User login
Irregular, Smooth, Pink Plaque on the Back
The Diagnosis: Fibroepithelioma of Pinkus
Fibroepithelioma of Pinkus (FeP) was first described in 19531 and was thought to be premalignant as evidenced by the proposed name premalignant fibroepithelial tumor of the skin. This neoplasm now is largely believed to represent a rare form of basal cell carcinoma (BCC). Typical presentation is a smooth, flesh-colored or pink plaque or nodule.2 Fibroepithelioma of Pinkus has a predilection for the lumbosacral back, though the groin also has been reported as a common site of incidence.1,3 Similar to other BCCs, it is seen in older individuals, typically those older than 50 years.3,4
Clinical diagnosis of FeP can be difficult. The differential diagnosis of FeP can include acrochordon, amelanotic melanoma, compound nevus, hemangioma, neurofibroma, nevus sebaceous, pyogenic granuloma, and seborrheic keratosis.5 Dermoscopic evaluation can aid in the diagnosis. A vascular network composed of fine arborizing vessels with or without dotted vessels and white streaks are characteristic findings of FeP. Patients with pigment also demonstrate structureless gray-brown areas and gray-blue dots.6
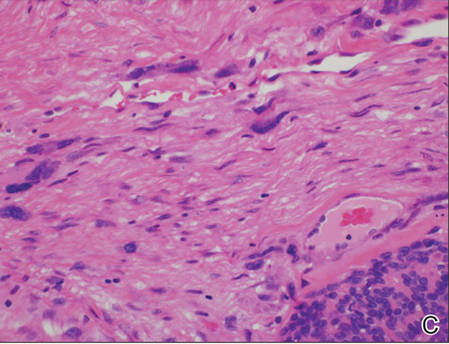
Biopsy with subsequent histopathologic evaluation confirms the diagnosis of FeP. The characteristic microscopic findings of thin eosinophilic epithelial strands with eccrine ducts anastomosing in an abundant fibromyxoid stroma with collections of basophilic cells located at the ends of the epithelial strands were demonstrated in our patient’s histopathologic specimen (Figure). The histologic appearance is similar to syringofibroadenoma of Mascaro. Recognition of basaloid nests, which often demonstrate retraction, and mitotic activity can differentiate FeP from syringofibroadenoma of Mascaro.7
Treatment of FeP is largely the same as other BCCs including destruction by electrodesiccation and curettage or complete removal by surgical excision. Several studies have demonstrated effective treatment of nonaggressive BCCs with curettage alone and subjectively reported improved cosmesis compared to electrodesiccation and curettage.8-10 Although methyl aminolevulinate photodynamic therapy has demonstrated some therapeutic efficacy for superficial and nodular BCCs,11 a case report utilizing the same modality for FeP did not provide adequate response.12 However, adequate data are not available to assess potential use of this less invasive therapy.
- Pinkus H. Premalignant fibroepithelial tumors of skin. AMA Arch Derm Syphilol. 1953;67:598-615.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
- Barr RJ, Herten RJ, Stone OJ. Multiple premalignant fibroepitheliomas of Pinkus: a case report and review of the literature. Cutis. 1978;21:335-337.
- Betti R, Inselvini E, Carducci M, et al. Age and site prevalence of histologic subtypes of basal cell carcinomas. Int J Dermatol. 1995;34:174-176.
- Cohen PR, Tschen JA. Fibroepithelioma of Pinkus presenting as a sessile thigh nodule. Skinmed. 2003;2:385-387.
- Zalaudek I, Ferrara G, Broganelli P, et al. Dermoscopy patterns of fibroepithelioma of Pinkus. Arch Dermatol. 2006;142:1318-1322.
- Schadt CR, Boyd AS. Eccrine syringofibroadenoma with co-existent squamous cell carcinoma. J Cutan Pathol. 2007;34(suppl 1):71-74.
- Barlow JO, Zalla MJ, Kyle A, et al. Treatment of basal cell carcinoma with curettage alone. J Am Acad Dermatol. 2006;54:1039-1045.
- McDaniel WE. Therapy for basal cell epitheliomas by curettage only. further study. Arch Dermatol. 1983;119:901-903.
- Reymann F. 15 Years’ experience with treatment of basal cell carcinomas of the skin with curettage. Acta Derm Venereol Suppl (Stockh). 1985;120:56-59.
- Fai D, Arpaia N, Romano I, et al. Methyl-aminolevulinate photodynamic therapy for the treatment of actinic keratoses and non-melanoma skin cancers: a retrospective analysis of response in 462 patients. G Ital Dermatol Venereol. 2009;144:281-285.
- Park MY, Kim YC. Fibroepithelioma of Pinkus: poor response to topical photodynamic therapy. Eur J Dermatol. 2010;20:133-134.
The Diagnosis: Fibroepithelioma of Pinkus
Fibroepithelioma of Pinkus (FeP) was first described in 19531 and was thought to be premalignant as evidenced by the proposed name premalignant fibroepithelial tumor of the skin. This neoplasm now is largely believed to represent a rare form of basal cell carcinoma (BCC). Typical presentation is a smooth, flesh-colored or pink plaque or nodule.2 Fibroepithelioma of Pinkus has a predilection for the lumbosacral back, though the groin also has been reported as a common site of incidence.1,3 Similar to other BCCs, it is seen in older individuals, typically those older than 50 years.3,4
Clinical diagnosis of FeP can be difficult. The differential diagnosis of FeP can include acrochordon, amelanotic melanoma, compound nevus, hemangioma, neurofibroma, nevus sebaceous, pyogenic granuloma, and seborrheic keratosis.5 Dermoscopic evaluation can aid in the diagnosis. A vascular network composed of fine arborizing vessels with or without dotted vessels and white streaks are characteristic findings of FeP. Patients with pigment also demonstrate structureless gray-brown areas and gray-blue dots.6
Biopsy with subsequent histopathologic evaluation confirms the diagnosis of FeP. The characteristic microscopic findings of thin eosinophilic epithelial strands with eccrine ducts anastomosing in an abundant fibromyxoid stroma with collections of basophilic cells located at the ends of the epithelial strands were demonstrated in our patient’s histopathologic specimen (Figure). The histologic appearance is similar to syringofibroadenoma of Mascaro. Recognition of basaloid nests, which often demonstrate retraction, and mitotic activity can differentiate FeP from syringofibroadenoma of Mascaro.7
Treatment of FeP is largely the same as other BCCs including destruction by electrodesiccation and curettage or complete removal by surgical excision. Several studies have demonstrated effective treatment of nonaggressive BCCs with curettage alone and subjectively reported improved cosmesis compared to electrodesiccation and curettage.8-10 Although methyl aminolevulinate photodynamic therapy has demonstrated some therapeutic efficacy for superficial and nodular BCCs,11 a case report utilizing the same modality for FeP did not provide adequate response.12 However, adequate data are not available to assess potential use of this less invasive therapy.
The Diagnosis: Fibroepithelioma of Pinkus
Fibroepithelioma of Pinkus (FeP) was first described in 19531 and was thought to be premalignant as evidenced by the proposed name premalignant fibroepithelial tumor of the skin. This neoplasm now is largely believed to represent a rare form of basal cell carcinoma (BCC). Typical presentation is a smooth, flesh-colored or pink plaque or nodule.2 Fibroepithelioma of Pinkus has a predilection for the lumbosacral back, though the groin also has been reported as a common site of incidence.1,3 Similar to other BCCs, it is seen in older individuals, typically those older than 50 years.3,4
Clinical diagnosis of FeP can be difficult. The differential diagnosis of FeP can include acrochordon, amelanotic melanoma, compound nevus, hemangioma, neurofibroma, nevus sebaceous, pyogenic granuloma, and seborrheic keratosis.5 Dermoscopic evaluation can aid in the diagnosis. A vascular network composed of fine arborizing vessels with or without dotted vessels and white streaks are characteristic findings of FeP. Patients with pigment also demonstrate structureless gray-brown areas and gray-blue dots.6
Biopsy with subsequent histopathologic evaluation confirms the diagnosis of FeP. The characteristic microscopic findings of thin eosinophilic epithelial strands with eccrine ducts anastomosing in an abundant fibromyxoid stroma with collections of basophilic cells located at the ends of the epithelial strands were demonstrated in our patient’s histopathologic specimen (Figure). The histologic appearance is similar to syringofibroadenoma of Mascaro. Recognition of basaloid nests, which often demonstrate retraction, and mitotic activity can differentiate FeP from syringofibroadenoma of Mascaro.7
Treatment of FeP is largely the same as other BCCs including destruction by electrodesiccation and curettage or complete removal by surgical excision. Several studies have demonstrated effective treatment of nonaggressive BCCs with curettage alone and subjectively reported improved cosmesis compared to electrodesiccation and curettage.8-10 Although methyl aminolevulinate photodynamic therapy has demonstrated some therapeutic efficacy for superficial and nodular BCCs,11 a case report utilizing the same modality for FeP did not provide adequate response.12 However, adequate data are not available to assess potential use of this less invasive therapy.
- Pinkus H. Premalignant fibroepithelial tumors of skin. AMA Arch Derm Syphilol. 1953;67:598-615.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
- Barr RJ, Herten RJ, Stone OJ. Multiple premalignant fibroepitheliomas of Pinkus: a case report and review of the literature. Cutis. 1978;21:335-337.
- Betti R, Inselvini E, Carducci M, et al. Age and site prevalence of histologic subtypes of basal cell carcinomas. Int J Dermatol. 1995;34:174-176.
- Cohen PR, Tschen JA. Fibroepithelioma of Pinkus presenting as a sessile thigh nodule. Skinmed. 2003;2:385-387.
- Zalaudek I, Ferrara G, Broganelli P, et al. Dermoscopy patterns of fibroepithelioma of Pinkus. Arch Dermatol. 2006;142:1318-1322.
- Schadt CR, Boyd AS. Eccrine syringofibroadenoma with co-existent squamous cell carcinoma. J Cutan Pathol. 2007;34(suppl 1):71-74.
- Barlow JO, Zalla MJ, Kyle A, et al. Treatment of basal cell carcinoma with curettage alone. J Am Acad Dermatol. 2006;54:1039-1045.
- McDaniel WE. Therapy for basal cell epitheliomas by curettage only. further study. Arch Dermatol. 1983;119:901-903.
- Reymann F. 15 Years’ experience with treatment of basal cell carcinomas of the skin with curettage. Acta Derm Venereol Suppl (Stockh). 1985;120:56-59.
- Fai D, Arpaia N, Romano I, et al. Methyl-aminolevulinate photodynamic therapy for the treatment of actinic keratoses and non-melanoma skin cancers: a retrospective analysis of response in 462 patients. G Ital Dermatol Venereol. 2009;144:281-285.
- Park MY, Kim YC. Fibroepithelioma of Pinkus: poor response to topical photodynamic therapy. Eur J Dermatol. 2010;20:133-134.
- Pinkus H. Premalignant fibroepithelial tumors of skin. AMA Arch Derm Syphilol. 1953;67:598-615.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
- Barr RJ, Herten RJ, Stone OJ. Multiple premalignant fibroepitheliomas of Pinkus: a case report and review of the literature. Cutis. 1978;21:335-337.
- Betti R, Inselvini E, Carducci M, et al. Age and site prevalence of histologic subtypes of basal cell carcinomas. Int J Dermatol. 1995;34:174-176.
- Cohen PR, Tschen JA. Fibroepithelioma of Pinkus presenting as a sessile thigh nodule. Skinmed. 2003;2:385-387.
- Zalaudek I, Ferrara G, Broganelli P, et al. Dermoscopy patterns of fibroepithelioma of Pinkus. Arch Dermatol. 2006;142:1318-1322.
- Schadt CR, Boyd AS. Eccrine syringofibroadenoma with co-existent squamous cell carcinoma. J Cutan Pathol. 2007;34(suppl 1):71-74.
- Barlow JO, Zalla MJ, Kyle A, et al. Treatment of basal cell carcinoma with curettage alone. J Am Acad Dermatol. 2006;54:1039-1045.
- McDaniel WE. Therapy for basal cell epitheliomas by curettage only. further study. Arch Dermatol. 1983;119:901-903.
- Reymann F. 15 Years’ experience with treatment of basal cell carcinomas of the skin with curettage. Acta Derm Venereol Suppl (Stockh). 1985;120:56-59.
- Fai D, Arpaia N, Romano I, et al. Methyl-aminolevulinate photodynamic therapy for the treatment of actinic keratoses and non-melanoma skin cancers: a retrospective analysis of response in 462 patients. G Ital Dermatol Venereol. 2009;144:281-285.
- Park MY, Kim YC. Fibroepithelioma of Pinkus: poor response to topical photodynamic therapy. Eur J Dermatol. 2010;20:133-134.
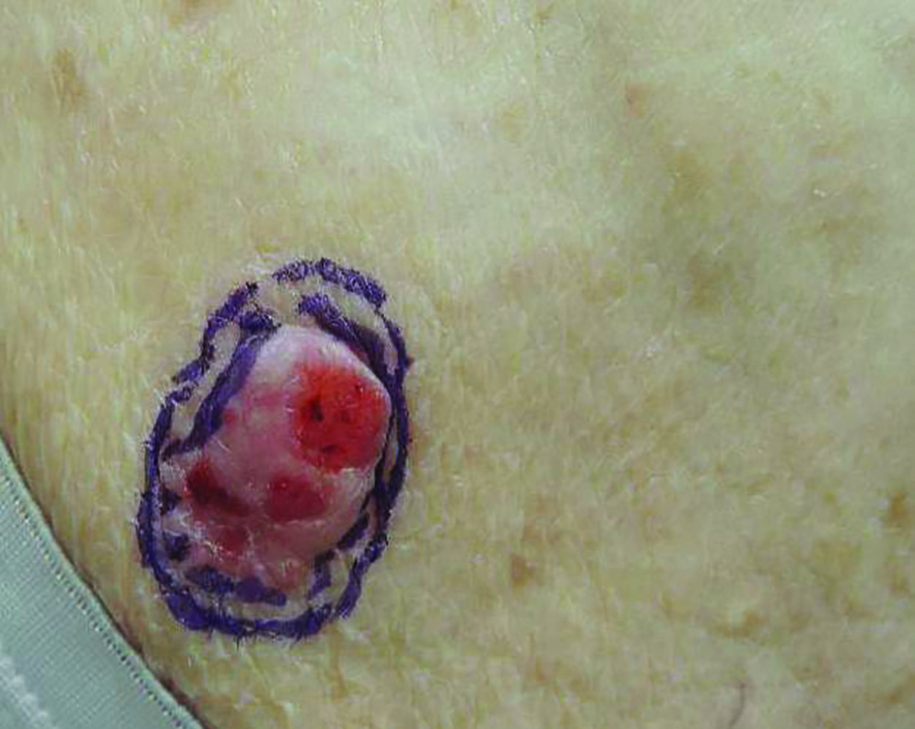
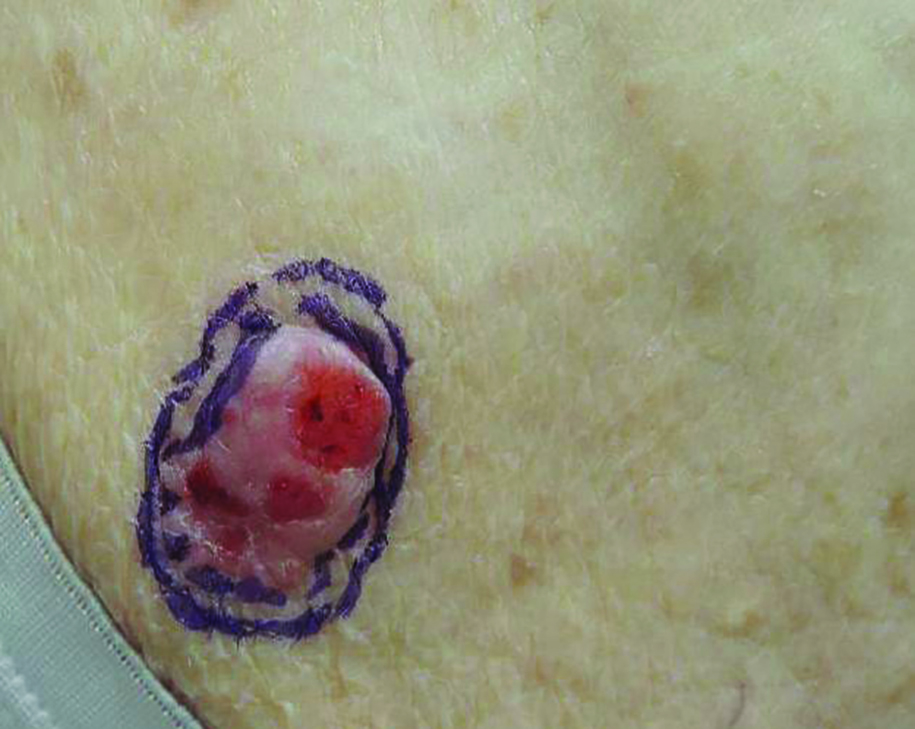
Exophytic Scalp Tumor
The Diagnosis: Primary Cutaneous Carcinosarcoma
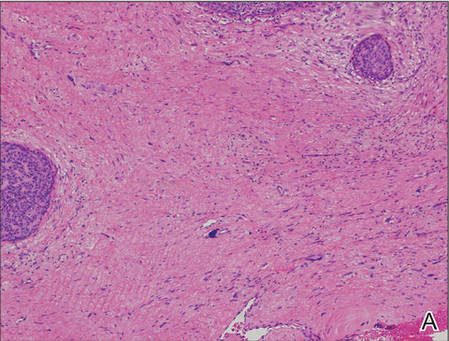
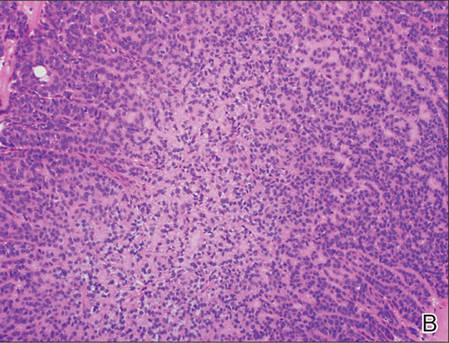
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
The Diagnosis: Primary Cutaneous Carcinosarcoma
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
|
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
The Diagnosis: Primary Cutaneous Carcinosarcoma
A generous shave biopsy and debulking performed on the initial visit revealed an infiltrating tumor consisting of malignant epithelial and stromal components (Figure). The basaloid and squamoid epithelial cells were keratin positive. The stromal cells demonstrated positivity for CD10 but were keratin negative. The epithelial portion of the tumor was composed mostly of basaloid islands of cells with nuclear pleomorphism, scattered mitoses, and focal sebaceous differentiation. The mesenchymal portion of the tumor displayed florid pleomorphism and polymorphism, with many large atypical cells and proliferation. A diagnosis of primary cutaneous carcinosarcoma (PCC) was rendered. Head and neck computed tomography showed tumor penetration of less than 1 cm into scalp soft tissues with no involvement of the underlying bone. There was some evidence of swelling of the supragaleal soft tissues without indication of perineural spread. An 11-mm hyperlucent lower cervical lymph node on the left side that likely represented an incidental finding was noted. Surgical excision with margin evaluation was recommended, but the patient declined. He instead received radiation therapy to the left side of the posterior scalp with a total dose of 30 Gy at 6 Gy per fraction and 1 fraction daily. The patient was found to have a well-healed scar with no evidence of recurrence at 4-week follow-up and again at 5 months after radiation therapy.
