User login
Antinuclear antibodies: When to test and how to interpret findings
› Reserve antinuclear antibody testing for instances of clinically suggestive connective tissue diseases (CTD) and for assessing CTD prognosis. It can also be useful in monitoring disease progression. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Antinuclear antibodies (ANA) are a spectrum of autoantibodies that react with various nuclear and cytoplasmic components of normal human cells. Their detection is important in the diagnosis of some connective tissue diseases (CTD)—eg, systemic lupus erythematosus (SLE), Sjögren’s syndrome (SS), scleroderma, polymyositis, or mixed connective tissue disease (MCTD). Unfortunately, ANA tests are often used indiscriminately in daily clinical practice.1
When is ANA testing warranted?
Indiscriminate use of ANA testing can yield positive results that falsely point to CTD in a high proportion of patients and thereby lead to further inappropriate testing and errant management decisions. To wit: The presence of ANA in the serum can be associated with any number of factors, such as genetic predisposition (eg, through histocompatibility locus DR3), environmental agents (viruses, drugs), chronic infections, neoplasms, and advancing age.1 Therefore, the test should not be ordered in a patient with low pre-test probability of CTD. Moreover, higher titers of ANA are more clinically significant than lower titers. In one multicenter study, 31.7% of healthy individuals were ANA-positive at a serum dilution of 1:40, but only 5% were ANA-positive at a dilution of 1:160.2
What is the clinical significance of different immunofluorescent patterns?
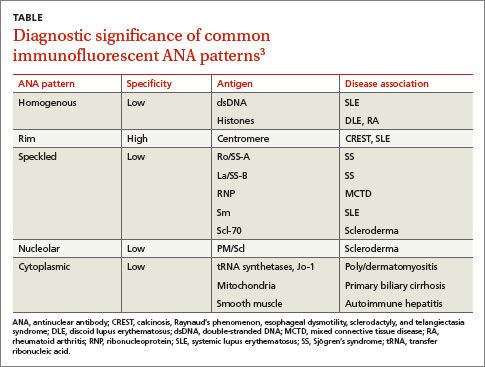
Immunofluorescent ANA testing not only determines if such antibodies are present in a patient’s serum but also reveals informative antibody patterns. Five distinct patterns of fluorescence are possible and can help differentiate between various CTDs (TABLE3):
1. Homogenous, in which the entire nucleus fluoresces, is seen in SLE and discoid lupus erythematosus (DLE).
2. Rim, in which the nuclear perimeter fluoresces, is seen most often in CREST (calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) syndrome and SLE.
3. Speckled, in which the nucleus fluoresces in a speckled pattern, can be seen in a variety of CTDs, including Sjögren’s syndrome, MCTD, SLE, and scleroderma.
4. Nucleolar, in which the nucleolus fluoresces, is associated with scleroderma.
5. Cytoplasmic, in which fluorescence occurs outside the nucleus, typically occurs with poly/dermatomyositis, primary biliary cirrhosis, or autoimmune hepatitis.
What is the next step if ANA is positive?
A positive ANA result warrants additional studies to identify specific autoantibodies suggested by the fluorescence pattern and by a patient’s signs and symptoms.
Following up diagnostic clues
Most systemic autoimmune diseases have a highly characteristic profile of autoantibodies to cellular antigens. A patient’s clinical features and ANA fluorescence pattern should direct additional testing.
Photosensitive butterfly rash, arthralgias/arthritis, pleuritic chest pain, fever of unknown cause, and urine sediment consistent with nephritis point to a diagnosis of SLE. Order an assay for anti-double-stranded DNA (dsDNA) antibodies, which, if present, confirm the diagnosis.4 Also order an assay for anti-Sm antibodies, which are highly specific for SLE but found only in 30% to 40% of SLE patients.4
Raynaud’s phenomenon, skin hardening or thickening, stiffness and tightening of the skin on the fingers, hands and forearms, tight and mask-like skin on the face, dry cough, shortness of breath, and difficulty in swallowing are features of scleroderma. If you suspect this disorder, order an assay for anti-Scl-70 antibodies. These antibodies are highly specific for scleroderma, but sensitivity of the assay is only 15% to 20%.5
Calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia indicate CREST syndrome. Anti-centromere antibodies are highly specific for CREST syndrome; sensitivity on assay is 50% to 90%.6
MCTD combines features of rheumatoid arthritis, SLE, myositis, and scleroderma. Order an assay of anti-RNP (ribonucleoprotein) antibodies. Although anti-RNP antibodies are also found in 25% to 30% of patients with SLE, they typically appear in the company of anti-Sm antibodies.7 Isolated high titers of anti-RNP antibodies point to MCTD, and sensitivity on assay is 100%.8 Their absence on testing, therefore, excludes the diagnosis of MCTD.
RNP, anti-Ro/SS-A, La/SS-B, and Sm are also referred to as extractable nuclear antigens (ENA). Assays of antibodies to ENA and anti-dsDNA are warranted only if the ANA assay result is positive. It is rare to have a positive anti-ENA antibody test (with the exception of antibodies to cytoplasmic antigens) in the absence of a positive ANA test.9
Dry eyes, dry mouth, joint pain and swelling, and swelling of parotid glands point to Sjögren’s syndrome. Anti-Ro/SS-A and La/SS-B antibodies are associated with Sjögren’s syndrome, but are also found in seronegative SLE.10 Therefore, if patients with features suggestive of SLE have a negative result on a dsDNA antibody assay, test for anti-Ro/SS-A and La/SS-B antibodies.
Muscle weakness and soreness, purplish discoloration of the upper eyelids, and purplish-red discoloration of the knuckles suggest dermatomyositis. Muscle biopsy and electromyography will clinch the diagnosis. Also test for anti–Jo-1 antibodies, which are associated with pulmonary involvement in polymyositis.11
ANA’s continuing role—prognosis and disease activity
Besides confirming a diagnosis of CTD in patients with suggestive clinical features, ANA testing serves 2 additional purposes: to help determine a patient’s prognosis and to monitor CTD activity. Consider the following:
- Patients with Sjögren’s syndrome who test positive for anti-Ro/SS-A antibodies have aggressive, extra-glandular disease that can cause vasculitis, purpura, lymphadenopathy, leukopenia, and thrombocytopenia.12
- The presence of anti-Ro/SS-A in the circulation of pregnant women with SLE confers a higher risk of neonatal lupus erythematosus and of congenital heart block in their newborns.13
- Severe interstitial lung disease is frequently found in scleroderma patients who test positive for anti-Scl-70.14 Antibodies to aminoacyl-tRNA synthetases—including anti–Jo-1, as mentioned earlier—are associated with pulmonary involvement in polymyositis patients.11
- A positive ANA test result in Raynaud’s phenomenon increases the likelihood that the patient will develop a systemic rheumatic disease; a negative result reduces this likelihood.15
- While the ANA test is not useful for diagnosing juvenile chronic arthritis (JCA), it is useful to test for ANA in patients with known JCA. A positive test result should prompt screening for uveitis.16
- An ANA test is not necessary for diagnosing antiphospholipid antibody syndrome (APS). However, the presence of ANA in a patient with APS increases the likelihood that APS is secondary to SLE.17
Monitoring disease activity
Documenting titers of anti-dsDNA antibodies may help in monitoring the disease activity of SLE in some patients. However, changes in titers of anti-dsDNA should be interpreted in the clinical context of the SLE Disease Activity Index.18
CORRESPONDENCE
Habib U. Rehman, MB, Department of Medicine, Regina Qu’Appelle Health Region, Regina General Hospital, 1440–14th Avenue, Regina, SK, S4P 0W5, Canada; habib31@sasktel.net
1. Volkmann ER, Taylor M, Ben-Artzi A. Using the antinuclear antibody test to diagnose rheumatic disease: when does a positive test warrant further investigation? South Med J. 2012;105:100-104.
2. Giannouli E, Chatzidimitriou D, Gerou S, et al. Frequency and specificity of antibodies against nuclear and cytoplasmic antigens in healthy individuals by classic and new methods. Clin Rheumatol. 2013;32:1541-1546.
3. O’Sullivan M, McLean-Tooke A, Loh RK. Antinuclear antibody test. Aust Fam Physician. 2013;42:718-721.
4. Kurien BT, Scofield RH. Autoantibody determination in the diagnosis of systemic lupus erythematosus. Scand J Immunol. 2006;64:227-235.
5. Basu D, Reveille JD. Ant-scl-70. Autoimmunity. 2005;38:65-72.
6. Caramaschi P, Biasi D, Manzo T, et al. Anticentromere antibody—clinical associations. A study of 44 patients. Rheumatol Int. 1995;14:253-255.
7. Migliorini P, Baldini C, Rocchi V, et al. Anti-Sm and anti-RNP antibodies. Autoimmunity. 2005;38:47-54.
8. Alarcón-Segovia D, Cardiel MH. Comparison between 3 diagnostic criteria for mixed connective tissue disease. Study of 593 patients. J Rheumatol. 1989;16:328-334.
9. Phan TG, Wong RC, Adelstein S. Autoantibodies to extractable nuclear antigens: making detection and interpretation more meaningful. Clin Diagn Lab Immunol. 2002;9:1-7.
10. Cross LS, Aslam A, Misbah SA. Antinuclear antibody-negative lupus as a distinct diagnostic entity—does it no longer exist? QJM. 2004;97:303-308.
11. Miller FW, Waite KA, Biswat T, et al. The role of an autoantigen, histidyl-tRNA synthetase, in the induction and maintenance of autoimmunity. Proc Natl Acad Sci USA. 1990;87:9933-9937.
12. Brito-Zerón P, Ramos-Casals M, Bove A, et al. Predicting adverse outcomes in primary Sjogren’s syndrome: identification of prognostic factors. Rheumatology. 2007;46:1359-1362.
13. Lindop R, Arentz G, Thurgood LA, et al. Pathogenicity and proteomic signatures of autoantibodies to Ro and La. Immunol Cell Biol. 2012;90:304-309.
14. Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum. 2005;35:35-42.
15. Spencer-Green G. Outcomes in primary Raynaud phenomenon: a meta-analysis of the frequency, rates, and predictors of transition to secondary diseases. Arch Intern Med. 1998;158:595-600.
16. Grassi A, Corona F, Casellato A, et al. Prevalence and outcome of juvenile idiopathic arthritis-associated uveitis and relation to articular disease. J Rheumatol. 2007;34:1139-1145.
17. Petri M. Diagnosis of antiphospholipid antibody syndrome. Rheum Dis Clin North Am. 1994;20:443.
18. Kavanaugh A, Tomar R, Reveille J, et al. Guidelines for clinical use of the antinuclear antibody test and tests for specific autoantibodies to nuclear antigens. American College of Pathologists. Arch Pathol Lab Med. 2000;124:71-81.
› Reserve antinuclear antibody testing for instances of clinically suggestive connective tissue diseases (CTD) and for assessing CTD prognosis. It can also be useful in monitoring disease progression. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Antinuclear antibodies (ANA) are a spectrum of autoantibodies that react with various nuclear and cytoplasmic components of normal human cells. Their detection is important in the diagnosis of some connective tissue diseases (CTD)—eg, systemic lupus erythematosus (SLE), Sjögren’s syndrome (SS), scleroderma, polymyositis, or mixed connective tissue disease (MCTD). Unfortunately, ANA tests are often used indiscriminately in daily clinical practice.1
When is ANA testing warranted?
Indiscriminate use of ANA testing can yield positive results that falsely point to CTD in a high proportion of patients and thereby lead to further inappropriate testing and errant management decisions. To wit: The presence of ANA in the serum can be associated with any number of factors, such as genetic predisposition (eg, through histocompatibility locus DR3), environmental agents (viruses, drugs), chronic infections, neoplasms, and advancing age.1 Therefore, the test should not be ordered in a patient with low pre-test probability of CTD. Moreover, higher titers of ANA are more clinically significant than lower titers. In one multicenter study, 31.7% of healthy individuals were ANA-positive at a serum dilution of 1:40, but only 5% were ANA-positive at a dilution of 1:160.2
What is the clinical significance of different immunofluorescent patterns?
Immunofluorescent ANA testing not only determines if such antibodies are present in a patient’s serum but also reveals informative antibody patterns. Five distinct patterns of fluorescence are possible and can help differentiate between various CTDs (TABLE3):
1. Homogenous, in which the entire nucleus fluoresces, is seen in SLE and discoid lupus erythematosus (DLE).
2. Rim, in which the nuclear perimeter fluoresces, is seen most often in CREST (calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) syndrome and SLE.
3. Speckled, in which the nucleus fluoresces in a speckled pattern, can be seen in a variety of CTDs, including Sjögren’s syndrome, MCTD, SLE, and scleroderma.
4. Nucleolar, in which the nucleolus fluoresces, is associated with scleroderma.
5. Cytoplasmic, in which fluorescence occurs outside the nucleus, typically occurs with poly/dermatomyositis, primary biliary cirrhosis, or autoimmune hepatitis.
What is the next step if ANA is positive?
A positive ANA result warrants additional studies to identify specific autoantibodies suggested by the fluorescence pattern and by a patient’s signs and symptoms.
Following up diagnostic clues
Most systemic autoimmune diseases have a highly characteristic profile of autoantibodies to cellular antigens. A patient’s clinical features and ANA fluorescence pattern should direct additional testing.
Photosensitive butterfly rash, arthralgias/arthritis, pleuritic chest pain, fever of unknown cause, and urine sediment consistent with nephritis point to a diagnosis of SLE. Order an assay for anti-double-stranded DNA (dsDNA) antibodies, which, if present, confirm the diagnosis.4 Also order an assay for anti-Sm antibodies, which are highly specific for SLE but found only in 30% to 40% of SLE patients.4
Raynaud’s phenomenon, skin hardening or thickening, stiffness and tightening of the skin on the fingers, hands and forearms, tight and mask-like skin on the face, dry cough, shortness of breath, and difficulty in swallowing are features of scleroderma. If you suspect this disorder, order an assay for anti-Scl-70 antibodies. These antibodies are highly specific for scleroderma, but sensitivity of the assay is only 15% to 20%.5
Calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia indicate CREST syndrome. Anti-centromere antibodies are highly specific for CREST syndrome; sensitivity on assay is 50% to 90%.6
MCTD combines features of rheumatoid arthritis, SLE, myositis, and scleroderma. Order an assay of anti-RNP (ribonucleoprotein) antibodies. Although anti-RNP antibodies are also found in 25% to 30% of patients with SLE, they typically appear in the company of anti-Sm antibodies.7 Isolated high titers of anti-RNP antibodies point to MCTD, and sensitivity on assay is 100%.8 Their absence on testing, therefore, excludes the diagnosis of MCTD.
RNP, anti-Ro/SS-A, La/SS-B, and Sm are also referred to as extractable nuclear antigens (ENA). Assays of antibodies to ENA and anti-dsDNA are warranted only if the ANA assay result is positive. It is rare to have a positive anti-ENA antibody test (with the exception of antibodies to cytoplasmic antigens) in the absence of a positive ANA test.9
Dry eyes, dry mouth, joint pain and swelling, and swelling of parotid glands point to Sjögren’s syndrome. Anti-Ro/SS-A and La/SS-B antibodies are associated with Sjögren’s syndrome, but are also found in seronegative SLE.10 Therefore, if patients with features suggestive of SLE have a negative result on a dsDNA antibody assay, test for anti-Ro/SS-A and La/SS-B antibodies.
Muscle weakness and soreness, purplish discoloration of the upper eyelids, and purplish-red discoloration of the knuckles suggest dermatomyositis. Muscle biopsy and electromyography will clinch the diagnosis. Also test for anti–Jo-1 antibodies, which are associated with pulmonary involvement in polymyositis.11
ANA’s continuing role—prognosis and disease activity
Besides confirming a diagnosis of CTD in patients with suggestive clinical features, ANA testing serves 2 additional purposes: to help determine a patient’s prognosis and to monitor CTD activity. Consider the following:
- Patients with Sjögren’s syndrome who test positive for anti-Ro/SS-A antibodies have aggressive, extra-glandular disease that can cause vasculitis, purpura, lymphadenopathy, leukopenia, and thrombocytopenia.12
- The presence of anti-Ro/SS-A in the circulation of pregnant women with SLE confers a higher risk of neonatal lupus erythematosus and of congenital heart block in their newborns.13
- Severe interstitial lung disease is frequently found in scleroderma patients who test positive for anti-Scl-70.14 Antibodies to aminoacyl-tRNA synthetases—including anti–Jo-1, as mentioned earlier—are associated with pulmonary involvement in polymyositis patients.11
- A positive ANA test result in Raynaud’s phenomenon increases the likelihood that the patient will develop a systemic rheumatic disease; a negative result reduces this likelihood.15
- While the ANA test is not useful for diagnosing juvenile chronic arthritis (JCA), it is useful to test for ANA in patients with known JCA. A positive test result should prompt screening for uveitis.16
- An ANA test is not necessary for diagnosing antiphospholipid antibody syndrome (APS). However, the presence of ANA in a patient with APS increases the likelihood that APS is secondary to SLE.17
Monitoring disease activity
Documenting titers of anti-dsDNA antibodies may help in monitoring the disease activity of SLE in some patients. However, changes in titers of anti-dsDNA should be interpreted in the clinical context of the SLE Disease Activity Index.18
CORRESPONDENCE
Habib U. Rehman, MB, Department of Medicine, Regina Qu’Appelle Health Region, Regina General Hospital, 1440–14th Avenue, Regina, SK, S4P 0W5, Canada; habib31@sasktel.net
› Reserve antinuclear antibody testing for instances of clinically suggestive connective tissue diseases (CTD) and for assessing CTD prognosis. It can also be useful in monitoring disease progression. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Antinuclear antibodies (ANA) are a spectrum of autoantibodies that react with various nuclear and cytoplasmic components of normal human cells. Their detection is important in the diagnosis of some connective tissue diseases (CTD)—eg, systemic lupus erythematosus (SLE), Sjögren’s syndrome (SS), scleroderma, polymyositis, or mixed connective tissue disease (MCTD). Unfortunately, ANA tests are often used indiscriminately in daily clinical practice.1
When is ANA testing warranted?
Indiscriminate use of ANA testing can yield positive results that falsely point to CTD in a high proportion of patients and thereby lead to further inappropriate testing and errant management decisions. To wit: The presence of ANA in the serum can be associated with any number of factors, such as genetic predisposition (eg, through histocompatibility locus DR3), environmental agents (viruses, drugs), chronic infections, neoplasms, and advancing age.1 Therefore, the test should not be ordered in a patient with low pre-test probability of CTD. Moreover, higher titers of ANA are more clinically significant than lower titers. In one multicenter study, 31.7% of healthy individuals were ANA-positive at a serum dilution of 1:40, but only 5% were ANA-positive at a dilution of 1:160.2
What is the clinical significance of different immunofluorescent patterns?
Immunofluorescent ANA testing not only determines if such antibodies are present in a patient’s serum but also reveals informative antibody patterns. Five distinct patterns of fluorescence are possible and can help differentiate between various CTDs (TABLE3):
1. Homogenous, in which the entire nucleus fluoresces, is seen in SLE and discoid lupus erythematosus (DLE).
2. Rim, in which the nuclear perimeter fluoresces, is seen most often in CREST (calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) syndrome and SLE.
3. Speckled, in which the nucleus fluoresces in a speckled pattern, can be seen in a variety of CTDs, including Sjögren’s syndrome, MCTD, SLE, and scleroderma.
4. Nucleolar, in which the nucleolus fluoresces, is associated with scleroderma.
5. Cytoplasmic, in which fluorescence occurs outside the nucleus, typically occurs with poly/dermatomyositis, primary biliary cirrhosis, or autoimmune hepatitis.
What is the next step if ANA is positive?
A positive ANA result warrants additional studies to identify specific autoantibodies suggested by the fluorescence pattern and by a patient’s signs and symptoms.
Following up diagnostic clues
Most systemic autoimmune diseases have a highly characteristic profile of autoantibodies to cellular antigens. A patient’s clinical features and ANA fluorescence pattern should direct additional testing.
Photosensitive butterfly rash, arthralgias/arthritis, pleuritic chest pain, fever of unknown cause, and urine sediment consistent with nephritis point to a diagnosis of SLE. Order an assay for anti-double-stranded DNA (dsDNA) antibodies, which, if present, confirm the diagnosis.4 Also order an assay for anti-Sm antibodies, which are highly specific for SLE but found only in 30% to 40% of SLE patients.4
Raynaud’s phenomenon, skin hardening or thickening, stiffness and tightening of the skin on the fingers, hands and forearms, tight and mask-like skin on the face, dry cough, shortness of breath, and difficulty in swallowing are features of scleroderma. If you suspect this disorder, order an assay for anti-Scl-70 antibodies. These antibodies are highly specific for scleroderma, but sensitivity of the assay is only 15% to 20%.5
Calcinosis, Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia indicate CREST syndrome. Anti-centromere antibodies are highly specific for CREST syndrome; sensitivity on assay is 50% to 90%.6
MCTD combines features of rheumatoid arthritis, SLE, myositis, and scleroderma. Order an assay of anti-RNP (ribonucleoprotein) antibodies. Although anti-RNP antibodies are also found in 25% to 30% of patients with SLE, they typically appear in the company of anti-Sm antibodies.7 Isolated high titers of anti-RNP antibodies point to MCTD, and sensitivity on assay is 100%.8 Their absence on testing, therefore, excludes the diagnosis of MCTD.
RNP, anti-Ro/SS-A, La/SS-B, and Sm are also referred to as extractable nuclear antigens (ENA). Assays of antibodies to ENA and anti-dsDNA are warranted only if the ANA assay result is positive. It is rare to have a positive anti-ENA antibody test (with the exception of antibodies to cytoplasmic antigens) in the absence of a positive ANA test.9
Dry eyes, dry mouth, joint pain and swelling, and swelling of parotid glands point to Sjögren’s syndrome. Anti-Ro/SS-A and La/SS-B antibodies are associated with Sjögren’s syndrome, but are also found in seronegative SLE.10 Therefore, if patients with features suggestive of SLE have a negative result on a dsDNA antibody assay, test for anti-Ro/SS-A and La/SS-B antibodies.
Muscle weakness and soreness, purplish discoloration of the upper eyelids, and purplish-red discoloration of the knuckles suggest dermatomyositis. Muscle biopsy and electromyography will clinch the diagnosis. Also test for anti–Jo-1 antibodies, which are associated with pulmonary involvement in polymyositis.11
ANA’s continuing role—prognosis and disease activity
Besides confirming a diagnosis of CTD in patients with suggestive clinical features, ANA testing serves 2 additional purposes: to help determine a patient’s prognosis and to monitor CTD activity. Consider the following:
- Patients with Sjögren’s syndrome who test positive for anti-Ro/SS-A antibodies have aggressive, extra-glandular disease that can cause vasculitis, purpura, lymphadenopathy, leukopenia, and thrombocytopenia.12
- The presence of anti-Ro/SS-A in the circulation of pregnant women with SLE confers a higher risk of neonatal lupus erythematosus and of congenital heart block in their newborns.13
- Severe interstitial lung disease is frequently found in scleroderma patients who test positive for anti-Scl-70.14 Antibodies to aminoacyl-tRNA synthetases—including anti–Jo-1, as mentioned earlier—are associated with pulmonary involvement in polymyositis patients.11
- A positive ANA test result in Raynaud’s phenomenon increases the likelihood that the patient will develop a systemic rheumatic disease; a negative result reduces this likelihood.15
- While the ANA test is not useful for diagnosing juvenile chronic arthritis (JCA), it is useful to test for ANA in patients with known JCA. A positive test result should prompt screening for uveitis.16
- An ANA test is not necessary for diagnosing antiphospholipid antibody syndrome (APS). However, the presence of ANA in a patient with APS increases the likelihood that APS is secondary to SLE.17
Monitoring disease activity
Documenting titers of anti-dsDNA antibodies may help in monitoring the disease activity of SLE in some patients. However, changes in titers of anti-dsDNA should be interpreted in the clinical context of the SLE Disease Activity Index.18
CORRESPONDENCE
Habib U. Rehman, MB, Department of Medicine, Regina Qu’Appelle Health Region, Regina General Hospital, 1440–14th Avenue, Regina, SK, S4P 0W5, Canada; habib31@sasktel.net
1. Volkmann ER, Taylor M, Ben-Artzi A. Using the antinuclear antibody test to diagnose rheumatic disease: when does a positive test warrant further investigation? South Med J. 2012;105:100-104.
2. Giannouli E, Chatzidimitriou D, Gerou S, et al. Frequency and specificity of antibodies against nuclear and cytoplasmic antigens in healthy individuals by classic and new methods. Clin Rheumatol. 2013;32:1541-1546.
3. O’Sullivan M, McLean-Tooke A, Loh RK. Antinuclear antibody test. Aust Fam Physician. 2013;42:718-721.
4. Kurien BT, Scofield RH. Autoantibody determination in the diagnosis of systemic lupus erythematosus. Scand J Immunol. 2006;64:227-235.
5. Basu D, Reveille JD. Ant-scl-70. Autoimmunity. 2005;38:65-72.
6. Caramaschi P, Biasi D, Manzo T, et al. Anticentromere antibody—clinical associations. A study of 44 patients. Rheumatol Int. 1995;14:253-255.
7. Migliorini P, Baldini C, Rocchi V, et al. Anti-Sm and anti-RNP antibodies. Autoimmunity. 2005;38:47-54.
8. Alarcón-Segovia D, Cardiel MH. Comparison between 3 diagnostic criteria for mixed connective tissue disease. Study of 593 patients. J Rheumatol. 1989;16:328-334.
9. Phan TG, Wong RC, Adelstein S. Autoantibodies to extractable nuclear antigens: making detection and interpretation more meaningful. Clin Diagn Lab Immunol. 2002;9:1-7.
10. Cross LS, Aslam A, Misbah SA. Antinuclear antibody-negative lupus as a distinct diagnostic entity—does it no longer exist? QJM. 2004;97:303-308.
11. Miller FW, Waite KA, Biswat T, et al. The role of an autoantigen, histidyl-tRNA synthetase, in the induction and maintenance of autoimmunity. Proc Natl Acad Sci USA. 1990;87:9933-9937.
12. Brito-Zerón P, Ramos-Casals M, Bove A, et al. Predicting adverse outcomes in primary Sjogren’s syndrome: identification of prognostic factors. Rheumatology. 2007;46:1359-1362.
13. Lindop R, Arentz G, Thurgood LA, et al. Pathogenicity and proteomic signatures of autoantibodies to Ro and La. Immunol Cell Biol. 2012;90:304-309.
14. Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum. 2005;35:35-42.
15. Spencer-Green G. Outcomes in primary Raynaud phenomenon: a meta-analysis of the frequency, rates, and predictors of transition to secondary diseases. Arch Intern Med. 1998;158:595-600.
16. Grassi A, Corona F, Casellato A, et al. Prevalence and outcome of juvenile idiopathic arthritis-associated uveitis and relation to articular disease. J Rheumatol. 2007;34:1139-1145.
17. Petri M. Diagnosis of antiphospholipid antibody syndrome. Rheum Dis Clin North Am. 1994;20:443.
18. Kavanaugh A, Tomar R, Reveille J, et al. Guidelines for clinical use of the antinuclear antibody test and tests for specific autoantibodies to nuclear antigens. American College of Pathologists. Arch Pathol Lab Med. 2000;124:71-81.
1. Volkmann ER, Taylor M, Ben-Artzi A. Using the antinuclear antibody test to diagnose rheumatic disease: when does a positive test warrant further investigation? South Med J. 2012;105:100-104.
2. Giannouli E, Chatzidimitriou D, Gerou S, et al. Frequency and specificity of antibodies against nuclear and cytoplasmic antigens in healthy individuals by classic and new methods. Clin Rheumatol. 2013;32:1541-1546.
3. O’Sullivan M, McLean-Tooke A, Loh RK. Antinuclear antibody test. Aust Fam Physician. 2013;42:718-721.
4. Kurien BT, Scofield RH. Autoantibody determination in the diagnosis of systemic lupus erythematosus. Scand J Immunol. 2006;64:227-235.
5. Basu D, Reveille JD. Ant-scl-70. Autoimmunity. 2005;38:65-72.
6. Caramaschi P, Biasi D, Manzo T, et al. Anticentromere antibody—clinical associations. A study of 44 patients. Rheumatol Int. 1995;14:253-255.
7. Migliorini P, Baldini C, Rocchi V, et al. Anti-Sm and anti-RNP antibodies. Autoimmunity. 2005;38:47-54.
8. Alarcón-Segovia D, Cardiel MH. Comparison between 3 diagnostic criteria for mixed connective tissue disease. Study of 593 patients. J Rheumatol. 1989;16:328-334.
9. Phan TG, Wong RC, Adelstein S. Autoantibodies to extractable nuclear antigens: making detection and interpretation more meaningful. Clin Diagn Lab Immunol. 2002;9:1-7.
10. Cross LS, Aslam A, Misbah SA. Antinuclear antibody-negative lupus as a distinct diagnostic entity—does it no longer exist? QJM. 2004;97:303-308.
11. Miller FW, Waite KA, Biswat T, et al. The role of an autoantigen, histidyl-tRNA synthetase, in the induction and maintenance of autoimmunity. Proc Natl Acad Sci USA. 1990;87:9933-9937.
12. Brito-Zerón P, Ramos-Casals M, Bove A, et al. Predicting adverse outcomes in primary Sjogren’s syndrome: identification of prognostic factors. Rheumatology. 2007;46:1359-1362.
13. Lindop R, Arentz G, Thurgood LA, et al. Pathogenicity and proteomic signatures of autoantibodies to Ro and La. Immunol Cell Biol. 2012;90:304-309.
14. Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum. 2005;35:35-42.
15. Spencer-Green G. Outcomes in primary Raynaud phenomenon: a meta-analysis of the frequency, rates, and predictors of transition to secondary diseases. Arch Intern Med. 1998;158:595-600.
16. Grassi A, Corona F, Casellato A, et al. Prevalence and outcome of juvenile idiopathic arthritis-associated uveitis and relation to articular disease. J Rheumatol. 2007;34:1139-1145.
17. Petri M. Diagnosis of antiphospholipid antibody syndrome. Rheum Dis Clin North Am. 1994;20:443.
18. Kavanaugh A, Tomar R, Reveille J, et al. Guidelines for clinical use of the antinuclear antibody test and tests for specific autoantibodies to nuclear antigens. American College of Pathologists. Arch Pathol Lab Med. 2000;124:71-81.
Tuberculosis: Which drug regimen and when
› Obtain a problem-focused history and physical, as well as chest radiography, to rule out active pulmonary tuberculosis (TB) before initiating treatment for latent tuberculosis infection (LTBI). B
› Prescribe isoniazid 5 mg/kg/d (10 mg/kg/d in children) up to a maximum dose of 300 mg/d for 9 months for most patients with LTBI. B
› Ensure that directly observed therapy is used for all patients with active TB, as well as for select high-risk cases of LTBI. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Mitchell J, age 62, comes to see you because he’s had a cough with increasing dyspnea for a month. Mr. J has never smoked but has type 2 diabetes mellitus. He also tells you that over the past month, he’s had occasional night sweats and has lost 8 pounds, although he’s not changed his diet. During the past week, he’s noticed blood-tinged sputum. Physical examination reveals a thin, chronically ill appearing man with an oral temperature of 100.6°F and mild tachypnea. You order a complete blood count, chest x-ray, and metabolic profile, administer a tuberculin skin test (TST), and initiate levofloxacin 500 mg/d for a presumed bacterial pneumonia. His lab work reveals mild leukocytosis and hyperglycemia, and the chest x-ray shows a left upper lobe infiltrate. The TST reaction—4 mm 50 hours after placement—was negative.
Mr. J returns a week later and says he feels worse. Your examination reveals worsened tachypnea, with tachycardia and crackles over the left upper lung fields.
How would you proceed with his care?
More people die of tuberculosis (TB) each year than any other infectious disease except human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome. In 2013, an estimated 9 million people worldwide developed active TB and 1.5 million died of the disease.1 Many of these deaths could have been prevented if patients had received a diagnosis and treatment during the latent phase (when the patient was infected, but had no active disease), or as soon as the patient developed active disease. In this article we describe treatment for both latent and active TB.
Before treating latent TB infection, first rule out active TB
Patients with latent tuberculosis infection (LTBI) have a 5% to 10% lifetime risk of developing active TB disease.2 Treatment of LTBI can reduce this risk to 1% to 2%.3
Although not the focus of this article, diagnosis of LTBI is made by using either a TST, in which the patient receives an intradermal injection of purified protein derivative and the size of the skin induration is measured 48 to 72 hours after administration, or an interferon-gamma release assay (IGRA), which requires a blood draw. After receiving a positive test result for LTBI, the next step is to rule out active TB.4 This is necessary because the primary treatment regimen for LTBI involves only one drug, whereas treating active TB with one drug is strongly associated with treatment failure and future resistance to that drug.5
To rule out active TB, perform a brief, problem-focused history and physical, and obtain a chest x-ray.4 Pertinent findings that suggest active disease include:
- any history of recent weight loss, unexplained fever, night sweats, cough or hemoptysis
- fever or any unexpected lung findings on physical exam
- any parenchymal infiltrates on chest x-ray. (Granulomas and scarring may be signs of previously healed TB infection, but do not indicate active TB.)
Any of these findings should prompt a further investigation to either confirm or definitively rule out active TB disease. In the absence of these findings, the physician may proceed with treatment for LTBI.
Latent TB infection treatment: Isoniazid alone, or another regimen?
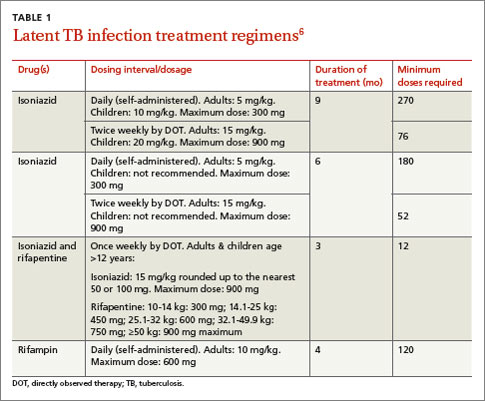
The current preferred regimen for most patients with LTBI is 9 months of isoniazid (INH) 5 mg/kg/d (10 mg/kg/d in children) up to a maximum of 300 mg/d. This regimen has been recommended by the Centers for Disease Control and Prevention (CDC), the American Thoracic Society, and the Infectious Diseases Society of America.3 However, there are 3 other CDC-recommended LTBI treatment regimens that include INH, INH plus rifapentine (RPT), or rifampin (RIF) for 6, 3, or 4 months, respectively (TABLE 1).6 These other regimens may be considered under certain circumstances. For example, INH and rifapentine might be used to treat an otherwise healthy patient who has had recent exposure to an individual with active, contagious TB.
If the patient is pregnant. INH is a pregnancy category C drug. Treatment for LTBI during pregnancy is generally regarded as safe and should be strongly considered if the patient has risk factors for progression to active TB, such as a recent exposure to someone with active TB.7 In otherwise healthy patients, treatment for LTBI may be deferred until after delivery.
Take steps to avoid complications of drug therapy
Drug-induced hepatitis is the primary adverse effect of INH treatment. Risk increases with age, previous hepatic injury, or concomitant use of other hepatotoxic medications. The risk is very small (<0.1%) for healthy children but may be over 10% for adults with multiple risk factors.8 Hepatitis is generally preceded by asymptomatic elevation of liver function tests (LFTs), which is much more common than clinical hepatitis.
Baseline LFTs should be obtained in patients who:
- have underlying liver disease, such as hepatitis B or C9
- consume ≥2 alcoholic drinks daily or >5 drinks at a time on any occasion
- take other medications with potential hepatotoxicity, such as statins
- have HIV infection10
- are pregnant or postpartum.
If a patient being considered for INH treatment has not had serologic testing for HIV, hepatitis B, or hepatitis C, these tests should be done prior to initiating INH. LFTs should be monitored every 1 to 2 months during INH therapy for patients who have ≥1 of these conditions and normal baseline LFTs. If baseline transaminases are >3 times the upper limit of normal, treatment for LTBI should probably be withheld, though might be considered in those whose LFTs return to normal after withdrawal of a modifiable risk factor, such as alcohol or a statin medication.
After beginning LTBI treatment, patients should be monitored regularly for signs and symptoms of hepatitis, including anorexia, nausea, abdominal pain, icterus, and dark urine, and LFTs performed if these develop. If during treatment transaminases increase to >3 times normal in a symptomatic patient (or >5 times normal in an asymptomatic patient), INH should be stopped and generally not resumed, even after LFTs return to normal. (Such patients would be considered to have partially treated LTBI, and their physicians should be alert to signs and symptoms of active TB, such as unexplained fever, weight loss, or blood-tinged sputum, during subsequent patient encounters.)
Peripheral neuropathy is a less common adverse effect of INH. It occurs in up to 2% of patients and is caused by interference with vitamin B6 (pyridoxine) metabolism. It can be prevented by supplementation with pyridoxine 25 to 50 mg/d. Vitamin B6, however, does not prevent INH-induced hepatotoxicity.
Noncompliance is a concern with INH therapy because treatment typically requires a 9-month course of daily medication.11 Patients for whom compliance is likely to be an issue might be considered for a 3-month, 12-dose course of once-weekly, directly-observed therapy (DOT) with INH and RPT administered by a public health agency. (See “Which patients with TB should receive directly observed therapy?” on page 32.12-14) A randomized, open-label trial involving nearly 8000 patients in 4 low-risk countries found this regimen was as effective as 9 months of self-administered INH.15 The CDC has published recommendations for using this regimen.16
Suspect active TB? Don’t wait for cultures to begin Tx
Unlike LTBI, for which the results of diagnostic testing are available within a few days, active TB is diagnosed by culture, which may take as long as 6 to 8 weeks. However, if you suspect your patient has active TB, do not delay treatment while waiting for culture results, or defer treatment for a patient who has a negative acid-fast bacilli (AFB) smear or rapid nucleic acid amplification test.17 These 2 tests, which are routinely performed during TB cultures, look for other evidence of the presence of TB bacilli; they are not as accurate as cultures, but results are available within days. Likewise, a negative TST or IGRA should not prevent empiric treatment for active TB. Treatment for active TB should be begun empirically based on risk factors and clinical presentation, and can be modified or stopped if cultures are negative, the patient fails to improve, or an alternative diagnosis is found to explain the patient’s symptoms.
Rapid testing for evidence of active TB disease—as well as resistance to medications commonly used to treat TB—can be performed using newer modalities such as MODS (Microscopic-Observation Drug-Susceptibility)18,19 or Xpert MTB/RIF20 testing. However, these tests are not available in many hospitals, and culture and drug sensitivity testing remain the gold standard.21
CASE › Mr. J’s clinical history and chest x-ray findings are highly suggestive of active TB. It was not unreasonable to initially treat him for a bacterial pneumonia, although fluoroquinolones should be used cautiously in this setting, because they are one of the most effective second-line drugs for TB, and using them as a single agent will often invoke drug resistance. Because he failed to respond to treatment for bacterial pneumonia and his presentation suggests TB or another serious cause of nonresponsiveness to standard treatment for community-acquired pneumonia (CAP), you admit him to the hospital.

Treatment for active TB requires multiple drugs in 2 phases
While all family physicians should suspect active TB in appropriate clinical situations and be comfortable with obtaining cultures and initiating empiric treatment, most will want to seek consultation with an infectious disease (ID) specialist especially in the scenarios listed in TABLE 2.5,22 Delayed or inappropriate treatment of active TB remains a major public health problem and cause of multidrug-resistant TB. Inappropriate treatment has been shown to be associated with a 27-fold increase in treatment failure.23 TB treatment guidelines are available from the CDC,24 World Health Organization,25 and International Union Against Tuberculosis and Lung Disease.26
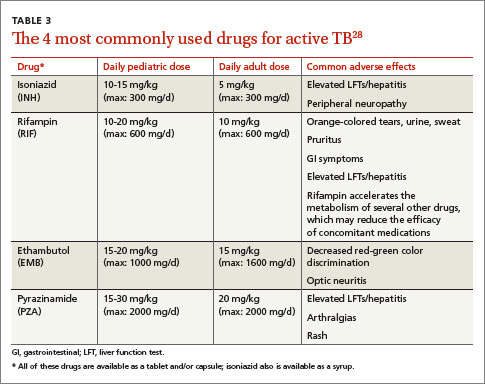
Appropriate treatment requires the use of multiple medications administered in 2 phases. In the initial phase, a patient with suspected TB should begin 4 drugs—usually INH, RIF, ethambutol (EMB), and pyrazinamide (PZA)—for 2 months.1,2,27 The daily pediatric and adult doses and common adverse effects of these medications are summarized in TABLE 3.28 Although most cases of TB can be adequately treated with 2 drugs to which the organism is susceptible, 4 drugs are used initially while awaiting drug sensitivity test results because of the risk of inadequately treating a strain of drug-resistant TB. Before beginning these medications, a chest x-ray, LFTs, HIV antibody test, hepatitis B and C serologies, a serum creatinine, and complete blood count should be obtained in all patients.5 If EMB is prescribed, the patient should also undergo testing for red-green color discrimination, because red-green color vision disturbance is a potential adverse effect of this medication.
All 4 drugs may be administered as a single daily dose, and may be taken together.29 They are ordinarily given either daily for 8 weeks, or daily for 2 weeks followed by a twice-weekly schedule for the remaining 6 weeks in higher doses, although the twice-weekly dose of RIF is the same as the daily dose. All are pregnancy category C, although for active TB, the benefit of treatment is almost always greater than the potential harm.
The continuation phase of treatment starts at 8 weeks, when the results of initial cultures and drug sensitivity tests should be available to guide therapy. A second set of cultures and AFB smears is obtained at 8 weeks to document clearing of the initial infection and guide duration of the continuation phase. If the initial culture was positive for Mycobacterium tuberculosis and the organism was sensitive to both INH and RIF, these 2 drugs should be continued for another 4 months (for a total of 6 months of treatment). PZA and EMB may be stopped at 2 months if the organism is sensitive to both INH and RIF. Thus, for most patients with active TB, the standard regimen will be 4 drugs for 2 months, then 2 drugs for 4 months.2
When should the standard treatment regimen be modified?
If the second set of cultures obtained 2 months after beginning drug treatment is positive and there was cavitary disease on the initial chest x-ray, the continuation phase should be extended by 7 months (for a total of 9 months of treatment).30 If a patient has either cavitary disease or persistently positive cultures (but not both), then the length of therapy is determined on an individual basis in consultation with an ID specialist.
Should a patient’s cultures show resistance to any of the first-line drugs, obtain consultation with an ID specialist. Treatment of multidrug-resistant TB (resistant to INH and RIF) and its subset, extensively drug-resistant TB (resistant to INH and RIF, plus any fluoroquinolone, plus either an aminoglycoside or capreomycin) requires prolonged courses of therapy with multiple drugs administered by DOT.31,32
If at any point during treatment a patient shows clinical deterioration that’s believed to be due to a resurgence of his or her TB disease, obtain a new set of cultures and, in consultation with an ID specialist, add at least 2 drugs to which the patient has not been exposed. Never add only one drug to a failing regimen; active TB always requires 2 drugs to cure, and the patient may have developed resistance to all of the drugs he or she is currently receiving.
If initial cultures are negative for Mycobacterium tuberculosis but the patient responds to treatment, he or she is considered to have “culture-negative TB,” and should generally be continued on INH and RIF for 2 more months after completion of the initial treatment phase (for a total of 4 months of INH and RIF).33
Remember to report. In the United States, active TB must be reported to your local health department, which can be invaluable in coordinating care and administering DOT.
Directly observed therapy (DOT) is preferred for certain high-risk patients with latent tuberculosis infection (LTBI), including those who are younger than 5 years of age, test positive for human immunodeficiency virus, are receiving immunosuppressive therapy, have chest radiography evidence of healed TB, have recently converted to active TB status while receiving serial TB testing, or have recently been exposed to active TB.12
Treatment for active TB should always be given by DOT.13 Because DOT is labor-intensive, twice-weekly dosing is usually preferred.14
CASE › In the hospital, Mr. J was placed in respiratory isolation, had prompt sputum cultures for TB, and was started on empiric treatment for active TB with INH, RIF, PZA, and EMB in standard doses. A search for other causes of nonresponsiveness to CAP showed no evidence of malignancy or HIV infection. He improved steadily and was discharged from the hospital after 2 weeks to complete 2 months of 4-drug therapy, with follow-up care coordinated by the local health department, including a home health nurse experienced in administering DOT. Cultures were positive for Mycobacterium tuberculosis sensitive to all drugs tested. After his initial 2 months of 4-drug therapy, he completed 4 months of additional treatment with INH and RIF, given by DOT, and recovered completely.
CORRESPONDENCE
Jeff Hall, MD, University of South Carolina Department of Family and Preventive Medicine, 3209 Colonial Drive, Columbia, SC 29203; jeff.hall@uscmed.sc.edu
1. World Health Organization. Global tuberculosis report 2014. World Health Organization Web site. Available at: http://www.who.int/tb/publications/global_report/en/. Accessed December 15, 2014.
2. Zumla AI, Raviglione M, Hafner R, et al. Tuberculosis. N Engl J Med. 2013;368:745-755.
3. American Thoracic Society. Targeted tuberculin testing and treatment of latent tuberculosis infection. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr4906a1.htm. Accessed December 15, 2014.
4. Hauck FR, Neese BH, Panchal AS, et al. Identification and management of latent tuberculosis infection. Am Fam Physician. 2009;79:879-886.
5. American Thoracic Society; CDC; Infectious Diseases Society of America. Treatment of tuberculosis. MMWR Recomm Rep. 2003;52:1-77.
6. Centers for Disease Control and Prevention. Latent tuberculosis infection: A guide for primary health care providers. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/tb/publications/LTBI/default.htm. Accessed December 11, 2014.
7. Centers for Disease Control and Prevention. Fact sheet: tuberculosis and pregnancy. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/TB/publications/factsheets/specpop/pregnancy.htm. Accessed September 6, 2014.
8. Kunst H, Khan KS. Age-related risk of hepatotoxicity in the treatment of latent tuberculosis infection: a systematic review. Int J Tuberc Lung Dis. 2010;14:1374-1381.
9. Bliven EE, Podewils LJ. The role of chronic hepatitis in isoniazid hepatotoxicity during treatment for latent tuberculosis infection. Int J Tuberc Lung Dis. 2009;13:1054-1060.
10. Akolo C, Adetifa I, Shepperd S, et al. Treatment of latent tuberculosis infection in HIV infected persons. Cochrane Database Syst Rev. 2010;1:CD000171.
11. Horsburgh CR Jr, Goldberg S, Bethel J, et al; Tuberculosis Epidemiologic Studies Consortium. Latent TB infection treatment acceptance and completion in the United States and Canada. Chest. 2010;137:401-409.
12. Horsburgh CR Jr. Priorities for the treatment of latent tuberculosis infection in the United States. N Engl J Med. 2004;350:2060-2070.
13. Potter B, Rindfleisch K, Kraus CK. Management of active tuberculosis. Am Fam Physician. 2005;72:2225-2232.
14. Volmink J, Garner P. Directly observed therapy for treating tuberculosis. Cochrane Database Syst Rev. 2007;4:CD003343.
15. Sterling TR, Villarina ME, Borisov AS, et al; TB Trials Consortium PREVENT TB Study Team. Three months of rifapentine and isoniazid for latent tuberculosis infection. N Engl J Med. 2011;365:2155-2166.
16. Centers for Disease Control and Prevention (CDC). Recommendations for use of an isoniazid-rifapentine regimen with direct observation to treat latent Mycobacterium tuberculosis infection. MMWR Morb Mortal Wkly Rep. 2011;60:1650-1653.
17. Inge LD, Wilson JW. Update on the treatment of tuberculosis. Am Fam Physician. 2008;78:457-465.
18. Moore DA, Evans CA, Gilman RH, et al. Microscopic-observation drug-susceptibility assay for the diagnosis of TB. N Engl J Med. 2006;355:1539-1550.
19. Minion J, Leung E, Menzies D, et al. Microscopic-observation drug susceptibility and thin layer agar assays for the detection of drug resistant tuberculosis: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10:688-698.
20. Boehme CC, Nabeta P, Hilleman D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363:1005-1015.
21. Arentz M, Sorensen B, Horne DJ, et al. Systematic review of the performance of rapid rifampicin resistance testing for drug-resistant tuberculosis. PLoS One. 2013;8:e76533.
22. Sia IG, Wieland ML. Current concepts in the management of tuberculosis. Mayo Clin Proc. 2011;86:348-361.
23. van der Werf MJ, Langendam MW, Huitric E, et al. Multidrug resistance after inappropriate tuberculosis treatment: a meta-analysis. Eur Respir J. 2012;39:1511-1519.
24. Centers for Disease Control and Prevention. Tuberculosis (TB). Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/TB/publications/guidelines/default.htm. Accessed September 6, 2014.
25. World Health Organization. Treatment of tuberculosis guidelines. 4th ed. World Health Organization Web site. Available at: http://whqlibdoc.who.int/publications/2010/9789241547833_eng.pdf?ua=1. Accessed September 6, 2014.
26. International Union Against Tuberculosis and Lung Disease. Management of tuberculosis: A guide to the essentials of good clinical practice. 6th ed. 2010. International Union Against Tuberculosis and Lung Disease Web site. Available at: http://www.theunion.org/what-we-do/publications/technical/management-of-tuberculosis-a-guide-to-the-essentials-of-good-clinical-practice. Accessed September 6, 2014.
27. Combs DL, O’Brien RJ, Geiter LJ. USPHS Tuberculosis Short-Course Chemotherapy Trial 21: effectiveness, toxicity and acceptability. The report of the final results. Ann Intern Med. 1990;112:397-406.
28. Drugs for tuberculosis. Treat Guidel Med Lett. 2012;10:29-36.
29. Chang KC, Leung CC, Grosset J, et al. Treatment of tuberculosis and optimal dosing schedules. Thorax. 2011;66:997-1007.
30. Blumberg HM, Burman WJ, Chaisson RE, et al; American Thoracic Society, Centers for Disease Control and Prevention and the Infectious Diseases Society. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167:603-662.
31. Lynch JB. Multidrug-resistant tuberculosis. Med Clin North Am. 2013;97:553-579,ix-x.
32. Keshavjee S, Farmer PE. Tuberculosis, drug resistance, and the history of modern medicine. N Engl J Med. 2012;367:931-936.
33. Dutt AK, Moers D, Stead WW. Smear- and culture-negative pulmonary tuberculosis: four-month short-course chemotherapy. Am Rev Respir Dis. 1989;139:867-870.
› Obtain a problem-focused history and physical, as well as chest radiography, to rule out active pulmonary tuberculosis (TB) before initiating treatment for latent tuberculosis infection (LTBI). B
› Prescribe isoniazid 5 mg/kg/d (10 mg/kg/d in children) up to a maximum dose of 300 mg/d for 9 months for most patients with LTBI. B
› Ensure that directly observed therapy is used for all patients with active TB, as well as for select high-risk cases of LTBI. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Mitchell J, age 62, comes to see you because he’s had a cough with increasing dyspnea for a month. Mr. J has never smoked but has type 2 diabetes mellitus. He also tells you that over the past month, he’s had occasional night sweats and has lost 8 pounds, although he’s not changed his diet. During the past week, he’s noticed blood-tinged sputum. Physical examination reveals a thin, chronically ill appearing man with an oral temperature of 100.6°F and mild tachypnea. You order a complete blood count, chest x-ray, and metabolic profile, administer a tuberculin skin test (TST), and initiate levofloxacin 500 mg/d for a presumed bacterial pneumonia. His lab work reveals mild leukocytosis and hyperglycemia, and the chest x-ray shows a left upper lobe infiltrate. The TST reaction—4 mm 50 hours after placement—was negative.
Mr. J returns a week later and says he feels worse. Your examination reveals worsened tachypnea, with tachycardia and crackles over the left upper lung fields.
How would you proceed with his care?
More people die of tuberculosis (TB) each year than any other infectious disease except human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome. In 2013, an estimated 9 million people worldwide developed active TB and 1.5 million died of the disease.1 Many of these deaths could have been prevented if patients had received a diagnosis and treatment during the latent phase (when the patient was infected, but had no active disease), or as soon as the patient developed active disease. In this article we describe treatment for both latent and active TB.
Before treating latent TB infection, first rule out active TB
Patients with latent tuberculosis infection (LTBI) have a 5% to 10% lifetime risk of developing active TB disease.2 Treatment of LTBI can reduce this risk to 1% to 2%.3
Although not the focus of this article, diagnosis of LTBI is made by using either a TST, in which the patient receives an intradermal injection of purified protein derivative and the size of the skin induration is measured 48 to 72 hours after administration, or an interferon-gamma release assay (IGRA), which requires a blood draw. After receiving a positive test result for LTBI, the next step is to rule out active TB.4 This is necessary because the primary treatment regimen for LTBI involves only one drug, whereas treating active TB with one drug is strongly associated with treatment failure and future resistance to that drug.5
To rule out active TB, perform a brief, problem-focused history and physical, and obtain a chest x-ray.4 Pertinent findings that suggest active disease include:
- any history of recent weight loss, unexplained fever, night sweats, cough or hemoptysis
- fever or any unexpected lung findings on physical exam
- any parenchymal infiltrates on chest x-ray. (Granulomas and scarring may be signs of previously healed TB infection, but do not indicate active TB.)
Any of these findings should prompt a further investigation to either confirm or definitively rule out active TB disease. In the absence of these findings, the physician may proceed with treatment for LTBI.
Latent TB infection treatment: Isoniazid alone, or another regimen?
The current preferred regimen for most patients with LTBI is 9 months of isoniazid (INH) 5 mg/kg/d (10 mg/kg/d in children) up to a maximum of 300 mg/d. This regimen has been recommended by the Centers for Disease Control and Prevention (CDC), the American Thoracic Society, and the Infectious Diseases Society of America.3 However, there are 3 other CDC-recommended LTBI treatment regimens that include INH, INH plus rifapentine (RPT), or rifampin (RIF) for 6, 3, or 4 months, respectively (TABLE 1).6 These other regimens may be considered under certain circumstances. For example, INH and rifapentine might be used to treat an otherwise healthy patient who has had recent exposure to an individual with active, contagious TB.
If the patient is pregnant. INH is a pregnancy category C drug. Treatment for LTBI during pregnancy is generally regarded as safe and should be strongly considered if the patient has risk factors for progression to active TB, such as a recent exposure to someone with active TB.7 In otherwise healthy patients, treatment for LTBI may be deferred until after delivery.
Take steps to avoid complications of drug therapy
Drug-induced hepatitis is the primary adverse effect of INH treatment. Risk increases with age, previous hepatic injury, or concomitant use of other hepatotoxic medications. The risk is very small (<0.1%) for healthy children but may be over 10% for adults with multiple risk factors.8 Hepatitis is generally preceded by asymptomatic elevation of liver function tests (LFTs), which is much more common than clinical hepatitis.
Baseline LFTs should be obtained in patients who:
- have underlying liver disease, such as hepatitis B or C9
- consume ≥2 alcoholic drinks daily or >5 drinks at a time on any occasion
- take other medications with potential hepatotoxicity, such as statins
- have HIV infection10
- are pregnant or postpartum.
If a patient being considered for INH treatment has not had serologic testing for HIV, hepatitis B, or hepatitis C, these tests should be done prior to initiating INH. LFTs should be monitored every 1 to 2 months during INH therapy for patients who have ≥1 of these conditions and normal baseline LFTs. If baseline transaminases are >3 times the upper limit of normal, treatment for LTBI should probably be withheld, though might be considered in those whose LFTs return to normal after withdrawal of a modifiable risk factor, such as alcohol or a statin medication.
After beginning LTBI treatment, patients should be monitored regularly for signs and symptoms of hepatitis, including anorexia, nausea, abdominal pain, icterus, and dark urine, and LFTs performed if these develop. If during treatment transaminases increase to >3 times normal in a symptomatic patient (or >5 times normal in an asymptomatic patient), INH should be stopped and generally not resumed, even after LFTs return to normal. (Such patients would be considered to have partially treated LTBI, and their physicians should be alert to signs and symptoms of active TB, such as unexplained fever, weight loss, or blood-tinged sputum, during subsequent patient encounters.)
Peripheral neuropathy is a less common adverse effect of INH. It occurs in up to 2% of patients and is caused by interference with vitamin B6 (pyridoxine) metabolism. It can be prevented by supplementation with pyridoxine 25 to 50 mg/d. Vitamin B6, however, does not prevent INH-induced hepatotoxicity.
Noncompliance is a concern with INH therapy because treatment typically requires a 9-month course of daily medication.11 Patients for whom compliance is likely to be an issue might be considered for a 3-month, 12-dose course of once-weekly, directly-observed therapy (DOT) with INH and RPT administered by a public health agency. (See “Which patients with TB should receive directly observed therapy?” on page 32.12-14) A randomized, open-label trial involving nearly 8000 patients in 4 low-risk countries found this regimen was as effective as 9 months of self-administered INH.15 The CDC has published recommendations for using this regimen.16
Suspect active TB? Don’t wait for cultures to begin Tx
Unlike LTBI, for which the results of diagnostic testing are available within a few days, active TB is diagnosed by culture, which may take as long as 6 to 8 weeks. However, if you suspect your patient has active TB, do not delay treatment while waiting for culture results, or defer treatment for a patient who has a negative acid-fast bacilli (AFB) smear or rapid nucleic acid amplification test.17 These 2 tests, which are routinely performed during TB cultures, look for other evidence of the presence of TB bacilli; they are not as accurate as cultures, but results are available within days. Likewise, a negative TST or IGRA should not prevent empiric treatment for active TB. Treatment for active TB should be begun empirically based on risk factors and clinical presentation, and can be modified or stopped if cultures are negative, the patient fails to improve, or an alternative diagnosis is found to explain the patient’s symptoms.
Rapid testing for evidence of active TB disease—as well as resistance to medications commonly used to treat TB—can be performed using newer modalities such as MODS (Microscopic-Observation Drug-Susceptibility)18,19 or Xpert MTB/RIF20 testing. However, these tests are not available in many hospitals, and culture and drug sensitivity testing remain the gold standard.21
CASE › Mr. J’s clinical history and chest x-ray findings are highly suggestive of active TB. It was not unreasonable to initially treat him for a bacterial pneumonia, although fluoroquinolones should be used cautiously in this setting, because they are one of the most effective second-line drugs for TB, and using them as a single agent will often invoke drug resistance. Because he failed to respond to treatment for bacterial pneumonia and his presentation suggests TB or another serious cause of nonresponsiveness to standard treatment for community-acquired pneumonia (CAP), you admit him to the hospital.
Treatment for active TB requires multiple drugs in 2 phases
While all family physicians should suspect active TB in appropriate clinical situations and be comfortable with obtaining cultures and initiating empiric treatment, most will want to seek consultation with an infectious disease (ID) specialist especially in the scenarios listed in TABLE 2.5,22 Delayed or inappropriate treatment of active TB remains a major public health problem and cause of multidrug-resistant TB. Inappropriate treatment has been shown to be associated with a 27-fold increase in treatment failure.23 TB treatment guidelines are available from the CDC,24 World Health Organization,25 and International Union Against Tuberculosis and Lung Disease.26
Appropriate treatment requires the use of multiple medications administered in 2 phases. In the initial phase, a patient with suspected TB should begin 4 drugs—usually INH, RIF, ethambutol (EMB), and pyrazinamide (PZA)—for 2 months.1,2,27 The daily pediatric and adult doses and common adverse effects of these medications are summarized in TABLE 3.28 Although most cases of TB can be adequately treated with 2 drugs to which the organism is susceptible, 4 drugs are used initially while awaiting drug sensitivity test results because of the risk of inadequately treating a strain of drug-resistant TB. Before beginning these medications, a chest x-ray, LFTs, HIV antibody test, hepatitis B and C serologies, a serum creatinine, and complete blood count should be obtained in all patients.5 If EMB is prescribed, the patient should also undergo testing for red-green color discrimination, because red-green color vision disturbance is a potential adverse effect of this medication.
All 4 drugs may be administered as a single daily dose, and may be taken together.29 They are ordinarily given either daily for 8 weeks, or daily for 2 weeks followed by a twice-weekly schedule for the remaining 6 weeks in higher doses, although the twice-weekly dose of RIF is the same as the daily dose. All are pregnancy category C, although for active TB, the benefit of treatment is almost always greater than the potential harm.
The continuation phase of treatment starts at 8 weeks, when the results of initial cultures and drug sensitivity tests should be available to guide therapy. A second set of cultures and AFB smears is obtained at 8 weeks to document clearing of the initial infection and guide duration of the continuation phase. If the initial culture was positive for Mycobacterium tuberculosis and the organism was sensitive to both INH and RIF, these 2 drugs should be continued for another 4 months (for a total of 6 months of treatment). PZA and EMB may be stopped at 2 months if the organism is sensitive to both INH and RIF. Thus, for most patients with active TB, the standard regimen will be 4 drugs for 2 months, then 2 drugs for 4 months.2
When should the standard treatment regimen be modified?
If the second set of cultures obtained 2 months after beginning drug treatment is positive and there was cavitary disease on the initial chest x-ray, the continuation phase should be extended by 7 months (for a total of 9 months of treatment).30 If a patient has either cavitary disease or persistently positive cultures (but not both), then the length of therapy is determined on an individual basis in consultation with an ID specialist.
Should a patient’s cultures show resistance to any of the first-line drugs, obtain consultation with an ID specialist. Treatment of multidrug-resistant TB (resistant to INH and RIF) and its subset, extensively drug-resistant TB (resistant to INH and RIF, plus any fluoroquinolone, plus either an aminoglycoside or capreomycin) requires prolonged courses of therapy with multiple drugs administered by DOT.31,32
If at any point during treatment a patient shows clinical deterioration that’s believed to be due to a resurgence of his or her TB disease, obtain a new set of cultures and, in consultation with an ID specialist, add at least 2 drugs to which the patient has not been exposed. Never add only one drug to a failing regimen; active TB always requires 2 drugs to cure, and the patient may have developed resistance to all of the drugs he or she is currently receiving.
If initial cultures are negative for Mycobacterium tuberculosis but the patient responds to treatment, he or she is considered to have “culture-negative TB,” and should generally be continued on INH and RIF for 2 more months after completion of the initial treatment phase (for a total of 4 months of INH and RIF).33
Remember to report. In the United States, active TB must be reported to your local health department, which can be invaluable in coordinating care and administering DOT.
Directly observed therapy (DOT) is preferred for certain high-risk patients with latent tuberculosis infection (LTBI), including those who are younger than 5 years of age, test positive for human immunodeficiency virus, are receiving immunosuppressive therapy, have chest radiography evidence of healed TB, have recently converted to active TB status while receiving serial TB testing, or have recently been exposed to active TB.12
Treatment for active TB should always be given by DOT.13 Because DOT is labor-intensive, twice-weekly dosing is usually preferred.14
CASE › In the hospital, Mr. J was placed in respiratory isolation, had prompt sputum cultures for TB, and was started on empiric treatment for active TB with INH, RIF, PZA, and EMB in standard doses. A search for other causes of nonresponsiveness to CAP showed no evidence of malignancy or HIV infection. He improved steadily and was discharged from the hospital after 2 weeks to complete 2 months of 4-drug therapy, with follow-up care coordinated by the local health department, including a home health nurse experienced in administering DOT. Cultures were positive for Mycobacterium tuberculosis sensitive to all drugs tested. After his initial 2 months of 4-drug therapy, he completed 4 months of additional treatment with INH and RIF, given by DOT, and recovered completely.
CORRESPONDENCE
Jeff Hall, MD, University of South Carolina Department of Family and Preventive Medicine, 3209 Colonial Drive, Columbia, SC 29203; jeff.hall@uscmed.sc.edu
› Obtain a problem-focused history and physical, as well as chest radiography, to rule out active pulmonary tuberculosis (TB) before initiating treatment for latent tuberculosis infection (LTBI). B
› Prescribe isoniazid 5 mg/kg/d (10 mg/kg/d in children) up to a maximum dose of 300 mg/d for 9 months for most patients with LTBI. B
› Ensure that directly observed therapy is used for all patients with active TB, as well as for select high-risk cases of LTBI. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Mitchell J, age 62, comes to see you because he’s had a cough with increasing dyspnea for a month. Mr. J has never smoked but has type 2 diabetes mellitus. He also tells you that over the past month, he’s had occasional night sweats and has lost 8 pounds, although he’s not changed his diet. During the past week, he’s noticed blood-tinged sputum. Physical examination reveals a thin, chronically ill appearing man with an oral temperature of 100.6°F and mild tachypnea. You order a complete blood count, chest x-ray, and metabolic profile, administer a tuberculin skin test (TST), and initiate levofloxacin 500 mg/d for a presumed bacterial pneumonia. His lab work reveals mild leukocytosis and hyperglycemia, and the chest x-ray shows a left upper lobe infiltrate. The TST reaction—4 mm 50 hours after placement—was negative.
Mr. J returns a week later and says he feels worse. Your examination reveals worsened tachypnea, with tachycardia and crackles over the left upper lung fields.
How would you proceed with his care?
More people die of tuberculosis (TB) each year than any other infectious disease except human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome. In 2013, an estimated 9 million people worldwide developed active TB and 1.5 million died of the disease.1 Many of these deaths could have been prevented if patients had received a diagnosis and treatment during the latent phase (when the patient was infected, but had no active disease), or as soon as the patient developed active disease. In this article we describe treatment for both latent and active TB.
Before treating latent TB infection, first rule out active TB
Patients with latent tuberculosis infection (LTBI) have a 5% to 10% lifetime risk of developing active TB disease.2 Treatment of LTBI can reduce this risk to 1% to 2%.3
Although not the focus of this article, diagnosis of LTBI is made by using either a TST, in which the patient receives an intradermal injection of purified protein derivative and the size of the skin induration is measured 48 to 72 hours after administration, or an interferon-gamma release assay (IGRA), which requires a blood draw. After receiving a positive test result for LTBI, the next step is to rule out active TB.4 This is necessary because the primary treatment regimen for LTBI involves only one drug, whereas treating active TB with one drug is strongly associated with treatment failure and future resistance to that drug.5
To rule out active TB, perform a brief, problem-focused history and physical, and obtain a chest x-ray.4 Pertinent findings that suggest active disease include:
- any history of recent weight loss, unexplained fever, night sweats, cough or hemoptysis
- fever or any unexpected lung findings on physical exam
- any parenchymal infiltrates on chest x-ray. (Granulomas and scarring may be signs of previously healed TB infection, but do not indicate active TB.)
Any of these findings should prompt a further investigation to either confirm or definitively rule out active TB disease. In the absence of these findings, the physician may proceed with treatment for LTBI.
Latent TB infection treatment: Isoniazid alone, or another regimen?
The current preferred regimen for most patients with LTBI is 9 months of isoniazid (INH) 5 mg/kg/d (10 mg/kg/d in children) up to a maximum of 300 mg/d. This regimen has been recommended by the Centers for Disease Control and Prevention (CDC), the American Thoracic Society, and the Infectious Diseases Society of America.3 However, there are 3 other CDC-recommended LTBI treatment regimens that include INH, INH plus rifapentine (RPT), or rifampin (RIF) for 6, 3, or 4 months, respectively (TABLE 1).6 These other regimens may be considered under certain circumstances. For example, INH and rifapentine might be used to treat an otherwise healthy patient who has had recent exposure to an individual with active, contagious TB.
