User login
Red hair in women linked to elevated CRP levels in Nurses’ Health Study
Red-haired women were significantly more likely than were women with nonred hair to have elevated levels of C-reactive protein that may increase risk for cardiovascular conditions, according to data from nearly 9,000 women participating in the Nurses’ Health Study.
“Positive associations between red hair and cardiovascular disease and cancer in women, but not men, have been reported,” wrote Rebecca I. Hartman, MD, of Brigham and Women’s Hospital, Harvard Medical School, Boston, and colleagues.
In a study published in the Journal of Investigative Dermatology, they reviewed data from the Nurses’ Health Study, a 1976 cohort study of 121,700 women registered nurses in the United States. They analyzed blood specimens from 8,994 women that were collected between 1989 and 1990. Participants’ natural hair color was determined by asking them their natural hair color at age 21 years, with choices of red, blonde, light brown, dark brown, or black. Overall, dark brown/black hair was the most common color (45%) and 390 of the women (4.3%) had red hair.
The average CRP levels were significantly higher for women with red hair (3.7 mg/L), compared with those with blonde (3.3 mg/L), light brown (3.0 mg/mL), or dark brown/black (3.2 mg/L).
Using the CRP levels for red-haired women as a reference, women with blond, light brown, and dark brown/black hair averaged significantly lower CRP levels than those of red-haired women in an age-adjusted model (–15.2%, –18/1%, and –14.2%, respectively) and in a multivariate analysis (–12.7%, –14.1%, and –10.9%, respectively).
Non-red-haired women had significantly lower odds of high CRP levels compared with red-haired women, with odds ratios of 0.62, 0.60, and 0.67 for women with blonde, light brown, and dark brown/black hair, respectively, in multivariate analysis, the researchers found.
The study was limited by several factors including the use of self-reports for hair color and the relative homogeneity of the Nurses’ Health Study, which has a population of mostly white, female health professionals, the researchers noted.
However, the findings of significantly increased CRP levels “could potentially explain a prior report of increased risks of cardiovascular disease and cancer in red-haired women,” they said. “Although, we observed similar associations in the NHS between red hair and cardiovascular disease and cancer, they were not statistically significant,” they added.
Additional studies are needed to validate and examine the clinical significance of the results, they concluded.
“Elevated CRP levels, a marker of inflammation, have been associated with increased risk for several diseases, including colon cancer and heart disease,” lead author Dr. Hartman said in an interview. “Another study suggested red-haired women have elevated risks of cardiovascular disease and cancer. We wanted to see if different levels of inflammation in red-haired women could possibly explain these findings.”
She said she was not surprised by the findings, “as they were in line with our hypothesis.” In addition, “animal studies suggest that the gene most responsible for red hair, MC1R, may be linked to inflammation,” she said.
While red-haired women were found to have higher CRP levels in the study, “the underlying mechanism and clinical significance remain unknown,” and more research is needed, Dr. Hartman emphasized. “First, our findings need to be validated in women and also examined in men. If our findings are validated, future studies should examine the mechanism of CRP elevation in red-haired women, and whether these women have elevated risks of colon cancer and heart disease,” she said.
“If red-haired women do have increased levels of inflammation, and as a result have elevated risks of colon cancer and heart disease, then future interventions can focus on enhanced screening and possibly chemoprevention in this population,” she added.
The study was supported by the National Institutes of Health. Lead author Dr. Hartman was supported by an American Skin Association Research Grant.
SOURCE: Hartman RI et al. J Invest Dermatol. 2020 Oct 12. doi: 10.1016/j.jid.2020.09.015.
Red-haired women were significantly more likely than were women with nonred hair to have elevated levels of C-reactive protein that may increase risk for cardiovascular conditions, according to data from nearly 9,000 women participating in the Nurses’ Health Study.
“Positive associations between red hair and cardiovascular disease and cancer in women, but not men, have been reported,” wrote Rebecca I. Hartman, MD, of Brigham and Women’s Hospital, Harvard Medical School, Boston, and colleagues.
In a study published in the Journal of Investigative Dermatology, they reviewed data from the Nurses’ Health Study, a 1976 cohort study of 121,700 women registered nurses in the United States. They analyzed blood specimens from 8,994 women that were collected between 1989 and 1990. Participants’ natural hair color was determined by asking them their natural hair color at age 21 years, with choices of red, blonde, light brown, dark brown, or black. Overall, dark brown/black hair was the most common color (45%) and 390 of the women (4.3%) had red hair.
The average CRP levels were significantly higher for women with red hair (3.7 mg/L), compared with those with blonde (3.3 mg/L), light brown (3.0 mg/mL), or dark brown/black (3.2 mg/L).
Using the CRP levels for red-haired women as a reference, women with blond, light brown, and dark brown/black hair averaged significantly lower CRP levels than those of red-haired women in an age-adjusted model (–15.2%, –18/1%, and –14.2%, respectively) and in a multivariate analysis (–12.7%, –14.1%, and –10.9%, respectively).
Non-red-haired women had significantly lower odds of high CRP levels compared with red-haired women, with odds ratios of 0.62, 0.60, and 0.67 for women with blonde, light brown, and dark brown/black hair, respectively, in multivariate analysis, the researchers found.
The study was limited by several factors including the use of self-reports for hair color and the relative homogeneity of the Nurses’ Health Study, which has a population of mostly white, female health professionals, the researchers noted.
However, the findings of significantly increased CRP levels “could potentially explain a prior report of increased risks of cardiovascular disease and cancer in red-haired women,” they said. “Although, we observed similar associations in the NHS between red hair and cardiovascular disease and cancer, they were not statistically significant,” they added.
Additional studies are needed to validate and examine the clinical significance of the results, they concluded.
“Elevated CRP levels, a marker of inflammation, have been associated with increased risk for several diseases, including colon cancer and heart disease,” lead author Dr. Hartman said in an interview. “Another study suggested red-haired women have elevated risks of cardiovascular disease and cancer. We wanted to see if different levels of inflammation in red-haired women could possibly explain these findings.”
She said she was not surprised by the findings, “as they were in line with our hypothesis.” In addition, “animal studies suggest that the gene most responsible for red hair, MC1R, may be linked to inflammation,” she said.
While red-haired women were found to have higher CRP levels in the study, “the underlying mechanism and clinical significance remain unknown,” and more research is needed, Dr. Hartman emphasized. “First, our findings need to be validated in women and also examined in men. If our findings are validated, future studies should examine the mechanism of CRP elevation in red-haired women, and whether these women have elevated risks of colon cancer and heart disease,” she said.
“If red-haired women do have increased levels of inflammation, and as a result have elevated risks of colon cancer and heart disease, then future interventions can focus on enhanced screening and possibly chemoprevention in this population,” she added.
The study was supported by the National Institutes of Health. Lead author Dr. Hartman was supported by an American Skin Association Research Grant.
SOURCE: Hartman RI et al. J Invest Dermatol. 2020 Oct 12. doi: 10.1016/j.jid.2020.09.015.
Red-haired women were significantly more likely than were women with nonred hair to have elevated levels of C-reactive protein that may increase risk for cardiovascular conditions, according to data from nearly 9,000 women participating in the Nurses’ Health Study.
“Positive associations between red hair and cardiovascular disease and cancer in women, but not men, have been reported,” wrote Rebecca I. Hartman, MD, of Brigham and Women’s Hospital, Harvard Medical School, Boston, and colleagues.
In a study published in the Journal of Investigative Dermatology, they reviewed data from the Nurses’ Health Study, a 1976 cohort study of 121,700 women registered nurses in the United States. They analyzed blood specimens from 8,994 women that were collected between 1989 and 1990. Participants’ natural hair color was determined by asking them their natural hair color at age 21 years, with choices of red, blonde, light brown, dark brown, or black. Overall, dark brown/black hair was the most common color (45%) and 390 of the women (4.3%) had red hair.
The average CRP levels were significantly higher for women with red hair (3.7 mg/L), compared with those with blonde (3.3 mg/L), light brown (3.0 mg/mL), or dark brown/black (3.2 mg/L).
Using the CRP levels for red-haired women as a reference, women with blond, light brown, and dark brown/black hair averaged significantly lower CRP levels than those of red-haired women in an age-adjusted model (–15.2%, –18/1%, and –14.2%, respectively) and in a multivariate analysis (–12.7%, –14.1%, and –10.9%, respectively).
Non-red-haired women had significantly lower odds of high CRP levels compared with red-haired women, with odds ratios of 0.62, 0.60, and 0.67 for women with blonde, light brown, and dark brown/black hair, respectively, in multivariate analysis, the researchers found.
The study was limited by several factors including the use of self-reports for hair color and the relative homogeneity of the Nurses’ Health Study, which has a population of mostly white, female health professionals, the researchers noted.
However, the findings of significantly increased CRP levels “could potentially explain a prior report of increased risks of cardiovascular disease and cancer in red-haired women,” they said. “Although, we observed similar associations in the NHS between red hair and cardiovascular disease and cancer, they were not statistically significant,” they added.
Additional studies are needed to validate and examine the clinical significance of the results, they concluded.
“Elevated CRP levels, a marker of inflammation, have been associated with increased risk for several diseases, including colon cancer and heart disease,” lead author Dr. Hartman said in an interview. “Another study suggested red-haired women have elevated risks of cardiovascular disease and cancer. We wanted to see if different levels of inflammation in red-haired women could possibly explain these findings.”
She said she was not surprised by the findings, “as they were in line with our hypothesis.” In addition, “animal studies suggest that the gene most responsible for red hair, MC1R, may be linked to inflammation,” she said.
While red-haired women were found to have higher CRP levels in the study, “the underlying mechanism and clinical significance remain unknown,” and more research is needed, Dr. Hartman emphasized. “First, our findings need to be validated in women and also examined in men. If our findings are validated, future studies should examine the mechanism of CRP elevation in red-haired women, and whether these women have elevated risks of colon cancer and heart disease,” she said.
“If red-haired women do have increased levels of inflammation, and as a result have elevated risks of colon cancer and heart disease, then future interventions can focus on enhanced screening and possibly chemoprevention in this population,” she added.
The study was supported by the National Institutes of Health. Lead author Dr. Hartman was supported by an American Skin Association Research Grant.
SOURCE: Hartman RI et al. J Invest Dermatol. 2020 Oct 12. doi: 10.1016/j.jid.2020.09.015.
FROM THE JOURNAL OF INVESTIGATIVE DERMATOLOGY
Eyebrow hair loss

Although it appeared that the hair loss was preceded by some dry skin, the patch of hair loss was smooth and nonscarring, consistent with a diagnosis of alopecia areata (AA).
AA typically is found on the scalp in solitary areas but can affect the whole body, including the eyelashes and eyebrows. Affected hair typically is narrower at the proximal end, resembling an exclamation point, as the hair fails to grow and falls out. Sometimes, patches of alopecia may coalesce into a larger area. Nail changes may be noted, as well. Nails may become brittle, with pitting and/or longitudinal ridges. Patients are usually asymptomatic but may complain of pruritus.
AA is believed to be an autoimmune disorder. It affects males and females of all ages but is more common in children and young adults. AA is believed to result from a T-cell–mediated immune response that transitions the hair follicles from the growth phase to the resting phase. This leads to sudden hair loss and inhibition of regrowth of the hair. However, the hair follicle is not permanently destroyed as in other processes of alopecia. There is also an association between AA and other autoimmune disorders such as vitiligo, thyroid disease, and lupus.
The diagnosis usually is made clinically, as in this patient, but a definitive diagnosis can be made by biopsy and pathology. Other differential diagnoses to consider are trichotillomania, tinea, traumatic alopecia, and lupus.
In almost half of cases, AA is self-resolving; therefore, in first episodes of localized disease, watchful waiting is appropriate. Treatment is tailored to the individual patient and may be difficult. Intralesional corticosteroids often are used for mild cases. Typically, 10 mg/mL of a glucocorticoid is injected into the mid-dermis every 4 to 6 weeks; however, this treatment carries a risk of transient or even permanent atrophy of the injection site. Potent topical corticosteroids often are used, especially in children who do not tolerate intralesional injections, and success can be variable. Other topical treatments to consider are photochemotherapy (psoralen plus UVA), or an irritant agent such as anthralin and a vasodilator such as minoxidil. Regrowth can be expected in a few months to a year, but recurrence is common.
Image courtesy of Stacy Nguy, MD, and text courtesy of Stacy Nguy, MD, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Papadopoulos AJ, Schwartz RA, Janniger C. Alopecia areata: pathogenesis, diagnosis, and therapy. Am J Clin Dermatol. 2000;1:101-105
Although it appeared that the hair loss was preceded by some dry skin, the patch of hair loss was smooth and nonscarring, consistent with a diagnosis of alopecia areata (AA).
AA typically is found on the scalp in solitary areas but can affect the whole body, including the eyelashes and eyebrows. Affected hair typically is narrower at the proximal end, resembling an exclamation point, as the hair fails to grow and falls out. Sometimes, patches of alopecia may coalesce into a larger area. Nail changes may be noted, as well. Nails may become brittle, with pitting and/or longitudinal ridges. Patients are usually asymptomatic but may complain of pruritus.
AA is believed to be an autoimmune disorder. It affects males and females of all ages but is more common in children and young adults. AA is believed to result from a T-cell–mediated immune response that transitions the hair follicles from the growth phase to the resting phase. This leads to sudden hair loss and inhibition of regrowth of the hair. However, the hair follicle is not permanently destroyed as in other processes of alopecia. There is also an association between AA and other autoimmune disorders such as vitiligo, thyroid disease, and lupus.
The diagnosis usually is made clinically, as in this patient, but a definitive diagnosis can be made by biopsy and pathology. Other differential diagnoses to consider are trichotillomania, tinea, traumatic alopecia, and lupus.
In almost half of cases, AA is self-resolving; therefore, in first episodes of localized disease, watchful waiting is appropriate. Treatment is tailored to the individual patient and may be difficult. Intralesional corticosteroids often are used for mild cases. Typically, 10 mg/mL of a glucocorticoid is injected into the mid-dermis every 4 to 6 weeks; however, this treatment carries a risk of transient or even permanent atrophy of the injection site. Potent topical corticosteroids often are used, especially in children who do not tolerate intralesional injections, and success can be variable. Other topical treatments to consider are photochemotherapy (psoralen plus UVA), or an irritant agent such as anthralin and a vasodilator such as minoxidil. Regrowth can be expected in a few months to a year, but recurrence is common.
Image courtesy of Stacy Nguy, MD, and text courtesy of Stacy Nguy, MD, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Although it appeared that the hair loss was preceded by some dry skin, the patch of hair loss was smooth and nonscarring, consistent with a diagnosis of alopecia areata (AA).
AA typically is found on the scalp in solitary areas but can affect the whole body, including the eyelashes and eyebrows. Affected hair typically is narrower at the proximal end, resembling an exclamation point, as the hair fails to grow and falls out. Sometimes, patches of alopecia may coalesce into a larger area. Nail changes may be noted, as well. Nails may become brittle, with pitting and/or longitudinal ridges. Patients are usually asymptomatic but may complain of pruritus.
AA is believed to be an autoimmune disorder. It affects males and females of all ages but is more common in children and young adults. AA is believed to result from a T-cell–mediated immune response that transitions the hair follicles from the growth phase to the resting phase. This leads to sudden hair loss and inhibition of regrowth of the hair. However, the hair follicle is not permanently destroyed as in other processes of alopecia. There is also an association between AA and other autoimmune disorders such as vitiligo, thyroid disease, and lupus.
The diagnosis usually is made clinically, as in this patient, but a definitive diagnosis can be made by biopsy and pathology. Other differential diagnoses to consider are trichotillomania, tinea, traumatic alopecia, and lupus.
In almost half of cases, AA is self-resolving; therefore, in first episodes of localized disease, watchful waiting is appropriate. Treatment is tailored to the individual patient and may be difficult. Intralesional corticosteroids often are used for mild cases. Typically, 10 mg/mL of a glucocorticoid is injected into the mid-dermis every 4 to 6 weeks; however, this treatment carries a risk of transient or even permanent atrophy of the injection site. Potent topical corticosteroids often are used, especially in children who do not tolerate intralesional injections, and success can be variable. Other topical treatments to consider are photochemotherapy (psoralen plus UVA), or an irritant agent such as anthralin and a vasodilator such as minoxidil. Regrowth can be expected in a few months to a year, but recurrence is common.
Image courtesy of Stacy Nguy, MD, and text courtesy of Stacy Nguy, MD, and Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Papadopoulos AJ, Schwartz RA, Janniger C. Alopecia areata: pathogenesis, diagnosis, and therapy. Am J Clin Dermatol. 2000;1:101-105
Papadopoulos AJ, Schwartz RA, Janniger C. Alopecia areata: pathogenesis, diagnosis, and therapy. Am J Clin Dermatol. 2000;1:101-105

Systemic sclerosis patients share their perspectives and needs in treatment trials
Patients with systemic sclerosis have variable disease progression but often experience debilitating fatigue, pain, and digestive issues – and they’re extremely concerned about progressive organ damage, according to those who spoke at and provided input at a public meeting on patient-focused drug development for the disease.

The virtual meeting was part of the Food and Drug Administration’s Patient-Focused Drug Development (PFDD) initiative, which began in 2012 and aims to provide a systematic way for patients’ experiences, needs, and priorities to be “captured and meaningfully incorporated” into drug development and evaluation.
Patients rate their most impactful symptoms
Dinesh Khanna, MBBS, MSc, a rheumatologist who directs a scleroderma research program at the University of Michigan, Ann Arbor, attended the meeting after giving an opening presentation on the disease to FDA officials, patients, and other participants. In a later interview, he said that patients’ ratings of their most impactful symptoms was especially striking.

Raynaud’s phenomenon, digestive symptoms, and fatigue were the top three answers to a poll question that asked patients what symptom had the most significant impact on daily life, he noted, “and none of these are being [strongly] addressed right now [in clinical trials] apart from Raynaud’s phenomenon, for which there are some trials ongoing.”
He and other researchers are “struggling with what outcomes measures to use [in their studies],” said Dr. Khanna, the Frederick G.L. Huetwell Professor of Rheumatology at the University. “My takeaway from the meeting as a clinical trialist is that we should be paying close attention to the symptoms that patients tell us are the most important. We should be including these in our trial designs as secondary endpoints, if not primary endpoints. We have not done that [thus far], really.”
Approximately 200,000 patients in the United States have scleroderma, and approximately 75,000-80,000 of these patients have systemic scleroderma, or systemic sclerosis, Dr. Khanna said in his opening presentation. Each year, he estimates, about 6,000 new diagnoses of systemic sclerosis are made.
More than 200 people – patients, FDA officials, and others – participated in the PFDD meeting. Patients participated in one of two panels – one focused on health effects and daily impacts, and the other on treatments – or submitted input electronically. All were invited to answer poll questions.
Raj Nair, MD, one of eight FDA leaders attending the meeting, noted in closing remarks that the pain experienced by patients with systemic sclerosis includes severe pain from Raynaud’s phenomenon and pain caused by digital ulcers and by calcinosis. “We heard about how paralyzing the pain from calcinosis is, and that there are very few options for alleviating this pain,” said Dr. Nair, of the division of rheumatology and transplant medicine.
Another takeaway, he said, is that the “fatigue can be severe and debilitating, leading to days where it is impossible to get out of bed,” and that digestive symptoms can also be severe. “Reflux,” he noted, “requires significant medical intervention.”
Patients describe their experiences
Rosemary Lyons, diagnosed with scleroderma 35 years ago, explained that while her skin is no longer hardened, she is overly sensitive to fabrics and skin care products and has difficulty with sleeping and eating. She moved away from family in the Northeast to live in the South where the climate is warmer, but even on a 90-degree night she needs a blanket and two comforters to curb the cold and attempt to sleep.
Impaired gastrointestinal motility has made food her “biggest problem” for the past 10 years, and because of GI symptoms, she can eat only one meal a day. She also experiences fainting, brain fog, and severe fatigue. On a good day, Ms. Lyons noted, she sometimes opts to do some house chores “knowing that I’ll have 1-3 days of recovery.”
