User login
Lessons abound for dermatologists when animal health and human health intersect
NEW YORK – We share more than affection with our dogs and cats. We also share diseases – about which our four-legged furry friends can teach us plenty.
That was the conclusion of speakers at a session on “cases at the intersection of human and veterinary dermatology,” presented at the summer meeting of the American Academy of Dermatology.
“Human health is intimately connected to animal health,” said Jennifer Gardner, MD, of the division of dermatology, University of Washington, Seattle, and a collaborating member of the school’s Center for One Health Research. The One Health framework looks at factors involved in the human, environmental, and animal sectors from the molecular level to the individual level and even to the planetary level.
Dr. Gardner challenged her audience to think beyond their individual areas of expertise. “How does the work you’re doing with a patient or test tube connect up the line and make an impact to levels higher up?” she asked.
The One Health framework also challenges practitioners to look horizontally, at how work done in the human world connects to what’s going on in the veterinary world – that is, how treatments for dermatologic conditions in dogs may one day affect how dermatologists treat the same or similar disorders in humans.
Learning from the mighty mite
For example, the study of mites that live on the skin of animals could eventually shed light on how dermatologists treat mite-related conditions in humans.
Dirk M. Elston, MD, professor and chair of the department of dermatology at the Medical University of South Carolina, Charleston, noted that Demodex mites occur in humans and in pets.

In such cases, “sulfur tends to be my most reliable” treatment, he said, noting that it releases a rotten egg smell. “You’re basically gassing the organism.” Dr. Elston said he frequently gets calls from fellow dermatologists whose antimite efforts have failed with ivermectin and permethrin and does not hesitate to give his advice. “I’m like a broken record,” he said. “Sulfur, sulfur, sulfur, sulfur.”
The Demodex mite affects dogs to varying degrees, depending on where they live, said Kathryn Rook, VMD, of the department of dermatology at the University of Pennsylvania School of Veterinary Medicine, Philadelphia. In North America, demodicosis occurs in 0.38%-0.58% of dogs, and in 25% of dogs in Mexico, she said.
Amitraz, the only Food and Drug Administration–approved treatment for canine demodicosis, is available only as a dip. But it has fallen from favor as a result of sometimes serious side effects, which can include sedation, bradycardia, ataxia, vomiting, diarrhea, and hyperglycemia.
Daily administration of oral ivermectin – often for months – also carries a risk of side effects, including dilated pupils, ataxia, sedation, stupor, coma, hypersalivation, vomiting, diarrhea, blindness, tremors, seizures, and respiratory depression.

But the discovery of isoxazoline has “revolutionized” the treatment of demodicosis and other parasitic infestations in dogs, Dr. Rook said, citing quicker resolution of disease and improved quality of life for both the patient and its owner.
Isoxazoline, which Dr. Rook said carries little risk for side effects, is licensed in the United States only as a flea and tick preventive.
Atopic dermatitis
Atopic dermatitis (AD) tends to be similar in people and dogs, according to Charles W. Bradley, DVM, of the University of Pennsylvania School of Veterinary Medicine, Philadelphia. About 10%-30% of children and up to 10% of adults have the disorder, the prevalence of which has more than doubled in recent years, he said.
In dogs, the prevalence is 10%-20%, making it “an extraordinarily common disorder,” he said. Lesions tend to be located on the feet, face, pinnae, ventrum, and axilla/inguinum. Additional sites vary by breed, with Dalmatians tending to get AD on the lips, French Bulldogs on the eyelids, German Shepherds on the elbows, Shar-Peis on the thorax, and Boxers on the ears.
In humans, Staphylococcus aureus is the chief microorganism of concern, said Elizabeth Grice, PhD, of the department of dermatology at the University of Pennsylvania, Philadelphia, who copresented the topic with Dr. Bradley.

“My true love is anything to do with the skin microbiome,” she said. “The more severe the disease, the lower the skin microbiome diversity.”
Though most studies of AD use mice as animal models, dogs would be better, according to Dr. Grice and Dr. Bradley.
That’s because canine AD occurs spontaneously and exhibits immunologic and clinical features similar to those of human AD. They include prevalence, environmental triggers, immunologic profiles, genetic predispositions, lesion distribution, and frequent colonization by Staphylococcus species. In addition, dogs and their owners tend to share the same environment.
A rash of itches
Among dermatology patients – man or beast – itch can outweigh rash as a key focus of concern, according to Brian Kim, MD, of the division of dermatology at Washington University in St. Louis, and codirector for the University’s Center for the Study of Itch. “The problem is my patients don’t complain about their rash; they complain about their itch,” he said. “But we don’t understand the basic question of itch.” In fact, the FDA has not approved any drugs for the treatment of chronic itch, he said.

For dogs, advances have been made with Janus kinase (JAK) inhibitors, which “may function as immunomodulators,” Dr. Kim said. And JAK-1 selective inhibition “may be more effective than broad JAK blockade for itch.”
‘The perfect culture plate’
Lessons can be learned from studying canine AD, which “is immunophysiologically homologous to human AD,” said Daniel O. Morris, DVM, MPH, professor of dermatology, at the University of Pennsylvania School of Veterinary Medicine, Philadelphia. “The main difference: My patients are covered in dense hair coats.” Because of that, systemic treatment is necessary, he said.
Canine AD primarily affects areas where hair is sparse or where the surface microclimate is moist, he said. A dog’s ear canal, which can be 10 times longer than a human’s, harbors plenty of moisture and heat, he said. “It’s the perfect culture plate.”
But, he added, the owners of his patients tend to resist using topical therapies “that could be potentially smeared on the babies and grandma’s diabetic foot ulcer.” So he has long relied on systemic treatments, initially steroids and cyclosporine. But they can have major side effects, and cyclosporine can take 60-90 days before it exerts maximum effect.
A faster-acting compound called oclacitinib has shown promise based on its high affinity for inhibiting JAK-1 enzyme-mediated activation of cytokine expression, including interleukin (IL)-31, he said. “Clinical trials demonstrate an antipruritic efficacy equivalent to both prednisolone and cyclosporine,” he noted. Contraindications include a history of neoplasia, the presence of severe infection, and age under 1 year.
Monoclonal antibody targets IL-31
The latest promising arrival is lokivetmab, a monoclonal antibody that targets canine IL-31, according to Dr. Morris. It acts rapidly (within 1 day for many dogs) and prevents binding of IL-31 to its neuronal receptor for at least a month, thereby interrupting neurotransmission of itch.
But side effects can be serious and common. Equal efficacy with a reduced side effect is the holy grail, he said.
Some doctors are not waiting. “People are throwing these two products at anything that itches,” he said. Unfortunately, they tend to “work miserably” for causes other than AD, he added.
Dr. Gardner, Dr. Elston, Dr. Rook, Dr. Bradley, and Dr. Morris reported no financial conflicts. Dr. Grice’s disclosures include having served as a speaker for GlaxoSmithKline and for L’Oreal France, and having received grants/research funding from Janssen Research & Development. Dr. Kim has served as a consultant to biotechnology and pharmaceutical companies.
NEW YORK – We share more than affection with our dogs and cats. We also share diseases – about which our four-legged furry friends can teach us plenty.
That was the conclusion of speakers at a session on “cases at the intersection of human and veterinary dermatology,” presented at the summer meeting of the American Academy of Dermatology.
“Human health is intimately connected to animal health,” said Jennifer Gardner, MD, of the division of dermatology, University of Washington, Seattle, and a collaborating member of the school’s Center for One Health Research. The One Health framework looks at factors involved in the human, environmental, and animal sectors from the molecular level to the individual level and even to the planetary level.
Dr. Gardner challenged her audience to think beyond their individual areas of expertise. “How does the work you’re doing with a patient or test tube connect up the line and make an impact to levels higher up?” she asked.
The One Health framework also challenges practitioners to look horizontally, at how work done in the human world connects to what’s going on in the veterinary world – that is, how treatments for dermatologic conditions in dogs may one day affect how dermatologists treat the same or similar disorders in humans.
Learning from the mighty mite
For example, the study of mites that live on the skin of animals could eventually shed light on how dermatologists treat mite-related conditions in humans.
Dirk M. Elston, MD, professor and chair of the department of dermatology at the Medical University of South Carolina, Charleston, noted that Demodex mites occur in humans and in pets.
In such cases, “sulfur tends to be my most reliable” treatment, he said, noting that it releases a rotten egg smell. “You’re basically gassing the organism.” Dr. Elston said he frequently gets calls from fellow dermatologists whose antimite efforts have failed with ivermectin and permethrin and does not hesitate to give his advice. “I’m like a broken record,” he said. “Sulfur, sulfur, sulfur, sulfur.”
The Demodex mite affects dogs to varying degrees, depending on where they live, said Kathryn Rook, VMD, of the department of dermatology at the University of Pennsylvania School of Veterinary Medicine, Philadelphia. In North America, demodicosis occurs in 0.38%-0.58% of dogs, and in 25% of dogs in Mexico, she said.
Amitraz, the only Food and Drug Administration–approved treatment for canine demodicosis, is available only as a dip. But it has fallen from favor as a result of sometimes serious side effects, which can include sedation, bradycardia, ataxia, vomiting, diarrhea, and hyperglycemia.
Daily administration of oral ivermectin – often for months – also carries a risk of side effects, including dilated pupils, ataxia, sedation, stupor, coma, hypersalivation, vomiting, diarrhea, blindness, tremors, seizures, and respiratory depression.
But the discovery of isoxazoline has “revolutionized” the treatment of demodicosis and other parasitic infestations in dogs, Dr. Rook said, citing quicker resolution of disease and improved quality of life for both the patient and its owner.
Isoxazoline, which Dr. Rook said carries little risk for side effects, is licensed in the United States only as a flea and tick preventive.
Atopic dermatitis
Atopic dermatitis (AD) tends to be similar in people and dogs, according to Charles W. Bradley, DVM, of the University of Pennsylvania School of Veterinary Medicine, Philadelphia. About 10%-30% of children and up to 10% of adults have the disorder, the prevalence of which has more than doubled in recent years, he said.
In dogs, the prevalence is 10%-20%, making it “an extraordinarily common disorder,” he said. Lesions tend to be located on the feet, face, pinnae, ventrum, and axilla/inguinum. Additional sites vary by breed, with Dalmatians tending to get AD on the lips, French Bulldogs on the eyelids, German Shepherds on the elbows, Shar-Peis on the thorax, and Boxers on the ears.
In humans, Staphylococcus aureus is the chief microorganism of concern, said Elizabeth Grice, PhD, of the department of dermatology at the University of Pennsylvania, Philadelphia, who copresented the topic with Dr. Bradley.
“My true love is anything to do with the skin microbiome,” she said. “The more severe the disease, the lower the skin microbiome diversity.”
Though most studies of AD use mice as animal models, dogs would be better, according to Dr. Grice and Dr. Bradley.
That’s because canine AD occurs spontaneously and exhibits immunologic and clinical features similar to those of human AD. They include prevalence, environmental triggers, immunologic profiles, genetic predispositions, lesion distribution, and frequent colonization by Staphylococcus species. In addition, dogs and their owners tend to share the same environment.
A rash of itches
Among dermatology patients – man or beast – itch can outweigh rash as a key focus of concern, according to Brian Kim, MD, of the division of dermatology at Washington University in St. Louis, and codirector for the University’s Center for the Study of Itch. “The problem is my patients don’t complain about their rash; they complain about their itch,” he said. “But we don’t understand the basic question of itch.” In fact, the FDA has not approved any drugs for the treatment of chronic itch, he said.
For dogs, advances have been made with Janus kinase (JAK) inhibitors, which “may function as immunomodulators,” Dr. Kim said. And JAK-1 selective inhibition “may be more effective than broad JAK blockade for itch.”
‘The perfect culture plate’
Lessons can be learned from studying canine AD, which “is immunophysiologically homologous to human AD,” said Daniel O. Morris, DVM, MPH, professor of dermatology, at the University of Pennsylvania School of Veterinary Medicine, Philadelphia. “The main difference: My patients are covered in dense hair coats.” Because of that, systemic treatment is necessary, he said.
Canine AD primarily affects areas where hair is sparse or where the surface microclimate is moist, he said. A dog’s ear canal, which can be 10 times longer than a human’s, harbors plenty of moisture and heat, he said. “It’s the perfect culture plate.”
But, he added, the owners of his patients tend to resist using topical therapies “that could be potentially smeared on the babies and grandma’s diabetic foot ulcer.” So he has long relied on systemic treatments, initially steroids and cyclosporine. But they can have major side effects, and cyclosporine can take 60-90 days before it exerts maximum effect.
A faster-acting compound called oclacitinib has shown promise based on its high affinity for inhibiting JAK-1 enzyme-mediated activation of cytokine expression, including interleukin (IL)-31, he said. “Clinical trials demonstrate an antipruritic efficacy equivalent to both prednisolone and cyclosporine,” he noted. Contraindications include a history of neoplasia, the presence of severe infection, and age under 1 year.
Monoclonal antibody targets IL-31
The latest promising arrival is lokivetmab, a monoclonal antibody that targets canine IL-31, according to Dr. Morris. It acts rapidly (within 1 day for many dogs) and prevents binding of IL-31 to its neuronal receptor for at least a month, thereby interrupting neurotransmission of itch.
But side effects can be serious and common. Equal efficacy with a reduced side effect is the holy grail, he said.
Some doctors are not waiting. “People are throwing these two products at anything that itches,” he said. Unfortunately, they tend to “work miserably” for causes other than AD, he added.
Dr. Gardner, Dr. Elston, Dr. Rook, Dr. Bradley, and Dr. Morris reported no financial conflicts. Dr. Grice’s disclosures include having served as a speaker for GlaxoSmithKline and for L’Oreal France, and having received grants/research funding from Janssen Research & Development. Dr. Kim has served as a consultant to biotechnology and pharmaceutical companies.
NEW YORK – We share more than affection with our dogs and cats. We also share diseases – about which our four-legged furry friends can teach us plenty.
That was the conclusion of speakers at a session on “cases at the intersection of human and veterinary dermatology,” presented at the summer meeting of the American Academy of Dermatology.
“Human health is intimately connected to animal health,” said Jennifer Gardner, MD, of the division of dermatology, University of Washington, Seattle, and a collaborating member of the school’s Center for One Health Research. The One Health framework looks at factors involved in the human, environmental, and animal sectors from the molecular level to the individual level and even to the planetary level.
Dr. Gardner challenged her audience to think beyond their individual areas of expertise. “How does the work you’re doing with a patient or test tube connect up the line and make an impact to levels higher up?” she asked.
The One Health framework also challenges practitioners to look horizontally, at how work done in the human world connects to what’s going on in the veterinary world – that is, how treatments for dermatologic conditions in dogs may one day affect how dermatologists treat the same or similar disorders in humans.
Learning from the mighty mite
For example, the study of mites that live on the skin of animals could eventually shed light on how dermatologists treat mite-related conditions in humans.
Dirk M. Elston, MD, professor and chair of the department of dermatology at the Medical University of South Carolina, Charleston, noted that Demodex mites occur in humans and in pets.
In such cases, “sulfur tends to be my most reliable” treatment, he said, noting that it releases a rotten egg smell. “You’re basically gassing the organism.” Dr. Elston said he frequently gets calls from fellow dermatologists whose antimite efforts have failed with ivermectin and permethrin and does not hesitate to give his advice. “I’m like a broken record,” he said. “Sulfur, sulfur, sulfur, sulfur.”
The Demodex mite affects dogs to varying degrees, depending on where they live, said Kathryn Rook, VMD, of the department of dermatology at the University of Pennsylvania School of Veterinary Medicine, Philadelphia. In North America, demodicosis occurs in 0.38%-0.58% of dogs, and in 25% of dogs in Mexico, she said.
Amitraz, the only Food and Drug Administration–approved treatment for canine demodicosis, is available only as a dip. But it has fallen from favor as a result of sometimes serious side effects, which can include sedation, bradycardia, ataxia, vomiting, diarrhea, and hyperglycemia.
Daily administration of oral ivermectin – often for months – also carries a risk of side effects, including dilated pupils, ataxia, sedation, stupor, coma, hypersalivation, vomiting, diarrhea, blindness, tremors, seizures, and respiratory depression.
But the discovery of isoxazoline has “revolutionized” the treatment of demodicosis and other parasitic infestations in dogs, Dr. Rook said, citing quicker resolution of disease and improved quality of life for both the patient and its owner.
Isoxazoline, which Dr. Rook said carries little risk for side effects, is licensed in the United States only as a flea and tick preventive.
Atopic dermatitis
Atopic dermatitis (AD) tends to be similar in people and dogs, according to Charles W. Bradley, DVM, of the University of Pennsylvania School of Veterinary Medicine, Philadelphia. About 10%-30% of children and up to 10% of adults have the disorder, the prevalence of which has more than doubled in recent years, he said.
In dogs, the prevalence is 10%-20%, making it “an extraordinarily common disorder,” he said. Lesions tend to be located on the feet, face, pinnae, ventrum, and axilla/inguinum. Additional sites vary by breed, with Dalmatians tending to get AD on the lips, French Bulldogs on the eyelids, German Shepherds on the elbows, Shar-Peis on the thorax, and Boxers on the ears.
In humans, Staphylococcus aureus is the chief microorganism of concern, said Elizabeth Grice, PhD, of the department of dermatology at the University of Pennsylvania, Philadelphia, who copresented the topic with Dr. Bradley.
“My true love is anything to do with the skin microbiome,” she said. “The more severe the disease, the lower the skin microbiome diversity.”
Though most studies of AD use mice as animal models, dogs would be better, according to Dr. Grice and Dr. Bradley.
That’s because canine AD occurs spontaneously and exhibits immunologic and clinical features similar to those of human AD. They include prevalence, environmental triggers, immunologic profiles, genetic predispositions, lesion distribution, and frequent colonization by Staphylococcus species. In addition, dogs and their owners tend to share the same environment.
A rash of itches
Among dermatology patients – man or beast – itch can outweigh rash as a key focus of concern, according to Brian Kim, MD, of the division of dermatology at Washington University in St. Louis, and codirector for the University’s Center for the Study of Itch. “The problem is my patients don’t complain about their rash; they complain about their itch,” he said. “But we don’t understand the basic question of itch.” In fact, the FDA has not approved any drugs for the treatment of chronic itch, he said.
For dogs, advances have been made with Janus kinase (JAK) inhibitors, which “may function as immunomodulators,” Dr. Kim said. And JAK-1 selective inhibition “may be more effective than broad JAK blockade for itch.”
‘The perfect culture plate’
Lessons can be learned from studying canine AD, which “is immunophysiologically homologous to human AD,” said Daniel O. Morris, DVM, MPH, professor of dermatology, at the University of Pennsylvania School of Veterinary Medicine, Philadelphia. “The main difference: My patients are covered in dense hair coats.” Because of that, systemic treatment is necessary, he said.
Canine AD primarily affects areas where hair is sparse or where the surface microclimate is moist, he said. A dog’s ear canal, which can be 10 times longer than a human’s, harbors plenty of moisture and heat, he said. “It’s the perfect culture plate.”
But, he added, the owners of his patients tend to resist using topical therapies “that could be potentially smeared on the babies and grandma’s diabetic foot ulcer.” So he has long relied on systemic treatments, initially steroids and cyclosporine. But they can have major side effects, and cyclosporine can take 60-90 days before it exerts maximum effect.
A faster-acting compound called oclacitinib has shown promise based on its high affinity for inhibiting JAK-1 enzyme-mediated activation of cytokine expression, including interleukin (IL)-31, he said. “Clinical trials demonstrate an antipruritic efficacy equivalent to both prednisolone and cyclosporine,” he noted. Contraindications include a history of neoplasia, the presence of severe infection, and age under 1 year.
Monoclonal antibody targets IL-31
The latest promising arrival is lokivetmab, a monoclonal antibody that targets canine IL-31, according to Dr. Morris. It acts rapidly (within 1 day for many dogs) and prevents binding of IL-31 to its neuronal receptor for at least a month, thereby interrupting neurotransmission of itch.
But side effects can be serious and common. Equal efficacy with a reduced side effect is the holy grail, he said.
Some doctors are not waiting. “People are throwing these two products at anything that itches,” he said. Unfortunately, they tend to “work miserably” for causes other than AD, he added.
Dr. Gardner, Dr. Elston, Dr. Rook, Dr. Bradley, and Dr. Morris reported no financial conflicts. Dr. Grice’s disclosures include having served as a speaker for GlaxoSmithKline and for L’Oreal France, and having received grants/research funding from Janssen Research & Development. Dr. Kim has served as a consultant to biotechnology and pharmaceutical companies.
AT THE 2017 AAD SUMMER MEETING
Recalcitrant Ulcer on the Lower Leg
The Diagnosis: Nonuremic Calciphylaxis
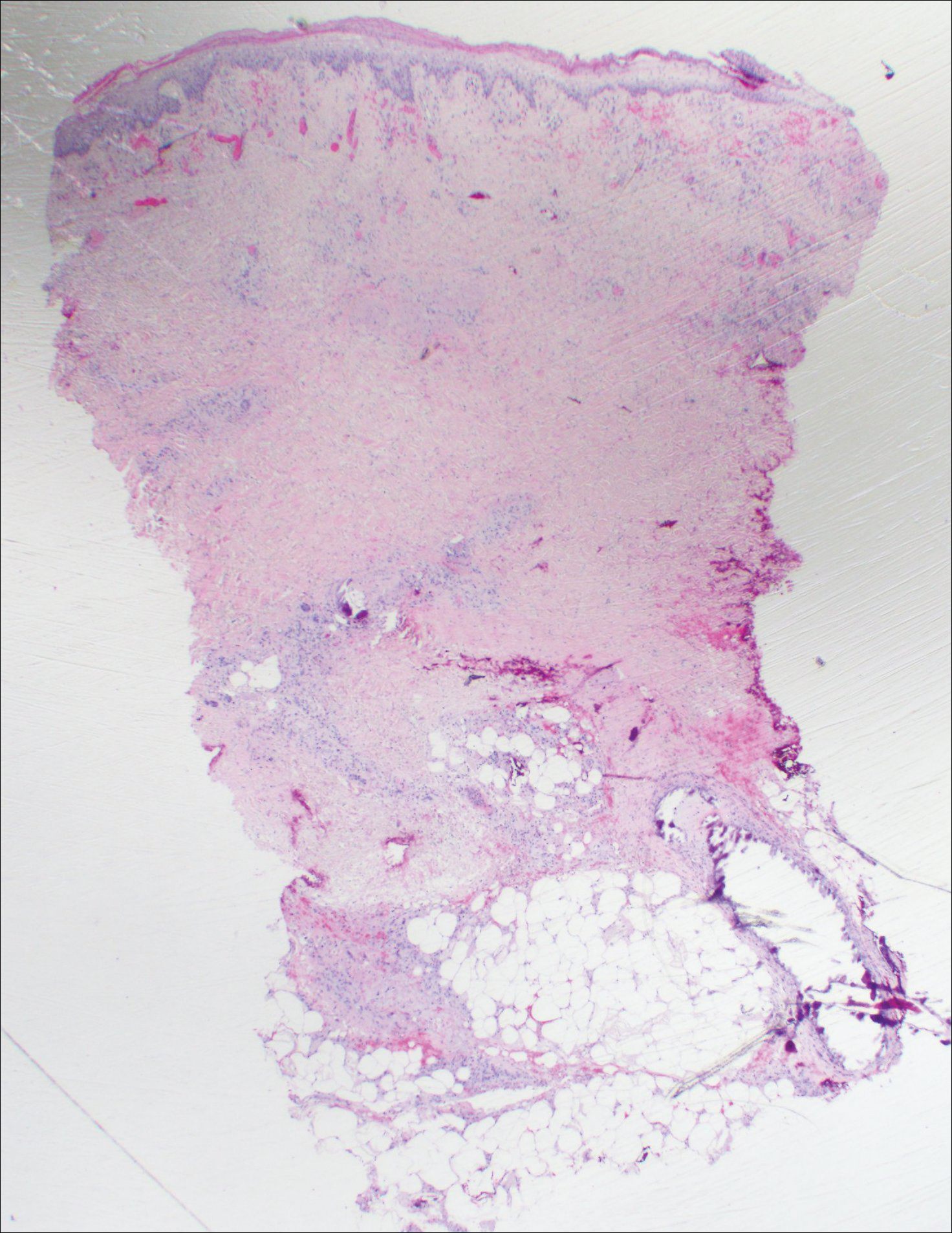
Histopathologic findings revealed ischemic necrosis and a subepidermal blister (Figure 1) with arteriosclerotic changes and fat necrosis. Foci of calcification were noted within the fat lobules. Arterioles within the deeper dermis and subcutis showed thickened hyalinized walls, narrowed lumina, and medial calcification (Figure 2). Multiple sections did not reveal any granulomatous inflammation. Periodic acid-Schiff and Gram stains were negative for fungal and bacterial elements, respectively. No dense neutrophilic infiltrate was seen. Multifocal calcific deposits within fat lobules and vessel walls (endothelium highlighted by the CD31 stain) suggested calciphylaxis.
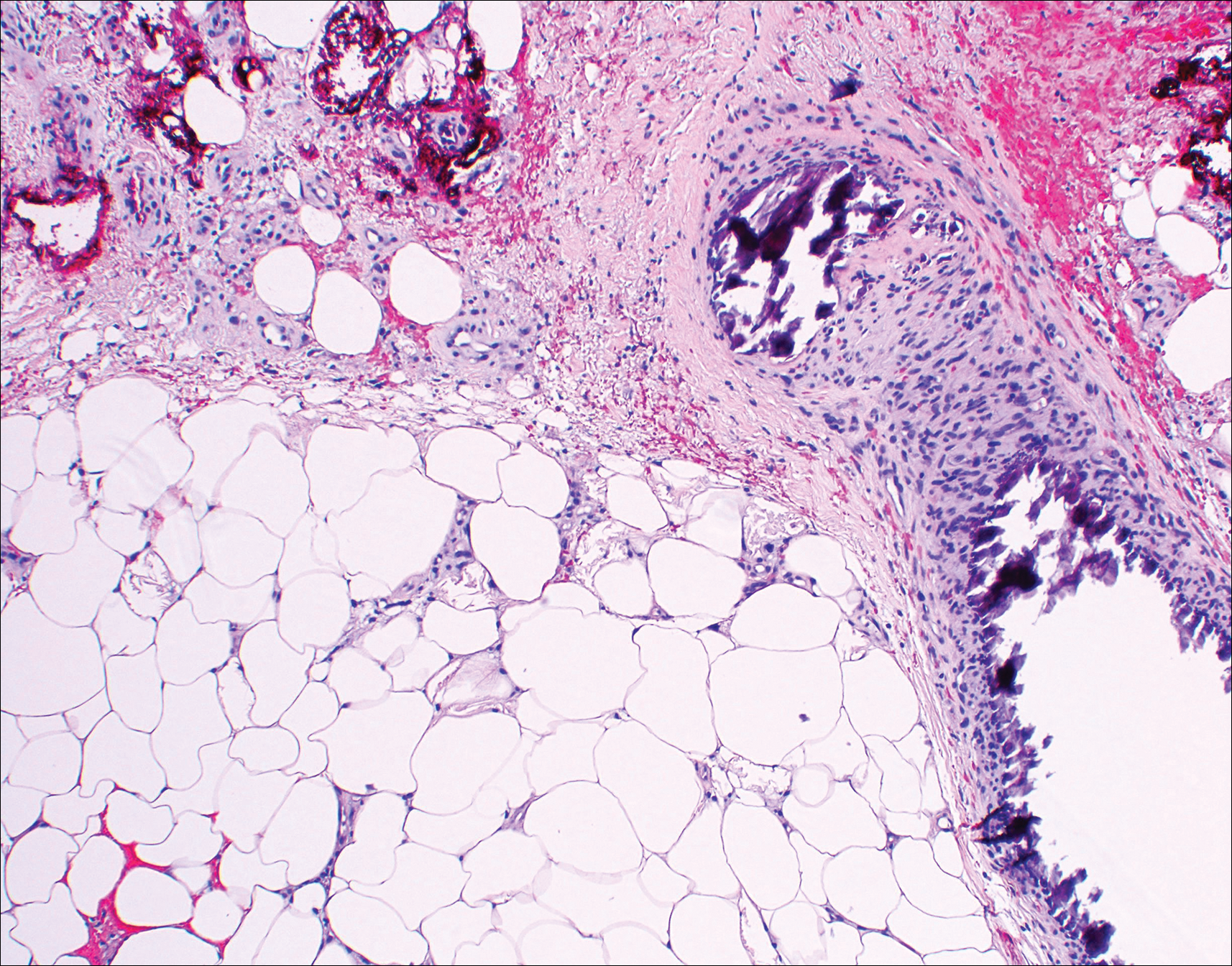
Laboratory test results revealed a normal white blood cell count, international normalized ratio level of 4 (on warfarin), and an elevated sedimentation rate at 72 mm/h (reference range, 0-20 mm/h). Serum creatinine was 1.1 mg/dL (reference range, 0.6-1.2 mg/dL) and the calcium-phosphorous product was 40.8 mg2/dL (reference range, <55 mg2/dL). Hemoglobin A1C (glycated hemoglobin) was 8.2% (reference range, 4%-7%). Wound cultures grew Proteus mirabilis sensitive to cefazolin. Acid-fast bacilli and fungal cultures were negative. Computed tomography of the left lower leg without contrast showed no evidence of osteomyelitis. Of note, the popliteal arteries and distal vessels showed moderate vascular calcification.
Histopathology findings as well as a clinical picture of painful ulceration on the distal extremities and uncontrolled diabetes with normal renal function favored a diagnosis of nonuremic calciphylaxis (NUC). The patient was treated with intravenous infusions of sodium thiosulfate 25 mg 3 times weekly and oral cefazolin for superadded bacterial infection. Local wound care included collagenase dressings with light compression. Warfarin was discontinued, as it can worsen calciphylaxis. Complete reepithelialization of the ulcer along with substantial reduction in pain was noted within 4 weeks.
Ulceration of the lower legs is a relatively common condition in the Western world, the prevalence of which increases up to 5% in patients older than 65 years.1 Of the myriad of causes that lead to ulceration of the distal aspect of the leg, NUC is a rare but known phenomenon. The pathogenesis of NUC is complicated based on theories of derangement of receptor activator of nuclear factor κβ, receptor activator of nuclear factor κβ ligand, and osteoprotegerin, leading to calcium deposits in the media of the arteries.2 This deposition precipitates vascular occlusion coupled with ischemic necrosis of the subcutaneous tissue and skin.3 Some of the more common causes of NUC are primary hyperparathyroidism, malignancy, and rheumatoid arthritis. Type 2 diabetes mellitus is a less common cause but often is found in association with NUC, as noted by Nigwekar et al.2 According to their study, the laboratory parameters commonly found in NUC included a calcium-phosphorous product greater than 50 mg2/dL and serum creatinine of 1.2 mg/dL or less.2
Our patient displayed these laboratory findings. However, distinguishing NUC from other atypical lower extremity ulcers such as Martorell hypertensive ischemic ulcer, pyoderma gangrenosum, and warfarin necrosis can pose a challenge to the dermatologist. Martorell hypertensive ischemic ulcer is excruciatingly painful and occurs more frequently near the Achilles tendon, responding well to surgical debridement. Histopathologically, medial calcinosis and arteriosclerosis are seen.4
Pyoderma gangrenosum is a neutrophilic dermatosis wherein the classical ulcerative variant is painful. It occurs mostly on the pretibial area and worsens after debridement.5 Clinically and histopathologically, it is a diagnosis of exclusion in which a dense neutrophilic to mixed lymphocytic infiltrate is seen with necrosis of dermal vessels.6
Warfarin necrosis is extremely rare, affecting 0.01% to 0.1% of patients on warfarin-derived anticoagulant therapy.7 Necrosis occurs mostly on fat-bearing areas such as the breasts, abdomen, and thighs 3 to 5 days after initiating treatment. Histologically, fibrin deposits occlude dermal vessels without perivascular inflammation.8
Necrobiosis lipoidica is a rare cutaneous entity seen in 0.3% of diabetic patients.9 The exact pathogenesis is unknown; however, microangiopathy in collaboration with cross-linking of abnormal collagen fibers play a role. These lesions appear as erythematous plaques with a slightly depressed to atrophic center, ultimately taking on a waxy porcelain appearance. Although most of these lesions either resolve or become chronically persistent, approximately 15% undergo ulceration, which can be painful. Histologically, with hematoxylin and eosin staining, areas of necrobiosis are seen surrounded by an inflammatory infiltrate comprised mainly of histiocytes along with lymphocytes and plasma cells.9
Nonuremic calciphylaxis can mimic the aforementioned conditions to a greater extent in female patients with obesity, diabetes mellitus, and hypertension. However, microscopic calcium deposition in the media of dermal arterioles, extravascular calcification within fat lobules, and cutaneous necrosis, along with remarkable response to intravenous sodium thiosulfate, confirmed a diagnosis of NUC in our patient. Sodium thiosulfate scavenges reactive oxygen species and promotes nitric oxygen generation, thereby reducing endothelial damage.10 Although there are no randomized controlled trials to support its use, sodium thiosulfate has been successfully used to treat established cases of NUC.11
- Spentzouris G, Labropoulos N. The evaluation of lower-extremity ulcers. Semin Intervent Radiol. 2009;26:286-295.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Bardin T. Musculoskeletal manifestations of chronic renal failure. Curr Opin Rheumatol. 2003;15:48-54.
- Hafner J, Nobbe S, Partsch H, et al. Martorell hypertensive ischemic leg ulcer: a model of ischemic subcutaneous arteriolosclerosis. Arch Dermatol. 2010;146:961-968.
- Sedda S, Caruso R, Marafini I, et al. Pyoderma gangrenosum in refractory celiac disease: a case report. BMC Gastroenterol. 2013;13:162.
- Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Breakey W, Hall C, Vann Jones S, et al. Warfarin-induced skin necrosis progressing to calciphylaxis. J Plast Reconstr Aesthet Surg. 2014;67:244-246.
- Kakagia DD, Papanas N, Karadimas E, et al. Warfarin-induced skin necrosis. Ann Dermatol. 2014;26:96-98.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23:258-262.
- Ning MS, Dahir KM, Castellanos EH, et al. Sodium thiosulfate in the treatment of non-uremic calciphylaxis. J Dermatol. 2013;40:649-652.
The Diagnosis: Nonuremic Calciphylaxis
Histopathologic findings revealed ischemic necrosis and a subepidermal blister (Figure 1) with arteriosclerotic changes and fat necrosis. Foci of calcification were noted within the fat lobules. Arterioles within the deeper dermis and subcutis showed thickened hyalinized walls, narrowed lumina, and medial calcification (Figure 2). Multiple sections did not reveal any granulomatous inflammation. Periodic acid-Schiff and Gram stains were negative for fungal and bacterial elements, respectively. No dense neutrophilic infiltrate was seen. Multifocal calcific deposits within fat lobules and vessel walls (endothelium highlighted by the CD31 stain) suggested calciphylaxis.
Laboratory test results revealed a normal white blood cell count, international normalized ratio level of 4 (on warfarin), and an elevated sedimentation rate at 72 mm/h (reference range, 0-20 mm/h). Serum creatinine was 1.1 mg/dL (reference range, 0.6-1.2 mg/dL) and the calcium-phosphorous product was 40.8 mg2/dL (reference range, <55 mg2/dL). Hemoglobin A1C (glycated hemoglobin) was 8.2% (reference range, 4%-7%). Wound cultures grew Proteus mirabilis sensitive to cefazolin. Acid-fast bacilli and fungal cultures were negative. Computed tomography of the left lower leg without contrast showed no evidence of osteomyelitis. Of note, the popliteal arteries and distal vessels showed moderate vascular calcification.
Histopathology findings as well as a clinical picture of painful ulceration on the distal extremities and uncontrolled diabetes with normal renal function favored a diagnosis of nonuremic calciphylaxis (NUC). The patient was treated with intravenous infusions of sodium thiosulfate 25 mg 3 times weekly and oral cefazolin for superadded bacterial infection. Local wound care included collagenase dressings with light compression. Warfarin was discontinued, as it can worsen calciphylaxis. Complete reepithelialization of the ulcer along with substantial reduction in pain was noted within 4 weeks.
Ulceration of the lower legs is a relatively common condition in the Western world, the prevalence of which increases up to 5% in patients older than 65 years.1 Of the myriad of causes that lead to ulceration of the distal aspect of the leg, NUC is a rare but known phenomenon. The pathogenesis of NUC is complicated based on theories of derangement of receptor activator of nuclear factor κβ, receptor activator of nuclear factor κβ ligand, and osteoprotegerin, leading to calcium deposits in the media of the arteries.2 This deposition precipitates vascular occlusion coupled with ischemic necrosis of the subcutaneous tissue and skin.3 Some of the more common causes of NUC are primary hyperparathyroidism, malignancy, and rheumatoid arthritis. Type 2 diabetes mellitus is a less common cause but often is found in association with NUC, as noted by Nigwekar et al.2 According to their study, the laboratory parameters commonly found in NUC included a calcium-phosphorous product greater than 50 mg2/dL and serum creatinine of 1.2 mg/dL or less.2
Our patient displayed these laboratory findings. However, distinguishing NUC from other atypical lower extremity ulcers such as Martorell hypertensive ischemic ulcer, pyoderma gangrenosum, and warfarin necrosis can pose a challenge to the dermatologist. Martorell hypertensive ischemic ulcer is excruciatingly painful and occurs more frequently near the Achilles tendon, responding well to surgical debridement. Histopathologically, medial calcinosis and arteriosclerosis are seen.4
Pyoderma gangrenosum is a neutrophilic dermatosis wherein the classical ulcerative variant is painful. It occurs mostly on the pretibial area and worsens after debridement.5 Clinically and histopathologically, it is a diagnosis of exclusion in which a dense neutrophilic to mixed lymphocytic infiltrate is seen with necrosis of dermal vessels.6
Warfarin necrosis is extremely rare, affecting 0.01% to 0.1% of patients on warfarin-derived anticoagulant therapy.7 Necrosis occurs mostly on fat-bearing areas such as the breasts, abdomen, and thighs 3 to 5 days after initiating treatment. Histologically, fibrin deposits occlude dermal vessels without perivascular inflammation.8
Necrobiosis lipoidica is a rare cutaneous entity seen in 0.3% of diabetic patients.9 The exact pathogenesis is unknown; however, microangiopathy in collaboration with cross-linking of abnormal collagen fibers play a role. These lesions appear as erythematous plaques with a slightly depressed to atrophic center, ultimately taking on a waxy porcelain appearance. Although most of these lesions either resolve or become chronically persistent, approximately 15% undergo ulceration, which can be painful. Histologically, with hematoxylin and eosin staining, areas of necrobiosis are seen surrounded by an inflammatory infiltrate comprised mainly of histiocytes along with lymphocytes and plasma cells.9
Nonuremic calciphylaxis can mimic the aforementioned conditions to a greater extent in female patients with obesity, diabetes mellitus, and hypertension. However, microscopic calcium deposition in the media of dermal arterioles, extravascular calcification within fat lobules, and cutaneous necrosis, along with remarkable response to intravenous sodium thiosulfate, confirmed a diagnosis of NUC in our patient. Sodium thiosulfate scavenges reactive oxygen species and promotes nitric oxygen generation, thereby reducing endothelial damage.10 Although there are no randomized controlled trials to support its use, sodium thiosulfate has been successfully used to treat established cases of NUC.11
The Diagnosis: Nonuremic Calciphylaxis
Histopathologic findings revealed ischemic necrosis and a subepidermal blister (Figure 1) with arteriosclerotic changes and fat necrosis. Foci of calcification were noted within the fat lobules. Arterioles within the deeper dermis and subcutis showed thickened hyalinized walls, narrowed lumina, and medial calcification (Figure 2). Multiple sections did not reveal any granulomatous inflammation. Periodic acid-Schiff and Gram stains were negative for fungal and bacterial elements, respectively. No dense neutrophilic infiltrate was seen. Multifocal calcific deposits within fat lobules and vessel walls (endothelium highlighted by the CD31 stain) suggested calciphylaxis.