|
|
|
|
| A generous shave biopsy and debulking performed on the initial visit revealed an inflitrating tumor consisting on malignant epithelial and stromal components (A-C)(H&E; original magnifications ×10, ×20, and ×40, respectively). |
Primary cutaneous carcinosarcoma is a rare biphasic neoplasm of unknown etiology that is characterized by the presence of both malignant epithelial and mesenchymal components.1 Carcinosarcomas have been reported in both the male and female reproductive tracts, urinary tract, gastrointestinal tract, lungs, breasts, larynx, thymus, and thyroid but is uncommon as a primary neoplasm of the skin.2 Epidermal PCC occurs with greater frequency in males than in females and typically presents in the eighth or ninth decades of life.3 These tumors tend to arise in sun-exposed regions, most commonly on the face and scalp.2
Morphologically, PCCs typically are exophytic growths that often feature surface ulceration and may or may not bleed upon palpation.4 Primary cutaneous carcinosarcomas may present as long-standing lesions that have undergone rapid transformation in the weeks preceding presentation.4 It is not uncommon for PCC lesions to carry the clinical diagnosis of squamous cell carcinoma, which suggests notable morphologic overlap between these entities. Histopathologically, PCC shows a basal cell carcinoma and/or a squamous cell carcinoma epithelial component intimately admixed with a sarcomatous component.5 The mesenchymal component of PCC typically resembles a superficial malignant fibrous histiocytoma characterized by pleomorphic nuclei and cytoplasm, necrosis, and an increased number of mitotic figures.2 Immunohistochemistry can be beneficial in the diagnosis of PCC. A combination of p63 and AE1/AE3 stains can be used to confirm cells of epithelial origin. Staining with vimentin, CD10, or caldesmon can help to delineate the mesenchymal component of PCC.
Epidermal PCC most commonly affects elderly individuals with a history of extensive sun exposure. It has been suggested that p53 mutations due to UV damage are key in tumor formation for both epithelial and mesenchymal elements.5 Literature supports a monoclonal origin for the epithelial and mesenchymal components of this tumor; however, there is insufficient evidence.6 Surgical excision is the primary treatment modality for epidermal PCC, but adjuvant or substitutive radiotherapy has been used in some cases.4 The prognosis of PCC is notably better than its visceral counterpart due to early diagnosis and treatment of easily visible lesions. Epidermal PCC has a 70% 5-year disease-free survival rate, while adnexal PCC tends to occur in younger patients and has a 25% 5-year disease-free survival rate.3 Due to the rarity of reported cases and limited follow-up, the long-term prognosis for PCC remains unclear.
We report an unusual case of PCC on the scalp that was successfully treated with radiation therapy alone. This modality should be considered in patients with large tumors who refuse surgery or are not good surgical candidates.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
1. El Harroudi T, Ech-Charif S, Amrani M, et al. Primary carcinosarcoma of the skin. J Hand Microsurg. 2010;2:79-81.
2. Patel NK, McKee PH, Smith NP. Primary metaplastic carcinoma (carcinosarcoma) of the skin: a clinicopathologic study of four cases and review of the literature. Am J Dermatopathol. 1997;19:363-372.
3. Hong SH, Hong SJ, Lee Y, et al. Primary cutaneous carcinosarcoma of the shoulder: case report with literature review. Dermatol Surg. 2013;39:338-340.
4. Syme-Grant J, Syme-Grant NJ, Motta L, et al. Are primary cutaneous carcinosarcomas underdiagnosed? five cases and a review of the literature. J Plast Reconstr Aesthet Surg. 2006;59:1402-1408.
5. Tran TA, Muller S, Chaudahri PJ, et al. Cutaneous carcinosarcoma: adnexal vs. epidermal types define high- and low-risk tumors. results of a meta-analysis. J Cutan Pathol. 2005;32:2-11.
6. Paniz Mondolfi AE, Jour G, Johnson M, et al. Primary cutaneous carcinosarcoma: insights into its clonal origin and mutational pattern expression analysis through next-generation sequencing. Hum Pathol. 2013;44:2853-2860.
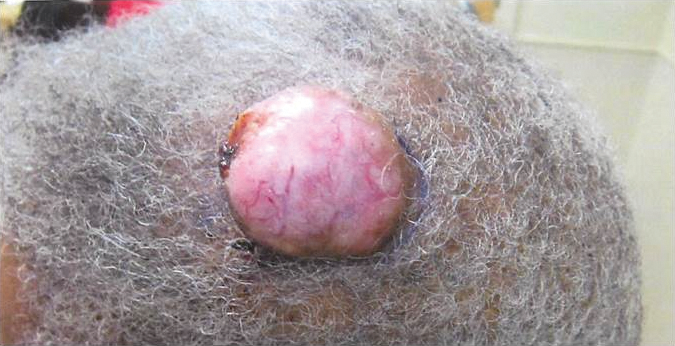
An 81-year-old man presented with a 3.5×3.0-cm pink exophytic tumor with an eroded surface and prominent vascularity on the left side of the parietal scalp. The patient reported that the tumor had been present for more than 30 years but recently had grown larger in size. He denied pain or pruritus in association with the lesion and did not report any systemic symptoms. He had received no prior treatments for the tumor.

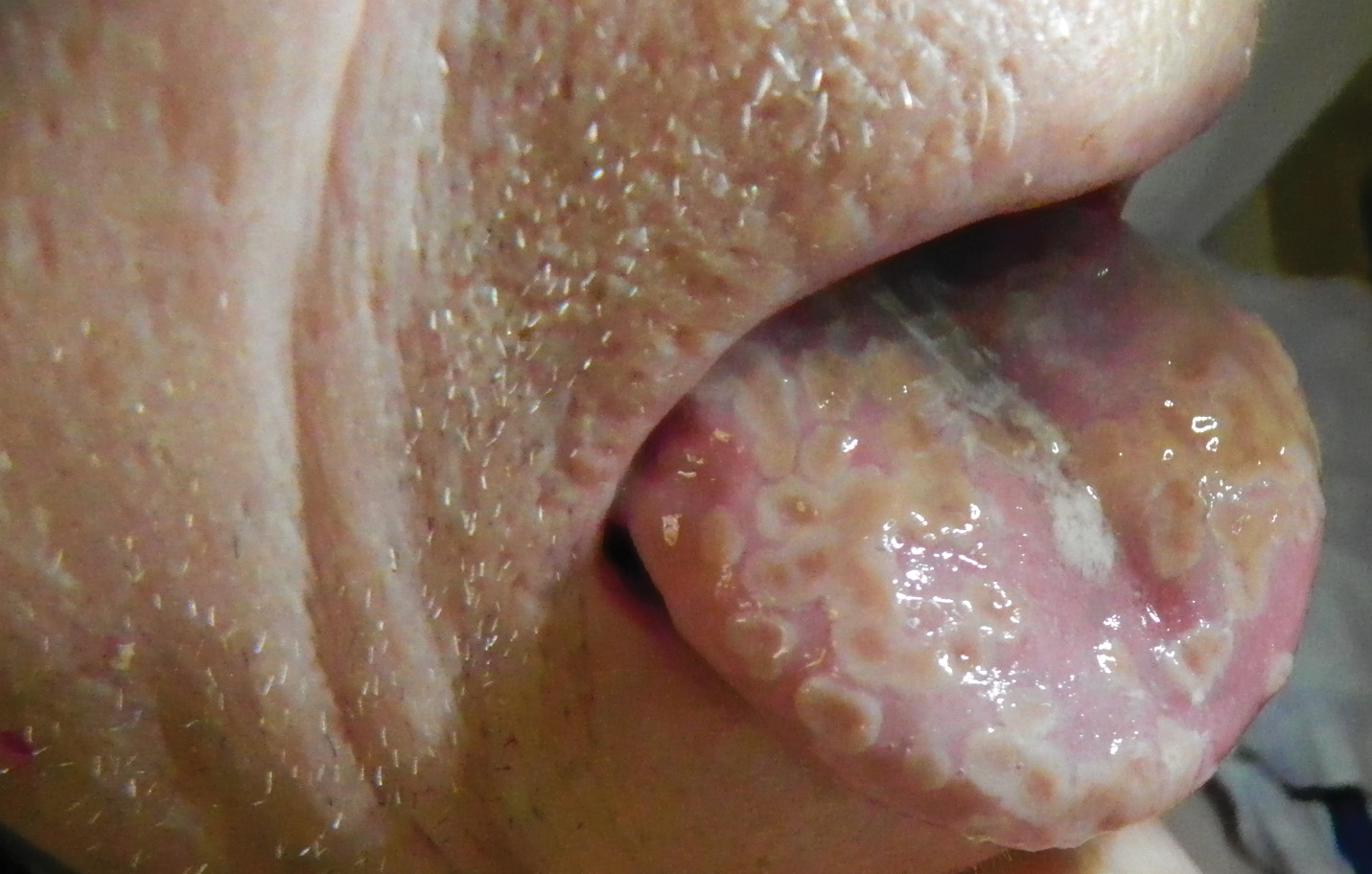
Painful Lesions on the Tongue
The Diagnosis: Herpetic Glossitis
Oral lesions of the tongue are common during primary herpetic gingivostomatitis, though most primary oral herpes simplex virus (HSV) infections occur during childhood or early adulthood. Reactivation of HSV type 1 most commonly manifests as herpes labialis.1 When recurrent HSV involves intraoral lesions, they are typically confined to the gingiva and palate, sparing the tongue.
Clinical presentation of herpetic glossitis varies. Recurrent herpetic glossitis has been described in immunocompromised patients, particularly those with hematologic malignancies and organ transplants.2 In addition, immunocompromised and human immunodeficiency virus–infected patients may present with deep and/or broad ulcers. A case of herpes infection presenting with nodules on the tongue has been reported in Hodgkin disease.3 Herpetic geometric glossitis also has been described, which is a linear, crosshatched, or sharply angled branching with painful fissuring of the tongue. Herpetic geometric glossitis has been reported to occur in both immunocompetent and immunocompromised individuals.4 Tongue involvement during oral reactivation of HSV is exceedingly rare and the pathogenesis remains elusive, though one hypothesis proposes a protective role of salivary-specific IgA and lysozyme.5 Here, we report a case in which a patient developed similar lingual HSV lesions following recent immunosuppression.
1. Arduino PG, Porter SR. Herpes simplex virus type 1 infection: overview on relevant clinico-pathological features. J Oral Pathol Med. 2008;37:107-121.
2. Nikkels AF, Piérard GE. Chronic herpes simplex virus type I glossitis in an immunocompromised man. Br J Dermatol. 1999;140:343-346.
3. Leming PD, Martin SE, Zwelling LA. Atypical herpes simplex (HSV) infection in a patient with Hodgkin’s disease. Cancer. 1984;54:3043-3047.
4. Mirowski GW, Goddard A. Herpetic geometric glossitis in an immunocompetent patient with pneumonia. J Am Acad Dermatol. 2009;61:139-142.
5. Heineman HS, Greenberg MS. Cell protective effect of human saliva specific for herpes simplex virus. Arch Oral Biol. 1980;25:257-261.
The Diagnosis: Herpetic Glossitis
Oral lesions of the tongue are common during primary herpetic gingivostomatitis, though most primary oral herpes simplex virus (HSV) infections occur during childhood or early adulthood. Reactivation of HSV type 1 most commonly manifests as herpes labialis.1 When recurrent HSV involves intraoral lesions, they are typically confined to the gingiva and palate, sparing the tongue.
Clinical presentation of herpetic glossitis varies. Recurrent herpetic glossitis has been described in immunocompromised patients, particularly those with hematologic malignancies and organ transplants.2 In addition, immunocompromised and human immunodeficiency virus–infected patients may present with deep and/or broad ulcers. A case of herpes infection presenting with nodules on the tongue has been reported in Hodgkin disease.3 Herpetic geometric glossitis also has been described, which is a linear, crosshatched, or sharply angled branching with painful fissuring of the tongue. Herpetic geometric glossitis has been reported to occur in both immunocompetent and immunocompromised individuals.4 Tongue involvement during oral reactivation of HSV is exceedingly rare and the pathogenesis remains elusive, though one hypothesis proposes a protective role of salivary-specific IgA and lysozyme.5 Here, we report a case in which a patient developed similar lingual HSV lesions following recent immunosuppression.
The Diagnosis: Herpetic Glossitis
Oral lesions of the tongue are common during primary herpetic gingivostomatitis, though most primary oral herpes simplex virus (HSV) infections occur during childhood or early adulthood. Reactivation of HSV type 1 most commonly manifests as herpes labialis.1 When recurrent HSV involves intraoral lesions, they are typically confined to the gingiva and palate, sparing the tongue.
Clinical presentation of herpetic glossitis varies. Recurrent herpetic glossitis has been described in immunocompromised patients, particularly those with hematologic malignancies and organ transplants.2 In addition, immunocompromised and human immunodeficiency virus–infected patients may present with deep and/or broad ulcers. A case of herpes infection presenting with nodules on the tongue has been reported in Hodgkin disease.3 Herpetic geometric glossitis also has been described, which is a linear, crosshatched, or sharply angled branching with painful fissuring of the tongue. Herpetic geometric glossitis has been reported to occur in both immunocompetent and immunocompromised individuals.4 Tongue involvement during oral reactivation of HSV is exceedingly rare and the pathogenesis remains elusive, though one hypothesis proposes a protective role of salivary-specific IgA and lysozyme.5 Here, we report a case in which a patient developed similar lingual HSV lesions following recent immunosuppression.
1. Arduino PG, Porter SR. Herpes simplex virus type 1 infection: overview on relevant clinico-pathological features. J Oral Pathol Med. 2008;37:107-121.
2. Nikkels AF, Piérard GE. Chronic herpes simplex virus type I glossitis in an immunocompromised man. Br J Dermatol. 1999;140:343-346.
3. Leming PD, Martin SE, Zwelling LA. Atypical herpes simplex (HSV) infection in a patient with Hodgkin’s disease. Cancer. 1984;54:3043-3047.
4. Mirowski GW, Goddard A. Herpetic geometric glossitis in an immunocompetent patient with pneumonia. J Am Acad Dermatol. 2009;61:139-142.
5. Heineman HS, Greenberg MS. Cell protective effect of human saliva specific for herpes simplex virus. Arch Oral Biol. 1980;25:257-261.
1. Arduino PG, Porter SR. Herpes simplex virus type 1 infection: overview on relevant clinico-pathological features. J Oral Pathol Med. 2008;37:107-121.
2. Nikkels AF, Piérard GE. Chronic herpes simplex virus type I glossitis in an immunocompromised man. Br J Dermatol. 1999;140:343-346.
3. Leming PD, Martin SE, Zwelling LA. Atypical herpes simplex (HSV) infection in a patient with Hodgkin’s disease. Cancer. 1984;54:3043-3047.
4. Mirowski GW, Goddard A. Herpetic geometric glossitis in an immunocompetent patient with pneumonia. J Am Acad Dermatol. 2009;61:139-142.
5. Heineman HS, Greenberg MS. Cell protective effect of human saliva specific for herpes simplex virus. Arch Oral Biol. 1980;25:257-261.

A 77-year-old man with a history of chronic obstructive pulmonary disease and recent pneumonia was treated with oral prednisone 40 mg daily, antibiotics, and a fluticasone-salmeterol inhaler. One week into treatment, the patient developed painful lesions limited to the oral cavity. Physical examination revealed many fixed, umbilicated, white-tan plaques on the lower lips, tongue, and posterior aspect of the oropharynx. The dermatology department was consulted because the lesions failed to respond to nystatin oral suspension.