Another patient, Amy Harding, said that 22 years after her scleroderma diagnosis, “the calcinosis I get in my fingers, elbows, toes, and ears tops all the prior symptoms.” The skin tightening and digital ulcers that she experienced in the first 10 years have tapered off, and while Raynaud’s symptoms and heartburn have worsened, they are at least partly manageable with medications, unlike the pain from calcinosis.
Treating symptoms vs. disease may be key in risk-benefit analysis
In questions after patient presentations, FDA officials probed for more perspective on issues such as how fatigue should be assessed, the differences between fatigue and brain fog, the impact of calcinosis on functioning, and how much risk patients would be willing to assume from treatments that have side effects and that may or may not modulate the disease and slow disease progression.
Most patients said in response to an FDA poll question that they definitely (almost 40%) or possibly (almost 50%) would be willing to try a hypothetical new self-injectable medication if it were shown to reduce their most impactful symptoms but had side effects.
“I think what [we’ve been hearing] today is that whether we’re working on the symptoms or the disease itself is [the key]” to patients’ risk-benefit analysis, said meeting moderator Capt. Robyn Bent, RN, MS, of the U.S. Public Health Service, and director of the PFDD.
Anita Devine, diagnosed 13 years ago with systemic sclerosis, was one of several panel members who said she would accept more bothersome treatment side effects and risks “if the gain was control of disease progression and overall quality of life ... and organ preservation.” Ms. Devine, who has needed kidney dialysis and multiple hand surgeries, noted that she previously took anti-neoplastic and anti-inflammatory agents “to try to stem the course of my disease, but unfortunately the disease did not abate.”
Treatments for systemic sclerosis include vasodilators, immunosuppressive medications, antifibrotic therapies, and stem cell transplants, Dr. Khanna said in his opening remarks.
Trials of drugs for scleroderma have focused on early disease that may be amenable to treatment, with the exception of trials for pulmonary arterial hypertension, which affects some patients with systemic sclerosis. There are multiple FDA-approved drugs for pulmonary arterial hypertension and more trials are underway.
Outcomes such as pain and fatigue are included in many of the trials currently underway, but they tend to be lower-level secondary outcomes measures that cannot be incorporated into drug labeling or are more “exploratory in nature,” Dr. Khanna said in the interview.
Dr. Khanna disclosed that he is the chief medical officer (an equity position) for CiVi Biopharma/Eicos Sciences Inc., which is developing a drug for Raynaud’s, and serves as a consultant and grant recipient for numerous companies that make or are developing drugs for systemic sclerosis.
The FDA will accept patient comments until Dec. 15, 2020, at which time comments will be compiled into a summary report, Ms. Bent said.
Patients with systemic sclerosis have variable disease progression but often experience debilitating fatigue, pain, and digestive issues – and they’re extremely concerned about progressive organ damage, according to those who spoke at and provided input at a public meeting on patient-focused drug development for the disease.
The virtual meeting was part of the Food and Drug Administration’s Patient-Focused Drug Development (PFDD) initiative, which began in 2012 and aims to provide a systematic way for patients’ experiences, needs, and priorities to be “captured and meaningfully incorporated” into drug development and evaluation.
Patients rate their most impactful symptoms
Dinesh Khanna, MBBS, MSc, a rheumatologist who directs a scleroderma research program at the University of Michigan, Ann Arbor, attended the meeting after giving an opening presentation on the disease to FDA officials, patients, and other participants. In a later interview, he said that patients’ ratings of their most impactful symptoms was especially striking.
Raynaud’s phenomenon, digestive symptoms, and fatigue were the top three answers to a poll question that asked patients what symptom had the most significant impact on daily life, he noted, “and none of these are being [strongly] addressed right now [in clinical trials] apart from Raynaud’s phenomenon, for which there are some trials ongoing.”
He and other researchers are “struggling with what outcomes measures to use [in their studies],” said Dr. Khanna, the Frederick G.L. Huetwell Professor of Rheumatology at the University. “My takeaway from the meeting as a clinical trialist is that we should be paying close attention to the symptoms that patients tell us are the most important. We should be including these in our trial designs as secondary endpoints, if not primary endpoints. We have not done that [thus far], really.”
Approximately 200,000 patients in the United States have scleroderma, and approximately 75,000-80,000 of these patients have systemic scleroderma, or systemic sclerosis, Dr. Khanna said in his opening presentation. Each year, he estimates, about 6,000 new diagnoses of systemic sclerosis are made.
More than 200 people – patients, FDA officials, and others – participated in the PFDD meeting. Patients participated in one of two panels – one focused on health effects and daily impacts, and the other on treatments – or submitted input electronically. All were invited to answer poll questions.
Raj Nair, MD, one of eight FDA leaders attending the meeting, noted in closing remarks that the pain experienced by patients with systemic sclerosis includes severe pain from Raynaud’s phenomenon and pain caused by digital ulcers and by calcinosis. “We heard about how paralyzing the pain from calcinosis is, and that there are very few options for alleviating this pain,” said Dr. Nair, of the division of rheumatology and transplant medicine.
Another takeaway, he said, is that the “fatigue can be severe and debilitating, leading to days where it is impossible to get out of bed,” and that digestive symptoms can also be severe. “Reflux,” he noted, “requires significant medical intervention.”
Patients describe their experiences
Rosemary Lyons, diagnosed with scleroderma 35 years ago, explained that while her skin is no longer hardened, she is overly sensitive to fabrics and skin care products and has difficulty with sleeping and eating. She moved away from family in the Northeast to live in the South where the climate is warmer, but even on a 90-degree night she needs a blanket and two comforters to curb the cold and attempt to sleep.
Impaired gastrointestinal motility has made food her “biggest problem” for the past 10 years, and because of GI symptoms, she can eat only one meal a day. She also experiences fainting, brain fog, and severe fatigue. On a good day, Ms. Lyons noted, she sometimes opts to do some house chores “knowing that I’ll have 1-3 days of recovery.”
Another patient, Amy Harding, said that 22 years after her scleroderma diagnosis, “the calcinosis I get in my fingers, elbows, toes, and ears tops all the prior symptoms.” The skin tightening and digital ulcers that she experienced in the first 10 years have tapered off, and while Raynaud’s symptoms and heartburn have worsened, they are at least partly manageable with medications, unlike the pain from calcinosis.
Treating symptoms vs. disease may be key in risk-benefit analysis
In questions after patient presentations, FDA officials probed for more perspective on issues such as how fatigue should be assessed, the differences between fatigue and brain fog, the impact of calcinosis on functioning, and how much risk patients would be willing to assume from treatments that have side effects and that may or may not modulate the disease and slow disease progression.
Most patients said in response to an FDA poll question that they definitely (almost 40%) or possibly (almost 50%) would be willing to try a hypothetical new self-injectable medication if it were shown to reduce their most impactful symptoms but had side effects.
“I think what [we’ve been hearing] today is that whether we’re working on the symptoms or the disease itself is [the key]” to patients’ risk-benefit analysis, said meeting moderator Capt. Robyn Bent, RN, MS, of the U.S. Public Health Service, and director of the PFDD.
Anita Devine, diagnosed 13 years ago with systemic sclerosis, was one of several panel members who said she would accept more bothersome treatment side effects and risks “if the gain was control of disease progression and overall quality of life ... and organ preservation.” Ms. Devine, who has needed kidney dialysis and multiple hand surgeries, noted that she previously took anti-neoplastic and anti-inflammatory agents “to try to stem the course of my disease, but unfortunately the disease did not abate.”
Treatments for systemic sclerosis include vasodilators, immunosuppressive medications, antifibrotic therapies, and stem cell transplants, Dr. Khanna said in his opening remarks.
Trials of drugs for scleroderma have focused on early disease that may be amenable to treatment, with the exception of trials for pulmonary arterial hypertension, which affects some patients with systemic sclerosis. There are multiple FDA-approved drugs for pulmonary arterial hypertension and more trials are underway.
Outcomes such as pain and fatigue are included in many of the trials currently underway, but they tend to be lower-level secondary outcomes measures that cannot be incorporated into drug labeling or are more “exploratory in nature,” Dr. Khanna said in the interview.
Dr. Khanna disclosed that he is the chief medical officer (an equity position) for CiVi Biopharma/Eicos Sciences Inc., which is developing a drug for Raynaud’s, and serves as a consultant and grant recipient for numerous companies that make or are developing drugs for systemic sclerosis.
The FDA will accept patient comments until Dec. 15, 2020, at which time comments will be compiled into a summary report, Ms. Bent said.
Patients with systemic sclerosis have variable disease progression but often experience debilitating fatigue, pain, and digestive issues – and they’re extremely concerned about progressive organ damage, according to those who spoke at and provided input at a public meeting on patient-focused drug development for the disease.
The virtual meeting was part of the Food and Drug Administration’s Patient-Focused Drug Development (PFDD) initiative, which began in 2012 and aims to provide a systematic way for patients’ experiences, needs, and priorities to be “captured and meaningfully incorporated” into drug development and evaluation.
Patients rate their most impactful symptoms
Dinesh Khanna, MBBS, MSc, a rheumatologist who directs a scleroderma research program at the University of Michigan, Ann Arbor, attended the meeting after giving an opening presentation on the disease to FDA officials, patients, and other participants. In a later interview, he said that patients’ ratings of their most impactful symptoms was especially striking.
Raynaud’s phenomenon, digestive symptoms, and fatigue were the top three answers to a poll question that asked patients what symptom had the most significant impact on daily life, he noted, “and none of these are being [strongly] addressed right now [in clinical trials] apart from Raynaud’s phenomenon, for which there are some trials ongoing.”
He and other researchers are “struggling with what outcomes measures to use [in their studies],” said Dr. Khanna, the Frederick G.L. Huetwell Professor of Rheumatology at the University. “My takeaway from the meeting as a clinical trialist is that we should be paying close attention to the symptoms that patients tell us are the most important. We should be including these in our trial designs as secondary endpoints, if not primary endpoints. We have not done that [thus far], really.”
Approximately 200,000 patients in the United States have scleroderma, and approximately 75,000-80,000 of these patients have systemic scleroderma, or systemic sclerosis, Dr. Khanna said in his opening presentation. Each year, he estimates, about 6,000 new diagnoses of systemic sclerosis are made.
More than 200 people – patients, FDA officials, and others – participated in the PFDD meeting. Patients participated in one of two panels – one focused on health effects and daily impacts, and the other on treatments – or submitted input electronically. All were invited to answer poll questions.
Raj Nair, MD, one of eight FDA leaders attending the meeting, noted in closing remarks that the pain experienced by patients with systemic sclerosis includes severe pain from Raynaud’s phenomenon and pain caused by digital ulcers and by calcinosis. “We heard about how paralyzing the pain from calcinosis is, and that there are very few options for alleviating this pain,” said Dr. Nair, of the division of rheumatology and transplant medicine.
Another takeaway, he said, is that the “fatigue can be severe and debilitating, leading to days where it is impossible to get out of bed,” and that digestive symptoms can also be severe. “Reflux,” he noted, “requires significant medical intervention.”
Patients describe their experiences
Rosemary Lyons, diagnosed with scleroderma 35 years ago, explained that while her skin is no longer hardened, she is overly sensitive to fabrics and skin care products and has difficulty with sleeping and eating. She moved away from family in the Northeast to live in the South where the climate is warmer, but even on a 90-degree night she needs a blanket and two comforters to curb the cold and attempt to sleep.
Impaired gastrointestinal motility has made food her “biggest problem” for the past 10 years, and because of GI symptoms, she can eat only one meal a day. She also experiences fainting, brain fog, and severe fatigue. On a good day, Ms. Lyons noted, she sometimes opts to do some house chores “knowing that I’ll have 1-3 days of recovery.”
Another patient, Amy Harding, said that 22 years after her scleroderma diagnosis, “the calcinosis I get in my fingers, elbows, toes, and ears tops all the prior symptoms.” The skin tightening and digital ulcers that she experienced in the first 10 years have tapered off, and while Raynaud’s symptoms and heartburn have worsened, they are at least partly manageable with medications, unlike the pain from calcinosis.
Treating symptoms vs. disease may be key in risk-benefit analysis
In questions after patient presentations, FDA officials probed for more perspective on issues such as how fatigue should be assessed, the differences between fatigue and brain fog, the impact of calcinosis on functioning, and how much risk patients would be willing to assume from treatments that have side effects and that may or may not modulate the disease and slow disease progression.
Most patients said in response to an FDA poll question that they definitely (almost 40%) or possibly (almost 50%) would be willing to try a hypothetical new self-injectable medication if it were shown to reduce their most impactful symptoms but had side effects.
“I think what [we’ve been hearing] today is that whether we’re working on the symptoms or the disease itself is [the key]” to patients’ risk-benefit analysis, said meeting moderator Capt. Robyn Bent, RN, MS, of the U.S. Public Health Service, and director of the PFDD.
Anita Devine, diagnosed 13 years ago with systemic sclerosis, was one of several panel members who said she would accept more bothersome treatment side effects and risks “if the gain was control of disease progression and overall quality of life ... and organ preservation.” Ms. Devine, who has needed kidney dialysis and multiple hand surgeries, noted that she previously took anti-neoplastic and anti-inflammatory agents “to try to stem the course of my disease, but unfortunately the disease did not abate.”
Treatments for systemic sclerosis include vasodilators, immunosuppressive medications, antifibrotic therapies, and stem cell transplants, Dr. Khanna said in his opening remarks.
Trials of drugs for scleroderma have focused on early disease that may be amenable to treatment, with the exception of trials for pulmonary arterial hypertension, which affects some patients with systemic sclerosis. There are multiple FDA-approved drugs for pulmonary arterial hypertension and more trials are underway.
Outcomes such as pain and fatigue are included in many of the trials currently underway, but they tend to be lower-level secondary outcomes measures that cannot be incorporated into drug labeling or are more “exploratory in nature,” Dr. Khanna said in the interview.
Dr. Khanna disclosed that he is the chief medical officer (an equity position) for CiVi Biopharma/Eicos Sciences Inc., which is developing a drug for Raynaud’s, and serves as a consultant and grant recipient for numerous companies that make or are developing drugs for systemic sclerosis.
The FDA will accept patient comments until Dec. 15, 2020, at which time comments will be compiled into a summary report, Ms. Bent said.
FROM AN FDA PATIENT-FOCUSED DRUG DEVELOPMENT MEETING
Baricitinib reduces adult atopic dermatitis severity in phase 3 study
in the phase 3, double-blind, placebo-controlled, BREEZE-AD7 study.
The study enrolled patients with inadequate responses to topical corticosteroids, according to Kristian Reich, MD, University Medical Center Hamburg-Eppendorf, Hamburg, Germany, and his coauthors.
First test of baricitinib plus topical steroids
Baricitinib, an oral selective Janus kinase (JAK)1/JAK2 inhibitor, inhibits several cytokines in AD pathogenesis, and in two monotherapy studies (BREEZE-AD1 and BREEZE-AD2), it was superior to placebo for reducing several AD clinical signs and symptoms. The current BREEZE-AD7 study is the first to test baricitinib plus background topical corticosteroid therapy, more closely mirroring clinical practice, the authors noted.
BREEZE-AD7 was conducted at 68 centers in 10 countries in Asia, Australia, Europe, and South America. It included 329 adults with moderate to severe AD (mean age around 34 years, and around 34% were female) with inadequate responses to topical corticosteroids documented within the last 6 months. They were randomized 1:1:1 to daily baricitinib 4 mg, daily baricitinib 2 mg, or placebo for 16 weeks. All patients received moderate- and/or low-potency topical corticosteroids (such as 0.1%triamcinolone cream and 2.5% hydrocortisone ointment, respectively) for active lesions.
Significant benefit at 4 mg
At week 16, 31% of AD patients receiving baricitinib 4 mg achieved Validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD) scores of 0 (clear) or 1 (almost clear) versus 15% in the placebo group (odds ratio, 2.8; 95% confidence interval, 1.4-5.6; P = .004). Among patients receiving baricitinib 2 mg, 24% achieved vI-GA-AD scores of 0 or 1 (OR, 1.9; 95% CI, 0.9-3.9; P = .08).
The same pattern of improving scores from placebo to baricitinib 2 mg to baricitinib 4 mg persisted, as reflected with secondary endpoints at week 16. Among patients receiving baricitinib 4 mg, 48% achieved Eczema Area Severity Index (EASI) 75 responses, versus 43% and 23% in 2 mg and placebo groups, respectively. Percent changes from baseline in total EASI score were –67%, –58%, –45% for baricitinib 4 mg, baricitinib 2 mg, and placebo, respectively; the proportion of patients achieving 4-point or greater improvements in Itch Numeric Rating Scale (NRS) was 44%, 38%, and 20% for baricitinib 4 mg, baricitinib 2 mg and placebo, respectively.
Similarly, mean change from baseline on the Skin Pain numeric rating scale was –3.7, –3.2, and –2.1 for baricitinib 4 mg, baricitinib 2 mg and placebo. Nighttime itch awakenings were also reduced in a similar progression from placebo to the higher baricitinib dose.
Adverse events dose related
Treatment-related adverse events were reported more frequently in the baricitinib groups (58% baricitinib 4 mg, 56% baricitinib 2 mg) versus placebo 38%. Nasopharyngitis was most common, followed by oral herpes, upper respiratory tract infection, acne, diarrhea, and back pain. Serious adverse event rates were similar across treatment groups. Permanent discontinuation rates were low at 5% for baricitinib 4 mg, 0% for baricitinib 2 mg, and 1% for placebo. The side-effect profile for baricitinib was consistent with prior studies, Dr. Reich and his coauthors reported.
The authors noted further, “data in this study suggest that patients with AD treated with baricitinib may be able to reduce the frequency and total quantity of concomitant TCSs [topical corticosteroids] used, thus mitigating concerns associated with continual or sustained application of topical treatments.”
“Overall, this study provides further evidence to support the efficacy and safety profile of baricitinib for the treatment of moderate-severe AD,” commented one of the authors, Jonathan I. Silverberg, MD, PhD, MPH, of the department of dermatology at George Washington University in Washington.
“In particular, this study shows that adding topical corticosteroids to baricitinib increases the rate of treatment success compared with the efficacy seen in baricitinib monotherapy studies. These data will be important to guide the use of baricitinib with topical corticosteroids in clinical practice. I think these data are also important because they show that baricitinib 4 mg may be more effective than 2 mg in some patients,” he said in an interview.
In late September, the European Medicines Agency’s Committee for Medicinal Products for Human Use recommended approval of oral baricitinib for adults with moderate to severe AD who are candidates for systemic therapy. Baricitinib is approved in the European Union and the United States to treat moderate to severe active rheumatoid arthritis. If approved in Europe, it will be the first JAK inhibitor and first oral medication indicated to treat patients with AD.
The study was funded by Eli Lilly and Company under license from Incyte Corporation. Dr. Reich reported receiving fees to the institution for participation in clinical trials from Eli Lilly and Company during the conduct of the study and personal fees for lectures. Dr. Silverberg reported receiving fees from Eli Lilly and Company during the conduct of the study, and fees from companies outside of this work. Other authors also reported disclosures related to Eli Lilly and other pharmaceutical companies, and several authors were Eli Lilly employees.
SOURCE: Reich K et al. JAMA Dermatol. 2020 Sep 30. doi: 10.1001/jamadermatol.2020.3260.
in the phase 3, double-blind, placebo-controlled, BREEZE-AD7 study.
The study enrolled patients with inadequate responses to topical corticosteroids, according to Kristian Reich, MD, University Medical Center Hamburg-Eppendorf, Hamburg, Germany, and his coauthors.
First test of baricitinib plus topical steroids
Baricitinib, an oral selective Janus kinase (JAK)1/JAK2 inhibitor, inhibits several cytokines in AD pathogenesis, and in two monotherapy studies (BREEZE-AD1 and BREEZE-AD2), it was superior to placebo for reducing several AD clinical signs and symptoms. The current BREEZE-AD7 study is the first to test baricitinib plus background topical corticosteroid therapy, more closely mirroring clinical practice, the authors noted.
