User login
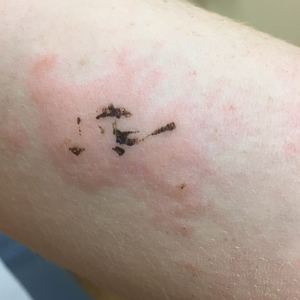
Blanchable Erythematous Patches on the Fingers
The Diagnosis: Irritant Contact Dermatitis
The diagnosis of irritant contact dermatitis secondary to skateboarding is similar to pool palms, a benign, self-limiting irritant contact dermatitis.1 We propose that contact with concrete surfaces during skateboarding can lead to a presentation similar to pool palms. In our case, it was likely that the finger pulpitis noted in the physical examination was due to daily skateboarding rather than once-weekly swimming. Furthermore, the fingertip contact with concrete in pool palms is similar to the rough surface exposure on the skateboard.
Pool palms is more commonly reported in children due to their participation in sports and other activities with recent exposure to rough surfaces, most commonly the floor of swimming pools.2 The condition resolves after eliminating exposures.3 The frequency and duration of exposure to rough surfaces in swimming pools leading to development of this condition is unknown.
There have been mixed reports on the pathogenesis of pool palms. Some literature supports the idea that it is a wet dermatitis, a combination of prolonged water contact, friction, chemicals, and microbes leading to a chronic dermatitis. This theory states that the primary factor influencing the development of erythematous patches on the fingers, palms, and soles is the hyperhydration of the corneal layer at these sites.4 A different theory attributes pool palms to a mechanical origin, such as repeated microtrauma from contact with the rough concrete surfaces of swimming pools.5 This theory further states that the chemicals in pool water, such as chlorine and sodium hypochlorite, rarely produce irritant, allergic, or urticarial reactions.3
Based on these theories, we hypothesized that fingertip pulpitis can result from activities other than swimming (eg, skateboarding). Our case supports the latter theory on fingertip pulpitis in pool palms being a result of frictional dermatitis rather than wet dermatitis because we attributed our patient’s findings to contact with rough surfaces during skateboarding. Although the patient did swim, he only did so once weekly in the summer months, and the lesions had been persistent for 2 years consistently. His skateboarding hobby was more frequent, and he endorsed contact of the pads of the bilateral second to fifth fingers to the rough surfaces of the road and skateboard. The patient did not have lesions on the toes, further supporting the hypothesis that skateboarding led to the current presentation.
In children, hand-foot-and-mouth disease classically presents with oval-shaped, erythematous vesicles on the palmar surfaces of the hands and feet and generally is accompanied by fever and sore throat.6 Furthermore, unlike in our case, the viral exanthem usually would be present for up to 3 weeks and would not persist for more than 2 years. Erythema multiforme has an erythematous color and can present on the palms; however, the lesions have a classic targetoid appearance. It would be unique for erythema multiforme to present only on the fingertips rather than more diffusely on the palms or in other areas such as the face.7 Limited cutaneous sclerosis (scleroderma) initially can present with edematous pitted scars on the digital tips; however, with time the fingers will have a taut, white, shiny appearance that can develop into contractures and debilitating ulcerations.8 In our patient, the plaques did not advance to any further disease. Lastly, in contrast to our patient, punctate palmoplantar keratoderma presents as hyperkeratotic, firm, translucent, or opaque papules on the palms and soles. Over time, the papules can appear verrucous or callouslike.9 In our case, the plaques on the fingertips were erythematous rather than translucent or opaque papules.
Our case raises questions on whether prior reports of pool palms can be attributed to other activities involving contact with rough surfaces. More research is needed on the frequency and duration of rough surface exposure resulting in fingertip pulpitis.
- Lopez-Neyra A, Vano-Galvan S, Alvarez-Twose I, et al. Pool palms [in Spanish]. Dermatol Online J. 2009;15:17.
- Wong LC, Rogers M. Pool palms. Pediatr Dermatol. 2007;24:95.
- Mandojana RM. Pool palms. J Am Acad Dermatol. 1993;28(2 pt 1):280-281.
- Novoa A, Klear S. Pool palms [published online September 30, 2015]. Arch Dis Child. 2016;101:41.
- Martín JM, Martín JM, Ricart JM. Erythematous-violaceous lesions on the palms [in Spanish]. Actas Dermosifiliogr. 2009;100:507-508.
- Marcini AJ, Shani-Adir A. Other viral diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:1345-1366.
- French LE, Prins C. Erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrosis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:319-334.
- Connoly MK. Systemic sclerosis (scleroderma) and related disorders. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:643-646.
- Krol AL, Siegel D. Keratodermas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:871-886.
The Diagnosis: Irritant Contact Dermatitis
The diagnosis of irritant contact dermatitis secondary to skateboarding is similar to pool palms, a benign, self-limiting irritant contact dermatitis.1 We propose that contact with concrete surfaces during skateboarding can lead to a presentation similar to pool palms. In our case, it was likely that the finger pulpitis noted in the physical examination was due to daily skateboarding rather than once-weekly swimming. Furthermore, the fingertip contact with concrete in pool palms is similar to the rough surface exposure on the skateboard.
Pool palms is more commonly reported in children due to their participation in sports and other activities with recent exposure to rough surfaces, most commonly the floor of swimming pools.2 The condition resolves after eliminating exposures.3 The frequency and duration of exposure to rough surfaces in swimming pools leading to development of this condition is unknown.
There have been mixed reports on the pathogenesis of pool palms. Some literature supports the idea that it is a wet dermatitis, a combination of prolonged water contact, friction, chemicals, and microbes leading to a chronic dermatitis. This theory states that the primary factor influencing the development of erythematous patches on the fingers, palms, and soles is the hyperhydration of the corneal layer at these sites.4 A different theory attributes pool palms to a mechanical origin, such as repeated microtrauma from contact with the rough concrete surfaces of swimming pools.5 This theory further states that the chemicals in pool water, such as chlorine and sodium hypochlorite, rarely produce irritant, allergic, or urticarial reactions.3
Based on these theories, we hypothesized that fingertip pulpitis can result from activities other than swimming (eg, skateboarding). Our case supports the latter theory on fingertip pulpitis in pool palms being a result of frictional dermatitis rather than wet dermatitis because we attributed our patient’s findings to contact with rough surfaces during skateboarding. Although the patient did swim, he only did so once weekly in the summer months, and the lesions had been persistent for 2 years consistently. His skateboarding hobby was more frequent, and he endorsed contact of the pads of the bilateral second to fifth fingers to the rough surfaces of the road and skateboard. The patient did not have lesions on the toes, further supporting the hypothesis that skateboarding led to the current presentation.
In children, hand-foot-and-mouth disease classically presents with oval-shaped, erythematous vesicles on the palmar surfaces of the hands and feet and generally is accompanied by fever and sore throat.6 Furthermore, unlike in our case, the viral exanthem usually would be present for up to 3 weeks and would not persist for more than 2 years. Erythema multiforme has an erythematous color and can present on the palms; however, the lesions have a classic targetoid appearance. It would be unique for erythema multiforme to present only on the fingertips rather than more diffusely on the palms or in other areas such as the face.7 Limited cutaneous sclerosis (scleroderma) initially can present with edematous pitted scars on the digital tips; however, with time the fingers will have a taut, white, shiny appearance that can develop into contractures and debilitating ulcerations.8 In our patient, the plaques did not advance to any further disease. Lastly, in contrast to our patient, punctate palmoplantar keratoderma presents as hyperkeratotic, firm, translucent, or opaque papules on the palms and soles. Over time, the papules can appear verrucous or callouslike.9 In our case, the plaques on the fingertips were erythematous rather than translucent or opaque papules.
Our case raises questions on whether prior reports of pool palms can be attributed to other activities involving contact with rough surfaces. More research is needed on the frequency and duration of rough surface exposure resulting in fingertip pulpitis.
The Diagnosis: Irritant Contact Dermatitis
The diagnosis of irritant contact dermatitis secondary to skateboarding is similar to pool palms, a benign, self-limiting irritant contact dermatitis.1 We propose that contact with concrete surfaces during skateboarding can lead to a presentation similar to pool palms. In our case, it was likely that the finger pulpitis noted in the physical examination was due to daily skateboarding rather than once-weekly swimming. Furthermore, the fingertip contact with concrete in pool palms is similar to the rough surface exposure on the skateboard.
Pool palms is more commonly reported in children due to their participation in sports and other activities with recent exposure to rough surfaces, most commonly the floor of swimming pools.2 The condition resolves after eliminating exposures.3 The frequency and duration of exposure to rough surfaces in swimming pools leading to development of this condition is unknown.
There have been mixed reports on the pathogenesis of pool palms. Some literature supports the idea that it is a wet dermatitis, a combination of prolonged water contact, friction, chemicals, and microbes leading to a chronic dermatitis. This theory states that the primary factor influencing the development of erythematous patches on the fingers, palms, and soles is the hyperhydration of the corneal layer at these sites.4 A different theory attributes pool palms to a mechanical origin, such as repeated microtrauma from contact with the rough concrete surfaces of swimming pools.5 This theory further states that the chemicals in pool water, such as chlorine and sodium hypochlorite, rarely produce irritant, allergic, or urticarial reactions.3
Based on these theories, we hypothesized that fingertip pulpitis can result from activities other than swimming (eg, skateboarding). Our case supports the latter theory on fingertip pulpitis in pool palms being a result of frictional dermatitis rather than wet dermatitis because we attributed our patient’s findings to contact with rough surfaces during skateboarding. Although the patient did swim, he only did so once weekly in the summer months, and the lesions had been persistent for 2 years consistently. His skateboarding hobby was more frequent, and he endorsed contact of the pads of the bilateral second to fifth fingers to the rough surfaces of the road and skateboard. The patient did not have lesions on the toes, further supporting the hypothesis that skateboarding led to the current presentation.
In children, hand-foot-and-mouth disease classically presents with oval-shaped, erythematous vesicles on the palmar surfaces of the hands and feet and generally is accompanied by fever and sore throat.6 Furthermore, unlike in our case, the viral exanthem usually would be present for up to 3 weeks and would not persist for more than 2 years. Erythema multiforme has an erythematous color and can present on the palms; however, the lesions have a classic targetoid appearance. It would be unique for erythema multiforme to present only on the fingertips rather than more diffusely on the palms or in other areas such as the face.7 Limited cutaneous sclerosis (scleroderma) initially can present with edematous pitted scars on the digital tips; however, with time the fingers will have a taut, white, shiny appearance that can develop into contractures and debilitating ulcerations.8 In our patient, the plaques did not advance to any further disease. Lastly, in contrast to our patient, punctate palmoplantar keratoderma presents as hyperkeratotic, firm, translucent, or opaque papules on the palms and soles. Over time, the papules can appear verrucous or callouslike.9 In our case, the plaques on the fingertips were erythematous rather than translucent or opaque papules.
Our case raises questions on whether prior reports of pool palms can be attributed to other activities involving contact with rough surfaces. More research is needed on the frequency and duration of rough surface exposure resulting in fingertip pulpitis.
- Lopez-Neyra A, Vano-Galvan S, Alvarez-Twose I, et al. Pool palms [in Spanish]. Dermatol Online J. 2009;15:17.
- Wong LC, Rogers M. Pool palms. Pediatr Dermatol. 2007;24:95.
- Mandojana RM. Pool palms. J Am Acad Dermatol. 1993;28(2 pt 1):280-281.
- Novoa A, Klear S. Pool palms [published online September 30, 2015]. Arch Dis Child. 2016;101:41.
- Martín JM, Martín JM, Ricart JM. Erythematous-violaceous lesions on the palms [in Spanish]. Actas Dermosifiliogr. 2009;100:507-508.
- Marcini AJ, Shani-Adir A. Other viral diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:1345-1366.
- French LE, Prins C. Erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrosis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:319-334.
- Connoly MK. Systemic sclerosis (scleroderma) and related disorders. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:643-646.
- Krol AL, Siegel D. Keratodermas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:871-886.
- Lopez-Neyra A, Vano-Galvan S, Alvarez-Twose I, et al. Pool palms [in Spanish]. Dermatol Online J. 2009;15:17.
- Wong LC, Rogers M. Pool palms. Pediatr Dermatol. 2007;24:95.
- Mandojana RM. Pool palms. J Am Acad Dermatol. 1993;28(2 pt 1):280-281.
- Novoa A, Klear S. Pool palms [published online September 30, 2015]. Arch Dis Child. 2016;101:41.
- Martín JM, Martín JM, Ricart JM. Erythematous-violaceous lesions on the palms [in Spanish]. Actas Dermosifiliogr. 2009;100:507-508.
- Marcini AJ, Shani-Adir A. Other viral diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:1345-1366.
- French LE, Prins C. Erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrosis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:319-334.
- Connoly MK. Systemic sclerosis (scleroderma) and related disorders. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:643-646.
- Krol AL, Siegel D. Keratodermas. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Ltd; 2012:871-886.

A 12-year-old boy presented with well-defined, blanchable, erythematous patches on the distal bilateral palmar aspects of the second to fifth fingers of 2 years’ duration. The patient stated that he skateboarded daily throughout the year and swam once weekly in the summer months. Furthermore, the patient cited frequent contact with the rough undersurface of the skateboard and concrete road surfaces while skateboarding. He stated that the lesions were always present and worsened in the summer months. The lesions had an occasional burning sensation when they were more prominently erythematous, and the patient denied any pattern of exacerbation, numbness, bleeding, or itching. There was no notable family history or evidence of systemic disease.

Verrucous Coalescing Dry Papules and Plaques on the Hip and Flank
The Diagnosis: Blaschkitis
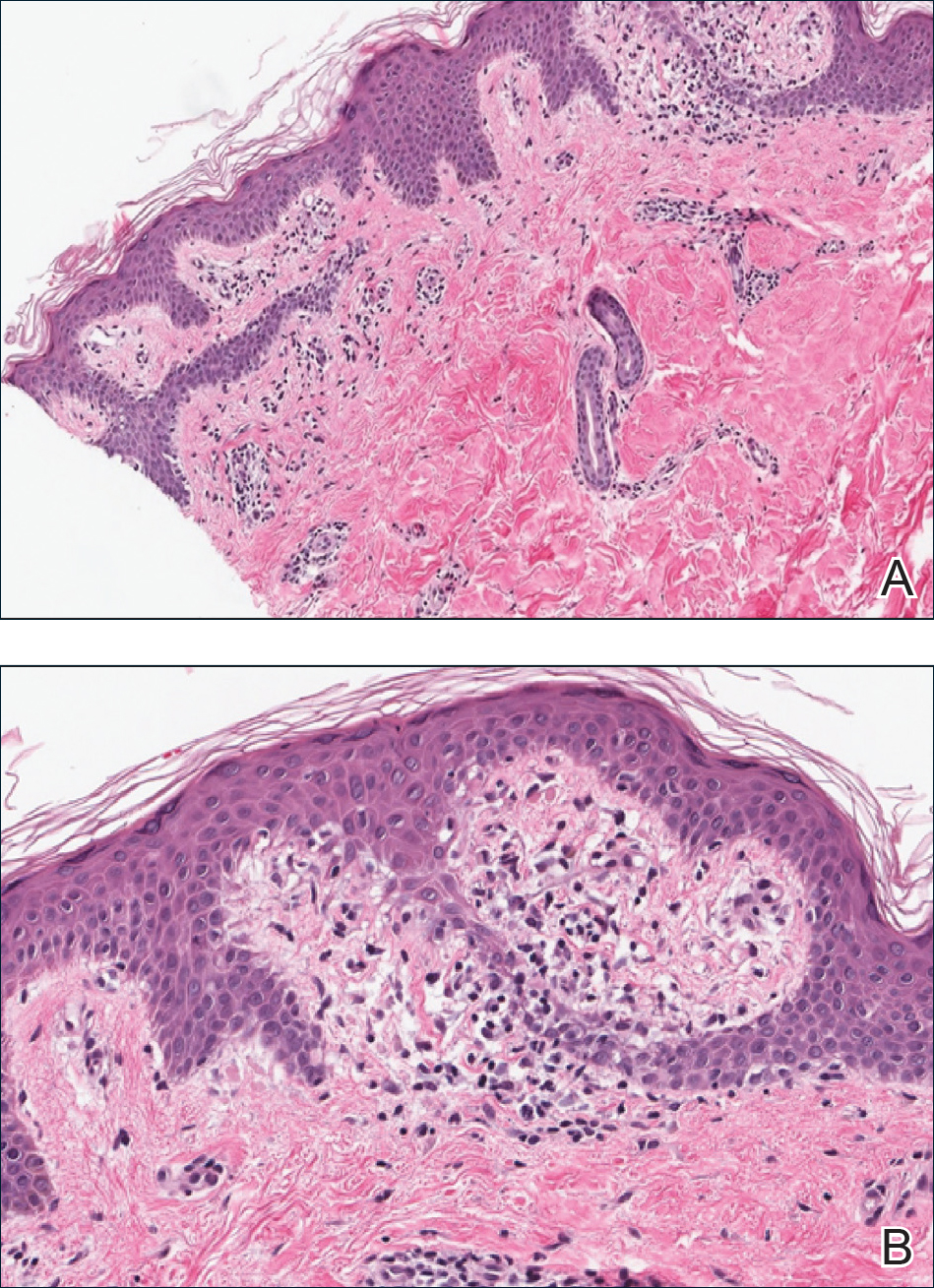
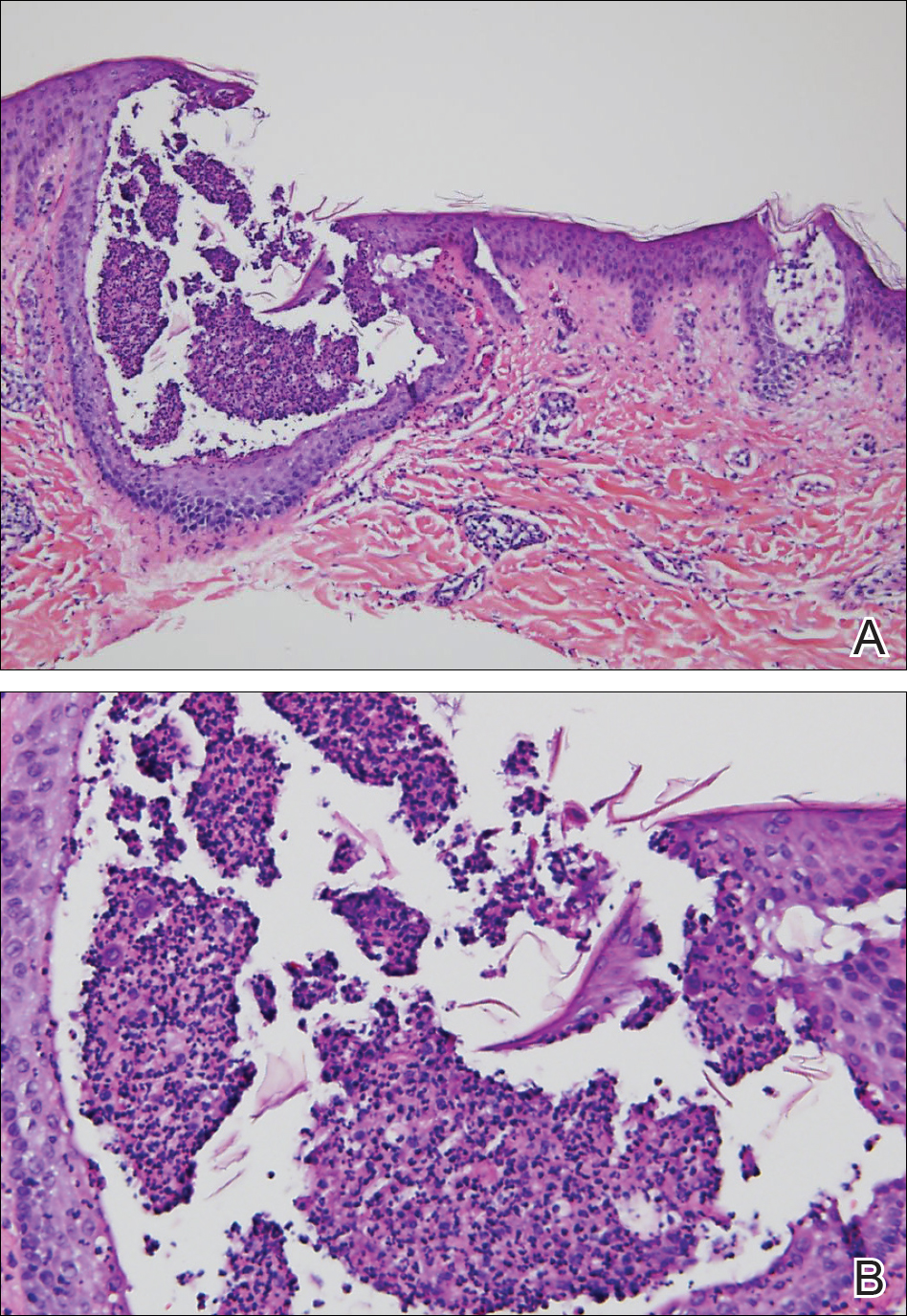
A punch biopsy from the right lateral hip was performed. Histopathologic examination revealed orthokeratosis overlying mild psoriasiform epidermal hyperplasia associated with a lichenoid infiltrate composed almost entirely of lymphocytes (Figure). The infiltrate did not entirely obscure the dermoepidermal junction and spared the adnexal structures. The clinical presentation along with histopathologic analysis confirmed a diagnosis of blaschkitis. The lesions were treated with triamcinolone ointment twice daily as needed, and the patient reported symptomatic and clinical improvement with this intervention at 4-week follow-up.
Described by Grosshans and Marot1 in 1990, blaschkitis is an acquired inflammatory dermatosis following the lines of Blaschko. It predominantly is seen on the trunk and typically presents with pruritic papules and vesicles. It frequently has a relapsing course and is more commonly found in adults. Blaschkitis is considered a form of cutaneous mosaicism representing somatic mutations affecting epidermal cell growth and migration during embryogenesis. It has been proposed that these aberrant cells are not clinically apparent at birth; however, viral infection and drug or other environmental triggers can induce antigen presentation of the clone cells activating a T cell–mediated inflammatory response.2-4
The differential diagnosis includes other acquired Blaschko-linear dermatoses such as lichen striatus, inflammatory linear verrucous epidermal nevus, unilateral lichen planus, linear lichen sclerosus, linear psoriasis, linear fixed drug reaction, lichen nitidus, and others.1,4 Given the overlap in clinical and histopathological presentations of the entities in the differential, it often is difficult to discern the etiology of the papular and vesicular eruption in question. Discrimination of one etiology from the others is further confounded by the fact that these lesions can all be pruritic and initially are treated with topical corticosteroids. A degree of clinical suspicion for blaschkitis coupled with prior understanding of lesional manifestations is helpful in this circumstance. Although classic lichen planus often affects the arms, legs, flexor surfaces, and occasionally the oral mucosa, blaschkitis generally is limited to the trunk. The lesions of lichen planus generally are violaceous, flattopped, polygonal papules that tend to coalesce. They often have a thin, transparent, and adherent scale overlying the papular lesions, and they occasionally demonstrate Wickham striae, which are faint white reticulated networks typically seen in oral mucosal lesions. In the case of lichen nitidus, lesions often follow a geometric line due to the Köbner response, whereas physical trauma from scratching or injury causes lesions to form along the line of insult. Assessing patients for any newly initiated medications can help eliminate lichenoid drug eruptions. Lichen striatus perhaps has the most overlap with blaschkitis, having been described as also following the lines of Blaschko but occurring in children rather than adults. Inflammatory linear verrucous epidermal nevi also can be distinguished from blaschkitis on this premise, as these lesions arise during the first 5 years of life and generally affect the lower extremities.4,5
Histopathology is somewhat variable but generally includes spongiotic dermatitis with concomitant interface
dermatitis characterized by T-cell infiltration. Spongiosis is a feature less commonly seen in lichen striatus. Lesions can progress over time from spongiotic dermatitis to spongiotic psoriasiform dermatitis and later to spongiotic psoriasiform lichenoid dermatitis.4 Treatment of blaschkitis should begin with reassurance of the benign nature of the dermatosis. Pruritic symptoms can be managed with a course of topical steroids.
Blaschkitis is a rare and self-limiting acquired dermatosis that should be incorporated into the differential diagnosis of Blaschko-linear dermatoses. Further investigation is needed to determine if blaschkitis and lichen striatus represent the ends of a disease spectrum or completely distinct entities.
- Grosshans E, Marot L. Blaschkitis in adults. Ann Dermatol Venereol. 1990;117:9-15.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis: reappraisal of the concept of blaschkolinear dermatoses [published online November 29, 2010]. Br J Dermatol. 2011;164:257-262.
- Sun BK, Tsao H. X-chromosome inactivation and skin disease. J Invest Dermatol. 2008;128:2753-2759.
- Keegan BR, Kamino H, Fangman W, et al. “Pediatric blaschkitis”: expanding spectrum of childhood acquired Blaschko-linear dermatoses. Pediatr Dermatol. 2007;24:261-267.
- Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
The Diagnosis: Blaschkitis
A punch biopsy from the right lateral hip was performed. Histopathologic examination revealed orthokeratosis overlying mild psoriasiform epidermal hyperplasia associated with a lichenoid infiltrate composed almost entirely of lymphocytes (Figure). The infiltrate did not entirely obscure the dermoepidermal junction and spared the adnexal structures. The clinical presentation along with histopathologic analysis confirmed a diagnosis of blaschkitis. The lesions were treated with triamcinolone ointment twice daily as needed, and the patient reported symptomatic and clinical improvement with this intervention at 4-week follow-up.
Described by Grosshans and Marot1 in 1990, blaschkitis is an acquired inflammatory dermatosis following the lines of Blaschko. It predominantly is seen on the trunk and typically presents with pruritic papules and vesicles. It frequently has a relapsing course and is more commonly found in adults. Blaschkitis is considered a form of cutaneous mosaicism representing somatic mutations affecting epidermal cell growth and migration during embryogenesis. It has been proposed that these aberrant cells are not clinically apparent at birth; however, viral infection and drug or other environmental triggers can induce antigen presentation of the clone cells activating a T cell–mediated inflammatory response.2-4
The differential diagnosis includes other acquired Blaschko-linear dermatoses such as lichen striatus, inflammatory linear verrucous epidermal nevus, unilateral lichen planus, linear lichen sclerosus, linear psoriasis, linear fixed drug reaction, lichen nitidus, and others.1,4 Given the overlap in clinical and histopathological presentations of the entities in the differential, it often is difficult to discern the etiology of the papular and vesicular eruption in question. Discrimination of one etiology from the others is further confounded by the fact that these lesions can all be pruritic and initially are treated with topical corticosteroids. A degree of clinical suspicion for blaschkitis coupled with prior understanding of lesional manifestations is helpful in this circumstance. Although classic lichen planus often affects the arms, legs, flexor surfaces, and occasionally the oral mucosa, blaschkitis generally is limited to the trunk. The lesions of lichen planus generally are violaceous, flattopped, polygonal papules that tend to coalesce. They often have a thin, transparent, and adherent scale overlying the papular lesions, and they occasionally demonstrate Wickham striae, which are faint white reticulated networks typically seen in oral mucosal lesions. In the case of lichen nitidus, lesions often follow a geometric line due to the Köbner response, whereas physical trauma from scratching or injury causes lesions to form along the line of insult. Assessing patients for any newly initiated medications can help eliminate lichenoid drug eruptions. Lichen striatus perhaps has the most overlap with blaschkitis, having been described as also following the lines of Blaschko but occurring in children rather than adults. Inflammatory linear verrucous epidermal nevi also can be distinguished from blaschkitis on this premise, as these lesions arise during the first 5 years of life and generally affect the lower extremities.4,5
Histopathology is somewhat variable but generally includes spongiotic dermatitis with concomitant interface
dermatitis characterized by T-cell infiltration. Spongiosis is a feature less commonly seen in lichen striatus. Lesions can progress over time from spongiotic dermatitis to spongiotic psoriasiform dermatitis and later to spongiotic psoriasiform lichenoid dermatitis.4 Treatment of blaschkitis should begin with reassurance of the benign nature of the dermatosis. Pruritic symptoms can be managed with a course of topical steroids.
