User login
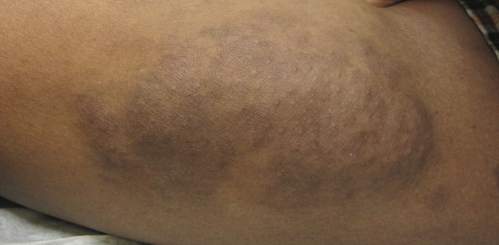
Erythematous Eruption on the Left Leg
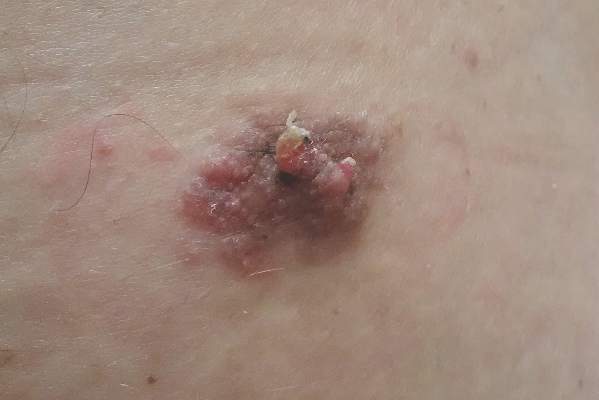
The Diagnosis: Bullous Henoch-Schönlein Purpura
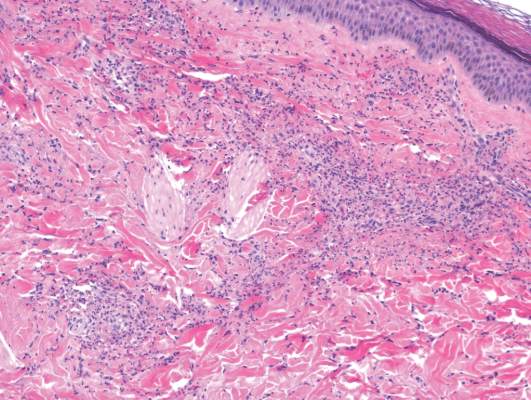
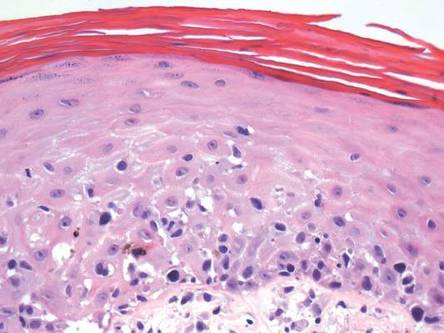
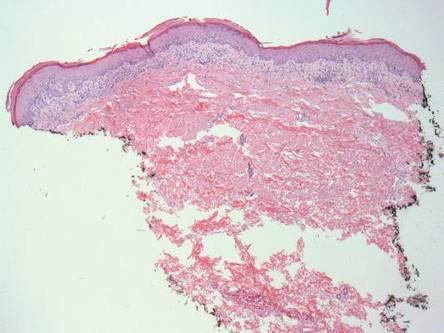
Laboratory tests in this patient showed no abnormalities for complete blood cell count, immunoglobulins, anti–double-stranded DNA, antinuclear antibody, p–antineutrophil cytoplasmic antibodies, lupus anticoagulant, Sjögren antibodies, liver enzymes, and erythrocyte sedimentation rate. Urinalysis was normal. Punch biopsies were obtained and a histologic examination showed an intense inflammatory infiltrate of neutrophils around blood vessels within the dermis (Figure). These blood vessels showed swollen endothelium and narrowing of the vessel lumina with leukocytoclasia. Direct immunofluorescence revealed granular IgA, C3, fibrin, and weak IgM deposits in blood vessels in the papillary dermis consistent with Henoch-Schönlein purpura (HSP).
Henoch-Schönlein purpura is the most common vasculitis in children.1-6 However, its bullous variant is rare, with few pediatric cases reported. Bullous HSP affects arterioles through an IgA-mediated pathway.1-6 It is believed that the bullae are formed secondary to neutrophilic release of matrix metalloproteinase 9 (MMP-9), which degrades extracellular collagen.2 Additionally, bullous fluid from HSP has been noted to have markedly elevated levels of soluble CD23, a form of the CD23 B-cell surface receptor used in antibody feedback regulation and B-cell recruitment, which also has been found to be elevated in the fluid of bullous pemphigoid, suggesting a similar pathogenesis of exaggerated humoral immunity.3
The most common sign of HSP is palpable purpura; however, other cutaneous findings can be present including targetoid plaques, macules, papules, petechiae, and bullae that may become hemorrhagic, ulcerated, necrotic, or scarred.1-6 Bullae appear in the most dependent parts of the body, such as the feet and lower legs. Hydrostatic pressure may play a role in the pathogenesis of this phenomenon.1 When other classic signs of HSP are absent, the presence of bullae clouds the diagnosis and creates controversy regarding treatment, as there is a dearth of literature on proper therapy for severe cutaneous manifestations of HSP.6
Our patient was treated with morphine for pain management along with topical mupirocin and nonadherent dressings for wound care. She also received pulse intravenous methylprednisolone 2 mg/kg daily for 3 days and then was transitioned to oral prednisone 1 mg/kg daily, which was tapered over 3 weeks after discharge. This regimen resulted in resolution of symptoms with rapid regression of bullae and subsequent postinflammatory hyperpigmentation. Prior reports have noted that the presence of bullae does not alter the prognosis or predict probability of renal involvement of this self-limited disease, leading to controversy in determining if treatment offers more favorable outcomes.1,3 One study suggested that steroids only improve symptoms, arthralgia, and abdominal pain, but they do not aid in the resolution of cutaneous lesions or prevent the progression of renal disease.3 Contrarily, others have suggested that the presence of bullae and renal disease is an indication to start treatment.6 This claim is based on the mechanistic finding that immunosuppression with corticosteroids decreases inflammation by inhibiting activator protein 1, a transcription factor for MMP-9, thereby reducing MMP-9 activity and the formation of bullae.4 Clinical anecdotes, including our own, that demonstrate dramatic improvement of hemorrhagic bullae with the administration of corticosteroids substantiate this mechanism. Through the inhibition of neutrophil interactions and IgA production, other anti-inflammatory and immunosuppressive medications such as colchicine, dapsone, and azathioprine also have been reported to aid in resolution of the cutaneous lesions.1,5,6 Although there is a clear drawback to the lack of controlled trials and prospective studies regarding the treatment of bullous HSP, it is nearly impossible to expect such studies to be carried out given the rare and unpredictable nature of the disease. For now, claims derived from case series and case reports guide our understanding of treatment efficacy.
Acknowledgment—Quiz photograph courtesy of Steve Taylor, BS, Phoenix, Arizona.
- Trapani S, Mariotti P, Resti M, et al. Severe hemorrhagic bullous lesions in Henoch Schönlein purpura: three pediatric cases and review of the literature [published online July 16, 2009]. Rheumatol Int. 2010;30:1355-1359. doi:10.1007/s00296-009-1055-8.
- Kobayashi T, Sakuraoka K, Iwamoto M, et al. A case of anaphylactoid purpura with multiple blister formation: possible pathophysiologic role of gelatinate (MMP-9). Dermatology. 1990;197:62-64.
- Bansal AS, Dwivedi N, Adsett M. Serum and blister fluid cytokines and complement proteins in a patient with Henoch Schönlein purpura associated with a bullous skin rash. Australas J Dermatol. 1997;38:190-192.
- Aljada A, Ghanim H, Mohanty P, et al. Hydrocortisone suppresses intranuclear activator-protein-1 (AP-1) binding activity in mononuclear cells and plasma matrix metalloproteinase 2 and 9 (MMP-2 and MMP-9). J Clin Endocrinol Metab. 2001;86:5988-5991.
- Iqbal H, Evans A. Dapsone therapy for Henoch-Schönlein purpura: a case series. Arch Dis Child. 2005;90:985-986.
- den Boer SL, Pasmans SG, Wulffraat NM, et al. Bullous lesions in Henoch Schönlein purpura as indication to start systemic prednisone [published online January 5, 2009]. Acta Paediatr. 2010;99:781-783. doi:10.1111/j.1651-2227.2009.01650.x.
The Diagnosis: Bullous Henoch-Schönlein Purpura
Laboratory tests in this patient showed no abnormalities for complete blood cell count, immunoglobulins, anti–double-stranded DNA, antinuclear antibody, p–antineutrophil cytoplasmic antibodies, lupus anticoagulant, Sjögren antibodies, liver enzymes, and erythrocyte sedimentation rate. Urinalysis was normal. Punch biopsies were obtained and a histologic examination showed an intense inflammatory infiltrate of neutrophils around blood vessels within the dermis (Figure). These blood vessels showed swollen endothelium and narrowing of the vessel lumina with leukocytoclasia. Direct immunofluorescence revealed granular IgA, C3, fibrin, and weak IgM deposits in blood vessels in the papillary dermis consistent with Henoch-Schönlein purpura (HSP).
Henoch-Schönlein purpura is the most common vasculitis in children.1-6 However, its bullous variant is rare, with few pediatric cases reported. Bullous HSP affects arterioles through an IgA-mediated pathway.1-6 It is believed that the bullae are formed secondary to neutrophilic release of matrix metalloproteinase 9 (MMP-9), which degrades extracellular collagen.2 Additionally, bullous fluid from HSP has been noted to have markedly elevated levels of soluble CD23, a form of the CD23 B-cell surface receptor used in antibody feedback regulation and B-cell recruitment, which also has been found to be elevated in the fluid of bullous pemphigoid, suggesting a similar pathogenesis of exaggerated humoral immunity.3
The most common sign of HSP is palpable purpura; however, other cutaneous findings can be present including targetoid plaques, macules, papules, petechiae, and bullae that may become hemorrhagic, ulcerated, necrotic, or scarred.1-6 Bullae appear in the most dependent parts of the body, such as the feet and lower legs. Hydrostatic pressure may play a role in the pathogenesis of this phenomenon.1 When other classic signs of HSP are absent, the presence of bullae clouds the diagnosis and creates controversy regarding treatment, as there is a dearth of literature on proper therapy for severe cutaneous manifestations of HSP.6
Our patient was treated with morphine for pain management along with topical mupirocin and nonadherent dressings for wound care. She also received pulse intravenous methylprednisolone 2 mg/kg daily for 3 days and then was transitioned to oral prednisone 1 mg/kg daily, which was tapered over 3 weeks after discharge. This regimen resulted in resolution of symptoms with rapid regression of bullae and subsequent postinflammatory hyperpigmentation. Prior reports have noted that the presence of bullae does not alter the prognosis or predict probability of renal involvement of this self-limited disease, leading to controversy in determining if treatment offers more favorable outcomes.1,3 One study suggested that steroids only improve symptoms, arthralgia, and abdominal pain, but they do not aid in the resolution of cutaneous lesions or prevent the progression of renal disease.3 Contrarily, others have suggested that the presence of bullae and renal disease is an indication to start treatment.6 This claim is based on the mechanistic finding that immunosuppression with corticosteroids decreases inflammation by inhibiting activator protein 1, a transcription factor for MMP-9, thereby reducing MMP-9 activity and the formation of bullae.4 Clinical anecdotes, including our own, that demonstrate dramatic improvement of hemorrhagic bullae with the administration of corticosteroids substantiate this mechanism. Through the inhibition of neutrophil interactions and IgA production, other anti-inflammatory and immunosuppressive medications such as colchicine, dapsone, and azathioprine also have been reported to aid in resolution of the cutaneous lesions.1,5,6 Although there is a clear drawback to the lack of controlled trials and prospective studies regarding the treatment of bullous HSP, it is nearly impossible to expect such studies to be carried out given the rare and unpredictable nature of the disease. For now, claims derived from case series and case reports guide our understanding of treatment efficacy.
Acknowledgment—Quiz photograph courtesy of Steve Taylor, BS, Phoenix, Arizona.
The Diagnosis: Bullous Henoch-Schönlein Purpura
Laboratory tests in this patient showed no abnormalities for complete blood cell count, immunoglobulins, anti–double-stranded DNA, antinuclear antibody, p–antineutrophil cytoplasmic antibodies, lupus anticoagulant, Sjögren antibodies, liver enzymes, and erythrocyte sedimentation rate. Urinalysis was normal. Punch biopsies were obtained and a histologic examination showed an intense inflammatory infiltrate of neutrophils around blood vessels within the dermis (Figure). These blood vessels showed swollen endothelium and narrowing of the vessel lumina with leukocytoclasia. Direct immunofluorescence revealed granular IgA, C3, fibrin, and weak IgM deposits in blood vessels in the papillary dermis consistent with Henoch-Schönlein purpura (HSP).
Henoch-Schönlein purpura is the most common vasculitis in children.1-6 However, its bullous variant is rare, with few pediatric cases reported. Bullous HSP affects arterioles through an IgA-mediated pathway.1-6 It is believed that the bullae are formed secondary to neutrophilic release of matrix metalloproteinase 9 (MMP-9), which degrades extracellular collagen.2 Additionally, bullous fluid from HSP has been noted to have markedly elevated levels of soluble CD23, a form of the CD23 B-cell surface receptor used in antibody feedback regulation and B-cell recruitment, which also has been found to be elevated in the fluid of bullous pemphigoid, suggesting a similar pathogenesis of exaggerated humoral immunity.3
The most common sign of HSP is palpable purpura; however, other cutaneous findings can be present including targetoid plaques, macules, papules, petechiae, and bullae that may become hemorrhagic, ulcerated, necrotic, or scarred.1-6 Bullae appear in the most dependent parts of the body, such as the feet and lower legs. Hydrostatic pressure may play a role in the pathogenesis of this phenomenon.1 When other classic signs of HSP are absent, the presence of bullae clouds the diagnosis and creates controversy regarding treatment, as there is a dearth of literature on proper therapy for severe cutaneous manifestations of HSP.6
Our patient was treated with morphine for pain management along with topical mupirocin and nonadherent dressings for wound care. She also received pulse intravenous methylprednisolone 2 mg/kg daily for 3 days and then was transitioned to oral prednisone 1 mg/kg daily, which was tapered over 3 weeks after discharge. This regimen resulted in resolution of symptoms with rapid regression of bullae and subsequent postinflammatory hyperpigmentation. Prior reports have noted that the presence of bullae does not alter the prognosis or predict probability of renal involvement of this self-limited disease, leading to controversy in determining if treatment offers more favorable outcomes.1,3 One study suggested that steroids only improve symptoms, arthralgia, and abdominal pain, but they do not aid in the resolution of cutaneous lesions or prevent the progression of renal disease.3 Contrarily, others have suggested that the presence of bullae and renal disease is an indication to start treatment.6 This claim is based on the mechanistic finding that immunosuppression with corticosteroids decreases inflammation by inhibiting activator protein 1, a transcription factor for MMP-9, thereby reducing MMP-9 activity and the formation of bullae.4 Clinical anecdotes, including our own, that demonstrate dramatic improvement of hemorrhagic bullae with the administration of corticosteroids substantiate this mechanism. Through the inhibition of neutrophil interactions and IgA production, other anti-inflammatory and immunosuppressive medications such as colchicine, dapsone, and azathioprine also have been reported to aid in resolution of the cutaneous lesions.1,5,6 Although there is a clear drawback to the lack of controlled trials and prospective studies regarding the treatment of bullous HSP, it is nearly impossible to expect such studies to be carried out given the rare and unpredictable nature of the disease. For now, claims derived from case series and case reports guide our understanding of treatment efficacy.
Acknowledgment—Quiz photograph courtesy of Steve Taylor, BS, Phoenix, Arizona.
- Trapani S, Mariotti P, Resti M, et al. Severe hemorrhagic bullous lesions in Henoch Schönlein purpura: three pediatric cases and review of the literature [published online July 16, 2009]. Rheumatol Int. 2010;30:1355-1359. doi:10.1007/s00296-009-1055-8.
- Kobayashi T, Sakuraoka K, Iwamoto M, et al. A case of anaphylactoid purpura with multiple blister formation: possible pathophysiologic role of gelatinate (MMP-9). Dermatology. 1990;197:62-64.
- Bansal AS, Dwivedi N, Adsett M. Serum and blister fluid cytokines and complement proteins in a patient with Henoch Schönlein purpura associated with a bullous skin rash. Australas J Dermatol. 1997;38:190-192.
- Aljada A, Ghanim H, Mohanty P, et al. Hydrocortisone suppresses intranuclear activator-protein-1 (AP-1) binding activity in mononuclear cells and plasma matrix metalloproteinase 2 and 9 (MMP-2 and MMP-9). J Clin Endocrinol Metab. 2001;86:5988-5991.
- Iqbal H, Evans A. Dapsone therapy for Henoch-Schönlein purpura: a case series. Arch Dis Child. 2005;90:985-986.
- den Boer SL, Pasmans SG, Wulffraat NM, et al. Bullous lesions in Henoch Schönlein purpura as indication to start systemic prednisone [published online January 5, 2009]. Acta Paediatr. 2010;99:781-783. doi:10.1111/j.1651-2227.2009.01650.x.
- Trapani S, Mariotti P, Resti M, et al. Severe hemorrhagic bullous lesions in Henoch Schönlein purpura: three pediatric cases and review of the literature [published online July 16, 2009]. Rheumatol Int. 2010;30:1355-1359. doi:10.1007/s00296-009-1055-8.
- Kobayashi T, Sakuraoka K, Iwamoto M, et al. A case of anaphylactoid purpura with multiple blister formation: possible pathophysiologic role of gelatinate (MMP-9). Dermatology. 1990;197:62-64.
- Bansal AS, Dwivedi N, Adsett M. Serum and blister fluid cytokines and complement proteins in a patient with Henoch Schönlein purpura associated with a bullous skin rash. Australas J Dermatol. 1997;38:190-192.
- Aljada A, Ghanim H, Mohanty P, et al. Hydrocortisone suppresses intranuclear activator-protein-1 (AP-1) binding activity in mononuclear cells and plasma matrix metalloproteinase 2 and 9 (MMP-2 and MMP-9). J Clin Endocrinol Metab. 2001;86:5988-5991.
- Iqbal H, Evans A. Dapsone therapy for Henoch-Schönlein purpura: a case series. Arch Dis Child. 2005;90:985-986.
- den Boer SL, Pasmans SG, Wulffraat NM, et al. Bullous lesions in Henoch Schönlein purpura as indication to start systemic prednisone [published online January 5, 2009]. Acta Paediatr. 2010;99:781-783. doi:10.1111/j.1651-2227.2009.01650.x.

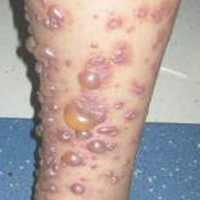
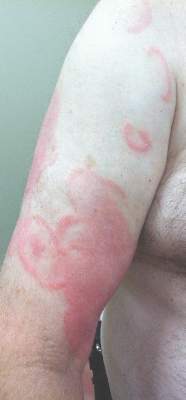
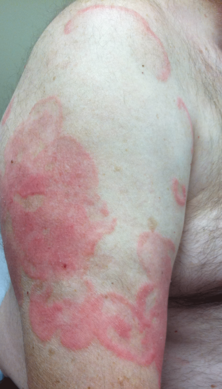
A 12-year-old girl presented with an erythematous eruption that had started on the left leg approximately 1 week prior with subsequent spread to the abdomen and arms. She had associated knee pain, myalgia, abdominal pain, nausea, and nonbloody and nonbilious emesis. Her medical history was notable for methicillin-resistant Staphylococcus aureus abscesses, the most recent of which was treated with trimethoprim-sulfamethoxazole; treatment was completed 5 days before the onset of the rash. Family history was notable for her paternal aunt who died of systemic lupus erythematosus. Physical examination showed erythematous macules and purpuric papules with central vesiculation extending up the thighs and lower abdomen associated with edema of the lower extremities and pain after palpation. Tense bullae also were present.
Subcutaneous Tortuous Nodules on the Posterior Lower Extremity
The Diagnosis: Plexiform Neurofibroma as a Manifestation of Neurofibromatosis Type I
Physical examination revealed a large 10×8-cm subcutaneous nodule that was boggy and resembled a bag of worms on palpation. It was covered by slightly hyperpigmented skin. He also had numerous (>20) café au lait spots measuring 2 to 3 cm across the body and several others on the axillae. There were no gross eye findings. Otherwise the examination was unremarkable on the rest of the body. The patient’s paternal grandfather and aunt had similar macules and multiple nodules. The patient had mild to moderate learning difficulties. He was subsequently referred for genetic and ophthalmology evaluation.
