.
The four nationally notifiable STDs are chlamydia, gonorrhea, syphilis, and chancroid.
The rise in newborn syphilis cases, currently at 23.3 cases per 100,000 live births, mirrors the increased U.S. prevalence of both primary and secondary syphilis in 2017, with 9.5 cases per 100,000 people. Syphilis has increased every year since 2000-2001, when prevalence was at a record low.
Chlamydia and gonorrhea rates climb too
The report also noted increases in the prevalence of other STDs. Chlamydia, the most common STD, increased 6.9% as compared to 2016, with 528.8 cases per 100,000 people. This increase occurred in all U.S. regions and independently of sex, race, or ethnicity, though rates were highest in teens and young adults. Nearly two-thirds of chlamydia cases in 2017 occurred in people ages 15-24 years old.
Reported rates were higher in women than in men, likely due to women’s increased likelihood of undergoing screening, the report suggested. Better surveillance may also partly explain the climb in men’s cases.
“Increases in rates among men may reflect an increased number of men, including gay, bisexual and other men who have sex with men (collectively referred to as MSM) being tested and diagnosed with a chlamydial infection due to increased availability of urine testing and extragenital screening,” according to the report.
The CDC received reports of more than a half million gonorrhea infections in 2017 (555,608 cases), an increase of 18.6% since the previous year, including a 19.3% increase among men and a 17.8% increase among women.
“The magnitude of the increase among men suggests either increased transmission, increased case ascertainment (e.g., through increased extra-genital screening among MSM), or both,” the authors wrote. “The concurrent increase in cases reported among women suggests parallel increases in heterosexual transmission, increased screening among women, or both.”
Overall, gonorrhea cases have skyrocketed 75.2% since their historic low in 2009, compounding the problem of antibiotic resistance that has limited CDC-recommended treatment to just ceftriaxone and azithromycin.
The report was supported by the Centers for Disease Control and Prevention. The authors did not report having any disclosures.
SOURCE: Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance 2017; https://www.cdc.gov/std/stats
Key clinical point: Newborn syphilis cases have more than doubled in 5 years along with substantial increases in chlamydia, gonorrhea, and syphilis.
Major finding: 918 cases of newborn syphilis were reported in 37 states in 2017.
Study details: The findings are based on data from public health notifiable disease reports and multiple federal and private surveillance projects.
Disclosures: The report was supported by the Centers for Disease Control and Prevention. The authors did not report having any disclosures.
Source: Centers for Disease Control and Prevention. Sexually Transmitted Disease Surveillance 2017.
FDA grants OBI-3424 orphan designation for ALL
The Food and Drug Administration has granted orphan drug designation to OBI-3424 for the treatment of acute lymphoblastic leukemia (ALL).
OBI-3424 is a small-molecule prodrug that targets cancers overexpressing aldo-keto reductase 1C3 (AKR1C3) and selectively releases a DNA alkylating agent in the presence of the AKR1C3 enzyme.
AKR1C3 overexpression has been observed in ALL, particularly T-cell ALL.
OBI-3424 demonstrated activity against T-ALL in preclinical research presented as a poster at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics in October 2017.
Researchers reported that OBI-3424 “exerted profound in vivo efficacy” against T-ALL xenografts derived mainly from patients with aggressive and fatal T-ALL. In addition, OBI-3424 significantly reduced leukemia bone marrow infiltration in four of six evaluable T-ALL xenografts, and OBI-3424 was considered well tolerated.
The poster presentation describing this research is available for download from the website of OBI Pharma, the company developing OBI-3424 in cooperation with Ascenta Pharma.
OBI-3424 also has orphan drug designation from the FDA as a treatment for hepatocellular carcinoma.
The Food and Drug Administration has granted orphan drug designation to OBI-3424 for the treatment of acute lymphoblastic leukemia (ALL).
OBI-3424 is a small-molecule prodrug that targets cancers overexpressing aldo-keto reductase 1C3 (AKR1C3) and selectively releases a DNA alkylating agent in the presence of the AKR1C3 enzyme.
AKR1C3 overexpression has been observed in ALL, particularly T-cell ALL.
OBI-3424 demonstrated activity against T-ALL in preclinical research presented as a poster at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics in October 2017.
