User login
Macitentan approved for pulmonary arterial hypertension
Another endothelin receptor blocker, macitentan, has been approved for treating pulmonary arterial hypertension, the Food and Drug Administration announced on Oct. 18.
In a study of 742 patients with pulmonary arterial hypertension (PAH), macitentan over an average of 2 years was "effective in delaying disease progression, a finding that included a decline in exercise ability, worsening symptoms of PAH or need for additional PAH medication," according to the FDA statement issued on Oct. 18. Anemia, nasopharyngitis, sore throat, bronchitis, headache, and urinary tract infection were among the common side effects associated with treatment, the statement said.
It will be marketed as Opsumit by Actelion Pharmaceuticals US. The approved indication is for the treatment of PAH, WHO Group I, to delay disease progression, which includes death, initiation of intravenous or subcutaneous prostanoids, or clinical worsening of PAH (decreased 6-minute walk distance, worsened PAH symptoms and need for additional PAH treatment, according to Actelion). The approved dose is 10 mg daily; it is an oral medication.
Like the other endothelin receptor blockers, macitentan’s label includes a boxed warning that it is a teratogen and should not be used in pregnant women, and that women can receive the drug only through a REMS (Risk Evaluation and Mitigation Strategy) program that will restrict the drug’s distribution. Under the Opsumit REMS, distribution of the drug will be restricted and prescribers will have to enroll in the REMS and become certified to prescribe the drug. Female patients will also need to be enrolled and must comply with pregnancy testing and contraception requirements before starting treatment. Pharmacies that dispense the drug will need to be certified and will dispense the drug only to authorized patients, the FDA said.
The study of 742 patients was SERAPHIN (Study With an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome), which compared treatment with 3 mg or 10 mg of macitentan once a day, or placebo, and were allowed to be treated with phosphodiesterase-5 inhibitors or oral or inhaled prostanoids, according to Actelion.
The primary end point, a composite of a PAH event or death from any cause, was reached by 38% of patients receiving 3-mg macitentan and 31% of those receiving 10-mg macitentan, compared with 46% of patients receiving placebo. The hazard ratios of 0.7 for the 3-mg dose and 0.55 for the 10-mg dose were statistically significant.
Macitentan is under review in Europe, Canada, Switzerland, Australia, Taiwan, Korea and Mexico, according to Actelion.
Actelion also markets bosentan (Tracleer), another endothelin receptor antagonist, which was approved for treating PAH in 2001. Ambrisentan (Letairis; Gilead), also an endothelin receptor antagonist, was approved in 2007.
Another endothelin receptor blocker, macitentan, has been approved for treating pulmonary arterial hypertension, the Food and Drug Administration announced on Oct. 18.
In a study of 742 patients with pulmonary arterial hypertension (PAH), macitentan over an average of 2 years was "effective in delaying disease progression, a finding that included a decline in exercise ability, worsening symptoms of PAH or need for additional PAH medication," according to the FDA statement issued on Oct. 18. Anemia, nasopharyngitis, sore throat, bronchitis, headache, and urinary tract infection were among the common side effects associated with treatment, the statement said.
It will be marketed as Opsumit by Actelion Pharmaceuticals US. The approved indication is for the treatment of PAH, WHO Group I, to delay disease progression, which includes death, initiation of intravenous or subcutaneous prostanoids, or clinical worsening of PAH (decreased 6-minute walk distance, worsened PAH symptoms and need for additional PAH treatment, according to Actelion). The approved dose is 10 mg daily; it is an oral medication.
Like the other endothelin receptor blockers, macitentan’s label includes a boxed warning that it is a teratogen and should not be used in pregnant women, and that women can receive the drug only through a REMS (Risk Evaluation and Mitigation Strategy) program that will restrict the drug’s distribution. Under the Opsumit REMS, distribution of the drug will be restricted and prescribers will have to enroll in the REMS and become certified to prescribe the drug. Female patients will also need to be enrolled and must comply with pregnancy testing and contraception requirements before starting treatment. Pharmacies that dispense the drug will need to be certified and will dispense the drug only to authorized patients, the FDA said.
The study of 742 patients was SERAPHIN (Study With an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome), which compared treatment with 3 mg or 10 mg of macitentan once a day, or placebo, and were allowed to be treated with phosphodiesterase-5 inhibitors or oral or inhaled prostanoids, according to Actelion.
The primary end point, a composite of a PAH event or death from any cause, was reached by 38% of patients receiving 3-mg macitentan and 31% of those receiving 10-mg macitentan, compared with 46% of patients receiving placebo. The hazard ratios of 0.7 for the 3-mg dose and 0.55 for the 10-mg dose were statistically significant.
Macitentan is under review in Europe, Canada, Switzerland, Australia, Taiwan, Korea and Mexico, according to Actelion.
Actelion also markets bosentan (Tracleer), another endothelin receptor antagonist, which was approved for treating PAH in 2001. Ambrisentan (Letairis; Gilead), also an endothelin receptor antagonist, was approved in 2007.
Another endothelin receptor blocker, macitentan, has been approved for treating pulmonary arterial hypertension, the Food and Drug Administration announced on Oct. 18.
In a study of 742 patients with pulmonary arterial hypertension (PAH), macitentan over an average of 2 years was "effective in delaying disease progression, a finding that included a decline in exercise ability, worsening symptoms of PAH or need for additional PAH medication," according to the FDA statement issued on Oct. 18. Anemia, nasopharyngitis, sore throat, bronchitis, headache, and urinary tract infection were among the common side effects associated with treatment, the statement said.
It will be marketed as Opsumit by Actelion Pharmaceuticals US. The approved indication is for the treatment of PAH, WHO Group I, to delay disease progression, which includes death, initiation of intravenous or subcutaneous prostanoids, or clinical worsening of PAH (decreased 6-minute walk distance, worsened PAH symptoms and need for additional PAH treatment, according to Actelion). The approved dose is 10 mg daily; it is an oral medication.
Like the other endothelin receptor blockers, macitentan’s label includes a boxed warning that it is a teratogen and should not be used in pregnant women, and that women can receive the drug only through a REMS (Risk Evaluation and Mitigation Strategy) program that will restrict the drug’s distribution. Under the Opsumit REMS, distribution of the drug will be restricted and prescribers will have to enroll in the REMS and become certified to prescribe the drug. Female patients will also need to be enrolled and must comply with pregnancy testing and contraception requirements before starting treatment. Pharmacies that dispense the drug will need to be certified and will dispense the drug only to authorized patients, the FDA said.
The study of 742 patients was SERAPHIN (Study With an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome), which compared treatment with 3 mg or 10 mg of macitentan once a day, or placebo, and were allowed to be treated with phosphodiesterase-5 inhibitors or oral or inhaled prostanoids, according to Actelion.
The primary end point, a composite of a PAH event or death from any cause, was reached by 38% of patients receiving 3-mg macitentan and 31% of those receiving 10-mg macitentan, compared with 46% of patients receiving placebo. The hazard ratios of 0.7 for the 3-mg dose and 0.55 for the 10-mg dose were statistically significant.
Macitentan is under review in Europe, Canada, Switzerland, Australia, Taiwan, Korea and Mexico, according to Actelion.
Actelion also markets bosentan (Tracleer), another endothelin receptor antagonist, which was approved for treating PAH in 2001. Ambrisentan (Letairis; Gilead), also an endothelin receptor antagonist, was approved in 2007.
Bariatric surgery may prevent heart failure
AMSTERDAM – New findings from the landmark Swedish Obese Subjects study "strongly suggest" that bariatric surgery reduces by roughly half the long-term risk of developing heart failure, Dr. Kristjan Karason reported at the annual congress of the European Society of Cardiology.
"This is the first study to look at the effect of bariatric surgery on heart failure risk," observed Dr. Karason of the University of Gothenburg (Sweden). "Our findings support a modulating role of excess body fat in the pathogenesis of heart failure."
The SOS (Swedish Obese Subjects) study has been a major driver of the growing enthusiasm for bariatric surgery, not only as a means of achieving sustained weight loss far beyond what can typically be accomplished medically, but also as a means of reducing obese patients’ elevated risks for a variety of serious chronic comorbid diseases.

For example, the SOS investigators have previously reported that bariatric surgery reduced the long-term risk of developing type 2 diabetes by 87% compared to matched obese controls who didn’t undergo weight-loss surgery (N. Engl. J. Med. 2012;367:695-704), that it reduced the risk of fatal or nonfatal acute MI or stroke by one-third during a median 14.7 years of follow-up (JAMA 2012;307:56-65), and it resulted in a 42% decrease in cancer incidence in women (Lancet Oncol. 2009;10:653-62).
The SOS is a nonrandomized, prospective, observational study involving 2,010 obese subjects who underwent bariatric surgery in 1987-2001, when they were 37-60 years old. A total of 68% of the bariatric surgery recipients had vertical band gastroplasty, 19% underwent gastric banding, and 13% had a Roux en-Y gastric bypass. They were extensively matched by 18 variables to 2,037 obese controls. The SOS study is being conducted at 25 surgical departments and 480 primary care clinics across Sweden. Follow-up is ongoing.
It has been known for more than a decade that increased body mass index is associated with greater risk of developing heart failure. The mechanism involved is not well defined but is probably multifactorial. Obesity imposes a greater hemodynamic load on the heart, both preload and afterload, with resultant left ventricular hypertrophy and diastolic dysfunction. Obesity is also associated with higher levels of cardiovascular risk factors and an increased risk of atrial fibrillation, Dr. Karason noted.
Mean weight loss after a median of 14.7 years of prospective follow-up in the SOS study was 18% in the bariatric surgery group and 1% in controls. During follow-up, 91 bariatric surgery patients and 152 controls were diagnosed with heart failure, for an incidence rate of 3.1 as compared with 5.2 cases per 1,000 person-years in the control subjects.
This translated to a 48% relative risk reduction in new-onset heart failure in a multivariate regression analysis adjusted for age, sex, baseline body mass index, waist circumference, blood glucose, lipids, prior cardiovascular disease, and smoking status.
One audience member asked how to interpret the new SOS findings in light of the heart failure obesity paradox, which is the observation in multiple studies that overweight and obese heart failure patients tends to fare better than leaner ones.
"There have been no studies of weight loss in obese patients with heart failure. There really should be," Dr. Karason replied. "But my feeling is that they would reduce their risk because they will improve several risk factors, and their hemodynamic situation is also improved. So my recommendation would be for those heart failure patients to lose weight. I don’t have any studies to support that."
The SOS study is funded by the Swedish Research Council. Dr. Karason reported having no germane financial interests.
AMSTERDAM – New findings from the landmark Swedish Obese Subjects study "strongly suggest" that bariatric surgery reduces by roughly half the long-term risk of developing heart failure, Dr. Kristjan Karason reported at the annual congress of the European Society of Cardiology.
"This is the first study to look at the effect of bariatric surgery on heart failure risk," observed Dr. Karason of the University of Gothenburg (Sweden). "Our findings support a modulating role of excess body fat in the pathogenesis of heart failure."
The SOS (Swedish Obese Subjects) study has been a major driver of the growing enthusiasm for bariatric surgery, not only as a means of achieving sustained weight loss far beyond what can typically be accomplished medically, but also as a means of reducing obese patients’ elevated risks for a variety of serious chronic comorbid diseases.
For example, the SOS investigators have previously reported that bariatric surgery reduced the long-term risk of developing type 2 diabetes by 87% compared to matched obese controls who didn’t undergo weight-loss surgery (N. Engl. J. Med. 2012;367:695-704), that it reduced the risk of fatal or nonfatal acute MI or stroke by one-third during a median 14.7 years of follow-up (JAMA 2012;307:56-65), and it resulted in a 42% decrease in cancer incidence in women (Lancet Oncol. 2009;10:653-62).
The SOS is a nonrandomized, prospective, observational study involving 2,010 obese subjects who underwent bariatric surgery in 1987-2001, when they were 37-60 years old. A total of 68% of the bariatric surgery recipients had vertical band gastroplasty, 19% underwent gastric banding, and 13% had a Roux en-Y gastric bypass. They were extensively matched by 18 variables to 2,037 obese controls. The SOS study is being conducted at 25 surgical departments and 480 primary care clinics across Sweden. Follow-up is ongoing.
It has been known for more than a decade that increased body mass index is associated with greater risk of developing heart failure. The mechanism involved is not well defined but is probably multifactorial. Obesity imposes a greater hemodynamic load on the heart, both preload and afterload, with resultant left ventricular hypertrophy and diastolic dysfunction. Obesity is also associated with higher levels of cardiovascular risk factors and an increased risk of atrial fibrillation, Dr. Karason noted.
Mean weight loss after a median of 14.7 years of prospective follow-up in the SOS study was 18% in the bariatric surgery group and 1% in controls. During follow-up, 91 bariatric surgery patients and 152 controls were diagnosed with heart failure, for an incidence rate of 3.1 as compared with 5.2 cases per 1,000 person-years in the control subjects.
This translated to a 48% relative risk reduction in new-onset heart failure in a multivariate regression analysis adjusted for age, sex, baseline body mass index, waist circumference, blood glucose, lipids, prior cardiovascular disease, and smoking status.
One audience member asked how to interpret the new SOS findings in light of the heart failure obesity paradox, which is the observation in multiple studies that overweight and obese heart failure patients tends to fare better than leaner ones.
"There have been no studies of weight loss in obese patients with heart failure. There really should be," Dr. Karason replied. "But my feeling is that they would reduce their risk because they will improve several risk factors, and their hemodynamic situation is also improved. So my recommendation would be for those heart failure patients to lose weight. I don’t have any studies to support that."
