User login
Take this simplified approach to correcting exposure of vaginal mesh
CASE: Pain during intercourse, well after mesh implantation
Your patient, 61 years old, para 3, has come to your office by referral with a complaint of dyspareunia. The history includes placement of a synthetic vaginal mesh kit 14 months earlier for prolapse.
The medical record shows that the referring physician performed a “mesh excision” 1 year after the original procedure.
The woman reports that she is “very frustrated” that she is still dealing with this problem so long after the original procedure.
On examination, you note a 2.5-cm diameter area of exposed mesh in the anterior vagina, with healthy surrounding tissue and without inflammation or purulence (FIGURE 1). You are unable to reproduce her complaint of pain on vaginal examination.
What options can you offer to this woman? And will those options meet her therapeutic expectations?
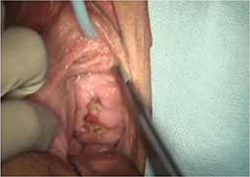
FIGURE 1 Examination of your referred patient: Mesh is noticeably exposedThe recent increase in the use of mesh grafts to reconstruct pelvic anatomy has been directed mainly at improving surgical outcomes. Yet, at the same time, gynecologic surgeons find themselves facing a rise in associated complications of such surgery that they did not see previously.
Among the most troublesome and concerning of those complications are 1) exposure of mesh through the vaginal epithelium and 2) contraction or hardening of mesh (or both) that can result in dyspareunia and chronic pelvic pain. Other, rare complications include infection and fistula.
Our goal in this article is to address the management of graft-healing abnormalities in which a segment of the mesh is palpable or visible, or both, within the vaginal canal. Our focus is on simple abnormalities that can be managed by most generalist gynecologists; to be clear, more complex abnormalities, and those that provoke more serious or lasting symptoms, belong under the care of a specialist.
A recent shift in terminology is significant
Early on, this complication was called “erosion” as understanding of the mechanism of its development grew, however, terminology applied to the problem has changed.
In fact, mesh itself very rarely erodes into the vagina or an underlying viscus. Instead, the complication occurs most commonly as a result of disruption of a suture line—most likely the result of a hematoma or localized inflammation that develops postoperatively.
“Exposure” (our preference here) and “extrusion” are now the recommended terms, based on a consensus terminology document published this year jointly by the International Urogynecological Association and the International Continence Society.1
Exposure of implanted mesh is considered a “simple” healing abnormality because it typically
- occurs along the suture line and early in the course of healing
- is not associated with infection of the graft.2
The typical physical appearance is one of visible mesh along an open suture line without granulation tissue or purulence—again, see FIGURE 1. The mesh is firmly adherent to the vaginal epithelial edges and underlying fascia.
The reported incidence of mesh exposures—in regard to currently used meshes, which are all Type-1, monofilament, macroporous polypropylene grafts—is approximately 10% but as high as 15% to 20% in some reported series.3,4 The higher rates of exposure are usually seen in series in which some patients have had a synthetic graft implanted as an overlay to fascial midline plication. When the graft is implanted in the subfascial layer of the vaginal wall (i.e., without midline plication), however, the reported rate of exposure falls—to 5% to 10%.5-7
Recommendations for management
Initially, recommendations for “erosion” management were based on concerns about underlying mesh infection or rejection, and included a need to remove the entire graft. That recommendation still applies to multifilament, microporous grafts that present with inflammatory infiltrates, granulation tissue, and purulence. Although these kinds of grafts (known as “Type-2/3 grafts”—e.g., GoreTex, IVS) have not been marketed for pelvic reconstruction over the past 3 to 5 years, their behavior post-implantation is less predictable—and patients who have delayed healing abnormalities are, therefore, still being seen. It’s fortunate that development of an overlying biofilm prevents tissue incorporation into these types of graft, allowing them to be removed easily.
Exposures related to Type-1 mesh—currently used in pelvic reconstruction—that occur without surrounding infection do not require extensive removal. Rather, they can be managed conservatively or, when necessary, with outpatient surgery. In patients who are not sexually active, exposures are usually asymptomatic; they might only be observed by the physician on vaginal examination and are amenable to simple monitoring. In sexually active patients, exposure of Type-1 mesh usually results in dyspareunia or a complaint that the partner “can feel the mesh.” Depending on the size and the nature of symptoms and the extent of the defect, these commonly seen exposures can be managed by following a simple algorithm.
Palpable or visible mesh fibrils can be trimmed in the office; they might even respond to local estrogen alone. Consider these options if the patient displays vaginal atrophy.
Typically, vaginal estrogen is prescribed as 1 g nightly for 2 weeks and then 1 g two or three nights a week. Re-examine the patient in 3 months; if symptoms of mesh exposure persist, it’s unlikely that continued conservative therapy will be successful, and outpatient surgery is recommended.
When exposure is asymptomatic, you can simply monitor the condition for 3 to 6 months; if complaints or findings arise, consider intervention.
Small (<0.5 cm in diameter) exposures can also be managed in the office, including excision of exposed mesh and local estrogen. If the exposure is easily reachable, we recommend grasping the exposed area with pick-ups or a hemostat and with gentle traction, using Metzenbaum scissors to trim exposed mesh as close to the vaginal epithelium as possible. Local topical or injected anesthesia may be needed. Bleeding should be minimal because no dissection is necessary. Silver nitrate can be applied for any minor bleeding. Larger (0.5–4.0 cm) exposures are unlikely to heal on their own. They require outpatient excision in the operating room.
Preoperative tissue preparation with local estrogen is key to successful repair of these exposures. Vaginal estrogen increases blood flow to the epithelium; as tissue becomes well-estrogenized, risk of recurrence diminishes.
The technique we employ includes:
- circumferential infiltration of vaginal epithelium surrounding the exposed mesh with 1% lidocaine with epinephrine
- sharp circumscription of the area of exposure, using a scalpel, with a 0.5-cm margin of vaginal epithelium (FIGURE 2)
- wide dissection, with undermining and mobilization of surrounding healthy vaginal epithelium around the exposure (FIGURE 3)
- excision of the exposed mesh and attached vaginal mucosa, with careful dissection of the mesh off underlying tissues with Metzenbaum scissors—being careful to avoid injury to underlying bladder or rectum (FIGURE 4)
- reapproximation of mesh edges, using 2-0 polypropylene suture to close the resulting defect so that prolapse does not recur (FIGURE 5)
- closing of the previously mobilized vaginal epithelium with 2-0 Vicryl suture, without tension, to cover the reapproximated mesh edges—after irrigation and assurance of adequate hemostasis (FIGURE 6).
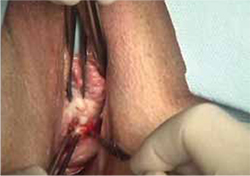
FIGURE 2 Incision of vaginal epithelium
Allow for a 0.5-cm margin.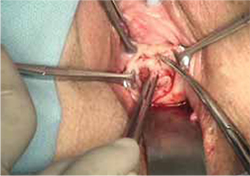
FIGURE 3 Undermining and mobilization of epithelium
Perform wide dissection.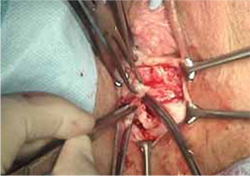
FIGURE 4 Dissection of mesh from underlying tissue
Keep clear of underlying bladder and rectum!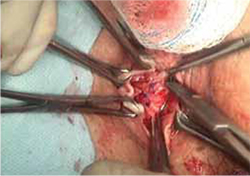
FIGURE 5 Reapproximation of edges to re-establish support
Our choice of suture is 2-0 polypropylene.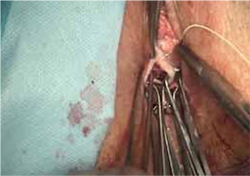
FIGURE 6 Irrigation of vaginal epithelium, followed by closure
Before you close, ensure that hemostasis is adequate.The choice of closure—vertical or horizontal—depends on the nature of the original defect.
You can watch a video of this technique that we’ve provided.
Several cautions should be taken with this technique, including:
- avoiding narrowing the vaginal canal
- minimizing trauma to healthy vaginal epithelium that will be used for closure
- maintaining hemostasis to avoid formation of hematomas.
Largest (>4 cm) exposures are likely the result of devascularized sloughing of vaginal epithelium. They are, fortunately, uncommon.
It’s unlikely that, after excision of exposed mesh, the vaginal epithelial edges can be approximated without significantly narrowing or shortening the vaginal canal. Proposed techniques for managing these large exposures include covering the defect with a biologic graft, such as small intestinal submucosa, to allow epithelium to re-grow. Regrettably, prolapse is likely to recur in the unprotected area that results.
Contraction and localized pain
Hardening and contraction typically occur along the fixation arms of the mesh. These complications might result from mesh shrinkage or from mesh being placed too tight, so to speak, at implantation. Rarely does the entire implanted mesh contract.
Severe mesh contraction can result in localized pain and de novo dyspareunia. Symptoms usually resolve after identification of the painful area and removal of the involved mesh segment.8
Diagnostic maneuver. In-office trigger-point injection of bupivacaine with triamcinolone is useful to accurately identify the location of pain that is causing dyspareunia. After injection, the patient is asked to return home and resume sexual intercourse; if dyspareunia diminishes significantly, surgical removal of the involved mesh segment is likely to ameliorate symptoms.
If dyspareunia persists after injection, however, the problem either 1) originates in a different location along the graft or 2) may not be related to the mesh—that is, it may be introital pain or preexisting vaginal pain.
The findings of trigger-point injection and a subsequent trial of sexual intercourse are useful for counseling the patient and developing realistic expectations that surgery will be successful.
Management note: Mesh contraction should be managed by a surgeon who is experienced in extensive deep pelvic dissection, which is necessary to remove the mesh arms.
Chronic pain
Diffuse vaginal pain after mesh implantation is unusual; typically, the patient’s report of pain has been preceded by recognition of another, underlying pelvic pain syndrome. Management of such pain is controversial, and many patients will not be satisfied until the entire graft is removed. Whether such drastic intervention actually resolves the pain is unclear; again, work with the patient to create realistic expectations before surgery—including the risk that prolapse will recur and that reoperation will be necessary.
Management note: An existing pelvic pain syndrome should be considered a relative contraindication to implantation of mesh.
Infection of the graft
Rarely, infection has been reported after implantation of Type-1 mesh—the result of either multi-microbial colonization or isolated infection by Bacteriodes melaninogenicus, Actinomyces spp, or Staphylococcus aureus. Untreated preoperative bacterial vaginitis is likely the underlying cause, and should be considered a contraindication to mesh implantation.
Typically, these patients complain of vaginal discharge and bleeding early postoperatively. Vaginal exposure of the mesh results from local inflammation and necrosis of tissue.
Management note: In these cases, it is necessary to 1) prescribe antimicrobial therapy that covers gram-negative and anaerobic bacteria and 2) undertake surgical removal of the exposed mesh, as we outlined above.9
Visceral erosion or fistula
Many experts believe that what is recorded as “erosion” of synthetic mesh into bladder or rectum is, in fact, a result of unrecognized visceral perforation at original implantation. This is a rare complication of mesh implantation.
Patients who experience mesh erosion into the bladder may have lower urinary-tract symptoms (LUTS) of urgency, frequency, dysuria, and hematuria. Any patient who reports de novo LUTS in the early postoperative period after a vaginal mesh procedure should receive office cystourethroscopy to ensure that no foreign body is present in the bladder or urethra.
Management note: Operative cystourethroscopy, with removal of exposed mesh, is the management of choice when mesh is found in the bladder or urethra.
Patients who have constant urinary or fecal incontinence immediately after surgery should be evaluated for vesicovaginal or rectovaginal fistula.
The presence of any of these complications necessitates removal of the involved mesh in its entirety, with concomitant repair of fistula. Typically, the procedures are performed by a specialist.
Our experience with correcting simple mesh exposures
During the past year at our tertiary referral center, 26 patients have undergone mesh revision because of exposure, using the technique we described above (FIGURE 2-6). The problem resolved in all; none had persistent dyspareunia. Many of these patients had already undergone attempts at correction of the exposure elsewhere—mostly, in the office, using techniques appropriate for that setting. Prolapse has not recurred in the 10 patients who required reapproximation of mesh edges because of a defect >2.5 cm.
CASE RESOLVED: Treatment, improvement
Under your care, the patient undergoes simplified outpatient excision of the exposed area of mesh. Mesh edges are reapproximated to support the resulting 3-cm defect.
At a 12-week postop visit, you note complete resolution of the exposure and normal vaginal caliber. The patient continues to apply estrogen cream and reports sustained improvement in sexual function.
- Preoperatively, prepare the vaginal epithelium with local estrogen cream (recommended dosage: 1 g, two nights every week for a trial of at least 6 weeks)
- Use hydrodissection to facilitate placement of the graft deep to the vaginal epithelial fibromuscular fascial layer
- Do not place a synthetic mesh as an overlay to a midline fascial plication
- Be fastidious about hemostasis
- Close the vaginal epithelium without tension
- Leave vaginal packing in place for 24 hours
- Consider using biologic grafts when appropriate (as an overlay to midline plication when used on the anterior vaginal wall).
For simple presentations, success is within reach
Simple mesh exposure can (as in the case we described) be managed by most gynecologists, utilizing the simple stepwise approach that we outlined above (for additional tips based on our experience, see “Pearls for avoiding mesh exposures”). In the case of more significant symptoms, de novo dyspareunia, visceral erosion, or fistula, however, referral to a specialist is warranted.
Transvaginal mesh surgery reduces pelvic organ prolapse
But dyspareunia may develop in premenopausal women
Transvaginal mesh (TVM) surgery is effective in treating pelvic organ prolapse (POP) in both pre- and postmenopausal women but dyspareunia may worsen in premenopausal women, according to a study published online May 23 in the Journal of Sexual Medicine.
Cheng-Yu Long, MD, PhD, from Kaohsiung Medical University in Taiwan, and colleagues compared the changes in sexual function of premenopausal and postmenopausal women after TVM surgery. A total of 68 sexually active women, categorized as premenopausal (36) and postmenopausal (32), with symptomatic POP stages II to IV were referred for TVM surgery. Preoperative and postoperative assessments included pelvic examination using the POP quantification (POP-Q) system, and completing the Female Sexual Function Index (FSFI), Urogenital Distress Inventory (UDI-6), and Incontinence Impact Questionnaire (IIQ-7).
The investigators found significant improvement in the POP-Q analysis at points Aa, Ba, C, Ap, and Bp in both groups but not in total vaginal length. The UDI-6 and IIQ-7 scores decreased significantly after TVM surgery. The dyspareunia domain score decreased significantly after surgery only in the premenopausal group. Reports of diminished scores of the dyspareunia domain and total scores were more common among women in the premenopausal group, but there were no significant differences in FSFI domains or total scores between the groups.
Copyright © 2011 HealthDay. All rights reserved.
We want to hear from you! Tell us what you think.
1. Haylen BT, Freeman RM, Swift SE, et al. An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint terminology and classification of the complications related directly to the insertion of prosthesis (meshes, implants, tapes) and grafts in female pelvic floor surgery. Int Urogynecol J Pelvic Floor Dysfunct. 2011;22(1):3-15.
2. Davila GW, Drutz H, Deprest J. Clinical implications of the biology of grafts: conclusions of the 2005 IUGA Grafts Roundtable. Int Urogynecol J Pelvic Floor Dysfunct. 2006;17(suppl 1):S51-55.
3. Iglesia CB, Sokol AI, Sokol ER, et al. Vaginal mesh for prolapse: a randomized controlled trial. Obstet Gynecol. 2010;116(2 pt 1):293-303.
4. Hiltunen R, Nieminen K, Takala T, et al. Low-weight polypropylene mesh for anterior vaginal wall prolapse: a randomized controlled trial. Obstet Gynecol. 2007;110(2 pt 2):455-462.
5. Fatton B, Amblard J, Debodiance P, Cosson M, Jacquetin B. Transvaginal repair of genital prolapse: preliminary results of a new tension-free vaginal mesh (Prolift technique)—a case series multicentric study. Int Urogynecol J Pelvic Floor Dysfunct. 2007;18(7):743-752.
6. Diwadkar GB, Barber MD, Feiner B, Maher C, Jelovsek JE. Complication and reoperation rates after apical vaginal prolapse surgical repair. Obstet Gynecol. 2009;113(2):367-373.
7. Nguyen JN, Burchette RJ. Outcome after anterior vaginal prolapse repair: a randomized controlled trial. Obstet Gynecol. 2008;111(4):891-898.
8. Feiner B, Maher C. Vaginal mesh contraction: definition clinical presentation, and management. Obstet Gynecol. 2010;115(2 pt 2):325-330.
9. Athanasiou S, Matthaiou DK, Falagas ME. Vaginal mesh infection due to Bacteroides melaninogenicus: a case report of another emerging foreign body related infection. Scand J Infect Dis. 2006;38(11-12):1108-1110.
| Watch “simplified managment of mesh exposure along the anterior vaginal wall”, by Dr. Davila and colleagues |
Aimee L. Smith, MD
Dr. Smith is a clinical fellow in the Section of Urogynecology and Reconstructive Pelvic Surgery, Department of Gynecology, Cleveland Clinic Florida, Weston, Fla.
Willy G. Davila, MD
Dr. Davila is Chair, Department of Gynecology, Section of Urogynecology and Reconstructive Pelvic Surgery, Cleveland Clinic Florida, Weston, Fla.
Dr. Smith reports no financial relationships relevant to this article. Dr. Davila receives grant support from American Medical Systems; is a consultant to American Medical Systems, Astellas, CL Medical, and NovaSys; and is a speaker for American Medical Systems, Astellas, CL Medical, NovaSys, and Watson.
| Watch “simplified managment of mesh exposure along the anterior vaginal wall”, by Dr. Davila and colleagues |
Aimee L. Smith, MD
Dr. Smith is a clinical fellow in the Section of Urogynecology and Reconstructive Pelvic Surgery, Department of Gynecology, Cleveland Clinic Florida, Weston, Fla.
Willy G. Davila, MD
Dr. Davila is Chair, Department of Gynecology, Section of Urogynecology and Reconstructive Pelvic Surgery, Cleveland Clinic Florida, Weston, Fla.
Dr. Smith reports no financial relationships relevant to this article. Dr. Davila receives grant support from American Medical Systems; is a consultant to American Medical Systems, Astellas, CL Medical, and NovaSys; and is a speaker for American Medical Systems, Astellas, CL Medical, NovaSys, and Watson.
| Watch “simplified managment of mesh exposure along the anterior vaginal wall”, by Dr. Davila and colleagues |
Aimee L. Smith, MD
Dr. Smith is a clinical fellow in the Section of Urogynecology and Reconstructive Pelvic Surgery, Department of Gynecology, Cleveland Clinic Florida, Weston, Fla.
Willy G. Davila, MD
Dr. Davila is Chair, Department of Gynecology, Section of Urogynecology and Reconstructive Pelvic Surgery, Cleveland Clinic Florida, Weston, Fla.
Dr. Smith reports no financial relationships relevant to this article. Dr. Davila receives grant support from American Medical Systems; is a consultant to American Medical Systems, Astellas, CL Medical, and NovaSys; and is a speaker for American Medical Systems, Astellas, CL Medical, NovaSys, and Watson.
CASE: Pain during intercourse, well after mesh implantation
Your patient, 61 years old, para 3, has come to your office by referral with a complaint of dyspareunia. The history includes placement of a synthetic vaginal mesh kit 14 months earlier for prolapse.
The medical record shows that the referring physician performed a “mesh excision” 1 year after the original procedure.
The woman reports that she is “very frustrated” that she is still dealing with this problem so long after the original procedure.
On examination, you note a 2.5-cm diameter area of exposed mesh in the anterior vagina, with healthy surrounding tissue and without inflammation or purulence (FIGURE 1). You are unable to reproduce her complaint of pain on vaginal examination.
What options can you offer to this woman? And will those options meet her therapeutic expectations?
FIGURE 1 Examination of your referred patient: Mesh is noticeably exposedThe recent increase in the use of mesh grafts to reconstruct pelvic anatomy has been directed mainly at improving surgical outcomes. Yet, at the same time, gynecologic surgeons find themselves facing a rise in associated complications of such surgery that they did not see previously.
Among the most troublesome and concerning of those complications are 1) exposure of mesh through the vaginal epithelium and 2) contraction or hardening of mesh (or both) that can result in dyspareunia and chronic pelvic pain. Other, rare complications include infection and fistula.
Our goal in this article is to address the management of graft-healing abnormalities in which a segment of the mesh is palpable or visible, or both, within the vaginal canal. Our focus is on simple abnormalities that can be managed by most generalist gynecologists; to be clear, more complex abnormalities, and those that provoke more serious or lasting symptoms, belong under the care of a specialist.
A recent shift in terminology is significant
Early on, this complication was called “erosion” as understanding of the mechanism of its development grew, however, terminology applied to the problem has changed.
In fact, mesh itself very rarely erodes into the vagina or an underlying viscus. Instead, the complication occurs most commonly as a result of disruption of a suture line—most likely the result of a hematoma or localized inflammation that develops postoperatively.
“Exposure” (our preference here) and “extrusion” are now the recommended terms, based on a consensus terminology document published this year jointly by the International Urogynecological Association and the International Continence Society.1
Exposure of implanted mesh is considered a “simple” healing abnormality because it typically
- occurs along the suture line and early in the course of healing
- is not associated with infection of the graft.2
The typical physical appearance is one of visible mesh along an open suture line without granulation tissue or purulence—again, see FIGURE 1. The mesh is firmly adherent to the vaginal epithelial edges and underlying fascia.
The reported incidence of mesh exposures—in regard to currently used meshes, which are all Type-1, monofilament, macroporous polypropylene grafts—is approximately 10% but as high as 15% to 20% in some reported series.3,4 The higher rates of exposure are usually seen in series in which some patients have had a synthetic graft implanted as an overlay to fascial midline plication. When the graft is implanted in the subfascial layer of the vaginal wall (i.e., without midline plication), however, the reported rate of exposure falls—to 5% to 10%.5-7
Recommendations for management
Initially, recommendations for “erosion” management were based on concerns about underlying mesh infection or rejection, and included a need to remove the entire graft. That recommendation still applies to multifilament, microporous grafts that present with inflammatory infiltrates, granulation tissue, and purulence. Although these kinds of grafts (known as “Type-2/3 grafts”—e.g., GoreTex, IVS) have not been marketed for pelvic reconstruction over the past 3 to 5 years, their behavior post-implantation is less predictable—and patients who have delayed healing abnormalities are, therefore, still being seen. It’s fortunate that development of an overlying biofilm prevents tissue incorporation into these types of graft, allowing them to be removed easily.
Exposures related to Type-1 mesh—currently used in pelvic reconstruction—that occur without surrounding infection do not require extensive removal. Rather, they can be managed conservatively or, when necessary, with outpatient surgery. In patients who are not sexually active, exposures are usually asymptomatic; they might only be observed by the physician on vaginal examination and are amenable to simple monitoring. In sexually active patients, exposure of Type-1 mesh usually results in dyspareunia or a complaint that the partner “can feel the mesh.” Depending on the size and the nature of symptoms and the extent of the defect, these commonly seen exposures can be managed by following a simple algorithm.
Palpable or visible mesh fibrils can be trimmed in the office; they might even respond to local estrogen alone. Consider these options if the patient displays vaginal atrophy.
Typically, vaginal estrogen is prescribed as 1 g nightly for 2 weeks and then 1 g two or three nights a week. Re-examine the patient in 3 months; if symptoms of mesh exposure persist, it’s unlikely that continued conservative therapy will be successful, and outpatient surgery is recommended.
When exposure is asymptomatic, you can simply monitor the condition for 3 to 6 months; if complaints or findings arise, consider intervention.
Small (<0.5 cm in diameter) exposures can also be managed in the office, including excision of exposed mesh and local estrogen. If the exposure is easily reachable, we recommend grasping the exposed area with pick-ups or a hemostat and with gentle traction, using Metzenbaum scissors to trim exposed mesh as close to the vaginal epithelium as possible. Local topical or injected anesthesia may be needed. Bleeding should be minimal because no dissection is necessary. Silver nitrate can be applied for any minor bleeding. Larger (0.5–4.0 cm) exposures are unlikely to heal on their own. They require outpatient excision in the operating room.
Preoperative tissue preparation with local estrogen is key to successful repair of these exposures. Vaginal estrogen increases blood flow to the epithelium; as tissue becomes well-estrogenized, risk of recurrence diminishes.
The technique we employ includes:
- circumferential infiltration of vaginal epithelium surrounding the exposed mesh with 1% lidocaine with epinephrine
- sharp circumscription of the area of exposure, using a scalpel, with a 0.5-cm margin of vaginal epithelium (FIGURE 2)
- wide dissection, with undermining and mobilization of surrounding healthy vaginal epithelium around the exposure (FIGURE 3)
- excision of the exposed mesh and attached vaginal mucosa, with careful dissection of the mesh off underlying tissues with Metzenbaum scissors—being careful to avoid injury to underlying bladder or rectum (FIGURE 4)
- reapproximation of mesh edges, using 2-0 polypropylene suture to close the resulting defect so that prolapse does not recur (FIGURE 5)
- closing of the previously mobilized vaginal epithelium with 2-0 Vicryl suture, without tension, to cover the reapproximated mesh edges—after irrigation and assurance of adequate hemostasis (FIGURE 6).
FIGURE 2 Incision of vaginal epithelium
Allow for a 0.5-cm margin.
FIGURE 3 Undermining and mobilization of epithelium
Perform wide dissection.
FIGURE 4 Dissection of mesh from underlying tissue
Keep clear of underlying bladder and rectum!
FIGURE 5 Reapproximation of edges to re-establish support
Our choice of suture is 2-0 polypropylene.
FIGURE 6 Irrigation of vaginal epithelium, followed by closure
Before you close, ensure that hemostasis is adequate.The choice of closure—vertical or horizontal—depends on the nature of the original defect.
You can watch a video of this technique that we’ve provided.
Several cautions should be taken with this technique, including:
- avoiding narrowing the vaginal canal
- minimizing trauma to healthy vaginal epithelium that will be used for closure
- maintaining hemostasis to avoid formation of hematomas.
Largest (>4 cm) exposures are likely the result of devascularized sloughing of vaginal epithelium. They are, fortunately, uncommon.
It’s unlikely that, after excision of exposed mesh, the vaginal epithelial edges can be approximated without significantly narrowing or shortening the vaginal canal. Proposed techniques for managing these large exposures include covering the defect with a biologic graft, such as small intestinal submucosa, to allow epithelium to re-grow. Regrettably, prolapse is likely to recur in the unprotected area that results.
Contraction and localized pain
Hardening and contraction typically occur along the fixation arms of the mesh. These complications might result from mesh shrinkage or from mesh being placed too tight, so to speak, at implantation. Rarely does the entire implanted mesh contract.
Severe mesh contraction can result in localized pain and de novo dyspareunia. Symptoms usually resolve after identification of the painful area and removal of the involved mesh segment.8
Diagnostic maneuver. In-office trigger-point injection of bupivacaine with triamcinolone is useful to accurately identify the location of pain that is causing dyspareunia. After injection, the patient is asked to return home and resume sexual intercourse; if dyspareunia diminishes significantly, surgical removal of the involved mesh segment is likely to ameliorate symptoms.
If dyspareunia persists after injection, however, the problem either 1) originates in a different location along the graft or 2) may not be related to the mesh—that is, it may be introital pain or preexisting vaginal pain.
The findings of trigger-point injection and a subsequent trial of sexual intercourse are useful for counseling the patient and developing realistic expectations that surgery will be successful.
Management note: Mesh contraction should be managed by a surgeon who is experienced in extensive deep pelvic dissection, which is necessary to remove the mesh arms.
Chronic pain
Diffuse vaginal pain after mesh implantation is unusual; typically, the patient’s report of pain has been preceded by recognition of another, underlying pelvic pain syndrome. Management of such pain is controversial, and many patients will not be satisfied until the entire graft is removed. Whether such drastic intervention actually resolves the pain is unclear; again, work with the patient to create realistic expectations before surgery—including the risk that prolapse will recur and that reoperation will be necessary.
Management note: An existing pelvic pain syndrome should be considered a relative contraindication to implantation of mesh.
Infection of the graft
Rarely, infection has been reported after implantation of Type-1 mesh—the result of either multi-microbial colonization or isolated infection by Bacteriodes melaninogenicus, Actinomyces spp, or Staphylococcus aureus. Untreated preoperative bacterial vaginitis is likely the underlying cause, and should be considered a contraindication to mesh implantation.
Typically, these patients complain of vaginal discharge and bleeding early postoperatively. Vaginal exposure of the mesh results from local inflammation and necrosis of tissue.
Management note: In these cases, it is necessary to 1) prescribe antimicrobial therapy that covers gram-negative and anaerobic bacteria and 2) undertake surgical removal of the exposed mesh, as we outlined above.9
Visceral erosion or fistula
Many experts believe that what is recorded as “erosion” of synthetic mesh into bladder or rectum is, in fact, a result of unrecognized visceral perforation at original implantation. This is a rare complication of mesh implantation.
Patients who experience mesh erosion into the bladder may have lower urinary-tract symptoms (LUTS) of urgency, frequency, dysuria, and hematuria. Any patient who reports de novo LUTS in the early postoperative period after a vaginal mesh procedure should receive office cystourethroscopy to ensure that no foreign body is present in the bladder or urethra.
Management note: Operative cystourethroscopy, with removal of exposed mesh, is the management of choice when mesh is found in the bladder or urethra.
Patients who have constant urinary or fecal incontinence immediately after surgery should be evaluated for vesicovaginal or rectovaginal fistula.
The presence of any of these complications necessitates removal of the involved mesh in its entirety, with concomitant repair of fistula. Typically, the procedures are performed by a specialist.
Our experience with correcting simple mesh exposures
During the past year at our tertiary referral center, 26 patients have undergone mesh revision because of exposure, using the technique we described above (FIGURE 2-6). The problem resolved in all; none had persistent dyspareunia. Many of these patients had already undergone attempts at correction of the exposure elsewhere—mostly, in the office, using techniques appropriate for that setting. Prolapse has not recurred in the 10 patients who required reapproximation of mesh edges because of a defect >2.5 cm.
CASE RESOLVED: Treatment, improvement
Under your care, the patient undergoes simplified outpatient excision of the exposed area of mesh. Mesh edges are reapproximated to support the resulting 3-cm defect.
At a 12-week postop visit, you note complete resolution of the exposure and normal vaginal caliber. The patient continues to apply estrogen cream and reports sustained improvement in sexual function.
- Preoperatively, prepare the vaginal epithelium with local estrogen cream (recommended dosage: 1 g, two nights every week for a trial of at least 6 weeks)
- Use hydrodissection to facilitate placement of the graft deep to the vaginal epithelial fibromuscular fascial layer
- Do not place a synthetic mesh as an overlay to a midline fascial plication
- Be fastidious about hemostasis
- Close the vaginal epithelium without tension
- Leave vaginal packing in place for 24 hours
- Consider using biologic grafts when appropriate (as an overlay to midline plication when used on the anterior vaginal wall).
For simple presentations, success is within reach
Simple mesh exposure can (as in the case we described) be managed by most gynecologists, utilizing the simple stepwise approach that we outlined above (for additional tips based on our experience, see “Pearls for avoiding mesh exposures”). In the case of more significant symptoms, de novo dyspareunia, visceral erosion, or fistula, however, referral to a specialist is warranted.
Transvaginal mesh surgery reduces pelvic organ prolapse
But dyspareunia may develop in premenopausal women
Transvaginal mesh (TVM) surgery is effective in treating pelvic organ prolapse (POP) in both pre- and postmenopausal women but dyspareunia may worsen in premenopausal women, according to a study published online May 23 in the Journal of Sexual Medicine.
Cheng-Yu Long, MD, PhD, from Kaohsiung Medical University in Taiwan, and colleagues compared the changes in sexual function of premenopausal and postmenopausal women after TVM surgery. A total of 68 sexually active women, categorized as premenopausal (36) and postmenopausal (32), with symptomatic POP stages II to IV were referred for TVM surgery. Preoperative and postoperative assessments included pelvic examination using the POP quantification (POP-Q) system, and completing the Female Sexual Function Index (FSFI), Urogenital Distress Inventory (UDI-6), and Incontinence Impact Questionnaire (IIQ-7).
The investigators found significant improvement in the POP-Q analysis at points Aa, Ba, C, Ap, and Bp in both groups but not in total vaginal length. The UDI-6 and IIQ-7 scores decreased significantly after TVM surgery. The dyspareunia domain score decreased significantly after surgery only in the premenopausal group. Reports of diminished scores of the dyspareunia domain and total scores were more common among women in the premenopausal group, but there were no significant differences in FSFI domains or total scores between the groups.
Copyright © 2011 HealthDay. All rights reserved.
We want to hear from you! Tell us what you think.
CASE: Pain during intercourse, well after mesh implantation
Your patient, 61 years old, para 3, has come to your office by referral with a complaint of dyspareunia. The history includes placement of a synthetic vaginal mesh kit 14 months earlier for prolapse.
The medical record shows that the referring physician performed a “mesh excision” 1 year after the original procedure.
The woman reports that she is “very frustrated” that she is still dealing with this problem so long after the original procedure.
On examination, you note a 2.5-cm diameter area of exposed mesh in the anterior vagina, with healthy surrounding tissue and without inflammation or purulence (FIGURE 1). You are unable to reproduce her complaint of pain on vaginal examination.
What options can you offer to this woman? And will those options meet her therapeutic expectations?
FIGURE 1 Examination of your referred patient: Mesh is noticeably exposedThe recent increase in the use of mesh grafts to reconstruct pelvic anatomy has been directed mainly at improving surgical outcomes. Yet, at the same time, gynecologic surgeons find themselves facing a rise in associated complications of such surgery that they did not see previously.
Among the most troublesome and concerning of those complications are 1) exposure of mesh through the vaginal epithelium and 2) contraction or hardening of mesh (or both) that can result in dyspareunia and chronic pelvic pain. Other, rare complications include infection and fistula.
Our goal in this article is to address the management of graft-healing abnormalities in which a segment of the mesh is palpable or visible, or both, within the vaginal canal. Our focus is on simple abnormalities that can be managed by most generalist gynecologists; to be clear, more complex abnormalities, and those that provoke more serious or lasting symptoms, belong under the care of a specialist.
A recent shift in terminology is significant
Early on, this complication was called “erosion” as understanding of the mechanism of its development grew, however, terminology applied to the problem has changed.
In fact, mesh itself very rarely erodes into the vagina or an underlying viscus. Instead, the complication occurs most commonly as a result of disruption of a suture line—most likely the result of a hematoma or localized inflammation that develops postoperatively.
“Exposure” (our preference here) and “extrusion” are now the recommended terms, based on a consensus terminology document published this year jointly by the International Urogynecological Association and the International Continence Society.1
Exposure of implanted mesh is considered a “simple” healing abnormality because it typically
- occurs along the suture line and early in the course of healing
- is not associated with infection of the graft.2
The typical physical appearance is one of visible mesh along an open suture line without granulation tissue or purulence—again, see FIGURE 1. The mesh is firmly adherent to the vaginal epithelial edges and underlying fascia.
The reported incidence of mesh exposures—in regard to currently used meshes, which are all Type-1, monofilament, macroporous polypropylene grafts—is approximately 10% but as high as 15% to 20% in some reported series.3,4 The higher rates of exposure are usually seen in series in which some patients have had a synthetic graft implanted as an overlay to fascial midline plication. When the graft is implanted in the subfascial layer of the vaginal wall (i.e., without midline plication), however, the reported rate of exposure falls—to 5% to 10%.5-7
Recommendations for management
Initially, recommendations for “erosion” management were based on concerns about underlying mesh infection or rejection, and included a need to remove the entire graft. That recommendation still applies to multifilament, microporous grafts that present with inflammatory infiltrates, granulation tissue, and purulence. Although these kinds of grafts (known as “Type-2/3 grafts”—e.g., GoreTex, IVS) have not been marketed for pelvic reconstruction over the past 3 to 5 years, their behavior post-implantation is less predictable—and patients who have delayed healing abnormalities are, therefore, still being seen. It’s fortunate that development of an overlying biofilm prevents tissue incorporation into these types of graft, allowing them to be removed easily.
Exposures related to Type-1 mesh—currently used in pelvic reconstruction—that occur without surrounding infection do not require extensive removal. Rather, they can be managed conservatively or, when necessary, with outpatient surgery. In patients who are not sexually active, exposures are usually asymptomatic; they might only be observed by the physician on vaginal examination and are amenable to simple monitoring. In sexually active patients, exposure of Type-1 mesh usually results in dyspareunia or a complaint that the partner “can feel the mesh.” Depending on the size and the nature of symptoms and the extent of the defect, these commonly seen exposures can be managed by following a simple algorithm.