Researchers reported that OBI-3424 “exerted profound in vivo efficacy” against T-ALL xenografts derived mainly from patients with aggressive and fatal T-ALL. In addition, OBI-3424 significantly reduced leukemia bone marrow infiltration in four of six evaluable T-ALL xenografts, and OBI-3424 was considered well tolerated.
The poster presentation describing this research is available for download from the website of OBI Pharma, the company developing OBI-3424 in cooperation with Ascenta Pharma.
OBI-3424 also has orphan drug designation from the FDA as a treatment for hepatocellular carcinoma.
The Food and Drug Administration has granted orphan drug designation to OBI-3424 for the treatment of acute lymphoblastic leukemia (ALL).
OBI-3424 is a small-molecule prodrug that targets cancers overexpressing aldo-keto reductase 1C3 (AKR1C3) and selectively releases a DNA alkylating agent in the presence of the AKR1C3 enzyme.
AKR1C3 overexpression has been observed in ALL, particularly T-cell ALL.
OBI-3424 demonstrated activity against T-ALL in preclinical research presented as a poster at the AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics in October 2017.
Researchers reported that OBI-3424 “exerted profound in vivo efficacy” against T-ALL xenografts derived mainly from patients with aggressive and fatal T-ALL. In addition, OBI-3424 significantly reduced leukemia bone marrow infiltration in four of six evaluable T-ALL xenografts, and OBI-3424 was considered well tolerated.
The poster presentation describing this research is available for download from the website of OBI Pharma, the company developing OBI-3424 in cooperation with Ascenta Pharma.
OBI-3424 also has orphan drug designation from the FDA as a treatment for hepatocellular carcinoma.
FDA review supports Nuplazid’s safety
Pimavanserin (Nuplazid) remains an acceptable treatment for the hallucinations and delusions associated with Parkinson’s disease, according to a statement issued by the Food and Drug Administration after the agency conducted a postmarketing review of deaths and serious adverse events associated with the drug.

“Based on an analysis of all available data, FDA did not identify any new or unexpected safety findings” associated with the drug, according to the Sept. 20 statement.
However, the FDA researchers identified prescribing patterns that might increase the risk of serious adverse events, such as the concomitant use of pimavanserin and other antipsychotic drugs or drugs that can cause QT prolongation. The QT prolongation risk is listed on the drug label, which also includes a Boxed Warning about increased mortality risk in elderly patients.
The FDA statement reminds clinicians to know the risks described in the label and to be aware that no other antipsychotics are currently approved for psychosis in Parkinson’s patients.
The review was prompted by the number of reports of serious adverse events and deaths associated with pimavanserin, based on data obtained from multiple sources including the FDA Adverse Event Reporting System (FAERS), drug utilization data, safety data from the new drug application, the sponsor’s Periodic Adverse Drug Experience Reports, the sponsor’s analysis of fatal adverse event reports, and data from published medical literature.
When conducting the review, the FDA considered several factors, including the fact that Parkinson’s disease patients have higher mortality in general because of older age, advanced disease, and other medical comorbidities. In addition, pimavanserin adverse events and deaths are more likely to be reported because the drug is distributed mainly through a patient-support program and specialty pharmacy. The FDA also found no pattern suggestive of a drug effect on causes of death in patients whose deaths were reported through FAERS.
“Overall, the postmarketing data were consistent with the safety data obtained from the premarketing controlled clinical trials of Nuplazid for Parkinson’s disease psychosis,” according to the FDA statement.
Pimavanserin (Nuplazid) remains an acceptable treatment for the hallucinations and delusions associated with Parkinson’s disease, according to a statement issued by the Food and Drug Administration after the agency conducted a postmarketing review of deaths and serious adverse events associated with the drug.
“Based on an analysis of all available data, FDA did not identify any new or unexpected safety findings” associated with the drug, according to the Sept. 20 statement.
However, the FDA researchers identified prescribing patterns that might increase the risk of serious adverse events, such as the concomitant use of pimavanserin and other antipsychotic drugs or drugs that can cause QT prolongation. The QT prolongation risk is listed on the drug label, which also includes a Boxed Warning about increased mortality risk in elderly patients.
