User login
Safety-net hospitals would be hurt by hospital-wide 30-day readmission penalties
Considering all readmissions within 30 days of discharge in the Hospital Readmissions Reduction Program would modestly increase the number of hospitals eligible for penalties and would have a bigger impact on safety-net hospitals, based on a study of two years of Medicare claims data from 3,443 hospitals.
“Transition to a hospital-wide measure would require an adjustment in the penalty formula to keep penalties in the same range for most hospitals and without a change in procedures would have a deleterious effect on safety-net hospitals,” according to Rachael B. Zuckerman, PhD, from the Department of Health and Human Services, Washington, and her co-authors.
Analyzing 6,807,899 admissions for hospital-wide readmission measures and 4,392,658 admissions for condition-specific measures, the researchers found that a condition-specific approach would result in 3,238 hospitals being eligible for penalties for at least one condition. A hospital-wide measure of readmissions would result in 76 additional hospitals being eligible for penalties based on one year of admissions data, and 128 additional hospitals based on 3 years of admissions data (NEJM 2017, 377:1551-58. DOI: 10.1056/NEJMsa1701791).
Moving to a hospital-wide measure of readmissions also would significantly increase mean annual penalty rates across all hospitals by 0.89% of base diagnosis-related group (DRG) payments or $393,000; 43% of hospitals would be penalized under this standard.
“Moving to the hospital-wide readmission measure would also substantially increase the disparity between safety-net and other hospitals: the mean penalty as a percentage of base DRG payments would be 0.41 percentage points ($198,000) higher among safety net hospitals,” the authors wrote.
“Since safety-net hospitals tend to perform slightly worse on the hospital-wide measure, they are more likely to receive a penalty, which would increase the disparity in penalties between the two groups.”
The study was supported by the Department of Health and Human Services. One author declared grants from funding bodies and universities outside the submitted work. One author is an associate editor of the New England Journal of Medicine. One author was an employee of the Department of Health and Human Services at the time of the study. No other conflicts of interest were declared.
Considering all readmissions within 30 days of discharge in the Hospital Readmissions Reduction Program would modestly increase the number of hospitals eligible for penalties and would have a bigger impact on safety-net hospitals, based on a study of two years of Medicare claims data from 3,443 hospitals.
“Transition to a hospital-wide measure would require an adjustment in the penalty formula to keep penalties in the same range for most hospitals and without a change in procedures would have a deleterious effect on safety-net hospitals,” according to Rachael B. Zuckerman, PhD, from the Department of Health and Human Services, Washington, and her co-authors.
Analyzing 6,807,899 admissions for hospital-wide readmission measures and 4,392,658 admissions for condition-specific measures, the researchers found that a condition-specific approach would result in 3,238 hospitals being eligible for penalties for at least one condition. A hospital-wide measure of readmissions would result in 76 additional hospitals being eligible for penalties based on one year of admissions data, and 128 additional hospitals based on 3 years of admissions data (NEJM 2017, 377:1551-58. DOI: 10.1056/NEJMsa1701791).
Moving to a hospital-wide measure of readmissions also would significantly increase mean annual penalty rates across all hospitals by 0.89% of base diagnosis-related group (DRG) payments or $393,000; 43% of hospitals would be penalized under this standard.
“Moving to the hospital-wide readmission measure would also substantially increase the disparity between safety-net and other hospitals: the mean penalty as a percentage of base DRG payments would be 0.41 percentage points ($198,000) higher among safety net hospitals,” the authors wrote.
“Since safety-net hospitals tend to perform slightly worse on the hospital-wide measure, they are more likely to receive a penalty, which would increase the disparity in penalties between the two groups.”
The study was supported by the Department of Health and Human Services. One author declared grants from funding bodies and universities outside the submitted work. One author is an associate editor of the New England Journal of Medicine. One author was an employee of the Department of Health and Human Services at the time of the study. No other conflicts of interest were declared.
Considering all readmissions within 30 days of discharge in the Hospital Readmissions Reduction Program would modestly increase the number of hospitals eligible for penalties and would have a bigger impact on safety-net hospitals, based on a study of two years of Medicare claims data from 3,443 hospitals.
“Transition to a hospital-wide measure would require an adjustment in the penalty formula to keep penalties in the same range for most hospitals and without a change in procedures would have a deleterious effect on safety-net hospitals,” according to Rachael B. Zuckerman, PhD, from the Department of Health and Human Services, Washington, and her co-authors.
Analyzing 6,807,899 admissions for hospital-wide readmission measures and 4,392,658 admissions for condition-specific measures, the researchers found that a condition-specific approach would result in 3,238 hospitals being eligible for penalties for at least one condition. A hospital-wide measure of readmissions would result in 76 additional hospitals being eligible for penalties based on one year of admissions data, and 128 additional hospitals based on 3 years of admissions data (NEJM 2017, 377:1551-58. DOI: 10.1056/NEJMsa1701791).
Moving to a hospital-wide measure of readmissions also would significantly increase mean annual penalty rates across all hospitals by 0.89% of base diagnosis-related group (DRG) payments or $393,000; 43% of hospitals would be penalized under this standard.
“Moving to the hospital-wide readmission measure would also substantially increase the disparity between safety-net and other hospitals: the mean penalty as a percentage of base DRG payments would be 0.41 percentage points ($198,000) higher among safety net hospitals,” the authors wrote.
“Since safety-net hospitals tend to perform slightly worse on the hospital-wide measure, they are more likely to receive a penalty, which would increase the disparity in penalties between the two groups.”
The study was supported by the Department of Health and Human Services. One author declared grants from funding bodies and universities outside the submitted work. One author is an associate editor of the New England Journal of Medicine. One author was an employee of the Department of Health and Human Services at the time of the study. No other conflicts of interest were declared.
FROM NEJM
Key clinical point: Adopting a hospital-wide measure of 30-day readmissions for the Hospital Readmissions Reduction Program would modestly increase the number of hospitals eligible for penalties and would have a bigger impact on safety-net hospitals.
Major finding: With a hospital-wide measure of readmissions in the Hospital Readmissions Reduction Program, the mean penalty as a percentage of base DRG payments would be 0.41 percentage points ($198,000) higher among safety net hospitals.
Data source: Analysis of two years of Medicare claims data from 3,443 hospitals.
Disclosures: The study was supported by the Department of Health and Human Services. One author declared grants from funding bodies and universities outside the submitted work. One author is an associated editor of the New England Journal of Medicine. One author was an employee of the Department of Health and Human Services at the time of the study. No other conflicts of interest were declared.
Robotic Nissen fundoplication has teaching advantages
Training level affected operative time for laparoscopic Nissen fundoplication, but not for the robotic procedure, according to Maureen D. Moore, MD, and her associates.
Further, in laparoscopic and robotic procedures, junior and senior assistant cohorts had similar outcome measures for estimated blood loss, length of stay, postoperative complications, and 30-day readmission rate.
“The robotic technique offers unique advantages as an educational platform and potentially allows for increased trainee participation without compromising perioperative outcomes,” researchers concluded.
They evaluated surgical times and outcomes for 105 patients; junior assistants (postgraduate year-3 surgery residents) were present in 29 laparoscopic and 44 robotic procedures and senior assistants (minimally invasive surgery fellows) assisted in 18 laparoscopic and 14 robotic procedures.
Median operative time was significantly shorter for the laparoscopic procedures, 112 minutes vs. 157 minutes for the robotic procedures (P less than 0.001). Plus, the median operative time was significantly lower when senior assistants were involved in the laparoscopic procedures, 85 minutes vs. 129 minutes for the junior assistants (P = 0.02).
For the robotic procedures, the median operative times were not significantly different based on the assistant’s level of training, 154 minutes vs. 158 minutes.
Read the full study in the Journal of Surgical Research (doi: 10.1016/j.jss.2017.05.127).
Training level affected operative time for laparoscopic Nissen fundoplication, but not for the robotic procedure, according to Maureen D. Moore, MD, and her associates.
Further, in laparoscopic and robotic procedures, junior and senior assistant cohorts had similar outcome measures for estimated blood loss, length of stay, postoperative complications, and 30-day readmission rate.
“The robotic technique offers unique advantages as an educational platform and potentially allows for increased trainee participation without compromising perioperative outcomes,” researchers concluded.
They evaluated surgical times and outcomes for 105 patients; junior assistants (postgraduate year-3 surgery residents) were present in 29 laparoscopic and 44 robotic procedures and senior assistants (minimally invasive surgery fellows) assisted in 18 laparoscopic and 14 robotic procedures.
Median operative time was significantly shorter for the laparoscopic procedures, 112 minutes vs. 157 minutes for the robotic procedures (P less than 0.001). Plus, the median operative time was significantly lower when senior assistants were involved in the laparoscopic procedures, 85 minutes vs. 129 minutes for the junior assistants (P = 0.02).
For the robotic procedures, the median operative times were not significantly different based on the assistant’s level of training, 154 minutes vs. 158 minutes.
Read the full study in the Journal of Surgical Research (doi: 10.1016/j.jss.2017.05.127).
Training level affected operative time for laparoscopic Nissen fundoplication, but not for the robotic procedure, according to Maureen D. Moore, MD, and her associates.
Further, in laparoscopic and robotic procedures, junior and senior assistant cohorts had similar outcome measures for estimated blood loss, length of stay, postoperative complications, and 30-day readmission rate.
“The robotic technique offers unique advantages as an educational platform and potentially allows for increased trainee participation without compromising perioperative outcomes,” researchers concluded.
They evaluated surgical times and outcomes for 105 patients; junior assistants (postgraduate year-3 surgery residents) were present in 29 laparoscopic and 44 robotic procedures and senior assistants (minimally invasive surgery fellows) assisted in 18 laparoscopic and 14 robotic procedures.
Median operative time was significantly shorter for the laparoscopic procedures, 112 minutes vs. 157 minutes for the robotic procedures (P less than 0.001). Plus, the median operative time was significantly lower when senior assistants were involved in the laparoscopic procedures, 85 minutes vs. 129 minutes for the junior assistants (P = 0.02).
For the robotic procedures, the median operative times were not significantly different based on the assistant’s level of training, 154 minutes vs. 158 minutes.
Read the full study in the Journal of Surgical Research (doi: 10.1016/j.jss.2017.05.127).
FROM THE JOURNAL OF SURGICAL RESEARCH
Antimalarial might fight Zika virus

Preclinical research suggests a drug used to prevent and treat malaria may also be effective against Zika virus.
Investigators found that chloroquine can protect human neural progenitors from infection with Zika virus.
The drug also decreased Zika-induced mortality in one mouse model and hindered transmission of Zika virus from mother to fetus in another mouse model.
Alexey Terskikh, PhD, of Sanford Burnham Prebys Medical Discovery Institute in La Jolla, California, and his colleagues conducted this research and disclosed the results in Scientific Reports.
“There is still an urgent need to bolster our preparedness and capacity to respond to the next Zika outbreak,” Dr Terskikh said. “Our latest research suggests the antimalaria drug chloroquine may be an effective drug to treat and prevent Zika infections.”
The investigators first found that chloroquine reduced Zika infection in primary human fetal neural precursor cells.
The team also discovered that chloroquine reduced the percentage of Zika-positive cells and the level of apoptosis in neurospheres derived from human induced pluripotent stem cells.
