User login
Primary Mucinous Carcinoma of the Eyelid Treated With Mohs Micrographic Surgery
To the Editor:
Primary mucinous carcinoma (PMC) is an exceedingly rare adnexal tumor with an incidence of 0.07 cases per million individuals.1,2 First described by Lennox et al3 in 1952, this entity often presents as slow-growing, solitary nodules that often are soft on palpation but may have an indurated quality and range in color from reddish blue to flesh colored to white.4 Primary mucinous carcinoma most commonly is found on the eyelid (38%) but may affect other sites on the face (20.3%), scalp (16%), and axilla (10%).5 Historically, it has been thought to be more common among men; however, a 2005 large case series by Kazakov et al5 found that women were twice as likely to be affected. Primary mucinous carcinoma most frequently is diagnosed in the fifth through seventh decades of life, with a median age at onset of 63 years.6,7 Because of its rarity, PMC is most frequently confused clinically with basal cell carcinoma, keratoacanthoma, apocrine hidrocystoma, epidermoid cyst, Kaposi sarcoma, neuroma, lacrimal sac tumor, squamous cell carcinoma, granulomatous tumors, and metastatic adenocarcinoma.1,8-10
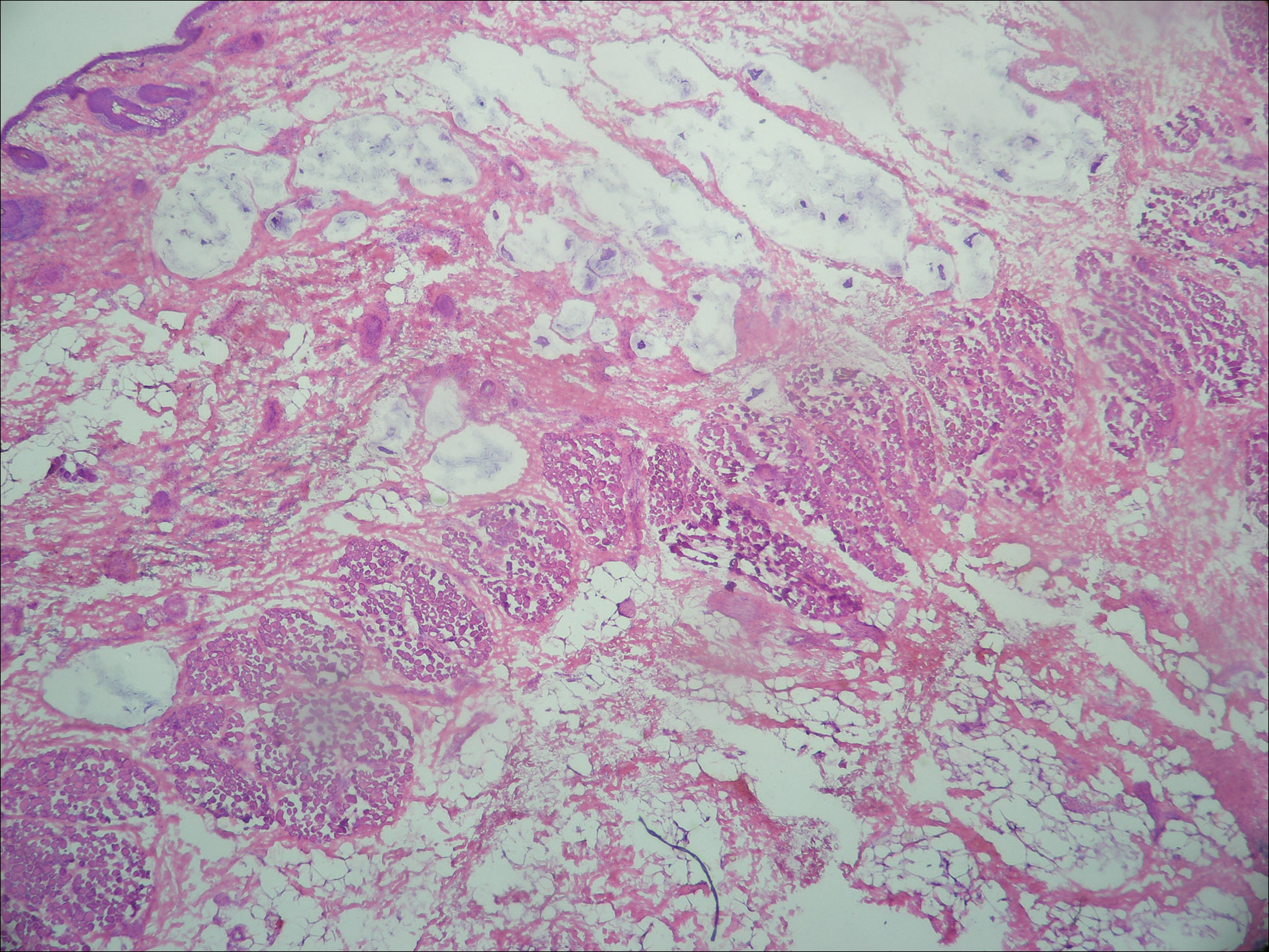
Primary mucinous carcinoma is thought to be derived from sweat glands, and select features such as decapitation secretion are more suggestive of apocrine than eccrine differentiation.5,8 On histopathology, PMC classically is described as nests of epithelial cells floating in lakes of extracellular mucin, primarily in the dermis and subcutis. The nests are composed of basaloid cells in solid to cribriform arrangements, usually with a low mitotic count and little nuclear atypia. These nests are suspended within periodic acid–Schiff positive mucinous pools partitioned by delicate fibrous septa. The mucin produced by PMC is sialomucin, and as such it is hyaluronidase resistant and sialidase labile.6 At least 1 report has been made of the presence of psammoma bodies in PMC.11
The neoplasm is characterized by an indolent course with frequent recurrence but rare metastasis.5,12 Treatment is primarily surgical, with Mohs micrographic surgery (MMS) offering improved tissue conservation and reduced recurrence rates.12 The diagnostic challenge lies in distinguishing PMC from a variety of metastatic mucinous internal malignancies that portend a notably greater morbidity and mortality to the patient. We describe a case of PMC, discuss the differentiation of PMC from metastatic mucinous carcinoma, and review the literature regarding treatment of this rare neoplasm.
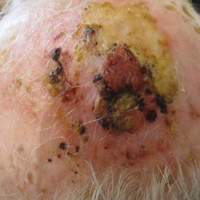
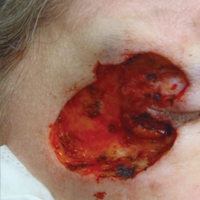
A 65-year-old white woman was referred to our tertiary-care dermatologic surgery clinic for treatment of an incompletely excised mucinous carcinoma of the right lateral canthus (Figure 1). The clinically evident scar measured 0.5×0.5 cm. Although difficult to appreciate in Figure 1, a slight textural change of the surrounding skin, including the upper and lower eyelid, was apparent. Prior to her arrival to our clinic, the referring physician had completed a thorough review of systems and physical examination, which did not suggest an underlying malignancy. Computed tomography of the head, neck, chest, abdomen, and pelvis revealed a mass in the thyroid that was removed and found to be benign. The patient’s cutaneous lesion was therefore considered to be a PMC of the skin.
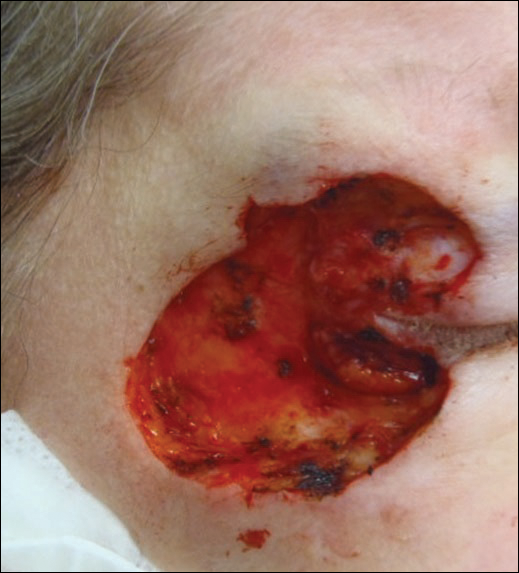
Given the prior incomplete excision of the lesion and its periocular location, we treated the patient with MMS. After 6 surgical stages, we continued to see evidence of the neoplasm as it tracked medially along the orbicularis oculi muscle (Figure 2). Due to the patient’s physical and emotional exhaustion at this point, we discontinued MMS and referred her to a colleague in plastic surgery for further excision of the remaining focus of positivity as well as repair. The final Mohs defect measured 4.2×4.0 cm (Figure 3). Approximately 2.3×1.0 cm of tissue in the area of remaining tumor was excised by plastic surgery, and the defect was repaired with a cervicofacial advancement flap closure of the right cheek and lower eyelid and full-thickness skin graft of the left upper eyelid. Histopathologic investigation found the additional tissue resected to be free of residual tumor.
To diagnose a patient with PMC, one must first rule out cutaneous metastasis of various internal malignancies that may appear similar on histopathology. A full clinical investigation consisting of a thorough history, physical examination, and appropriate radiographic imaging is required. Cutaneous metastases most commonly arise from the breast or gastrointestinal tract (GIT) but also can originate from the prostate, lungs, ovaries, pancreas, and kidneys.5 Histologically, PMC may be identical to metastatic adenocarcinoma.13 Location on the body may be a clue to a lesion’s origin, as metastases from a mucinous adenocarcinoma of the breast typically occur on the chest, breast, or axilla,5 whereas PMC primarily is found on the head and neck.
Certain histopathologic features may be suggestive of either a primary or metastatic etiology. Lesions arising in the skin may reveal an in situ component representing ductal hyperplasia, atypical ductal hyperplasia, or ductal carcinoma in situ. Identification of an in situ component defines a cutaneous primary neoplasm, but its absence does not exclude PMC.5 Additionally, metastatic lesions from the GIT typically have greater pleomorphism and “dirty” necrosis defined as eosinophilic foci containing nuclear debris.5
The expression pattern of cytokeratins (CKs) also can be suggestive. Primary mucinous carcinoma and metastatic breast adenocarcinoma are both CK7+ and CK20−. By contrast, mucinous adenocarcinoma of the GIT stains CK20+ and CK7−.14 Another marker that stains PMC is CK5 and CK6, though infrequently present. Levy et al15 reported positive staining for CK5 and CK6 in only 1 of 5 PMC cases. Positive staining for CK5 and CK6 has not been reported in any metastatic mucinous carcinoma.
The role of p63 immunostaining in the setting of mucinous carcinoma is controversial.16-18 Some practi-tioners have reported using p63 immunostaining to assist in establishing the diagnosis of PMC but only after performing a clinical workup to search for any primary sites of mucinous carcinoma in other organs.11 Other studies, however, have found select metastatic lesions from the breast17,18 and GIT18 to stain positively with p63. It is important to remember that these clinical and pathologic features are only suggestive of the primary etiology and are not replacement for a full clinical investigation.
Primary mucinous carcinoma is considered an indolent tumor with the majority of patient morbidity attributable to local recurrence and regional metastasis. Although uncommon, regional and distant metastasis rates have been reported to be 11% and 3%, respectively.19 Direct lymphatic invasion has been reported and indicates a more aggressive tumor with shorter recurrence-free intervals and predicts nodal metastases. Paradela et al20 recommended the use of D2-40, a monoclonal antibody and specific marker for lymphatic endothelium, to detect lymphatic invasion, particularly in node-negative primary tumors.
In one case of PMC on the jaw of a 39-year-old Japanese man, no recurrence or metastases were discovered until the 11th year of follow-up. At that time, he was found to have lung and bone metastases and died after 3 years.21 Other investigators report death occurring 4 to 24 months following diagnosis of distant metastases.7,22 Direct extension of the tumor into skeletal muscle, periosteum, bone, and dura also has been documented.7
Treatment principally is surgical, with PMC known to be resistant to both chemotherapy and radiation therapy.19,22 The recommended margins for simple excision range from 1 to 2 cm, but this method of treatment yields recurrence rates upward of 30% to 40%, especially for lesions located on the eyelid.12,13 First utilized in PMC of the eyelid to conserve tissue, MMS is rapidly becoming the treatment of choice because of its notably improved recurrence rate. A case series of 4 PMCs of the eyelid treated via MMS or frozen section control found the recurrence rate to be 7%.23 Another report of 2 cases of PMC treated by MMS reported no recurrence after 42 and 26 months.13 Ortiz et al7 reported an additional case of a patient treated by MMS that was recurrence free for 30 months at the time of publication. Further investigation is required to definitively recommend MMS on the basis of improved recurrence rate but should now be considered standard of care in recurrent, sizeable, or eyelid PMC.
Despite its ascension as treatment of choice in many cases of PMC, MMS is not without its risk of metastasis and recurrence. Tam et al24 reported a case of PMC with multiple recurrences and metastases following 3 simple excisions and 2 excisions via MMS. Although the lesion’s previously recurrent nature increased the likelihood of failure of MMS, this case demonstrates that all patients should be followed periodically after the treatment of PMC.
We presented a case of PMC in which standard surgical margins would have been insufficient to clear the lesion. Mohs micrographic surgery was used to remove the majority of the tumor. As is common in PMC, the lesion was indolent and periocular in location. It also was incompletely excised due to notable subclinical extension, which is common for PMC. The distinction of PMC from metastatic mucinous carcinoma is paramount but sometimes difficult. Randomized controlled trials are lacking with regards to preferred method of treatment, but MMS has shown benefit and should be considered for recurrent lesions and lesions in cosmetically sensitive areas.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Martinez SR, Young SE. Primary mucinous carcinoma of the skin: a review. Int J Oncol. 2005;2:432-437.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Marra DE, Schanbacher CF, Torres A. Mohs micrographic surgery of primary cutaneous mucinous carcinoma using immunohistochemistry for margin control. Dermatol Surg. 2004;30:799-802.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
- Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
- Ortiz KJ, Gaughan MD, Bang RH, et al. A case of primary mucinous carcinoma of the scalp treated with Mohs surgery. Dermatol Surg. 2002;28:751-754.
- Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
- Teng P, Muir J. Small primary cutaneous mucinous carcinoma mimicking an early basal cell carcinoma. Dermatol Online J. 2013;19:3.
- Terada T, Sato Y, Furukawa K, et al. Primary cutaneous mucinous carcinoma initially diagnosed as metastatic adenocarcinoma. Tohoku J Exp Med. 2004;203:345-348.
- Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
- Cabell CE, Helm KF, Sakol PJ, et al. Primary mucinous carcinoma in a 54-year-old man. J Am Acad Dermatol. 2003;49:941-943.
- Cecchi R, Rapicano V. Primary cutaneous mucinous carcinoma: report of two cases treated with Mohs’ micrographic surgery. Australas J Dermatol. 2006;47:192-194.
- Eckert F, Schmid U, Hardmeier T, et al. Cytokeratin expression in mucinous sweat gland carcinomas: an immunohistochemical analysis of four cases. Histopathology. 1992;21:161-165.
- Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin. J Cutan Pathol. 2010;37:411-415.
- Ivan D, Hafeez Diwan A, Prieto VG. Expression of p63 in primary cutaneous adnexal neoplasms and adenocarcinoma metastatic to the skin. Mod Pathol. 2005;18:137-142.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Snow SN, Reizner GT. Mucinous eccrine carcinoma of the eyelid. Cancer. 1992;70:2099-2104.
- Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
- Miyasaka M, Tanaka R, Hirabayashi K, et al. Primary mucinous carcinoma of the skin: a case of metastasis after 10 years of disease-free interval. Eur J Plast Surg. 2009;32:189-193.
- Yeung KY, Stinson JC. Mucinous (adenocystic) carcinoma of sweat glands with widespread metastasis. case report with ultrastructural study. Cancer. 1977;39:2556-2562.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Tam CC, Dare DM, DiGiovanni JJ, et al. Recurrent and metastatic primary cutaneous mucinous carcinoma after excision and Mohs micrographic surgery. Cutis. 2011;87:245-248.
To the Editor:
Primary mucinous carcinoma (PMC) is an exceedingly rare adnexal tumor with an incidence of 0.07 cases per million individuals.1,2 First described by Lennox et al3 in 1952, this entity often presents as slow-growing, solitary nodules that often are soft on palpation but may have an indurated quality and range in color from reddish blue to flesh colored to white.4 Primary mucinous carcinoma most commonly is found on the eyelid (38%) but may affect other sites on the face (20.3%), scalp (16%), and axilla (10%).5 Historically, it has been thought to be more common among men; however, a 2005 large case series by Kazakov et al5 found that women were twice as likely to be affected. Primary mucinous carcinoma most frequently is diagnosed in the fifth through seventh decades of life, with a median age at onset of 63 years.6,7 Because of its rarity, PMC is most frequently confused clinically with basal cell carcinoma, keratoacanthoma, apocrine hidrocystoma, epidermoid cyst, Kaposi sarcoma, neuroma, lacrimal sac tumor, squamous cell carcinoma, granulomatous tumors, and metastatic adenocarcinoma.1,8-10
Primary mucinous carcinoma is thought to be derived from sweat glands, and select features such as decapitation secretion are more suggestive of apocrine than eccrine differentiation.5,8 On histopathology, PMC classically is described as nests of epithelial cells floating in lakes of extracellular mucin, primarily in the dermis and subcutis. The nests are composed of basaloid cells in solid to cribriform arrangements, usually with a low mitotic count and little nuclear atypia. These nests are suspended within periodic acid–Schiff positive mucinous pools partitioned by delicate fibrous septa. The mucin produced by PMC is sialomucin, and as such it is hyaluronidase resistant and sialidase labile.6 At least 1 report has been made of the presence of psammoma bodies in PMC.11
The neoplasm is characterized by an indolent course with frequent recurrence but rare metastasis.5,12 Treatment is primarily surgical, with Mohs micrographic surgery (MMS) offering improved tissue conservation and reduced recurrence rates.12 The diagnostic challenge lies in distinguishing PMC from a variety of metastatic mucinous internal malignancies that portend a notably greater morbidity and mortality to the patient. We describe a case of PMC, discuss the differentiation of PMC from metastatic mucinous carcinoma, and review the literature regarding treatment of this rare neoplasm.
A 65-year-old white woman was referred to our tertiary-care dermatologic surgery clinic for treatment of an incompletely excised mucinous carcinoma of the right lateral canthus (Figure 1). The clinically evident scar measured 0.5×0.5 cm. Although difficult to appreciate in Figure 1, a slight textural change of the surrounding skin, including the upper and lower eyelid, was apparent. Prior to her arrival to our clinic, the referring physician had completed a thorough review of systems and physical examination, which did not suggest an underlying malignancy. Computed tomography of the head, neck, chest, abdomen, and pelvis revealed a mass in the thyroid that was removed and found to be benign. The patient’s cutaneous lesion was therefore considered to be a PMC of the skin.
Given the prior incomplete excision of the lesion and its periocular location, we treated the patient with MMS. After 6 surgical stages, we continued to see evidence of the neoplasm as it tracked medially along the orbicularis oculi muscle (Figure 2). Due to the patient’s physical and emotional exhaustion at this point, we discontinued MMS and referred her to a colleague in plastic surgery for further excision of the remaining focus of positivity as well as repair. The final Mohs defect measured 4.2×4.0 cm (Figure 3). Approximately 2.3×1.0 cm of tissue in the area of remaining tumor was excised by plastic surgery, and the defect was repaired with a cervicofacial advancement flap closure of the right cheek and lower eyelid and full-thickness skin graft of the left upper eyelid. Histopathologic investigation found the additional tissue resected to be free of residual tumor.
To diagnose a patient with PMC, one must first rule out cutaneous metastasis of various internal malignancies that may appear similar on histopathology. A full clinical investigation consisting of a thorough history, physical examination, and appropriate radiographic imaging is required. Cutaneous metastases most commonly arise from the breast or gastrointestinal tract (GIT) but also can originate from the prostate, lungs, ovaries, pancreas, and kidneys.5 Histologically, PMC may be identical to metastatic adenocarcinoma.13 Location on the body may be a clue to a lesion’s origin, as metastases from a mucinous adenocarcinoma of the breast typically occur on the chest, breast, or axilla,5 whereas PMC primarily is found on the head and neck.
Certain histopathologic features may be suggestive of either a primary or metastatic etiology. Lesions arising in the skin may reveal an in situ component representing ductal hyperplasia, atypical ductal hyperplasia, or ductal carcinoma in situ. Identification of an in situ component defines a cutaneous primary neoplasm, but its absence does not exclude PMC.5 Additionally, metastatic lesions from the GIT typically have greater pleomorphism and “dirty” necrosis defined as eosinophilic foci containing nuclear debris.5
The expression pattern of cytokeratins (CKs) also can be suggestive. Primary mucinous carcinoma and metastatic breast adenocarcinoma are both CK7+ and CK20−. By contrast, mucinous adenocarcinoma of the GIT stains CK20+ and CK7−.14 Another marker that stains PMC is CK5 and CK6, though infrequently present. Levy et al15 reported positive staining for CK5 and CK6 in only 1 of 5 PMC cases. Positive staining for CK5 and CK6 has not been reported in any metastatic mucinous carcinoma.
The role of p63 immunostaining in the setting of mucinous carcinoma is controversial.16-18 Some practi-tioners have reported using p63 immunostaining to assist in establishing the diagnosis of PMC but only after performing a clinical workup to search for any primary sites of mucinous carcinoma in other organs.11 Other studies, however, have found select metastatic lesions from the breast17,18 and GIT18 to stain positively with p63. It is important to remember that these clinical and pathologic features are only suggestive of the primary etiology and are not replacement for a full clinical investigation.
Primary mucinous carcinoma is considered an indolent tumor with the majority of patient morbidity attributable to local recurrence and regional metastasis. Although uncommon, regional and distant metastasis rates have been reported to be 11% and 3%, respectively.19 Direct lymphatic invasion has been reported and indicates a more aggressive tumor with shorter recurrence-free intervals and predicts nodal metastases. Paradela et al20 recommended the use of D2-40, a monoclonal antibody and specific marker for lymphatic endothelium, to detect lymphatic invasion, particularly in node-negative primary tumors.
In one case of PMC on the jaw of a 39-year-old Japanese man, no recurrence or metastases were discovered until the 11th year of follow-up. At that time, he was found to have lung and bone metastases and died after 3 years.21 Other investigators report death occurring 4 to 24 months following diagnosis of distant metastases.7,22 Direct extension of the tumor into skeletal muscle, periosteum, bone, and dura also has been documented.7
Treatment principally is surgical, with PMC known to be resistant to both chemotherapy and radiation therapy.19,22 The recommended margins for simple excision range from 1 to 2 cm, but this method of treatment yields recurrence rates upward of 30% to 40%, especially for lesions located on the eyelid.12,13 First utilized in PMC of the eyelid to conserve tissue, MMS is rapidly becoming the treatment of choice because of its notably improved recurrence rate. A case series of 4 PMCs of the eyelid treated via MMS or frozen section control found the recurrence rate to be 7%.23 Another report of 2 cases of PMC treated by MMS reported no recurrence after 42 and 26 months.13 Ortiz et al7 reported an additional case of a patient treated by MMS that was recurrence free for 30 months at the time of publication. Further investigation is required to definitively recommend MMS on the basis of improved recurrence rate but should now be considered standard of care in recurrent, sizeable, or eyelid PMC.
Despite its ascension as treatment of choice in many cases of PMC, MMS is not without its risk of metastasis and recurrence. Tam et al24 reported a case of PMC with multiple recurrences and metastases following 3 simple excisions and 2 excisions via MMS. Although the lesion’s previously recurrent nature increased the likelihood of failure of MMS, this case demonstrates that all patients should be followed periodically after the treatment of PMC.
We presented a case of PMC in which standard surgical margins would have been insufficient to clear the lesion. Mohs micrographic surgery was used to remove the majority of the tumor. As is common in PMC, the lesion was indolent and periocular in location. It also was incompletely excised due to notable subclinical extension, which is common for PMC. The distinction of PMC from metastatic mucinous carcinoma is paramount but sometimes difficult. Randomized controlled trials are lacking with regards to preferred method of treatment, but MMS has shown benefit and should be considered for recurrent lesions and lesions in cosmetically sensitive areas.
To the Editor:
Primary mucinous carcinoma (PMC) is an exceedingly rare adnexal tumor with an incidence of 0.07 cases per million individuals.1,2 First described by Lennox et al3 in 1952, this entity often presents as slow-growing, solitary nodules that often are soft on palpation but may have an indurated quality and range in color from reddish blue to flesh colored to white.4 Primary mucinous carcinoma most commonly is found on the eyelid (38%) but may affect other sites on the face (20.3%), scalp (16%), and axilla (10%).5 Historically, it has been thought to be more common among men; however, a 2005 large case series by Kazakov et al5 found that women were twice as likely to be affected. Primary mucinous carcinoma most frequently is diagnosed in the fifth through seventh decades of life, with a median age at onset of 63 years.6,7 Because of its rarity, PMC is most frequently confused clinically with basal cell carcinoma, keratoacanthoma, apocrine hidrocystoma, epidermoid cyst, Kaposi sarcoma, neuroma, lacrimal sac tumor, squamous cell carcinoma, granulomatous tumors, and metastatic adenocarcinoma.1,8-10
Primary mucinous carcinoma is thought to be derived from sweat glands, and select features such as decapitation secretion are more suggestive of apocrine than eccrine differentiation.5,8 On histopathology, PMC classically is described as nests of epithelial cells floating in lakes of extracellular mucin, primarily in the dermis and subcutis. The nests are composed of basaloid cells in solid to cribriform arrangements, usually with a low mitotic count and little nuclear atypia. These nests are suspended within periodic acid–Schiff positive mucinous pools partitioned by delicate fibrous septa. The mucin produced by PMC is sialomucin, and as such it is hyaluronidase resistant and sialidase labile.6 At least 1 report has been made of the presence of psammoma bodies in PMC.11
The neoplasm is characterized by an indolent course with frequent recurrence but rare metastasis.5,12 Treatment is primarily surgical, with Mohs micrographic surgery (MMS) offering improved tissue conservation and reduced recurrence rates.12 The diagnostic challenge lies in distinguishing PMC from a variety of metastatic mucinous internal malignancies that portend a notably greater morbidity and mortality to the patient. We describe a case of PMC, discuss the differentiation of PMC from metastatic mucinous carcinoma, and review the literature regarding treatment of this rare neoplasm.
A 65-year-old white woman was referred to our tertiary-care dermatologic surgery clinic for treatment of an incompletely excised mucinous carcinoma of the right lateral canthus (Figure 1). The clinically evident scar measured 0.5×0.5 cm. Although difficult to appreciate in Figure 1, a slight textural change of the surrounding skin, including the upper and lower eyelid, was apparent. Prior to her arrival to our clinic, the referring physician had completed a thorough review of systems and physical examination, which did not suggest an underlying malignancy. Computed tomography of the head, neck, chest, abdomen, and pelvis revealed a mass in the thyroid that was removed and found to be benign. The patient’s cutaneous lesion was therefore considered to be a PMC of the skin.
Given the prior incomplete excision of the lesion and its periocular location, we treated the patient with MMS. After 6 surgical stages, we continued to see evidence of the neoplasm as it tracked medially along the orbicularis oculi muscle (Figure 2). Due to the patient’s physical and emotional exhaustion at this point, we discontinued MMS and referred her to a colleague in plastic surgery for further excision of the remaining focus of positivity as well as repair. The final Mohs defect measured 4.2×4.0 cm (Figure 3). Approximately 2.3×1.0 cm of tissue in the area of remaining tumor was excised by plastic surgery, and the defect was repaired with a cervicofacial advancement flap closure of the right cheek and lower eyelid and full-thickness skin graft of the left upper eyelid. Histopathologic investigation found the additional tissue resected to be free of residual tumor.
To diagnose a patient with PMC, one must first rule out cutaneous metastasis of various internal malignancies that may appear similar on histopathology. A full clinical investigation consisting of a thorough history, physical examination, and appropriate radiographic imaging is required. Cutaneous metastases most commonly arise from the breast or gastrointestinal tract (GIT) but also can originate from the prostate, lungs, ovaries, pancreas, and kidneys.5 Histologically, PMC may be identical to metastatic adenocarcinoma.13 Location on the body may be a clue to a lesion’s origin, as metastases from a mucinous adenocarcinoma of the breast typically occur on the chest, breast, or axilla,5 whereas PMC primarily is found on the head and neck.
Certain histopathologic features may be suggestive of either a primary or metastatic etiology. Lesions arising in the skin may reveal an in situ component representing ductal hyperplasia, atypical ductal hyperplasia, or ductal carcinoma in situ. Identification of an in situ component defines a cutaneous primary neoplasm, but its absence does not exclude PMC.5 Additionally, metastatic lesions from the GIT typically have greater pleomorphism and “dirty” necrosis defined as eosinophilic foci containing nuclear debris.5
The expression pattern of cytokeratins (CKs) also can be suggestive. Primary mucinous carcinoma and metastatic breast adenocarcinoma are both CK7+ and CK20−. By contrast, mucinous adenocarcinoma of the GIT stains CK20+ and CK7−.14 Another marker that stains PMC is CK5 and CK6, though infrequently present. Levy et al15 reported positive staining for CK5 and CK6 in only 1 of 5 PMC cases. Positive staining for CK5 and CK6 has not been reported in any metastatic mucinous carcinoma.
The role of p63 immunostaining in the setting of mucinous carcinoma is controversial.16-18 Some practi-tioners have reported using p63 immunostaining to assist in establishing the diagnosis of PMC but only after performing a clinical workup to search for any primary sites of mucinous carcinoma in other organs.11 Other studies, however, have found select metastatic lesions from the breast17,18 and GIT18 to stain positively with p63. It is important to remember that these clinical and pathologic features are only suggestive of the primary etiology and are not replacement for a full clinical investigation.
Primary mucinous carcinoma is considered an indolent tumor with the majority of patient morbidity attributable to local recurrence and regional metastasis. Although uncommon, regional and distant metastasis rates have been reported to be 11% and 3%, respectively.19 Direct lymphatic invasion has been reported and indicates a more aggressive tumor with shorter recurrence-free intervals and predicts nodal metastases. Paradela et al20 recommended the use of D2-40, a monoclonal antibody and specific marker for lymphatic endothelium, to detect lymphatic invasion, particularly in node-negative primary tumors.
In one case of PMC on the jaw of a 39-year-old Japanese man, no recurrence or metastases were discovered until the 11th year of follow-up. At that time, he was found to have lung and bone metastases and died after 3 years.21 Other investigators report death occurring 4 to 24 months following diagnosis of distant metastases.7,22 Direct extension of the tumor into skeletal muscle, periosteum, bone, and dura also has been documented.7
Treatment principally is surgical, with PMC known to be resistant to both chemotherapy and radiation therapy.19,22 The recommended margins for simple excision range from 1 to 2 cm, but this method of treatment yields recurrence rates upward of 30% to 40%, especially for lesions located on the eyelid.12,13 First utilized in PMC of the eyelid to conserve tissue, MMS is rapidly becoming the treatment of choice because of its notably improved recurrence rate. A case series of 4 PMCs of the eyelid treated via MMS or frozen section control found the recurrence rate to be 7%.23 Another report of 2 cases of PMC treated by MMS reported no recurrence after 42 and 26 months.13 Ortiz et al7 reported an additional case of a patient treated by MMS that was recurrence free for 30 months at the time of publication. Further investigation is required to definitively recommend MMS on the basis of improved recurrence rate but should now be considered standard of care in recurrent, sizeable, or eyelid PMC.
Despite its ascension as treatment of choice in many cases of PMC, MMS is not without its risk of metastasis and recurrence. Tam et al24 reported a case of PMC with multiple recurrences and metastases following 3 simple excisions and 2 excisions via MMS. Although the lesion’s previously recurrent nature increased the likelihood of failure of MMS, this case demonstrates that all patients should be followed periodically after the treatment of PMC.
We presented a case of PMC in which standard surgical margins would have been insufficient to clear the lesion. Mohs micrographic surgery was used to remove the majority of the tumor. As is common in PMC, the lesion was indolent and periocular in location. It also was incompletely excised due to notable subclinical extension, which is common for PMC. The distinction of PMC from metastatic mucinous carcinoma is paramount but sometimes difficult. Randomized controlled trials are lacking with regards to preferred method of treatment, but MMS has shown benefit and should be considered for recurrent lesions and lesions in cosmetically sensitive areas.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Martinez SR, Young SE. Primary mucinous carcinoma of the skin: a review. Int J Oncol. 2005;2:432-437.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Marra DE, Schanbacher CF, Torres A. Mohs micrographic surgery of primary cutaneous mucinous carcinoma using immunohistochemistry for margin control. Dermatol Surg. 2004;30:799-802.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
- Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
- Ortiz KJ, Gaughan MD, Bang RH, et al. A case of primary mucinous carcinoma of the scalp treated with Mohs surgery. Dermatol Surg. 2002;28:751-754.
- Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
- Teng P, Muir J. Small primary cutaneous mucinous carcinoma mimicking an early basal cell carcinoma. Dermatol Online J. 2013;19:3.
- Terada T, Sato Y, Furukawa K, et al. Primary cutaneous mucinous carcinoma initially diagnosed as metastatic adenocarcinoma. Tohoku J Exp Med. 2004;203:345-348.
- Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
- Cabell CE, Helm KF, Sakol PJ, et al. Primary mucinous carcinoma in a 54-year-old man. J Am Acad Dermatol. 2003;49:941-943.
- Cecchi R, Rapicano V. Primary cutaneous mucinous carcinoma: report of two cases treated with Mohs’ micrographic surgery. Australas J Dermatol. 2006;47:192-194.
- Eckert F, Schmid U, Hardmeier T, et al. Cytokeratin expression in mucinous sweat gland carcinomas: an immunohistochemical analysis of four cases. Histopathology. 1992;21:161-165.
- Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin. J Cutan Pathol. 2010;37:411-415.
- Ivan D, Hafeez Diwan A, Prieto VG. Expression of p63 in primary cutaneous adnexal neoplasms and adenocarcinoma metastatic to the skin. Mod Pathol. 2005;18:137-142.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Snow SN, Reizner GT. Mucinous eccrine carcinoma of the eyelid. Cancer. 1992;70:2099-2104.
- Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
- Miyasaka M, Tanaka R, Hirabayashi K, et al. Primary mucinous carcinoma of the skin: a case of metastasis after 10 years of disease-free interval. Eur J Plast Surg. 2009;32:189-193.
- Yeung KY, Stinson JC. Mucinous (adenocystic) carcinoma of sweat glands with widespread metastasis. case report with ultrastructural study. Cancer. 1977;39:2556-2562.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Tam CC, Dare DM, DiGiovanni JJ, et al. Recurrent and metastatic primary cutaneous mucinous carcinoma after excision and Mohs micrographic surgery. Cutis. 2011;87:245-248.
- Breiting L, Christensen L, Dahlstrom K, et al. Primary mucinous carcinoma of the skin: a population-based study. Int J Dermatol. 2008;47:242-245.
- Martinez SR, Young SE. Primary mucinous carcinoma of the skin: a review. Int J Oncol. 2005;2:432-437.
- Lennox B, Pearse AG, Richards HG. Mucin-secreting tumours of the skin with special reference to the so-called mixed-salivary tumour of the skin and its relation to hidradenoma. J Pathol Bacteriol. 1952;64:865-880.
- Marra DE, Schanbacher CF, Torres A. Mohs micrographic surgery of primary cutaneous mucinous carcinoma using immunohistochemistry for margin control. Dermatol Surg. 2004;30:799-802.
- Kazakov DV, Suster S, LeBoit PE, et al. Mucinous carcinoma of the skin, primary, and secondary: a clinicopathologic study of 63 cases with emphasis on the morphologic spectrum of primary cutaneous forms: homologies with mucinous lesions in the breast. Am J Surg Pathol. 2005;29:764-782.
- Mendoza S, Helwig EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103:68-78.
- Ortiz KJ, Gaughan MD, Bang RH, et al. A case of primary mucinous carcinoma of the scalp treated with Mohs surgery. Dermatol Surg. 2002;28:751-754.
- Bellezza G, Sidoni A, Bucciarelli E. Primary mucinous carcinoma of the skin. Am J Dermatopathol. 2000;22:166-170.
- Teng P, Muir J. Small primary cutaneous mucinous carcinoma mimicking an early basal cell carcinoma. Dermatol Online J. 2013;19:3.
- Terada T, Sato Y, Furukawa K, et al. Primary cutaneous mucinous carcinoma initially diagnosed as metastatic adenocarcinoma. Tohoku J Exp Med. 2004;203:345-348.
- Kalebi A, Hale M. Primary mucinous carcinoma of the skin: usefulness of p63 in excluding metastasis and first report of psammoma bodies. Am J Dermatopathol. 2008;30:510.
- Cabell CE, Helm KF, Sakol PJ, et al. Primary mucinous carcinoma in a 54-year-old man. J Am Acad Dermatol. 2003;49:941-943.
- Cecchi R, Rapicano V. Primary cutaneous mucinous carcinoma: report of two cases treated with Mohs’ micrographic surgery. Australas J Dermatol. 2006;47:192-194.
- Eckert F, Schmid U, Hardmeier T, et al. Cytokeratin expression in mucinous sweat gland carcinomas: an immunohistochemical analysis of four cases. Histopathology. 1992;21:161-165.
- Levy G, Finkelstein A, McNiff JM. Immunohistochemical techniques to compare primary vs. metastatic mucinous carcinoma of the skin. J Cutan Pathol. 2010;37:411-415.
