User login
One-year data support dupilumab’s efficacy and safety in adolescents with AD
A study of and continued evidence of efficacy for up to 52 weeks, reported the authors of the study, published online Oct. 9 in the British Journal of Dermatology.
The phase 2a open-label, ascending-dose cohort study of dupilumab in 40 adolescents with moderate to severe AD was followed by a 48-week phase 3 open-label extension study in 36 of those participants. Dupilumab is a monoclonal antibody that inhibits signaling of interleukin (IL)-4 and IL-13.
In the phase 2a study, participants were treated with a single subcutaneous dose of dupilumab – either 2 mg/kg or 4 mg/kg – and had 8 weeks of pharmacokinetic sampling. They subsequently received that same dose weekly for 4 weeks, with an 8-week-long safety follow-up period. Those who participated in the open-label extension continued their weekly dose to a maximum of 300 mg. per kg
The most common treatment-emergent adverse events (a primary endpoint) seen in both the phase 2a and phase 3 studies were nasopharyngitis and exacerbation of AD – in the phase 2a study, exacerbations were seen in the period when patients weren’t taking the treatment. In the 2-mg and 4-mg groups, the incidence of skin infections was 29% and 42%, respectively, and the incidence of injection site reactions – which were mostly mild – were 18% and 11%, respectively. Researchers also noted conjunctivitis in 18% and 16% of the patients in the 2-mg and 4-mg groups, respectively, but none of the cases were considered serious and all resolved over the course of the study. In the phase 2a study, 50% of patients on the 2-mg/kg dose and 65% of those on the 4-mg/kg dose experienced an adverse event, while in the open-label extension all reported at least one adverse event.
There was one case of suicidal behavior and one case of systemic or severe hypersensitivity reported in the 2-mg/kg groups, both of which were considered adverse events of special interest. There were no deaths.
However none of the serious adverse events – which included infected AD, palpitations, patent ductus arteriosus, and food allergy – were linked to the study treatment, and no adverse events led to study discontinuation, the authors reported.
By week 12, 70% of participants in the 2-mg/kg group and 75% in the 4-mg/kg group had achieved a 50% or greater improvement in their Eczema Area and Severity Index (EASI) scores, which was a secondary outcome. By week 52, that had increased to 100% and 89% respectively.
More than half the patients (55%) in the 2-mg/kg group, and 40% of those in the 4-mg/kg group achieved a 75% or more improvement in their EASI scores by week 12, which increased to 88% and 78%, respectively, by week 52 in the open label phase.
“The results from these studies support use of dupilumab for the long-term management of moderate to severe AD in adolescents,” wrote Michael J. Cork, MD, professor of dermatology, University of Sheffield, England, and coauthors. No new safety signals were identified, “compared with the known safety profile of dupilumab in adults with moderate to severe AD,” and “the PK profile was characterized by nonlinear, target-mediated kinetics, consistent with the profile in adults with moderate to severe AD,” they added.
Dupilumab was approved in the United States in March 2019 for adolescents with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.
The study was sponsored by dupilumab manufacturers Sanofi and Regeneron Pharmaceuticals, which market dupilumab as Dupixent in the United States. Dr. Cork disclosures included those related to Sanofi Genzyme and Regeneron; other authors included employees of the companies.
SOURCE: Cork M et al. Br J Dermatol. 2019 Oct 9. doi: 10.1111/bjd.18476.
A study of and continued evidence of efficacy for up to 52 weeks, reported the authors of the study, published online Oct. 9 in the British Journal of Dermatology.
The phase 2a open-label, ascending-dose cohort study of dupilumab in 40 adolescents with moderate to severe AD was followed by a 48-week phase 3 open-label extension study in 36 of those participants. Dupilumab is a monoclonal antibody that inhibits signaling of interleukin (IL)-4 and IL-13.
In the phase 2a study, participants were treated with a single subcutaneous dose of dupilumab – either 2 mg/kg or 4 mg/kg – and had 8 weeks of pharmacokinetic sampling. They subsequently received that same dose weekly for 4 weeks, with an 8-week-long safety follow-up period. Those who participated in the open-label extension continued their weekly dose to a maximum of 300 mg. per kg
The most common treatment-emergent adverse events (a primary endpoint) seen in both the phase 2a and phase 3 studies were nasopharyngitis and exacerbation of AD – in the phase 2a study, exacerbations were seen in the period when patients weren’t taking the treatment. In the 2-mg and 4-mg groups, the incidence of skin infections was 29% and 42%, respectively, and the incidence of injection site reactions – which were mostly mild – were 18% and 11%, respectively. Researchers also noted conjunctivitis in 18% and 16% of the patients in the 2-mg and 4-mg groups, respectively, but none of the cases were considered serious and all resolved over the course of the study. In the phase 2a study, 50% of patients on the 2-mg/kg dose and 65% of those on the 4-mg/kg dose experienced an adverse event, while in the open-label extension all reported at least one adverse event.
There was one case of suicidal behavior and one case of systemic or severe hypersensitivity reported in the 2-mg/kg groups, both of which were considered adverse events of special interest. There were no deaths.
However none of the serious adverse events – which included infected AD, palpitations, patent ductus arteriosus, and food allergy – were linked to the study treatment, and no adverse events led to study discontinuation, the authors reported.
By week 12, 70% of participants in the 2-mg/kg group and 75% in the 4-mg/kg group had achieved a 50% or greater improvement in their Eczema Area and Severity Index (EASI) scores, which was a secondary outcome. By week 52, that had increased to 100% and 89% respectively.
More than half the patients (55%) in the 2-mg/kg group, and 40% of those in the 4-mg/kg group achieved a 75% or more improvement in their EASI scores by week 12, which increased to 88% and 78%, respectively, by week 52 in the open label phase.
“The results from these studies support use of dupilumab for the long-term management of moderate to severe AD in adolescents,” wrote Michael J. Cork, MD, professor of dermatology, University of Sheffield, England, and coauthors. No new safety signals were identified, “compared with the known safety profile of dupilumab in adults with moderate to severe AD,” and “the PK profile was characterized by nonlinear, target-mediated kinetics, consistent with the profile in adults with moderate to severe AD,” they added.
Dupilumab was approved in the United States in March 2019 for adolescents with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.
The study was sponsored by dupilumab manufacturers Sanofi and Regeneron Pharmaceuticals, which market dupilumab as Dupixent in the United States. Dr. Cork disclosures included those related to Sanofi Genzyme and Regeneron; other authors included employees of the companies.
SOURCE: Cork M et al. Br J Dermatol. 2019 Oct 9. doi: 10.1111/bjd.18476.
A study of and continued evidence of efficacy for up to 52 weeks, reported the authors of the study, published online Oct. 9 in the British Journal of Dermatology.
The phase 2a open-label, ascending-dose cohort study of dupilumab in 40 adolescents with moderate to severe AD was followed by a 48-week phase 3 open-label extension study in 36 of those participants. Dupilumab is a monoclonal antibody that inhibits signaling of interleukin (IL)-4 and IL-13.
In the phase 2a study, participants were treated with a single subcutaneous dose of dupilumab – either 2 mg/kg or 4 mg/kg – and had 8 weeks of pharmacokinetic sampling. They subsequently received that same dose weekly for 4 weeks, with an 8-week-long safety follow-up period. Those who participated in the open-label extension continued their weekly dose to a maximum of 300 mg. per kg
The most common treatment-emergent adverse events (a primary endpoint) seen in both the phase 2a and phase 3 studies were nasopharyngitis and exacerbation of AD – in the phase 2a study, exacerbations were seen in the period when patients weren’t taking the treatment. In the 2-mg and 4-mg groups, the incidence of skin infections was 29% and 42%, respectively, and the incidence of injection site reactions – which were mostly mild – were 18% and 11%, respectively. Researchers also noted conjunctivitis in 18% and 16% of the patients in the 2-mg and 4-mg groups, respectively, but none of the cases were considered serious and all resolved over the course of the study. In the phase 2a study, 50% of patients on the 2-mg/kg dose and 65% of those on the 4-mg/kg dose experienced an adverse event, while in the open-label extension all reported at least one adverse event.
There was one case of suicidal behavior and one case of systemic or severe hypersensitivity reported in the 2-mg/kg groups, both of which were considered adverse events of special interest. There were no deaths.
However none of the serious adverse events – which included infected AD, palpitations, patent ductus arteriosus, and food allergy – were linked to the study treatment, and no adverse events led to study discontinuation, the authors reported.
By week 12, 70% of participants in the 2-mg/kg group and 75% in the 4-mg/kg group had achieved a 50% or greater improvement in their Eczema Area and Severity Index (EASI) scores, which was a secondary outcome. By week 52, that had increased to 100% and 89% respectively.
More than half the patients (55%) in the 2-mg/kg group, and 40% of those in the 4-mg/kg group achieved a 75% or more improvement in their EASI scores by week 12, which increased to 88% and 78%, respectively, by week 52 in the open label phase.
“The results from these studies support use of dupilumab for the long-term management of moderate to severe AD in adolescents,” wrote Michael J. Cork, MD, professor of dermatology, University of Sheffield, England, and coauthors. No new safety signals were identified, “compared with the known safety profile of dupilumab in adults with moderate to severe AD,” and “the PK profile was characterized by nonlinear, target-mediated kinetics, consistent with the profile in adults with moderate to severe AD,” they added.
Dupilumab was approved in the United States in March 2019 for adolescents with moderate to severe AD whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.
The study was sponsored by dupilumab manufacturers Sanofi and Regeneron Pharmaceuticals, which market dupilumab as Dupixent in the United States. Dr. Cork disclosures included those related to Sanofi Genzyme and Regeneron; other authors included employees of the companies.
SOURCE: Cork M et al. Br J Dermatol. 2019 Oct 9. doi: 10.1111/bjd.18476.
FROM THE BRITISH JOURNAL OF DERMATOLOGY
FDA approves afamelanotide for treatment of rare condition with light-induced pain
The Food and Drug Administration has approved , a rare condition that causes extremely painful reactions when skin is exposed to light, according to an FDA announcement.

This is the first treatment approved to help patients with this condition increase their exposure to light, according to the release.
Afamelanotide, administered in a subcutaneous implant, is a melanocortin-1 receptor (MC1-R) agonist, which “increases the production of eumelanin in the skin independent of exposure to sunlight or artificial light sources,” the release says.
Approval is based on a pair of parallel-group clinical trials that compared the number of hours spent in sunlight in the treatment and placebo groups. The first trial enrolled 93 patients; 48 received afamelanotide. The treated patients spent a median of 61 hours in total over 180 days in direct sunlight between 10 a.m. and 6 p.m. on days with no pain, compared with 41 hours for patients taking placebo.
The second trial assessed the total number of hours over 270 days spent outdoors between 10 a.m. and 3 p.m. on days with no pain for which “most of the day” was spent in direct sunlight. In this study, 38 patients treated with afamelanotide spent a median total of 6 hours, compared with 0.75 hours among the remaining 36 who were taking a placebo.
The most common side effects include implant site reaction, nausea, and oropharyngeal pain. The implant should be administered only by trained professionals. Because afamelanotide may cause skin darkening, it’s recommended that patients should undergo twice-yearly skin examinations. Patients are also encouraged to maintain sun protection measures to help prevent phototoxic reactions.
“Today’s approval is one example of the FDA’s ongoing commitment to encourage industry innovation of therapies to treat rare diseases, and work with drug developers to make promising new therapies available to patients as safely and efficiently as possible,” said Julie Beitz, MD, director of FDA’s Center for Drug Evaluation and Research Office of Drug Evaluation III in the FDA release.
The Food and Drug Administration has approved , a rare condition that causes extremely painful reactions when skin is exposed to light, according to an FDA announcement.
This is the first treatment approved to help patients with this condition increase their exposure to light, according to the release.
Afamelanotide, administered in a subcutaneous implant, is a melanocortin-1 receptor (MC1-R) agonist, which “increases the production of eumelanin in the skin independent of exposure to sunlight or artificial light sources,” the release says.
Approval is based on a pair of parallel-group clinical trials that compared the number of hours spent in sunlight in the treatment and placebo groups. The first trial enrolled 93 patients; 48 received afamelanotide. The treated patients spent a median of 61 hours in total over 180 days in direct sunlight between 10 a.m. and 6 p.m. on days with no pain, compared with 41 hours for patients taking placebo.
The second trial assessed the total number of hours over 270 days spent outdoors between 10 a.m. and 3 p.m. on days with no pain for which “most of the day” was spent in direct sunlight. In this study, 38 patients treated with afamelanotide spent a median total of 6 hours, compared with 0.75 hours among the remaining 36 who were taking a placebo.
The most common side effects include implant site reaction, nausea, and oropharyngeal pain. The implant should be administered only by trained professionals. Because afamelanotide may cause skin darkening, it’s recommended that patients should undergo twice-yearly skin examinations. Patients are also encouraged to maintain sun protection measures to help prevent phototoxic reactions.
“Today’s approval is one example of the FDA’s ongoing commitment to encourage industry innovation of therapies to treat rare diseases, and work with drug developers to make promising new therapies available to patients as safely and efficiently as possible,” said Julie Beitz, MD, director of FDA’s Center for Drug Evaluation and Research Office of Drug Evaluation III in the FDA release.
The Food and Drug Administration has approved , a rare condition that causes extremely painful reactions when skin is exposed to light, according to an FDA announcement.
This is the first treatment approved to help patients with this condition increase their exposure to light, according to the release.
Afamelanotide, administered in a subcutaneous implant, is a melanocortin-1 receptor (MC1-R) agonist, which “increases the production of eumelanin in the skin independent of exposure to sunlight or artificial light sources,” the release says.
Approval is based on a pair of parallel-group clinical trials that compared the number of hours spent in sunlight in the treatment and placebo groups. The first trial enrolled 93 patients; 48 received afamelanotide. The treated patients spent a median of 61 hours in total over 180 days in direct sunlight between 10 a.m. and 6 p.m. on days with no pain, compared with 41 hours for patients taking placebo.
The second trial assessed the total number of hours over 270 days spent outdoors between 10 a.m. and 3 p.m. on days with no pain for which “most of the day” was spent in direct sunlight. In this study, 38 patients treated with afamelanotide spent a median total of 6 hours, compared with 0.75 hours among the remaining 36 who were taking a placebo.
The most common side effects include implant site reaction, nausea, and oropharyngeal pain. The implant should be administered only by trained professionals. Because afamelanotide may cause skin darkening, it’s recommended that patients should undergo twice-yearly skin examinations. Patients are also encouraged to maintain sun protection measures to help prevent phototoxic reactions.
“Today’s approval is one example of the FDA’s ongoing commitment to encourage industry innovation of therapies to treat rare diseases, and work with drug developers to make promising new therapies available to patients as safely and efficiently as possible,” said Julie Beitz, MD, director of FDA’s Center for Drug Evaluation and Research Office of Drug Evaluation III in the FDA release.
Persistent rash on the sole
A 52-year-old Chinese woman presented to a tertiary hospital in Singapore with a 3-month history of persistent and intermittently painful rashes over her right calf and foot (FIGURE). The patient had pancytopenia due to ongoing chemotherapy for metastatic nasopharyngeal carcinoma. She was systemically well and denied other dermatoses. Examination demonstrated scattered crops of tense hemorrhagic vesicles, each surrounded by a livid purpuric base, over the right plantar aspect of the foot, with areas of eschar over the right medial hallux. No allodynia, hyperaesthesia, or lymphadenopathy was noted.
A punch biopsy of an intact vesicle was performed.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis:
Herpes zoster
Histopathologic examination showed full-thickness epidermal necrosis with ballooning degeneration resulting in an intra-epidermal blister. Multinucleated keratinocytes with nuclear moulding were seen within the blister cavity. Grocott-Gomori methenamine-silver (GMS), acid-fast, and Gram stains were negative. Granular immunoglobulin (Ig) G, IgM, and C3 were seen intramurally. DNA analysis of vesicular fluid was positive for varicella zoster virus (VZV). A diagnosis of herpes zoster (HZ) of the right S1 dermatome with primary obliterative vasculitis was established.
Immunocompromised people—those who have impaired T-cell immunity (eg, recipients of organ or hematopoietic stem-cell transplants), take immunosuppressive therapy, or have lymphoma, leukemia, or human immunodeficiency virus (HIV) infection—have an increased risk for HZ. For example, in patients with acquired immunodeficiency syndrome (AIDS), HZ uniquely manifests as recurrent shingles. An estimated 20% to 30% of HIV-infected patients will have more than 1 episode of HZ, which may involve the same or different dermatomes.1,2 Furthermore, HZ in this population is more commonly associated with atypical presentations.3
What an atypical presentation may look like
In immunocompromised patients, HZ may present with atypical cutaneous manifestations or with atypical generalized symptoms.
Atypical cutaneous manifestations, as in disseminated zoster, manifest with multiple hyperkeratotic papules (3-20 mm in diameter) that follow no dermatomal pattern. These lesions may be chronic, persisting for months or years, and may be associated with acyclovir-resistant strains of VZV.2,3 Another dermatologic variant is ecthymatous VZV, which manifests with multiple large (10-30 mm) punched-out ulcerations with a central black eschar and a peripheral rim of vesicles.4 Viral folliculitis—in which infection is limited to the hair follicle, with no associated blisters—has also been reported in atypical HZ.5
Our patient presented with hemorrhagic vesicles mimicking vasculitic lesions, which had persisted over a 3-month period with intermittent localized pain. It has been proposed that in atypical presentations, the reactivated VZV spreads transaxonally from adjacent nerves to the outermost adventitial layer of the arterial wall, leading to a vasculitic appearance of the vesicles.6 Viral-induced vasculitis may also result either directly from infection of the blood vessels or secondary to vascular damage from an inflammatory immune complex–mediated reaction, cell-mediated hypersensitivity, or inflammation due to immune dysregulation.7,8
Continue to: Differential includes vesiculobullous conditions
Differential includes vesiculobullous conditions
There are several important items to consider in the differential.
Cutaneous vasculitis, in severe cases, may manifest with vesicles or bullae that resemble the lesions seen in HZ. However, its unilateral nature and distribution distinguish it.
Angioinvasive fungal infections in immunocompromised patients may manifest with scattered ulceronecrotic lesions to purpuric vesiculobullous dermatoses.9 However, no fungal organisms were seen on GMS staining of the biopsied tissue.
Atypical hand-foot-and-mouth disease tends to affect adults and is associated with Coxsackievirus A6 infection.10 It may manifest as generalized vesiculobullous exanthem resembling varicella. The chronic nature and restricted extent of the patient’s rash made this diagnosis unlikely.
Successful management depends on timely identification
Although most cases of HZ can be diagnosed clinically, atypical rashes may require a biopsy and direct immunofluorescence assay for VZV antigen or a polymerase-chain-reaction (PCR) assay for VZV DNA in cells from the base of blisters. Therefore, it is important to consider the diagnosis of HZ in immunocompromised patients presenting with an atypical rash to avoid misdiagnosis and costly testing.
Continue to: Our patient was treated...
Our patient was treated with oral acyclovir 800 mg 5 times/day for 10 days, with prompt resolution of her rash.
CORRESPONDENCE
Joel Hua-Liang Lim, MBBS, MRCP, MMed, 1 Mandalay Road, Singapore 308205; joellimhl@nsc.com.sg
1. LeBoit PE, Limova M, Yen TS, et al. Chronic verrucous varicella-zoster virus infection in patients with the acquired immunodeficiency syndrome (AIDS): histologic and molecular biologic findings. Am J Dermatopathol. 1992;14:1-7.
2. Gnann JW Jr. Varicella-zoster virus: atypical presentations and unusual complications. J Infect Dis. 2002;186(suppl 1):S91-S98.
3. Weinberg JM, Mysliwiec A, Turiansky GW, et al. Viral folliculitis: atypical presentations of herpes simplex, herpes zoster, and molluscum contagiosum. Arch Dermatol. 1997;133:983-986.
4. Gilson IH, Barnett JH, Conant MA, et al. Disseminated ecthymatous herpes varicella zoster virus infection in patients with acquired immunodeficiency syndrome. J Am Acad Dermatol. 1989;20:637-642.
5. Løkke BJ, Weismann K, Mathiesen L, et al. Atypical varicella-zoster infection in AIDS. Acta Derm Venereol. 1993;73:123-125.
6. Uhoda I, Piérard-Franchimont C, Piérard GE. Varicella-zoster virus vasculitis: a case of recurrent varicella without epidermal involvement. Dermatology. 2000;200:173-175.
7. Teng GG, Chatham WW. Vasculitis related to viral and other microbial agents. Best Pract Res Clin Rheumatol. 2015;29:226-243.
8. Nagel MA, Gilden D. Developments in varicella zoster virus vasculopathy. Curr Neurol Neurosci Rep. 2016;16:12.
9. Pfaller MA, Diekema DJ. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol. 2010;36:1-53.
10. Lott JP, Liu K, Landry M-L, et al. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J Am Acad Dermatol. 2013;69:736-741.
A 52-year-old Chinese woman presented to a tertiary hospital in Singapore with a 3-month history of persistent and intermittently painful rashes over her right calf and foot (FIGURE). The patient had pancytopenia due to ongoing chemotherapy for metastatic nasopharyngeal carcinoma. She was systemically well and denied other dermatoses. Examination demonstrated scattered crops of tense hemorrhagic vesicles, each surrounded by a livid purpuric base, over the right plantar aspect of the foot, with areas of eschar over the right medial hallux. No allodynia, hyperaesthesia, or lymphadenopathy was noted.
A punch biopsy of an intact vesicle was performed.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis:
Herpes zoster
Histopathologic examination showed full-thickness epidermal necrosis with ballooning degeneration resulting in an intra-epidermal blister. Multinucleated keratinocytes with nuclear moulding were seen within the blister cavity. Grocott-Gomori methenamine-silver (GMS), acid-fast, and Gram stains were negative. Granular immunoglobulin (Ig) G, IgM, and C3 were seen intramurally. DNA analysis of vesicular fluid was positive for varicella zoster virus (VZV). A diagnosis of herpes zoster (HZ) of the right S1 dermatome with primary obliterative vasculitis was established.
Immunocompromised people—those who have impaired T-cell immunity (eg, recipients of organ or hematopoietic stem-cell transplants), take immunosuppressive therapy, or have lymphoma, leukemia, or human immunodeficiency virus (HIV) infection—have an increased risk for HZ. For example, in patients with acquired immunodeficiency syndrome (AIDS), HZ uniquely manifests as recurrent shingles. An estimated 20% to 30% of HIV-infected patients will have more than 1 episode of HZ, which may involve the same or different dermatomes.1,2 Furthermore, HZ in this population is more commonly associated with atypical presentations.3
What an atypical presentation may look like
In immunocompromised patients, HZ may present with atypical cutaneous manifestations or with atypical generalized symptoms.
Atypical cutaneous manifestations, as in disseminated zoster, manifest with multiple hyperkeratotic papules (3-20 mm in diameter) that follow no dermatomal pattern. These lesions may be chronic, persisting for months or years, and may be associated with acyclovir-resistant strains of VZV.2,3 Another dermatologic variant is ecthymatous VZV, which manifests with multiple large (10-30 mm) punched-out ulcerations with a central black eschar and a peripheral rim of vesicles.4 Viral folliculitis—in which infection is limited to the hair follicle, with no associated blisters—has also been reported in atypical HZ.5
Our patient presented with hemorrhagic vesicles mimicking vasculitic lesions, which had persisted over a 3-month period with intermittent localized pain. It has been proposed that in atypical presentations, the reactivated VZV spreads transaxonally from adjacent nerves to the outermost adventitial layer of the arterial wall, leading to a vasculitic appearance of the vesicles.6 Viral-induced vasculitis may also result either directly from infection of the blood vessels or secondary to vascular damage from an inflammatory immune complex–mediated reaction, cell-mediated hypersensitivity, or inflammation due to immune dysregulation.7,8
Continue to: Differential includes vesiculobullous conditions
Differential includes vesiculobullous conditions
There are several important items to consider in the differential.
Cutaneous vasculitis, in severe cases, may manifest with vesicles or bullae that resemble the lesions seen in HZ. However, its unilateral nature and distribution distinguish it.
Angioinvasive fungal infections in immunocompromised patients may manifest with scattered ulceronecrotic lesions to purpuric vesiculobullous dermatoses.9 However, no fungal organisms were seen on GMS staining of the biopsied tissue.
Atypical hand-foot-and-mouth disease tends to affect adults and is associated with Coxsackievirus A6 infection.10 It may manifest as generalized vesiculobullous exanthem resembling varicella. The chronic nature and restricted extent of the patient’s rash made this diagnosis unlikely.
Successful management depends on timely identification
Although most cases of HZ can be diagnosed clinically, atypical rashes may require a biopsy and direct immunofluorescence assay for VZV antigen or a polymerase-chain-reaction (PCR) assay for VZV DNA in cells from the base of blisters. Therefore, it is important to consider the diagnosis of HZ in immunocompromised patients presenting with an atypical rash to avoid misdiagnosis and costly testing.
Continue to: Our patient was treated...
Our patient was treated with oral acyclovir 800 mg 5 times/day for 10 days, with prompt resolution of her rash.
CORRESPONDENCE
Joel Hua-Liang Lim, MBBS, MRCP, MMed, 1 Mandalay Road, Singapore 308205; joellimhl@nsc.com.sg
A 52-year-old Chinese woman presented to a tertiary hospital in Singapore with a 3-month history of persistent and intermittently painful rashes over her right calf and foot (FIGURE). The patient had pancytopenia due to ongoing chemotherapy for metastatic nasopharyngeal carcinoma. She was systemically well and denied other dermatoses. Examination demonstrated scattered crops of tense hemorrhagic vesicles, each surrounded by a livid purpuric base, over the right plantar aspect of the foot, with areas of eschar over the right medial hallux. No allodynia, hyperaesthesia, or lymphadenopathy was noted.
A punch biopsy of an intact vesicle was performed.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis:
Herpes zoster
Histopathologic examination showed full-thickness epidermal necrosis with ballooning degeneration resulting in an intra-epidermal blister. Multinucleated keratinocytes with nuclear moulding were seen within the blister cavity. Grocott-Gomori methenamine-silver (GMS), acid-fast, and Gram stains were negative. Granular immunoglobulin (Ig) G, IgM, and C3 were seen intramurally. DNA analysis of vesicular fluid was positive for varicella zoster virus (VZV). A diagnosis of herpes zoster (HZ) of the right S1 dermatome with primary obliterative vasculitis was established.
Immunocompromised people—those who have impaired T-cell immunity (eg, recipients of organ or hematopoietic stem-cell transplants), take immunosuppressive therapy, or have lymphoma, leukemia, or human immunodeficiency virus (HIV) infection—have an increased risk for HZ. For example, in patients with acquired immunodeficiency syndrome (AIDS), HZ uniquely manifests as recurrent shingles. An estimated 20% to 30% of HIV-infected patients will have more than 1 episode of HZ, which may involve the same or different dermatomes.1,2 Furthermore, HZ in this population is more commonly associated with atypical presentations.3
What an atypical presentation may look like
In immunocompromised patients, HZ may present with atypical cutaneous manifestations or with atypical generalized symptoms.