Blaschkitis is a rare and self-limiting acquired dermatosis that should be incorporated into the differential diagnosis of Blaschko-linear dermatoses. Further investigation is needed to determine if blaschkitis and lichen striatus represent the ends of a disease spectrum or completely distinct entities.
The Diagnosis: Blaschkitis
A punch biopsy from the right lateral hip was performed. Histopathologic examination revealed orthokeratosis overlying mild psoriasiform epidermal hyperplasia associated with a lichenoid infiltrate composed almost entirely of lymphocytes (Figure). The infiltrate did not entirely obscure the dermoepidermal junction and spared the adnexal structures. The clinical presentation along with histopathologic analysis confirmed a diagnosis of blaschkitis. The lesions were treated with triamcinolone ointment twice daily as needed, and the patient reported symptomatic and clinical improvement with this intervention at 4-week follow-up.
Described by Grosshans and Marot1 in 1990, blaschkitis is an acquired inflammatory dermatosis following the lines of Blaschko. It predominantly is seen on the trunk and typically presents with pruritic papules and vesicles. It frequently has a relapsing course and is more commonly found in adults. Blaschkitis is considered a form of cutaneous mosaicism representing somatic mutations affecting epidermal cell growth and migration during embryogenesis. It has been proposed that these aberrant cells are not clinically apparent at birth; however, viral infection and drug or other environmental triggers can induce antigen presentation of the clone cells activating a T cell–mediated inflammatory response.2-4
The differential diagnosis includes other acquired Blaschko-linear dermatoses such as lichen striatus, inflammatory linear verrucous epidermal nevus, unilateral lichen planus, linear lichen sclerosus, linear psoriasis, linear fixed drug reaction, lichen nitidus, and others.1,4 Given the overlap in clinical and histopathological presentations of the entities in the differential, it often is difficult to discern the etiology of the papular and vesicular eruption in question. Discrimination of one etiology from the others is further confounded by the fact that these lesions can all be pruritic and initially are treated with topical corticosteroids. A degree of clinical suspicion for blaschkitis coupled with prior understanding of lesional manifestations is helpful in this circumstance. Although classic lichen planus often affects the arms, legs, flexor surfaces, and occasionally the oral mucosa, blaschkitis generally is limited to the trunk. The lesions of lichen planus generally are violaceous, flattopped, polygonal papules that tend to coalesce. They often have a thin, transparent, and adherent scale overlying the papular lesions, and they occasionally demonstrate Wickham striae, which are faint white reticulated networks typically seen in oral mucosal lesions. In the case of lichen nitidus, lesions often follow a geometric line due to the Köbner response, whereas physical trauma from scratching or injury causes lesions to form along the line of insult. Assessing patients for any newly initiated medications can help eliminate lichenoid drug eruptions. Lichen striatus perhaps has the most overlap with blaschkitis, having been described as also following the lines of Blaschko but occurring in children rather than adults. Inflammatory linear verrucous epidermal nevi also can be distinguished from blaschkitis on this premise, as these lesions arise during the first 5 years of life and generally affect the lower extremities.4,5
Histopathology is somewhat variable but generally includes spongiotic dermatitis with concomitant interface
dermatitis characterized by T-cell infiltration. Spongiosis is a feature less commonly seen in lichen striatus. Lesions can progress over time from spongiotic dermatitis to spongiotic psoriasiform dermatitis and later to spongiotic psoriasiform lichenoid dermatitis.4 Treatment of blaschkitis should begin with reassurance of the benign nature of the dermatosis. Pruritic symptoms can be managed with a course of topical steroids.
Blaschkitis is a rare and self-limiting acquired dermatosis that should be incorporated into the differential diagnosis of Blaschko-linear dermatoses. Further investigation is needed to determine if blaschkitis and lichen striatus represent the ends of a disease spectrum or completely distinct entities.
- Grosshans E, Marot L. Blaschkitis in adults. Ann Dermatol Venereol. 1990;117:9-15.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis: reappraisal of the concept of blaschkolinear dermatoses [published online November 29, 2010]. Br J Dermatol. 2011;164:257-262.
- Sun BK, Tsao H. X-chromosome inactivation and skin disease. J Invest Dermatol. 2008;128:2753-2759.
- Keegan BR, Kamino H, Fangman W, et al. “Pediatric blaschkitis”: expanding spectrum of childhood acquired Blaschko-linear dermatoses. Pediatr Dermatol. 2007;24:261-267.
- Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
- Grosshans E, Marot L. Blaschkitis in adults. Ann Dermatol Venereol. 1990;117:9-15.
- Müller CS, Schmaltz R, Vogt T, et al. Lichen striatus and blaschkitis: reappraisal of the concept of blaschkolinear dermatoses [published online November 29, 2010]. Br J Dermatol. 2011;164:257-262.
- Sun BK, Tsao H. X-chromosome inactivation and skin disease. J Invest Dermatol. 2008;128:2753-2759.
- Keegan BR, Kamino H, Fangman W, et al. “Pediatric blaschkitis”: expanding spectrum of childhood acquired Blaschko-linear dermatoses. Pediatr Dermatol. 2007;24:261-267.
- Goldsmith LA, Katz SI, Gilchrest BA, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
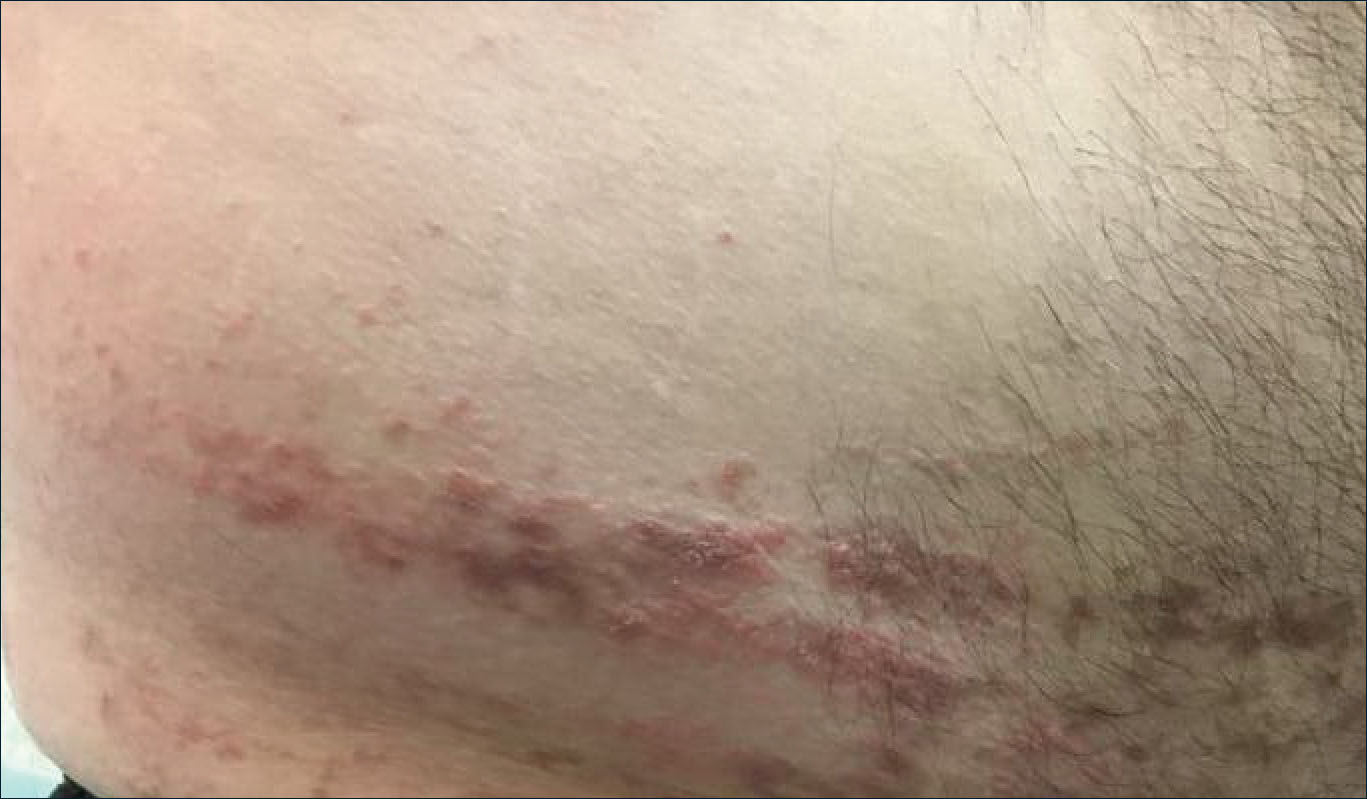
A 31-year-old man presented with a recurring pruritic rash on the right lateral flank and hip of 2 years’ duration. Physical examination revealed erythematous, verrucous, dry papules and plaques coalescing into larger plaques on the right flank and hip in dermatomal distributions involving the T10 and T11 dermatomes. A few papules were scattered in a linear eruption along the right flank and right upper thigh. Some postinflammatory changes were noted. No rash was noted over any other area of the body. Physical examination was otherwise unremarkable.

Erythematous Pruritic Plaque on the Cheek
The Diagnosis: Tinea Faciei
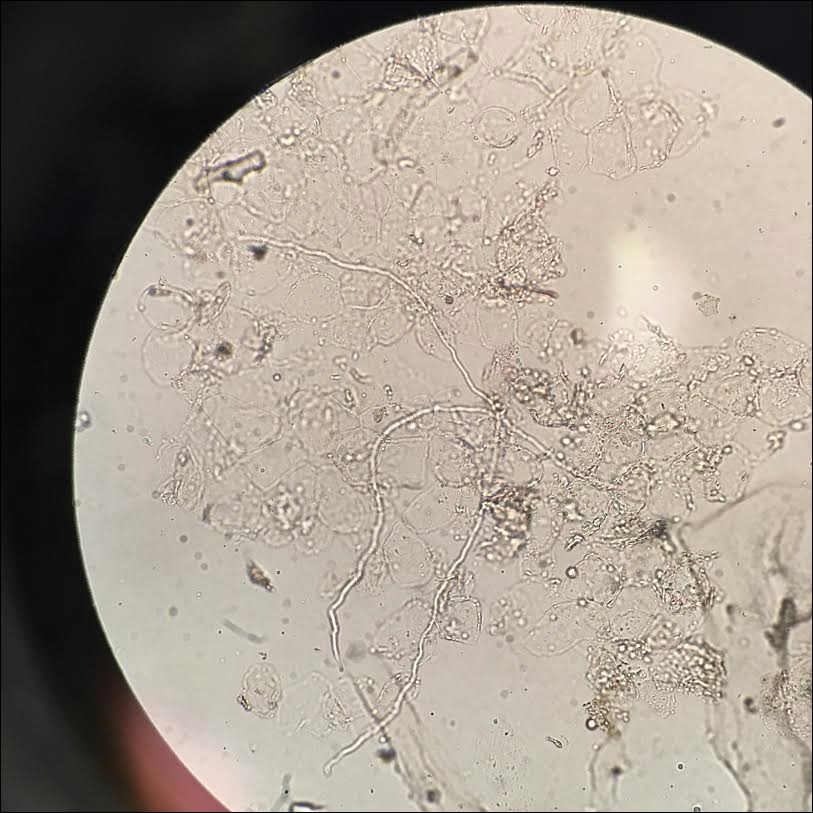
Given the morphology of the plaque, a potassium hydroxide preparation was performed and was positive for hyphal elements consistent with dermatophyte infection (Figure).
Tinea faciei is a fungal infection of the face caused by a dermatophyte that invades the stratum corneum.1 It is transmitted through direct contact with an infected individual or fomite.2 Infections typically are characterized by annular or serpiginous erythematous plaques with a scaly appearance and advancing edge. There may be associated vesicles, papules, or pustules with crusting around the advancing border.3 Tinea faciei can occur concomitantly with other dermatophytic infections and frequently presents atypically due to different characteristics of facial anatomy when compared to other tinea infections. As a result, it often is misdiagnosed.1
Tinea faciei represents roughly 19% of all superficial fungal infections and occurs more commonly in temperate humid regions.4 It can occur at any age but has bimodal peaks in incidence during childhood and early adulthood.5 The most common causative dermatophytes are Trichophyton tonsurans, Microsporum canis, Trichophyton mentagrophytes, and Trichophyton rubrum.1 Transmission is mainly through direct contact with infected individuals, animals, or soil, which likely occurred during the close quarters and exercises our patient experienced during basic training in the military.
Tinea faciei often is misdiagnosed and treated with topical corticosteroids. The steroids can give a false impression that the rash is resolving by initially decreasing the inflammatory component and reducing scale, which is referred to as tinea incognito. Once the steroid is stopped, however, the fungal infection often returns worse than the original presentation. The differential diagnosis includes subacute cutaneous lupus erythematosus, periorificial dermatitis, seborrheic dermatitis, psoriasis, rosacea, erythema annulare centrifugum, granuloma annulare, sarcoidosis, and contact dermatitis.1,3,6
Diagnosis of tinea faciei is best made with skin scraping of the active border of the lesion. The scraping is treated with potassium hydroxide 10%. Visualizing branching or curving hyphae confirms the diagnosis. Fungal speciation often is not performed due to the long time needed to culture. Wood lamp may fluoresce blue-green if tinea faciei is caused by Microsporum species; however, diagnosis in this manner is limited because other common species do not fluoresce.7
Options for treatment of tinea faciei include topical antifungals for 2 to 6 weeks for localized disease or oral antifungals for more extensive or unresponsive infections for 1 to 8 weeks depending on the agent that is used. If fungal folliculitis is present, oral medication should be given.1 Our patient was treated with oral terbinafine 250 mg once daily for 4 weeks with follow-up after that time to ensure resolution.
- Lin RL, Szepietowski JC, Schwartz RA. Tinea faciei, an often deceptive facial eruption. Int J Dermatol. 2004;43:437-440.
- Raimer SS, Beightler EL, Hebert AA, et al. Tinea faciei in infants caused by Trichophyton tonsurans. Pediatr Dermatol. 1986;3:452-454.
- Shapiro L, Cohen HJ. Tinea faciei simulating other dermatoses. JAMA. 1971;215:2106-2107.
- Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51(suppl 4):2-15.
- Jorquera E, Moreno JC, Camacho F. Tinea faciei: epidemiology. Ann Dermatol Venereol. 1991;119:101-104.
- Hsu S, Le EH, Khoshevis MR. Differential diagnosis of annular lesions. Am Fam Physician. 2001;64:289-296.
- Ponka D, Baddar F. Wood lamp examination. Can Fam Physician. 2012;58:976.
The Diagnosis: Tinea Faciei
Given the morphology of the plaque, a potassium hydroxide preparation was performed and was positive for hyphal elements consistent with dermatophyte infection (Figure).
Tinea faciei is a fungal infection of the face caused by a dermatophyte that invades the stratum corneum.1 It is transmitted through direct contact with an infected individual or fomite.2 Infections typically are characterized by annular or serpiginous erythematous plaques with a scaly appearance and advancing edge. There may be associated vesicles, papules, or pustules with crusting around the advancing border.3 Tinea faciei can occur concomitantly with other dermatophytic infections and frequently presents atypically due to different characteristics of facial anatomy when compared to other tinea infections. As a result, it often is misdiagnosed.1
Tinea faciei represents roughly 19% of all superficial fungal infections and occurs more commonly in temperate humid regions.4 It can occur at any age but has bimodal peaks in incidence during childhood and early adulthood.5 The most common causative dermatophytes are Trichophyton tonsurans, Microsporum canis, Trichophyton mentagrophytes, and Trichophyton rubrum.1 Transmission is mainly through direct contact with infected individuals, animals, or soil, which likely occurred during the close quarters and exercises our patient experienced during basic training in the military.
Tinea faciei often is misdiagnosed and treated with topical corticosteroids. The steroids can give a false impression that the rash is resolving by initially decreasing the inflammatory component and reducing scale, which is referred to as tinea incognito. Once the steroid is stopped, however, the fungal infection often returns worse than the original presentation. The differential diagnosis includes subacute cutaneous lupus erythematosus, periorificial dermatitis, seborrheic dermatitis, psoriasis, rosacea, erythema annulare centrifugum, granuloma annulare, sarcoidosis, and contact dermatitis.1,3,6
Diagnosis of tinea faciei is best made with skin scraping of the active border of the lesion. The scraping is treated with potassium hydroxide 10%. Visualizing branching or curving hyphae confirms the diagnosis. Fungal speciation often is not performed due to the long time needed to culture. Wood lamp may fluoresce blue-green if tinea faciei is caused by Microsporum species; however, diagnosis in this manner is limited because other common species do not fluoresce.7
Options for treatment of tinea faciei include topical antifungals for 2 to 6 weeks for localized disease or oral antifungals for more extensive or unresponsive infections for 1 to 8 weeks depending on the agent that is used. If fungal folliculitis is present, oral medication should be given.1 Our patient was treated with oral terbinafine 250 mg once daily for 4 weeks with follow-up after that time to ensure resolution.
The Diagnosis: Tinea Faciei
Given the morphology of the plaque, a potassium hydroxide preparation was performed and was positive for hyphal elements consistent with dermatophyte infection (Figure).
Tinea faciei is a fungal infection of the face caused by a dermatophyte that invades the stratum corneum.1 It is transmitted through direct contact with an infected individual or fomite.2 Infections typically are characterized by annular or serpiginous erythematous plaques with a scaly appearance and advancing edge. There may be associated vesicles, papules, or pustules with crusting around the advancing border.3 Tinea faciei can occur concomitantly with other dermatophytic infections and frequently presents atypically due to different characteristics of facial anatomy when compared to other tinea infections. As a result, it often is misdiagnosed.1
Tinea faciei represents roughly 19% of all superficial fungal infections and occurs more commonly in temperate humid regions.4 It can occur at any age but has bimodal peaks in incidence during childhood and early adulthood.5 The most common causative dermatophytes are Trichophyton tonsurans, Microsporum canis, Trichophyton mentagrophytes, and Trichophyton rubrum.1 Transmission is mainly through direct contact with infected individuals, animals, or soil, which likely occurred during the close quarters and exercises our patient experienced during basic training in the military.
Tinea faciei often is misdiagnosed and treated with topical corticosteroids. The steroids can give a false impression that the rash is resolving by initially decreasing the inflammatory component and reducing scale, which is referred to as tinea incognito. Once the steroid is stopped, however, the fungal infection often returns worse than the original presentation. The differential diagnosis includes subacute cutaneous lupus erythematosus, periorificial dermatitis, seborrheic dermatitis, psoriasis, rosacea, erythema annulare centrifugum, granuloma annulare, sarcoidosis, and contact dermatitis.1,3,6
Diagnosis of tinea faciei is best made with skin scraping of the active border of the lesion. The scraping is treated with potassium hydroxide 10%. Visualizing branching or curving hyphae confirms the diagnosis. Fungal speciation often is not performed due to the long time needed to culture. Wood lamp may fluoresce blue-green if tinea faciei is caused by Microsporum species; however, diagnosis in this manner is limited because other common species do not fluoresce.7
Options for treatment of tinea faciei include topical antifungals for 2 to 6 weeks for localized disease or oral antifungals for more extensive or unresponsive infections for 1 to 8 weeks depending on the agent that is used. If fungal folliculitis is present, oral medication should be given.1 Our patient was treated with oral terbinafine 250 mg once daily for 4 weeks with follow-up after that time to ensure resolution.
- Lin RL, Szepietowski JC, Schwartz RA. Tinea faciei, an often deceptive facial eruption. Int J Dermatol. 2004;43:437-440.
- Raimer SS, Beightler EL, Hebert AA, et al. Tinea faciei in infants caused by Trichophyton tonsurans. Pediatr Dermatol. 1986;3:452-454.
- Shapiro L, Cohen HJ. Tinea faciei simulating other dermatoses. JAMA. 1971;215:2106-2107.
- Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51(suppl 4):2-15.
- Jorquera E, Moreno JC, Camacho F. Tinea faciei: epidemiology. Ann Dermatol Venereol. 1991;119:101-104.
- Hsu S, Le EH, Khoshevis MR. Differential diagnosis of annular lesions. Am Fam Physician. 2001;64:289-296.
- Ponka D, Baddar F. Wood lamp examination. Can Fam Physician. 2012;58:976.
- Lin RL, Szepietowski JC, Schwartz RA. Tinea faciei, an often deceptive facial eruption. Int J Dermatol. 2004;43:437-440.
- Raimer SS, Beightler EL, Hebert AA, et al. Tinea faciei in infants caused by Trichophyton tonsurans. Pediatr Dermatol. 1986;3:452-454.
- Shapiro L, Cohen HJ. Tinea faciei simulating other dermatoses. JAMA. 1971;215:2106-2107.
- Havlickova B, Czaika VA, Friedrich M. Epidemiological trends in skin mycoses worldwide. Mycoses. 2008;51(suppl 4):2-15.
- Jorquera E, Moreno JC, Camacho F. Tinea faciei: epidemiology. Ann Dermatol Venereol. 1991;119:101-104.
- Hsu S, Le EH, Khoshevis MR. Differential diagnosis of annular lesions. Am Fam Physician. 2001;64:289-296.
- Ponka D, Baddar F. Wood lamp examination. Can Fam Physician. 2012;58:976.
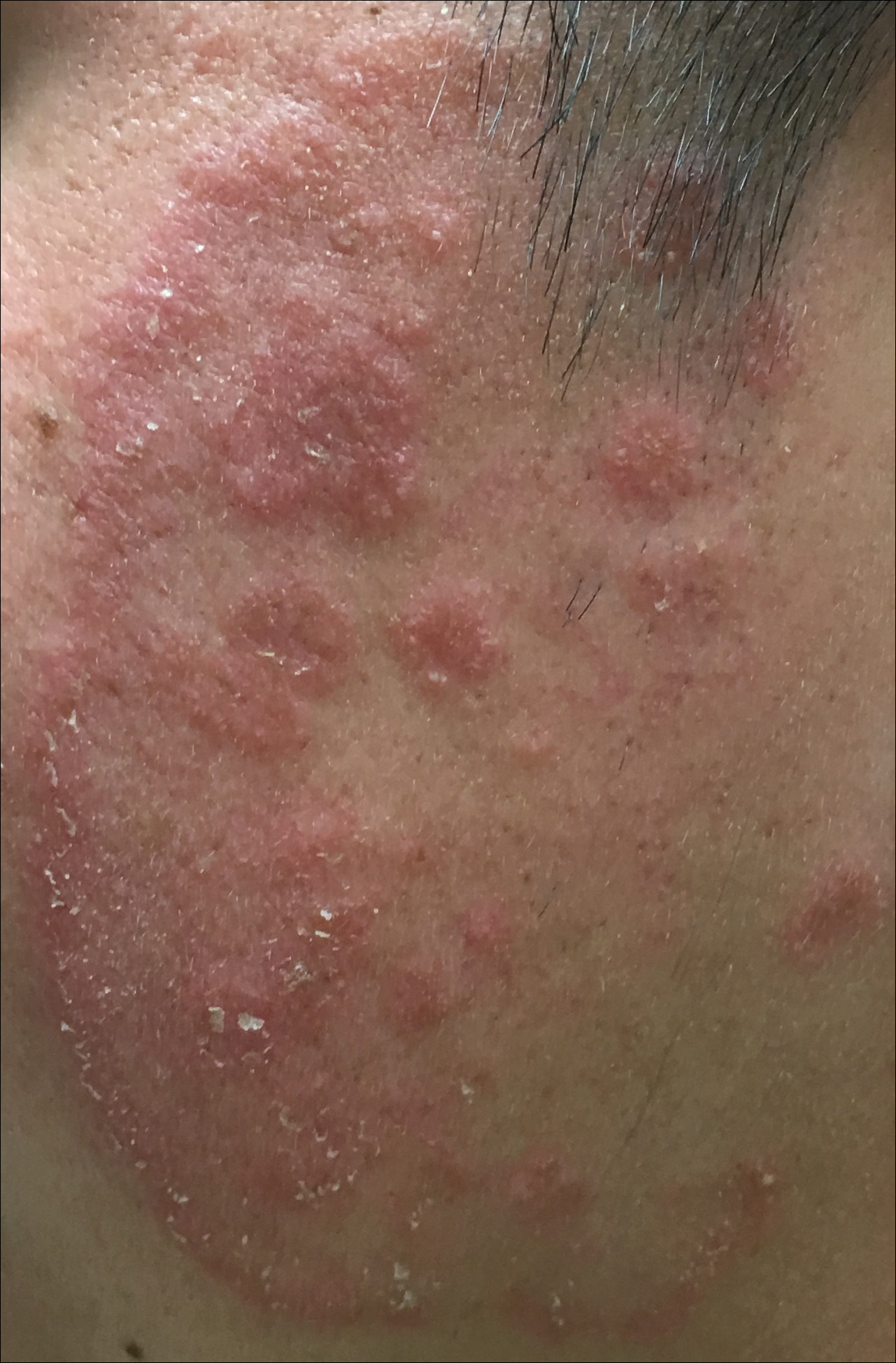
A 19-year-old man with a medical history of keloids presented with a slowly enlarging, red, itchy plaque on the left cheek of 1 year's duration that first began to develop during basic training in the military. The patient denied other pain, pruritus, or separate dermatitis. He initially was treated with triamcinolone cream 0.1%, which he used for 8 days prior to referral to the dermatology department. The patient denied other acute concerns. On physical examination, multiple erythematous papules coalescing into a large, 10-cm, papulosquamous, arciform plaque were noted on the left preauricular cheek.
Coalescing Hyperkeratotic Plaques and Papules
The Diagnosis: X-Linked Ichthyosis
Immunohistochemical staining of a punch biopsy specimen from the left foot with cytokeratin markers AE1/3, 5/6, and 19 showed normal positive uptake. Further workup was recommended, and the patient was referred to genetics for an ichthyosis gene panel. DNA sequencing revealed a c.1121G>A transition in exon 10 of the steroid sulfatase gene, STS, consistent with X-linked ichthyosis (XLI).