Laboratory test results revealed a normal white blood cell count, international normalized ratio level of 4 (on warfarin), and an elevated sedimentation rate at 72 mm/h (reference range, 0-20 mm/h). Serum creatinine was 1.1 mg/dL (reference range, 0.6-1.2 mg/dL) and the calcium-phosphorous product was 40.8 mg2/dL (reference range, <55 mg2/dL). Hemoglobin A1C (glycated hemoglobin) was 8.2% (reference range, 4%-7%). Wound cultures grew Proteus mirabilis sensitive to cefazolin. Acid-fast bacilli and fungal cultures were negative. Computed tomography of the left lower leg without contrast showed no evidence of osteomyelitis. Of note, the popliteal arteries and distal vessels showed moderate vascular calcification.
Histopathology findings as well as a clinical picture of painful ulceration on the distal extremities and uncontrolled diabetes with normal renal function favored a diagnosis of nonuremic calciphylaxis (NUC). The patient was treated with intravenous infusions of sodium thiosulfate 25 mg 3 times weekly and oral cefazolin for superadded bacterial infection. Local wound care included collagenase dressings with light compression. Warfarin was discontinued, as it can worsen calciphylaxis. Complete reepithelialization of the ulcer along with substantial reduction in pain was noted within 4 weeks.
Ulceration of the lower legs is a relatively common condition in the Western world, the prevalence of which increases up to 5% in patients older than 65 years.1 Of the myriad of causes that lead to ulceration of the distal aspect of the leg, NUC is a rare but known phenomenon. The pathogenesis of NUC is complicated based on theories of derangement of receptor activator of nuclear factor κβ, receptor activator of nuclear factor κβ ligand, and osteoprotegerin, leading to calcium deposits in the media of the arteries.2 This deposition precipitates vascular occlusion coupled with ischemic necrosis of the subcutaneous tissue and skin.3 Some of the more common causes of NUC are primary hyperparathyroidism, malignancy, and rheumatoid arthritis. Type 2 diabetes mellitus is a less common cause but often is found in association with NUC, as noted by Nigwekar et al.2 According to their study, the laboratory parameters commonly found in NUC included a calcium-phosphorous product greater than 50 mg2/dL and serum creatinine of 1.2 mg/dL or less.2
Our patient displayed these laboratory findings. However, distinguishing NUC from other atypical lower extremity ulcers such as Martorell hypertensive ischemic ulcer, pyoderma gangrenosum, and warfarin necrosis can pose a challenge to the dermatologist. Martorell hypertensive ischemic ulcer is excruciatingly painful and occurs more frequently near the Achilles tendon, responding well to surgical debridement. Histopathologically, medial calcinosis and arteriosclerosis are seen.4
Pyoderma gangrenosum is a neutrophilic dermatosis wherein the classical ulcerative variant is painful. It occurs mostly on the pretibial area and worsens after debridement.5 Clinically and histopathologically, it is a diagnosis of exclusion in which a dense neutrophilic to mixed lymphocytic infiltrate is seen with necrosis of dermal vessels.6
Warfarin necrosis is extremely rare, affecting 0.01% to 0.1% of patients on warfarin-derived anticoagulant therapy.7 Necrosis occurs mostly on fat-bearing areas such as the breasts, abdomen, and thighs 3 to 5 days after initiating treatment. Histologically, fibrin deposits occlude dermal vessels without perivascular inflammation.8
Necrobiosis lipoidica is a rare cutaneous entity seen in 0.3% of diabetic patients.9 The exact pathogenesis is unknown; however, microangiopathy in collaboration with cross-linking of abnormal collagen fibers play a role. These lesions appear as erythematous plaques with a slightly depressed to atrophic center, ultimately taking on a waxy porcelain appearance. Although most of these lesions either resolve or become chronically persistent, approximately 15% undergo ulceration, which can be painful. Histologically, with hematoxylin and eosin staining, areas of necrobiosis are seen surrounded by an inflammatory infiltrate comprised mainly of histiocytes along with lymphocytes and plasma cells.9
Nonuremic calciphylaxis can mimic the aforementioned conditions to a greater extent in female patients with obesity, diabetes mellitus, and hypertension. However, microscopic calcium deposition in the media of dermal arterioles, extravascular calcification within fat lobules, and cutaneous necrosis, along with remarkable response to intravenous sodium thiosulfate, confirmed a diagnosis of NUC in our patient. Sodium thiosulfate scavenges reactive oxygen species and promotes nitric oxygen generation, thereby reducing endothelial damage.10 Although there are no randomized controlled trials to support its use, sodium thiosulfate has been successfully used to treat established cases of NUC.11
- Spentzouris G, Labropoulos N. The evaluation of lower-extremity ulcers. Semin Intervent Radiol. 2009;26:286-295.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Bardin T. Musculoskeletal manifestations of chronic renal failure. Curr Opin Rheumatol. 2003;15:48-54.
- Hafner J, Nobbe S, Partsch H, et al. Martorell hypertensive ischemic leg ulcer: a model of ischemic subcutaneous arteriolosclerosis. Arch Dermatol. 2010;146:961-968.
- Sedda S, Caruso R, Marafini I, et al. Pyoderma gangrenosum in refractory celiac disease: a case report. BMC Gastroenterol. 2013;13:162.
- Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Breakey W, Hall C, Vann Jones S, et al. Warfarin-induced skin necrosis progressing to calciphylaxis. J Plast Reconstr Aesthet Surg. 2014;67:244-246.
- Kakagia DD, Papanas N, Karadimas E, et al. Warfarin-induced skin necrosis. Ann Dermatol. 2014;26:96-98.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23:258-262.
- Ning MS, Dahir KM, Castellanos EH, et al. Sodium thiosulfate in the treatment of non-uremic calciphylaxis. J Dermatol. 2013;40:649-652.
- Spentzouris G, Labropoulos N. The evaluation of lower-extremity ulcers. Semin Intervent Radiol. 2009;26:286-295.
- Nigwekar SU, Wolf M, Sterns RH, et al. Calciphylaxis from nonuremic causes: a systematic review. Clin J Am Soc Nephrol. 2008;3:1139-1143.
- Bardin T. Musculoskeletal manifestations of chronic renal failure. Curr Opin Rheumatol. 2003;15:48-54.
- Hafner J, Nobbe S, Partsch H, et al. Martorell hypertensive ischemic leg ulcer: a model of ischemic subcutaneous arteriolosclerosis. Arch Dermatol. 2010;146:961-968.
- Sedda S, Caruso R, Marafini I, et al. Pyoderma gangrenosum in refractory celiac disease: a case report. BMC Gastroenterol. 2013;13:162.
- Su WP, Davis MD, Weenig RH, et al. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43:790-800.
- Breakey W, Hall C, Vann Jones S, et al. Warfarin-induced skin necrosis progressing to calciphylaxis. J Plast Reconstr Aesthet Surg. 2014;67:244-246.
- Kakagia DD, Papanas N, Karadimas E, et al. Warfarin-induced skin necrosis. Ann Dermatol. 2014;26:96-98.
- Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
- Hayden MR, Goldsmith DJ. Sodium thiosulfate: new hope for the treatment of calciphylaxis. Semin Dial. 2010;23:258-262.
- Ning MS, Dahir KM, Castellanos EH, et al. Sodium thiosulfate in the treatment of non-uremic calciphylaxis. J Dermatol. 2013;40:649-652.
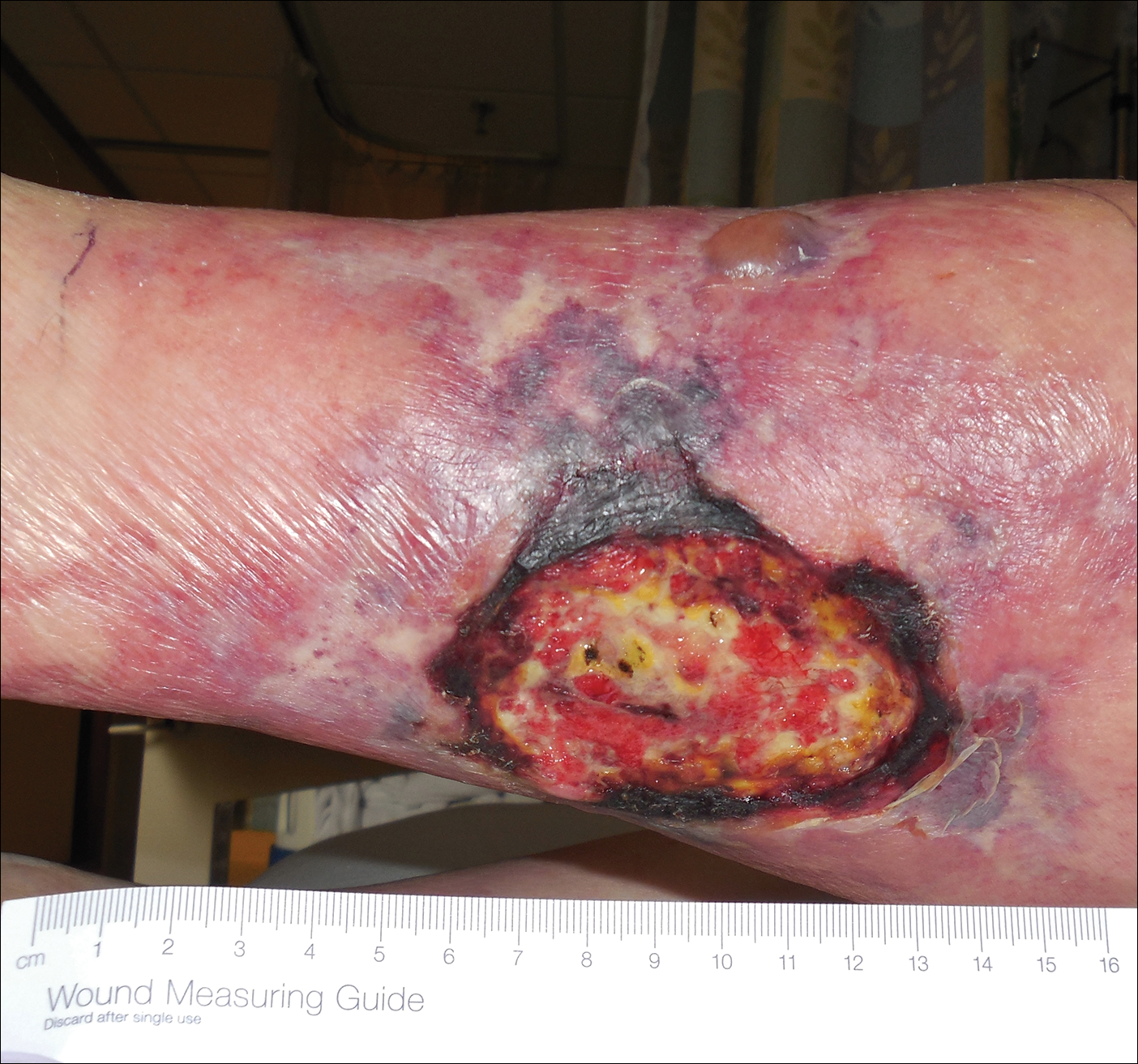
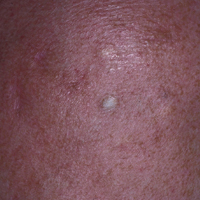
An 80-year-old woman with a medical history notable for obesity (body mass index, 31.2), type 2 diabetes mellitus, hypertension, and chronic atrial fibrillation treated with warfarin presented with a chronic painful wound on the left lower calf of 1 month's duration. A 7×7-cm ulcer on the posterior aspect of the left calf with necrotic debris was seen surrounded by skin of mottled purple discoloration. The edge of the ulcer was not undermined. There were tense nonhemorrhagic bullae on the medial aspect of the left leg and on bilateral anterior tibial areas. Two punch biopsy specimens were obtained from the anterior tibial bulla and the edge of the ulcer.
Orange Nodules on the Scalp
The Diagnosis: Rosai-Dorfman Disease
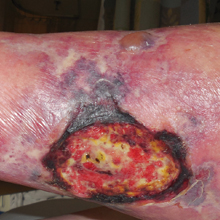
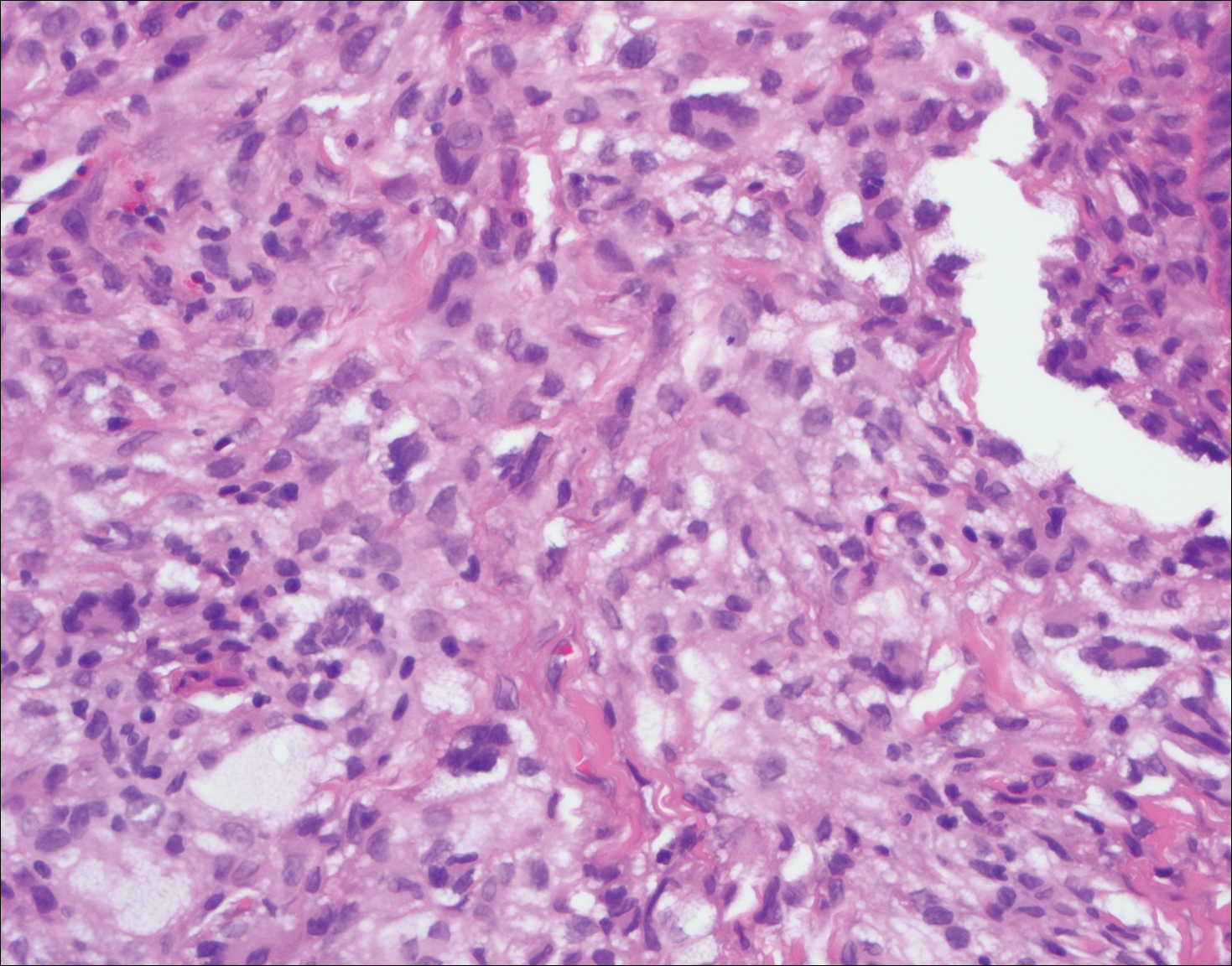
Rosai-Dorfman disease is a rare histiocytic proliferative disorder of unknown etiology. It has 2 forms: limited cutaneous and systemic. The systemic form, also known as sinus histiocytosis with massive lymphadenopathy, affects the lymph nodes and other organs at times. The disease is characterized by a proliferation of histiocytes in the lymph nodes, most commonly in the cervical basin1; however, the inguinal, axillary, mediastinal, or para-aortic nodes also may be affected.1,2 The skin is the most common site of extranodal disease, seen in approximately 10% of cases.1 Cutaneous involvement often is in the facial area but also can be found on the trunk, ears, neck, arms, legs, and genitals. Clinically, skin lesions appear as papules, plaques, and/or nodules.2
Histopathologic examination of Rosai-Dorfman disease generally shows a dense sheetlike dermal infiltrate of large polygonal histiocytes (Figure 1). Histiocytes may display pale pink or clear cytoplasm. The pathognomonic finding is emperipolesis, which consists of histiocytes with engulfed lymphocytes, erythrocytes, plasma cells, and/or granulocytes surrounded by a clear halo. Immunohistochemical staining also is characteristic, with lesional histiocytes showing expression of S-100 protein (Figure 1, inset) and CD68. The associated inflammatory infiltrate is mixed, containing primarily plasma cells but also lymphocytes, neutrophils, and eosinophils.
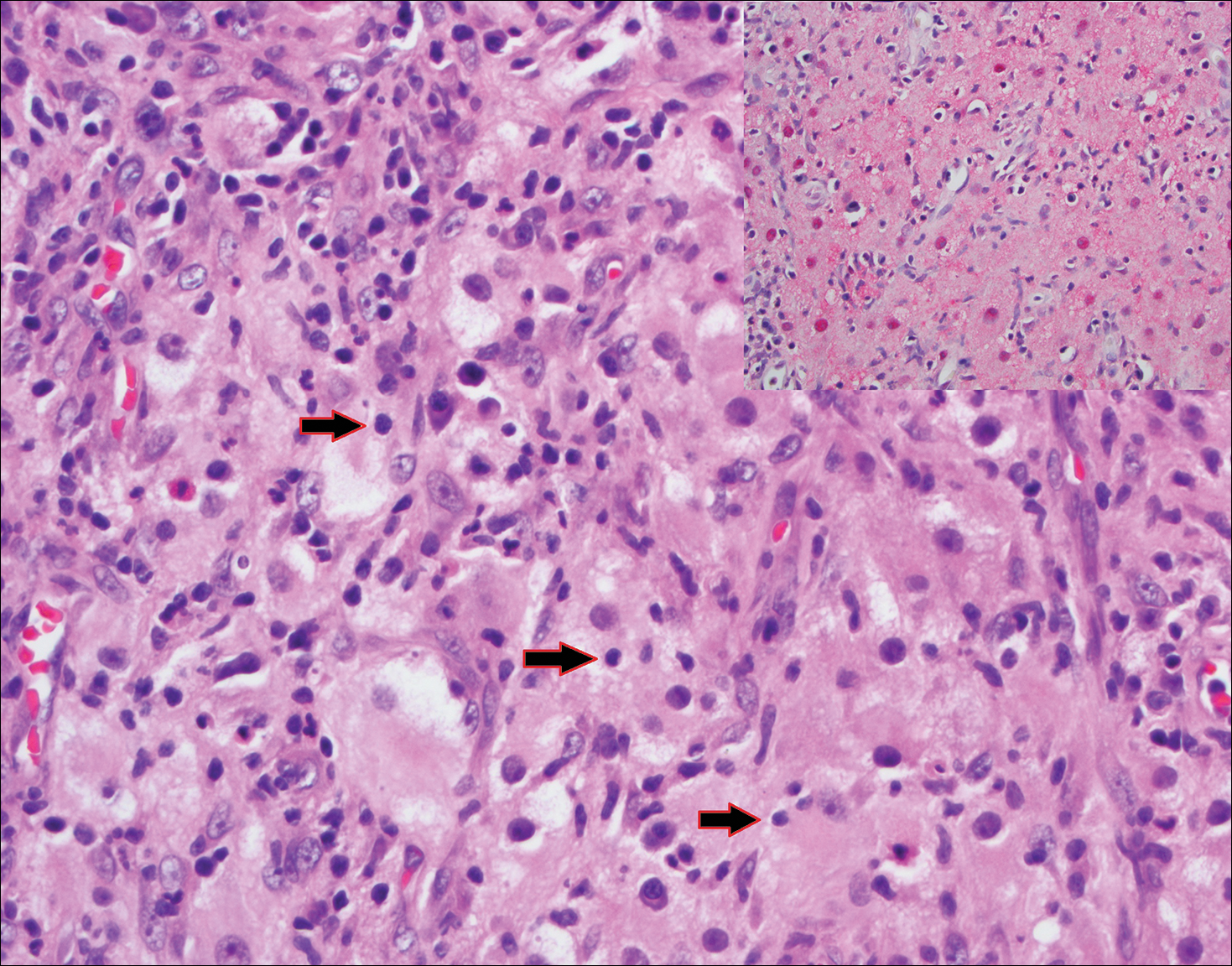
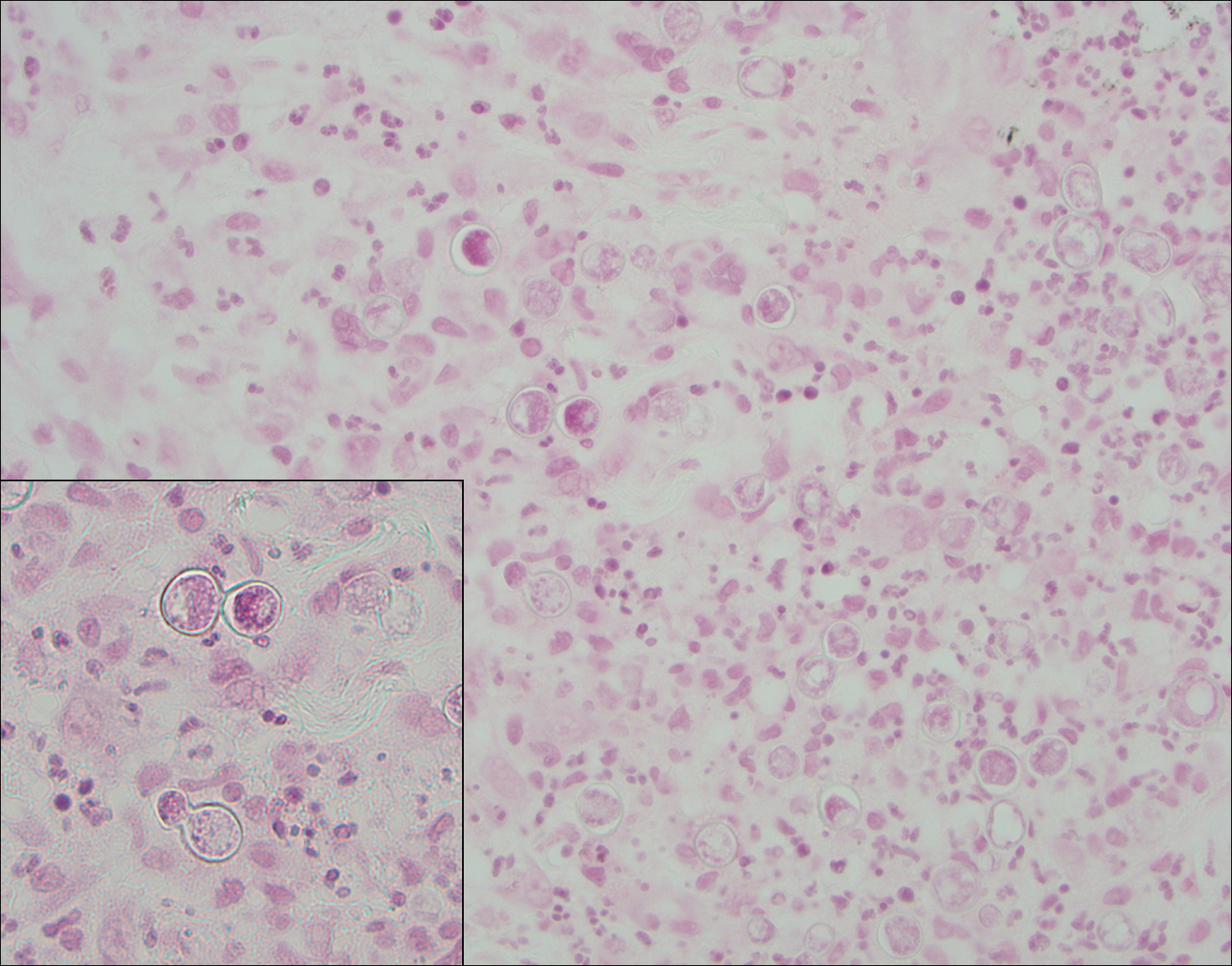
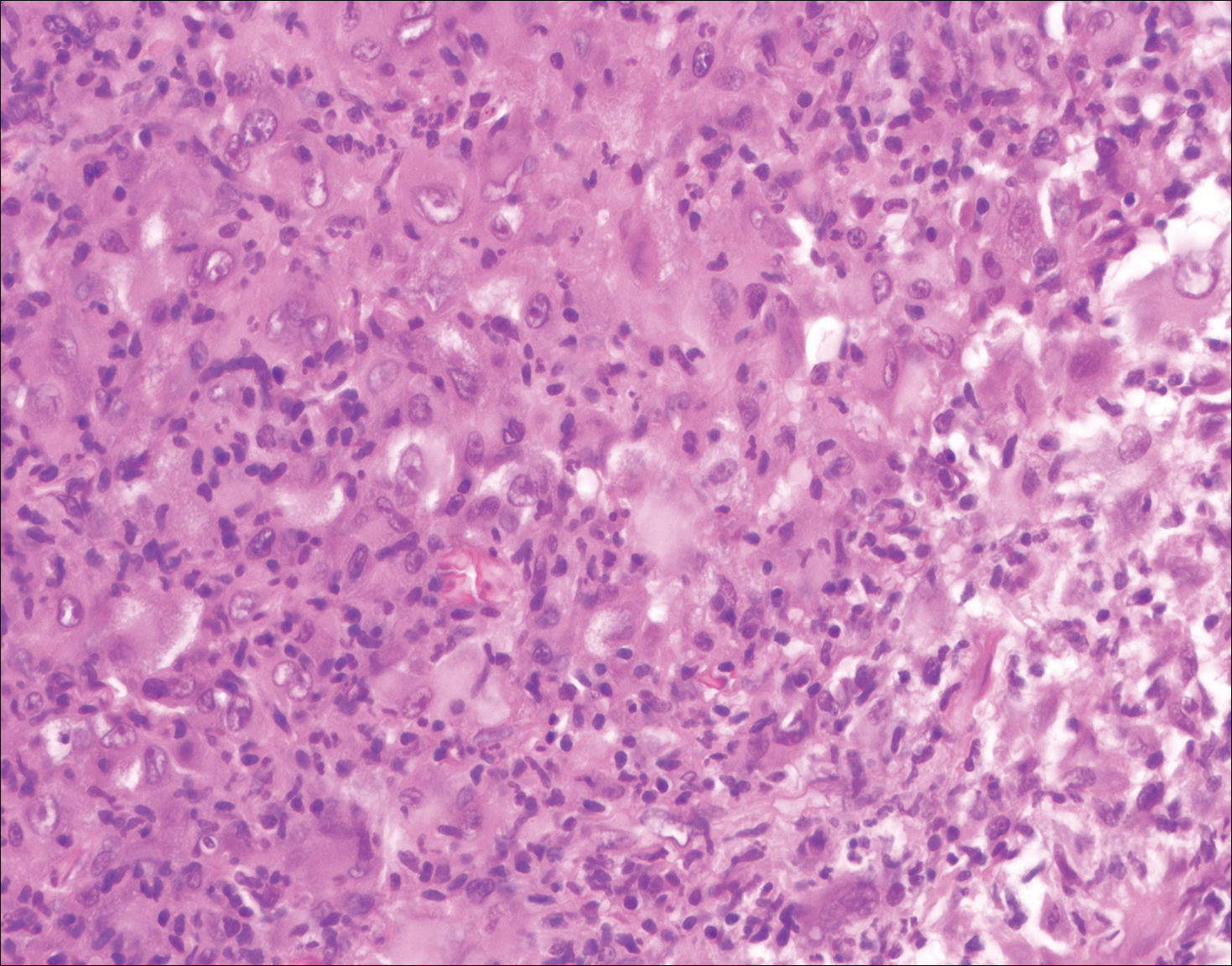
Blastomycosis (Figure 2) is a systemic infection due to inhalation of Blastomyces dermatitidis conidia. Primary infection occurs in the lungs, and with dissemination the skin is the most common subsequently involved organ.3 Cutaneous blastomycosis shows pseudoepitheliomatous hyperplasia with neutrophilic microabscesses and a dense dermal infiltrate containing suppurative granulomatous inflammation. The nonpigmented yeast phase typically is 8 to 15 µm in length with a refractile cell wall and characteristic single, broad-based budding.3
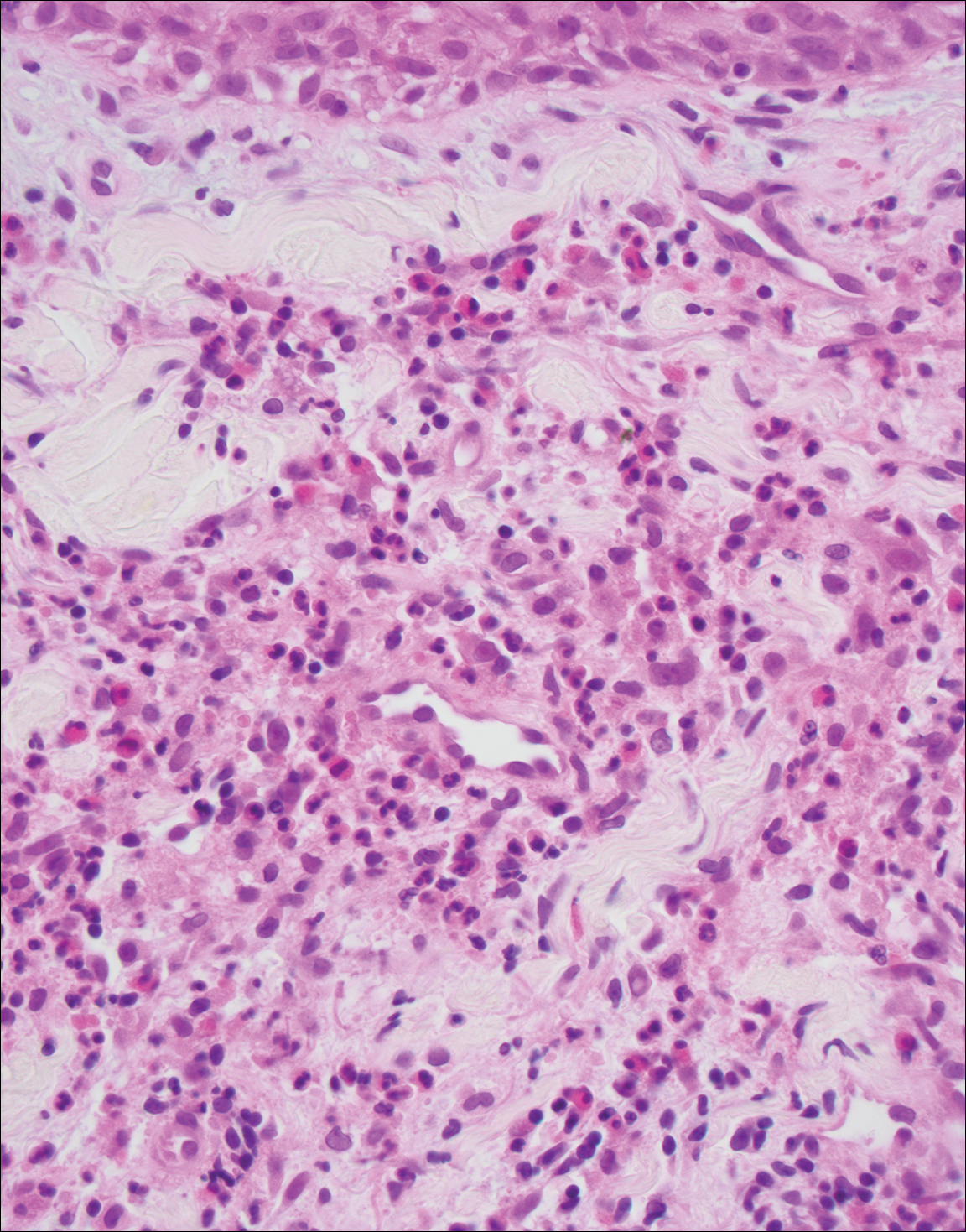
Granuloma faciale (Figure 3) is a rare disease with unknown etiology characterized by reddish brown plaques or nodules most commonly occurring on the face.4,5 Histology shows a dense nodular dermal infiltrate with a grenz zone. The infiltrate is mixed, containing mostly neutrophils with leukocytoclasis and eosinophils. Leukocytoclastic vasculitis is present with associated extravasated erythrocytes. In chronic fibrosing granuloma faciale, lesions can demonstrate fibrosis and hemosiderin deposition, similar to erythema elevatum diutinum.
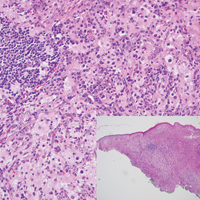
Juvenile xanthogranuloma (Figure 4) is a common histiocytic disease of early childhood, though adult cases have been reported.6 Tumors are found on the head and trunk and are typically firm, reddish yellow papules or nodules.6,7 Histologic examination shows a nodular infiltrate of foamy histiocytes in the superficial dermis. Touton-type multinucleated giant cells with a peripheral rim of xanthomatized foamy cytoplasm and a wreathlike arrangement of nuclei are characteristic. Associated eosinophils are seen. No emperipolesis is present.
Reticulohistiocytoma (Figure 5) is a benign dermal lesion that presents as solitary or less commonly multiple red-brown papules or nodules.8 Lesions consist of well-delineated nodular aggregates of histiocytes containing a finely granular eosinophilic ground glass cytoplasm. Few, if any, eosinophils are found. The lack of Touton multinucleated giant cells or emperipolesis and lack of expression of S-100 protein helps to distinguish reticulohistiocytoma from other entities in the differential diagnosis.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Wolff K, Johnson R, Saavedra AP. Fitzpatrick's Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Marcoval J, Moreno A, Peyrí J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Tanz WS, Schwartz RA, Janniger CK. Juvenile xanthogranuloma. Cutis. 1994;54:241-245.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatology Online J. 2014;20. pii:doj_21725.
The Diagnosis: Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare histiocytic proliferative disorder of unknown etiology. It has 2 forms: limited cutaneous and systemic. The systemic form, also known as sinus histiocytosis with massive lymphadenopathy, affects the lymph nodes and other organs at times. The disease is characterized by a proliferation of histiocytes in the lymph nodes, most commonly in the cervical basin1; however, the inguinal, axillary, mediastinal, or para-aortic nodes also may be affected.1,2 The skin is the most common site of extranodal disease, seen in approximately 10% of cases.1 Cutaneous involvement often is in the facial area but also can be found on the trunk, ears, neck, arms, legs, and genitals. Clinically, skin lesions appear as papules, plaques, and/or nodules.2
Histopathologic examination of Rosai-Dorfman disease generally shows a dense sheetlike dermal infiltrate of large polygonal histiocytes (Figure 1). Histiocytes may display pale pink or clear cytoplasm. The pathognomonic finding is emperipolesis, which consists of histiocytes with engulfed lymphocytes, erythrocytes, plasma cells, and/or granulocytes surrounded by a clear halo. Immunohistochemical staining also is characteristic, with lesional histiocytes showing expression of S-100 protein (Figure 1, inset) and CD68. The associated inflammatory infiltrate is mixed, containing primarily plasma cells but also lymphocytes, neutrophils, and eosinophils.
Blastomycosis (Figure 2) is a systemic infection due to inhalation of Blastomyces dermatitidis conidia. Primary infection occurs in the lungs, and with dissemination the skin is the most common subsequently involved organ.3 Cutaneous blastomycosis shows pseudoepitheliomatous hyperplasia with neutrophilic microabscesses and a dense dermal infiltrate containing suppurative granulomatous inflammation. The nonpigmented yeast phase typically is 8 to 15 µm in length with a refractile cell wall and characteristic single, broad-based budding.3
Granuloma faciale (Figure 3) is a rare disease with unknown etiology characterized by reddish brown plaques or nodules most commonly occurring on the face.4,5 Histology shows a dense nodular dermal infiltrate with a grenz zone. The infiltrate is mixed, containing mostly neutrophils with leukocytoclasis and eosinophils. Leukocytoclastic vasculitis is present with associated extravasated erythrocytes. In chronic fibrosing granuloma faciale, lesions can demonstrate fibrosis and hemosiderin deposition, similar to erythema elevatum diutinum.
Juvenile xanthogranuloma (Figure 4) is a common histiocytic disease of early childhood, though adult cases have been reported.6 Tumors are found on the head and trunk and are typically firm, reddish yellow papules or nodules.6,7 Histologic examination shows a nodular infiltrate of foamy histiocytes in the superficial dermis. Touton-type multinucleated giant cells with a peripheral rim of xanthomatized foamy cytoplasm and a wreathlike arrangement of nuclei are characteristic. Associated eosinophils are seen. No emperipolesis is present.
Reticulohistiocytoma (Figure 5) is a benign dermal lesion that presents as solitary or less commonly multiple red-brown papules or nodules.8 Lesions consist of well-delineated nodular aggregates of histiocytes containing a finely granular eosinophilic ground glass cytoplasm. Few, if any, eosinophils are found. The lack of Touton multinucleated giant cells or emperipolesis and lack of expression of S-100 protein helps to distinguish reticulohistiocytoma from other entities in the differential diagnosis.
The Diagnosis: Rosai-Dorfman Disease
Rosai-Dorfman disease is a rare histiocytic proliferative disorder of unknown etiology. It has 2 forms: limited cutaneous and systemic. The systemic form, also known as sinus histiocytosis with massive lymphadenopathy, affects the lymph nodes and other organs at times. The disease is characterized by a proliferation of histiocytes in the lymph nodes, most commonly in the cervical basin1; however, the inguinal, axillary, mediastinal, or para-aortic nodes also may be affected.1,2 The skin is the most common site of extranodal disease, seen in approximately 10% of cases.1 Cutaneous involvement often is in the facial area but also can be found on the trunk, ears, neck, arms, legs, and genitals. Clinically, skin lesions appear as papules, plaques, and/or nodules.2
Histopathologic examination of Rosai-Dorfman disease generally shows a dense sheetlike dermal infiltrate of large polygonal histiocytes (Figure 1). Histiocytes may display pale pink or clear cytoplasm. The pathognomonic finding is emperipolesis, which consists of histiocytes with engulfed lymphocytes, erythrocytes, plasma cells, and/or granulocytes surrounded by a clear halo. Immunohistochemical staining also is characteristic, with lesional histiocytes showing expression of S-100 protein (Figure 1, inset) and CD68. The associated inflammatory infiltrate is mixed, containing primarily plasma cells but also lymphocytes, neutrophils, and eosinophils.