Atypical cutaneous manifestations, as in disseminated zoster, manifest with multiple hyperkeratotic papules (3-20 mm in diameter) that follow no dermatomal pattern. These lesions may be chronic, persisting for months or years, and may be associated with acyclovir-resistant strains of VZV.2,3 Another dermatologic variant is ecthymatous VZV, which manifests with multiple large (10-30 mm) punched-out ulcerations with a central black eschar and a peripheral rim of vesicles.4 Viral folliculitis—in which infection is limited to the hair follicle, with no associated blisters—has also been reported in atypical HZ.5
Our patient presented with hemorrhagic vesicles mimicking vasculitic lesions, which had persisted over a 3-month period with intermittent localized pain. It has been proposed that in atypical presentations, the reactivated VZV spreads transaxonally from adjacent nerves to the outermost adventitial layer of the arterial wall, leading to a vasculitic appearance of the vesicles.6 Viral-induced vasculitis may also result either directly from infection of the blood vessels or secondary to vascular damage from an inflammatory immune complex–mediated reaction, cell-mediated hypersensitivity, or inflammation due to immune dysregulation.7,8
Continue to: Differential includes vesiculobullous conditions
Differential includes vesiculobullous conditions
There are several important items to consider in the differential.
Cutaneous vasculitis, in severe cases, may manifest with vesicles or bullae that resemble the lesions seen in HZ. However, its unilateral nature and distribution distinguish it.
Angioinvasive fungal infections in immunocompromised patients may manifest with scattered ulceronecrotic lesions to purpuric vesiculobullous dermatoses.9 However, no fungal organisms were seen on GMS staining of the biopsied tissue.
Atypical hand-foot-and-mouth disease tends to affect adults and is associated with Coxsackievirus A6 infection.10 It may manifest as generalized vesiculobullous exanthem resembling varicella. The chronic nature and restricted extent of the patient’s rash made this diagnosis unlikely.
Successful management depends on timely identification
Although most cases of HZ can be diagnosed clinically, atypical rashes may require a biopsy and direct immunofluorescence assay for VZV antigen or a polymerase-chain-reaction (PCR) assay for VZV DNA in cells from the base of blisters. Therefore, it is important to consider the diagnosis of HZ in immunocompromised patients presenting with an atypical rash to avoid misdiagnosis and costly testing.
Continue to: Our patient was treated...
Our patient was treated with oral acyclovir 800 mg 5 times/day for 10 days, with prompt resolution of her rash.
CORRESPONDENCE
Joel Hua-Liang Lim, MBBS, MRCP, MMed, 1 Mandalay Road, Singapore 308205; joellimhl@nsc.com.sg
1. LeBoit PE, Limova M, Yen TS, et al. Chronic verrucous varicella-zoster virus infection in patients with the acquired immunodeficiency syndrome (AIDS): histologic and molecular biologic findings. Am J Dermatopathol. 1992;14:1-7.
2. Gnann JW Jr. Varicella-zoster virus: atypical presentations and unusual complications. J Infect Dis. 2002;186(suppl 1):S91-S98.
3. Weinberg JM, Mysliwiec A, Turiansky GW, et al. Viral folliculitis: atypical presentations of herpes simplex, herpes zoster, and molluscum contagiosum. Arch Dermatol. 1997;133:983-986.
4. Gilson IH, Barnett JH, Conant MA, et al. Disseminated ecthymatous herpes varicella zoster virus infection in patients with acquired immunodeficiency syndrome. J Am Acad Dermatol. 1989;20:637-642.
5. Løkke BJ, Weismann K, Mathiesen L, et al. Atypical varicella-zoster infection in AIDS. Acta Derm Venereol. 1993;73:123-125.
6. Uhoda I, Piérard-Franchimont C, Piérard GE. Varicella-zoster virus vasculitis: a case of recurrent varicella without epidermal involvement. Dermatology. 2000;200:173-175.
7. Teng GG, Chatham WW. Vasculitis related to viral and other microbial agents. Best Pract Res Clin Rheumatol. 2015;29:226-243.
8. Nagel MA, Gilden D. Developments in varicella zoster virus vasculopathy. Curr Neurol Neurosci Rep. 2016;16:12.
9. Pfaller MA, Diekema DJ. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol. 2010;36:1-53.
10. Lott JP, Liu K, Landry M-L, et al. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J Am Acad Dermatol. 2013;69:736-741.
1. LeBoit PE, Limova M, Yen TS, et al. Chronic verrucous varicella-zoster virus infection in patients with the acquired immunodeficiency syndrome (AIDS): histologic and molecular biologic findings. Am J Dermatopathol. 1992;14:1-7.
2. Gnann JW Jr. Varicella-zoster virus: atypical presentations and unusual complications. J Infect Dis. 2002;186(suppl 1):S91-S98.
3. Weinberg JM, Mysliwiec A, Turiansky GW, et al. Viral folliculitis: atypical presentations of herpes simplex, herpes zoster, and molluscum contagiosum. Arch Dermatol. 1997;133:983-986.
4. Gilson IH, Barnett JH, Conant MA, et al. Disseminated ecthymatous herpes varicella zoster virus infection in patients with acquired immunodeficiency syndrome. J Am Acad Dermatol. 1989;20:637-642.
5. Løkke BJ, Weismann K, Mathiesen L, et al. Atypical varicella-zoster infection in AIDS. Acta Derm Venereol. 1993;73:123-125.
6. Uhoda I, Piérard-Franchimont C, Piérard GE. Varicella-zoster virus vasculitis: a case of recurrent varicella without epidermal involvement. Dermatology. 2000;200:173-175.
7. Teng GG, Chatham WW. Vasculitis related to viral and other microbial agents. Best Pract Res Clin Rheumatol. 2015;29:226-243.
8. Nagel MA, Gilden D. Developments in varicella zoster virus vasculopathy. Curr Neurol Neurosci Rep. 2016;16:12.
9. Pfaller MA, Diekema DJ. Epidemiology of invasive mycoses in North America. Crit Rev Microbiol. 2010;36:1-53.
10. Lott JP, Liu K, Landry M-L, et al. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J Am Acad Dermatol. 2013;69:736-741.

Painful ulcers on gingiva, tongue, and buccal mucosa
A 29-year-old man with no prior history of mouth sores abruptly developed many 1- to 1.5-mm blisters on the gingiva (FIGURE 1A),tongue (FIGURE 1B), and buccal mucosa (FIGURE 1C), which evolved into small erosions accompanied by a low-grade fever 5 days prior to presentation. The patient had no history of any dermatologic conditions or systemic illnesses and was taking no medication.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Acute primary herpetic gingivostomatitis
Herpes simplex virus (HSV) is the causative agent for acute primary herpetic gingivostomatitis.1 HSV-1 is primarily responsible for oral mucosal infections, while HSV-2 is implicated in most genital and cutaneous lower body lesions.1 Herpetic gingivostomatitis often presents as a sudden vesiculoulcerative eruption anywhere in the mouth, including the perioral skin, vermillion border, gingiva, tongue, or buccal mucosa.2 Associated symptoms include malaise, headache, fever, and cervical lymphadenopathy; however, most occurrences are subclinical or asymptomatic.2
A diagnosis that’s more common in children. Primary HSV occurs in people who have not previously been exposed to the virus. While it is an infection that classically presents in childhood, it is not limited to this group. Manifestations often are more severe in adults.1
Following an incubation period of a few days to 3 weeks, the primary infection typically lasts 10 to 14 days.1,2 Recurrence is highly variable and generally less severe than primary infection, with grouped vesicles often recurring in the same spot with each recurrence on the vermillion border of the lip. Triggers for reactivation include immunosuppression, pregnancy, fever, UV radiation, or trauma.1,2
Differential includes other conditions with mucosal lesions
Acute herpetic gingivostomatitis must be distinguished from other disease processes that cause ulcerative mucosal lesions.
Aphthous stomatitis (canker sores) is the most common ulcerative disease of the oral mucosa.3 It presents as painful, punched-out, shallow ulcers with a yellowish gray pseudomembranous center and surrounding erythema.3 No definitive etiology has been established; however, aphthae often occur after trauma.
Continue to: Herpangina...
Herpangina is caused by coxsackie A virus and primarily is seen in infants and children younger than 5.4 The papulovesicular lesions primarily affect the posterior oral cavity, including the soft palate, anterior tonsillar pillars, and uvula.4
Allergic contact dermatitis is precipitated by contact with an allergen and presents with pain or pruritus. Lesions are erythematous with vesicles, erosions, ulcers, or hyperkeratosis that gradually resolve after withdrawal of the causative allergen.5
Pemphigus vulgaris. Oral ulcerations of the buccal mucosa and gingiva are the first manifestation of pemphigus vulgaris in the majority of patients, with skin blisters occurring months to years later over areas exposed to frictional stress.6 Skin sloughs may be seen in response to frictional stress (Nikolsky sign).6
The new Dx gold standard is PCR
Acute herpetic gingivostomatitis usually is diagnosed by history and hallmark clinical signs and symptoms.1 In this case, our patient presented with a sudden eruption of painful blisters on multiple areas of the oral mucosa associated with fever. The diagnosis can be confirmed by viral culture, serology with anti-HSV IgM and IgG, Tzanck preparation, immunofluorescence, and polymerase chain reaction (PCR).1 Viral culture has been the gold standard for mucosal HSV diagnosis; however, PCR is emerging as the new gold standard because of its unrivaled sensitivity, specificity, and rapid turnaround time.7,8 Specimens for PCR are submitted using a swab of infected cells placed in the same viral transport medium used for HSV cultures.
Our patient’s culture was positive for HSV-1.
Continue to: Prompt use of antivirals is key
Prompt use of antivirals is key
Treatment of acute HSV gingivostomatitis involves symptomatic management with topical anesthetics, oral analgesics, and normal saline rinses.1 Acyclovir is an established therapy; however, it has poor bioavailability and gastrointestinal absorption.1 Valacyclovir has improved bioavailability and is well tolerated.1 For primary herpes gingivostomatitis, we favor 1 g twice daily for 7 days.1 Our patient responded well to this valacyclovir regimen and healed completely in 1 week.
CORRESPONDENCE
Robert T. Brodell, MD, 2500 N State St, Jackson, MS 39216; rbrodell@umc.edu
1. Ajar AH, Chauvin PJ. Acute herpetic gingivostomatitis in adults: a review of 13 cases, including diagnosis and management. J Can Dent Assoc. 2002;68:247-251.
2. George AK, Anil S. Acute herpetic gingivostomatitis associated with herpes simplex virus 2: report of a case. J Int Oral Health. 2014;6:99-102.
3. Akintoye SO, Greenburg MS. Recurrent aphthous stomatitis. Dent Clin N Am. 2014;58:281-297.
4. Scott LA, Stone MS. Viral exanthems. Dermatol Online J. 2003;9:4.
5. Feller L, Wood NH, Khammissa RA, et al. Review: allergic contact stomatitis. Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;123:559-565.
6. Bascones-Martinez A, Munoz-Corcuera M, Bascones-Ilundain C, et al. Oral manifestations of pemphigus vulgaris: clinical presentation, differential diagnosis and management. J Clin Exp Dermatol Res. 2010;1:112.
7. LeGoff J, Péré H, Bélec L. Diagnosis of genital herpes simplex virus infection in the clinical laboratory. Virol J. 2014;11:83.
8. Centers for Disease Control and Prevention. Genital HSV infections. www.cdc.gov/std/tg2015/herpes.htm. Updated June 4, 2015. Accessed September 26, 2019.
A 29-year-old man with no prior history of mouth sores abruptly developed many 1- to 1.5-mm blisters on the gingiva (FIGURE 1A),tongue (FIGURE 1B), and buccal mucosa (FIGURE 1C), which evolved into small erosions accompanied by a low-grade fever 5 days prior to presentation. The patient had no history of any dermatologic conditions or systemic illnesses and was taking no medication.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Acute primary herpetic gingivostomatitis
Herpes simplex virus (HSV) is the causative agent for acute primary herpetic gingivostomatitis.1 HSV-1 is primarily responsible for oral mucosal infections, while HSV-2 is implicated in most genital and cutaneous lower body lesions.1 Herpetic gingivostomatitis often presents as a sudden vesiculoulcerative eruption anywhere in the mouth, including the perioral skin, vermillion border, gingiva, tongue, or buccal mucosa.2 Associated symptoms include malaise, headache, fever, and cervical lymphadenopathy; however, most occurrences are subclinical or asymptomatic.2
A diagnosis that’s more common in children. Primary HSV occurs in people who have not previously been exposed to the virus. While it is an infection that classically presents in childhood, it is not limited to this group. Manifestations often are more severe in adults.1
Following an incubation period of a few days to 3 weeks, the primary infection typically lasts 10 to 14 days.1,2 Recurrence is highly variable and generally less severe than primary infection, with grouped vesicles often recurring in the same spot with each recurrence on the vermillion border of the lip. Triggers for reactivation include immunosuppression, pregnancy, fever, UV radiation, or trauma.1,2
Differential includes other conditions with mucosal lesions
Acute herpetic gingivostomatitis must be distinguished from other disease processes that cause ulcerative mucosal lesions.
Aphthous stomatitis (canker sores) is the most common ulcerative disease of the oral mucosa.3 It presents as painful, punched-out, shallow ulcers with a yellowish gray pseudomembranous center and surrounding erythema.3 No definitive etiology has been established; however, aphthae often occur after trauma.
Continue to: Herpangina...
Herpangina is caused by coxsackie A virus and primarily is seen in infants and children younger than 5.4 The papulovesicular lesions primarily affect the posterior oral cavity, including the soft palate, anterior tonsillar pillars, and uvula.4
Allergic contact dermatitis is precipitated by contact with an allergen and presents with pain or pruritus. Lesions are erythematous with vesicles, erosions, ulcers, or hyperkeratosis that gradually resolve after withdrawal of the causative allergen.5
Pemphigus vulgaris. Oral ulcerations of the buccal mucosa and gingiva are the first manifestation of pemphigus vulgaris in the majority of patients, with skin blisters occurring months to years later over areas exposed to frictional stress.6 Skin sloughs may be seen in response to frictional stress (Nikolsky sign).6
The new Dx gold standard is PCR
Acute herpetic gingivostomatitis usually is diagnosed by history and hallmark clinical signs and symptoms.1 In this case, our patient presented with a sudden eruption of painful blisters on multiple areas of the oral mucosa associated with fever. The diagnosis can be confirmed by viral culture, serology with anti-HSV IgM and IgG, Tzanck preparation, immunofluorescence, and polymerase chain reaction (PCR).1 Viral culture has been the gold standard for mucosal HSV diagnosis; however, PCR is emerging as the new gold standard because of its unrivaled sensitivity, specificity, and rapid turnaround time.7,8 Specimens for PCR are submitted using a swab of infected cells placed in the same viral transport medium used for HSV cultures.
Our patient’s culture was positive for HSV-1.
Continue to: Prompt use of antivirals is key
Prompt use of antivirals is key
Treatment of acute HSV gingivostomatitis involves symptomatic management with topical anesthetics, oral analgesics, and normal saline rinses.1 Acyclovir is an established therapy; however, it has poor bioavailability and gastrointestinal absorption.1 Valacyclovir has improved bioavailability and is well tolerated.1 For primary herpes gingivostomatitis, we favor 1 g twice daily for 7 days.1 Our patient responded well to this valacyclovir regimen and healed completely in 1 week.
CORRESPONDENCE
Robert T. Brodell, MD, 2500 N State St, Jackson, MS 39216; rbrodell@umc.edu
A 29-year-old man with no prior history of mouth sores abruptly developed many 1- to 1.5-mm blisters on the gingiva (FIGURE 1A),tongue (FIGURE 1B), and buccal mucosa (FIGURE 1C), which evolved into small erosions accompanied by a low-grade fever 5 days prior to presentation. The patient had no history of any dermatologic conditions or systemic illnesses and was taking no medication.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Acute primary herpetic gingivostomatitis
Herpes simplex virus (HSV) is the causative agent for acute primary herpetic gingivostomatitis.1 HSV-1 is primarily responsible for oral mucosal infections, while HSV-2 is implicated in most genital and cutaneous lower body lesions.1 Herpetic gingivostomatitis often presents as a sudden vesiculoulcerative eruption anywhere in the mouth, including the perioral skin, vermillion border, gingiva, tongue, or buccal mucosa.2 Associated symptoms include malaise, headache, fever, and cervical lymphadenopathy; however, most occurrences are subclinical or asymptomatic.2
A diagnosis that’s more common in children. Primary HSV occurs in people who have not previously been exposed to the virus. While it is an infection that classically presents in childhood, it is not limited to this group. Manifestations often are more severe in adults.1
Following an incubation period of a few days to 3 weeks, the primary infection typically lasts 10 to 14 days.1,2 Recurrence is highly variable and generally less severe than primary infection, with grouped vesicles often recurring in the same spot with each recurrence on the vermillion border of the lip. Triggers for reactivation include immunosuppression, pregnancy, fever, UV radiation, or trauma.1,2
Differential includes other conditions with mucosal lesions
Acute herpetic gingivostomatitis must be distinguished from other disease processes that cause ulcerative mucosal lesions.
Aphthous stomatitis (canker sores) is the most common ulcerative disease of the oral mucosa.3 It presents as painful, punched-out, shallow ulcers with a yellowish gray pseudomembranous center and surrounding erythema.3 No definitive etiology has been established; however, aphthae often occur after trauma.
Continue to: Herpangina...
Herpangina is caused by coxsackie A virus and primarily is seen in infants and children younger than 5.4 The papulovesicular lesions primarily affect the posterior oral cavity, including the soft palate, anterior tonsillar pillars, and uvula.4
Allergic contact dermatitis is precipitated by contact with an allergen and presents with pain or pruritus. Lesions are erythematous with vesicles, erosions, ulcers, or hyperkeratosis that gradually resolve after withdrawal of the causative allergen.5
Pemphigus vulgaris. Oral ulcerations of the buccal mucosa and gingiva are the first manifestation of pemphigus vulgaris in the majority of patients, with skin blisters occurring months to years later over areas exposed to frictional stress.6 Skin sloughs may be seen in response to frictional stress (Nikolsky sign).6
The new Dx gold standard is PCR
Acute herpetic gingivostomatitis usually is diagnosed by history and hallmark clinical signs and symptoms.1 In this case, our patient presented with a sudden eruption of painful blisters on multiple areas of the oral mucosa associated with fever. The diagnosis can be confirmed by viral culture, serology with anti-HSV IgM and IgG, Tzanck preparation, immunofluorescence, and polymerase chain reaction (PCR).1 Viral culture has been the gold standard for mucosal HSV diagnosis; however, PCR is emerging as the new gold standard because of its unrivaled sensitivity, specificity, and rapid turnaround time.7,8 Specimens for PCR are submitted using a swab of infected cells placed in the same viral transport medium used for HSV cultures.
Our patient’s culture was positive for HSV-1.
Continue to: Prompt use of antivirals is key
Prompt use of antivirals is key
Treatment of acute HSV gingivostomatitis involves symptomatic management with topical anesthetics, oral analgesics, and normal saline rinses.1 Acyclovir is an established therapy; however, it has poor bioavailability and gastrointestinal absorption.1 Valacyclovir has improved bioavailability and is well tolerated.1 For primary herpes gingivostomatitis, we favor 1 g twice daily for 7 days.1 Our patient responded well to this valacyclovir regimen and healed completely in 1 week.
CORRESPONDENCE
Robert T. Brodell, MD, 2500 N State St, Jackson, MS 39216; rbrodell@umc.edu
1. Ajar AH, Chauvin PJ. Acute herpetic gingivostomatitis in adults: a review of 13 cases, including diagnosis and management. J Can Dent Assoc. 2002;68:247-251.
2. George AK, Anil S. Acute herpetic gingivostomatitis associated with herpes simplex virus 2: report of a case. J Int Oral Health. 2014;6:99-102.
3. Akintoye SO, Greenburg MS. Recurrent aphthous stomatitis. Dent Clin N Am. 2014;58:281-297.
4. Scott LA, Stone MS. Viral exanthems. Dermatol Online J. 2003;9:4.
5. Feller L, Wood NH, Khammissa RA, et al. Review: allergic contact stomatitis. Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;123:559-565.
6. Bascones-Martinez A, Munoz-Corcuera M, Bascones-Ilundain C, et al. Oral manifestations of pemphigus vulgaris: clinical presentation, differential diagnosis and management. J Clin Exp Dermatol Res. 2010;1:112.
7. LeGoff J, Péré H, Bélec L. Diagnosis of genital herpes simplex virus infection in the clinical laboratory. Virol J. 2014;11:83.
8. Centers for Disease Control and Prevention. Genital HSV infections. www.cdc.gov/std/tg2015/herpes.htm. Updated June 4, 2015. Accessed September 26, 2019.
1. Ajar AH, Chauvin PJ. Acute herpetic gingivostomatitis in adults: a review of 13 cases, including diagnosis and management. J Can Dent Assoc. 2002;68:247-251.
2. George AK, Anil S. Acute herpetic gingivostomatitis associated with herpes simplex virus 2: report of a case. J Int Oral Health. 2014;6:99-102.
3. Akintoye SO, Greenburg MS. Recurrent aphthous stomatitis. Dent Clin N Am. 2014;58:281-297.
4. Scott LA, Stone MS. Viral exanthems. Dermatol Online J. 2003;9:4.
5. Feller L, Wood NH, Khammissa RA, et al. Review: allergic contact stomatitis. Oral Surg Oral Med Oral Pathol Oral Radiol. 2017;123:559-565.
6. Bascones-Martinez A, Munoz-Corcuera M, Bascones-Ilundain C, et al. Oral manifestations of pemphigus vulgaris: clinical presentation, differential diagnosis and management. J Clin Exp Dermatol Res. 2010;1:112.
7. LeGoff J, Péré H, Bélec L. Diagnosis of genital herpes simplex virus infection in the clinical laboratory. Virol J. 2014;11:83.
8. Centers for Disease Control and Prevention. Genital HSV infections. www.cdc.gov/std/tg2015/herpes.htm. Updated June 4, 2015. Accessed September 26, 2019.

Rash with weakness

The FP diagnosed dermatomyositis based on the patient’s proximal muscle weakness along with the typical shawl sign (erythema and scale over the shawl distribution) seen in the top image and the Gottron papules (erythematous and scaling papules over the dorsum of the fingers) and holster sign (erythema and scale over the lateral hip region) seen in the bottom image.
The FP started the patient on prednisone 60 mg/d and topical steroids for the affected areas. Laboratory exams showed an elevated creatinine kinase and mildly elevated antinuclear antibodies (typical for dermatomyositis). The FP also started the patient on oral prednisone 40 mg/d while putting in referrals for Dermatology and Rheumatology.
Two weeks later, the patient felt stronger and the rash had faded. Dermatology saw her first and started her on a steroid-sparing agent, methotrexate, with plans to taper her prednisone to lower doses over time. As underlying malignancy may precipitate dermatomyositis, the patient was screened for internal cancers—especially ovarian cancer. Fortunately, the mammogram, Pap smear, colonoscopy, and thoracic, abdominal, and pelvic computed tomography scans all were normal.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Buckley J, Burks M, Allred A, Usatine R. Dermatomyositis. In: Usatine R, Smith M, Mayeaux EJ, et al. eds. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1194-1203.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the 3rd edition of the Color Atlas and Synopsis of Family Medicine as an app by clicking on this link: https://usatinemedia.com/app/color-atlas-of-family-medicine/
The FP diagnosed dermatomyositis based on the patient’s proximal muscle weakness along with the typical shawl sign (erythema and scale over the shawl distribution) seen in the top image and the Gottron papules (erythematous and scaling papules over the dorsum of the fingers) and holster sign (erythema and scale over the lateral hip region) seen in the bottom image.
The FP started the patient on prednisone 60 mg/d and topical steroids for the affected areas. Laboratory exams showed an elevated creatinine kinase and mildly elevated antinuclear antibodies (typical for dermatomyositis). The FP also started the patient on oral prednisone 40 mg/d while putting in referrals for Dermatology and Rheumatology.
Two weeks later, the patient felt stronger and the rash had faded. Dermatology saw her first and started her on a steroid-sparing agent, methotrexate, with plans to taper her prednisone to lower doses over time. As underlying malignancy may precipitate dermatomyositis, the patient was screened for internal cancers—especially ovarian cancer. Fortunately, the mammogram, Pap smear, colonoscopy, and thoracic, abdominal, and pelvic computed tomography scans all were normal.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Buckley J, Burks M, Allred A, Usatine R. Dermatomyositis. In: Usatine R, Smith M, Mayeaux EJ, et al. eds. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1194-1203.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the 3rd edition of the Color Atlas and Synopsis of Family Medicine as an app by clicking on this link: https://usatinemedia.com/app/color-atlas-of-family-medicine/
The FP diagnosed dermatomyositis based on the patient’s proximal muscle weakness along with the typical shawl sign (erythema and scale over the shawl distribution) seen in the top image and the Gottron papules (erythematous and scaling papules over the dorsum of the fingers) and holster sign (erythema and scale over the lateral hip region) seen in the bottom image.
The FP started the patient on prednisone 60 mg/d and topical steroids for the affected areas. Laboratory exams showed an elevated creatinine kinase and mildly elevated antinuclear antibodies (typical for dermatomyositis). The FP also started the patient on oral prednisone 40 mg/d while putting in referrals for Dermatology and Rheumatology.
Two weeks later, the patient felt stronger and the rash had faded. Dermatology saw her first and started her on a steroid-sparing agent, methotrexate, with plans to taper her prednisone to lower doses over time. As underlying malignancy may precipitate dermatomyositis, the patient was screened for internal cancers—especially ovarian cancer. Fortunately, the mammogram, Pap smear, colonoscopy, and thoracic, abdominal, and pelvic computed tomography scans all were normal.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Buckley J, Burks M, Allred A, Usatine R. Dermatomyositis. In: Usatine R, Smith M, Mayeaux EJ, et al. eds. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1194-1203.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the 3rd edition of the Color Atlas and Synopsis of Family Medicine as an app by clicking on this link: https://usatinemedia.com/app/color-atlas-of-family-medicine/

Scars and color changes on face

The FP recognized the scarring, skin atrophy, erythema and hyperpigmentation in the malar distribution as discoid lupus erythematosus (DLE). Upon examination, the FP noted similar patterns (with the addition of hypopigmentation) in the conchal bowls of both pinna. This was confirmatory for the DLE diagnosis (AKA chronic cutaneous lupus). (If in doubt, a 4-mm punch biopsy could be performed on the lesions of the face.) Without the lesions in the ears, sarcoidosis might have been considered as part of the differential diagnosis.
DLE lesions are characterized by discrete, erythematous, slightly infiltrated papules or plaques. Hypopigmentation develops in the central area and hyperpigmentation develops at the active border. Resolution of the active lesion results in atrophy and scarring.
In this case, the FP explained the diagnosis of DLE to the patient and ordered testing for antinuclear antibodies (ANA), a complete blood count, and comprehensive metabolic panel. All the labs were normal, and the ANA was negative, which confirmed that this was not a case of systemic lupus erythematosus. Patients with only DLE generally have negative or low-titer ANA titers.