Palpable or visible mesh fibrils can be trimmed in the office; they might even respond to local estrogen alone. Consider these options if the patient displays vaginal atrophy.
Typically, vaginal estrogen is prescribed as 1 g nightly for 2 weeks and then 1 g two or three nights a week. Re-examine the patient in 3 months; if symptoms of mesh exposure persist, it’s unlikely that continued conservative therapy will be successful, and outpatient surgery is recommended.
When exposure is asymptomatic, you can simply monitor the condition for 3 to 6 months; if complaints or findings arise, consider intervention.
Small (<0.5 cm in diameter) exposures can also be managed in the office, including excision of exposed mesh and local estrogen. If the exposure is easily reachable, we recommend grasping the exposed area with pick-ups or a hemostat and with gentle traction, using Metzenbaum scissors to trim exposed mesh as close to the vaginal epithelium as possible. Local topical or injected anesthesia may be needed. Bleeding should be minimal because no dissection is necessary. Silver nitrate can be applied for any minor bleeding. Larger (0.5–4.0 cm) exposures are unlikely to heal on their own. They require outpatient excision in the operating room.
Preoperative tissue preparation with local estrogen is key to successful repair of these exposures. Vaginal estrogen increases blood flow to the epithelium; as tissue becomes well-estrogenized, risk of recurrence diminishes.
The technique we employ includes:
- circumferential infiltration of vaginal epithelium surrounding the exposed mesh with 1% lidocaine with epinephrine
- sharp circumscription of the area of exposure, using a scalpel, with a 0.5-cm margin of vaginal epithelium (FIGURE 2)
- wide dissection, with undermining and mobilization of surrounding healthy vaginal epithelium around the exposure (FIGURE 3)
- excision of the exposed mesh and attached vaginal mucosa, with careful dissection of the mesh off underlying tissues with Metzenbaum scissors—being careful to avoid injury to underlying bladder or rectum (FIGURE 4)
- reapproximation of mesh edges, using 2-0 polypropylene suture to close the resulting defect so that prolapse does not recur (FIGURE 5)
- closing of the previously mobilized vaginal epithelium with 2-0 Vicryl suture, without tension, to cover the reapproximated mesh edges—after irrigation and assurance of adequate hemostasis (FIGURE 6).
FIGURE 2 Incision of vaginal epithelium
Allow for a 0.5-cm margin.
FIGURE 3 Undermining and mobilization of epithelium
Perform wide dissection.
FIGURE 4 Dissection of mesh from underlying tissue
Keep clear of underlying bladder and rectum!
FIGURE 5 Reapproximation of edges to re-establish support
Our choice of suture is 2-0 polypropylene.
FIGURE 6 Irrigation of vaginal epithelium, followed by closure
Before you close, ensure that hemostasis is adequate.The choice of closure—vertical or horizontal—depends on the nature of the original defect.
You can watch a video of this technique that we’ve provided.
Several cautions should be taken with this technique, including:
- avoiding narrowing the vaginal canal
- minimizing trauma to healthy vaginal epithelium that will be used for closure
- maintaining hemostasis to avoid formation of hematomas.
Largest (>4 cm) exposures are likely the result of devascularized sloughing of vaginal epithelium. They are, fortunately, uncommon.
It’s unlikely that, after excision of exposed mesh, the vaginal epithelial edges can be approximated without significantly narrowing or shortening the vaginal canal. Proposed techniques for managing these large exposures include covering the defect with a biologic graft, such as small intestinal submucosa, to allow epithelium to re-grow. Regrettably, prolapse is likely to recur in the unprotected area that results.
Contraction and localized pain
Hardening and contraction typically occur along the fixation arms of the mesh. These complications might result from mesh shrinkage or from mesh being placed too tight, so to speak, at implantation. Rarely does the entire implanted mesh contract.
Severe mesh contraction can result in localized pain and de novo dyspareunia. Symptoms usually resolve after identification of the painful area and removal of the involved mesh segment.8
Diagnostic maneuver. In-office trigger-point injection of bupivacaine with triamcinolone is useful to accurately identify the location of pain that is causing dyspareunia. After injection, the patient is asked to return home and resume sexual intercourse; if dyspareunia diminishes significantly, surgical removal of the involved mesh segment is likely to ameliorate symptoms.
If dyspareunia persists after injection, however, the problem either 1) originates in a different location along the graft or 2) may not be related to the mesh—that is, it may be introital pain or preexisting vaginal pain.
The findings of trigger-point injection and a subsequent trial of sexual intercourse are useful for counseling the patient and developing realistic expectations that surgery will be successful.
Management note: Mesh contraction should be managed by a surgeon who is experienced in extensive deep pelvic dissection, which is necessary to remove the mesh arms.
Chronic pain
Diffuse vaginal pain after mesh implantation is unusual; typically, the patient’s report of pain has been preceded by recognition of another, underlying pelvic pain syndrome. Management of such pain is controversial, and many patients will not be satisfied until the entire graft is removed. Whether such drastic intervention actually resolves the pain is unclear; again, work with the patient to create realistic expectations before surgery—including the risk that prolapse will recur and that reoperation will be necessary.
Management note: An existing pelvic pain syndrome should be considered a relative contraindication to implantation of mesh.
Infection of the graft
Rarely, infection has been reported after implantation of Type-1 mesh—the result of either multi-microbial colonization or isolated infection by Bacteriodes melaninogenicus, Actinomyces spp, or Staphylococcus aureus. Untreated preoperative bacterial vaginitis is likely the underlying cause, and should be considered a contraindication to mesh implantation.
Typically, these patients complain of vaginal discharge and bleeding early postoperatively. Vaginal exposure of the mesh results from local inflammation and necrosis of tissue.
Management note: In these cases, it is necessary to 1) prescribe antimicrobial therapy that covers gram-negative and anaerobic bacteria and 2) undertake surgical removal of the exposed mesh, as we outlined above.9
Visceral erosion or fistula
Many experts believe that what is recorded as “erosion” of synthetic mesh into bladder or rectum is, in fact, a result of unrecognized visceral perforation at original implantation. This is a rare complication of mesh implantation.
Patients who experience mesh erosion into the bladder may have lower urinary-tract symptoms (LUTS) of urgency, frequency, dysuria, and hematuria. Any patient who reports de novo LUTS in the early postoperative period after a vaginal mesh procedure should receive office cystourethroscopy to ensure that no foreign body is present in the bladder or urethra.
Management note: Operative cystourethroscopy, with removal of exposed mesh, is the management of choice when mesh is found in the bladder or urethra.
Patients who have constant urinary or fecal incontinence immediately after surgery should be evaluated for vesicovaginal or rectovaginal fistula.
The presence of any of these complications necessitates removal of the involved mesh in its entirety, with concomitant repair of fistula. Typically, the procedures are performed by a specialist.
Our experience with correcting simple mesh exposures
During the past year at our tertiary referral center, 26 patients have undergone mesh revision because of exposure, using the technique we described above (FIGURE 2-6). The problem resolved in all; none had persistent dyspareunia. Many of these patients had already undergone attempts at correction of the exposure elsewhere—mostly, in the office, using techniques appropriate for that setting. Prolapse has not recurred in the 10 patients who required reapproximation of mesh edges because of a defect >2.5 cm.
CASE RESOLVED: Treatment, improvement
Under your care, the patient undergoes simplified outpatient excision of the exposed area of mesh. Mesh edges are reapproximated to support the resulting 3-cm defect.
At a 12-week postop visit, you note complete resolution of the exposure and normal vaginal caliber. The patient continues to apply estrogen cream and reports sustained improvement in sexual function.
- Preoperatively, prepare the vaginal epithelium with local estrogen cream (recommended dosage: 1 g, two nights every week for a trial of at least 6 weeks)
- Use hydrodissection to facilitate placement of the graft deep to the vaginal epithelial fibromuscular fascial layer
- Do not place a synthetic mesh as an overlay to a midline fascial plication
- Be fastidious about hemostasis
- Close the vaginal epithelium without tension
- Leave vaginal packing in place for 24 hours
- Consider using biologic grafts when appropriate (as an overlay to midline plication when used on the anterior vaginal wall).
For simple presentations, success is within reach
Simple mesh exposure can (as in the case we described) be managed by most gynecologists, utilizing the simple stepwise approach that we outlined above (for additional tips based on our experience, see “Pearls for avoiding mesh exposures”). In the case of more significant symptoms, de novo dyspareunia, visceral erosion, or fistula, however, referral to a specialist is warranted.
Transvaginal mesh surgery reduces pelvic organ prolapse
But dyspareunia may develop in premenopausal women
Transvaginal mesh (TVM) surgery is effective in treating pelvic organ prolapse (POP) in both pre- and postmenopausal women but dyspareunia may worsen in premenopausal women, according to a study published online May 23 in the Journal of Sexual Medicine.
Cheng-Yu Long, MD, PhD, from Kaohsiung Medical University in Taiwan, and colleagues compared the changes in sexual function of premenopausal and postmenopausal women after TVM surgery. A total of 68 sexually active women, categorized as premenopausal (36) and postmenopausal (32), with symptomatic POP stages II to IV were referred for TVM surgery. Preoperative and postoperative assessments included pelvic examination using the POP quantification (POP-Q) system, and completing the Female Sexual Function Index (FSFI), Urogenital Distress Inventory (UDI-6), and Incontinence Impact Questionnaire (IIQ-7).
The investigators found significant improvement in the POP-Q analysis at points Aa, Ba, C, Ap, and Bp in both groups but not in total vaginal length. The UDI-6 and IIQ-7 scores decreased significantly after TVM surgery. The dyspareunia domain score decreased significantly after surgery only in the premenopausal group. Reports of diminished scores of the dyspareunia domain and total scores were more common among women in the premenopausal group, but there were no significant differences in FSFI domains or total scores between the groups.
Copyright © 2011 HealthDay. All rights reserved.
We want to hear from you! Tell us what you think.
1. Haylen BT, Freeman RM, Swift SE, et al. An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint terminology and classification of the complications related directly to the insertion of prosthesis (meshes, implants, tapes) and grafts in female pelvic floor surgery. Int Urogynecol J Pelvic Floor Dysfunct. 2011;22(1):3-15.
2. Davila GW, Drutz H, Deprest J. Clinical implications of the biology of grafts: conclusions of the 2005 IUGA Grafts Roundtable. Int Urogynecol J Pelvic Floor Dysfunct. 2006;17(suppl 1):S51-55.
3. Iglesia CB, Sokol AI, Sokol ER, et al. Vaginal mesh for prolapse: a randomized controlled trial. Obstet Gynecol. 2010;116(2 pt 1):293-303.
4. Hiltunen R, Nieminen K, Takala T, et al. Low-weight polypropylene mesh for anterior vaginal wall prolapse: a randomized controlled trial. Obstet Gynecol. 2007;110(2 pt 2):455-462.
5. Fatton B, Amblard J, Debodiance P, Cosson M, Jacquetin B. Transvaginal repair of genital prolapse: preliminary results of a new tension-free vaginal mesh (Prolift technique)—a case series multicentric study. Int Urogynecol J Pelvic Floor Dysfunct. 2007;18(7):743-752.
6. Diwadkar GB, Barber MD, Feiner B, Maher C, Jelovsek JE. Complication and reoperation rates after apical vaginal prolapse surgical repair. Obstet Gynecol. 2009;113(2):367-373.
7. Nguyen JN, Burchette RJ. Outcome after anterior vaginal prolapse repair: a randomized controlled trial. Obstet Gynecol. 2008;111(4):891-898.
8. Feiner B, Maher C. Vaginal mesh contraction: definition clinical presentation, and management. Obstet Gynecol. 2010;115(2 pt 2):325-330.
9. Athanasiou S, Matthaiou DK, Falagas ME. Vaginal mesh infection due to Bacteroides melaninogenicus: a case report of another emerging foreign body related infection. Scand J Infect Dis. 2006;38(11-12):1108-1110.
1. Haylen BT, Freeman RM, Swift SE, et al. An International Urogynecological Association (IUGA)/International Continence Society (ICS) joint terminology and classification of the complications related directly to the insertion of prosthesis (meshes, implants, tapes) and grafts in female pelvic floor surgery. Int Urogynecol J Pelvic Floor Dysfunct. 2011;22(1):3-15.
2. Davila GW, Drutz H, Deprest J. Clinical implications of the biology of grafts: conclusions of the 2005 IUGA Grafts Roundtable. Int Urogynecol J Pelvic Floor Dysfunct. 2006;17(suppl 1):S51-55.
3. Iglesia CB, Sokol AI, Sokol ER, et al. Vaginal mesh for prolapse: a randomized controlled trial. Obstet Gynecol. 2010;116(2 pt 1):293-303.
4. Hiltunen R, Nieminen K, Takala T, et al. Low-weight polypropylene mesh for anterior vaginal wall prolapse: a randomized controlled trial. Obstet Gynecol. 2007;110(2 pt 2):455-462.
5. Fatton B, Amblard J, Debodiance P, Cosson M, Jacquetin B. Transvaginal repair of genital prolapse: preliminary results of a new tension-free vaginal mesh (Prolift technique)—a case series multicentric study. Int Urogynecol J Pelvic Floor Dysfunct. 2007;18(7):743-752.
6. Diwadkar GB, Barber MD, Feiner B, Maher C, Jelovsek JE. Complication and reoperation rates after apical vaginal prolapse surgical repair. Obstet Gynecol. 2009;113(2):367-373.
7. Nguyen JN, Burchette RJ. Outcome after anterior vaginal prolapse repair: a randomized controlled trial. Obstet Gynecol. 2008;111(4):891-898.
8. Feiner B, Maher C. Vaginal mesh contraction: definition clinical presentation, and management. Obstet Gynecol. 2010;115(2 pt 2):325-330.
9. Athanasiou S, Matthaiou DK, Falagas ME. Vaginal mesh infection due to Bacteroides melaninogenicus: a case report of another emerging foreign body related infection. Scand J Infect Dis. 2006;38(11-12):1108-1110.
The ABCDEs of identifying eating disorders
Although eating disorders can be life-threatening,1 many patients remain undiagnosed until late in the disease course. Early identification and treatment may reduce the risk of chronic health consequences and mortality.
Based on the DSM-IV-TR categorical approach, many clinicians think of anorexia nervosa and bulimia nervosa as the primary eating disorders. However, eating disorder not otherwise specified tends to be the most common diagnosis.2 Several authors have suggested that combining categorical and dimensional approaches may be useful in diagnosing these patients.2
It is easy to suspect an eating disorder in patients of very low weight, but patients who are of normal weight or obese also may have an eating disorder. In addition to measuring body mass index, inquire about patients’ lowest and highest adult, nonpregnant weights and what they consider to be their “ideal” weight. The mnemonic ABCDE can help you remember key components of assessing patients who might have an eating disorder.
Associated health problems. Anorexia patients commonly present with emaciation, skin and hair dryness, cold intolerance, bradycardia, and orthostatic hypotension. Look for calluses on dorsum of the hands, parotid enlargement, mouth ulcers, dental caries, and edema, which may be found in bulimia patients.
Body image. Determine whether your patients’ self esteem is correlated with body weight and shape, how often they weigh themselves, and if they are satisfied with the way their body is proportioned. Ask if they fear weight gain or are driven to be thin.
Compensatory behaviors may include self-induced vomiting or excessive use of diet pills, diuretics, or laxatives. Other examples are restricting food, chewing and spitting out food, and over-exercising (especially lengthy cardiovascular workouts).
Diet. Inquire whether your patients have ever been on a diet, reduced the amount of food they consume, or used medications indicated for obesity. Patients may avoid entire categories of foods (lipids, carbohydrates), which may lead to malnutrition and vitamin deficiencies.
Eating behaviors. Patients may eat very small meals or excessive portions of certain foods. They might skip meals or eat only alone or at night. Finishing meals very slowly or very quickly, mixing food on their plate, eating with tiny bites, or drinking a lot of water with and between meals also may be clues to disordered eating. None of these behaviors by itself indicate an eating disorder if not accompanied by other symptoms.
Disclosure
The authors report no financial relationship with any manufacturer whose products are mentioned in this article or with manufacturers of competing products.
Although eating disorders can be life-threatening,1 many patients remain undiagnosed until late in the disease course. Early identification and treatment may reduce the risk of chronic health consequences and mortality.
Based on the DSM-IV-TR categorical approach, many clinicians think of anorexia nervosa and bulimia nervosa as the primary eating disorders. However, eating disorder not otherwise specified tends to be the most common diagnosis.2 Several authors have suggested that combining categorical and dimensional approaches may be useful in diagnosing these patients.2
It is easy to suspect an eating disorder in patients of very low weight, but patients who are of normal weight or obese also may have an eating disorder. In addition to measuring body mass index, inquire about patients’ lowest and highest adult, nonpregnant weights and what they consider to be their “ideal” weight. The mnemonic ABCDE can help you remember key components of assessing patients who might have an eating disorder.
Associated health problems. Anorexia patients commonly present with emaciation, skin and hair dryness, cold intolerance, bradycardia, and orthostatic hypotension. Look for calluses on dorsum of the hands, parotid enlargement, mouth ulcers, dental caries, and edema, which may be found in bulimia patients.
Body image. Determine whether your patients’ self esteem is correlated with body weight and shape, how often they weigh themselves, and if they are satisfied with the way their body is proportioned. Ask if they fear weight gain or are driven to be thin.
Compensatory behaviors may include self-induced vomiting or excessive use of diet pills, diuretics, or laxatives. Other examples are restricting food, chewing and spitting out food, and over-exercising (especially lengthy cardiovascular workouts).
Diet. Inquire whether your patients have ever been on a diet, reduced the amount of food they consume, or used medications indicated for obesity. Patients may avoid entire categories of foods (lipids, carbohydrates), which may lead to malnutrition and vitamin deficiencies.
Eating behaviors. Patients may eat very small meals or excessive portions of certain foods. They might skip meals or eat only alone or at night. Finishing meals very slowly or very quickly, mixing food on their plate, eating with tiny bites, or drinking a lot of water with and between meals also may be clues to disordered eating. None of these behaviors by itself indicate an eating disorder if not accompanied by other symptoms.
Disclosure
The authors report no financial relationship with any manufacturer whose products are mentioned in this article or with manufacturers of competing products.
Although eating disorders can be life-threatening,1 many patients remain undiagnosed until late in the disease course. Early identification and treatment may reduce the risk of chronic health consequences and mortality.
Based on the DSM-IV-TR categorical approach, many clinicians think of anorexia nervosa and bulimia nervosa as the primary eating disorders. However, eating disorder not otherwise specified tends to be the most common diagnosis.2 Several authors have suggested that combining categorical and dimensional approaches may be useful in diagnosing these patients.2
It is easy to suspect an eating disorder in patients of very low weight, but patients who are of normal weight or obese also may have an eating disorder. In addition to measuring body mass index, inquire about patients’ lowest and highest adult, nonpregnant weights and what they consider to be their “ideal” weight. The mnemonic ABCDE can help you remember key components of assessing patients who might have an eating disorder.
Associated health problems. Anorexia patients commonly present with emaciation, skin and hair dryness, cold intolerance, bradycardia, and orthostatic hypotension. Look for calluses on dorsum of the hands, parotid enlargement, mouth ulcers, dental caries, and edema, which may be found in bulimia patients.
Body image. Determine whether your patients’ self esteem is correlated with body weight and shape, how often they weigh themselves, and if they are satisfied with the way their body is proportioned. Ask if they fear weight gain or are driven to be thin.
Compensatory behaviors may include self-induced vomiting or excessive use of diet pills, diuretics, or laxatives. Other examples are restricting food, chewing and spitting out food, and over-exercising (especially lengthy cardiovascular workouts).
Diet. Inquire whether your patients have ever been on a diet, reduced the amount of food they consume, or used medications indicated for obesity. Patients may avoid entire categories of foods (lipids, carbohydrates), which may lead to malnutrition and vitamin deficiencies.
Eating behaviors. Patients may eat very small meals or excessive portions of certain foods. They might skip meals or eat only alone or at night. Finishing meals very slowly or very quickly, mixing food on their plate, eating with tiny bites, or drinking a lot of water with and between meals also may be clues to disordered eating. None of these behaviors by itself indicate an eating disorder if not accompanied by other symptoms.
Disclosure
The authors report no financial relationship with any manufacturer whose products are mentioned in this article or with manufacturers of competing products.
AIM-HIGH and HDL
When the National Heart, Lung, and Blood Institute announced that the Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health (AIM-HIGH) trial was being terminated because of the futility of showing benefit, the study's failure made front page news.
The medical community, patient population, and interested public ask “why?” at this unexpected result of the study that compared extended-release niacin (Niaspan) plus simvastatin with placebo plus simvastatin.
Cardiologists and lipidologists may be better able to answer this question when the full results are published. More information will certainly be required concerning the reported increase in stroke in the niacin arm (28 vs 12). At this stage, a hypothesis regarding the lack of benefit must be considered tentative at best.
However, the first thing that comes to mind is whether the study was sufficiently powered. A clinical trial currently underway at Oxford (England) University, HPS2-THRIVE, is comparing extended-release niacin and laropiprant (a prostaglandin receptor antagonist to decrease flushing approved in Europe but not the United States) against a background of simvastatin. HPS2-THRIVE aims to enroll 25,000 subjects, so this difference in size compared with AIM-HIGH must indicate large differences in expected event rate.
One should also bear in mind that the decrease in events reported with niacin in the Coronary Drug Project (J. Am. Coll. Cardiol. 1986;8:1245-55) was against a background of placebo. Statins have set a very high bar for efficacy, and it can be difficult to demonstrate incremental benefit with an add-on to statin therapy, as was the case in the ENHANCE trial with ezetimibe. In fact, there was substantial use of ezetimibe in the placebo group of AIM-HIGH, which therefore was not a true placebo group, in order to reach a target LDL cholesterol level of less than 80 mg/dL. This is somewhat ironic since niacin plus statin was reported to be more effective than ezetimibe plus statin in reducing carotid intima-media thickness in the ARBITER 6-HALTS study (N. Engl. J. Med. 2009;361:2113-22).
The National Lipid Association has recommended that physicians should wait until the full results of AIM-HIGH are reported before integrating the findings into their clinical practice, and that patients should not stop taking niacin without the advice of their physician.
I agree with this recommendation. It remains to be seen whether we will ever know the full explanation for the futility results of AIM-HIGH. We certainly will know much more when the results are analyzed and when HPS2-THRIVE is reported.
In the meantime, there are alternative ways of raising HDL currently being tested, including with the cholesteryl ester transfer protein (CETP) inhibitors anacetrapib and dalcetrapib, so the final results are far from complete concerning the benefit of raising HDL cholesterol, particularly in patients with low HDL cholesterol. AIM-HIGH does not disprove the theory that raising HDL will be beneficial, and there are abundant data showing that low HDL increases cardiovascular risk.
Hence, AIM-HIGH does not provide support for the “HDL hypothesis,” but neither does it drive the final nail in the coffin.
When the National Heart, Lung, and Blood Institute announced that the Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health (AIM-HIGH) trial was being terminated because of the futility of showing benefit, the study's failure made front page news.
The medical community, patient population, and interested public ask “why?” at this unexpected result of the study that compared extended-release niacin (Niaspan) plus simvastatin with placebo plus simvastatin.
Cardiologists and lipidologists may be better able to answer this question when the full results are published. More information will certainly be required concerning the reported increase in stroke in the niacin arm (28 vs 12). At this stage, a hypothesis regarding the lack of benefit must be considered tentative at best.
However, the first thing that comes to mind is whether the study was sufficiently powered. A clinical trial currently underway at Oxford (England) University, HPS2-THRIVE, is comparing extended-release niacin and laropiprant (a prostaglandin receptor antagonist to decrease flushing approved in Europe but not the United States) against a background of simvastatin. HPS2-THRIVE aims to enroll 25,000 subjects, so this difference in size compared with AIM-HIGH must indicate large differences in expected event rate.
One should also bear in mind that the decrease in events reported with niacin in the Coronary Drug Project (J. Am. Coll. Cardiol. 1986;8:1245-55) was against a background of placebo. Statins have set a very high bar for efficacy, and it can be difficult to demonstrate incremental benefit with an add-on to statin therapy, as was the case in the ENHANCE trial with ezetimibe. In fact, there was substantial use of ezetimibe in the placebo group of AIM-HIGH, which therefore was not a true placebo group, in order to reach a target LDL cholesterol level of less than 80 mg/dL. This is somewhat ironic since niacin plus statin was reported to be more effective than ezetimibe plus statin in reducing carotid intima-media thickness in the ARBITER 6-HALTS study (N. Engl. J. Med. 2009;361:2113-22).
The National Lipid Association has recommended that physicians should wait until the full results of AIM-HIGH are reported before integrating the findings into their clinical practice, and that patients should not stop taking niacin without the advice of their physician.
I agree with this recommendation. It remains to be seen whether we will ever know the full explanation for the futility results of AIM-HIGH. We certainly will know much more when the results are analyzed and when HPS2-THRIVE is reported.
In the meantime, there are alternative ways of raising HDL currently being tested, including with the cholesteryl ester transfer protein (CETP) inhibitors anacetrapib and dalcetrapib, so the final results are far from complete concerning the benefit of raising HDL cholesterol, particularly in patients with low HDL cholesterol. AIM-HIGH does not disprove the theory that raising HDL will be beneficial, and there are abundant data showing that low HDL increases cardiovascular risk.
Hence, AIM-HIGH does not provide support for the “HDL hypothesis,” but neither does it drive the final nail in the coffin.
When the National Heart, Lung, and Blood Institute announced that the Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides: Impact on Global Health (AIM-HIGH) trial was being terminated because of the futility of showing benefit, the study's failure made front page news.
The medical community, patient population, and interested public ask “why?” at this unexpected result of the study that compared extended-release niacin (Niaspan) plus simvastatin with placebo plus simvastatin.
Cardiologists and lipidologists may be better able to answer this question when the full results are published. More information will certainly be required concerning the reported increase in stroke in the niacin arm (28 vs 12). At this stage, a hypothesis regarding the lack of benefit must be considered tentative at best.
However, the first thing that comes to mind is whether the study was sufficiently powered. A clinical trial currently underway at Oxford (England) University, HPS2-THRIVE, is comparing extended-release niacin and laropiprant (a prostaglandin receptor antagonist to decrease flushing approved in Europe but not the United States) against a background of simvastatin. HPS2-THRIVE aims to enroll 25,000 subjects, so this difference in size compared with AIM-HIGH must indicate large differences in expected event rate.
One should also bear in mind that the decrease in events reported with niacin in the Coronary Drug Project (J. Am. Coll. Cardiol. 1986;8:1245-55) was against a background of placebo. Statins have set a very high bar for efficacy, and it can be difficult to demonstrate incremental benefit with an add-on to statin therapy, as was the case in the ENHANCE trial with ezetimibe. In fact, there was substantial use of ezetimibe in the placebo group of AIM-HIGH, which therefore was not a true placebo group, in order to reach a target LDL cholesterol level of less than 80 mg/dL. This is somewhat ironic since niacin plus statin was reported to be more effective than ezetimibe plus statin in reducing carotid intima-media thickness in the ARBITER 6-HALTS study (N. Engl. J. Med. 2009;361:2113-22).
The National Lipid Association has recommended that physicians should wait until the full results of AIM-HIGH are reported before integrating the findings into their clinical practice, and that patients should not stop taking niacin without the advice of their physician.
I agree with this recommendation. It remains to be seen whether we will ever know the full explanation for the futility results of AIM-HIGH. We certainly will know much more when the results are analyzed and when HPS2-THRIVE is reported.
In the meantime, there are alternative ways of raising HDL currently being tested, including with the cholesteryl ester transfer protein (CETP) inhibitors anacetrapib and dalcetrapib, so the final results are far from complete concerning the benefit of raising HDL cholesterol, particularly in patients with low HDL cholesterol. AIM-HIGH does not disprove the theory that raising HDL will be beneficial, and there are abundant data showing that low HDL increases cardiovascular risk.
Hence, AIM-HIGH does not provide support for the “HDL hypothesis,” but neither does it drive the final nail in the coffin.
Underreporting of neutropenic toxicity associated with current treatment regimens for selected hematologic malignancies
Stephanie A. Gregory, MD,1 Steve Abella, MD,2 and Tim Moore, MD3
1 Section of Hematology, Rush University Medical Center, Chicago, IL; 2 Global Clinical Development, Hematology/Oncology, Amgen Inc., Thousand Oaks, CA; and 3 Zangmeister Center, Columbus, OH
Most chemotherapy regimens considered standard of care for treating hematologic malignancies are myelosuppressive. They include chemotherapy regimens recommended by the National Comprehensive Cancer Network (NCCN),1 such as cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) to treat non-Hodgkin lymphoma (NHL) 2,3; fludarabine plus cyclophosphamide (FC) to treat chronic lymphocytic leukemia (CLL)4,5; and escalated-dose bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone (BEACOPP) or doxorubicin, vinblastine, mechlorethamine, etoposide, vincristine, bleomycin, and prednisone (Stanford V) to treat Hodgkin lymphoma (HL).6–8
Emerging regimens that incorporate targeted therapies or other novel agents (eg, rituximab [Rituxan], lenalidomide [Revlimid], or bendamustine [Treanda]) have also been shown to be myelosuppressive, mainly because they are generally combined with myelosuppressive chemotherapy to achieve optimal efficacy. Examples include CHOP plus rituximab (R-CHOP) to treat NHL9,10; FC plus rituximab (FCR) to treat CLL11,12; or bortezomib plus melphalan-prednisone (MPB) to treat multiple myeloma.13,14 Additionally, some agents show toxicity when used as monotherapies, including bendamustine15–17 and alemtuzumab (Campath) 18 to treat CLL. Therefore, improved clinical outcomes may be achieved with concurrent increased myelosuppression.
Patients receiving myelosuppresive chemotherapy are at risk for developing chemotherapy-induced neutropenia, including severe or prolonged neutropenia and febrile neutropenia (FN). This condition often leads to treatment delays/interruptions, dose reductions, or treatment discontinuations, which can result in suboptimal treatment delivery and compromised patient outcomes.19–22 Colony-stimulating factor (CSF) has thus become an important component of many current treatment regimens for hematologic malignancies. International clinical guidelines, including those from the NCCN,1 the American Society of Clinical Oncology (ASCO),22 the European Society for Medical Oncology (ESMO),23 and the European Organization for Research and Treatment of Cancer (EORTC),24 recommend CSF use when the risk of FN is ≥ 20% and consideration of CSF use when the risk of FN is between 10% and 20%.
Numerous studies have demonstrated CSF effectiveness in decreasing the incidence of severe neutropenia and/or FN.25–34 A meta-analysis of 17 randomized controlled trials, which enrolled 3,493 cancer patients receiving chemotherapy, demonstrated that primary prophylaxis with CSF was associated with a decreased incidence of FN and reduced rates of infection-related mortality and early mortality across different tumor types.35 The occurrence of FN was associated with a 35% increase in the hazard of early mortality, and prophylactic granulocyte (G)-CSF use decreased this number by 45%.36 In a separate analysis of 25 trials (total n = 12,804), CSF support in cancer patients receiving chemotherapy was associated with a significant increase in overall survival (OS).37 Furthermore, a meta-analysis of results from 12 randomized controlled trials, which enrolled 1,823 patients with malignant lymphoma, showed that CSF prophylaxis, compared with no prophylaxis, significantly reduced the relative risk of severe neutropenia, FN, and infection.38
Evidence-based data that could guide the use of CSF in the setting of current treatment regimens for hematologic malignancies are not always readily available. Publications that report clinical trial results focus on overall efficacy and safety parameters of treatment regimens and often do not report the incidence or severity of neutropenia and/or FN.39 Similarly, these publications often do not include information on supportive care measures, including prophylaxis with antibiotics and/or CSF (primary or secondary).40,41 Also, when CSF support is reported, often the agent and dosing schedule are not provided. Many trials permit the use of CSF at the investigator’s discretion; however, the proportion of patients treated or supported with CSF and related outcomes is often not reported. These gaps in reporting neutropenic toxicity and related outcomes may result in an underestimation of the degree of significant toxicity associated with current treatment regimens for hematologic malignancies.
We conducted a comprehensive review of English-language reports published after January 2005. From the retrieved list of publications, we identified studies reporting data from trials (including phase II and III) that evaluated regimens considered NCCN Guideline recommendations for treating selected hematologic malignancies. 1 We excluded trials that enrolled patients with acute leukemia or chronic myelogenous leukemia; trials with the primary objective of assessing radiotherapy, radioimmunotherapy, stem cell transplantation, or patient-reported outcomes; and trials that described the study design but not the results. If multiple publications reported results of the same trial, we selected the publication with the most complete data on hematologic toxicity. Publications that met the inclusion criteria were retrieved and reviewed for neutropenic toxicity outcomes and the reported use of CSF or antibiotics.
Neutropenic toxicity associated with current treatment regimens for NHL
Diffuse large B-cell lymphoma
Diffuse large B-cell lymphoma (DLBCL) is an aggressive type of lymphoma generally treated with curative intent in the frontline setting. Beginning in the 1970s, the standard of care for DLBCL was CHOP, administered every 21 days (CHOP- 21).9 However, approximately half of patients > 60 years of age do not benefit from this regimen. In a study by Coiffier et al,42 3-year OS in this patient population was less than 40%. The addition of rituximab to CHOP-21 (R-CHOP-21) or CHOP-21–like regimens was subsequently shown to improve OS significantly across patient populations, with no increased neutropenic toxicity (Table 1).10 The R-CHOP regimen is now considered the standard of care for DLBCL when the goal of treatment is cure.9Another randomized study by Pfreundschuh et al compared dose-dense CHOP (given every 14 days, CHOP-14) with CHOP-21 in NHL patients ≥ 60 years of age.2 The CHOP-14 dosedense regimen required support with primary prophylactic CSF in all cycles (CHOP-14-G), whereas prophylactic CSF use with CHOP-21 was at the discretion of the treating physician, based on patient characteristics. CHOP-14-G significantly improved event-free survival (EFS) and OS. Grade 4 neutropenia was less frequent with CHOP-14-G than with CHOP-21 (24% vs 44%; P < 0.001), demonstrating that CSF support could adequately protect patients from neutropenic toxicity associated with CHOP.2
The RICOVER-60 study43 evaluated 6 or 8 cycles of dose-dense CHOP (CHOP-14-G) with or without rituximab in patients 61– 80 years of age who had aggressive B-cell lymphoma and were receiving primary prophylaxis with CSF (R-CHOP-14-G vs CHOP-14-G). R-CHOP-14-G significantly improved EFS (66.5% vs 47.2%) and OS (78.1% vs 67.7%). Leukopenia was the most common grade 3/4 toxicity, with grade 4 events occurring in 48%–52% across treatment arms. However, the incidence of leukopenia and the incidence of grade 3/4 infection were similar across the regimens (Table 1).
The Groupe d’Etude des Lymphomes de l’Adulte intergroup (GELA) study,44 compared RCHOP- 14 with R-CHOP-21 in DLBCL patients 60–80 years of age. Results from a 24-month interim analysis showed similar efficacy for R-CHOP-14 and R-CHOP-21 (2-year EFS of 48% vs 61%; P = not significant [NS]). Typically, trials of dose-dense regimens are evaluated with CSF support for all patients1,24; however, in the GELA study, patients received CSF at the physician’s discretion. Even though CSF use was higher with R-CHOP-14 than with R-CHOP-21 (90% vs 66%; Table 1), more patients in the R-CHOP-14 than in the R-CHOP-21 arm experienced grade 3/4 hematologic toxicity and FN (percentages were not reported).
Follicular lymphoma
Follicular lymphoma (FL) is usually diagnosed at an advanced stage and is incurable with current therapy.1 As shown in Table 1, current regimens for treating FL, including rituximab- and bendamustine-based regimens, are associated with neutropenic toxicity.
Rituximab-based treatment/consolidation regimens: The NCCN recommends R-CHOP and rituximab, cyclophosphamide, vincristine, and prednisone (R-CVP) for treating FL.1 A randomized phase III study by the German Low-Grade Lymphoma Study Group (GLSG) showed the superiority of first-line R-CHOP compared with CHOP in patients with untreated advanced FL.45 R-CHOP reduced the relative risk of treatment failure by 60% (28 of 223 patients vs 61 of 205 patients; P < 0.001), improved the overall response rate (ORR; 96% vs 90%; P = 0.011), and improved OS (6 deaths vs 17 deaths within the first 3 years; P = 0.016). Severe neutropenia was the most common treatment-related adverse event and occurred more often with R-CHOP than with CHOP (63% vs 53%; P = 0.01; Table 1).45 However, the incidence of severe infections was similar in the two groups (5% vs 7%; P = NS). Details of CSF use in this study were not reported.
