User login
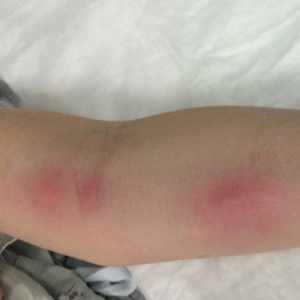
Annular Atrophic Plaques on the Forearm
Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology. The skin is the second most common location for disease manifestation following the lungs.1 Cutaneous sarcoidosis is present in 35% of patients with sarcoidosis and may be further subtyped by its morphologic characteristics (eg, hyperpigmented, papular, nodular, atrophic, ulcerative, psoriasiform). Cutaneous sarcoidosis has an increased tendency to occur at areas of prior injury such as surgeries or tattoos.2 Although sarcoidosis affects all races and sexes, it is more prevalent in women and in the black population.3
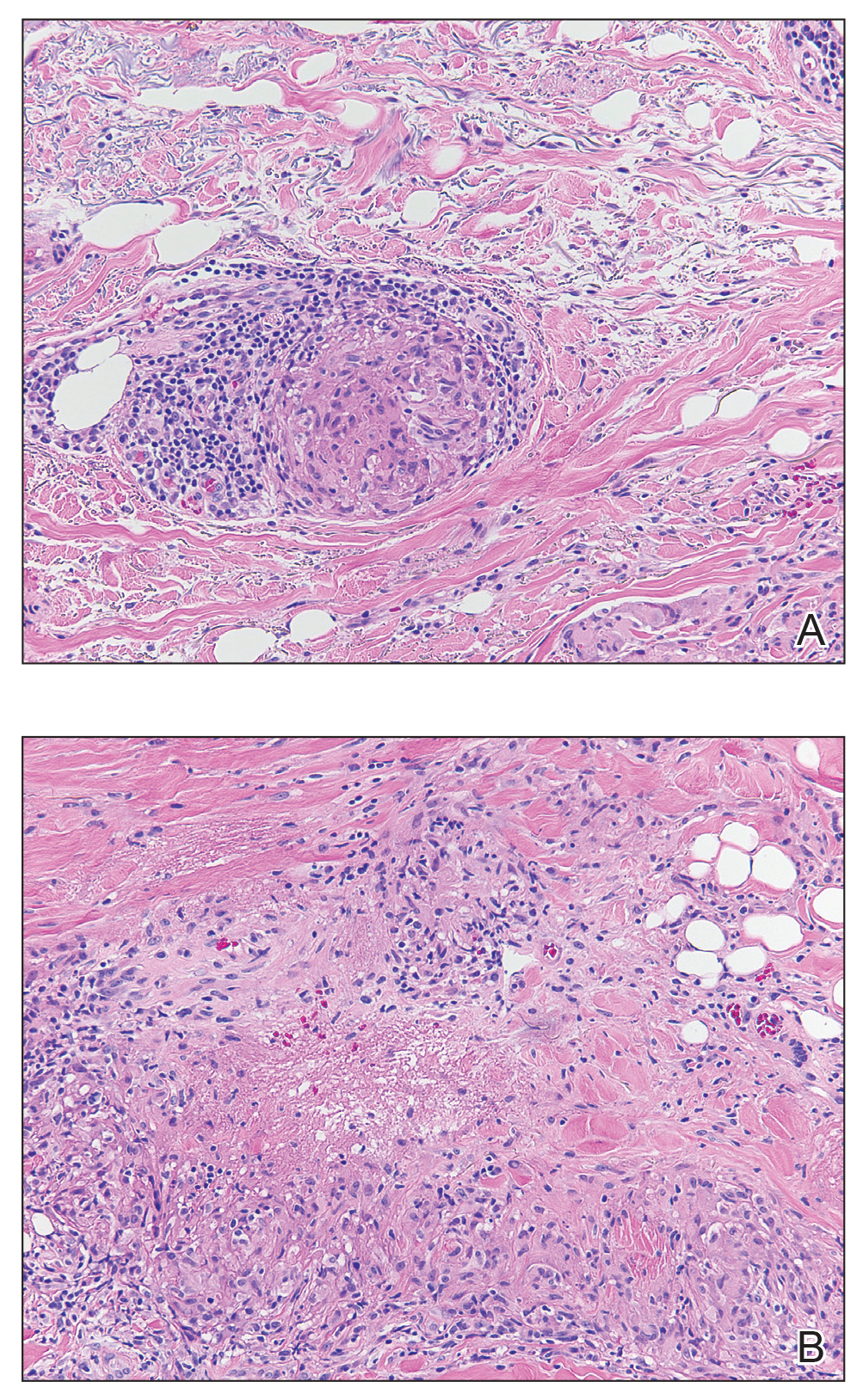
The clinical presentation of sarcoidosis is difficult due to its morphologic variation, allowing for a wide differential diagnosis. With our patient’s presentation of atrophic plaques, the differential diagnosis included granuloma annulare, necrobiosis lipoidica, tumid lupus erythematosus, leprosy, and sarcoidosis; however, biopsy is required for definitive diagnosis. The characteristic histopathology for cutaneous sarcoidosis includes noncaseating granulomas (Figure, A) composed of epithelioid histiocytes with giant cells surrounded by a lymphocytic infiltrate. Noncaseating granulomas are considered specific to sarcoidosis and are present in 71% to 89% of biopsied lesions.4 Interestingly, our patient presented with a rare subtype of atrophic ulcerative cutaneous sarcoidosis, necrobiosis lipoidica–like sarcoidosis, which is more common in females and in the black population. It is characterized by pink to violaceous plaques with depressed centers and prominent necrotizing granuloma (Figure, B) on histopathology. In a small case series, all 3 patients with necrobiosis lipoidica–like sarcoidosis were female and had systemic involvement at the time of diagnosis.5
Sarcoidosis typically is a systemic disease with only a limited number of cases presenting with isolated cutaneous findings. Therefore, patients require a systemic evaluation, which may include a chest radiograph, complete blood cell count, ophthalmologic examinations, thyroid testing, and vitamin D monitoring, as well as an echocardiogram and electrocardiogram.2
Treatment is guided by the severity of disease. For isolated cutaneous lesions, topical or intralesional high-potency steroids have been shown to be effective.6,7 Several studies also have shown phototherapy and laser therapy as well as surgical excision to be beneficial.8-10 Once cutaneous lesions become disfiguring or systemic involvement is found, systemic corticosteroids or other immunomodulatory medications may be warranted.11 Our patient was started on intralesional and topical high-potency steroids, which failed, and she was transitioned to methotrexate and adalimumab. Unfortunately, even with advanced therapies, our patient did not have notableresolution of the lesions.
- Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med. 2012;41 (6, pt 2): E355-E374.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med.2015; 36:685-702.
- Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics ofpatients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10, pt 1):1885-1889.
- Ball NJ, Kho GT, Martinka M. The histologic spectrum of cutaneous sarcoidosis: a study of twenty-eight cases. J Cutan Pathol. 2004; 31:160-168.
- Mendoza V, Vahid B, Kozic H, et al. Clinical and pathologic manifestations of necrobiosis lipoidica-like skin involvement in sarcoidosis. Joint Bone Spine. 2007; 74:647-649.
- Khatri KA, Chotzen VA, Burrall BA. Lupus pernio: successful treatment with a potent topical corticosteroid. Arch Dermatol. 1995; 131:617-618.
- Singh SK, Singh S, Pandey SS. Cutaneous sarcoidosis without systemic involvement: response to intralesional corticosteroid. Indian J Dermatol Venereol Leprol. 1996; 62:273-274.
- Karrer S, Abels C, Wimmershoff MB, et al. Successful treatment of cutaneous sarcoidosis using topical photodynamic therapy. Arch Dermatol. 2002; 138:581-584.
- Mahnke N, Medve-koenigs K, Berneburg M, et al. Cutaneous sarcoidosis treated with medium-dose UVA1. J Am Acad Dermatol. 2004; 50:978-979.
- Frederiksen LG, Jørgensen K. Sarcoidosis of the nose treated with laser surgery. Rhinology. 1996; 34:245-246.
- Baughman RP, Lower EE. Evidence-based therapy for cutaneous sarcoidosis. Clin Dermatol. 2007; 25:334-340.
Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology. The skin is the second most common location for disease manifestation following the lungs.1 Cutaneous sarcoidosis is present in 35% of patients with sarcoidosis and may be further subtyped by its morphologic characteristics (eg, hyperpigmented, papular, nodular, atrophic, ulcerative, psoriasiform). Cutaneous sarcoidosis has an increased tendency to occur at areas of prior injury such as surgeries or tattoos.2 Although sarcoidosis affects all races and sexes, it is more prevalent in women and in the black population.3
The clinical presentation of sarcoidosis is difficult due to its morphologic variation, allowing for a wide differential diagnosis. With our patient’s presentation of atrophic plaques, the differential diagnosis included granuloma annulare, necrobiosis lipoidica, tumid lupus erythematosus, leprosy, and sarcoidosis; however, biopsy is required for definitive diagnosis. The characteristic histopathology for cutaneous sarcoidosis includes noncaseating granulomas (Figure, A) composed of epithelioid histiocytes with giant cells surrounded by a lymphocytic infiltrate. Noncaseating granulomas are considered specific to sarcoidosis and are present in 71% to 89% of biopsied lesions.4 Interestingly, our patient presented with a rare subtype of atrophic ulcerative cutaneous sarcoidosis, necrobiosis lipoidica–like sarcoidosis, which is more common in females and in the black population. It is characterized by pink to violaceous plaques with depressed centers and prominent necrotizing granuloma (Figure, B) on histopathology. In a small case series, all 3 patients with necrobiosis lipoidica–like sarcoidosis were female and had systemic involvement at the time of diagnosis.5
Sarcoidosis typically is a systemic disease with only a limited number of cases presenting with isolated cutaneous findings. Therefore, patients require a systemic evaluation, which may include a chest radiograph, complete blood cell count, ophthalmologic examinations, thyroid testing, and vitamin D monitoring, as well as an echocardiogram and electrocardiogram.2
Treatment is guided by the severity of disease. For isolated cutaneous lesions, topical or intralesional high-potency steroids have been shown to be effective.6,7 Several studies also have shown phototherapy and laser therapy as well as surgical excision to be beneficial.8-10 Once cutaneous lesions become disfiguring or systemic involvement is found, systemic corticosteroids or other immunomodulatory medications may be warranted.11 Our patient was started on intralesional and topical high-potency steroids, which failed, and she was transitioned to methotrexate and adalimumab. Unfortunately, even with advanced therapies, our patient did not have notableresolution of the lesions.
Sarcoidosis is a systemic noncaseating granulomatous disease of unknown etiology. The skin is the second most common location for disease manifestation following the lungs.1 Cutaneous sarcoidosis is present in 35% of patients with sarcoidosis and may be further subtyped by its morphologic characteristics (eg, hyperpigmented, papular, nodular, atrophic, ulcerative, psoriasiform). Cutaneous sarcoidosis has an increased tendency to occur at areas of prior injury such as surgeries or tattoos.2 Although sarcoidosis affects all races and sexes, it is more prevalent in women and in the black population.3
The clinical presentation of sarcoidosis is difficult due to its morphologic variation, allowing for a wide differential diagnosis. With our patient’s presentation of atrophic plaques, the differential diagnosis included granuloma annulare, necrobiosis lipoidica, tumid lupus erythematosus, leprosy, and sarcoidosis; however, biopsy is required for definitive diagnosis. The characteristic histopathology for cutaneous sarcoidosis includes noncaseating granulomas (Figure, A) composed of epithelioid histiocytes with giant cells surrounded by a lymphocytic infiltrate. Noncaseating granulomas are considered specific to sarcoidosis and are present in 71% to 89% of biopsied lesions.4 Interestingly, our patient presented with a rare subtype of atrophic ulcerative cutaneous sarcoidosis, necrobiosis lipoidica–like sarcoidosis, which is more common in females and in the black population. It is characterized by pink to violaceous plaques with depressed centers and prominent necrotizing granuloma (Figure, B) on histopathology. In a small case series, all 3 patients with necrobiosis lipoidica–like sarcoidosis were female and had systemic involvement at the time of diagnosis.5
Sarcoidosis typically is a systemic disease with only a limited number of cases presenting with isolated cutaneous findings. Therefore, patients require a systemic evaluation, which may include a chest radiograph, complete blood cell count, ophthalmologic examinations, thyroid testing, and vitamin D monitoring, as well as an echocardiogram and electrocardiogram.2
Treatment is guided by the severity of disease. For isolated cutaneous lesions, topical or intralesional high-potency steroids have been shown to be effective.6,7 Several studies also have shown phototherapy and laser therapy as well as surgical excision to be beneficial.8-10 Once cutaneous lesions become disfiguring or systemic involvement is found, systemic corticosteroids or other immunomodulatory medications may be warranted.11 Our patient was started on intralesional and topical high-potency steroids, which failed, and she was transitioned to methotrexate and adalimumab. Unfortunately, even with advanced therapies, our patient did not have notableresolution of the lesions.
- Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med. 2012;41 (6, pt 2): E355-E374.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med.2015; 36:685-702.
- Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics ofpatients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10, pt 1):1885-1889.
- Ball NJ, Kho GT, Martinka M. The histologic spectrum of cutaneous sarcoidosis: a study of twenty-eight cases. J Cutan Pathol. 2004; 31:160-168.
- Mendoza V, Vahid B, Kozic H, et al. Clinical and pathologic manifestations of necrobiosis lipoidica-like skin involvement in sarcoidosis. Joint Bone Spine. 2007; 74:647-649.
- Khatri KA, Chotzen VA, Burrall BA. Lupus pernio: successful treatment with a potent topical corticosteroid. Arch Dermatol. 1995; 131:617-618.
- Singh SK, Singh S, Pandey SS. Cutaneous sarcoidosis without systemic involvement: response to intralesional corticosteroid. Indian J Dermatol Venereol Leprol. 1996; 62:273-274.
- Karrer S, Abels C, Wimmershoff MB, et al. Successful treatment of cutaneous sarcoidosis using topical photodynamic therapy. Arch Dermatol. 2002; 138:581-584.
- Mahnke N, Medve-koenigs K, Berneburg M, et al. Cutaneous sarcoidosis treated with medium-dose UVA1. J Am Acad Dermatol. 2004; 50:978-979.
- Frederiksen LG, Jørgensen K. Sarcoidosis of the nose treated with laser surgery. Rhinology. 1996; 34:245-246.
- Baughman RP, Lower EE. Evidence-based therapy for cutaneous sarcoidosis. Clin Dermatol. 2007; 25:334-340.
- Mañá J, Marcoval J. Skin manifestations of sarcoidosis. Presse Med. 2012;41 (6, pt 2): E355-E374.
- Wanat KA, Rosenbach M. Cutaneous sarcoidosis. Clin Chest Med.2015; 36:685-702.
- Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics ofpatients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10, pt 1):1885-1889.
- Ball NJ, Kho GT, Martinka M. The histologic spectrum of cutaneous sarcoidosis: a study of twenty-eight cases. J Cutan Pathol. 2004; 31:160-168.
- Mendoza V, Vahid B, Kozic H, et al. Clinical and pathologic manifestations of necrobiosis lipoidica-like skin involvement in sarcoidosis. Joint Bone Spine. 2007; 74:647-649.
- Khatri KA, Chotzen VA, Burrall BA. Lupus pernio: successful treatment with a potent topical corticosteroid. Arch Dermatol. 1995; 131:617-618.
- Singh SK, Singh S, Pandey SS. Cutaneous sarcoidosis without systemic involvement: response to intralesional corticosteroid. Indian J Dermatol Venereol Leprol. 1996; 62:273-274.
- Karrer S, Abels C, Wimmershoff MB, et al. Successful treatment of cutaneous sarcoidosis using topical photodynamic therapy. Arch Dermatol. 2002; 138:581-584.
- Mahnke N, Medve-koenigs K, Berneburg M, et al. Cutaneous sarcoidosis treated with medium-dose UVA1. J Am Acad Dermatol. 2004; 50:978-979.
- Frederiksen LG, Jørgensen K. Sarcoidosis of the nose treated with laser surgery. Rhinology. 1996; 34:245-246.
- Baughman RP, Lower EE. Evidence-based therapy for cutaneous sarcoidosis. Clin Dermatol. 2007; 25:334-340.
A 57-year-old woman presented with several lesions on the left extensor forearm of 10 years’ duration. A single annular indurated lesion with central atrophy initially developed near a prior surgical site. The lesions were pruritic with no associated pain or bleeding. Over 5 years, similar lesions had developed extending up the arm. No benefit was seen with low-potency topical steroid application. Biopsy for histopathologic examination was performed to confirm the diagnosis.
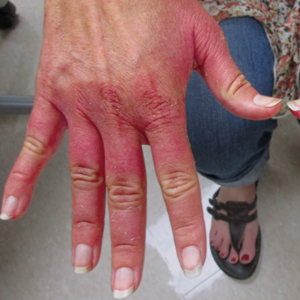
Erythematous Edematous Plaques on the Dorsal Aspects of the Hands
The Diagnosis: Phytophotodermatitis
Initially, there was concern for autoimmune or connective tissue disease because of the edematous plaques localized over sun-exposed regions of the hands with marked sparing of the knuckles. Lupus erythematosus (LE), mixed connective tissue disease, CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, telangiectasia) syndrome, dermatomyositis (DM), and erythromelalgia all were considered. Common disorders such as contact dermatitis and phytophotodermatitis remained in the differential diagnosis, though the patient adamantly denied any recent exposures. As part of the initial workup, laboratory studies including a complete blood cell count, comprehensive metabolic panel, serum lactate dehydrogenase, serum creatinine kinase, erythrocyte sedimentation rate, and an antinuclear antibody panel were performed. Additionally, a punch biopsy at the border of the lesion was performed.
Lupus erythematosus was considered given the patient’s age and sex and the photoexposed location of the plaques. The photosensitive rash of LE classically affects the dorsal aspects of the hands while sparing the interphalangeal joints.1,2 However, the patient had no nail fold findings consistent with systemic LE with no evidence of erythema or dilated tortuous vessels.3 Furthermore, there were no other cutaneous symptoms, and there was a negative review of systems, including malar/discoid rash, oral ulcers, photosensitivity, history of hematologic abnormalities, and end organ damage.4,5 A negative antinuclear antibody serologic panel combined with a negative review of systems made the diagnosis of LE less likely.
Given the presenting clinical appearance, DM also was considered. Dermatomyositis traditionally displays ragged cuticular dystrophy with nail fold telangiectasia, mechanic hands, and involvement of the dorsal aspects of the hands with violaceous accentuation of the knuckles.6 The patient reported pruritus, which is common among DM patients; however, the nail folds were unaffected.7 Finally, she demonstrated sparing rather than involvement of the knuckles, which would be an unlikely presentation for DM.6
CREST syndrome, systemic sclerosis, and syndromes with overlapping features such as mixed connective tissue disease also were considered. The cutaneous features of CREST syndrome are characterized by initial edema of the digits with a subsequent taut and shiny indurated phase. Flexion contractures, ulceration, tapering of the digits, and loss of cutaneous fat pads can progressively occur.8,9 Raynaud phenomenon is a common early finding in CREST syndrome or systemic sclerosis, and patients may develop ice pick digital infarcts and calcinosis in progressed disease.8 Common nail fold findings include periungual telangiectasia with dropout areas.10,11 The marked edema and white discoloration of the knuckles in this patient could be mistaken for Raynaud phenomenon; however, she lacked pain or cold sensitivity and her discoloration was static.12 Without sclerodermoid changes, nail fold findings, matted telangiectasia, taut skin, or systemic findings, a diagnosis of CREST syndrome, scleroderma, or other mixed connective tissue disease would be unlikely.8
Erythromelalgia is a clinical syndrome characterized by burning pain, erythema, and increased skin temperature that intermittently affects both the arms and legs. This rare disorder can be further classified into type 1 (associated with thrombocytopenia), type 2 (primary or idiopathic), and type 3 (associated with other medical cause excluding thrombocytopenia).1,13 The patient endorsed some discomfort from the lesions but denied any subjective feeling of burning pain or increased skin temperature. Additionally, she had no family history of inheritable skin disorders and no personal history of polycythemia. Consequently, erythromelalgia remained less likely on the differential diagnosis.
The histology of the acral skin revealed mild focal spongiosis with no increase in dermal mucin on colloidal iron or mucopolysaccharide stains (Figure). After receiving the biopsy results and additional questioning of the patient, it was discovered that 2 days prior to her initial presentation she had juiced numerous limes by hand and subsequently spent a long period of time outside with sunlight exposure. Upon discovery of this additional historical information, the diagnosis of phytophotodermatitis was made.
Phytophotodermatitis is an erythematous inflammatory reaction that occurs on the skin after exposure to a plant-derived photosensitizer followed by UVA light radiation.14 This phenomenon was first described by the ancient Egyptians as a treatment for vitiligo.1 The most common plant families that can cause this nonimmune cutaneous reaction include Apiaceae eg, hogweed, celery, dill, fennel) and Rutaceae (eg, citrus plants, rue).14 The psoralens or furocoumarins found in these plants bind loosely to DNA at their ground state but covalently bond to pyrimidine bases during photoexcitation with UVA, resulting in DNA damage and subsequent local inflammation.14 Given the patient’s clinical examination, pathology findings, and history, phytophotodermatitis secondary to lime juice exposure was confirmed. Two weeks after applying clobetasol ointment twice daily, the patient’s hands had returned to baseline with complete resolution of the erythematous lesions.
Although lime phytophotodermatitis is a routine diagnosis, this clinical case stands as an important reminder to demonstrate how common diseases can masquerade as more exotic cutaneous disorders. There often is a clinical desire to seek out more complicated diagnoses, particularly during residency training; however, this case reinforces the invaluable importance of collecting a thorough patient history, as it can ultimately minimize excessive testing and in some cases prevent unnecessary therapy.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China:Elsevier Saunders; 2012.
- Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemiclupus erythematosus. Autoimmune Dis. 2012;2012:834291.
- Furtado R, Pucinelli M, Cristo V, et al. Scleroderma-like nailfold capillaroscopicabnormalities are associated with anti-U1-RNP antibodies and Raynaud’s phenomenon in SLE patients. Lupus. 2002;11:35-41.
- Wenzel J, Zahn S, Tuting T. Pathogenesis of cutaneous lupus erythematosus:common and different features in distinct subsets. Lupus. 2010;19:1020-1028.