The FDA statement reminds clinicians to know the risks described in the label and to be aware that no other antipsychotics are currently approved for psychosis in Parkinson’s patients.
The review was prompted by the number of reports of serious adverse events and deaths associated with pimavanserin, based on data obtained from multiple sources including the FDA Adverse Event Reporting System (FAERS), drug utilization data, safety data from the new drug application, the sponsor’s Periodic Adverse Drug Experience Reports, the sponsor’s analysis of fatal adverse event reports, and data from published medical literature.
When conducting the review, the FDA considered several factors, including the fact that Parkinson’s disease patients have higher mortality in general because of older age, advanced disease, and other medical comorbidities. In addition, pimavanserin adverse events and deaths are more likely to be reported because the drug is distributed mainly through a patient-support program and specialty pharmacy. The FDA also found no pattern suggestive of a drug effect on causes of death in patients whose deaths were reported through FAERS.
“Overall, the postmarketing data were consistent with the safety data obtained from the premarketing controlled clinical trials of Nuplazid for Parkinson’s disease psychosis,” according to the FDA statement.
Pimavanserin (Nuplazid) remains an acceptable treatment for the hallucinations and delusions associated with Parkinson’s disease, according to a statement issued by the Food and Drug Administration after the agency conducted a postmarketing review of deaths and serious adverse events associated with the drug.
“Based on an analysis of all available data, FDA did not identify any new or unexpected safety findings” associated with the drug, according to the Sept. 20 statement.
However, the FDA researchers identified prescribing patterns that might increase the risk of serious adverse events, such as the concomitant use of pimavanserin and other antipsychotic drugs or drugs that can cause QT prolongation. The QT prolongation risk is listed on the drug label, which also includes a Boxed Warning about increased mortality risk in elderly patients.
The FDA statement reminds clinicians to know the risks described in the label and to be aware that no other antipsychotics are currently approved for psychosis in Parkinson’s patients.
The review was prompted by the number of reports of serious adverse events and deaths associated with pimavanserin, based on data obtained from multiple sources including the FDA Adverse Event Reporting System (FAERS), drug utilization data, safety data from the new drug application, the sponsor’s Periodic Adverse Drug Experience Reports, the sponsor’s analysis of fatal adverse event reports, and data from published medical literature.
When conducting the review, the FDA considered several factors, including the fact that Parkinson’s disease patients have higher mortality in general because of older age, advanced disease, and other medical comorbidities. In addition, pimavanserin adverse events and deaths are more likely to be reported because the drug is distributed mainly through a patient-support program and specialty pharmacy. The FDA also found no pattern suggestive of a drug effect on causes of death in patients whose deaths were reported through FAERS.
“Overall, the postmarketing data were consistent with the safety data obtained from the premarketing controlled clinical trials of Nuplazid for Parkinson’s disease psychosis,” according to the FDA statement.
FDA clears gold microparticles for acne vulgaris
The Food and Drug Administration has granted clearance to “Sebacia Microparticles” for the treatment of acne vulgaris, for use with 1,064 nm lasers , according to a press release from Sebacia, the company developing the product.
The product, gold-coated silica microparticles, in a topical suspension, is cleared for use “as an accessory to 1064 nm lasers to facilitate photothermal heating of sebaceous glands for the treatment of mild to moderate inflammatory acne vulgaris,” the release said.
FDA clearance is based on a randomized, blinded, controlled trial of 168 patients with mild to moderate acne vulgaris, according to the company. Patients were treated with either laser and microparticles or with laser alone. The primary endpoint – noninferiority – was met with a median 53% reduction in inflammatory lesion count in the group receiving microparticles plus laser versus 45% in the laser-only group, at 12 weeks. All adverse events were mild to moderate, with none that were serious, the company statement said.
The Food and Drug Administration has granted clearance to “Sebacia Microparticles” for the treatment of acne vulgaris, for use with 1,064 nm lasers , according to a press release from Sebacia, the company developing the product.
The product, gold-coated silica microparticles, in a topical suspension, is cleared for use “as an accessory to 1064 nm lasers to facilitate photothermal heating of sebaceous glands for the treatment of mild to moderate inflammatory acne vulgaris,” the release said.