The SOS study is funded by the Swedish Research Council. Dr. Karason reported having no germane financial interests.
AMSTERDAM – New findings from the landmark Swedish Obese Subjects study "strongly suggest" that bariatric surgery reduces by roughly half the long-term risk of developing heart failure, Dr. Kristjan Karason reported at the annual congress of the European Society of Cardiology.
"This is the first study to look at the effect of bariatric surgery on heart failure risk," observed Dr. Karason of the University of Gothenburg (Sweden). "Our findings support a modulating role of excess body fat in the pathogenesis of heart failure."
The SOS (Swedish Obese Subjects) study has been a major driver of the growing enthusiasm for bariatric surgery, not only as a means of achieving sustained weight loss far beyond what can typically be accomplished medically, but also as a means of reducing obese patients’ elevated risks for a variety of serious chronic comorbid diseases.
For example, the SOS investigators have previously reported that bariatric surgery reduced the long-term risk of developing type 2 diabetes by 87% compared to matched obese controls who didn’t undergo weight-loss surgery (N. Engl. J. Med. 2012;367:695-704), that it reduced the risk of fatal or nonfatal acute MI or stroke by one-third during a median 14.7 years of follow-up (JAMA 2012;307:56-65), and it resulted in a 42% decrease in cancer incidence in women (Lancet Oncol. 2009;10:653-62).
The SOS is a nonrandomized, prospective, observational study involving 2,010 obese subjects who underwent bariatric surgery in 1987-2001, when they were 37-60 years old. A total of 68% of the bariatric surgery recipients had vertical band gastroplasty, 19% underwent gastric banding, and 13% had a Roux en-Y gastric bypass. They were extensively matched by 18 variables to 2,037 obese controls. The SOS study is being conducted at 25 surgical departments and 480 primary care clinics across Sweden. Follow-up is ongoing.
It has been known for more than a decade that increased body mass index is associated with greater risk of developing heart failure. The mechanism involved is not well defined but is probably multifactorial. Obesity imposes a greater hemodynamic load on the heart, both preload and afterload, with resultant left ventricular hypertrophy and diastolic dysfunction. Obesity is also associated with higher levels of cardiovascular risk factors and an increased risk of atrial fibrillation, Dr. Karason noted.
Mean weight loss after a median of 14.7 years of prospective follow-up in the SOS study was 18% in the bariatric surgery group and 1% in controls. During follow-up, 91 bariatric surgery patients and 152 controls were diagnosed with heart failure, for an incidence rate of 3.1 as compared with 5.2 cases per 1,000 person-years in the control subjects.
This translated to a 48% relative risk reduction in new-onset heart failure in a multivariate regression analysis adjusted for age, sex, baseline body mass index, waist circumference, blood glucose, lipids, prior cardiovascular disease, and smoking status.
One audience member asked how to interpret the new SOS findings in light of the heart failure obesity paradox, which is the observation in multiple studies that overweight and obese heart failure patients tends to fare better than leaner ones.
"There have been no studies of weight loss in obese patients with heart failure. There really should be," Dr. Karason replied. "But my feeling is that they would reduce their risk because they will improve several risk factors, and their hemodynamic situation is also improved. So my recommendation would be for those heart failure patients to lose weight. I don’t have any studies to support that."
The SOS study is funded by the Swedish Research Council. Dr. Karason reported having no germane financial interests.
AT THE ESC CONGRESS 2013
Major finding: The incidence rate of heart failure during a median 15 years of prospective follow-up after bariatric surgery was 3.1 cases per 1,000 person-years, compared with 5.2/1,000 person-years in obese controls.
Data source: The Swedish Obese Subjects study included 2,010 obese subjects who underwent bariatric surgery in 1987-2001 and 2,037 closely matched obese controls. It is a nonrandomized, prospective, observational study.
Disclosures: The study is funded by the Swedish Research Council. The presenter reported having no financial conflicts.

Hunting for cardiovascular signals from diabetes drugs
Diabetes drug development entered a new era in 2007 when a meta-analysis of 42 studies that had compared rosiglitazone with other drugs in a total of nearly 28,000 patients suggested that rosiglitazone treatment led to significantly increased rates of cardiovascular death and myocardial infarction.
Based largely on that, the Food and Drug Administration in 2008 issued recommendations to companies developing new diabetes drugs to run large-scale trials aimed at assessing their cardiovascular effects.

In 2011, the FDA placed restrictions on prescribing rosiglitazone, and U.S. use of the drug plummeted. (An FDA panel voted in June to relax some of the restrictions the agency had imposed.)
Some consequences of the long shadow cast by the rosiglitazone experience played out in talks at the annual congress of the European Society for the Study of Diabetes in September, as well as at the annual congress of the European Society of Cardiology a few weeks before that. The new data show just how painstaking investigators have become in trying to parse out hints of cardiovascular danger lurking in masses of megatrial data.
Heart failure haunts DPP-4 inhibitors
At ESC, as well as in a pair of simultaneously published articles, researchers reported results from two large trials designed to follow the FDA recommendations and assess cardiovascular safety for two new selective dipeptidyl peptidase-4 (DPP-4) inhibitors, saxagliptin (Onglyza) and alogliptin, in patients with type 2 diabetes.
Saxagliptin was the drug with the red-flag signal, while alogliptin faced guilt by association. The saxagliptin study, SAVOR-TIMI 53, used patients with either an established history of cardiovascular disease (CVD) or multiple CVD risk factors. The study’s primary safety endpoint, the combined rate of CVD death, nonfatal myocardial infarction, or nonfatal stroke during 2 years of follow-up, was virtually identical in the two groups, 7.3% in the saxagliptin patients and 7.2% in the controls.
The study’s secondary safety endpoint, which included the primary combined plus other endpoints such as need for revascularization and hospitalization for heart failure, was also similar in the two study arms, with this expanded combined endpoint occurring in 12.8% of the saxagliptin patients and 12.4% of the controls (N. Engl. J. Med. 2013;369:1317-26).
The whisper of a saxagliptin problem was in just one secondary, individual safety endpoint, hospitalizations for heart failure. These occurred in 3.5% of the saxagliptin patients and in 2.8% of controls, a statistically significant difference. Further data presented at EASD showed that this difference clustered predominantly in the subgroup of patients who entered the study with presumed heart failure, based on having a blood level of NT-pro brain natriuretic peptide of at least 333, about 25% of patients in both treatment arms (although the investigators identified heart failure in only 13% of enrolled patients). Within this subgroup, heart failure–associated hospitalizations occurred in 10.9% of the saxagliptin patients and 8.9% of the controls, a statistically significant difference, reported Dr. Deepak L. Bhatt, one of the investigators for the SAVOR-TIMI 53 study, which randomized more than 16,000 patients.
In contrast, the alogliptin study, EXAMINE, which randomized more than 5,000 patients with type 2 diabetes and a recent acute coronary syndrome event to treatment with alogliptin or placebo, found no real confirmation of this problem (N. Engl. J. Med. 2013;369:1327-35).
After the early-September report of this saxagliptin and heart failure hospitalization signal, the EXAMINE researchers went back to their data to specifically look at this issue, Dr. William B. White, the lead investigator, said when he presented the new findings at EASD. During a median of 18 months of follow-up, the rate of hospitalizations for heart failure was 3.1% in the alogliptin group and 2.9% in the placebo group, a difference that was not statistically significant, although it trended just slightly toward an alogliptin problem. Among the subgroup of patients who entered EXAMINE with a history of heart failure, the rate of the combined cardiovascular safety endpoint of CV death, nonfatal MI, or nonfatal stroke was 18% in the alogliptin group and 22% in the placebo group, a difference that was not statistically significant.
Despite the absence of any apparent heart failure–hospitalization effect in EXAMINE, Dr. Naveed Sattar, the invited discussant for the two reports at EASD, meta-analyzed the heart failure–hospitalization results from both studies, and found a combined, 24% relative increase in this outcome for the two DPP-4 inhibitors combined, a statistically significant effect largely driven toward significance by the underlying statistical significance in the saxagliptin study; the alogliptin results couldn’t resolve this difference since they trended in the same direction. Dr. Sattar concluded that it raised enough doubt about the safety of this entire drug class in patients with pre-existing heart failure to recommend not using drugs from this class in patients with any indication of heart failure – regardless of severity – until the apparent link received further scrutiny. Dr. Bhatt promised a report with further information on the heart failure issue at the American Heart Association Scientific Sessions in November.
Signals for canagliflozin, too?
SAVOR-TIMI 53 and EXAMINE provide the first results from what are more than a dozen large studies – involving more than 100,000 patients – launched recently to look for adverse cardiovascular effects from drugs in six different diabetes drug classes, according to Dr. Sanjay Kaul, who also spoke at EASD. He presented results from a meta-analysis he performed on results from nine phase III and phase II trials of canagliflozin (Invokana), an agent from the sodium-glucose cotransporter 2 (SGLT2) inhibitor class and approved by the FDA last March for treatment of type 2 diabetes, the first drug in that class to get U.S. marketing approval.
In the largest of these trials, CANVAS – the cardiovascular-assessment study for canagliflozin set up by the drug’s developer, Janssen – treatment with canagliflozin linked with a statistically significant, sixfold increase in cardiovascular events during the first 30 days of treatment, a link that then disappeared with longer-term treatment in patients with high CV risk. (Complete results from CANVAS have not yet been reported.) But Dr. Kaul did not see a similar excess in the other eight studies.
The canagliflozin meta-analysis also showed a nonsignificant, 46% increased risk of stroke in patients on the drug, a signal not seen in any other individual cardiovascular endpoint. Dr. Kaul analyzed data from 14 phase II and III trials for a second SGLT2 inhibitor drug, dapagliflozin, which showed no suggestion of causing any cardiovascular risk.
The canagliflozin risk signals are not reasons to stop using the drug or to withdraw it from the market, said Dr. Kaul, a member of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. The early signal of risk in CANVAS was not seen in any other study, and it may have been a statistical artifact, he said.
He said that signals of risk must be considered relative to a drug’s benefit. Applying the same standard of cardiovascular harm to drugs that are highly effective and to drugs that are not very effective "doesn’t make sense," he said. "A risk-benefit trade-off depends on the benefit. If you have a diabetes drug that lowers hemoglobin A1c by 1.5%-2% I’m willing to accept greater harm than I would from a drug that produced a 0.5% drop in hemoglobin A1c."
A final rosiglitazone thought
Dr. Kaul also made this observation about the rosiglitazone experience, which in retrospect produced "insufficient evidence to either incriminate or exonerate rosiglitazone," he said.
"That a diabetes drug [rosiglitazone] could have become a blockbuster without any established outcome benefits does not reflect well on the drug development and approval process. Equally lamentable is that this drug was virtually killed based on insufficient evidence and entrenched opinions."
Dr. Bhatt has received research grants from several companies, including AstraZeneca and Bristol-Myers Squibb (BMS), which market saxagliptin, and Takeda, maker of alogliptin. Dr. White is a safety consultant for several companies, including AstraZeneca and Takeda. Dr. Sattar is a consultant to several pharmaceutical firms, including AstraZeneca and BMS. Dr. Kaul is a consultant to Novo Nordisk, Sanofi, and Boehringer Ingelheim, and owns stock in Johnson & Johnson.
On Twitter @mitchelzoler
Diabetes drug development entered a new era in 2007 when a meta-analysis of 42 studies that had compared rosiglitazone with other drugs in a total of nearly 28,000 patients suggested that rosiglitazone treatment led to significantly increased rates of cardiovascular death and myocardial infarction.
Based largely on that, the Food and Drug Administration in 2008 issued recommendations to companies developing new diabetes drugs to run large-scale trials aimed at assessing their cardiovascular effects.
In 2011, the FDA placed restrictions on prescribing rosiglitazone, and U.S. use of the drug plummeted. (An FDA panel voted in June to relax some of the restrictions the agency had imposed.)
Some consequences of the long shadow cast by the rosiglitazone experience played out in talks at the annual congress of the European Society for the Study of Diabetes in September, as well as at the annual congress of the European Society of Cardiology a few weeks before that. The new data show just how painstaking investigators have become in trying to parse out hints of cardiovascular danger lurking in masses of megatrial data.
Heart failure haunts DPP-4 inhibitors
At ESC, as well as in a pair of simultaneously published articles, researchers reported results from two large trials designed to follow the FDA recommendations and assess cardiovascular safety for two new selective dipeptidyl peptidase-4 (DPP-4) inhibitors, saxagliptin (Onglyza) and alogliptin, in patients with type 2 diabetes.
Saxagliptin was the drug with the red-flag signal, while alogliptin faced guilt by association. The saxagliptin study, SAVOR-TIMI 53, used patients with either an established history of cardiovascular disease (CVD) or multiple CVD risk factors. The study’s primary safety endpoint, the combined rate of CVD death, nonfatal myocardial infarction, or nonfatal stroke during 2 years of follow-up, was virtually identical in the two groups, 7.3% in the saxagliptin patients and 7.2% in the controls.
The study’s secondary safety endpoint, which included the primary combined plus other endpoints such as need for revascularization and hospitalization for heart failure, was also similar in the two study arms, with this expanded combined endpoint occurring in 12.8% of the saxagliptin patients and 12.4% of the controls (N. Engl. J. Med. 2013;369:1317-26).