- Avilés Izquierdo JA, Cano Martínez N, Lázaro Ochaita P. Epidemiologicalcharacteristics of patients with cutaneous lupus erythematosus.Actas Dermosifiliogr. 2014;105:69-73.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Shirani Z, Kucenic MJ, Carroll CL, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004;29:273-276.
- Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48(suppl 3):14-18.
- Mizutani H, Mizutani T, Okada H, et al. Round fingerpad sign: an early sign of scleroderma. J Am Acad Dermatol. 1991;24:67-69.
- Baran R, Dawber RP, Haneke E, et al, eds. A Text Atlas of Nail Disorders Techniques in Investigation and Diagnosis. 3rd ed. Boca Raton, FL: CRC Press; 2005.
- Ghali FE, Stein LD, Fine J, et al. Gingival telangiectases: an underappreciated physical sign of juvenile dermatomyositis. Arch Dermatol. 1999;135:1370-1374.
- Grader-Beck T, Wigley FM. Raynaud’s phenomenon in mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31:465-481.
- Davis MD, Weenig RH, Genebriera J, et al. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55:519-522.
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
The Diagnosis: Phytophotodermatitis
Initially, there was concern for autoimmune or connective tissue disease because of the edematous plaques localized over sun-exposed regions of the hands with marked sparing of the knuckles. Lupus erythematosus (LE), mixed connective tissue disease, CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, telangiectasia) syndrome, dermatomyositis (DM), and erythromelalgia all were considered. Common disorders such as contact dermatitis and phytophotodermatitis remained in the differential diagnosis, though the patient adamantly denied any recent exposures. As part of the initial workup, laboratory studies including a complete blood cell count, comprehensive metabolic panel, serum lactate dehydrogenase, serum creatinine kinase, erythrocyte sedimentation rate, and an antinuclear antibody panel were performed. Additionally, a punch biopsy at the border of the lesion was performed.
Lupus erythematosus was considered given the patient’s age and sex and the photoexposed location of the plaques. The photosensitive rash of LE classically affects the dorsal aspects of the hands while sparing the interphalangeal joints.1,2 However, the patient had no nail fold findings consistent with systemic LE with no evidence of erythema or dilated tortuous vessels.3 Furthermore, there were no other cutaneous symptoms, and there was a negative review of systems, including malar/discoid rash, oral ulcers, photosensitivity, history of hematologic abnormalities, and end organ damage.4,5 A negative antinuclear antibody serologic panel combined with a negative review of systems made the diagnosis of LE less likely.
Given the presenting clinical appearance, DM also was considered. Dermatomyositis traditionally displays ragged cuticular dystrophy with nail fold telangiectasia, mechanic hands, and involvement of the dorsal aspects of the hands with violaceous accentuation of the knuckles.6 The patient reported pruritus, which is common among DM patients; however, the nail folds were unaffected.7 Finally, she demonstrated sparing rather than involvement of the knuckles, which would be an unlikely presentation for DM.6
CREST syndrome, systemic sclerosis, and syndromes with overlapping features such as mixed connective tissue disease also were considered. The cutaneous features of CREST syndrome are characterized by initial edema of the digits with a subsequent taut and shiny indurated phase. Flexion contractures, ulceration, tapering of the digits, and loss of cutaneous fat pads can progressively occur.8,9 Raynaud phenomenon is a common early finding in CREST syndrome or systemic sclerosis, and patients may develop ice pick digital infarcts and calcinosis in progressed disease.8 Common nail fold findings include periungual telangiectasia with dropout areas.10,11 The marked edema and white discoloration of the knuckles in this patient could be mistaken for Raynaud phenomenon; however, she lacked pain or cold sensitivity and her discoloration was static.12 Without sclerodermoid changes, nail fold findings, matted telangiectasia, taut skin, or systemic findings, a diagnosis of CREST syndrome, scleroderma, or other mixed connective tissue disease would be unlikely.8
Erythromelalgia is a clinical syndrome characterized by burning pain, erythema, and increased skin temperature that intermittently affects both the arms and legs. This rare disorder can be further classified into type 1 (associated with thrombocytopenia), type 2 (primary or idiopathic), and type 3 (associated with other medical cause excluding thrombocytopenia).1,13 The patient endorsed some discomfort from the lesions but denied any subjective feeling of burning pain or increased skin temperature. Additionally, she had no family history of inheritable skin disorders and no personal history of polycythemia. Consequently, erythromelalgia remained less likely on the differential diagnosis.
The histology of the acral skin revealed mild focal spongiosis with no increase in dermal mucin on colloidal iron or mucopolysaccharide stains (Figure). After receiving the biopsy results and additional questioning of the patient, it was discovered that 2 days prior to her initial presentation she had juiced numerous limes by hand and subsequently spent a long period of time outside with sunlight exposure. Upon discovery of this additional historical information, the diagnosis of phytophotodermatitis was made.
Phytophotodermatitis is an erythematous inflammatory reaction that occurs on the skin after exposure to a plant-derived photosensitizer followed by UVA light radiation.14 This phenomenon was first described by the ancient Egyptians as a treatment for vitiligo.1 The most common plant families that can cause this nonimmune cutaneous reaction include Apiaceae eg, hogweed, celery, dill, fennel) and Rutaceae (eg, citrus plants, rue).14 The psoralens or furocoumarins found in these plants bind loosely to DNA at their ground state but covalently bond to pyrimidine bases during photoexcitation with UVA, resulting in DNA damage and subsequent local inflammation.14 Given the patient’s clinical examination, pathology findings, and history, phytophotodermatitis secondary to lime juice exposure was confirmed. Two weeks after applying clobetasol ointment twice daily, the patient’s hands had returned to baseline with complete resolution of the erythematous lesions.
Although lime phytophotodermatitis is a routine diagnosis, this clinical case stands as an important reminder to demonstrate how common diseases can masquerade as more exotic cutaneous disorders. There often is a clinical desire to seek out more complicated diagnoses, particularly during residency training; however, this case reinforces the invaluable importance of collecting a thorough patient history, as it can ultimately minimize excessive testing and in some cases prevent unnecessary therapy.
The Diagnosis: Phytophotodermatitis
Initially, there was concern for autoimmune or connective tissue disease because of the edematous plaques localized over sun-exposed regions of the hands with marked sparing of the knuckles. Lupus erythematosus (LE), mixed connective tissue disease, CREST (calcinosis, Raynaud phenomenon, esophageal motility disorders, sclerodactyly, telangiectasia) syndrome, dermatomyositis (DM), and erythromelalgia all were considered. Common disorders such as contact dermatitis and phytophotodermatitis remained in the differential diagnosis, though the patient adamantly denied any recent exposures. As part of the initial workup, laboratory studies including a complete blood cell count, comprehensive metabolic panel, serum lactate dehydrogenase, serum creatinine kinase, erythrocyte sedimentation rate, and an antinuclear antibody panel were performed. Additionally, a punch biopsy at the border of the lesion was performed.
Lupus erythematosus was considered given the patient’s age and sex and the photoexposed location of the plaques. The photosensitive rash of LE classically affects the dorsal aspects of the hands while sparing the interphalangeal joints.1,2 However, the patient had no nail fold findings consistent with systemic LE with no evidence of erythema or dilated tortuous vessels.3 Furthermore, there were no other cutaneous symptoms, and there was a negative review of systems, including malar/discoid rash, oral ulcers, photosensitivity, history of hematologic abnormalities, and end organ damage.4,5 A negative antinuclear antibody serologic panel combined with a negative review of systems made the diagnosis of LE less likely.
Given the presenting clinical appearance, DM also was considered. Dermatomyositis traditionally displays ragged cuticular dystrophy with nail fold telangiectasia, mechanic hands, and involvement of the dorsal aspects of the hands with violaceous accentuation of the knuckles.6 The patient reported pruritus, which is common among DM patients; however, the nail folds were unaffected.7 Finally, she demonstrated sparing rather than involvement of the knuckles, which would be an unlikely presentation for DM.6
CREST syndrome, systemic sclerosis, and syndromes with overlapping features such as mixed connective tissue disease also were considered. The cutaneous features of CREST syndrome are characterized by initial edema of the digits with a subsequent taut and shiny indurated phase. Flexion contractures, ulceration, tapering of the digits, and loss of cutaneous fat pads can progressively occur.8,9 Raynaud phenomenon is a common early finding in CREST syndrome or systemic sclerosis, and patients may develop ice pick digital infarcts and calcinosis in progressed disease.8 Common nail fold findings include periungual telangiectasia with dropout areas.10,11 The marked edema and white discoloration of the knuckles in this patient could be mistaken for Raynaud phenomenon; however, she lacked pain or cold sensitivity and her discoloration was static.12 Without sclerodermoid changes, nail fold findings, matted telangiectasia, taut skin, or systemic findings, a diagnosis of CREST syndrome, scleroderma, or other mixed connective tissue disease would be unlikely.8
Erythromelalgia is a clinical syndrome characterized by burning pain, erythema, and increased skin temperature that intermittently affects both the arms and legs. This rare disorder can be further classified into type 1 (associated with thrombocytopenia), type 2 (primary or idiopathic), and type 3 (associated with other medical cause excluding thrombocytopenia).1,13 The patient endorsed some discomfort from the lesions but denied any subjective feeling of burning pain or increased skin temperature. Additionally, she had no family history of inheritable skin disorders and no personal history of polycythemia. Consequently, erythromelalgia remained less likely on the differential diagnosis.
The histology of the acral skin revealed mild focal spongiosis with no increase in dermal mucin on colloidal iron or mucopolysaccharide stains (Figure). After receiving the biopsy results and additional questioning of the patient, it was discovered that 2 days prior to her initial presentation she had juiced numerous limes by hand and subsequently spent a long period of time outside with sunlight exposure. Upon discovery of this additional historical information, the diagnosis of phytophotodermatitis was made.
Phytophotodermatitis is an erythematous inflammatory reaction that occurs on the skin after exposure to a plant-derived photosensitizer followed by UVA light radiation.14 This phenomenon was first described by the ancient Egyptians as a treatment for vitiligo.1 The most common plant families that can cause this nonimmune cutaneous reaction include Apiaceae eg, hogweed, celery, dill, fennel) and Rutaceae (eg, citrus plants, rue).14 The psoralens or furocoumarins found in these plants bind loosely to DNA at their ground state but covalently bond to pyrimidine bases during photoexcitation with UVA, resulting in DNA damage and subsequent local inflammation.14 Given the patient’s clinical examination, pathology findings, and history, phytophotodermatitis secondary to lime juice exposure was confirmed. Two weeks after applying clobetasol ointment twice daily, the patient’s hands had returned to baseline with complete resolution of the erythematous lesions.
Although lime phytophotodermatitis is a routine diagnosis, this clinical case stands as an important reminder to demonstrate how common diseases can masquerade as more exotic cutaneous disorders. There often is a clinical desire to seek out more complicated diagnoses, particularly during residency training; however, this case reinforces the invaluable importance of collecting a thorough patient history, as it can ultimately minimize excessive testing and in some cases prevent unnecessary therapy.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China:Elsevier Saunders; 2012.
- Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemiclupus erythematosus. Autoimmune Dis. 2012;2012:834291.
- Furtado R, Pucinelli M, Cristo V, et al. Scleroderma-like nailfold capillaroscopicabnormalities are associated with anti-U1-RNP antibodies and Raynaud’s phenomenon in SLE patients. Lupus. 2002;11:35-41.
- Wenzel J, Zahn S, Tuting T. Pathogenesis of cutaneous lupus erythematosus:common and different features in distinct subsets. Lupus. 2010;19:1020-1028.
- Avilés Izquierdo JA, Cano Martínez N, Lázaro Ochaita P. Epidemiologicalcharacteristics of patients with cutaneous lupus erythematosus.Actas Dermosifiliogr. 2014;105:69-73.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Shirani Z, Kucenic MJ, Carroll CL, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004;29:273-276.
- Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48(suppl 3):14-18.
- Mizutani H, Mizutani T, Okada H, et al. Round fingerpad sign: an early sign of scleroderma. J Am Acad Dermatol. 1991;24:67-69.
- Baran R, Dawber RP, Haneke E, et al, eds. A Text Atlas of Nail Disorders Techniques in Investigation and Diagnosis. 3rd ed. Boca Raton, FL: CRC Press; 2005.
- Ghali FE, Stein LD, Fine J, et al. Gingival telangiectases: an underappreciated physical sign of juvenile dermatomyositis. Arch Dermatol. 1999;135:1370-1374.
- Grader-Beck T, Wigley FM. Raynaud’s phenomenon in mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31:465-481.
- Davis MD, Weenig RH, Genebriera J, et al. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55:519-522.
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China:Elsevier Saunders; 2012.
- Uva L, Miguel D, Pinheiro C, et al. Cutaneous manifestations of systemiclupus erythematosus. Autoimmune Dis. 2012;2012:834291.
- Furtado R, Pucinelli M, Cristo V, et al. Scleroderma-like nailfold capillaroscopicabnormalities are associated with anti-U1-RNP antibodies and Raynaud’s phenomenon in SLE patients. Lupus. 2002;11:35-41.
- Wenzel J, Zahn S, Tuting T. Pathogenesis of cutaneous lupus erythematosus:common and different features in distinct subsets. Lupus. 2010;19:1020-1028.
- Avilés Izquierdo JA, Cano Martínez N, Lázaro Ochaita P. Epidemiologicalcharacteristics of patients with cutaneous lupus erythematosus.Actas Dermosifiliogr. 2014;105:69-73.
- Marvi U, Chung L, Fiorentino DF. Clinical presentation and evaluation of dermatomyositis. Indian J Dermatol. 2012;57:375-381.
- Shirani Z, Kucenic MJ, Carroll CL, et al. Pruritus in adult dermatomyositis. Clin Exp Dermatol. 2004;29:273-276.
- Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology (Oxford). 2009;48(suppl 3):14-18.
- Mizutani H, Mizutani T, Okada H, et al. Round fingerpad sign: an early sign of scleroderma. J Am Acad Dermatol. 1991;24:67-69.
- Baran R, Dawber RP, Haneke E, et al, eds. A Text Atlas of Nail Disorders Techniques in Investigation and Diagnosis. 3rd ed. Boca Raton, FL: CRC Press; 2005.
- Ghali FE, Stein LD, Fine J, et al. Gingival telangiectases: an underappreciated physical sign of juvenile dermatomyositis. Arch Dermatol. 1999;135:1370-1374.
- Grader-Beck T, Wigley FM. Raynaud’s phenomenon in mixed connective tissue disease. Rheum Dis Clin North Am. 2005;31:465-481.
- Davis MD, Weenig RH, Genebriera J, et al. Histopathologic findings in primary erythromelalgia are nonspecific: special studies show a decrease in small nerve fiber density. J Am Acad Dermatol. 2006;55:519-522.
- Sasseville D. Clinical patterns of phytophotodermatitis. Dermatol Clin. 2009;27:299-308.
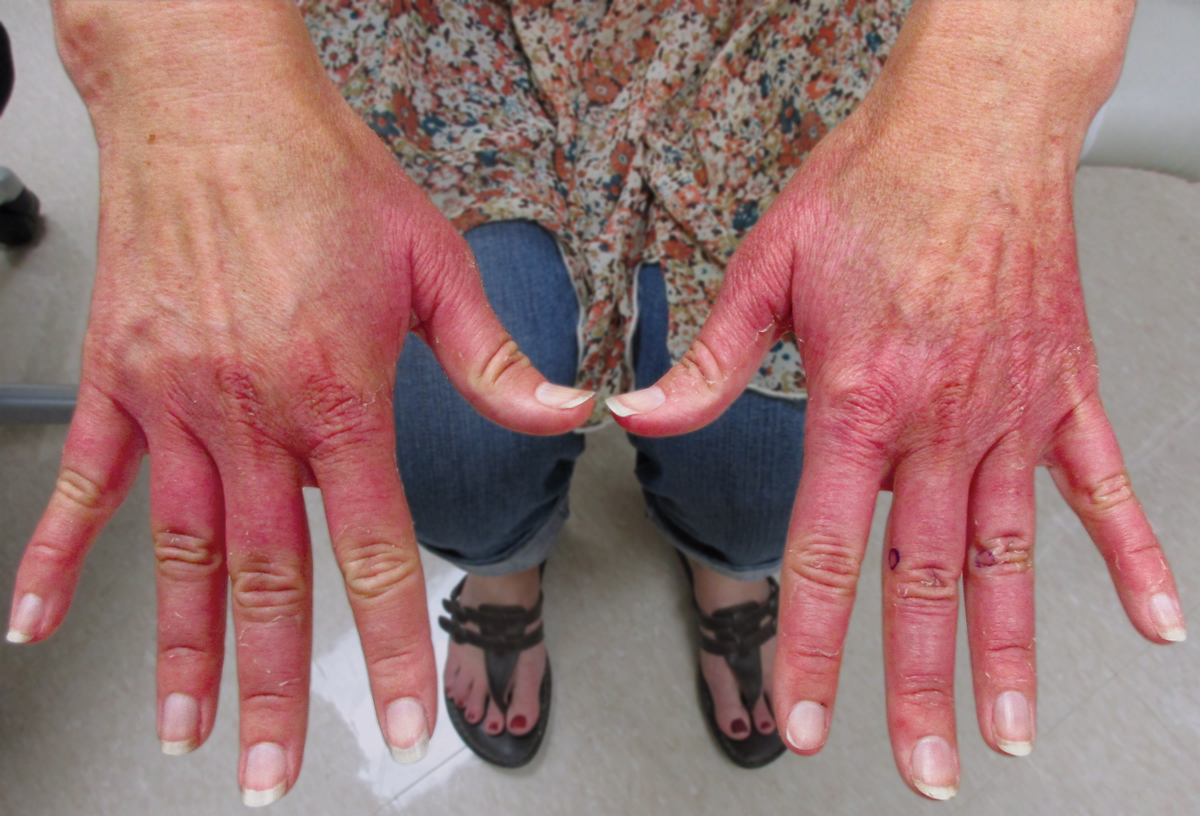
A 48-year-old woman presented with erythematous swelling of the dorsal aspects of the bilateral hands followed by desquamation and pruritus of 2 weeks’ duration. She denied any recent contact with plants, chemicals, or topical products or use of over-the-counter medications. A 6-day course of prednisone provided by her primary care physician relieved the swelling and pruritus; however, the erythema persisted. Physical examination revealed clearly demarcated, erythematous to violaceous, edematous plaques with peripheral scaling that involved all digits. There was notable sparing of the proximal interphalangeal joints and volar aspects of the hands extending proximally to the metacarpophalangeal joints.
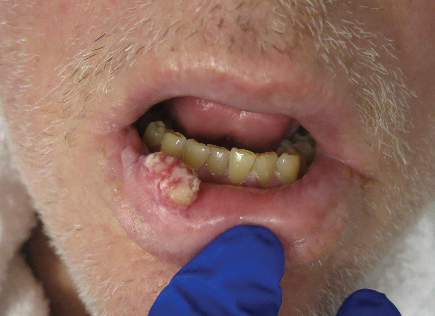
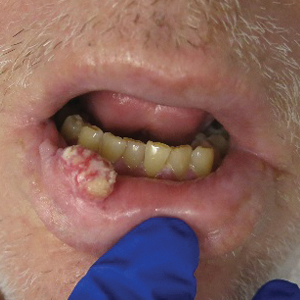
Growing Painful Nodule on the Lower Lip
The Diagnosis: Verrucous Carcinoma
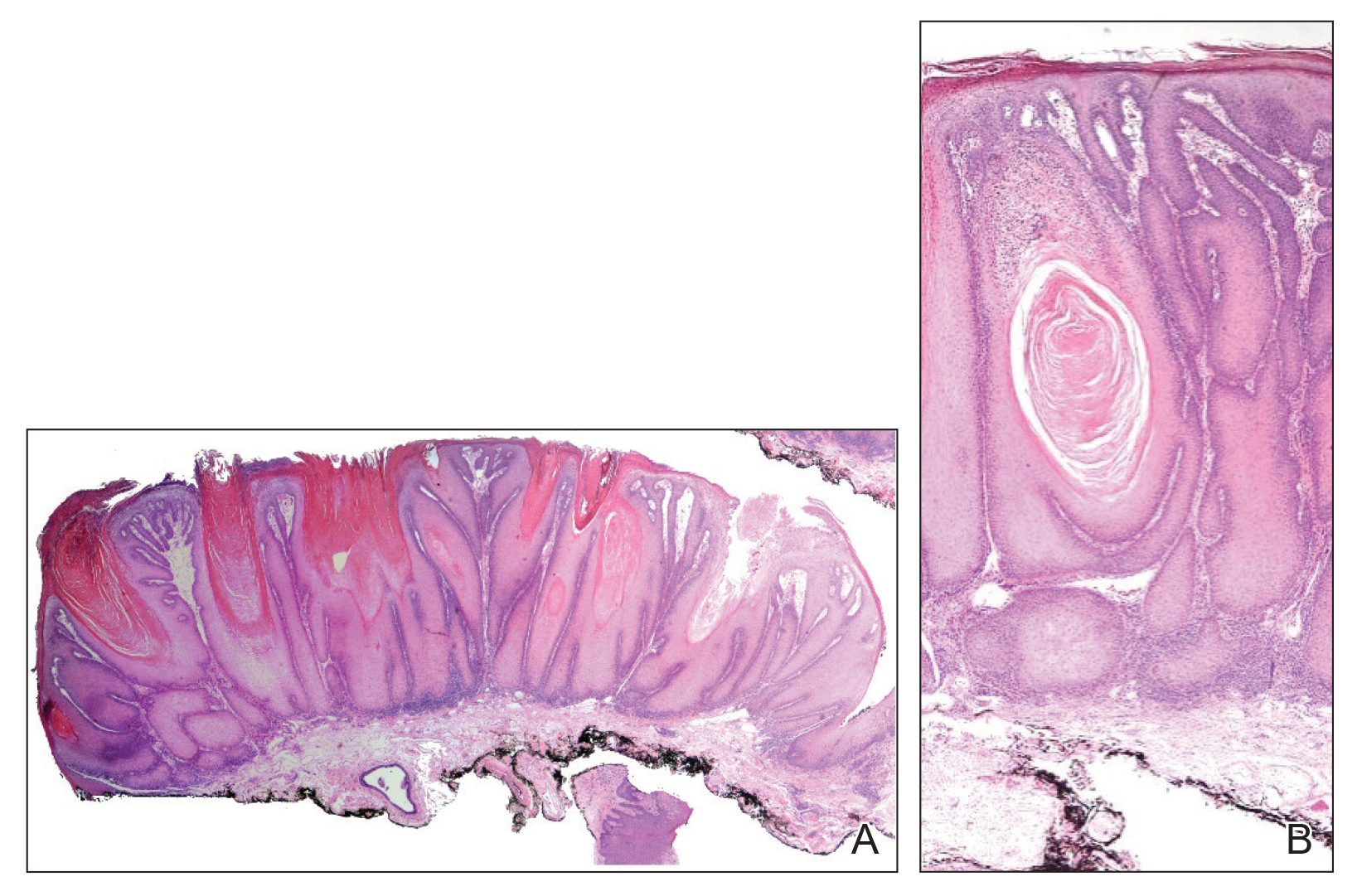
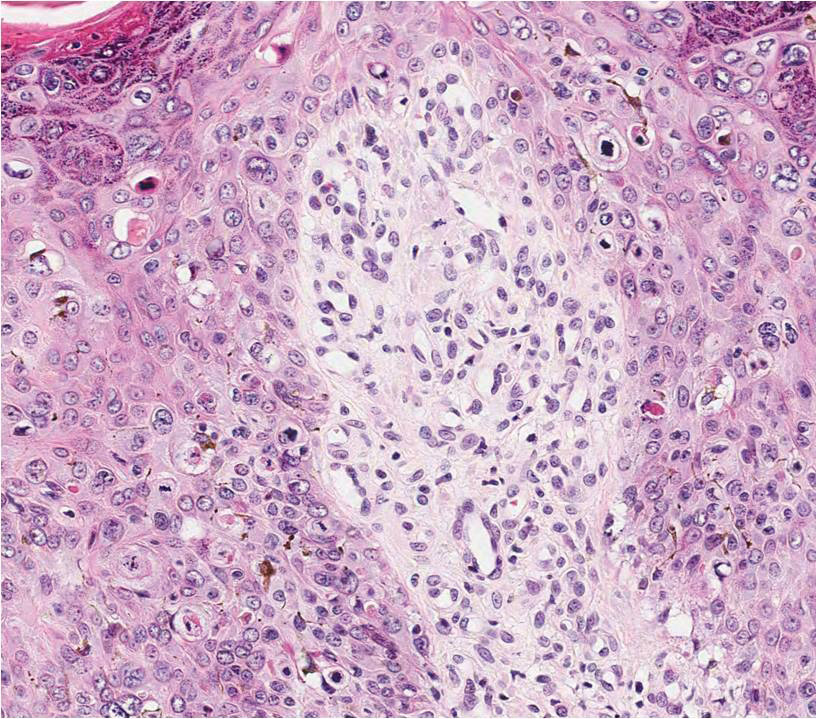
An excisional biopsy revealed an endophytic and exophytic squamous proliferation with a papillomatous growth pattern, bulbous pushing border, and confluent parakeratosis (Figure). No fungal organisms were seen. Due to clinical and histological findings, a diagnosis of verrucous carcinoma (VC) was made.