- Ivan D, Hafeez Diwan A, Prieto VG. Expression of p63 in primary cutaneous adnexal neoplasms and adenocarcinoma metastatic to the skin. Mod Pathol. 2005;18:137-142.
- Kanitakis J, Chouvet B. Expression of p63 in cutaneous metastases. Am J Clin Pathol. 2007;128:753-758.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Snow SN, Reizner GT. Mucinous eccrine carcinoma of the eyelid. Cancer. 1992;70:2099-2104.
- Paradela S, Castiñeiras I, Cuevas J, et al. Mucinous carcinoma of the skin: evaluation of lymphatic invasion with D2-40. Am J Dermatopathol. 2008;30:504-508.
- Miyasaka M, Tanaka R, Hirabayashi K, et al. Primary mucinous carcinoma of the skin: a case of metastasis after 10 years of disease-free interval. Eur J Plast Surg. 2009;32:189-193.
- Yeung KY, Stinson JC. Mucinous (adenocystic) carcinoma of sweat glands with widespread metastasis. case report with ultrastructural study. Cancer. 1977;39:2556-2562.
- Papalas JA, Proia AD. Primary mucinous carcinoma of the eyelid: a clinicopathologic and immunohistochemical study of 4 cases and an update on recurrence rates. Arch Ophthalmol. 2010;128:1160-1165.
- Tam CC, Dare DM, DiGiovanni JJ, et al. Recurrent and metastatic primary cutaneous mucinous carcinoma after excision and Mohs micrographic surgery. Cutis. 2011;87:245-248.
Practice Points
- Primary mucinous carcinoma (PMC) of the skin is a rare adnexal tumor.
- Prior to treatment, the diagnostic importance lies in distinguishing PMC from metastatic mucinous malignancies, which portend a poorer prognosis.
- Treatment primarily is surgical, with Mohs micrographic surgery offering improved tissue conservation and reduced recurrence rates.
Acrodermatitis Enteropathica in a Patient With Short Bowel Syndrome
To the Editor:
Acrodermatitis enteropathica (AE) is an inherited defect in zinc absorption that leads to hypozincemia. Its clinical presentation can vary based on serum zinc level and ranges from periorificial erosive dermatitis to psoriasiform dermatitis.1 Recognition of the cutaneous manifestations of zinc deficiency can lead to early intervention with zinc supplementation and prevention of long-term morbidity and even mortality. In our case, the coexistence of a bullous acral dermatosis with the additional feature of extensor digital dermatitis with fissuring suggests a diagnosis of AE and can alert the astute clinician to the need for testing of serum zinc levels and/or treatment with zinc supplementation. Causes of acquired zinc deficiency that have been reported in the literature include eating disorders such as anorexia nervosa and bulimia nervosa, Crohn disease, food allergy, intestinal parasitic infestations, and an inborn error of metabolism known as nonketotic hyperglycemia (Table).2-4
RELATED ARTICLE: Acquired Acrodermatitis Enteropathica Secondary to Alcoholism

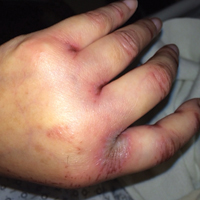
A 42-year-old woman with a medical history of rheumatoid arthritis and short bowel syndrome due to multiple small bowel obstructions with subsequent bowel resections who was on chronic total parenteral nutrition (TPN) presented with bullae on the hands, shins, and feet. The patient initially noticed small erythematous macules on the hands and feet months prior to presentation. Three weeks prior to presentation, bullae started to form on the hands, mostly between the web spaces; dorsal aspects of the feet; and anterior aspects of the shins. The patient denied any oral ulcers. One day prior to presentation the patient was seen at an outside hospital and was started on prednisone 5 mg daily, oral clindamycin, mupirocin ointment, and nystatin-triamcinolone cream. These medications failed to improve her condition. On review of systems, the patient denied any fever, chills, eye pain, or dysuria.
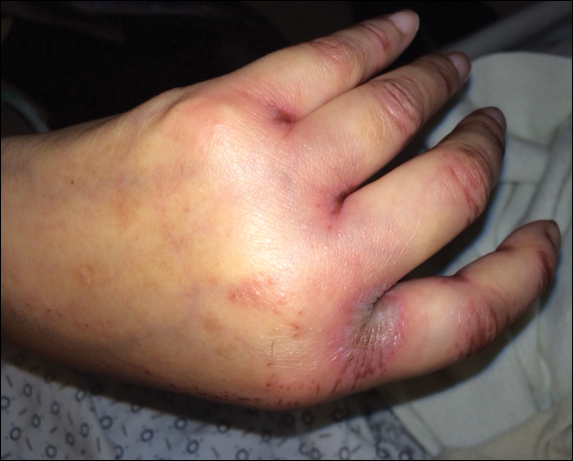
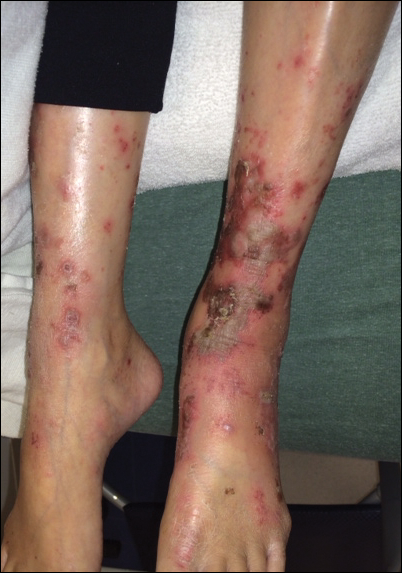
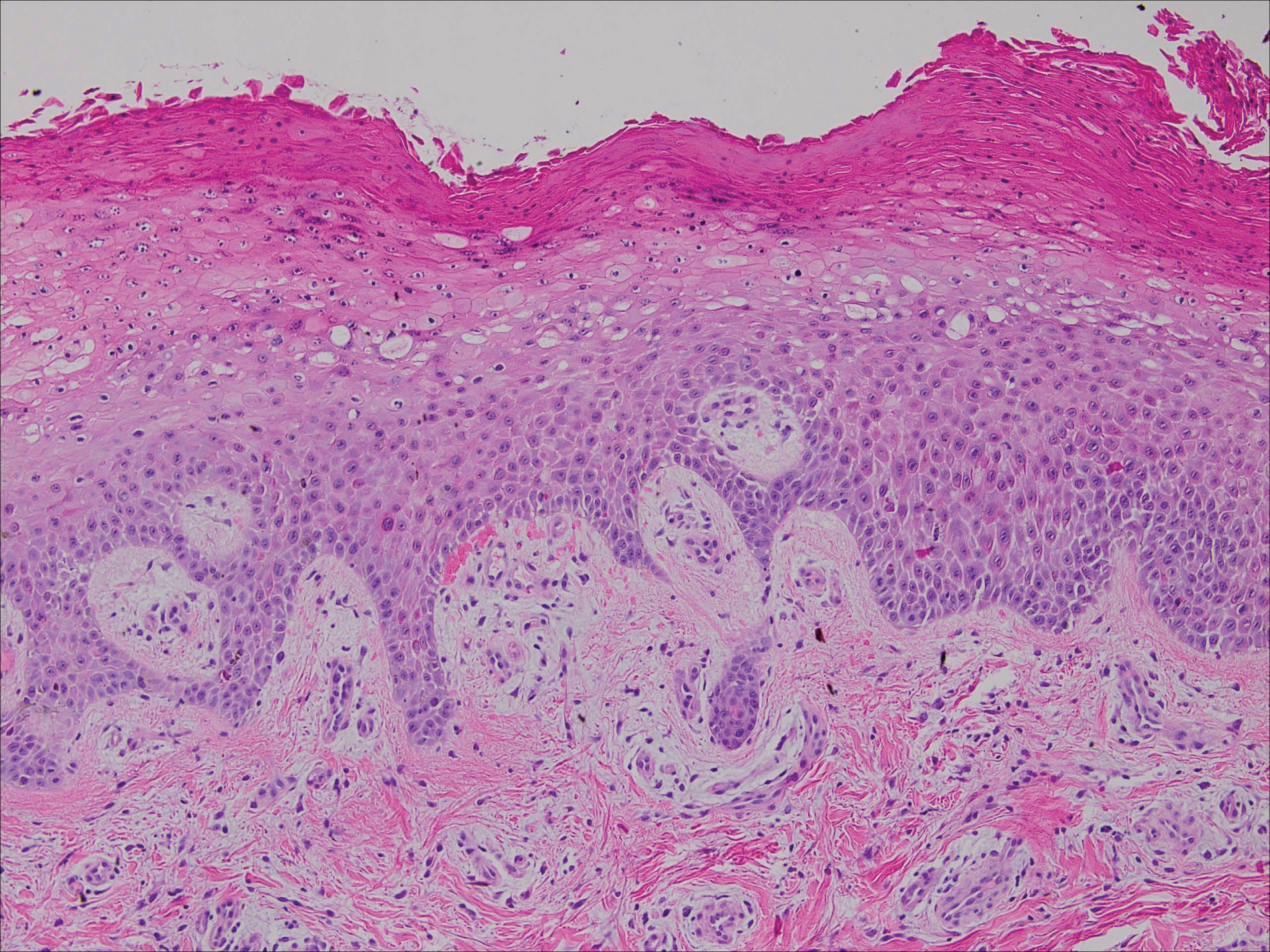
Upon initial presentation the patient appeared weak and fatigued, though vital signs were normal. Physical examination revealed multiple flaccid bullae in the web spaces of the hands and shallow erosions with hemorrhagic crusts on the bilateral wrists. She also had violaceous patches in the extensor creases of the metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints, which were strikingly symmetric (Figure 1). Prominent flaccid bullae and shallow erosions with hemorrhagic crusts also were present on the bilateral shins and dorsal aspects of the feet (Figure 2). No oral ulcers were present. A punch biopsy from the dorsal aspect of the left foot revealed psoriasiform hyperplasia of the epidermis with prominent ballooning degeneration and hyperkeratosis/parakeratosis (Figure 3); a periodic acid–Schiff stain was negative for fungal organisms.
Given the biopsy results and clinical presentation, a nutritional deficiency was suspected and serum levels of zinc, vitamin B1, vitamin B2, and vitamin B3 were assessed. Vitamins B1, B2
Zinc is an essential trace element and can be found in high concentration in foods such as shellfish, green vegetables, legumes, nuts, and whole grains.6 The majority of zinc is absorbed in the jejunum; as such, many cases of acquired zinc deficiency leading to AE are dueto disorders that affect the small intestine.2 Conditions that may lead to poor gastrointestinal zinc absorption include alcoholism, eating disorders, TPN, burns, surgery, and malignancies.2,7
Diagnosis typically is made based on characteristic clinical features, biopsy results, and a measurement of the serum zinc concentration. Although a low serum zinc level supports the diagnosis, serum zinc concentration is not a reliable indicator of body zinc stores and a normal serum zinc concentration does not rule out AE. The gold standard for diagnosis is the resolution of lesions after zinc supplementation.1 Notably, because the production of alkaline phosphatase is dependent on zinc, levels of this enzyme also may be low in cases of AE,6 as in our patient.
The clinical manifestations of AE can vary greatly; patients may initially present with eczematous pink scaly plaques, which may subsequently become vesicular, bullous, pustular, or desquamative. The lesions may develop over the arms and legs as well as the anogenital and periorificial areas.5 Other notable manifestations that may present early in the course of AE include angular cheilitis followed by paronychia. In patients who are not promptly treated, long-term zinc deficiency may lead to growth delay, mental slowing, poor wound healing, anemia, and anorexia.5 Of note, deficiencies of branched-chain amino acids and essential fatty acids may appear clinically similar to AE.2
Zinc replacement is the treatment of choice for patients with AE due to dietary deficiency, and replacement therapy should begin with 0.5 to 1 mg/kg daily of elemental zinc.5 Response to acquired AE with zinc supplementation often is rapid. Lesions tend to resolve within days to weeks depending on the degree of deficiency.2
Although AE is an uncommon dermatosis in the United States, it is an important diagnosis to make because its clinical features are fairly specific and early zinc supplementation allows for full resolution of the disease without permanent sequelae. The diagnosis of AE should be strongly considered when features of an acral bullous dermatosis are combined with a fissured dermatitis of extensor joints of the hands or elbows. It is particularly important to recognize that alcoholics, burn victims, postsurgical patients, and those with malignancies and eating disorders are at an increased risk for developing this nutritional deficiency.
- Kumar P, Lal NR, Mondal AK, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Suchithra N, Sreejith P, Pappachan JM, et al. Acrodermatitis enteropathica-like skin eruption in a case of short bowel syndrome following jejuno-transverse colon anastomosis. Dermatol Online J. 2007;13:20.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol. 2002;34:207-220.
- Griffin IJ, Kim SC, Hicks PD, et al. Zinc metabolism in adolescents with Crohn’s disease. Pediatr Res. 2004;56:235-239.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism [published online October 30, 2006]. J Am Acad Dermatol. 2007;56:116-124.
- Cheshire H, Stather P, Vorster J. Acquired acrodermatitis enteropathica due to zinc deficiency in a patient with pre-existing Darier’s disease. J Dermatol Case Rep. 2009;3:41-43.
- Strumia R. Dermatologic signs in patients with eating disorders. Am J Clin Dermatol. 2005;6:165-173.
To the Editor:
Acrodermatitis enteropathica (AE) is an inherited defect in zinc absorption that leads to hypozincemia. Its clinical presentation can vary based on serum zinc level and ranges from periorificial erosive dermatitis to psoriasiform dermatitis.1 Recognition of the cutaneous manifestations of zinc deficiency can lead to early intervention with zinc supplementation and prevention of long-term morbidity and even mortality. In our case, the coexistence of a bullous acral dermatosis with the additional feature of extensor digital dermatitis with fissuring suggests a diagnosis of AE and can alert the astute clinician to the need for testing of serum zinc levels and/or treatment with zinc supplementation. Causes of acquired zinc deficiency that have been reported in the literature include eating disorders such as anorexia nervosa and bulimia nervosa, Crohn disease, food allergy, intestinal parasitic infestations, and an inborn error of metabolism known as nonketotic hyperglycemia (Table).2-4
RELATED ARTICLE: Acquired Acrodermatitis Enteropathica Secondary to Alcoholism
A 42-year-old woman with a medical history of rheumatoid arthritis and short bowel syndrome due to multiple small bowel obstructions with subsequent bowel resections who was on chronic total parenteral nutrition (TPN) presented with bullae on the hands, shins, and feet. The patient initially noticed small erythematous macules on the hands and feet months prior to presentation. Three weeks prior to presentation, bullae started to form on the hands, mostly between the web spaces; dorsal aspects of the feet; and anterior aspects of the shins. The patient denied any oral ulcers. One day prior to presentation the patient was seen at an outside hospital and was started on prednisone 5 mg daily, oral clindamycin, mupirocin ointment, and nystatin-triamcinolone cream. These medications failed to improve her condition. On review of systems, the patient denied any fever, chills, eye pain, or dysuria.
Upon initial presentation the patient appeared weak and fatigued, though vital signs were normal. Physical examination revealed multiple flaccid bullae in the web spaces of the hands and shallow erosions with hemorrhagic crusts on the bilateral wrists. She also had violaceous patches in the extensor creases of the metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints, which were strikingly symmetric (Figure 1). Prominent flaccid bullae and shallow erosions with hemorrhagic crusts also were present on the bilateral shins and dorsal aspects of the feet (Figure 2). No oral ulcers were present. A punch biopsy from the dorsal aspect of the left foot revealed psoriasiform hyperplasia of the epidermis with prominent ballooning degeneration and hyperkeratosis/parakeratosis (Figure 3); a periodic acid–Schiff stain was negative for fungal organisms.
Given the biopsy results and clinical presentation, a nutritional deficiency was suspected and serum levels of zinc, vitamin B1, vitamin B2, and vitamin B3 were assessed. Vitamins B1, B2
Zinc is an essential trace element and can be found in high concentration in foods such as shellfish, green vegetables, legumes, nuts, and whole grains.6 The majority of zinc is absorbed in the jejunum; as such, many cases of acquired zinc deficiency leading to AE are dueto disorders that affect the small intestine.2 Conditions that may lead to poor gastrointestinal zinc absorption include alcoholism, eating disorders, TPN, burns, surgery, and malignancies.2,7
Diagnosis typically is made based on characteristic clinical features, biopsy results, and a measurement of the serum zinc concentration. Although a low serum zinc level supports the diagnosis, serum zinc concentration is not a reliable indicator of body zinc stores and a normal serum zinc concentration does not rule out AE. The gold standard for diagnosis is the resolution of lesions after zinc supplementation.1 Notably, because the production of alkaline phosphatase is dependent on zinc, levels of this enzyme also may be low in cases of AE,6 as in our patient.
The clinical manifestations of AE can vary greatly; patients may initially present with eczematous pink scaly plaques, which may subsequently become vesicular, bullous, pustular, or desquamative. The lesions may develop over the arms and legs as well as the anogenital and periorificial areas.5 Other notable manifestations that may present early in the course of AE include angular cheilitis followed by paronychia. In patients who are not promptly treated, long-term zinc deficiency may lead to growth delay, mental slowing, poor wound healing, anemia, and anorexia.5 Of note, deficiencies of branched-chain amino acids and essential fatty acids may appear clinically similar to AE.2
Zinc replacement is the treatment of choice for patients with AE due to dietary deficiency, and replacement therapy should begin with 0.5 to 1 mg/kg daily of elemental zinc.5 Response to acquired AE with zinc supplementation often is rapid. Lesions tend to resolve within days to weeks depending on the degree of deficiency.2
Although AE is an uncommon dermatosis in the United States, it is an important diagnosis to make because its clinical features are fairly specific and early zinc supplementation allows for full resolution of the disease without permanent sequelae. The diagnosis of AE should be strongly considered when features of an acral bullous dermatosis are combined with a fissured dermatitis of extensor joints of the hands or elbows. It is particularly important to recognize that alcoholics, burn victims, postsurgical patients, and those with malignancies and eating disorders are at an increased risk for developing this nutritional deficiency.
To the Editor:
Acrodermatitis enteropathica (AE) is an inherited defect in zinc absorption that leads to hypozincemia. Its clinical presentation can vary based on serum zinc level and ranges from periorificial erosive dermatitis to psoriasiform dermatitis.1 Recognition of the cutaneous manifestations of zinc deficiency can lead to early intervention with zinc supplementation and prevention of long-term morbidity and even mortality. In our case, the coexistence of a bullous acral dermatosis with the additional feature of extensor digital dermatitis with fissuring suggests a diagnosis of AE and can alert the astute clinician to the need for testing of serum zinc levels and/or treatment with zinc supplementation. Causes of acquired zinc deficiency that have been reported in the literature include eating disorders such as anorexia nervosa and bulimia nervosa, Crohn disease, food allergy, intestinal parasitic infestations, and an inborn error of metabolism known as nonketotic hyperglycemia (Table).2-4
RELATED ARTICLE: Acquired Acrodermatitis Enteropathica Secondary to Alcoholism
A 42-year-old woman with a medical history of rheumatoid arthritis and short bowel syndrome due to multiple small bowel obstructions with subsequent bowel resections who was on chronic total parenteral nutrition (TPN) presented with bullae on the hands, shins, and feet. The patient initially noticed small erythematous macules on the hands and feet months prior to presentation. Three weeks prior to presentation, bullae started to form on the hands, mostly between the web spaces; dorsal aspects of the feet; and anterior aspects of the shins. The patient denied any oral ulcers. One day prior to presentation the patient was seen at an outside hospital and was started on prednisone 5 mg daily, oral clindamycin, mupirocin ointment, and nystatin-triamcinolone cream. These medications failed to improve her condition. On review of systems, the patient denied any fever, chills, eye pain, or dysuria.
Upon initial presentation the patient appeared weak and fatigued, though vital signs were normal. Physical examination revealed multiple flaccid bullae in the web spaces of the hands and shallow erosions with hemorrhagic crusts on the bilateral wrists. She also had violaceous patches in the extensor creases of the metacarpophalangeal, proximal interphalangeal, and distal interphalangeal joints, which were strikingly symmetric (Figure 1). Prominent flaccid bullae and shallow erosions with hemorrhagic crusts also were present on the bilateral shins and dorsal aspects of the feet (Figure 2). No oral ulcers were present. A punch biopsy from the dorsal aspect of the left foot revealed psoriasiform hyperplasia of the epidermis with prominent ballooning degeneration and hyperkeratosis/parakeratosis (Figure 3); a periodic acid–Schiff stain was negative for fungal organisms.
Given the biopsy results and clinical presentation, a nutritional deficiency was suspected and serum levels of zinc, vitamin B1, vitamin B2, and vitamin B3 were assessed. Vitamins B1, B2
Zinc is an essential trace element and can be found in high concentration in foods such as shellfish, green vegetables, legumes, nuts, and whole grains.6 The majority of zinc is absorbed in the jejunum; as such, many cases of acquired zinc deficiency leading to AE are dueto disorders that affect the small intestine.2 Conditions that may lead to poor gastrointestinal zinc absorption include alcoholism, eating disorders, TPN, burns, surgery, and malignancies.2,7
Diagnosis typically is made based on characteristic clinical features, biopsy results, and a measurement of the serum zinc concentration. Although a low serum zinc level supports the diagnosis, serum zinc concentration is not a reliable indicator of body zinc stores and a normal serum zinc concentration does not rule out AE. The gold standard for diagnosis is the resolution of lesions after zinc supplementation.1 Notably, because the production of alkaline phosphatase is dependent on zinc, levels of this enzyme also may be low in cases of AE,6 as in our patient.
The clinical manifestations of AE can vary greatly; patients may initially present with eczematous pink scaly plaques, which may subsequently become vesicular, bullous, pustular, or desquamative. The lesions may develop over the arms and legs as well as the anogenital and periorificial areas.5 Other notable manifestations that may present early in the course of AE include angular cheilitis followed by paronychia. In patients who are not promptly treated, long-term zinc deficiency may lead to growth delay, mental slowing, poor wound healing, anemia, and anorexia.5 Of note, deficiencies of branched-chain amino acids and essential fatty acids may appear clinically similar to AE.2
Zinc replacement is the treatment of choice for patients with AE due to dietary deficiency, and replacement therapy should begin with 0.5 to 1 mg/kg daily of elemental zinc.5 Response to acquired AE with zinc supplementation often is rapid. Lesions tend to resolve within days to weeks depending on the degree of deficiency.2
Although AE is an uncommon dermatosis in the United States, it is an important diagnosis to make because its clinical features are fairly specific and early zinc supplementation allows for full resolution of the disease without permanent sequelae. The diagnosis of AE should be strongly considered when features of an acral bullous dermatosis are combined with a fissured dermatitis of extensor joints of the hands or elbows. It is particularly important to recognize that alcoholics, burn victims, postsurgical patients, and those with malignancies and eating disorders are at an increased risk for developing this nutritional deficiency.
- Kumar P, Lal NR, Mondal AK, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Suchithra N, Sreejith P, Pappachan JM, et al. Acrodermatitis enteropathica-like skin eruption in a case of short bowel syndrome following jejuno-transverse colon anastomosis. Dermatol Online J. 2007;13:20.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol. 2002;34:207-220.
- Griffin IJ, Kim SC, Hicks PD, et al. Zinc metabolism in adolescents with Crohn’s disease. Pediatr Res. 2004;56:235-239.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism [published online October 30, 2006]. J Am Acad Dermatol. 2007;56:116-124.
- Cheshire H, Stather P, Vorster J. Acquired acrodermatitis enteropathica due to zinc deficiency in a patient with pre-existing Darier’s disease. J Dermatol Case Rep. 2009;3:41-43.
- Strumia R. Dermatologic signs in patients with eating disorders. Am J Clin Dermatol. 2005;6:165-173.
- Kumar P, Lal NR, Mondal AK, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Suchithra N, Sreejith P, Pappachan JM, et al. Acrodermatitis enteropathica-like skin eruption in a case of short bowel syndrome following jejuno-transverse colon anastomosis. Dermatol Online J. 2007;13:20.
- Sundaram A, Koutkia P, Apovian CM. Nutritional management of short bowel syndrome in adults. J Clin Gastroenterol. 2002;34:207-220.
- Griffin IJ, Kim SC, Hicks PD, et al. Zinc metabolism in adolescents with Crohn’s disease. Pediatr Res. 2004;56:235-239.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism [published online October 30, 2006]. J Am Acad Dermatol. 2007;56:116-124.
- Cheshire H, Stather P, Vorster J. Acquired acrodermatitis enteropathica due to zinc deficiency in a patient with pre-existing Darier’s disease. J Dermatol Case Rep. 2009;3:41-43.
- Strumia R. Dermatologic signs in patients with eating disorders. Am J Clin Dermatol. 2005;6:165-173.
Practice Points
- Acrodermatitis enteropathica can be a manifestation of zinc deficiency.
- Acrodermatitis enteropathica should be considered in patients with poor intestinal absorption of nutrients.
Solitary Tender Nodule on the Back
The Diagnosis: Solitary Fibrous Tumor
Solitary fibrous tumors (SFTs), as first described by Klemperer and Rabin1 in 1931, are relatively uncommon mesenchymal neoplasms that occur primarily in the pleura. This lesion is now known to affect many other extrathoracic sites, such as the liver, kidney, adrenal glands, thyroid, central nervous system, and soft tissue, with rare examples originating from the skin.2 Okamura et al3 reported the first known case of cutaneous SFT in 1997, with most of the literature limited to case reports. Erdag et al2 described one of the largest case series of primary cutaneous SFTs. These lesions can occur across a wide age range but tend to primarily affect middle-aged adults. Solitary fibrous tumors have been known to have no sex predilection; however, Erdag et al2 found a male predominance with a male to female ratio of 4 to 1.
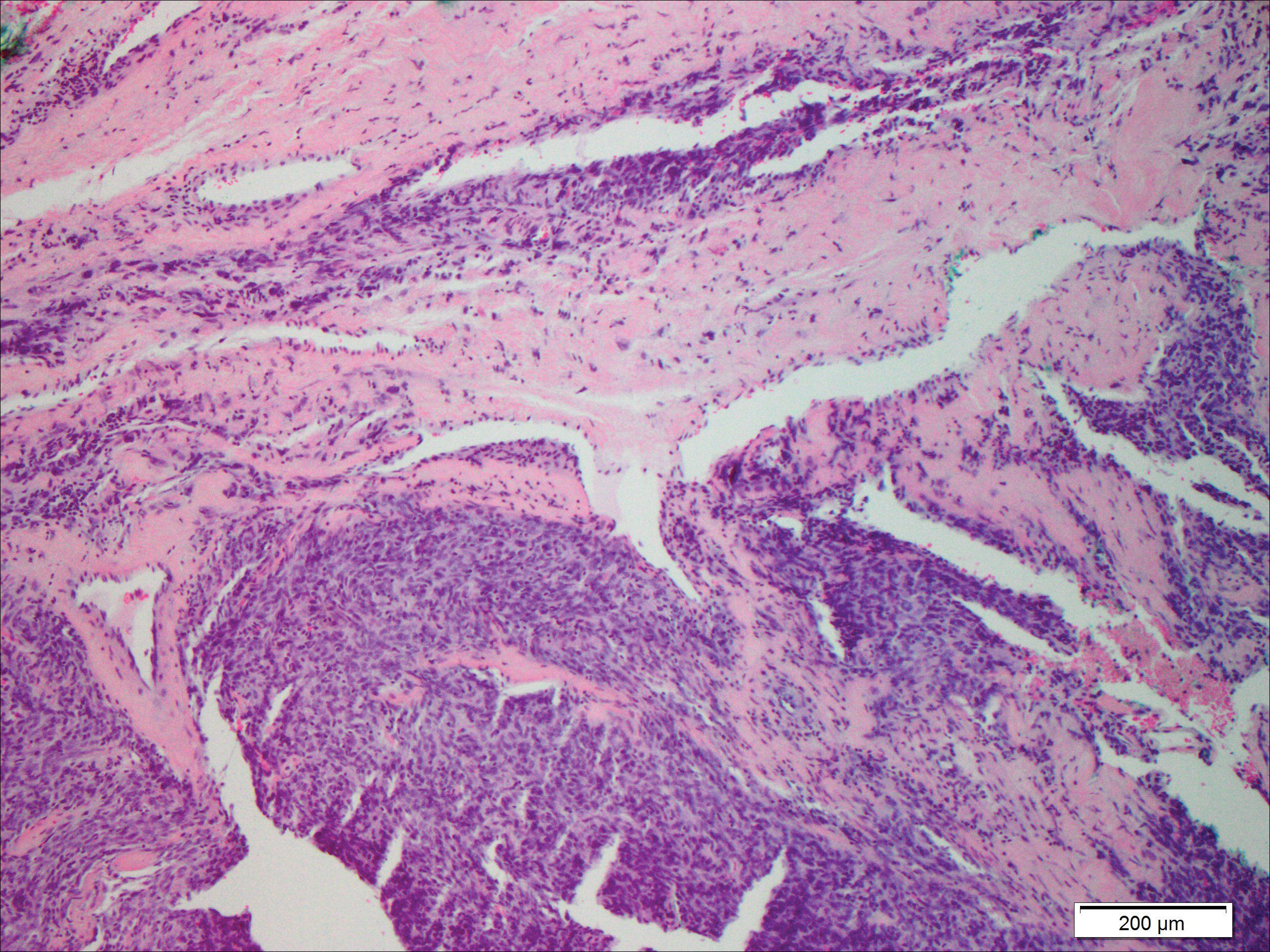
Histopathologically, a cutaneous SFT is known to appear as a well-circumscribed nodular spindle cell proliferation arranged in interlacing fascicles with an abundant hyalinized collagen stroma (quiz image). Alternating hypocellular and hypercellular areas can be seen. Supporting vasculature often is relatively prominent, represented by angulated and branching staghorn blood vessels (Figure 1).2 A common histopathologic finding of SFTs is a patternless pattern, which suggests that the tumor can have a variety of morphologic appearances (eg, storiform, fascicular, neural, herringbone growth patterns), making histologic diagnosis difficult (quiz image).4 Therefore, immunohistochemistry plays a large role in the diagnosis of this tumor. The most important positive markers include CD34, CD99, B-cell lymphoma 2 (BCL-2), and signal transducer and activator of transcription 6 (STAT6).5 Nuclear STAT6 staining is an immunomarker for NGFI-A binding protein 2 (NAB2)-STAT6 gene fusion, which is specific for SFT.5,6 Vivero et al7 also reported glutamate receptor, inotropic, AMPA 2 (GRIA2) as a useful immunostain in SFT, though it is also expressed in dermatofibrosarcoma protuberans (DFSP). In this case, the clinical and histopathologic findings best supported a diagnosis of SFT. Some consider hemangiopericytomas to be examples of SFTs; however, true hemangiopericytomas lack the thick hyalinized collagen and hypercellular areas seen in SFT.
A cellular dermatofibroma generally presents as a single round, reddish brown papule or nodule approximately 0.5 to 1 cm in diameter that is firm to palpation with a central depression or dimple created over the lesion from the lateral pressure. Cellular dermatofibromas mostly occur in middle-aged adults, with the most common locations on the legs and on the sides of the trunk. They are thought to arise after injuries to the skin. On histopathologic examination, cellular dermatofibromas typically exhibit a proliferation of fibrohistiocytic cells with collagen trapping, often at the periphery of the tumor (Figure 2). Although cellular dermatofibromas appear clinically different than SFTs, they often mimic SFTs histopathologically. Immunostaining also can be helpful in differentiating cellular dermatofibromas in which cells stain positive for factor XIIIa. CD34 staining is negative.