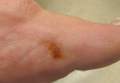
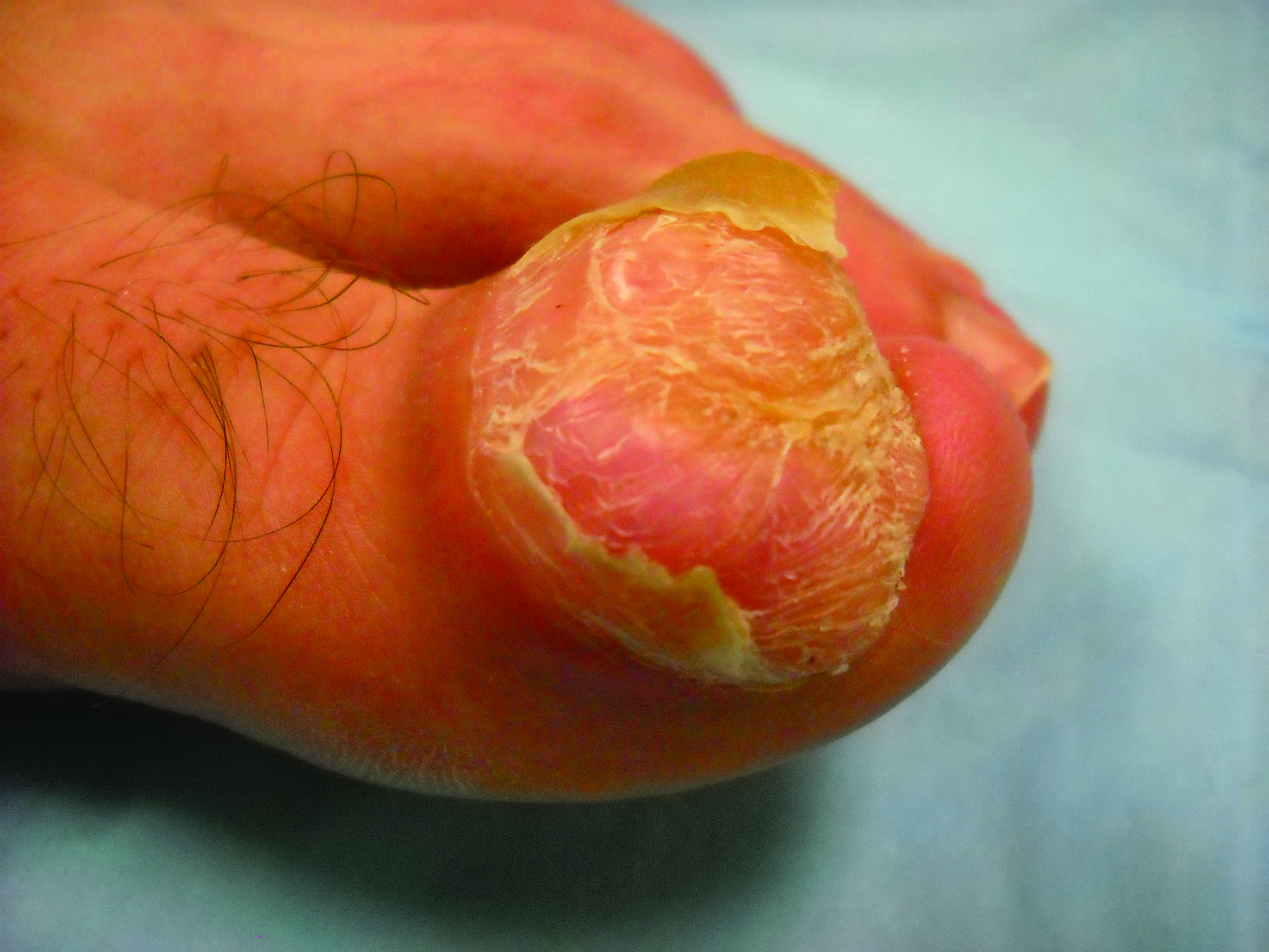
Toe Nodule Obliterating the Nail Bed
The Diagnosis: Superficial Acral Fibromyxoma
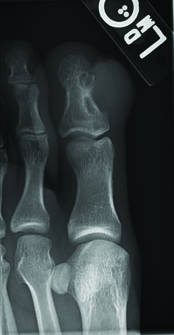
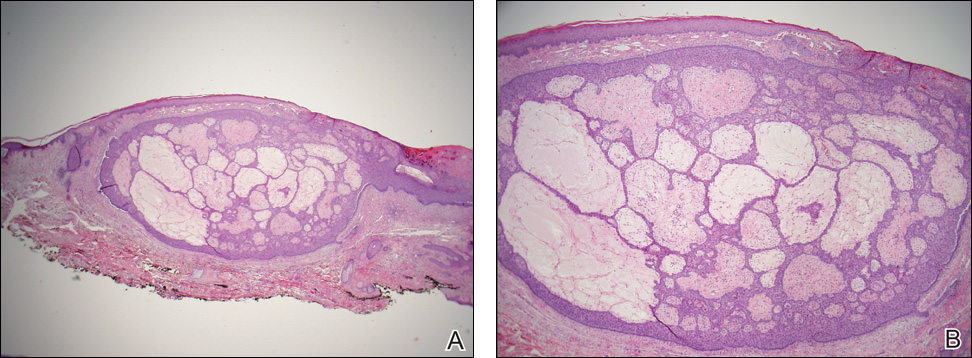
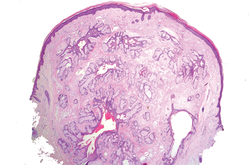
Superficial acral fibromyxoma (SAF) was first described in 2001 by Fetsch et al.1 Subsequently, the term digital fibromyxoma was proposed in 2012 by Hollmann et al2 to describe a distinctive, slow-growing, soft-tissue tumor with a predilection for the periungual or subungual regions of the fingers and toes. The benign growth typically presents as a painless or tender nodule in middle-aged adults with a slight male predominance (1.3:1 ratio).1,2 In a case series (N=124) described by Hollmann et al,2 9 of 25 patients (36%) who had imaging studies showed bone involvement by an erosive or lytic lesion. Reports of SAF with bone involvement also have been described in the radiologic and orthopedic surgery literature.3,4 Radiographically, the soft-tissue invasion of the bone is demonstrated by scalloping on plain radiographs (Figure 1).3
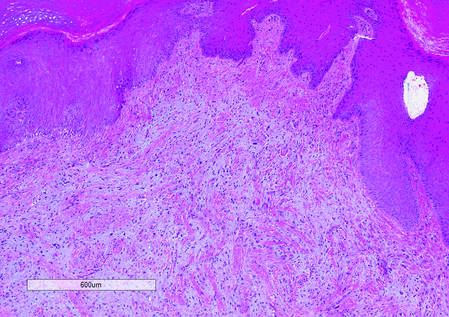
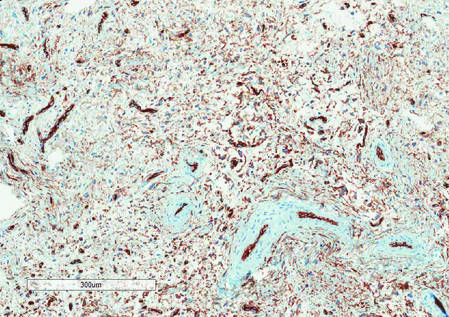
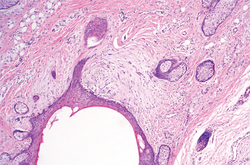
Histologically, SAFs are moderately cellular with spindled or stellate fibroblastlike cells within a myxoid or collagenous matrix (Figure 2).1 The vasculature is mildly accentuated and an increase in mast cells usually is observed. The nuclei have a low degree of atypia with few mitotic figures, and the stellate cells exhibit positive immunohistochemical staining for CD34 (Figure 3), epithelial membrane antigen, and CD99.1 Hollmann et al2 found that 66 of 95 tumors (69.5%) infiltrated the dermal collagen, 26 (27.4%) infiltrated fat, and 3 (3.2%) invaded bone. Of the 47 cases that were evaluated on follow-up, 10 tumors (21.3%) recurred locally (all near the nail unit of the fingers or toes) after a mean interval of 27 months. Although invasion of underlying tissues and recurrence of the tumor has been demonstrated, this growth is considered benign. The histologic differential diagnosis includes neurofibroma, myxoma, fibroma, low-grade fibromyxoid sarcoma, dermatofibroma, superficial angiomyxoma, and dermatofibrosarcoma protuberans.2
The primary treatment of SAF is local excision. The incidence of local recurrence found in the case series by Hollmann et al2 was directly linked to positive margins after the first excision (10/47 [21.3%] recurrent lesions had positive margins). To date, there are no known reports of metastatic disease in SAF.2 Our case manifested with a late recurrence of the tumor and bone involvement requiring surgical excision, which illustrates the role of adjuvant imaging and close follow-up following excision of any soft-tissue tumors of the fingers and toes that have been histologically confirmed as SAF, particularly those of the periungual region.
|
|
|
|
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma (a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes.) Hum Pathol. 2001;32:704-714.
2. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
3. Varikatt W, Soper J, Simmon G, et al. Superficial acral fibromyxoma: a report of two cases with radiological findings. Skeletal Radiol. 2008;37:499-503.
4. Oteo-Alvaro A, Meizoso T, Scarpellini A, et al. Superficial acral fibromyxoma of the toe, with erosion of the distal phalanx. a clinical report. Arch Orthop Trauma Surg. 2008;128:271-274.
The Diagnosis: Superficial Acral Fibromyxoma
Superficial acral fibromyxoma (SAF) was first described in 2001 by Fetsch et al.1 Subsequently, the term digital fibromyxoma was proposed in 2012 by Hollmann et al2 to describe a distinctive, slow-growing, soft-tissue tumor with a predilection for the periungual or subungual regions of the fingers and toes. The benign growth typically presents as a painless or tender nodule in middle-aged adults with a slight male predominance (1.3:1 ratio).1,2 In a case series (N=124) described by Hollmann et al,2 9 of 25 patients (36%) who had imaging studies showed bone involvement by an erosive or lytic lesion. Reports of SAF with bone involvement also have been described in the radiologic and orthopedic surgery literature.3,4 Radiographically, the soft-tissue invasion of the bone is demonstrated by scalloping on plain radiographs (Figure 1).3
Histologically, SAFs are moderately cellular with spindled or stellate fibroblastlike cells within a myxoid or collagenous matrix (Figure 2).1 The vasculature is mildly accentuated and an increase in mast cells usually is observed. The nuclei have a low degree of atypia with few mitotic figures, and the stellate cells exhibit positive immunohistochemical staining for CD34 (Figure 3), epithelial membrane antigen, and CD99.1 Hollmann et al2 found that 66 of 95 tumors (69.5%) infiltrated the dermal collagen, 26 (27.4%) infiltrated fat, and 3 (3.2%) invaded bone. Of the 47 cases that were evaluated on follow-up, 10 tumors (21.3%) recurred locally (all near the nail unit of the fingers or toes) after a mean interval of 27 months. Although invasion of underlying tissues and recurrence of the tumor has been demonstrated, this growth is considered benign. The histologic differential diagnosis includes neurofibroma, myxoma, fibroma, low-grade fibromyxoid sarcoma, dermatofibroma, superficial angiomyxoma, and dermatofibrosarcoma protuberans.2
The primary treatment of SAF is local excision. The incidence of local recurrence found in the case series by Hollmann et al2 was directly linked to positive margins after the first excision (10/47 [21.3%] recurrent lesions had positive margins). To date, there are no known reports of metastatic disease in SAF.2 Our case manifested with a late recurrence of the tumor and bone involvement requiring surgical excision, which illustrates the role of adjuvant imaging and close follow-up following excision of any soft-tissue tumors of the fingers and toes that have been histologically confirmed as SAF, particularly those of the periungual region.
|
|
|
|
|
|
The Diagnosis: Superficial Acral Fibromyxoma
Superficial acral fibromyxoma (SAF) was first described in 2001 by Fetsch et al.1 Subsequently, the term digital fibromyxoma was proposed in 2012 by Hollmann et al2 to describe a distinctive, slow-growing, soft-tissue tumor with a predilection for the periungual or subungual regions of the fingers and toes. The benign growth typically presents as a painless or tender nodule in middle-aged adults with a slight male predominance (1.3:1 ratio).1,2 In a case series (N=124) described by Hollmann et al,2 9 of 25 patients (36%) who had imaging studies showed bone involvement by an erosive or lytic lesion. Reports of SAF with bone involvement also have been described in the radiologic and orthopedic surgery literature.3,4 Radiographically, the soft-tissue invasion of the bone is demonstrated by scalloping on plain radiographs (Figure 1).3
Histologically, SAFs are moderately cellular with spindled or stellate fibroblastlike cells within a myxoid or collagenous matrix (Figure 2).1 The vasculature is mildly accentuated and an increase in mast cells usually is observed. The nuclei have a low degree of atypia with few mitotic figures, and the stellate cells exhibit positive immunohistochemical staining for CD34 (Figure 3), epithelial membrane antigen, and CD99.1 Hollmann et al2 found that 66 of 95 tumors (69.5%) infiltrated the dermal collagen, 26 (27.4%) infiltrated fat, and 3 (3.2%) invaded bone. Of the 47 cases that were evaluated on follow-up, 10 tumors (21.3%) recurred locally (all near the nail unit of the fingers or toes) after a mean interval of 27 months. Although invasion of underlying tissues and recurrence of the tumor has been demonstrated, this growth is considered benign. The histologic differential diagnosis includes neurofibroma, myxoma, fibroma, low-grade fibromyxoid sarcoma, dermatofibroma, superficial angiomyxoma, and dermatofibrosarcoma protuberans.2
The primary treatment of SAF is local excision. The incidence of local recurrence found in the case series by Hollmann et al2 was directly linked to positive margins after the first excision (10/47 [21.3%] recurrent lesions had positive margins). To date, there are no known reports of metastatic disease in SAF.2 Our case manifested with a late recurrence of the tumor and bone involvement requiring surgical excision, which illustrates the role of adjuvant imaging and close follow-up following excision of any soft-tissue tumors of the fingers and toes that have been histologically confirmed as SAF, particularly those of the periungual region.
|
|
|
|
|
|
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma (a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes.) Hum Pathol. 2001;32:704-714.
2. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
3. Varikatt W, Soper J, Simmon G, et al. Superficial acral fibromyxoma: a report of two cases with radiological findings. Skeletal Radiol. 2008;37:499-503.
4. Oteo-Alvaro A, Meizoso T, Scarpellini A, et al. Superficial acral fibromyxoma of the toe, with erosion of the distal phalanx. a clinical report. Arch Orthop Trauma Surg. 2008;128:271-274.
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma (a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes.) Hum Pathol. 2001;32:704-714.
2. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
3. Varikatt W, Soper J, Simmon G, et al. Superficial acral fibromyxoma: a report of two cases with radiological findings. Skeletal Radiol. 2008;37:499-503.
4. Oteo-Alvaro A, Meizoso T, Scarpellini A, et al. Superficial acral fibromyxoma of the toe, with erosion of the distal phalanx. a clinical report. Arch Orthop Trauma Surg. 2008;128:271-274.
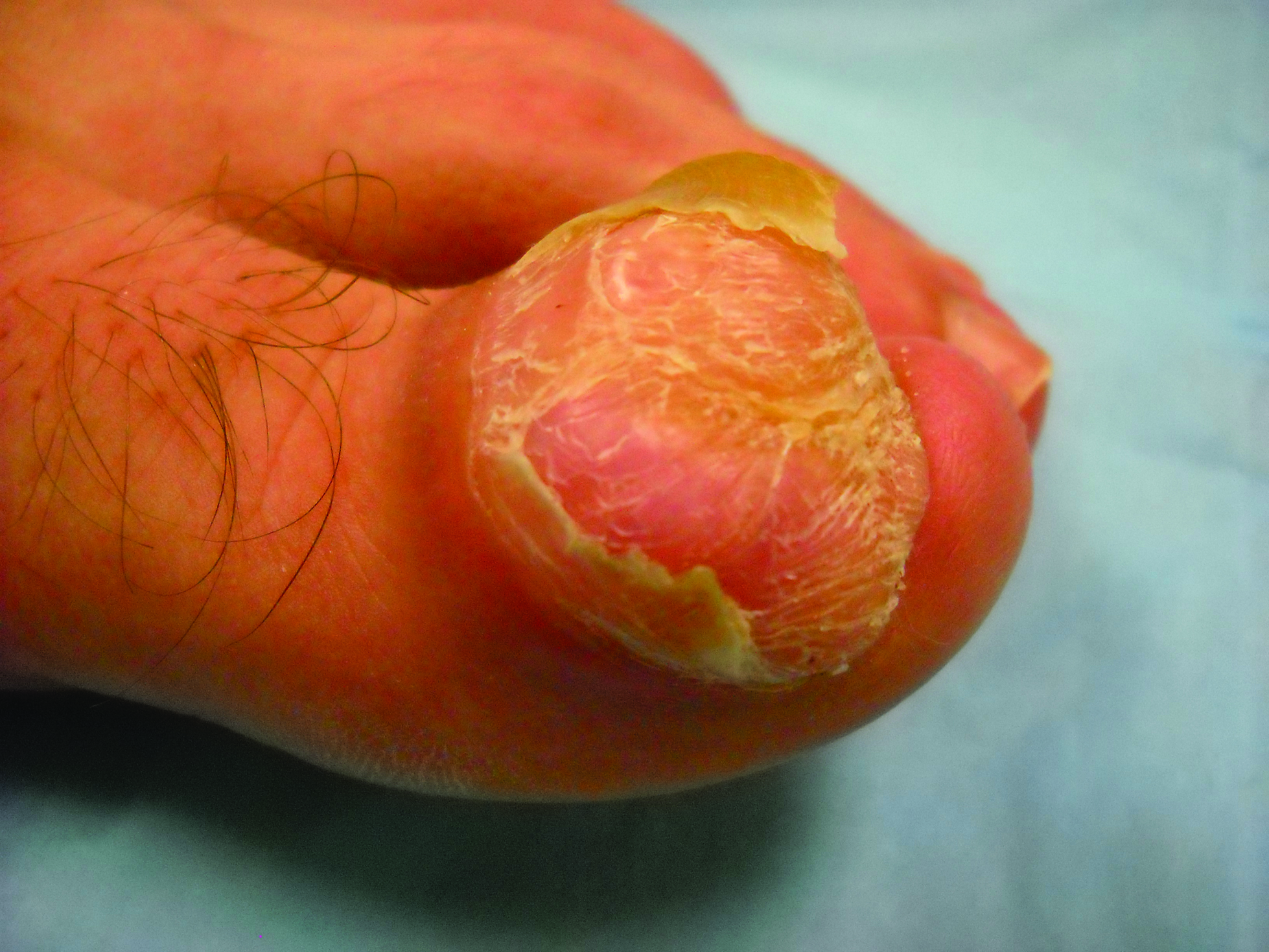
A generally healthy 30-year-old man presented with a 3-cm exophytic, yellowish red, subungual nodule of the left great toe of 1 year’s duration that was obliterating the nail plate. Ten years prior, a similar nodule in the same location was removed via laser by a podiatrist. Medical records were not retrievable, but the patient reported that he was told the excised lesion was a benign tumor. Plain radiographs were performed at the current presentation and demonstrated an inferior cortical lucency of the distal phalanx as well as a lucency over the nail bed region with extension of calcification to the soft tissues. Magnetic resonance imaging showed a mass with a proximal to distal maximum dimension of 2.1 cm that involved the dorsal surface of the proximal phalanx. Magnetic resonance imaging also demonstrated bone erosion from the overlying mass. A 4-mm incisional punch biopsy was performed prior to surgical excision.