FDA clearance is based on a randomized, blinded, controlled trial of 168 patients with mild to moderate acne vulgaris, according to the company. Patients were treated with either laser and microparticles or with laser alone. The primary endpoint – noninferiority – was met with a median 53% reduction in inflammatory lesion count in the group receiving microparticles plus laser versus 45% in the laser-only group, at 12 weeks. All adverse events were mild to moderate, with none that were serious, the company statement said.
The Food and Drug Administration has granted clearance to “Sebacia Microparticles” for the treatment of acne vulgaris, for use with 1,064 nm lasers , according to a press release from Sebacia, the company developing the product.
The product, gold-coated silica microparticles, in a topical suspension, is cleared for use “as an accessory to 1064 nm lasers to facilitate photothermal heating of sebaceous glands for the treatment of mild to moderate inflammatory acne vulgaris,” the release said.
FDA clearance is based on a randomized, blinded, controlled trial of 168 patients with mild to moderate acne vulgaris, according to the company. Patients were treated with either laser and microparticles or with laser alone. The primary endpoint – noninferiority – was met with a median 53% reduction in inflammatory lesion count in the group receiving microparticles plus laser versus 45% in the laser-only group, at 12 weeks. All adverse events were mild to moderate, with none that were serious, the company statement said.
FDA approves device for coronary artery perforations
according to an announcement from the agency.
These small tears in the arterial wall are rare but life-threatening complications of certain procedures, such as percutaneous coronary interventions. The device uses a balloon catheter like that used with PCI and, when successful, it can spare patients more invasive procedures, such as open heart surgery.
The approval is based on survey data from 80 patients treated with this device. In 76 of the patients (95%), the device was successfully delivered to the site of the acute coronary arterial perforation, and it successfully healed the tear in 73 patients (91%). Two patients died during the procedure, and five whose perforations were sealed successfully died in the hospital after the procedure, as did one whose perforation was not sealed.
The agency noted that this is the first device approved for the indication in 17 years. More information can be found in the full FDA announcement.
according to an announcement from the agency.
These small tears in the arterial wall are rare but life-threatening complications of certain procedures, such as percutaneous coronary interventions. The device uses a balloon catheter like that used with PCI and, when successful, it can spare patients more invasive procedures, such as open heart surgery.
The approval is based on survey data from 80 patients treated with this device. In 76 of the patients (95%), the device was successfully delivered to the site of the acute coronary arterial perforation, and it successfully healed the tear in 73 patients (91%). Two patients died during the procedure, and five whose perforations were sealed successfully died in the hospital after the procedure, as did one whose perforation was not sealed.
The agency noted that this is the first device approved for the indication in 17 years. More information can be found in the full FDA announcement.
according to an announcement from the agency.
These small tears in the arterial wall are rare but life-threatening complications of certain procedures, such as percutaneous coronary interventions. The device uses a balloon catheter like that used with PCI and, when successful, it can spare patients more invasive procedures, such as open heart surgery.
The approval is based on survey data from 80 patients treated with this device. In 76 of the patients (95%), the device was successfully delivered to the site of the acute coronary arterial perforation, and it successfully healed the tear in 73 patients (91%). Two patients died during the procedure, and five whose perforations were sealed successfully died in the hospital after the procedure, as did one whose perforation was not sealed.
The agency noted that this is the first device approved for the indication in 17 years. More information can be found in the full FDA announcement.
FDA approves Ajovy for migraine prevention
The .
Fremanezumab-vfrm is an injectable anti–calcitonin gene-related peptide monoclonal antibody, the first ever approved by the FDA. It can be administered at a 225-mg dose every month or at a 675-mg dose every 3 months, according to a press release from Teva, the drug’s manufacturer.
FDA approval was based on results from two, phase 3 trials in patients with disabling migraines, one of which involved fremanezumab-vfrm alone, with the other involving the injection in tandem with an oral treatment.
In both trials, patients experienced a reduction of monthly migraine days over a 12-week period. The most common adverse event in both trials was injection site reactions.
“Migraine is a disabling neurological disease that affects more than 36 million people in the United States. About 40 percent of people living with migraine may be appropriate candidates for preventive treatment, yet the majority of them are untreated. I am pleased to have another treatment option that may allow my patients to experience fewer monthly migraine days,” Stephen Silberstein, MD, director of the Jefferson Headache Center at Thomas Jefferson University Hospital in Philadelphia, said in the Teva press release.