Plexiform neurofibromas are usually benign nerve sheath tumors that are elongated and are multinodular, forming when the tumor involves either multiple trunks of a plexus or multiple fascicles of a large nerve such as the sciatic. Some plexiform neurofibromas resemble a bag of worms; others produce a massive ropy enlargement of the nerve.1,2 Plexiform neurofibromas are associated with cases of neurofibromatosis type I (NFI) and are themselves one of the diagnostic criteria for NFI.1
Plexiform neurofibromas are benign tumors that are the result of a genetic mutation in which loss of heterozygosity occurs, as is the case with the other predominant neoplasms of NFI, that results in unrestricted cell growth.3,4 Some patients have a loss of heterozygosity of this tumor suppression gene with overgrowth of neurofibromatosis on a Blaschko segment. One study in mouse models showed that stromal mast cells were involved in promoting inflammation and increasing tumor growth by mediation of mitogenic signals involved in vascular ingrowth, collagen deposition, and cellular proliferation.5 Plexiform neurofibromas are a presenting feature in 30% of NFI cases within the first year of life. They are extensive nerve sheath tumors with an unpredictable growth pattern that can involve multiple fascicles (ie, large nerves and their branches). Five percent become malignant and the transformation is often heralded by rapid growth and pain.6 If malignant transformation is suspected, biopsy is diagnostic. Magnetic resonance imaging with and without contrast can categorize them into 3 growth categories: superficial, displacing, and invasive.7 Because plexiform neurofibromas are rare tumors, it previously was common practice to delay surgical intervention until disfigurement or disability arose. Complete surgical resection at more advanced stages is nearly impossible given the networklike growth pattern that commonly encapsulates vital structures.8,9 Therefore, surgery has been used in the past for debulking the large growths that eventually will recur. A study of 9 small superficial plexiform neurofibromas in children aged 3 to 15 years documented treatment with early surgical resection, which showed complete resection and no relapse at 4 years. This study showed a promising strategy to prevent future extension of these fast-growing tumors into vital structures.8 There also are current clinical trials investigating sirolimus and peginterferon alfa-2b in patients with more invasive plexiform neurofibromas that are unable to undergo surgical resection due to encapsulation or proximity to essential anatomical structures (registered at www.clinicaltrials.gov with the identifiers NCT00652990 and NCT00678951, respectively).
Pain, development of a neurologic deficit, or enlargement of a preexisting plexiform neurofibroma may signal a malignant peripheral nerve sheath tumor (MPNST) and require immediate evaluation.10 Examination by magnetic resonance imaging and positron emission tomography is useful in distinguishing benign and MPNSTs,8,11,12 but definitive differentiation can only be made by histologic examination of the tumor. Complete surgical excision, when possible, is the only treatment that offers the possibility of cure of MPNSTs. Adjuvant chemotherapy or radiotherapy also is sometimes used, though benefit has not been clearly established.8,9,13,14
Death certificate and population-based studies have shown that approximately 10% of patients with NFI have a reduced life expectancy due to MPNSTs; indeed, these tumors arising from plexiform neurofibromas are the main cause of death in adults with NFI. In 2003, Mautner et al7 studied 50 individuals with NFI. The objective was to establish magnetic resonance imaging criteria for MPNST and to test their usefulness in detecting early malignant change in plexiform neurofibromas. This study found that MPNST in patients with NFI frequently showed inhomogeneous contrast enhancement. This inhomogeneity was due to necrosis and hemorrhage, as shown by macroscopic and histologic analysis of amputated limbs in 2 patients within the study. The investigators found it to be possible to detect malignant transformation at an early stage in patients with no overt clinical signs of progression.7 Careful follow-up will determine how frequently early malignancy can be detected and if it is worthwhile carrying out magnetic resonance imaging at defined intervals.2,7,10,15,16
- Friedman JM. Neurofibromatosis 1. In: Pagon RA, ed. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1109/. Updated September 4, 2014. Accessed April 6, 2015.
- Friedman JM, Riccardi VM. Clinical epidemiological features. In: Friedman JM, Gutmann DH, MacCollin M, et al, eds. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. Baltimore, MD: Johns Hopkins University Press; 1999:29-86.
- Bausch B, Borozdin W, Mautner VF, et al; European-American Phaeochromocytoma Registry Study Group. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with phaeochromocytoma and neurofibromatosis type 1. J Clin Endocrinol Metab. 2007;92:2784-2792.
- Bottillo I, Ahlquist T, Brekke H, et al. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J Pathol. 2009;217:693-701.
- Staser K, Yang FC, Clapp DW. Pathogenesis of plexiform neurofibroma: tumor-stromal/hematopoietic interactions in tumor progression. Ann Rev Pathol. 2012;7:469-495.
- Murphey MD, Smith WS, Smith SE, et al. From the archives of the AFIP: imaging of musculoskeletal neurogenic tumors: radiologic-pathologic correlation. Radiographics. 1999;19:1253-1280.
- Mautner VF, Friedrich RE, von Deimling A, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis type 1: MRI supports the diagnosis of malignant plexiform neurofibroma. Neuroradiology. 2003;45:618-625.
- Friedrich RE, Schmelzle R, Hartmann M, et al. Resection of small plexiform neurofibromas in neurofibromatosis type 1 children. World J Surg Oncol. 2005;3:6.
- Gottfried ON, Viskochil DH, Fults DW, et al. Molecular, genetic, and cellular pathogenesis of neurofibromas and surgical implications. Neurosurgery. 2006;58:1-16.
- Valeyrie-Allanore L, Ismaïli N, Bastuji-Garin S, et al. Symptoms associated with malignancy of peripheral nerve sheath tumours: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153:79-82.
- Bensaid B, Giammarile F, Mognetti T, et al. Utility of 18 FDG positron emission tomography in detection of sarcomatous transformation in neurofibromatosis type 1. Ann Dermatol Venereol. 2007;134:735-741.
- Ferner RE, Lucas JD, O’Doherty MJ, et al. Evaluation of (18)fluorodeoxyglucose positron emission tomography ((18)FDG PET) in the detection of malignant peripheral nerve sheath tumours arising from within plexiform neurofibromas in neurofibromatosis 1. J Neurol Neurosurg Psychiatry. 2000;68:3353-3357.
- Baujat B, Krastinova-Lolov D, Blumen M, et al. Radiofrequency in the treatment of craniofacial plexiform neurofibromatosis: a pilot study. Plast Reconstr Surg. 2006;117:1261-1268.
- Hummel T, Anyane-Yeboa A, Mo J, et al. Response of NF1-related plexiform neurofibroma to high-dose carboplatin. Pediatr Blood Cancer. 2011;56:488-490.
- Feldmann R, Schuierer G, Wessel A, et al. Development of MRI T2 hyperintensities and cognitive functioning in patients with neurofibromatosis type 1. Acta Paediatr. 2010;99:1657-1660.
- Blazo MA, Lewis RA, Chintagumpala MM, et al. Outcomes of systematic screening for optic pathway tumors in children with neurofibromatosis type 1. Am J Med Genet A. 2004;127A:224-229.
The Diagnosis: Plexiform Neurofibroma as a Manifestation of Neurofibromatosis Type I
Physical examination revealed a large 10×8-cm subcutaneous nodule that was boggy and resembled a bag of worms on palpation. It was covered by slightly hyperpigmented skin. He also had numerous (>20) café au lait spots measuring 2 to 3 cm across the body and several others on the axillae. There were no gross eye findings. Otherwise the examination was unremarkable on the rest of the body. The patient’s paternal grandfather and aunt had similar macules and multiple nodules. The patient had mild to moderate learning difficulties. He was subsequently referred for genetic and ophthalmology evaluation.
Plexiform neurofibromas are usually benign nerve sheath tumors that are elongated and are multinodular, forming when the tumor involves either multiple trunks of a plexus or multiple fascicles of a large nerve such as the sciatic. Some plexiform neurofibromas resemble a bag of worms; others produce a massive ropy enlargement of the nerve.1,2 Plexiform neurofibromas are associated with cases of neurofibromatosis type I (NFI) and are themselves one of the diagnostic criteria for NFI.1
Plexiform neurofibromas are benign tumors that are the result of a genetic mutation in which loss of heterozygosity occurs, as is the case with the other predominant neoplasms of NFI, that results in unrestricted cell growth.3,4 Some patients have a loss of heterozygosity of this tumor suppression gene with overgrowth of neurofibromatosis on a Blaschko segment. One study in mouse models showed that stromal mast cells were involved in promoting inflammation and increasing tumor growth by mediation of mitogenic signals involved in vascular ingrowth, collagen deposition, and cellular proliferation.5 Plexiform neurofibromas are a presenting feature in 30% of NFI cases within the first year of life. They are extensive nerve sheath tumors with an unpredictable growth pattern that can involve multiple fascicles (ie, large nerves and their branches). Five percent become malignant and the transformation is often heralded by rapid growth and pain.6 If malignant transformation is suspected, biopsy is diagnostic. Magnetic resonance imaging with and without contrast can categorize them into 3 growth categories: superficial, displacing, and invasive.7 Because plexiform neurofibromas are rare tumors, it previously was common practice to delay surgical intervention until disfigurement or disability arose. Complete surgical resection at more advanced stages is nearly impossible given the networklike growth pattern that commonly encapsulates vital structures.8,9 Therefore, surgery has been used in the past for debulking the large growths that eventually will recur. A study of 9 small superficial plexiform neurofibromas in children aged 3 to 15 years documented treatment with early surgical resection, which showed complete resection and no relapse at 4 years. This study showed a promising strategy to prevent future extension of these fast-growing tumors into vital structures.8 There also are current clinical trials investigating sirolimus and peginterferon alfa-2b in patients with more invasive plexiform neurofibromas that are unable to undergo surgical resection due to encapsulation or proximity to essential anatomical structures (registered at www.clinicaltrials.gov with the identifiers NCT00652990 and NCT00678951, respectively).
Pain, development of a neurologic deficit, or enlargement of a preexisting plexiform neurofibroma may signal a malignant peripheral nerve sheath tumor (MPNST) and require immediate evaluation.10 Examination by magnetic resonance imaging and positron emission tomography is useful in distinguishing benign and MPNSTs,8,11,12 but definitive differentiation can only be made by histologic examination of the tumor. Complete surgical excision, when possible, is the only treatment that offers the possibility of cure of MPNSTs. Adjuvant chemotherapy or radiotherapy also is sometimes used, though benefit has not been clearly established.8,9,13,14
Death certificate and population-based studies have shown that approximately 10% of patients with NFI have a reduced life expectancy due to MPNSTs; indeed, these tumors arising from plexiform neurofibromas are the main cause of death in adults with NFI. In 2003, Mautner et al7 studied 50 individuals with NFI. The objective was to establish magnetic resonance imaging criteria for MPNST and to test their usefulness in detecting early malignant change in plexiform neurofibromas. This study found that MPNST in patients with NFI frequently showed inhomogeneous contrast enhancement. This inhomogeneity was due to necrosis and hemorrhage, as shown by macroscopic and histologic analysis of amputated limbs in 2 patients within the study. The investigators found it to be possible to detect malignant transformation at an early stage in patients with no overt clinical signs of progression.7 Careful follow-up will determine how frequently early malignancy can be detected and if it is worthwhile carrying out magnetic resonance imaging at defined intervals.2,7,10,15,16
The Diagnosis: Plexiform Neurofibroma as a Manifestation of Neurofibromatosis Type I
Physical examination revealed a large 10×8-cm subcutaneous nodule that was boggy and resembled a bag of worms on palpation. It was covered by slightly hyperpigmented skin. He also had numerous (>20) café au lait spots measuring 2 to 3 cm across the body and several others on the axillae. There were no gross eye findings. Otherwise the examination was unremarkable on the rest of the body. The patient’s paternal grandfather and aunt had similar macules and multiple nodules. The patient had mild to moderate learning difficulties. He was subsequently referred for genetic and ophthalmology evaluation.
Plexiform neurofibromas are usually benign nerve sheath tumors that are elongated and are multinodular, forming when the tumor involves either multiple trunks of a plexus or multiple fascicles of a large nerve such as the sciatic. Some plexiform neurofibromas resemble a bag of worms; others produce a massive ropy enlargement of the nerve.1,2 Plexiform neurofibromas are associated with cases of neurofibromatosis type I (NFI) and are themselves one of the diagnostic criteria for NFI.1
Plexiform neurofibromas are benign tumors that are the result of a genetic mutation in which loss of heterozygosity occurs, as is the case with the other predominant neoplasms of NFI, that results in unrestricted cell growth.3,4 Some patients have a loss of heterozygosity of this tumor suppression gene with overgrowth of neurofibromatosis on a Blaschko segment. One study in mouse models showed that stromal mast cells were involved in promoting inflammation and increasing tumor growth by mediation of mitogenic signals involved in vascular ingrowth, collagen deposition, and cellular proliferation.5 Plexiform neurofibromas are a presenting feature in 30% of NFI cases within the first year of life. They are extensive nerve sheath tumors with an unpredictable growth pattern that can involve multiple fascicles (ie, large nerves and their branches). Five percent become malignant and the transformation is often heralded by rapid growth and pain.6 If malignant transformation is suspected, biopsy is diagnostic. Magnetic resonance imaging with and without contrast can categorize them into 3 growth categories: superficial, displacing, and invasive.7 Because plexiform neurofibromas are rare tumors, it previously was common practice to delay surgical intervention until disfigurement or disability arose. Complete surgical resection at more advanced stages is nearly impossible given the networklike growth pattern that commonly encapsulates vital structures.8,9 Therefore, surgery has been used in the past for debulking the large growths that eventually will recur. A study of 9 small superficial plexiform neurofibromas in children aged 3 to 15 years documented treatment with early surgical resection, which showed complete resection and no relapse at 4 years. This study showed a promising strategy to prevent future extension of these fast-growing tumors into vital structures.8 There also are current clinical trials investigating sirolimus and peginterferon alfa-2b in patients with more invasive plexiform neurofibromas that are unable to undergo surgical resection due to encapsulation or proximity to essential anatomical structures (registered at www.clinicaltrials.gov with the identifiers NCT00652990 and NCT00678951, respectively).
Pain, development of a neurologic deficit, or enlargement of a preexisting plexiform neurofibroma may signal a malignant peripheral nerve sheath tumor (MPNST) and require immediate evaluation.10 Examination by magnetic resonance imaging and positron emission tomography is useful in distinguishing benign and MPNSTs,8,11,12 but definitive differentiation can only be made by histologic examination of the tumor. Complete surgical excision, when possible, is the only treatment that offers the possibility of cure of MPNSTs. Adjuvant chemotherapy or radiotherapy also is sometimes used, though benefit has not been clearly established.8,9,13,14
Death certificate and population-based studies have shown that approximately 10% of patients with NFI have a reduced life expectancy due to MPNSTs; indeed, these tumors arising from plexiform neurofibromas are the main cause of death in adults with NFI. In 2003, Mautner et al7 studied 50 individuals with NFI. The objective was to establish magnetic resonance imaging criteria for MPNST and to test their usefulness in detecting early malignant change in plexiform neurofibromas. This study found that MPNST in patients with NFI frequently showed inhomogeneous contrast enhancement. This inhomogeneity was due to necrosis and hemorrhage, as shown by macroscopic and histologic analysis of amputated limbs in 2 patients within the study. The investigators found it to be possible to detect malignant transformation at an early stage in patients with no overt clinical signs of progression.7 Careful follow-up will determine how frequently early malignancy can be detected and if it is worthwhile carrying out magnetic resonance imaging at defined intervals.2,7,10,15,16
- Friedman JM. Neurofibromatosis 1. In: Pagon RA, ed. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1109/. Updated September 4, 2014. Accessed April 6, 2015.
- Friedman JM, Riccardi VM. Clinical epidemiological features. In: Friedman JM, Gutmann DH, MacCollin M, et al, eds. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. Baltimore, MD: Johns Hopkins University Press; 1999:29-86.
- Bausch B, Borozdin W, Mautner VF, et al; European-American Phaeochromocytoma Registry Study Group. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with phaeochromocytoma and neurofibromatosis type 1. J Clin Endocrinol Metab. 2007;92:2784-2792.
- Bottillo I, Ahlquist T, Brekke H, et al. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J Pathol. 2009;217:693-701.
- Staser K, Yang FC, Clapp DW. Pathogenesis of plexiform neurofibroma: tumor-stromal/hematopoietic interactions in tumor progression. Ann Rev Pathol. 2012;7:469-495.
- Murphey MD, Smith WS, Smith SE, et al. From the archives of the AFIP: imaging of musculoskeletal neurogenic tumors: radiologic-pathologic correlation. Radiographics. 1999;19:1253-1280.
- Mautner VF, Friedrich RE, von Deimling A, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis type 1: MRI supports the diagnosis of malignant plexiform neurofibroma. Neuroradiology. 2003;45:618-625.
- Friedrich RE, Schmelzle R, Hartmann M, et al. Resection of small plexiform neurofibromas in neurofibromatosis type 1 children. World J Surg Oncol. 2005;3:6.
- Gottfried ON, Viskochil DH, Fults DW, et al. Molecular, genetic, and cellular pathogenesis of neurofibromas and surgical implications. Neurosurgery. 2006;58:1-16.
- Valeyrie-Allanore L, Ismaïli N, Bastuji-Garin S, et al. Symptoms associated with malignancy of peripheral nerve sheath tumours: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153:79-82.
- Bensaid B, Giammarile F, Mognetti T, et al. Utility of 18 FDG positron emission tomography in detection of sarcomatous transformation in neurofibromatosis type 1. Ann Dermatol Venereol. 2007;134:735-741.
- Ferner RE, Lucas JD, O’Doherty MJ, et al. Evaluation of (18)fluorodeoxyglucose positron emission tomography ((18)FDG PET) in the detection of malignant peripheral nerve sheath tumours arising from within plexiform neurofibromas in neurofibromatosis 1. J Neurol Neurosurg Psychiatry. 2000;68:3353-3357.
- Baujat B, Krastinova-Lolov D, Blumen M, et al. Radiofrequency in the treatment of craniofacial plexiform neurofibromatosis: a pilot study. Plast Reconstr Surg. 2006;117:1261-1268.
- Hummel T, Anyane-Yeboa A, Mo J, et al. Response of NF1-related plexiform neurofibroma to high-dose carboplatin. Pediatr Blood Cancer. 2011;56:488-490.
- Feldmann R, Schuierer G, Wessel A, et al. Development of MRI T2 hyperintensities and cognitive functioning in patients with neurofibromatosis type 1. Acta Paediatr. 2010;99:1657-1660.
- Blazo MA, Lewis RA, Chintagumpala MM, et al. Outcomes of systematic screening for optic pathway tumors in children with neurofibromatosis type 1. Am J Med Genet A. 2004;127A:224-229.
- Friedman JM. Neurofibromatosis 1. In: Pagon RA, ed. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1109/. Updated September 4, 2014. Accessed April 6, 2015.
- Friedman JM, Riccardi VM. Clinical epidemiological features. In: Friedman JM, Gutmann DH, MacCollin M, et al, eds. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. Baltimore, MD: Johns Hopkins University Press; 1999:29-86.
- Bausch B, Borozdin W, Mautner VF, et al; European-American Phaeochromocytoma Registry Study Group. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with phaeochromocytoma and neurofibromatosis type 1. J Clin Endocrinol Metab. 2007;92:2784-2792.
- Bottillo I, Ahlquist T, Brekke H, et al. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J Pathol. 2009;217:693-701.
- Staser K, Yang FC, Clapp DW. Pathogenesis of plexiform neurofibroma: tumor-stromal/hematopoietic interactions in tumor progression. Ann Rev Pathol. 2012;7:469-495.
- Murphey MD, Smith WS, Smith SE, et al. From the archives of the AFIP: imaging of musculoskeletal neurogenic tumors: radiologic-pathologic correlation. Radiographics. 1999;19:1253-1280.
- Mautner VF, Friedrich RE, von Deimling A, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis type 1: MRI supports the diagnosis of malignant plexiform neurofibroma. Neuroradiology. 2003;45:618-625.
- Friedrich RE, Schmelzle R, Hartmann M, et al. Resection of small plexiform neurofibromas in neurofibromatosis type 1 children. World J Surg Oncol. 2005;3:6.
- Gottfried ON, Viskochil DH, Fults DW, et al. Molecular, genetic, and cellular pathogenesis of neurofibromas and surgical implications. Neurosurgery. 2006;58:1-16.
- Valeyrie-Allanore L, Ismaïli N, Bastuji-Garin S, et al. Symptoms associated with malignancy of peripheral nerve sheath tumours: a retrospective study of 69 patients with neurofibromatosis 1. Br J Dermatol. 2005;153:79-82.
- Bensaid B, Giammarile F, Mognetti T, et al. Utility of 18 FDG positron emission tomography in detection of sarcomatous transformation in neurofibromatosis type 1. Ann Dermatol Venereol. 2007;134:735-741.
- Ferner RE, Lucas JD, O’Doherty MJ, et al. Evaluation of (18)fluorodeoxyglucose positron emission tomography ((18)FDG PET) in the detection of malignant peripheral nerve sheath tumours arising from within plexiform neurofibromas in neurofibromatosis 1. J Neurol Neurosurg Psychiatry. 2000;68:3353-3357.
- Baujat B, Krastinova-Lolov D, Blumen M, et al. Radiofrequency in the treatment of craniofacial plexiform neurofibromatosis: a pilot study. Plast Reconstr Surg. 2006;117:1261-1268.
- Hummel T, Anyane-Yeboa A, Mo J, et al. Response of NF1-related plexiform neurofibroma to high-dose carboplatin. Pediatr Blood Cancer. 2011;56:488-490.