DLE therapy includes corticosteroids (topical or intralesional) and oral antimalarials, such as hydroxychloroquine. The FP in this case started the patient on hydroxychloroquine 200 mg bid and topical triamcinolone 0.1% applied twice daily to the erythematous portions of the cheeks for 2 weeks. At the 2-week follow-up, the patient’s skin looked less red and inflamed and she was happy with the improved appearance of her skin. Patients on hydroxychloroquine need a baseline eye exam by an ophthalmologist and then yearly exams after 5 years of therapy to detect any retinal or visual field problems.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Pye A, Mayeaux, EJ, Mishra V, et al. Lupus. In: Usatine R, Smith M, Mayeaux EJ, et al. eds. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1183-1193.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the 3rd edition of the Color Atlas and Synopsis of Family Medicine as an app by clicking on this link: https://usatinemedia.com/app/color-atlas-of-family-medicine/
The FP recognized the scarring, skin atrophy, erythema and hyperpigmentation in the malar distribution as discoid lupus erythematosus (DLE). Upon examination, the FP noted similar patterns (with the addition of hypopigmentation) in the conchal bowls of both pinna. This was confirmatory for the DLE diagnosis (AKA chronic cutaneous lupus). (If in doubt, a 4-mm punch biopsy could be performed on the lesions of the face.) Without the lesions in the ears, sarcoidosis might have been considered as part of the differential diagnosis.
DLE lesions are characterized by discrete, erythematous, slightly infiltrated papules or plaques. Hypopigmentation develops in the central area and hyperpigmentation develops at the active border. Resolution of the active lesion results in atrophy and scarring.
In this case, the FP explained the diagnosis of DLE to the patient and ordered testing for antinuclear antibodies (ANA), a complete blood count, and comprehensive metabolic panel. All the labs were normal, and the ANA was negative, which confirmed that this was not a case of systemic lupus erythematosus. Patients with only DLE generally have negative or low-titer ANA titers.
DLE therapy includes corticosteroids (topical or intralesional) and oral antimalarials, such as hydroxychloroquine. The FP in this case started the patient on hydroxychloroquine 200 mg bid and topical triamcinolone 0.1% applied twice daily to the erythematous portions of the cheeks for 2 weeks. At the 2-week follow-up, the patient’s skin looked less red and inflamed and she was happy with the improved appearance of her skin. Patients on hydroxychloroquine need a baseline eye exam by an ophthalmologist and then yearly exams after 5 years of therapy to detect any retinal or visual field problems.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Pye A, Mayeaux, EJ, Mishra V, et al. Lupus. In: Usatine R, Smith M, Mayeaux EJ, et al. eds. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1183-1193.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the 3rd edition of the Color Atlas and Synopsis of Family Medicine as an app by clicking on this link: https://usatinemedia.com/app/color-atlas-of-family-medicine/
The FP recognized the scarring, skin atrophy, erythema and hyperpigmentation in the malar distribution as discoid lupus erythematosus (DLE). Upon examination, the FP noted similar patterns (with the addition of hypopigmentation) in the conchal bowls of both pinna. This was confirmatory for the DLE diagnosis (AKA chronic cutaneous lupus). (If in doubt, a 4-mm punch biopsy could be performed on the lesions of the face.) Without the lesions in the ears, sarcoidosis might have been considered as part of the differential diagnosis.
DLE lesions are characterized by discrete, erythematous, slightly infiltrated papules or plaques. Hypopigmentation develops in the central area and hyperpigmentation develops at the active border. Resolution of the active lesion results in atrophy and scarring.
In this case, the FP explained the diagnosis of DLE to the patient and ordered testing for antinuclear antibodies (ANA), a complete blood count, and comprehensive metabolic panel. All the labs were normal, and the ANA was negative, which confirmed that this was not a case of systemic lupus erythematosus. Patients with only DLE generally have negative or low-titer ANA titers.
DLE therapy includes corticosteroids (topical or intralesional) and oral antimalarials, such as hydroxychloroquine. The FP in this case started the patient on hydroxychloroquine 200 mg bid and topical triamcinolone 0.1% applied twice daily to the erythematous portions of the cheeks for 2 weeks. At the 2-week follow-up, the patient’s skin looked less red and inflamed and she was happy with the improved appearance of her skin. Patients on hydroxychloroquine need a baseline eye exam by an ophthalmologist and then yearly exams after 5 years of therapy to detect any retinal or visual field problems.
Photos and text for Photo Rounds Friday courtesy of Richard P. Usatine, MD. This case was adapted from: Pye A, Mayeaux, EJ, Mishra V, et al. Lupus. In: Usatine R, Smith M, Mayeaux EJ, et al. eds. Color Atlas and Synopsis of Family Medicine. 3rd ed. New York, NY: McGraw-Hill; 2019:1183-1193.
To learn more about the newest 3rd edition of the Color Atlas and Synopsis of Family Medicine, see: https://www.amazon.com/Color-Atlas-Synopsis-Family-Medicine/dp/1259862046/
You can get the 3rd edition of the Color Atlas and Synopsis of Family Medicine as an app by clicking on this link: https://usatinemedia.com/app/color-atlas-of-family-medicine/

Long-term opioid use more common in hidradenitis suppurativa
, in a retrospective cohort study.
“These results suggest that periodic assessment of pain and screening for long-term opioid use may be warranted, particularly among patients who are older, who smoke tobacco, or who have depression and other medical comorbidities,” wrote the authors of the study (JAMA Dermatol. 2019 Sep 11. doi: 10.1001/jamadermatol.2019.2610).
Researchers led by Sarah Reddy, BA, of the Zucker School of Medicine at Hofstra/ Northwell, New Hyde Park, N.Y., used data from a health-care database that represents an estimated 17% of the U.S. population. They focused on opioid-naive adults who were in the database for at least 3 years from 2008-2018 and monitored whether they began opioid use and then maintained use for at least 1 year.
Nearly 829,000 patients were in the control group, and 22,277 were in the HS group. The mean age of those with HS was 41 years, 76% were women, and 59% were white.
Over 1 year, the crude incidence of long-term opioid use among HS patients who were opioid naive was 0.33%, compared with 0.14% of controls (P less than .001).
An analysis, adjusted for potential confounding factors, found that compared with controls, those with HS were more likely to develop long-term opioid use (odds ratio [OR], 1.53, 95% confidence interval, 1.20-1.95; P less than .001). In the adjusted analysis, long-term opioid use was increased among those in the HS group who had ever smoked tobacco (OR, 3.64, 95% CI, 2.06-6.41; P less than .001), compared with patients with HS who had never smoked; and those who had a history of depression (OR, 1.97, 95% CI, 1.21-3.19; P = .006), compared with HS patients who had not had depression.
The risk of long-term opioid use among those with HS increased by 2% with each additional year in age.
In addition, 5% of patients with HS and long-term opioid use were diagnosed with opioid use disorder over the study period. “Sex, race/ethnicity, disease duration, established dermatologic care, alcohol abuse, and nonopioid substance abuse were not associated with increased risk of long-term opioid use among patients with HS,” the authors wrote.
Emphasizing that these results “should not further stigmatize” people with HS, they said, “our hope is that the medical community, including dermatologists, will further embrace and engage in an integrated care plan that comprehensively supports the needs of patients with HS, including pain management.”
Future research, they added, “should include evaluating the association between disease severity and risk of opioid use, the role of disease-modifying therapies in reducing opioid use, and the development of effective and appropriate multimodal pain management strategies for HS.”
An educational grant to a study author from AbbVie partially funded the study. No other study funding was reported. Ms. Reddy had no disclosures; one author disclosed having received grants and personal fees from AbbVie and UCB during the study.
SOURCE: Reddy S et al. JAMA Dermatol. 2019 Sep 11. doi: 10.1001/jamadermatol.2019.2610.
, in a retrospective cohort study.
“These results suggest that periodic assessment of pain and screening for long-term opioid use may be warranted, particularly among patients who are older, who smoke tobacco, or who have depression and other medical comorbidities,” wrote the authors of the study (JAMA Dermatol. 2019 Sep 11. doi: 10.1001/jamadermatol.2019.2610).
Researchers led by Sarah Reddy, BA, of the Zucker School of Medicine at Hofstra/ Northwell, New Hyde Park, N.Y., used data from a health-care database that represents an estimated 17% of the U.S. population. They focused on opioid-naive adults who were in the database for at least 3 years from 2008-2018 and monitored whether they began opioid use and then maintained use for at least 1 year.
Nearly 829,000 patients were in the control group, and 22,277 were in the HS group. The mean age of those with HS was 41 years, 76% were women, and 59% were white.
Over 1 year, the crude incidence of long-term opioid use among HS patients who were opioid naive was 0.33%, compared with 0.14% of controls (P less than .001).
An analysis, adjusted for potential confounding factors, found that compared with controls, those with HS were more likely to develop long-term opioid use (odds ratio [OR], 1.53, 95% confidence interval, 1.20-1.95; P less than .001). In the adjusted analysis, long-term opioid use was increased among those in the HS group who had ever smoked tobacco (OR, 3.64, 95% CI, 2.06-6.41; P less than .001), compared with patients with HS who had never smoked; and those who had a history of depression (OR, 1.97, 95% CI, 1.21-3.19; P = .006), compared with HS patients who had not had depression.
The risk of long-term opioid use among those with HS increased by 2% with each additional year in age.
In addition, 5% of patients with HS and long-term opioid use were diagnosed with opioid use disorder over the study period. “Sex, race/ethnicity, disease duration, established dermatologic care, alcohol abuse, and nonopioid substance abuse were not associated with increased risk of long-term opioid use among patients with HS,” the authors wrote.
Emphasizing that these results “should not further stigmatize” people with HS, they said, “our hope is that the medical community, including dermatologists, will further embrace and engage in an integrated care plan that comprehensively supports the needs of patients with HS, including pain management.”
Future research, they added, “should include evaluating the association between disease severity and risk of opioid use, the role of disease-modifying therapies in reducing opioid use, and the development of effective and appropriate multimodal pain management strategies for HS.”
An educational grant to a study author from AbbVie partially funded the study. No other study funding was reported. Ms. Reddy had no disclosures; one author disclosed having received grants and personal fees from AbbVie and UCB during the study.
SOURCE: Reddy S et al. JAMA Dermatol. 2019 Sep 11. doi: 10.1001/jamadermatol.2019.2610.
, in a retrospective cohort study.
“These results suggest that periodic assessment of pain and screening for long-term opioid use may be warranted, particularly among patients who are older, who smoke tobacco, or who have depression and other medical comorbidities,” wrote the authors of the study (JAMA Dermatol. 2019 Sep 11. doi: 10.1001/jamadermatol.2019.2610).
Researchers led by Sarah Reddy, BA, of the Zucker School of Medicine at Hofstra/ Northwell, New Hyde Park, N.Y., used data from a health-care database that represents an estimated 17% of the U.S. population. They focused on opioid-naive adults who were in the database for at least 3 years from 2008-2018 and monitored whether they began opioid use and then maintained use for at least 1 year.
Nearly 829,000 patients were in the control group, and 22,277 were in the HS group. The mean age of those with HS was 41 years, 76% were women, and 59% were white.
Over 1 year, the crude incidence of long-term opioid use among HS patients who were opioid naive was 0.33%, compared with 0.14% of controls (P less than .001).
An analysis, adjusted for potential confounding factors, found that compared with controls, those with HS were more likely to develop long-term opioid use (odds ratio [OR], 1.53, 95% confidence interval, 1.20-1.95; P less than .001). In the adjusted analysis, long-term opioid use was increased among those in the HS group who had ever smoked tobacco (OR, 3.64, 95% CI, 2.06-6.41; P less than .001), compared with patients with HS who had never smoked; and those who had a history of depression (OR, 1.97, 95% CI, 1.21-3.19; P = .006), compared with HS patients who had not had depression.
The risk of long-term opioid use among those with HS increased by 2% with each additional year in age.
In addition, 5% of patients with HS and long-term opioid use were diagnosed with opioid use disorder over the study period. “Sex, race/ethnicity, disease duration, established dermatologic care, alcohol abuse, and nonopioid substance abuse were not associated with increased risk of long-term opioid use among patients with HS,” the authors wrote.
Emphasizing that these results “should not further stigmatize” people with HS, they said, “our hope is that the medical community, including dermatologists, will further embrace and engage in an integrated care plan that comprehensively supports the needs of patients with HS, including pain management.”
Future research, they added, “should include evaluating the association between disease severity and risk of opioid use, the role of disease-modifying therapies in reducing opioid use, and the development of effective and appropriate multimodal pain management strategies for HS.”
An educational grant to a study author from AbbVie partially funded the study. No other study funding was reported. Ms. Reddy had no disclosures; one author disclosed having received grants and personal fees from AbbVie and UCB during the study.
SOURCE: Reddy S et al. JAMA Dermatol. 2019 Sep 11. doi: 10.1001/jamadermatol.2019.2610.
FROM JAMA DERMATOLOGY
Cardiovascular complications of systemic sclerosis: What to look for
Autoimmune rheumatic diseases increase the risk of cardiovascular disease. In rheumatoid arthritis and systemic lupus erythematosus, the risk is driven primarily by the inflammatory milieu, leading to accelerated coronary and cerebrovascular atherosclerosis independent of traditional atherosclerotic risk factors.1–3 The extent of cardiovascular involvement in other rheumatologic diseases has been less well characterized but is an area of growing interest.
In this review, we focus on the cardiovascular complications of systemic sclerosis and review recommendations for monitoring these patients in clinical practice.
SYSTEMIC SCLEROSIS, AN AUTOIMMUNE RHEUMATIC DISEASE
Systemic sclerosis is an autoimmune rheumatic disease characterized by excessive extracellular matrix deposition leading to diffuse fibrosis, endothelial dysfunction, and microvascular injury. It is most common in North America, Southern Europe, and Australia,4,5 and it affects women more than men in ratios ranging from 3:1 to 14:1.6 The mean age at diagnosis is around 50.
The disease can affect the lungs (interstitial lung disease and pulmonary hypertension), the heart, the kidneys, and the gastrointestinal tract.
Systemic sclerosis has 2 main subtypes: limited cutaneous systemic sclerosis, formerly called CREST syndrome) and diffuse cutaneous systemic sclerosis. The limited cutaneous subtype is characterized by tightening of the skin of the distal extremities (below the elbows and knees) and face, while diffuse cutaneous systemic sclerosis can manifest as more extensive skin tightening also involving proximal extremities and the trunk. Both subtypes can have an effect on the cardiovascular system.
Some cardiovascular risk factors such as dyslipidemia, diabetes mellitus, and high body mass index are less common in patients with systemic sclerosis than in patients with rheumatoid arthritis, while the rates of arterial hypertension, smoking, chronic obstructive pulmonary disease, osteoporosis, and neoplasms are similar between the 2 groups.7
HEART INVOLVEMENT HAS SERIOUS CONSEQUENCES
Overt cardiac involvement in systemic sclerosis is associated with a mortality rate of up to 70% over 5 years,8,9 and about one-fourth of deaths in patients with systemic sclerosis are from cardiac causes.10,11 Studies in Europe10,12 showed that many patients with systemic sclerosis have cardiac involvement detectable by magnetic resonance imaging even if they do not have clinical disease. Pulmonary arterial hypertension (PAH) is a complication of both subtypes of systemic sclerosis and portends a higher risk of death.8
Thus, it is critical for clinicians to understand the potential comorbid conditions associated with systemic sclerosis, particularly the cardiovascular ones, and to work closely with cardiologists to help optimize the evaluation and management.
MECHANISMS OF CARDIAC DISEASE IN SYSTEMIC SCLEROSIS

Abnormal vasoreactivity, a consequence of an imbalance between endothelium-derived vasoconstrictors and vasodilators, defective angiogenesis, and endothelial injury, leads to tissue ischemia and vascular endothelial growth factor expression, which initiates injury and fibrosis in the myocardium and in other organs.14–17 Fibrosis involves the myocardium, pericardium, and conduction system.13,18
Myocardial involvement in systemic sclerosis is thought to be due mainly to abnormal vasoreactivity and microvascular abnormalities such as transient coronary artery spasm leading to repeated focal ischemia.19,20 Abnormal vasoreactivity has been demonstrated during cardiac catheterization21: while mean coronary sinus blood flow in systemic sclerosis patients was normal at rest, vasodilator reserve was significantly reduced in patients with diffuse cutaneous systemic sclerosis after maximal vasodilation with dipyridamole. Additionally, endomyocardial biopsy showed fibrosis and concentric intimal hypertrophy with normal epicardial coronary arteries.21
More research into other mechanisms of cardiovascular disease in systemic sclerosis is needed to allow for better preventive care for these patients.
PULMONARY ARTERIAL HYPERTENSION
Systemic sclerosis can be associated with World Health Organization (WHO) groups 1, 2, 3, and 4 pulmonary hypertension. WHO group 1, called pulmonary arterial hypertension or PAH, is one of the most common cardiac complications of systemic sclerosis, with a reported prevalence as high as 12%.22 Systemic sclerosis-associated PAH carries a high mortality rate, with a mean survival of only 3 years.23
With advances in treatments for other complications of systemic sclerosis, the percentage of systemic sclerosis patients who die of PAH has increased from 6% to 33%.24
Compared with patients with idiopathic PAH, those with systemic sclerosis get less of a response from therapy and have poorer outcomes despite lower mean pulmonary artery pressures and similar reductions in cardiac index. However, recent studies have suggested that with aggressive treatment, patients with systemic sclerosis-related PAH can achieve outcomes similar to those with idiopathic PAH.25 Thus, recognizing this condition early is imperative.
Pulmonary arterial hypertension defined
PAH is defined as the combination of all of the following26:
- Mean pulmonary artery pressure > 20 mm Hg at rest
- Normal pulmonary capillary wedge pressure (≤ 15 mm Hg)
- Pulmonary vascular resistance ≥ 3 Wood units on right heart catheterization.
Other causes of pulmonary hypertension such as interstitial lung disease, chronic pulmonary thromboembolic disease, and left heart disease must be excluded.24,27
Remodeling in the pulmonary arteries
The events that lead to PAH in systemic sclerosis remain unclear but are believed to involve initial inflammation or endothelial injury that leads to a dysequilibrium between proliferative mediators and antiproliferative vasodilators. This dysequilibrium, along with endothelial dysfunction, causes an obliterative vasculopathy in the pulmonary artery branches and arterioles. Sympathetic overactivity, hypoxemia, and ischemia-reperfusion injury additionally promote vascular proliferation, fibrosis, and remodeling, leading to increased pulmonary vascular resistance, PAH, and increased right ventricular pressures.23,27
The subtype of systemic sclerosis is an important factor in the development and progression of PAH. PAH appears to be the major cause of death in limited cutaneous systemic sclerosis, while interstitial lung disease is the major cause of death in diffuse cutaneous systemic sclerosis.28
Pulmonary arterial hypertension is a late complication of systemic sclerosis
Data from the South Australian Scleroderma Registry29 revealed that PAH tends to be a late complication of systemic sclerosis, occurring around 20 years after disease onset. In this study of 608 patients, no patient with diffuse cutaneous systemic sclerosis developed PAH.
Systemic sclerosis-related PAH initially follows an indolent course with few symptoms until right ventricular function deteriorates. Early in the disease, patients may experience nonspecific symptoms of fatigue, lightheadedness, and dyspnea on exertion.23 As it progresses, they tend to have worsening dyspnea and may experience exertional syncope, palpitations, and chest pain.
Physical findings may suggest elevated right ventricular pressure and right ventricular failure; these include a loud P2, a prominent jugular a wave, a tricuspid regurgitant murmur, jugular venous distention, and lower-extremity edema.27
Screening for pulmonary arterial hypertension in systemic sclerosis
Significant signs and symptoms usually occur late in the disease; thus, it is important to appropriately screen patients who are at risk so that they can begin aggressive treatment.
Doppler echocardiography is recommended by European and American guidelines to screen for PAH in patients who have systemic sclerosis, and most agree that screening is appropriate even if the patient has no symptoms.30 European consensus documents recommend that transthoracic echocardiography be done annually for the first 5 years of disease and be continued every year in patients at high risk, ie, those with anticentromere antibodies, anti-Th/To antibodies, or interstitial lung disease. Patients not at high risk of developing pulmonary hypertension should also have regular transthoracic echocardiography, though the exact timing is not defined.31 While American societies have not issued corresponding recommendations, many experts follow the European recommendations.
Worrisome features on echocardiography in asymptomatic patients should be followed up with right heart catheterization to assess mean right ventricular pressure. These include:
- Estimated right ventricular systolic pressure ≥ 40 mm Hg
- Tricuspid regurgitant jet velocity > 2.8 m/s
- Right atrial enlargement > 53 mm
- Right ventricular enlargement (mid-cavity dimension > 35 mm).32
Although echocardiography is the most common form of screening, it gives only an estimate of right ventricular systolic pressure, which is imprecise. Other noninvasive markers are helpful and necessary to appropriately screen this population.
Diffusion capacity. The Itinerair study33 found that a diffusing capacity for carbon monoxide (DLCO) of 60% or higher has a high specificity in excluding PAH.
Uric acid has been found to be elevated in patients with systemic sclerosis-related PAH, and levels inversely correlate with 6-minute walking distance.34
Other predictors. N-terminal pro-B-type natriuretic peptide (NT-proBNP), left atrial volume, and the right ventricular myocardial performance index have also been shown to be independent predictors of PAH in patients with systemic sclerosis.35
An algorithm. The DETECT study36 enrolled patients at increased risk who had had systemic sclerosis longer than 3 years and a DLCO less than 60%. The investigators developed a 2-step algorithm to determine which patients should be referred for right heart catheterization to try to detect PAH earlier while minimizing the number of missed diagnoses and optimizing the use of invasive diagnostic right heart catheterization.
The first step was to assess serum values of anticentromere antibodies, NT-proBNP, and urate, and clinical features (telangiectasias), forced vital capacity, and electrocardiographic changes of right axis deviation to derive a prediction score. The second step was to assess surface echocardiographic features of the right atrial area and tricuspid regurgitation velocity.
This approach led to right heart catheterization in 62% of patients and was associated with a false-negative rate of 4%. Importantly, of the patients with PAH, 1 in 5 had no symptoms, and 33% had tricuspid regurgitation velocity less than 2.8 m/s. No single measurement performed well in isolation in this study.37
Thus, we recommend that, in addition to routine surface echocardiography, a multimodal approach be used that includes laboratory testing, clinical features, and electrocardiographic findings when screening this high-risk patient population.
ATHEROSCLEROTIC DISEASES
Although macrovascular disease has not typically been regarded as a significant systemic feature in systemic sclerosis, myocardial infarction and stroke are more common in patients with systemic sclerosis than in controls.38,39
Coronary artery disease in systemic sclerosis
Man et al38 reported that the incidence of myocardial infarction in patients with systemic sclerosis was 4.4 per 1,000 persons per year, and the incidence of stroke was 4.8 per 1,000 persons per year, compared with 2.5 per 1,000 persons per year for both myocardial infarction and stroke in healthy controls matched for age, sex, and time of entry.
The Australian Scleroderma Cohort Study39 found a 3-fold higher prevalence of coronary artery disease in systemic sclerosis patients than in controls after factoring in traditional risk factors.
Aviña-Zubieta et al,40 in a cohort of 1,239 systemic sclerosis patients, estimated a hazard ratio (HR) of 3.49 for myocardial infarction and 2.35 for stroke compared with age- and sex-matched controls. Not all of these events were related to macrovascular atherosclerosis—vasospasm and microvascular ischemia may have played significant roles in the etiology of clinical manifestations.
Studies of coronary atherosclerosis in systemic sclerosis are limited. An autopsy study41 of 58 patients with systemic sclerosis and 58 controls matched for age, sex, and ethnicity found that the prevalence of atherosclerosis of small coronary arteries and arterioles was significantly higher in systemic sclerosis patients than in controls (17% vs 2%, P < .01). However, the prevalence of medium-vessel coronary atherosclerosis was similar (48% vs 43%).
Why patients with systemic sclerosis develop atherosclerosis has not yet been determined. Traditional risk factors such as hypertension, dyslipidemia, diabetes mellitus, and obesity are typically no more prevalent in systemic sclerosis patients than in controls,38,42 and thus do not explain the increased risk of atherosclerotic cardiovascular disease. There is some evidence that novel markers of atherosclerotic risk such as homocysteine,43 lipoprotein[a],44 and oxidized low-density lipoprotein45 are more prevalent in systemic sclerosis, but these results have not been substantiated in more extensive studies.
Peripheral artery disease
It remains unclear whether peripheral artery disease is more prevalent in systemic sclerosis patients than in controls.
Individual studies have shown mixed results in comparing carotid artery stenosis between systemic sclerosis patients and controls using carotid duplex ultrasonography,46 the ankle-brachial index,46–48 carotid intima-media thickness,49–54 and brachial flow-mediated dilation.51,53,55–58 A meta-analysis found that the carotid intima and media are significantly thicker in systemic sclerosis patients than in controls,59 and the magnitude of difference is similar to that in other groups at increased cardiovascular risk, such as those with rheumatoid arthritis, diabetes, and familial hypercholesterolemia.60–63
A meta-analysis of brachial artery findings showed significantly lower flow-mediated dilation in systemic sclerosis patients than in controls.64
Overall, given the inconsistency of study results, systemic sclerosis patients should be screened and managed as in other patients with peripheral artery disease, but the clinician should be aware that there may be a higher risk of peripheral artery disease in these patients.
RIGHT AND LEFT VENTRICULAR DYSFUNCTION
Many patients with systemic sclerosis have right ventricular dysfunction as a consequence of PAH.65 It is important to detect diastolic dysfunction in this population, as it may be an even stronger predictor of death than pulmonary hypertension on right heart catheterization (HR 3.7 vs 2.0).66
Fewer patients have left ventricular dysfunction. In a multicenter study of 570 systemic sclerosis patients, only 1.4% had left ventricular systolic dysfunction on echocardiography, though 22.6% had left ventricular hypertrophy and 17.7% had left ventricular diastolic dysfunction.67 In the European League Against Rheumatism (EULAR) database, the prevalence of reduced left ventricular ejection fraction was 5.4%.68
Though traditional echocardiographic screening suggests the prevalence of left ventricular dysfunction in systemic sclerosis patients is low, cardiac magnetic resonance imaging (MRI) may be more sensitive than echocardiography for detecting subclinical myocardial involvement. Cardiac MRI has been shown to detect evidence of myocardial pathology (increased T2 signal, left ventricular thinning, pericardial effusion, reduced left ventricular and right ventricular ejection fraction, left ventricular diastolic dysfunction, and delayed myocardial contrast enhancement) in up to 75% of systemic sclerosis cases studied.69
Patients with systemic sclerosis should already be undergoing echocardiography every year to screen for PAH, and screening should also include tissue Doppler imaging to detect various forms of left and right ventricular systolic and diastolic dysfunction that may not be clinically apparent.
Though cardiac MRI can provide useful additional information, it is not currently recommended for routine screening in patients with systemic sclerosis.
ARRHYTHMIAS AND CONDUCTION DEFECTS
Patients with systemic sclerosis are prone to arrhythmias due to both conduction system fibrosis and myocardial damage.
Arrhythmias accounted for 6% of the deaths in the EULAR Scleroderma Trials and Research (EUSTAR) database.11
In the Genetics Versus Environment in Scleroderma Outcome Study (GENISOS),70 250 patients who had had systemic sclerosis for at least 3 years were studied during a period of approximately 6 years, during which there were 52 deaths, 29 of which were directly attributable to systemic sclerosis. Multivariable Cox modeling showed that 7 variables predicted mortality:
- Body mass index < 18.5 kg/m2
- Age ≥ 65
- Forced vital capacity < 50% predicted
- Systolic blood pressure ≥ 140 or diastolic blood pressure ≥ 90 mm Hg
- Pulmonary fibrosis
- Positive anticentromere antibodies
- Cardiac arrhythmias.