Dome-Shaped Papule With a Bloody Crust
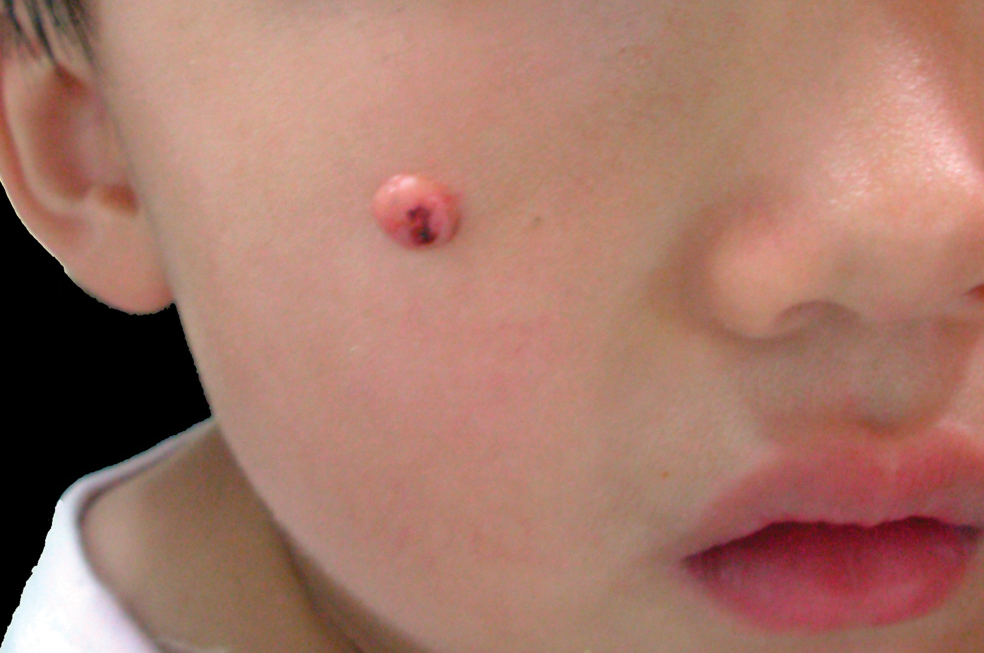
The Diagnosis: Congenital Folliculosebaceous Cystic Hamartoma
Folliculosebaceous cystic hamartoma (FSCH) is a rare skin condition that is either congenital or acquired. It presents as a slow-growing and flesh-colored papulonodular lesion1 that mainly occurs on the head and neck. Involvement of the nipples, perineum, back, forearms, genital areas, and subcutaneous tissue also has been reported but usually indicates a larger lesion.1,2
Histologically, FSCH is considered a hamartoma composed of both ectodermal and mesodermal elements.1 Folliculosebaceous cystic hamartoma is a more complex lesion composed of infundibulocystic structures connected to maloriented folliculosebaceous units surrounded by whorls of highly vascularized fibrous stroma and adipocytes. Clefts between fibroepithelial units and surrounding stroma usually are present.1
Epithelial components contribute to the adnexal and folliculosebaceous cystic proliferations, and mesenchymal elements include vascular tissue, adipose tissue, and fibroblast-rich stroma.1,2 Acquired lesions arising in adults have been described,1-5 but the congenital presentation of FSCH in infancy is rare.
Histopathologically, some variations of FSCH are mainly composed of epithelial components while others are composed of nonepithelial components. Nonepithelial components include neural proliferation, muscle components, vascular proliferation, and mucin deposition.1-4 In some cases, FSCH may coexist with other diseases, such as nevus lipomatosus cutaneous superficialis and neurofibromatosis type I.4,5
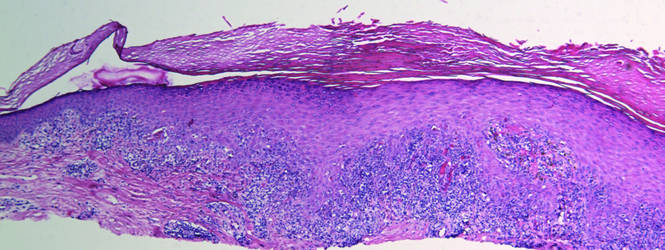
In our case, histopathology showed several dermal infundibulocystic structures that were lined by stratified squamous epithelium and contained horny material (Figure 1). Numerous immature sebaceous lobules and rudimentary hair follicles emanated from some of the cyst walls. Mesenchymal changes around the fibroepithelial units included fibrillary bundles of collagen, clusters of adipocytes, and an increased number of small venules (Figure 2). In addition, the stroma adjacent to the malformed perifollicle contained some amount of mucin. Prominent clefts formed between fibroepithelial units and the surrounding altered stroma.
| 
| |
|
The differential diagnosis mainly includes sebaceous trichofolliculoma, molluscum contagiosum, dermoid cysts, pilomatrixoma, Spitz nevus, and nevus lipomatosus superficialis. The differential diagnosis between FSCH and sebaceous trichofolliculoma is challenging. Both lesions show an infundibular cyst and surrounding sebaceous nodules. According to Plewig,6 trichofolliculoma has a wide spectrum ranging from low to high differentiation represented by trichofolliculoma, sebaceous trichofolliculoma, and FSCH, respectively. It is not difficult to distinguish FSCH from other diseases according to its peculiar histopathologic features.
The clinicopathologic features of our case were similar to those of reported FSCH cases, except for the following unique characteristics: congenital lesion, lack of terminal hair, and no sebaceous material extrusion. These features of hair and sebaceous material may be correlated with the patient’s age and hormonal level.1 Androgen may play a key role in sebaceous gland development at puberty, which leads to sebaceous gland hyperplasia and hypertrophy. Therefore, slight pressure from the lesions can make ivory-white sebaceous material discharge. Hence, the dermatologist and pediatrician must be poised and sensitive while making an initial diagnosis of FSCH.
1. Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma: a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
2. Moriki M, Ito T, Hirakawa S, et al. Folliculosebaceous cystic hamartoma presenting as a subcutaneous nodule on the thigh. J Dermatol. 2013;40:483-484.
3. Aloi F, Tomasini C, Pippione M. Folliculosebaceous cystic hamartoma with perifollicular mucinosis. Am J Dermatopathol. 1996;18:58-62.
4. Brasanac D, Boricic I. Giant nevus lipomatosus superficialis with multiple folliculosebaceous cystic hamartomas and dermoid cysts. J Eur Acad Dermatol Venereol. 2005;19:84-86.
5. Noh S, Kwon JE, Lee KG, et al. Folliculosebaceous cystic hamartoma in a patient with neurofibromatosis type I. Ann Dermatol. 2011;23(suppl 2):S185-S187.
6. Plewig G. In discussion of: Leserbrief zu Zheng LQ, Han XC, Huang Y, Li HW. Several acneiform papules and nodules on the neck. diagnosis: folliculosebaceous cystic hamartoma. J Dtsch Dermatol Ges. 2014;12:824-825.
The Diagnosis: Congenital Folliculosebaceous Cystic Hamartoma
Folliculosebaceous cystic hamartoma (FSCH) is a rare skin condition that is either congenital or acquired. It presents as a slow-growing and flesh-colored papulonodular lesion1 that mainly occurs on the head and neck. Involvement of the nipples, perineum, back, forearms, genital areas, and subcutaneous tissue also has been reported but usually indicates a larger lesion.1,2
Histologically, FSCH is considered a hamartoma composed of both ectodermal and mesodermal elements.1 Folliculosebaceous cystic hamartoma is a more complex lesion composed of infundibulocystic structures connected to maloriented folliculosebaceous units surrounded by whorls of highly vascularized fibrous stroma and adipocytes. Clefts between fibroepithelial units and surrounding stroma usually are present.1
Epithelial components contribute to the adnexal and folliculosebaceous cystic proliferations, and mesenchymal elements include vascular tissue, adipose tissue, and fibroblast-rich stroma.1,2 Acquired lesions arising in adults have been described,1-5 but the congenital presentation of FSCH in infancy is rare.
Histopathologically, some variations of FSCH are mainly composed of epithelial components while others are composed of nonepithelial components. Nonepithelial components include neural proliferation, muscle components, vascular proliferation, and mucin deposition.1-4 In some cases, FSCH may coexist with other diseases, such as nevus lipomatosus cutaneous superficialis and neurofibromatosis type I.4,5
In our case, histopathology showed several dermal infundibulocystic structures that were lined by stratified squamous epithelium and contained horny material (Figure 1). Numerous immature sebaceous lobules and rudimentary hair follicles emanated from some of the cyst walls. Mesenchymal changes around the fibroepithelial units included fibrillary bundles of collagen, clusters of adipocytes, and an increased number of small venules (Figure 2). In addition, the stroma adjacent to the malformed perifollicle contained some amount of mucin. Prominent clefts formed between fibroepithelial units and the surrounding altered stroma.
|
|
| |
|
The differential diagnosis mainly includes sebaceous trichofolliculoma, molluscum contagiosum, dermoid cysts, pilomatrixoma, Spitz nevus, and nevus lipomatosus superficialis. The differential diagnosis between FSCH and sebaceous trichofolliculoma is challenging. Both lesions show an infundibular cyst and surrounding sebaceous nodules. According to Plewig,6 trichofolliculoma has a wide spectrum ranging from low to high differentiation represented by trichofolliculoma, sebaceous trichofolliculoma, and FSCH, respectively. It is not difficult to distinguish FSCH from other diseases according to its peculiar histopathologic features.
The clinicopathologic features of our case were similar to those of reported FSCH cases, except for the following unique characteristics: congenital lesion, lack of terminal hair, and no sebaceous material extrusion. These features of hair and sebaceous material may be correlated with the patient’s age and hormonal level.1 Androgen may play a key role in sebaceous gland development at puberty, which leads to sebaceous gland hyperplasia and hypertrophy. Therefore, slight pressure from the lesions can make ivory-white sebaceous material discharge. Hence, the dermatologist and pediatrician must be poised and sensitive while making an initial diagnosis of FSCH.
The Diagnosis: Congenital Folliculosebaceous Cystic Hamartoma
Folliculosebaceous cystic hamartoma (FSCH) is a rare skin condition that is either congenital or acquired. It presents as a slow-growing and flesh-colored papulonodular lesion1 that mainly occurs on the head and neck. Involvement of the nipples, perineum, back, forearms, genital areas, and subcutaneous tissue also has been reported but usually indicates a larger lesion.1,2
Histologically, FSCH is considered a hamartoma composed of both ectodermal and mesodermal elements.1 Folliculosebaceous cystic hamartoma is a more complex lesion composed of infundibulocystic structures connected to maloriented folliculosebaceous units surrounded by whorls of highly vascularized fibrous stroma and adipocytes. Clefts between fibroepithelial units and surrounding stroma usually are present.1
Epithelial components contribute to the adnexal and folliculosebaceous cystic proliferations, and mesenchymal elements include vascular tissue, adipose tissue, and fibroblast-rich stroma.1,2 Acquired lesions arising in adults have been described,1-5 but the congenital presentation of FSCH in infancy is rare.
Histopathologically, some variations of FSCH are mainly composed of epithelial components while others are composed of nonepithelial components. Nonepithelial components include neural proliferation, muscle components, vascular proliferation, and mucin deposition.1-4 In some cases, FSCH may coexist with other diseases, such as nevus lipomatosus cutaneous superficialis and neurofibromatosis type I.4,5
In our case, histopathology showed several dermal infundibulocystic structures that were lined by stratified squamous epithelium and contained horny material (Figure 1). Numerous immature sebaceous lobules and rudimentary hair follicles emanated from some of the cyst walls. Mesenchymal changes around the fibroepithelial units included fibrillary bundles of collagen, clusters of adipocytes, and an increased number of small venules (Figure 2). In addition, the stroma adjacent to the malformed perifollicle contained some amount of mucin. Prominent clefts formed between fibroepithelial units and the surrounding altered stroma.
|
|
| |
|
The differential diagnosis mainly includes sebaceous trichofolliculoma, molluscum contagiosum, dermoid cysts, pilomatrixoma, Spitz nevus, and nevus lipomatosus superficialis. The differential diagnosis between FSCH and sebaceous trichofolliculoma is challenging. Both lesions show an infundibular cyst and surrounding sebaceous nodules. According to Plewig,6 trichofolliculoma has a wide spectrum ranging from low to high differentiation represented by trichofolliculoma, sebaceous trichofolliculoma, and FSCH, respectively. It is not difficult to distinguish FSCH from other diseases according to its peculiar histopathologic features.
The clinicopathologic features of our case were similar to those of reported FSCH cases, except for the following unique characteristics: congenital lesion, lack of terminal hair, and no sebaceous material extrusion. These features of hair and sebaceous material may be correlated with the patient’s age and hormonal level.1 Androgen may play a key role in sebaceous gland development at puberty, which leads to sebaceous gland hyperplasia and hypertrophy. Therefore, slight pressure from the lesions can make ivory-white sebaceous material discharge. Hence, the dermatologist and pediatrician must be poised and sensitive while making an initial diagnosis of FSCH.
1. Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma: a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
2. Moriki M, Ito T, Hirakawa S, et al. Folliculosebaceous cystic hamartoma presenting as a subcutaneous nodule on the thigh. J Dermatol. 2013;40:483-484.
3. Aloi F, Tomasini C, Pippione M. Folliculosebaceous cystic hamartoma with perifollicular mucinosis. Am J Dermatopathol. 1996;18:58-62.
4. Brasanac D, Boricic I. Giant nevus lipomatosus superficialis with multiple folliculosebaceous cystic hamartomas and dermoid cysts. J Eur Acad Dermatol Venereol. 2005;19:84-86.
5. Noh S, Kwon JE, Lee KG, et al. Folliculosebaceous cystic hamartoma in a patient with neurofibromatosis type I. Ann Dermatol. 2011;23(suppl 2):S185-S187.
6. Plewig G. In discussion of: Leserbrief zu Zheng LQ, Han XC, Huang Y, Li HW. Several acneiform papules and nodules on the neck. diagnosis: folliculosebaceous cystic hamartoma. J Dtsch Dermatol Ges. 2014;12:824-825.
1. Kimura T, Miyazawa H, Aoyagi T, et al. Folliculosebaceous cystic hamartoma: a distinctive malformation of the skin. Am J Dermatopathol. 1991;13:213-220.
2. Moriki M, Ito T, Hirakawa S, et al. Folliculosebaceous cystic hamartoma presenting as a subcutaneous nodule on the thigh. J Dermatol. 2013;40:483-484.
3. Aloi F, Tomasini C, Pippione M. Folliculosebaceous cystic hamartoma with perifollicular mucinosis. Am J Dermatopathol. 1996;18:58-62.
4. Brasanac D, Boricic I. Giant nevus lipomatosus superficialis with multiple folliculosebaceous cystic hamartomas and dermoid cysts. J Eur Acad Dermatol Venereol. 2005;19:84-86.
5. Noh S, Kwon JE, Lee KG, et al. Folliculosebaceous cystic hamartoma in a patient with neurofibromatosis type I. Ann Dermatol. 2011;23(suppl 2):S185-S187.
6. Plewig G. In discussion of: Leserbrief zu Zheng LQ, Han XC, Huang Y, Li HW. Several acneiform papules and nodules on the neck. diagnosis: folliculosebaceous cystic hamartoma. J Dtsch Dermatol Ges. 2014;12:824-825.

A 3-year-old girl was referred to our clinic for a lesion on the face that had been present since birth and had enlarged slowly with slight itching. Physical examination revealed a 1.0×1.0-cm, sessile, flesh-colored, sharply demarcated, and dome-shaped papule with a bloody crust. It was firm and slightly painful to palpation. Dilated hair follicle–like orifices and thick central terminal hair were not found. Sebaceous material was not discharged. There was no notable family history or evidence of systemic disease. The lesion was surgically removed for cosmetic reasons and further histopathologic examination was performed.