Blastomycosis (Figure 2) is a systemic infection due to inhalation of Blastomyces dermatitidis conidia. Primary infection occurs in the lungs, and with dissemination the skin is the most common subsequently involved organ.3 Cutaneous blastomycosis shows pseudoepitheliomatous hyperplasia with neutrophilic microabscesses and a dense dermal infiltrate containing suppurative granulomatous inflammation. The nonpigmented yeast phase typically is 8 to 15 µm in length with a refractile cell wall and characteristic single, broad-based budding.3
Granuloma faciale (Figure 3) is a rare disease with unknown etiology characterized by reddish brown plaques or nodules most commonly occurring on the face.4,5 Histology shows a dense nodular dermal infiltrate with a grenz zone. The infiltrate is mixed, containing mostly neutrophils with leukocytoclasis and eosinophils. Leukocytoclastic vasculitis is present with associated extravasated erythrocytes. In chronic fibrosing granuloma faciale, lesions can demonstrate fibrosis and hemosiderin deposition, similar to erythema elevatum diutinum.
Juvenile xanthogranuloma (Figure 4) is a common histiocytic disease of early childhood, though adult cases have been reported.6 Tumors are found on the head and trunk and are typically firm, reddish yellow papules or nodules.6,7 Histologic examination shows a nodular infiltrate of foamy histiocytes in the superficial dermis. Touton-type multinucleated giant cells with a peripheral rim of xanthomatized foamy cytoplasm and a wreathlike arrangement of nuclei are characteristic. Associated eosinophils are seen. No emperipolesis is present.
Reticulohistiocytoma (Figure 5) is a benign dermal lesion that presents as solitary or less commonly multiple red-brown papules or nodules.8 Lesions consist of well-delineated nodular aggregates of histiocytes containing a finely granular eosinophilic ground glass cytoplasm. Few, if any, eosinophils are found. The lack of Touton multinucleated giant cells or emperipolesis and lack of expression of S-100 protein helps to distinguish reticulohistiocytoma from other entities in the differential diagnosis.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Wolff K, Johnson R, Saavedra AP. Fitzpatrick's Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Marcoval J, Moreno A, Peyrí J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Tanz WS, Schwartz RA, Janniger CK. Juvenile xanthogranuloma. Cutis. 1994;54:241-245.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatology Online J. 2014;20. pii:doj_21725.
- Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol. 1990;7:19-73.
- Kutlubay Z, Bairamov O, Sevim A, et al. Rosai-Dorfman disease: a case report with nodal and cutaneous involvement and review of the literature. Am J Dermatopathol. 2014;36:353-357.
- James WD, Berger TG, Elston DM, eds. Andrews' Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: Elsevier; 2015.
- Wolff K, Johnson R, Saavedra AP. Fitzpatrick's Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill; 2013.
- Marcoval J, Moreno A, Peyrí J. Granuloma faciale: a clinicopathological study of 11 cases. J Am Acad Dermatol. 2004;51:269-273.
- Rodriguez J, Ackerman AB. Xanthogranuloma in adults. Arch Dermatol. 1976;112:43-44.
- Tanz WS, Schwartz RA, Janniger CK. Juvenile xanthogranuloma. Cutis. 1994;54:241-245.
- Cohen PR, Lee RA. Adult-onset reticulohistiocytoma presenting as a solitary asymptomatic red knee nodule: report and review of clinical presentations and immunohistochemistry staining features of reticulohistiocytosis. Dermatology Online J. 2014;20. pii:doj_21725.
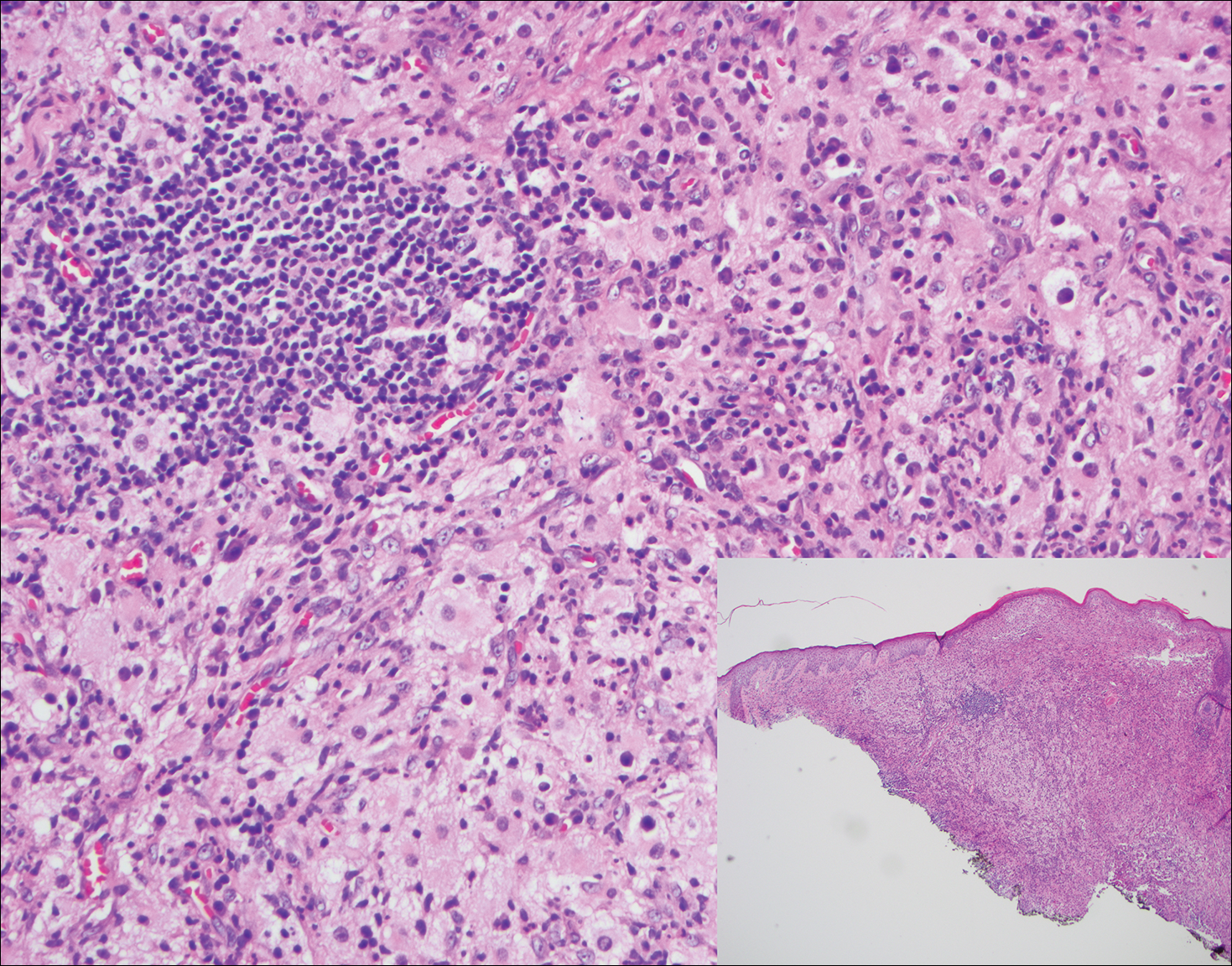
A 59-year-old man presented with itchy and mildly painful nodules on the head and neck of 7 months' duration. The patient denied fever, chills, unintentional weight loss, night sweats, and other systemic symptoms. Physical examination revealed multiple firm pink-orange nodules of varying sizes distributed on the scalp, face, and neck. Right-sided, painless, bulky cervical lymphadenopathy also was noted. An incisional biopsy was performed.
Hyperpigmented Patch on the Leg
The Diagnosis: Lichen Aureus
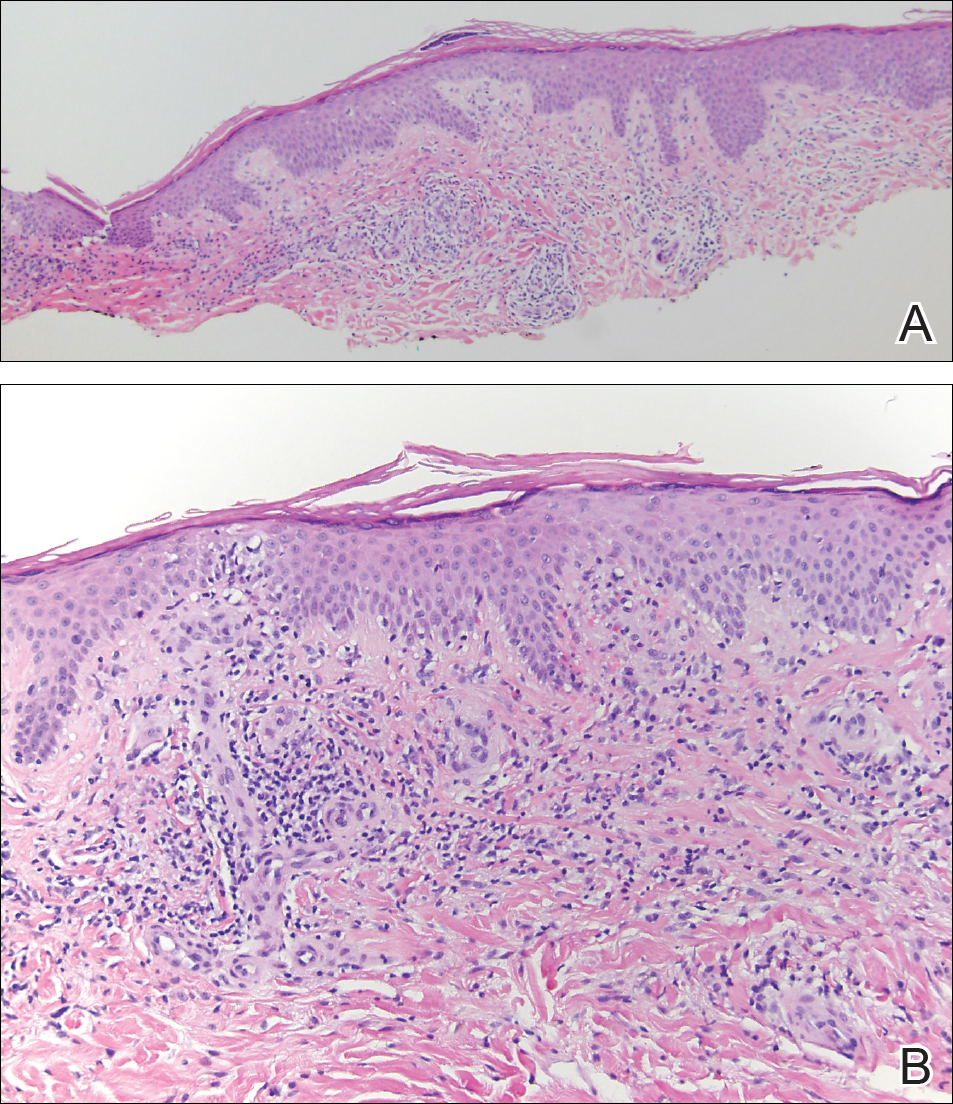
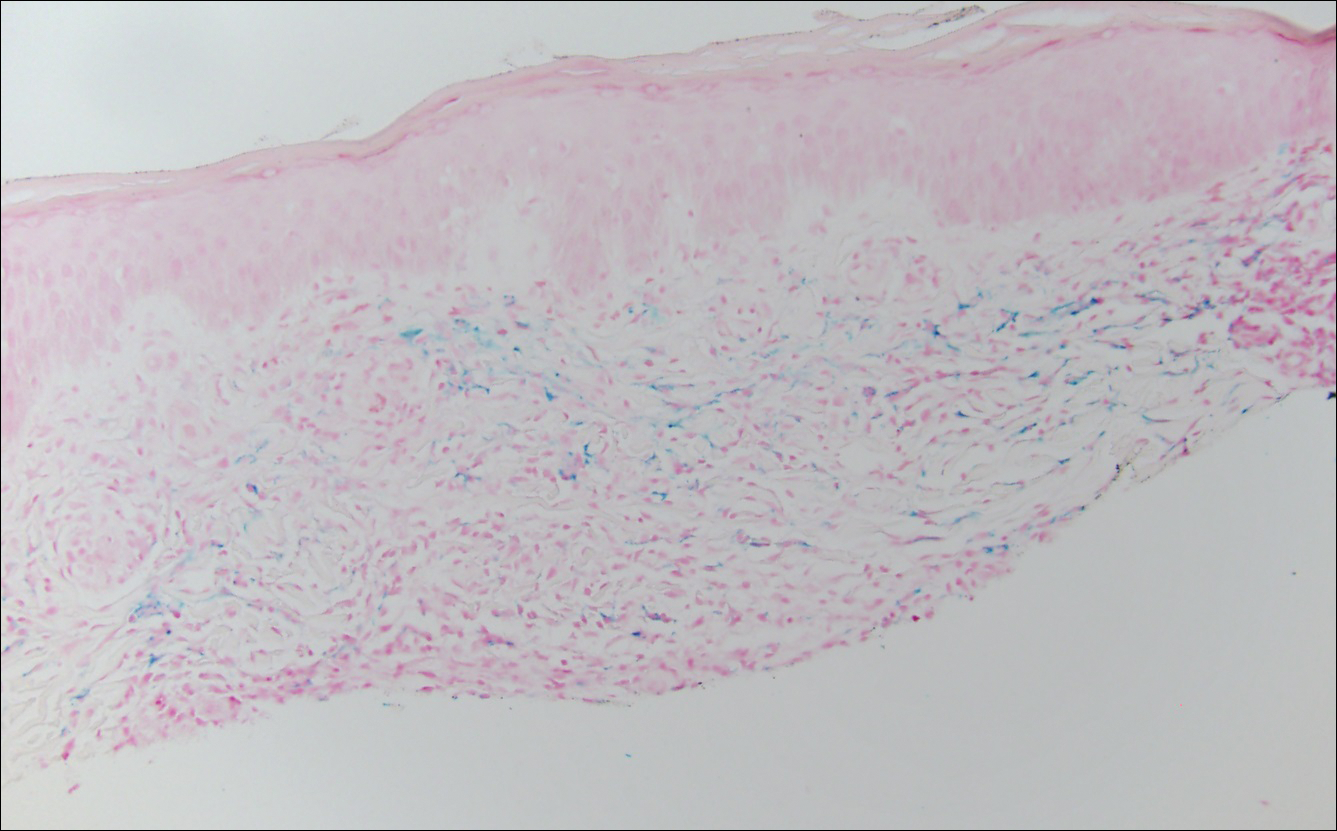
The clinicopathological findings were diagnostic of lichen aureus (LA). Microscopic examination revealed a relatively sparse, superficial, perivascular and interstitial lymphohistiocytic infiltrate with scattered siderophages in the upper dermis. Extravasation of red blood cells also was noted (Figure 1). An immunohistochemical stain for Melan-A highlighted a normal number and distribution of single melanocytes at the dermoepidermal junction with no evidence of pagetoid scatter. A Perls Prussian blue stain for iron demonstrated abundant hemosiderin in the dermis (Figure 2).
Pigmented purpuric dermatosis (PPD) describes a group of cutaneous lesions that are characterized by petechiae and pigmentary changes. These lesions most commonly present on the lower limbs; however, other sites have been reported.1 This group includes several major clinical forms such as Schamberg disease, LA, purpura annularis telangiectodes of Majocchi, eczematidlike purpura of Doucas and Kapetanakis, and lichenoid PPD of Gougerot and Blum. Lesions typically demonstrate a striking golden brown color clinically and by definition occur in the absence of platelet defects or vasculitis.1
Factors implicated in the pathogenesis of pigmented purpura include gravitational dependency, venous stasis, infection, and drugs.2 It is suggested that cellular immunity may play a role in the development of the disease based on the presence of CD4+ T lymphocytes in the infiltrate and the expression of HLA-DR by these lymphocytes and the keratinocytes.3 Lichen aureus differs in that it relates to increased intravascular pressure from an incompetent valve in an underlying perforating vein.4
Lichen aureus, also referred to as lichen purpuricus, is one major variant of PPD. The name reflects both the characteristic golden brown color and the histopathologic pattern of inflammation.1 Lichen aureus usually presents as a unilateral, asymptomatic, confined single lesion located mainly on the leg,1 though it can develop at other sites or as a localized group of lesions. Extensive lesions have been reported5 and cases with a segmental distribution have been described.6 In contrast, Schamberg disease demonstrates pinhead-sized reddish lesions giving the characteristic cayenne pepper pigmentation. These lesions coalesce to form thumbprint patches that progress proximally.1 Majocchi purpura is annular and telangiectatic, while lichenoid purpura of Gougerot and Blum presents with flat-topped, polygonal, violaceous papules that turn brown over time.
Some authors have championed a role for dermoscopy in diagnosis of LA.7 By dermoscopy, LA demonstrates a diffuse copper background reflecting the lymphohistiocytic dermal infiltrate, red dots and globules representing the extravasated red blood cells and the dilated swollen vessels, and grey dots that reflect the hemosiderin present in the dermis.8
Histologically, LA demonstrates a superficial perivascular infiltrate composed mainly of CD4+ lymphocytes surrounding the superficial capillaries. Over time, red cell extravasation leads to the formation of hemosiderin-laden macrophages, which can be highlighted with Perls Prussian blue stain. A bandlike infiltrate with thin strands of collagen separating it from the epidermis also may be noted.9
An important consideration in the differential diagnosis of PPD is mycosis fungoides (MF). Mycosis fungoides is a cutaneous T-cell lymphoma that clinically presents as a single or multiple hypopigmented or hyperpigmented patches or as erythematous scaly lesions in the patch or plaque stage. These lesions eventually may evolve into tumor stage.10 Mycosis fungoides may mimic PPD clinically and/or histopathologically, and rarely PPD also may precede MF.11 Involvement of the trunk, especially the lower abdomen and buttock region, favors a diagnosis of MF. Typically, histopathologic examination of MF demonstrates an epidermotropic lymphocytic infiltrate composed of atypical cerebriform lymphocytes overlying papillary dermal fibrosis. Although classic MF would be difficult to confuse with PPD, the atrophic lichenoid pattern of MF may show remarkable overlap with PPD.12 Such cases require clinicopathologic correlation, immunophenotyping of the epidermotropic lymphocytes, and occasionally T-cell clonality studies.
Lichen aureus is a chronic persistent disease unless the underlying incompetent perforator vessel is ligated. Various treatments have been used for other forms of pigmented purpura including topical corticosteroids, topical tacrolimus, systemic vasodilators such as prostacyclin and pentoxifylline, and phototherapy.1 Clinical follow-up is recommended for lesions that show some clinical or histopathological overlap with MF. Additional biopsies also may prove useful in establishing a definitive diagnosis in ambiguous cases.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Aiba S, Tagami H. Immunohistologic studies in Schamberg's disease. evidence for cellular immune reaction in lesional skin. Arch Dermatol. 1988;124:1058-1062.
- English J. Lichen aureus. J Am Acad Dermatol. 1985;12(2, pt 1):377-379.
- Duhra P, Tan CY. Lichen aureus. Br J Dermatol. 1986;114:395.
- Moche J, Glassman S, Modi D, et al. Segmental lichen aureus: a report of two cases treated with methylprednisolone aceponate. Australas J Dermatol. 2011;52:E15-E18.
- Zaballos P, Puig S, Malvehy J. Dermoscopy of pigmented purpuric dermatoses (lichen aureus): a useful tool for clinical diagnosis. Arch Dermatol. 2004;140:1290-1291.
- Portela PS, Melo DF, Ormiga P, et al. Dermoscopy of lichen aureus. An Bras Dermatol. 2013;88:253-255.
- Smoller BR, Kamel OW. Pigmented purpuric eruptions: immunopathologic studies supportive of a common immunophenotype. J Cutan Pathol. 1991;18:423-427.
- Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. a progress report. Am J Clin Pathol. 1999;111(1 suppl 1):S8-S12.
- Hanna S, Walsh N, D'Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
- Toro JR, Sander CA, LeBoit PE. Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
The Diagnosis: Lichen Aureus
The clinicopathological findings were diagnostic of lichen aureus (LA). Microscopic examination revealed a relatively sparse, superficial, perivascular and interstitial lymphohistiocytic infiltrate with scattered siderophages in the upper dermis. Extravasation of red blood cells also was noted (Figure 1). An immunohistochemical stain for Melan-A highlighted a normal number and distribution of single melanocytes at the dermoepidermal junction with no evidence of pagetoid scatter. A Perls Prussian blue stain for iron demonstrated abundant hemosiderin in the dermis (Figure 2).
Pigmented purpuric dermatosis (PPD) describes a group of cutaneous lesions that are characterized by petechiae and pigmentary changes. These lesions most commonly present on the lower limbs; however, other sites have been reported.1 This group includes several major clinical forms such as Schamberg disease, LA, purpura annularis telangiectodes of Majocchi, eczematidlike purpura of Doucas and Kapetanakis, and lichenoid PPD of Gougerot and Blum. Lesions typically demonstrate a striking golden brown color clinically and by definition occur in the absence of platelet defects or vasculitis.1
Factors implicated in the pathogenesis of pigmented purpura include gravitational dependency, venous stasis, infection, and drugs.2 It is suggested that cellular immunity may play a role in the development of the disease based on the presence of CD4+ T lymphocytes in the infiltrate and the expression of HLA-DR by these lymphocytes and the keratinocytes.3 Lichen aureus differs in that it relates to increased intravascular pressure from an incompetent valve in an underlying perforating vein.4
Lichen aureus, also referred to as lichen purpuricus, is one major variant of PPD. The name reflects both the characteristic golden brown color and the histopathologic pattern of inflammation.1 Lichen aureus usually presents as a unilateral, asymptomatic, confined single lesion located mainly on the leg,1 though it can develop at other sites or as a localized group of lesions. Extensive lesions have been reported5 and cases with a segmental distribution have been described.6 In contrast, Schamberg disease demonstrates pinhead-sized reddish lesions giving the characteristic cayenne pepper pigmentation. These lesions coalesce to form thumbprint patches that progress proximally.1 Majocchi purpura is annular and telangiectatic, while lichenoid purpura of Gougerot and Blum presents with flat-topped, polygonal, violaceous papules that turn brown over time.
Some authors have championed a role for dermoscopy in diagnosis of LA.7 By dermoscopy, LA demonstrates a diffuse copper background reflecting the lymphohistiocytic dermal infiltrate, red dots and globules representing the extravasated red blood cells and the dilated swollen vessels, and grey dots that reflect the hemosiderin present in the dermis.8
Histologically, LA demonstrates a superficial perivascular infiltrate composed mainly of CD4+ lymphocytes surrounding the superficial capillaries. Over time, red cell extravasation leads to the formation of hemosiderin-laden macrophages, which can be highlighted with Perls Prussian blue stain. A bandlike infiltrate with thin strands of collagen separating it from the epidermis also may be noted.9
An important consideration in the differential diagnosis of PPD is mycosis fungoides (MF). Mycosis fungoides is a cutaneous T-cell lymphoma that clinically presents as a single or multiple hypopigmented or hyperpigmented patches or as erythematous scaly lesions in the patch or plaque stage. These lesions eventually may evolve into tumor stage.10 Mycosis fungoides may mimic PPD clinically and/or histopathologically, and rarely PPD also may precede MF.11 Involvement of the trunk, especially the lower abdomen and buttock region, favors a diagnosis of MF. Typically, histopathologic examination of MF demonstrates an epidermotropic lymphocytic infiltrate composed of atypical cerebriform lymphocytes overlying papillary dermal fibrosis. Although classic MF would be difficult to confuse with PPD, the atrophic lichenoid pattern of MF may show remarkable overlap with PPD.12 Such cases require clinicopathologic correlation, immunophenotyping of the epidermotropic lymphocytes, and occasionally T-cell clonality studies.
Lichen aureus is a chronic persistent disease unless the underlying incompetent perforator vessel is ligated. Various treatments have been used for other forms of pigmented purpura including topical corticosteroids, topical tacrolimus, systemic vasodilators such as prostacyclin and pentoxifylline, and phototherapy.1 Clinical follow-up is recommended for lesions that show some clinical or histopathological overlap with MF. Additional biopsies also may prove useful in establishing a definitive diagnosis in ambiguous cases.
The Diagnosis: Lichen Aureus
The clinicopathological findings were diagnostic of lichen aureus (LA). Microscopic examination revealed a relatively sparse, superficial, perivascular and interstitial lymphohistiocytic infiltrate with scattered siderophages in the upper dermis. Extravasation of red blood cells also was noted (Figure 1). An immunohistochemical stain for Melan-A highlighted a normal number and distribution of single melanocytes at the dermoepidermal junction with no evidence of pagetoid scatter. A Perls Prussian blue stain for iron demonstrated abundant hemosiderin in the dermis (Figure 2).
Pigmented purpuric dermatosis (PPD) describes a group of cutaneous lesions that are characterized by petechiae and pigmentary changes. These lesions most commonly present on the lower limbs; however, other sites have been reported.1 This group includes several major clinical forms such as Schamberg disease, LA, purpura annularis telangiectodes of Majocchi, eczematidlike purpura of Doucas and Kapetanakis, and lichenoid PPD of Gougerot and Blum. Lesions typically demonstrate a striking golden brown color clinically and by definition occur in the absence of platelet defects or vasculitis.1
Factors implicated in the pathogenesis of pigmented purpura include gravitational dependency, venous stasis, infection, and drugs.2 It is suggested that cellular immunity may play a role in the development of the disease based on the presence of CD4+ T lymphocytes in the infiltrate and the expression of HLA-DR by these lymphocytes and the keratinocytes.3 Lichen aureus differs in that it relates to increased intravascular pressure from an incompetent valve in an underlying perforating vein.4
Lichen aureus, also referred to as lichen purpuricus, is one major variant of PPD. The name reflects both the characteristic golden brown color and the histopathologic pattern of inflammation.1 Lichen aureus usually presents as a unilateral, asymptomatic, confined single lesion located mainly on the leg,1 though it can develop at other sites or as a localized group of lesions. Extensive lesions have been reported5 and cases with a segmental distribution have been described.6 In contrast, Schamberg disease demonstrates pinhead-sized reddish lesions giving the characteristic cayenne pepper pigmentation. These lesions coalesce to form thumbprint patches that progress proximally.1 Majocchi purpura is annular and telangiectatic, while lichenoid purpura of Gougerot and Blum presents with flat-topped, polygonal, violaceous papules that turn brown over time.
Some authors have championed a role for dermoscopy in diagnosis of LA.7 By dermoscopy, LA demonstrates a diffuse copper background reflecting the lymphohistiocytic dermal infiltrate, red dots and globules representing the extravasated red blood cells and the dilated swollen vessels, and grey dots that reflect the hemosiderin present in the dermis.8
Histologically, LA demonstrates a superficial perivascular infiltrate composed mainly of CD4+ lymphocytes surrounding the superficial capillaries. Over time, red cell extravasation leads to the formation of hemosiderin-laden macrophages, which can be highlighted with Perls Prussian blue stain. A bandlike infiltrate with thin strands of collagen separating it from the epidermis also may be noted.9
An important consideration in the differential diagnosis of PPD is mycosis fungoides (MF). Mycosis fungoides is a cutaneous T-cell lymphoma that clinically presents as a single or multiple hypopigmented or hyperpigmented patches or as erythematous scaly lesions in the patch or plaque stage. These lesions eventually may evolve into tumor stage.10 Mycosis fungoides may mimic PPD clinically and/or histopathologically, and rarely PPD also may precede MF.11 Involvement of the trunk, especially the lower abdomen and buttock region, favors a diagnosis of MF. Typically, histopathologic examination of MF demonstrates an epidermotropic lymphocytic infiltrate composed of atypical cerebriform lymphocytes overlying papillary dermal fibrosis. Although classic MF would be difficult to confuse with PPD, the atrophic lichenoid pattern of MF may show remarkable overlap with PPD.12 Such cases require clinicopathologic correlation, immunophenotyping of the epidermotropic lymphocytes, and occasionally T-cell clonality studies.
Lichen aureus is a chronic persistent disease unless the underlying incompetent perforator vessel is ligated. Various treatments have been used for other forms of pigmented purpura including topical corticosteroids, topical tacrolimus, systemic vasodilators such as prostacyclin and pentoxifylline, and phototherapy.1 Clinical follow-up is recommended for lesions that show some clinical or histopathological overlap with MF. Additional biopsies also may prove useful in establishing a definitive diagnosis in ambiguous cases.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Aiba S, Tagami H. Immunohistologic studies in Schamberg's disease. evidence for cellular immune reaction in lesional skin. Arch Dermatol. 1988;124:1058-1062.
- English J. Lichen aureus. J Am Acad Dermatol. 1985;12(2, pt 1):377-379.
- Duhra P, Tan CY. Lichen aureus. Br J Dermatol. 1986;114:395.
- Moche J, Glassman S, Modi D, et al. Segmental lichen aureus: a report of two cases treated with methylprednisolone aceponate. Australas J Dermatol. 2011;52:E15-E18.
- Zaballos P, Puig S, Malvehy J. Dermoscopy of pigmented purpuric dermatoses (lichen aureus): a useful tool for clinical diagnosis. Arch Dermatol. 2004;140:1290-1291.
- Portela PS, Melo DF, Ormiga P, et al. Dermoscopy of lichen aureus. An Bras Dermatol. 2013;88:253-255.
- Smoller BR, Kamel OW. Pigmented purpuric eruptions: immunopathologic studies supportive of a common immunophenotype. J Cutan Pathol. 1991;18:423-427.
- Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. a progress report. Am J Clin Pathol. 1999;111(1 suppl 1):S8-S12.
- Hanna S, Walsh N, D'Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
- Toro JR, Sander CA, LeBoit PE. Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
- Sardana K, Sarkar R, Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Newton RC, Raimer SS. Pigmented purpuric eruptions. Dermatol Clin. 1985;3:165-169.
- Aiba S, Tagami H. Immunohistologic studies in Schamberg's disease. evidence for cellular immune reaction in lesional skin. Arch Dermatol. 1988;124:1058-1062.
- English J. Lichen aureus. J Am Acad Dermatol. 1985;12(2, pt 1):377-379.
- Duhra P, Tan CY. Lichen aureus. Br J Dermatol. 1986;114:395.
- Moche J, Glassman S, Modi D, et al. Segmental lichen aureus: a report of two cases treated with methylprednisolone aceponate. Australas J Dermatol. 2011;52:E15-E18.
- Zaballos P, Puig S, Malvehy J. Dermoscopy of pigmented purpuric dermatoses (lichen aureus): a useful tool for clinical diagnosis. Arch Dermatol. 2004;140:1290-1291.
- Portela PS, Melo DF, Ormiga P, et al. Dermoscopy of lichen aureus. An Bras Dermatol. 2013;88:253-255.
- Smoller BR, Kamel OW. Pigmented purpuric eruptions: immunopathologic studies supportive of a common immunophenotype. J Cutan Pathol. 1991;18:423-427.
- Jaffe ES, Harris NL, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues. a progress report. Am J Clin Pathol. 1999;111(1 suppl 1):S8-S12.
- Hanna S, Walsh N, D'Intino Y, et al. Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350-354.
- Toro JR, Sander CA, LeBoit PE. Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? a study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108-118.
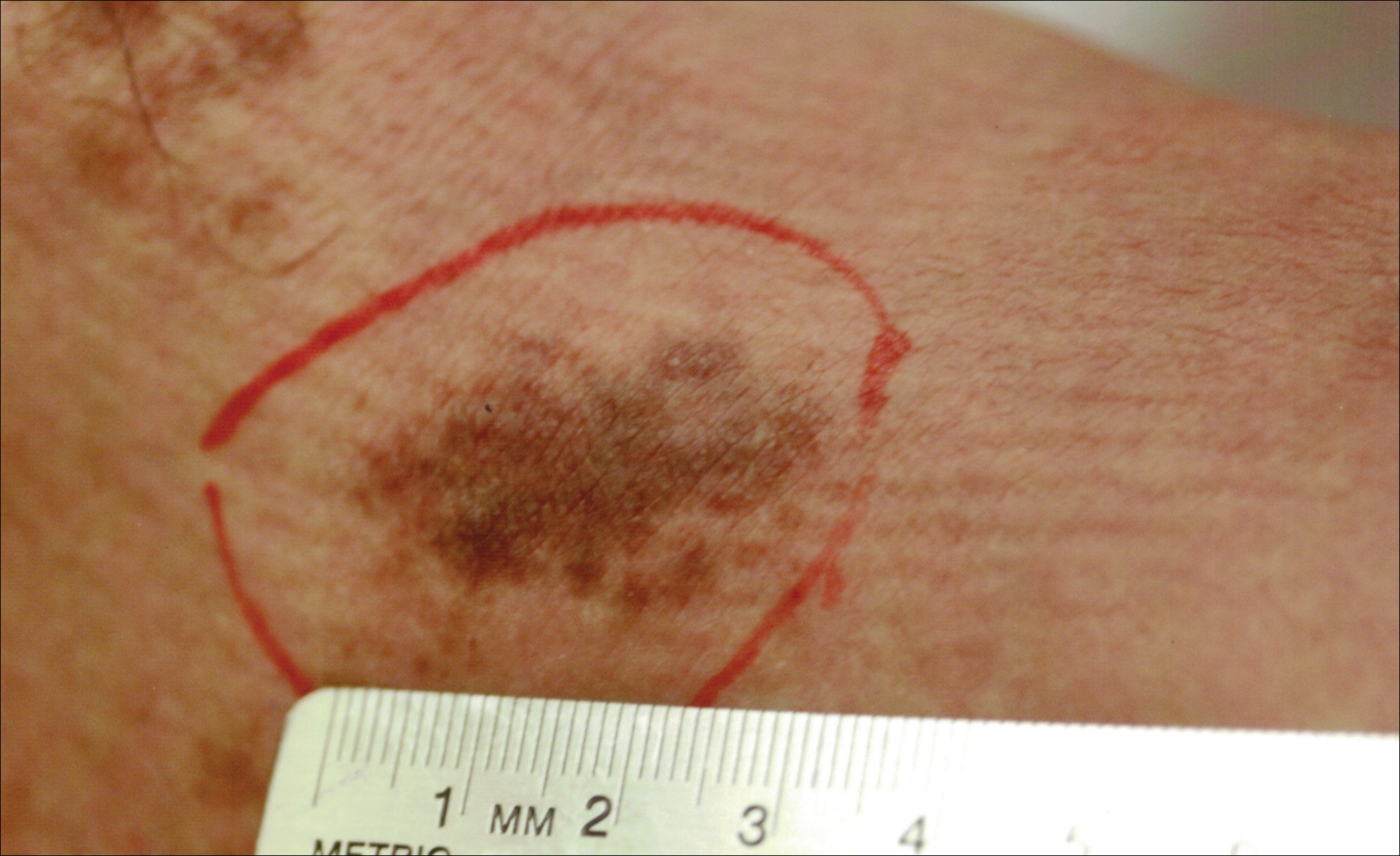
A 32-year-old man presented with an asymptomatic pigmented lesion on the left foot that developed over the course of 4 months. Physical examination revealed a 4-cm asymmetrical, deeply pigmented macule on the left foot. A shave biopsy of the lesion was performed.
Traumatic Ulcerative Granuloma With Stromal Eosinophilia: A Malignant-Appearing Benign Lesion
Traumatic ulcerative granuloma with stromal eosinophilia (TUGSE) is an uncommon, benign, self-limited condition that is restricted to the oral mucosa, most commonly seen in the fifth to seventh decades of life.1-3 The pathogenesis of TUGSE is unknown, but current theory suggests trauma is the instigating factor. The presence of CD30+ mononuclear cells within TUGSE raises the possibility of a CD30+ lymphoproliferative disorder in some cases.4 However, because CD30+ cells are not uncommon in other benign reactive processes, they may simply represent a reactive phenomenon.3
Traumatic ulcerative granuloma with stromal eosinophilia traverses multiple disciplines, including dermatology, oral surgery, dentistry, and pathology, resulting in a diverse nomenclature including traumatic granuloma of the tongue, traumatic eosinophilic granuloma of the oral mucosa, ulcerated granuloma eosinophilicum diutinum, and eosinophilic ulcer of the oral mucosa.1,4-6 It is important to differentiate eosinophilic granuloma of the oral mucosa from the eosinophilic granuloma that is associated with Langerhans cell histiocytosis. Although both may present with oral ulceration, Langerhans cell–associated eosinophilic granuloma typically develops from underlying bone, whereas eosinophilic granuloma of the oral mucosa (TUGSE) is described as nonosseous.7,8 Furthermore, the gingiva is the most common oral site in Langerhans cell–associated eosinophilic granuloma, whereas the tongue is most commonly involved in TUGSE.8 Shapiro and Juhlin9 clearly distinguished TUGSE from Langerhans cell–associated eosinophilic granuloma in 1970. Histologically, the 2 conditions are completely different.
When ulcerative granulomas develop in the pediatric population, usually in children younger than 2 years, it is termed Riga-Fede disease.10 These children were typically breastfeeding, suckling, or teething, suggesting trauma as a triggering event. In 1961, Hjorting-Hansen and Schmidt5 described 3 separate lesions similar to Riga-Fede disease in an adult patient. Subsequently, Riga-Fede disease was grouped under TUGSE.3
Histologically, TUGSE shows an ulcerated epithelium with a polymorphic inflammatory cell infiltrate that has a large predominance of eosinophils. The infiltrate affects the superficial and deep layers of the muscle tissue and penetrates into the salivary glands. Large atypical mononuclear cells with an ovoid and pale-appearing nucleus often are present. These cells may be mitotically active and stain positively for CD30.1,4,11 CD68+ macrophages, T lymphocytes, and factor XIIIa–positive dendritic cells commonly are present.12
Given the presence of large atypical CD30+ cells in many lesions, the possibility of a CD30+ lymphoproliferative disorder has been postulated by some authors. Indeed, lymphomatoid papulosis (LyP) has been documented to involve the oral mucosa.2,4
Case Report
An 81-year-old man presented with a rapidly enlarging, 1.7×1.3-cm, vascular-appearing nodule with a collarette of mucosal epithelium on the left side of the dorsal surface of the tongue of 2 weeks’ duration (Figure 1). He denied any history of trauma, tobacco chewing, weight change, fever, or fatigue; however, he did report a 30 pack-year smoking history. There was no other pertinent medical history to include medications or allergies.

The differential diagnosis included pyogenic granuloma, granular cell tumor, squamous cell carcinoma, other neoplasms (eg, oral lymphoma, salivary gland tumors), and a traumatic blood blister from tongue biting. The patient was referred to the oral maxillofacial surgery department for an excisional biopsy, which showed a solitary ulcerated nodule with associated granulation tissue, thrombus, and fibrinoid debris (Figure 2). A surrounding dense mixed inflammatory cell infiltrate composed of lymphocytes, histiocytes, and numerous eosinophils was noted extending through the submucosal tissue and underlying striated muscle fibers (Figure 3). The adjacent mucosal epithelium appeared normal. CD30 staining showed only rare positive cells. These findings were consistent with TUGSE.


Due to the benign nature of TUGSE, the patient was released with symptomatic care and instructed to return for any new growth. The growth spontaneously resolved over 1 month and no recurrence or new lesions were reported 1 year later.
Comment
Despite encompassing multiple disciplines of medicine, TUGSE has minimal exposure in the dermatologic literature. It is an important clinical and histologic diagnosis that will provide reassurance to the patient when accurately identified and reduce potentially harmful treatments.