If the patient is pregnant. INH is a pregnancy category C drug. Treatment for LTBI during pregnancy is generally regarded as safe and should be strongly considered if the patient has risk factors for progression to active TB, such as a recent exposure to someone with active TB.7 In otherwise healthy patients, treatment for LTBI may be deferred until after delivery.
Take steps to avoid complications of drug therapy
Drug-induced hepatitis is the primary adverse effect of INH treatment. Risk increases with age, previous hepatic injury, or concomitant use of other hepatotoxic medications. The risk is very small (<0.1%) for healthy children but may be over 10% for adults with multiple risk factors.8 Hepatitis is generally preceded by asymptomatic elevation of liver function tests (LFTs), which is much more common than clinical hepatitis.
Baseline LFTs should be obtained in patients who:
- have underlying liver disease, such as hepatitis B or C9
- consume ≥2 alcoholic drinks daily or >5 drinks at a time on any occasion
- take other medications with potential hepatotoxicity, such as statins
- have HIV infection10
- are pregnant or postpartum.
If a patient being considered for INH treatment has not had serologic testing for HIV, hepatitis B, or hepatitis C, these tests should be done prior to initiating INH. LFTs should be monitored every 1 to 2 months during INH therapy for patients who have ≥1 of these conditions and normal baseline LFTs. If baseline transaminases are >3 times the upper limit of normal, treatment for LTBI should probably be withheld, though might be considered in those whose LFTs return to normal after withdrawal of a modifiable risk factor, such as alcohol or a statin medication.
After beginning LTBI treatment, patients should be monitored regularly for signs and symptoms of hepatitis, including anorexia, nausea, abdominal pain, icterus, and dark urine, and LFTs performed if these develop. If during treatment transaminases increase to >3 times normal in a symptomatic patient (or >5 times normal in an asymptomatic patient), INH should be stopped and generally not resumed, even after LFTs return to normal. (Such patients would be considered to have partially treated LTBI, and their physicians should be alert to signs and symptoms of active TB, such as unexplained fever, weight loss, or blood-tinged sputum, during subsequent patient encounters.)
Peripheral neuropathy is a less common adverse effect of INH. It occurs in up to 2% of patients and is caused by interference with vitamin B6 (pyridoxine) metabolism. It can be prevented by supplementation with pyridoxine 25 to 50 mg/d. Vitamin B6, however, does not prevent INH-induced hepatotoxicity.
Noncompliance is a concern with INH therapy because treatment typically requires a 9-month course of daily medication.11 Patients for whom compliance is likely to be an issue might be considered for a 3-month, 12-dose course of once-weekly, directly-observed therapy (DOT) with INH and RPT administered by a public health agency. (See “Which patients with TB should receive directly observed therapy?” on page 32.12-14) A randomized, open-label trial involving nearly 8000 patients in 4 low-risk countries found this regimen was as effective as 9 months of self-administered INH.15 The CDC has published recommendations for using this regimen.16
Suspect active TB? Don’t wait for cultures to begin Tx
Unlike LTBI, for which the results of diagnostic testing are available within a few days, active TB is diagnosed by culture, which may take as long as 6 to 8 weeks. However, if you suspect your patient has active TB, do not delay treatment while waiting for culture results, or defer treatment for a patient who has a negative acid-fast bacilli (AFB) smear or rapid nucleic acid amplification test.17 These 2 tests, which are routinely performed during TB cultures, look for other evidence of the presence of TB bacilli; they are not as accurate as cultures, but results are available within days. Likewise, a negative TST or IGRA should not prevent empiric treatment for active TB. Treatment for active TB should be begun empirically based on risk factors and clinical presentation, and can be modified or stopped if cultures are negative, the patient fails to improve, or an alternative diagnosis is found to explain the patient’s symptoms.
Rapid testing for evidence of active TB disease—as well as resistance to medications commonly used to treat TB—can be performed using newer modalities such as MODS (Microscopic-Observation Drug-Susceptibility)18,19 or Xpert MTB/RIF20 testing. However, these tests are not available in many hospitals, and culture and drug sensitivity testing remain the gold standard.21
CASE › Mr. J’s clinical history and chest x-ray findings are highly suggestive of active TB. It was not unreasonable to initially treat him for a bacterial pneumonia, although fluoroquinolones should be used cautiously in this setting, because they are one of the most effective second-line drugs for TB, and using them as a single agent will often invoke drug resistance. Because he failed to respond to treatment for bacterial pneumonia and his presentation suggests TB or another serious cause of nonresponsiveness to standard treatment for community-acquired pneumonia (CAP), you admit him to the hospital.
Treatment for active TB requires multiple drugs in 2 phases
While all family physicians should suspect active TB in appropriate clinical situations and be comfortable with obtaining cultures and initiating empiric treatment, most will want to seek consultation with an infectious disease (ID) specialist especially in the scenarios listed in TABLE 2.5,22 Delayed or inappropriate treatment of active TB remains a major public health problem and cause of multidrug-resistant TB. Inappropriate treatment has been shown to be associated with a 27-fold increase in treatment failure.23 TB treatment guidelines are available from the CDC,24 World Health Organization,25 and International Union Against Tuberculosis and Lung Disease.26
Appropriate treatment requires the use of multiple medications administered in 2 phases. In the initial phase, a patient with suspected TB should begin 4 drugs—usually INH, RIF, ethambutol (EMB), and pyrazinamide (PZA)—for 2 months.1,2,27 The daily pediatric and adult doses and common adverse effects of these medications are summarized in TABLE 3.28 Although most cases of TB can be adequately treated with 2 drugs to which the organism is susceptible, 4 drugs are used initially while awaiting drug sensitivity test results because of the risk of inadequately treating a strain of drug-resistant TB. Before beginning these medications, a chest x-ray, LFTs, HIV antibody test, hepatitis B and C serologies, a serum creatinine, and complete blood count should be obtained in all patients.5 If EMB is prescribed, the patient should also undergo testing for red-green color discrimination, because red-green color vision disturbance is a potential adverse effect of this medication.
All 4 drugs may be administered as a single daily dose, and may be taken together.29 They are ordinarily given either daily for 8 weeks, or daily for 2 weeks followed by a twice-weekly schedule for the remaining 6 weeks in higher doses, although the twice-weekly dose of RIF is the same as the daily dose. All are pregnancy category C, although for active TB, the benefit of treatment is almost always greater than the potential harm.
The continuation phase of treatment starts at 8 weeks, when the results of initial cultures and drug sensitivity tests should be available to guide therapy. A second set of cultures and AFB smears is obtained at 8 weeks to document clearing of the initial infection and guide duration of the continuation phase. If the initial culture was positive for Mycobacterium tuberculosis and the organism was sensitive to both INH and RIF, these 2 drugs should be continued for another 4 months (for a total of 6 months of treatment). PZA and EMB may be stopped at 2 months if the organism is sensitive to both INH and RIF. Thus, for most patients with active TB, the standard regimen will be 4 drugs for 2 months, then 2 drugs for 4 months.2
When should the standard treatment regimen be modified?
If the second set of cultures obtained 2 months after beginning drug treatment is positive and there was cavitary disease on the initial chest x-ray, the continuation phase should be extended by 7 months (for a total of 9 months of treatment).30 If a patient has either cavitary disease or persistently positive cultures (but not both), then the length of therapy is determined on an individual basis in consultation with an ID specialist.
Should a patient’s cultures show resistance to any of the first-line drugs, obtain consultation with an ID specialist. Treatment of multidrug-resistant TB (resistant to INH and RIF) and its subset, extensively drug-resistant TB (resistant to INH and RIF, plus any fluoroquinolone, plus either an aminoglycoside or capreomycin) requires prolonged courses of therapy with multiple drugs administered by DOT.31,32
If at any point during treatment a patient shows clinical deterioration that’s believed to be due to a resurgence of his or her TB disease, obtain a new set of cultures and, in consultation with an ID specialist, add at least 2 drugs to which the patient has not been exposed. Never add only one drug to a failing regimen; active TB always requires 2 drugs to cure, and the patient may have developed resistance to all of the drugs he or she is currently receiving.
If initial cultures are negative for Mycobacterium tuberculosis but the patient responds to treatment, he or she is considered to have “culture-negative TB,” and should generally be continued on INH and RIF for 2 more months after completion of the initial treatment phase (for a total of 4 months of INH and RIF).33
Remember to report. In the United States, active TB must be reported to your local health department, which can be invaluable in coordinating care and administering DOT.
Directly observed therapy (DOT) is preferred for certain high-risk patients with latent tuberculosis infection (LTBI), including those who are younger than 5 years of age, test positive for human immunodeficiency virus, are receiving immunosuppressive therapy, have chest radiography evidence of healed TB, have recently converted to active TB status while receiving serial TB testing, or have recently been exposed to active TB.12
Treatment for active TB should always be given by DOT.13 Because DOT is labor-intensive, twice-weekly dosing is usually preferred.14
CASE › In the hospital, Mr. J was placed in respiratory isolation, had prompt sputum cultures for TB, and was started on empiric treatment for active TB with INH, RIF, PZA, and EMB in standard doses. A search for other causes of nonresponsiveness to CAP showed no evidence of malignancy or HIV infection. He improved steadily and was discharged from the hospital after 2 weeks to complete 2 months of 4-drug therapy, with follow-up care coordinated by the local health department, including a home health nurse experienced in administering DOT. Cultures were positive for Mycobacterium tuberculosis sensitive to all drugs tested. After his initial 2 months of 4-drug therapy, he completed 4 months of additional treatment with INH and RIF, given by DOT, and recovered completely.
CORRESPONDENCE
Jeff Hall, MD, University of South Carolina Department of Family and Preventive Medicine, 3209 Colonial Drive, Columbia, SC 29203; jeff.hall@uscmed.sc.edu
1. World Health Organization. Global tuberculosis report 2014. World Health Organization Web site. Available at: http://www.who.int/tb/publications/global_report/en/. Accessed December 15, 2014.
2. Zumla AI, Raviglione M, Hafner R, et al. Tuberculosis. N Engl J Med. 2013;368:745-755.
3. American Thoracic Society. Targeted tuberculin testing and treatment of latent tuberculosis infection. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr4906a1.htm. Accessed December 15, 2014.
4. Hauck FR, Neese BH, Panchal AS, et al. Identification and management of latent tuberculosis infection. Am Fam Physician. 2009;79:879-886.
5. American Thoracic Society; CDC; Infectious Diseases Society of America. Treatment of tuberculosis. MMWR Recomm Rep. 2003;52:1-77.
6. Centers for Disease Control and Prevention. Latent tuberculosis infection: A guide for primary health care providers. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/tb/publications/LTBI/default.htm. Accessed December 11, 2014.
7. Centers for Disease Control and Prevention. Fact sheet: tuberculosis and pregnancy. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/TB/publications/factsheets/specpop/pregnancy.htm. Accessed September 6, 2014.
8. Kunst H, Khan KS. Age-related risk of hepatotoxicity in the treatment of latent tuberculosis infection: a systematic review. Int J Tuberc Lung Dis. 2010;14:1374-1381.
9. Bliven EE, Podewils LJ. The role of chronic hepatitis in isoniazid hepatotoxicity during treatment for latent tuberculosis infection. Int J Tuberc Lung Dis. 2009;13:1054-1060.
10. Akolo C, Adetifa I, Shepperd S, et al. Treatment of latent tuberculosis infection in HIV infected persons. Cochrane Database Syst Rev. 2010;1:CD000171.
11. Horsburgh CR Jr, Goldberg S, Bethel J, et al; Tuberculosis Epidemiologic Studies Consortium. Latent TB infection treatment acceptance and completion in the United States and Canada. Chest. 2010;137:401-409.
12. Horsburgh CR Jr. Priorities for the treatment of latent tuberculosis infection in the United States. N Engl J Med. 2004;350:2060-2070.
13. Potter B, Rindfleisch K, Kraus CK. Management of active tuberculosis. Am Fam Physician. 2005;72:2225-2232.
14. Volmink J, Garner P. Directly observed therapy for treating tuberculosis. Cochrane Database Syst Rev. 2007;4:CD003343.
15. Sterling TR, Villarina ME, Borisov AS, et al; TB Trials Consortium PREVENT TB Study Team. Three months of rifapentine and isoniazid for latent tuberculosis infection. N Engl J Med. 2011;365:2155-2166.
16. Centers for Disease Control and Prevention (CDC). Recommendations for use of an isoniazid-rifapentine regimen with direct observation to treat latent Mycobacterium tuberculosis infection. MMWR Morb Mortal Wkly Rep. 2011;60:1650-1653.
17. Inge LD, Wilson JW. Update on the treatment of tuberculosis. Am Fam Physician. 2008;78:457-465.
18. Moore DA, Evans CA, Gilman RH, et al. Microscopic-observation drug-susceptibility assay for the diagnosis of TB. N Engl J Med. 2006;355:1539-1550.
19. Minion J, Leung E, Menzies D, et al. Microscopic-observation drug susceptibility and thin layer agar assays for the detection of drug resistant tuberculosis: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10:688-698.
20. Boehme CC, Nabeta P, Hilleman D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363:1005-1015.
21. Arentz M, Sorensen B, Horne DJ, et al. Systematic review of the performance of rapid rifampicin resistance testing for drug-resistant tuberculosis. PLoS One. 2013;8:e76533.
22. Sia IG, Wieland ML. Current concepts in the management of tuberculosis. Mayo Clin Proc. 2011;86:348-361.
23. van der Werf MJ, Langendam MW, Huitric E, et al. Multidrug resistance after inappropriate tuberculosis treatment: a meta-analysis. Eur Respir J. 2012;39:1511-1519.
24. Centers for Disease Control and Prevention. Tuberculosis (TB). Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/TB/publications/guidelines/default.htm. Accessed September 6, 2014.
25. World Health Organization. Treatment of tuberculosis guidelines. 4th ed. World Health Organization Web site. Available at: http://whqlibdoc.who.int/publications/2010/9789241547833_eng.pdf?ua=1. Accessed September 6, 2014.
26. International Union Against Tuberculosis and Lung Disease. Management of tuberculosis: A guide to the essentials of good clinical practice. 6th ed. 2010. International Union Against Tuberculosis and Lung Disease Web site. Available at: http://www.theunion.org/what-we-do/publications/technical/management-of-tuberculosis-a-guide-to-the-essentials-of-good-clinical-practice. Accessed September 6, 2014.
27. Combs DL, O’Brien RJ, Geiter LJ. USPHS Tuberculosis Short-Course Chemotherapy Trial 21: effectiveness, toxicity and acceptability. The report of the final results. Ann Intern Med. 1990;112:397-406.
28. Drugs for tuberculosis. Treat Guidel Med Lett. 2012;10:29-36.
29. Chang KC, Leung CC, Grosset J, et al. Treatment of tuberculosis and optimal dosing schedules. Thorax. 2011;66:997-1007.
30. Blumberg HM, Burman WJ, Chaisson RE, et al; American Thoracic Society, Centers for Disease Control and Prevention and the Infectious Diseases Society. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167:603-662.
31. Lynch JB. Multidrug-resistant tuberculosis. Med Clin North Am. 2013;97:553-579,ix-x.
32. Keshavjee S, Farmer PE. Tuberculosis, drug resistance, and the history of modern medicine. N Engl J Med. 2012;367:931-936.
33. Dutt AK, Moers D, Stead WW. Smear- and culture-negative pulmonary tuberculosis: four-month short-course chemotherapy. Am Rev Respir Dis. 1989;139:867-870.
1. World Health Organization. Global tuberculosis report 2014. World Health Organization Web site. Available at: http://www.who.int/tb/publications/global_report/en/. Accessed December 15, 2014.
2. Zumla AI, Raviglione M, Hafner R, et al. Tuberculosis. N Engl J Med. 2013;368:745-755.
3. American Thoracic Society. Targeted tuberculin testing and treatment of latent tuberculosis infection. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/mmwr/preview/mmwrhtml/rr4906a1.htm. Accessed December 15, 2014.
4. Hauck FR, Neese BH, Panchal AS, et al. Identification and management of latent tuberculosis infection. Am Fam Physician. 2009;79:879-886.
5. American Thoracic Society; CDC; Infectious Diseases Society of America. Treatment of tuberculosis. MMWR Recomm Rep. 2003;52:1-77.
6. Centers for Disease Control and Prevention. Latent tuberculosis infection: A guide for primary health care providers. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/tb/publications/LTBI/default.htm. Accessed December 11, 2014.
7. Centers for Disease Control and Prevention. Fact sheet: tuberculosis and pregnancy. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/TB/publications/factsheets/specpop/pregnancy.htm. Accessed September 6, 2014.
8. Kunst H, Khan KS. Age-related risk of hepatotoxicity in the treatment of latent tuberculosis infection: a systematic review. Int J Tuberc Lung Dis. 2010;14:1374-1381.
9. Bliven EE, Podewils LJ. The role of chronic hepatitis in isoniazid hepatotoxicity during treatment for latent tuberculosis infection. Int J Tuberc Lung Dis. 2009;13:1054-1060.
10. Akolo C, Adetifa I, Shepperd S, et al. Treatment of latent tuberculosis infection in HIV infected persons. Cochrane Database Syst Rev. 2010;1:CD000171.
11. Horsburgh CR Jr, Goldberg S, Bethel J, et al; Tuberculosis Epidemiologic Studies Consortium. Latent TB infection treatment acceptance and completion in the United States and Canada. Chest. 2010;137:401-409.
12. Horsburgh CR Jr. Priorities for the treatment of latent tuberculosis infection in the United States. N Engl J Med. 2004;350:2060-2070.
13. Potter B, Rindfleisch K, Kraus CK. Management of active tuberculosis. Am Fam Physician. 2005;72:2225-2232.
14. Volmink J, Garner P. Directly observed therapy for treating tuberculosis. Cochrane Database Syst Rev. 2007;4:CD003343.
15. Sterling TR, Villarina ME, Borisov AS, et al; TB Trials Consortium PREVENT TB Study Team. Three months of rifapentine and isoniazid for latent tuberculosis infection. N Engl J Med. 2011;365:2155-2166.
16. Centers for Disease Control and Prevention (CDC). Recommendations for use of an isoniazid-rifapentine regimen with direct observation to treat latent Mycobacterium tuberculosis infection. MMWR Morb Mortal Wkly Rep. 2011;60:1650-1653.
17. Inge LD, Wilson JW. Update on the treatment of tuberculosis. Am Fam Physician. 2008;78:457-465.
18. Moore DA, Evans CA, Gilman RH, et al. Microscopic-observation drug-susceptibility assay for the diagnosis of TB. N Engl J Med. 2006;355:1539-1550.
19. Minion J, Leung E, Menzies D, et al. Microscopic-observation drug susceptibility and thin layer agar assays for the detection of drug resistant tuberculosis: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10:688-698.
20. Boehme CC, Nabeta P, Hilleman D, et al. Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med. 2010;363:1005-1015.
21. Arentz M, Sorensen B, Horne DJ, et al. Systematic review of the performance of rapid rifampicin resistance testing for drug-resistant tuberculosis. PLoS One. 2013;8:e76533.
22. Sia IG, Wieland ML. Current concepts in the management of tuberculosis. Mayo Clin Proc. 2011;86:348-361.
23. van der Werf MJ, Langendam MW, Huitric E, et al. Multidrug resistance after inappropriate tuberculosis treatment: a meta-analysis. Eur Respir J. 2012;39:1511-1519.
24. Centers for Disease Control and Prevention. Tuberculosis (TB). Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/TB/publications/guidelines/default.htm. Accessed September 6, 2014.
25. World Health Organization. Treatment of tuberculosis guidelines. 4th ed. World Health Organization Web site. Available at: http://whqlibdoc.who.int/publications/2010/9789241547833_eng.pdf?ua=1. Accessed September 6, 2014.
26. International Union Against Tuberculosis and Lung Disease. Management of tuberculosis: A guide to the essentials of good clinical practice. 6th ed. 2010. International Union Against Tuberculosis and Lung Disease Web site. Available at: http://www.theunion.org/what-we-do/publications/technical/management-of-tuberculosis-a-guide-to-the-essentials-of-good-clinical-practice. Accessed September 6, 2014.
27. Combs DL, O’Brien RJ, Geiter LJ. USPHS Tuberculosis Short-Course Chemotherapy Trial 21: effectiveness, toxicity and acceptability. The report of the final results. Ann Intern Med. 1990;112:397-406.
28. Drugs for tuberculosis. Treat Guidel Med Lett. 2012;10:29-36.
29. Chang KC, Leung CC, Grosset J, et al. Treatment of tuberculosis and optimal dosing schedules. Thorax. 2011;66:997-1007.
30. Blumberg HM, Burman WJ, Chaisson RE, et al; American Thoracic Society, Centers for Disease Control and Prevention and the Infectious Diseases Society. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167:603-662.
31. Lynch JB. Multidrug-resistant tuberculosis. Med Clin North Am. 2013;97:553-579,ix-x.
32. Keshavjee S, Farmer PE. Tuberculosis, drug resistance, and the history of modern medicine. N Engl J Med. 2012;367:931-936.
33. Dutt AK, Moers D, Stead WW. Smear- and culture-negative pulmonary tuberculosis: four-month short-course chemotherapy. Am Rev Respir Dis. 1989;139:867-870.
Nontraumatic knee pain: A diagnostic & treatment guide
› Consider radiography for
a patient with patellofemoral pain syndrome if examination reveals an effusion, the patient is age
50 years or older, or the condition does not improve after 8 to 12 weeks of treatment. C
› Order plain radiography
for all patients with patellofemoral instability to assess for osseous trauma/deformity; consider magnetic resonance imaging if you suspect significant soft tissue damage or the patient does not respond to conservative therapy. C
› Perform joint aspiration with synovial fluid analysis for patients with painful knee effusion, and provide an orthopedic referral without delay when an infectious joint is suspected. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Jane T, age 42, comes to see you because of right knee pain that she’s had for about 6 months. She denies any trauma. Ms. T describes the pain as vague and poorly localized, but worse with activity. She says she started a walking/running program 9 months ago, when she was told she was overweight (body mass index, 29). She has lost 10 pounds since then, Ms. T says, and hopes to lose more by continuing to exercise. upon further review, you find that Ms. T has had increasing pain while ascending and descending stairs and that the pain is also exacerbated when she stands after prolonged sitting.
If Ms. T were your patient, what would you include in a physical examination and how would you diagnose and treat her?
Knee pain is a common presentation in primary care. While traumatic knee pain is frequently addressed in the medical literature, little has been written about chronic nontraumatic nonarthritic knee pain like that of Ms. T. Thus, while physical exam tests often lead to the correct diagnosis for traumatic knee pain, there is limited information on the use of such tests to determine the etiology of chronic knee pain.
This review was developed to fill that gap. In the pages that follow, we provide general guidance on the diagnosis and treatment of chronic nontraumatic knee pain. The conditions are presented anatomically—anterior, lateral, medial, or posterior—with common etiologies, history and physical exam findings, and diagnosis and treatment options for each (TABLE).1-31
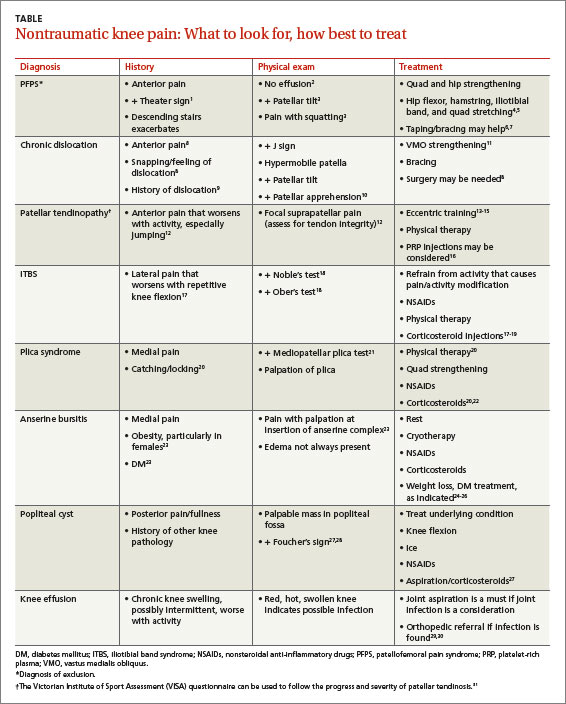
Anterior knee pain
Patellofemoral pain syndrome
Patellofemoral pain syndrome (PFPS), the most common cause of anterior knee pain, is a complex entity with an etiology that has not been well described.2 The quadriceps tendon, medial and lateral retinacula, iliotibial band (ITB), vastus medialis and lateralis, and the insertion of the patellar tendon on the anterior tibial tubercle all play a role in proper tracking of the patellofemoral joint; an imbalance in any of these forces leads to abnormal patellar tracking over the femoral condyles, and pain ensues. PFPS can also be secondary to joint overload, in which excessive physical activity (eg, running, lunges, or squats) overloads the patellofemoral joint and causes pain.
Risk factors for PFPS include strength imbalances in the quadriceps, hamstring, and hip muscle groups, and increased training, such as running longer distances.4,32 A recent review showed no relationship between an increased quadriceps (Q)-angle and PFPS, so that is no longer considered a major risk factor.5
Diagnosis. PFPS is a diagnosis of exclusion, and is primarily based on history and physical exam. Anterior knee pain that is exacerbated when seated for long periods of time (the “theater sign”) or by descending stairs is a classic indication of PFPS.1 Patients may complain of knee stiffness or “giving out” secondary to sharp knee pain and a sensation of popping or crepitus in the joint. Swelling is not a common finding.2
A recent meta-analysis revealed limited evidence for the use of any specific physical exam tests to diagnose PFPS. But pain during squatting and pain with a patellar tilt test were most consistent with a diagnosis of PFPS. (The patellar tilt test involves lifting the lateral edge of the patella superiorly while the patient lies supine with knee extended; pain with <20° of lift suggests a tight lateral retinaculum). Conversely, the absence of pain during squatting or the absence of lateral retinacular pain helps rule it out.2 A physical exam of the cruciate and collateral ligaments should be performed in a patient with a history of instability. Radiography is not needed for a diagnosis, but may be considered if examination reveals an effusion, the patient is age 50 years or older, or no improvement occurs after 8 to 12 weeks of treatment.33
Treatment. The most effective and strongly supported treatment for PFPS is a 6-week physiotherapy program focusing on strengthening the quadriceps and hip muscles and stretching the quadriceps, ITB, hamstrings, and hip flexors.4,5 There is limited information about the use of nonsteroidal anti-inflammatory drugs (NSAIDs), but they can be considered for short-term management.2
Patellar taping and bracing have shown some promise as adjunct therapies for PFPS, although the data for both are non-conclusive. There is a paucity of prospective randomized trials of patellar bracing and a 2012 Cochrane review found limited evidence of its efficacy.34 But a 2014 meta-analysis revealed moderate evidence in support of patellar taping early on to help decrease pain,6 and a recent review suggests that it can be helpful in both the short and long term.7
Taping or bracing may be useful when combined with a tailored physical therapy program. Evidence for treatments such as biofeedback, chiropractic manipulation, and orthotics is limited, and they should be used only as adjunctive therapy.4
CASE › When you examine Ms. T, you find no swelling of the affected knee. You perform the tilt test, which elicits pain. Squatting causes some pain, as well. You diagnose PFPS and provide a referral for 6 weeks of physiotherapy.
Patellar subluxation or chronic dislocation
Patellofemoral instability (PFI) occurs when the patella disengages completely from the trochlear groove.11 PFI’s etiology also relates to the complexity of the patellofemoral joint. Here, too, stability of the joint is achieved with a combination of soft tissue and bony restraints. At full extension and early flexion of the knee, however, the mechanisms of stability are limited, resulting in increased instability. Other associated factors include Q-angle, lateral pull from a tight ITB, and opposing forces from the vastus lateralis and vastus medialis obliquus (VMO).8-10
Risk factors for PFI. The most common predisposing factors for PFI are trochlear dysplasia, patella alta, and lateralization of the tibial tuberosity or patella.10,11 Older patients, predominately women, have an increased risk for PFI.9 Patients usually have a history of patellar subluxation or dislocation in their youth, with approximately 17% of those who had a first dislocation experiencing a recurrence.9 A family history of PFI is common, as well.10
Diagnosis. Patients with PFI often present with nonspecific anterior knee pain secondary to recurrent dislocation.13 Notable physical exam findings are:
- a positive J sign (noted if the patella suddenly shifts medially during early knee flexion or laterally during full extension)
- decreased quadriceps (specifically VMO) and hamstring strength and flexibility
- patellar hypermobility, which should be no more than a quarter to a half of the patellar diameter bilaterally
- pain during a patellar tilt test
- a positive patellar apprehension test.10 (With the patient lying with the knee flexed to 20°, place thumbs on the medial patella and push laterally; the patient will straighten leg with pain or “apprehension” prior to patellar dislocation.)
Plain radiography should be ordered in all cases to assess for osseous trauma/ deformity and to help guide surgical consideration. Magnetic resonance imaging (MRI) can provide additional information when significant soft tissue damage is suspected or the patient does not improve with conservative therapy.8,11
Treatment. A recent Cochrane review showed that conservative treatment (VMO strengthening, bracing, and proprioceptive therapy) prevented future dislocations more effectively than surgical intervention.11 However, surgery is indicated when obvious predisposing anatomic conditions (osteochondral fracture, intra-articular deformity, or a major tear of a medial soft tissue stabilizer) are clearly shown on imaging.8,11
Patellar tendinopathy (jumper’s knee)
Patellar tendinopathy, an overuse injury often called “jumper’s knee” because it is associated with high-intensity jumping sports like volleyball and basketball, is an insertional tendinopathy with pain most commonly at the proximal patellar tendon.10 The pathology of the injury is poorly understood, but is believed to be the result of an impaired healing response to microtears.12,14
Diagnosis. Patients with patellar tendinopathy typically present with anterior suprapatellar pain aggravated by activity. Classically, the pain can occur in any of 4 phases:12 1. pain isolated after activity; 2. pain that occurs during activity but does not impede activity; 3. pain that occurs both during and after the activity and interferes with competition ; 4. a complete tendon disruption.
Examination should include an assessment of the patellar tendon for localized thickening, nodularity, crepitus, and focal suprapatellar tenderness. The muscle-tendon function should be evaluated by assessing knee mobility and strength of the quads via straight leg raise, decline squat, or single leg squats.12 The Victorian Institute of Sport Assessment (VISA) questionnaire can be used to quantify the symptoms and to help track the patient’s progress throughout therapy.31 There are no proven special tests or radiologic studies to aid in the diagnosis of patellar tendinopathy,14 but magnetic resonance imaging (MRI) can be used for further evaluation when findings are equivocal.35
Treatment. A wide range of options, from eccentric training—eg, 3 sets of 15 repetitions performed twice a day for 12 weeks—and physical therapy to platelet-rich plasma (PRP) injections, sclerosing injections, and surgery, are available for the treatment of patellar tendinopathy.13-15 While no specific data have proven the superiority of any one therapy, expert consensus recommends eccentric exercise as initial therapy, performed for 12 weeks.14,15
It’s also interesting to note that a recently published study showed that 3 weekly PRP injections helped 75% of patients—all of whom failed to respond to 4 months of eccentric therapy—return to their pre-symptom activity level within 90 days.16 Corticosteroid injections should not be used to treat patellar tendinopathy due to the risk of tendon rupture.15 Orthopedic referral for surgical intervention should be considered for patients who fail to respond after 3 to 6 months of conservative therapy.14
Lateral knee pain
Iliotibial band tendinopathy
Iliotibial band syndrome (ITBS) is a common source of lateral knee pain, particularly in runners, cyclists, and endurance athletes.17-19,36,37 The exact pathophysiology behind this diagnosis is debatable, but the most accepted etiology is inflammation generated from micro trauma to the soft tissues with inadequate healing time, resulting in persistent inflammation. ITBS is often associated with excessive overall running mileage, a sudden increase in mileage, or an abrupt change in training.18,37
Diagnosis. Patients often complain of persistent nontraumatic lateral knee pain that worsens with repetitive knee flexion (eg, running or cycling).17-19,37 A physical exam will often reveal pain over the lateral femoral condyle and a positive Noble’s test (FIGURE 1). A positive Ober’s test (FIGURE 2) is suggestive of ITBS, as well. The sensitivity and specificity of these tests are not well established, but in patients performing repetitive knee flexion activities with subjective lateral knee pain, pain over the lateral femoral condyle and a positive Ober’s and/or Noble’s test suggest an ITBS diagnosis.18 Imaging is not indicated initially, but MRI should be used in refractory cases to rule out other etiologies.17,19
Treatment. First-line therapy for ITBS is conservative,17-19,36,37 often involving a combination of techniques such as refraining from the activity that triggers the pain, NSAIDs, activity modification to reduce the strain over the ITB, myofascial release via foam rollers, and physical therapy focused on stretching the iliotibial band, tensor fasciae latae, and gluteus medius while strengthening the gluteus medius and core muscles.17 No single program has been shown to be better than another.
Corticosteroid injections are second-line therapy and have been shown to improve pain compared with placebo up to 2 weeks post injection.17,19 When symptoms persist for more than 6 months despite conservative treatment, surgical intervention may be indicated.18,19 Patients who experience temporary pain relief with corticosteroid injections often respond best to surgery.36
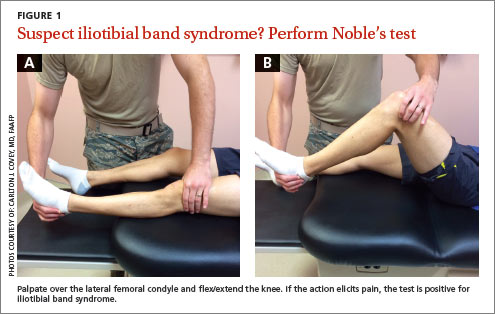
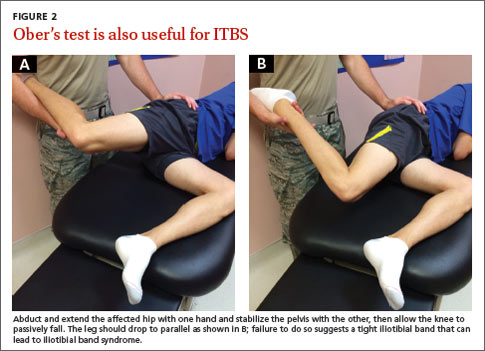
Medial knee pain
Medial plica syndrome
Because of its anatomic location, the medial plica—which can be palpated in up to 84% of the population20—is susceptible to impingement by the medial femoral condyle or the patellofemoral joint. Trauma with repetitive knee movement leads to inflammation and thickening of the plica, resulting in medial plica syndrome.20,38 Initial inflammation may be triggered by blunt trauma, a sudden increase in activity, or transient synovitis.22
Diagnosis. Medial plica syndrome is a challenging diagnosis. Patients generally have nonspecific complaints of aching medial knee pain, locking, and catching similar to complaints of a medial meniscal injury.20
Evaluation should include the mediopatellar plica test, which is performed with the patient lying supine with the knee fully extended. Pressure is placed over the inferomedial patellofemoral joint, creating an impingement of the medial plica between the finger and the medial femoral condyle. Elimination or marked diminishing of pain with knee flexion to 90° is considered a positive test.21
A recent systematic review found this test to be more diagnostically accurate than an MRI (sensitivity of the test is 90% and specificity is 89%, vs 77% and 58%, respectively, for MRI) for detection of medial plica syndrome. Ultrasound is almost as accurate, with a sensitivity of 90% and specificity of 83%.39
Treatment of medial plica syndrome centers on physiotherapy and quadriceps strengthening,20 augmented with NSAIDs. Intra-articular corticosteroid injections are considered second-line treatment.20,22 An orthopedics referral is indicated to consider arthroscopic plica removal for refractory cases.20,22
Pes anserine bursitis
The anserine bursal complex, located approximately 5 cm distal to the medial joint line, is formed by the combined insertion of the sartorius, gracilis, and semitendinosus tendons,39 but the exact mechanism of pain is not well understood. Whether the pathophysiology is from an insertional tendonitis or overt bursitis is unknown, and no studies have focused on prevalence or risk factors. What is known is that overweight individuals and women with a wide pelvis seem to have a greater predilection and those with pes planus, diabetes, or knee osteoarthritis are at increased risk.23
Diagnosis. Medial knee pain reproduced on palpation of the anatomical site of insertion of the pes anserine tendon complex supports a diagnosis of pes anserine bursitis, with or without edema. Radiologic studies are not needed, but may be helpful if significant bony pathology is suspected. Ultrasound, computed tomography (CT), and MRI are not recommended.23
Treatment. Resting the affected knee, cryotherapy, NSAIDs, and using a pillow at night to relieve direct bursal pressure are recommended.33 Weight loss in obese patients, treatment of pes planus, and control of diabetes may be helpful, as well. Although the literature is limited and dated, corticosteroid injection has been found to reduce the pain and may be considered as second-line treatment.24-26
Posterior knee pain
Popliteal (Baker’s) cyst
The popliteal fossa contains 6 of the numerous bursa of the knee; the bursa beneath the medial head of the gastrocnemius muscle and the semimembranosus tendon is most commonly involved in the formation of a popliteal cyst.40 It is postulated that increased intra-articular pressure forces fluid into the bursa, leading to expansion and pain. This can be idiopathic or secondary to internal derangement or trauma to the knee.41 Older age, a remote history of knee trauma, or a coexisting joint disease such as osteoarthritis, meniscal pathology, or rheumatoid arthritis are significant risk factors for the development of popliteal cysts.27
Diagnosis. Most popliteal cysts are asymptomatic in adults and discovered incidentally after routine imaging to evaluate other knee pathology. However, symptomatic popliteal cysts present as a palpable mass in the popliteal fossa, resulting in pain and limited range of motion.
During the physical exam with the patient lying supine, a medial popliteal mass that is most prominent with the knee fully extended is common. A positive Foucher’s sign (the painful mass is palpated posteriorly in the popliteal fossa with the knee fully extended; pain is relieved and/or the mass reduced in size with knee flexion to 45°) suggests a diagnosis of popliteal cyst.27,28
Radiologic studies are generally not needed to diagnose a popliteal cyst. However, if diagnostic uncertainty remains after the history and physical exam, plain knee radiographs and ultrasound should be obtained. This combination provides complementary information and helps rule out a fracture, arthritis, and thrombosis as the cause of the pain.27 MRI is helpful if the diagnosis is still in doubt and for patients suspected of having significant internal derangement leading to cyst formation. Arthrography or CT is generally not needed.27,41
Treatment. As popliteal cysts are often associated with other knee pathology, management of the underlying condition often leads to cyst regression. Keeping the knee in flexion can decrease the available space and assist in pain control in the acute phase.27 Cold packs and NSAIDs can also be used initially. Cyst aspiration and intra-articular steroid injection have been shown to be effective for cysts that do not respond to this conservative approach.27 However, addressing and managing the underlying knee pathology (eg, osteoarthritis, meniscal pathology, or rheumatoid arthritis) will prevent popliteal cysts from recurring.
When the problem is painful knee effusion
Nontraumatic knee effusion can be the primary source of knee pain or the result of underlying pathology. We mention it here because clinical suspicion is paramount in diagnosing a septic joint, a serious cause of painful knee effusion that warrants prompt treatment.
As in other causes of knee pain, a detailed history of the character of the pain is essential. Septic arthritis and crystalline disease (gout, pseudogout) should be suspected in patients without a history of trauma who cannot bear weight. Systemic complaints point to an infection and, with the exception of a possible low-grade fever, are not typically seen in crystalline disease. Notable findings include an erythematous, hot, swollen knee and pain with both active and passive movement.
Plain radiographs of the knee should be ordered to rule out significant trauma or arthritis as the etiology. It is important to perform joint aspiration with synovial fluid analysis. Fluid analysis should include a white blood cell (WBC) count with differential, Gram stain and cultures, and polarized light microscopy (not readily available in an outpatient setting).29
Synovial fluid analysis characteristics suggestive of a septic joint include turbid quality, WBC >50,000 per mm3, an elevated protein content, and a low glucose concentration.30 Gram stain and culture will help identify the infectious agent. Orthopedic referral should not be delayed in patients with a suspected infectious joint. Corticosteroids should not be injected during aspiration if infection is being ruled out.
CASE › When Ms. T returns for a follow-up visit 8 weeks later, she states that the knee pain has resolved and that she has returned to running. She has lost an additional 8 pounds and continues to diet. And, at the advice of her physical therapist, she is continuing her physiotherapy regimen at home to prevent a recurrence of PFPS.
CORRESPONDENCE
Carlton J. Covey, MD, FAAFP, Nellis Family Medicine Residency Program, 4700 Las Vegas Boulevard North, Nellis Air Force Base, NV 89191; carlton.covey@us.af.mil
1. Earl JE, Vetter CS. Patellofemoral pain. Phys Med Rehabil Clin N Am. 2007;18:439-458,viii.
2. McGowan HJ, Beutler A. Patellofemoral syndrome. Essential Evidence Plus Web site. Available at: http://www.essentialevidenceplus.com. Accessed: March 20, 2014.
3. Nunes GS, Stapait EL, Kirsten MH, et al. Clinical test for diagnosis of patellofemoral pain syndrome: Systematic review with meta-analysis. Phys Ther Sport. 2013;14:54-59.
4. Rixe JA, Glick JE, Brady J, et al. A review of the management of patellofemoral pain syndrome. Phys Sportsmed. 2013;41: 19-28.
5. Bolgla LA, Boling MC. An update for the conservative management of patellofemoral pain syndrome: a systematic review of the literature from 2000 to 2010. Int J Sports Phys Ther. 2011;6:112-125.
6. Barton C, Balachandar V, Lack S, et al. Patellar taping for patellofemoral pain: a systematic review and meta-analysis to evaluate clinical outcomes and biomechanical mechanisms. Br J Sports Med. 2014;48:417-424.
7. Dutton RA, Khadavi MJ, Fredericson M. Update on rehabilitation of patellofemoral pain. Curr Sports Med Rep. 2014;13: 172-178.
8. Kapur S, Wissman RD, Robertson M, et al. Acute knee dislocation: review of an elusive entity. Curr Probl Diagn Radiol. 2009;38:237-250.
9. Colvin AC, West RV. Patellar instability. J Bone Joint Surg Am. 2008;90:2751-2762.
10. Tscholl PM, Koch PP, Fucentese SF. Treatment options for patellofemoral instability in sports traumatology. Orthop Rev (Pavia). 2013;5:e23.
11. Earhart C, Patel DB, White EA, et al. Transient lateral patellar dislocation: review of imaging findings, patellofemoral anatomy, and treatment options. Emerg Radiol. 2013;20:11-23.
12. Tan SC, Chan O. Achilles and patellar tendinopathy: current understanding of pathophysiology and management. Disabil Rehabil. 2008;30:1608-1615.
13. Gaida JE, Cook J. Treatment options for patellar tendinopathy: critical review. Curr Sports Med Rep. 2011;10:255-270.
14. Rodriguez-Merchan EC. The treatment of patellar tendinopathy. J Orthop Traumatol. 2013;14:77-81.
15. Childress MA, Beutler A. Management of chronic tendon injuries. Am Fam Physician. 2013;87:486-490.
16. Charousset C, Zaoui A, Bellaiche L, et al. Are multiple platelet-rich plasma injections useful for treatment of chronic patellar tendinopathy in athletes? A prospective study. Am J Sports Med. 2014;42:906-911.
17. Strauss EJ, Kim S, Calcei JG, et al. Iliotibial band syndrome: evaluation and management. J Am Acad Orthop Surg. 2011;19:728-736.
18. Bellary SS, Lynch G, Housman B, et al. Medial plica syndrome: a review of the literature. Clin Anat. 2012;25:423-428.
19. Hong JH, Kim JS. Diagnosis of iliotibial band friction syndrome and ultrasound guided steroid injection. Korean J Pain. 2013;26:387-391.
20. Bellary SS, Lynch G, Housman B, et al. Medial plica syndrome: a review of the literature. Clin Anat. 2012;25:423-428.
21. Kim SJ, Jeong JH, Cheon YM, et al. MPP test in the diagnosis of medial patellar plica syndrome. Arthroscopy. 2004;20: 1101-1103.
22. Schindler OS. ‘The Sneaky Plica’ revisited: morphology, pathophysiology and treatment of synovial plicae of the knee. Knee Surg Sports Traumatol Arthrosc. 2014;22:247-262.
23. Helfenstein M Jr, Kuromoto J. Anserine syndrome. Rev Bras Rheumatol. 2010;50:313-327.
24. Abeles M. Osteoarthritis of the knee: anserine bursitis as an extra-articular cause of pain. Clin Res. 1983;31:4471-4476.
25. Kang I, Han SW. Anserine bursitis in patients with osteoarthritis of the knee. South Med J. 2000;93:207-209.
26. Yoon HS, Kim SE, Suh YR, et al. Correlation between ultrasonographic findings and the response to corticosteroid injection in pes anserinus tendinobursitis syndrome in knee osteoarthritis patients. J Korean Med Sci. 2005;20:109-112.
27. Stein D, Cantlon M, MacKay B, et al. Cysts about the knee: evaluation and management. J Am Acad Orthop Surg. 2013;21: 469-479.
28. Canoso JJ, Goldsmith MR, Gerzof SG, et al. Foucher’s sign of the Baker’s cyst. Ann Rheum Dis. 1987;46:228-232.
29. Palmer T. Knee pain. Essential Evidence Plus Web site. Available at: http://www.essentialevidenceplus.com. Accessed: December 12, 2013.
30. Franks AG Jr. Rheumatologic aspects of knee disorders. In: Scott WN, ed. The Knee. St. Louis: Mosby; 1994:315-329.
31. Visentini PJ, Khan KM, Cook JL, et al. The VISA score: an index of severity of symptoms in patients with jumper’s knee (patellar tendinosis). Victorian Institute of Sport Tendon Study Group. J Sci Med Sport. 1998;1:22-28.
32. Halabchi F, Mazaheri R, Seif-Barghi T. Patellofemoral pain syndrome and modifiable intrinsic risk factors; how to assess and address? Asian J Sports Med. 2013;4:85-100.
33. Dixit S, DiFiori JP, Burton M, et al. Management of patellofemoral pain syndrome. Am Fam Physician. 2007;75:194-202.
34. Callaghan MJ, Selfe J. Patellar taping for patellofemoral pain syndrome in adults. Cochrane Database Syst Rev. 2012;4:CD006717.
35. Atanda AJ Jr, Ruiz D, Dodson CC, et al. Approach to the active patient with chronic anterior knee pain. Phys Sportsmed. 2012;40:41-50.
36. Ellis R, Hing W, Reid D. Iliotibial band friction syndrome—a systematic review. Man Ther. 2007;12:200-208.
37. Kirk KL, Kuklo T, Klemme W. Iliotibial band friction syndrome. Orthopedics. 2000;23:1209-1217.
38. Stubbings N, Smith T. Diagnostic test accuracy of clinical and radiological assessments for medial patella plica syndrome: a systematic review and meta-analysis. Knee. 2014;21: 486-490.
39. Alvarez-Nemegyei J, Canoso JJ. Evidence-based soft tissue rheumatology IV: anserine bursitis. J Clin Rheumatol. 2004;10:205-206.
40. Fritschy D, Fasel J, Imbert JC, et al. The popliteal cyst. Knee Surg Sports Traumatol Arthrosc. 2006;14:623-628.
41. Handy JR. Popliteal cysts in adults: a review. Semin Arthritis Rheum. 2001;31:108-118.
› Consider radiography for
a patient with patellofemoral pain syndrome if examination reveals an effusion, the patient is age
50 years or older, or the condition does not improve after 8 to 12 weeks of treatment. C
› Order plain radiography
for all patients with patellofemoral instability to assess for osseous trauma/deformity; consider magnetic resonance imaging if you suspect significant soft tissue damage or the patient does not respond to conservative therapy. C
› Perform joint aspiration with synovial fluid analysis for patients with painful knee effusion, and provide an orthopedic referral without delay when an infectious joint is suspected. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Jane T, age 42, comes to see you because of right knee pain that she’s had for about 6 months. She denies any trauma. Ms. T describes the pain as vague and poorly localized, but worse with activity. She says she started a walking/running program 9 months ago, when she was told she was overweight (body mass index, 29). She has lost 10 pounds since then, Ms. T says, and hopes to lose more by continuing to exercise. upon further review, you find that Ms. T has had increasing pain while ascending and descending stairs and that the pain is also exacerbated when she stands after prolonged sitting.
If Ms. T were your patient, what would you include in a physical examination and how would you diagnose and treat her?
Knee pain is a common presentation in primary care. While traumatic knee pain is frequently addressed in the medical literature, little has been written about chronic nontraumatic nonarthritic knee pain like that of Ms. T. Thus, while physical exam tests often lead to the correct diagnosis for traumatic knee pain, there is limited information on the use of such tests to determine the etiology of chronic knee pain.
This review was developed to fill that gap. In the pages that follow, we provide general guidance on the diagnosis and treatment of chronic nontraumatic knee pain. The conditions are presented anatomically—anterior, lateral, medial, or posterior—with common etiologies, history and physical exam findings, and diagnosis and treatment options for each (TABLE).1-31
Anterior knee pain
Patellofemoral pain syndrome
Patellofemoral pain syndrome (PFPS), the most common cause of anterior knee pain, is a complex entity with an etiology that has not been well described.2 The quadriceps tendon, medial and lateral retinacula, iliotibial band (ITB), vastus medialis and lateralis, and the insertion of the patellar tendon on the anterior tibial tubercle all play a role in proper tracking of the patellofemoral joint; an imbalance in any of these forces leads to abnormal patellar tracking over the femoral condyles, and pain ensues. PFPS can also be secondary to joint overload, in which excessive physical activity (eg, running, lunges, or squats) overloads the patellofemoral joint and causes pain.
Risk factors for PFPS include strength imbalances in the quadriceps, hamstring, and hip muscle groups, and increased training, such as running longer distances.4,32 A recent review showed no relationship between an increased quadriceps (Q)-angle and PFPS, so that is no longer considered a major risk factor.5
Diagnosis. PFPS is a diagnosis of exclusion, and is primarily based on history and physical exam. Anterior knee pain that is exacerbated when seated for long periods of time (the “theater sign”) or by descending stairs is a classic indication of PFPS.1 Patients may complain of knee stiffness or “giving out” secondary to sharp knee pain and a sensation of popping or crepitus in the joint. Swelling is not a common finding.2
A recent meta-analysis revealed limited evidence for the use of any specific physical exam tests to diagnose PFPS. But pain during squatting and pain with a patellar tilt test were most consistent with a diagnosis of PFPS. (The patellar tilt test involves lifting the lateral edge of the patella superiorly while the patient lies supine with knee extended; pain with <20° of lift suggests a tight lateral retinaculum). Conversely, the absence of pain during squatting or the absence of lateral retinacular pain helps rule it out.2 A physical exam of the cruciate and collateral ligaments should be performed in a patient with a history of instability. Radiography is not needed for a diagnosis, but may be considered if examination reveals an effusion, the patient is age 50 years or older, or no improvement occurs after 8 to 12 weeks of treatment.33
Treatment. The most effective and strongly supported treatment for PFPS is a 6-week physiotherapy program focusing on strengthening the quadriceps and hip muscles and stretching the quadriceps, ITB, hamstrings, and hip flexors.4,5 There is limited information about the use of nonsteroidal anti-inflammatory drugs (NSAIDs), but they can be considered for short-term management.2
Patellar taping and bracing have shown some promise as adjunct therapies for PFPS, although the data for both are non-conclusive. There is a paucity of prospective randomized trials of patellar bracing and a 2012 Cochrane review found limited evidence of its efficacy.34 But a 2014 meta-analysis revealed moderate evidence in support of patellar taping early on to help decrease pain,6 and a recent review suggests that it can be helpful in both the short and long term.7
Taping or bracing may be useful when combined with a tailored physical therapy program. Evidence for treatments such as biofeedback, chiropractic manipulation, and orthotics is limited, and they should be used only as adjunctive therapy.4
CASE › When you examine Ms. T, you find no swelling of the affected knee. You perform the tilt test, which elicits pain. Squatting causes some pain, as well. You diagnose PFPS and provide a referral for 6 weeks of physiotherapy.
Patellar subluxation or chronic dislocation
Patellofemoral instability (PFI) occurs when the patella disengages completely from the trochlear groove.11 PFI’s etiology also relates to the complexity of the patellofemoral joint. Here, too, stability of the joint is achieved with a combination of soft tissue and bony restraints. At full extension and early flexion of the knee, however, the mechanisms of stability are limited, resulting in increased instability. Other associated factors include Q-angle, lateral pull from a tight ITB, and opposing forces from the vastus lateralis and vastus medialis obliquus (VMO).8-10
Risk factors for PFI. The most common predisposing factors for PFI are trochlear dysplasia, patella alta, and lateralization of the tibial tuberosity or patella.10,11 Older patients, predominately women, have an increased risk for PFI.9 Patients usually have a history of patellar subluxation or dislocation in their youth, with approximately 17% of those who had a first dislocation experiencing a recurrence.9 A family history of PFI is common, as well.10
Diagnosis. Patients with PFI often present with nonspecific anterior knee pain secondary to recurrent dislocation.13 Notable physical exam findings are:
- a positive J sign (noted if the patella suddenly shifts medially during early knee flexion or laterally during full extension)
- decreased quadriceps (specifically VMO) and hamstring strength and flexibility
- patellar hypermobility, which should be no more than a quarter to a half of the patellar diameter bilaterally
- pain during a patellar tilt test
- a positive patellar apprehension test.10 (With the patient lying with the knee flexed to 20°, place thumbs on the medial patella and push laterally; the patient will straighten leg with pain or “apprehension” prior to patellar dislocation.)
Plain radiography should be ordered in all cases to assess for osseous trauma/ deformity and to help guide surgical consideration. Magnetic resonance imaging (MRI) can provide additional information when significant soft tissue damage is suspected or the patient does not improve with conservative therapy.8,11
Treatment. A recent Cochrane review showed that conservative treatment (VMO strengthening, bracing, and proprioceptive therapy) prevented future dislocations more effectively than surgical intervention.11 However, surgery is indicated when obvious predisposing anatomic conditions (osteochondral fracture, intra-articular deformity, or a major tear of a medial soft tissue stabilizer) are clearly shown on imaging.8,11
Patellar tendinopathy (jumper’s knee)
Patellar tendinopathy, an overuse injury often called “jumper’s knee” because it is associated with high-intensity jumping sports like volleyball and basketball, is an insertional tendinopathy with pain most commonly at the proximal patellar tendon.10 The pathology of the injury is poorly understood, but is believed to be the result of an impaired healing response to microtears.12,14
Diagnosis. Patients with patellar tendinopathy typically present with anterior suprapatellar pain aggravated by activity. Classically, the pain can occur in any of 4 phases:12 1. pain isolated after activity; 2. pain that occurs during activity but does not impede activity; 3. pain that occurs both during and after the activity and interferes with competition ; 4. a complete tendon disruption.
Examination should include an assessment of the patellar tendon for localized thickening, nodularity, crepitus, and focal suprapatellar tenderness. The muscle-tendon function should be evaluated by assessing knee mobility and strength of the quads via straight leg raise, decline squat, or single leg squats.12 The Victorian Institute of Sport Assessment (VISA) questionnaire can be used to quantify the symptoms and to help track the patient’s progress throughout therapy.31 There are no proven special tests or radiologic studies to aid in the diagnosis of patellar tendinopathy,14 but magnetic resonance imaging (MRI) can be used for further evaluation when findings are equivocal.35
Treatment. A wide range of options, from eccentric training—eg, 3 sets of 15 repetitions performed twice a day for 12 weeks—and physical therapy to platelet-rich plasma (PRP) injections, sclerosing injections, and surgery, are available for the treatment of patellar tendinopathy.13-15 While no specific data have proven the superiority of any one therapy, expert consensus recommends eccentric exercise as initial therapy, performed for 12 weeks.14,15
It’s also interesting to note that a recently published study showed that 3 weekly PRP injections helped 75% of patients—all of whom failed to respond to 4 months of eccentric therapy—return to their pre-symptom activity level within 90 days.16 Corticosteroid injections should not be used to treat patellar tendinopathy due to the risk of tendon rupture.15 Orthopedic referral for surgical intervention should be considered for patients who fail to respond after 3 to 6 months of conservative therapy.14
Lateral knee pain
Iliotibial band tendinopathy
Iliotibial band syndrome (ITBS) is a common source of lateral knee pain, particularly in runners, cyclists, and endurance athletes.17-19,36,37 The exact pathophysiology behind this diagnosis is debatable, but the most accepted etiology is inflammation generated from micro trauma to the soft tissues with inadequate healing time, resulting in persistent inflammation. ITBS is often associated with excessive overall running mileage, a sudden increase in mileage, or an abrupt change in training.18,37
Diagnosis. Patients often complain of persistent nontraumatic lateral knee pain that worsens with repetitive knee flexion (eg, running or cycling).17-19,37 A physical exam will often reveal pain over the lateral femoral condyle and a positive Noble’s test (FIGURE 1). A positive Ober’s test (FIGURE 2) is suggestive of ITBS, as well. The sensitivity and specificity of these tests are not well established, but in patients performing repetitive knee flexion activities with subjective lateral knee pain, pain over the lateral femoral condyle and a positive Ober’s and/or Noble’s test suggest an ITBS diagnosis.18 Imaging is not indicated initially, but MRI should be used in refractory cases to rule out other etiologies.17,19
Treatment. First-line therapy for ITBS is conservative,17-19,36,37 often involving a combination of techniques such as refraining from the activity that triggers the pain, NSAIDs, activity modification to reduce the strain over the ITB, myofascial release via foam rollers, and physical therapy focused on stretching the iliotibial band, tensor fasciae latae, and gluteus medius while strengthening the gluteus medius and core muscles.17 No single program has been shown to be better than another.
Corticosteroid injections are second-line therapy and have been shown to improve pain compared with placebo up to 2 weeks post injection.17,19 When symptoms persist for more than 6 months despite conservative treatment, surgical intervention may be indicated.18,19 Patients who experience temporary pain relief with corticosteroid injections often respond best to surgery.36
Medial knee pain
Medial plica syndrome
Because of its anatomic location, the medial plica—which can be palpated in up to 84% of the population20—is susceptible to impingement by the medial femoral condyle or the patellofemoral joint. Trauma with repetitive knee movement leads to inflammation and thickening of the plica, resulting in medial plica syndrome.20,38 Initial inflammation may be triggered by blunt trauma, a sudden increase in activity, or transient synovitis.22
Diagnosis. Medial plica syndrome is a challenging diagnosis. Patients generally have nonspecific complaints of aching medial knee pain, locking, and catching similar to complaints of a medial meniscal injury.20
Evaluation should include the mediopatellar plica test, which is performed with the patient lying supine with the knee fully extended. Pressure is placed over the inferomedial patellofemoral joint, creating an impingement of the medial plica between the finger and the medial femoral condyle. Elimination or marked diminishing of pain with knee flexion to 90° is considered a positive test.21
A recent systematic review found this test to be more diagnostically accurate than an MRI (sensitivity of the test is 90% and specificity is 89%, vs 77% and 58%, respectively, for MRI) for detection of medial plica syndrome. Ultrasound is almost as accurate, with a sensitivity of 90% and specificity of 83%.39
Treatment of medial plica syndrome centers on physiotherapy and quadriceps strengthening,20 augmented with NSAIDs. Intra-articular corticosteroid injections are considered second-line treatment.20,22 An orthopedics referral is indicated to consider arthroscopic plica removal for refractory cases.20,22
Pes anserine bursitis
The anserine bursal complex, located approximately 5 cm distal to the medial joint line, is formed by the combined insertion of the sartorius, gracilis, and semitendinosus tendons,39 but the exact mechanism of pain is not well understood. Whether the pathophysiology is from an insertional tendonitis or overt bursitis is unknown, and no studies have focused on prevalence or risk factors. What is known is that overweight individuals and women with a wide pelvis seem to have a greater predilection and those with pes planus, diabetes, or knee osteoarthritis are at increased risk.23
Diagnosis. Medial knee pain reproduced on palpation of the anatomical site of insertion of the pes anserine tendon complex supports a diagnosis of pes anserine bursitis, with or without edema. Radiologic studies are not needed, but may be helpful if significant bony pathology is suspected. Ultrasound, computed tomography (CT), and MRI are not recommended.23
Treatment. Resting the affected knee, cryotherapy, NSAIDs, and using a pillow at night to relieve direct bursal pressure are recommended.33 Weight loss in obese patients, treatment of pes planus, and control of diabetes may be helpful, as well. Although the literature is limited and dated, corticosteroid injection has been found to reduce the pain and may be considered as second-line treatment.24-26
Posterior knee pain
Popliteal (Baker’s) cyst
The popliteal fossa contains 6 of the numerous bursa of the knee; the bursa beneath the medial head of the gastrocnemius muscle and the semimembranosus tendon is most commonly involved in the formation of a popliteal cyst.40 It is postulated that increased intra-articular pressure forces fluid into the bursa, leading to expansion and pain. This can be idiopathic or secondary to internal derangement or trauma to the knee.41 Older age, a remote history of knee trauma, or a coexisting joint disease such as osteoarthritis, meniscal pathology, or rheumatoid arthritis are significant risk factors for the development of popliteal cysts.27
Diagnosis. Most popliteal cysts are asymptomatic in adults and discovered incidentally after routine imaging to evaluate other knee pathology. However, symptomatic popliteal cysts present as a palpable mass in the popliteal fossa, resulting in pain and limited range of motion.
During the physical exam with the patient lying supine, a medial popliteal mass that is most prominent with the knee fully extended is common. A positive Foucher’s sign (the painful mass is palpated posteriorly in the popliteal fossa with the knee fully extended; pain is relieved and/or the mass reduced in size with knee flexion to 45°) suggests a diagnosis of popliteal cyst.27,28
Radiologic studies are generally not needed to diagnose a popliteal cyst. However, if diagnostic uncertainty remains after the history and physical exam, plain knee radiographs and ultrasound should be obtained. This combination provides complementary information and helps rule out a fracture, arthritis, and thrombosis as the cause of the pain.27 MRI is helpful if the diagnosis is still in doubt and for patients suspected of having significant internal derangement leading to cyst formation. Arthrography or CT is generally not needed.27,41
Treatment. As popliteal cysts are often associated with other knee pathology, management of the underlying condition often leads to cyst regression. Keeping the knee in flexion can decrease the available space and assist in pain control in the acute phase.27 Cold packs and NSAIDs can also be used initially. Cyst aspiration and intra-articular steroid injection have been shown to be effective for cysts that do not respond to this conservative approach.27 However, addressing and managing the underlying knee pathology (eg, osteoarthritis, meniscal pathology, or rheumatoid arthritis) will prevent popliteal cysts from recurring.
When the problem is painful knee effusion
Nontraumatic knee effusion can be the primary source of knee pain or the result of underlying pathology. We mention it here because clinical suspicion is paramount in diagnosing a septic joint, a serious cause of painful knee effusion that warrants prompt treatment.
As in other causes of knee pain, a detailed history of the character of the pain is essential. Septic arthritis and crystalline disease (gout, pseudogout) should be suspected in patients without a history of trauma who cannot bear weight. Systemic complaints point to an infection and, with the exception of a possible low-grade fever, are not typically seen in crystalline disease. Notable findings include an erythematous, hot, swollen knee and pain with both active and passive movement.
Plain radiographs of the knee should be ordered to rule out significant trauma or arthritis as the etiology. It is important to perform joint aspiration with synovial fluid analysis. Fluid analysis should include a white blood cell (WBC) count with differential, Gram stain and cultures, and polarized light microscopy (not readily available in an outpatient setting).29
Synovial fluid analysis characteristics suggestive of a septic joint include turbid quality, WBC >50,000 per mm3, an elevated protein content, and a low glucose concentration.30 Gram stain and culture will help identify the infectious agent. Orthopedic referral should not be delayed in patients with a suspected infectious joint. Corticosteroids should not be injected during aspiration if infection is being ruled out.
CASE › When Ms. T returns for a follow-up visit 8 weeks later, she states that the knee pain has resolved and that she has returned to running. She has lost an additional 8 pounds and continues to diet. And, at the advice of her physical therapist, she is continuing her physiotherapy regimen at home to prevent a recurrence of PFPS.
CORRESPONDENCE
Carlton J. Covey, MD, FAAFP, Nellis Family Medicine Residency Program, 4700 Las Vegas Boulevard North, Nellis Air Force Base, NV 89191; carlton.covey@us.af.mil
› Consider radiography for
a patient with patellofemoral pain syndrome if examination reveals an effusion, the patient is age
50 years or older, or the condition does not improve after 8 to 12 weeks of treatment. C
› Order plain radiography
for all patients with patellofemoral instability to assess for osseous trauma/deformity; consider magnetic resonance imaging if you suspect significant soft tissue damage or the patient does not respond to conservative therapy. C
› Perform joint aspiration with synovial fluid analysis for patients with painful knee effusion, and provide an orthopedic referral without delay when an infectious joint is suspected. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Jane T, age 42, comes to see you because of right knee pain that she’s had for about 6 months. She denies any trauma. Ms. T describes the pain as vague and poorly localized, but worse with activity. She says she started a walking/running program 9 months ago, when she was told she was overweight (body mass index, 29). She has lost 10 pounds since then, Ms. T says, and hopes to lose more by continuing to exercise. upon further review, you find that Ms. T has had increasing pain while ascending and descending stairs and that the pain is also exacerbated when she stands after prolonged sitting.
If Ms. T were your patient, what would you include in a physical examination and how would you diagnose and treat her?
Knee pain is a common presentation in primary care. While traumatic knee pain is frequently addressed in the medical literature, little has been written about chronic nontraumatic nonarthritic knee pain like that of Ms. T. Thus, while physical exam tests often lead to the correct diagnosis for traumatic knee pain, there is limited information on the use of such tests to determine the etiology of chronic knee pain.
This review was developed to fill that gap. In the pages that follow, we provide general guidance on the diagnosis and treatment of chronic nontraumatic knee pain. The conditions are presented anatomically—anterior, lateral, medial, or posterior—with common etiologies, history and physical exam findings, and diagnosis and treatment options for each (TABLE).1-31
Anterior knee pain
Patellofemoral pain syndrome
Patellofemoral pain syndrome (PFPS), the most common cause of anterior knee pain, is a complex entity with an etiology that has not been well described.2 The quadriceps tendon, medial and lateral retinacula, iliotibial band (ITB), vastus medialis and lateralis, and the insertion of the patellar tendon on the anterior tibial tubercle all play a role in proper tracking of the patellofemoral joint; an imbalance in any of these forces leads to abnormal patellar tracking over the femoral condyles, and pain ensues. PFPS can also be secondary to joint overload, in which excessive physical activity (eg, running, lunges, or squats) overloads the patellofemoral joint and causes pain.