Confluent Erythematous Plaques on the Palm
The Diagnosis: Palmoplantar Lichen Planus
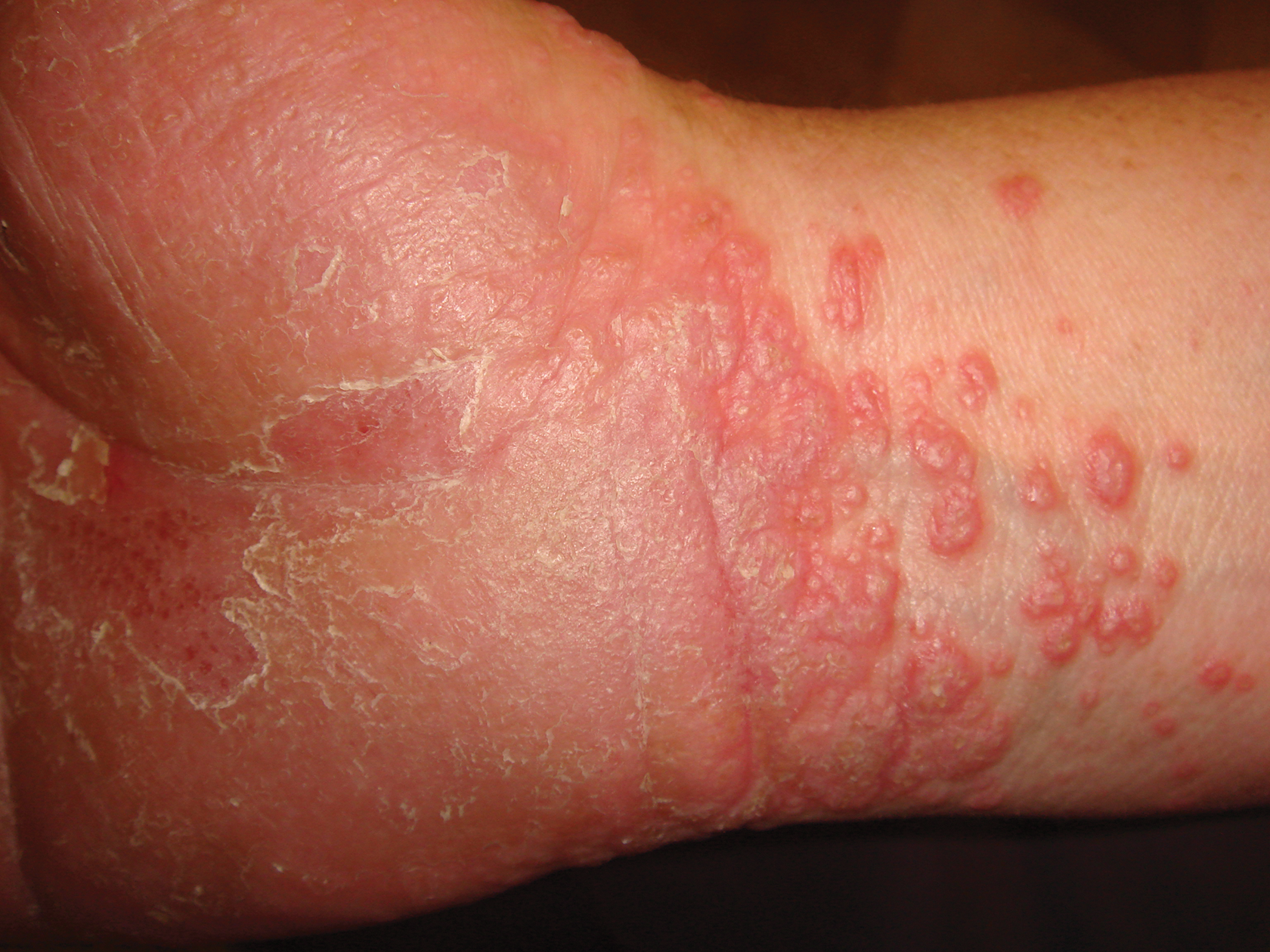
A skin biopsy from a lesion on the inner wrist showed an interface pattern with a dense bandlike infiltrate obscuring the dermoepidermal junction coupled with a superficial perivascular infiltrate (Figure). At higher magnification (×10), the histologic features included compact orthokeratosis, wedge-shaped hypergranulosis, vacuolar degeneration of the basal layer, basal dyskeratosis, a dense lymphohistiocytic infiltrate obscuring the basement membrane, and melanophages in the papillary dermis.
Lichen planus (LP) is a common inflammatory disease of the skin presenting with flat-topped, violaceous, polygonal papules with fine white lines (Wickham striae) on the surface. It is recognized and diagnosed clinically by its characteristic appearance. Common areas of LP presentation include the shins, inner thighs, genitalia, trunk, volar aspect of the wrists, and oral mucosa.
Palmoplantar LP can present as erythematous plaques, punctuate keratosis, diffuse keratoderma, or ulcerated lesions. The most common concern among patients with LP is pruritus. One-fourth of patients with LP may present with lesions on the palms and soles, but diffuse palmoplantar hyperkeratosis is rare.1 Lesions typically heal in 1 to 8 months, with an average of 3 months. Palmoplantar LP recurs within 1 year after stopping treatment in one-third of patients.1
The cause of LP is unknown, but the pathophysiology is beginning to be understood. Cytotoxic CD8+ T cells stimulate apoptosis of the keratinocytes. The induction of this mechanism may be due to a self-antigen in a genetically predisposed patient. The evidence for LP being an autoimmune disease is supported by the high female predominance and the association of LP with other autoimmune diseases.2 Patients with LP have an increased chance of coexisting hepatitis C virus. In a cross-sectional study of 303 patients, Lodi et al3 found that approximately 20% of LP patients were hepatitis C virus seropositive.
Treatment options for LP include topical and systemic steroids, tazarotene, acitretin, and immunosuppressive agents.4 Our patient initially was treated with oral cyclosporine 100 mg every morning and oral methotrexate at a dose of 7.5 mg weekly. She also was treated with clobetasol ointment 0.05%. After 3 months, cyclosporine was discontinued. Methotrexate was maintained. At 5 months’ followup there was marked improvement of both clinical and symptomatic concerns with only residual palmoplantar erythema.
The differential diagnosis for pruritic palmoplantar hyperkeratosis is large. The most common differential diagnoses include hyperkeratotic eczema, psoriasis, secondary syphilis, and hereditary palmoplantar keratoderma. Lichen planus should be considered in the differential diagnosis of palmoplantar hyperkeratosis. A skin biopsy may be needed, as palmoplantar LP often has an atypical presentation.5
1. Sánchez-Pérez J, Rios Buceta L, Fraga J, et al. Lichen planus with lesions on the palms and/or soles: prevalence and clinicopathological study of 36 patients. Br J Dermatol. 2000;142:310-314.
2. Farhi D, Dupin N. Pathophysiology, etiologic factors, and clinical management of oral lichen planus, part I: facts and controversies. Clin Dermatol. 2010;28:100-108.
3. Lodi G, Giuliani M, Majorana A, et al. Lichen planus and hepatitis C virus: a multicentre study of patients with oral lesions and a systematic review. Br J Dermatol. 2004;151:1172-1181.
4. Karakatsanis G, Patsatsi A, Kastoridou C, et al. Palmoplantar lichen planus with umbilicated papules: an atypical case with rapid therapeutic response to cyclosporin. J Eur Acad Dermatol Venereol. 2007;21:1006-1007.
5. Rotunda AM, Craft N, Haley JC. Hyperkeratotic plaques on the palms and soles. palmoplantar lichen planus, hyperkeratotic variant. Arch Dermatol. 2004;140:1275-1280.
The Diagnosis: Palmoplantar Lichen Planus
A skin biopsy from a lesion on the inner wrist showed an interface pattern with a dense bandlike infiltrate obscuring the dermoepidermal junction coupled with a superficial perivascular infiltrate (Figure). At higher magnification (×10), the histologic features included compact orthokeratosis, wedge-shaped hypergranulosis, vacuolar degeneration of the basal layer, basal dyskeratosis, a dense lymphohistiocytic infiltrate obscuring the basement membrane, and melanophages in the papillary dermis.
Lichen planus (LP) is a common inflammatory disease of the skin presenting with flat-topped, violaceous, polygonal papules with fine white lines (Wickham striae) on the surface. It is recognized and diagnosed clinically by its characteristic appearance. Common areas of LP presentation include the shins, inner thighs, genitalia, trunk, volar aspect of the wrists, and oral mucosa.
Palmoplantar LP can present as erythematous plaques, punctuate keratosis, diffuse keratoderma, or ulcerated lesions. The most common concern among patients with LP is pruritus. One-fourth of patients with LP may present with lesions on the palms and soles, but diffuse palmoplantar hyperkeratosis is rare.1 Lesions typically heal in 1 to 8 months, with an average of 3 months. Palmoplantar LP recurs within 1 year after stopping treatment in one-third of patients.1
The cause of LP is unknown, but the pathophysiology is beginning to be understood. Cytotoxic CD8+ T cells stimulate apoptosis of the keratinocytes. The induction of this mechanism may be due to a self-antigen in a genetically predisposed patient. The evidence for LP being an autoimmune disease is supported by the high female predominance and the association of LP with other autoimmune diseases.2 Patients with LP have an increased chance of coexisting hepatitis C virus. In a cross-sectional study of 303 patients, Lodi et al3 found that approximately 20% of LP patients were hepatitis C virus seropositive.
Treatment options for LP include topical and systemic steroids, tazarotene, acitretin, and immunosuppressive agents.4 Our patient initially was treated with oral cyclosporine 100 mg every morning and oral methotrexate at a dose of 7.5 mg weekly. She also was treated with clobetasol ointment 0.05%. After 3 months, cyclosporine was discontinued. Methotrexate was maintained. At 5 months’ followup there was marked improvement of both clinical and symptomatic concerns with only residual palmoplantar erythema.
The differential diagnosis for pruritic palmoplantar hyperkeratosis is large. The most common differential diagnoses include hyperkeratotic eczema, psoriasis, secondary syphilis, and hereditary palmoplantar keratoderma. Lichen planus should be considered in the differential diagnosis of palmoplantar hyperkeratosis. A skin biopsy may be needed, as palmoplantar LP often has an atypical presentation.5
The Diagnosis: Palmoplantar Lichen Planus
A skin biopsy from a lesion on the inner wrist showed an interface pattern with a dense bandlike infiltrate obscuring the dermoepidermal junction coupled with a superficial perivascular infiltrate (Figure). At higher magnification (×10), the histologic features included compact orthokeratosis, wedge-shaped hypergranulosis, vacuolar degeneration of the basal layer, basal dyskeratosis, a dense lymphohistiocytic infiltrate obscuring the basement membrane, and melanophages in the papillary dermis.
Lichen planus (LP) is a common inflammatory disease of the skin presenting with flat-topped, violaceous, polygonal papules with fine white lines (Wickham striae) on the surface. It is recognized and diagnosed clinically by its characteristic appearance. Common areas of LP presentation include the shins, inner thighs, genitalia, trunk, volar aspect of the wrists, and oral mucosa.
Palmoplantar LP can present as erythematous plaques, punctuate keratosis, diffuse keratoderma, or ulcerated lesions. The most common concern among patients with LP is pruritus. One-fourth of patients with LP may present with lesions on the palms and soles, but diffuse palmoplantar hyperkeratosis is rare.1 Lesions typically heal in 1 to 8 months, with an average of 3 months. Palmoplantar LP recurs within 1 year after stopping treatment in one-third of patients.1
The cause of LP is unknown, but the pathophysiology is beginning to be understood. Cytotoxic CD8+ T cells stimulate apoptosis of the keratinocytes. The induction of this mechanism may be due to a self-antigen in a genetically predisposed patient. The evidence for LP being an autoimmune disease is supported by the high female predominance and the association of LP with other autoimmune diseases.2 Patients with LP have an increased chance of coexisting hepatitis C virus. In a cross-sectional study of 303 patients, Lodi et al3 found that approximately 20% of LP patients were hepatitis C virus seropositive.
Treatment options for LP include topical and systemic steroids, tazarotene, acitretin, and immunosuppressive agents.4 Our patient initially was treated with oral cyclosporine 100 mg every morning and oral methotrexate at a dose of 7.5 mg weekly. She also was treated with clobetasol ointment 0.05%. After 3 months, cyclosporine was discontinued. Methotrexate was maintained. At 5 months’ followup there was marked improvement of both clinical and symptomatic concerns with only residual palmoplantar erythema.
The differential diagnosis for pruritic palmoplantar hyperkeratosis is large. The most common differential diagnoses include hyperkeratotic eczema, psoriasis, secondary syphilis, and hereditary palmoplantar keratoderma. Lichen planus should be considered in the differential diagnosis of palmoplantar hyperkeratosis. A skin biopsy may be needed, as palmoplantar LP often has an atypical presentation.5
1. Sánchez-Pérez J, Rios Buceta L, Fraga J, et al. Lichen planus with lesions on the palms and/or soles: prevalence and clinicopathological study of 36 patients. Br J Dermatol. 2000;142:310-314.
2. Farhi D, Dupin N. Pathophysiology, etiologic factors, and clinical management of oral lichen planus, part I: facts and controversies. Clin Dermatol. 2010;28:100-108.
3. Lodi G, Giuliani M, Majorana A, et al. Lichen planus and hepatitis C virus: a multicentre study of patients with oral lesions and a systematic review. Br J Dermatol. 2004;151:1172-1181.
4. Karakatsanis G, Patsatsi A, Kastoridou C, et al. Palmoplantar lichen planus with umbilicated papules: an atypical case with rapid therapeutic response to cyclosporin. J Eur Acad Dermatol Venereol. 2007;21:1006-1007.
5. Rotunda AM, Craft N, Haley JC. Hyperkeratotic plaques on the palms and soles. palmoplantar lichen planus, hyperkeratotic variant. Arch Dermatol. 2004;140:1275-1280.
1. Sánchez-Pérez J, Rios Buceta L, Fraga J, et al. Lichen planus with lesions on the palms and/or soles: prevalence and clinicopathological study of 36 patients. Br J Dermatol. 2000;142:310-314.
2. Farhi D, Dupin N. Pathophysiology, etiologic factors, and clinical management of oral lichen planus, part I: facts and controversies. Clin Dermatol. 2010;28:100-108.
3. Lodi G, Giuliani M, Majorana A, et al. Lichen planus and hepatitis C virus: a multicentre study of patients with oral lesions and a systematic review. Br J Dermatol. 2004;151:1172-1181.
4. Karakatsanis G, Patsatsi A, Kastoridou C, et al. Palmoplantar lichen planus with umbilicated papules: an atypical case with rapid therapeutic response to cyclosporin. J Eur Acad Dermatol Venereol. 2007;21:1006-1007.
5. Rotunda AM, Craft N, Haley JC. Hyperkeratotic plaques on the palms and soles. palmoplantar lichen planus, hyperkeratotic variant. Arch Dermatol. 2004;140:1275-1280.
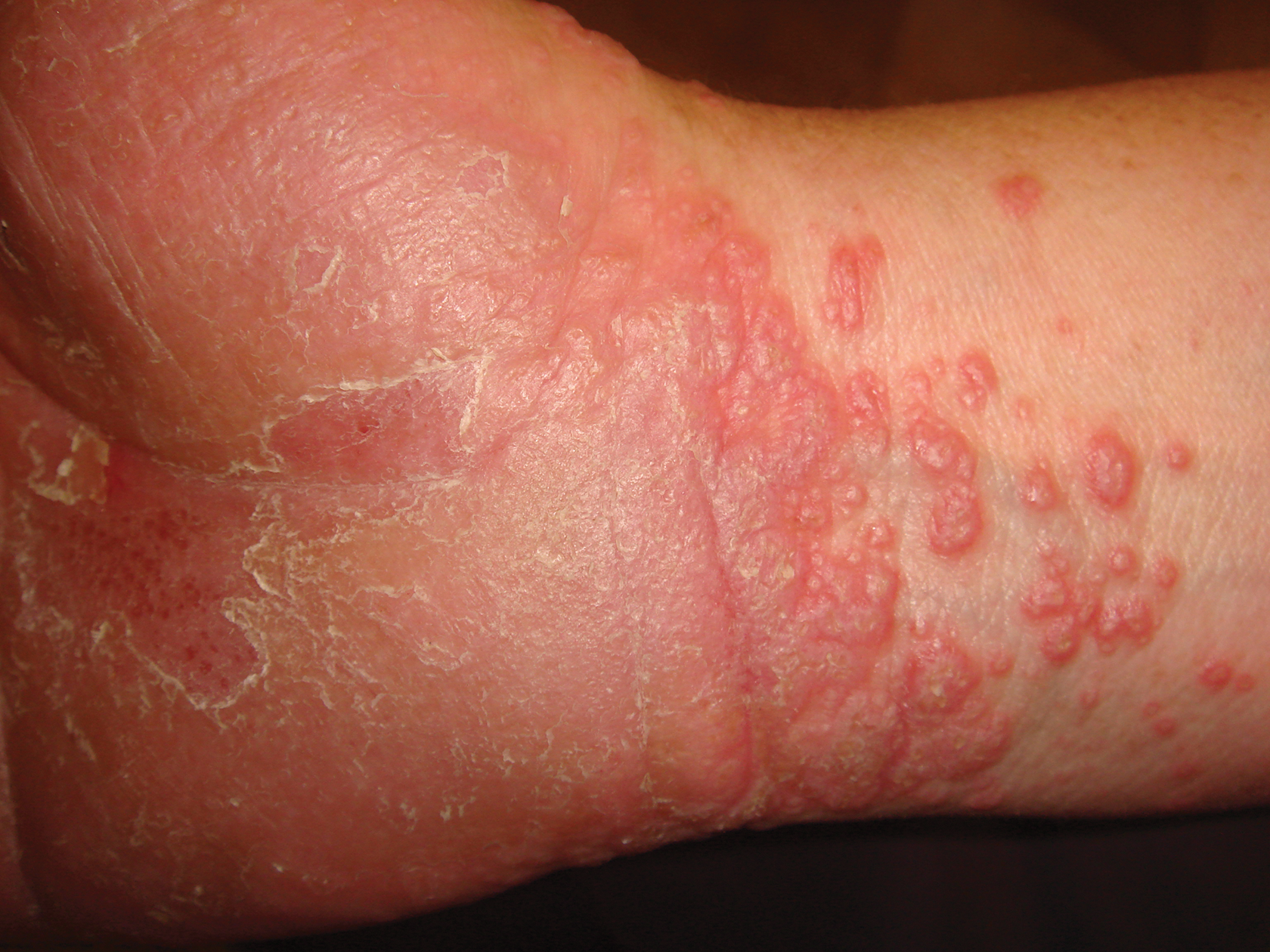
A 45-year-old woman was referred to dermatology by her general internist for the management of a pruritic rash on the hands and feet that was unresponsive to topical steroid creams. The pruritus also was unresponsive to hydroxyzine and aspirin. Erythematous plaques were present on the palms and soles. Physical examination revealed thickened volar skin with a yellowish surface. There were individual papules with atrophic tops at the edge of the plaques on the inner wrists. The patient’s medical history was otherwise unremarkable. Blood tests for glucose and liver function did not reveal any abnormalities.