BREEZE-AD7 was conducted at 68 centers in 10 countries in Asia, Australia, Europe, and South America. It included 329 adults with moderate to severe AD (mean age around 34 years, and around 34% were female) with inadequate responses to topical corticosteroids documented within the last 6 months. They were randomized 1:1:1 to daily baricitinib 4 mg, daily baricitinib 2 mg, or placebo for 16 weeks. All patients received moderate- and/or low-potency topical corticosteroids (such as 0.1%triamcinolone cream and 2.5% hydrocortisone ointment, respectively) for active lesions.
Significant benefit at 4 mg
At week 16, 31% of AD patients receiving baricitinib 4 mg achieved Validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD) scores of 0 (clear) or 1 (almost clear) versus 15% in the placebo group (odds ratio, 2.8; 95% confidence interval, 1.4-5.6; P = .004). Among patients receiving baricitinib 2 mg, 24% achieved vI-GA-AD scores of 0 or 1 (OR, 1.9; 95% CI, 0.9-3.9; P = .08).
The same pattern of improving scores from placebo to baricitinib 2 mg to baricitinib 4 mg persisted, as reflected with secondary endpoints at week 16. Among patients receiving baricitinib 4 mg, 48% achieved Eczema Area Severity Index (EASI) 75 responses, versus 43% and 23% in 2 mg and placebo groups, respectively. Percent changes from baseline in total EASI score were –67%, –58%, –45% for baricitinib 4 mg, baricitinib 2 mg, and placebo, respectively; the proportion of patients achieving 4-point or greater improvements in Itch Numeric Rating Scale (NRS) was 44%, 38%, and 20% for baricitinib 4 mg, baricitinib 2 mg and placebo, respectively.
Similarly, mean change from baseline on the Skin Pain numeric rating scale was –3.7, –3.2, and –2.1 for baricitinib 4 mg, baricitinib 2 mg and placebo. Nighttime itch awakenings were also reduced in a similar progression from placebo to the higher baricitinib dose.
Adverse events dose related
Treatment-related adverse events were reported more frequently in the baricitinib groups (58% baricitinib 4 mg, 56% baricitinib 2 mg) versus placebo 38%. Nasopharyngitis was most common, followed by oral herpes, upper respiratory tract infection, acne, diarrhea, and back pain. Serious adverse event rates were similar across treatment groups. Permanent discontinuation rates were low at 5% for baricitinib 4 mg, 0% for baricitinib 2 mg, and 1% for placebo. The side-effect profile for baricitinib was consistent with prior studies, Dr. Reich and his coauthors reported.
The authors noted further, “data in this study suggest that patients with AD treated with baricitinib may be able to reduce the frequency and total quantity of concomitant TCSs [topical corticosteroids] used, thus mitigating concerns associated with continual or sustained application of topical treatments.”
“Overall, this study provides further evidence to support the efficacy and safety profile of baricitinib for the treatment of moderate-severe AD,” commented one of the authors, Jonathan I. Silverberg, MD, PhD, MPH, of the department of dermatology at George Washington University in Washington.
“In particular, this study shows that adding topical corticosteroids to baricitinib increases the rate of treatment success compared with the efficacy seen in baricitinib monotherapy studies. These data will be important to guide the use of baricitinib with topical corticosteroids in clinical practice. I think these data are also important because they show that baricitinib 4 mg may be more effective than 2 mg in some patients,” he said in an interview.
In late September, the European Medicines Agency’s Committee for Medicinal Products for Human Use recommended approval of oral baricitinib for adults with moderate to severe AD who are candidates for systemic therapy. Baricitinib is approved in the European Union and the United States to treat moderate to severe active rheumatoid arthritis. If approved in Europe, it will be the first JAK inhibitor and first oral medication indicated to treat patients with AD.
The study was funded by Eli Lilly and Company under license from Incyte Corporation. Dr. Reich reported receiving fees to the institution for participation in clinical trials from Eli Lilly and Company during the conduct of the study and personal fees for lectures. Dr. Silverberg reported receiving fees from Eli Lilly and Company during the conduct of the study, and fees from companies outside of this work. Other authors also reported disclosures related to Eli Lilly and other pharmaceutical companies, and several authors were Eli Lilly employees.
SOURCE: Reich K et al. JAMA Dermatol. 2020 Sep 30. doi: 10.1001/jamadermatol.2020.3260.
in the phase 3, double-blind, placebo-controlled, BREEZE-AD7 study.
The study enrolled patients with inadequate responses to topical corticosteroids, according to Kristian Reich, MD, University Medical Center Hamburg-Eppendorf, Hamburg, Germany, and his coauthors.
First test of baricitinib plus topical steroids
Baricitinib, an oral selective Janus kinase (JAK)1/JAK2 inhibitor, inhibits several cytokines in AD pathogenesis, and in two monotherapy studies (BREEZE-AD1 and BREEZE-AD2), it was superior to placebo for reducing several AD clinical signs and symptoms. The current BREEZE-AD7 study is the first to test baricitinib plus background topical corticosteroid therapy, more closely mirroring clinical practice, the authors noted.
BREEZE-AD7 was conducted at 68 centers in 10 countries in Asia, Australia, Europe, and South America. It included 329 adults with moderate to severe AD (mean age around 34 years, and around 34% were female) with inadequate responses to topical corticosteroids documented within the last 6 months. They were randomized 1:1:1 to daily baricitinib 4 mg, daily baricitinib 2 mg, or placebo for 16 weeks. All patients received moderate- and/or low-potency topical corticosteroids (such as 0.1%triamcinolone cream and 2.5% hydrocortisone ointment, respectively) for active lesions.
Significant benefit at 4 mg
At week 16, 31% of AD patients receiving baricitinib 4 mg achieved Validated Investigator Global Assessment for Atopic Dermatitis (vIGA-AD) scores of 0 (clear) or 1 (almost clear) versus 15% in the placebo group (odds ratio, 2.8; 95% confidence interval, 1.4-5.6; P = .004). Among patients receiving baricitinib 2 mg, 24% achieved vI-GA-AD scores of 0 or 1 (OR, 1.9; 95% CI, 0.9-3.9; P = .08).
The same pattern of improving scores from placebo to baricitinib 2 mg to baricitinib 4 mg persisted, as reflected with secondary endpoints at week 16. Among patients receiving baricitinib 4 mg, 48% achieved Eczema Area Severity Index (EASI) 75 responses, versus 43% and 23% in 2 mg and placebo groups, respectively. Percent changes from baseline in total EASI score were –67%, –58%, –45% for baricitinib 4 mg, baricitinib 2 mg, and placebo, respectively; the proportion of patients achieving 4-point or greater improvements in Itch Numeric Rating Scale (NRS) was 44%, 38%, and 20% for baricitinib 4 mg, baricitinib 2 mg and placebo, respectively.
Similarly, mean change from baseline on the Skin Pain numeric rating scale was –3.7, –3.2, and –2.1 for baricitinib 4 mg, baricitinib 2 mg and placebo. Nighttime itch awakenings were also reduced in a similar progression from placebo to the higher baricitinib dose.
Adverse events dose related
Treatment-related adverse events were reported more frequently in the baricitinib groups (58% baricitinib 4 mg, 56% baricitinib 2 mg) versus placebo 38%. Nasopharyngitis was most common, followed by oral herpes, upper respiratory tract infection, acne, diarrhea, and back pain. Serious adverse event rates were similar across treatment groups. Permanent discontinuation rates were low at 5% for baricitinib 4 mg, 0% for baricitinib 2 mg, and 1% for placebo. The side-effect profile for baricitinib was consistent with prior studies, Dr. Reich and his coauthors reported.
The authors noted further, “data in this study suggest that patients with AD treated with baricitinib may be able to reduce the frequency and total quantity of concomitant TCSs [topical corticosteroids] used, thus mitigating concerns associated with continual or sustained application of topical treatments.”
“Overall, this study provides further evidence to support the efficacy and safety profile of baricitinib for the treatment of moderate-severe AD,” commented one of the authors, Jonathan I. Silverberg, MD, PhD, MPH, of the department of dermatology at George Washington University in Washington.
“In particular, this study shows that adding topical corticosteroids to baricitinib increases the rate of treatment success compared with the efficacy seen in baricitinib monotherapy studies. These data will be important to guide the use of baricitinib with topical corticosteroids in clinical practice. I think these data are also important because they show that baricitinib 4 mg may be more effective than 2 mg in some patients,” he said in an interview.
In late September, the European Medicines Agency’s Committee for Medicinal Products for Human Use recommended approval of oral baricitinib for adults with moderate to severe AD who are candidates for systemic therapy. Baricitinib is approved in the European Union and the United States to treat moderate to severe active rheumatoid arthritis. If approved in Europe, it will be the first JAK inhibitor and first oral medication indicated to treat patients with AD.
The study was funded by Eli Lilly and Company under license from Incyte Corporation. Dr. Reich reported receiving fees to the institution for participation in clinical trials from Eli Lilly and Company during the conduct of the study and personal fees for lectures. Dr. Silverberg reported receiving fees from Eli Lilly and Company during the conduct of the study, and fees from companies outside of this work. Other authors also reported disclosures related to Eli Lilly and other pharmaceutical companies, and several authors were Eli Lilly employees.
SOURCE: Reich K et al. JAMA Dermatol. 2020 Sep 30. doi: 10.1001/jamadermatol.2020.3260.
FROM JAMA DERMATOLOGY
Was This Tattoo a Rash Choice?
ANSWER
The correct answer is koebnerization of pre-existing psoriasis (choice “c”).
DISCUSSION
Tattoos have been known to cause bacterial infection (choice “a”), but this was unlikely given the diffuse nature of the rash and the lack of pain or adenopathy. Allergic reactions to tattoo dyes (choice “b”) are certainly common, but usually red or yellow dyes—which were not used for this tattoo—provoke the worst reactions. Furthermore, itching would have been a more prominent feature of the patient's complaint. Had it been fungal infection (choice “d”), the steroid cream would have made it worse.
One possibility remained: the so-called isomorphic phenomenon (otherwise known as koebnerization). First described by Heinrich Koebner in the mid-19th century, koebnerization is characterized by the appearance of psoriasis in traumatized skin such as surgical wounds, abrasions, burns, or even tattoos. Several other conditions also exhibit this same linear response to trauma, including warts, molluscum, and lichen planus.
To test for this diagnosis, corroborative findings of psoriasis were sought and found in the patient’s nails. His history of rashes on the knees and elbows also contributed to establishing the diagnosis. Moreover, his complaint of arthritis was quite suggestive of psoriatic arthropathy, which afflicts about 25% of patients with psoriasis and has little to do with the severity of the skin disease itself. Once the diagnosis became more apparent, the patient recalled a family history of psoriasis. Had any question remained, a biopsy could remove doubt.
TREATMENT
For the patient, twice-daily application of a stronger steroid cream (augmented betamethasone) was prescribed. Though this quickly cleared the koebnerizing psoriasis, it is likely we haven’t seen the last of this disease.
ANSWER
The correct answer is koebnerization of pre-existing psoriasis (choice “c”).
DISCUSSION
Tattoos have been known to cause bacterial infection (choice “a”), but this was unlikely given the diffuse nature of the rash and the lack of pain or adenopathy. Allergic reactions to tattoo dyes (choice “b”) are certainly common, but usually red or yellow dyes—which were not used for this tattoo—provoke the worst reactions. Furthermore, itching would have been a more prominent feature of the patient's complaint. Had it been fungal infection (choice “d”), the steroid cream would have made it worse.
One possibility remained: the so-called isomorphic phenomenon (otherwise known as koebnerization). First described by Heinrich Koebner in the mid-19th century, koebnerization is characterized by the appearance of psoriasis in traumatized skin such as surgical wounds, abrasions, burns, or even tattoos. Several other conditions also exhibit this same linear response to trauma, including warts, molluscum, and lichen planus.
To test for this diagnosis, corroborative findings of psoriasis were sought and found in the patient’s nails. His history of rashes on the knees and elbows also contributed to establishing the diagnosis. Moreover, his complaint of arthritis was quite suggestive of psoriatic arthropathy, which afflicts about 25% of patients with psoriasis and has little to do with the severity of the skin disease itself. Once the diagnosis became more apparent, the patient recalled a family history of psoriasis. Had any question remained, a biopsy could remove doubt.
TREATMENT
For the patient, twice-daily application of a stronger steroid cream (augmented betamethasone) was prescribed. Though this quickly cleared the koebnerizing psoriasis, it is likely we haven’t seen the last of this disease.
ANSWER
The correct answer is koebnerization of pre-existing psoriasis (choice “c”).
DISCUSSION
Tattoos have been known to cause bacterial infection (choice “a”), but this was unlikely given the diffuse nature of the rash and the lack of pain or adenopathy. Allergic reactions to tattoo dyes (choice “b”) are certainly common, but usually red or yellow dyes—which were not used for this tattoo—provoke the worst reactions. Furthermore, itching would have been a more prominent feature of the patient's complaint. Had it been fungal infection (choice “d”), the steroid cream would have made it worse.
One possibility remained: the so-called isomorphic phenomenon (otherwise known as koebnerization). First described by Heinrich Koebner in the mid-19th century, koebnerization is characterized by the appearance of psoriasis in traumatized skin such as surgical wounds, abrasions, burns, or even tattoos. Several other conditions also exhibit this same linear response to trauma, including warts, molluscum, and lichen planus.
To test for this diagnosis, corroborative findings of psoriasis were sought and found in the patient’s nails. His history of rashes on the knees and elbows also contributed to establishing the diagnosis. Moreover, his complaint of arthritis was quite suggestive of psoriatic arthropathy, which afflicts about 25% of patients with psoriasis and has little to do with the severity of the skin disease itself. Once the diagnosis became more apparent, the patient recalled a family history of psoriasis. Had any question remained, a biopsy could remove doubt.
TREATMENT
For the patient, twice-daily application of a stronger steroid cream (augmented betamethasone) was prescribed. Though this quickly cleared the koebnerizing psoriasis, it is likely we haven’t seen the last of this disease.

Weeks ago, a 43-year-old man received a birthday tattoo of his choice: a geometric pattern etched in blue ink on his wrist. Unfortunately, a rash began to develop within the lines of the tattoo. The rash itches, but its appearance is of greater concern to the patient. He’s gotten some relief from topical creams, although the rash quickly returns with cessation of treatment.
Past medical history is notable for arthritis affecting his left elbow and right heel. He also has intermittent rashes that manifest on his elbows and knees, but these are partially relieved by a steroid cream (triamcinolone 0.1%).
His tattoo is located on the extensor right wrist. The affected areas show a brisk, red, inflammatory response, which—in several locations—is also scaly. There is no tenderness or induration on palpation of the rash and no palpable adenopathy in local nodal locations (epitrochlear and axillary). Elsewhere, 5 of his 10 fingernails demonstrate pitting; 2 show onycholysis and oil spotting. His scalp, knees, and elbows are free of any notable changes.

A teen presents with a severe, tender rash on the extremities
“There’s rue for you, and here’s some for me; we may call it herb of grace o’ Sundays. O, you must wear your rue with a difference.”
— Ophelia in Hamlet by William Shakespeare
The patient was admitted to the hospital for IV fluids, pain control, and observation. The following day she admitted using the leaves of a plant on the trail as a bug repellent, as one time was taught by her grandfather. She rubbed some of the leaves on the brother as well. The grandfather shared some pictures of the bushes, and the plant was identified as Ruta graveolens.
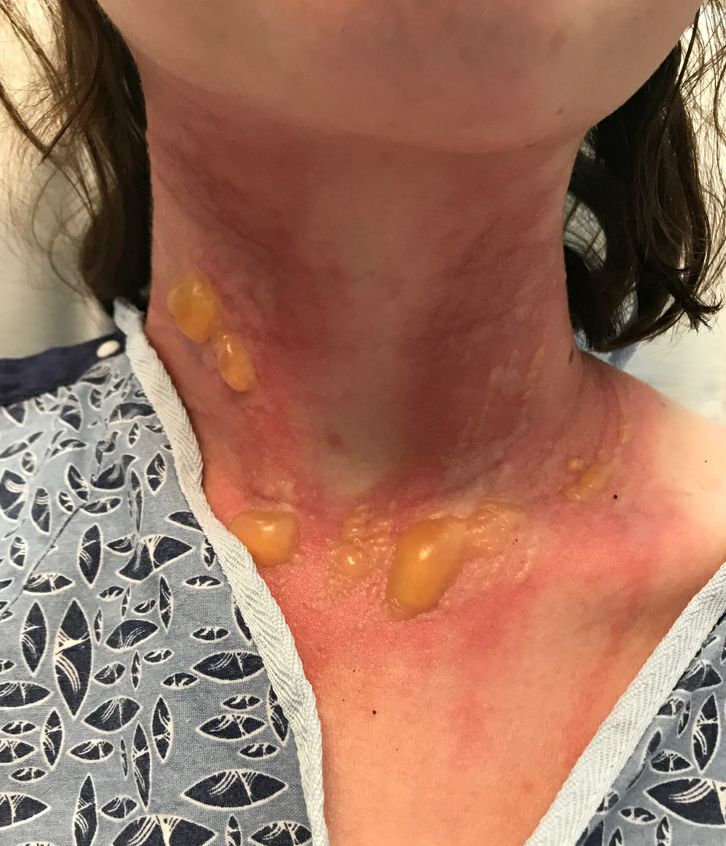
The blisters were deroofed, cleaned with saline, and wrapped with triamcinolone ointment and petrolatum. The patient was also started on a prednisone taper and received analgesics for the severe pain.
Ruta graveolens also known as common rue or herb of grace, is an ornamental plant from the Rutaceae family. This plant is also used as a medicinal herb, condiment, and as an insect repellent. If ingested in large doses, it can cause severe abdominal pain and vomiting. It also can be hepatotoxic.
The herb contains furocumarines, such as 8-methoxypsoralen and 5-methoxypsoralen and furoquinoline alkaloids. These chemicals when exposed to UVA radiation cause cell injury and inflammation of the skin. This is considered a phototoxic reaction of the skin, compared with allergic reactions, such as poison ivy dermatitis, which need a prior sensitization to the allergen for the T cells to be activated and cause injury in the skin. Other common plants and fruits that can cause phytophotodermatitis include citrus fruits, figs, carrots, celery, parsnips, parsley, and other wildflowers like hogweed.
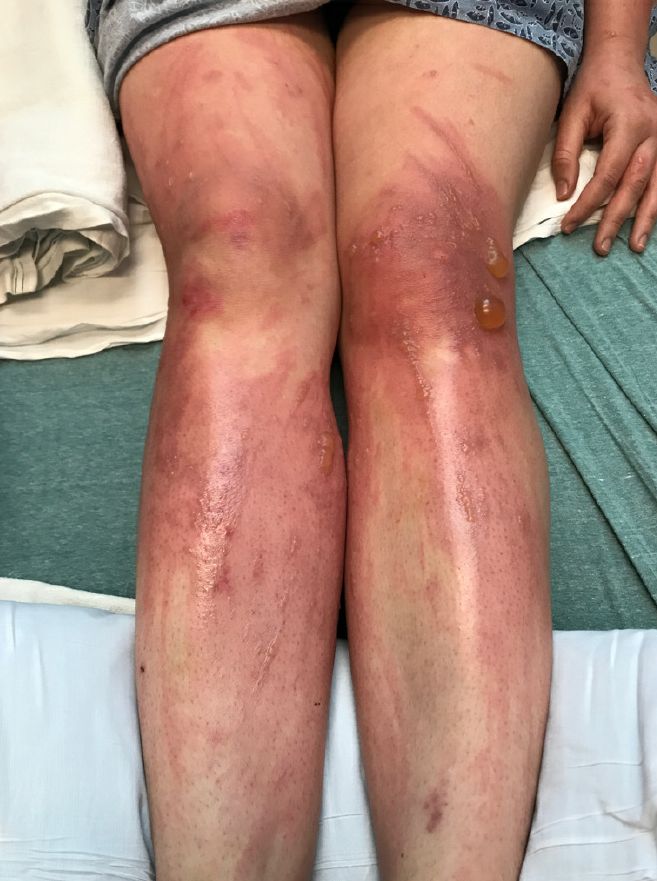
Depending on the degree of injury, the patients can be treated with topical corticosteroids, petrolatum wraps, and pain control. In severe cases like our patient, systemic prednisone may help stop the progression of the lesions and help with the inflammation. Skin hyperpigmentation after the initial injury may take months to clear, and some patient can develop scars.