The investigators then tested chloroquine in AG129 mice, which lack receptors for type I and II interferons and have been used to model Zika virus infection.
Some of these mice received chloroquine (50 mg/kg/day) in their drinking water for 2 days and were then infected with Zika virus. The mice continued to receive chloroquine at the same dose for 5 days and then received 5 mg/kg/day until the end of the experiment.
Control AG129 mice were infected with Zika virus and received regular drinking water.
Compared to controls, chloroquine-treated mice had significantly prolonged survival and a significant reduction in Zika-induced weight loss (P<0.01 for both).
Next, the investigators used SJL mice to study horizontal and vertical transmission of Zika. They observed successful transmission of the virus from males to females and from mothers to pups.
The team then analyzed the effects of chloroquine on fetal transmission of Zika. Pregnant SJL mice were infected with Zika and given drinking water containing chloroquine (30 mg/kg/day) starting on day E13.5. The mice were euthanized on E18.5, and maternal blood and fetal brain samples were collected.
Chloroquine treatment resulted in a roughly 20-fold reduction in Zika virus titers in the maternal blood and fetal brain.
“Although chloroquine didn’t completely clear Zika from infected mice, it did reduce the viral load, suggesting it could limit the neurological damage found in newborns infected by the virus,” Dr Terskikh said.
“Chloroquine has a long history of successfully treating malaria, and there are no reports of it causing birth defects. Additional studies are certainly needed to determine the precise details of how it works. But given its low cost, availability, and safety history, further study in a clinical trial to test its effectiveness against Zika virus in humans is merited.” ![]()
Preclinical research suggests a drug used to prevent and treat malaria may also be effective against Zika virus.
Investigators found that chloroquine can protect human neural progenitors from infection with Zika virus.
The drug also decreased Zika-induced mortality in one mouse model and hindered transmission of Zika virus from mother to fetus in another mouse model.
Alexey Terskikh, PhD, of Sanford Burnham Prebys Medical Discovery Institute in La Jolla, California, and his colleagues conducted this research and disclosed the results in Scientific Reports.
“There is still an urgent need to bolster our preparedness and capacity to respond to the next Zika outbreak,” Dr Terskikh said. “Our latest research suggests the antimalaria drug chloroquine may be an effective drug to treat and prevent Zika infections.”
The investigators first found that chloroquine reduced Zika infection in primary human fetal neural precursor cells.
The team also discovered that chloroquine reduced the percentage of Zika-positive cells and the level of apoptosis in neurospheres derived from human induced pluripotent stem cells.
The investigators then tested chloroquine in AG129 mice, which lack receptors for type I and II interferons and have been used to model Zika virus infection.
Some of these mice received chloroquine (50 mg/kg/day) in their drinking water for 2 days and were then infected with Zika virus. The mice continued to receive chloroquine at the same dose for 5 days and then received 5 mg/kg/day until the end of the experiment.
Control AG129 mice were infected with Zika virus and received regular drinking water.
Compared to controls, chloroquine-treated mice had significantly prolonged survival and a significant reduction in Zika-induced weight loss (P<0.01 for both).
Next, the investigators used SJL mice to study horizontal and vertical transmission of Zika. They observed successful transmission of the virus from males to females and from mothers to pups.
The team then analyzed the effects of chloroquine on fetal transmission of Zika. Pregnant SJL mice were infected with Zika and given drinking water containing chloroquine (30 mg/kg/day) starting on day E13.5. The mice were euthanized on E18.5, and maternal blood and fetal brain samples were collected.
Chloroquine treatment resulted in a roughly 20-fold reduction in Zika virus titers in the maternal blood and fetal brain.
“Although chloroquine didn’t completely clear Zika from infected mice, it did reduce the viral load, suggesting it could limit the neurological damage found in newborns infected by the virus,” Dr Terskikh said.
“Chloroquine has a long history of successfully treating malaria, and there are no reports of it causing birth defects. Additional studies are certainly needed to determine the precise details of how it works. But given its low cost, availability, and safety history, further study in a clinical trial to test its effectiveness against Zika virus in humans is merited.” ![]()
Preclinical research suggests a drug used to prevent and treat malaria may also be effective against Zika virus.
Investigators found that chloroquine can protect human neural progenitors from infection with Zika virus.
The drug also decreased Zika-induced mortality in one mouse model and hindered transmission of Zika virus from mother to fetus in another mouse model.
Alexey Terskikh, PhD, of Sanford Burnham Prebys Medical Discovery Institute in La Jolla, California, and his colleagues conducted this research and disclosed the results in Scientific Reports.
“There is still an urgent need to bolster our preparedness and capacity to respond to the next Zika outbreak,” Dr Terskikh said. “Our latest research suggests the antimalaria drug chloroquine may be an effective drug to treat and prevent Zika infections.”
The investigators first found that chloroquine reduced Zika infection in primary human fetal neural precursor cells.
The team also discovered that chloroquine reduced the percentage of Zika-positive cells and the level of apoptosis in neurospheres derived from human induced pluripotent stem cells.
The investigators then tested chloroquine in AG129 mice, which lack receptors for type I and II interferons and have been used to model Zika virus infection.
Some of these mice received chloroquine (50 mg/kg/day) in their drinking water for 2 days and were then infected with Zika virus. The mice continued to receive chloroquine at the same dose for 5 days and then received 5 mg/kg/day until the end of the experiment.
Control AG129 mice were infected with Zika virus and received regular drinking water.
Compared to controls, chloroquine-treated mice had significantly prolonged survival and a significant reduction in Zika-induced weight loss (P<0.01 for both).
Next, the investigators used SJL mice to study horizontal and vertical transmission of Zika. They observed successful transmission of the virus from males to females and from mothers to pups.
The team then analyzed the effects of chloroquine on fetal transmission of Zika. Pregnant SJL mice were infected with Zika and given drinking water containing chloroquine (30 mg/kg/day) starting on day E13.5. The mice were euthanized on E18.5, and maternal blood and fetal brain samples were collected.
Chloroquine treatment resulted in a roughly 20-fold reduction in Zika virus titers in the maternal blood and fetal brain.
“Although chloroquine didn’t completely clear Zika from infected mice, it did reduce the viral load, suggesting it could limit the neurological damage found in newborns infected by the virus,” Dr Terskikh said.
“Chloroquine has a long history of successfully treating malaria, and there are no reports of it causing birth defects. Additional studies are certainly needed to determine the precise details of how it works. But given its low cost, availability, and safety history, further study in a clinical trial to test its effectiveness against Zika virus in humans is merited.” ![]()
Educational innovation
My son’s in medical school,” Fred told me. “First year.”
“Which school?”
Fred named a newly-chartered one.
“Really innovative curriculum,” he said.
“He got into a different school, too,” he said, “but he didn’t like how that school had canceled all lectures. Students just watch the lecture videos on their devices, so they scratched all the live ones.”
“Now that they don’t need lecturers, did they cut tuition?” I asked.
We both chuckled.

I was exposed to educational innovation on Day One of medical school, in the fall of 1968. (Go ahead, do the math.) The Dean addressed our entering class. “We’ve abolished tests,” he said. “We cut preclinical study from 2 years to 18 months, followed by one comprehensive exam.
"We want to get you into the clinic right away," he said, "because you chose to be doctors to help people. Even during the preclinical period, you’ll be getting not just dry frontal teaching but exposure to actual patients.”
We guessed that sounded good, especially the part about no tests. Less day-to-day studying. A lot less.
We still had lectures, of course, on biochemistry, anatomy, and so forth (they ran out of time and did all four extremities in 1 hour), but we felt little need to pay close attention. After all, any details we’d have to memorize would be diluted over three semesters, and 18 months was so far away.
As to the lectures themselves, I’ve never been diagnosed with actual narcolepsy, but when they turned out the lights and started showing slides, I fell fast asleep. Always have.
Most of us passed the comprehensive exam and moved on to the clinic. (We didn’t get into med school without knowing how to take tests, did we?)
The basic science faculty hated this educational innovation and recognized – correctly – that without tests we would take their classes less seriously. A couple of years later, the students themselves demanded that the tests be reinstated; lack of regular, numerical feedback made them anxious. So it was back to exams every 2 weeks. Poor devils.
Fast forward 40 years. Over lunch at a friend’s house, I recently met a young woman in her second year at a medical school in Chicago. “The school has an exciting, innovative curriculum,” she told me.
“No kidding,” I said. “What’s that?”
“They want to get us into the clinic as soon as possible,” she replied. “So they cut preclinical years to 18 months. And no regular tests. Just a comprehensive exam at the end. Much less day-to-day pressure.”
“Very innovative,” I observed.
***
Once in practice, I joined the clinical faculty of a local medical school. For 35 years, I hosted senior medical students for a month-long elective. During that time, I tried to pass on some of the things that weren’t on the standard academic curriculum. For instance, that patients have their own ideas about what is wrong with them, how it got that way, and what to do about it. That medical advice is less an order than a negotiation. Students seemed to find such notions – and their daily illustrations in the office – of some interest.
I put these and related deep thoughts between the covers of a book and sent a copy to the medical school registrar, from whom I had heard little over the years unless my student evaluations were 2 days late. My cover letter suggested that the school’s educators might be curious about what I’d been teaching their charges for 35 years.

The registrar replied by e-mail. “Thank you for your book,” she wrote. “I showed it to our dean of education, who told me that we would definitely not be changing our curriculum on the basis of your book. You might contact the medical librarian to see if they want to display it.”
I didn’t recall thinking, much less saying, that my ruminations ought to overturn the medical curriculum. I just thought they might be of some passing interest, coming as they did from an outside perspective.
Not so much, it turns out.
There are, in any event, so many exciting vistas of educational innovation to develop: genomics, precision medicine, and folding doctors into health care teams, and replacing clinical judgment with algorithms. The next generation of physicians, innovatively educated, will surpass all predecessors, just as we did.
Anyhow, they'll be bound to know more about the origins and insertions of the leg muscles. They could hardly know any less.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@frontlinemedcom.com.
My son’s in medical school,” Fred told me. “First year.”
“Which school?”
Fred named a newly-chartered one.
“Really innovative curriculum,” he said.
“He got into a different school, too,” he said, “but he didn’t like how that school had canceled all lectures. Students just watch the lecture videos on their devices, so they scratched all the live ones.”
“Now that they don’t need lecturers, did they cut tuition?” I asked.
We both chuckled.
I was exposed to educational innovation on Day One of medical school, in the fall of 1968. (Go ahead, do the math.) The Dean addressed our entering class. “We’ve abolished tests,” he said. “We cut preclinical study from 2 years to 18 months, followed by one comprehensive exam.
"We want to get you into the clinic right away," he said, "because you chose to be doctors to help people. Even during the preclinical period, you’ll be getting not just dry frontal teaching but exposure to actual patients.”
We guessed that sounded good, especially the part about no tests. Less day-to-day studying. A lot less.
We still had lectures, of course, on biochemistry, anatomy, and so forth (they ran out of time and did all four extremities in 1 hour), but we felt little need to pay close attention. After all, any details we’d have to memorize would be diluted over three semesters, and 18 months was so far away.
As to the lectures themselves, I’ve never been diagnosed with actual narcolepsy, but when they turned out the lights and started showing slides, I fell fast asleep. Always have.