A randomized phase III study in treatment-naive patients with advanced FL compared R-CVP with CVP.46 This study demonstrated that R-CVP significantly improved the ORR (81% vs 57%; P = 0.001), significantly prolonged the time to treatment failure (TTF; 27 months vs 7 months; P < 0.0001), and more than doubled the time to disease progression (TTP; 32 months vs 15 months; P < 0.001).46 The incidence of grade 3/4 neutropenia was higher with RCVP than with CVP (24% vs 14%), but the rates of infection and neutropenic sepsis were similar in the two treatment arms (Table 1).46 Details of CSF use were not provided in this report.
Rituximab-based maintenance regimens: Recent studies, including trials in frontline and relapsed settings, have demonstrated the benefits of rituximab maintenance after induction chemotherapy in patients with lymphoma.47–50
Two studies, one in the United States and one in Europe, randomized patients with relapsed/refractory FL to receive induction therapy with R-CHOP or CHOP; then those with a compete response (CR) or a partial response (PR) were randomized to receive rituximab maintenance (375 mg/m2 intravenously once every 3 months for up to 2 years) or no further treatment (observation group).48 Rituximab maintenance improved progression-free survival (PFS; 51.5 months vs 15.0 months; P < 0.001) and the 3-year OS rate (85% vs 77%; P = 0.011). The PFS benefit of rituximab maintenance was confirmed at a median follow-up of 6 years (3.7 years vs 1.3 years; P < 0.001; hazard ratio [HR] = 0.55), but the 5-year OS was not significantly different between the groups (74% vs 64%; P = 0.07).49 During the maintenance period, the frequency of grade 3/4 neutropenia and grade 3/4 infection was higher with rituximab than with no treatment: 12% vs 6% and 9% vs 2% (P = 0.009), respectively (Table 1).48,49 Details of CSF use during induction or maintenance therapy were not provided in the report.
A study by the GLSG group compared rituximab maintenance with no treatment following salvage therapy for patients with refractory or recurrent FL or mantle cell lymphoma.47 The maintenance regimen consisted of two courses of rituximab (4 doses of 375 mg/m2/day for 4 consecutive weeks) administered 3 months and 9 months after patients achieved a CR or a PR to induction chemotherapy with fludarabine, cyclophosphamide, and mitoxantrone (FCM) alone or in combination with rituximab (FCM-R). Rituximab maintenance significantly improved the response duration; the median response duration had not been reached in the rituximab arm vs an estimated median of 16 months in the observation arm (P < 0.001). During the maintenance period, grade 3/4 neutropenia was more common in the rituximab arm than in the observation arm (13% vs 6%; P = NS), but the incidence of grade 3/4 infection was similar in the two treatment arms (4% vs 3%; Table 1).47 Details of CSF use in both the induction and maintenance periods were not provided.
In the first-line setting, a randomized phase III study by the Eastern Cooperative Oncology Group (ECOG) evaluated the benefits of rituximab maintenance in patients with FL or small lymphocytic lymphoma following CVP treatment.50 Four weeks after the last CVP cycle, patients with responding or stable disease were randomized to receive rituximab (375 mg/m2 once per week for 4 weeks every 6 months for 2 years) or observation. Rituximab maintenance improved the 3-year PFS (68% vs 33%; HR = 0.4; P < 0.0001) and the 3-year OS (92% vs 86%; HR = 0.6; P = 0.05). During maintenance therapy, grade 3 neutropenia and grade 3 infection rates appeared to be similar in the two treatment groups (Table 1).50 Secondary CSF prophylaxis was permitted during induction chemotherapy in response to neutropenic events but not specified for the maintenance phase.
The Primary Rituximab and Maintenance (PRIMA) trial conducted by the GELA group evaluated the benefits of rituximab maintenance in previously untreated patients with indolent NHL.51 Patients who responded to one of three immunochemotherapy regimens (R-CHOP, R-CVP, or FCM with rituximab) were randomized to receive rituximab (375 mg/m2 given once every 8 weeks for 2 years) or observation. At a median followup of 2 years, maintenance rituximab significantly improved PFS (75% vs 58%; HR = 0.55; P < 0.0001). More patients in the rituximab arm than in the observation arm experienced grade 2 or higher infections (39% vs 24%), grade 3/4 infections (4% vs 1%), and grade 3/4 neutropenia (4% vs 1%). Rates of grade 3/4 FN were similar between treatment arms (< 1%); the definition of FN used in the trial was not provided.51 Details on CSF use during induction and maintenance therapies were not reported in the publication.
Ital Bendamustine-based regimens: Bendamustine, a novel bifunctional alkylating agent, was recently approved by the US Food and Drug Administration (FDA) to treat indolent NHL that has progressed after rituximab treatment.52 In a pivotal multicenter, open-label, single-arm trial, bendamustine (120 mg/m2) was administered to rituximab-refractory patients on days 1 and 2 every 21 days for 6–8 cycles.15 This study is included here because bendamustine has become an important component of regimens for the management of FL (either as monotherapy or in combination with other agents). In this study, the ORR was 74% (95% confidence interval [CI], 65%–83%), and the duration of response was 9.2 months (95% CI, 7.1–10.8 months), based on a median follow-up of 11.4 months. In 38 patients who had no objective response to their latest chemotherapy regimen, the ORR was 64%, and the median PFS was 7.5 months.
Primary CSF prophylaxis was not allowed in this study. Secondary CSF use was permitted if patients had grade 4 neutropenia that lasted at least 1 week, persistent leukopenia (grade > 2) at the next scheduled dose, or FN in any treatment cycle.15 The incidence of neutropenic complications was high (grade 3/4 neutropenia, 61%; grade 3/4 FN, 6%; and grade 3/4 infection, 21%). These findings demonstrate that when administered at the approved dose of 120 mg/m2 in the absence of primary CSF prophylaxis, bendamustine is associated with a high risk of neutropenic toxicity.
A randomized phase III trial compared bendamustine (90 mg/m2) plus rituximab (BR) with R-CHOP in patients with previously untreated indolent NHL.53 After a median observation period of 32 months, the BR regimen improved the CR rate (40% vs 31%; P = 0.03), PFS (55 vs 35 months; P = 0.0002), EFS (54 months vs 31 months; P = 0.0002), and time to next treatment (not reached vs 41 months; P = 0.0002). The rate of grade 3/4 neutropenia and number of infectious complications were significantly lower with the BR regimen than with R-CHOP: 11% vs 47% (P < 0.001) and 95 vs 121 (P < 0.04), respectively. 53 CSF was administered at the discretion of the treating physician and was used less frequently with the BR regimen than with R-CHOP (4% vs 20%).
Neutropenic toxicity associated with current treatment regimens for CLL
The NCCN recommends chemotherapy, primarily combinations containing alkylating agents and chemoimmunotherapy, as the standard of care for advanced CLL.1 Monotherapy or combination regimens with an alkylating agent or purine analog are preferred first-line therapies for elderly patients (≥ 70 years of age) and for frail patients with significant comorbidity. However, a more aggressive approach with rituximab-containing chemoimmunotherapy regimens is recommended for patients < 70 years old and for older patients with no significant comorbidities.1
Chemotherapy regimens
Two large randomized controlled trials4,5 showed that FC compared with fludarabine alone increased ORR, CR, and PFS in patients with CLL. The neutropenic toxicity of these regimens appeared similar in both studies. In Flinn et al,5 rates of grade 3/4 neutropenia, grade 3/4 FN, and grade 3–5 infection with grade 3/4 FN were similar (Table 1). CSF use was higher in the FC arm than in the fludarabine arm; however, CSF use was required in the FC arm only and not in the fludarabine arm. In Catovsky et al,4 rates of grade 3/4 neutropenia and all febrile episodes were similar (Table 1). In this study, CSF support was used according to local guidelines; however, the proportion of patients who required CSF support in the different treatment arms was not reported.
Chemoimmunotherapy regimens
In two large randomized controlled trials, FCR improved survival in patients with CLL compared with FC alone.11,12 In the CLL8 trial in chemotherapy-naive patients with advanced CLL,12 FCR was more efficacious than FC, as measured by CR rate (44% vs 22%; P < 0.001), PFS (52 vs 33 months; P < 0.001), and OS at 38 months (84% vs 79%; P = 0.01). The median OS had not been reached in either treatment arm at the time these data were published in abstract form. Hematologic adverse events, including neutropenia, were more common with FCR (percentages not reported) than with FC, but the infection rates were similar in the two treatment arms (Table 1).12 CSF use in this study was not reported.
In the REACH study, which compared FCR and FC in previously treated patients with CLL,11 FCR improved PFS (median, 31 months vs 21 months; HR = 0.65; P < 0.001) at a median follow-up of 25 months. Rates of grade 3/4 neutropenia and grade 3/4 infection were similar in the two groups (Table 1). In this study, 58% of patients in the FCR arm and 49% in the FC arm received CSF, administered at the discretion of the investigator.
Other chemoimmunotherapy regimens for CLL recommended by the NCCN include pentostatin, cyclophosphamide, and rituximab; and oxaliplatin, fludarabine, cytarabine, and rituximab.1 This recommendation was made on the basis of safety and efficacy results from nonrandomized trials.
Alemtuzumab-based regimens
In 2001, the FDA approved alemtuzumab to treat patients with CLL who had failed to respond to prior fludarabine-containing chemotherapy. 54 In an open-label, randomized controlled trial comparing alemtuzumab with chlorambucil (Leukeran) in previously untreated patients with CLL, alemtuzumab improved the ORR (83% vs 55%; P < 0.0001), PFS (15 vs 12 months; P < 0.0001), CR (24% vs 2%; P < 0.0001), and time to next treatment (23 vs 15 months; P < 0.0001).18 Grade 3/4 neutropenia was significantly more common with alemtuzumab than with chlorambucil (Table 1), but the rates of FN and serious infections were low in both treatment arms. In that study, CSF was administered to more than twice as many patients receiving alemtuzumab as receiving chlorambucil (Table 1)18; however, no further details were provided. Alemtuzumab-fludarabine and alemtuzumab with or without rituximab are regimens also recommended by the NCCN for relapsed or refractory CLL based on the results of nonrandomized trials.1
Bendamustine-based regimens
Bendamustine is recommended by the NCCN as a single agent for firstline therapy and as a single agent or in combination with rituximab for second-line therapy in patients with CLL.1 An open-label, multicenter, randomized phase III study compared bendamustine (100 mg/m2 on days 1–2 of each 28-day cycle) with chlorambucil in patients with untreated advanced CLL.16 Bendamustine significantly improved PFS (22 vs 8 months; P < 0.0001) and CR or PR (68% vs 31%; P < 0.0001). Grade 3/4 neutropenia occurred in twice as many bendamustine-treated patients as chlorambucil-treated patients (Table 1). The authors of this study report that even though the use of hematopoietic growth factors was discouraged in this study, CSF was administered in the bendamustine arm at the discretion of the treating investigator (Table 1).16
Bendamustine in combination with rituximab is also recommended for relapsed CLL.1 In a phase II study, patients with CLL were treated with bendamustine (70 mg/m2 on days 1 and 2 of each 28-day cycle) and rituximab (375 mg/m2 for the first cycle and 500 mg/m2 for subsequent cycles). 55 This single-arm study is included here because bendamustine is an important component of regimens for treating CLL. After a mean of 4.5 cycles, the ORR was 77%. Myelosup pression and infections were the most frequent severe adverse events reported, with grade 3/4 leukopenia or neutropenia observed in 12% of patients. Grade 3 or greater infections were documented in 5% of patients, and infection-related mortality occurred in 4% of patients. CSF use was not documented in this article.
Ofatumumab
Ofatumumab (Arzerra), a human monoclonal antibody directed against CD20, was recently approved by the FDA for the treatment of CLL refractory to fludarabine and alemtuzumab. 56 The NCCN recommends ofatumumab for relapsed or refractory disease.1 The registrational trial was a nonrandomized phase II study that evaluated safety and efficacy of ofatumumab in patients with fludarabineand alemtuzumab-refractory CLL (group A) and in patients with fludarabine- refractory CLL who were not candidates for alemtuzumab treatment because of bulky lymphadenopathy (group B).57 The study is included here because ofatumumab is a relatively new treatment option available to patients who fail to respond to other therapies. A planned interim analysis demonstrated benefits with ofatumumab in the two treatment groups (ORR, 58% and 47%; duration of response, 7.1 months and 5.6 months; PFS, 5.7 months and 5.9 months; and OS, 13.7 months and 15.4 months, respectively). 57 Grade 3/4 neutropenia was 14% in group A and 6% in group B; grade 3/4 infection was 12% and 8%, respectively. Of the 189 infectious events (all grades) with onset during treatment reported in this study, 13 (7%) were fatal. No information about CSF use was provided.
Neutropenic toxicity associated with current treatment regimens for HLThe NCCN recommends doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD); Stanford V; and escalated-dose BEACOPP for the treatment of HL. ABVD was introduced in the 1990s, and Stanford V and BEACOPP were introduced in the early 2000s.8,58–61 These regimens are known to be highly myelotoxic.
For the ABVD regimen, an 18% rate of severe neutropenia was reported in one study,61 and a 57% rate of grade 3/4 neutropenia was reported in another study.58 With the Stanford V regimen, the incidence of grade 4 neutropenia and FN was as high as 82% and 14%, respectively.60 It should be noted that despite the high level of myelosuppression associated with regimens for HL, the NCCN does not recommend the routine use of CSF because neutropenia is not considered a major factor for dose reductions or dose delays.1
Trials have compared the ABVD and Stanford V regimens in patients with HL. One trial in patients with advanced disease demonstrated comparable efficacy of the two regimens.6 However, another trial in patients with intermediate- and advancedstage disease demonstrated the superiority of ABVD combined with optional limited radiotherapy over the Stanford V regimen, as measured by response rate and PFS.7 Both studies reported comparable neutropenic toxicity of the ABVD and Stanford V regimens when secondary CSF prophylaxis was permitted (Table 1).6,7
The BEACOPP regimen, which incorporates chemotherapy dose intensification and frequent scheduling, has been shown to improve patient outcomes in advanced disease.8 A relatively recent trial directly compared ABVD vs BEACOPP (four escalated-dose schedules followed by two standard-dose schedules) vs cyclophosphamide, lomustine, vindesine, melphalan, prednisone, epidoxirubicin, vincristine, procarbazine, vinblastine, and bleomycin (CEC).62 At a median follow-up of 41 months, BEACOPP compared with ABVD significantly improved the 5-year PFS (81% vs 68%; P = 0.038) but showed no significant differences with CEC. Both the BEACOPP and CEC regimens were associated with higher rates of grade 3/4 neutropenia than ABVD; BEACOPP was also associated with higher rates of severe infections than ABVD and CEC (Table 1).62 Daily CSF was incorporated into the BEACOPP regimen and administered for at least 8 days, until an absolute neutrophil count of 500/ mm3 was reached.62 Routine CSF prophylaxis was not required with the ABVD and CEC regimens but was used at the discretion of the treating physician.
Neutropenic toxicity associated with current treatment regimens for multiple myeloma
A variety of regimens that incorporate the novel agents bortezomib (Velcade), lenalidomide (Revlimid), or thalidomide (Thalomid) have been evaluated for the treatment of multiple myeloma. These agents directly target the myeloma cells and can also interfere with the interaction of tumor cells with the bone marrow microenvironment. 63 The NCCN recommends these agents as components of combination regimens for induction chemotherapy (whether or not stem cell transplantation is indicated), as maintenance treatment after transplantation, or as salvage therapy for patients with multiple myeloma.1
Bortezomib-based regimens
Bortezomib, a member of a new class of drugs called proteasome inhibitors, is FDA approved to treat multiple myeloma.64 Patients with previously untreated myeloma are treated with bortezomib in combination with melphalan and prednisone (MPB). Results from the Velcade as Initial Standard Therapy in Multiple Myeloma trial compared MPB wit melphalan and prednisone (MP) in patients who were ineligible for transplant therapy.13,14 At a median follow-up of 37 months, MPB reduced the risk of death by 35% (HR, 0.653; P < 0.001) and improved the 3-year OS (69% vs 54%).13 The incidence of grade 3/4 neutropenia was comparable for MPB and MP (40% vs 38%; Table 1), suggesting that the MP component of the regimen is primarily responsible for the neutropenic toxicity. Information on CSF use in this study was not provided. The APEX trial compared bortezomib with high-dose dexamethasone as salvage therapy in patients with recurrent myeloma.65,66 At a median follow-up of 22 months, bortezomib significantly improved the ORR (43% vs 18%; P < 0.0001) and the 1-year survival rates (80% vs 67%; P = 0.00002).66 Bortezomib was associated with a higher incidence of grade 3/4 neutropenia than was highdose dexamethasone (14% vs 1%; P < 0.01). However, the incidence of grade 3/4 infections was similar between the arms (13% vs 16%; P = 0.19).65 CSF use was permitted at the physician’s discretion; however, details were not provided.
Bortezomib in combination with pegylated liposomal doxorubicin (Doxil; B + PLD) is FDA approved for salvage therapy for multiple myeloma, with a category 1 recommendation from the NCCN. Interim data from a randomized phase III study67 demonstrated the superiority of B + PLD to bortezomib monotherapy (TTP, 9.3 months vs 6.5 months; P < 0.0001; PFS, 9.0 months vs 6.5 months; P < 0.0001; duration of response, 10 months vs 7 months; P < 0.001; and 15-month OS rates, 76% vs 65%; P = 0.03). Grade 3/4 neutropenia was significantly more common with the combination regimen; however, the rate of FN was similar (Table 1).67 CSF use was allowed in this study, but details were not provided.
Lenalidomide-based regimens
Lenalidomide is an immunomodulatory agent that is FDA approved for use in combination with dexamethasone to treat patients with multiple myeloma who have received at least one prior therapy.68 Lenalidomide is taken orally once daily on days 1–21 of 28-day cycles as a part of the lenalidomide-dexamethasone regimen.68
A phase III trial conducted in the US and Canada69 and a companion trial conducted in Europe, Israel, and Australia70 compared the lenalidomide- dexamethasone regimen with placebo-dexamethasone in patients with refractory or recurrent myeloma. In both trials, lenalidomidedexamethasone significantly improved the ORR, TTP, and OS.69,70 In both studies, neutropenic toxicity (including grade 3/4 neutropenia, FN, or grade 3/4 infection) was higher in the lenalidomide-dexamethasone arm than in the dexamethasone alone arm (Table 1).
Secondary CSF prophylaxis in response to neutropenic toxicity was permitted in both studies. In the Weber at al study,69 60 of the 177 patients (33.9%) in the lenalidomide- dexamethasone group received CSF support; 28 of the 60 patients (46.7%) received CSF to maintain the full lenalidomide dose, and 12 of these 28 patients (43%) were able to continue at the 25-mg dose level. In the Dimopoulos et al study,70 38 of 176 patients (22%) in the lenalidomide- dexamethasone group received CSF support; 23 of these patients (61%) needed CSF to maintain the lenalidomide dose, and 12 (52%) were able to continue on 25 mg of lenalidomide.
A recent trial evaluated lenalidomide- dexamethasone as initial therapy for patients with newly diagnosed multiple myeloma.71 In this open-label study with a noninferiority design, lenalidomide plus low-dose dexamethasone was compared with lenalidomide plus high-dose dexamethasone. The trial was stopped early because of the superior survival results with the low-dose dexamethasone regimen at a 1-year interim analysis (OS, 96% vs 87%; P = 0.0002). The NCCN now recommends lenalidomide with low-dose dexamethasone for previously untreated patients who are not candidates for transplant therapy.1 The low-dose dexamethasone regimen was associated with fewer infections than the high-dose dexamethasome regimen (9% vs 16%; P = 0.04), even though it was associated with a higher incidence of grade 3/4 neutropenia (20% vs 12%; P = 0.02). Details of CSF use were not reported for this study.
Thalidomide-based regimens
Thalidomide is also an immunomodulator that is FDA approved for use in combination with dexamethasone to treat patients with newly diagnosed multiple myeloma. FDA approval of this regimen was supported by results from the Eastern Cooperative Oncology Group (ECOG) study, which compared thalidomidedexamethasone with dexamethasone alone.72 The response rate with thalidomide- dexamethasone was significantly higher than with dexamethasone alone (63% vs 41%; P = 0.017). The incidence of neutropenia and infection was similar between the arms (Table 1).72 Details of CSF use in this study were not provided.
Thalidomide in combination with MP (MPT) is recommended by the NCCN as a primary induction therapy for transplant-ineligible myeloma patients. The Intergroup Francophone du Myélome 01/01 Trial of MPT in patients with untreated multiple myeloma compared MPT with MP-placebo.73 MPT improved OS (44 vs 29 months; P = 0.03) and PFS (24 vs 18.5 months; P = 0.001), at a median follow-up of 47.5 months. Grade 3/4 neutropenia was significantly more common with MPT, but the incidence of severe infection was similar in the two treatment arms (Table 1). CSF use was permitted in this study; however, details were not provided.
Of note, unlike conventional chemotherapeutic agents, novel agents used to treat multiple myeloma are not administered in 14- or 21-day cycles. For example, bortezomib is initially administered twice-weekly (with rest periods) followed by weekly dosing as a component of the MPB regimen.13,14 Lenalidomide is taken orally once daily on days 1–21 of 28-day cycles as part of the lenalidomide-dexamethasone regimen. 69,70 Similarly, thalidomide is administered daily as an oral tablet.72 Furthermore, although clinical trials have integrated CSF use, no studies specifically address it with these novel agents (ie, whether CSF should be given concurrently or sequentially with the therapy). Therefore, clinical trials evaluating the safety of CSF use with these novel agents are warranted.
Quantitative analysis of underreporting of neutropenic toxicity
As previously discussed, most reports of trials evaluating therapies for treating hematologic malignancies include information about the frequency of severe neutropenia. However, our literature review showed that data on the incidence of FN and the use of CSF are frequently not provided. The omission of this information limits the comparison of results across trials and the ability to make informed decisions on the true risk of FN for a treatment modality. The objective of this quantitative analysis was to evaluate the reporting of FN and other neutropenic outcomes, as well as related CSF or antibiotic use, in randomized controlled trials that evaluated regimens for the treatment of NHL, CLL, HL, or multiple myeloma.
Selection criteria for articles included For this quantitative analysis, phase III trials published between January 2005 and June 2009 were identified from the original list of trials retrieved through the comprehensive literature search, as previously discussed. We included phase III trials only for this analysis, because most are designed to capture both safety and efficacy associated with a treatment modality, compared with phase II trials, which may sometimes primarily focus on safety parameters. We also included all articles that met the specified criteria, whether or not the treatment regimen reported in the article was recommended by the NCCN.
Articles that met the inclusion criteria were retrieved and data on myelotoxic outcomes were abstracted by two reviewers and reconciled by a third reviewer. The neutropenic outcomes included were grade 3/4 neutropenia or granulocytopenia, FN, leukopenia, all-cause hospitalization, neutropenia-related hospitalization, infection or sepsis, and infection-related mortality. Outcomes on chemotherapy delivery included dose delays, dose reductions, and dose intensity or relative dose intensity. We also collected data on CSF use defined in the methods section, CSF use presented in the results section, and antibiotic use defined in the methods and/or results section.
Results
Table 2 summarizes our findings on the reporting of neutropenic toxicity outcomes. Of the 57 trials that met the inclusion criteria, 86% reported results of at least one neutropenic endpoint. Across tumor types, 68% of trials reported on the incidence of grade 3/4 neutropenia (80%, multiple myeloma; 71%, CLL; 63%, NHL, 50%, HL). However, a few trials (19%) reported on the incidence of FN (57%, CLL; 20%, multiple myeloma; 12%, NHL). Similarly, only a few trials (4%) reported on neutropenia- related hospitalizations (8%, NHL). The incidence of infection or sepsis and infection-related mortality was reported in 79% and 60% of publications, respectively. Dose delays/interruptions were reported in 21% of trials overall. Dose reductions were reported in 30% of articles overall.
Data on the reporting of CSF and antibiotic use are shown in Table 3. About half (49%) of the publications reported planned use of CSF in the methods section (71%, CLL; 67%, HL; 50%, NHL; 35%, multiple myeloma). However, overall, only 25% of publications reported CSF use in the results section (43%, CLL; 29%, NHL; 17%, HL; 15%, multiple myeloma). Overall reporting on prophylactic antibiotic use was also low. Antibiotic use was discussed in the methods sections of only 21% of papers (71%, CLL; 17%, HL; 15%, multiple myeloma; 13%, NHL), and actual use of antibiotics was not reported in the results section of any of the publications.
Discussion
Our review shows that many phase III trials of current treatment regimens for hematologic malignancies omit important outcome data on the incidence of FN, neutropenia-related hospitalization, infection-related mortality, chemotherapy dose delays/ interruptions or dose reductions, use of primary or secondary CSF prophylaxis, or use of antibiotics. These findings are similar to recent observations by others.
For instance, Duff and colleagues40 reported that publications describing results from phase III trials fail to consistently report details that would enable clinicians in the community to translate findings to clinical practice. When these researchers asked medical oncologists and oncology pharmacists to identify the most important information necessary for clinical application of an oncology drug, 3 of the 10 most common responses were premedication, growth factor support, and dose adjustments for hematologic toxicity.
The researchers then reviewed 262 articles published in five journals (Blood, Cancer, the Journal of Clinical Oncology, the Journal of the National Cancer Institute, and the New England Journal of Medicine) between 2005 and 2008. They found that each of these elements (premedication, growth factor support, and dose adjustments for hematologic toxicity) was reported fewer than half the time (P < 0.0001) compared with the name of the drug, which was reported 100% of the time. Duff and colleagues40 recommend that journal editors require reporting of these and other highly ranked elements in the article or in an online appendix and provide Internet- open access to the clinical trial protocol.
Dale and colleagues39 examined 58 reports on NHL therapy trials published between 1990 and 2000. They found that 34% did not include data on neutropenic toxicity and 3% included only details on clinical consequences, such as fatal infection. In the other trials, hematologic toxicity was reported 18 different ways. These authors recommend that certain details about hematologic toxicity should routinely be documented in reports on cancer chemotherapy: rates of leukopenia and neutropenia; the timing of blood cell counts used to determine these rates; protocols for antibiotics and CSF use; actual use of antibiotics and CSF; rates of all infectious complications, including hospitalizations and bacteremias; and relative dose intensity. 39
Conclusion
In addition to efficacy data, reports on clinical trials should provide details on the toxicity of treatment and requirements for supportive care. A standardized approach to collecting and reporting neutropenic outcomes and the related use of supportive care measures can assist clinicians in prospectively managing the relevant toxicities associated with treatment regimens for hematologic malignancies. This information is essential for the safe and effective transition of these regimens into broad clinical practice. These data should include all grade 3 or greater hematologic and nonhematologic toxicities in phase II, III, or IV clinical trials, as well as details on prophylactic and interventional CSF and antibiotic use. Armed with knowledge of the risk of neutropenic toxicity associated with each treatment regimen, oncologists can then focus on the patient-related risks when making decisions regarding appropriate supportive care. Mitigation of neutropenic toxicity associated with treatment regimens is important to decrease patients’ risk for treatment delays/interruptions, dose reductions, or discontinuations, which can compromise patient outcomes.19–22
Acknowledgments
Amgen sponsored an external agency for data abstraction and analysis. The authors thank Beverly A. Caley and Leta Shy for data abstraction; Supriya Srinivasan for data reconciliation; and Supriya Srinivasan and Martha Mutomba for writing assistance. The sponsor played a role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the manuscript for publication. The corresponding author had full access to all data and had final responsibility for the decision to submit the article for publication. All authors provided comments during manuscript development and have approved the final version of the submitted article.
Conflicts of interest
Dr. Gregory has served as a consultant or in an advisory role with Amgen Inc, Genentech (Roche), Novartis, and Spectrum Pharmaceuticals; and her institution has received research funding from Astellas, Celgene, Cephalon, Genentech (Roche), GlaxoSmithKline, Immunomedics, NCIC–CTG, and Novartis. Dr. Abella is an employee and stock owner of Amgen Inc. Dr. Moore has served as a consultant or in an advisory role with Amgen Inc and is on the speakers’ bureaus of Amgen Inc, sanofi-aventis, and GlaxoSmithKline
References
1. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology, 2010. http://www.nccn.org/professionals/ physician_gls/f_guidelines.asp. Accessed December 17, 2010.
2. Pfreundschuh M, Trümper L, Kloess M, et al. Two-weekly or 3-weekly CHOP chemotherapy with or without etoposide for the treatment of elderly patients with aggressive lymphomas: results of the NHL-B2 trial of the DSHNHL. Blood 2004;104:634–641.
3. Wunderlich A, Kloess M, Reiser M, et al. Practicability and acute haematological toxicity of 2- and 3-weekly CHOP and CHOEP chemotherapy for aggressive non-Hodgkin’s lymphoma: results from the NHL-B trial of the German High-Grade Non-Hodgkin’s Lymphoma Study Group (DSHNHL). Ann Oncol 2003;14:881–893.
4. Catovsky D, Richards S, Matutes E, et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 2007;370:230–239.
5. Flinn IW, Neuberg DS, Grever MR, et al. Phase III trial of fludarabine plus cyclophosphamide compared with fludarabine for patients with previously untreated chronic lymphocytic leukemia: US Intergroup Trial E2997. J Clin Oncol 2007;25:793–798.
6. Hoskin PJ, Lowry L, Horwich A, et al. Randomized comparison of the stanford V regimen and ABVD in the treatment of advanced Hodgkin’s Lymphoma: United Kingdom National Cancer Research Institute Lymphoma Group Study ISRC TN 64141244. J Clin Oncol 2009;27:5390–5396.
7. Gobbi PG, Levis A, Chisesi T, et al. ABVD versus modified Stanford V versus MOPPEBVCAD with optional and limited radiotherapy in intermediate- and advanced- stage Hodgkin’s lymphoma: final results of a multicenter randomized trial by the Intergruppo Italiano Linfomi. J Clin Oncol 2005;23:9198–9207.
8. Diehl V, Franklin J, Pfreundschuh M, et al. Standard and increased-dose BEACOPP chemotherapy compared with COPP-ABVD for advanced Hodgkin’s disease. N Engl J Med 2003;348:2386–2395.
9. Michallet AS, Coiffier B. Recent developments in the treatment of aggressive non- Hodgkin lymphoma. Blood Rev 2009;23:11– 23.
10. Pfreundschuh M, Trümper L, Osterborg A, et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol 2006;7:379–391.
11. Robak T, Dmoszynska A, Solal-Céligny P, et al. Rituximab plus fludarabine and cyclophosphamide prolongs progression-free survival compared with fludarabine and cyclophosphamide alone in previously treated chronic lymphocytic leukemia. J Clin Oncol 2010;28:1756–1765.
12. Hallek M, Fingerle-Rowson G, Fink AM, et al. First-line treatment with fludarabine (F), cyclophosphamide (C), and rituximab (R) (FCR) improves overall survival (OS) in previously untreated patients (pts) with advanced chronic lymphocytic leukemia (CLL): results of a randomized phase III trial on behalf of an international group of investigators and the German CLL Study Group. Blood 2009;114:535.
13. Mateos MV, Oriol A, Martinez-López J, et al. Bortezomib, melphalan, and prednisone versus bortezomib, thalidomide, and prednisone as induction therapy followed by maintenance treatment with bortezomib and thalidomide versus bortezomib and prednisone in elderly patients with untreated multiple myeloma: a randomised trial. Lancet Oncol 2010;11:934–941.
14. San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med 2008;359:906–917.
15. Kahl BS, Bartlett NL, Leonard JP, et al. Bendamustine is effective therapy in patients with rituximab-refractory, indolent B-cell non- Hodgkin lymphoma: results from a Multicenter Study. Cancer 2010;116:106–114.
16. Knauf WU, Lissichkov T, Aldaoud A, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:4378–4384.
17. Fischer K, Cramer P, Stilgenbauer S, et al. Bendamustine combined with rituximab (BR) in first-line therapy of advanced CLL: a multicenter phase II trial of the German CLL Study Group (GCLLSG). Blood 2009;114:205.
18. Hillmen P, Skotnicki AB, Robak T, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol 2007;25:5616–5623
19. Mayordomo JI, López A, Viñolas N, et al. Retrospective cost analysis of management of febrile neutropenia in cancer patients in Spain. Curr Med Res Opin 2009;25:2533–2542.
20. Pettengell R, Schwenkglenks M, Leonard R, et al. Neutropenia occurrence and predictors of reduced chemotherapy delivery: results from the INC-EU prospective observational European neutropenia study. Support Care Cancer 2008;16:1299–1309.
21. Lyman GH, Kleiner JM. Summary and comparison of myeloid growth factor guidelines in patients receiving cancer chemotherapy. J Natl Compr Canc Netw 2007;5:217–228.
22. Smith TJ, Khatcheressian J, Lyman GH, et al. 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol 2006;24:3187–3205.
23. Crawford J, Caserta C, Roila F; ESMO Guidelines Working Group. Hematopoietic growth factors: ESMO Clinical Practice Guidelines for the applications. Ann Oncol 2010;21(suppl 5):v248–v251.
24. Aapro MS, Cameron DA, Pettengell R, et al. EORTC guidelines for the use of granulocyte- colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphomas and solid tumours. Eur J Cancer 2006;42:2433–2453.
25. Crawford J, Ozer H, Stoller R, et al. Reduction by granulocyte colony-stimulating factor of fever and neutropenia induced by chemotherapy in patients with small-cell lung cancer. N Engl J Med 1991;325:164–170.
26. Gabrilove JL, Jakubowski A, Scher H, et al. Effect of granulocyte colony-stimulating factor on neutropenia and associated morbidity due to chemotherapy for transitional-cell carcinoma of the urothelium. N Engl J Med 1988;318:1414–1422.
27. Grigg A, Solal-Celigny P, Hoskin P, et al. Open-label, randomized study of pegfilgrastim vs. daily filgrastim as an adjunct to chemotherapy in elderly patients with non-Hodgkin’s lymphoma. Leuk Lymphoma 2003;44:1503–1508.
28. Holmes FA, Jones SE, O’Shaughnessy J, et al. Comparable efficacy and safety profiles of once-per-cycle pegfilgrastim and daily injection filgrastim in chemotherapy-induced neutropenia: a multicenter dose-finding study in women with breast cancer. Ann Oncol 2002;13:903–909.
29. Holmes FA, O’Shaughnessy JA, Vukelja S, et al. Blinded, randomized, multicenter study to evaluate single administration pegfilgrastim once per cycle versus daily filgrastim as an adjunct to chemotherapy in patients with highrisk stage II or stage III/IV breast cancer. J Clin Oncol 2002;20:727–731.
30. Johnston E, Crawford J, Blackwell S, et al. Randomized, dose-escalation study of SD/01 compared with daily filgrastim in patients receiving chemotherapy. J Clin Oncol 2000;18:2522–2528.
31. Trillet-Lenoir V, Green J, Manegold C, et al. Recombinant granulocyte colony stimulating factor reduces the infectious complications of cytotoxic chemotherapy. Eur J Cancer 1993;29A:319–324.
32. Vogel CL, Wojtukiewicz MZ, Carroll RR, et al. First and subsequent cycle use of pegfilgrastim prevents febrile neutropenia in patients with breast cancer: a multicenter, double- blind, placebo-controlled phase III study. J Clin Oncol 2005;23:1178–1184.
33. Green MD, Koelbl H, Baselga J, et al. A randomized double-blind multicenter phase III study of fixed-dose single-administration pegfilgrastim versus daily filgrastim in patients receiving myelosuppressive chemotherapy. Ann Oncol 2003;14:29–35.
34. Vose JM, Crump M, Lazarus H, et al. Randomized, multicenter, open-label study of pegfilgrastim compared with daily filgrastim after chemotherapy for lymphoma. J Clin Oncol 2003;21:514–519.
35. Kuderer NM, Dale DC, Crawford J, Lyman GH. Impact of primary prophylaxis with granulocyte colony-stimulating factor on febrile neutropenia and mortality in adult cancer patients receiving chemotherapy: a systematic review. J Clin Oncol 2007;25:3158–3167.
36. Lyman GH, Michels SL, Reynolds MW, Barron R, Tomic KS, Yu J. Risk of mortality in patients with cancer who experience febrile neutropenia. Cancer 2010;116:5555– 5563.
37. Lyman GH, Dale DC, Wolff DA, et al. Acute myeloid leukemia or myelodysplastic syndrome in randomized controlled clinical trials of cancer chemotherapy with granulocyte colony-stimulating factor: a systematic review. J Clin Oncol 2010;28:2914–2924.