Multiple Superficial White Nodules on the Bilateral Helical Rims
The Diagnosis: Bilateral Auricular Tophaceous Gout
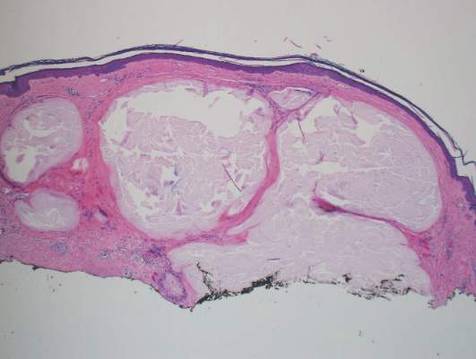
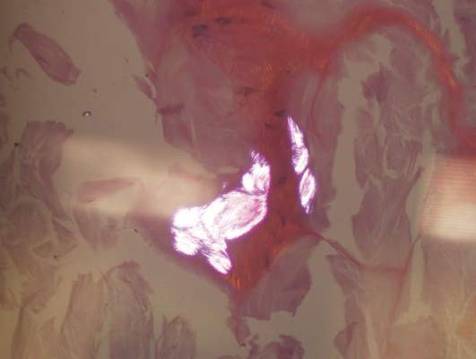
Histopathologic evaluation with hematoxylin and eosin staining demonstrated clusters of abundant granular amorphous material within the subcutaneous tissue (Figure 1). The overlying epidermis and dermis were unremarkable. The granular amorphous material demonstrated numerous monosodium urate crystals under polarized light (Figure 2). At a return visit following the biopsy results, the patient reported a history of a single episode of monoarticular gouty arthritis involving the right hallux approximately 6 months after the onset of the skin lesions. With the added clinical history and the biopsy results, his serum uric acid level was obtained and was found to be elevated at 9.2 mg/dL (reference range, 3.5–8 mg/dL).
|
|
In our patient, the clinical differential diagnosis included calcium deposits, weathering nodules, and tophaceous gout. The differential diagnosis of auricular lesions is broad, and benign lesions may mimic cancerous entities such as basal cell carcinoma and squamous cell carcinoma.1 Therefore a detailed history, thorough physical examination, and tissue sampling are key to establishing the correct diagnosis. Our patient’s history of monoarticular gouty arthritis was only elucidated after a diagnosis of bilateral auricular tophaceous gout was made based on the biopsy results.
Subcutaneous tophi represent a chronic state of hyperuricemia and tend to manifest after long-standing polyarthritis and repeated acute gout attacks.2-5 These lesions develop in approximately 50% of gout patients and usually occur an average of 11.6 years after the onset of disease.2 There is a subset of individuals that are at higher risk for developing tophi, including elderly and female patients, diuretic and chronic nonsteroidal anti-inflammatory drug users, patients with a history of cyclosporine therapy, and patients with underlying chronic renal insufficiency.2,6,7 The most commonly affected tissues are those with poor blood supply and lower temperatures, such as the ear helix and first metacarpal joint.4 The auricle is the most common site of tophi on the head and neck. Tophi of the helices are generally asymptomatic and nontender; however, tophi can become large, inflamed, and ulcerated, causing pressure and discomfort.2 Combination treatment with dietary modification and antihyperuricemic therapy (eg, allopurinol) has been shown to reduce the size of lesions and prevent future tophi formation. However, these results may take months, warranting excision of large and symptomatic lesions.4,8
Our case is unusual in that the onset of the auricular lesions predated the articular gout by 6 months. Gouty tophi as the initial presentation of hyperuricemia is rare; however, tophi formation without concomitant arthritis has been reported.2,3,7,9 Wernick et al7 described 6 patients presenting with tophi before the onset of inflammatory arthritis that they attributed to changes in active inflammation by age (eg, elderly patients were more commonly immunosuppressed), chronic illnesses, and anti-inflammatory medications (eg, nonsteroidal anti-inflammatory drugs). Another possible explanation for this atypical presentation is misdiagnosis caused by other forms of arthritis (eg, rheumatoid arthritis, osteoarthritis) masking acute gout episodes. It also has been reported that monosodium urate crystals can be found in synovial fluid with no inflammation and therefore no symptoms.7
Tophi, although rare, may be the sole clinical manifestation of underlying gouty disease. It is important to be aware of this atypical presentation to prevent misdiagnosis and provide appropriate treatment.
- Dompmartin A. Nodules of the external ear [in French]. Ann Dermatol Venereol. 1999;126:261-266.
- Griffin G, Munns J, Fullen D, et al. Auricular tophi as the initial presentation of gout. Otolaryngol Head Neck Surg. 2009;141:153-154.
- Koley S, Salodkar A, Choudhary S, et al. Tophi as first manifestation of gout. Indian J Dermatol Venerol. 2010;76:393-393-396.
- Moriwaki Y. Tophaceous gout [in Japanese]. Nihon Rinsho. 2008;66:711-716.
- Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76:801-808.
- Hollingworth P, Scott JT, Burry HC. Nonarticular gout: hyperuricemia and tophus formation without gouty arthritis. Arthritis Rheum. 1983;26:98-101.
- Wernick R, Winkler C, Campbell S. Tophi as the initial manifestation of gout. report of six cases and review of the literature. Arch Intern Med. 1992;152:873-876.
- Caldas CA, Fuller R. Excellent response to the clinical treatment of tophaceous gout. Clin Rheumatol. 2009;26:1553-1555.
- Iglesias A, Londono JC, Saaibi DL, et al. Gout nodulosis: widespread subcutaneous deposits without gout. Arthritis Care Res. 1996;9:74-77.
The Diagnosis: Bilateral Auricular Tophaceous Gout
Histopathologic evaluation with hematoxylin and eosin staining demonstrated clusters of abundant granular amorphous material within the subcutaneous tissue (Figure 1). The overlying epidermis and dermis were unremarkable. The granular amorphous material demonstrated numerous monosodium urate crystals under polarized light (Figure 2). At a return visit following the biopsy results, the patient reported a history of a single episode of monoarticular gouty arthritis involving the right hallux approximately 6 months after the onset of the skin lesions. With the added clinical history and the biopsy results, his serum uric acid level was obtained and was found to be elevated at 9.2 mg/dL (reference range, 3.5–8 mg/dL).
|
|
|
|
In our patient, the clinical differential diagnosis included calcium deposits, weathering nodules, and tophaceous gout. The differential diagnosis of auricular lesions is broad, and benign lesions may mimic cancerous entities such as basal cell carcinoma and squamous cell carcinoma.1 Therefore a detailed history, thorough physical examination, and tissue sampling are key to establishing the correct diagnosis. Our patient’s history of monoarticular gouty arthritis was only elucidated after a diagnosis of bilateral auricular tophaceous gout was made based on the biopsy results.
Subcutaneous tophi represent a chronic state of hyperuricemia and tend to manifest after long-standing polyarthritis and repeated acute gout attacks.2-5 These lesions develop in approximately 50% of gout patients and usually occur an average of 11.6 years after the onset of disease.2 There is a subset of individuals that are at higher risk for developing tophi, including elderly and female patients, diuretic and chronic nonsteroidal anti-inflammatory drug users, patients with a history of cyclosporine therapy, and patients with underlying chronic renal insufficiency.2,6,7 The most commonly affected tissues are those with poor blood supply and lower temperatures, such as the ear helix and first metacarpal joint.4 The auricle is the most common site of tophi on the head and neck. Tophi of the helices are generally asymptomatic and nontender; however, tophi can become large, inflamed, and ulcerated, causing pressure and discomfort.2 Combination treatment with dietary modification and antihyperuricemic therapy (eg, allopurinol) has been shown to reduce the size of lesions and prevent future tophi formation. However, these results may take months, warranting excision of large and symptomatic lesions.4,8
Our case is unusual in that the onset of the auricular lesions predated the articular gout by 6 months. Gouty tophi as the initial presentation of hyperuricemia is rare; however, tophi formation without concomitant arthritis has been reported.2,3,7,9 Wernick et al7 described 6 patients presenting with tophi before the onset of inflammatory arthritis that they attributed to changes in active inflammation by age (eg, elderly patients were more commonly immunosuppressed), chronic illnesses, and anti-inflammatory medications (eg, nonsteroidal anti-inflammatory drugs). Another possible explanation for this atypical presentation is misdiagnosis caused by other forms of arthritis (eg, rheumatoid arthritis, osteoarthritis) masking acute gout episodes. It also has been reported that monosodium urate crystals can be found in synovial fluid with no inflammation and therefore no symptoms.7
Tophi, although rare, may be the sole clinical manifestation of underlying gouty disease. It is important to be aware of this atypical presentation to prevent misdiagnosis and provide appropriate treatment.
The Diagnosis: Bilateral Auricular Tophaceous Gout
Histopathologic evaluation with hematoxylin and eosin staining demonstrated clusters of abundant granular amorphous material within the subcutaneous tissue (Figure 1). The overlying epidermis and dermis were unremarkable. The granular amorphous material demonstrated numerous monosodium urate crystals under polarized light (Figure 2). At a return visit following the biopsy results, the patient reported a history of a single episode of monoarticular gouty arthritis involving the right hallux approximately 6 months after the onset of the skin lesions. With the added clinical history and the biopsy results, his serum uric acid level was obtained and was found to be elevated at 9.2 mg/dL (reference range, 3.5–8 mg/dL).
|
|
|
|
In our patient, the clinical differential diagnosis included calcium deposits, weathering nodules, and tophaceous gout. The differential diagnosis of auricular lesions is broad, and benign lesions may mimic cancerous entities such as basal cell carcinoma and squamous cell carcinoma.1 Therefore a detailed history, thorough physical examination, and tissue sampling are key to establishing the correct diagnosis. Our patient’s history of monoarticular gouty arthritis was only elucidated after a diagnosis of bilateral auricular tophaceous gout was made based on the biopsy results.
Subcutaneous tophi represent a chronic state of hyperuricemia and tend to manifest after long-standing polyarthritis and repeated acute gout attacks.2-5 These lesions develop in approximately 50% of gout patients and usually occur an average of 11.6 years after the onset of disease.2 There is a subset of individuals that are at higher risk for developing tophi, including elderly and female patients, diuretic and chronic nonsteroidal anti-inflammatory drug users, patients with a history of cyclosporine therapy, and patients with underlying chronic renal insufficiency.2,6,7 The most commonly affected tissues are those with poor blood supply and lower temperatures, such as the ear helix and first metacarpal joint.4 The auricle is the most common site of tophi on the head and neck. Tophi of the helices are generally asymptomatic and nontender; however, tophi can become large, inflamed, and ulcerated, causing pressure and discomfort.2 Combination treatment with dietary modification and antihyperuricemic therapy (eg, allopurinol) has been shown to reduce the size of lesions and prevent future tophi formation. However, these results may take months, warranting excision of large and symptomatic lesions.4,8
Our case is unusual in that the onset of the auricular lesions predated the articular gout by 6 months. Gouty tophi as the initial presentation of hyperuricemia is rare; however, tophi formation without concomitant arthritis has been reported.2,3,7,9 Wernick et al7 described 6 patients presenting with tophi before the onset of inflammatory arthritis that they attributed to changes in active inflammation by age (eg, elderly patients were more commonly immunosuppressed), chronic illnesses, and anti-inflammatory medications (eg, nonsteroidal anti-inflammatory drugs). Another possible explanation for this atypical presentation is misdiagnosis caused by other forms of arthritis (eg, rheumatoid arthritis, osteoarthritis) masking acute gout episodes. It also has been reported that monosodium urate crystals can be found in synovial fluid with no inflammation and therefore no symptoms.7
Tophi, although rare, may be the sole clinical manifestation of underlying gouty disease. It is important to be aware of this atypical presentation to prevent misdiagnosis and provide appropriate treatment.
- Dompmartin A. Nodules of the external ear [in French]. Ann Dermatol Venereol. 1999;126:261-266.
- Griffin G, Munns J, Fullen D, et al. Auricular tophi as the initial presentation of gout. Otolaryngol Head Neck Surg. 2009;141:153-154.
- Koley S, Salodkar A, Choudhary S, et al. Tophi as first manifestation of gout. Indian J Dermatol Venerol. 2010;76:393-393-396.
- Moriwaki Y. Tophaceous gout [in Japanese]. Nihon Rinsho. 2008;66:711-716.
- Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76:801-808.
- Hollingworth P, Scott JT, Burry HC. Nonarticular gout: hyperuricemia and tophus formation without gouty arthritis. Arthritis Rheum. 1983;26:98-101.
- Wernick R, Winkler C, Campbell S. Tophi as the initial manifestation of gout. report of six cases and review of the literature. Arch Intern Med. 1992;152:873-876.
- Caldas CA, Fuller R. Excellent response to the clinical treatment of tophaceous gout. Clin Rheumatol. 2009;26:1553-1555.
- Iglesias A, Londono JC, Saaibi DL, et al. Gout nodulosis: widespread subcutaneous deposits without gout. Arthritis Care Res. 1996;9:74-77.
- Dompmartin A. Nodules of the external ear [in French]. Ann Dermatol Venereol. 1999;126:261-266.
- Griffin G, Munns J, Fullen D, et al. Auricular tophi as the initial presentation of gout. Otolaryngol Head Neck Surg. 2009;141:153-154.
- Koley S, Salodkar A, Choudhary S, et al. Tophi as first manifestation of gout. Indian J Dermatol Venerol. 2010;76:393-393-396.
- Moriwaki Y. Tophaceous gout [in Japanese]. Nihon Rinsho. 2008;66:711-716.
- Eggebeen AT. Gout: an update. Am Fam Physician. 2007;76:801-808.
- Hollingworth P, Scott JT, Burry HC. Nonarticular gout: hyperuricemia and tophus formation without gouty arthritis. Arthritis Rheum. 1983;26:98-101.
- Wernick R, Winkler C, Campbell S. Tophi as the initial manifestation of gout. report of six cases and review of the literature. Arch Intern Med. 1992;152:873-876.
- Caldas CA, Fuller R. Excellent response to the clinical treatment of tophaceous gout. Clin Rheumatol. 2009;26:1553-1555.
- Iglesias A, Londono JC, Saaibi DL, et al. Gout nodulosis: widespread subcutaneous deposits without gout. Arthritis Care Res. 1996;9:74-77.
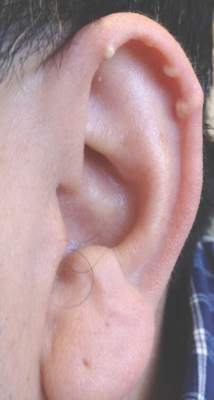
A 40-year-old man presented for evaluation of multiple small nodules on the bilateral auricles primarily involving the helices of 1 year’s duration. The lesions were nontender with no associated bleeding, burning, or pruritus. He denied any trauma to these sites and denied any systemic symptoms including fever, chills, joint pain, or weight loss. His medical history was remarkable for type 2 diabetes mellitus. He had no history of similar skin lesions or renal disease and denied any alcohol intake. He also denied taking any over-the-counter or prescription medications. Physical examination revealed several 1- to 4-mm superficial white dermal nodules located on the bilateral helical rims. The lesions were firm and well circumscribed and the surrounding skin showed mild erythema. Shave biopsies of the nodules were performed.
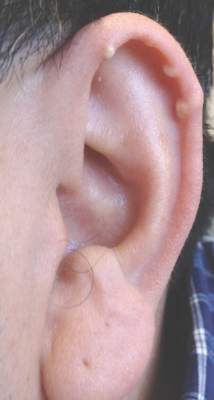
An Eruption While on Total Parenteral Nutrition
The Diagnosis: Acquired Acrodermatitis Enteropathica
Acquired acrodermatitis enteropathica (AAE) is a rare disorder caused by severe zinc deficiency. Although acrodermatitis enteropathica is an autosomal-recessive disorder that typically manifests in infancy, AAE also can result from poor zinc intake, impaired absorption, or accelerated losses. There are reports of AAE in patients with zinc-deficient diets,1 eating disorders,2 bariatric and other gastrointestinal surgeries,3 malabsorptive diseases,4 and nephrotic syndrome.5
Zinc plays an important role in DNA and RNA synthesis, reactive oxygen species attenuation, and energy metabolism, allowing for proper wound healing, skin differentiation, and proliferation.6 Zinc is found in most foods, but animal protein contains higher concentrations (Table).7 Approximately 85% of zinc is stored in muscles and bones, with only a small amount of accessible zinc available in the liver. Liver stores can be depleted as quickly as 1 week.8 Total parenteral nutrition without trace element supplementation can quickly predispose patients to AAE.
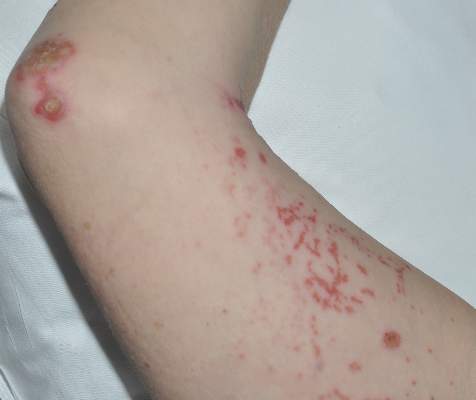
|
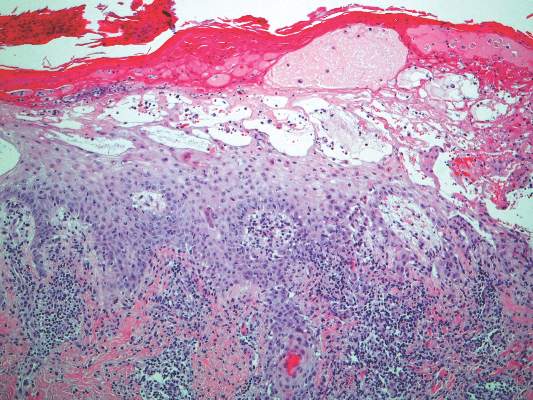
|
Diagnosis of this condition requires triangulation of clinical presentation, histopathology examination, and laboratory findings. Acrodermatitis enteropathica typically is characterized by dermatitis, diarrhea, and epidermal appendage findings. In its early stages, the dermatitis often manifests with angular cheilitis and paronychia.9 Patients then develop erythema, erosions, and occasionally vesicles or psoriasiform plaques in periorificial, perineal, and acral sites (Figure 1). Epidermal appendage effects include generalized alopecia and thinning nails with white transverse ridges. Although dermatologic and gastrointestinal manifestations are the most obvious, severe AAE may cause other symptoms, including mental slowing, hypogonadism, and impaired immune function.9
Histopathology of AAE skin lesions is similar to other nutritional deficiencies. Early changes are more specific to deficiency dermatitis and include cytoplasmic pallor and ballooning degeneration of keratinocytes in the stratum spinosum and granulosum.9 Necrolysis results in confluent keratinocyte necrosis developing into subcorneal bulla. Later in the disease course, the presentation becomes psoriasiform with keratinocyte dyskeratosis and confluent parakeratosis10 (Figure 2). Dermal edema with dilated tortuous vessels and a neutrophilic infiltrate may be present throughout disease progression.