Most of us passed the comprehensive exam and moved on to the clinic. (We didn’t get into med school without knowing how to take tests, did we?)
The basic science faculty hated this educational innovation and recognized – correctly – that without tests we would take their classes less seriously. A couple of years later, the students themselves demanded that the tests be reinstated; lack of regular, numerical feedback made them anxious. So it was back to exams every 2 weeks. Poor devils.
Fast forward 40 years. Over lunch at a friend’s house, I recently met a young woman in her second year at a medical school in Chicago. “The school has an exciting, innovative curriculum,” she told me.
“No kidding,” I said. “What’s that?”
“They want to get us into the clinic as soon as possible,” she replied. “So they cut preclinical years to 18 months. And no regular tests. Just a comprehensive exam at the end. Much less day-to-day pressure.”
“Very innovative,” I observed.
***
Once in practice, I joined the clinical faculty of a local medical school. For 35 years, I hosted senior medical students for a month-long elective. During that time, I tried to pass on some of the things that weren’t on the standard academic curriculum. For instance, that patients have their own ideas about what is wrong with them, how it got that way, and what to do about it. That medical advice is less an order than a negotiation. Students seemed to find such notions – and their daily illustrations in the office – of some interest.
I put these and related deep thoughts between the covers of a book and sent a copy to the medical school registrar, from whom I had heard little over the years unless my student evaluations were 2 days late. My cover letter suggested that the school’s educators might be curious about what I’d been teaching their charges for 35 years.
The registrar replied by e-mail. “Thank you for your book,” she wrote. “I showed it to our dean of education, who told me that we would definitely not be changing our curriculum on the basis of your book. You might contact the medical librarian to see if they want to display it.”
I didn’t recall thinking, much less saying, that my ruminations ought to overturn the medical curriculum. I just thought they might be of some passing interest, coming as they did from an outside perspective.
Not so much, it turns out.
There are, in any event, so many exciting vistas of educational innovation to develop: genomics, precision medicine, and folding doctors into health care teams, and replacing clinical judgment with algorithms. The next generation of physicians, innovatively educated, will surpass all predecessors, just as we did.
Anyhow, they'll be bound to know more about the origins and insertions of the leg muscles. They could hardly know any less.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@frontlinemedcom.com.
My son’s in medical school,” Fred told me. “First year.”
“Which school?”
Fred named a newly-chartered one.
“Really innovative curriculum,” he said.
“He got into a different school, too,” he said, “but he didn’t like how that school had canceled all lectures. Students just watch the lecture videos on their devices, so they scratched all the live ones.”
“Now that they don’t need lecturers, did they cut tuition?” I asked.
We both chuckled.
I was exposed to educational innovation on Day One of medical school, in the fall of 1968. (Go ahead, do the math.) The Dean addressed our entering class. “We’ve abolished tests,” he said. “We cut preclinical study from 2 years to 18 months, followed by one comprehensive exam.
"We want to get you into the clinic right away," he said, "because you chose to be doctors to help people. Even during the preclinical period, you’ll be getting not just dry frontal teaching but exposure to actual patients.”
We guessed that sounded good, especially the part about no tests. Less day-to-day studying. A lot less.
We still had lectures, of course, on biochemistry, anatomy, and so forth (they ran out of time and did all four extremities in 1 hour), but we felt little need to pay close attention. After all, any details we’d have to memorize would be diluted over three semesters, and 18 months was so far away.
As to the lectures themselves, I’ve never been diagnosed with actual narcolepsy, but when they turned out the lights and started showing slides, I fell fast asleep. Always have.
Most of us passed the comprehensive exam and moved on to the clinic. (We didn’t get into med school without knowing how to take tests, did we?)
The basic science faculty hated this educational innovation and recognized – correctly – that without tests we would take their classes less seriously. A couple of years later, the students themselves demanded that the tests be reinstated; lack of regular, numerical feedback made them anxious. So it was back to exams every 2 weeks. Poor devils.
Fast forward 40 years. Over lunch at a friend’s house, I recently met a young woman in her second year at a medical school in Chicago. “The school has an exciting, innovative curriculum,” she told me.
“No kidding,” I said. “What’s that?”
“They want to get us into the clinic as soon as possible,” she replied. “So they cut preclinical years to 18 months. And no regular tests. Just a comprehensive exam at the end. Much less day-to-day pressure.”
“Very innovative,” I observed.
***
Once in practice, I joined the clinical faculty of a local medical school. For 35 years, I hosted senior medical students for a month-long elective. During that time, I tried to pass on some of the things that weren’t on the standard academic curriculum. For instance, that patients have their own ideas about what is wrong with them, how it got that way, and what to do about it. That medical advice is less an order than a negotiation. Students seemed to find such notions – and their daily illustrations in the office – of some interest.
I put these and related deep thoughts between the covers of a book and sent a copy to the medical school registrar, from whom I had heard little over the years unless my student evaluations were 2 days late. My cover letter suggested that the school’s educators might be curious about what I’d been teaching their charges for 35 years.
The registrar replied by e-mail. “Thank you for your book,” she wrote. “I showed it to our dean of education, who told me that we would definitely not be changing our curriculum on the basis of your book. You might contact the medical librarian to see if they want to display it.”
I didn’t recall thinking, much less saying, that my ruminations ought to overturn the medical curriculum. I just thought they might be of some passing interest, coming as they did from an outside perspective.
Not so much, it turns out.
There are, in any event, so many exciting vistas of educational innovation to develop: genomics, precision medicine, and folding doctors into health care teams, and replacing clinical judgment with algorithms. The next generation of physicians, innovatively educated, will surpass all predecessors, just as we did.
Anyhow, they'll be bound to know more about the origins and insertions of the leg muscles. They could hardly know any less.
Dr. Rockoff practices dermatology in Brookline, Mass., and is a longtime contributor to Dermatology News. He serves on the clinical faculty at Tufts University, Boston, and has taught senior medical students and other trainees for 30 years. His second book, “Act Like a Doctor, Think Like a Patient,” is available at amazon.com and barnesandnoble.com. Write to him at dermnews@frontlinemedcom.com.
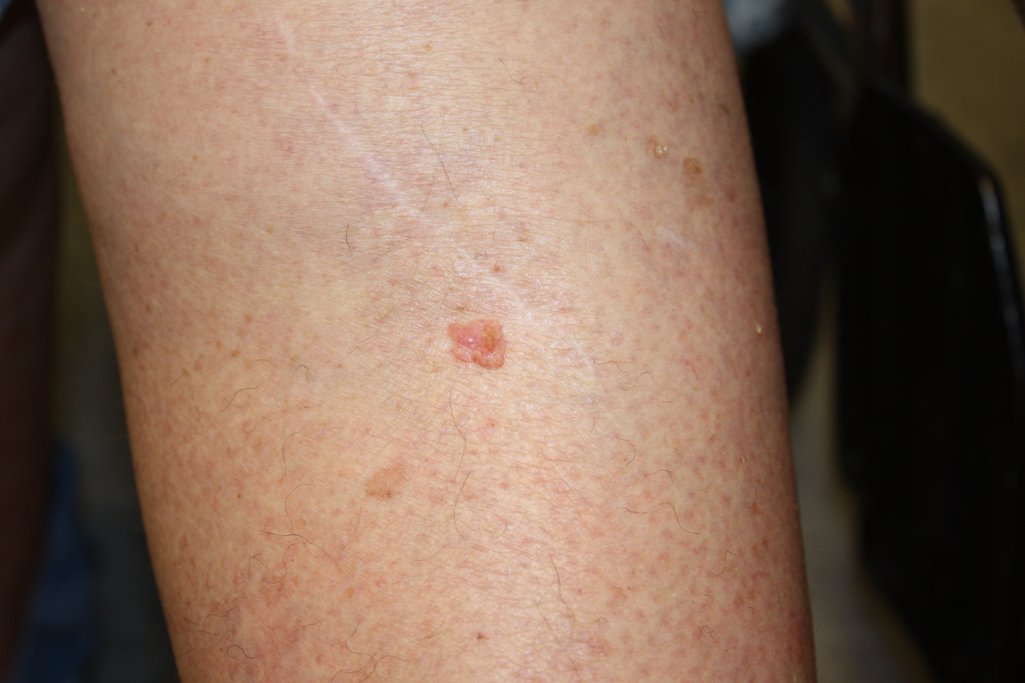
Squamous cell carcinoma linked to 25% increase in all-cause mortality
FROM JAAD
Squamous cell carcinomas (SCC), but not basal cell carcinomas (BCC), were associated with a risk of death from any cause that was 25% higher than that seen in the general population, based on a systematic literature review and meta-analysis published in the Journal of American Academy of Dermatology (2017. doi: 10.1016/j.jaad.2017.11.026).
“Because these tumors often occur in the same patients and are both often caused by exposure to ultraviolet radiation, patients with BCC and SCC are often grouped together,” Mackenzie R. Wehner, MD, of the University of Pennsylvania, Philadelphia, and co-authors wrote. “Our data contributes to the argument that the carcinogenesis of these tumors and long-term outcomes for patients with these tumors may be distinct.”
Patients with SCC “may need additional education and age-appropriate screening to prevent deaths from major diseases,” the authors concluded.
Dr. Wehner and colleagues systematically searched the medical literature and found four studies encompassing a total of 175,849 patients with SCC and 464,230 patients with BCC.
Relative to the general population, mortality for those with an SCC was 1.25 (95% CI, 1.17-1.32). At 0.92 (95% CI 0.83-1.02), there was no significant difference in mortality for patients with a BCC.
Collectively and individually, the studies found a statistically significant increased relative mortality for having SCC.
There are clear distinctions between BCC and SCC with regard to histology, pathophysiology, survival, and other parameters, the study authors said. “While many patients get both BCC and SCC, future research should take into account that these cancers may have different long-term risks and outcomes.”
FROM JAAD
Squamous cell carcinomas (SCC), but not basal cell carcinomas (BCC), were associated with a risk of death from any cause that was 25% higher than that seen in the general population, based on a systematic literature review and meta-analysis published in the Journal of American Academy of Dermatology (2017. doi: 10.1016/j.jaad.2017.11.026).
“Because these tumors often occur in the same patients and are both often caused by exposure to ultraviolet radiation, patients with BCC and SCC are often grouped together,” Mackenzie R. Wehner, MD, of the University of Pennsylvania, Philadelphia, and co-authors wrote. “Our data contributes to the argument that the carcinogenesis of these tumors and long-term outcomes for patients with these tumors may be distinct.”
Patients with SCC “may need additional education and age-appropriate screening to prevent deaths from major diseases,” the authors concluded.
Dr. Wehner and colleagues systematically searched the medical literature and found four studies encompassing a total of 175,849 patients with SCC and 464,230 patients with BCC.
Relative to the general population, mortality for those with an SCC was 1.25 (95% CI, 1.17-1.32). At 0.92 (95% CI 0.83-1.02), there was no significant difference in mortality for patients with a BCC.
Collectively and individually, the studies found a statistically significant increased relative mortality for having SCC.
There are clear distinctions between BCC and SCC with regard to histology, pathophysiology, survival, and other parameters, the study authors said. “While many patients get both BCC and SCC, future research should take into account that these cancers may have different long-term risks and outcomes.”