X-linked ichthyosis, also known as steroid sulfatase deficiency and X-linked recessive ichthyosis, is a congenital skin disorder classified in 1965 by Wells and Kerr.1 Ichthyoses are a heterogenous group of acquired and congenital disorders of keratinization that manifest with xerosis, hyperkeratosis, and scaling.2 Of more than 20 ichthyoses, XLI is the second most common ichthyosis, with a prevalence of 1 in 6000 males.3 X-linked ichthyosis occurs almost exclusively in males, and although females can be carriers, they rarely exhibit skin manifestations.4
X-linked ichthyosis is caused by either a partial or full deletion or mutation in the STS gene on the X chromosome.2 The absence of STS activity results in the accumulation of cholesterol sulfate in the stratum corneum, leading to corneocyte cohesion, hyperkeratosis, and impaired skin permeability. The most common clinical phenotype is characterized by polygonal scales concentrated on the upper and lower extremities as well as the trunk (Figure), consistent with our patient's clinical presentation.5
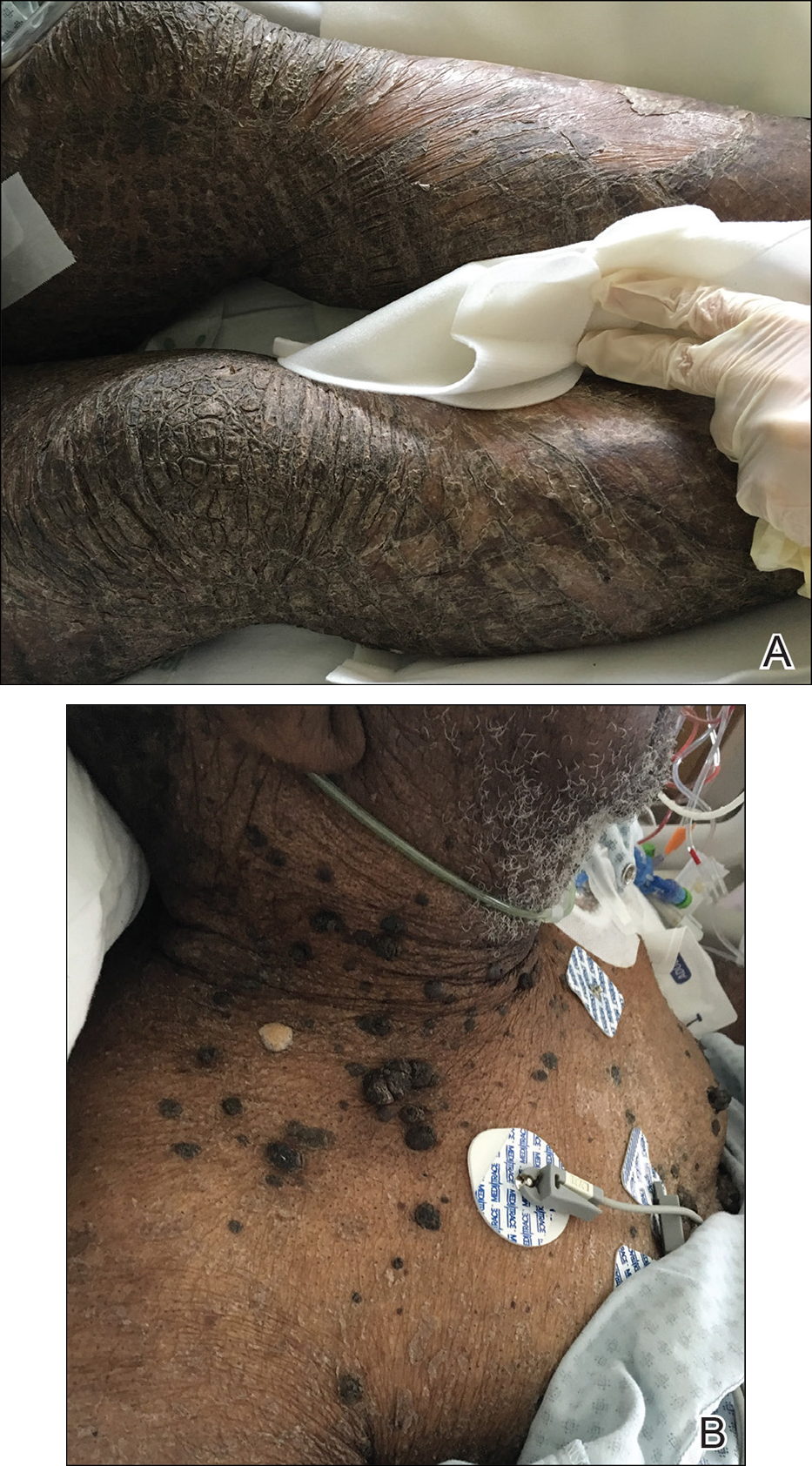
anterior knees (A) as well as large exophytic papules on the upper
chest and neck (B).
X-linked ichthyosis typically presents in the first 6 months of life as generalized desquamation and xerosis that progresses to fine scaling on the trunk and extremities, more commonly and heavily involving the legs; however, the extensor surfaces of the arms also may be affected.6 After the neonatal period, fine scaling persists on the trunk and extremities, but scales often become coarser and darker over time. Although scaling is generalized, it typically spares the antecubital and popliteal fossae, palms, soles, and midface. The lateral face, axillae, and neck always remain involved.4 The most common extracutaneous manifestations of XLI affect the ocular, genitourinary, and cognitive/behavioral systems. Patients can develop corneal comma-shaped opacities, hypogonadism, cryptorchidism, and an increased risk for testicular cancer. Female carriers may have prolonged delivery of affected neonates.2,5,7-9 Given the unrelated debilitating neurologic consequences of our patient's presenting subarachnoid hemorrhage, further workup was not pursued into these associations.
Although XLI is most commonly diagnosed in early childhood, it also must be considered in adult patients presenting with severe scaling of the trunk, arms, and legs who have not had prior dermatologic workup. Given the similarity of XLI presentation to other ichthyoses, particularly ichthyosis vulgaris, lamellar ichthyosis, and ichthyosis bullosa of Siemens, genetic analysis is the most accurate diagnostic tool and should be considered in patients with an atypical presentation. Rupioid psoriasis also may be considered and can be confirmed on biopsy. Diagnosis of XLI should prompt symptomatic treatment, genetic counseling, and workup for extracutaneous manifestations.
- Wells RS, Kerr CB. Genetic classification of ichthyosis. Arch Dermatol. 1965;92:1-6.
- Fernandes NF, Janniger CK, Schwartz RA. X-linked ichthyosis: an oculocutaneous genodermatosis. J Am Acad Dermatol. 2010;62:480-485.
- Hernández-Martín A, González-Sarmiento R, De Unamuno P. X-linked ichthyosis: an update. Br J Dermatol. 1999;141:617-627.
- Elias PM, Williams ML, Choi EH, et al. Role of cholesterol sulfate in epidermal structure and function: lessons from X-linked ichthyosis [published online November 27, 2013]. Biochim Biophys Acta. 2014;1841:353-361.
- Wu B, Paller AS. Ichthyosis, X-Linked. Treasure Island, FL: StatPearls Publishing LLC; 2019.
- Marukian NV, Choate KA. Recent advances in understanding ichthyosis pathogenesis. F1000Res. 2016;5. doi:10.12688/f1000research.8584.1.
- Baek WS, Aypar U. Case report neurological manifestations of X-linked ichthyosis: case report and review of the literature [published online August 13, 2017]. 2017;2017:9086408.
- Brookes KJ, Hawi Z, Park J, et al. Polymorphisms of the steroid sulfatase (STS) gene are associated with attention deficit hyperactivity disorder and influence brain tissue mRNA expression. Am J Med Genet Part B Neuropsychiatr Genet. 2010;153:1417-1424.
- Kent L, Emerton J, Bhadravathi V, et al. X-linked ichthyosis (steroid sulfatase deficiency) is associated with increased risk of attention deficit hyperactivity disorder, autism and social communication deficits. J Med Genet. 2008;45:519-524.
The Diagnosis: X-Linked Ichthyosis
Immunohistochemical staining of a punch biopsy specimen from the left foot with cytokeratin markers AE1/3, 5/6, and 19 showed normal positive uptake. Further workup was recommended, and the patient was referred to genetics for an ichthyosis gene panel. DNA sequencing revealed a c.1121G>A transition in exon 10 of the steroid sulfatase gene, STS, consistent with X-linked ichthyosis (XLI).
X-linked ichthyosis, also known as steroid sulfatase deficiency and X-linked recessive ichthyosis, is a congenital skin disorder classified in 1965 by Wells and Kerr.1 Ichthyoses are a heterogenous group of acquired and congenital disorders of keratinization that manifest with xerosis, hyperkeratosis, and scaling.2 Of more than 20 ichthyoses, XLI is the second most common ichthyosis, with a prevalence of 1 in 6000 males.3 X-linked ichthyosis occurs almost exclusively in males, and although females can be carriers, they rarely exhibit skin manifestations.4
X-linked ichthyosis is caused by either a partial or full deletion or mutation in the STS gene on the X chromosome.2 The absence of STS activity results in the accumulation of cholesterol sulfate in the stratum corneum, leading to corneocyte cohesion, hyperkeratosis, and impaired skin permeability. The most common clinical phenotype is characterized by polygonal scales concentrated on the upper and lower extremities as well as the trunk (Figure), consistent with our patient's clinical presentation.5
anterior knees (A) as well as large exophytic papules on the upper
chest and neck (B).
X-linked ichthyosis typically presents in the first 6 months of life as generalized desquamation and xerosis that progresses to fine scaling on the trunk and extremities, more commonly and heavily involving the legs; however, the extensor surfaces of the arms also may be affected.6 After the neonatal period, fine scaling persists on the trunk and extremities, but scales often become coarser and darker over time. Although scaling is generalized, it typically spares the antecubital and popliteal fossae, palms, soles, and midface. The lateral face, axillae, and neck always remain involved.4 The most common extracutaneous manifestations of XLI affect the ocular, genitourinary, and cognitive/behavioral systems. Patients can develop corneal comma-shaped opacities, hypogonadism, cryptorchidism, and an increased risk for testicular cancer. Female carriers may have prolonged delivery of affected neonates.2,5,7-9 Given the unrelated debilitating neurologic consequences of our patient's presenting subarachnoid hemorrhage, further workup was not pursued into these associations.
Although XLI is most commonly diagnosed in early childhood, it also must be considered in adult patients presenting with severe scaling of the trunk, arms, and legs who have not had prior dermatologic workup. Given the similarity of XLI presentation to other ichthyoses, particularly ichthyosis vulgaris, lamellar ichthyosis, and ichthyosis bullosa of Siemens, genetic analysis is the most accurate diagnostic tool and should be considered in patients with an atypical presentation. Rupioid psoriasis also may be considered and can be confirmed on biopsy. Diagnosis of XLI should prompt symptomatic treatment, genetic counseling, and workup for extracutaneous manifestations.
The Diagnosis: X-Linked Ichthyosis
Immunohistochemical staining of a punch biopsy specimen from the left foot with cytokeratin markers AE1/3, 5/6, and 19 showed normal positive uptake. Further workup was recommended, and the patient was referred to genetics for an ichthyosis gene panel. DNA sequencing revealed a c.1121G>A transition in exon 10 of the steroid sulfatase gene, STS, consistent with X-linked ichthyosis (XLI).
X-linked ichthyosis, also known as steroid sulfatase deficiency and X-linked recessive ichthyosis, is a congenital skin disorder classified in 1965 by Wells and Kerr.1 Ichthyoses are a heterogenous group of acquired and congenital disorders of keratinization that manifest with xerosis, hyperkeratosis, and scaling.2 Of more than 20 ichthyoses, XLI is the second most common ichthyosis, with a prevalence of 1 in 6000 males.3 X-linked ichthyosis occurs almost exclusively in males, and although females can be carriers, they rarely exhibit skin manifestations.4
X-linked ichthyosis is caused by either a partial or full deletion or mutation in the STS gene on the X chromosome.2 The absence of STS activity results in the accumulation of cholesterol sulfate in the stratum corneum, leading to corneocyte cohesion, hyperkeratosis, and impaired skin permeability. The most common clinical phenotype is characterized by polygonal scales concentrated on the upper and lower extremities as well as the trunk (Figure), consistent with our patient's clinical presentation.5
anterior knees (A) as well as large exophytic papules on the upper
chest and neck (B).
X-linked ichthyosis typically presents in the first 6 months of life as generalized desquamation and xerosis that progresses to fine scaling on the trunk and extremities, more commonly and heavily involving the legs; however, the extensor surfaces of the arms also may be affected.6 After the neonatal period, fine scaling persists on the trunk and extremities, but scales often become coarser and darker over time. Although scaling is generalized, it typically spares the antecubital and popliteal fossae, palms, soles, and midface. The lateral face, axillae, and neck always remain involved.4 The most common extracutaneous manifestations of XLI affect the ocular, genitourinary, and cognitive/behavioral systems. Patients can develop corneal comma-shaped opacities, hypogonadism, cryptorchidism, and an increased risk for testicular cancer. Female carriers may have prolonged delivery of affected neonates.2,5,7-9 Given the unrelated debilitating neurologic consequences of our patient's presenting subarachnoid hemorrhage, further workup was not pursued into these associations.
Although XLI is most commonly diagnosed in early childhood, it also must be considered in adult patients presenting with severe scaling of the trunk, arms, and legs who have not had prior dermatologic workup. Given the similarity of XLI presentation to other ichthyoses, particularly ichthyosis vulgaris, lamellar ichthyosis, and ichthyosis bullosa of Siemens, genetic analysis is the most accurate diagnostic tool and should be considered in patients with an atypical presentation. Rupioid psoriasis also may be considered and can be confirmed on biopsy. Diagnosis of XLI should prompt symptomatic treatment, genetic counseling, and workup for extracutaneous manifestations.
- Wells RS, Kerr CB. Genetic classification of ichthyosis. Arch Dermatol. 1965;92:1-6.
- Fernandes NF, Janniger CK, Schwartz RA. X-linked ichthyosis: an oculocutaneous genodermatosis. J Am Acad Dermatol. 2010;62:480-485.
- Hernández-Martín A, González-Sarmiento R, De Unamuno P. X-linked ichthyosis: an update. Br J Dermatol. 1999;141:617-627.
- Elias PM, Williams ML, Choi EH, et al. Role of cholesterol sulfate in epidermal structure and function: lessons from X-linked ichthyosis [published online November 27, 2013]. Biochim Biophys Acta. 2014;1841:353-361.
- Wu B, Paller AS. Ichthyosis, X-Linked. Treasure Island, FL: StatPearls Publishing LLC; 2019.
- Marukian NV, Choate KA. Recent advances in understanding ichthyosis pathogenesis. F1000Res. 2016;5. doi:10.12688/f1000research.8584.1.
- Baek WS, Aypar U. Case report neurological manifestations of X-linked ichthyosis: case report and review of the literature [published online August 13, 2017]. 2017;2017:9086408.
- Brookes KJ, Hawi Z, Park J, et al. Polymorphisms of the steroid sulfatase (STS) gene are associated with attention deficit hyperactivity disorder and influence brain tissue mRNA expression. Am J Med Genet Part B Neuropsychiatr Genet. 2010;153:1417-1424.
- Kent L, Emerton J, Bhadravathi V, et al. X-linked ichthyosis (steroid sulfatase deficiency) is associated with increased risk of attention deficit hyperactivity disorder, autism and social communication deficits. J Med Genet. 2008;45:519-524.
- Wells RS, Kerr CB. Genetic classification of ichthyosis. Arch Dermatol. 1965;92:1-6.
- Fernandes NF, Janniger CK, Schwartz RA. X-linked ichthyosis: an oculocutaneous genodermatosis. J Am Acad Dermatol. 2010;62:480-485.
- Hernández-Martín A, González-Sarmiento R, De Unamuno P. X-linked ichthyosis: an update. Br J Dermatol. 1999;141:617-627.
- Elias PM, Williams ML, Choi EH, et al. Role of cholesterol sulfate in epidermal structure and function: lessons from X-linked ichthyosis [published online November 27, 2013]. Biochim Biophys Acta. 2014;1841:353-361.
- Wu B, Paller AS. Ichthyosis, X-Linked. Treasure Island, FL: StatPearls Publishing LLC; 2019.
- Marukian NV, Choate KA. Recent advances in understanding ichthyosis pathogenesis. F1000Res. 2016;5. doi:10.12688/f1000research.8584.1.
- Baek WS, Aypar U. Case report neurological manifestations of X-linked ichthyosis: case report and review of the literature [published online August 13, 2017]. 2017;2017:9086408.
- Brookes KJ, Hawi Z, Park J, et al. Polymorphisms of the steroid sulfatase (STS) gene are associated with attention deficit hyperactivity disorder and influence brain tissue mRNA expression. Am J Med Genet Part B Neuropsychiatr Genet. 2010;153:1417-1424.
- Kent L, Emerton J, Bhadravathi V, et al. X-linked ichthyosis (steroid sulfatase deficiency) is associated with increased risk of attention deficit hyperactivity disorder, autism and social communication deficits. J Med Genet. 2008;45:519-524.
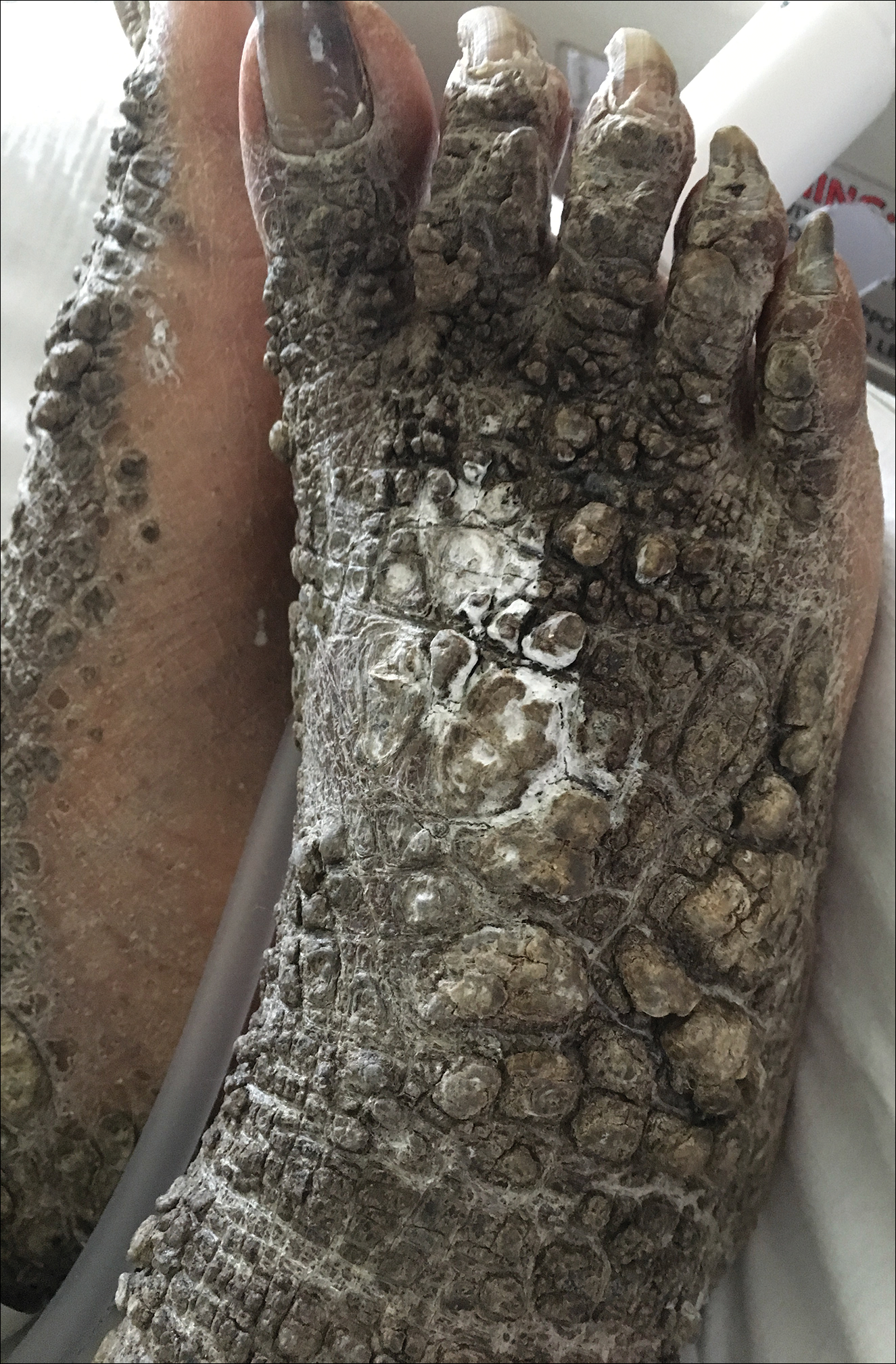
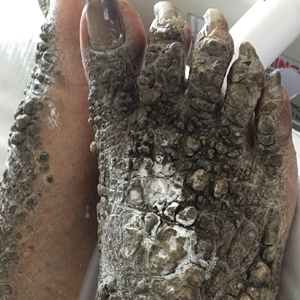
A 67-year-old man with a history of congestive heart failure, type 2 diabetes mellitus, hypertension, and schizophrenia was admitted to the hospital for subarachnoid hemorrhage and was noted to have heavy scaling on the bilateral legs. Given recent medical events, the patient was nonconversant at the time of consultation, but his daughter provided his medical history at bedside. The patient usually wore long-sleeved clothing and pants, thus no one had seen his skin in many years, and it was unclear how long the scaling had been present. His family history was notable for eczema in distant relatives but negative for comparable conditions. Physical examination revealed thick lichenified skin with many large, exophytic, brown papules (largest measured 1.5×1×1 cm) and platelike scaling on the anterior chest, abdomen, lateral arms, and forearms. Extensive coalescing hyperkeratotic plaques and papules (up to 1 cm in thickness) were present on the anterior legs and feet, and scattered verrucous brown papules were noted on the plantar aspects of the bilateral feet. A punch biopsy of the left foot revealed extensive, dense, compact, orthokeratotic hyperkeratosis with a preserved granular layer with no epidermolysis.
Lesions With a Distinct Black Pigment
The Diagnosis: Black-Spot Poison Ivy
Due to the detailed account of the patient's history including acuity of current presentation, history of recent activities, travel history, and recent exposures, as well as a thorough skin examination, a diagnosis of black-spot poison ivy was made. In this case, the linear distribution of the lesions with overlying black pigment that could not be removed (Figures 1 and 2) provided important clues to diagnosis.