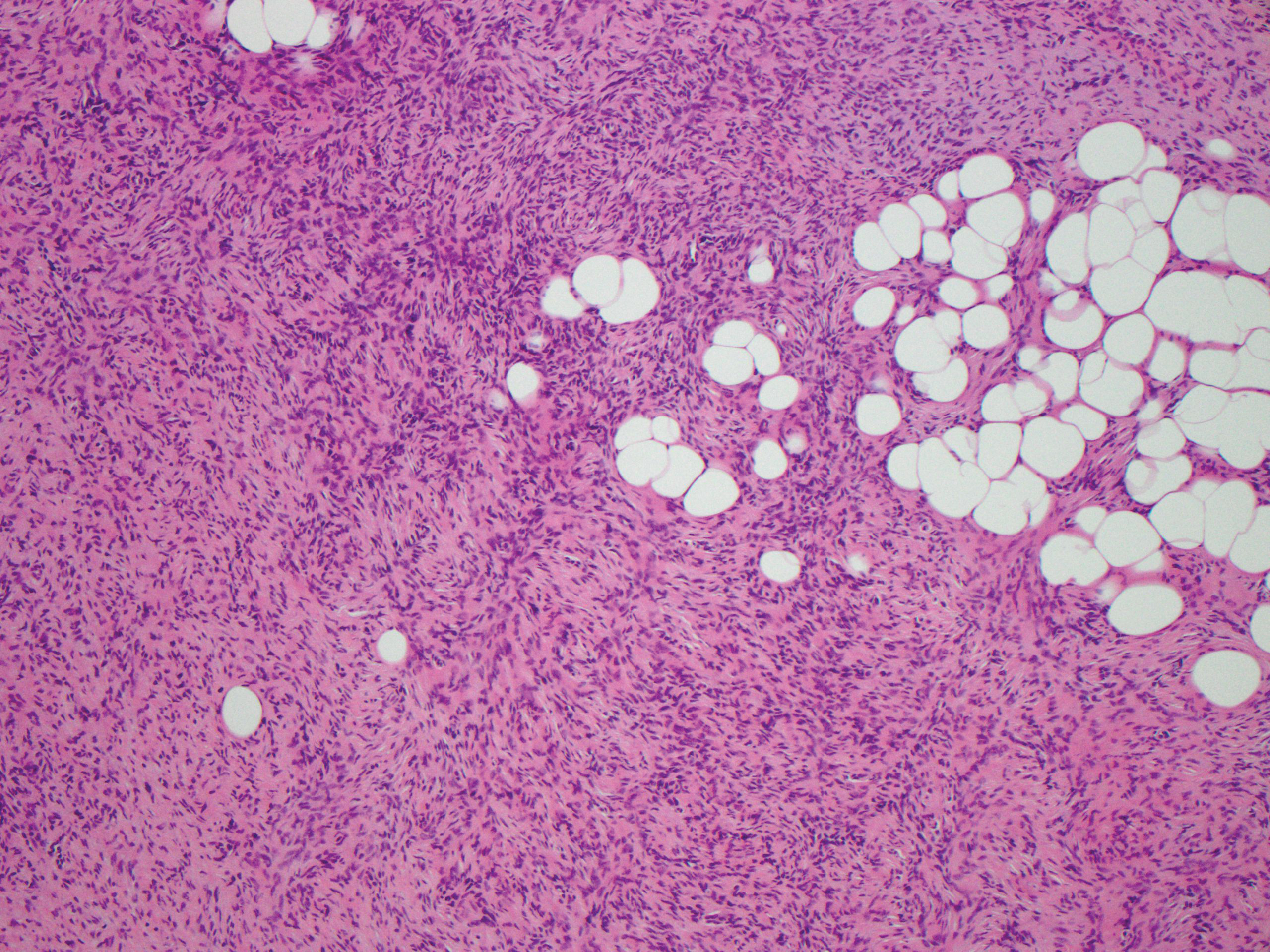
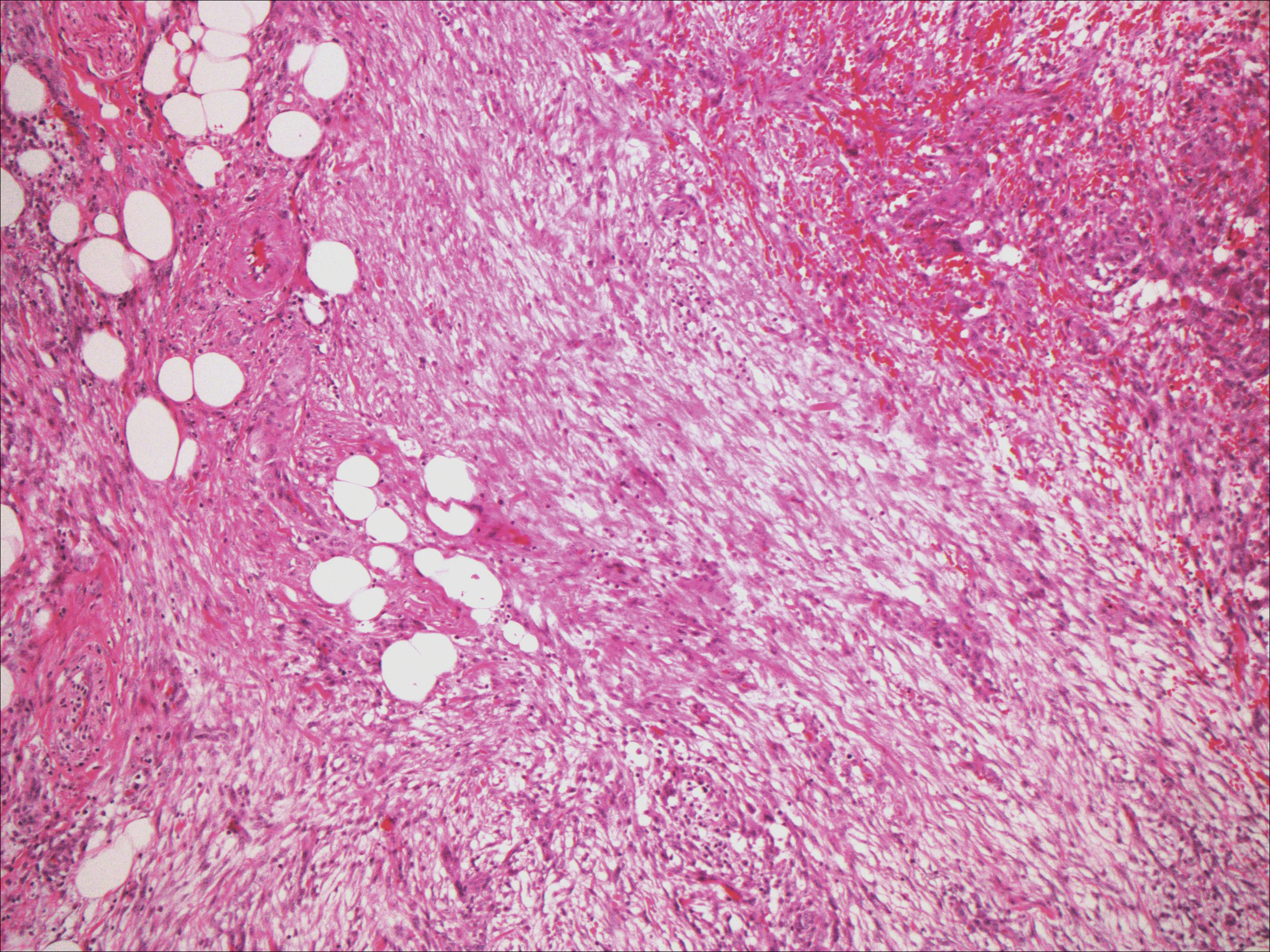
Dermatofibrosarcoma protuberans usually appears as one or multiple firm, red to violaceous nodules or plaques. They most often occur on the trunk in middle-aged adults. Histopathologically, DFSP presents with a dense, hypercellular, spindle cell proliferation that demonstrates a typical storiform pattern. The tumor generally infiltrates into the deep dermis and subcutaneous adipose layer with characteristic adipocyte entrapment (Figure 3). Positive CD34 and negative factor XIIIa staining helps to differentiate DFSP from a cellular dermatofibroma. Immunohistochemically, it is more difficult to distinguish DFSP from SFT, as both are CD34+ spindle cell neoplasms that also stain positive for CD99 and BCL-2.2 GRIA2 positivity also is seen in both SFT and DFSP.7 However, differentiation can be made on morphologic grounds alone, as DFSP has ill-defined tumor borders with adnexal and fat entrapment and SFT tends to be more circumscribed with prominent arborizing hyalinized vessels.8
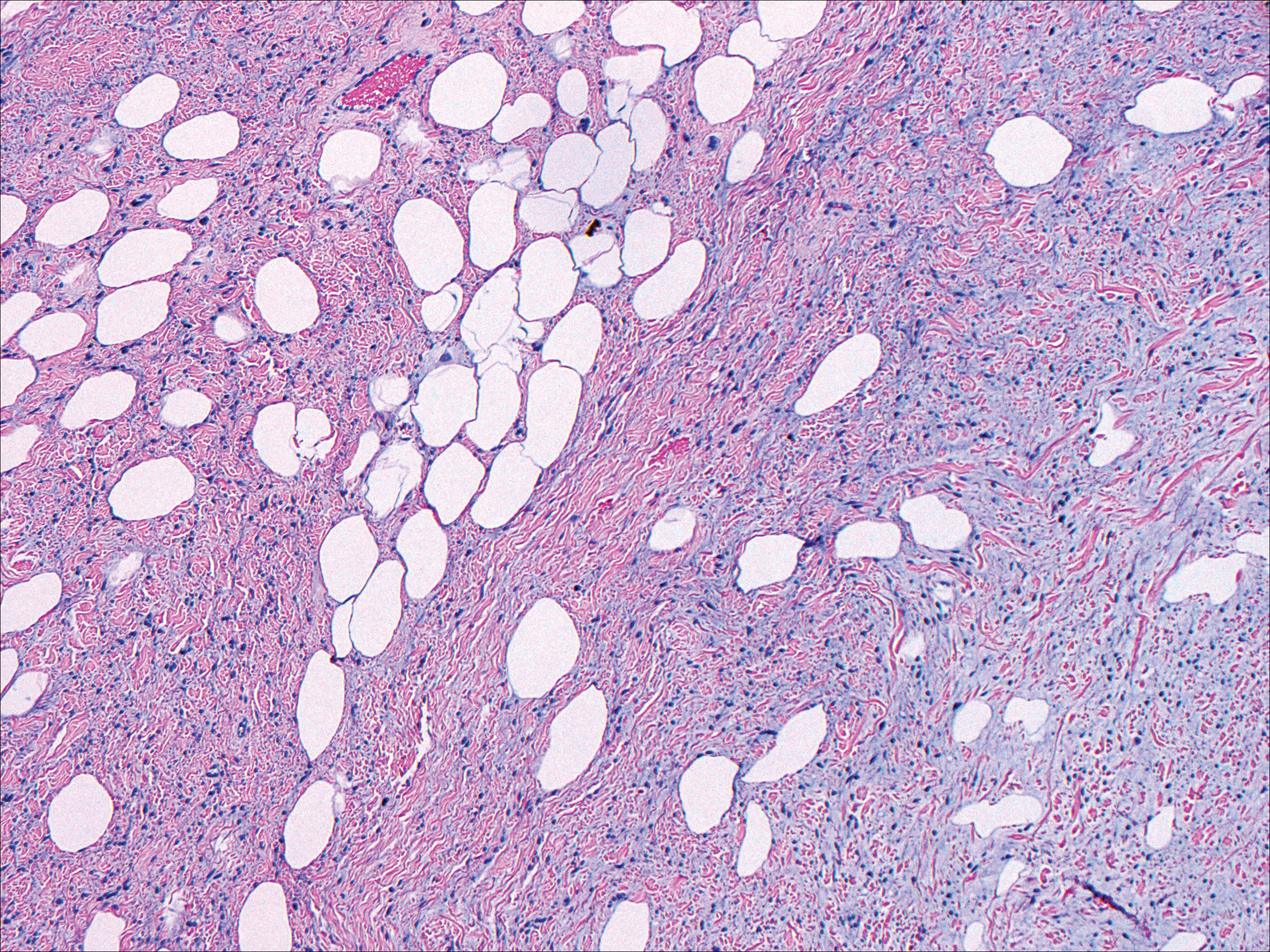
Spindle cell lipoma (SCL) is an asymptomatic subcutaneous tumor commonly located on the back, neck, and shoulders in older patients, typically men. It often presents as a solitary lesion, though multiple lesions may occur. It is a well-circumscribed tumor of mature adipose tissue with areas of spindle cell proliferation and ropey collagen bundles (Figure 4). In early lesions, the spindle cell areas are myxoid with the presence of many mast cells.9 The spindle cells stain positive for CD34. Although spindle cell lipoma would be included in both the clinical and histopathologic differential diagnosis for SFT, its histopathologic features often are enough to differentiate SCL, which is highlighted by the aforementioned features as well as a relatively low cellularity and lack of ectatic vessels.8 However, discerning tumor variants, such as low-fat pseudoangiomatous SCL and lipomatous or myxoid SFT, might prove more challenging.
Nodular fasciitis typically presents as a rapidly growing subcutaneous nodule that may be tender. It is a benign reactive process usually affecting the arms and trunk of young to middle-aged adults, though it commonly involves the head and neck region in children.10 The tumor histopathologically appears as a well-circumscribed subcutaneous or fascial nodule with an angulated appearance. Spindle-shaped and stellate fibroblasts are loosely arranged in an edematous myxomatous stroma with a feathered appearance (Figure 5). Extravasated erythrocytes often are present. With time, collagen bundles become thicker and hyalinized. Immunohistochemical studies demonstrate positivity for vimentin, calponin, muscle-specific actin, and smooth muscle actin. Desmin, CD34, cytokeratin, and S-100 typically are negative.10-12 Therefore, CD34 staining is one of the main differentiating factors between nodular fasciitis and SFTs.
- Klemperer P, Rabin CB. Primary neoplasms of the pleura: a report of five cases. Arch Pathol. 1931;11:385-412.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Okamura JM, Barr RJ, Battifora H. Solitary fibrous tumor of the skin. Am J Dermatopathol. 1997;19:515-518.
- Lee JY, Park SE, Shin SJ, et al. Solitary fibrous tumor with myxoid stromal change. Am J Dermatopathol. 2015;37:570-573.
- Geramizadeh B, Marzban M, Churg A. Role of immunohistochemistry in the diagnosis of solitary fibrous tumor, a review. Iran J Pathol. 2016;11:195-293.
- Creytens D, Ferdinande L, Dorpe JV. Histopathologically malignant solitary fibrous tumor of the skin: a report of an unusual case. J Cutan Pathol. 2016;43:629-631.
- Vivero M, Doyle LA, Fletcher CD, et al. GRIA2 is a novel diagnostic marker for solitary fibrous tumour identified through gene expression profiling. Histopathology. 2014;65:71-80.
- Wood L, Fountaine TJ, Rosamilia L, et al. Cutaneous CD34 spindle cell neoplasms: histopathologic features distinguish spindle cell lipoma, solitary fibrous tumor, and dermatofibrosarcoma protuberans. Am J Dermatopathol. 2010;32:764-768.
- Khatib Y, Khade AL, Shah VB, et al. Cytohistological features of spindle cell lipoma--a case report with differential diagnosis. J Clin Diagn Res. 2017;11:10-11.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Bracey TS, Wharton S, Smith ME. Nodular 'fasciitis' presenting as a cutaneous polyp. J Cutan Pathol. 2009;36:980-982.
- Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
The Diagnosis: Solitary Fibrous Tumor
Solitary fibrous tumors (SFTs), as first described by Klemperer and Rabin1 in 1931, are relatively uncommon mesenchymal neoplasms that occur primarily in the pleura. This lesion is now known to affect many other extrathoracic sites, such as the liver, kidney, adrenal glands, thyroid, central nervous system, and soft tissue, with rare examples originating from the skin.2 Okamura et al3 reported the first known case of cutaneous SFT in 1997, with most of the literature limited to case reports. Erdag et al2 described one of the largest case series of primary cutaneous SFTs. These lesions can occur across a wide age range but tend to primarily affect middle-aged adults. Solitary fibrous tumors have been known to have no sex predilection; however, Erdag et al2 found a male predominance with a male to female ratio of 4 to 1.
Histopathologically, a cutaneous SFT is known to appear as a well-circumscribed nodular spindle cell proliferation arranged in interlacing fascicles with an abundant hyalinized collagen stroma (quiz image). Alternating hypocellular and hypercellular areas can be seen. Supporting vasculature often is relatively prominent, represented by angulated and branching staghorn blood vessels (Figure 1).2 A common histopathologic finding of SFTs is a patternless pattern, which suggests that the tumor can have a variety of morphologic appearances (eg, storiform, fascicular, neural, herringbone growth patterns), making histologic diagnosis difficult (quiz image).4 Therefore, immunohistochemistry plays a large role in the diagnosis of this tumor. The most important positive markers include CD34, CD99, B-cell lymphoma 2 (BCL-2), and signal transducer and activator of transcription 6 (STAT6).5 Nuclear STAT6 staining is an immunomarker for NGFI-A binding protein 2 (NAB2)-STAT6 gene fusion, which is specific for SFT.5,6 Vivero et al7 also reported glutamate receptor, inotropic, AMPA 2 (GRIA2) as a useful immunostain in SFT, though it is also expressed in dermatofibrosarcoma protuberans (DFSP). In this case, the clinical and histopathologic findings best supported a diagnosis of SFT. Some consider hemangiopericytomas to be examples of SFTs; however, true hemangiopericytomas lack the thick hyalinized collagen and hypercellular areas seen in SFT.
A cellular dermatofibroma generally presents as a single round, reddish brown papule or nodule approximately 0.5 to 1 cm in diameter that is firm to palpation with a central depression or dimple created over the lesion from the lateral pressure. Cellular dermatofibromas mostly occur in middle-aged adults, with the most common locations on the legs and on the sides of the trunk. They are thought to arise after injuries to the skin. On histopathologic examination, cellular dermatofibromas typically exhibit a proliferation of fibrohistiocytic cells with collagen trapping, often at the periphery of the tumor (Figure 2). Although cellular dermatofibromas appear clinically different than SFTs, they often mimic SFTs histopathologically. Immunostaining also can be helpful in differentiating cellular dermatofibromas in which cells stain positive for factor XIIIa. CD34 staining is negative.
Dermatofibrosarcoma protuberans usually appears as one or multiple firm, red to violaceous nodules or plaques. They most often occur on the trunk in middle-aged adults. Histopathologically, DFSP presents with a dense, hypercellular, spindle cell proliferation that demonstrates a typical storiform pattern. The tumor generally infiltrates into the deep dermis and subcutaneous adipose layer with characteristic adipocyte entrapment (Figure 3). Positive CD34 and negative factor XIIIa staining helps to differentiate DFSP from a cellular dermatofibroma. Immunohistochemically, it is more difficult to distinguish DFSP from SFT, as both are CD34+ spindle cell neoplasms that also stain positive for CD99 and BCL-2.2 GRIA2 positivity also is seen in both SFT and DFSP.7 However, differentiation can be made on morphologic grounds alone, as DFSP has ill-defined tumor borders with adnexal and fat entrapment and SFT tends to be more circumscribed with prominent arborizing hyalinized vessels.8
Spindle cell lipoma (SCL) is an asymptomatic subcutaneous tumor commonly located on the back, neck, and shoulders in older patients, typically men. It often presents as a solitary lesion, though multiple lesions may occur. It is a well-circumscribed tumor of mature adipose tissue with areas of spindle cell proliferation and ropey collagen bundles (Figure 4). In early lesions, the spindle cell areas are myxoid with the presence of many mast cells.9 The spindle cells stain positive for CD34. Although spindle cell lipoma would be included in both the clinical and histopathologic differential diagnosis for SFT, its histopathologic features often are enough to differentiate SCL, which is highlighted by the aforementioned features as well as a relatively low cellularity and lack of ectatic vessels.8 However, discerning tumor variants, such as low-fat pseudoangiomatous SCL and lipomatous or myxoid SFT, might prove more challenging.
Nodular fasciitis typically presents as a rapidly growing subcutaneous nodule that may be tender. It is a benign reactive process usually affecting the arms and trunk of young to middle-aged adults, though it commonly involves the head and neck region in children.10 The tumor histopathologically appears as a well-circumscribed subcutaneous or fascial nodule with an angulated appearance. Spindle-shaped and stellate fibroblasts are loosely arranged in an edematous myxomatous stroma with a feathered appearance (Figure 5). Extravasated erythrocytes often are present. With time, collagen bundles become thicker and hyalinized. Immunohistochemical studies demonstrate positivity for vimentin, calponin, muscle-specific actin, and smooth muscle actin. Desmin, CD34, cytokeratin, and S-100 typically are negative.10-12 Therefore, CD34 staining is one of the main differentiating factors between nodular fasciitis and SFTs.
The Diagnosis: Solitary Fibrous Tumor
Solitary fibrous tumors (SFTs), as first described by Klemperer and Rabin1 in 1931, are relatively uncommon mesenchymal neoplasms that occur primarily in the pleura. This lesion is now known to affect many other extrathoracic sites, such as the liver, kidney, adrenal glands, thyroid, central nervous system, and soft tissue, with rare examples originating from the skin.2 Okamura et al3 reported the first known case of cutaneous SFT in 1997, with most of the literature limited to case reports. Erdag et al2 described one of the largest case series of primary cutaneous SFTs. These lesions can occur across a wide age range but tend to primarily affect middle-aged adults. Solitary fibrous tumors have been known to have no sex predilection; however, Erdag et al2 found a male predominance with a male to female ratio of 4 to 1.
Histopathologically, a cutaneous SFT is known to appear as a well-circumscribed nodular spindle cell proliferation arranged in interlacing fascicles with an abundant hyalinized collagen stroma (quiz image). Alternating hypocellular and hypercellular areas can be seen. Supporting vasculature often is relatively prominent, represented by angulated and branching staghorn blood vessels (Figure 1).2 A common histopathologic finding of SFTs is a patternless pattern, which suggests that the tumor can have a variety of morphologic appearances (eg, storiform, fascicular, neural, herringbone growth patterns), making histologic diagnosis difficult (quiz image).4 Therefore, immunohistochemistry plays a large role in the diagnosis of this tumor. The most important positive markers include CD34, CD99, B-cell lymphoma 2 (BCL-2), and signal transducer and activator of transcription 6 (STAT6).5 Nuclear STAT6 staining is an immunomarker for NGFI-A binding protein 2 (NAB2)-STAT6 gene fusion, which is specific for SFT.5,6 Vivero et al7 also reported glutamate receptor, inotropic, AMPA 2 (GRIA2) as a useful immunostain in SFT, though it is also expressed in dermatofibrosarcoma protuberans (DFSP). In this case, the clinical and histopathologic findings best supported a diagnosis of SFT. Some consider hemangiopericytomas to be examples of SFTs; however, true hemangiopericytomas lack the thick hyalinized collagen and hypercellular areas seen in SFT.
A cellular dermatofibroma generally presents as a single round, reddish brown papule or nodule approximately 0.5 to 1 cm in diameter that is firm to palpation with a central depression or dimple created over the lesion from the lateral pressure. Cellular dermatofibromas mostly occur in middle-aged adults, with the most common locations on the legs and on the sides of the trunk. They are thought to arise after injuries to the skin. On histopathologic examination, cellular dermatofibromas typically exhibit a proliferation of fibrohistiocytic cells with collagen trapping, often at the periphery of the tumor (Figure 2). Although cellular dermatofibromas appear clinically different than SFTs, they often mimic SFTs histopathologically. Immunostaining also can be helpful in differentiating cellular dermatofibromas in which cells stain positive for factor XIIIa. CD34 staining is negative.
Dermatofibrosarcoma protuberans usually appears as one or multiple firm, red to violaceous nodules or plaques. They most often occur on the trunk in middle-aged adults. Histopathologically, DFSP presents with a dense, hypercellular, spindle cell proliferation that demonstrates a typical storiform pattern. The tumor generally infiltrates into the deep dermis and subcutaneous adipose layer with characteristic adipocyte entrapment (Figure 3). Positive CD34 and negative factor XIIIa staining helps to differentiate DFSP from a cellular dermatofibroma. Immunohistochemically, it is more difficult to distinguish DFSP from SFT, as both are CD34+ spindle cell neoplasms that also stain positive for CD99 and BCL-2.2 GRIA2 positivity also is seen in both SFT and DFSP.7 However, differentiation can be made on morphologic grounds alone, as DFSP has ill-defined tumor borders with adnexal and fat entrapment and SFT tends to be more circumscribed with prominent arborizing hyalinized vessels.8
Spindle cell lipoma (SCL) is an asymptomatic subcutaneous tumor commonly located on the back, neck, and shoulders in older patients, typically men. It often presents as a solitary lesion, though multiple lesions may occur. It is a well-circumscribed tumor of mature adipose tissue with areas of spindle cell proliferation and ropey collagen bundles (Figure 4). In early lesions, the spindle cell areas are myxoid with the presence of many mast cells.9 The spindle cells stain positive for CD34. Although spindle cell lipoma would be included in both the clinical and histopathologic differential diagnosis for SFT, its histopathologic features often are enough to differentiate SCL, which is highlighted by the aforementioned features as well as a relatively low cellularity and lack of ectatic vessels.8 However, discerning tumor variants, such as low-fat pseudoangiomatous SCL and lipomatous or myxoid SFT, might prove more challenging.
Nodular fasciitis typically presents as a rapidly growing subcutaneous nodule that may be tender. It is a benign reactive process usually affecting the arms and trunk of young to middle-aged adults, though it commonly involves the head and neck region in children.10 The tumor histopathologically appears as a well-circumscribed subcutaneous or fascial nodule with an angulated appearance. Spindle-shaped and stellate fibroblasts are loosely arranged in an edematous myxomatous stroma with a feathered appearance (Figure 5). Extravasated erythrocytes often are present. With time, collagen bundles become thicker and hyalinized. Immunohistochemical studies demonstrate positivity for vimentin, calponin, muscle-specific actin, and smooth muscle actin. Desmin, CD34, cytokeratin, and S-100 typically are negative.10-12 Therefore, CD34 staining is one of the main differentiating factors between nodular fasciitis and SFTs.
- Klemperer P, Rabin CB. Primary neoplasms of the pleura: a report of five cases. Arch Pathol. 1931;11:385-412.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Okamura JM, Barr RJ, Battifora H. Solitary fibrous tumor of the skin. Am J Dermatopathol. 1997;19:515-518.
- Lee JY, Park SE, Shin SJ, et al. Solitary fibrous tumor with myxoid stromal change. Am J Dermatopathol. 2015;37:570-573.
- Geramizadeh B, Marzban M, Churg A. Role of immunohistochemistry in the diagnosis of solitary fibrous tumor, a review. Iran J Pathol. 2016;11:195-293.
- Creytens D, Ferdinande L, Dorpe JV. Histopathologically malignant solitary fibrous tumor of the skin: a report of an unusual case. J Cutan Pathol. 2016;43:629-631.
- Vivero M, Doyle LA, Fletcher CD, et al. GRIA2 is a novel diagnostic marker for solitary fibrous tumour identified through gene expression profiling. Histopathology. 2014;65:71-80.
- Wood L, Fountaine TJ, Rosamilia L, et al. Cutaneous CD34 spindle cell neoplasms: histopathologic features distinguish spindle cell lipoma, solitary fibrous tumor, and dermatofibrosarcoma protuberans. Am J Dermatopathol. 2010;32:764-768.
- Khatib Y, Khade AL, Shah VB, et al. Cytohistological features of spindle cell lipoma--a case report with differential diagnosis. J Clin Diagn Res. 2017;11:10-11.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Bracey TS, Wharton S, Smith ME. Nodular 'fasciitis' presenting as a cutaneous polyp. J Cutan Pathol. 2009;36:980-982.
- Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
- Klemperer P, Rabin CB. Primary neoplasms of the pleura: a report of five cases. Arch Pathol. 1931;11:385-412.
- Erdag G, Qureshi HS, Patterson JW, et al. Solitary fibrous tumors of the skin: a clinicopathologic study of 10 cases and review of the literature. J Cutan Pathol. 2007;34:844-850.
- Okamura JM, Barr RJ, Battifora H. Solitary fibrous tumor of the skin. Am J Dermatopathol. 1997;19:515-518.
- Lee JY, Park SE, Shin SJ, et al. Solitary fibrous tumor with myxoid stromal change. Am J Dermatopathol. 2015;37:570-573.
- Geramizadeh B, Marzban M, Churg A. Role of immunohistochemistry in the diagnosis of solitary fibrous tumor, a review. Iran J Pathol. 2016;11:195-293.
- Creytens D, Ferdinande L, Dorpe JV. Histopathologically malignant solitary fibrous tumor of the skin: a report of an unusual case. J Cutan Pathol. 2016;43:629-631.
- Vivero M, Doyle LA, Fletcher CD, et al. GRIA2 is a novel diagnostic marker for solitary fibrous tumour identified through gene expression profiling. Histopathology. 2014;65:71-80.
- Wood L, Fountaine TJ, Rosamilia L, et al. Cutaneous CD34 spindle cell neoplasms: histopathologic features distinguish spindle cell lipoma, solitary fibrous tumor, and dermatofibrosarcoma protuberans. Am J Dermatopathol. 2010;32:764-768.
- Khatib Y, Khade AL, Shah VB, et al. Cytohistological features of spindle cell lipoma--a case report with differential diagnosis. J Clin Diagn Res. 2017;11:10-11.
- Kumar E, Patel NR, Demicco EG, et al. Cutaneous nodular fasciitis with genetic analysis: a case series. J Cutan Pathol. 2016;43:1143-1149.
- Bracey TS, Wharton S, Smith ME. Nodular 'fasciitis' presenting as a cutaneous polyp. J Cutan Pathol. 2009;36:980-982.
- Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
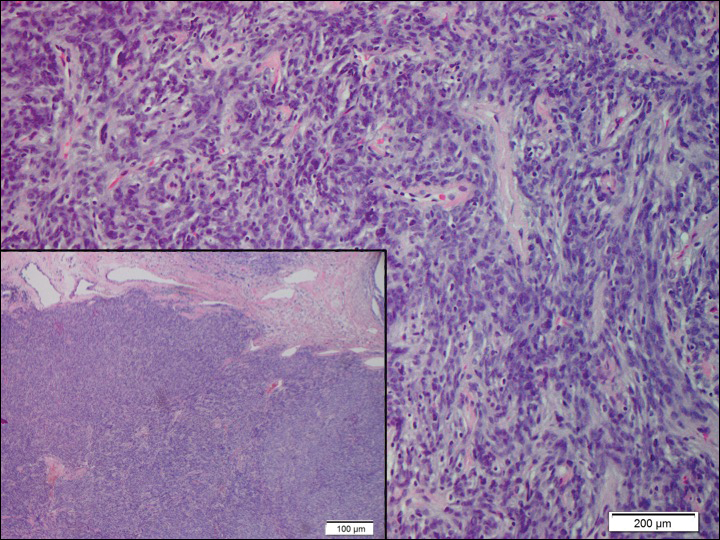
A 73-year-old man presented with a tender nodule on the back that had recently increased in size. On physical examination, a solitary 4-cm nodule was noted in the right trapezius region. The patient denied any personal or family history of similar lesions or a penchant for cysts. Due to the symptomatic nature of the lesion, surgical excision was performed.
Black Eschars on the Face and Body
The Diagnosis: Lymphomatoid Papulosis
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
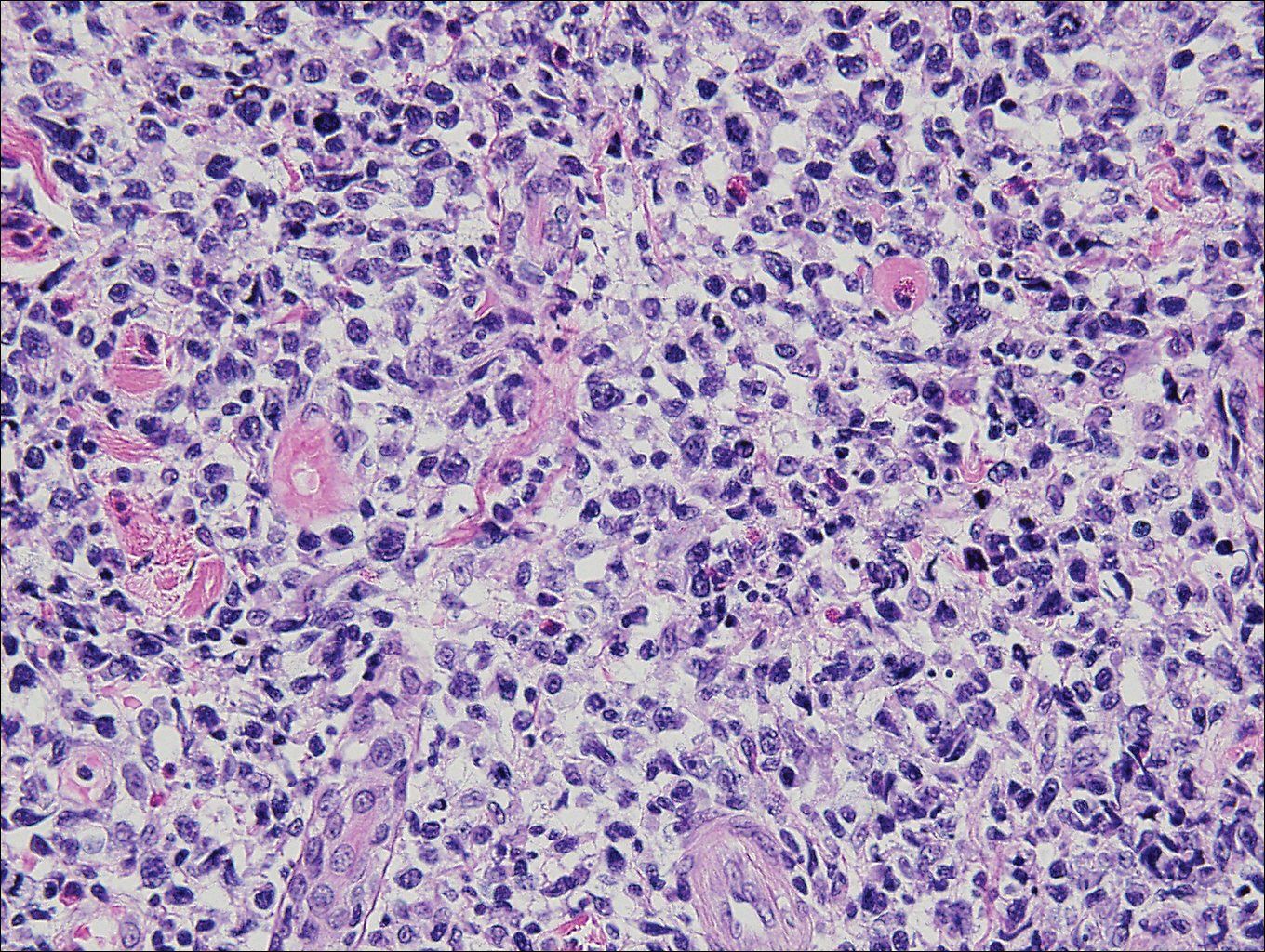
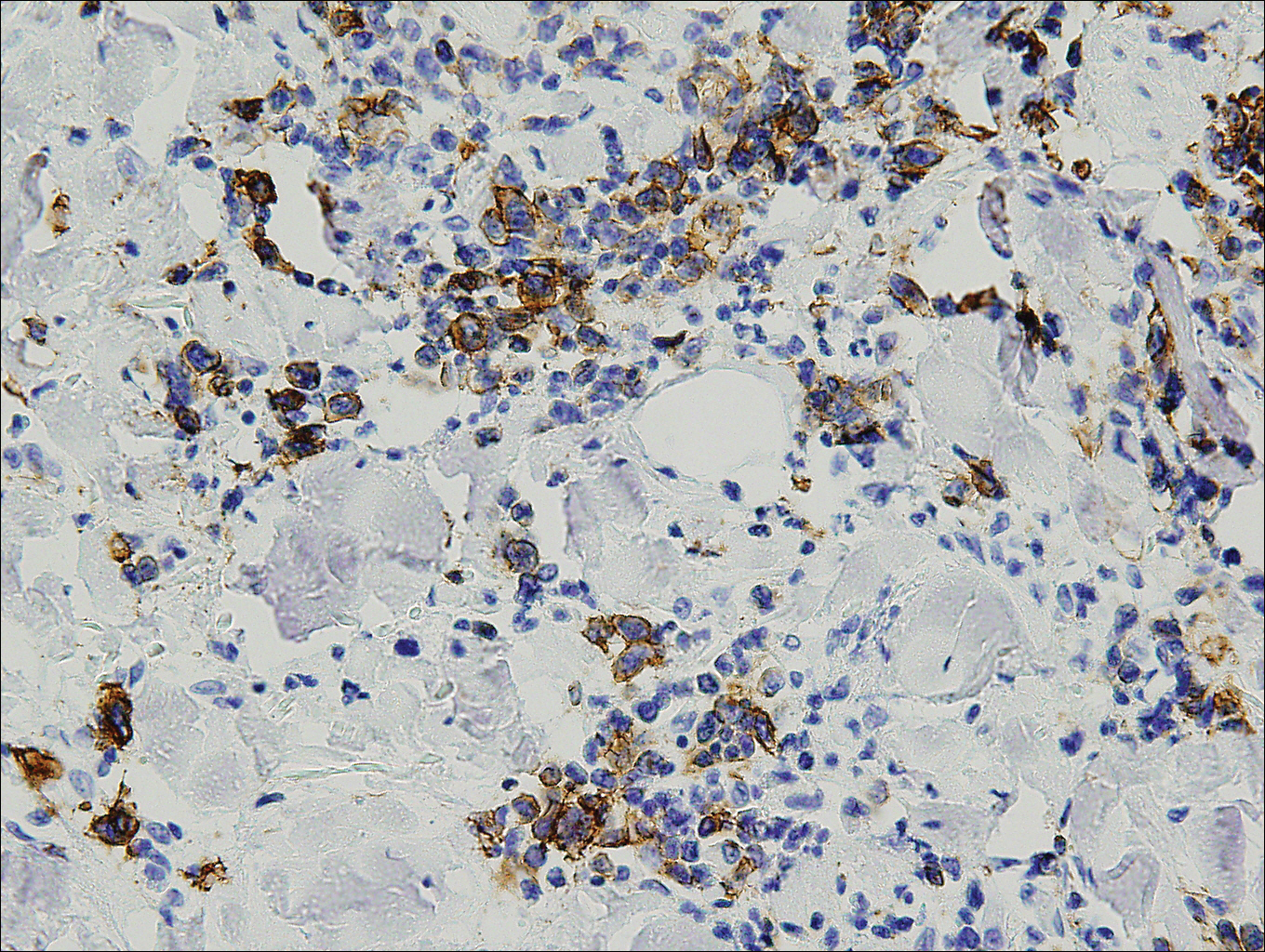
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
The Diagnosis: Lymphomatoid Papulosis
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
The Diagnosis: Lymphomatoid Papulosis
Histopathologic and immunohistochemical examination of the ulcer revealed a dense nodular and diffuse infiltrate in the papillary and reticular dermis comprised predominantly of atypical, CD30+, small T cells and large lymphoid cells admixed with neutrophils and eosinophils (Figures 1 and 2). Tissue cultures and infectious stains were negative. The complete blood cell count, metabolic panel, serum lactate dehydrogenase level, and peripheral blood flow cytometry were normal. Correlation of the lesions' self-healing nature with the histopathologic and immunohistochemical findings led to a diagnosis of lymphomatoid papulosis (LyP). In light of this diagnosis, a shave biopsy was obtained of one of the patient's poikilodermatous patches and was found to be consistent with poikilodermatous mycosis fungoides (MF).
At 4-month follow-up, the patient reported that she continued to develop crops of 1 to 3 LyP lesions each month. She continued to deny systemic concerns, and the poikilodermatous MF appeared unchanged. As part of a hematologic workup, a positron emission tomography-computed tomography scan revealed glucose-avid lymph nodes in the axillary, supraclavicular, abdominal, and inguinal regions. These findings raised concern for possible lymphomatous involvement of the patient's MF. Systemic therapy may be required pending further surveillance.
Lymphomatoid papulosis is a chronic papulonecrotic disease characterized clinically by recurrent crops of self-healing papules. Histopathologically, LyP features a perivascular infiltrate with atypical dermal T cells. Macaulay1 first described LyP in 1968 in a 41-year-old woman with a several-year history of continuously self-resolving crops of necrotic papules, noting the paradox between the patient's benign clinical course and malignant-seeming histology featuring "an alarming infiltrate of anaplastic cells." Since this report, LyP has continued to spur debate regarding its malignant potential but is now recognized as an indolent cutaneous T-cell lymphoma with an excellent prognosis.2
There are several histopathologic subtypes of LyP, the most common of which are type A, resembling Hodgkin lymphoma; type B, resembling MF; type C, resembling primary cutaneous anaplastic large cell lymphoma (C-ALCL); and type D, resembling aggressive epidermotropic CD8+ cutaneous T-cell lymphoma.2
The multifocal ulcers and eschars of LyP may appropriately raise suspicion for an infectious process, as in the present case. Numerous reports show that LyP may be initially misdiagnosed as an infection, such as cellulitis,3 furunculosis,4 parapoxvirus Orf,5 and ecthyma.6 Furthermore, several cutaneous infections have histopathologic features indistinguishable from LyP.7 For example, herpes simplex virus infection, molluscum contagiosum, Milker nodule, syphilis, and leishmaniasis may contain an appreciable number of large CD30+ T cells, which is compatible with both LyP type C and C-ALCL.7 As in the present case, the final diagnosis rests on clinicopathologic correlation, with LyP often distinguished by its invariable self-resolution, unlike its numerous infectious mimickers. The self-regressing nature of LyP also helps differentiate LyP occurring in the setting of MF from MF that has underwent CD30+ large cell transformation. In addition, the diagnosis of MF-associated LyP is favored over transformed MF when, as in the present case, CD30+ lesions develop on skin distinct from MF-affected skin.
Although isolated LyP is benign, 18% (11/61) of patients will subsequently develop lymphoma. More commonly, lymphomas may precede or occur concomitantly with the onset of LyP. In a retrospective study of 84 LyP patients, for example, 40% (34/84) had prior or concomitant lymphoma.8 Owing to the well-established link between LyP and lymphoma, there is appropriate emphasis on close monitoring of these patients. In addition, a careful history and physical examination are necessary to evaluate for a preceding, previously undiagnosed lymphoma. In point of fact, our patient had undiagnosed poikilodermatous MF prior to developing LyP, which was proven by biopsy at the time of LyP diagnosis. A distinct clinical variant of MF, poikilodermatous MF is characterized by hyperpigmented and hypopigmented patches, atrophy, and telangiectasia. A study of 49 patients with poikilodermatous MF found that this variant had an earlier age of onset compared with other types of MF. The study also showed that 18% (9/49) of patients had coexistent LyP, suggesting that poikilodermatous MF and LyP may be more frequently associated than previously believed.9
Treatment of LyP is unnecessary beyond basic wound care to avoid bacterial superinfection.2,10 Therapy for poikilodermatous MF, similar to other types of MF, is based on disease stage. Topical therapy may be utilized for localized disease, while systemic therapies are reserved for recalcitrant cases and internal involvement.9
Acknowledgments
We thank David L. Ramsay, MD, for obtaining aspects of the patient's history, and Shane A. Meehan, MD, and Adnan Mir, MD, PhD, as well as Cynthia M. Magro, MD, (all from New York, New York) for performing the histopathologic and immunohistochemical analyses.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
- Macaulay WL. Lymphomatoid papulosis. a continuing self-healing eruption, clinically benign--histologically malignant. Arch Dermatol. 1968;97:23-30.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Meena M, Martin PA, Abouseif C, et al. Lymphomatoid papulosis type C of the eyelid in a young girl: a case report and review of literature. Orbit. 2014;3:395-398.