Verrucous carcinoma is a rare variant of squamous cell carcinoma (SCC) with specific clinical and histological features.1 These tumors have a slow and localized growth pattern but can be locally aggressive. Metastasis of VC is rare, giving VC an overall good prognosis, with a 5-year survival rate greater than 75%.2 Verrucous carcinoma typically occurs in 1 of 3 locations: the oropharynx, genitals, or soles of the feet. Depending on the site of involvement, various names have been used in the literature to describe this entity, including Ackerman tumor (solitary oral mucosal lesion), Buschke-Lowenstein tumor (genital involvement), florid oral papillomatosis (multiple oral lesions), and carcinoma cuniculatum (sole of the foot).3 The most common sites for VC in the oral cavity are the buccal mucosa and gingiva.4
Verrucous carcinoma occurs more often among men in the sixth decade of life.3 The etiology of oral VC remains unclear; however, use of chewing tobacco, chemical carcinogens, chronic irritation, human papillomavirus (HPV), and poor oral hygiene have been reported as predisposing risk factors.4,5 The role of HPV in the pathogenesis of VC remains controversial, but both low-risk types HPV-6 and HPV-11 and high-risk types HPV-16 and HPV-18 have been found in association with VC.5,6
Clinically, oral VC lesions most often present as pink-white erythematous papules or plaques with exophytic cauliflowerlike surface alterations. Although the tumors are slow growing with little risk for metastasis, they may be locally invasive with deep involvement of the surrounding
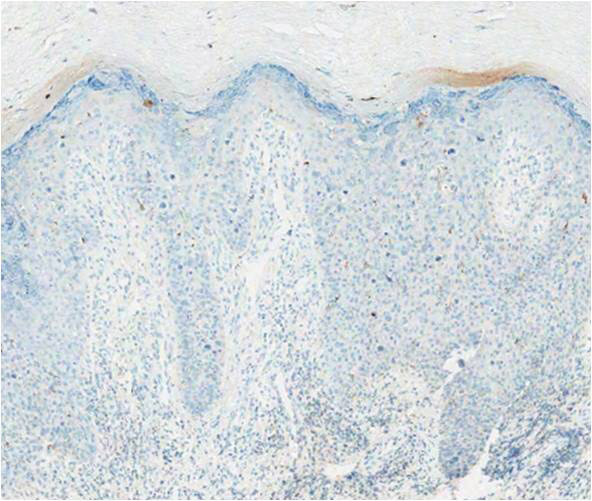
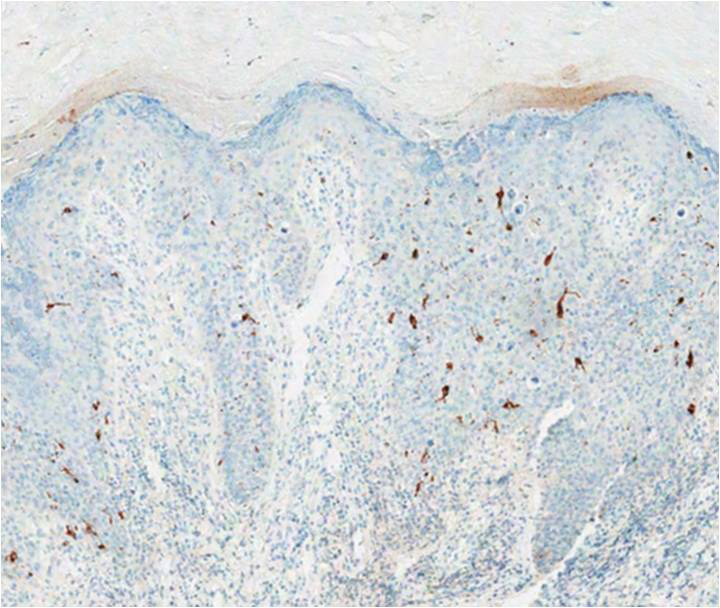
structures.1 Histopathologically, VC displays proliferation of the epithelium with downward growth into the connective tissue but usually without a pattern of true invasion. The epithelium is well differentiated and displays little pleomorphism or mitoses.5,7 Obtaining a generous biopsy specimen is essential to view the diagnostic architecture of VC and rule out other entities, such as viral verruca, blastomycosis, SCC, and verruciform xanthoma. Squamous cell carcinoma characteristically has a more infiltrative border as opposed to the bulbous border of VC. In addition, the distribution of p53 and Ki-67 staining differs between SCC and VC. Squamous cell carcinoma shows positive p53 and Ki-67 staining for the full thickness of the epidermis, while VC has positive staining only in the lower third of the epidermis.5
Surgical resection is considered the first-line treatment of VC through excision or Mohs micrographic surgery. Radiation therapy is controversial due to the risk for anaplastic transformation. When surgery is not ideal due to the tumor size or location or the patient’s preference, other treatment modalities with reported success include intralesional interferon alfa; cryosurgery; topical imiquimod; and topical or systemic cytostatic agents such as bleomycin, 5-fluorouracil, cisplatin, or methotrexate.1,2
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2006;56:506-508.
- Nikkels AF, Thirion L, Quatresooz P, et al. Photodynamic therapy for cutaneous verrucous carcinoma. J Am Acad Dermatol. 2007;57:516-519.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. Arch Dermatol. 2000;136:547-552.
- Sonalika WG, Anand T. Oral verrucous carcinoma: a retrospective analysis for clinicopathologic features. J Cancer Res Ther. 2016;12:142-145.
- Dubina M, Goldenberg G. Viral-associated nonmelanoma skin cancers: a review. Am J Dermatopathol. 2009;31:561-573.
- Geusau A, Heinz-Peer G, Volc-Platzer B, et al. Regression of deeply infiltrating giant condyloma (Buschke-Lowenstein tumor) following long-term intralesional interferon alpha therapy. Arch Dermatol. 2000;136:707-710.
- Ansai S, Kimura T, Hayashi M. Fatal genital verrucous carcinoma. Am J Dermatopathol. 2007;29:68-71.
The Diagnosis: Verrucous Carcinoma
An excisional biopsy revealed an endophytic and exophytic squamous proliferation with a papillomatous growth pattern, bulbous pushing border, and confluent parakeratosis (Figure). No fungal organisms were seen. Due to clinical and histological findings, a diagnosis of verrucous carcinoma (VC) was made.
Verrucous carcinoma is a rare variant of squamous cell carcinoma (SCC) with specific clinical and histological features.1 These tumors have a slow and localized growth pattern but can be locally aggressive. Metastasis of VC is rare, giving VC an overall good prognosis, with a 5-year survival rate greater than 75%.2 Verrucous carcinoma typically occurs in 1 of 3 locations: the oropharynx, genitals, or soles of the feet. Depending on the site of involvement, various names have been used in the literature to describe this entity, including Ackerman tumor (solitary oral mucosal lesion), Buschke-Lowenstein tumor (genital involvement), florid oral papillomatosis (multiple oral lesions), and carcinoma cuniculatum (sole of the foot).3 The most common sites for VC in the oral cavity are the buccal mucosa and gingiva.4
Verrucous carcinoma occurs more often among men in the sixth decade of life.3 The etiology of oral VC remains unclear; however, use of chewing tobacco, chemical carcinogens, chronic irritation, human papillomavirus (HPV), and poor oral hygiene have been reported as predisposing risk factors.4,5 The role of HPV in the pathogenesis of VC remains controversial, but both low-risk types HPV-6 and HPV-11 and high-risk types HPV-16 and HPV-18 have been found in association with VC.5,6
Clinically, oral VC lesions most often present as pink-white erythematous papules or plaques with exophytic cauliflowerlike surface alterations. Although the tumors are slow growing with little risk for metastasis, they may be locally invasive with deep involvement of the surrounding
structures.1 Histopathologically, VC displays proliferation of the epithelium with downward growth into the connective tissue but usually without a pattern of true invasion. The epithelium is well differentiated and displays little pleomorphism or mitoses.5,7 Obtaining a generous biopsy specimen is essential to view the diagnostic architecture of VC and rule out other entities, such as viral verruca, blastomycosis, SCC, and verruciform xanthoma. Squamous cell carcinoma characteristically has a more infiltrative border as opposed to the bulbous border of VC. In addition, the distribution of p53 and Ki-67 staining differs between SCC and VC. Squamous cell carcinoma shows positive p53 and Ki-67 staining for the full thickness of the epidermis, while VC has positive staining only in the lower third of the epidermis.5
Surgical resection is considered the first-line treatment of VC through excision or Mohs micrographic surgery. Radiation therapy is controversial due to the risk for anaplastic transformation. When surgery is not ideal due to the tumor size or location or the patient’s preference, other treatment modalities with reported success include intralesional interferon alfa; cryosurgery; topical imiquimod; and topical or systemic cytostatic agents such as bleomycin, 5-fluorouracil, cisplatin, or methotrexate.1,2
The Diagnosis: Verrucous Carcinoma
An excisional biopsy revealed an endophytic and exophytic squamous proliferation with a papillomatous growth pattern, bulbous pushing border, and confluent parakeratosis (Figure). No fungal organisms were seen. Due to clinical and histological findings, a diagnosis of verrucous carcinoma (VC) was made.
Verrucous carcinoma is a rare variant of squamous cell carcinoma (SCC) with specific clinical and histological features.1 These tumors have a slow and localized growth pattern but can be locally aggressive. Metastasis of VC is rare, giving VC an overall good prognosis, with a 5-year survival rate greater than 75%.2 Verrucous carcinoma typically occurs in 1 of 3 locations: the oropharynx, genitals, or soles of the feet. Depending on the site of involvement, various names have been used in the literature to describe this entity, including Ackerman tumor (solitary oral mucosal lesion), Buschke-Lowenstein tumor (genital involvement), florid oral papillomatosis (multiple oral lesions), and carcinoma cuniculatum (sole of the foot).3 The most common sites for VC in the oral cavity are the buccal mucosa and gingiva.4
Verrucous carcinoma occurs more often among men in the sixth decade of life.3 The etiology of oral VC remains unclear; however, use of chewing tobacco, chemical carcinogens, chronic irritation, human papillomavirus (HPV), and poor oral hygiene have been reported as predisposing risk factors.4,5 The role of HPV in the pathogenesis of VC remains controversial, but both low-risk types HPV-6 and HPV-11 and high-risk types HPV-16 and HPV-18 have been found in association with VC.5,6
Clinically, oral VC lesions most often present as pink-white erythematous papules or plaques with exophytic cauliflowerlike surface alterations. Although the tumors are slow growing with little risk for metastasis, they may be locally invasive with deep involvement of the surrounding
structures.1 Histopathologically, VC displays proliferation of the epithelium with downward growth into the connective tissue but usually without a pattern of true invasion. The epithelium is well differentiated and displays little pleomorphism or mitoses.5,7 Obtaining a generous biopsy specimen is essential to view the diagnostic architecture of VC and rule out other entities, such as viral verruca, blastomycosis, SCC, and verruciform xanthoma. Squamous cell carcinoma characteristically has a more infiltrative border as opposed to the bulbous border of VC. In addition, the distribution of p53 and Ki-67 staining differs between SCC and VC. Squamous cell carcinoma shows positive p53 and Ki-67 staining for the full thickness of the epidermis, while VC has positive staining only in the lower third of the epidermis.5
Surgical resection is considered the first-line treatment of VC through excision or Mohs micrographic surgery. Radiation therapy is controversial due to the risk for anaplastic transformation. When surgery is not ideal due to the tumor size or location or the patient’s preference, other treatment modalities with reported success include intralesional interferon alfa; cryosurgery; topical imiquimod; and topical or systemic cytostatic agents such as bleomycin, 5-fluorouracil, cisplatin, or methotrexate.1,2
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2006;56:506-508.
- Nikkels AF, Thirion L, Quatresooz P, et al. Photodynamic therapy for cutaneous verrucous carcinoma. J Am Acad Dermatol. 2007;57:516-519.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. Arch Dermatol. 2000;136:547-552.
- Sonalika WG, Anand T. Oral verrucous carcinoma: a retrospective analysis for clinicopathologic features. J Cancer Res Ther. 2016;12:142-145.
- Dubina M, Goldenberg G. Viral-associated nonmelanoma skin cancers: a review. Am J Dermatopathol. 2009;31:561-573.
- Geusau A, Heinz-Peer G, Volc-Platzer B, et al. Regression of deeply infiltrating giant condyloma (Buschke-Lowenstein tumor) following long-term intralesional interferon alpha therapy. Arch Dermatol. 2000;136:707-710.
- Ansai S, Kimura T, Hayashi M. Fatal genital verrucous carcinoma. Am J Dermatopathol. 2007;29:68-71.
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2006;56:506-508.
- Nikkels AF, Thirion L, Quatresooz P, et al. Photodynamic therapy for cutaneous verrucous carcinoma. J Am Acad Dermatol. 2007;57:516-519.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. Arch Dermatol. 2000;136:547-552.
- Sonalika WG, Anand T. Oral verrucous carcinoma: a retrospective analysis for clinicopathologic features. J Cancer Res Ther. 2016;12:142-145.
- Dubina M, Goldenberg G. Viral-associated nonmelanoma skin cancers: a review. Am J Dermatopathol. 2009;31:561-573.
- Geusau A, Heinz-Peer G, Volc-Platzer B, et al. Regression of deeply infiltrating giant condyloma (Buschke-Lowenstein tumor) following long-term intralesional interferon alpha therapy. Arch Dermatol. 2000;136:707-710.
- Ansai S, Kimura T, Hayashi M. Fatal genital verrucous carcinoma. Am J Dermatopathol. 2007;29:68-71.
Irregularly Hyperpigmented Plaque on the Right Heel
The Diagnosis: Pigmented Bowen Disease
A biopsy of the lesion was performed for suspected acral malignant melanoma. Hematoxylin and eosin staining revealed acanthosis, elongation of rete ridges, and keratinocytes in complete disorder with atypical mitoses and pleomorphism affecting the full layer of the epidermis (Figure 1). The basement membrane was intact. Melanin pigmentation was increased in the lower epidermis and the upper dermis, and a lymphohistiocytic inflammatory infiltrate was present in the dermis. Staining for carcinoembryonic antigen (Figure 2) and melanoma
antigen (Figure 3) recognized by T cells (melan-A) both revealed negative results. Histopathologic findings led to the diagnosis of pigmented Bowen disease (BD).
Pigmented BD is a rare variant that accounts for 1.7% (N=420) to 5.5% (N=951) of all cases of BD.1,2 It is reported to affect men more than women and to be more prevalent in individuals with higher Fitzpatrick skin types.3 Furthermore, exposure to UV radiation, chemicals (eg, arsenic), or human papillomavirus, as well as immunosuppression, are known to be related to pigmented BD.2,4 Clinically, pigmented BD commonly involves nonexposed areas such as the anogenital area, trunk, and extremities, unlike typical BD that involves sun-exposed areas.5 In addition, it most frequently presents as a well-delineated, irregularly pigmented, asymptomatic
plaque and not as a scaly erythematous plaque. Therefore, the clinical diagnosis may be challenging. The differential diagnosis includes malignant melanoma, pigmented extramammary Paget disease, pigmented basal cell carcinoma, seborrheic keratosis, pigmented actinic keratosis, solar lentigo, and melanocytic nevi.
Histopathologically, a varying amount of melanin deposit is noted on hematoxylin and eosin staining, along with features of BD, including disarrayed atypical keratinocytes involving the full epidermis but not the basement membrane, with atypical individual cell keratinization.3,5,6 Pigmented extramammary Paget disease can mimic pigmented BD clinically and pathologically, but Paget cells stain positive for anticytokeratin (CAM 5.2), carcinoembryonic antigen, and mucicarmine, whereas cells in pigmented BD stain negative.7 Moreover, negative staining for human melanoma black, melan-A, and S-100 helps differentiate malignant melanoma from pigmented BD.8
The prognosis of pigmented BD is similar to classic BD and is independent of the presence of melanin pigment.6 Therefore, the treatment options do not differ from those for typical BD and include surgical excision, cryotherapy, laser ablation, topical imiquimod or 5-fluorouracil, curettage, electrosurgery, and photodynamic therapy (PDT).
In our case, the patient and her family did not want surgical removal; therefore, 1 course of fractional laser-assisted PDT and 2 courses of ablative laser-assisted PDT were performed. Unfortunately, the lesion persisted, possibly because it was too large and pigmented. Two months later, ingenol mebutate gel 0.05% was applied (4 courses) after using an ablative laser over 3 consecutive days with a 1-month interval between courses. The lesion resolved without any adverse events.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease [published online January 15, 2010]. J Am Acad Dermatol. 2010;62:597-604.
- Ragi G, Turner MS, Klein LE, et al. Pigmented Bowen’s disease and review of 420 Bowen’s disease lesions. J Dermatol Surg Oncol. 1988;14:765-769.
- Hernandez C, Ivkovic A, Fowler A. Growing plaque on foot. J Fam Pract. 2008;57:603-605.
- Hwang SW, Kim JW, Park SW, et al. Two cases of pigmented Bowen’s disease. Ann Dermatol 2002;14:127-129.
- Wilmer EM, Lee KC, Higgins W 2nd, et al. Hyperpigmented palmar plaque: an unexpected diagnosis of Bowen disease. Dermatol Online J. 2013;19:18573.
- Brinca A, Teixeira V, Gonçalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-884.
- Hilliard NJ, Huang C, Andea A. Pigmented extramammary Paget’s disease of the axilla mimicking melanoma: case report and review of the literature. J Cutan Pathol. 2009;36:995-1000.
- Öztürk Durmaz E, Dog˘ an Ekici I, Ozian F, et al. Pigmented Bowen’s disease of the genitalia masquerading as malignant melanoma. Acta Dermatovenerol Croat. 2015;23:130-133.
The Diagnosis: Pigmented Bowen Disease
A biopsy of the lesion was performed for suspected acral malignant melanoma. Hematoxylin and eosin staining revealed acanthosis, elongation of rete ridges, and keratinocytes in complete disorder with atypical mitoses and pleomorphism affecting the full layer of the epidermis (Figure 1). The basement membrane was intact. Melanin pigmentation was increased in the lower epidermis and the upper dermis, and a lymphohistiocytic inflammatory infiltrate was present in the dermis. Staining for carcinoembryonic antigen (Figure 2) and melanoma
antigen (Figure 3) recognized by T cells (melan-A) both revealed negative results. Histopathologic findings led to the diagnosis of pigmented Bowen disease (BD).
Pigmented BD is a rare variant that accounts for 1.7% (N=420) to 5.5% (N=951) of all cases of BD.1,2 It is reported to affect men more than women and to be more prevalent in individuals with higher Fitzpatrick skin types.3 Furthermore, exposure to UV radiation, chemicals (eg, arsenic), or human papillomavirus, as well as immunosuppression, are known to be related to pigmented BD.2,4 Clinically, pigmented BD commonly involves nonexposed areas such as the anogenital area, trunk, and extremities, unlike typical BD that involves sun-exposed areas.5 In addition, it most frequently presents as a well-delineated, irregularly pigmented, asymptomatic
plaque and not as a scaly erythematous plaque. Therefore, the clinical diagnosis may be challenging. The differential diagnosis includes malignant melanoma, pigmented extramammary Paget disease, pigmented basal cell carcinoma, seborrheic keratosis, pigmented actinic keratosis, solar lentigo, and melanocytic nevi.
Histopathologically, a varying amount of melanin deposit is noted on hematoxylin and eosin staining, along with features of BD, including disarrayed atypical keratinocytes involving the full epidermis but not the basement membrane, with atypical individual cell keratinization.3,5,6 Pigmented extramammary Paget disease can mimic pigmented BD clinically and pathologically, but Paget cells stain positive for anticytokeratin (CAM 5.2), carcinoembryonic antigen, and mucicarmine, whereas cells in pigmented BD stain negative.7 Moreover, negative staining for human melanoma black, melan-A, and S-100 helps differentiate malignant melanoma from pigmented BD.8
The prognosis of pigmented BD is similar to classic BD and is independent of the presence of melanin pigment.6 Therefore, the treatment options do not differ from those for typical BD and include surgical excision, cryotherapy, laser ablation, topical imiquimod or 5-fluorouracil, curettage, electrosurgery, and photodynamic therapy (PDT).