FROM JAAD
Squamous cell carcinomas (SCC), but not basal cell carcinomas (BCC), were associated with a risk of death from any cause that was 25% higher than that seen in the general population, based on a systematic literature review and meta-analysis published in the Journal of American Academy of Dermatology (2017. doi: 10.1016/j.jaad.2017.11.026).
“Because these tumors often occur in the same patients and are both often caused by exposure to ultraviolet radiation, patients with BCC and SCC are often grouped together,” Mackenzie R. Wehner, MD, of the University of Pennsylvania, Philadelphia, and co-authors wrote. “Our data contributes to the argument that the carcinogenesis of these tumors and long-term outcomes for patients with these tumors may be distinct.”
Patients with SCC “may need additional education and age-appropriate screening to prevent deaths from major diseases,” the authors concluded.
Dr. Wehner and colleagues systematically searched the medical literature and found four studies encompassing a total of 175,849 patients with SCC and 464,230 patients with BCC.
Relative to the general population, mortality for those with an SCC was 1.25 (95% CI, 1.17-1.32). At 0.92 (95% CI 0.83-1.02), there was no significant difference in mortality for patients with a BCC.
Collectively and individually, the studies found a statistically significant increased relative mortality for having SCC.
There are clear distinctions between BCC and SCC with regard to histology, pathophysiology, survival, and other parameters, the study authors said. “While many patients get both BCC and SCC, future research should take into account that these cancers may have different long-term risks and outcomes.”
Studies need to address best follow-on therapy to ibrutinib in CLL
Reporting AT LYMPHOMA & MYELOMA 2017
NEW YORK – Clinical trials are needed to determine the best follow-on therapies when patients discontinue the ibrutinib due to adverse events or disease progression, according to a leading expert on chronic lymphocytic leukemia (CLL).
Anthony Mato, MD, MSCE, from the Abramson Cancer Center at the University of Pennsylvania in Philadelphia, discussed how real-world experience with the use of ibrutinib (Imbruvica) can fill the gaps in knowledge left by clinical trials and point to the need for further study.
“Regulatory bodies around the world are more and more interested in what’s going on in the clinic, and there is a question about whether or not the experiences for patients that we take care of might actually answer some important questions that aren’t easily answered in the context of clinical research,” he said at the annual Lymphoma & Myeloma International Congress on Hematologic Malignancies here.
“Are the experiences in practice with novel agents similar to experiences from clinical trials? I think that’s very important,” he added.
Other important questions that real-world experience may help to answer include whether it’s possible to refine adverse event profiles and reasons for ibrutinib discontinuation, what therapies should be prescribed after ibrutinib, and what is the optimal sequencing of therapies for CLL.
For example, in the RESONATE-2 trial, an open-label, international phase 3 study comparing ibrutinib with chlorambucil in previously untreated patients 65 and older, ibrutinib was found to be superior to chlorambucil in terms of progression-free survival (PFS), overall survival (OS), response rate, and improvements in hematologic variables.
However, this trial excluded patients with the deleterious chromosome 17p deletion (del17p) and included only patients 65 and older, a population that does not necessarily reflect clinical experience.
To get a better sense of how ibrutinib is used to treat CLL in the front-line setting Dr. Mato and colleagues conducted a retrospective cohort study of 391 patients treated in 19 US and international academic and community centers.
The median age of the sample was 68 years, but 41% of the patients were younger than 65. In all, 62% were male, and 80% had Rai stage 2 or greater disease. Genetic analyses showed that 30% of the patients were positive for del17p, and 17% had both del17p and the 11q deletion (del11q). Mutations in TP53 were seen in 20% of patients, 23% had a complex karyotype, and 67% had an unmutated immuglobulin heavy chain variable region (IGHV). Only 57 patients (14.5%) were classified as genetically low risk.

Additionally, only 79 of the 391 patients had complete data for CLL International Prognostic Index (CLL-IPI) scoring, “which goes, I think, to show how often this is actually being tested and utilized in clinical practice,” Dr. Mato said.
Off-label use of ibrutinib in combination therapy was given to 16% of patients, most commonly with an anti-CD20 inhibitor such as rituximab.
In all, 17% of patients required permanent dose reductions; and 42% had a dose interruption, with a median hiatus of 12 days.
Grade 3 or 4 adverse events were uncommon, but more than 20% of patients experienced arthralgias or myalgias of any kind, about 19% reported fatigue, 18% had dermatologic toxicities, 18% reported bruising, 17% had diarrhea or colitis and 15% had infections.
The toxicities seen in RESONATE-2 were somewhat similar, but generally occurred in higher frequencies in the trial than in real-world practice.
Dr. Mato and colleagues found that at a median of 12 months of follow-up, 24% of patients had discontinued ibrutinib. In contrast, in RESONATE-2, after 18 months of follow-up, 13% of patients had discontinued the drug.
The most common reasons for discontinuation in clinical practice were for toxicities (59.5% of 94 discontinuations) including atrial fibrillation in 20% of the patients who discontinued, arthralgias/myalgias and skin toxicities in 14.5% each, and bleeding in 9.1%.
Other reasons for discontinuation included Richter’s transformation in 9.6%, doctor or patient preference in 7.4%, and deaths that were not secondary to CLL progression in 3.2%.
“We also tried to get a sense of whether or not cost was a factor for patients, and in this series and the relapsed refractory setting, 1% or less of patients discontinued due to financial issues,” Dr. Mato said.
Outcomes in the real word were quite good, he noted, with an overall response rate (ORR) of 81.7%, which included 17.4% complete responses (CR), Neither median PFS nor OS have been reached and the respective PFS and OS at 12 months were 92% and 95%. The respective PFS and OS rates for patients with del17p were 87% and 89%. An analysis of predictors of survival showed that only the presence of del17p was associated with inferior PFS (odds ratio 1.91, P = .035)
Dr. Mato noted that there was no clear standard treatment approach for patients who discontinued ibrutinib or for whom ibrutinib did not work. The top three second-line approaches used included an anti-CD20 agent combined with chlorambucil, venetoclax (Venclexta), or a different kinase inhibitor. Chemoimmunotherapy with either fludarabine, cyclophosphamide, and rituximab or bendamustine and rituximab was given to only 5 patients as a second line therapy.
Dr. Mato disclosed serving as a consultant for AbbVie, AstraZeneca, Janssen/Pharmacyclics, and TG Therapeutics.
Reporting AT LYMPHOMA & MYELOMA 2017
NEW YORK – Clinical trials are needed to determine the best follow-on therapies when patients discontinue the ibrutinib due to adverse events or disease progression, according to a leading expert on chronic lymphocytic leukemia (CLL).
Anthony Mato, MD, MSCE, from the Abramson Cancer Center at the University of Pennsylvania in Philadelphia, discussed how real-world experience with the use of ibrutinib (Imbruvica) can fill the gaps in knowledge left by clinical trials and point to the need for further study.
“Regulatory bodies around the world are more and more interested in what’s going on in the clinic, and there is a question about whether or not the experiences for patients that we take care of might actually answer some important questions that aren’t easily answered in the context of clinical research,” he said at the annual Lymphoma & Myeloma International Congress on Hematologic Malignancies here.
“Are the experiences in practice with novel agents similar to experiences from clinical trials? I think that’s very important,” he added.
Other important questions that real-world experience may help to answer include whether it’s possible to refine adverse event profiles and reasons for ibrutinib discontinuation, what therapies should be prescribed after ibrutinib, and what is the optimal sequencing of therapies for CLL.
For example, in the RESONATE-2 trial, an open-label, international phase 3 study comparing ibrutinib with chlorambucil in previously untreated patients 65 and older, ibrutinib was found to be superior to chlorambucil in terms of progression-free survival (PFS), overall survival (OS), response rate, and improvements in hematologic variables.
However, this trial excluded patients with the deleterious chromosome 17p deletion (del17p) and included only patients 65 and older, a population that does not necessarily reflect clinical experience.
To get a better sense of how ibrutinib is used to treat CLL in the front-line setting Dr. Mato and colleagues conducted a retrospective cohort study of 391 patients treated in 19 US and international academic and community centers.
The median age of the sample was 68 years, but 41% of the patients were younger than 65. In all, 62% were male, and 80% had Rai stage 2 or greater disease. Genetic analyses showed that 30% of the patients were positive for del17p, and 17% had both del17p and the 11q deletion (del11q). Mutations in TP53 were seen in 20% of patients, 23% had a complex karyotype, and 67% had an unmutated immuglobulin heavy chain variable region (IGHV). Only 57 patients (14.5%) were classified as genetically low risk.
Additionally, only 79 of the 391 patients had complete data for CLL International Prognostic Index (CLL-IPI) scoring, “which goes, I think, to show how often this is actually being tested and utilized in clinical practice,” Dr. Mato said.
Off-label use of ibrutinib in combination therapy was given to 16% of patients, most commonly with an anti-CD20 inhibitor such as rituximab.
In all, 17% of patients required permanent dose reductions; and 42% had a dose interruption, with a median hiatus of 12 days.
Grade 3 or 4 adverse events were uncommon, but more than 20% of patients experienced arthralgias or myalgias of any kind, about 19% reported fatigue, 18% had dermatologic toxicities, 18% reported bruising, 17% had diarrhea or colitis and 15% had infections.
The toxicities seen in RESONATE-2 were somewhat similar, but generally occurred in higher frequencies in the trial than in real-world practice.
Dr. Mato and colleagues found that at a median of 12 months of follow-up, 24% of patients had discontinued ibrutinib. In contrast, in RESONATE-2, after 18 months of follow-up, 13% of patients had discontinued the drug.
The most common reasons for discontinuation in clinical practice were for toxicities (59.5% of 94 discontinuations) including atrial fibrillation in 20% of the patients who discontinued, arthralgias/myalgias and skin toxicities in 14.5% each, and bleeding in 9.1%.
Other reasons for discontinuation included Richter’s transformation in 9.6%, doctor or patient preference in 7.4%, and deaths that were not secondary to CLL progression in 3.2%.
“We also tried to get a sense of whether or not cost was a factor for patients, and in this series and the relapsed refractory setting, 1% or less of patients discontinued due to financial issues,” Dr. Mato said.
Outcomes in the real word were quite good, he noted, with an overall response rate (ORR) of 81.7%, which included 17.4% complete responses (CR), Neither median PFS nor OS have been reached and the respective PFS and OS at 12 months were 92% and 95%. The respective PFS and OS rates for patients with del17p were 87% and 89%. An analysis of predictors of survival showed that only the presence of del17p was associated with inferior PFS (odds ratio 1.91, P = .035)
Dr. Mato noted that there was no clear standard treatment approach for patients who discontinued ibrutinib or for whom ibrutinib did not work. The top three second-line approaches used included an anti-CD20 agent combined with chlorambucil, venetoclax (Venclexta), or a different kinase inhibitor. Chemoimmunotherapy with either fludarabine, cyclophosphamide, and rituximab or bendamustine and rituximab was given to only 5 patients as a second line therapy.
Dr. Mato disclosed serving as a consultant for AbbVie, AstraZeneca, Janssen/Pharmacyclics, and TG Therapeutics.