Risk factors for PFPS include strength imbalances in the quadriceps, hamstring, and hip muscle groups, and increased training, such as running longer distances.4,32 A recent review showed no relationship between an increased quadriceps (Q)-angle and PFPS, so that is no longer considered a major risk factor.5
Diagnosis. PFPS is a diagnosis of exclusion, and is primarily based on history and physical exam. Anterior knee pain that is exacerbated when seated for long periods of time (the “theater sign”) or by descending stairs is a classic indication of PFPS.1 Patients may complain of knee stiffness or “giving out” secondary to sharp knee pain and a sensation of popping or crepitus in the joint. Swelling is not a common finding.2
A recent meta-analysis revealed limited evidence for the use of any specific physical exam tests to diagnose PFPS. But pain during squatting and pain with a patellar tilt test were most consistent with a diagnosis of PFPS. (The patellar tilt test involves lifting the lateral edge of the patella superiorly while the patient lies supine with knee extended; pain with <20° of lift suggests a tight lateral retinaculum). Conversely, the absence of pain during squatting or the absence of lateral retinacular pain helps rule it out.2 A physical exam of the cruciate and collateral ligaments should be performed in a patient with a history of instability. Radiography is not needed for a diagnosis, but may be considered if examination reveals an effusion, the patient is age 50 years or older, or no improvement occurs after 8 to 12 weeks of treatment.33
Treatment. The most effective and strongly supported treatment for PFPS is a 6-week physiotherapy program focusing on strengthening the quadriceps and hip muscles and stretching the quadriceps, ITB, hamstrings, and hip flexors.4,5 There is limited information about the use of nonsteroidal anti-inflammatory drugs (NSAIDs), but they can be considered for short-term management.2
Patellar taping and bracing have shown some promise as adjunct therapies for PFPS, although the data for both are non-conclusive. There is a paucity of prospective randomized trials of patellar bracing and a 2012 Cochrane review found limited evidence of its efficacy.34 But a 2014 meta-analysis revealed moderate evidence in support of patellar taping early on to help decrease pain,6 and a recent review suggests that it can be helpful in both the short and long term.7
Taping or bracing may be useful when combined with a tailored physical therapy program. Evidence for treatments such as biofeedback, chiropractic manipulation, and orthotics is limited, and they should be used only as adjunctive therapy.4
CASE › When you examine Ms. T, you find no swelling of the affected knee. You perform the tilt test, which elicits pain. Squatting causes some pain, as well. You diagnose PFPS and provide a referral for 6 weeks of physiotherapy.
Patellar subluxation or chronic dislocation
Patellofemoral instability (PFI) occurs when the patella disengages completely from the trochlear groove.11 PFI’s etiology also relates to the complexity of the patellofemoral joint. Here, too, stability of the joint is achieved with a combination of soft tissue and bony restraints. At full extension and early flexion of the knee, however, the mechanisms of stability are limited, resulting in increased instability. Other associated factors include Q-angle, lateral pull from a tight ITB, and opposing forces from the vastus lateralis and vastus medialis obliquus (VMO).8-10
Risk factors for PFI. The most common predisposing factors for PFI are trochlear dysplasia, patella alta, and lateralization of the tibial tuberosity or patella.10,11 Older patients, predominately women, have an increased risk for PFI.9 Patients usually have a history of patellar subluxation or dislocation in their youth, with approximately 17% of those who had a first dislocation experiencing a recurrence.9 A family history of PFI is common, as well.10
Diagnosis. Patients with PFI often present with nonspecific anterior knee pain secondary to recurrent dislocation.13 Notable physical exam findings are:
- a positive J sign (noted if the patella suddenly shifts medially during early knee flexion or laterally during full extension)
- decreased quadriceps (specifically VMO) and hamstring strength and flexibility
- patellar hypermobility, which should be no more than a quarter to a half of the patellar diameter bilaterally
- pain during a patellar tilt test
- a positive patellar apprehension test.10 (With the patient lying with the knee flexed to 20°, place thumbs on the medial patella and push laterally; the patient will straighten leg with pain or “apprehension” prior to patellar dislocation.)
Plain radiography should be ordered in all cases to assess for osseous trauma/ deformity and to help guide surgical consideration. Magnetic resonance imaging (MRI) can provide additional information when significant soft tissue damage is suspected or the patient does not improve with conservative therapy.8,11
Treatment. A recent Cochrane review showed that conservative treatment (VMO strengthening, bracing, and proprioceptive therapy) prevented future dislocations more effectively than surgical intervention.11 However, surgery is indicated when obvious predisposing anatomic conditions (osteochondral fracture, intra-articular deformity, or a major tear of a medial soft tissue stabilizer) are clearly shown on imaging.8,11
Patellar tendinopathy (jumper’s knee)
Patellar tendinopathy, an overuse injury often called “jumper’s knee” because it is associated with high-intensity jumping sports like volleyball and basketball, is an insertional tendinopathy with pain most commonly at the proximal patellar tendon.10 The pathology of the injury is poorly understood, but is believed to be the result of an impaired healing response to microtears.12,14
Diagnosis. Patients with patellar tendinopathy typically present with anterior suprapatellar pain aggravated by activity. Classically, the pain can occur in any of 4 phases:12 1. pain isolated after activity; 2. pain that occurs during activity but does not impede activity; 3. pain that occurs both during and after the activity and interferes with competition ; 4. a complete tendon disruption.
Examination should include an assessment of the patellar tendon for localized thickening, nodularity, crepitus, and focal suprapatellar tenderness. The muscle-tendon function should be evaluated by assessing knee mobility and strength of the quads via straight leg raise, decline squat, or single leg squats.12 The Victorian Institute of Sport Assessment (VISA) questionnaire can be used to quantify the symptoms and to help track the patient’s progress throughout therapy.31 There are no proven special tests or radiologic studies to aid in the diagnosis of patellar tendinopathy,14 but magnetic resonance imaging (MRI) can be used for further evaluation when findings are equivocal.35
Treatment. A wide range of options, from eccentric training—eg, 3 sets of 15 repetitions performed twice a day for 12 weeks—and physical therapy to platelet-rich plasma (PRP) injections, sclerosing injections, and surgery, are available for the treatment of patellar tendinopathy.13-15 While no specific data have proven the superiority of any one therapy, expert consensus recommends eccentric exercise as initial therapy, performed for 12 weeks.14,15
It’s also interesting to note that a recently published study showed that 3 weekly PRP injections helped 75% of patients—all of whom failed to respond to 4 months of eccentric therapy—return to their pre-symptom activity level within 90 days.16 Corticosteroid injections should not be used to treat patellar tendinopathy due to the risk of tendon rupture.15 Orthopedic referral for surgical intervention should be considered for patients who fail to respond after 3 to 6 months of conservative therapy.14
Lateral knee pain
Iliotibial band tendinopathy
Iliotibial band syndrome (ITBS) is a common source of lateral knee pain, particularly in runners, cyclists, and endurance athletes.17-19,36,37 The exact pathophysiology behind this diagnosis is debatable, but the most accepted etiology is inflammation generated from micro trauma to the soft tissues with inadequate healing time, resulting in persistent inflammation. ITBS is often associated with excessive overall running mileage, a sudden increase in mileage, or an abrupt change in training.18,37
Diagnosis. Patients often complain of persistent nontraumatic lateral knee pain that worsens with repetitive knee flexion (eg, running or cycling).17-19,37 A physical exam will often reveal pain over the lateral femoral condyle and a positive Noble’s test (FIGURE 1). A positive Ober’s test (FIGURE 2) is suggestive of ITBS, as well. The sensitivity and specificity of these tests are not well established, but in patients performing repetitive knee flexion activities with subjective lateral knee pain, pain over the lateral femoral condyle and a positive Ober’s and/or Noble’s test suggest an ITBS diagnosis.18 Imaging is not indicated initially, but MRI should be used in refractory cases to rule out other etiologies.17,19
Treatment. First-line therapy for ITBS is conservative,17-19,36,37 often involving a combination of techniques such as refraining from the activity that triggers the pain, NSAIDs, activity modification to reduce the strain over the ITB, myofascial release via foam rollers, and physical therapy focused on stretching the iliotibial band, tensor fasciae latae, and gluteus medius while strengthening the gluteus medius and core muscles.17 No single program has been shown to be better than another.
Corticosteroid injections are second-line therapy and have been shown to improve pain compared with placebo up to 2 weeks post injection.17,19 When symptoms persist for more than 6 months despite conservative treatment, surgical intervention may be indicated.18,19 Patients who experience temporary pain relief with corticosteroid injections often respond best to surgery.36
Medial knee pain
Medial plica syndrome
Because of its anatomic location, the medial plica—which can be palpated in up to 84% of the population20—is susceptible to impingement by the medial femoral condyle or the patellofemoral joint. Trauma with repetitive knee movement leads to inflammation and thickening of the plica, resulting in medial plica syndrome.20,38 Initial inflammation may be triggered by blunt trauma, a sudden increase in activity, or transient synovitis.22
Diagnosis. Medial plica syndrome is a challenging diagnosis. Patients generally have nonspecific complaints of aching medial knee pain, locking, and catching similar to complaints of a medial meniscal injury.20
Evaluation should include the mediopatellar plica test, which is performed with the patient lying supine with the knee fully extended. Pressure is placed over the inferomedial patellofemoral joint, creating an impingement of the medial plica between the finger and the medial femoral condyle. Elimination or marked diminishing of pain with knee flexion to 90° is considered a positive test.21
A recent systematic review found this test to be more diagnostically accurate than an MRI (sensitivity of the test is 90% and specificity is 89%, vs 77% and 58%, respectively, for MRI) for detection of medial plica syndrome. Ultrasound is almost as accurate, with a sensitivity of 90% and specificity of 83%.39
Treatment of medial plica syndrome centers on physiotherapy and quadriceps strengthening,20 augmented with NSAIDs. Intra-articular corticosteroid injections are considered second-line treatment.20,22 An orthopedics referral is indicated to consider arthroscopic plica removal for refractory cases.20,22
Pes anserine bursitis
The anserine bursal complex, located approximately 5 cm distal to the medial joint line, is formed by the combined insertion of the sartorius, gracilis, and semitendinosus tendons,39 but the exact mechanism of pain is not well understood. Whether the pathophysiology is from an insertional tendonitis or overt bursitis is unknown, and no studies have focused on prevalence or risk factors. What is known is that overweight individuals and women with a wide pelvis seem to have a greater predilection and those with pes planus, diabetes, or knee osteoarthritis are at increased risk.23
Diagnosis. Medial knee pain reproduced on palpation of the anatomical site of insertion of the pes anserine tendon complex supports a diagnosis of pes anserine bursitis, with or without edema. Radiologic studies are not needed, but may be helpful if significant bony pathology is suspected. Ultrasound, computed tomography (CT), and MRI are not recommended.23
Treatment. Resting the affected knee, cryotherapy, NSAIDs, and using a pillow at night to relieve direct bursal pressure are recommended.33 Weight loss in obese patients, treatment of pes planus, and control of diabetes may be helpful, as well. Although the literature is limited and dated, corticosteroid injection has been found to reduce the pain and may be considered as second-line treatment.24-26
Posterior knee pain
Popliteal (Baker’s) cyst
The popliteal fossa contains 6 of the numerous bursa of the knee; the bursa beneath the medial head of the gastrocnemius muscle and the semimembranosus tendon is most commonly involved in the formation of a popliteal cyst.40 It is postulated that increased intra-articular pressure forces fluid into the bursa, leading to expansion and pain. This can be idiopathic or secondary to internal derangement or trauma to the knee.41 Older age, a remote history of knee trauma, or a coexisting joint disease such as osteoarthritis, meniscal pathology, or rheumatoid arthritis are significant risk factors for the development of popliteal cysts.27
Diagnosis. Most popliteal cysts are asymptomatic in adults and discovered incidentally after routine imaging to evaluate other knee pathology. However, symptomatic popliteal cysts present as a palpable mass in the popliteal fossa, resulting in pain and limited range of motion.
During the physical exam with the patient lying supine, a medial popliteal mass that is most prominent with the knee fully extended is common. A positive Foucher’s sign (the painful mass is palpated posteriorly in the popliteal fossa with the knee fully extended; pain is relieved and/or the mass reduced in size with knee flexion to 45°) suggests a diagnosis of popliteal cyst.27,28
Radiologic studies are generally not needed to diagnose a popliteal cyst. However, if diagnostic uncertainty remains after the history and physical exam, plain knee radiographs and ultrasound should be obtained. This combination provides complementary information and helps rule out a fracture, arthritis, and thrombosis as the cause of the pain.27 MRI is helpful if the diagnosis is still in doubt and for patients suspected of having significant internal derangement leading to cyst formation. Arthrography or CT is generally not needed.27,41
Treatment. As popliteal cysts are often associated with other knee pathology, management of the underlying condition often leads to cyst regression. Keeping the knee in flexion can decrease the available space and assist in pain control in the acute phase.27 Cold packs and NSAIDs can also be used initially. Cyst aspiration and intra-articular steroid injection have been shown to be effective for cysts that do not respond to this conservative approach.27 However, addressing and managing the underlying knee pathology (eg, osteoarthritis, meniscal pathology, or rheumatoid arthritis) will prevent popliteal cysts from recurring.
When the problem is painful knee effusion
Nontraumatic knee effusion can be the primary source of knee pain or the result of underlying pathology. We mention it here because clinical suspicion is paramount in diagnosing a septic joint, a serious cause of painful knee effusion that warrants prompt treatment.
As in other causes of knee pain, a detailed history of the character of the pain is essential. Septic arthritis and crystalline disease (gout, pseudogout) should be suspected in patients without a history of trauma who cannot bear weight. Systemic complaints point to an infection and, with the exception of a possible low-grade fever, are not typically seen in crystalline disease. Notable findings include an erythematous, hot, swollen knee and pain with both active and passive movement.
Plain radiographs of the knee should be ordered to rule out significant trauma or arthritis as the etiology. It is important to perform joint aspiration with synovial fluid analysis. Fluid analysis should include a white blood cell (WBC) count with differential, Gram stain and cultures, and polarized light microscopy (not readily available in an outpatient setting).29
Synovial fluid analysis characteristics suggestive of a septic joint include turbid quality, WBC >50,000 per mm3, an elevated protein content, and a low glucose concentration.30 Gram stain and culture will help identify the infectious agent. Orthopedic referral should not be delayed in patients with a suspected infectious joint. Corticosteroids should not be injected during aspiration if infection is being ruled out.
CASE › When Ms. T returns for a follow-up visit 8 weeks later, she states that the knee pain has resolved and that she has returned to running. She has lost an additional 8 pounds and continues to diet. And, at the advice of her physical therapist, she is continuing her physiotherapy regimen at home to prevent a recurrence of PFPS.
CORRESPONDENCE
Carlton J. Covey, MD, FAAFP, Nellis Family Medicine Residency Program, 4700 Las Vegas Boulevard North, Nellis Air Force Base, NV 89191; carlton.covey@us.af.mil
1. Earl JE, Vetter CS. Patellofemoral pain. Phys Med Rehabil Clin N Am. 2007;18:439-458,viii.
2. McGowan HJ, Beutler A. Patellofemoral syndrome. Essential Evidence Plus Web site. Available at: http://www.essentialevidenceplus.com. Accessed: March 20, 2014.
3. Nunes GS, Stapait EL, Kirsten MH, et al. Clinical test for diagnosis of patellofemoral pain syndrome: Systematic review with meta-analysis. Phys Ther Sport. 2013;14:54-59.
4. Rixe JA, Glick JE, Brady J, et al. A review of the management of patellofemoral pain syndrome. Phys Sportsmed. 2013;41: 19-28.
5. Bolgla LA, Boling MC. An update for the conservative management of patellofemoral pain syndrome: a systematic review of the literature from 2000 to 2010. Int J Sports Phys Ther. 2011;6:112-125.
6. Barton C, Balachandar V, Lack S, et al. Patellar taping for patellofemoral pain: a systematic review and meta-analysis to evaluate clinical outcomes and biomechanical mechanisms. Br J Sports Med. 2014;48:417-424.
7. Dutton RA, Khadavi MJ, Fredericson M. Update on rehabilitation of patellofemoral pain. Curr Sports Med Rep. 2014;13: 172-178.
8. Kapur S, Wissman RD, Robertson M, et al. Acute knee dislocation: review of an elusive entity. Curr Probl Diagn Radiol. 2009;38:237-250.
9. Colvin AC, West RV. Patellar instability. J Bone Joint Surg Am. 2008;90:2751-2762.
10. Tscholl PM, Koch PP, Fucentese SF. Treatment options for patellofemoral instability in sports traumatology. Orthop Rev (Pavia). 2013;5:e23.
11. Earhart C, Patel DB, White EA, et al. Transient lateral patellar dislocation: review of imaging findings, patellofemoral anatomy, and treatment options. Emerg Radiol. 2013;20:11-23.
12. Tan SC, Chan O. Achilles and patellar tendinopathy: current understanding of pathophysiology and management. Disabil Rehabil. 2008;30:1608-1615.
13. Gaida JE, Cook J. Treatment options for patellar tendinopathy: critical review. Curr Sports Med Rep. 2011;10:255-270.
14. Rodriguez-Merchan EC. The treatment of patellar tendinopathy. J Orthop Traumatol. 2013;14:77-81.
15. Childress MA, Beutler A. Management of chronic tendon injuries. Am Fam Physician. 2013;87:486-490.
16. Charousset C, Zaoui A, Bellaiche L, et al. Are multiple platelet-rich plasma injections useful for treatment of chronic patellar tendinopathy in athletes? A prospective study. Am J Sports Med. 2014;42:906-911.
17. Strauss EJ, Kim S, Calcei JG, et al. Iliotibial band syndrome: evaluation and management. J Am Acad Orthop Surg. 2011;19:728-736.
18. Bellary SS, Lynch G, Housman B, et al. Medial plica syndrome: a review of the literature. Clin Anat. 2012;25:423-428.
19. Hong JH, Kim JS. Diagnosis of iliotibial band friction syndrome and ultrasound guided steroid injection. Korean J Pain. 2013;26:387-391.
20. Bellary SS, Lynch G, Housman B, et al. Medial plica syndrome: a review of the literature. Clin Anat. 2012;25:423-428.
21. Kim SJ, Jeong JH, Cheon YM, et al. MPP test in the diagnosis of medial patellar plica syndrome. Arthroscopy. 2004;20: 1101-1103.
22. Schindler OS. ‘The Sneaky Plica’ revisited: morphology, pathophysiology and treatment of synovial plicae of the knee. Knee Surg Sports Traumatol Arthrosc. 2014;22:247-262.
23. Helfenstein M Jr, Kuromoto J. Anserine syndrome. Rev Bras Rheumatol. 2010;50:313-327.
24. Abeles M. Osteoarthritis of the knee: anserine bursitis as an extra-articular cause of pain. Clin Res. 1983;31:4471-4476.
25. Kang I, Han SW. Anserine bursitis in patients with osteoarthritis of the knee. South Med J. 2000;93:207-209.
26. Yoon HS, Kim SE, Suh YR, et al. Correlation between ultrasonographic findings and the response to corticosteroid injection in pes anserinus tendinobursitis syndrome in knee osteoarthritis patients. J Korean Med Sci. 2005;20:109-112.
27. Stein D, Cantlon M, MacKay B, et al. Cysts about the knee: evaluation and management. J Am Acad Orthop Surg. 2013;21: 469-479.
28. Canoso JJ, Goldsmith MR, Gerzof SG, et al. Foucher’s sign of the Baker’s cyst. Ann Rheum Dis. 1987;46:228-232.
29. Palmer T. Knee pain. Essential Evidence Plus Web site. Available at: http://www.essentialevidenceplus.com. Accessed: December 12, 2013.
30. Franks AG Jr. Rheumatologic aspects of knee disorders. In: Scott WN, ed. The Knee. St. Louis: Mosby; 1994:315-329.
31. Visentini PJ, Khan KM, Cook JL, et al. The VISA score: an index of severity of symptoms in patients with jumper’s knee (patellar tendinosis). Victorian Institute of Sport Tendon Study Group. J Sci Med Sport. 1998;1:22-28.
32. Halabchi F, Mazaheri R, Seif-Barghi T. Patellofemoral pain syndrome and modifiable intrinsic risk factors; how to assess and address? Asian J Sports Med. 2013;4:85-100.
33. Dixit S, DiFiori JP, Burton M, et al. Management of patellofemoral pain syndrome. Am Fam Physician. 2007;75:194-202.
34. Callaghan MJ, Selfe J. Patellar taping for patellofemoral pain syndrome in adults. Cochrane Database Syst Rev. 2012;4:CD006717.
35. Atanda AJ Jr, Ruiz D, Dodson CC, et al. Approach to the active patient with chronic anterior knee pain. Phys Sportsmed. 2012;40:41-50.
36. Ellis R, Hing W, Reid D. Iliotibial band friction syndrome—a systematic review. Man Ther. 2007;12:200-208.
37. Kirk KL, Kuklo T, Klemme W. Iliotibial band friction syndrome. Orthopedics. 2000;23:1209-1217.
38. Stubbings N, Smith T. Diagnostic test accuracy of clinical and radiological assessments for medial patella plica syndrome: a systematic review and meta-analysis. Knee. 2014;21: 486-490.
39. Alvarez-Nemegyei J, Canoso JJ. Evidence-based soft tissue rheumatology IV: anserine bursitis. J Clin Rheumatol. 2004;10:205-206.
40. Fritschy D, Fasel J, Imbert JC, et al. The popliteal cyst. Knee Surg Sports Traumatol Arthrosc. 2006;14:623-628.
41. Handy JR. Popliteal cysts in adults: a review. Semin Arthritis Rheum. 2001;31:108-118.
1. Earl JE, Vetter CS. Patellofemoral pain. Phys Med Rehabil Clin N Am. 2007;18:439-458,viii.
2. McGowan HJ, Beutler A. Patellofemoral syndrome. Essential Evidence Plus Web site. Available at: http://www.essentialevidenceplus.com. Accessed: March 20, 2014.
3. Nunes GS, Stapait EL, Kirsten MH, et al. Clinical test for diagnosis of patellofemoral pain syndrome: Systematic review with meta-analysis. Phys Ther Sport. 2013;14:54-59.
4. Rixe JA, Glick JE, Brady J, et al. A review of the management of patellofemoral pain syndrome. Phys Sportsmed. 2013;41: 19-28.
5. Bolgla LA, Boling MC. An update for the conservative management of patellofemoral pain syndrome: a systematic review of the literature from 2000 to 2010. Int J Sports Phys Ther. 2011;6:112-125.
6. Barton C, Balachandar V, Lack S, et al. Patellar taping for patellofemoral pain: a systematic review and meta-analysis to evaluate clinical outcomes and biomechanical mechanisms. Br J Sports Med. 2014;48:417-424.
7. Dutton RA, Khadavi MJ, Fredericson M. Update on rehabilitation of patellofemoral pain. Curr Sports Med Rep. 2014;13: 172-178.
8. Kapur S, Wissman RD, Robertson M, et al. Acute knee dislocation: review of an elusive entity. Curr Probl Diagn Radiol. 2009;38:237-250.
9. Colvin AC, West RV. Patellar instability. J Bone Joint Surg Am. 2008;90:2751-2762.
10. Tscholl PM, Koch PP, Fucentese SF. Treatment options for patellofemoral instability in sports traumatology. Orthop Rev (Pavia). 2013;5:e23.
11. Earhart C, Patel DB, White EA, et al. Transient lateral patellar dislocation: review of imaging findings, patellofemoral anatomy, and treatment options. Emerg Radiol. 2013;20:11-23.
12. Tan SC, Chan O. Achilles and patellar tendinopathy: current understanding of pathophysiology and management. Disabil Rehabil. 2008;30:1608-1615.
13. Gaida JE, Cook J. Treatment options for patellar tendinopathy: critical review. Curr Sports Med Rep. 2011;10:255-270.
14. Rodriguez-Merchan EC. The treatment of patellar tendinopathy. J Orthop Traumatol. 2013;14:77-81.
15. Childress MA, Beutler A. Management of chronic tendon injuries. Am Fam Physician. 2013;87:486-490.
16. Charousset C, Zaoui A, Bellaiche L, et al. Are multiple platelet-rich plasma injections useful for treatment of chronic patellar tendinopathy in athletes? A prospective study. Am J Sports Med. 2014;42:906-911.
17. Strauss EJ, Kim S, Calcei JG, et al. Iliotibial band syndrome: evaluation and management. J Am Acad Orthop Surg. 2011;19:728-736.
18. Bellary SS, Lynch G, Housman B, et al. Medial plica syndrome: a review of the literature. Clin Anat. 2012;25:423-428.
19. Hong JH, Kim JS. Diagnosis of iliotibial band friction syndrome and ultrasound guided steroid injection. Korean J Pain. 2013;26:387-391.
20. Bellary SS, Lynch G, Housman B, et al. Medial plica syndrome: a review of the literature. Clin Anat. 2012;25:423-428.
21. Kim SJ, Jeong JH, Cheon YM, et al. MPP test in the diagnosis of medial patellar plica syndrome. Arthroscopy. 2004;20: 1101-1103.
22. Schindler OS. ‘The Sneaky Plica’ revisited: morphology, pathophysiology and treatment of synovial plicae of the knee. Knee Surg Sports Traumatol Arthrosc. 2014;22:247-262.
23. Helfenstein M Jr, Kuromoto J. Anserine syndrome. Rev Bras Rheumatol. 2010;50:313-327.
24. Abeles M. Osteoarthritis of the knee: anserine bursitis as an extra-articular cause of pain. Clin Res. 1983;31:4471-4476.
25. Kang I, Han SW. Anserine bursitis in patients with osteoarthritis of the knee. South Med J. 2000;93:207-209.
26. Yoon HS, Kim SE, Suh YR, et al. Correlation between ultrasonographic findings and the response to corticosteroid injection in pes anserinus tendinobursitis syndrome in knee osteoarthritis patients. J Korean Med Sci. 2005;20:109-112.
27. Stein D, Cantlon M, MacKay B, et al. Cysts about the knee: evaluation and management. J Am Acad Orthop Surg. 2013;21: 469-479.
28. Canoso JJ, Goldsmith MR, Gerzof SG, et al. Foucher’s sign of the Baker’s cyst. Ann Rheum Dis. 1987;46:228-232.
29. Palmer T. Knee pain. Essential Evidence Plus Web site. Available at: http://www.essentialevidenceplus.com. Accessed: December 12, 2013.
30. Franks AG Jr. Rheumatologic aspects of knee disorders. In: Scott WN, ed. The Knee. St. Louis: Mosby; 1994:315-329.
31. Visentini PJ, Khan KM, Cook JL, et al. The VISA score: an index of severity of symptoms in patients with jumper’s knee (patellar tendinosis). Victorian Institute of Sport Tendon Study Group. J Sci Med Sport. 1998;1:22-28.
32. Halabchi F, Mazaheri R, Seif-Barghi T. Patellofemoral pain syndrome and modifiable intrinsic risk factors; how to assess and address? Asian J Sports Med. 2013;4:85-100.
33. Dixit S, DiFiori JP, Burton M, et al. Management of patellofemoral pain syndrome. Am Fam Physician. 2007;75:194-202.
34. Callaghan MJ, Selfe J. Patellar taping for patellofemoral pain syndrome in adults. Cochrane Database Syst Rev. 2012;4:CD006717.
35. Atanda AJ Jr, Ruiz D, Dodson CC, et al. Approach to the active patient with chronic anterior knee pain. Phys Sportsmed. 2012;40:41-50.
36. Ellis R, Hing W, Reid D. Iliotibial band friction syndrome—a systematic review. Man Ther. 2007;12:200-208.
37. Kirk KL, Kuklo T, Klemme W. Iliotibial band friction syndrome. Orthopedics. 2000;23:1209-1217.
38. Stubbings N, Smith T. Diagnostic test accuracy of clinical and radiological assessments for medial patella plica syndrome: a systematic review and meta-analysis. Knee. 2014;21: 486-490.
39. Alvarez-Nemegyei J, Canoso JJ. Evidence-based soft tissue rheumatology IV: anserine bursitis. J Clin Rheumatol. 2004;10:205-206.
40. Fritschy D, Fasel J, Imbert JC, et al. The popliteal cyst. Knee Surg Sports Traumatol Arthrosc. 2006;14:623-628.
41. Handy JR. Popliteal cysts in adults: a review. Semin Arthritis Rheum. 2001;31:108-118.
When vaccine misconceptions jeopardize public health
› Reassure parents that vaccines are some of the safest and most effective interventions we have to prevent infectious disease. A
› Advise parents that there are multiple systems in place to monitor vaccine safety. C
› Educate parents that lapses in immunization rates can put children at risk of resurgent cases of previously well-controlled diseases, like measles and Haemophilus influenza type b. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
When a public health intervention succeeds and achieves long-term suppression of the target problem, an unfortunate irony is that, with time, the intervention can seem less vital. So it is with vaccines. Many patients and physicians today have never experienced the infectious diseases that once caused millions of deaths and much disability each year, and they therefore do not appreciate the impact these diseases had when they were prevalent.
It is estimated that just 9 of the routinely recommended vaccines prevent 42,000 deaths and 20 million cases of disease in every birth cohort.1 With many of these diseases thus held at bay, attention shifted instead to the supposed risks of vaccines. Many people mistakenly believe a vaccine’s potential for harm is more likely than the chance of acquiring the disease it prevents, and they therefore refuse vaccines for themselves and their children, with little chance in the short term of suffering an adverse outcome for their decision.
In this review—which can inform primary care physicians’ discussions with vaccine-hesitant patients—we first highlight 2 preventable diseases, measles and Haemophilus influenzae type b (Hib) infection. Recent residency graduates may never see these diseases thanks to sustained vaccination programs. However, the risk of acquiring these infections has not disappeared entirely. After considering these examples, we examine the totality of the morbidity and mortality prevented by vaccination and describe the safety of current vaccines and the systems in place to assure their continued safety.
Measles: No longer endemic to the United States, but still a risk from importation
In the pre-vaccine era, measles (rubeola) infected more than 500,000 Americans annually and killed roughly 500.2 This highly communicable systemic acute viral infection was once considered universal in childhood. After vaccine licensure in 1963, widespread immunization reduced the incidence by more than 98%, and by 2000 it had eliminated endemic measles from the United States. However, the disease has now reappeared—largely due to international travel and neglect in becoming vaccinated. As of October 31, 2014, the United States had 20 outbreaks and 603 cases of measles reported in 2014—a dramatic increase over recent years.3
Clinical appearance. Acute measles infection is characterized by high fever, cough, coryza, conjunctivitis, and rash. Koplik spots are a 24- to 72-hour pathognomonic exanthem of blue-white spots 1 to 3 mm in diameter on an erythematous base along the buccal mucosa. The resolving exanthem coincides with the eruption of a blanching, maculopapular exanthem originating at the hairline, progressing down the trunk and out to the limbs (sparing the palms and soles), coalescing, and then fading with a fine desquamation in the same order of appearance over 7 days. Additional associated symptoms include anorexia, diarrhea, and generalized lymphadenopathy.2,4
Complications are common with measles. Acute measles infection is rarely fatal. However, serious complications occur in nearly one-third of reported cases.2 During the 1989-1991 measles resurgence in the United States, more than 100 deaths occurred among the 55,000 cases reported.5-7 In early 2011, the United States saw the highest reported number of measles cases since 1996 due to importation. Of the 118 reported cases, 105 (89%) occurred in unvaccinated people, 47 (40%) required hospitalization, and 9 individuals developed pneumonia.8
Complications of measles infection are shown in TABLE 1.2,4 Pneumonia (viral or superimposed bacterial) accounts for 60% of measles-related deaths.2 Neurologic complications, while less frequent, can be severe.
Acute encephalitis occurs in 1 in 1000 to 2000 cases and presents within a week following the exanthem with fever, headache, vomiting, meningismus, change in mental status, convulsions, and coma.2 Encephalitis has a fatality rate of 15%, leaving another 25% with residual neurologic damage.2 Subacute sclerosing panencephalitis (SSPE) occurs in 5 to 10 cases per million (in the United States), on average 7 years after the initial measles infection.9,10 After an insidious onset, behavior and intellect deteriorate, followed by ataxia, myoclonic seizures, and ultimately death. In the United States, the number of reported cases of SSPE has declined with the reduction in measles cases. However, in countries with less robust measles immunization eradication programs, the risk of developing SSPE remains.9,10
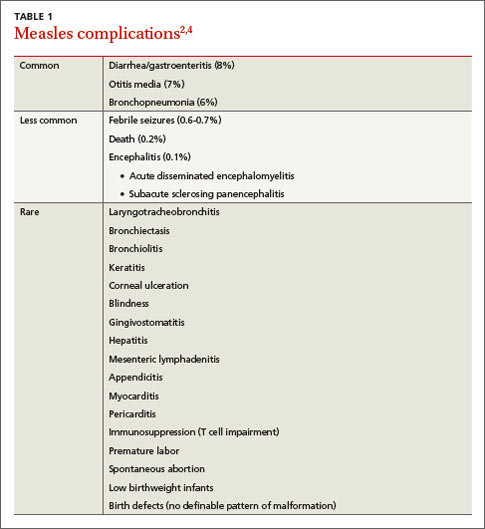
Hib: Contained but not eradicated
Hib was once the leading cause of meningitis and a major cause of other invasive bacterial diseases, but it has been greatly controlled since the advent of routine Hib vaccination in 1990.11 Hib is an encapsulated, gram-negative coccobacillus. There are 6 major capsular serotypes of Haemophilus influenzae, but serotype b was linked to major invasive disease in humans 95% of the time.12 The spectrum of diseases caused by Hib is seen in TABLE 2.13-15 Hib is transmitted by respiratory droplets from noninfected as well as infected carriers. Asymptomatic nasal carriage in the pre-vaccine era varied from 0.5% to 5%.12
Hib is primarily a disease of young children, with almost all cases occurring in children younger than 5 years of age (66% in those younger than 18 months). Other risk factors for invasive disease are those that increase the spread of respiratory droplets: crowding, lower socioeconomic status, day care attendance, large household size, and school-aged siblings. American Indian and Alaskan Native populations remain at higher risk due to incomplete vaccination rates and the sociodemographic risk factors noted above. Breastfeeding is protective.12
Three percent to 6% of cases of invasive Hib disease are fatal; another 20% can have long-term sequelae such as hearing loss. In the early 1990s, the peak incidence of Hib disease reached 41 cases per 100,000 population.12 The reduction in incidence of Hib disease brought about by universal vaccination has been attributed to individual immunity, decreased asymptomatic nasal carriage, and herd immunity.12
Despite this progress, Hib continues to evade eradication. In Minnesota in 2008, 5 children, ages 5 months to 3 years, contracted invasive Hib disease (3 with meningitis, 1 with pneumonia, 1 with epiglottitis).16 Of the 5, only one was up to date with Hib vaccination; the others had not received vaccine because of shortages or parent refusal. These children were unrelated and had not been in contact with each other.
In a daycare outbreak in the United Kingdom, 2 cases of Hib disease (meningitis and septic arthritis) were identified in fully immunized children younger than 18 months, presumably due to a lack of complete vaccine efficacy.17 A study of nasal carriage (performed just prior to rifampin prophylaxis) among other attendees and caregivers revealed 3 asymptomatic carriers.17 Although Hib is largely well-contained in developed countries due to vaccination policies, the burden of disease in developing countries is estimated to be approximately 8.1 million serious illnesses with 371,000 deaths annually.13

The totality of morbidity and mortality prevented by vaccines
Measles and Hib are 2 examples of vaccine-preventable diseases and the reduction in morbidity and mortality achievable with vaccines. TABLE 318 summarizes the number of pre-vaccine era cases for selected diseases. Routine vaccination against 7 common childhood diseases not only prevents many thousands of deaths, as mentioned earlier,1 but it saves $13.5 billion in direct costs in each birth cohort and saves society $68.8 billion in costs that include disability and lost productivity of both patients and caregivers.1
Put simply, every dollar spent on the vaccination program saves $10 in direct and indirect costs to society.1 Sustaining these successes and averting the resurgence of contained diseases requires a commitment to high immunization rates without delays and lapses—an effort made more challenging in light of misinformation about vaccine safety and resultant parental vaccine hesitancy.
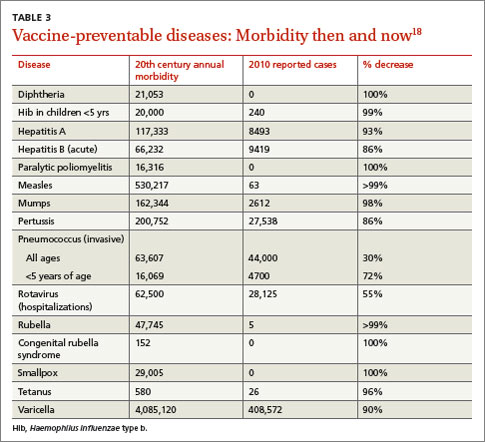
Vaccine safety is ensured by rigorous systems
Despite an impressive record of safety, vaccines still cause anxiety among patients and parents in family practices. A recent survey identified concerns of long-term complications, autism, and thimerosal effects to be foremost on the minds of parents, whereas short-term effects were of much less concern.19 Causation of autism related to vaccines has been dismissed; the initial linkages have been shown to be fraudulent.20 With the exception of some influenza vaccine preparations, thimerosal is no longer present in routinely administered children’s vaccines and has been shown not to be associated with autism.21,22 To address parents’ and patients’ concerns about vaccine safety, and especially those surrounding short- and long-term complications, physicians should have a general understanding of the pre- and post-licensure mechanisms in the United States.
Pre-licensure safety is under the purview of vaccine manufacturers and the Center for Biologics Evaluation and Research at the US Food and Drug Administration (http://www.fda.gov/biologicsbloodvaccines/vaccines/default.htm). For licensure, manufacturers must provide clinical data to demonstrate sufficient safety and efficacy. Accordingly, pre-licensure assessments are conducted in a “closed system” under a research protocol. The vaccine recipients are volitional research subjects selected according to inclusion and exclusion criteria. They are also compensated. However, sample sizes are rarely large enough to exclude rare serious adverse events.
Once licensure has been granted, the focus of safety then shifts to the “open system” of usual clinical practice. Vaccine recipients are unselected members of the general population and may have underlying medical conditions, and sometimes—such as with school entry mandates—are less volitional. In this sphere, the responsible parties for safety include the government, manufacturers, and health care systems.
Three ongoing systems function to assure vaccine safety: the Vaccine Adverse Event Reporting System (VAERS), the Vaccine Safety Datalink (VSD), and the Clinical Immunization Safety Assessment (CISA) Network.23,24
VAERS serves as an early warning system for coincidental safety signals and can generate hypotheses for further investigation.24,25 It is characterized by high sensitivity but low specificity as it relies on voluntary reporting from health care personnel, parents, and others. This system was instrumental in identifying the initial cases of intussusception attributable to the rotavirus vaccine, RotaShield.26
The VSD is a network of 10 large, geographically diverse and linked health maintenance organizations that cover about 3% of the US population. Within this “real time” network, vaccination (exposure) can be compared with outpatient, emergency department, hospital, and laboratory data (health outcomes), while accounting for demographic variables (confounders).27,28 VSD studies linked the measles, mumps, rubella, and varicella vaccine to febrile seizures29 and showed no relationship between cumulative vaccine antigen exposure and autism.30
The CISA was established in 2001 to investigate the pathophysiologic mechanisms and biologic risks of adverse effects following immunization and to provide evidence-based vaccine safety assessments.
Based on all available evidence, routinely recommended vaccines have attained a very high level of safety. As with other preventive services, immunizations are generally provided to healthy individuals to maintain good health; thus, a low tolerance for significant adverse events exists. Well over 100 million doses of vaccines are given each year, yet the VAERS receives, on average, only 28,000 adverse event reports per year.
These reports comprise mild, moderate, and severe reactions to vaccines, but also adverse events that may not be related in any way other than chronologically to the vaccine’s administration. Despite this relatively low number of real safety concerns, it is still more likely that patients will know someone who has had a vaccine-related adverse event than someone who has had some of the diseases the vaccines prevent.31
Final thoughts
Current anti-vaccine sentiments appear to arise from varying perspectives. Some are held by parents of children who have allegedly suffered a severe vaccine related adverse event; others by those opposed to government-mandated school immunization requirements; and some from those who have a dislike of vaccine manufacturers. These sentiments persist in part because of a low level of vaccine preventable diseases: When such illnesses are no longer deemed a threat, those who have concerns about vaccine safety, no matter how invalid, believe their concerns should trump all other considerations.
To appreciate the true benefit of vaccine acceptance, we need only look to Europe, where the anti-vaccine movement has led to high levels of vaccine refusal and a resurgence of vaccine-preventable diseases, such as measles, with their associated morbidity and mortality.32 In advocating for continued acceptance and widespread use of vaccines, family physicians can convey to patients and parents the magnitude of associated health benefits while confidently attesting to the effectiveness and safety of vaccines.
CORRESPONDENCE
John Epling, MD, MSEd, Department of Family Medicine, SUNY Upstate Medical University, 475 Irving Ave, Suite 200, Syracuse, NY 13210; eplingj@upstate.edu
1. Zhou F, Shefer A, Wenger J, et al. Economic evaluation of the routine childhood immunization program in the United States, 2009. Pediatrics. 2014;133:577-585.
2. Centers for Disease Control and Prevention. Measles. Epidemiology and prevention of vaccine-preventable diseases. The Pink Book: Course Textbook. 12th ed. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/pubs/pinkbook/meas.html. Accessed March 4, 2012.
3. Centers for Disease Control and Prevention. Measles (Rubeola). Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/measles/. Accessed November 19, 2014.
4. Measles. In: Pickering LK, Baker CJ, Kimberlin DW, et al, eds. Red Book: 2009 Report of the Committee on Infectious Diseases. 28th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2009:444-455.
5. Atkinson WL, Orenstein WA, Krugman S. The resurgence of measles in the United States, 1989-1990. Annu Rev Med. 1992;43:451-463.
6. Centers for Disease Control and Prevention (CDC). Measles--United States, 1990. MMWR Morb Mortal Wkly Rep. 1991;40:369-372.
7. Gindler J, Tinker S, Markowitz L, et al. Acute measles mortality in the United States, 1987-2002. J Infect Dis. 2004;189(suppl 1): S69-S77.
8. Centers for Disease Control and Prevention (CDC). Measles: United States, January-May 20, 2011. MMWR Morb Mortal Wkly Rep. 2011;60:666-668.
9. Bernstein DI, Reuman PD, Schiff GM. Rubeola (measles) and subacute sclerosing panencephalitis virus. In: Gorbach SL, Bartlett JG, Blacklow NR (eds). Infectious Diseases. Philadelphia, PA: WB Saunders; 1998:2135.
10. Bellini WJ, Rota JS, Lowe LE, et al. Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than was previously recognized. J Infect Dis. 2005;192:1686-1693.
11. Centers for Disease Control and Prevention (CDC). Progress toward elimination of Haemophilus influenza type b invasive disease among infants and children—United States, 1998-2000. MMWR Morb Mortal Wkly Rep. 2002;51:234-237.
12. Centers for Disease Control and Prevention (CDC). Epidemiology and Prevention of Vaccine-Preventable Diseases. The Pink Book, Course Textbook, 12th ed. Haemophilus influenzae type b. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/pubs/pinkbook/hib.html. Accessed March 4, 2012.
13. Watt JP, Wolfson LJ, O’Brien KL, et al; Hib and Pneumococcal Global Burden of Disease Study Team. Burden of disease caused by Haemophilus influenzae type b in children younger than 5 years: global estimates. Lancet. 2009;374:903-911.
14. Agrawal A, Murphy TF. Haemophilus influenzae infections in the H. influenzae type b conjugate vaccine era. J Clin Microbiol. 2011;49:3728-3732.
15. Chandran A, Watt JP, Santosham M. Prevention of Haemophilus influenza type b disease: past successes and future challenges. Informa Healthcare. 2005;4:819-827.
16. Centers for Disease Control and Prevention (CDC). Invasive Haemophilus influenza Type B disease in five young children--Minnesota, 2008. MMWR Morb Mortal Wkly Rep. 2009;58:58-60.
17. McVernon J, Morgan P, Mallaghan C, et al. Outbreak of Haemophilus influenzae type b disease among fully vaccinated children in a day-care center. Pediatr Infect Dis J. 2004;23:38-41.
18. Hinman AR, Orenstein WA, Schuchat A; Centers for Disease Control and Prevention (CDC). Vaccine-preventable diseases, immunizations, and MMWR—1961-2011. MMWR Morb Mortal Wkly Rep. 2011;60 suppl 4:49-57.
19. Kempe A, Daley MF, McCauley MM, et al. Prevalence of parental concerns about childhood vaccines: the experience of primary care physicians. Am J Prev Med. 2011;40:548-555.
20. Godlee F, Smith J, Marcovitch H. Wakefield’s article linking MMR vaccine and autism was fraudulent. BMJ. 2011;342:c7452.
21. Institute for Vaccine Safety. Thimerosal content in some US Licensed vaccines. Institute for Vaccine Safety Web site. Available at: http://www.vaccinesafety.edu/thi-table.htm. Accessed March 8, 2012.
22. Centers for Disease Control and Prevention. Centers for Disease Control and Prevention: Immunization safety and autism. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccinesafety/00_pdf/CDCStudiesonVaccinesandAutism.pdf. Accessed September 23, 2013.
23. Centers for Disease Control and Prevention. Ensuring the safety of vaccines in the United States. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/hcp/patient-ed/conversations/downloads/vacsafe-ensuring-color-office.pdf. Accessed November 18, 2014.
24. Wharton M. Vaccine safety: current systems and recent findings. Curr Opin Pediatr. 2010;22:88-93.
25. US Food and Drug Administration. Understanding the Vaccine Adverse Event Reporting System (VAERS). US Food and Drug Administration Web site. Available at: http://www.fda.gov/downloads/BiologicsBloodVaccines/SafetyAvailability/VaccineSafety/UCM298183.pdf. Accessed October 15, 2014.
26. Centers for Disease Control and Prevention (CDC). Suspension of rotavirus vaccine after reports of intussusception--United States, 1999. MMWR Morb Mortal Wkly Rep. 2004;53:786-789.
27. Greene SK, Kulldorff M, Lewis EM, et al. Near real-time surveillance for influenza vaccine safety: proof-of-concept in the Vaccine Safety Datalink Project. Am J Epidemiol. 2010;171:177-188.
28. Iskander J, Broder K. Monitoring the safety of annual and pandemic influenza vaccines: lessons from the US experience. Expert Rev Vaccines. 2008;7:75-82.
29. Klein NP, Fireman B, Yih WK, et al. Measles-mumps-rubella-varicella combination vaccine and the risk of febrile seizures. Pediatrics. 2010;126:e1-e8.
30. DeStefano F, Price CS, Weintraub ES. Increasing exposure to antibody-stimulating proteins and polysaccharides in vaccines is not associated with risk of autism. J Pediatr. 2013;163:561-567.
31. Centers for Disease Control and Prevention (CDC). Epidemiology and prevention of vaccine-preventable diseases. The Pink Book, Course Textbook, 12th ed. Available at: http://www.cdc.gov/vaccines/pubs/pinkbook/safety.html. Accessed November 17, 2014.
32. World Health Organization (WHO). Measles outbreak in Europe. Global alert and response. Available at: http://www.who.int/csr/don/2011_04_21/en/index.html. Accessed March 8, 2012.
› Reassure parents that vaccines are some of the safest and most effective interventions we have to prevent infectious disease. A
› Advise parents that there are multiple systems in place to monitor vaccine safety. C
› Educate parents that lapses in immunization rates can put children at risk of resurgent cases of previously well-controlled diseases, like measles and Haemophilus influenza type b. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
When a public health intervention succeeds and achieves long-term suppression of the target problem, an unfortunate irony is that, with time, the intervention can seem less vital. So it is with vaccines. Many patients and physicians today have never experienced the infectious diseases that once caused millions of deaths and much disability each year, and they therefore do not appreciate the impact these diseases had when they were prevalent.
It is estimated that just 9 of the routinely recommended vaccines prevent 42,000 deaths and 20 million cases of disease in every birth cohort.1 With many of these diseases thus held at bay, attention shifted instead to the supposed risks of vaccines. Many people mistakenly believe a vaccine’s potential for harm is more likely than the chance of acquiring the disease it prevents, and they therefore refuse vaccines for themselves and their children, with little chance in the short term of suffering an adverse outcome for their decision.
In this review—which can inform primary care physicians’ discussions with vaccine-hesitant patients—we first highlight 2 preventable diseases, measles and Haemophilus influenzae type b (Hib) infection. Recent residency graduates may never see these diseases thanks to sustained vaccination programs. However, the risk of acquiring these infections has not disappeared entirely. After considering these examples, we examine the totality of the morbidity and mortality prevented by vaccination and describe the safety of current vaccines and the systems in place to assure their continued safety.
Measles: No longer endemic to the United States, but still a risk from importation
In the pre-vaccine era, measles (rubeola) infected more than 500,000 Americans annually and killed roughly 500.2 This highly communicable systemic acute viral infection was once considered universal in childhood. After vaccine licensure in 1963, widespread immunization reduced the incidence by more than 98%, and by 2000 it had eliminated endemic measles from the United States. However, the disease has now reappeared—largely due to international travel and neglect in becoming vaccinated. As of October 31, 2014, the United States had 20 outbreaks and 603 cases of measles reported in 2014—a dramatic increase over recent years.3
Clinical appearance. Acute measles infection is characterized by high fever, cough, coryza, conjunctivitis, and rash. Koplik spots are a 24- to 72-hour pathognomonic exanthem of blue-white spots 1 to 3 mm in diameter on an erythematous base along the buccal mucosa. The resolving exanthem coincides with the eruption of a blanching, maculopapular exanthem originating at the hairline, progressing down the trunk and out to the limbs (sparing the palms and soles), coalescing, and then fading with a fine desquamation in the same order of appearance over 7 days. Additional associated symptoms include anorexia, diarrhea, and generalized lymphadenopathy.2,4
Complications are common with measles. Acute measles infection is rarely fatal. However, serious complications occur in nearly one-third of reported cases.2 During the 1989-1991 measles resurgence in the United States, more than 100 deaths occurred among the 55,000 cases reported.5-7 In early 2011, the United States saw the highest reported number of measles cases since 1996 due to importation. Of the 118 reported cases, 105 (89%) occurred in unvaccinated people, 47 (40%) required hospitalization, and 9 individuals developed pneumonia.8
Complications of measles infection are shown in TABLE 1.2,4 Pneumonia (viral or superimposed bacterial) accounts for 60% of measles-related deaths.2 Neurologic complications, while less frequent, can be severe.
Acute encephalitis occurs in 1 in 1000 to 2000 cases and presents within a week following the exanthem with fever, headache, vomiting, meningismus, change in mental status, convulsions, and coma.2 Encephalitis has a fatality rate of 15%, leaving another 25% with residual neurologic damage.2 Subacute sclerosing panencephalitis (SSPE) occurs in 5 to 10 cases per million (in the United States), on average 7 years after the initial measles infection.9,10 After an insidious onset, behavior and intellect deteriorate, followed by ataxia, myoclonic seizures, and ultimately death. In the United States, the number of reported cases of SSPE has declined with the reduction in measles cases. However, in countries with less robust measles immunization eradication programs, the risk of developing SSPE remains.9,10
Hib: Contained but not eradicated
Hib was once the leading cause of meningitis and a major cause of other invasive bacterial diseases, but it has been greatly controlled since the advent of routine Hib vaccination in 1990.11 Hib is an encapsulated, gram-negative coccobacillus. There are 6 major capsular serotypes of Haemophilus influenzae, but serotype b was linked to major invasive disease in humans 95% of the time.12 The spectrum of diseases caused by Hib is seen in TABLE 2.13-15 Hib is transmitted by respiratory droplets from noninfected as well as infected carriers. Asymptomatic nasal carriage in the pre-vaccine era varied from 0.5% to 5%.12
Hib is primarily a disease of young children, with almost all cases occurring in children younger than 5 years of age (66% in those younger than 18 months). Other risk factors for invasive disease are those that increase the spread of respiratory droplets: crowding, lower socioeconomic status, day care attendance, large household size, and school-aged siblings. American Indian and Alaskan Native populations remain at higher risk due to incomplete vaccination rates and the sociodemographic risk factors noted above. Breastfeeding is protective.12
Three percent to 6% of cases of invasive Hib disease are fatal; another 20% can have long-term sequelae such as hearing loss. In the early 1990s, the peak incidence of Hib disease reached 41 cases per 100,000 population.12 The reduction in incidence of Hib disease brought about by universal vaccination has been attributed to individual immunity, decreased asymptomatic nasal carriage, and herd immunity.12
Despite this progress, Hib continues to evade eradication. In Minnesota in 2008, 5 children, ages 5 months to 3 years, contracted invasive Hib disease (3 with meningitis, 1 with pneumonia, 1 with epiglottitis).16 Of the 5, only one was up to date with Hib vaccination; the others had not received vaccine because of shortages or parent refusal. These children were unrelated and had not been in contact with each other.
In a daycare outbreak in the United Kingdom, 2 cases of Hib disease (meningitis and septic arthritis) were identified in fully immunized children younger than 18 months, presumably due to a lack of complete vaccine efficacy.17 A study of nasal carriage (performed just prior to rifampin prophylaxis) among other attendees and caregivers revealed 3 asymptomatic carriers.17 Although Hib is largely well-contained in developed countries due to vaccination policies, the burden of disease in developing countries is estimated to be approximately 8.1 million serious illnesses with 371,000 deaths annually.13
The totality of morbidity and mortality prevented by vaccines
Measles and Hib are 2 examples of vaccine-preventable diseases and the reduction in morbidity and mortality achievable with vaccines. TABLE 318 summarizes the number of pre-vaccine era cases for selected diseases. Routine vaccination against 7 common childhood diseases not only prevents many thousands of deaths, as mentioned earlier,1 but it saves $13.5 billion in direct costs in each birth cohort and saves society $68.8 billion in costs that include disability and lost productivity of both patients and caregivers.1
Put simply, every dollar spent on the vaccination program saves $10 in direct and indirect costs to society.1 Sustaining these successes and averting the resurgence of contained diseases requires a commitment to high immunization rates without delays and lapses—an effort made more challenging in light of misinformation about vaccine safety and resultant parental vaccine hesitancy.
Vaccine safety is ensured by rigorous systems
Despite an impressive record of safety, vaccines still cause anxiety among patients and parents in family practices. A recent survey identified concerns of long-term complications, autism, and thimerosal effects to be foremost on the minds of parents, whereas short-term effects were of much less concern.19 Causation of autism related to vaccines has been dismissed; the initial linkages have been shown to be fraudulent.20 With the exception of some influenza vaccine preparations, thimerosal is no longer present in routinely administered children’s vaccines and has been shown not to be associated with autism.21,22 To address parents’ and patients’ concerns about vaccine safety, and especially those surrounding short- and long-term complications, physicians should have a general understanding of the pre- and post-licensure mechanisms in the United States.
Pre-licensure safety is under the purview of vaccine manufacturers and the Center for Biologics Evaluation and Research at the US Food and Drug Administration (http://www.fda.gov/biologicsbloodvaccines/vaccines/default.htm). For licensure, manufacturers must provide clinical data to demonstrate sufficient safety and efficacy. Accordingly, pre-licensure assessments are conducted in a “closed system” under a research protocol. The vaccine recipients are volitional research subjects selected according to inclusion and exclusion criteria. They are also compensated. However, sample sizes are rarely large enough to exclude rare serious adverse events.
Once licensure has been granted, the focus of safety then shifts to the “open system” of usual clinical practice. Vaccine recipients are unselected members of the general population and may have underlying medical conditions, and sometimes—such as with school entry mandates—are less volitional. In this sphere, the responsible parties for safety include the government, manufacturers, and health care systems.
Three ongoing systems function to assure vaccine safety: the Vaccine Adverse Event Reporting System (VAERS), the Vaccine Safety Datalink (VSD), and the Clinical Immunization Safety Assessment (CISA) Network.23,24
VAERS serves as an early warning system for coincidental safety signals and can generate hypotheses for further investigation.24,25 It is characterized by high sensitivity but low specificity as it relies on voluntary reporting from health care personnel, parents, and others. This system was instrumental in identifying the initial cases of intussusception attributable to the rotavirus vaccine, RotaShield.26
The VSD is a network of 10 large, geographically diverse and linked health maintenance organizations that cover about 3% of the US population. Within this “real time” network, vaccination (exposure) can be compared with outpatient, emergency department, hospital, and laboratory data (health outcomes), while accounting for demographic variables (confounders).27,28 VSD studies linked the measles, mumps, rubella, and varicella vaccine to febrile seizures29 and showed no relationship between cumulative vaccine antigen exposure and autism.30
The CISA was established in 2001 to investigate the pathophysiologic mechanisms and biologic risks of adverse effects following immunization and to provide evidence-based vaccine safety assessments.
Based on all available evidence, routinely recommended vaccines have attained a very high level of safety. As with other preventive services, immunizations are generally provided to healthy individuals to maintain good health; thus, a low tolerance for significant adverse events exists. Well over 100 million doses of vaccines are given each year, yet the VAERS receives, on average, only 28,000 adverse event reports per year.
These reports comprise mild, moderate, and severe reactions to vaccines, but also adverse events that may not be related in any way other than chronologically to the vaccine’s administration. Despite this relatively low number of real safety concerns, it is still more likely that patients will know someone who has had a vaccine-related adverse event than someone who has had some of the diseases the vaccines prevent.31
Final thoughts
Current anti-vaccine sentiments appear to arise from varying perspectives. Some are held by parents of children who have allegedly suffered a severe vaccine related adverse event; others by those opposed to government-mandated school immunization requirements; and some from those who have a dislike of vaccine manufacturers. These sentiments persist in part because of a low level of vaccine preventable diseases: When such illnesses are no longer deemed a threat, those who have concerns about vaccine safety, no matter how invalid, believe their concerns should trump all other considerations.
To appreciate the true benefit of vaccine acceptance, we need only look to Europe, where the anti-vaccine movement has led to high levels of vaccine refusal and a resurgence of vaccine-preventable diseases, such as measles, with their associated morbidity and mortality.32 In advocating for continued acceptance and widespread use of vaccines, family physicians can convey to patients and parents the magnitude of associated health benefits while confidently attesting to the effectiveness and safety of vaccines.
CORRESPONDENCE
John Epling, MD, MSEd, Department of Family Medicine, SUNY Upstate Medical University, 475 Irving Ave, Suite 200, Syracuse, NY 13210; eplingj@upstate.edu
› Reassure parents that vaccines are some of the safest and most effective interventions we have to prevent infectious disease. A
› Advise parents that there are multiple systems in place to monitor vaccine safety. C
› Educate parents that lapses in immunization rates can put children at risk of resurgent cases of previously well-controlled diseases, like measles and Haemophilus influenza type b. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
When a public health intervention succeeds and achieves long-term suppression of the target problem, an unfortunate irony is that, with time, the intervention can seem less vital. So it is with vaccines. Many patients and physicians today have never experienced the infectious diseases that once caused millions of deaths and much disability each year, and they therefore do not appreciate the impact these diseases had when they were prevalent.
It is estimated that just 9 of the routinely recommended vaccines prevent 42,000 deaths and 20 million cases of disease in every birth cohort.1 With many of these diseases thus held at bay, attention shifted instead to the supposed risks of vaccines. Many people mistakenly believe a vaccine’s potential for harm is more likely than the chance of acquiring the disease it prevents, and they therefore refuse vaccines for themselves and their children, with little chance in the short term of suffering an adverse outcome for their decision.
In this review—which can inform primary care physicians’ discussions with vaccine-hesitant patients—we first highlight 2 preventable diseases, measles and Haemophilus influenzae type b (Hib) infection. Recent residency graduates may never see these diseases thanks to sustained vaccination programs. However, the risk of acquiring these infections has not disappeared entirely. After considering these examples, we examine the totality of the morbidity and mortality prevented by vaccination and describe the safety of current vaccines and the systems in place to assure their continued safety.
Measles: No longer endemic to the United States, but still a risk from importation
In the pre-vaccine era, measles (rubeola) infected more than 500,000 Americans annually and killed roughly 500.2 This highly communicable systemic acute viral infection was once considered universal in childhood. After vaccine licensure in 1963, widespread immunization reduced the incidence by more than 98%, and by 2000 it had eliminated endemic measles from the United States. However, the disease has now reappeared—largely due to international travel and neglect in becoming vaccinated. As of October 31, 2014, the United States had 20 outbreaks and 603 cases of measles reported in 2014—a dramatic increase over recent years.3
Clinical appearance. Acute measles infection is characterized by high fever, cough, coryza, conjunctivitis, and rash. Koplik spots are a 24- to 72-hour pathognomonic exanthem of blue-white spots 1 to 3 mm in diameter on an erythematous base along the buccal mucosa. The resolving exanthem coincides with the eruption of a blanching, maculopapular exanthem originating at the hairline, progressing down the trunk and out to the limbs (sparing the palms and soles), coalescing, and then fading with a fine desquamation in the same order of appearance over 7 days. Additional associated symptoms include anorexia, diarrhea, and generalized lymphadenopathy.2,4
Complications are common with measles. Acute measles infection is rarely fatal. However, serious complications occur in nearly one-third of reported cases.2 During the 1989-1991 measles resurgence in the United States, more than 100 deaths occurred among the 55,000 cases reported.5-7 In early 2011, the United States saw the highest reported number of measles cases since 1996 due to importation. Of the 118 reported cases, 105 (89%) occurred in unvaccinated people, 47 (40%) required hospitalization, and 9 individuals developed pneumonia.8
Complications of measles infection are shown in TABLE 1.2,4 Pneumonia (viral or superimposed bacterial) accounts for 60% of measles-related deaths.2 Neurologic complications, while less frequent, can be severe.
Acute encephalitis occurs in 1 in 1000 to 2000 cases and presents within a week following the exanthem with fever, headache, vomiting, meningismus, change in mental status, convulsions, and coma.2 Encephalitis has a fatality rate of 15%, leaving another 25% with residual neurologic damage.2 Subacute sclerosing panencephalitis (SSPE) occurs in 5 to 10 cases per million (in the United States), on average 7 years after the initial measles infection.9,10 After an insidious onset, behavior and intellect deteriorate, followed by ataxia, myoclonic seizures, and ultimately death. In the United States, the number of reported cases of SSPE has declined with the reduction in measles cases. However, in countries with less robust measles immunization eradication programs, the risk of developing SSPE remains.9,10
Hib: Contained but not eradicated
Hib was once the leading cause of meningitis and a major cause of other invasive bacterial diseases, but it has been greatly controlled since the advent of routine Hib vaccination in 1990.11 Hib is an encapsulated, gram-negative coccobacillus. There are 6 major capsular serotypes of Haemophilus influenzae, but serotype b was linked to major invasive disease in humans 95% of the time.12 The spectrum of diseases caused by Hib is seen in TABLE 2.13-15 Hib is transmitted by respiratory droplets from noninfected as well as infected carriers. Asymptomatic nasal carriage in the pre-vaccine era varied from 0.5% to 5%.12
Hib is primarily a disease of young children, with almost all cases occurring in children younger than 5 years of age (66% in those younger than 18 months). Other risk factors for invasive disease are those that increase the spread of respiratory droplets: crowding, lower socioeconomic status, day care attendance, large household size, and school-aged siblings. American Indian and Alaskan Native populations remain at higher risk due to incomplete vaccination rates and the sociodemographic risk factors noted above. Breastfeeding is protective.12
Three percent to 6% of cases of invasive Hib disease are fatal; another 20% can have long-term sequelae such as hearing loss. In the early 1990s, the peak incidence of Hib disease reached 41 cases per 100,000 population.12 The reduction in incidence of Hib disease brought about by universal vaccination has been attributed to individual immunity, decreased asymptomatic nasal carriage, and herd immunity.12
Despite this progress, Hib continues to evade eradication. In Minnesota in 2008, 5 children, ages 5 months to 3 years, contracted invasive Hib disease (3 with meningitis, 1 with pneumonia, 1 with epiglottitis).16 Of the 5, only one was up to date with Hib vaccination; the others had not received vaccine because of shortages or parent refusal. These children were unrelated and had not been in contact with each other.
In a daycare outbreak in the United Kingdom, 2 cases of Hib disease (meningitis and septic arthritis) were identified in fully immunized children younger than 18 months, presumably due to a lack of complete vaccine efficacy.17 A study of nasal carriage (performed just prior to rifampin prophylaxis) among other attendees and caregivers revealed 3 asymptomatic carriers.17 Although Hib is largely well-contained in developed countries due to vaccination policies, the burden of disease in developing countries is estimated to be approximately 8.1 million serious illnesses with 371,000 deaths annually.13
The totality of morbidity and mortality prevented by vaccines
Measles and Hib are 2 examples of vaccine-preventable diseases and the reduction in morbidity and mortality achievable with vaccines. TABLE 318 summarizes the number of pre-vaccine era cases for selected diseases. Routine vaccination against 7 common childhood diseases not only prevents many thousands of deaths, as mentioned earlier,1 but it saves $13.5 billion in direct costs in each birth cohort and saves society $68.8 billion in costs that include disability and lost productivity of both patients and caregivers.1
Put simply, every dollar spent on the vaccination program saves $10 in direct and indirect costs to society.1 Sustaining these successes and averting the resurgence of contained diseases requires a commitment to high immunization rates without delays and lapses—an effort made more challenging in light of misinformation about vaccine safety and resultant parental vaccine hesitancy.
Vaccine safety is ensured by rigorous systems
Despite an impressive record of safety, vaccines still cause anxiety among patients and parents in family practices. A recent survey identified concerns of long-term complications, autism, and thimerosal effects to be foremost on the minds of parents, whereas short-term effects were of much less concern.19 Causation of autism related to vaccines has been dismissed; the initial linkages have been shown to be fraudulent.20 With the exception of some influenza vaccine preparations, thimerosal is no longer present in routinely administered children’s vaccines and has been shown not to be associated with autism.21,22 To address parents’ and patients’ concerns about vaccine safety, and especially those surrounding short- and long-term complications, physicians should have a general understanding of the pre- and post-licensure mechanisms in the United States.
Pre-licensure safety is under the purview of vaccine manufacturers and the Center for Biologics Evaluation and Research at the US Food and Drug Administration (http://www.fda.gov/biologicsbloodvaccines/vaccines/default.htm). For licensure, manufacturers must provide clinical data to demonstrate sufficient safety and efficacy. Accordingly, pre-licensure assessments are conducted in a “closed system” under a research protocol. The vaccine recipients are volitional research subjects selected according to inclusion and exclusion criteria. They are also compensated. However, sample sizes are rarely large enough to exclude rare serious adverse events.
Once licensure has been granted, the focus of safety then shifts to the “open system” of usual clinical practice. Vaccine recipients are unselected members of the general population and may have underlying medical conditions, and sometimes—such as with school entry mandates—are less volitional. In this sphere, the responsible parties for safety include the government, manufacturers, and health care systems.