The .
Fremanezumab-vfrm is an injectable anti–calcitonin gene-related peptide monoclonal antibody, the first ever approved by the FDA. It can be administered at a 225-mg dose every month or at a 675-mg dose every 3 months, according to a press release from Teva, the drug’s manufacturer.
FDA approval was based on results from two, phase 3 trials in patients with disabling migraines, one of which involved fremanezumab-vfrm alone, with the other involving the injection in tandem with an oral treatment.
In both trials, patients experienced a reduction of monthly migraine days over a 12-week period. The most common adverse event in both trials was injection site reactions.
“Migraine is a disabling neurological disease that affects more than 36 million people in the United States. About 40 percent of people living with migraine may be appropriate candidates for preventive treatment, yet the majority of them are untreated. I am pleased to have another treatment option that may allow my patients to experience fewer monthly migraine days,” Stephen Silberstein, MD, director of the Jefferson Headache Center at Thomas Jefferson University Hospital in Philadelphia, said in the Teva press release.
The .
Fremanezumab-vfrm is an injectable anti–calcitonin gene-related peptide monoclonal antibody, the first ever approved by the FDA. It can be administered at a 225-mg dose every month or at a 675-mg dose every 3 months, according to a press release from Teva, the drug’s manufacturer.
FDA approval was based on results from two, phase 3 trials in patients with disabling migraines, one of which involved fremanezumab-vfrm alone, with the other involving the injection in tandem with an oral treatment.
In both trials, patients experienced a reduction of monthly migraine days over a 12-week period. The most common adverse event in both trials was injection site reactions.
“Migraine is a disabling neurological disease that affects more than 36 million people in the United States. About 40 percent of people living with migraine may be appropriate candidates for preventive treatment, yet the majority of them are untreated. I am pleased to have another treatment option that may allow my patients to experience fewer monthly migraine days,” Stephen Silberstein, MD, director of the Jefferson Headache Center at Thomas Jefferson University Hospital in Philadelphia, said in the Teva press release.
FDA attacks antibiotic resistance with new strategy
WASHINGTON – A strategy combining stewardship and science is needed to help combat antimicrobial resistance, and updated plans from the U.S. Food and Drug Administration include four key components to address all aspects of product development and use, FDA commissioner Scott Gottlieb, MD, said in a press briefing in Washington on Sept. 14.
“The FDA plays a unique role in advancing human and animal health” that provides a unique vantage point for coordinating all aspects of product development and application, he said.
The FDA’s comprehensive approach to the challenge of antimicrobial resistance (AMR) includes:
- Facilitating product development.
- Promoting antimicrobial stewardship.
- Supporting the development of new tools for surveillance.
- Advancing scientific initiatives, including research for the development of alternative treatments.

The FDA’s product development plan to combat AMR includes the creation of incentives for companies to develop new antibiotic products and create a robust pipeline, which is a challenge because of the lack of immediate economic gain, Dr. Gottlieb said.
“It necessary to change the perception that the costs and risks of antibiotic innovation are too high relative to their expected gains,” he emphasized.
Strategies to incentivize companies include fast track designation, priority review, and breakthrough therapy designation. In addition, the Limited Population Pathway for Antibacterial and Antifungal Drugs (LPAD) is designed to promote development of antimicrobial drugs for limited and underserved populations, Dr. Gottlieb said. The FDA plan also calls for pursuing reimbursement options with the Centers for Medicare & Medicaid Services.
Promoting antimicrobial stewardship remains an ongoing element of the FDA’s plan to reduce AMR. In conjunction with the release of the FDA’s updated approach to AMR, the FDA’s Center for Veterinary Medicine CVM released a 5-year action plan to promote and support antimicrobial stewardship in not only the agricultural arena, but in companion animals as well.
The FDA plans to bring all antimicrobials of medical importance that are approved for use in animals under the oversight of CVM, which will pursue the improve labeling on antimicrobial drugs used in the feed and water of food-producing animals, including defining durations of use, Dr. Gottlieb noted.
Supporting the development and improvement of surveillance tools is “essential to understanding the drivers of resistance in human and veterinary settings and formulating appropriate responses” to outbreaks, Dr. Gottlieb said.