Reporting AT LYMPHOMA & MYELOMA 2017
NEW YORK – Clinical trials are needed to determine the best follow-on therapies when patients discontinue the ibrutinib due to adverse events or disease progression, according to a leading expert on chronic lymphocytic leukemia (CLL).
Anthony Mato, MD, MSCE, from the Abramson Cancer Center at the University of Pennsylvania in Philadelphia, discussed how real-world experience with the use of ibrutinib (Imbruvica) can fill the gaps in knowledge left by clinical trials and point to the need for further study.
“Regulatory bodies around the world are more and more interested in what’s going on in the clinic, and there is a question about whether or not the experiences for patients that we take care of might actually answer some important questions that aren’t easily answered in the context of clinical research,” he said at the annual Lymphoma & Myeloma International Congress on Hematologic Malignancies here.
“Are the experiences in practice with novel agents similar to experiences from clinical trials? I think that’s very important,” he added.
Other important questions that real-world experience may help to answer include whether it’s possible to refine adverse event profiles and reasons for ibrutinib discontinuation, what therapies should be prescribed after ibrutinib, and what is the optimal sequencing of therapies for CLL.
For example, in the RESONATE-2 trial, an open-label, international phase 3 study comparing ibrutinib with chlorambucil in previously untreated patients 65 and older, ibrutinib was found to be superior to chlorambucil in terms of progression-free survival (PFS), overall survival (OS), response rate, and improvements in hematologic variables.
However, this trial excluded patients with the deleterious chromosome 17p deletion (del17p) and included only patients 65 and older, a population that does not necessarily reflect clinical experience.
To get a better sense of how ibrutinib is used to treat CLL in the front-line setting Dr. Mato and colleagues conducted a retrospective cohort study of 391 patients treated in 19 US and international academic and community centers.
The median age of the sample was 68 years, but 41% of the patients were younger than 65. In all, 62% were male, and 80% had Rai stage 2 or greater disease. Genetic analyses showed that 30% of the patients were positive for del17p, and 17% had both del17p and the 11q deletion (del11q). Mutations in TP53 were seen in 20% of patients, 23% had a complex karyotype, and 67% had an unmutated immuglobulin heavy chain variable region (IGHV). Only 57 patients (14.5%) were classified as genetically low risk.
Additionally, only 79 of the 391 patients had complete data for CLL International Prognostic Index (CLL-IPI) scoring, “which goes, I think, to show how often this is actually being tested and utilized in clinical practice,” Dr. Mato said.
Off-label use of ibrutinib in combination therapy was given to 16% of patients, most commonly with an anti-CD20 inhibitor such as rituximab.
In all, 17% of patients required permanent dose reductions; and 42% had a dose interruption, with a median hiatus of 12 days.
Grade 3 or 4 adverse events were uncommon, but more than 20% of patients experienced arthralgias or myalgias of any kind, about 19% reported fatigue, 18% had dermatologic toxicities, 18% reported bruising, 17% had diarrhea or colitis and 15% had infections.
The toxicities seen in RESONATE-2 were somewhat similar, but generally occurred in higher frequencies in the trial than in real-world practice.
Dr. Mato and colleagues found that at a median of 12 months of follow-up, 24% of patients had discontinued ibrutinib. In contrast, in RESONATE-2, after 18 months of follow-up, 13% of patients had discontinued the drug.
The most common reasons for discontinuation in clinical practice were for toxicities (59.5% of 94 discontinuations) including atrial fibrillation in 20% of the patients who discontinued, arthralgias/myalgias and skin toxicities in 14.5% each, and bleeding in 9.1%.
Other reasons for discontinuation included Richter’s transformation in 9.6%, doctor or patient preference in 7.4%, and deaths that were not secondary to CLL progression in 3.2%.
“We also tried to get a sense of whether or not cost was a factor for patients, and in this series and the relapsed refractory setting, 1% or less of patients discontinued due to financial issues,” Dr. Mato said.
Outcomes in the real word were quite good, he noted, with an overall response rate (ORR) of 81.7%, which included 17.4% complete responses (CR), Neither median PFS nor OS have been reached and the respective PFS and OS at 12 months were 92% and 95%. The respective PFS and OS rates for patients with del17p were 87% and 89%. An analysis of predictors of survival showed that only the presence of del17p was associated with inferior PFS (odds ratio 1.91, P = .035)
Dr. Mato noted that there was no clear standard treatment approach for patients who discontinued ibrutinib or for whom ibrutinib did not work. The top three second-line approaches used included an anti-CD20 agent combined with chlorambucil, venetoclax (Venclexta), or a different kinase inhibitor. Chemoimmunotherapy with either fludarabine, cyclophosphamide, and rituximab or bendamustine and rituximab was given to only 5 patients as a second line therapy.
Dr. Mato disclosed serving as a consultant for AbbVie, AstraZeneca, Janssen/Pharmacyclics, and TG Therapeutics.
Heart failure readmission penalties linked with rise in deaths
ANAHEIM, CALIF. – Evidence continues to mount that Medicare’s penalization of hospitals with excess heart failure readmissions has cut readmissions but at the apparent price of more deaths.
During the penalty phase of the Hospital Readmission Reduction Program (HRRP), which started in Oct. 2012, 30-day all-cause mortality following a heart failure hospitalization was 18% higher compared with the adjusted rate during 2006-2010, based on Medicare data from 2006-2014 that underwent “extensive” risk adjustment using prospectively-collected clinical data, Gregg C. Fonarow, MD, and his associates reported in a poster at the American Heart Association scientific sessions. During the same 2012-2014 period with imposed penalties, 30-day all-cause readmissions following an index heart failure hospitalization fell by a risk-adjusted 9% compared to the era just before the HRRP. Both the drop in readmissions and rise in deaths were statistically significant.
A similar pattern existed for the risk-adjusted readmissions and mortality rates during the year following the index hospitalization: readmissions fell by 8% compared with the time before the program but deaths rose by a relative 10%, also statistically significant differences.
“This is urgent and alarming. The Centers for Medicare & Medicaid Services needs to revamp the program to exclude heart failure patients and take steps to mitigate the damage,” Dr. Fonarow said in an interview. He estimated that the uptick in mortality following heart failure hospitalizations is causing 5,000-10,000 excess annual deaths among U.S. heart failure patients that are directly attributable to the HRRP. Similar effects have not been seen for patients with an index hospitalization of pneumonia or acute MI, two other targets of the HRRP, he noted.
The HRRP “currently has penalties for readmissions that are 15-fold higher than for mortality. They need to penalize equally, and they need to get at the gaming that hospitals are doing” to shift outcomes away from readmissions even if it means more patients will die. Heart failure patients “who need hospitalization are being denied admission by hospitals out of fear of the readmissions penalty,” said Dr. Fonarow, professor and co-chief of cardiology at the University of California, Los Angeles. “Seeing increased mortality linked with implementation of the penalty is completely unacceptable.”
Although a prior report used similar Medicare data from 2008-2014 to initially find this inverse association, that analysis relied entirely on administrative data collected in Medicare records to perform risk adjustments (JAMA. 2017 July 17;318[3]:270-8). The new analysis reported by Dr. Fonarow and his associates combined the Medicare data with detailed clinical records for the same patients collected by the Get With the Guidelines--Heart Failure program. The extensive clinical data that the researchers used for risk-adjustment allowed for a more reliable attribution to the HRRP of readmission and mortality differences between the two time periods. Despite the extensive risk adjustment “we see exactly the same result” as initially reported, Dr. Fonarow said.
The findings “remind us that it is very important to look at the unintended consequences” of interventions that might initially seem reasonable, commented Lynne Warner Stevenson, MD, professor and director of cardiomyopathy at Vanderbilt University in Nashville, Tenn.
Concurrent with the presentation at the meeting the results also appeared in an article published online (JAMA Cardiol. 2017 Nov 12;doi:10.1001/jamacardio.2017.4265).
A separate analysis of data collected in the Get With the Guidelines--Heart Failure during 2005-2009 showed that within the past decade the 5-year survival of U.S. hospitalized heart failure patients has remained dismally low, and similar regardless of whether patients had heart failure with reduced ejection fraction (HFrEF, 46% of all heart failure patients in the analysis), heart failure with preserved ejection fraction (HFpEF, also 46% of patients), or the in-between patients who had heart failure with borderline ejection fraction (HFbEF, an ejection fraction of 41%-49%, in 8% of patients).
The results, from 39,982 patients, showed a 75% mortality rate during 5-years of follow-up, with similar mortality rates regardless of the patient’s ejection-fraction level, reported Dr. Fonarow and his associates in a separate poster. In every age group examined, patients with heart failure had dramatically reduced life expectancies compared with the general population. For example, among heart failure patients aged 65-69 years in the study, median survival was less than 4 years compared with a 19-year expected median survival for people in the general U.S. population in the same age range.
These very low survival rates of heart failure patients initially hospitalized for heart failure during the relatively recent era of 2005-2009 “is a call to action to prevent heart failure,” said Dr. Fonarow.
The poor prognosis most heart failure patients face should also spur aggressive treatment of HFrEF patients with all proven treatments, Dr. Fonarow said. It should also spur more effort to find effective treatments for HFpEF, which currently has no clearly-proven effective treatment.
These results also appeared in a report simultaneously published online (J Amer Coll Cardiol. 2017 Nov 12;doi: 10.1016/j.jacc.2017.08.074).
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
ANAHEIM, CALIF. – Evidence continues to mount that Medicare’s penalization of hospitals with excess heart failure readmissions has cut readmissions but at the apparent price of more deaths.
During the penalty phase of the Hospital Readmission Reduction Program (HRRP), which started in Oct. 2012, 30-day all-cause mortality following a heart failure hospitalization was 18% higher compared with the adjusted rate during 2006-2010, based on Medicare data from 2006-2014 that underwent “extensive” risk adjustment using prospectively-collected clinical data, Gregg C. Fonarow, MD, and his associates reported in a poster at the American Heart Association scientific sessions. During the same 2012-2014 period with imposed penalties, 30-day all-cause readmissions following an index heart failure hospitalization fell by a risk-adjusted 9% compared to the era just before the HRRP. Both the drop in readmissions and rise in deaths were statistically significant.
A similar pattern existed for the risk-adjusted readmissions and mortality rates during the year following the index hospitalization: readmissions fell by 8% compared with the time before the program but deaths rose by a relative 10%, also statistically significant differences.
“This is urgent and alarming. The Centers for Medicare & Medicaid Services needs to revamp the program to exclude heart failure patients and take steps to mitigate the damage,” Dr. Fonarow said in an interview. He estimated that the uptick in mortality following heart failure hospitalizations is causing 5,000-10,000 excess annual deaths among U.S. heart failure patients that are directly attributable to the HRRP. Similar effects have not been seen for patients with an index hospitalization of pneumonia or acute MI, two other targets of the HRRP, he noted.
The HRRP “currently has penalties for readmissions that are 15-fold higher than for mortality. They need to penalize equally, and they need to get at the gaming that hospitals are doing” to shift outcomes away from readmissions even if it means more patients will die. Heart failure patients “who need hospitalization are being denied admission by hospitals out of fear of the readmissions penalty,” said Dr. Fonarow, professor and co-chief of cardiology at the University of California, Los Angeles. “Seeing increased mortality linked with implementation of the penalty is completely unacceptable.”