- Feldmann R, Schuierer G, Wessel A, et al. Development of MRI T2 hyperintensities and cognitive functioning in patients with neurofibromatosis type 1. Acta Paediatr. 2010;99:1657-1660.
- Blazo MA, Lewis RA, Chintagumpala MM, et al. Outcomes of systematic screening for optic pathway tumors in children with neurofibromatosis type 1. Am J Med Genet A. 2004;127A:224-229.
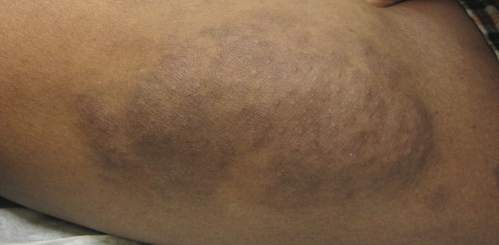
An 11-year-old boy presented for evaluation of a 10×8-cm tortuous lesion on the right posterior leg. Although it had been present since birth, the patient’s mother reported recent growth of the lesion. The lesion was noted to occasionally become irritated and pruritic. The patient’s history was remarkable for asthma.
What Is Your Diagnosis? Mycosis Fungoides
The Diagnosis: Mycosis Fungoides
Physical examination revealed erythematous polycyclic and arcuate plaques with fine overlying scale on the right arm and shoulder (Figure 1). Mild wrinkling and telangiectasias were noted on the skin surrounding the lesions. Laboratory tests showed normal values for antinuclear antibodies, anti–Sjögren syndrome–related antigen A, and anti–Sjögren syndrome–related antigen B.
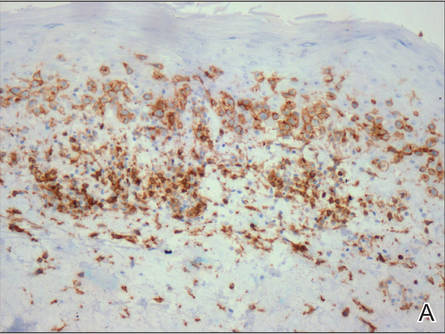
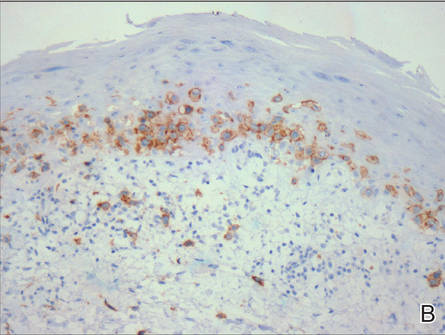
A skin biopsy of a plaque on the right upper arm showed enlarged pleomorphic lymphocytes arranged along the basal layer and in focal collections within the epidermis (Figure 2). Within the dermis were wiry bundles of collagen, a sparse superficial and patchy infiltrate of lymphocytes, and scattered large mononuclear cells (Figure 3). Immunoperoxidase staining revealed large intraepidermal lymphocytes positive for CD4 (Figure 4A) and CD5. Notably, these lymphocytes also stained positive for CD30 (Figure 4B). Staining for CD8, CD1a, CD56, and anaplastic lymphoma kinase was negative, with aberrant loss of CD3. The morphology and pattern of immunoreactivity supported the diagnosis of mycosis fungoides (MF).
Mycosis fungoides is the most common form of cutaneous T-cell lymphoma.1 Its progression is classified in 3 stages: (1) early (patch) stage, (2) plaque stage, and (3) tumor stage. Conclusive diagnosis of early stage MF often is difficult due to its clinical features that are similar to more common benign dermatoses (eg, atopic dermatitis, psoriasis, lichen planus), leading to shortcomings in determining prognosis and selecting an appropriate treatment regimen. With this diagnositic difficulty in mind, guidelines have been created to aid in the diagnosis of early stage MF.2
Clinical features consistent with early stage MF include multiple erythematous, well-demarcated lesions with varying shapes that typically are greater than 5 cm in diameter.2 Lesions usually are flat or thinly elevated and may exhibit slight scaling. As was noted in our patient, poikiloderma of the surrounding skin is fairly specific for early stage MF, as it is not a feature associated with common clinical mimics of MF (eg, atopic dermatitis, psoriasis, lichen planus). The distribution of skin lesions in non–sun-exposed areas is common. The eruption is persistent, though it may wax and wane in severity.2
| 
| |
|
|
Histopathologic examination is necessary to confirm a diagnosis of MF. Typically, early stage MF is marked by enlarged T lymphocytes within the epidermis as well as the papillary and superficial reticular dermis. Cerebriform nuclei are a key finding in the diagnosis of MF. Lymphocytes frequently are arranged linearly along the basal layer of the epidermis. Within the epidermis, clusters of atypical lymphocytes (Pautrier microabscesses) without spongiosis are uncommon but are a characteristic finding of MF if present.1 Papillary dermal fibrosis also may be evident.2
| 
| |
Figure 4. Large intraepidermal lymphocytes were highlighted on CD4 (A) and CD30 immunostaining (B)(original magnification ×200 and ×200). | ||
Immunostaining typically reveals positivity for CD3 and CD4, as well as for lymphocyte antigens CD2 and CD5.1 CD30 positivity in early stage MF rarely has been reported in the literature.3,4 Such cases appear histologically similarly to CD30‒negative cases in other respects. One study showed that the presence of CD30-positive lymphocytes does not alter the clinical course of MF.3 Another study found that, while epidermal CD30-postive lymphocytes had no prognostic relevance, an increased percentage of dermal CD30-positive cells was linked to a higher stage at diagnosis and worse overall prognosis.5 Pathogenesis underlying CD30 positivity in early MF is unknown. It is important to note that CD30-positive cells commonly are seen in lymphomatoid papulosis and anaplastic large cell lymphoma, as well as a variety of nonneoplastic conditions.3,6,7
- Smoller BR. Mycosis fungoides: what do/do not we know? J Cutan Pathol. 2008;35(suppl 2):35-39.
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
- Wu H, Telang GH, Lessin SR, et al. Mycosis fungoides with CD30-positive cells in the epidermis. Am J Dermatopathol. 2000;22:212-216.
- Ohtani T, Kikuchi K, Koizumi H, et al. A case of CD30+ large-cell transformation in a patient with unilesional patch-stage mycosis fungoides. Int J Dermatol. 2009;48:623-626.
- Edinger JT, Clark BZ, Pucevich BE, et al. CD30 expression and proliferative fraction in nontransformed mycosis fungoides. Am J Surg Pathol. 2009;33:1860-1868.
- Resnik KS, Kutzner H. Of lymphocytes and cutaneous epithelium: keratoacanthomatous hyperplasia in CD30+ lymphoproliferative disorders and CD30+ cells associated with keratoacanthoma. Am J Dermatopathol. 2010;32:314-315.
- Kempf W. CD30+ lymphoproliferative disorders: histopathology, differential diagnosis, new variants, and simulators. J Cutan Pathol. 2006;33(suppl 1):58-70.
The Diagnosis: Mycosis Fungoides
Physical examination revealed erythematous polycyclic and arcuate plaques with fine overlying scale on the right arm and shoulder (Figure 1). Mild wrinkling and telangiectasias were noted on the skin surrounding the lesions. Laboratory tests showed normal values for antinuclear antibodies, anti–Sjögren syndrome–related antigen A, and anti–Sjögren syndrome–related antigen B.
A skin biopsy of a plaque on the right upper arm showed enlarged pleomorphic lymphocytes arranged along the basal layer and in focal collections within the epidermis (Figure 2). Within the dermis were wiry bundles of collagen, a sparse superficial and patchy infiltrate of lymphocytes, and scattered large mononuclear cells (Figure 3). Immunoperoxidase staining revealed large intraepidermal lymphocytes positive for CD4 (Figure 4A) and CD5. Notably, these lymphocytes also stained positive for CD30 (Figure 4B). Staining for CD8, CD1a, CD56, and anaplastic lymphoma kinase was negative, with aberrant loss of CD3. The morphology and pattern of immunoreactivity supported the diagnosis of mycosis fungoides (MF).
Mycosis fungoides is the most common form of cutaneous T-cell lymphoma.1 Its progression is classified in 3 stages: (1) early (patch) stage, (2) plaque stage, and (3) tumor stage. Conclusive diagnosis of early stage MF often is difficult due to its clinical features that are similar to more common benign dermatoses (eg, atopic dermatitis, psoriasis, lichen planus), leading to shortcomings in determining prognosis and selecting an appropriate treatment regimen. With this diagnositic difficulty in mind, guidelines have been created to aid in the diagnosis of early stage MF.2
Clinical features consistent with early stage MF include multiple erythematous, well-demarcated lesions with varying shapes that typically are greater than 5 cm in diameter.2 Lesions usually are flat or thinly elevated and may exhibit slight scaling. As was noted in our patient, poikiloderma of the surrounding skin is fairly specific for early stage MF, as it is not a feature associated with common clinical mimics of MF (eg, atopic dermatitis, psoriasis, lichen planus). The distribution of skin lesions in non–sun-exposed areas is common. The eruption is persistent, though it may wax and wane in severity.2
|
|
| |
|
|
Histopathologic examination is necessary to confirm a diagnosis of MF. Typically, early stage MF is marked by enlarged T lymphocytes within the epidermis as well as the papillary and superficial reticular dermis. Cerebriform nuclei are a key finding in the diagnosis of MF. Lymphocytes frequently are arranged linearly along the basal layer of the epidermis. Within the epidermis, clusters of atypical lymphocytes (Pautrier microabscesses) without spongiosis are uncommon but are a characteristic finding of MF if present.1 Papillary dermal fibrosis also may be evident.2
|
|
| |
Figure 4. Large intraepidermal lymphocytes were highlighted on CD4 (A) and CD30 immunostaining (B)(original magnification ×200 and ×200). | ||
Immunostaining typically reveals positivity for CD3 and CD4, as well as for lymphocyte antigens CD2 and CD5.1 CD30 positivity in early stage MF rarely has been reported in the literature.3,4 Such cases appear histologically similarly to CD30‒negative cases in other respects. One study showed that the presence of CD30-positive lymphocytes does not alter the clinical course of MF.3 Another study found that, while epidermal CD30-postive lymphocytes had no prognostic relevance, an increased percentage of dermal CD30-positive cells was linked to a higher stage at diagnosis and worse overall prognosis.5 Pathogenesis underlying CD30 positivity in early MF is unknown. It is important to note that CD30-positive cells commonly are seen in lymphomatoid papulosis and anaplastic large cell lymphoma, as well as a variety of nonneoplastic conditions.3,6,7
The Diagnosis: Mycosis Fungoides
Physical examination revealed erythematous polycyclic and arcuate plaques with fine overlying scale on the right arm and shoulder (Figure 1). Mild wrinkling and telangiectasias were noted on the skin surrounding the lesions. Laboratory tests showed normal values for antinuclear antibodies, anti–Sjögren syndrome–related antigen A, and anti–Sjögren syndrome–related antigen B.
A skin biopsy of a plaque on the right upper arm showed enlarged pleomorphic lymphocytes arranged along the basal layer and in focal collections within the epidermis (Figure 2). Within the dermis were wiry bundles of collagen, a sparse superficial and patchy infiltrate of lymphocytes, and scattered large mononuclear cells (Figure 3). Immunoperoxidase staining revealed large intraepidermal lymphocytes positive for CD4 (Figure 4A) and CD5. Notably, these lymphocytes also stained positive for CD30 (Figure 4B). Staining for CD8, CD1a, CD56, and anaplastic lymphoma kinase was negative, with aberrant loss of CD3. The morphology and pattern of immunoreactivity supported the diagnosis of mycosis fungoides (MF).
Mycosis fungoides is the most common form of cutaneous T-cell lymphoma.1 Its progression is classified in 3 stages: (1) early (patch) stage, (2) plaque stage, and (3) tumor stage. Conclusive diagnosis of early stage MF often is difficult due to its clinical features that are similar to more common benign dermatoses (eg, atopic dermatitis, psoriasis, lichen planus), leading to shortcomings in determining prognosis and selecting an appropriate treatment regimen. With this diagnositic difficulty in mind, guidelines have been created to aid in the diagnosis of early stage MF.2
Clinical features consistent with early stage MF include multiple erythematous, well-demarcated lesions with varying shapes that typically are greater than 5 cm in diameter.2 Lesions usually are flat or thinly elevated and may exhibit slight scaling. As was noted in our patient, poikiloderma of the surrounding skin is fairly specific for early stage MF, as it is not a feature associated with common clinical mimics of MF (eg, atopic dermatitis, psoriasis, lichen planus). The distribution of skin lesions in non–sun-exposed areas is common. The eruption is persistent, though it may wax and wane in severity.2
|
|
| |
|
|
Histopathologic examination is necessary to confirm a diagnosis of MF. Typically, early stage MF is marked by enlarged T lymphocytes within the epidermis as well as the papillary and superficial reticular dermis. Cerebriform nuclei are a key finding in the diagnosis of MF. Lymphocytes frequently are arranged linearly along the basal layer of the epidermis. Within the epidermis, clusters of atypical lymphocytes (Pautrier microabscesses) without spongiosis are uncommon but are a characteristic finding of MF if present.1 Papillary dermal fibrosis also may be evident.2
|
|
| |
Figure 4. Large intraepidermal lymphocytes were highlighted on CD4 (A) and CD30 immunostaining (B)(original magnification ×200 and ×200). | ||
Immunostaining typically reveals positivity for CD3 and CD4, as well as for lymphocyte antigens CD2 and CD5.1 CD30 positivity in early stage MF rarely has been reported in the literature.3,4 Such cases appear histologically similarly to CD30‒negative cases in other respects. One study showed that the presence of CD30-positive lymphocytes does not alter the clinical course of MF.3 Another study found that, while epidermal CD30-postive lymphocytes had no prognostic relevance, an increased percentage of dermal CD30-positive cells was linked to a higher stage at diagnosis and worse overall prognosis.5 Pathogenesis underlying CD30 positivity in early MF is unknown. It is important to note that CD30-positive cells commonly are seen in lymphomatoid papulosis and anaplastic large cell lymphoma, as well as a variety of nonneoplastic conditions.3,6,7
- Smoller BR. Mycosis fungoides: what do/do not we know? J Cutan Pathol. 2008;35(suppl 2):35-39.
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
- Wu H, Telang GH, Lessin SR, et al. Mycosis fungoides with CD30-positive cells in the epidermis. Am J Dermatopathol. 2000;22:212-216.
- Ohtani T, Kikuchi K, Koizumi H, et al. A case of CD30+ large-cell transformation in a patient with unilesional patch-stage mycosis fungoides. Int J Dermatol. 2009;48:623-626.
- Edinger JT, Clark BZ, Pucevich BE, et al. CD30 expression and proliferative fraction in nontransformed mycosis fungoides. Am J Surg Pathol. 2009;33:1860-1868.
- Resnik KS, Kutzner H. Of lymphocytes and cutaneous epithelium: keratoacanthomatous hyperplasia in CD30+ lymphoproliferative disorders and CD30+ cells associated with keratoacanthoma. Am J Dermatopathol. 2010;32:314-315.
- Kempf W. CD30+ lymphoproliferative disorders: histopathology, differential diagnosis, new variants, and simulators. J Cutan Pathol. 2006;33(suppl 1):58-70.
- Smoller BR. Mycosis fungoides: what do/do not we know? J Cutan Pathol. 2008;35(suppl 2):35-39.
- Pimpinelli N, Olsen EA, Santucci M, et al. Defining early mycosis fungoides. J Am Acad Dermatol. 2005;53:1053-1063.
- Wu H, Telang GH, Lessin SR, et al. Mycosis fungoides with CD30-positive cells in the epidermis. Am J Dermatopathol. 2000;22:212-216.
- Ohtani T, Kikuchi K, Koizumi H, et al. A case of CD30+ large-cell transformation in a patient with unilesional patch-stage mycosis fungoides. Int J Dermatol. 2009;48:623-626.
- Edinger JT, Clark BZ, Pucevich BE, et al. CD30 expression and proliferative fraction in nontransformed mycosis fungoides. Am J Surg Pathol. 2009;33:1860-1868.
- Resnik KS, Kutzner H. Of lymphocytes and cutaneous epithelium: keratoacanthomatous hyperplasia in CD30+ lymphoproliferative disorders and CD30+ cells associated with keratoacanthoma. Am J Dermatopathol. 2010;32:314-315.
- Kempf W. CD30+ lymphoproliferative disorders: histopathology, differential diagnosis, new variants, and simulators. J Cutan Pathol. 2006;33(suppl 1):58-70.
An otherwise healthy 62-year-old man presented for evaluation of multiple scaly erythematous plaques on the right upper arm and shoulder of 10 years’ duration. The patient reported a burning sensation but no exacerbation of the lesions upon sun exposure. He previously had been treated for a presumed clinical diagnosis of erythema annulare centrifugum but experienced only modest improvement with topical corticosteroids and tacrolimus ointment 0.1%. Previous trials of systemic antifungals also yielded minimal benefit.
Itchy Papules and Plaques on the Dorsal Hands
The Diagnosis: Neutrophilic Dermatosis of the Dorsal Hands
Neutrophilic dermatosis of the dorsal hands (NDDH) is considered to be an uncommon localized variant of Sweet syndrome (SS). The term pustular vasculitis originally was used to describe this condition by Strutton et al1 in 1995 due to the presence of leukocytoclastic vasculitis on histology. In 2000, Galaria et al2 suggested this eruption was a localized variant of SS based on clinical presentations that demonstrated associated fever and lack of necrotizing vasculitis and proposed the term neutrophilic dermatosis of the dorsal hands to describe the condition. Cases of similar cutaneous eruptions on the hands associated with fever, leukocytosis, elevated erythrocyte sedimentation rate, and leukocytoclasis have since been reported.3-5 Some authors have concluded that these eruptions, previously termed atypical pyoderma gangrenosum and pustular vasculitis of the hands, represent a single disease entity and should be designated as NDDH.3,4
Neutrophilic dermatosis of the dorsal hands characteristically presents with hemorrhagic pustular ulcerations limited to or predominantly located on the dorsal hands, as seen in our patient. Histopathologically, NDDH demonstrates a neutrophil-predominant infiltrate of the upper dermis and marked papillary dermal edema; a punch biopsy specimen from our patient was consistent with these features (Figure). Two punch biopsies were performed and were negative for fungus and acid-fast bacteria and positive for methicillin-sensitive Staphylococcus aureus. Vasculitis, if present, is more commonly seen in eruptions of longer duration (ie, months to years) and is thought to be secondary to the dense neutrophilic infiltrate and not a primary vasculitis.3,6,7 Similar to classic SS, NDDH is inherently responsive to corticosteroid therapy. Successful treatment also has been reported with dapsone, colchicine, sulfapyridine, potassium iodide, intralesional and topical corticosteroids, and topical tacrolimus.2-8 Oral minocycline has shown variable results.3,4
Numerous case series have demonstrated that a majority of cases of NDDH are associated with hematologic or solid organ malignancies, myelodysplastic syndrome (MDS), inflammatory bowel disease, or other underlying systemic diseases.3,5,9 It is important for dermatologists to recognize NDDH, distinguish it from localized infection, and perform the appropriate workup (eg, basic laboratory tests [complete blood count, complete metabolic panel], age-appropriate malignancy screening, colonoscopy, bone marrow biopsy) to exclude associated systemic diseases.
Our patient demonstrated characteristic clinical and histopathologic findings of NDDH in association with early MDS and possible common bile duct (CBD) malignancy. The lesions showed a rapid response to topical corticosteroid therapy. The initial differential diagnoses included NDDH or other neutrophilic dermatosis, phototoxic drug eruption, and atypical mycobacterial or fungal infection (cultures were negative in our patient). Physical examination and histopathologic findings along with the patient’s clinical course and rapid response to topical corticosteroid therapy supported the diagnosis of NDDH. Our patient’s multiple comorbidities, including macrocytic anemia, MDS, and potential CBD malignancy, presented a therapeutic challenge. Oral dapsone, an ideal steroid-sparing agent for neutrophilic dermatoses including NDDH, was avoided given its associated hematologic side effects including hemolysis, methemoglobinemia, and possible agranulocytosis. To date, the patient has not received any further treatment for MDS or the CBD mass and continues regular follow-up with hematology, gastroenterology, and dermatology.
This case highlights the importance of including NDDH in the differential diagnosis of papules and plaques on the hands, especially in patients with known malignancies, and emphasizes the association of neutrophilic dermatoses with malignancy and systemic disease.
- Strutton G, Weedon D, Robertson I. Pustular vasculitis of the hands. J Am Acad Dermatol. 1995;32:192-198.
- Galaria NA, Junkins-Hopkins JM, Kligman D, et al. Neutrophilic dermatosis of the dorsal hands: pustular vasculitis revisited. J Am Acad Dermatol. 2000;43:870-874.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- DiCaudo DJ, Connolly SM. Neutrophilic dermatosis (pustular vasculitis) of the dorsal hands. Arch Dermatol. 2002;138:361-365.
- Weening RH, Bruce AJ, McEvoy MT, et al. Neutrophilic dermatosis of the hands: four new cases and review of the literature. Int J Dermatol. 2004;43:95-102.