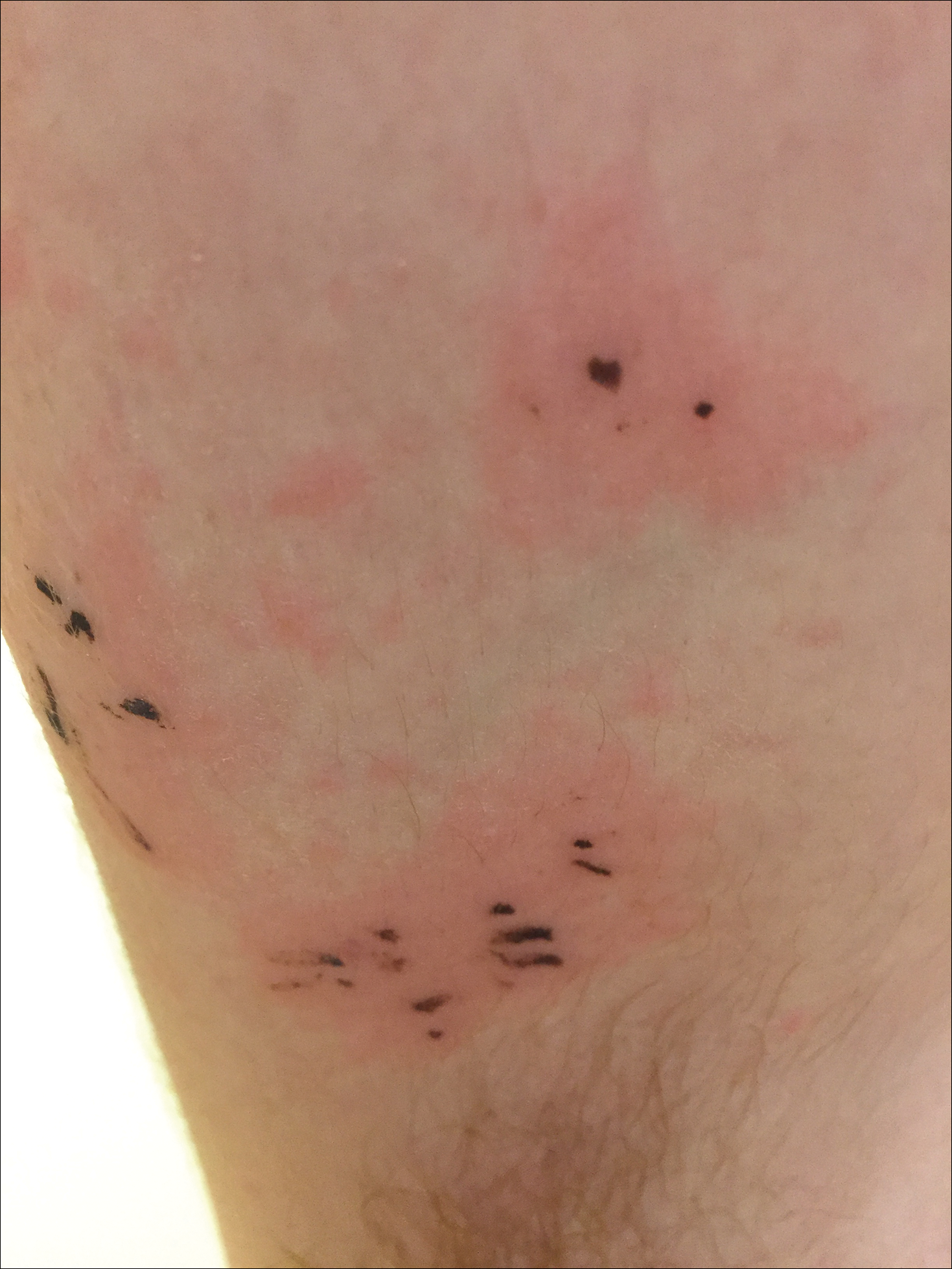
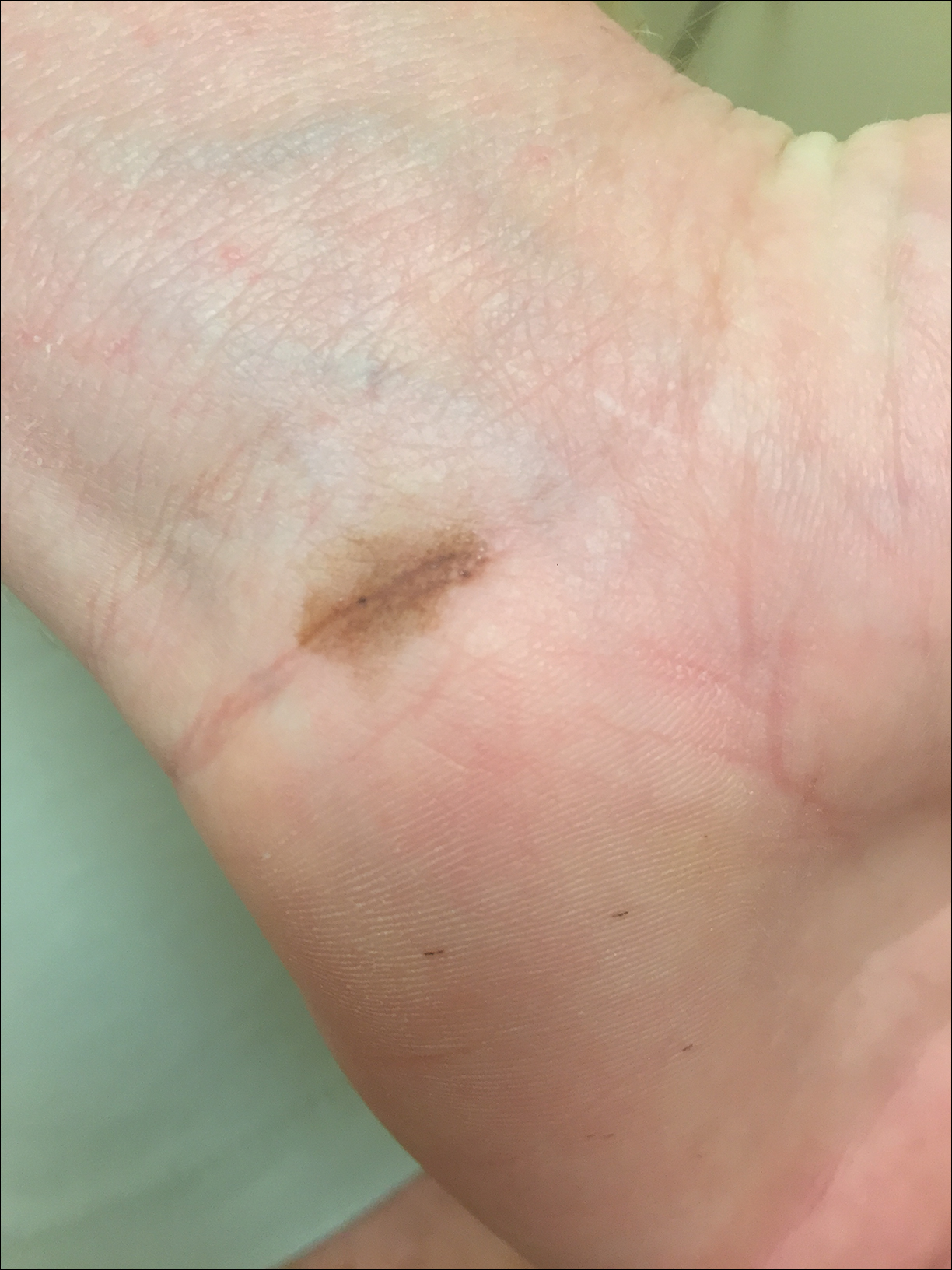
Poison ivy is an allergic contact dermatitis that affects an estimated 25 to 40 million Americans annually who are exposed to its resin. Poison ivy is a plant from the Toxicodendron genus, and an estimated 85% of the North American population report sensitivity to these plants, of which poison ivy (Toxicodendron radicans) is the most common.1 Other related plants include poison sumac and poison oak. Poison ivy and other Toxicodendron plants produce urushiol, the oleoresin responsible for one of the most common allergic contact dermatitides in the United States.2 Black-spot poison ivy is an uncommon presentation following exposure to urushiol or oleoresin,3 as sufficient concentration of urushiol on the skin rarely is achieved.3,4 This plant's resin oxidizes and turns coal black when exposed to air.5 Contact with enough of this oleoresin will produce black-spot poison ivy.6 Patients with sufficient concentrations of oleoresin on their skin to cause this black oxidation usually have similar black spots on their clothing.7 Interestingly, some Toxicodendron species, such as the Japanese lacquer tree, Toxicodendron vernicifluum, have a black lacquer sap that was historically used as ink.8 This ink was used on Chinese and Japanese jars and has caused contact dermatitis hundreds of years after they were created.7
Poison ivy is characterized by a generalized, pruritic, erythematous rash with vesicles and papules in a linear distribution.9 Black-spot poison ivy presents the same with the addition of black lacquer-like macules with surrounding erythema.10 The skin lesions usually appear on exposed areas 24 to 48 hours after contact.11 Histology of black-spot poison ivy lesions should reveal yellow material in the stratum corneum with epidermal necrosis, in addition to classic features of acute allergic contact dermatitis.3 Interestingly, because these lesions occur with the first exposure to poison ivy, a patient may not develop the typical itchy eczematous eruption characteristic of poison ivy dermatitis. Differential diagnosis includes superficial purpura; exogenous pigment such as marker, ink, or tattoo pigment; tinea nigra; purpuric allergic contact dermatitis to resins or dyes; arthropod assault; irritant contact dermatitis; and infectious and noninfectious vasculitis.11
Similar to poison ivy, treatment of black-spot poison ivy involves oral and topical steroids combined with antihistamines if the patient continues to experience pruritus.6,12 It was recommended to our patient to apply cool compresses with water or Burow solution to alleviate itching and promote drying of the lesions. Calamine lotion can provide similar outcomes.13 Once the oleoresin is oxidized and bound to skin, the black spots cannot be removed with soap, water, or alcohol. The black spots gradually desquamate 1 to 2 weeks after formation without scarring,11 and patients do not require further monitoring.1 Patients should clean or discard clothing and evaluate for possible sources of poison ivy exposure. Because this type of poison ivy dermatitis is rare, most health care workers likely have never seen black-spot poison ivy, and it is an important diagnosis to consider.13
- Baer RL. Poison ivy dermatitis. Cutis. 1990;46:34-36.
- Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
- Hurwitz RM, Rivera HP, Guin JD. Black-spot poison ivy dermatitis. an acute irritant contact dermatitis superimposed upon an allergic contact dermatitis. Am J Dermatopathol. 1984;6:319-322.
- Kurlan JG, Lucky AW. Black spot poison ivy: a report of 5 cases and a review of the literature. J Am Acad Dermatol. 2001;45:246-249.
- Guin JD. The black spot test for recognizing poison ivy and related species. J Am Acad Dermatol. 1980;2:332-333.
- Mallory SB, Hurwitz RM. Black-spot poison-ivy dermatitis. Clin Dermatol. 1986;4:149-151.
- Mallory SB, Miller OF, Tyler WB. Toxicodendron radicans dermatitis with black lacquer deposit on the skin. J Am Acad Dermatol. 1982;6:363-368.
- Rietschel R, Fowler J. Toxicodendron plants and species. Fisher's Contact Dermatitis. 4th ed. Baltimore, MD: Williams & Wilkins; 1995:469-472.
- Fisher AA. Poison ivy/oak dermatitis. part I: prevention--soap and water, topical barriers, hyposensitization. Cutis. 1996;57:384-386.
- McClanahan C, Asarch A, Swick BL. Black spot poison ivy. Int J Dermatol. 2014;53:752-753.
- Mu EW, Capell BC, Castelo-Soccio L. Black spots on a toddler's skin. Contemp Pediatr. 2013;30:31-32.
- Schram SE, Willey A, Lee PK, et al. Black-spot poison ivy. Dermatitis. 2008;19:48-51.
- Paniagua CT, Bean AS. Black-spot poison ivy: a rare phenomenon. J Am Acad Nurse Pract. 2011;23:275-277.
The Diagnosis: Black-Spot Poison Ivy
Due to the detailed account of the patient's history including acuity of current presentation, history of recent activities, travel history, and recent exposures, as well as a thorough skin examination, a diagnosis of black-spot poison ivy was made. In this case, the linear distribution of the lesions with overlying black pigment that could not be removed (Figures 1 and 2) provided important clues to diagnosis.
Poison ivy is an allergic contact dermatitis that affects an estimated 25 to 40 million Americans annually who are exposed to its resin. Poison ivy is a plant from the Toxicodendron genus, and an estimated 85% of the North American population report sensitivity to these plants, of which poison ivy (Toxicodendron radicans) is the most common.1 Other related plants include poison sumac and poison oak. Poison ivy and other Toxicodendron plants produce urushiol, the oleoresin responsible for one of the most common allergic contact dermatitides in the United States.2 Black-spot poison ivy is an uncommon presentation following exposure to urushiol or oleoresin,3 as sufficient concentration of urushiol on the skin rarely is achieved.3,4 This plant's resin oxidizes and turns coal black when exposed to air.5 Contact with enough of this oleoresin will produce black-spot poison ivy.6 Patients with sufficient concentrations of oleoresin on their skin to cause this black oxidation usually have similar black spots on their clothing.7 Interestingly, some Toxicodendron species, such as the Japanese lacquer tree, Toxicodendron vernicifluum, have a black lacquer sap that was historically used as ink.8 This ink was used on Chinese and Japanese jars and has caused contact dermatitis hundreds of years after they were created.7
Poison ivy is characterized by a generalized, pruritic, erythematous rash with vesicles and papules in a linear distribution.9 Black-spot poison ivy presents the same with the addition of black lacquer-like macules with surrounding erythema.10 The skin lesions usually appear on exposed areas 24 to 48 hours after contact.11 Histology of black-spot poison ivy lesions should reveal yellow material in the stratum corneum with epidermal necrosis, in addition to classic features of acute allergic contact dermatitis.3 Interestingly, because these lesions occur with the first exposure to poison ivy, a patient may not develop the typical itchy eczematous eruption characteristic of poison ivy dermatitis. Differential diagnosis includes superficial purpura; exogenous pigment such as marker, ink, or tattoo pigment; tinea nigra; purpuric allergic contact dermatitis to resins or dyes; arthropod assault; irritant contact dermatitis; and infectious and noninfectious vasculitis.11
Similar to poison ivy, treatment of black-spot poison ivy involves oral and topical steroids combined with antihistamines if the patient continues to experience pruritus.6,12 It was recommended to our patient to apply cool compresses with water or Burow solution to alleviate itching and promote drying of the lesions. Calamine lotion can provide similar outcomes.13 Once the oleoresin is oxidized and bound to skin, the black spots cannot be removed with soap, water, or alcohol. The black spots gradually desquamate 1 to 2 weeks after formation without scarring,11 and patients do not require further monitoring.1 Patients should clean or discard clothing and evaluate for possible sources of poison ivy exposure. Because this type of poison ivy dermatitis is rare, most health care workers likely have never seen black-spot poison ivy, and it is an important diagnosis to consider.13
The Diagnosis: Black-Spot Poison Ivy
Due to the detailed account of the patient's history including acuity of current presentation, history of recent activities, travel history, and recent exposures, as well as a thorough skin examination, a diagnosis of black-spot poison ivy was made. In this case, the linear distribution of the lesions with overlying black pigment that could not be removed (Figures 1 and 2) provided important clues to diagnosis.
Poison ivy is an allergic contact dermatitis that affects an estimated 25 to 40 million Americans annually who are exposed to its resin. Poison ivy is a plant from the Toxicodendron genus, and an estimated 85% of the North American population report sensitivity to these plants, of which poison ivy (Toxicodendron radicans) is the most common.1 Other related plants include poison sumac and poison oak. Poison ivy and other Toxicodendron plants produce urushiol, the oleoresin responsible for one of the most common allergic contact dermatitides in the United States.2 Black-spot poison ivy is an uncommon presentation following exposure to urushiol or oleoresin,3 as sufficient concentration of urushiol on the skin rarely is achieved.3,4 This plant's resin oxidizes and turns coal black when exposed to air.5 Contact with enough of this oleoresin will produce black-spot poison ivy.6 Patients with sufficient concentrations of oleoresin on their skin to cause this black oxidation usually have similar black spots on their clothing.7 Interestingly, some Toxicodendron species, such as the Japanese lacquer tree, Toxicodendron vernicifluum, have a black lacquer sap that was historically used as ink.8 This ink was used on Chinese and Japanese jars and has caused contact dermatitis hundreds of years after they were created.7
Poison ivy is characterized by a generalized, pruritic, erythematous rash with vesicles and papules in a linear distribution.9 Black-spot poison ivy presents the same with the addition of black lacquer-like macules with surrounding erythema.10 The skin lesions usually appear on exposed areas 24 to 48 hours after contact.11 Histology of black-spot poison ivy lesions should reveal yellow material in the stratum corneum with epidermal necrosis, in addition to classic features of acute allergic contact dermatitis.3 Interestingly, because these lesions occur with the first exposure to poison ivy, a patient may not develop the typical itchy eczematous eruption characteristic of poison ivy dermatitis. Differential diagnosis includes superficial purpura; exogenous pigment such as marker, ink, or tattoo pigment; tinea nigra; purpuric allergic contact dermatitis to resins or dyes; arthropod assault; irritant contact dermatitis; and infectious and noninfectious vasculitis.11
Similar to poison ivy, treatment of black-spot poison ivy involves oral and topical steroids combined with antihistamines if the patient continues to experience pruritus.6,12 It was recommended to our patient to apply cool compresses with water or Burow solution to alleviate itching and promote drying of the lesions. Calamine lotion can provide similar outcomes.13 Once the oleoresin is oxidized and bound to skin, the black spots cannot be removed with soap, water, or alcohol. The black spots gradually desquamate 1 to 2 weeks after formation without scarring,11 and patients do not require further monitoring.1 Patients should clean or discard clothing and evaluate for possible sources of poison ivy exposure. Because this type of poison ivy dermatitis is rare, most health care workers likely have never seen black-spot poison ivy, and it is an important diagnosis to consider.13
- Baer RL. Poison ivy dermatitis. Cutis. 1990;46:34-36.
- Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
- Hurwitz RM, Rivera HP, Guin JD. Black-spot poison ivy dermatitis. an acute irritant contact dermatitis superimposed upon an allergic contact dermatitis. Am J Dermatopathol. 1984;6:319-322.
- Kurlan JG, Lucky AW. Black spot poison ivy: a report of 5 cases and a review of the literature. J Am Acad Dermatol. 2001;45:246-249.
- Guin JD. The black spot test for recognizing poison ivy and related species. J Am Acad Dermatol. 1980;2:332-333.
- Mallory SB, Hurwitz RM. Black-spot poison-ivy dermatitis. Clin Dermatol. 1986;4:149-151.
- Mallory SB, Miller OF, Tyler WB. Toxicodendron radicans dermatitis with black lacquer deposit on the skin. J Am Acad Dermatol. 1982;6:363-368.
- Rietschel R, Fowler J. Toxicodendron plants and species. Fisher's Contact Dermatitis. 4th ed. Baltimore, MD: Williams & Wilkins; 1995:469-472.
- Fisher AA. Poison ivy/oak dermatitis. part I: prevention--soap and water, topical barriers, hyposensitization. Cutis. 1996;57:384-386.
- McClanahan C, Asarch A, Swick BL. Black spot poison ivy. Int J Dermatol. 2014;53:752-753.
- Mu EW, Capell BC, Castelo-Soccio L. Black spots on a toddler's skin. Contemp Pediatr. 2013;30:31-32.
- Schram SE, Willey A, Lee PK, et al. Black-spot poison ivy. Dermatitis. 2008;19:48-51.
- Paniagua CT, Bean AS. Black-spot poison ivy: a rare phenomenon. J Am Acad Nurse Pract. 2011;23:275-277.
- Baer RL. Poison ivy dermatitis. Cutis. 1990;46:34-36.
- Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician. 2010;82:249-255.
- Hurwitz RM, Rivera HP, Guin JD. Black-spot poison ivy dermatitis. an acute irritant contact dermatitis superimposed upon an allergic contact dermatitis. Am J Dermatopathol. 1984;6:319-322.
- Kurlan JG, Lucky AW. Black spot poison ivy: a report of 5 cases and a review of the literature. J Am Acad Dermatol. 2001;45:246-249.
- Guin JD. The black spot test for recognizing poison ivy and related species. J Am Acad Dermatol. 1980;2:332-333.
- Mallory SB, Hurwitz RM. Black-spot poison-ivy dermatitis. Clin Dermatol. 1986;4:149-151.
- Mallory SB, Miller OF, Tyler WB. Toxicodendron radicans dermatitis with black lacquer deposit on the skin. J Am Acad Dermatol. 1982;6:363-368.
- Rietschel R, Fowler J. Toxicodendron plants and species. Fisher's Contact Dermatitis. 4th ed. Baltimore, MD: Williams & Wilkins; 1995:469-472.
- Fisher AA. Poison ivy/oak dermatitis. part I: prevention--soap and water, topical barriers, hyposensitization. Cutis. 1996;57:384-386.
- McClanahan C, Asarch A, Swick BL. Black spot poison ivy. Int J Dermatol. 2014;53:752-753.
- Mu EW, Capell BC, Castelo-Soccio L. Black spots on a toddler's skin. Contemp Pediatr. 2013;30:31-32.
- Schram SE, Willey A, Lee PK, et al. Black-spot poison ivy. Dermatitis. 2008;19:48-51.
- Paniagua CT, Bean AS. Black-spot poison ivy: a rare phenomenon. J Am Acad Nurse Pract. 2011;23:275-277.
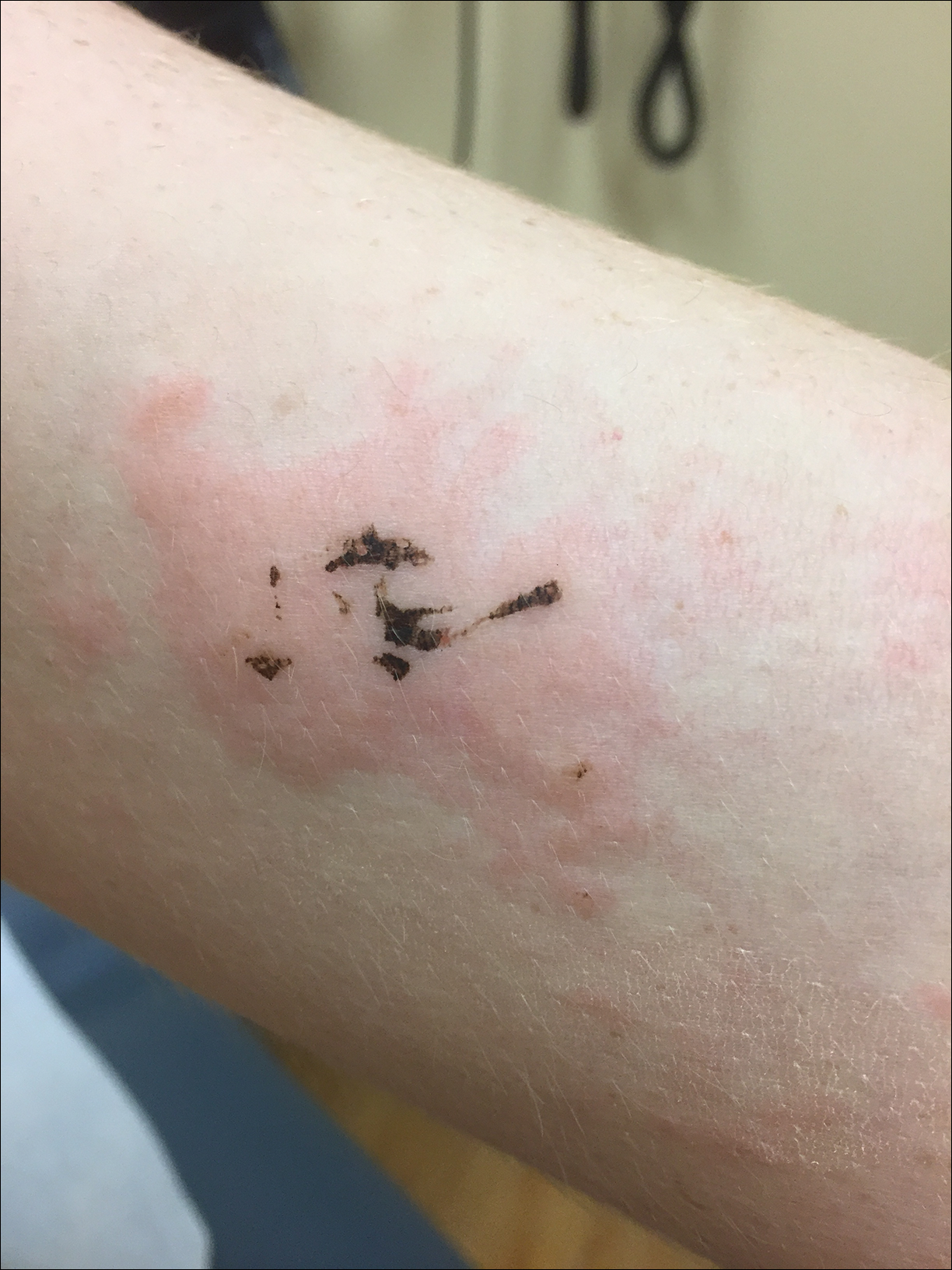
A 17-year-old adolescent boy presented to urgent care with a pruritic eruption on the bilateral arms of 1 day's duration. He was camping in the woods the night prior to presentation. On physical examination linear, erythematous, edematous plaques were observed bilaterally with overlying brown and black pigment on the arms. The pigment could not be removed with alcohol or vigorous scrubbing. The patient's condition improved with prednisone.
Diffuse Pustular Eruption Following Computed Tomography
The Diagnosis: Acute Generalized Exanthematous Pustulosis
Histopathology demonstrated spongiosis with subcorneal pustules and an overlying basket-weave pattern stratum corneum. There was mild papillary dermal edema with scattered dermal neutrophils and rare eosinophils (Figure). The patient's clinical presentation and histopathology were consistent with acute generalized exanthematous pustulosis (AGEP). The inciting agent in this case was the contrast medium iopamidol. The patient was treated with a short course of prednisone, triamcinolone cream, diphenhydramine, and acetaminophen. Within 1 week the pustules and erythema had resolved.
Acute generalized exanthematous pustulosis is an uncommon T cell-mediated cutaneous reaction characterized by widespread progressive erythema with numerous nonfollicular pinpoint pustules. The patient usually is well appearing; however, he/she often will have concurrent fever and facial edema. Mucous membranes rarely are involved. Laboratory results typically are notable only for leukocytosis with neutrophilia.
The pustular eruption typically occurs within 1 to 2 days after exposure to an inciting agent1; however, this latent period can range from 1 hour to nearly 4 weeks in some studies.2 Systemic medications are the cause in approximately 90% of cases, with antibiotics being the most common category. Frequently implicated medications include β-lactams, macrolides, quinolones, sulfonamides, proton pump inhibitors, hydroxychloroquine, terbinafine, nonsteroidal anti-inflammatory drugs, diltiazem, ketoconazole, and fluconazole. Acute generalized exanthematous pustulosis also has been rarely reported following contact with mercury, viral and bacterial infections, and spider bites.3
Iodinated contrast agents have long been known to cause immediate and delayed adverse cutaneous reactions. However, one consensus study indicated that these reactions occur in only 0.05% to 0.10% of patients.4 Although rare, iodinated contrast media (eg, iopamidol, iohexol, ioversol, iodixanol, iomeprol, iobitridol, iopromide) have been reported as a cause of AGEP. A PubMed search of articles indexed for MEDLINE using the terms acute generalized exanthematous pustulosis, contrast, iodine, and iodinated revealed 10 adult cases reported in 6 articles in the English-language literature.1,5-9 The most recent articles focus on methods to identify the causative agent. If the etiology of the reaction is unclear, patch or intradermal testing can help to confirm the causative agent. These tests also can help determine similar agents to which the patient may cross-react.4,5
It can be difficult to differentiate AGEP from other cutaneous drug reactions and other nonfollicular pustular conditions. Drug-induced hypersensitivity syndrome typically presents with facial edema and a morbilliform rash. Although it can present with pustules, the latent period is longer (2-6 weeks), and there frequently are signs of multiorgan involvement including hepatic dysfunction, eosinophilia, atypical lymphocytosis, and lymphadenopathy. Patients with generalized pustular psoriasis often have a history of plaque psoriasis; the pustules are more concentrated in flexural sites; the eruption is gradual in onset; and histologically there tends to be features of psoriasis including parakeratosis, Munro microabscesses, and dilated blood vessels.10 Subcorneal pustular dermatosis also is more concentrated in flexural sites and frequently has an annular or serpiginous configuration. The onset also is gradual, and it follows a more chronic course than AGEP. Exfoliative erythroderma presents with widespread erythema and superficial desquamating scale. It often occurs in association with systemic symptoms and can be the result of a drug reaction or underlying inflammatory dermatosis such as psoriasis, mycosis fungoides, or pityriasis rubra pilaris.
Acute generalized exanthematous pustulosis usually resolves spontaneously within 2 weeks and is associated with a superficial desquamation as it clears. Appropriate treatment includes discontinuing the offending agent; monitoring for systemic involvement; and treating the patient's symptoms with antihistamines, analgesics, topical steroids, and emollients. In more severe or persistent cases, treatment with systemic steroids and tumor necrosis factor α inhibitors has been attempted, though their efficacy remains unclear. We report a case of iopamidol-induced AGEP that highlights the importance of eliciting a history of contrast exposure from a patient with suspected AGEP.
- Hammerbeck AA, Daniels NH, Callen JP. Ioversol-induced acute generalized exanthematous pustulosis. Arch Dermatol. 2009;145:683-687.
- Thienvibul C, Vachiramon V, Chanprapaph K. Five-year retrospective review of acute generalized exanthematous pustulosis. Dermatol Res Pract. 2015;2015:1-8.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2016;73:843-848.
- Rosado Ingelmo A, Doña Diaz I, Cabañas Moreno R, et al. Clinical practice guidelines for diagnosis and management of hypersensitivity reactions to contrast media. J Investig Allergol Clin Immunol. 2016;26:144-155.
- Grandvuillemin A, Ripert C, Sgro C, et al. Iodinated contrast media-induced acute generalized exanthematous pustulosis confirmed by delayed skin tests. J Allergy Clin Immunol Pract. 2014;2:805-806.
- Bavbek S, Sözener ZÇ, Aydin Ö, et al. First case report of acute generalized exanthematous pustulosis due to intravenous iopromide. J Investig Allergol Clin Immunol. 2014;24:66-67.
- Kim SJ, Lee T, Lee YS, et al. Acute generalized exanthematous pustulosis caused by radiocontrast media. Ann Allergy Asthma Immunol. 2010;105:492-493.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. Am J Roentgenol. 2006;187:198-201.
- Atasoy M, Erdem T, Sari RA. A case of acute generalized exanthematous pustulosis (AGEP) possibly induced by iohexol. J Dermatol. 2003;30:723-726.