Three ongoing systems function to assure vaccine safety: the Vaccine Adverse Event Reporting System (VAERS), the Vaccine Safety Datalink (VSD), and the Clinical Immunization Safety Assessment (CISA) Network.23,24
VAERS serves as an early warning system for coincidental safety signals and can generate hypotheses for further investigation.24,25 It is characterized by high sensitivity but low specificity as it relies on voluntary reporting from health care personnel, parents, and others. This system was instrumental in identifying the initial cases of intussusception attributable to the rotavirus vaccine, RotaShield.26
The VSD is a network of 10 large, geographically diverse and linked health maintenance organizations that cover about 3% of the US population. Within this “real time” network, vaccination (exposure) can be compared with outpatient, emergency department, hospital, and laboratory data (health outcomes), while accounting for demographic variables (confounders).27,28 VSD studies linked the measles, mumps, rubella, and varicella vaccine to febrile seizures29 and showed no relationship between cumulative vaccine antigen exposure and autism.30
The CISA was established in 2001 to investigate the pathophysiologic mechanisms and biologic risks of adverse effects following immunization and to provide evidence-based vaccine safety assessments.
Based on all available evidence, routinely recommended vaccines have attained a very high level of safety. As with other preventive services, immunizations are generally provided to healthy individuals to maintain good health; thus, a low tolerance for significant adverse events exists. Well over 100 million doses of vaccines are given each year, yet the VAERS receives, on average, only 28,000 adverse event reports per year.
These reports comprise mild, moderate, and severe reactions to vaccines, but also adverse events that may not be related in any way other than chronologically to the vaccine’s administration. Despite this relatively low number of real safety concerns, it is still more likely that patients will know someone who has had a vaccine-related adverse event than someone who has had some of the diseases the vaccines prevent.31
Final thoughts
Current anti-vaccine sentiments appear to arise from varying perspectives. Some are held by parents of children who have allegedly suffered a severe vaccine related adverse event; others by those opposed to government-mandated school immunization requirements; and some from those who have a dislike of vaccine manufacturers. These sentiments persist in part because of a low level of vaccine preventable diseases: When such illnesses are no longer deemed a threat, those who have concerns about vaccine safety, no matter how invalid, believe their concerns should trump all other considerations.
To appreciate the true benefit of vaccine acceptance, we need only look to Europe, where the anti-vaccine movement has led to high levels of vaccine refusal and a resurgence of vaccine-preventable diseases, such as measles, with their associated morbidity and mortality.32 In advocating for continued acceptance and widespread use of vaccines, family physicians can convey to patients and parents the magnitude of associated health benefits while confidently attesting to the effectiveness and safety of vaccines.
CORRESPONDENCE
John Epling, MD, MSEd, Department of Family Medicine, SUNY Upstate Medical University, 475 Irving Ave, Suite 200, Syracuse, NY 13210; eplingj@upstate.edu
1. Zhou F, Shefer A, Wenger J, et al. Economic evaluation of the routine childhood immunization program in the United States, 2009. Pediatrics. 2014;133:577-585.
2. Centers for Disease Control and Prevention. Measles. Epidemiology and prevention of vaccine-preventable diseases. The Pink Book: Course Textbook. 12th ed. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/pubs/pinkbook/meas.html. Accessed March 4, 2012.
3. Centers for Disease Control and Prevention. Measles (Rubeola). Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/measles/. Accessed November 19, 2014.
4. Measles. In: Pickering LK, Baker CJ, Kimberlin DW, et al, eds. Red Book: 2009 Report of the Committee on Infectious Diseases. 28th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2009:444-455.
5. Atkinson WL, Orenstein WA, Krugman S. The resurgence of measles in the United States, 1989-1990. Annu Rev Med. 1992;43:451-463.
6. Centers for Disease Control and Prevention (CDC). Measles--United States, 1990. MMWR Morb Mortal Wkly Rep. 1991;40:369-372.
7. Gindler J, Tinker S, Markowitz L, et al. Acute measles mortality in the United States, 1987-2002. J Infect Dis. 2004;189(suppl 1): S69-S77.
8. Centers for Disease Control and Prevention (CDC). Measles: United States, January-May 20, 2011. MMWR Morb Mortal Wkly Rep. 2011;60:666-668.
9. Bernstein DI, Reuman PD, Schiff GM. Rubeola (measles) and subacute sclerosing panencephalitis virus. In: Gorbach SL, Bartlett JG, Blacklow NR (eds). Infectious Diseases. Philadelphia, PA: WB Saunders; 1998:2135.
10. Bellini WJ, Rota JS, Lowe LE, et al. Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than was previously recognized. J Infect Dis. 2005;192:1686-1693.
11. Centers for Disease Control and Prevention (CDC). Progress toward elimination of Haemophilus influenza type b invasive disease among infants and children—United States, 1998-2000. MMWR Morb Mortal Wkly Rep. 2002;51:234-237.
12. Centers for Disease Control and Prevention (CDC). Epidemiology and Prevention of Vaccine-Preventable Diseases. The Pink Book, Course Textbook, 12th ed. Haemophilus influenzae type b. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/pubs/pinkbook/hib.html. Accessed March 4, 2012.
13. Watt JP, Wolfson LJ, O’Brien KL, et al; Hib and Pneumococcal Global Burden of Disease Study Team. Burden of disease caused by Haemophilus influenzae type b in children younger than 5 years: global estimates. Lancet. 2009;374:903-911.
14. Agrawal A, Murphy TF. Haemophilus influenzae infections in the H. influenzae type b conjugate vaccine era. J Clin Microbiol. 2011;49:3728-3732.
15. Chandran A, Watt JP, Santosham M. Prevention of Haemophilus influenza type b disease: past successes and future challenges. Informa Healthcare. 2005;4:819-827.
16. Centers for Disease Control and Prevention (CDC). Invasive Haemophilus influenza Type B disease in five young children--Minnesota, 2008. MMWR Morb Mortal Wkly Rep. 2009;58:58-60.
17. McVernon J, Morgan P, Mallaghan C, et al. Outbreak of Haemophilus influenzae type b disease among fully vaccinated children in a day-care center. Pediatr Infect Dis J. 2004;23:38-41.
18. Hinman AR, Orenstein WA, Schuchat A; Centers for Disease Control and Prevention (CDC). Vaccine-preventable diseases, immunizations, and MMWR—1961-2011. MMWR Morb Mortal Wkly Rep. 2011;60 suppl 4:49-57.
19. Kempe A, Daley MF, McCauley MM, et al. Prevalence of parental concerns about childhood vaccines: the experience of primary care physicians. Am J Prev Med. 2011;40:548-555.
20. Godlee F, Smith J, Marcovitch H. Wakefield’s article linking MMR vaccine and autism was fraudulent. BMJ. 2011;342:c7452.
21. Institute for Vaccine Safety. Thimerosal content in some US Licensed vaccines. Institute for Vaccine Safety Web site. Available at: http://www.vaccinesafety.edu/thi-table.htm. Accessed March 8, 2012.
22. Centers for Disease Control and Prevention. Centers for Disease Control and Prevention: Immunization safety and autism. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccinesafety/00_pdf/CDCStudiesonVaccinesandAutism.pdf. Accessed September 23, 2013.
23. Centers for Disease Control and Prevention. Ensuring the safety of vaccines in the United States. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/hcp/patient-ed/conversations/downloads/vacsafe-ensuring-color-office.pdf. Accessed November 18, 2014.
24. Wharton M. Vaccine safety: current systems and recent findings. Curr Opin Pediatr. 2010;22:88-93.
25. US Food and Drug Administration. Understanding the Vaccine Adverse Event Reporting System (VAERS). US Food and Drug Administration Web site. Available at: http://www.fda.gov/downloads/BiologicsBloodVaccines/SafetyAvailability/VaccineSafety/UCM298183.pdf. Accessed October 15, 2014.
26. Centers for Disease Control and Prevention (CDC). Suspension of rotavirus vaccine after reports of intussusception--United States, 1999. MMWR Morb Mortal Wkly Rep. 2004;53:786-789.
27. Greene SK, Kulldorff M, Lewis EM, et al. Near real-time surveillance for influenza vaccine safety: proof-of-concept in the Vaccine Safety Datalink Project. Am J Epidemiol. 2010;171:177-188.
28. Iskander J, Broder K. Monitoring the safety of annual and pandemic influenza vaccines: lessons from the US experience. Expert Rev Vaccines. 2008;7:75-82.
29. Klein NP, Fireman B, Yih WK, et al. Measles-mumps-rubella-varicella combination vaccine and the risk of febrile seizures. Pediatrics. 2010;126:e1-e8.
30. DeStefano F, Price CS, Weintraub ES. Increasing exposure to antibody-stimulating proteins and polysaccharides in vaccines is not associated with risk of autism. J Pediatr. 2013;163:561-567.
31. Centers for Disease Control and Prevention (CDC). Epidemiology and prevention of vaccine-preventable diseases. The Pink Book, Course Textbook, 12th ed. Available at: http://www.cdc.gov/vaccines/pubs/pinkbook/safety.html. Accessed November 17, 2014.
32. World Health Organization (WHO). Measles outbreak in Europe. Global alert and response. Available at: http://www.who.int/csr/don/2011_04_21/en/index.html. Accessed March 8, 2012.
1. Zhou F, Shefer A, Wenger J, et al. Economic evaluation of the routine childhood immunization program in the United States, 2009. Pediatrics. 2014;133:577-585.
2. Centers for Disease Control and Prevention. Measles. Epidemiology and prevention of vaccine-preventable diseases. The Pink Book: Course Textbook. 12th ed. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/pubs/pinkbook/meas.html. Accessed March 4, 2012.
3. Centers for Disease Control and Prevention. Measles (Rubeola). Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/measles/. Accessed November 19, 2014.
4. Measles. In: Pickering LK, Baker CJ, Kimberlin DW, et al, eds. Red Book: 2009 Report of the Committee on Infectious Diseases. 28th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2009:444-455.
5. Atkinson WL, Orenstein WA, Krugman S. The resurgence of measles in the United States, 1989-1990. Annu Rev Med. 1992;43:451-463.
6. Centers for Disease Control and Prevention (CDC). Measles--United States, 1990. MMWR Morb Mortal Wkly Rep. 1991;40:369-372.
7. Gindler J, Tinker S, Markowitz L, et al. Acute measles mortality in the United States, 1987-2002. J Infect Dis. 2004;189(suppl 1): S69-S77.
8. Centers for Disease Control and Prevention (CDC). Measles: United States, January-May 20, 2011. MMWR Morb Mortal Wkly Rep. 2011;60:666-668.
9. Bernstein DI, Reuman PD, Schiff GM. Rubeola (measles) and subacute sclerosing panencephalitis virus. In: Gorbach SL, Bartlett JG, Blacklow NR (eds). Infectious Diseases. Philadelphia, PA: WB Saunders; 1998:2135.
10. Bellini WJ, Rota JS, Lowe LE, et al. Subacute sclerosing panencephalitis: more cases of this fatal disease are prevented by measles immunization than was previously recognized. J Infect Dis. 2005;192:1686-1693.
11. Centers for Disease Control and Prevention (CDC). Progress toward elimination of Haemophilus influenza type b invasive disease among infants and children—United States, 1998-2000. MMWR Morb Mortal Wkly Rep. 2002;51:234-237.
12. Centers for Disease Control and Prevention (CDC). Epidemiology and Prevention of Vaccine-Preventable Diseases. The Pink Book, Course Textbook, 12th ed. Haemophilus influenzae type b. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/pubs/pinkbook/hib.html. Accessed March 4, 2012.
13. Watt JP, Wolfson LJ, O’Brien KL, et al; Hib and Pneumococcal Global Burden of Disease Study Team. Burden of disease caused by Haemophilus influenzae type b in children younger than 5 years: global estimates. Lancet. 2009;374:903-911.
14. Agrawal A, Murphy TF. Haemophilus influenzae infections in the H. influenzae type b conjugate vaccine era. J Clin Microbiol. 2011;49:3728-3732.
15. Chandran A, Watt JP, Santosham M. Prevention of Haemophilus influenza type b disease: past successes and future challenges. Informa Healthcare. 2005;4:819-827.
16. Centers for Disease Control and Prevention (CDC). Invasive Haemophilus influenza Type B disease in five young children--Minnesota, 2008. MMWR Morb Mortal Wkly Rep. 2009;58:58-60.
17. McVernon J, Morgan P, Mallaghan C, et al. Outbreak of Haemophilus influenzae type b disease among fully vaccinated children in a day-care center. Pediatr Infect Dis J. 2004;23:38-41.
18. Hinman AR, Orenstein WA, Schuchat A; Centers for Disease Control and Prevention (CDC). Vaccine-preventable diseases, immunizations, and MMWR—1961-2011. MMWR Morb Mortal Wkly Rep. 2011;60 suppl 4:49-57.
19. Kempe A, Daley MF, McCauley MM, et al. Prevalence of parental concerns about childhood vaccines: the experience of primary care physicians. Am J Prev Med. 2011;40:548-555.
20. Godlee F, Smith J, Marcovitch H. Wakefield’s article linking MMR vaccine and autism was fraudulent. BMJ. 2011;342:c7452.
21. Institute for Vaccine Safety. Thimerosal content in some US Licensed vaccines. Institute for Vaccine Safety Web site. Available at: http://www.vaccinesafety.edu/thi-table.htm. Accessed March 8, 2012.
22. Centers for Disease Control and Prevention. Centers for Disease Control and Prevention: Immunization safety and autism. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccinesafety/00_pdf/CDCStudiesonVaccinesandAutism.pdf. Accessed September 23, 2013.
23. Centers for Disease Control and Prevention. Ensuring the safety of vaccines in the United States. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/hcp/patient-ed/conversations/downloads/vacsafe-ensuring-color-office.pdf. Accessed November 18, 2014.
24. Wharton M. Vaccine safety: current systems and recent findings. Curr Opin Pediatr. 2010;22:88-93.
25. US Food and Drug Administration. Understanding the Vaccine Adverse Event Reporting System (VAERS). US Food and Drug Administration Web site. Available at: http://www.fda.gov/downloads/BiologicsBloodVaccines/SafetyAvailability/VaccineSafety/UCM298183.pdf. Accessed October 15, 2014.
26. Centers for Disease Control and Prevention (CDC). Suspension of rotavirus vaccine after reports of intussusception--United States, 1999. MMWR Morb Mortal Wkly Rep. 2004;53:786-789.
27. Greene SK, Kulldorff M, Lewis EM, et al. Near real-time surveillance for influenza vaccine safety: proof-of-concept in the Vaccine Safety Datalink Project. Am J Epidemiol. 2010;171:177-188.
28. Iskander J, Broder K. Monitoring the safety of annual and pandemic influenza vaccines: lessons from the US experience. Expert Rev Vaccines. 2008;7:75-82.
29. Klein NP, Fireman B, Yih WK, et al. Measles-mumps-rubella-varicella combination vaccine and the risk of febrile seizures. Pediatrics. 2010;126:e1-e8.
30. DeStefano F, Price CS, Weintraub ES. Increasing exposure to antibody-stimulating proteins and polysaccharides in vaccines is not associated with risk of autism. J Pediatr. 2013;163:561-567.
31. Centers for Disease Control and Prevention (CDC). Epidemiology and prevention of vaccine-preventable diseases. The Pink Book, Course Textbook, 12th ed. Available at: http://www.cdc.gov/vaccines/pubs/pinkbook/safety.html. Accessed November 17, 2014.
32. World Health Organization (WHO). Measles outbreak in Europe. Global alert and response. Available at: http://www.who.int/csr/don/2011_04_21/en/index.html. Accessed March 8, 2012.
Managing gout: There’s more we can do
› Prescribe an anti-inflammatory drug whenever you initiate urate-lowering therapy (ULT). A
› Do not initiate ULT during an acute gout attack; if a patient on an established ULT regimen has an acute attack, however, therapy should not be stopped. C
› Increase the dosage of ULT to achieve a lower target if gout symptoms persist despite a serum urate level <6 mg/dL. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
From the 1960s to the ’90s, the prevalence of gout more than doubled among US residents.1 In the years since,1-5 gout has become increasingly prevalent worldwide. The key causes—an aging population, poor diet, widespread use of diuretics to treat cardiovascular disease, and comorbidities that promote hyperuricemia—have made the presentation of gout more complex and harder to manage, as well.4-6
In fact, gout is frequently poorly managed. Initiation and maintenance of urate-lowering therapy (ULT)—as well as monitoring of serum urate—is not done often enough, and there is significant variation in medications used to treat gout. As a result, recommended serum urate targets commonly remain unattained.7-9
We can do better. Enhanced understanding of risk factors for gout, augmented by recent research and the approval of 2 new pharmacologic agents (febuxostat in 200910 and pegloticase in 201011), led to the American College of Rheumatology (ACR)’s first edition of gout guidelines, published in 2012.12,13 The key components of gout management—patient education, lifestyle modifications, and pharmacologic therapy—are detailed in the text and tables that follow.
Understanding gout
Gout is actually a heterogeneous spectrum of diseases. It is characterized by an elevated serum urate concentration, with recurrent attacks of acute arthritis associated with monosodium urate crystals in synovial fluid leukocytes, but may also include tophi— typically painless nodular deposits of monosodium urate crystals in tissues in and around the joints—interstitial renal disease, and uric acid nephrolithiasis.14 Symptoms occur when the excess uric acid, the result of inefficient excretion rather than overproduction,2,12,14,15 is deposited in restricted joint spaces.
Who’s at risk?
Risk factors include numerous cardiovascular and metabolic conditions, such as increased adiposity, hypertension, dyslipidemia, heart failure, insulin resistance, hyperglycemia, and renal disease.3,6,12 Older age, genetics, poor diet, alcohol consumption, and medications associated with hyperuricemia, such as loop and thiazide diuretics and low-dose acetylsalicylic acid, are risk factors, as well.
Defined as a serum urate level ≥6.8 mg/dL—the point at which urate becomes insoluble in extracellular fluids12,16,17—hyperuricemia is the most important modifiable risk factor for the development of gout. It can precipitate painful episodic attacks and complications such as chronic arthritis, urolithiasis, and tophi.12
What you’ll see
Patients often present with acute onset of pain and inflammation of a single joint, usually the first metatarsophalangeal. Other joints and soft tissues that may be involved to a lesser extent include (in order of frequency) the insteps, ankles, heels, knees, fingers, and elbows.14 Polyarticular attacks are an atypical manifestation and are sometimes confused with rheumatoid arthritis or osteoarthritis, particularly in the elderly.
Clinical evaluation should include a history of symptom severity, disease burden, and comorbidities, and a thorough physical examination focused on findings such as tophi and acute and chronic synovitis.12 Imaging studies are not recommended for the evaluation of gout because therapy is guided by symptoms.14
Asymptomatic hyperuricemia alone does not establish a diagnosis of gout, and there is no evidence to support ULT for isolated hyperuricemia. However, advice regarding lifestyle modifications and treatment of associated comorbidities may be warranted for such patients.18
How best to manage gout
Optimal gout management encompasses nonpharmacologic therapy, symptom management of acute attacks, and combination anti-inflammatory and ULT prophylaxis for patients with chronic gout.12,13 It is important to work with patients to track and document both the number and the severity of acute attacks occurring over a 12-month period so that those who qualify for ULT can begin it without delay.12 It is important to discuss treatment objectives and management of comorbidities, as well.
Review the medications the patient is taking, and consider eliminating prescription drugs associated with hyperuricemia if the risks outweigh the benefits.19-21 In many cases, however, lifestyle modification—ie, eating a heart-healthy diet, exercising regularly, and losing weight—may do more to prevent gout attacks and manage complications than stopping medications that provide cardioprotection.6 The ACR divides food and beverages into 3 simple categories—avoid, limit, or encourage (TABLE 1.)12

Responding to an acute attack
Whenever possible, initiate pharmacologic therapy within 24 hours of symptom onset, because this has been associated with decreased pain and shorter duration of an acute attack.8,13 The choice of drug should be guided by the severity of the attack, as determined by both a pain score on a visual analog scale (VAS) and the number of affected joints; patient preference, prior response, and associated comorbidities are also important considerations (TABLE 28,13,14). When medications are prescribed for acute attacks or chronic gout, a discussion of adverse effects, drug interactions, contraindications, cost, and the importance of adherence is needed, as well.
For mild to moderate pain (≤6 out of 10 on a VAS) involving a few small joints or one or 2 large joints, monotherapy with a nonsteroidal anti-inflammatory drug (NSAID), a corticosteroid, or colchicine is recommended. For severe pain (>6 out of 10) and/or polyarticular involvement (≥4 joints in more than one region of the body), combination therapy is recommended (eg, colchicine and either an NSAID or a corticosteroid).13 Prednisone, methylprednisolone, and adrenocorticotropic hormone are options for patients who are NPO. Acute gout therapy should be continued until the attack resolves, which can range from 5 to 14 days.13
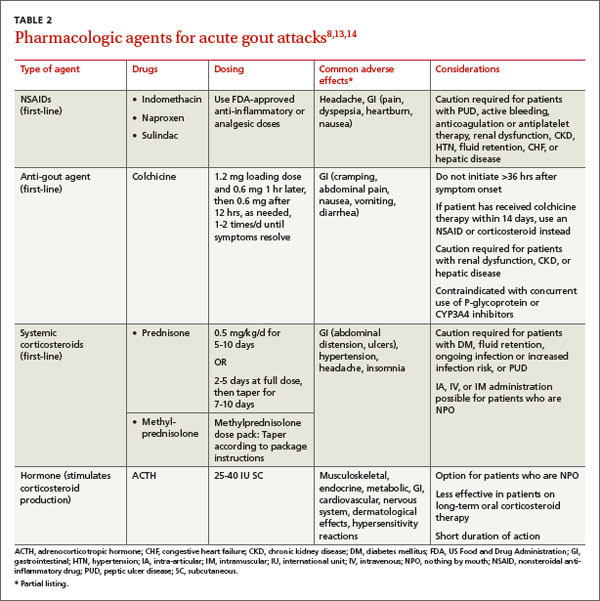
Colchicine considerations. The dose of colchicine recommended by the ACR for an acute gout attack (1.2 mg loading dose, followed by 0.6 mg one hour later, then followed after 12 hours, as needed, by up to 0.6 mg once or twice a day) is substantially lower than the dosing schedule used historically (1.0 mg loading dose, followed by 0.5 mg every 2-3 hours). Higher doses have not proven to be more effective, however, and typically led to gastrointestinal toxicity, causing patients to stop taking the drug before acute symptoms resolved.8,13,22
Keep in mind, too, that colchicine therapy should not be initiated more than 36 hours after symptom onset, as therapy is less effective beyond this time frame.8,13 In addition, concurrent use with P-glycoprotein and CYP3A4 inhibitors—eg, clarithromycin and erythromycin and some antifungals, antiretrovirals, calcium-channel blockers, immunosuppressants, and statins—may increase the risk of colchicine toxicity and should be avoided.
Treating chronic gout
Management of recurrent or progressive gout is aimed at reducing and maintaining serum urate levels <6.0 mg/dL, using ULT (TABLE 312,23-25) combined with anti-inflammatory prophylaxis to reduce the frequency of gout flares and the size and number of tophi.12,23 Patients who meet one or more of the following criteria qualify for ULT:
- the presence of tophi
- ≥2 acute attacks per year
- chronic kidney disease (CKD) stages 2 through 5
- a history of urolithiasis.12
Both ULT and anti-inflammatory therapy should be started after an acute gout attack resolves, but patients already on prophylactic therapy should continue the regimen both during and after acute attacks to avoid more frequent exacerbations.9,12 If gout symptoms persist despite a serum urate level of <6.0 mg/dL, increase the dose of ULT to achieve a target of <5 mg/dL to reduce the frequency of flares and the size and number of tophi.12,26
Allopurinol, a xanthine oxidase inhibitor, is typically used as first-line ULT due to efficacy and low cost.13 Febuxostat, also a xanthine oxidase inhibitor, is an additional first-line option, although the US Food and Drug Administration issued a warning based on postmarketing reports of hepatic failure.25 In the case of a xanthine oxidase allergy or intolerance, probenecid may be used as an alternative first-line therapy. First-line agents for anti-inflammatory prophylaxis include low-dose colchicine (0.6 mg once or twice daily) and low-dose NSAIDs. Oral corticosteroids (<10 mg/d) are considered second-line therapy.13
Allopurinol hypersensitivity. Although allopurinol is generally well tolerated, about 2% of patients develop a mild rash and up to 5% of patients stop taking it because of an adverse effect.25 More importantly, allopurinol hypersensitivity syndrome (AHS) is rare but potentially fatal; in the United States, it is estimated that one in every 1000 patients treated with allopurinol will develop AHS.12,27
AHS is characterized by a rash (eg, Stevens-Johnson syndrome or toxic epidermal necrolysis), eosinophilia, leukocytosis, fever, hepatitis, and renal failure.12,25 There is no cure; the mainstay of treatment is early diagnosis, withdrawal of allopurinol, and supportive care.25 Because of the high mortality rate (20%-25%),12,27 genetic screening for allele HLA-B*5801 prior to starting allopurinol therapy is recommended for patients in high-risk groups: Koreans with CKD (stage 3 or worse) and all Han Chinese and Thai patients, regardless of kidney function.12 Alternative therapies should be used for patients who test positive for the allele.
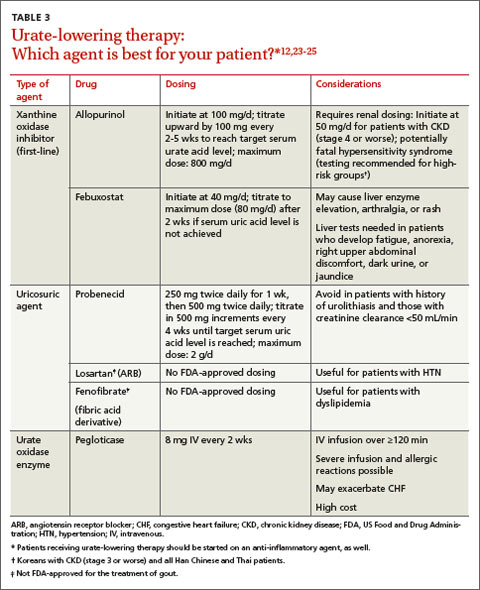
Duration of therapy
Pharmacologic treatment of an acute gout attack should continue until the attack resolves, which can range from 5 to 14 days. The duration of treatment for chronic gout is far longer.
Anti-inflammatory prophylaxis should continue for whichever is greater: 3 months after the target serum urate level is achieved for patients with no evidence of tophi; or 6 months after the target serum urate level is achieved and previously detected tophi have resolved.13
ULT should continue indefinitely,12 with monitoring of serum urate levels every 2 to 5 weeks until the target is achieved and every 6 months thereafter.
Not responding to therapy? Consider nonadherence, refractory gout
If a patient is not responding as expected, consider whether he or she is taking the medication as prescribed. Gout therapy has one of the lowest adherence rates of any chronic disorder.7,8,12,28-30 Studies have found that less than half of patients started on ULT take their medication as prescribed for the entire first year of therapy.9,28
Evidence suggests that nonadherence is especially likely among younger and healthier individuals, possibly because they have little experience managing chronic conditions or needing ongoing care.28-30 Such patients may also be unsure of when and how to take their medication. To promote adherence, physicians should schedule more frequent follow-up appointments after initiating ULT to assess management of the disease and stress the importance of following the medication regimen as prescribed.9,28
Not all patients who don’t respond to ULT are nonadherent, of course. Some have refractory gout. If uric acid levels do not reach the goal of <6 mg/dL (or <5 mg/dL) at the maximum dose of a first-line xanthine oxidase inhibitor, add a uricosuric agent such as probenecid, fenofibrate, or losartan.12
Pegloticase, a pegylated recombinant form of urate oxidase enzyme that converts uric acid to allantoin31 (a water-soluble metabolite of uric acid), is a possible therapeutic option for patients who do not achieve adequate serum urate levels and continue to have symptoms of gout.12 Candidates for pegloticase therapy, which is administered intravenously, include adult patients with gout refractory to conventional ULT or excessive uric acid accumulation due to chemotherapy and those with contraindications to conventional ULT.
Pegloticase is associated with anaphylactic and infusion reactions, requires extensive monitoring, and costs thousands of dollars per month, however. Thus, it is important to carefully evaluate the extent of disease burden (ie, gout symptoms and effect on quality of life) and determine whether the patient has taken ULT and uricosuric drugs as prescribed before considering this option. Pegloticase requires the same anti-inflammatory prophylaxis as other forms of ULT, but there is no consensus on the duration of use.12
CORRESPONDENCE
Tatum Mead, PharmD, University of Missouri-Kansas City School of Pharmacy, Health Sciences Building, Room 2243, 2464 Charlotte Street, Kansas City, MO 64108-2792; meadt@umkc.edu
1. Lawrence RC, Felson DT, Helmick CG, et al; National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58:26-35.
2. Brook RA, Forsythe A, Smeeding JE, et al. Chronic gout: epidemiology, disease progression, treatment and disease burden. Curr Med Res Opin. 2010;26:2813-2821.
3. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63: 3136-3141.
4. Wallace KL, Riedel AA, Joseph-Ridge N, et al. Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004;31: 1582-1587.
5. Roddy E, Zhang W, Doherty M. The changing epidemiology of gout. Nat Clin Pract Rheumatol. 2007;3:443-449.
6. Choi HK. A prescription for lifestyle change in patients with hyperuricemia and gout. Curr Opin Rheumatol. 2010;22: 165-172.
7. Dalbeth N, Lindsay K. The patient’s experience of gout: new insights to optimize management. Curr Rheumatol Rep. 2012;14:173-178.
8. Edwards NL. Quality of care in patients with gout: why is management suboptimal and what can be done about it? Curr Rheumatol Rep. 2011;13:154-159.
9. Singh JA, Hodges JS, Asch SM. Opportunities for improving medication use and monitoring in gout. Ann Rheum Dis. 2009;68: 1265-1270.
10. Drugs.com. FDA approves Uloric (febuxostat) for the chronic management of hyperuricemia in patients with gout [press release]. February 13, 2009. Drugs.com Web site. Available at: http://www.drugs.com/newdrugs/fda-approves-uloric-febuxostat-chronic-management-hyperuricemia-patients-gout-1266.html. Accessed October 29, 2014.
11. US Food and Drug Administration. FDA approves new drug for gout [press release]. September 14, 2010. US Food and Drug Administration Web site. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm225810.htm. Accessed October 29, 2014.
12. Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64:1431-1446.
13. Khanna D, Khanna PP, Fitzgerald JD, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64:1447-1461.
14. Fravel MA, Ernst ME, Clark EC. Gout and hyperuricemia. In: DiPiro JT, Talbert RL, Yee GC, et al. Pharmacotherapy: A Pathophysiologic Approach. 9th ed. New York, NY: McGraw Hill; 2014: 1505-1523.
15. Neogi T. Clinical practice. Gout. N Engl J Med. 2011;364:443-452.
16. Loeb JN. The influence of temperature on the solubility of monosodium urate. Arthritis Rheum. 1972;15:189-192. 17. Terkeltaub R. Update on gout: new therapeutic strategies and options. Nat Rev Rheumatol. 2010;6:30-38.
18. Zhang W, Doherty M, Bardin T, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65:1312-1324.
19. Roddy E, Doherty M. Epidemiology of gout. Arthritis Res Ther. 2010;12:223.
20. McAdams DeMarco MA, Maynard JW, Baer AN, et al. Diuretic use, increased serum urate levels, and risk of incident gout in a population-based study of adults with hypertension: the Atherosclerosis Risk in Communities cohort study. Arthritis Rheum. 2012;64:121-129.
21. Caspi D, Lubart E, Graff E, et al. The effect of mini-dose aspirin on renal function and uric acid handling in elderly patients. Arthritis Rheum. 2000;43:103-108.
22. Terkeltaub RA, Furst DE, Bennett K, et al. High versus low dosing of oral colchicine for early acute gout flare: Twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010;62:1060-1068.
23. Wortmann RL, Macdonald PA, Hunt B, et al. Effect of prophylaxis on gout flares after the initiation of urate-lowering therapy: analysis of data from three phase III trials. Clin Ther. 2010;32:2386-2397.
24. US Food and Drug Administration. Uloric (febuxostat tablets). US Food and Drug Administration Web site. Available at: http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm243770.htm. Accessed October 29, 2014.
25. Stamp LK, Taylor WJ, Jones PB, et al. Starting dose is a risk factor for allopurinol hypersensitivity syndrome: a proposed safe starting dose of allopurinol. Arthritis Rheum. 2012;64:2529-2536.
26. Becker MA, Schumacher HR, Benjamin KL, et al; Gout National Study Group. Quality of life and disability in patients with treatment-failure gout. J Rheumatol. 2009;36:1041-1048.
27. Lupton GP, Odom RB. The allopurinol hypersensitivity syndrome. J Am Acad Dermatol. 1979;1:365-374.
28. Harrold LR, Andrade SE, Briesacher BA, et al. Adherence with urate-lowering therapies for the treatment of gout. Arthritis Res Ther. 2009;11:R46.
29. Reach G. Treatment adherence in patients with gout. Joint Bone Spine. 2011;78:456-459.
30. Briesacher BA, Andrade SE, Fouayzi H, et al. Comparison of drug adherence rates among patients with seven different medical conditions. Pharmacotherapy. 2008;28:437-443.
31. Lexicomp. News from the world of pharmacology. Lexicomp Web site. Available at: https://www.lexi.com/individuals/pharmacists/newsletters.jsp?id=october_10. Accessed January 13, 2014.
› Prescribe an anti-inflammatory drug whenever you initiate urate-lowering therapy (ULT). A
› Do not initiate ULT during an acute gout attack; if a patient on an established ULT regimen has an acute attack, however, therapy should not be stopped. C
› Increase the dosage of ULT to achieve a lower target if gout symptoms persist despite a serum urate level <6 mg/dL. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
From the 1960s to the ’90s, the prevalence of gout more than doubled among US residents.1 In the years since,1-5 gout has become increasingly prevalent worldwide. The key causes—an aging population, poor diet, widespread use of diuretics to treat cardiovascular disease, and comorbidities that promote hyperuricemia—have made the presentation of gout more complex and harder to manage, as well.4-6
In fact, gout is frequently poorly managed. Initiation and maintenance of urate-lowering therapy (ULT)—as well as monitoring of serum urate—is not done often enough, and there is significant variation in medications used to treat gout. As a result, recommended serum urate targets commonly remain unattained.7-9
We can do better. Enhanced understanding of risk factors for gout, augmented by recent research and the approval of 2 new pharmacologic agents (febuxostat in 200910 and pegloticase in 201011), led to the American College of Rheumatology (ACR)’s first edition of gout guidelines, published in 2012.12,13 The key components of gout management—patient education, lifestyle modifications, and pharmacologic therapy—are detailed in the text and tables that follow.
Understanding gout
Gout is actually a heterogeneous spectrum of diseases. It is characterized by an elevated serum urate concentration, with recurrent attacks of acute arthritis associated with monosodium urate crystals in synovial fluid leukocytes, but may also include tophi— typically painless nodular deposits of monosodium urate crystals in tissues in and around the joints—interstitial renal disease, and uric acid nephrolithiasis.14 Symptoms occur when the excess uric acid, the result of inefficient excretion rather than overproduction,2,12,14,15 is deposited in restricted joint spaces.
Who’s at risk?
Risk factors include numerous cardiovascular and metabolic conditions, such as increased adiposity, hypertension, dyslipidemia, heart failure, insulin resistance, hyperglycemia, and renal disease.3,6,12 Older age, genetics, poor diet, alcohol consumption, and medications associated with hyperuricemia, such as loop and thiazide diuretics and low-dose acetylsalicylic acid, are risk factors, as well.
Defined as a serum urate level ≥6.8 mg/dL—the point at which urate becomes insoluble in extracellular fluids12,16,17—hyperuricemia is the most important modifiable risk factor for the development of gout. It can precipitate painful episodic attacks and complications such as chronic arthritis, urolithiasis, and tophi.12
What you’ll see
Patients often present with acute onset of pain and inflammation of a single joint, usually the first metatarsophalangeal. Other joints and soft tissues that may be involved to a lesser extent include (in order of frequency) the insteps, ankles, heels, knees, fingers, and elbows.14 Polyarticular attacks are an atypical manifestation and are sometimes confused with rheumatoid arthritis or osteoarthritis, particularly in the elderly.
Clinical evaluation should include a history of symptom severity, disease burden, and comorbidities, and a thorough physical examination focused on findings such as tophi and acute and chronic synovitis.12 Imaging studies are not recommended for the evaluation of gout because therapy is guided by symptoms.14
Asymptomatic hyperuricemia alone does not establish a diagnosis of gout, and there is no evidence to support ULT for isolated hyperuricemia. However, advice regarding lifestyle modifications and treatment of associated comorbidities may be warranted for such patients.18
How best to manage gout
Optimal gout management encompasses nonpharmacologic therapy, symptom management of acute attacks, and combination anti-inflammatory and ULT prophylaxis for patients with chronic gout.12,13 It is important to work with patients to track and document both the number and the severity of acute attacks occurring over a 12-month period so that those who qualify for ULT can begin it without delay.12 It is important to discuss treatment objectives and management of comorbidities, as well.
Review the medications the patient is taking, and consider eliminating prescription drugs associated with hyperuricemia if the risks outweigh the benefits.19-21 In many cases, however, lifestyle modification—ie, eating a heart-healthy diet, exercising regularly, and losing weight—may do more to prevent gout attacks and manage complications than stopping medications that provide cardioprotection.6 The ACR divides food and beverages into 3 simple categories—avoid, limit, or encourage (TABLE 1.)12
Responding to an acute attack
Whenever possible, initiate pharmacologic therapy within 24 hours of symptom onset, because this has been associated with decreased pain and shorter duration of an acute attack.8,13 The choice of drug should be guided by the severity of the attack, as determined by both a pain score on a visual analog scale (VAS) and the number of affected joints; patient preference, prior response, and associated comorbidities are also important considerations (TABLE 28,13,14). When medications are prescribed for acute attacks or chronic gout, a discussion of adverse effects, drug interactions, contraindications, cost, and the importance of adherence is needed, as well.
For mild to moderate pain (≤6 out of 10 on a VAS) involving a few small joints or one or 2 large joints, monotherapy with a nonsteroidal anti-inflammatory drug (NSAID), a corticosteroid, or colchicine is recommended. For severe pain (>6 out of 10) and/or polyarticular involvement (≥4 joints in more than one region of the body), combination therapy is recommended (eg, colchicine and either an NSAID or a corticosteroid).13 Prednisone, methylprednisolone, and adrenocorticotropic hormone are options for patients who are NPO. Acute gout therapy should be continued until the attack resolves, which can range from 5 to 14 days.13
Colchicine considerations. The dose of colchicine recommended by the ACR for an acute gout attack (1.2 mg loading dose, followed by 0.6 mg one hour later, then followed after 12 hours, as needed, by up to 0.6 mg once or twice a day) is substantially lower than the dosing schedule used historically (1.0 mg loading dose, followed by 0.5 mg every 2-3 hours). Higher doses have not proven to be more effective, however, and typically led to gastrointestinal toxicity, causing patients to stop taking the drug before acute symptoms resolved.8,13,22
Keep in mind, too, that colchicine therapy should not be initiated more than 36 hours after symptom onset, as therapy is less effective beyond this time frame.8,13 In addition, concurrent use with P-glycoprotein and CYP3A4 inhibitors—eg, clarithromycin and erythromycin and some antifungals, antiretrovirals, calcium-channel blockers, immunosuppressants, and statins—may increase the risk of colchicine toxicity and should be avoided.
Treating chronic gout
Management of recurrent or progressive gout is aimed at reducing and maintaining serum urate levels <6.0 mg/dL, using ULT (TABLE 312,23-25) combined with anti-inflammatory prophylaxis to reduce the frequency of gout flares and the size and number of tophi.12,23 Patients who meet one or more of the following criteria qualify for ULT:
- the presence of tophi
- ≥2 acute attacks per year
- chronic kidney disease (CKD) stages 2 through 5
- a history of urolithiasis.12
Both ULT and anti-inflammatory therapy should be started after an acute gout attack resolves, but patients already on prophylactic therapy should continue the regimen both during and after acute attacks to avoid more frequent exacerbations.9,12 If gout symptoms persist despite a serum urate level of <6.0 mg/dL, increase the dose of ULT to achieve a target of <5 mg/dL to reduce the frequency of flares and the size and number of tophi.12,26
Allopurinol, a xanthine oxidase inhibitor, is typically used as first-line ULT due to efficacy and low cost.13 Febuxostat, also a xanthine oxidase inhibitor, is an additional first-line option, although the US Food and Drug Administration issued a warning based on postmarketing reports of hepatic failure.25 In the case of a xanthine oxidase allergy or intolerance, probenecid may be used as an alternative first-line therapy. First-line agents for anti-inflammatory prophylaxis include low-dose colchicine (0.6 mg once or twice daily) and low-dose NSAIDs. Oral corticosteroids (<10 mg/d) are considered second-line therapy.13
Allopurinol hypersensitivity. Although allopurinol is generally well tolerated, about 2% of patients develop a mild rash and up to 5% of patients stop taking it because of an adverse effect.25 More importantly, allopurinol hypersensitivity syndrome (AHS) is rare but potentially fatal; in the United States, it is estimated that one in every 1000 patients treated with allopurinol will develop AHS.12,27
AHS is characterized by a rash (eg, Stevens-Johnson syndrome or toxic epidermal necrolysis), eosinophilia, leukocytosis, fever, hepatitis, and renal failure.12,25 There is no cure; the mainstay of treatment is early diagnosis, withdrawal of allopurinol, and supportive care.25 Because of the high mortality rate (20%-25%),12,27 genetic screening for allele HLA-B*5801 prior to starting allopurinol therapy is recommended for patients in high-risk groups: Koreans with CKD (stage 3 or worse) and all Han Chinese and Thai patients, regardless of kidney function.12 Alternative therapies should be used for patients who test positive for the allele.
Duration of therapy
Pharmacologic treatment of an acute gout attack should continue until the attack resolves, which can range from 5 to 14 days. The duration of treatment for chronic gout is far longer.
Anti-inflammatory prophylaxis should continue for whichever is greater: 3 months after the target serum urate level is achieved for patients with no evidence of tophi; or 6 months after the target serum urate level is achieved and previously detected tophi have resolved.13
ULT should continue indefinitely,12 with monitoring of serum urate levels every 2 to 5 weeks until the target is achieved and every 6 months thereafter.
Not responding to therapy? Consider nonadherence, refractory gout
If a patient is not responding as expected, consider whether he or she is taking the medication as prescribed. Gout therapy has one of the lowest adherence rates of any chronic disorder.7,8,12,28-30 Studies have found that less than half of patients started on ULT take their medication as prescribed for the entire first year of therapy.9,28
Evidence suggests that nonadherence is especially likely among younger and healthier individuals, possibly because they have little experience managing chronic conditions or needing ongoing care.28-30 Such patients may also be unsure of when and how to take their medication. To promote adherence, physicians should schedule more frequent follow-up appointments after initiating ULT to assess management of the disease and stress the importance of following the medication regimen as prescribed.9,28
Not all patients who don’t respond to ULT are nonadherent, of course. Some have refractory gout. If uric acid levels do not reach the goal of <6 mg/dL (or <5 mg/dL) at the maximum dose of a first-line xanthine oxidase inhibitor, add a uricosuric agent such as probenecid, fenofibrate, or losartan.12
Pegloticase, a pegylated recombinant form of urate oxidase enzyme that converts uric acid to allantoin31 (a water-soluble metabolite of uric acid), is a possible therapeutic option for patients who do not achieve adequate serum urate levels and continue to have symptoms of gout.12 Candidates for pegloticase therapy, which is administered intravenously, include adult patients with gout refractory to conventional ULT or excessive uric acid accumulation due to chemotherapy and those with contraindications to conventional ULT.
Pegloticase is associated with anaphylactic and infusion reactions, requires extensive monitoring, and costs thousands of dollars per month, however. Thus, it is important to carefully evaluate the extent of disease burden (ie, gout symptoms and effect on quality of life) and determine whether the patient has taken ULT and uricosuric drugs as prescribed before considering this option. Pegloticase requires the same anti-inflammatory prophylaxis as other forms of ULT, but there is no consensus on the duration of use.12
CORRESPONDENCE
Tatum Mead, PharmD, University of Missouri-Kansas City School of Pharmacy, Health Sciences Building, Room 2243, 2464 Charlotte Street, Kansas City, MO 64108-2792; meadt@umkc.edu
› Prescribe an anti-inflammatory drug whenever you initiate urate-lowering therapy (ULT). A
› Do not initiate ULT during an acute gout attack; if a patient on an established ULT regimen has an acute attack, however, therapy should not be stopped. C
› Increase the dosage of ULT to achieve a lower target if gout symptoms persist despite a serum urate level <6 mg/dL. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
From the 1960s to the ’90s, the prevalence of gout more than doubled among US residents.1 In the years since,1-5 gout has become increasingly prevalent worldwide. The key causes—an aging population, poor diet, widespread use of diuretics to treat cardiovascular disease, and comorbidities that promote hyperuricemia—have made the presentation of gout more complex and harder to manage, as well.4-6
In fact, gout is frequently poorly managed. Initiation and maintenance of urate-lowering therapy (ULT)—as well as monitoring of serum urate—is not done often enough, and there is significant variation in medications used to treat gout. As a result, recommended serum urate targets commonly remain unattained.7-9
We can do better. Enhanced understanding of risk factors for gout, augmented by recent research and the approval of 2 new pharmacologic agents (febuxostat in 200910 and pegloticase in 201011), led to the American College of Rheumatology (ACR)’s first edition of gout guidelines, published in 2012.12,13 The key components of gout management—patient education, lifestyle modifications, and pharmacologic therapy—are detailed in the text and tables that follow.
Understanding gout
Gout is actually a heterogeneous spectrum of diseases. It is characterized by an elevated serum urate concentration, with recurrent attacks of acute arthritis associated with monosodium urate crystals in synovial fluid leukocytes, but may also include tophi— typically painless nodular deposits of monosodium urate crystals in tissues in and around the joints—interstitial renal disease, and uric acid nephrolithiasis.14 Symptoms occur when the excess uric acid, the result of inefficient excretion rather than overproduction,2,12,14,15 is deposited in restricted joint spaces.
Who’s at risk?
Risk factors include numerous cardiovascular and metabolic conditions, such as increased adiposity, hypertension, dyslipidemia, heart failure, insulin resistance, hyperglycemia, and renal disease.3,6,12 Older age, genetics, poor diet, alcohol consumption, and medications associated with hyperuricemia, such as loop and thiazide diuretics and low-dose acetylsalicylic acid, are risk factors, as well.
Defined as a serum urate level ≥6.8 mg/dL—the point at which urate becomes insoluble in extracellular fluids12,16,17—hyperuricemia is the most important modifiable risk factor for the development of gout. It can precipitate painful episodic attacks and complications such as chronic arthritis, urolithiasis, and tophi.12
What you’ll see
Patients often present with acute onset of pain and inflammation of a single joint, usually the first metatarsophalangeal. Other joints and soft tissues that may be involved to a lesser extent include (in order of frequency) the insteps, ankles, heels, knees, fingers, and elbows.14 Polyarticular attacks are an atypical manifestation and are sometimes confused with rheumatoid arthritis or osteoarthritis, particularly in the elderly.
Clinical evaluation should include a history of symptom severity, disease burden, and comorbidities, and a thorough physical examination focused on findings such as tophi and acute and chronic synovitis.12 Imaging studies are not recommended for the evaluation of gout because therapy is guided by symptoms.14
Asymptomatic hyperuricemia alone does not establish a diagnosis of gout, and there is no evidence to support ULT for isolated hyperuricemia. However, advice regarding lifestyle modifications and treatment of associated comorbidities may be warranted for such patients.18
How best to manage gout
Optimal gout management encompasses nonpharmacologic therapy, symptom management of acute attacks, and combination anti-inflammatory and ULT prophylaxis for patients with chronic gout.12,13 It is important to work with patients to track and document both the number and the severity of acute attacks occurring over a 12-month period so that those who qualify for ULT can begin it without delay.12 It is important to discuss treatment objectives and management of comorbidities, as well.
Review the medications the patient is taking, and consider eliminating prescription drugs associated with hyperuricemia if the risks outweigh the benefits.19-21 In many cases, however, lifestyle modification—ie, eating a heart-healthy diet, exercising regularly, and losing weight—may do more to prevent gout attacks and manage complications than stopping medications that provide cardioprotection.6 The ACR divides food and beverages into 3 simple categories—avoid, limit, or encourage (TABLE 1.)12
Responding to an acute attack
Whenever possible, initiate pharmacologic therapy within 24 hours of symptom onset, because this has been associated with decreased pain and shorter duration of an acute attack.8,13 The choice of drug should be guided by the severity of the attack, as determined by both a pain score on a visual analog scale (VAS) and the number of affected joints; patient preference, prior response, and associated comorbidities are also important considerations (TABLE 28,13,14). When medications are prescribed for acute attacks or chronic gout, a discussion of adverse effects, drug interactions, contraindications, cost, and the importance of adherence is needed, as well.
For mild to moderate pain (≤6 out of 10 on a VAS) involving a few small joints or one or 2 large joints, monotherapy with a nonsteroidal anti-inflammatory drug (NSAID), a corticosteroid, or colchicine is recommended. For severe pain (>6 out of 10) and/or polyarticular involvement (≥4 joints in more than one region of the body), combination therapy is recommended (eg, colchicine and either an NSAID or a corticosteroid).13 Prednisone, methylprednisolone, and adrenocorticotropic hormone are options for patients who are NPO. Acute gout therapy should be continued until the attack resolves, which can range from 5 to 14 days.13
Colchicine considerations. The dose of colchicine recommended by the ACR for an acute gout attack (1.2 mg loading dose, followed by 0.6 mg one hour later, then followed after 12 hours, as needed, by up to 0.6 mg once or twice a day) is substantially lower than the dosing schedule used historically (1.0 mg loading dose, followed by 0.5 mg every 2-3 hours). Higher doses have not proven to be more effective, however, and typically led to gastrointestinal toxicity, causing patients to stop taking the drug before acute symptoms resolved.8,13,22
Keep in mind, too, that colchicine therapy should not be initiated more than 36 hours after symptom onset, as therapy is less effective beyond this time frame.8,13 In addition, concurrent use with P-glycoprotein and CYP3A4 inhibitors—eg, clarithromycin and erythromycin and some antifungals, antiretrovirals, calcium-channel blockers, immunosuppressants, and statins—may increase the risk of colchicine toxicity and should be avoided.
Treating chronic gout
Management of recurrent or progressive gout is aimed at reducing and maintaining serum urate levels <6.0 mg/dL, using ULT (TABLE 312,23-25) combined with anti-inflammatory prophylaxis to reduce the frequency of gout flares and the size and number of tophi.12,23 Patients who meet one or more of the following criteria qualify for ULT:
- the presence of tophi
- ≥2 acute attacks per year
- chronic kidney disease (CKD) stages 2 through 5
- a history of urolithiasis.12
Both ULT and anti-inflammatory therapy should be started after an acute gout attack resolves, but patients already on prophylactic therapy should continue the regimen both during and after acute attacks to avoid more frequent exacerbations.9,12 If gout symptoms persist despite a serum urate level of <6.0 mg/dL, increase the dose of ULT to achieve a target of <5 mg/dL to reduce the frequency of flares and the size and number of tophi.12,26
Allopurinol, a xanthine oxidase inhibitor, is typically used as first-line ULT due to efficacy and low cost.13 Febuxostat, also a xanthine oxidase inhibitor, is an additional first-line option, although the US Food and Drug Administration issued a warning based on postmarketing reports of hepatic failure.25 In the case of a xanthine oxidase allergy or intolerance, probenecid may be used as an alternative first-line therapy. First-line agents for anti-inflammatory prophylaxis include low-dose colchicine (0.6 mg once or twice daily) and low-dose NSAIDs. Oral corticosteroids (<10 mg/d) are considered second-line therapy.13
Allopurinol hypersensitivity. Although allopurinol is generally well tolerated, about 2% of patients develop a mild rash and up to 5% of patients stop taking it because of an adverse effect.25 More importantly, allopurinol hypersensitivity syndrome (AHS) is rare but potentially fatal; in the United States, it is estimated that one in every 1000 patients treated with allopurinol will develop AHS.12,27
AHS is characterized by a rash (eg, Stevens-Johnson syndrome or toxic epidermal necrolysis), eosinophilia, leukocytosis, fever, hepatitis, and renal failure.12,25 There is no cure; the mainstay of treatment is early diagnosis, withdrawal of allopurinol, and supportive care.25 Because of the high mortality rate (20%-25%),12,27 genetic screening for allele HLA-B*5801 prior to starting allopurinol therapy is recommended for patients in high-risk groups: Koreans with CKD (stage 3 or worse) and all Han Chinese and Thai patients, regardless of kidney function.12 Alternative therapies should be used for patients who test positive for the allele.
Duration of therapy
Pharmacologic treatment of an acute gout attack should continue until the attack resolves, which can range from 5 to 14 days. The duration of treatment for chronic gout is far longer.
Anti-inflammatory prophylaxis should continue for whichever is greater: 3 months after the target serum urate level is achieved for patients with no evidence of tophi; or 6 months after the target serum urate level is achieved and previously detected tophi have resolved.13
ULT should continue indefinitely,12 with monitoring of serum urate levels every 2 to 5 weeks until the target is achieved and every 6 months thereafter.
Not responding to therapy? Consider nonadherence, refractory gout
If a patient is not responding as expected, consider whether he or she is taking the medication as prescribed. Gout therapy has one of the lowest adherence rates of any chronic disorder.7,8,12,28-30 Studies have found that less than half of patients started on ULT take their medication as prescribed for the entire first year of therapy.9,28
Evidence suggests that nonadherence is especially likely among younger and healthier individuals, possibly because they have little experience managing chronic conditions or needing ongoing care.28-30 Such patients may also be unsure of when and how to take their medication. To promote adherence, physicians should schedule more frequent follow-up appointments after initiating ULT to assess management of the disease and stress the importance of following the medication regimen as prescribed.9,28
Not all patients who don’t respond to ULT are nonadherent, of course. Some have refractory gout. If uric acid levels do not reach the goal of <6 mg/dL (or <5 mg/dL) at the maximum dose of a first-line xanthine oxidase inhibitor, add a uricosuric agent such as probenecid, fenofibrate, or losartan.12
Pegloticase, a pegylated recombinant form of urate oxidase enzyme that converts uric acid to allantoin31 (a water-soluble metabolite of uric acid), is a possible therapeutic option for patients who do not achieve adequate serum urate levels and continue to have symptoms of gout.12 Candidates for pegloticase therapy, which is administered intravenously, include adult patients with gout refractory to conventional ULT or excessive uric acid accumulation due to chemotherapy and those with contraindications to conventional ULT.
Pegloticase is associated with anaphylactic and infusion reactions, requires extensive monitoring, and costs thousands of dollars per month, however. Thus, it is important to carefully evaluate the extent of disease burden (ie, gout symptoms and effect on quality of life) and determine whether the patient has taken ULT and uricosuric drugs as prescribed before considering this option. Pegloticase requires the same anti-inflammatory prophylaxis as other forms of ULT, but there is no consensus on the duration of use.12
CORRESPONDENCE
Tatum Mead, PharmD, University of Missouri-Kansas City School of Pharmacy, Health Sciences Building, Room 2243, 2464 Charlotte Street, Kansas City, MO 64108-2792; meadt@umkc.edu
1. Lawrence RC, Felson DT, Helmick CG, et al; National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58:26-35.
2. Brook RA, Forsythe A, Smeeding JE, et al. Chronic gout: epidemiology, disease progression, treatment and disease burden. Curr Med Res Opin. 2010;26:2813-2821.
3. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63: 3136-3141.
4. Wallace KL, Riedel AA, Joseph-Ridge N, et al. Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004;31: 1582-1587.
5. Roddy E, Zhang W, Doherty M. The changing epidemiology of gout. Nat Clin Pract Rheumatol. 2007;3:443-449.
6. Choi HK. A prescription for lifestyle change in patients with hyperuricemia and gout. Curr Opin Rheumatol. 2010;22: 165-172.
7. Dalbeth N, Lindsay K. The patient’s experience of gout: new insights to optimize management. Curr Rheumatol Rep. 2012;14:173-178.
8. Edwards NL. Quality of care in patients with gout: why is management suboptimal and what can be done about it? Curr Rheumatol Rep. 2011;13:154-159.
9. Singh JA, Hodges JS, Asch SM. Opportunities for improving medication use and monitoring in gout. Ann Rheum Dis. 2009;68: 1265-1270.
10. Drugs.com. FDA approves Uloric (febuxostat) for the chronic management of hyperuricemia in patients with gout [press release]. February 13, 2009. Drugs.com Web site. Available at: http://www.drugs.com/newdrugs/fda-approves-uloric-febuxostat-chronic-management-hyperuricemia-patients-gout-1266.html. Accessed October 29, 2014.
11. US Food and Drug Administration. FDA approves new drug for gout [press release]. September 14, 2010. US Food and Drug Administration Web site. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm225810.htm. Accessed October 29, 2014.
12. Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64:1431-1446.
13. Khanna D, Khanna PP, Fitzgerald JD, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64:1447-1461.
14. Fravel MA, Ernst ME, Clark EC. Gout and hyperuricemia. In: DiPiro JT, Talbert RL, Yee GC, et al. Pharmacotherapy: A Pathophysiologic Approach. 9th ed. New York, NY: McGraw Hill; 2014: 1505-1523.
15. Neogi T. Clinical practice. Gout. N Engl J Med. 2011;364:443-452.
16. Loeb JN. The influence of temperature on the solubility of monosodium urate. Arthritis Rheum. 1972;15:189-192. 17. Terkeltaub R. Update on gout: new therapeutic strategies and options. Nat Rev Rheumatol. 2010;6:30-38.
18. Zhang W, Doherty M, Bardin T, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65:1312-1324.
19. Roddy E, Doherty M. Epidemiology of gout. Arthritis Res Ther. 2010;12:223.
20. McAdams DeMarco MA, Maynard JW, Baer AN, et al. Diuretic use, increased serum urate levels, and risk of incident gout in a population-based study of adults with hypertension: the Atherosclerosis Risk in Communities cohort study. Arthritis Rheum. 2012;64:121-129.
21. Caspi D, Lubart E, Graff E, et al. The effect of mini-dose aspirin on renal function and uric acid handling in elderly patients. Arthritis Rheum. 2000;43:103-108.
22. Terkeltaub RA, Furst DE, Bennett K, et al. High versus low dosing of oral colchicine for early acute gout flare: Twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010;62:1060-1068.
23. Wortmann RL, Macdonald PA, Hunt B, et al. Effect of prophylaxis on gout flares after the initiation of urate-lowering therapy: analysis of data from three phase III trials. Clin Ther. 2010;32:2386-2397.
24. US Food and Drug Administration. Uloric (febuxostat tablets). US Food and Drug Administration Web site. Available at: http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm243770.htm. Accessed October 29, 2014.
25. Stamp LK, Taylor WJ, Jones PB, et al. Starting dose is a risk factor for allopurinol hypersensitivity syndrome: a proposed safe starting dose of allopurinol. Arthritis Rheum. 2012;64:2529-2536.
26. Becker MA, Schumacher HR, Benjamin KL, et al; Gout National Study Group. Quality of life and disability in patients with treatment-failure gout. J Rheumatol. 2009;36:1041-1048.
27. Lupton GP, Odom RB. The allopurinol hypersensitivity syndrome. J Am Acad Dermatol. 1979;1:365-374.
28. Harrold LR, Andrade SE, Briesacher BA, et al. Adherence with urate-lowering therapies for the treatment of gout. Arthritis Res Ther. 2009;11:R46.
29. Reach G. Treatment adherence in patients with gout. Joint Bone Spine. 2011;78:456-459.
30. Briesacher BA, Andrade SE, Fouayzi H, et al. Comparison of drug adherence rates among patients with seven different medical conditions. Pharmacotherapy. 2008;28:437-443.
31. Lexicomp. News from the world of pharmacology. Lexicomp Web site. Available at: https://www.lexi.com/individuals/pharmacists/newsletters.jsp?id=october_10. Accessed January 13, 2014.
1. Lawrence RC, Felson DT, Helmick CG, et al; National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58:26-35.
2. Brook RA, Forsythe A, Smeeding JE, et al. Chronic gout: epidemiology, disease progression, treatment and disease burden. Curr Med Res Opin. 2010;26:2813-2821.
3. Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007-2008. Arthritis Rheum. 2011;63: 3136-3141.
4. Wallace KL, Riedel AA, Joseph-Ridge N, et al. Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004;31: 1582-1587.
5. Roddy E, Zhang W, Doherty M. The changing epidemiology of gout. Nat Clin Pract Rheumatol. 2007;3:443-449.
6. Choi HK. A prescription for lifestyle change in patients with hyperuricemia and gout. Curr Opin Rheumatol. 2010;22: 165-172.
7. Dalbeth N, Lindsay K. The patient’s experience of gout: new insights to optimize management. Curr Rheumatol Rep. 2012;14:173-178.
8. Edwards NL. Quality of care in patients with gout: why is management suboptimal and what can be done about it? Curr Rheumatol Rep. 2011;13:154-159.
9. Singh JA, Hodges JS, Asch SM. Opportunities for improving medication use and monitoring in gout. Ann Rheum Dis. 2009;68: 1265-1270.
10. Drugs.com. FDA approves Uloric (febuxostat) for the chronic management of hyperuricemia in patients with gout [press release]. February 13, 2009. Drugs.com Web site. Available at: http://www.drugs.com/newdrugs/fda-approves-uloric-febuxostat-chronic-management-hyperuricemia-patients-gout-1266.html. Accessed October 29, 2014.
11. US Food and Drug Administration. FDA approves new drug for gout [press release]. September 14, 2010. US Food and Drug Administration Web site. Available at: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm225810.htm. Accessed October 29, 2014.
12. Khanna D, Fitzgerald JD, Khanna PP, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken). 2012;64:1431-1446.
13. Khanna D, Khanna PP, Fitzgerald JD, et al; American College of Rheumatology. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64:1447-1461.
14. Fravel MA, Ernst ME, Clark EC. Gout and hyperuricemia. In: DiPiro JT, Talbert RL, Yee GC, et al. Pharmacotherapy: A Pathophysiologic Approach. 9th ed. New York, NY: McGraw Hill; 2014: 1505-1523.
15. Neogi T. Clinical practice. Gout. N Engl J Med. 2011;364:443-452.
16. Loeb JN. The influence of temperature on the solubility of monosodium urate. Arthritis Rheum. 1972;15:189-192. 17. Terkeltaub R. Update on gout: new therapeutic strategies and options. Nat Rev Rheumatol. 2010;6:30-38.
18. Zhang W, Doherty M, Bardin T, et al; EULAR Standing Committee for International Clinical Studies Including Therapeutics. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics (ESCISIT). Ann Rheum Dis. 2006;65:1312-1324.
19. Roddy E, Doherty M. Epidemiology of gout. Arthritis Res Ther. 2010;12:223.
20. McAdams DeMarco MA, Maynard JW, Baer AN, et al. Diuretic use, increased serum urate levels, and risk of incident gout in a population-based study of adults with hypertension: the Atherosclerosis Risk in Communities cohort study. Arthritis Rheum. 2012;64:121-129.
21. Caspi D, Lubart E, Graff E, et al. The effect of mini-dose aspirin on renal function and uric acid handling in elderly patients. Arthritis Rheum. 2000;43:103-108.
22. Terkeltaub RA, Furst DE, Bennett K, et al. High versus low dosing of oral colchicine for early acute gout flare: Twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010;62:1060-1068.
23. Wortmann RL, Macdonald PA, Hunt B, et al. Effect of prophylaxis on gout flares after the initiation of urate-lowering therapy: analysis of data from three phase III trials. Clin Ther. 2010;32:2386-2397.
24. US Food and Drug Administration. Uloric (febuxostat tablets). US Food and Drug Administration Web site. Available at: http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm243770.htm. Accessed October 29, 2014.
25. Stamp LK, Taylor WJ, Jones PB, et al. Starting dose is a risk factor for allopurinol hypersensitivity syndrome: a proposed safe starting dose of allopurinol. Arthritis Rheum. 2012;64:2529-2536.
26. Becker MA, Schumacher HR, Benjamin KL, et al; Gout National Study Group. Quality of life and disability in patients with treatment-failure gout. J Rheumatol. 2009;36:1041-1048.
27. Lupton GP, Odom RB. The allopurinol hypersensitivity syndrome. J Am Acad Dermatol. 1979;1:365-374.
28. Harrold LR, Andrade SE, Briesacher BA, et al. Adherence with urate-lowering therapies for the treatment of gout. Arthritis Res Ther. 2009;11:R46.
29. Reach G. Treatment adherence in patients with gout. Joint Bone Spine. 2011;78:456-459.
30. Briesacher BA, Andrade SE, Fouayzi H, et al. Comparison of drug adherence rates among patients with seven different medical conditions. Pharmacotherapy. 2008;28:437-443.
31. Lexicomp. News from the world of pharmacology. Lexicomp Web site. Available at: https://www.lexi.com/individuals/pharmacists/newsletters.jsp?id=october_10. Accessed January 13, 2014.
“Difficult” patient? Or does he have a personality disorder?
› Evaluate a patient’s sense of identity and interpersonal relationships for clues of a personality disorder (PD). A
› Use validation, promote mentalization, and
manage countertransference to help patients with PDs. A
› Consider medications such as antidepressants or antipsychotics for patients with PDs, but only as adjuncts to psychotherapy, and only to target specific symptoms, such as impulsive aggression. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Bob A, age 48, comes to his family physician (FP) to ask for authorization for extended medical leave from his job as an electrician. He frequently misses days at work and complains of stress on the job, saying his coworkers look down on him and make cruel jokes at his expense. He reports having chronic interpersonal conflicts and no significant relationships with family members or friends. Mr. A refuses a referral to a psychiatrist because he fears he will be “locked up and forced to take medications.”
If Mr. A were your patient, how would you proceed?
Personality disorders (PDs) are patterns of inflexible and maladaptive personality traits and behaviors that cause subjective distress and significant social or occupational impairment.1 An individual with a PD tends to have a limited repertoire of responses to the rough-and-tumble of life, with coping mechanisms that often perpetuate difficulty and distress. Examples include distrust and suspiciousness of others’ motives (paranoid PD); disregard and violation of the rights of others (antisocial PD); instability in interpersonal relationships, self-image, and affect (borderline PD); and social inhibition, feelings of inadequacy, and hypersensitivity to negative evaluation (avoidant PD).1
FPs may view patients with PDs as “difficult patients” because of their frequent crises and the interpersonal problems they bring into the physician-patient relationship.2,3 Help, of course, can come in the way of a referral to a psychotherapist who specializes in treating PDs. But you can also make use of some evidence-based psychotherapy techniques to improve your patients’ lives and the quality of the physician-patient relationship. This article focuses on identifying and managing PDs in family practice, using practical strategies drawn from empirically supported therapies.