38. Bohlius J, Reiser M, Schwarzer G, Engert A. Granulopoiesis-stimulating factors to prevent adverse effects in the treatment of malignant lymphoma. Cochrane Database Syst Rev 2004:CD003189.
39. Dale DC, McCarter GC, Crawford J, Lyman GH. Myelotoxicity and dose intensity of chemotherapy: reporting practices from randomized clinical trials. J Natl Compr Canc Netw 2003;1:440–454.
40. Duff JM, Leather H, Walden EO, LaPlant KD, George TJ Jr. Adequacy of published oncology randomized controlled trials to provide therapeutic details needed for clinical application. J Natl Cancer Inst 2010;102:702– 705.
41. Freedman OC, Zimmermann C, Clemons MJ: Interpreting the results of clinical trials of cancer chemotherapy: the importance of reporting concurrent supportive care. Proceedings from 31st Annual San Antonio Breast Cancer Symposium; December 14, 2008; San Antonio, TX. Abstract 6138.
42. Coiffier B, Lepage E, Briere J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235–242.
43. Pfreundschuh M, Schubert J, Ziepert M, et al. Six versus eight cycles of bi-weekly CHOP-14 with or without rituximab in elderly patients with aggressive CD20+ B-cell lymphomas: a randomised controlled trial (RICOVER-60). Lancet Oncol 2008;9:105– 116.
44. Delarue R, Tilly H, Salles G, et al. RCHOP14 compared to R-CHOP21 in elderly patients with diffuse large B-cell lymphoma: results of the interim analysis of the LNH03- 6B GELA study. Blood 2009;114:406.
45. Hiddemann W, Kneba M, Dreyling M, et al. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 2005;106:3725–3732.
46. Marcus R, Imrie K, Belch A, et al. CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood 2005;105:1417–1423.
47. Forstpointner R, Unterhalt M, Dreyling M, et al. Maintenance therapy with rituximab leads to a significant prolongation of response duration after salvage therapy with a combination of rituximab, fludarabine, cyclophosphamide, and mitoxantrone (R-FCM) in patients with recurring and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German Low Grade Lymphoma Study Group (GLSG). Blood 2006;108:4003–4008.
48. van Oers MH, Klasa R, Marcus RE, et al. Rituximab maintenance improves clinical outcome of relapsed/resistant follicular non- Hodgkin lymphoma in patients both with and without rituximab during induction: results of a prospective randomized phase 3 intergroup trial. Blood 2006;108:3295-3301.
49. van Oers MH, Van Glabbeke M, Giurgea L, et al. Rituximab maintenance treatment of relapsed/resistant follicular non-Hodgkin’s lymphoma: long-term outcome of the EORTC 20981 phase III randomized intergroup study. J Clin Oncol 2010;28:2853–2858.
50. Hochster H, Weller E, Gascoyne RD, et al. Maintenance rituximab after cyclophosphamide, vincristine, and prednisone prolongs progression-free survival in advanced indolent lymphoma: results of the randomized phase III ECOG1496 Study. J Clin Oncol 2009;27:1607–1614.
51. Salles GA, Seymour JF, Feugier P, et al. Rituximab maintenance for 2 years in patients with untreated high tumor burden follicular lymphoma after response to immunochemotherapy. J Clin Oncol 2010;28[15S]:8004.
52. Treanda [prescribing information]. Frazer, PA: Cephalon, Inc.; 2010.
53. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab is superior in respect of progression free survival and CR rate when compared to CHOP plus ritux imab as first-line treatment of patients with advanced follicular, indolent, and mantle cell lymphomas: final results of a randomized phase III study of the StiL (Study Group Indolent Lymphomas, Germany). Blood 2009;114:405.
54. Demko S, Summers J, Keegan P, Pazdur R. FDA drug approval summary: alemtuzumab as single-agent treatment for B-cell chronic lymphocytic leukemia. Oncologist 2008;13:167–174.
55. Fischer K, Stilgenbauer S, Schweighofer CD, et al. Bendamustine in combination with rituximab (BR) for patients with relapsed chronic lymphocytic leukemia (CLL): a multicentre phase II trial of the German CLL Study Group (GCLLSG). Blood 2008;112:330.
56. Arzerra [prescribing information]. Research Triangle Park, NC: GlaxoSmithKline; 2010.
57. Wierda WG, Kipps TJ, Mayer J, et al. Ofatumumab as single-agent CD20 immunotherapy in fludarabine-refractory chronic lymphocytic leukemia. J Clin Oncol 2010;28:1749– 1755.
58. Straus DJ, Portlock CS, Qin J, et al. Results of a prospective randomized clinical trial of doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) followed by radiation therapy (RT) versus ABVD alone for stages I, II, and IIIA nonbulky Hodgkin disease. Blood 2004;104:3483–3489.
59. Duggan DB, Petroni GR, Johnson JL, et al. Randomized comparison of ABVD and MOPP/ABV hybrid for the treatment of advanced Hodgkin’s disease: report of an intergroup trial. J Clin Oncol 2003;21:607–614.
60. Horning SJ, Hoppe RT, Breslin S, Bartlett NL, Brown BW, Rosenberg SA. Stanford V and radiotherapy for locally extensive and advanced Hodgkin’s disease: mature results of a prospective clinical trial. J Clin Oncol 2002;20:630–637.
61. Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin’s disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med 1992;327:1478–1484.
62. Federico M, Luminari S, Iannitto E, et al. ABVD compared with BEACOPP compared with CEC for the initial treatment of patients with advanced Hodgkin’s lymphoma: results from the HD2000 Gruppo Italiano per lo Studio dei Linfomi Trial. J Clin Oncol 2009;27:805–811.
63. Hideshima T, Bergsagel PL, Kuehl WM, Anderson KC. Advances in biology of multiple myeloma: clinical applications. Blood 2004;104:607–618.
64. Thalomid [prescribing information]. Summit, NJ: Celgene Corporation; 2009.
65. Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005;352:2487–2498.
66. Richardson PG, Sonneveld P, Schuster M, et al. Extended follow-up of a phase 3 trial in relapsed multiple myeloma: final timeto- event results of the APEX trial. Blood 2007;110:3557–3560.
67. Orlowski RZ, Nagler A, Sonneveld P, et al. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. J Clin Oncol 2007;25:3892–3901.
68. Revlimid [prescribing information]. Summit, NJ: Celgene Corporation; 2009.
69. Weber DM, Chen C, Niesvizky R, et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med 2007;357:2133–2142.
70. Dimopoulos M, Spencer A, Attal M, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med 2007;357:2123–2132.
71. Rajkumar SV, Jacobus S, Callander NS, et al. Lenalidomide plus high-dose dexamethasone versus lenalidomide plus low-dose dexamethasone as initial therapy for newly diagnosed multiple myeloma: an open-label randomised controlled trial. Lancet Oncol 2010;11:29–37.
72. Rajkumar SV, Blood E, Vesole D, Fonseca R, Greipp PR. Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: a clinical trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol 2006;24:431–436.
73. Hulin C, Facon T, Rodon P, et al. Efficacy of melphalan and prednisone plus thalidomide in patients older than 75 years with newly diagnosed multiple myeloma: IFM 01/01 trial. J Clin Oncol 2009;27:3664–3670.
Stephanie A. Gregory, MD,1 Steve Abella, MD,2 and Tim Moore, MD3
1 Section of Hematology, Rush University Medical Center, Chicago, IL; 2 Global Clinical Development, Hematology/Oncology, Amgen Inc., Thousand Oaks, CA; and 3 Zangmeister Center, Columbus, OH
Most chemotherapy regimens considered standard of care for treating hematologic malignancies are myelosuppressive. They include chemotherapy regimens recommended by the National Comprehensive Cancer Network (NCCN),1 such as cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) to treat non-Hodgkin lymphoma (NHL) 2,3; fludarabine plus cyclophosphamide (FC) to treat chronic lymphocytic leukemia (CLL)4,5; and escalated-dose bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone (BEACOPP) or doxorubicin, vinblastine, mechlorethamine, etoposide, vincristine, bleomycin, and prednisone (Stanford V) to treat Hodgkin lymphoma (HL).6–8
Emerging regimens that incorporate targeted therapies or other novel agents (eg, rituximab [Rituxan], lenalidomide [Revlimid], or bendamustine [Treanda]) have also been shown to be myelosuppressive, mainly because they are generally combined with myelosuppressive chemotherapy to achieve optimal efficacy. Examples include CHOP plus rituximab (R-CHOP) to treat NHL9,10; FC plus rituximab (FCR) to treat CLL11,12; or bortezomib plus melphalan-prednisone (MPB) to treat multiple myeloma.13,14 Additionally, some agents show toxicity when used as monotherapies, including bendamustine15–17 and alemtuzumab (Campath) 18 to treat CLL. Therefore, improved clinical outcomes may be achieved with concurrent increased myelosuppression.
Patients receiving myelosuppresive chemotherapy are at risk for developing chemotherapy-induced neutropenia, including severe or prolonged neutropenia and febrile neutropenia (FN). This condition often leads to treatment delays/interruptions, dose reductions, or treatment discontinuations, which can result in suboptimal treatment delivery and compromised patient outcomes.19–22 Colony-stimulating factor (CSF) has thus become an important component of many current treatment regimens for hematologic malignancies. International clinical guidelines, including those from the NCCN,1 the American Society of Clinical Oncology (ASCO),22 the European Society for Medical Oncology (ESMO),23 and the European Organization for Research and Treatment of Cancer (EORTC),24 recommend CSF use when the risk of FN is ≥ 20% and consideration of CSF use when the risk of FN is between 10% and 20%.
Numerous studies have demonstrated CSF effectiveness in decreasing the incidence of severe neutropenia and/or FN.25–34 A meta-analysis of 17 randomized controlled trials, which enrolled 3,493 cancer patients receiving chemotherapy, demonstrated that primary prophylaxis with CSF was associated with a decreased incidence of FN and reduced rates of infection-related mortality and early mortality across different tumor types.35 The occurrence of FN was associated with a 35% increase in the hazard of early mortality, and prophylactic granulocyte (G)-CSF use decreased this number by 45%.36 In a separate analysis of 25 trials (total n = 12,804), CSF support in cancer patients receiving chemotherapy was associated with a significant increase in overall survival (OS).37 Furthermore, a meta-analysis of results from 12 randomized controlled trials, which enrolled 1,823 patients with malignant lymphoma, showed that CSF prophylaxis, compared with no prophylaxis, significantly reduced the relative risk of severe neutropenia, FN, and infection.38
Evidence-based data that could guide the use of CSF in the setting of current treatment regimens for hematologic malignancies are not always readily available. Publications that report clinical trial results focus on overall efficacy and safety parameters of treatment regimens and often do not report the incidence or severity of neutropenia and/or FN.39 Similarly, these publications often do not include information on supportive care measures, including prophylaxis with antibiotics and/or CSF (primary or secondary).40,41 Also, when CSF support is reported, often the agent and dosing schedule are not provided. Many trials permit the use of CSF at the investigator’s discretion; however, the proportion of patients treated or supported with CSF and related outcomes is often not reported. These gaps in reporting neutropenic toxicity and related outcomes may result in an underestimation of the degree of significant toxicity associated with current treatment regimens for hematologic malignancies.
We conducted a comprehensive review of English-language reports published after January 2005. From the retrieved list of publications, we identified studies reporting data from trials (including phase II and III) that evaluated regimens considered NCCN Guideline recommendations for treating selected hematologic malignancies. 1 We excluded trials that enrolled patients with acute leukemia or chronic myelogenous leukemia; trials with the primary objective of assessing radiotherapy, radioimmunotherapy, stem cell transplantation, or patient-reported outcomes; and trials that described the study design but not the results. If multiple publications reported results of the same trial, we selected the publication with the most complete data on hematologic toxicity. Publications that met the inclusion criteria were retrieved and reviewed for neutropenic toxicity outcomes and the reported use of CSF or antibiotics.
Neutropenic toxicity associated with current treatment regimens for NHL
Diffuse large B-cell lymphoma
Diffuse large B-cell lymphoma (DLBCL) is an aggressive type of lymphoma generally treated with curative intent in the frontline setting. Beginning in the 1970s, the standard of care for DLBCL was CHOP, administered every 21 days (CHOP- 21).9 However, approximately half of patients > 60 years of age do not benefit from this regimen. In a study by Coiffier et al,42 3-year OS in this patient population was less than 40%. The addition of rituximab to CHOP-21 (R-CHOP-21) or CHOP-21–like regimens was subsequently shown to improve OS significantly across patient populations, with no increased neutropenic toxicity (Table 1).10 The R-CHOP regimen is now considered the standard of care for DLBCL when the goal of treatment is cure.9Another randomized study by Pfreundschuh et al compared dose-dense CHOP (given every 14 days, CHOP-14) with CHOP-21 in NHL patients ≥ 60 years of age.2 The CHOP-14 dosedense regimen required support with primary prophylactic CSF in all cycles (CHOP-14-G), whereas prophylactic CSF use with CHOP-21 was at the discretion of the treating physician, based on patient characteristics. CHOP-14-G significantly improved event-free survival (EFS) and OS. Grade 4 neutropenia was less frequent with CHOP-14-G than with CHOP-21 (24% vs 44%; P < 0.001), demonstrating that CSF support could adequately protect patients from neutropenic toxicity associated with CHOP.2
The RICOVER-60 study43 evaluated 6 or 8 cycles of dose-dense CHOP (CHOP-14-G) with or without rituximab in patients 61– 80 years of age who had aggressive B-cell lymphoma and were receiving primary prophylaxis with CSF (R-CHOP-14-G vs CHOP-14-G). R-CHOP-14-G significantly improved EFS (66.5% vs 47.2%) and OS (78.1% vs 67.7%). Leukopenia was the most common grade 3/4 toxicity, with grade 4 events occurring in 48%–52% across treatment arms. However, the incidence of leukopenia and the incidence of grade 3/4 infection were similar across the regimens (Table 1).
The Groupe d’Etude des Lymphomes de l’Adulte intergroup (GELA) study,44 compared RCHOP- 14 with R-CHOP-21 in DLBCL patients 60–80 years of age. Results from a 24-month interim analysis showed similar efficacy for R-CHOP-14 and R-CHOP-21 (2-year EFS of 48% vs 61%; P = not significant [NS]). Typically, trials of dose-dense regimens are evaluated with CSF support for all patients1,24; however, in the GELA study, patients received CSF at the physician’s discretion. Even though CSF use was higher with R-CHOP-14 than with R-CHOP-21 (90% vs 66%; Table 1), more patients in the R-CHOP-14 than in the R-CHOP-21 arm experienced grade 3/4 hematologic toxicity and FN (percentages were not reported).
Follicular lymphoma
Follicular lymphoma (FL) is usually diagnosed at an advanced stage and is incurable with current therapy.1 As shown in Table 1, current regimens for treating FL, including rituximab- and bendamustine-based regimens, are associated with neutropenic toxicity.
Rituximab-based treatment/consolidation regimens: The NCCN recommends R-CHOP and rituximab, cyclophosphamide, vincristine, and prednisone (R-CVP) for treating FL.1 A randomized phase III study by the German Low-Grade Lymphoma Study Group (GLSG) showed the superiority of first-line R-CHOP compared with CHOP in patients with untreated advanced FL.45 R-CHOP reduced the relative risk of treatment failure by 60% (28 of 223 patients vs 61 of 205 patients; P < 0.001), improved the overall response rate (ORR; 96% vs 90%; P = 0.011), and improved OS (6 deaths vs 17 deaths within the first 3 years; P = 0.016). Severe neutropenia was the most common treatment-related adverse event and occurred more often with R-CHOP than with CHOP (63% vs 53%; P = 0.01; Table 1).45 However, the incidence of severe infections was similar in the two groups (5% vs 7%; P = NS). Details of CSF use in this study were not reported.
A randomized phase III study in treatment-naive patients with advanced FL compared R-CVP with CVP.46 This study demonstrated that R-CVP significantly improved the ORR (81% vs 57%; P = 0.001), significantly prolonged the time to treatment failure (TTF; 27 months vs 7 months; P < 0.0001), and more than doubled the time to disease progression (TTP; 32 months vs 15 months; P < 0.001).46 The incidence of grade 3/4 neutropenia was higher with RCVP than with CVP (24% vs 14%), but the rates of infection and neutropenic sepsis were similar in the two treatment arms (Table 1).46 Details of CSF use were not provided in this report.
Rituximab-based maintenance regimens: Recent studies, including trials in frontline and relapsed settings, have demonstrated the benefits of rituximab maintenance after induction chemotherapy in patients with lymphoma.47–50
Two studies, one in the United States and one in Europe, randomized patients with relapsed/refractory FL to receive induction therapy with R-CHOP or CHOP; then those with a compete response (CR) or a partial response (PR) were randomized to receive rituximab maintenance (375 mg/m2 intravenously once every 3 months for up to 2 years) or no further treatment (observation group).48 Rituximab maintenance improved progression-free survival (PFS; 51.5 months vs 15.0 months; P < 0.001) and the 3-year OS rate (85% vs 77%; P = 0.011). The PFS benefit of rituximab maintenance was confirmed at a median follow-up of 6 years (3.7 years vs 1.3 years; P < 0.001; hazard ratio [HR] = 0.55), but the 5-year OS was not significantly different between the groups (74% vs 64%; P = 0.07).49 During the maintenance period, the frequency of grade 3/4 neutropenia and grade 3/4 infection was higher with rituximab than with no treatment: 12% vs 6% and 9% vs 2% (P = 0.009), respectively (Table 1).48,49 Details of CSF use during induction or maintenance therapy were not provided in the report.
A study by the GLSG group compared rituximab maintenance with no treatment following salvage therapy for patients with refractory or recurrent FL or mantle cell lymphoma.47 The maintenance regimen consisted of two courses of rituximab (4 doses of 375 mg/m2/day for 4 consecutive weeks) administered 3 months and 9 months after patients achieved a CR or a PR to induction chemotherapy with fludarabine, cyclophosphamide, and mitoxantrone (FCM) alone or in combination with rituximab (FCM-R). Rituximab maintenance significantly improved the response duration; the median response duration had not been reached in the rituximab arm vs an estimated median of 16 months in the observation arm (P < 0.001). During the maintenance period, grade 3/4 neutropenia was more common in the rituximab arm than in the observation arm (13% vs 6%; P = NS), but the incidence of grade 3/4 infection was similar in the two treatment arms (4% vs 3%; Table 1).47 Details of CSF use in both the induction and maintenance periods were not provided.
In the first-line setting, a randomized phase III study by the Eastern Cooperative Oncology Group (ECOG) evaluated the benefits of rituximab maintenance in patients with FL or small lymphocytic lymphoma following CVP treatment.50 Four weeks after the last CVP cycle, patients with responding or stable disease were randomized to receive rituximab (375 mg/m2 once per week for 4 weeks every 6 months for 2 years) or observation. Rituximab maintenance improved the 3-year PFS (68% vs 33%; HR = 0.4; P < 0.0001) and the 3-year OS (92% vs 86%; HR = 0.6; P = 0.05). During maintenance therapy, grade 3 neutropenia and grade 3 infection rates appeared to be similar in the two treatment groups (Table 1).50 Secondary CSF prophylaxis was permitted during induction chemotherapy in response to neutropenic events but not specified for the maintenance phase.
The Primary Rituximab and Maintenance (PRIMA) trial conducted by the GELA group evaluated the benefits of rituximab maintenance in previously untreated patients with indolent NHL.51 Patients who responded to one of three immunochemotherapy regimens (R-CHOP, R-CVP, or FCM with rituximab) were randomized to receive rituximab (375 mg/m2 given once every 8 weeks for 2 years) or observation. At a median followup of 2 years, maintenance rituximab significantly improved PFS (75% vs 58%; HR = 0.55; P < 0.0001). More patients in the rituximab arm than in the observation arm experienced grade 2 or higher infections (39% vs 24%), grade 3/4 infections (4% vs 1%), and grade 3/4 neutropenia (4% vs 1%). Rates of grade 3/4 FN were similar between treatment arms (< 1%); the definition of FN used in the trial was not provided.51 Details on CSF use during induction and maintenance therapies were not reported in the publication.
Ital Bendamustine-based regimens: Bendamustine, a novel bifunctional alkylating agent, was recently approved by the US Food and Drug Administration (FDA) to treat indolent NHL that has progressed after rituximab treatment.52 In a pivotal multicenter, open-label, single-arm trial, bendamustine (120 mg/m2) was administered to rituximab-refractory patients on days 1 and 2 every 21 days for 6–8 cycles.15 This study is included here because bendamustine has become an important component of regimens for the management of FL (either as monotherapy or in combination with other agents). In this study, the ORR was 74% (95% confidence interval [CI], 65%–83%), and the duration of response was 9.2 months (95% CI, 7.1–10.8 months), based on a median follow-up of 11.4 months. In 38 patients who had no objective response to their latest chemotherapy regimen, the ORR was 64%, and the median PFS was 7.5 months.
Primary CSF prophylaxis was not allowed in this study. Secondary CSF use was permitted if patients had grade 4 neutropenia that lasted at least 1 week, persistent leukopenia (grade > 2) at the next scheduled dose, or FN in any treatment cycle.15 The incidence of neutropenic complications was high (grade 3/4 neutropenia, 61%; grade 3/4 FN, 6%; and grade 3/4 infection, 21%). These findings demonstrate that when administered at the approved dose of 120 mg/m2 in the absence of primary CSF prophylaxis, bendamustine is associated with a high risk of neutropenic toxicity.
A randomized phase III trial compared bendamustine (90 mg/m2) plus rituximab (BR) with R-CHOP in patients with previously untreated indolent NHL.53 After a median observation period of 32 months, the BR regimen improved the CR rate (40% vs 31%; P = 0.03), PFS (55 vs 35 months; P = 0.0002), EFS (54 months vs 31 months; P = 0.0002), and time to next treatment (not reached vs 41 months; P = 0.0002). The rate of grade 3/4 neutropenia and number of infectious complications were significantly lower with the BR regimen than with R-CHOP: 11% vs 47% (P < 0.001) and 95 vs 121 (P < 0.04), respectively. 53 CSF was administered at the discretion of the treating physician and was used less frequently with the BR regimen than with R-CHOP (4% vs 20%).
Neutropenic toxicity associated with current treatment regimens for CLL
The NCCN recommends chemotherapy, primarily combinations containing alkylating agents and chemoimmunotherapy, as the standard of care for advanced CLL.1 Monotherapy or combination regimens with an alkylating agent or purine analog are preferred first-line therapies for elderly patients (≥ 70 years of age) and for frail patients with significant comorbidity. However, a more aggressive approach with rituximab-containing chemoimmunotherapy regimens is recommended for patients < 70 years old and for older patients with no significant comorbidities.1
Chemotherapy regimens
Two large randomized controlled trials4,5 showed that FC compared with fludarabine alone increased ORR, CR, and PFS in patients with CLL. The neutropenic toxicity of these regimens appeared similar in both studies. In Flinn et al,5 rates of grade 3/4 neutropenia, grade 3/4 FN, and grade 3–5 infection with grade 3/4 FN were similar (Table 1). CSF use was higher in the FC arm than in the fludarabine arm; however, CSF use was required in the FC arm only and not in the fludarabine arm. In Catovsky et al,4 rates of grade 3/4 neutropenia and all febrile episodes were similar (Table 1). In this study, CSF support was used according to local guidelines; however, the proportion of patients who required CSF support in the different treatment arms was not reported.
Chemoimmunotherapy regimens
In two large randomized controlled trials, FCR improved survival in patients with CLL compared with FC alone.11,12 In the CLL8 trial in chemotherapy-naive patients with advanced CLL,12 FCR was more efficacious than FC, as measured by CR rate (44% vs 22%; P < 0.001), PFS (52 vs 33 months; P < 0.001), and OS at 38 months (84% vs 79%; P = 0.01). The median OS had not been reached in either treatment arm at the time these data were published in abstract form. Hematologic adverse events, including neutropenia, were more common with FCR (percentages not reported) than with FC, but the infection rates were similar in the two treatment arms (Table 1).12 CSF use in this study was not reported.
In the REACH study, which compared FCR and FC in previously treated patients with CLL,11 FCR improved PFS (median, 31 months vs 21 months; HR = 0.65; P < 0.001) at a median follow-up of 25 months. Rates of grade 3/4 neutropenia and grade 3/4 infection were similar in the two groups (Table 1). In this study, 58% of patients in the FCR arm and 49% in the FC arm received CSF, administered at the discretion of the investigator.
Other chemoimmunotherapy regimens for CLL recommended by the NCCN include pentostatin, cyclophosphamide, and rituximab; and oxaliplatin, fludarabine, cytarabine, and rituximab.1 This recommendation was made on the basis of safety and efficacy results from nonrandomized trials.
Alemtuzumab-based regimens
In 2001, the FDA approved alemtuzumab to treat patients with CLL who had failed to respond to prior fludarabine-containing chemotherapy. 54 In an open-label, randomized controlled trial comparing alemtuzumab with chlorambucil (Leukeran) in previously untreated patients with CLL, alemtuzumab improved the ORR (83% vs 55%; P < 0.0001), PFS (15 vs 12 months; P < 0.0001), CR (24% vs 2%; P < 0.0001), and time to next treatment (23 vs 15 months; P < 0.0001).18 Grade 3/4 neutropenia was significantly more common with alemtuzumab than with chlorambucil (Table 1), but the rates of FN and serious infections were low in both treatment arms. In that study, CSF was administered to more than twice as many patients receiving alemtuzumab as receiving chlorambucil (Table 1)18; however, no further details were provided. Alemtuzumab-fludarabine and alemtuzumab with or without rituximab are regimens also recommended by the NCCN for relapsed or refractory CLL based on the results of nonrandomized trials.1
Bendamustine-based regimens
Bendamustine is recommended by the NCCN as a single agent for firstline therapy and as a single agent or in combination with rituximab for second-line therapy in patients with CLL.1 An open-label, multicenter, randomized phase III study compared bendamustine (100 mg/m2 on days 1–2 of each 28-day cycle) with chlorambucil in patients with untreated advanced CLL.16 Bendamustine significantly improved PFS (22 vs 8 months; P < 0.0001) and CR or PR (68% vs 31%; P < 0.0001). Grade 3/4 neutropenia occurred in twice as many bendamustine-treated patients as chlorambucil-treated patients (Table 1). The authors of this study report that even though the use of hematopoietic growth factors was discouraged in this study, CSF was administered in the bendamustine arm at the discretion of the treating investigator (Table 1).16
Bendamustine in combination with rituximab is also recommended for relapsed CLL.1 In a phase II study, patients with CLL were treated with bendamustine (70 mg/m2 on days 1 and 2 of each 28-day cycle) and rituximab (375 mg/m2 for the first cycle and 500 mg/m2 for subsequent cycles). 55 This single-arm study is included here because bendamustine is an important component of regimens for treating CLL. After a mean of 4.5 cycles, the ORR was 77%. Myelosup pression and infections were the most frequent severe adverse events reported, with grade 3/4 leukopenia or neutropenia observed in 12% of patients. Grade 3 or greater infections were documented in 5% of patients, and infection-related mortality occurred in 4% of patients. CSF use was not documented in this article.
Ofatumumab
Ofatumumab (Arzerra), a human monoclonal antibody directed against CD20, was recently approved by the FDA for the treatment of CLL refractory to fludarabine and alemtuzumab. 56 The NCCN recommends ofatumumab for relapsed or refractory disease.1 The registrational trial was a nonrandomized phase II study that evaluated safety and efficacy of ofatumumab in patients with fludarabineand alemtuzumab-refractory CLL (group A) and in patients with fludarabine- refractory CLL who were not candidates for alemtuzumab treatment because of bulky lymphadenopathy (group B).57 The study is included here because ofatumumab is a relatively new treatment option available to patients who fail to respond to other therapies. A planned interim analysis demonstrated benefits with ofatumumab in the two treatment groups (ORR, 58% and 47%; duration of response, 7.1 months and 5.6 months; PFS, 5.7 months and 5.9 months; and OS, 13.7 months and 15.4 months, respectively). 57 Grade 3/4 neutropenia was 14% in group A and 6% in group B; grade 3/4 infection was 12% and 8%, respectively. Of the 189 infectious events (all grades) with onset during treatment reported in this study, 13 (7%) were fatal. No information about CSF use was provided.
Neutropenic toxicity associated with current treatment regimens for HLThe NCCN recommends doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD); Stanford V; and escalated-dose BEACOPP for the treatment of HL. ABVD was introduced in the 1990s, and Stanford V and BEACOPP were introduced in the early 2000s.8,58–61 These regimens are known to be highly myelotoxic.
For the ABVD regimen, an 18% rate of severe neutropenia was reported in one study,61 and a 57% rate of grade 3/4 neutropenia was reported in another study.58 With the Stanford V regimen, the incidence of grade 4 neutropenia and FN was as high as 82% and 14%, respectively.60 It should be noted that despite the high level of myelosuppression associated with regimens for HL, the NCCN does not recommend the routine use of CSF because neutropenia is not considered a major factor for dose reductions or dose delays.1
Trials have compared the ABVD and Stanford V regimens in patients with HL. One trial in patients with advanced disease demonstrated comparable efficacy of the two regimens.6 However, another trial in patients with intermediate- and advancedstage disease demonstrated the superiority of ABVD combined with optional limited radiotherapy over the Stanford V regimen, as measured by response rate and PFS.7 Both studies reported comparable neutropenic toxicity of the ABVD and Stanford V regimens when secondary CSF prophylaxis was permitted (Table 1).6,7
The BEACOPP regimen, which incorporates chemotherapy dose intensification and frequent scheduling, has been shown to improve patient outcomes in advanced disease.8 A relatively recent trial directly compared ABVD vs BEACOPP (four escalated-dose schedules followed by two standard-dose schedules) vs cyclophosphamide, lomustine, vindesine, melphalan, prednisone, epidoxirubicin, vincristine, procarbazine, vinblastine, and bleomycin (CEC).62 At a median follow-up of 41 months, BEACOPP compared with ABVD significantly improved the 5-year PFS (81% vs 68%; P = 0.038) but showed no significant differences with CEC. Both the BEACOPP and CEC regimens were associated with higher rates of grade 3/4 neutropenia than ABVD; BEACOPP was also associated with higher rates of severe infections than ABVD and CEC (Table 1).62 Daily CSF was incorporated into the BEACOPP regimen and administered for at least 8 days, until an absolute neutrophil count of 500/ mm3 was reached.62 Routine CSF prophylaxis was not required with the ABVD and CEC regimens but was used at the discretion of the treating physician.
Neutropenic toxicity associated with current treatment regimens for multiple myeloma
A variety of regimens that incorporate the novel agents bortezomib (Velcade), lenalidomide (Revlimid), or thalidomide (Thalomid) have been evaluated for the treatment of multiple myeloma. These agents directly target the myeloma cells and can also interfere with the interaction of tumor cells with the bone marrow microenvironment. 63 The NCCN recommends these agents as components of combination regimens for induction chemotherapy (whether or not stem cell transplantation is indicated), as maintenance treatment after transplantation, or as salvage therapy for patients with multiple myeloma.1
Bortezomib-based regimens
Bortezomib, a member of a new class of drugs called proteasome inhibitors, is FDA approved to treat multiple myeloma.64 Patients with previously untreated myeloma are treated with bortezomib in combination with melphalan and prednisone (MPB). Results from the Velcade as Initial Standard Therapy in Multiple Myeloma trial compared MPB wit melphalan and prednisone (MP) in patients who were ineligible for transplant therapy.13,14 At a median follow-up of 37 months, MPB reduced the risk of death by 35% (HR, 0.653; P < 0.001) and improved the 3-year OS (69% vs 54%).13 The incidence of grade 3/4 neutropenia was comparable for MPB and MP (40% vs 38%; Table 1), suggesting that the MP component of the regimen is primarily responsible for the neutropenic toxicity. Information on CSF use in this study was not provided. The APEX trial compared bortezomib with high-dose dexamethasone as salvage therapy in patients with recurrent myeloma.65,66 At a median follow-up of 22 months, bortezomib significantly improved the ORR (43% vs 18%; P < 0.0001) and the 1-year survival rates (80% vs 67%; P = 0.00002).66 Bortezomib was associated with a higher incidence of grade 3/4 neutropenia than was highdose dexamethasone (14% vs 1%; P < 0.01). However, the incidence of grade 3/4 infections was similar between the arms (13% vs 16%; P = 0.19).65 CSF use was permitted at the physician’s discretion; however, details were not provided.
Bortezomib in combination with pegylated liposomal doxorubicin (Doxil; B + PLD) is FDA approved for salvage therapy for multiple myeloma, with a category 1 recommendation from the NCCN. Interim data from a randomized phase III study67 demonstrated the superiority of B + PLD to bortezomib monotherapy (TTP, 9.3 months vs 6.5 months; P < 0.0001; PFS, 9.0 months vs 6.5 months; P < 0.0001; duration of response, 10 months vs 7 months; P < 0.001; and 15-month OS rates, 76% vs 65%; P = 0.03). Grade 3/4 neutropenia was significantly more common with the combination regimen; however, the rate of FN was similar (Table 1).67 CSF use was allowed in this study, but details were not provided.
Lenalidomide-based regimens
Lenalidomide is an immunomodulatory agent that is FDA approved for use in combination with dexamethasone to treat patients with multiple myeloma who have received at least one prior therapy.68 Lenalidomide is taken orally once daily on days 1–21 of 28-day cycles as a part of the lenalidomide-dexamethasone regimen.68
A phase III trial conducted in the US and Canada69 and a companion trial conducted in Europe, Israel, and Australia70 compared the lenalidomide- dexamethasone regimen with placebo-dexamethasone in patients with refractory or recurrent myeloma. In both trials, lenalidomidedexamethasone significantly improved the ORR, TTP, and OS.69,70 In both studies, neutropenic toxicity (including grade 3/4 neutropenia, FN, or grade 3/4 infection) was higher in the lenalidomide-dexamethasone arm than in the dexamethasone alone arm (Table 1).
Secondary CSF prophylaxis in response to neutropenic toxicity was permitted in both studies. In the Weber at al study,69 60 of the 177 patients (33.9%) in the lenalidomide- dexamethasone group received CSF support; 28 of the 60 patients (46.7%) received CSF to maintain the full lenalidomide dose, and 12 of these 28 patients (43%) were able to continue at the 25-mg dose level. In the Dimopoulos et al study,70 38 of 176 patients (22%) in the lenalidomide- dexamethasone group received CSF support; 23 of these patients (61%) needed CSF to maintain the lenalidomide dose, and 12 (52%) were able to continue on 25 mg of lenalidomide.
A recent trial evaluated lenalidomide- dexamethasone as initial therapy for patients with newly diagnosed multiple myeloma.71 In this open-label study with a noninferiority design, lenalidomide plus low-dose dexamethasone was compared with lenalidomide plus high-dose dexamethasone. The trial was stopped early because of the superior survival results with the low-dose dexamethasone regimen at a 1-year interim analysis (OS, 96% vs 87%; P = 0.0002). The NCCN now recommends lenalidomide with low-dose dexamethasone for previously untreated patients who are not candidates for transplant therapy.1 The low-dose dexamethasone regimen was associated with fewer infections than the high-dose dexamethasome regimen (9% vs 16%; P = 0.04), even though it was associated with a higher incidence of grade 3/4 neutropenia (20% vs 12%; P = 0.02). Details of CSF use were not reported for this study.
Thalidomide-based regimens
Thalidomide is also an immunomodulator that is FDA approved for use in combination with dexamethasone to treat patients with newly diagnosed multiple myeloma. FDA approval of this regimen was supported by results from the Eastern Cooperative Oncology Group (ECOG) study, which compared thalidomidedexamethasone with dexamethasone alone.72 The response rate with thalidomide- dexamethasone was significantly higher than with dexamethasone alone (63% vs 41%; P = 0.017). The incidence of neutropenia and infection was similar between the arms (Table 1).72 Details of CSF use in this study were not provided.
Thalidomide in combination with MP (MPT) is recommended by the NCCN as a primary induction therapy for transplant-ineligible myeloma patients. The Intergroup Francophone du Myélome 01/01 Trial of MPT in patients with untreated multiple myeloma compared MPT with MP-placebo.73 MPT improved OS (44 vs 29 months; P = 0.03) and PFS (24 vs 18.5 months; P = 0.001), at a median follow-up of 47.5 months. Grade 3/4 neutropenia was significantly more common with MPT, but the incidence of severe infection was similar in the two treatment arms (Table 1). CSF use was permitted in this study; however, details were not provided.