Although a prior report used similar Medicare data from 2008-2014 to initially find this inverse association, that analysis relied entirely on administrative data collected in Medicare records to perform risk adjustments (JAMA. 2017 July 17;318[3]:270-8). The new analysis reported by Dr. Fonarow and his associates combined the Medicare data with detailed clinical records for the same patients collected by the Get With the Guidelines--Heart Failure program. The extensive clinical data that the researchers used for risk-adjustment allowed for a more reliable attribution to the HRRP of readmission and mortality differences between the two time periods. Despite the extensive risk adjustment “we see exactly the same result” as initially reported, Dr. Fonarow said.
The findings “remind us that it is very important to look at the unintended consequences” of interventions that might initially seem reasonable, commented Lynne Warner Stevenson, MD, professor and director of cardiomyopathy at Vanderbilt University in Nashville, Tenn.
Concurrent with the presentation at the meeting the results also appeared in an article published online (JAMA Cardiol. 2017 Nov 12;doi:10.1001/jamacardio.2017.4265).
A separate analysis of data collected in the Get With the Guidelines--Heart Failure during 2005-2009 showed that within the past decade the 5-year survival of U.S. hospitalized heart failure patients has remained dismally low, and similar regardless of whether patients had heart failure with reduced ejection fraction (HFrEF, 46% of all heart failure patients in the analysis), heart failure with preserved ejection fraction (HFpEF, also 46% of patients), or the in-between patients who had heart failure with borderline ejection fraction (HFbEF, an ejection fraction of 41%-49%, in 8% of patients).
The results, from 39,982 patients, showed a 75% mortality rate during 5-years of follow-up, with similar mortality rates regardless of the patient’s ejection-fraction level, reported Dr. Fonarow and his associates in a separate poster. In every age group examined, patients with heart failure had dramatically reduced life expectancies compared with the general population. For example, among heart failure patients aged 65-69 years in the study, median survival was less than 4 years compared with a 19-year expected median survival for people in the general U.S. population in the same age range.
These very low survival rates of heart failure patients initially hospitalized for heart failure during the relatively recent era of 2005-2009 “is a call to action to prevent heart failure,” said Dr. Fonarow.
The poor prognosis most heart failure patients face should also spur aggressive treatment of HFrEF patients with all proven treatments, Dr. Fonarow said. It should also spur more effort to find effective treatments for HFpEF, which currently has no clearly-proven effective treatment.
These results also appeared in a report simultaneously published online (J Amer Coll Cardiol. 2017 Nov 12;doi: 10.1016/j.jacc.2017.08.074).
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
ANAHEIM, CALIF. – Evidence continues to mount that Medicare’s penalization of hospitals with excess heart failure readmissions has cut readmissions but at the apparent price of more deaths.
During the penalty phase of the Hospital Readmission Reduction Program (HRRP), which started in Oct. 2012, 30-day all-cause mortality following a heart failure hospitalization was 18% higher compared with the adjusted rate during 2006-2010, based on Medicare data from 2006-2014 that underwent “extensive” risk adjustment using prospectively-collected clinical data, Gregg C. Fonarow, MD, and his associates reported in a poster at the American Heart Association scientific sessions. During the same 2012-2014 period with imposed penalties, 30-day all-cause readmissions following an index heart failure hospitalization fell by a risk-adjusted 9% compared to the era just before the HRRP. Both the drop in readmissions and rise in deaths were statistically significant.
A similar pattern existed for the risk-adjusted readmissions and mortality rates during the year following the index hospitalization: readmissions fell by 8% compared with the time before the program but deaths rose by a relative 10%, also statistically significant differences.
“This is urgent and alarming. The Centers for Medicare & Medicaid Services needs to revamp the program to exclude heart failure patients and take steps to mitigate the damage,” Dr. Fonarow said in an interview. He estimated that the uptick in mortality following heart failure hospitalizations is causing 5,000-10,000 excess annual deaths among U.S. heart failure patients that are directly attributable to the HRRP. Similar effects have not been seen for patients with an index hospitalization of pneumonia or acute MI, two other targets of the HRRP, he noted.
The HRRP “currently has penalties for readmissions that are 15-fold higher than for mortality. They need to penalize equally, and they need to get at the gaming that hospitals are doing” to shift outcomes away from readmissions even if it means more patients will die. Heart failure patients “who need hospitalization are being denied admission by hospitals out of fear of the readmissions penalty,” said Dr. Fonarow, professor and co-chief of cardiology at the University of California, Los Angeles. “Seeing increased mortality linked with implementation of the penalty is completely unacceptable.”
Although a prior report used similar Medicare data from 2008-2014 to initially find this inverse association, that analysis relied entirely on administrative data collected in Medicare records to perform risk adjustments (JAMA. 2017 July 17;318[3]:270-8). The new analysis reported by Dr. Fonarow and his associates combined the Medicare data with detailed clinical records for the same patients collected by the Get With the Guidelines--Heart Failure program. The extensive clinical data that the researchers used for risk-adjustment allowed for a more reliable attribution to the HRRP of readmission and mortality differences between the two time periods. Despite the extensive risk adjustment “we see exactly the same result” as initially reported, Dr. Fonarow said.
The findings “remind us that it is very important to look at the unintended consequences” of interventions that might initially seem reasonable, commented Lynne Warner Stevenson, MD, professor and director of cardiomyopathy at Vanderbilt University in Nashville, Tenn.
Concurrent with the presentation at the meeting the results also appeared in an article published online (JAMA Cardiol. 2017 Nov 12;doi:10.1001/jamacardio.2017.4265).
A separate analysis of data collected in the Get With the Guidelines--Heart Failure during 2005-2009 showed that within the past decade the 5-year survival of U.S. hospitalized heart failure patients has remained dismally low, and similar regardless of whether patients had heart failure with reduced ejection fraction (HFrEF, 46% of all heart failure patients in the analysis), heart failure with preserved ejection fraction (HFpEF, also 46% of patients), or the in-between patients who had heart failure with borderline ejection fraction (HFbEF, an ejection fraction of 41%-49%, in 8% of patients).
The results, from 39,982 patients, showed a 75% mortality rate during 5-years of follow-up, with similar mortality rates regardless of the patient’s ejection-fraction level, reported Dr. Fonarow and his associates in a separate poster. In every age group examined, patients with heart failure had dramatically reduced life expectancies compared with the general population. For example, among heart failure patients aged 65-69 years in the study, median survival was less than 4 years compared with a 19-year expected median survival for people in the general U.S. population in the same age range.
These very low survival rates of heart failure patients initially hospitalized for heart failure during the relatively recent era of 2005-2009 “is a call to action to prevent heart failure,” said Dr. Fonarow.
The poor prognosis most heart failure patients face should also spur aggressive treatment of HFrEF patients with all proven treatments, Dr. Fonarow said. It should also spur more effort to find effective treatments for HFpEF, which currently has no clearly-proven effective treatment.
These results also appeared in a report simultaneously published online (J Amer Coll Cardiol. 2017 Nov 12;doi: 10.1016/j.jacc.2017.08.074).
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point:
Major finding: Risk-adjusted 30-day readmissions fell by a relative 9% during the reduction program but relative mortality rose by 18%.
Data source: Review of 115,245 Medicare beneficiaries with heart failure treated at hospitals in the Get With the Guidelines--Heart Failure program.
Disclosures: Dr. Fonarow has been a consultant to Amgen, Janssen, Medtronic, Novartis, and St. Jude. Dr. Stevenson has been a consultant to Abbott, has received travel support from St. Jude, and has received research funding and food from Novartis.
FDA approves first-in-class drug for hemophilia A with Factor VIII
The Food and Drug Administration has approved emicizumab-kxwh (Hemlibra) for the prevention or reduction of bleeding episodes for adult and pediatric patients with hemophilia A with Factor VIII inhibitors.

The drug received the FDA’s Priority Review, Breakthrough Therapy, and Orphan Drug designations.
It was shown to be safe and effective in two clinical trials, one of boys and men aged 12 years and older and the other of boys younger than 12 years. In the first trial, patients taking emicizumab-kxwh had an 87% reduction in the rate of treated bleeding episodes per year, compared with patients not receiving prophylactic treatment (2.9 vs. 23.3; P less than .0001).
In the second trial, 87% of the children receiving emicizumab-kxwh did not experience a bleeding episode that required treatment.
The most common adverse events were injection site reactions, headache, and arthralgia. The drug labeling includes a boxed warning about the possibility of thrombotic microangiopathy and thromboembolism in patients given an activated prothrombin complex concentrate rescue treatment for 24 hours or more while taking emicizumab-kxwh.
The Food and Drug Administration has approved emicizumab-kxwh (Hemlibra) for the prevention or reduction of bleeding episodes for adult and pediatric patients with hemophilia A with Factor VIII inhibitors.
The drug received the FDA’s Priority Review, Breakthrough Therapy, and Orphan Drug designations.
It was shown to be safe and effective in two clinical trials, one of boys and men aged 12 years and older and the other of boys younger than 12 years. In the first trial, patients taking emicizumab-kxwh had an 87% reduction in the rate of treated bleeding episodes per year, compared with patients not receiving prophylactic treatment (2.9 vs. 23.3; P less than .0001).
In the second trial, 87% of the children receiving emicizumab-kxwh did not experience a bleeding episode that required treatment.
The most common adverse events were injection site reactions, headache, and arthralgia. The drug labeling includes a boxed warning about the possibility of thrombotic microangiopathy and thromboembolism in patients given an activated prothrombin complex concentrate rescue treatment for 24 hours or more while taking emicizumab-kxwh.
The Food and Drug Administration has approved emicizumab-kxwh (Hemlibra) for the prevention or reduction of bleeding episodes for adult and pediatric patients with hemophilia A with Factor VIII inhibitors.
The drug received the FDA’s Priority Review, Breakthrough Therapy, and Orphan Drug designations.
It was shown to be safe and effective in two clinical trials, one of boys and men aged 12 years and older and the other of boys younger than 12 years. In the first trial, patients taking emicizumab-kxwh had an 87% reduction in the rate of treated bleeding episodes per year, compared with patients not receiving prophylactic treatment (2.9 vs. 23.3; P less than .0001).
In the second trial, 87% of the children receiving emicizumab-kxwh did not experience a bleeding episode that required treatment.
The most common adverse events were injection site reactions, headache, and arthralgia. The drug labeling includes a boxed warning about the possibility of thrombotic microangiopathy and thromboembolism in patients given an activated prothrombin complex concentrate rescue treatment for 24 hours or more while taking emicizumab-kxwh.
Scandinavian registries answer key questions about ADHD
PARIS – Huge longitudinal Scandinavian population registries constitute a unique data source that, in recent years, has provided new insights into attention-deficit/hyperactivity disorder and its associated risks of suicidal behavior, accidents, and early mortality, Henrik Larsson, PhD, said at the annual congress of the European College of Neuropsychopharmacology.
“Randomized, controlled trials have provided little information about real-world effectiveness of ADHD medications, such as their potential effects on adverse health outcomes,” said Dr. Larsson of the Karolinska Institute in Stockholm.

Suicidality
Dr. Larsson was senior author of a Swedish national registry study that identified 51,707 patients with ADHD matched by sex and birth year to 258,535 controls. The ADHD patients had significantly higher rates of both attempted and completed suicide.