Clinical Presentation
Typically, TUGSE presents as a painful solitary nodule with a central ulcer and yellow fibrinous base. The margins of the ulcer typically have an indurated and rolled appearance.1,4 More than 50% of the lesions develop on the tongue, specifically the dorsal or lateral surfaces, but they may present anywhere in the oral mucosa.7 Traumatic ulcerative granuloma with stromal eosinophilia is a fast-growing lesion, typically developing in days to weeks. Although it spontaneously regresses, the lesion may take weeks or months to resolve. In one case, it resolved 1 year later.1 Traumatic ulcerative granuloma with stromal eosinophilia has a bimodal age distribution, generally appearing in the first 2 years of life and later in the fifth through seventh decades. The male-to-female predominance is equal.1,7,11 Reoccurrence is rare, but some reports have shown patients with multiple episodes of TUGSE.13,14
Differential Diagnosis
The clinical differential diagnosis for TUGSE includes squamous cell carcinoma, pyogenic granuloma, lymphoproliferative disorder, traumatic neuroma, Langerhans cell histiocytosis, granulomatous disorders, and oral lymphoma. Inflammatory disorders such as syphilis, Behçet’s disease, herpes, histoplasmosis, Wegener granulomatosis, and others also should be considered.
Immunohistochemistry
Immunohistochemical analysis of TUGSE lesions recently has revealed the presence of CD30+ cells. These cells are associated with cutaneous lymphoproliferative disorders including LyP, anaplastic large cell lymphoma (ALCL), and borderline CD30+ lesions, among others. Systemic diseases with CD30+ cells include mycosis fungoides, other T-cell lymphomas, and Hodgkin lymphoma.15,16 Once CD30+ cells were recognized, multiple authors began speculating there was a correlation between TUGSE and the CD30+ lymphoproliferative disorders.1,2,13 Anaplastic large cell lymphoma and LyP of the oral mucosa have been reported in several cases.17-20 One report described 2 cases of ulcerated CD30+ T-cell non-Hodgkin lymphoma of the oral mucosa, one of which showed eosinophilic infiltrates and was initially thought to be TUGSE. Based on these overlapping clinical and histologic features, the authors hypothesized there was a correlation between oral ALCL, LyP, and TUGSE.17 In one report, a patient developed multiple TUGSE lesions throughout his life, suggesting a pathologic process similar to LyP. The lesion biopsied showed that 70% of the T cells expressed CD30 (Ki-1) antigen.13
Underlying Causes
In support of an underlying immunologic process that augments the growth of these lesions, 2 separate case reports of TUGSE in the presence of human T-lymphotropic virus 1 (HTLV-1) and Epstein-Barr virus have been documented.2,21 Concurrent presentation of TUGSE and HTLV-1 in one report demonstrated eosinophilia in both the oral lesion and peripheral blood, suggesting an immunologic relationship. Furthermore, the authors postulated that local trauma initiated the development of TUGSE, providing the catalyst for the HTLV-1 carrier to develop peripheral eosinophilia.21
In the second case, a 12-year-old boy developed TUGSE in the presence of Epstein-Barr virus.2 Immunologically, this virus can be reactivated from its latent stage during immunosuppression. Epstein-Barr virus has been implicated in lymphoproliferative diseases of both B- and T-cell origin, including CD30+ ALCL and LyP.22,23 The authors in this report again hypothesized there was a correlation between lymphoproliferative disorders and TUGSE lesions.2,24
Alternatively, TUGSE may simply be a reactive process to trauma or another underlying trigger. It has been speculated that the presence of eosinophils correlates with antigen insertion into the oral mucosa, whereas other ulcers of the oral mucosa are devoid of eosinophils.1 These antigens may include microorganisms, endogenous degradation products, or foreign proteins.7,25 Additionally, the presence of CD30+ lymphocytes is not isolated to lymphoproliferative disorders. CD30+ cells have been documented in arthropod bite reactions, atopic dermatitis, drug reactions, molluscum contagiosum, and scabies, among others.1,26
Healing and Management
The length of healing in TUGSE ulcers has substantial variability, from days to up to 1 year in an isolated case.1,24 Sequential expression of transforming growth factor (TGF) α and TGF-β expressed by tissue eosinophils may be underlying factors associated with a quicker healing response as demonstrated by similar ulcers in hamsters.27 Chronic nonhealing oral ulcers, particularly TUGSE lesions that demonstrated the typical increase in eosinophils in 11 of 12 cases, showed minimal TGF-α or TGF-β expression by eosinophils, perhaps indicating a possible mechanism leading to delayed wound healing in some cases. Interestingly, incisional biopsies often led to rapid wound healing, suggesting that the biopsy itself allowed for a transition back to the regular wound-healing processes.28
Traumatic ulcerative granuloma with stromal eosinophilia spontaneously resolves on its own in most cases; however, because of the concern for malignancy, it has the potential to be overtreated.26 Symptomatic treatment only is the mainstay of therapy. The patient should be instructed to avoid trauma, and referral to a dental professional is indicated when associated with dentures or other periprosthetic devices. Diet should consist of soft foods while avoiding spicy foods. Topical or oral analgesics may be necessary if substantial pain is associated with the lesion.2 Oral prednisolone was used in a patient with concurrent HTLV-1 and TUGSE to treat peripheral eosinophilia.21 The patient’s peripheral eosinophils dropped to 1% in 1 day, and the patient’s oral lesion began to improve at day 3 and disappeared by day 10. Although TUGSE may spontaneously resolve within a 10-day period without steroids, it may be a reasonable treatment to improve healing time in an otherwise healthy individual.21,26 If there is concern for malignancy, the patient should have the lesion biopsied to provide reassurance and for the added benefit of a transition to normal healing response and decreased healing time.28
Clinical Recognition
The clinician should be aware of the possibility of a CD30+ lymphoproliferative disorder, which has been associated with TUGSE in some cases, or may simulate TUGSE both clinically and histologically. Further studies are needed to clarify the relationship between these 2 entities. Whether it is a true relationship, simple coincidence, or simply overlapping clinical and histologic features remains to be determined.
- Hirshberg A, Amariglio N, Akrish S, et al. Traumatic ulcerative granuloma with stromal eosinophilia: reactive lesion of the oral mucosa. Am J Clin Pathol. 2006;126:522-529.
- Abdel-Naser MB, Tsatsou F, Hippe S, et al. Oral eosinophilic ulcer, an Epstein-Barr virus-associated CD30+ lymphoproliferation? [published online April 5, 2011]. Dermatology. 2011;222:113-118.
- Fonseca FP, Benevenuto de Andrade BA, Coletta RD, et al. Clinicopathological and immunohistochemical analysis of 19 cases of oral eosinophilic ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:532-540.
- Alobeid B, Pan LX, Milligan L, et al. Eosinophil-rich CD30+ lymphoproliferative disorder of the oral mucosa. Am J Clin Pathol. 2004;121:43-50.
- Hjorting-Hansen E, Schmidt H. Ulcerated granuloma eosinophilicum diutinum of the tongue. report of a case. Acta Derm Venereol. 1961;41:235-239.
- Velez A, Alamillos FJ, Dean A, et al. Eosinophilic ulcer of the oral mucosa: report of a recurrent case on the tongue. Clin Exp Dermatol. 1997;22:154-156.
- Elzay RP. Traumatic ulcerative granuloma with stromal eosinophilia (Riga-Fede’s disease and traumatic eosinophilic granuloma). Oral Surg Oral Med Oral Pathol. 1983;55:497-506.
- Val-Bernal JF, Gonzalez-Vela MC, Sanchez-Santolino S, et al. Localized eosinophilic (Langerhans’ cell) granuloma of the lower lip. a lesion that may cause diagnostic error. J Cutan Pathol. 2009;36:1109-1113.
- Shapiro L, Juhlin EA. Eosinophilic ulcer of the tongue report of two cases and review of the literature. Dermatologica. 1970;140:242-250.
- Amberg S. Sublingual growth in infants. Am J Med Sci. 1902;126:257-269.
- EI-Mofty SK, Swanson PE, Wick MR, et al. Eosinophilic ulcer of the oral mucosa: report of 38 new cases with immunohistochemical observations. Oral Surg Oral Med Oral Pathol. 1993;75:716-722.
- Regezi JA, Zarbo RJ, Daniels TE, et al. Oral traumatic granuloma: characterization of the cellular infiltrate. Oral Surg Oral Med Oral Pathol. 1993;75:723-727.
- Ficarra G, Prignano F, Romagnoli P. Traumatic eosinophilic granuloma of the oral mucosa: a CD30+ (Ki-1) lymphoproliferative disorder? Oral Oncol. 1997;33:375-379.
- Doyle JL, Geary W, Baden E. Eosinophilic ulcer. J Oral Maxillofac Surg. 1989;47:349-352.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985;66:848-858.
- Rosenberg A, Biesma DH, Sie-Go DMDS, et al. Primary extranodal CD30-positive T-cell non-Hodgkin’s lymphoma of the oral mucosa. report of two cases. Int J Oral Maxillofac Surg. 1996;25:57-59.
- Kato N, Tomita Y, Yoshida K, et al. Involvement of the tongue by lymphomatoid papulosis. Am J Dermatopathol. 1998;20:522-526.
- Savarrio L, Gibson J, Dunlop DJ, et al. Spontaneous regression of an anaplastic large cell lymphoma in the oral cavity: first reported case and review of the literature. Oral Oncol. 1999;35:609-613.
- Sciubba J, Said-Al-Naief N, Fantasia J. Critical review of lymphomatoid papulosis of the oral cavity with case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:195-204.
- Yamazaki H, Shirasugi Y, Kajiwara H, et al. Concurrent onset of eosinophilic ulcer of the oral mucosa with peripheral eosinophilia in a human T-cell leukemia virus type I carrier. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;114:E43-E48.
- Dojcinov SD, Venkataram G, Raffeld M, et al. EBV positive mucocutaneous ulcer—a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol. 2010;34:405-417.
- Kim YC, Yang WI, Lee MG, et al. Epstein-Barr virus in CD30 anaplastic large cell lymphoma involving the skin and lymphomatoid papulosis in South Korea. Int J Dermatol. 2006;45:1312-1316.
- Pietersma F, Piriou E, van Baarle D. Immune surveillance of EBV-infected B cells and the development of non-Hodgkin lymphomas in immunocompromised patients. Leuk Lymphoma. 2008;49:1028-1041.
- Salisbury CL, Budnick SD, Li S. T cell receptor gene rearrangement and CD 30 immunoreactivity in traumatic ulcerative granuloma with stromal eosinophilia of oral cavity. Am J Clin Pathol. 2009;132:722-727.
- Marszalek A, Neska-Dlugosz I. Traumatic ulcerative granuloma with stromal eosinophilia. a case report and short literature review. Pol J Pathol. 2011;3:172-175.
- Wong DT, Donoff RB, Yang J, et al. Sequential expression of transforming growth factors alpha and beta 1 by eosinophils during cutaneous wound healing in the hamster. Am J Pathol. 1993;143:130-142.
- Elovic AE, Gallagher GT, Kabani S, et al. Lack of TGF-alpha and TGF-beta synthesis by human eosinophils in chronic oral ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:672-681.
Traumatic ulcerative granuloma with stromal eosinophilia (TUGSE) is an uncommon, benign, self-limited condition that is restricted to the oral mucosa, most commonly seen in the fifth to seventh decades of life.1-3 The pathogenesis of TUGSE is unknown, but current theory suggests trauma is the instigating factor. The presence of CD30+ mononuclear cells within TUGSE raises the possibility of a CD30+ lymphoproliferative disorder in some cases.4 However, because CD30+ cells are not uncommon in other benign reactive processes, they may simply represent a reactive phenomenon.3
Traumatic ulcerative granuloma with stromal eosinophilia traverses multiple disciplines, including dermatology, oral surgery, dentistry, and pathology, resulting in a diverse nomenclature including traumatic granuloma of the tongue, traumatic eosinophilic granuloma of the oral mucosa, ulcerated granuloma eosinophilicum diutinum, and eosinophilic ulcer of the oral mucosa.1,4-6 It is important to differentiate eosinophilic granuloma of the oral mucosa from the eosinophilic granuloma that is associated with Langerhans cell histiocytosis. Although both may present with oral ulceration, Langerhans cell–associated eosinophilic granuloma typically develops from underlying bone, whereas eosinophilic granuloma of the oral mucosa (TUGSE) is described as nonosseous.7,8 Furthermore, the gingiva is the most common oral site in Langerhans cell–associated eosinophilic granuloma, whereas the tongue is most commonly involved in TUGSE.8 Shapiro and Juhlin9 clearly distinguished TUGSE from Langerhans cell–associated eosinophilic granuloma in 1970. Histologically, the 2 conditions are completely different.
When ulcerative granulomas develop in the pediatric population, usually in children younger than 2 years, it is termed Riga-Fede disease.10 These children were typically breastfeeding, suckling, or teething, suggesting trauma as a triggering event. In 1961, Hjorting-Hansen and Schmidt5 described 3 separate lesions similar to Riga-Fede disease in an adult patient. Subsequently, Riga-Fede disease was grouped under TUGSE.3
Histologically, TUGSE shows an ulcerated epithelium with a polymorphic inflammatory cell infiltrate that has a large predominance of eosinophils. The infiltrate affects the superficial and deep layers of the muscle tissue and penetrates into the salivary glands. Large atypical mononuclear cells with an ovoid and pale-appearing nucleus often are present. These cells may be mitotically active and stain positively for CD30.1,4,11 CD68+ macrophages, T lymphocytes, and factor XIIIa–positive dendritic cells commonly are present.12
Given the presence of large atypical CD30+ cells in many lesions, the possibility of a CD30+ lymphoproliferative disorder has been postulated by some authors. Indeed, lymphomatoid papulosis (LyP) has been documented to involve the oral mucosa.2,4
Case Report
An 81-year-old man presented with a rapidly enlarging, 1.7×1.3-cm, vascular-appearing nodule with a collarette of mucosal epithelium on the left side of the dorsal surface of the tongue of 2 weeks’ duration (Figure 1). He denied any history of trauma, tobacco chewing, weight change, fever, or fatigue; however, he did report a 30 pack-year smoking history. There was no other pertinent medical history to include medications or allergies.
The differential diagnosis included pyogenic granuloma, granular cell tumor, squamous cell carcinoma, other neoplasms (eg, oral lymphoma, salivary gland tumors), and a traumatic blood blister from tongue biting. The patient was referred to the oral maxillofacial surgery department for an excisional biopsy, which showed a solitary ulcerated nodule with associated granulation tissue, thrombus, and fibrinoid debris (Figure 2). A surrounding dense mixed inflammatory cell infiltrate composed of lymphocytes, histiocytes, and numerous eosinophils was noted extending through the submucosal tissue and underlying striated muscle fibers (Figure 3). The adjacent mucosal epithelium appeared normal. CD30 staining showed only rare positive cells. These findings were consistent with TUGSE.
Due to the benign nature of TUGSE, the patient was released with symptomatic care and instructed to return for any new growth. The growth spontaneously resolved over 1 month and no recurrence or new lesions were reported 1 year later.
Comment
Despite encompassing multiple disciplines of medicine, TUGSE has minimal exposure in the dermatologic literature. It is an important clinical and histologic diagnosis that will provide reassurance to the patient when accurately identified and reduce potentially harmful treatments.
Clinical Presentation
Typically, TUGSE presents as a painful solitary nodule with a central ulcer and yellow fibrinous base. The margins of the ulcer typically have an indurated and rolled appearance.1,4 More than 50% of the lesions develop on the tongue, specifically the dorsal or lateral surfaces, but they may present anywhere in the oral mucosa.7 Traumatic ulcerative granuloma with stromal eosinophilia is a fast-growing lesion, typically developing in days to weeks. Although it spontaneously regresses, the lesion may take weeks or months to resolve. In one case, it resolved 1 year later.1 Traumatic ulcerative granuloma with stromal eosinophilia has a bimodal age distribution, generally appearing in the first 2 years of life and later in the fifth through seventh decades. The male-to-female predominance is equal.1,7,11 Reoccurrence is rare, but some reports have shown patients with multiple episodes of TUGSE.13,14
Differential Diagnosis
The clinical differential diagnosis for TUGSE includes squamous cell carcinoma, pyogenic granuloma, lymphoproliferative disorder, traumatic neuroma, Langerhans cell histiocytosis, granulomatous disorders, and oral lymphoma. Inflammatory disorders such as syphilis, Behçet’s disease, herpes, histoplasmosis, Wegener granulomatosis, and others also should be considered.
Immunohistochemistry
Immunohistochemical analysis of TUGSE lesions recently has revealed the presence of CD30+ cells. These cells are associated with cutaneous lymphoproliferative disorders including LyP, anaplastic large cell lymphoma (ALCL), and borderline CD30+ lesions, among others. Systemic diseases with CD30+ cells include mycosis fungoides, other T-cell lymphomas, and Hodgkin lymphoma.15,16 Once CD30+ cells were recognized, multiple authors began speculating there was a correlation between TUGSE and the CD30+ lymphoproliferative disorders.1,2,13 Anaplastic large cell lymphoma and LyP of the oral mucosa have been reported in several cases.17-20 One report described 2 cases of ulcerated CD30+ T-cell non-Hodgkin lymphoma of the oral mucosa, one of which showed eosinophilic infiltrates and was initially thought to be TUGSE. Based on these overlapping clinical and histologic features, the authors hypothesized there was a correlation between oral ALCL, LyP, and TUGSE.17 In one report, a patient developed multiple TUGSE lesions throughout his life, suggesting a pathologic process similar to LyP. The lesion biopsied showed that 70% of the T cells expressed CD30 (Ki-1) antigen.13
Underlying Causes
In support of an underlying immunologic process that augments the growth of these lesions, 2 separate case reports of TUGSE in the presence of human T-lymphotropic virus 1 (HTLV-1) and Epstein-Barr virus have been documented.2,21 Concurrent presentation of TUGSE and HTLV-1 in one report demonstrated eosinophilia in both the oral lesion and peripheral blood, suggesting an immunologic relationship. Furthermore, the authors postulated that local trauma initiated the development of TUGSE, providing the catalyst for the HTLV-1 carrier to develop peripheral eosinophilia.21
In the second case, a 12-year-old boy developed TUGSE in the presence of Epstein-Barr virus.2 Immunologically, this virus can be reactivated from its latent stage during immunosuppression. Epstein-Barr virus has been implicated in lymphoproliferative diseases of both B- and T-cell origin, including CD30+ ALCL and LyP.22,23 The authors in this report again hypothesized there was a correlation between lymphoproliferative disorders and TUGSE lesions.2,24
Alternatively, TUGSE may simply be a reactive process to trauma or another underlying trigger. It has been speculated that the presence of eosinophils correlates with antigen insertion into the oral mucosa, whereas other ulcers of the oral mucosa are devoid of eosinophils.1 These antigens may include microorganisms, endogenous degradation products, or foreign proteins.7,25 Additionally, the presence of CD30+ lymphocytes is not isolated to lymphoproliferative disorders. CD30+ cells have been documented in arthropod bite reactions, atopic dermatitis, drug reactions, molluscum contagiosum, and scabies, among others.1,26
Healing and Management
The length of healing in TUGSE ulcers has substantial variability, from days to up to 1 year in an isolated case.1,24 Sequential expression of transforming growth factor (TGF) α and TGF-β expressed by tissue eosinophils may be underlying factors associated with a quicker healing response as demonstrated by similar ulcers in hamsters.27 Chronic nonhealing oral ulcers, particularly TUGSE lesions that demonstrated the typical increase in eosinophils in 11 of 12 cases, showed minimal TGF-α or TGF-β expression by eosinophils, perhaps indicating a possible mechanism leading to delayed wound healing in some cases. Interestingly, incisional biopsies often led to rapid wound healing, suggesting that the biopsy itself allowed for a transition back to the regular wound-healing processes.28
Traumatic ulcerative granuloma with stromal eosinophilia spontaneously resolves on its own in most cases; however, because of the concern for malignancy, it has the potential to be overtreated.26 Symptomatic treatment only is the mainstay of therapy. The patient should be instructed to avoid trauma, and referral to a dental professional is indicated when associated with dentures or other periprosthetic devices. Diet should consist of soft foods while avoiding spicy foods. Topical or oral analgesics may be necessary if substantial pain is associated with the lesion.2 Oral prednisolone was used in a patient with concurrent HTLV-1 and TUGSE to treat peripheral eosinophilia.21 The patient’s peripheral eosinophils dropped to 1% in 1 day, and the patient’s oral lesion began to improve at day 3 and disappeared by day 10. Although TUGSE may spontaneously resolve within a 10-day period without steroids, it may be a reasonable treatment to improve healing time in an otherwise healthy individual.21,26 If there is concern for malignancy, the patient should have the lesion biopsied to provide reassurance and for the added benefit of a transition to normal healing response and decreased healing time.28
Clinical Recognition
The clinician should be aware of the possibility of a CD30+ lymphoproliferative disorder, which has been associated with TUGSE in some cases, or may simulate TUGSE both clinically and histologically. Further studies are needed to clarify the relationship between these 2 entities. Whether it is a true relationship, simple coincidence, or simply overlapping clinical and histologic features remains to be determined.
Traumatic ulcerative granuloma with stromal eosinophilia (TUGSE) is an uncommon, benign, self-limited condition that is restricted to the oral mucosa, most commonly seen in the fifth to seventh decades of life.1-3 The pathogenesis of TUGSE is unknown, but current theory suggests trauma is the instigating factor. The presence of CD30+ mononuclear cells within TUGSE raises the possibility of a CD30+ lymphoproliferative disorder in some cases.4 However, because CD30+ cells are not uncommon in other benign reactive processes, they may simply represent a reactive phenomenon.3
Traumatic ulcerative granuloma with stromal eosinophilia traverses multiple disciplines, including dermatology, oral surgery, dentistry, and pathology, resulting in a diverse nomenclature including traumatic granuloma of the tongue, traumatic eosinophilic granuloma of the oral mucosa, ulcerated granuloma eosinophilicum diutinum, and eosinophilic ulcer of the oral mucosa.1,4-6 It is important to differentiate eosinophilic granuloma of the oral mucosa from the eosinophilic granuloma that is associated with Langerhans cell histiocytosis. Although both may present with oral ulceration, Langerhans cell–associated eosinophilic granuloma typically develops from underlying bone, whereas eosinophilic granuloma of the oral mucosa (TUGSE) is described as nonosseous.7,8 Furthermore, the gingiva is the most common oral site in Langerhans cell–associated eosinophilic granuloma, whereas the tongue is most commonly involved in TUGSE.8 Shapiro and Juhlin9 clearly distinguished TUGSE from Langerhans cell–associated eosinophilic granuloma in 1970. Histologically, the 2 conditions are completely different.
When ulcerative granulomas develop in the pediatric population, usually in children younger than 2 years, it is termed Riga-Fede disease.10 These children were typically breastfeeding, suckling, or teething, suggesting trauma as a triggering event. In 1961, Hjorting-Hansen and Schmidt5 described 3 separate lesions similar to Riga-Fede disease in an adult patient. Subsequently, Riga-Fede disease was grouped under TUGSE.3
Histologically, TUGSE shows an ulcerated epithelium with a polymorphic inflammatory cell infiltrate that has a large predominance of eosinophils. The infiltrate affects the superficial and deep layers of the muscle tissue and penetrates into the salivary glands. Large atypical mononuclear cells with an ovoid and pale-appearing nucleus often are present. These cells may be mitotically active and stain positively for CD30.1,4,11 CD68+ macrophages, T lymphocytes, and factor XIIIa–positive dendritic cells commonly are present.12
Given the presence of large atypical CD30+ cells in many lesions, the possibility of a CD30+ lymphoproliferative disorder has been postulated by some authors. Indeed, lymphomatoid papulosis (LyP) has been documented to involve the oral mucosa.2,4
Case Report
An 81-year-old man presented with a rapidly enlarging, 1.7×1.3-cm, vascular-appearing nodule with a collarette of mucosal epithelium on the left side of the dorsal surface of the tongue of 2 weeks’ duration (Figure 1). He denied any history of trauma, tobacco chewing, weight change, fever, or fatigue; however, he did report a 30 pack-year smoking history. There was no other pertinent medical history to include medications or allergies.
The differential diagnosis included pyogenic granuloma, granular cell tumor, squamous cell carcinoma, other neoplasms (eg, oral lymphoma, salivary gland tumors), and a traumatic blood blister from tongue biting. The patient was referred to the oral maxillofacial surgery department for an excisional biopsy, which showed a solitary ulcerated nodule with associated granulation tissue, thrombus, and fibrinoid debris (Figure 2). A surrounding dense mixed inflammatory cell infiltrate composed of lymphocytes, histiocytes, and numerous eosinophils was noted extending through the submucosal tissue and underlying striated muscle fibers (Figure 3). The adjacent mucosal epithelium appeared normal. CD30 staining showed only rare positive cells. These findings were consistent with TUGSE.
Due to the benign nature of TUGSE, the patient was released with symptomatic care and instructed to return for any new growth. The growth spontaneously resolved over 1 month and no recurrence or new lesions were reported 1 year later.
Comment
Despite encompassing multiple disciplines of medicine, TUGSE has minimal exposure in the dermatologic literature. It is an important clinical and histologic diagnosis that will provide reassurance to the patient when accurately identified and reduce potentially harmful treatments.
Clinical Presentation
Typically, TUGSE presents as a painful solitary nodule with a central ulcer and yellow fibrinous base. The margins of the ulcer typically have an indurated and rolled appearance.1,4 More than 50% of the lesions develop on the tongue, specifically the dorsal or lateral surfaces, but they may present anywhere in the oral mucosa.7 Traumatic ulcerative granuloma with stromal eosinophilia is a fast-growing lesion, typically developing in days to weeks. Although it spontaneously regresses, the lesion may take weeks or months to resolve. In one case, it resolved 1 year later.1 Traumatic ulcerative granuloma with stromal eosinophilia has a bimodal age distribution, generally appearing in the first 2 years of life and later in the fifth through seventh decades. The male-to-female predominance is equal.1,7,11 Reoccurrence is rare, but some reports have shown patients with multiple episodes of TUGSE.13,14
Differential Diagnosis
The clinical differential diagnosis for TUGSE includes squamous cell carcinoma, pyogenic granuloma, lymphoproliferative disorder, traumatic neuroma, Langerhans cell histiocytosis, granulomatous disorders, and oral lymphoma. Inflammatory disorders such as syphilis, Behçet’s disease, herpes, histoplasmosis, Wegener granulomatosis, and others also should be considered.
Immunohistochemistry
Immunohistochemical analysis of TUGSE lesions recently has revealed the presence of CD30+ cells. These cells are associated with cutaneous lymphoproliferative disorders including LyP, anaplastic large cell lymphoma (ALCL), and borderline CD30+ lesions, among others. Systemic diseases with CD30+ cells include mycosis fungoides, other T-cell lymphomas, and Hodgkin lymphoma.15,16 Once CD30+ cells were recognized, multiple authors began speculating there was a correlation between TUGSE and the CD30+ lymphoproliferative disorders.1,2,13 Anaplastic large cell lymphoma and LyP of the oral mucosa have been reported in several cases.17-20 One report described 2 cases of ulcerated CD30+ T-cell non-Hodgkin lymphoma of the oral mucosa, one of which showed eosinophilic infiltrates and was initially thought to be TUGSE. Based on these overlapping clinical and histologic features, the authors hypothesized there was a correlation between oral ALCL, LyP, and TUGSE.17 In one report, a patient developed multiple TUGSE lesions throughout his life, suggesting a pathologic process similar to LyP. The lesion biopsied showed that 70% of the T cells expressed CD30 (Ki-1) antigen.13
Underlying Causes
In support of an underlying immunologic process that augments the growth of these lesions, 2 separate case reports of TUGSE in the presence of human T-lymphotropic virus 1 (HTLV-1) and Epstein-Barr virus have been documented.2,21 Concurrent presentation of TUGSE and HTLV-1 in one report demonstrated eosinophilia in both the oral lesion and peripheral blood, suggesting an immunologic relationship. Furthermore, the authors postulated that local trauma initiated the development of TUGSE, providing the catalyst for the HTLV-1 carrier to develop peripheral eosinophilia.21
In the second case, a 12-year-old boy developed TUGSE in the presence of Epstein-Barr virus.2 Immunologically, this virus can be reactivated from its latent stage during immunosuppression. Epstein-Barr virus has been implicated in lymphoproliferative diseases of both B- and T-cell origin, including CD30+ ALCL and LyP.22,23 The authors in this report again hypothesized there was a correlation between lymphoproliferative disorders and TUGSE lesions.2,24
Alternatively, TUGSE may simply be a reactive process to trauma or another underlying trigger. It has been speculated that the presence of eosinophils correlates with antigen insertion into the oral mucosa, whereas other ulcers of the oral mucosa are devoid of eosinophils.1 These antigens may include microorganisms, endogenous degradation products, or foreign proteins.7,25 Additionally, the presence of CD30+ lymphocytes is not isolated to lymphoproliferative disorders. CD30+ cells have been documented in arthropod bite reactions, atopic dermatitis, drug reactions, molluscum contagiosum, and scabies, among others.1,26
Healing and Management
The length of healing in TUGSE ulcers has substantial variability, from days to up to 1 year in an isolated case.1,24 Sequential expression of transforming growth factor (TGF) α and TGF-β expressed by tissue eosinophils may be underlying factors associated with a quicker healing response as demonstrated by similar ulcers in hamsters.27 Chronic nonhealing oral ulcers, particularly TUGSE lesions that demonstrated the typical increase in eosinophils in 11 of 12 cases, showed minimal TGF-α or TGF-β expression by eosinophils, perhaps indicating a possible mechanism leading to delayed wound healing in some cases. Interestingly, incisional biopsies often led to rapid wound healing, suggesting that the biopsy itself allowed for a transition back to the regular wound-healing processes.28
Traumatic ulcerative granuloma with stromal eosinophilia spontaneously resolves on its own in most cases; however, because of the concern for malignancy, it has the potential to be overtreated.26 Symptomatic treatment only is the mainstay of therapy. The patient should be instructed to avoid trauma, and referral to a dental professional is indicated when associated with dentures or other periprosthetic devices. Diet should consist of soft foods while avoiding spicy foods. Topical or oral analgesics may be necessary if substantial pain is associated with the lesion.2 Oral prednisolone was used in a patient with concurrent HTLV-1 and TUGSE to treat peripheral eosinophilia.21 The patient’s peripheral eosinophils dropped to 1% in 1 day, and the patient’s oral lesion began to improve at day 3 and disappeared by day 10. Although TUGSE may spontaneously resolve within a 10-day period without steroids, it may be a reasonable treatment to improve healing time in an otherwise healthy individual.21,26 If there is concern for malignancy, the patient should have the lesion biopsied to provide reassurance and for the added benefit of a transition to normal healing response and decreased healing time.28
Clinical Recognition
The clinician should be aware of the possibility of a CD30+ lymphoproliferative disorder, which has been associated with TUGSE in some cases, or may simulate TUGSE both clinically and histologically. Further studies are needed to clarify the relationship between these 2 entities. Whether it is a true relationship, simple coincidence, or simply overlapping clinical and histologic features remains to be determined.
- Hirshberg A, Amariglio N, Akrish S, et al. Traumatic ulcerative granuloma with stromal eosinophilia: reactive lesion of the oral mucosa. Am J Clin Pathol. 2006;126:522-529.
- Abdel-Naser MB, Tsatsou F, Hippe S, et al. Oral eosinophilic ulcer, an Epstein-Barr virus-associated CD30+ lymphoproliferation? [published online April 5, 2011]. Dermatology. 2011;222:113-118.
- Fonseca FP, Benevenuto de Andrade BA, Coletta RD, et al. Clinicopathological and immunohistochemical analysis of 19 cases of oral eosinophilic ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:532-540.
- Alobeid B, Pan LX, Milligan L, et al. Eosinophil-rich CD30+ lymphoproliferative disorder of the oral mucosa. Am J Clin Pathol. 2004;121:43-50.
- Hjorting-Hansen E, Schmidt H. Ulcerated granuloma eosinophilicum diutinum of the tongue. report of a case. Acta Derm Venereol. 1961;41:235-239.
- Velez A, Alamillos FJ, Dean A, et al. Eosinophilic ulcer of the oral mucosa: report of a recurrent case on the tongue. Clin Exp Dermatol. 1997;22:154-156.
- Elzay RP. Traumatic ulcerative granuloma with stromal eosinophilia (Riga-Fede’s disease and traumatic eosinophilic granuloma). Oral Surg Oral Med Oral Pathol. 1983;55:497-506.
- Val-Bernal JF, Gonzalez-Vela MC, Sanchez-Santolino S, et al. Localized eosinophilic (Langerhans’ cell) granuloma of the lower lip. a lesion that may cause diagnostic error. J Cutan Pathol. 2009;36:1109-1113.
- Shapiro L, Juhlin EA. Eosinophilic ulcer of the tongue report of two cases and review of the literature. Dermatologica. 1970;140:242-250.
- Amberg S. Sublingual growth in infants. Am J Med Sci. 1902;126:257-269.
- EI-Mofty SK, Swanson PE, Wick MR, et al. Eosinophilic ulcer of the oral mucosa: report of 38 new cases with immunohistochemical observations. Oral Surg Oral Med Oral Pathol. 1993;75:716-722.
- Regezi JA, Zarbo RJ, Daniels TE, et al. Oral traumatic granuloma: characterization of the cellular infiltrate. Oral Surg Oral Med Oral Pathol. 1993;75:723-727.
- Ficarra G, Prignano F, Romagnoli P. Traumatic eosinophilic granuloma of the oral mucosa: a CD30+ (Ki-1) lymphoproliferative disorder? Oral Oncol. 1997;33:375-379.
- Doyle JL, Geary W, Baden E. Eosinophilic ulcer. J Oral Maxillofac Surg. 1989;47:349-352.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985;66:848-858.
- Rosenberg A, Biesma DH, Sie-Go DMDS, et al. Primary extranodal CD30-positive T-cell non-Hodgkin’s lymphoma of the oral mucosa. report of two cases. Int J Oral Maxillofac Surg. 1996;25:57-59.
- Kato N, Tomita Y, Yoshida K, et al. Involvement of the tongue by lymphomatoid papulosis. Am J Dermatopathol. 1998;20:522-526.
- Savarrio L, Gibson J, Dunlop DJ, et al. Spontaneous regression of an anaplastic large cell lymphoma in the oral cavity: first reported case and review of the literature. Oral Oncol. 1999;35:609-613.
- Sciubba J, Said-Al-Naief N, Fantasia J. Critical review of lymphomatoid papulosis of the oral cavity with case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:195-204.
- Yamazaki H, Shirasugi Y, Kajiwara H, et al. Concurrent onset of eosinophilic ulcer of the oral mucosa with peripheral eosinophilia in a human T-cell leukemia virus type I carrier. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;114:E43-E48.
- Dojcinov SD, Venkataram G, Raffeld M, et al. EBV positive mucocutaneous ulcer—a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol. 2010;34:405-417.
- Kim YC, Yang WI, Lee MG, et al. Epstein-Barr virus in CD30 anaplastic large cell lymphoma involving the skin and lymphomatoid papulosis in South Korea. Int J Dermatol. 2006;45:1312-1316.
- Pietersma F, Piriou E, van Baarle D. Immune surveillance of EBV-infected B cells and the development of non-Hodgkin lymphomas in immunocompromised patients. Leuk Lymphoma. 2008;49:1028-1041.
- Salisbury CL, Budnick SD, Li S. T cell receptor gene rearrangement and CD 30 immunoreactivity in traumatic ulcerative granuloma with stromal eosinophilia of oral cavity. Am J Clin Pathol. 2009;132:722-727.
- Marszalek A, Neska-Dlugosz I. Traumatic ulcerative granuloma with stromal eosinophilia. a case report and short literature review. Pol J Pathol. 2011;3:172-175.
- Wong DT, Donoff RB, Yang J, et al. Sequential expression of transforming growth factors alpha and beta 1 by eosinophils during cutaneous wound healing in the hamster. Am J Pathol. 1993;143:130-142.
- Elovic AE, Gallagher GT, Kabani S, et al. Lack of TGF-alpha and TGF-beta synthesis by human eosinophils in chronic oral ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:672-681.
- Hirshberg A, Amariglio N, Akrish S, et al. Traumatic ulcerative granuloma with stromal eosinophilia: reactive lesion of the oral mucosa. Am J Clin Pathol. 2006;126:522-529.
- Abdel-Naser MB, Tsatsou F, Hippe S, et al. Oral eosinophilic ulcer, an Epstein-Barr virus-associated CD30+ lymphoproliferation? [published online April 5, 2011]. Dermatology. 2011;222:113-118.
- Fonseca FP, Benevenuto de Andrade BA, Coletta RD, et al. Clinicopathological and immunohistochemical analysis of 19 cases of oral eosinophilic ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:532-540.
- Alobeid B, Pan LX, Milligan L, et al. Eosinophil-rich CD30+ lymphoproliferative disorder of the oral mucosa. Am J Clin Pathol. 2004;121:43-50.
- Hjorting-Hansen E, Schmidt H. Ulcerated granuloma eosinophilicum diutinum of the tongue. report of a case. Acta Derm Venereol. 1961;41:235-239.
- Velez A, Alamillos FJ, Dean A, et al. Eosinophilic ulcer of the oral mucosa: report of a recurrent case on the tongue. Clin Exp Dermatol. 1997;22:154-156.
- Elzay RP. Traumatic ulcerative granuloma with stromal eosinophilia (Riga-Fede’s disease and traumatic eosinophilic granuloma). Oral Surg Oral Med Oral Pathol. 1983;55:497-506.
- Val-Bernal JF, Gonzalez-Vela MC, Sanchez-Santolino S, et al. Localized eosinophilic (Langerhans’ cell) granuloma of the lower lip. a lesion that may cause diagnostic error. J Cutan Pathol. 2009;36:1109-1113.
- Shapiro L, Juhlin EA. Eosinophilic ulcer of the tongue report of two cases and review of the literature. Dermatologica. 1970;140:242-250.
- Amberg S. Sublingual growth in infants. Am J Med Sci. 1902;126:257-269.
- EI-Mofty SK, Swanson PE, Wick MR, et al. Eosinophilic ulcer of the oral mucosa: report of 38 new cases with immunohistochemical observations. Oral Surg Oral Med Oral Pathol. 1993;75:716-722.
- Regezi JA, Zarbo RJ, Daniels TE, et al. Oral traumatic granuloma: characterization of the cellular infiltrate. Oral Surg Oral Med Oral Pathol. 1993;75:723-727.
- Ficarra G, Prignano F, Romagnoli P. Traumatic eosinophilic granuloma of the oral mucosa: a CD30+ (Ki-1) lymphoproliferative disorder? Oral Oncol. 1997;33:375-379.
- Doyle JL, Geary W, Baden E. Eosinophilic ulcer. J Oral Maxillofac Surg. 1989;47:349-352.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985;66:848-858.
- Rosenberg A, Biesma DH, Sie-Go DMDS, et al. Primary extranodal CD30-positive T-cell non-Hodgkin’s lymphoma of the oral mucosa. report of two cases. Int J Oral Maxillofac Surg. 1996;25:57-59.
- Kato N, Tomita Y, Yoshida K, et al. Involvement of the tongue by lymphomatoid papulosis. Am J Dermatopathol. 1998;20:522-526.
- Savarrio L, Gibson J, Dunlop DJ, et al. Spontaneous regression of an anaplastic large cell lymphoma in the oral cavity: first reported case and review of the literature. Oral Oncol. 1999;35:609-613.
- Sciubba J, Said-Al-Naief N, Fantasia J. Critical review of lymphomatoid papulosis of the oral cavity with case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90:195-204.
- Yamazaki H, Shirasugi Y, Kajiwara H, et al. Concurrent onset of eosinophilic ulcer of the oral mucosa with peripheral eosinophilia in a human T-cell leukemia virus type I carrier. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;114:E43-E48.
