User login
New guideline for managing MCL
Rituximab should be included in first-line chemotherapy when treating mantle cell lymphoma, according to a new management guideline from the British Society for Haematology.
The best outcome data is for the R-CHOP regimen (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) followed by maintenance treatment with rituximab, wrote Pamela McKay, MD, of Beatson West of Scotland Cancer Centre in Glasgow, and her colleagues. The report was published in the British Journal of Haematology. But the combination of rituximab and bendamustine is also effective and a more favorable safety profile, according to the guideline. Single agent rituximab is not recommended.
At relapse, the guideline calls on physicians to take an individualized approach based on age, comorbidities, performance status, and response to prior therapy. Some options to consider include ibrutinib as a single agent or rituximab plus chemotherapy. The authors cautioned that there is little evidence to support maintenance rituximab after relapse treatment.
The guideline also explores the role of autologous stem cell transplantation (ASCT) and allogeneic SCT (alloSCT). The authors recommend that ASCT be considered as consolidation of first-line therapy for patients who are fit for intensive therapy. AlloSCT is a viable option in second remission among fit patients who have an appropriate donor and it may also be effective as a rescue therapy for patients who relapse after ASCT. But alloSCT is appropriate only as a first-line therapy for high-risk patients and is best used as part of a clinical trial, according to the recommendations.
The British Society of Haematology previously issued guidance on mantle cell lymphoma in 2012, but the updated document includes new drug therapeutic options and transplant data. The guideline includes a therapeutic algorithm to assist physicians in choosing first-line therapy, options after first relapse, and management in the case of higher relapse.
The guideline authors reported having no conflicts of interest.
SOURCE: McKay P et al. Br J Haematol. 2018 Jul;182(1):46-62.
Rituximab should be included in first-line chemotherapy when treating mantle cell lymphoma, according to a new management guideline from the British Society for Haematology.
The best outcome data is for the R-CHOP regimen (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) followed by maintenance treatment with rituximab, wrote Pamela McKay, MD, of Beatson West of Scotland Cancer Centre in Glasgow, and her colleagues. The report was published in the British Journal of Haematology. But the combination of rituximab and bendamustine is also effective and a more favorable safety profile, according to the guideline. Single agent rituximab is not recommended.
At relapse, the guideline calls on physicians to take an individualized approach based on age, comorbidities, performance status, and response to prior therapy. Some options to consider include ibrutinib as a single agent or rituximab plus chemotherapy. The authors cautioned that there is little evidence to support maintenance rituximab after relapse treatment.
The guideline also explores the role of autologous stem cell transplantation (ASCT) and allogeneic SCT (alloSCT). The authors recommend that ASCT be considered as consolidation of first-line therapy for patients who are fit for intensive therapy. AlloSCT is a viable option in second remission among fit patients who have an appropriate donor and it may also be effective as a rescue therapy for patients who relapse after ASCT. But alloSCT is appropriate only as a first-line therapy for high-risk patients and is best used as part of a clinical trial, according to the recommendations.
The British Society of Haematology previously issued guidance on mantle cell lymphoma in 2012, but the updated document includes new drug therapeutic options and transplant data. The guideline includes a therapeutic algorithm to assist physicians in choosing first-line therapy, options after first relapse, and management in the case of higher relapse.
The guideline authors reported having no conflicts of interest.
SOURCE: McKay P et al. Br J Haematol. 2018 Jul;182(1):46-62.
Rituximab should be included in first-line chemotherapy when treating mantle cell lymphoma, according to a new management guideline from the British Society for Haematology.
The best outcome data is for the R-CHOP regimen (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone) followed by maintenance treatment with rituximab, wrote Pamela McKay, MD, of Beatson West of Scotland Cancer Centre in Glasgow, and her colleagues. The report was published in the British Journal of Haematology. But the combination of rituximab and bendamustine is also effective and a more favorable safety profile, according to the guideline. Single agent rituximab is not recommended.
At relapse, the guideline calls on physicians to take an individualized approach based on age, comorbidities, performance status, and response to prior therapy. Some options to consider include ibrutinib as a single agent or rituximab plus chemotherapy. The authors cautioned that there is little evidence to support maintenance rituximab after relapse treatment.
The guideline also explores the role of autologous stem cell transplantation (ASCT) and allogeneic SCT (alloSCT). The authors recommend that ASCT be considered as consolidation of first-line therapy for patients who are fit for intensive therapy. AlloSCT is a viable option in second remission among fit patients who have an appropriate donor and it may also be effective as a rescue therapy for patients who relapse after ASCT. But alloSCT is appropriate only as a first-line therapy for high-risk patients and is best used as part of a clinical trial, according to the recommendations.
The British Society of Haematology previously issued guidance on mantle cell lymphoma in 2012, but the updated document includes new drug therapeutic options and transplant data. The guideline includes a therapeutic algorithm to assist physicians in choosing first-line therapy, options after first relapse, and management in the case of higher relapse.
The guideline authors reported having no conflicts of interest.
SOURCE: McKay P et al. Br J Haematol. 2018 Jul;182(1):46-62.
FROM THE BRITISH JOURNAL OF HAEMATOLOGY
SAMHSA Releases Money for Opioid Treatment
The Targeted Capacity Expansion: Medication Assisted Treatment-Prescription Drug Opioid Addiction grant program will expand access to treatment and recovery support services in states, tribes, and tribal organizations with the highest per-capita rates of primary treatment admissions for heroin and prescription opioids. The funding includes the areas with the “most dramatic increases” for heroin and prescription opioids, as identified by SAMHSA’s 2015 Treatment Episode Data Set.