- Malone JC, Slone SP, Wills-Frank LA, et al. Vascular inflammation (vasculitis) in Sweet syndrome: a clinicopathologic study of 28 biopsy specimens from 21 patients. Arch Dermatol. 2002;138:345-349.
- Cohen PR. Skin lesions of Sweet syndrome and its dorsal hand variant contain vasculitis: an oxymoron or an epiphenomenon? Arch Dermatol. 2002;138:400-403.
- Del Pozo J, Sacristán F, Martínez W, et al. Neutrophilic dermatosis of the hands: presentation of eight cases and review of the literature. J Dermatol. 2007;34:243-247.
- Callen JP. Neutrophilic dermatoses. Dermatol Clin. 2002;20:409-419.
The Diagnosis: Neutrophilic Dermatosis of the Dorsal Hands
Neutrophilic dermatosis of the dorsal hands (NDDH) is considered to be an uncommon localized variant of Sweet syndrome (SS). The term pustular vasculitis originally was used to describe this condition by Strutton et al1 in 1995 due to the presence of leukocytoclastic vasculitis on histology. In 2000, Galaria et al2 suggested this eruption was a localized variant of SS based on clinical presentations that demonstrated associated fever and lack of necrotizing vasculitis and proposed the term neutrophilic dermatosis of the dorsal hands to describe the condition. Cases of similar cutaneous eruptions on the hands associated with fever, leukocytosis, elevated erythrocyte sedimentation rate, and leukocytoclasis have since been reported.3-5 Some authors have concluded that these eruptions, previously termed atypical pyoderma gangrenosum and pustular vasculitis of the hands, represent a single disease entity and should be designated as NDDH.3,4
Neutrophilic dermatosis of the dorsal hands characteristically presents with hemorrhagic pustular ulcerations limited to or predominantly located on the dorsal hands, as seen in our patient. Histopathologically, NDDH demonstrates a neutrophil-predominant infiltrate of the upper dermis and marked papillary dermal edema; a punch biopsy specimen from our patient was consistent with these features (Figure). Two punch biopsies were performed and were negative for fungus and acid-fast bacteria and positive for methicillin-sensitive Staphylococcus aureus. Vasculitis, if present, is more commonly seen in eruptions of longer duration (ie, months to years) and is thought to be secondary to the dense neutrophilic infiltrate and not a primary vasculitis.3,6,7 Similar to classic SS, NDDH is inherently responsive to corticosteroid therapy. Successful treatment also has been reported with dapsone, colchicine, sulfapyridine, potassium iodide, intralesional and topical corticosteroids, and topical tacrolimus.2-8 Oral minocycline has shown variable results.3,4
Numerous case series have demonstrated that a majority of cases of NDDH are associated with hematologic or solid organ malignancies, myelodysplastic syndrome (MDS), inflammatory bowel disease, or other underlying systemic diseases.3,5,9 It is important for dermatologists to recognize NDDH, distinguish it from localized infection, and perform the appropriate workup (eg, basic laboratory tests [complete blood count, complete metabolic panel], age-appropriate malignancy screening, colonoscopy, bone marrow biopsy) to exclude associated systemic diseases.
Our patient demonstrated characteristic clinical and histopathologic findings of NDDH in association with early MDS and possible common bile duct (CBD) malignancy. The lesions showed a rapid response to topical corticosteroid therapy. The initial differential diagnoses included NDDH or other neutrophilic dermatosis, phototoxic drug eruption, and atypical mycobacterial or fungal infection (cultures were negative in our patient). Physical examination and histopathologic findings along with the patient’s clinical course and rapid response to topical corticosteroid therapy supported the diagnosis of NDDH. Our patient’s multiple comorbidities, including macrocytic anemia, MDS, and potential CBD malignancy, presented a therapeutic challenge. Oral dapsone, an ideal steroid-sparing agent for neutrophilic dermatoses including NDDH, was avoided given its associated hematologic side effects including hemolysis, methemoglobinemia, and possible agranulocytosis. To date, the patient has not received any further treatment for MDS or the CBD mass and continues regular follow-up with hematology, gastroenterology, and dermatology.
This case highlights the importance of including NDDH in the differential diagnosis of papules and plaques on the hands, especially in patients with known malignancies, and emphasizes the association of neutrophilic dermatoses with malignancy and systemic disease.
The Diagnosis: Neutrophilic Dermatosis of the Dorsal Hands
Neutrophilic dermatosis of the dorsal hands (NDDH) is considered to be an uncommon localized variant of Sweet syndrome (SS). The term pustular vasculitis originally was used to describe this condition by Strutton et al1 in 1995 due to the presence of leukocytoclastic vasculitis on histology. In 2000, Galaria et al2 suggested this eruption was a localized variant of SS based on clinical presentations that demonstrated associated fever and lack of necrotizing vasculitis and proposed the term neutrophilic dermatosis of the dorsal hands to describe the condition. Cases of similar cutaneous eruptions on the hands associated with fever, leukocytosis, elevated erythrocyte sedimentation rate, and leukocytoclasis have since been reported.3-5 Some authors have concluded that these eruptions, previously termed atypical pyoderma gangrenosum and pustular vasculitis of the hands, represent a single disease entity and should be designated as NDDH.3,4
Neutrophilic dermatosis of the dorsal hands characteristically presents with hemorrhagic pustular ulcerations limited to or predominantly located on the dorsal hands, as seen in our patient. Histopathologically, NDDH demonstrates a neutrophil-predominant infiltrate of the upper dermis and marked papillary dermal edema; a punch biopsy specimen from our patient was consistent with these features (Figure). Two punch biopsies were performed and were negative for fungus and acid-fast bacteria and positive for methicillin-sensitive Staphylococcus aureus. Vasculitis, if present, is more commonly seen in eruptions of longer duration (ie, months to years) and is thought to be secondary to the dense neutrophilic infiltrate and not a primary vasculitis.3,6,7 Similar to classic SS, NDDH is inherently responsive to corticosteroid therapy. Successful treatment also has been reported with dapsone, colchicine, sulfapyridine, potassium iodide, intralesional and topical corticosteroids, and topical tacrolimus.2-8 Oral minocycline has shown variable results.3,4
Numerous case series have demonstrated that a majority of cases of NDDH are associated with hematologic or solid organ malignancies, myelodysplastic syndrome (MDS), inflammatory bowel disease, or other underlying systemic diseases.3,5,9 It is important for dermatologists to recognize NDDH, distinguish it from localized infection, and perform the appropriate workup (eg, basic laboratory tests [complete blood count, complete metabolic panel], age-appropriate malignancy screening, colonoscopy, bone marrow biopsy) to exclude associated systemic diseases.
Our patient demonstrated characteristic clinical and histopathologic findings of NDDH in association with early MDS and possible common bile duct (CBD) malignancy. The lesions showed a rapid response to topical corticosteroid therapy. The initial differential diagnoses included NDDH or other neutrophilic dermatosis, phototoxic drug eruption, and atypical mycobacterial or fungal infection (cultures were negative in our patient). Physical examination and histopathologic findings along with the patient’s clinical course and rapid response to topical corticosteroid therapy supported the diagnosis of NDDH. Our patient’s multiple comorbidities, including macrocytic anemia, MDS, and potential CBD malignancy, presented a therapeutic challenge. Oral dapsone, an ideal steroid-sparing agent for neutrophilic dermatoses including NDDH, was avoided given its associated hematologic side effects including hemolysis, methemoglobinemia, and possible agranulocytosis. To date, the patient has not received any further treatment for MDS or the CBD mass and continues regular follow-up with hematology, gastroenterology, and dermatology.
This case highlights the importance of including NDDH in the differential diagnosis of papules and plaques on the hands, especially in patients with known malignancies, and emphasizes the association of neutrophilic dermatoses with malignancy and systemic disease.
- Strutton G, Weedon D, Robertson I. Pustular vasculitis of the hands. J Am Acad Dermatol. 1995;32:192-198.
- Galaria NA, Junkins-Hopkins JM, Kligman D, et al. Neutrophilic dermatosis of the dorsal hands: pustular vasculitis revisited. J Am Acad Dermatol. 2000;43:870-874.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- DiCaudo DJ, Connolly SM. Neutrophilic dermatosis (pustular vasculitis) of the dorsal hands. Arch Dermatol. 2002;138:361-365.
- Weening RH, Bruce AJ, McEvoy MT, et al. Neutrophilic dermatosis of the hands: four new cases and review of the literature. Int J Dermatol. 2004;43:95-102.
- Malone JC, Slone SP, Wills-Frank LA, et al. Vascular inflammation (vasculitis) in Sweet syndrome: a clinicopathologic study of 28 biopsy specimens from 21 patients. Arch Dermatol. 2002;138:345-349.
- Cohen PR. Skin lesions of Sweet syndrome and its dorsal hand variant contain vasculitis: an oxymoron or an epiphenomenon? Arch Dermatol. 2002;138:400-403.
- Del Pozo J, Sacristán F, Martínez W, et al. Neutrophilic dermatosis of the hands: presentation of eight cases and review of the literature. J Dermatol. 2007;34:243-247.
- Callen JP. Neutrophilic dermatoses. Dermatol Clin. 2002;20:409-419.
- Strutton G, Weedon D, Robertson I. Pustular vasculitis of the hands. J Am Acad Dermatol. 1995;32:192-198.
- Galaria NA, Junkins-Hopkins JM, Kligman D, et al. Neutrophilic dermatosis of the dorsal hands: pustular vasculitis revisited. J Am Acad Dermatol. 2000;43:870-874.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and Sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- DiCaudo DJ, Connolly SM. Neutrophilic dermatosis (pustular vasculitis) of the dorsal hands. Arch Dermatol. 2002;138:361-365.
- Weening RH, Bruce AJ, McEvoy MT, et al. Neutrophilic dermatosis of the hands: four new cases and review of the literature. Int J Dermatol. 2004;43:95-102.
- Malone JC, Slone SP, Wills-Frank LA, et al. Vascular inflammation (vasculitis) in Sweet syndrome: a clinicopathologic study of 28 biopsy specimens from 21 patients. Arch Dermatol. 2002;138:345-349.
- Cohen PR. Skin lesions of Sweet syndrome and its dorsal hand variant contain vasculitis: an oxymoron or an epiphenomenon? Arch Dermatol. 2002;138:400-403.
- Del Pozo J, Sacristán F, Martínez W, et al. Neutrophilic dermatosis of the hands: presentation of eight cases and review of the literature. J Dermatol. 2007;34:243-247.
- Callen JP. Neutrophilic dermatoses. Dermatol Clin. 2002;20:409-419.
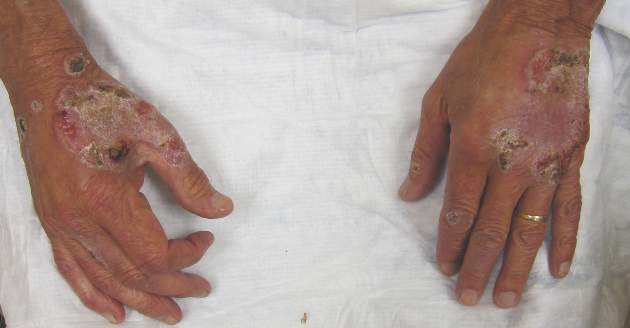
A 69-year-old man presented with tender, itchy papules and plaques on the bilateral dorsal hands of 2 months’ duration. The plaques had started as small papules that gradually enlarged and then became ulcerated. The patient denied prior trauma or constitutional symptoms. Laboratory testing revealed macrocytic anemia, thrombocytosis, and hypoalbuminemia. A complete blood count and complete metabolic panel were otherwise unremarkable. A recent bone marrow biopsy for macrocytic anemia performed prior to the current presentation suggested early myelodysplastic syndrome, and endoscopic retrograde cholangiopancreatography revealed a large mass in the common bile duct that was suspicious for malignancy. Two punch biopsies were performed and were negative for fungus and acid-fast bacteria and positive for methicillin-sensitive Staphylococcus aureus. Treatment with topical clobetasol 0.05% twice daily was initiated with complete healing of the plaques on the hands after 2 weeks of use; however, the patient continued to develop new ulcerated papulonodules distally.
Erythematous Scaly Papules on the Shins and Calves
The Diagnosis: Hyperkeratosis Lenticularis Perstans
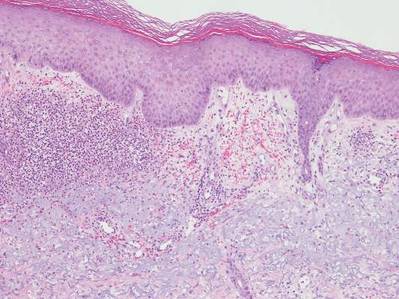
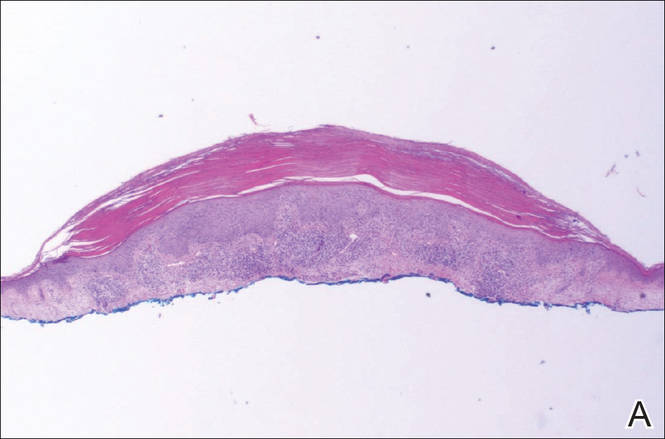
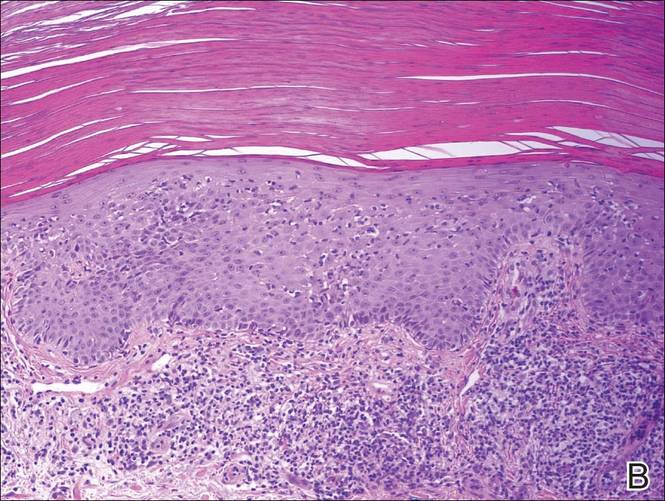
A shave biopsy of a lesion on the right leg was performed. Histopathology revealed a discrete papule with overlying compact hyperkeratosis. There was parakeratosis with an absent granular layer and a lichenoid lymphocytic infiltrate within the papillary dermis (Figure). Given the clinical context, these changes were consistent with a diagnosis of hyperkeratosis lenticularis perstans (HLP), also known as Flegel disease.
The patient was started on tretinoin cream 0.1% nightly for 3 months and triamcinolone ointment 0.1% as needed for pruritus but showed no clinical response. Given the benign nature of the condition and because the lesions were asymptomatic, additional treatment options were not pursued.
Originally described by Flegel1 in 1958, HLP is a rare skin disorder commonly seen in white individuals with onset in the fourth or fifth decades of life.1,2 While most cases are sporadic,3-6 HLP also has been associated with autosomal dominant inheritance.7-10
Patients with HLP typically present with multiple 1- to 5-mm reddish-brown, hyperkeratotic, scaly papules that reveal a moist, erythematous base with pinpoint bleeding upon removal of the scale. Lesions usually are distributed symmetrically and most commonly present on the extensor surfaces of the lower legs and dorsal feet.1,2,7 Lesions also may appear on the extensor surfaces of the arms, pinna, periocular region, antecubital and popliteal fossae, and oral mucosa and also may present as pits on the palms and soles.2,4,7,8 Furthermore, unilateral and localized variants of HLP have been described.11,12 Hyperkeratosis lenticularis perstans usually is asymptomatic but can present with mild pruritus or burning.3,5,13
The etiology and pathogenesis of HLP are unknown. Exposure to UV light has been implicated as an inciting factor14; however, reports of spontaneous resolution in the summer13 and upon treatment with psoralen plus UVA therapy15 make the role of UV light unclear. Furthermore, investigators disagree as to whether the primary pathogenic event in HLP is an inflammatory process or one of abnormal keratinization.1,3,7,10 Fernandez-Flores and Manjon16 suggested HLP is an inflammatory process with periods of exacerbations and remissions after finding mounds of parakeratosis with neutrophils arranged in different strata in the stratum corneum.
Histologically, compact hyperkeratosis usually is noted, often with associated parakeratosis, epidermal atrophy with thinning or absence of the granular layer, and a bandlike lymphohistiocytic infiltrate in the papillary dermis.1-3 Histopathologic differences between recent-onset versus longstanding lesions have been found, with old lesions lacking an inflammatory infiltrate.3 Furthermore, new lesions often show abnormalities in quantity and/or morphology of membrane-coating granules, also known as Odland bodies, in keratinocytes on electron microscopy,3,10,17 while old lesions do not.3 Odland bodies are involved in normal desquamation, leading some to speculate on their role in HLP.10 Currently, it is unclear whether abnormalities in these organelles cause the retention hyperkeratosis seen in HLP or if such abnormalities are a secondary phenomenon.3,17
There are questionable associations between HLP and diabetes mellitus type 2, hyperthyroidism, basal and squamous cell carcinomas of the skin, and gastrointestinal malignancy.4,9,18 Our patient had a history of basal cell carcinoma on the face, diet-controlled diabetes mellitus, and hypothyroidism. Given the high prevalence of these diseases in the general population, however, it is difficult to ascertain whether a true association with HLP exists.
While HLP can slowly progress to involve additional body sites, it is overall a benign condition that does not require treatment. Therapeutic options are based on case reports, with no single treatment showing a consistent response. From review of the literature, therapies that have been most effective include dermabrasion, excision,19 topical 5-fluorouracil,2,17,20 and oral retinoids.8 Hyperkeratosis lenticularis perstans generally is resistant to topical steroids, retinoids, and vitamin D3 analogs, although success with betamethasone dipropionate,5 isotretinoin
gel 0.05%,11 and calcipotriol have been reported.6 A case of HLP with clinical response to psoralen plus UVA therapy also has been described.15
- Flegel H. Hyperkeratosis lenticularis perstans. Hautarzt. 1958;9:363-364.
- Pearson LH, Smith JG, Chalker DK. Hyperkeratosis lenticularis perstans (Flegel’s disease). J Am Acad Dermatol. 1987;16:190-195.
- Ando K, Hattori H, Yamauchi Y. Histopathological differences between early and old lesions of hyperkeratosis lenticularis perstans (Flegel’s disease). Am J Dermatopathol. 2006;28:122-126.
- Fernández-Crehuet P, Rodríguez-Rey E, Ríos-Martín JJ, et al. Hyperkeratosis lenticularis perstans, or Flegel disease, with palmoplantar involvement. Actas Dermosifiliogr. 2009;100:157-159.
- Sterneberg-Vos H, van Marion AM, Frank J, et al. Hyperkeratosis lenticularis perstans (Flegel’s disease)—successful treatment with topical corticosteroids. Int J Dermatol. 2008;47:38-41.
- Bayramgürler D, Apaydin R, Dökmeci S, et al. Flegel’s disease: treatment with topical calcipotriol. Clin Exp Dermatol. 2002;27:161-162.
- Price ML, Jones EW, MacDonald DM. A clinicopathological study of Flegel’s disease (hyperkeratosis lenticularis perstans). Br J Dermatol. 1987;116:681-691.
- Krishnan A, Kar S. Photoletter to the editor: hyperkeratosis lenticularis perstans (Flegel’s disease) with unusual clinical presentation. response to isotretinoin therapy. J Dermatol Case Rep. 2012;6:93-95.
- Beveridge GW, Langlands AO. Familial hyperkeratosis lenticularis perstans associated with tumours of the skin. Br J Dermatol. 1973;88:453-458.
- Frenk E, Tapernoux B. Hyperkeratosis lenticularis perstans (Flegel): a biological model for keratinization occurring in the absence of Odland bodies? Dermatologica. 1976;153:253-262.
- Miranda-Romero A, Sánchez Sambucety P, Bajo del Pozo C, et al. Unilateral hyperkeratosis lenticularis perstans (Flegel's disease). J Am Acad Dermatol. 1998;39:655-657.
- Gutiérrez MC, Hasson A, Arias MD, et al. Localized hyperkeratosis lenticularis perstans (Flegel's disease). Cutis. 1991;48:201-204.
- Fathy S, Azadeh B. Hyperkeratosis lenticularis perstans. Int J Dermatol. 1988;27:120-121.
- Rosdahl I, Rosen K. Hyperkeratosis lenticularis perstans: report on two cases. Acta Derm Venerol. 1985;65:562-564.
- Cooper SM, George S. Flegel's disease treated with psoralen ultraviolet A. Br J Dermatol. 2000;142:340-342.
- Fernandez-Flores A, Manjon JA. Morphological evidence of periodical exacerbation of hyperkeratosis lenticularis perstans. Acta Dermatovenerol Croat. 2009;17:16-19.