In our case, the patient and her family did not want surgical removal; therefore, 1 course of fractional laser-assisted PDT and 2 courses of ablative laser-assisted PDT were performed. Unfortunately, the lesion persisted, possibly because it was too large and pigmented. Two months later, ingenol mebutate gel 0.05% was applied (4 courses) after using an ablative laser over 3 consecutive days with a 1-month interval between courses. The lesion resolved without any adverse events.
The Diagnosis: Pigmented Bowen Disease
A biopsy of the lesion was performed for suspected acral malignant melanoma. Hematoxylin and eosin staining revealed acanthosis, elongation of rete ridges, and keratinocytes in complete disorder with atypical mitoses and pleomorphism affecting the full layer of the epidermis (Figure 1). The basement membrane was intact. Melanin pigmentation was increased in the lower epidermis and the upper dermis, and a lymphohistiocytic inflammatory infiltrate was present in the dermis. Staining for carcinoembryonic antigen (Figure 2) and melanoma
antigen (Figure 3) recognized by T cells (melan-A) both revealed negative results. Histopathologic findings led to the diagnosis of pigmented Bowen disease (BD).
Pigmented BD is a rare variant that accounts for 1.7% (N=420) to 5.5% (N=951) of all cases of BD.1,2 It is reported to affect men more than women and to be more prevalent in individuals with higher Fitzpatrick skin types.3 Furthermore, exposure to UV radiation, chemicals (eg, arsenic), or human papillomavirus, as well as immunosuppression, are known to be related to pigmented BD.2,4 Clinically, pigmented BD commonly involves nonexposed areas such as the anogenital area, trunk, and extremities, unlike typical BD that involves sun-exposed areas.5 In addition, it most frequently presents as a well-delineated, irregularly pigmented, asymptomatic
plaque and not as a scaly erythematous plaque. Therefore, the clinical diagnosis may be challenging. The differential diagnosis includes malignant melanoma, pigmented extramammary Paget disease, pigmented basal cell carcinoma, seborrheic keratosis, pigmented actinic keratosis, solar lentigo, and melanocytic nevi.
Histopathologically, a varying amount of melanin deposit is noted on hematoxylin and eosin staining, along with features of BD, including disarrayed atypical keratinocytes involving the full epidermis but not the basement membrane, with atypical individual cell keratinization.3,5,6 Pigmented extramammary Paget disease can mimic pigmented BD clinically and pathologically, but Paget cells stain positive for anticytokeratin (CAM 5.2), carcinoembryonic antigen, and mucicarmine, whereas cells in pigmented BD stain negative.7 Moreover, negative staining for human melanoma black, melan-A, and S-100 helps differentiate malignant melanoma from pigmented BD.8
The prognosis of pigmented BD is similar to classic BD and is independent of the presence of melanin pigment.6 Therefore, the treatment options do not differ from those for typical BD and include surgical excision, cryotherapy, laser ablation, topical imiquimod or 5-fluorouracil, curettage, electrosurgery, and photodynamic therapy (PDT).
In our case, the patient and her family did not want surgical removal; therefore, 1 course of fractional laser-assisted PDT and 2 courses of ablative laser-assisted PDT were performed. Unfortunately, the lesion persisted, possibly because it was too large and pigmented. Two months later, ingenol mebutate gel 0.05% was applied (4 courses) after using an ablative laser over 3 consecutive days with a 1-month interval between courses. The lesion resolved without any adverse events.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease [published online January 15, 2010]. J Am Acad Dermatol. 2010;62:597-604.
- Ragi G, Turner MS, Klein LE, et al. Pigmented Bowen’s disease and review of 420 Bowen’s disease lesions. J Dermatol Surg Oncol. 1988;14:765-769.
- Hernandez C, Ivkovic A, Fowler A. Growing plaque on foot. J Fam Pract. 2008;57:603-605.
- Hwang SW, Kim JW, Park SW, et al. Two cases of pigmented Bowen’s disease. Ann Dermatol 2002;14:127-129.
- Wilmer EM, Lee KC, Higgins W 2nd, et al. Hyperpigmented palmar plaque: an unexpected diagnosis of Bowen disease. Dermatol Online J. 2013;19:18573.
- Brinca A, Teixeira V, Gonçalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-884.
- Hilliard NJ, Huang C, Andea A. Pigmented extramammary Paget’s disease of the axilla mimicking melanoma: case report and review of the literature. J Cutan Pathol. 2009;36:995-1000.
- Öztürk Durmaz E, Dog˘ an Ekici I, Ozian F, et al. Pigmented Bowen’s disease of the genitalia masquerading as malignant melanoma. Acta Dermatovenerol Croat. 2015;23:130-133.
- Cameron A, Rosendahl C, Tschandl P, et al. Dermatoscopy of pigmented Bowen’s disease [published online January 15, 2010]. J Am Acad Dermatol. 2010;62:597-604.
- Ragi G, Turner MS, Klein LE, et al. Pigmented Bowen’s disease and review of 420 Bowen’s disease lesions. J Dermatol Surg Oncol. 1988;14:765-769.
- Hernandez C, Ivkovic A, Fowler A. Growing plaque on foot. J Fam Pract. 2008;57:603-605.
- Hwang SW, Kim JW, Park SW, et al. Two cases of pigmented Bowen’s disease. Ann Dermatol 2002;14:127-129.
- Wilmer EM, Lee KC, Higgins W 2nd, et al. Hyperpigmented palmar plaque: an unexpected diagnosis of Bowen disease. Dermatol Online J. 2013;19:18573.
- Brinca A, Teixeira V, Gonçalo M, et al. A large pigmented lesion mimicking malignant melanoma. Clin Exp Dermatol. 2012;37:817-884.
- Hilliard NJ, Huang C, Andea A. Pigmented extramammary Paget’s disease of the axilla mimicking melanoma: case report and review of the literature. J Cutan Pathol. 2009;36:995-1000.
- Öztürk Durmaz E, Dog˘ an Ekici I, Ozian F, et al. Pigmented Bowen’s disease of the genitalia masquerading as malignant melanoma. Acta Dermatovenerol Croat. 2015;23:130-133.
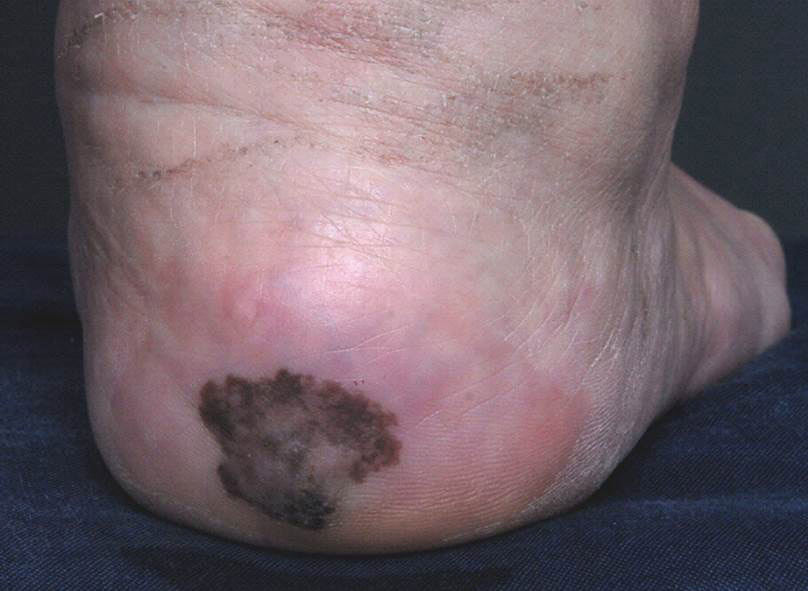
A 56-year-old woman presented with an asymptomatic plaque on the right heel that had grown
steadily over the last year. Pigmented lesions were not appreciated on other sites, and lymph nodes were not enlarged. Her medical history was otherwise normal, except for bilateral hearing loss due to encephalitis at the age of 5 years. None of her family members had similar symptoms. Physical examination revealed a well-defined, irregularly hyperpigmented plaque on the right heel.
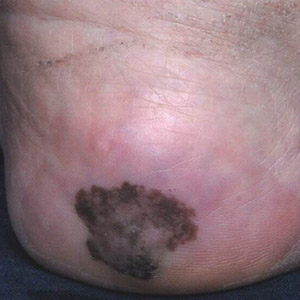
Large Hemorrhagic Plaque With Central Crusting
The Diagnosis: Bullous/Hemorrhagic Lichen Sclerosus et Atrophicus
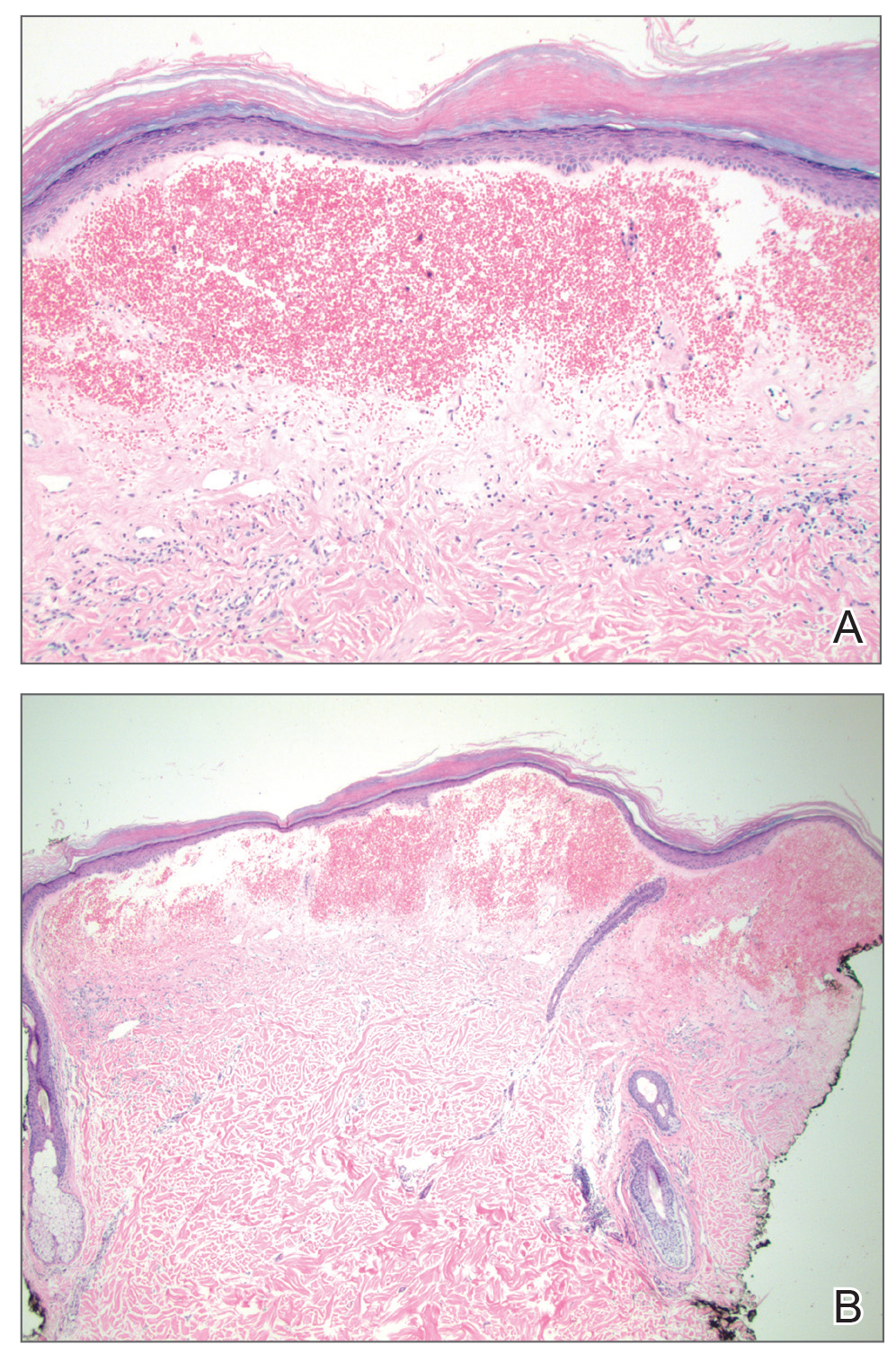
Histopathologic examination revealed hyperkeratosis of the stratum corneum and thinning of the epidermis (Figure). Subepidermal edema and hemorrhage in the papillary dermis were seen. There were dilated vessels beneath the edema in the reticular dermis, as well as perivascular, perifollicular, and interstitial lymphocytic inflammation. No cytologic atypia characteristic of squamous cell carcinoma (SCC) and angiosarcoma or large lymphatic channels characteristic of lymphangioma were noted. Based on clinicopathologic correlation, the diagnosis of the bullous/hemorrhagic form of lichen sclerosus et atrophicus (LS&A) was made. The patient was treated with high-potency topical steroids with notable symptomatic improvement and rapid resolution of the hemorrhagic lesion.
Lichen sclerosus et atrophicus is a chronic inflammatory condition with a predilection for the anogenital region, though rare cases of extragenital involvement have been reported. It is seen in both sexes and across all age groups, with notably higher prevalence in females in the fifth and sixth decades of life.1,2 Lichen sclerosus et atrophicus can be difficult to diagnose, as these patients may present to a variety of specialists, may be embarrassed by the condition and reluctant for full evaluation, or may have asymptomatic lesions.2,3 Rare cases of isolated extragenital involvement and hemorrhagic or bullous lesions further complicate the diagnosis.1,2 Despite these difficulties, diagnosis is essential, as there is potential for cosmetically and functionally detrimental scarring as well as atrophy and development of overlying malignancies. Lichen sclerosus et atrophicus is not curable and rarely remits spontaneously, but appropriate treatment strategies can help control the symptoms of the condition as well as its most devastating sequelae.3
For females, classic LS&A is most common in theprepubertal, perimenopausal, or postmenopausal periods, commonly involving the vulva or perineum. Symptoms include pruritus, burning sensation, dysuria, dyspareunia, and labial stenosis, among others. For males, most cases involve the glans penis in prepubertal boys or middleaged men, and symptoms include pruritus, new-onset phimosis, decreased sensation, painful erections, dysuria, and urinary obstruction.1-3 An estimated 97% of patients have some form of genital involvement with only 2.5% showing isolated extragenital involvement, though the latter may be underdiagnosed, as this area is more likely to be asymptomatic.3-6 Extragenital LS&A most often involves the neck and shoulders. The classic appearance of LS&A includes shiny, white-red macules and papules that ultimately coalesce into atrophic plaques and can be accompanied by fissuring or scarring, especially in the genital area.2 There is an increased risk for SCC associated with genital LS&A.1
Bullous/hemorrhagic LS&A has been described as a rare phenotype. One case report cited an increased incidence of this subtype in patients with exclusively extragenital lesions, and the authors considered blister formation to be a characteristic feature of extragenital LS&A. The pathogenesis of blister formation and hemorrhage in LS&A is not completely understood, but trauma is thought to play a role due to decreased stress tolerance from atrophic skin.4 Furthermore, distortion of blood vessel architecture in LS&A has been described with loss of the capillary network and enlargement of vessels along the dermoepidermal junction, which also could play a role in hemorrhage. Differential diagnosis of the bullous/hemorrhagic type of LS&A includes bullous pemphigoid, bullous lichen planus, or bullous scleroderma.7 In our more exophytic hemorrhagic case, malignancies such as SCC or angiosarcoma also had to be considered. Unlike genital LS&A, extragenital LS&A including the bullous/hemorrhagic variant has not been linked to an increasedrisk for malignancy.1,5
The mainstay of treatment of all forms of LS&A is high-potency topical steroids, but topical retinoids, tacrolimus, and UVA phototherapy also have been used. Bullous/hemorrhagic lesions often resolve quickly with topical steroids, leaving behind more classic plaques in their place, which can be more refractory to treatment.5,7
- Meffert JJ, Davis BM, Grimwood RE. Lichen sclerosus. J Am Acad Dermatol. 1995;32:393-416.
- Pugliese JM, Morey AF, Peterson AC. Lichen sclerosus: review of the literature and current recommendations for management. J Urol. 2007;178:2268-2276.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Kimura A, Kambe N, Satoh T, et al. Follicular keratosis and bullous formation are typical signs of extragenital lichen sclerosus. J Dermatol. 2011;38:834-836.
- Khatu S, Vasani R. Isolated, localised extragenital bullous lichen sclerosus et atrophicus: a rare entity. Indian J Dermatol. 2013;58:409.
- Luzar B, Neil SM, Calonje E. Angiokeratoma-like changes in extragenital and genital lichen sclerosus. J Cutan Pathol. 2009;36:540-542.
- Lima RS, Maquine GA, Schettini AP, et al. Bullous and hemorrhagic lichen sclerosus—case report. An Bras Dermatol. 2015;90 (3 suppl 1):118-120.
The Diagnosis: Bullous/Hemorrhagic Lichen Sclerosus et Atrophicus
Histopathologic examination revealed hyperkeratosis of the stratum corneum and thinning of the epidermis (Figure). Subepidermal edema and hemorrhage in the papillary dermis were seen. There were dilated vessels beneath the edema in the reticular dermis, as well as perivascular, perifollicular, and interstitial lymphocytic inflammation. No cytologic atypia characteristic of squamous cell carcinoma (SCC) and angiosarcoma or large lymphatic channels characteristic of lymphangioma were noted. Based on clinicopathologic correlation, the diagnosis of the bullous/hemorrhagic form of lichen sclerosus et atrophicus (LS&A) was made. The patient was treated with high-potency topical steroids with notable symptomatic improvement and rapid resolution of the hemorrhagic lesion.
Lichen sclerosus et atrophicus is a chronic inflammatory condition with a predilection for the anogenital region, though rare cases of extragenital involvement have been reported. It is seen in both sexes and across all age groups, with notably higher prevalence in females in the fifth and sixth decades of life.1,2 Lichen sclerosus et atrophicus can be difficult to diagnose, as these patients may present to a variety of specialists, may be embarrassed by the condition and reluctant for full evaluation, or may have asymptomatic lesions.2,3 Rare cases of isolated extragenital involvement and hemorrhagic or bullous lesions further complicate the diagnosis.1,2 Despite these difficulties, diagnosis is essential, as there is potential for cosmetically and functionally detrimental scarring as well as atrophy and development of overlying malignancies. Lichen sclerosus et atrophicus is not curable and rarely remits spontaneously, but appropriate treatment strategies can help control the symptoms of the condition as well as its most devastating sequelae.3
For females, classic LS&A is most common in theprepubertal, perimenopausal, or postmenopausal periods, commonly involving the vulva or perineum. Symptoms include pruritus, burning sensation, dysuria, dyspareunia, and labial stenosis, among others. For males, most cases involve the glans penis in prepubertal boys or middleaged men, and symptoms include pruritus, new-onset phimosis, decreased sensation, painful erections, dysuria, and urinary obstruction.1-3 An estimated 97% of patients have some form of genital involvement with only 2.5% showing isolated extragenital involvement, though the latter may be underdiagnosed, as this area is more likely to be asymptomatic.3-6 Extragenital LS&A most often involves the neck and shoulders. The classic appearance of LS&A includes shiny, white-red macules and papules that ultimately coalesce into atrophic plaques and can be accompanied by fissuring or scarring, especially in the genital area.2 There is an increased risk for SCC associated with genital LS&A.1
Bullous/hemorrhagic LS&A has been described as a rare phenotype. One case report cited an increased incidence of this subtype in patients with exclusively extragenital lesions, and the authors considered blister formation to be a characteristic feature of extragenital LS&A. The pathogenesis of blister formation and hemorrhage in LS&A is not completely understood, but trauma is thought to play a role due to decreased stress tolerance from atrophic skin.4 Furthermore, distortion of blood vessel architecture in LS&A has been described with loss of the capillary network and enlargement of vessels along the dermoepidermal junction, which also could play a role in hemorrhage. Differential diagnosis of the bullous/hemorrhagic type of LS&A includes bullous pemphigoid, bullous lichen planus, or bullous scleroderma.7 In our more exophytic hemorrhagic case, malignancies such as SCC or angiosarcoma also had to be considered. Unlike genital LS&A, extragenital LS&A including the bullous/hemorrhagic variant has not been linked to an increasedrisk for malignancy.1,5
The mainstay of treatment of all forms of LS&A is high-potency topical steroids, but topical retinoids, tacrolimus, and UVA phototherapy also have been used. Bullous/hemorrhagic lesions often resolve quickly with topical steroids, leaving behind more classic plaques in their place, which can be more refractory to treatment.5,7
The Diagnosis: Bullous/Hemorrhagic Lichen Sclerosus et Atrophicus
Histopathologic examination revealed hyperkeratosis of the stratum corneum and thinning of the epidermis (Figure). Subepidermal edema and hemorrhage in the papillary dermis were seen. There were dilated vessels beneath the edema in the reticular dermis, as well as perivascular, perifollicular, and interstitial lymphocytic inflammation. No cytologic atypia characteristic of squamous cell carcinoma (SCC) and angiosarcoma or large lymphatic channels characteristic of lymphangioma were noted. Based on clinicopathologic correlation, the diagnosis of the bullous/hemorrhagic form of lichen sclerosus et atrophicus (LS&A) was made. The patient was treated with high-potency topical steroids with notable symptomatic improvement and rapid resolution of the hemorrhagic lesion.