The differential diagnosis should include severe bullous contact dermatitis like exposure to urushiol in poison ivy; second- and third-degree burns; severe medications reactions such Stevens-Johnson syndrome or toxic epidermal necrolysis, and inmunobullous diseases such as bullous lupus erythematosus, pemphigus vulgaris, or bullous pemphigoid. If there is no history of exposure or there are any other systemic symptoms, consider performing a skin biopsy of one of the lesions.
In this patient’s case, the history of exposure and skin findings helped the dermatologist on call make the right diagnosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
J Burn Care Res. 2018 Oct 23;39(6):1064-6.
Dermatitis. 2007 Mar;18(1):52-5.
BMJ Case Rep. 2015 Dec 23;2015:bcr2015213388.
“There’s rue for you, and here’s some for me; we may call it herb of grace o’ Sundays. O, you must wear your rue with a difference.”
— Ophelia in Hamlet by William Shakespeare
The patient was admitted to the hospital for IV fluids, pain control, and observation. The following day she admitted using the leaves of a plant on the trail as a bug repellent, as one time was taught by her grandfather. She rubbed some of the leaves on the brother as well. The grandfather shared some pictures of the bushes, and the plant was identified as Ruta graveolens.
The blisters were deroofed, cleaned with saline, and wrapped with triamcinolone ointment and petrolatum. The patient was also started on a prednisone taper and received analgesics for the severe pain.
Ruta graveolens also known as common rue or herb of grace, is an ornamental plant from the Rutaceae family. This plant is also used as a medicinal herb, condiment, and as an insect repellent. If ingested in large doses, it can cause severe abdominal pain and vomiting. It also can be hepatotoxic.
The herb contains furocumarines, such as 8-methoxypsoralen and 5-methoxypsoralen and furoquinoline alkaloids. These chemicals when exposed to UVA radiation cause cell injury and inflammation of the skin. This is considered a phototoxic reaction of the skin, compared with allergic reactions, such as poison ivy dermatitis, which need a prior sensitization to the allergen for the T cells to be activated and cause injury in the skin. Other common plants and fruits that can cause phytophotodermatitis include citrus fruits, figs, carrots, celery, parsnips, parsley, and other wildflowers like hogweed.
Depending on the degree of injury, the patients can be treated with topical corticosteroids, petrolatum wraps, and pain control. In severe cases like our patient, systemic prednisone may help stop the progression of the lesions and help with the inflammation. Skin hyperpigmentation after the initial injury may take months to clear, and some patient can develop scars.
The differential diagnosis should include severe bullous contact dermatitis like exposure to urushiol in poison ivy; second- and third-degree burns; severe medications reactions such Stevens-Johnson syndrome or toxic epidermal necrolysis, and inmunobullous diseases such as bullous lupus erythematosus, pemphigus vulgaris, or bullous pemphigoid. If there is no history of exposure or there are any other systemic symptoms, consider performing a skin biopsy of one of the lesions.
In this patient’s case, the history of exposure and skin findings helped the dermatologist on call make the right diagnosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
J Burn Care Res. 2018 Oct 23;39(6):1064-6.
Dermatitis. 2007 Mar;18(1):52-5.
BMJ Case Rep. 2015 Dec 23;2015:bcr2015213388.
“There’s rue for you, and here’s some for me; we may call it herb of grace o’ Sundays. O, you must wear your rue with a difference.”
— Ophelia in Hamlet by William Shakespeare
The patient was admitted to the hospital for IV fluids, pain control, and observation. The following day she admitted using the leaves of a plant on the trail as a bug repellent, as one time was taught by her grandfather. She rubbed some of the leaves on the brother as well. The grandfather shared some pictures of the bushes, and the plant was identified as Ruta graveolens.
The blisters were deroofed, cleaned with saline, and wrapped with triamcinolone ointment and petrolatum. The patient was also started on a prednisone taper and received analgesics for the severe pain.
Ruta graveolens also known as common rue or herb of grace, is an ornamental plant from the Rutaceae family. This plant is also used as a medicinal herb, condiment, and as an insect repellent. If ingested in large doses, it can cause severe abdominal pain and vomiting. It also can be hepatotoxic.
The herb contains furocumarines, such as 8-methoxypsoralen and 5-methoxypsoralen and furoquinoline alkaloids. These chemicals when exposed to UVA radiation cause cell injury and inflammation of the skin. This is considered a phototoxic reaction of the skin, compared with allergic reactions, such as poison ivy dermatitis, which need a prior sensitization to the allergen for the T cells to be activated and cause injury in the skin. Other common plants and fruits that can cause phytophotodermatitis include citrus fruits, figs, carrots, celery, parsnips, parsley, and other wildflowers like hogweed.
Depending on the degree of injury, the patients can be treated with topical corticosteroids, petrolatum wraps, and pain control. In severe cases like our patient, systemic prednisone may help stop the progression of the lesions and help with the inflammation. Skin hyperpigmentation after the initial injury may take months to clear, and some patient can develop scars.
The differential diagnosis should include severe bullous contact dermatitis like exposure to urushiol in poison ivy; second- and third-degree burns; severe medications reactions such Stevens-Johnson syndrome or toxic epidermal necrolysis, and inmunobullous diseases such as bullous lupus erythematosus, pemphigus vulgaris, or bullous pemphigoid. If there is no history of exposure or there are any other systemic symptoms, consider performing a skin biopsy of one of the lesions.
In this patient’s case, the history of exposure and skin findings helped the dermatologist on call make the right diagnosis.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. Email her at pdnews@mdedge.com.
References
J Burn Care Res. 2018 Oct 23;39(6):1064-6.
Dermatitis. 2007 Mar;18(1):52-5.
BMJ Case Rep. 2015 Dec 23;2015:bcr2015213388.
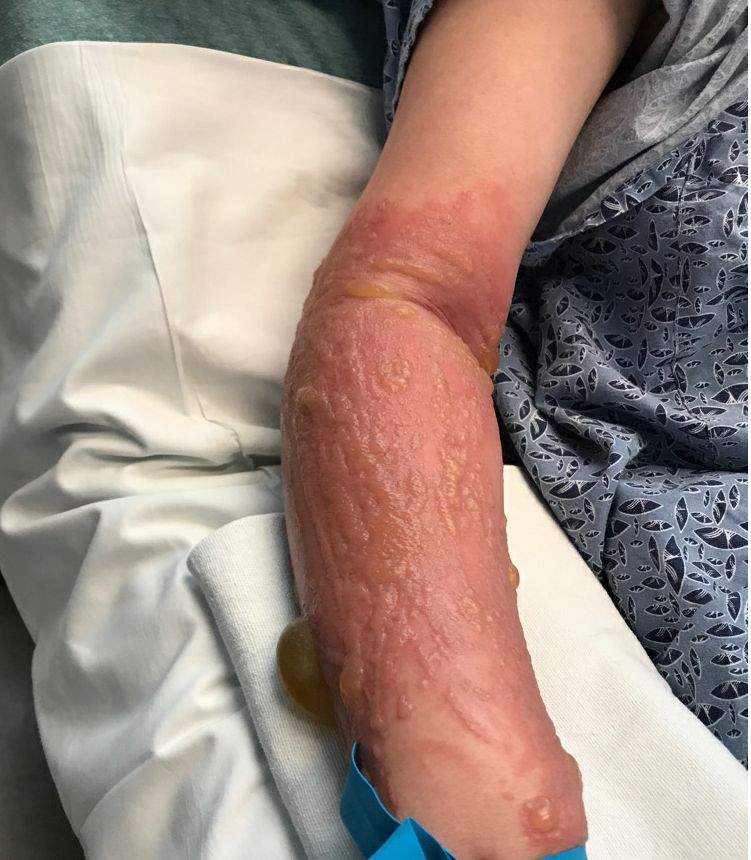
She started taking lithium for depression and anxiety 3 weeks prior to her developing the rash. She denies taking any other medications, supplements, or recreational drugs.
She denied any prior history of photosensitivity, no history of mouth ulcers, joint pain, muscle weakness, hair loss, or any other symptoms.
Besides her brother, there are no other affected family members, and no history of immune bullous disorders or other skin conditions.
On physical exam, the girl appears in a lot of pain and is uncomfortable. The skin is red and hot, and there are tense bullae on the neck, arms, and legs. There are no ocular or mucosal lesions.
Rapidly developing vesicular eruption
A 23-month-old girl with a history of well-controlled atopic dermatitis was admitted to the hospital with fever and a widespread vesicular eruption of 2 days’ duration. Two days prior to admission, the patient had 3 episodes of nonbloody diarrhea and redness in the diaper area. The child’s parents reported that the red areas spread to her arms and legs later that day, and that she subsequently developed a fever, cough, and rhinorrhea. She was taken to an urgent care facility where she was diagnosed with vulvovaginitis and an upper respiratory infection; amoxicillin was prescribed. Shortly thereafter, the patient developed more lesions in and around the mouth, as well as on the trunk, prompting the parents to bring her to the emergency department.
The history revealed that the patient had spent time with her aunt and cousins who had “red spots” on their palms and soles. The patient’s sister had a flare of “cold sores,” about 2 weeks prior to the current presentation. The patient had received a varicella zoster virus (VZV) vaccine several months earlier.
Physical examination was notable for an uncomfortable infant with erythematous macules on the bilateral palms and soles and an erythematous hard palate. The child also had scattered vesicles on an erythematous base with confluent crusted plaques on her lips, perioral skin (FIGURE 1A), abdomen, back, buttocks, arms, legs (FIGURE 1B), and dorsal aspects of her hands and feet.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Eczema coxsackium
Given the history of atopic dermatitis; prodromal diarrhea/rhinorrhea; papulovesicular eruption involving areas of prior dermatitis as well as the palms, soles, and mouth; recent contacts with suspected hand-foot-mouth disease (HFMD); and history of VZV vaccination, the favored diagnosis was eczema coxsackium.
Eczema coxsackium is an atypical form of HFMD that occurs in patients with a history of eczema. Classic HFMD usually is caused by coxsackievirus A16 or enterovirus 71, while atypical HFMD often is caused by coxsackievirus A6.1,2,3 Patients with HFMD present with painful oral vesicles and ulcers and a papulovesicular eruption on the palms, soles, and sometimes the buttocks and genitalia. Patients may have prodromal fever, fussiness, and diarrhea. Painful oral lesions may result in poor oral intake.1,2
Differential includes viral eruptions
Other conditions may manifest similarly to eczema coxsackium and must be ruled out before initiating proper treatment.
Eczema herpeticum (EH). In atypical HFMD, the virus can show tropism for active or previously inflamed areas of eczematous skin, leading to a widespread vesicular eruption, which can be difficult to distinguish from EH.1 Similar to EH, eczema coxsackium does not exclusively affect children with atopic dermatitis. It also has been described in adults and patients with Darier disease, incontinentia pigmenti, and epidermolytic ichthyosis.4-6
In cases of vesicular eruptions in eczema patients, it is imperative to rule out EH. One prospective study of atypical HFMD compared similarities of the conditions. Both have a predilection for mucosa during primary infection and develop vesicular eruptions on cutaneous eczematous skin.1 One key difference between eczema coxsackium and EH is that EH tends to produce intraoral vesicles beyond simple erythema; it also tends to predominate in the area of the head and neck.7
Continue to: Eczema varicellicum
Eczema varicellicum has been reported, and it has been suggested that some cases of EH may actually be caused by VZV as the 2 are clinically indistinguishable and less than half of EH cases are diagnosed with laboratory confirmation.8
Confirm Dx before you treat
To guide management, cases of suspected eczema coxsackium should be confirmed, and HSV/VZV should be ruled out.9 Testing modalities include swabbing vesicular fluid for enterovirus polymerase chain reaction (PCR) analysis (preferred modality), oropharyngeal swab up to 2 weeks after infection, or viral isolate from stool samples up to 3 months after infection.2,3
Treatment for eczema coxsackium involves supportive care such as intravenous (IV) hydration and antipyretics. Some studies show potential benefit with IV immunoglobulin in treating severe HFMD, while other studies show the exacerbation of widespread HFMD with this treatment.7,10
Prompt diagnosis and treatment for eczema coxsackium is critical to prevent unnecessary antiviral therapy and to help guide monitoring for associated morbidities including Gianotti-Crosti syndrome–like eruptions, purpuric eruptions, and onychomadesis.
Our patient. Because EH was in the differential, our patient was started on empiric IV acyclovir 10 mg/kg every 8 hours while test results were pending. In addition, she received acetaminophen, IV fluids, gentle sponge baths, and diligent emollient application. Scraping from a vesicle revealed negative herpes simplex virus 1/2 PCR, negative VZV direct fluorescent antibody, and a positive enterovirus PCR—confirming the diagnosis of eczema coxsackium. Interestingly, a viral culture was negative in our patient, consistent with prior reports of enterovirus being difficult to culture.11
With confirmation of the diagnosis of eczema coxsackium, the IV acyclovir was discontinued, and symptoms resolved after 7 days.
CORRESPONDENCE
Shane M. Swink, DO, MS, Division of Dermatology, 1200 South Cedar Crest Boulevard, Allentown, PA 18103; shanesw@pcom.edu
1. Neri I, Dondi A, Wollenberg A, et al. Atypical forms of hand, foot, and mouth disease: a prospective study of 47 Italian children. Pediatr Dermatol. 2016;33:429-437.
2. Nassef C, Ziemer C, Morrell DS. Hand-foot-and-mouth disease: a new look at a classic viral rash. Curr Opin Pediatr. 2015;27:486-491.
3. Horsten H, Fisker N, Bygu, A. Eczema coxsackium caused by coxsackievirus A6. Pediatr Dermatol. 2016;33:230-231.
4. Jefferson J, Grossberg A. Incontinentia pigmenti coxsackium. Pediatr Dermatol. 2016;33:E280-E281.
5. Ganguly S, Kuruvila S. Eczema coxsackium. Indian J Dermatol. 2016;61:682-683.
6. Harris P, Wang AD, Yin M, et al. Atypical hand, foot, and mouth disease: eczema coxsackium can also occur in adults. Lancet Infect Dis. 2014;14:1043.
7. Wollenberg A, Zoch C, Wetzel S, et al. Predisposing factors and clinical features of eczema herpeticum: a retrospective analysis of 100 cases. J Am Acad Dermatol. 2003;49:198-205.
8. Austin TA, Steele RW. Eczema varicella/zoster (varicellicum). Clin Pediatr. 2017;56:579-581.
9. Leung DYM. Why is eczema herpeticum unexpectedly rare? Antiviral Res. 2013;98:153-157.
10. Cao RY, Dong DY, Liu RJ, et al. Human IgG subclasses against enterovirus type 71: neutralization versus antibody dependent enhancement of infection. PLoS One. 2013;8:E64024.
11. Mathes EF, Oza V, Frieden IJ, et al. Eczema coxsackium and unusual cutaneous findings in an enterovirus outbreak. Pediatrics. 2013;132:149-157.
A 23-month-old girl with a history of well-controlled atopic dermatitis was admitted to the hospital with fever and a widespread vesicular eruption of 2 days’ duration. Two days prior to admission, the patient had 3 episodes of nonbloody diarrhea and redness in the diaper area. The child’s parents reported that the red areas spread to her arms and legs later that day, and that she subsequently developed a fever, cough, and rhinorrhea. She was taken to an urgent care facility where she was diagnosed with vulvovaginitis and an upper respiratory infection; amoxicillin was prescribed. Shortly thereafter, the patient developed more lesions in and around the mouth, as well as on the trunk, prompting the parents to bring her to the emergency department.
The history revealed that the patient had spent time with her aunt and cousins who had “red spots” on their palms and soles. The patient’s sister had a flare of “cold sores,” about 2 weeks prior to the current presentation. The patient had received a varicella zoster virus (VZV) vaccine several months earlier.
Physical examination was notable for an uncomfortable infant with erythematous macules on the bilateral palms and soles and an erythematous hard palate. The child also had scattered vesicles on an erythematous base with confluent crusted plaques on her lips, perioral skin (FIGURE 1A), abdomen, back, buttocks, arms, legs (FIGURE 1B), and dorsal aspects of her hands and feet.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Eczema coxsackium
Given the history of atopic dermatitis; prodromal diarrhea/rhinorrhea; papulovesicular eruption involving areas of prior dermatitis as well as the palms, soles, and mouth; recent contacts with suspected hand-foot-mouth disease (HFMD); and history of VZV vaccination, the favored diagnosis was eczema coxsackium.
Eczema coxsackium is an atypical form of HFMD that occurs in patients with a history of eczema. Classic HFMD usually is caused by coxsackievirus A16 or enterovirus 71, while atypical HFMD often is caused by coxsackievirus A6.1,2,3 Patients with HFMD present with painful oral vesicles and ulcers and a papulovesicular eruption on the palms, soles, and sometimes the buttocks and genitalia. Patients may have prodromal fever, fussiness, and diarrhea. Painful oral lesions may result in poor oral intake.1,2
Differential includes viral eruptions
Other conditions may manifest similarly to eczema coxsackium and must be ruled out before initiating proper treatment.
Eczema herpeticum (EH). In atypical HFMD, the virus can show tropism for active or previously inflamed areas of eczematous skin, leading to a widespread vesicular eruption, which can be difficult to distinguish from EH.1 Similar to EH, eczema coxsackium does not exclusively affect children with atopic dermatitis. It also has been described in adults and patients with Darier disease, incontinentia pigmenti, and epidermolytic ichthyosis.4-6
In cases of vesicular eruptions in eczema patients, it is imperative to rule out EH. One prospective study of atypical HFMD compared similarities of the conditions. Both have a predilection for mucosa during primary infection and develop vesicular eruptions on cutaneous eczematous skin.1 One key difference between eczema coxsackium and EH is that EH tends to produce intraoral vesicles beyond simple erythema; it also tends to predominate in the area of the head and neck.7
Continue to: Eczema varicellicum
Eczema varicellicum has been reported, and it has been suggested that some cases of EH may actually be caused by VZV as the 2 are clinically indistinguishable and less than half of EH cases are diagnosed with laboratory confirmation.8
Confirm Dx before you treat
To guide management, cases of suspected eczema coxsackium should be confirmed, and HSV/VZV should be ruled out.9 Testing modalities include swabbing vesicular fluid for enterovirus polymerase chain reaction (PCR) analysis (preferred modality), oropharyngeal swab up to 2 weeks after infection, or viral isolate from stool samples up to 3 months after infection.2,3
Treatment for eczema coxsackium involves supportive care such as intravenous (IV) hydration and antipyretics. Some studies show potential benefit with IV immunoglobulin in treating severe HFMD, while other studies show the exacerbation of widespread HFMD with this treatment.7,10
Prompt diagnosis and treatment for eczema coxsackium is critical to prevent unnecessary antiviral therapy and to help guide monitoring for associated morbidities including Gianotti-Crosti syndrome–like eruptions, purpuric eruptions, and onychomadesis.
Our patient. Because EH was in the differential, our patient was started on empiric IV acyclovir 10 mg/kg every 8 hours while test results were pending. In addition, she received acetaminophen, IV fluids, gentle sponge baths, and diligent emollient application. Scraping from a vesicle revealed negative herpes simplex virus 1/2 PCR, negative VZV direct fluorescent antibody, and a positive enterovirus PCR—confirming the diagnosis of eczema coxsackium. Interestingly, a viral culture was negative in our patient, consistent with prior reports of enterovirus being difficult to culture.11
With confirmation of the diagnosis of eczema coxsackium, the IV acyclovir was discontinued, and symptoms resolved after 7 days.