- Langer K, Zonzits E, Konrad K. Hyperkeratosis lenticularis perstans (Flegel's disease). ultrastructural study of lesional and perilesional skin and therapeutic trial of topical tretinoin versus 5-fluorouracil. J Am Acad Dermatol. 1992;27:812-816.
- Ishibashi A, Tsuboi R, Fujita K. Familial hyperkeratosis lenticularis perstans. associated with cancers of the digestive organs. J Dermatol. 1984;11:407-409.
- Cunha Filho RR, Almeida Jr HL. Hyperkeratosis lenticularis perstans. An Bras Dermatol. 2011;86(4 suppl 1):S76-S77.
- Blaheta HJ, Metzler G, Rassner G, et al. Hyperkeratosis lenticularis perstans (Flegel's disease)—lack of response to treatment with tacalcitol and calcipotriol. Dermatology. 2001;202:255-258.
The Diagnosis: Hyperkeratosis Lenticularis Perstans
A shave biopsy of a lesion on the right leg was performed. Histopathology revealed a discrete papule with overlying compact hyperkeratosis. There was parakeratosis with an absent granular layer and a lichenoid lymphocytic infiltrate within the papillary dermis (Figure). Given the clinical context, these changes were consistent with a diagnosis of hyperkeratosis lenticularis perstans (HLP), also known as Flegel disease.
The patient was started on tretinoin cream 0.1% nightly for 3 months and triamcinolone ointment 0.1% as needed for pruritus but showed no clinical response. Given the benign nature of the condition and because the lesions were asymptomatic, additional treatment options were not pursued.
Originally described by Flegel1 in 1958, HLP is a rare skin disorder commonly seen in white individuals with onset in the fourth or fifth decades of life.1,2 While most cases are sporadic,3-6 HLP also has been associated with autosomal dominant inheritance.7-10
Patients with HLP typically present with multiple 1- to 5-mm reddish-brown, hyperkeratotic, scaly papules that reveal a moist, erythematous base with pinpoint bleeding upon removal of the scale. Lesions usually are distributed symmetrically and most commonly present on the extensor surfaces of the lower legs and dorsal feet.1,2,7 Lesions also may appear on the extensor surfaces of the arms, pinna, periocular region, antecubital and popliteal fossae, and oral mucosa and also may present as pits on the palms and soles.2,4,7,8 Furthermore, unilateral and localized variants of HLP have been described.11,12 Hyperkeratosis lenticularis perstans usually is asymptomatic but can present with mild pruritus or burning.3,5,13
The etiology and pathogenesis of HLP are unknown. Exposure to UV light has been implicated as an inciting factor14; however, reports of spontaneous resolution in the summer13 and upon treatment with psoralen plus UVA therapy15 make the role of UV light unclear. Furthermore, investigators disagree as to whether the primary pathogenic event in HLP is an inflammatory process or one of abnormal keratinization.1,3,7,10 Fernandez-Flores and Manjon16 suggested HLP is an inflammatory process with periods of exacerbations and remissions after finding mounds of parakeratosis with neutrophils arranged in different strata in the stratum corneum.
Histologically, compact hyperkeratosis usually is noted, often with associated parakeratosis, epidermal atrophy with thinning or absence of the granular layer, and a bandlike lymphohistiocytic infiltrate in the papillary dermis.1-3 Histopathologic differences between recent-onset versus longstanding lesions have been found, with old lesions lacking an inflammatory infiltrate.3 Furthermore, new lesions often show abnormalities in quantity and/or morphology of membrane-coating granules, also known as Odland bodies, in keratinocytes on electron microscopy,3,10,17 while old lesions do not.3 Odland bodies are involved in normal desquamation, leading some to speculate on their role in HLP.10 Currently, it is unclear whether abnormalities in these organelles cause the retention hyperkeratosis seen in HLP or if such abnormalities are a secondary phenomenon.3,17
There are questionable associations between HLP and diabetes mellitus type 2, hyperthyroidism, basal and squamous cell carcinomas of the skin, and gastrointestinal malignancy.4,9,18 Our patient had a history of basal cell carcinoma on the face, diet-controlled diabetes mellitus, and hypothyroidism. Given the high prevalence of these diseases in the general population, however, it is difficult to ascertain whether a true association with HLP exists.
While HLP can slowly progress to involve additional body sites, it is overall a benign condition that does not require treatment. Therapeutic options are based on case reports, with no single treatment showing a consistent response. From review of the literature, therapies that have been most effective include dermabrasion, excision,19 topical 5-fluorouracil,2,17,20 and oral retinoids.8 Hyperkeratosis lenticularis perstans generally is resistant to topical steroids, retinoids, and vitamin D3 analogs, although success with betamethasone dipropionate,5 isotretinoin
gel 0.05%,11 and calcipotriol have been reported.6 A case of HLP with clinical response to psoralen plus UVA therapy also has been described.15
The Diagnosis: Hyperkeratosis Lenticularis Perstans
A shave biopsy of a lesion on the right leg was performed. Histopathology revealed a discrete papule with overlying compact hyperkeratosis. There was parakeratosis with an absent granular layer and a lichenoid lymphocytic infiltrate within the papillary dermis (Figure). Given the clinical context, these changes were consistent with a diagnosis of hyperkeratosis lenticularis perstans (HLP), also known as Flegel disease.
The patient was started on tretinoin cream 0.1% nightly for 3 months and triamcinolone ointment 0.1% as needed for pruritus but showed no clinical response. Given the benign nature of the condition and because the lesions were asymptomatic, additional treatment options were not pursued.
Originally described by Flegel1 in 1958, HLP is a rare skin disorder commonly seen in white individuals with onset in the fourth or fifth decades of life.1,2 While most cases are sporadic,3-6 HLP also has been associated with autosomal dominant inheritance.7-10
Patients with HLP typically present with multiple 1- to 5-mm reddish-brown, hyperkeratotic, scaly papules that reveal a moist, erythematous base with pinpoint bleeding upon removal of the scale. Lesions usually are distributed symmetrically and most commonly present on the extensor surfaces of the lower legs and dorsal feet.1,2,7 Lesions also may appear on the extensor surfaces of the arms, pinna, periocular region, antecubital and popliteal fossae, and oral mucosa and also may present as pits on the palms and soles.2,4,7,8 Furthermore, unilateral and localized variants of HLP have been described.11,12 Hyperkeratosis lenticularis perstans usually is asymptomatic but can present with mild pruritus or burning.3,5,13
The etiology and pathogenesis of HLP are unknown. Exposure to UV light has been implicated as an inciting factor14; however, reports of spontaneous resolution in the summer13 and upon treatment with psoralen plus UVA therapy15 make the role of UV light unclear. Furthermore, investigators disagree as to whether the primary pathogenic event in HLP is an inflammatory process or one of abnormal keratinization.1,3,7,10 Fernandez-Flores and Manjon16 suggested HLP is an inflammatory process with periods of exacerbations and remissions after finding mounds of parakeratosis with neutrophils arranged in different strata in the stratum corneum.
Histologically, compact hyperkeratosis usually is noted, often with associated parakeratosis, epidermal atrophy with thinning or absence of the granular layer, and a bandlike lymphohistiocytic infiltrate in the papillary dermis.1-3 Histopathologic differences between recent-onset versus longstanding lesions have been found, with old lesions lacking an inflammatory infiltrate.3 Furthermore, new lesions often show abnormalities in quantity and/or morphology of membrane-coating granules, also known as Odland bodies, in keratinocytes on electron microscopy,3,10,17 while old lesions do not.3 Odland bodies are involved in normal desquamation, leading some to speculate on their role in HLP.10 Currently, it is unclear whether abnormalities in these organelles cause the retention hyperkeratosis seen in HLP or if such abnormalities are a secondary phenomenon.3,17
There are questionable associations between HLP and diabetes mellitus type 2, hyperthyroidism, basal and squamous cell carcinomas of the skin, and gastrointestinal malignancy.4,9,18 Our patient had a history of basal cell carcinoma on the face, diet-controlled diabetes mellitus, and hypothyroidism. Given the high prevalence of these diseases in the general population, however, it is difficult to ascertain whether a true association with HLP exists.
While HLP can slowly progress to involve additional body sites, it is overall a benign condition that does not require treatment. Therapeutic options are based on case reports, with no single treatment showing a consistent response. From review of the literature, therapies that have been most effective include dermabrasion, excision,19 topical 5-fluorouracil,2,17,20 and oral retinoids.8 Hyperkeratosis lenticularis perstans generally is resistant to topical steroids, retinoids, and vitamin D3 analogs, although success with betamethasone dipropionate,5 isotretinoin
gel 0.05%,11 and calcipotriol have been reported.6 A case of HLP with clinical response to psoralen plus UVA therapy also has been described.15
- Flegel H. Hyperkeratosis lenticularis perstans. Hautarzt. 1958;9:363-364.
- Pearson LH, Smith JG, Chalker DK. Hyperkeratosis lenticularis perstans (Flegel’s disease). J Am Acad Dermatol. 1987;16:190-195.
- Ando K, Hattori H, Yamauchi Y. Histopathological differences between early and old lesions of hyperkeratosis lenticularis perstans (Flegel’s disease). Am J Dermatopathol. 2006;28:122-126.
- Fernández-Crehuet P, Rodríguez-Rey E, Ríos-Martín JJ, et al. Hyperkeratosis lenticularis perstans, or Flegel disease, with palmoplantar involvement. Actas Dermosifiliogr. 2009;100:157-159.
- Sterneberg-Vos H, van Marion AM, Frank J, et al. Hyperkeratosis lenticularis perstans (Flegel’s disease)—successful treatment with topical corticosteroids. Int J Dermatol. 2008;47:38-41.
- Bayramgürler D, Apaydin R, Dökmeci S, et al. Flegel’s disease: treatment with topical calcipotriol. Clin Exp Dermatol. 2002;27:161-162.
- Price ML, Jones EW, MacDonald DM. A clinicopathological study of Flegel’s disease (hyperkeratosis lenticularis perstans). Br J Dermatol. 1987;116:681-691.
- Krishnan A, Kar S. Photoletter to the editor: hyperkeratosis lenticularis perstans (Flegel’s disease) with unusual clinical presentation. response to isotretinoin therapy. J Dermatol Case Rep. 2012;6:93-95.
- Beveridge GW, Langlands AO. Familial hyperkeratosis lenticularis perstans associated with tumours of the skin. Br J Dermatol. 1973;88:453-458.
- Frenk E, Tapernoux B. Hyperkeratosis lenticularis perstans (Flegel): a biological model for keratinization occurring in the absence of Odland bodies? Dermatologica. 1976;153:253-262.
- Miranda-Romero A, Sánchez Sambucety P, Bajo del Pozo C, et al. Unilateral hyperkeratosis lenticularis perstans (Flegel's disease). J Am Acad Dermatol. 1998;39:655-657.
- Gutiérrez MC, Hasson A, Arias MD, et al. Localized hyperkeratosis lenticularis perstans (Flegel's disease). Cutis. 1991;48:201-204.
- Fathy S, Azadeh B. Hyperkeratosis lenticularis perstans. Int J Dermatol. 1988;27:120-121.
- Rosdahl I, Rosen K. Hyperkeratosis lenticularis perstans: report on two cases. Acta Derm Venerol. 1985;65:562-564.
- Cooper SM, George S. Flegel's disease treated with psoralen ultraviolet A. Br J Dermatol. 2000;142:340-342.
- Fernandez-Flores A, Manjon JA. Morphological evidence of periodical exacerbation of hyperkeratosis lenticularis perstans. Acta Dermatovenerol Croat. 2009;17:16-19.
- Langer K, Zonzits E, Konrad K. Hyperkeratosis lenticularis perstans (Flegel's disease). ultrastructural study of lesional and perilesional skin and therapeutic trial of topical tretinoin versus 5-fluorouracil. J Am Acad Dermatol. 1992;27:812-816.
- Ishibashi A, Tsuboi R, Fujita K. Familial hyperkeratosis lenticularis perstans. associated with cancers of the digestive organs. J Dermatol. 1984;11:407-409.
- Cunha Filho RR, Almeida Jr HL. Hyperkeratosis lenticularis perstans. An Bras Dermatol. 2011;86(4 suppl 1):S76-S77.
- Blaheta HJ, Metzler G, Rassner G, et al. Hyperkeratosis lenticularis perstans (Flegel's disease)—lack of response to treatment with tacalcitol and calcipotriol. Dermatology. 2001;202:255-258.
- Flegel H. Hyperkeratosis lenticularis perstans. Hautarzt. 1958;9:363-364.
- Pearson LH, Smith JG, Chalker DK. Hyperkeratosis lenticularis perstans (Flegel’s disease). J Am Acad Dermatol. 1987;16:190-195.
- Ando K, Hattori H, Yamauchi Y. Histopathological differences between early and old lesions of hyperkeratosis lenticularis perstans (Flegel’s disease). Am J Dermatopathol. 2006;28:122-126.
- Fernández-Crehuet P, Rodríguez-Rey E, Ríos-Martín JJ, et al. Hyperkeratosis lenticularis perstans, or Flegel disease, with palmoplantar involvement. Actas Dermosifiliogr. 2009;100:157-159.
- Sterneberg-Vos H, van Marion AM, Frank J, et al. Hyperkeratosis lenticularis perstans (Flegel’s disease)—successful treatment with topical corticosteroids. Int J Dermatol. 2008;47:38-41.
- Bayramgürler D, Apaydin R, Dökmeci S, et al. Flegel’s disease: treatment with topical calcipotriol. Clin Exp Dermatol. 2002;27:161-162.
- Price ML, Jones EW, MacDonald DM. A clinicopathological study of Flegel’s disease (hyperkeratosis lenticularis perstans). Br J Dermatol. 1987;116:681-691.
- Krishnan A, Kar S. Photoletter to the editor: hyperkeratosis lenticularis perstans (Flegel’s disease) with unusual clinical presentation. response to isotretinoin therapy. J Dermatol Case Rep. 2012;6:93-95.
- Beveridge GW, Langlands AO. Familial hyperkeratosis lenticularis perstans associated with tumours of the skin. Br J Dermatol. 1973;88:453-458.
- Frenk E, Tapernoux B. Hyperkeratosis lenticularis perstans (Flegel): a biological model for keratinization occurring in the absence of Odland bodies? Dermatologica. 1976;153:253-262.
- Miranda-Romero A, Sánchez Sambucety P, Bajo del Pozo C, et al. Unilateral hyperkeratosis lenticularis perstans (Flegel's disease). J Am Acad Dermatol. 1998;39:655-657.
- Gutiérrez MC, Hasson A, Arias MD, et al. Localized hyperkeratosis lenticularis perstans (Flegel's disease). Cutis. 1991;48:201-204.
- Fathy S, Azadeh B. Hyperkeratosis lenticularis perstans. Int J Dermatol. 1988;27:120-121.
- Rosdahl I, Rosen K. Hyperkeratosis lenticularis perstans: report on two cases. Acta Derm Venerol. 1985;65:562-564.
- Cooper SM, George S. Flegel's disease treated with psoralen ultraviolet A. Br J Dermatol. 2000;142:340-342.
- Fernandez-Flores A, Manjon JA. Morphological evidence of periodical exacerbation of hyperkeratosis lenticularis perstans. Acta Dermatovenerol Croat. 2009;17:16-19.
- Langer K, Zonzits E, Konrad K. Hyperkeratosis lenticularis perstans (Flegel's disease). ultrastructural study of lesional and perilesional skin and therapeutic trial of topical tretinoin versus 5-fluorouracil. J Am Acad Dermatol. 1992;27:812-816.
- Ishibashi A, Tsuboi R, Fujita K. Familial hyperkeratosis lenticularis perstans. associated with cancers of the digestive organs. J Dermatol. 1984;11:407-409.
- Cunha Filho RR, Almeida Jr HL. Hyperkeratosis lenticularis perstans. An Bras Dermatol. 2011;86(4 suppl 1):S76-S77.
- Blaheta HJ, Metzler G, Rassner G, et al. Hyperkeratosis lenticularis perstans (Flegel's disease)—lack of response to treatment with tacalcitol and calcipotriol. Dermatology. 2001;202:255-258.

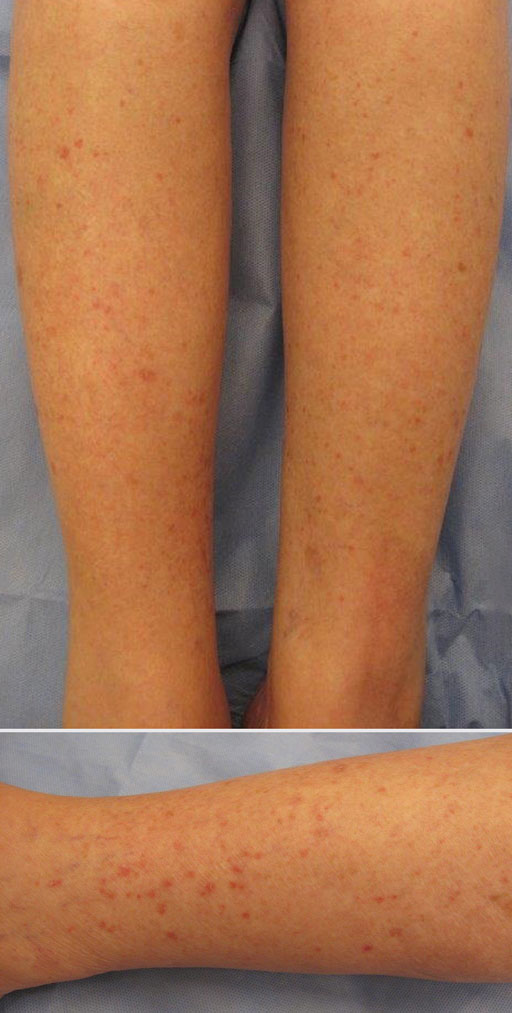
What Is Your Diagnosis? Herpes Zoster
The Diagnosis: Herpes Zoster
Herpes zoster is a common infection that clinically presents with moderate to intense pain in the involved dermatome (1–3 days before the outbreak1) followed by a unilateral dermatomal eruption. The most common sites of involvement include the trunk (dermatomes T3–L2) and upper face (dermatome V1).2 Herpes zoster characteristically begins as erythematous macules and papules that progress to vesicles and sometimes pustules, which subsequently crust over (7–10 days after the initial outbreak).1 Regional lymphadenopathy is present in most cases. Due to the classic presentation of herpes zoster, clinical diagnosis is the mainstay for most cases. Although it can present in any age group, herpes zoster is most commonly associated with advancing age.3
During the course of primary varicella infection, the herpes virus spreads from infected lesions to the contiguous endings of sensory nerves and travels to the dorsal root ganglion cells where it remains latent.1 It is hypothesized that cell-mediated immunity suppresses viral activity and maintains viral latency. A decline in varicella zoster virus–specific cell-mediated immunity can result in reactivation of the latent virus.1 The virus is then transported to the skin from the dorsal root ganglion via myelinated nerves, which terminate at the isthmus of hair follicles, and subsequent infection of the folliculosebaceous unit occurs.4
Laboratory tests that can assist in the diagnosis of herpes zoster, especially atypical cases, include Tzanck smear and viral culture of the vesicle fluid.1 When vesicles are not present, biopsy of the lesion followed by immunohistochemical staining and polymerase chain reaction assay can aid in the diagnosis. The differential diagnosis for our patient included pseudolymphoma, herpes simplex virus, lymphomatoid papulosis, sarcoidosis, trigeminal trophic syndrome, and Sweet syndrome.
Vesicles were not present in our patient, but the dermatomal nature of the eruption and the pain she experienced made the clinical scenario suspicious for herpes zoster. A 4-mm punch biopsy of a single folliculosebaceous unit revealed herpetic, cytopathic features including prominent keratinocyte necrosis involving sebaceous, isthmic, and infundibular epithelium; ballooning of epithelial cells with steel gray nuclei; and multinucleation with nuclear molding (Figure 1). Strong nuclear and cytoplasmic staining was seen in the affected keratinocytes under anti–varicella zoster virus immunohistochemical analysis (Figure 2). Staining for herpes simplex virus types 1 and 2 was negative. Within days of starting valacyclovir 1000 mg (every 8 hours for 1 week), the patient’s symptoms resolved.
| 
| |
|
|
Atypical presentations of herpes zoster (eg, presentations that are not completely dermatomal) are becoming more common. Herpetic infections should always be in the differential diagnosis for cutaneous ulcerations. Misdiagnosis of herpes zoster due to atypical features is common and can delay prompt and adequate treatment.
- Rockley PF, Tyring SK. Pathophysiology and clinical manifestations of varicella zoster virus infections. Int J Dermatol. 1994;33:227-232.
- Chen TM, George S, Woodruff CA, et al. Clinical manifestations of varicella-zoster virus infection. Dermatol Clin. 2002;20:267-282.
- Hope-Simpson RE. The nature of herpes zoster: a long-term study and a new hypothesis. Proc R Soc Med. 1965;58:9-20.
- Walsh N, Boutilier R, Glasgow D, et al. Exclusive involvement of folliculosebaceous units by herpes: a reflection of early herpes zoster. Am J Dermatopathol. 2005;27:189-194.