Lichen sclerosus et atrophicus is a chronic inflammatory condition with a predilection for the anogenital region, though rare cases of extragenital involvement have been reported. It is seen in both sexes and across all age groups, with notably higher prevalence in females in the fifth and sixth decades of life.1,2 Lichen sclerosus et atrophicus can be difficult to diagnose, as these patients may present to a variety of specialists, may be embarrassed by the condition and reluctant for full evaluation, or may have asymptomatic lesions.2,3 Rare cases of isolated extragenital involvement and hemorrhagic or bullous lesions further complicate the diagnosis.1,2 Despite these difficulties, diagnosis is essential, as there is potential for cosmetically and functionally detrimental scarring as well as atrophy and development of overlying malignancies. Lichen sclerosus et atrophicus is not curable and rarely remits spontaneously, but appropriate treatment strategies can help control the symptoms of the condition as well as its most devastating sequelae.3
For females, classic LS&A is most common in theprepubertal, perimenopausal, or postmenopausal periods, commonly involving the vulva or perineum. Symptoms include pruritus, burning sensation, dysuria, dyspareunia, and labial stenosis, among others. For males, most cases involve the glans penis in prepubertal boys or middleaged men, and symptoms include pruritus, new-onset phimosis, decreased sensation, painful erections, dysuria, and urinary obstruction.1-3 An estimated 97% of patients have some form of genital involvement with only 2.5% showing isolated extragenital involvement, though the latter may be underdiagnosed, as this area is more likely to be asymptomatic.3-6 Extragenital LS&A most often involves the neck and shoulders. The classic appearance of LS&A includes shiny, white-red macules and papules that ultimately coalesce into atrophic plaques and can be accompanied by fissuring or scarring, especially in the genital area.2 There is an increased risk for SCC associated with genital LS&A.1
Bullous/hemorrhagic LS&A has been described as a rare phenotype. One case report cited an increased incidence of this subtype in patients with exclusively extragenital lesions, and the authors considered blister formation to be a characteristic feature of extragenital LS&A. The pathogenesis of blister formation and hemorrhage in LS&A is not completely understood, but trauma is thought to play a role due to decreased stress tolerance from atrophic skin.4 Furthermore, distortion of blood vessel architecture in LS&A has been described with loss of the capillary network and enlargement of vessels along the dermoepidermal junction, which also could play a role in hemorrhage. Differential diagnosis of the bullous/hemorrhagic type of LS&A includes bullous pemphigoid, bullous lichen planus, or bullous scleroderma.7 In our more exophytic hemorrhagic case, malignancies such as SCC or angiosarcoma also had to be considered. Unlike genital LS&A, extragenital LS&A including the bullous/hemorrhagic variant has not been linked to an increasedrisk for malignancy.1,5
The mainstay of treatment of all forms of LS&A is high-potency topical steroids, but topical retinoids, tacrolimus, and UVA phototherapy also have been used. Bullous/hemorrhagic lesions often resolve quickly with topical steroids, leaving behind more classic plaques in their place, which can be more refractory to treatment.5,7
- Meffert JJ, Davis BM, Grimwood RE. Lichen sclerosus. J Am Acad Dermatol. 1995;32:393-416.
- Pugliese JM, Morey AF, Peterson AC. Lichen sclerosus: review of the literature and current recommendations for management. J Urol. 2007;178:2268-2276.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Kimura A, Kambe N, Satoh T, et al. Follicular keratosis and bullous formation are typical signs of extragenital lichen sclerosus. J Dermatol. 2011;38:834-836.
- Khatu S, Vasani R. Isolated, localised extragenital bullous lichen sclerosus et atrophicus: a rare entity. Indian J Dermatol. 2013;58:409.
- Luzar B, Neil SM, Calonje E. Angiokeratoma-like changes in extragenital and genital lichen sclerosus. J Cutan Pathol. 2009;36:540-542.
- Lima RS, Maquine GA, Schettini AP, et al. Bullous and hemorrhagic lichen sclerosus—case report. An Bras Dermatol. 2015;90 (3 suppl 1):118-120.
- Meffert JJ, Davis BM, Grimwood RE. Lichen sclerosus. J Am Acad Dermatol. 1995;32:393-416.
- Pugliese JM, Morey AF, Peterson AC. Lichen sclerosus: review of the literature and current recommendations for management. J Urol. 2007;178:2268-2276.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Kimura A, Kambe N, Satoh T, et al. Follicular keratosis and bullous formation are typical signs of extragenital lichen sclerosus. J Dermatol. 2011;38:834-836.
- Khatu S, Vasani R. Isolated, localised extragenital bullous lichen sclerosus et atrophicus: a rare entity. Indian J Dermatol. 2013;58:409.
- Luzar B, Neil SM, Calonje E. Angiokeratoma-like changes in extragenital and genital lichen sclerosus. J Cutan Pathol. 2009;36:540-542.
- Lima RS, Maquine GA, Schettini AP, et al. Bullous and hemorrhagic lichen sclerosus—case report. An Bras Dermatol. 2015;90 (3 suppl 1):118-120.

A 54-year-old woman with no notable medical history was referred to dermatology by her primary care provider for evaluation of a hematoma on the posterior neck that had developed gradually over 5 months. The lesion initially was asymptomatic but more recently had started to be painful and bleed intermittently. The patient denied any personal or family history of skin cancer. Physical examination revealed a large hemorrhagic plaque on the left side of the posterior neck with central brown-yellow crusting. There were few smaller, white, thin, sclerotic plaques with crinkling atrophy at the periphery of and inferolateral to the lesion. A punch biopsy specimen was obtained from the hemorrhagic plaque.

Erythematous Periumbilical Papules and Plaques
The Diagnosis: Metastatic Cancer
Further workup of patient 1 revealed an alkaline phosphatase level of 743 U/L (reference range, 30–120 U/L), total bilirubin level of 8.5 mg/dL (reference range, 0.3–1.2 mg/dL), and a white blood cell count of 14,000/μL (reference range, 4500–11,000/μL). Computed tomography of the abdomen and pelvis demonstrated cancer of unknown primary site that had metastasized to the colon, liver, and lungs. There was suspicion for potential colon cancer as the primary disease; however, based on the cutaneous findings, a skin biopsy was performed to confirm the diagnosis. Histology and immunohistochemistry revealed adenocarcinoma tumor cells positive for CDX2 (caudal type homeobox 2) and cytokeratin (CK) 7 with a subset positive for CK-20. The cells were negative for estrogen receptor, progesterone receptor, mammaglobin, gross cystic disease fluid protein, and GATA3 (GATA binding protein 3). Immunohistochemistry was most consistent with pancreatic cancer. During palliative percutaneous transhepatic biliary drainage placement, a liver biopsy confirmed the skin biopsy results.
Further workup of patient 2 revealed a white blood cell count of 13,000/μL (reference range, 4500–11,000/μL). Computed tomography of the chest, abdomen, and pelvis revealed metastatic disease to the lungs with a suspicion for colon cancer as the primary site. Biopsy of the skin lesion revealed a mucin-producing adenocarcinoma, and immunohistochemistry was positive for keratin (AE1/AE3), CK-20, and CDX2, consistent with metastatic colon carcinoma. Immunohistochemistry of the biopsied skin lesion was nonreactive for CK-7. The patient had a colonoscopy that revealed a fungating, partially obstructing, circumferential large mass in the ascending colon.
Metastasis to the skin from visceral malignancies is not uncommon and may represent the first evidence of widespread disease, particularly in breast cancer or mucosal cancers of the head and neck.1 Cutaneous metastasis of colon cancer is uncommon and cutaneous metastasis of pancreatic cancer is rare. Furthermore, nonumbilical sites are much more common than umbilical sites for cutaneous metastatic disease.2 Pancreatic cancer is estimated to be the origin of a cutaneous umbilical metastasis, frequently termed Sister Mary Joseph nodule, in 7% to 9% of cases; colon cancer is estimated to account for 13% to 15% of cases.3 Sister Mary Joseph nodule or sign refers to a nodule often bulging into the umbilicus, signifying metastasis from a
malignant cancer.
In a study of cutaneous metastases, 10% (42/420) of patients with metastatic disease had cutaneous metastasis; 0.48% (2/420) were due to pancreatic cancer and 4.3% (18/420) were due to colon cancer.4 In another review, 63 cases of cutaneous metastasis of pancreatic cancer were found, 43 of which were nonumbilical.2
On immunohistochemistry, CK-7 positivity is highly specific for pancreatic cancer.2 Cytokeratin 7 often is used in conjunction with CK-20 to differentiate various types of glandular tumors. CDX2 is a highly sensitive and specific marker for adenocarcinomas of intestinal origin.5 The negative estrogen receptor, progesterone receptor, mammaglobin, gross cystic disease fluid protein, and GATA3 stains are useful in excluding breast cancer (patient 1 had history of breast cancer).
When cutaneous involvement is present in pancreatic cancer, the disease usually is widespread. Multiple studies have reported involvement of other organs with cutaneous metastasis at rates of 88.9%,6 90.3%,7 and 93.5%.2 However, early recognition of metastatic cancerous lesions can lead to earlier diagnosis and earlier palliative treatment, perhaps prolonging median survival time in patients. In a review of 63 patients with cutaneous metastatic pancreatic cancer, the authors found a median survival time of 5 months, with surgery, chemotherapy, radiation therapy, or a combination helping to improve survival time from a median of 3.0 to 8.3 months.2
The location of lesions and duration of disease in both patients was atypical for arthropod assault. Acyclovir-resistant herpes zoster rarely is reported outside of human immunodeficiency patients; in addition, there was a lack of clear dermatomal distribution. Although cutaneous Crohn disease can manifest as pink papules, it is rare and unlikely as a presenting symptom. Cutaneous sarcoidosis can take many different skin manifestations, and patients can have cutaneous involvement without systemic manifestation. In both patients, medical history was more indicative of metastatic cancer than the other options in the differential diagnosis.
Cutaneous metastasis from colon cancer and pancreatic cancer is rare, and the prognosis is poor in these cases; however, in the appropriate clinical scenario, especially in a patient with a history of cancer, sinister etiologies should be considered for firm red papules of the umbilicus. Skin biopsy coupled with immunohistochemical staining can assist in identifying the primary malignancy.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33:161-165.
- Zhou HY, Wang XB, Gao F, et al. Cutaneous metastasis from pancreatic cancer: a case report and systematic review of the literature [published online October 10, 2014]. Oncol Lett. 2014;8:2654-2660.
- Galvañ VG. Sister Mary Joseph's nodule. Ann Intern Med. 1998;128:410.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Werling RW, Yaziji H, Bacchi CE, et al. CDX2, a highly sensitive and specific marker of adenocarcinomas of intestinal origin: an immnohistochemical survey of 476 primary and metastatic carcinomas. Am J Surg Pathol. 2003;27:303-310.
- Takeuchi H, Kawano T, Toda T, et al. Cutaneous metastasis from pancreatic adenocarcinoma: a case report and a review of the literature. Hepatogastroenterology. 2003;50:275-277.
- Horino K, Hiraoka T, Kanemitsu K, et al. Subcutaneous metastases after curative resection for pancreatic carcinoma: a case report and review of the literature. Pancreas. 1999;19:406-408.
The Diagnosis: Metastatic Cancer
Further workup of patient 1 revealed an alkaline phosphatase level of 743 U/L (reference range, 30–120 U/L), total bilirubin level of 8.5 mg/dL (reference range, 0.3–1.2 mg/dL), and a white blood cell count of 14,000/μL (reference range, 4500–11,000/μL). Computed tomography of the abdomen and pelvis demonstrated cancer of unknown primary site that had metastasized to the colon, liver, and lungs. There was suspicion for potential colon cancer as the primary disease; however, based on the cutaneous findings, a skin biopsy was performed to confirm the diagnosis. Histology and immunohistochemistry revealed adenocarcinoma tumor cells positive for CDX2 (caudal type homeobox 2) and cytokeratin (CK) 7 with a subset positive for CK-20. The cells were negative for estrogen receptor, progesterone receptor, mammaglobin, gross cystic disease fluid protein, and GATA3 (GATA binding protein 3). Immunohistochemistry was most consistent with pancreatic cancer. During palliative percutaneous transhepatic biliary drainage placement, a liver biopsy confirmed the skin biopsy results.
Further workup of patient 2 revealed a white blood cell count of 13,000/μL (reference range, 4500–11,000/μL). Computed tomography of the chest, abdomen, and pelvis revealed metastatic disease to the lungs with a suspicion for colon cancer as the primary site. Biopsy of the skin lesion revealed a mucin-producing adenocarcinoma, and immunohistochemistry was positive for keratin (AE1/AE3), CK-20, and CDX2, consistent with metastatic colon carcinoma. Immunohistochemistry of the biopsied skin lesion was nonreactive for CK-7. The patient had a colonoscopy that revealed a fungating, partially obstructing, circumferential large mass in the ascending colon.
Metastasis to the skin from visceral malignancies is not uncommon and may represent the first evidence of widespread disease, particularly in breast cancer or mucosal cancers of the head and neck.1 Cutaneous metastasis of colon cancer is uncommon and cutaneous metastasis of pancreatic cancer is rare. Furthermore, nonumbilical sites are much more common than umbilical sites for cutaneous metastatic disease.2 Pancreatic cancer is estimated to be the origin of a cutaneous umbilical metastasis, frequently termed Sister Mary Joseph nodule, in 7% to 9% of cases; colon cancer is estimated to account for 13% to 15% of cases.3 Sister Mary Joseph nodule or sign refers to a nodule often bulging into the umbilicus, signifying metastasis from a
malignant cancer.
In a study of cutaneous metastases, 10% (42/420) of patients with metastatic disease had cutaneous metastasis; 0.48% (2/420) were due to pancreatic cancer and 4.3% (18/420) were due to colon cancer.4 In another review, 63 cases of cutaneous metastasis of pancreatic cancer were found, 43 of which were nonumbilical.2
On immunohistochemistry, CK-7 positivity is highly specific for pancreatic cancer.2 Cytokeratin 7 often is used in conjunction with CK-20 to differentiate various types of glandular tumors. CDX2 is a highly sensitive and specific marker for adenocarcinomas of intestinal origin.5 The negative estrogen receptor, progesterone receptor, mammaglobin, gross cystic disease fluid protein, and GATA3 stains are useful in excluding breast cancer (patient 1 had history of breast cancer).
When cutaneous involvement is present in pancreatic cancer, the disease usually is widespread. Multiple studies have reported involvement of other organs with cutaneous metastasis at rates of 88.9%,6 90.3%,7 and 93.5%.2 However, early recognition of metastatic cancerous lesions can lead to earlier diagnosis and earlier palliative treatment, perhaps prolonging median survival time in patients. In a review of 63 patients with cutaneous metastatic pancreatic cancer, the authors found a median survival time of 5 months, with surgery, chemotherapy, radiation therapy, or a combination helping to improve survival time from a median of 3.0 to 8.3 months.2
The location of lesions and duration of disease in both patients was atypical for arthropod assault. Acyclovir-resistant herpes zoster rarely is reported outside of human immunodeficiency patients; in addition, there was a lack of clear dermatomal distribution. Although cutaneous Crohn disease can manifest as pink papules, it is rare and unlikely as a presenting symptom. Cutaneous sarcoidosis can take many different skin manifestations, and patients can have cutaneous involvement without systemic manifestation. In both patients, medical history was more indicative of metastatic cancer than the other options in the differential diagnosis.
Cutaneous metastasis from colon cancer and pancreatic cancer is rare, and the prognosis is poor in these cases; however, in the appropriate clinical scenario, especially in a patient with a history of cancer, sinister etiologies should be considered for firm red papules of the umbilicus. Skin biopsy coupled with immunohistochemical staining can assist in identifying the primary malignancy.
The Diagnosis: Metastatic Cancer
Further workup of patient 1 revealed an alkaline phosphatase level of 743 U/L (reference range, 30–120 U/L), total bilirubin level of 8.5 mg/dL (reference range, 0.3–1.2 mg/dL), and a white blood cell count of 14,000/μL (reference range, 4500–11,000/μL). Computed tomography of the abdomen and pelvis demonstrated cancer of unknown primary site that had metastasized to the colon, liver, and lungs. There was suspicion for potential colon cancer as the primary disease; however, based on the cutaneous findings, a skin biopsy was performed to confirm the diagnosis. Histology and immunohistochemistry revealed adenocarcinoma tumor cells positive for CDX2 (caudal type homeobox 2) and cytokeratin (CK) 7 with a subset positive for CK-20. The cells were negative for estrogen receptor, progesterone receptor, mammaglobin, gross cystic disease fluid protein, and GATA3 (GATA binding protein 3). Immunohistochemistry was most consistent with pancreatic cancer. During palliative percutaneous transhepatic biliary drainage placement, a liver biopsy confirmed the skin biopsy results.
Further workup of patient 2 revealed a white blood cell count of 13,000/μL (reference range, 4500–11,000/μL). Computed tomography of the chest, abdomen, and pelvis revealed metastatic disease to the lungs with a suspicion for colon cancer as the primary site. Biopsy of the skin lesion revealed a mucin-producing adenocarcinoma, and immunohistochemistry was positive for keratin (AE1/AE3), CK-20, and CDX2, consistent with metastatic colon carcinoma. Immunohistochemistry of the biopsied skin lesion was nonreactive for CK-7. The patient had a colonoscopy that revealed a fungating, partially obstructing, circumferential large mass in the ascending colon.
Metastasis to the skin from visceral malignancies is not uncommon and may represent the first evidence of widespread disease, particularly in breast cancer or mucosal cancers of the head and neck.1 Cutaneous metastasis of colon cancer is uncommon and cutaneous metastasis of pancreatic cancer is rare. Furthermore, nonumbilical sites are much more common than umbilical sites for cutaneous metastatic disease.2 Pancreatic cancer is estimated to be the origin of a cutaneous umbilical metastasis, frequently termed Sister Mary Joseph nodule, in 7% to 9% of cases; colon cancer is estimated to account for 13% to 15% of cases.3 Sister Mary Joseph nodule or sign refers to a nodule often bulging into the umbilicus, signifying metastasis from a
malignant cancer.
In a study of cutaneous metastases, 10% (42/420) of patients with metastatic disease had cutaneous metastasis; 0.48% (2/420) were due to pancreatic cancer and 4.3% (18/420) were due to colon cancer.4 In another review, 63 cases of cutaneous metastasis of pancreatic cancer were found, 43 of which were nonumbilical.2
On immunohistochemistry, CK-7 positivity is highly specific for pancreatic cancer.2 Cytokeratin 7 often is used in conjunction with CK-20 to differentiate various types of glandular tumors. CDX2 is a highly sensitive and specific marker for adenocarcinomas of intestinal origin.5 The negative estrogen receptor, progesterone receptor, mammaglobin, gross cystic disease fluid protein, and GATA3 stains are useful in excluding breast cancer (patient 1 had history of breast cancer).
When cutaneous involvement is present in pancreatic cancer, the disease usually is widespread. Multiple studies have reported involvement of other organs with cutaneous metastasis at rates of 88.9%,6 90.3%,7 and 93.5%.2 However, early recognition of metastatic cancerous lesions can lead to earlier diagnosis and earlier palliative treatment, perhaps prolonging median survival time in patients. In a review of 63 patients with cutaneous metastatic pancreatic cancer, the authors found a median survival time of 5 months, with surgery, chemotherapy, radiation therapy, or a combination helping to improve survival time from a median of 3.0 to 8.3 months.2
The location of lesions and duration of disease in both patients was atypical for arthropod assault. Acyclovir-resistant herpes zoster rarely is reported outside of human immunodeficiency patients; in addition, there was a lack of clear dermatomal distribution. Although cutaneous Crohn disease can manifest as pink papules, it is rare and unlikely as a presenting symptom. Cutaneous sarcoidosis can take many different skin manifestations, and patients can have cutaneous involvement without systemic manifestation. In both patients, medical history was more indicative of metastatic cancer than the other options in the differential diagnosis.
Cutaneous metastasis from colon cancer and pancreatic cancer is rare, and the prognosis is poor in these cases; however, in the appropriate clinical scenario, especially in a patient with a history of cancer, sinister etiologies should be considered for firm red papules of the umbilicus. Skin biopsy coupled with immunohistochemical staining can assist in identifying the primary malignancy.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33:161-165.
- Zhou HY, Wang XB, Gao F, et al. Cutaneous metastasis from pancreatic cancer: a case report and systematic review of the literature [published online October 10, 2014]. Oncol Lett. 2014;8:2654-2660.
- Galvañ VG. Sister Mary Joseph's nodule. Ann Intern Med. 1998;128:410.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Werling RW, Yaziji H, Bacchi CE, et al. CDX2, a highly sensitive and specific marker of adenocarcinomas of intestinal origin: an immnohistochemical survey of 476 primary and metastatic carcinomas. Am J Surg Pathol. 2003;27:303-310.
- Takeuchi H, Kawano T, Toda T, et al. Cutaneous metastasis from pancreatic adenocarcinoma: a case report and a review of the literature. Hepatogastroenterology. 2003;50:275-277.
- Horino K, Hiraoka T, Kanemitsu K, et al. Subcutaneous metastases after curative resection for pancreatic carcinoma: a case report and review of the literature. Pancreas. 1999;19:406-408.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33:161-165.
- Zhou HY, Wang XB, Gao F, et al. Cutaneous metastasis from pancreatic cancer: a case report and systematic review of the literature [published online October 10, 2014]. Oncol Lett. 2014;8:2654-2660.
- Galvañ VG. Sister Mary Joseph's nodule. Ann Intern Med. 1998;128:410.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Werling RW, Yaziji H, Bacchi CE, et al. CDX2, a highly sensitive and specific marker of adenocarcinomas of intestinal origin: an immnohistochemical survey of 476 primary and metastatic carcinomas. Am J Surg Pathol. 2003;27:303-310.
- Takeuchi H, Kawano T, Toda T, et al. Cutaneous metastasis from pancreatic adenocarcinoma: a case report and a review of the literature. Hepatogastroenterology. 2003;50:275-277.
- Horino K, Hiraoka T, Kanemitsu K, et al. Subcutaneous metastases after curative resection for pancreatic carcinoma: a case report and review of the literature. Pancreas. 1999;19:406-408.

A 75-year-old woman (patient 1) with a history of localized invasive ductal breast cancer treated definitively with lumpectomy and radiation therapy more than a decade ago presented to the emergency department with jaundice, abdominal pain, weakness, and multiple periumbilical pink-red papules (top) of 2 weeks’ duration. Prior to presentation, the skin lesions did not improve with 10 days of acyclovir treatment prescribed by her primary care physician for presumed herpes zoster.
An 86-year-old man (patient 2) with chronic lymphocytic leukemia treated with ibrutinib presented to the emergency department with jaundice, abdominal pain, weakness, and multiple pink periumbilical papules (bottom) of 6 weeks’ duration. Prior to presentation, the skin lesions did not improve with 21 days of valacyclovir treatment prescribed by his oncologist for presumed herpes zoster.