Of note, unlike conventional chemotherapeutic agents, novel agents used to treat multiple myeloma are not administered in 14- or 21-day cycles. For example, bortezomib is initially administered twice-weekly (with rest periods) followed by weekly dosing as a component of the MPB regimen.13,14 Lenalidomide is taken orally once daily on days 1–21 of 28-day cycles as part of the lenalidomide-dexamethasone regimen. 69,70 Similarly, thalidomide is administered daily as an oral tablet.72 Furthermore, although clinical trials have integrated CSF use, no studies specifically address it with these novel agents (ie, whether CSF should be given concurrently or sequentially with the therapy). Therefore, clinical trials evaluating the safety of CSF use with these novel agents are warranted.
Quantitative analysis of underreporting of neutropenic toxicity
As previously discussed, most reports of trials evaluating therapies for treating hematologic malignancies include information about the frequency of severe neutropenia. However, our literature review showed that data on the incidence of FN and the use of CSF are frequently not provided. The omission of this information limits the comparison of results across trials and the ability to make informed decisions on the true risk of FN for a treatment modality. The objective of this quantitative analysis was to evaluate the reporting of FN and other neutropenic outcomes, as well as related CSF or antibiotic use, in randomized controlled trials that evaluated regimens for the treatment of NHL, CLL, HL, or multiple myeloma.
Selection criteria for articles included For this quantitative analysis, phase III trials published between January 2005 and June 2009 were identified from the original list of trials retrieved through the comprehensive literature search, as previously discussed. We included phase III trials only for this analysis, because most are designed to capture both safety and efficacy associated with a treatment modality, compared with phase II trials, which may sometimes primarily focus on safety parameters. We also included all articles that met the specified criteria, whether or not the treatment regimen reported in the article was recommended by the NCCN.
Articles that met the inclusion criteria were retrieved and data on myelotoxic outcomes were abstracted by two reviewers and reconciled by a third reviewer. The neutropenic outcomes included were grade 3/4 neutropenia or granulocytopenia, FN, leukopenia, all-cause hospitalization, neutropenia-related hospitalization, infection or sepsis, and infection-related mortality. Outcomes on chemotherapy delivery included dose delays, dose reductions, and dose intensity or relative dose intensity. We also collected data on CSF use defined in the methods section, CSF use presented in the results section, and antibiotic use defined in the methods and/or results section.
Results
Table 2 summarizes our findings on the reporting of neutropenic toxicity outcomes. Of the 57 trials that met the inclusion criteria, 86% reported results of at least one neutropenic endpoint. Across tumor types, 68% of trials reported on the incidence of grade 3/4 neutropenia (80%, multiple myeloma; 71%, CLL; 63%, NHL, 50%, HL). However, a few trials (19%) reported on the incidence of FN (57%, CLL; 20%, multiple myeloma; 12%, NHL). Similarly, only a few trials (4%) reported on neutropenia- related hospitalizations (8%, NHL). The incidence of infection or sepsis and infection-related mortality was reported in 79% and 60% of publications, respectively. Dose delays/interruptions were reported in 21% of trials overall. Dose reductions were reported in 30% of articles overall.
Data on the reporting of CSF and antibiotic use are shown in Table 3. About half (49%) of the publications reported planned use of CSF in the methods section (71%, CLL; 67%, HL; 50%, NHL; 35%, multiple myeloma). However, overall, only 25% of publications reported CSF use in the results section (43%, CLL; 29%, NHL; 17%, HL; 15%, multiple myeloma). Overall reporting on prophylactic antibiotic use was also low. Antibiotic use was discussed in the methods sections of only 21% of papers (71%, CLL; 17%, HL; 15%, multiple myeloma; 13%, NHL), and actual use of antibiotics was not reported in the results section of any of the publications.
Discussion
Our review shows that many phase III trials of current treatment regimens for hematologic malignancies omit important outcome data on the incidence of FN, neutropenia-related hospitalization, infection-related mortality, chemotherapy dose delays/ interruptions or dose reductions, use of primary or secondary CSF prophylaxis, or use of antibiotics. These findings are similar to recent observations by others.
For instance, Duff and colleagues40 reported that publications describing results from phase III trials fail to consistently report details that would enable clinicians in the community to translate findings to clinical practice. When these researchers asked medical oncologists and oncology pharmacists to identify the most important information necessary for clinical application of an oncology drug, 3 of the 10 most common responses were premedication, growth factor support, and dose adjustments for hematologic toxicity.
The researchers then reviewed 262 articles published in five journals (Blood, Cancer, the Journal of Clinical Oncology, the Journal of the National Cancer Institute, and the New England Journal of Medicine) between 2005 and 2008. They found that each of these elements (premedication, growth factor support, and dose adjustments for hematologic toxicity) was reported fewer than half the time (P < 0.0001) compared with the name of the drug, which was reported 100% of the time. Duff and colleagues40 recommend that journal editors require reporting of these and other highly ranked elements in the article or in an online appendix and provide Internet- open access to the clinical trial protocol.
Dale and colleagues39 examined 58 reports on NHL therapy trials published between 1990 and 2000. They found that 34% did not include data on neutropenic toxicity and 3% included only details on clinical consequences, such as fatal infection. In the other trials, hematologic toxicity was reported 18 different ways. These authors recommend that certain details about hematologic toxicity should routinely be documented in reports on cancer chemotherapy: rates of leukopenia and neutropenia; the timing of blood cell counts used to determine these rates; protocols for antibiotics and CSF use; actual use of antibiotics and CSF; rates of all infectious complications, including hospitalizations and bacteremias; and relative dose intensity. 39
Conclusion
In addition to efficacy data, reports on clinical trials should provide details on the toxicity of treatment and requirements for supportive care. A standardized approach to collecting and reporting neutropenic outcomes and the related use of supportive care measures can assist clinicians in prospectively managing the relevant toxicities associated with treatment regimens for hematologic malignancies. This information is essential for the safe and effective transition of these regimens into broad clinical practice. These data should include all grade 3 or greater hematologic and nonhematologic toxicities in phase II, III, or IV clinical trials, as well as details on prophylactic and interventional CSF and antibiotic use. Armed with knowledge of the risk of neutropenic toxicity associated with each treatment regimen, oncologists can then focus on the patient-related risks when making decisions regarding appropriate supportive care. Mitigation of neutropenic toxicity associated with treatment regimens is important to decrease patients’ risk for treatment delays/interruptions, dose reductions, or discontinuations, which can compromise patient outcomes.19–22
Acknowledgments
Amgen sponsored an external agency for data abstraction and analysis. The authors thank Beverly A. Caley and Leta Shy for data abstraction; Supriya Srinivasan for data reconciliation; and Supriya Srinivasan and Martha Mutomba for writing assistance. The sponsor played a role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the manuscript for publication. The corresponding author had full access to all data and had final responsibility for the decision to submit the article for publication. All authors provided comments during manuscript development and have approved the final version of the submitted article.
Conflicts of interest
Dr. Gregory has served as a consultant or in an advisory role with Amgen Inc, Genentech (Roche), Novartis, and Spectrum Pharmaceuticals; and her institution has received research funding from Astellas, Celgene, Cephalon, Genentech (Roche), GlaxoSmithKline, Immunomedics, NCIC–CTG, and Novartis. Dr. Abella is an employee and stock owner of Amgen Inc. Dr. Moore has served as a consultant or in an advisory role with Amgen Inc and is on the speakers’ bureaus of Amgen Inc, sanofi-aventis, and GlaxoSmithKline
References
1. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology, 2010. http://www.nccn.org/professionals/ physician_gls/f_guidelines.asp. Accessed December 17, 2010.
2. Pfreundschuh M, Trümper L, Kloess M, et al. Two-weekly or 3-weekly CHOP chemotherapy with or without etoposide for the treatment of elderly patients with aggressive lymphomas: results of the NHL-B2 trial of the DSHNHL. Blood 2004;104:634–641.
3. Wunderlich A, Kloess M, Reiser M, et al. Practicability and acute haematological toxicity of 2- and 3-weekly CHOP and CHOEP chemotherapy for aggressive non-Hodgkin’s lymphoma: results from the NHL-B trial of the German High-Grade Non-Hodgkin’s Lymphoma Study Group (DSHNHL). Ann Oncol 2003;14:881–893.
4. Catovsky D, Richards S, Matutes E, et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 2007;370:230–239.
5. Flinn IW, Neuberg DS, Grever MR, et al. Phase III trial of fludarabine plus cyclophosphamide compared with fludarabine for patients with previously untreated chronic lymphocytic leukemia: US Intergroup Trial E2997. J Clin Oncol 2007;25:793–798.
6. Hoskin PJ, Lowry L, Horwich A, et al. Randomized comparison of the stanford V regimen and ABVD in the treatment of advanced Hodgkin’s Lymphoma: United Kingdom National Cancer Research Institute Lymphoma Group Study ISRC TN 64141244. J Clin Oncol 2009;27:5390–5396.
7. Gobbi PG, Levis A, Chisesi T, et al. ABVD versus modified Stanford V versus MOPPEBVCAD with optional and limited radiotherapy in intermediate- and advanced- stage Hodgkin’s lymphoma: final results of a multicenter randomized trial by the Intergruppo Italiano Linfomi. J Clin Oncol 2005;23:9198–9207.
8. Diehl V, Franklin J, Pfreundschuh M, et al. Standard and increased-dose BEACOPP chemotherapy compared with COPP-ABVD for advanced Hodgkin’s disease. N Engl J Med 2003;348:2386–2395.
9. Michallet AS, Coiffier B. Recent developments in the treatment of aggressive non- Hodgkin lymphoma. Blood Rev 2009;23:11– 23.
10. Pfreundschuh M, Trümper L, Osterborg A, et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol 2006;7:379–391.
11. Robak T, Dmoszynska A, Solal-Céligny P, et al. Rituximab plus fludarabine and cyclophosphamide prolongs progression-free survival compared with fludarabine and cyclophosphamide alone in previously treated chronic lymphocytic leukemia. J Clin Oncol 2010;28:1756–1765.
12. Hallek M, Fingerle-Rowson G, Fink AM, et al. First-line treatment with fludarabine (F), cyclophosphamide (C), and rituximab (R) (FCR) improves overall survival (OS) in previously untreated patients (pts) with advanced chronic lymphocytic leukemia (CLL): results of a randomized phase III trial on behalf of an international group of investigators and the German CLL Study Group. Blood 2009;114:535.
13. Mateos MV, Oriol A, Martinez-López J, et al. Bortezomib, melphalan, and prednisone versus bortezomib, thalidomide, and prednisone as induction therapy followed by maintenance treatment with bortezomib and thalidomide versus bortezomib and prednisone in elderly patients with untreated multiple myeloma: a randomised trial. Lancet Oncol 2010;11:934–941.
14. San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med 2008;359:906–917.
15. Kahl BS, Bartlett NL, Leonard JP, et al. Bendamustine is effective therapy in patients with rituximab-refractory, indolent B-cell non- Hodgkin lymphoma: results from a Multicenter Study. Cancer 2010;116:106–114.
16. Knauf WU, Lissichkov T, Aldaoud A, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:4378–4384.
17. Fischer K, Cramer P, Stilgenbauer S, et al. Bendamustine combined with rituximab (BR) in first-line therapy of advanced CLL: a multicenter phase II trial of the German CLL Study Group (GCLLSG). Blood 2009;114:205.
18. Hillmen P, Skotnicki AB, Robak T, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol 2007;25:5616–5623
19. Mayordomo JI, López A, Viñolas N, et al. Retrospective cost analysis of management of febrile neutropenia in cancer patients in Spain. Curr Med Res Opin 2009;25:2533–2542.
20. Pettengell R, Schwenkglenks M, Leonard R, et al. Neutropenia occurrence and predictors of reduced chemotherapy delivery: results from the INC-EU prospective observational European neutropenia study. Support Care Cancer 2008;16:1299–1309.
21. Lyman GH, Kleiner JM. Summary and comparison of myeloid growth factor guidelines in patients receiving cancer chemotherapy. J Natl Compr Canc Netw 2007;5:217–228.
22. Smith TJ, Khatcheressian J, Lyman GH, et al. 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol 2006;24:3187–3205.
23. Crawford J, Caserta C, Roila F; ESMO Guidelines Working Group. Hematopoietic growth factors: ESMO Clinical Practice Guidelines for the applications. Ann Oncol 2010;21(suppl 5):v248–v251.
24. Aapro MS, Cameron DA, Pettengell R, et al. EORTC guidelines for the use of granulocyte- colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphomas and solid tumours. Eur J Cancer 2006;42:2433–2453.
25. Crawford J, Ozer H, Stoller R, et al. Reduction by granulocyte colony-stimulating factor of fever and neutropenia induced by chemotherapy in patients with small-cell lung cancer. N Engl J Med 1991;325:164–170.
26. Gabrilove JL, Jakubowski A, Scher H, et al. Effect of granulocyte colony-stimulating factor on neutropenia and associated morbidity due to chemotherapy for transitional-cell carcinoma of the urothelium. N Engl J Med 1988;318:1414–1422.
27. Grigg A, Solal-Celigny P, Hoskin P, et al. Open-label, randomized study of pegfilgrastim vs. daily filgrastim as an adjunct to chemotherapy in elderly patients with non-Hodgkin’s lymphoma. Leuk Lymphoma 2003;44:1503–1508.
28. Holmes FA, Jones SE, O’Shaughnessy J, et al. Comparable efficacy and safety profiles of once-per-cycle pegfilgrastim and daily injection filgrastim in chemotherapy-induced neutropenia: a multicenter dose-finding study in women with breast cancer. Ann Oncol 2002;13:903–909.
29. Holmes FA, O’Shaughnessy JA, Vukelja S, et al. Blinded, randomized, multicenter study to evaluate single administration pegfilgrastim once per cycle versus daily filgrastim as an adjunct to chemotherapy in patients with highrisk stage II or stage III/IV breast cancer. J Clin Oncol 2002;20:727–731.
30. Johnston E, Crawford J, Blackwell S, et al. Randomized, dose-escalation study of SD/01 compared with daily filgrastim in patients receiving chemotherapy. J Clin Oncol 2000;18:2522–2528.
31. Trillet-Lenoir V, Green J, Manegold C, et al. Recombinant granulocyte colony stimulating factor reduces the infectious complications of cytotoxic chemotherapy. Eur J Cancer 1993;29A:319–324.
32. Vogel CL, Wojtukiewicz MZ, Carroll RR, et al. First and subsequent cycle use of pegfilgrastim prevents febrile neutropenia in patients with breast cancer: a multicenter, double- blind, placebo-controlled phase III study. J Clin Oncol 2005;23:1178–1184.
33. Green MD, Koelbl H, Baselga J, et al. A randomized double-blind multicenter phase III study of fixed-dose single-administration pegfilgrastim versus daily filgrastim in patients receiving myelosuppressive chemotherapy. Ann Oncol 2003;14:29–35.
34. Vose JM, Crump M, Lazarus H, et al. Randomized, multicenter, open-label study of pegfilgrastim compared with daily filgrastim after chemotherapy for lymphoma. J Clin Oncol 2003;21:514–519.
35. Kuderer NM, Dale DC, Crawford J, Lyman GH. Impact of primary prophylaxis with granulocyte colony-stimulating factor on febrile neutropenia and mortality in adult cancer patients receiving chemotherapy: a systematic review. J Clin Oncol 2007;25:3158–3167.
36. Lyman GH, Michels SL, Reynolds MW, Barron R, Tomic KS, Yu J. Risk of mortality in patients with cancer who experience febrile neutropenia. Cancer 2010;116:5555– 5563.
37. Lyman GH, Dale DC, Wolff DA, et al. Acute myeloid leukemia or myelodysplastic syndrome in randomized controlled clinical trials of cancer chemotherapy with granulocyte colony-stimulating factor: a systematic review. J Clin Oncol 2010;28:2914–2924.
38. Bohlius J, Reiser M, Schwarzer G, Engert A. Granulopoiesis-stimulating factors to prevent adverse effects in the treatment of malignant lymphoma. Cochrane Database Syst Rev 2004:CD003189.
39. Dale DC, McCarter GC, Crawford J, Lyman GH. Myelotoxicity and dose intensity of chemotherapy: reporting practices from randomized clinical trials. J Natl Compr Canc Netw 2003;1:440–454.
40. Duff JM, Leather H, Walden EO, LaPlant KD, George TJ Jr. Adequacy of published oncology randomized controlled trials to provide therapeutic details needed for clinical application. J Natl Cancer Inst 2010;102:702– 705.
41. Freedman OC, Zimmermann C, Clemons MJ: Interpreting the results of clinical trials of cancer chemotherapy: the importance of reporting concurrent supportive care. Proceedings from 31st Annual San Antonio Breast Cancer Symposium; December 14, 2008; San Antonio, TX. Abstract 6138.
42. Coiffier B, Lepage E, Briere J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235–242.
43. Pfreundschuh M, Schubert J, Ziepert M, et al. Six versus eight cycles of bi-weekly CHOP-14 with or without rituximab in elderly patients with aggressive CD20+ B-cell lymphomas: a randomised controlled trial (RICOVER-60). Lancet Oncol 2008;9:105– 116.
44. Delarue R, Tilly H, Salles G, et al. RCHOP14 compared to R-CHOP21 in elderly patients with diffuse large B-cell lymphoma: results of the interim analysis of the LNH03- 6B GELA study. Blood 2009;114:406.
45. Hiddemann W, Kneba M, Dreyling M, et al. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 2005;106:3725–3732.
46. Marcus R, Imrie K, Belch A, et al. CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood 2005;105:1417–1423.
47. Forstpointner R, Unterhalt M, Dreyling M, et al. Maintenance therapy with rituximab leads to a significant prolongation of response duration after salvage therapy with a combination of rituximab, fludarabine, cyclophosphamide, and mitoxantrone (R-FCM) in patients with recurring and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German Low Grade Lymphoma Study Group (GLSG). Blood 2006;108:4003–4008.
48. van Oers MH, Klasa R, Marcus RE, et al. Rituximab maintenance improves clinical outcome of relapsed/resistant follicular non- Hodgkin lymphoma in patients both with and without rituximab during induction: results of a prospective randomized phase 3 intergroup trial. Blood 2006;108:3295-3301.
49. van Oers MH, Van Glabbeke M, Giurgea L, et al. Rituximab maintenance treatment of relapsed/resistant follicular non-Hodgkin’s lymphoma: long-term outcome of the EORTC 20981 phase III randomized intergroup study. J Clin Oncol 2010;28:2853–2858.
50. Hochster H, Weller E, Gascoyne RD, et al. Maintenance rituximab after cyclophosphamide, vincristine, and prednisone prolongs progression-free survival in advanced indolent lymphoma: results of the randomized phase III ECOG1496 Study. J Clin Oncol 2009;27:1607–1614.
51. Salles GA, Seymour JF, Feugier P, et al. Rituximab maintenance for 2 years in patients with untreated high tumor burden follicular lymphoma after response to immunochemotherapy. J Clin Oncol 2010;28[15S]:8004.
52. Treanda [prescribing information]. Frazer, PA: Cephalon, Inc.; 2010.
53. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab is superior in respect of progression free survival and CR rate when compared to CHOP plus ritux imab as first-line treatment of patients with advanced follicular, indolent, and mantle cell lymphomas: final results of a randomized phase III study of the StiL (Study Group Indolent Lymphomas, Germany). Blood 2009;114:405.
54. Demko S, Summers J, Keegan P, Pazdur R. FDA drug approval summary: alemtuzumab as single-agent treatment for B-cell chronic lymphocytic leukemia. Oncologist 2008;13:167–174.
55. Fischer K, Stilgenbauer S, Schweighofer CD, et al. Bendamustine in combination with rituximab (BR) for patients with relapsed chronic lymphocytic leukemia (CLL): a multicentre phase II trial of the German CLL Study Group (GCLLSG). Blood 2008;112:330.
56. Arzerra [prescribing information]. Research Triangle Park, NC: GlaxoSmithKline; 2010.
57. Wierda WG, Kipps TJ, Mayer J, et al. Ofatumumab as single-agent CD20 immunotherapy in fludarabine-refractory chronic lymphocytic leukemia. J Clin Oncol 2010;28:1749– 1755.
58. Straus DJ, Portlock CS, Qin J, et al. Results of a prospective randomized clinical trial of doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) followed by radiation therapy (RT) versus ABVD alone for stages I, II, and IIIA nonbulky Hodgkin disease. Blood 2004;104:3483–3489.
59. Duggan DB, Petroni GR, Johnson JL, et al. Randomized comparison of ABVD and MOPP/ABV hybrid for the treatment of advanced Hodgkin’s disease: report of an intergroup trial. J Clin Oncol 2003;21:607–614.
60. Horning SJ, Hoppe RT, Breslin S, Bartlett NL, Brown BW, Rosenberg SA. Stanford V and radiotherapy for locally extensive and advanced Hodgkin’s disease: mature results of a prospective clinical trial. J Clin Oncol 2002;20:630–637.
61. Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin’s disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med 1992;327:1478–1484.
62. Federico M, Luminari S, Iannitto E, et al. ABVD compared with BEACOPP compared with CEC for the initial treatment of patients with advanced Hodgkin’s lymphoma: results from the HD2000 Gruppo Italiano per lo Studio dei Linfomi Trial. J Clin Oncol 2009;27:805–811.
63. Hideshima T, Bergsagel PL, Kuehl WM, Anderson KC. Advances in biology of multiple myeloma: clinical applications. Blood 2004;104:607–618.
64. Thalomid [prescribing information]. Summit, NJ: Celgene Corporation; 2009.
65. Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005;352:2487–2498.
66. Richardson PG, Sonneveld P, Schuster M, et al. Extended follow-up of a phase 3 trial in relapsed multiple myeloma: final timeto- event results of the APEX trial. Blood 2007;110:3557–3560.
67. Orlowski RZ, Nagler A, Sonneveld P, et al. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. J Clin Oncol 2007;25:3892–3901.
68. Revlimid [prescribing information]. Summit, NJ: Celgene Corporation; 2009.
69. Weber DM, Chen C, Niesvizky R, et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med 2007;357:2133–2142.
70. Dimopoulos M, Spencer A, Attal M, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med 2007;357:2123–2132.
71. Rajkumar SV, Jacobus S, Callander NS, et al. Lenalidomide plus high-dose dexamethasone versus lenalidomide plus low-dose dexamethasone as initial therapy for newly diagnosed multiple myeloma: an open-label randomised controlled trial. Lancet Oncol 2010;11:29–37.
72. Rajkumar SV, Blood E, Vesole D, Fonseca R, Greipp PR. Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: a clinical trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol 2006;24:431–436.
73. Hulin C, Facon T, Rodon P, et al. Efficacy of melphalan and prednisone plus thalidomide in patients older than 75 years with newly diagnosed multiple myeloma: IFM 01/01 trial. J Clin Oncol 2009;27:3664–3670.
Stephanie A. Gregory, MD,1 Steve Abella, MD,2 and Tim Moore, MD3
1 Section of Hematology, Rush University Medical Center, Chicago, IL; 2 Global Clinical Development, Hematology/Oncology, Amgen Inc., Thousand Oaks, CA; and 3 Zangmeister Center, Columbus, OH
Most chemotherapy regimens considered standard of care for treating hematologic malignancies are myelosuppressive. They include chemotherapy regimens recommended by the National Comprehensive Cancer Network (NCCN),1 such as cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) to treat non-Hodgkin lymphoma (NHL) 2,3; fludarabine plus cyclophosphamide (FC) to treat chronic lymphocytic leukemia (CLL)4,5; and escalated-dose bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone (BEACOPP) or doxorubicin, vinblastine, mechlorethamine, etoposide, vincristine, bleomycin, and prednisone (Stanford V) to treat Hodgkin lymphoma (HL).6–8
Emerging regimens that incorporate targeted therapies or other novel agents (eg, rituximab [Rituxan], lenalidomide [Revlimid], or bendamustine [Treanda]) have also been shown to be myelosuppressive, mainly because they are generally combined with myelosuppressive chemotherapy to achieve optimal efficacy. Examples include CHOP plus rituximab (R-CHOP) to treat NHL9,10; FC plus rituximab (FCR) to treat CLL11,12; or bortezomib plus melphalan-prednisone (MPB) to treat multiple myeloma.13,14 Additionally, some agents show toxicity when used as monotherapies, including bendamustine15–17 and alemtuzumab (Campath) 18 to treat CLL. Therefore, improved clinical outcomes may be achieved with concurrent increased myelosuppression.
Patients receiving myelosuppresive chemotherapy are at risk for developing chemotherapy-induced neutropenia, including severe or prolonged neutropenia and febrile neutropenia (FN). This condition often leads to treatment delays/interruptions, dose reductions, or treatment discontinuations, which can result in suboptimal treatment delivery and compromised patient outcomes.19–22 Colony-stimulating factor (CSF) has thus become an important component of many current treatment regimens for hematologic malignancies. International clinical guidelines, including those from the NCCN,1 the American Society of Clinical Oncology (ASCO),22 the European Society for Medical Oncology (ESMO),23 and the European Organization for Research and Treatment of Cancer (EORTC),24 recommend CSF use when the risk of FN is ≥ 20% and consideration of CSF use when the risk of FN is between 10% and 20%.
Numerous studies have demonstrated CSF effectiveness in decreasing the incidence of severe neutropenia and/or FN.25–34 A meta-analysis of 17 randomized controlled trials, which enrolled 3,493 cancer patients receiving chemotherapy, demonstrated that primary prophylaxis with CSF was associated with a decreased incidence of FN and reduced rates of infection-related mortality and early mortality across different tumor types.35 The occurrence of FN was associated with a 35% increase in the hazard of early mortality, and prophylactic granulocyte (G)-CSF use decreased this number by 45%.36 In a separate analysis of 25 trials (total n = 12,804), CSF support in cancer patients receiving chemotherapy was associated with a significant increase in overall survival (OS).37 Furthermore, a meta-analysis of results from 12 randomized controlled trials, which enrolled 1,823 patients with malignant lymphoma, showed that CSF prophylaxis, compared with no prophylaxis, significantly reduced the relative risk of severe neutropenia, FN, and infection.38
Evidence-based data that could guide the use of CSF in the setting of current treatment regimens for hematologic malignancies are not always readily available. Publications that report clinical trial results focus on overall efficacy and safety parameters of treatment regimens and often do not report the incidence or severity of neutropenia and/or FN.39 Similarly, these publications often do not include information on supportive care measures, including prophylaxis with antibiotics and/or CSF (primary or secondary).40,41 Also, when CSF support is reported, often the agent and dosing schedule are not provided. Many trials permit the use of CSF at the investigator’s discretion; however, the proportion of patients treated or supported with CSF and related outcomes is often not reported. These gaps in reporting neutropenic toxicity and related outcomes may result in an underestimation of the degree of significant toxicity associated with current treatment regimens for hematologic malignancies.
We conducted a comprehensive review of English-language reports published after January 2005. From the retrieved list of publications, we identified studies reporting data from trials (including phase II and III) that evaluated regimens considered NCCN Guideline recommendations for treating selected hematologic malignancies. 1 We excluded trials that enrolled patients with acute leukemia or chronic myelogenous leukemia; trials with the primary objective of assessing radiotherapy, radioimmunotherapy, stem cell transplantation, or patient-reported outcomes; and trials that described the study design but not the results. If multiple publications reported results of the same trial, we selected the publication with the most complete data on hematologic toxicity. Publications that met the inclusion criteria were retrieved and reviewed for neutropenic toxicity outcomes and the reported use of CSF or antibiotics.
Neutropenic toxicity associated with current treatment regimens for NHL
Diffuse large B-cell lymphoma
Diffuse large B-cell lymphoma (DLBCL) is an aggressive type of lymphoma generally treated with curative intent in the frontline setting. Beginning in the 1970s, the standard of care for DLBCL was CHOP, administered every 21 days (CHOP- 21).9 However, approximately half of patients > 60 years of age do not benefit from this regimen. In a study by Coiffier et al,42 3-year OS in this patient population was less than 40%. The addition of rituximab to CHOP-21 (R-CHOP-21) or CHOP-21–like regimens was subsequently shown to improve OS significantly across patient populations, with no increased neutropenic toxicity (Table 1).10 The R-CHOP regimen is now considered the standard of care for DLBCL when the goal of treatment is cure.9Another randomized study by Pfreundschuh et al compared dose-dense CHOP (given every 14 days, CHOP-14) with CHOP-21 in NHL patients ≥ 60 years of age.2 The CHOP-14 dosedense regimen required support with primary prophylactic CSF in all cycles (CHOP-14-G), whereas prophylactic CSF use with CHOP-21 was at the discretion of the treating physician, based on patient characteristics. CHOP-14-G significantly improved event-free survival (EFS) and OS. Grade 4 neutropenia was less frequent with CHOP-14-G than with CHOP-21 (24% vs 44%; P < 0.001), demonstrating that CSF support could adequately protect patients from neutropenic toxicity associated with CHOP.2
The RICOVER-60 study43 evaluated 6 or 8 cycles of dose-dense CHOP (CHOP-14-G) with or without rituximab in patients 61– 80 years of age who had aggressive B-cell lymphoma and were receiving primary prophylaxis with CSF (R-CHOP-14-G vs CHOP-14-G). R-CHOP-14-G significantly improved EFS (66.5% vs 47.2%) and OS (78.1% vs 67.7%). Leukopenia was the most common grade 3/4 toxicity, with grade 4 events occurring in 48%–52% across treatment arms. However, the incidence of leukopenia and the incidence of grade 3/4 infection were similar across the regimens (Table 1).
The Groupe d’Etude des Lymphomes de l’Adulte intergroup (GELA) study,44 compared RCHOP- 14 with R-CHOP-21 in DLBCL patients 60–80 years of age. Results from a 24-month interim analysis showed similar efficacy for R-CHOP-14 and R-CHOP-21 (2-year EFS of 48% vs 61%; P = not significant [NS]). Typically, trials of dose-dense regimens are evaluated with CSF support for all patients1,24; however, in the GELA study, patients received CSF at the physician’s discretion. Even though CSF use was higher with R-CHOP-14 than with R-CHOP-21 (90% vs 66%; Table 1), more patients in the R-CHOP-14 than in the R-CHOP-21 arm experienced grade 3/4 hematologic toxicity and FN (percentages were not reported).
Follicular lymphoma
Follicular lymphoma (FL) is usually diagnosed at an advanced stage and is incurable with current therapy.1 As shown in Table 1, current regimens for treating FL, including rituximab- and bendamustine-based regimens, are associated with neutropenic toxicity.
Rituximab-based treatment/consolidation regimens: The NCCN recommends R-CHOP and rituximab, cyclophosphamide, vincristine, and prednisone (R-CVP) for treating FL.1 A randomized phase III study by the German Low-Grade Lymphoma Study Group (GLSG) showed the superiority of first-line R-CHOP compared with CHOP in patients with untreated advanced FL.45 R-CHOP reduced the relative risk of treatment failure by 60% (28 of 223 patients vs 61 of 205 patients; P < 0.001), improved the overall response rate (ORR; 96% vs 90%; P = 0.011), and improved OS (6 deaths vs 17 deaths within the first 3 years; P = 0.016). Severe neutropenia was the most common treatment-related adverse event and occurred more often with R-CHOP than with CHOP (63% vs 53%; P = 0.01; Table 1).45 However, the incidence of severe infections was similar in the two groups (5% vs 7%; P = NS). Details of CSF use in this study were not reported.
A randomized phase III study in treatment-naive patients with advanced FL compared R-CVP with CVP.46 This study demonstrated that R-CVP significantly improved the ORR (81% vs 57%; P = 0.001), significantly prolonged the time to treatment failure (TTF; 27 months vs 7 months; P < 0.0001), and more than doubled the time to disease progression (TTP; 32 months vs 15 months; P < 0.001).46 The incidence of grade 3/4 neutropenia was higher with RCVP than with CVP (24% vs 14%), but the rates of infection and neutropenic sepsis were similar in the two treatment arms (Table 1).46 Details of CSF use were not provided in this report.
Rituximab-based maintenance regimens: Recent studies, including trials in frontline and relapsed settings, have demonstrated the benefits of rituximab maintenance after induction chemotherapy in patients with lymphoma.47–50
Two studies, one in the United States and one in Europe, randomized patients with relapsed/refractory FL to receive induction therapy with R-CHOP or CHOP; then those with a compete response (CR) or a partial response (PR) were randomized to receive rituximab maintenance (375 mg/m2 intravenously once every 3 months for up to 2 years) or no further treatment (observation group).48 Rituximab maintenance improved progression-free survival (PFS; 51.5 months vs 15.0 months; P < 0.001) and the 3-year OS rate (85% vs 77%; P = 0.011). The PFS benefit of rituximab maintenance was confirmed at a median follow-up of 6 years (3.7 years vs 1.3 years; P < 0.001; hazard ratio [HR] = 0.55), but the 5-year OS was not significantly different between the groups (74% vs 64%; P = 0.07).49 During the maintenance period, the frequency of grade 3/4 neutropenia and grade 3/4 infection was higher with rituximab than with no treatment: 12% vs 6% and 9% vs 2% (P = 0.009), respectively (Table 1).48,49 Details of CSF use during induction or maintenance therapy were not provided in the report.
A study by the GLSG group compared rituximab maintenance with no treatment following salvage therapy for patients with refractory or recurrent FL or mantle cell lymphoma.47 The maintenance regimen consisted of two courses of rituximab (4 doses of 375 mg/m2/day for 4 consecutive weeks) administered 3 months and 9 months after patients achieved a CR or a PR to induction chemotherapy with fludarabine, cyclophosphamide, and mitoxantrone (FCM) alone or in combination with rituximab (FCM-R). Rituximab maintenance significantly improved the response duration; the median response duration had not been reached in the rituximab arm vs an estimated median of 16 months in the observation arm (P < 0.001). During the maintenance period, grade 3/4 neutropenia was more common in the rituximab arm than in the observation arm (13% vs 6%; P = NS), but the incidence of grade 3/4 infection was similar in the two treatment arms (4% vs 3%; Table 1).47 Details of CSF use in both the induction and maintenance periods were not provided.
In the first-line setting, a randomized phase III study by the Eastern Cooperative Oncology Group (ECOG) evaluated the benefits of rituximab maintenance in patients with FL or small lymphocytic lymphoma following CVP treatment.50 Four weeks after the last CVP cycle, patients with responding or stable disease were randomized to receive rituximab (375 mg/m2 once per week for 4 weeks every 6 months for 2 years) or observation. Rituximab maintenance improved the 3-year PFS (68% vs 33%; HR = 0.4; P < 0.0001) and the 3-year OS (92% vs 86%; HR = 0.6; P = 0.05). During maintenance therapy, grade 3 neutropenia and grade 3 infection rates appeared to be similar in the two treatment groups (Table 1).50 Secondary CSF prophylaxis was permitted during induction chemotherapy in response to neutropenic events but not specified for the maintenance phase.
The Primary Rituximab and Maintenance (PRIMA) trial conducted by the GELA group evaluated the benefits of rituximab maintenance in previously untreated patients with indolent NHL.51 Patients who responded to one of three immunochemotherapy regimens (R-CHOP, R-CVP, or FCM with rituximab) were randomized to receive rituximab (375 mg/m2 given once every 8 weeks for 2 years) or observation. At a median followup of 2 years, maintenance rituximab significantly improved PFS (75% vs 58%; HR = 0.55; P < 0.0001). More patients in the rituximab arm than in the observation arm experienced grade 2 or higher infections (39% vs 24%), grade 3/4 infections (4% vs 1%), and grade 3/4 neutropenia (4% vs 1%). Rates of grade 3/4 FN were similar between treatment arms (< 1%); the definition of FN used in the trial was not provided.51 Details on CSF use during induction and maintenance therapies were not reported in the publication.
Ital Bendamustine-based regimens: Bendamustine, a novel bifunctional alkylating agent, was recently approved by the US Food and Drug Administration (FDA) to treat indolent NHL that has progressed after rituximab treatment.52 In a pivotal multicenter, open-label, single-arm trial, bendamustine (120 mg/m2) was administered to rituximab-refractory patients on days 1 and 2 every 21 days for 6–8 cycles.15 This study is included here because bendamustine has become an important component of regimens for the management of FL (either as monotherapy or in combination with other agents). In this study, the ORR was 74% (95% confidence interval [CI], 65%–83%), and the duration of response was 9.2 months (95% CI, 7.1–10.8 months), based on a median follow-up of 11.4 months. In 38 patients who had no objective response to their latest chemotherapy regimen, the ORR was 64%, and the median PFS was 7.5 months.
Primary CSF prophylaxis was not allowed in this study. Secondary CSF use was permitted if patients had grade 4 neutropenia that lasted at least 1 week, persistent leukopenia (grade > 2) at the next scheduled dose, or FN in any treatment cycle.15 The incidence of neutropenic complications was high (grade 3/4 neutropenia, 61%; grade 3/4 FN, 6%; and grade 3/4 infection, 21%). These findings demonstrate that when administered at the approved dose of 120 mg/m2 in the absence of primary CSF prophylaxis, bendamustine is associated with a high risk of neutropenic toxicity.