The Diagnosis: Herpes Zoster
Herpes zoster is a common infection that clinically presents with moderate to intense pain in the involved dermatome (1–3 days before the outbreak1) followed by a unilateral dermatomal eruption. The most common sites of involvement include the trunk (dermatomes T3–L2) and upper face (dermatome V1).2 Herpes zoster characteristically begins as erythematous macules and papules that progress to vesicles and sometimes pustules, which subsequently crust over (7–10 days after the initial outbreak).1 Regional lymphadenopathy is present in most cases. Due to the classic presentation of herpes zoster, clinical diagnosis is the mainstay for most cases. Although it can present in any age group, herpes zoster is most commonly associated with advancing age.3
During the course of primary varicella infection, the herpes virus spreads from infected lesions to the contiguous endings of sensory nerves and travels to the dorsal root ganglion cells where it remains latent.1 It is hypothesized that cell-mediated immunity suppresses viral activity and maintains viral latency. A decline in varicella zoster virus–specific cell-mediated immunity can result in reactivation of the latent virus.1 The virus is then transported to the skin from the dorsal root ganglion via myelinated nerves, which terminate at the isthmus of hair follicles, and subsequent infection of the folliculosebaceous unit occurs.4
Laboratory tests that can assist in the diagnosis of herpes zoster, especially atypical cases, include Tzanck smear and viral culture of the vesicle fluid.1 When vesicles are not present, biopsy of the lesion followed by immunohistochemical staining and polymerase chain reaction assay can aid in the diagnosis. The differential diagnosis for our patient included pseudolymphoma, herpes simplex virus, lymphomatoid papulosis, sarcoidosis, trigeminal trophic syndrome, and Sweet syndrome.
Vesicles were not present in our patient, but the dermatomal nature of the eruption and the pain she experienced made the clinical scenario suspicious for herpes zoster. A 4-mm punch biopsy of a single folliculosebaceous unit revealed herpetic, cytopathic features including prominent keratinocyte necrosis involving sebaceous, isthmic, and infundibular epithelium; ballooning of epithelial cells with steel gray nuclei; and multinucleation with nuclear molding (Figure 1). Strong nuclear and cytoplasmic staining was seen in the affected keratinocytes under anti–varicella zoster virus immunohistochemical analysis (Figure 2). Staining for herpes simplex virus types 1 and 2 was negative. Within days of starting valacyclovir 1000 mg (every 8 hours for 1 week), the patient’s symptoms resolved.
|
|
| |
|
|
Atypical presentations of herpes zoster (eg, presentations that are not completely dermatomal) are becoming more common. Herpetic infections should always be in the differential diagnosis for cutaneous ulcerations. Misdiagnosis of herpes zoster due to atypical features is common and can delay prompt and adequate treatment.
The Diagnosis: Herpes Zoster
Herpes zoster is a common infection that clinically presents with moderate to intense pain in the involved dermatome (1–3 days before the outbreak1) followed by a unilateral dermatomal eruption. The most common sites of involvement include the trunk (dermatomes T3–L2) and upper face (dermatome V1).2 Herpes zoster characteristically begins as erythematous macules and papules that progress to vesicles and sometimes pustules, which subsequently crust over (7–10 days after the initial outbreak).1 Regional lymphadenopathy is present in most cases. Due to the classic presentation of herpes zoster, clinical diagnosis is the mainstay for most cases. Although it can present in any age group, herpes zoster is most commonly associated with advancing age.3
During the course of primary varicella infection, the herpes virus spreads from infected lesions to the contiguous endings of sensory nerves and travels to the dorsal root ganglion cells where it remains latent.1 It is hypothesized that cell-mediated immunity suppresses viral activity and maintains viral latency. A decline in varicella zoster virus–specific cell-mediated immunity can result in reactivation of the latent virus.1 The virus is then transported to the skin from the dorsal root ganglion via myelinated nerves, which terminate at the isthmus of hair follicles, and subsequent infection of the folliculosebaceous unit occurs.4
Laboratory tests that can assist in the diagnosis of herpes zoster, especially atypical cases, include Tzanck smear and viral culture of the vesicle fluid.1 When vesicles are not present, biopsy of the lesion followed by immunohistochemical staining and polymerase chain reaction assay can aid in the diagnosis. The differential diagnosis for our patient included pseudolymphoma, herpes simplex virus, lymphomatoid papulosis, sarcoidosis, trigeminal trophic syndrome, and Sweet syndrome.
Vesicles were not present in our patient, but the dermatomal nature of the eruption and the pain she experienced made the clinical scenario suspicious for herpes zoster. A 4-mm punch biopsy of a single folliculosebaceous unit revealed herpetic, cytopathic features including prominent keratinocyte necrosis involving sebaceous, isthmic, and infundibular epithelium; ballooning of epithelial cells with steel gray nuclei; and multinucleation with nuclear molding (Figure 1). Strong nuclear and cytoplasmic staining was seen in the affected keratinocytes under anti–varicella zoster virus immunohistochemical analysis (Figure 2). Staining for herpes simplex virus types 1 and 2 was negative. Within days of starting valacyclovir 1000 mg (every 8 hours for 1 week), the patient’s symptoms resolved.
|
|
| |
|
|
Atypical presentations of herpes zoster (eg, presentations that are not completely dermatomal) are becoming more common. Herpetic infections should always be in the differential diagnosis for cutaneous ulcerations. Misdiagnosis of herpes zoster due to atypical features is common and can delay prompt and adequate treatment.
- Rockley PF, Tyring SK. Pathophysiology and clinical manifestations of varicella zoster virus infections. Int J Dermatol. 1994;33:227-232.
- Chen TM, George S, Woodruff CA, et al. Clinical manifestations of varicella-zoster virus infection. Dermatol Clin. 2002;20:267-282.
- Hope-Simpson RE. The nature of herpes zoster: a long-term study and a new hypothesis. Proc R Soc Med. 1965;58:9-20.
- Walsh N, Boutilier R, Glasgow D, et al. Exclusive involvement of folliculosebaceous units by herpes: a reflection of early herpes zoster. Am J Dermatopathol. 2005;27:189-194.
- Rockley PF, Tyring SK. Pathophysiology and clinical manifestations of varicella zoster virus infections. Int J Dermatol. 1994;33:227-232.
- Chen TM, George S, Woodruff CA, et al. Clinical manifestations of varicella-zoster virus infection. Dermatol Clin. 2002;20:267-282.
- Hope-Simpson RE. The nature of herpes zoster: a long-term study and a new hypothesis. Proc R Soc Med. 1965;58:9-20.
- Walsh N, Boutilier R, Glasgow D, et al. Exclusive involvement of folliculosebaceous units by herpes: a reflection of early herpes zoster. Am J Dermatopathol. 2005;27:189-194.
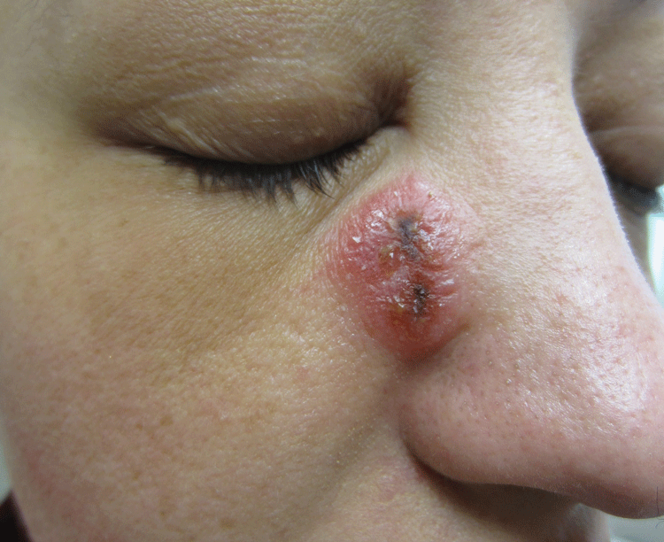
A 32-year-old woman with no remarkable medical history presented with a progressively worsening erythematous and edematous plaque on the right cheek of 8 days’ duration. She had previously been treated by her primary care physician with cephalexin for 3 days and trimethoprim-sulfamethoxazole for 2 days, which resulted in “flattening” of the plaque, but the lesion did not resolve. She was referred to our dermatology clinic for further evaluation. She denied any trauma to the cheek or scratching of the lesion. On physical examination, a 2-cm pink, erythematous, edematous plaque with a central eschar was noted on the right cheek with crusting of the right nasal wall.
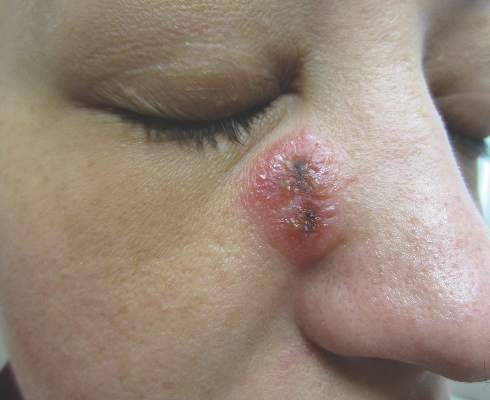
Scaly Plaque With Pustules and Anonychia on the Middle Finger
The Diagnosis: Acrodermatitis Continua of Hallopeau
Acrodermatitis continua of Hallopeau (ACH) is considered to be a form of acropustular psoriasis that presents as a sterile, pustular eruption initially affecting the fingertips and/or toes.1 The slow-growing pustules typically progress locally and can lead to onychodystrophy and/or osteolysis of the underlying bone.2,3 Most commonly affecting adult women, ACH often begins following local trauma to or infection of a single digit.4 As the disease progresses proximally, the small pustules burst, leaving a shiny, erythematous surface on which new pustules can develop. These pustules have a tendency to amalgamate, leading to the characteristic clinical finding of lakes of pus. Pustules frequently appear on the nail matrix and nail bed presenting as severe onychodystrophy and ultimately anonychia.5,6 Rarely, ACH can be associated with generalized pustular psoriasis as well as conjunctivitis, balanitis, and fissuring or annulus migrans of the tongue.2,7
Diagnosis can be established based on clinical findings, biopsy, and bacterial and fungal cultures revealing sterile pustules.8,9 Histologic findings are similar to those seen in pustular psoriasis, demonstrating subcorneal neutrophilic pustules, Munro microabscesses, and dilated blood vessels with lymphocytic infiltrate in the papillary dermis.10
Due to the refractory nature of the disease, there are no recommended guidelines for treatment of ACH. Most successful treatment regimens consist of topical psoriasis medications combined with systemic psoriatic therapies such as cyclosporine, methotrexate, acitretin, or biologic therapy.8,11-16 Our patient achieved satisfactory clinical improvement with clobetasol propionate ointment 0.05% twice daily alternating with calcipotriene cream 0.005% twice daily.
- Suchanek J. Relation of Hallopeau’s acrodermatitis continua to psoriasis. Przegl Dermatol. 1951;1:165-181.
- Adam BA, Loh CL. Acropustulosis (acrodermatitis continua) with resorption of terminal phalanges. Med J Malaysia. 1972;27:30-32.
- Mrowietz U. Pustular eruptions of palms and soles. In: Wolff K, Goldsmith LS, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2007:215-218.
- Yerushalmi J, Grunwald MH, Hallel-Halevy D, et al. Chronic pustular eruption of the thumbs. diagnosis: acrodermatitis continue of Hallopeau (ACH). Arch Dermatol. 2000:136:925-930.
- Granelli U. Impetigo herpetiformis; acrodermatitis continue of Hallopeau and pustular psoriasis; etiology and pathogenesis and differential diagnosis. Minerva Dermatol. 1956;31:120-126.
- Mobini N, Toussaint S, Kamino H. Noninfectious erythematous, papular, and squamous diseases. In: Elder DE, Elenitsas R, Johnson B, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2005:174-210.
- Radcliff-Crocker H. Diseases of the Skin: Their Descriptions, Pathology, Diagnosis and Treatment. Philadelphia, PA: P. Blakiston, Son, & Co; 1888.
- Sehgal VN, Verma P, Sharma S, et al. Review: acrodermatitis continua of Hallopeau: evolution of treatment options. Int J Dermatol. 2011;50:1195-1211.
- Post CF, Hopper ME. Dermatitis repens: a report of two cases with bacteriologic studies. AMA Arc Derm Syphilol. 1951;63:220-223.
- Sehgal VN, Sharma S. The significance of Gram’s stain smear, potassium hydroxide mount, culture and microscopic pathology in the diagnosis of acrodermatitis continua of Hallopeau. Skinmed. 2011;9:260-261.
- Mosser G, Pillekamp H, Peter RU. Suppurative acrodermatitis continua of Hallopeau. a differential diagnosis of paronychia. Dtsch Med Wochenschr. 1998;123:386-390.
- Piquero-Casals J, Fonseca de Mello AP, Dal Coleto C, et al. Using oral tetracycline and topical betamethasone valerate to treat acrodermatitis continua of Hallopeau. Cutis. 2002;70:106-108.
- Tsuji T, Nishimura M. Topically administered fluorouracil in acrodermatitis continua of Hallopeau. Arch Dermatol. 1991;127:27-28.
- Van de Kerkhof PCM. In vivo effects of vitamin D3 analogs. J Dermatolog Treat. 1998;(suppl 3):S25-S29.
- Kokelj F, Plozzer C, Trevisan G. Uselessness of topical calcipotriol as monotherapy for acrodermatitis continua of Hallopeau. Acta Derm Venereol. 2001;81:153.
- Schneider LA, Hinrichs R, Scharffetter-Kochanek K. Phototherapy and photochemotherapy. Clin Dermatol. 2008;26:464-476.
The Diagnosis: Acrodermatitis Continua of Hallopeau
Acrodermatitis continua of Hallopeau (ACH) is considered to be a form of acropustular psoriasis that presents as a sterile, pustular eruption initially affecting the fingertips and/or toes.1 The slow-growing pustules typically progress locally and can lead to onychodystrophy and/or osteolysis of the underlying bone.2,3 Most commonly affecting adult women, ACH often begins following local trauma to or infection of a single digit.4 As the disease progresses proximally, the small pustules burst, leaving a shiny, erythematous surface on which new pustules can develop. These pustules have a tendency to amalgamate, leading to the characteristic clinical finding of lakes of pus. Pustules frequently appear on the nail matrix and nail bed presenting as severe onychodystrophy and ultimately anonychia.5,6 Rarely, ACH can be associated with generalized pustular psoriasis as well as conjunctivitis, balanitis, and fissuring or annulus migrans of the tongue.2,7
Diagnosis can be established based on clinical findings, biopsy, and bacterial and fungal cultures revealing sterile pustules.8,9 Histologic findings are similar to those seen in pustular psoriasis, demonstrating subcorneal neutrophilic pustules, Munro microabscesses, and dilated blood vessels with lymphocytic infiltrate in the papillary dermis.10
Due to the refractory nature of the disease, there are no recommended guidelines for treatment of ACH. Most successful treatment regimens consist of topical psoriasis medications combined with systemic psoriatic therapies such as cyclosporine, methotrexate, acitretin, or biologic therapy.8,11-16 Our patient achieved satisfactory clinical improvement with clobetasol propionate ointment 0.05% twice daily alternating with calcipotriene cream 0.005% twice daily.
The Diagnosis: Acrodermatitis Continua of Hallopeau
Acrodermatitis continua of Hallopeau (ACH) is considered to be a form of acropustular psoriasis that presents as a sterile, pustular eruption initially affecting the fingertips and/or toes.1 The slow-growing pustules typically progress locally and can lead to onychodystrophy and/or osteolysis of the underlying bone.2,3 Most commonly affecting adult women, ACH often begins following local trauma to or infection of a single digit.4 As the disease progresses proximally, the small pustules burst, leaving a shiny, erythematous surface on which new pustules can develop. These pustules have a tendency to amalgamate, leading to the characteristic clinical finding of lakes of pus. Pustules frequently appear on the nail matrix and nail bed presenting as severe onychodystrophy and ultimately anonychia.5,6 Rarely, ACH can be associated with generalized pustular psoriasis as well as conjunctivitis, balanitis, and fissuring or annulus migrans of the tongue.2,7
Diagnosis can be established based on clinical findings, biopsy, and bacterial and fungal cultures revealing sterile pustules.8,9 Histologic findings are similar to those seen in pustular psoriasis, demonstrating subcorneal neutrophilic pustules, Munro microabscesses, and dilated blood vessels with lymphocytic infiltrate in the papillary dermis.10
Due to the refractory nature of the disease, there are no recommended guidelines for treatment of ACH. Most successful treatment regimens consist of topical psoriasis medications combined with systemic psoriatic therapies such as cyclosporine, methotrexate, acitretin, or biologic therapy.8,11-16 Our patient achieved satisfactory clinical improvement with clobetasol propionate ointment 0.05% twice daily alternating with calcipotriene cream 0.005% twice daily.
- Suchanek J. Relation of Hallopeau’s acrodermatitis continua to psoriasis. Przegl Dermatol. 1951;1:165-181.
- Adam BA, Loh CL. Acropustulosis (acrodermatitis continua) with resorption of terminal phalanges. Med J Malaysia. 1972;27:30-32.
- Mrowietz U. Pustular eruptions of palms and soles. In: Wolff K, Goldsmith LS, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2007:215-218.
- Yerushalmi J, Grunwald MH, Hallel-Halevy D, et al. Chronic pustular eruption of the thumbs. diagnosis: acrodermatitis continue of Hallopeau (ACH). Arch Dermatol. 2000:136:925-930.
- Granelli U. Impetigo herpetiformis; acrodermatitis continue of Hallopeau and pustular psoriasis; etiology and pathogenesis and differential diagnosis. Minerva Dermatol. 1956;31:120-126.
- Mobini N, Toussaint S, Kamino H. Noninfectious erythematous, papular, and squamous diseases. In: Elder DE, Elenitsas R, Johnson B, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2005:174-210.
- Radcliff-Crocker H. Diseases of the Skin: Their Descriptions, Pathology, Diagnosis and Treatment. Philadelphia, PA: P. Blakiston, Son, & Co; 1888.
- Sehgal VN, Verma P, Sharma S, et al. Review: acrodermatitis continua of Hallopeau: evolution of treatment options. Int J Dermatol. 2011;50:1195-1211.
- Post CF, Hopper ME. Dermatitis repens: a report of two cases with bacteriologic studies. AMA Arc Derm Syphilol. 1951;63:220-223.
- Sehgal VN, Sharma S. The significance of Gram’s stain smear, potassium hydroxide mount, culture and microscopic pathology in the diagnosis of acrodermatitis continua of Hallopeau. Skinmed. 2011;9:260-261.
- Mosser G, Pillekamp H, Peter RU. Suppurative acrodermatitis continua of Hallopeau. a differential diagnosis of paronychia. Dtsch Med Wochenschr. 1998;123:386-390.
- Piquero-Casals J, Fonseca de Mello AP, Dal Coleto C, et al. Using oral tetracycline and topical betamethasone valerate to treat acrodermatitis continua of Hallopeau. Cutis. 2002;70:106-108.
- Tsuji T, Nishimura M. Topically administered fluorouracil in acrodermatitis continua of Hallopeau. Arch Dermatol. 1991;127:27-28.
- Van de Kerkhof PCM. In vivo effects of vitamin D3 analogs. J Dermatolog Treat. 1998;(suppl 3):S25-S29.
- Kokelj F, Plozzer C, Trevisan G. Uselessness of topical calcipotriol as monotherapy for acrodermatitis continua of Hallopeau. Acta Derm Venereol. 2001;81:153.
- Schneider LA, Hinrichs R, Scharffetter-Kochanek K. Phototherapy and photochemotherapy. Clin Dermatol. 2008;26:464-476.
- Suchanek J. Relation of Hallopeau’s acrodermatitis continua to psoriasis. Przegl Dermatol. 1951;1:165-181.
- Adam BA, Loh CL. Acropustulosis (acrodermatitis continua) with resorption of terminal phalanges. Med J Malaysia. 1972;27:30-32.
- Mrowietz U. Pustular eruptions of palms and soles. In: Wolff K, Goldsmith LS, Katz SI, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw-Hill; 2007:215-218.
- Yerushalmi J, Grunwald MH, Hallel-Halevy D, et al. Chronic pustular eruption of the thumbs. diagnosis: acrodermatitis continue of Hallopeau (ACH). Arch Dermatol. 2000:136:925-930.
- Granelli U. Impetigo herpetiformis; acrodermatitis continue of Hallopeau and pustular psoriasis; etiology and pathogenesis and differential diagnosis. Minerva Dermatol. 1956;31:120-126.
- Mobini N, Toussaint S, Kamino H. Noninfectious erythematous, papular, and squamous diseases. In: Elder DE, Elenitsas R, Johnson B, et al, eds. Lever’s Histopathology of the Skin. 9th ed. Philadelphia, PA: Lippincott, Williams & Wilkins; 2005:174-210.
- Radcliff-Crocker H. Diseases of the Skin: Their Descriptions, Pathology, Diagnosis and Treatment. Philadelphia, PA: P. Blakiston, Son, & Co; 1888.