The whisper of a saxagliptin problem was in just one secondary, individual safety endpoint, hospitalizations for heart failure. These occurred in 3.5% of the saxagliptin patients and in 2.8% of controls, a statistically significant difference. Further data presented at EASD showed that this difference clustered predominantly in the subgroup of patients who entered the study with presumed heart failure, based on having a blood level of NT-pro brain natriuretic peptide of at least 333, about 25% of patients in both treatment arms (although the investigators identified heart failure in only 13% of enrolled patients). Within this subgroup, heart failure–associated hospitalizations occurred in 10.9% of the saxagliptin patients and 8.9% of the controls, a statistically significant difference, reported Dr. Deepak L. Bhatt, one of the investigators for the SAVOR-TIMI 53 study, which randomized more than 16,000 patients.
In contrast, the alogliptin study, EXAMINE, which randomized more than 5,000 patients with type 2 diabetes and a recent acute coronary syndrome event to treatment with alogliptin or placebo, found no real confirmation of this problem (N. Engl. J. Med. 2013;369:1327-35).
After the early-September report of this saxagliptin and heart failure hospitalization signal, the EXAMINE researchers went back to their data to specifically look at this issue, Dr. William B. White, the lead investigator, said when he presented the new findings at EASD. During a median of 18 months of follow-up, the rate of hospitalizations for heart failure was 3.1% in the alogliptin group and 2.9% in the placebo group, a difference that was not statistically significant, although it trended just slightly toward an alogliptin problem. Among the subgroup of patients who entered EXAMINE with a history of heart failure, the rate of the combined cardiovascular safety endpoint of CV death, nonfatal MI, or nonfatal stroke was 18% in the alogliptin group and 22% in the placebo group, a difference that was not statistically significant.
Despite the absence of any apparent heart failure–hospitalization effect in EXAMINE, Dr. Naveed Sattar, the invited discussant for the two reports at EASD, meta-analyzed the heart failure–hospitalization results from both studies, and found a combined, 24% relative increase in this outcome for the two DPP-4 inhibitors combined, a statistically significant effect largely driven toward significance by the underlying statistical significance in the saxagliptin study; the alogliptin results couldn’t resolve this difference since they trended in the same direction. Dr. Sattar concluded that it raised enough doubt about the safety of this entire drug class in patients with pre-existing heart failure to recommend not using drugs from this class in patients with any indication of heart failure – regardless of severity – until the apparent link received further scrutiny. Dr. Bhatt promised a report with further information on the heart failure issue at the American Heart Association Scientific Sessions in November.
Signals for canagliflozin, too?
SAVOR-TIMI 53 and EXAMINE provide the first results from what are more than a dozen large studies – involving more than 100,000 patients – launched recently to look for adverse cardiovascular effects from drugs in six different diabetes drug classes, according to Dr. Sanjay Kaul, who also spoke at EASD. He presented results from a meta-analysis he performed on results from nine phase III and phase II trials of canagliflozin (Invokana), an agent from the sodium-glucose cotransporter 2 (SGLT2) inhibitor class and approved by the FDA last March for treatment of type 2 diabetes, the first drug in that class to get U.S. marketing approval.
In the largest of these trials, CANVAS – the cardiovascular-assessment study for canagliflozin set up by the drug’s developer, Janssen – treatment with canagliflozin linked with a statistically significant, sixfold increase in cardiovascular events during the first 30 days of treatment, a link that then disappeared with longer-term treatment in patients with high CV risk. (Complete results from CANVAS have not yet been reported.) But Dr. Kaul did not see a similar excess in the other eight studies.
The canagliflozin meta-analysis also showed a nonsignificant, 46% increased risk of stroke in patients on the drug, a signal not seen in any other individual cardiovascular endpoint. Dr. Kaul analyzed data from 14 phase II and III trials for a second SGLT2 inhibitor drug, dapagliflozin, which showed no suggestion of causing any cardiovascular risk.
The canagliflozin risk signals are not reasons to stop using the drug or to withdraw it from the market, said Dr. Kaul, a member of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. The early signal of risk in CANVAS was not seen in any other study, and it may have been a statistical artifact, he said.
He said that signals of risk must be considered relative to a drug’s benefit. Applying the same standard of cardiovascular harm to drugs that are highly effective and to drugs that are not very effective "doesn’t make sense," he said. "A risk-benefit trade-off depends on the benefit. If you have a diabetes drug that lowers hemoglobin A1c by 1.5%-2% I’m willing to accept greater harm than I would from a drug that produced a 0.5% drop in hemoglobin A1c."
A final rosiglitazone thought
Dr. Kaul also made this observation about the rosiglitazone experience, which in retrospect produced "insufficient evidence to either incriminate or exonerate rosiglitazone," he said.
"That a diabetes drug [rosiglitazone] could have become a blockbuster without any established outcome benefits does not reflect well on the drug development and approval process. Equally lamentable is that this drug was virtually killed based on insufficient evidence and entrenched opinions."
Dr. Bhatt has received research grants from several companies, including AstraZeneca and Bristol-Myers Squibb (BMS), which market saxagliptin, and Takeda, maker of alogliptin. Dr. White is a safety consultant for several companies, including AstraZeneca and Takeda. Dr. Sattar is a consultant to several pharmaceutical firms, including AstraZeneca and BMS. Dr. Kaul is a consultant to Novo Nordisk, Sanofi, and Boehringer Ingelheim, and owns stock in Johnson & Johnson.
On Twitter @mitchelzoler
Diabetes drug development entered a new era in 2007 when a meta-analysis of 42 studies that had compared rosiglitazone with other drugs in a total of nearly 28,000 patients suggested that rosiglitazone treatment led to significantly increased rates of cardiovascular death and myocardial infarction.
Based largely on that, the Food and Drug Administration in 2008 issued recommendations to companies developing new diabetes drugs to run large-scale trials aimed at assessing their cardiovascular effects.
In 2011, the FDA placed restrictions on prescribing rosiglitazone, and U.S. use of the drug plummeted. (An FDA panel voted in June to relax some of the restrictions the agency had imposed.)
Some consequences of the long shadow cast by the rosiglitazone experience played out in talks at the annual congress of the European Society for the Study of Diabetes in September, as well as at the annual congress of the European Society of Cardiology a few weeks before that. The new data show just how painstaking investigators have become in trying to parse out hints of cardiovascular danger lurking in masses of megatrial data.
Heart failure haunts DPP-4 inhibitors
At ESC, as well as in a pair of simultaneously published articles, researchers reported results from two large trials designed to follow the FDA recommendations and assess cardiovascular safety for two new selective dipeptidyl peptidase-4 (DPP-4) inhibitors, saxagliptin (Onglyza) and alogliptin, in patients with type 2 diabetes.
Saxagliptin was the drug with the red-flag signal, while alogliptin faced guilt by association. The saxagliptin study, SAVOR-TIMI 53, used patients with either an established history of cardiovascular disease (CVD) or multiple CVD risk factors. The study’s primary safety endpoint, the combined rate of CVD death, nonfatal myocardial infarction, or nonfatal stroke during 2 years of follow-up, was virtually identical in the two groups, 7.3% in the saxagliptin patients and 7.2% in the controls.
The study’s secondary safety endpoint, which included the primary combined plus other endpoints such as need for revascularization and hospitalization for heart failure, was also similar in the two study arms, with this expanded combined endpoint occurring in 12.8% of the saxagliptin patients and 12.4% of the controls (N. Engl. J. Med. 2013;369:1317-26).
The whisper of a saxagliptin problem was in just one secondary, individual safety endpoint, hospitalizations for heart failure. These occurred in 3.5% of the saxagliptin patients and in 2.8% of controls, a statistically significant difference. Further data presented at EASD showed that this difference clustered predominantly in the subgroup of patients who entered the study with presumed heart failure, based on having a blood level of NT-pro brain natriuretic peptide of at least 333, about 25% of patients in both treatment arms (although the investigators identified heart failure in only 13% of enrolled patients). Within this subgroup, heart failure–associated hospitalizations occurred in 10.9% of the saxagliptin patients and 8.9% of the controls, a statistically significant difference, reported Dr. Deepak L. Bhatt, one of the investigators for the SAVOR-TIMI 53 study, which randomized more than 16,000 patients.
In contrast, the alogliptin study, EXAMINE, which randomized more than 5,000 patients with type 2 diabetes and a recent acute coronary syndrome event to treatment with alogliptin or placebo, found no real confirmation of this problem (N. Engl. J. Med. 2013;369:1327-35).
After the early-September report of this saxagliptin and heart failure hospitalization signal, the EXAMINE researchers went back to their data to specifically look at this issue, Dr. William B. White, the lead investigator, said when he presented the new findings at EASD. During a median of 18 months of follow-up, the rate of hospitalizations for heart failure was 3.1% in the alogliptin group and 2.9% in the placebo group, a difference that was not statistically significant, although it trended just slightly toward an alogliptin problem. Among the subgroup of patients who entered EXAMINE with a history of heart failure, the rate of the combined cardiovascular safety endpoint of CV death, nonfatal MI, or nonfatal stroke was 18% in the alogliptin group and 22% in the placebo group, a difference that was not statistically significant.
Despite the absence of any apparent heart failure–hospitalization effect in EXAMINE, Dr. Naveed Sattar, the invited discussant for the two reports at EASD, meta-analyzed the heart failure–hospitalization results from both studies, and found a combined, 24% relative increase in this outcome for the two DPP-4 inhibitors combined, a statistically significant effect largely driven toward significance by the underlying statistical significance in the saxagliptin study; the alogliptin results couldn’t resolve this difference since they trended in the same direction. Dr. Sattar concluded that it raised enough doubt about the safety of this entire drug class in patients with pre-existing heart failure to recommend not using drugs from this class in patients with any indication of heart failure – regardless of severity – until the apparent link received further scrutiny. Dr. Bhatt promised a report with further information on the heart failure issue at the American Heart Association Scientific Sessions in November.
Signals for canagliflozin, too?
SAVOR-TIMI 53 and EXAMINE provide the first results from what are more than a dozen large studies – involving more than 100,000 patients – launched recently to look for adverse cardiovascular effects from drugs in six different diabetes drug classes, according to Dr. Sanjay Kaul, who also spoke at EASD. He presented results from a meta-analysis he performed on results from nine phase III and phase II trials of canagliflozin (Invokana), an agent from the sodium-glucose cotransporter 2 (SGLT2) inhibitor class and approved by the FDA last March for treatment of type 2 diabetes, the first drug in that class to get U.S. marketing approval.
In the largest of these trials, CANVAS – the cardiovascular-assessment study for canagliflozin set up by the drug’s developer, Janssen – treatment with canagliflozin linked with a statistically significant, sixfold increase in cardiovascular events during the first 30 days of treatment, a link that then disappeared with longer-term treatment in patients with high CV risk. (Complete results from CANVAS have not yet been reported.) But Dr. Kaul did not see a similar excess in the other eight studies.
The canagliflozin meta-analysis also showed a nonsignificant, 46% increased risk of stroke in patients on the drug, a signal not seen in any other individual cardiovascular endpoint. Dr. Kaul analyzed data from 14 phase II and III trials for a second SGLT2 inhibitor drug, dapagliflozin, which showed no suggestion of causing any cardiovascular risk.
The canagliflozin risk signals are not reasons to stop using the drug or to withdraw it from the market, said Dr. Kaul, a member of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. The early signal of risk in CANVAS was not seen in any other study, and it may have been a statistical artifact, he said.
He said that signals of risk must be considered relative to a drug’s benefit. Applying the same standard of cardiovascular harm to drugs that are highly effective and to drugs that are not very effective "doesn’t make sense," he said. "A risk-benefit trade-off depends on the benefit. If you have a diabetes drug that lowers hemoglobin A1c by 1.5%-2% I’m willing to accept greater harm than I would from a drug that produced a 0.5% drop in hemoglobin A1c."
A final rosiglitazone thought
Dr. Kaul also made this observation about the rosiglitazone experience, which in retrospect produced "insufficient evidence to either incriminate or exonerate rosiglitazone," he said.
"That a diabetes drug [rosiglitazone] could have become a blockbuster without any established outcome benefits does not reflect well on the drug development and approval process. Equally lamentable is that this drug was virtually killed based on insufficient evidence and entrenched opinions."
Dr. Bhatt has received research grants from several companies, including AstraZeneca and Bristol-Myers Squibb (BMS), which market saxagliptin, and Takeda, maker of alogliptin. Dr. White is a safety consultant for several companies, including AstraZeneca and Takeda. Dr. Sattar is a consultant to several pharmaceutical firms, including AstraZeneca and BMS. Dr. Kaul is a consultant to Novo Nordisk, Sanofi, and Boehringer Ingelheim, and owns stock in Johnson & Johnson.
On Twitter @mitchelzoler
Marked BPN elevations found in a third of diastolic HF patients
VANCOUVER, B.C. – B-type natriuretic peptide elevations above 1,000 pg/mL may herald worse prognosis in patients with diastolic heart failure but normal renal function, according to a retrospective database analysis from the University of California, Davis.
Among 421 hospitalized patients with diastolic heart failure – heart failure (HF) with ejection fractions above 45% – investigators found that 117 (28%) patients had B-type natriuretic peptide (BNP) levels above 1,000 pg/mL.
Fifty of those patients (43%) had stage IV or V chronic kidney disease, compared with 7 (13%) of the 54 diastolic HF patients with BNPs below 100 pg/mL. The team also found a significant relationship between estimated glomerular filtration rate (eGFR) and BNP (R = 0.39). In most cases, BNP levels above 1,000 pg/mL "were likely due to reduced [renal] clearance and associated factors. The degree of renal dysfunction is related to the degree of BNP elevation," said senior investigator Dr. Saul Schaefer, professor at the university and also chief of cardiology at the Veterans Affairs Medical Center in Sacramento.