The hazard ratio for death in patients with arrhythmias in this model was 2.18 (95% CI 1.05–4.50, P = .035). Thus, finding arrhythmias in systemic sclerosis patients can provide important prognostic information.
While resting electrocardiography in patients with systemic sclerosis most commonly shows sinus rhythm, 24-hour electrocardiographic monitoring has revealed nonsustained supraventricular and ventricular arrhythmias in a significant percentage.71,72 Although difficult to quantify in routine practice, parameters controlled by the autonomic nervous system including heart rate variability and heart rate turbulence have been shown to be impaired in systemic sclerosis, and these measures are associated with an increased risk of malignant arrhythmias and sudden cardiac death.73,74
Conduction abnormalities
Conduction abnormalities occur in one-fifth to one-third of patients with systemic sclerosis.75,76 The most common abnormal conduction finding is left bundle branch block, followed by first-degree atrioventricular block. High-degree atrioventricular block is uncommon,76 though a few case reports of complete heart block thought to be related to systemic sclerosis have been published.77–79 An autopsy study showed that the conduction system is relatively spared from myocardial changes seen in systemic sclerosis patients, and thus it is speculated that the conduction disturbances are a consequence of damaged myocardium rather than damage to conduction tissue.80
Given the array of electrophysiologic abnormalities that systemic sclerosis patients can have, it is critical to monitor all patients with routine (annual or biannual) electrocardiography; to take possible arrhythmia-related symptoms seriously; and to evaluate them with further workup such as Holter monitoring for 24 hours or even longer, event monitoring, exercise testing, or tilt-table testing.
PERICARDIAL DISEASE
Pericardial disease is clinically apparent in 5% to 16% of patients with systemic sclerosis81; patients with limited cutaneous systemic sclerosis have more pericardial disease than those with diffuse cutaneous systemic sclerosis (30% vs 16%).82 Forty-one percent of systemic sclerosis patients have been shown to have pericardial effusion by echocardiography,81 but the effusions are typically small and rarely cause tamponade, though tamponade is associated with a poor prognosis.
Large pericardial effusions can develop before skin thickening and diagnosis of systemic sclerosis.81,83,84 Thus, systemic sclerosis should be considered in patients with pericardial effusions of unknown etiology.
In a small study,85 the pericardial fluid in systemic sclerosis was typically exudative, with lactate dehydrogenase greater than 200 U/L, a fluid-serum lactate dehydrogenase ratio greater than 0.6, and a fluid-serum total protein ratio greater than 0.5.
Pericardial effusion can be a sign of impending scleroderma renal crisis,86 and thus renal function should be carefully monitored in systemic sclerosis patients with pericardial effusion. Constrictive pericarditis and restrictive cardiomyopathy can rarely occur in systemic sclerosis and may more commonly present with symptoms.
Pericardial disease in systemic sclerosis should be treated in a standard fashion with nonsteroidal anti-inflammatory drugs. Corticosteroids are generally of limited benefit and should be avoided, especially in the setting of scleroderma renal crisis.81
VALVULAR HEART DISEASE
Based on limited studies, the prevalence of significant valvular heart disease in systemic sclerosis patients does not seem to be higher than that in the general population. While patients with systemic sclerosis and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) have been shown to have a higher frequency of mitral valve prolapse and mild mitral regurgitation,87,88 these abnormalities do not often progress in severity, and thus their clinical significance is limited.
RECOMMENDATIONS FOR CARE OF SYSTEMIC SCLEROSIS PATIENTS
It is important for physicians caring for patients with systemic sclerosis to be aware of its most common cardiac manifestations, including left and right ventricular systolic and diastolic dysfunction, pulmonary hypertension, conduction abnormalities, arrhythmias, and cardiomyopathy.
Look for volume overload
On clinical examination, assess for clinical markers of volume overload such as distended neck veins, peripheral edema, or an abnormal blood pressure response to the Valsalva maneuver. These findings should prompt measurement of NT-proBNP,89 and may warrant prescription of a diuretic.
Electrocardiography to investigate arrhythmias
Electrocardiography should be done if patients describe symptoms of palpitations, and should also include continuous rhythm monitoring with Holter or event monitoring, depending on the frequency of symptoms. Otherwise, patients should routinely undergo electrocardiography once or twice a year.
Q waves are common in systemic sclerosis patients (especially those with diffuse cutaneous systemic sclerosis), notably in the precordial leads, and can occur without coronary artery disease.90 Symptoms such as presyncope should be further investigated with Holter monitoring and tilt-table testing.
Assess, modify traditional risk factors
Subclinical atherosclerosis as detected by carotid intima-media thickness is as common in systemic sclerosis as in rheumatoid arthritis.61 However, traditional risk indices such as SCORE (Systematic Coronary Risk Evaluation), QRISK2, and the American College of Cardiology/American Heart Association indices may underestimate risk in patients who have systemic sclerosis.
Strict hypertension control should be the goal for all systemic sclerosis patients. Though there are no specific guidelines on which antihypertensive medications are preferred, calcium channel blockers or angiotensin II receptor blockers, which are typically used to treat systemic sclerosis-related Raynaud phenomenon, may be appropriate.
Statins reduce vascular complications and are generally well tolerated in patients with systemic sclerosis.91,92
Aspirin is not recommended for routine primary prevention in view of data suggesting that its benefits in diabetic patients are counterbalanced by increased bleeding risk.93
Echocardiography to detect pulmonary arterial hypertension
At this time, guidelines for monitoring for cardiovascular manifestations in systemic sclerosis patients are limited. The only well-defined ones are European consensus guidelines, which suggest annual transthoracic echocardiography for the first 5 years after systemic sclerosis is diagnosed and continued annual screening in patients at risk of developing PAH.31
We support this strategy, with annual screening for the first 5 years followed by surveillance echocardiography every 2 to 3 years unless there is a high risk of PAH. Specific attention should be paid to right ventricular diastolic function, right atrial volume, and right ventricular myocardial performance index.
Emerging data suggest that the addition of global longitudinal strain of ventricles to routine echocardiography can help detect subclinical cardiac risk.94 Although further study is needed into the predictive value of global longitudinal strain, it is a low-cost and noninvasive addition to standard echocardiography that can help guide risk stratification, and thus we recommend that it be part of the echocardiographic examination for all systemic sclerosis patients.
Pulmonary function testing. In addition to screening for PAH with echocardiography, we recommend obtaining baseline pulmonary function tests, including DLCO, at the time systemic sclerosis is diagnosed, with repeat testing annually.
Magnetic resonance imaging
While echocardiography is the gold standard for monitoring systemic sclerosis patients, cardiovascular MRI may have a role in identifying those at higher risk of dangerous arrhythmias such as ventricular tachycardia and ventricular fibrillation. In addition to assessing ventricular function, MRI can detect myocardial inflammation, ischemia, and fibrosis that may predispose a patient to develop ventricular tachycardia or fibrillation.95 Variables such as T1/T2 mapping, extracellular volume fraction, T2 signal ratio, and early vs late gadolinium enhancement can help identify patients who had past ventricular tachycardia or fibrillation.96
Finding an increased risk of arrhythmias may prompt a conversation between the patient and the physician about the need for an implantable cardiac defibrillator.
If cardiac MRI is available and is reimbursed by the patient’s insurance carrier, physicians should strongly consider obtaining at least one baseline scan in systemic sclerosis patients to identify those at risk of highly fatal arrhythmias.
Teamwork is needed
Systemic sclerosis has not traditionally been associated with cardiovascular disease to the extent of other rheumatic conditions, but the cardiovascular system can be affected in various ways that can ultimately lead to an early death. These manifestations may be asymptomatic for long periods, and overt clinical disease portends a poorer prognosis.
Primary care physicians managing these patients should be aware of the cardiovascular complications of systemic sclerosis and should implement appropriate screening tests in conjunction with rheumatologists and cardiologists. It is also essential for general and subspecialty cardiologists to understand the broad spectrum of organ system involvement that can affect systemic sclerosis patients and to tailor their investigation and management recommendations accordingly. By designing a multidisciplinary approach to the treatment of systemic sclerosis patients, physicians can help to optimize cardiovascular risk modification in this vulnerable population.
- Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum 2005; 52(2):402–411. doi:10.1002/art.20853
- Naranjo A, Sokka T, Descalzo MA, et al; QUEST-RA Group. Cardiovascular disease in patients with rheumatoid arthritis: results from the QUEST-RA study. Arthritis Res Ther 2008; 10(2):R30. doi:10.1186/ar2383
- Innala L, Möller B, Ljung L, et al. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: a five year prospective study. Arthritis Res Ther 2011; 13(4):R131. doi:10.1186/ar3442
- Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol 2012; 24(2):165–170. doi:10.1097/BOR.0b013e32834ff2e8
- Chifflot H, Fautrel B, Sordet C, Chatelus E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum 2008; 37(4):223–235 doi:10.1016/j.semarthrit.2007.05.003
- Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med 2009; 360(19):1989–2003. doi:10.1056/NEJMra0806188
- Panopoulos S, Tektonidou M, Drosos AA, et al. Prevalence of comorbidities in systemic sclerosis versus rheumatoid arthritis: a comparative, multicenter, matched-cohort study. Arthritis Res Ther 2018; 20(1):267. doi:10.1186/s13075-018-1771-0
- Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002; 81(8):139–153. doi:10.1097/00005792-200203000-00004
- Steen VD, Medsger TA Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum 2000; 43(11):2437–2444. doi:10.1002/1529-0131(200011)43:11<2437::AID-ANR10>3.0.CO;2-U
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69(10):1809–1815. doi:10.1136/ard.2009.114264
- Nassenstein K, Breuckmann F, Huger M, et al. Detection of myocardial fibrosis in systemic sclerosis by contrast-enhanced magnetic resonance imaging. Rofo 2008; 180(12):1054–1060. doi:10.1055/s-2008-1027864
- Psarras A, Soulaidopoulos S, Garyfallos A, Kitas G, Dimitroulas T. A critical view on cardiovascular risk in systemic sclerosis. Rheumatol Int 2017; 37(1):85–95. doi:10.1007/s00296-016-3530-3
- Lekakis J, Mavrikakis M, Emmanuel M, et al. Cold-induced coronary Raynaud’s phenomenon in patients with systemic sclerosis. Clin Exp Rheumatol 1998; 16(2):135–140. pmid:9536388
- Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol 2014; 26(6):615–620. doi:10.1097/BOR.0000000000000112
- Fleming JN, Nash RA, Mahoney WM Jr, Schwartz SM. Is scleroderma a vasculopathy? Curr Rheumatol Rep 2009; 11(2):103–110. pmid:19296882
- Maurer B, Distler A, Suliman YA, et al. Vascular endothelial growth factor aggravates fibrosis and vasculopathy in experimental models of systemic sclerosis. Ann Rheum Dis 2014; 73(10):1880–1887. doi:10.1136/annrheumdis-2013-203535
- Meune C, Vignaux O, Kahan A, Allanore Y. Heart involvement in systemic sclerosis: evolving concept and diagnostic methodologies. Arch Cardiovasc Dis 2010; 103(1):46–52. doi:10.1016/j.acvd.2009.06.009
- Dimitroulas T, Giannakoulas G, Karvounis H, Garyfallos A, Settas L, Kitas GD. Micro- and macrovascular treatment targets in scleroderma heart disease. Curr Pharm Des 2014; 20(4):536–544. pmid:23565639
- Allanore Y, Meune C. Primary myocardial involvement in systemic sclerosis: evidence for a microvascular origin. Clin Exp Rheumatol 2010; 28(5 suppl 62):S48–S53. pmid:21050545
- Kahan A, Nitenberg A, Foult JM, et al. Decreased coronary reserve in primary scleroderma myocardial disease. Arthritis Rheum 1985; 28(6):637–646. pmid:4004974
- Morrisroe K, Stevens W, Sahhar J, et al. Epidemiology and disease characteristics of systemic sclerosis-related pulmonary arterial hypertension: results from a real-life screening program. Arthritis Res Ther 2017; 19(1):42. doi:10.1186/s13075-017-1250-z
- Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest 2013; 144(4):1346–1356. doi:10.1378/chest.12-2396
- Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 2007; 66(7):940–944. doi:10.1136/ard.2006.066068
- Coghlan JG, Galiè N, Barberà JA, et al; AMBITION investigators. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis 2017; 76(7):1219–1227. doi:10.1136/annrheumdis-2016-210236
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53(1):1801913. doi:10.1183/13993003.01913-2018
- Chatterjee S. Pulmonary hypertension in systemic sclerosis. Semin Arthritis Rheum 2011; 41(1):19–37. doi:10.1016/j.semarthrit.2010.08.004
- Sweiss NJ, Hushaw L, Thenappan T, et al. Diagnosis and management of pulmonary hypertension in systemic sclerosis. Curr Rheumatol Rep 2010; 12(1):8–18. doi:10.1007/s11926-009-0078-1
- Cox SR, Walker JG, Coleman M, et al. Isolated pulmonary hypertension in scleroderma. Intern Med J 2005; 35(1):28–33. doi:10.1111/j.1445-5994.2004.00646.x
- Sánchez-Román J, Opitz CF, Kowal-Bielecka O, García-Hernández FJ, Castillo-Palma MJ, Pittrow D; EPOSS-OMERACT Group. Screening for PAH in patients with systemic sclerosis: focus on Doppler echocardiography. Rheumatology (Oxford) 2008; 47(suppl 5):v33–v35. doi:10.1093/rheumatology/ken306
- Walker KM, Pope J; Scleroderma Clinical Trials Consortium; Canadian Scleroderma Research Group. Expert agreement on EULAR/EUSTAR recommendations for the management of systemic sclerosis. J Rheumatol 2011; 38(7):1326–1328. doi:10.3899/jrheum.101262
- Khanna D, Gladue H, Channick R, et al; Scleroderma Foundation and Pulmonary Hypertension Association. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheum 2013; 65(12):3194–3201. doi:10.1002/art.38172
- Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum 2005; 52(12):3792–3800. doi:10.1002/art.21433
- Dimitroulas T, Giannakoulas G, Dimitroula H, et al. Significance of serum uric acid in pulmonary hypertension due to systemic sclerosis: a pilot study. Rheumatol Int 2011; 31(2):263–267. doi:10.1007/s00296-010-1557-4
- Dimitroulas T, Giannakoulas G, Papadopoulou K, et al. Left atrial volume and N-terminal pro-B type natriuretic peptide are associated with elevated pulmonary artery pressure in patients with systemic sclerosis. Clin Rheumatol 2010; 29(9):957–964. doi:10.1007/s10067-010-1494-3
- Coghlan JG, Denton CP, Grünig E, et al; DETECT study group. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73(7):1340–1349. doi:10.1136/annrheumdis-2013-203301
- Schwaiger JP, Khanna D, Gerry Coghlan J. Screening patients with scleroderma for pulmonary arterial hypertension and implications for other at-risk populations. Eur Respir Rev 2013; 22(130):515–525. doi:10.1183/09059180.00006013
- Man A, Zhu Y, Zhang Y, et al. The risk of cardiovascular disease in systemic sclerosis: a population-based cohort study. Ann Rheum Dis 2013; 72(7):1188–1193. doi:10.1136/annrheumdis-2012-202007
- Ngian G-S, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Aviña-Zubieta JA, Man A, Yurkovich M, Huang K, Sayre EC, Choi HK. Early cardiovascular disease after the diagnosis of systemic sclerosis. Am J Med 2016; 29(3):324–331. doi:10.1016/j.amjmed.2015.10.037
- D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med 1969; 46(3):428–440. doi:10.1016/0002-9343(69)90044-8
- Ngian GS, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Khurma V, Meyer C, Park GS, et al. A pilot study of subclinical coronary atherosclerosis in systemic sclerosis: coronary artery calcification in cases and controls. Arthritis Rheum 2008; 59(4):591–597. doi:10.1002/art.23540
- Lippi G, Caramaschi P, Montagnana M, Salvagno GL, Volpe A, Guidi G. Lipoprotein[a] and the lipid profile in patients with systemic sclerosis. Clin Chim Acta 2006; 364(1–2):345–348. doi:10.1016/j.cca.2005.07.015
- Palinski W, Hörkkö S, Miller E, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest 1996; 98(3):800–814. doi:10.1172/JCI118853
- Ho M, Veale D, Eastmond C, Nuki G, Belch J. Macrovascular disease and systemic sclerosis. Ann Rheum Dis 2000; 59(1):39–43. doi:10.1136/ard.59.1.39
- Kaloudi O, Basta G, Perfetto F, et al. Circulating levels of Ne-(carboxymethyl)lysine are increased in systemic sclerosis. Rheumatology (Oxford) 2007; 46(3):412–416. doi:10.1093/rheumatology/kel076
- Muro Y, Sugiura K, Morita Y, Tomita Y. An evaluation of the efficacy of the toe brachial index measuring vascular involvement in systemic sclerosis and other connective tissue diseases. Clin Exp Rheumatol 2009; 27(3 suppl 54):26–31. pmid:19796558
- Cheng K-S, Tiwari A, Boutin A, et al. Differentiation of primary and secondary Raynaud’s disease by carotid arterial stiffness. Eur J Vasc Endovasc Surg 2003; 25(4):336–341. doi:10.1053/ejvs.2002.1845
- Kawasaki M, Ito Y, Yokoyama H, et al. Assessment of arterial medial characteristics in human carotid arteries using integrated backscatter ultrasound and its histological implications. Atherosclerosis 2005; 180(1):145–154. doi:10.1016/j.atherosclerosis.2004.11.018
- Szucs G, Tímár O, Szekanecz Z, et al. Endothelial dysfunction precedes atherosclerosis in systemic sclerosis—relevance for prevention of vascular complications. Rheumatology (Oxford) 2007; 46(5):759–762. doi:10.1093/rheumatology/kel426
- Hettema ME, Zhang D, de Leeuw K, et al. Early atherosclerosis in systemic sclerosis and its relation to disease or traditional risk factors. Arthritis Res Ther 2008;10(2):R49. doi:10.1186/ar2408
- Roustit M, Simmons GH, Baguet JP, Carpentier P, Cracowski JL. Discrepancy between simultaneous digital skin microvascular and brachial artery macrovascular post-occlusive hyperemia in systemic sclerosis. J Rheumatol 2008; 35(8):1576–1583. pmid:18597404
- Vettori S, Maresca L, Cuomo G, Abbadessa S, Leonardo G, Valentini G. Clinical and subclinical atherosclerosis in systemic sclerosis: consequences of previous corticosteroid treatment. Scand J Rheumatol 2010; 39(6):485–489. doi:10.3109/03009741003781985
- Lekakis J, Mavrikakis M, Papamichael C, et al. Short-term estrogen administration improves abnormal endothelial function in women with systemic sclerosis and Raynaud’s phenomenon. Am Heart J 1998; 136(5):905–912. doi:10.1016/s0002-8703(98)70137-1
- Bartoli F, Blagojevic J, Bacci M, et al. Flow-mediated vasodilation and carotid intima-media thickness in systemic sclerosis. Ann N Y Acad Sci 2007; 1108:283–290. doi:10.1196/annals.1422.030
- Rollando D, Bezante GP, Sulli A, et al. Brachial artery endothelial-dependent flow-mediated dilation identifies early-stage endothelial dysfunction in systemic sclerosis and correlates with nailfold microvascular impairment. J Rheumatol 2010; 37(6):1168–1173. doi:10.3899/jrheum.091116
- Andersen GN, Mincheva-Nilsson L, Kazzam E, et al. Assessment of vascular function in systemic sclerosis: indications of the development of nitrate tolerance as a result of enhanced endothelial nitric oxide production. Arthritis Rheum 2002; 46(5):1324–1332. doi:10.1002/art.10191
- Au K, Singh MK, Bodukam V, et al. Atherosclerosis in systemic sclerosis: a systematic review and meta-analysis. Arthritis Rheum 2011; 63(7):2078–2090. doi:10.1002/art.30380
- van Sijl AM, Peters MJ, Knol DK, et al. Carotid intima media thickness in rheumatoid arthritis as compared to control subjects: a meta-analysis. Semin Arthritis Rheum 2011; 40(5):389–397. doi:10.1016/j.semarthrit.2010.06.006
- Brohall G, Odén A, Fagerberg B. Carotid artery intima-media thickness in patients with type 2 diabetes mellitus and impaired glucose tolerance: a systematic review. Diabet Med 2006; 23(6):609–616. doi:10.1111/j.1464-5491.2005.01725.x
- Masoura C, Pitsavos C, Aznaouridis K, Skoumas I, Vlachopoulos C, Stefanadis C. Arterial endothelial function and wall thickness in familial hypercholesterolemia and familial combined hyperlipidemia and the effect of statins. A systematic review and meta-analysis. Atherosclerosis 2011; 214(1):129–138. doi:10.1016/j.atherosclerosis.2010.10.008
- Ozen G, Inanc N, Unal AU, et al. Subclinical atherosclerosis in systemic sclerosis: not less frequent than rheumatoid arthritis and not detected with cardiovascular risk indices. Arthritis Care Res (Hoboken) 2016; 68(10):1538–1546. doi:10.1002/acr.22852
- Inaba Y, Chen JA, Bergmann SR. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: a meta-analysis. Int J Cardiovasc Imaging 2010; 26(6):631–640. doi:10.1007/s10554-010-9616-1
- Meune C, Avouac J, Wahbi K, et al. Cardiac involvement in systemic sclerosis assessed by tissue-doppler echocardiography during routine care: a controlled study of 100 consecutive patients. Arthritis Rheum 2008; 58(6):1803–1809. doi:10.1002/art.23463
- Tennøe AH, Murbræch K, Andreassen JC, et al. Left ventricular diastolic dysfunction predicts mortality in patients with systemic sclerosis. J Am Coll Cardiol 2018; 72(15):1804–1813. doi:10.1016/j.jacc.2018.07.068
- de Groote P, Gressin V, Hachulla E, et al; ItinerAIR-Scleroderma Investigators. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis 2008; 67(1):31–36. doi:10.1136/ard.2006.057760
- Allanore Y, Meune C, Vonk MC, et al; EUSTAR co-authors. Prevalence and factors associated with left ventricular dysfunction in the EULAR Scleroderma Trial and Research group (EUSTAR) database of patients with systemic sclerosis. Ann Rheum Dis 2010; 69(1):218–221. doi:10.1136/ard.2008.103382
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Assassi S, Del Junco D, Sutter K, et al. Clinical and genetic factors predictive of mortality in early systemic sclerosis. Arthritis Rheum 2009; 61(10):1403–1411. doi:10.1002/art.24734
- Rokas S, Mavrikakis M, Agrios N, Mylonas D, Antoniadou L, Moulopoulos S. Electrophysiologic abnormalities of cardiac function in progressive systemic sclerosis. J Electrocardiol 1996; 29(1):17–25. pmid:8808521
- Kostis JB, Seibold JR, Turkevich D, et al. Prognostic importance of cardiac arrhythmias in systemic sclerosis. Am J Med 1988; 84(6):1007–1015. doi:10.1016/0002-9343(88)90305-1
- Biełous-Wilk A, Poreba M, Staniszewska-Marszałek E, et al. Electrocardiographic evaluation in patients with systemic scleroderma and without clinically evident heart disease. Ann Noninvasive Electrocardiol 2009; 14(3):251–257. doi:10.1111/j.1542-474X.2009.00306.x
- Bienias P, Ciurzynski M, Glinska-Wielochowska M, et al. Heart rate turbulence assessment in systemic sclerosis: the role for the detection of cardiac autonomic nervous system dysfunction. Rheumatology (Oxford) 2010; 49(2):355–360. doi:10.1093/rheumatology/kep394
- Ferri C, Bernini L, Bongiorni MG, et al. Noninvasive evaluation of cardiac dysrhythmias, and their relationship with multisystemic symptoms, in progressive systemic sclerosis patients. Arthritis Rheum 1985; 28(11):1259–1266. pmid:4063000
- Roberts NK, Cabeen WR, Moss J, Clements PJ, Furst DE. The prevalence of conduction defects and cardiac arrhythmias in progressive systemic sclerosis. Ann Intern Med 1981; 94(1):38–40. doi:10.7326/0003-4819-94-1-38
- Wang Q, Shang Y, Li S, Wu Y, Wang C, Yan X. Complete heart block in systemic sclerosis: a case report and literature review. Medicine (Baltimore) 2018; 97(46):e13226. doi:10.1097/MD.0000000000013226
- Summerfield BJ. Progressive systemic sclerosis with complete heart block. Br Heart J 1975; 37(12):1308–1310. doi:10.1136/hrt.37.12.1308
- Moyssakis I, Papadopoulos DP, Tzioufas AG, Votteas V. Complete heart block in a patient with systemic sclerosis. Clin Rheumatol 2006; 25(4):551–552. doi:10.1007/s10067-005-0068-2
- Ridolfi RL, Bulkley BH, Hutchins GM. The cardiac conduction system in progressive systemic sclerosis. Clinical and pathologic features of 35 patients. Am J Med 1976; 61(3):361–366. doi:10.1016/0002-9343(76)90373-9
- Champion HC. The heart in scleroderma. Rheum Dis Clin North Am 2008; 34(1):181–190. doi:10.1016/j.rdc.2007.12.002
- Gowda RM, Khan IA, Sacchi TJ, Vasavada BC. Scleroderma pericardial disease presented with a large pericardial effusion—a case report. Angiology 2001; 52(1):59–62. doi:10.1177/000331970105200108
- Meier FMP, Frommer KW, Dinser R, et al; EUSTAR Co-authors. Update on the profile of the EUSTAR cohort: an analysis of the EULAR scleroderma trials and research group database. Ann Rheum Dis 2012; 71(8):1355–1360. doi:10.1136/annrheumdis-2011-200742
- Subramanian SR, Akram R, Velayati A, Chadow H. New development of cardiac tamponade on underlying effusive-constrictive pericarditis: an uncommon initial presentation of scleroderma. BMJ Case Rep 2013; 2013. doi:10.1136/bcr-2013-010254
- Kitchongcharoenying P, Foocharoen C, Mahakkanukrauh A, Suwannaroj S, Nanagara R. Pericardial fluid profiles of pericardial effusion in systemic sclerosis patients. Asian Pac J Allergy Immunol 2013; 31(4):314–319. doi:10.12932/AP0305.31.4.2013
- McWhorter JE, LeRoy EC. Pericardial disease in scleroderma (systemic sclerosis). Am J Med 1974; 57(4):566–575. doi:10.1016/0002-9343(74)90008-4
- Comens SM, Alpert MA, Sharp GC, et al. Frequency of mitral valve prolapse in systemic lupus erythematosus, progressive systemic sclerosis and mixed connective tissue disease. Am J Cardiol 1989; 63(5):369–370. doi:10.1016/0002-9149(89)90351-2
- Candell-Riera J, Armadans-Gil L, Simeón CP, et al. Comprehensive noninvasive assessment of cardiac involvement in limited systemic sclerosis. Arthritis Rheum 1996; 39(7):1138–1145. pmid:8670322
- Caforio ALP, Adler Y, Agostini C, et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Disease. Eur Heart J 2017; 38(35):2649–2662. doi:10.1093/eurheartj/ehx321
- Mavrogeni S, Karabela G, Koutsogeorgopoulou L, et al. Pseudo-infarction pattern in diffuse systemic sclerosis. Evaluation using cardiovascular magnetic resonance. Int J Cardiol 2016; 214:465–468. doi:10.1016/j.ijcard.2016.03.235
- Ladak K, Pope JE. A review of the effects of statins in systemic sclerosis. Semin Arthritis Rheum 2016; 45(6):698–705. doi:10.1016/j.semarthrit.2015.10.013
- Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35(9):1801–1808. pmid:18709692
- ASCEND Study Collaborative Group; Bowman L, Mafham M, Wallendszus K, et al. Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med 2018; 379(16):1529–1539. doi:10.1056/NEJMoa1804988
- Guerra F, Stronati G, Fischietti C, et al. Global longitudinal strain measured by speckle tracking identifies subclinical heart involvement in patients with systemic sclerosis. Eur J Prev Cardiol 2018; 25(15):1598–1606. doi:10.1177/2047487318786315
- Mavrogeni SI, Sfikakis PP, Dimitroulas T, et al. Prospects of using cardiovascular magnetic resonance in the identification of arrhythmogenic substrate in autoimmune rheumatic diseases. Rheumatol Int 2018; 38(9):1615–1621. doi:10.1007/s00296-018-4110-5
- Mavrogeni SI, Sfikakis PP, Markousis-Mavrogenis G, et al. Cardiovascular magnetic resonance imaging pattern in patients with autoimmune rheumatic diseases and ventricular tachycardia with preserved ejection fraction. Int J Cardiol 2019; 284:105–109. doi:10.1016/j.ijcard.2018.10.067
Autoimmune rheumatic diseases increase the risk of cardiovascular disease. In rheumatoid arthritis and systemic lupus erythematosus, the risk is driven primarily by the inflammatory milieu, leading to accelerated coronary and cerebrovascular atherosclerosis independent of traditional atherosclerotic risk factors.1–3 The extent of cardiovascular involvement in other rheumatologic diseases has been less well characterized but is an area of growing interest.