- Sehgal VN, Verma P, Sharma S, et al. Review: acrodermatitis continua of Hallopeau: evolution of treatment options. Int J Dermatol. 2011;50:1195-1211.
- Post CF, Hopper ME. Dermatitis repens: a report of two cases with bacteriologic studies. AMA Arc Derm Syphilol. 1951;63:220-223.
- Sehgal VN, Sharma S. The significance of Gram’s stain smear, potassium hydroxide mount, culture and microscopic pathology in the diagnosis of acrodermatitis continua of Hallopeau. Skinmed. 2011;9:260-261.
- Mosser G, Pillekamp H, Peter RU. Suppurative acrodermatitis continua of Hallopeau. a differential diagnosis of paronychia. Dtsch Med Wochenschr. 1998;123:386-390.
- Piquero-Casals J, Fonseca de Mello AP, Dal Coleto C, et al. Using oral tetracycline and topical betamethasone valerate to treat acrodermatitis continua of Hallopeau. Cutis. 2002;70:106-108.
- Tsuji T, Nishimura M. Topically administered fluorouracil in acrodermatitis continua of Hallopeau. Arch Dermatol. 1991;127:27-28.
- Van de Kerkhof PCM. In vivo effects of vitamin D3 analogs. J Dermatolog Treat. 1998;(suppl 3):S25-S29.
- Kokelj F, Plozzer C, Trevisan G. Uselessness of topical calcipotriol as monotherapy for acrodermatitis continua of Hallopeau. Acta Derm Venereol. 2001;81:153.
- Schneider LA, Hinrichs R, Scharffetter-Kochanek K. Phototherapy and photochemotherapy. Clin Dermatol. 2008;26:464-476.
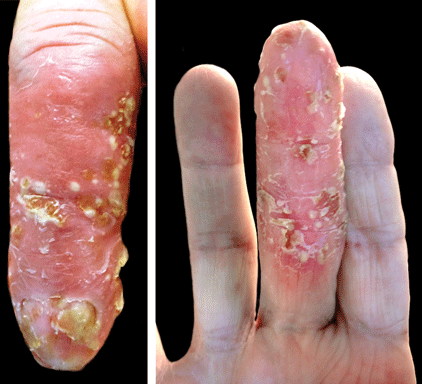
A 69-year-old man presented to our dermatology clinic with a persistent rash on the right middle finger of 5 years’ duration (left). Physical examination revealed a well-demarcated scaly plaque with pustules and anonychia localized to the right middle finger (right). Fungal and bacterial cultures revealed sterile pustules. The patient was successfully treated with an occluded superpotent topical steroid alternating with a topical vitamin D analogue.
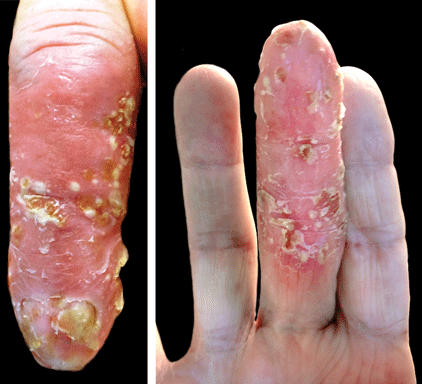
What Is Your Diagnosis? Tinea Corporis
The Diagnosis: Tinea Corporis
Although contact dermatitis from the metal on the back of the watch was suspected, many modern wrist watches are made with stainless steel rather than nickel, which is a common contact allergen; therefore, other diagnoses were considered in the differential including irritant contact dermatitis, psoriasis, and tinea infection. A potassium hydroxide (KOH) preparation was performed to rule out tinea infection. Unexpectedly, the KOH preparation was positive for fungal hyphae, confirming a diagnosis of tinea corporis. The patient was treated with clotrimazole cream 1% twice daily for 3 weeks during which time the rash completely resolved.
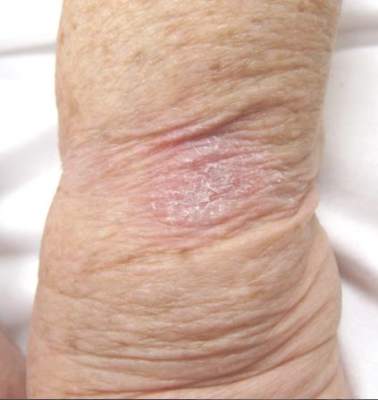
We present this case to stress the importance of performing KOH preparations even when the likelihood of tinea infection seems remote. At our institution, we teach our residents, “If it’s scaly, scrape it.” This adage has served us well. Tinea corporis may be mistaken for many other skin diseases, including eczema, psoriasis, and seborrheic dermatitis.1 A KOH preparation often is a helpful tool in confirming the diagnosis and should be performed when a dermatophyte infection is suspected. The KOH preparation is the most sensitive diagnostic test used to confirm dermatophyte infection, with 90% of infections showing positive results.2,3
Tinea infections may occur anywhere on the body, but areas that are prone to excessive heat and/or moisture are particularly susceptible.4 Dermatophyte infections typically present as annular, scaly, pruritic patches or plaques often with central clearing and an active border.1 In our patient, the lesion showed characteristics that were suggestive of a dermatophyte infection but was somewhat atypical in appearance, as it lacked central clearing (Figure). The 3 genera of dermatophytes—Trichophyton, Microsporum, and Epidermophyton—are common causes of fungal infections.2 The pathogenesis of dermatophytosis is the synthesis of keratinases that digest keratin and sustain the presence of the fungi. Local factors such as sweating and occlusion facilitate the activity of these organisms.2 In our case, the pathogenesis was believed to be due to the entrapment of moisture behind the patient’s watch, creating a favorable environment for fungal growth.
- Ely JW, Rosenfeld S, Seabury SM. Diagnosis and management of tinea infections. Am Fam Physician. 2014:90:702-710.
- Wolff K, Saavedra AP, Fitzpatrick TB. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill Medical; 2013.
- Levitt JO, Levitt BH, Akhavan A, et al. The sensitivity and specificity of potassium hydroxide smear and fungal culture relative to clinical assessment in the evaluation of tinea pedis: a pooled analysis (published online ahead of print June 22, 2010). Dermatol Res Pract. doi:10.1155/2010/764843.
- Gupta AK, Chaudhry M, Elewski B. Tinea corporis, tinea cruris, tinea nigra and piedra. Dermatol Clin. 2003;21:395-400.
The Diagnosis: Tinea Corporis
Although contact dermatitis from the metal on the back of the watch was suspected, many modern wrist watches are made with stainless steel rather than nickel, which is a common contact allergen; therefore, other diagnoses were considered in the differential including irritant contact dermatitis, psoriasis, and tinea infection. A potassium hydroxide (KOH) preparation was performed to rule out tinea infection. Unexpectedly, the KOH preparation was positive for fungal hyphae, confirming a diagnosis of tinea corporis. The patient was treated with clotrimazole cream 1% twice daily for 3 weeks during which time the rash completely resolved.
We present this case to stress the importance of performing KOH preparations even when the likelihood of tinea infection seems remote. At our institution, we teach our residents, “If it’s scaly, scrape it.” This adage has served us well. Tinea corporis may be mistaken for many other skin diseases, including eczema, psoriasis, and seborrheic dermatitis.1 A KOH preparation often is a helpful tool in confirming the diagnosis and should be performed when a dermatophyte infection is suspected. The KOH preparation is the most sensitive diagnostic test used to confirm dermatophyte infection, with 90% of infections showing positive results.2,3
Tinea infections may occur anywhere on the body, but areas that are prone to excessive heat and/or moisture are particularly susceptible.4 Dermatophyte infections typically present as annular, scaly, pruritic patches or plaques often with central clearing and an active border.1 In our patient, the lesion showed characteristics that were suggestive of a dermatophyte infection but was somewhat atypical in appearance, as it lacked central clearing (Figure). The 3 genera of dermatophytes—Trichophyton, Microsporum, and Epidermophyton—are common causes of fungal infections.2 The pathogenesis of dermatophytosis is the synthesis of keratinases that digest keratin and sustain the presence of the fungi. Local factors such as sweating and occlusion facilitate the activity of these organisms.2 In our case, the pathogenesis was believed to be due to the entrapment of moisture behind the patient’s watch, creating a favorable environment for fungal growth.
The Diagnosis: Tinea Corporis
Although contact dermatitis from the metal on the back of the watch was suspected, many modern wrist watches are made with stainless steel rather than nickel, which is a common contact allergen; therefore, other diagnoses were considered in the differential including irritant contact dermatitis, psoriasis, and tinea infection. A potassium hydroxide (KOH) preparation was performed to rule out tinea infection. Unexpectedly, the KOH preparation was positive for fungal hyphae, confirming a diagnosis of tinea corporis. The patient was treated with clotrimazole cream 1% twice daily for 3 weeks during which time the rash completely resolved.
We present this case to stress the importance of performing KOH preparations even when the likelihood of tinea infection seems remote. At our institution, we teach our residents, “If it’s scaly, scrape it.” This adage has served us well. Tinea corporis may be mistaken for many other skin diseases, including eczema, psoriasis, and seborrheic dermatitis.1 A KOH preparation often is a helpful tool in confirming the diagnosis and should be performed when a dermatophyte infection is suspected. The KOH preparation is the most sensitive diagnostic test used to confirm dermatophyte infection, with 90% of infections showing positive results.2,3
Tinea infections may occur anywhere on the body, but areas that are prone to excessive heat and/or moisture are particularly susceptible.4 Dermatophyte infections typically present as annular, scaly, pruritic patches or plaques often with central clearing and an active border.1 In our patient, the lesion showed characteristics that were suggestive of a dermatophyte infection but was somewhat atypical in appearance, as it lacked central clearing (Figure). The 3 genera of dermatophytes—Trichophyton, Microsporum, and Epidermophyton—are common causes of fungal infections.2 The pathogenesis of dermatophytosis is the synthesis of keratinases that digest keratin and sustain the presence of the fungi. Local factors such as sweating and occlusion facilitate the activity of these organisms.2 In our case, the pathogenesis was believed to be due to the entrapment of moisture behind the patient’s watch, creating a favorable environment for fungal growth.
- Ely JW, Rosenfeld S, Seabury SM. Diagnosis and management of tinea infections. Am Fam Physician. 2014:90:702-710.
- Wolff K, Saavedra AP, Fitzpatrick TB. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill Medical; 2013.
- Levitt JO, Levitt BH, Akhavan A, et al. The sensitivity and specificity of potassium hydroxide smear and fungal culture relative to clinical assessment in the evaluation of tinea pedis: a pooled analysis (published online ahead of print June 22, 2010). Dermatol Res Pract. doi:10.1155/2010/764843.
- Gupta AK, Chaudhry M, Elewski B. Tinea corporis, tinea cruris, tinea nigra and piedra. Dermatol Clin. 2003;21:395-400.
- Ely JW, Rosenfeld S, Seabury SM. Diagnosis and management of tinea infections. Am Fam Physician. 2014:90:702-710.
- Wolff K, Saavedra AP, Fitzpatrick TB. Fitzpatrick’s Color Atlas and Synopsis of Clinical Dermatology. 7th ed. New York, NY: McGraw-Hill Medical; 2013.
- Levitt JO, Levitt BH, Akhavan A, et al. The sensitivity and specificity of potassium hydroxide smear and fungal culture relative to clinical assessment in the evaluation of tinea pedis: a pooled analysis (published online ahead of print June 22, 2010). Dermatol Res Pract. doi:10.1155/2010/764843.
- Gupta AK, Chaudhry M, Elewski B. Tinea corporis, tinea cruris, tinea nigra and piedra. Dermatol Clin. 2003;21:395-400.
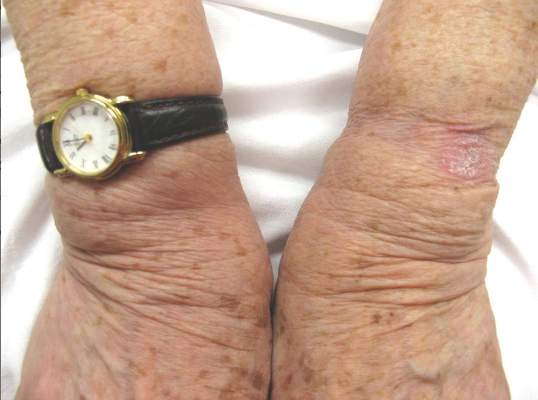
An 81-year-old woman presented with a 2-cm erythematous, scaly, pruritic rash on the left dorsal wrist localized to the skin under her watch. The patient first noticed the lesion 2 months prior. She moved the watch to the right wrist a few days prior to presentation and no symptoms developed in that location. No other areas of the skin were affected. She had no known allergies and was otherwise in good health.
Linear Bluish Black Papules on the Shoulder
The Diagnosis: Agminated Blue Nevus
Agminated blue nevus is a rare melanocytic nevus that characteristically presents as a group of multiple small, bluish papules occurring in a well-circumscribed area.1 It also has been referred to as a plaque-type nevus2,3 and an eruptive blue nevus.4 Originally described by Upshaw et al2 in 1947, agminated blue nevi may be congenital or arise in early childhood and almost always occur on the trunk. The skin between the papules often is unaffected or sometimes may show bluish or brown pigmentation.4 Agminated blue nevi usually are smaller than 10 cm in diameter; however, rare cases have measured up to 24 cm.1,4-6 The incidence of agminated blue nevi is 2 times higher in males than in females.1
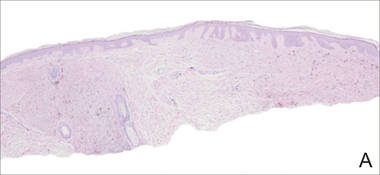
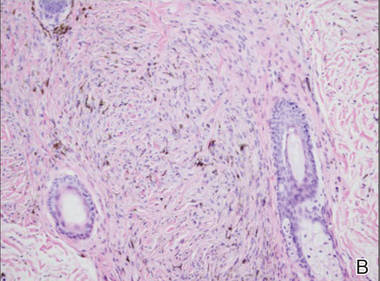
|
A biopsy of the lesion revealed pigmented dendritic melanocytes admixed with melanophages, forming fascicles and bundles (A)(H&E, original magnification ×10). In some areas the dermis was uninvolved. Higher magnification showed dendritic and epithelioid melanocytes extending down along the adnexal structures (B)(H&E, original magnification ×40). |
Histopathologically, agminated blue nevi typically demonstrate the features of common and/or cellular blue nevi. Cytologic atypia and mitoses are rare.1 The degree of cellularity and pigmentation of the lesions is variable, and the presence of subcutaneous cellular nodules also has been described.5
In our patient, histologic evaluation revealed foci of diffuse dermal spindle cell proliferation composed of heavily pigmented dendritic melanocytes admixed with melanophages in a fibrotic stroma (Figure, A). The dermis was uninvolved in some areas and the melanocytes were epithelioid and formed fascicles and bundles that extended down adnexal structures in other areas (Figure, B). Junctional involvement of melanocytes, cellular atypia, and mitoses were not identified. Our case demonstrated a combination of histologic findings of a cellular blue nevus as well as features reminiscent of a deep penetrating nevus. The differential diagnosis of agminated blue nevus includes agminated Spitz nevus arising in a speckled lentiginous nevus,7 dermal melanocytosis, melanoma, and pilar neurocristic hamartoma. Pilar neurocristic hamartomas may resemble plaque-type blue nevi; however, the former show a predilection for the scalp, histologically demonstrate features that overlap with blue nevi and congenital nevi, and are associated with neural structures that show Schwannian differentiation.8 Agminated blue nevi usually are characterized by a benign clinical course, but few cases describing malignant changes with development of malignant melanoma have been reported.9,10 Therefore, recognition of the clinical and histopathologic spectrum of agminated blue nevus is critical in order to avoid diagnostic pitfalls and confusion with melanoma.
- Vélez A, del-Río E, Martín-de-Hijas C, et al. Agminated blue nevi: case report and review of the literature. Dermatology. 1993;186:144-148.
- Upshaw BY, Ghormley RK, Montgomery H. Extensive blue nevus of Jadassohn-Tièche; report of a case. Surgery. 1947;22:761-765.
- Pittman JL, Fisher BK. Plaque-type blue nevus. Arch Dermatol. 1976;112:1127-1128.
- Hendricks WM. Eruptive blue nevi. J Am Acad Dermatol.1981;4:50-53.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Shenfield HT, Maize JC. Multiple and agminated blue nevi. J Dermatol Surg Oncol. 1980;6:725-728.
- Misago N, Narisawa Y, Kohda H. A combination of speckled lentiginous nevus with patch-type blue nevus. J Dermatol. 1993;20:643-647.
- Bevona C, Tannous Z, Tsao H. Dermal melanocytic proliferation with features of a plaque-type blue nevus and neurocristic hamartoma. J Am Acad Dermatol. 2003;49:924-929.
- Yeh I, Fang Y, Busam KJ. Melanoma arising in a large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2012;36:1258-1263.
- Zattra E, Salmaso R, Montesco MC, et al. Large plaque type blue nevus with subcutaneous cellular nodules. Eur J Dermatol. 2009;19:287-288.
The Diagnosis: Agminated Blue Nevus
Agminated blue nevus is a rare melanocytic nevus that characteristically presents as a group of multiple small, bluish papules occurring in a well-circumscribed area.1 It also has been referred to as a plaque-type nevus2,3 and an eruptive blue nevus.4 Originally described by Upshaw et al2 in 1947, agminated blue nevi may be congenital or arise in early childhood and almost always occur on the trunk. The skin between the papules often is unaffected or sometimes may show bluish or brown pigmentation.4 Agminated blue nevi usually are smaller than 10 cm in diameter; however, rare cases have measured up to 24 cm.1,4-6 The incidence of agminated blue nevi is 2 times higher in males than in females.1
|
|
A biopsy of the lesion revealed pigmented dendritic melanocytes admixed with melanophages, forming fascicles and bundles (A)(H&E, original magnification ×10). In some areas the dermis was uninvolved. Higher magnification showed dendritic and epithelioid melanocytes extending down along the adnexal structures (B)(H&E, original magnification ×40). |
Histopathologically, agminated blue nevi typically demonstrate the features of common and/or cellular blue nevi. Cytologic atypia and mitoses are rare.1 The degree of cellularity and pigmentation of the lesions is variable, and the presence of subcutaneous cellular nodules also has been described.5
In our patient, histologic evaluation revealed foci of diffuse dermal spindle cell proliferation composed of heavily pigmented dendritic melanocytes admixed with melanophages in a fibrotic stroma (Figure, A). The dermis was uninvolved in some areas and the melanocytes were epithelioid and formed fascicles and bundles that extended down adnexal structures in other areas (Figure, B). Junctional involvement of melanocytes, cellular atypia, and mitoses were not identified. Our case demonstrated a combination of histologic findings of a cellular blue nevus as well as features reminiscent of a deep penetrating nevus. The differential diagnosis of agminated blue nevus includes agminated Spitz nevus arising in a speckled lentiginous nevus,7 dermal melanocytosis, melanoma, and pilar neurocristic hamartoma. Pilar neurocristic hamartomas may resemble plaque-type blue nevi; however, the former show a predilection for the scalp, histologically demonstrate features that overlap with blue nevi and congenital nevi, and are associated with neural structures that show Schwannian differentiation.8 Agminated blue nevi usually are characterized by a benign clinical course, but few cases describing malignant changes with development of malignant melanoma have been reported.9,10 Therefore, recognition of the clinical and histopathologic spectrum of agminated blue nevus is critical in order to avoid diagnostic pitfalls and confusion with melanoma.
The Diagnosis: Agminated Blue Nevus
Agminated blue nevus is a rare melanocytic nevus that characteristically presents as a group of multiple small, bluish papules occurring in a well-circumscribed area.1 It also has been referred to as a plaque-type nevus2,3 and an eruptive blue nevus.4 Originally described by Upshaw et al2 in 1947, agminated blue nevi may be congenital or arise in early childhood and almost always occur on the trunk. The skin between the papules often is unaffected or sometimes may show bluish or brown pigmentation.4 Agminated blue nevi usually are smaller than 10 cm in diameter; however, rare cases have measured up to 24 cm.1,4-6 The incidence of agminated blue nevi is 2 times higher in males than in females.1
|
|
A biopsy of the lesion revealed pigmented dendritic melanocytes admixed with melanophages, forming fascicles and bundles (A)(H&E, original magnification ×10). In some areas the dermis was uninvolved. Higher magnification showed dendritic and epithelioid melanocytes extending down along the adnexal structures (B)(H&E, original magnification ×40). |
Histopathologically, agminated blue nevi typically demonstrate the features of common and/or cellular blue nevi. Cytologic atypia and mitoses are rare.1 The degree of cellularity and pigmentation of the lesions is variable, and the presence of subcutaneous cellular nodules also has been described.5
In our patient, histologic evaluation revealed foci of diffuse dermal spindle cell proliferation composed of heavily pigmented dendritic melanocytes admixed with melanophages in a fibrotic stroma (Figure, A). The dermis was uninvolved in some areas and the melanocytes were epithelioid and formed fascicles and bundles that extended down adnexal structures in other areas (Figure, B). Junctional involvement of melanocytes, cellular atypia, and mitoses were not identified. Our case demonstrated a combination of histologic findings of a cellular blue nevus as well as features reminiscent of a deep penetrating nevus. The differential diagnosis of agminated blue nevus includes agminated Spitz nevus arising in a speckled lentiginous nevus,7 dermal melanocytosis, melanoma, and pilar neurocristic hamartoma. Pilar neurocristic hamartomas may resemble plaque-type blue nevi; however, the former show a predilection for the scalp, histologically demonstrate features that overlap with blue nevi and congenital nevi, and are associated with neural structures that show Schwannian differentiation.8 Agminated blue nevi usually are characterized by a benign clinical course, but few cases describing malignant changes with development of malignant melanoma have been reported.9,10 Therefore, recognition of the clinical and histopathologic spectrum of agminated blue nevus is critical in order to avoid diagnostic pitfalls and confusion with melanoma.