But 25 patients (21%) with BNPs above 1,000 pg/mL "actually had normal renal function; that is, an [eGFR] greater than 60 mL/min per 1.73 m2. It’s unclear why these patients would have such an elevated BNP. They were older [mean age 70.2 years vs. 67.7 years] and had slightly reduced ejection fractions in comparison to the overall group," he said at the 18th World Congress on Heart Disease.
"My guess is that these patients with high BNPs and normal renal function have more severe abnormalities and diastolic failure. It’s a potential marker of higher risk. We could potentially follow these patients more closely and treat them more aggressively," he said.
Before that happens, however, "we need to prospectively look at these patients and see if they can be distinguished from the lower-BNP patients, and track them to see whether their prognosis is adversely affected," Dr. Schaefer said.
Compared with diastolic HF patients in the study with BNPs below 100 pg/mL, the 117 with levels above 1,000 pg/mL tended to be older (68 vs. 61.4 years), have higher troponin levels (0.2 vs. 0 ng/mL), and have greater use of antihypertensive medications, suggesting that they had more chronic hypertension. There were no associations between markedly elevated BNP and gender, diabetes, or ischemic heart disease. Echo parameters did not predict BNP.
Dr. Schaefer said that he had no relevant disclosures.
VANCOUVER, B.C. – B-type natriuretic peptide elevations above 1,000 pg/mL may herald worse prognosis in patients with diastolic heart failure but normal renal function, according to a retrospective database analysis from the University of California, Davis.
Among 421 hospitalized patients with diastolic heart failure – heart failure (HF) with ejection fractions above 45% – investigators found that 117 (28%) patients had B-type natriuretic peptide (BNP) levels above 1,000 pg/mL.
Fifty of those patients (43%) had stage IV or V chronic kidney disease, compared with 7 (13%) of the 54 diastolic HF patients with BNPs below 100 pg/mL. The team also found a significant relationship between estimated glomerular filtration rate (eGFR) and BNP (R = 0.39). In most cases, BNP levels above 1,000 pg/mL "were likely due to reduced [renal] clearance and associated factors. The degree of renal dysfunction is related to the degree of BNP elevation," said senior investigator Dr. Saul Schaefer, professor at the university and also chief of cardiology at the Veterans Affairs Medical Center in Sacramento.
But 25 patients (21%) with BNPs above 1,000 pg/mL "actually had normal renal function; that is, an [eGFR] greater than 60 mL/min per 1.73 m2. It’s unclear why these patients would have such an elevated BNP. They were older [mean age 70.2 years vs. 67.7 years] and had slightly reduced ejection fractions in comparison to the overall group," he said at the 18th World Congress on Heart Disease.
"My guess is that these patients with high BNPs and normal renal function have more severe abnormalities and diastolic failure. It’s a potential marker of higher risk. We could potentially follow these patients more closely and treat them more aggressively," he said.
Before that happens, however, "we need to prospectively look at these patients and see if they can be distinguished from the lower-BNP patients, and track them to see whether their prognosis is adversely affected," Dr. Schaefer said.
Compared with diastolic HF patients in the study with BNPs below 100 pg/mL, the 117 with levels above 1,000 pg/mL tended to be older (68 vs. 61.4 years), have higher troponin levels (0.2 vs. 0 ng/mL), and have greater use of antihypertensive medications, suggesting that they had more chronic hypertension. There were no associations between markedly elevated BNP and gender, diabetes, or ischemic heart disease. Echo parameters did not predict BNP.
Dr. Schaefer said that he had no relevant disclosures.
VANCOUVER, B.C. – B-type natriuretic peptide elevations above 1,000 pg/mL may herald worse prognosis in patients with diastolic heart failure but normal renal function, according to a retrospective database analysis from the University of California, Davis.
Among 421 hospitalized patients with diastolic heart failure – heart failure (HF) with ejection fractions above 45% – investigators found that 117 (28%) patients had B-type natriuretic peptide (BNP) levels above 1,000 pg/mL.
Fifty of those patients (43%) had stage IV or V chronic kidney disease, compared with 7 (13%) of the 54 diastolic HF patients with BNPs below 100 pg/mL. The team also found a significant relationship between estimated glomerular filtration rate (eGFR) and BNP (R = 0.39). In most cases, BNP levels above 1,000 pg/mL "were likely due to reduced [renal] clearance and associated factors. The degree of renal dysfunction is related to the degree of BNP elevation," said senior investigator Dr. Saul Schaefer, professor at the university and also chief of cardiology at the Veterans Affairs Medical Center in Sacramento.
But 25 patients (21%) with BNPs above 1,000 pg/mL "actually had normal renal function; that is, an [eGFR] greater than 60 mL/min per 1.73 m2. It’s unclear why these patients would have such an elevated BNP. They were older [mean age 70.2 years vs. 67.7 years] and had slightly reduced ejection fractions in comparison to the overall group," he said at the 18th World Congress on Heart Disease.
"My guess is that these patients with high BNPs and normal renal function have more severe abnormalities and diastolic failure. It’s a potential marker of higher risk. We could potentially follow these patients more closely and treat them more aggressively," he said.
Before that happens, however, "we need to prospectively look at these patients and see if they can be distinguished from the lower-BNP patients, and track them to see whether their prognosis is adversely affected," Dr. Schaefer said.
Compared with diastolic HF patients in the study with BNPs below 100 pg/mL, the 117 with levels above 1,000 pg/mL tended to be older (68 vs. 61.4 years), have higher troponin levels (0.2 vs. 0 ng/mL), and have greater use of antihypertensive medications, suggesting that they had more chronic hypertension. There were no associations between markedly elevated BNP and gender, diabetes, or ischemic heart disease. Echo parameters did not predict BNP.
Dr. Schaefer said that he had no relevant disclosures.
AT THE 18TH WORLD CONGRESS ON HEART DISEASE
Major finding: Among 421 hospitalized patients with diastolic heart failure, 117 (28%) had BNP levels above 1,000 pg/mL.
Data Source: Record review.
Disclosures: Dr. Schaefer said that he had no relevant disclosures.

Advisers support FDA approval of wireless HF monitoring device
GAITHERSBURG, MD. – An implantable wireless device that provides measurements of pulmonary arterial pressure and heart rate in patients with heart failure was supported for approval by a majority of a Food and Drug Administration advisory panel, almost 2 years after it was rejected for approval.
The Champion Heart Failure Monitoring System, a permanent implantable pressure-measurement system, includes a wireless sensor that is implanted in a small branch of the pulmonary artery during a right-heart cardiac catheterization, providing wireless PA measurements.
At a meeting of the FDA’s Circulatory System Devices Panel, the panel voted 6-4, with 1 abstention, that the benefits of the device outweighed its risks for monitoring patients who meet the criteria in the proposed indication, which is for wirelessly measuring and monitoring PA pressures and heart rate in patients with New York Heart Association (NYHA) class III heart failure who have been hospitalized for HF in the previous year.
The proposed indication includes the statement that the hemodynamic data "are used by physicians for heart failure management and to reduce heart failure hospitalizations" in hospital or office settings, and that it can also be used by patients at home or another location "to wirelessly obtain and send hemodynamic and PA pressure measurements" to a database to be reviewed and evaluated by their physicians. The manufacturer is CardioMEMS.
The panel was divided regarding the strength of the data supporting the effect the device had on reducing HF hospitalizations, the primary endpoint of studies, and this was reflected in the vote on the device’s effectiveness. The panel voted 7-4 that there was not reasonable assurance that the device was effective for use for the proposed indication. But they unanimously voted that there was reasonable assurance it was safe, based on the finding that most complications occurred in the 30 days after implantation.
However, no benefit was seen on HF hospitalizations in women, which panelists thought may have been due to the low number of women enrolled in the studies, and panelists recommended that the device needed to be studied further in women. Another recommendation was to drop the specific HF class in the indication because patients with HF tend to move from one class to the other.
The original study enrolled 550 patients with NYHA class III HF and a recent hospitalization for HF. All patients had the device implanted, but physicians had access to daily PA measurements for those randomized to the treatment group, and were able to adjust HF medications based on the values provided.
At 6 months, there were 84 HF hospitalizations in the treatment group (0.32 per patient), compared with 120 (0.44 per patient) in the control group, a highly significant difference that represented a 28% reduction in the rate of HF hospitalizations. In addition, 5.6% of the patients in the treatment group died, versus 7.1% in the control group, a nonsignificant 23% reduction in the relative risk of death.
In December 2011, the FDA’s Circulatory System Devices Panel met for the first time to review the device, and voted 6-4 that the risks outweighed the benefits. Like the FDA reviewers, the panel raised concerns that the nurse communications in this study may have influenced treatment decisions and biased the results, and the FDA did not approve the device.
In response, the company did not conduct another trial but conducted various analyses of HF hospitalizations and deaths during the open-access period of the study, which eliminated nurse input. Patients in the control and treatment groups in the randomized study were included in the study’s open-access period, where the PA pressures on all patients were provided to physicians.
Among the results was that HF hospitalization rates among those patients who were controls in the first part of the study dropped to a rate comparable to the rate among those who were in the original treatment group, when access to the PA measurements of the original controls were available. A third-party analysis showed that the possible effect the nurses’ input had on the outcomes in the randomized study was minimal, according to the company.
On the basis of these analyses, the company concluded that the effect on HF hospitalizations was a result of physicians knowing the PA measurements provided by the device, and was not related to the communications from nurses during the randomized segment of the study or during the open-access period.
Panelist Dr. Jeffrey Borer, chief of the division of cardiovascular medicine at the State University of New York Downstate Medical Center, Brooklyn, who voted yes on all three questions, said he found the ancillary studies very helpful in clarifying the utility of the device seen in the original study, and that this was "an approvable and probably very useful device." He, like other panelists, remained concerned about the use in women, noting that there was no benefit on HF hospitalizations in women who had the device implanted, but this may have been due to the low number of women enrolled in the study.
Dr. David Yuh, chief of cardiac surgery at Yale University, New Haven, Conn., said that while he was not surprised by the split vote on risk-benefit, he voted positively on this question. "I did feel this was an effective monitoring device that did facilitate closer monitoring of these very difficult patients for heart failure cardiologists."
Dr. Valluvan Jeevanandam, chief of cardiac and thoracic surgery at the University of Chicago, was one of two panelists who were not convinced by efficacy in terms of reducing HF hospitalizations; however, he voted in favor of the risk-benefit profile because of what he considered the device’s value as a diagnostic tool, which he said he would find useful in "innumerable" patients.
"I just got paged for two people who need right-heart catheterizations tomorrow, who wouldn’t need them" if they had this device, he said.
Voting no on the effectiveness and risk-benefit questions, Dr. David Milan, a cardiac electrophysiologist at Massachusetts General Hospital, Boston, said he believed the device was safe, but there were not enough data on effectiveness. The data analyses presented by the company were not valid or convincing, and the company should have conducted another randomized controlled study, he said.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. If approved, CardioMEMS has planned a postapproval study that would prospectively follow the safety and effectiveness of the device in a real-world setting in about 1,200 patients, including 420 women.
GAITHERSBURG, MD. – An implantable wireless device that provides measurements of pulmonary arterial pressure and heart rate in patients with heart failure was supported for approval by a majority of a Food and Drug Administration advisory panel, almost 2 years after it was rejected for approval.
The Champion Heart Failure Monitoring System, a permanent implantable pressure-measurement system, includes a wireless sensor that is implanted in a small branch of the pulmonary artery during a right-heart cardiac catheterization, providing wireless PA measurements.
At a meeting of the FDA’s Circulatory System Devices Panel, the panel voted 6-4, with 1 abstention, that the benefits of the device outweighed its risks for monitoring patients who meet the criteria in the proposed indication, which is for wirelessly measuring and monitoring PA pressures and heart rate in patients with New York Heart Association (NYHA) class III heart failure who have been hospitalized for HF in the previous year.
The proposed indication includes the statement that the hemodynamic data "are used by physicians for heart failure management and to reduce heart failure hospitalizations" in hospital or office settings, and that it can also be used by patients at home or another location "to wirelessly obtain and send hemodynamic and PA pressure measurements" to a database to be reviewed and evaluated by their physicians. The manufacturer is CardioMEMS.
The panel was divided regarding the strength of the data supporting the effect the device had on reducing HF hospitalizations, the primary endpoint of studies, and this was reflected in the vote on the device’s effectiveness. The panel voted 7-4 that there was not reasonable assurance that the device was effective for use for the proposed indication. But they unanimously voted that there was reasonable assurance it was safe, based on the finding that most complications occurred in the 30 days after implantation.
However, no benefit was seen on HF hospitalizations in women, which panelists thought may have been due to the low number of women enrolled in the studies, and panelists recommended that the device needed to be studied further in women. Another recommendation was to drop the specific HF class in the indication because patients with HF tend to move from one class to the other.
The original study enrolled 550 patients with NYHA class III HF and a recent hospitalization for HF. All patients had the device implanted, but physicians had access to daily PA measurements for those randomized to the treatment group, and were able to adjust HF medications based on the values provided.
At 6 months, there were 84 HF hospitalizations in the treatment group (0.32 per patient), compared with 120 (0.44 per patient) in the control group, a highly significant difference that represented a 28% reduction in the rate of HF hospitalizations. In addition, 5.6% of the patients in the treatment group died, versus 7.1% in the control group, a nonsignificant 23% reduction in the relative risk of death.
In December 2011, the FDA’s Circulatory System Devices Panel met for the first time to review the device, and voted 6-4 that the risks outweighed the benefits. Like the FDA reviewers, the panel raised concerns that the nurse communications in this study may have influenced treatment decisions and biased the results, and the FDA did not approve the device.