After adjustment for socioeconomic status, these individuals were at 8.46-fold increased risk for attempted suicide. However, after further adjustment for comorbid psychiatric disorders, this dropped substantially to a 3.62-fold increased risk.
The same pattern pertained to completed suicide: The risk after adjustment for socioeconomic status was increased 12.3-fold in individuals with ADHD, compared with controls, but the risk dropped to 5.91-fold after further adjustment for comorbid psychiatric disorders (JAMA Psychiatry. 2014 Aug;71[8]:958-64).
The clinical take home point: “Detection and treatment of comorbid conditions probably will help reduce suicidal behavior in ADHD,” Dr. Larsson said.
This study also showed that increased familial risk also is a key factor in the increased risk of suicidal behavior in the ADHD population. Parents of individuals with ADHD were at 2.42-fold increased risk of attempted suicide, compared with controls, and full siblings were at 2.28-fold increased risk. In contrast, the risk in half-siblings, while significantly greater than in controls, was lower than in the genetically closer first-degree relatives: Maternal half-siblings were at 1.57-fold increased risk, and paternal half-siblings were at 1.57-fold greater risk. Cousins were at 1.39-fold increased risk.
The same held true for completed suicide risk. And the familial associations remained significant even after excluding relatives with ADHD.
“Regarding the shared familial factors, I’m tempted to hypothesize that this might involve pleiotropic effects reflecting genetic variants associated with impulsivity,” said Dr. Larsson. To further understand the biological mechanisms underlying ADHD and associated adverse health outcomes requires multiple disciplines to work together. For such work, Dr. Larsson collaborates with international colleagues in several consortia.
ADHD medications and suicidality
Concerns regarding this question were raised by a meta-analysis based on clinical trials data that suggested patients’ increased suicidality might be caused by the effects of ADHD medications. But the meta-analysis was seriously flawed by what epidemiologists call confounding by indication, which is the potential for bias to be introduced when a group of patients on medication is compared with another group off medication. The confounding results from the fact that ADHD patients on medication are different from those who aren’t: They are likely to be more symptomatic and have more comorbidities.
To bypass the confounding issue, Dr. Larsson and his coinvestigators turned to the Swedish registries and identified 37,936 patients with ADHD with a total of 7,019 suicide-related events during nearly 151,000 person-years of follow-up. When they compared patients on drug treatment with those who were not, they found – as in the other investigators’ meta-analysis – that drug treatment was associated with a statistically significant 1.31-fold increased risk of suicide-related events. However, when they performed a more appropriate between-individual analysis, Dr. Larsson and his colleagues found that, when patients with ADHD were using stimulant medications, they had a significant 19% lower risk of suicide-related events than when they were off medication. While on nonstimulant ADHD medications, their suicidality risk was no different from when off medication (BMJ. 2014 Jun 18;348:g3769. doi: 10.1136/bmj.g3769).
ADHD and early mortality risk
Prior studies have established that ADHD is associated with a proclivity to engage in risk-taking behaviors, including substance abuse, criminality, risky sexual behavior, and accidents, which are themselves associated with early mortality.
Sure enough, when Danish investigators turned to their national registries, identified 32,061 individuals with ADHD born during 1981-2011, and followed them through 2013, they found that, during nearly 25 million person-years of follow-up, the mortality rate was 5.85 deaths per 10,000 person-years in individuals with ADHD, compared with 2.21 deaths per 10,000 person-years in controls, resulting in a fully adjusted mortality rate ratio of 2.07. The rate ratio was 1.86 for ADHD patients under age 6 years, 1.58 in those aged 6-17 years, and 4.25 for patients aged 18 years and older (Lancet. 2015 May 30;385[9983]:2190-6).
Accidents were the most common cause of death. Could ADHD medications modify this risk of fatal accidents?
Serious motor vehicle accidents
Dr. Larsson and his coinvestigators used registry data to follow 17,408 Swedish adults with ADHD for serious transport accidents involving a trip to the emergency room or death during 2006-2009. The risk was increased by an adjusted 1.47-fold in men with ADHD and by 1.45-fold in women with the disorder. However, in the within-individual analysis, men were 58% less likely to have a serious transport accident when they were on ADHD medication than when off medication. There was no statistically significant effect of ADHD medications on the risk in women with ADHD.
The investigators estimated that 41%-49% of transport accidents in men with ADHD could have been avoided had they been on drug therapy continuously throughout the follow-up period (JAMA Psychiatry. 2014 Mar;71[3]:319-25).
Similar results – that is, data showing that being on ADHD medication reduces the elevated risk of serious accidents – have been reported in four other independent studies conducted in Denmark, Germany, Hong Kong, and most recently in a U.S. analysis by Dr. Larsson and coinvestigators of more than 2.3 million patients with ADHD in a U.S. commercial health insurance claims database (JAMA Psychiatry. 2017 Jun 1;74[6]:597-603).
These findings collectively highlight the public health importance of diagnosing and treating ADHD.
But Dr. Larsson wanted his audience to take home another key lesson: “ADHD is a disorder that can be associated with serious outcomes, including suicide and accidents. It’s nevertheless important to remember that the absolute risks here are very low for any of these outcomes, so the majority of individuals with ADHD will never suffer from any of these outcomes. It’s important to keep that in mind.”
Dr. Larsson’s research is funded by the Swedish Research Council, the National Institute of Mental Health, FORTE, Horizon 2020, and Shire.
bjancin@frontlinemedcom.com
PARIS – Huge longitudinal Scandinavian population registries constitute a unique data source that, in recent years, has provided new insights into attention-deficit/hyperactivity disorder and its associated risks of suicidal behavior, accidents, and early mortality, Henrik Larsson, PhD, said at the annual congress of the European College of Neuropsychopharmacology.
“Randomized, controlled trials have provided little information about real-world effectiveness of ADHD medications, such as their potential effects on adverse health outcomes,” said Dr. Larsson of the Karolinska Institute in Stockholm.
Suicidality
Dr. Larsson was senior author of a Swedish national registry study that identified 51,707 patients with ADHD matched by sex and birth year to 258,535 controls. The ADHD patients had significantly higher rates of both attempted and completed suicide.
After adjustment for socioeconomic status, these individuals were at 8.46-fold increased risk for attempted suicide. However, after further adjustment for comorbid psychiatric disorders, this dropped substantially to a 3.62-fold increased risk.
The same pattern pertained to completed suicide: The risk after adjustment for socioeconomic status was increased 12.3-fold in individuals with ADHD, compared with controls, but the risk dropped to 5.91-fold after further adjustment for comorbid psychiatric disorders (JAMA Psychiatry. 2014 Aug;71[8]:958-64).
The clinical take home point: “Detection and treatment of comorbid conditions probably will help reduce suicidal behavior in ADHD,” Dr. Larsson said.
This study also showed that increased familial risk also is a key factor in the increased risk of suicidal behavior in the ADHD population. Parents of individuals with ADHD were at 2.42-fold increased risk of attempted suicide, compared with controls, and full siblings were at 2.28-fold increased risk. In contrast, the risk in half-siblings, while significantly greater than in controls, was lower than in the genetically closer first-degree relatives: Maternal half-siblings were at 1.57-fold increased risk, and paternal half-siblings were at 1.57-fold greater risk. Cousins were at 1.39-fold increased risk.
The same held true for completed suicide risk. And the familial associations remained significant even after excluding relatives with ADHD.
“Regarding the shared familial factors, I’m tempted to hypothesize that this might involve pleiotropic effects reflecting genetic variants associated with impulsivity,” said Dr. Larsson. To further understand the biological mechanisms underlying ADHD and associated adverse health outcomes requires multiple disciplines to work together. For such work, Dr. Larsson collaborates with international colleagues in several consortia.
ADHD medications and suicidality
Concerns regarding this question were raised by a meta-analysis based on clinical trials data that suggested patients’ increased suicidality might be caused by the effects of ADHD medications. But the meta-analysis was seriously flawed by what epidemiologists call confounding by indication, which is the potential for bias to be introduced when a group of patients on medication is compared with another group off medication. The confounding results from the fact that ADHD patients on medication are different from those who aren’t: They are likely to be more symptomatic and have more comorbidities.
To bypass the confounding issue, Dr. Larsson and his coinvestigators turned to the Swedish registries and identified 37,936 patients with ADHD with a total of 7,019 suicide-related events during nearly 151,000 person-years of follow-up. When they compared patients on drug treatment with those who were not, they found – as in the other investigators’ meta-analysis – that drug treatment was associated with a statistically significant 1.31-fold increased risk of suicide-related events. However, when they performed a more appropriate between-individual analysis, Dr. Larsson and his colleagues found that, when patients with ADHD were using stimulant medications, they had a significant 19% lower risk of suicide-related events than when they were off medication. While on nonstimulant ADHD medications, their suicidality risk was no different from when off medication (BMJ. 2014 Jun 18;348:g3769. doi: 10.1136/bmj.g3769).
ADHD and early mortality risk
Prior studies have established that ADHD is associated with a proclivity to engage in risk-taking behaviors, including substance abuse, criminality, risky sexual behavior, and accidents, which are themselves associated with early mortality.
Sure enough, when Danish investigators turned to their national registries, identified 32,061 individuals with ADHD born during 1981-2011, and followed them through 2013, they found that, during nearly 25 million person-years of follow-up, the mortality rate was 5.85 deaths per 10,000 person-years in individuals with ADHD, compared with 2.21 deaths per 10,000 person-years in controls, resulting in a fully adjusted mortality rate ratio of 2.07. The rate ratio was 1.86 for ADHD patients under age 6 years, 1.58 in those aged 6-17 years, and 4.25 for patients aged 18 years and older (Lancet. 2015 May 30;385[9983]:2190-6).
Accidents were the most common cause of death. Could ADHD medications modify this risk of fatal accidents?
Serious motor vehicle accidents
Dr. Larsson and his coinvestigators used registry data to follow 17,408 Swedish adults with ADHD for serious transport accidents involving a trip to the emergency room or death during 2006-2009. The risk was increased by an adjusted 1.47-fold in men with ADHD and by 1.45-fold in women with the disorder. However, in the within-individual analysis, men were 58% less likely to have a serious transport accident when they were on ADHD medication than when off medication. There was no statistically significant effect of ADHD medications on the risk in women with ADHD.
The investigators estimated that 41%-49% of transport accidents in men with ADHD could have been avoided had they been on drug therapy continuously throughout the follow-up period (JAMA Psychiatry. 2014 Mar;71[3]:319-25).
Similar results – that is, data showing that being on ADHD medication reduces the elevated risk of serious accidents – have been reported in four other independent studies conducted in Denmark, Germany, Hong Kong, and most recently in a U.S. analysis by Dr. Larsson and coinvestigators of more than 2.3 million patients with ADHD in a U.S. commercial health insurance claims database (JAMA Psychiatry. 2017 Jun 1;74[6]:597-603).
These findings collectively highlight the public health importance of diagnosing and treating ADHD.
But Dr. Larsson wanted his audience to take home another key lesson: “ADHD is a disorder that can be associated with serious outcomes, including suicide and accidents. It’s nevertheless important to remember that the absolute risks here are very low for any of these outcomes, so the majority of individuals with ADHD will never suffer from any of these outcomes. It’s important to keep that in mind.”