- Dojcinov SD, Venkataram G, Raffeld M, et al. EBV positive mucocutaneous ulcer—a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol. 2010;34:405-417.
- Kim YC, Yang WI, Lee MG, et al. Epstein-Barr virus in CD30 anaplastic large cell lymphoma involving the skin and lymphomatoid papulosis in South Korea. Int J Dermatol. 2006;45:1312-1316.
- Pietersma F, Piriou E, van Baarle D. Immune surveillance of EBV-infected B cells and the development of non-Hodgkin lymphomas in immunocompromised patients. Leuk Lymphoma. 2008;49:1028-1041.
- Salisbury CL, Budnick SD, Li S. T cell receptor gene rearrangement and CD 30 immunoreactivity in traumatic ulcerative granuloma with stromal eosinophilia of oral cavity. Am J Clin Pathol. 2009;132:722-727.
- Marszalek A, Neska-Dlugosz I. Traumatic ulcerative granuloma with stromal eosinophilia. a case report and short literature review. Pol J Pathol. 2011;3:172-175.
- Wong DT, Donoff RB, Yang J, et al. Sequential expression of transforming growth factors alpha and beta 1 by eosinophils during cutaneous wound healing in the hamster. Am J Pathol. 1993;143:130-142.
- Elovic AE, Gallagher GT, Kabani S, et al. Lack of TGF-alpha and TGF-beta synthesis by human eosinophils in chronic oral ulcers. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:672-681.
Practice Points
- Immunohistochemical staining of traumatic ulcerative granuloma with stromal eosinophilia (TUGSE) may suggest an underlying lymphoproliferative disorder.
- Early recognition of TUGSE, which often is malignant appearing, is key, with watchful waiting as the mainstay therapy.
- Adjunctive therapy for TUGSE includes prednisolone and oral analgesics.
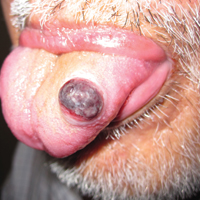
Pruritic Eruption on the Chest
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
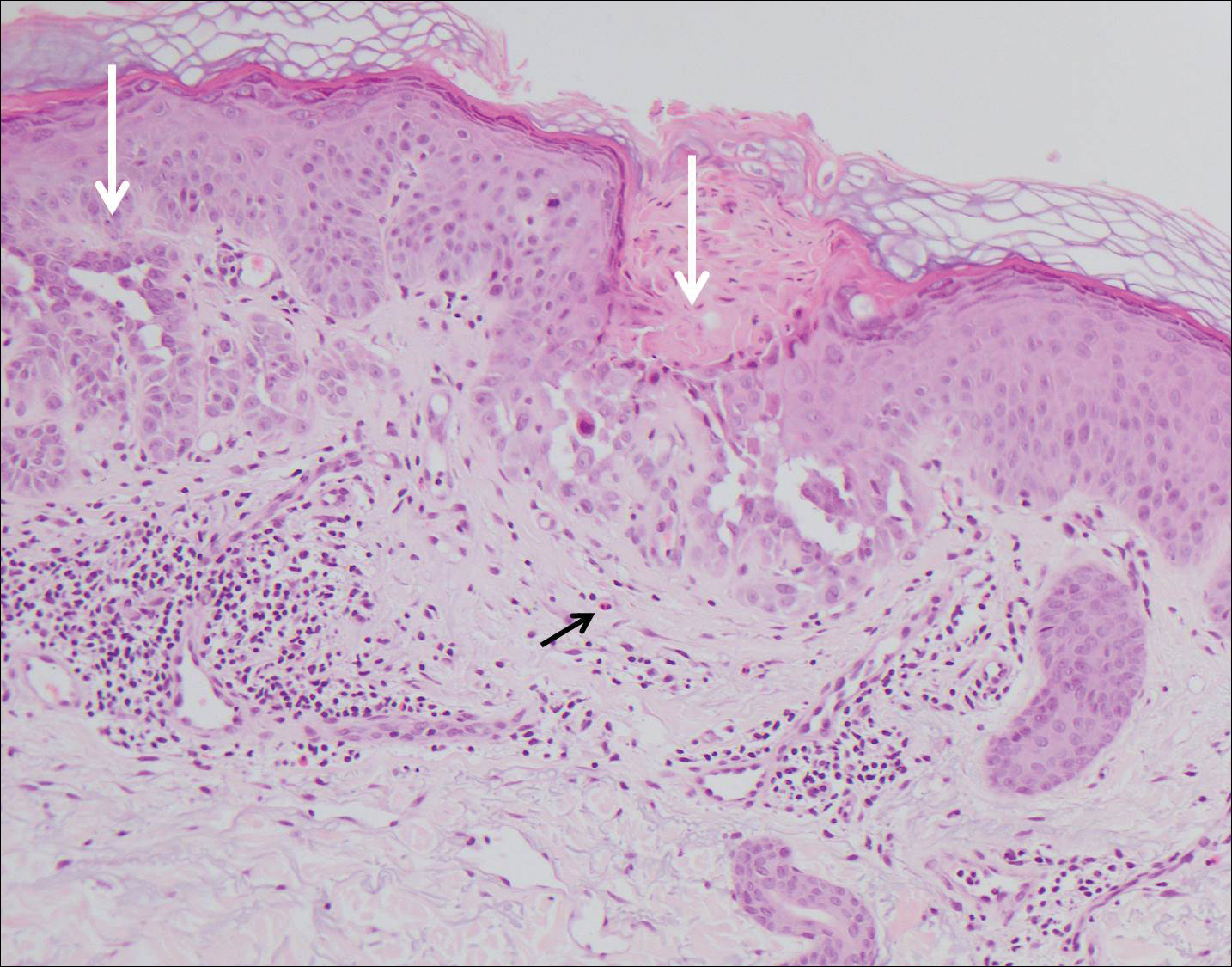
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
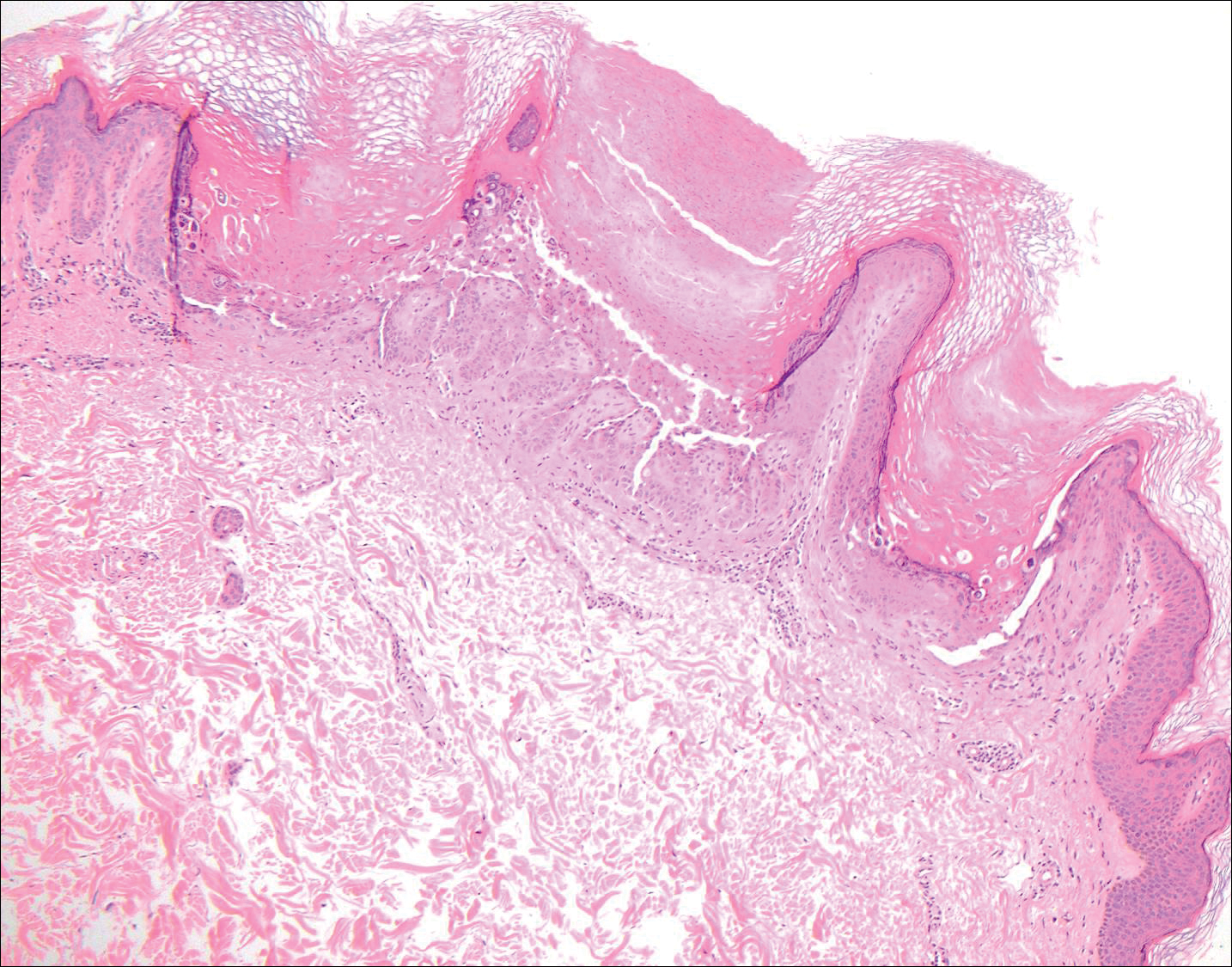
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
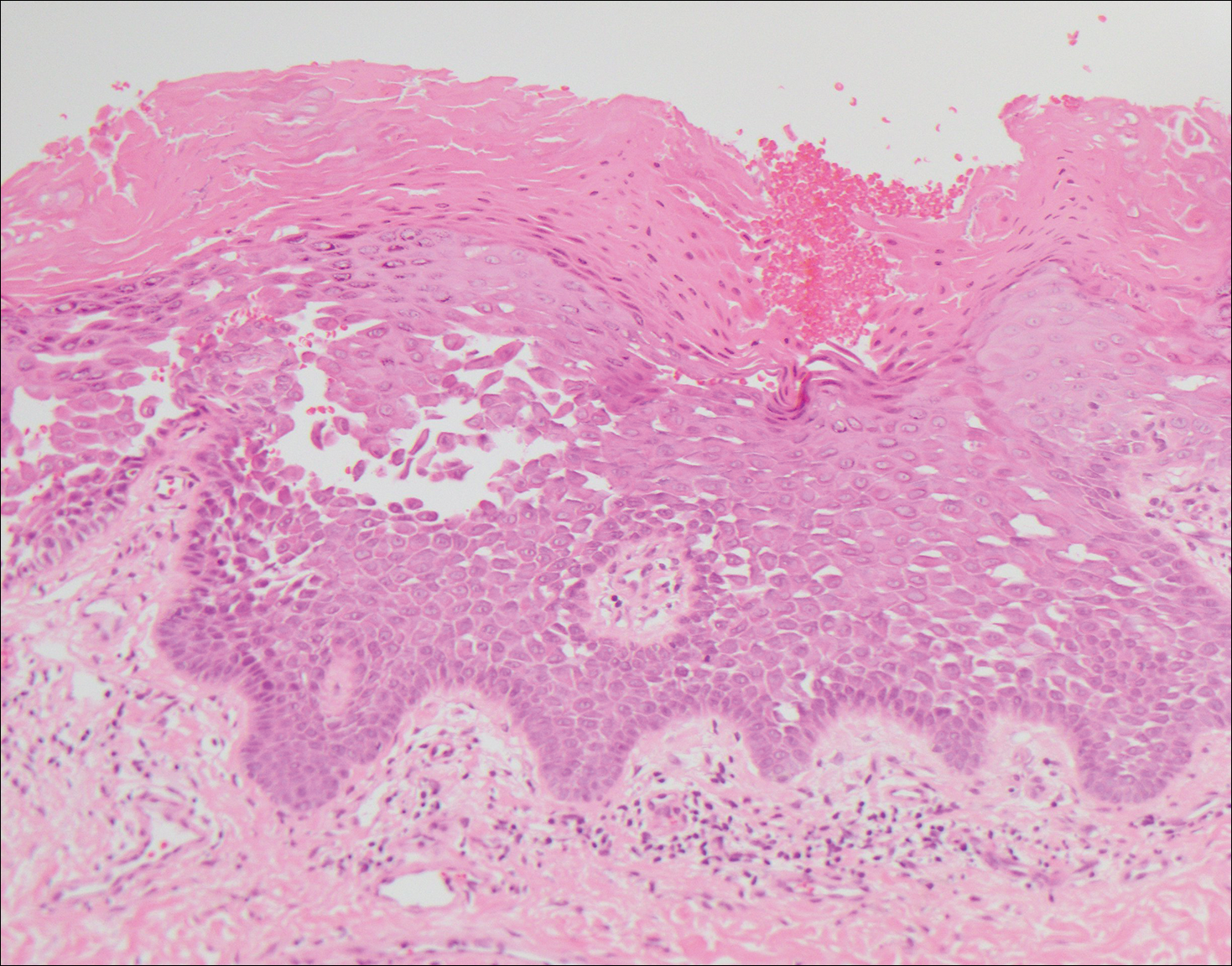
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
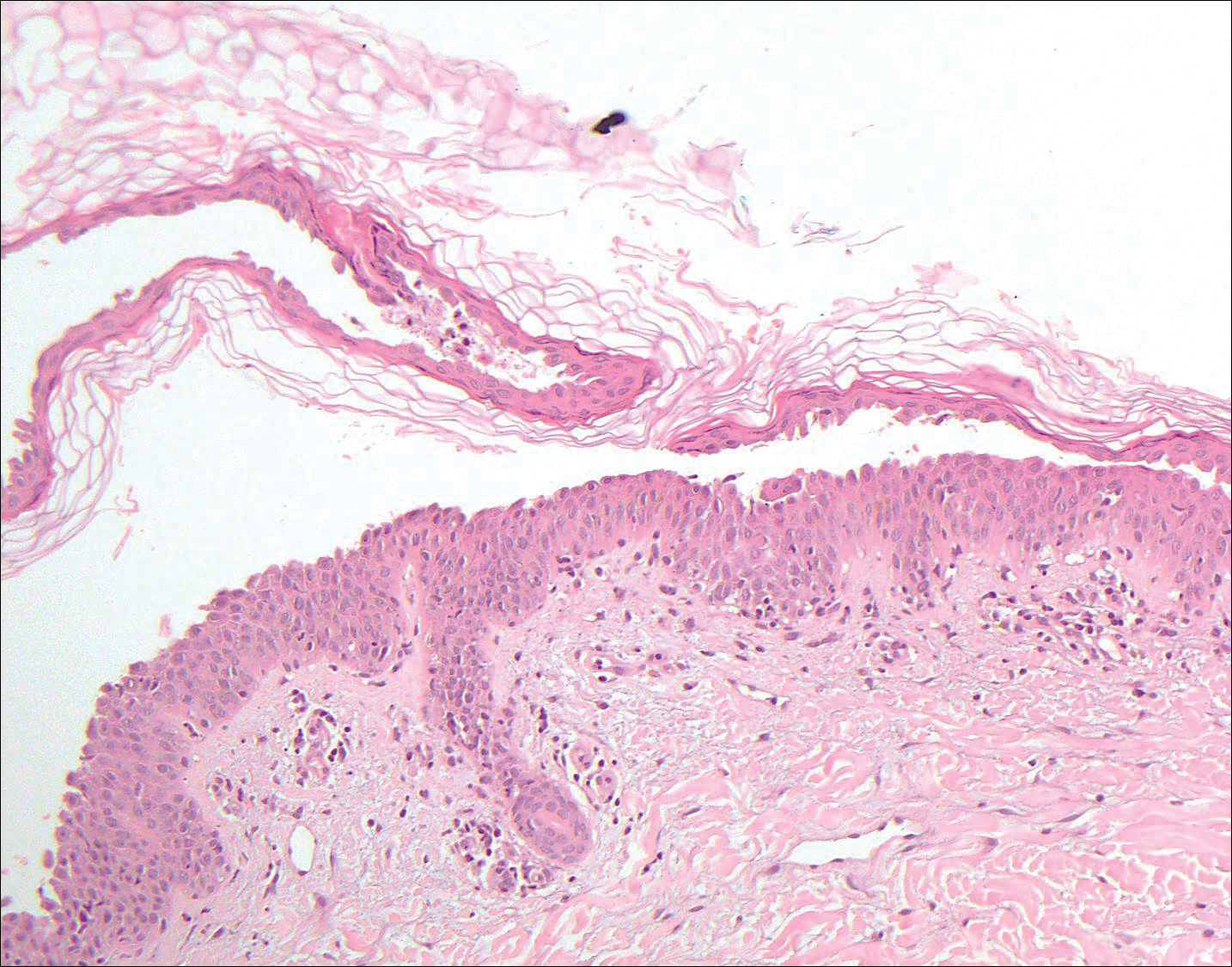
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
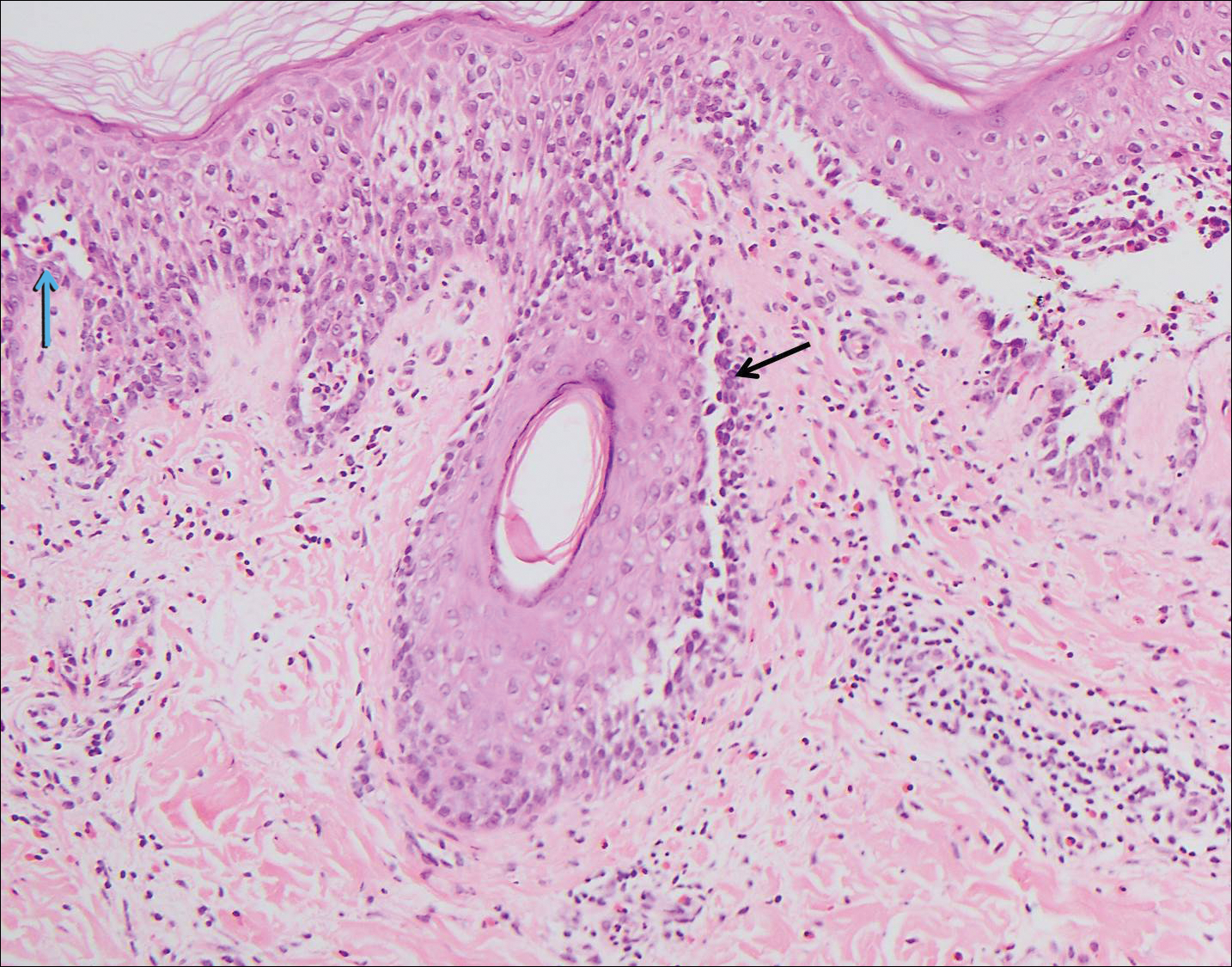
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
The Diagnosis: Grover Disease
Grover disease (also known as transient acantholytic dermatosis) was first described by Ralph W. Grover in 1970 as an idiopathic, acquired, monomorphous, papulovesicular eruption. Although originally characterized by solely transient acantholytic dermatosis, over time the term Grover disease has been expanded to include persistent acantholytic dermatoses. Grover disease chiefly affects white adults older than 40 years and is more prevalent in males than females. Cases generally are self-limited but correlate with age, as older adults are more likely to have prolonged eruptions.1
Grover disease typically erupts with discrete, erythematous, edematous, acneform, red-brown or flesh-colored papules, papulovesicles, or keratotic papules that primarily are seen on the trunk and anterior portion of the chest. As the rash spreads, it can erupt on the neck and thighs. The etiology of Grover disease is unknown, but many factors have been associated with the condition in a limited number of patients, including exposure to UV radiation, excessive heat or sweating, use of sulfadoxine-pyrimethamine and recombinant human IL-4, and infection with Malassezia furfur and Demodex folliculorum.1 Grover disease also has been associated with other conditions such as asteatotic eczema, allergic contact dermatitis, and atopic dermatitis.2
Histologically, Grover disease (Figure 1) is an acantholytic process that can exhibit dyskeratosis (corps ronds and grains). Foci often are small and multiple foci are seen on shave biopsy. There also may be spongiotic changes when associated with an eczematous element. A perivascular lymphohistiocytic infiltrate with eosinophils usually is seen.3 Basket weave keratin may be seen; however, as the lesions cause pruritus, erosions and ulcerations often are present.4
Grover disease has multiple histologic variants that may resemble Darier disease, Hailey-Hailey disease, pemphigus foliaceus, pemphigus vulgaris, and spongiotic dermatitis and can present in combination.5
The variant of Grover disease that has a Darier-like pattern is difficult to distinguish from Darier disease, an autosomal-dominant-inherited disorder classified by small papules that emerge in seborrheic areas during childhood and adolescence. Histologically, Darier disease (Figure 2) shows broad areas of dyskeratosis and acantholysis that lead to suprabasal cleavage. Follicular extension may be present. In addition, there often is prominent vertical parakeratosis in Darier disease.6 Histologic features that favor Darier disease over the Darier-like variant of Grover disease include a broad focus of acanthotic dyskeratosis with follicular extension; the presence of a hyperkeratotic stratum corneum; and a lack of spongiosis and eosinophils, which are notably absent in Darier disease but may be present in Grover disease.4
Another variant of Grover disease has a Hailey-Hailey-like pattern, which is characterized by Hailey-Hailey disease's dilapidated brick wall appearance or the diffuse suprabasal acantholysis of all epidermal layers without notable dyskeratosis.4 Hailey-Hailey disease, also known as familial benign pemphigus, is an autosomal-dominant disorder that presents with erythematous vesicular plaques in flexural areas. The plaques progress to flaccid bullae with rupture and crusting and spread peripherally.7 Pathology shows suprabasilar clefts and numerous acantholytic cells (Figure 3). Dyskeratotic keratinocytes are rare with infrequent corps ronds and rare grains. The epidermis also is less hyperplastic in Grover disease than in Hailey-Hailey disease.1
Grover disease also may present histologically with a pemphiguslike pattern, mimicking pemphigus foliaceus and pemphigus vulgaris; however, direct immunofluorescence studies are negative in Grover disease.
Pemphigus foliaceus is an autoimmune disorder caused by autoantibodies to desmoglein 1, which are present on the surfaces of keratinocytes, and is characterized by scaly crusts and blisters.8 Histologically, pemphigus foliaceus (Figure 4) shows a superficial epidermal blistering process. The acantholysis may be subtle and is commonly localized to the stratum granulosum, extending into the stratum corneum. Complete loss of the stratum corneum can be seen, resulting in only scattered acantholytic cells. Spongiosis also may be seen. The dermis shows a perivascular infiltrate that often contains eosinophils. Pemphigus foliaceus is confirmed by direct immunofluorescence.9
Pemphigus vulgaris is an autoimmune blistering disorder that is characterized by IgG autoantibodies to desmoglein 3, a component of desmosomes that are involved in keratinocyte-to-keratinocyte adhesion. Clinically, patients present with flaccid fragile blisters on the skin and mucous membranes that rupture easily, leading to painful erosions.10 Intraepidermal blisters are seen histologically (Figure 5) with the loss of cohesion (acantholysis) seen classically in the lower portions of the epidermis where desmoglein 3 is most prominent. When only the basal layer remains, the histology has been likened to a tombstone row.11 Extension of the blister along the adnexa is common. The underlying dermis shows a perivascular infiltrate with eosinophils. Early lesions may show only eosinophilic spongiosis. Direct immunofluorescence studies show IgG and C3 in an intercellular pattern that resembles a fish net or chicken wire.4,11
The spongioticlike pattern of Grover disease is marked by epidermal edema with separation of the keratinocytes and the revelation of their intracellular bridges,4 which manifests as vesiculation in the stratum corneum or upper layers of the epidermis.12
Grover disease is self-limited and may spontaneously resolve; however, the disease may be responsive to topical and systemic steroids. Additionally, avoidance of aggravating factors such as sunlight, heat, and sweating can improve symptoms.2
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
- Parsons JM. Transient acantholytic dermatosis (Grover's disease): a global perspective. J Am Acad Dermatol. 1996;35(5, pt 1):653-666; quiz 667-670.
- Quirk CJ, Heenan PJ. Grover's disease: 34 years on. Australas J Dermatol. 2004;45:83-86.
- Davis MD, Dinneen AM, Landa N, et al. Grover's disease: clinicopathologic review of 72 cases. Mayo Clin Proc. 1999;74:229-234.
- Weaver J, Bergfeld WF. Grover disease (transient acantholytic dermatosis). Arch Pathol Lab Med. 2009;133:1490-1494.
- Chalet M, Grover R, Ackerman AB. Transient acantholytic dermatosis: a reevaluation. Arch Dermatol. 1977;133:431-435.
- Takagi A, Kamijo M, Ikeda S. Darier disease. J Dermatol. 2016;43:275-279.
- Engin B, Kutlubay Z, Celik U, et al. Hailey-Hailey disease: a fold (intertriginous) dermatosis. Clin Dermatol. 2015;33:452-455.
- de Sena Nogueira Maehara L, Huizinga J, Jonkman MF. Rituximab therapy in pemphigus foliaceus: report of 12 cases and review of recent literature [published online March 31, 2015]. Br J Dermatol. 2015;172:1420-1423.
- James KA, Culton DA, Diaz LA. Diagnosis and clinical features of pemphigus foliaceus. Dermatol Clin. 2011;29:405-412.
- Black M, Mignogna MD, Scully C. Number II. pemphigus vulgaris. Oral Dis. 2005;11:119-130.
- Madke B, Doshi B, Khopkar U, et al. Appearances in dermatopathology: the diagnostic and the deceptive. Indian J Dermatol Venerol Leprol. 2013;79:338-348.
- Motaparthi K. Pseudoherpetic transient acantholytic dermatosis (Grover disease): case series and review of the literature [published online February 16, 2017]. J Cutan Pathol. 2017;44:486-489.
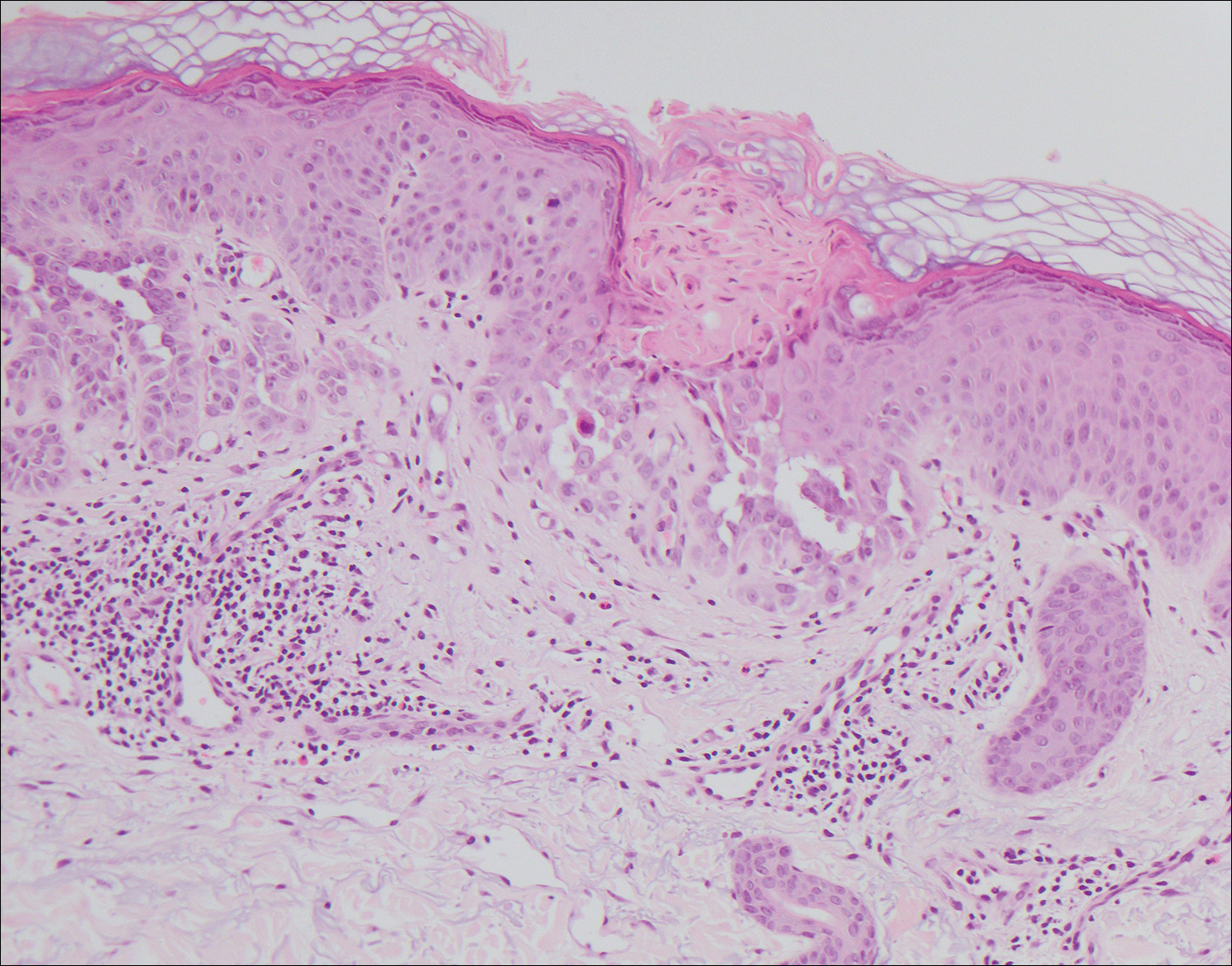
A 55-year-old man presented with small, erythematous, nonfollicular, pruritic papules on the mid chest.
Asymptomatic Cutaneous Polyarteritis Nodosa: Treatment Options and Therapeutic Guidelines
In 1931, Lindberg1 described a cutaneous variant of polyarteritis nodosa, which lacked visceral involvement and possessed a more favorable prognosis.2 Cutaneous polyarteritis nodosa (CPAN) is a localized small- to medium-vessel vasculitis restricted to the skin. Both benign and chronic courses have been described, and systemic involvement does not occur.3 Diagnostic criteria proposed by Nakamura et al3 in 2009 included cutaneous nodules, livedo reticularis, purpura, or ulcers; histopathologic fibrinoid necrotizing vasculitis of small- to medium-sized vessels; and exclusion of systemic symptoms (eg, fever, hypertension, weight loss, renal failure, cerebral hemorrhage, neuropathy, myocardial infarction, ischemic heart disease, pericarditis, pleuritis, arthralgia/myalgia). Nodules occur in 30% to 50% of cases and can remain for years if left untreated. Ulcerations occur in up to 30% of patients. Myositis, arthritis, and weakness also have been reported with this condition.4 Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibody, as well as elevated anti–phosphatidylserine-prothrombin complex antibody.5 These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. The distinction of this entity from systemic polyartertitis nodosa is key when determining treatment options and monitoring parameters.
Case Report
A 66-year-old woman was referred to our facility by an outside dermatologist with a mildly pruritic, blanchable, reticulated erythema on the chest and bilateral arms and legs of 3 months’ duration consistent with livedo reticularis (Figure 1). Prior systemic therapy included prednisone 10 mg 3 times daily, fexofenadine, loratadine, and hydroxyzine. When the systemic steroid was tapered, the patient developed an asymptomatic flare of her eruption. On presentation, the lesions had waxed and waned, and the patient was taking only vitamin B12 and vitamin C. Her medical history was notable for an unknown-type lymphoma of the chest wall diagnosed at 46 years of age that was treated with an unknown chemotherapeutic agent, chronic pancreatitis that resulted in a duodenectomy at 61 years of age, chronic cholecystitis, and 1 first-trimester miscarriage. Outside laboratory tests, including a comprehensive metabolic panel, complete blood cell count, urinalysis, renal function, and liver function tests were within reference range, except for the finding of mild leukocytosis (11,000/µL)(reference range, 3800–10,800/µL), which resolved after steroids were discontinued, with otherwise normal results. Punch biopsy of a specimen from the right thigh revealed medium-vessel vasculitis consistent with polyarteritis nodosa (Figure 2). Laboratory workup by our facility including hepatitis panel, perinuclear antineutrophil cytoplasmic antibody, cytoplasmic antineutrophil cytoplasmic antibody, factor V Leiden, prothrombin time/international normalized ratio, anticardiolipin antibody, and proteins C and S were all within reference range. Abnormal values included a low positive but nondiagnostic antinuclear antibody screen with negative titers, and the lupus anticoagulant titer was mildly elevated at 44 IgG binding units (reference range, <40 IgG binding units). Serum protein electrophoresis (SPEP) and urine protein electrophoresis also were performed, and SPEP was low positive for elevated κ and γ light chains. The patient was referred to oncology, and further testing revealed no underlying malignancy. The patient was monitored and no treatment was initiated; her rash completely resolved within 3 months. Laboratory monitoring at 6 months including SPEP, urine protein electrophoresis, lupus anticoagulant, and clotting studies all were within reference range.
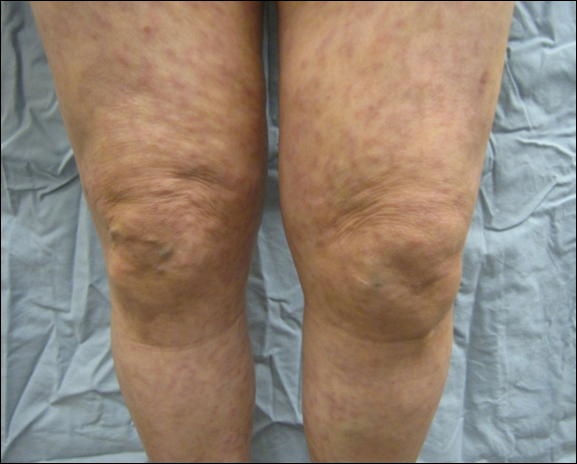
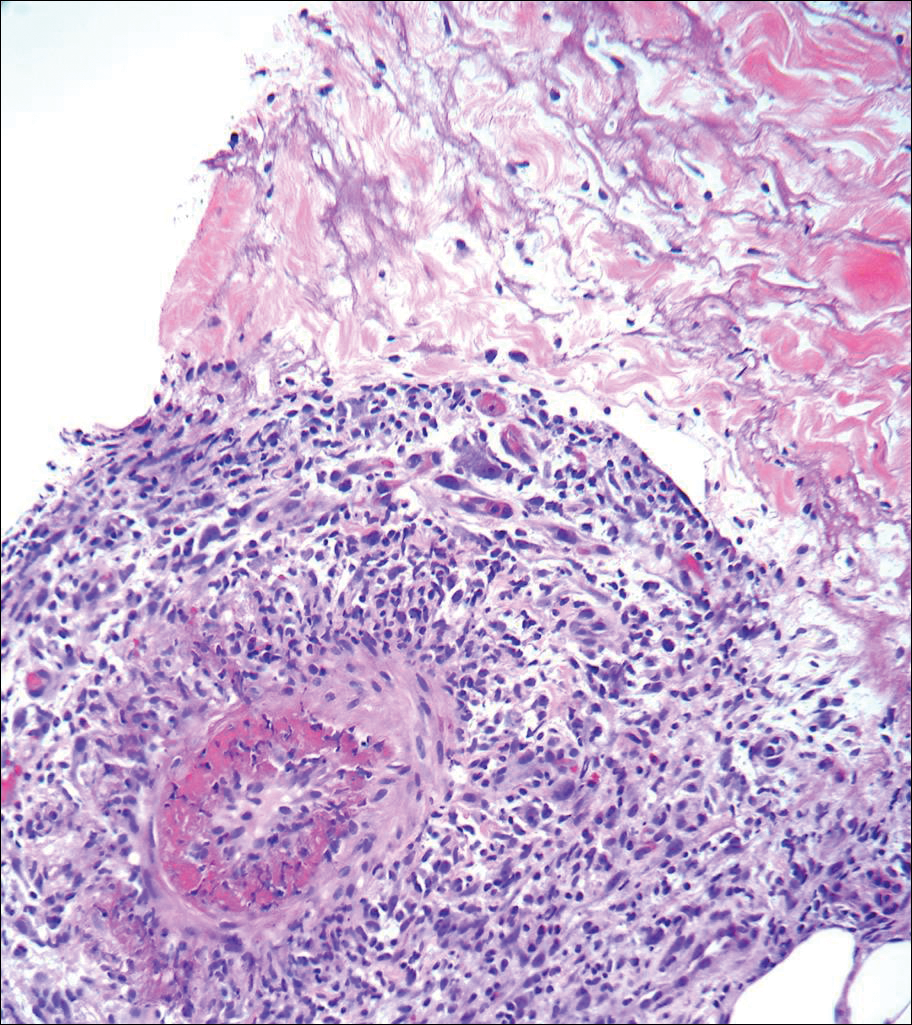
Comment
Although the treatment of systemic polyarteritis nodosa often is necessary and typically involves high-dose corticosteroids and cyclophosphamide, the treatment of CPAN initially is less aggressive. Of the options available for treatment of CPAN, each has associated risks and side effects. Chen6 classified CPAN into 3 groups: 1 (mild), 2 (severe with no systemic involvement), and 3 (severe with progression to systemic disease)(Table). The authors performed a review of all the published treatments and their respective side effects to evaluate if treatment should be instituted for asymptomatic (group 1) disease presenting with abnormal antibody findings as demonstrated in our case.

First-line treatment of CPAN includes nonsteroidal anti-inflammatory drugs (NSAIDS) and colchicine.7 Nonsteroidal anti-inflammatory drugs are preferred; however, they also have been associated with gastrointestinal tract upset and increased risk for peptic ulcer disease with long-term use. Although colchicine often is used in conjunction with NSAIDS8 for its anti-inflammatory activity, no studies have been performed on this drug as monotherapy, and the side effect of diarrhea often limits its use.