In response, the company did not conduct another trial but conducted various analyses of HF hospitalizations and deaths during the open-access period of the study, which eliminated nurse input. Patients in the control and treatment groups in the randomized study were included in the study’s open-access period, where the PA pressures on all patients were provided to physicians.
Among the results was that HF hospitalization rates among those patients who were controls in the first part of the study dropped to a rate comparable to the rate among those who were in the original treatment group, when access to the PA measurements of the original controls were available. A third-party analysis showed that the possible effect the nurses’ input had on the outcomes in the randomized study was minimal, according to the company.
On the basis of these analyses, the company concluded that the effect on HF hospitalizations was a result of physicians knowing the PA measurements provided by the device, and was not related to the communications from nurses during the randomized segment of the study or during the open-access period.
Panelist Dr. Jeffrey Borer, chief of the division of cardiovascular medicine at the State University of New York Downstate Medical Center, Brooklyn, who voted yes on all three questions, said he found the ancillary studies very helpful in clarifying the utility of the device seen in the original study, and that this was "an approvable and probably very useful device." He, like other panelists, remained concerned about the use in women, noting that there was no benefit on HF hospitalizations in women who had the device implanted, but this may have been due to the low number of women enrolled in the study.
Dr. David Yuh, chief of cardiac surgery at Yale University, New Haven, Conn., said that while he was not surprised by the split vote on risk-benefit, he voted positively on this question. "I did feel this was an effective monitoring device that did facilitate closer monitoring of these very difficult patients for heart failure cardiologists."
Dr. Valluvan Jeevanandam, chief of cardiac and thoracic surgery at the University of Chicago, was one of two panelists who were not convinced by efficacy in terms of reducing HF hospitalizations; however, he voted in favor of the risk-benefit profile because of what he considered the device’s value as a diagnostic tool, which he said he would find useful in "innumerable" patients.
"I just got paged for two people who need right-heart catheterizations tomorrow, who wouldn’t need them" if they had this device, he said.
Voting no on the effectiveness and risk-benefit questions, Dr. David Milan, a cardiac electrophysiologist at Massachusetts General Hospital, Boston, said he believed the device was safe, but there were not enough data on effectiveness. The data analyses presented by the company were not valid or convincing, and the company should have conducted another randomized controlled study, he said.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. If approved, CardioMEMS has planned a postapproval study that would prospectively follow the safety and effectiveness of the device in a real-world setting in about 1,200 patients, including 420 women.
GAITHERSBURG, MD. – An implantable wireless device that provides measurements of pulmonary arterial pressure and heart rate in patients with heart failure was supported for approval by a majority of a Food and Drug Administration advisory panel, almost 2 years after it was rejected for approval.
The Champion Heart Failure Monitoring System, a permanent implantable pressure-measurement system, includes a wireless sensor that is implanted in a small branch of the pulmonary artery during a right-heart cardiac catheterization, providing wireless PA measurements.
At a meeting of the FDA’s Circulatory System Devices Panel, the panel voted 6-4, with 1 abstention, that the benefits of the device outweighed its risks for monitoring patients who meet the criteria in the proposed indication, which is for wirelessly measuring and monitoring PA pressures and heart rate in patients with New York Heart Association (NYHA) class III heart failure who have been hospitalized for HF in the previous year.
The proposed indication includes the statement that the hemodynamic data "are used by physicians for heart failure management and to reduce heart failure hospitalizations" in hospital or office settings, and that it can also be used by patients at home or another location "to wirelessly obtain and send hemodynamic and PA pressure measurements" to a database to be reviewed and evaluated by their physicians. The manufacturer is CardioMEMS.
The panel was divided regarding the strength of the data supporting the effect the device had on reducing HF hospitalizations, the primary endpoint of studies, and this was reflected in the vote on the device’s effectiveness. The panel voted 7-4 that there was not reasonable assurance that the device was effective for use for the proposed indication. But they unanimously voted that there was reasonable assurance it was safe, based on the finding that most complications occurred in the 30 days after implantation.
However, no benefit was seen on HF hospitalizations in women, which panelists thought may have been due to the low number of women enrolled in the studies, and panelists recommended that the device needed to be studied further in women. Another recommendation was to drop the specific HF class in the indication because patients with HF tend to move from one class to the other.
The original study enrolled 550 patients with NYHA class III HF and a recent hospitalization for HF. All patients had the device implanted, but physicians had access to daily PA measurements for those randomized to the treatment group, and were able to adjust HF medications based on the values provided.
At 6 months, there were 84 HF hospitalizations in the treatment group (0.32 per patient), compared with 120 (0.44 per patient) in the control group, a highly significant difference that represented a 28% reduction in the rate of HF hospitalizations. In addition, 5.6% of the patients in the treatment group died, versus 7.1% in the control group, a nonsignificant 23% reduction in the relative risk of death.
In December 2011, the FDA’s Circulatory System Devices Panel met for the first time to review the device, and voted 6-4 that the risks outweighed the benefits. Like the FDA reviewers, the panel raised concerns that the nurse communications in this study may have influenced treatment decisions and biased the results, and the FDA did not approve the device.
In response, the company did not conduct another trial but conducted various analyses of HF hospitalizations and deaths during the open-access period of the study, which eliminated nurse input. Patients in the control and treatment groups in the randomized study were included in the study’s open-access period, where the PA pressures on all patients were provided to physicians.
Among the results was that HF hospitalization rates among those patients who were controls in the first part of the study dropped to a rate comparable to the rate among those who were in the original treatment group, when access to the PA measurements of the original controls were available. A third-party analysis showed that the possible effect the nurses’ input had on the outcomes in the randomized study was minimal, according to the company.
On the basis of these analyses, the company concluded that the effect on HF hospitalizations was a result of physicians knowing the PA measurements provided by the device, and was not related to the communications from nurses during the randomized segment of the study or during the open-access period.
Panelist Dr. Jeffrey Borer, chief of the division of cardiovascular medicine at the State University of New York Downstate Medical Center, Brooklyn, who voted yes on all three questions, said he found the ancillary studies very helpful in clarifying the utility of the device seen in the original study, and that this was "an approvable and probably very useful device." He, like other panelists, remained concerned about the use in women, noting that there was no benefit on HF hospitalizations in women who had the device implanted, but this may have been due to the low number of women enrolled in the study.
Dr. David Yuh, chief of cardiac surgery at Yale University, New Haven, Conn., said that while he was not surprised by the split vote on risk-benefit, he voted positively on this question. "I did feel this was an effective monitoring device that did facilitate closer monitoring of these very difficult patients for heart failure cardiologists."
Dr. Valluvan Jeevanandam, chief of cardiac and thoracic surgery at the University of Chicago, was one of two panelists who were not convinced by efficacy in terms of reducing HF hospitalizations; however, he voted in favor of the risk-benefit profile because of what he considered the device’s value as a diagnostic tool, which he said he would find useful in "innumerable" patients.
"I just got paged for two people who need right-heart catheterizations tomorrow, who wouldn’t need them" if they had this device, he said.
Voting no on the effectiveness and risk-benefit questions, Dr. David Milan, a cardiac electrophysiologist at Massachusetts General Hospital, Boston, said he believed the device was safe, but there were not enough data on effectiveness. The data analyses presented by the company were not valid or convincing, and the company should have conducted another randomized controlled study, he said.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. If approved, CardioMEMS has planned a postapproval study that would prospectively follow the safety and effectiveness of the device in a real-world setting in about 1,200 patients, including 420 women.
AT AN FDA ADVISORY PANEL MEETING
FDA panel narrowly backs expanding CRT devices’ indications
GAITHERSBURG, MD. – A Food and Drug Administration advisory panel narrowly voted to support expanding the approval of Medtronic’s Cardiac Resynchronization Therapy (CRT) devices to include patients with atrioventricular dysfunction and left ventricular systolic dysfunction.
At a meeting on Oct. 8, the Circulatory System Devices Panel voted 4-3, with one abstention, that the benefits of treatment with Medtronic’s CRT defibrillators and CRT pacemakers outweigh the risks for patients who meet the criteria in the company’s proposed indication for use – which the panel slightly modified before the vote.
Medtronic has proposed that approval of the company’s CRT-pacemaker (CRT-P) and CRT-defibrillator (CRT-D) devices be expanded to include treatment of patients who meet the following criteria: class I or class IIa indications for pacemaker implantation in accordance with American College of Cardiology/American Heart Association/Heart Rhythm Society guidelines, have New York Heart Association (NYHA) class I, II, or III heart failure, left ventricular ejection fraction (LVEF) less than or equal to 50%, with at least one of the following: third- or second-degree AV block; first-degree AV block with symptoms similar to pacemaker syndrome; or documented Wenckebach or PR interval greater than 300 ms when paced at 100 ppm. (Patients eligible for CRT-D also would need to be at risk of a life-threatening ventricular arrhythmia.)
The panel modified the proposed indication by dropping the segments on first-degree AV block and the documented Wenckebach or PR interval in response to issues that included uncertainty over whether this group of patients would benefit from treatment. This was changed to patients with first-degree AV block who are judged, with reliable confidence, to be in need of pacing most of the time.
The company’s proposed indication is based on the results of the BLOCK HF (Biventricular versus Right Ventricular Pacing in Heart Failure Patients with Atrioventricular Block) study, which randomized 691 patients with an indication for pacing with AV block; NYHA class I, II, or III heart failure; and a LVEF of 50% or less to right ventricular or biventricular pacing with a CRT-P or CRT-D.
The primary endpoint, a composite of death, an urgent care visit for HF that required intravenous therapy, or at least a 15% increase in the left ventricular end-systolic volume index (LVESVI), was met by 45.8% of those in the biventricular pacing arm vs. 55.8% of those in the right ventricular pacing arm, a statistically significant difference and a 27% reduction in risk of the primary outcome. The rate of left ventricular lead complications was 6.4% (N. Engl. J. Med. 2013;368:1585-93).
Currently, the CRT-P devices are approved for NYHA class III and IV patients who remain symptomatic despite stable, optimal medical therapy and have an LVEF of 35% or less and a prolonged QRS duration.
The CRT-D devices are approved for ventricular antitachycardia pacing and ventricular defibrillation for automated treatment of life-threatening ventricular arrhythmias, and for providing CRT in heart failure patients who remain symptomatic despite optimal medical therapy and meet any of the following classifications: NYHA class III or IV heart failure with an LVEF of 35% or less and a prolonged QRS duration; left bundle branch block with a QRS duration of 130 ms or more; an LVEF of 30% or less; and NYHA class II heart failure.
Panelists who said that the benefits did not outweigh the risks cited the potential long-term left ventricular lead complications as a concern. Those voting in favor also wrestled with this issue but agreed BLOCK HF was a well-conducted trial with results that some panelists found more compelling than others.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
GAITHERSBURG, MD. – A Food and Drug Administration advisory panel narrowly voted to support expanding the approval of Medtronic’s Cardiac Resynchronization Therapy (CRT) devices to include patients with atrioventricular dysfunction and left ventricular systolic dysfunction.
At a meeting on Oct. 8, the Circulatory System Devices Panel voted 4-3, with one abstention, that the benefits of treatment with Medtronic’s CRT defibrillators and CRT pacemakers outweigh the risks for patients who meet the criteria in the company’s proposed indication for use – which the panel slightly modified before the vote.
Medtronic has proposed that approval of the company’s CRT-pacemaker (CRT-P) and CRT-defibrillator (CRT-D) devices be expanded to include treatment of patients who meet the following criteria: class I or class IIa indications for pacemaker implantation in accordance with American College of Cardiology/American Heart Association/Heart Rhythm Society guidelines, have New York Heart Association (NYHA) class I, II, or III heart failure, left ventricular ejection fraction (LVEF) less than or equal to 50%, with at least one of the following: third- or second-degree AV block; first-degree AV block with symptoms similar to pacemaker syndrome; or documented Wenckebach or PR interval greater than 300 ms when paced at 100 ppm. (Patients eligible for CRT-D also would need to be at risk of a life-threatening ventricular arrhythmia.)
The panel modified the proposed indication by dropping the segments on first-degree AV block and the documented Wenckebach or PR interval in response to issues that included uncertainty over whether this group of patients would benefit from treatment. This was changed to patients with first-degree AV block who are judged, with reliable confidence, to be in need of pacing most of the time.
The company’s proposed indication is based on the results of the BLOCK HF (Biventricular versus Right Ventricular Pacing in Heart Failure Patients with Atrioventricular Block) study, which randomized 691 patients with an indication for pacing with AV block; NYHA class I, II, or III heart failure; and a LVEF of 50% or less to right ventricular or biventricular pacing with a CRT-P or CRT-D.
The primary endpoint, a composite of death, an urgent care visit for HF that required intravenous therapy, or at least a 15% increase in the left ventricular end-systolic volume index (LVESVI), was met by 45.8% of those in the biventricular pacing arm vs. 55.8% of those in the right ventricular pacing arm, a statistically significant difference and a 27% reduction in risk of the primary outcome. The rate of left ventricular lead complications was 6.4% (N. Engl. J. Med. 2013;368:1585-93).
Currently, the CRT-P devices are approved for NYHA class III and IV patients who remain symptomatic despite stable, optimal medical therapy and have an LVEF of 35% or less and a prolonged QRS duration.