Small White Spots on the Lips
The Diagnosis: Fordyce Granules
Fordyce granules are prevalent benign anatomic variations that occur in approximately 80% of the population.1 The spots usually present as multiple (usually >10) 1- to 2-mm, painless, yellow-white papules in a symmetric bilateral distribution. They are normal superficial sebaceous glands seen on mucosal surfaces including the oral mucosa, lips, and genitalia. The papules are asymptomatic, and patients often are unaware of their presence. They can appear at any age and can last for months to years. No treatment is indicated, and patients need only reassurance.1
There are several differential diagnoses.2 Granular cell tumors present as solitary, yellowish or pink, slightly indurated, nonmobile, firm masses that usually measure less than 2 cm in diameter and can be associated with local paresthesia. The oral cavity is the second most common site after the skin and usually involves the dorsum of the tongue; however, granular cell tumors also may develop in the substance of the buccal mucosa, lips, or floor of the mouth. On histopathology, the neoplasm is composed of cells with granular cytoplasm that is of neural origin. Granular cell tumors are slow growing and may be present for months. The mean age of onset is in the fourth decade, and females are more likely to be affected. Excisional biopsy is diagnostic and curative.2
Mucoceles of the mouth are solitary, bluish clear, fluctuant, dome-shaped, well-demarcated nodules that usually appear on the lower lip.3 They are caused by rupture of a salivary gland duct due to minor trauma. Mucin is excreted into the surrounding soft tissues, leading to abrupt nontender swelling over the next several weeks. If they originate deeper within the lip they may appear normal in color. Most range from 1 to 2 mm in diameter but can grow to up to several centimeters in size. Other affected sites may include the ventral tongue, posterior buccal mucosa, or soft palate. Excisional biopsy and conservative surgical excision are recommended for diagnosis and management, respectively.3
Oral leukoplakia is a sharply demarcated, white, mucosal plaque that represents either epithelial dysplasia, carcinoma in situ, invasive carcinoma, or hyperkeratosis of unknown etiology. It is a clinical diagnosis of exclusion. The patient may present with a hoarse voice and history of tobacco use. The risk for malignant transformation to squamous cell carcinoma varies from 0% to 20% over the course of 30
years.4 The lesions occur on any mucosal surface, cannot be rubbed off, and usually are asymptomatic.5 The ventral tongue, floor of the mouth, and soft palate are associated with epithelial dysplasia and invasive carcinoma more often than other mucosal sites. There are 2 main types of leukoplakia: localized (unilateral plaque) and proliferative. Because of the risk for cancer, biopsy always is indicated and should be taken from different areas of the lesion (ie, red, verrucous, or nodular areas) if the lesion is nonhomogeneous. Treatment involves excision in the setting of dysplasia or invasive carcinoma. Photodynamic therapy has been shown to reduce the size of oral leukoplakia lesions and is being studied as an alternative therapy.5
Herpes simplex virus type 1 is a common infection of the oral mucosa that classically causes multiple vesicular lesions with an inflammatory erythematous base.6 The lesions are painful and may last for 10 to 14 days. Patients also may develop systemic symptoms such as fever and malaise. Once primary infection with herpes simplex virus has occurred, the virus lives in a latent state in ganglion neurons and can reactivate.6
- Massmanian A, Sorni Valls G, Vera Sempere FJ. Fordyce spots on the glans penis. Br J Dermatol. 1995;133:498-500.
- Lerman M, Freedman PD. Nonneural granular cell tumor of the oral cavity: a case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103:382-384.
- Oka M, Nishioka E, Miyachi R, et al. Case of superficial mucocele of the lower lip. J Dermatol. 2007;34:754-756.
- Lodi G, Sardella A, Bez C, et al. Interventions for treating oral leukoplakia. Cochrane Database Syst Rev. 2006:CD001829.
- Selvam NP, Sadaksharam J, Singaravelu G, et al. Treatment of oral leukoplakia with photodynamic therapy: a pilot study. J Cancer Res Ther. 2015;11:464-467.
- Klein RS. Clinical manifestations and diagnosis of herpes simplex virus type 1 infection. UpToDate website. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-herpes-simplex-virus-type-1-infection.
The Diagnosis: Fordyce Granules
Fordyce granules are prevalent benign anatomic variations that occur in approximately 80% of the population.1 The spots usually present as multiple (usually >10) 1- to 2-mm, painless, yellow-white papules in a symmetric bilateral distribution. They are normal superficial sebaceous glands seen on mucosal surfaces including the oral mucosa, lips, and genitalia. The papules are asymptomatic, and patients often are unaware of their presence. They can appear at any age and can last for months to years. No treatment is indicated, and patients need only reassurance.1
There are several differential diagnoses.2 Granular cell tumors present as solitary, yellowish or pink, slightly indurated, nonmobile, firm masses that usually measure less than 2 cm in diameter and can be associated with local paresthesia. The oral cavity is the second most common site after the skin and usually involves the dorsum of the tongue; however, granular cell tumors also may develop in the substance of the buccal mucosa, lips, or floor of the mouth. On histopathology, the neoplasm is composed of cells with granular cytoplasm that is of neural origin. Granular cell tumors are slow growing and may be present for months. The mean age of onset is in the fourth decade, and females are more likely to be affected. Excisional biopsy is diagnostic and curative.2
Mucoceles of the mouth are solitary, bluish clear, fluctuant, dome-shaped, well-demarcated nodules that usually appear on the lower lip.3 They are caused by rupture of a salivary gland duct due to minor trauma. Mucin is excreted into the surrounding soft tissues, leading to abrupt nontender swelling over the next several weeks. If they originate deeper within the lip they may appear normal in color. Most range from 1 to 2 mm in diameter but can grow to up to several centimeters in size. Other affected sites may include the ventral tongue, posterior buccal mucosa, or soft palate. Excisional biopsy and conservative surgical excision are recommended for diagnosis and management, respectively.3
Oral leukoplakia is a sharply demarcated, white, mucosal plaque that represents either epithelial dysplasia, carcinoma in situ, invasive carcinoma, or hyperkeratosis of unknown etiology. It is a clinical diagnosis of exclusion. The patient may present with a hoarse voice and history of tobacco use. The risk for malignant transformation to squamous cell carcinoma varies from 0% to 20% over the course of 30
years.4 The lesions occur on any mucosal surface, cannot be rubbed off, and usually are asymptomatic.5 The ventral tongue, floor of the mouth, and soft palate are associated with epithelial dysplasia and invasive carcinoma more often than other mucosal sites. There are 2 main types of leukoplakia: localized (unilateral plaque) and proliferative. Because of the risk for cancer, biopsy always is indicated and should be taken from different areas of the lesion (ie, red, verrucous, or nodular areas) if the lesion is nonhomogeneous. Treatment involves excision in the setting of dysplasia or invasive carcinoma. Photodynamic therapy has been shown to reduce the size of oral leukoplakia lesions and is being studied as an alternative therapy.5
Herpes simplex virus type 1 is a common infection of the oral mucosa that classically causes multiple vesicular lesions with an inflammatory erythematous base.6 The lesions are painful and may last for 10 to 14 days. Patients also may develop systemic symptoms such as fever and malaise. Once primary infection with herpes simplex virus has occurred, the virus lives in a latent state in ganglion neurons and can reactivate.6
The Diagnosis: Fordyce Granules
Fordyce granules are prevalent benign anatomic variations that occur in approximately 80% of the population.1 The spots usually present as multiple (usually >10) 1- to 2-mm, painless, yellow-white papules in a symmetric bilateral distribution. They are normal superficial sebaceous glands seen on mucosal surfaces including the oral mucosa, lips, and genitalia. The papules are asymptomatic, and patients often are unaware of their presence. They can appear at any age and can last for months to years. No treatment is indicated, and patients need only reassurance.1
There are several differential diagnoses.2 Granular cell tumors present as solitary, yellowish or pink, slightly indurated, nonmobile, firm masses that usually measure less than 2 cm in diameter and can be associated with local paresthesia. The oral cavity is the second most common site after the skin and usually involves the dorsum of the tongue; however, granular cell tumors also may develop in the substance of the buccal mucosa, lips, or floor of the mouth. On histopathology, the neoplasm is composed of cells with granular cytoplasm that is of neural origin. Granular cell tumors are slow growing and may be present for months. The mean age of onset is in the fourth decade, and females are more likely to be affected. Excisional biopsy is diagnostic and curative.2
Mucoceles of the mouth are solitary, bluish clear, fluctuant, dome-shaped, well-demarcated nodules that usually appear on the lower lip.3 They are caused by rupture of a salivary gland duct due to minor trauma. Mucin is excreted into the surrounding soft tissues, leading to abrupt nontender swelling over the next several weeks. If they originate deeper within the lip they may appear normal in color. Most range from 1 to 2 mm in diameter but can grow to up to several centimeters in size. Other affected sites may include the ventral tongue, posterior buccal mucosa, or soft palate. Excisional biopsy and conservative surgical excision are recommended for diagnosis and management, respectively.3
Oral leukoplakia is a sharply demarcated, white, mucosal plaque that represents either epithelial dysplasia, carcinoma in situ, invasive carcinoma, or hyperkeratosis of unknown etiology. It is a clinical diagnosis of exclusion. The patient may present with a hoarse voice and history of tobacco use. The risk for malignant transformation to squamous cell carcinoma varies from 0% to 20% over the course of 30
years.4 The lesions occur on any mucosal surface, cannot be rubbed off, and usually are asymptomatic.5 The ventral tongue, floor of the mouth, and soft palate are associated with epithelial dysplasia and invasive carcinoma more often than other mucosal sites. There are 2 main types of leukoplakia: localized (unilateral plaque) and proliferative. Because of the risk for cancer, biopsy always is indicated and should be taken from different areas of the lesion (ie, red, verrucous, or nodular areas) if the lesion is nonhomogeneous. Treatment involves excision in the setting of dysplasia or invasive carcinoma. Photodynamic therapy has been shown to reduce the size of oral leukoplakia lesions and is being studied as an alternative therapy.5
Herpes simplex virus type 1 is a common infection of the oral mucosa that classically causes multiple vesicular lesions with an inflammatory erythematous base.6 The lesions are painful and may last for 10 to 14 days. Patients also may develop systemic symptoms such as fever and malaise. Once primary infection with herpes simplex virus has occurred, the virus lives in a latent state in ganglion neurons and can reactivate.6
- Massmanian A, Sorni Valls G, Vera Sempere FJ. Fordyce spots on the glans penis. Br J Dermatol. 1995;133:498-500.
- Lerman M, Freedman PD. Nonneural granular cell tumor of the oral cavity: a case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103:382-384.
- Oka M, Nishioka E, Miyachi R, et al. Case of superficial mucocele of the lower lip. J Dermatol. 2007;34:754-756.
- Lodi G, Sardella A, Bez C, et al. Interventions for treating oral leukoplakia. Cochrane Database Syst Rev. 2006:CD001829.
- Selvam NP, Sadaksharam J, Singaravelu G, et al. Treatment of oral leukoplakia with photodynamic therapy: a pilot study. J Cancer Res Ther. 2015;11:464-467.
- Klein RS. Clinical manifestations and diagnosis of herpes simplex virus type 1 infection. UpToDate website. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-herpes-simplex-virus-type-1-infection.
- Massmanian A, Sorni Valls G, Vera Sempere FJ. Fordyce spots on the glans penis. Br J Dermatol. 1995;133:498-500.
- Lerman M, Freedman PD. Nonneural granular cell tumor of the oral cavity: a case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103:382-384.
- Oka M, Nishioka E, Miyachi R, et al. Case of superficial mucocele of the lower lip. J Dermatol. 2007;34:754-756.
- Lodi G, Sardella A, Bez C, et al. Interventions for treating oral leukoplakia. Cochrane Database Syst Rev. 2006:CD001829.
- Selvam NP, Sadaksharam J, Singaravelu G, et al. Treatment of oral leukoplakia with photodynamic therapy: a pilot study. J Cancer Res Ther. 2015;11:464-467.
- Klein RS. Clinical manifestations and diagnosis of herpes simplex virus type 1 infection. UpToDate website. https://www.uptodate.com/contents/clinical-manifestations-and-diagnosis-of-herpes-simplex-virus-type-1-infection.
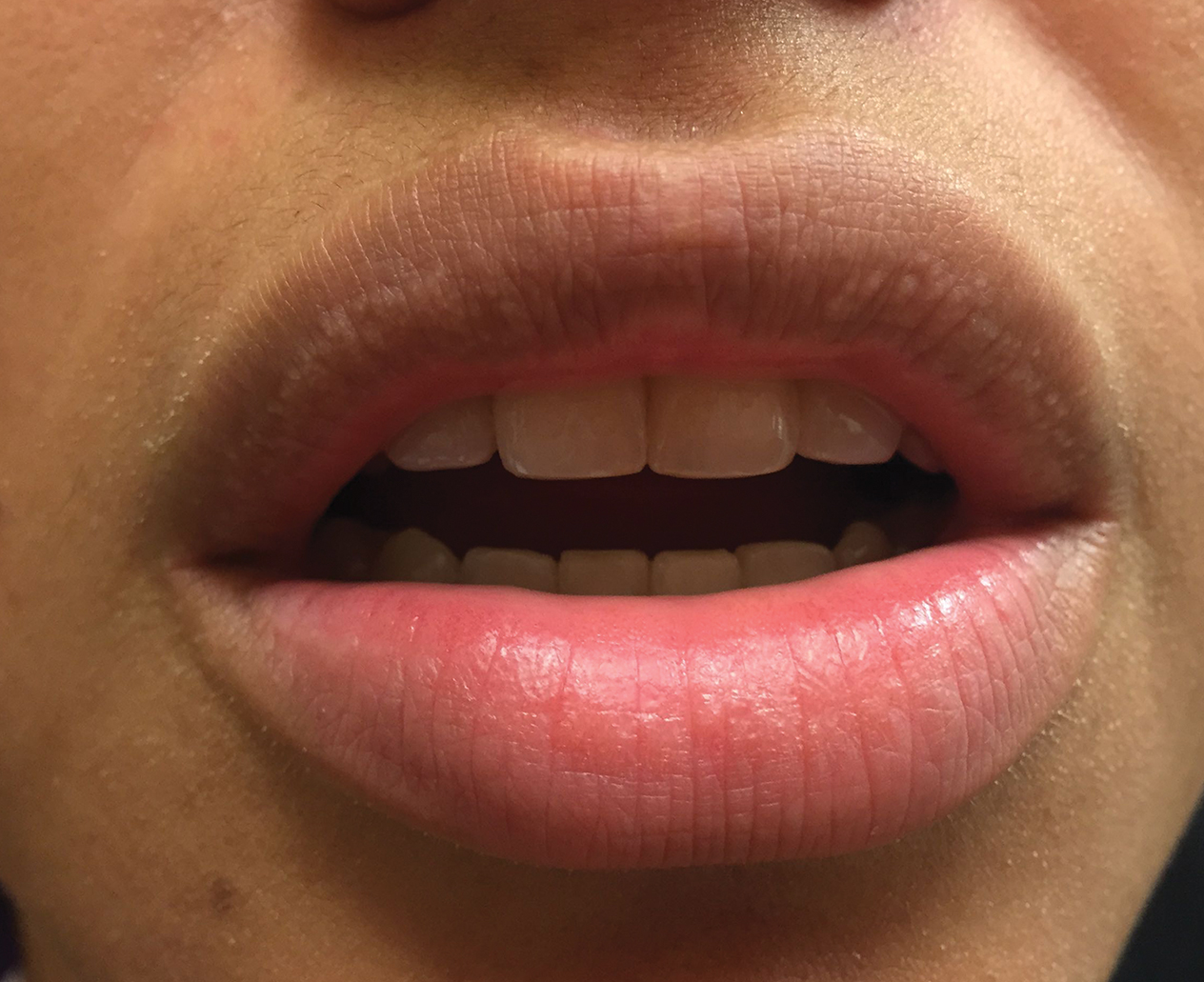
White Concretions on the Hair Shaft
The Diagnosis: White Piedra
A fungal culture demonstrated a filamentous fungus that was identified as Trichosporon inkin via DNA sequencing, which confirmed the diagnosis of white piedra (WP).
Piedra refers to a group of fungal infections presenting as gritty nodules adherent to the hair shaft.1 It is further categorized into black piedra, which occurs more commonly in tropical climates and is caused by Piedraia hortae, and WP, which occurs in tropical and temperate climates and is caused by the Trichosporon genus.1-3 Among the Trichosporon genus, clinical manifestations have varied based on species; for example, T inkin commonly causes genital WP, Trichosporon ovoides commonly causes scalp WP, and Trichosporon asahii and Trichosporon mucoides have been described to cause systemic fungal infections in immunocompromised hosts.1,4 Scalp WP most commonly occurs in children and young adults, and females are at greater risk than males.1,2,5,6
Clinically, WP presents with pale irregular nodules along the hair shaft that are not fluorescent on Wood lamp examination.1,6,7 Nodules are soft and easily detached from the hair shaft, unlike the hard, tightly adherent nodules seen in black piedra.1,7 White piedra affects hair in a variety of areas including the scalp, beard, eyebrows, eyelashes, axillae, and genitals.1,7 Affected hair may become brittle and break at points of invasion.1 Alternatively, WP may resemble tinea capitis with scalp hyperkeratosis and alopecia, though tinea typically affects the base of the hair shaft.1 Immunocompromised patients can develop disseminated WP, and cases of progressive pneumonia, lung abscess, peritonitis, vascular access infection, and endocarditis have been reported.2
Diagnosis of WP is made through a combination of clinical findings and culture of infected hair. Potassium hydroxide preparation demonstrates sleevelike concretions formed of masses of septate hyphae with dense zones of arthrospores and blastospores.1,2 Culture on Sabouraud agar demonstrates creamy colonies that develop a dull, gray, wrinkled surface.1,2 Differential diagnosis includes pediculosis; however, the concretions of WP are circumferential around the hair shaft on microscopy.1 Notably, a case of concomitant WP and pediculosis has been reported.8 In cases of potential pediculosis resistant to therapy, consider hair casts, which are asymptomatic, white, cylindrical concretions that encircle the hair without adherence and can therefore be differentiated from pediculosis via dermoscopy.9 Because this phenomenon is more commonly observed in preadolescent girls, it is hypothesized that scalp inflammation due to traction from hairstyles or atopic dermatitis contributes to the development of hair casts.9,10 Thus, when a potassium hydroxide mount is equivocal for nits and dermoscopy demonstrates concretions that completely encircle the hair shaft, it is important to perform a microbiologic culture to rule out piedra of the hair or scalp. Other differential diagnoses include tinea capitis, black piedra, trichobacteriosis, and hair shaft abnormalities.
Transmission of WP is thought to result from a combination of poor hygiene; humidity due to climate; personal care practices such as habitually tying wet hair, applying hair oils and conditioners, or covering hair according to social customs; and close contact with an infected individual.1,3,6 Long scalp hair potentially correlates with increased risk.1,6 Finally, WP has been described in animals and has been isolated from soil, vegetable matter, and water.3,10
Treatment of WP generally involves removal of infected hair, antifungal agents, and improved hygienic habits to avoid relapses. The American Academy of Dermatology’s Guidelines/Outcomes Committee recommends complete removal of infected hair; however, patients may desire hair-preserving treatments.11 Kiken et al1 reported success with the combination of an oral azole antifungal agent for 3 weeks to 1 month and an antifungal shampoo for 2 to 3 months. The authors proposed that oral medication eliminates scalp carriage while antifungal shampoo eliminates hair shaft concretions.1
1. Kiken DA, Sekaran A, Antaya RJ, et al. White piedra in children. J Am Acad Dermatol. 2006;55:956-961.
2. Bonifaz A, Gómez-Daza F, Paredes V, et al. Tinea versicolor, tinea nigra, white piedra, and black piedra. Clin Dermatol. 2010;28:140-145.
3. Shivaprakash MR, Singh G, Gupta P, et al. Extensive white piedra of the scalp caused by Trichosporon inkin: a case report and review of literature. Mycopathologia. 2011;172:481-486.
4. Goldberg LJ, Wise EM, Miller NS. White piedra caused by Trichosporon inkin: a report of two cases in a northern climate. Br J Dermatol. 2015;173:866-868.
5. Schwartz RA. Superficial fungal infections. Lancet. 2004;364:1173-1182.
6. Fischman O, Bezerra FC, Francisco EC, et al. Trichosporon inkin: an uncommon agent of scalp white piedra. report of four cases in Brazilian children. Mycopathologia. 2014;178:85-89.
7. Pontes ZB, Ramos AL, Lima Ede O, et al. Clinical and mycological study of scalp white piedra in the State of Paraíba, Brazil. Mem Inst Oswaldo Cruz. 2002;97:747-750.
8. Marques SA, Richini-Pereira VB, Camargo RM. White piedra and pediculosis capitis in the same patient. An Bras Dermatol. 2012;87:786-787.
9. Gnarra M, Saraceni P, Rossi A, et al. Challenging diagnosis of peripillous sheaths. Pediatr Dermatol. 2014;31:E112-E113.
10. França K, Villa RT, Silva IR, et al. Hair casts or pseudonits. Int J Trichology. 2011;3:121-122.
11. Guidelines of care for superficial mycotic infections of the skin: piedra. Guidelines/Outcomes Committee. American Academy of Dermatology. J Am Acad Dermatol. 1996;34:122-124.
The Diagnosis: White Piedra
A fungal culture demonstrated a filamentous fungus that was identified as Trichosporon inkin via DNA sequencing, which confirmed the diagnosis of white piedra (WP).