CORRESPONDENCE
Shane M. Swink, DO, MS, Division of Dermatology, 1200 South Cedar Crest Boulevard, Allentown, PA 18103; shanesw@pcom.edu
A 23-month-old girl with a history of well-controlled atopic dermatitis was admitted to the hospital with fever and a widespread vesicular eruption of 2 days’ duration. Two days prior to admission, the patient had 3 episodes of nonbloody diarrhea and redness in the diaper area. The child’s parents reported that the red areas spread to her arms and legs later that day, and that she subsequently developed a fever, cough, and rhinorrhea. She was taken to an urgent care facility where she was diagnosed with vulvovaginitis and an upper respiratory infection; amoxicillin was prescribed. Shortly thereafter, the patient developed more lesions in and around the mouth, as well as on the trunk, prompting the parents to bring her to the emergency department.
The history revealed that the patient had spent time with her aunt and cousins who had “red spots” on their palms and soles. The patient’s sister had a flare of “cold sores,” about 2 weeks prior to the current presentation. The patient had received a varicella zoster virus (VZV) vaccine several months earlier.
Physical examination was notable for an uncomfortable infant with erythematous macules on the bilateral palms and soles and an erythematous hard palate. The child also had scattered vesicles on an erythematous base with confluent crusted plaques on her lips, perioral skin (FIGURE 1A), abdomen, back, buttocks, arms, legs (FIGURE 1B), and dorsal aspects of her hands and feet.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Eczema coxsackium
Given the history of atopic dermatitis; prodromal diarrhea/rhinorrhea; papulovesicular eruption involving areas of prior dermatitis as well as the palms, soles, and mouth; recent contacts with suspected hand-foot-mouth disease (HFMD); and history of VZV vaccination, the favored diagnosis was eczema coxsackium.
Eczema coxsackium is an atypical form of HFMD that occurs in patients with a history of eczema. Classic HFMD usually is caused by coxsackievirus A16 or enterovirus 71, while atypical HFMD often is caused by coxsackievirus A6.1,2,3 Patients with HFMD present with painful oral vesicles and ulcers and a papulovesicular eruption on the palms, soles, and sometimes the buttocks and genitalia. Patients may have prodromal fever, fussiness, and diarrhea. Painful oral lesions may result in poor oral intake.1,2
Differential includes viral eruptions
Other conditions may manifest similarly to eczema coxsackium and must be ruled out before initiating proper treatment.
Eczema herpeticum (EH). In atypical HFMD, the virus can show tropism for active or previously inflamed areas of eczematous skin, leading to a widespread vesicular eruption, which can be difficult to distinguish from EH.1 Similar to EH, eczema coxsackium does not exclusively affect children with atopic dermatitis. It also has been described in adults and patients with Darier disease, incontinentia pigmenti, and epidermolytic ichthyosis.4-6
In cases of vesicular eruptions in eczema patients, it is imperative to rule out EH. One prospective study of atypical HFMD compared similarities of the conditions. Both have a predilection for mucosa during primary infection and develop vesicular eruptions on cutaneous eczematous skin.1 One key difference between eczema coxsackium and EH is that EH tends to produce intraoral vesicles beyond simple erythema; it also tends to predominate in the area of the head and neck.7
Continue to: Eczema varicellicum
Eczema varicellicum has been reported, and it has been suggested that some cases of EH may actually be caused by VZV as the 2 are clinically indistinguishable and less than half of EH cases are diagnosed with laboratory confirmation.8
Confirm Dx before you treat
To guide management, cases of suspected eczema coxsackium should be confirmed, and HSV/VZV should be ruled out.9 Testing modalities include swabbing vesicular fluid for enterovirus polymerase chain reaction (PCR) analysis (preferred modality), oropharyngeal swab up to 2 weeks after infection, or viral isolate from stool samples up to 3 months after infection.2,3
Treatment for eczema coxsackium involves supportive care such as intravenous (IV) hydration and antipyretics. Some studies show potential benefit with IV immunoglobulin in treating severe HFMD, while other studies show the exacerbation of widespread HFMD with this treatment.7,10
Prompt diagnosis and treatment for eczema coxsackium is critical to prevent unnecessary antiviral therapy and to help guide monitoring for associated morbidities including Gianotti-Crosti syndrome–like eruptions, purpuric eruptions, and onychomadesis.
Our patient. Because EH was in the differential, our patient was started on empiric IV acyclovir 10 mg/kg every 8 hours while test results were pending. In addition, she received acetaminophen, IV fluids, gentle sponge baths, and diligent emollient application. Scraping from a vesicle revealed negative herpes simplex virus 1/2 PCR, negative VZV direct fluorescent antibody, and a positive enterovirus PCR—confirming the diagnosis of eczema coxsackium. Interestingly, a viral culture was negative in our patient, consistent with prior reports of enterovirus being difficult to culture.11
With confirmation of the diagnosis of eczema coxsackium, the IV acyclovir was discontinued, and symptoms resolved after 7 days.
CORRESPONDENCE
Shane M. Swink, DO, MS, Division of Dermatology, 1200 South Cedar Crest Boulevard, Allentown, PA 18103; shanesw@pcom.edu
1. Neri I, Dondi A, Wollenberg A, et al. Atypical forms of hand, foot, and mouth disease: a prospective study of 47 Italian children. Pediatr Dermatol. 2016;33:429-437.
2. Nassef C, Ziemer C, Morrell DS. Hand-foot-and-mouth disease: a new look at a classic viral rash. Curr Opin Pediatr. 2015;27:486-491.
3. Horsten H, Fisker N, Bygu, A. Eczema coxsackium caused by coxsackievirus A6. Pediatr Dermatol. 2016;33:230-231.
4. Jefferson J, Grossberg A. Incontinentia pigmenti coxsackium. Pediatr Dermatol. 2016;33:E280-E281.
5. Ganguly S, Kuruvila S. Eczema coxsackium. Indian J Dermatol. 2016;61:682-683.
6. Harris P, Wang AD, Yin M, et al. Atypical hand, foot, and mouth disease: eczema coxsackium can also occur in adults. Lancet Infect Dis. 2014;14:1043.
7. Wollenberg A, Zoch C, Wetzel S, et al. Predisposing factors and clinical features of eczema herpeticum: a retrospective analysis of 100 cases. J Am Acad Dermatol. 2003;49:198-205.
8. Austin TA, Steele RW. Eczema varicella/zoster (varicellicum). Clin Pediatr. 2017;56:579-581.
9. Leung DYM. Why is eczema herpeticum unexpectedly rare? Antiviral Res. 2013;98:153-157.
10. Cao RY, Dong DY, Liu RJ, et al. Human IgG subclasses against enterovirus type 71: neutralization versus antibody dependent enhancement of infection. PLoS One. 2013;8:E64024.
11. Mathes EF, Oza V, Frieden IJ, et al. Eczema coxsackium and unusual cutaneous findings in an enterovirus outbreak. Pediatrics. 2013;132:149-157.
1. Neri I, Dondi A, Wollenberg A, et al. Atypical forms of hand, foot, and mouth disease: a prospective study of 47 Italian children. Pediatr Dermatol. 2016;33:429-437.
2. Nassef C, Ziemer C, Morrell DS. Hand-foot-and-mouth disease: a new look at a classic viral rash. Curr Opin Pediatr. 2015;27:486-491.
3. Horsten H, Fisker N, Bygu, A. Eczema coxsackium caused by coxsackievirus A6. Pediatr Dermatol. 2016;33:230-231.
4. Jefferson J, Grossberg A. Incontinentia pigmenti coxsackium. Pediatr Dermatol. 2016;33:E280-E281.
5. Ganguly S, Kuruvila S. Eczema coxsackium. Indian J Dermatol. 2016;61:682-683.
6. Harris P, Wang AD, Yin M, et al. Atypical hand, foot, and mouth disease: eczema coxsackium can also occur in adults. Lancet Infect Dis. 2014;14:1043.
7. Wollenberg A, Zoch C, Wetzel S, et al. Predisposing factors and clinical features of eczema herpeticum: a retrospective analysis of 100 cases. J Am Acad Dermatol. 2003;49:198-205.
8. Austin TA, Steele RW. Eczema varicella/zoster (varicellicum). Clin Pediatr. 2017;56:579-581.
9. Leung DYM. Why is eczema herpeticum unexpectedly rare? Antiviral Res. 2013;98:153-157.
10. Cao RY, Dong DY, Liu RJ, et al. Human IgG subclasses against enterovirus type 71: neutralization versus antibody dependent enhancement of infection. PLoS One. 2013;8:E64024.
11. Mathes EF, Oza V, Frieden IJ, et al. Eczema coxsackium and unusual cutaneous findings in an enterovirus outbreak. Pediatrics. 2013;132:149-157.

Index finger plaque

The characteristic finding of small, scattered vesicular lesions on the hands that sometimes coalesce, and often are itchy or irritated led to the diagnosis of vesicular hand dermatitis, a form of eczema. It also is referred to as dyshidrotic eczema or pompholyx. (Worth noting is the fact that common warts and flat warts usually present as raised papular—not vesicular—lesions on the hands.)
The exact etiology of vesicular hand dermatitis is unknown. It is more common in women than men and often occurs in patients 20 to 40 years of age who tend to have a positive family history of eczema. It usually develops acutely and often is triggered by topical irritants or frequent hand washing. Treatment during the acute phase includes topical steroids. Avoidance of topical irritants, use of mild cleansers instead of harsh soaps, reduction of hand washing frequency (if possible), and frequent application of emollients can reduce recurrence.
This patient’s eczema had been successfully treated with betamethasone dipropionate ointment 0.05% in the past. Since she still had some at home, she was instructed to use it twice daily along with topical emmolients. She reported great improvement within 1 week.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Sobering G, Dika C. Vesicular hand dermatitis. Nurse Pract. 2018;43:33-37.
The characteristic finding of small, scattered vesicular lesions on the hands that sometimes coalesce, and often are itchy or irritated led to the diagnosis of vesicular hand dermatitis, a form of eczema. It also is referred to as dyshidrotic eczema or pompholyx. (Worth noting is the fact that common warts and flat warts usually present as raised papular—not vesicular—lesions on the hands.)
The exact etiology of vesicular hand dermatitis is unknown. It is more common in women than men and often occurs in patients 20 to 40 years of age who tend to have a positive family history of eczema. It usually develops acutely and often is triggered by topical irritants or frequent hand washing. Treatment during the acute phase includes topical steroids. Avoidance of topical irritants, use of mild cleansers instead of harsh soaps, reduction of hand washing frequency (if possible), and frequent application of emollients can reduce recurrence.
This patient’s eczema had been successfully treated with betamethasone dipropionate ointment 0.05% in the past. Since she still had some at home, she was instructed to use it twice daily along with topical emmolients. She reported great improvement within 1 week.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
The characteristic finding of small, scattered vesicular lesions on the hands that sometimes coalesce, and often are itchy or irritated led to the diagnosis of vesicular hand dermatitis, a form of eczema. It also is referred to as dyshidrotic eczema or pompholyx. (Worth noting is the fact that common warts and flat warts usually present as raised papular—not vesicular—lesions on the hands.)
The exact etiology of vesicular hand dermatitis is unknown. It is more common in women than men and often occurs in patients 20 to 40 years of age who tend to have a positive family history of eczema. It usually develops acutely and often is triggered by topical irritants or frequent hand washing. Treatment during the acute phase includes topical steroids. Avoidance of topical irritants, use of mild cleansers instead of harsh soaps, reduction of hand washing frequency (if possible), and frequent application of emollients can reduce recurrence.
This patient’s eczema had been successfully treated with betamethasone dipropionate ointment 0.05% in the past. Since she still had some at home, she was instructed to use it twice daily along with topical emmolients. She reported great improvement within 1 week.
Photo and text courtesy of Daniel Stulberg, MD, FAAFP, Department of Family and Community Medicine, University of New Mexico School of Medicine, Albuquerque.
Sobering G, Dika C. Vesicular hand dermatitis. Nurse Pract. 2018;43:33-37.
Sobering G, Dika C. Vesicular hand dermatitis. Nurse Pract. 2018;43:33-37.

Does early introduction of peanuts to an infant’s diet reduce the risk for peanut allergy?
EVIDENCE SUMMARY
A 2016 systematic review identified 2 RCTs that examined whether early introduction of peanuts affects subsequent allergies.1 The first RCT recruited 1303 3-month-old infants from the general population in the United Kingdom.2 All patients had either a negative skin prick test (SPT) to peanuts or a negative oral peanut challenge (if an initial SPT was positive). The control group breastfed exclusively until age 6 months, at which time allergenic foods could be introduced at parental discretion.
Timing doesn’t affect peanut allergy in nonallergic patients
The intervention group received 6 common allergenic foods (peanuts, eggs, cow’s milk, wheat, sesame, and whitefish) twice weekly between ages 3 and 6 months. Researchers then performed double-blinded, placebo-controlled oral food challenges at ages 12 and 36 months.
More patients in the late-introduction group demonstrated peanut allergies by age 36 months than in the early-introduction group, but the difference wasn’t significant (2.5% vs 1.2%; P = 0.11).A key weakness of the study was combining peanuts with other common food allergens.2
Children with eczema, egg allergy benefit from earlier peanut introduction
The second RCT divided 640 infants with severe eczema, egg allergy, or both into 2 groups according to their response to an SPT to peanuts: patients with no wheal and patients with a positive wheal measuring 1 to 4 mm.3 Researchers then randomized patients to either early exposure (peanut products given from ages 4 to 11 months) or avoidance (no peanuts until age 60 months). The primary endpoint was a positive clinical response to oral peanut allergen at age 60 months.
In the negative SPT group (atopic children expected to have a lower risk for allergy), patients introduced to peanuts later had a higher rate of subsequent allergy than children exposed earlier (14% vs 2%; absolute risk reduction [ARR] = 12%; 95% confidence interval [CI], 3%-20%; number needed to treat [NNT] = 9).3
In the positive SPT group (atopic children expected to have a higher risk for allergy), later peanut introduction likewise increased risk compared to earlier introduction (35% vs 11%; ARR = 24%; 95% CI, 5%-43%; NNT = 5). Children in the early-exposure group, however, had more URIs, viral exanthems, gastroenteritis, urticaria, and conjunctivitis (4527 events in the early-exposure group vs 4287 in the avoidance group, P = 0.02; about 1 more event per patient over the course of the study).3
The authors of the systematic review performed a meta-analysis of the 2 RCTs (1793 patients). They concluded that early introduction of peanuts to an infant’s diet (between ages 3 and 11 months) decreased the risk for eventual peanut allergy (relative risk [RR] = 0.29; 95% CI, 0.11-0.74), compared with introduction at or after age 1 year.1 A key weakness, however, was the researchers’ choice to combine trials with very different inclusion criteria (infants with severe eczema and a general population).
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
A 2017 National Institute of Allergy and Infectious Diseases guideline recommends a 3-tiered approach to peanut introduction: 4
- For children with severe eczema or egg allergy who aren’t currently allergic to peanuts (per SPT or immunoglobulin E [IgE] test), the guideline advises adding peanuts to the diet between ages 4 and 6 months. (Patients with positive SPT or IgE should be referred to an allergy specialist.)
- Children with mild or moderate eczema can be introduced to peanuts around age 6 months “in accordance with family preferences and cultural practices.”
- Children with no evidence of allergy or eczema can be “freely introduced” to peanut-containing foods with no specific guidance on age.
Editor’s takeaway
Good-quality evidence supports family physicians encouraging introduction of foods containing peanuts at age 4 to 6 months for children at increased risk because of atopy, allergies, or eczema.
1. Ierodiakonou D, Garcia-Larsen V, Logan A, et al. Timing of allergenic food introduction to the infant diet and risk of allergic or autoimmune disease: a systematic review and meta-analysis. JAMA. 2016;316:1181-1192.
2. Perkin MR, Logan K, Tseng A, et al. Randomized trial of introduction of allergenic foods in breast-fed infants. N Engl J Med. 2016;374:1733-1743.
3. Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372:803-813.
4. Togias A, Cooper SF, Acebal ML, et al. Addendum guidelines for the prevention of peanut allergy in the United States: report of the National Institute of Allergy and Infectious Diseases–sponsored expert panel. J Allergy Clin Immunol. 2017;139:29-44.
EVIDENCE SUMMARY
A 2016 systematic review identified 2 RCTs that examined whether early introduction of peanuts affects subsequent allergies.1 The first RCT recruited 1303 3-month-old infants from the general population in the United Kingdom.2 All patients had either a negative skin prick test (SPT) to peanuts or a negative oral peanut challenge (if an initial SPT was positive). The control group breastfed exclusively until age 6 months, at which time allergenic foods could be introduced at parental discretion.
Timing doesn’t affect peanut allergy in nonallergic patients
The intervention group received 6 common allergenic foods (peanuts, eggs, cow’s milk, wheat, sesame, and whitefish) twice weekly between ages 3 and 6 months. Researchers then performed double-blinded, placebo-controlled oral food challenges at ages 12 and 36 months.
More patients in the late-introduction group demonstrated peanut allergies by age 36 months than in the early-introduction group, but the difference wasn’t significant (2.5% vs 1.2%; P = 0.11).A key weakness of the study was combining peanuts with other common food allergens.2
Children with eczema, egg allergy benefit from earlier peanut introduction
The second RCT divided 640 infants with severe eczema, egg allergy, or both into 2 groups according to their response to an SPT to peanuts: patients with no wheal and patients with a positive wheal measuring 1 to 4 mm.3 Researchers then randomized patients to either early exposure (peanut products given from ages 4 to 11 months) or avoidance (no peanuts until age 60 months). The primary endpoint was a positive clinical response to oral peanut allergen at age 60 months.
In the negative SPT group (atopic children expected to have a lower risk for allergy), patients introduced to peanuts later had a higher rate of subsequent allergy than children exposed earlier (14% vs 2%; absolute risk reduction [ARR] = 12%; 95% confidence interval [CI], 3%-20%; number needed to treat [NNT] = 9).3
In the positive SPT group (atopic children expected to have a higher risk for allergy), later peanut introduction likewise increased risk compared to earlier introduction (35% vs 11%; ARR = 24%; 95% CI, 5%-43%; NNT = 5). Children in the early-exposure group, however, had more URIs, viral exanthems, gastroenteritis, urticaria, and conjunctivitis (4527 events in the early-exposure group vs 4287 in the avoidance group, P = 0.02; about 1 more event per patient over the course of the study).3
The authors of the systematic review performed a meta-analysis of the 2 RCTs (1793 patients). They concluded that early introduction of peanuts to an infant’s diet (between ages 3 and 11 months) decreased the risk for eventual peanut allergy (relative risk [RR] = 0.29; 95% CI, 0.11-0.74), compared with introduction at or after age 1 year.1 A key weakness, however, was the researchers’ choice to combine trials with very different inclusion criteria (infants with severe eczema and a general population).
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
A 2017 National Institute of Allergy and Infectious Diseases guideline recommends a 3-tiered approach to peanut introduction: 4
- For children with severe eczema or egg allergy who aren’t currently allergic to peanuts (per SPT or immunoglobulin E [IgE] test), the guideline advises adding peanuts to the diet between ages 4 and 6 months. (Patients with positive SPT or IgE should be referred to an allergy specialist.)
- Children with mild or moderate eczema can be introduced to peanuts around age 6 months “in accordance with family preferences and cultural practices.”
- Children with no evidence of allergy or eczema can be “freely introduced” to peanut-containing foods with no specific guidance on age.
Editor’s takeaway
Good-quality evidence supports family physicians encouraging introduction of foods containing peanuts at age 4 to 6 months for children at increased risk because of atopy, allergies, or eczema.