To help meet this goal, the FDA will expand sampling via the National Antimicrobial Resistance Monitoring System (NARMS) database, he said. Other surveillance goals include supporting genomics research and expanding AMR monitoring to include pathogens associated with animal feed and companion animals, he added.
As part of the final component of the FDA’s AMR strategy to advance scientific initiatives, the FDA has released a new Request for Information “to obtain additional, external input on how best to develop an annual list of regulatory science initiatives specific for antimicrobial products,” Dr. Gottlieb announced. The FDA intends to use the information gained from clinicians and others in its creation of guidance documents and recommendations to streamline the antibiotic development process. He also cited the FDA’s ongoing support of partnerships with public and private organizations such as the Clinical Trials Transformation Initiative, which focuses on drug development for severe bacterial infections with current unmet medical need.
“We need to harness science and policy to help our public health systems and researchers become nimbler in the battle against drug-resistant pathogens,” Dr. Gottlieb concluded.
In a panel discussion following the briefing, several experts offered perspective on the FDA’s goals and on the challenges of AMR.
William Flynn, DVM, deputy director of science policy for the Center of Veterinary Medicine, noted some goals for reducing the use of antibiotics in the veterinary arena.
“We are trying to focus on the driver: What are the disease conditions that drive use of the product,” he said. Ideally, better management of disease conditions can reduce reliance on antibiotics, he added.
Also in the panel discussion, Steven Gitterman, MD, deputy director of the division of microbiology devices at the Center for Devices and Radiological Health, emphasized the value of sustainable trial databases so AMR research can continue on an ongoing basis. Finally, Carolyn Wilson, PhD, associate director of research at the Center for Biologics Evaluation and Research, noted that the FDA’s research and development efforts include antibiotic alternatives, including live biotherapeutic products, fecal microbiota transplantation, and bacteriophage therapy.
Visit www.fda.gov for a transcript of Dr. Gottlieb’s talk, and for the updated FDA website page with more details on the agency’s plans to combat antimicrobial resistance.
Dr. Gottlieb and the panelists had no financial conflicts to disclose.
WASHINGTON – A strategy combining stewardship and science is needed to help combat antimicrobial resistance, and updated plans from the U.S. Food and Drug Administration include four key components to address all aspects of product development and use, FDA commissioner Scott Gottlieb, MD, said in a press briefing in Washington on Sept. 14.
“The FDA plays a unique role in advancing human and animal health” that provides a unique vantage point for coordinating all aspects of product development and application, he said.
The FDA’s comprehensive approach to the challenge of antimicrobial resistance (AMR) includes:
- Facilitating product development.
- Promoting antimicrobial stewardship.
- Supporting the development of new tools for surveillance.
- Advancing scientific initiatives, including research for the development of alternative treatments.
The FDA’s product development plan to combat AMR includes the creation of incentives for companies to develop new antibiotic products and create a robust pipeline, which is a challenge because of the lack of immediate economic gain, Dr. Gottlieb said.
“It necessary to change the perception that the costs and risks of antibiotic innovation are too high relative to their expected gains,” he emphasized.
Strategies to incentivize companies include fast track designation, priority review, and breakthrough therapy designation. In addition, the Limited Population Pathway for Antibacterial and Antifungal Drugs (LPAD) is designed to promote development of antimicrobial drugs for limited and underserved populations, Dr. Gottlieb said. The FDA plan also calls for pursuing reimbursement options with the Centers for Medicare & Medicaid Services.
Promoting antimicrobial stewardship remains an ongoing element of the FDA’s plan to reduce AMR. In conjunction with the release of the FDA’s updated approach to AMR, the FDA’s Center for Veterinary Medicine CVM released a 5-year action plan to promote and support antimicrobial stewardship in not only the agricultural arena, but in companion animals as well.
The FDA plans to bring all antimicrobials of medical importance that are approved for use in animals under the oversight of CVM, which will pursue the improve labeling on antimicrobial drugs used in the feed and water of food-producing animals, including defining durations of use, Dr. Gottlieb noted.
Supporting the development and improvement of surveillance tools is “essential to understanding the drivers of resistance in human and veterinary settings and formulating appropriate responses” to outbreaks, Dr. Gottlieb said.