- Halevy S, Kardaun S, Davidovici B, et al; EuroSCAR and RegiSCAR Study Group. The spectrum of histopathological features in acute generalized exanthematous pustulosis: a study of 102 cases. Br J Dermatol. 2010:163:1245-1252.
The Diagnosis: Acute Generalized Exanthematous Pustulosis
Histopathology demonstrated spongiosis with subcorneal pustules and an overlying basket-weave pattern stratum corneum. There was mild papillary dermal edema with scattered dermal neutrophils and rare eosinophils (Figure). The patient's clinical presentation and histopathology were consistent with acute generalized exanthematous pustulosis (AGEP). The inciting agent in this case was the contrast medium iopamidol. The patient was treated with a short course of prednisone, triamcinolone cream, diphenhydramine, and acetaminophen. Within 1 week the pustules and erythema had resolved.
Acute generalized exanthematous pustulosis is an uncommon T cell-mediated cutaneous reaction characterized by widespread progressive erythema with numerous nonfollicular pinpoint pustules. The patient usually is well appearing; however, he/she often will have concurrent fever and facial edema. Mucous membranes rarely are involved. Laboratory results typically are notable only for leukocytosis with neutrophilia.
The pustular eruption typically occurs within 1 to 2 days after exposure to an inciting agent1; however, this latent period can range from 1 hour to nearly 4 weeks in some studies.2 Systemic medications are the cause in approximately 90% of cases, with antibiotics being the most common category. Frequently implicated medications include β-lactams, macrolides, quinolones, sulfonamides, proton pump inhibitors, hydroxychloroquine, terbinafine, nonsteroidal anti-inflammatory drugs, diltiazem, ketoconazole, and fluconazole. Acute generalized exanthematous pustulosis also has been rarely reported following contact with mercury, viral and bacterial infections, and spider bites.3
Iodinated contrast agents have long been known to cause immediate and delayed adverse cutaneous reactions. However, one consensus study indicated that these reactions occur in only 0.05% to 0.10% of patients.4 Although rare, iodinated contrast media (eg, iopamidol, iohexol, ioversol, iodixanol, iomeprol, iobitridol, iopromide) have been reported as a cause of AGEP. A PubMed search of articles indexed for MEDLINE using the terms acute generalized exanthematous pustulosis, contrast, iodine, and iodinated revealed 10 adult cases reported in 6 articles in the English-language literature.1,5-9 The most recent articles focus on methods to identify the causative agent. If the etiology of the reaction is unclear, patch or intradermal testing can help to confirm the causative agent. These tests also can help determine similar agents to which the patient may cross-react.4,5
It can be difficult to differentiate AGEP from other cutaneous drug reactions and other nonfollicular pustular conditions. Drug-induced hypersensitivity syndrome typically presents with facial edema and a morbilliform rash. Although it can present with pustules, the latent period is longer (2-6 weeks), and there frequently are signs of multiorgan involvement including hepatic dysfunction, eosinophilia, atypical lymphocytosis, and lymphadenopathy. Patients with generalized pustular psoriasis often have a history of plaque psoriasis; the pustules are more concentrated in flexural sites; the eruption is gradual in onset; and histologically there tends to be features of psoriasis including parakeratosis, Munro microabscesses, and dilated blood vessels.10 Subcorneal pustular dermatosis also is more concentrated in flexural sites and frequently has an annular or serpiginous configuration. The onset also is gradual, and it follows a more chronic course than AGEP. Exfoliative erythroderma presents with widespread erythema and superficial desquamating scale. It often occurs in association with systemic symptoms and can be the result of a drug reaction or underlying inflammatory dermatosis such as psoriasis, mycosis fungoides, or pityriasis rubra pilaris.
Acute generalized exanthematous pustulosis usually resolves spontaneously within 2 weeks and is associated with a superficial desquamation as it clears. Appropriate treatment includes discontinuing the offending agent; monitoring for systemic involvement; and treating the patient's symptoms with antihistamines, analgesics, topical steroids, and emollients. In more severe or persistent cases, treatment with systemic steroids and tumor necrosis factor α inhibitors has been attempted, though their efficacy remains unclear. We report a case of iopamidol-induced AGEP that highlights the importance of eliciting a history of contrast exposure from a patient with suspected AGEP.
The Diagnosis: Acute Generalized Exanthematous Pustulosis
Histopathology demonstrated spongiosis with subcorneal pustules and an overlying basket-weave pattern stratum corneum. There was mild papillary dermal edema with scattered dermal neutrophils and rare eosinophils (Figure). The patient's clinical presentation and histopathology were consistent with acute generalized exanthematous pustulosis (AGEP). The inciting agent in this case was the contrast medium iopamidol. The patient was treated with a short course of prednisone, triamcinolone cream, diphenhydramine, and acetaminophen. Within 1 week the pustules and erythema had resolved.
Acute generalized exanthematous pustulosis is an uncommon T cell-mediated cutaneous reaction characterized by widespread progressive erythema with numerous nonfollicular pinpoint pustules. The patient usually is well appearing; however, he/she often will have concurrent fever and facial edema. Mucous membranes rarely are involved. Laboratory results typically are notable only for leukocytosis with neutrophilia.
The pustular eruption typically occurs within 1 to 2 days after exposure to an inciting agent1; however, this latent period can range from 1 hour to nearly 4 weeks in some studies.2 Systemic medications are the cause in approximately 90% of cases, with antibiotics being the most common category. Frequently implicated medications include β-lactams, macrolides, quinolones, sulfonamides, proton pump inhibitors, hydroxychloroquine, terbinafine, nonsteroidal anti-inflammatory drugs, diltiazem, ketoconazole, and fluconazole. Acute generalized exanthematous pustulosis also has been rarely reported following contact with mercury, viral and bacterial infections, and spider bites.3
Iodinated contrast agents have long been known to cause immediate and delayed adverse cutaneous reactions. However, one consensus study indicated that these reactions occur in only 0.05% to 0.10% of patients.4 Although rare, iodinated contrast media (eg, iopamidol, iohexol, ioversol, iodixanol, iomeprol, iobitridol, iopromide) have been reported as a cause of AGEP. A PubMed search of articles indexed for MEDLINE using the terms acute generalized exanthematous pustulosis, contrast, iodine, and iodinated revealed 10 adult cases reported in 6 articles in the English-language literature.1,5-9 The most recent articles focus on methods to identify the causative agent. If the etiology of the reaction is unclear, patch or intradermal testing can help to confirm the causative agent. These tests also can help determine similar agents to which the patient may cross-react.4,5
It can be difficult to differentiate AGEP from other cutaneous drug reactions and other nonfollicular pustular conditions. Drug-induced hypersensitivity syndrome typically presents with facial edema and a morbilliform rash. Although it can present with pustules, the latent period is longer (2-6 weeks), and there frequently are signs of multiorgan involvement including hepatic dysfunction, eosinophilia, atypical lymphocytosis, and lymphadenopathy. Patients with generalized pustular psoriasis often have a history of plaque psoriasis; the pustules are more concentrated in flexural sites; the eruption is gradual in onset; and histologically there tends to be features of psoriasis including parakeratosis, Munro microabscesses, and dilated blood vessels.10 Subcorneal pustular dermatosis also is more concentrated in flexural sites and frequently has an annular or serpiginous configuration. The onset also is gradual, and it follows a more chronic course than AGEP. Exfoliative erythroderma presents with widespread erythema and superficial desquamating scale. It often occurs in association with systemic symptoms and can be the result of a drug reaction or underlying inflammatory dermatosis such as psoriasis, mycosis fungoides, or pityriasis rubra pilaris.
Acute generalized exanthematous pustulosis usually resolves spontaneously within 2 weeks and is associated with a superficial desquamation as it clears. Appropriate treatment includes discontinuing the offending agent; monitoring for systemic involvement; and treating the patient's symptoms with antihistamines, analgesics, topical steroids, and emollients. In more severe or persistent cases, treatment with systemic steroids and tumor necrosis factor α inhibitors has been attempted, though their efficacy remains unclear. We report a case of iopamidol-induced AGEP that highlights the importance of eliciting a history of contrast exposure from a patient with suspected AGEP.
- Hammerbeck AA, Daniels NH, Callen JP. Ioversol-induced acute generalized exanthematous pustulosis. Arch Dermatol. 2009;145:683-687.
- Thienvibul C, Vachiramon V, Chanprapaph K. Five-year retrospective review of acute generalized exanthematous pustulosis. Dermatol Res Pract. 2015;2015:1-8.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2016;73:843-848.
- Rosado Ingelmo A, Doña Diaz I, Cabañas Moreno R, et al. Clinical practice guidelines for diagnosis and management of hypersensitivity reactions to contrast media. J Investig Allergol Clin Immunol. 2016;26:144-155.
- Grandvuillemin A, Ripert C, Sgro C, et al. Iodinated contrast media-induced acute generalized exanthematous pustulosis confirmed by delayed skin tests. J Allergy Clin Immunol Pract. 2014;2:805-806.
- Bavbek S, Sözener ZÇ, Aydin Ö, et al. First case report of acute generalized exanthematous pustulosis due to intravenous iopromide. J Investig Allergol Clin Immunol. 2014;24:66-67.
- Kim SJ, Lee T, Lee YS, et al. Acute generalized exanthematous pustulosis caused by radiocontrast media. Ann Allergy Asthma Immunol. 2010;105:492-493.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. Am J Roentgenol. 2006;187:198-201.
- Atasoy M, Erdem T, Sari RA. A case of acute generalized exanthematous pustulosis (AGEP) possibly induced by iohexol. J Dermatol. 2003;30:723-726.
- Halevy S, Kardaun S, Davidovici B, et al; EuroSCAR and RegiSCAR Study Group. The spectrum of histopathological features in acute generalized exanthematous pustulosis: a study of 102 cases. Br J Dermatol. 2010:163:1245-1252.
- Hammerbeck AA, Daniels NH, Callen JP. Ioversol-induced acute generalized exanthematous pustulosis. Arch Dermatol. 2009;145:683-687.
- Thienvibul C, Vachiramon V, Chanprapaph K. Five-year retrospective review of acute generalized exanthematous pustulosis. Dermatol Res Pract. 2015;2015:1-8.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2016;73:843-848.
- Rosado Ingelmo A, Doña Diaz I, Cabañas Moreno R, et al. Clinical practice guidelines for diagnosis and management of hypersensitivity reactions to contrast media. J Investig Allergol Clin Immunol. 2016;26:144-155.
- Grandvuillemin A, Ripert C, Sgro C, et al. Iodinated contrast media-induced acute generalized exanthematous pustulosis confirmed by delayed skin tests. J Allergy Clin Immunol Pract. 2014;2:805-806.
- Bavbek S, Sözener ZÇ, Aydin Ö, et al. First case report of acute generalized exanthematous pustulosis due to intravenous iopromide. J Investig Allergol Clin Immunol. 2014;24:66-67.
- Kim SJ, Lee T, Lee YS, et al. Acute generalized exanthematous pustulosis caused by radiocontrast media. Ann Allergy Asthma Immunol. 2010;105:492-493.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. Am J Roentgenol. 2006;187:198-201.
- Atasoy M, Erdem T, Sari RA. A case of acute generalized exanthematous pustulosis (AGEP) possibly induced by iohexol. J Dermatol. 2003;30:723-726.
- Halevy S, Kardaun S, Davidovici B, et al; EuroSCAR and RegiSCAR Study Group. The spectrum of histopathological features in acute generalized exanthematous pustulosis: a study of 102 cases. Br J Dermatol. 2010:163:1245-1252.
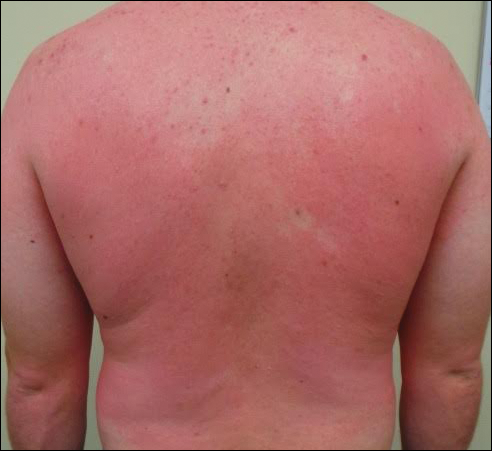
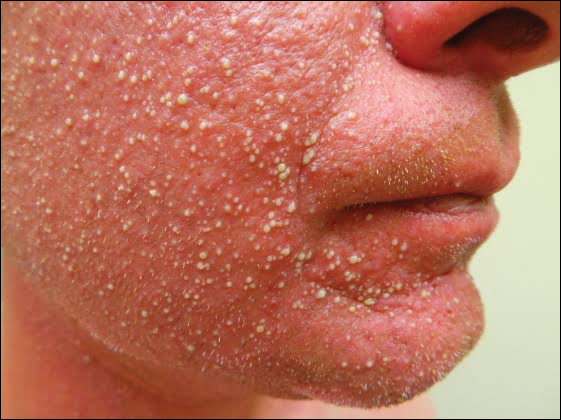
A 31-year-old man presented with a rapidly progressive, burning rash of 1 day's duration, along with malaise, nausea, and dizziness. At the time of presentation, he was hemodynamically stable and afebrile. Laboratory analysis revealed mild leukocytosis with neutrophilia. A complete metabolic panel was within normal limits. He had no chronic medical conditions and was taking no medications or supplements. One day prior to onset of the rash, he underwent contrast-enhanced (iopamidol) computed tomography of the abdomen. Physical examination revealed large edematous plaques on the face, neck, and trunk (top) that were studded with numerous pinpoint pustules (bottom). He also had subtle facial edema. There was relative sparing of the flexural sites and no involvement of the palms, soles, or mucous membranes. A shave biopsy was obtained from a pustular area on the neck.
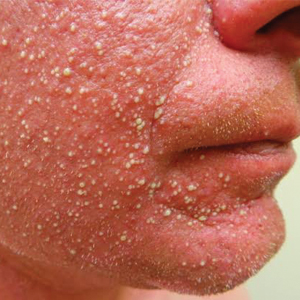
Hemorrhagic Crusted Papule on the Arm
The Diagnosis: Self-healing Langerhans Cell Histiocytosis
Histopathologic examination showed an infiltrate of mononuclear cells with indented nuclei admixed with a variable dermal inflammatory infiltrate. Immunohistochemistry demonstrated cells that were strongly positive for CD1a (Figure, A) and langerin (Figure, B) antigens as well as S-100 protein (Figure, C), which was consistent with Langerhans cell histiocytosis (LCH).
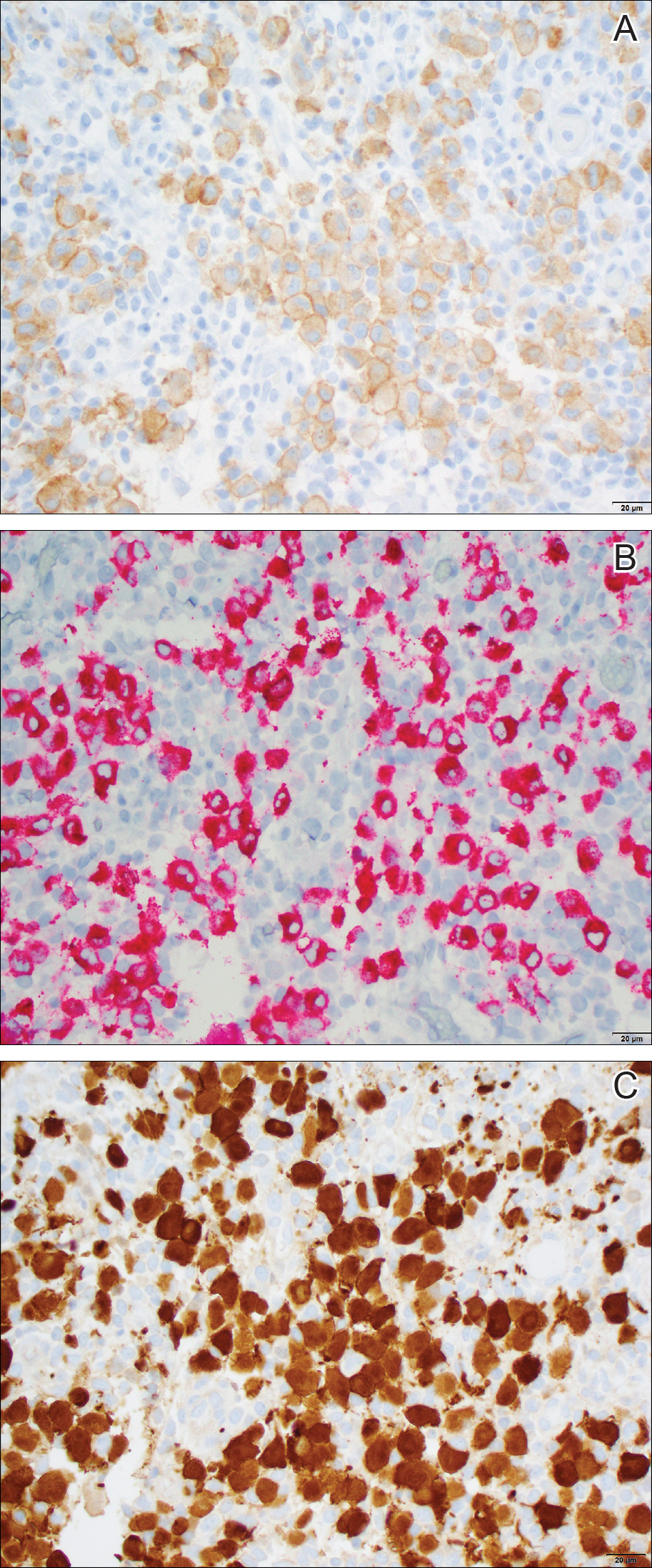
Histiocytoses are a heterogeneous group of disorders in which the infiltrating cells belong to the mononuclear phagocyte system.1,2 Langerhans cell histiocytosis is the most common dendritic cell-related histiocytosis, occurring in approximately 5 per 1 million children annually, giving it an incidence comparable to pediatric Hodgkin lymphoma and acute myeloid leukemia.1,2
Historically, there has been much debate about the pathogenesis of the disease.2 Until recently it was unknown whether LCH was primarily a neoplastic or an inflammatory disorder. Although the condition initially was thought to have a reactive etiology,1 more recent evidence suggests a clonal neoplastic process. Langerhans cell histiocytosis lesions are clonal and display malignancy-associated mechanisms such as immune evasion. Genome sequencing has revealed several mutations in precursor myeloid cells that result in the common downstream hyperactivation of the mitogen-activated protein kinase signaling pathway that regulates cell proliferation and differentiation.1
Langerhans cell histiocytosis displays a wide spectrum of clinical phenotypes, which historically were subclassified as eosinophilic granulomas (localized lesions in bone), Hand-Schüller-Christian disease (multiple organ involvement with the classic triad of skull defects, diabetes insipidus, and exophthalmos), and Letterer-Siwe disease (visceral lesions involving multiple organs).3 However, in 1997 the Reclassification Working Group of the Histiocyte Society redefined LCH as single-system single site (SS-s) LCH, single-system multisite LCH, and multisystem LCH.4
In SS-s LCH, the most common site is bone (82%), followed by the skin (12%).5 Skin SS-s LCH classically presents as multiple skin lesions at birth without systemic manifestations; the lesions spontaneously involute within a few months.6 Less commonly, skin SS-s LCH can present as a single lesion. Berger et al7 described 4 neonates with unilesional skin SS-s LCH. Since then, more than 30 cases have been reported in the literature,8 and we report herein another unilesional self-healing LCH.
The morphology of skin lesions in self-healing LCH is highly variable, with the most common being multiple erythematous crusted papules (50%), followed by eczematous scaly lesions resembling seborrheic dermatitis in intertriginous areas (37.5%).3,6 Unilesional self-healing LCH typically presents as an ulcerated or crusted nodule or papule on the trunk. This variability results in a large differential diagnosis. Self-healing LCH is easily mistaken for infectious processes including neonatal herpes simplex and varicella-zoster virus infection.9 Often, the dermatology department is consulted to rule out LCH when the asymptomatic neonate does not respond to parenteral acyclovir.
Less commonly, the magenta-colored papulonodules of self-healing LCH can mimic blueberry muffin rash and mandate a workup for intrauterine infections, especially cytomegalovirus, rubella, and blood dyscrasia.10 Other noninfectious processes in the differential of self-healing LCH include congenital infantile hemangioma, neonatal lupus erythematosus, seborrheic dermatitis (cradle cap), pyogenic granuloma, and psoriasis.3,10 Definitive diagnosis requires histopathology.
Because unilesional self-healing LCH has an excellent prognosis and usually resolves on its own, therapy is unnecessary.3,8 One large retrospective study (N=146) found that of all patients with skin lesions, 56% were managed with biopsy only.5 Other options include watchful waiting and topical corticosteroids. If the skin lesions are large, ulcerated, and/or painful, alkylating antitumor agents have been used. For extensive cutaneous disease, systemic corticosteroids combined with chemotherapy and psoralen plus UVA can be effective.6
The primary concern in the management of self-healing LCH is that the solitary skin lesion may be the harbinger of an aggressive disorder that can progress to systemic disease.5 Moreover, recurrent visceral or disseminated disease may occur months to years after resolution of solitary skin lesions.9 Studies have shown that localized and disseminated disease cannot be differentiated on the basis of clinical findings, histology, immunohistochemistry, or biomarkers.3,11 As a result, an evaluation for systemic disease should be performed at the time of diagnosis for cutaneous LCH.3,9 Minimum baseline studies recommended by the Writing Group of the Histiocyte Society include a complete blood cell count, liver function tests, coagulation studies, chest radiography, skeletal surveys, and urine osmolality testing.12 Periodic clinical follow-up is recommended for all variants of LCH.9
Our case was diagnosed as self-healing LCH based on histologic findings. No treatment was required, and at 3-month follow-up the infant was asymptomatic without recurrence and was meeting all developmental milestones.
- Berres ML, Merad M, Allen CE. Progress in understanding the pathogenesis of Langerhans cell histiocytosis: back to histiocytosis X? Br J Haematol. 2015;169:3-13.
- Jordan MB, Filipovich AH. Histiocytic disorders. In: Hoffman R, Benz EJ Jr, Silberstein LE, eds. Hematology: Basic Principles and Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:686-700.
- Stein SL, Paller AS, Haut PR, et al. Langerhans cell histiocytosis presenting in the neonatal period: a retrospective case series. Arch Pediatr Adolesc Med. 2001;155:778-783.
- Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. Pediatr Blood Cancer. 1997;29:157-166.
- Morimoto A, Ishida Y, Suzuki N, et al. Nationwide survey of single-system single site Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer. 2010;54:98-102.
- Morren MA, Broecke KV, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492.
- Berger TG, Lane AT, Headington JT, et al. A solitary variant of congenital self-healing reticulohistiocytosis: solitary Hashimoto-Pritzker disease. Pediatr Dermatol. 1986;3:230.
- Wheller L, Carman N, Butler G. Unilesional self-limited Langerhans cell histiocytosis: a case report and review of the literature. J Cutan Pathol. 2013;40:595-599.
- Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156.
- Mehta V, Balachandran C, Lonikar V. Blueberry muffin baby: a pictoral differential diagnosis. Dermatol Online J. 2008;14:8.
- Kapur P, Erickson C, Rakheja D, et al. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children's Medical Center. J Am Acad Dermatol. 2007;56:290-294.
- Writing Group of the Histiocyte Society. Histiocytosis syndromes in children. Lancet. 1987;24:208-209.
The Diagnosis: Self-healing Langerhans Cell Histiocytosis
Histopathologic examination showed an infiltrate of mononuclear cells with indented nuclei admixed with a variable dermal inflammatory infiltrate. Immunohistochemistry demonstrated cells that were strongly positive for CD1a (Figure, A) and langerin (Figure, B) antigens as well as S-100 protein (Figure, C), which was consistent with Langerhans cell histiocytosis (LCH).
Histiocytoses are a heterogeneous group of disorders in which the infiltrating cells belong to the mononuclear phagocyte system.1,2 Langerhans cell histiocytosis is the most common dendritic cell-related histiocytosis, occurring in approximately 5 per 1 million children annually, giving it an incidence comparable to pediatric Hodgkin lymphoma and acute myeloid leukemia.1,2
Historically, there has been much debate about the pathogenesis of the disease.2 Until recently it was unknown whether LCH was primarily a neoplastic or an inflammatory disorder. Although the condition initially was thought to have a reactive etiology,1 more recent evidence suggests a clonal neoplastic process. Langerhans cell histiocytosis lesions are clonal and display malignancy-associated mechanisms such as immune evasion. Genome sequencing has revealed several mutations in precursor myeloid cells that result in the common downstream hyperactivation of the mitogen-activated protein kinase signaling pathway that regulates cell proliferation and differentiation.1
Langerhans cell histiocytosis displays a wide spectrum of clinical phenotypes, which historically were subclassified as eosinophilic granulomas (localized lesions in bone), Hand-Schüller-Christian disease (multiple organ involvement with the classic triad of skull defects, diabetes insipidus, and exophthalmos), and Letterer-Siwe disease (visceral lesions involving multiple organs).3 However, in 1997 the Reclassification Working Group of the Histiocyte Society redefined LCH as single-system single site (SS-s) LCH, single-system multisite LCH, and multisystem LCH.4
In SS-s LCH, the most common site is bone (82%), followed by the skin (12%).5 Skin SS-s LCH classically presents as multiple skin lesions at birth without systemic manifestations; the lesions spontaneously involute within a few months.6 Less commonly, skin SS-s LCH can present as a single lesion. Berger et al7 described 4 neonates with unilesional skin SS-s LCH. Since then, more than 30 cases have been reported in the literature,8 and we report herein another unilesional self-healing LCH.
The morphology of skin lesions in self-healing LCH is highly variable, with the most common being multiple erythematous crusted papules (50%), followed by eczematous scaly lesions resembling seborrheic dermatitis in intertriginous areas (37.5%).3,6 Unilesional self-healing LCH typically presents as an ulcerated or crusted nodule or papule on the trunk. This variability results in a large differential diagnosis. Self-healing LCH is easily mistaken for infectious processes including neonatal herpes simplex and varicella-zoster virus infection.9 Often, the dermatology department is consulted to rule out LCH when the asymptomatic neonate does not respond to parenteral acyclovir.
Less commonly, the magenta-colored papulonodules of self-healing LCH can mimic blueberry muffin rash and mandate a workup for intrauterine infections, especially cytomegalovirus, rubella, and blood dyscrasia.10 Other noninfectious processes in the differential of self-healing LCH include congenital infantile hemangioma, neonatal lupus erythematosus, seborrheic dermatitis (cradle cap), pyogenic granuloma, and psoriasis.3,10 Definitive diagnosis requires histopathology.