- Dinotta F, Lacarrubba F, Micali G. Sixteen-year-old girl with papules and nodules on the face and upper limbs. Pediatr Dermatol. 2014;31:103-104.
- Eminger LA, Shinohara MM, Kim EJ, et al. Clinicopathologic challenge: acral lymphomatoid papulosis. Int J Dermatol. 2012;51:531-534.
- Harder D, Kuhn A, Mahrle G. Lymphomatoid papulosis resembling ecthyma. a case report. Z Hautkr. 1989;64:593-595.
- Werner B, Massone C, Kerl H, et al. Large CD30-positive cells in benign, atypical lymphoid infiltrates of the skin. J Cutan Pathol. 2008;35:1100-1107.
- Kunishige JH, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-581.
- Abbott RA, Sahni D, Robson A, et al. Poikilodermatous mycosis fungoides: a study of its clinicopathological, immunophenotypic, and prognostic features. J Am Acad Dermatol. 2011;65:313-319.
- Kempf W, Pfaltz K, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011;118:4024-4035.
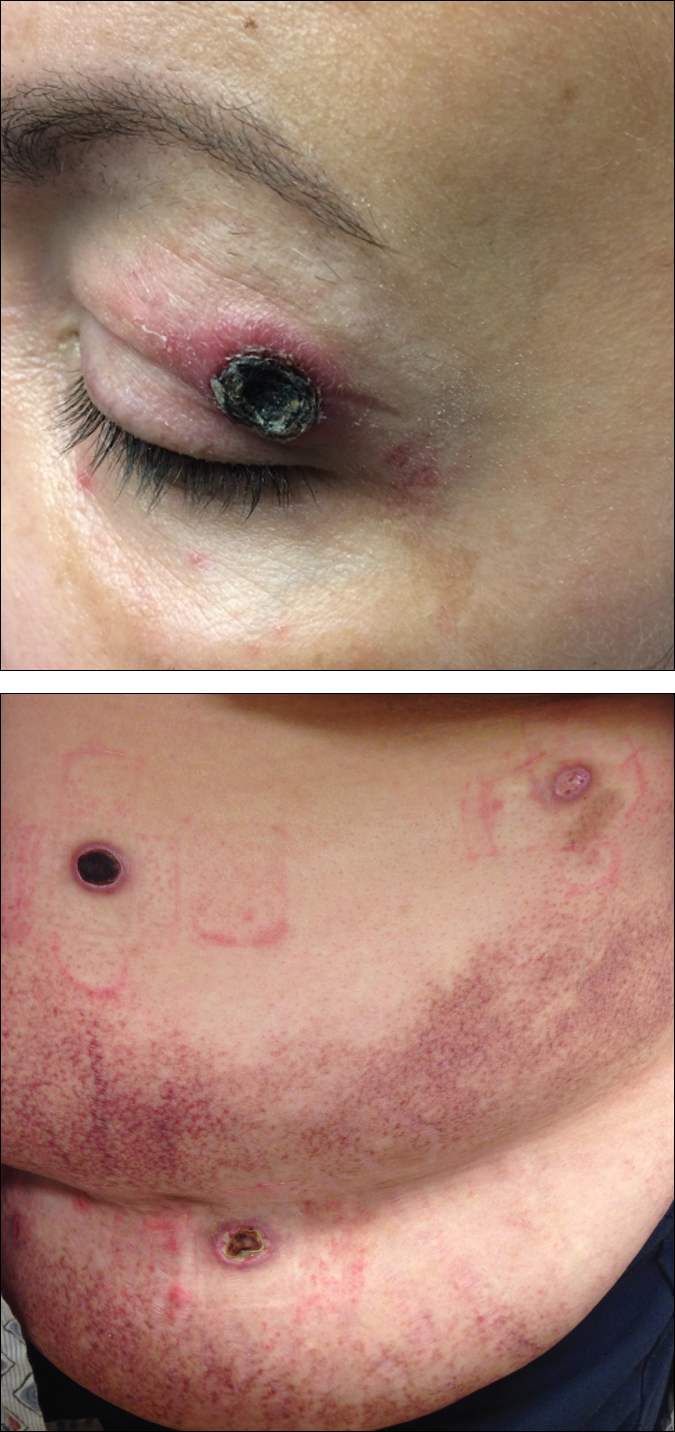
A 50-year-old woman presented for evaluation of black eschars on the face and body. Over the preceding 8 weeks she had developed several asymptomatic papules that gradually enlarged, ulcerated, and formed a black eschar, prior to gradually self-resolving over the course of several weeks. During this time, new lesions were forming. The resulting skin revealed dyspigmentation and scar formation. Prior to presentation, antimicrobial therapy had been initiated for a presumed infectious etiology; however, the eruption continued to progress. The patient denied sick contacts, livestock exposure, or recent travel. A complete review of systems, including fever, chills, or lymphadenopathy, was negative. Physical examination revealed 6 circular necrotic ulcers with an overlying black eschar on the face (top), trunk (bottom), hands, and thighs, all in various stages of healing. In addition, large, reticulated, poikilodermatous patches were incidentally noted in areas free of ulcers and eschars on the trunk (bottom) and bilateral arms and legs. Upon questioning, the patient said these patches had been present for more than 30 years. A punch biopsy from an ulcer on the chest was obtained and sent for histopathologic and immunohistochemical examination.
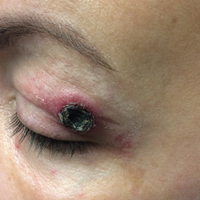
Chromoblastomycosis Infection From a House Plant
To the Editor:
A 69-year-old woman with no history of immunodeficiency presented 1 month after a thorn from her locally grown Madagascar palm plant (Pachypodium lamerei) pierced the skin. The patient developed a painful nodule at the site on the left elbow (Figure 1). An excisional biopsy by an outside dermatologist was performed, which showed granulomatous inflammation within the dermis with epidermal hyperplasia and the presence of golden brown spherules (medlar bodies). The diagnosis was a dermal fungal infection consistent with chromoblastomycosis. A curative surgical excision was performed, and medlar bodies were seen adjacent to a polarizable foreign body consistent with plant material on histology (Figure 2). Because the lesion was localized, adjuvant medical treatment was not deemed necessary. The patient has not had any recurrence in the last 1.5 years since the resection.
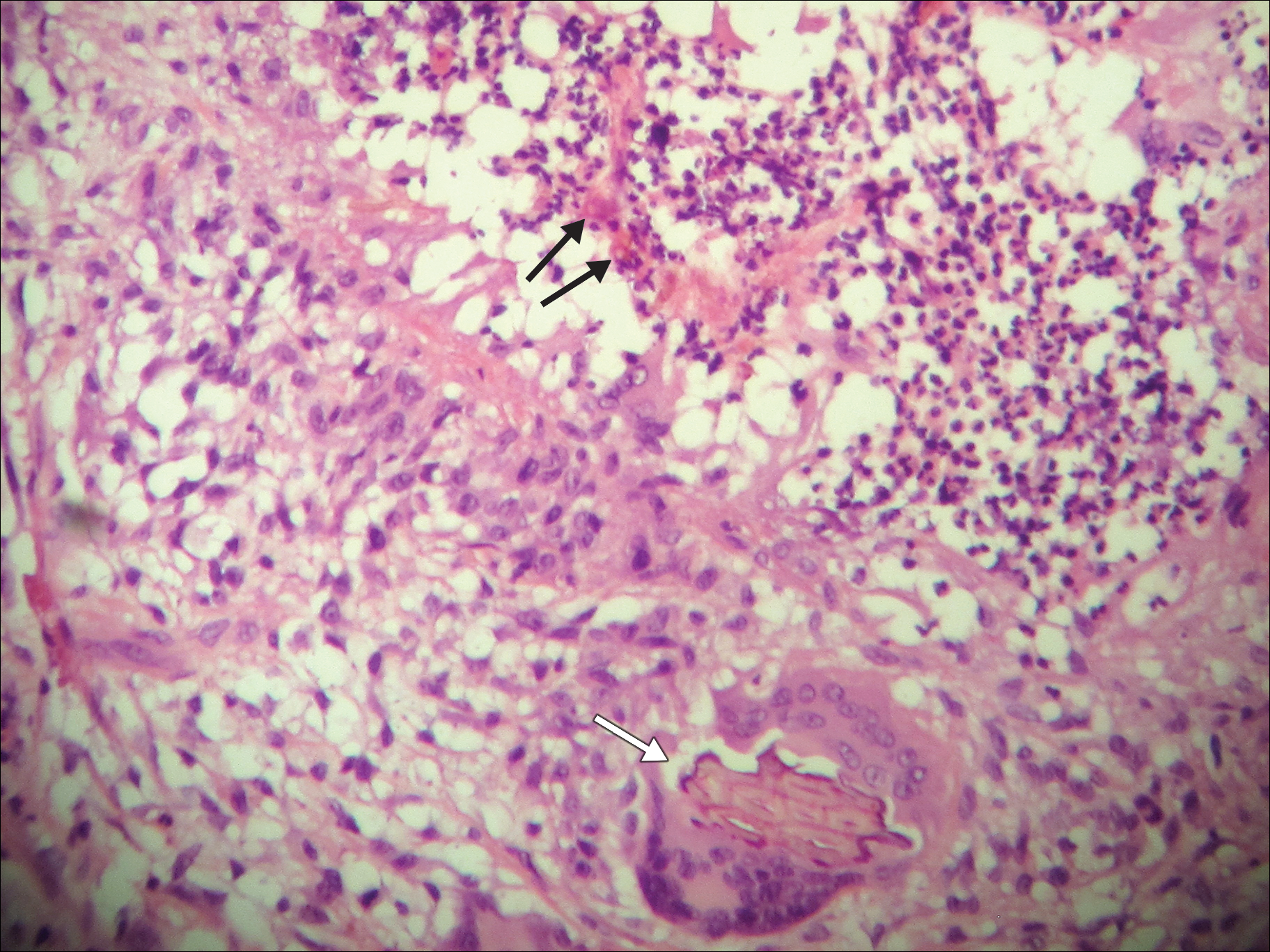
The categorization of chromoblastomycosis includes a chronic fungal infection of the cutaneous and subcutaneous tissues by dematiaceous (pigmented) fungi. This definition is such that there are a multitude of organisms that can be the primary cause of this diagnosis. Generally, infection follows a traumatic permeation of the skin by a foreign body contaminated by the causative organism in agricultural workers. The most common dematiaceous pathogens are Fonsecaea pedrosoi, Phialophora verrucosa, and Cladosporium carrionii; however, the specific causative organism varies heavily on geographic location. With inoculation by a foreign body, a small papule develops at the site of the lesion. Several years after the primary infection, nodules and verrucous erythematous plaques develop in the same area, and patients present with concerns of pain and pruritus.1 Lesions usually are localized to the initial area of inoculation, generally a break in the skin by the offending foreign body, on the legs, arms, or hands, but hematogenous or lymphatic dissemination with distant transmission due to scratching also can occur. Ulceration due to secondary bacterial infection is another possible manifestation, resulting in a foul odor and less commonly lymphedema. Rarely, squamous cell carcinoma is a complication.2
RELATED ARTICLE: Fungal Foes: Presentations of Chromoblastomycosis Post–Hurricane Ike
On histopathology, thick-walled sclerotic bodies termed medlar bodies or copper pennies are pathognomonic for chromoblastomycosis and represent the fungal elements. Grossly, black dots can be seen on the skin in the affected areas from the transepidermal elimination of the fungi.1,2 However, there is no specificity for determining the causative organism in this manner, or even with culture, as it is difficult to differentiate the species morphologically. More advanced tests can help, such as polymerase chain reaction or enzyme-linked immunosorbent assay, where available.2 Hematoxylin and eosin stain also shows epidermal hyperplasia and dermal mononuclear infiltrate.
Treatment modalities include surgical excision, cryotherapy, pharmacologic treatment, and combination therapy. Localized lesions often can be resected, but more severe infections can require pharmacologic treatment. Unfortunately, there tends to be a high risk for relapse with most antifungal modalities. The combination of itraconazole and terbinafine has been shown to offer the best medical therapy with lower risk for refractoriness to treatment by producing a synergistic effect between the 2 antifungals.2,3 Many surgical treatments often are combined with oral antifungals to try to attain complete eradication in deep or extensive lesions, as seen in a case in which oral terbinafine was used prior to surgery to reduce the size of the lesion, followed by complete resection.4 With localized lesions that are resectable, a wide and deep incision often can be curative. Cryotherapy also may be coupled with surgical excision or pharmacologic therapy. Most literature suggests that cryotherapy or the use of antifungals prior to excision offers improved outcomes.2,5 Prognosis tends to be good for chromoblastomycoses, particularly with smaller lesions. Complete eradication varies greatly on the size and depth of the lesion, independent of the causative pathogen.
Our patient’s presentation with chromoblastomycosis is unique because of the source of infection, which was a plant grown from seeds in a local nursery in South Florida and then sold to the patient. The majority of chromoblastomycosis infections occur in agricultural workers, typically in tropical climates such as South and Central America, the Caribbean, and Mexico.1,2 Historically, infections in the United States have been uncommon, with the majority presenting in patients on prolonged corticosteroid therapy or with other immunosuppressive conditions.6,7
- Torres-Guerrero E, Isa-Isa R, Isa M, et al. Chromoblastomycosis. Clin Dermatol. 2012;30:403-408.
- Ameen M. Managing chromoblastomycosis. Trop Doct. 2010;40:65-67.
- Zhang J, Xi L, Lu C, et al. Successful treatment for chromoblastomycosis caused by Fonsecaea monophora: a report of three cases in Guangdong, China. Mycoses. 2009;52:176-181.
- Tamura K, Matsuyama T, Yahagi E, et al. A case of chromomycosis treated by surgical therapy combined with preceded oral administration of terbinafine to reduce the size of the lesion. Tokai J Exp Clin Med. 2012;37:6-10.
- Patel U, Chu J, Patel R, et al. Subcutaneous dematiaceous fungal infection. Dermatol Online J. 2011;17:19.
- Basílio FM, Hammerschmidt M, Mukai MM, et al. Mucormycosis and chromoblastomycosis occurring in a patient with leprosy type 2 reaction under prolonged corticosteroid and thalidomide therapy. An Bras Dermatol. 2012;87:767-771.
- Parente JN, Talhari C, Ginter-Hanselmayer G, et al. Subcutaneous phaeohyphomycosis in immunocompetent patients: two new cases caused by Exophiala jeanselmei and Cladophialophora carrionii. Mycoses. 2001;54:265-269.
To the Editor:
A 69-year-old woman with no history of immunodeficiency presented 1 month after a thorn from her locally grown Madagascar palm plant (Pachypodium lamerei) pierced the skin. The patient developed a painful nodule at the site on the left elbow (Figure 1). An excisional biopsy by an outside dermatologist was performed, which showed granulomatous inflammation within the dermis with epidermal hyperplasia and the presence of golden brown spherules (medlar bodies). The diagnosis was a dermal fungal infection consistent with chromoblastomycosis. A curative surgical excision was performed, and medlar bodies were seen adjacent to a polarizable foreign body consistent with plant material on histology (Figure 2). Because the lesion was localized, adjuvant medical treatment was not deemed necessary. The patient has not had any recurrence in the last 1.5 years since the resection.
The categorization of chromoblastomycosis includes a chronic fungal infection of the cutaneous and subcutaneous tissues by dematiaceous (pigmented) fungi. This definition is such that there are a multitude of organisms that can be the primary cause of this diagnosis. Generally, infection follows a traumatic permeation of the skin by a foreign body contaminated by the causative organism in agricultural workers. The most common dematiaceous pathogens are Fonsecaea pedrosoi, Phialophora verrucosa, and Cladosporium carrionii; however, the specific causative organism varies heavily on geographic location. With inoculation by a foreign body, a small papule develops at the site of the lesion. Several years after the primary infection, nodules and verrucous erythematous plaques develop in the same area, and patients present with concerns of pain and pruritus.1 Lesions usually are localized to the initial area of inoculation, generally a break in the skin by the offending foreign body, on the legs, arms, or hands, but hematogenous or lymphatic dissemination with distant transmission due to scratching also can occur. Ulceration due to secondary bacterial infection is another possible manifestation, resulting in a foul odor and less commonly lymphedema. Rarely, squamous cell carcinoma is a complication.2
RELATED ARTICLE: Fungal Foes: Presentations of Chromoblastomycosis Post–Hurricane Ike
On histopathology, thick-walled sclerotic bodies termed medlar bodies or copper pennies are pathognomonic for chromoblastomycosis and represent the fungal elements. Grossly, black dots can be seen on the skin in the affected areas from the transepidermal elimination of the fungi.1,2 However, there is no specificity for determining the causative organism in this manner, or even with culture, as it is difficult to differentiate the species morphologically. More advanced tests can help, such as polymerase chain reaction or enzyme-linked immunosorbent assay, where available.2 Hematoxylin and eosin stain also shows epidermal hyperplasia and dermal mononuclear infiltrate.
Treatment modalities include surgical excision, cryotherapy, pharmacologic treatment, and combination therapy. Localized lesions often can be resected, but more severe infections can require pharmacologic treatment. Unfortunately, there tends to be a high risk for relapse with most antifungal modalities. The combination of itraconazole and terbinafine has been shown to offer the best medical therapy with lower risk for refractoriness to treatment by producing a synergistic effect between the 2 antifungals.2,3 Many surgical treatments often are combined with oral antifungals to try to attain complete eradication in deep or extensive lesions, as seen in a case in which oral terbinafine was used prior to surgery to reduce the size of the lesion, followed by complete resection.4 With localized lesions that are resectable, a wide and deep incision often can be curative. Cryotherapy also may be coupled with surgical excision or pharmacologic therapy. Most literature suggests that cryotherapy or the use of antifungals prior to excision offers improved outcomes.2,5 Prognosis tends to be good for chromoblastomycoses, particularly with smaller lesions. Complete eradication varies greatly on the size and depth of the lesion, independent of the causative pathogen.
Our patient’s presentation with chromoblastomycosis is unique because of the source of infection, which was a plant grown from seeds in a local nursery in South Florida and then sold to the patient. The majority of chromoblastomycosis infections occur in agricultural workers, typically in tropical climates such as South and Central America, the Caribbean, and Mexico.1,2 Historically, infections in the United States have been uncommon, with the majority presenting in patients on prolonged corticosteroid therapy or with other immunosuppressive conditions.6,7
To the Editor:
A 69-year-old woman with no history of immunodeficiency presented 1 month after a thorn from her locally grown Madagascar palm plant (Pachypodium lamerei) pierced the skin. The patient developed a painful nodule at the site on the left elbow (Figure 1). An excisional biopsy by an outside dermatologist was performed, which showed granulomatous inflammation within the dermis with epidermal hyperplasia and the presence of golden brown spherules (medlar bodies). The diagnosis was a dermal fungal infection consistent with chromoblastomycosis. A curative surgical excision was performed, and medlar bodies were seen adjacent to a polarizable foreign body consistent with plant material on histology (Figure 2). Because the lesion was localized, adjuvant medical treatment was not deemed necessary. The patient has not had any recurrence in the last 1.5 years since the resection.
The categorization of chromoblastomycosis includes a chronic fungal infection of the cutaneous and subcutaneous tissues by dematiaceous (pigmented) fungi. This definition is such that there are a multitude of organisms that can be the primary cause of this diagnosis. Generally, infection follows a traumatic permeation of the skin by a foreign body contaminated by the causative organism in agricultural workers. The most common dematiaceous pathogens are Fonsecaea pedrosoi, Phialophora verrucosa, and Cladosporium carrionii; however, the specific causative organism varies heavily on geographic location. With inoculation by a foreign body, a small papule develops at the site of the lesion. Several years after the primary infection, nodules and verrucous erythematous plaques develop in the same area, and patients present with concerns of pain and pruritus.1 Lesions usually are localized to the initial area of inoculation, generally a break in the skin by the offending foreign body, on the legs, arms, or hands, but hematogenous or lymphatic dissemination with distant transmission due to scratching also can occur. Ulceration due to secondary bacterial infection is another possible manifestation, resulting in a foul odor and less commonly lymphedema. Rarely, squamous cell carcinoma is a complication.2
RELATED ARTICLE: Fungal Foes: Presentations of Chromoblastomycosis Post–Hurricane Ike
On histopathology, thick-walled sclerotic bodies termed medlar bodies or copper pennies are pathognomonic for chromoblastomycosis and represent the fungal elements. Grossly, black dots can be seen on the skin in the affected areas from the transepidermal elimination of the fungi.1,2 However, there is no specificity for determining the causative organism in this manner, or even with culture, as it is difficult to differentiate the species morphologically. More advanced tests can help, such as polymerase chain reaction or enzyme-linked immunosorbent assay, where available.2 Hematoxylin and eosin stain also shows epidermal hyperplasia and dermal mononuclear infiltrate.
Treatment modalities include surgical excision, cryotherapy, pharmacologic treatment, and combination therapy. Localized lesions often can be resected, but more severe infections can require pharmacologic treatment. Unfortunately, there tends to be a high risk for relapse with most antifungal modalities. The combination of itraconazole and terbinafine has been shown to offer the best medical therapy with lower risk for refractoriness to treatment by producing a synergistic effect between the 2 antifungals.2,3 Many surgical treatments often are combined with oral antifungals to try to attain complete eradication in deep or extensive lesions, as seen in a case in which oral terbinafine was used prior to surgery to reduce the size of the lesion, followed by complete resection.4 With localized lesions that are resectable, a wide and deep incision often can be curative. Cryotherapy also may be coupled with surgical excision or pharmacologic therapy. Most literature suggests that cryotherapy or the use of antifungals prior to excision offers improved outcomes.2,5 Prognosis tends to be good for chromoblastomycoses, particularly with smaller lesions. Complete eradication varies greatly on the size and depth of the lesion, independent of the causative pathogen.
Our patient’s presentation with chromoblastomycosis is unique because of the source of infection, which was a plant grown from seeds in a local nursery in South Florida and then sold to the patient. The majority of chromoblastomycosis infections occur in agricultural workers, typically in tropical climates such as South and Central America, the Caribbean, and Mexico.1,2 Historically, infections in the United States have been uncommon, with the majority presenting in patients on prolonged corticosteroid therapy or with other immunosuppressive conditions.6,7
- Torres-Guerrero E, Isa-Isa R, Isa M, et al. Chromoblastomycosis. Clin Dermatol. 2012;30:403-408.
- Ameen M. Managing chromoblastomycosis. Trop Doct. 2010;40:65-67.
- Zhang J, Xi L, Lu C, et al. Successful treatment for chromoblastomycosis caused by Fonsecaea monophora: a report of three cases in Guangdong, China. Mycoses. 2009;52:176-181.
- Tamura K, Matsuyama T, Yahagi E, et al. A case of chromomycosis treated by surgical therapy combined with preceded oral administration of terbinafine to reduce the size of the lesion. Tokai J Exp Clin Med. 2012;37:6-10.
- Patel U, Chu J, Patel R, et al. Subcutaneous dematiaceous fungal infection. Dermatol Online J. 2011;17:19.
- Basílio FM, Hammerschmidt M, Mukai MM, et al. Mucormycosis and chromoblastomycosis occurring in a patient with leprosy type 2 reaction under prolonged corticosteroid and thalidomide therapy. An Bras Dermatol. 2012;87:767-771.
- Parente JN, Talhari C, Ginter-Hanselmayer G, et al. Subcutaneous phaeohyphomycosis in immunocompetent patients: two new cases caused by Exophiala jeanselmei and Cladophialophora carrionii. Mycoses. 2001;54:265-269.
- Torres-Guerrero E, Isa-Isa R, Isa M, et al. Chromoblastomycosis. Clin Dermatol. 2012;30:403-408.
- Ameen M. Managing chromoblastomycosis. Trop Doct. 2010;40:65-67.
- Zhang J, Xi L, Lu C, et al. Successful treatment for chromoblastomycosis caused by Fonsecaea monophora: a report of three cases in Guangdong, China. Mycoses. 2009;52:176-181.
- Tamura K, Matsuyama T, Yahagi E, et al. A case of chromomycosis treated by surgical therapy combined with preceded oral administration of terbinafine to reduce the size of the lesion. Tokai J Exp Clin Med. 2012;37:6-10.
- Patel U, Chu J, Patel R, et al. Subcutaneous dematiaceous fungal infection. Dermatol Online J. 2011;17:19.
- Basílio FM, Hammerschmidt M, Mukai MM, et al. Mucormycosis and chromoblastomycosis occurring in a patient with leprosy type 2 reaction under prolonged corticosteroid and thalidomide therapy. An Bras Dermatol. 2012;87:767-771.
- Parente JN, Talhari C, Ginter-Hanselmayer G, et al. Subcutaneous phaeohyphomycosis in immunocompetent patients: two new cases caused by Exophiala jeanselmei and Cladophialophora carrionii. Mycoses. 2001;54:265-269.
Practice Points
- Chromoblastomycosis is an uncommon fungal infection that should be considered in cases of traumatic injuries to the skin.
- Biopsies of growing or nonhealing nodules will demonstrate characteristic golden brown spherules (medlar bodies).
- In localized cases, surgical excision may be curative.
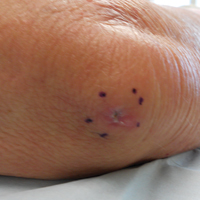
Verrucoid Lesion on the Eyelid
The Diagnosis: Inverted Follicular Keratosis
The differential diagnosis for endophytic squamous neoplasms encompasses benign and malignant entities. The histologic findings of our patient's lesion were compatible with the diagnosis of inverted follicular keratosis (IFK), a benign neoplasm that usually presents as a keratotic papule on the head or neck. Histologically, IFK is characterized by an endophytic growth pattern with squamous eddies (quiz images). Inverted follicular keratosis may represent an irritated seborrheic keratosis or a distinct neoplasm derived from the infundibular portion of the hair follicle; the exact etiology is uncertain.1,2 No relationship between IFK and human papillomavirus (HPV) has been established.3 Inverted follicular keratosis can mimic squamous cell carcinoma (SCC). Important clues to the diagnosis of IFK are the presence of squamous eddies and the lack of squamous pearls or cytologic atypia.4 Squamous eddies consist of whorled keratinocytes without keratinization or atypia. Superficial shave biopsies may fail to demonstrate the characteristic well-circumscribed architecture and may lead to an erroneous diagnosis.
Acantholytic SCC is characterized by atypical keratinocytes that have lost cohesive properties, resulting in acantholysis (Figure 1).5 This histologic variant was once categorized as an aggressive variant of SCC, but studies have failed to support this assertion.5,6 Acantholytic SCC has a discohesive nature producing a pseudoglandular appearance sometimes mistaken for adenosquamous carcinoma or metastatic carcinoma. Recent literature has suggested that acantholytic SCCs, similar to IFKs, are derived from the follicular infundibulum.5,6 Also similar to IFKs, acantholytic SCCs often are located on the face. The invasive architecture and atypical cytology of acantholytic SCCs can differentiate them from IFKs. Acantholytic SCCs can contain keratin pearls with concentric keratinocytes showing incomplete keratinization centrally, often with retained nuclei, but rare to no squamous eddies unless irritated.

Trichilemmoma is an endophytic benign neoplasm derived from the outer sheath of the pilosebaceous follicle characterized by lobules of clear cells hanging from the epidermis.7 A study investigating the relationship between HPV and trichilemmomas failed to definitively detect HPV in trichilemmomas and this relationship remains unclear.8 Desmoplastic trichilemmoma is a subtype histologically characterized by jagged islands of epithelial cells separated by dense pink stroma and encased in a glassy basement membrane (Figure 2). The presence of desmoplasia and a jagged growth pattern can mimic invasive SCC, but the absence of cytologic atypia and the surrounding basement membrane differs from SCC.4,7 Trichilemmomas typically are solitary, but multiple lesions are associated with Cowden syndrome. Cowden syndrome is a rare autosomal-dominant condition characterized by the presence of benign hamartomas and a predisposition to the development of malignancies including breast, endometrial, and thyroid cancers.9,10 There is no such association with desmoplastic trichilemmomas.11

Pilar sheath acanthoma is a benign neoplasm that clinically presents as a solitary flesh-colored nodule with a central pore containing keratin.12 Histologically, pilar sheath acanthoma is similar to a dilated pore of Winer with the addition of acanthotic epidermal projections (Figure 3).

Warty dyskeratoma (WD) is a benign endophytic neoplasm traditionally seen as a solitary lesion histologically similar to Darier disease. Warty dyskeratomas are known to occur both on the skin and oral mucosa.13 Histologically, WD is characterized as a cup-shaped lesion with numerous villi at the base of the lesion along with acantholysis and dyskeratosis (Figure 4). The dyskeratotic cells in WD consist of corps ronds, which are cells with abundant pink cytoplasm, and small nuclei along with grains, which are flattened basophilic cells. These dyskeratotic cells help differentiate WD from IFK. Although they are endophytic neoplasms, WDs are well circumscribed and should not be confused with SCC. Despite this entity's name and histologic similarity to verrucae, no relationship with HPV has been established.14
- Ruhoy SM, Thomas D, Nuovo GJ. Multiple inverted follicular keratoses as a presenting sign of Cowden's syndrome: case report with human papillomavirus studies. J Am Acad Dermatol. 2004;51:411-415.
- Lever WF. Inverted follicular keratosis is an irritated seborrheic keratosis. Am J Dermatopathol. 1983;5:474.
- Kambiz KH, Kaveh D, Maede D, et al. Human papillomavirus deoxyribonucleic acid may not be detected in non-genital benign papillomatous skin lesions by polymerase chain reaction. Indian J Dermatol. 2014;59:334-338.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- Ogawa T, Kiuru M, Konia TH, et al. Acantholytic squamous cell carcinoma is usually associated with hair follicles, not acantholytic actinic keratosis, and is not "high risk": diagnosis, management, and clinical outcomes in a series of 115 cases. J Am Acad Dermatol. 2017;76:327-333.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer staging guidelines, prognostic factors, and histopathologic variants. Adv Anat Pathol. 2017;24:171-194.
- Sano DT, Yang JJ, Tebcherani AJ, et al. A rare clinical presentation of desmoplastic trichilemmoma mimicking invasive carcinoma. An Bras Dermatol. 2014;89:796-798.
- Stierman S, Chen S, Nuovo G, et al. Detection of human papillomavirus infection in trichilemmomas and verrucae using in situ hybridization. J Cutan Pathol. 2010;37:75-80.
- Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol [published online October 22, 2014]. Methods. 2015;77-78:11-19.
- Molvi M, Sharma YK, Dash K. Cowden syndrome: case report, update and proposed diagnostic and surveillance routines. Indian J Dermatol. 2015;60:255-259.
- Jin M, Hampel H, Pilarski R, et al. Phosphatase and tensin homolog immunohistochemical staining and clinical criteria for Cowden syndrome in patients with trichilemmoma or associated lesions. Am J Dermatopathol. 2013;35:637-640.
- Mehregan AH, Brownstein MH. Pilar sheath acanthoma. Arch Dermatol. 1978;114:1495-1497.
- Newland JR, Leventon GS. Warty dyskeratoma of the oral mucosa. correlated light and electron microscopic study. Oral Surg Oral Med Oral Pathol. 1984;58:176-183.
- Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma--"follicular dyskeratoma": analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
The Diagnosis: Inverted Follicular Keratosis
The differential diagnosis for endophytic squamous neoplasms encompasses benign and malignant entities. The histologic findings of our patient's lesion were compatible with the diagnosis of inverted follicular keratosis (IFK), a benign neoplasm that usually presents as a keratotic papule on the head or neck. Histologically, IFK is characterized by an endophytic growth pattern with squamous eddies (quiz images). Inverted follicular keratosis may represent an irritated seborrheic keratosis or a distinct neoplasm derived from the infundibular portion of the hair follicle; the exact etiology is uncertain.1,2 No relationship between IFK and human papillomavirus (HPV) has been established.3 Inverted follicular keratosis can mimic squamous cell carcinoma (SCC). Important clues to the diagnosis of IFK are the presence of squamous eddies and the lack of squamous pearls or cytologic atypia.4 Squamous eddies consist of whorled keratinocytes without keratinization or atypia. Superficial shave biopsies may fail to demonstrate the characteristic well-circumscribed architecture and may lead to an erroneous diagnosis.
Acantholytic SCC is characterized by atypical keratinocytes that have lost cohesive properties, resulting in acantholysis (Figure 1).5 This histologic variant was once categorized as an aggressive variant of SCC, but studies have failed to support this assertion.5,6 Acantholytic SCC has a discohesive nature producing a pseudoglandular appearance sometimes mistaken for adenosquamous carcinoma or metastatic carcinoma. Recent literature has suggested that acantholytic SCCs, similar to IFKs, are derived from the follicular infundibulum.5,6 Also similar to IFKs, acantholytic SCCs often are located on the face. The invasive architecture and atypical cytology of acantholytic SCCs can differentiate them from IFKs. Acantholytic SCCs can contain keratin pearls with concentric keratinocytes showing incomplete keratinization centrally, often with retained nuclei, but rare to no squamous eddies unless irritated.
Trichilemmoma is an endophytic benign neoplasm derived from the outer sheath of the pilosebaceous follicle characterized by lobules of clear cells hanging from the epidermis.7 A study investigating the relationship between HPV and trichilemmomas failed to definitively detect HPV in trichilemmomas and this relationship remains unclear.8 Desmoplastic trichilemmoma is a subtype histologically characterized by jagged islands of epithelial cells separated by dense pink stroma and encased in a glassy basement membrane (Figure 2). The presence of desmoplasia and a jagged growth pattern can mimic invasive SCC, but the absence of cytologic atypia and the surrounding basement membrane differs from SCC.4,7 Trichilemmomas typically are solitary, but multiple lesions are associated with Cowden syndrome. Cowden syndrome is a rare autosomal-dominant condition characterized by the presence of benign hamartomas and a predisposition to the development of malignancies including breast, endometrial, and thyroid cancers.9,10 There is no such association with desmoplastic trichilemmomas.11
Pilar sheath acanthoma is a benign neoplasm that clinically presents as a solitary flesh-colored nodule with a central pore containing keratin.12 Histologically, pilar sheath acanthoma is similar to a dilated pore of Winer with the addition of acanthotic epidermal projections (Figure 3).
Warty dyskeratoma (WD) is a benign endophytic neoplasm traditionally seen as a solitary lesion histologically similar to Darier disease. Warty dyskeratomas are known to occur both on the skin and oral mucosa.13 Histologically, WD is characterized as a cup-shaped lesion with numerous villi at the base of the lesion along with acantholysis and dyskeratosis (Figure 4). The dyskeratotic cells in WD consist of corps ronds, which are cells with abundant pink cytoplasm, and small nuclei along with grains, which are flattened basophilic cells. These dyskeratotic cells help differentiate WD from IFK. Although they are endophytic neoplasms, WDs are well circumscribed and should not be confused with SCC. Despite this entity's name and histologic similarity to verrucae, no relationship with HPV has been established.14
The Diagnosis: Inverted Follicular Keratosis
The differential diagnosis for endophytic squamous neoplasms encompasses benign and malignant entities. The histologic findings of our patient's lesion were compatible with the diagnosis of inverted follicular keratosis (IFK), a benign neoplasm that usually presents as a keratotic papule on the head or neck. Histologically, IFK is characterized by an endophytic growth pattern with squamous eddies (quiz images). Inverted follicular keratosis may represent an irritated seborrheic keratosis or a distinct neoplasm derived from the infundibular portion of the hair follicle; the exact etiology is uncertain.1,2 No relationship between IFK and human papillomavirus (HPV) has been established.3 Inverted follicular keratosis can mimic squamous cell carcinoma (SCC). Important clues to the diagnosis of IFK are the presence of squamous eddies and the lack of squamous pearls or cytologic atypia.4 Squamous eddies consist of whorled keratinocytes without keratinization or atypia. Superficial shave biopsies may fail to demonstrate the characteristic well-circumscribed architecture and may lead to an erroneous diagnosis.
Acantholytic SCC is characterized by atypical keratinocytes that have lost cohesive properties, resulting in acantholysis (Figure 1).5 This histologic variant was once categorized as an aggressive variant of SCC, but studies have failed to support this assertion.5,6 Acantholytic SCC has a discohesive nature producing a pseudoglandular appearance sometimes mistaken for adenosquamous carcinoma or metastatic carcinoma. Recent literature has suggested that acantholytic SCCs, similar to IFKs, are derived from the follicular infundibulum.5,6 Also similar to IFKs, acantholytic SCCs often are located on the face. The invasive architecture and atypical cytology of acantholytic SCCs can differentiate them from IFKs. Acantholytic SCCs can contain keratin pearls with concentric keratinocytes showing incomplete keratinization centrally, often with retained nuclei, but rare to no squamous eddies unless irritated.