A randomized phase III trial compared bendamustine (90 mg/m2) plus rituximab (BR) with R-CHOP in patients with previously untreated indolent NHL.53 After a median observation period of 32 months, the BR regimen improved the CR rate (40% vs 31%; P = 0.03), PFS (55 vs 35 months; P = 0.0002), EFS (54 months vs 31 months; P = 0.0002), and time to next treatment (not reached vs 41 months; P = 0.0002). The rate of grade 3/4 neutropenia and number of infectious complications were significantly lower with the BR regimen than with R-CHOP: 11% vs 47% (P < 0.001) and 95 vs 121 (P < 0.04), respectively. 53 CSF was administered at the discretion of the treating physician and was used less frequently with the BR regimen than with R-CHOP (4% vs 20%).
Neutropenic toxicity associated with current treatment regimens for CLL
The NCCN recommends chemotherapy, primarily combinations containing alkylating agents and chemoimmunotherapy, as the standard of care for advanced CLL.1 Monotherapy or combination regimens with an alkylating agent or purine analog are preferred first-line therapies for elderly patients (≥ 70 years of age) and for frail patients with significant comorbidity. However, a more aggressive approach with rituximab-containing chemoimmunotherapy regimens is recommended for patients < 70 years old and for older patients with no significant comorbidities.1
Chemotherapy regimens
Two large randomized controlled trials4,5 showed that FC compared with fludarabine alone increased ORR, CR, and PFS in patients with CLL. The neutropenic toxicity of these regimens appeared similar in both studies. In Flinn et al,5 rates of grade 3/4 neutropenia, grade 3/4 FN, and grade 3–5 infection with grade 3/4 FN were similar (Table 1). CSF use was higher in the FC arm than in the fludarabine arm; however, CSF use was required in the FC arm only and not in the fludarabine arm. In Catovsky et al,4 rates of grade 3/4 neutropenia and all febrile episodes were similar (Table 1). In this study, CSF support was used according to local guidelines; however, the proportion of patients who required CSF support in the different treatment arms was not reported.
Chemoimmunotherapy regimens
In two large randomized controlled trials, FCR improved survival in patients with CLL compared with FC alone.11,12 In the CLL8 trial in chemotherapy-naive patients with advanced CLL,12 FCR was more efficacious than FC, as measured by CR rate (44% vs 22%; P < 0.001), PFS (52 vs 33 months; P < 0.001), and OS at 38 months (84% vs 79%; P = 0.01). The median OS had not been reached in either treatment arm at the time these data were published in abstract form. Hematologic adverse events, including neutropenia, were more common with FCR (percentages not reported) than with FC, but the infection rates were similar in the two treatment arms (Table 1).12 CSF use in this study was not reported.
In the REACH study, which compared FCR and FC in previously treated patients with CLL,11 FCR improved PFS (median, 31 months vs 21 months; HR = 0.65; P < 0.001) at a median follow-up of 25 months. Rates of grade 3/4 neutropenia and grade 3/4 infection were similar in the two groups (Table 1). In this study, 58% of patients in the FCR arm and 49% in the FC arm received CSF, administered at the discretion of the investigator.
Other chemoimmunotherapy regimens for CLL recommended by the NCCN include pentostatin, cyclophosphamide, and rituximab; and oxaliplatin, fludarabine, cytarabine, and rituximab.1 This recommendation was made on the basis of safety and efficacy results from nonrandomized trials.
Alemtuzumab-based regimens
In 2001, the FDA approved alemtuzumab to treat patients with CLL who had failed to respond to prior fludarabine-containing chemotherapy. 54 In an open-label, randomized controlled trial comparing alemtuzumab with chlorambucil (Leukeran) in previously untreated patients with CLL, alemtuzumab improved the ORR (83% vs 55%; P < 0.0001), PFS (15 vs 12 months; P < 0.0001), CR (24% vs 2%; P < 0.0001), and time to next treatment (23 vs 15 months; P < 0.0001).18 Grade 3/4 neutropenia was significantly more common with alemtuzumab than with chlorambucil (Table 1), but the rates of FN and serious infections were low in both treatment arms. In that study, CSF was administered to more than twice as many patients receiving alemtuzumab as receiving chlorambucil (Table 1)18; however, no further details were provided. Alemtuzumab-fludarabine and alemtuzumab with or without rituximab are regimens also recommended by the NCCN for relapsed or refractory CLL based on the results of nonrandomized trials.1
Bendamustine-based regimens
Bendamustine is recommended by the NCCN as a single agent for firstline therapy and as a single agent or in combination with rituximab for second-line therapy in patients with CLL.1 An open-label, multicenter, randomized phase III study compared bendamustine (100 mg/m2 on days 1–2 of each 28-day cycle) with chlorambucil in patients with untreated advanced CLL.16 Bendamustine significantly improved PFS (22 vs 8 months; P < 0.0001) and CR or PR (68% vs 31%; P < 0.0001). Grade 3/4 neutropenia occurred in twice as many bendamustine-treated patients as chlorambucil-treated patients (Table 1). The authors of this study report that even though the use of hematopoietic growth factors was discouraged in this study, CSF was administered in the bendamustine arm at the discretion of the treating investigator (Table 1).16
Bendamustine in combination with rituximab is also recommended for relapsed CLL.1 In a phase II study, patients with CLL were treated with bendamustine (70 mg/m2 on days 1 and 2 of each 28-day cycle) and rituximab (375 mg/m2 for the first cycle and 500 mg/m2 for subsequent cycles). 55 This single-arm study is included here because bendamustine is an important component of regimens for treating CLL. After a mean of 4.5 cycles, the ORR was 77%. Myelosup pression and infections were the most frequent severe adverse events reported, with grade 3/4 leukopenia or neutropenia observed in 12% of patients. Grade 3 or greater infections were documented in 5% of patients, and infection-related mortality occurred in 4% of patients. CSF use was not documented in this article.
Ofatumumab
Ofatumumab (Arzerra), a human monoclonal antibody directed against CD20, was recently approved by the FDA for the treatment of CLL refractory to fludarabine and alemtuzumab. 56 The NCCN recommends ofatumumab for relapsed or refractory disease.1 The registrational trial was a nonrandomized phase II study that evaluated safety and efficacy of ofatumumab in patients with fludarabineand alemtuzumab-refractory CLL (group A) and in patients with fludarabine- refractory CLL who were not candidates for alemtuzumab treatment because of bulky lymphadenopathy (group B).57 The study is included here because ofatumumab is a relatively new treatment option available to patients who fail to respond to other therapies. A planned interim analysis demonstrated benefits with ofatumumab in the two treatment groups (ORR, 58% and 47%; duration of response, 7.1 months and 5.6 months; PFS, 5.7 months and 5.9 months; and OS, 13.7 months and 15.4 months, respectively). 57 Grade 3/4 neutropenia was 14% in group A and 6% in group B; grade 3/4 infection was 12% and 8%, respectively. Of the 189 infectious events (all grades) with onset during treatment reported in this study, 13 (7%) were fatal. No information about CSF use was provided.
Neutropenic toxicity associated with current treatment regimens for HLThe NCCN recommends doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD); Stanford V; and escalated-dose BEACOPP for the treatment of HL. ABVD was introduced in the 1990s, and Stanford V and BEACOPP were introduced in the early 2000s.8,58–61 These regimens are known to be highly myelotoxic.
For the ABVD regimen, an 18% rate of severe neutropenia was reported in one study,61 and a 57% rate of grade 3/4 neutropenia was reported in another study.58 With the Stanford V regimen, the incidence of grade 4 neutropenia and FN was as high as 82% and 14%, respectively.60 It should be noted that despite the high level of myelosuppression associated with regimens for HL, the NCCN does not recommend the routine use of CSF because neutropenia is not considered a major factor for dose reductions or dose delays.1
Trials have compared the ABVD and Stanford V regimens in patients with HL. One trial in patients with advanced disease demonstrated comparable efficacy of the two regimens.6 However, another trial in patients with intermediate- and advancedstage disease demonstrated the superiority of ABVD combined with optional limited radiotherapy over the Stanford V regimen, as measured by response rate and PFS.7 Both studies reported comparable neutropenic toxicity of the ABVD and Stanford V regimens when secondary CSF prophylaxis was permitted (Table 1).6,7
The BEACOPP regimen, which incorporates chemotherapy dose intensification and frequent scheduling, has been shown to improve patient outcomes in advanced disease.8 A relatively recent trial directly compared ABVD vs BEACOPP (four escalated-dose schedules followed by two standard-dose schedules) vs cyclophosphamide, lomustine, vindesine, melphalan, prednisone, epidoxirubicin, vincristine, procarbazine, vinblastine, and bleomycin (CEC).62 At a median follow-up of 41 months, BEACOPP compared with ABVD significantly improved the 5-year PFS (81% vs 68%; P = 0.038) but showed no significant differences with CEC. Both the BEACOPP and CEC regimens were associated with higher rates of grade 3/4 neutropenia than ABVD; BEACOPP was also associated with higher rates of severe infections than ABVD and CEC (Table 1).62 Daily CSF was incorporated into the BEACOPP regimen and administered for at least 8 days, until an absolute neutrophil count of 500/ mm3 was reached.62 Routine CSF prophylaxis was not required with the ABVD and CEC regimens but was used at the discretion of the treating physician.
Neutropenic toxicity associated with current treatment regimens for multiple myeloma
A variety of regimens that incorporate the novel agents bortezomib (Velcade), lenalidomide (Revlimid), or thalidomide (Thalomid) have been evaluated for the treatment of multiple myeloma. These agents directly target the myeloma cells and can also interfere with the interaction of tumor cells with the bone marrow microenvironment. 63 The NCCN recommends these agents as components of combination regimens for induction chemotherapy (whether or not stem cell transplantation is indicated), as maintenance treatment after transplantation, or as salvage therapy for patients with multiple myeloma.1
Bortezomib-based regimens
Bortezomib, a member of a new class of drugs called proteasome inhibitors, is FDA approved to treat multiple myeloma.64 Patients with previously untreated myeloma are treated with bortezomib in combination with melphalan and prednisone (MPB). Results from the Velcade as Initial Standard Therapy in Multiple Myeloma trial compared MPB wit melphalan and prednisone (MP) in patients who were ineligible for transplant therapy.13,14 At a median follow-up of 37 months, MPB reduced the risk of death by 35% (HR, 0.653; P < 0.001) and improved the 3-year OS (69% vs 54%).13 The incidence of grade 3/4 neutropenia was comparable for MPB and MP (40% vs 38%; Table 1), suggesting that the MP component of the regimen is primarily responsible for the neutropenic toxicity. Information on CSF use in this study was not provided. The APEX trial compared bortezomib with high-dose dexamethasone as salvage therapy in patients with recurrent myeloma.65,66 At a median follow-up of 22 months, bortezomib significantly improved the ORR (43% vs 18%; P < 0.0001) and the 1-year survival rates (80% vs 67%; P = 0.00002).66 Bortezomib was associated with a higher incidence of grade 3/4 neutropenia than was highdose dexamethasone (14% vs 1%; P < 0.01). However, the incidence of grade 3/4 infections was similar between the arms (13% vs 16%; P = 0.19).65 CSF use was permitted at the physician’s discretion; however, details were not provided.
Bortezomib in combination with pegylated liposomal doxorubicin (Doxil; B + PLD) is FDA approved for salvage therapy for multiple myeloma, with a category 1 recommendation from the NCCN. Interim data from a randomized phase III study67 demonstrated the superiority of B + PLD to bortezomib monotherapy (TTP, 9.3 months vs 6.5 months; P < 0.0001; PFS, 9.0 months vs 6.5 months; P < 0.0001; duration of response, 10 months vs 7 months; P < 0.001; and 15-month OS rates, 76% vs 65%; P = 0.03). Grade 3/4 neutropenia was significantly more common with the combination regimen; however, the rate of FN was similar (Table 1).67 CSF use was allowed in this study, but details were not provided.
Lenalidomide-based regimens
Lenalidomide is an immunomodulatory agent that is FDA approved for use in combination with dexamethasone to treat patients with multiple myeloma who have received at least one prior therapy.68 Lenalidomide is taken orally once daily on days 1–21 of 28-day cycles as a part of the lenalidomide-dexamethasone regimen.68
A phase III trial conducted in the US and Canada69 and a companion trial conducted in Europe, Israel, and Australia70 compared the lenalidomide- dexamethasone regimen with placebo-dexamethasone in patients with refractory or recurrent myeloma. In both trials, lenalidomidedexamethasone significantly improved the ORR, TTP, and OS.69,70 In both studies, neutropenic toxicity (including grade 3/4 neutropenia, FN, or grade 3/4 infection) was higher in the lenalidomide-dexamethasone arm than in the dexamethasone alone arm (Table 1).
Secondary CSF prophylaxis in response to neutropenic toxicity was permitted in both studies. In the Weber at al study,69 60 of the 177 patients (33.9%) in the lenalidomide- dexamethasone group received CSF support; 28 of the 60 patients (46.7%) received CSF to maintain the full lenalidomide dose, and 12 of these 28 patients (43%) were able to continue at the 25-mg dose level. In the Dimopoulos et al study,70 38 of 176 patients (22%) in the lenalidomide- dexamethasone group received CSF support; 23 of these patients (61%) needed CSF to maintain the lenalidomide dose, and 12 (52%) were able to continue on 25 mg of lenalidomide.
A recent trial evaluated lenalidomide- dexamethasone as initial therapy for patients with newly diagnosed multiple myeloma.71 In this open-label study with a noninferiority design, lenalidomide plus low-dose dexamethasone was compared with lenalidomide plus high-dose dexamethasone. The trial was stopped early because of the superior survival results with the low-dose dexamethasone regimen at a 1-year interim analysis (OS, 96% vs 87%; P = 0.0002). The NCCN now recommends lenalidomide with low-dose dexamethasone for previously untreated patients who are not candidates for transplant therapy.1 The low-dose dexamethasone regimen was associated with fewer infections than the high-dose dexamethasome regimen (9% vs 16%; P = 0.04), even though it was associated with a higher incidence of grade 3/4 neutropenia (20% vs 12%; P = 0.02). Details of CSF use were not reported for this study.
Thalidomide-based regimens
Thalidomide is also an immunomodulator that is FDA approved for use in combination with dexamethasone to treat patients with newly diagnosed multiple myeloma. FDA approval of this regimen was supported by results from the Eastern Cooperative Oncology Group (ECOG) study, which compared thalidomidedexamethasone with dexamethasone alone.72 The response rate with thalidomide- dexamethasone was significantly higher than with dexamethasone alone (63% vs 41%; P = 0.017). The incidence of neutropenia and infection was similar between the arms (Table 1).72 Details of CSF use in this study were not provided.
Thalidomide in combination with MP (MPT) is recommended by the NCCN as a primary induction therapy for transplant-ineligible myeloma patients. The Intergroup Francophone du Myélome 01/01 Trial of MPT in patients with untreated multiple myeloma compared MPT with MP-placebo.73 MPT improved OS (44 vs 29 months; P = 0.03) and PFS (24 vs 18.5 months; P = 0.001), at a median follow-up of 47.5 months. Grade 3/4 neutropenia was significantly more common with MPT, but the incidence of severe infection was similar in the two treatment arms (Table 1). CSF use was permitted in this study; however, details were not provided.
Of note, unlike conventional chemotherapeutic agents, novel agents used to treat multiple myeloma are not administered in 14- or 21-day cycles. For example, bortezomib is initially administered twice-weekly (with rest periods) followed by weekly dosing as a component of the MPB regimen.13,14 Lenalidomide is taken orally once daily on days 1–21 of 28-day cycles as part of the lenalidomide-dexamethasone regimen. 69,70 Similarly, thalidomide is administered daily as an oral tablet.72 Furthermore, although clinical trials have integrated CSF use, no studies specifically address it with these novel agents (ie, whether CSF should be given concurrently or sequentially with the therapy). Therefore, clinical trials evaluating the safety of CSF use with these novel agents are warranted.
Quantitative analysis of underreporting of neutropenic toxicity
As previously discussed, most reports of trials evaluating therapies for treating hematologic malignancies include information about the frequency of severe neutropenia. However, our literature review showed that data on the incidence of FN and the use of CSF are frequently not provided. The omission of this information limits the comparison of results across trials and the ability to make informed decisions on the true risk of FN for a treatment modality. The objective of this quantitative analysis was to evaluate the reporting of FN and other neutropenic outcomes, as well as related CSF or antibiotic use, in randomized controlled trials that evaluated regimens for the treatment of NHL, CLL, HL, or multiple myeloma.
Selection criteria for articles included For this quantitative analysis, phase III trials published between January 2005 and June 2009 were identified from the original list of trials retrieved through the comprehensive literature search, as previously discussed. We included phase III trials only for this analysis, because most are designed to capture both safety and efficacy associated with a treatment modality, compared with phase II trials, which may sometimes primarily focus on safety parameters. We also included all articles that met the specified criteria, whether or not the treatment regimen reported in the article was recommended by the NCCN.
Articles that met the inclusion criteria were retrieved and data on myelotoxic outcomes were abstracted by two reviewers and reconciled by a third reviewer. The neutropenic outcomes included were grade 3/4 neutropenia or granulocytopenia, FN, leukopenia, all-cause hospitalization, neutropenia-related hospitalization, infection or sepsis, and infection-related mortality. Outcomes on chemotherapy delivery included dose delays, dose reductions, and dose intensity or relative dose intensity. We also collected data on CSF use defined in the methods section, CSF use presented in the results section, and antibiotic use defined in the methods and/or results section.
Results
Table 2 summarizes our findings on the reporting of neutropenic toxicity outcomes. Of the 57 trials that met the inclusion criteria, 86% reported results of at least one neutropenic endpoint. Across tumor types, 68% of trials reported on the incidence of grade 3/4 neutropenia (80%, multiple myeloma; 71%, CLL; 63%, NHL, 50%, HL). However, a few trials (19%) reported on the incidence of FN (57%, CLL; 20%, multiple myeloma; 12%, NHL). Similarly, only a few trials (4%) reported on neutropenia- related hospitalizations (8%, NHL). The incidence of infection or sepsis and infection-related mortality was reported in 79% and 60% of publications, respectively. Dose delays/interruptions were reported in 21% of trials overall. Dose reductions were reported in 30% of articles overall.
Data on the reporting of CSF and antibiotic use are shown in Table 3. About half (49%) of the publications reported planned use of CSF in the methods section (71%, CLL; 67%, HL; 50%, NHL; 35%, multiple myeloma). However, overall, only 25% of publications reported CSF use in the results section (43%, CLL; 29%, NHL; 17%, HL; 15%, multiple myeloma). Overall reporting on prophylactic antibiotic use was also low. Antibiotic use was discussed in the methods sections of only 21% of papers (71%, CLL; 17%, HL; 15%, multiple myeloma; 13%, NHL), and actual use of antibiotics was not reported in the results section of any of the publications.
Discussion
Our review shows that many phase III trials of current treatment regimens for hematologic malignancies omit important outcome data on the incidence of FN, neutropenia-related hospitalization, infection-related mortality, chemotherapy dose delays/ interruptions or dose reductions, use of primary or secondary CSF prophylaxis, or use of antibiotics. These findings are similar to recent observations by others.
For instance, Duff and colleagues40 reported that publications describing results from phase III trials fail to consistently report details that would enable clinicians in the community to translate findings to clinical practice. When these researchers asked medical oncologists and oncology pharmacists to identify the most important information necessary for clinical application of an oncology drug, 3 of the 10 most common responses were premedication, growth factor support, and dose adjustments for hematologic toxicity.
The researchers then reviewed 262 articles published in five journals (Blood, Cancer, the Journal of Clinical Oncology, the Journal of the National Cancer Institute, and the New England Journal of Medicine) between 2005 and 2008. They found that each of these elements (premedication, growth factor support, and dose adjustments for hematologic toxicity) was reported fewer than half the time (P < 0.0001) compared with the name of the drug, which was reported 100% of the time. Duff and colleagues40 recommend that journal editors require reporting of these and other highly ranked elements in the article or in an online appendix and provide Internet- open access to the clinical trial protocol.
Dale and colleagues39 examined 58 reports on NHL therapy trials published between 1990 and 2000. They found that 34% did not include data on neutropenic toxicity and 3% included only details on clinical consequences, such as fatal infection. In the other trials, hematologic toxicity was reported 18 different ways. These authors recommend that certain details about hematologic toxicity should routinely be documented in reports on cancer chemotherapy: rates of leukopenia and neutropenia; the timing of blood cell counts used to determine these rates; protocols for antibiotics and CSF use; actual use of antibiotics and CSF; rates of all infectious complications, including hospitalizations and bacteremias; and relative dose intensity. 39
Conclusion
In addition to efficacy data, reports on clinical trials should provide details on the toxicity of treatment and requirements for supportive care. A standardized approach to collecting and reporting neutropenic outcomes and the related use of supportive care measures can assist clinicians in prospectively managing the relevant toxicities associated with treatment regimens for hematologic malignancies. This information is essential for the safe and effective transition of these regimens into broad clinical practice. These data should include all grade 3 or greater hematologic and nonhematologic toxicities in phase II, III, or IV clinical trials, as well as details on prophylactic and interventional CSF and antibiotic use. Armed with knowledge of the risk of neutropenic toxicity associated with each treatment regimen, oncologists can then focus on the patient-related risks when making decisions regarding appropriate supportive care. Mitigation of neutropenic toxicity associated with treatment regimens is important to decrease patients’ risk for treatment delays/interruptions, dose reductions, or discontinuations, which can compromise patient outcomes.19–22
Acknowledgments
Amgen sponsored an external agency for data abstraction and analysis. The authors thank Beverly A. Caley and Leta Shy for data abstraction; Supriya Srinivasan for data reconciliation; and Supriya Srinivasan and Martha Mutomba for writing assistance. The sponsor played a role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the manuscript for publication. The corresponding author had full access to all data and had final responsibility for the decision to submit the article for publication. All authors provided comments during manuscript development and have approved the final version of the submitted article.
Conflicts of interest
Dr. Gregory has served as a consultant or in an advisory role with Amgen Inc, Genentech (Roche), Novartis, and Spectrum Pharmaceuticals; and her institution has received research funding from Astellas, Celgene, Cephalon, Genentech (Roche), GlaxoSmithKline, Immunomedics, NCIC–CTG, and Novartis. Dr. Abella is an employee and stock owner of Amgen Inc. Dr. Moore has served as a consultant or in an advisory role with Amgen Inc and is on the speakers’ bureaus of Amgen Inc, sanofi-aventis, and GlaxoSmithKline
References
1. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology, 2010. http://www.nccn.org/professionals/ physician_gls/f_guidelines.asp. Accessed December 17, 2010.
2. Pfreundschuh M, Trümper L, Kloess M, et al. Two-weekly or 3-weekly CHOP chemotherapy with or without etoposide for the treatment of elderly patients with aggressive lymphomas: results of the NHL-B2 trial of the DSHNHL. Blood 2004;104:634–641.
3. Wunderlich A, Kloess M, Reiser M, et al. Practicability and acute haematological toxicity of 2- and 3-weekly CHOP and CHOEP chemotherapy for aggressive non-Hodgkin’s lymphoma: results from the NHL-B trial of the German High-Grade Non-Hodgkin’s Lymphoma Study Group (DSHNHL). Ann Oncol 2003;14:881–893.
4. Catovsky D, Richards S, Matutes E, et al. Assessment of fludarabine plus cyclophosphamide for patients with chronic lymphocytic leukaemia (the LRF CLL4 Trial): a randomised controlled trial. Lancet 2007;370:230–239.
5. Flinn IW, Neuberg DS, Grever MR, et al. Phase III trial of fludarabine plus cyclophosphamide compared with fludarabine for patients with previously untreated chronic lymphocytic leukemia: US Intergroup Trial E2997. J Clin Oncol 2007;25:793–798.
6. Hoskin PJ, Lowry L, Horwich A, et al. Randomized comparison of the stanford V regimen and ABVD in the treatment of advanced Hodgkin’s Lymphoma: United Kingdom National Cancer Research Institute Lymphoma Group Study ISRC TN 64141244. J Clin Oncol 2009;27:5390–5396.
7. Gobbi PG, Levis A, Chisesi T, et al. ABVD versus modified Stanford V versus MOPPEBVCAD with optional and limited radiotherapy in intermediate- and advanced- stage Hodgkin’s lymphoma: final results of a multicenter randomized trial by the Intergruppo Italiano Linfomi. J Clin Oncol 2005;23:9198–9207.
8. Diehl V, Franklin J, Pfreundschuh M, et al. Standard and increased-dose BEACOPP chemotherapy compared with COPP-ABVD for advanced Hodgkin’s disease. N Engl J Med 2003;348:2386–2395.
9. Michallet AS, Coiffier B. Recent developments in the treatment of aggressive non- Hodgkin lymphoma. Blood Rev 2009;23:11– 23.
10. Pfreundschuh M, Trümper L, Osterborg A, et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol 2006;7:379–391.
11. Robak T, Dmoszynska A, Solal-Céligny P, et al. Rituximab plus fludarabine and cyclophosphamide prolongs progression-free survival compared with fludarabine and cyclophosphamide alone in previously treated chronic lymphocytic leukemia. J Clin Oncol 2010;28:1756–1765.
12. Hallek M, Fingerle-Rowson G, Fink AM, et al. First-line treatment with fludarabine (F), cyclophosphamide (C), and rituximab (R) (FCR) improves overall survival (OS) in previously untreated patients (pts) with advanced chronic lymphocytic leukemia (CLL): results of a randomized phase III trial on behalf of an international group of investigators and the German CLL Study Group. Blood 2009;114:535.
13. Mateos MV, Oriol A, Martinez-López J, et al. Bortezomib, melphalan, and prednisone versus bortezomib, thalidomide, and prednisone as induction therapy followed by maintenance treatment with bortezomib and thalidomide versus bortezomib and prednisone in elderly patients with untreated multiple myeloma: a randomised trial. Lancet Oncol 2010;11:934–941.
14. San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med 2008;359:906–917.
15. Kahl BS, Bartlett NL, Leonard JP, et al. Bendamustine is effective therapy in patients with rituximab-refractory, indolent B-cell non- Hodgkin lymphoma: results from a Multicenter Study. Cancer 2010;116:106–114.
16. Knauf WU, Lissichkov T, Aldaoud A, et al. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:4378–4384.
17. Fischer K, Cramer P, Stilgenbauer S, et al. Bendamustine combined with rituximab (BR) in first-line therapy of advanced CLL: a multicenter phase II trial of the German CLL Study Group (GCLLSG). Blood 2009;114:205.
18. Hillmen P, Skotnicki AB, Robak T, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol 2007;25:5616–5623
19. Mayordomo JI, López A, Viñolas N, et al. Retrospective cost analysis of management of febrile neutropenia in cancer patients in Spain. Curr Med Res Opin 2009;25:2533–2542.
20. Pettengell R, Schwenkglenks M, Leonard R, et al. Neutropenia occurrence and predictors of reduced chemotherapy delivery: results from the INC-EU prospective observational European neutropenia study. Support Care Cancer 2008;16:1299–1309.
21. Lyman GH, Kleiner JM. Summary and comparison of myeloid growth factor guidelines in patients receiving cancer chemotherapy. J Natl Compr Canc Netw 2007;5:217–228.
22. Smith TJ, Khatcheressian J, Lyman GH, et al. 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol 2006;24:3187–3205.
23. Crawford J, Caserta C, Roila F; ESMO Guidelines Working Group. Hematopoietic growth factors: ESMO Clinical Practice Guidelines for the applications. Ann Oncol 2010;21(suppl 5):v248–v251.
24. Aapro MS, Cameron DA, Pettengell R, et al. EORTC guidelines for the use of granulocyte- colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphomas and solid tumours. Eur J Cancer 2006;42:2433–2453.
25. Crawford J, Ozer H, Stoller R, et al. Reduction by granulocyte colony-stimulating factor of fever and neutropenia induced by chemotherapy in patients with small-cell lung cancer. N Engl J Med 1991;325:164–170.
26. Gabrilove JL, Jakubowski A, Scher H, et al. Effect of granulocyte colony-stimulating factor on neutropenia and associated morbidity due to chemotherapy for transitional-cell carcinoma of the urothelium. N Engl J Med 1988;318:1414–1422.
27. Grigg A, Solal-Celigny P, Hoskin P, et al. Open-label, randomized study of pegfilgrastim vs. daily filgrastim as an adjunct to chemotherapy in elderly patients with non-Hodgkin’s lymphoma. Leuk Lymphoma 2003;44:1503–1508.
28. Holmes FA, Jones SE, O’Shaughnessy J, et al. Comparable efficacy and safety profiles of once-per-cycle pegfilgrastim and daily injection filgrastim in chemotherapy-induced neutropenia: a multicenter dose-finding study in women with breast cancer. Ann Oncol 2002;13:903–909.
29. Holmes FA, O’Shaughnessy JA, Vukelja S, et al. Blinded, randomized, multicenter study to evaluate single administration pegfilgrastim once per cycle versus daily filgrastim as an adjunct to chemotherapy in patients with highrisk stage II or stage III/IV breast cancer. J Clin Oncol 2002;20:727–731.
30. Johnston E, Crawford J, Blackwell S, et al. Randomized, dose-escalation study of SD/01 compared with daily filgrastim in patients receiving chemotherapy. J Clin Oncol 2000;18:2522–2528.
31. Trillet-Lenoir V, Green J, Manegold C, et al. Recombinant granulocyte colony stimulating factor reduces the infectious complications of cytotoxic chemotherapy. Eur J Cancer 1993;29A:319–324.
32. Vogel CL, Wojtukiewicz MZ, Carroll RR, et al. First and subsequent cycle use of pegfilgrastim prevents febrile neutropenia in patients with breast cancer: a multicenter, double- blind, placebo-controlled phase III study. J Clin Oncol 2005;23:1178–1184.
33. Green MD, Koelbl H, Baselga J, et al. A randomized double-blind multicenter phase III study of fixed-dose single-administration pegfilgrastim versus daily filgrastim in patients receiving myelosuppressive chemotherapy. Ann Oncol 2003;14:29–35.
34. Vose JM, Crump M, Lazarus H, et al. Randomized, multicenter, open-label study of pegfilgrastim compared with daily filgrastim after chemotherapy for lymphoma. J Clin Oncol 2003;21:514–519.
35. Kuderer NM, Dale DC, Crawford J, Lyman GH. Impact of primary prophylaxis with granulocyte colony-stimulating factor on febrile neutropenia and mortality in adult cancer patients receiving chemotherapy: a systematic review. J Clin Oncol 2007;25:3158–3167.
36. Lyman GH, Michels SL, Reynolds MW, Barron R, Tomic KS, Yu J. Risk of mortality in patients with cancer who experience febrile neutropenia. Cancer 2010;116:5555– 5563.
37. Lyman GH, Dale DC, Wolff DA, et al. Acute myeloid leukemia or myelodysplastic syndrome in randomized controlled clinical trials of cancer chemotherapy with granulocyte colony-stimulating factor: a systematic review. J Clin Oncol 2010;28:2914–2924.
38. Bohlius J, Reiser M, Schwarzer G, Engert A. Granulopoiesis-stimulating factors to prevent adverse effects in the treatment of malignant lymphoma. Cochrane Database Syst Rev 2004:CD003189.
39. Dale DC, McCarter GC, Crawford J, Lyman GH. Myelotoxicity and dose intensity of chemotherapy: reporting practices from randomized clinical trials. J Natl Compr Canc Netw 2003;1:440–454.
40. Duff JM, Leather H, Walden EO, LaPlant KD, George TJ Jr. Adequacy of published oncology randomized controlled trials to provide therapeutic details needed for clinical application. J Natl Cancer Inst 2010;102:702– 705.
41. Freedman OC, Zimmermann C, Clemons MJ: Interpreting the results of clinical trials of cancer chemotherapy: the importance of reporting concurrent supportive care. Proceedings from 31st Annual San Antonio Breast Cancer Symposium; December 14, 2008; San Antonio, TX. Abstract 6138.
42. Coiffier B, Lepage E, Briere J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med 2002;346:235–242.
43. Pfreundschuh M, Schubert J, Ziepert M, et al. Six versus eight cycles of bi-weekly CHOP-14 with or without rituximab in elderly patients with aggressive CD20+ B-cell lymphomas: a randomised controlled trial (RICOVER-60). Lancet Oncol 2008;9:105– 116.
44. Delarue R, Tilly H, Salles G, et al. RCHOP14 compared to R-CHOP21 in elderly patients with diffuse large B-cell lymphoma: results of the interim analysis of the LNH03- 6B GELA study. Blood 2009;114:406.
45. Hiddemann W, Kneba M, Dreyling M, et al. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood 2005;106:3725–3732.
46. Marcus R, Imrie K, Belch A, et al. CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood 2005;105:1417–1423.
47. Forstpointner R, Unterhalt M, Dreyling M, et al. Maintenance therapy with rituximab leads to a significant prolongation of response duration after salvage therapy with a combination of rituximab, fludarabine, cyclophosphamide, and mitoxantrone (R-FCM) in patients with recurring and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German Low Grade Lymphoma Study Group (GLSG). Blood 2006;108:4003–4008.
48. van Oers MH, Klasa R, Marcus RE, et al. Rituximab maintenance improves clinical outcome of relapsed/resistant follicular non- Hodgkin lymphoma in patients both with and without rituximab during induction: results of a prospective randomized phase 3 intergroup trial. Blood 2006;108:3295-3301.
49. van Oers MH, Van Glabbeke M, Giurgea L, et al. Rituximab maintenance treatment of relapsed/resistant follicular non-Hodgkin’s lymphoma: long-term outcome of the EORTC 20981 phase III randomized intergroup study. J Clin Oncol 2010;28:2853–2858.
50. Hochster H, Weller E, Gascoyne RD, et al. Maintenance rituximab after cyclophosphamide, vincristine, and prednisone prolongs progression-free survival in advanced indolent lymphoma: results of the randomized phase III ECOG1496 Study. J Clin Oncol 2009;27:1607–1614.
51. Salles GA, Seymour JF, Feugier P, et al. Rituximab maintenance for 2 years in patients with untreated high tumor burden follicular lymphoma after response to immunochemotherapy. J Clin Oncol 2010;28[15S]:8004.
52. Treanda [prescribing information]. Frazer, PA: Cephalon, Inc.; 2010.
53. Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab is superior in respect of progression free survival and CR rate when compared to CHOP plus ritux imab as first-line treatment of patients with advanced follicular, indolent, and mantle cell lymphomas: final results of a randomized phase III study of the StiL (Study Group Indolent Lymphomas, Germany). Blood 2009;114:405.
54. Demko S, Summers J, Keegan P, Pazdur R. FDA drug approval summary: alemtuzumab as single-agent treatment for B-cell chronic lymphocytic leukemia. Oncologist 2008;13:167–174.
55. Fischer K, Stilgenbauer S, Schweighofer CD, et al. Bendamustine in combination with rituximab (BR) for patients with relapsed chronic lymphocytic leukemia (CLL): a multicentre phase II trial of the German CLL Study Group (GCLLSG). Blood 2008;112:330.
56. Arzerra [prescribing information]. Research Triangle Park, NC: GlaxoSmithKline; 2010.
57. Wierda WG, Kipps TJ, Mayer J, et al. Ofatumumab as single-agent CD20 immunotherapy in fludarabine-refractory chronic lymphocytic leukemia. J Clin Oncol 2010;28:1749– 1755.
58. Straus DJ, Portlock CS, Qin J, et al. Results of a prospective randomized clinical trial of doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) followed by radiation therapy (RT) versus ABVD alone for stages I, II, and IIIA nonbulky Hodgkin disease. Blood 2004;104:3483–3489.
59. Duggan DB, Petroni GR, Johnson JL, et al. Randomized comparison of ABVD and MOPP/ABV hybrid for the treatment of advanced Hodgkin’s disease: report of an intergroup trial. J Clin Oncol 2003;21:607–614.
60. Horning SJ, Hoppe RT, Breslin S, Bartlett NL, Brown BW, Rosenberg SA. Stanford V and radiotherapy for locally extensive and advanced Hodgkin’s disease: mature results of a prospective clinical trial. J Clin Oncol 2002;20:630–637.
61. Canellos GP, Anderson JR, Propert KJ, et al. Chemotherapy of advanced Hodgkin’s disease with MOPP, ABVD, or MOPP alternating with ABVD. N Engl J Med 1992;327:1478–1484.
62. Federico M, Luminari S, Iannitto E, et al. ABVD compared with BEACOPP compared with CEC for the initial treatment of patients with advanced Hodgkin’s lymphoma: results from the HD2000 Gruppo Italiano per lo Studio dei Linfomi Trial. J Clin Oncol 2009;27:805–811.