Other therapies include dapsone, which should be monitored carefully due to the risk for dapsone hypersensitivity syndrome.8,9 Topical corticosteroids have been proven effective for mild cases of confluent erythema with remission occurring as early as 4 weeks.4 Some reports emphasize the role of streptococcal infections in CPAN, especially in children.8,10-12 Consequently it is recommended that anti–streptolysin O titers should be included in the workup for CPAN. Long-term penicillin prophylaxis and tonsillectomy have been used to prevent disease flares with limited success.8,10-12
For more severe disease, especially with neuromuscular involvement, oral methylprednisolone up to 1 mg/kg daily has been used and has proven effective in the control of acute exacerbations.7,13 However, the many adverse effects of systemic steroids limit their use long-term, and taper will often result in flare of disease.4,7 Medications used in conjunction with steroids include hydroxychloroquine, dapsone, azathioprine, cyclophosphamide, methotrexate, sulfapyridine, pentoxifylline, infliximab, etanercept, and intravenous immunoglobulin.4,9,12-17
Low-dose methotrexate has shown some improvement in skin disease with CPAN, but other case reports suggest that complete remission is not achieved with this drug.15,18 More studies are needed to assess the use of methotrexate for CPAN.
Immunomodulators have been used in multiple case reports with varying levels of success. Rogalski and Sticherling4 reported 3 cases that cleared with methylprednisolone plus azathioprine ranging from 4 weeks to 6 months; nausea limited tolerance of azathioprine in 1 case. Mycophenolate mofetil also was successfully used in 2 cases with clearance at 17 weeks and 6 months. In this series of cases, cyclosporine was ineffective for CPAN.4 Two case reports documented cutaneous clearance with cyclophosphamide in conjunction with prednisolone.9,10 No prospective trials have been performed on these medications, and immunosuppressants should only be considered in steroid-resistant cases.
The use of intravenous immunoglobulin has been reported effective in prior cases that showed resistance to more conventional trials of steroids, azathioprine, and/or cyclophosphamide.12,14 Intravenous immunoglobulin may be regarded as a treatment option for severe resistant disease. Several case reports also have documented success using tumor necrosis factor α blockers, particularly infliximab, as an adjunct to steroids and etanercept as both a steroid adjunct and monotherapy.16,17,19 More studies are necessary to evaluate these treatments.
Additionally, single case reports have outlined the use of other therapeutic agents, including tamoxifen (10 mg twice daily increased to 20 mg twice daily during episodes of breakthrough lesions),20 hyperbaric oxygen therapy (100% oxygen for 90 minutes 5 times weekly at 1.5 atm absolute followed by 2 weeks of 2 atm absolute),21 and granulocyte-macrophage colony-stimulating factor (300 µg injection in small portion to ulcer edges twice monthly for 2 months).22 All of these treatments show promise, but data are limited.
Because thrombosis is postulated to be a potential mechanism leading to CPAN, agents such as pentoxifylline, clopidogrel, and warfarin have been examined as treatment options. Pentoxifylline in combination with mycophenolate mofetil has been successful in treating a case that was resistant to other immunosuppressants.23 Clopidogrel blocks the adenosine diphosphate pathway and impairs clot retraction. Clopidogrel was reported effective in an acute flare of CPAN for clearance of skin lesions and normalization of lupus anticoagulant.24 It also was used successfully in recurrent CPAN after steroid treatments in a patient with neuromuscular symptoms. There was no recurrence in either of the patients in this case report series. Warfarin therapy at an international normalized ratio of 3.0 also has demonstrated success in halting disease progression and in facilitating the resolution of skin changes and normalization of anti–phosphatidylserine-prothrombin complex antibodies.24 Our review of the literature did not reveal evidence of a standardized length of treatment following symptom resolution or if treatment is indicated in asymptomatic disease, or as in our case, with only mild elevations of antiphospholipid antibodies.
Conclusion
Multiple treatment options exist for CPAN, but the data on their efficacies is limited and based only on anecdotal evidence, not prospective analysis. We believe that it seems reasonable to initiate treatment only for symptomatic disease or cases in which the antibody titers suggest that the patient may be at high risk for thrombosis. Mild symptoms and mild cutaneous changes would suggest the likely choice of NSAIDs, colchicine, or dapsone as treatment options versus no treatment. In patients with antibody titers, pentoxifylline, clopidogrel, or warfarin may be considered first-line therapies. With severe ulcerative lesions and neuromuscular involvement, steroids, immunosuppressants, and other investigative agents should be contemplated. In our patient, the laboratory studies were repeated and normalized on complete resolution of her livedo eruption. She remained asymptomatic and clear for 8 months without any treatment. The incidence of this presentation of CPAN is unknown and is likely underreported, as we would not expect most patients to present to their physicians for the evaluation of otherwise asymptomatic livedo reticularis. In essence, our case report suggests that it may be prudent to simply monitor patients with asymptomatic CPAN.
- Lindberg K. Ein Beitrag zur Kenntnis der Periarteritis nodosa. Acta Med Scand. 1931;76:183-225.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa [published online August 26, 2005]. J Neurol. 2005;252:1155-1166.
- Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res. 2009;301:117-121.
- Rogalski C, Sticherling M. Panateritis cutanea benigna—an entity limited to the skin or cutaneous presentation of a systemic necrotizing vasculitis? report of seven cases and review of the literature. Int J Dermatol. 2007;46:817-821.
- Kawakami T, Yamazaki M, Mizoguchi M, et al. High titer of anti-phosphatidylserine-prothrombin complex antibodies in patients with cutaneous polyarteritis nodosa. Arthritis Rheum. 2007;57:1507-1513.
- Chen KR. Cutaneous polyarteritis nodosa: a clinical and histopathological study of 20 cases. J Dermatol. 1989;6:429-442.
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756.
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathologic analysis and review of the published work. J Dermatol. 2010;37:85-93.
- Flanagan N, Casey EB, Watson R, et al. Cutaneous polyartertitis nodosa with seronegative arthritis. Rheumatology (Oxford). 1999;38:1161-1162.
- Fathalla B, Miller L, Brady S, et al. Cutaneous polyarteritis nodosa in children. J Am Acad Dermatol. 2005;53:724-728.
- Misago N, Mochizuki Y, Sekiyama-Kodera H, et al. Cutaneous polyarteritis nodosa: therapy and clinical course in four cases. J Dermatol. 2001;28:719-727.
- Breda L, Franchini S, Marzetti V, et al. Intravenous immunoglobulins for cutaneous polyarteritis nodosa resistant to conventional treatment. Scand J Rheumatol. 2016;45:169-170.
- Maillard H, Szczesniak S, Martin L. Cutaneous periarteritis nodosa: diagnostic and therapeutic aspects of 9 cases. Ann Dermatol Venereol. 1999;26:125-129.
- Lobo I, Ferreira M, Silva E. Cutaneous polyarteritis nodosa treated with intravenous immunoglobulin. J Eur Acad Dermatol Venereol. 2007;22:880-882.
- Boehm I, Bauer R. Low-dose methotrexate controls a severe form of polyarteritis nodosa. Arch Dermatol. 2000;136:167-169.
- Campanilho-Marques R, Ramos F, Canhão H, et al. Remission induced by infliximab in a childhood polyarteritis nodosa refractory to conventional immunosuppression and rituximab. Joint Bone Spine. 2014;81:277-278.
- Inoue N, Shimizu M, Mizuta M, et al. Refractory cutaneous polyarteritis nodosa: successful treatment with etanercept. Pediatr Int. 2017;59:751-752.
- Schartz NE. Successful treatment in two cases of steroid dependent cutaneous polyarteritis nodosa with low-dose methotrexate. Dermatology. 2001;203:336-338.
- Valor L, Monteagudo I, de la Torre I, et al. Young male patient diagnosed with cutaneous polyarteritis nodosa successfully treated with etanercept. Mod Rheumatol. 2014;24:688-689.
- Cvancara JL, Meffert JJ, Elston DM. Estrogen sensitive cutaneous polyarteritis nodosa: response to tamoxifen. J Am Acad Dermatol. 1998;39:643-646.
- Mazokopakis E, Milkas A, Tsartsalis A, et al. Improvement of cutaneous polyarteritis nodosa with hyperbaric oxygen. Int J Dermatol. 2009;48:1017-1029.
- Tursen U, Api H, Kaya TI, et al. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony stimulating factor in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol. 2006;20:1341-1343.
- Kluger N, Guillot B, Bessis D. Ulcerative cutaneous polyarteritis nodosa treated with mycophenolate mofetil and pentoxifylline. J Dermatolog Treat. 2011;22:175-177.
- Kawakami T, Soma Y. Use of warfarin therapy at a target international normalized ratio of 3.0 for cutaneous polyarteritis nodosa. J Am Acad Dermatol. 2010;63:602-606.
In 1931, Lindberg1 described a cutaneous variant of polyarteritis nodosa, which lacked visceral involvement and possessed a more favorable prognosis.2 Cutaneous polyarteritis nodosa (CPAN) is a localized small- to medium-vessel vasculitis restricted to the skin. Both benign and chronic courses have been described, and systemic involvement does not occur.3 Diagnostic criteria proposed by Nakamura et al3 in 2009 included cutaneous nodules, livedo reticularis, purpura, or ulcers; histopathologic fibrinoid necrotizing vasculitis of small- to medium-sized vessels; and exclusion of systemic symptoms (eg, fever, hypertension, weight loss, renal failure, cerebral hemorrhage, neuropathy, myocardial infarction, ischemic heart disease, pericarditis, pleuritis, arthralgia/myalgia). Nodules occur in 30% to 50% of cases and can remain for years if left untreated. Ulcerations occur in up to 30% of patients. Myositis, arthritis, and weakness also have been reported with this condition.4 Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibody, as well as elevated anti–phosphatidylserine-prothrombin complex antibody.5 These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. The distinction of this entity from systemic polyartertitis nodosa is key when determining treatment options and monitoring parameters.
Case Report
A 66-year-old woman was referred to our facility by an outside dermatologist with a mildly pruritic, blanchable, reticulated erythema on the chest and bilateral arms and legs of 3 months’ duration consistent with livedo reticularis (Figure 1). Prior systemic therapy included prednisone 10 mg 3 times daily, fexofenadine, loratadine, and hydroxyzine. When the systemic steroid was tapered, the patient developed an asymptomatic flare of her eruption. On presentation, the lesions had waxed and waned, and the patient was taking only vitamin B12 and vitamin C. Her medical history was notable for an unknown-type lymphoma of the chest wall diagnosed at 46 years of age that was treated with an unknown chemotherapeutic agent, chronic pancreatitis that resulted in a duodenectomy at 61 years of age, chronic cholecystitis, and 1 first-trimester miscarriage. Outside laboratory tests, including a comprehensive metabolic panel, complete blood cell count, urinalysis, renal function, and liver function tests were within reference range, except for the finding of mild leukocytosis (11,000/µL)(reference range, 3800–10,800/µL), which resolved after steroids were discontinued, with otherwise normal results. Punch biopsy of a specimen from the right thigh revealed medium-vessel vasculitis consistent with polyarteritis nodosa (Figure 2). Laboratory workup by our facility including hepatitis panel, perinuclear antineutrophil cytoplasmic antibody, cytoplasmic antineutrophil cytoplasmic antibody, factor V Leiden, prothrombin time/international normalized ratio, anticardiolipin antibody, and proteins C and S were all within reference range. Abnormal values included a low positive but nondiagnostic antinuclear antibody screen with negative titers, and the lupus anticoagulant titer was mildly elevated at 44 IgG binding units (reference range, <40 IgG binding units). Serum protein electrophoresis (SPEP) and urine protein electrophoresis also were performed, and SPEP was low positive for elevated κ and γ light chains. The patient was referred to oncology, and further testing revealed no underlying malignancy. The patient was monitored and no treatment was initiated; her rash completely resolved within 3 months. Laboratory monitoring at 6 months including SPEP, urine protein electrophoresis, lupus anticoagulant, and clotting studies all were within reference range.
Comment
Although the treatment of systemic polyarteritis nodosa often is necessary and typically involves high-dose corticosteroids and cyclophosphamide, the treatment of CPAN initially is less aggressive. Of the options available for treatment of CPAN, each has associated risks and side effects. Chen6 classified CPAN into 3 groups: 1 (mild), 2 (severe with no systemic involvement), and 3 (severe with progression to systemic disease)(Table). The authors performed a review of all the published treatments and their respective side effects to evaluate if treatment should be instituted for asymptomatic (group 1) disease presenting with abnormal antibody findings as demonstrated in our case.
First-line treatment of CPAN includes nonsteroidal anti-inflammatory drugs (NSAIDS) and colchicine.7 Nonsteroidal anti-inflammatory drugs are preferred; however, they also have been associated with gastrointestinal tract upset and increased risk for peptic ulcer disease with long-term use. Although colchicine often is used in conjunction with NSAIDS8 for its anti-inflammatory activity, no studies have been performed on this drug as monotherapy, and the side effect of diarrhea often limits its use.
Other therapies include dapsone, which should be monitored carefully due to the risk for dapsone hypersensitivity syndrome.8,9 Topical corticosteroids have been proven effective for mild cases of confluent erythema with remission occurring as early as 4 weeks.4 Some reports emphasize the role of streptococcal infections in CPAN, especially in children.8,10-12 Consequently it is recommended that anti–streptolysin O titers should be included in the workup for CPAN. Long-term penicillin prophylaxis and tonsillectomy have been used to prevent disease flares with limited success.8,10-12
For more severe disease, especially with neuromuscular involvement, oral methylprednisolone up to 1 mg/kg daily has been used and has proven effective in the control of acute exacerbations.7,13 However, the many adverse effects of systemic steroids limit their use long-term, and taper will often result in flare of disease.4,7 Medications used in conjunction with steroids include hydroxychloroquine, dapsone, azathioprine, cyclophosphamide, methotrexate, sulfapyridine, pentoxifylline, infliximab, etanercept, and intravenous immunoglobulin.4,9,12-17
Low-dose methotrexate has shown some improvement in skin disease with CPAN, but other case reports suggest that complete remission is not achieved with this drug.15,18 More studies are needed to assess the use of methotrexate for CPAN.
Immunomodulators have been used in multiple case reports with varying levels of success. Rogalski and Sticherling4 reported 3 cases that cleared with methylprednisolone plus azathioprine ranging from 4 weeks to 6 months; nausea limited tolerance of azathioprine in 1 case. Mycophenolate mofetil also was successfully used in 2 cases with clearance at 17 weeks and 6 months. In this series of cases, cyclosporine was ineffective for CPAN.4 Two case reports documented cutaneous clearance with cyclophosphamide in conjunction with prednisolone.9,10 No prospective trials have been performed on these medications, and immunosuppressants should only be considered in steroid-resistant cases.
The use of intravenous immunoglobulin has been reported effective in prior cases that showed resistance to more conventional trials of steroids, azathioprine, and/or cyclophosphamide.12,14 Intravenous immunoglobulin may be regarded as a treatment option for severe resistant disease. Several case reports also have documented success using tumor necrosis factor α blockers, particularly infliximab, as an adjunct to steroids and etanercept as both a steroid adjunct and monotherapy.16,17,19 More studies are necessary to evaluate these treatments.
Additionally, single case reports have outlined the use of other therapeutic agents, including tamoxifen (10 mg twice daily increased to 20 mg twice daily during episodes of breakthrough lesions),20 hyperbaric oxygen therapy (100% oxygen for 90 minutes 5 times weekly at 1.5 atm absolute followed by 2 weeks of 2 atm absolute),21 and granulocyte-macrophage colony-stimulating factor (300 µg injection in small portion to ulcer edges twice monthly for 2 months).22 All of these treatments show promise, but data are limited.
Because thrombosis is postulated to be a potential mechanism leading to CPAN, agents such as pentoxifylline, clopidogrel, and warfarin have been examined as treatment options. Pentoxifylline in combination with mycophenolate mofetil has been successful in treating a case that was resistant to other immunosuppressants.23 Clopidogrel blocks the adenosine diphosphate pathway and impairs clot retraction. Clopidogrel was reported effective in an acute flare of CPAN for clearance of skin lesions and normalization of lupus anticoagulant.24 It also was used successfully in recurrent CPAN after steroid treatments in a patient with neuromuscular symptoms. There was no recurrence in either of the patients in this case report series. Warfarin therapy at an international normalized ratio of 3.0 also has demonstrated success in halting disease progression and in facilitating the resolution of skin changes and normalization of anti–phosphatidylserine-prothrombin complex antibodies.24 Our review of the literature did not reveal evidence of a standardized length of treatment following symptom resolution or if treatment is indicated in asymptomatic disease, or as in our case, with only mild elevations of antiphospholipid antibodies.
Conclusion
Multiple treatment options exist for CPAN, but the data on their efficacies is limited and based only on anecdotal evidence, not prospective analysis. We believe that it seems reasonable to initiate treatment only for symptomatic disease or cases in which the antibody titers suggest that the patient may be at high risk for thrombosis. Mild symptoms and mild cutaneous changes would suggest the likely choice of NSAIDs, colchicine, or dapsone as treatment options versus no treatment. In patients with antibody titers, pentoxifylline, clopidogrel, or warfarin may be considered first-line therapies. With severe ulcerative lesions and neuromuscular involvement, steroids, immunosuppressants, and other investigative agents should be contemplated. In our patient, the laboratory studies were repeated and normalized on complete resolution of her livedo eruption. She remained asymptomatic and clear for 8 months without any treatment. The incidence of this presentation of CPAN is unknown and is likely underreported, as we would not expect most patients to present to their physicians for the evaluation of otherwise asymptomatic livedo reticularis. In essence, our case report suggests that it may be prudent to simply monitor patients with asymptomatic CPAN.
In 1931, Lindberg1 described a cutaneous variant of polyarteritis nodosa, which lacked visceral involvement and possessed a more favorable prognosis.2 Cutaneous polyarteritis nodosa (CPAN) is a localized small- to medium-vessel vasculitis restricted to the skin. Both benign and chronic courses have been described, and systemic involvement does not occur.3 Diagnostic criteria proposed by Nakamura et al3 in 2009 included cutaneous nodules, livedo reticularis, purpura, or ulcers; histopathologic fibrinoid necrotizing vasculitis of small- to medium-sized vessels; and exclusion of systemic symptoms (eg, fever, hypertension, weight loss, renal failure, cerebral hemorrhage, neuropathy, myocardial infarction, ischemic heart disease, pericarditis, pleuritis, arthralgia/myalgia). Nodules occur in 30% to 50% of cases and can remain for years if left untreated. Ulcerations occur in up to 30% of patients. Myositis, arthritis, and weakness also have been reported with this condition.4 Cutaneous polyarteritis nodosa has been associated with abnormal antibody testing with elevations of antiphospholipid cofactor antibody, lupus anticoagulant, anticardiolipin antibody, and anti-β2-glycoprotein I–dependent cardiolipin antibody, as well as elevated anti–phosphatidylserine-prothrombin complex antibody.5 These antibodies suggest increased risk for thrombosis and systemic diseases such as lupus or other autoimmune connective tissue disease. The distinction of this entity from systemic polyartertitis nodosa is key when determining treatment options and monitoring parameters.
Case Report
A 66-year-old woman was referred to our facility by an outside dermatologist with a mildly pruritic, blanchable, reticulated erythema on the chest and bilateral arms and legs of 3 months’ duration consistent with livedo reticularis (Figure 1). Prior systemic therapy included prednisone 10 mg 3 times daily, fexofenadine, loratadine, and hydroxyzine. When the systemic steroid was tapered, the patient developed an asymptomatic flare of her eruption. On presentation, the lesions had waxed and waned, and the patient was taking only vitamin B12 and vitamin C. Her medical history was notable for an unknown-type lymphoma of the chest wall diagnosed at 46 years of age that was treated with an unknown chemotherapeutic agent, chronic pancreatitis that resulted in a duodenectomy at 61 years of age, chronic cholecystitis, and 1 first-trimester miscarriage. Outside laboratory tests, including a comprehensive metabolic panel, complete blood cell count, urinalysis, renal function, and liver function tests were within reference range, except for the finding of mild leukocytosis (11,000/µL)(reference range, 3800–10,800/µL), which resolved after steroids were discontinued, with otherwise normal results. Punch biopsy of a specimen from the right thigh revealed medium-vessel vasculitis consistent with polyarteritis nodosa (Figure 2). Laboratory workup by our facility including hepatitis panel, perinuclear antineutrophil cytoplasmic antibody, cytoplasmic antineutrophil cytoplasmic antibody, factor V Leiden, prothrombin time/international normalized ratio, anticardiolipin antibody, and proteins C and S were all within reference range. Abnormal values included a low positive but nondiagnostic antinuclear antibody screen with negative titers, and the lupus anticoagulant titer was mildly elevated at 44 IgG binding units (reference range, <40 IgG binding units). Serum protein electrophoresis (SPEP) and urine protein electrophoresis also were performed, and SPEP was low positive for elevated κ and γ light chains. The patient was referred to oncology, and further testing revealed no underlying malignancy. The patient was monitored and no treatment was initiated; her rash completely resolved within 3 months. Laboratory monitoring at 6 months including SPEP, urine protein electrophoresis, lupus anticoagulant, and clotting studies all were within reference range.
Comment
Although the treatment of systemic polyarteritis nodosa often is necessary and typically involves high-dose corticosteroids and cyclophosphamide, the treatment of CPAN initially is less aggressive. Of the options available for treatment of CPAN, each has associated risks and side effects. Chen6 classified CPAN into 3 groups: 1 (mild), 2 (severe with no systemic involvement), and 3 (severe with progression to systemic disease)(Table). The authors performed a review of all the published treatments and their respective side effects to evaluate if treatment should be instituted for asymptomatic (group 1) disease presenting with abnormal antibody findings as demonstrated in our case.
First-line treatment of CPAN includes nonsteroidal anti-inflammatory drugs (NSAIDS) and colchicine.7 Nonsteroidal anti-inflammatory drugs are preferred; however, they also have been associated with gastrointestinal tract upset and increased risk for peptic ulcer disease with long-term use. Although colchicine often is used in conjunction with NSAIDS8 for its anti-inflammatory activity, no studies have been performed on this drug as monotherapy, and the side effect of diarrhea often limits its use.
Other therapies include dapsone, which should be monitored carefully due to the risk for dapsone hypersensitivity syndrome.8,9 Topical corticosteroids have been proven effective for mild cases of confluent erythema with remission occurring as early as 4 weeks.4 Some reports emphasize the role of streptococcal infections in CPAN, especially in children.8,10-12 Consequently it is recommended that anti–streptolysin O titers should be included in the workup for CPAN. Long-term penicillin prophylaxis and tonsillectomy have been used to prevent disease flares with limited success.8,10-12
For more severe disease, especially with neuromuscular involvement, oral methylprednisolone up to 1 mg/kg daily has been used and has proven effective in the control of acute exacerbations.7,13 However, the many adverse effects of systemic steroids limit their use long-term, and taper will often result in flare of disease.4,7 Medications used in conjunction with steroids include hydroxychloroquine, dapsone, azathioprine, cyclophosphamide, methotrexate, sulfapyridine, pentoxifylline, infliximab, etanercept, and intravenous immunoglobulin.4,9,12-17
Low-dose methotrexate has shown some improvement in skin disease with CPAN, but other case reports suggest that complete remission is not achieved with this drug.15,18 More studies are needed to assess the use of methotrexate for CPAN.
Immunomodulators have been used in multiple case reports with varying levels of success. Rogalski and Sticherling4 reported 3 cases that cleared with methylprednisolone plus azathioprine ranging from 4 weeks to 6 months; nausea limited tolerance of azathioprine in 1 case. Mycophenolate mofetil also was successfully used in 2 cases with clearance at 17 weeks and 6 months. In this series of cases, cyclosporine was ineffective for CPAN.4 Two case reports documented cutaneous clearance with cyclophosphamide in conjunction with prednisolone.9,10 No prospective trials have been performed on these medications, and immunosuppressants should only be considered in steroid-resistant cases.
The use of intravenous immunoglobulin has been reported effective in prior cases that showed resistance to more conventional trials of steroids, azathioprine, and/or cyclophosphamide.12,14 Intravenous immunoglobulin may be regarded as a treatment option for severe resistant disease. Several case reports also have documented success using tumor necrosis factor α blockers, particularly infliximab, as an adjunct to steroids and etanercept as both a steroid adjunct and monotherapy.16,17,19 More studies are necessary to evaluate these treatments.
Additionally, single case reports have outlined the use of other therapeutic agents, including tamoxifen (10 mg twice daily increased to 20 mg twice daily during episodes of breakthrough lesions),20 hyperbaric oxygen therapy (100% oxygen for 90 minutes 5 times weekly at 1.5 atm absolute followed by 2 weeks of 2 atm absolute),21 and granulocyte-macrophage colony-stimulating factor (300 µg injection in small portion to ulcer edges twice monthly for 2 months).22 All of these treatments show promise, but data are limited.
Because thrombosis is postulated to be a potential mechanism leading to CPAN, agents such as pentoxifylline, clopidogrel, and warfarin have been examined as treatment options. Pentoxifylline in combination with mycophenolate mofetil has been successful in treating a case that was resistant to other immunosuppressants.23 Clopidogrel blocks the adenosine diphosphate pathway and impairs clot retraction. Clopidogrel was reported effective in an acute flare of CPAN for clearance of skin lesions and normalization of lupus anticoagulant.24 It also was used successfully in recurrent CPAN after steroid treatments in a patient with neuromuscular symptoms. There was no recurrence in either of the patients in this case report series. Warfarin therapy at an international normalized ratio of 3.0 also has demonstrated success in halting disease progression and in facilitating the resolution of skin changes and normalization of anti–phosphatidylserine-prothrombin complex antibodies.24 Our review of the literature did not reveal evidence of a standardized length of treatment following symptom resolution or if treatment is indicated in asymptomatic disease, or as in our case, with only mild elevations of antiphospholipid antibodies.
Conclusion
Multiple treatment options exist for CPAN, but the data on their efficacies is limited and based only on anecdotal evidence, not prospective analysis. We believe that it seems reasonable to initiate treatment only for symptomatic disease or cases in which the antibody titers suggest that the patient may be at high risk for thrombosis. Mild symptoms and mild cutaneous changes would suggest the likely choice of NSAIDs, colchicine, or dapsone as treatment options versus no treatment. In patients with antibody titers, pentoxifylline, clopidogrel, or warfarin may be considered first-line therapies. With severe ulcerative lesions and neuromuscular involvement, steroids, immunosuppressants, and other investigative agents should be contemplated. In our patient, the laboratory studies were repeated and normalized on complete resolution of her livedo eruption. She remained asymptomatic and clear for 8 months without any treatment. The incidence of this presentation of CPAN is unknown and is likely underreported, as we would not expect most patients to present to their physicians for the evaluation of otherwise asymptomatic livedo reticularis. In essence, our case report suggests that it may be prudent to simply monitor patients with asymptomatic CPAN.
- Lindberg K. Ein Beitrag zur Kenntnis der Periarteritis nodosa. Acta Med Scand. 1931;76:183-225.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa [published online August 26, 2005]. J Neurol. 2005;252:1155-1166.
- Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res. 2009;301:117-121.
- Rogalski C, Sticherling M. Panateritis cutanea benigna—an entity limited to the skin or cutaneous presentation of a systemic necrotizing vasculitis? report of seven cases and review of the literature. Int J Dermatol. 2007;46:817-821.
- Kawakami T, Yamazaki M, Mizoguchi M, et al. High titer of anti-phosphatidylserine-prothrombin complex antibodies in patients with cutaneous polyarteritis nodosa. Arthritis Rheum. 2007;57:1507-1513.
- Chen KR. Cutaneous polyarteritis nodosa: a clinical and histopathological study of 20 cases. J Dermatol. 1989;6:429-442.
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756.
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathologic analysis and review of the published work. J Dermatol. 2010;37:85-93.
- Flanagan N, Casey EB, Watson R, et al. Cutaneous polyartertitis nodosa with seronegative arthritis. Rheumatology (Oxford). 1999;38:1161-1162.
- Fathalla B, Miller L, Brady S, et al. Cutaneous polyarteritis nodosa in children. J Am Acad Dermatol. 2005;53:724-728.
- Misago N, Mochizuki Y, Sekiyama-Kodera H, et al. Cutaneous polyarteritis nodosa: therapy and clinical course in four cases. J Dermatol. 2001;28:719-727.
- Breda L, Franchini S, Marzetti V, et al. Intravenous immunoglobulins for cutaneous polyarteritis nodosa resistant to conventional treatment. Scand J Rheumatol. 2016;45:169-170.
- Maillard H, Szczesniak S, Martin L. Cutaneous periarteritis nodosa: diagnostic and therapeutic aspects of 9 cases. Ann Dermatol Venereol. 1999;26:125-129.
- Lobo I, Ferreira M, Silva E. Cutaneous polyarteritis nodosa treated with intravenous immunoglobulin. J Eur Acad Dermatol Venereol. 2007;22:880-882.
- Boehm I, Bauer R. Low-dose methotrexate controls a severe form of polyarteritis nodosa. Arch Dermatol. 2000;136:167-169.
- Campanilho-Marques R, Ramos F, Canhão H, et al. Remission induced by infliximab in a childhood polyarteritis nodosa refractory to conventional immunosuppression and rituximab. Joint Bone Spine. 2014;81:277-278.
- Inoue N, Shimizu M, Mizuta M, et al. Refractory cutaneous polyarteritis nodosa: successful treatment with etanercept. Pediatr Int. 2017;59:751-752.
- Schartz NE. Successful treatment in two cases of steroid dependent cutaneous polyarteritis nodosa with low-dose methotrexate. Dermatology. 2001;203:336-338.
- Valor L, Monteagudo I, de la Torre I, et al. Young male patient diagnosed with cutaneous polyarteritis nodosa successfully treated with etanercept. Mod Rheumatol. 2014;24:688-689.
- Cvancara JL, Meffert JJ, Elston DM. Estrogen sensitive cutaneous polyarteritis nodosa: response to tamoxifen. J Am Acad Dermatol. 1998;39:643-646.
- Mazokopakis E, Milkas A, Tsartsalis A, et al. Improvement of cutaneous polyarteritis nodosa with hyperbaric oxygen. Int J Dermatol. 2009;48:1017-1029.
- Tursen U, Api H, Kaya TI, et al. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony stimulating factor in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol. 2006;20:1341-1343.
- Kluger N, Guillot B, Bessis D. Ulcerative cutaneous polyarteritis nodosa treated with mycophenolate mofetil and pentoxifylline. J Dermatolog Treat. 2011;22:175-177.
- Kawakami T, Soma Y. Use of warfarin therapy at a target international normalized ratio of 3.0 for cutaneous polyarteritis nodosa. J Am Acad Dermatol. 2010;63:602-606.
- Lindberg K. Ein Beitrag zur Kenntnis der Periarteritis nodosa. Acta Med Scand. 1931;76:183-225.
- Kraemer M, Linden D, Berlit P. The spectrum of differential diagnosis in neurological patients with livedo reticularis and livedo racemosa [published online August 26, 2005]. J Neurol. 2005;252:1155-1166.
- Nakamura T, Kanazawa N, Ikeda T, et al. Cutaneous polyarteritis nodosa: revisiting its definition and diagnostic criteria. Arch Dermatol Res. 2009;301:117-121.
- Rogalski C, Sticherling M. Panateritis cutanea benigna—an entity limited to the skin or cutaneous presentation of a systemic necrotizing vasculitis? report of seven cases and review of the literature. Int J Dermatol. 2007;46:817-821.
- Kawakami T, Yamazaki M, Mizoguchi M, et al. High titer of anti-phosphatidylserine-prothrombin complex antibodies in patients with cutaneous polyarteritis nodosa. Arthritis Rheum. 2007;57:1507-1513.
- Chen KR. Cutaneous polyarteritis nodosa: a clinical and histopathological study of 20 cases. J Dermatol. 1989;6:429-442.
- Morgan AJ, Schwartz RA. Cutaneous polyarteritis nodosa: a comprehensive review. Int J Dermatol. 2010;49:750-756.
- Ishiguro N, Kawashima M. Cutaneous polyarteritis nodosa: a report of 16 cases with clinical and histopathologic analysis and review of the published work. J Dermatol. 2010;37:85-93.
- Flanagan N, Casey EB, Watson R, et al. Cutaneous polyartertitis nodosa with seronegative arthritis. Rheumatology (Oxford). 1999;38:1161-1162.
- Fathalla B, Miller L, Brady S, et al. Cutaneous polyarteritis nodosa in children. J Am Acad Dermatol. 2005;53:724-728.
- Misago N, Mochizuki Y, Sekiyama-Kodera H, et al. Cutaneous polyarteritis nodosa: therapy and clinical course in four cases. J Dermatol. 2001;28:719-727.
- Breda L, Franchini S, Marzetti V, et al. Intravenous immunoglobulins for cutaneous polyarteritis nodosa resistant to conventional treatment. Scand J Rheumatol. 2016;45:169-170.
- Maillard H, Szczesniak S, Martin L. Cutaneous periarteritis nodosa: diagnostic and therapeutic aspects of 9 cases. Ann Dermatol Venereol. 1999;26:125-129.
- Lobo I, Ferreira M, Silva E. Cutaneous polyarteritis nodosa treated with intravenous immunoglobulin. J Eur Acad Dermatol Venereol. 2007;22:880-882.
- Boehm I, Bauer R. Low-dose methotrexate controls a severe form of polyarteritis nodosa. Arch Dermatol. 2000;136:167-169.
- Campanilho-Marques R, Ramos F, Canhão H, et al. Remission induced by infliximab in a childhood polyarteritis nodosa refractory to conventional immunosuppression and rituximab. Joint Bone Spine. 2014;81:277-278.
- Inoue N, Shimizu M, Mizuta M, et al. Refractory cutaneous polyarteritis nodosa: successful treatment with etanercept. Pediatr Int. 2017;59:751-752.
- Schartz NE. Successful treatment in two cases of steroid dependent cutaneous polyarteritis nodosa with low-dose methotrexate. Dermatology. 2001;203:336-338.
- Valor L, Monteagudo I, de la Torre I, et al. Young male patient diagnosed with cutaneous polyarteritis nodosa successfully treated with etanercept. Mod Rheumatol. 2014;24:688-689.
- Cvancara JL, Meffert JJ, Elston DM. Estrogen sensitive cutaneous polyarteritis nodosa: response to tamoxifen. J Am Acad Dermatol. 1998;39:643-646.
- Mazokopakis E, Milkas A, Tsartsalis A, et al. Improvement of cutaneous polyarteritis nodosa with hyperbaric oxygen. Int J Dermatol. 2009;48:1017-1029.
- Tursen U, Api H, Kaya TI, et al. Rapid healing of chronic leg ulcers during perilesional injections of granulocyte-macrophage colony stimulating factor in a patient with cutaneous polyarteritis nodosa. J Eur Acad Dermatol Venereol. 2006;20:1341-1343.
- Kluger N, Guillot B, Bessis D. Ulcerative cutaneous polyarteritis nodosa treated with mycophenolate mofetil and pentoxifylline. J Dermatolog Treat. 2011;22:175-177.
- Kawakami T, Soma Y. Use of warfarin therapy at a target international normalized ratio of 3.0 for cutaneous polyarteritis nodosa. J Am Acad Dermatol. 2010;63:602-606.
Practice Points
- Cutaneous polyarteritis nodosa should be in the differential of new-onset livedo reticularis.
- Workup with biopsy and specific blood work is important.
- Treatment options at this time are limited.
Necrotic Ulcer on the Thigh
The Diagnosis: Disseminated Cryptococcosis
Histopathologic examination of a 3-mm punch biopsy showed a diffuse dermal neutrophilic infiltrate with necrosis and subcutaneous tissue with round yeast surrounded by a prominent halo staining bright red with mucicarmine, representing a thick mucinous capsule (Figure). Grocott-Gomori methenamine-silver and periodic acid-Schiff stains also demonstrated fungal spores morphologically. Cerebrospinal fluid culture grew Cryptococcus neoformans, and cryptococcal antigen titers were positive in both serum and cerebrospinal fluid samples (>1:4096). The patient had autolytic debridement of the ulcer after completing a 4-week induction course of intravenous liposomal amphotericin B with oral flucytosine. He was transitioned to oral fluconazole for the consolidation phase of treatment.
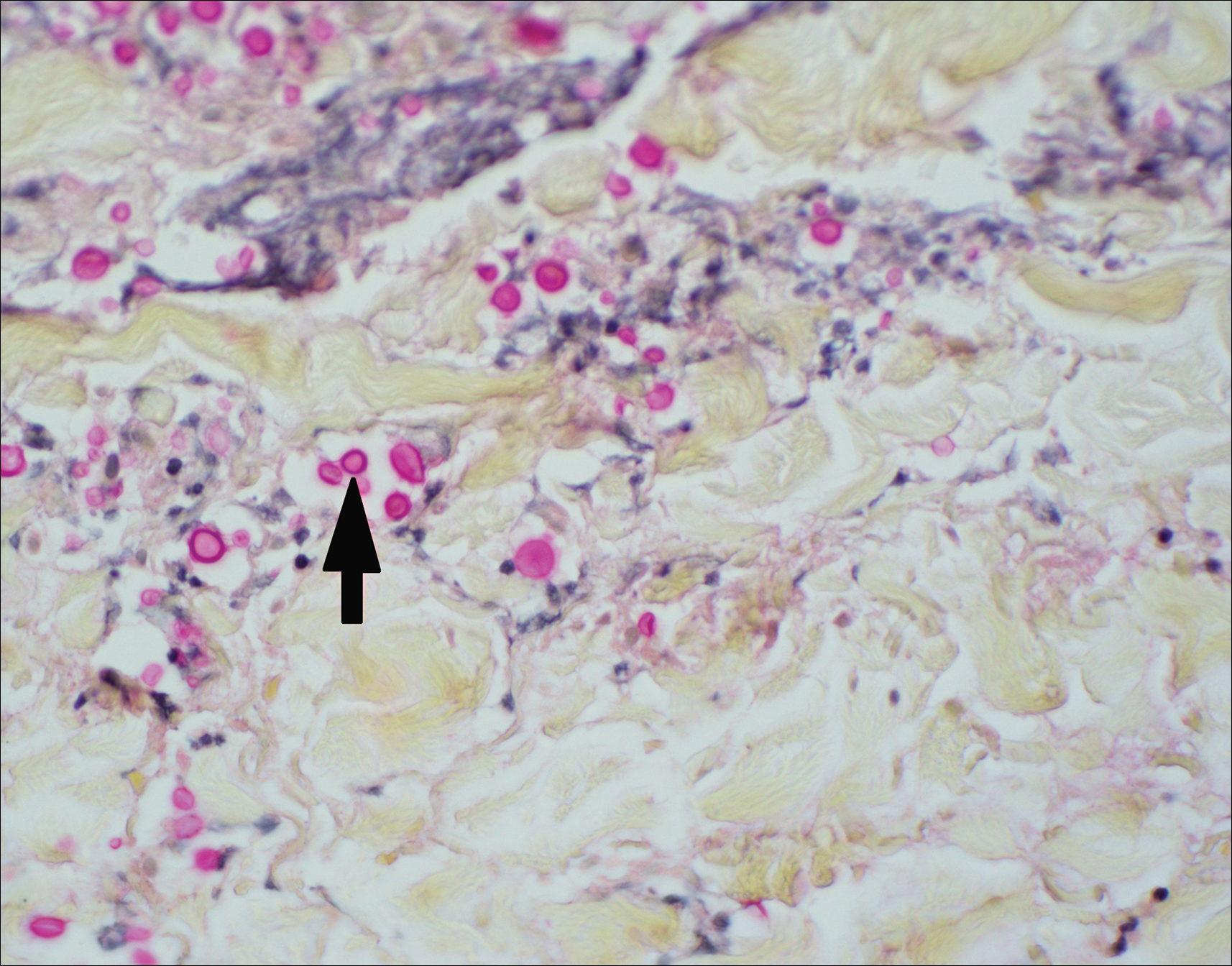
Cryptococcus is an opportunistic basidiomycetous yeast with worldwide distribution and 2 primary pathogenic species in humans: C neoformans and Cryptococcus gattii. It is associated with bird feces, composted food, and decayed wood.1,2 A predilection toward an immunosuppressed host is recognized in 70% to 90% of the infections caused by C neoformans; however, C gattii commonly affects individuals with apparently intact immune systems.1,3 Risk factors for infection include advanced human immunodeficiency virus infection, solid organ transplantation, chronic liver disease, autoimmune disease, hematological malignancy, and underlying genetic susceptibility.1,2
Initial exposure is through the respiratory tract with formation of latent reservoirs in the pulmonary lymph nodes with subsequent reactivation that can result in hematogenous dissemination.1,2 Cutaneous involvement was described in 108 patients (5%) in a large review of 1974 cases in France.4 Among those with cutaneous involvement, disseminated disease was diagnosed in 80 cases (74%), and 28 cases (26%) were considered primary cutaneous cryptococcosis. Primary cutaneous cryptococcosis typically presents as a single lesion, predominantly on the hand, with whitlow and more rarely with extensive cellulitis or necrotizing fasciitis.4 In disseminated cutaneous disease, there is no pathognomonic single lesion; however, it is commonly associated with multiple cutaneous lesions predominantly involving the head and neck. Plaques, abscesses, nodules, and pustular or umbilicated papules have been reported.1,5 There are few case reports that describe a single isolated necrotic ulcer with disseminated disease similar to our presented case, and more typically the necrotic ulcer is seen in transplanted patients.6 The differential diagnosis of a necrotic thigh ulcer includes pseudomonal ecthyma gangrenosum, cutaneous anthrax and aspergillosis, fusariosis, and a bite from the brown recluse spider.7 Our patient had an increased susceptibility to infection from his ongoing chemotherapy, a risk previously described in oncology patients with cell-mediated immunosuppression.8
Management for disseminated cryptococcosis is a 3-phase therapy including induction with intravenous amphotericin B and oral flucytosine for a minimum of 2 weeks, with consolidation and maintenance phases both with oral fluconazole for a length depending on underlying immunosuppression.9
- Chen SC, Meyer W, Sorrell TC. Cryptococcus gattii infections. Clin Microbiol Rev. 2014;27:980-1024.