The CRT-D devices are approved for ventricular antitachycardia pacing and ventricular defibrillation for automated treatment of life-threatening ventricular arrhythmias, and for providing CRT in heart failure patients who remain symptomatic despite optimal medical therapy and meet any of the following classifications: NYHA class III or IV heart failure with an LVEF of 35% or less and a prolonged QRS duration; left bundle branch block with a QRS duration of 130 ms or more; an LVEF of 30% or less; and NYHA class II heart failure.
Panelists who said that the benefits did not outweigh the risks cited the potential long-term left ventricular lead complications as a concern. Those voting in favor also wrestled with this issue but agreed BLOCK HF was a well-conducted trial with results that some panelists found more compelling than others.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
GAITHERSBURG, MD. – A Food and Drug Administration advisory panel narrowly voted to support expanding the approval of Medtronic’s Cardiac Resynchronization Therapy (CRT) devices to include patients with atrioventricular dysfunction and left ventricular systolic dysfunction.
At a meeting on Oct. 8, the Circulatory System Devices Panel voted 4-3, with one abstention, that the benefits of treatment with Medtronic’s CRT defibrillators and CRT pacemakers outweigh the risks for patients who meet the criteria in the company’s proposed indication for use – which the panel slightly modified before the vote.
Medtronic has proposed that approval of the company’s CRT-pacemaker (CRT-P) and CRT-defibrillator (CRT-D) devices be expanded to include treatment of patients who meet the following criteria: class I or class IIa indications for pacemaker implantation in accordance with American College of Cardiology/American Heart Association/Heart Rhythm Society guidelines, have New York Heart Association (NYHA) class I, II, or III heart failure, left ventricular ejection fraction (LVEF) less than or equal to 50%, with at least one of the following: third- or second-degree AV block; first-degree AV block with symptoms similar to pacemaker syndrome; or documented Wenckebach or PR interval greater than 300 ms when paced at 100 ppm. (Patients eligible for CRT-D also would need to be at risk of a life-threatening ventricular arrhythmia.)
The panel modified the proposed indication by dropping the segments on first-degree AV block and the documented Wenckebach or PR interval in response to issues that included uncertainty over whether this group of patients would benefit from treatment. This was changed to patients with first-degree AV block who are judged, with reliable confidence, to be in need of pacing most of the time.
The company’s proposed indication is based on the results of the BLOCK HF (Biventricular versus Right Ventricular Pacing in Heart Failure Patients with Atrioventricular Block) study, which randomized 691 patients with an indication for pacing with AV block; NYHA class I, II, or III heart failure; and a LVEF of 50% or less to right ventricular or biventricular pacing with a CRT-P or CRT-D.
The primary endpoint, a composite of death, an urgent care visit for HF that required intravenous therapy, or at least a 15% increase in the left ventricular end-systolic volume index (LVESVI), was met by 45.8% of those in the biventricular pacing arm vs. 55.8% of those in the right ventricular pacing arm, a statistically significant difference and a 27% reduction in risk of the primary outcome. The rate of left ventricular lead complications was 6.4% (N. Engl. J. Med. 2013;368:1585-93).
Currently, the CRT-P devices are approved for NYHA class III and IV patients who remain symptomatic despite stable, optimal medical therapy and have an LVEF of 35% or less and a prolonged QRS duration.
The CRT-D devices are approved for ventricular antitachycardia pacing and ventricular defibrillation for automated treatment of life-threatening ventricular arrhythmias, and for providing CRT in heart failure patients who remain symptomatic despite optimal medical therapy and meet any of the following classifications: NYHA class III or IV heart failure with an LVEF of 35% or less and a prolonged QRS duration; left bundle branch block with a QRS duration of 130 ms or more; an LVEF of 30% or less; and NYHA class II heart failure.
Panelists who said that the benefits did not outweigh the risks cited the potential long-term left ventricular lead complications as a concern. Those voting in favor also wrestled with this issue but agreed BLOCK HF was a well-conducted trial with results that some panelists found more compelling than others.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting.
AT AN FDA ADVISORY COMMITTEE MEETING
Novel oral vasodilator approved to treat pulmonary hypertension
Riociguat, a potent vasodilator that is the first in a new class of drugs, has been approved by the Food and Drug Administration for treating two types of pulmonary hypertension in adults.
The drug, which will be marketed as Adempas, was approved Oct. 8 to treat chronic thromboembolic pulmonary hypertension (CTEPH) and pulmonary arterial hypertension (PAH), the agency said in a statement.
Orally administered in tablet form, riociguat is a soluble guanylate cyclase (sGC) stimulator, the first drug in this class to be approved for pulmonary hypertension. It is also "the first drug of any class to be shown to be effective for patients with CTEPH," Dr. Norman Stockbridge, director of the Division of Cardiovascular and Renal Drug Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
Riociguat is a pregnancy category X drug and is available to women only through a Risk Evaluation and Mitigation Strategy (REMS) program.
The approved indications for the drug, which will be marketed by Bayer HealthCare Pharmaceuticals, are for persistent/recurrent CTEPH (WHO group 4) "after surgical treatment or inoperable CTEPH to improve exercise capacity and WHO functional class" or PAH (WHO group 1) "to improve exercise capacity, improve WHO functional class, and to delay clinical worsening."
Approval was based on the results of two international studies that found treatment resulted in significant improvements over placebo in the 6 minute walk test. Side effects of treatment include headache, dizziness, dyspepsia, peripheral edema, nausea, diarrhea, and vomiting, according to the FDA. Based on these results, an FDA advisory panel unanimously recommended approval of the two indications in August.
In the study of 380 patients with PAH, the change from baseline in the 6-minute walk test at 12 weeks improved by a mean of 30 m among those treated with riociguat, vs. a mean drop of 6 m in the placebo group. The WHO functional class improved in 21% of those on riociguat, compared with 14% in the placebo group, deteriorating in 4% and 14%, respectively.
In the study of 261 patients with CTEPH, the change from baseline in the 6-minute walk test at 16 weeks improved by a mean of 39 m among treated patients vs. a mean 6 m reduction in the placebo group at 16 weeks. WHO functional class improved in 33% of those on riociguat and 15% of those on placebo, deteriorating in 5% and 7%, respectively.
The two studies were published in the July 25 issue of the New England Journal of Medicine (2013;369:330-40; 2013;369:319-29).
The prescribing information includes a boxed warning about embryo-fetal toxicity. Women can receive the drug only through the REMS program.
Because of the risk of hypotension, the drug is contraindicated for use with nitrates or nitric oxide donors, such as amyl nitrate, and with phosphodiesterase inhibitors or nonspecific PDE inhibitors.
The wholesale cost of the drug is $7,500 for 30 days of treatment, with one tablet taken three times a day, according to a Bayer spokesperson. The company has set up a patient assistance program to help with coverage.
To date, riociguat has been approved in Canada for the treatment of inoperable or persistent/recurrent CTEPH after surgery in adults with WHO functional class II or III pulmonary hypertension, and it is under review in the European Union, according to the company spokesperson.
Riociguat, a potent vasodilator that is the first in a new class of drugs, has been approved by the Food and Drug Administration for treating two types of pulmonary hypertension in adults.
The drug, which will be marketed as Adempas, was approved Oct. 8 to treat chronic thromboembolic pulmonary hypertension (CTEPH) and pulmonary arterial hypertension (PAH), the agency said in a statement.
Orally administered in tablet form, riociguat is a soluble guanylate cyclase (sGC) stimulator, the first drug in this class to be approved for pulmonary hypertension. It is also "the first drug of any class to be shown to be effective for patients with CTEPH," Dr. Norman Stockbridge, director of the Division of Cardiovascular and Renal Drug Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
Riociguat is a pregnancy category X drug and is available to women only through a Risk Evaluation and Mitigation Strategy (REMS) program.
The approved indications for the drug, which will be marketed by Bayer HealthCare Pharmaceuticals, are for persistent/recurrent CTEPH (WHO group 4) "after surgical treatment or inoperable CTEPH to improve exercise capacity and WHO functional class" or PAH (WHO group 1) "to improve exercise capacity, improve WHO functional class, and to delay clinical worsening."
Approval was based on the results of two international studies that found treatment resulted in significant improvements over placebo in the 6 minute walk test. Side effects of treatment include headache, dizziness, dyspepsia, peripheral edema, nausea, diarrhea, and vomiting, according to the FDA. Based on these results, an FDA advisory panel unanimously recommended approval of the two indications in August.
In the study of 380 patients with PAH, the change from baseline in the 6-minute walk test at 12 weeks improved by a mean of 30 m among those treated with riociguat, vs. a mean drop of 6 m in the placebo group. The WHO functional class improved in 21% of those on riociguat, compared with 14% in the placebo group, deteriorating in 4% and 14%, respectively.
In the study of 261 patients with CTEPH, the change from baseline in the 6-minute walk test at 16 weeks improved by a mean of 39 m among treated patients vs. a mean 6 m reduction in the placebo group at 16 weeks. WHO functional class improved in 33% of those on riociguat and 15% of those on placebo, deteriorating in 5% and 7%, respectively.
The two studies were published in the July 25 issue of the New England Journal of Medicine (2013;369:330-40; 2013;369:319-29).
The prescribing information includes a boxed warning about embryo-fetal toxicity. Women can receive the drug only through the REMS program.
Because of the risk of hypotension, the drug is contraindicated for use with nitrates or nitric oxide donors, such as amyl nitrate, and with phosphodiesterase inhibitors or nonspecific PDE inhibitors.
The wholesale cost of the drug is $7,500 for 30 days of treatment, with one tablet taken three times a day, according to a Bayer spokesperson. The company has set up a patient assistance program to help with coverage.
To date, riociguat has been approved in Canada for the treatment of inoperable or persistent/recurrent CTEPH after surgery in adults with WHO functional class II or III pulmonary hypertension, and it is under review in the European Union, according to the company spokesperson.
Riociguat, a potent vasodilator that is the first in a new class of drugs, has been approved by the Food and Drug Administration for treating two types of pulmonary hypertension in adults.
The drug, which will be marketed as Adempas, was approved Oct. 8 to treat chronic thromboembolic pulmonary hypertension (CTEPH) and pulmonary arterial hypertension (PAH), the agency said in a statement.
Orally administered in tablet form, riociguat is a soluble guanylate cyclase (sGC) stimulator, the first drug in this class to be approved for pulmonary hypertension. It is also "the first drug of any class to be shown to be effective for patients with CTEPH," Dr. Norman Stockbridge, director of the Division of Cardiovascular and Renal Drug Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
Riociguat is a pregnancy category X drug and is available to women only through a Risk Evaluation and Mitigation Strategy (REMS) program.
The approved indications for the drug, which will be marketed by Bayer HealthCare Pharmaceuticals, are for persistent/recurrent CTEPH (WHO group 4) "after surgical treatment or inoperable CTEPH to improve exercise capacity and WHO functional class" or PAH (WHO group 1) "to improve exercise capacity, improve WHO functional class, and to delay clinical worsening."
Approval was based on the results of two international studies that found treatment resulted in significant improvements over placebo in the 6 minute walk test. Side effects of treatment include headache, dizziness, dyspepsia, peripheral edema, nausea, diarrhea, and vomiting, according to the FDA. Based on these results, an FDA advisory panel unanimously recommended approval of the two indications in August.
In the study of 380 patients with PAH, the change from baseline in the 6-minute walk test at 12 weeks improved by a mean of 30 m among those treated with riociguat, vs. a mean drop of 6 m in the placebo group. The WHO functional class improved in 21% of those on riociguat, compared with 14% in the placebo group, deteriorating in 4% and 14%, respectively.
In the study of 261 patients with CTEPH, the change from baseline in the 6-minute walk test at 16 weeks improved by a mean of 39 m among treated patients vs. a mean 6 m reduction in the placebo group at 16 weeks. WHO functional class improved in 33% of those on riociguat and 15% of those on placebo, deteriorating in 5% and 7%, respectively.
The two studies were published in the July 25 issue of the New England Journal of Medicine (2013;369:330-40; 2013;369:319-29).
The prescribing information includes a boxed warning about embryo-fetal toxicity. Women can receive the drug only through the REMS program.
Because of the risk of hypotension, the drug is contraindicated for use with nitrates or nitric oxide donors, such as amyl nitrate, and with phosphodiesterase inhibitors or nonspecific PDE inhibitors.
The wholesale cost of the drug is $7,500 for 30 days of treatment, with one tablet taken three times a day, according to a Bayer spokesperson. The company has set up a patient assistance program to help with coverage.
To date, riociguat has been approved in Canada for the treatment of inoperable or persistent/recurrent CTEPH after surgery in adults with WHO functional class II or III pulmonary hypertension, and it is under review in the European Union, according to the company spokesperson.
Full-dose beta-blockers still show heart failure benefit
AMSTERDAM – Patients with heart failure appeared to continue to benefit from full-dose beta-blocker therapy against a background of optimal, contemporary treatment, based on an analysis of data collected from an 1,800-patient trial of cardiac resynchronization therapy.
Dr. L. Brent Mitchell, who presented this finding at the annual congress of the European Society of Cardiology, acknowledged that assessing the efficacy of drugs in a study that did not randomize patients based on their use of those drugs had questionable validity and was vulnerable to unidentified confounding, but he also noted that data on the role of beta-blockers in contemporary heart failure management were unlikely to come from any other source.
"You need some tool to ask whether beta-blockers are still useful," said Dr. Mitchell, professor of cardiac sciences at the University of Calgary (Alta.). "The purpose of this analysis was to ask if beta-blockers at full dosages are still better than not using a full-dose beta-blocker despite all the other treatments" that patients received while in the study during 2003-2009. The analysis showed that those receiving full-dosage or nearly full-dosage beta-blocker regimens had a one-third cut in all-cause death or heart failure hospitalization, compared with patients receiving less than half the recommended dosage, after adjustment for many baseline differences between these two subgroups.