Common laboratory abnormalities used to confirm zinc deficiency are decreased plasma zinc and alkaline phosphatase levels. Plasma zinc levels should be drawn after fasting because zinc levels decrease after food intake.9 Concurrent albumin levels should be drawn to correct for low levels caused by hypoalbuminemia. Acquired acrodermatitis enteropathica has been seen in patients with only mildly decreased plasma zinc levels or even zinc levels within reference range.11 Alkaline phosphatase metalloenzyme synthesis requires zinc and a decreased level suggests zinc deficiency even with a plasma zinc level within reference range. Alkaline phosphatase levels usually can be ascertained in a matter of hours, while the zinc levels take much longer to result.

Acquired acrodermatitis enteropathica is treated with oral elemental zinc supplementation at 1 to 2 mg/kg daily.12 Diarrhea typically resolves within 24 hours, but skin lesions heal in 1 to 2 weeks or longer. Although there is no consensus on when to discontinue zinc replacement therapy, therapy generally is not lifelong. Once the patient is zinc replete and the inciting factor has resolved, patients can discontinue supplementation without risk for recurrence.
Trace elements had not been added to our patient’s total parenteral nutrition prior to admission. Basic nutrition laboratory results and zinc levels returned markedly low: 14 μg/dL (reference range, 60–120 μg/dL). Alkaline phosphatase, a zinc-dependent protein, also was low at 12 U/L (reference range, 40–150 U/L). We added trace elements and vitamins and began empiric zinc replacement with 440 mg oral zinc sulfate daily (100 mg elemental zinc). Cephalexin was prescribed for impetiginized skin lesions. The patient noted skin improvement after 3 days on zinc replacement therapy.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Kim ST, Kang JS, Baek JW, et al. Acrodermatitis enteropathica with anorexia nervosa. J Dermatol. 2010;37:726-729.
- Bae-Harboe YS, Solky A, Masterpol KS. A case of acquired zinc deficiency. Dermatol Online J. 2012;18:1.
- Krasovec M, Frenk E. Acrodermatitis enteropathica secondary to Crohn’s disease. Dermatol Basel Switz. 1996;193:361-363.
- Reichel M, Mauro TM, Ziboh VA, et al. Acrodermatitis enteropathica in a patient with the acquired immunodeficiency syndrome. Arch Dermatol. 1992;128:415-417.
- Perafan-Riveros C, Franca LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- National Nutrient Database for Standard Reference, Release 28. United States Department of Agriculture, Agricultural Research Service website. http://ndb.nal.usda.gov/ndb/nutrients/report/nutrientsfrm?max=25&offset=0&totCount=0&nutrient1=309&nutrient2=&nutrient3=&subset=0&fg=&sort=f&measureby=m. Accessed December 14, 2015.
- McPherson RA, Pincus MR. Henry’s Clinical Diagnosis and Management by Laboratory Methods. 22nd ed. Philadelphia, PA: Saunders Elsevier; 2011.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Kumar P, Lal NR, Mondal A, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
The Diagnosis: Acquired Acrodermatitis Enteropathica
Acquired acrodermatitis enteropathica (AAE) is a rare disorder caused by severe zinc deficiency. Although acrodermatitis enteropathica is an autosomal-recessive disorder that typically manifests in infancy, AAE also can result from poor zinc intake, impaired absorption, or accelerated losses. There are reports of AAE in patients with zinc-deficient diets,1 eating disorders,2 bariatric and other gastrointestinal surgeries,3 malabsorptive diseases,4 and nephrotic syndrome.5
Zinc plays an important role in DNA and RNA synthesis, reactive oxygen species attenuation, and energy metabolism, allowing for proper wound healing, skin differentiation, and proliferation.6 Zinc is found in most foods, but animal protein contains higher concentrations (Table).7 Approximately 85% of zinc is stored in muscles and bones, with only a small amount of accessible zinc available in the liver. Liver stores can be depleted as quickly as 1 week.8 Total parenteral nutrition without trace element supplementation can quickly predispose patients to AAE.
|
|
|
|
Diagnosis of this condition requires triangulation of clinical presentation, histopathology examination, and laboratory findings. Acrodermatitis enteropathica typically is characterized by dermatitis, diarrhea, and epidermal appendage findings. In its early stages, the dermatitis often manifests with angular cheilitis and paronychia.9 Patients then develop erythema, erosions, and occasionally vesicles or psoriasiform plaques in periorificial, perineal, and acral sites (Figure 1). Epidermal appendage effects include generalized alopecia and thinning nails with white transverse ridges. Although dermatologic and gastrointestinal manifestations are the most obvious, severe AAE may cause other symptoms, including mental slowing, hypogonadism, and impaired immune function.9
Histopathology of AAE skin lesions is similar to other nutritional deficiencies. Early changes are more specific to deficiency dermatitis and include cytoplasmic pallor and ballooning degeneration of keratinocytes in the stratum spinosum and granulosum.9 Necrolysis results in confluent keratinocyte necrosis developing into subcorneal bulla. Later in the disease course, the presentation becomes psoriasiform with keratinocyte dyskeratosis and confluent parakeratosis10 (Figure 2). Dermal edema with dilated tortuous vessels and a neutrophilic infiltrate may be present throughout disease progression.
Common laboratory abnormalities used to confirm zinc deficiency are decreased plasma zinc and alkaline phosphatase levels. Plasma zinc levels should be drawn after fasting because zinc levels decrease after food intake.9 Concurrent albumin levels should be drawn to correct for low levels caused by hypoalbuminemia. Acquired acrodermatitis enteropathica has been seen in patients with only mildly decreased plasma zinc levels or even zinc levels within reference range.11 Alkaline phosphatase metalloenzyme synthesis requires zinc and a decreased level suggests zinc deficiency even with a plasma zinc level within reference range. Alkaline phosphatase levels usually can be ascertained in a matter of hours, while the zinc levels take much longer to result.
Acquired acrodermatitis enteropathica is treated with oral elemental zinc supplementation at 1 to 2 mg/kg daily.12 Diarrhea typically resolves within 24 hours, but skin lesions heal in 1 to 2 weeks or longer. Although there is no consensus on when to discontinue zinc replacement therapy, therapy generally is not lifelong. Once the patient is zinc replete and the inciting factor has resolved, patients can discontinue supplementation without risk for recurrence.
Trace elements had not been added to our patient’s total parenteral nutrition prior to admission. Basic nutrition laboratory results and zinc levels returned markedly low: 14 μg/dL (reference range, 60–120 μg/dL). Alkaline phosphatase, a zinc-dependent protein, also was low at 12 U/L (reference range, 40–150 U/L). We added trace elements and vitamins and began empiric zinc replacement with 440 mg oral zinc sulfate daily (100 mg elemental zinc). Cephalexin was prescribed for impetiginized skin lesions. The patient noted skin improvement after 3 days on zinc replacement therapy.
The Diagnosis: Acquired Acrodermatitis Enteropathica
Acquired acrodermatitis enteropathica (AAE) is a rare disorder caused by severe zinc deficiency. Although acrodermatitis enteropathica is an autosomal-recessive disorder that typically manifests in infancy, AAE also can result from poor zinc intake, impaired absorption, or accelerated losses. There are reports of AAE in patients with zinc-deficient diets,1 eating disorders,2 bariatric and other gastrointestinal surgeries,3 malabsorptive diseases,4 and nephrotic syndrome.5
Zinc plays an important role in DNA and RNA synthesis, reactive oxygen species attenuation, and energy metabolism, allowing for proper wound healing, skin differentiation, and proliferation.6 Zinc is found in most foods, but animal protein contains higher concentrations (Table).7 Approximately 85% of zinc is stored in muscles and bones, with only a small amount of accessible zinc available in the liver. Liver stores can be depleted as quickly as 1 week.8 Total parenteral nutrition without trace element supplementation can quickly predispose patients to AAE.
|
|
|
|
Diagnosis of this condition requires triangulation of clinical presentation, histopathology examination, and laboratory findings. Acrodermatitis enteropathica typically is characterized by dermatitis, diarrhea, and epidermal appendage findings. In its early stages, the dermatitis often manifests with angular cheilitis and paronychia.9 Patients then develop erythema, erosions, and occasionally vesicles or psoriasiform plaques in periorificial, perineal, and acral sites (Figure 1). Epidermal appendage effects include generalized alopecia and thinning nails with white transverse ridges. Although dermatologic and gastrointestinal manifestations are the most obvious, severe AAE may cause other symptoms, including mental slowing, hypogonadism, and impaired immune function.9
Histopathology of AAE skin lesions is similar to other nutritional deficiencies. Early changes are more specific to deficiency dermatitis and include cytoplasmic pallor and ballooning degeneration of keratinocytes in the stratum spinosum and granulosum.9 Necrolysis results in confluent keratinocyte necrosis developing into subcorneal bulla. Later in the disease course, the presentation becomes psoriasiform with keratinocyte dyskeratosis and confluent parakeratosis10 (Figure 2). Dermal edema with dilated tortuous vessels and a neutrophilic infiltrate may be present throughout disease progression.
Common laboratory abnormalities used to confirm zinc deficiency are decreased plasma zinc and alkaline phosphatase levels. Plasma zinc levels should be drawn after fasting because zinc levels decrease after food intake.9 Concurrent albumin levels should be drawn to correct for low levels caused by hypoalbuminemia. Acquired acrodermatitis enteropathica has been seen in patients with only mildly decreased plasma zinc levels or even zinc levels within reference range.11 Alkaline phosphatase metalloenzyme synthesis requires zinc and a decreased level suggests zinc deficiency even with a plasma zinc level within reference range. Alkaline phosphatase levels usually can be ascertained in a matter of hours, while the zinc levels take much longer to result.
Acquired acrodermatitis enteropathica is treated with oral elemental zinc supplementation at 1 to 2 mg/kg daily.12 Diarrhea typically resolves within 24 hours, but skin lesions heal in 1 to 2 weeks or longer. Although there is no consensus on when to discontinue zinc replacement therapy, therapy generally is not lifelong. Once the patient is zinc replete and the inciting factor has resolved, patients can discontinue supplementation without risk for recurrence.
Trace elements had not been added to our patient’s total parenteral nutrition prior to admission. Basic nutrition laboratory results and zinc levels returned markedly low: 14 μg/dL (reference range, 60–120 μg/dL). Alkaline phosphatase, a zinc-dependent protein, also was low at 12 U/L (reference range, 40–150 U/L). We added trace elements and vitamins and began empiric zinc replacement with 440 mg oral zinc sulfate daily (100 mg elemental zinc). Cephalexin was prescribed for impetiginized skin lesions. The patient noted skin improvement after 3 days on zinc replacement therapy.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Kim ST, Kang JS, Baek JW, et al. Acrodermatitis enteropathica with anorexia nervosa. J Dermatol. 2010;37:726-729.
- Bae-Harboe YS, Solky A, Masterpol KS. A case of acquired zinc deficiency. Dermatol Online J. 2012;18:1.
- Krasovec M, Frenk E. Acrodermatitis enteropathica secondary to Crohn’s disease. Dermatol Basel Switz. 1996;193:361-363.
- Reichel M, Mauro TM, Ziboh VA, et al. Acrodermatitis enteropathica in a patient with the acquired immunodeficiency syndrome. Arch Dermatol. 1992;128:415-417.
- Perafan-Riveros C, Franca LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- National Nutrient Database for Standard Reference, Release 28. United States Department of Agriculture, Agricultural Research Service website. http://ndb.nal.usda.gov/ndb/nutrients/report/nutrientsfrm?max=25&offset=0&totCount=0&nutrient1=309&nutrient2=&nutrient3=&subset=0&fg=&sort=f&measureby=m. Accessed December 14, 2015.
- McPherson RA, Pincus MR. Henry’s Clinical Diagnosis and Management by Laboratory Methods. 22nd ed. Philadelphia, PA: Saunders Elsevier; 2011.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Kumar P, Lal NR, Mondal A, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Kim ST, Kang JS, Baek JW, et al. Acrodermatitis enteropathica with anorexia nervosa. J Dermatol. 2010;37:726-729.
- Bae-Harboe YS, Solky A, Masterpol KS. A case of acquired zinc deficiency. Dermatol Online J. 2012;18:1.
- Krasovec M, Frenk E. Acrodermatitis enteropathica secondary to Crohn’s disease. Dermatol Basel Switz. 1996;193:361-363.
- Reichel M, Mauro TM, Ziboh VA, et al. Acrodermatitis enteropathica in a patient with the acquired immunodeficiency syndrome. Arch Dermatol. 1992;128:415-417.
- Perafan-Riveros C, Franca LFS, Alves ACF, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- National Nutrient Database for Standard Reference, Release 28. United States Department of Agriculture, Agricultural Research Service website. http://ndb.nal.usda.gov/ndb/nutrients/report/nutrientsfrm?max=25&offset=0&totCount=0&nutrient1=309&nutrient2=&nutrient3=&subset=0&fg=&sort=f&measureby=m. Accessed December 14, 2015.
- McPherson RA, Pincus MR. Henry’s Clinical Diagnosis and Management by Laboratory Methods. 22nd ed. Philadelphia, PA: Saunders Elsevier; 2011.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Gonzalez JR, Botet MV, Sanchez JL. The histopathology of acrodermatitis enteropathica. Am J Dermatopathol. 1982;4:303-311.
- Macdonald JB, Connolly SM, DiCaudo DJ. Think zinc deficiency: acquired acrodermatitis enteropathica due to poor diet and common medications. Arch Dermatol. 2012;148:961-963.
- Kumar P, Lal NR, Mondal A, et al. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
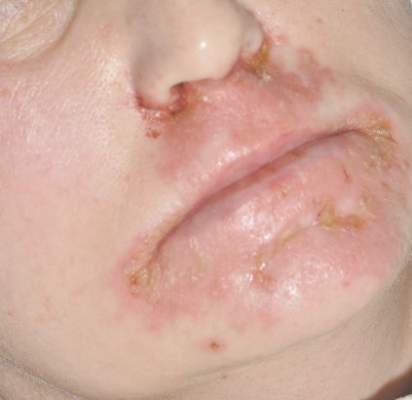
A 47-year-old woman with a history of bulimia and gastroparesis who had been on total parenteral nutrition for 8 weeks presented with a painful, perioral, perineal, and acral eruption of 7 weeks’ duration. Additionally, she had experienced diarrhea, vomiting, and a 13.5-kg weight loss in the last 4 months. Physical examination revealed perioral and perineal, well-demarcated, erythematous, scaly plaques with yellow crusting. She had edematous crusted erosions on the bilateral palms and soles and psoriasiform plaques along the right arm and flank. Punch biopsies (4 mm) from the right inguinal fold and right elbow were obtained.
Acute Serpiginous Rash
The Diagnosis: Cutaneous Larva Migrans
Three punch biopsies were obtained. Spongiotic dermatitis with eosinophils was seen. There was a single specimen of tissue that showed a possible intraepidermal larva with a tract in the epidermis. The differential diagnosis included allergic contact dermatitis and arthropod bite eruption, among others, but clinical correlation made cutaneous larva migrans (CLM) the likely diagnosis.
The patient was treated empirically with albendazole 400 mg once daily for 3 days. In addition, he was prescribed triamcinolone for symptomatic relief and remained asymptomatic for 8 weeks at which time he presented again to the dermatology clinic with a similar rash in the same distribution. He was treated with a repeat course of albendazole and further educated on the etiology of the infection. The patient has not exhibited a recurrence after treatment of the second episode of CLM.
Cutaneous larva migrans is a dermatosis of the skin caused by the larvae of parasitic nematodes from the hookworm family, most commonly Ancylostoma caninum and Ancylostoma braziliense.1,2 These hookworms thrive in warm moist climates and are most frequently found in tropical coastal regions. They normally inhabit the intestines of animals such as dogs and cats and are transmitted to soil and sand via feces. Humans become accidental hosts through contact with the contaminated sand or soil3; however, the larvae are unable to penetrate deeper than the upper dermis of the skin in humans, subsequently limiting the infection. Because humans are accidental hosts, the larvae are unable to complete their life cycle and larval death occurs within weeks to months after the initial infection3; thus treatment may be unnecessary unless complications arise.