Piedra refers to a group of fungal infections presenting as gritty nodules adherent to the hair shaft.1 It is further categorized into black piedra, which occurs more commonly in tropical climates and is caused by Piedraia hortae, and WP, which occurs in tropical and temperate climates and is caused by the Trichosporon genus.1-3 Among the Trichosporon genus, clinical manifestations have varied based on species; for example, T inkin commonly causes genital WP, Trichosporon ovoides commonly causes scalp WP, and Trichosporon asahii and Trichosporon mucoides have been described to cause systemic fungal infections in immunocompromised hosts.1,4 Scalp WP most commonly occurs in children and young adults, and females are at greater risk than males.1,2,5,6
Clinically, WP presents with pale irregular nodules along the hair shaft that are not fluorescent on Wood lamp examination.1,6,7 Nodules are soft and easily detached from the hair shaft, unlike the hard, tightly adherent nodules seen in black piedra.1,7 White piedra affects hair in a variety of areas including the scalp, beard, eyebrows, eyelashes, axillae, and genitals.1,7 Affected hair may become brittle and break at points of invasion.1 Alternatively, WP may resemble tinea capitis with scalp hyperkeratosis and alopecia, though tinea typically affects the base of the hair shaft.1 Immunocompromised patients can develop disseminated WP, and cases of progressive pneumonia, lung abscess, peritonitis, vascular access infection, and endocarditis have been reported.2
Diagnosis of WP is made through a combination of clinical findings and culture of infected hair. Potassium hydroxide preparation demonstrates sleevelike concretions formed of masses of septate hyphae with dense zones of arthrospores and blastospores.1,2 Culture on Sabouraud agar demonstrates creamy colonies that develop a dull, gray, wrinkled surface.1,2 Differential diagnosis includes pediculosis; however, the concretions of WP are circumferential around the hair shaft on microscopy.1 Notably, a case of concomitant WP and pediculosis has been reported.8 In cases of potential pediculosis resistant to therapy, consider hair casts, which are asymptomatic, white, cylindrical concretions that encircle the hair without adherence and can therefore be differentiated from pediculosis via dermoscopy.9 Because this phenomenon is more commonly observed in preadolescent girls, it is hypothesized that scalp inflammation due to traction from hairstyles or atopic dermatitis contributes to the development of hair casts.9,10 Thus, when a potassium hydroxide mount is equivocal for nits and dermoscopy demonstrates concretions that completely encircle the hair shaft, it is important to perform a microbiologic culture to rule out piedra of the hair or scalp. Other differential diagnoses include tinea capitis, black piedra, trichobacteriosis, and hair shaft abnormalities.
Transmission of WP is thought to result from a combination of poor hygiene; humidity due to climate; personal care practices such as habitually tying wet hair, applying hair oils and conditioners, or covering hair according to social customs; and close contact with an infected individual.1,3,6 Long scalp hair potentially correlates with increased risk.1,6 Finally, WP has been described in animals and has been isolated from soil, vegetable matter, and water.3,10
Treatment of WP generally involves removal of infected hair, antifungal agents, and improved hygienic habits to avoid relapses. The American Academy of Dermatology’s Guidelines/Outcomes Committee recommends complete removal of infected hair; however, patients may desire hair-preserving treatments.11 Kiken et al1 reported success with the combination of an oral azole antifungal agent for 3 weeks to 1 month and an antifungal shampoo for 2 to 3 months. The authors proposed that oral medication eliminates scalp carriage while antifungal shampoo eliminates hair shaft concretions.1
The Diagnosis: White Piedra
A fungal culture demonstrated a filamentous fungus that was identified as Trichosporon inkin via DNA sequencing, which confirmed the diagnosis of white piedra (WP).
Piedra refers to a group of fungal infections presenting as gritty nodules adherent to the hair shaft.1 It is further categorized into black piedra, which occurs more commonly in tropical climates and is caused by Piedraia hortae, and WP, which occurs in tropical and temperate climates and is caused by the Trichosporon genus.1-3 Among the Trichosporon genus, clinical manifestations have varied based on species; for example, T inkin commonly causes genital WP, Trichosporon ovoides commonly causes scalp WP, and Trichosporon asahii and Trichosporon mucoides have been described to cause systemic fungal infections in immunocompromised hosts.1,4 Scalp WP most commonly occurs in children and young adults, and females are at greater risk than males.1,2,5,6
Clinically, WP presents with pale irregular nodules along the hair shaft that are not fluorescent on Wood lamp examination.1,6,7 Nodules are soft and easily detached from the hair shaft, unlike the hard, tightly adherent nodules seen in black piedra.1,7 White piedra affects hair in a variety of areas including the scalp, beard, eyebrows, eyelashes, axillae, and genitals.1,7 Affected hair may become brittle and break at points of invasion.1 Alternatively, WP may resemble tinea capitis with scalp hyperkeratosis and alopecia, though tinea typically affects the base of the hair shaft.1 Immunocompromised patients can develop disseminated WP, and cases of progressive pneumonia, lung abscess, peritonitis, vascular access infection, and endocarditis have been reported.2
Diagnosis of WP is made through a combination of clinical findings and culture of infected hair. Potassium hydroxide preparation demonstrates sleevelike concretions formed of masses of septate hyphae with dense zones of arthrospores and blastospores.1,2 Culture on Sabouraud agar demonstrates creamy colonies that develop a dull, gray, wrinkled surface.1,2 Differential diagnosis includes pediculosis; however, the concretions of WP are circumferential around the hair shaft on microscopy.1 Notably, a case of concomitant WP and pediculosis has been reported.8 In cases of potential pediculosis resistant to therapy, consider hair casts, which are asymptomatic, white, cylindrical concretions that encircle the hair without adherence and can therefore be differentiated from pediculosis via dermoscopy.9 Because this phenomenon is more commonly observed in preadolescent girls, it is hypothesized that scalp inflammation due to traction from hairstyles or atopic dermatitis contributes to the development of hair casts.9,10 Thus, when a potassium hydroxide mount is equivocal for nits and dermoscopy demonstrates concretions that completely encircle the hair shaft, it is important to perform a microbiologic culture to rule out piedra of the hair or scalp. Other differential diagnoses include tinea capitis, black piedra, trichobacteriosis, and hair shaft abnormalities.
Transmission of WP is thought to result from a combination of poor hygiene; humidity due to climate; personal care practices such as habitually tying wet hair, applying hair oils and conditioners, or covering hair according to social customs; and close contact with an infected individual.1,3,6 Long scalp hair potentially correlates with increased risk.1,6 Finally, WP has been described in animals and has been isolated from soil, vegetable matter, and water.3,10
Treatment of WP generally involves removal of infected hair, antifungal agents, and improved hygienic habits to avoid relapses. The American Academy of Dermatology’s Guidelines/Outcomes Committee recommends complete removal of infected hair; however, patients may desire hair-preserving treatments.11 Kiken et al1 reported success with the combination of an oral azole antifungal agent for 3 weeks to 1 month and an antifungal shampoo for 2 to 3 months. The authors proposed that oral medication eliminates scalp carriage while antifungal shampoo eliminates hair shaft concretions.1
1. Kiken DA, Sekaran A, Antaya RJ, et al. White piedra in children. J Am Acad Dermatol. 2006;55:956-961.
2. Bonifaz A, Gómez-Daza F, Paredes V, et al. Tinea versicolor, tinea nigra, white piedra, and black piedra. Clin Dermatol. 2010;28:140-145.
3. Shivaprakash MR, Singh G, Gupta P, et al. Extensive white piedra of the scalp caused by Trichosporon inkin: a case report and review of literature. Mycopathologia. 2011;172:481-486.
4. Goldberg LJ, Wise EM, Miller NS. White piedra caused by Trichosporon inkin: a report of two cases in a northern climate. Br J Dermatol. 2015;173:866-868.
5. Schwartz RA. Superficial fungal infections. Lancet. 2004;364:1173-1182.
6. Fischman O, Bezerra FC, Francisco EC, et al. Trichosporon inkin: an uncommon agent of scalp white piedra. report of four cases in Brazilian children. Mycopathologia. 2014;178:85-89.
7. Pontes ZB, Ramos AL, Lima Ede O, et al. Clinical and mycological study of scalp white piedra in the State of Paraíba, Brazil. Mem Inst Oswaldo Cruz. 2002;97:747-750.
8. Marques SA, Richini-Pereira VB, Camargo RM. White piedra and pediculosis capitis in the same patient. An Bras Dermatol. 2012;87:786-787.
9. Gnarra M, Saraceni P, Rossi A, et al. Challenging diagnosis of peripillous sheaths. Pediatr Dermatol. 2014;31:E112-E113.
10. França K, Villa RT, Silva IR, et al. Hair casts or pseudonits. Int J Trichology. 2011;3:121-122.
11. Guidelines of care for superficial mycotic infections of the skin: piedra. Guidelines/Outcomes Committee. American Academy of Dermatology. J Am Acad Dermatol. 1996;34:122-124.
1. Kiken DA, Sekaran A, Antaya RJ, et al. White piedra in children. J Am Acad Dermatol. 2006;55:956-961.
2. Bonifaz A, Gómez-Daza F, Paredes V, et al. Tinea versicolor, tinea nigra, white piedra, and black piedra. Clin Dermatol. 2010;28:140-145.
3. Shivaprakash MR, Singh G, Gupta P, et al. Extensive white piedra of the scalp caused by Trichosporon inkin: a case report and review of literature. Mycopathologia. 2011;172:481-486.
4. Goldberg LJ, Wise EM, Miller NS. White piedra caused by Trichosporon inkin: a report of two cases in a northern climate. Br J Dermatol. 2015;173:866-868.
5. Schwartz RA. Superficial fungal infections. Lancet. 2004;364:1173-1182.
6. Fischman O, Bezerra FC, Francisco EC, et al. Trichosporon inkin: an uncommon agent of scalp white piedra. report of four cases in Brazilian children. Mycopathologia. 2014;178:85-89.
7. Pontes ZB, Ramos AL, Lima Ede O, et al. Clinical and mycological study of scalp white piedra in the State of Paraíba, Brazil. Mem Inst Oswaldo Cruz. 2002;97:747-750.
8. Marques SA, Richini-Pereira VB, Camargo RM. White piedra and pediculosis capitis in the same patient. An Bras Dermatol. 2012;87:786-787.
9. Gnarra M, Saraceni P, Rossi A, et al. Challenging diagnosis of peripillous sheaths. Pediatr Dermatol. 2014;31:E112-E113.
10. França K, Villa RT, Silva IR, et al. Hair casts or pseudonits. Int J Trichology. 2011;3:121-122.
11. Guidelines of care for superficial mycotic infections of the skin: piedra. Guidelines/Outcomes Committee. American Academy of Dermatology. J Am Acad Dermatol. 1996;34:122-124.

A 35-year-old woman presented with possible nits on the hair of 1 year’s duration. She was previously evaluated by several outside medical providers and was unsuccessfully treated with topical and systemic medications for pediculosis. She reported sporadic scalp pruritus but denied hair loss, breakage, close contacts with similar symptoms, or recent travel outside the United States. She was otherwise healthy and was not taking any medications. Physical examination revealed small 1- to 2-mm, generalized, somewhat detachable, white concretions randomly distributed on the hair shafts. No broken hairs were observed. The eyebrows, eyelash hairs, and surrounding skin were normal. Potassium hydroxide mount was equivocal for nits.

Nonhealing Eroded Plaque on an Interdigital Web Space of the Foot
The Diagnosis: Basal Cell Nevus Syndrome
Given the patient’s history of numerous basal cell carcinomas (BCCs), odontogenic keratocysts, palmar pits, and a nonhealing ulcer, the clinical presentation was highly suggestive of interdigital BCC in the setting of basal cell nevus syndrome (BCNS). A shave biopsy was performed revealing islands of basaloid cells with peripheral palisading and a retraction artifact surrounded by fibromyxoid stroma, consistent with nodular and infiltrative BCC (Figure 1).
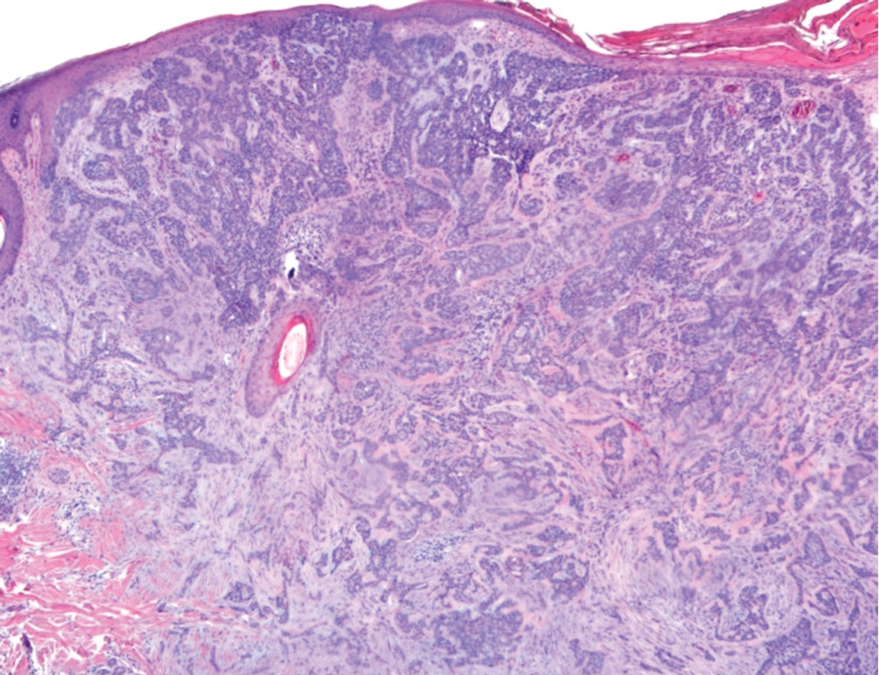
Basal cell nevus syndrome (also known as Gorlin syndrome) is a rare neurocutaneous syndrome that manifests with multiple BCCs; palmar and plantar pits (Figure 2); central nervous system tumors; and skeletal anomalies including jaw cysts, macrocephaly, frontal bossing, and bifid ribs.1 It is an autosomal-dominant condition caused by mutations in the PTCH1 gene, a tumor suppressor gene involved in the Hedgehog signaling pathway.2 Basal cell carcinoma is the most distinctive feature of BCNS, causing notable morbidity. Tumors typically present between puberty and 35 years of age, and patients can have anywhere from a few to thousands of tumors. They rarely become locally aggressive; however, with radiation therapy, proliferation and local invasion may occur within a few years. Therefore, radiotherapy should be avoided in these patients.1
Although the most common sites for BCCs in BCNS are the head, neck, and back, there is a higher rate of occurrence on sun-protected areas in BCNS compared to the general population.3 Our patient presented with interdigital BCC of the foot, which is an extremely rare occurrence. PubMed and Ovid searches using the terms basal cell carcinoma, BCC, foot, interdigital, and nonmelanoma skin cancer revealed only 3 cases of interdigital BCC of the foot. One case was associated with prior surgical trauma, the second presented as a junctional nevus, and the third did not appear to have any associated inciting factors.4-6 Dermatologists need to have a low threshold for biopsy for any unusual nonhealing lesions, especially in the setting of BCNS. Basal cell carcinomas in BCNS cannot be histologically differentiated from sporadic BCCs, and management largely depends on the size, location, recurrence, and number of lesions. Treatment methods range from topical agents to Mohs micrographic surgery.1
Nonhealing lesions of the foot may give an initial clinical impression of infection overlying peripheral vascular disease or diabetes mellitus with the possibility of associated osteomyelitis. Our patient had no clinical history to suggest peripheral vascular disease or diabetes mellitus, and he had palpable dorsalis pedis pulses as well as a normal neurologic examination. Clinicians also may consider fungal infection in the differential diagnosis. Erosio interdigitalis blastomycetica is a superficial yeast infection described as a well-defined, red, shiny plaque found in chronically wet areas, usually affecting the third or fourth interdigital spaces of the fingers.7 However, the lack of improvement with antibiotics and antifungals argued against bacterial or fungal infection in our patient. Although BCC also is a common feature of Bazex Dupré-Christol syndrome, it also is characterized by follicular atrophoderma, milia, hypohidrosis, and hypotrichosis,8 which were not evident in our patient. Pseudomonas hot foot syndrome is characterized by painful, plantar, erythematous nodules after exposure to ontaminated water that typically is self-limited but does respond to antibiotics for Pseudomonas.9
Our patient underwent Mohs micrographic surgery with a complex repair utilizing a full-thickness skin graft. There were no signs of recurrence at 3-month follow-up, and he was counseled on the importance of sun-protective behaviors along with regular dermatologic follow-up.
1. Gorlin RJ. Nevoid basal cell (Gorlin) syndrome. Genet Med. 2004; 6:530-539.
2. Bale A. The nevoid basal cell carcinoma syndrome: genetics and mechanism of carcinogenesis. Cancer Invest. 1997;15:180-186.
3. Goldstein AM, Bale SJ, Peck GL, et al. Sun exposure and basal cell carcinomas in nevoid basal cell carcinoma syndrome. J Am Acad Dermatol. 1993;29:34-41.
4. Silvers SH. Interdigital pedal basal cell carcinoma. Cutis. 1983;31:199-200.
5. Weitzner S. Basal cell carcinoma of toeweb presenting as a junctional nevus. Southwest Med. 1968;49:175.
6. Niwa A, Pimentel E. Basal cell carcinoma in unusual locations. An Bras Dermatol. 2006;81:281-284.
7. Mitchell JH. Erosio interdigitalis blastomycetica. Arch Derm Syphilol. 1922;6:675-679.
8. Kidd A, Carson L, Gregory DW, et al. A Scottish family with Bazex-Dupré-Christol syndrome: follicular atrophoderma, congenital hypotrichosis, and basal cell carcinoma. J Med Genet. 1996;33:493-497.
9. Yu Y, Cheng AS, Wang L, et al. Hot tub folliculitis or hot hand-foot syndrome caused by Pseudomonas aeruginosa. J Am Acad Dermatol. 2007;57:596-600.
The Diagnosis: Basal Cell Nevus Syndrome
Given the patient’s history of numerous basal cell carcinomas (BCCs), odontogenic keratocysts, palmar pits, and a nonhealing ulcer, the clinical presentation was highly suggestive of interdigital BCC in the setting of basal cell nevus syndrome (BCNS). A shave biopsy was performed revealing islands of basaloid cells with peripheral palisading and a retraction artifact surrounded by fibromyxoid stroma, consistent with nodular and infiltrative BCC (Figure 1).
Basal cell nevus syndrome (also known as Gorlin syndrome) is a rare neurocutaneous syndrome that manifests with multiple BCCs; palmar and plantar pits (Figure 2); central nervous system tumors; and skeletal anomalies including jaw cysts, macrocephaly, frontal bossing, and bifid ribs.1 It is an autosomal-dominant condition caused by mutations in the PTCH1 gene, a tumor suppressor gene involved in the Hedgehog signaling pathway.2 Basal cell carcinoma is the most distinctive feature of BCNS, causing notable morbidity. Tumors typically present between puberty and 35 years of age, and patients can have anywhere from a few to thousands of tumors. They rarely become locally aggressive; however, with radiation therapy, proliferation and local invasion may occur within a few years. Therefore, radiotherapy should be avoided in these patients.1
Although the most common sites for BCCs in BCNS are the head, neck, and back, there is a higher rate of occurrence on sun-protected areas in BCNS compared to the general population.3 Our patient presented with interdigital BCC of the foot, which is an extremely rare occurrence. PubMed and Ovid searches using the terms basal cell carcinoma, BCC, foot, interdigital, and nonmelanoma skin cancer revealed only 3 cases of interdigital BCC of the foot. One case was associated with prior surgical trauma, the second presented as a junctional nevus, and the third did not appear to have any associated inciting factors.4-6 Dermatologists need to have a low threshold for biopsy for any unusual nonhealing lesions, especially in the setting of BCNS. Basal cell carcinomas in BCNS cannot be histologically differentiated from sporadic BCCs, and management largely depends on the size, location, recurrence, and number of lesions. Treatment methods range from topical agents to Mohs micrographic surgery.1
Nonhealing lesions of the foot may give an initial clinical impression of infection overlying peripheral vascular disease or diabetes mellitus with the possibility of associated osteomyelitis. Our patient had no clinical history to suggest peripheral vascular disease or diabetes mellitus, and he had palpable dorsalis pedis pulses as well as a normal neurologic examination. Clinicians also may consider fungal infection in the differential diagnosis. Erosio interdigitalis blastomycetica is a superficial yeast infection described as a well-defined, red, shiny plaque found in chronically wet areas, usually affecting the third or fourth interdigital spaces of the fingers.7 However, the lack of improvement with antibiotics and antifungals argued against bacterial or fungal infection in our patient. Although BCC also is a common feature of Bazex Dupré-Christol syndrome, it also is characterized by follicular atrophoderma, milia, hypohidrosis, and hypotrichosis,8 which were not evident in our patient. Pseudomonas hot foot syndrome is characterized by painful, plantar, erythematous nodules after exposure to ontaminated water that typically is self-limited but does respond to antibiotics for Pseudomonas.9
Our patient underwent Mohs micrographic surgery with a complex repair utilizing a full-thickness skin graft. There were no signs of recurrence at 3-month follow-up, and he was counseled on the importance of sun-protective behaviors along with regular dermatologic follow-up.
The Diagnosis: Basal Cell Nevus Syndrome
Given the patient’s history of numerous basal cell carcinomas (BCCs), odontogenic keratocysts, palmar pits, and a nonhealing ulcer, the clinical presentation was highly suggestive of interdigital BCC in the setting of basal cell nevus syndrome (BCNS). A shave biopsy was performed revealing islands of basaloid cells with peripheral palisading and a retraction artifact surrounded by fibromyxoid stroma, consistent with nodular and infiltrative BCC (Figure 1).
Basal cell nevus syndrome (also known as Gorlin syndrome) is a rare neurocutaneous syndrome that manifests with multiple BCCs; palmar and plantar pits (Figure 2); central nervous system tumors; and skeletal anomalies including jaw cysts, macrocephaly, frontal bossing, and bifid ribs.1 It is an autosomal-dominant condition caused by mutations in the PTCH1 gene, a tumor suppressor gene involved in the Hedgehog signaling pathway.2 Basal cell carcinoma is the most distinctive feature of BCNS, causing notable morbidity. Tumors typically present between puberty and 35 years of age, and patients can have anywhere from a few to thousands of tumors. They rarely become locally aggressive; however, with radiation therapy, proliferation and local invasion may occur within a few years. Therefore, radiotherapy should be avoided in these patients.1
Although the most common sites for BCCs in BCNS are the head, neck, and back, there is a higher rate of occurrence on sun-protected areas in BCNS compared to the general population.3 Our patient presented with interdigital BCC of the foot, which is an extremely rare occurrence. PubMed and Ovid searches using the terms basal cell carcinoma, BCC, foot, interdigital, and nonmelanoma skin cancer revealed only 3 cases of interdigital BCC of the foot. One case was associated with prior surgical trauma, the second presented as a junctional nevus, and the third did not appear to have any associated inciting factors.4-6 Dermatologists need to have a low threshold for biopsy for any unusual nonhealing lesions, especially in the setting of BCNS. Basal cell carcinomas in BCNS cannot be histologically differentiated from sporadic BCCs, and management largely depends on the size, location, recurrence, and number of lesions. Treatment methods range from topical agents to Mohs micrographic surgery.1
Nonhealing lesions of the foot may give an initial clinical impression of infection overlying peripheral vascular disease or diabetes mellitus with the possibility of associated osteomyelitis. Our patient had no clinical history to suggest peripheral vascular disease or diabetes mellitus, and he had palpable dorsalis pedis pulses as well as a normal neurologic examination. Clinicians also may consider fungal infection in the differential diagnosis. Erosio interdigitalis blastomycetica is a superficial yeast infection described as a well-defined, red, shiny plaque found in chronically wet areas, usually affecting the third or fourth interdigital spaces of the fingers.7 However, the lack of improvement with antibiotics and antifungals argued against bacterial or fungal infection in our patient. Although BCC also is a common feature of Bazex Dupré-Christol syndrome, it also is characterized by follicular atrophoderma, milia, hypohidrosis, and hypotrichosis,8 which were not evident in our patient. Pseudomonas hot foot syndrome is characterized by painful, plantar, erythematous nodules after exposure to ontaminated water that typically is self-limited but does respond to antibiotics for Pseudomonas.9
Our patient underwent Mohs micrographic surgery with a complex repair utilizing a full-thickness skin graft. There were no signs of recurrence at 3-month follow-up, and he was counseled on the importance of sun-protective behaviors along with regular dermatologic follow-up.