- Williamson PR, Jarvis JN, Panackal AA, et al. Cryptococcal meningitis: epidemiology, immunology, diagnosis, and therapy [published online November 25, 2016]. Nat Rev Neurol. 2017;13:13-24.
- Speed B, Dunt D. Clinical and host differences between infections with the two varieties of Cryptococcus neoformans. Clin Infect Dis. 1995;21:28-34.
- Neuville S, Dromer F, Morin O, et al; French Cryptococcosis Study Group. Primary cutaneous cryptococcosis: a distinct clinical entity [published online January 17, 2003]. Clin Infect Dis. 2003;36:337-347.
- Murakawa GJ, Kerschmann R, Berger T. Cutaneous cryptococcus infection and AIDS: report of 12 cases and review of the literature. JAMA Dermatol. 1996;132:545-548.
- Sun HY, Alexander BD, Lortholary O, et al. Cutaneous cryptococcosis in solid organ transplant recipients. Med Mycol. 2010;48:785-791.
- Grossman ME, Fox LP, Kovarik C, et al. Cutaneous Manifestations of Infection in the Immunocompromised Host. Baltimore, MD: Williams & Wilkins; 2012.
- Korfel A, Menssen HD, Schwartz S, et al. Cryptococcosis in Hodgkin's disease: description of two cases and review of the literature. Ann Hematol. 1998;76:283-286.
- Perfect JR, Dismukes WE, Dromer F. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2010;50:291-322.
The Diagnosis: Disseminated Cryptococcosis
Histopathologic examination of a 3-mm punch biopsy showed a diffuse dermal neutrophilic infiltrate with necrosis and subcutaneous tissue with round yeast surrounded by a prominent halo staining bright red with mucicarmine, representing a thick mucinous capsule (Figure). Grocott-Gomori methenamine-silver and periodic acid-Schiff stains also demonstrated fungal spores morphologically. Cerebrospinal fluid culture grew Cryptococcus neoformans, and cryptococcal antigen titers were positive in both serum and cerebrospinal fluid samples (>1:4096). The patient had autolytic debridement of the ulcer after completing a 4-week induction course of intravenous liposomal amphotericin B with oral flucytosine. He was transitioned to oral fluconazole for the consolidation phase of treatment.
Cryptococcus is an opportunistic basidiomycetous yeast with worldwide distribution and 2 primary pathogenic species in humans: C neoformans and Cryptococcus gattii. It is associated with bird feces, composted food, and decayed wood.1,2 A predilection toward an immunosuppressed host is recognized in 70% to 90% of the infections caused by C neoformans; however, C gattii commonly affects individuals with apparently intact immune systems.1,3 Risk factors for infection include advanced human immunodeficiency virus infection, solid organ transplantation, chronic liver disease, autoimmune disease, hematological malignancy, and underlying genetic susceptibility.1,2
Initial exposure is through the respiratory tract with formation of latent reservoirs in the pulmonary lymph nodes with subsequent reactivation that can result in hematogenous dissemination.1,2 Cutaneous involvement was described in 108 patients (5%) in a large review of 1974 cases in France.4 Among those with cutaneous involvement, disseminated disease was diagnosed in 80 cases (74%), and 28 cases (26%) were considered primary cutaneous cryptococcosis. Primary cutaneous cryptococcosis typically presents as a single lesion, predominantly on the hand, with whitlow and more rarely with extensive cellulitis or necrotizing fasciitis.4 In disseminated cutaneous disease, there is no pathognomonic single lesion; however, it is commonly associated with multiple cutaneous lesions predominantly involving the head and neck. Plaques, abscesses, nodules, and pustular or umbilicated papules have been reported.1,5 There are few case reports that describe a single isolated necrotic ulcer with disseminated disease similar to our presented case, and more typically the necrotic ulcer is seen in transplanted patients.6 The differential diagnosis of a necrotic thigh ulcer includes pseudomonal ecthyma gangrenosum, cutaneous anthrax and aspergillosis, fusariosis, and a bite from the brown recluse spider.7 Our patient had an increased susceptibility to infection from his ongoing chemotherapy, a risk previously described in oncology patients with cell-mediated immunosuppression.8
Management for disseminated cryptococcosis is a 3-phase therapy including induction with intravenous amphotericin B and oral flucytosine for a minimum of 2 weeks, with consolidation and maintenance phases both with oral fluconazole for a length depending on underlying immunosuppression.9
The Diagnosis: Disseminated Cryptococcosis
Histopathologic examination of a 3-mm punch biopsy showed a diffuse dermal neutrophilic infiltrate with necrosis and subcutaneous tissue with round yeast surrounded by a prominent halo staining bright red with mucicarmine, representing a thick mucinous capsule (Figure). Grocott-Gomori methenamine-silver and periodic acid-Schiff stains also demonstrated fungal spores morphologically. Cerebrospinal fluid culture grew Cryptococcus neoformans, and cryptococcal antigen titers were positive in both serum and cerebrospinal fluid samples (>1:4096). The patient had autolytic debridement of the ulcer after completing a 4-week induction course of intravenous liposomal amphotericin B with oral flucytosine. He was transitioned to oral fluconazole for the consolidation phase of treatment.
Cryptococcus is an opportunistic basidiomycetous yeast with worldwide distribution and 2 primary pathogenic species in humans: C neoformans and Cryptococcus gattii. It is associated with bird feces, composted food, and decayed wood.1,2 A predilection toward an immunosuppressed host is recognized in 70% to 90% of the infections caused by C neoformans; however, C gattii commonly affects individuals with apparently intact immune systems.1,3 Risk factors for infection include advanced human immunodeficiency virus infection, solid organ transplantation, chronic liver disease, autoimmune disease, hematological malignancy, and underlying genetic susceptibility.1,2
Initial exposure is through the respiratory tract with formation of latent reservoirs in the pulmonary lymph nodes with subsequent reactivation that can result in hematogenous dissemination.1,2 Cutaneous involvement was described in 108 patients (5%) in a large review of 1974 cases in France.4 Among those with cutaneous involvement, disseminated disease was diagnosed in 80 cases (74%), and 28 cases (26%) were considered primary cutaneous cryptococcosis. Primary cutaneous cryptococcosis typically presents as a single lesion, predominantly on the hand, with whitlow and more rarely with extensive cellulitis or necrotizing fasciitis.4 In disseminated cutaneous disease, there is no pathognomonic single lesion; however, it is commonly associated with multiple cutaneous lesions predominantly involving the head and neck. Plaques, abscesses, nodules, and pustular or umbilicated papules have been reported.1,5 There are few case reports that describe a single isolated necrotic ulcer with disseminated disease similar to our presented case, and more typically the necrotic ulcer is seen in transplanted patients.6 The differential diagnosis of a necrotic thigh ulcer includes pseudomonal ecthyma gangrenosum, cutaneous anthrax and aspergillosis, fusariosis, and a bite from the brown recluse spider.7 Our patient had an increased susceptibility to infection from his ongoing chemotherapy, a risk previously described in oncology patients with cell-mediated immunosuppression.8
Management for disseminated cryptococcosis is a 3-phase therapy including induction with intravenous amphotericin B and oral flucytosine for a minimum of 2 weeks, with consolidation and maintenance phases both with oral fluconazole for a length depending on underlying immunosuppression.9
- Chen SC, Meyer W, Sorrell TC. Cryptococcus gattii infections. Clin Microbiol Rev. 2014;27:980-1024.
- Williamson PR, Jarvis JN, Panackal AA, et al. Cryptococcal meningitis: epidemiology, immunology, diagnosis, and therapy [published online November 25, 2016]. Nat Rev Neurol. 2017;13:13-24.
- Speed B, Dunt D. Clinical and host differences between infections with the two varieties of Cryptococcus neoformans. Clin Infect Dis. 1995;21:28-34.
- Neuville S, Dromer F, Morin O, et al; French Cryptococcosis Study Group. Primary cutaneous cryptococcosis: a distinct clinical entity [published online January 17, 2003]. Clin Infect Dis. 2003;36:337-347.
- Murakawa GJ, Kerschmann R, Berger T. Cutaneous cryptococcus infection and AIDS: report of 12 cases and review of the literature. JAMA Dermatol. 1996;132:545-548.
- Sun HY, Alexander BD, Lortholary O, et al. Cutaneous cryptococcosis in solid organ transplant recipients. Med Mycol. 2010;48:785-791.
- Grossman ME, Fox LP, Kovarik C, et al. Cutaneous Manifestations of Infection in the Immunocompromised Host. Baltimore, MD: Williams & Wilkins; 2012.
- Korfel A, Menssen HD, Schwartz S, et al. Cryptococcosis in Hodgkin's disease: description of two cases and review of the literature. Ann Hematol. 1998;76:283-286.
- Perfect JR, Dismukes WE, Dromer F. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2010;50:291-322.
- Chen SC, Meyer W, Sorrell TC. Cryptococcus gattii infections. Clin Microbiol Rev. 2014;27:980-1024.
- Williamson PR, Jarvis JN, Panackal AA, et al. Cryptococcal meningitis: epidemiology, immunology, diagnosis, and therapy [published online November 25, 2016]. Nat Rev Neurol. 2017;13:13-24.
- Speed B, Dunt D. Clinical and host differences between infections with the two varieties of Cryptococcus neoformans. Clin Infect Dis. 1995;21:28-34.
- Neuville S, Dromer F, Morin O, et al; French Cryptococcosis Study Group. Primary cutaneous cryptococcosis: a distinct clinical entity [published online January 17, 2003]. Clin Infect Dis. 2003;36:337-347.
- Murakawa GJ, Kerschmann R, Berger T. Cutaneous cryptococcus infection and AIDS: report of 12 cases and review of the literature. JAMA Dermatol. 1996;132:545-548.
- Sun HY, Alexander BD, Lortholary O, et al. Cutaneous cryptococcosis in solid organ transplant recipients. Med Mycol. 2010;48:785-791.
- Grossman ME, Fox LP, Kovarik C, et al. Cutaneous Manifestations of Infection in the Immunocompromised Host. Baltimore, MD: Williams & Wilkins; 2012.
- Korfel A, Menssen HD, Schwartz S, et al. Cryptococcosis in Hodgkin's disease: description of two cases and review of the literature. Ann Hematol. 1998;76:283-286.
- Perfect JR, Dismukes WE, Dromer F. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis. 2010;50:291-322.
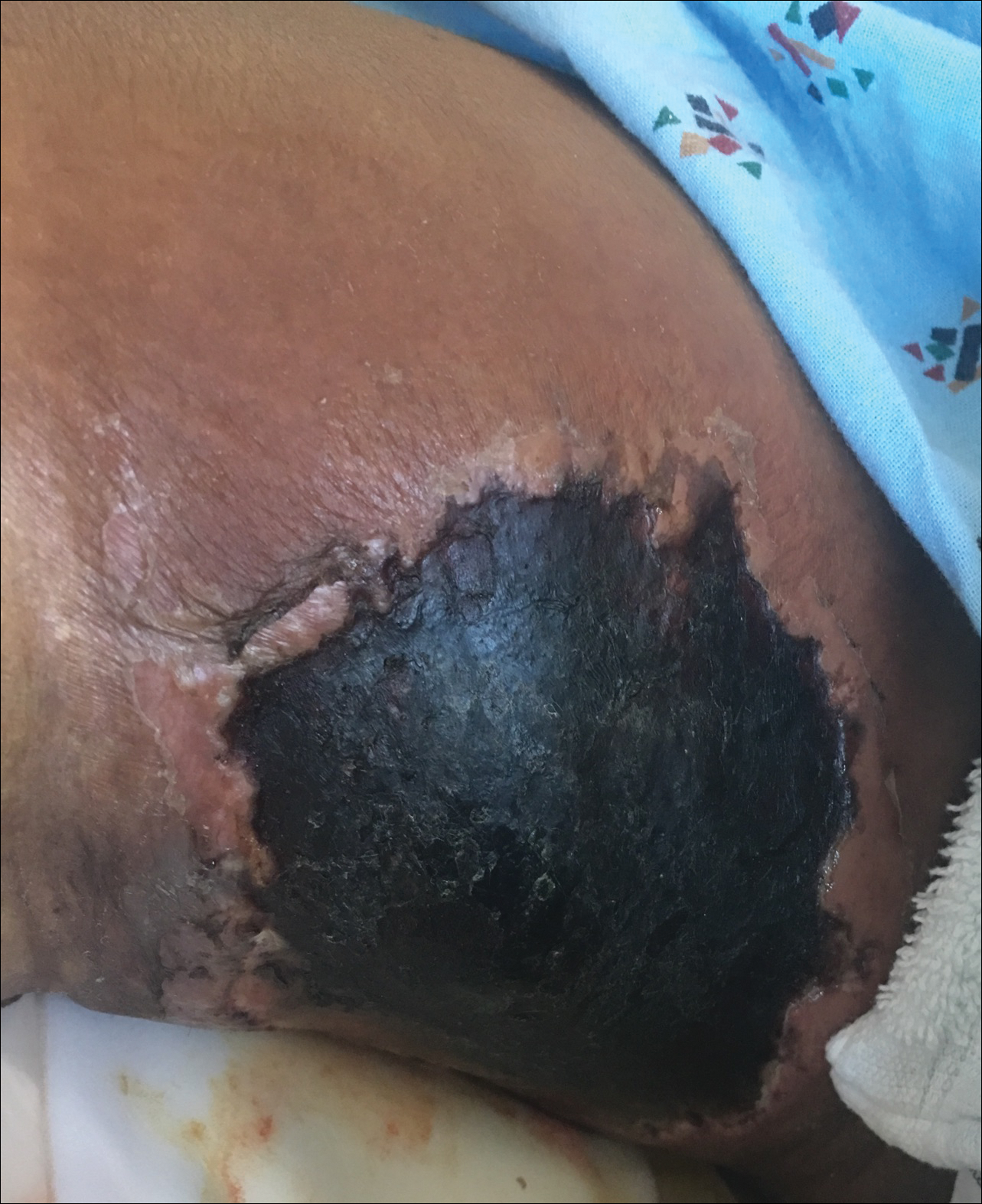
A 29-year-old man with a history of acute lymphoblastic leukemia was admitted for acute encephalopathy and a necrotic ulcer on the right thigh of 2 weeks' duration. He had received chemotherapy with pegaspargase and vincristine 6 weeks prior to admission. He reported headache with nausea and vomiting of 2 weeks' duration and had sustained a fall in the bathtub a week prior that initially resulted in a right thigh abrasion. He denied recent travel, unusual food consumption, animal exposure, exposure to sick persons, and alcohol or other drug use. On examination the patient was alert but was not oriented to person, place, or time. A 10.2 ×10-cm necrotic ulcer with surrounding mild erythema and tenderness was noted on the right inner thigh.
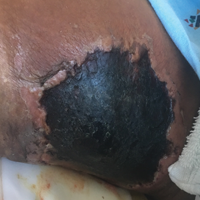
When the painful ‘bumps’ are calciphylaxis, what’s next?
EXPERT ANALYSIS FROM THE 2017 AAD SUMMER MEETING
NEW YORK – When patients come to the office with painful “bumps” on the legs or elsewhere, panniculitis should be in the differential. And for some patients, said Alina Bridges, DO, the panniculitis may come with the dire diagnosis of calciphylaxis.
Calciphylaxis is an underrecognized crystal deposition disease that’s associated with panniculitis, said Dr. Bridges, speaking at the American Academy of Dermatology summer meeting. When calcium accumulates in small subcutaneous vessels, an occlusive vasculopathy is created within the dermis.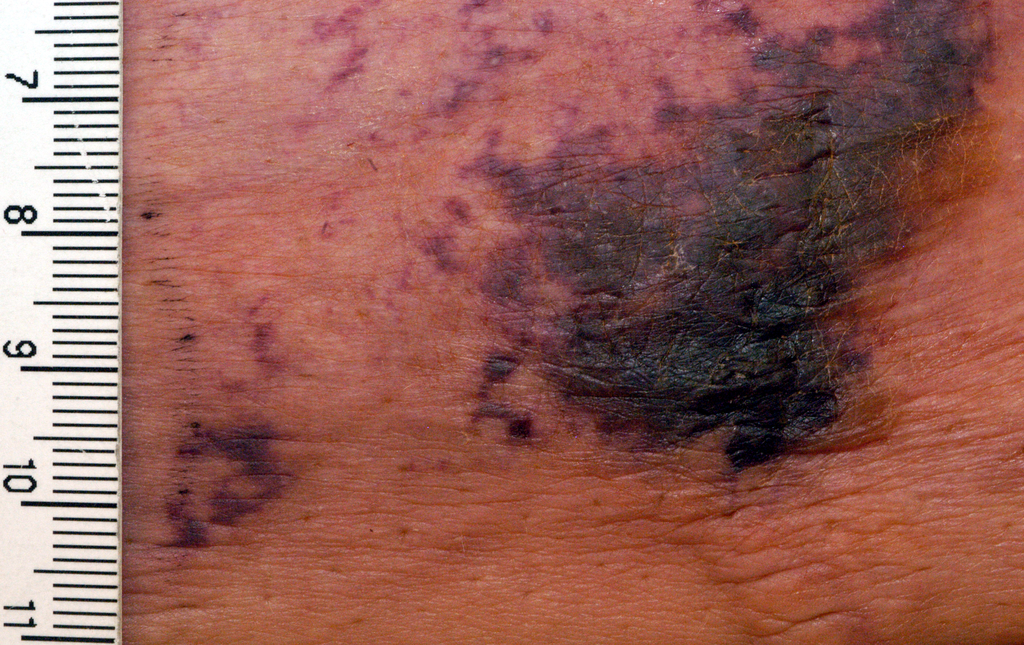
A soft-tissue radiograph of the affected area may also be helpful. Calciphylaxis shows as a fine netlike pattern of calcification, a finding that Dr. Bridges said has 90% specificity for the condition.
However, Dr. Bridges said, patients with panniculitis need a biopsy. “Careful selection of biopsy site and a deep specimen containing abundant fat obtained by incisional or excisional biopsy” is the best approach, allowing the pathologist to see the complete picture. In some cases, she said, a double-punch biopsy could also produce adequate specimens.
In addition to the calcium deposition, other pathologic findings may be lobular fat necrosis, with a pannicular vascular thrombosis. Though extravascular calcification can be seen in the panniculus, it’s not uncommon also to see intravascular calcification, said Dr. Bridges, who is the dermatopathology fellowship program director at the Mayo Clinic, Rochester, Minn.
Dr. Bridges said that the patients with calciphylaxis can present with predominant panniculitis or vasculitis, or a mixed picture; patients can also have bullae, ulcers, or livedo reticularis.
The lesions are extremely painful and become increasingly violaceous, with firm subcutaneous nodules. They are variably necrotic, and become more ulcerated over time.
Calciphylaxis is multifactorial and progressive. The prognosis is very poor for individuals with the condition, Dr. Bridges said. The median survival is 10 months, with 1-year survival rates of 46%, and just 20% of individuals with calciphylaxis surviving 2 years after diagnosis.
Gangrene is a frequent complication, and multisystem organ failure often occurs as well, she said.
Calciphylaxis most commonly occurs in individuals with chronic kidney disease and is seen in 4% of hemodialysis patients. However, it may also occur in individuals without uremia. In associations that are incompletely understood, calciphylaxis has been associated with warfarin therapy, connective tissue disorders, Crohn’s disease, liver disease, diabetes, hematologic malignancies, factor V Leiden deficiency, and protein C and S deficiency.
There’s a need for clinical suspicion of calciphylaxis when individuals with any of these conditions present with painful erythematous nodules, or with a vasculitic picture, she said.
Other, more common crystal deposition diseases can also be associated with panniculitis and can be in the differential, Dr. Bridges said. In patients with gout, sodium urate crystal deposition can occur in subcutaneous tissues.
Cutaneous oxalosis can occur as a primary disorder, when patients have metabolic errors and lack alanine-glyoxylate aminotransferase or D-glycerate dehydrogenase. Oxalosis can also be an acquired syndrome in patients with chronic renal failure who have been on long-term hemodialysis.
Although there is not a clearly effective treatment for calciphylaxis, a multitargeted, multidisciplinary approach is needed to help improve tissue health and patient quality of life. Since the primary mechanism of tissue damage is thrombotic tissue ischemia, strategies are aimed at existing clots and at preventing further clot formation.
To correct the calcium-phosphate balance, several medications have been used, including sodium thiosulfate and cinacalcet. For individuals on hemodialysis, a low-calcium dialysate may be used.

Tissue perfusion and oxygenation can be improved using tissue plasminogen activator, hyperbaric oxygen therapy, and the avoidance of warfarin if the patient requires anticoagulation.
To address wounds directly, debridement can begin with whirlpool time for patients. Surgical debridement may be required, and maggots can also help clean up wound beds.
Palliative care for patients should always include optimizing pain control and improving quality of life for patients with this serious and often life-limiting condition, Dr. Bridges said.
She reported no relevant conflicts of interest.
koakes@frontlinemedcom.com
On Twitter @karioakes
EXPERT ANALYSIS FROM THE 2017 AAD SUMMER MEETING
NEW YORK – When patients come to the office with painful “bumps” on the legs or elsewhere, panniculitis should be in the differential. And for some patients, said Alina Bridges, DO, the panniculitis may come with the dire diagnosis of calciphylaxis.
Calciphylaxis is an underrecognized crystal deposition disease that’s associated with panniculitis, said Dr. Bridges, speaking at the American Academy of Dermatology summer meeting. When calcium accumulates in small subcutaneous vessels, an occlusive vasculopathy is created within the dermis.
A soft-tissue radiograph of the affected area may also be helpful. Calciphylaxis shows as a fine netlike pattern of calcification, a finding that Dr. Bridges said has 90% specificity for the condition.
However, Dr. Bridges said, patients with panniculitis need a biopsy. “Careful selection of biopsy site and a deep specimen containing abundant fat obtained by incisional or excisional biopsy” is the best approach, allowing the pathologist to see the complete picture. In some cases, she said, a double-punch biopsy could also produce adequate specimens.
In addition to the calcium deposition, other pathologic findings may be lobular fat necrosis, with a pannicular vascular thrombosis. Though extravascular calcification can be seen in the panniculus, it’s not uncommon also to see intravascular calcification, said Dr. Bridges, who is the dermatopathology fellowship program director at the Mayo Clinic, Rochester, Minn.
Dr. Bridges said that the patients with calciphylaxis can present with predominant panniculitis or vasculitis, or a mixed picture; patients can also have bullae, ulcers, or livedo reticularis.
The lesions are extremely painful and become increasingly violaceous, with firm subcutaneous nodules. They are variably necrotic, and become more ulcerated over time.
Calciphylaxis is multifactorial and progressive. The prognosis is very poor for individuals with the condition, Dr. Bridges said. The median survival is 10 months, with 1-year survival rates of 46%, and just 20% of individuals with calciphylaxis surviving 2 years after diagnosis.
Gangrene is a frequent complication, and multisystem organ failure often occurs as well, she said.
Calciphylaxis most commonly occurs in individuals with chronic kidney disease and is seen in 4% of hemodialysis patients. However, it may also occur in individuals without uremia. In associations that are incompletely understood, calciphylaxis has been associated with warfarin therapy, connective tissue disorders, Crohn’s disease, liver disease, diabetes, hematologic malignancies, factor V Leiden deficiency, and protein C and S deficiency.
There’s a need for clinical suspicion of calciphylaxis when individuals with any of these conditions present with painful erythematous nodules, or with a vasculitic picture, she said.
Other, more common crystal deposition diseases can also be associated with panniculitis and can be in the differential, Dr. Bridges said. In patients with gout, sodium urate crystal deposition can occur in subcutaneous tissues.
Cutaneous oxalosis can occur as a primary disorder, when patients have metabolic errors and lack alanine-glyoxylate aminotransferase or D-glycerate dehydrogenase. Oxalosis can also be an acquired syndrome in patients with chronic renal failure who have been on long-term hemodialysis.
Although there is not a clearly effective treatment for calciphylaxis, a multitargeted, multidisciplinary approach is needed to help improve tissue health and patient quality of life. Since the primary mechanism of tissue damage is thrombotic tissue ischemia, strategies are aimed at existing clots and at preventing further clot formation.
To correct the calcium-phosphate balance, several medications have been used, including sodium thiosulfate and cinacalcet. For individuals on hemodialysis, a low-calcium dialysate may be used.
Tissue perfusion and oxygenation can be improved using tissue plasminogen activator, hyperbaric oxygen therapy, and the avoidance of warfarin if the patient requires anticoagulation.
To address wounds directly, debridement can begin with whirlpool time for patients. Surgical debridement may be required, and maggots can also help clean up wound beds.
Palliative care for patients should always include optimizing pain control and improving quality of life for patients with this serious and often life-limiting condition, Dr. Bridges said.
She reported no relevant conflicts of interest.
koakes@frontlinemedcom.com
On Twitter @karioakes
EXPERT ANALYSIS FROM THE 2017 AAD SUMMER MEETING
NEW YORK – When patients come to the office with painful “bumps” on the legs or elsewhere, panniculitis should be in the differential. And for some patients, said Alina Bridges, DO, the panniculitis may come with the dire diagnosis of calciphylaxis.
Calciphylaxis is an underrecognized crystal deposition disease that’s associated with panniculitis, said Dr. Bridges, speaking at the American Academy of Dermatology summer meeting. When calcium accumulates in small subcutaneous vessels, an occlusive vasculopathy is created within the dermis.
A soft-tissue radiograph of the affected area may also be helpful. Calciphylaxis shows as a fine netlike pattern of calcification, a finding that Dr. Bridges said has 90% specificity for the condition.
However, Dr. Bridges said, patients with panniculitis need a biopsy. “Careful selection of biopsy site and a deep specimen containing abundant fat obtained by incisional or excisional biopsy” is the best approach, allowing the pathologist to see the complete picture. In some cases, she said, a double-punch biopsy could also produce adequate specimens.
In addition to the calcium deposition, other pathologic findings may be lobular fat necrosis, with a pannicular vascular thrombosis. Though extravascular calcification can be seen in the panniculus, it’s not uncommon also to see intravascular calcification, said Dr. Bridges, who is the dermatopathology fellowship program director at the Mayo Clinic, Rochester, Minn.
Dr. Bridges said that the patients with calciphylaxis can present with predominant panniculitis or vasculitis, or a mixed picture; patients can also have bullae, ulcers, or livedo reticularis.
The lesions are extremely painful and become increasingly violaceous, with firm subcutaneous nodules. They are variably necrotic, and become more ulcerated over time.
Calciphylaxis is multifactorial and progressive. The prognosis is very poor for individuals with the condition, Dr. Bridges said. The median survival is 10 months, with 1-year survival rates of 46%, and just 20% of individuals with calciphylaxis surviving 2 years after diagnosis.
Gangrene is a frequent complication, and multisystem organ failure often occurs as well, she said.
Calciphylaxis most commonly occurs in individuals with chronic kidney disease and is seen in 4% of hemodialysis patients. However, it may also occur in individuals without uremia. In associations that are incompletely understood, calciphylaxis has been associated with warfarin therapy, connective tissue disorders, Crohn’s disease, liver disease, diabetes, hematologic malignancies, factor V Leiden deficiency, and protein C and S deficiency.
There’s a need for clinical suspicion of calciphylaxis when individuals with any of these conditions present with painful erythematous nodules, or with a vasculitic picture, she said.
Other, more common crystal deposition diseases can also be associated with panniculitis and can be in the differential, Dr. Bridges said. In patients with gout, sodium urate crystal deposition can occur in subcutaneous tissues.
Cutaneous oxalosis can occur as a primary disorder, when patients have metabolic errors and lack alanine-glyoxylate aminotransferase or D-glycerate dehydrogenase. Oxalosis can also be an acquired syndrome in patients with chronic renal failure who have been on long-term hemodialysis.
Although there is not a clearly effective treatment for calciphylaxis, a multitargeted, multidisciplinary approach is needed to help improve tissue health and patient quality of life. Since the primary mechanism of tissue damage is thrombotic tissue ischemia, strategies are aimed at existing clots and at preventing further clot formation.
To correct the calcium-phosphate balance, several medications have been used, including sodium thiosulfate and cinacalcet. For individuals on hemodialysis, a low-calcium dialysate may be used.
Tissue perfusion and oxygenation can be improved using tissue plasminogen activator, hyperbaric oxygen therapy, and the avoidance of warfarin if the patient requires anticoagulation.
To address wounds directly, debridement can begin with whirlpool time for patients. Surgical debridement may be required, and maggots can also help clean up wound beds.
Palliative care for patients should always include optimizing pain control and improving quality of life for patients with this serious and often life-limiting condition, Dr. Bridges said.
She reported no relevant conflicts of interest.
koakes@frontlinemedcom.com
On Twitter @karioakes
Co-occurrence of Steatocystoma Multiplex, Eruptive Vellus Hair Cysts, and Trichofolliculomas
An association between steatocystoma multiplex (SCM) and eruptive vellus hair cysts (EVHCs) has been recognized. They are related conditions representing nevoid malformations of the pilosebaceous junctions1-10 that have similar clinical features but distinctive histologic features. Both conditions most commonly involve the anterior aspect of the chest. Six cases of a rare facial variant of SCM have been reported,11-16 3 involving lesions limited to the forehead.13-15 Two patients with a rare facial variant of EVHC also have been reported.17 The development of separate lesions of SCM and EVHC on the trunk can uncommonly occur.5,6,10 One case of SCM and EVHC on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. The simultaneous occurrence of multiple trichoblastomas, trichoepitheliomas, and SCM on the face and trunk has been reported in 1 case.18 Milia, SCM, and EVHC on the face and trunk have been reported in 1 family.4 A report of facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus also has occurred in 1 patient.19 Here, we report the simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead.
Case Report
A 37-year-old man had an increasing number of flesh-colored to yellow papules on the forehead that had been present since puberty. Although the lesions were asymptomatic, some had recently become tender, which led him to seek medical care. There was no history of trauma, burns, irradiation, or application of topical agents to the area or use of eyeglasses or goggles. The patient’s father had similar lesions limited to the forehead, which developed during adolescence.
On evaluation at our clinic, skin examination revealed 16 discrete, 0.3- to 1-cm, flesh-colored, yellow to blue, mobile, smooth papules, as well as flesh-colored papules with a central black punctum, on the forehead (Figure 1). Similar lesions were not present on the rest of the face; around the ears; or on the scalp, neck, chest, back, abdomen, genitalia, buttocks, palms, soles, axillae, arms, or legs. There were no nail abnormalities.
Multiple 3-, 4-, and 6-mm punch and excisional biopsies were performed to remove all 16 lesions on the forehead. Histologic examination revealed a collapsed cystic structure in the mid dermis in 10 lesions. The cysts were lined with a squamous epithelium without a granular layer but with an eosinophilic corrugated lining, and the cyst cavity contained scant homogeneous eosinophilic secretion. Mature sebaceous glands were adjacent to the outer portion of the cyst wall. These histologic findings were consistent with SCM (Figure 2).
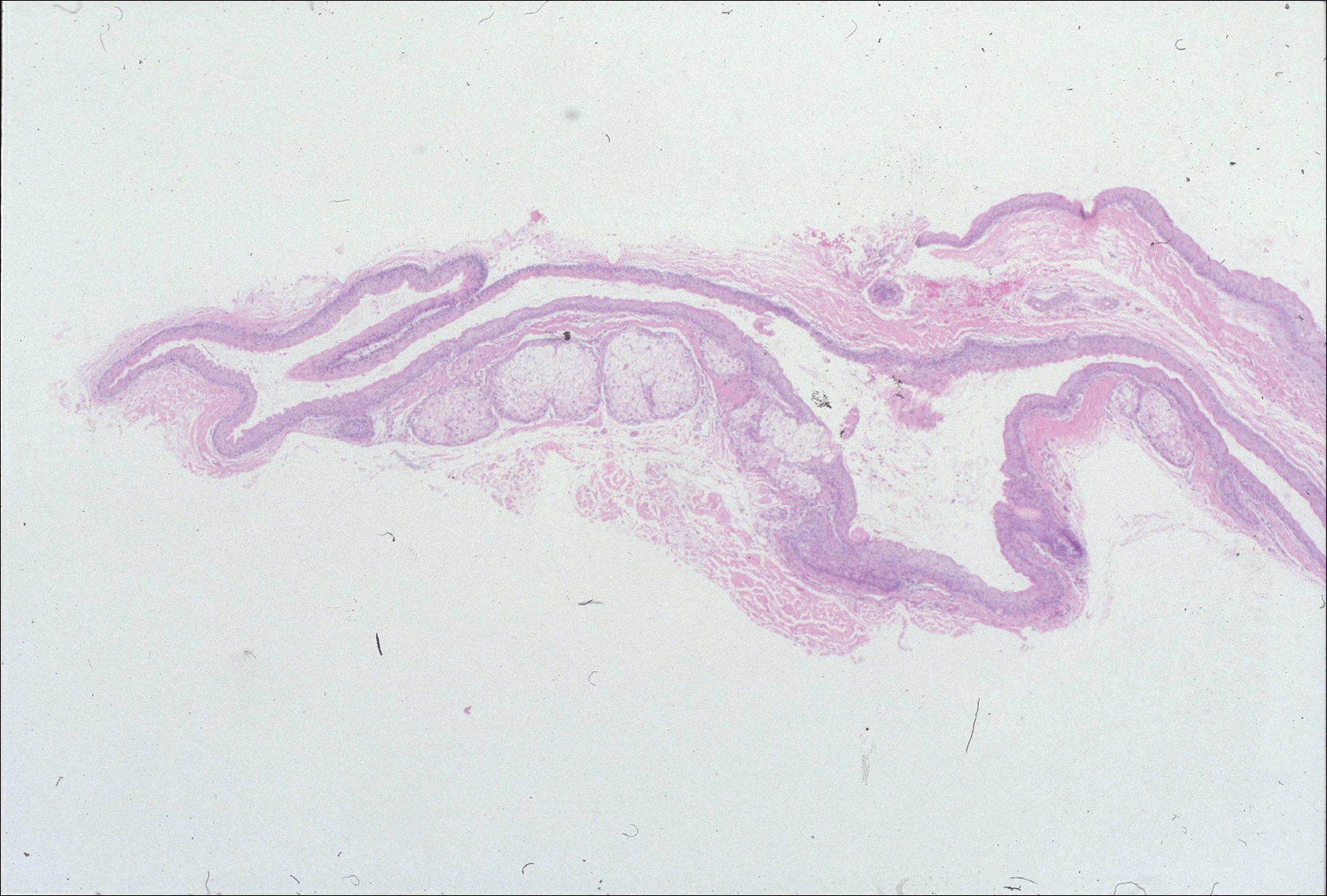
In 3 lesions, histologic examination revealed a cystic structure lined by a few layers of stratified squamous epithelium in the mid dermis. The cyst cavity contained numerous small vellus hairs and laminated keratin. These histologic findings were consistent with EVHC (Figure 3).