In this review, we focus on the cardiovascular complications of systemic sclerosis and review recommendations for monitoring these patients in clinical practice.
SYSTEMIC SCLEROSIS, AN AUTOIMMUNE RHEUMATIC DISEASE
Systemic sclerosis is an autoimmune rheumatic disease characterized by excessive extracellular matrix deposition leading to diffuse fibrosis, endothelial dysfunction, and microvascular injury. It is most common in North America, Southern Europe, and Australia,4,5 and it affects women more than men in ratios ranging from 3:1 to 14:1.6 The mean age at diagnosis is around 50.
The disease can affect the lungs (interstitial lung disease and pulmonary hypertension), the heart, the kidneys, and the gastrointestinal tract.
Systemic sclerosis has 2 main subtypes: limited cutaneous systemic sclerosis, formerly called CREST syndrome) and diffuse cutaneous systemic sclerosis. The limited cutaneous subtype is characterized by tightening of the skin of the distal extremities (below the elbows and knees) and face, while diffuse cutaneous systemic sclerosis can manifest as more extensive skin tightening also involving proximal extremities and the trunk. Both subtypes can have an effect on the cardiovascular system.
Some cardiovascular risk factors such as dyslipidemia, diabetes mellitus, and high body mass index are less common in patients with systemic sclerosis than in patients with rheumatoid arthritis, while the rates of arterial hypertension, smoking, chronic obstructive pulmonary disease, osteoporosis, and neoplasms are similar between the 2 groups.7
HEART INVOLVEMENT HAS SERIOUS CONSEQUENCES
Overt cardiac involvement in systemic sclerosis is associated with a mortality rate of up to 70% over 5 years,8,9 and about one-fourth of deaths in patients with systemic sclerosis are from cardiac causes.10,11 Studies in Europe10,12 showed that many patients with systemic sclerosis have cardiac involvement detectable by magnetic resonance imaging even if they do not have clinical disease. Pulmonary arterial hypertension (PAH) is a complication of both subtypes of systemic sclerosis and portends a higher risk of death.8
Thus, it is critical for clinicians to understand the potential comorbid conditions associated with systemic sclerosis, particularly the cardiovascular ones, and to work closely with cardiologists to help optimize the evaluation and management.
MECHANISMS OF CARDIAC DISEASE IN SYSTEMIC SCLEROSIS
Abnormal vasoreactivity, a consequence of an imbalance between endothelium-derived vasoconstrictors and vasodilators, defective angiogenesis, and endothelial injury, leads to tissue ischemia and vascular endothelial growth factor expression, which initiates injury and fibrosis in the myocardium and in other organs.14–17 Fibrosis involves the myocardium, pericardium, and conduction system.13,18
Myocardial involvement in systemic sclerosis is thought to be due mainly to abnormal vasoreactivity and microvascular abnormalities such as transient coronary artery spasm leading to repeated focal ischemia.19,20 Abnormal vasoreactivity has been demonstrated during cardiac catheterization21: while mean coronary sinus blood flow in systemic sclerosis patients was normal at rest, vasodilator reserve was significantly reduced in patients with diffuse cutaneous systemic sclerosis after maximal vasodilation with dipyridamole. Additionally, endomyocardial biopsy showed fibrosis and concentric intimal hypertrophy with normal epicardial coronary arteries.21
More research into other mechanisms of cardiovascular disease in systemic sclerosis is needed to allow for better preventive care for these patients.
PULMONARY ARTERIAL HYPERTENSION
Systemic sclerosis can be associated with World Health Organization (WHO) groups 1, 2, 3, and 4 pulmonary hypertension. WHO group 1, called pulmonary arterial hypertension or PAH, is one of the most common cardiac complications of systemic sclerosis, with a reported prevalence as high as 12%.22 Systemic sclerosis-associated PAH carries a high mortality rate, with a mean survival of only 3 years.23
With advances in treatments for other complications of systemic sclerosis, the percentage of systemic sclerosis patients who die of PAH has increased from 6% to 33%.24
Compared with patients with idiopathic PAH, those with systemic sclerosis get less of a response from therapy and have poorer outcomes despite lower mean pulmonary artery pressures and similar reductions in cardiac index. However, recent studies have suggested that with aggressive treatment, patients with systemic sclerosis-related PAH can achieve outcomes similar to those with idiopathic PAH.25 Thus, recognizing this condition early is imperative.
Pulmonary arterial hypertension defined
PAH is defined as the combination of all of the following26:
- Mean pulmonary artery pressure > 20 mm Hg at rest
- Normal pulmonary capillary wedge pressure (≤ 15 mm Hg)
- Pulmonary vascular resistance ≥ 3 Wood units on right heart catheterization.
Other causes of pulmonary hypertension such as interstitial lung disease, chronic pulmonary thromboembolic disease, and left heart disease must be excluded.24,27
Remodeling in the pulmonary arteries
The events that lead to PAH in systemic sclerosis remain unclear but are believed to involve initial inflammation or endothelial injury that leads to a dysequilibrium between proliferative mediators and antiproliferative vasodilators. This dysequilibrium, along with endothelial dysfunction, causes an obliterative vasculopathy in the pulmonary artery branches and arterioles. Sympathetic overactivity, hypoxemia, and ischemia-reperfusion injury additionally promote vascular proliferation, fibrosis, and remodeling, leading to increased pulmonary vascular resistance, PAH, and increased right ventricular pressures.23,27
The subtype of systemic sclerosis is an important factor in the development and progression of PAH. PAH appears to be the major cause of death in limited cutaneous systemic sclerosis, while interstitial lung disease is the major cause of death in diffuse cutaneous systemic sclerosis.28
Pulmonary arterial hypertension is a late complication of systemic sclerosis
Data from the South Australian Scleroderma Registry29 revealed that PAH tends to be a late complication of systemic sclerosis, occurring around 20 years after disease onset. In this study of 608 patients, no patient with diffuse cutaneous systemic sclerosis developed PAH.
Systemic sclerosis-related PAH initially follows an indolent course with few symptoms until right ventricular function deteriorates. Early in the disease, patients may experience nonspecific symptoms of fatigue, lightheadedness, and dyspnea on exertion.23 As it progresses, they tend to have worsening dyspnea and may experience exertional syncope, palpitations, and chest pain.
Physical findings may suggest elevated right ventricular pressure and right ventricular failure; these include a loud P2, a prominent jugular a wave, a tricuspid regurgitant murmur, jugular venous distention, and lower-extremity edema.27
Screening for pulmonary arterial hypertension in systemic sclerosis
Significant signs and symptoms usually occur late in the disease; thus, it is important to appropriately screen patients who are at risk so that they can begin aggressive treatment.
Doppler echocardiography is recommended by European and American guidelines to screen for PAH in patients who have systemic sclerosis, and most agree that screening is appropriate even if the patient has no symptoms.30 European consensus documents recommend that transthoracic echocardiography be done annually for the first 5 years of disease and be continued every year in patients at high risk, ie, those with anticentromere antibodies, anti-Th/To antibodies, or interstitial lung disease. Patients not at high risk of developing pulmonary hypertension should also have regular transthoracic echocardiography, though the exact timing is not defined.31 While American societies have not issued corresponding recommendations, many experts follow the European recommendations.
Worrisome features on echocardiography in asymptomatic patients should be followed up with right heart catheterization to assess mean right ventricular pressure. These include:
- Estimated right ventricular systolic pressure ≥ 40 mm Hg
- Tricuspid regurgitant jet velocity > 2.8 m/s
- Right atrial enlargement > 53 mm
- Right ventricular enlargement (mid-cavity dimension > 35 mm).32
Although echocardiography is the most common form of screening, it gives only an estimate of right ventricular systolic pressure, which is imprecise. Other noninvasive markers are helpful and necessary to appropriately screen this population.
Diffusion capacity. The Itinerair study33 found that a diffusing capacity for carbon monoxide (DLCO) of 60% or higher has a high specificity in excluding PAH.
Uric acid has been found to be elevated in patients with systemic sclerosis-related PAH, and levels inversely correlate with 6-minute walking distance.34
Other predictors. N-terminal pro-B-type natriuretic peptide (NT-proBNP), left atrial volume, and the right ventricular myocardial performance index have also been shown to be independent predictors of PAH in patients with systemic sclerosis.35
An algorithm. The DETECT study36 enrolled patients at increased risk who had had systemic sclerosis longer than 3 years and a DLCO less than 60%. The investigators developed a 2-step algorithm to determine which patients should be referred for right heart catheterization to try to detect PAH earlier while minimizing the number of missed diagnoses and optimizing the use of invasive diagnostic right heart catheterization.
The first step was to assess serum values of anticentromere antibodies, NT-proBNP, and urate, and clinical features (telangiectasias), forced vital capacity, and electrocardiographic changes of right axis deviation to derive a prediction score. The second step was to assess surface echocardiographic features of the right atrial area and tricuspid regurgitation velocity.
This approach led to right heart catheterization in 62% of patients and was associated with a false-negative rate of 4%. Importantly, of the patients with PAH, 1 in 5 had no symptoms, and 33% had tricuspid regurgitation velocity less than 2.8 m/s. No single measurement performed well in isolation in this study.37
Thus, we recommend that, in addition to routine surface echocardiography, a multimodal approach be used that includes laboratory testing, clinical features, and electrocardiographic findings when screening this high-risk patient population.
ATHEROSCLEROTIC DISEASES
Although macrovascular disease has not typically been regarded as a significant systemic feature in systemic sclerosis, myocardial infarction and stroke are more common in patients with systemic sclerosis than in controls.38,39
Coronary artery disease in systemic sclerosis
Man et al38 reported that the incidence of myocardial infarction in patients with systemic sclerosis was 4.4 per 1,000 persons per year, and the incidence of stroke was 4.8 per 1,000 persons per year, compared with 2.5 per 1,000 persons per year for both myocardial infarction and stroke in healthy controls matched for age, sex, and time of entry.
The Australian Scleroderma Cohort Study39 found a 3-fold higher prevalence of coronary artery disease in systemic sclerosis patients than in controls after factoring in traditional risk factors.
Aviña-Zubieta et al,40 in a cohort of 1,239 systemic sclerosis patients, estimated a hazard ratio (HR) of 3.49 for myocardial infarction and 2.35 for stroke compared with age- and sex-matched controls. Not all of these events were related to macrovascular atherosclerosis—vasospasm and microvascular ischemia may have played significant roles in the etiology of clinical manifestations.
Studies of coronary atherosclerosis in systemic sclerosis are limited. An autopsy study41 of 58 patients with systemic sclerosis and 58 controls matched for age, sex, and ethnicity found that the prevalence of atherosclerosis of small coronary arteries and arterioles was significantly higher in systemic sclerosis patients than in controls (17% vs 2%, P < .01). However, the prevalence of medium-vessel coronary atherosclerosis was similar (48% vs 43%).
Why patients with systemic sclerosis develop atherosclerosis has not yet been determined. Traditional risk factors such as hypertension, dyslipidemia, diabetes mellitus, and obesity are typically no more prevalent in systemic sclerosis patients than in controls,38,42 and thus do not explain the increased risk of atherosclerotic cardiovascular disease. There is some evidence that novel markers of atherosclerotic risk such as homocysteine,43 lipoprotein[a],44 and oxidized low-density lipoprotein45 are more prevalent in systemic sclerosis, but these results have not been substantiated in more extensive studies.
Peripheral artery disease
It remains unclear whether peripheral artery disease is more prevalent in systemic sclerosis patients than in controls.
Individual studies have shown mixed results in comparing carotid artery stenosis between systemic sclerosis patients and controls using carotid duplex ultrasonography,46 the ankle-brachial index,46–48 carotid intima-media thickness,49–54 and brachial flow-mediated dilation.51,53,55–58 A meta-analysis found that the carotid intima and media are significantly thicker in systemic sclerosis patients than in controls,59 and the magnitude of difference is similar to that in other groups at increased cardiovascular risk, such as those with rheumatoid arthritis, diabetes, and familial hypercholesterolemia.60–63
A meta-analysis of brachial artery findings showed significantly lower flow-mediated dilation in systemic sclerosis patients than in controls.64
Overall, given the inconsistency of study results, systemic sclerosis patients should be screened and managed as in other patients with peripheral artery disease, but the clinician should be aware that there may be a higher risk of peripheral artery disease in these patients.
RIGHT AND LEFT VENTRICULAR DYSFUNCTION
Many patients with systemic sclerosis have right ventricular dysfunction as a consequence of PAH.65 It is important to detect diastolic dysfunction in this population, as it may be an even stronger predictor of death than pulmonary hypertension on right heart catheterization (HR 3.7 vs 2.0).66
Fewer patients have left ventricular dysfunction. In a multicenter study of 570 systemic sclerosis patients, only 1.4% had left ventricular systolic dysfunction on echocardiography, though 22.6% had left ventricular hypertrophy and 17.7% had left ventricular diastolic dysfunction.67 In the European League Against Rheumatism (EULAR) database, the prevalence of reduced left ventricular ejection fraction was 5.4%.68
Though traditional echocardiographic screening suggests the prevalence of left ventricular dysfunction in systemic sclerosis patients is low, cardiac magnetic resonance imaging (MRI) may be more sensitive than echocardiography for detecting subclinical myocardial involvement. Cardiac MRI has been shown to detect evidence of myocardial pathology (increased T2 signal, left ventricular thinning, pericardial effusion, reduced left ventricular and right ventricular ejection fraction, left ventricular diastolic dysfunction, and delayed myocardial contrast enhancement) in up to 75% of systemic sclerosis cases studied.69
Patients with systemic sclerosis should already be undergoing echocardiography every year to screen for PAH, and screening should also include tissue Doppler imaging to detect various forms of left and right ventricular systolic and diastolic dysfunction that may not be clinically apparent.
Though cardiac MRI can provide useful additional information, it is not currently recommended for routine screening in patients with systemic sclerosis.
ARRHYTHMIAS AND CONDUCTION DEFECTS
Patients with systemic sclerosis are prone to arrhythmias due to both conduction system fibrosis and myocardial damage.
Arrhythmias accounted for 6% of the deaths in the EULAR Scleroderma Trials and Research (EUSTAR) database.11
In the Genetics Versus Environment in Scleroderma Outcome Study (GENISOS),70 250 patients who had had systemic sclerosis for at least 3 years were studied during a period of approximately 6 years, during which there were 52 deaths, 29 of which were directly attributable to systemic sclerosis. Multivariable Cox modeling showed that 7 variables predicted mortality:
- Body mass index < 18.5 kg/m2
- Age ≥ 65
- Forced vital capacity < 50% predicted
- Systolic blood pressure ≥ 140 or diastolic blood pressure ≥ 90 mm Hg
- Pulmonary fibrosis
- Positive anticentromere antibodies
- Cardiac arrhythmias.
The hazard ratio for death in patients with arrhythmias in this model was 2.18 (95% CI 1.05–4.50, P = .035). Thus, finding arrhythmias in systemic sclerosis patients can provide important prognostic information.
While resting electrocardiography in patients with systemic sclerosis most commonly shows sinus rhythm, 24-hour electrocardiographic monitoring has revealed nonsustained supraventricular and ventricular arrhythmias in a significant percentage.71,72 Although difficult to quantify in routine practice, parameters controlled by the autonomic nervous system including heart rate variability and heart rate turbulence have been shown to be impaired in systemic sclerosis, and these measures are associated with an increased risk of malignant arrhythmias and sudden cardiac death.73,74
Conduction abnormalities
Conduction abnormalities occur in one-fifth to one-third of patients with systemic sclerosis.75,76 The most common abnormal conduction finding is left bundle branch block, followed by first-degree atrioventricular block. High-degree atrioventricular block is uncommon,76 though a few case reports of complete heart block thought to be related to systemic sclerosis have been published.77–79 An autopsy study showed that the conduction system is relatively spared from myocardial changes seen in systemic sclerosis patients, and thus it is speculated that the conduction disturbances are a consequence of damaged myocardium rather than damage to conduction tissue.80
Given the array of electrophysiologic abnormalities that systemic sclerosis patients can have, it is critical to monitor all patients with routine (annual or biannual) electrocardiography; to take possible arrhythmia-related symptoms seriously; and to evaluate them with further workup such as Holter monitoring for 24 hours or even longer, event monitoring, exercise testing, or tilt-table testing.
PERICARDIAL DISEASE
Pericardial disease is clinically apparent in 5% to 16% of patients with systemic sclerosis81; patients with limited cutaneous systemic sclerosis have more pericardial disease than those with diffuse cutaneous systemic sclerosis (30% vs 16%).82 Forty-one percent of systemic sclerosis patients have been shown to have pericardial effusion by echocardiography,81 but the effusions are typically small and rarely cause tamponade, though tamponade is associated with a poor prognosis.
Large pericardial effusions can develop before skin thickening and diagnosis of systemic sclerosis.81,83,84 Thus, systemic sclerosis should be considered in patients with pericardial effusions of unknown etiology.
In a small study,85 the pericardial fluid in systemic sclerosis was typically exudative, with lactate dehydrogenase greater than 200 U/L, a fluid-serum lactate dehydrogenase ratio greater than 0.6, and a fluid-serum total protein ratio greater than 0.5.
Pericardial effusion can be a sign of impending scleroderma renal crisis,86 and thus renal function should be carefully monitored in systemic sclerosis patients with pericardial effusion. Constrictive pericarditis and restrictive cardiomyopathy can rarely occur in systemic sclerosis and may more commonly present with symptoms.
Pericardial disease in systemic sclerosis should be treated in a standard fashion with nonsteroidal anti-inflammatory drugs. Corticosteroids are generally of limited benefit and should be avoided, especially in the setting of scleroderma renal crisis.81
VALVULAR HEART DISEASE
Based on limited studies, the prevalence of significant valvular heart disease in systemic sclerosis patients does not seem to be higher than that in the general population. While patients with systemic sclerosis and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) have been shown to have a higher frequency of mitral valve prolapse and mild mitral regurgitation,87,88 these abnormalities do not often progress in severity, and thus their clinical significance is limited.
RECOMMENDATIONS FOR CARE OF SYSTEMIC SCLEROSIS PATIENTS
It is important for physicians caring for patients with systemic sclerosis to be aware of its most common cardiac manifestations, including left and right ventricular systolic and diastolic dysfunction, pulmonary hypertension, conduction abnormalities, arrhythmias, and cardiomyopathy.
Look for volume overload
On clinical examination, assess for clinical markers of volume overload such as distended neck veins, peripheral edema, or an abnormal blood pressure response to the Valsalva maneuver. These findings should prompt measurement of NT-proBNP,89 and may warrant prescription of a diuretic.
Electrocardiography to investigate arrhythmias
Electrocardiography should be done if patients describe symptoms of palpitations, and should also include continuous rhythm monitoring with Holter or event monitoring, depending on the frequency of symptoms. Otherwise, patients should routinely undergo electrocardiography once or twice a year.
Q waves are common in systemic sclerosis patients (especially those with diffuse cutaneous systemic sclerosis), notably in the precordial leads, and can occur without coronary artery disease.90 Symptoms such as presyncope should be further investigated with Holter monitoring and tilt-table testing.
Assess, modify traditional risk factors
Subclinical atherosclerosis as detected by carotid intima-media thickness is as common in systemic sclerosis as in rheumatoid arthritis.61 However, traditional risk indices such as SCORE (Systematic Coronary Risk Evaluation), QRISK2, and the American College of Cardiology/American Heart Association indices may underestimate risk in patients who have systemic sclerosis.
Strict hypertension control should be the goal for all systemic sclerosis patients. Though there are no specific guidelines on which antihypertensive medications are preferred, calcium channel blockers or angiotensin II receptor blockers, which are typically used to treat systemic sclerosis-related Raynaud phenomenon, may be appropriate.
Statins reduce vascular complications and are generally well tolerated in patients with systemic sclerosis.91,92
Aspirin is not recommended for routine primary prevention in view of data suggesting that its benefits in diabetic patients are counterbalanced by increased bleeding risk.93
Echocardiography to detect pulmonary arterial hypertension
At this time, guidelines for monitoring for cardiovascular manifestations in systemic sclerosis patients are limited. The only well-defined ones are European consensus guidelines, which suggest annual transthoracic echocardiography for the first 5 years after systemic sclerosis is diagnosed and continued annual screening in patients at risk of developing PAH.31
We support this strategy, with annual screening for the first 5 years followed by surveillance echocardiography every 2 to 3 years unless there is a high risk of PAH. Specific attention should be paid to right ventricular diastolic function, right atrial volume, and right ventricular myocardial performance index.
Emerging data suggest that the addition of global longitudinal strain of ventricles to routine echocardiography can help detect subclinical cardiac risk.94 Although further study is needed into the predictive value of global longitudinal strain, it is a low-cost and noninvasive addition to standard echocardiography that can help guide risk stratification, and thus we recommend that it be part of the echocardiographic examination for all systemic sclerosis patients.
Pulmonary function testing. In addition to screening for PAH with echocardiography, we recommend obtaining baseline pulmonary function tests, including DLCO, at the time systemic sclerosis is diagnosed, with repeat testing annually.
Magnetic resonance imaging
While echocardiography is the gold standard for monitoring systemic sclerosis patients, cardiovascular MRI may have a role in identifying those at higher risk of dangerous arrhythmias such as ventricular tachycardia and ventricular fibrillation. In addition to assessing ventricular function, MRI can detect myocardial inflammation, ischemia, and fibrosis that may predispose a patient to develop ventricular tachycardia or fibrillation.95 Variables such as T1/T2 mapping, extracellular volume fraction, T2 signal ratio, and early vs late gadolinium enhancement can help identify patients who had past ventricular tachycardia or fibrillation.96
Finding an increased risk of arrhythmias may prompt a conversation between the patient and the physician about the need for an implantable cardiac defibrillator.
If cardiac MRI is available and is reimbursed by the patient’s insurance carrier, physicians should strongly consider obtaining at least one baseline scan in systemic sclerosis patients to identify those at risk of highly fatal arrhythmias.
Teamwork is needed
Systemic sclerosis has not traditionally been associated with cardiovascular disease to the extent of other rheumatic conditions, but the cardiovascular system can be affected in various ways that can ultimately lead to an early death. These manifestations may be asymptomatic for long periods, and overt clinical disease portends a poorer prognosis.
Primary care physicians managing these patients should be aware of the cardiovascular complications of systemic sclerosis and should implement appropriate screening tests in conjunction with rheumatologists and cardiologists. It is also essential for general and subspecialty cardiologists to understand the broad spectrum of organ system involvement that can affect systemic sclerosis patients and to tailor their investigation and management recommendations accordingly. By designing a multidisciplinary approach to the treatment of systemic sclerosis patients, physicians can help to optimize cardiovascular risk modification in this vulnerable population.
Autoimmune rheumatic diseases increase the risk of cardiovascular disease. In rheumatoid arthritis and systemic lupus erythematosus, the risk is driven primarily by the inflammatory milieu, leading to accelerated coronary and cerebrovascular atherosclerosis independent of traditional atherosclerotic risk factors.1–3 The extent of cardiovascular involvement in other rheumatologic diseases has been less well characterized but is an area of growing interest.
In this review, we focus on the cardiovascular complications of systemic sclerosis and review recommendations for monitoring these patients in clinical practice.
SYSTEMIC SCLEROSIS, AN AUTOIMMUNE RHEUMATIC DISEASE
Systemic sclerosis is an autoimmune rheumatic disease characterized by excessive extracellular matrix deposition leading to diffuse fibrosis, endothelial dysfunction, and microvascular injury. It is most common in North America, Southern Europe, and Australia,4,5 and it affects women more than men in ratios ranging from 3:1 to 14:1.6 The mean age at diagnosis is around 50.
The disease can affect the lungs (interstitial lung disease and pulmonary hypertension), the heart, the kidneys, and the gastrointestinal tract.
Systemic sclerosis has 2 main subtypes: limited cutaneous systemic sclerosis, formerly called CREST syndrome) and diffuse cutaneous systemic sclerosis. The limited cutaneous subtype is characterized by tightening of the skin of the distal extremities (below the elbows and knees) and face, while diffuse cutaneous systemic sclerosis can manifest as more extensive skin tightening also involving proximal extremities and the trunk. Both subtypes can have an effect on the cardiovascular system.
Some cardiovascular risk factors such as dyslipidemia, diabetes mellitus, and high body mass index are less common in patients with systemic sclerosis than in patients with rheumatoid arthritis, while the rates of arterial hypertension, smoking, chronic obstructive pulmonary disease, osteoporosis, and neoplasms are similar between the 2 groups.7
HEART INVOLVEMENT HAS SERIOUS CONSEQUENCES
Overt cardiac involvement in systemic sclerosis is associated with a mortality rate of up to 70% over 5 years,8,9 and about one-fourth of deaths in patients with systemic sclerosis are from cardiac causes.10,11 Studies in Europe10,12 showed that many patients with systemic sclerosis have cardiac involvement detectable by magnetic resonance imaging even if they do not have clinical disease. Pulmonary arterial hypertension (PAH) is a complication of both subtypes of systemic sclerosis and portends a higher risk of death.8
Thus, it is critical for clinicians to understand the potential comorbid conditions associated with systemic sclerosis, particularly the cardiovascular ones, and to work closely with cardiologists to help optimize the evaluation and management.