Cutaneous larva migrans is most commonly observed in travelers or inhabitants of tropical coastal regions but can occur anywhere in the world.1 Clinically, CLM presents as an enlarging, intensely pruritic, erythematous linear or serpiginous tract,3 most commonly on the hands, feet, abdomen, and buttocks.1 Complications may include allergic reactions, secondary bacterial infections, and hookworm folliculitis.4 Although rare, migration to the intestinal tract5 and/or hematological spread with Löffler syndrome has been described.6 Although this dermatological disease has been well described in the medical literature, it is not well recognized by Western physicians and is consequently either not diagnosed or misdiagnosed, leading to delays in treatment.4 Although the infection is usually self-limiting without treatment, the risk for prolonged active disease may occur, with 1 reported case lasting up to 18 months.4,5 The first indicator of CLM is intense pruritus localized to the site of infection.4 As the larvae migrate or creep, they create a lesion that may appear edematous with vesiculobullous lesions that are either serpiginous or linear.4 The differential diagnosis may include fungal infection, bacterial infection, and atypical herpes simplex infections; however, the key finding in CLM is the presence of undulating tracts localized to the borders of the lesion.2 Patients may report experiencing a stinging sensation prior to the formation of the erythematous scaly papule,5 which is attributed to the initial penetration of the larva into the skin. This development, accompanied with a history of travel to tropical or subtropical regions, should elicit CLM as a likely diagnosis. Because hookworms are a type of helminth, they likely elicit an eosinophilic immune response and thus peripheral eosinophilia may be present.5
Effective treatment of CLM is accomplished with oral albendazole 400 mg once daily for 3 to 7 days.2,7 Alternatively, oral ivermectin, topical thiabendazole, and cryosurgery can be used,2 though albendazole currently is the preferred treatment of CLM.7
- Hotez PJ, Brooker S, Bethony JM, et al. Hookworm infection. N Engl J Med. 2004;351:799-807.
- Roest MA, Ratnavel R. Cutaneous larva migrans contracted in England: a reminder. Clin Exp Dermatol. 2001;26:389-390.
- Blackwell V, Vega-Lopez F. Cutaneous larva migrans: clinical features and management of 44 cases presenting in the returning traveller. Br J Dermatol. 2001;145:434-437.
- Hochedez P, Caumes E. Hookworm-related cutaneous larva migrans. J Travel Med. 2007;14:326-333.
- Bravo F, Sanchez MR. New and re-emerging cutaneous infectious diseases in Latin America and other geographic areas. Dermatol Clin. 2003;21:655-668, viii.
- Guill MA, Odom RB. Larva migrans complicated by Loeffler’s syndrome. Arch Dermatol. 1978;114:1525-1526.
- Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814.
The Diagnosis: Cutaneous Larva Migrans
Three punch biopsies were obtained. Spongiotic dermatitis with eosinophils was seen. There was a single specimen of tissue that showed a possible intraepidermal larva with a tract in the epidermis. The differential diagnosis included allergic contact dermatitis and arthropod bite eruption, among others, but clinical correlation made cutaneous larva migrans (CLM) the likely diagnosis.
The patient was treated empirically with albendazole 400 mg once daily for 3 days. In addition, he was prescribed triamcinolone for symptomatic relief and remained asymptomatic for 8 weeks at which time he presented again to the dermatology clinic with a similar rash in the same distribution. He was treated with a repeat course of albendazole and further educated on the etiology of the infection. The patient has not exhibited a recurrence after treatment of the second episode of CLM.
Cutaneous larva migrans is a dermatosis of the skin caused by the larvae of parasitic nematodes from the hookworm family, most commonly Ancylostoma caninum and Ancylostoma braziliense.1,2 These hookworms thrive in warm moist climates and are most frequently found in tropical coastal regions. They normally inhabit the intestines of animals such as dogs and cats and are transmitted to soil and sand via feces. Humans become accidental hosts through contact with the contaminated sand or soil3; however, the larvae are unable to penetrate deeper than the upper dermis of the skin in humans, subsequently limiting the infection. Because humans are accidental hosts, the larvae are unable to complete their life cycle and larval death occurs within weeks to months after the initial infection3; thus treatment may be unnecessary unless complications arise.
Cutaneous larva migrans is most commonly observed in travelers or inhabitants of tropical coastal regions but can occur anywhere in the world.1 Clinically, CLM presents as an enlarging, intensely pruritic, erythematous linear or serpiginous tract,3 most commonly on the hands, feet, abdomen, and buttocks.1 Complications may include allergic reactions, secondary bacterial infections, and hookworm folliculitis.4 Although rare, migration to the intestinal tract5 and/or hematological spread with Löffler syndrome has been described.6 Although this dermatological disease has been well described in the medical literature, it is not well recognized by Western physicians and is consequently either not diagnosed or misdiagnosed, leading to delays in treatment.4 Although the infection is usually self-limiting without treatment, the risk for prolonged active disease may occur, with 1 reported case lasting up to 18 months.4,5 The first indicator of CLM is intense pruritus localized to the site of infection.4 As the larvae migrate or creep, they create a lesion that may appear edematous with vesiculobullous lesions that are either serpiginous or linear.4 The differential diagnosis may include fungal infection, bacterial infection, and atypical herpes simplex infections; however, the key finding in CLM is the presence of undulating tracts localized to the borders of the lesion.2 Patients may report experiencing a stinging sensation prior to the formation of the erythematous scaly papule,5 which is attributed to the initial penetration of the larva into the skin. This development, accompanied with a history of travel to tropical or subtropical regions, should elicit CLM as a likely diagnosis. Because hookworms are a type of helminth, they likely elicit an eosinophilic immune response and thus peripheral eosinophilia may be present.5
Effective treatment of CLM is accomplished with oral albendazole 400 mg once daily for 3 to 7 days.2,7 Alternatively, oral ivermectin, topical thiabendazole, and cryosurgery can be used,2 though albendazole currently is the preferred treatment of CLM.7
The Diagnosis: Cutaneous Larva Migrans
Three punch biopsies were obtained. Spongiotic dermatitis with eosinophils was seen. There was a single specimen of tissue that showed a possible intraepidermal larva with a tract in the epidermis. The differential diagnosis included allergic contact dermatitis and arthropod bite eruption, among others, but clinical correlation made cutaneous larva migrans (CLM) the likely diagnosis.
The patient was treated empirically with albendazole 400 mg once daily for 3 days. In addition, he was prescribed triamcinolone for symptomatic relief and remained asymptomatic for 8 weeks at which time he presented again to the dermatology clinic with a similar rash in the same distribution. He was treated with a repeat course of albendazole and further educated on the etiology of the infection. The patient has not exhibited a recurrence after treatment of the second episode of CLM.
Cutaneous larva migrans is a dermatosis of the skin caused by the larvae of parasitic nematodes from the hookworm family, most commonly Ancylostoma caninum and Ancylostoma braziliense.1,2 These hookworms thrive in warm moist climates and are most frequently found in tropical coastal regions. They normally inhabit the intestines of animals such as dogs and cats and are transmitted to soil and sand via feces. Humans become accidental hosts through contact with the contaminated sand or soil3; however, the larvae are unable to penetrate deeper than the upper dermis of the skin in humans, subsequently limiting the infection. Because humans are accidental hosts, the larvae are unable to complete their life cycle and larval death occurs within weeks to months after the initial infection3; thus treatment may be unnecessary unless complications arise.
Cutaneous larva migrans is most commonly observed in travelers or inhabitants of tropical coastal regions but can occur anywhere in the world.1 Clinically, CLM presents as an enlarging, intensely pruritic, erythematous linear or serpiginous tract,3 most commonly on the hands, feet, abdomen, and buttocks.1 Complications may include allergic reactions, secondary bacterial infections, and hookworm folliculitis.4 Although rare, migration to the intestinal tract5 and/or hematological spread with Löffler syndrome has been described.6 Although this dermatological disease has been well described in the medical literature, it is not well recognized by Western physicians and is consequently either not diagnosed or misdiagnosed, leading to delays in treatment.4 Although the infection is usually self-limiting without treatment, the risk for prolonged active disease may occur, with 1 reported case lasting up to 18 months.4,5 The first indicator of CLM is intense pruritus localized to the site of infection.4 As the larvae migrate or creep, they create a lesion that may appear edematous with vesiculobullous lesions that are either serpiginous or linear.4 The differential diagnosis may include fungal infection, bacterial infection, and atypical herpes simplex infections; however, the key finding in CLM is the presence of undulating tracts localized to the borders of the lesion.2 Patients may report experiencing a stinging sensation prior to the formation of the erythematous scaly papule,5 which is attributed to the initial penetration of the larva into the skin. This development, accompanied with a history of travel to tropical or subtropical regions, should elicit CLM as a likely diagnosis. Because hookworms are a type of helminth, they likely elicit an eosinophilic immune response and thus peripheral eosinophilia may be present.5
Effective treatment of CLM is accomplished with oral albendazole 400 mg once daily for 3 to 7 days.2,7 Alternatively, oral ivermectin, topical thiabendazole, and cryosurgery can be used,2 though albendazole currently is the preferred treatment of CLM.7
- Hotez PJ, Brooker S, Bethony JM, et al. Hookworm infection. N Engl J Med. 2004;351:799-807.
- Roest MA, Ratnavel R. Cutaneous larva migrans contracted in England: a reminder. Clin Exp Dermatol. 2001;26:389-390.
- Blackwell V, Vega-Lopez F. Cutaneous larva migrans: clinical features and management of 44 cases presenting in the returning traveller. Br J Dermatol. 2001;145:434-437.
- Hochedez P, Caumes E. Hookworm-related cutaneous larva migrans. J Travel Med. 2007;14:326-333.
- Bravo F, Sanchez MR. New and re-emerging cutaneous infectious diseases in Latin America and other geographic areas. Dermatol Clin. 2003;21:655-668, viii.
- Guill MA, Odom RB. Larva migrans complicated by Loeffler’s syndrome. Arch Dermatol. 1978;114:1525-1526.
- Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814.
- Hotez PJ, Brooker S, Bethony JM, et al. Hookworm infection. N Engl J Med. 2004;351:799-807.
- Roest MA, Ratnavel R. Cutaneous larva migrans contracted in England: a reminder. Clin Exp Dermatol. 2001;26:389-390.
- Blackwell V, Vega-Lopez F. Cutaneous larva migrans: clinical features and management of 44 cases presenting in the returning traveller. Br J Dermatol. 2001;145:434-437.
- Hochedez P, Caumes E. Hookworm-related cutaneous larva migrans. J Travel Med. 2007;14:326-333.
- Bravo F, Sanchez MR. New and re-emerging cutaneous infectious diseases in Latin America and other geographic areas. Dermatol Clin. 2003;21:655-668, viii.
- Guill MA, Odom RB. Larva migrans complicated by Loeffler’s syndrome. Arch Dermatol. 1978;114:1525-1526.
- Caumes E. Treatment of cutaneous larva migrans. Clin Infect Dis. 2000;30:811-814.
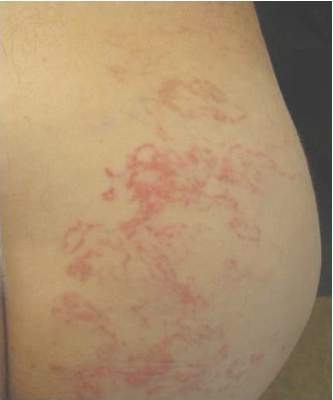
A 62-year-old man presented to the dermatology clinic with a severely pruritic and painful rash of 1 week’s duration. The rash began as an erythematous papule on the right buttock but had spread in a serpiginous manner to the groin and left buttock. The patient stated that he could see the rash spreading in a serpiginous manner over a matter of hours. The patient’s medical history was unremarkable and a review of symptoms was otherwise negative. Physical examination revealed an erythematous serpiginous eruption that was most prominent on the right buttock but extended to the left buttock and down the right leg. He also exhibited several erythematous papules with excoriations in that region.
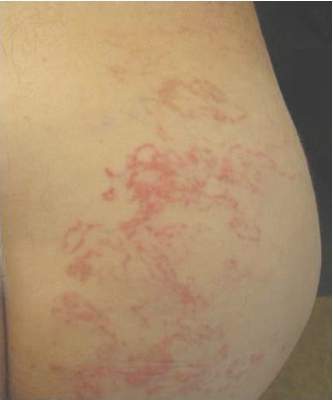
What Is Your Diagnosis? Stinkbug Staining
The Diagnosis: Stinkbug Staining
After discussing management options with the patient including biopsy, we decided that we would photograph the lesion and follow-up in clinic. While dressing, the patient discovered the source of the pigment, a stinkbug, stuck to the corresponding area of the sock.
The brown marmorated stinkbug (Halyomorpha halys)(Figure) is a member of the Pentatomidae family. This insect is native to East Asia and has become an invasive species in the United States. Their presence has recently increased in the eastern United States and they have become an important agricultural pest as well as a household nuisance. Stinkbugs most commonly interact with humans during the fall and winter months when they enter homes because of cooler temperatures outdoors. They can fit into many unexpected places because of their thin profile.1

Stinkbugs earned their name because of their defensive release of a malodorous chemical. This chemical is comprised of trans-2-decenal and trans-2-octenal, which are both aldehydes and are chemically related to formaldehyde. Based on the material safety data sheet, trans-2-decenal also may be responsible for the orange-brown color seen on the patient’s skin.2 Contact dermatitis caused by direct excretion of this chemical onto human skin has been reported3; anecdotal reports of irritation in agricultural workers have been noted. Stinkbugs are becoming a more common household and agricultural pest and should be recognized as possible causes of some presentations in the dermatology clinic.
- Nielsen AL, Hamilton GC. Seasonal occurrence and impact of Halyomorpha halys (Hemiptera: Pentatomidae) in tree fruit. J Econ Entomol. 2009;102:1133-1140.
- Material safety data sheet: trans-2-Decenal. https://fscimage.fishersci.com/msds/45077.htm. Published October 24, 1998. Updated November 20, 2008. Accessed January 11, 2016.
- Anderson BE, Miller JJ, Adams DR. Irritant contact dermatitis to the brown marmorated stink bug, Halyomorpha halys. Dermatitis. 2012;23:170-172.
The Diagnosis: Stinkbug Staining
After discussing management options with the patient including biopsy, we decided that we would photograph the lesion and follow-up in clinic. While dressing, the patient discovered the source of the pigment, a stinkbug, stuck to the corresponding area of the sock.
The brown marmorated stinkbug (Halyomorpha halys)(Figure) is a member of the Pentatomidae family. This insect is native to East Asia and has become an invasive species in the United States. Their presence has recently increased in the eastern United States and they have become an important agricultural pest as well as a household nuisance. Stinkbugs most commonly interact with humans during the fall and winter months when they enter homes because of cooler temperatures outdoors. They can fit into many unexpected places because of their thin profile.1
Stinkbugs earned their name because of their defensive release of a malodorous chemical. This chemical is comprised of trans-2-decenal and trans-2-octenal, which are both aldehydes and are chemically related to formaldehyde. Based on the material safety data sheet, trans-2-decenal also may be responsible for the orange-brown color seen on the patient’s skin.2 Contact dermatitis caused by direct excretion of this chemical onto human skin has been reported3; anecdotal reports of irritation in agricultural workers have been noted. Stinkbugs are becoming a more common household and agricultural pest and should be recognized as possible causes of some presentations in the dermatology clinic.
The Diagnosis: Stinkbug Staining
After discussing management options with the patient including biopsy, we decided that we would photograph the lesion and follow-up in clinic. While dressing, the patient discovered the source of the pigment, a stinkbug, stuck to the corresponding area of the sock.
The brown marmorated stinkbug (Halyomorpha halys)(Figure) is a member of the Pentatomidae family. This insect is native to East Asia and has become an invasive species in the United States. Their presence has recently increased in the eastern United States and they have become an important agricultural pest as well as a household nuisance. Stinkbugs most commonly interact with humans during the fall and winter months when they enter homes because of cooler temperatures outdoors. They can fit into many unexpected places because of their thin profile.1
Stinkbugs earned their name because of their defensive release of a malodorous chemical. This chemical is comprised of trans-2-decenal and trans-2-octenal, which are both aldehydes and are chemically related to formaldehyde. Based on the material safety data sheet, trans-2-decenal also may be responsible for the orange-brown color seen on the patient’s skin.2 Contact dermatitis caused by direct excretion of this chemical onto human skin has been reported3; anecdotal reports of irritation in agricultural workers have been noted. Stinkbugs are becoming a more common household and agricultural pest and should be recognized as possible causes of some presentations in the dermatology clinic.
- Nielsen AL, Hamilton GC. Seasonal occurrence and impact of Halyomorpha halys (Hemiptera: Pentatomidae) in tree fruit. J Econ Entomol. 2009;102:1133-1140.
- Material safety data sheet: trans-2-Decenal. https://fscimage.fishersci.com/msds/45077.htm. Published October 24, 1998. Updated November 20, 2008. Accessed January 11, 2016.
- Anderson BE, Miller JJ, Adams DR. Irritant contact dermatitis to the brown marmorated stink bug, Halyomorpha halys. Dermatitis. 2012;23:170-172.
- Nielsen AL, Hamilton GC. Seasonal occurrence and impact of Halyomorpha halys (Hemiptera: Pentatomidae) in tree fruit. J Econ Entomol. 2009;102:1133-1140.
- Material safety data sheet: trans-2-Decenal. https://fscimage.fishersci.com/msds/45077.htm. Published October 24, 1998. Updated November 20, 2008. Accessed January 11, 2016.
- Anderson BE, Miller JJ, Adams DR. Irritant contact dermatitis to the brown marmorated stink bug, Halyomorpha halys. Dermatitis. 2012;23:170-172.
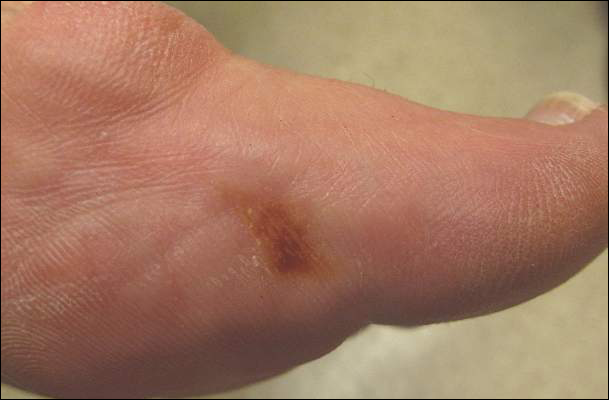
A 56-year-old woman presented at the clinic for a total-body skin examination. A pigmented lesion was found on the medial aspect of the left first toe during the examination. The patient did not recognize this spot as a long-standing nevus. The area was scrubbed vigorously with an alcohol swab, which did not change the pigment. Clinically the lesion was concerning for an atypical nevus. Dermoscopic examination showed an unusual pattern with pigment deposition in ridges and on furrows.