Trichilemmoma is an endophytic benign neoplasm derived from the outer sheath of the pilosebaceous follicle characterized by lobules of clear cells hanging from the epidermis.7 A study investigating the relationship between HPV and trichilemmomas failed to definitively detect HPV in trichilemmomas and this relationship remains unclear.8 Desmoplastic trichilemmoma is a subtype histologically characterized by jagged islands of epithelial cells separated by dense pink stroma and encased in a glassy basement membrane (Figure 2). The presence of desmoplasia and a jagged growth pattern can mimic invasive SCC, but the absence of cytologic atypia and the surrounding basement membrane differs from SCC.4,7 Trichilemmomas typically are solitary, but multiple lesions are associated with Cowden syndrome. Cowden syndrome is a rare autosomal-dominant condition characterized by the presence of benign hamartomas and a predisposition to the development of malignancies including breast, endometrial, and thyroid cancers.9,10 There is no such association with desmoplastic trichilemmomas.11
Pilar sheath acanthoma is a benign neoplasm that clinically presents as a solitary flesh-colored nodule with a central pore containing keratin.12 Histologically, pilar sheath acanthoma is similar to a dilated pore of Winer with the addition of acanthotic epidermal projections (Figure 3).
Warty dyskeratoma (WD) is a benign endophytic neoplasm traditionally seen as a solitary lesion histologically similar to Darier disease. Warty dyskeratomas are known to occur both on the skin and oral mucosa.13 Histologically, WD is characterized as a cup-shaped lesion with numerous villi at the base of the lesion along with acantholysis and dyskeratosis (Figure 4). The dyskeratotic cells in WD consist of corps ronds, which are cells with abundant pink cytoplasm, and small nuclei along with grains, which are flattened basophilic cells. These dyskeratotic cells help differentiate WD from IFK. Although they are endophytic neoplasms, WDs are well circumscribed and should not be confused with SCC. Despite this entity's name and histologic similarity to verrucae, no relationship with HPV has been established.14
- Ruhoy SM, Thomas D, Nuovo GJ. Multiple inverted follicular keratoses as a presenting sign of Cowden's syndrome: case report with human papillomavirus studies. J Am Acad Dermatol. 2004;51:411-415.
- Lever WF. Inverted follicular keratosis is an irritated seborrheic keratosis. Am J Dermatopathol. 1983;5:474.
- Kambiz KH, Kaveh D, Maede D, et al. Human papillomavirus deoxyribonucleic acid may not be detected in non-genital benign papillomatous skin lesions by polymerase chain reaction. Indian J Dermatol. 2014;59:334-338.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- Ogawa T, Kiuru M, Konia TH, et al. Acantholytic squamous cell carcinoma is usually associated with hair follicles, not acantholytic actinic keratosis, and is not "high risk": diagnosis, management, and clinical outcomes in a series of 115 cases. J Am Acad Dermatol. 2017;76:327-333.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer staging guidelines, prognostic factors, and histopathologic variants. Adv Anat Pathol. 2017;24:171-194.
- Sano DT, Yang JJ, Tebcherani AJ, et al. A rare clinical presentation of desmoplastic trichilemmoma mimicking invasive carcinoma. An Bras Dermatol. 2014;89:796-798.
- Stierman S, Chen S, Nuovo G, et al. Detection of human papillomavirus infection in trichilemmomas and verrucae using in situ hybridization. J Cutan Pathol. 2010;37:75-80.
- Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol [published online October 22, 2014]. Methods. 2015;77-78:11-19.
- Molvi M, Sharma YK, Dash K. Cowden syndrome: case report, update and proposed diagnostic and surveillance routines. Indian J Dermatol. 2015;60:255-259.
- Jin M, Hampel H, Pilarski R, et al. Phosphatase and tensin homolog immunohistochemical staining and clinical criteria for Cowden syndrome in patients with trichilemmoma or associated lesions. Am J Dermatopathol. 2013;35:637-640.
- Mehregan AH, Brownstein MH. Pilar sheath acanthoma. Arch Dermatol. 1978;114:1495-1497.
- Newland JR, Leventon GS. Warty dyskeratoma of the oral mucosa. correlated light and electron microscopic study. Oral Surg Oral Med Oral Pathol. 1984;58:176-183.
- Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma--"follicular dyskeratoma": analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
- Ruhoy SM, Thomas D, Nuovo GJ. Multiple inverted follicular keratoses as a presenting sign of Cowden's syndrome: case report with human papillomavirus studies. J Am Acad Dermatol. 2004;51:411-415.
- Lever WF. Inverted follicular keratosis is an irritated seborrheic keratosis. Am J Dermatopathol. 1983;5:474.
- Kambiz KH, Kaveh D, Maede D, et al. Human papillomavirus deoxyribonucleic acid may not be detected in non-genital benign papillomatous skin lesions by polymerase chain reaction. Indian J Dermatol. 2014;59:334-338.
- Tan KB, Tan SH, Aw DC, et al. Simulators of squamous cell carcinoma of the skin: diagnostic challenges on small biopsies and clinicopathological correlation [published online June 25, 2013]. J Skin Cancer. 2013;2013:752864.
- Ogawa T, Kiuru M, Konia TH, et al. Acantholytic squamous cell carcinoma is usually associated with hair follicles, not acantholytic actinic keratosis, and is not "high risk": diagnosis, management, and clinical outcomes in a series of 115 cases. J Am Acad Dermatol. 2017;76:327-333.
- Motaparthi K, Kapil JP, Velazquez EF. Cutaneous squamous cell carcinoma: review of the eighth edition of the American Joint Committee on Cancer staging guidelines, prognostic factors, and histopathologic variants. Adv Anat Pathol. 2017;24:171-194.
- Sano DT, Yang JJ, Tebcherani AJ, et al. A rare clinical presentation of desmoplastic trichilemmoma mimicking invasive carcinoma. An Bras Dermatol. 2014;89:796-798.
- Stierman S, Chen S, Nuovo G, et al. Detection of human papillomavirus infection in trichilemmomas and verrucae using in situ hybridization. J Cutan Pathol. 2010;37:75-80.
- Ngeow J, Eng C. PTEN hamartoma tumor syndrome: clinical risk assessment and management protocol [published online October 22, 2014]. Methods. 2015;77-78:11-19.
- Molvi M, Sharma YK, Dash K. Cowden syndrome: case report, update and proposed diagnostic and surveillance routines. Indian J Dermatol. 2015;60:255-259.
- Jin M, Hampel H, Pilarski R, et al. Phosphatase and tensin homolog immunohistochemical staining and clinical criteria for Cowden syndrome in patients with trichilemmoma or associated lesions. Am J Dermatopathol. 2013;35:637-640.
- Mehregan AH, Brownstein MH. Pilar sheath acanthoma. Arch Dermatol. 1978;114:1495-1497.
- Newland JR, Leventon GS. Warty dyskeratoma of the oral mucosa. correlated light and electron microscopic study. Oral Surg Oral Med Oral Pathol. 1984;58:176-183.
- Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma--"follicular dyskeratoma": analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
A 60-year-old man presented with a 3-mm verrucous papule on the right upper eyelid of 2 years' duration.
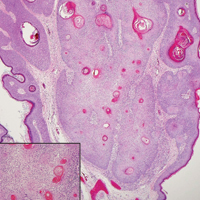
Cyanosis of the Foot
The Diagnosis: Antiphospholipid Antibody Syndrome
A biopsy demonstrated scattered intravascular thrombi in the dermis and subcutis, intact vascular walls, and scant lymphocytic inflammation in a background of stasis (Figure 1). A periodic acid-Schiff stain was negative for fungal elements and highlighted the intravascular thrombi. Histologic findings were consistent with thrombotic vasculopathy. On further laboratory workup, lupus anticoagulant studies, including a mixing study, diluted Russell viper venom test, and hexagonal phase phospholipid neutralization test, were abnormal. Titers of anticardiolipin and β2-glycoprotein I antibodies were elevated (anticardiolipin IgG, 137.7 calculated units [normal, <15 calculated units]; β2-glycoprotein I IgG, 256.4 calculated units [normal, <20 calculated units]). Tissue cultures showed no growth of microorganisms and studies for cryoglobulinemia were negative.

The patient was diagnosed with primary antiphospholipid syndrome (APS). He remained on anticoagulation therapy with fondaparinux as an inpatient and was treated with pulse-dose intravenous (IV) corticosteroids followed by a slow oral taper, daily plasmapheresis for 1 week, IV immunoglobulin (0.5 g/kg) for 3 doses, and 4 weekly doses of rituximab (375 mg/m2). His cutaneous findings slowly improved over the next several weeks (Figure 2).
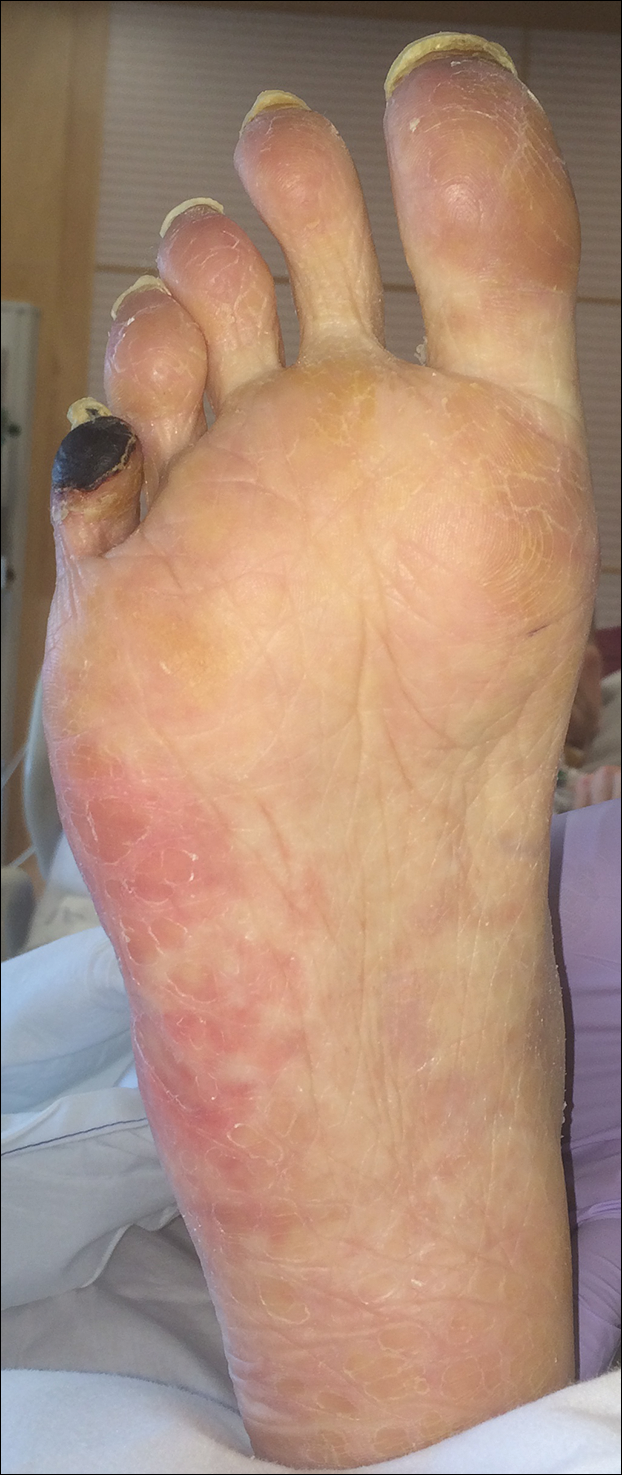
Antiphospholipid syndrome is an autoimmune disorder characterized by thrombotic events and the presence of autoantibodies. The syndrome is defined by 2 major criteria: (1) the occurrence of at least 1 clinical feature of either an episode of vascular thrombosis or pregnancy morbidity such as unexplained fetal death beyond 10 weeks of gestation or recurrent unexplained pregnancy losses; and (2) the presence of at least 1 type of autoantibody, including lupus anticoagulant, anticardiolipin, or β2-glycoprotein antibodies, on 2 separate occasions at least 12 weeks apart.1 Antiphospholipid syndrome can either be primary with no identifiable associated rheumatologic disease or secondary to another autoimmune disease such as systemic lupus erythematosus. Cutaneous manifestations are common and frequently are the first sign of disease in 30% to 40% of patients.2 The most common skin finding is persistent livedo reticularis, which can be seen in 20% to 25% of patients. Patients also may develop skin necrosis, ulcerations, digital gangrene, splinter hemorrhages, and livedoid vasculopathy.2 Systemic manifestations of APS include thrombocytopenia, nephropathy, cognitive dysfunction, and cardiac valve abnormalities.
The exact pathogenesis of APS remains unknown. It is thought to be due to the combination of an inflammatory stimulus that has yet to be characterized in conjunction with autoantibodies that affect multiple target cells including monocytes, platelets, and endothelial cells, which results in activation of the complement system and clotting cascade.3 In rare cases, the disorder can progress to catastrophic antiphospholipid syndrome (CAPS), which requires fulfillment of 4 criteria: (1) evidence of involvement of 3 organs, tissues, or systems; (2) development of manifestations simultaneously or in less than 1 week; (3) laboratory confirmation of the presence of antiphospholipid antibodies; and (4) confirmation by histopathology of small vessel occlusion.4 Probable CAPS is diagnosed when 3 of 4 criteria are present. Our patient met criteria for probable CAPS, as his antibody titers remained elevated 15 weeks after initial presentation. Precipitating factors that can lead to CAPS are thought to include infection, surgical procedures, medications, or discontinuation of anticoagulation drugs.2 Although the mainstay of management of APS is anticoagulation therapy with warfarin and antiplatelet agents such as aspirin, first-line treatment of CAPS involves high-dose systemic glucocorticoids and plasma exchange. Intravenous immunoglobulin also may be employed in treatment. Data from the CAPS registry demonstrate a role for rituximab, an anti-CD20 antibody, at 375 mg/m2 weekly for 4 weeks (the regimen described in our case) or 1 g every 14 days for 2 sessions.5 A majority of the registry patients treated with rituximab recovered (75% [15/20]) and had no recurrent thrombosis (87% [13/15]) at follow-up.5 Data also are emerging on the role of eculizumab, an anti-C5 antibody that inhibits the terminal complement cascade, as a therapy in difficult-to-treat or refractory CAPS.6-8 The prognosis for CAPS patients without treatment is poor, and mortality has been reported in up to 44% of patients. However, with intervention mortality is reduced by more than 2-fold.9,10
It is important to recognize that acral cyanosis with persistent livedo reticularis and digital gangrene can be a presenting manifestation of APS. These cutaneous manifestations should prompt histologic evaluation for thrombotic vasculopathy in addition to serologic tests for APS autoantibodies. Although APS may be treated with anticoagulants and antiplatelet agents, CAPS may require more aggressive therapy with systemic steroids, plasma exchange, IV immunoglobulin, rituximab, and/or eculizumab.
- Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309-1311.
- Pinto-Almeida T, Caetano M, Sanches M, et al. Cutaneous manifestations of antiphospholipid syndrome: a review of the clinical features, diagnosis and management. Acta Reumatol Port. 2013;38:10-18.
- Meroni PL, Chighizola CB, Rovelli F, et al. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16:209.
- Asherson RA, Cervera R, de Grott PG, et al; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12:530-534.
- Berman H, Rodríguez-Pintó I, Cervera R, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab [published online June 15, 2013]. Autoimmun Rev. 2013;12:1085-1090.
- Shapira I, Andrade D, Allen SL, et al. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64:2719-2723.
- Strakhan M, Hurtado-Sbordoni M, Galeas N, et al. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. 2014;2014:704371.
- Lonze BE, Zachary AA, Magro CM, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459-465.
- Bucciarelli S, Espinosa G, Cervera R, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54:2568-2576.
- Asherson RA, Cervera R, Piette JC, et al. Catastrophic antiphospholipid syndrome. clinical and laboratory features of 50 patients. Medicine (Baltimore). 1998;77:195-207.
The Diagnosis: Antiphospholipid Antibody Syndrome
A biopsy demonstrated scattered intravascular thrombi in the dermis and subcutis, intact vascular walls, and scant lymphocytic inflammation in a background of stasis (Figure 1). A periodic acid-Schiff stain was negative for fungal elements and highlighted the intravascular thrombi. Histologic findings were consistent with thrombotic vasculopathy. On further laboratory workup, lupus anticoagulant studies, including a mixing study, diluted Russell viper venom test, and hexagonal phase phospholipid neutralization test, were abnormal. Titers of anticardiolipin and β2-glycoprotein I antibodies were elevated (anticardiolipin IgG, 137.7 calculated units [normal, <15 calculated units]; β2-glycoprotein I IgG, 256.4 calculated units [normal, <20 calculated units]). Tissue cultures showed no growth of microorganisms and studies for cryoglobulinemia were negative.
The patient was diagnosed with primary antiphospholipid syndrome (APS). He remained on anticoagulation therapy with fondaparinux as an inpatient and was treated with pulse-dose intravenous (IV) corticosteroids followed by a slow oral taper, daily plasmapheresis for 1 week, IV immunoglobulin (0.5 g/kg) for 3 doses, and 4 weekly doses of rituximab (375 mg/m2). His cutaneous findings slowly improved over the next several weeks (Figure 2).
Antiphospholipid syndrome is an autoimmune disorder characterized by thrombotic events and the presence of autoantibodies. The syndrome is defined by 2 major criteria: (1) the occurrence of at least 1 clinical feature of either an episode of vascular thrombosis or pregnancy morbidity such as unexplained fetal death beyond 10 weeks of gestation or recurrent unexplained pregnancy losses; and (2) the presence of at least 1 type of autoantibody, including lupus anticoagulant, anticardiolipin, or β2-glycoprotein antibodies, on 2 separate occasions at least 12 weeks apart.1 Antiphospholipid syndrome can either be primary with no identifiable associated rheumatologic disease or secondary to another autoimmune disease such as systemic lupus erythematosus. Cutaneous manifestations are common and frequently are the first sign of disease in 30% to 40% of patients.2 The most common skin finding is persistent livedo reticularis, which can be seen in 20% to 25% of patients. Patients also may develop skin necrosis, ulcerations, digital gangrene, splinter hemorrhages, and livedoid vasculopathy.2 Systemic manifestations of APS include thrombocytopenia, nephropathy, cognitive dysfunction, and cardiac valve abnormalities.
The exact pathogenesis of APS remains unknown. It is thought to be due to the combination of an inflammatory stimulus that has yet to be characterized in conjunction with autoantibodies that affect multiple target cells including monocytes, platelets, and endothelial cells, which results in activation of the complement system and clotting cascade.3 In rare cases, the disorder can progress to catastrophic antiphospholipid syndrome (CAPS), which requires fulfillment of 4 criteria: (1) evidence of involvement of 3 organs, tissues, or systems; (2) development of manifestations simultaneously or in less than 1 week; (3) laboratory confirmation of the presence of antiphospholipid antibodies; and (4) confirmation by histopathology of small vessel occlusion.4 Probable CAPS is diagnosed when 3 of 4 criteria are present. Our patient met criteria for probable CAPS, as his antibody titers remained elevated 15 weeks after initial presentation. Precipitating factors that can lead to CAPS are thought to include infection, surgical procedures, medications, or discontinuation of anticoagulation drugs.2 Although the mainstay of management of APS is anticoagulation therapy with warfarin and antiplatelet agents such as aspirin, first-line treatment of CAPS involves high-dose systemic glucocorticoids and plasma exchange. Intravenous immunoglobulin also may be employed in treatment. Data from the CAPS registry demonstrate a role for rituximab, an anti-CD20 antibody, at 375 mg/m2 weekly for 4 weeks (the regimen described in our case) or 1 g every 14 days for 2 sessions.5 A majority of the registry patients treated with rituximab recovered (75% [15/20]) and had no recurrent thrombosis (87% [13/15]) at follow-up.5 Data also are emerging on the role of eculizumab, an anti-C5 antibody that inhibits the terminal complement cascade, as a therapy in difficult-to-treat or refractory CAPS.6-8 The prognosis for CAPS patients without treatment is poor, and mortality has been reported in up to 44% of patients. However, with intervention mortality is reduced by more than 2-fold.9,10
It is important to recognize that acral cyanosis with persistent livedo reticularis and digital gangrene can be a presenting manifestation of APS. These cutaneous manifestations should prompt histologic evaluation for thrombotic vasculopathy in addition to serologic tests for APS autoantibodies. Although APS may be treated with anticoagulants and antiplatelet agents, CAPS may require more aggressive therapy with systemic steroids, plasma exchange, IV immunoglobulin, rituximab, and/or eculizumab.
The Diagnosis: Antiphospholipid Antibody Syndrome
A biopsy demonstrated scattered intravascular thrombi in the dermis and subcutis, intact vascular walls, and scant lymphocytic inflammation in a background of stasis (Figure 1). A periodic acid-Schiff stain was negative for fungal elements and highlighted the intravascular thrombi. Histologic findings were consistent with thrombotic vasculopathy. On further laboratory workup, lupus anticoagulant studies, including a mixing study, diluted Russell viper venom test, and hexagonal phase phospholipid neutralization test, were abnormal. Titers of anticardiolipin and β2-glycoprotein I antibodies were elevated (anticardiolipin IgG, 137.7 calculated units [normal, <15 calculated units]; β2-glycoprotein I IgG, 256.4 calculated units [normal, <20 calculated units]). Tissue cultures showed no growth of microorganisms and studies for cryoglobulinemia were negative.
The patient was diagnosed with primary antiphospholipid syndrome (APS). He remained on anticoagulation therapy with fondaparinux as an inpatient and was treated with pulse-dose intravenous (IV) corticosteroids followed by a slow oral taper, daily plasmapheresis for 1 week, IV immunoglobulin (0.5 g/kg) for 3 doses, and 4 weekly doses of rituximab (375 mg/m2). His cutaneous findings slowly improved over the next several weeks (Figure 2).
Antiphospholipid syndrome is an autoimmune disorder characterized by thrombotic events and the presence of autoantibodies. The syndrome is defined by 2 major criteria: (1) the occurrence of at least 1 clinical feature of either an episode of vascular thrombosis or pregnancy morbidity such as unexplained fetal death beyond 10 weeks of gestation or recurrent unexplained pregnancy losses; and (2) the presence of at least 1 type of autoantibody, including lupus anticoagulant, anticardiolipin, or β2-glycoprotein antibodies, on 2 separate occasions at least 12 weeks apart.1 Antiphospholipid syndrome can either be primary with no identifiable associated rheumatologic disease or secondary to another autoimmune disease such as systemic lupus erythematosus. Cutaneous manifestations are common and frequently are the first sign of disease in 30% to 40% of patients.2 The most common skin finding is persistent livedo reticularis, which can be seen in 20% to 25% of patients. Patients also may develop skin necrosis, ulcerations, digital gangrene, splinter hemorrhages, and livedoid vasculopathy.2 Systemic manifestations of APS include thrombocytopenia, nephropathy, cognitive dysfunction, and cardiac valve abnormalities.
The exact pathogenesis of APS remains unknown. It is thought to be due to the combination of an inflammatory stimulus that has yet to be characterized in conjunction with autoantibodies that affect multiple target cells including monocytes, platelets, and endothelial cells, which results in activation of the complement system and clotting cascade.3 In rare cases, the disorder can progress to catastrophic antiphospholipid syndrome (CAPS), which requires fulfillment of 4 criteria: (1) evidence of involvement of 3 organs, tissues, or systems; (2) development of manifestations simultaneously or in less than 1 week; (3) laboratory confirmation of the presence of antiphospholipid antibodies; and (4) confirmation by histopathology of small vessel occlusion.4 Probable CAPS is diagnosed when 3 of 4 criteria are present. Our patient met criteria for probable CAPS, as his antibody titers remained elevated 15 weeks after initial presentation. Precipitating factors that can lead to CAPS are thought to include infection, surgical procedures, medications, or discontinuation of anticoagulation drugs.2 Although the mainstay of management of APS is anticoagulation therapy with warfarin and antiplatelet agents such as aspirin, first-line treatment of CAPS involves high-dose systemic glucocorticoids and plasma exchange. Intravenous immunoglobulin also may be employed in treatment. Data from the CAPS registry demonstrate a role for rituximab, an anti-CD20 antibody, at 375 mg/m2 weekly for 4 weeks (the regimen described in our case) or 1 g every 14 days for 2 sessions.5 A majority of the registry patients treated with rituximab recovered (75% [15/20]) and had no recurrent thrombosis (87% [13/15]) at follow-up.5 Data also are emerging on the role of eculizumab, an anti-C5 antibody that inhibits the terminal complement cascade, as a therapy in difficult-to-treat or refractory CAPS.6-8 The prognosis for CAPS patients without treatment is poor, and mortality has been reported in up to 44% of patients. However, with intervention mortality is reduced by more than 2-fold.9,10
It is important to recognize that acral cyanosis with persistent livedo reticularis and digital gangrene can be a presenting manifestation of APS. These cutaneous manifestations should prompt histologic evaluation for thrombotic vasculopathy in addition to serologic tests for APS autoantibodies. Although APS may be treated with anticoagulants and antiplatelet agents, CAPS may require more aggressive therapy with systemic steroids, plasma exchange, IV immunoglobulin, rituximab, and/or eculizumab.
- Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309-1311.
- Pinto-Almeida T, Caetano M, Sanches M, et al. Cutaneous manifestations of antiphospholipid syndrome: a review of the clinical features, diagnosis and management. Acta Reumatol Port. 2013;38:10-18.
- Meroni PL, Chighizola CB, Rovelli F, et al. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16:209.
- Asherson RA, Cervera R, de Grott PG, et al; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12:530-534.
- Berman H, Rodríguez-Pintó I, Cervera R, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab [published online June 15, 2013]. Autoimmun Rev. 2013;12:1085-1090.
- Shapira I, Andrade D, Allen SL, et al. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64:2719-2723.
- Strakhan M, Hurtado-Sbordoni M, Galeas N, et al. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. 2014;2014:704371.
- Lonze BE, Zachary AA, Magro CM, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459-465.
- Bucciarelli S, Espinosa G, Cervera R, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54:2568-2576.
- Asherson RA, Cervera R, Piette JC, et al. Catastrophic antiphospholipid syndrome. clinical and laboratory features of 50 patients. Medicine (Baltimore). 1998;77:195-207.
- Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309-1311.
- Pinto-Almeida T, Caetano M, Sanches M, et al. Cutaneous manifestations of antiphospholipid syndrome: a review of the clinical features, diagnosis and management. Acta Reumatol Port. 2013;38:10-18.
- Meroni PL, Chighizola CB, Rovelli F, et al. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16:209.
- Asherson RA, Cervera R, de Grott PG, et al; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12:530-534.
- Berman H, Rodríguez-Pintó I, Cervera R, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab [published online June 15, 2013]. Autoimmun Rev. 2013;12:1085-1090.
- Shapira I, Andrade D, Allen SL, et al. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64:2719-2723.
- Strakhan M, Hurtado-Sbordoni M, Galeas N, et al. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. 2014;2014:704371.
- Lonze BE, Zachary AA, Magro CM, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459-465.
- Bucciarelli S, Espinosa G, Cervera R, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54:2568-2576.
- Asherson RA, Cervera R, Piette JC, et al. Catastrophic antiphospholipid syndrome. clinical and laboratory features of 50 patients. Medicine (Baltimore). 1998;77:195-207.
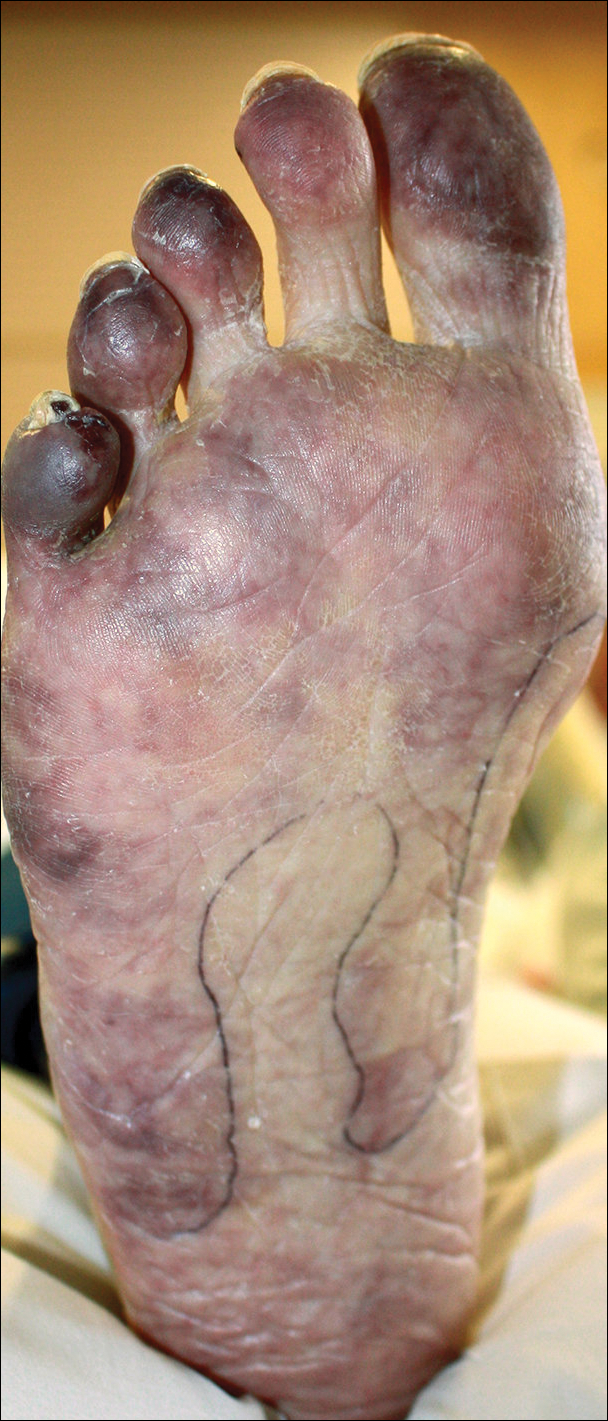
A man in his 50s with a medical history of arterial thrombosis of the right arm, multiple deep vein thromboses (DVTs) of the legs on long-term warfarin, ischemic stroke, atrial fibrillation, and peripheral arterial disease presented with discoloration of the right foot and increasing tenderness of 1 month's duration. There was no history of trauma or recent change in outpatient medications. A family history was notable for an aunt and 2 cousins with DVTs and protein S deficiency. Physical examination revealed livedo reticularis on the sole and lateral aspect of the right foot. There was violaceous discoloration of the volar aspects of all 5 toes and a focal area of ulceration on the fifth toe. Pulses were palpable bilaterally. Initial laboratory evaluation was notable for thrombocytopenia, and preliminary blood cultures revealed no growth of bacterial or fungal organisms. Imaging studies revealed increased arterial stenosis of the right leg as well as DVT of the right great saphenous vein. A punch biopsy of the right medial foot was performed for hematoxylin and eosin stain as well as tissue culture.
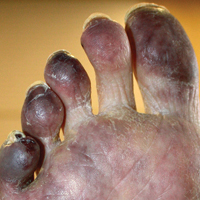
Sweet Syndrome Induced by Oral Acetaminophen-Codeine Following Repair of a Facial Fracture
In 1964, Sweet1 described 8 women with acute onset of fever and erythematous plaques associated with a nonspecific infection of the respiratory or gastrointestinal tract. The lesions were histologically characterized by a neutrophilic infiltrate, and the author named the constellation of findings acute febrile neutrophilic dermatosis.1 In 1968, Whittle et al2 reported on similar cases and coined the term Sweet syndrome (SS).
Although the etiology and pathogenesis of SS remain unknown, several theories have been proposed. Because SS often is preceded by a respiratory or gastrointestinal tract infection, it has been postulated that it may represent a hypersensitivity reaction or may be related to local or systemic dysregulation of cytokine secretion.3,4 In addition to respiratory or gastrointestinal tract infections, SS has been reported in association with malignancies, autoimmune diseases, drugs, vaccines, pregnancy, inflammatory bowel disease, and chemotherapy. It also may be idiopathic.5
The eruption of SS manifests as erythematous, indurated, and sharply demarcated plaques or nodules that typically favor the head, neck, and arms, with a particularly strong predilection for the dorsal aspects of the hands.6 Plaques and nodules are histologically characterized by a diffuse dermal neutrophilic infiltrate, papillary dermal edema, neutrophilic spongiosis, subcorneal pustules, and leukocytoclasia. Vasculitic features are not seen.7 The eruption typically resolves spontaneously in 5 to 12 weeks but recurs in approximately 30% of cases.8 Relatively common extracutaneous findings include ocular involvement, arthralgia, myalgia, and arthritis.4,9 Both cutaneous and extracutaneous findings typically are responsive to prednisone at a dosage of 0.5 to 1 mg/kg daily for 4 to 6 weeks. Prolonged low-dose prednisone for 2 to 3 additional months may be necessary to suppress recurrence.8 Potassium iodide at 900 mg daily may be used as an alternative regimen.3,8
Sweet syndrome is divided into 5 subcategories based on the underlying etiology: (1) classic or idiopathic, (2) paraneoplastic, (3) inflammatory and/or autoimmune disease related, (4) pregnancy related, and (5) drug induced.3 Although drug-induced SS comprises the minority of total cases (<5%), its reported incidence has been rising in recent years and has been associated with an escalating number of medications.10 We report a rare case of SS induced by administration of oral acetaminophen-codeine.
Case Report
A 32-year-old man with a history of diabetes mellitus underwent postoperative repair of a facial fracture. The patient was administered an oral acetaminophen-codeine suspension for postoperative pain control. One week later, he developed a painful eruption on the forehead and presented to the emergency department. He was prescribed acetaminophen-codeine 300/30-mg tablets every 6 hours in addition to hydrocortisone cream 1% applied every 6 hours. After this reintroduction of oral acetaminophen-codeine, he experienced intermittent fevers and an exacerbation of the initial cutaneous eruption. The patient presented for a second time 2 days after being seen in the emergency department and a dermatology consultation was obtained.
At the time of consultation, the patient was noted to have injected conjunctiva and erythematous, well-demarcated, and indurated plaques on the forehead with associated pain and burning (Figures 1A and 1B). Additional erythematous annular plaques were found on the palms, arms, and right knee. Laboratory workup revealed only mild anemia on complete blood cell count with a white blood cell count of 10.1×109/L (reference range, 4.5–11.0×109/L), hemoglobin of 12.9 g/dL (reference range, 14.0–17.4 g/dL), and hematocrit of 37.3% (reference range, 41%–50%). The platelet count was 284×103/µL (reference range, 150–350×103/µL). Basic metabolic panel was notable for an elevated glucose level of 418 mg/dL (reference range, 70–110 mg/dL). The most recent hemoglobin A1C (several months prior) was notable at 14.7% of total hemoglobin (reference range, 4%–7% of total hemoglobin). A 4-mm punch biopsy of the right side of the forehead demonstrated minimal to mild papillary dermal edema and a diffuse dermal neutrophilic infiltrate spanning the upper, middle, and lower dermis with evidence of mild leukocytoclasia and no evidence of leukocytoclastic vasculitis (Figure 2). These histologic features together with the clinical presentation were consistent with a diagnosis of SS.