Because unilesional self-healing LCH has an excellent prognosis and usually resolves on its own, therapy is unnecessary.3,8 One large retrospective study (N=146) found that of all patients with skin lesions, 56% were managed with biopsy only.5 Other options include watchful waiting and topical corticosteroids. If the skin lesions are large, ulcerated, and/or painful, alkylating antitumor agents have been used. For extensive cutaneous disease, systemic corticosteroids combined with chemotherapy and psoralen plus UVA can be effective.6
The primary concern in the management of self-healing LCH is that the solitary skin lesion may be the harbinger of an aggressive disorder that can progress to systemic disease.5 Moreover, recurrent visceral or disseminated disease may occur months to years after resolution of solitary skin lesions.9 Studies have shown that localized and disseminated disease cannot be differentiated on the basis of clinical findings, histology, immunohistochemistry, or biomarkers.3,11 As a result, an evaluation for systemic disease should be performed at the time of diagnosis for cutaneous LCH.3,9 Minimum baseline studies recommended by the Writing Group of the Histiocyte Society include a complete blood cell count, liver function tests, coagulation studies, chest radiography, skeletal surveys, and urine osmolality testing.12 Periodic clinical follow-up is recommended for all variants of LCH.9
Our case was diagnosed as self-healing LCH based on histologic findings. No treatment was required, and at 3-month follow-up the infant was asymptomatic without recurrence and was meeting all developmental milestones.
The Diagnosis: Self-healing Langerhans Cell Histiocytosis
Histopathologic examination showed an infiltrate of mononuclear cells with indented nuclei admixed with a variable dermal inflammatory infiltrate. Immunohistochemistry demonstrated cells that were strongly positive for CD1a (Figure, A) and langerin (Figure, B) antigens as well as S-100 protein (Figure, C), which was consistent with Langerhans cell histiocytosis (LCH).
Histiocytoses are a heterogeneous group of disorders in which the infiltrating cells belong to the mononuclear phagocyte system.1,2 Langerhans cell histiocytosis is the most common dendritic cell-related histiocytosis, occurring in approximately 5 per 1 million children annually, giving it an incidence comparable to pediatric Hodgkin lymphoma and acute myeloid leukemia.1,2
Historically, there has been much debate about the pathogenesis of the disease.2 Until recently it was unknown whether LCH was primarily a neoplastic or an inflammatory disorder. Although the condition initially was thought to have a reactive etiology,1 more recent evidence suggests a clonal neoplastic process. Langerhans cell histiocytosis lesions are clonal and display malignancy-associated mechanisms such as immune evasion. Genome sequencing has revealed several mutations in precursor myeloid cells that result in the common downstream hyperactivation of the mitogen-activated protein kinase signaling pathway that regulates cell proliferation and differentiation.1
Langerhans cell histiocytosis displays a wide spectrum of clinical phenotypes, which historically were subclassified as eosinophilic granulomas (localized lesions in bone), Hand-Schüller-Christian disease (multiple organ involvement with the classic triad of skull defects, diabetes insipidus, and exophthalmos), and Letterer-Siwe disease (visceral lesions involving multiple organs).3 However, in 1997 the Reclassification Working Group of the Histiocyte Society redefined LCH as single-system single site (SS-s) LCH, single-system multisite LCH, and multisystem LCH.4
In SS-s LCH, the most common site is bone (82%), followed by the skin (12%).5 Skin SS-s LCH classically presents as multiple skin lesions at birth without systemic manifestations; the lesions spontaneously involute within a few months.6 Less commonly, skin SS-s LCH can present as a single lesion. Berger et al7 described 4 neonates with unilesional skin SS-s LCH. Since then, more than 30 cases have been reported in the literature,8 and we report herein another unilesional self-healing LCH.
The morphology of skin lesions in self-healing LCH is highly variable, with the most common being multiple erythematous crusted papules (50%), followed by eczematous scaly lesions resembling seborrheic dermatitis in intertriginous areas (37.5%).3,6 Unilesional self-healing LCH typically presents as an ulcerated or crusted nodule or papule on the trunk. This variability results in a large differential diagnosis. Self-healing LCH is easily mistaken for infectious processes including neonatal herpes simplex and varicella-zoster virus infection.9 Often, the dermatology department is consulted to rule out LCH when the asymptomatic neonate does not respond to parenteral acyclovir.
Less commonly, the magenta-colored papulonodules of self-healing LCH can mimic blueberry muffin rash and mandate a workup for intrauterine infections, especially cytomegalovirus, rubella, and blood dyscrasia.10 Other noninfectious processes in the differential of self-healing LCH include congenital infantile hemangioma, neonatal lupus erythematosus, seborrheic dermatitis (cradle cap), pyogenic granuloma, and psoriasis.3,10 Definitive diagnosis requires histopathology.
Because unilesional self-healing LCH has an excellent prognosis and usually resolves on its own, therapy is unnecessary.3,8 One large retrospective study (N=146) found that of all patients with skin lesions, 56% were managed with biopsy only.5 Other options include watchful waiting and topical corticosteroids. If the skin lesions are large, ulcerated, and/or painful, alkylating antitumor agents have been used. For extensive cutaneous disease, systemic corticosteroids combined with chemotherapy and psoralen plus UVA can be effective.6
The primary concern in the management of self-healing LCH is that the solitary skin lesion may be the harbinger of an aggressive disorder that can progress to systemic disease.5 Moreover, recurrent visceral or disseminated disease may occur months to years after resolution of solitary skin lesions.9 Studies have shown that localized and disseminated disease cannot be differentiated on the basis of clinical findings, histology, immunohistochemistry, or biomarkers.3,11 As a result, an evaluation for systemic disease should be performed at the time of diagnosis for cutaneous LCH.3,9 Minimum baseline studies recommended by the Writing Group of the Histiocyte Society include a complete blood cell count, liver function tests, coagulation studies, chest radiography, skeletal surveys, and urine osmolality testing.12 Periodic clinical follow-up is recommended for all variants of LCH.9
Our case was diagnosed as self-healing LCH based on histologic findings. No treatment was required, and at 3-month follow-up the infant was asymptomatic without recurrence and was meeting all developmental milestones.
- Berres ML, Merad M, Allen CE. Progress in understanding the pathogenesis of Langerhans cell histiocytosis: back to histiocytosis X? Br J Haematol. 2015;169:3-13.
- Jordan MB, Filipovich AH. Histiocytic disorders. In: Hoffman R, Benz EJ Jr, Silberstein LE, eds. Hematology: Basic Principles and Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:686-700.
- Stein SL, Paller AS, Haut PR, et al. Langerhans cell histiocytosis presenting in the neonatal period: a retrospective case series. Arch Pediatr Adolesc Med. 2001;155:778-783.
- Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. Pediatr Blood Cancer. 1997;29:157-166.
- Morimoto A, Ishida Y, Suzuki N, et al. Nationwide survey of single-system single site Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer. 2010;54:98-102.
- Morren MA, Broecke KV, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492.
- Berger TG, Lane AT, Headington JT, et al. A solitary variant of congenital self-healing reticulohistiocytosis: solitary Hashimoto-Pritzker disease. Pediatr Dermatol. 1986;3:230.
- Wheller L, Carman N, Butler G. Unilesional self-limited Langerhans cell histiocytosis: a case report and review of the literature. J Cutan Pathol. 2013;40:595-599.
- Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156.
- Mehta V, Balachandran C, Lonikar V. Blueberry muffin baby: a pictoral differential diagnosis. Dermatol Online J. 2008;14:8.
- Kapur P, Erickson C, Rakheja D, et al. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children's Medical Center. J Am Acad Dermatol. 2007;56:290-294.
- Writing Group of the Histiocyte Society. Histiocytosis syndromes in children. Lancet. 1987;24:208-209.
- Berres ML, Merad M, Allen CE. Progress in understanding the pathogenesis of Langerhans cell histiocytosis: back to histiocytosis X? Br J Haematol. 2015;169:3-13.
- Jordan MB, Filipovich AH. Histiocytic disorders. In: Hoffman R, Benz EJ Jr, Silberstein LE, eds. Hematology: Basic Principles and Practice. 6th ed. Philadelphia, PA: Elsevier Saunders; 2013:686-700.
- Stein SL, Paller AS, Haut PR, et al. Langerhans cell histiocytosis presenting in the neonatal period: a retrospective case series. Arch Pediatr Adolesc Med. 2001;155:778-783.
- Favara BE, Feller AC, Pauli M, et al. Contemporary classification of histiocytic disorders. Pediatr Blood Cancer. 1997;29:157-166.
- Morimoto A, Ishida Y, Suzuki N, et al. Nationwide survey of single-system single site Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer. 2010;54:98-102.
- Morren MA, Broecke KV, Vangeebergen L, et al. Diverse cutaneous presentations of Langerhans cell histiocytosis in children: a retrospective cohort study. Pediatr Blood Cancer. 2016;63:486-492.
- Berger TG, Lane AT, Headington JT, et al. A solitary variant of congenital self-healing reticulohistiocytosis: solitary Hashimoto-Pritzker disease. Pediatr Dermatol. 1986;3:230.
- Wheller L, Carman N, Butler G. Unilesional self-limited Langerhans cell histiocytosis: a case report and review of the literature. J Cutan Pathol. 2013;40:595-599.
- Battistella M, Fraitag S, Teillac DH, et al. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol. 2010;146:149-156.
- Mehta V, Balachandran C, Lonikar V. Blueberry muffin baby: a pictoral differential diagnosis. Dermatol Online J. 2008;14:8.
- Kapur P, Erickson C, Rakheja D, et al. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children's Medical Center. J Am Acad Dermatol. 2007;56:290-294.
- Writing Group of the Histiocyte Society. Histiocytosis syndromes in children. Lancet. 1987;24:208-209.
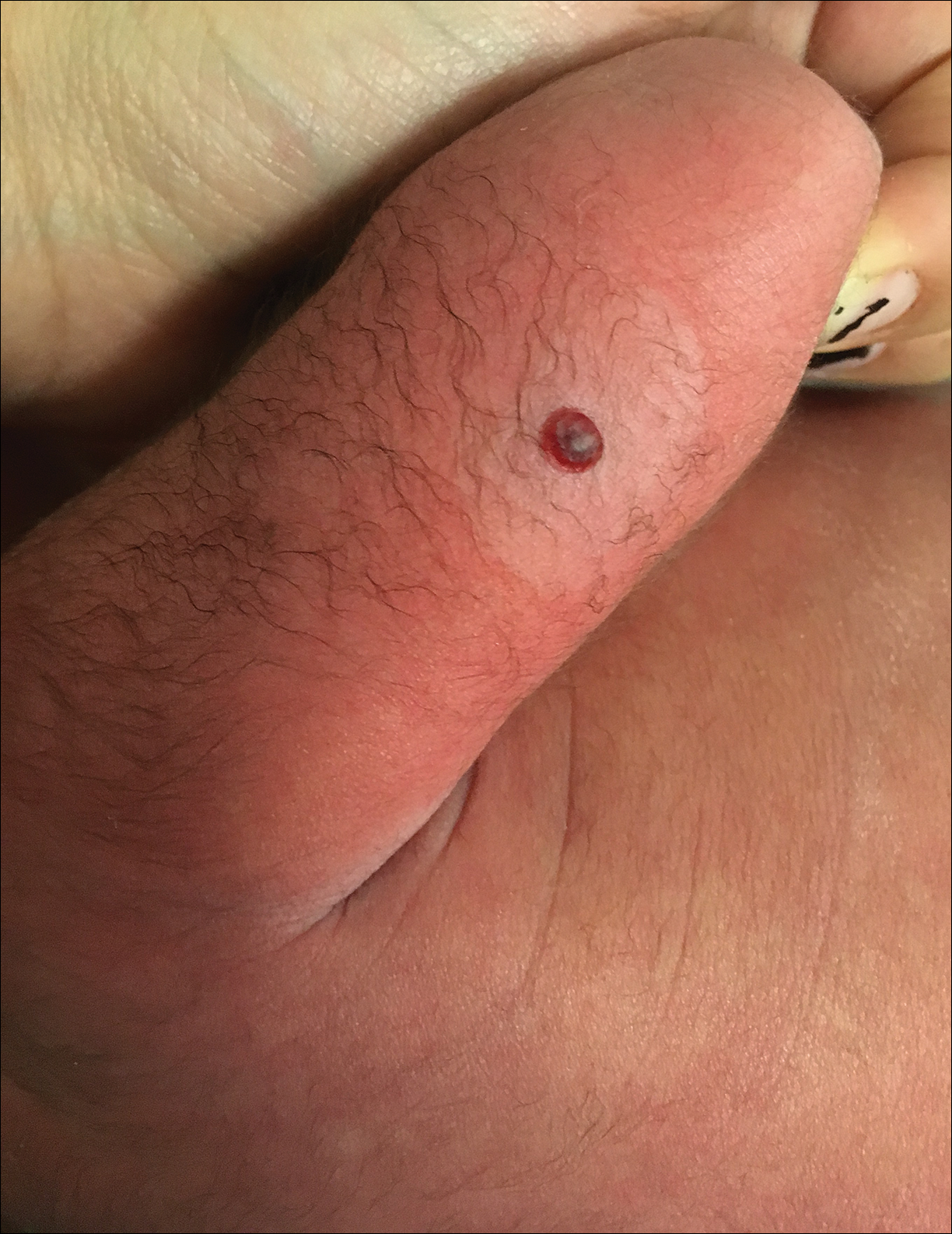
Dermatology consultation was called to the delivery room to evaluate a red, hemorrhagic, crusted, 5-mm papule on the right lateral upper arm of a preterm newborn. He appeared vigorous with an Apgar score of 7 at 1 minute and 8 at 5 minutes. Physical examination was otherwise normal. Of note, the mother presented late to prenatal care. Her herpes simplex and varicella-zoster virus status was unknown. A shave biopsy of the papule was performed at 3 days of age.
Pigmented Pruritic Macules in the Genital Area
The Diagnosis: Pediculosis Pubis
Dermoscopy of the pubic hair demonstrated a louse clutching multiple shafts of hairs (Figure) as well as scattered nits, confirming the presence of Phthirus pubis and diagnosis of pediculosis pubis. The key clinical diagnostic feature in this case was severe itching of the pubic area, with visible nits present on pubic hairs. Itching and infestation also can involve other hair-bearing areas such as the chest, legs, and axillae. The patient was treated with permethrin cream 5% applied to the pubic area and chest. Symptoms and infestation were resolved at 1-week follow-up.
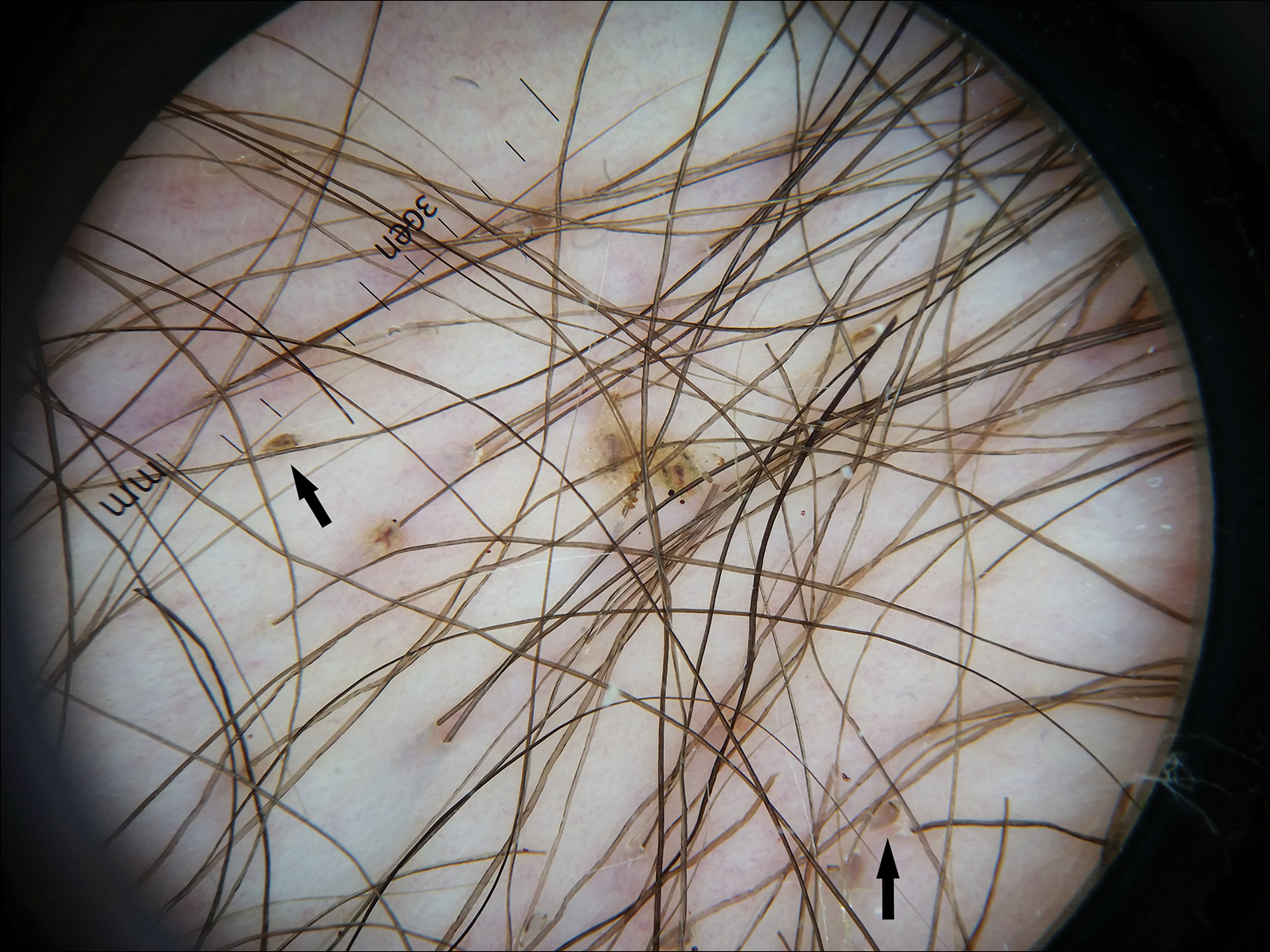
Pediculosis pubis is an infestation of pubic hairs by the pubic (crab) louse P pubis, which feeds on host blood. Other body areas covered with dense hair also may be involved; 60% of patients are infested in at least 2 different sites.1 Pediculosis pubis is most commonly sexually transmitted through direct contact.2 Worldwide prevalence has been estimated at approximately 2% of the adult population, and a survey of 817 US college students in 2009 indicated a lifetime prevalence of 1.3%.3 The prevalence is slightly higher in men, highest in men who have sex with men, and rare in individuals with shaved pubic hair.1 The most common symptom is pruritus of the genital area. Infested patients also may develop asymptomatic bluish gray macules (maculae ceruleae) secondary to hemosiderin deposition from louse bites.4
The diagnosis of pediculosis pubis is made by identification of P pubis, either by examination with the naked eye or confirmation with dermoscopy or microscopy. Although the 0.8- to 1.2-mm lice are visible to the naked eye, they can be difficult to see if not filled with blood, and nits on the hairs can be mistaken for white piedra (fungal infection of the hair shafts) or trichomycosis pubis (bacterial infection of the hair shafts).4 Scabies and tinea cruris do not present with attachments to the hairs. Scabies may present with papules and burrows and tinea cruris with scaly erythematous plaques. Small numbers of lice and nits may be missed by the naked eye or a traditional magnifying glass.5 The use of dermoscopy allows for fast and accurate identification of the characteristic lice and nits, even in these more challenging cases.5 Accurate diagnosis is important, as approximately 31% of infested patients have other concurrent sexually transmitted infections that warrant screening.6
The Centers for Disease Control and Prevention recommends first-line treatment with permethrin cream (1% or 5%) or pyrethrin with piperonyl butoxide applied to all affected areas and washed off after 10 minutes.7 Patients should be reevaluated after 1 week if symptoms persist and re-treated if lice are found on examination.7,8 Malathion lotion 0.5% (applied and washed off after 8-12 hours) or oral ivermectin (250 µg/kg, repeated after 2 weeks) may be used for alternative therapies or cases of permethrin or pyrethrin resistance.7 Ivermectin also is effective for involvement of eyelashes where topical insecticides should not be used.9 Sexual partners should be treated to prevent repeat transmission.7,8 Bedding and clothing can be decontaminated by machine wash on hot cycle or isolating from body contact for 72 hours.7 Patients also should be screened for other sexually transmitted infections, including human immunodeficiency virus.6
- Burkhart CN, Burkhart CG, Morrell DS. Infestations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier/Saunders; 2012:1429-1430.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Anderson AL, Chaney E. Pubic lice (Pthirus pubis): history, biology and treatment vs. knowledge and beliefs of US college students. Int J Environ Res Public Health. 2009;6:592-600.
- Ko CJ, Elston DM. Pediculosis. J Am Acad Dermatol. 2004;50:1-12; quiz 13-14.
- Chuh A, Lee A, Wong W, et al. Diagnosis of pediculosis pubis: a novel application of digital epiluminescence dermatoscopy. J Eur Acad Dermatol Venereol. 2007;21:837-838.
- Chapel TA, Katta T, Kuszmar T, et al. Pediculosis pubis in a clinic for treatment of sexually transmitted diseases. Sex Transm Dis. 1979;6:257-260.
- Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
- Leone PA. Scabies and pediculosis pubis: an update of treatment regimens and general review. Clin Infect Dis. 2007;44(suppl 3):S153-S159.
- Burkhart CN, Burkhart CG. Oral ivermectin therapy for phthiriasis palpebrum. Arch Ophthalmol. 2000;118:134-135.
The Diagnosis: Pediculosis Pubis
Dermoscopy of the pubic hair demonstrated a louse clutching multiple shafts of hairs (Figure) as well as scattered nits, confirming the presence of Phthirus pubis and diagnosis of pediculosis pubis. The key clinical diagnostic feature in this case was severe itching of the pubic area, with visible nits present on pubic hairs. Itching and infestation also can involve other hair-bearing areas such as the chest, legs, and axillae. The patient was treated with permethrin cream 5% applied to the pubic area and chest. Symptoms and infestation were resolved at 1-week follow-up.
Pediculosis pubis is an infestation of pubic hairs by the pubic (crab) louse P pubis, which feeds on host blood. Other body areas covered with dense hair also may be involved; 60% of patients are infested in at least 2 different sites.1 Pediculosis pubis is most commonly sexually transmitted through direct contact.2 Worldwide prevalence has been estimated at approximately 2% of the adult population, and a survey of 817 US college students in 2009 indicated a lifetime prevalence of 1.3%.3 The prevalence is slightly higher in men, highest in men who have sex with men, and rare in individuals with shaved pubic hair.1 The most common symptom is pruritus of the genital area. Infested patients also may develop asymptomatic bluish gray macules (maculae ceruleae) secondary to hemosiderin deposition from louse bites.4
The diagnosis of pediculosis pubis is made by identification of P pubis, either by examination with the naked eye or confirmation with dermoscopy or microscopy. Although the 0.8- to 1.2-mm lice are visible to the naked eye, they can be difficult to see if not filled with blood, and nits on the hairs can be mistaken for white piedra (fungal infection of the hair shafts) or trichomycosis pubis (bacterial infection of the hair shafts).4 Scabies and tinea cruris do not present with attachments to the hairs. Scabies may present with papules and burrows and tinea cruris with scaly erythematous plaques. Small numbers of lice and nits may be missed by the naked eye or a traditional magnifying glass.5 The use of dermoscopy allows for fast and accurate identification of the characteristic lice and nits, even in these more challenging cases.5 Accurate diagnosis is important, as approximately 31% of infested patients have other concurrent sexually transmitted infections that warrant screening.6
The Centers for Disease Control and Prevention recommends first-line treatment with permethrin cream (1% or 5%) or pyrethrin with piperonyl butoxide applied to all affected areas and washed off after 10 minutes.7 Patients should be reevaluated after 1 week if symptoms persist and re-treated if lice are found on examination.7,8 Malathion lotion 0.5% (applied and washed off after 8-12 hours) or oral ivermectin (250 µg/kg, repeated after 2 weeks) may be used for alternative therapies or cases of permethrin or pyrethrin resistance.7 Ivermectin also is effective for involvement of eyelashes where topical insecticides should not be used.9 Sexual partners should be treated to prevent repeat transmission.7,8 Bedding and clothing can be decontaminated by machine wash on hot cycle or isolating from body contact for 72 hours.7 Patients also should be screened for other sexually transmitted infections, including human immunodeficiency virus.6
The Diagnosis: Pediculosis Pubis
Dermoscopy of the pubic hair demonstrated a louse clutching multiple shafts of hairs (Figure) as well as scattered nits, confirming the presence of Phthirus pubis and diagnosis of pediculosis pubis. The key clinical diagnostic feature in this case was severe itching of the pubic area, with visible nits present on pubic hairs. Itching and infestation also can involve other hair-bearing areas such as the chest, legs, and axillae. The patient was treated with permethrin cream 5% applied to the pubic area and chest. Symptoms and infestation were resolved at 1-week follow-up.
Pediculosis pubis is an infestation of pubic hairs by the pubic (crab) louse P pubis, which feeds on host blood. Other body areas covered with dense hair also may be involved; 60% of patients are infested in at least 2 different sites.1 Pediculosis pubis is most commonly sexually transmitted through direct contact.2 Worldwide prevalence has been estimated at approximately 2% of the adult population, and a survey of 817 US college students in 2009 indicated a lifetime prevalence of 1.3%.3 The prevalence is slightly higher in men, highest in men who have sex with men, and rare in individuals with shaved pubic hair.1 The most common symptom is pruritus of the genital area. Infested patients also may develop asymptomatic bluish gray macules (maculae ceruleae) secondary to hemosiderin deposition from louse bites.4
The diagnosis of pediculosis pubis is made by identification of P pubis, either by examination with the naked eye or confirmation with dermoscopy or microscopy. Although the 0.8- to 1.2-mm lice are visible to the naked eye, they can be difficult to see if not filled with blood, and nits on the hairs can be mistaken for white piedra (fungal infection of the hair shafts) or trichomycosis pubis (bacterial infection of the hair shafts).4 Scabies and tinea cruris do not present with attachments to the hairs. Scabies may present with papules and burrows and tinea cruris with scaly erythematous plaques. Small numbers of lice and nits may be missed by the naked eye or a traditional magnifying glass.5 The use of dermoscopy allows for fast and accurate identification of the characteristic lice and nits, even in these more challenging cases.5 Accurate diagnosis is important, as approximately 31% of infested patients have other concurrent sexually transmitted infections that warrant screening.6
The Centers for Disease Control and Prevention recommends first-line treatment with permethrin cream (1% or 5%) or pyrethrin with piperonyl butoxide applied to all affected areas and washed off after 10 minutes.7 Patients should be reevaluated after 1 week if symptoms persist and re-treated if lice are found on examination.7,8 Malathion lotion 0.5% (applied and washed off after 8-12 hours) or oral ivermectin (250 µg/kg, repeated after 2 weeks) may be used for alternative therapies or cases of permethrin or pyrethrin resistance.7 Ivermectin also is effective for involvement of eyelashes where topical insecticides should not be used.9 Sexual partners should be treated to prevent repeat transmission.7,8 Bedding and clothing can be decontaminated by machine wash on hot cycle or isolating from body contact for 72 hours.7 Patients also should be screened for other sexually transmitted infections, including human immunodeficiency virus.6
- Burkhart CN, Burkhart CG, Morrell DS. Infestations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier/Saunders; 2012:1429-1430.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Anderson AL, Chaney E. Pubic lice (Pthirus pubis): history, biology and treatment vs. knowledge and beliefs of US college students. Int J Environ Res Public Health. 2009;6:592-600.