EVIDENCE SUMMARY
A 2016 systematic review identified 2 RCTs that examined whether early introduction of peanuts affects subsequent allergies.1 The first RCT recruited 1303 3-month-old infants from the general population in the United Kingdom.2 All patients had either a negative skin prick test (SPT) to peanuts or a negative oral peanut challenge (if an initial SPT was positive). The control group breastfed exclusively until age 6 months, at which time allergenic foods could be introduced at parental discretion.
Timing doesn’t affect peanut allergy in nonallergic patients
The intervention group received 6 common allergenic foods (peanuts, eggs, cow’s milk, wheat, sesame, and whitefish) twice weekly between ages 3 and 6 months. Researchers then performed double-blinded, placebo-controlled oral food challenges at ages 12 and 36 months.
More patients in the late-introduction group demonstrated peanut allergies by age 36 months than in the early-introduction group, but the difference wasn’t significant (2.5% vs 1.2%; P = 0.11).A key weakness of the study was combining peanuts with other common food allergens.2
Children with eczema, egg allergy benefit from earlier peanut introduction
The second RCT divided 640 infants with severe eczema, egg allergy, or both into 2 groups according to their response to an SPT to peanuts: patients with no wheal and patients with a positive wheal measuring 1 to 4 mm.3 Researchers then randomized patients to either early exposure (peanut products given from ages 4 to 11 months) or avoidance (no peanuts until age 60 months). The primary endpoint was a positive clinical response to oral peanut allergen at age 60 months.
In the negative SPT group (atopic children expected to have a lower risk for allergy), patients introduced to peanuts later had a higher rate of subsequent allergy than children exposed earlier (14% vs 2%; absolute risk reduction [ARR] = 12%; 95% confidence interval [CI], 3%-20%; number needed to treat [NNT] = 9).3
In the positive SPT group (atopic children expected to have a higher risk for allergy), later peanut introduction likewise increased risk compared to earlier introduction (35% vs 11%; ARR = 24%; 95% CI, 5%-43%; NNT = 5). Children in the early-exposure group, however, had more URIs, viral exanthems, gastroenteritis, urticaria, and conjunctivitis (4527 events in the early-exposure group vs 4287 in the avoidance group, P = 0.02; about 1 more event per patient over the course of the study).3
The authors of the systematic review performed a meta-analysis of the 2 RCTs (1793 patients). They concluded that early introduction of peanuts to an infant’s diet (between ages 3 and 11 months) decreased the risk for eventual peanut allergy (relative risk [RR] = 0.29; 95% CI, 0.11-0.74), compared with introduction at or after age 1 year.1 A key weakness, however, was the researchers’ choice to combine trials with very different inclusion criteria (infants with severe eczema and a general population).
Continue to: RECOMMENDATIONS
RECOMMENDATIONS
A 2017 National Institute of Allergy and Infectious Diseases guideline recommends a 3-tiered approach to peanut introduction: 4
- For children with severe eczema or egg allergy who aren’t currently allergic to peanuts (per SPT or immunoglobulin E [IgE] test), the guideline advises adding peanuts to the diet between ages 4 and 6 months. (Patients with positive SPT or IgE should be referred to an allergy specialist.)
- Children with mild or moderate eczema can be introduced to peanuts around age 6 months “in accordance with family preferences and cultural practices.”
- Children with no evidence of allergy or eczema can be “freely introduced” to peanut-containing foods with no specific guidance on age.
Editor’s takeaway
Good-quality evidence supports family physicians encouraging introduction of foods containing peanuts at age 4 to 6 months for children at increased risk because of atopy, allergies, or eczema.
1. Ierodiakonou D, Garcia-Larsen V, Logan A, et al. Timing of allergenic food introduction to the infant diet and risk of allergic or autoimmune disease: a systematic review and meta-analysis. JAMA. 2016;316:1181-1192.
2. Perkin MR, Logan K, Tseng A, et al. Randomized trial of introduction of allergenic foods in breast-fed infants. N Engl J Med. 2016;374:1733-1743.
3. Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372:803-813.
4. Togias A, Cooper SF, Acebal ML, et al. Addendum guidelines for the prevention of peanut allergy in the United States: report of the National Institute of Allergy and Infectious Diseases–sponsored expert panel. J Allergy Clin Immunol. 2017;139:29-44.
1. Ierodiakonou D, Garcia-Larsen V, Logan A, et al. Timing of allergenic food introduction to the infant diet and risk of allergic or autoimmune disease: a systematic review and meta-analysis. JAMA. 2016;316:1181-1192.
2. Perkin MR, Logan K, Tseng A, et al. Randomized trial of introduction of allergenic foods in breast-fed infants. N Engl J Med. 2016;374:1733-1743.
3. Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372:803-813.
4. Togias A, Cooper SF, Acebal ML, et al. Addendum guidelines for the prevention of peanut allergy in the United States: report of the National Institute of Allergy and Infectious Diseases–sponsored expert panel. J Allergy Clin Immunol. 2017;139:29-44.
EVIDENCE-BASED ANSWER:
Probably not, unless the child has severe eczema or egg allergy. In a general pediatric population, introducing peanuts early (at age 3 to 6 months) doesn’t appear to alter rates of subsequent peanut allergy compared with introduction after age 6 months (strength of recommendation [SOR]: B, randomized clinical trial [RCT] using multiple potential food allergens).
In children with severe eczema, egg allergy, or both, however, the risk for a peanut allergy is 12% to 24% lower when peanut-containing foods are introduced at age 4 to 11 months than after age 1 year. Early introduction of peanuts is associated with about 1 additional mild virus-associated syndrome (upper respiratory infection [URI], exanthem, conjunctivitis, or gastroenteritis) per patient (SOR: B, RCT).
Introducing peanuts before age 1 year is recommended for atopic children without evidence of pre-existing peanut allergy; an earlier start, at age 4 to 6 months, is advised for infants with severe eczema or egg allergy (SOR: C, expert opinion).

Hidradenitis Suppurativa in the Military
Case Report
A 19-year-old female marine with a 10-year history of hidradenitis suppurativa (HS) presented with hyperpigmented nodules in the inguinal folds and a recurrent cyst in the right groin area of 2 to 3 weeks’ duration. She denied axillary or inframammary involvement. She underwent several incision and drainage procedures 1 year prior to her enlistment in the US Marine Corps at 18 years of age. She previously had been treated by dermatology with doxycycline 100-mg tablets twice daily, benzoyl peroxide wash 5% applied to affected areas and rinsed daily, and clindamycin solution 1% with minimal improvement. She denied smoking or alcohol intake and said she typically wore a loose-fitting uniform to work. As a marine, she was expected to participate in daily physical training and exercises with her military unit, during which she wore a standardized physical training uniform, including nylon shorts and a cotton T-shirt. She requested light duty—military duty status with physical limitations or restrictions—to avoid physical training that would cause further friction and irritation to the inguinal region.
Physical examination demonstrated a woman with Fitzpatrick skin type III and normal body mass index. There were hyperpigmented nodules and scarring in the inguinal folds, most consistent with Hurley stage 2. A single, 0.5-cm, draining lesion was visualized. No hyperhidrosis was noted. The patient was placed on light duty for 7 days, with physical training only at her own pace and discretion. Moreover, she was restricted from field training, rifle range training, and other situations where she may excessively sweat or not be able to adequately maintain personal hygiene. She was encouraged to continue clindamycin solution 1% to the affected area twice daily and was prescribed chlorhexidine solution 4% to use when washing affected areas in the shower. The patient also was referred to the dermatology department at the Naval Hospital Camp Pendleton (Oceanside, California), where she was treated with laser hair removal in the inguinal region, thus avoiding waxing and further aggravation of HS flares. Due to the combination of topical therapies along with laser hair removal and duty restrictions, the patient had a dramatic decrease in development of severe nodular lesions.
Comment
Presentation
Historically, the incidence of HS is estimated at 0.5% to 4% of the general population with female predominance.1 Predisposing factors include obesity, smoking, genetic predisposition to acne, apocrine duct obstruction, and secondary bacterial infection.2 During acute flares, patients generally present with tender subcutaneous nodules that drain malodorous purulent material.3,4 Acute flares are unpredictable, and patients deal with chronic, recurrent, draining wounds, leading to a poor quality of life with resulting physical, psychological, financial, social, and emotional distress.3-5 The negative impact of HS on a patient’s quality of life has been reported to be greater than other dermatologic conditions.6 Lesions can be particularly painful and can cause disfiguration to the surface of the skin.7 Lesion severity is described using the Hurley staging system. Patient quality of life is directly correlated with disease severity and Hurley stage. In stage 1, abscesses develop, but no sinus tracts or cicatrization is present. In stage 2, recurrent abscesses will form tracts and cicatrization. In stage 3, the abscesses become diffuse or near diffuse, with multiple interconnected tracts and abscesses across the entire area of the body.8,9
Severe or refractory HS within the physically active military population may require consideration of light or limited duty or even separation from service. Similarly, severe HS may pose challenges with other physically demanding occupations, such as the police force and firefighters.
Prevention Focus
Prevention of flares is key for patients with HS; secondary prevention aims to reduce impact of the disease or injury that has already occurred,10,11 which includes prevention of the infundibulofolliculitis from becoming a deep folliculitis, nodule, or fistula, as well as Hurley stage progression. Prompt diagnosis with appropriate treatment can decrease the severity of lesions, pain, and scarring. Globally, HS patients continue to experience considerable diagnostic delays of 8 to 12 years after onset of initial symptoms.11,12 Earlier accurate diagnosis and initiation of treatment from the primary care provider or general medical officer is imperative. Initial accurate management may help keep symptoms from progressing to more severe painful lesions. Similarly, patients should be educated on how to prevent HS flares. Patients should avoid known triggers, including smoking, obesity, sweating, mechanical irritation, stress, and poor hygiene.11
Shaving for hair reduction creates ingrown hair shafts, which may lead to folliculitis in mechanically stressed areas in skin folds, thus initiating the inflammatory cascade of HS.11,13 Therefore, shaving along with any other mechanical stress should be avoided in patients with HS. Laser hair removal has been shown to be quite helpful in both the prevention and treatment of HS. In one study, 22 patients with Hurley stage 2 to 3 disease were treated with an Nd:YAG laser once monthly. Results demonstrated a 65% decrease in disease severity after 3 monthly treatments.11 Similarly, other lasers have been used with success in several small case series; an 800-nm diode laser, intense pulsed light therapy, and a ruby laser have each demonstrated efficacy.14 Given these results, hair removal should be recommended to patients with HS. Military servicemembers (SMs) with certain conditions, such as polycystic ovary syndrome, pseudofolliculitis barbae, and HS, are eligible for laser hair removal when available at local military treatment facilities. Primary care providers for military SMs must have a working understanding of the disease process of HS and awareness of what resources are available for treatment, which allows for more streamlined care and improved outcomes.
Treatment Options
Treatment options are diverse and depend on the severity of HS. Typically, treatment begins with medical therapy followed by escalation to surgical intervention. Medical therapies often include antibiotics, acne treatments, antiandrogen therapy, immunosuppressive agents, and biologic therapy.15,16 If first-line medical interventions fail to control HS, surgical interventions should be considered. Surgical intervention in conjunction with medical therapy decreases the chance for recurrence.3,15,16
Although HS is internationally recognized as an inflammatory disease and not an infectious process, topical antibiotics can help to prevent and improve formation of abscesses, nodules, and pustules.11 Agents such as clindamycin and chlorhexidine wash have proven effective in preventing flares.11,16 Other antibiotics used alone or in combination also are efficacious. Tetracyclines are recommended as monotherapy for mild stages of HS.17-19 Doxycycline is the most commonly used tetracycline in HS patients and has been demonstrated to penetrate Staphylococcus aureus biofilm in high enough concentrations to maintain its antibacterial activity.20 Moreover, doxycycline, as with other tetracyclines, has a multitude of anti-inflammatory and immunomodulatory properties21 and can reduce the production of IL-1, IL-6, tumor necrosis factor α, and IL-8; downregulate chemotaxis; and promote lipo-oxygenase, matrix metalloproteinase, and nuclear factor κB (NF-κB) signaling inhibition.17
Clindamycin is the only known agent that has been studied for topical treatment and utilization in milder cases of HS.17,22 Systemic combination of clindamycin and rifampicin is the most studied, with well-established efficacy in managing HS.17,23,24 Clindamycin has bacteriostatic activity toward both aerobic and anaerobic gram-positive bacteria by binding irreversibly to the 50S ribosomal subunit, thereby inhibiting bacterial protein synthesis. Rifampicin binds to the beta subunit of DNA-dependent RNA polymerase, inhibiting bacterial DNA-dependent RNA synthesis. Rifampicin has broad-spectrum activity, mostly against gram-positive as well as some gram-negative bacteria. Moreover, rifampicin has anti-inflammatory and immunomodulatory properties, including evidence that it inhibits excessive helper T cell (TH17) responses by reducing inducible nitric oxide synthase transcription and NF-κB activity.25,26
Metronidazole, moxifloxacin, and rifampicin as triple combination therapy has been shown to be effective in reducing HS activity in moderate to severe cases that were refractory to other treatments.27 Research suggests that moxifloxacin has anti-inflammatory properties, mainly by reducing IL-1β, IL-8, and tumor necrosis factor α; stabilizing IXb protein; suppressing NF-κB signaling; and reducing IL-17A.28,29
Ertapenem can be utilized as a single 6-week antibiotic course during surgical planning or rescue therapy.18 Moreover, ertapenem can be used to treat complicated skin and soft tissue infections and has been shown to substantially improve clinical aspects of severe HS.17,27
Disease-modifying antirheumatic drugs are effective in the treatment of moderate to severe HS.17-19 In 2 phase 3 trials (PIONEER I and II), adalimumab was used as monotherapy or in conjunction with antibiotics in patients with moderate to severe HS compared to placebo.30 Results demonstrated a disease burden reduction of greater than 50%. Antibiotic dual therapy was not noted to significantly affect disease burden.30 Of note, use of immunosuppressants in the military affects an SM’s availability for worldwide deployment and duty station assignment.
Antiandrogen therapies have demonstrated some reduction in HS flares. Although recommendations for use in HS is based on limited evidence, one randomized controlled trial compared ethinyl estradiol–norgestrel to ethinyl estradiol and cyproterone acetate. Both therapies resulted in similar efficacy, with 12 of 24 (50%) patients reporting HS symptoms improving or completely resolved.31 In another retrospective study of women treated with antiandrogen therapies, including ethinyl estriol, cyproterone acetate, and spironolactone, 16 of 29 (55%) patients reported improvement.32 In another study, daily doses of 100 to 150 mg of spironolactone resulted in improvement in 17 of 20 (85%) patients, including complete remission in 11 of 20 (55%) patients. Of the 3 patients with severe HS, none had complete clearing of disease burden.33 Patients with polycystic ovary syndrome or HS flares that occur around menstruation are more likely to benefit from treatment with spironolactone.18,32,34
Retinoids frequently have been utilized in the management of HS. In some retrospective studies and other prospective studies with 5 or more patients, isotretinoin monotherapy was utilized for a 4- to 10-month period.18,35-38 In the Alikhan et al18 study, 85 of 207 patients demonstrated improvement of HS symptoms, with more remarkable improvements in milder cases. Isotretinoin for management of patients with HS who have concomitant nodulocystic acne would have two-fold benefits.18
Wound Care
Given the purulent nodular formation in HS, adequate wound care management is vital. There is an abundance of HS wound care management strategies utilized by clinicians and patients. When selecting the appropriate dressing, consideration for the type of HS wound, cost, ease of application, patient comfort, absorbency, and odor management is important.3 However, living arrangements for military SMs can create difficulties applying and maintaining HS dressings, especially if deployed or in a field setting. Active-duty SMs often find themselves in austere living conditions in the field, aboard ships, or in other scenarios where they may or may not have running water or showers. Maintaining adequate hygiene may be difficult, and additional education about how to keep wounds clean must be imparted. Ideal dressings for HS should be highly absorbent, comfortable when applied to the anatomic locations of the HS lesions, and easily self-applied. Ideally, dressings would have atraumatic adhesion and antimicrobial properties.3 Cost-effective dressing options that have good absorption capability include sanitary napkins, adult briefs, infant diapers, and gauze.3 These dressings help to wick moisture, thus protecting the wound from maceration, which is a common patient concern. Although gauze dressings are easier to obtain, they are not as absorbent. Abdominal pads can be utilized, but they are moderately absorbent, bulky, and more challenging to obtain over-the-counter. Hydrofiber and calcium alginate dressings with silver are not accessible to the common consumer and are more expensive than the aforementioned dressings, but they do have some antimicrobial activity. Silver-impregnated foam dressings are moldable to intertriginous areas, easy to self-apply, and have moderate-heavy absorption abilities.
Final Thoughts
Hidradenitis suppurativa poses cumbersome and uncomfortable symptoms for all patients and may pose additional hardships for military SMs or those with physically demanding occupations who work in austere environments. Severe HS can restrict a military SM from certain duty stations, positions, or deployments. Early identification of HS can help reduce HS flares, disfigurement, and placement on limited duty status, therefore rendering the SM more able to engage in his/her operational responsibilities. Hidradenitis suppurativa should be discussed with the patient, with the goal to prevent flares for SMs that will be in the field, placed in austere environments, or be deployed. Use of immunosuppressants in active-duty SMs may affect their deployability, duty assignment, and retention.
For a military SM with HS, all aspects of prevention and treatment need to be balanced with his/her ability to remain deployable and complete his/her daily duties. Military SMs are not guaranteed the ideal scenario for treatment and prevention of HS. Unsanitary environments and occlusive uniforms undoubtedly contribute to disease process and make treatment more challenging. If a military SM is in a field setting or deployed, frequent daily dressing changes should still be attempted.
- Dufour DN, Emtestam L, Jemec GB. Hidradenitis suppurativa: a common and burdensome, yet under-recognised, inflammatory skin disease. Postgrad Med J. 2014;90:216-221.
- Beshara MA. Hidradenitis suppurativa: a clinician’s tool for early diagnosis and treatment. Nurse Pract. 2010;35:24-28.
- Kazemi A, Carnaggio K, Clark M, et al. Optimal wound care management in hidradenitis suppurativa. J Dermatolog Treat. 2017;29:165-167.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003:49:96-98.
- Blattner C, Polley DC, Ferrito F, et al. Central centrifugal cicatricial alopecia. Indian Dermatol Online J. 2013:4:50.
- Wolkenstein P, Loundou A, Barrau K, et al. Quality of life impairment in hidradenitis suppurativa: a study of 61 cases. J Am Acad Dermatol. 2007;56:621-623.
- Smith HS, Chao JD, Teitelbaum J. Painful hidradenitis suppurativa. Clin J Pain. 2010;26:435-444.
- Alavi A, Anooshirvani N, Kim WB, et al. Quality-of-life impairment in patients with hidradenitis suppurativa: a Canadian study. Am J Clin Dermatol. 2015;16:61-65.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH Jr, eds. Dermatologic Surgery: Principles and Practice. 2nd ed. New York, NY: Marcel Dekker; 1996:623-645.
- Kligman AM. Welcome letter. 2nd International Conference on the Sebaceous Gland, Acne, Rosacea and Related Disorders; September 13-16, 2008; Rome Italy.
- Kurzen H, Kurzen M. Secondary prevention of hidradenitis suppurativa. Dermatol Reports. 2019;11:8243.
- Sabat R, Tsaousi A, Rossbacher J, et al. Acne inversa/hidradenitis suppurativa: an update [in German]. Hautarzt. 2017;68:999-1006.
- Boer J, Nazary M, Riis PT. The role of mechanical stress in hidradenitis suppurativa. Dermatol Clin. 2016;34:37-43.
- Hamzavi IH, Griffith JL, Riyaz F, et al. Laser and light-based treatment options for hidradenitis suppurativa. J Am Acad Dermatol. 2015;73(5 suppl 1):S78-S81.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Michel C, DiBianco JM, Sabarwal V, et al. The treatment of genitoperineal hidradenitis suppurativa: a review of the literature. Urology. 2019;124:1-5.