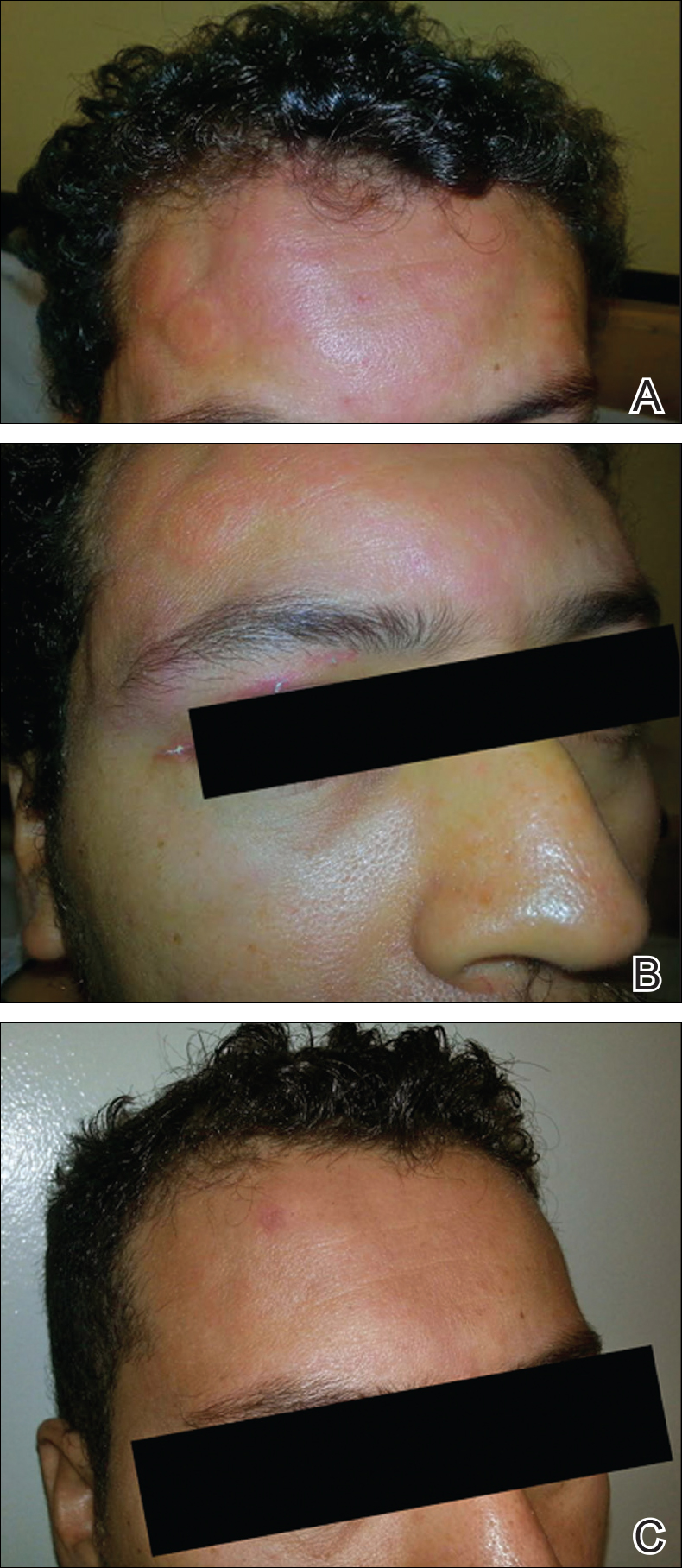
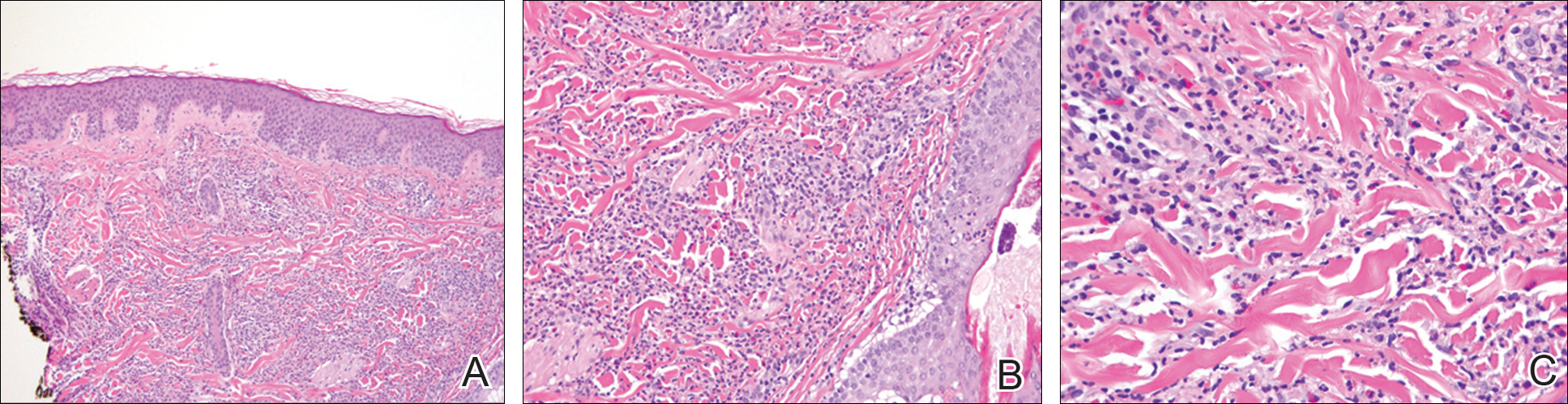
After an initial dose of intravenous methylprednisolone sodium succinate 125 mg in the emergency department, the patient was admitted for additional intravenous steroid administration in the context of uncontrolled hyperglycemia and history of poor glucose control. Upon admission, acetaminophen-codeine was discontinued and the patient was transitioned to intravenous methylprednisolone sodium succinate 60 mg every 8 hours. The patient also was given intravenous diphenhydramine 25 mg every 6 hours and desonide ointment 0.05% was applied to facial lesions. The inpatient medication regimen resulted in notable improvement of
Comment
Although SS itself is relatively rare, there has been an increasing incidence of the drug-induced subtype, most often in association with use of granulocyte colony-stimulating factor and granulocyte monocyte-stimulating factor. There also have been reported associations with a growing number of medications that include antibiotics, antiepileptic drugs, furosemide, hydralazine, and all-trans retinoic acid.11-19 Moghim
Several therapies for advanced melanoma also have been reviewed in the literature, including ipilimumab and vemurafenib,27-30 as have several medications for the treatment of myelodysplastic syndrome including azacitidine.31,32 A seve
Additional medications more recently involved in the pathogenesis of drug-induced SS include the chemotherapeutic agents topetecan, mitoxantrone, gemcitabine, and vorinostat.34-37 The antimalarial medication chloroquine also has been implicated, as have selective cyclooxygenase-2 inhibitors, hypomethylating agents, the tumor necrosis factor inhibitor adalimumab, IL-2 therapies, aripiprazole, and several other medications.38-49
Despite drug-induced SS being reported in association with an increasing number of medications, there had been a lack of appropriate diagnostic criteria. To tha
Conclusion
The number of cases of drug-induced SS in the literature continues to climb; however, the association with acetaminophen-codeine is unique. The importance of this case lies in educating both physicians and pharmacists alike regarding a newly recognized adverse effect of acetaminophen-codeine. Because acetaminophen-codeine often is used for its analgesic properties, and the predominant symptom of the cutaneous eruption of SS is pain, the therapeutic value of acetaminophen-codeine is substantially diminished in acetaminophen-codeine–induced SS. Accordingly, in these cases, the medication may be discontinued or substituted upon recognition of this adverse reaction to reduce patient morbidity.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Whittle CH, Back GA, Champion RH. Recurrent neutrophilic dermatosis of the face—a variant of Sweet’s syndrome. Br J Dermatol. 1968;80:806-810.
- Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-536.
- Honigsmann H, Cohen PR, Wolff K. Acute febrile neutrophilic dermatosis (Sweet’s syndrome). Wien Klin Wochenschr. 1979;91:842-847.
- Limdiwala PG, Parikh SJ, Shah JS. Sweet’s Syndrome. Indian J Dent Res. 2014;25:401-405.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Moschella SL, Davis MDP. Neutrophilic dermatoses. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier; 2012:423-428.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: signs and symptoms and associated disorders. Mayo Clinic Proc. 1995;70:234-240.
- Carvalho R, Fernandes C, Afonso A, et al. Drug-induced Sweet’s syndrome by alclofenac. Cutan Ocul Toxicol. 2011;30:315-316.
- Moghimi J, Pahlevan D, Azizzadeh M, et al. Isotretinoin-associated Sweet’s syndrome: a case report. Daru. 2014;22:69.
- Cholongitas E, Pipili C, Dasenaki M, et al. Piperacillin/tazobactam-induced Sweet syndrome in a patient with chronic lymphocytic leukemia and autoimmune cholangitis. Am J Dermatopathol. 2008;30:203-204.
- Kandula S, Burke WS, Goldfarb JN. Clindamycin-induced Sweet syndrome. J Am Acad Dermatol. 2010;62:898-900.
- Jamet A, Lagarce L, Le Clec’h C, et al. Doxycycline-induced Sweet’s syndrome. Eur J Dermatol. 2008;18:595-596.
- Cartee TV, Chen SC. Sweet syndrome associated with hydralazine-induced lupus erythematosus. Cutis. 2012;89:121-124.
- Baybay H, Elhatimi A, Idrissi R, et al. Sweet’s syndrome following oral ciprofloxacin therapy. Ann Dermatol Venereol. 2011;138:606-607.
- Khaled A, Kharfi M, Fazaa B, et al. A first case of trimethoprim-sulfamethoxazole induced Sweet’s syndrome in a child. Pediatr Dermatol. 2009;26:744-746.
- Calixto R, Menezes Y, Ostronoff M, et al. Favorable outcome of severe, extensive, granulocyte colony-stimulating factor-induced, corticosteroid-resistant Sweet’s syndrome treated with high-dose intravenous immunoglobulin. J Clin Oncol. 2014;32:E1-E2.
- Margaretten ME, Ruben BS, Fye K. Systemic sulfa-induced Sweet’s syndrome. Arthritis Rheum. 2008;59:1044-1046.
- Tanguy-Schmidt A, Avenel-Audran M, Croué A, et al. Bortezomib-induced acute neutrophilic dermatosis. Ann Dermatol Venereol. 2009;136:443-446.
- Choonhakarn C, Chaowattanapanit S. Azathioprine-induced Sweet’s syndrome and published work review. J Dermatol. 2013;40:267-271.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- Valentine MC, Walsh JS. Neutrophilic dermatosis caused by azathioprine. Skinmed. 2011;9:386-388.
- Kim JS, Roh HS, Lee JW, et al. Distinct variant of Sweet’s syndrome: bortezomib-induced histiocytoid Sweet’s syndrome in a patient with multiple myeloma. Int J Dermatol. 2012;51:1491-1493.
- Ozlem C, Deram B, Mustafa S, et al. Propylthiouracil-induced anti-neutrophil cytoplasmic antibodies and agranulocytosis together with granulocyte colony-stimulating factor induced Sweet’s syndrome in a patient with Graves’ disease. Intern Med. 2011;50:1973-1976.
- Kyllo RL, Parker MK, Rosman I, et al. Ipilimumab-associated Sweet syndrome in a patient with high-risk melanoma. J Am Acad Dermatol. 2014;70:E85-E86.
- Pintova S, Sidhu H, Friedlander PA, et al. Sweet’s syndrome in a patient with metastatic melanoma after ipilimumab therapy. Melanoma Res. 2013;23:498-501.
- Yorio JT, Mays SR, Ciurea AM, et al. Case of vemurafenib-induced Sweet’s syndrome. J Dermatol. 2014;41:817-820.
- Pattanaprichakul P, Tetzlaff MT, Lapolla WJ, et al. Sweet syndrome following vemurafenib therapy for recurrent cholangiocarcinoma. J Cutan Pathol. 2014;41:326-328.
- Trickett HB, Cumpston A, Craig M. Azacitidine-associated Sweet’s syndrome. Am J Health Syst Pharm. 2012;69:869-871.
- Tintle S, Patel V, Ruskin A, et al. Azacitidine: a new medication associated with Sweet syndrome. J Am Acad Dermatol. 2011;64:E77-E79.
- Thieu KP, Rosenbach M, Xu X, et al. Neutrophilic dermatosis complicating lenalidomide therapy. J Am Acad Dermatol. 2009;61:709-710.
- Dickson EL, Bakhru A, Chan MP. Topotecan-induced Sweet’s syndrome: a case report. Gynecol Oncol Case Rep. 2013;4:50-52.
- Kümpfel T, Gerdes LA, Flaig M, et al. Drug-induced Sweet’s syndrome after mitoxantrone therapy in a patient with multiple sclerosis. Mult Scler. 2011;17:495-497.
- Martorell-Calatayud A, Requena C, Sanmartin O, et al. Gemcitabine-associated sweet syndrome-like eruption. J Am Acad Dermatol. 2011;65:1236-1238.
- Pang A, Tan KB, Aw D, et al. A case of Sweet’s syndrome due to 5-azacytidine and vorinostat in a patient with NK/T cell lymphoma. Cutan Ocul Toxicol. 2012;31:64-66.
- El Moutaoui L, Zouhair K, Benchikhi H. Sweet syndrome induced by chloroquine. Ann Dermatol Venereol. 2009;136:56-57.
- Rosmaninho A, Lobo I, Selores M. Sweet’s syndrome associated with the intake of a selective cyclooxygenase-2 (COX-2) inhibitor. Cutan Ocul Toxicol. 2011;30:298-301.
- Alencar C, Abramowtiz M, Parekh S, et al. Atypical presentations of Sweet’s syndrome in patients with MDS/AML receiving combinations of hypomethylating agents with histone deacetylase inhibitors. Am J Hematol. 2009;84:688-689.
- Keidel S, McColl A, Edmonds S. Sweet’s syndrome after adalimumab therapy for refractory relapsing polychondritis. BMJ Case Rep. 2011;2011.
- Rondina A, Watson AC. Bullous Sweet’s syndrome and pseudolymphoma precipitated by IL-2 therapy. Cutis. 2010;85:206-213.
- Gheorghe L, Cotruta B, Trifu V, et al. Drug-induced Sweet’s syndrome secondary to hepatitis C antiviral therapy. Int J Dermatol. 2008;47:957-959.
- Zobniw CM, Saad SA, Kostoff D, et al. Bortezomib-induced Sweet’s syndrome confirmed by rechallenge. Pharmacotherapy. 2014;34:E18-E21.
- Kolb-Mäurer A, Kneitz H, Goebeler M. Sweet-like syndrome induced by bortezomib. J Dtsch Dermatol Ges. 2013;11:1200-1202.
- Thuillier D, Lenglet A, Chaby G, et al. Bortezomib-induced eruption: Sweet syndrome? two case reports [in French]. Ann Dermatol Venereol. 2009;136:427-430.
- Kim MJ, Jang KT, Choe YH. Azathioprine hypersensitivity presenting as sweet syndrome in a child with ulcerative colitis. Indian Pediatr. 2011;48:969-971.
- Truchuelo M, Bagazgoitia L, Alcántara J, et al. Sweet-like lesions induced by bortezomib: a review of the literature and a report of 2 cases. Actas Dermosifiliogr. 2012;103:829-831.
- Hoelt P, Fattouh K, Villani AP. Dermpath & clinic: drug-induced Sweet syndrome. Eur J Dermatol. 2016;26:641-642.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
In 1964, Sweet1 described 8 women with acute onset of fever and erythematous plaques associated with a nonspecific infection of the respiratory or gastrointestinal tract. The lesions were histologically characterized by a neutrophilic infiltrate, and the author named the constellation of findings acute febrile neutrophilic dermatosis.1 In 1968, Whittle et al2 reported on similar cases and coined the term Sweet syndrome (SS).
Although the etiology and pathogenesis of SS remain unknown, several theories have been proposed. Because SS often is preceded by a respiratory or gastrointestinal tract infection, it has been postulated that it may represent a hypersensitivity reaction or may be related to local or systemic dysregulation of cytokine secretion.3,4 In addition to respiratory or gastrointestinal tract infections, SS has been reported in association with malignancies, autoimmune diseases, drugs, vaccines, pregnancy, inflammatory bowel disease, and chemotherapy. It also may be idiopathic.5
The eruption of SS manifests as erythematous, indurated, and sharply demarcated plaques or nodules that typically favor the head, neck, and arms, with a particularly strong predilection for the dorsal aspects of the hands.6 Plaques and nodules are histologically characterized by a diffuse dermal neutrophilic infiltrate, papillary dermal edema, neutrophilic spongiosis, subcorneal pustules, and leukocytoclasia. Vasculitic features are not seen.7 The eruption typically resolves spontaneously in 5 to 12 weeks but recurs in approximately 30% of cases.8 Relatively common extracutaneous findings include ocular involvement, arthralgia, myalgia, and arthritis.4,9 Both cutaneous and extracutaneous findings typically are responsive to prednisone at a dosage of 0.5 to 1 mg/kg daily for 4 to 6 weeks. Prolonged low-dose prednisone for 2 to 3 additional months may be necessary to suppress recurrence.8 Potassium iodide at 900 mg daily may be used as an alternative regimen.3,8
Sweet syndrome is divided into 5 subcategories based on the underlying etiology: (1) classic or idiopathic, (2) paraneoplastic, (3) inflammatory and/or autoimmune disease related, (4) pregnancy related, and (5) drug induced.3 Although drug-induced SS comprises the minority of total cases (<5%), its reported incidence has been rising in recent years and has been associated with an escalating number of medications.10 We report a rare case of SS induced by administration of oral acetaminophen-codeine.
Case Report
A 32-year-old man with a history of diabetes mellitus underwent postoperative repair of a facial fracture. The patient was administered an oral acetaminophen-codeine suspension for postoperative pain control. One week later, he developed a painful eruption on the forehead and presented to the emergency department. He was prescribed acetaminophen-codeine 300/30-mg tablets every 6 hours in addition to hydrocortisone cream 1% applied every 6 hours. After this reintroduction of oral acetaminophen-codeine, he experienced intermittent fevers and an exacerbation of the initial cutaneous eruption. The patient presented for a second time 2 days after being seen in the emergency department and a dermatology consultation was obtained.
At the time of consultation, the patient was noted to have injected conjunctiva and erythematous, well-demarcated, and indurated plaques on the forehead with associated pain and burning (Figures 1A and 1B). Additional erythematous annular plaques were found on the palms, arms, and right knee. Laboratory workup revealed only mild anemia on complete blood cell count with a white blood cell count of 10.1×109/L (reference range, 4.5–11.0×109/L), hemoglobin of 12.9 g/dL (reference range, 14.0–17.4 g/dL), and hematocrit of 37.3% (reference range, 41%–50%). The platelet count was 284×103/µL (reference range, 150–350×103/µL). Basic metabolic panel was notable for an elevated glucose level of 418 mg/dL (reference range, 70–110 mg/dL). The most recent hemoglobin A1C (several months prior) was notable at 14.7% of total hemoglobin (reference range, 4%–7% of total hemoglobin). A 4-mm punch biopsy of the right side of the forehead demonstrated minimal to mild papillary dermal edema and a diffuse dermal neutrophilic infiltrate spanning the upper, middle, and lower dermis with evidence of mild leukocytoclasia and no evidence of leukocytoclastic vasculitis (Figure 2). These histologic features together with the clinical presentation were consistent with a diagnosis of SS.
After an initial dose of intravenous methylprednisolone sodium succinate 125 mg in the emergency department, the patient was admitted for additional intravenous steroid administration in the context of uncontrolled hyperglycemia and history of poor glucose control. Upon admission, acetaminophen-codeine was discontinued and the patient was transitioned to intravenous methylprednisolone sodium succinate 60 mg every 8 hours. The patient also was given intravenous diphenhydramine 25 mg every 6 hours and desonide ointment 0.05% was applied to facial lesions. The inpatient medication regimen resulted in notable improvement of
Comment
Although SS itself is relatively rare, there has been an increasing incidence of the drug-induced subtype, most often in association with use of granulocyte colony-stimulating factor and granulocyte monocyte-stimulating factor. There also have been reported associations with a growing number of medications that include antibiotics, antiepileptic drugs, furosemide, hydralazine, and all-trans retinoic acid.11-19 Moghim
Several therapies for advanced melanoma also have been reviewed in the literature, including ipilimumab and vemurafenib,27-30 as have several medications for the treatment of myelodysplastic syndrome including azacitidine.31,32 A seve
Additional medications more recently involved in the pathogenesis of drug-induced SS include the chemotherapeutic agents topetecan, mitoxantrone, gemcitabine, and vorinostat.34-37 The antimalarial medication chloroquine also has been implicated, as have selective cyclooxygenase-2 inhibitors, hypomethylating agents, the tumor necrosis factor inhibitor adalimumab, IL-2 therapies, aripiprazole, and several other medications.38-49
Despite drug-induced SS being reported in association with an increasing number of medications, there had been a lack of appropriate diagnostic criteria. To tha
Conclusion
The number of cases of drug-induced SS in the literature continues to climb; however, the association with acetaminophen-codeine is unique. The importance of this case lies in educating both physicians and pharmacists alike regarding a newly recognized adverse effect of acetaminophen-codeine. Because acetaminophen-codeine often is used for its analgesic properties, and the predominant symptom of the cutaneous eruption of SS is pain, the therapeutic value of acetaminophen-codeine is substantially diminished in acetaminophen-codeine–induced SS. Accordingly, in these cases, the medication may be discontinued or substituted upon recognition of this adverse reaction to reduce patient morbidity.
In 1964, Sweet1 described 8 women with acute onset of fever and erythematous plaques associated with a nonspecific infection of the respiratory or gastrointestinal tract. The lesions were histologically characterized by a neutrophilic infiltrate, and the author named the constellation of findings acute febrile neutrophilic dermatosis.1 In 1968, Whittle et al2 reported on similar cases and coined the term Sweet syndrome (SS).
Although the etiology and pathogenesis of SS remain unknown, several theories have been proposed. Because SS often is preceded by a respiratory or gastrointestinal tract infection, it has been postulated that it may represent a hypersensitivity reaction or may be related to local or systemic dysregulation of cytokine secretion.3,4 In addition to respiratory or gastrointestinal tract infections, SS has been reported in association with malignancies, autoimmune diseases, drugs, vaccines, pregnancy, inflammatory bowel disease, and chemotherapy. It also may be idiopathic.5
The eruption of SS manifests as erythematous, indurated, and sharply demarcated plaques or nodules that typically favor the head, neck, and arms, with a particularly strong predilection for the dorsal aspects of the hands.6 Plaques and nodules are histologically characterized by a diffuse dermal neutrophilic infiltrate, papillary dermal edema, neutrophilic spongiosis, subcorneal pustules, and leukocytoclasia. Vasculitic features are not seen.7 The eruption typically resolves spontaneously in 5 to 12 weeks but recurs in approximately 30% of cases.8 Relatively common extracutaneous findings include ocular involvement, arthralgia, myalgia, and arthritis.4,9 Both cutaneous and extracutaneous findings typically are responsive to prednisone at a dosage of 0.5 to 1 mg/kg daily for 4 to 6 weeks. Prolonged low-dose prednisone for 2 to 3 additional months may be necessary to suppress recurrence.8 Potassium iodide at 900 mg daily may be used as an alternative regimen.3,8
Sweet syndrome is divided into 5 subcategories based on the underlying etiology: (1) classic or idiopathic, (2) paraneoplastic, (3) inflammatory and/or autoimmune disease related, (4) pregnancy related, and (5) drug induced.3 Although drug-induced SS comprises the minority of total cases (<5%), its reported incidence has been rising in recent years and has been associated with an escalating number of medications.10 We report a rare case of SS induced by administration of oral acetaminophen-codeine.
Case Report
A 32-year-old man with a history of diabetes mellitus underwent postoperative repair of a facial fracture. The patient was administered an oral acetaminophen-codeine suspension for postoperative pain control. One week later, he developed a painful eruption on the forehead and presented to the emergency department. He was prescribed acetaminophen-codeine 300/30-mg tablets every 6 hours in addition to hydrocortisone cream 1% applied every 6 hours. After this reintroduction of oral acetaminophen-codeine, he experienced intermittent fevers and an exacerbation of the initial cutaneous eruption. The patient presented for a second time 2 days after being seen in the emergency department and a dermatology consultation was obtained.
At the time of consultation, the patient was noted to have injected conjunctiva and erythematous, well-demarcated, and indurated plaques on the forehead with associated pain and burning (Figures 1A and 1B). Additional erythematous annular plaques were found on the palms, arms, and right knee. Laboratory workup revealed only mild anemia on complete blood cell count with a white blood cell count of 10.1×109/L (reference range, 4.5–11.0×109/L), hemoglobin of 12.9 g/dL (reference range, 14.0–17.4 g/dL), and hematocrit of 37.3% (reference range, 41%–50%). The platelet count was 284×103/µL (reference range, 150–350×103/µL). Basic metabolic panel was notable for an elevated glucose level of 418 mg/dL (reference range, 70–110 mg/dL). The most recent hemoglobin A1C (several months prior) was notable at 14.7% of total hemoglobin (reference range, 4%–7% of total hemoglobin). A 4-mm punch biopsy of the right side of the forehead demonstrated minimal to mild papillary dermal edema and a diffuse dermal neutrophilic infiltrate spanning the upper, middle, and lower dermis with evidence of mild leukocytoclasia and no evidence of leukocytoclastic vasculitis (Figure 2). These histologic features together with the clinical presentation were consistent with a diagnosis of SS.
After an initial dose of intravenous methylprednisolone sodium succinate 125 mg in the emergency department, the patient was admitted for additional intravenous steroid administration in the context of uncontrolled hyperglycemia and history of poor glucose control. Upon admission, acetaminophen-codeine was discontinued and the patient was transitioned to intravenous methylprednisolone sodium succinate 60 mg every 8 hours. The patient also was given intravenous diphenhydramine 25 mg every 6 hours and desonide ointment 0.05% was applied to facial lesions. The inpatient medication regimen resulted in notable improvement of
Comment
Although SS itself is relatively rare, there has been an increasing incidence of the drug-induced subtype, most often in association with use of granulocyte colony-stimulating factor and granulocyte monocyte-stimulating factor. There also have been reported associations with a growing number of medications that include antibiotics, antiepileptic drugs, furosemide, hydralazine, and all-trans retinoic acid.11-19 Moghim
Several therapies for advanced melanoma also have been reviewed in the literature, including ipilimumab and vemurafenib,27-30 as have several medications for the treatment of myelodysplastic syndrome including azacitidine.31,32 A seve
Additional medications more recently involved in the pathogenesis of drug-induced SS include the chemotherapeutic agents topetecan, mitoxantrone, gemcitabine, and vorinostat.34-37 The antimalarial medication chloroquine also has been implicated, as have selective cyclooxygenase-2 inhibitors, hypomethylating agents, the tumor necrosis factor inhibitor adalimumab, IL-2 therapies, aripiprazole, and several other medications.38-49
Despite drug-induced SS being reported in association with an increasing number of medications, there had been a lack of appropriate diagnostic criteria. To tha
Conclusion
The number of cases of drug-induced SS in the literature continues to climb; however, the association with acetaminophen-codeine is unique. The importance of this case lies in educating both physicians and pharmacists alike regarding a newly recognized adverse effect of acetaminophen-codeine. Because acetaminophen-codeine often is used for its analgesic properties, and the predominant symptom of the cutaneous eruption of SS is pain, the therapeutic value of acetaminophen-codeine is substantially diminished in acetaminophen-codeine–induced SS. Accordingly, in these cases, the medication may be discontinued or substituted upon recognition of this adverse reaction to reduce patient morbidity.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Whittle CH, Back GA, Champion RH. Recurrent neutrophilic dermatosis of the face—a variant of Sweet’s syndrome. Br J Dermatol. 1968;80:806-810.
- Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-536.
- Honigsmann H, Cohen PR, Wolff K. Acute febrile neutrophilic dermatosis (Sweet’s syndrome). Wien Klin Wochenschr. 1979;91:842-847.
- Limdiwala PG, Parikh SJ, Shah JS. Sweet’s Syndrome. Indian J Dent Res. 2014;25:401-405.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Moschella SL, Davis MDP. Neutrophilic dermatoses. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier; 2012:423-428.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: signs and symptoms and associated disorders. Mayo Clinic Proc. 1995;70:234-240.
- Carvalho R, Fernandes C, Afonso A, et al. Drug-induced Sweet’s syndrome by alclofenac. Cutan Ocul Toxicol. 2011;30:315-316.
- Moghimi J, Pahlevan D, Azizzadeh M, et al. Isotretinoin-associated Sweet’s syndrome: a case report. Daru. 2014;22:69.
- Cholongitas E, Pipili C, Dasenaki M, et al. Piperacillin/tazobactam-induced Sweet syndrome in a patient with chronic lymphocytic leukemia and autoimmune cholangitis. Am J Dermatopathol. 2008;30:203-204.
- Kandula S, Burke WS, Goldfarb JN. Clindamycin-induced Sweet syndrome. J Am Acad Dermatol. 2010;62:898-900.
- Jamet A, Lagarce L, Le Clec’h C, et al. Doxycycline-induced Sweet’s syndrome. Eur J Dermatol. 2008;18:595-596.
- Cartee TV, Chen SC. Sweet syndrome associated with hydralazine-induced lupus erythematosus. Cutis. 2012;89:121-124.
- Baybay H, Elhatimi A, Idrissi R, et al. Sweet’s syndrome following oral ciprofloxacin therapy. Ann Dermatol Venereol. 2011;138:606-607.
- Khaled A, Kharfi M, Fazaa B, et al. A first case of trimethoprim-sulfamethoxazole induced Sweet’s syndrome in a child. Pediatr Dermatol. 2009;26:744-746.
- Calixto R, Menezes Y, Ostronoff M, et al. Favorable outcome of severe, extensive, granulocyte colony-stimulating factor-induced, corticosteroid-resistant Sweet’s syndrome treated with high-dose intravenous immunoglobulin. J Clin Oncol. 2014;32:E1-E2.
- Margaretten ME, Ruben BS, Fye K. Systemic sulfa-induced Sweet’s syndrome. Arthritis Rheum. 2008;59:1044-1046.
- Tanguy-Schmidt A, Avenel-Audran M, Croué A, et al. Bortezomib-induced acute neutrophilic dermatosis. Ann Dermatol Venereol. 2009;136:443-446.
- Choonhakarn C, Chaowattanapanit S. Azathioprine-induced Sweet’s syndrome and published work review. J Dermatol. 2013;40:267-271.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- Valentine MC, Walsh JS. Neutrophilic dermatosis caused by azathioprine. Skinmed. 2011;9:386-388.
- Kim JS, Roh HS, Lee JW, et al. Distinct variant of Sweet’s syndrome: bortezomib-induced histiocytoid Sweet’s syndrome in a patient with multiple myeloma. Int J Dermatol. 2012;51:1491-1493.
- Ozlem C, Deram B, Mustafa S, et al. Propylthiouracil-induced anti-neutrophil cytoplasmic antibodies and agranulocytosis together with granulocyte colony-stimulating factor induced Sweet’s syndrome in a patient with Graves’ disease. Intern Med. 2011;50:1973-1976.
- Kyllo RL, Parker MK, Rosman I, et al. Ipilimumab-associated Sweet syndrome in a patient with high-risk melanoma. J Am Acad Dermatol. 2014;70:E85-E86.
- Pintova S, Sidhu H, Friedlander PA, et al. Sweet’s syndrome in a patient with metastatic melanoma after ipilimumab therapy. Melanoma Res. 2013;23:498-501.
- Yorio JT, Mays SR, Ciurea AM, et al. Case of vemurafenib-induced Sweet’s syndrome. J Dermatol. 2014;41:817-820.
- Pattanaprichakul P, Tetzlaff MT, Lapolla WJ, et al. Sweet syndrome following vemurafenib therapy for recurrent cholangiocarcinoma. J Cutan Pathol. 2014;41:326-328.
- Trickett HB, Cumpston A, Craig M. Azacitidine-associated Sweet’s syndrome. Am J Health Syst Pharm. 2012;69:869-871.
- Tintle S, Patel V, Ruskin A, et al. Azacitidine: a new medication associated with Sweet syndrome. J Am Acad Dermatol. 2011;64:E77-E79.
- Thieu KP, Rosenbach M, Xu X, et al. Neutrophilic dermatosis complicating lenalidomide therapy. J Am Acad Dermatol. 2009;61:709-710.
- Dickson EL, Bakhru A, Chan MP. Topotecan-induced Sweet’s syndrome: a case report. Gynecol Oncol Case Rep. 2013;4:50-52.
- Kümpfel T, Gerdes LA, Flaig M, et al. Drug-induced Sweet’s syndrome after mitoxantrone therapy in a patient with multiple sclerosis. Mult Scler. 2011;17:495-497.
- Martorell-Calatayud A, Requena C, Sanmartin O, et al. Gemcitabine-associated sweet syndrome-like eruption. J Am Acad Dermatol. 2011;65:1236-1238.
- Pang A, Tan KB, Aw D, et al. A case of Sweet’s syndrome due to 5-azacytidine and vorinostat in a patient with NK/T cell lymphoma. Cutan Ocul Toxicol. 2012;31:64-66.
- El Moutaoui L, Zouhair K, Benchikhi H. Sweet syndrome induced by chloroquine. Ann Dermatol Venereol. 2009;136:56-57.
- Rosmaninho A, Lobo I, Selores M. Sweet’s syndrome associated with the intake of a selective cyclooxygenase-2 (COX-2) inhibitor. Cutan Ocul Toxicol. 2011;30:298-301.
- Alencar C, Abramowtiz M, Parekh S, et al. Atypical presentations of Sweet’s syndrome in patients with MDS/AML receiving combinations of hypomethylating agents with histone deacetylase inhibitors. Am J Hematol. 2009;84:688-689.
- Keidel S, McColl A, Edmonds S. Sweet’s syndrome after adalimumab therapy for refractory relapsing polychondritis. BMJ Case Rep. 2011;2011.
- Rondina A, Watson AC. Bullous Sweet’s syndrome and pseudolymphoma precipitated by IL-2 therapy. Cutis. 2010;85:206-213.
- Gheorghe L, Cotruta B, Trifu V, et al. Drug-induced Sweet’s syndrome secondary to hepatitis C antiviral therapy. Int J Dermatol. 2008;47:957-959.
- Zobniw CM, Saad SA, Kostoff D, et al. Bortezomib-induced Sweet’s syndrome confirmed by rechallenge. Pharmacotherapy. 2014;34:E18-E21.
- Kolb-Mäurer A, Kneitz H, Goebeler M. Sweet-like syndrome induced by bortezomib. J Dtsch Dermatol Ges. 2013;11:1200-1202.
- Thuillier D, Lenglet A, Chaby G, et al. Bortezomib-induced eruption: Sweet syndrome? two case reports [in French]. Ann Dermatol Venereol. 2009;136:427-430.
- Kim MJ, Jang KT, Choe YH. Azathioprine hypersensitivity presenting as sweet syndrome in a child with ulcerative colitis. Indian Pediatr. 2011;48:969-971.
- Truchuelo M, Bagazgoitia L, Alcántara J, et al. Sweet-like lesions induced by bortezomib: a review of the literature and a report of 2 cases. Actas Dermosifiliogr. 2012;103:829-831.
- Hoelt P, Fattouh K, Villani AP. Dermpath & clinic: drug-induced Sweet syndrome. Eur J Dermatol. 2016;26:641-642.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Whittle CH, Back GA, Champion RH. Recurrent neutrophilic dermatosis of the face—a variant of Sweet’s syndrome. Br J Dermatol. 1968;80:806-810.
- Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-536.
- Honigsmann H, Cohen PR, Wolff K. Acute febrile neutrophilic dermatosis (Sweet’s syndrome). Wien Klin Wochenschr. 1979;91:842-847.
- Limdiwala PG, Parikh SJ, Shah JS. Sweet’s Syndrome. Indian J Dent Res. 2014;25:401-405.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Moschella SL, Davis MDP. Neutrophilic dermatoses. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier; 2012:423-428.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: signs and symptoms and associated disorders. Mayo Clinic Proc. 1995;70:234-240.
- Carvalho R, Fernandes C, Afonso A, et al. Drug-induced Sweet’s syndrome by alclofenac. Cutan Ocul Toxicol. 2011;30:315-316.
- Moghimi J, Pahlevan D, Azizzadeh M, et al. Isotretinoin-associated Sweet’s syndrome: a case report. Daru. 2014;22:69.
- Cholongitas E, Pipili C, Dasenaki M, et al. Piperacillin/tazobactam-induced Sweet syndrome in a patient with chronic lymphocytic leukemia and autoimmune cholangitis. Am J Dermatopathol. 2008;30:203-204.
- Kandula S, Burke WS, Goldfarb JN. Clindamycin-induced Sweet syndrome. J Am Acad Dermatol. 2010;62:898-900.
- Jamet A, Lagarce L, Le Clec’h C, et al. Doxycycline-induced Sweet’s syndrome. Eur J Dermatol. 2008;18:595-596.
- Cartee TV, Chen SC. Sweet syndrome associated with hydralazine-induced lupus erythematosus. Cutis. 2012;89:121-124.
- Baybay H, Elhatimi A, Idrissi R, et al. Sweet’s syndrome following oral ciprofloxacin therapy. Ann Dermatol Venereol. 2011;138:606-607.
- Khaled A, Kharfi M, Fazaa B, et al. A first case of trimethoprim-sulfamethoxazole induced Sweet’s syndrome in a child. Pediatr Dermatol. 2009;26:744-746.
- Calixto R, Menezes Y, Ostronoff M, et al. Favorable outcome of severe, extensive, granulocyte colony-stimulating factor-induced, corticosteroid-resistant Sweet’s syndrome treated with high-dose intravenous immunoglobulin. J Clin Oncol. 2014;32:E1-E2.
- Margaretten ME, Ruben BS, Fye K. Systemic sulfa-induced Sweet’s syndrome. Arthritis Rheum. 2008;59:1044-1046.
- Tanguy-Schmidt A, Avenel-Audran M, Croué A, et al. Bortezomib-induced acute neutrophilic dermatosis. Ann Dermatol Venereol. 2009;136:443-446.
- Choonhakarn C, Chaowattanapanit S. Azathioprine-induced Sweet’s syndrome and published work review. J Dermatol. 2013;40:267-271.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- Valentine MC, Walsh JS. Neutrophilic dermatosis caused by azathioprine. Skinmed. 2011;9:386-388.
- Kim JS, Roh HS, Lee JW, et al. Distinct variant of Sweet’s syndrome: bortezomib-induced histiocytoid Sweet’s syndrome in a patient with multiple myeloma. Int J Dermatol. 2012;51:1491-1493.
- Ozlem C, Deram B, Mustafa S, et al. Propylthiouracil-induced anti-neutrophil cytoplasmic antibodies and agranulocytosis together with granulocyte colony-stimulating factor induced Sweet’s syndrome in a patient with Graves’ disease. Intern Med. 2011;50:1973-1976.
- Kyllo RL, Parker MK, Rosman I, et al. Ipilimumab-associated Sweet syndrome in a patient with high-risk melanoma. J Am Acad Dermatol. 2014;70:E85-E86.
- Pintova S, Sidhu H, Friedlander PA, et al. Sweet’s syndrome in a patient with metastatic melanoma after ipilimumab therapy. Melanoma Res. 2013;23:498-501.
- Yorio JT, Mays SR, Ciurea AM, et al. Case of vemurafenib-induced Sweet’s syndrome. J Dermatol. 2014;41:817-820.
- Pattanaprichakul P, Tetzlaff MT, Lapolla WJ, et al. Sweet syndrome following vemurafenib therapy for recurrent cholangiocarcinoma. J Cutan Pathol. 2014;41:326-328.