63. Hideshima T, Bergsagel PL, Kuehl WM, Anderson KC. Advances in biology of multiple myeloma: clinical applications. Blood 2004;104:607–618.
64. Thalomid [prescribing information]. Summit, NJ: Celgene Corporation; 2009.
65. Richardson PG, Sonneveld P, Schuster MW, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005;352:2487–2498.
66. Richardson PG, Sonneveld P, Schuster M, et al. Extended follow-up of a phase 3 trial in relapsed multiple myeloma: final timeto- event results of the APEX trial. Blood 2007;110:3557–3560.
67. Orlowski RZ, Nagler A, Sonneveld P, et al. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. J Clin Oncol 2007;25:3892–3901.
68. Revlimid [prescribing information]. Summit, NJ: Celgene Corporation; 2009.
69. Weber DM, Chen C, Niesvizky R, et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med 2007;357:2133–2142.
70. Dimopoulos M, Spencer A, Attal M, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med 2007;357:2123–2132.
71. Rajkumar SV, Jacobus S, Callander NS, et al. Lenalidomide plus high-dose dexamethasone versus lenalidomide plus low-dose dexamethasone as initial therapy for newly diagnosed multiple myeloma: an open-label randomised controlled trial. Lancet Oncol 2010;11:29–37.
72. Rajkumar SV, Blood E, Vesole D, Fonseca R, Greipp PR. Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: a clinical trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol 2006;24:431–436.
73. Hulin C, Facon T, Rodon P, et al. Efficacy of melphalan and prednisone plus thalidomide in patients older than 75 years with newly diagnosed multiple myeloma: IFM 01/01 trial. J Clin Oncol 2009;27:3664–3670.
Landing Helicopter Parents
The recently coined term “helicopter parents” refers to parents who hover close to their children with the impression that this will keep them safe or ensure their success. We more formally call these parents “overprotective,” “overintrusive,” or “facilitators” for their kids. The new term may make this behavior seem more normal or even more amusing than it really is.
When you see helicopter parents in your practice, it's hard to know exactly what your role should be. How intrusive should you be to try to change their ways? Is helicopter parenting just a trendy societal or cultural phenomenon? Or do you really have enough information that it is a problem to justify your offering advice?
Even though answers to those questions may still be “up in the air,” overall such overprotection can have significant side effects and should be “on our radar”!
There are some pretty obvious reasons for the increase in helicopter parenting. One is cell phones. Everybody has cell phones, even some 6-year-old children. All the calling and texting back and forth makes it too easy to know every move the child makes. These days, if the children are not out of the house with their cell phones, they likely are at home playing video games or in a sport to which they were driven by the parent.
And these activity choices are part of a vicious cycle being selected – if not to promote entry to Harvard then to keep kids busy. Once one family puts their kids in planned activities, there are fewer peers available to play with after school, so other parents do the same. Smaller family sizes also can encourage helicopter parenting. If parents have five children, there is no time for hovering! Families with fewer children also may have more psychologically invested in each child.
I think that it is no accident that helicopter parenting has emerged at the same time most women are in the workplace full time. When home, working mothers, full of guilt, “up their efforts” to make sure they are being good parents.
Other reasons for a heightened level of hovering is parents' perception that they need to keep their kids safe in what feels like a threatening world. Families also are responding to increased competitiveness for college entry and jobs by doing all they can to position their children to achieve these goals. Of course, parents also want to show their love and concern. In some cases, high levels of protectiveness are appropriate: The child may attend a school where kids are carrying knives, or Dad is coming home drunk and the child needs to be protected. Such circumstances are the exception, though. More common pathology is that a parent is overanxious in general or even has an anxiety disorder.
While overprotectiveness may be understandable, it has significant developmental consequences. When parents dictate most of the children's activities, this can preclude the children from discovering, pursuing, and “owning” their interests. Overall, it can lessen the children's self-esteem because they have fewer opportunities to achieve things they consider to be all their own. Also, when a parent participates in the child's activities, they, as adults, will likely be more competent than the child. That can make a child feel less competent, whereas a kid doing things with peers has a decent chance of being the best. Helicopter parents also typically are aiming to avoid all kinds of risk for their precious child. The protected child may be physically safe, but can become risk aversive and miss the chance to learn how to appropriately assess real dangers. If parents “helicopter” because they are always afraid something bad is going to happen – like a pedophile is going to jump out of the bushes or the child is going to be abducted – they also transmit these fears to the child.
Children who are too restricted may even have health effects from sitting at home, eating too much, and not getting enough exercise.
But what can you do when you think problems may be developing from a helicopter parent? Parents may not perceive any problem at all. In this case, motivational interviewing can help. This technique can be used to move many different types of behavior and can fit into a primary care visit.
A motivational interview is a dialogue between the clinician and patient with specific steps. First, find out if the parent or child perceives any problem at all. You might say, “Wow, you certainly have your kid in all kinds of activities and are working hard to provide all these opportunities. Is your level of involvement a problem for you or your child in any way?” Watching the child's face when you ask this can be very revealing, and can also be used as a point of reflection for the interview.
You might also say: “Have you considered whether you really need to or want to do all of these things?” or “Have you considered backing off?” If the response is, “No, I've never thought about this before,” the parent may ask you in return: “Do you think it's a problem?” Then you have the opportunity to go over the potential pros and cons I've already outlined.
You might ask: “What are the good things about being this involved with your child?” And when you ask this, push them to include not only the effects (he's learned to play the violin or is now state ranked in tennis) but the way they feel about it as well. Parents might say things that reveal their reasons such as: “It makes me feel that I'm a good parent because I've done all these things for her” or “I feel more comfortable when I'm at work because I know he is safe at his karate class.”
Then you might ask them: “Is there any downside to being so involved in all of your child's activities?” You might get this response: “I'm beginning to resent it. I signed him up for all these activities, and now I don't have any free time any more.”
The next step in motivational interviewing is to ask about their readiness for change in a gentle way. “Do you think you might consider backing off?” If they say yes, you can ask, “What would be one of the things you could back off on now?” Make sure it is specific and also includes a time frame: “When would you be able to make this change?” In one family I was helping, the child had been talking to his parents on the phone 20 times a day. For him, a goal-setting question was, “What would it take to cut that down to 15 times?” Don't set an unrealistic goal such as stopping altogether.
Some parents initially will not be amenable to changing their behavior. For example, if they say, “I don't know. I never thought about this before,” you may need to be more circumspect. You might say, “Is there something else about this way of relating to your child that is making you want to continue?” Or use other parents as an example: “Some parents find when they back off the child becomes more relaxed, gets right on his homework by himself, and is happier.”
Garnering support for a change in behavior is an important component. You might ask: “Who could help you back off?” Finding other parents to have as friends who are not so intense, who don't feel the need to have a perfect child, or who are willing to let their kids be more autonomous may be key. Some websites and social networks developing to help parents back off from helicoptering promote “slow parenting,” “free-range parenting,” or “simplicity parenting.”
One important goal of the motivational interview is to come away with a time-based action plan. For a parent who says: “I don't want to change anything” or “This is the most important thing I'm doing for my kid,” you can keep change on the agenda by saying something like: “OK, perhaps we can talk about it when you bring her back for her vaccine in 2 months” and then make a note in the chart so you remember.
Inability to follow an agreed-upon plan can reveal where the parents or child is getting stuck, so this can be subsequently addressed. On a follow-up contact ask how it went and praise them, especially if they exceeded the goal. Or the parent may say, “When I tried to do that, he had a panic attack” or “I got depressed. I felt worthless, like I was not protecting my child.” That will help you understand the barriers for these parents and help you arrange appropriate treatment.
Even though helicopter parenting sounds like something new, addressing it employs your same old clinical skills.
---Barbara J. Howard, M.D.
The recently coined term “helicopter parents” refers to parents who hover close to their children with the impression that this will keep them safe or ensure their success. We more formally call these parents “overprotective,” “overintrusive,” or “facilitators” for their kids. The new term may make this behavior seem more normal or even more amusing than it really is.
When you see helicopter parents in your practice, it's hard to know exactly what your role should be. How intrusive should you be to try to change their ways? Is helicopter parenting just a trendy societal or cultural phenomenon? Or do you really have enough information that it is a problem to justify your offering advice?
Even though answers to those questions may still be “up in the air,” overall such overprotection can have significant side effects and should be “on our radar”!
There are some pretty obvious reasons for the increase in helicopter parenting. One is cell phones. Everybody has cell phones, even some 6-year-old children. All the calling and texting back and forth makes it too easy to know every move the child makes. These days, if the children are not out of the house with their cell phones, they likely are at home playing video games or in a sport to which they were driven by the parent.
And these activity choices are part of a vicious cycle being selected – if not to promote entry to Harvard then to keep kids busy. Once one family puts their kids in planned activities, there are fewer peers available to play with after school, so other parents do the same. Smaller family sizes also can encourage helicopter parenting. If parents have five children, there is no time for hovering! Families with fewer children also may have more psychologically invested in each child.
I think that it is no accident that helicopter parenting has emerged at the same time most women are in the workplace full time. When home, working mothers, full of guilt, “up their efforts” to make sure they are being good parents.
Other reasons for a heightened level of hovering is parents' perception that they need to keep their kids safe in what feels like a threatening world. Families also are responding to increased competitiveness for college entry and jobs by doing all they can to position their children to achieve these goals. Of course, parents also want to show their love and concern. In some cases, high levels of protectiveness are appropriate: The child may attend a school where kids are carrying knives, or Dad is coming home drunk and the child needs to be protected. Such circumstances are the exception, though. More common pathology is that a parent is overanxious in general or even has an anxiety disorder.
While overprotectiveness may be understandable, it has significant developmental consequences. When parents dictate most of the children's activities, this can preclude the children from discovering, pursuing, and “owning” their interests. Overall, it can lessen the children's self-esteem because they have fewer opportunities to achieve things they consider to be all their own. Also, when a parent participates in the child's activities, they, as adults, will likely be more competent than the child. That can make a child feel less competent, whereas a kid doing things with peers has a decent chance of being the best. Helicopter parents also typically are aiming to avoid all kinds of risk for their precious child. The protected child may be physically safe, but can become risk aversive and miss the chance to learn how to appropriately assess real dangers. If parents “helicopter” because they are always afraid something bad is going to happen – like a pedophile is going to jump out of the bushes or the child is going to be abducted – they also transmit these fears to the child.
Children who are too restricted may even have health effects from sitting at home, eating too much, and not getting enough exercise.
But what can you do when you think problems may be developing from a helicopter parent? Parents may not perceive any problem at all. In this case, motivational interviewing can help. This technique can be used to move many different types of behavior and can fit into a primary care visit.
A motivational interview is a dialogue between the clinician and patient with specific steps. First, find out if the parent or child perceives any problem at all. You might say, “Wow, you certainly have your kid in all kinds of activities and are working hard to provide all these opportunities. Is your level of involvement a problem for you or your child in any way?” Watching the child's face when you ask this can be very revealing, and can also be used as a point of reflection for the interview.
You might also say: “Have you considered whether you really need to or want to do all of these things?” or “Have you considered backing off?” If the response is, “No, I've never thought about this before,” the parent may ask you in return: “Do you think it's a problem?” Then you have the opportunity to go over the potential pros and cons I've already outlined.
You might ask: “What are the good things about being this involved with your child?” And when you ask this, push them to include not only the effects (he's learned to play the violin or is now state ranked in tennis) but the way they feel about it as well. Parents might say things that reveal their reasons such as: “It makes me feel that I'm a good parent because I've done all these things for her” or “I feel more comfortable when I'm at work because I know he is safe at his karate class.”
Then you might ask them: “Is there any downside to being so involved in all of your child's activities?” You might get this response: “I'm beginning to resent it. I signed him up for all these activities, and now I don't have any free time any more.”
The next step in motivational interviewing is to ask about their readiness for change in a gentle way. “Do you think you might consider backing off?” If they say yes, you can ask, “What would be one of the things you could back off on now?” Make sure it is specific and also includes a time frame: “When would you be able to make this change?” In one family I was helping, the child had been talking to his parents on the phone 20 times a day. For him, a goal-setting question was, “What would it take to cut that down to 15 times?” Don't set an unrealistic goal such as stopping altogether.
Some parents initially will not be amenable to changing their behavior. For example, if they say, “I don't know. I never thought about this before,” you may need to be more circumspect. You might say, “Is there something else about this way of relating to your child that is making you want to continue?” Or use other parents as an example: “Some parents find when they back off the child becomes more relaxed, gets right on his homework by himself, and is happier.”
Garnering support for a change in behavior is an important component. You might ask: “Who could help you back off?” Finding other parents to have as friends who are not so intense, who don't feel the need to have a perfect child, or who are willing to let their kids be more autonomous may be key. Some websites and social networks developing to help parents back off from helicoptering promote “slow parenting,” “free-range parenting,” or “simplicity parenting.”
One important goal of the motivational interview is to come away with a time-based action plan. For a parent who says: “I don't want to change anything” or “This is the most important thing I'm doing for my kid,” you can keep change on the agenda by saying something like: “OK, perhaps we can talk about it when you bring her back for her vaccine in 2 months” and then make a note in the chart so you remember.
Inability to follow an agreed-upon plan can reveal where the parents or child is getting stuck, so this can be subsequently addressed. On a follow-up contact ask how it went and praise them, especially if they exceeded the goal. Or the parent may say, “When I tried to do that, he had a panic attack” or “I got depressed. I felt worthless, like I was not protecting my child.” That will help you understand the barriers for these parents and help you arrange appropriate treatment.
Even though helicopter parenting sounds like something new, addressing it employs your same old clinical skills.
---Barbara J. Howard, M.D.
The recently coined term “helicopter parents” refers to parents who hover close to their children with the impression that this will keep them safe or ensure their success. We more formally call these parents “overprotective,” “overintrusive,” or “facilitators” for their kids. The new term may make this behavior seem more normal or even more amusing than it really is.
When you see helicopter parents in your practice, it's hard to know exactly what your role should be. How intrusive should you be to try to change their ways? Is helicopter parenting just a trendy societal or cultural phenomenon? Or do you really have enough information that it is a problem to justify your offering advice?
Even though answers to those questions may still be “up in the air,” overall such overprotection can have significant side effects and should be “on our radar”!
There are some pretty obvious reasons for the increase in helicopter parenting. One is cell phones. Everybody has cell phones, even some 6-year-old children. All the calling and texting back and forth makes it too easy to know every move the child makes. These days, if the children are not out of the house with their cell phones, they likely are at home playing video games or in a sport to which they were driven by the parent.
And these activity choices are part of a vicious cycle being selected – if not to promote entry to Harvard then to keep kids busy. Once one family puts their kids in planned activities, there are fewer peers available to play with after school, so other parents do the same. Smaller family sizes also can encourage helicopter parenting. If parents have five children, there is no time for hovering! Families with fewer children also may have more psychologically invested in each child.
I think that it is no accident that helicopter parenting has emerged at the same time most women are in the workplace full time. When home, working mothers, full of guilt, “up their efforts” to make sure they are being good parents.
Other reasons for a heightened level of hovering is parents' perception that they need to keep their kids safe in what feels like a threatening world. Families also are responding to increased competitiveness for college entry and jobs by doing all they can to position their children to achieve these goals. Of course, parents also want to show their love and concern. In some cases, high levels of protectiveness are appropriate: The child may attend a school where kids are carrying knives, or Dad is coming home drunk and the child needs to be protected. Such circumstances are the exception, though. More common pathology is that a parent is overanxious in general or even has an anxiety disorder.
While overprotectiveness may be understandable, it has significant developmental consequences. When parents dictate most of the children's activities, this can preclude the children from discovering, pursuing, and “owning” their interests. Overall, it can lessen the children's self-esteem because they have fewer opportunities to achieve things they consider to be all their own. Also, when a parent participates in the child's activities, they, as adults, will likely be more competent than the child. That can make a child feel less competent, whereas a kid doing things with peers has a decent chance of being the best. Helicopter parents also typically are aiming to avoid all kinds of risk for their precious child. The protected child may be physically safe, but can become risk aversive and miss the chance to learn how to appropriately assess real dangers. If parents “helicopter” because they are always afraid something bad is going to happen – like a pedophile is going to jump out of the bushes or the child is going to be abducted – they also transmit these fears to the child.
Children who are too restricted may even have health effects from sitting at home, eating too much, and not getting enough exercise.
But what can you do when you think problems may be developing from a helicopter parent? Parents may not perceive any problem at all. In this case, motivational interviewing can help. This technique can be used to move many different types of behavior and can fit into a primary care visit.
A motivational interview is a dialogue between the clinician and patient with specific steps. First, find out if the parent or child perceives any problem at all. You might say, “Wow, you certainly have your kid in all kinds of activities and are working hard to provide all these opportunities. Is your level of involvement a problem for you or your child in any way?” Watching the child's face when you ask this can be very revealing, and can also be used as a point of reflection for the interview.
You might also say: “Have you considered whether you really need to or want to do all of these things?” or “Have you considered backing off?” If the response is, “No, I've never thought about this before,” the parent may ask you in return: “Do you think it's a problem?” Then you have the opportunity to go over the potential pros and cons I've already outlined.
You might ask: “What are the good things about being this involved with your child?” And when you ask this, push them to include not only the effects (he's learned to play the violin or is now state ranked in tennis) but the way they feel about it as well. Parents might say things that reveal their reasons such as: “It makes me feel that I'm a good parent because I've done all these things for her” or “I feel more comfortable when I'm at work because I know he is safe at his karate class.”
Then you might ask them: “Is there any downside to being so involved in all of your child's activities?” You might get this response: “I'm beginning to resent it. I signed him up for all these activities, and now I don't have any free time any more.”
The next step in motivational interviewing is to ask about their readiness for change in a gentle way. “Do you think you might consider backing off?” If they say yes, you can ask, “What would be one of the things you could back off on now?” Make sure it is specific and also includes a time frame: “When would you be able to make this change?” In one family I was helping, the child had been talking to his parents on the phone 20 times a day. For him, a goal-setting question was, “What would it take to cut that down to 15 times?” Don't set an unrealistic goal such as stopping altogether.
Some parents initially will not be amenable to changing their behavior. For example, if they say, “I don't know. I never thought about this before,” you may need to be more circumspect. You might say, “Is there something else about this way of relating to your child that is making you want to continue?” Or use other parents as an example: “Some parents find when they back off the child becomes more relaxed, gets right on his homework by himself, and is happier.”
Garnering support for a change in behavior is an important component. You might ask: “Who could help you back off?” Finding other parents to have as friends who are not so intense, who don't feel the need to have a perfect child, or who are willing to let their kids be more autonomous may be key. Some websites and social networks developing to help parents back off from helicoptering promote “slow parenting,” “free-range parenting,” or “simplicity parenting.”
One important goal of the motivational interview is to come away with a time-based action plan. For a parent who says: “I don't want to change anything” or “This is the most important thing I'm doing for my kid,” you can keep change on the agenda by saying something like: “OK, perhaps we can talk about it when you bring her back for her vaccine in 2 months” and then make a note in the chart so you remember.
Inability to follow an agreed-upon plan can reveal where the parents or child is getting stuck, so this can be subsequently addressed. On a follow-up contact ask how it went and praise them, especially if they exceeded the goal. Or the parent may say, “When I tried to do that, he had a panic attack” or “I got depressed. I felt worthless, like I was not protecting my child.” That will help you understand the barriers for these parents and help you arrange appropriate treatment.
Even though helicopter parenting sounds like something new, addressing it employs your same old clinical skills.
---Barbara J. Howard, M.D.
Management of the Child With Cystic Fibrosis
All 50 states require newborn screening for cystic fibrosis, and pediatricians play an important role when a newborn is diagnosed. The positive screens from your state laboratory will include referral options for confirmatory testing and specialty care at an accredited Cystic Fibrosis Center or CF center affiliate in your area. Pediatricians remain the patient's “medical home,” help to coordinate this specialty care, and remain in close communication with the patient, families, and specialists at a CF center as these children grow.
Optimal outcomes come from assessment and treatment by a multidisciplinary team at an accredited CF center. Encourage your CF patient and families to be seen there regularly. You want those specialists on board to help you take optimal care of the child. Monitoring at least every 3 months is recommended and is called “expectant” or “proactive management” for an individual with CF. A more thorough assessment occurs at least annually.
Pediatricians are essential, in particular, when patients live far from a CF center. We have patients who live a 3-hour drive from our center at the University of Chicago. In some states, patients may live hundreds of miles from a center and can be seen for specialty care only once per year.
There are some specialist outreach efforts as well for children who live in extremely remote, rural areas: Clinicians in Seattle fly to Anchorage, Alaska, to monitor and treat children with CF.
If a sick child with CF comes as a new patient to your office and the records are not available, you should always feel free to call the closest CF center to speak to one of the specialists there and eventually refer them for care. Physicians at CF centers work in collaboration with primary care physicians who are the “go to” professionals, often treating other members of a CF child's family.
Consultation and referral are important. Pediatricians tend to be very, very busy and see 20-40 patients a day in their office for a wide range of indications, including earaches, stomach aches, and well-child exams. But if you are taking care of a sick child with CF who is just not getting better with antibiotic or other treatment, definitely communicate with a CF care provider.
Many pediatricians we know well in the Chicago area will call us up and say, “I have so-and-so in my office. They are coughing a little bit more. I know you sent a letter before saying I should try this particular antibiotic. Is there anything else I should do at this point?” They stay in close touch, and we always have a center physician or nurse available to speak with them.
Regular assessment by a pediatric pulmonologist is part of the CF center care. If a child has an increased cough and they have certain bacteria such as Pseudomonas, we aggressively treat them so they don't get worse. We monitor lung function and obtain frequent respiratory cultures.
In addition, up to 90% of these children and adolescents develop digestive problems and may benefit from consultation with a gastroenterologist. Some patients develop sinus-related problems and referral to an ENT may be warranted. Accredited centers are required to provide these specialists, as well as dieticians, social workers, and respiratory therapists, who are experienced and knowledgeable about CF and who are involved in providing a care plan for children with CF.
Pediatricians play an important role in the facilitation of regular testing of children and adolescents with cystic fibrosis. At a minimum of once a year, they require blood work and chest x-rays. Many patients do their annual visits in the summer because testing takes almost a whole day. The lab work and chest x-ray copy (on a CD) can then be brought to CF physicians on the day of that “annual” visit. Depending on the patient's insurance, it may be less expensive and more convenient for the family for the pediatrician to coordinate this annual testing at the local community hospital. Lung function testing typically needs to be done at the center.
We also now do an oral glucose tolerance test for CF-related diabetes in any patient older than 10 years. That often can be done locally as well. Patients with CF may develop a specific type of diabetes. Most patients with CF have pancreatic insufficiency, which results in problems with digestion and the need to take medication (pancreatic enzymes) with every meal. Over time, scarring of the pancreas results and insufficient insulin may be produced.
The Cystic Fibrosis Foundation, which approves and accredits CF centers, is a great source of information for patients and families. Advise parents to visit www.cff.org
All 50 states require newborn screening for cystic fibrosis, and pediatricians play an important role when a newborn is diagnosed. The positive screens from your state laboratory will include referral options for confirmatory testing and specialty care at an accredited Cystic Fibrosis Center or CF center affiliate in your area. Pediatricians remain the patient's “medical home,” help to coordinate this specialty care, and remain in close communication with the patient, families, and specialists at a CF center as these children grow.
Optimal outcomes come from assessment and treatment by a multidisciplinary team at an accredited CF center. Encourage your CF patient and families to be seen there regularly. You want those specialists on board to help you take optimal care of the child. Monitoring at least every 3 months is recommended and is called “expectant” or “proactive management” for an individual with CF. A more thorough assessment occurs at least annually.
Pediatricians are essential, in particular, when patients live far from a CF center. We have patients who live a 3-hour drive from our center at the University of Chicago. In some states, patients may live hundreds of miles from a center and can be seen for specialty care only once per year.
There are some specialist outreach efforts as well for children who live in extremely remote, rural areas: Clinicians in Seattle fly to Anchorage, Alaska, to monitor and treat children with CF.
If a sick child with CF comes as a new patient to your office and the records are not available, you should always feel free to call the closest CF center to speak to one of the specialists there and eventually refer them for care. Physicians at CF centers work in collaboration with primary care physicians who are the “go to” professionals, often treating other members of a CF child's family.
Consultation and referral are important. Pediatricians tend to be very, very busy and see 20-40 patients a day in their office for a wide range of indications, including earaches, stomach aches, and well-child exams. But if you are taking care of a sick child with CF who is just not getting better with antibiotic or other treatment, definitely communicate with a CF care provider.
Many pediatricians we know well in the Chicago area will call us up and say, “I have so-and-so in my office. They are coughing a little bit more. I know you sent a letter before saying I should try this particular antibiotic. Is there anything else I should do at this point?” They stay in close touch, and we always have a center physician or nurse available to speak with them.
Regular assessment by a pediatric pulmonologist is part of the CF center care. If a child has an increased cough and they have certain bacteria such as Pseudomonas, we aggressively treat them so they don't get worse. We monitor lung function and obtain frequent respiratory cultures.
In addition, up to 90% of these children and adolescents develop digestive problems and may benefit from consultation with a gastroenterologist. Some patients develop sinus-related problems and referral to an ENT may be warranted. Accredited centers are required to provide these specialists, as well as dieticians, social workers, and respiratory therapists, who are experienced and knowledgeable about CF and who are involved in providing a care plan for children with CF.
Pediatricians play an important role in the facilitation of regular testing of children and adolescents with cystic fibrosis. At a minimum of once a year, they require blood work and chest x-rays. Many patients do their annual visits in the summer because testing takes almost a whole day. The lab work and chest x-ray copy (on a CD) can then be brought to CF physicians on the day of that “annual” visit. Depending on the patient's insurance, it may be less expensive and more convenient for the family for the pediatrician to coordinate this annual testing at the local community hospital. Lung function testing typically needs to be done at the center.
We also now do an oral glucose tolerance test for CF-related diabetes in any patient older than 10 years. That often can be done locally as well. Patients with CF may develop a specific type of diabetes. Most patients with CF have pancreatic insufficiency, which results in problems with digestion and the need to take medication (pancreatic enzymes) with every meal. Over time, scarring of the pancreas results and insufficient insulin may be produced.
The Cystic Fibrosis Foundation, which approves and accredits CF centers, is a great source of information for patients and families. Advise parents to visit www.cff.org
All 50 states require newborn screening for cystic fibrosis, and pediatricians play an important role when a newborn is diagnosed. The positive screens from your state laboratory will include referral options for confirmatory testing and specialty care at an accredited Cystic Fibrosis Center or CF center affiliate in your area. Pediatricians remain the patient's “medical home,” help to coordinate this specialty care, and remain in close communication with the patient, families, and specialists at a CF center as these children grow.
Optimal outcomes come from assessment and treatment by a multidisciplinary team at an accredited CF center. Encourage your CF patient and families to be seen there regularly. You want those specialists on board to help you take optimal care of the child. Monitoring at least every 3 months is recommended and is called “expectant” or “proactive management” for an individual with CF. A more thorough assessment occurs at least annually.
Pediatricians are essential, in particular, when patients live far from a CF center. We have patients who live a 3-hour drive from our center at the University of Chicago. In some states, patients may live hundreds of miles from a center and can be seen for specialty care only once per year.
There are some specialist outreach efforts as well for children who live in extremely remote, rural areas: Clinicians in Seattle fly to Anchorage, Alaska, to monitor and treat children with CF.
If a sick child with CF comes as a new patient to your office and the records are not available, you should always feel free to call the closest CF center to speak to one of the specialists there and eventually refer them for care. Physicians at CF centers work in collaboration with primary care physicians who are the “go to” professionals, often treating other members of a CF child's family.
Consultation and referral are important. Pediatricians tend to be very, very busy and see 20-40 patients a day in their office for a wide range of indications, including earaches, stomach aches, and well-child exams. But if you are taking care of a sick child with CF who is just not getting better with antibiotic or other treatment, definitely communicate with a CF care provider.
Many pediatricians we know well in the Chicago area will call us up and say, “I have so-and-so in my office. They are coughing a little bit more. I know you sent a letter before saying I should try this particular antibiotic. Is there anything else I should do at this point?” They stay in close touch, and we always have a center physician or nurse available to speak with them.
Regular assessment by a pediatric pulmonologist is part of the CF center care. If a child has an increased cough and they have certain bacteria such as Pseudomonas, we aggressively treat them so they don't get worse. We monitor lung function and obtain frequent respiratory cultures.
In addition, up to 90% of these children and adolescents develop digestive problems and may benefit from consultation with a gastroenterologist. Some patients develop sinus-related problems and referral to an ENT may be warranted. Accredited centers are required to provide these specialists, as well as dieticians, social workers, and respiratory therapists, who are experienced and knowledgeable about CF and who are involved in providing a care plan for children with CF.
Pediatricians play an important role in the facilitation of regular testing of children and adolescents with cystic fibrosis. At a minimum of once a year, they require blood work and chest x-rays. Many patients do their annual visits in the summer because testing takes almost a whole day. The lab work and chest x-ray copy (on a CD) can then be brought to CF physicians on the day of that “annual” visit. Depending on the patient's insurance, it may be less expensive and more convenient for the family for the pediatrician to coordinate this annual testing at the local community hospital. Lung function testing typically needs to be done at the center.
We also now do an oral glucose tolerance test for CF-related diabetes in any patient older than 10 years. That often can be done locally as well. Patients with CF may develop a specific type of diabetes. Most patients with CF have pancreatic insufficiency, which results in problems with digestion and the need to take medication (pancreatic enzymes) with every meal. Over time, scarring of the pancreas results and insufficient insulin may be produced.
The Cystic Fibrosis Foundation, which approves and accredits CF centers, is a great source of information for patients and families. Advise parents to visit www.cff.org
Hospitalists Can Be Champions of Clinical Documentation
To improve your hospital’s reimbursements and key indicators, including observed and expected (O/E) mortality ratios, personalize your collaboration with clinical documentation specialists (CDSs). That’s what has worked at two medical centers where hospitalists teamed up with CDSs to improve their hospitals’ claims processes.
At Northwestern Memorial Hospital in Chicago, the hematology/oncology service was selected for a pilot program to focus on improving expected mortality rates. The specialists needed to ensure that coded data sent to state- and hospital-associated databases (as well as payor claims, such as those submitted to Medicare) accurately represented the severity of patients’ conditions upon admission. To do that, they needed buy-in from hospitalists to make their notes as complete as possible.
Hospitalists usually encounter CDSs anonymously, through an electronic query in the electronic health record (EHR). Kristine Green, RN, a CDS, quality leader, and interim manager at Northwestern’s clinical documentation program, approached hospitalist Charlotta Weaver, MD, medical director of the oncology HM service and a clinical instructor at Northwestern University’s Feinberg School of Medicine. Green suggested she shadow Dr. Weaver on rounds.
“We had implemented this technique in a couple of our other service lines, with good results,” Green says. She compared her notes on patient visits with Dr. Weaver’s notes and was able to catch conditions that were being undercoded. They generated a list, now posted in the work room, disseminated via email, and included in the orientation binder, of frequently missed coding diagnoses.
For example, Dr. Weaver explains, “instead of writing ‘AKI-obstructive,’ we now write ‘AKI due to ureteral obstruction from peritoneal carcinomatosis from metastatic gastric cancer.’ ”
Such specificity in physicians’ notes translates to a more accurate level of billing for the hospital and a more accurate reflection of patients’ acuity in comparative databases. With Dr. Weaver paving the way, Green has forged “a nice rapport” with the other hospitalists in the oncology medicine service.
Audiences for Your Notes
CDS and hospitalists might initially view notes differently. Most physicians train in programs where the “primary intent of a note” is to communicate to the rest of the medical team what’s happening with the patient, Dr. Weaver says.

“When we train, we’re always thinking about communicating with each other,” says Theodore (Ted) Tsomides, MD, PhD, an attending physician on the hospital medicine service at WakeMed Hospital and assistant professor of medicine at the University of North Carolina’s School of Medicine in Raleigh. “But as we get into the system, we realize that there are a lot of eyes on those documents. And whether we think about it or not, those are all our different audiences.”
Once hospitalists develop confidence and comfort on the job, Dr. Tsomides says, they can move on to aligning themselves with the hospital’s interests. Dr. Weaver thinks hospitalists are uniquely positioned to help champion the CDI efforts. “We’re here to improve the mission of the hospital,” she says.
As physician liaison for quality programs, Dr. Tsomides began working on clinical documentation improvement. He became a resource for the department, and then worked to achieve a financial incentive plan for hospital physicians when their documentation improved. He’s also been pushing his institution to make the documentation process easier by using electronic queries, and by introducing residents to the “real world” of clinical documentation in their curriculum. (Click here to listen to more of Dr. Tsomides’ ideas to improve clinical documentation.)
He advises hospitalists meet their clinical documentation specialists face to face. “Once you know there are people who are doing their part, and have a relationship with them, you approach the whole problem differently,” Dr. Tsomides says, “as opposed to [viewing them] as anonymous reviewers breathing down your neck and giving you yet another thing to worry about.”
Gretchen Henkel is a freelance writer based in California.
To improve your hospital’s reimbursements and key indicators, including observed and expected (O/E) mortality ratios, personalize your collaboration with clinical documentation specialists (CDSs). That’s what has worked at two medical centers where hospitalists teamed up with CDSs to improve their hospitals’ claims processes.
At Northwestern Memorial Hospital in Chicago, the hematology/oncology service was selected for a pilot program to focus on improving expected mortality rates. The specialists needed to ensure that coded data sent to state- and hospital-associated databases (as well as payor claims, such as those submitted to Medicare) accurately represented the severity of patients’ conditions upon admission. To do that, they needed buy-in from hospitalists to make their notes as complete as possible.
Hospitalists usually encounter CDSs anonymously, through an electronic query in the electronic health record (EHR). Kristine Green, RN, a CDS, quality leader, and interim manager at Northwestern’s clinical documentation program, approached hospitalist Charlotta Weaver, MD, medical director of the oncology HM service and a clinical instructor at Northwestern University’s Feinberg School of Medicine. Green suggested she shadow Dr. Weaver on rounds.
“We had implemented this technique in a couple of our other service lines, with good results,” Green says. She compared her notes on patient visits with Dr. Weaver’s notes and was able to catch conditions that were being undercoded. They generated a list, now posted in the work room, disseminated via email, and included in the orientation binder, of frequently missed coding diagnoses.
For example, Dr. Weaver explains, “instead of writing ‘AKI-obstructive,’ we now write ‘AKI due to ureteral obstruction from peritoneal carcinomatosis from metastatic gastric cancer.’ ”
Such specificity in physicians’ notes translates to a more accurate level of billing for the hospital and a more accurate reflection of patients’ acuity in comparative databases. With Dr. Weaver paving the way, Green has forged “a nice rapport” with the other hospitalists in the oncology medicine service.
Audiences for Your Notes
CDS and hospitalists might initially view notes differently. Most physicians train in programs where the “primary intent of a note” is to communicate to the rest of the medical team what’s happening with the patient, Dr. Weaver says.
“When we train, we’re always thinking about communicating with each other,” says Theodore (Ted) Tsomides, MD, PhD, an attending physician on the hospital medicine service at WakeMed Hospital and assistant professor of medicine at the University of North Carolina’s School of Medicine in Raleigh. “But as we get into the system, we realize that there are a lot of eyes on those documents. And whether we think about it or not, those are all our different audiences.”
Once hospitalists develop confidence and comfort on the job, Dr. Tsomides says, they can move on to aligning themselves with the hospital’s interests. Dr. Weaver thinks hospitalists are uniquely positioned to help champion the CDI efforts. “We’re here to improve the mission of the hospital,” she says.
As physician liaison for quality programs, Dr. Tsomides began working on clinical documentation improvement. He became a resource for the department, and then worked to achieve a financial incentive plan for hospital physicians when their documentation improved. He’s also been pushing his institution to make the documentation process easier by using electronic queries, and by introducing residents to the “real world” of clinical documentation in their curriculum. (Click here to listen to more of Dr. Tsomides’ ideas to improve clinical documentation.)
He advises hospitalists meet their clinical documentation specialists face to face. “Once you know there are people who are doing their part, and have a relationship with them, you approach the whole problem differently,” Dr. Tsomides says, “as opposed to [viewing them] as anonymous reviewers breathing down your neck and giving you yet another thing to worry about.”
Gretchen Henkel is a freelance writer based in California.
To improve your hospital’s reimbursements and key indicators, including observed and expected (O/E) mortality ratios, personalize your collaboration with clinical documentation specialists (CDSs). That’s what has worked at two medical centers where hospitalists teamed up with CDSs to improve their hospitals’ claims processes.