MECHANISMS OF CARDIAC DISEASE IN SYSTEMIC SCLEROSIS
Abnormal vasoreactivity, a consequence of an imbalance between endothelium-derived vasoconstrictors and vasodilators, defective angiogenesis, and endothelial injury, leads to tissue ischemia and vascular endothelial growth factor expression, which initiates injury and fibrosis in the myocardium and in other organs.14–17 Fibrosis involves the myocardium, pericardium, and conduction system.13,18
Myocardial involvement in systemic sclerosis is thought to be due mainly to abnormal vasoreactivity and microvascular abnormalities such as transient coronary artery spasm leading to repeated focal ischemia.19,20 Abnormal vasoreactivity has been demonstrated during cardiac catheterization21: while mean coronary sinus blood flow in systemic sclerosis patients was normal at rest, vasodilator reserve was significantly reduced in patients with diffuse cutaneous systemic sclerosis after maximal vasodilation with dipyridamole. Additionally, endomyocardial biopsy showed fibrosis and concentric intimal hypertrophy with normal epicardial coronary arteries.21
More research into other mechanisms of cardiovascular disease in systemic sclerosis is needed to allow for better preventive care for these patients.
PULMONARY ARTERIAL HYPERTENSION
Systemic sclerosis can be associated with World Health Organization (WHO) groups 1, 2, 3, and 4 pulmonary hypertension. WHO group 1, called pulmonary arterial hypertension or PAH, is one of the most common cardiac complications of systemic sclerosis, with a reported prevalence as high as 12%.22 Systemic sclerosis-associated PAH carries a high mortality rate, with a mean survival of only 3 years.23
With advances in treatments for other complications of systemic sclerosis, the percentage of systemic sclerosis patients who die of PAH has increased from 6% to 33%.24
Compared with patients with idiopathic PAH, those with systemic sclerosis get less of a response from therapy and have poorer outcomes despite lower mean pulmonary artery pressures and similar reductions in cardiac index. However, recent studies have suggested that with aggressive treatment, patients with systemic sclerosis-related PAH can achieve outcomes similar to those with idiopathic PAH.25 Thus, recognizing this condition early is imperative.
Pulmonary arterial hypertension defined
PAH is defined as the combination of all of the following26:
- Mean pulmonary artery pressure > 20 mm Hg at rest
- Normal pulmonary capillary wedge pressure (≤ 15 mm Hg)
- Pulmonary vascular resistance ≥ 3 Wood units on right heart catheterization.
Other causes of pulmonary hypertension such as interstitial lung disease, chronic pulmonary thromboembolic disease, and left heart disease must be excluded.24,27
Remodeling in the pulmonary arteries
The events that lead to PAH in systemic sclerosis remain unclear but are believed to involve initial inflammation or endothelial injury that leads to a dysequilibrium between proliferative mediators and antiproliferative vasodilators. This dysequilibrium, along with endothelial dysfunction, causes an obliterative vasculopathy in the pulmonary artery branches and arterioles. Sympathetic overactivity, hypoxemia, and ischemia-reperfusion injury additionally promote vascular proliferation, fibrosis, and remodeling, leading to increased pulmonary vascular resistance, PAH, and increased right ventricular pressures.23,27
The subtype of systemic sclerosis is an important factor in the development and progression of PAH. PAH appears to be the major cause of death in limited cutaneous systemic sclerosis, while interstitial lung disease is the major cause of death in diffuse cutaneous systemic sclerosis.28
Pulmonary arterial hypertension is a late complication of systemic sclerosis
Data from the South Australian Scleroderma Registry29 revealed that PAH tends to be a late complication of systemic sclerosis, occurring around 20 years after disease onset. In this study of 608 patients, no patient with diffuse cutaneous systemic sclerosis developed PAH.
Systemic sclerosis-related PAH initially follows an indolent course with few symptoms until right ventricular function deteriorates. Early in the disease, patients may experience nonspecific symptoms of fatigue, lightheadedness, and dyspnea on exertion.23 As it progresses, they tend to have worsening dyspnea and may experience exertional syncope, palpitations, and chest pain.
Physical findings may suggest elevated right ventricular pressure and right ventricular failure; these include a loud P2, a prominent jugular a wave, a tricuspid regurgitant murmur, jugular venous distention, and lower-extremity edema.27
Screening for pulmonary arterial hypertension in systemic sclerosis
Significant signs and symptoms usually occur late in the disease; thus, it is important to appropriately screen patients who are at risk so that they can begin aggressive treatment.
Doppler echocardiography is recommended by European and American guidelines to screen for PAH in patients who have systemic sclerosis, and most agree that screening is appropriate even if the patient has no symptoms.30 European consensus documents recommend that transthoracic echocardiography be done annually for the first 5 years of disease and be continued every year in patients at high risk, ie, those with anticentromere antibodies, anti-Th/To antibodies, or interstitial lung disease. Patients not at high risk of developing pulmonary hypertension should also have regular transthoracic echocardiography, though the exact timing is not defined.31 While American societies have not issued corresponding recommendations, many experts follow the European recommendations.
Worrisome features on echocardiography in asymptomatic patients should be followed up with right heart catheterization to assess mean right ventricular pressure. These include:
- Estimated right ventricular systolic pressure ≥ 40 mm Hg
- Tricuspid regurgitant jet velocity > 2.8 m/s
- Right atrial enlargement > 53 mm
- Right ventricular enlargement (mid-cavity dimension > 35 mm).32
Although echocardiography is the most common form of screening, it gives only an estimate of right ventricular systolic pressure, which is imprecise. Other noninvasive markers are helpful and necessary to appropriately screen this population.
Diffusion capacity. The Itinerair study33 found that a diffusing capacity for carbon monoxide (DLCO) of 60% or higher has a high specificity in excluding PAH.
Uric acid has been found to be elevated in patients with systemic sclerosis-related PAH, and levels inversely correlate with 6-minute walking distance.34
Other predictors. N-terminal pro-B-type natriuretic peptide (NT-proBNP), left atrial volume, and the right ventricular myocardial performance index have also been shown to be independent predictors of PAH in patients with systemic sclerosis.35
An algorithm. The DETECT study36 enrolled patients at increased risk who had had systemic sclerosis longer than 3 years and a DLCO less than 60%. The investigators developed a 2-step algorithm to determine which patients should be referred for right heart catheterization to try to detect PAH earlier while minimizing the number of missed diagnoses and optimizing the use of invasive diagnostic right heart catheterization.
The first step was to assess serum values of anticentromere antibodies, NT-proBNP, and urate, and clinical features (telangiectasias), forced vital capacity, and electrocardiographic changes of right axis deviation to derive a prediction score. The second step was to assess surface echocardiographic features of the right atrial area and tricuspid regurgitation velocity.
This approach led to right heart catheterization in 62% of patients and was associated with a false-negative rate of 4%. Importantly, of the patients with PAH, 1 in 5 had no symptoms, and 33% had tricuspid regurgitation velocity less than 2.8 m/s. No single measurement performed well in isolation in this study.37
Thus, we recommend that, in addition to routine surface echocardiography, a multimodal approach be used that includes laboratory testing, clinical features, and electrocardiographic findings when screening this high-risk patient population.
ATHEROSCLEROTIC DISEASES
Although macrovascular disease has not typically been regarded as a significant systemic feature in systemic sclerosis, myocardial infarction and stroke are more common in patients with systemic sclerosis than in controls.38,39
Coronary artery disease in systemic sclerosis
Man et al38 reported that the incidence of myocardial infarction in patients with systemic sclerosis was 4.4 per 1,000 persons per year, and the incidence of stroke was 4.8 per 1,000 persons per year, compared with 2.5 per 1,000 persons per year for both myocardial infarction and stroke in healthy controls matched for age, sex, and time of entry.
The Australian Scleroderma Cohort Study39 found a 3-fold higher prevalence of coronary artery disease in systemic sclerosis patients than in controls after factoring in traditional risk factors.
Aviña-Zubieta et al,40 in a cohort of 1,239 systemic sclerosis patients, estimated a hazard ratio (HR) of 3.49 for myocardial infarction and 2.35 for stroke compared with age- and sex-matched controls. Not all of these events were related to macrovascular atherosclerosis—vasospasm and microvascular ischemia may have played significant roles in the etiology of clinical manifestations.
Studies of coronary atherosclerosis in systemic sclerosis are limited. An autopsy study41 of 58 patients with systemic sclerosis and 58 controls matched for age, sex, and ethnicity found that the prevalence of atherosclerosis of small coronary arteries and arterioles was significantly higher in systemic sclerosis patients than in controls (17% vs 2%, P < .01). However, the prevalence of medium-vessel coronary atherosclerosis was similar (48% vs 43%).
Why patients with systemic sclerosis develop atherosclerosis has not yet been determined. Traditional risk factors such as hypertension, dyslipidemia, diabetes mellitus, and obesity are typically no more prevalent in systemic sclerosis patients than in controls,38,42 and thus do not explain the increased risk of atherosclerotic cardiovascular disease. There is some evidence that novel markers of atherosclerotic risk such as homocysteine,43 lipoprotein[a],44 and oxidized low-density lipoprotein45 are more prevalent in systemic sclerosis, but these results have not been substantiated in more extensive studies.
Peripheral artery disease
It remains unclear whether peripheral artery disease is more prevalent in systemic sclerosis patients than in controls.
Individual studies have shown mixed results in comparing carotid artery stenosis between systemic sclerosis patients and controls using carotid duplex ultrasonography,46 the ankle-brachial index,46–48 carotid intima-media thickness,49–54 and brachial flow-mediated dilation.51,53,55–58 A meta-analysis found that the carotid intima and media are significantly thicker in systemic sclerosis patients than in controls,59 and the magnitude of difference is similar to that in other groups at increased cardiovascular risk, such as those with rheumatoid arthritis, diabetes, and familial hypercholesterolemia.60–63
A meta-analysis of brachial artery findings showed significantly lower flow-mediated dilation in systemic sclerosis patients than in controls.64
Overall, given the inconsistency of study results, systemic sclerosis patients should be screened and managed as in other patients with peripheral artery disease, but the clinician should be aware that there may be a higher risk of peripheral artery disease in these patients.
RIGHT AND LEFT VENTRICULAR DYSFUNCTION
Many patients with systemic sclerosis have right ventricular dysfunction as a consequence of PAH.65 It is important to detect diastolic dysfunction in this population, as it may be an even stronger predictor of death than pulmonary hypertension on right heart catheterization (HR 3.7 vs 2.0).66
Fewer patients have left ventricular dysfunction. In a multicenter study of 570 systemic sclerosis patients, only 1.4% had left ventricular systolic dysfunction on echocardiography, though 22.6% had left ventricular hypertrophy and 17.7% had left ventricular diastolic dysfunction.67 In the European League Against Rheumatism (EULAR) database, the prevalence of reduced left ventricular ejection fraction was 5.4%.68
Though traditional echocardiographic screening suggests the prevalence of left ventricular dysfunction in systemic sclerosis patients is low, cardiac magnetic resonance imaging (MRI) may be more sensitive than echocardiography for detecting subclinical myocardial involvement. Cardiac MRI has been shown to detect evidence of myocardial pathology (increased T2 signal, left ventricular thinning, pericardial effusion, reduced left ventricular and right ventricular ejection fraction, left ventricular diastolic dysfunction, and delayed myocardial contrast enhancement) in up to 75% of systemic sclerosis cases studied.69
Patients with systemic sclerosis should already be undergoing echocardiography every year to screen for PAH, and screening should also include tissue Doppler imaging to detect various forms of left and right ventricular systolic and diastolic dysfunction that may not be clinically apparent.
Though cardiac MRI can provide useful additional information, it is not currently recommended for routine screening in patients with systemic sclerosis.
ARRHYTHMIAS AND CONDUCTION DEFECTS
Patients with systemic sclerosis are prone to arrhythmias due to both conduction system fibrosis and myocardial damage.
Arrhythmias accounted for 6% of the deaths in the EULAR Scleroderma Trials and Research (EUSTAR) database.11
In the Genetics Versus Environment in Scleroderma Outcome Study (GENISOS),70 250 patients who had had systemic sclerosis for at least 3 years were studied during a period of approximately 6 years, during which there were 52 deaths, 29 of which were directly attributable to systemic sclerosis. Multivariable Cox modeling showed that 7 variables predicted mortality:
- Body mass index < 18.5 kg/m2
- Age ≥ 65
- Forced vital capacity < 50% predicted
- Systolic blood pressure ≥ 140 or diastolic blood pressure ≥ 90 mm Hg
- Pulmonary fibrosis
- Positive anticentromere antibodies
- Cardiac arrhythmias.
The hazard ratio for death in patients with arrhythmias in this model was 2.18 (95% CI 1.05–4.50, P = .035). Thus, finding arrhythmias in systemic sclerosis patients can provide important prognostic information.
While resting electrocardiography in patients with systemic sclerosis most commonly shows sinus rhythm, 24-hour electrocardiographic monitoring has revealed nonsustained supraventricular and ventricular arrhythmias in a significant percentage.71,72 Although difficult to quantify in routine practice, parameters controlled by the autonomic nervous system including heart rate variability and heart rate turbulence have been shown to be impaired in systemic sclerosis, and these measures are associated with an increased risk of malignant arrhythmias and sudden cardiac death.73,74
Conduction abnormalities
Conduction abnormalities occur in one-fifth to one-third of patients with systemic sclerosis.75,76 The most common abnormal conduction finding is left bundle branch block, followed by first-degree atrioventricular block. High-degree atrioventricular block is uncommon,76 though a few case reports of complete heart block thought to be related to systemic sclerosis have been published.77–79 An autopsy study showed that the conduction system is relatively spared from myocardial changes seen in systemic sclerosis patients, and thus it is speculated that the conduction disturbances are a consequence of damaged myocardium rather than damage to conduction tissue.80
Given the array of electrophysiologic abnormalities that systemic sclerosis patients can have, it is critical to monitor all patients with routine (annual or biannual) electrocardiography; to take possible arrhythmia-related symptoms seriously; and to evaluate them with further workup such as Holter monitoring for 24 hours or even longer, event monitoring, exercise testing, or tilt-table testing.
PERICARDIAL DISEASE
Pericardial disease is clinically apparent in 5% to 16% of patients with systemic sclerosis81; patients with limited cutaneous systemic sclerosis have more pericardial disease than those with diffuse cutaneous systemic sclerosis (30% vs 16%).82 Forty-one percent of systemic sclerosis patients have been shown to have pericardial effusion by echocardiography,81 but the effusions are typically small and rarely cause tamponade, though tamponade is associated with a poor prognosis.
Large pericardial effusions can develop before skin thickening and diagnosis of systemic sclerosis.81,83,84 Thus, systemic sclerosis should be considered in patients with pericardial effusions of unknown etiology.
In a small study,85 the pericardial fluid in systemic sclerosis was typically exudative, with lactate dehydrogenase greater than 200 U/L, a fluid-serum lactate dehydrogenase ratio greater than 0.6, and a fluid-serum total protein ratio greater than 0.5.
Pericardial effusion can be a sign of impending scleroderma renal crisis,86 and thus renal function should be carefully monitored in systemic sclerosis patients with pericardial effusion. Constrictive pericarditis and restrictive cardiomyopathy can rarely occur in systemic sclerosis and may more commonly present with symptoms.
Pericardial disease in systemic sclerosis should be treated in a standard fashion with nonsteroidal anti-inflammatory drugs. Corticosteroids are generally of limited benefit and should be avoided, especially in the setting of scleroderma renal crisis.81
VALVULAR HEART DISEASE
Based on limited studies, the prevalence of significant valvular heart disease in systemic sclerosis patients does not seem to be higher than that in the general population. While patients with systemic sclerosis and CREST syndrome (calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia) have been shown to have a higher frequency of mitral valve prolapse and mild mitral regurgitation,87,88 these abnormalities do not often progress in severity, and thus their clinical significance is limited.
RECOMMENDATIONS FOR CARE OF SYSTEMIC SCLEROSIS PATIENTS
It is important for physicians caring for patients with systemic sclerosis to be aware of its most common cardiac manifestations, including left and right ventricular systolic and diastolic dysfunction, pulmonary hypertension, conduction abnormalities, arrhythmias, and cardiomyopathy.
Look for volume overload
On clinical examination, assess for clinical markers of volume overload such as distended neck veins, peripheral edema, or an abnormal blood pressure response to the Valsalva maneuver. These findings should prompt measurement of NT-proBNP,89 and may warrant prescription of a diuretic.
Electrocardiography to investigate arrhythmias
Electrocardiography should be done if patients describe symptoms of palpitations, and should also include continuous rhythm monitoring with Holter or event monitoring, depending on the frequency of symptoms. Otherwise, patients should routinely undergo electrocardiography once or twice a year.
Q waves are common in systemic sclerosis patients (especially those with diffuse cutaneous systemic sclerosis), notably in the precordial leads, and can occur without coronary artery disease.90 Symptoms such as presyncope should be further investigated with Holter monitoring and tilt-table testing.
Assess, modify traditional risk factors
Subclinical atherosclerosis as detected by carotid intima-media thickness is as common in systemic sclerosis as in rheumatoid arthritis.61 However, traditional risk indices such as SCORE (Systematic Coronary Risk Evaluation), QRISK2, and the American College of Cardiology/American Heart Association indices may underestimate risk in patients who have systemic sclerosis.
Strict hypertension control should be the goal for all systemic sclerosis patients. Though there are no specific guidelines on which antihypertensive medications are preferred, calcium channel blockers or angiotensin II receptor blockers, which are typically used to treat systemic sclerosis-related Raynaud phenomenon, may be appropriate.
Statins reduce vascular complications and are generally well tolerated in patients with systemic sclerosis.91,92
Aspirin is not recommended for routine primary prevention in view of data suggesting that its benefits in diabetic patients are counterbalanced by increased bleeding risk.93
Echocardiography to detect pulmonary arterial hypertension
At this time, guidelines for monitoring for cardiovascular manifestations in systemic sclerosis patients are limited. The only well-defined ones are European consensus guidelines, which suggest annual transthoracic echocardiography for the first 5 years after systemic sclerosis is diagnosed and continued annual screening in patients at risk of developing PAH.31
We support this strategy, with annual screening for the first 5 years followed by surveillance echocardiography every 2 to 3 years unless there is a high risk of PAH. Specific attention should be paid to right ventricular diastolic function, right atrial volume, and right ventricular myocardial performance index.
Emerging data suggest that the addition of global longitudinal strain of ventricles to routine echocardiography can help detect subclinical cardiac risk.94 Although further study is needed into the predictive value of global longitudinal strain, it is a low-cost and noninvasive addition to standard echocardiography that can help guide risk stratification, and thus we recommend that it be part of the echocardiographic examination for all systemic sclerosis patients.
Pulmonary function testing. In addition to screening for PAH with echocardiography, we recommend obtaining baseline pulmonary function tests, including DLCO, at the time systemic sclerosis is diagnosed, with repeat testing annually.
Magnetic resonance imaging
While echocardiography is the gold standard for monitoring systemic sclerosis patients, cardiovascular MRI may have a role in identifying those at higher risk of dangerous arrhythmias such as ventricular tachycardia and ventricular fibrillation. In addition to assessing ventricular function, MRI can detect myocardial inflammation, ischemia, and fibrosis that may predispose a patient to develop ventricular tachycardia or fibrillation.95 Variables such as T1/T2 mapping, extracellular volume fraction, T2 signal ratio, and early vs late gadolinium enhancement can help identify patients who had past ventricular tachycardia or fibrillation.96
Finding an increased risk of arrhythmias may prompt a conversation between the patient and the physician about the need for an implantable cardiac defibrillator.
If cardiac MRI is available and is reimbursed by the patient’s insurance carrier, physicians should strongly consider obtaining at least one baseline scan in systemic sclerosis patients to identify those at risk of highly fatal arrhythmias.
Teamwork is needed
Systemic sclerosis has not traditionally been associated with cardiovascular disease to the extent of other rheumatic conditions, but the cardiovascular system can be affected in various ways that can ultimately lead to an early death. These manifestations may be asymptomatic for long periods, and overt clinical disease portends a poorer prognosis.
Primary care physicians managing these patients should be aware of the cardiovascular complications of systemic sclerosis and should implement appropriate screening tests in conjunction with rheumatologists and cardiologists. It is also essential for general and subspecialty cardiologists to understand the broad spectrum of organ system involvement that can affect systemic sclerosis patients and to tailor their investigation and management recommendations accordingly. By designing a multidisciplinary approach to the treatment of systemic sclerosis patients, physicians can help to optimize cardiovascular risk modification in this vulnerable population.
- Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum 2005; 52(2):402–411. doi:10.1002/art.20853
- Naranjo A, Sokka T, Descalzo MA, et al; QUEST-RA Group. Cardiovascular disease in patients with rheumatoid arthritis: results from the QUEST-RA study. Arthritis Res Ther 2008; 10(2):R30. doi:10.1186/ar2383
- Innala L, Möller B, Ljung L, et al. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: a five year prospective study. Arthritis Res Ther 2011; 13(4):R131. doi:10.1186/ar3442
- Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol 2012; 24(2):165–170. doi:10.1097/BOR.0b013e32834ff2e8
- Chifflot H, Fautrel B, Sordet C, Chatelus E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum 2008; 37(4):223–235 doi:10.1016/j.semarthrit.2007.05.003
- Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med 2009; 360(19):1989–2003. doi:10.1056/NEJMra0806188
- Panopoulos S, Tektonidou M, Drosos AA, et al. Prevalence of comorbidities in systemic sclerosis versus rheumatoid arthritis: a comparative, multicenter, matched-cohort study. Arthritis Res Ther 2018; 20(1):267. doi:10.1186/s13075-018-1771-0
- Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002; 81(8):139–153. doi:10.1097/00005792-200203000-00004
- Steen VD, Medsger TA Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum 2000; 43(11):2437–2444. doi:10.1002/1529-0131(200011)43:11<2437::AID-ANR10>3.0.CO;2-U
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69(10):1809–1815. doi:10.1136/ard.2009.114264
- Nassenstein K, Breuckmann F, Huger M, et al. Detection of myocardial fibrosis in systemic sclerosis by contrast-enhanced magnetic resonance imaging. Rofo 2008; 180(12):1054–1060. doi:10.1055/s-2008-1027864
- Psarras A, Soulaidopoulos S, Garyfallos A, Kitas G, Dimitroulas T. A critical view on cardiovascular risk in systemic sclerosis. Rheumatol Int 2017; 37(1):85–95. doi:10.1007/s00296-016-3530-3
- Lekakis J, Mavrikakis M, Emmanuel M, et al. Cold-induced coronary Raynaud’s phenomenon in patients with systemic sclerosis. Clin Exp Rheumatol 1998; 16(2):135–140. pmid:9536388
- Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol 2014; 26(6):615–620. doi:10.1097/BOR.0000000000000112
- Fleming JN, Nash RA, Mahoney WM Jr, Schwartz SM. Is scleroderma a vasculopathy? Curr Rheumatol Rep 2009; 11(2):103–110. pmid:19296882
- Maurer B, Distler A, Suliman YA, et al. Vascular endothelial growth factor aggravates fibrosis and vasculopathy in experimental models of systemic sclerosis. Ann Rheum Dis 2014; 73(10):1880–1887. doi:10.1136/annrheumdis-2013-203535
- Meune C, Vignaux O, Kahan A, Allanore Y. Heart involvement in systemic sclerosis: evolving concept and diagnostic methodologies. Arch Cardiovasc Dis 2010; 103(1):46–52. doi:10.1016/j.acvd.2009.06.009
- Dimitroulas T, Giannakoulas G, Karvounis H, Garyfallos A, Settas L, Kitas GD. Micro- and macrovascular treatment targets in scleroderma heart disease. Curr Pharm Des 2014; 20(4):536–544. pmid:23565639
- Allanore Y, Meune C. Primary myocardial involvement in systemic sclerosis: evidence for a microvascular origin. Clin Exp Rheumatol 2010; 28(5 suppl 62):S48–S53. pmid:21050545
- Kahan A, Nitenberg A, Foult JM, et al. Decreased coronary reserve in primary scleroderma myocardial disease. Arthritis Rheum 1985; 28(6):637–646. pmid:4004974
- Morrisroe K, Stevens W, Sahhar J, et al. Epidemiology and disease characteristics of systemic sclerosis-related pulmonary arterial hypertension: results from a real-life screening program. Arthritis Res Ther 2017; 19(1):42. doi:10.1186/s13075-017-1250-z
- Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest 2013; 144(4):1346–1356. doi:10.1378/chest.12-2396
- Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 2007; 66(7):940–944. doi:10.1136/ard.2006.066068
- Coghlan JG, Galiè N, Barberà JA, et al; AMBITION investigators. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis 2017; 76(7):1219–1227. doi:10.1136/annrheumdis-2016-210236
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53(1):1801913. doi:10.1183/13993003.01913-2018
- Chatterjee S. Pulmonary hypertension in systemic sclerosis. Semin Arthritis Rheum 2011; 41(1):19–37. doi:10.1016/j.semarthrit.2010.08.004
- Sweiss NJ, Hushaw L, Thenappan T, et al. Diagnosis and management of pulmonary hypertension in systemic sclerosis. Curr Rheumatol Rep 2010; 12(1):8–18. doi:10.1007/s11926-009-0078-1
- Cox SR, Walker JG, Coleman M, et al. Isolated pulmonary hypertension in scleroderma. Intern Med J 2005; 35(1):28–33. doi:10.1111/j.1445-5994.2004.00646.x
- Sánchez-Román J, Opitz CF, Kowal-Bielecka O, García-Hernández FJ, Castillo-Palma MJ, Pittrow D; EPOSS-OMERACT Group. Screening for PAH in patients with systemic sclerosis: focus on Doppler echocardiography. Rheumatology (Oxford) 2008; 47(suppl 5):v33–v35. doi:10.1093/rheumatology/ken306
- Walker KM, Pope J; Scleroderma Clinical Trials Consortium; Canadian Scleroderma Research Group. Expert agreement on EULAR/EUSTAR recommendations for the management of systemic sclerosis. J Rheumatol 2011; 38(7):1326–1328. doi:10.3899/jrheum.101262
- Khanna D, Gladue H, Channick R, et al; Scleroderma Foundation and Pulmonary Hypertension Association. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheum 2013; 65(12):3194–3201. doi:10.1002/art.38172
- Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum 2005; 52(12):3792–3800. doi:10.1002/art.21433
- Dimitroulas T, Giannakoulas G, Dimitroula H, et al. Significance of serum uric acid in pulmonary hypertension due to systemic sclerosis: a pilot study. Rheumatol Int 2011; 31(2):263–267. doi:10.1007/s00296-010-1557-4
- Dimitroulas T, Giannakoulas G, Papadopoulou K, et al. Left atrial volume and N-terminal pro-B type natriuretic peptide are associated with elevated pulmonary artery pressure in patients with systemic sclerosis. Clin Rheumatol 2010; 29(9):957–964. doi:10.1007/s10067-010-1494-3
- Coghlan JG, Denton CP, Grünig E, et al; DETECT study group. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73(7):1340–1349. doi:10.1136/annrheumdis-2013-203301
- Schwaiger JP, Khanna D, Gerry Coghlan J. Screening patients with scleroderma for pulmonary arterial hypertension and implications for other at-risk populations. Eur Respir Rev 2013; 22(130):515–525. doi:10.1183/09059180.00006013
- Man A, Zhu Y, Zhang Y, et al. The risk of cardiovascular disease in systemic sclerosis: a population-based cohort study. Ann Rheum Dis 2013; 72(7):1188–1193. doi:10.1136/annrheumdis-2012-202007
- Ngian G-S, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Aviña-Zubieta JA, Man A, Yurkovich M, Huang K, Sayre EC, Choi HK. Early cardiovascular disease after the diagnosis of systemic sclerosis. Am J Med 2016; 29(3):324–331. doi:10.1016/j.amjmed.2015.10.037
- D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med 1969; 46(3):428–440. doi:10.1016/0002-9343(69)90044-8
- Ngian GS, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Khurma V, Meyer C, Park GS, et al. A pilot study of subclinical coronary atherosclerosis in systemic sclerosis: coronary artery calcification in cases and controls. Arthritis Rheum 2008; 59(4):591–597. doi:10.1002/art.23540
- Lippi G, Caramaschi P, Montagnana M, Salvagno GL, Volpe A, Guidi G. Lipoprotein[a] and the lipid profile in patients with systemic sclerosis. Clin Chim Acta 2006; 364(1–2):345–348. doi:10.1016/j.cca.2005.07.015
- Palinski W, Hörkkö S, Miller E, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest 1996; 98(3):800–814. doi:10.1172/JCI118853
- Ho M, Veale D, Eastmond C, Nuki G, Belch J. Macrovascular disease and systemic sclerosis. Ann Rheum Dis 2000; 59(1):39–43. doi:10.1136/ard.59.1.39
- Kaloudi O, Basta G, Perfetto F, et al. Circulating levels of Ne-(carboxymethyl)lysine are increased in systemic sclerosis. Rheumatology (Oxford) 2007; 46(3):412–416. doi:10.1093/rheumatology/kel076
- Muro Y, Sugiura K, Morita Y, Tomita Y. An evaluation of the efficacy of the toe brachial index measuring vascular involvement in systemic sclerosis and other connective tissue diseases. Clin Exp Rheumatol 2009; 27(3 suppl 54):26–31. pmid:19796558
- Cheng K-S, Tiwari A, Boutin A, et al. Differentiation of primary and secondary Raynaud’s disease by carotid arterial stiffness. Eur J Vasc Endovasc Surg 2003; 25(4):336–341. doi:10.1053/ejvs.2002.1845
- Kawasaki M, Ito Y, Yokoyama H, et al. Assessment of arterial medial characteristics in human carotid arteries using integrated backscatter ultrasound and its histological implications. Atherosclerosis 2005; 180(1):145–154. doi:10.1016/j.atherosclerosis.2004.11.018
- Szucs G, Tímár O, Szekanecz Z, et al. Endothelial dysfunction precedes atherosclerosis in systemic sclerosis—relevance for prevention of vascular complications. Rheumatology (Oxford) 2007; 46(5):759–762. doi:10.1093/rheumatology/kel426
- Hettema ME, Zhang D, de Leeuw K, et al. Early atherosclerosis in systemic sclerosis and its relation to disease or traditional risk factors. Arthritis Res Ther 2008;10(2):R49. doi:10.1186/ar2408
- Roustit M, Simmons GH, Baguet JP, Carpentier P, Cracowski JL. Discrepancy between simultaneous digital skin microvascular and brachial artery macrovascular post-occlusive hyperemia in systemic sclerosis. J Rheumatol 2008; 35(8):1576–1583. pmid:18597404
- Vettori S, Maresca L, Cuomo G, Abbadessa S, Leonardo G, Valentini G. Clinical and subclinical atherosclerosis in systemic sclerosis: consequences of previous corticosteroid treatment. Scand J Rheumatol 2010; 39(6):485–489. doi:10.3109/03009741003781985
- Lekakis J, Mavrikakis M, Papamichael C, et al. Short-term estrogen administration improves abnormal endothelial function in women with systemic sclerosis and Raynaud’s phenomenon. Am Heart J 1998; 136(5):905–912. doi:10.1016/s0002-8703(98)70137-1
- Bartoli F, Blagojevic J, Bacci M, et al. Flow-mediated vasodilation and carotid intima-media thickness in systemic sclerosis. Ann N Y Acad Sci 2007; 1108:283–290. doi:10.1196/annals.1422.030
- Rollando D, Bezante GP, Sulli A, et al. Brachial artery endothelial-dependent flow-mediated dilation identifies early-stage endothelial dysfunction in systemic sclerosis and correlates with nailfold microvascular impairment. J Rheumatol 2010; 37(6):1168–1173. doi:10.3899/jrheum.091116
- Andersen GN, Mincheva-Nilsson L, Kazzam E, et al. Assessment of vascular function in systemic sclerosis: indications of the development of nitrate tolerance as a result of enhanced endothelial nitric oxide production. Arthritis Rheum 2002; 46(5):1324–1332. doi:10.1002/art.10191
- Au K, Singh MK, Bodukam V, et al. Atherosclerosis in systemic sclerosis: a systematic review and meta-analysis. Arthritis Rheum 2011; 63(7):2078–2090. doi:10.1002/art.30380
- van Sijl AM, Peters MJ, Knol DK, et al. Carotid intima media thickness in rheumatoid arthritis as compared to control subjects: a meta-analysis. Semin Arthritis Rheum 2011; 40(5):389–397. doi:10.1016/j.semarthrit.2010.06.006
- Brohall G, Odén A, Fagerberg B. Carotid artery intima-media thickness in patients with type 2 diabetes mellitus and impaired glucose tolerance: a systematic review. Diabet Med 2006; 23(6):609–616. doi:10.1111/j.1464-5491.2005.01725.x
- Masoura C, Pitsavos C, Aznaouridis K, Skoumas I, Vlachopoulos C, Stefanadis C. Arterial endothelial function and wall thickness in familial hypercholesterolemia and familial combined hyperlipidemia and the effect of statins. A systematic review and meta-analysis. Atherosclerosis 2011; 214(1):129–138. doi:10.1016/j.atherosclerosis.2010.10.008
- Ozen G, Inanc N, Unal AU, et al. Subclinical atherosclerosis in systemic sclerosis: not less frequent than rheumatoid arthritis and not detected with cardiovascular risk indices. Arthritis Care Res (Hoboken) 2016; 68(10):1538–1546. doi:10.1002/acr.22852
- Inaba Y, Chen JA, Bergmann SR. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: a meta-analysis. Int J Cardiovasc Imaging 2010; 26(6):631–640. doi:10.1007/s10554-010-9616-1
- Meune C, Avouac J, Wahbi K, et al. Cardiac involvement in systemic sclerosis assessed by tissue-doppler echocardiography during routine care: a controlled study of 100 consecutive patients. Arthritis Rheum 2008; 58(6):1803–1809. doi:10.1002/art.23463
- Tennøe AH, Murbræch K, Andreassen JC, et al. Left ventricular diastolic dysfunction predicts mortality in patients with systemic sclerosis. J Am Coll Cardiol 2018; 72(15):1804–1813. doi:10.1016/j.jacc.2018.07.068
- de Groote P, Gressin V, Hachulla E, et al; ItinerAIR-Scleroderma Investigators. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis 2008; 67(1):31–36. doi:10.1136/ard.2006.057760
- Allanore Y, Meune C, Vonk MC, et al; EUSTAR co-authors. Prevalence and factors associated with left ventricular dysfunction in the EULAR Scleroderma Trial and Research group (EUSTAR) database of patients with systemic sclerosis. Ann Rheum Dis 2010; 69(1):218–221. doi:10.1136/ard.2008.103382
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Assassi S, Del Junco D, Sutter K, et al. Clinical and genetic factors predictive of mortality in early systemic sclerosis. Arthritis Rheum 2009; 61(10):1403–1411. doi:10.1002/art.24734
- Rokas S, Mavrikakis M, Agrios N, Mylonas D, Antoniadou L, Moulopoulos S. Electrophysiologic abnormalities of cardiac function in progressive systemic sclerosis. J Electrocardiol 1996; 29(1):17–25. pmid:8808521
- Kostis JB, Seibold JR, Turkevich D, et al. Prognostic importance of cardiac arrhythmias in systemic sclerosis. Am J Med 1988; 84(6):1007–1015. doi:10.1016/0002-9343(88)90305-1
- Biełous-Wilk A, Poreba M, Staniszewska-Marszałek E, et al. Electrocardiographic evaluation in patients with systemic scleroderma and without clinically evident heart disease. Ann Noninvasive Electrocardiol 2009; 14(3):251–257. doi:10.1111/j.1542-474X.2009.00306.x
- Bienias P, Ciurzynski M, Glinska-Wielochowska M, et al. Heart rate turbulence assessment in systemic sclerosis: the role for the detection of cardiac autonomic nervous system dysfunction. Rheumatology (Oxford) 2010; 49(2):355–360. doi:10.1093/rheumatology/kep394
- Ferri C, Bernini L, Bongiorni MG, et al. Noninvasive evaluation of cardiac dysrhythmias, and their relationship with multisystemic symptoms, in progressive systemic sclerosis patients. Arthritis Rheum 1985; 28(11):1259–1266. pmid:4063000
- Roberts NK, Cabeen WR, Moss J, Clements PJ, Furst DE. The prevalence of conduction defects and cardiac arrhythmias in progressive systemic sclerosis. Ann Intern Med 1981; 94(1):38–40. doi:10.7326/0003-4819-94-1-38
- Wang Q, Shang Y, Li S, Wu Y, Wang C, Yan X. Complete heart block in systemic sclerosis: a case report and literature review. Medicine (Baltimore) 2018; 97(46):e13226. doi:10.1097/MD.0000000000013226
- Summerfield BJ. Progressive systemic sclerosis with complete heart block. Br Heart J 1975; 37(12):1308–1310. doi:10.1136/hrt.37.12.1308
- Moyssakis I, Papadopoulos DP, Tzioufas AG, Votteas V. Complete heart block in a patient with systemic sclerosis. Clin Rheumatol 2006; 25(4):551–552. doi:10.1007/s10067-005-0068-2
- Ridolfi RL, Bulkley BH, Hutchins GM. The cardiac conduction system in progressive systemic sclerosis. Clinical and pathologic features of 35 patients. Am J Med 1976; 61(3):361–366. doi:10.1016/0002-9343(76)90373-9
- Champion HC. The heart in scleroderma. Rheum Dis Clin North Am 2008; 34(1):181–190. doi:10.1016/j.rdc.2007.12.002
- Gowda RM, Khan IA, Sacchi TJ, Vasavada BC. Scleroderma pericardial disease presented with a large pericardial effusion—a case report. Angiology 2001; 52(1):59–62. doi:10.1177/000331970105200108
- Meier FMP, Frommer KW, Dinser R, et al; EUSTAR Co-authors. Update on the profile of the EUSTAR cohort: an analysis of the EULAR scleroderma trials and research group database. Ann Rheum Dis 2012; 71(8):1355–1360. doi:10.1136/annrheumdis-2011-200742
- Subramanian SR, Akram R, Velayati A, Chadow H. New development of cardiac tamponade on underlying effusive-constrictive pericarditis: an uncommon initial presentation of scleroderma. BMJ Case Rep 2013; 2013. doi:10.1136/bcr-2013-010254
- Kitchongcharoenying P, Foocharoen C, Mahakkanukrauh A, Suwannaroj S, Nanagara R. Pericardial fluid profiles of pericardial effusion in systemic sclerosis patients. Asian Pac J Allergy Immunol 2013; 31(4):314–319. doi:10.12932/AP0305.31.4.2013
- McWhorter JE, LeRoy EC. Pericardial disease in scleroderma (systemic sclerosis). Am J Med 1974; 57(4):566–575. doi:10.1016/0002-9343(74)90008-4
- Comens SM, Alpert MA, Sharp GC, et al. Frequency of mitral valve prolapse in systemic lupus erythematosus, progressive systemic sclerosis and mixed connective tissue disease. Am J Cardiol 1989; 63(5):369–370. doi:10.1016/0002-9149(89)90351-2
- Candell-Riera J, Armadans-Gil L, Simeón CP, et al. Comprehensive noninvasive assessment of cardiac involvement in limited systemic sclerosis. Arthritis Rheum 1996; 39(7):1138–1145. pmid:8670322
- Caforio ALP, Adler Y, Agostini C, et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Disease. Eur Heart J 2017; 38(35):2649–2662. doi:10.1093/eurheartj/ehx321
- Mavrogeni S, Karabela G, Koutsogeorgopoulou L, et al. Pseudo-infarction pattern in diffuse systemic sclerosis. Evaluation using cardiovascular magnetic resonance. Int J Cardiol 2016; 214:465–468. doi:10.1016/j.ijcard.2016.03.235
- Ladak K, Pope JE. A review of the effects of statins in systemic sclerosis. Semin Arthritis Rheum 2016; 45(6):698–705. doi:10.1016/j.semarthrit.2015.10.013
- Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35(9):1801–1808. pmid:18709692
- ASCEND Study Collaborative Group; Bowman L, Mafham M, Wallendszus K, et al. Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med 2018; 379(16):1529–1539. doi:10.1056/NEJMoa1804988
- Guerra F, Stronati G, Fischietti C, et al. Global longitudinal strain measured by speckle tracking identifies subclinical heart involvement in patients with systemic sclerosis. Eur J Prev Cardiol 2018; 25(15):1598–1606. doi:10.1177/2047487318786315
- Mavrogeni SI, Sfikakis PP, Dimitroulas T, et al. Prospects of using cardiovascular magnetic resonance in the identification of arrhythmogenic substrate in autoimmune rheumatic diseases. Rheumatol Int 2018; 38(9):1615–1621. doi:10.1007/s00296-018-4110-5
- Mavrogeni SI, Sfikakis PP, Markousis-Mavrogenis G, et al. Cardiovascular magnetic resonance imaging pattern in patients with autoimmune rheumatic diseases and ventricular tachycardia with preserved ejection fraction. Int J Cardiol 2019; 284:105–109. doi:10.1016/j.ijcard.2018.10.067
- Maradit-Kremers H, Crowson CS, Nicola PJ, et al. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: a population-based cohort study. Arthritis Rheum 2005; 52(2):402–411. doi:10.1002/art.20853
- Naranjo A, Sokka T, Descalzo MA, et al; QUEST-RA Group. Cardiovascular disease in patients with rheumatoid arthritis: results from the QUEST-RA study. Arthritis Res Ther 2008; 10(2):R30. doi:10.1186/ar2383
- Innala L, Möller B, Ljung L, et al. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: a five year prospective study. Arthritis Res Ther 2011; 13(4):R131. doi:10.1186/ar3442
- Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol 2012; 24(2):165–170. doi:10.1097/BOR.0b013e32834ff2e8
- Chifflot H, Fautrel B, Sordet C, Chatelus E, Sibilia J. Incidence and prevalence of systemic sclerosis: a systematic literature review. Semin Arthritis Rheum 2008; 37(4):223–235 doi:10.1016/j.semarthrit.2007.05.003
- Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med 2009; 360(19):1989–2003. doi:10.1056/NEJMra0806188
- Panopoulos S, Tektonidou M, Drosos AA, et al. Prevalence of comorbidities in systemic sclerosis versus rheumatoid arthritis: a comparative, multicenter, matched-cohort study. Arthritis Res Ther 2018; 20(1):267. doi:10.1186/s13075-018-1771-0
- Ferri C, Valentini G, Cozzi F, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002; 81(8):139–153. doi:10.1097/00005792-200203000-00004
- Steen VD, Medsger TA Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum 2000; 43(11):2437–2444. doi:10.1002/1529-0131(200011)43:11<2437::AID-ANR10>3.0.CO;2-U
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis 2010; 69(10):1809–1815. doi:10.1136/ard.2009.114264
- Nassenstein K, Breuckmann F, Huger M, et al. Detection of myocardial fibrosis in systemic sclerosis by contrast-enhanced magnetic resonance imaging. Rofo 2008; 180(12):1054–1060. doi:10.1055/s-2008-1027864
- Psarras A, Soulaidopoulos S, Garyfallos A, Kitas G, Dimitroulas T. A critical view on cardiovascular risk in systemic sclerosis. Rheumatol Int 2017; 37(1):85–95. doi:10.1007/s00296-016-3530-3
- Lekakis J, Mavrikakis M, Emmanuel M, et al. Cold-induced coronary Raynaud’s phenomenon in patients with systemic sclerosis. Clin Exp Rheumatol 1998; 16(2):135–140. pmid:9536388
- Altorok N, Wang Y, Kahaleh B. Endothelial dysfunction in systemic sclerosis. Curr Opin Rheumatol 2014; 26(6):615–620. doi:10.1097/BOR.0000000000000112
- Fleming JN, Nash RA, Mahoney WM Jr, Schwartz SM. Is scleroderma a vasculopathy? Curr Rheumatol Rep 2009; 11(2):103–110. pmid:19296882
- Maurer B, Distler A, Suliman YA, et al. Vascular endothelial growth factor aggravates fibrosis and vasculopathy in experimental models of systemic sclerosis. Ann Rheum Dis 2014; 73(10):1880–1887. doi:10.1136/annrheumdis-2013-203535
- Meune C, Vignaux O, Kahan A, Allanore Y. Heart involvement in systemic sclerosis: evolving concept and diagnostic methodologies. Arch Cardiovasc Dis 2010; 103(1):46–52. doi:10.1016/j.acvd.2009.06.009
- Dimitroulas T, Giannakoulas G, Karvounis H, Garyfallos A, Settas L, Kitas GD. Micro- and macrovascular treatment targets in scleroderma heart disease. Curr Pharm Des 2014; 20(4):536–544. pmid:23565639
- Allanore Y, Meune C. Primary myocardial involvement in systemic sclerosis: evidence for a microvascular origin. Clin Exp Rheumatol 2010; 28(5 suppl 62):S48–S53. pmid:21050545
- Kahan A, Nitenberg A, Foult JM, et al. Decreased coronary reserve in primary scleroderma myocardial disease. Arthritis Rheum 1985; 28(6):637–646. pmid:4004974
- Morrisroe K, Stevens W, Sahhar J, et al. Epidemiology and disease characteristics of systemic sclerosis-related pulmonary arterial hypertension: results from a real-life screening program. Arthritis Res Ther 2017; 19(1):42. doi:10.1186/s13075-017-1250-z
- Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. Chest 2013; 144(4):1346–1356. doi:10.1378/chest.12-2396
- Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 2007; 66(7):940–944. doi:10.1136/ard.2006.066068
- Coghlan JG, Galiè N, Barberà JA, et al; AMBITION investigators. Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial. Ann Rheum Dis 2017; 76(7):1219–1227. doi:10.1136/annrheumdis-2016-210236
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53(1):1801913. doi:10.1183/13993003.01913-2018
- Chatterjee S. Pulmonary hypertension in systemic sclerosis. Semin Arthritis Rheum 2011; 41(1):19–37. doi:10.1016/j.semarthrit.2010.08.004
- Sweiss NJ, Hushaw L, Thenappan T, et al. Diagnosis and management of pulmonary hypertension in systemic sclerosis. Curr Rheumatol Rep 2010; 12(1):8–18. doi:10.1007/s11926-009-0078-1
- Cox SR, Walker JG, Coleman M, et al. Isolated pulmonary hypertension in scleroderma. Intern Med J 2005; 35(1):28–33. doi:10.1111/j.1445-5994.2004.00646.x
- Sánchez-Román J, Opitz CF, Kowal-Bielecka O, García-Hernández FJ, Castillo-Palma MJ, Pittrow D; EPOSS-OMERACT Group. Screening for PAH in patients with systemic sclerosis: focus on Doppler echocardiography. Rheumatology (Oxford) 2008; 47(suppl 5):v33–v35. doi:10.1093/rheumatology/ken306
- Walker KM, Pope J; Scleroderma Clinical Trials Consortium; Canadian Scleroderma Research Group. Expert agreement on EULAR/EUSTAR recommendations for the management of systemic sclerosis. J Rheumatol 2011; 38(7):1326–1328. doi:10.3899/jrheum.101262
- Khanna D, Gladue H, Channick R, et al; Scleroderma Foundation and Pulmonary Hypertension Association. Recommendations for screening and detection of connective tissue disease-associated pulmonary arterial hypertension. Arthritis Rheum 2013; 65(12):3194–3201. doi:10.1002/art.38172
- Hachulla E, Gressin V, Guillevin L, et al. Early detection of pulmonary arterial hypertension in systemic sclerosis: a French nationwide prospective multicenter study. Arthritis Rheum 2005; 52(12):3792–3800. doi:10.1002/art.21433
- Dimitroulas T, Giannakoulas G, Dimitroula H, et al. Significance of serum uric acid in pulmonary hypertension due to systemic sclerosis: a pilot study. Rheumatol Int 2011; 31(2):263–267. doi:10.1007/s00296-010-1557-4
- Dimitroulas T, Giannakoulas G, Papadopoulou K, et al. Left atrial volume and N-terminal pro-B type natriuretic peptide are associated with elevated pulmonary artery pressure in patients with systemic sclerosis. Clin Rheumatol 2010; 29(9):957–964. doi:10.1007/s10067-010-1494-3
- Coghlan JG, Denton CP, Grünig E, et al; DETECT study group. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73(7):1340–1349. doi:10.1136/annrheumdis-2013-203301
- Schwaiger JP, Khanna D, Gerry Coghlan J. Screening patients with scleroderma for pulmonary arterial hypertension and implications for other at-risk populations. Eur Respir Rev 2013; 22(130):515–525. doi:10.1183/09059180.00006013
- Man A, Zhu Y, Zhang Y, et al. The risk of cardiovascular disease in systemic sclerosis: a population-based cohort study. Ann Rheum Dis 2013; 72(7):1188–1193. doi:10.1136/annrheumdis-2012-202007
- Ngian G-S, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Aviña-Zubieta JA, Man A, Yurkovich M, Huang K, Sayre EC, Choi HK. Early cardiovascular disease after the diagnosis of systemic sclerosis. Am J Med 2016; 29(3):324–331. doi:10.1016/j.amjmed.2015.10.037
- D’Angelo WA, Fries JF, Masi AT, Shulman LE. Pathologic observations in systemic sclerosis (scleroderma). A study of fifty-eight autopsy cases and fifty-eight matched controls. Am J Med 1969; 46(3):428–440. doi:10.1016/0002-9343(69)90044-8
- Ngian GS, Sahhar J, Proudman SM, Stevens W, Wicks IP, Van Doornum S. Prevalence of coronary heart disease and cardiovascular risk factors in a national cross-sectional cohort study of systemic sclerosis. Ann Rheum Dis 2012; 71(12):1980–1983. doi:10.1136/annrheumdis-2011-201176
- Khurma V, Meyer C, Park GS, et al. A pilot study of subclinical coronary atherosclerosis in systemic sclerosis: coronary artery calcification in cases and controls. Arthritis Rheum 2008; 59(4):591–597. doi:10.1002/art.23540
- Lippi G, Caramaschi P, Montagnana M, Salvagno GL, Volpe A, Guidi G. Lipoprotein[a] and the lipid profile in patients with systemic sclerosis. Clin Chim Acta 2006; 364(1–2):345–348. doi:10.1016/j.cca.2005.07.015
- Palinski W, Hörkkö S, Miller E, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest 1996; 98(3):800–814. doi:10.1172/JCI118853
- Ho M, Veale D, Eastmond C, Nuki G, Belch J. Macrovascular disease and systemic sclerosis. Ann Rheum Dis 2000; 59(1):39–43. doi:10.1136/ard.59.1.39
- Kaloudi O, Basta G, Perfetto F, et al. Circulating levels of Ne-(carboxymethyl)lysine are increased in systemic sclerosis. Rheumatology (Oxford) 2007; 46(3):412–416. doi:10.1093/rheumatology/kel076
- Muro Y, Sugiura K, Morita Y, Tomita Y. An evaluation of the efficacy of the toe brachial index measuring vascular involvement in systemic sclerosis and other connective tissue diseases. Clin Exp Rheumatol 2009; 27(3 suppl 54):26–31. pmid:19796558
- Cheng K-S, Tiwari A, Boutin A, et al. Differentiation of primary and secondary Raynaud’s disease by carotid arterial stiffness. Eur J Vasc Endovasc Surg 2003; 25(4):336–341. doi:10.1053/ejvs.2002.1845
- Kawasaki M, Ito Y, Yokoyama H, et al. Assessment of arterial medial characteristics in human carotid arteries using integrated backscatter ultrasound and its histological implications. Atherosclerosis 2005; 180(1):145–154. doi:10.1016/j.atherosclerosis.2004.11.018
- Szucs G, Tímár O, Szekanecz Z, et al. Endothelial dysfunction precedes atherosclerosis in systemic sclerosis—relevance for prevention of vascular complications. Rheumatology (Oxford) 2007; 46(5):759–762. doi:10.1093/rheumatology/kel426
- Hettema ME, Zhang D, de Leeuw K, et al. Early atherosclerosis in systemic sclerosis and its relation to disease or traditional risk factors. Arthritis Res Ther 2008;10(2):R49. doi:10.1186/ar2408
- Roustit M, Simmons GH, Baguet JP, Carpentier P, Cracowski JL. Discrepancy between simultaneous digital skin microvascular and brachial artery macrovascular post-occlusive hyperemia in systemic sclerosis. J Rheumatol 2008; 35(8):1576–1583. pmid:18597404