To help meet this goal, the FDA will expand sampling via the National Antimicrobial Resistance Monitoring System (NARMS) database, he said. Other surveillance goals include supporting genomics research and expanding AMR monitoring to include pathogens associated with animal feed and companion animals, he added.
As part of the final component of the FDA’s AMR strategy to advance scientific initiatives, the FDA has released a new Request for Information “to obtain additional, external input on how best to develop an annual list of regulatory science initiatives specific for antimicrobial products,” Dr. Gottlieb announced. The FDA intends to use the information gained from clinicians and others in its creation of guidance documents and recommendations to streamline the antibiotic development process. He also cited the FDA’s ongoing support of partnerships with public and private organizations such as the Clinical Trials Transformation Initiative, which focuses on drug development for severe bacterial infections with current unmet medical need.
“We need to harness science and policy to help our public health systems and researchers become nimbler in the battle against drug-resistant pathogens,” Dr. Gottlieb concluded.
In a panel discussion following the briefing, several experts offered perspective on the FDA’s goals and on the challenges of AMR.
William Flynn, DVM, deputy director of science policy for the Center of Veterinary Medicine, noted some goals for reducing the use of antibiotics in the veterinary arena.
“We are trying to focus on the driver: What are the disease conditions that drive use of the product,” he said. Ideally, better management of disease conditions can reduce reliance on antibiotics, he added.
Also in the panel discussion, Steven Gitterman, MD, deputy director of the division of microbiology devices at the Center for Devices and Radiological Health, emphasized the value of sustainable trial databases so AMR research can continue on an ongoing basis. Finally, Carolyn Wilson, PhD, associate director of research at the Center for Biologics Evaluation and Research, noted that the FDA’s research and development efforts include antibiotic alternatives, including live biotherapeutic products, fecal microbiota transplantation, and bacteriophage therapy.
Visit www.fda.gov for a transcript of Dr. Gottlieb’s talk, and for the updated FDA website page with more details on the agency’s plans to combat antimicrobial resistance.
Dr. Gottlieb and the panelists had no financial conflicts to disclose.
WASHINGTON – A strategy combining stewardship and science is needed to help combat antimicrobial resistance, and updated plans from the U.S. Food and Drug Administration include four key components to address all aspects of product development and use, FDA commissioner Scott Gottlieb, MD, said in a press briefing in Washington on Sept. 14.
“The FDA plays a unique role in advancing human and animal health” that provides a unique vantage point for coordinating all aspects of product development and application, he said.
The FDA’s comprehensive approach to the challenge of antimicrobial resistance (AMR) includes:
- Facilitating product development.
- Promoting antimicrobial stewardship.
- Supporting the development of new tools for surveillance.
- Advancing scientific initiatives, including research for the development of alternative treatments.
The FDA’s product development plan to combat AMR includes the creation of incentives for companies to develop new antibiotic products and create a robust pipeline, which is a challenge because of the lack of immediate economic gain, Dr. Gottlieb said.
“It necessary to change the perception that the costs and risks of antibiotic innovation are too high relative to their expected gains,” he emphasized.
Strategies to incentivize companies include fast track designation, priority review, and breakthrough therapy designation. In addition, the Limited Population Pathway for Antibacterial and Antifungal Drugs (LPAD) is designed to promote development of antimicrobial drugs for limited and underserved populations, Dr. Gottlieb said. The FDA plan also calls for pursuing reimbursement options with the Centers for Medicare & Medicaid Services.
Promoting antimicrobial stewardship remains an ongoing element of the FDA’s plan to reduce AMR. In conjunction with the release of the FDA’s updated approach to AMR, the FDA’s Center for Veterinary Medicine CVM released a 5-year action plan to promote and support antimicrobial stewardship in not only the agricultural arena, but in companion animals as well.
The FDA plans to bring all antimicrobials of medical importance that are approved for use in animals under the oversight of CVM, which will pursue the improve labeling on antimicrobial drugs used in the feed and water of food-producing animals, including defining durations of use, Dr. Gottlieb noted.
Supporting the development and improvement of surveillance tools is “essential to understanding the drivers of resistance in human and veterinary settings and formulating appropriate responses” to outbreaks, Dr. Gottlieb said.