- Trickett HB, Cumpston A, Craig M. Azacitidine-associated Sweet’s syndrome. Am J Health Syst Pharm. 2012;69:869-871.
- Tintle S, Patel V, Ruskin A, et al. Azacitidine: a new medication associated with Sweet syndrome. J Am Acad Dermatol. 2011;64:E77-E79.
- Thieu KP, Rosenbach M, Xu X, et al. Neutrophilic dermatosis complicating lenalidomide therapy. J Am Acad Dermatol. 2009;61:709-710.
- Dickson EL, Bakhru A, Chan MP. Topotecan-induced Sweet’s syndrome: a case report. Gynecol Oncol Case Rep. 2013;4:50-52.
- Kümpfel T, Gerdes LA, Flaig M, et al. Drug-induced Sweet’s syndrome after mitoxantrone therapy in a patient with multiple sclerosis. Mult Scler. 2011;17:495-497.
- Martorell-Calatayud A, Requena C, Sanmartin O, et al. Gemcitabine-associated sweet syndrome-like eruption. J Am Acad Dermatol. 2011;65:1236-1238.
- Pang A, Tan KB, Aw D, et al. A case of Sweet’s syndrome due to 5-azacytidine and vorinostat in a patient with NK/T cell lymphoma. Cutan Ocul Toxicol. 2012;31:64-66.
- El Moutaoui L, Zouhair K, Benchikhi H. Sweet syndrome induced by chloroquine. Ann Dermatol Venereol. 2009;136:56-57.
- Rosmaninho A, Lobo I, Selores M. Sweet’s syndrome associated with the intake of a selective cyclooxygenase-2 (COX-2) inhibitor. Cutan Ocul Toxicol. 2011;30:298-301.
- Alencar C, Abramowtiz M, Parekh S, et al. Atypical presentations of Sweet’s syndrome in patients with MDS/AML receiving combinations of hypomethylating agents with histone deacetylase inhibitors. Am J Hematol. 2009;84:688-689.
- Keidel S, McColl A, Edmonds S. Sweet’s syndrome after adalimumab therapy for refractory relapsing polychondritis. BMJ Case Rep. 2011;2011.
- Rondina A, Watson AC. Bullous Sweet’s syndrome and pseudolymphoma precipitated by IL-2 therapy. Cutis. 2010;85:206-213.
- Gheorghe L, Cotruta B, Trifu V, et al. Drug-induced Sweet’s syndrome secondary to hepatitis C antiviral therapy. Int J Dermatol. 2008;47:957-959.
- Zobniw CM, Saad SA, Kostoff D, et al. Bortezomib-induced Sweet’s syndrome confirmed by rechallenge. Pharmacotherapy. 2014;34:E18-E21.
- Kolb-Mäurer A, Kneitz H, Goebeler M. Sweet-like syndrome induced by bortezomib. J Dtsch Dermatol Ges. 2013;11:1200-1202.
- Thuillier D, Lenglet A, Chaby G, et al. Bortezomib-induced eruption: Sweet syndrome? two case reports [in French]. Ann Dermatol Venereol. 2009;136:427-430.
- Kim MJ, Jang KT, Choe YH. Azathioprine hypersensitivity presenting as sweet syndrome in a child with ulcerative colitis. Indian Pediatr. 2011;48:969-971.
- Truchuelo M, Bagazgoitia L, Alcántara J, et al. Sweet-like lesions induced by bortezomib: a review of the literature and a report of 2 cases. Actas Dermosifiliogr. 2012;103:829-831.
- Hoelt P, Fattouh K, Villani AP. Dermpath & clinic: drug-induced Sweet syndrome. Eur J Dermatol. 2016;26:641-642.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
Practice Points
- The rate of medication-induced Sweet syndrome is on the rise.
- Oral acetaminophen-codeine may induce Sweet syndrome.

Melanotrichoblastoma: A Rare Pigmented Variant of Trichoblastoma
Trichoblastomas are rare cutaneous tumors that recapitulate the germinative hair bulb and the surrounding mesenchyme. Although benign, they can present diagnostic difficulties for both the clinician and pathologist because of their rarity and overlap both clinically and microscopically with other follicular neoplasms as well as basal cell carcinoma (BCC). Several classification schemes for hair follicle neoplasms have been established based on the relative proportions of epithelial and mesenchymal components as well as stromal inductive change, but nomenclature continues to be problematic, as individual neoplasms show varying degrees of differentiation that do not always uniformly fit within these categories.1,2 One of these established categories is a pigmented trichoblastoma.3 An exceedingly rare variant of a pigmented trichoblastoma referred to as melanotrichoblastoma was first described in 20024 and has only been documented in 3 cases, according to a PubMed search of articles indexed for MEDLINE using the term melanotrichoblastoma.4-6 We report another case of this rare tumor and review the literature on this unique group of tumors.
Case Report
A 25-year-old white woman with a medical history of chronic migraines, myofascial syndrome, and Arnold-Chiari malformation type I presented to dermatology with a 1.5-cm, pedunculated, well-circumscribed tumor on the left side of the scalp (Figure 1). The tumor was grossly flesh colored with heterogeneous areas of dark pigmentation. Microscopic examination demonstrated that within the superficial and deep dermis were variable-sized nests of basaloid cells. Some of the nests had large central cystic spaces with brown pigment within some of these spaces and focal pigmentation of the basaloid cells (Figure 2A). Focal areas of keratinization were present. Mitotic figures were easily identified; however, no atypical mitotic figures were present. Areas of peripheral palisading were present but there was no retraction artifact. Connection to the overlying epidermis was not identified. Surrounding the basaloid nodules was a mildly cellular proliferation of cytologically bland spindle cells. Occasional pigment-laden macrophages were present in the dermis. Focal areas suggestive of papillary mesenchymal body formation were present (Figure 2B). Immunohistochemical staining for Melan-A was performed and demonstrated the presence of a prominent number of melanocytes in some of the nests (Figure 3) and minimal to no melanocytes in other nests. There was no evidence of a melanocytic lesion involving the overlying epidermis. Features of nevus sebaceus were not present. Immunohistochemical staining for cytokeratin (CK) 20 was performed and demonstrated no notable number of Merkel cells within the lesion.
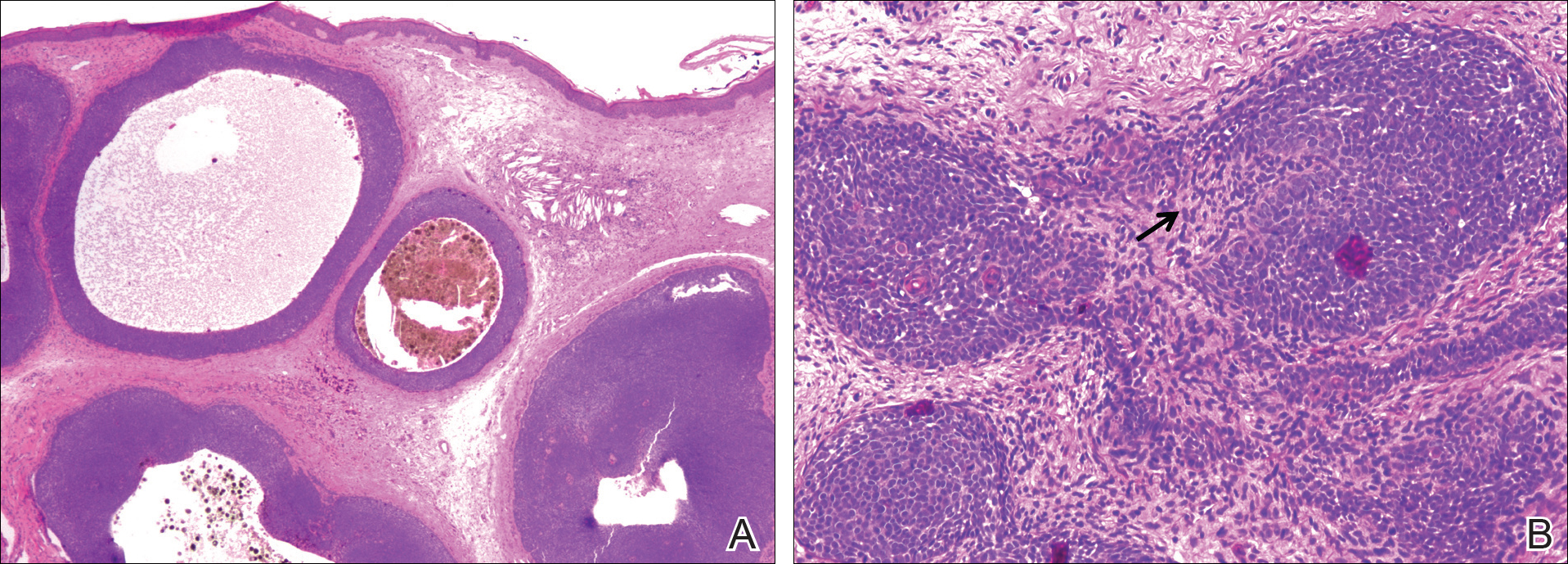
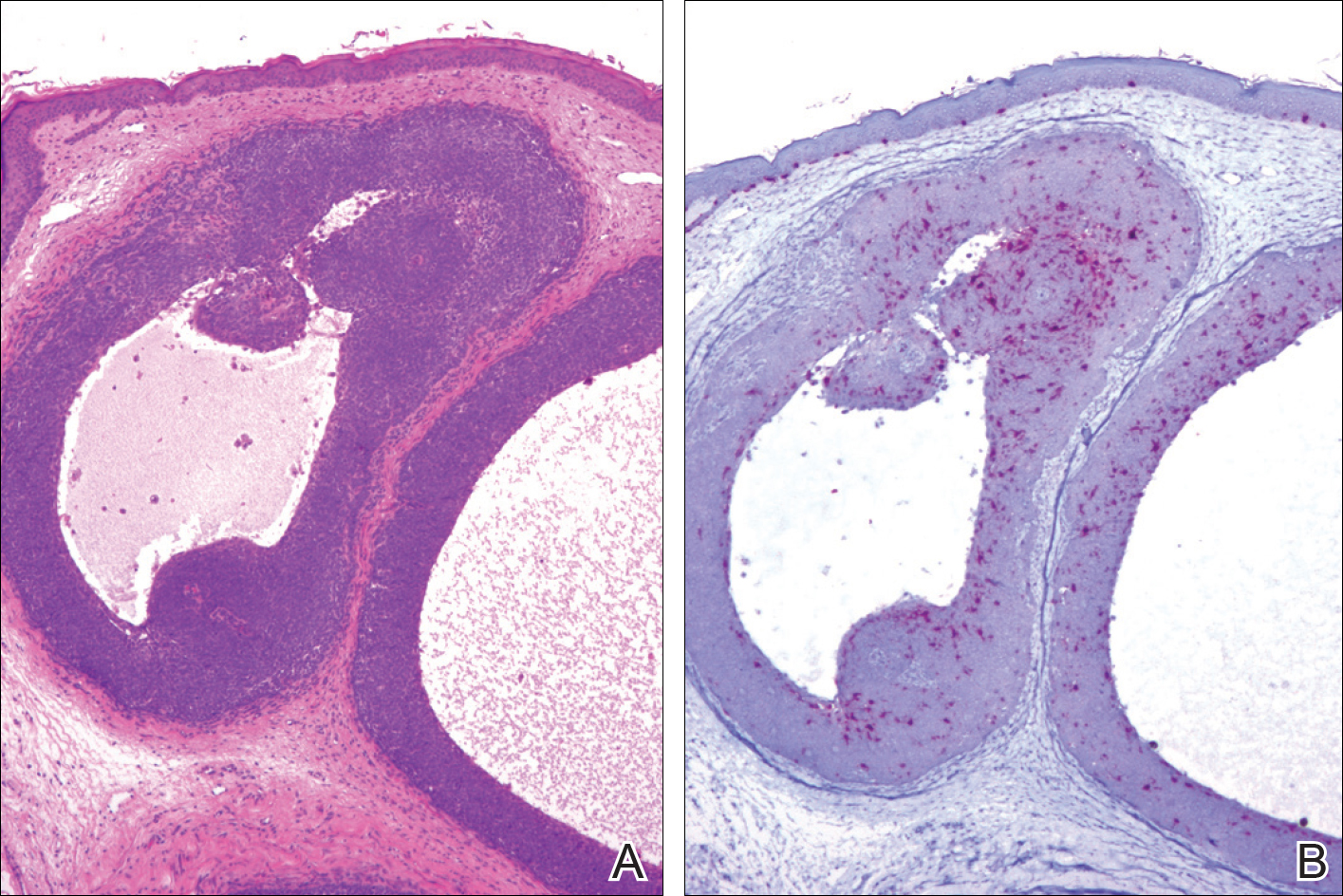
Comment
Overview of Trichoblastomas
Trichoblastomas most often present as solitary, flesh-colored, well-circumscribed, slow-growing tumors that usually progress in size over months to years. Although they may be present at any age, they most commonly occur in adults in the fifth to seventh decades of life and are equally distributed between males and females.7,8 They most often occur on the head and neck with a predilection for the scalp. Although they behave in a benign fashion, cases of malignant trichoblastomas have been reported.9
Histopathology
Histologically, these tumors are well circumscribed but unencapsulated and usually located within the deep dermis, often with extension into the subcutaneous tissue. An epidermal connection is not identified. The tumor typically is composed of variable-sized nests of basaloid cells surrounded by a variable cellular stromal component. Although peripheral palisading is present in the basaloid component, retraction artifact is not present. Several histologic variants of trichoblastomas have been reported including cribriform, racemiform, retiform, pigmented, giant, subcutaneous, rippled pattern, and clear cell.5 Pigmented trichoblastomas are histologically similar to typical trichoblastomas, except for the presence of large amounts of melanin deposited within and around the tumor nests.6 A melanotrichoblastoma is a rare variant of a pigmented trichoblastoma; pigment is present in the lesion and melanocytes are identified within the basaloid nests.
The stromal component of trichoblastomas may show areas of condensation associated with some of the basaloid cells, resembling an attempt at hair bulb formation. Staining for CD10 will be positive in these areas of papillary mesenchymal bodies.10
In an immunohistochemical study of 13 cases of trichoblastomas, there was diffuse positive staining for CK14 and CK17 in all cases (similar to BCC) and positive staining for CK19 in 70% (9/13) of cases compared to 21% (4/19) of BCC cases. Staining for CK8 and CK20 demonstrated the presence of numerous Merkel cells in all trichoblastomas but in none of the 19 cases of BCC tested.11 However, other studies have reported the presence of Merkel cells in only 42% to 70% of trichoblastomas.12,13 Despite the lack of Merkel cells in our case, the lesion was interpreted as a melanotrichoblastoma based on the histologic features in conjunction with the presence of the melanocytes.
Differential Diagnosis
The clinical and histologic differential diagnosis of trichoblastomas includes both trichoepithelioma and BCC. Clinically, all 3 lesions often are slow growing, dome shaped, and small in size (several millimeters), and are observed in the same anatomic distribution of the head and neck region. Furthermore, they often affect middle-aged to older individuals and those of Caucasian descent, though other ethnicities can be affected. Histologic evaluation often is necessary to differentiate between these 3 entities.
Histologically, trichoepitheliomas are composed of nodules of basaloid cells encircled by stromal spindle cells. Although there can be histologic overlap between trichoepitheliomas and trichoblastoma, trichoepitheliomas typically will display obvious features of hair follicle differentiation with the presence of small keratinous cysts and hair bulb structures, while trichoblastomas tend to display minimal changes suggestive of its hair follicle origin. Similar to trichoblastomas, BCC is composed of nests of basaloid cells; however, BCCs often demonstrate retraction artifact and connection to the overlying epidermis. In addition, BCCs typically demonstrate a fibromucinous stromal component that is distinct from the cellular stroma of trichoblastic tumors. Immunoperoxidase staining for androgen receptors has been reported to be positive in 78% (25/32) of BCCs and negative in trichoblastic tumors.14
Melanotrichoblastoma Differentiating Characteristics
An exceedingly rare variant of pigmented trichoblastoma is the melanotrichoblastoma. There are clinical and histologic similarities and differences between the reported cases. The first case, described by Kanitakis et al,4 reported a 32-year-old black woman with a 2-cm scalp mass that slowly enlarged over the course of 2 years. The second case, presented by Kim et al,5 described a 51-year-old Korean man with a subcutaneous 6-cm mass on the back that had been present and slowly enlarging over the course of 5 years. The third case, reported by Hung et al,6 described a 34-year-old Taiwanese man with a 1-cm, left-sided, temporal scalp mass present for 3 years, arising from a nevus sebaceous. Comparing these clinical findings with our case of a 25-year-old white woman with a 1.5-cm mass on the left side of the scalp, melanotrichoblastomas demonstrate a relatively similar age of onset in the early to middle-aged adult years. All 4 tumors were slow growing. Additionally, 3 of 4 cases demonstrated a predilection for the head, particularly the scalp, and grossly showed well-circumscribed lesions with notable pigmentation. Although age, size, location, and gross appearance were similar, a comparable ethnic and gender demographic was not identified.
Microscopic similarities between the 4 cases were present. Each case was characterized by a large, well-circumscribed, unencapsulated, basaloid tumor present in the lower dermis, with only 1 case having tumor cells occasionally reaching the undersurface of the epidermis. The tumor cells were monomorphic round-ovoid in appearance with scant cytoplasm. There was melanin pigment in the basaloid nests. The basaloid nests were surrounded by a proliferation of stromal cells. The mitotic rate was sparse in 2 cases, brisk in 1 case, and not discussed in 1 case. Melanocytes were identified in the basaloid nests in all 4 cases; however, in the current case, the melanocytes were seen in only some of the nests. None of the cases exhibited an overlying junctional melanocytic lesion, which would argue against a possible collision tumor or colonization of an epithelial lesion by a melanocytic lesion.
Although the histologic features of our cases are consistent with prior reports of melanotrichoblastoma, there is some question as to whether it represents a true variant of a pigmented trichoblastoma. There are relatively few articles in the literature that describe pigmented trichoblastomas, and of those, immunohistochemistry staining for melanocytes is uncommon. In one of the earliest descriptions of a pigmented trichoblastoma, dendritic melanocytes were present within the tumor lobules; however, the lesion was reported as a pigmented trichoblastoma and not a melanotrichoblastoma.3 It is possible that all pigmented trichoblastomas may contain some number of dendritic melanocytes, thus negating the existence of a melanotrichoblastoma as a true subtype of pigmented trichoblastomas. Additional study looking at multiple examples of pigmented trichoblastomas would be required to more definitively classify melanotrichoblastomas. It is important to appreciate that at least some cases of pigmented trichoblastomas may contain melanocytes and not to confuse the lesion as representing an example of colonization or collision tumor. A rare case of melanoma possibly arising from these dendritic melanocytes has been reported.15
Conclusion
Trichoblastomas are uncommon tumors of germinative hair bulb origin that can have several histologic variants. A well-documented subtype of trichoblastoma characterized by melanin deposits within and around tumor nests has been identified and classified as a pigmented trichoblastoma. Four cases of melanotrichoblastoma have been reported and represent a variant of a pigmented trichoblastoma characterized by the presence of melanocytes within the lesion. Whether they represent a true variant is of some debate and additional study is required. Although these tumors are exceedingly rare, it is important for the clinician and pathologist to be aware of this entity to prevent confusion with other similarly appearing follicular lesions, most notably BCCs, because of the difference in treatment and follow-up.
- Headington JT. Tumors of the hair follicle: a review. Am J Pathol. 1976; 85 : 479- 514 .
- Wong TY, Reed JA, Suster S, et al. Benign trichogenic tumors: a report of two cases supporting a simplified nomenclature. Histopathology. 1993;22:575-580.
- Aloi F, Tomasini C, Pippione M. Pigmented trichoblastoma. Am J Dermatopathol. 1992;14:345-349.
- Kanitakis J, Brutzkus A, Butnaru AC, et al. Melanotrichoblastoma: immunohistochemical study of a variant of pigmented trichoblastoma. Am J Dermatopathol. 2002;24:498-501.
- Kim DW, Lee JH, Kim I. Giant melanotrichoblastoma. Am J Dermatopathol. 2011;33:E37-E40.
- Hung CT, Chiang CP, Gao HW, et al. Ripple-pattern melanotrichoblastoma arising within nevus sebaceous. Indian J Dermatol Venereol Leprol. 2012;78:665.
- Sau P, Lupton GP, Graham JH. Trichogerminoma: report of 14 cases. J Cutan Pathol. 1992;19:357-365.
- Johnson TV, Wojno TH, Grossniklaus HE. Trichoblastoma of the eyelid. Ophthal Plast Reconstr Surg. 2011;27:E148-E149.
- Schulz T, Proske S, Hartschuh W, et al. High-grade trichoblasticcarcinoma arising in trichoblastoma: a rare adnexal neoplasm often showing metastatic spread. Am J Dermatopathol. 2005;27:9-16.
- Aslani FS, Akbarzadeh-Jahromi M, Jowkar F. Value of CD10 expression in differentiating cutaneous basal from squamous cell carcinomas and basal cell carcinoma from trichoepithelioma. Iran J Med Sci. 2013;38:100-106.
- Kurzen H, Esposito L, Langbein L, et al. Cytokeratins as markers of follicular differentiation: an immunohistochemical study of trichoblastoma and basal cell carcinoma. Am J Dermatopathol. 2001;23:501-509.
- Schulz T, Hartschuh W. Merkel cells are absent in basal cell carcinoma but frequently found in trichoblastomas. an immunohistochemical study. J Cutan Pathol. 1997;24:14-24.
- McNiff JM, Eisen RN, Glusac EJ. Immunohistochemical comparison of cutaneous lymphadenoma, trichoblastoma, and basal cell carcinoma: support for classification of lymphadenoma as a variant of trichoblastoma. J Cutan Pathol. 1999;26:119-124.
- Izikson L, Bhan A, Zembowicz A. Androgen receptor expression helps to differentiate basal cell carcinoma from benign trichoblastic tumors. Am J Dermatopathol. 2005;27:91-95.
- Benaim G, Castillo C, Houang M, et al. Melanoma arising from a long standing pigmented trichoblastoma: clinicopathologic study with complementary aCGH/mutation analysis. Am J Dermatopathol. 2014;36:E146-E151.
Trichoblastomas are rare cutaneous tumors that recapitulate the germinative hair bulb and the surrounding mesenchyme. Although benign, they can present diagnostic difficulties for both the clinician and pathologist because of their rarity and overlap both clinically and microscopically with other follicular neoplasms as well as basal cell carcinoma (BCC). Several classification schemes for hair follicle neoplasms have been established based on the relative proportions of epithelial and mesenchymal components as well as stromal inductive change, but nomenclature continues to be problematic, as individual neoplasms show varying degrees of differentiation that do not always uniformly fit within these categories.1,2 One of these established categories is a pigmented trichoblastoma.3 An exceedingly rare variant of a pigmented trichoblastoma referred to as melanotrichoblastoma was first described in 20024 and has only been documented in 3 cases, according to a PubMed search of articles indexed for MEDLINE using the term melanotrichoblastoma.4-6 We report another case of this rare tumor and review the literature on this unique group of tumors.
Case Report
A 25-year-old white woman with a medical history of chronic migraines, myofascial syndrome, and Arnold-Chiari malformation type I presented to dermatology with a 1.5-cm, pedunculated, well-circumscribed tumor on the left side of the scalp (Figure 1). The tumor was grossly flesh colored with heterogeneous areas of dark pigmentation. Microscopic examination demonstrated that within the superficial and deep dermis were variable-sized nests of basaloid cells. Some of the nests had large central cystic spaces with brown pigment within some of these spaces and focal pigmentation of the basaloid cells (Figure 2A). Focal areas of keratinization were present. Mitotic figures were easily identified; however, no atypical mitotic figures were present. Areas of peripheral palisading were present but there was no retraction artifact. Connection to the overlying epidermis was not identified. Surrounding the basaloid nodules was a mildly cellular proliferation of cytologically bland spindle cells. Occasional pigment-laden macrophages were present in the dermis. Focal areas suggestive of papillary mesenchymal body formation were present (Figure 2B). Immunohistochemical staining for Melan-A was performed and demonstrated the presence of a prominent number of melanocytes in some of the nests (Figure 3) and minimal to no melanocytes in other nests. There was no evidence of a melanocytic lesion involving the overlying epidermis. Features of nevus sebaceus were not present. Immunohistochemical staining for cytokeratin (CK) 20 was performed and demonstrated no notable number of Merkel cells within the lesion.
Comment
Overview of Trichoblastomas
Trichoblastomas most often present as solitary, flesh-colored, well-circumscribed, slow-growing tumors that usually progress in size over months to years. Although they may be present at any age, they most commonly occur in adults in the fifth to seventh decades of life and are equally distributed between males and females.7,8 They most often occur on the head and neck with a predilection for the scalp. Although they behave in a benign fashion, cases of malignant trichoblastomas have been reported.9
Histopathology
Histologically, these tumors are well circumscribed but unencapsulated and usually located within the deep dermis, often with extension into the subcutaneous tissue. An epidermal connection is not identified. The tumor typically is composed of variable-sized nests of basaloid cells surrounded by a variable cellular stromal component. Although peripheral palisading is present in the basaloid component, retraction artifact is not present. Several histologic variants of trichoblastomas have been reported including cribriform, racemiform, retiform, pigmented, giant, subcutaneous, rippled pattern, and clear cell.5 Pigmented trichoblastomas are histologically similar to typical trichoblastomas, except for the presence of large amounts of melanin deposited within and around the tumor nests.6 A melanotrichoblastoma is a rare variant of a pigmented trichoblastoma; pigment is present in the lesion and melanocytes are identified within the basaloid nests.
The stromal component of trichoblastomas may show areas of condensation associated with some of the basaloid cells, resembling an attempt at hair bulb formation. Staining for CD10 will be positive in these areas of papillary mesenchymal bodies.10
In an immunohistochemical study of 13 cases of trichoblastomas, there was diffuse positive staining for CK14 and CK17 in all cases (similar to BCC) and positive staining for CK19 in 70% (9/13) of cases compared to 21% (4/19) of BCC cases. Staining for CK8 and CK20 demonstrated the presence of numerous Merkel cells in all trichoblastomas but in none of the 19 cases of BCC tested.11 However, other studies have reported the presence of Merkel cells in only 42% to 70% of trichoblastomas.12,13 Despite the lack of Merkel cells in our case, the lesion was interpreted as a melanotrichoblastoma based on the histologic features in conjunction with the presence of the melanocytes.
Differential Diagnosis
The clinical and histologic differential diagnosis of trichoblastomas includes both trichoepithelioma and BCC. Clinically, all 3 lesions often are slow growing, dome shaped, and small in size (several millimeters), and are observed in the same anatomic distribution of the head and neck region. Furthermore, they often affect middle-aged to older individuals and those of Caucasian descent, though other ethnicities can be affected. Histologic evaluation often is necessary to differentiate between these 3 entities.
Histologically, trichoepitheliomas are composed of nodules of basaloid cells encircled by stromal spindle cells. Although there can be histologic overlap between trichoepitheliomas and trichoblastoma, trichoepitheliomas typically will display obvious features of hair follicle differentiation with the presence of small keratinous cysts and hair bulb structures, while trichoblastomas tend to display minimal changes suggestive of its hair follicle origin. Similar to trichoblastomas, BCC is composed of nests of basaloid cells; however, BCCs often demonstrate retraction artifact and connection to the overlying epidermis. In addition, BCCs typically demonstrate a fibromucinous stromal component that is distinct from the cellular stroma of trichoblastic tumors. Immunoperoxidase staining for androgen receptors has been reported to be positive in 78% (25/32) of BCCs and negative in trichoblastic tumors.14
Melanotrichoblastoma Differentiating Characteristics
An exceedingly rare variant of pigmented trichoblastoma is the melanotrichoblastoma. There are clinical and histologic similarities and differences between the reported cases. The first case, described by Kanitakis et al,4 reported a 32-year-old black woman with a 2-cm scalp mass that slowly enlarged over the course of 2 years. The second case, presented by Kim et al,5 described a 51-year-old Korean man with a subcutaneous 6-cm mass on the back that had been present and slowly enlarging over the course of 5 years. The third case, reported by Hung et al,6 described a 34-year-old Taiwanese man with a 1-cm, left-sided, temporal scalp mass present for 3 years, arising from a nevus sebaceous. Comparing these clinical findings with our case of a 25-year-old white woman with a 1.5-cm mass on the left side of the scalp, melanotrichoblastomas demonstrate a relatively similar age of onset in the early to middle-aged adult years. All 4 tumors were slow growing. Additionally, 3 of 4 cases demonstrated a predilection for the head, particularly the scalp, and grossly showed well-circumscribed lesions with notable pigmentation. Although age, size, location, and gross appearance were similar, a comparable ethnic and gender demographic was not identified.
Microscopic similarities between the 4 cases were present. Each case was characterized by a large, well-circumscribed, unencapsulated, basaloid tumor present in the lower dermis, with only 1 case having tumor cells occasionally reaching the undersurface of the epidermis. The tumor cells were monomorphic round-ovoid in appearance with scant cytoplasm. There was melanin pigment in the basaloid nests. The basaloid nests were surrounded by a proliferation of stromal cells. The mitotic rate was sparse in 2 cases, brisk in 1 case, and not discussed in 1 case. Melanocytes were identified in the basaloid nests in all 4 cases; however, in the current case, the melanocytes were seen in only some of the nests. None of the cases exhibited an overlying junctional melanocytic lesion, which would argue against a possible collision tumor or colonization of an epithelial lesion by a melanocytic lesion.
Although the histologic features of our cases are consistent with prior reports of melanotrichoblastoma, there is some question as to whether it represents a true variant of a pigmented trichoblastoma. There are relatively few articles in the literature that describe pigmented trichoblastomas, and of those, immunohistochemistry staining for melanocytes is uncommon. In one of the earliest descriptions of a pigmented trichoblastoma, dendritic melanocytes were present within the tumor lobules; however, the lesion was reported as a pigmented trichoblastoma and not a melanotrichoblastoma.3 It is possible that all pigmented trichoblastomas may contain some number of dendritic melanocytes, thus negating the existence of a melanotrichoblastoma as a true subtype of pigmented trichoblastomas. Additional study looking at multiple examples of pigmented trichoblastomas would be required to more definitively classify melanotrichoblastomas. It is important to appreciate that at least some cases of pigmented trichoblastomas may contain melanocytes and not to confuse the lesion as representing an example of colonization or collision tumor. A rare case of melanoma possibly arising from these dendritic melanocytes has been reported.15
Conclusion
Trichoblastomas are uncommon tumors of germinative hair bulb origin that can have several histologic variants. A well-documented subtype of trichoblastoma characterized by melanin deposits within and around tumor nests has been identified and classified as a pigmented trichoblastoma. Four cases of melanotrichoblastoma have been reported and represent a variant of a pigmented trichoblastoma characterized by the presence of melanocytes within the lesion. Whether they represent a true variant is of some debate and additional study is required. Although these tumors are exceedingly rare, it is important for the clinician and pathologist to be aware of this entity to prevent confusion with other similarly appearing follicular lesions, most notably BCCs, because of the difference in treatment and follow-up.
Trichoblastomas are rare cutaneous tumors that recapitulate the germinative hair bulb and the surrounding mesenchyme. Although benign, they can present diagnostic difficulties for both the clinician and pathologist because of their rarity and overlap both clinically and microscopically with other follicular neoplasms as well as basal cell carcinoma (BCC). Several classification schemes for hair follicle neoplasms have been established based on the relative proportions of epithelial and mesenchymal components as well as stromal inductive change, but nomenclature continues to be problematic, as individual neoplasms show varying degrees of differentiation that do not always uniformly fit within these categories.1,2 One of these established categories is a pigmented trichoblastoma.3 An exceedingly rare variant of a pigmented trichoblastoma referred to as melanotrichoblastoma was first described in 20024 and has only been documented in 3 cases, according to a PubMed search of articles indexed for MEDLINE using the term melanotrichoblastoma.4-6 We report another case of this rare tumor and review the literature on this unique group of tumors.
Case Report
A 25-year-old white woman with a medical history of chronic migraines, myofascial syndrome, and Arnold-Chiari malformation type I presented to dermatology with a 1.5-cm, pedunculated, well-circumscribed tumor on the left side of the scalp (Figure 1). The tumor was grossly flesh colored with heterogeneous areas of dark pigmentation. Microscopic examination demonstrated that within the superficial and deep dermis were variable-sized nests of basaloid cells. Some of the nests had large central cystic spaces with brown pigment within some of these spaces and focal pigmentation of the basaloid cells (Figure 2A). Focal areas of keratinization were present. Mitotic figures were easily identified; however, no atypical mitotic figures were present. Areas of peripheral palisading were present but there was no retraction artifact. Connection to the overlying epidermis was not identified. Surrounding the basaloid nodules was a mildly cellular proliferation of cytologically bland spindle cells. Occasional pigment-laden macrophages were present in the dermis. Focal areas suggestive of papillary mesenchymal body formation were present (Figure 2B). Immunohistochemical staining for Melan-A was performed and demonstrated the presence of a prominent number of melanocytes in some of the nests (Figure 3) and minimal to no melanocytes in other nests. There was no evidence of a melanocytic lesion involving the overlying epidermis. Features of nevus sebaceus were not present. Immunohistochemical staining for cytokeratin (CK) 20 was performed and demonstrated no notable number of Merkel cells within the lesion.
Comment
Overview of Trichoblastomas
Trichoblastomas most often present as solitary, flesh-colored, well-circumscribed, slow-growing tumors that usually progress in size over months to years. Although they may be present at any age, they most commonly occur in adults in the fifth to seventh decades of life and are equally distributed between males and females.7,8 They most often occur on the head and neck with a predilection for the scalp. Although they behave in a benign fashion, cases of malignant trichoblastomas have been reported.9
Histopathology
Histologically, these tumors are well circumscribed but unencapsulated and usually located within the deep dermis, often with extension into the subcutaneous tissue. An epidermal connection is not identified. The tumor typically is composed of variable-sized nests of basaloid cells surrounded by a variable cellular stromal component. Although peripheral palisading is present in the basaloid component, retraction artifact is not present. Several histologic variants of trichoblastomas have been reported including cribriform, racemiform, retiform, pigmented, giant, subcutaneous, rippled pattern, and clear cell.5 Pigmented trichoblastomas are histologically similar to typical trichoblastomas, except for the presence of large amounts of melanin deposited within and around the tumor nests.6 A melanotrichoblastoma is a rare variant of a pigmented trichoblastoma; pigment is present in the lesion and melanocytes are identified within the basaloid nests.
The stromal component of trichoblastomas may show areas of condensation associated with some of the basaloid cells, resembling an attempt at hair bulb formation. Staining for CD10 will be positive in these areas of papillary mesenchymal bodies.10
In an immunohistochemical study of 13 cases of trichoblastomas, there was diffuse positive staining for CK14 and CK17 in all cases (similar to BCC) and positive staining for CK19 in 70% (9/13) of cases compared to 21% (4/19) of BCC cases. Staining for CK8 and CK20 demonstrated the presence of numerous Merkel cells in all trichoblastomas but in none of the 19 cases of BCC tested.11 However, other studies have reported the presence of Merkel cells in only 42% to 70% of trichoblastomas.12,13 Despite the lack of Merkel cells in our case, the lesion was interpreted as a melanotrichoblastoma based on the histologic features in conjunction with the presence of the melanocytes.
Differential Diagnosis
The clinical and histologic differential diagnosis of trichoblastomas includes both trichoepithelioma and BCC. Clinically, all 3 lesions often are slow growing, dome shaped, and small in size (several millimeters), and are observed in the same anatomic distribution of the head and neck region. Furthermore, they often affect middle-aged to older individuals and those of Caucasian descent, though other ethnicities can be affected. Histologic evaluation often is necessary to differentiate between these 3 entities.
Histologically, trichoepitheliomas are composed of nodules of basaloid cells encircled by stromal spindle cells. Although there can be histologic overlap between trichoepitheliomas and trichoblastoma, trichoepitheliomas typically will display obvious features of hair follicle differentiation with the presence of small keratinous cysts and hair bulb structures, while trichoblastomas tend to display minimal changes suggestive of its hair follicle origin. Similar to trichoblastomas, BCC is composed of nests of basaloid cells; however, BCCs often demonstrate retraction artifact and connection to the overlying epidermis. In addition, BCCs typically demonstrate a fibromucinous stromal component that is distinct from the cellular stroma of trichoblastic tumors. Immunoperoxidase staining for androgen receptors has been reported to be positive in 78% (25/32) of BCCs and negative in trichoblastic tumors.14
Melanotrichoblastoma Differentiating Characteristics
An exceedingly rare variant of pigmented trichoblastoma is the melanotrichoblastoma. There are clinical and histologic similarities and differences between the reported cases. The first case, described by Kanitakis et al,4 reported a 32-year-old black woman with a 2-cm scalp mass that slowly enlarged over the course of 2 years. The second case, presented by Kim et al,5 described a 51-year-old Korean man with a subcutaneous 6-cm mass on the back that had been present and slowly enlarging over the course of 5 years. The third case, reported by Hung et al,6 described a 34-year-old Taiwanese man with a 1-cm, left-sided, temporal scalp mass present for 3 years, arising from a nevus sebaceous. Comparing these clinical findings with our case of a 25-year-old white woman with a 1.5-cm mass on the left side of the scalp, melanotrichoblastomas demonstrate a relatively similar age of onset in the early to middle-aged adult years. All 4 tumors were slow growing. Additionally, 3 of 4 cases demonstrated a predilection for the head, particularly the scalp, and grossly showed well-circumscribed lesions with notable pigmentation. Although age, size, location, and gross appearance were similar, a comparable ethnic and gender demographic was not identified.