- Constantinou CA, Fragoulis GE, Nikiphorou E. Hidradenitis suppurativa: infection, autoimmunity, or both [published online December 30, 2019]? Ther Adv Musculoskelet Dis. doi:10.1177/1759720x19895488.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Zouboulis CC, Desai N, Emtestam, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29:619-644.
- Mandell JB, Orr S, Koch J, et al. Large variations in clinical antibiotic activity against Staphylococcus aureus biofilms of periprosthetic joint infection isolates. J Orthop Res. 2019;37:1604-1609.
- Sun J, Shigemi H, Tanaka Y, et al. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. 2015;4:397-404.
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. Int J Dermatol. 1983;22:325-328.
- Gener G, Canoui-Poitrine F, Revuz JE, et al. Combination therapy with clindamycin and rifampicin for hidradenitis suppurativa: a series of 116 consecutive patients. Dermatology. 2009;219:148-154.
- Griffiths CEM. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. 2006;154:977-978.
- Ma K, Chen X, Chen J-C, et al. Rifampicin attenuates experimental autoimmune encephalomyelitis by inhibiting pathogenic Th17 cells responses. J Neurochem. 2016;139:1151-1162.
- Yuhas Y, Berent E, Ovadiah H, et al. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob Agents Chemother. 2006;50:396-398.
- Join-Lambert O, Coignard H, Jais J-P, et al. Efficacy of rifampin-moxifloxacin-metronidazole combination therapy in hidradenitis suppurativa. Dermatology. 2011;222:49-58.
- Choi J-H, Song M-J, Kim S-H, et al. Effect of moxifloxacin on production of proinflammatory cytokines from human peripheral blood mononuclear cells. Antimicrob Agents Chemother. 2003;47:3704-3707.
- Weiss T, Shalit I, Blau H, et al. Anti-inflammatory effects of moxifloxacin on activated human monocytic cells: inhibition of NF-kappaB and mitogen-activated protein kinase activation and of synthesis of proinflammatory cytokines.” Antimicrob Agents Chemother. 2004;48:1974-1982.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Mortimer PS, Dawber RP, Gales MA, et al. A double-blind controlled cross-over trial of cyproterone acetate in females with hidradenitis suppurativa. Br J Dermatol. 1986;115:263-268.
- Kraft JN, Searles GE. Hidradenitis suppurativa in 64 female patients: retrospective study comparing oral antibiotics and antiandrogen therapy. J Cutan Med Surg. 2007;11:125-131.
- Lee A, Fischer G. A case series of 20 women with hidradenitis suppurativa treated with spironolactone. Australas J Dermatol. 2015;56:192-196.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a “male” therapy for a predominantly “female” disease. J Clin Aesthet Dermatol. 2016;9:44-50.
- Dicken CH, Powell ST, Spear KL. Evaluation of isotretinoin treatment of hidradenitis suppurativa. J Am Acad Dermatol. 1984;11:500-502.
- Huang CM, Kirchof MG. A new perspective on isotretinoin treatment of hidradenitis suppurativa: a retrospective chart review of patient outcomes. Dermatology. 2017;233:120-125.
- Norris JF, Cunliffe WJ. Failure of treatment of familial widespread hidradenitis suppurativa with isotretinoin. Clin Exp Dermatol. 1986;11:579-583.
- Soria A, Canoui-Poitrine F, Wolkenstein P, et al. Absence of efficacy of oral isotretinoin in hidradenitis suppurativa: a retrospective study based on patients’ outcome assessment. Dermatology. 2009;218:134-135.
Case Report
A 19-year-old female marine with a 10-year history of hidradenitis suppurativa (HS) presented with hyperpigmented nodules in the inguinal folds and a recurrent cyst in the right groin area of 2 to 3 weeks’ duration. She denied axillary or inframammary involvement. She underwent several incision and drainage procedures 1 year prior to her enlistment in the US Marine Corps at 18 years of age. She previously had been treated by dermatology with doxycycline 100-mg tablets twice daily, benzoyl peroxide wash 5% applied to affected areas and rinsed daily, and clindamycin solution 1% with minimal improvement. She denied smoking or alcohol intake and said she typically wore a loose-fitting uniform to work. As a marine, she was expected to participate in daily physical training and exercises with her military unit, during which she wore a standardized physical training uniform, including nylon shorts and a cotton T-shirt. She requested light duty—military duty status with physical limitations or restrictions—to avoid physical training that would cause further friction and irritation to the inguinal region.
Physical examination demonstrated a woman with Fitzpatrick skin type III and normal body mass index. There were hyperpigmented nodules and scarring in the inguinal folds, most consistent with Hurley stage 2. A single, 0.5-cm, draining lesion was visualized. No hyperhidrosis was noted. The patient was placed on light duty for 7 days, with physical training only at her own pace and discretion. Moreover, she was restricted from field training, rifle range training, and other situations where she may excessively sweat or not be able to adequately maintain personal hygiene. She was encouraged to continue clindamycin solution 1% to the affected area twice daily and was prescribed chlorhexidine solution 4% to use when washing affected areas in the shower. The patient also was referred to the dermatology department at the Naval Hospital Camp Pendleton (Oceanside, California), where she was treated with laser hair removal in the inguinal region, thus avoiding waxing and further aggravation of HS flares. Due to the combination of topical therapies along with laser hair removal and duty restrictions, the patient had a dramatic decrease in development of severe nodular lesions.
Comment
Presentation
Historically, the incidence of HS is estimated at 0.5% to 4% of the general population with female predominance.1 Predisposing factors include obesity, smoking, genetic predisposition to acne, apocrine duct obstruction, and secondary bacterial infection.2 During acute flares, patients generally present with tender subcutaneous nodules that drain malodorous purulent material.3,4 Acute flares are unpredictable, and patients deal with chronic, recurrent, draining wounds, leading to a poor quality of life with resulting physical, psychological, financial, social, and emotional distress.3-5 The negative impact of HS on a patient’s quality of life has been reported to be greater than other dermatologic conditions.6 Lesions can be particularly painful and can cause disfiguration to the surface of the skin.7 Lesion severity is described using the Hurley staging system. Patient quality of life is directly correlated with disease severity and Hurley stage. In stage 1, abscesses develop, but no sinus tracts or cicatrization is present. In stage 2, recurrent abscesses will form tracts and cicatrization. In stage 3, the abscesses become diffuse or near diffuse, with multiple interconnected tracts and abscesses across the entire area of the body.8,9
Severe or refractory HS within the physically active military population may require consideration of light or limited duty or even separation from service. Similarly, severe HS may pose challenges with other physically demanding occupations, such as the police force and firefighters.
Prevention Focus
Prevention of flares is key for patients with HS; secondary prevention aims to reduce impact of the disease or injury that has already occurred,10,11 which includes prevention of the infundibulofolliculitis from becoming a deep folliculitis, nodule, or fistula, as well as Hurley stage progression. Prompt diagnosis with appropriate treatment can decrease the severity of lesions, pain, and scarring. Globally, HS patients continue to experience considerable diagnostic delays of 8 to 12 years after onset of initial symptoms.11,12 Earlier accurate diagnosis and initiation of treatment from the primary care provider or general medical officer is imperative. Initial accurate management may help keep symptoms from progressing to more severe painful lesions. Similarly, patients should be educated on how to prevent HS flares. Patients should avoid known triggers, including smoking, obesity, sweating, mechanical irritation, stress, and poor hygiene.11
Shaving for hair reduction creates ingrown hair shafts, which may lead to folliculitis in mechanically stressed areas in skin folds, thus initiating the inflammatory cascade of HS.11,13 Therefore, shaving along with any other mechanical stress should be avoided in patients with HS. Laser hair removal has been shown to be quite helpful in both the prevention and treatment of HS. In one study, 22 patients with Hurley stage 2 to 3 disease were treated with an Nd:YAG laser once monthly. Results demonstrated a 65% decrease in disease severity after 3 monthly treatments.11 Similarly, other lasers have been used with success in several small case series; an 800-nm diode laser, intense pulsed light therapy, and a ruby laser have each demonstrated efficacy.14 Given these results, hair removal should be recommended to patients with HS. Military servicemembers (SMs) with certain conditions, such as polycystic ovary syndrome, pseudofolliculitis barbae, and HS, are eligible for laser hair removal when available at local military treatment facilities. Primary care providers for military SMs must have a working understanding of the disease process of HS and awareness of what resources are available for treatment, which allows for more streamlined care and improved outcomes.
Treatment Options
Treatment options are diverse and depend on the severity of HS. Typically, treatment begins with medical therapy followed by escalation to surgical intervention. Medical therapies often include antibiotics, acne treatments, antiandrogen therapy, immunosuppressive agents, and biologic therapy.15,16 If first-line medical interventions fail to control HS, surgical interventions should be considered. Surgical intervention in conjunction with medical therapy decreases the chance for recurrence.3,15,16
Although HS is internationally recognized as an inflammatory disease and not an infectious process, topical antibiotics can help to prevent and improve formation of abscesses, nodules, and pustules.11 Agents such as clindamycin and chlorhexidine wash have proven effective in preventing flares.11,16 Other antibiotics used alone or in combination also are efficacious. Tetracyclines are recommended as monotherapy for mild stages of HS.17-19 Doxycycline is the most commonly used tetracycline in HS patients and has been demonstrated to penetrate Staphylococcus aureus biofilm in high enough concentrations to maintain its antibacterial activity.20 Moreover, doxycycline, as with other tetracyclines, has a multitude of anti-inflammatory and immunomodulatory properties21 and can reduce the production of IL-1, IL-6, tumor necrosis factor α, and IL-8; downregulate chemotaxis; and promote lipo-oxygenase, matrix metalloproteinase, and nuclear factor κB (NF-κB) signaling inhibition.17
Clindamycin is the only known agent that has been studied for topical treatment and utilization in milder cases of HS.17,22 Systemic combination of clindamycin and rifampicin is the most studied, with well-established efficacy in managing HS.17,23,24 Clindamycin has bacteriostatic activity toward both aerobic and anaerobic gram-positive bacteria by binding irreversibly to the 50S ribosomal subunit, thereby inhibiting bacterial protein synthesis. Rifampicin binds to the beta subunit of DNA-dependent RNA polymerase, inhibiting bacterial DNA-dependent RNA synthesis. Rifampicin has broad-spectrum activity, mostly against gram-positive as well as some gram-negative bacteria. Moreover, rifampicin has anti-inflammatory and immunomodulatory properties, including evidence that it inhibits excessive helper T cell (TH17) responses by reducing inducible nitric oxide synthase transcription and NF-κB activity.25,26
Metronidazole, moxifloxacin, and rifampicin as triple combination therapy has been shown to be effective in reducing HS activity in moderate to severe cases that were refractory to other treatments.27 Research suggests that moxifloxacin has anti-inflammatory properties, mainly by reducing IL-1β, IL-8, and tumor necrosis factor α; stabilizing IXb protein; suppressing NF-κB signaling; and reducing IL-17A.28,29
Ertapenem can be utilized as a single 6-week antibiotic course during surgical planning or rescue therapy.18 Moreover, ertapenem can be used to treat complicated skin and soft tissue infections and has been shown to substantially improve clinical aspects of severe HS.17,27
Disease-modifying antirheumatic drugs are effective in the treatment of moderate to severe HS.17-19 In 2 phase 3 trials (PIONEER I and II), adalimumab was used as monotherapy or in conjunction with antibiotics in patients with moderate to severe HS compared to placebo.30 Results demonstrated a disease burden reduction of greater than 50%. Antibiotic dual therapy was not noted to significantly affect disease burden.30 Of note, use of immunosuppressants in the military affects an SM’s availability for worldwide deployment and duty station assignment.
Antiandrogen therapies have demonstrated some reduction in HS flares. Although recommendations for use in HS is based on limited evidence, one randomized controlled trial compared ethinyl estradiol–norgestrel to ethinyl estradiol and cyproterone acetate. Both therapies resulted in similar efficacy, with 12 of 24 (50%) patients reporting HS symptoms improving or completely resolved.31 In another retrospective study of women treated with antiandrogen therapies, including ethinyl estriol, cyproterone acetate, and spironolactone, 16 of 29 (55%) patients reported improvement.32 In another study, daily doses of 100 to 150 mg of spironolactone resulted in improvement in 17 of 20 (85%) patients, including complete remission in 11 of 20 (55%) patients. Of the 3 patients with severe HS, none had complete clearing of disease burden.33 Patients with polycystic ovary syndrome or HS flares that occur around menstruation are more likely to benefit from treatment with spironolactone.18,32,34
Retinoids frequently have been utilized in the management of HS. In some retrospective studies and other prospective studies with 5 or more patients, isotretinoin monotherapy was utilized for a 4- to 10-month period.18,35-38 In the Alikhan et al18 study, 85 of 207 patients demonstrated improvement of HS symptoms, with more remarkable improvements in milder cases. Isotretinoin for management of patients with HS who have concomitant nodulocystic acne would have two-fold benefits.18
Wound Care
Given the purulent nodular formation in HS, adequate wound care management is vital. There is an abundance of HS wound care management strategies utilized by clinicians and patients. When selecting the appropriate dressing, consideration for the type of HS wound, cost, ease of application, patient comfort, absorbency, and odor management is important.3 However, living arrangements for military SMs can create difficulties applying and maintaining HS dressings, especially if deployed or in a field setting. Active-duty SMs often find themselves in austere living conditions in the field, aboard ships, or in other scenarios where they may or may not have running water or showers. Maintaining adequate hygiene may be difficult, and additional education about how to keep wounds clean must be imparted. Ideal dressings for HS should be highly absorbent, comfortable when applied to the anatomic locations of the HS lesions, and easily self-applied. Ideally, dressings would have atraumatic adhesion and antimicrobial properties.3 Cost-effective dressing options that have good absorption capability include sanitary napkins, adult briefs, infant diapers, and gauze.3 These dressings help to wick moisture, thus protecting the wound from maceration, which is a common patient concern. Although gauze dressings are easier to obtain, they are not as absorbent. Abdominal pads can be utilized, but they are moderately absorbent, bulky, and more challenging to obtain over-the-counter. Hydrofiber and calcium alginate dressings with silver are not accessible to the common consumer and are more expensive than the aforementioned dressings, but they do have some antimicrobial activity. Silver-impregnated foam dressings are moldable to intertriginous areas, easy to self-apply, and have moderate-heavy absorption abilities.
Final Thoughts
Hidradenitis suppurativa poses cumbersome and uncomfortable symptoms for all patients and may pose additional hardships for military SMs or those with physically demanding occupations who work in austere environments. Severe HS can restrict a military SM from certain duty stations, positions, or deployments. Early identification of HS can help reduce HS flares, disfigurement, and placement on limited duty status, therefore rendering the SM more able to engage in his/her operational responsibilities. Hidradenitis suppurativa should be discussed with the patient, with the goal to prevent flares for SMs that will be in the field, placed in austere environments, or be deployed. Use of immunosuppressants in active-duty SMs may affect their deployability, duty assignment, and retention.
For a military SM with HS, all aspects of prevention and treatment need to be balanced with his/her ability to remain deployable and complete his/her daily duties. Military SMs are not guaranteed the ideal scenario for treatment and prevention of HS. Unsanitary environments and occlusive uniforms undoubtedly contribute to disease process and make treatment more challenging. If a military SM is in a field setting or deployed, frequent daily dressing changes should still be attempted.
Case Report
A 19-year-old female marine with a 10-year history of hidradenitis suppurativa (HS) presented with hyperpigmented nodules in the inguinal folds and a recurrent cyst in the right groin area of 2 to 3 weeks’ duration. She denied axillary or inframammary involvement. She underwent several incision and drainage procedures 1 year prior to her enlistment in the US Marine Corps at 18 years of age. She previously had been treated by dermatology with doxycycline 100-mg tablets twice daily, benzoyl peroxide wash 5% applied to affected areas and rinsed daily, and clindamycin solution 1% with minimal improvement. She denied smoking or alcohol intake and said she typically wore a loose-fitting uniform to work. As a marine, she was expected to participate in daily physical training and exercises with her military unit, during which she wore a standardized physical training uniform, including nylon shorts and a cotton T-shirt. She requested light duty—military duty status with physical limitations or restrictions—to avoid physical training that would cause further friction and irritation to the inguinal region.
Physical examination demonstrated a woman with Fitzpatrick skin type III and normal body mass index. There were hyperpigmented nodules and scarring in the inguinal folds, most consistent with Hurley stage 2. A single, 0.5-cm, draining lesion was visualized. No hyperhidrosis was noted. The patient was placed on light duty for 7 days, with physical training only at her own pace and discretion. Moreover, she was restricted from field training, rifle range training, and other situations where she may excessively sweat or not be able to adequately maintain personal hygiene. She was encouraged to continue clindamycin solution 1% to the affected area twice daily and was prescribed chlorhexidine solution 4% to use when washing affected areas in the shower. The patient also was referred to the dermatology department at the Naval Hospital Camp Pendleton (Oceanside, California), where she was treated with laser hair removal in the inguinal region, thus avoiding waxing and further aggravation of HS flares. Due to the combination of topical therapies along with laser hair removal and duty restrictions, the patient had a dramatic decrease in development of severe nodular lesions.
Comment
Presentation
Historically, the incidence of HS is estimated at 0.5% to 4% of the general population with female predominance.1 Predisposing factors include obesity, smoking, genetic predisposition to acne, apocrine duct obstruction, and secondary bacterial infection.2 During acute flares, patients generally present with tender subcutaneous nodules that drain malodorous purulent material.3,4 Acute flares are unpredictable, and patients deal with chronic, recurrent, draining wounds, leading to a poor quality of life with resulting physical, psychological, financial, social, and emotional distress.3-5 The negative impact of HS on a patient’s quality of life has been reported to be greater than other dermatologic conditions.6 Lesions can be particularly painful and can cause disfiguration to the surface of the skin.7 Lesion severity is described using the Hurley staging system. Patient quality of life is directly correlated with disease severity and Hurley stage. In stage 1, abscesses develop, but no sinus tracts or cicatrization is present. In stage 2, recurrent abscesses will form tracts and cicatrization. In stage 3, the abscesses become diffuse or near diffuse, with multiple interconnected tracts and abscesses across the entire area of the body.8,9
Severe or refractory HS within the physically active military population may require consideration of light or limited duty or even separation from service. Similarly, severe HS may pose challenges with other physically demanding occupations, such as the police force and firefighters.
Prevention Focus
Prevention of flares is key for patients with HS; secondary prevention aims to reduce impact of the disease or injury that has already occurred,10,11 which includes prevention of the infundibulofolliculitis from becoming a deep folliculitis, nodule, or fistula, as well as Hurley stage progression. Prompt diagnosis with appropriate treatment can decrease the severity of lesions, pain, and scarring. Globally, HS patients continue to experience considerable diagnostic delays of 8 to 12 years after onset of initial symptoms.11,12 Earlier accurate diagnosis and initiation of treatment from the primary care provider or general medical officer is imperative. Initial accurate management may help keep symptoms from progressing to more severe painful lesions. Similarly, patients should be educated on how to prevent HS flares. Patients should avoid known triggers, including smoking, obesity, sweating, mechanical irritation, stress, and poor hygiene.11
Shaving for hair reduction creates ingrown hair shafts, which may lead to folliculitis in mechanically stressed areas in skin folds, thus initiating the inflammatory cascade of HS.11,13 Therefore, shaving along with any other mechanical stress should be avoided in patients with HS. Laser hair removal has been shown to be quite helpful in both the prevention and treatment of HS. In one study, 22 patients with Hurley stage 2 to 3 disease were treated with an Nd:YAG laser once monthly. Results demonstrated a 65% decrease in disease severity after 3 monthly treatments.11 Similarly, other lasers have been used with success in several small case series; an 800-nm diode laser, intense pulsed light therapy, and a ruby laser have each demonstrated efficacy.14 Given these results, hair removal should be recommended to patients with HS. Military servicemembers (SMs) with certain conditions, such as polycystic ovary syndrome, pseudofolliculitis barbae, and HS, are eligible for laser hair removal when available at local military treatment facilities. Primary care providers for military SMs must have a working understanding of the disease process of HS and awareness of what resources are available for treatment, which allows for more streamlined care and improved outcomes.