- Ko CJ, Elston DM. Pediculosis. J Am Acad Dermatol. 2004;50:1-12; quiz 13-14.
- Chuh A, Lee A, Wong W, et al. Diagnosis of pediculosis pubis: a novel application of digital epiluminescence dermatoscopy. J Eur Acad Dermatol Venereol. 2007;21:837-838.
- Chapel TA, Katta T, Kuszmar T, et al. Pediculosis pubis in a clinic for treatment of sexually transmitted diseases. Sex Transm Dis. 1979;6:257-260.
- Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
- Leone PA. Scabies and pediculosis pubis: an update of treatment regimens and general review. Clin Infect Dis. 2007;44(suppl 3):S153-S159.
- Burkhart CN, Burkhart CG. Oral ivermectin therapy for phthiriasis palpebrum. Arch Ophthalmol. 2000;118:134-135.
- Burkhart CN, Burkhart CG, Morrell DS. Infestations. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. Vol 2. 3rd ed. China: Elsevier/Saunders; 2012:1429-1430.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Anderson AL, Chaney E. Pubic lice (Pthirus pubis): history, biology and treatment vs. knowledge and beliefs of US college students. Int J Environ Res Public Health. 2009;6:592-600.
- Ko CJ, Elston DM. Pediculosis. J Am Acad Dermatol. 2004;50:1-12; quiz 13-14.
- Chuh A, Lee A, Wong W, et al. Diagnosis of pediculosis pubis: a novel application of digital epiluminescence dermatoscopy. J Eur Acad Dermatol Venereol. 2007;21:837-838.
- Chapel TA, Katta T, Kuszmar T, et al. Pediculosis pubis in a clinic for treatment of sexually transmitted diseases. Sex Transm Dis. 1979;6:257-260.
- Workowski KA, Bolan GA. Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64:1-137.
- Leone PA. Scabies and pediculosis pubis: an update of treatment regimens and general review. Clin Infect Dis. 2007;44(suppl 3):S153-S159.
- Burkhart CN, Burkhart CG. Oral ivermectin therapy for phthiriasis palpebrum. Arch Ophthalmol. 2000;118:134-135.
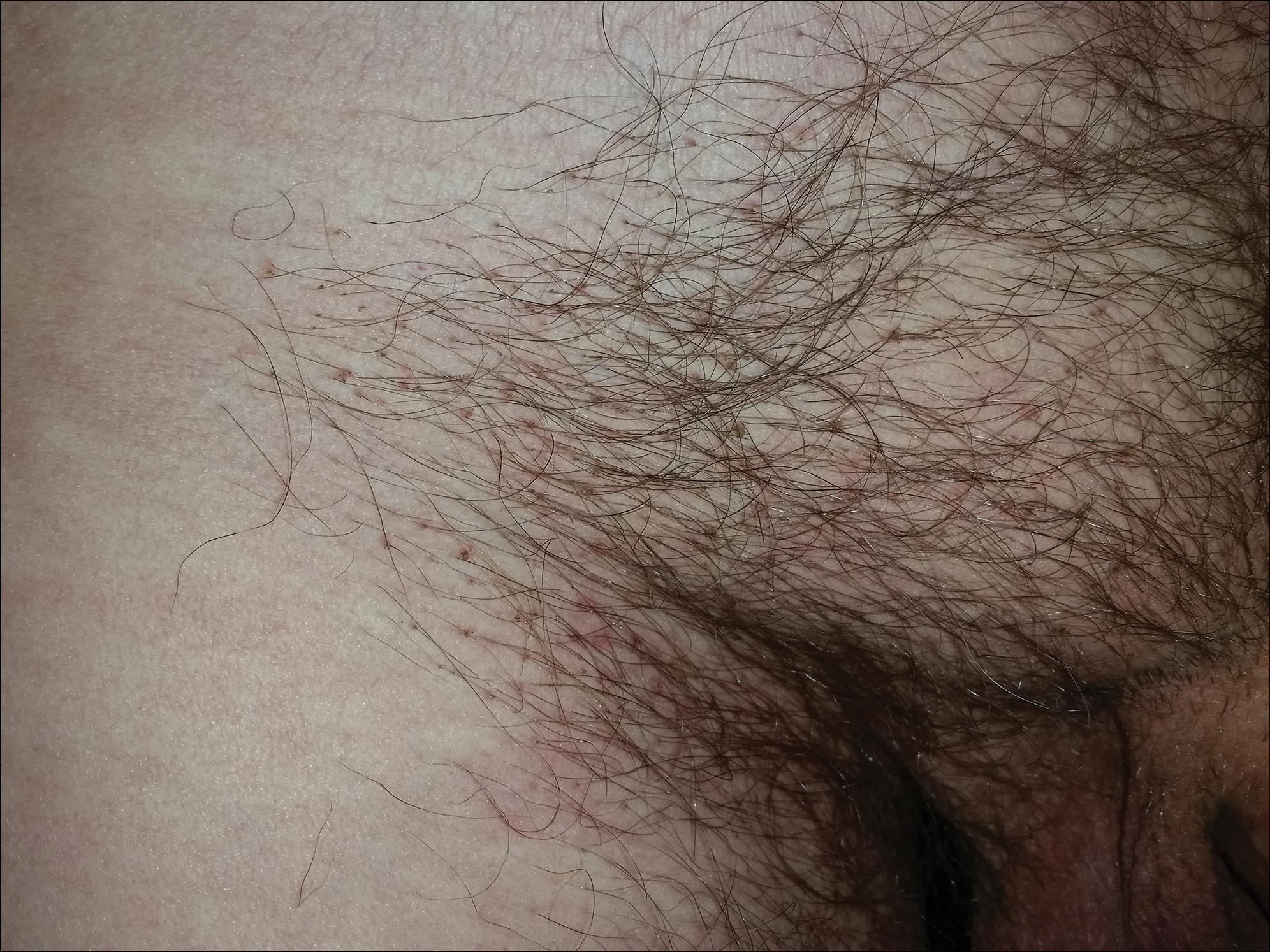
A 50-year-old man with a history of cerebrovascular accident presented with severe itching along the inguinal folds and over the chest of 2 months' duration. His last sexual encounter was 5 months prior. He had previously seen a primary care physician who told him he needed to clean the hair better. Examination of the genital area revealed pigmented macules and overlying particles among the pubic hair.
Solitary Exophytic Plaque on the Left Groin
The Diagnosis: Pemphigus Vegetans
A punch biopsy was taken from the verrucous plaque, and microscopic examination demonstrated prominent epidermal hyperplasia with intraepidermal eosinophilic microabscesses and a superficial dermatitis with abundant eosinophils (Figure 1A). Suprabasal acantholytic cleft formation was noted in a focal area (Figure 1B). Another punch biopsy was performed from the perilesional skin for direct immunofluorescence examination, which revealed intercellular deposits of IgG and C3 throughout the lower half of the epidermis (Figure 1C). Indirect immunofluorescence performed on monkey esophagus substrate showed circulating intercellular IgG antibodies in all the titers of up to 1/160 and an elevated level of IgG antidesmoglein 3 (anti-Dsg3) antibody (enzyme-linked immunosorbent assay index value, >200 RU/mL [reference range, <20 RU/mL]).
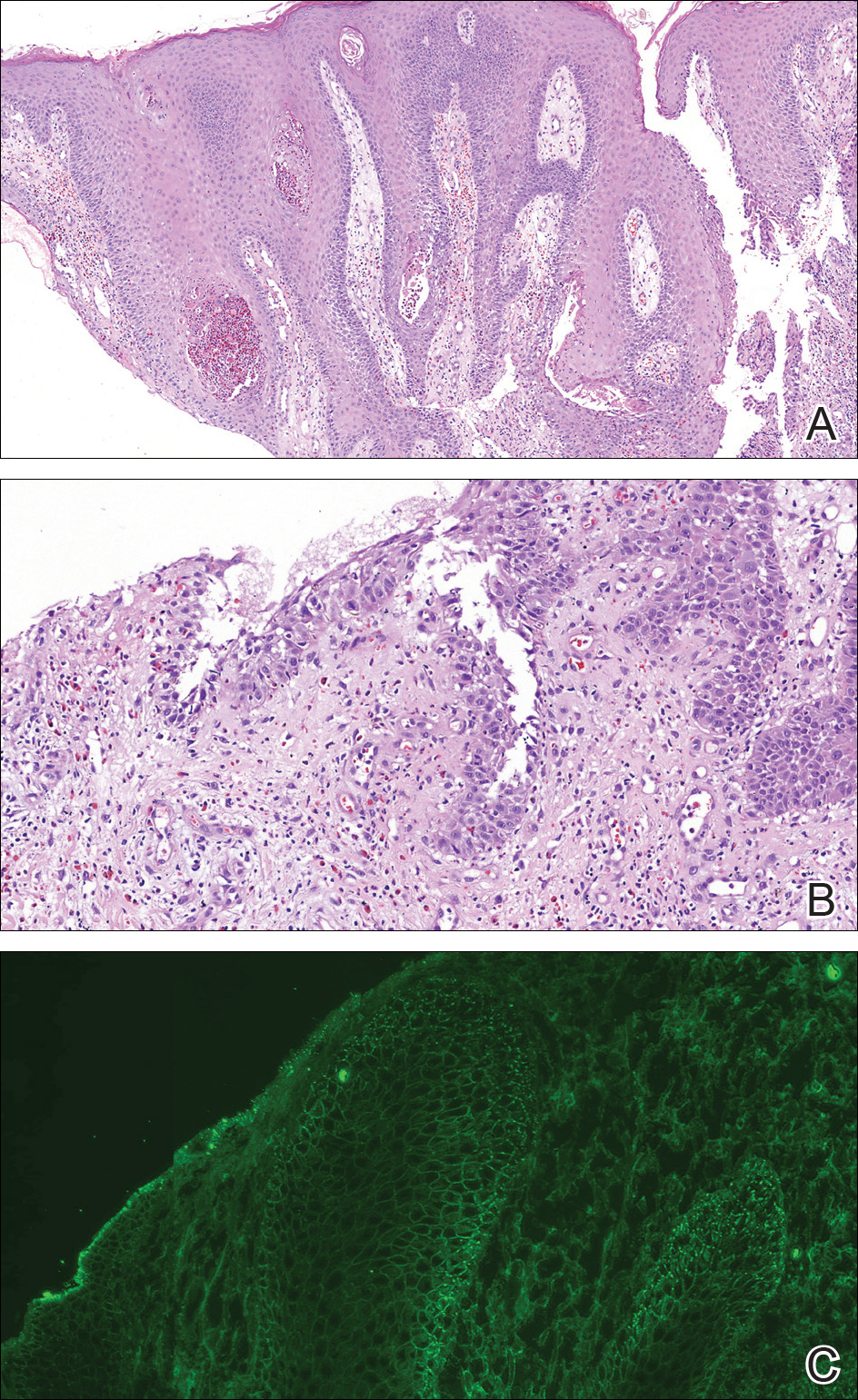
Because there was a solitary lesion, the decision was made to perform local treatment. One intralesional triamcinolone acetonide injection (20 mg/mL) resulted in remarkable flattening of the lesion (Figure 2). Subsequently, treatment was continued with clobetasol propionate ointment 3 times weekly for 1 month. During a follow-up period of 2 years, no signs of local relapse or new lesions elsewhere were noted, and the patient continued to be on long-term longitudinal evaluation.
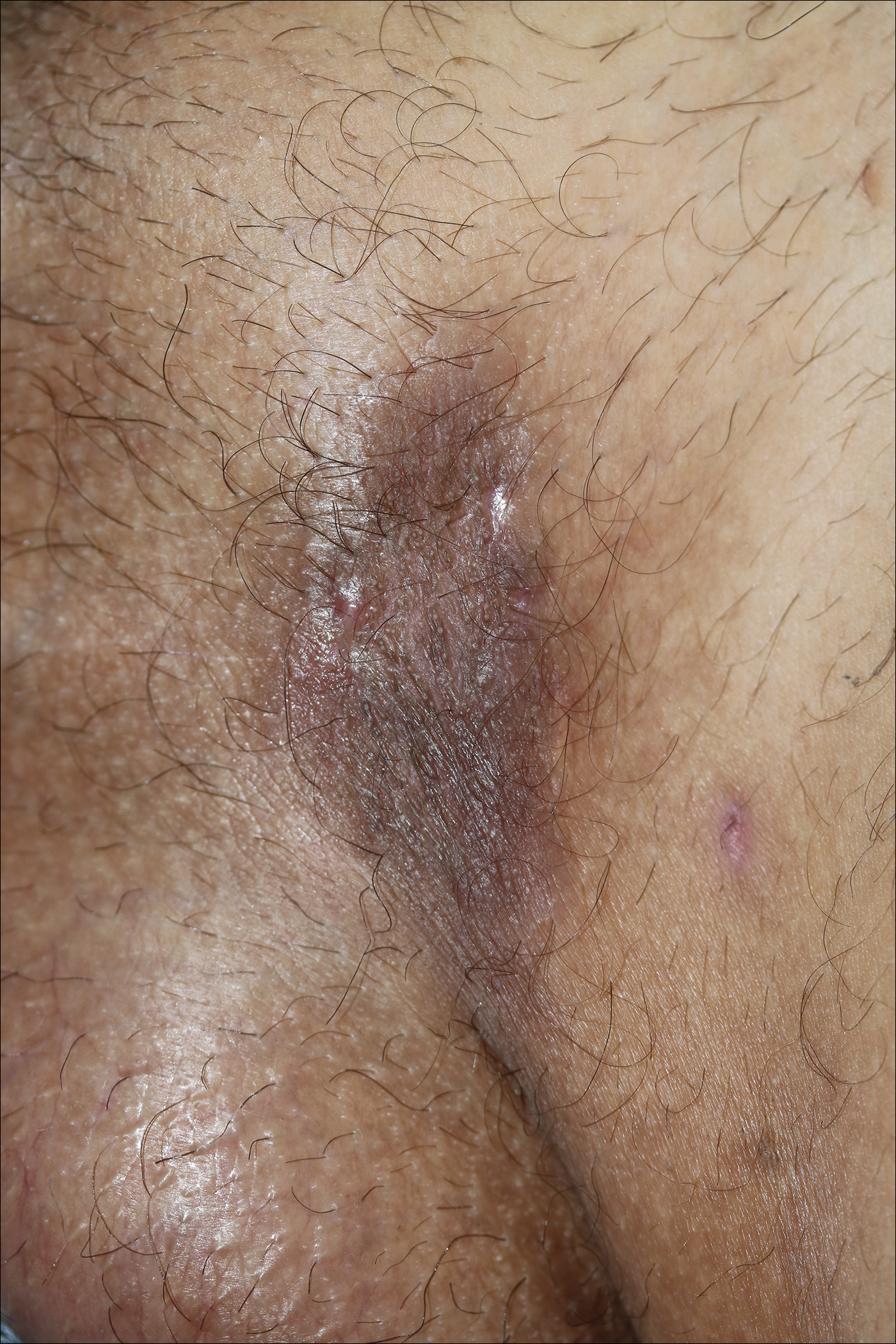
Pemphigus vegetans (PV) is an uncommon variant of pemphigus, typically manifesting with vegetating erosions and plaques localized to the intertriginous areas of the body. Local factors such as semiocclusion, maceration, and/or bacterial or fungal colonization have been hypothesized to account for the distinctive localization and vegetation of the lesions.1,2 Traditionally, 2 clinical subtypes of PV have been described: (1) Hallopeau type presenting with pustules that later evolve into vegetating plaques, and (2) Neumann type that initially manifests as vesicles and bullae with a more disseminated distribution, transforming into hypertrophic masses with erosions.1-5 However, this distinction may not always be clear, and patients with features of both forms have been reported.2,5
At present, our case would best be regarded as a localized form of PV presenting with a solitary lesion. It may progress to more disseminated disease or remain localized during its course; the literature contains reports exemplifying both possibilities. In a large retrospective study from Tunisia encompassing almost 3 decades, the majority of the patients initially presented with unifocal involvement; however, the disease eventually became multifocal in almost all patients during the study period, emphasizing the need for long-term follow-up.2 There also are reports of PV confined to a single anatomic site, such as the scalp, sole, or vulva, that remained localized for years.2,4,6,7 Involvement of the oral mucosa is an important finding of PV and the presenting concern in approximately three-quarters of patients.2 Interestingly, the oral mucosa was not involved in our patient despite the high titer of anti-Dsg3 antibody, which suggests the need for the presence of other factors for clinical expression of the disease.
Although PV is considered a vegetating clinicomorphologic variant of pemphigus vulgaris, PV is histopathologically distinguished from pemphigus vulgaris by the presence of epidermal hyperplasia and intraepidermal eosinophilic microabscesses. Importantly, the epidermis displays signs of exuberant proliferation such as pseudoepitheliomatous hyperplasia and/or papillomatosis of a varying degree.1,2,5 Of note, suprabasal acantholysis is usually overshadowed by the changes in PV and presents only focally, as in our patient. The most common autoantibody profile is IgG targeting Dsg3; however, a spectrum of other autoantibodies has been identified, such as IgG antidesmocollin 3, IgA anti-Dsg3, and IgG anti-Dsg1.8,9
The most important differential diagnosis of PV is pyodermatitis-pyostomatitis vegetans. These 2 entities share many clinical and histopathological features; however, direct immunofluorescence is helpfulfor differentiation because it generally is negative in pyodermatitis-pyostomatitis vegetans.2,10 Furthermore, there is a well-established association between pyodermatitis-pyostomatitis vegetans and inflammatory bowel disorders, whereas PV has anecdotally been linked to malignancy, human immunodeficiency virus infection, and heroin abuse.1,2,10 Our patient was seronegative for human immunodeficiency virus and denied weight loss or loss of appetite. For those cases of PV involving a single anatomic site, the differential diagnosis is broader and encompasses dermatoses such as verrucae, syphilitic chancre, condylomata lata, granuloma inguinale, herpes simplex virus infection, and Kaposi sarcoma.
Treatment of PV is similar to pemphigus vulgaris and consists of a combination of systemic corticosteroids and steroid-sparing agents.1,5 On the other hand, more limited presentations of PV may be suitable for intralesional treatment with triamcinolone acetonide, thus avoiding potential adverse effects of systemic therapy.1,2 In our case with localized involvement, a favorable response was obtained with intralesional triamcinolone acetonide, and we plan to utilize systemic corticosteroids if the disease becomes generalized during follow-up.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Zaraa I, Sellami A, Bouguerra C, et al. Pemphigus vegetans: a clinical, histological, immunopathological and prognostic study. J Eur Acad Dermatol Venereol. 2011;25:1160-1167.
- Madan V, August PJ. Exophytic plaques, blisters, and mouth ulcers. pemphigus vegetans (PV), Neumann type. Arch Dermatol. 2009;145:715-720.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Monshi B, Marker M, Feichtinger H, et al. Pemphigus vegetans--immunopathological findings in a rare variant of pemphigus vulgaris. J Dtsch Dermatol Ges. 2010;8:179-183.
- Jain VK, Dixit VB, Mohan H. Pemphigus vegetans in an unusual site. Int J Dermatol. 1989;28:352-353.
- Wong KT, Wong KK. A case of acantholytic dermatosis of the vulva with features of pemphigus vegetans. J Cutan Pathol. 1994;21:453-456.
- Morizane S, Yamamoto T, Hisamatsu Y, et al. Pemphigus vegetans with IgG and IgA antidesmoglein 3 antibodies. Br J Dermatol. 2005;153:1236-1237.
- Saruta H, Ishii N, Teye K, et al. Two cases of pemphigus vegetans with IgG anti-desmocollin 3 antibodies. JAMA Dermatol. 2013;149:1209-1213.
- Mehravaran M, Kemény L, Husz S, et al. Pyodermatitis-pyostomatitis vegetans. Br J Dermatol. 1997;137:266-269.
The Diagnosis: Pemphigus Vegetans
A punch biopsy was taken from the verrucous plaque, and microscopic examination demonstrated prominent epidermal hyperplasia with intraepidermal eosinophilic microabscesses and a superficial dermatitis with abundant eosinophils (Figure 1A). Suprabasal acantholytic cleft formation was noted in a focal area (Figure 1B). Another punch biopsy was performed from the perilesional skin for direct immunofluorescence examination, which revealed intercellular deposits of IgG and C3 throughout the lower half of the epidermis (Figure 1C). Indirect immunofluorescence performed on monkey esophagus substrate showed circulating intercellular IgG antibodies in all the titers of up to 1/160 and an elevated level of IgG antidesmoglein 3 (anti-Dsg3) antibody (enzyme-linked immunosorbent assay index value, >200 RU/mL [reference range, <20 RU/mL]).
Because there was a solitary lesion, the decision was made to perform local treatment. One intralesional triamcinolone acetonide injection (20 mg/mL) resulted in remarkable flattening of the lesion (Figure 2). Subsequently, treatment was continued with clobetasol propionate ointment 3 times weekly for 1 month. During a follow-up period of 2 years, no signs of local relapse or new lesions elsewhere were noted, and the patient continued to be on long-term longitudinal evaluation.
Pemphigus vegetans (PV) is an uncommon variant of pemphigus, typically manifesting with vegetating erosions and plaques localized to the intertriginous areas of the body. Local factors such as semiocclusion, maceration, and/or bacterial or fungal colonization have been hypothesized to account for the distinctive localization and vegetation of the lesions.1,2 Traditionally, 2 clinical subtypes of PV have been described: (1) Hallopeau type presenting with pustules that later evolve into vegetating plaques, and (2) Neumann type that initially manifests as vesicles and bullae with a more disseminated distribution, transforming into hypertrophic masses with erosions.1-5 However, this distinction may not always be clear, and patients with features of both forms have been reported.2,5
At present, our case would best be regarded as a localized form of PV presenting with a solitary lesion. It may progress to more disseminated disease or remain localized during its course; the literature contains reports exemplifying both possibilities. In a large retrospective study from Tunisia encompassing almost 3 decades, the majority of the patients initially presented with unifocal involvement; however, the disease eventually became multifocal in almost all patients during the study period, emphasizing the need for long-term follow-up.2 There also are reports of PV confined to a single anatomic site, such as the scalp, sole, or vulva, that remained localized for years.2,4,6,7 Involvement of the oral mucosa is an important finding of PV and the presenting concern in approximately three-quarters of patients.2 Interestingly, the oral mucosa was not involved in our patient despite the high titer of anti-Dsg3 antibody, which suggests the need for the presence of other factors for clinical expression of the disease.
Although PV is considered a vegetating clinicomorphologic variant of pemphigus vulgaris, PV is histopathologically distinguished from pemphigus vulgaris by the presence of epidermal hyperplasia and intraepidermal eosinophilic microabscesses. Importantly, the epidermis displays signs of exuberant proliferation such as pseudoepitheliomatous hyperplasia and/or papillomatosis of a varying degree.1,2,5 Of note, suprabasal acantholysis is usually overshadowed by the changes in PV and presents only focally, as in our patient. The most common autoantibody profile is IgG targeting Dsg3; however, a spectrum of other autoantibodies has been identified, such as IgG antidesmocollin 3, IgA anti-Dsg3, and IgG anti-Dsg1.8,9
The most important differential diagnosis of PV is pyodermatitis-pyostomatitis vegetans. These 2 entities share many clinical and histopathological features; however, direct immunofluorescence is helpfulfor differentiation because it generally is negative in pyodermatitis-pyostomatitis vegetans.2,10 Furthermore, there is a well-established association between pyodermatitis-pyostomatitis vegetans and inflammatory bowel disorders, whereas PV has anecdotally been linked to malignancy, human immunodeficiency virus infection, and heroin abuse.1,2,10 Our patient was seronegative for human immunodeficiency virus and denied weight loss or loss of appetite. For those cases of PV involving a single anatomic site, the differential diagnosis is broader and encompasses dermatoses such as verrucae, syphilitic chancre, condylomata lata, granuloma inguinale, herpes simplex virus infection, and Kaposi sarcoma.
Treatment of PV is similar to pemphigus vulgaris and consists of a combination of systemic corticosteroids and steroid-sparing agents.1,5 On the other hand, more limited presentations of PV may be suitable for intralesional treatment with triamcinolone acetonide, thus avoiding potential adverse effects of systemic therapy.1,2 In our case with localized involvement, a favorable response was obtained with intralesional triamcinolone acetonide, and we plan to utilize systemic corticosteroids if the disease becomes generalized during follow-up.
The Diagnosis: Pemphigus Vegetans
A punch biopsy was taken from the verrucous plaque, and microscopic examination demonstrated prominent epidermal hyperplasia with intraepidermal eosinophilic microabscesses and a superficial dermatitis with abundant eosinophils (Figure 1A). Suprabasal acantholytic cleft formation was noted in a focal area (Figure 1B). Another punch biopsy was performed from the perilesional skin for direct immunofluorescence examination, which revealed intercellular deposits of IgG and C3 throughout the lower half of the epidermis (Figure 1C). Indirect immunofluorescence performed on monkey esophagus substrate showed circulating intercellular IgG antibodies in all the titers of up to 1/160 and an elevated level of IgG antidesmoglein 3 (anti-Dsg3) antibody (enzyme-linked immunosorbent assay index value, >200 RU/mL [reference range, <20 RU/mL]).
Because there was a solitary lesion, the decision was made to perform local treatment. One intralesional triamcinolone acetonide injection (20 mg/mL) resulted in remarkable flattening of the lesion (Figure 2). Subsequently, treatment was continued with clobetasol propionate ointment 3 times weekly for 1 month. During a follow-up period of 2 years, no signs of local relapse or new lesions elsewhere were noted, and the patient continued to be on long-term longitudinal evaluation.
Pemphigus vegetans (PV) is an uncommon variant of pemphigus, typically manifesting with vegetating erosions and plaques localized to the intertriginous areas of the body. Local factors such as semiocclusion, maceration, and/or bacterial or fungal colonization have been hypothesized to account for the distinctive localization and vegetation of the lesions.1,2 Traditionally, 2 clinical subtypes of PV have been described: (1) Hallopeau type presenting with pustules that later evolve into vegetating plaques, and (2) Neumann type that initially manifests as vesicles and bullae with a more disseminated distribution, transforming into hypertrophic masses with erosions.1-5 However, this distinction may not always be clear, and patients with features of both forms have been reported.2,5
At present, our case would best be regarded as a localized form of PV presenting with a solitary lesion. It may progress to more disseminated disease or remain localized during its course; the literature contains reports exemplifying both possibilities. In a large retrospective study from Tunisia encompassing almost 3 decades, the majority of the patients initially presented with unifocal involvement; however, the disease eventually became multifocal in almost all patients during the study period, emphasizing the need for long-term follow-up.2 There also are reports of PV confined to a single anatomic site, such as the scalp, sole, or vulva, that remained localized for years.2,4,6,7 Involvement of the oral mucosa is an important finding of PV and the presenting concern in approximately three-quarters of patients.2 Interestingly, the oral mucosa was not involved in our patient despite the high titer of anti-Dsg3 antibody, which suggests the need for the presence of other factors for clinical expression of the disease.