To help meet this goal, the FDA will expand sampling via the National Antimicrobial Resistance Monitoring System (NARMS) database, he said. Other surveillance goals include supporting genomics research and expanding AMR monitoring to include pathogens associated with animal feed and companion animals, he added.
As part of the final component of the FDA’s AMR strategy to advance scientific initiatives, the FDA has released a new Request for Information “to obtain additional, external input on how best to develop an annual list of regulatory science initiatives specific for antimicrobial products,” Dr. Gottlieb announced. The FDA intends to use the information gained from clinicians and others in its creation of guidance documents and recommendations to streamline the antibiotic development process. He also cited the FDA’s ongoing support of partnerships with public and private organizations such as the Clinical Trials Transformation Initiative, which focuses on drug development for severe bacterial infections with current unmet medical need.
“We need to harness science and policy to help our public health systems and researchers become nimbler in the battle against drug-resistant pathogens,” Dr. Gottlieb concluded.
In a panel discussion following the briefing, several experts offered perspective on the FDA’s goals and on the challenges of AMR.
William Flynn, DVM, deputy director of science policy for the Center of Veterinary Medicine, noted some goals for reducing the use of antibiotics in the veterinary arena.
“We are trying to focus on the driver: What are the disease conditions that drive use of the product,” he said. Ideally, better management of disease conditions can reduce reliance on antibiotics, he added.
Also in the panel discussion, Steven Gitterman, MD, deputy director of the division of microbiology devices at the Center for Devices and Radiological Health, emphasized the value of sustainable trial databases so AMR research can continue on an ongoing basis. Finally, Carolyn Wilson, PhD, associate director of research at the Center for Biologics Evaluation and Research, noted that the FDA’s research and development efforts include antibiotic alternatives, including live biotherapeutic products, fecal microbiota transplantation, and bacteriophage therapy.
Visit www.fda.gov for a transcript of Dr. Gottlieb’s talk, and for the updated FDA website page with more details on the agency’s plans to combat antimicrobial resistance.
Dr. Gottlieb and the panelists had no financial conflicts to disclose.
FDA grants praliciguat Fast Track Designation for HFpEF
fraction (HFpEF), according to its developer Ironwood.
A phase 2, randomized, double-blind, placebo-controlled trial is currently enrolling patients to evaluate praliciguat as a treatment for HFpEF. The trial aims to enroll about 175 patients and intends to evaluate safety and efficacy, and topline data is expected later in 2019.
Praliciguat is an oral, once-daily, soluble guanylate cyclase (sGC) stimulator. It is being studied in patients with diabetic nephropathy and in patients with HFpEF. The condition affects an estimated 3 million Americans, but there are no approved therapies at this time to treat it; however, praliciguat may have the potential to treat the underlying causes by improving nitric oxide signaling, according to the press release from Ironwood.
fraction (HFpEF), according to its developer Ironwood.
A phase 2, randomized, double-blind, placebo-controlled trial is currently enrolling patients to evaluate praliciguat as a treatment for HFpEF. The trial aims to enroll about 175 patients and intends to evaluate safety and efficacy, and topline data is expected later in 2019.
Praliciguat is an oral, once-daily, soluble guanylate cyclase (sGC) stimulator. It is being studied in patients with diabetic nephropathy and in patients with HFpEF. The condition affects an estimated 3 million Americans, but there are no approved therapies at this time to treat it; however, praliciguat may have the potential to treat the underlying causes by improving nitric oxide signaling, according to the press release from Ironwood.
fraction (HFpEF), according to its developer Ironwood.
A phase 2, randomized, double-blind, placebo-controlled trial is currently enrolling patients to evaluate praliciguat as a treatment for HFpEF. The trial aims to enroll about 175 patients and intends to evaluate safety and efficacy, and topline data is expected later in 2019.
Praliciguat is an oral, once-daily, soluble guanylate cyclase (sGC) stimulator. It is being studied in patients with diabetic nephropathy and in patients with HFpEF. The condition affects an estimated 3 million Americans, but there are no approved therapies at this time to treat it; however, praliciguat may have the potential to treat the underlying causes by improving nitric oxide signaling, according to the press release from Ironwood.