The analysis also showed that just half of the nearly 1,500 patients who received treatment with a beta-blocker in this cohort received an adequate dosage, defined as at least 50% of the level recommended by the European Society of Cardiology in heart failure treatment guidelines issued last year (Euro. Heart J. 2012;33:1787-847). Two-thirds of the patients on carvedilol received at least half of the recommended target dosage of 50 mg/day. In contrast, about a third of patients being treated with either bisoprolol or metoprolol received a dosage that was at least half the target dosage (10 mg/day for bisoprolol, 200 mg/day for metoprolol).
The analysis used data collected in the Resynchronization-Defibrillation for Ambulatory Heart Failure Trial (RAFT), which enrolled 1,798 patients primarily with New York Heart Association class II heart failure at 34 centers worldwide. When the primary endpoint of RAFT was evaluated, treatment with cardiac resynchronization therapy (CRT) with defibrillator capability was more effective at preventing death or heart failure hospitalization in the study population than was an implanted defibrillator alone against a background of optimal medical treatment (N. Engl. J. Med. 2010;363:2385-95).
Dr. Mitchell and his associates focused on 1,474 patients who all received beta-blocker treatment while in RAFT. Like the entire RAFT population, 97% of these patients also received treatment with an ACE inhibitor or angiotensin receptor blocker, and 42% received treatment with spironolactone, and their average left ventricular ejection fraction was 23%.
The patients in this group who were undertreated with beta-blockers were significantly older and had a significantly greater prevalence of New York Heart Association class III heart failure and heart failure with an ischemic etiology compared with those who received at least 50% of the target dosage.
In a regression model that adjusted for all measured baseline differences, patients treated with at least half of the recommended dosage had an incidence of mortality or heart failure hospitalization during an average 40 months of follow-up that was 33% below the rate among patients on lower beta-blocker dosages, a statistically significant difference. This 33% relative risk reduction with adequate beta-blocker treatment is "indistinguishable" from the risk reductions seen with placebo in the major beta-blocker efficacy trials in heart failure patients during the 1990s, Dr. Mitchell noted. "This suggests that beta-blocker treatment still has potency," he said.
Other significant, independent predictors of the primary outcome in this model were prior coronary artery bypass, which boosted the risk by 63%; a history of ischemic heart disease, which raised the risk by 39%; having peripheral vascular disease, which increased the risk by 36%; and receiving CRT, which cut the risk by 33%. In addition, adverse outcomes rose 10% for each reduction of 5 mL/min per 1.73 m2 in estimated glomerular filtration rate.
The impact of underdosing was roughly similar across all three beta-blockers, bisoprolol, carvedilol, and metoprolol. The adverse effect of underdosing was mitigated to some extent when patients also received CRT.
RAFT was partially funded by Medtronic of Canada. Dr. Mitchell said he had no relevant financial disclosures.
On Twitter @mitchelzoler
A post hoc analysis of the nonrandomized interventions that patients receive during randomized clinical trials is generally discouraged. Patients treated with lower dosages of hemodynamically active drugs like beta-blockers are often sicker and have a worse prognosis. This problem can be seen in the baseline characteristics of the two subgroups in this analysis, which revealed many significant differences between the two. Performing statistical compensations to counterbalance these differences is very uncertain.
In addition, a few years ago, my associates and I ran the Systolic Heart Failure Treatment With the If inhibitor Ivabradine Trial (SHIFT) (Lancet 2010;376:875-85) to assess the efficacy and safety of ivabradine to lower heart rate and improve outcomes in optimally treated heart failure patients. In SHIFT we saw a relationship similar to the one seen in RAFT by the current investigators: Patients on lower dosages of beta-blockers were sicker and at higher risk than patients on higher beta-blocker dosages. In our analyses, adjusting for differences in heart rate eliminated the link between beta-blocker dosage and outcomes. We found that the entire treatment effect could be explained by differences in patient heart rate.
Dr. Karl Swedberg is a professor of medicine at the University of Gothenburg (Sweden). He was lead investigator for the SHIFT trial, which was funded by Servier, the company that markets ivabradine (Procoralan). He made these comments as designated discussant for Dr. Mitchell’s report.
A post hoc analysis of the nonrandomized interventions that patients receive during randomized clinical trials is generally discouraged. Patients treated with lower dosages of hemodynamically active drugs like beta-blockers are often sicker and have a worse prognosis. This problem can be seen in the baseline characteristics of the two subgroups in this analysis, which revealed many significant differences between the two. Performing statistical compensations to counterbalance these differences is very uncertain.
In addition, a few years ago, my associates and I ran the Systolic Heart Failure Treatment With the If inhibitor Ivabradine Trial (SHIFT) (Lancet 2010;376:875-85) to assess the efficacy and safety of ivabradine to lower heart rate and improve outcomes in optimally treated heart failure patients. In SHIFT we saw a relationship similar to the one seen in RAFT by the current investigators: Patients on lower dosages of beta-blockers were sicker and at higher risk than patients on higher beta-blocker dosages. In our analyses, adjusting for differences in heart rate eliminated the link between beta-blocker dosage and outcomes. We found that the entire treatment effect could be explained by differences in patient heart rate.
Dr. Karl Swedberg is a professor of medicine at the University of Gothenburg (Sweden). He was lead investigator for the SHIFT trial, which was funded by Servier, the company that markets ivabradine (Procoralan). He made these comments as designated discussant for Dr. Mitchell’s report.
A post hoc analysis of the nonrandomized interventions that patients receive during randomized clinical trials is generally discouraged. Patients treated with lower dosages of hemodynamically active drugs like beta-blockers are often sicker and have a worse prognosis. This problem can be seen in the baseline characteristics of the two subgroups in this analysis, which revealed many significant differences between the two. Performing statistical compensations to counterbalance these differences is very uncertain.
In addition, a few years ago, my associates and I ran the Systolic Heart Failure Treatment With the If inhibitor Ivabradine Trial (SHIFT) (Lancet 2010;376:875-85) to assess the efficacy and safety of ivabradine to lower heart rate and improve outcomes in optimally treated heart failure patients. In SHIFT we saw a relationship similar to the one seen in RAFT by the current investigators: Patients on lower dosages of beta-blockers were sicker and at higher risk than patients on higher beta-blocker dosages. In our analyses, adjusting for differences in heart rate eliminated the link between beta-blocker dosage and outcomes. We found that the entire treatment effect could be explained by differences in patient heart rate.
Dr. Karl Swedberg is a professor of medicine at the University of Gothenburg (Sweden). He was lead investigator for the SHIFT trial, which was funded by Servier, the company that markets ivabradine (Procoralan). He made these comments as designated discussant for Dr. Mitchell’s report.
AMSTERDAM – Patients with heart failure appeared to continue to benefit from full-dose beta-blocker therapy against a background of optimal, contemporary treatment, based on an analysis of data collected from an 1,800-patient trial of cardiac resynchronization therapy.
Dr. L. Brent Mitchell, who presented this finding at the annual congress of the European Society of Cardiology, acknowledged that assessing the efficacy of drugs in a study that did not randomize patients based on their use of those drugs had questionable validity and was vulnerable to unidentified confounding, but he also noted that data on the role of beta-blockers in contemporary heart failure management were unlikely to come from any other source.
"You need some tool to ask whether beta-blockers are still useful," said Dr. Mitchell, professor of cardiac sciences at the University of Calgary (Alta.). "The purpose of this analysis was to ask if beta-blockers at full dosages are still better than not using a full-dose beta-blocker despite all the other treatments" that patients received while in the study during 2003-2009. The analysis showed that those receiving full-dosage or nearly full-dosage beta-blocker regimens had a one-third cut in all-cause death or heart failure hospitalization, compared with patients receiving less than half the recommended dosage, after adjustment for many baseline differences between these two subgroups.
The analysis also showed that just half of the nearly 1,500 patients who received treatment with a beta-blocker in this cohort received an adequate dosage, defined as at least 50% of the level recommended by the European Society of Cardiology in heart failure treatment guidelines issued last year (Euro. Heart J. 2012;33:1787-847). Two-thirds of the patients on carvedilol received at least half of the recommended target dosage of 50 mg/day. In contrast, about a third of patients being treated with either bisoprolol or metoprolol received a dosage that was at least half the target dosage (10 mg/day for bisoprolol, 200 mg/day for metoprolol).
The analysis used data collected in the Resynchronization-Defibrillation for Ambulatory Heart Failure Trial (RAFT), which enrolled 1,798 patients primarily with New York Heart Association class II heart failure at 34 centers worldwide. When the primary endpoint of RAFT was evaluated, treatment with cardiac resynchronization therapy (CRT) with defibrillator capability was more effective at preventing death or heart failure hospitalization in the study population than was an implanted defibrillator alone against a background of optimal medical treatment (N. Engl. J. Med. 2010;363:2385-95).
Dr. Mitchell and his associates focused on 1,474 patients who all received beta-blocker treatment while in RAFT. Like the entire RAFT population, 97% of these patients also received treatment with an ACE inhibitor or angiotensin receptor blocker, and 42% received treatment with spironolactone, and their average left ventricular ejection fraction was 23%.
The patients in this group who were undertreated with beta-blockers were significantly older and had a significantly greater prevalence of New York Heart Association class III heart failure and heart failure with an ischemic etiology compared with those who received at least 50% of the target dosage.
In a regression model that adjusted for all measured baseline differences, patients treated with at least half of the recommended dosage had an incidence of mortality or heart failure hospitalization during an average 40 months of follow-up that was 33% below the rate among patients on lower beta-blocker dosages, a statistically significant difference. This 33% relative risk reduction with adequate beta-blocker treatment is "indistinguishable" from the risk reductions seen with placebo in the major beta-blocker efficacy trials in heart failure patients during the 1990s, Dr. Mitchell noted. "This suggests that beta-blocker treatment still has potency," he said.
Other significant, independent predictors of the primary outcome in this model were prior coronary artery bypass, which boosted the risk by 63%; a history of ischemic heart disease, which raised the risk by 39%; having peripheral vascular disease, which increased the risk by 36%; and receiving CRT, which cut the risk by 33%. In addition, adverse outcomes rose 10% for each reduction of 5 mL/min per 1.73 m2 in estimated glomerular filtration rate.
The impact of underdosing was roughly similar across all three beta-blockers, bisoprolol, carvedilol, and metoprolol. The adverse effect of underdosing was mitigated to some extent when patients also received CRT.
RAFT was partially funded by Medtronic of Canada. Dr. Mitchell said he had no relevant financial disclosures.
On Twitter @mitchelzoler
AMSTERDAM – Patients with heart failure appeared to continue to benefit from full-dose beta-blocker therapy against a background of optimal, contemporary treatment, based on an analysis of data collected from an 1,800-patient trial of cardiac resynchronization therapy.
Dr. L. Brent Mitchell, who presented this finding at the annual congress of the European Society of Cardiology, acknowledged that assessing the efficacy of drugs in a study that did not randomize patients based on their use of those drugs had questionable validity and was vulnerable to unidentified confounding, but he also noted that data on the role of beta-blockers in contemporary heart failure management were unlikely to come from any other source.
"You need some tool to ask whether beta-blockers are still useful," said Dr. Mitchell, professor of cardiac sciences at the University of Calgary (Alta.). "The purpose of this analysis was to ask if beta-blockers at full dosages are still better than not using a full-dose beta-blocker despite all the other treatments" that patients received while in the study during 2003-2009. The analysis showed that those receiving full-dosage or nearly full-dosage beta-blocker regimens had a one-third cut in all-cause death or heart failure hospitalization, compared with patients receiving less than half the recommended dosage, after adjustment for many baseline differences between these two subgroups.
The analysis also showed that just half of the nearly 1,500 patients who received treatment with a beta-blocker in this cohort received an adequate dosage, defined as at least 50% of the level recommended by the European Society of Cardiology in heart failure treatment guidelines issued last year (Euro. Heart J. 2012;33:1787-847). Two-thirds of the patients on carvedilol received at least half of the recommended target dosage of 50 mg/day. In contrast, about a third of patients being treated with either bisoprolol or metoprolol received a dosage that was at least half the target dosage (10 mg/day for bisoprolol, 200 mg/day for metoprolol).
The analysis used data collected in the Resynchronization-Defibrillation for Ambulatory Heart Failure Trial (RAFT), which enrolled 1,798 patients primarily with New York Heart Association class II heart failure at 34 centers worldwide. When the primary endpoint of RAFT was evaluated, treatment with cardiac resynchronization therapy (CRT) with defibrillator capability was more effective at preventing death or heart failure hospitalization in the study population than was an implanted defibrillator alone against a background of optimal medical treatment (N. Engl. J. Med. 2010;363:2385-95).
Dr. Mitchell and his associates focused on 1,474 patients who all received beta-blocker treatment while in RAFT. Like the entire RAFT population, 97% of these patients also received treatment with an ACE inhibitor or angiotensin receptor blocker, and 42% received treatment with spironolactone, and their average left ventricular ejection fraction was 23%.
The patients in this group who were undertreated with beta-blockers were significantly older and had a significantly greater prevalence of New York Heart Association class III heart failure and heart failure with an ischemic etiology compared with those who received at least 50% of the target dosage.
In a regression model that adjusted for all measured baseline differences, patients treated with at least half of the recommended dosage had an incidence of mortality or heart failure hospitalization during an average 40 months of follow-up that was 33% below the rate among patients on lower beta-blocker dosages, a statistically significant difference. This 33% relative risk reduction with adequate beta-blocker treatment is "indistinguishable" from the risk reductions seen with placebo in the major beta-blocker efficacy trials in heart failure patients during the 1990s, Dr. Mitchell noted. "This suggests that beta-blocker treatment still has potency," he said.