Although PV is considered a vegetating clinicomorphologic variant of pemphigus vulgaris, PV is histopathologically distinguished from pemphigus vulgaris by the presence of epidermal hyperplasia and intraepidermal eosinophilic microabscesses. Importantly, the epidermis displays signs of exuberant proliferation such as pseudoepitheliomatous hyperplasia and/or papillomatosis of a varying degree.1,2,5 Of note, suprabasal acantholysis is usually overshadowed by the changes in PV and presents only focally, as in our patient. The most common autoantibody profile is IgG targeting Dsg3; however, a spectrum of other autoantibodies has been identified, such as IgG antidesmocollin 3, IgA anti-Dsg3, and IgG anti-Dsg1.8,9
The most important differential diagnosis of PV is pyodermatitis-pyostomatitis vegetans. These 2 entities share many clinical and histopathological features; however, direct immunofluorescence is helpfulfor differentiation because it generally is negative in pyodermatitis-pyostomatitis vegetans.2,10 Furthermore, there is a well-established association between pyodermatitis-pyostomatitis vegetans and inflammatory bowel disorders, whereas PV has anecdotally been linked to malignancy, human immunodeficiency virus infection, and heroin abuse.1,2,10 Our patient was seronegative for human immunodeficiency virus and denied weight loss or loss of appetite. For those cases of PV involving a single anatomic site, the differential diagnosis is broader and encompasses dermatoses such as verrucae, syphilitic chancre, condylomata lata, granuloma inguinale, herpes simplex virus infection, and Kaposi sarcoma.
Treatment of PV is similar to pemphigus vulgaris and consists of a combination of systemic corticosteroids and steroid-sparing agents.1,5 On the other hand, more limited presentations of PV may be suitable for intralesional treatment with triamcinolone acetonide, thus avoiding potential adverse effects of systemic therapy.1,2 In our case with localized involvement, a favorable response was obtained with intralesional triamcinolone acetonide, and we plan to utilize systemic corticosteroids if the disease becomes generalized during follow-up.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Zaraa I, Sellami A, Bouguerra C, et al. Pemphigus vegetans: a clinical, histological, immunopathological and prognostic study. J Eur Acad Dermatol Venereol. 2011;25:1160-1167.
- Madan V, August PJ. Exophytic plaques, blisters, and mouth ulcers. pemphigus vegetans (PV), Neumann type. Arch Dermatol. 2009;145:715-720.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Monshi B, Marker M, Feichtinger H, et al. Pemphigus vegetans--immunopathological findings in a rare variant of pemphigus vulgaris. J Dtsch Dermatol Ges. 2010;8:179-183.
- Jain VK, Dixit VB, Mohan H. Pemphigus vegetans in an unusual site. Int J Dermatol. 1989;28:352-353.
- Wong KT, Wong KK. A case of acantholytic dermatosis of the vulva with features of pemphigus vegetans. J Cutan Pathol. 1994;21:453-456.
- Morizane S, Yamamoto T, Hisamatsu Y, et al. Pemphigus vegetans with IgG and IgA antidesmoglein 3 antibodies. Br J Dermatol. 2005;153:1236-1237.
- Saruta H, Ishii N, Teye K, et al. Two cases of pemphigus vegetans with IgG anti-desmocollin 3 antibodies. JAMA Dermatol. 2013;149:1209-1213.
- Mehravaran M, Kemény L, Husz S, et al. Pyodermatitis-pyostomatitis vegetans. Br J Dermatol. 1997;137:266-269.
- Ruocco V, Ruocco E, Caccavale S, et al. Pemphigus vegetans of the folds (intertriginous areas). Clin Dermatol. 2015;33:471-476.
- Zaraa I, Sellami A, Bouguerra C, et al. Pemphigus vegetans: a clinical, histological, immunopathological and prognostic study. J Eur Acad Dermatol Venereol. 2011;25:1160-1167.
- Madan V, August PJ. Exophytic plaques, blisters, and mouth ulcers. pemphigus vegetans (PV), Neumann type. Arch Dermatol. 2009;145:715-720.
- Mori M, Mariotti G, Grandi V, et al. Pemphigus vegetans of the scalp. J Eur Acad Dermatol Venereol. 2016;30:368-370.
- Monshi B, Marker M, Feichtinger H, et al. Pemphigus vegetans--immunopathological findings in a rare variant of pemphigus vulgaris. J Dtsch Dermatol Ges. 2010;8:179-183.
- Jain VK, Dixit VB, Mohan H. Pemphigus vegetans in an unusual site. Int J Dermatol. 1989;28:352-353.
- Wong KT, Wong KK. A case of acantholytic dermatosis of the vulva with features of pemphigus vegetans. J Cutan Pathol. 1994;21:453-456.
- Morizane S, Yamamoto T, Hisamatsu Y, et al. Pemphigus vegetans with IgG and IgA antidesmoglein 3 antibodies. Br J Dermatol. 2005;153:1236-1237.
- Saruta H, Ishii N, Teye K, et al. Two cases of pemphigus vegetans with IgG anti-desmocollin 3 antibodies. JAMA Dermatol. 2013;149:1209-1213.
- Mehravaran M, Kemény L, Husz S, et al. Pyodermatitis-pyostomatitis vegetans. Br J Dermatol. 1997;137:266-269.
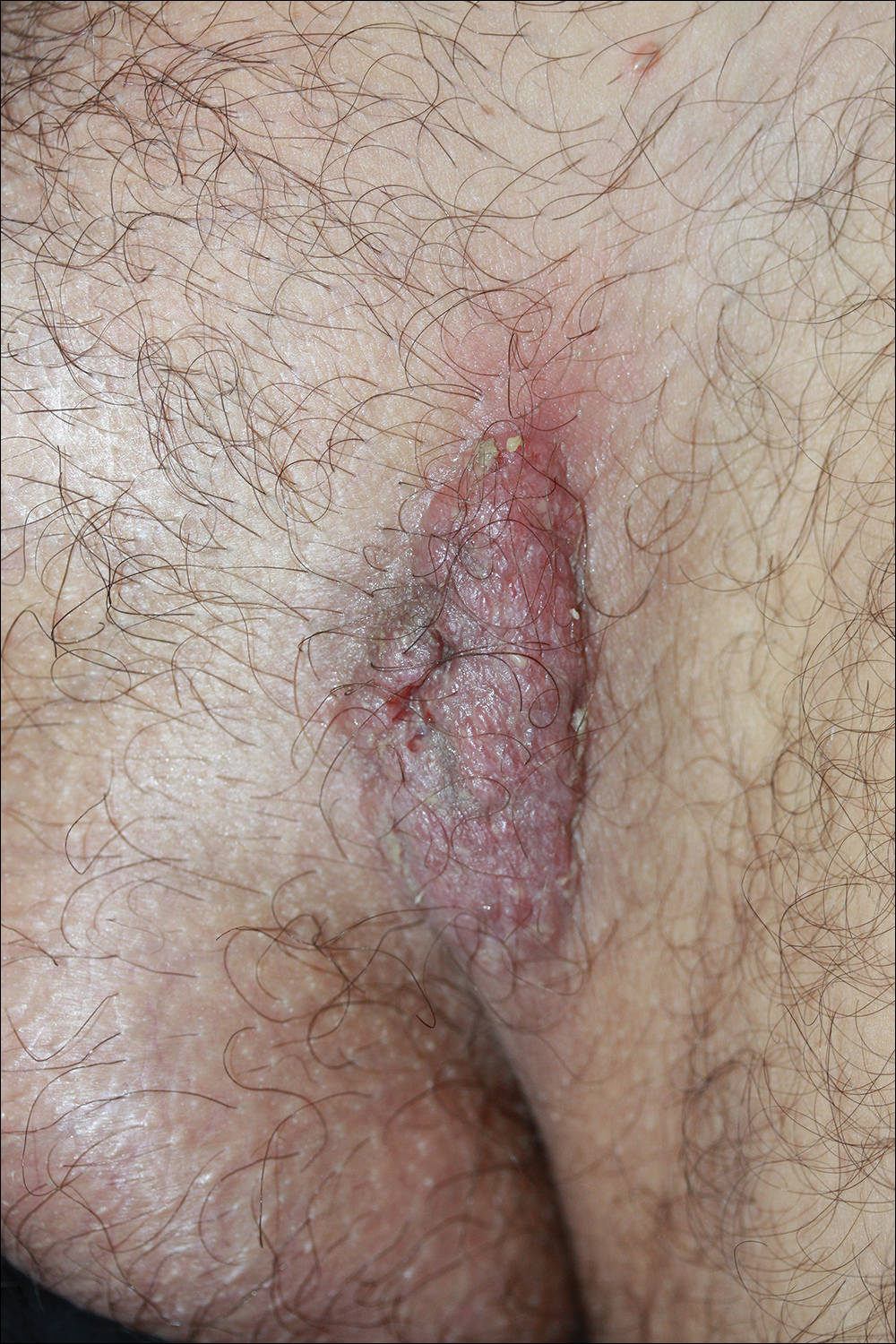
A 40-year-old otherwise healthy man presented with an exophytic plaque on the left groin of 1 month's duration. The lesion reportedly emerged as pustules that slowly expanded and coalesced. At an outside institution, cryotherapy was planned for a presumed diagnosis of condyloma acuminatum; however, the patient decided to get a second opinion. He denied recent intake of new drugs. Six months prior he had traveled to China and engaged in unprotected sexual intercourse. Physical examination revealed an approximately 4×2-cm exophytic plaque with a partially eroded and exudative surface on the left inguinal fold. Dermatologic examination, including the oral mucosa, was otherwise normal. Complete blood cell count and sexually transmitted disease panel were unremarkable.
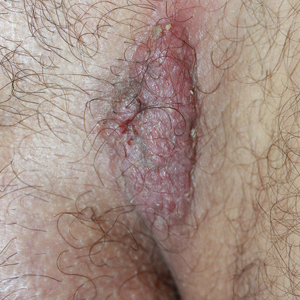
Diffuse Nonscarring Alopecia
The Diagnosis: Trichotillomania
A scalp punch biopsy revealed pigmented hair casts, an increase in catagen and telogen follicles, and a lack of perifollicular inflammation (Figure). Based on the clinical and histopathological findings, a diagnosis of trichotillomania (TTM) was established.
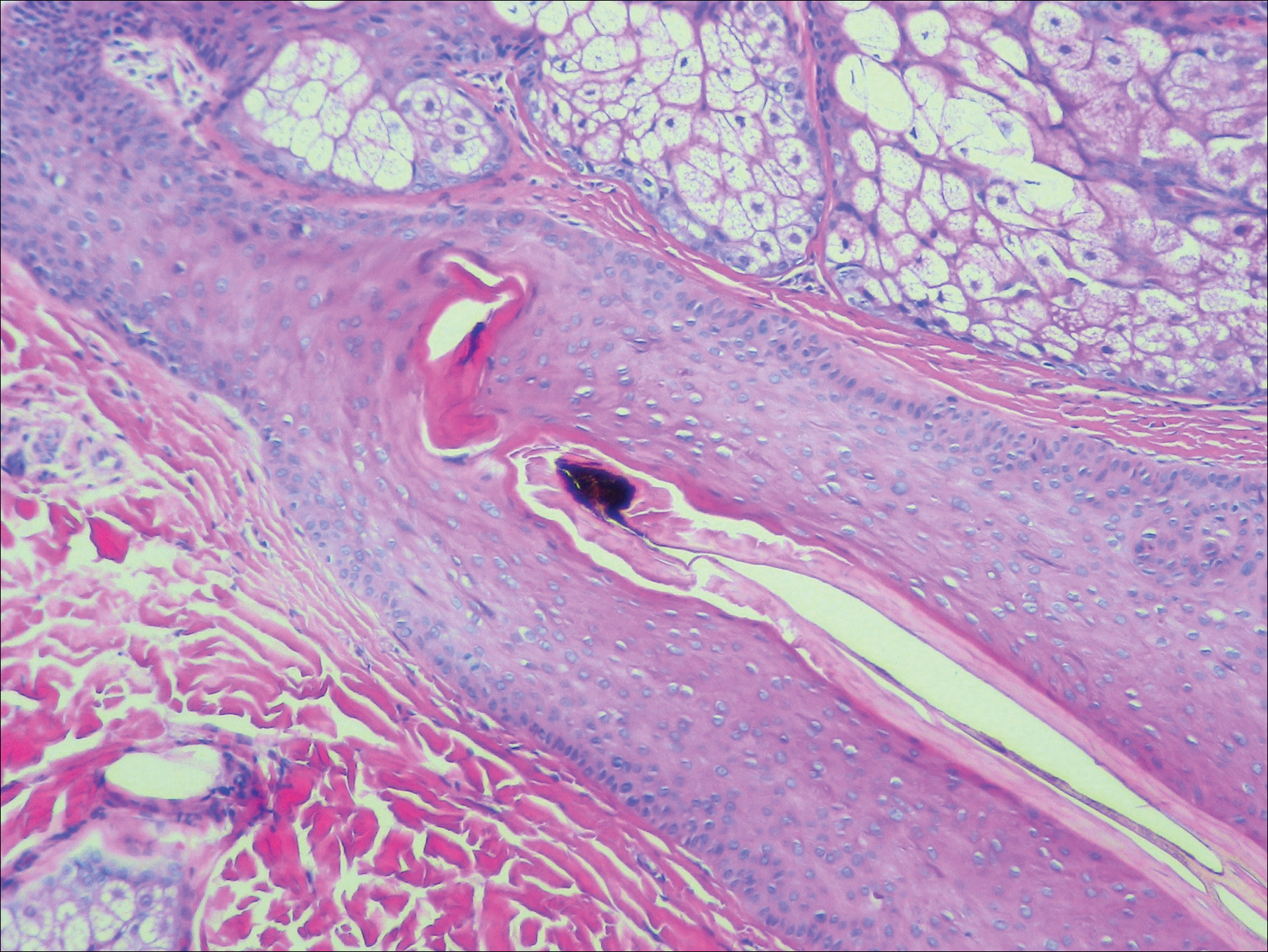
Trichotillomania is a hairpulling disorder with notable dermatologic and psychiatric overlap. Although previously considered an impulse control disorder, the Diagnostic and Statistical Manual of Mental Disorders (Fifth Edition) reclassified it within obsessive-compulsive and related disorders, which also include body dysmorphic disorder and excoriation (skin-picking) disorder. Diagnostic criteria for TTM include the following: the patient must have recurrent pulling out of his/her hair resulting in hair loss despite repeated attempts to stop; underlying medical conditions and other psychiatric diagnoses must be excluded; and the patient must experience distress or impairment in social, occupational, or other areas of functioning from the hairpulling.1 Trichotillomania mainly occurs in children and young adults, with a lifetime prevalence of approximately 1% to 2%.2 The coexistence of a mood or anxiety disorder is common, as seen in our patient.
The diagnosis of TTM requires strong clinical suspicion because patients and their parents/guardians usually deny hairpulling. The main clinical differential diagnosis often is alopecia areata (AA) because both conditions can present as well-defined patches of nonscarring hair loss. Trichoscopy provides an invaluable noninvasive diagnostic tool that can be particularly useful in pediatric patients who may be reluctant to have a scalp biopsy. There are many overlapping trichoscopic findings of TTM and AA, including yellow dots, black dots, broken hairs, coiled hairs, and exclamation mark hairs.3 More specific trichoscopy findings for TTM include flame hairs (wavy proximal hair residue), V-sign (2 shafts within 1 follicle broken at the same length), and tulip hairs (dark, tulip-shaped ends of broken hairs).4 Hair breakage of varying lengths and trichoptilosis (split ends) can be better visualized using trichoscopy and support a diagnosis of TTM over AA.
Androgenetic alopecia (female pattern hair loss) presents with gradual thinning around the part line of the frontal and parietal scalp with trichoscopy showing miniaturization of hairs and decreased follicle density. The moth-eaten-like appearance of alopecia due to secondary syphilis may mimic alopecia areata clinically, but serologic testing can confirm the diagnosis of syphilis. Telogen effluvium does not have the trichoscopic features that are seen in TTM and is clinically distinguished by hair shedding and a positive hair pull test.
Biopsy can provide objective yet nonspecific support for the diagnosis, demonstrating trichomalacia, pigmented hair casts, empty follicles, and an increase in catagen hairs with a lack of inflammation. Normal and damaged hair follicles may be seen in close proximity, and hemorrhage may be seen secondary to trauma. Pigmented hair casts are not specific to TTM and are present in other traumatic hair disorders, such as traction alopecia; therefore, clinical correlation is essential for diagnosis.
Habit reversal training is the most effective treatment of TTM and involves 3 major components: awareness training with self-monitoring, stimulus control, and competing response procedures.5 Although numerous pharmacotherapies have been reported as effective treatments for TTM, a 2013 Cochrane review of 8 randomized controlled trials concluded that no medication has demonstrated reliable efficacy. Reported therapies included selective serotonin reuptake inhibitors, naltrexone, olanzapine, N-acetylcysteine, and clomipramine.6
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington, DC: American Psychiatric Association; 2013.
- Schumer MC, Panza KE, Mulqueen JM, et al. Long-term outcome in pediatric trichotillomania. Depress Anxiety. 2015;32:737-743.
- Lencastre A, Tosti A. Role of trichoscopy on children's scalp and hair disorders. Pediatr Dermatol. 2013;30:674-682.
- Rakowska A, Slowinska M, Olszewska M, et al. New trichoscopy findings in trichotillomania: flame hairs, V-sign, hook hairs, hair powder, tulip hairs. Acta Derm Venereol. 2014;94:303-306.
- Morris S, Zickgraf H, Dingfelder H, et al. Habit reversal training in trichotillomania: guide for the clinician. Expert Rev Neurother. 2013;13:1069-1177.
- Rothbart R, Amos T, Siegfried N, et al. Pharmacotherapy for trichotillomania. Cochrane Database Syst Rev. 2013;11:CD007662.
The Diagnosis: Trichotillomania
A scalp punch biopsy revealed pigmented hair casts, an increase in catagen and telogen follicles, and a lack of perifollicular inflammation (Figure). Based on the clinical and histopathological findings, a diagnosis of trichotillomania (TTM) was established.
Trichotillomania is a hairpulling disorder with notable dermatologic and psychiatric overlap. Although previously considered an impulse control disorder, the Diagnostic and Statistical Manual of Mental Disorders (Fifth Edition) reclassified it within obsessive-compulsive and related disorders, which also include body dysmorphic disorder and excoriation (skin-picking) disorder. Diagnostic criteria for TTM include the following: the patient must have recurrent pulling out of his/her hair resulting in hair loss despite repeated attempts to stop; underlying medical conditions and other psychiatric diagnoses must be excluded; and the patient must experience distress or impairment in social, occupational, or other areas of functioning from the hairpulling.1 Trichotillomania mainly occurs in children and young adults, with a lifetime prevalence of approximately 1% to 2%.2 The coexistence of a mood or anxiety disorder is common, as seen in our patient.
The diagnosis of TTM requires strong clinical suspicion because patients and their parents/guardians usually deny hairpulling. The main clinical differential diagnosis often is alopecia areata (AA) because both conditions can present as well-defined patches of nonscarring hair loss. Trichoscopy provides an invaluable noninvasive diagnostic tool that can be particularly useful in pediatric patients who may be reluctant to have a scalp biopsy. There are many overlapping trichoscopic findings of TTM and AA, including yellow dots, black dots, broken hairs, coiled hairs, and exclamation mark hairs.3 More specific trichoscopy findings for TTM include flame hairs (wavy proximal hair residue), V-sign (2 shafts within 1 follicle broken at the same length), and tulip hairs (dark, tulip-shaped ends of broken hairs).4 Hair breakage of varying lengths and trichoptilosis (split ends) can be better visualized using trichoscopy and support a diagnosis of TTM over AA.
Androgenetic alopecia (female pattern hair loss) presents with gradual thinning around the part line of the frontal and parietal scalp with trichoscopy showing miniaturization of hairs and decreased follicle density. The moth-eaten-like appearance of alopecia due to secondary syphilis may mimic alopecia areata clinically, but serologic testing can confirm the diagnosis of syphilis. Telogen effluvium does not have the trichoscopic features that are seen in TTM and is clinically distinguished by hair shedding and a positive hair pull test.
Biopsy can provide objective yet nonspecific support for the diagnosis, demonstrating trichomalacia, pigmented hair casts, empty follicles, and an increase in catagen hairs with a lack of inflammation. Normal and damaged hair follicles may be seen in close proximity, and hemorrhage may be seen secondary to trauma. Pigmented hair casts are not specific to TTM and are present in other traumatic hair disorders, such as traction alopecia; therefore, clinical correlation is essential for diagnosis.
Habit reversal training is the most effective treatment of TTM and involves 3 major components: awareness training with self-monitoring, stimulus control, and competing response procedures.5 Although numerous pharmacotherapies have been reported as effective treatments for TTM, a 2013 Cochrane review of 8 randomized controlled trials concluded that no medication has demonstrated reliable efficacy. Reported therapies included selective serotonin reuptake inhibitors, naltrexone, olanzapine, N-acetylcysteine, and clomipramine.6
The Diagnosis: Trichotillomania
A scalp punch biopsy revealed pigmented hair casts, an increase in catagen and telogen follicles, and a lack of perifollicular inflammation (Figure). Based on the clinical and histopathological findings, a diagnosis of trichotillomania (TTM) was established.
Trichotillomania is a hairpulling disorder with notable dermatologic and psychiatric overlap. Although previously considered an impulse control disorder, the Diagnostic and Statistical Manual of Mental Disorders (Fifth Edition) reclassified it within obsessive-compulsive and related disorders, which also include body dysmorphic disorder and excoriation (skin-picking) disorder. Diagnostic criteria for TTM include the following: the patient must have recurrent pulling out of his/her hair resulting in hair loss despite repeated attempts to stop; underlying medical conditions and other psychiatric diagnoses must be excluded; and the patient must experience distress or impairment in social, occupational, or other areas of functioning from the hairpulling.1 Trichotillomania mainly occurs in children and young adults, with a lifetime prevalence of approximately 1% to 2%.2 The coexistence of a mood or anxiety disorder is common, as seen in our patient.
The diagnosis of TTM requires strong clinical suspicion because patients and their parents/guardians usually deny hairpulling. The main clinical differential diagnosis often is alopecia areata (AA) because both conditions can present as well-defined patches of nonscarring hair loss. Trichoscopy provides an invaluable noninvasive diagnostic tool that can be particularly useful in pediatric patients who may be reluctant to have a scalp biopsy. There are many overlapping trichoscopic findings of TTM and AA, including yellow dots, black dots, broken hairs, coiled hairs, and exclamation mark hairs.3 More specific trichoscopy findings for TTM include flame hairs (wavy proximal hair residue), V-sign (2 shafts within 1 follicle broken at the same length), and tulip hairs (dark, tulip-shaped ends of broken hairs).4 Hair breakage of varying lengths and trichoptilosis (split ends) can be better visualized using trichoscopy and support a diagnosis of TTM over AA.
Androgenetic alopecia (female pattern hair loss) presents with gradual thinning around the part line of the frontal and parietal scalp with trichoscopy showing miniaturization of hairs and decreased follicle density. The moth-eaten-like appearance of alopecia due to secondary syphilis may mimic alopecia areata clinically, but serologic testing can confirm the diagnosis of syphilis. Telogen effluvium does not have the trichoscopic features that are seen in TTM and is clinically distinguished by hair shedding and a positive hair pull test.
Biopsy can provide objective yet nonspecific support for the diagnosis, demonstrating trichomalacia, pigmented hair casts, empty follicles, and an increase in catagen hairs with a lack of inflammation. Normal and damaged hair follicles may be seen in close proximity, and hemorrhage may be seen secondary to trauma. Pigmented hair casts are not specific to TTM and are present in other traumatic hair disorders, such as traction alopecia; therefore, clinical correlation is essential for diagnosis.
Habit reversal training is the most effective treatment of TTM and involves 3 major components: awareness training with self-monitoring, stimulus control, and competing response procedures.5 Although numerous pharmacotherapies have been reported as effective treatments for TTM, a 2013 Cochrane review of 8 randomized controlled trials concluded that no medication has demonstrated reliable efficacy. Reported therapies included selective serotonin reuptake inhibitors, naltrexone, olanzapine, N-acetylcysteine, and clomipramine.6
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington, DC: American Psychiatric Association; 2013.
- Schumer MC, Panza KE, Mulqueen JM, et al. Long-term outcome in pediatric trichotillomania. Depress Anxiety. 2015;32:737-743.
- Lencastre A, Tosti A. Role of trichoscopy on children's scalp and hair disorders. Pediatr Dermatol. 2013;30:674-682.
- Rakowska A, Slowinska M, Olszewska M, et al. New trichoscopy findings in trichotillomania: flame hairs, V-sign, hook hairs, hair powder, tulip hairs. Acta Derm Venereol. 2014;94:303-306.
- Morris S, Zickgraf H, Dingfelder H, et al. Habit reversal training in trichotillomania: guide for the clinician. Expert Rev Neurother. 2013;13:1069-1177.
- Rothbart R, Amos T, Siegfried N, et al. Pharmacotherapy for trichotillomania. Cochrane Database Syst Rev. 2013;11:CD007662.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Washington, DC: American Psychiatric Association; 2013.
- Schumer MC, Panza KE, Mulqueen JM, et al. Long-term outcome in pediatric trichotillomania. Depress Anxiety. 2015;32:737-743.
- Lencastre A, Tosti A. Role of trichoscopy on children's scalp and hair disorders. Pediatr Dermatol. 2013;30:674-682.
- Rakowska A, Slowinska M, Olszewska M, et al. New trichoscopy findings in trichotillomania: flame hairs, V-sign, hook hairs, hair powder, tulip hairs. Acta Derm Venereol. 2014;94:303-306.
- Morris S, Zickgraf H, Dingfelder H, et al. Habit reversal training in trichotillomania: guide for the clinician. Expert Rev Neurother. 2013;13:1069-1177.
- Rothbart R, Amos T, Siegfried N, et al. Pharmacotherapy for trichotillomania. Cochrane Database Syst Rev. 2013;11:CD007662.
A 19-year-old woman with attention deficit hyperactivity disorder and an anxiety disorder presented with hair loss of 2 years' duration. She initially had small circular bald areas throughout the scalp that had progressed to diffuse hair loss of the entire scalp. She denied recent hairpulling but admitted to a remote prior history of eyelash and eyebrow pulling. She denied any voice changes, acne, or menstrual irregularities. Physical examination revealed short hairs of varying lengths throughout the scalp with no loss of follicles, erythema, scale, or exclamation point hairs. Eyebrows and eyelashes were normal. A hair-pull test was negative. Trichoscopy illuminated variation in hair shaft diameters, as well as short, irregularly broken hairs of different lengths (inset).