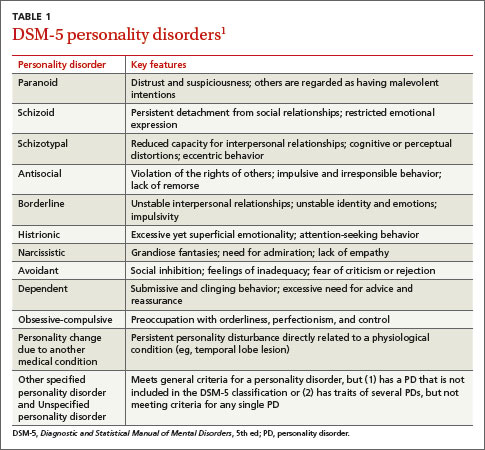
PDs are more common than you might suspect
The overall prevalence of PD in the community ranges from 4.4% to 14.8%, with no consistent pattern of sex differences.4 Between 31.4% and 45.5% of psychiatric outpatients and up to 24% of primary care patients likely meet criteria for at least one PD.5-7 PDs impede recovery from other mental disorders,8 increase the risk for suicide,9 and are associated with substance abuse, impulsivity, and violence.10,11 Personality pathology also is associated with greater incidence of serious medical illness12,13 and reduced social functioning.14 Not surprisingly, patients with PDs frequently use medical and social services.15
PDs tend to be underdiagnosed, perhaps partly because of concern about stigmatization, but also due to difficulties in identifying and classifying these disorders. Published in 2013, the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM-5) originally was to include a major revision of PDs—reflecting concern about the limitations of PD categories—but ultimately the existing categories were retained (TABLE 1).1 There is considerable overlap among PD categories; many patients meet the criteria for more than one PD, but it is unlikely that they actually suffer from several distinct PDs. Other patients—perhaps even the majority—are best diagnosed with “unspecified personality disorder” because they do not neatly fit into one of these categories.
Suspect your patient has a PD? Evaluate these 2 areas
Identifying patients who have PDs in primary care is useful for 2 reasons: to explore the option of specialty treatment for patients who may be amenable to it, and to improve management of the patient’s complaints in the primary care setting, including a smoother doctor-patient interaction. In either case, determining the specific DSM-5 diagnosis is less important than recognizing core personality impairment: an ingrained disturbance in one’s perceptions of self and others. This can be done by paying attention to how the patient adapts to life’s challenges and if he or she has problematic interpersonal tendencies, including difficulties in the doctor-patient relationship.
Unfortunately, assessing and diagnosing PDs in the primary care setting can be challenging. Limited time doesn’t allow for extensive, personality-focused interviews. Self-report screening tools are limited, because patients may underreport key interpersonal problems such as lack of empathy. Furthermore, very few patients seek help from their FP in addressing personality dysfunction; PDs typically are identified while investigating other complaints.
The most reliable and useful areas to evaluate in a patient you suspect may have a PD are identity (one’s sense of who one is and can be) and interpersonal relationships, including the capacity for empathy and intimacy.16,17 These should be considered longitudinally and in the context of the individual’s stage of development. For example, identity is generally less stable among adolescents compared to middle-aged adults.
A cohesive sense of identity allows one to embrace life’s tasks and challenges, to develop and strive toward personal goals, and to handle setbacks and disappointments. A person with a stable identity may develop a depressive reaction to difficult life circumstances, but with some assistance can generally bounce back and re-engage in his or her personal goals. By contrast, an individual with an unstable sense of self may feel chronically insecure and empty, with limited capacity to constructively deal with life’s ups and downs. Patients with borderline PD, for example, try to manage a fragmented identity by frantically clinging to others, while narcissistic patients tend to suppress a fragile sense of self by putting forth an arrogant and entitled attitude.
How does the patient interact with others? As is the case with identity, an individual’s capacity for interpersonal functioning is developed early in life, through interactions with primary caregivers. Mental maps of who we are and what we can expect from others are formed and reinforced in attachment relationships, such as those with our parents; traumatic attachments, including abuse or neglect by a caregiver or loved one, are strongly associated with PD.18,19 The resulting belief structures guide subsequent interpersonal functioning, and become interactively reinforced. For example, a person whose internal map of relationships includes others abandoning him might behave in a clingy manner, which may ultimately induce others to reject him, thus creating a self-fulfilling prophecy.
Distorted interpersonal expectations can impair a person’s capacity for sustained intimate connections (a troubled relationship history is characteristic of PDs) and limit empathic functioning.20 Other people’s actions may be interpreted according to the patient’s belief structures rather than with an open mind about the other person’s experience.
Focus on the physician-patient relationship
The interpersonal dysfunction of patients with PDs will often surface in the physician-patient relationship, serving as a clue to broader interpersonal dysfunction. An FP’s relatively innocuous oversight, for example, might be taken as proof of suspected incompetence in the eyes of a patient with paranoid or narcissistic tendencies. Or a patient with a recurrent complaint who repeatedly rejects the physician’s interventions probably oscillates between seeking and rejecting nurturance in other relationships, as well. A patient who tends to make sarcastic remarks regarding the doctor’s earnest efforts likely holds negative views of others and sabotages potentially positive interactions.
So what strategies are best for managing these types of scenarios?
Bringing up a potential diagnosis of PD may be a delicate matter for the FP; patients might experience this as a jarring diagnosis in the absence of a thorough psychiatric evaluation. If the FP decides to explore whether the patient is open to discussing the relationship between moods, behaviors, and personality features, he or she can begin this conversation by noting that, as with physical health, we all have our vulnerabilities, and that these vulnerabilities may be strengthened through specialist consultation and support. In this way, the patient can view a referral as an opportunity to explore herself with professional support. If a psychiatrist or psychotherapist colleague does become involved, it is important to clarify the roles of treatment providers and to communicate with one another, should difficulties arise.
Evidence supports 2 forms of psychotherapy
Treatment for PDs has seen considerable growth over the past decade, largely due to research on therapies that target the troubling self-injurious and suicidal features of borderline PD. Considerable evidence shows that specialized psychotherapy can significantly reduce suffering and improve functioning among these patients. The 2 major evidence-based treatments for patients with borderline PD are dialectical behavior therapy (DBT) and psychodynamic therapy.
DBT is an intensive cognitive-behavioral approach that teaches patients how to regulate their emotions and develop an accepting, mindful attitude toward their mental experience.21 Several randomized controlled trials (RCTs) have demonstrated the effectiveness of DBT in reducing hospitalizations and self-injurious and suicidal behavior in patients with borderline PD.22
Psychodynamic therapy, which focuses on helping patients discover how unconscious conflicts influence their present moods and behaviors, has also been validated by multiple RCTs for patients with borderline PD.23-25 Like DBT, empirically supported psychodynamic therapy tends to be structured, long-term (>12 months), and often intensively delivered in multiple sessions per week. However, a recent study found that a less-intensive, general psychodynamic therapy, along with occasional medication management, was equivalent to intensive DBT.26
Although the research has focused primarily on borderline PD, these approaches can be applied to other PDs. These therapies focus on understanding one’s emotional and behavioral patterns, developing a healthy self-concept, and improving interpersonal relationships—areas that are relevant treatment targets across all PD types.
Indeed, studies of day treatment programs that explicitly welcome patients with a range of PD types have had promising findings.27 Day treatment involves an intensive array of therapies, mostly in a group format; patients work together to support and embolden one another to make positive changes. Unfortunately, FPs may be challenged to find appropriate services for patients who are amenable to psychotherapy; public mental health resources tend to lag far behind best practices in the case of PD.
Medication might improve symptoms, not personality deficits
Most research on pharmacotherapy for PDs has focused on borderline PD; findings have been mixed and fairly limited.28 Medication cannot address underlying identity and relational deficits, and will not result in remission of PD. Nonetheless, judicious, circumscribed use of medications to target specific symptoms may be helpful for some patients. Selective serotonin reuptake inhibitors can reduce anger and impulsive aggression in patients with borderline PD.28,29
Atypical antipsychotics may help reduce impulsive aggression or transient psychotic symptoms.28-30 For example, olanzapine and aripiprazole can reduce anxiety, anger/aggression, paranoia, and interpersonal sensitivity in borderline PD.31,32 Mood stabilizers such as valproate, lamotrigine, and topiramate may also help some borderline patients, although they do so by reducing impulsivity and aggression rather than improving core unstable identity and affect.28,29
Carefully obtained informed consent is necessary because of the danger of adverse effects with many of these medications; for example, antipsychotics have been associated with metabolic syndrome and weight gain that can threaten a patient’s already fragile self-image.33 Polypharmacy is also a potential problem: Well-intentioned physicians may be prompted to offer multiple medications in response to patients’ unremitting complaints of distress, when a psychotherapeutic approach may need to be the primary treatment. The bottom line is that medications do not resolve personality dysfunction, and are best used symptomatically as adjuncts to psychotherapy.28,30
Steps you can take during the office visit
Although it is not feasible for most FPs to provide comprehensive treatment for PD, key elements from specialized therapies can be integrated into your management of these patients. Steps you can take include using validation, promoting mentalization, and managing countertransference.
Validation, which is a component of DBT, is providing the expressed acknowledgement that the patient is entitled to her feelings. This is not the same as agreeing with a position the patient has taken on an issue, but rather conveying the sense that one sees how the patient might feel the way she does. A study of women with borderline PD and substance abuse found a validation intervention by itself was significantly helpful.34 Validation can contribute to a “corrective emotional experience.” For instance, your supportive acknowledgement of a patient with a history of abuse or neglect may counter the patient’s expectation of being invalidated, and over time this can reduce the patient’s defensive rigidity.
Mentalization. Psychodynamic treatment involves a similar tack; clinicians empathize with the patient’s emotional state while also demonstrating a degree of separateness from the emotion.23-25 This promotes mentalization in the patient—the ability to contemplate one’s own and others’ subjective mental states.18 Mentalization is often impaired in PD patients, who presume to “know” what others are thinking. A patient, for instance, “just knows” that her friend secretly hates her, based on a vaguely worded text message.
You can help patients with mentalization by taking an inquisitive “not knowing” stance and by emphasizing a collaborative and reflective approach toward a given problem—to examine the issue together, from all sides. You can point out that while a patient is entitled to feel whatever he is feeling, it may not be in his best interest to act on the feelings without adequately considering the potential consequences of the action. This helps the patient to distinguish thoughts, feelings, and impulses from behavior. It also teaches the value of anticipatory thinking, impulse control, and affect regulation.
Countertransference. Managing your emotional reactions to a patient with PD is a well-documented challenge.35 Your feelings about the patient, known as countertransference, can range from considerable concern and sympathy to severe frustration, bewilderment, and frank hostility. A common reaction is the sense that one must “do something” to respond to the patient’s emotional distress or interpersonal pressure. This may trigger an impulse to give advice or offer tests or medications despite knowing that these are unlikely to be helpful. A more useful response may be to tolerate such feelings and listen empathically to the patient’s frustration. Recognizing subtle countertransference can guard against extreme reactions and maintain an appropriate clinical focus. Discussion with a trusted colleague can be helpful.
Psychodynamic approaches consider managing countertransference to be a therapeutic intervention, even when psychotherapy is not explicitly being carried out. Strong emotional responses may reflect something that the patient needs the physician to experience, as the patient cannot bear to experience it himself. The patient needs to see—and learn from—the physician’s handling of unbearable (for the patient) feelings. This occurs at a level of unconscious communication and may be repeated over time. Although not discussed with the patient, a physician’s capacity for self-containment and provision of undisrupted, good medical care is in itself a psychotherapeutic accomplishment.
CASE › Based on Mr. A’s history of interpersonal conflicts and perceived persecution by coworkers, the FP consults with a psychotherapist colleague, who says Mr. A’s chronic mistrust and social isolation suggest he may have a severe identity disturbance and unspecified PD with paranoid and schizoid features. Because Mr. A refuses to see a therapist, his FP decides to focus on promoting small improvements in Mr. A’s interpersonal interactions and reducing absenteeism at work.
The FP validates Mr. A’s feelings (“it can be very stressful to constantly feel like others are at odds with you”) and tries to promote mentalizing (“I want to understand more about what you think regarding your work situation and your coworkers. Let’s try to look at this from all perspectives—maybe we can come up with some new ideas.”)
Despite wanting to help his patient, the FP feels uneasy and reluctant to engage with Mr. A, who likely evokes such feelings to keep others at a distance. The FP tactfully seeks to remain Mr. A’s ally without endorsing his distorted interpretation of events. Given Mr. A’s paranoid rejection of therapy, the FP refrains from making further such recommendations. The FP’s interventions, however, may help Mr. A warm to the idea of further help over time, and the FP’s supportive stance will help to ameliorate the patient’s distress. (For 2 additional examples of how FPs can use the strategies described in this article to help patients with PDs, see TABLE 2.)
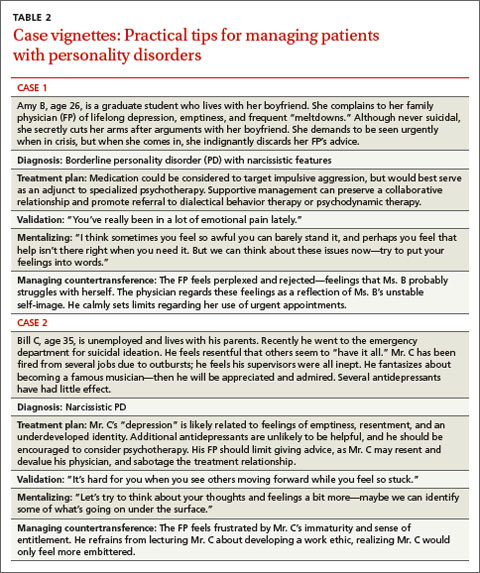
CORRESPONDENCE
David Kealy, MSW, Psychotherapy Program, Department
of Psychiatry, University of British Columbia, #420-5950 University Boulevard, Vancouver, BC Canada V6T 1Z3; david.kealy@ubc.ca
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
2. Hahn SR, Thompson KS, Wills TA, et al. The difficult doctor-patient relationship: somatization, personality and psychopathology. J Clin Epidemiol. 1994:47:647-657.
3. Schafer S, Nowlis DP. Personality disorders among difficult patients. Arch Fam Med. 1998;7:126-129.
4. Paris J. Estimating the prevalence of personality disorders in the community. J Pers Disord. 2010;24:405-411.
5. Newton-Howes G, Tyrer P, Anagnostakis K, et al. The prevalence of personality disorder, its comorbidity with mental state disorders, and its clinical significance in community mental health teams. Soc Psychiatry Psychiatr Epidemiol. 2010;45:453-460.
6. Zimmerman M, Rothschild L, Chelminski I. The prevalence of DSM-IV personality disorders in psychiatric outpatients. Am J Psychiatry. 2005;162:1911-1918.
7. Moran P, Jenkins R, Tylee A, et al. The prevalence of personality disorder among UK primary care attenders. Acta Psychiatr Scand. 2000;102:52-57.
8. Newton-Howes G, Tyrer P, Johnson T. Personality disorder and the outcome of depression: Meta-analysis of published studies. Br J Psychiatry. 2006;188:13-20.
9. Blasco-Fontecilla H, Baca-Garcia E, Dervic K, et al. Severity of personality disorders and suicide attempt. Acta Psychiatr Scand. 2009;119:149-155.
10. Colpaert K, Vanderplasschen W, De Maeyer J, et al. Prevalence and determinants of personality disorders in a clinical sample of alcohol-, drug-, and dual-dependent patients. Subst Use Misuse. 2012;47:649-661.
11. Yu R, Geddes JR, Fazel S. Personality disorders, violence, and antisocial behavior: A systematic review and meta-regression analysis. J Pers Disord. 2012;26:775-792.
12. Frankenburg FR, Zanarini MC. The association between borderline personality disorder and chronic medical illnesses, poor health-related lifestyle choices, and costly forms of health care utilization. J Clin Psychiatry. 2004;65:1660-1665.
13. Lee HB, Bienvenu OJ, Cho SJ, et al. Personality disorders and traits as predictors of incident cardiovascular disease: Findings from the 23-year follow-up of the Baltimore ECA Study. Psychosomatics. 2010;51:289-296.
14. Skodol AE, Gunderson JG, McGlashan TH, et al. Functional impairment in patients with schizotypal, borderline, avoidant, or obsessive-compulsive personality disorder. Am J Psychiatry. 2002;159:276-283.
15. Bender DS, Dolan RT, Skodol AE, et al. Treatment utilization by patients with personality disorders. Am J Psychiatry. 2001;158:295-302.
16. Livesley WJ. An empirically-based classification of personality disorder. J Pers Disord. 2011;25:397-420.
17. Bender DS, Morey LC, Skodol AE. Toward a model for assessing personality functioning in DSM-5, part I: a review of theory and methods. J Pers Assess. 2011;93:332-346.
18. Fonagy P, Gergely G, Jurist EL, et al. Affect Regulation, Mentalization, and the Development of the Self. New York, NY: Other Press; 2002.
19. Yen S, Shea MT, Battle CL, et al. Traumatic exposure and posttraumatic stress disorder in borderline, schizotypal, avoidant, and obsessive-compulsive personality disorders: findings from the collaborative longitudinal personality disorders study. J Nerv Ment Dis. 2002;190:510-518.
20. Morey LC, Stagner BH. Narcissistic pathology as core personality dysfunction: comparing DSM-IV and the DSM-5 proposal for narcissistic personality disorder. J Clin Psychol. 2012;68:908-921.
21. Lynch TR, Chapman AL, Rosenthal MZ, et al. Mechanisms of change in dialectical behaviour therapy: theoretical and empirical observations. J Clin Psychol. 2006;62:459-480.
22. Kliem S, Kröger C, Kosfelder J. Dialectical behavior therapy for borderline personality disorder: a meta-analysis using mixed-effects modeling. J Consult Clin Psychol. 2010;78:936-951.
23. Clarkin JF, Levy KN, Lenzenweger MF, et al. Evaluating three treatments for borderline personality disorder: a multiwave study. Am J Psychiatry. 2007;164:922-928.
24. Gregory RJ, DeLucia-Deranja E, Mogle JA. Dynamic deconstructive psychotherapy versus optimized community care for borderline personality disorder co-occurring with alcohol use disorders: a 30-month follow-up. J Nerv Ment Dis. 2010;198:292-298.
25. Bateman A, Fonagy P. Randomized controlled trial of outpatient mentalization-based treatment versus structured clinical management for borderline personality disorder. Am J Psychiatry. 2009;166:1355-1364.
26. McMain SF, Links PS, Gnam WH, et al. A randomized trial of dialectical behavior therapy versus general psychiatric management for borderline personality disorder. Am J Psychiatry. 2009;166:1365-1374.
27. Ogrodniczuk JS, Piper WE. Day treatment for personality disorders: a review of research findings. Harv Rev Psychiatry. 2001;9:105-117.
28. Paris J. Pharmacological treatments for personality disorders. Int Rev Psychiatry. 2011;23:303-309.
29. Ripoll LH, Triebwasser J, Siever LJ. Evidence-based pharmacotherapy for personality disorders. Int J Neuropsychopharmacol. 2011;14:1257-1288.
30. Steinberg PI. The use of low-dose neuroleptics in the treatment of patients with severe personality disorder: An adjunct to psychotherapy. BCMJ. 2007;49:306-310.
31. Zanarini MC, Frankenburg FR. Olanzapine treatment of female borderline personality disorder patients: a double-blind, placebo controlled pilot study. J Clin Psychiatry. 2001;62:849-854.
32. Nickel MK, Loew TH, Pedrosa Gil F. Aripiprazole in treatment of borderline patients, part II: an 18-month follow up. Psychopharmacology (Berl). 2007;191:1023-1026.
33. Silk KR. The process of managing medications in patients with borderline personality disorder. J Psychiatr Pract. 2011;17:311-319.
34. Linehan MM, Dimeff LA, Reynolds SK, et al. Dialectal behavior therapy versus comprehensive validation therapy plus 12-step for the treatment of opioid dependent women meeting criteria for borderline personality disorder. Drug Alcohol Depend. 2002;67:13-26.
35. Rossberg JI, Karterud S, Pedersen G, et al. An empirical study of countertransference reactions toward patients with personality disorders. Compr Psychiatry. 2007;48:225-230.
› Evaluate a patient’s sense of identity and interpersonal relationships for clues of a personality disorder (PD). A
› Use validation, promote mentalization, and
manage countertransference to help patients with PDs. A
› Consider medications such as antidepressants or antipsychotics for patients with PDs, but only as adjuncts to psychotherapy, and only to target specific symptoms, such as impulsive aggression. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Bob A, age 48, comes to his family physician (FP) to ask for authorization for extended medical leave from his job as an electrician. He frequently misses days at work and complains of stress on the job, saying his coworkers look down on him and make cruel jokes at his expense. He reports having chronic interpersonal conflicts and no significant relationships with family members or friends. Mr. A refuses a referral to a psychiatrist because he fears he will be “locked up and forced to take medications.”
If Mr. A were your patient, how would you proceed?
Personality disorders (PDs) are patterns of inflexible and maladaptive personality traits and behaviors that cause subjective distress and significant social or occupational impairment.1 An individual with a PD tends to have a limited repertoire of responses to the rough-and-tumble of life, with coping mechanisms that often perpetuate difficulty and distress. Examples include distrust and suspiciousness of others’ motives (paranoid PD); disregard and violation of the rights of others (antisocial PD); instability in interpersonal relationships, self-image, and affect (borderline PD); and social inhibition, feelings of inadequacy, and hypersensitivity to negative evaluation (avoidant PD).1
FPs may view patients with PDs as “difficult patients” because of their frequent crises and the interpersonal problems they bring into the physician-patient relationship.2,3 Help, of course, can come in the way of a referral to a psychotherapist who specializes in treating PDs. But you can also make use of some evidence-based psychotherapy techniques to improve your patients’ lives and the quality of the physician-patient relationship. This article focuses on identifying and managing PDs in family practice, using practical strategies drawn from empirically supported therapies.
PDs are more common than you might suspect
The overall prevalence of PD in the community ranges from 4.4% to 14.8%, with no consistent pattern of sex differences.4 Between 31.4% and 45.5% of psychiatric outpatients and up to 24% of primary care patients likely meet criteria for at least one PD.5-7 PDs impede recovery from other mental disorders,8 increase the risk for suicide,9 and are associated with substance abuse, impulsivity, and violence.10,11 Personality pathology also is associated with greater incidence of serious medical illness12,13 and reduced social functioning.14 Not surprisingly, patients with PDs frequently use medical and social services.15
PDs tend to be underdiagnosed, perhaps partly because of concern about stigmatization, but also due to difficulties in identifying and classifying these disorders. Published in 2013, the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM-5) originally was to include a major revision of PDs—reflecting concern about the limitations of PD categories—but ultimately the existing categories were retained (TABLE 1).1 There is considerable overlap among PD categories; many patients meet the criteria for more than one PD, but it is unlikely that they actually suffer from several distinct PDs. Other patients—perhaps even the majority—are best diagnosed with “unspecified personality disorder” because they do not neatly fit into one of these categories.
Suspect your patient has a PD? Evaluate these 2 areas
Identifying patients who have PDs in primary care is useful for 2 reasons: to explore the option of specialty treatment for patients who may be amenable to it, and to improve management of the patient’s complaints in the primary care setting, including a smoother doctor-patient interaction. In either case, determining the specific DSM-5 diagnosis is less important than recognizing core personality impairment: an ingrained disturbance in one’s perceptions of self and others. This can be done by paying attention to how the patient adapts to life’s challenges and if he or she has problematic interpersonal tendencies, including difficulties in the doctor-patient relationship.
Unfortunately, assessing and diagnosing PDs in the primary care setting can be challenging. Limited time doesn’t allow for extensive, personality-focused interviews. Self-report screening tools are limited, because patients may underreport key interpersonal problems such as lack of empathy. Furthermore, very few patients seek help from their FP in addressing personality dysfunction; PDs typically are identified while investigating other complaints.
The most reliable and useful areas to evaluate in a patient you suspect may have a PD are identity (one’s sense of who one is and can be) and interpersonal relationships, including the capacity for empathy and intimacy.16,17 These should be considered longitudinally and in the context of the individual’s stage of development. For example, identity is generally less stable among adolescents compared to middle-aged adults.
A cohesive sense of identity allows one to embrace life’s tasks and challenges, to develop and strive toward personal goals, and to handle setbacks and disappointments. A person with a stable identity may develop a depressive reaction to difficult life circumstances, but with some assistance can generally bounce back and re-engage in his or her personal goals. By contrast, an individual with an unstable sense of self may feel chronically insecure and empty, with limited capacity to constructively deal with life’s ups and downs. Patients with borderline PD, for example, try to manage a fragmented identity by frantically clinging to others, while narcissistic patients tend to suppress a fragile sense of self by putting forth an arrogant and entitled attitude.
How does the patient interact with others? As is the case with identity, an individual’s capacity for interpersonal functioning is developed early in life, through interactions with primary caregivers. Mental maps of who we are and what we can expect from others are formed and reinforced in attachment relationships, such as those with our parents; traumatic attachments, including abuse or neglect by a caregiver or loved one, are strongly associated with PD.18,19 The resulting belief structures guide subsequent interpersonal functioning, and become interactively reinforced. For example, a person whose internal map of relationships includes others abandoning him might behave in a clingy manner, which may ultimately induce others to reject him, thus creating a self-fulfilling prophecy.
Distorted interpersonal expectations can impair a person’s capacity for sustained intimate connections (a troubled relationship history is characteristic of PDs) and limit empathic functioning.20 Other people’s actions may be interpreted according to the patient’s belief structures rather than with an open mind about the other person’s experience.
Focus on the physician-patient relationship
The interpersonal dysfunction of patients with PDs will often surface in the physician-patient relationship, serving as a clue to broader interpersonal dysfunction. An FP’s relatively innocuous oversight, for example, might be taken as proof of suspected incompetence in the eyes of a patient with paranoid or narcissistic tendencies. Or a patient with a recurrent complaint who repeatedly rejects the physician’s interventions probably oscillates between seeking and rejecting nurturance in other relationships, as well. A patient who tends to make sarcastic remarks regarding the doctor’s earnest efforts likely holds negative views of others and sabotages potentially positive interactions.
So what strategies are best for managing these types of scenarios?
Bringing up a potential diagnosis of PD may be a delicate matter for the FP; patients might experience this as a jarring diagnosis in the absence of a thorough psychiatric evaluation. If the FP decides to explore whether the patient is open to discussing the relationship between moods, behaviors, and personality features, he or she can begin this conversation by noting that, as with physical health, we all have our vulnerabilities, and that these vulnerabilities may be strengthened through specialist consultation and support. In this way, the patient can view a referral as an opportunity to explore herself with professional support. If a psychiatrist or psychotherapist colleague does become involved, it is important to clarify the roles of treatment providers and to communicate with one another, should difficulties arise.
Evidence supports 2 forms of psychotherapy
Treatment for PDs has seen considerable growth over the past decade, largely due to research on therapies that target the troubling self-injurious and suicidal features of borderline PD. Considerable evidence shows that specialized psychotherapy can significantly reduce suffering and improve functioning among these patients. The 2 major evidence-based treatments for patients with borderline PD are dialectical behavior therapy (DBT) and psychodynamic therapy.
DBT is an intensive cognitive-behavioral approach that teaches patients how to regulate their emotions and develop an accepting, mindful attitude toward their mental experience.21 Several randomized controlled trials (RCTs) have demonstrated the effectiveness of DBT in reducing hospitalizations and self-injurious and suicidal behavior in patients with borderline PD.22
Psychodynamic therapy, which focuses on helping patients discover how unconscious conflicts influence their present moods and behaviors, has also been validated by multiple RCTs for patients with borderline PD.23-25 Like DBT, empirically supported psychodynamic therapy tends to be structured, long-term (>12 months), and often intensively delivered in multiple sessions per week. However, a recent study found that a less-intensive, general psychodynamic therapy, along with occasional medication management, was equivalent to intensive DBT.26
Although the research has focused primarily on borderline PD, these approaches can be applied to other PDs. These therapies focus on understanding one’s emotional and behavioral patterns, developing a healthy self-concept, and improving interpersonal relationships—areas that are relevant treatment targets across all PD types.
Indeed, studies of day treatment programs that explicitly welcome patients with a range of PD types have had promising findings.27 Day treatment involves an intensive array of therapies, mostly in a group format; patients work together to support and embolden one another to make positive changes. Unfortunately, FPs may be challenged to find appropriate services for patients who are amenable to psychotherapy; public mental health resources tend to lag far behind best practices in the case of PD.
Medication might improve symptoms, not personality deficits
Most research on pharmacotherapy for PDs has focused on borderline PD; findings have been mixed and fairly limited.28 Medication cannot address underlying identity and relational deficits, and will not result in remission of PD. Nonetheless, judicious, circumscribed use of medications to target specific symptoms may be helpful for some patients. Selective serotonin reuptake inhibitors can reduce anger and impulsive aggression in patients with borderline PD.28,29
Atypical antipsychotics may help reduce impulsive aggression or transient psychotic symptoms.28-30 For example, olanzapine and aripiprazole can reduce anxiety, anger/aggression, paranoia, and interpersonal sensitivity in borderline PD.31,32 Mood stabilizers such as valproate, lamotrigine, and topiramate may also help some borderline patients, although they do so by reducing impulsivity and aggression rather than improving core unstable identity and affect.28,29
Carefully obtained informed consent is necessary because of the danger of adverse effects with many of these medications; for example, antipsychotics have been associated with metabolic syndrome and weight gain that can threaten a patient’s already fragile self-image.33 Polypharmacy is also a potential problem: Well-intentioned physicians may be prompted to offer multiple medications in response to patients’ unremitting complaints of distress, when a psychotherapeutic approach may need to be the primary treatment. The bottom line is that medications do not resolve personality dysfunction, and are best used symptomatically as adjuncts to psychotherapy.28,30
Steps you can take during the office visit
Although it is not feasible for most FPs to provide comprehensive treatment for PD, key elements from specialized therapies can be integrated into your management of these patients. Steps you can take include using validation, promoting mentalization, and managing countertransference.
Validation, which is a component of DBT, is providing the expressed acknowledgement that the patient is entitled to her feelings. This is not the same as agreeing with a position the patient has taken on an issue, but rather conveying the sense that one sees how the patient might feel the way she does. A study of women with borderline PD and substance abuse found a validation intervention by itself was significantly helpful.34 Validation can contribute to a “corrective emotional experience.” For instance, your supportive acknowledgement of a patient with a history of abuse or neglect may counter the patient’s expectation of being invalidated, and over time this can reduce the patient’s defensive rigidity.
Mentalization. Psychodynamic treatment involves a similar tack; clinicians empathize with the patient’s emotional state while also demonstrating a degree of separateness from the emotion.23-25 This promotes mentalization in the patient—the ability to contemplate one’s own and others’ subjective mental states.18 Mentalization is often impaired in PD patients, who presume to “know” what others are thinking. A patient, for instance, “just knows” that her friend secretly hates her, based on a vaguely worded text message.
You can help patients with mentalization by taking an inquisitive “not knowing” stance and by emphasizing a collaborative and reflective approach toward a given problem—to examine the issue together, from all sides. You can point out that while a patient is entitled to feel whatever he is feeling, it may not be in his best interest to act on the feelings without adequately considering the potential consequences of the action. This helps the patient to distinguish thoughts, feelings, and impulses from behavior. It also teaches the value of anticipatory thinking, impulse control, and affect regulation.
Countertransference. Managing your emotional reactions to a patient with PD is a well-documented challenge.35 Your feelings about the patient, known as countertransference, can range from considerable concern and sympathy to severe frustration, bewilderment, and frank hostility. A common reaction is the sense that one must “do something” to respond to the patient’s emotional distress or interpersonal pressure. This may trigger an impulse to give advice or offer tests or medications despite knowing that these are unlikely to be helpful. A more useful response may be to tolerate such feelings and listen empathically to the patient’s frustration. Recognizing subtle countertransference can guard against extreme reactions and maintain an appropriate clinical focus. Discussion with a trusted colleague can be helpful.
Psychodynamic approaches consider managing countertransference to be a therapeutic intervention, even when psychotherapy is not explicitly being carried out. Strong emotional responses may reflect something that the patient needs the physician to experience, as the patient cannot bear to experience it himself. The patient needs to see—and learn from—the physician’s handling of unbearable (for the patient) feelings. This occurs at a level of unconscious communication and may be repeated over time. Although not discussed with the patient, a physician’s capacity for self-containment and provision of undisrupted, good medical care is in itself a psychotherapeutic accomplishment.
CASE › Based on Mr. A’s history of interpersonal conflicts and perceived persecution by coworkers, the FP consults with a psychotherapist colleague, who says Mr. A’s chronic mistrust and social isolation suggest he may have a severe identity disturbance and unspecified PD with paranoid and schizoid features. Because Mr. A refuses to see a therapist, his FP decides to focus on promoting small improvements in Mr. A’s interpersonal interactions and reducing absenteeism at work.
The FP validates Mr. A’s feelings (“it can be very stressful to constantly feel like others are at odds with you”) and tries to promote mentalizing (“I want to understand more about what you think regarding your work situation and your coworkers. Let’s try to look at this from all perspectives—maybe we can come up with some new ideas.”)
Despite wanting to help his patient, the FP feels uneasy and reluctant to engage with Mr. A, who likely evokes such feelings to keep others at a distance. The FP tactfully seeks to remain Mr. A’s ally without endorsing his distorted interpretation of events. Given Mr. A’s paranoid rejection of therapy, the FP refrains from making further such recommendations. The FP’s interventions, however, may help Mr. A warm to the idea of further help over time, and the FP’s supportive stance will help to ameliorate the patient’s distress. (For 2 additional examples of how FPs can use the strategies described in this article to help patients with PDs, see TABLE 2.)
CORRESPONDENCE
David Kealy, MSW, Psychotherapy Program, Department
of Psychiatry, University of British Columbia, #420-5950 University Boulevard, Vancouver, BC Canada V6T 1Z3; david.kealy@ubc.ca
› Evaluate a patient’s sense of identity and interpersonal relationships for clues of a personality disorder (PD). A
› Use validation, promote mentalization, and
manage countertransference to help patients with PDs. A
› Consider medications such as antidepressants or antipsychotics for patients with PDs, but only as adjuncts to psychotherapy, and only to target specific symptoms, such as impulsive aggression. B
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Bob A, age 48, comes to his family physician (FP) to ask for authorization for extended medical leave from his job as an electrician. He frequently misses days at work and complains of stress on the job, saying his coworkers look down on him and make cruel jokes at his expense. He reports having chronic interpersonal conflicts and no significant relationships with family members or friends. Mr. A refuses a referral to a psychiatrist because he fears he will be “locked up and forced to take medications.”
If Mr. A were your patient, how would you proceed?
Personality disorders (PDs) are patterns of inflexible and maladaptive personality traits and behaviors that cause subjective distress and significant social or occupational impairment.1 An individual with a PD tends to have a limited repertoire of responses to the rough-and-tumble of life, with coping mechanisms that often perpetuate difficulty and distress. Examples include distrust and suspiciousness of others’ motives (paranoid PD); disregard and violation of the rights of others (antisocial PD); instability in interpersonal relationships, self-image, and affect (borderline PD); and social inhibition, feelings of inadequacy, and hypersensitivity to negative evaluation (avoidant PD).1
FPs may view patients with PDs as “difficult patients” because of their frequent crises and the interpersonal problems they bring into the physician-patient relationship.2,3 Help, of course, can come in the way of a referral to a psychotherapist who specializes in treating PDs. But you can also make use of some evidence-based psychotherapy techniques to improve your patients’ lives and the quality of the physician-patient relationship. This article focuses on identifying and managing PDs in family practice, using practical strategies drawn from empirically supported therapies.
PDs are more common than you might suspect
The overall prevalence of PD in the community ranges from 4.4% to 14.8%, with no consistent pattern of sex differences.4 Between 31.4% and 45.5% of psychiatric outpatients and up to 24% of primary care patients likely meet criteria for at least one PD.5-7 PDs impede recovery from other mental disorders,8 increase the risk for suicide,9 and are associated with substance abuse, impulsivity, and violence.10,11 Personality pathology also is associated with greater incidence of serious medical illness12,13 and reduced social functioning.14 Not surprisingly, patients with PDs frequently use medical and social services.15
PDs tend to be underdiagnosed, perhaps partly because of concern about stigmatization, but also due to difficulties in identifying and classifying these disorders. Published in 2013, the American Psychiatric Association’s Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM-5) originally was to include a major revision of PDs—reflecting concern about the limitations of PD categories—but ultimately the existing categories were retained (TABLE 1).1 There is considerable overlap among PD categories; many patients meet the criteria for more than one PD, but it is unlikely that they actually suffer from several distinct PDs. Other patients—perhaps even the majority—are best diagnosed with “unspecified personality disorder” because they do not neatly fit into one of these categories.
Suspect your patient has a PD? Evaluate these 2 areas
Identifying patients who have PDs in primary care is useful for 2 reasons: to explore the option of specialty treatment for patients who may be amenable to it, and to improve management of the patient’s complaints in the primary care setting, including a smoother doctor-patient interaction. In either case, determining the specific DSM-5 diagnosis is less important than recognizing core personality impairment: an ingrained disturbance in one’s perceptions of self and others. This can be done by paying attention to how the patient adapts to life’s challenges and if he or she has problematic interpersonal tendencies, including difficulties in the doctor-patient relationship.
Unfortunately, assessing and diagnosing PDs in the primary care setting can be challenging. Limited time doesn’t allow for extensive, personality-focused interviews. Self-report screening tools are limited, because patients may underreport key interpersonal problems such as lack of empathy. Furthermore, very few patients seek help from their FP in addressing personality dysfunction; PDs typically are identified while investigating other complaints.
The most reliable and useful areas to evaluate in a patient you suspect may have a PD are identity (one’s sense of who one is and can be) and interpersonal relationships, including the capacity for empathy and intimacy.16,17 These should be considered longitudinally and in the context of the individual’s stage of development. For example, identity is generally less stable among adolescents compared to middle-aged adults.
A cohesive sense of identity allows one to embrace life’s tasks and challenges, to develop and strive toward personal goals, and to handle setbacks and disappointments. A person with a stable identity may develop a depressive reaction to difficult life circumstances, but with some assistance can generally bounce back and re-engage in his or her personal goals. By contrast, an individual with an unstable sense of self may feel chronically insecure and empty, with limited capacity to constructively deal with life’s ups and downs. Patients with borderline PD, for example, try to manage a fragmented identity by frantically clinging to others, while narcissistic patients tend to suppress a fragile sense of self by putting forth an arrogant and entitled attitude.
How does the patient interact with others? As is the case with identity, an individual’s capacity for interpersonal functioning is developed early in life, through interactions with primary caregivers. Mental maps of who we are and what we can expect from others are formed and reinforced in attachment relationships, such as those with our parents; traumatic attachments, including abuse or neglect by a caregiver or loved one, are strongly associated with PD.18,19 The resulting belief structures guide subsequent interpersonal functioning, and become interactively reinforced. For example, a person whose internal map of relationships includes others abandoning him might behave in a clingy manner, which may ultimately induce others to reject him, thus creating a self-fulfilling prophecy.
Distorted interpersonal expectations can impair a person’s capacity for sustained intimate connections (a troubled relationship history is characteristic of PDs) and limit empathic functioning.20 Other people’s actions may be interpreted according to the patient’s belief structures rather than with an open mind about the other person’s experience.
Focus on the physician-patient relationship
The interpersonal dysfunction of patients with PDs will often surface in the physician-patient relationship, serving as a clue to broader interpersonal dysfunction. An FP’s relatively innocuous oversight, for example, might be taken as proof of suspected incompetence in the eyes of a patient with paranoid or narcissistic tendencies. Or a patient with a recurrent complaint who repeatedly rejects the physician’s interventions probably oscillates between seeking and rejecting nurturance in other relationships, as well. A patient who tends to make sarcastic remarks regarding the doctor’s earnest efforts likely holds negative views of others and sabotages potentially positive interactions.
So what strategies are best for managing these types of scenarios?
Bringing up a potential diagnosis of PD may be a delicate matter for the FP; patients might experience this as a jarring diagnosis in the absence of a thorough psychiatric evaluation. If the FP decides to explore whether the patient is open to discussing the relationship between moods, behaviors, and personality features, he or she can begin this conversation by noting that, as with physical health, we all have our vulnerabilities, and that these vulnerabilities may be strengthened through specialist consultation and support. In this way, the patient can view a referral as an opportunity to explore herself with professional support. If a psychiatrist or psychotherapist colleague does become involved, it is important to clarify the roles of treatment providers and to communicate with one another, should difficulties arise.
Evidence supports 2 forms of psychotherapy
Treatment for PDs has seen considerable growth over the past decade, largely due to research on therapies that target the troubling self-injurious and suicidal features of borderline PD. Considerable evidence shows that specialized psychotherapy can significantly reduce suffering and improve functioning among these patients. The 2 major evidence-based treatments for patients with borderline PD are dialectical behavior therapy (DBT) and psychodynamic therapy.
DBT is an intensive cognitive-behavioral approach that teaches patients how to regulate their emotions and develop an accepting, mindful attitude toward their mental experience.21 Several randomized controlled trials (RCTs) have demonstrated the effectiveness of DBT in reducing hospitalizations and self-injurious and suicidal behavior in patients with borderline PD.22
Psychodynamic therapy, which focuses on helping patients discover how unconscious conflicts influence their present moods and behaviors, has also been validated by multiple RCTs for patients with borderline PD.23-25 Like DBT, empirically supported psychodynamic therapy tends to be structured, long-term (>12 months), and often intensively delivered in multiple sessions per week. However, a recent study found that a less-intensive, general psychodynamic therapy, along with occasional medication management, was equivalent to intensive DBT.26
Although the research has focused primarily on borderline PD, these approaches can be applied to other PDs. These therapies focus on understanding one’s emotional and behavioral patterns, developing a healthy self-concept, and improving interpersonal relationships—areas that are relevant treatment targets across all PD types.
Indeed, studies of day treatment programs that explicitly welcome patients with a range of PD types have had promising findings.27 Day treatment involves an intensive array of therapies, mostly in a group format; patients work together to support and embolden one another to make positive changes. Unfortunately, FPs may be challenged to find appropriate services for patients who are amenable to psychotherapy; public mental health resources tend to lag far behind best practices in the case of PD.
Medication might improve symptoms, not personality deficits
Most research on pharmacotherapy for PDs has focused on borderline PD; findings have been mixed and fairly limited.28 Medication cannot address underlying identity and relational deficits, and will not result in remission of PD. Nonetheless, judicious, circumscribed use of medications to target specific symptoms may be helpful for some patients. Selective serotonin reuptake inhibitors can reduce anger and impulsive aggression in patients with borderline PD.28,29
Atypical antipsychotics may help reduce impulsive aggression or transient psychotic symptoms.28-30 For example, olanzapine and aripiprazole can reduce anxiety, anger/aggression, paranoia, and interpersonal sensitivity in borderline PD.31,32 Mood stabilizers such as valproate, lamotrigine, and topiramate may also help some borderline patients, although they do so by reducing impulsivity and aggression rather than improving core unstable identity and affect.28,29
Carefully obtained informed consent is necessary because of the danger of adverse effects with many of these medications; for example, antipsychotics have been associated with metabolic syndrome and weight gain that can threaten a patient’s already fragile self-image.33 Polypharmacy is also a potential problem: Well-intentioned physicians may be prompted to offer multiple medications in response to patients’ unremitting complaints of distress, when a psychotherapeutic approach may need to be the primary treatment. The bottom line is that medications do not resolve personality dysfunction, and are best used symptomatically as adjuncts to psychotherapy.28,30
Steps you can take during the office visit
Although it is not feasible for most FPs to provide comprehensive treatment for PD, key elements from specialized therapies can be integrated into your management of these patients. Steps you can take include using validation, promoting mentalization, and managing countertransference.
Validation, which is a component of DBT, is providing the expressed acknowledgement that the patient is entitled to her feelings. This is not the same as agreeing with a position the patient has taken on an issue, but rather conveying the sense that one sees how the patient might feel the way she does. A study of women with borderline PD and substance abuse found a validation intervention by itself was significantly helpful.34 Validation can contribute to a “corrective emotional experience.” For instance, your supportive acknowledgement of a patient with a history of abuse or neglect may counter the patient’s expectation of being invalidated, and over time this can reduce the patient’s defensive rigidity.
Mentalization. Psychodynamic treatment involves a similar tack; clinicians empathize with the patient’s emotional state while also demonstrating a degree of separateness from the emotion.23-25 This promotes mentalization in the patient—the ability to contemplate one’s own and others’ subjective mental states.18 Mentalization is often impaired in PD patients, who presume to “know” what others are thinking. A patient, for instance, “just knows” that her friend secretly hates her, based on a vaguely worded text message.
You can help patients with mentalization by taking an inquisitive “not knowing” stance and by emphasizing a collaborative and reflective approach toward a given problem—to examine the issue together, from all sides. You can point out that while a patient is entitled to feel whatever he is feeling, it may not be in his best interest to act on the feelings without adequately considering the potential consequences of the action. This helps the patient to distinguish thoughts, feelings, and impulses from behavior. It also teaches the value of anticipatory thinking, impulse control, and affect regulation.
Countertransference. Managing your emotional reactions to a patient with PD is a well-documented challenge.35 Your feelings about the patient, known as countertransference, can range from considerable concern and sympathy to severe frustration, bewilderment, and frank hostility. A common reaction is the sense that one must “do something” to respond to the patient’s emotional distress or interpersonal pressure. This may trigger an impulse to give advice or offer tests or medications despite knowing that these are unlikely to be helpful. A more useful response may be to tolerate such feelings and listen empathically to the patient’s frustration. Recognizing subtle countertransference can guard against extreme reactions and maintain an appropriate clinical focus. Discussion with a trusted colleague can be helpful.
Psychodynamic approaches consider managing countertransference to be a therapeutic intervention, even when psychotherapy is not explicitly being carried out. Strong emotional responses may reflect something that the patient needs the physician to experience, as the patient cannot bear to experience it himself. The patient needs to see—and learn from—the physician’s handling of unbearable (for the patient) feelings. This occurs at a level of unconscious communication and may be repeated over time. Although not discussed with the patient, a physician’s capacity for self-containment and provision of undisrupted, good medical care is in itself a psychotherapeutic accomplishment.
CASE › Based on Mr. A’s history of interpersonal conflicts and perceived persecution by coworkers, the FP consults with a psychotherapist colleague, who says Mr. A’s chronic mistrust and social isolation suggest he may have a severe identity disturbance and unspecified PD with paranoid and schizoid features. Because Mr. A refuses to see a therapist, his FP decides to focus on promoting small improvements in Mr. A’s interpersonal interactions and reducing absenteeism at work.
The FP validates Mr. A’s feelings (“it can be very stressful to constantly feel like others are at odds with you”) and tries to promote mentalizing (“I want to understand more about what you think regarding your work situation and your coworkers. Let’s try to look at this from all perspectives—maybe we can come up with some new ideas.”)
Despite wanting to help his patient, the FP feels uneasy and reluctant to engage with Mr. A, who likely evokes such feelings to keep others at a distance. The FP tactfully seeks to remain Mr. A’s ally without endorsing his distorted interpretation of events. Given Mr. A’s paranoid rejection of therapy, the FP refrains from making further such recommendations. The FP’s interventions, however, may help Mr. A warm to the idea of further help over time, and the FP’s supportive stance will help to ameliorate the patient’s distress. (For 2 additional examples of how FPs can use the strategies described in this article to help patients with PDs, see TABLE 2.)
CORRESPONDENCE
David Kealy, MSW, Psychotherapy Program, Department
of Psychiatry, University of British Columbia, #420-5950 University Boulevard, Vancouver, BC Canada V6T 1Z3; david.kealy@ubc.ca
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
2. Hahn SR, Thompson KS, Wills TA, et al. The difficult doctor-patient relationship: somatization, personality and psychopathology. J Clin Epidemiol. 1994:47:647-657.
3. Schafer S, Nowlis DP. Personality disorders among difficult patients. Arch Fam Med. 1998;7:126-129.
4. Paris J. Estimating the prevalence of personality disorders in the community. J Pers Disord. 2010;24:405-411.
5. Newton-Howes G, Tyrer P, Anagnostakis K, et al. The prevalence of personality disorder, its comorbidity with mental state disorders, and its clinical significance in community mental health teams. Soc Psychiatry Psychiatr Epidemiol. 2010;45:453-460.
6. Zimmerman M, Rothschild L, Chelminski I. The prevalence of DSM-IV personality disorders in psychiatric outpatients. Am J Psychiatry. 2005;162:1911-1918.
7. Moran P, Jenkins R, Tylee A, et al. The prevalence of personality disorder among UK primary care attenders. Acta Psychiatr Scand. 2000;102:52-57.
8. Newton-Howes G, Tyrer P, Johnson T. Personality disorder and the outcome of depression: Meta-analysis of published studies. Br J Psychiatry. 2006;188:13-20.
9. Blasco-Fontecilla H, Baca-Garcia E, Dervic K, et al. Severity of personality disorders and suicide attempt. Acta Psychiatr Scand. 2009;119:149-155.
10. Colpaert K, Vanderplasschen W, De Maeyer J, et al. Prevalence and determinants of personality disorders in a clinical sample of alcohol-, drug-, and dual-dependent patients. Subst Use Misuse. 2012;47:649-661.
11. Yu R, Geddes JR, Fazel S. Personality disorders, violence, and antisocial behavior: A systematic review and meta-regression analysis. J Pers Disord. 2012;26:775-792.
12. Frankenburg FR, Zanarini MC. The association between borderline personality disorder and chronic medical illnesses, poor health-related lifestyle choices, and costly forms of health care utilization. J Clin Psychiatry. 2004;65:1660-1665.
13. Lee HB, Bienvenu OJ, Cho SJ, et al. Personality disorders and traits as predictors of incident cardiovascular disease: Findings from the 23-year follow-up of the Baltimore ECA Study. Psychosomatics. 2010;51:289-296.
14. Skodol AE, Gunderson JG, McGlashan TH, et al. Functional impairment in patients with schizotypal, borderline, avoidant, or obsessive-compulsive personality disorder. Am J Psychiatry. 2002;159:276-283.
15. Bender DS, Dolan RT, Skodol AE, et al. Treatment utilization by patients with personality disorders. Am J Psychiatry. 2001;158:295-302.
16. Livesley WJ. An empirically-based classification of personality disorder. J Pers Disord. 2011;25:397-420.
17. Bender DS, Morey LC, Skodol AE. Toward a model for assessing personality functioning in DSM-5, part I: a review of theory and methods. J Pers Assess. 2011;93:332-346.
18. Fonagy P, Gergely G, Jurist EL, et al. Affect Regulation, Mentalization, and the Development of the Self. New York, NY: Other Press; 2002.
19. Yen S, Shea MT, Battle CL, et al. Traumatic exposure and posttraumatic stress disorder in borderline, schizotypal, avoidant, and obsessive-compulsive personality disorders: findings from the collaborative longitudinal personality disorders study. J Nerv Ment Dis. 2002;190:510-518.
20. Morey LC, Stagner BH. Narcissistic pathology as core personality dysfunction: comparing DSM-IV and the DSM-5 proposal for narcissistic personality disorder. J Clin Psychol. 2012;68:908-921.
21. Lynch TR, Chapman AL, Rosenthal MZ, et al. Mechanisms of change in dialectical behaviour therapy: theoretical and empirical observations. J Clin Psychol. 2006;62:459-480.
22. Kliem S, Kröger C, Kosfelder J. Dialectical behavior therapy for borderline personality disorder: a meta-analysis using mixed-effects modeling. J Consult Clin Psychol. 2010;78:936-951.
23. Clarkin JF, Levy KN, Lenzenweger MF, et al. Evaluating three treatments for borderline personality disorder: a multiwave study. Am J Psychiatry. 2007;164:922-928.
24. Gregory RJ, DeLucia-Deranja E, Mogle JA. Dynamic deconstructive psychotherapy versus optimized community care for borderline personality disorder co-occurring with alcohol use disorders: a 30-month follow-up. J Nerv Ment Dis. 2010;198:292-298.
25. Bateman A, Fonagy P. Randomized controlled trial of outpatient mentalization-based treatment versus structured clinical management for borderline personality disorder. Am J Psychiatry. 2009;166:1355-1364.
26. McMain SF, Links PS, Gnam WH, et al. A randomized trial of dialectical behavior therapy versus general psychiatric management for borderline personality disorder. Am J Psychiatry. 2009;166:1365-1374.
27. Ogrodniczuk JS, Piper WE. Day treatment for personality disorders: a review of research findings. Harv Rev Psychiatry. 2001;9:105-117.
28. Paris J. Pharmacological treatments for personality disorders. Int Rev Psychiatry. 2011;23:303-309.
29. Ripoll LH, Triebwasser J, Siever LJ. Evidence-based pharmacotherapy for personality disorders. Int J Neuropsychopharmacol. 2011;14:1257-1288.
30. Steinberg PI. The use of low-dose neuroleptics in the treatment of patients with severe personality disorder: An adjunct to psychotherapy. BCMJ. 2007;49:306-310.
31. Zanarini MC, Frankenburg FR. Olanzapine treatment of female borderline personality disorder patients: a double-blind, placebo controlled pilot study. J Clin Psychiatry. 2001;62:849-854.
32. Nickel MK, Loew TH, Pedrosa Gil F. Aripiprazole in treatment of borderline patients, part II: an 18-month follow up. Psychopharmacology (Berl). 2007;191:1023-1026.
33. Silk KR. The process of managing medications in patients with borderline personality disorder. J Psychiatr Pract. 2011;17:311-319.
34. Linehan MM, Dimeff LA, Reynolds SK, et al. Dialectal behavior therapy versus comprehensive validation therapy plus 12-step for the treatment of opioid dependent women meeting criteria for borderline personality disorder. Drug Alcohol Depend. 2002;67:13-26.
35. Rossberg JI, Karterud S, Pedersen G, et al. An empirical study of countertransference reactions toward patients with personality disorders. Compr Psychiatry. 2007;48:225-230.
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association; 2013.
2. Hahn SR, Thompson KS, Wills TA, et al. The difficult doctor-patient relationship: somatization, personality and psychopathology. J Clin Epidemiol. 1994:47:647-657.
3. Schafer S, Nowlis DP. Personality disorders among difficult patients. Arch Fam Med. 1998;7:126-129.
4. Paris J. Estimating the prevalence of personality disorders in the community. J Pers Disord. 2010;24:405-411.
5. Newton-Howes G, Tyrer P, Anagnostakis K, et al. The prevalence of personality disorder, its comorbidity with mental state disorders, and its clinical significance in community mental health teams. Soc Psychiatry Psychiatr Epidemiol. 2010;45:453-460.
6. Zimmerman M, Rothschild L, Chelminski I. The prevalence of DSM-IV personality disorders in psychiatric outpatients. Am J Psychiatry. 2005;162:1911-1918.
7. Moran P, Jenkins R, Tylee A, et al. The prevalence of personality disorder among UK primary care attenders. Acta Psychiatr Scand. 2000;102:52-57.
8. Newton-Howes G, Tyrer P, Johnson T. Personality disorder and the outcome of depression: Meta-analysis of published studies. Br J Psychiatry. 2006;188:13-20.
9. Blasco-Fontecilla H, Baca-Garcia E, Dervic K, et al. Severity of personality disorders and suicide attempt. Acta Psychiatr Scand. 2009;119:149-155.
10. Colpaert K, Vanderplasschen W, De Maeyer J, et al. Prevalence and determinants of personality disorders in a clinical sample of alcohol-, drug-, and dual-dependent patients. Subst Use Misuse. 2012;47:649-661.
11. Yu R, Geddes JR, Fazel S. Personality disorders, violence, and antisocial behavior: A systematic review and meta-regression analysis. J Pers Disord. 2012;26:775-792.
12. Frankenburg FR, Zanarini MC. The association between borderline personality disorder and chronic medical illnesses, poor health-related lifestyle choices, and costly forms of health care utilization. J Clin Psychiatry. 2004;65:1660-1665.
13. Lee HB, Bienvenu OJ, Cho SJ, et al. Personality disorders and traits as predictors of incident cardiovascular disease: Findings from the 23-year follow-up of the Baltimore ECA Study. Psychosomatics. 2010;51:289-296.
14. Skodol AE, Gunderson JG, McGlashan TH, et al. Functional impairment in patients with schizotypal, borderline, avoidant, or obsessive-compulsive personality disorder. Am J Psychiatry. 2002;159:276-283.
15. Bender DS, Dolan RT, Skodol AE, et al. Treatment utilization by patients with personality disorders. Am J Psychiatry. 2001;158:295-302.
16. Livesley WJ. An empirically-based classification of personality disorder. J Pers Disord. 2011;25:397-420.
17. Bender DS, Morey LC, Skodol AE. Toward a model for assessing personality functioning in DSM-5, part I: a review of theory and methods. J Pers Assess. 2011;93:332-346.
18. Fonagy P, Gergely G, Jurist EL, et al. Affect Regulation, Mentalization, and the Development of the Self. New York, NY: Other Press; 2002.
19. Yen S, Shea MT, Battle CL, et al. Traumatic exposure and posttraumatic stress disorder in borderline, schizotypal, avoidant, and obsessive-compulsive personality disorders: findings from the collaborative longitudinal personality disorders study. J Nerv Ment Dis. 2002;190:510-518.
20. Morey LC, Stagner BH. Narcissistic pathology as core personality dysfunction: comparing DSM-IV and the DSM-5 proposal for narcissistic personality disorder. J Clin Psychol. 2012;68:908-921.
21. Lynch TR, Chapman AL, Rosenthal MZ, et al. Mechanisms of change in dialectical behaviour therapy: theoretical and empirical observations. J Clin Psychol. 2006;62:459-480.
22. Kliem S, Kröger C, Kosfelder J. Dialectical behavior therapy for borderline personality disorder: a meta-analysis using mixed-effects modeling. J Consult Clin Psychol. 2010;78:936-951.
23. Clarkin JF, Levy KN, Lenzenweger MF, et al. Evaluating three treatments for borderline personality disorder: a multiwave study. Am J Psychiatry. 2007;164:922-928.
24. Gregory RJ, DeLucia-Deranja E, Mogle JA. Dynamic deconstructive psychotherapy versus optimized community care for borderline personality disorder co-occurring with alcohol use disorders: a 30-month follow-up. J Nerv Ment Dis. 2010;198:292-298.
25. Bateman A, Fonagy P. Randomized controlled trial of outpatient mentalization-based treatment versus structured clinical management for borderline personality disorder. Am J Psychiatry. 2009;166:1355-1364.
26. McMain SF, Links PS, Gnam WH, et al. A randomized trial of dialectical behavior therapy versus general psychiatric management for borderline personality disorder. Am J Psychiatry. 2009;166:1365-1374.
27. Ogrodniczuk JS, Piper WE. Day treatment for personality disorders: a review of research findings. Harv Rev Psychiatry. 2001;9:105-117.
28. Paris J. Pharmacological treatments for personality disorders. Int Rev Psychiatry. 2011;23:303-309.
29. Ripoll LH, Triebwasser J, Siever LJ. Evidence-based pharmacotherapy for personality disorders. Int J Neuropsychopharmacol. 2011;14:1257-1288.
30. Steinberg PI. The use of low-dose neuroleptics in the treatment of patients with severe personality disorder: An adjunct to psychotherapy. BCMJ. 2007;49:306-310.
31. Zanarini MC, Frankenburg FR. Olanzapine treatment of female borderline personality disorder patients: a double-blind, placebo controlled pilot study. J Clin Psychiatry. 2001;62:849-854.
32. Nickel MK, Loew TH, Pedrosa Gil F. Aripiprazole in treatment of borderline patients, part II: an 18-month follow up. Psychopharmacology (Berl). 2007;191:1023-1026.
33. Silk KR. The process of managing medications in patients with borderline personality disorder. J Psychiatr Pract. 2011;17:311-319.
34. Linehan MM, Dimeff LA, Reynolds SK, et al. Dialectal behavior therapy versus comprehensive validation therapy plus 12-step for the treatment of opioid dependent women meeting criteria for borderline personality disorder. Drug Alcohol Depend. 2002;67:13-26.
35. Rossberg JI, Karterud S, Pedersen G, et al. An empirical study of countertransference reactions toward patients with personality disorders. Compr Psychiatry. 2007;48:225-230.
How to do a 3-minute diabetic foot exam
› Screen for lower
extremity complications at every visit for all patients with a suspected or confirmed diagnosis of diabetes. A
› Consider implementing a risk-based referral system to connect primary screening with a specialist's care. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Foot ulcers and other lower-limb complications secondary to diabetes are common, complex, costly, and associated with increased morbidity and mortality.1-6 Unfortunately, patients often have difficulty recognizing the heightened risk status that accompanies the diagnosis of diabetes, particularly the substantial risk for lower limb complications.7 In addition, loss of protective sensation (LOPS) can render patients unable to recognize damage to their lower extremities, thus creating a cycle of tissue damage and other foot complications. Strong evidence suggests that consistent provision of foot-care services and preventive care can reduce amputations among patients with diabetes.7-9 However, routine foot examination and rapid risk stratification is often difficult to incorporate into busy primary care settings. Data suggest that the diabetic foot is adequately evaluated only 12% to 20% of the time.10
In response to the need for more consistent foot exams, an American Diabetes Association (ADA) task force lead by 2 of the authors of this article (AB and DA) created the Comprehensive Foot Examination and Risk Assessment.5 This set the standard for the detailed investigation of lower limb pathology by a specialist, but was not well suited for other practice settings, including primary care. One reason is that it would be difficult to complete the comprehensive examination during a typical 15-minute primary care office visit. In addition, certain examination parameters require the use of neurologic and vascular assessment equipment and training not available in all health care settings.11
With these thoughts in mind, we set out to develop an exam that could be done by a wide range of health care providers—one that takes substantially less time to complete than a comprehensive exam and eliminates common barriers to frequent assessment. The exam, which we’ll describe here, consists of 3 components: taking a patient history, performing a physical exam, and providing patient education. And best of all, it should only take 3 minutes.
The patient history (1 minute)
Patients may present with concerns about their feet, but may not be able to differentiate between benign and threatening symptoms. A thorough medical history can identify factors that may increase patients’ risk of developing lower-limb complications. Reviewing the patient’s medical history also can help guide the physical exam.
Review the patient’s diabetic history, blood glucose control, and previous diabetic complications. Ask patients about their history of peripheral vascular disease, quality of peripheral protective sensation, and previous lower-limb interventions and operations (TABLE 15,12). Patients with diabetes and suboptimal glycemic control have an increased risk for LOPS, chronic and recalcitrant ulcers, and wound infections.2 Additionally, patients with diabetes and a previous lower extremity amputation are at high risk for reulceration.5,12 Lastly, nicotine use and smoking are common pathogenic risk factors that contribute to peripheral artery disease (PAD).13

Physical examination (1 minute)
Careful inspection of the feet should be performed at every visit for patients with confirmed or suspected diabetes. Because up to 50% of patients with significant sensory loss due to neuropathy may be completely asymptomatic,14 failing to search for early signs of infection (FIGURE 1), skin breakdown, ulcer formation (FIGURE 2), skin temperature changes, and inadequate vascular perfusion may allow complications to develop.5 TABLE 25,15,16 outlines the essential components—dermatologic, neurologic, musculoskeletal, and vascular—of a rapid lower limb physical exam.

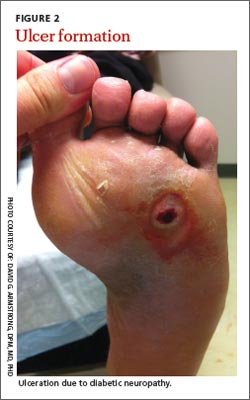
The dermatologic exam. This serves as a barometer for early intervention, and often results in a limb-saving referral to a specialist. It should begin with a global inspection for discolorations, calluses, wounds, fissures, macerations, nail dystrophy, or paronychia.5 Skin discoloration or loss of hair growth may be the first signs of vascular insufficiency, while calluses and hypertrophic skin often are precursors to ulcers.5,17-19 Inspection of the toes should include a search for fungal, ingrown, or elongated nails. Carefully examine the areas between the toes, where deeper lesions may go unnoticed.5
The neurologic exam. Without protective sensation, patients with neuropathy are at a heightened risk of unrecognized injury and are unlikely to mention their deformities to medical staff.20-23 Consequently, skin deterioration may unknowingly progress to ulceration that requires extensive medical intervention or amputation.
Neuropathic LOPS is easily detectable, yet it is linked to at least 75% of all nontraumatic diabetic amputations.20-23 Adiminished vibratory perception threshold (VPT) is one of the earliest indicators of neuropathic LOPS and is the best predictor of long-term lower extremity complications.1,24,25 However, VPT devices are expensive and time-consuming to operate, and they require training to ensure proper use. The Semmes-Weinstein monofilament is a well-documented alternative to VPT for predicting ulcer risk26-28 and has long been advocated as an essential component of a thorough foot exam.5 The 128 Hz tuning fork is another regularly used alternative.5 However, physicians would need to purchase one of these devices and receive training on how to use it, and, in the case of the monofilament, to regularly stock replacements to maintain accurate results.16
The Ipswich Touch Test (IpTT) is an alternative neurologic test that requires only the physician’s index finger. During the IpTT, the physician instructs the patient to close his or her eyes while the physician lightly rests his or her finger on each of the patient’s first, third, and fifth toes for 1 to 2 seconds (FIGURE 3). Patients are instructed to respond with a “yes” when they feel the physician’s touch. In a head-to-head trial, diagnostic results of the IpTT directly paralleled those of the monofilament in detecting LOPS; IpTT was also equally sensitive and specific (k=.88, indicating almost perfect agreement; P<.0001).29 The IpTT’s use of only 6 palpation points, constant availability, and accuracy make it a first-line neurologic test for rapidly screening the feet of a patient with diabetes.
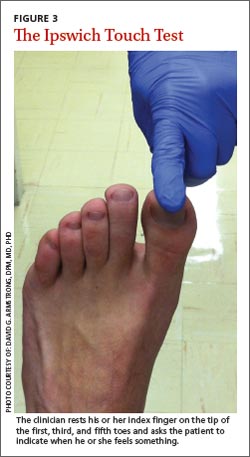
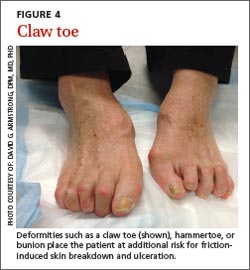
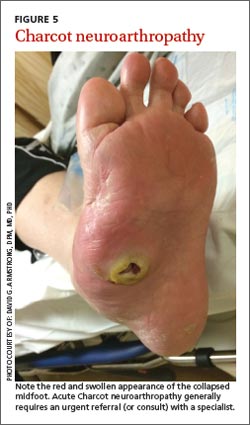
Neuromuscular/musculoskeletal exam. Neuromuscular disturbances, such as a reduction in the strength of dorsiflexion and plantar flexion, may indicate a complicated neurologic compromise.5 In addition to being aesthetically problematic, musculoskeletal deformities such as a hammer toe, claw toe (FIGURE 4), or bunion can cause significant pain and/or gait disturbance, and can increase patients’ risk for ulceration.30 These deformities also may compromise patients’ general health and grossly escalate their risk of falls and resultant injuries.5,31 Therefore, patients who present with previously unreported musculoskeletal deformities should be referred to a specialist.31
Also screen patients for Charcot neuroarthropathy (FIGURE 5), a devastating complication that classically presents as a hot, red, swollen foot; the redness resolves upon elevation.32 Charcot neuroarthropathy is hypothesized to be a dysregulation of normal bone metabolism typically occurring secondary to diabetic neuropathy and repetitive minor trauma.33,34 This dysregulation leads to joint instability and disorganization of normal midfoot bone architecture.31,32 Charcot neuroarthropathy is an urgent pathology that requires management by a foot specialist.35
Vascular exam. PAD is particularly common in patients with diabetes and contributes to the development of impaired healing in up to half of foot ulcers.13,18,36-39 Bilateral femoral, popliteal, posterior tibial, or dorsalis pedis pulses should be assessed by palpation; a diminished or absent pulse is a key indicator of vascular compromise.40,41 An integrated care approach between foot specialists and vascular surgeons results in optimal treatment.