Treatment Options
Treatment options are diverse and depend on the severity of HS. Typically, treatment begins with medical therapy followed by escalation to surgical intervention. Medical therapies often include antibiotics, acne treatments, antiandrogen therapy, immunosuppressive agents, and biologic therapy.15,16 If first-line medical interventions fail to control HS, surgical interventions should be considered. Surgical intervention in conjunction with medical therapy decreases the chance for recurrence.3,15,16
Although HS is internationally recognized as an inflammatory disease and not an infectious process, topical antibiotics can help to prevent and improve formation of abscesses, nodules, and pustules.11 Agents such as clindamycin and chlorhexidine wash have proven effective in preventing flares.11,16 Other antibiotics used alone or in combination also are efficacious. Tetracyclines are recommended as monotherapy for mild stages of HS.17-19 Doxycycline is the most commonly used tetracycline in HS patients and has been demonstrated to penetrate Staphylococcus aureus biofilm in high enough concentrations to maintain its antibacterial activity.20 Moreover, doxycycline, as with other tetracyclines, has a multitude of anti-inflammatory and immunomodulatory properties21 and can reduce the production of IL-1, IL-6, tumor necrosis factor α, and IL-8; downregulate chemotaxis; and promote lipo-oxygenase, matrix metalloproteinase, and nuclear factor κB (NF-κB) signaling inhibition.17
Clindamycin is the only known agent that has been studied for topical treatment and utilization in milder cases of HS.17,22 Systemic combination of clindamycin and rifampicin is the most studied, with well-established efficacy in managing HS.17,23,24 Clindamycin has bacteriostatic activity toward both aerobic and anaerobic gram-positive bacteria by binding irreversibly to the 50S ribosomal subunit, thereby inhibiting bacterial protein synthesis. Rifampicin binds to the beta subunit of DNA-dependent RNA polymerase, inhibiting bacterial DNA-dependent RNA synthesis. Rifampicin has broad-spectrum activity, mostly against gram-positive as well as some gram-negative bacteria. Moreover, rifampicin has anti-inflammatory and immunomodulatory properties, including evidence that it inhibits excessive helper T cell (TH17) responses by reducing inducible nitric oxide synthase transcription and NF-κB activity.25,26
Metronidazole, moxifloxacin, and rifampicin as triple combination therapy has been shown to be effective in reducing HS activity in moderate to severe cases that were refractory to other treatments.27 Research suggests that moxifloxacin has anti-inflammatory properties, mainly by reducing IL-1β, IL-8, and tumor necrosis factor α; stabilizing IXb protein; suppressing NF-κB signaling; and reducing IL-17A.28,29
Ertapenem can be utilized as a single 6-week antibiotic course during surgical planning or rescue therapy.18 Moreover, ertapenem can be used to treat complicated skin and soft tissue infections and has been shown to substantially improve clinical aspects of severe HS.17,27
Disease-modifying antirheumatic drugs are effective in the treatment of moderate to severe HS.17-19 In 2 phase 3 trials (PIONEER I and II), adalimumab was used as monotherapy or in conjunction with antibiotics in patients with moderate to severe HS compared to placebo.30 Results demonstrated a disease burden reduction of greater than 50%. Antibiotic dual therapy was not noted to significantly affect disease burden.30 Of note, use of immunosuppressants in the military affects an SM’s availability for worldwide deployment and duty station assignment.
Antiandrogen therapies have demonstrated some reduction in HS flares. Although recommendations for use in HS is based on limited evidence, one randomized controlled trial compared ethinyl estradiol–norgestrel to ethinyl estradiol and cyproterone acetate. Both therapies resulted in similar efficacy, with 12 of 24 (50%) patients reporting HS symptoms improving or completely resolved.31 In another retrospective study of women treated with antiandrogen therapies, including ethinyl estriol, cyproterone acetate, and spironolactone, 16 of 29 (55%) patients reported improvement.32 In another study, daily doses of 100 to 150 mg of spironolactone resulted in improvement in 17 of 20 (85%) patients, including complete remission in 11 of 20 (55%) patients. Of the 3 patients with severe HS, none had complete clearing of disease burden.33 Patients with polycystic ovary syndrome or HS flares that occur around menstruation are more likely to benefit from treatment with spironolactone.18,32,34
Retinoids frequently have been utilized in the management of HS. In some retrospective studies and other prospective studies with 5 or more patients, isotretinoin monotherapy was utilized for a 4- to 10-month period.18,35-38 In the Alikhan et al18 study, 85 of 207 patients demonstrated improvement of HS symptoms, with more remarkable improvements in milder cases. Isotretinoin for management of patients with HS who have concomitant nodulocystic acne would have two-fold benefits.18
Wound Care
Given the purulent nodular formation in HS, adequate wound care management is vital. There is an abundance of HS wound care management strategies utilized by clinicians and patients. When selecting the appropriate dressing, consideration for the type of HS wound, cost, ease of application, patient comfort, absorbency, and odor management is important.3 However, living arrangements for military SMs can create difficulties applying and maintaining HS dressings, especially if deployed or in a field setting. Active-duty SMs often find themselves in austere living conditions in the field, aboard ships, or in other scenarios where they may or may not have running water or showers. Maintaining adequate hygiene may be difficult, and additional education about how to keep wounds clean must be imparted. Ideal dressings for HS should be highly absorbent, comfortable when applied to the anatomic locations of the HS lesions, and easily self-applied. Ideally, dressings would have atraumatic adhesion and antimicrobial properties.3 Cost-effective dressing options that have good absorption capability include sanitary napkins, adult briefs, infant diapers, and gauze.3 These dressings help to wick moisture, thus protecting the wound from maceration, which is a common patient concern. Although gauze dressings are easier to obtain, they are not as absorbent. Abdominal pads can be utilized, but they are moderately absorbent, bulky, and more challenging to obtain over-the-counter. Hydrofiber and calcium alginate dressings with silver are not accessible to the common consumer and are more expensive than the aforementioned dressings, but they do have some antimicrobial activity. Silver-impregnated foam dressings are moldable to intertriginous areas, easy to self-apply, and have moderate-heavy absorption abilities.
Final Thoughts
Hidradenitis suppurativa poses cumbersome and uncomfortable symptoms for all patients and may pose additional hardships for military SMs or those with physically demanding occupations who work in austere environments. Severe HS can restrict a military SM from certain duty stations, positions, or deployments. Early identification of HS can help reduce HS flares, disfigurement, and placement on limited duty status, therefore rendering the SM more able to engage in his/her operational responsibilities. Hidradenitis suppurativa should be discussed with the patient, with the goal to prevent flares for SMs that will be in the field, placed in austere environments, or be deployed. Use of immunosuppressants in active-duty SMs may affect their deployability, duty assignment, and retention.
For a military SM with HS, all aspects of prevention and treatment need to be balanced with his/her ability to remain deployable and complete his/her daily duties. Military SMs are not guaranteed the ideal scenario for treatment and prevention of HS. Unsanitary environments and occlusive uniforms undoubtedly contribute to disease process and make treatment more challenging. If a military SM is in a field setting or deployed, frequent daily dressing changes should still be attempted.
- Dufour DN, Emtestam L, Jemec GB. Hidradenitis suppurativa: a common and burdensome, yet under-recognised, inflammatory skin disease. Postgrad Med J. 2014;90:216-221.
- Beshara MA. Hidradenitis suppurativa: a clinician’s tool for early diagnosis and treatment. Nurse Pract. 2010;35:24-28.
- Kazemi A, Carnaggio K, Clark M, et al. Optimal wound care management in hidradenitis suppurativa. J Dermatolog Treat. 2017;29:165-167.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003:49:96-98.
- Blattner C, Polley DC, Ferrito F, et al. Central centrifugal cicatricial alopecia. Indian Dermatol Online J. 2013:4:50.
- Wolkenstein P, Loundou A, Barrau K, et al. Quality of life impairment in hidradenitis suppurativa: a study of 61 cases. J Am Acad Dermatol. 2007;56:621-623.
- Smith HS, Chao JD, Teitelbaum J. Painful hidradenitis suppurativa. Clin J Pain. 2010;26:435-444.
- Alavi A, Anooshirvani N, Kim WB, et al. Quality-of-life impairment in patients with hidradenitis suppurativa: a Canadian study. Am J Clin Dermatol. 2015;16:61-65.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH Jr, eds. Dermatologic Surgery: Principles and Practice. 2nd ed. New York, NY: Marcel Dekker; 1996:623-645.
- Kligman AM. Welcome letter. 2nd International Conference on the Sebaceous Gland, Acne, Rosacea and Related Disorders; September 13-16, 2008; Rome Italy.
- Kurzen H, Kurzen M. Secondary prevention of hidradenitis suppurativa. Dermatol Reports. 2019;11:8243.
- Sabat R, Tsaousi A, Rossbacher J, et al. Acne inversa/hidradenitis suppurativa: an update [in German]. Hautarzt. 2017;68:999-1006.
- Boer J, Nazary M, Riis PT. The role of mechanical stress in hidradenitis suppurativa. Dermatol Clin. 2016;34:37-43.
- Hamzavi IH, Griffith JL, Riyaz F, et al. Laser and light-based treatment options for hidradenitis suppurativa. J Am Acad Dermatol. 2015;73(5 suppl 1):S78-S81.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Michel C, DiBianco JM, Sabarwal V, et al. The treatment of genitoperineal hidradenitis suppurativa: a review of the literature. Urology. 2019;124:1-5.
- Constantinou CA, Fragoulis GE, Nikiphorou E. Hidradenitis suppurativa: infection, autoimmunity, or both [published online December 30, 2019]? Ther Adv Musculoskelet Dis. doi:10.1177/1759720x19895488.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Zouboulis CC, Desai N, Emtestam, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29:619-644.
- Mandell JB, Orr S, Koch J, et al. Large variations in clinical antibiotic activity against Staphylococcus aureus biofilms of periprosthetic joint infection isolates. J Orthop Res. 2019;37:1604-1609.
- Sun J, Shigemi H, Tanaka Y, et al. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. 2015;4:397-404.
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. Int J Dermatol. 1983;22:325-328.
- Gener G, Canoui-Poitrine F, Revuz JE, et al. Combination therapy with clindamycin and rifampicin for hidradenitis suppurativa: a series of 116 consecutive patients. Dermatology. 2009;219:148-154.
- Griffiths CEM. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. 2006;154:977-978.
- Ma K, Chen X, Chen J-C, et al. Rifampicin attenuates experimental autoimmune encephalomyelitis by inhibiting pathogenic Th17 cells responses. J Neurochem. 2016;139:1151-1162.
- Yuhas Y, Berent E, Ovadiah H, et al. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob Agents Chemother. 2006;50:396-398.
- Join-Lambert O, Coignard H, Jais J-P, et al. Efficacy of rifampin-moxifloxacin-metronidazole combination therapy in hidradenitis suppurativa. Dermatology. 2011;222:49-58.
- Choi J-H, Song M-J, Kim S-H, et al. Effect of moxifloxacin on production of proinflammatory cytokines from human peripheral blood mononuclear cells. Antimicrob Agents Chemother. 2003;47:3704-3707.
- Weiss T, Shalit I, Blau H, et al. Anti-inflammatory effects of moxifloxacin on activated human monocytic cells: inhibition of NF-kappaB and mitogen-activated protein kinase activation and of synthesis of proinflammatory cytokines.” Antimicrob Agents Chemother. 2004;48:1974-1982.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Mortimer PS, Dawber RP, Gales MA, et al. A double-blind controlled cross-over trial of cyproterone acetate in females with hidradenitis suppurativa. Br J Dermatol. 1986;115:263-268.
- Kraft JN, Searles GE. Hidradenitis suppurativa in 64 female patients: retrospective study comparing oral antibiotics and antiandrogen therapy. J Cutan Med Surg. 2007;11:125-131.
- Lee A, Fischer G. A case series of 20 women with hidradenitis suppurativa treated with spironolactone. Australas J Dermatol. 2015;56:192-196.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a “male” therapy for a predominantly “female” disease. J Clin Aesthet Dermatol. 2016;9:44-50.
- Dicken CH, Powell ST, Spear KL. Evaluation of isotretinoin treatment of hidradenitis suppurativa. J Am Acad Dermatol. 1984;11:500-502.
- Huang CM, Kirchof MG. A new perspective on isotretinoin treatment of hidradenitis suppurativa: a retrospective chart review of patient outcomes. Dermatology. 2017;233:120-125.
- Norris JF, Cunliffe WJ. Failure of treatment of familial widespread hidradenitis suppurativa with isotretinoin. Clin Exp Dermatol. 1986;11:579-583.
- Soria A, Canoui-Poitrine F, Wolkenstein P, et al. Absence of efficacy of oral isotretinoin in hidradenitis suppurativa: a retrospective study based on patients’ outcome assessment. Dermatology. 2009;218:134-135.
- Dufour DN, Emtestam L, Jemec GB. Hidradenitis suppurativa: a common and burdensome, yet under-recognised, inflammatory skin disease. Postgrad Med J. 2014;90:216-221.
- Beshara MA. Hidradenitis suppurativa: a clinician’s tool for early diagnosis and treatment. Nurse Pract. 2010;35:24-28.
- Kazemi A, Carnaggio K, Clark M, et al. Optimal wound care management in hidradenitis suppurativa. J Dermatolog Treat. 2017;29:165-167.
- Tosti A, Piraccini BM, Pazzaglia M, et al. Clobetasol propionate 0.05% under occlusion in the treatment of alopecia totalis/universalis. J Am Acad Dermatol. 2003:49:96-98.
- Blattner C, Polley DC, Ferrito F, et al. Central centrifugal cicatricial alopecia. Indian Dermatol Online J. 2013:4:50.
- Wolkenstein P, Loundou A, Barrau K, et al. Quality of life impairment in hidradenitis suppurativa: a study of 61 cases. J Am Acad Dermatol. 2007;56:621-623.
- Smith HS, Chao JD, Teitelbaum J. Painful hidradenitis suppurativa. Clin J Pain. 2010;26:435-444.
- Alavi A, Anooshirvani N, Kim WB, et al. Quality-of-life impairment in patients with hidradenitis suppurativa: a Canadian study. Am J Clin Dermatol. 2015;16:61-65.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH Jr, eds. Dermatologic Surgery: Principles and Practice. 2nd ed. New York, NY: Marcel Dekker; 1996:623-645.
- Kligman AM. Welcome letter. 2nd International Conference on the Sebaceous Gland, Acne, Rosacea and Related Disorders; September 13-16, 2008; Rome Italy.
- Kurzen H, Kurzen M. Secondary prevention of hidradenitis suppurativa. Dermatol Reports. 2019;11:8243.
- Sabat R, Tsaousi A, Rossbacher J, et al. Acne inversa/hidradenitis suppurativa: an update [in German]. Hautarzt. 2017;68:999-1006.
- Boer J, Nazary M, Riis PT. The role of mechanical stress in hidradenitis suppurativa. Dermatol Clin. 2016;34:37-43.
- Hamzavi IH, Griffith JL, Riyaz F, et al. Laser and light-based treatment options for hidradenitis suppurativa. J Am Acad Dermatol. 2015;73(5 suppl 1):S78-S81.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Michel C, DiBianco JM, Sabarwal V, et al. The treatment of genitoperineal hidradenitis suppurativa: a review of the literature. Urology. 2019;124:1-5.
- Constantinou CA, Fragoulis GE, Nikiphorou E. Hidradenitis suppurativa: infection, autoimmunity, or both [published online December 30, 2019]? Ther Adv Musculoskelet Dis. doi:10.1177/1759720x19895488.
- Alikhan A, Sayed C, Alavi A, et al. North American clinical management guidelines for hidradenitis suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations: part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Zouboulis CC, Desai N, Emtestam, et al. European S1 guideline for the treatment of hidradenitis suppurativa/acne inversa. J Eur Acad Dermatol Venereol. 2015;29:619-644.
- Mandell JB, Orr S, Koch J, et al. Large variations in clinical antibiotic activity against Staphylococcus aureus biofilms of periprosthetic joint infection isolates. J Orthop Res. 2019;37:1604-1609.
- Sun J, Shigemi H, Tanaka Y, et al. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. 2015;4:397-404.
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. Int J Dermatol. 1983;22:325-328.
- Gener G, Canoui-Poitrine F, Revuz JE, et al. Combination therapy with clindamycin and rifampicin for hidradenitis suppurativa: a series of 116 consecutive patients. Dermatology. 2009;219:148-154.
- Griffiths CEM. Clindamycin and rifampicin combination therapy for hidradenitis suppurativa. Br J Dermatol. 2006;154:977-978.
- Ma K, Chen X, Chen J-C, et al. Rifampicin attenuates experimental autoimmune encephalomyelitis by inhibiting pathogenic Th17 cells responses. J Neurochem. 2016;139:1151-1162.
- Yuhas Y, Berent E, Ovadiah H, et al. Rifampin augments cytokine-induced nitric oxide production in human alveolar epithelial cells. Antimicrob Agents Chemother. 2006;50:396-398.
- Join-Lambert O, Coignard H, Jais J-P, et al. Efficacy of rifampin-moxifloxacin-metronidazole combination therapy in hidradenitis suppurativa. Dermatology. 2011;222:49-58.
- Choi J-H, Song M-J, Kim S-H, et al. Effect of moxifloxacin on production of proinflammatory cytokines from human peripheral blood mononuclear cells. Antimicrob Agents Chemother. 2003;47:3704-3707.
- Weiss T, Shalit I, Blau H, et al. Anti-inflammatory effects of moxifloxacin on activated human monocytic cells: inhibition of NF-kappaB and mitogen-activated protein kinase activation and of synthesis of proinflammatory cytokines.” Antimicrob Agents Chemother. 2004;48:1974-1982.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Mortimer PS, Dawber RP, Gales MA, et al. A double-blind controlled cross-over trial of cyproterone acetate in females with hidradenitis suppurativa. Br J Dermatol. 1986;115:263-268.
- Kraft JN, Searles GE. Hidradenitis suppurativa in 64 female patients: retrospective study comparing oral antibiotics and antiandrogen therapy. J Cutan Med Surg. 2007;11:125-131.
- Lee A, Fischer G. A case series of 20 women with hidradenitis suppurativa treated with spironolactone. Australas J Dermatol. 2015;56:192-196.
- Khandalavala BN, Do MV. Finasteride in hidradenitis suppurativa: a “male” therapy for a predominantly “female” disease. J Clin Aesthet Dermatol. 2016;9:44-50.
- Dicken CH, Powell ST, Spear KL. Evaluation of isotretinoin treatment of hidradenitis suppurativa. J Am Acad Dermatol. 1984;11:500-502.
- Huang CM, Kirchof MG. A new perspective on isotretinoin treatment of hidradenitis suppurativa: a retrospective chart review of patient outcomes. Dermatology. 2017;233:120-125.
- Norris JF, Cunliffe WJ. Failure of treatment of familial widespread hidradenitis suppurativa with isotretinoin. Clin Exp Dermatol. 1986;11:579-583.
- Soria A, Canoui-Poitrine F, Wolkenstein P, et al. Absence of efficacy of oral isotretinoin in hidradenitis suppurativa: a retrospective study based on patients’ outcome assessment. Dermatology. 2009;218:134-135.
Practice Points
- Hidradenitis suppurativa (HS) can be more difficult to treat in physically active military servicemembers (SMs).
- Patient education and primary care physician awareness of HS is critical to initial diagnosis and long-term management.
- Primary care physician knowledge of HS as well as an understanding of the capabilities at local military medical facilities is important for optimal treatment of HS in military SMs.