Dr. Larsson’s research is funded by the Swedish Research Council, the National Institute of Mental Health, FORTE, Horizon 2020, and Shire.
bjancin@frontlinemedcom.com
PARIS – Huge longitudinal Scandinavian population registries constitute a unique data source that, in recent years, has provided new insights into attention-deficit/hyperactivity disorder and its associated risks of suicidal behavior, accidents, and early mortality, Henrik Larsson, PhD, said at the annual congress of the European College of Neuropsychopharmacology.
“Randomized, controlled trials have provided little information about real-world effectiveness of ADHD medications, such as their potential effects on adverse health outcomes,” said Dr. Larsson of the Karolinska Institute in Stockholm.
Suicidality
Dr. Larsson was senior author of a Swedish national registry study that identified 51,707 patients with ADHD matched by sex and birth year to 258,535 controls. The ADHD patients had significantly higher rates of both attempted and completed suicide.
After adjustment for socioeconomic status, these individuals were at 8.46-fold increased risk for attempted suicide. However, after further adjustment for comorbid psychiatric disorders, this dropped substantially to a 3.62-fold increased risk.
The same pattern pertained to completed suicide: The risk after adjustment for socioeconomic status was increased 12.3-fold in individuals with ADHD, compared with controls, but the risk dropped to 5.91-fold after further adjustment for comorbid psychiatric disorders (JAMA Psychiatry. 2014 Aug;71[8]:958-64).
The clinical take home point: “Detection and treatment of comorbid conditions probably will help reduce suicidal behavior in ADHD,” Dr. Larsson said.
This study also showed that increased familial risk also is a key factor in the increased risk of suicidal behavior in the ADHD population. Parents of individuals with ADHD were at 2.42-fold increased risk of attempted suicide, compared with controls, and full siblings were at 2.28-fold increased risk. In contrast, the risk in half-siblings, while significantly greater than in controls, was lower than in the genetically closer first-degree relatives: Maternal half-siblings were at 1.57-fold increased risk, and paternal half-siblings were at 1.57-fold greater risk. Cousins were at 1.39-fold increased risk.
The same held true for completed suicide risk. And the familial associations remained significant even after excluding relatives with ADHD.
“Regarding the shared familial factors, I’m tempted to hypothesize that this might involve pleiotropic effects reflecting genetic variants associated with impulsivity,” said Dr. Larsson. To further understand the biological mechanisms underlying ADHD and associated adverse health outcomes requires multiple disciplines to work together. For such work, Dr. Larsson collaborates with international colleagues in several consortia.
ADHD medications and suicidality
Concerns regarding this question were raised by a meta-analysis based on clinical trials data that suggested patients’ increased suicidality might be caused by the effects of ADHD medications. But the meta-analysis was seriously flawed by what epidemiologists call confounding by indication, which is the potential for bias to be introduced when a group of patients on medication is compared with another group off medication. The confounding results from the fact that ADHD patients on medication are different from those who aren’t: They are likely to be more symptomatic and have more comorbidities.
To bypass the confounding issue, Dr. Larsson and his coinvestigators turned to the Swedish registries and identified 37,936 patients with ADHD with a total of 7,019 suicide-related events during nearly 151,000 person-years of follow-up. When they compared patients on drug treatment with those who were not, they found – as in the other investigators’ meta-analysis – that drug treatment was associated with a statistically significant 1.31-fold increased risk of suicide-related events. However, when they performed a more appropriate between-individual analysis, Dr. Larsson and his colleagues found that, when patients with ADHD were using stimulant medications, they had a significant 19% lower risk of suicide-related events than when they were off medication. While on nonstimulant ADHD medications, their suicidality risk was no different from when off medication (BMJ. 2014 Jun 18;348:g3769. doi: 10.1136/bmj.g3769).
ADHD and early mortality risk
Prior studies have established that ADHD is associated with a proclivity to engage in risk-taking behaviors, including substance abuse, criminality, risky sexual behavior, and accidents, which are themselves associated with early mortality.
Sure enough, when Danish investigators turned to their national registries, identified 32,061 individuals with ADHD born during 1981-2011, and followed them through 2013, they found that, during nearly 25 million person-years of follow-up, the mortality rate was 5.85 deaths per 10,000 person-years in individuals with ADHD, compared with 2.21 deaths per 10,000 person-years in controls, resulting in a fully adjusted mortality rate ratio of 2.07. The rate ratio was 1.86 for ADHD patients under age 6 years, 1.58 in those aged 6-17 years, and 4.25 for patients aged 18 years and older (Lancet. 2015 May 30;385[9983]:2190-6).
Accidents were the most common cause of death. Could ADHD medications modify this risk of fatal accidents?
Serious motor vehicle accidents
Dr. Larsson and his coinvestigators used registry data to follow 17,408 Swedish adults with ADHD for serious transport accidents involving a trip to the emergency room or death during 2006-2009. The risk was increased by an adjusted 1.47-fold in men with ADHD and by 1.45-fold in women with the disorder. However, in the within-individual analysis, men were 58% less likely to have a serious transport accident when they were on ADHD medication than when off medication. There was no statistically significant effect of ADHD medications on the risk in women with ADHD.
The investigators estimated that 41%-49% of transport accidents in men with ADHD could have been avoided had they been on drug therapy continuously throughout the follow-up period (JAMA Psychiatry. 2014 Mar;71[3]:319-25).
Similar results – that is, data showing that being on ADHD medication reduces the elevated risk of serious accidents – have been reported in four other independent studies conducted in Denmark, Germany, Hong Kong, and most recently in a U.S. analysis by Dr. Larsson and coinvestigators of more than 2.3 million patients with ADHD in a U.S. commercial health insurance claims database (JAMA Psychiatry. 2017 Jun 1;74[6]:597-603).
These findings collectively highlight the public health importance of diagnosing and treating ADHD.
But Dr. Larsson wanted his audience to take home another key lesson: “ADHD is a disorder that can be associated with serious outcomes, including suicide and accidents. It’s nevertheless important to remember that the absolute risks here are very low for any of these outcomes, so the majority of individuals with ADHD will never suffer from any of these outcomes. It’s important to keep that in mind.”
Dr. Larsson’s research is funded by the Swedish Research Council, the National Institute of Mental Health, FORTE, Horizon 2020, and Shire.
bjancin@frontlinemedcom.com
expert analysis from THE ECNP CONGRESS
A program to increase flu vaccine compliance
. It won’t hurt your bottom line either and actually will help it. A flu shot program potentially can be run by a licensed practical nurse, registered nurse, physician’s assistant, or pediatric nurse practitioner, depending on your state’s law regarding vaccine administration by other than a physician, thus freeing up the physician to see well-child and sick-call patients.
It’s easy to set up a flu shot program and run it. Start preparing in June, preceding the upcoming flu season. Designate several Saturdays and or Sundays in September, October, November, December, and January as flu shot Saturdays and/or Sundays. And if Columbus day falls on a weekday, consider adding Columbus Day to your program dates as the kids often are off from school that day (check the local school calendar).
Next, prepare a postcard to be mailed to all patients on the lists your EMR produced for you. Keep the postcard simple. Announce the program, and state the dates the flu shot program is running. Ask parents to call to make an appointment for a flu vaccine only by appointment “with the program.” In addition to mailing a postcard, announce the flu shot program by sending out automated telephone calls and emails to all three lists the EMR has produced for you. The postcard mailing is your first contact, essentially announcing the program with dates and times. An automated phone call may be used to announce a specific date for which you are “now booking.” A good option when using automated phone calls is to allow the caller to press “zero” to be connected to the office to schedule a “flu shot only” appointment! Finally, emails announcing the dates of the program simply will reinforce information about the program.

Mr. Berman has been providing practice management services to physicians and other medical providers since 1983. He is the CEO of a pediatrics practice with locations in Staten Island and Brooklyn, N.Y. He holds a faculty appointment at State University of New York, Brooklyn, as a lecturer for the department of family medicine’s residency training program. He has no disclosures to report. Email him at pdnews@frontlinemedcom.com.
. It won’t hurt your bottom line either and actually will help it. A flu shot program potentially can be run by a licensed practical nurse, registered nurse, physician’s assistant, or pediatric nurse practitioner, depending on your state’s law regarding vaccine administration by other than a physician, thus freeing up the physician to see well-child and sick-call patients.
It’s easy to set up a flu shot program and run it. Start preparing in June, preceding the upcoming flu season. Designate several Saturdays and or Sundays in September, October, November, December, and January as flu shot Saturdays and/or Sundays. And if Columbus day falls on a weekday, consider adding Columbus Day to your program dates as the kids often are off from school that day (check the local school calendar).
Next, prepare a postcard to be mailed to all patients on the lists your EMR produced for you. Keep the postcard simple. Announce the program, and state the dates the flu shot program is running. Ask parents to call to make an appointment for a flu vaccine only by appointment “with the program.” In addition to mailing a postcard, announce the flu shot program by sending out automated telephone calls and emails to all three lists the EMR has produced for you. The postcard mailing is your first contact, essentially announcing the program with dates and times. An automated phone call may be used to announce a specific date for which you are “now booking.” A good option when using automated phone calls is to allow the caller to press “zero” to be connected to the office to schedule a “flu shot only” appointment! Finally, emails announcing the dates of the program simply will reinforce information about the program.
Mr. Berman has been providing practice management services to physicians and other medical providers since 1983. He is the CEO of a pediatrics practice with locations in Staten Island and Brooklyn, N.Y. He holds a faculty appointment at State University of New York, Brooklyn, as a lecturer for the department of family medicine’s residency training program. He has no disclosures to report. Email him at pdnews@frontlinemedcom.com.
. It won’t hurt your bottom line either and actually will help it. A flu shot program potentially can be run by a licensed practical nurse, registered nurse, physician’s assistant, or pediatric nurse practitioner, depending on your state’s law regarding vaccine administration by other than a physician, thus freeing up the physician to see well-child and sick-call patients.
It’s easy to set up a flu shot program and run it. Start preparing in June, preceding the upcoming flu season. Designate several Saturdays and or Sundays in September, October, November, December, and January as flu shot Saturdays and/or Sundays. And if Columbus day falls on a weekday, consider adding Columbus Day to your program dates as the kids often are off from school that day (check the local school calendar).
Next, prepare a postcard to be mailed to all patients on the lists your EMR produced for you. Keep the postcard simple. Announce the program, and state the dates the flu shot program is running. Ask parents to call to make an appointment for a flu vaccine only by appointment “with the program.” In addition to mailing a postcard, announce the flu shot program by sending out automated telephone calls and emails to all three lists the EMR has produced for you. The postcard mailing is your first contact, essentially announcing the program with dates and times. An automated phone call may be used to announce a specific date for which you are “now booking.” A good option when using automated phone calls is to allow the caller to press “zero” to be connected to the office to schedule a “flu shot only” appointment! Finally, emails announcing the dates of the program simply will reinforce information about the program.
Mr. Berman has been providing practice management services to physicians and other medical providers since 1983. He is the CEO of a pediatrics practice with locations in Staten Island and Brooklyn, N.Y. He holds a faculty appointment at State University of New York, Brooklyn, as a lecturer for the department of family medicine’s residency training program. He has no disclosures to report. Email him at pdnews@frontlinemedcom.com.