Other significant, independent predictors of the primary outcome in this model were prior coronary artery bypass, which boosted the risk by 63%; a history of ischemic heart disease, which raised the risk by 39%; having peripheral vascular disease, which increased the risk by 36%; and receiving CRT, which cut the risk by 33%. In addition, adverse outcomes rose 10% for each reduction of 5 mL/min per 1.73 m2 in estimated glomerular filtration rate.
The impact of underdosing was roughly similar across all three beta-blockers, bisoprolol, carvedilol, and metoprolol. The adverse effect of underdosing was mitigated to some extent when patients also received CRT.
RAFT was partially funded by Medtronic of Canada. Dr. Mitchell said he had no relevant financial disclosures.
On Twitter @mitchelzoler
AT THE ESC CONGRESS 2013
Major finding: Heart failure patients on recommended beta-blocker dosages had 33% fewer deaths or hospitalizations than did patients on lower dosages.
Data source: A post hoc subgroup analysis of results from RAFT, with 1,798 patients with heart failure at 34 worldwide sites.
Disclosures: RAFT was partially funded by Medtronic of Canada. Dr. Mitchell said he had no relevant financial disclosures.
HFSA13: Health reform, mechanical support devices take center stage
At the 2013 Heart Failure Society of America, held in Orlando, hotly debated topics will include multidisciplinary approaches to treating heart failure patients, health reform, better selection of mechanical support devices for heart failure patients, and heart failure readmissions.
Dr. David DeNofrio, the scientific program's co-chair, and director of heart failure and cardiac transplantation at Tufts Medical Center, Boston, discusses the issues in an interview with IMNG Medical Media.
At the 2013 Heart Failure Society of America, held in Orlando, hotly debated topics will include multidisciplinary approaches to treating heart failure patients, health reform, better selection of mechanical support devices for heart failure patients, and heart failure readmissions.
Dr. David DeNofrio, the scientific program's co-chair, and director of heart failure and cardiac transplantation at Tufts Medical Center, Boston, discusses the issues in an interview with IMNG Medical Media.
At the 2013 Heart Failure Society of America, held in Orlando, hotly debated topics will include multidisciplinary approaches to treating heart failure patients, health reform, better selection of mechanical support devices for heart failure patients, and heart failure readmissions.
Dr. David DeNofrio, the scientific program's co-chair, and director of heart failure and cardiac transplantation at Tufts Medical Center, Boston, discusses the issues in an interview with IMNG Medical Media.

Losartan shown effective in Marfan syndrome
AMSTERDAM – Daily losartan significantly slowed the aortic root dilatation rate in adults with Marfan syndrome in a 3-year randomized clinical trial.
"I think we can be positive about this treatment. We can now recommend losartan in clinical practice," Dr. Maarten Groenink said at the annual congress of the European Society of Cardiology.
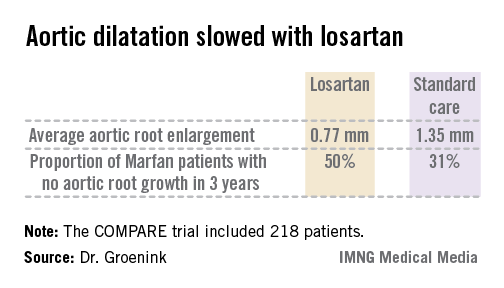
The COMPARE (Cozaar in Marfan Patients Reduces Aortic Enlargement) trial included 218 patients at all four university Marfan centers in the Netherlands. Patients were randomized to oral losartan at a target dose of 100 mg/day or no losartan in addition to standard-of-care treatment with beta-blockers. Roughly half of the patients in the losartan group were unable to tolerate the full dose of losartan in addition to a beta-blocker; those patients were maintained on losartan at 50 mg/day. Aortic root diameter was measured by MRI at enrollment and after 3 years of prospective follow-up. The aortic dilatation rate was significantly lower in the losartan group than in controls both in the patients with a native root and in those who had undergone aortic root replacement surgery, reported Dr. Groenink, a cardiologist at the Academic Medical Center, Amsterdam.

There were no aortic dissections in the losartan group and two in the control arm. Elective aortic replacement surgery was performed in a similar number of patients in both groups.
Blood pressure was lower in the losartan group, yet blood pressure didn’t correlate with the aortic dilatation rate. Dr. Groenink speculated that losartan’s chief mechanism of benefit in Marfan syndrome is its ability to curb overexpression of transforming growth factor-beta, which weakens the structure of the media layer of the aortic wall.
Dr. Groenink said it’s unknown whether losartan’s benefits are specific to that drug or are a class effect obtainable with other angiotensin II receptor antagonists, though he suspects it’s a class effect.
Ongoing clinical trials are evaluating losartan in children and adolescents with Marfan syndrome, he said, adding that there is a solid rationale for beginning treatment as early in life as possible.
"I believe the adverse effects on the aortic wall in Marfan syndrome are caused by the fibrillin defect but also by wear and tear due to cyclic stress by the beating heart. So you can hypothesize that the earlier you start treatment, the better the results," he explained.
Marfan syndrome is a genetic connective tissue disorder affecting multiple organ systems. The prognosis is mainly determined by the aortic complications, including dilatation, aneurysm formation, and possible acute dissection. Affected individuals tend to be tall, long-limbed, and have distinctively long, thin fingers. The prevalence of Marfan syndrome has been estimated at 1 in 5,000, but Dr. Groenink suspects the syndrome may actually be more common than that.
Simultaneous with Dr. Groenink’s presentation at the ESC, the COMPARE results were published online (Eur. Heart J. 2013 [doi:10.1093/eurheartj/eht334]).
The COMPARE trial was funded by the Dutch Heart Association. Dr. Groenink reported having no relevant financial interests.
*CORRECTION 11/14/13: The first version of this story had Dr. Groenink's name misspelled.

|
Bruce Jancin/IMNG Medical Media
|
COMPARE is a very important study whose results are going to mean a paradigm shift for the management of Marfan syndrome.
It is intriguing to consider that the benefits of losartan might possibly also extend to patients with thoracic aortic disease in general, a worthy topic for future investigation.
Dr. John Gordon Harold is with Cedars-Sinai Heart Institute, Los Angeles, and president of the American College of Cardiology. He had no relevant financial disclosures.
|
|
Bruce Jancin/IMNG Medical Media
|
COMPARE is a very important study whose results are going to mean a paradigm shift for the management of Marfan syndrome.
It is intriguing to consider that the benefits of losartan might possibly also extend to patients with thoracic aortic disease in general, a worthy topic for future investigation.
Dr. John Gordon Harold is with Cedars-Sinai Heart Institute, Los Angeles, and president of the American College of Cardiology. He had no relevant financial disclosures.
|
|
Bruce Jancin/IMNG Medical Media
|
COMPARE is a very important study whose results are going to mean a paradigm shift for the management of Marfan syndrome.
It is intriguing to consider that the benefits of losartan might possibly also extend to patients with thoracic aortic disease in general, a worthy topic for future investigation.
Dr. John Gordon Harold is with Cedars-Sinai Heart Institute, Los Angeles, and president of the American College of Cardiology. He had no relevant financial disclosures.
AMSTERDAM – Daily losartan significantly slowed the aortic root dilatation rate in adults with Marfan syndrome in a 3-year randomized clinical trial.
"I think we can be positive about this treatment. We can now recommend losartan in clinical practice," Dr. Maarten Groenink said at the annual congress of the European Society of Cardiology.
The COMPARE (Cozaar in Marfan Patients Reduces Aortic Enlargement) trial included 218 patients at all four university Marfan centers in the Netherlands. Patients were randomized to oral losartan at a target dose of 100 mg/day or no losartan in addition to standard-of-care treatment with beta-blockers. Roughly half of the patients in the losartan group were unable to tolerate the full dose of losartan in addition to a beta-blocker; those patients were maintained on losartan at 50 mg/day. Aortic root diameter was measured by MRI at enrollment and after 3 years of prospective follow-up. The aortic dilatation rate was significantly lower in the losartan group than in controls both in the patients with a native root and in those who had undergone aortic root replacement surgery, reported Dr. Groenink, a cardiologist at the Academic Medical Center, Amsterdam.
There were no aortic dissections in the losartan group and two in the control arm. Elective aortic replacement surgery was performed in a similar number of patients in both groups.
Blood pressure was lower in the losartan group, yet blood pressure didn’t correlate with the aortic dilatation rate. Dr. Groenink speculated that losartan’s chief mechanism of benefit in Marfan syndrome is its ability to curb overexpression of transforming growth factor-beta, which weakens the structure of the media layer of the aortic wall.
Dr. Groenink said it’s unknown whether losartan’s benefits are specific to that drug or are a class effect obtainable with other angiotensin II receptor antagonists, though he suspects it’s a class effect.
Ongoing clinical trials are evaluating losartan in children and adolescents with Marfan syndrome, he said, adding that there is a solid rationale for beginning treatment as early in life as possible.
"I believe the adverse effects on the aortic wall in Marfan syndrome are caused by the fibrillin defect but also by wear and tear due to cyclic stress by the beating heart. So you can hypothesize that the earlier you start treatment, the better the results," he explained.
Marfan syndrome is a genetic connective tissue disorder affecting multiple organ systems. The prognosis is mainly determined by the aortic complications, including dilatation, aneurysm formation, and possible acute dissection. Affected individuals tend to be tall, long-limbed, and have distinctively long, thin fingers. The prevalence of Marfan syndrome has been estimated at 1 in 5,000, but Dr. Groenink suspects the syndrome may actually be more common than that.
Simultaneous with Dr. Groenink’s presentation at the ESC, the COMPARE results were published online (Eur. Heart J. 2013 [doi:10.1093/eurheartj/eht334]).
The COMPARE trial was funded by the Dutch Heart Association. Dr. Groenink reported having no relevant financial interests.
*CORRECTION 11/14/13: The first version of this story had Dr. Groenink's name misspelled.
AMSTERDAM – Daily losartan significantly slowed the aortic root dilatation rate in adults with Marfan syndrome in a 3-year randomized clinical trial.
"I think we can be positive about this treatment. We can now recommend losartan in clinical practice," Dr. Maarten Groenink said at the annual congress of the European Society of Cardiology.
The COMPARE (Cozaar in Marfan Patients Reduces Aortic Enlargement) trial included 218 patients at all four university Marfan centers in the Netherlands. Patients were randomized to oral losartan at a target dose of 100 mg/day or no losartan in addition to standard-of-care treatment with beta-blockers. Roughly half of the patients in the losartan group were unable to tolerate the full dose of losartan in addition to a beta-blocker; those patients were maintained on losartan at 50 mg/day. Aortic root diameter was measured by MRI at enrollment and after 3 years of prospective follow-up. The aortic dilatation rate was significantly lower in the losartan group than in controls both in the patients with a native root and in those who had undergone aortic root replacement surgery, reported Dr. Groenink, a cardiologist at the Academic Medical Center, Amsterdam.
There were no aortic dissections in the losartan group and two in the control arm. Elective aortic replacement surgery was performed in a similar number of patients in both groups.
Blood pressure was lower in the losartan group, yet blood pressure didn’t correlate with the aortic dilatation rate. Dr. Groenink speculated that losartan’s chief mechanism of benefit in Marfan syndrome is its ability to curb overexpression of transforming growth factor-beta, which weakens the structure of the media layer of the aortic wall.
Dr. Groenink said it’s unknown whether losartan’s benefits are specific to that drug or are a class effect obtainable with other angiotensin II receptor antagonists, though he suspects it’s a class effect.
Ongoing clinical trials are evaluating losartan in children and adolescents with Marfan syndrome, he said, adding that there is a solid rationale for beginning treatment as early in life as possible.
"I believe the adverse effects on the aortic wall in Marfan syndrome are caused by the fibrillin defect but also by wear and tear due to cyclic stress by the beating heart. So you can hypothesize that the earlier you start treatment, the better the results," he explained.
Marfan syndrome is a genetic connective tissue disorder affecting multiple organ systems. The prognosis is mainly determined by the aortic complications, including dilatation, aneurysm formation, and possible acute dissection. Affected individuals tend to be tall, long-limbed, and have distinctively long, thin fingers. The prevalence of Marfan syndrome has been estimated at 1 in 5,000, but Dr. Groenink suspects the syndrome may actually be more common than that.
Simultaneous with Dr. Groenink’s presentation at the ESC, the COMPARE results were published online (Eur. Heart J. 2013 [doi:10.1093/eurheartj/eht334]).
The COMPARE trial was funded by the Dutch Heart Association. Dr. Groenink reported having no relevant financial interests.
*CORRECTION 11/14/13: The first version of this story had Dr. Groenink's name misspelled.
AT THE ESC CONGRESS 2013
Major finding: The rate of aortic root enlargement during 3 years of prospective follow-up was 0.77 mm in losartan-treated patients with Marfan syndrome, significantly less than the 1.35 mm in patients on standard-of-care treatment with no losartan.
Data source: The COMPARE trial was a randomized, prospective, open-label multicenter study in which 218 patients with Marfan syndrome were randomized to losartan at a target dose of 100 mg or to no losartan and followed for 3 years with the aortic root dilatation rate as measured by MRI the primary endpoint.
Disclosures: The COMPARE trial was supported by the Dutch Heart Association. Dr. Groenink reported having no financial conflicts.