Patient education (1 minute)
It is imperative to include patients in their treatment process to reduce the likelihood of complications and, ultimately, decrease the incidence of amputations.12,42 Patient education improves patients’ self-reported home care behaviors, even at the most fundamental levels.43,44 TABLE 35,15,45 lists topics to cover during patient education.
Patients’ lack of understanding about self-care for diabetes is a common barrier to prevention.23 El-Nahas et al46 found a lack of appropriate education regarding diabetes was a factor in more than 90% of recurrent ulcers, which emphasizes the need for repeated education for at-risk patients.47,48 Involve all levels of medical staff in the effort to educate patients on the importance of foot screenings, both at home and in-office. Even with proper patient education, many patients may be in various stages of coping with this all-consuming yet frequently asymptomatic condition, which makes the need for repeated patient education even more critical.

Who to refer, and when
After completing the 3-minute foot exam, create a treatment and follow-up plan, focusing on the need for referral to a specialist. TABLE 4 outlines suggested indications, priorities, and timelines for referral based on ADA guidelines.5 It incorporates the ADA’s patient risk categories (very low, low, moderate, and high risk) and also provides a recommended frequency for patient follow-ups.
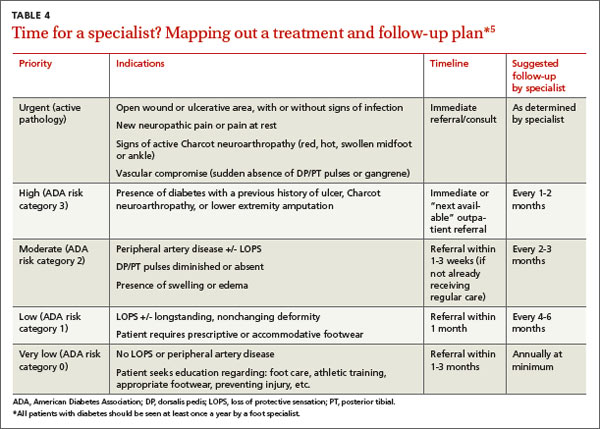
Care for patients with lower extremity complications of diabetes mellitus is time-consuming and expensive. The brief exam described here can help you to rapidly identify patients at risk for these complications and prompt you to provide timely referrals to appropriate specialists.
CORRESPONDENCE
David G. Armstrong, DPM, MD, PhD, Professor, Department of Surgery, Director, Southern Arizona Limb Salvage Alliance (SALSA), 1501 N. Campbell Avenue, Tucson, AZ 85724-5072; armstrong@usa.net
1. Shearer A, Scuffham P, Gordois A, et al. Predicted costs and outcomes from reduced vibration detection in people with diabetes in the U.S. Diabetes Care. 2003;26:2305-2310.
2. Apelqvist J, Larsson J. What is the most effective way to reduce incidence of amputation in the diabetic foot? Diabetes Metab Res Rev. 2000;16 suppl 1:S75-S83.
3. Armstrong DG, Kanda VA, Lavery LA, et al. Mind the gap: disparity between research funding and costs of care for diabetic foot ulcers. Diabetes Care. 2013;36:1815-1817.
4. Driver VR, Fabbi M, Lavery LA, et al. The costs of diabetic foot: the economic case for the limb salvage team. J Vasc Surg. 2010;52(3 suppl):17S-22S.
5. Boulton AJ, Armstrong DG, Albert SF, et al; American Diabetes Association; American Association of Clinical Endocrinologists. Comprehensive foot examination and risk assessment: a report of the Task Force of the Foot Care Interest Group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care. 2008;31:1679-1685.
6. American Diabetes Association. Standards of medical care in diabetes—2014. Diabetes Care. 2014;37 suppl 1:S14-S80.
7. Sloan FA, Feinglos MN, Grossman DS. Receipt of care and reduction of lower extremity amputations in a nationally representative sample of U.S. Elderly. Health Serv Res. 2010;45(6 pt 1):1740-1762.
8. Carls GS, Gibson TB, Driver VR, et al. The economic value of specialized lower-extremity medical care by podiatric physicians in the treatment of diabetic foot ulcers. J Am Podiatr Med Assoc. 2011;101:93-115.
9. McCabe CJ, Stevenson RC, Dolan AM. Evaluation of a diabetic foot screening and protection programme. Diabet Med. 1998;15:80-84.
10. Bailey TS, Yu HM, Rayfield EJ. Patterns of foot examination in a diabetes clinic. Am J Med. 1985;78:371-374.
11. Chin MH, Cook S, Jin L, et al. Barriers to providing diabetes care in community health centers. Diabetes Care. 2001;24:268-274.
12. Abbott CA, Carrington AL, Ashe H, et al; North-West Diabetes Foot Care Study. The North-West Diabetes Foot Care Study: incidence of, and risk factors for, new diabetic foot ulceration in a community-based patient cohort. Diabet Med. 2002;19:377-384.
13. Fowkes FG, Rudan D, Rudan I, et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet. 2013;382:1329-1340.
14. Boulton A, Vinik AI, Arezzo JC, et al; American Diabetes Association. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes. 2005;28:956-962.
15. Singh N, Armstrong DG, Lipsky BA. Preventing foot ulcers in patients with diabetes. JAMA. 2005;293:217-228.
16. Pham H, Armstrong DG, Harvey C, et al. Screening techniques to identify people at high risk for diabetic foot ulceration: a prospective multicenter trial. Diabetes Care. 2000;23:606-611.
17. Marso SP, Hiatt WR. Peripheral arterial disease in patients with diabetes. J Am Coll Cardiol. 2006;47:921-929.
18. American Diabetes Association. Peripheral arterial disease in people with diabetes. JAPMA. 2005;95:309-319.
19. Pataky Z, Golay A, Faravel L, et al. The impact of callosities on the magnitude and duration of plantar pressure in patients with diabetes mellitus. A callus may cause 18,600 kilograms of excess plantar pressure per day. Diabetes Metab. 2002;28: 356-361.
20. Holzer SE, Camerota A, Martens L, et al. Costs and duration of care for lower extremity ulcers in patients with diabetes. Clin Ther. 1998;20:169-181.
21. Boulton AJ, Gries FA, Jervell JA. Guidelines for the diagnosis and outpatient management of diabetic peripheral neuropathy. Diabet Med. 1998;15:508-514.
22. Malay DS, Margolis DJ, Hoffstad OJ, et al. The incidence and risks of failure to heal after lower extremity amputation for the treatment of diabetic neuropathic foot ulcer. J Foot Ankle Surg. 2006;45:366-374.
23. van Houtum WH. Barriers to implementing foot care. Diabetes Metab Res Rev. 2012;28 suppl 1:112-115.
24. Jayaprakash P, Bhansali A, Bhansali S, et al. Validation of bedside methods in evaluation of diabetic peripheral neuropathy. Indian J Med Res. 2011;133:645-649.
25. Young MJ, Breddy JL, Veves A, et al. The prediction of diabetic neuropathic foot ulceration using vibration perception thresholds. A prospective study. Diabetes Care. 1994;17:557-560.
26. Leese GP, Reid F, Green V, et al. Stratification of foot ulcer risk in patients with diabetes: a population-based study. Int J Clin Pract. 2006;60:541-545.
27. Adler AI, Boyko EJ, Ahroni JH, et al. Risk factors for diabetic peripheral sensory neuropathy. Results of the Seattle Prospective Diabetic Foot Study. Diabetes Care. 1997;20:1162-1167.
28. Armstrong DG, Lavery LA, Vela SA, et al. Choosing a practical screening instrument to identify patients at risk for diabetic foot ulceration. Arch Intern Med. 1998;158:289-292.
29. Rayman G, Vas PR, Baker N, et al. The Ipswich Touch Test: a simple and novel method to identify inpatients with diabetes at risk of foot ulceration. Diabetes Care. 2011;34:1517-1518.
30. Lavery LA, Armstrong DG, Vela SA, et al. Practical criteria for screening patients at high risk for diabetic foot ulceration. Arch Intern Med. 1998;158:157-162.
31. Frykberg RG, Zgonis T, Armstrong DG, et al; American College of Foot and Ankle Surgeons. Diabetic foot disorders. A clinical practice guideline (2006 revision). J Foot Ankle Surg. 2006;45(5 suppl):S1-S66.
32. Nielson DL, Armstrong DG. The natural history of Charcot’s neuroarthropathy. Clin Podiatr Med Surg. 2008;25:53-62,vi.
33. Jeffcoate W, Lima J, Nobrega L. The Charcot foot. Diabet Med. 2000;17:253-258.
34. Blume PA, Sumpio B, Schmidt B, et al. Charcot neuroarthropathy of the foot and ankle: diagnosis and management strategies. Clin Podiatr Med Surg. 2014;31:151-172.
35. Petrova NL, Edmonds ME. Medical management of Charcot arthropathy. Diabetes Obes Metab. 2012;15:193-197.
36. Prompers L, Huijberts M, Apelqvist J, et al. Delivery of care to diabetic patients with foot ulcers in daily practice: results of the Eurodiale Study, a prospective cohort study. Diabet Med. 2008;25:700-707.
37. Armstrong DG, Bharara M, White M, et al. The impact and outcomes of establishing an integrated interdisciplinary surgical team to care for the diabetic foot. Diabetes Metab Res Rev. 2012;28:514-518.
38. Rogers LC, Andros G, Caporusso J, et al. Toe and flow: essential components and structure of the amputation prevention team. J Vasc Surg. 2010;52:23S-27S.
39. Mills JL Sr, Conte MS, Armstrong DG, et al; Society for Vascular Surgery Lower Extremity Guidelines Committee. The Society for Vascular Surgery Lower Extremity Threatened Limb Classification System: risk stratification based on wound, ischemia, and foot infection (WIfI). J Vasc Surg. 2014;59:220-34.e1-2.
40. Khan NA, Rahim SA, Anand SS, et al. Does the clinical examination predict lower extremity peripheral arterial disease? JAMA. 2006;295:536-546.
41. Sumpio BE, Lee T, Blume PA. Vascular evaluation and arterial reconstruction of the diabetic foot. Clin Podiatr Med Surg. 2003;20:689-708.
42. Dorresteijn JAN, Valk GD. Patient education for preventing diabetic foot ulceration. Diabetes Metab Res Rev. 2012;28 Suppl 1:101-106.
43. Lincoln NB, Radford KA, Game FL, et al. Education for secondary prevention of foot ulcers in people with diabetes: a randomised controlled trial. Diabetologia. 2008;51:1954-1961.
44. McMurray SD, Johnson G, Davis S, et al. Diabetes education and care management significantly improve patient outcomes in the dialysis unit. Am J Kidney Dis. 2002;40:566-575.
45. Armstrong DG, Lavery LA. Diabetic foot ulcers: prevention, diagnosis and classification. Am Fam Physician. 1998;57:1325-1332,1337-1338.
46. El-Nahas MR, Gawish HMS, Tarshoby MM, et al. The prevalence of risk factors for foot ulceration in Egyptian diabetic patients. Practical Diabetes Int. 2008;25:362-366.
47. Hämäläinen H, Rönnemaa T, Toikka T, et al. Long-term effects of one year of intensified podiatric activities on foot-care knowledge and self-care habits in patients with diabetes. Diabetes Educ. 1998;24:734-740.
48. Rönnemaa T, Hämäläinen H, Toikka T, et al. Evaluation of the impact of podiatrist care in the primary prevention of foot problems in diabetic subjects. Diabetes Care. 1997;20:1833-1837.
› Screen for lower
extremity complications at every visit for all patients with a suspected or confirmed diagnosis of diabetes. A
› Consider implementing a risk-based referral system to connect primary screening with a specialist's care. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Foot ulcers and other lower-limb complications secondary to diabetes are common, complex, costly, and associated with increased morbidity and mortality.1-6 Unfortunately, patients often have difficulty recognizing the heightened risk status that accompanies the diagnosis of diabetes, particularly the substantial risk for lower limb complications.7 In addition, loss of protective sensation (LOPS) can render patients unable to recognize damage to their lower extremities, thus creating a cycle of tissue damage and other foot complications. Strong evidence suggests that consistent provision of foot-care services and preventive care can reduce amputations among patients with diabetes.7-9 However, routine foot examination and rapid risk stratification is often difficult to incorporate into busy primary care settings. Data suggest that the diabetic foot is adequately evaluated only 12% to 20% of the time.10
In response to the need for more consistent foot exams, an American Diabetes Association (ADA) task force lead by 2 of the authors of this article (AB and DA) created the Comprehensive Foot Examination and Risk Assessment.5 This set the standard for the detailed investigation of lower limb pathology by a specialist, but was not well suited for other practice settings, including primary care. One reason is that it would be difficult to complete the comprehensive examination during a typical 15-minute primary care office visit. In addition, certain examination parameters require the use of neurologic and vascular assessment equipment and training not available in all health care settings.11
With these thoughts in mind, we set out to develop an exam that could be done by a wide range of health care providers—one that takes substantially less time to complete than a comprehensive exam and eliminates common barriers to frequent assessment. The exam, which we’ll describe here, consists of 3 components: taking a patient history, performing a physical exam, and providing patient education. And best of all, it should only take 3 minutes.
The patient history (1 minute)
Patients may present with concerns about their feet, but may not be able to differentiate between benign and threatening symptoms. A thorough medical history can identify factors that may increase patients’ risk of developing lower-limb complications. Reviewing the patient’s medical history also can help guide the physical exam.
Review the patient’s diabetic history, blood glucose control, and previous diabetic complications. Ask patients about their history of peripheral vascular disease, quality of peripheral protective sensation, and previous lower-limb interventions and operations (TABLE 15,12). Patients with diabetes and suboptimal glycemic control have an increased risk for LOPS, chronic and recalcitrant ulcers, and wound infections.2 Additionally, patients with diabetes and a previous lower extremity amputation are at high risk for reulceration.5,12 Lastly, nicotine use and smoking are common pathogenic risk factors that contribute to peripheral artery disease (PAD).13
Physical examination (1 minute)
Careful inspection of the feet should be performed at every visit for patients with confirmed or suspected diabetes. Because up to 50% of patients with significant sensory loss due to neuropathy may be completely asymptomatic,14 failing to search for early signs of infection (FIGURE 1), skin breakdown, ulcer formation (FIGURE 2), skin temperature changes, and inadequate vascular perfusion may allow complications to develop.5 TABLE 25,15,16 outlines the essential components—dermatologic, neurologic, musculoskeletal, and vascular—of a rapid lower limb physical exam.
The dermatologic exam. This serves as a barometer for early intervention, and often results in a limb-saving referral to a specialist. It should begin with a global inspection for discolorations, calluses, wounds, fissures, macerations, nail dystrophy, or paronychia.5 Skin discoloration or loss of hair growth may be the first signs of vascular insufficiency, while calluses and hypertrophic skin often are precursors to ulcers.5,17-19 Inspection of the toes should include a search for fungal, ingrown, or elongated nails. Carefully examine the areas between the toes, where deeper lesions may go unnoticed.5
The neurologic exam. Without protective sensation, patients with neuropathy are at a heightened risk of unrecognized injury and are unlikely to mention their deformities to medical staff.20-23 Consequently, skin deterioration may unknowingly progress to ulceration that requires extensive medical intervention or amputation.
Neuropathic LOPS is easily detectable, yet it is linked to at least 75% of all nontraumatic diabetic amputations.20-23 Adiminished vibratory perception threshold (VPT) is one of the earliest indicators of neuropathic LOPS and is the best predictor of long-term lower extremity complications.1,24,25 However, VPT devices are expensive and time-consuming to operate, and they require training to ensure proper use. The Semmes-Weinstein monofilament is a well-documented alternative to VPT for predicting ulcer risk26-28 and has long been advocated as an essential component of a thorough foot exam.5 The 128 Hz tuning fork is another regularly used alternative.5 However, physicians would need to purchase one of these devices and receive training on how to use it, and, in the case of the monofilament, to regularly stock replacements to maintain accurate results.16
The Ipswich Touch Test (IpTT) is an alternative neurologic test that requires only the physician’s index finger. During the IpTT, the physician instructs the patient to close his or her eyes while the physician lightly rests his or her finger on each of the patient’s first, third, and fifth toes for 1 to 2 seconds (FIGURE 3). Patients are instructed to respond with a “yes” when they feel the physician’s touch. In a head-to-head trial, diagnostic results of the IpTT directly paralleled those of the monofilament in detecting LOPS; IpTT was also equally sensitive and specific (k=.88, indicating almost perfect agreement; P<.0001).29 The IpTT’s use of only 6 palpation points, constant availability, and accuracy make it a first-line neurologic test for rapidly screening the feet of a patient with diabetes.
Neuromuscular/musculoskeletal exam. Neuromuscular disturbances, such as a reduction in the strength of dorsiflexion and plantar flexion, may indicate a complicated neurologic compromise.5 In addition to being aesthetically problematic, musculoskeletal deformities such as a hammer toe, claw toe (FIGURE 4), or bunion can cause significant pain and/or gait disturbance, and can increase patients’ risk for ulceration.30 These deformities also may compromise patients’ general health and grossly escalate their risk of falls and resultant injuries.5,31 Therefore, patients who present with previously unreported musculoskeletal deformities should be referred to a specialist.31
Also screen patients for Charcot neuroarthropathy (FIGURE 5), a devastating complication that classically presents as a hot, red, swollen foot; the redness resolves upon elevation.32 Charcot neuroarthropathy is hypothesized to be a dysregulation of normal bone metabolism typically occurring secondary to diabetic neuropathy and repetitive minor trauma.33,34 This dysregulation leads to joint instability and disorganization of normal midfoot bone architecture.31,32 Charcot neuroarthropathy is an urgent pathology that requires management by a foot specialist.35
Vascular exam. PAD is particularly common in patients with diabetes and contributes to the development of impaired healing in up to half of foot ulcers.13,18,36-39 Bilateral femoral, popliteal, posterior tibial, or dorsalis pedis pulses should be assessed by palpation; a diminished or absent pulse is a key indicator of vascular compromise.40,41 An integrated care approach between foot specialists and vascular surgeons results in optimal treatment.
Patient education (1 minute)
It is imperative to include patients in their treatment process to reduce the likelihood of complications and, ultimately, decrease the incidence of amputations.12,42 Patient education improves patients’ self-reported home care behaviors, even at the most fundamental levels.43,44 TABLE 35,15,45 lists topics to cover during patient education.
Patients’ lack of understanding about self-care for diabetes is a common barrier to prevention.23 El-Nahas et al46 found a lack of appropriate education regarding diabetes was a factor in more than 90% of recurrent ulcers, which emphasizes the need for repeated education for at-risk patients.47,48 Involve all levels of medical staff in the effort to educate patients on the importance of foot screenings, both at home and in-office. Even with proper patient education, many patients may be in various stages of coping with this all-consuming yet frequently asymptomatic condition, which makes the need for repeated patient education even more critical.
Who to refer, and when
After completing the 3-minute foot exam, create a treatment and follow-up plan, focusing on the need for referral to a specialist. TABLE 4 outlines suggested indications, priorities, and timelines for referral based on ADA guidelines.5 It incorporates the ADA’s patient risk categories (very low, low, moderate, and high risk) and also provides a recommended frequency for patient follow-ups.
Care for patients with lower extremity complications of diabetes mellitus is time-consuming and expensive. The brief exam described here can help you to rapidly identify patients at risk for these complications and prompt you to provide timely referrals to appropriate specialists.
CORRESPONDENCE
David G. Armstrong, DPM, MD, PhD, Professor, Department of Surgery, Director, Southern Arizona Limb Salvage Alliance (SALSA), 1501 N. Campbell Avenue, Tucson, AZ 85724-5072; armstrong@usa.net
› Screen for lower
extremity complications at every visit for all patients with a suspected or confirmed diagnosis of diabetes. A
› Consider implementing a risk-based referral system to connect primary screening with a specialist's care. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Foot ulcers and other lower-limb complications secondary to diabetes are common, complex, costly, and associated with increased morbidity and mortality.1-6 Unfortunately, patients often have difficulty recognizing the heightened risk status that accompanies the diagnosis of diabetes, particularly the substantial risk for lower limb complications.7 In addition, loss of protective sensation (LOPS) can render patients unable to recognize damage to their lower extremities, thus creating a cycle of tissue damage and other foot complications. Strong evidence suggests that consistent provision of foot-care services and preventive care can reduce amputations among patients with diabetes.7-9 However, routine foot examination and rapid risk stratification is often difficult to incorporate into busy primary care settings. Data suggest that the diabetic foot is adequately evaluated only 12% to 20% of the time.10
In response to the need for more consistent foot exams, an American Diabetes Association (ADA) task force lead by 2 of the authors of this article (AB and DA) created the Comprehensive Foot Examination and Risk Assessment.5 This set the standard for the detailed investigation of lower limb pathology by a specialist, but was not well suited for other practice settings, including primary care. One reason is that it would be difficult to complete the comprehensive examination during a typical 15-minute primary care office visit. In addition, certain examination parameters require the use of neurologic and vascular assessment equipment and training not available in all health care settings.11
With these thoughts in mind, we set out to develop an exam that could be done by a wide range of health care providers—one that takes substantially less time to complete than a comprehensive exam and eliminates common barriers to frequent assessment. The exam, which we’ll describe here, consists of 3 components: taking a patient history, performing a physical exam, and providing patient education. And best of all, it should only take 3 minutes.
The patient history (1 minute)
Patients may present with concerns about their feet, but may not be able to differentiate between benign and threatening symptoms. A thorough medical history can identify factors that may increase patients’ risk of developing lower-limb complications. Reviewing the patient’s medical history also can help guide the physical exam.
Review the patient’s diabetic history, blood glucose control, and previous diabetic complications. Ask patients about their history of peripheral vascular disease, quality of peripheral protective sensation, and previous lower-limb interventions and operations (TABLE 15,12). Patients with diabetes and suboptimal glycemic control have an increased risk for LOPS, chronic and recalcitrant ulcers, and wound infections.2 Additionally, patients with diabetes and a previous lower extremity amputation are at high risk for reulceration.5,12 Lastly, nicotine use and smoking are common pathogenic risk factors that contribute to peripheral artery disease (PAD).13
Physical examination (1 minute)
Careful inspection of the feet should be performed at every visit for patients with confirmed or suspected diabetes. Because up to 50% of patients with significant sensory loss due to neuropathy may be completely asymptomatic,14 failing to search for early signs of infection (FIGURE 1), skin breakdown, ulcer formation (FIGURE 2), skin temperature changes, and inadequate vascular perfusion may allow complications to develop.5 TABLE 25,15,16 outlines the essential components—dermatologic, neurologic, musculoskeletal, and vascular—of a rapid lower limb physical exam.
The dermatologic exam. This serves as a barometer for early intervention, and often results in a limb-saving referral to a specialist. It should begin with a global inspection for discolorations, calluses, wounds, fissures, macerations, nail dystrophy, or paronychia.5 Skin discoloration or loss of hair growth may be the first signs of vascular insufficiency, while calluses and hypertrophic skin often are precursors to ulcers.5,17-19 Inspection of the toes should include a search for fungal, ingrown, or elongated nails. Carefully examine the areas between the toes, where deeper lesions may go unnoticed.5
The neurologic exam. Without protective sensation, patients with neuropathy are at a heightened risk of unrecognized injury and are unlikely to mention their deformities to medical staff.20-23 Consequently, skin deterioration may unknowingly progress to ulceration that requires extensive medical intervention or amputation.
Neuropathic LOPS is easily detectable, yet it is linked to at least 75% of all nontraumatic diabetic amputations.20-23 Adiminished vibratory perception threshold (VPT) is one of the earliest indicators of neuropathic LOPS and is the best predictor of long-term lower extremity complications.1,24,25 However, VPT devices are expensive and time-consuming to operate, and they require training to ensure proper use. The Semmes-Weinstein monofilament is a well-documented alternative to VPT for predicting ulcer risk26-28 and has long been advocated as an essential component of a thorough foot exam.5 The 128 Hz tuning fork is another regularly used alternative.5 However, physicians would need to purchase one of these devices and receive training on how to use it, and, in the case of the monofilament, to regularly stock replacements to maintain accurate results.16
The Ipswich Touch Test (IpTT) is an alternative neurologic test that requires only the physician’s index finger. During the IpTT, the physician instructs the patient to close his or her eyes while the physician lightly rests his or her finger on each of the patient’s first, third, and fifth toes for 1 to 2 seconds (FIGURE 3). Patients are instructed to respond with a “yes” when they feel the physician’s touch. In a head-to-head trial, diagnostic results of the IpTT directly paralleled those of the monofilament in detecting LOPS; IpTT was also equally sensitive and specific (k=.88, indicating almost perfect agreement; P<.0001).29 The IpTT’s use of only 6 palpation points, constant availability, and accuracy make it a first-line neurologic test for rapidly screening the feet of a patient with diabetes.
Neuromuscular/musculoskeletal exam. Neuromuscular disturbances, such as a reduction in the strength of dorsiflexion and plantar flexion, may indicate a complicated neurologic compromise.5 In addition to being aesthetically problematic, musculoskeletal deformities such as a hammer toe, claw toe (FIGURE 4), or bunion can cause significant pain and/or gait disturbance, and can increase patients’ risk for ulceration.30 These deformities also may compromise patients’ general health and grossly escalate their risk of falls and resultant injuries.5,31 Therefore, patients who present with previously unreported musculoskeletal deformities should be referred to a specialist.31
Also screen patients for Charcot neuroarthropathy (FIGURE 5), a devastating complication that classically presents as a hot, red, swollen foot; the redness resolves upon elevation.32 Charcot neuroarthropathy is hypothesized to be a dysregulation of normal bone metabolism typically occurring secondary to diabetic neuropathy and repetitive minor trauma.33,34 This dysregulation leads to joint instability and disorganization of normal midfoot bone architecture.31,32 Charcot neuroarthropathy is an urgent pathology that requires management by a foot specialist.35
Vascular exam. PAD is particularly common in patients with diabetes and contributes to the development of impaired healing in up to half of foot ulcers.13,18,36-39 Bilateral femoral, popliteal, posterior tibial, or dorsalis pedis pulses should be assessed by palpation; a diminished or absent pulse is a key indicator of vascular compromise.40,41 An integrated care approach between foot specialists and vascular surgeons results in optimal treatment.
Patient education (1 minute)
It is imperative to include patients in their treatment process to reduce the likelihood of complications and, ultimately, decrease the incidence of amputations.12,42 Patient education improves patients’ self-reported home care behaviors, even at the most fundamental levels.43,44 TABLE 35,15,45 lists topics to cover during patient education.
Patients’ lack of understanding about self-care for diabetes is a common barrier to prevention.23 El-Nahas et al46 found a lack of appropriate education regarding diabetes was a factor in more than 90% of recurrent ulcers, which emphasizes the need for repeated education for at-risk patients.47,48 Involve all levels of medical staff in the effort to educate patients on the importance of foot screenings, both at home and in-office. Even with proper patient education, many patients may be in various stages of coping with this all-consuming yet frequently asymptomatic condition, which makes the need for repeated patient education even more critical.
Who to refer, and when
After completing the 3-minute foot exam, create a treatment and follow-up plan, focusing on the need for referral to a specialist. TABLE 4 outlines suggested indications, priorities, and timelines for referral based on ADA guidelines.5 It incorporates the ADA’s patient risk categories (very low, low, moderate, and high risk) and also provides a recommended frequency for patient follow-ups.
Care for patients with lower extremity complications of diabetes mellitus is time-consuming and expensive. The brief exam described here can help you to rapidly identify patients at risk for these complications and prompt you to provide timely referrals to appropriate specialists.
CORRESPONDENCE
David G. Armstrong, DPM, MD, PhD, Professor, Department of Surgery, Director, Southern Arizona Limb Salvage Alliance (SALSA), 1501 N. Campbell Avenue, Tucson, AZ 85724-5072; armstrong@usa.net
1. Shearer A, Scuffham P, Gordois A, et al. Predicted costs and outcomes from reduced vibration detection in people with diabetes in the U.S. Diabetes Care. 2003;26:2305-2310.
2. Apelqvist J, Larsson J. What is the most effective way to reduce incidence of amputation in the diabetic foot? Diabetes Metab Res Rev. 2000;16 suppl 1:S75-S83.
3. Armstrong DG, Kanda VA, Lavery LA, et al. Mind the gap: disparity between research funding and costs of care for diabetic foot ulcers. Diabetes Care. 2013;36:1815-1817.
4. Driver VR, Fabbi M, Lavery LA, et al. The costs of diabetic foot: the economic case for the limb salvage team. J Vasc Surg. 2010;52(3 suppl):17S-22S.
5. Boulton AJ, Armstrong DG, Albert SF, et al; American Diabetes Association; American Association of Clinical Endocrinologists. Comprehensive foot examination and risk assessment: a report of the Task Force of the Foot Care Interest Group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care. 2008;31:1679-1685.
6. American Diabetes Association. Standards of medical care in diabetes—2014. Diabetes Care. 2014;37 suppl 1:S14-S80.
7. Sloan FA, Feinglos MN, Grossman DS. Receipt of care and reduction of lower extremity amputations in a nationally representative sample of U.S. Elderly. Health Serv Res. 2010;45(6 pt 1):1740-1762.
8. Carls GS, Gibson TB, Driver VR, et al. The economic value of specialized lower-extremity medical care by podiatric physicians in the treatment of diabetic foot ulcers. J Am Podiatr Med Assoc. 2011;101:93-115.
9. McCabe CJ, Stevenson RC, Dolan AM. Evaluation of a diabetic foot screening and protection programme. Diabet Med. 1998;15:80-84.
10. Bailey TS, Yu HM, Rayfield EJ. Patterns of foot examination in a diabetes clinic. Am J Med. 1985;78:371-374.
11. Chin MH, Cook S, Jin L, et al. Barriers to providing diabetes care in community health centers. Diabetes Care. 2001;24:268-274.
12. Abbott CA, Carrington AL, Ashe H, et al; North-West Diabetes Foot Care Study. The North-West Diabetes Foot Care Study: incidence of, and risk factors for, new diabetic foot ulceration in a community-based patient cohort. Diabet Med. 2002;19:377-384.
13. Fowkes FG, Rudan D, Rudan I, et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet. 2013;382:1329-1340.
14. Boulton A, Vinik AI, Arezzo JC, et al; American Diabetes Association. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes. 2005;28:956-962.
15. Singh N, Armstrong DG, Lipsky BA. Preventing foot ulcers in patients with diabetes. JAMA. 2005;293:217-228.
16. Pham H, Armstrong DG, Harvey C, et al. Screening techniques to identify people at high risk for diabetic foot ulceration: a prospective multicenter trial. Diabetes Care. 2000;23:606-611.
17. Marso SP, Hiatt WR. Peripheral arterial disease in patients with diabetes. J Am Coll Cardiol. 2006;47:921-929.
18. American Diabetes Association. Peripheral arterial disease in people with diabetes. JAPMA. 2005;95:309-319.
19. Pataky Z, Golay A, Faravel L, et al. The impact of callosities on the magnitude and duration of plantar pressure in patients with diabetes mellitus. A callus may cause 18,600 kilograms of excess plantar pressure per day. Diabetes Metab. 2002;28: 356-361.
20. Holzer SE, Camerota A, Martens L, et al. Costs and duration of care for lower extremity ulcers in patients with diabetes. Clin Ther. 1998;20:169-181.
21. Boulton AJ, Gries FA, Jervell JA. Guidelines for the diagnosis and outpatient management of diabetic peripheral neuropathy. Diabet Med. 1998;15:508-514.
22. Malay DS, Margolis DJ, Hoffstad OJ, et al. The incidence and risks of failure to heal after lower extremity amputation for the treatment of diabetic neuropathic foot ulcer. J Foot Ankle Surg. 2006;45:366-374.
23. van Houtum WH. Barriers to implementing foot care. Diabetes Metab Res Rev. 2012;28 suppl 1:112-115.
24. Jayaprakash P, Bhansali A, Bhansali S, et al. Validation of bedside methods in evaluation of diabetic peripheral neuropathy. Indian J Med Res. 2011;133:645-649.
25. Young MJ, Breddy JL, Veves A, et al. The prediction of diabetic neuropathic foot ulceration using vibration perception thresholds. A prospective study. Diabetes Care. 1994;17:557-560.
26. Leese GP, Reid F, Green V, et al. Stratification of foot ulcer risk in patients with diabetes: a population-based study. Int J Clin Pract. 2006;60:541-545.
27. Adler AI, Boyko EJ, Ahroni JH, et al. Risk factors for diabetic peripheral sensory neuropathy. Results of the Seattle Prospective Diabetic Foot Study. Diabetes Care. 1997;20:1162-1167.
28. Armstrong DG, Lavery LA, Vela SA, et al. Choosing a practical screening instrument to identify patients at risk for diabetic foot ulceration. Arch Intern Med. 1998;158:289-292.
29. Rayman G, Vas PR, Baker N, et al. The Ipswich Touch Test: a simple and novel method to identify inpatients with diabetes at risk of foot ulceration. Diabetes Care. 2011;34:1517-1518.
30. Lavery LA, Armstrong DG, Vela SA, et al. Practical criteria for screening patients at high risk for diabetic foot ulceration. Arch Intern Med. 1998;158:157-162.
31. Frykberg RG, Zgonis T, Armstrong DG, et al; American College of Foot and Ankle Surgeons. Diabetic foot disorders. A clinical practice guideline (2006 revision). J Foot Ankle Surg. 2006;45(5 suppl):S1-S66.
32. Nielson DL, Armstrong DG. The natural history of Charcot’s neuroarthropathy. Clin Podiatr Med Surg. 2008;25:53-62,vi.
33. Jeffcoate W, Lima J, Nobrega L. The Charcot foot. Diabet Med. 2000;17:253-258.
34. Blume PA, Sumpio B, Schmidt B, et al. Charcot neuroarthropathy of the foot and ankle: diagnosis and management strategies. Clin Podiatr Med Surg. 2014;31:151-172.
35. Petrova NL, Edmonds ME. Medical management of Charcot arthropathy. Diabetes Obes Metab. 2012;15:193-197.
36. Prompers L, Huijberts M, Apelqvist J, et al. Delivery of care to diabetic patients with foot ulcers in daily practice: results of the Eurodiale Study, a prospective cohort study. Diabet Med. 2008;25:700-707.
37. Armstrong DG, Bharara M, White M, et al. The impact and outcomes of establishing an integrated interdisciplinary surgical team to care for the diabetic foot. Diabetes Metab Res Rev. 2012;28:514-518.
38. Rogers LC, Andros G, Caporusso J, et al. Toe and flow: essential components and structure of the amputation prevention team. J Vasc Surg. 2010;52:23S-27S.
39. Mills JL Sr, Conte MS, Armstrong DG, et al; Society for Vascular Surgery Lower Extremity Guidelines Committee. The Society for Vascular Surgery Lower Extremity Threatened Limb Classification System: risk stratification based on wound, ischemia, and foot infection (WIfI). J Vasc Surg. 2014;59:220-34.e1-2.
40. Khan NA, Rahim SA, Anand SS, et al. Does the clinical examination predict lower extremity peripheral arterial disease? JAMA. 2006;295:536-546.
41. Sumpio BE, Lee T, Blume PA. Vascular evaluation and arterial reconstruction of the diabetic foot. Clin Podiatr Med Surg. 2003;20:689-708.
42. Dorresteijn JAN, Valk GD. Patient education for preventing diabetic foot ulceration. Diabetes Metab Res Rev. 2012;28 Suppl 1:101-106.
43. Lincoln NB, Radford KA, Game FL, et al. Education for secondary prevention of foot ulcers in people with diabetes: a randomised controlled trial. Diabetologia. 2008;51:1954-1961.
44. McMurray SD, Johnson G, Davis S, et al. Diabetes education and care management significantly improve patient outcomes in the dialysis unit. Am J Kidney Dis. 2002;40:566-575.
45. Armstrong DG, Lavery LA. Diabetic foot ulcers: prevention, diagnosis and classification. Am Fam Physician. 1998;57:1325-1332,1337-1338.
46. El-Nahas MR, Gawish HMS, Tarshoby MM, et al. The prevalence of risk factors for foot ulceration in Egyptian diabetic patients. Practical Diabetes Int. 2008;25:362-366.
47. Hämäläinen H, Rönnemaa T, Toikka T, et al. Long-term effects of one year of intensified podiatric activities on foot-care knowledge and self-care habits in patients with diabetes. Diabetes Educ. 1998;24:734-740.
48. Rönnemaa T, Hämäläinen H, Toikka T, et al. Evaluation of the impact of podiatrist care in the primary prevention of foot problems in diabetic subjects. Diabetes Care. 1997;20:1833-1837.
1. Shearer A, Scuffham P, Gordois A, et al. Predicted costs and outcomes from reduced vibration detection in people with diabetes in the U.S. Diabetes Care. 2003;26:2305-2310.
2. Apelqvist J, Larsson J. What is the most effective way to reduce incidence of amputation in the diabetic foot? Diabetes Metab Res Rev. 2000;16 suppl 1:S75-S83.
3. Armstrong DG, Kanda VA, Lavery LA, et al. Mind the gap: disparity between research funding and costs of care for diabetic foot ulcers. Diabetes Care. 2013;36:1815-1817.
4. Driver VR, Fabbi M, Lavery LA, et al. The costs of diabetic foot: the economic case for the limb salvage team. J Vasc Surg. 2010;52(3 suppl):17S-22S.
5. Boulton AJ, Armstrong DG, Albert SF, et al; American Diabetes Association; American Association of Clinical Endocrinologists. Comprehensive foot examination and risk assessment: a report of the Task Force of the Foot Care Interest Group of the American Diabetes Association, with endorsement by the American Association of Clinical Endocrinologists. Diabetes Care. 2008;31:1679-1685.
6. American Diabetes Association. Standards of medical care in diabetes—2014. Diabetes Care. 2014;37 suppl 1:S14-S80.
7. Sloan FA, Feinglos MN, Grossman DS. Receipt of care and reduction of lower extremity amputations in a nationally representative sample of U.S. Elderly. Health Serv Res. 2010;45(6 pt 1):1740-1762.
8. Carls GS, Gibson TB, Driver VR, et al. The economic value of specialized lower-extremity medical care by podiatric physicians in the treatment of diabetic foot ulcers. J Am Podiatr Med Assoc. 2011;101:93-115.
9. McCabe CJ, Stevenson RC, Dolan AM. Evaluation of a diabetic foot screening and protection programme. Diabet Med. 1998;15:80-84.
10. Bailey TS, Yu HM, Rayfield EJ. Patterns of foot examination in a diabetes clinic. Am J Med. 1985;78:371-374.
11. Chin MH, Cook S, Jin L, et al. Barriers to providing diabetes care in community health centers. Diabetes Care. 2001;24:268-274.
12. Abbott CA, Carrington AL, Ashe H, et al; North-West Diabetes Foot Care Study. The North-West Diabetes Foot Care Study: incidence of, and risk factors for, new diabetic foot ulceration in a community-based patient cohort. Diabet Med. 2002;19:377-384.
13. Fowkes FG, Rudan D, Rudan I, et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet. 2013;382:1329-1340.
14. Boulton A, Vinik AI, Arezzo JC, et al; American Diabetes Association. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes. 2005;28:956-962.
15. Singh N, Armstrong DG, Lipsky BA. Preventing foot ulcers in patients with diabetes. JAMA. 2005;293:217-228.
16. Pham H, Armstrong DG, Harvey C, et al. Screening techniques to identify people at high risk for diabetic foot ulceration: a prospective multicenter trial. Diabetes Care. 2000;23:606-611.
17. Marso SP, Hiatt WR. Peripheral arterial disease in patients with diabetes. J Am Coll Cardiol. 2006;47:921-929.
18. American Diabetes Association. Peripheral arterial disease in people with diabetes. JAPMA. 2005;95:309-319.
19. Pataky Z, Golay A, Faravel L, et al. The impact of callosities on the magnitude and duration of plantar pressure in patients with diabetes mellitus. A callus may cause 18,600 kilograms of excess plantar pressure per day. Diabetes Metab. 2002;28: 356-361.
20. Holzer SE, Camerota A, Martens L, et al. Costs and duration of care for lower extremity ulcers in patients with diabetes. Clin Ther. 1998;20:169-181.
21. Boulton AJ, Gries FA, Jervell JA. Guidelines for the diagnosis and outpatient management of diabetic peripheral neuropathy. Diabet Med. 1998;15:508-514.
22. Malay DS, Margolis DJ, Hoffstad OJ, et al. The incidence and risks of failure to heal after lower extremity amputation for the treatment of diabetic neuropathic foot ulcer. J Foot Ankle Surg. 2006;45:366-374.
23. van Houtum WH. Barriers to implementing foot care. Diabetes Metab Res Rev. 2012;28 suppl 1:112-115.
24. Jayaprakash P, Bhansali A, Bhansali S, et al. Validation of bedside methods in evaluation of diabetic peripheral neuropathy. Indian J Med Res. 2011;133:645-649.
25. Young MJ, Breddy JL, Veves A, et al. The prediction of diabetic neuropathic foot ulceration using vibration perception thresholds. A prospective study. Diabetes Care. 1994;17:557-560.
26. Leese GP, Reid F, Green V, et al. Stratification of foot ulcer risk in patients with diabetes: a population-based study. Int J Clin Pract. 2006;60:541-545.
27. Adler AI, Boyko EJ, Ahroni JH, et al. Risk factors for diabetic peripheral sensory neuropathy. Results of the Seattle Prospective Diabetic Foot Study. Diabetes Care. 1997;20:1162-1167.
28. Armstrong DG, Lavery LA, Vela SA, et al. Choosing a practical screening instrument to identify patients at risk for diabetic foot ulceration. Arch Intern Med. 1998;158:289-292.
29. Rayman G, Vas PR, Baker N, et al. The Ipswich Touch Test: a simple and novel method to identify inpatients with diabetes at risk of foot ulceration. Diabetes Care. 2011;34:1517-1518.
30. Lavery LA, Armstrong DG, Vela SA, et al. Practical criteria for screening patients at high risk for diabetic foot ulceration. Arch Intern Med. 1998;158:157-162.
31. Frykberg RG, Zgonis T, Armstrong DG, et al; American College of Foot and Ankle Surgeons. Diabetic foot disorders. A clinical practice guideline (2006 revision). J Foot Ankle Surg. 2006;45(5 suppl):S1-S66.
32. Nielson DL, Armstrong DG. The natural history of Charcot’s neuroarthropathy. Clin Podiatr Med Surg. 2008;25:53-62,vi.
33. Jeffcoate W, Lima J, Nobrega L. The Charcot foot. Diabet Med. 2000;17:253-258.
34. Blume PA, Sumpio B, Schmidt B, et al. Charcot neuroarthropathy of the foot and ankle: diagnosis and management strategies. Clin Podiatr Med Surg. 2014;31:151-172.
35. Petrova NL, Edmonds ME. Medical management of Charcot arthropathy. Diabetes Obes Metab. 2012;15:193-197.
36. Prompers L, Huijberts M, Apelqvist J, et al. Delivery of care to diabetic patients with foot ulcers in daily practice: results of the Eurodiale Study, a prospective cohort study. Diabet Med. 2008;25:700-707.
37. Armstrong DG, Bharara M, White M, et al. The impact and outcomes of establishing an integrated interdisciplinary surgical team to care for the diabetic foot. Diabetes Metab Res Rev. 2012;28:514-518.
38. Rogers LC, Andros G, Caporusso J, et al. Toe and flow: essential components and structure of the amputation prevention team. J Vasc Surg. 2010;52:23S-27S.
39. Mills JL Sr, Conte MS, Armstrong DG, et al; Society for Vascular Surgery Lower Extremity Guidelines Committee. The Society for Vascular Surgery Lower Extremity Threatened Limb Classification System: risk stratification based on wound, ischemia, and foot infection (WIfI). J Vasc Surg. 2014;59:220-34.e1-2.
40. Khan NA, Rahim SA, Anand SS, et al. Does the clinical examination predict lower extremity peripheral arterial disease? JAMA. 2006;295:536-546.
41. Sumpio BE, Lee T, Blume PA. Vascular evaluation and arterial reconstruction of the diabetic foot. Clin Podiatr Med Surg. 2003;20:689-708.
42. Dorresteijn JAN, Valk GD. Patient education for preventing diabetic foot ulceration. Diabetes Metab Res Rev. 2012;28 Suppl 1:101-106.
43. Lincoln NB, Radford KA, Game FL, et al. Education for secondary prevention of foot ulcers in people with diabetes: a randomised controlled trial. Diabetologia. 2008;51:1954-1961.
44. McMurray SD, Johnson G, Davis S, et al. Diabetes education and care management significantly improve patient outcomes in the dialysis unit. Am J Kidney Dis. 2002;40:566-575.
45. Armstrong DG, Lavery LA. Diabetic foot ulcers: prevention, diagnosis and classification. Am Fam Physician. 1998;57:1325-1332,1337-1338.
46. El-Nahas MR, Gawish HMS, Tarshoby MM, et al. The prevalence of risk factors for foot ulceration in Egyptian diabetic patients. Practical Diabetes Int. 2008;25:362-366.
47. Hämäläinen H, Rönnemaa T, Toikka T, et al. Long-term effects of one year of intensified podiatric activities on foot-care knowledge and self-care habits in patients with diabetes. Diabetes Educ. 1998;24:734-740.
48. Rönnemaa T, Hämäläinen H, Toikka T, et al. Evaluation of the impact of podiatrist care in the primary prevention of foot problems in diabetic subjects. Diabetes Care. 1997;20:1833-1837.
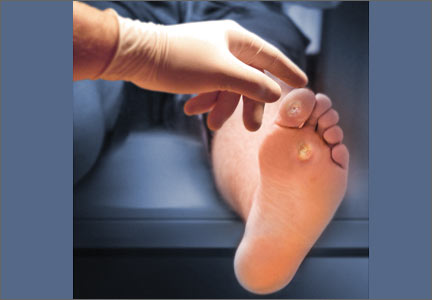
Staying ahead of pertussis
› Recommend a
one-time Tdap (tetanus-diphtheria-acellular pertussis) combination vaccine for adults younger than age 64 who need tetanus booster vaccination. A
› Suspect pertussis in a patient who presents with a persistent, paroxysmal cough, with an inspiratory “whoop,” that has lasted for at least
2 weeks. B
› Prescribe a macrolide antibiotic as a first-line treatment for infants, children, and adults who have pertussis. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Despite a high vaccination rate, pertussis is the only vaccine-preventable disease whose incidence is on the rise.1-3 The Centers for Disease Control and Prevention (CDC) reported 48,277 laboratory-confirmed cases in 2012—the most since 1955—and 20 pertussis-related deaths.4 And while only 28,639 pertussis cases were reported in 2013, more than 17,000 cases had already been reported through August 15, 2014, suggesting that the incidence may again be on the rise this year.4
This uptick is likely due to a combination of factors, including a growing awareness of pertussis, and therefore a lower threshold for physicians to test for it. In addition, there’s some evidence that the immunity provided by the currently used pertussis vaccines may wane over time. Recently reported epidemics, including those in California this year and in 2010, as well as in Washington in 2011, have added to this concern.5 This article outlines what you can do to improve prevention, diagnosis, and treatment of pertussis.
A 3-stage course of disease
Bordetella pertussis is an aerobic, gram-negative bacterium that causes symptoms by producing multiple antigenic and biologically active components, including pertussis toxin, filamentous hemagglutinin, and agglutinogens. The bacteria adhere to the cilia in the respiratory tract and initiate an inflammatory cascade that paralyzes the cilia and inhibits the respiratory functions responsible for clearing secretions, largely through an immune-mediated response.
Pertussis has an incubation period of approximately 7 days, but this can last as long as 3 to 6 weeks. The 3 stages in the course of the disease are:6
- Catarrhal. This stage lasts 1 to 2 weeks and is characterized by coryza, sneezing, and a mild, occasional cough.
- Paroxysmal. This stage lasts 1 to 6 weeks, and is characterized by periods of severe coughing “fits” that include the inspiratory "whoop." These coughing episodes may occur more often at night and may worsen in intensity and frequency in the first 2 to 3 weeks and then gradually decrease. This stage also may include posttussive vomiting.
- Convalescent. During this stage, the cough begins to wane.
Vaccination: Don’t forget adults
The 2 vaccines used to prevent pertussis are DTaP (diphtheria-tetanus-acellular pertussis) and Tdap (tetanus-diphtheria-acellular pertussis). The difference between the 2 is that the Tdap vaccine contains a reduced dose of the diphtheria and acellular pertussis vaccines. DTaP is designed primarily for children younger than 7 years of age. Tdap is given to older children and adults. The CDC and Advisory Committee on Immunization Practices recommend that children receive 5 doses of DTaP, one dose at each of the following ages: 2, 4, 6, and 15 to 18 months and at 4 to 6 years.7 All adults 19 years of age and older who have not yet received a dose of Tdap should receive a single dose regardless of when they last received any immunization for tetanus or diphtheria.7-10 A one-time Tdap booster should be given to all adults in place of a tetanus booster (TABLE 1).7-10

What about pregnant women? Tdap should be administered to every pregnant woman between 27 to 36 weeks gestation regardless of Tdap history.7,11 This strategy allows maternal antibodies to transfer to the infant, thus providing some protection to the newborn prior to pediatric vaccinations.
Is the vaccine becoming less effective? Since 1991, the number of cases of pertussis reported in previously vaccinated adolescents and adults has increased, which suggests waning immunity.12,13 Another recent trial investigating the acellular pertussis vaccine found that immunity decreases dramatically 5 years after the fifth dose.14
Recommendations on who should receive pertussis vaccination have been expanded to include adolescents and adults, including pregnant women and those ages 65 and older in close contact with infants, and this should decrease the overall incidence of disease through decreased communicability.15 Current recommendations call for a single adult vaccination; however, ongoing studies are evaluating whether a booster later in life might be necessary.15
Diagnosis needs to be confirmed by lab testing
Any patient who reports having a persistent cough should be considered for pertussis testing and treatment, and any clinician who triages such patients should ask detailed questions about the characteristics and duration of the patient’s symptoms. However, while a prolonged cough is the hallmark of pertussis, there are many other potential causes of this symptom. Therefore, diagnosis of pertussis requires a combination of clinical and laboratory testing, because clinical parameters alone are neither sensitive nor specific enough for pertussis infection.
TABLE 216 outlines the clinical and laboratory diagnostic criteria for pertussis from the CDC and the World Health Organization. Suspect pertussis in a patient who’s had a cough for more than 14 days that includes an inspiratory “whoop.” In infants, pertussis should be suspected in those with symptoms that suggest cough and associated apnea.16 Order laboratory testing for any patients who have clinical signs or symptoms of pertussis.

Four methods of lab testing for pertussis infection are polymerase chain reaction (PCR), direct fluorescent antibody (DFA) testing, serologic testing, and culture (TABLE 3).17-19 The sensitivity of these tests is as follows: PCR, 90% to 95%; DFA, 50% to 60%; serologic testing, 70% to 80%; and culture, 50% to 70%. The specificity is: PCR, 95% to 98%; DFA, 90% to 100%; serologic testing, 90% to 100%; and culture, 100%.
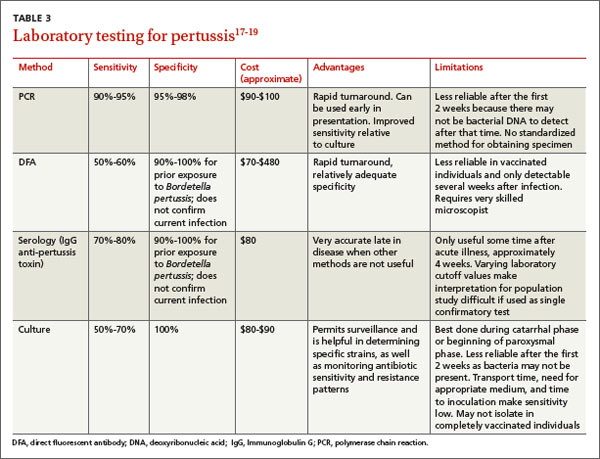
PCR is the preferred method because of its rapid turnaround and fairly high sensitivity. The reliability of PCR decreases, however, for a patient who’s had a cough for more than 2 weeks because the individual may have transitioned to the convalescent phase, when less bacterial DNA remain.
Results from DFA testing also are rapidly available, but the need for specialized equipment and a well-trained examiner of the specimen limits widespread use of this test. It also is not particularly sensitive for pertussis.
Serologic testing is less reliable in patients who have received an acellular pertussis vaccine and is not helpful in the first few weeks of infection.
The sensitivity of culture is best if the sample is collected appropriately (more on this in a bit) and within the first 2 weeks of symptoms (catarrhal stage). Culture is also very specific.
Given the strengths and weakness of the different tests, an acceptable method of laboratory confirmation is to obtain PCR and/or culture within the first 2 weeks of symptoms in all age groups.17-20 Testing after 2 weeks should include a combination of PCR and serology.17 It is essential that the clinical specimen used for PCR or culture testing for pertussis is properly collected. (See “Collecting a swab for pertussis testing” below.21)
The illustration below shows the correct swab and sampling method. Swab tips may be polyester (such as Dacron or rayon) or they may be nylon-flocked. Cotton-tipped or calcium alginate swabs are not acceptable because the residue will inhibit DNA assays.21 The specimen must be obtained from the posterior nasopharynx and not the nares or oropharynx. The Centers for Disease Control and Prevention offers a video that demonstrates how to properly collect a specimen for testing at http://www.cdc.gov/pertussis/clinical/diagnostic-testing/specimen-collection.html.
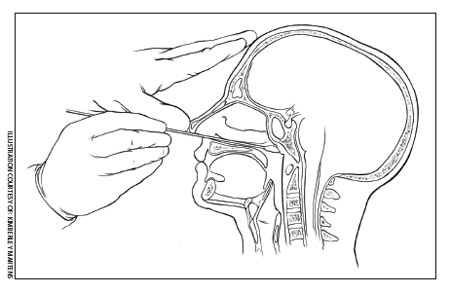
Tx is effective when started early
Antibiotics are an effective treatment for pertussis, but they need to be started within the first few weeks of developing symptoms. Studies have not found evidence that antibiotics shorten the duration of pertussis symptoms unless they are given in the catarrhal phase.22,23 It can be challenging to get treatment started during this window, however, because patients may put off seeking care for symptoms they perceive as only minor, such as a cough, until the disease progresses. In addition, physicians may not suspect pertussis in patients who present with a cough they have had for only a short time, and therefore may not test for it.
It may be necessary to rely on clinical suspicion when deciding whether to initiate treatment for pertussis before testing to confirm the diagnosis. For patients in whom clinical suspicion of pertussis is high and who may be in contact with high-risk individuals, it may be acceptable to begin treatment before receiving lab test results.24,25 A recent Cochrane meta-analysis26 recommended initiating treatment to render a patient who has pertussis “noninfectious” but without an expectation of diminishing symptoms.
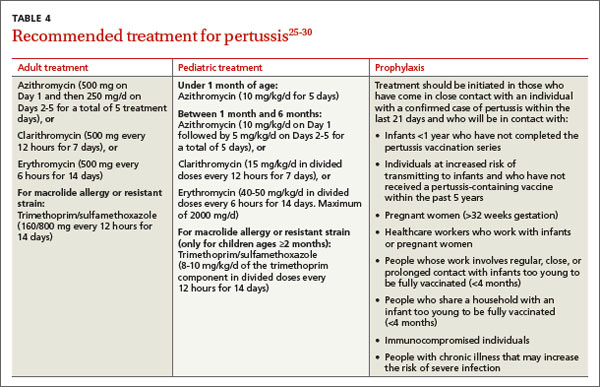
Limited role for prophylaxis. There is little evidence that prophylactic treatment for pertussis can decrease the spread of the disease. Studies that investigated potential benefits of prophylactic treatment for pertussis have been inconclusive, except for individuals who are in close contact with an infant younger than 6 months of age who has not been fully immunized.27,28
A macrolide antibiotic is generally used to treat pertussis (TABLE 4).25-30 Erythromycin had been the drug of choice, but recent studies have found similar efficacy for azithromycin and clarithromycin.29 For infants younger than one month of age, azithromycin is preferred because in addition to being as effective as other macrolides, it has a better adverse effect profile.29 For patients who are at least 2 months of age, trimethoprim-sulfamethoxazole is an acceptable alternative to a macrolide.
The CDC recommends that any adolescent or adult who has a cough and has had close contact with an individual with a laboratory-confirmed case of pertussis within the past 21 days should be treated.30 Close contacts younger than 7 years of age who have not received the first 4 doses of the pertussis vaccine should be offered treatment.
CORRESPONDENCE
Gary Rivard, DO, Family Medicine Residency Program, Central Maine Medical Center, 76 High Street, Lewiston, ME 04282; rivardga@cmhc.org
1. Orenstein WA. Pertussis in adults: epidemiology, signs, symptoms, and implications for vaccination. Clin Infect Dis. 1999;28 suppl 2:S147-S150.
2. Tanaka M, Vitek CR, Pascual FB, et al. Trends in pertussis among infants in the United States, 1980-1999. JAMA. 2003;290:2968-2975.
3. Vitek CR, Pascual FB, Baughman AL, et al. Increase in deaths from pertussis among young infants in the United States in the 1990s. Pediatr Infect Dis J. 2003;22:628-634.
4. Centers for Disease Control and Prevention. Pertussis outbreak trends. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/pertussis/outbreaks/trends.html. Accessed October 10, 2014.
5. Shapiro ED. Acellular vaccines and resurgence of pertussis. JAMA. 2012;308:2149-2150.
6. Centers for Disease Control and Prevention. Pertussis. In: Epidemiology and Prevention of Vaccine-Preventable Diseases. The Pink Book. 2012. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/pubs/pinkbook/pert.html. Accessed October 10, 2014.
7. Centers for Disease Control and Prevention. Pertussis: Summary of vaccine recommendations. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/vpd-vac/pertussis/recs-summary.htm. Accessed October 10, 2014.
8. Lee GM, Murphy TV, Lett S, et al. Cost effectiveness of pertussis vaccination in adults. Am J Prev Med. 2007;32:186-193.
9. Pertussis vaccines: WHO position paper. Wkly Epidemiol Rec. 2010;85:385-400.
10. Kretsinger K, Broder KR, Cortese MM, et al; Centers for Disease Control and Prevention; Advisory Committee on Immunization Practices; Healthcare Infection Control Practices Advisory Committee. Preventing tetanus, diphtheria, and pertussis among adults: use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccine recommendations of the Advisory Committee on Immunization Practices (ACIP) and recommendation of ACIP, supported by the Healthcare Infection Control Practices Advisory Committee (HICPAC), for use of Tdap among healthcare personnel. MMWR Recomm Rep. 2006;55(RR-17):1-37.
11. English P. Pertussis vaccination in pregnant women will protect neonates. Practitioner. 2012;256:5.
12. Winter K, Harriman K, Zipprich J, et al. California pertussis epidemic, 2010. J Pediatr. 2012;161:1091-1096.
13. Centers for Disease Control and Prevention (CDC). Pertussis epidemic—Washington, 2012. MMWR Morb Mortal Wkly Rep. 2012;61:517-522.
14. Klein NP, Bartlett J, Rowhani-Rahbar A, et al. Waning protection after fifth dose of acellular pertussis vaccine in children. N Engl J Med. 2012;367:1012-1019.
15. Centers for Disease Control and Prevention (CDC). Updated recommendations for use of tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis (Tdap) vaccine in adults aged 65 years and older - Advisory Committee on Immunization Practices (ACIP), 2012. MMWR Morb Mortal Wkly Rep. 2012;61:468-470.
16. Cherry JD, Tan T, Wirsing von Konig C, et al. Clinical definitions of pertussis: Summary of a global pertussis initiative roundtable meeting, February 2011. Clin Infect Dis. 2012;54:1756-1764.
17. Zouari A, Smaoui H, Kechrid A. The diagnosis of pertussis: which method to choose?. Crit Rev Microbiol. 2012;38:111-121.
18. Loeffelholz MJ, Thompson CJ, Long KS, et al. Comparison of PCR, culture, and direct fluorescent-antibody testing for detection of Bordetella pertussis. J Clin Microbiol. 1999;37:2872-2876.
19. Tozzi A, Celentano L, Ciofi degli Atti ML, et al. Diagnosis and management of pertussis. CMAJ. 2005;172:509-515.
20. von König CH, Halperin S, Riffelmann M, et al. Pertussis of adults and infants. Lancet Infect Dis. 2002;2:744-750.
21. Cattaneo LA, Edwards KM. Bordetella pertussis (whooping cough). Semin Pediatr Infect Dis. 1995;6:107-117.
22. Hoppe JE, Eichhorn A. Activity of new macrolides against Bordetella pertussis and Bordetella parapertussis. Eur J Clin Microbiol Infect Dis. 1989;8:653-654.
23. Bass JW. Erythromycin for treatment and prevention of pertussis. Pediatr Infect Dis. 1986;5:154-157.
24. Health Protection Surveillance Centre. Guidelines for the Public Health Management of Pertussis: Public Health Medicine Communicable Disease Group HSE—October 2013. Health Protection Surveillance Centre Web site. Available at: http://www.hpsc.ie/A-Z/VaccinePreventable/PertussisWhoopingCough/InformationforHealthcareWorkers/File,13577,en.pdf. Accessed October 2, 2014.
25. Dodhia H, Miller E. Review of the evidence for the use of erythromycin in the management of persons exposed to pertussis. Epidemiol Infect. 1998;120:143-149.
26. Altunaiji S, Kukuruzovic R, Curtis N, et al. Antibiotics for whooping cough (pertussis). Cochrane Database Syst Rev. 2007;(3):CD004404.
27. Prophylactic erythromycin for whooping-cough contacts. Lancet. 1981;1:772.
28. Halperin SA, Bortolussi R, Langley JM, et al. A randomized, placebo-controlled trial of erythromycin estolate chemoprophylaxis for household contacts of children with culture-positive bordetella pertussis infection. Pediatrics. 1999;104:e42.
29. Langley JM, Halperin SA, Boucher FD, et al; Pediatric Investigators Collaborative Network on Infections in Canada (PICNIC). Azithromycin is as effective as and better tolerated than erythromycin estolate for the treatment of pertussis. Pediatrics. 2004;114:e96-e101.
30. Tiwari T, Murphy TV, Moran J; National Immunization Program, CDC. Recommended antimicrobial agents for the treatment and postexposure prophylaxis of pertussis: 2005 CDC Guidelines. MMWR Recomm Rep. 2005;54(RR-14):1-16.
› Recommend a
one-time Tdap (tetanus-diphtheria-acellular pertussis) combination vaccine for adults younger than age 64 who need tetanus booster vaccination. A
› Suspect pertussis in a patient who presents with a persistent, paroxysmal cough, with an inspiratory “whoop,” that has lasted for at least
2 weeks. B
› Prescribe a macrolide antibiotic as a first-line treatment for infants, children, and adults who have pertussis. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Despite a high vaccination rate, pertussis is the only vaccine-preventable disease whose incidence is on the rise.1-3 The Centers for Disease Control and Prevention (CDC) reported 48,277 laboratory-confirmed cases in 2012—the most since 1955—and 20 pertussis-related deaths.4 And while only 28,639 pertussis cases were reported in 2013, more than 17,000 cases had already been reported through August 15, 2014, suggesting that the incidence may again be on the rise this year.4
This uptick is likely due to a combination of factors, including a growing awareness of pertussis, and therefore a lower threshold for physicians to test for it. In addition, there’s some evidence that the immunity provided by the currently used pertussis vaccines may wane over time. Recently reported epidemics, including those in California this year and in 2010, as well as in Washington in 2011, have added to this concern.5 This article outlines what you can do to improve prevention, diagnosis, and treatment of pertussis.
A 3-stage course of disease
Bordetella pertussis is an aerobic, gram-negative bacterium that causes symptoms by producing multiple antigenic and biologically active components, including pertussis toxin, filamentous hemagglutinin, and agglutinogens. The bacteria adhere to the cilia in the respiratory tract and initiate an inflammatory cascade that paralyzes the cilia and inhibits the respiratory functions responsible for clearing secretions, largely through an immune-mediated response.
Pertussis has an incubation period of approximately 7 days, but this can last as long as 3 to 6 weeks. The 3 stages in the course of the disease are:6
- Catarrhal. This stage lasts 1 to 2 weeks and is characterized by coryza, sneezing, and a mild, occasional cough.
- Paroxysmal. This stage lasts 1 to 6 weeks, and is characterized by periods of severe coughing “fits” that include the inspiratory "whoop." These coughing episodes may occur more often at night and may worsen in intensity and frequency in the first 2 to 3 weeks and then gradually decrease. This stage also may include posttussive vomiting.
- Convalescent. During this stage, the cough begins to wane.
Vaccination: Don’t forget adults
The 2 vaccines used to prevent pertussis are DTaP (diphtheria-tetanus-acellular pertussis) and Tdap (tetanus-diphtheria-acellular pertussis). The difference between the 2 is that the Tdap vaccine contains a reduced dose of the diphtheria and acellular pertussis vaccines. DTaP is designed primarily for children younger than 7 years of age. Tdap is given to older children and adults. The CDC and Advisory Committee on Immunization Practices recommend that children receive 5 doses of DTaP, one dose at each of the following ages: 2, 4, 6, and 15 to 18 months and at 4 to 6 years.7 All adults 19 years of age and older who have not yet received a dose of Tdap should receive a single dose regardless of when they last received any immunization for tetanus or diphtheria.7-10 A one-time Tdap booster should be given to all adults in place of a tetanus booster (TABLE 1).7-10
What about pregnant women? Tdap should be administered to every pregnant woman between 27 to 36 weeks gestation regardless of Tdap history.7,11 This strategy allows maternal antibodies to transfer to the infant, thus providing some protection to the newborn prior to pediatric vaccinations.
Is the vaccine becoming less effective? Since 1991, the number of cases of pertussis reported in previously vaccinated adolescents and adults has increased, which suggests waning immunity.12,13 Another recent trial investigating the acellular pertussis vaccine found that immunity decreases dramatically 5 years after the fifth dose.14
Recommendations on who should receive pertussis vaccination have been expanded to include adolescents and adults, including pregnant women and those ages 65 and older in close contact with infants, and this should decrease the overall incidence of disease through decreased communicability.15 Current recommendations call for a single adult vaccination; however, ongoing studies are evaluating whether a booster later in life might be necessary.15
Diagnosis needs to be confirmed by lab testing
Any patient who reports having a persistent cough should be considered for pertussis testing and treatment, and any clinician who triages such patients should ask detailed questions about the characteristics and duration of the patient’s symptoms. However, while a prolonged cough is the hallmark of pertussis, there are many other potential causes of this symptom. Therefore, diagnosis of pertussis requires a combination of clinical and laboratory testing, because clinical parameters alone are neither sensitive nor specific enough for pertussis infection.