- Vélez A, del-Río E, Martín-de-Hijas C, et al. Agminated blue nevi: case report and review of the literature. Dermatology. 1993;186:144-148.
- Upshaw BY, Ghormley RK, Montgomery H. Extensive blue nevus of Jadassohn-Tièche; report of a case. Surgery. 1947;22:761-765.
- Pittman JL, Fisher BK. Plaque-type blue nevus. Arch Dermatol. 1976;112:1127-1128.
- Hendricks WM. Eruptive blue nevi. J Am Acad Dermatol.1981;4:50-53.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Shenfield HT, Maize JC. Multiple and agminated blue nevi. J Dermatol Surg Oncol. 1980;6:725-728.
- Misago N, Narisawa Y, Kohda H. A combination of speckled lentiginous nevus with patch-type blue nevus. J Dermatol. 1993;20:643-647.
- Bevona C, Tannous Z, Tsao H. Dermal melanocytic proliferation with features of a plaque-type blue nevus and neurocristic hamartoma. J Am Acad Dermatol. 2003;49:924-929.
- Yeh I, Fang Y, Busam KJ. Melanoma arising in a large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2012;36:1258-1263.
- Zattra E, Salmaso R, Montesco MC, et al. Large plaque type blue nevus with subcutaneous cellular nodules. Eur J Dermatol. 2009;19:287-288.
- Vélez A, del-Río E, Martín-de-Hijas C, et al. Agminated blue nevi: case report and review of the literature. Dermatology. 1993;186:144-148.
- Upshaw BY, Ghormley RK, Montgomery H. Extensive blue nevus of Jadassohn-Tièche; report of a case. Surgery. 1947;22:761-765.
- Pittman JL, Fisher BK. Plaque-type blue nevus. Arch Dermatol. 1976;112:1127-1128.
- Hendricks WM. Eruptive blue nevi. J Am Acad Dermatol.1981;4:50-53.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Shenfield HT, Maize JC. Multiple and agminated blue nevi. J Dermatol Surg Oncol. 1980;6:725-728.
- Misago N, Narisawa Y, Kohda H. A combination of speckled lentiginous nevus with patch-type blue nevus. J Dermatol. 1993;20:643-647.
- Bevona C, Tannous Z, Tsao H. Dermal melanocytic proliferation with features of a plaque-type blue nevus and neurocristic hamartoma. J Am Acad Dermatol. 2003;49:924-929.
- Yeh I, Fang Y, Busam KJ. Melanoma arising in a large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2012;36:1258-1263.
- Zattra E, Salmaso R, Montesco MC, et al. Large plaque type blue nevus with subcutaneous cellular nodules. Eur J Dermatol. 2009;19:287-288.
A 57-year-old woman presented with an asymptomatic, unchanging, 3.5×0.7-cm linear plaque on the right shoulder composed of dozens of clustered, bluish black papules that had been present for several decades. The skin between the papules was unaffected. The patient’s medical and family histories were unremarkable. A deep shave biopsy from the center of the plaque was performed.
Sessile Pink Plaque on the Lower Back
The Diagnosis: Eccrine Poroma
A shave biopsy of the lesion was performed for definitive diagnosis and demonstrated a well-circumscribed tumor with cords and broad columns composed of uniform basaloid cells extending into the dermis and in areas connecting to the overlying epidermis (Figure). There also were small ducts and cysts admixed in the tumor columns that were embedded in a tumor stroma rich in blood vessels. A diagnosis of eccrine poroma was made based on these characteristic histologic features.
|
Biopsy revealed a basaloid tumor originating from the epidermis and extending into the dermis (A)(H&E, original magnification ×4). On higher magnification, ducts were evident amongst the tumor cells and a vascular rich stroma was revealed (B)(H&E, original magnification ×10). |
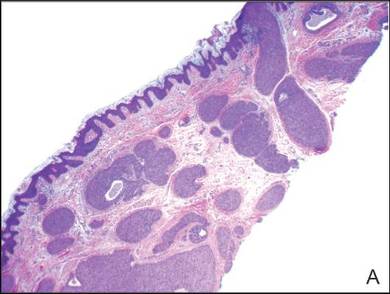

First described by Pinkus et al1 in 1956, eccrine poroma is a benign neoplasm of cells from the intraepidermal ductal portion of the eccrine sweat gland. Eccrine poroma (along with hidroacanthoma simplex, dermal duct tumor, and poroid hidradenoma) is one of the poroid neoplasms, which account for approximately 10% of all primary sweat gland tumors.2 Eccrine poroma usually is seen in patients over 40 years of age without any predilection for race or sex.
Characteristically, eccrine poromas clinically manifest as solitary, firm, sharply demarcated papules or nodules that may be sessile or pedunculated and rarely exceed 2 cm in diameter. This entity classically presents on acral, non–hair-bearing areas (eg, palms and soles). Eccrine poromas have a wide range of clinical appearances that can lead to broad differential diagnoses3 and have been described as flesh-colored,3 pink to red,4 purple,5 and pigmented3,4 papules or nodules depending on features such as blood vessel proliferation and pigment deposition.
Eccrine poromas also have been reported on hair-bearing areas of the body, including the head,3 neck,3,6 chest,4,6 hip,7 and pubic area,8 despite the paucity of eccrine glands in these areas on the body. These findings suggest that these neoplasms may not be purely eccrine in origin. The wide range of clinical presentations of eccrine poromas has prompted investigation into further classification and delineation of this neoplasm.3 The occurrence of eccrine poromas on areas of the skin known to have few eccrine glands suggests that eccrine poromas may not be purely comprised of eccrine ducts and instead may be of apocrine origin.3,9,10 Histologic features of eccrine poromas that suggest apocrine origination include sebaceous and follicular differentiation (eg, folliculocentric distribution), the association with the follicular infundibulum, and the presence of follicular germ cells.3,9,10 Thus, apocrine gland involvement in eccrine poromas may account for their appearance in anatomic areas that do not have high concentrations of eccrine glands, such as the trunk and pubic area.
Based on these findings, eccrine poromas may therefore be of eccrine and/or apocrine origin; however, the nomenclature of this neoplasm remains confusing and possibly misleading, as the term eccrine poroma continues to be accepted even in instances in which the differentiation appears to be largely apocrine. The terms poroma and eccrine poroma often are used interchangeably, which contributes to the confusion by failing to acknowledge the possibility of apocrine influence and possibly causing the clinician to exclude eccrine poromas from the differential diagnosis in areas that do not have high concentrations of eccrine glands.
Because of their high degree of clinical variability, characteristic acral location, and misleading nomenclature, eccrine poromas often are mistakenly confused with a long list of other cutaneous neoplasms, including hemangiomas, pyogenic granulomas, melanocytic nevi, warts, cysts, and other adnexal neoplasms.3 In our case, the lesion was abnormally large and was clinically concerning for an unusual sebaceous nevus. Its location on the lower back is not commonly noted and should remind the clinician of the possibility of apocrine differentiation. Clinicians should be aware of the wide phenotypic diversity of eccrine poromas, and therefore they should consider this diagnosis in their differential diagnosis for solitary papules or nodules occurring in any anatomic area.
- Pinkus H, Rogin JR, Goldman P. Eccrine poroma: tumors exhibiting features of the epidermal sweat duct unit. Arch Dermatol. 1956;74:511-521.
- Pylyser K, Dewolf-Peeters C, Marlen K. The histology of eccrine poromas: a study of 14 cases. Dermatologica. 1983;167:243-249.
- Moore TO, Orman HL, Orman SK, et al. Poromas of the head and neck. J Am Acad Dermatol. 2001;44:48-52.
- Agarwal S, Kumar B, Sharma N. Nodule on the chest. eccrine poroma. Indian J Dermatol Venereol Leprol. 2009;75:639.
- Ackerman AB, Abenoza P. Neoplasms With Eccrine Differentiation. Philadelphia, PA: Lea & Febinger; 1990:113-185.
- Okun M, Ansell H. Eccrine poroma. report of three cases, two with an unusual location. Arch Dermatol. 1963;88:561-566.
- Sarma DP, Zaman SU, Santos EE, et al. Poroma of the hip and buttock. Dermatol Online J. 2009;15:10.
- Altamura D, Piccolo D, Lozzi GP, et al. Eccrine poroma in an unusual site: a clinical and dermoscopic simulator of amelanotic melanoma. J Am Acad Dermatol. 2005;53:539-541.
- Groben PA, Hitchcock MG, Leshin B, et al. Apocrine poroma, a distinctive case in a patient with nevoid BCC. Am J Dermatopathol. 1992;21:31-33.
- Harvell JD, Kerschmann RL, LeBoit PE. Eccrine or apocrine poroma? six poromas with divergent adnexal differentiation. Am J Dermatopathol. 1996;18:1-9.
The Diagnosis: Eccrine Poroma
A shave biopsy of the lesion was performed for definitive diagnosis and demonstrated a well-circumscribed tumor with cords and broad columns composed of uniform basaloid cells extending into the dermis and in areas connecting to the overlying epidermis (Figure). There also were small ducts and cysts admixed in the tumor columns that were embedded in a tumor stroma rich in blood vessels. A diagnosis of eccrine poroma was made based on these characteristic histologic features.
|
Biopsy revealed a basaloid tumor originating from the epidermis and extending into the dermis (A)(H&E, original magnification ×4). On higher magnification, ducts were evident amongst the tumor cells and a vascular rich stroma was revealed (B)(H&E, original magnification ×10). |
First described by Pinkus et al1 in 1956, eccrine poroma is a benign neoplasm of cells from the intraepidermal ductal portion of the eccrine sweat gland. Eccrine poroma (along with hidroacanthoma simplex, dermal duct tumor, and poroid hidradenoma) is one of the poroid neoplasms, which account for approximately 10% of all primary sweat gland tumors.2 Eccrine poroma usually is seen in patients over 40 years of age without any predilection for race or sex.
Characteristically, eccrine poromas clinically manifest as solitary, firm, sharply demarcated papules or nodules that may be sessile or pedunculated and rarely exceed 2 cm in diameter. This entity classically presents on acral, non–hair-bearing areas (eg, palms and soles). Eccrine poromas have a wide range of clinical appearances that can lead to broad differential diagnoses3 and have been described as flesh-colored,3 pink to red,4 purple,5 and pigmented3,4 papules or nodules depending on features such as blood vessel proliferation and pigment deposition.
Eccrine poromas also have been reported on hair-bearing areas of the body, including the head,3 neck,3,6 chest,4,6 hip,7 and pubic area,8 despite the paucity of eccrine glands in these areas on the body. These findings suggest that these neoplasms may not be purely eccrine in origin. The wide range of clinical presentations of eccrine poromas has prompted investigation into further classification and delineation of this neoplasm.3 The occurrence of eccrine poromas on areas of the skin known to have few eccrine glands suggests that eccrine poromas may not be purely comprised of eccrine ducts and instead may be of apocrine origin.3,9,10 Histologic features of eccrine poromas that suggest apocrine origination include sebaceous and follicular differentiation (eg, folliculocentric distribution), the association with the follicular infundibulum, and the presence of follicular germ cells.3,9,10 Thus, apocrine gland involvement in eccrine poromas may account for their appearance in anatomic areas that do not have high concentrations of eccrine glands, such as the trunk and pubic area.
Based on these findings, eccrine poromas may therefore be of eccrine and/or apocrine origin; however, the nomenclature of this neoplasm remains confusing and possibly misleading, as the term eccrine poroma continues to be accepted even in instances in which the differentiation appears to be largely apocrine. The terms poroma and eccrine poroma often are used interchangeably, which contributes to the confusion by failing to acknowledge the possibility of apocrine influence and possibly causing the clinician to exclude eccrine poromas from the differential diagnosis in areas that do not have high concentrations of eccrine glands.
Because of their high degree of clinical variability, characteristic acral location, and misleading nomenclature, eccrine poromas often are mistakenly confused with a long list of other cutaneous neoplasms, including hemangiomas, pyogenic granulomas, melanocytic nevi, warts, cysts, and other adnexal neoplasms.3 In our case, the lesion was abnormally large and was clinically concerning for an unusual sebaceous nevus. Its location on the lower back is not commonly noted and should remind the clinician of the possibility of apocrine differentiation. Clinicians should be aware of the wide phenotypic diversity of eccrine poromas, and therefore they should consider this diagnosis in their differential diagnosis for solitary papules or nodules occurring in any anatomic area.
The Diagnosis: Eccrine Poroma
A shave biopsy of the lesion was performed for definitive diagnosis and demonstrated a well-circumscribed tumor with cords and broad columns composed of uniform basaloid cells extending into the dermis and in areas connecting to the overlying epidermis (Figure). There also were small ducts and cysts admixed in the tumor columns that were embedded in a tumor stroma rich in blood vessels. A diagnosis of eccrine poroma was made based on these characteristic histologic features.
|
Biopsy revealed a basaloid tumor originating from the epidermis and extending into the dermis (A)(H&E, original magnification ×4). On higher magnification, ducts were evident amongst the tumor cells and a vascular rich stroma was revealed (B)(H&E, original magnification ×10). |
First described by Pinkus et al1 in 1956, eccrine poroma is a benign neoplasm of cells from the intraepidermal ductal portion of the eccrine sweat gland. Eccrine poroma (along with hidroacanthoma simplex, dermal duct tumor, and poroid hidradenoma) is one of the poroid neoplasms, which account for approximately 10% of all primary sweat gland tumors.2 Eccrine poroma usually is seen in patients over 40 years of age without any predilection for race or sex.
Characteristically, eccrine poromas clinically manifest as solitary, firm, sharply demarcated papules or nodules that may be sessile or pedunculated and rarely exceed 2 cm in diameter. This entity classically presents on acral, non–hair-bearing areas (eg, palms and soles). Eccrine poromas have a wide range of clinical appearances that can lead to broad differential diagnoses3 and have been described as flesh-colored,3 pink to red,4 purple,5 and pigmented3,4 papules or nodules depending on features such as blood vessel proliferation and pigment deposition.
Eccrine poromas also have been reported on hair-bearing areas of the body, including the head,3 neck,3,6 chest,4,6 hip,7 and pubic area,8 despite the paucity of eccrine glands in these areas on the body. These findings suggest that these neoplasms may not be purely eccrine in origin. The wide range of clinical presentations of eccrine poromas has prompted investigation into further classification and delineation of this neoplasm.3 The occurrence of eccrine poromas on areas of the skin known to have few eccrine glands suggests that eccrine poromas may not be purely comprised of eccrine ducts and instead may be of apocrine origin.3,9,10 Histologic features of eccrine poromas that suggest apocrine origination include sebaceous and follicular differentiation (eg, folliculocentric distribution), the association with the follicular infundibulum, and the presence of follicular germ cells.3,9,10 Thus, apocrine gland involvement in eccrine poromas may account for their appearance in anatomic areas that do not have high concentrations of eccrine glands, such as the trunk and pubic area.
Based on these findings, eccrine poromas may therefore be of eccrine and/or apocrine origin; however, the nomenclature of this neoplasm remains confusing and possibly misleading, as the term eccrine poroma continues to be accepted even in instances in which the differentiation appears to be largely apocrine. The terms poroma and eccrine poroma often are used interchangeably, which contributes to the confusion by failing to acknowledge the possibility of apocrine influence and possibly causing the clinician to exclude eccrine poromas from the differential diagnosis in areas that do not have high concentrations of eccrine glands.
Because of their high degree of clinical variability, characteristic acral location, and misleading nomenclature, eccrine poromas often are mistakenly confused with a long list of other cutaneous neoplasms, including hemangiomas, pyogenic granulomas, melanocytic nevi, warts, cysts, and other adnexal neoplasms.3 In our case, the lesion was abnormally large and was clinically concerning for an unusual sebaceous nevus. Its location on the lower back is not commonly noted and should remind the clinician of the possibility of apocrine differentiation. Clinicians should be aware of the wide phenotypic diversity of eccrine poromas, and therefore they should consider this diagnosis in their differential diagnosis for solitary papules or nodules occurring in any anatomic area.
- Pinkus H, Rogin JR, Goldman P. Eccrine poroma: tumors exhibiting features of the epidermal sweat duct unit. Arch Dermatol. 1956;74:511-521.
- Pylyser K, Dewolf-Peeters C, Marlen K. The histology of eccrine poromas: a study of 14 cases. Dermatologica. 1983;167:243-249.
- Moore TO, Orman HL, Orman SK, et al. Poromas of the head and neck. J Am Acad Dermatol. 2001;44:48-52.
- Agarwal S, Kumar B, Sharma N. Nodule on the chest. eccrine poroma. Indian J Dermatol Venereol Leprol. 2009;75:639.
- Ackerman AB, Abenoza P. Neoplasms With Eccrine Differentiation. Philadelphia, PA: Lea & Febinger; 1990:113-185.
- Okun M, Ansell H. Eccrine poroma. report of three cases, two with an unusual location. Arch Dermatol. 1963;88:561-566.
- Sarma DP, Zaman SU, Santos EE, et al. Poroma of the hip and buttock. Dermatol Online J. 2009;15:10.
- Altamura D, Piccolo D, Lozzi GP, et al. Eccrine poroma in an unusual site: a clinical and dermoscopic simulator of amelanotic melanoma. J Am Acad Dermatol. 2005;53:539-541.
- Groben PA, Hitchcock MG, Leshin B, et al. Apocrine poroma, a distinctive case in a patient with nevoid BCC. Am J Dermatopathol. 1992;21:31-33.
- Harvell JD, Kerschmann RL, LeBoit PE. Eccrine or apocrine poroma? six poromas with divergent adnexal differentiation. Am J Dermatopathol. 1996;18:1-9.
- Pinkus H, Rogin JR, Goldman P. Eccrine poroma: tumors exhibiting features of the epidermal sweat duct unit. Arch Dermatol. 1956;74:511-521.
- Pylyser K, Dewolf-Peeters C, Marlen K. The histology of eccrine poromas: a study of 14 cases. Dermatologica. 1983;167:243-249.
- Moore TO, Orman HL, Orman SK, et al. Poromas of the head and neck. J Am Acad Dermatol. 2001;44:48-52.
- Agarwal S, Kumar B, Sharma N. Nodule on the chest. eccrine poroma. Indian J Dermatol Venereol Leprol. 2009;75:639.
- Ackerman AB, Abenoza P. Neoplasms With Eccrine Differentiation. Philadelphia, PA: Lea & Febinger; 1990:113-185.
- Okun M, Ansell H. Eccrine poroma. report of three cases, two with an unusual location. Arch Dermatol. 1963;88:561-566.
- Sarma DP, Zaman SU, Santos EE, et al. Poroma of the hip and buttock. Dermatol Online J. 2009;15:10.
- Altamura D, Piccolo D, Lozzi GP, et al. Eccrine poroma in an unusual site: a clinical and dermoscopic simulator of amelanotic melanoma. J Am Acad Dermatol. 2005;53:539-541.
- Groben PA, Hitchcock MG, Leshin B, et al. Apocrine poroma, a distinctive case in a patient with nevoid BCC. Am J Dermatopathol. 1992;21:31-33.
- Harvell JD, Kerschmann RL, LeBoit PE. Eccrine or apocrine poroma? six poromas with divergent adnexal differentiation. Am J Dermatopathol. 1996;18:1-9.
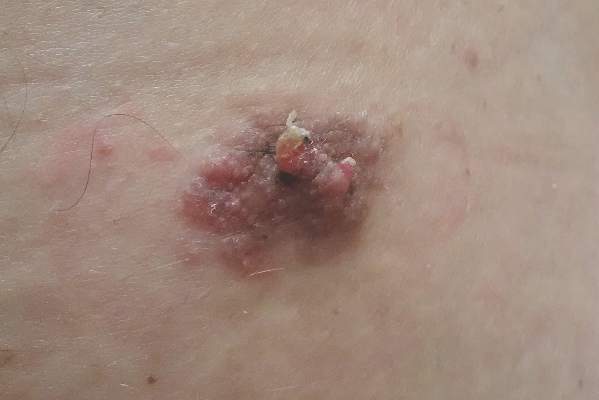
A 47-year-old man presented with an asymptomatic, 2.5×1.5-cm, sessile pink plaque with a coalescing papular texture on the lower back of 30 years’ duration. The patient reported that 2 papillated papules with peripheral rims of dark crust had developed in the center of the lesion over the past 6 months. His personal and family histories were unremarkable.