At Northwestern Memorial Hospital in Chicago, the hematology/oncology service was selected for a pilot program to focus on improving expected mortality rates. The specialists needed to ensure that coded data sent to state- and hospital-associated databases (as well as payor claims, such as those submitted to Medicare) accurately represented the severity of patients’ conditions upon admission. To do that, they needed buy-in from hospitalists to make their notes as complete as possible.
Hospitalists usually encounter CDSs anonymously, through an electronic query in the electronic health record (EHR). Kristine Green, RN, a CDS, quality leader, and interim manager at Northwestern’s clinical documentation program, approached hospitalist Charlotta Weaver, MD, medical director of the oncology HM service and a clinical instructor at Northwestern University’s Feinberg School of Medicine. Green suggested she shadow Dr. Weaver on rounds.
“We had implemented this technique in a couple of our other service lines, with good results,” Green says. She compared her notes on patient visits with Dr. Weaver’s notes and was able to catch conditions that were being undercoded. They generated a list, now posted in the work room, disseminated via email, and included in the orientation binder, of frequently missed coding diagnoses.
For example, Dr. Weaver explains, “instead of writing ‘AKI-obstructive,’ we now write ‘AKI due to ureteral obstruction from peritoneal carcinomatosis from metastatic gastric cancer.’ ”
Such specificity in physicians’ notes translates to a more accurate level of billing for the hospital and a more accurate reflection of patients’ acuity in comparative databases. With Dr. Weaver paving the way, Green has forged “a nice rapport” with the other hospitalists in the oncology medicine service.
Audiences for Your Notes
CDS and hospitalists might initially view notes differently. Most physicians train in programs where the “primary intent of a note” is to communicate to the rest of the medical team what’s happening with the patient, Dr. Weaver says.
“When we train, we’re always thinking about communicating with each other,” says Theodore (Ted) Tsomides, MD, PhD, an attending physician on the hospital medicine service at WakeMed Hospital and assistant professor of medicine at the University of North Carolina’s School of Medicine in Raleigh. “But as we get into the system, we realize that there are a lot of eyes on those documents. And whether we think about it or not, those are all our different audiences.”
Once hospitalists develop confidence and comfort on the job, Dr. Tsomides says, they can move on to aligning themselves with the hospital’s interests. Dr. Weaver thinks hospitalists are uniquely positioned to help champion the CDI efforts. “We’re here to improve the mission of the hospital,” she says.
As physician liaison for quality programs, Dr. Tsomides began working on clinical documentation improvement. He became a resource for the department, and then worked to achieve a financial incentive plan for hospital physicians when their documentation improved. He’s also been pushing his institution to make the documentation process easier by using electronic queries, and by introducing residents to the “real world” of clinical documentation in their curriculum. (Click here to listen to more of Dr. Tsomides’ ideas to improve clinical documentation.)
He advises hospitalists meet their clinical documentation specialists face to face. “Once you know there are people who are doing their part, and have a relationship with them, you approach the whole problem differently,” Dr. Tsomides says, “as opposed to [viewing them] as anonymous reviewers breathing down your neck and giving you yet another thing to worry about.”
Gretchen Henkel is a freelance writer based in California.
CT Screening Cuts Lung Cancer Mortality; Raises Policy Questions
Final results from the National Lung Screening Trial show a significant reduction in lung cancer mortality with the use of annual low-dose CT screening, compared with standard chest x-rays among former heavy smokers at high risk for lung cancer.
Low-dose CT screening led to a relative reduction of 20% in the rate of death from lung cancer, according to findings released online by the New England Journal of Medicine on June 29 (doi: 10.1056/NEJMoa1102873).The number needed to screen with low-dose CT to prevent one death from lung cancer was 320.
Although preliminary study results were announced in November 2010, the article by the National Lung Screening Trial (NLST) research team marks the first time that the results appear in a peer-reviewed journal. Acknowledging that the earlier announcement has led to calls for lung cancer screening, the authors urge rigorous analysis of cost-effectiveness before public policy recommendations are made.
"The reduction in lung-cancer mortality must be weighed against the harms from positive screening results and overdiagnosis, as well as the costs," they wrote.
In the study, 53,454 men and women aged 55-74 years – who were current or former smokers with a smoking history of at least 30 pack-years – were recruited at 33 U.S. medical centers. A total of 26,722 participants were randomized to receive three annual screens with low-dose helical CT; 26,732 were randomized to three annual screens using chest x-ray. The two groups were virtually identical in demographics and smoking history.
In all three screening rounds, positive screening tests were substantially more common in the low-dose CT group than in the radiography group (27.3% vs. 9.2% in the first round; 27.9% vs. 6.2% in the second; and 16.8% vs. 5% in the third). All told, 39.1% of the CT group and 16% of the radiography group had at least one positive result.
The percentage of screening tests that identified a clinically significant abnormality -- other than an abnormality suspicious for lung cancer – also was more than three times as high in the low-dose CT group as in the radiography group (7.5% vs. 2.1%).
More than 90% of positive screenings in the first round of the study led to a diagnostic evaluation, though the follow-up rates were lower in the later rounds. Diagnostic evaluation most often consisted of additional imaging with invasive procedures being performed infrequently.
Across the three screenings, most of the positive results were false positives – 96.4% in the CT group and 94.5% in the radiography group. Of the total number of low-dose CT screening tests, 24.2% were classified as positive and 23.4% had false-positive results; of the total number of radiographic screening tests, 6.9% were classified as positive and 6.5% were false-positive results.
In all, 1,060 lung cancers were diagnosed in the low-dose CT group (645/100,000 person-years) vs. 941 in the radiography group (572/100,000 person-years). Of these cancers, 649 in the low-dose CT group were diagnosed after a positive screening test and 44 were diagnosed after a negative screening test. In the radiography group, 279 cancers were diagnosed after a positive screening test and 137 were diagnosed after a negative screening test.
In both groups, the remaining cases were among participants who missed screening or were diagnosed after their trial screening phase was over
Analysis of lung cancer-specific mortality showed that in the CT group 356 lung cancer deaths occurred after 144,103 person-years; in the radiography group 443 lung cancer deaths occurred after 143,368 person-years. This corresponded to 247 and 309 lung cancer deaths, respectively, per 100,000 person-years in the CT and radiography groups.
There were 1,877 and 2,000 deaths from all causes in the CT and radiography groups, respectively, "representing a significant reduction with low-dose CT screening of 6.7% ... in the rate of death from any cause," the investigators wrote. While lung cancer accounted for 24.1% of all the deaths in the trial, 60.3% of the excess deaths in the radiography group were due to lung cancer.
The authors concluded that "although some agencies and organizations are contemplating the establishment of lung-cancer screening recommendations on the basis of the findings of the NLST, the current NLST data alone are, in our opinion, insufficient to fully inform such important decisions."
They noted that "The observation that low-dose CT screening can reduce the rate of death from lung cancer has generated many questions." Among these they listed: Will populations with different risk profiles benefit from screening? Could less frequent screening programs be equally effective? Would the use of different criteria for a positive screening result translate to similar benefit? For how long should people be screened?
In an accompanying editorial, Dr. Harold C. Sox, professor of medicine at the Dartmouth Institute in Hanover, N. H., agreed with the investigators’ reservations. In particular, the cost effectiveness of low-dose CT screening for lung cancer must be analyzed, he said: "Policy makers should wait for cost-effectiveness analyses to determine the amount of overdiagnosis in the NLST, and, perhaps identification of biologic markers of cancers that do not progress."
In addition, "it may be possible to define subgroups of smokers who are at higher or lower risk for lung cancer and tailor the screening strategy accordingly," he said. "The findings of the NLST regarding lung-cancer mortality signal the beginning of the end of one era of research on lung-cancer screening and the start of another. The focus will shift to informing the difficult patient-centered and policy decisions that are yet to come."
Dr. Sox also noted that "overdiagnosis is a problem because predicting which early-stage cancers will not progress is in an early stage of development, so that everyone with screen-detected cancer receives treatment that some do not need," he wrote in an accompanying editorial (doi: 10.1056/NEJME1103776).
All but two of the NLST study authors reported that they have no relevant financial relationships. Jonathan D. Clap reported having financial interest in Human Genome Sciences. Constantine Gatsonis, Ph.D., is a consultant for Wilex AG, Mela Sciences, and Endocyte Inc., has received speaker fees from Bayer Health and payment for education development by the Radiologic Society of North America. He also has invested in the Vanguard Health Fund. Dr. Sox had no conflicts.
This is an exciting
study that does show an impact on mortality, which has not been a screening
result from previous studies. What it doesn’t tell us exactly is: What does
this mean from a policy standpoint?
We need to look at a lot
more to see what’s the best model with this kind of screening and when this is
screening appropriate. I think the authors of this study were right to say that
this is a very positive result and it’s helpful … but that the best way to
implement this in day-to-day practice still is not completely resolved. There’s
a lot more work to be done in that regard.
One message that is still
very clear is that if you don’t want to die from lung cancer, you need to stop
smoking or never start smoking. This still has to be foremost in our public
health preventative message.
The study results do help
by saying that screening can have a role in day-to-day practice. The fact that
these patients were treated in a community setting showed that … the process
for diagnosing lung cancer can be handled by community physicians. I’m a
pulmonary physician. So when I sit down with patients who have the risk of
smoking, and we talk about what is the role of getting a low-dose CT scan for
screening, I think I have a lot more information to help both me and the
patient to decide whether this is beneficial to them versus a risk.
In the past, with CT
screening there was certainly risk from the radiation and risks for having
unnecessary procedures done, but no real proven benefit that we were going to
impact mortality if we found an early cancer. The study results do add value on
a day-to-day basis.
We just don’t know whether
it’s something that should be applied to everybody. Another question is whether
there are there markers that might help in this group of individuals to
identify who is at high risk for fast-growing tumors or for slow-growing tumors
Are there biologic markers that we can find with a blood test that might add to
this information to help us sort out who would benefit from screening or not?
[Other questions to
answer] from these data or from other ongoing studies include: Are there
subgroups of this 55- to 74-year-old population that are at higher risk? Are
there individuals who with less frequent screening can do just as well? Are
there individuals for whom more screening is necessary? The population looked
at [in the study] was a narrow window of high-risk individuals … It represents
about 7 million people out of the 94 million current and former smokers that we
have in this country.
We may even be able to
look at genetic markers at some point in the near future to determine who is at
higher risk and that will help us better identify who needs to be screened. I
think biomarkers and genetic markers all could be added to the formula when
we’re trying to decide what the best risk population to be screened is.
Screening tools work best when the screening population is well defined.
So now we have evidence
that screening in general can have an impact on disease. Unfortunately, prior
to this, lung cancer was diagnosed too late to make a big impact for most
patients. In lung cancer, an earlier diagnosis hopefully impacts mortality.
Lung cancer could become a curable disease if it’s found early enough to be
completely resected.
Dr. Albert A. Rizzo is
chair-elect of the American Lung Association board and chief of Christiana
Care’s pulmonary and critical care medicine section in Newark, Del.
He has no conflicts of interest.
This is an exciting
study that does show an impact on mortality, which has not been a screening
result from previous studies. What it doesn’t tell us exactly is: What does
this mean from a policy standpoint?
We need to look at a lot
more to see what’s the best model with this kind of screening and when this is
screening appropriate. I think the authors of this study were right to say that
this is a very positive result and it’s helpful … but that the best way to
implement this in day-to-day practice still is not completely resolved. There’s
a lot more work to be done in that regard.
One message that is still
very clear is that if you don’t want to die from lung cancer, you need to stop
smoking or never start smoking. This still has to be foremost in our public
health preventative message.
The study results do help
by saying that screening can have a role in day-to-day practice. The fact that
these patients were treated in a community setting showed that … the process
for diagnosing lung cancer can be handled by community physicians. I’m a
pulmonary physician. So when I sit down with patients who have the risk of
smoking, and we talk about what is the role of getting a low-dose CT scan for
screening, I think I have a lot more information to help both me and the
patient to decide whether this is beneficial to them versus a risk.
In the past, with CT
screening there was certainly risk from the radiation and risks for having
unnecessary procedures done, but no real proven benefit that we were going to
impact mortality if we found an early cancer. The study results do add value on
a day-to-day basis.
We just don’t know whether
it’s something that should be applied to everybody. Another question is whether
there are there markers that might help in this group of individuals to
identify who is at high risk for fast-growing tumors or for slow-growing tumors
Are there biologic markers that we can find with a blood test that might add to
this information to help us sort out who would benefit from screening or not?
[Other questions to
answer] from these data or from other ongoing studies include: Are there
subgroups of this 55- to 74-year-old population that are at higher risk? Are
there individuals who with less frequent screening can do just as well? Are
there individuals for whom more screening is necessary? The population looked
at [in the study] was a narrow window of high-risk individuals … It represents
about 7 million people out of the 94 million current and former smokers that we
have in this country.
We may even be able to
look at genetic markers at some point in the near future to determine who is at
higher risk and that will help us better identify who needs to be screened. I
think biomarkers and genetic markers all could be added to the formula when
we’re trying to decide what the best risk population to be screened is.
Screening tools work best when the screening population is well defined.
So now we have evidence
that screening in general can have an impact on disease. Unfortunately, prior
to this, lung cancer was diagnosed too late to make a big impact for most
patients. In lung cancer, an earlier diagnosis hopefully impacts mortality.
Lung cancer could become a curable disease if it’s found early enough to be
completely resected.
Dr. Albert A. Rizzo is
chair-elect of the American Lung Association board and chief of Christiana
Care’s pulmonary and critical care medicine section in Newark, Del.
He has no conflicts of interest.
This is an exciting
study that does show an impact on mortality, which has not been a screening
result from previous studies. What it doesn’t tell us exactly is: What does
this mean from a policy standpoint?
We need to look at a lot
more to see what’s the best model with this kind of screening and when this is
screening appropriate. I think the authors of this study were right to say that
this is a very positive result and it’s helpful … but that the best way to
implement this in day-to-day practice still is not completely resolved. There’s
a lot more work to be done in that regard.
One message that is still
very clear is that if you don’t want to die from lung cancer, you need to stop
smoking or never start smoking. This still has to be foremost in our public
health preventative message.
The study results do help
by saying that screening can have a role in day-to-day practice. The fact that
these patients were treated in a community setting showed that … the process
for diagnosing lung cancer can be handled by community physicians. I’m a
pulmonary physician. So when I sit down with patients who have the risk of
smoking, and we talk about what is the role of getting a low-dose CT scan for
screening, I think I have a lot more information to help both me and the
patient to decide whether this is beneficial to them versus a risk.
In the past, with CT
screening there was certainly risk from the radiation and risks for having
unnecessary procedures done, but no real proven benefit that we were going to
impact mortality if we found an early cancer. The study results do add value on
a day-to-day basis.
We just don’t know whether
it’s something that should be applied to everybody. Another question is whether
there are there markers that might help in this group of individuals to
identify who is at high risk for fast-growing tumors or for slow-growing tumors
Are there biologic markers that we can find with a blood test that might add to
this information to help us sort out who would benefit from screening or not?
[Other questions to
answer] from these data or from other ongoing studies include: Are there
subgroups of this 55- to 74-year-old population that are at higher risk? Are
there individuals who with less frequent screening can do just as well? Are
there individuals for whom more screening is necessary? The population looked
at [in the study] was a narrow window of high-risk individuals … It represents
about 7 million people out of the 94 million current and former smokers that we
have in this country.
We may even be able to
look at genetic markers at some point in the near future to determine who is at
higher risk and that will help us better identify who needs to be screened. I
think biomarkers and genetic markers all could be added to the formula when
we’re trying to decide what the best risk population to be screened is.
Screening tools work best when the screening population is well defined.
So now we have evidence
that screening in general can have an impact on disease. Unfortunately, prior
to this, lung cancer was diagnosed too late to make a big impact for most
patients. In lung cancer, an earlier diagnosis hopefully impacts mortality.
Lung cancer could become a curable disease if it’s found early enough to be
completely resected.
Dr. Albert A. Rizzo is
chair-elect of the American Lung Association board and chief of Christiana
Care’s pulmonary and critical care medicine section in Newark, Del.
He has no conflicts of interest.
Final results from the National Lung Screening Trial show a significant reduction in lung cancer mortality with the use of annual low-dose CT screening, compared with standard chest x-rays among former heavy smokers at high risk for lung cancer.
Low-dose CT screening led to a relative reduction of 20% in the rate of death from lung cancer, according to findings released online by the New England Journal of Medicine on June 29 (doi: 10.1056/NEJMoa1102873).The number needed to screen with low-dose CT to prevent one death from lung cancer was 320.
Although preliminary study results were announced in November 2010, the article by the National Lung Screening Trial (NLST) research team marks the first time that the results appear in a peer-reviewed journal. Acknowledging that the earlier announcement has led to calls for lung cancer screening, the authors urge rigorous analysis of cost-effectiveness before public policy recommendations are made.
"The reduction in lung-cancer mortality must be weighed against the harms from positive screening results and overdiagnosis, as well as the costs," they wrote.
In the study, 53,454 men and women aged 55-74 years – who were current or former smokers with a smoking history of at least 30 pack-years – were recruited at 33 U.S. medical centers. A total of 26,722 participants were randomized to receive three annual screens with low-dose helical CT; 26,732 were randomized to three annual screens using chest x-ray. The two groups were virtually identical in demographics and smoking history.
In all three screening rounds, positive screening tests were substantially more common in the low-dose CT group than in the radiography group (27.3% vs. 9.2% in the first round; 27.9% vs. 6.2% in the second; and 16.8% vs. 5% in the third). All told, 39.1% of the CT group and 16% of the radiography group had at least one positive result.
The percentage of screening tests that identified a clinically significant abnormality -- other than an abnormality suspicious for lung cancer – also was more than three times as high in the low-dose CT group as in the radiography group (7.5% vs. 2.1%).
More than 90% of positive screenings in the first round of the study led to a diagnostic evaluation, though the follow-up rates were lower in the later rounds. Diagnostic evaluation most often consisted of additional imaging with invasive procedures being performed infrequently.
Across the three screenings, most of the positive results were false positives – 96.4% in the CT group and 94.5% in the radiography group. Of the total number of low-dose CT screening tests, 24.2% were classified as positive and 23.4% had false-positive results; of the total number of radiographic screening tests, 6.9% were classified as positive and 6.5% were false-positive results.
In all, 1,060 lung cancers were diagnosed in the low-dose CT group (645/100,000 person-years) vs. 941 in the radiography group (572/100,000 person-years). Of these cancers, 649 in the low-dose CT group were diagnosed after a positive screening test and 44 were diagnosed after a negative screening test. In the radiography group, 279 cancers were diagnosed after a positive screening test and 137 were diagnosed after a negative screening test.
In both groups, the remaining cases were among participants who missed screening or were diagnosed after their trial screening phase was over
Analysis of lung cancer-specific mortality showed that in the CT group 356 lung cancer deaths occurred after 144,103 person-years; in the radiography group 443 lung cancer deaths occurred after 143,368 person-years. This corresponded to 247 and 309 lung cancer deaths, respectively, per 100,000 person-years in the CT and radiography groups.
There were 1,877 and 2,000 deaths from all causes in the CT and radiography groups, respectively, "representing a significant reduction with low-dose CT screening of 6.7% ... in the rate of death from any cause," the investigators wrote. While lung cancer accounted for 24.1% of all the deaths in the trial, 60.3% of the excess deaths in the radiography group were due to lung cancer.
The authors concluded that "although some agencies and organizations are contemplating the establishment of lung-cancer screening recommendations on the basis of the findings of the NLST, the current NLST data alone are, in our opinion, insufficient to fully inform such important decisions."
They noted that "The observation that low-dose CT screening can reduce the rate of death from lung cancer has generated many questions." Among these they listed: Will populations with different risk profiles benefit from screening? Could less frequent screening programs be equally effective? Would the use of different criteria for a positive screening result translate to similar benefit? For how long should people be screened?
In an accompanying editorial, Dr. Harold C. Sox, professor of medicine at the Dartmouth Institute in Hanover, N. H., agreed with the investigators’ reservations. In particular, the cost effectiveness of low-dose CT screening for lung cancer must be analyzed, he said: "Policy makers should wait for cost-effectiveness analyses to determine the amount of overdiagnosis in the NLST, and, perhaps identification of biologic markers of cancers that do not progress."
In addition, "it may be possible to define subgroups of smokers who are at higher or lower risk for lung cancer and tailor the screening strategy accordingly," he said. "The findings of the NLST regarding lung-cancer mortality signal the beginning of the end of one era of research on lung-cancer screening and the start of another. The focus will shift to informing the difficult patient-centered and policy decisions that are yet to come."
Dr. Sox also noted that "overdiagnosis is a problem because predicting which early-stage cancers will not progress is in an early stage of development, so that everyone with screen-detected cancer receives treatment that some do not need," he wrote in an accompanying editorial (doi: 10.1056/NEJME1103776).
All but two of the NLST study authors reported that they have no relevant financial relationships. Jonathan D. Clap reported having financial interest in Human Genome Sciences. Constantine Gatsonis, Ph.D., is a consultant for Wilex AG, Mela Sciences, and Endocyte Inc., has received speaker fees from Bayer Health and payment for education development by the Radiologic Society of North America. He also has invested in the Vanguard Health Fund. Dr. Sox had no conflicts.
Final results from the National Lung Screening Trial show a significant reduction in lung cancer mortality with the use of annual low-dose CT screening, compared with standard chest x-rays among former heavy smokers at high risk for lung cancer.
Low-dose CT screening led to a relative reduction of 20% in the rate of death from lung cancer, according to findings released online by the New England Journal of Medicine on June 29 (doi: 10.1056/NEJMoa1102873).The number needed to screen with low-dose CT to prevent one death from lung cancer was 320.
Although preliminary study results were announced in November 2010, the article by the National Lung Screening Trial (NLST) research team marks the first time that the results appear in a peer-reviewed journal. Acknowledging that the earlier announcement has led to calls for lung cancer screening, the authors urge rigorous analysis of cost-effectiveness before public policy recommendations are made.
"The reduction in lung-cancer mortality must be weighed against the harms from positive screening results and overdiagnosis, as well as the costs," they wrote.
In the study, 53,454 men and women aged 55-74 years – who were current or former smokers with a smoking history of at least 30 pack-years – were recruited at 33 U.S. medical centers. A total of 26,722 participants were randomized to receive three annual screens with low-dose helical CT; 26,732 were randomized to three annual screens using chest x-ray. The two groups were virtually identical in demographics and smoking history.
In all three screening rounds, positive screening tests were substantially more common in the low-dose CT group than in the radiography group (27.3% vs. 9.2% in the first round; 27.9% vs. 6.2% in the second; and 16.8% vs. 5% in the third). All told, 39.1% of the CT group and 16% of the radiography group had at least one positive result.
The percentage of screening tests that identified a clinically significant abnormality -- other than an abnormality suspicious for lung cancer – also was more than three times as high in the low-dose CT group as in the radiography group (7.5% vs. 2.1%).
More than 90% of positive screenings in the first round of the study led to a diagnostic evaluation, though the follow-up rates were lower in the later rounds. Diagnostic evaluation most often consisted of additional imaging with invasive procedures being performed infrequently.
Across the three screenings, most of the positive results were false positives – 96.4% in the CT group and 94.5% in the radiography group. Of the total number of low-dose CT screening tests, 24.2% were classified as positive and 23.4% had false-positive results; of the total number of radiographic screening tests, 6.9% were classified as positive and 6.5% were false-positive results.
In all, 1,060 lung cancers were diagnosed in the low-dose CT group (645/100,000 person-years) vs. 941 in the radiography group (572/100,000 person-years). Of these cancers, 649 in the low-dose CT group were diagnosed after a positive screening test and 44 were diagnosed after a negative screening test. In the radiography group, 279 cancers were diagnosed after a positive screening test and 137 were diagnosed after a negative screening test.
In both groups, the remaining cases were among participants who missed screening or were diagnosed after their trial screening phase was over
Analysis of lung cancer-specific mortality showed that in the CT group 356 lung cancer deaths occurred after 144,103 person-years; in the radiography group 443 lung cancer deaths occurred after 143,368 person-years. This corresponded to 247 and 309 lung cancer deaths, respectively, per 100,000 person-years in the CT and radiography groups.
There were 1,877 and 2,000 deaths from all causes in the CT and radiography groups, respectively, "representing a significant reduction with low-dose CT screening of 6.7% ... in the rate of death from any cause," the investigators wrote. While lung cancer accounted for 24.1% of all the deaths in the trial, 60.3% of the excess deaths in the radiography group were due to lung cancer.
The authors concluded that "although some agencies and organizations are contemplating the establishment of lung-cancer screening recommendations on the basis of the findings of the NLST, the current NLST data alone are, in our opinion, insufficient to fully inform such important decisions."
They noted that "The observation that low-dose CT screening can reduce the rate of death from lung cancer has generated many questions." Among these they listed: Will populations with different risk profiles benefit from screening? Could less frequent screening programs be equally effective? Would the use of different criteria for a positive screening result translate to similar benefit? For how long should people be screened?
In an accompanying editorial, Dr. Harold C. Sox, professor of medicine at the Dartmouth Institute in Hanover, N. H., agreed with the investigators’ reservations. In particular, the cost effectiveness of low-dose CT screening for lung cancer must be analyzed, he said: "Policy makers should wait for cost-effectiveness analyses to determine the amount of overdiagnosis in the NLST, and, perhaps identification of biologic markers of cancers that do not progress."
In addition, "it may be possible to define subgroups of smokers who are at higher or lower risk for lung cancer and tailor the screening strategy accordingly," he said. "The findings of the NLST regarding lung-cancer mortality signal the beginning of the end of one era of research on lung-cancer screening and the start of another. The focus will shift to informing the difficult patient-centered and policy decisions that are yet to come."
Dr. Sox also noted that "overdiagnosis is a problem because predicting which early-stage cancers will not progress is in an early stage of development, so that everyone with screen-detected cancer receives treatment that some do not need," he wrote in an accompanying editorial (doi: 10.1056/NEJME1103776).
All but two of the NLST study authors reported that they have no relevant financial relationships. Jonathan D. Clap reported having financial interest in Human Genome Sciences. Constantine Gatsonis, Ph.D., is a consultant for Wilex AG, Mela Sciences, and Endocyte Inc., has received speaker fees from Bayer Health and payment for education development by the Radiologic Society of North America. He also has invested in the Vanguard Health Fund. Dr. Sox had no conflicts.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Major Finding: Low-dose CT screening reduced the relative rate of death from lung cancer by 20%, compared with chest x-ray screening.
Data Source: A study of 53,454 Americans aged 55-74 years, who were current or former smokers with a smoking history of at least 30 pack-years.
Disclosures: All but two of the NLST study authors reported that they have no relevant financial relationships. Jonathan Clap reported having financial interest in Human Genome Sciences Inc. Constantine Gatsonis is a consultant for Wilex AG, Mela Sciences, and Endocyte Inc., has received speaker fees from Bayer Health, and payment for education development by the Radiologic Society of North America. He also has invested in the Vanguard Health Fund. Dr. Sox had no conflicts.
The Government, Undercover
A quickly-scuttled plan by the Department of Health and Human Services (HHS) to use "mystery shoppers" to test the availability of primary-care physicians (PCPs) would likely never be extended to HM—but the federal tack has certainly touched a nerve with some practitioners. (Ed note: updated June 29, 2011)
"Using surrogate patients or a sneaky way to get the data feels a bit less straightforward," says James Levy, PA-C, vice president of personnel for Traverse City-based Hospitalists of Northern Michigan. "If I were the government, I'm sure it would occur to me to do it that way. Nobody is going to be comfortable with surreptitious observations. That would seem to carry the message that the government doesn’t trust the providers it's reimbursing."
The government proposed using people pretending to be patients, both insured and uninsured, to gauge how long it takes to get PCP appointments. The action was proposed in the April 28 Federal Register. Comments were solicited, but negative feedback prompted HHS to drop the plan in a June 29 announcement.
Levy and David Friar, MD, CEO of Northern Hospitalists, say that the logistics of hospital admission make it near impossible for the government to have anybody pose as something they're not. Still, in an email, Dr. Friar adds that, aside from "philosophical indignation," he would likely not object should HHS try a similar tack with hospitalist groups.
"In essence, every patient that sees a hospitalist is a mystery patient," he writes. "We don't control who comes in, or what their diagnosis is or when they'll arrive. We have little control over any of those things. Since payors are collecting data on every aspect of hospitalists’ performance, and what they aren't, the hospitals we work for are, we have little left to hide."
A quickly-scuttled plan by the Department of Health and Human Services (HHS) to use "mystery shoppers" to test the availability of primary-care physicians (PCPs) would likely never be extended to HM—but the federal tack has certainly touched a nerve with some practitioners. (Ed note: updated June 29, 2011)
"Using surrogate patients or a sneaky way to get the data feels a bit less straightforward," says James Levy, PA-C, vice president of personnel for Traverse City-based Hospitalists of Northern Michigan. "If I were the government, I'm sure it would occur to me to do it that way. Nobody is going to be comfortable with surreptitious observations. That would seem to carry the message that the government doesn’t trust the providers it's reimbursing."
The government proposed using people pretending to be patients, both insured and uninsured, to gauge how long it takes to get PCP appointments. The action was proposed in the April 28 Federal Register. Comments were solicited, but negative feedback prompted HHS to drop the plan in a June 29 announcement.
Levy and David Friar, MD, CEO of Northern Hospitalists, say that the logistics of hospital admission make it near impossible for the government to have anybody pose as something they're not. Still, in an email, Dr. Friar adds that, aside from "philosophical indignation," he would likely not object should HHS try a similar tack with hospitalist groups.
"In essence, every patient that sees a hospitalist is a mystery patient," he writes. "We don't control who comes in, or what their diagnosis is or when they'll arrive. We have little control over any of those things. Since payors are collecting data on every aspect of hospitalists’ performance, and what they aren't, the hospitals we work for are, we have little left to hide."
A quickly-scuttled plan by the Department of Health and Human Services (HHS) to use "mystery shoppers" to test the availability of primary-care physicians (PCPs) would likely never be extended to HM—but the federal tack has certainly touched a nerve with some practitioners. (Ed note: updated June 29, 2011)
"Using surrogate patients or a sneaky way to get the data feels a bit less straightforward," says James Levy, PA-C, vice president of personnel for Traverse City-based Hospitalists of Northern Michigan. "If I were the government, I'm sure it would occur to me to do it that way. Nobody is going to be comfortable with surreptitious observations. That would seem to carry the message that the government doesn’t trust the providers it's reimbursing."
The government proposed using people pretending to be patients, both insured and uninsured, to gauge how long it takes to get PCP appointments. The action was proposed in the April 28 Federal Register. Comments were solicited, but negative feedback prompted HHS to drop the plan in a June 29 announcement.
Levy and David Friar, MD, CEO of Northern Hospitalists, say that the logistics of hospital admission make it near impossible for the government to have anybody pose as something they're not. Still, in an email, Dr. Friar adds that, aside from "philosophical indignation," he would likely not object should HHS try a similar tack with hospitalist groups.
"In essence, every patient that sees a hospitalist is a mystery patient," he writes. "We don't control who comes in, or what their diagnosis is or when they'll arrive. We have little control over any of those things. Since payors are collecting data on every aspect of hospitalists’ performance, and what they aren't, the hospitals we work for are, we have little left to hide."
Statewide Initiative To Tackle Hospital Readmissions, Infections
The Quality Institute of the Ohio Hospital Association (OHA) recently launched its fifth regional quality collaborative in the state, bringing together hospital administrators, physicians, and other clinicians to tackle statewide goals of reducing infections, readmissions, and adverse events while increasing patient satisfaction. A hospitalist involved in the initiative says it sets an example for other states to follow.
"Hospitalists are on the front lines of quality," says Craig Cairns, MD, MPH, a hospitalist and vice president of medical affairs at Licking Memorial Health Systems in Newark, Ohio. "But it helps to get a statewide or regional group together to share problems and potential solutions."
Licking Memorial, for example, participates in OHA's statewide quality initiatives, including one on physician handwashing and STAAR (State Action on Avoidable Rehospitalizations), a multistate care-transitions initiative developed by the Institute for Healthcare Improvement.
Licking has set a goal of reducing its readmission rate to 10.5%, Dr. Cairns says. "Trying to get the patient back to the primary medical home as quickly as possible can be difficult," he adds. "We work with support people at medical offices to try to ensure a spot for our patients going home."
Because one of the risk factors for preventable rehospitalizations is heart failure, Licking also recently instituted a heart-failure clinic, staffed by two hospital cardiologists.
The first of the regional collaborations started in the Dayton area in 1998, according to David Engler, PhD, vice president of OHA's Quality Institute. The collaborative has posted a 36% reduction in heart-attack mortality over the past three years, the equivalent of 52 lives saved.
"We were brought in to help them on a specific issue: a higher-than-expected acute myocardial infarction mortality rate," Dr. Engler says. "We held collaborative meetings, developed risk management models, and began to track data across sites." Peer-review protocols developed by OHA make it possible to share quality data among the participating hospitals, with participants agreeing not to use these for marketing or competitive advantage.
A total of 133 hospitals participate in one of OHA's regional or statewide quality collaborations.
The Quality Institute of the Ohio Hospital Association (OHA) recently launched its fifth regional quality collaborative in the state, bringing together hospital administrators, physicians, and other clinicians to tackle statewide goals of reducing infections, readmissions, and adverse events while increasing patient satisfaction. A hospitalist involved in the initiative says it sets an example for other states to follow.
"Hospitalists are on the front lines of quality," says Craig Cairns, MD, MPH, a hospitalist and vice president of medical affairs at Licking Memorial Health Systems in Newark, Ohio. "But it helps to get a statewide or regional group together to share problems and potential solutions."
Licking Memorial, for example, participates in OHA's statewide quality initiatives, including one on physician handwashing and STAAR (State Action on Avoidable Rehospitalizations), a multistate care-transitions initiative developed by the Institute for Healthcare Improvement.
Licking has set a goal of reducing its readmission rate to 10.5%, Dr. Cairns says. "Trying to get the patient back to the primary medical home as quickly as possible can be difficult," he adds. "We work with support people at medical offices to try to ensure a spot for our patients going home."
Because one of the risk factors for preventable rehospitalizations is heart failure, Licking also recently instituted a heart-failure clinic, staffed by two hospital cardiologists.
The first of the regional collaborations started in the Dayton area in 1998, according to David Engler, PhD, vice president of OHA's Quality Institute. The collaborative has posted a 36% reduction in heart-attack mortality over the past three years, the equivalent of 52 lives saved.
"We were brought in to help them on a specific issue: a higher-than-expected acute myocardial infarction mortality rate," Dr. Engler says. "We held collaborative meetings, developed risk management models, and began to track data across sites." Peer-review protocols developed by OHA make it possible to share quality data among the participating hospitals, with participants agreeing not to use these for marketing or competitive advantage.
A total of 133 hospitals participate in one of OHA's regional or statewide quality collaborations.
The Quality Institute of the Ohio Hospital Association (OHA) recently launched its fifth regional quality collaborative in the state, bringing together hospital administrators, physicians, and other clinicians to tackle statewide goals of reducing infections, readmissions, and adverse events while increasing patient satisfaction. A hospitalist involved in the initiative says it sets an example for other states to follow.
"Hospitalists are on the front lines of quality," says Craig Cairns, MD, MPH, a hospitalist and vice president of medical affairs at Licking Memorial Health Systems in Newark, Ohio. "But it helps to get a statewide or regional group together to share problems and potential solutions."
Licking Memorial, for example, participates in OHA's statewide quality initiatives, including one on physician handwashing and STAAR (State Action on Avoidable Rehospitalizations), a multistate care-transitions initiative developed by the Institute for Healthcare Improvement.
Licking has set a goal of reducing its readmission rate to 10.5%, Dr. Cairns says. "Trying to get the patient back to the primary medical home as quickly as possible can be difficult," he adds. "We work with support people at medical offices to try to ensure a spot for our patients going home."
Because one of the risk factors for preventable rehospitalizations is heart failure, Licking also recently instituted a heart-failure clinic, staffed by two hospital cardiologists.
The first of the regional collaborations started in the Dayton area in 1998, according to David Engler, PhD, vice president of OHA's Quality Institute. The collaborative has posted a 36% reduction in heart-attack mortality over the past three years, the equivalent of 52 lives saved.
"We were brought in to help them on a specific issue: a higher-than-expected acute myocardial infarction mortality rate," Dr. Engler says. "We held collaborative meetings, developed risk management models, and began to track data across sites." Peer-review protocols developed by OHA make it possible to share quality data among the participating hospitals, with participants agreeing not to use these for marketing or competitive advantage.
A total of 133 hospitals participate in one of OHA's regional or statewide quality collaborations.