TABLE 216 outlines the clinical and laboratory diagnostic criteria for pertussis from the CDC and the World Health Organization. Suspect pertussis in a patient who’s had a cough for more than 14 days that includes an inspiratory “whoop.” In infants, pertussis should be suspected in those with symptoms that suggest cough and associated apnea.16 Order laboratory testing for any patients who have clinical signs or symptoms of pertussis.
Four methods of lab testing for pertussis infection are polymerase chain reaction (PCR), direct fluorescent antibody (DFA) testing, serologic testing, and culture (TABLE 3).17-19 The sensitivity of these tests is as follows: PCR, 90% to 95%; DFA, 50% to 60%; serologic testing, 70% to 80%; and culture, 50% to 70%. The specificity is: PCR, 95% to 98%; DFA, 90% to 100%; serologic testing, 90% to 100%; and culture, 100%.
PCR is the preferred method because of its rapid turnaround and fairly high sensitivity. The reliability of PCR decreases, however, for a patient who’s had a cough for more than 2 weeks because the individual may have transitioned to the convalescent phase, when less bacterial DNA remain.
Results from DFA testing also are rapidly available, but the need for specialized equipment and a well-trained examiner of the specimen limits widespread use of this test. It also is not particularly sensitive for pertussis.
Serologic testing is less reliable in patients who have received an acellular pertussis vaccine and is not helpful in the first few weeks of infection.
The sensitivity of culture is best if the sample is collected appropriately (more on this in a bit) and within the first 2 weeks of symptoms (catarrhal stage). Culture is also very specific.
Given the strengths and weakness of the different tests, an acceptable method of laboratory confirmation is to obtain PCR and/or culture within the first 2 weeks of symptoms in all age groups.17-20 Testing after 2 weeks should include a combination of PCR and serology.17 It is essential that the clinical specimen used for PCR or culture testing for pertussis is properly collected. (See “Collecting a swab for pertussis testing” below.21)
The illustration below shows the correct swab and sampling method. Swab tips may be polyester (such as Dacron or rayon) or they may be nylon-flocked. Cotton-tipped or calcium alginate swabs are not acceptable because the residue will inhibit DNA assays.21 The specimen must be obtained from the posterior nasopharynx and not the nares or oropharynx. The Centers for Disease Control and Prevention offers a video that demonstrates how to properly collect a specimen for testing at http://www.cdc.gov/pertussis/clinical/diagnostic-testing/specimen-collection.html.
Tx is effective when started early
Antibiotics are an effective treatment for pertussis, but they need to be started within the first few weeks of developing symptoms. Studies have not found evidence that antibiotics shorten the duration of pertussis symptoms unless they are given in the catarrhal phase.22,23 It can be challenging to get treatment started during this window, however, because patients may put off seeking care for symptoms they perceive as only minor, such as a cough, until the disease progresses. In addition, physicians may not suspect pertussis in patients who present with a cough they have had for only a short time, and therefore may not test for it.
It may be necessary to rely on clinical suspicion when deciding whether to initiate treatment for pertussis before testing to confirm the diagnosis. For patients in whom clinical suspicion of pertussis is high and who may be in contact with high-risk individuals, it may be acceptable to begin treatment before receiving lab test results.24,25 A recent Cochrane meta-analysis26 recommended initiating treatment to render a patient who has pertussis “noninfectious” but without an expectation of diminishing symptoms.
Limited role for prophylaxis. There is little evidence that prophylactic treatment for pertussis can decrease the spread of the disease. Studies that investigated potential benefits of prophylactic treatment for pertussis have been inconclusive, except for individuals who are in close contact with an infant younger than 6 months of age who has not been fully immunized.27,28
A macrolide antibiotic is generally used to treat pertussis (TABLE 4).25-30 Erythromycin had been the drug of choice, but recent studies have found similar efficacy for azithromycin and clarithromycin.29 For infants younger than one month of age, azithromycin is preferred because in addition to being as effective as other macrolides, it has a better adverse effect profile.29 For patients who are at least 2 months of age, trimethoprim-sulfamethoxazole is an acceptable alternative to a macrolide.
The CDC recommends that any adolescent or adult who has a cough and has had close contact with an individual with a laboratory-confirmed case of pertussis within the past 21 days should be treated.30 Close contacts younger than 7 years of age who have not received the first 4 doses of the pertussis vaccine should be offered treatment.
CORRESPONDENCE
Gary Rivard, DO, Family Medicine Residency Program, Central Maine Medical Center, 76 High Street, Lewiston, ME 04282; rivardga@cmhc.org
› Recommend a
one-time Tdap (tetanus-diphtheria-acellular pertussis) combination vaccine for adults younger than age 64 who need tetanus booster vaccination. A
› Suspect pertussis in a patient who presents with a persistent, paroxysmal cough, with an inspiratory “whoop,” that has lasted for at least
2 weeks. B
› Prescribe a macrolide antibiotic as a first-line treatment for infants, children, and adults who have pertussis. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Despite a high vaccination rate, pertussis is the only vaccine-preventable disease whose incidence is on the rise.1-3 The Centers for Disease Control and Prevention (CDC) reported 48,277 laboratory-confirmed cases in 2012—the most since 1955—and 20 pertussis-related deaths.4 And while only 28,639 pertussis cases were reported in 2013, more than 17,000 cases had already been reported through August 15, 2014, suggesting that the incidence may again be on the rise this year.4
This uptick is likely due to a combination of factors, including a growing awareness of pertussis, and therefore a lower threshold for physicians to test for it. In addition, there’s some evidence that the immunity provided by the currently used pertussis vaccines may wane over time. Recently reported epidemics, including those in California this year and in 2010, as well as in Washington in 2011, have added to this concern.5 This article outlines what you can do to improve prevention, diagnosis, and treatment of pertussis.
A 3-stage course of disease
Bordetella pertussis is an aerobic, gram-negative bacterium that causes symptoms by producing multiple antigenic and biologically active components, including pertussis toxin, filamentous hemagglutinin, and agglutinogens. The bacteria adhere to the cilia in the respiratory tract and initiate an inflammatory cascade that paralyzes the cilia and inhibits the respiratory functions responsible for clearing secretions, largely through an immune-mediated response.
Pertussis has an incubation period of approximately 7 days, but this can last as long as 3 to 6 weeks. The 3 stages in the course of the disease are:6
- Catarrhal. This stage lasts 1 to 2 weeks and is characterized by coryza, sneezing, and a mild, occasional cough.
- Paroxysmal. This stage lasts 1 to 6 weeks, and is characterized by periods of severe coughing “fits” that include the inspiratory "whoop." These coughing episodes may occur more often at night and may worsen in intensity and frequency in the first 2 to 3 weeks and then gradually decrease. This stage also may include posttussive vomiting.
- Convalescent. During this stage, the cough begins to wane.
Vaccination: Don’t forget adults
The 2 vaccines used to prevent pertussis are DTaP (diphtheria-tetanus-acellular pertussis) and Tdap (tetanus-diphtheria-acellular pertussis). The difference between the 2 is that the Tdap vaccine contains a reduced dose of the diphtheria and acellular pertussis vaccines. DTaP is designed primarily for children younger than 7 years of age. Tdap is given to older children and adults. The CDC and Advisory Committee on Immunization Practices recommend that children receive 5 doses of DTaP, one dose at each of the following ages: 2, 4, 6, and 15 to 18 months and at 4 to 6 years.7 All adults 19 years of age and older who have not yet received a dose of Tdap should receive a single dose regardless of when they last received any immunization for tetanus or diphtheria.7-10 A one-time Tdap booster should be given to all adults in place of a tetanus booster (TABLE 1).7-10
What about pregnant women? Tdap should be administered to every pregnant woman between 27 to 36 weeks gestation regardless of Tdap history.7,11 This strategy allows maternal antibodies to transfer to the infant, thus providing some protection to the newborn prior to pediatric vaccinations.
Is the vaccine becoming less effective? Since 1991, the number of cases of pertussis reported in previously vaccinated adolescents and adults has increased, which suggests waning immunity.12,13 Another recent trial investigating the acellular pertussis vaccine found that immunity decreases dramatically 5 years after the fifth dose.14
Recommendations on who should receive pertussis vaccination have been expanded to include adolescents and adults, including pregnant women and those ages 65 and older in close contact with infants, and this should decrease the overall incidence of disease through decreased communicability.15 Current recommendations call for a single adult vaccination; however, ongoing studies are evaluating whether a booster later in life might be necessary.15
Diagnosis needs to be confirmed by lab testing
Any patient who reports having a persistent cough should be considered for pertussis testing and treatment, and any clinician who triages such patients should ask detailed questions about the characteristics and duration of the patient’s symptoms. However, while a prolonged cough is the hallmark of pertussis, there are many other potential causes of this symptom. Therefore, diagnosis of pertussis requires a combination of clinical and laboratory testing, because clinical parameters alone are neither sensitive nor specific enough for pertussis infection.
TABLE 216 outlines the clinical and laboratory diagnostic criteria for pertussis from the CDC and the World Health Organization. Suspect pertussis in a patient who’s had a cough for more than 14 days that includes an inspiratory “whoop.” In infants, pertussis should be suspected in those with symptoms that suggest cough and associated apnea.16 Order laboratory testing for any patients who have clinical signs or symptoms of pertussis.
Four methods of lab testing for pertussis infection are polymerase chain reaction (PCR), direct fluorescent antibody (DFA) testing, serologic testing, and culture (TABLE 3).17-19 The sensitivity of these tests is as follows: PCR, 90% to 95%; DFA, 50% to 60%; serologic testing, 70% to 80%; and culture, 50% to 70%. The specificity is: PCR, 95% to 98%; DFA, 90% to 100%; serologic testing, 90% to 100%; and culture, 100%.
PCR is the preferred method because of its rapid turnaround and fairly high sensitivity. The reliability of PCR decreases, however, for a patient who’s had a cough for more than 2 weeks because the individual may have transitioned to the convalescent phase, when less bacterial DNA remain.
Results from DFA testing also are rapidly available, but the need for specialized equipment and a well-trained examiner of the specimen limits widespread use of this test. It also is not particularly sensitive for pertussis.
Serologic testing is less reliable in patients who have received an acellular pertussis vaccine and is not helpful in the first few weeks of infection.
The sensitivity of culture is best if the sample is collected appropriately (more on this in a bit) and within the first 2 weeks of symptoms (catarrhal stage). Culture is also very specific.
Given the strengths and weakness of the different tests, an acceptable method of laboratory confirmation is to obtain PCR and/or culture within the first 2 weeks of symptoms in all age groups.17-20 Testing after 2 weeks should include a combination of PCR and serology.17 It is essential that the clinical specimen used for PCR or culture testing for pertussis is properly collected. (See “Collecting a swab for pertussis testing” below.21)
The illustration below shows the correct swab and sampling method. Swab tips may be polyester (such as Dacron or rayon) or they may be nylon-flocked. Cotton-tipped or calcium alginate swabs are not acceptable because the residue will inhibit DNA assays.21 The specimen must be obtained from the posterior nasopharynx and not the nares or oropharynx. The Centers for Disease Control and Prevention offers a video that demonstrates how to properly collect a specimen for testing at http://www.cdc.gov/pertussis/clinical/diagnostic-testing/specimen-collection.html.
Tx is effective when started early
Antibiotics are an effective treatment for pertussis, but they need to be started within the first few weeks of developing symptoms. Studies have not found evidence that antibiotics shorten the duration of pertussis symptoms unless they are given in the catarrhal phase.22,23 It can be challenging to get treatment started during this window, however, because patients may put off seeking care for symptoms they perceive as only minor, such as a cough, until the disease progresses. In addition, physicians may not suspect pertussis in patients who present with a cough they have had for only a short time, and therefore may not test for it.
It may be necessary to rely on clinical suspicion when deciding whether to initiate treatment for pertussis before testing to confirm the diagnosis. For patients in whom clinical suspicion of pertussis is high and who may be in contact with high-risk individuals, it may be acceptable to begin treatment before receiving lab test results.24,25 A recent Cochrane meta-analysis26 recommended initiating treatment to render a patient who has pertussis “noninfectious” but without an expectation of diminishing symptoms.
Limited role for prophylaxis. There is little evidence that prophylactic treatment for pertussis can decrease the spread of the disease. Studies that investigated potential benefits of prophylactic treatment for pertussis have been inconclusive, except for individuals who are in close contact with an infant younger than 6 months of age who has not been fully immunized.27,28
A macrolide antibiotic is generally used to treat pertussis (TABLE 4).25-30 Erythromycin had been the drug of choice, but recent studies have found similar efficacy for azithromycin and clarithromycin.29 For infants younger than one month of age, azithromycin is preferred because in addition to being as effective as other macrolides, it has a better adverse effect profile.29 For patients who are at least 2 months of age, trimethoprim-sulfamethoxazole is an acceptable alternative to a macrolide.
The CDC recommends that any adolescent or adult who has a cough and has had close contact with an individual with a laboratory-confirmed case of pertussis within the past 21 days should be treated.30 Close contacts younger than 7 years of age who have not received the first 4 doses of the pertussis vaccine should be offered treatment.
CORRESPONDENCE
Gary Rivard, DO, Family Medicine Residency Program, Central Maine Medical Center, 76 High Street, Lewiston, ME 04282; rivardga@cmhc.org
1. Orenstein WA. Pertussis in adults: epidemiology, signs, symptoms, and implications for vaccination. Clin Infect Dis. 1999;28 suppl 2:S147-S150.
2. Tanaka M, Vitek CR, Pascual FB, et al. Trends in pertussis among infants in the United States, 1980-1999. JAMA. 2003;290:2968-2975.
3. Vitek CR, Pascual FB, Baughman AL, et al. Increase in deaths from pertussis among young infants in the United States in the 1990s. Pediatr Infect Dis J. 2003;22:628-634.
4. Centers for Disease Control and Prevention. Pertussis outbreak trends. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/pertussis/outbreaks/trends.html. Accessed October 10, 2014.
5. Shapiro ED. Acellular vaccines and resurgence of pertussis. JAMA. 2012;308:2149-2150.
6. Centers for Disease Control and Prevention. Pertussis. In: Epidemiology and Prevention of Vaccine-Preventable Diseases. The Pink Book. 2012. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/pubs/pinkbook/pert.html. Accessed October 10, 2014.
7. Centers for Disease Control and Prevention. Pertussis: Summary of vaccine recommendations. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/vpd-vac/pertussis/recs-summary.htm. Accessed October 10, 2014.
8. Lee GM, Murphy TV, Lett S, et al. Cost effectiveness of pertussis vaccination in adults. Am J Prev Med. 2007;32:186-193.
9. Pertussis vaccines: WHO position paper. Wkly Epidemiol Rec. 2010;85:385-400.
10. Kretsinger K, Broder KR, Cortese MM, et al; Centers for Disease Control and Prevention; Advisory Committee on Immunization Practices; Healthcare Infection Control Practices Advisory Committee. Preventing tetanus, diphtheria, and pertussis among adults: use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccine recommendations of the Advisory Committee on Immunization Practices (ACIP) and recommendation of ACIP, supported by the Healthcare Infection Control Practices Advisory Committee (HICPAC), for use of Tdap among healthcare personnel. MMWR Recomm Rep. 2006;55(RR-17):1-37.
11. English P. Pertussis vaccination in pregnant women will protect neonates. Practitioner. 2012;256:5.
12. Winter K, Harriman K, Zipprich J, et al. California pertussis epidemic, 2010. J Pediatr. 2012;161:1091-1096.
13. Centers for Disease Control and Prevention (CDC). Pertussis epidemic—Washington, 2012. MMWR Morb Mortal Wkly Rep. 2012;61:517-522.
14. Klein NP, Bartlett J, Rowhani-Rahbar A, et al. Waning protection after fifth dose of acellular pertussis vaccine in children. N Engl J Med. 2012;367:1012-1019.
15. Centers for Disease Control and Prevention (CDC). Updated recommendations for use of tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis (Tdap) vaccine in adults aged 65 years and older - Advisory Committee on Immunization Practices (ACIP), 2012. MMWR Morb Mortal Wkly Rep. 2012;61:468-470.
16. Cherry JD, Tan T, Wirsing von Konig C, et al. Clinical definitions of pertussis: Summary of a global pertussis initiative roundtable meeting, February 2011. Clin Infect Dis. 2012;54:1756-1764.
17. Zouari A, Smaoui H, Kechrid A. The diagnosis of pertussis: which method to choose?. Crit Rev Microbiol. 2012;38:111-121.
18. Loeffelholz MJ, Thompson CJ, Long KS, et al. Comparison of PCR, culture, and direct fluorescent-antibody testing for detection of Bordetella pertussis. J Clin Microbiol. 1999;37:2872-2876.
19. Tozzi A, Celentano L, Ciofi degli Atti ML, et al. Diagnosis and management of pertussis. CMAJ. 2005;172:509-515.
20. von König CH, Halperin S, Riffelmann M, et al. Pertussis of adults and infants. Lancet Infect Dis. 2002;2:744-750.
21. Cattaneo LA, Edwards KM. Bordetella pertussis (whooping cough). Semin Pediatr Infect Dis. 1995;6:107-117.
22. Hoppe JE, Eichhorn A. Activity of new macrolides against Bordetella pertussis and Bordetella parapertussis. Eur J Clin Microbiol Infect Dis. 1989;8:653-654.
23. Bass JW. Erythromycin for treatment and prevention of pertussis. Pediatr Infect Dis. 1986;5:154-157.
24. Health Protection Surveillance Centre. Guidelines for the Public Health Management of Pertussis: Public Health Medicine Communicable Disease Group HSE—October 2013. Health Protection Surveillance Centre Web site. Available at: http://www.hpsc.ie/A-Z/VaccinePreventable/PertussisWhoopingCough/InformationforHealthcareWorkers/File,13577,en.pdf. Accessed October 2, 2014.
25. Dodhia H, Miller E. Review of the evidence for the use of erythromycin in the management of persons exposed to pertussis. Epidemiol Infect. 1998;120:143-149.
26. Altunaiji S, Kukuruzovic R, Curtis N, et al. Antibiotics for whooping cough (pertussis). Cochrane Database Syst Rev. 2007;(3):CD004404.
27. Prophylactic erythromycin for whooping-cough contacts. Lancet. 1981;1:772.
28. Halperin SA, Bortolussi R, Langley JM, et al. A randomized, placebo-controlled trial of erythromycin estolate chemoprophylaxis for household contacts of children with culture-positive bordetella pertussis infection. Pediatrics. 1999;104:e42.
29. Langley JM, Halperin SA, Boucher FD, et al; Pediatric Investigators Collaborative Network on Infections in Canada (PICNIC). Azithromycin is as effective as and better tolerated than erythromycin estolate for the treatment of pertussis. Pediatrics. 2004;114:e96-e101.
30. Tiwari T, Murphy TV, Moran J; National Immunization Program, CDC. Recommended antimicrobial agents for the treatment and postexposure prophylaxis of pertussis: 2005 CDC Guidelines. MMWR Recomm Rep. 2005;54(RR-14):1-16.
1. Orenstein WA. Pertussis in adults: epidemiology, signs, symptoms, and implications for vaccination. Clin Infect Dis. 1999;28 suppl 2:S147-S150.
2. Tanaka M, Vitek CR, Pascual FB, et al. Trends in pertussis among infants in the United States, 1980-1999. JAMA. 2003;290:2968-2975.
3. Vitek CR, Pascual FB, Baughman AL, et al. Increase in deaths from pertussis among young infants in the United States in the 1990s. Pediatr Infect Dis J. 2003;22:628-634.
4. Centers for Disease Control and Prevention. Pertussis outbreak trends. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/pertussis/outbreaks/trends.html. Accessed October 10, 2014.
5. Shapiro ED. Acellular vaccines and resurgence of pertussis. JAMA. 2012;308:2149-2150.
6. Centers for Disease Control and Prevention. Pertussis. In: Epidemiology and Prevention of Vaccine-Preventable Diseases. The Pink Book. 2012. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/pubs/pinkbook/pert.html. Accessed October 10, 2014.
7. Centers for Disease Control and Prevention. Pertussis: Summary of vaccine recommendations. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/vaccines/vpd-vac/pertussis/recs-summary.htm. Accessed October 10, 2014.
8. Lee GM, Murphy TV, Lett S, et al. Cost effectiveness of pertussis vaccination in adults. Am J Prev Med. 2007;32:186-193.
9. Pertussis vaccines: WHO position paper. Wkly Epidemiol Rec. 2010;85:385-400.
10. Kretsinger K, Broder KR, Cortese MM, et al; Centers for Disease Control and Prevention; Advisory Committee on Immunization Practices; Healthcare Infection Control Practices Advisory Committee. Preventing tetanus, diphtheria, and pertussis among adults: use of tetanus toxoid, reduced diphtheria toxoid and acellular pertussis vaccine recommendations of the Advisory Committee on Immunization Practices (ACIP) and recommendation of ACIP, supported by the Healthcare Infection Control Practices Advisory Committee (HICPAC), for use of Tdap among healthcare personnel. MMWR Recomm Rep. 2006;55(RR-17):1-37.
11. English P. Pertussis vaccination in pregnant women will protect neonates. Practitioner. 2012;256:5.
12. Winter K, Harriman K, Zipprich J, et al. California pertussis epidemic, 2010. J Pediatr. 2012;161:1091-1096.
13. Centers for Disease Control and Prevention (CDC). Pertussis epidemic—Washington, 2012. MMWR Morb Mortal Wkly Rep. 2012;61:517-522.
14. Klein NP, Bartlett J, Rowhani-Rahbar A, et al. Waning protection after fifth dose of acellular pertussis vaccine in children. N Engl J Med. 2012;367:1012-1019.
15. Centers for Disease Control and Prevention (CDC). Updated recommendations for use of tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis (Tdap) vaccine in adults aged 65 years and older - Advisory Committee on Immunization Practices (ACIP), 2012. MMWR Morb Mortal Wkly Rep. 2012;61:468-470.
16. Cherry JD, Tan T, Wirsing von Konig C, et al. Clinical definitions of pertussis: Summary of a global pertussis initiative roundtable meeting, February 2011. Clin Infect Dis. 2012;54:1756-1764.
17. Zouari A, Smaoui H, Kechrid A. The diagnosis of pertussis: which method to choose?. Crit Rev Microbiol. 2012;38:111-121.
18. Loeffelholz MJ, Thompson CJ, Long KS, et al. Comparison of PCR, culture, and direct fluorescent-antibody testing for detection of Bordetella pertussis. J Clin Microbiol. 1999;37:2872-2876.
19. Tozzi A, Celentano L, Ciofi degli Atti ML, et al. Diagnosis and management of pertussis. CMAJ. 2005;172:509-515.
20. von König CH, Halperin S, Riffelmann M, et al. Pertussis of adults and infants. Lancet Infect Dis. 2002;2:744-750.
21. Cattaneo LA, Edwards KM. Bordetella pertussis (whooping cough). Semin Pediatr Infect Dis. 1995;6:107-117.
22. Hoppe JE, Eichhorn A. Activity of new macrolides against Bordetella pertussis and Bordetella parapertussis. Eur J Clin Microbiol Infect Dis. 1989;8:653-654.
23. Bass JW. Erythromycin for treatment and prevention of pertussis. Pediatr Infect Dis. 1986;5:154-157.
24. Health Protection Surveillance Centre. Guidelines for the Public Health Management of Pertussis: Public Health Medicine Communicable Disease Group HSE—October 2013. Health Protection Surveillance Centre Web site. Available at: http://www.hpsc.ie/A-Z/VaccinePreventable/PertussisWhoopingCough/InformationforHealthcareWorkers/File,13577,en.pdf. Accessed October 2, 2014.
25. Dodhia H, Miller E. Review of the evidence for the use of erythromycin in the management of persons exposed to pertussis. Epidemiol Infect. 1998;120:143-149.
26. Altunaiji S, Kukuruzovic R, Curtis N, et al. Antibiotics for whooping cough (pertussis). Cochrane Database Syst Rev. 2007;(3):CD004404.
27. Prophylactic erythromycin for whooping-cough contacts. Lancet. 1981;1:772.
28. Halperin SA, Bortolussi R, Langley JM, et al. A randomized, placebo-controlled trial of erythromycin estolate chemoprophylaxis for household contacts of children with culture-positive bordetella pertussis infection. Pediatrics. 1999;104:e42.
29. Langley JM, Halperin SA, Boucher FD, et al; Pediatric Investigators Collaborative Network on Infections in Canada (PICNIC). Azithromycin is as effective as and better tolerated than erythromycin estolate for the treatment of pertussis. Pediatrics. 2004;114:e96-e101.
30. Tiwari T, Murphy TV, Moran J; National Immunization Program, CDC. Recommended antimicrobial agents for the treatment and postexposure prophylaxis of pertussis: 2005 CDC Guidelines. MMWR Recomm Rep. 2005;54(RR-14):1-16.

Another risk to US travelers—malaria
› Assess the need for nonpharmacologic, behavioral interventions and for chemoprophylaxis based on a destination’s relative risk to travelers, planned and potential activities, and patient comorbidities. B
› Choose an antimalarial medication based on knowledge of area-specific drug effectiveness or resistance patterns, trip duration, drug cost, tolerance for adverse effects, and comorbidities. C
› Presume a diagnosis of malaria until proven otherwise in any traveler who is febrile after returning from a malaria-endemic region. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Although malaria was eradicated as an endemic disease in the United States in the early 1950s,1 it still returns yearly in approximately 1500 individuals who travel to foreign countries2—most of whom neglected to use prophylactic measures or use them properly.3 In more than 60 documented cases, these infected individuals have been the source of local transmission in their communities.2 To reduce the individual and public health risks associated with malaria, this article focuses on steps that international travelers can take to limit their risk of the disease.
What travelers need to know
In 2013, more than 61.5 million residents of the United States traveled abroad, approximately 30% of whom visited malaria-endemic regions: Mexico and equatorial nations in Central and South America; Africa; the Middle East; and South, East, and Southeast Asia.4 Counseling on appropriate preventive measures fits the Medical Home concept of comprehensive, preventive, patient-centered care, and pre-travel consultation—including a review of health data and itineraries, and patient education—can be a team-based effort.5
Begin your planning for malaria prophylaxis by assessing your patient’s individual risk. Key variables are a patient’s detailed itinerary, a credible and current source of information on location-specific malaria prevalence, personal risk factors, and risk tolerance. Shared decision-making is vital and enhances adherence to the prescribed regimen.
Endemicity varies regionally. Without chemoprophylaxis, risk of infection ranges from more than 20% in Papua New Guinea to 0.01% in Central America, with wide exposure risk variations likely, even within regions.6 Travel to areas of high endemicity requires more aggressive malaria prevention strategies than travel to low-endemicity regions.
Risk of exposure is lower with short visits,7 business-only travel, urban-only stays in some countries, day trips to endemic areas,7 and travel during seasons with lower mosquito burden. Likewise, travelers staying in a hotel with sealed windows will face lower nighttime Anopheles mosquito exposure. In these cases, nonpharmacologic measures alone may be appropriate.
Those at particularly high risk for complicated or lethal malarial infection are children, pregnant women, elderly individuals, and immunocompromised patients.7 In addition to counseling high-risk patients about prophylactic measures, consider advising against travel in certain circumstances. Among those at highest risk for acquiring malaria are immigrants and refugees traveling to their ancestral homelands to visit friends and relatives (VFR).2 Many VFR travelers fail to take appropriate prophylactic measures when “going home.”8 A significant number of cases of travel-acquired malaria occurs in VFR children.9
Individualizing prevention directives
The mainstays of malaria prevention include nonpharmacologic and behavioral interventions, as well as chemoprophylaxis. Most cases of malaria in travelers returning to the United States result from the improper implementation of prophylactic measures.3 Discussing individual risk with travelers is an easy way to bolster adherence to malaria prevention measures, and some evidence suggests it is effective10 (strength of recommendation [SOR]: C). Other limited studies have also shown that malaria education can improve knowledge about malaria transmission and increase the likelihood that preventive measures will be used.11,12
Recommend nonpharmacologic measures even for those using chemoprophylaxis
Nonpharmacologic interventions such as sleeping under permethrin-treated bednets, wearing long sleeves and full-length pants, treating clothes with permethrin, and applying DEET (N,N-diethyl-meta-toluamide) to exposed skin are effective and have the added benefit of preventing non-malarial arthropod-borne diseases4 (SOR: B). Studies have shown that, compared with sleeping without nets, the use of insecticide treated-nets can reduce child mortality by 17% and the incidence of uncomplicated malarial episodes by 50%.13 In areas with malaria transmission, 10% to 30% DEET—used alone or in combination with permethrin-treated clothing— can reduce bite load, although the American Academy of Pediatrics recommends against using DEET in children younger than 2 months of age.14,15
Using these measures in combination from dusk to dawn, when Anopheles mosquitoes are active, has been shown to be effective, although randomized, controlled studies are lacking.16 Remaining indoors during these peak biting periods is also advisable. In certain areas, and with the right itinerary, the traveler may only need to employ nonpharmacologic methods of preventing malarial infection. Recommend them to all patients traveling to malarial regions, even to individuals using pharmacologic prophylaxis.
Factors determining the need for, and selection of, chemoprophylaxis
When used properly, chemoprophylactic drugs are effective in preventing malaria (SOR: A). Atovaquone-proguanil achieves efficacy of 95% to 100%,17 while doxycycline, primaquine, and mefloquine are slightly less effective.18-20 Chloroquine is effective in 6 regions of the tropics and subtropics where Plasmodium falciparum resistance has not developed. Select a drug based on your assessment of an individual’s level of risk according to the personal itinerary, trip duration and accommodations, cost of medication, tolerance for adverse effects, and other factors (eg, comorbidities, concurrent drug usage, pregnancy).
Location matters. The risk of malaria transmission can vary considerably not only between countries, but also regionally within countries and even between a city and its immediate surroundings. Therefore, select a chemoprophylactic agent based on the specific itinerary, planned activities, the potential for unforeseen additional excursions, and local Plasmodium resistance patterns. For example, chloroquine is effective only in the Caribbean, Central America, and some countries in the Middle East.21 Mefloquine resistance has been reported in parts of Cambodia, Thailand, Vietnam, Burma, China, and Laos.21
On its Travelers’ Health Web site (www.cdc.gov/travel), the Centers for Disease Control and Prevention (CDC) reports for each country 1) the risk of malaria transmission, 2) areas within the country that pose a risk, 3) evidence of Plasmodium drug resistance, 4) which Plasmodium species are active, and 5) which chemoprophylactic medications are recommended.22 Additional Web sites, either free or subscription-based, allow users to view this same information on maps, advise on where insect precautions alone are sufficiently protective, and provide information about the traveler’s risk of contracting other diseases (TABLE 1).
TABLE 1
| Web resources on infectious diseases of concern to international travelers | |
| Resource | Notes |
Centers for Disease Control and Prevention | Free Site Go to Yellow Book » Contents » Chapter 3 » “Travel Vaccines & Malaria Information, by Country” for country-specific information about the risk of malaria transmission |
VHI Healthcare | Free Site Destination-specific information about travel alerts and vaccine recommendations Does not report malaria transmission data |
Gideon | Subscription only Online application that helps with diagnosing infectious diseases and keeping up to date with global health literature |
Travax | Subscription only Information about recommended vaccines and country-specific risk of malaria transmission |
Tropimed | Subscription only Information about recommended vaccines and country-specific risk of malaria transmission |
Comparative adverse effects of antimalarial agents. A Cochrane Review on the tolerability of chemoprophylactic agents concluded that atovaquone-proguanil and doxycycline were better tolerated than mefloquine (SOR: B). Compared with mefloquine, atovaquone-proguanil led to fewer reports of any adverse effects (relative risk [RR]=0.72), gastrointestinal adverse effects (RR=0.54), and neuropsychiatric adverse events (RR= 0.49-0.86, depending on the studies).23 Doxycycline users have reported fewer neuropsychiatric events (RR=0.84) than mefloquine users.23 These are relatively small differences, and the authors point out that these figures are based on low-quality evidence. Additional research is likely to have an impact on the confidence in the estimate of effect and to ultimately change the estimate.
Mefloquine is contraindicated in travelers with seizures, active or recent history of depression, generalized anxiety disorder, psychosis, schizophrenia, or other psychiatric disorders. Compared with mefloquine, atovaquone-proguanil and doxycycline cause fewer neuropsychiatric adverse effects (such as vivid dreams, dizziness, anxiety, depression, visual disturbance, or seizures).24 Caution is advised when prescribing chloroquine for patients with epilepsy because the medication has the potential to lower the seizure threshold.25
Use caution when prescribing mefloquine for patients with cardiac conduction disturbances. Electrocardiogram alterations such as sinus bradycardia, first-degree AV block, prolongation of QTc intervals, and abnormal T wave changes have been reported.26 Chloroquine can also prolong QTc intervals.26
Safety in pregnancy and breastfeeding. Malaria in pregnancy is associated with increased rates of anemia, low birth weight, prematurity, intrauterine growth restriction, and infant mortality.27 Chloroquine and mefloquine are considered safe during pregnancy and breastfeeding. Doxycycline has been associated with increased risk of harm to the fetus. Atovaquone-proguanil can be used in breastfeeding women if the child is ≥5 kg (≥11 lbs). Chemoprophylaxis taken by the mother while breastfeeding does not protect the infant from infection.
Dosing considerations. Mefloquine and chloroquine are dosed weekly; doxycycline and atovaquone-proguanil are taken daily. Travelers staying in a malaria-endemic region for longer periods (months rather than weeks) often prefer the weekly rather than daily medications; however, this may not be possible due to the adverse-effect profile of mefloquine or to traveling in an area with known chloroquine resistance. Some individuals prefer the routine of taking a medication daily, since remembering to take a single dose on the same day each week can be challenging. Others may not want to carry a large number of pills and therefore prefer weekly dosing. Have patients take medications before the trip, to assess tolerability and to ensure adequate blood concentrations before exposure.
Because mefloquine, doxycycline, and chloroquine target only the blood stages of Plasmodium, patients must continue these medications for 4 weeks following the exposure period to ensure adequate coverage as parasites are released from the liver. Because doxycycline is taken daily and has to be continued for 4 weeks following the exposure period, the total number of pills taken is higher for this regimen. Atovaquone-proguanil is active against hepatic and blood stages and can be discontinued a week following the exposure period.
With children, base dosing on body weight and do not exceed the recommended adult dose. When fractions of tablets are used (such as with mefloquine and atovaquone-proguanil dosing), pharmacists can crush tablets and place divided doses in capsules, to be sprinkled as needed into food such as applesauce or jelly. Mefloquine and chloroquine can be given to children of all ages and weights. Although atovaquone-proguanil is approved only for children ≥11 kg (24 lbs), dosing schedules have been calculated for children who weigh ≥5 kg.21 Doxycycline is recommended only for children who are at least 8 years of age.
Cost. For a 2-week exposure period, chloroquine is the least expensive medication (although regions in which it is recommended are limited due to resistance) (TABLE 27,25,26).
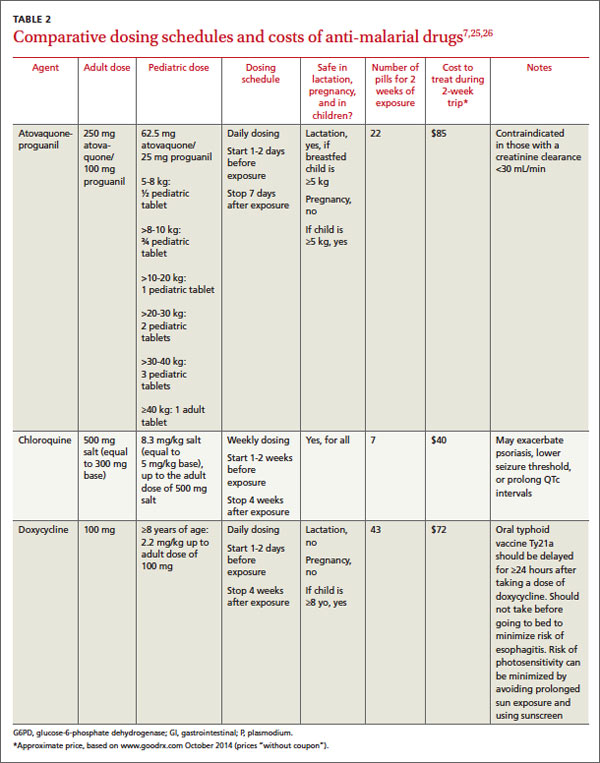
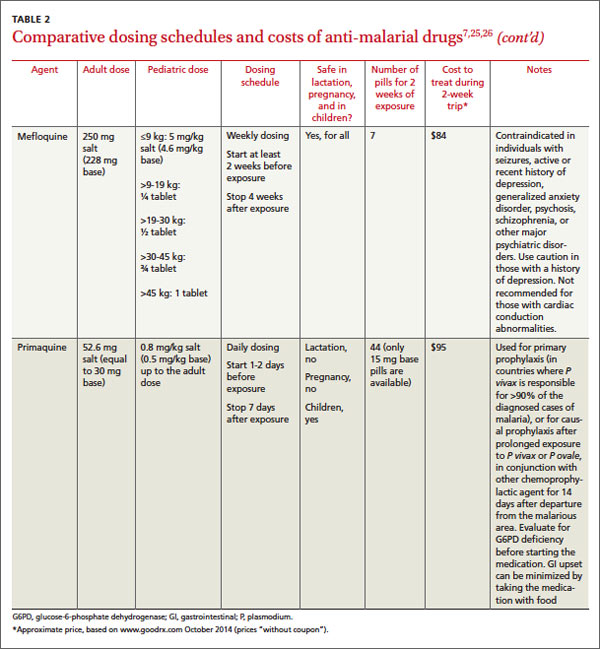
Ask about accommodations
Since Anopheles mosquitoes feed between dusk and dawn, inquiring about accommodations can further clarify a patient’s malaria risk. Staying in air-conditioned housing (implying that the interior can be sealed) or that has screened windows can reduce exposure to mosquitoes, although data are lacking regarding whether the latter practice reduces the incidence of malaria transmission28 (SOR: C).
Share decision making
After considering the key factors determining a patient’s level of risk, you may decide to recommend no specific interventions, to advise insect avoidance measures only, to combine insect avoidance with chemoprophylaxis, or to caution against traveling to a malaria-endemic region. The patient’s contribution to the final decision includes personal preferences, values, and risk tolerance—particularly when comorbidities are involved.
When preventive measures fail
Approximately 0.2% of travelers to malaria-endemic regions will become infected, despite proper pre-travel counseling and prophylaxis.29 In the United States, malaria is often misdiagnosed or improperly treated.30 The time from initial presentation to correct diagnosis of malaria has been reported as an astonishingly high 4 to 8.5 days, depending on the population.31,32
A high index of suspicion is needed and will ensure timely care when any febrile traveler returns from a malaria-endemic area.33 Be sure to advise patients to seek medical attention if they are feverish upon returning home.
Once suspected, the diagnosis of malaria can be readily confirmed through the use of antibody-, nucleic acid-, or microscopy-based techniques (the latter to directly visualize Plasmodium species in blood smears).
Although malaria chemoprophylaxis is relatively straightforward, malaria treatment—especially in cases of chemoprophylaxis failures—may not be, and the topic is beyond the scope of this article. For guidance on treating malaria, consult a knowledgeable physician or contact the CDC at www.cdc.gov/malaria/, or at (855) 856-4713 (weekdays, 9 am to 5 pm EST) or (770) 488-7100 (weekends or after normal business hours; ask for the Malaria Branch clinician on call).
CORRESPONDENCE
Mark K. Huntington, MD, PhD, Center for Family Medicine, 1115 East 20th Street, Sioux Falls, SD 57105; mark.huntington@usd.edu
1. Mali S, Steele S, Slutsker L, et al; Centers for Disease Control and Prevention (CDC). Malaria surveillance - United States, 2008. MMWR Surveill Summ. 2010;59:1-15.
2. Centers for Disease Control and Prevention. Malaria facts. Centers for Disease Control and Prevention Web site. Available at: www.cdc.gov/malaria/about/facts.html. Accessed September 29, 2014.
3. Huntington MK. Healthy people, malaria and South Dakota. S D Med. 2012;65:297-300.
4. Office of Travel and Tourism Industries. U.S. citizen travel to international regions, 2013. Office of Travel and Tourism Industries Web site. Available at: http://travel.trade.gov/view/m-2013-O-001/index.html. Accessed September 29, 2014.
5. Bazemore AW, Huntington M. The pretravel consultation. Am Fam Physician. 2009;80:583-590.
6. Bradley DJ, Warhurst DC, Blaze M, et al. Malaria imported into the United Kingdom in 1996. Euro Surveill. 1998;3:40-42.
7. Arguin PM, Tan KR, et al; Centers for Disease Control and Prevention. Infectious diseases related to travel. Centers for Disease Control and Prevention Web site. Available at: http://wwwnc.cdc.gov/travel/yellowbook/2014/chapter-3-infectious-diseases-related-to-travel/malaria. Accessed October 15, 2014.
8. Pavli A, Maltezou HC. Malaria and travellers visiting friends and relatives. Travel Med Infect Dis. 2010;8:161-168.
9. Stäger K, Legros F, Krause G, et al. Imported malaria in children in industrialized countries, 1992-2002. Emerg Infect Dis. 2009;15:185-191.
10. Hartjes LB, Baumann LC, Henriques JB. Travel health risk perceptions and prevention behaviors of US study abroad students. J Travel Med. 2009;16:338-343.
11. Kishore J, Gupta VK, Singh SV, et al. Impact of health education intervention on knowledge and community action for malaria control in Delhi. J Commun Dis. 2008;40:183-192.
12. Chirdan OO, Zoakah AI, Ejembi CL. Impact of health education on home treatment and prevention of malaria in Jengre, North Central Nigeria. Ann Afr Med. 2008;7:112-119.
13. Lengeler C. Insecticide-treated bed nets and curtains for preventing malaria. Cochrane Database Syst Rev. 2004;(2): CD000363.
14. Centers for Disease Control and Prevention. Fight the bite for protection from malaria: Guidelines for DEET insect repellent use. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/malaria/toolkit/DEET.pdf. Accessed September 29, 2014.
15. American Academy of Pediatrics. Safety & prevention. Healthychildren.org Web site. Available at: http://www.healthychildren. org/English/safety-prevention/at-play/Pages/Insect-Repellents. aspx. Accessed September 29, 2014.
16. Croft AM, Baker D, von Bertele MJ. An evidence-based vector control strategy for military deployments: the British Army experience. Med Trop (Mars). 2001;61:91-98.
17. Boggild AK, Parise ME, Lewis LS, et al. Atovaquone-proguanil: report from the CDC expert meeting on malaria chemoprophylaxis (II). Am J Trop Med Hyg. 2007;76:208-223.
18. Tan KR, Magill AJ, Parise ME, et al; Centers for Disease Control and Prevention. Doxycycline for malaria chemoprophylaxis and treatment: report from the CDC expert meeting on malaria chemoprophylaxis. Am J Trop Med Hyg. 2011;84:517-531.
19. Hill DR, Baird JK, Parise ME, et al. Primaquine: report from CDC expert meeting on malaria chemoprophylaxis I. Am J Trop Med Hyg. 2006;75:402-415.
20. Steffen R, Fuchs E, Schildknecht J, et al. Mefloquine compared with other malaria chemoprophylactic regimens in tourists visiting east Africa. Lancet. 1993;341:1299-1303.
21. Centers for Disease Control and Prevention. CDC Health Information for International Travel 2014. New York, NY: Oxford University Press; 2014.
22. Gershman MD, Jentes ES, Johnson KJ, et al; Centers for Disease Control and Prevention. Infectious diseases related to travel. Centers for Disease Control and Prevention Web site. Available at: http://wwwnc.cdc.gov/travel/yellowbook/2012/chapter-3- infectious-diseases-related-to-travel/yellow-fever-and-malaria- information-by-country.htm. Accessed September 29, 2014.
23. Jacquerioz FA, Croft AM. Drugs for preventing malaria in travellers. Cochrane Database Syst Rev. 2009;(4):CD006491.
24. Schlagenhauf P, Tschopp A, Johnson R, et al. Tolerability of malaria chemoprophylaxis in non-immune travellers to sub-Saharan Africa: multicentre, randomised, double blind, four arm study. BMJ. 2003;327:1078.
25. Chloroquine phosphate [package insert]. Eatontown, NJ: Westward Pharmaceutical Corp; 2010.
26. Lariam [package insert]. Roche Laboratories, Inc: Nutley, NJ; 2004.
27. Steketee RW, Nahlen BL, Parise ME, et al. The burden of malaria in pregnancy in malaria-endemic areas. Am J Trop Med Hyg. 2001;64(1-2 suppl):28-35.
28. Kirby MJ, Ameh D, Bottomley C, et al. Effect of two different house screening interventions on exposure to malaria vectors and on anaemia in children in The Gambia: a randomised controlled trial. Lancet. 2009;374:998-1009.
29. Steffen R, Amitirigala I, Mutsch M. Health risks among travelers--need for regular updates. J Travel Med. 2008;15:145-146.
30. Dorsey G, Gandhi M, Oyugi JH, et al. Difficulties in the prevention, diagnosis, and treatment of imported malaria. Arch Intern Med. 2000;160:2505-2510.
31. Newman RD, Parise ME, Barber AM, et al. Malaria-related deaths among U.S. travelers, 1963-2001. Ann Intern Med. 2004;141: 547-555.
32. Lesko CR, Arguin PM, Newman RD. Congenital malaria in the United States: a review of cases from 1966 to 2005. Arch Pediatr Adolesc Med. 2007;161:1062-1067.
33. Blair JE. Evaluation of fever in the international traveler. Unwanted ‘souvenir’ can have many causes. Postgrad Med. 2004;116: 13-20,29.
› Assess the need for nonpharmacologic, behavioral interventions and for chemoprophylaxis based on a destination’s relative risk to travelers, planned and potential activities, and patient comorbidities. B
› Choose an antimalarial medication based on knowledge of area-specific drug effectiveness or resistance patterns, trip duration, drug cost, tolerance for adverse effects, and comorbidities. C
› Presume a diagnosis of malaria until proven otherwise in any traveler who is febrile after returning from a malaria-endemic region. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Although malaria was eradicated as an endemic disease in the United States in the early 1950s,1 it still returns yearly in approximately 1500 individuals who travel to foreign countries2—most of whom neglected to use prophylactic measures or use them properly.3 In more than 60 documented cases, these infected individuals have been the source of local transmission in their communities.2 To reduce the individual and public health risks associated with malaria, this article focuses on steps that international travelers can take to limit their risk of the disease.
What travelers need to know
In 2013, more than 61.5 million residents of the United States traveled abroad, approximately 30% of whom visited malaria-endemic regions: Mexico and equatorial nations in Central and South America; Africa; the Middle East; and South, East, and Southeast Asia.4 Counseling on appropriate preventive measures fits the Medical Home concept of comprehensive, preventive, patient-centered care, and pre-travel consultation—including a review of health data and itineraries, and patient education—can be a team-based effort.5
Begin your planning for malaria prophylaxis by assessing your patient’s individual risk. Key variables are a patient’s detailed itinerary, a credible and current source of information on location-specific malaria prevalence, personal risk factors, and risk tolerance. Shared decision-making is vital and enhances adherence to the prescribed regimen.
Endemicity varies regionally. Without chemoprophylaxis, risk of infection ranges from more than 20% in Papua New Guinea to 0.01% in Central America, with wide exposure risk variations likely, even within regions.6 Travel to areas of high endemicity requires more aggressive malaria prevention strategies than travel to low-endemicity regions.
Risk of exposure is lower with short visits,7 business-only travel, urban-only stays in some countries, day trips to endemic areas,7 and travel during seasons with lower mosquito burden. Likewise, travelers staying in a hotel with sealed windows will face lower nighttime Anopheles mosquito exposure. In these cases, nonpharmacologic measures alone may be appropriate.
Those at particularly high risk for complicated or lethal malarial infection are children, pregnant women, elderly individuals, and immunocompromised patients.7 In addition to counseling high-risk patients about prophylactic measures, consider advising against travel in certain circumstances. Among those at highest risk for acquiring malaria are immigrants and refugees traveling to their ancestral homelands to visit friends and relatives (VFR).2 Many VFR travelers fail to take appropriate prophylactic measures when “going home.”8 A significant number of cases of travel-acquired malaria occurs in VFR children.9
Individualizing prevention directives
The mainstays of malaria prevention include nonpharmacologic and behavioral interventions, as well as chemoprophylaxis. Most cases of malaria in travelers returning to the United States result from the improper implementation of prophylactic measures.3 Discussing individual risk with travelers is an easy way to bolster adherence to malaria prevention measures, and some evidence suggests it is effective10 (strength of recommendation [SOR]: C). Other limited studies have also shown that malaria education can improve knowledge about malaria transmission and increase the likelihood that preventive measures will be used.11,12
Recommend nonpharmacologic measures even for those using chemoprophylaxis
Nonpharmacologic interventions such as sleeping under permethrin-treated bednets, wearing long sleeves and full-length pants, treating clothes with permethrin, and applying DEET (N,N-diethyl-meta-toluamide) to exposed skin are effective and have the added benefit of preventing non-malarial arthropod-borne diseases4 (SOR: B). Studies have shown that, compared with sleeping without nets, the use of insecticide treated-nets can reduce child mortality by 17% and the incidence of uncomplicated malarial episodes by 50%.13 In areas with malaria transmission, 10% to 30% DEET—used alone or in combination with permethrin-treated clothing— can reduce bite load, although the American Academy of Pediatrics recommends against using DEET in children younger than 2 months of age.14,15
Using these measures in combination from dusk to dawn, when Anopheles mosquitoes are active, has been shown to be effective, although randomized, controlled studies are lacking.16 Remaining indoors during these peak biting periods is also advisable. In certain areas, and with the right itinerary, the traveler may only need to employ nonpharmacologic methods of preventing malarial infection. Recommend them to all patients traveling to malarial regions, even to individuals using pharmacologic prophylaxis.
Factors determining the need for, and selection of, chemoprophylaxis
When used properly, chemoprophylactic drugs are effective in preventing malaria (SOR: A). Atovaquone-proguanil achieves efficacy of 95% to 100%,17 while doxycycline, primaquine, and mefloquine are slightly less effective.18-20 Chloroquine is effective in 6 regions of the tropics and subtropics where Plasmodium falciparum resistance has not developed. Select a drug based on your assessment of an individual’s level of risk according to the personal itinerary, trip duration and accommodations, cost of medication, tolerance for adverse effects, and other factors (eg, comorbidities, concurrent drug usage, pregnancy).
Location matters. The risk of malaria transmission can vary considerably not only between countries, but also regionally within countries and even between a city and its immediate surroundings. Therefore, select a chemoprophylactic agent based on the specific itinerary, planned activities, the potential for unforeseen additional excursions, and local Plasmodium resistance patterns. For example, chloroquine is effective only in the Caribbean, Central America, and some countries in the Middle East.21 Mefloquine resistance has been reported in parts of Cambodia, Thailand, Vietnam, Burma, China, and Laos.21
On its Travelers’ Health Web site (www.cdc.gov/travel), the Centers for Disease Control and Prevention (CDC) reports for each country 1) the risk of malaria transmission, 2) areas within the country that pose a risk, 3) evidence of Plasmodium drug resistance, 4) which Plasmodium species are active, and 5) which chemoprophylactic medications are recommended.22 Additional Web sites, either free or subscription-based, allow users to view this same information on maps, advise on where insect precautions alone are sufficiently protective, and provide information about the traveler’s risk of contracting other diseases (TABLE 1).
TABLE 1
| Web resources on infectious diseases of concern to international travelers | |
| Resource | Notes |
Centers for Disease Control and Prevention | Free Site Go to Yellow Book » Contents » Chapter 3 » “Travel Vaccines & Malaria Information, by Country” for country-specific information about the risk of malaria transmission |
VHI Healthcare | Free Site Destination-specific information about travel alerts and vaccine recommendations Does not report malaria transmission data |
Gideon | Subscription only Online application that helps with diagnosing infectious diseases and keeping up to date with global health literature |
Travax | Subscription only Information about recommended vaccines and country-specific risk of malaria transmission |
Tropimed | Subscription only Information about recommended vaccines and country-specific risk of malaria transmission |
Comparative adverse effects of antimalarial agents. A Cochrane Review on the tolerability of chemoprophylactic agents concluded that atovaquone-proguanil and doxycycline were better tolerated than mefloquine (SOR: B). Compared with mefloquine, atovaquone-proguanil led to fewer reports of any adverse effects (relative risk [RR]=0.72), gastrointestinal adverse effects (RR=0.54), and neuropsychiatric adverse events (RR= 0.49-0.86, depending on the studies).23 Doxycycline users have reported fewer neuropsychiatric events (RR=0.84) than mefloquine users.23 These are relatively small differences, and the authors point out that these figures are based on low-quality evidence. Additional research is likely to have an impact on the confidence in the estimate of effect and to ultimately change the estimate.
Mefloquine is contraindicated in travelers with seizures, active or recent history of depression, generalized anxiety disorder, psychosis, schizophrenia, or other psychiatric disorders. Compared with mefloquine, atovaquone-proguanil and doxycycline cause fewer neuropsychiatric adverse effects (such as vivid dreams, dizziness, anxiety, depression, visual disturbance, or seizures).24 Caution is advised when prescribing chloroquine for patients with epilepsy because the medication has the potential to lower the seizure threshold.25
Use caution when prescribing mefloquine for patients with cardiac conduction disturbances. Electrocardiogram alterations such as sinus bradycardia, first-degree AV block, prolongation of QTc intervals, and abnormal T wave changes have been reported.26 Chloroquine can also prolong QTc intervals.26
Safety in pregnancy and breastfeeding. Malaria in pregnancy is associated with increased rates of anemia, low birth weight, prematurity, intrauterine growth restriction, and infant mortality.27 Chloroquine and mefloquine are considered safe during pregnancy and breastfeeding. Doxycycline has been associated with increased risk of harm to the fetus. Atovaquone-proguanil can be used in breastfeeding women if the child is ≥5 kg (≥11 lbs). Chemoprophylaxis taken by the mother while breastfeeding does not protect the infant from infection.
Dosing considerations. Mefloquine and chloroquine are dosed weekly; doxycycline and atovaquone-proguanil are taken daily. Travelers staying in a malaria-endemic region for longer periods (months rather than weeks) often prefer the weekly rather than daily medications; however, this may not be possible due to the adverse-effect profile of mefloquine or to traveling in an area with known chloroquine resistance. Some individuals prefer the routine of taking a medication daily, since remembering to take a single dose on the same day each week can be challenging. Others may not want to carry a large number of pills and therefore prefer weekly dosing. Have patients take medications before the trip, to assess tolerability and to ensure adequate blood concentrations before exposure.
Because mefloquine, doxycycline, and chloroquine target only the blood stages of Plasmodium, patients must continue these medications for 4 weeks following the exposure period to ensure adequate coverage as parasites are released from the liver. Because doxycycline is taken daily and has to be continued for 4 weeks following the exposure period, the total number of pills taken is higher for this regimen. Atovaquone-proguanil is active against hepatic and blood stages and can be discontinued a week following the exposure period.
With children, base dosing on body weight and do not exceed the recommended adult dose. When fractions of tablets are used (such as with mefloquine and atovaquone-proguanil dosing), pharmacists can crush tablets and place divided doses in capsules, to be sprinkled as needed into food such as applesauce or jelly. Mefloquine and chloroquine can be given to children of all ages and weights. Although atovaquone-proguanil is approved only for children ≥11 kg (24 lbs), dosing schedules have been calculated for children who weigh ≥5 kg.21 Doxycycline is recommended only for children who are at least 8 years of age.
Cost. For a 2-week exposure period, chloroquine is the least expensive medication (although regions in which it is recommended are limited due to resistance) (TABLE 27,25,26).
Ask about accommodations
Since Anopheles mosquitoes feed between dusk and dawn, inquiring about accommodations can further clarify a patient’s malaria risk. Staying in air-conditioned housing (implying that the interior can be sealed) or that has screened windows can reduce exposure to mosquitoes, although data are lacking regarding whether the latter practice reduces the incidence of malaria transmission28 (SOR: C).
Share decision making
After considering the key factors determining a patient’s level of risk, you may decide to recommend no specific interventions, to advise insect avoidance measures only, to combine insect avoidance with chemoprophylaxis, or to caution against traveling to a malaria-endemic region. The patient’s contribution to the final decision includes personal preferences, values, and risk tolerance—particularly when comorbidities are involved.
When preventive measures fail
Approximately 0.2% of travelers to malaria-endemic regions will become infected, despite proper pre-travel counseling and prophylaxis.29 In the United States, malaria is often misdiagnosed or improperly treated.30 The time from initial presentation to correct diagnosis of malaria has been reported as an astonishingly high 4 to 8.5 days, depending on the population.31,32
A high index of suspicion is needed and will ensure timely care when any febrile traveler returns from a malaria-endemic area.33 Be sure to advise patients to seek medical attention if they are feverish upon returning home.
Once suspected, the diagnosis of malaria can be readily confirmed through the use of antibody-, nucleic acid-, or microscopy-based techniques (the latter to directly visualize Plasmodium species in blood smears).
Although malaria chemoprophylaxis is relatively straightforward, malaria treatment—especially in cases of chemoprophylaxis failures—may not be, and the topic is beyond the scope of this article. For guidance on treating malaria, consult a knowledgeable physician or contact the CDC at www.cdc.gov/malaria/, or at (855) 856-4713 (weekdays, 9 am to 5 pm EST) or (770) 488-7100 (weekends or after normal business hours; ask for the Malaria Branch clinician on call).
CORRESPONDENCE
Mark K. Huntington, MD, PhD, Center for Family Medicine, 1115 East 20th Street, Sioux Falls, SD 57105; mark.huntington@usd.edu
› Assess the need for nonpharmacologic, behavioral interventions and for chemoprophylaxis based on a destination’s relative risk to travelers, planned and potential activities, and patient comorbidities. B
› Choose an antimalarial medication based on knowledge of area-specific drug effectiveness or resistance patterns, trip duration, drug cost, tolerance for adverse effects, and comorbidities. C
› Presume a diagnosis of malaria until proven otherwise in any traveler who is febrile after returning from a malaria-endemic region. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
Although malaria was eradicated as an endemic disease in the United States in the early 1950s,1 it still returns yearly in approximately 1500 individuals who travel to foreign countries2—most of whom neglected to use prophylactic measures or use them properly.3 In more than 60 documented cases, these infected individuals have been the source of local transmission in their communities.2 To reduce the individual and public health risks associated with malaria, this article focuses on steps that international travelers can take to limit their risk of the disease.
What travelers need to know
In 2013, more than 61.5 million residents of the United States traveled abroad, approximately 30% of whom visited malaria-endemic regions: Mexico and equatorial nations in Central and South America; Africa; the Middle East; and South, East, and Southeast Asia.4 Counseling on appropriate preventive measures fits the Medical Home concept of comprehensive, preventive, patient-centered care, and pre-travel consultation—including a review of health data and itineraries, and patient education—can be a team-based effort.5
Begin your planning for malaria prophylaxis by assessing your patient’s individual risk. Key variables are a patient’s detailed itinerary, a credible and current source of information on location-specific malaria prevalence, personal risk factors, and risk tolerance. Shared decision-making is vital and enhances adherence to the prescribed regimen.
Endemicity varies regionally. Without chemoprophylaxis, risk of infection ranges from more than 20% in Papua New Guinea to 0.01% in Central America, with wide exposure risk variations likely, even within regions.6 Travel to areas of high endemicity requires more aggressive malaria prevention strategies than travel to low-endemicity regions.
Risk of exposure is lower with short visits,7 business-only travel, urban-only stays in some countries, day trips to endemic areas,7 and travel during seasons with lower mosquito burden. Likewise, travelers staying in a hotel with sealed windows will face lower nighttime Anopheles mosquito exposure. In these cases, nonpharmacologic measures alone may be appropriate.
Those at particularly high risk for complicated or lethal malarial infection are children, pregnant women, elderly individuals, and immunocompromised patients.7 In addition to counseling high-risk patients about prophylactic measures, consider advising against travel in certain circumstances. Among those at highest risk for acquiring malaria are immigrants and refugees traveling to their ancestral homelands to visit friends and relatives (VFR).2 Many VFR travelers fail to take appropriate prophylactic measures when “going home.”8 A significant number of cases of travel-acquired malaria occurs in VFR children.9
Individualizing prevention directives
The mainstays of malaria prevention include nonpharmacologic and behavioral interventions, as well as chemoprophylaxis. Most cases of malaria in travelers returning to the United States result from the improper implementation of prophylactic measures.3 Discussing individual risk with travelers is an easy way to bolster adherence to malaria prevention measures, and some evidence suggests it is effective10 (strength of recommendation [SOR]: C). Other limited studies have also shown that malaria education can improve knowledge about malaria transmission and increase the likelihood that preventive measures will be used.11,12
Recommend nonpharmacologic measures even for those using chemoprophylaxis
Nonpharmacologic interventions such as sleeping under permethrin-treated bednets, wearing long sleeves and full-length pants, treating clothes with permethrin, and applying DEET (N,N-diethyl-meta-toluamide) to exposed skin are effective and have the added benefit of preventing non-malarial arthropod-borne diseases4 (SOR: B). Studies have shown that, compared with sleeping without nets, the use of insecticide treated-nets can reduce child mortality by 17% and the incidence of uncomplicated malarial episodes by 50%.13 In areas with malaria transmission, 10% to 30% DEET—used alone or in combination with permethrin-treated clothing— can reduce bite load, although the American Academy of Pediatrics recommends against using DEET in children younger than 2 months of age.14,15
Using these measures in combination from dusk to dawn, when Anopheles mosquitoes are active, has been shown to be effective, although randomized, controlled studies are lacking.16 Remaining indoors during these peak biting periods is also advisable. In certain areas, and with the right itinerary, the traveler may only need to employ nonpharmacologic methods of preventing malarial infection. Recommend them to all patients traveling to malarial regions, even to individuals using pharmacologic prophylaxis.
Factors determining the need for, and selection of, chemoprophylaxis
When used properly, chemoprophylactic drugs are effective in preventing malaria (SOR: A). Atovaquone-proguanil achieves efficacy of 95% to 100%,17 while doxycycline, primaquine, and mefloquine are slightly less effective.18-20 Chloroquine is effective in 6 regions of the tropics and subtropics where Plasmodium falciparum resistance has not developed. Select a drug based on your assessment of an individual’s level of risk according to the personal itinerary, trip duration and accommodations, cost of medication, tolerance for adverse effects, and other factors (eg, comorbidities, concurrent drug usage, pregnancy).
Location matters. The risk of malaria transmission can vary considerably not only between countries, but also regionally within countries and even between a city and its immediate surroundings. Therefore, select a chemoprophylactic agent based on the specific itinerary, planned activities, the potential for unforeseen additional excursions, and local Plasmodium resistance patterns. For example, chloroquine is effective only in the Caribbean, Central America, and some countries in the Middle East.21 Mefloquine resistance has been reported in parts of Cambodia, Thailand, Vietnam, Burma, China, and Laos.21
On its Travelers’ Health Web site (www.cdc.gov/travel), the Centers for Disease Control and Prevention (CDC) reports for each country 1) the risk of malaria transmission, 2) areas within the country that pose a risk, 3) evidence of Plasmodium drug resistance, 4) which Plasmodium species are active, and 5) which chemoprophylactic medications are recommended.22 Additional Web sites, either free or subscription-based, allow users to view this same information on maps, advise on where insect precautions alone are sufficiently protective, and provide information about the traveler’s risk of contracting other diseases (TABLE 1).
TABLE 1
| Web resources on infectious diseases of concern to international travelers | |
| Resource | Notes |
Centers for Disease Control and Prevention | Free Site Go to Yellow Book » Contents » Chapter 3 » “Travel Vaccines & Malaria Information, by Country” for country-specific information about the risk of malaria transmission |
VHI Healthcare | Free Site Destination-specific information about travel alerts and vaccine recommendations Does not report malaria transmission data |
Gideon | Subscription only Online application that helps with diagnosing infectious diseases and keeping up to date with global health literature |
Travax | Subscription only Information about recommended vaccines and country-specific risk of malaria transmission |
Tropimed | Subscription only Information about recommended vaccines and country-specific risk of malaria transmission |
Comparative adverse effects of antimalarial agents. A Cochrane Review on the tolerability of chemoprophylactic agents concluded that atovaquone-proguanil and doxycycline were better tolerated than mefloquine (SOR: B). Compared with mefloquine, atovaquone-proguanil led to fewer reports of any adverse effects (relative risk [RR]=0.72), gastrointestinal adverse effects (RR=0.54), and neuropsychiatric adverse events (RR= 0.49-0.86, depending on the studies).23 Doxycycline users have reported fewer neuropsychiatric events (RR=0.84) than mefloquine users.23 These are relatively small differences, and the authors point out that these figures are based on low-quality evidence. Additional research is likely to have an impact on the confidence in the estimate of effect and to ultimately change the estimate.
Mefloquine is contraindicated in travelers with seizures, active or recent history of depression, generalized anxiety disorder, psychosis, schizophrenia, or other psychiatric disorders. Compared with mefloquine, atovaquone-proguanil and doxycycline cause fewer neuropsychiatric adverse effects (such as vivid dreams, dizziness, anxiety, depression, visual disturbance, or seizures).24 Caution is advised when prescribing chloroquine for patients with epilepsy because the medication has the potential to lower the seizure threshold.25
Use caution when prescribing mefloquine for patients with cardiac conduction disturbances. Electrocardiogram alterations such as sinus bradycardia, first-degree AV block, prolongation of QTc intervals, and abnormal T wave changes have been reported.26 Chloroquine can also prolong QTc intervals.26
Safety in pregnancy and breastfeeding. Malaria in pregnancy is associated with increased rates of anemia, low birth weight, prematurity, intrauterine growth restriction, and infant mortality.27 Chloroquine and mefloquine are considered safe during pregnancy and breastfeeding. Doxycycline has been associated with increased risk of harm to the fetus. Atovaquone-proguanil can be used in breastfeeding women if the child is ≥5 kg (≥11 lbs). Chemoprophylaxis taken by the mother while breastfeeding does not protect the infant from infection.
Dosing considerations. Mefloquine and chloroquine are dosed weekly; doxycycline and atovaquone-proguanil are taken daily. Travelers staying in a malaria-endemic region for longer periods (months rather than weeks) often prefer the weekly rather than daily medications; however, this may not be possible due to the adverse-effect profile of mefloquine or to traveling in an area with known chloroquine resistance. Some individuals prefer the routine of taking a medication daily, since remembering to take a single dose on the same day each week can be challenging. Others may not want to carry a large number of pills and therefore prefer weekly dosing. Have patients take medications before the trip, to assess tolerability and to ensure adequate blood concentrations before exposure.
Because mefloquine, doxycycline, and chloroquine target only the blood stages of Plasmodium, patients must continue these medications for 4 weeks following the exposure period to ensure adequate coverage as parasites are released from the liver. Because doxycycline is taken daily and has to be continued for 4 weeks following the exposure period, the total number of pills taken is higher for this regimen. Atovaquone-proguanil is active against hepatic and blood stages and can be discontinued a week following the exposure period.