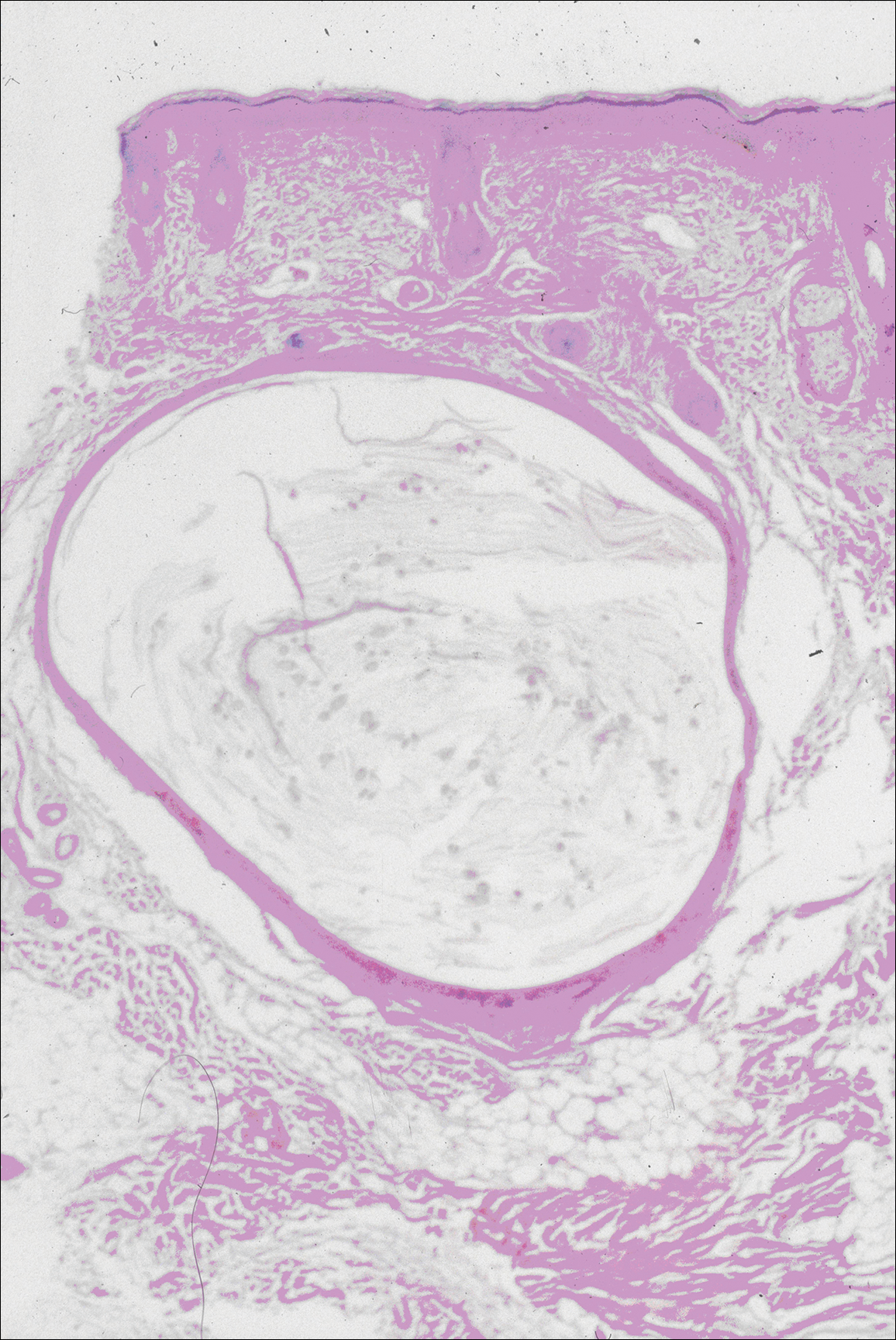
In the other 3 lesions, histologic examination revealed a dilated central cystic cavity filled with laminated keratin in the mid dermis. Multiple small follicles arose from the cysts and showed differentiation toward germinative epithelium. The surrounding stroma was fibrotic and contained a patchy lymphocytic infiltrate. These histologic findings were consistent with trichofolliculomas (Figure 4).
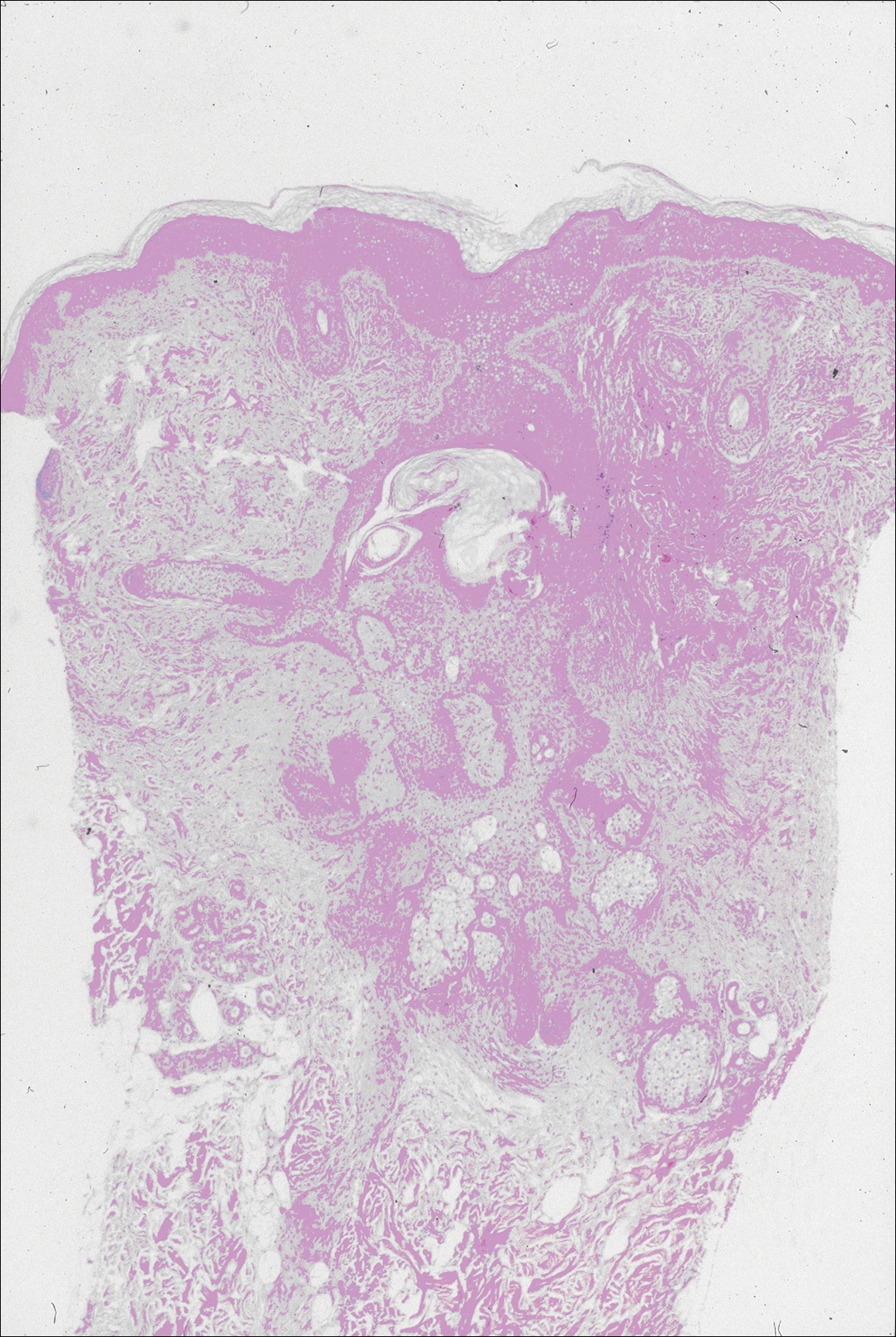
Comment
Characteristics of SCM
Steatocystoma multiplex is an uncommon condition characterized by the formation of asymptomatic, 0.2- to 2-cm, yellow to flesh-colored, soft, mobile papules or nodules on the trunk, extremities, axillae, genitalia, and/or chest. The lesions contain a clear or opaque, oily, milky or yellow, odorless fluid and most commonly are located on the anterior aspect of the chest. The face is not a commonly involved site in this condition. Six cases of a rare facial variant of SCM have been reported,11-16 with lesions limited to the forehead in 3 cases.13-15
In 1937, Mount20 credited Bozellini for describing the first case, though 3 cases reported in the late 1800s probably were SCM.21 In 1899, Pringle22 coined the term steatocystoma multiplex for this condition. It can be sporadic or have an autosomal-dominant inheritance pattern. Steatocystoma multiplex can occur at any age, though lesions develop most frequently in adolescence or young adulthood. There is no sex predilection.
Steatocystoma multiplex with pachyonychia congenita has been reported in a familial case.23 Other findings reported in patients with SCM include ichthyosis, koilonychia, acrokeratosis verruciformis of Hopf and hypertrophic lichen planus, hidradenitis suppurativa, hypotrichosis, multiple keratoacanthomas, and rheumatoid arthritis.12,24-26
Steatocystoma multiplex is a cyst lined by stratified squamous epithelium without a granular layer but with a thick eosinophilic cuticle. Mature sebaceous lobules are closely associated with the cyst wall. Steatocystoma multiplex arises from the sebaceous duct because the lining of the lumen is composed of undulating eosinophilic cuticle.
Characteristics of EVHCs
Eruptive vellus hair cysts, which were first described by Esterly et al,27 can occur at any age but develop most frequently in adolescents or young adults. Sometimes the lesions are congenital or appear in childhood. There is no sex predilection. They can be sporadic or have an autosomal-dominant inheritance pattern.
Eruptive vellus hair cysts are asymptomatic, 1- to 2-mm, smooth, crusted, or umbilicated papules on the chest or arms and legs. Eruptive vellus hair cysts most commonly involve the anterior aspect of the chest. The lesions are flesh-colored to yellow, though they have a slate gray color in darker-skinned individuals. A rare facial variant has been reported in 2 patients of Asian descent.17
Eruptive vellus hair cysts are small cystic structures lined by a stratified squamous epithelium with a granular layer. The cyst cavity contains numerous small vellus hair shafts and laminated keratin. Eruptive vellus hair cysts originate from the infundibulum or less frequently the isthmus or infundibular-isthmic junction of the hair follicle.
Characteristics of Trichofolliculomas
Trichofolliculomas are solitary, 3- to 5-mm, flesh-colored papules that occur on the face. They are highly differentiated, benign, neoplastic proliferations of an actively trichogenic epithelium, with structural components reflecting all portions of the pilosebaceous unit. Trichofolliculomas consist of a central dilated primary follicle contiguous with the surface epidermis embedded in a fibrous stroma. Multiple small secondary follicles with varying degrees of follicular differentiation arise from the primary follicle.
Co-occurrence of Lesions
An association between SCM and EVHC has been recognized.5-10 Steatocystoma multiplex and EVHC have similar clinical features but distinctive histologic features. They also have a similar age of onset, location/appearance of lesions, and mode of inheritance. Steatocystoma multiplex and EVHC can be distinguished by immunohistochemical techniques: SCM shows expression of keratin 10 and keratin 17, whereas EVHCs express only keratin 17.28
Steatocystoma multiplex and EVHC have only rarely been reported to occur together on the trunk. One case of SCM and EVHC occurring on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. Milia, SCM, and EVHC on the face and trunk have been reported in 1 family,4 and facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus was reported in 1 patient.19 Although trichofolliculomas have not been reported to occur with SCM or EVHC, 2 related follicular neoplasms—trichoepitheliomas and trichoblastomas—have been reported to occur in association with SCM on the face and chest and around the ears in 1 case.18
Differential Diagnosis
The clinical differential diagnosis includes multiple epidermoid cysts, dermoid cysts, Gardner syndrome, sebaceous adenomas, Muir-Torre syndrome, syringomas, milia, leiomyomas, lipomas, acneiform folliculitis, multiple familial and nonfamilial trichoepitheliomas, cylindromas, and angiofibromas.3,29
Conclusion
Our patient represents a rare case of simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead. The patient had multiple neoplasms involving differentiation toward various regions of the pilosebaceous unit. This case gives further support to the hypothesis that these benign follicular neoplasms are closely related but are distinct conditions within the spectrum of the same disease process. They represent nevoid malformations of the pilosebaceous unit that can be sporadic or inherited in an autosomal-dominant pattern. Pure types of these lesions may represent one end of the spectrum, but in some patients, there are overlapping features or hybrids of each condition. Several biopsies from patients with multiple lesions should be performed to establish an accurate diagnosis.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Ogawa Y, Nogita T, Kawashima M. The coexistence of eruptive vellus hair cysts and steatocystoma multiplex. J Dermatol. 1992;19:570-571.
- Sanchez Yus E, Requena L. Eruptive vellus hair cyst and steatocystoma multiplex. Am J Dermatopathol. 1990;12:536-537.
- Patrizi A, Neri I, Guerrini V, et al. Persistent milia, steatocystoma multiplex and eruptive vellus hair cysts: variable expression of multiple pilosebaceous cysts within an affected family. Dermatology. 1998;196:392-396.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Kiene P, Hauschild A, Christophers E. Eruptive vellus hair cysts and steatocystoma multiplex: variants of one entity? Br J Dermatol. 1996;134:365-367.
- Hurlimann AF, Panizzon RG, Burg G. Eruptive vellus hair cyst and steatocystoma multiplex: hybrid cysts. Dermatology. 1996;192:64-66.
- Sexton M, Murdock DK. Eruptive vellus hair cysts: a follicular cyst of the sebaceous duct (sometimes). Am J Dermatopathol. 1989;11:364-368.
- Sanchez-Yus E, Aguilar-Martinez A, Cristobal-Gil MC, et al. Eruptive vellus hair cyst and steatocystoma multiplex: two related conditions? J Cutan Pathol. 1988;15:40-42.
- Ahn SK, Chung J, Lee WS, et al. Hybrid cysts showing alternate combination of eruptive vellus hair cyst, steatocystoma multiplex, and epidermoid cyst, and an association among the three conditions. Am J Dermatopathol. 1996;18:645-649.
- Ahn SK, Hwang SM, Lee SH, et al. Steatocystoma multiplex localized only in the face. Int J Dermatol. 1997;36:372-373.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Hansen KK, Troy JL, Fairley JA. Multiple papules of the scalp and forehead. steatocystoma multiplex (facial papular variant). Arch Dermatol. 1995;131:835-838.
- Nishimura M, Kohda H, Urabe A. Steatocystoma multiplex: a facial popular variant. Arch Dermatol. 1986;122:205-207.
- Requena L, Martin L, Renedo G, et al. A facial variant of steatocystoma multiplex. Cutis. 1993;51:449-452.
- Holmes R, Black MM. Steatocystoma multiplex with unusually prominent cysts on the face. Br J Dermatol. 1980;102:711-713.
- Kumakiri M, Takashima I, Iju M, et al. Eruptive vellus hair cysts: a facial variant. J Am Acad Dermatol. 1982;7:461-467.
- Gianotti R, Cavicchini S, Alessi E. Simultaneous occurrence of multiple trichoblastomas and steatocystoma multiplex. Am J Dermatopathol. 1997;19:294-298.
- Sardana K, Sharma RC, Jain A, et al. Facial steatocystoma multiplex associated with pilar cyst and bilateral preauricular sinus. J Dermatol. 2002;29:157-159.
- Mount LB. Steatocystoma multiplex. Arch Dermatol Syphilol. 1937;36:31-39.
- Dubreuilh W, Auche B. Kystes grassieux sudoripares. Arch Clin de Bordeaux. 1896;5:387-391.
- Pringle JJ. A case of peculiar multiple sebaceous cysts (steatocystoma multiplex). Br J Dermatol. 1899;11:381-88.
- Vineyard WR, Scott RA. Steatocystoma multiplex with pachyonychia congenital: eight cases in four generations. Arch Dermatol. 1961;84:824-827.
- Contreras MA, Costello MJ. Steatocystoma multiplex with embryonal hair formation: case presentation and consideration of pathogenesis. AMA Arch Derm. 1957;76:720-725.
- Sohn D, Chin TC, Fellner MJ. Multiple keratoacanthomas associated with steatocystoma multiplex and rheumatoid arthritis: a case report. Arch Dermatol. 1980;116:913-915.
- Verbov J. Acrokeratosis verruciformis of Hopf with steatocystoma multiplex and hypertrophic lichen planus. Br J Dermatol. 1972;86:91-94.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Tomkova H, Fujimoto W, Arata J. Expression of keratins (K10 and K17) in steatocystoma multiplex, eruptive vellus hair cysts, and epidermoid and trichilemmal cysts. Am J Dermatopathol. 1997;19:250-253.
- Feinstein A, Trau H, Movshovitz M, et al. Steatocystoma multiplex. Cutis. 1983;31:425-427.
An association between steatocystoma multiplex (SCM) and eruptive vellus hair cysts (EVHCs) has been recognized. They are related conditions representing nevoid malformations of the pilosebaceous junctions1-10 that have similar clinical features but distinctive histologic features. Both conditions most commonly involve the anterior aspect of the chest. Six cases of a rare facial variant of SCM have been reported,11-16 3 involving lesions limited to the forehead.13-15 Two patients with a rare facial variant of EVHC also have been reported.17 The development of separate lesions of SCM and EVHC on the trunk can uncommonly occur.5,6,10 One case of SCM and EVHC on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. The simultaneous occurrence of multiple trichoblastomas, trichoepitheliomas, and SCM on the face and trunk has been reported in 1 case.18 Milia, SCM, and EVHC on the face and trunk have been reported in 1 family.4 A report of facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus also has occurred in 1 patient.19 Here, we report the simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead.
Case Report
A 37-year-old man had an increasing number of flesh-colored to yellow papules on the forehead that had been present since puberty. Although the lesions were asymptomatic, some had recently become tender, which led him to seek medical care. There was no history of trauma, burns, irradiation, or application of topical agents to the area or use of eyeglasses or goggles. The patient’s father had similar lesions limited to the forehead, which developed during adolescence.
On evaluation at our clinic, skin examination revealed 16 discrete, 0.3- to 1-cm, flesh-colored, yellow to blue, mobile, smooth papules, as well as flesh-colored papules with a central black punctum, on the forehead (Figure 1). Similar lesions were not present on the rest of the face; around the ears; or on the scalp, neck, chest, back, abdomen, genitalia, buttocks, palms, soles, axillae, arms, or legs. There were no nail abnormalities.
Multiple 3-, 4-, and 6-mm punch and excisional biopsies were performed to remove all 16 lesions on the forehead. Histologic examination revealed a collapsed cystic structure in the mid dermis in 10 lesions. The cysts were lined with a squamous epithelium without a granular layer but with an eosinophilic corrugated lining, and the cyst cavity contained scant homogeneous eosinophilic secretion. Mature sebaceous glands were adjacent to the outer portion of the cyst wall. These histologic findings were consistent with SCM (Figure 2).
In 3 lesions, histologic examination revealed a cystic structure lined by a few layers of stratified squamous epithelium in the mid dermis. The cyst cavity contained numerous small vellus hairs and laminated keratin. These histologic findings were consistent with EVHC (Figure 3).
In the other 3 lesions, histologic examination revealed a dilated central cystic cavity filled with laminated keratin in the mid dermis. Multiple small follicles arose from the cysts and showed differentiation toward germinative epithelium. The surrounding stroma was fibrotic and contained a patchy lymphocytic infiltrate. These histologic findings were consistent with trichofolliculomas (Figure 4).
Comment
Characteristics of SCM
Steatocystoma multiplex is an uncommon condition characterized by the formation of asymptomatic, 0.2- to 2-cm, yellow to flesh-colored, soft, mobile papules or nodules on the trunk, extremities, axillae, genitalia, and/or chest. The lesions contain a clear or opaque, oily, milky or yellow, odorless fluid and most commonly are located on the anterior aspect of the chest. The face is not a commonly involved site in this condition. Six cases of a rare facial variant of SCM have been reported,11-16 with lesions limited to the forehead in 3 cases.13-15
In 1937, Mount20 credited Bozellini for describing the first case, though 3 cases reported in the late 1800s probably were SCM.21 In 1899, Pringle22 coined the term steatocystoma multiplex for this condition. It can be sporadic or have an autosomal-dominant inheritance pattern. Steatocystoma multiplex can occur at any age, though lesions develop most frequently in adolescence or young adulthood. There is no sex predilection.
Steatocystoma multiplex with pachyonychia congenita has been reported in a familial case.23 Other findings reported in patients with SCM include ichthyosis, koilonychia, acrokeratosis verruciformis of Hopf and hypertrophic lichen planus, hidradenitis suppurativa, hypotrichosis, multiple keratoacanthomas, and rheumatoid arthritis.12,24-26
Steatocystoma multiplex is a cyst lined by stratified squamous epithelium without a granular layer but with a thick eosinophilic cuticle. Mature sebaceous lobules are closely associated with the cyst wall. Steatocystoma multiplex arises from the sebaceous duct because the lining of the lumen is composed of undulating eosinophilic cuticle.
Characteristics of EVHCs
Eruptive vellus hair cysts, which were first described by Esterly et al,27 can occur at any age but develop most frequently in adolescents or young adults. Sometimes the lesions are congenital or appear in childhood. There is no sex predilection. They can be sporadic or have an autosomal-dominant inheritance pattern.
Eruptive vellus hair cysts are asymptomatic, 1- to 2-mm, smooth, crusted, or umbilicated papules on the chest or arms and legs. Eruptive vellus hair cysts most commonly involve the anterior aspect of the chest. The lesions are flesh-colored to yellow, though they have a slate gray color in darker-skinned individuals. A rare facial variant has been reported in 2 patients of Asian descent.17
Eruptive vellus hair cysts are small cystic structures lined by a stratified squamous epithelium with a granular layer. The cyst cavity contains numerous small vellus hair shafts and laminated keratin. Eruptive vellus hair cysts originate from the infundibulum or less frequently the isthmus or infundibular-isthmic junction of the hair follicle.
Characteristics of Trichofolliculomas
Trichofolliculomas are solitary, 3- to 5-mm, flesh-colored papules that occur on the face. They are highly differentiated, benign, neoplastic proliferations of an actively trichogenic epithelium, with structural components reflecting all portions of the pilosebaceous unit. Trichofolliculomas consist of a central dilated primary follicle contiguous with the surface epidermis embedded in a fibrous stroma. Multiple small secondary follicles with varying degrees of follicular differentiation arise from the primary follicle.
Co-occurrence of Lesions
An association between SCM and EVHC has been recognized.5-10 Steatocystoma multiplex and EVHC have similar clinical features but distinctive histologic features. They also have a similar age of onset, location/appearance of lesions, and mode of inheritance. Steatocystoma multiplex and EVHC can be distinguished by immunohistochemical techniques: SCM shows expression of keratin 10 and keratin 17, whereas EVHCs express only keratin 17.28
Steatocystoma multiplex and EVHC have only rarely been reported to occur together on the trunk. One case of SCM and EVHC occurring on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. Milia, SCM, and EVHC on the face and trunk have been reported in 1 family,4 and facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus was reported in 1 patient.19 Although trichofolliculomas have not been reported to occur with SCM or EVHC, 2 related follicular neoplasms—trichoepitheliomas and trichoblastomas—have been reported to occur in association with SCM on the face and chest and around the ears in 1 case.18
Differential Diagnosis
The clinical differential diagnosis includes multiple epidermoid cysts, dermoid cysts, Gardner syndrome, sebaceous adenomas, Muir-Torre syndrome, syringomas, milia, leiomyomas, lipomas, acneiform folliculitis, multiple familial and nonfamilial trichoepitheliomas, cylindromas, and angiofibromas.3,29
Conclusion
Our patient represents a rare case of simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead. The patient had multiple neoplasms involving differentiation toward various regions of the pilosebaceous unit. This case gives further support to the hypothesis that these benign follicular neoplasms are closely related but are distinct conditions within the spectrum of the same disease process. They represent nevoid malformations of the pilosebaceous unit that can be sporadic or inherited in an autosomal-dominant pattern. Pure types of these lesions may represent one end of the spectrum, but in some patients, there are overlapping features or hybrids of each condition. Several biopsies from patients with multiple lesions should be performed to establish an accurate diagnosis.
An association between steatocystoma multiplex (SCM) and eruptive vellus hair cysts (EVHCs) has been recognized. They are related conditions representing nevoid malformations of the pilosebaceous junctions1-10 that have similar clinical features but distinctive histologic features. Both conditions most commonly involve the anterior aspect of the chest. Six cases of a rare facial variant of SCM have been reported,11-16 3 involving lesions limited to the forehead.13-15 Two patients with a rare facial variant of EVHC also have been reported.17 The development of separate lesions of SCM and EVHC on the trunk can uncommonly occur.5,6,10 One case of SCM and EVHC on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. The simultaneous occurrence of multiple trichoblastomas, trichoepitheliomas, and SCM on the face and trunk has been reported in 1 case.18 Milia, SCM, and EVHC on the face and trunk have been reported in 1 family.4 A report of facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus also has occurred in 1 patient.19 Here, we report the simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead.
Case Report
A 37-year-old man had an increasing number of flesh-colored to yellow papules on the forehead that had been present since puberty. Although the lesions were asymptomatic, some had recently become tender, which led him to seek medical care. There was no history of trauma, burns, irradiation, or application of topical agents to the area or use of eyeglasses or goggles. The patient’s father had similar lesions limited to the forehead, which developed during adolescence.
On evaluation at our clinic, skin examination revealed 16 discrete, 0.3- to 1-cm, flesh-colored, yellow to blue, mobile, smooth papules, as well as flesh-colored papules with a central black punctum, on the forehead (Figure 1). Similar lesions were not present on the rest of the face; around the ears; or on the scalp, neck, chest, back, abdomen, genitalia, buttocks, palms, soles, axillae, arms, or legs. There were no nail abnormalities.
Multiple 3-, 4-, and 6-mm punch and excisional biopsies were performed to remove all 16 lesions on the forehead. Histologic examination revealed a collapsed cystic structure in the mid dermis in 10 lesions. The cysts were lined with a squamous epithelium without a granular layer but with an eosinophilic corrugated lining, and the cyst cavity contained scant homogeneous eosinophilic secretion. Mature sebaceous glands were adjacent to the outer portion of the cyst wall. These histologic findings were consistent with SCM (Figure 2).
In 3 lesions, histologic examination revealed a cystic structure lined by a few layers of stratified squamous epithelium in the mid dermis. The cyst cavity contained numerous small vellus hairs and laminated keratin. These histologic findings were consistent with EVHC (Figure 3).
In the other 3 lesions, histologic examination revealed a dilated central cystic cavity filled with laminated keratin in the mid dermis. Multiple small follicles arose from the cysts and showed differentiation toward germinative epithelium. The surrounding stroma was fibrotic and contained a patchy lymphocytic infiltrate. These histologic findings were consistent with trichofolliculomas (Figure 4).
Comment
Characteristics of SCM
Steatocystoma multiplex is an uncommon condition characterized by the formation of asymptomatic, 0.2- to 2-cm, yellow to flesh-colored, soft, mobile papules or nodules on the trunk, extremities, axillae, genitalia, and/or chest. The lesions contain a clear or opaque, oily, milky or yellow, odorless fluid and most commonly are located on the anterior aspect of the chest. The face is not a commonly involved site in this condition. Six cases of a rare facial variant of SCM have been reported,11-16 with lesions limited to the forehead in 3 cases.13-15
In 1937, Mount20 credited Bozellini for describing the first case, though 3 cases reported in the late 1800s probably were SCM.21 In 1899, Pringle22 coined the term steatocystoma multiplex for this condition. It can be sporadic or have an autosomal-dominant inheritance pattern. Steatocystoma multiplex can occur at any age, though lesions develop most frequently in adolescence or young adulthood. There is no sex predilection.
Steatocystoma multiplex with pachyonychia congenita has been reported in a familial case.23 Other findings reported in patients with SCM include ichthyosis, koilonychia, acrokeratosis verruciformis of Hopf and hypertrophic lichen planus, hidradenitis suppurativa, hypotrichosis, multiple keratoacanthomas, and rheumatoid arthritis.12,24-26
Steatocystoma multiplex is a cyst lined by stratified squamous epithelium without a granular layer but with a thick eosinophilic cuticle. Mature sebaceous lobules are closely associated with the cyst wall. Steatocystoma multiplex arises from the sebaceous duct because the lining of the lumen is composed of undulating eosinophilic cuticle.
Characteristics of EVHCs
Eruptive vellus hair cysts, which were first described by Esterly et al,27 can occur at any age but develop most frequently in adolescents or young adults. Sometimes the lesions are congenital or appear in childhood. There is no sex predilection. They can be sporadic or have an autosomal-dominant inheritance pattern.
Eruptive vellus hair cysts are asymptomatic, 1- to 2-mm, smooth, crusted, or umbilicated papules on the chest or arms and legs. Eruptive vellus hair cysts most commonly involve the anterior aspect of the chest. The lesions are flesh-colored to yellow, though they have a slate gray color in darker-skinned individuals. A rare facial variant has been reported in 2 patients of Asian descent.17
Eruptive vellus hair cysts are small cystic structures lined by a stratified squamous epithelium with a granular layer. The cyst cavity contains numerous small vellus hair shafts and laminated keratin. Eruptive vellus hair cysts originate from the infundibulum or less frequently the isthmus or infundibular-isthmic junction of the hair follicle.
Characteristics of Trichofolliculomas
Trichofolliculomas are solitary, 3- to 5-mm, flesh-colored papules that occur on the face. They are highly differentiated, benign, neoplastic proliferations of an actively trichogenic epithelium, with structural components reflecting all portions of the pilosebaceous unit. Trichofolliculomas consist of a central dilated primary follicle contiguous with the surface epidermis embedded in a fibrous stroma. Multiple small secondary follicles with varying degrees of follicular differentiation arise from the primary follicle.
Co-occurrence of Lesions
An association between SCM and EVHC has been recognized.5-10 Steatocystoma multiplex and EVHC have similar clinical features but distinctive histologic features. They also have a similar age of onset, location/appearance of lesions, and mode of inheritance. Steatocystoma multiplex and EVHC can be distinguished by immunohistochemical techniques: SCM shows expression of keratin 10 and keratin 17, whereas EVHCs express only keratin 17.28
Steatocystoma multiplex and EVHC have only rarely been reported to occur together on the trunk. One case of SCM and EVHC occurring on the forehead has been described.3 Other types of benign follicular neoplasms simultaneously developing in association with SCM or EVHC also are rare. Milia, SCM, and EVHC on the face and trunk have been reported in 1 family,4 and facial steatocystoma associated with a pilar cyst and bilateral preauricular sinus was reported in 1 patient.19 Although trichofolliculomas have not been reported to occur with SCM or EVHC, 2 related follicular neoplasms—trichoepitheliomas and trichoblastomas—have been reported to occur in association with SCM on the face and chest and around the ears in 1 case.18
Differential Diagnosis
The clinical differential diagnosis includes multiple epidermoid cysts, dermoid cysts, Gardner syndrome, sebaceous adenomas, Muir-Torre syndrome, syringomas, milia, leiomyomas, lipomas, acneiform folliculitis, multiple familial and nonfamilial trichoepitheliomas, cylindromas, and angiofibromas.3,29
Conclusion
Our patient represents a rare case of simultaneous occurrence of SCM, EVHC, and trichofolliculomas localized to the forehead. The patient had multiple neoplasms involving differentiation toward various regions of the pilosebaceous unit. This case gives further support to the hypothesis that these benign follicular neoplasms are closely related but are distinct conditions within the spectrum of the same disease process. They represent nevoid malformations of the pilosebaceous unit that can be sporadic or inherited in an autosomal-dominant pattern. Pure types of these lesions may represent one end of the spectrum, but in some patients, there are overlapping features or hybrids of each condition. Several biopsies from patients with multiple lesions should be performed to establish an accurate diagnosis.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Ogawa Y, Nogita T, Kawashima M. The coexistence of eruptive vellus hair cysts and steatocystoma multiplex. J Dermatol. 1992;19:570-571.
- Sanchez Yus E, Requena L. Eruptive vellus hair cyst and steatocystoma multiplex. Am J Dermatopathol. 1990;12:536-537.
- Patrizi A, Neri I, Guerrini V, et al. Persistent milia, steatocystoma multiplex and eruptive vellus hair cysts: variable expression of multiple pilosebaceous cysts within an affected family. Dermatology. 1998;196:392-396.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Kiene P, Hauschild A, Christophers E. Eruptive vellus hair cysts and steatocystoma multiplex: variants of one entity? Br J Dermatol. 1996;134:365-367.
- Hurlimann AF, Panizzon RG, Burg G. Eruptive vellus hair cyst and steatocystoma multiplex: hybrid cysts. Dermatology. 1996;192:64-66.
- Sexton M, Murdock DK. Eruptive vellus hair cysts: a follicular cyst of the sebaceous duct (sometimes). Am J Dermatopathol. 1989;11:364-368.
- Sanchez-Yus E, Aguilar-Martinez A, Cristobal-Gil MC, et al. Eruptive vellus hair cyst and steatocystoma multiplex: two related conditions? J Cutan Pathol. 1988;15:40-42.
- Ahn SK, Chung J, Lee WS, et al. Hybrid cysts showing alternate combination of eruptive vellus hair cyst, steatocystoma multiplex, and epidermoid cyst, and an association among the three conditions. Am J Dermatopathol. 1996;18:645-649.
- Ahn SK, Hwang SM, Lee SH, et al. Steatocystoma multiplex localized only in the face. Int J Dermatol. 1997;36:372-373.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Hansen KK, Troy JL, Fairley JA. Multiple papules of the scalp and forehead. steatocystoma multiplex (facial papular variant). Arch Dermatol. 1995;131:835-838.
- Nishimura M, Kohda H, Urabe A. Steatocystoma multiplex: a facial popular variant. Arch Dermatol. 1986;122:205-207.
- Requena L, Martin L, Renedo G, et al. A facial variant of steatocystoma multiplex. Cutis. 1993;51:449-452.
- Holmes R, Black MM. Steatocystoma multiplex with unusually prominent cysts on the face. Br J Dermatol. 1980;102:711-713.
- Kumakiri M, Takashima I, Iju M, et al. Eruptive vellus hair cysts: a facial variant. J Am Acad Dermatol. 1982;7:461-467.
- Gianotti R, Cavicchini S, Alessi E. Simultaneous occurrence of multiple trichoblastomas and steatocystoma multiplex. Am J Dermatopathol. 1997;19:294-298.
- Sardana K, Sharma RC, Jain A, et al. Facial steatocystoma multiplex associated with pilar cyst and bilateral preauricular sinus. J Dermatol. 2002;29:157-159.
- Mount LB. Steatocystoma multiplex. Arch Dermatol Syphilol. 1937;36:31-39.
- Dubreuilh W, Auche B. Kystes grassieux sudoripares. Arch Clin de Bordeaux. 1896;5:387-391.
- Pringle JJ. A case of peculiar multiple sebaceous cysts (steatocystoma multiplex). Br J Dermatol. 1899;11:381-88.
- Vineyard WR, Scott RA. Steatocystoma multiplex with pachyonychia congenital: eight cases in four generations. Arch Dermatol. 1961;84:824-827.
- Contreras MA, Costello MJ. Steatocystoma multiplex with embryonal hair formation: case presentation and consideration of pathogenesis. AMA Arch Derm. 1957;76:720-725.
- Sohn D, Chin TC, Fellner MJ. Multiple keratoacanthomas associated with steatocystoma multiplex and rheumatoid arthritis: a case report. Arch Dermatol. 1980;116:913-915.
- Verbov J. Acrokeratosis verruciformis of Hopf with steatocystoma multiplex and hypertrophic lichen planus. Br J Dermatol. 1972;86:91-94.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Tomkova H, Fujimoto W, Arata J. Expression of keratins (K10 and K17) in steatocystoma multiplex, eruptive vellus hair cysts, and epidermoid and trichilemmal cysts. Am J Dermatopathol. 1997;19:250-253.
- Feinstein A, Trau H, Movshovitz M, et al. Steatocystoma multiplex. Cutis. 1983;31:425-427.
- Cho S, Chang SE, Choi JH, et al. Clinical and histologic features of 64 cases of steatocystoma multiplex. J Dermatol. 2002;29:152-156.
- Ogawa Y, Nogita T, Kawashima M. The coexistence of eruptive vellus hair cysts and steatocystoma multiplex. J Dermatol. 1992;19:570-571.
- Sanchez Yus E, Requena L. Eruptive vellus hair cyst and steatocystoma multiplex. Am J Dermatopathol. 1990;12:536-537.
- Patrizi A, Neri I, Guerrini V, et al. Persistent milia, steatocystoma multiplex and eruptive vellus hair cysts: variable expression of multiple pilosebaceous cysts within an affected family. Dermatology. 1998;196:392-396.
- Ohtake N, Kubota Y, Takayama O, et al. Relationship between steatocystoma multiplex and eruptive vellus hair cysts. J Am Acad Dermatol. 1992;26(5, pt 2):876-878.
- Kiene P, Hauschild A, Christophers E. Eruptive vellus hair cysts and steatocystoma multiplex: variants of one entity? Br J Dermatol. 1996;134:365-367.
- Hurlimann AF, Panizzon RG, Burg G. Eruptive vellus hair cyst and steatocystoma multiplex: hybrid cysts. Dermatology. 1996;192:64-66.
- Sexton M, Murdock DK. Eruptive vellus hair cysts: a follicular cyst of the sebaceous duct (sometimes). Am J Dermatopathol. 1989;11:364-368.
- Sanchez-Yus E, Aguilar-Martinez A, Cristobal-Gil MC, et al. Eruptive vellus hair cyst and steatocystoma multiplex: two related conditions? J Cutan Pathol. 1988;15:40-42.
- Ahn SK, Chung J, Lee WS, et al. Hybrid cysts showing alternate combination of eruptive vellus hair cyst, steatocystoma multiplex, and epidermoid cyst, and an association among the three conditions. Am J Dermatopathol. 1996;18:645-649.
- Ahn SK, Hwang SM, Lee SH, et al. Steatocystoma multiplex localized only in the face. Int J Dermatol. 1997;36:372-373.
- Cole LA. Steatocystoma multiplex. Arch Dermatol. 1976;112:1437-1439.
- Hansen KK, Troy JL, Fairley JA. Multiple papules of the scalp and forehead. steatocystoma multiplex (facial papular variant). Arch Dermatol. 1995;131:835-838.
- Nishimura M, Kohda H, Urabe A. Steatocystoma multiplex: a facial popular variant. Arch Dermatol. 1986;122:205-207.
- Requena L, Martin L, Renedo G, et al. A facial variant of steatocystoma multiplex. Cutis. 1993;51:449-452.
- Holmes R, Black MM. Steatocystoma multiplex with unusually prominent cysts on the face. Br J Dermatol. 1980;102:711-713.
- Kumakiri M, Takashima I, Iju M, et al. Eruptive vellus hair cysts: a facial variant. J Am Acad Dermatol. 1982;7:461-467.
- Gianotti R, Cavicchini S, Alessi E. Simultaneous occurrence of multiple trichoblastomas and steatocystoma multiplex. Am J Dermatopathol. 1997;19:294-298.
- Sardana K, Sharma RC, Jain A, et al. Facial steatocystoma multiplex associated with pilar cyst and bilateral preauricular sinus. J Dermatol. 2002;29:157-159.
- Mount LB. Steatocystoma multiplex. Arch Dermatol Syphilol. 1937;36:31-39.
- Dubreuilh W, Auche B. Kystes grassieux sudoripares. Arch Clin de Bordeaux. 1896;5:387-391.
- Pringle JJ. A case of peculiar multiple sebaceous cysts (steatocystoma multiplex). Br J Dermatol. 1899;11:381-88.
- Vineyard WR, Scott RA. Steatocystoma multiplex with pachyonychia congenital: eight cases in four generations. Arch Dermatol. 1961;84:824-827.
- Contreras MA, Costello MJ. Steatocystoma multiplex with embryonal hair formation: case presentation and consideration of pathogenesis. AMA Arch Derm. 1957;76:720-725.
- Sohn D, Chin TC, Fellner MJ. Multiple keratoacanthomas associated with steatocystoma multiplex and rheumatoid arthritis: a case report. Arch Dermatol. 1980;116:913-915.
- Verbov J. Acrokeratosis verruciformis of Hopf with steatocystoma multiplex and hypertrophic lichen planus. Br J Dermatol. 1972;86:91-94.
- Esterly NB, Fretzin DF, Pinkus H. Eruptive vellus hair cysts. Arch Dermatol. 1977;113:500-503.
- Tomkova H, Fujimoto W, Arata J. Expression of keratins (K10 and K17) in steatocystoma multiplex, eruptive vellus hair cysts, and epidermoid and trichilemmal cysts. Am J Dermatopathol. 1997;19:250-253.
- Feinstein A, Trau H, Movshovitz M, et al. Steatocystoma multiplex. Cutis. 1983;31:425-427.
Practice Points
- Steatocystoma multiplex (SCM) and eruptive vellus hair cysts (EVHCs) have similar clinical features but distinctive histologic features.
- Milia, pilar cyst, trichoepitheliomas, and trichoblastomas simultaneously developing in association with SCM or EVHC on the face are rare.
- This case supports the hypothesis that these benign follicular neoplasms are related but distinct nevoid malformations of the pilosebaceous unit within the same disease spectrum.