Microscopic similarities between the 4 cases were present. Each case was characterized by a large, well-circumscribed, unencapsulated, basaloid tumor present in the lower dermis, with only 1 case having tumor cells occasionally reaching the undersurface of the epidermis. The tumor cells were monomorphic round-ovoid in appearance with scant cytoplasm. There was melanin pigment in the basaloid nests. The basaloid nests were surrounded by a proliferation of stromal cells. The mitotic rate was sparse in 2 cases, brisk in 1 case, and not discussed in 1 case. Melanocytes were identified in the basaloid nests in all 4 cases; however, in the current case, the melanocytes were seen in only some of the nests. None of the cases exhibited an overlying junctional melanocytic lesion, which would argue against a possible collision tumor or colonization of an epithelial lesion by a melanocytic lesion.
Although the histologic features of our cases are consistent with prior reports of melanotrichoblastoma, there is some question as to whether it represents a true variant of a pigmented trichoblastoma. There are relatively few articles in the literature that describe pigmented trichoblastomas, and of those, immunohistochemistry staining for melanocytes is uncommon. In one of the earliest descriptions of a pigmented trichoblastoma, dendritic melanocytes were present within the tumor lobules; however, the lesion was reported as a pigmented trichoblastoma and not a melanotrichoblastoma.3 It is possible that all pigmented trichoblastomas may contain some number of dendritic melanocytes, thus negating the existence of a melanotrichoblastoma as a true subtype of pigmented trichoblastomas. Additional study looking at multiple examples of pigmented trichoblastomas would be required to more definitively classify melanotrichoblastomas. It is important to appreciate that at least some cases of pigmented trichoblastomas may contain melanocytes and not to confuse the lesion as representing an example of colonization or collision tumor. A rare case of melanoma possibly arising from these dendritic melanocytes has been reported.15
Conclusion
Trichoblastomas are uncommon tumors of germinative hair bulb origin that can have several histologic variants. A well-documented subtype of trichoblastoma characterized by melanin deposits within and around tumor nests has been identified and classified as a pigmented trichoblastoma. Four cases of melanotrichoblastoma have been reported and represent a variant of a pigmented trichoblastoma characterized by the presence of melanocytes within the lesion. Whether they represent a true variant is of some debate and additional study is required. Although these tumors are exceedingly rare, it is important for the clinician and pathologist to be aware of this entity to prevent confusion with other similarly appearing follicular lesions, most notably BCCs, because of the difference in treatment and follow-up.
- Headington JT. Tumors of the hair follicle: a review. Am J Pathol. 1976; 85 : 479- 514 .
- Wong TY, Reed JA, Suster S, et al. Benign trichogenic tumors: a report of two cases supporting a simplified nomenclature. Histopathology. 1993;22:575-580.
- Aloi F, Tomasini C, Pippione M. Pigmented trichoblastoma. Am J Dermatopathol. 1992;14:345-349.
- Kanitakis J, Brutzkus A, Butnaru AC, et al. Melanotrichoblastoma: immunohistochemical study of a variant of pigmented trichoblastoma. Am J Dermatopathol. 2002;24:498-501.
- Kim DW, Lee JH, Kim I. Giant melanotrichoblastoma. Am J Dermatopathol. 2011;33:E37-E40.
- Hung CT, Chiang CP, Gao HW, et al. Ripple-pattern melanotrichoblastoma arising within nevus sebaceous. Indian J Dermatol Venereol Leprol. 2012;78:665.
- Sau P, Lupton GP, Graham JH. Trichogerminoma: report of 14 cases. J Cutan Pathol. 1992;19:357-365.
- Johnson TV, Wojno TH, Grossniklaus HE. Trichoblastoma of the eyelid. Ophthal Plast Reconstr Surg. 2011;27:E148-E149.
- Schulz T, Proske S, Hartschuh W, et al. High-grade trichoblasticcarcinoma arising in trichoblastoma: a rare adnexal neoplasm often showing metastatic spread. Am J Dermatopathol. 2005;27:9-16.
- Aslani FS, Akbarzadeh-Jahromi M, Jowkar F. Value of CD10 expression in differentiating cutaneous basal from squamous cell carcinomas and basal cell carcinoma from trichoepithelioma. Iran J Med Sci. 2013;38:100-106.
- Kurzen H, Esposito L, Langbein L, et al. Cytokeratins as markers of follicular differentiation: an immunohistochemical study of trichoblastoma and basal cell carcinoma. Am J Dermatopathol. 2001;23:501-509.
- Schulz T, Hartschuh W. Merkel cells are absent in basal cell carcinoma but frequently found in trichoblastomas. an immunohistochemical study. J Cutan Pathol. 1997;24:14-24.
- McNiff JM, Eisen RN, Glusac EJ. Immunohistochemical comparison of cutaneous lymphadenoma, trichoblastoma, and basal cell carcinoma: support for classification of lymphadenoma as a variant of trichoblastoma. J Cutan Pathol. 1999;26:119-124.
- Izikson L, Bhan A, Zembowicz A. Androgen receptor expression helps to differentiate basal cell carcinoma from benign trichoblastic tumors. Am J Dermatopathol. 2005;27:91-95.
- Benaim G, Castillo C, Houang M, et al. Melanoma arising from a long standing pigmented trichoblastoma: clinicopathologic study with complementary aCGH/mutation analysis. Am J Dermatopathol. 2014;36:E146-E151.
- Headington JT. Tumors of the hair follicle: a review. Am J Pathol. 1976; 85 : 479- 514 .
- Wong TY, Reed JA, Suster S, et al. Benign trichogenic tumors: a report of two cases supporting a simplified nomenclature. Histopathology. 1993;22:575-580.
- Aloi F, Tomasini C, Pippione M. Pigmented trichoblastoma. Am J Dermatopathol. 1992;14:345-349.
- Kanitakis J, Brutzkus A, Butnaru AC, et al. Melanotrichoblastoma: immunohistochemical study of a variant of pigmented trichoblastoma. Am J Dermatopathol. 2002;24:498-501.
- Kim DW, Lee JH, Kim I. Giant melanotrichoblastoma. Am J Dermatopathol. 2011;33:E37-E40.
- Hung CT, Chiang CP, Gao HW, et al. Ripple-pattern melanotrichoblastoma arising within nevus sebaceous. Indian J Dermatol Venereol Leprol. 2012;78:665.
- Sau P, Lupton GP, Graham JH. Trichogerminoma: report of 14 cases. J Cutan Pathol. 1992;19:357-365.
- Johnson TV, Wojno TH, Grossniklaus HE. Trichoblastoma of the eyelid. Ophthal Plast Reconstr Surg. 2011;27:E148-E149.
- Schulz T, Proske S, Hartschuh W, et al. High-grade trichoblasticcarcinoma arising in trichoblastoma: a rare adnexal neoplasm often showing metastatic spread. Am J Dermatopathol. 2005;27:9-16.
- Aslani FS, Akbarzadeh-Jahromi M, Jowkar F. Value of CD10 expression in differentiating cutaneous basal from squamous cell carcinomas and basal cell carcinoma from trichoepithelioma. Iran J Med Sci. 2013;38:100-106.
- Kurzen H, Esposito L, Langbein L, et al. Cytokeratins as markers of follicular differentiation: an immunohistochemical study of trichoblastoma and basal cell carcinoma. Am J Dermatopathol. 2001;23:501-509.
- Schulz T, Hartschuh W. Merkel cells are absent in basal cell carcinoma but frequently found in trichoblastomas. an immunohistochemical study. J Cutan Pathol. 1997;24:14-24.
- McNiff JM, Eisen RN, Glusac EJ. Immunohistochemical comparison of cutaneous lymphadenoma, trichoblastoma, and basal cell carcinoma: support for classification of lymphadenoma as a variant of trichoblastoma. J Cutan Pathol. 1999;26:119-124.
- Izikson L, Bhan A, Zembowicz A. Androgen receptor expression helps to differentiate basal cell carcinoma from benign trichoblastic tumors. Am J Dermatopathol. 2005;27:91-95.
- Benaim G, Castillo C, Houang M, et al. Melanoma arising from a long standing pigmented trichoblastoma: clinicopathologic study with complementary aCGH/mutation analysis. Am J Dermatopathol. 2014;36:E146-E151.
Practice Points
- Pigmented trichoblastoma is a histologic variant of trichoblastoma characterized by the presence of melanin pigment.
- At least some pigmented trichoblastomas contain melanocytes and have been referred to as melanotrichoblastomas.
- The presence of melanocytes within pigmented trichoblastomas should not be confused as representing an example of colonization or a collision tumor.
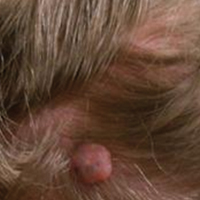
Atypical Fibroxanthoma Arising Within Erosive Pustular Dermatosis of the Scalp
Atypical fibroxanthoma (AFX) is a low-grade dermal malignancy comprised of atypical spindle cells.1 Classified as a superficial fibrohistiocytic tumor with intermediate malignant potential, AFX has an incidence of approximately 0.24% worldwide.2 The tumor appears mainly on the head and neck in sun-exposed areas but can occur less frequently on the trunk and limbs in non–sun-exposed areas. There is a 70% to 80% predominance in men aged 69 to 77 years, with lesions primarily occurring in sun-exposed areas of the head and neck.3 A median period of 4 months between time of onset and time of diagnosis has been previously established.4
When AFX does occur in non–sun-exposed areas, it tends to be in a younger patient population. Clinically, it presents as a rather nondescript, firm, erythematous papule or nodule less than 2 cm in diameter. Atypical fibroxanthoma most often presents asymptomatically, but the tumor may ulcerate and bleed, though pain and pruritus are uncommon.5 Findings are nonspecific, and the diagnosis must be confirmed with biopsy, as it can resemble other common dermatological lesions. The pathogenesis of AFX has been controversial. Two different studies looked at AFX using electron microscopy and concluded that the tumor most closely resembled a myofibroblast,6,7 which is consistent with current thinking today.
Atypical fibroxanthoma is believed to be associated with p53 mutation and is closely linked with exposure to UV radiation due to its predominance in sun-exposed areas. Other predisposing factors may include prior exposure to UV radiation, history of organ transplantation, immunosuppression, advanced age in men, and xeroderma pigmentosum. The differential diagnosis for AFX encompasses basal cell carcinoma, squamous cell carcinoma, Merkel cell carcinoma, adnexal tumor, and pyogenic granuloma.
Case Report
On physical examination, the lesions appeared erosive with crusting and granulation tissue (Figure 1A). The presentation was consistent with erosive pustular dermatosis of the scalp. Biopsy revealed granulation tissue. The patient underwent PDT and prednisone treatment with improvement. Additional biopsies revealed AKs. His condition improved with 2 PDT sessions but never fully cleared. During the PDT sessions, the patient reported intense unilateral headaches without visual changes. The headaches were intermittent and not apparently related to the treatments. He was referred for a temporal artery biopsy and rebiopsy of the remaining lesion on the scalp. The temporal artery biopsy was negative. The lesion that remained was a large nodule on the vertex scalp, and biopsy revealed AFX.
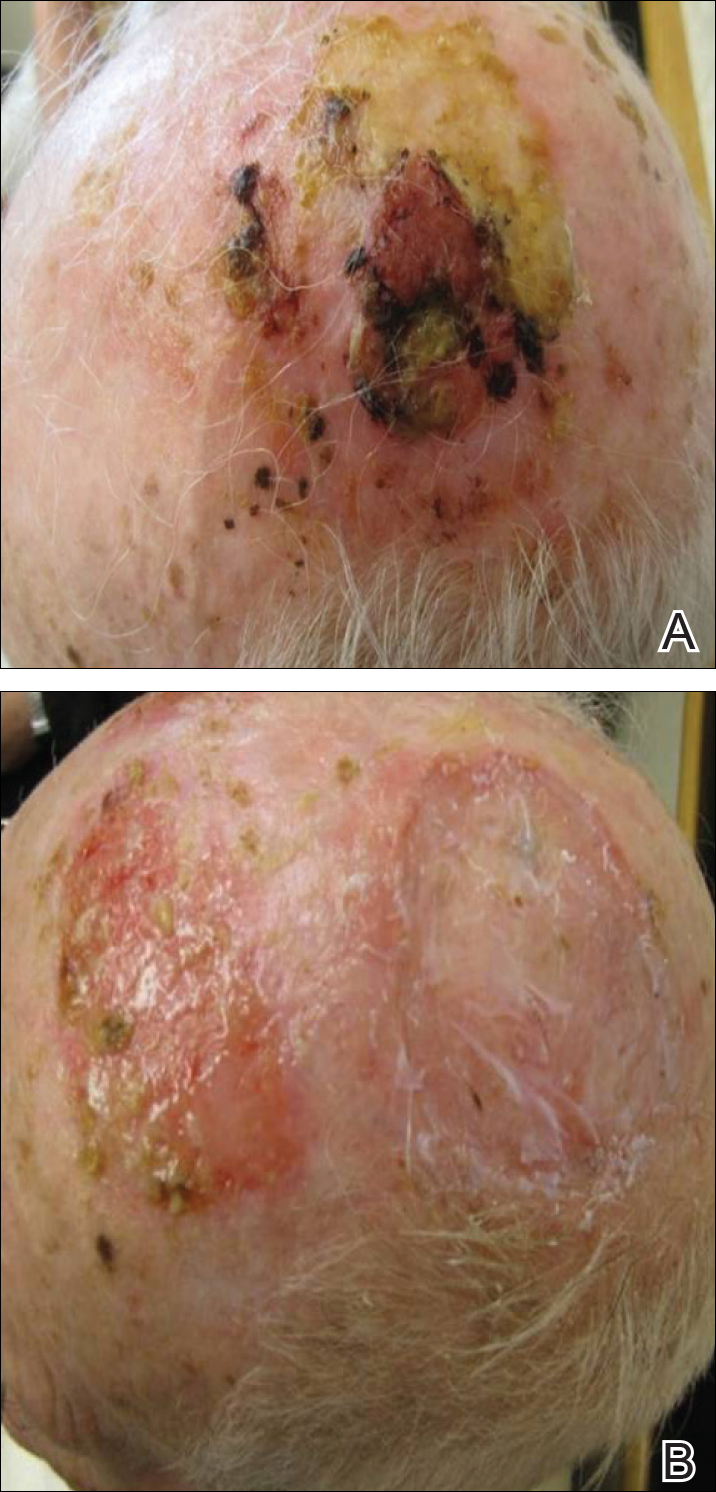
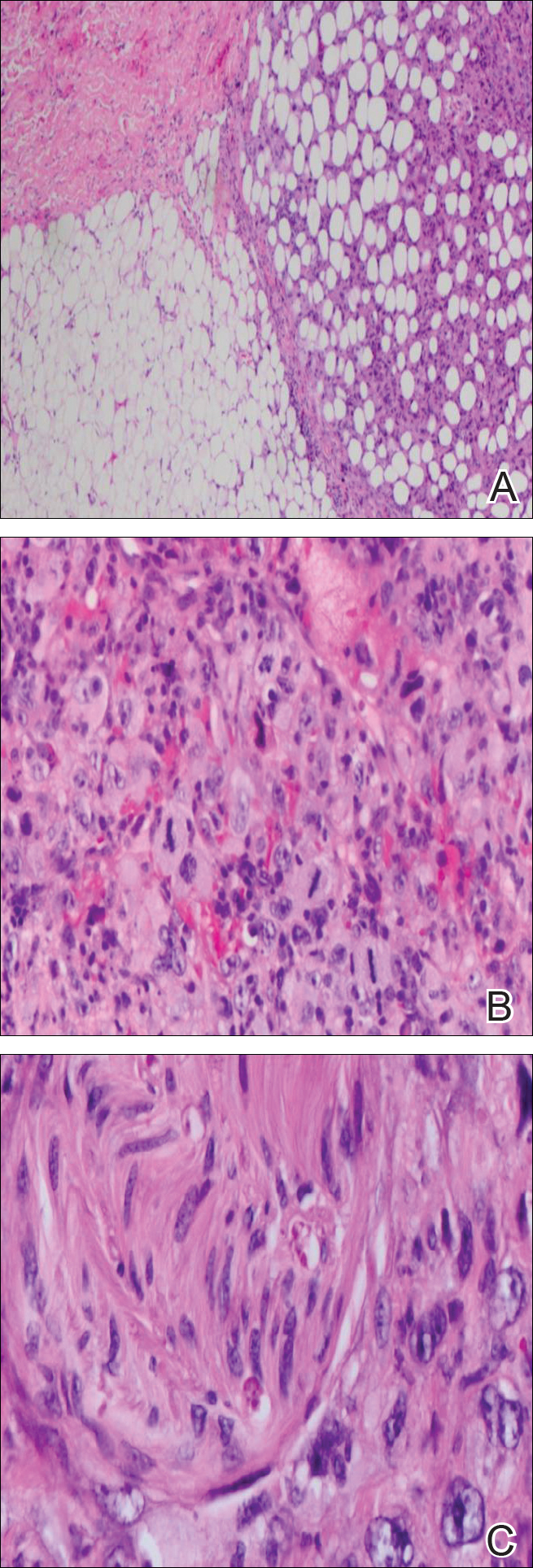
Immunohistochemical marker studies for S-100 and cytokeratin were negative. Invasion into subcutaneous fat was encountered (Figure 2A). Highly atypical spindle cells and mitoses were present (Figure 2B). Neoplastic cells were noted adjacent to nerve (Figure 2C). Excision of the lesion was curative, and his symptoms of pain and erosive pustular dermatosis resolved weeks thereafter (Figure 1B). The area of erosive pustular dermatosis was not excised, but symptoms resolved weeks following excision of the AFX.
Comment
Our case of AFX is unique due to the patient’s atypical presentation of severe pain. Because AFX usually presents asymptomatically, pain is an uncommon symptom. Based on the histologic findings in our case, we suspected that neural involvement of the tumor most likely explained the intense pain that our patient experienced.
The presence of erosive pustular dermatosis of the scalp also is interesting in our case. This elderly man had an extensive history of actinic damage and had reported pustules, scaling, itching, and scabbing of the scalp. It is possible that erosive pustular dermatosis was superimposed over the tumor and could have been the reason that multiple biopsies were needed to eventually arrive at a diagnosis. The coexistence of the 2 entities suggests that the chronic actinic damage played a role in the etiology of both.
Classification
There is a question regarding nomenclature when discussing AFX. Atypical fibroxanthoma has been referred to as a variant of undifferentiated pleomorphic sarcoma, which is a type of soft tissue sarcoma. Atypical fibroxanthoma can be referred to as undifferentiated pleomorphic sarcoma if it is more than 2 cm in diameter, if it involves the fascia or subcutaneous tissue, or if there is evidence of necrosis.3 Atypical fibroxanthoma generally is confined to the head and neck region and usually is less than 2 cm in diameter. In this patient, the presentation was consistent with AFX, as there was evidence of necrosis and invasion into the subcutaneous fat. The fact that the lesion also appeared on the scalp further supported the diagnosis of AFX.
Pathology
Biopsy of AFX typically reveals a spindle cell proliferation that usually arises in the setting of profound actinic damage. The epidermis may or may not be ulcerated, and in most cases, it is seen in close proximity to the overlying epidermis but not arising from it.8 Classic AFX is composed of highly atypical histiocytelike (epithelioid) cells admixed with pleomorphic spindle cells and giant cells, all showing frequent mitoses including atypical ones.9 Several histologic subtypes of AFX have been described, including clear cell, granular cell, pigmented cell, chondroid, osteoid, osteoclastic, and the most common spindle cell subtype.9 Features that indicate potential aggressive behavior include infiltration into the subcutaneous tissue, vascular invasion, and presence of necrosis. A diagnosis of AFX is made by exclusion of other malignant neoplasms with similar morphology, namely spindle cell squamous cell carcinoma, spindle cell melanoma, and leiomyoscarcoma.9 As such, immunohistochemistry plays a critical role in distinguishing these lesions, as they arise as part of the differential diagnosis. A panel of immunohistochemical stains is helpful for diagnosis and commonly includes but is not limited to S-100, Melan-A, smooth muscle actin, desmin, and cytokeratin.
Sampling error is an inherent flaw in any biopsy specimen. The eventual diagnosis of AFX in our case supports the argument for multiple biopsies of an unknown lesion, seeing as the affected area was interpreted as both granulation tissue and AK prior to the eventual diagnosis. Repeat biopsies, especially if a lesion is nonhealing, often can help clinicians arrive at a definitive diagnosis.
Treatment
Different treatment options have been used to manage AFX. Mohs micrographic surgery is most often used because of its tissue-sparing potential, often giving the most cosmetically appealing result. Wide local excision is another surgical technique utilized, generally with fixed margins of at least 1 cm.10 Radiation at the tumor site is used as a treatment method but most often during cases of reoccurrence. Cryotherapy as well as electrodesiccation and curettage are possible treatment options but are not the standard of care.
- Helwig EB. Atypical fibroxanthoma, in tumor seminar. proceedings of 18th Annual Seminar of San Antonio Society of Pathologists, 1961. Tex State J Med. 1963;59:664-667.
- Anderson HL, Joseph AK. A pilot feasibility study of a rare skin tumor database. Dermatol Surg. 2007;33:693-696.
- Iorizzo LJ 3rd, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Fretzin DF, Helwig EB. Atypical fibroxanthoma of the skin. a clinicopathologic study of 140 cases. Cancer. 1973;31:1541-1552.
- Vandergriff TW, Reed JA, Orengo IF. An unusual presentation of atypical fibroxanthoma. Dermatol Online J. 2008;14:6.
- Weedon D, Kerr JF. Atypical fibroxanthoma of skin: an electron microscope study. Pathology. 1975;7:173-177.
- Woyke S, Domagala W, Olszewski W, et al. Pseudosarcoma of the skin. an electron microscopic study and comparison with the fine structure of spindle-cell variant of squamous carcinoma. Cancer. 1974;33:970-980.
- Edward S, Yung A. Essential Dermatopathology. Philadelphia, PA: Lippincott Williams & Wilkins; 2012.
- Luzar B, Calonje E. Morphologic and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol. 2010;37:301-309.
- González-García R, Nam-Cha SH, Muñoz-Guerra MF, et al. Atypical fibroxanthoma of the head and neck: report of 5 cases. J Oral Maxillofac Surg. 2007;65:526-531.
Atypical fibroxanthoma (AFX) is a low-grade dermal malignancy comprised of atypical spindle cells.1 Classified as a superficial fibrohistiocytic tumor with intermediate malignant potential, AFX has an incidence of approximately 0.24% worldwide.2 The tumor appears mainly on the head and neck in sun-exposed areas but can occur less frequently on the trunk and limbs in non–sun-exposed areas. There is a 70% to 80% predominance in men aged 69 to 77 years, with lesions primarily occurring in sun-exposed areas of the head and neck.3 A median period of 4 months between time of onset and time of diagnosis has been previously established.4
When AFX does occur in non–sun-exposed areas, it tends to be in a younger patient population. Clinically, it presents as a rather nondescript, firm, erythematous papule or nodule less than 2 cm in diameter. Atypical fibroxanthoma most often presents asymptomatically, but the tumor may ulcerate and bleed, though pain and pruritus are uncommon.5 Findings are nonspecific, and the diagnosis must be confirmed with biopsy, as it can resemble other common dermatological lesions. The pathogenesis of AFX has been controversial. Two different studies looked at AFX using electron microscopy and concluded that the tumor most closely resembled a myofibroblast,6,7 which is consistent with current thinking today.
Atypical fibroxanthoma is believed to be associated with p53 mutation and is closely linked with exposure to UV radiation due to its predominance in sun-exposed areas. Other predisposing factors may include prior exposure to UV radiation, history of organ transplantation, immunosuppression, advanced age in men, and xeroderma pigmentosum. The differential diagnosis for AFX encompasses basal cell carcinoma, squamous cell carcinoma, Merkel cell carcinoma, adnexal tumor, and pyogenic granuloma.
Case Report
On physical examination, the lesions appeared erosive with crusting and granulation tissue (Figure 1A). The presentation was consistent with erosive pustular dermatosis of the scalp. Biopsy revealed granulation tissue. The patient underwent PDT and prednisone treatment with improvement. Additional biopsies revealed AKs. His condition improved with 2 PDT sessions but never fully cleared. During the PDT sessions, the patient reported intense unilateral headaches without visual changes. The headaches were intermittent and not apparently related to the treatments. He was referred for a temporal artery biopsy and rebiopsy of the remaining lesion on the scalp. The temporal artery biopsy was negative. The lesion that remained was a large nodule on the vertex scalp, and biopsy revealed AFX.
Immunohistochemical marker studies for S-100 and cytokeratin were negative. Invasion into subcutaneous fat was encountered (Figure 2A). Highly atypical spindle cells and mitoses were present (Figure 2B). Neoplastic cells were noted adjacent to nerve (Figure 2C). Excision of the lesion was curative, and his symptoms of pain and erosive pustular dermatosis resolved weeks thereafter (Figure 1B). The area of erosive pustular dermatosis was not excised, but symptoms resolved weeks following excision of the AFX.
Comment
Our case of AFX is unique due to the patient’s atypical presentation of severe pain. Because AFX usually presents asymptomatically, pain is an uncommon symptom. Based on the histologic findings in our case, we suspected that neural involvement of the tumor most likely explained the intense pain that our patient experienced.
The presence of erosive pustular dermatosis of the scalp also is interesting in our case. This elderly man had an extensive history of actinic damage and had reported pustules, scaling, itching, and scabbing of the scalp. It is possible that erosive pustular dermatosis was superimposed over the tumor and could have been the reason that multiple biopsies were needed to eventually arrive at a diagnosis. The coexistence of the 2 entities suggests that the chronic actinic damage played a role in the etiology of both.
Classification
There is a question regarding nomenclature when discussing AFX. Atypical fibroxanthoma has been referred to as a variant of undifferentiated pleomorphic sarcoma, which is a type of soft tissue sarcoma. Atypical fibroxanthoma can be referred to as undifferentiated pleomorphic sarcoma if it is more than 2 cm in diameter, if it involves the fascia or subcutaneous tissue, or if there is evidence of necrosis.3 Atypical fibroxanthoma generally is confined to the head and neck region and usually is less than 2 cm in diameter. In this patient, the presentation was consistent with AFX, as there was evidence of necrosis and invasion into the subcutaneous fat. The fact that the lesion also appeared on the scalp further supported the diagnosis of AFX.
Pathology
Biopsy of AFX typically reveals a spindle cell proliferation that usually arises in the setting of profound actinic damage. The epidermis may or may not be ulcerated, and in most cases, it is seen in close proximity to the overlying epidermis but not arising from it.8 Classic AFX is composed of highly atypical histiocytelike (epithelioid) cells admixed with pleomorphic spindle cells and giant cells, all showing frequent mitoses including atypical ones.9 Several histologic subtypes of AFX have been described, including clear cell, granular cell, pigmented cell, chondroid, osteoid, osteoclastic, and the most common spindle cell subtype.9 Features that indicate potential aggressive behavior include infiltration into the subcutaneous tissue, vascular invasion, and presence of necrosis. A diagnosis of AFX is made by exclusion of other malignant neoplasms with similar morphology, namely spindle cell squamous cell carcinoma, spindle cell melanoma, and leiomyoscarcoma.9 As such, immunohistochemistry plays a critical role in distinguishing these lesions, as they arise as part of the differential diagnosis. A panel of immunohistochemical stains is helpful for diagnosis and commonly includes but is not limited to S-100, Melan-A, smooth muscle actin, desmin, and cytokeratin.
Sampling error is an inherent flaw in any biopsy specimen. The eventual diagnosis of AFX in our case supports the argument for multiple biopsies of an unknown lesion, seeing as the affected area was interpreted as both granulation tissue and AK prior to the eventual diagnosis. Repeat biopsies, especially if a lesion is nonhealing, often can help clinicians arrive at a definitive diagnosis.
Treatment
Different treatment options have been used to manage AFX. Mohs micrographic surgery is most often used because of its tissue-sparing potential, often giving the most cosmetically appealing result. Wide local excision is another surgical technique utilized, generally with fixed margins of at least 1 cm.10 Radiation at the tumor site is used as a treatment method but most often during cases of reoccurrence. Cryotherapy as well as electrodesiccation and curettage are possible treatment options but are not the standard of care.
Atypical fibroxanthoma (AFX) is a low-grade dermal malignancy comprised of atypical spindle cells.1 Classified as a superficial fibrohistiocytic tumor with intermediate malignant potential, AFX has an incidence of approximately 0.24% worldwide.2 The tumor appears mainly on the head and neck in sun-exposed areas but can occur less frequently on the trunk and limbs in non–sun-exposed areas. There is a 70% to 80% predominance in men aged 69 to 77 years, with lesions primarily occurring in sun-exposed areas of the head and neck.3 A median period of 4 months between time of onset and time of diagnosis has been previously established.4
When AFX does occur in non–sun-exposed areas, it tends to be in a younger patient population. Clinically, it presents as a rather nondescript, firm, erythematous papule or nodule less than 2 cm in diameter. Atypical fibroxanthoma most often presents asymptomatically, but the tumor may ulcerate and bleed, though pain and pruritus are uncommon.5 Findings are nonspecific, and the diagnosis must be confirmed with biopsy, as it can resemble other common dermatological lesions. The pathogenesis of AFX has been controversial. Two different studies looked at AFX using electron microscopy and concluded that the tumor most closely resembled a myofibroblast,6,7 which is consistent with current thinking today.
Atypical fibroxanthoma is believed to be associated with p53 mutation and is closely linked with exposure to UV radiation due to its predominance in sun-exposed areas. Other predisposing factors may include prior exposure to UV radiation, history of organ transplantation, immunosuppression, advanced age in men, and xeroderma pigmentosum. The differential diagnosis for AFX encompasses basal cell carcinoma, squamous cell carcinoma, Merkel cell carcinoma, adnexal tumor, and pyogenic granuloma.
Case Report
On physical examination, the lesions appeared erosive with crusting and granulation tissue (Figure 1A). The presentation was consistent with erosive pustular dermatosis of the scalp. Biopsy revealed granulation tissue. The patient underwent PDT and prednisone treatment with improvement. Additional biopsies revealed AKs. His condition improved with 2 PDT sessions but never fully cleared. During the PDT sessions, the patient reported intense unilateral headaches without visual changes. The headaches were intermittent and not apparently related to the treatments. He was referred for a temporal artery biopsy and rebiopsy of the remaining lesion on the scalp. The temporal artery biopsy was negative. The lesion that remained was a large nodule on the vertex scalp, and biopsy revealed AFX.
Immunohistochemical marker studies for S-100 and cytokeratin were negative. Invasion into subcutaneous fat was encountered (Figure 2A). Highly atypical spindle cells and mitoses were present (Figure 2B). Neoplastic cells were noted adjacent to nerve (Figure 2C). Excision of the lesion was curative, and his symptoms of pain and erosive pustular dermatosis resolved weeks thereafter (Figure 1B). The area of erosive pustular dermatosis was not excised, but symptoms resolved weeks following excision of the AFX.
Comment
Our case of AFX is unique due to the patient’s atypical presentation of severe pain. Because AFX usually presents asymptomatically, pain is an uncommon symptom. Based on the histologic findings in our case, we suspected that neural involvement of the tumor most likely explained the intense pain that our patient experienced.
The presence of erosive pustular dermatosis of the scalp also is interesting in our case. This elderly man had an extensive history of actinic damage and had reported pustules, scaling, itching, and scabbing of the scalp. It is possible that erosive pustular dermatosis was superimposed over the tumor and could have been the reason that multiple biopsies were needed to eventually arrive at a diagnosis. The coexistence of the 2 entities suggests that the chronic actinic damage played a role in the etiology of both.
Classification
There is a question regarding nomenclature when discussing AFX. Atypical fibroxanthoma has been referred to as a variant of undifferentiated pleomorphic sarcoma, which is a type of soft tissue sarcoma. Atypical fibroxanthoma can be referred to as undifferentiated pleomorphic sarcoma if it is more than 2 cm in diameter, if it involves the fascia or subcutaneous tissue, or if there is evidence of necrosis.3 Atypical fibroxanthoma generally is confined to the head and neck region and usually is less than 2 cm in diameter. In this patient, the presentation was consistent with AFX, as there was evidence of necrosis and invasion into the subcutaneous fat. The fact that the lesion also appeared on the scalp further supported the diagnosis of AFX.
Pathology
Biopsy of AFX typically reveals a spindle cell proliferation that usually arises in the setting of profound actinic damage. The epidermis may or may not be ulcerated, and in most cases, it is seen in close proximity to the overlying epidermis but not arising from it.8 Classic AFX is composed of highly atypical histiocytelike (epithelioid) cells admixed with pleomorphic spindle cells and giant cells, all showing frequent mitoses including atypical ones.9 Several histologic subtypes of AFX have been described, including clear cell, granular cell, pigmented cell, chondroid, osteoid, osteoclastic, and the most common spindle cell subtype.9 Features that indicate potential aggressive behavior include infiltration into the subcutaneous tissue, vascular invasion, and presence of necrosis. A diagnosis of AFX is made by exclusion of other malignant neoplasms with similar morphology, namely spindle cell squamous cell carcinoma, spindle cell melanoma, and leiomyoscarcoma.9 As such, immunohistochemistry plays a critical role in distinguishing these lesions, as they arise as part of the differential diagnosis. A panel of immunohistochemical stains is helpful for diagnosis and commonly includes but is not limited to S-100, Melan-A, smooth muscle actin, desmin, and cytokeratin.
Sampling error is an inherent flaw in any biopsy specimen. The eventual diagnosis of AFX in our case supports the argument for multiple biopsies of an unknown lesion, seeing as the affected area was interpreted as both granulation tissue and AK prior to the eventual diagnosis. Repeat biopsies, especially if a lesion is nonhealing, often can help clinicians arrive at a definitive diagnosis.
Treatment
Different treatment options have been used to manage AFX. Mohs micrographic surgery is most often used because of its tissue-sparing potential, often giving the most cosmetically appealing result. Wide local excision is another surgical technique utilized, generally with fixed margins of at least 1 cm.10 Radiation at the tumor site is used as a treatment method but most often during cases of reoccurrence. Cryotherapy as well as electrodesiccation and curettage are possible treatment options but are not the standard of care.
- Helwig EB. Atypical fibroxanthoma, in tumor seminar. proceedings of 18th Annual Seminar of San Antonio Society of Pathologists, 1961. Tex State J Med. 1963;59:664-667.
- Anderson HL, Joseph AK. A pilot feasibility study of a rare skin tumor database. Dermatol Surg. 2007;33:693-696.
- Iorizzo LJ 3rd, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Fretzin DF, Helwig EB. Atypical fibroxanthoma of the skin. a clinicopathologic study of 140 cases. Cancer. 1973;31:1541-1552.
- Vandergriff TW, Reed JA, Orengo IF. An unusual presentation of atypical fibroxanthoma. Dermatol Online J. 2008;14:6.
- Weedon D, Kerr JF. Atypical fibroxanthoma of skin: an electron microscope study. Pathology. 1975;7:173-177.
- Woyke S, Domagala W, Olszewski W, et al. Pseudosarcoma of the skin. an electron microscopic study and comparison with the fine structure of spindle-cell variant of squamous carcinoma. Cancer. 1974;33:970-980.
- Edward S, Yung A. Essential Dermatopathology. Philadelphia, PA: Lippincott Williams & Wilkins; 2012.
- Luzar B, Calonje E. Morphologic and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol. 2010;37:301-309.
- González-García R, Nam-Cha SH, Muñoz-Guerra MF, et al. Atypical fibroxanthoma of the head and neck: report of 5 cases. J Oral Maxillofac Surg. 2007;65:526-531.
- Helwig EB. Atypical fibroxanthoma, in tumor seminar. proceedings of 18th Annual Seminar of San Antonio Society of Pathologists, 1961. Tex State J Med. 1963;59:664-667.
- Anderson HL, Joseph AK. A pilot feasibility study of a rare skin tumor database. Dermatol Surg. 2007;33:693-696.
- Iorizzo LJ 3rd, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
- Fretzin DF, Helwig EB. Atypical fibroxanthoma of the skin. a clinicopathologic study of 140 cases. Cancer. 1973;31:1541-1552.
- Vandergriff TW, Reed JA, Orengo IF. An unusual presentation of atypical fibroxanthoma. Dermatol Online J. 2008;14:6.
- Weedon D, Kerr JF. Atypical fibroxanthoma of skin: an electron microscope study. Pathology. 1975;7:173-177.
- Woyke S, Domagala W, Olszewski W, et al. Pseudosarcoma of the skin. an electron microscopic study and comparison with the fine structure of spindle-cell variant of squamous carcinoma. Cancer. 1974;33:970-980.
- Edward S, Yung A. Essential Dermatopathology. Philadelphia, PA: Lippincott Williams & Wilkins; 2012.
- Luzar B, Calonje E. Morphologic and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol. 2010;37:301-309.
- González-García R, Nam-Cha SH, Muñoz-Guerra MF, et al. Atypical fibroxanthoma of the head and neck: report of 5 cases. J Oral Maxillofac Surg. 2007;65:526-531.
Practice Points
- Atypical fibroxanthoma predominantly occurs in older men on the head and neck.
- Erosive pustular dermatosis may be a benign entity, but if it does not resolve, continue to rebiopsy, as rare tumors may mimic this condition.