With children, base dosing on body weight and do not exceed the recommended adult dose. When fractions of tablets are used (such as with mefloquine and atovaquone-proguanil dosing), pharmacists can crush tablets and place divided doses in capsules, to be sprinkled as needed into food such as applesauce or jelly. Mefloquine and chloroquine can be given to children of all ages and weights. Although atovaquone-proguanil is approved only for children ≥11 kg (24 lbs), dosing schedules have been calculated for children who weigh ≥5 kg.21 Doxycycline is recommended only for children who are at least 8 years of age.
Cost. For a 2-week exposure period, chloroquine is the least expensive medication (although regions in which it is recommended are limited due to resistance) (TABLE 27,25,26).
Ask about accommodations
Since Anopheles mosquitoes feed between dusk and dawn, inquiring about accommodations can further clarify a patient’s malaria risk. Staying in air-conditioned housing (implying that the interior can be sealed) or that has screened windows can reduce exposure to mosquitoes, although data are lacking regarding whether the latter practice reduces the incidence of malaria transmission28 (SOR: C).
Share decision making
After considering the key factors determining a patient’s level of risk, you may decide to recommend no specific interventions, to advise insect avoidance measures only, to combine insect avoidance with chemoprophylaxis, or to caution against traveling to a malaria-endemic region. The patient’s contribution to the final decision includes personal preferences, values, and risk tolerance—particularly when comorbidities are involved.
When preventive measures fail
Approximately 0.2% of travelers to malaria-endemic regions will become infected, despite proper pre-travel counseling and prophylaxis.29 In the United States, malaria is often misdiagnosed or improperly treated.30 The time from initial presentation to correct diagnosis of malaria has been reported as an astonishingly high 4 to 8.5 days, depending on the population.31,32
A high index of suspicion is needed and will ensure timely care when any febrile traveler returns from a malaria-endemic area.33 Be sure to advise patients to seek medical attention if they are feverish upon returning home.
Once suspected, the diagnosis of malaria can be readily confirmed through the use of antibody-, nucleic acid-, or microscopy-based techniques (the latter to directly visualize Plasmodium species in blood smears).
Although malaria chemoprophylaxis is relatively straightforward, malaria treatment—especially in cases of chemoprophylaxis failures—may not be, and the topic is beyond the scope of this article. For guidance on treating malaria, consult a knowledgeable physician or contact the CDC at www.cdc.gov/malaria/, or at (855) 856-4713 (weekdays, 9 am to 5 pm EST) or (770) 488-7100 (weekends or after normal business hours; ask for the Malaria Branch clinician on call).
CORRESPONDENCE
Mark K. Huntington, MD, PhD, Center for Family Medicine, 1115 East 20th Street, Sioux Falls, SD 57105; mark.huntington@usd.edu
1. Mali S, Steele S, Slutsker L, et al; Centers for Disease Control and Prevention (CDC). Malaria surveillance - United States, 2008. MMWR Surveill Summ. 2010;59:1-15.
2. Centers for Disease Control and Prevention. Malaria facts. Centers for Disease Control and Prevention Web site. Available at: www.cdc.gov/malaria/about/facts.html. Accessed September 29, 2014.
3. Huntington MK. Healthy people, malaria and South Dakota. S D Med. 2012;65:297-300.
4. Office of Travel and Tourism Industries. U.S. citizen travel to international regions, 2013. Office of Travel and Tourism Industries Web site. Available at: http://travel.trade.gov/view/m-2013-O-001/index.html. Accessed September 29, 2014.
5. Bazemore AW, Huntington M. The pretravel consultation. Am Fam Physician. 2009;80:583-590.
6. Bradley DJ, Warhurst DC, Blaze M, et al. Malaria imported into the United Kingdom in 1996. Euro Surveill. 1998;3:40-42.
7. Arguin PM, Tan KR, et al; Centers for Disease Control and Prevention. Infectious diseases related to travel. Centers for Disease Control and Prevention Web site. Available at: http://wwwnc.cdc.gov/travel/yellowbook/2014/chapter-3-infectious-diseases-related-to-travel/malaria. Accessed October 15, 2014.
8. Pavli A, Maltezou HC. Malaria and travellers visiting friends and relatives. Travel Med Infect Dis. 2010;8:161-168.
9. Stäger K, Legros F, Krause G, et al. Imported malaria in children in industrialized countries, 1992-2002. Emerg Infect Dis. 2009;15:185-191.
10. Hartjes LB, Baumann LC, Henriques JB. Travel health risk perceptions and prevention behaviors of US study abroad students. J Travel Med. 2009;16:338-343.
11. Kishore J, Gupta VK, Singh SV, et al. Impact of health education intervention on knowledge and community action for malaria control in Delhi. J Commun Dis. 2008;40:183-192.
12. Chirdan OO, Zoakah AI, Ejembi CL. Impact of health education on home treatment and prevention of malaria in Jengre, North Central Nigeria. Ann Afr Med. 2008;7:112-119.
13. Lengeler C. Insecticide-treated bed nets and curtains for preventing malaria. Cochrane Database Syst Rev. 2004;(2): CD000363.
14. Centers for Disease Control and Prevention. Fight the bite for protection from malaria: Guidelines for DEET insect repellent use. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/malaria/toolkit/DEET.pdf. Accessed September 29, 2014.
15. American Academy of Pediatrics. Safety & prevention. Healthychildren.org Web site. Available at: http://www.healthychildren. org/English/safety-prevention/at-play/Pages/Insect-Repellents. aspx. Accessed September 29, 2014.
16. Croft AM, Baker D, von Bertele MJ. An evidence-based vector control strategy for military deployments: the British Army experience. Med Trop (Mars). 2001;61:91-98.
17. Boggild AK, Parise ME, Lewis LS, et al. Atovaquone-proguanil: report from the CDC expert meeting on malaria chemoprophylaxis (II). Am J Trop Med Hyg. 2007;76:208-223.
18. Tan KR, Magill AJ, Parise ME, et al; Centers for Disease Control and Prevention. Doxycycline for malaria chemoprophylaxis and treatment: report from the CDC expert meeting on malaria chemoprophylaxis. Am J Trop Med Hyg. 2011;84:517-531.
19. Hill DR, Baird JK, Parise ME, et al. Primaquine: report from CDC expert meeting on malaria chemoprophylaxis I. Am J Trop Med Hyg. 2006;75:402-415.
20. Steffen R, Fuchs E, Schildknecht J, et al. Mefloquine compared with other malaria chemoprophylactic regimens in tourists visiting east Africa. Lancet. 1993;341:1299-1303.
21. Centers for Disease Control and Prevention. CDC Health Information for International Travel 2014. New York, NY: Oxford University Press; 2014.
22. Gershman MD, Jentes ES, Johnson KJ, et al; Centers for Disease Control and Prevention. Infectious diseases related to travel. Centers for Disease Control and Prevention Web site. Available at: http://wwwnc.cdc.gov/travel/yellowbook/2012/chapter-3- infectious-diseases-related-to-travel/yellow-fever-and-malaria- information-by-country.htm. Accessed September 29, 2014.
23. Jacquerioz FA, Croft AM. Drugs for preventing malaria in travellers. Cochrane Database Syst Rev. 2009;(4):CD006491.
24. Schlagenhauf P, Tschopp A, Johnson R, et al. Tolerability of malaria chemoprophylaxis in non-immune travellers to sub-Saharan Africa: multicentre, randomised, double blind, four arm study. BMJ. 2003;327:1078.
25. Chloroquine phosphate [package insert]. Eatontown, NJ: Westward Pharmaceutical Corp; 2010.
26. Lariam [package insert]. Roche Laboratories, Inc: Nutley, NJ; 2004.
27. Steketee RW, Nahlen BL, Parise ME, et al. The burden of malaria in pregnancy in malaria-endemic areas. Am J Trop Med Hyg. 2001;64(1-2 suppl):28-35.
28. Kirby MJ, Ameh D, Bottomley C, et al. Effect of two different house screening interventions on exposure to malaria vectors and on anaemia in children in The Gambia: a randomised controlled trial. Lancet. 2009;374:998-1009.
29. Steffen R, Amitirigala I, Mutsch M. Health risks among travelers--need for regular updates. J Travel Med. 2008;15:145-146.
30. Dorsey G, Gandhi M, Oyugi JH, et al. Difficulties in the prevention, diagnosis, and treatment of imported malaria. Arch Intern Med. 2000;160:2505-2510.
31. Newman RD, Parise ME, Barber AM, et al. Malaria-related deaths among U.S. travelers, 1963-2001. Ann Intern Med. 2004;141: 547-555.
32. Lesko CR, Arguin PM, Newman RD. Congenital malaria in the United States: a review of cases from 1966 to 2005. Arch Pediatr Adolesc Med. 2007;161:1062-1067.
33. Blair JE. Evaluation of fever in the international traveler. Unwanted ‘souvenir’ can have many causes. Postgrad Med. 2004;116: 13-20,29.
1. Mali S, Steele S, Slutsker L, et al; Centers for Disease Control and Prevention (CDC). Malaria surveillance - United States, 2008. MMWR Surveill Summ. 2010;59:1-15.
2. Centers for Disease Control and Prevention. Malaria facts. Centers for Disease Control and Prevention Web site. Available at: www.cdc.gov/malaria/about/facts.html. Accessed September 29, 2014.
3. Huntington MK. Healthy people, malaria and South Dakota. S D Med. 2012;65:297-300.
4. Office of Travel and Tourism Industries. U.S. citizen travel to international regions, 2013. Office of Travel and Tourism Industries Web site. Available at: http://travel.trade.gov/view/m-2013-O-001/index.html. Accessed September 29, 2014.
5. Bazemore AW, Huntington M. The pretravel consultation. Am Fam Physician. 2009;80:583-590.
6. Bradley DJ, Warhurst DC, Blaze M, et al. Malaria imported into the United Kingdom in 1996. Euro Surveill. 1998;3:40-42.
7. Arguin PM, Tan KR, et al; Centers for Disease Control and Prevention. Infectious diseases related to travel. Centers for Disease Control and Prevention Web site. Available at: http://wwwnc.cdc.gov/travel/yellowbook/2014/chapter-3-infectious-diseases-related-to-travel/malaria. Accessed October 15, 2014.
8. Pavli A, Maltezou HC. Malaria and travellers visiting friends and relatives. Travel Med Infect Dis. 2010;8:161-168.
9. Stäger K, Legros F, Krause G, et al. Imported malaria in children in industrialized countries, 1992-2002. Emerg Infect Dis. 2009;15:185-191.
10. Hartjes LB, Baumann LC, Henriques JB. Travel health risk perceptions and prevention behaviors of US study abroad students. J Travel Med. 2009;16:338-343.
11. Kishore J, Gupta VK, Singh SV, et al. Impact of health education intervention on knowledge and community action for malaria control in Delhi. J Commun Dis. 2008;40:183-192.
12. Chirdan OO, Zoakah AI, Ejembi CL. Impact of health education on home treatment and prevention of malaria in Jengre, North Central Nigeria. Ann Afr Med. 2008;7:112-119.
13. Lengeler C. Insecticide-treated bed nets and curtains for preventing malaria. Cochrane Database Syst Rev. 2004;(2): CD000363.
14. Centers for Disease Control and Prevention. Fight the bite for protection from malaria: Guidelines for DEET insect repellent use. Centers for Disease Control and Prevention Web site. Available at: http://www.cdc.gov/malaria/toolkit/DEET.pdf. Accessed September 29, 2014.
15. American Academy of Pediatrics. Safety & prevention. Healthychildren.org Web site. Available at: http://www.healthychildren. org/English/safety-prevention/at-play/Pages/Insect-Repellents. aspx. Accessed September 29, 2014.
16. Croft AM, Baker D, von Bertele MJ. An evidence-based vector control strategy for military deployments: the British Army experience. Med Trop (Mars). 2001;61:91-98.
17. Boggild AK, Parise ME, Lewis LS, et al. Atovaquone-proguanil: report from the CDC expert meeting on malaria chemoprophylaxis (II). Am J Trop Med Hyg. 2007;76:208-223.
18. Tan KR, Magill AJ, Parise ME, et al; Centers for Disease Control and Prevention. Doxycycline for malaria chemoprophylaxis and treatment: report from the CDC expert meeting on malaria chemoprophylaxis. Am J Trop Med Hyg. 2011;84:517-531.
19. Hill DR, Baird JK, Parise ME, et al. Primaquine: report from CDC expert meeting on malaria chemoprophylaxis I. Am J Trop Med Hyg. 2006;75:402-415.
20. Steffen R, Fuchs E, Schildknecht J, et al. Mefloquine compared with other malaria chemoprophylactic regimens in tourists visiting east Africa. Lancet. 1993;341:1299-1303.
21. Centers for Disease Control and Prevention. CDC Health Information for International Travel 2014. New York, NY: Oxford University Press; 2014.
22. Gershman MD, Jentes ES, Johnson KJ, et al; Centers for Disease Control and Prevention. Infectious diseases related to travel. Centers for Disease Control and Prevention Web site. Available at: http://wwwnc.cdc.gov/travel/yellowbook/2012/chapter-3- infectious-diseases-related-to-travel/yellow-fever-and-malaria- information-by-country.htm. Accessed September 29, 2014.
23. Jacquerioz FA, Croft AM. Drugs for preventing malaria in travellers. Cochrane Database Syst Rev. 2009;(4):CD006491.
24. Schlagenhauf P, Tschopp A, Johnson R, et al. Tolerability of malaria chemoprophylaxis in non-immune travellers to sub-Saharan Africa: multicentre, randomised, double blind, four arm study. BMJ. 2003;327:1078.
25. Chloroquine phosphate [package insert]. Eatontown, NJ: Westward Pharmaceutical Corp; 2010.
26. Lariam [package insert]. Roche Laboratories, Inc: Nutley, NJ; 2004.
27. Steketee RW, Nahlen BL, Parise ME, et al. The burden of malaria in pregnancy in malaria-endemic areas. Am J Trop Med Hyg. 2001;64(1-2 suppl):28-35.
28. Kirby MJ, Ameh D, Bottomley C, et al. Effect of two different house screening interventions on exposure to malaria vectors and on anaemia in children in The Gambia: a randomised controlled trial. Lancet. 2009;374:998-1009.
29. Steffen R, Amitirigala I, Mutsch M. Health risks among travelers--need for regular updates. J Travel Med. 2008;15:145-146.
30. Dorsey G, Gandhi M, Oyugi JH, et al. Difficulties in the prevention, diagnosis, and treatment of imported malaria. Arch Intern Med. 2000;160:2505-2510.
31. Newman RD, Parise ME, Barber AM, et al. Malaria-related deaths among U.S. travelers, 1963-2001. Ann Intern Med. 2004;141: 547-555.
32. Lesko CR, Arguin PM, Newman RD. Congenital malaria in the United States: a review of cases from 1966 to 2005. Arch Pediatr Adolesc Med. 2007;161:1062-1067.
33. Blair JE. Evaluation of fever in the international traveler. Unwanted ‘souvenir’ can have many causes. Postgrad Med. 2004;116: 13-20,29.
Suspect myopathy? Take this approach to the work-up
› Categorize patients with muscle complaints into suspected myositic, intrinsic, or toxic myopathy to help guide subsequent work-up. C
› Look for diffusely painful, swollen, or boggy-feeling muscles—as well as weakness and pain with exertion—in patients you suspect may have viral myopathy. C
› Consider electromyography and muscle biopsy for patients you suspect may have dermatomyositis. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Marie C, a 75-year-old Asian woman, reports weakness in her legs and arms with unsteadiness when walking. She has a vague but persistent ache in her large muscles. Her symptoms have developed slowly over the past 3 months. She denies recent signs or symptoms of infection or other illness. Her medical history includes hypertension, hyperlipidemia, osteopenia, and obesity. Ms. C takes lisinopril 10 mg/d and atorvastatin, which was recently increased from 10 to 20 mg/d.
What would your next steps be in caring for this patient?
Patients who experience muscle-related symptoms such as pain, fatigue, or weakness often seek help from their family physician (FP). The list of possible causes of these complaints can be lengthy and vary greatly, from nonmyopathic conditions such as fibromyalgia to worrisome forms of myopathy such as inclusion body myositis or polymyositis. This article will help you to quickly identify which patients with muscle-related complaints should be evaluated for myopathy and what your work-up should include.
Myopathy or not?
Distinguishing between myopathy and nonmyopathic muscle pain or weakness is the first step in evaluating patients with muscle-related complaints. Many conditions share muscle-related symptoms, but actual muscle damage is not always present (eg, fibromyalgia, chronic pain, and chronic fatigue syndromes).1 While there is some overlap in presentation between patients with myopathy and nonmyopathic conditions, there are important differences in symptoms, physical exam findings, and lab test results (TABLE 11-4). Notably, in myopathic disease, patients’ symptoms are usually progressive, vital signs are abnormal, and weakness is common, whereas patients with nonmyopathic disease typically have remitting and relapsing symptoms, normal vital signs, and no weakness.
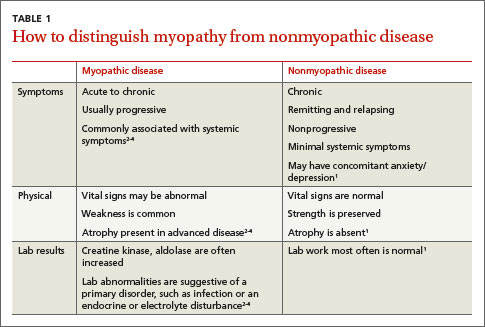
Myopathy itself is divided into 3 categories—myositic, intrinsic, and toxic—which reflect the condition, or medication, that brought on the muscle damage (TABLE 22,4-15). Placing patients into one of these categories based on their risk factors, history, and physical exam findings can help to focus the diagnostic work-up on areas most likely to provide useful information.
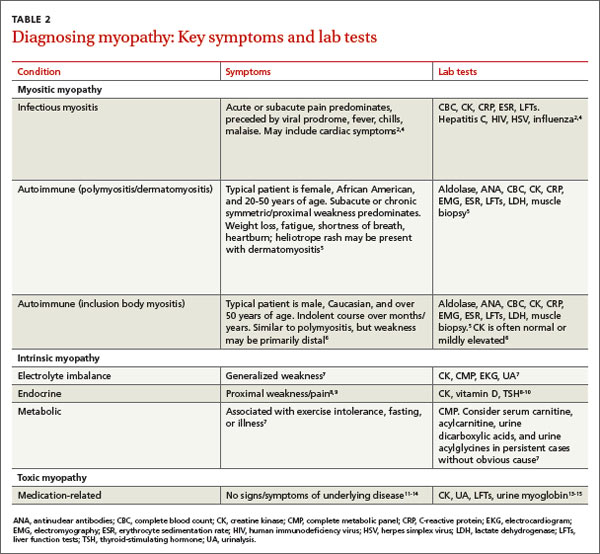
Myositic myopathy can be caused by infection or autoimmunity
Myositic myopathies result in inflammatory destruction of muscle tissue. Patients with myositic myopathy often exhibit fever, malaise, weight loss, and general fatigue. Though weakness and pain are common, both can be variable or even absent in myositic myopathy.2,5 Myositic myopathy can be caused by infectious agents or can develop from an autoimmune disease.
Infectious myositic myopathy is one of the more common types of myopathy that FPs will encounter.2 Viruses such as influenza, parainfluenza, coxsackievirus, human immunodeficiency virus, cytomegalovirus, echovirus, adenovirus, Epstein-Barr, and hepatitis C are common causes.2,4,16 Bacterial and fungal myositides are relatively rare. Both most often occur as the result of penetrating trauma or immunocompromise, and are generally not subtle.2 Parasitic myopathy can occur from the invasion of skeletal muscle by trichinella after ingesting undercooked, infected meat.2 Although previously a more common problem, currently only 10 to 20 cases of trichinellosis are reported in the United States each year.17 Due to their rarity, bacterial, fungal, and parasitic myositides are not reviewed here.
Patients with a viral myositis often report prodromal symptoms such as fever, upper respiratory illness, or gastrointestinal distress one to 2 weeks before the onset of muscle complaints. Muscle pain is usually multifocal, involving larger, bilateral muscle groups, and may be associated with swelling.
Patients with viral myositis may exhibit diffusely painful, swollen, or boggy-feeling muscles as well as weakness and pain with exertion. Other signs of viral infection such as rash, fever, upper respiratory symptoms, or meningeal signs may be present. Severe signs include arrhythmia or respiratory failure due to cardiac muscle or diaphragm involvement, or signs of renal failure due to precipitation of myoglobin in the renal system (ie, rhabdomyolysis).2 If the infection affects the heart, patients may develop palpitations, pleuritic chest pain, or shortness of breath.2
Diagnosis of viral myositis relies heavily on clinical suspicion in patients with a fitting history and physical exam findings. Helpful lab tests include a complete blood count (CBC), erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), creatine kinase (CK), and liver function tests (LFTs), all of which can be abnormal in viral myositis. Viral polymerase chain reaction, culture, or antigen testing may be helpful in severe or confusing cases, but in most cases such testing is unnecessary. Muscle biopsy is not recommended except in persistent cases, where definitive identification of the causative agent might alter treatment or when nonviral infection is suspected.2
Autoimmune myositic myopathy. Unlike infectious myopathies, autoimmune myopathies are usually chronic, subtle, and relatively rare. The 3 most common autoimmune myopathies—polymyositis, dermatomyositis, and inclusion body myositis—have a combined prevalence of approximately 10:100,000.6 Although these types of myopathies are uncommon, FPs will likely be the first to evaluate a patient with one of them.
Patients with an autoimmune myopathy typically complain of weakness and mild to moderate muscle pain, although pain may be absent. Compared to infectious myopathies, autoimmune myopathies usually exhibit a more indolent course. Patients with advanced disease may report fever, weight loss, shortness of breath from cardiomyopathy, heartburn from a weakened lower esophageal sphincter, and/or a rash.5
Physical examination may reveal symmetric, proximal muscle weakness. Atrophy is typically not seen until late in the disease. Skin exam usually is normal in patients with inclusion body myositis and polymyositis. The typical rash of dermatomyositis is a heliotrope (blue-purple) discoloration on the upper eyelids and a raised, violaceous, scaly eruption on the knuckles (Gottron’s papules).
Laboratory tests that can be helpful include CK, lactate dehydrogenase (LDH), aldolase, and LFTs (reflecting muscle injury, not liver involvement). For polymyositis and dermatomyositis, CK is the most sensitive lab test and often exhibits the highest elevation above normal.6 Conversely, CK is often normal or only mildly elevated in inclusion body myositis. Up to 80% of patients with autoimmune myopathy will have antinuclear antibodies.3,5 ESR and CRP levels are also often elevated.
Both electromyography (EMG) and muscle biopsy may be required to diagnose autoimmune myopathy, but these are typically done under the direction of a rheumatologist after an FP’s initial work-up is inconclusive.
Intrinsic myopathy: Suspect electrolyte problems, other causes
Intrinsic myopathy occurs in patients with electrolyte disorders, diseases of the endocrine system, or underlying metabolic dysfunction.
Electrolyte disorders. Muscle-related symptoms are unlikely to be the chief complaint of patients with severe electrolyte imbalance. However, a patient with mild to moderate electrolyte problems may develop muscle fatigue, weakness, or pain. TABLE 3 reviews other signs and symptoms of electrolyte abnormalities that may be helpful in establishing a diagnosis in a patient with muscle complaints.
Ordering a complete metabolic panel (CMP), CK, and urinalysis (UA) can help rule out electrolyte disorders. If electrolyte disorders are detected, an electrocardiogram is useful to evaluate for cardiac dysfunction. Once an electrolyte disorder is identified, investigate its underlying cause. Correcting the electrolyte disorder should help improve symptoms of myopathy.
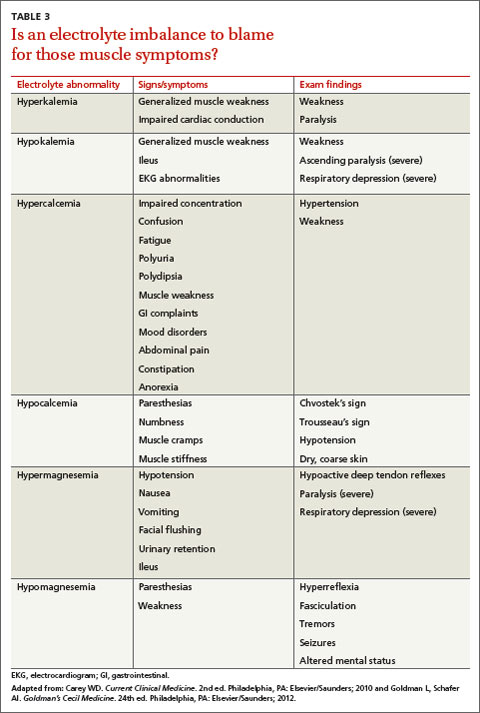
Endocrine myopathy can be associated with hypothyroidism, hyperthyroidism, parathyroid disease, vitamin D deficiency, or Cushing syndrome.8-10,18,19 Although less common than some other causes, identifying endocrine myopathy is crucial because correcting the underlying disease will often improve multiple aspects of the patient’s health.
The presentation of endocrine myopathy may be subtle. Patients with hypothyroidism may experience muscle pain or weakness, fatigue, cold sensitivity, constipation, and dry skin.20 Muscle-related symptoms may be the only sign of endocrine myopathy in a patient who would otherwise be considered to have subclinical hypothyroidism.8,18 Hyperthyroidism can present with weight loss, heat intolerance, frequent bowel movements, tachycardia, and muscle weakness.21
Patients with parathyroid disease— especially patients with chronic renal failure—may report proximal muscle weakness, often in the lower extremities.19 Complaints of muscle weakness or pain can occur with severe vitamin D deficiency.10 Patients with Cushing syndrome often experience proximal weakness and weight gain.9
Patients with a personal or family history of endocrine disorders, previous thyroid surgery, or those taking medications that can impair thyroid function, such as lithium, amiodarone, or interferon, are at risk for endocrine myopathy.18-20 Suspect hyperparathyroidism in patients with chronic kidney disease who complain of weakness.
Vitamin D deficiency is relatively common, with at minimum 20% of elderly adults estimated to be deficient.10 Patients at risk for Cushing disease are most likely receiving pharmacologic doses of glucocorticoids, which can increase their risk of myopathy, or to have ectopic adrenocorticotropic hormone secretion.
Metabolic myopathy results from a lack of sufficient energy production in the muscle. The 3 main groups of metabolic myopathy are impaired muscle glycogenoses, disorders of fatty acid oxidation, and mitochondrial myopathies.7
Because metabolic myopathy can occur at any age, a thorough history and physical is crucial for diagnosis. Proximal weakness in metabolic myopathy is often associated with exercise intolerance, stressful illness, or fasting. Patients often present with dynamic abnormalities such as fatigue, muscle cramping, and even rhabdomyolysis during exertion.7
When evaluating patients you suspect may have metabolic myopathy, a physical exam may reveal muscle contractures, muscle swelling, or proximal muscle weakness. Patients with certain types of fatty acid oxidation disorders or mitochondrial disorders may also exhibit cardiomyopathy, neuropathy, retinopathy, ataxia, hearing loss, or other systemic manifestations.7
Basic labs for investigating suspected metabolic myopathy include serum electrolytes, glucose, LFTs, CK (which may or may not be elevated), lactate, ammonia, and UA for myoglobinuria. More advanced labs, such as serum total carnitine and acylcarnitine as well as urinary levels of dicarboxylic acids and acylglycines, may be needed if a metabolic disorder is strongly suspected.7 Muscle biopsy, EMG, and genetic testing can also prove helpful in diagnosis. Definitive diagnosis and treatment of metabolic myopathy usually requires a multidisciplinary team of providers, including subspecialty referral.
Toxic myopathy
Toxic myopathy refers to muscle damage caused by an exogenous chemical agent, most often a drug. The mechanism of toxicity is not always clear and may result from the activation of inflammatory responses similar to autoimmune myopathy.22 Toxic myopathies may result from several commonly used medications; cholesterol-lowering medications are a common culprit.13-15,23-25 Drug-induced myopathies vary in frequency and severity. For instance, in patients taking statins, the rate of myalgias is 6%, while the incidence of rhabdomyolysis is estimated to be 4 per 100,000, and is found most often in patients taking concomitant fibrates.23
Drug-induced toxic myopathy differs from previously discussed myopathies in that symptoms are usually more insidious, findings on exam are more often mixed muscular and neurologic, and lab abnormalities are usually more subtle.11,12 Symptoms of myopathy typically occur weeks or months after initiating a drug and usually improve or resolve within weeks after discontinuing the offending agent. Knowing the patient’s medication list and which medications cause certain patterns of myopathy symptoms can help guide the differential diagnosis (TABLE 411-15,22-25).
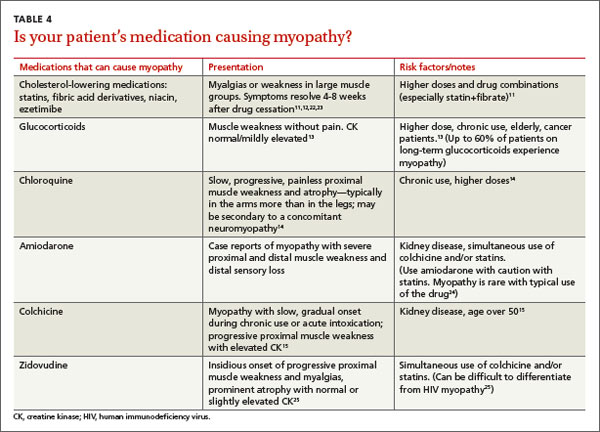
Risk factors for most medication-related myopathies are polypharmacy, renal or liver disease, and age over 50 years13-15,23-25 The physical exam for patients with drug- or toxin-related myopathy will most often reveal relatively minor abnormalities such as muscle tenderness and mild weakness, except for the most severe or advanced cases. Most patients will not have physical signs that suggest an underlying illness. CK levels and LFTs should be obtained. Basic chemistry and UA may also be helpful in patients with risk factors for renal disease.
CASE › Ms. C has been taking a statin for more than 10 years, and the dose was recently increased. You are aware that statin-related muscle injury can develop even after years of use, and suspect the statin may be causing her myopathy. You order a CK test, which is mildly elevated. You recommend discontinuing the statin. After 8 weeks off her statin, Ms. C’s Symptoms do not improve. Given her lack of systemic complaints, myositic myopathy from an infectious or rheumatologic cause seems unlikely. You begin to consider an intrinsic cause of myopathy, and order the following tests: a CMP, UA, thyroid-stimulating hormone, repeat CK, and vitamin D level. This testing reveals a vitamin D deficiency at 17 ng/ml (normal range: 30-74 ng/ml). You recommend vitamin D, 50,000 IU per week for 8 weeks. At follow-up, Ms. C's vitamin D level is 40. She says she feels better and her muscle complaints have resolved.
CORRESPONDENCE
Brent W. Smith, MD, Travis Air Force Base Family Medicine Residency, 101 Bodin Circle, Travis Air Force Base, CA 94535; smithb@smithnet.us
1. Huynh CN, Yanni LM, Morgan LA. Fibromyalgia: diagnosis and management for the primary healthcare provider. J Womens Health. 2008;8:1379-1387.
2. Crum-Cianflone NF. Bacterial, fungal, parasitic, and viral myositis. Clin Microbiol Rev. 2008;21:473-494.
3. Reichlin M, Arnett FC Jr. Multiplicity of antibodies in myositis sera. Arthritis Rheum. 1984;27:1150-1156.
4. Yoshino M, Suzuki S, Adachi K, et al. High incidence of acute myositis with type A influenza virus infection in the elderly. Intern Med. 2000;39:431-432.
5. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971-982.
6. Wilson FC, Ytterberg SR, St Sauver JL, et al. Epidemiology of sporadic inclusion body myositis and polymyositis in Olmsted County, Minnesota. J Rheumatol. 2008;35:445-447.
7. Smith EC, El-Gharbawy A, Koeberl DD. Metabolic myopathies: clinical features and diagnostic approach. Rheum Dis Clin N Am. 2011:37:201-217.
8. Reuters V, Teixeira Pde F, Vigário PS, et al. Functional capacity and muscular abnormalities in subclinical hypothyroidism. Am J Med Sci. 2009;338:259-263.
9. Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2008;93:1526-1540.
10. Holick MF, Binkley NC, Bischoff-Ferrari HA, et al; Endocrine Society. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:1911-1930.
11. Antons KA, Williams CD, Baker SK, et al. Clinical perspectives of statin-induced rhabdomyolysis. Am J Med. 2006;119:400-409.
12. Phillips PS, Haas RH, Bannykh S, et al; Scripps Mercy Clinical Research Center. Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med. 2002;137:581-585.
13. Pereira RM, Freire de Carvalho J. Glucocorticoid-induced myopathy. Joint Bone Spine. 2011;78:41-44.
14. Posada C, García-Cruz A, García-Doval I, et al. Chloroquine-induced myopathy. Lupus. 2011;20:773-774.
15. Uri DS, Biavis M. Colchicine neuromyopathy. J Clin Rheumatol. 1996;2:163-166.
16. Mannix R, Tan ML, Wright R, et al. Acute pediatric rhabdomyolysis: causes and rates of renal failure. Pediatrics. 2006;118:2119-2125.
17. Pozio E. World distribution of Trichinella spp. infections in animals and humans. Vet Parasitol. 2007;149:3-21.
18. Rodolico C, Toscano A, Benvenga S, et al. Myopathy as the persistently isolated symptomatology of primary autoimmune hypothyroidism. Thyroid.1998;8:1033-1038.
19. AACE/AAES Task Force on Primary Hyperparathyroidism. The American Association of Clinical Endocrinologists and The American Association of Endocrine Surgeons position statement on the diagnosis and management of primary hyperparathyroidism. Endocr Pract. 2005;11:49-54.
20. Garber JR, Cobin RH, Gharib H, et al; American Association of Clinical Endocrinologists and American Thyroid Association Taskforce on Hypothyroidism in Adults. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Endocrine Pract. 2012;18:988-1028.
21. Bahn Chair RS, Burch HB, Cooper DS, et al; American Thyroid Association; American Association of Clinical Endocrinologists. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Thyroid. 2011;21:593-646.
22. Mammen AL, Amato AA. Statin myopathy: a review of recent progress. Curr Opin Rheumatol. 2010;22:644-650.
23. Buettner C, Davis RB, Leveille SG, et al. Prevalence of musculoskeletal pain and statin use. J Gen Intern Med. 2008;23: 1182-1186.
24. Marot A, Morelle J, Chouinard VA, et al. Concomitant use of simvastatin and amiodarone resulting in severe rhabdomyolysis: a case report and review of the literature. Acta Clin Belg. 2011;66:134-136.
25. Peters BS, Winer J, Landon DN, et al. Mitochondrial myopathy associated with chronic zidovudine therapy in AIDS. Q J Med. 1993;86:5-15.
› Categorize patients with muscle complaints into suspected myositic, intrinsic, or toxic myopathy to help guide subsequent work-up. C
› Look for diffusely painful, swollen, or boggy-feeling muscles—as well as weakness and pain with exertion—in patients you suspect may have viral myopathy. C
› Consider electromyography and muscle biopsy for patients you suspect may have dermatomyositis. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Marie C, a 75-year-old Asian woman, reports weakness in her legs and arms with unsteadiness when walking. She has a vague but persistent ache in her large muscles. Her symptoms have developed slowly over the past 3 months. She denies recent signs or symptoms of infection or other illness. Her medical history includes hypertension, hyperlipidemia, osteopenia, and obesity. Ms. C takes lisinopril 10 mg/d and atorvastatin, which was recently increased from 10 to 20 mg/d.
What would your next steps be in caring for this patient?
Patients who experience muscle-related symptoms such as pain, fatigue, or weakness often seek help from their family physician (FP). The list of possible causes of these complaints can be lengthy and vary greatly, from nonmyopathic conditions such as fibromyalgia to worrisome forms of myopathy such as inclusion body myositis or polymyositis. This article will help you to quickly identify which patients with muscle-related complaints should be evaluated for myopathy and what your work-up should include.
Myopathy or not?
Distinguishing between myopathy and nonmyopathic muscle pain or weakness is the first step in evaluating patients with muscle-related complaints. Many conditions share muscle-related symptoms, but actual muscle damage is not always present (eg, fibromyalgia, chronic pain, and chronic fatigue syndromes).1 While there is some overlap in presentation between patients with myopathy and nonmyopathic conditions, there are important differences in symptoms, physical exam findings, and lab test results (TABLE 11-4). Notably, in myopathic disease, patients’ symptoms are usually progressive, vital signs are abnormal, and weakness is common, whereas patients with nonmyopathic disease typically have remitting and relapsing symptoms, normal vital signs, and no weakness.
Myopathy itself is divided into 3 categories—myositic, intrinsic, and toxic—which reflect the condition, or medication, that brought on the muscle damage (TABLE 22,4-15). Placing patients into one of these categories based on their risk factors, history, and physical exam findings can help to focus the diagnostic work-up on areas most likely to provide useful information.
Myositic myopathy can be caused by infection or autoimmunity
Myositic myopathies result in inflammatory destruction of muscle tissue. Patients with myositic myopathy often exhibit fever, malaise, weight loss, and general fatigue. Though weakness and pain are common, both can be variable or even absent in myositic myopathy.2,5 Myositic myopathy can be caused by infectious agents or can develop from an autoimmune disease.
Infectious myositic myopathy is one of the more common types of myopathy that FPs will encounter.2 Viruses such as influenza, parainfluenza, coxsackievirus, human immunodeficiency virus, cytomegalovirus, echovirus, adenovirus, Epstein-Barr, and hepatitis C are common causes.2,4,16 Bacterial and fungal myositides are relatively rare. Both most often occur as the result of penetrating trauma or immunocompromise, and are generally not subtle.2 Parasitic myopathy can occur from the invasion of skeletal muscle by trichinella after ingesting undercooked, infected meat.2 Although previously a more common problem, currently only 10 to 20 cases of trichinellosis are reported in the United States each year.17 Due to their rarity, bacterial, fungal, and parasitic myositides are not reviewed here.
Patients with a viral myositis often report prodromal symptoms such as fever, upper respiratory illness, or gastrointestinal distress one to 2 weeks before the onset of muscle complaints. Muscle pain is usually multifocal, involving larger, bilateral muscle groups, and may be associated with swelling.
Patients with viral myositis may exhibit diffusely painful, swollen, or boggy-feeling muscles as well as weakness and pain with exertion. Other signs of viral infection such as rash, fever, upper respiratory symptoms, or meningeal signs may be present. Severe signs include arrhythmia or respiratory failure due to cardiac muscle or diaphragm involvement, or signs of renal failure due to precipitation of myoglobin in the renal system (ie, rhabdomyolysis).2 If the infection affects the heart, patients may develop palpitations, pleuritic chest pain, or shortness of breath.2
Diagnosis of viral myositis relies heavily on clinical suspicion in patients with a fitting history and physical exam findings. Helpful lab tests include a complete blood count (CBC), erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), creatine kinase (CK), and liver function tests (LFTs), all of which can be abnormal in viral myositis. Viral polymerase chain reaction, culture, or antigen testing may be helpful in severe or confusing cases, but in most cases such testing is unnecessary. Muscle biopsy is not recommended except in persistent cases, where definitive identification of the causative agent might alter treatment or when nonviral infection is suspected.2
Autoimmune myositic myopathy. Unlike infectious myopathies, autoimmune myopathies are usually chronic, subtle, and relatively rare. The 3 most common autoimmune myopathies—polymyositis, dermatomyositis, and inclusion body myositis—have a combined prevalence of approximately 10:100,000.6 Although these types of myopathies are uncommon, FPs will likely be the first to evaluate a patient with one of them.
Patients with an autoimmune myopathy typically complain of weakness and mild to moderate muscle pain, although pain may be absent. Compared to infectious myopathies, autoimmune myopathies usually exhibit a more indolent course. Patients with advanced disease may report fever, weight loss, shortness of breath from cardiomyopathy, heartburn from a weakened lower esophageal sphincter, and/or a rash.5
Physical examination may reveal symmetric, proximal muscle weakness. Atrophy is typically not seen until late in the disease. Skin exam usually is normal in patients with inclusion body myositis and polymyositis. The typical rash of dermatomyositis is a heliotrope (blue-purple) discoloration on the upper eyelids and a raised, violaceous, scaly eruption on the knuckles (Gottron’s papules).
Laboratory tests that can be helpful include CK, lactate dehydrogenase (LDH), aldolase, and LFTs (reflecting muscle injury, not liver involvement). For polymyositis and dermatomyositis, CK is the most sensitive lab test and often exhibits the highest elevation above normal.6 Conversely, CK is often normal or only mildly elevated in inclusion body myositis. Up to 80% of patients with autoimmune myopathy will have antinuclear antibodies.3,5 ESR and CRP levels are also often elevated.
Both electromyography (EMG) and muscle biopsy may be required to diagnose autoimmune myopathy, but these are typically done under the direction of a rheumatologist after an FP’s initial work-up is inconclusive.
Intrinsic myopathy: Suspect electrolyte problems, other causes
Intrinsic myopathy occurs in patients with electrolyte disorders, diseases of the endocrine system, or underlying metabolic dysfunction.
Electrolyte disorders. Muscle-related symptoms are unlikely to be the chief complaint of patients with severe electrolyte imbalance. However, a patient with mild to moderate electrolyte problems may develop muscle fatigue, weakness, or pain. TABLE 3 reviews other signs and symptoms of electrolyte abnormalities that may be helpful in establishing a diagnosis in a patient with muscle complaints.
Ordering a complete metabolic panel (CMP), CK, and urinalysis (UA) can help rule out electrolyte disorders. If electrolyte disorders are detected, an electrocardiogram is useful to evaluate for cardiac dysfunction. Once an electrolyte disorder is identified, investigate its underlying cause. Correcting the electrolyte disorder should help improve symptoms of myopathy.
Endocrine myopathy can be associated with hypothyroidism, hyperthyroidism, parathyroid disease, vitamin D deficiency, or Cushing syndrome.8-10,18,19 Although less common than some other causes, identifying endocrine myopathy is crucial because correcting the underlying disease will often improve multiple aspects of the patient’s health.
The presentation of endocrine myopathy may be subtle. Patients with hypothyroidism may experience muscle pain or weakness, fatigue, cold sensitivity, constipation, and dry skin.20 Muscle-related symptoms may be the only sign of endocrine myopathy in a patient who would otherwise be considered to have subclinical hypothyroidism.8,18 Hyperthyroidism can present with weight loss, heat intolerance, frequent bowel movements, tachycardia, and muscle weakness.21
Patients with parathyroid disease— especially patients with chronic renal failure—may report proximal muscle weakness, often in the lower extremities.19 Complaints of muscle weakness or pain can occur with severe vitamin D deficiency.10 Patients with Cushing syndrome often experience proximal weakness and weight gain.9
Patients with a personal or family history of endocrine disorders, previous thyroid surgery, or those taking medications that can impair thyroid function, such as lithium, amiodarone, or interferon, are at risk for endocrine myopathy.18-20 Suspect hyperparathyroidism in patients with chronic kidney disease who complain of weakness.
Vitamin D deficiency is relatively common, with at minimum 20% of elderly adults estimated to be deficient.10 Patients at risk for Cushing disease are most likely receiving pharmacologic doses of glucocorticoids, which can increase their risk of myopathy, or to have ectopic adrenocorticotropic hormone secretion.
Metabolic myopathy results from a lack of sufficient energy production in the muscle. The 3 main groups of metabolic myopathy are impaired muscle glycogenoses, disorders of fatty acid oxidation, and mitochondrial myopathies.7
Because metabolic myopathy can occur at any age, a thorough history and physical is crucial for diagnosis. Proximal weakness in metabolic myopathy is often associated with exercise intolerance, stressful illness, or fasting. Patients often present with dynamic abnormalities such as fatigue, muscle cramping, and even rhabdomyolysis during exertion.7
When evaluating patients you suspect may have metabolic myopathy, a physical exam may reveal muscle contractures, muscle swelling, or proximal muscle weakness. Patients with certain types of fatty acid oxidation disorders or mitochondrial disorders may also exhibit cardiomyopathy, neuropathy, retinopathy, ataxia, hearing loss, or other systemic manifestations.7
Basic labs for investigating suspected metabolic myopathy include serum electrolytes, glucose, LFTs, CK (which may or may not be elevated), lactate, ammonia, and UA for myoglobinuria. More advanced labs, such as serum total carnitine and acylcarnitine as well as urinary levels of dicarboxylic acids and acylglycines, may be needed if a metabolic disorder is strongly suspected.7 Muscle biopsy, EMG, and genetic testing can also prove helpful in diagnosis. Definitive diagnosis and treatment of metabolic myopathy usually requires a multidisciplinary team of providers, including subspecialty referral.
Toxic myopathy
Toxic myopathy refers to muscle damage caused by an exogenous chemical agent, most often a drug. The mechanism of toxicity is not always clear and may result from the activation of inflammatory responses similar to autoimmune myopathy.22 Toxic myopathies may result from several commonly used medications; cholesterol-lowering medications are a common culprit.13-15,23-25 Drug-induced myopathies vary in frequency and severity. For instance, in patients taking statins, the rate of myalgias is 6%, while the incidence of rhabdomyolysis is estimated to be 4 per 100,000, and is found most often in patients taking concomitant fibrates.23
Drug-induced toxic myopathy differs from previously discussed myopathies in that symptoms are usually more insidious, findings on exam are more often mixed muscular and neurologic, and lab abnormalities are usually more subtle.11,12 Symptoms of myopathy typically occur weeks or months after initiating a drug and usually improve or resolve within weeks after discontinuing the offending agent. Knowing the patient’s medication list and which medications cause certain patterns of myopathy symptoms can help guide the differential diagnosis (TABLE 411-15,22-25).
Risk factors for most medication-related myopathies are polypharmacy, renal or liver disease, and age over 50 years13-15,23-25 The physical exam for patients with drug- or toxin-related myopathy will most often reveal relatively minor abnormalities such as muscle tenderness and mild weakness, except for the most severe or advanced cases. Most patients will not have physical signs that suggest an underlying illness. CK levels and LFTs should be obtained. Basic chemistry and UA may also be helpful in patients with risk factors for renal disease.
CASE › Ms. C has been taking a statin for more than 10 years, and the dose was recently increased. You are aware that statin-related muscle injury can develop even after years of use, and suspect the statin may be causing her myopathy. You order a CK test, which is mildly elevated. You recommend discontinuing the statin. After 8 weeks off her statin, Ms. C’s Symptoms do not improve. Given her lack of systemic complaints, myositic myopathy from an infectious or rheumatologic cause seems unlikely. You begin to consider an intrinsic cause of myopathy, and order the following tests: a CMP, UA, thyroid-stimulating hormone, repeat CK, and vitamin D level. This testing reveals a vitamin D deficiency at 17 ng/ml (normal range: 30-74 ng/ml). You recommend vitamin D, 50,000 IU per week for 8 weeks. At follow-up, Ms. C's vitamin D level is 40. She says she feels better and her muscle complaints have resolved.
CORRESPONDENCE
Brent W. Smith, MD, Travis Air Force Base Family Medicine Residency, 101 Bodin Circle, Travis Air Force Base, CA 94535; smithb@smithnet.us
› Categorize patients with muscle complaints into suspected myositic, intrinsic, or toxic myopathy to help guide subsequent work-up. C
› Look for diffusely painful, swollen, or boggy-feeling muscles—as well as weakness and pain with exertion—in patients you suspect may have viral myopathy. C
› Consider electromyography and muscle biopsy for patients you suspect may have dermatomyositis. C
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
CASE › Marie C, a 75-year-old Asian woman, reports weakness in her legs and arms with unsteadiness when walking. She has a vague but persistent ache in her large muscles. Her symptoms have developed slowly over the past 3 months. She denies recent signs or symptoms of infection or other illness. Her medical history includes hypertension, hyperlipidemia, osteopenia, and obesity. Ms. C takes lisinopril 10 mg/d and atorvastatin, which was recently increased from 10 to 20 mg/d.
What would your next steps be in caring for this patient?
Patients who experience muscle-related symptoms such as pain, fatigue, or weakness often seek help from their family physician (FP). The list of possible causes of these complaints can be lengthy and vary greatly, from nonmyopathic conditions such as fibromyalgia to worrisome forms of myopathy such as inclusion body myositis or polymyositis. This article will help you to quickly identify which patients with muscle-related complaints should be evaluated for myopathy and what your work-up should include.
Myopathy or not?
Distinguishing between myopathy and nonmyopathic muscle pain or weakness is the first step in evaluating patients with muscle-related complaints. Many conditions share muscle-related symptoms, but actual muscle damage is not always present (eg, fibromyalgia, chronic pain, and chronic fatigue syndromes).1 While there is some overlap in presentation between patients with myopathy and nonmyopathic conditions, there are important differences in symptoms, physical exam findings, and lab test results (TABLE 11-4). Notably, in myopathic disease, patients’ symptoms are usually progressive, vital signs are abnormal, and weakness is common, whereas patients with nonmyopathic disease typically have remitting and relapsing symptoms, normal vital signs, and no weakness.
Myopathy itself is divided into 3 categories—myositic, intrinsic, and toxic—which reflect the condition, or medication, that brought on the muscle damage (TABLE 22,4-15). Placing patients into one of these categories based on their risk factors, history, and physical exam findings can help to focus the diagnostic work-up on areas most likely to provide useful information.
Myositic myopathy can be caused by infection or autoimmunity
Myositic myopathies result in inflammatory destruction of muscle tissue. Patients with myositic myopathy often exhibit fever, malaise, weight loss, and general fatigue. Though weakness and pain are common, both can be variable or even absent in myositic myopathy.2,5 Myositic myopathy can be caused by infectious agents or can develop from an autoimmune disease.
Infectious myositic myopathy is one of the more common types of myopathy that FPs will encounter.2 Viruses such as influenza, parainfluenza, coxsackievirus, human immunodeficiency virus, cytomegalovirus, echovirus, adenovirus, Epstein-Barr, and hepatitis C are common causes.2,4,16 Bacterial and fungal myositides are relatively rare. Both most often occur as the result of penetrating trauma or immunocompromise, and are generally not subtle.2 Parasitic myopathy can occur from the invasion of skeletal muscle by trichinella after ingesting undercooked, infected meat.2 Although previously a more common problem, currently only 10 to 20 cases of trichinellosis are reported in the United States each year.17 Due to their rarity, bacterial, fungal, and parasitic myositides are not reviewed here.
Patients with a viral myositis often report prodromal symptoms such as fever, upper respiratory illness, or gastrointestinal distress one to 2 weeks before the onset of muscle complaints. Muscle pain is usually multifocal, involving larger, bilateral muscle groups, and may be associated with swelling.
Patients with viral myositis may exhibit diffusely painful, swollen, or boggy-feeling muscles as well as weakness and pain with exertion. Other signs of viral infection such as rash, fever, upper respiratory symptoms, or meningeal signs may be present. Severe signs include arrhythmia or respiratory failure due to cardiac muscle or diaphragm involvement, or signs of renal failure due to precipitation of myoglobin in the renal system (ie, rhabdomyolysis).2 If the infection affects the heart, patients may develop palpitations, pleuritic chest pain, or shortness of breath.2
Diagnosis of viral myositis relies heavily on clinical suspicion in patients with a fitting history and physical exam findings. Helpful lab tests include a complete blood count (CBC), erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), creatine kinase (CK), and liver function tests (LFTs), all of which can be abnormal in viral myositis. Viral polymerase chain reaction, culture, or antigen testing may be helpful in severe or confusing cases, but in most cases such testing is unnecessary. Muscle biopsy is not recommended except in persistent cases, where definitive identification of the causative agent might alter treatment or when nonviral infection is suspected.2
Autoimmune myositic myopathy. Unlike infectious myopathies, autoimmune myopathies are usually chronic, subtle, and relatively rare. The 3 most common autoimmune myopathies—polymyositis, dermatomyositis, and inclusion body myositis—have a combined prevalence of approximately 10:100,000.6 Although these types of myopathies are uncommon, FPs will likely be the first to evaluate a patient with one of them.
Patients with an autoimmune myopathy typically complain of weakness and mild to moderate muscle pain, although pain may be absent. Compared to infectious myopathies, autoimmune myopathies usually exhibit a more indolent course. Patients with advanced disease may report fever, weight loss, shortness of breath from cardiomyopathy, heartburn from a weakened lower esophageal sphincter, and/or a rash.5
Physical examination may reveal symmetric, proximal muscle weakness. Atrophy is typically not seen until late in the disease. Skin exam usually is normal in patients with inclusion body myositis and polymyositis. The typical rash of dermatomyositis is a heliotrope (blue-purple) discoloration on the upper eyelids and a raised, violaceous, scaly eruption on the knuckles (Gottron’s papules).
Laboratory tests that can be helpful include CK, lactate dehydrogenase (LDH), aldolase, and LFTs (reflecting muscle injury, not liver involvement). For polymyositis and dermatomyositis, CK is the most sensitive lab test and often exhibits the highest elevation above normal.6 Conversely, CK is often normal or only mildly elevated in inclusion body myositis. Up to 80% of patients with autoimmune myopathy will have antinuclear antibodies.3,5 ESR and CRP levels are also often elevated.
Both electromyography (EMG) and muscle biopsy may be required to diagnose autoimmune myopathy, but these are typically done under the direction of a rheumatologist after an FP’s initial work-up is inconclusive.
Intrinsic myopathy: Suspect electrolyte problems, other causes
Intrinsic myopathy occurs in patients with electrolyte disorders, diseases of the endocrine system, or underlying metabolic dysfunction.
Electrolyte disorders. Muscle-related symptoms are unlikely to be the chief complaint of patients with severe electrolyte imbalance. However, a patient with mild to moderate electrolyte problems may develop muscle fatigue, weakness, or pain. TABLE 3 reviews other signs and symptoms of electrolyte abnormalities that may be helpful in establishing a diagnosis in a patient with muscle complaints.
Ordering a complete metabolic panel (CMP), CK, and urinalysis (UA) can help rule out electrolyte disorders. If electrolyte disorders are detected, an electrocardiogram is useful to evaluate for cardiac dysfunction. Once an electrolyte disorder is identified, investigate its underlying cause. Correcting the electrolyte disorder should help improve symptoms of myopathy.
Endocrine myopathy can be associated with hypothyroidism, hyperthyroidism, parathyroid disease, vitamin D deficiency, or Cushing syndrome.8-10,18,19 Although less common than some other causes, identifying endocrine myopathy is crucial because correcting the underlying disease will often improve multiple aspects of the patient’s health.
The presentation of endocrine myopathy may be subtle. Patients with hypothyroidism may experience muscle pain or weakness, fatigue, cold sensitivity, constipation, and dry skin.20 Muscle-related symptoms may be the only sign of endocrine myopathy in a patient who would otherwise be considered to have subclinical hypothyroidism.8,18 Hyperthyroidism can present with weight loss, heat intolerance, frequent bowel movements, tachycardia, and muscle weakness.21
Patients with parathyroid disease— especially patients with chronic renal failure—may report proximal muscle weakness, often in the lower extremities.19 Complaints of muscle weakness or pain can occur with severe vitamin D deficiency.10 Patients with Cushing syndrome often experience proximal weakness and weight gain.9
Patients with a personal or family history of endocrine disorders, previous thyroid surgery, or those taking medications that can impair thyroid function, such as lithium, amiodarone, or interferon, are at risk for endocrine myopathy.18-20 Suspect hyperparathyroidism in patients with chronic kidney disease who complain of weakness.
Vitamin D deficiency is relatively common, with at minimum 20% of elderly adults estimated to be deficient.10 Patients at risk for Cushing disease are most likely receiving pharmacologic doses of glucocorticoids, which can increase their risk of myopathy, or to have ectopic adrenocorticotropic hormone secretion.
Metabolic myopathy results from a lack of sufficient energy production in the muscle. The 3 main groups of metabolic myopathy are impaired muscle glycogenoses, disorders of fatty acid oxidation, and mitochondrial myopathies.7
Because metabolic myopathy can occur at any age, a thorough history and physical is crucial for diagnosis. Proximal weakness in metabolic myopathy is often associated with exercise intolerance, stressful illness, or fasting. Patients often present with dynamic abnormalities such as fatigue, muscle cramping, and even rhabdomyolysis during exertion.7
When evaluating patients you suspect may have metabolic myopathy, a physical exam may reveal muscle contractures, muscle swelling, or proximal muscle weakness. Patients with certain types of fatty acid oxidation disorders or mitochondrial disorders may also exhibit cardiomyopathy, neuropathy, retinopathy, ataxia, hearing loss, or other systemic manifestations.7
Basic labs for investigating suspected metabolic myopathy include serum electrolytes, glucose, LFTs, CK (which may or may not be elevated), lactate, ammonia, and UA for myoglobinuria. More advanced labs, such as serum total carnitine and acylcarnitine as well as urinary levels of dicarboxylic acids and acylglycines, may be needed if a metabolic disorder is strongly suspected.7 Muscle biopsy, EMG, and genetic testing can also prove helpful in diagnosis. Definitive diagnosis and treatment of metabolic myopathy usually requires a multidisciplinary team of providers, including subspecialty referral.
Toxic myopathy
Toxic myopathy refers to muscle damage caused by an exogenous chemical agent, most often a drug. The mechanism of toxicity is not always clear and may result from the activation of inflammatory responses similar to autoimmune myopathy.22 Toxic myopathies may result from several commonly used medications; cholesterol-lowering medications are a common culprit.13-15,23-25 Drug-induced myopathies vary in frequency and severity. For instance, in patients taking statins, the rate of myalgias is 6%, while the incidence of rhabdomyolysis is estimated to be 4 per 100,000, and is found most often in patients taking concomitant fibrates.23
Drug-induced toxic myopathy differs from previously discussed myopathies in that symptoms are usually more insidious, findings on exam are more often mixed muscular and neurologic, and lab abnormalities are usually more subtle.11,12 Symptoms of myopathy typically occur weeks or months after initiating a drug and usually improve or resolve within weeks after discontinuing the offending agent. Knowing the patient’s medication list and which medications cause certain patterns of myopathy symptoms can help guide the differential diagnosis (TABLE 411-15,22-25).
Risk factors for most medication-related myopathies are polypharmacy, renal or liver disease, and age over 50 years13-15,23-25 The physical exam for patients with drug- or toxin-related myopathy will most often reveal relatively minor abnormalities such as muscle tenderness and mild weakness, except for the most severe or advanced cases. Most patients will not have physical signs that suggest an underlying illness. CK levels and LFTs should be obtained. Basic chemistry and UA may also be helpful in patients with risk factors for renal disease.
CASE › Ms. C has been taking a statin for more than 10 years, and the dose was recently increased. You are aware that statin-related muscle injury can develop even after years of use, and suspect the statin may be causing her myopathy. You order a CK test, which is mildly elevated. You recommend discontinuing the statin. After 8 weeks off her statin, Ms. C’s Symptoms do not improve. Given her lack of systemic complaints, myositic myopathy from an infectious or rheumatologic cause seems unlikely. You begin to consider an intrinsic cause of myopathy, and order the following tests: a CMP, UA, thyroid-stimulating hormone, repeat CK, and vitamin D level. This testing reveals a vitamin D deficiency at 17 ng/ml (normal range: 30-74 ng/ml). You recommend vitamin D, 50,000 IU per week for 8 weeks. At follow-up, Ms. C's vitamin D level is 40. She says she feels better and her muscle complaints have resolved.
CORRESPONDENCE
Brent W. Smith, MD, Travis Air Force Base Family Medicine Residency, 101 Bodin Circle, Travis Air Force Base, CA 94535; smithb@smithnet.us
1. Huynh CN, Yanni LM, Morgan LA. Fibromyalgia: diagnosis and management for the primary healthcare provider. J Womens Health. 2008;8:1379-1387.
2. Crum-Cianflone NF. Bacterial, fungal, parasitic, and viral myositis. Clin Microbiol Rev. 2008;21:473-494.
3. Reichlin M, Arnett FC Jr. Multiplicity of antibodies in myositis sera. Arthritis Rheum. 1984;27:1150-1156.
4. Yoshino M, Suzuki S, Adachi K, et al. High incidence of acute myositis with type A influenza virus infection in the elderly. Intern Med. 2000;39:431-432.
5. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971-982.
6. Wilson FC, Ytterberg SR, St Sauver JL, et al. Epidemiology of sporadic inclusion body myositis and polymyositis in Olmsted County, Minnesota. J Rheumatol. 2008;35:445-447.
7. Smith EC, El-Gharbawy A, Koeberl DD. Metabolic myopathies: clinical features and diagnostic approach. Rheum Dis Clin N Am. 2011:37:201-217.
8. Reuters V, Teixeira Pde F, Vigário PS, et al. Functional capacity and muscular abnormalities in subclinical hypothyroidism. Am J Med Sci. 2009;338:259-263.
9. Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2008;93:1526-1540.
10. Holick MF, Binkley NC, Bischoff-Ferrari HA, et al; Endocrine Society. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:1911-1930.
11. Antons KA, Williams CD, Baker SK, et al. Clinical perspectives of statin-induced rhabdomyolysis. Am J Med. 2006;119:400-409.
12. Phillips PS, Haas RH, Bannykh S, et al; Scripps Mercy Clinical Research Center. Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med. 2002;137:581-585.
13. Pereira RM, Freire de Carvalho J. Glucocorticoid-induced myopathy. Joint Bone Spine. 2011;78:41-44.
14. Posada C, García-Cruz A, García-Doval I, et al. Chloroquine-induced myopathy. Lupus. 2011;20:773-774.
15. Uri DS, Biavis M. Colchicine neuromyopathy. J Clin Rheumatol. 1996;2:163-166.
16. Mannix R, Tan ML, Wright R, et al. Acute pediatric rhabdomyolysis: causes and rates of renal failure. Pediatrics. 2006;118:2119-2125.
17. Pozio E. World distribution of Trichinella spp. infections in animals and humans. Vet Parasitol. 2007;149:3-21.
18. Rodolico C, Toscano A, Benvenga S, et al. Myopathy as the persistently isolated symptomatology of primary autoimmune hypothyroidism. Thyroid.1998;8:1033-1038.
19. AACE/AAES Task Force on Primary Hyperparathyroidism. The American Association of Clinical Endocrinologists and The American Association of Endocrine Surgeons position statement on the diagnosis and management of primary hyperparathyroidism. Endocr Pract. 2005;11:49-54.
20. Garber JR, Cobin RH, Gharib H, et al; American Association of Clinical Endocrinologists and American Thyroid Association Taskforce on Hypothyroidism in Adults. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Endocrine Pract. 2012;18:988-1028.
21. Bahn Chair RS, Burch HB, Cooper DS, et al; American Thyroid Association; American Association of Clinical Endocrinologists. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Thyroid. 2011;21:593-646.
22. Mammen AL, Amato AA. Statin myopathy: a review of recent progress. Curr Opin Rheumatol. 2010;22:644-650.
23. Buettner C, Davis RB, Leveille SG, et al. Prevalence of musculoskeletal pain and statin use. J Gen Intern Med. 2008;23: 1182-1186.
24. Marot A, Morelle J, Chouinard VA, et al. Concomitant use of simvastatin and amiodarone resulting in severe rhabdomyolysis: a case report and review of the literature. Acta Clin Belg. 2011;66:134-136.
25. Peters BS, Winer J, Landon DN, et al. Mitochondrial myopathy associated with chronic zidovudine therapy in AIDS. Q J Med. 1993;86:5-15.
1. Huynh CN, Yanni LM, Morgan LA. Fibromyalgia: diagnosis and management for the primary healthcare provider. J Womens Health. 2008;8:1379-1387.
2. Crum-Cianflone NF. Bacterial, fungal, parasitic, and viral myositis. Clin Microbiol Rev. 2008;21:473-494.
3. Reichlin M, Arnett FC Jr. Multiplicity of antibodies in myositis sera. Arthritis Rheum. 1984;27:1150-1156.
4. Yoshino M, Suzuki S, Adachi K, et al. High incidence of acute myositis with type A influenza virus infection in the elderly. Intern Med. 2000;39:431-432.
5. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362:971-982.
6. Wilson FC, Ytterberg SR, St Sauver JL, et al. Epidemiology of sporadic inclusion body myositis and polymyositis in Olmsted County, Minnesota. J Rheumatol. 2008;35:445-447.
7. Smith EC, El-Gharbawy A, Koeberl DD. Metabolic myopathies: clinical features and diagnostic approach. Rheum Dis Clin N Am. 2011:37:201-217.
8. Reuters V, Teixeira Pde F, Vigário PS, et al. Functional capacity and muscular abnormalities in subclinical hypothyroidism. Am J Med Sci. 2009;338:259-263.
9. Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2008;93:1526-1540.
10. Holick MF, Binkley NC, Bischoff-Ferrari HA, et al; Endocrine Society. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:1911-1930.
11. Antons KA, Williams CD, Baker SK, et al. Clinical perspectives of statin-induced rhabdomyolysis. Am J Med. 2006;119:400-409.
12. Phillips PS, Haas RH, Bannykh S, et al; Scripps Mercy Clinical Research Center. Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med. 2002;137:581-585.
13. Pereira RM, Freire de Carvalho J. Glucocorticoid-induced myopathy. Joint Bone Spine. 2011;78:41-44.
14. Posada C, García-Cruz A, García-Doval I, et al. Chloroquine-induced myopathy. Lupus. 2011;20:773-774.
15. Uri DS, Biavis M. Colchicine neuromyopathy. J Clin Rheumatol. 1996;2:163-166.
16. Mannix R, Tan ML, Wright R, et al. Acute pediatric rhabdomyolysis: causes and rates of renal failure. Pediatrics. 2006;118:2119-2125.
17. Pozio E. World distribution of Trichinella spp. infections in animals and humans. Vet Parasitol. 2007;149:3-21.
18. Rodolico C, Toscano A, Benvenga S, et al. Myopathy as the persistently isolated symptomatology of primary autoimmune hypothyroidism. Thyroid.1998;8:1033-1038.
19. AACE/AAES Task Force on Primary Hyperparathyroidism. The American Association of Clinical Endocrinologists and The American Association of Endocrine Surgeons position statement on the diagnosis and management of primary hyperparathyroidism. Endocr Pract. 2005;11:49-54.
20. Garber JR, Cobin RH, Gharib H, et al; American Association of Clinical Endocrinologists and American Thyroid Association Taskforce on Hypothyroidism in Adults. Clinical practice guidelines for hypothyroidism in adults: cosponsored by the American Association of Clinical Endocrinologists and the American Thyroid Association. Endocrine Pract. 2012;18:988-1028.
21. Bahn Chair RS, Burch HB, Cooper DS, et al; American Thyroid Association; American Association of Clinical Endocrinologists. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Thyroid. 2011;21:593-646.
22. Mammen AL, Amato AA. Statin myopathy: a review of recent progress. Curr Opin Rheumatol. 2010;22:644-650.
23. Buettner C, Davis RB, Leveille SG, et al. Prevalence of musculoskeletal pain and statin use. J Gen Intern Med. 2008;23: 1182-1186.
24. Marot A, Morelle J, Chouinard VA, et al. Concomitant use of simvastatin and amiodarone resulting in severe rhabdomyolysis: a case report and review of the literature. Acta Clin Belg. 2011;66:134-136.
25. Peters BS, Winer J, Landon DN, et al. Mitochondrial myopathy associated with chronic zidovudine therapy in AIDS. Q J Med. 1993;86:5-15.