1. Gorlin RJ. Nevoid basal cell (Gorlin) syndrome. Genet Med. 2004; 6:530-539.
2. Bale A. The nevoid basal cell carcinoma syndrome: genetics and mechanism of carcinogenesis. Cancer Invest. 1997;15:180-186.
3. Goldstein AM, Bale SJ, Peck GL, et al. Sun exposure and basal cell carcinomas in nevoid basal cell carcinoma syndrome. J Am Acad Dermatol. 1993;29:34-41.
4. Silvers SH. Interdigital pedal basal cell carcinoma. Cutis. 1983;31:199-200.
5. Weitzner S. Basal cell carcinoma of toeweb presenting as a junctional nevus. Southwest Med. 1968;49:175.
6. Niwa A, Pimentel E. Basal cell carcinoma in unusual locations. An Bras Dermatol. 2006;81:281-284.
7. Mitchell JH. Erosio interdigitalis blastomycetica. Arch Derm Syphilol. 1922;6:675-679.
8. Kidd A, Carson L, Gregory DW, et al. A Scottish family with Bazex-Dupré-Christol syndrome: follicular atrophoderma, congenital hypotrichosis, and basal cell carcinoma. J Med Genet. 1996;33:493-497.
9. Yu Y, Cheng AS, Wang L, et al. Hot tub folliculitis or hot hand-foot syndrome caused by Pseudomonas aeruginosa. J Am Acad Dermatol. 2007;57:596-600.
1. Gorlin RJ. Nevoid basal cell (Gorlin) syndrome. Genet Med. 2004; 6:530-539.
2. Bale A. The nevoid basal cell carcinoma syndrome: genetics and mechanism of carcinogenesis. Cancer Invest. 1997;15:180-186.
3. Goldstein AM, Bale SJ, Peck GL, et al. Sun exposure and basal cell carcinomas in nevoid basal cell carcinoma syndrome. J Am Acad Dermatol. 1993;29:34-41.
4. Silvers SH. Interdigital pedal basal cell carcinoma. Cutis. 1983;31:199-200.
5. Weitzner S. Basal cell carcinoma of toeweb presenting as a junctional nevus. Southwest Med. 1968;49:175.
6. Niwa A, Pimentel E. Basal cell carcinoma in unusual locations. An Bras Dermatol. 2006;81:281-284.
7. Mitchell JH. Erosio interdigitalis blastomycetica. Arch Derm Syphilol. 1922;6:675-679.
8. Kidd A, Carson L, Gregory DW, et al. A Scottish family with Bazex-Dupré-Christol syndrome: follicular atrophoderma, congenital hypotrichosis, and basal cell carcinoma. J Med Genet. 1996;33:493-497.
9. Yu Y, Cheng AS, Wang L, et al. Hot tub folliculitis or hot hand-foot syndrome caused by Pseudomonas aeruginosa. J Am Acad Dermatol. 2007;57:596-600.
A 53-year-old man with a history of numerous basal cell carcinomas and odontogenic keratocysts presented with a nonhealing erosion between the left second and third toes of several months’ duration. He was treated empirically with multiple courses of topical and systemic antibiotics as well as antifungals with minimal improvement. Physical examination revealed a 1.2×0.6-cm eroded plaque with rolled borders on the left second toe web; bilateral palmar pits; diffuse actinic damage; and several well-healed surgical scars on the head, neck, and back. Neurologic examination was normal, and dorsalis pedis pulses were equal and palpable bilaterally.
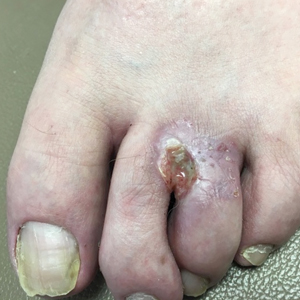
Ascending Erythematous Nodules on the Arm
The Diagnosis: Primary Cutaneous Nocardiosis
Comprehensive metabolic panel and complete blood cell count were unremarkable; human immunodeficiency virus screening was nonreactive. Punch biopsies were obtained for histopathology, as well as bacterial, fungal, and mycobacterial cultures. Histopathologic examination of a 4-mm punch biopsy of the forearm nodule showed a dermal abscess with neutrophilic infiltration in the dermis (Figure 1). No organisms were seen on Gram, methenamine-silver, periodic acid–Schiff, or acid-fast bacteria stains. Given the clinical suspicion for lymphocutaneous sporotrichosis, the patient was started on itraconazole. She reported modest improvement but subsequently developed a morbilliform eruption necessitating medication discontinuation.
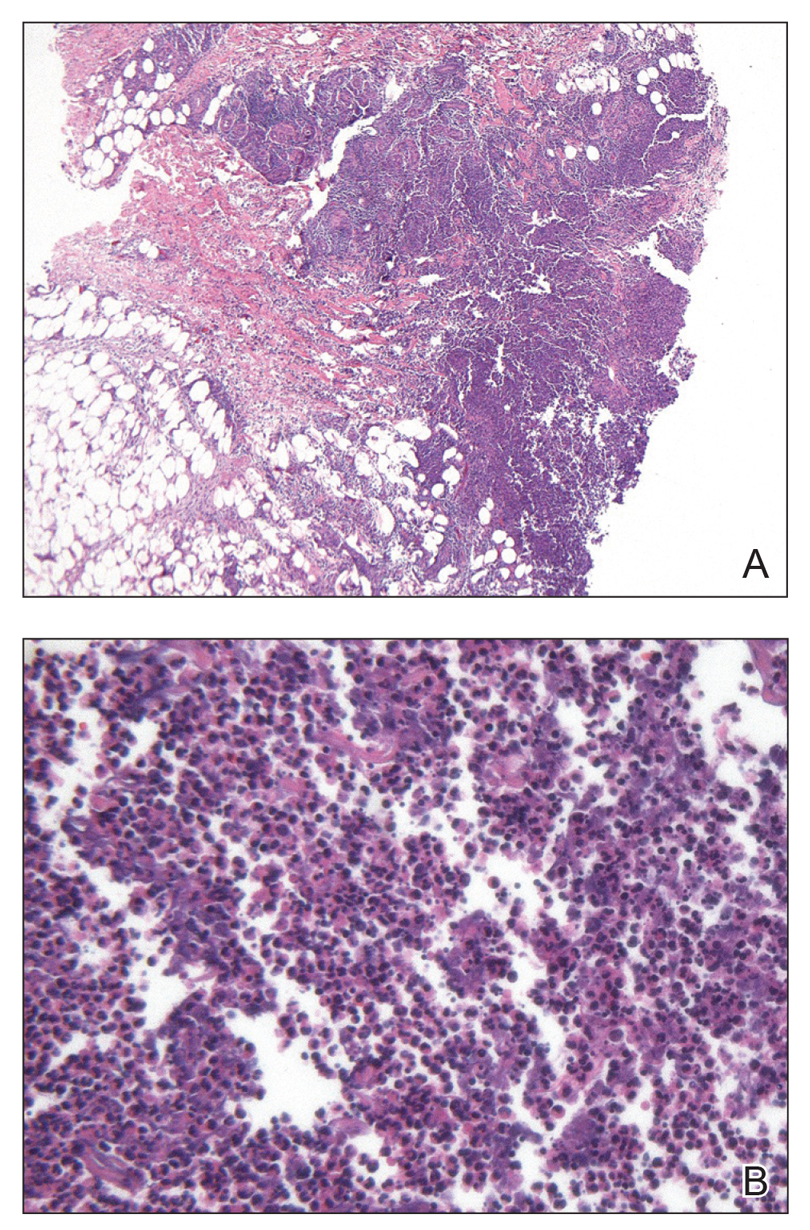
Eighteen days after obtaining the tissue culture, acid-fast organisms grew in culture. These organisms were subcultured on Middlebrook 7H11 agar (Sigma-Aldrich) with growth noted at 30°C and 37°C. Gram stain revealed filamentous gram-variable bacteria (Figure 2) that were identified as Nocardia brasiliensis by 16S ribosomal DNA analysis. Given the patient’s sulfonamide allergy, she started oral minocycline 100 mg twice daily. She responded to the therapy and subsequent testing confirmed susceptibility.
Nocardia brasiliensis, isolated from subculture on Gram stain (original
magnification ×1000).
The genus Nocardia consists of more than 50 species of gram-positive, weakly acid-fast, aerobic actinomycetes that can cause primary cutaneous infection via percutaneous inoculation. Nocardia brasiliensis is the leading cause (approximately 80% of cases) of primary cutaneous or subcutaneous nocardiosis and is found ubiquitously in soil and decaying vegetation.1 The clinical presentation varies, rendering definitive diagnosis a challenge without histopathologic and microbiologic testing.2 Patients presenting with nocardial cellulitis often are suspected to have Streptococcus pyogenes or Staphylococcus aureus infections. The differential diagnosis for patients presenting with nocardial nodular lymphangitis, also known as lymphocutaneous syndrome, includes atypical mycobacterial infections, leishmaniasis, and lymphocutaneous sporotrichosis.2
Histologic examination of nocardial nodules typically shows granulomatous or neutrophilic inflammation, and organisms may appear in small collections resembling sulfur granules.2 The organism itself is weakly positive on acid-fast stain, and useful stains include acid-fast bacteria, methenamine silver, and periodic acid–Schiff.2 Tissue culture often provides the definitive diagnosis, as the histology is nonspecific and organisms may not be visualized.
Oral trimethoprim-sulfamethoxazole 2.5 to 10 mg/kg and 12.5 to 50 mg/kg, respectively, twice daily is the treatment of choice for primary cutaneous nocardiosis. Minocycline 100 to 200 mg twice daily is an accepted alternative in case of sulfonamide allergy, as in our patient. Antibiotics should be tailored according to the susceptibility profile of the isolated organism.3
This case highlights the importance of forming a broad differential diagnosis for patients presenting with lymphocutaneous syndrome. The incidence and prevalence of N brasiliensis infection is difficult to determine due to its nonspecific clinical presentation and a lack of recent epidemiologic studies. Although primary cutaneous nocardiosis in the United States often is diagnosed in the South or Southwest, cases have been reported in other regions.4-6 Traumatic inoculation of contaminated soil, plants, and other organic matter, a well-known method of Sporothrix schenckii transmission, also is a method of N brasiliensis transmission. Because this organism may not be detected on histologic examination, empiric treatment should be considered if the diagnosis is suspected.
1. Brown-Eliot BA, Brown JM, Conville PS, et al. Clinical and laboratory features of the Nocardia spp. based on current molecular taxonomy. Clin Microbiol Rev. 2006;19:259-282.
2. Smego RA Jr, Castiglia M, Asperilla MO. Lymphocutaneous syndrome: a review of non-sporothrix causes. Medicine. 1999;78:38-63.
3. Lerner P. Nocardiosis. Clin Infect Dis. 1996;22:891-903.
4. Smego RA Jr, Gallis HA. The clinical spectrum of Nocardia brasiliensis infection in the United States. Rev Infect Dis. 1984;6:164-180.
5. Fukuda H, Saotome A, Usami N, et al. Lymphocutaneous type of nocardiosis caused by Nocardia brasiliensis: a case report and review of primary cutaneous nocardiosis caused by N. brasiliensis reported in Japan. J Dermatol. 2008;35:346-353.
6. Kil EH, Tsai CL, Kwark EH, et al. A case of nocardiosis with an uncharacteristically long incubation period. Cutis. 2005;76:33-36.
The Diagnosis: Primary Cutaneous Nocardiosis
Comprehensive metabolic panel and complete blood cell count were unremarkable; human immunodeficiency virus screening was nonreactive. Punch biopsies were obtained for histopathology, as well as bacterial, fungal, and mycobacterial cultures. Histopathologic examination of a 4-mm punch biopsy of the forearm nodule showed a dermal abscess with neutrophilic infiltration in the dermis (Figure 1). No organisms were seen on Gram, methenamine-silver, periodic acid–Schiff, or acid-fast bacteria stains. Given the clinical suspicion for lymphocutaneous sporotrichosis, the patient was started on itraconazole. She reported modest improvement but subsequently developed a morbilliform eruption necessitating medication discontinuation.
Eighteen days after obtaining the tissue culture, acid-fast organisms grew in culture. These organisms were subcultured on Middlebrook 7H11 agar (Sigma-Aldrich) with growth noted at 30°C and 37°C. Gram stain revealed filamentous gram-variable bacteria (Figure 2) that were identified as Nocardia brasiliensis by 16S ribosomal DNA analysis. Given the patient’s sulfonamide allergy, she started oral minocycline 100 mg twice daily. She responded to the therapy and subsequent testing confirmed susceptibility.
Nocardia brasiliensis, isolated from subculture on Gram stain (original
magnification ×1000).
The genus Nocardia consists of more than 50 species of gram-positive, weakly acid-fast, aerobic actinomycetes that can cause primary cutaneous infection via percutaneous inoculation. Nocardia brasiliensis is the leading cause (approximately 80% of cases) of primary cutaneous or subcutaneous nocardiosis and is found ubiquitously in soil and decaying vegetation.1 The clinical presentation varies, rendering definitive diagnosis a challenge without histopathologic and microbiologic testing.2 Patients presenting with nocardial cellulitis often are suspected to have Streptococcus pyogenes or Staphylococcus aureus infections. The differential diagnosis for patients presenting with nocardial nodular lymphangitis, also known as lymphocutaneous syndrome, includes atypical mycobacterial infections, leishmaniasis, and lymphocutaneous sporotrichosis.2
Histologic examination of nocardial nodules typically shows granulomatous or neutrophilic inflammation, and organisms may appear in small collections resembling sulfur granules.2 The organism itself is weakly positive on acid-fast stain, and useful stains include acid-fast bacteria, methenamine silver, and periodic acid–Schiff.2 Tissue culture often provides the definitive diagnosis, as the histology is nonspecific and organisms may not be visualized.
Oral trimethoprim-sulfamethoxazole 2.5 to 10 mg/kg and 12.5 to 50 mg/kg, respectively, twice daily is the treatment of choice for primary cutaneous nocardiosis. Minocycline 100 to 200 mg twice daily is an accepted alternative in case of sulfonamide allergy, as in our patient. Antibiotics should be tailored according to the susceptibility profile of the isolated organism.3
This case highlights the importance of forming a broad differential diagnosis for patients presenting with lymphocutaneous syndrome. The incidence and prevalence of N brasiliensis infection is difficult to determine due to its nonspecific clinical presentation and a lack of recent epidemiologic studies. Although primary cutaneous nocardiosis in the United States often is diagnosed in the South or Southwest, cases have been reported in other regions.4-6 Traumatic inoculation of contaminated soil, plants, and other organic matter, a well-known method of Sporothrix schenckii transmission, also is a method of N brasiliensis transmission. Because this organism may not be detected on histologic examination, empiric treatment should be considered if the diagnosis is suspected.
The Diagnosis: Primary Cutaneous Nocardiosis
Comprehensive metabolic panel and complete blood cell count were unremarkable; human immunodeficiency virus screening was nonreactive. Punch biopsies were obtained for histopathology, as well as bacterial, fungal, and mycobacterial cultures. Histopathologic examination of a 4-mm punch biopsy of the forearm nodule showed a dermal abscess with neutrophilic infiltration in the dermis (Figure 1). No organisms were seen on Gram, methenamine-silver, periodic acid–Schiff, or acid-fast bacteria stains. Given the clinical suspicion for lymphocutaneous sporotrichosis, the patient was started on itraconazole. She reported modest improvement but subsequently developed a morbilliform eruption necessitating medication discontinuation.
Eighteen days after obtaining the tissue culture, acid-fast organisms grew in culture. These organisms were subcultured on Middlebrook 7H11 agar (Sigma-Aldrich) with growth noted at 30°C and 37°C. Gram stain revealed filamentous gram-variable bacteria (Figure 2) that were identified as Nocardia brasiliensis by 16S ribosomal DNA analysis. Given the patient’s sulfonamide allergy, she started oral minocycline 100 mg twice daily. She responded to the therapy and subsequent testing confirmed susceptibility.
Nocardia brasiliensis, isolated from subculture on Gram stain (original
magnification ×1000).
The genus Nocardia consists of more than 50 species of gram-positive, weakly acid-fast, aerobic actinomycetes that can cause primary cutaneous infection via percutaneous inoculation. Nocardia brasiliensis is the leading cause (approximately 80% of cases) of primary cutaneous or subcutaneous nocardiosis and is found ubiquitously in soil and decaying vegetation.1 The clinical presentation varies, rendering definitive diagnosis a challenge without histopathologic and microbiologic testing.2 Patients presenting with nocardial cellulitis often are suspected to have Streptococcus pyogenes or Staphylococcus aureus infections. The differential diagnosis for patients presenting with nocardial nodular lymphangitis, also known as lymphocutaneous syndrome, includes atypical mycobacterial infections, leishmaniasis, and lymphocutaneous sporotrichosis.2
Histologic examination of nocardial nodules typically shows granulomatous or neutrophilic inflammation, and organisms may appear in small collections resembling sulfur granules.2 The organism itself is weakly positive on acid-fast stain, and useful stains include acid-fast bacteria, methenamine silver, and periodic acid–Schiff.2 Tissue culture often provides the definitive diagnosis, as the histology is nonspecific and organisms may not be visualized.
Oral trimethoprim-sulfamethoxazole 2.5 to 10 mg/kg and 12.5 to 50 mg/kg, respectively, twice daily is the treatment of choice for primary cutaneous nocardiosis. Minocycline 100 to 200 mg twice daily is an accepted alternative in case of sulfonamide allergy, as in our patient. Antibiotics should be tailored according to the susceptibility profile of the isolated organism.3
This case highlights the importance of forming a broad differential diagnosis for patients presenting with lymphocutaneous syndrome. The incidence and prevalence of N brasiliensis infection is difficult to determine due to its nonspecific clinical presentation and a lack of recent epidemiologic studies. Although primary cutaneous nocardiosis in the United States often is diagnosed in the South or Southwest, cases have been reported in other regions.4-6 Traumatic inoculation of contaminated soil, plants, and other organic matter, a well-known method of Sporothrix schenckii transmission, also is a method of N brasiliensis transmission. Because this organism may not be detected on histologic examination, empiric treatment should be considered if the diagnosis is suspected.
1. Brown-Eliot BA, Brown JM, Conville PS, et al. Clinical and laboratory features of the Nocardia spp. based on current molecular taxonomy. Clin Microbiol Rev. 2006;19:259-282.
2. Smego RA Jr, Castiglia M, Asperilla MO. Lymphocutaneous syndrome: a review of non-sporothrix causes. Medicine. 1999;78:38-63.
3. Lerner P. Nocardiosis. Clin Infect Dis. 1996;22:891-903.
4. Smego RA Jr, Gallis HA. The clinical spectrum of Nocardia brasiliensis infection in the United States. Rev Infect Dis. 1984;6:164-180.
5. Fukuda H, Saotome A, Usami N, et al. Lymphocutaneous type of nocardiosis caused by Nocardia brasiliensis: a case report and review of primary cutaneous nocardiosis caused by N. brasiliensis reported in Japan. J Dermatol. 2008;35:346-353.
6. Kil EH, Tsai CL, Kwark EH, et al. A case of nocardiosis with an uncharacteristically long incubation period. Cutis. 2005;76:33-36.
1. Brown-Eliot BA, Brown JM, Conville PS, et al. Clinical and laboratory features of the Nocardia spp. based on current molecular taxonomy. Clin Microbiol Rev. 2006;19:259-282.
2. Smego RA Jr, Castiglia M, Asperilla MO. Lymphocutaneous syndrome: a review of non-sporothrix causes. Medicine. 1999;78:38-63.
3. Lerner P. Nocardiosis. Clin Infect Dis. 1996;22:891-903.
4. Smego RA Jr, Gallis HA. The clinical spectrum of Nocardia brasiliensis infection in the United States. Rev Infect Dis. 1984;6:164-180.
5. Fukuda H, Saotome A, Usami N, et al. Lymphocutaneous type of nocardiosis caused by Nocardia brasiliensis: a case report and review of primary cutaneous nocardiosis caused by N. brasiliensis reported in Japan. J Dermatol. 2008;35:346-353.
6. Kil EH, Tsai CL, Kwark EH, et al. A case of nocardiosis with an uncharacteristically long incubation period. Cutis. 2005;76:33-36.
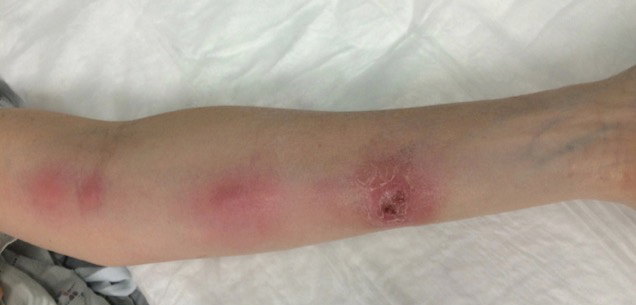
A 54-year-old woman called her primary care provider to report a painful pink nodule on the left wrist 1 week after sustaining thorn injuries while weeding in her garden. She started cephalexin and noted a pink streak with additional nodules extending up the arm over the next 2 days. She
was admitted to an outside hospital for incision and drainage of the wrist nodule and a 3-day course of intravenous vancomycin. Bacterial culture was negative, and she was discharged on oral clindamycin and doxycycline. Two days later, she presented to our emergency department with pain in the left axilla. Physical examination revealed 3 tender erythematous nodules in a linear distribution on the left arm with crusting at the incision and drainage site and painful left axillary lymphadenopathy. The patient was afebrile and otherwise asymptomatic.