- Vettori S, Maresca L, Cuomo G, Abbadessa S, Leonardo G, Valentini G. Clinical and subclinical atherosclerosis in systemic sclerosis: consequences of previous corticosteroid treatment. Scand J Rheumatol 2010; 39(6):485–489. doi:10.3109/03009741003781985
- Lekakis J, Mavrikakis M, Papamichael C, et al. Short-term estrogen administration improves abnormal endothelial function in women with systemic sclerosis and Raynaud’s phenomenon. Am Heart J 1998; 136(5):905–912. doi:10.1016/s0002-8703(98)70137-1
- Bartoli F, Blagojevic J, Bacci M, et al. Flow-mediated vasodilation and carotid intima-media thickness in systemic sclerosis. Ann N Y Acad Sci 2007; 1108:283–290. doi:10.1196/annals.1422.030
- Rollando D, Bezante GP, Sulli A, et al. Brachial artery endothelial-dependent flow-mediated dilation identifies early-stage endothelial dysfunction in systemic sclerosis and correlates with nailfold microvascular impairment. J Rheumatol 2010; 37(6):1168–1173. doi:10.3899/jrheum.091116
- Andersen GN, Mincheva-Nilsson L, Kazzam E, et al. Assessment of vascular function in systemic sclerosis: indications of the development of nitrate tolerance as a result of enhanced endothelial nitric oxide production. Arthritis Rheum 2002; 46(5):1324–1332. doi:10.1002/art.10191
- Au K, Singh MK, Bodukam V, et al. Atherosclerosis in systemic sclerosis: a systematic review and meta-analysis. Arthritis Rheum 2011; 63(7):2078–2090. doi:10.1002/art.30380
- van Sijl AM, Peters MJ, Knol DK, et al. Carotid intima media thickness in rheumatoid arthritis as compared to control subjects: a meta-analysis. Semin Arthritis Rheum 2011; 40(5):389–397. doi:10.1016/j.semarthrit.2010.06.006
- Brohall G, Odén A, Fagerberg B. Carotid artery intima-media thickness in patients with type 2 diabetes mellitus and impaired glucose tolerance: a systematic review. Diabet Med 2006; 23(6):609–616. doi:10.1111/j.1464-5491.2005.01725.x
- Masoura C, Pitsavos C, Aznaouridis K, Skoumas I, Vlachopoulos C, Stefanadis C. Arterial endothelial function and wall thickness in familial hypercholesterolemia and familial combined hyperlipidemia and the effect of statins. A systematic review and meta-analysis. Atherosclerosis 2011; 214(1):129–138. doi:10.1016/j.atherosclerosis.2010.10.008
- Ozen G, Inanc N, Unal AU, et al. Subclinical atherosclerosis in systemic sclerosis: not less frequent than rheumatoid arthritis and not detected with cardiovascular risk indices. Arthritis Care Res (Hoboken) 2016; 68(10):1538–1546. doi:10.1002/acr.22852
- Inaba Y, Chen JA, Bergmann SR. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: a meta-analysis. Int J Cardiovasc Imaging 2010; 26(6):631–640. doi:10.1007/s10554-010-9616-1
- Meune C, Avouac J, Wahbi K, et al. Cardiac involvement in systemic sclerosis assessed by tissue-doppler echocardiography during routine care: a controlled study of 100 consecutive patients. Arthritis Rheum 2008; 58(6):1803–1809. doi:10.1002/art.23463
- Tennøe AH, Murbræch K, Andreassen JC, et al. Left ventricular diastolic dysfunction predicts mortality in patients with systemic sclerosis. J Am Coll Cardiol 2018; 72(15):1804–1813. doi:10.1016/j.jacc.2018.07.068
- de Groote P, Gressin V, Hachulla E, et al; ItinerAIR-Scleroderma Investigators. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann Rheum Dis 2008; 67(1):31–36. doi:10.1136/ard.2006.057760
- Allanore Y, Meune C, Vonk MC, et al; EUSTAR co-authors. Prevalence and factors associated with left ventricular dysfunction in the EULAR Scleroderma Trial and Research group (EUSTAR) database of patients with systemic sclerosis. Ann Rheum Dis 2010; 69(1):218–221. doi:10.1136/ard.2008.103382
- Hachulla AL, Launay D, Gaxotte V, et al. Cardiac magnetic resonance imaging in systemic sclerosis: a cross-sectional observational study of 52 patients. Ann Rheum Dis 2009; 68(12):1878–1884. doi:10.1136/ard.2008.095836
- Assassi S, Del Junco D, Sutter K, et al. Clinical and genetic factors predictive of mortality in early systemic sclerosis. Arthritis Rheum 2009; 61(10):1403–1411. doi:10.1002/art.24734
- Rokas S, Mavrikakis M, Agrios N, Mylonas D, Antoniadou L, Moulopoulos S. Electrophysiologic abnormalities of cardiac function in progressive systemic sclerosis. J Electrocardiol 1996; 29(1):17–25. pmid:8808521
- Kostis JB, Seibold JR, Turkevich D, et al. Prognostic importance of cardiac arrhythmias in systemic sclerosis. Am J Med 1988; 84(6):1007–1015. doi:10.1016/0002-9343(88)90305-1
- Biełous-Wilk A, Poreba M, Staniszewska-Marszałek E, et al. Electrocardiographic evaluation in patients with systemic scleroderma and without clinically evident heart disease. Ann Noninvasive Electrocardiol 2009; 14(3):251–257. doi:10.1111/j.1542-474X.2009.00306.x
- Bienias P, Ciurzynski M, Glinska-Wielochowska M, et al. Heart rate turbulence assessment in systemic sclerosis: the role for the detection of cardiac autonomic nervous system dysfunction. Rheumatology (Oxford) 2010; 49(2):355–360. doi:10.1093/rheumatology/kep394
- Ferri C, Bernini L, Bongiorni MG, et al. Noninvasive evaluation of cardiac dysrhythmias, and their relationship with multisystemic symptoms, in progressive systemic sclerosis patients. Arthritis Rheum 1985; 28(11):1259–1266. pmid:4063000
- Roberts NK, Cabeen WR, Moss J, Clements PJ, Furst DE. The prevalence of conduction defects and cardiac arrhythmias in progressive systemic sclerosis. Ann Intern Med 1981; 94(1):38–40. doi:10.7326/0003-4819-94-1-38
- Wang Q, Shang Y, Li S, Wu Y, Wang C, Yan X. Complete heart block in systemic sclerosis: a case report and literature review. Medicine (Baltimore) 2018; 97(46):e13226. doi:10.1097/MD.0000000000013226
- Summerfield BJ. Progressive systemic sclerosis with complete heart block. Br Heart J 1975; 37(12):1308–1310. doi:10.1136/hrt.37.12.1308
- Moyssakis I, Papadopoulos DP, Tzioufas AG, Votteas V. Complete heart block in a patient with systemic sclerosis. Clin Rheumatol 2006; 25(4):551–552. doi:10.1007/s10067-005-0068-2
- Ridolfi RL, Bulkley BH, Hutchins GM. The cardiac conduction system in progressive systemic sclerosis. Clinical and pathologic features of 35 patients. Am J Med 1976; 61(3):361–366. doi:10.1016/0002-9343(76)90373-9
- Champion HC. The heart in scleroderma. Rheum Dis Clin North Am 2008; 34(1):181–190. doi:10.1016/j.rdc.2007.12.002
- Gowda RM, Khan IA, Sacchi TJ, Vasavada BC. Scleroderma pericardial disease presented with a large pericardial effusion—a case report. Angiology 2001; 52(1):59–62. doi:10.1177/000331970105200108
- Meier FMP, Frommer KW, Dinser R, et al; EUSTAR Co-authors. Update on the profile of the EUSTAR cohort: an analysis of the EULAR scleroderma trials and research group database. Ann Rheum Dis 2012; 71(8):1355–1360. doi:10.1136/annrheumdis-2011-200742
- Subramanian SR, Akram R, Velayati A, Chadow H. New development of cardiac tamponade on underlying effusive-constrictive pericarditis: an uncommon initial presentation of scleroderma. BMJ Case Rep 2013; 2013. doi:10.1136/bcr-2013-010254
- Kitchongcharoenying P, Foocharoen C, Mahakkanukrauh A, Suwannaroj S, Nanagara R. Pericardial fluid profiles of pericardial effusion in systemic sclerosis patients. Asian Pac J Allergy Immunol 2013; 31(4):314–319. doi:10.12932/AP0305.31.4.2013
- McWhorter JE, LeRoy EC. Pericardial disease in scleroderma (systemic sclerosis). Am J Med 1974; 57(4):566–575. doi:10.1016/0002-9343(74)90008-4
- Comens SM, Alpert MA, Sharp GC, et al. Frequency of mitral valve prolapse in systemic lupus erythematosus, progressive systemic sclerosis and mixed connective tissue disease. Am J Cardiol 1989; 63(5):369–370. doi:10.1016/0002-9149(89)90351-2
- Candell-Riera J, Armadans-Gil L, Simeón CP, et al. Comprehensive noninvasive assessment of cardiac involvement in limited systemic sclerosis. Arthritis Rheum 1996; 39(7):1138–1145. pmid:8670322
- Caforio ALP, Adler Y, Agostini C, et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Disease. Eur Heart J 2017; 38(35):2649–2662. doi:10.1093/eurheartj/ehx321
- Mavrogeni S, Karabela G, Koutsogeorgopoulou L, et al. Pseudo-infarction pattern in diffuse systemic sclerosis. Evaluation using cardiovascular magnetic resonance. Int J Cardiol 2016; 214:465–468. doi:10.1016/j.ijcard.2016.03.235
- Ladak K, Pope JE. A review of the effects of statins in systemic sclerosis. Semin Arthritis Rheum 2016; 45(6):698–705. doi:10.1016/j.semarthrit.2015.10.013
- Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35(9):1801–1808. pmid:18709692
- ASCEND Study Collaborative Group; Bowman L, Mafham M, Wallendszus K, et al. Effects of aspirin for primary prevention in persons with diabetes mellitus. N Engl J Med 2018; 379(16):1529–1539. doi:10.1056/NEJMoa1804988
- Guerra F, Stronati G, Fischietti C, et al. Global longitudinal strain measured by speckle tracking identifies subclinical heart involvement in patients with systemic sclerosis. Eur J Prev Cardiol 2018; 25(15):1598–1606. doi:10.1177/2047487318786315
- Mavrogeni SI, Sfikakis PP, Dimitroulas T, et al. Prospects of using cardiovascular magnetic resonance in the identification of arrhythmogenic substrate in autoimmune rheumatic diseases. Rheumatol Int 2018; 38(9):1615–1621. doi:10.1007/s00296-018-4110-5
- Mavrogeni SI, Sfikakis PP, Markousis-Mavrogenis G, et al. Cardiovascular magnetic resonance imaging pattern in patients with autoimmune rheumatic diseases and ventricular tachycardia with preserved ejection fraction. Int J Cardiol 2019; 284:105–109. doi:10.1016/j.ijcard.2018.10.067
KEY POINTS
- Pulmonary hypertension is common in systemic sclerosis and carries a poor prognosis. Patients with systemic sclerosis should be screened regularly with echocardiography, followed, when necessary, by right heart catheterization to detect it early.
- Myocardial infarction and stroke are more common in patients with systemic sclerosis, and preventive measures are the same as for the general population.
- Right ventricular dysfunction secondary to pulmonary hypertension is common in systemic sclerosis; left ventricular dysfunction is less so. Routine echocardiography should include assessment of right and left ventricular function.
- Electrocardiography should be performed periodically, and urgently when indicated, to look for potentially dangerous arrhythmias.
Vulvar and gluteal manifestations of Crohn disease
A 37-year-old woman presented with recurring painful swelling and erythema of the vulva over the last year. Despite a series of negative vaginal cultures, she was prescribed multiple courses of antifungal and antibacterial treatments, while her symptoms continued to worsen. She had no other relevant medical history except for occasional diarrhea and abdominal cramping, which were attributed to irritable bowel syndrome.
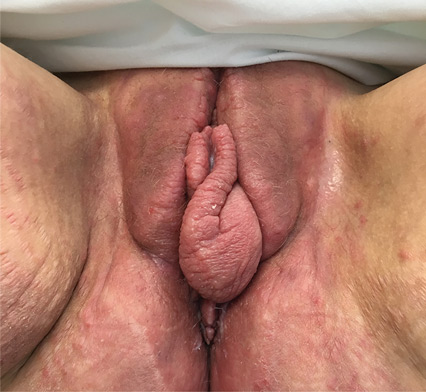
CROHN DISEASE OUTSIDE THE GASTROINTESTINAL TRACT
Crohn disease primarily affects the gastrointestinal tract but is associated with extraintestinal manifestations (in the oral cavity, eyes, skin, and joints) in up to 45% of patients.1
The most common mucocutaneous manifestations are granulomatous lesions that extend directly from the gastrointestinal tract, including perianal and peristomal skin tags, fistulas, and perineal ulcerations. In most cases, the onset of cutaneous manifestations follows intestinal disease, but vulvar Crohn disease may precede gastrointestinal symptoms in approximately 25% of patients, with the average age at onset in the mid-30s.1
The pathogenesis of vulvar Crohn disease remains unclear. One theory involves production of immune complexes from the gastrointestinal tract and a possible T-lymphocyte-mediated type IV hypersensitivity reaction.2

The diagnosis of vulvar Crohn disease should be considered in a patient who has vulvar pain, edema, and ulcerations not otherwise explained, whether or not gastrointestinal Crohn disease is present. The diagnosis is established with clinical history and characteristic histopathology on biopsy. Multiple biopsies may be needed, and early endoscopy is recommended to establish the diagnosis. The histologic features include noncaseating and nonnecrotizing granulomatous dermatitis or vulvitis with occasional reports of eosinophilic infiltrates and necrobiosis.5,6 An imaging study such as ultrasonography is sometimes used to differentiate between a specific cutaneous manifestation of Crohn disease and its complications such as perianal fistula or abscess.
Clinical vulvar lesions are nonspecific, and those of Crohn disease are frequently mistaken for infectious, inflammatory, or traumatic vulvitis. Diagnostic biopsy for histologic analysis is warranted.
- Andreani SM, Ratnasingham K, Dang HH, Gravante G, Giordano P. Crohn’s disease of the vulva. Int J Surg 2010; 8(1):2–5. doi:10.1016/j.ijsu.2009.09.012
- Siroy A, Wasman J. Metastatic Crohn disease: a rare cutaneous entity. Arch Pathol Lab Med 2012; 136(3):329–332. doi:10.5858/arpa.2010-0666-RS
- Foo WC, Papalas JA, Robboy SJ, Selim MA. Vulvar manifestations of Crohn’s disease. Am J Dermatopathol 2011; 33(6):588–593. doi:10.1097/DAD.0b013e31820a2635
- Amankwah Y, Haefner H. Vulvar edema. Dermatol Clin 2010; 28(4):765–777. doi:10.1016/j.det.2010.08.001
- Emanuel PO, Phelps RG. Metastatic Crohn’s disease: a histopathologic study of 12 cases. J Cutan Pathol 2008; 35(5):457–461. doi:10.1111/j.1600-0560.2007.00849.x
- Hackzell-Bradley M, Hedblad MA, Stephansson EA. Metastatic Crohn’s disease: report of 3 cases with special reference to histopathologic findings. Arch Dermatol 1996; 132(8):928–932.
A 37-year-old woman presented with recurring painful swelling and erythema of the vulva over the last year. Despite a series of negative vaginal cultures, she was prescribed multiple courses of antifungal and antibacterial treatments, while her symptoms continued to worsen. She had no other relevant medical history except for occasional diarrhea and abdominal cramping, which were attributed to irritable bowel syndrome.
CROHN DISEASE OUTSIDE THE GASTROINTESTINAL TRACT
Crohn disease primarily affects the gastrointestinal tract but is associated with extraintestinal manifestations (in the oral cavity, eyes, skin, and joints) in up to 45% of patients.1
The most common mucocutaneous manifestations are granulomatous lesions that extend directly from the gastrointestinal tract, including perianal and peristomal skin tags, fistulas, and perineal ulcerations. In most cases, the onset of cutaneous manifestations follows intestinal disease, but vulvar Crohn disease may precede gastrointestinal symptoms in approximately 25% of patients, with the average age at onset in the mid-30s.1
The pathogenesis of vulvar Crohn disease remains unclear. One theory involves production of immune complexes from the gastrointestinal tract and a possible T-lymphocyte-mediated type IV hypersensitivity reaction.2
The diagnosis of vulvar Crohn disease should be considered in a patient who has vulvar pain, edema, and ulcerations not otherwise explained, whether or not gastrointestinal Crohn disease is present. The diagnosis is established with clinical history and characteristic histopathology on biopsy. Multiple biopsies may be needed, and early endoscopy is recommended to establish the diagnosis. The histologic features include noncaseating and nonnecrotizing granulomatous dermatitis or vulvitis with occasional reports of eosinophilic infiltrates and necrobiosis.5,6 An imaging study such as ultrasonography is sometimes used to differentiate between a specific cutaneous manifestation of Crohn disease and its complications such as perianal fistula or abscess.
Clinical vulvar lesions are nonspecific, and those of Crohn disease are frequently mistaken for infectious, inflammatory, or traumatic vulvitis. Diagnostic biopsy for histologic analysis is warranted.
A 37-year-old woman presented with recurring painful swelling and erythema of the vulva over the last year. Despite a series of negative vaginal cultures, she was prescribed multiple courses of antifungal and antibacterial treatments, while her symptoms continued to worsen. She had no other relevant medical history except for occasional diarrhea and abdominal cramping, which were attributed to irritable bowel syndrome.
CROHN DISEASE OUTSIDE THE GASTROINTESTINAL TRACT
Crohn disease primarily affects the gastrointestinal tract but is associated with extraintestinal manifestations (in the oral cavity, eyes, skin, and joints) in up to 45% of patients.1
The most common mucocutaneous manifestations are granulomatous lesions that extend directly from the gastrointestinal tract, including perianal and peristomal skin tags, fistulas, and perineal ulcerations. In most cases, the onset of cutaneous manifestations follows intestinal disease, but vulvar Crohn disease may precede gastrointestinal symptoms in approximately 25% of patients, with the average age at onset in the mid-30s.1
The pathogenesis of vulvar Crohn disease remains unclear. One theory involves production of immune complexes from the gastrointestinal tract and a possible T-lymphocyte-mediated type IV hypersensitivity reaction.2
The diagnosis of vulvar Crohn disease should be considered in a patient who has vulvar pain, edema, and ulcerations not otherwise explained, whether or not gastrointestinal Crohn disease is present. The diagnosis is established with clinical history and characteristic histopathology on biopsy. Multiple biopsies may be needed, and early endoscopy is recommended to establish the diagnosis. The histologic features include noncaseating and nonnecrotizing granulomatous dermatitis or vulvitis with occasional reports of eosinophilic infiltrates and necrobiosis.5,6 An imaging study such as ultrasonography is sometimes used to differentiate between a specific cutaneous manifestation of Crohn disease and its complications such as perianal fistula or abscess.
Clinical vulvar lesions are nonspecific, and those of Crohn disease are frequently mistaken for infectious, inflammatory, or traumatic vulvitis. Diagnostic biopsy for histologic analysis is warranted.
- Andreani SM, Ratnasingham K, Dang HH, Gravante G, Giordano P. Crohn’s disease of the vulva. Int J Surg 2010; 8(1):2–5. doi:10.1016/j.ijsu.2009.09.012
- Siroy A, Wasman J. Metastatic Crohn disease: a rare cutaneous entity. Arch Pathol Lab Med 2012; 136(3):329–332. doi:10.5858/arpa.2010-0666-RS
- Foo WC, Papalas JA, Robboy SJ, Selim MA. Vulvar manifestations of Crohn’s disease. Am J Dermatopathol 2011; 33(6):588–593. doi:10.1097/DAD.0b013e31820a2635
- Amankwah Y, Haefner H. Vulvar edema. Dermatol Clin 2010; 28(4):765–777. doi:10.1016/j.det.2010.08.001
- Emanuel PO, Phelps RG. Metastatic Crohn’s disease: a histopathologic study of 12 cases. J Cutan Pathol 2008; 35(5):457–461. doi:10.1111/j.1600-0560.2007.00849.x
- Hackzell-Bradley M, Hedblad MA, Stephansson EA. Metastatic Crohn’s disease: report of 3 cases with special reference to histopathologic findings. Arch Dermatol 1996; 132(8):928–932.
- Andreani SM, Ratnasingham K, Dang HH, Gravante G, Giordano P. Crohn’s disease of the vulva. Int J Surg 2010; 8(1):2–5. doi:10.1016/j.ijsu.2009.09.012
- Siroy A, Wasman J. Metastatic Crohn disease: a rare cutaneous entity. Arch Pathol Lab Med 2012; 136(3):329–332. doi:10.5858/arpa.2010-0666-RS
- Foo WC, Papalas JA, Robboy SJ, Selim MA. Vulvar manifestations of Crohn’s disease. Am J Dermatopathol 2011; 33(6):588–593. doi:10.1097/DAD.0b013e31820a2635
- Amankwah Y, Haefner H. Vulvar edema. Dermatol Clin 2010; 28(4):765–777. doi:10.1016/j.det.2010.08.001
- Emanuel PO, Phelps RG. Metastatic Crohn’s disease: a histopathologic study of 12 cases. J Cutan Pathol 2008; 35(5):457–461. doi:10.1111/j.1600-0560.2007.00849.x
- Hackzell-Bradley M, Hedblad MA, Stephansson EA. Metastatic Crohn’s disease: report of 3 cases with special reference to histopathologic findings. Arch Dermatol 1996; 132(8):928–932.
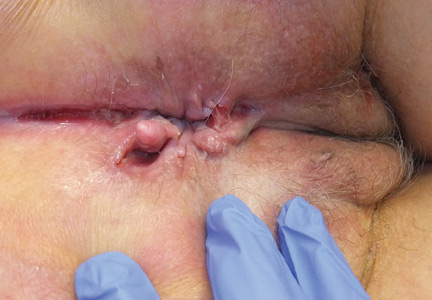
Apple cider vinegar soaks fall short in atopic dermatitis
, in a pilot split-arm study.

The aim of the study was to evaluate the effects of diluted apple cider vinegar application on transepidermal water loss (TEWL) and pH on skin affected by AD and on healthy skin, according to Lydia A. Luu of the department of dermatology at University of Virginia, Charlottesville, and colleagues. “Acetic acid, particularly apple cider vinegar, is prominent among emerging natural remedies used in AD. Therefore, determining the safety of this commonly used product is crucial,” they wrote in the study, published in Pediatric Dermatology.
In total, 11 patients with AD and 11 healthy controls were included; most of those with AD were considered mild (36.4%) or moderate (45.5%). Participants had not used systemic or topical antimicrobial treatments in the month preceding the study, and they were aged 12 years and older (mean ages were 20.6 years in the AD group and 28.8 years among controls). Those with AD had significantly elevated TEWL at baseline, compared with controls.
For 14 days, study participants soaked one forearm in dilute apple cider vinegar (0.5% acetic acid) and the other in tap water for 10 minutes daily. Changes in pH and TEWL before and after application were measured.
The researchers found that TEWL significantly increased immediately post treatment (at 0 and 15 minutes) in both groups, dropping to baseline at 30 minutes among those with AD and at 60 minutes among controls.
Skin pH was similar in both groups at baseline (4.86-4.88). After the cider vinegar soak, there was a transient reduction in skin pH among AD patients that lasted for 15 minutes among those with AD and 60 minutes in controls. This finding “suggests temporary acidification of the skin that has theoretical benefit of correcting disrupted skin pH in AD,” the authors wrote, noting that increased TEWL and alkaline skin pH is common among people with AD because of skin barrier dysfunction.
With respect to safety, 72.7% (16) of the participants experienced skin discomfort, mostly described as mild, limited to the vinegar-treated arm. After discontinuation, the majority of skin irritation resolved quickly, with no additional therapy.
The authors acknowledged two key limitations of the study were the homogeneous patient population and small sample size. “Although epidermal acidification would theoretically be beneficial in treating AD, our study shows that acidification by way of topical bathing in a 0.5% [apple cider vinegar] solution as performed in this study is not useful in AD treatment,” they wrote. “Further studies in a more diverse population will be necessary to fully characterize the risk/benefit profile of topical dilute apple cider vinegar treatments.”
The study was funded by the University of Virginia. The authors did not provide information on financial disclosures.
SOURCE: Luu LA et al. Pediatr Dermatol. 2019 Jul 22. doi: 10.1111/pde.13888.
, in a pilot split-arm study.
The aim of the study was to evaluate the effects of diluted apple cider vinegar application on transepidermal water loss (TEWL) and pH on skin affected by AD and on healthy skin, according to Lydia A. Luu of the department of dermatology at University of Virginia, Charlottesville, and colleagues. “Acetic acid, particularly apple cider vinegar, is prominent among emerging natural remedies used in AD. Therefore, determining the safety of this commonly used product is crucial,” they wrote in the study, published in Pediatric Dermatology.
In total, 11 patients with AD and 11 healthy controls were included; most of those with AD were considered mild (36.4%) or moderate (45.5%). Participants had not used systemic or topical antimicrobial treatments in the month preceding the study, and they were aged 12 years and older (mean ages were 20.6 years in the AD group and 28.8 years among controls). Those with AD had significantly elevated TEWL at baseline, compared with controls.
For 14 days, study participants soaked one forearm in dilute apple cider vinegar (0.5% acetic acid) and the other in tap water for 10 minutes daily. Changes in pH and TEWL before and after application were measured.
The researchers found that TEWL significantly increased immediately post treatment (at 0 and 15 minutes) in both groups, dropping to baseline at 30 minutes among those with AD and at 60 minutes among controls.
Skin pH was similar in both groups at baseline (4.86-4.88). After the cider vinegar soak, there was a transient reduction in skin pH among AD patients that lasted for 15 minutes among those with AD and 60 minutes in controls. This finding “suggests temporary acidification of the skin that has theoretical benefit of correcting disrupted skin pH in AD,” the authors wrote, noting that increased TEWL and alkaline skin pH is common among people with AD because of skin barrier dysfunction.
With respect to safety, 72.7% (16) of the participants experienced skin discomfort, mostly described as mild, limited to the vinegar-treated arm. After discontinuation, the majority of skin irritation resolved quickly, with no additional therapy.
The authors acknowledged two key limitations of the study were the homogeneous patient population and small sample size. “Although epidermal acidification would theoretically be beneficial in treating AD, our study shows that acidification by way of topical bathing in a 0.5% [apple cider vinegar] solution as performed in this study is not useful in AD treatment,” they wrote. “Further studies in a more diverse population will be necessary to fully characterize the risk/benefit profile of topical dilute apple cider vinegar treatments.”
The study was funded by the University of Virginia. The authors did not provide information on financial disclosures.
SOURCE: Luu LA et al. Pediatr Dermatol. 2019 Jul 22. doi: 10.1111/pde.13888.
, in a pilot split-arm study.
The aim of the study was to evaluate the effects of diluted apple cider vinegar application on transepidermal water loss (TEWL) and pH on skin affected by AD and on healthy skin, according to Lydia A. Luu of the department of dermatology at University of Virginia, Charlottesville, and colleagues. “Acetic acid, particularly apple cider vinegar, is prominent among emerging natural remedies used in AD. Therefore, determining the safety of this commonly used product is crucial,” they wrote in the study, published in Pediatric Dermatology.
In total, 11 patients with AD and 11 healthy controls were included; most of those with AD were considered mild (36.4%) or moderate (45.5%). Participants had not used systemic or topical antimicrobial treatments in the month preceding the study, and they were aged 12 years and older (mean ages were 20.6 years in the AD group and 28.8 years among controls). Those with AD had significantly elevated TEWL at baseline, compared with controls.
For 14 days, study participants soaked one forearm in dilute apple cider vinegar (0.5% acetic acid) and the other in tap water for 10 minutes daily. Changes in pH and TEWL before and after application were measured.
The researchers found that TEWL significantly increased immediately post treatment (at 0 and 15 minutes) in both groups, dropping to baseline at 30 minutes among those with AD and at 60 minutes among controls.
Skin pH was similar in both groups at baseline (4.86-4.88). After the cider vinegar soak, there was a transient reduction in skin pH among AD patients that lasted for 15 minutes among those with AD and 60 minutes in controls. This finding “suggests temporary acidification of the skin that has theoretical benefit of correcting disrupted skin pH in AD,” the authors wrote, noting that increased TEWL and alkaline skin pH is common among people with AD because of skin barrier dysfunction.
With respect to safety, 72.7% (16) of the participants experienced skin discomfort, mostly described as mild, limited to the vinegar-treated arm. After discontinuation, the majority of skin irritation resolved quickly, with no additional therapy.
The authors acknowledged two key limitations of the study were the homogeneous patient population and small sample size. “Although epidermal acidification would theoretically be beneficial in treating AD, our study shows that acidification by way of topical bathing in a 0.5% [apple cider vinegar] solution as performed in this study is not useful in AD treatment,” they wrote. “Further studies in a more diverse population will be necessary to fully characterize the risk/benefit profile of topical dilute apple cider vinegar treatments.”
The study was funded by the University of Virginia. The authors did not provide information on financial disclosures.
SOURCE: Luu LA et al. Pediatr Dermatol. 2019 Jul 22. doi: 10.1111/pde.13888.
FROM PEDIATRIC DERMATOLOGY