“We know medication-assisted treatment is an effective, essential tool in fighting the opioid crisis,” said HHS Secretary Alex Azar, “and HHS will continue working to expand access to it.”
The Targeted Capacity Expansion: Medication Assisted Treatment-Prescription Drug Opioid Addiction grant program will expand access to treatment and recovery support services in states, tribes, and tribal organizations with the highest per-capita rates of primary treatment admissions for heroin and prescription opioids. The funding includes the areas with the “most dramatic increases” for heroin and prescription opioids, as identified by SAMHSA’s 2015 Treatment Episode Data Set.
“We know medication-assisted treatment is an effective, essential tool in fighting the opioid crisis,” said HHS Secretary Alex Azar, “and HHS will continue working to expand access to it.”
The Targeted Capacity Expansion: Medication Assisted Treatment-Prescription Drug Opioid Addiction grant program will expand access to treatment and recovery support services in states, tribes, and tribal organizations with the highest per-capita rates of primary treatment admissions for heroin and prescription opioids. The funding includes the areas with the “most dramatic increases” for heroin and prescription opioids, as identified by SAMHSA’s 2015 Treatment Episode Data Set.
“We know medication-assisted treatment is an effective, essential tool in fighting the opioid crisis,” said HHS Secretary Alex Azar, “and HHS will continue working to expand access to it.”
Primary efficacy not met by new M. tuberculosis vaccine strategies
Vaccination may have reduced the rate of sustained Mycobacterium tuberculosis infection in a recent randomized, placebo-controlled clinical trial conducted in a high-risk setting for tuberculosis transmission, despite not meeting the primary endpoint of the study.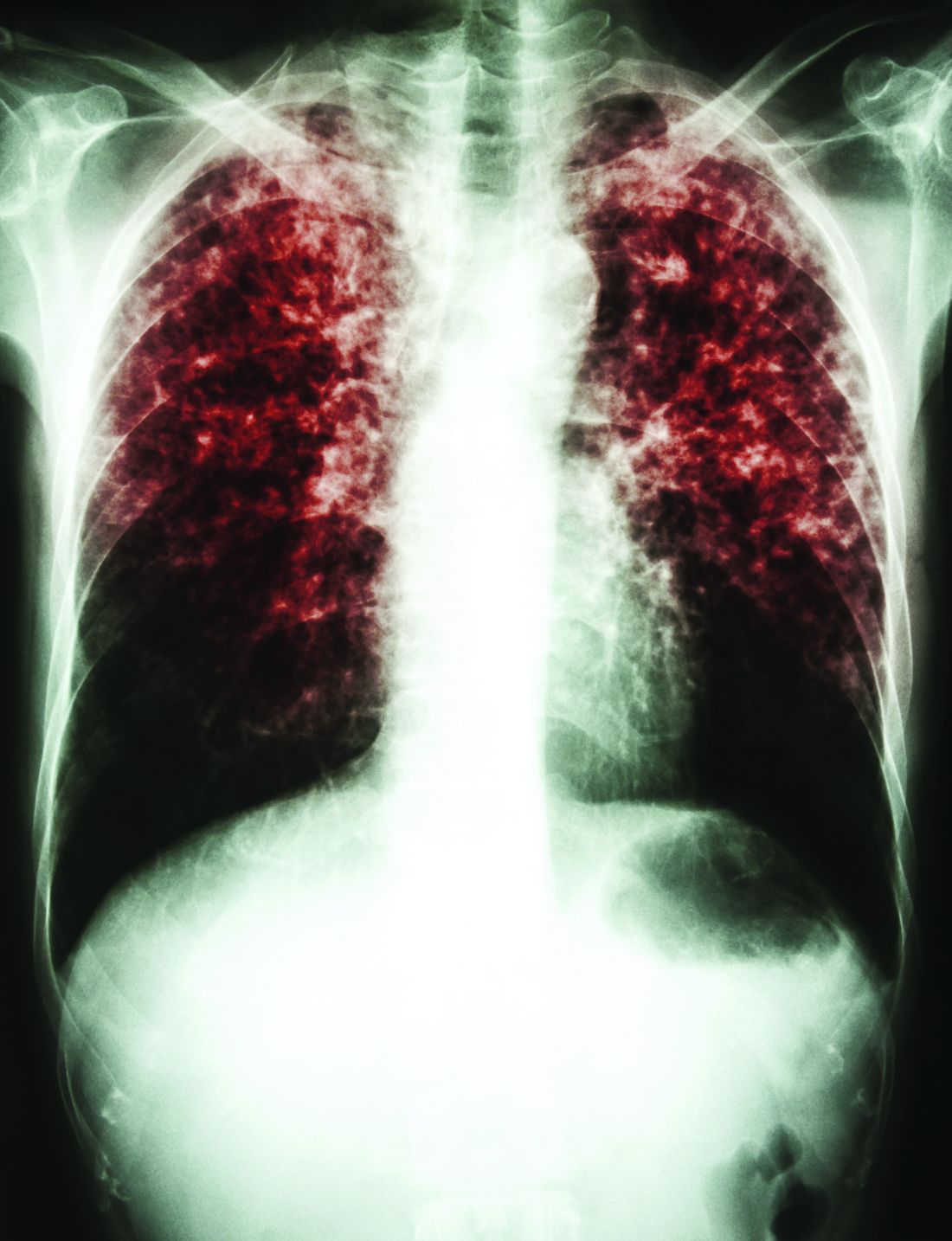
In adolescents who had received the bacille Calmette-Guérin (BCG) vaccine in infancy, BCG revaccination reduced the rate of sustained conversion of QuantiFERON-TB Gold In-Tube assay (QFT), a test that is thought to reflect sustained M. tuberculosis infection.
The study also evaluated a candidate subunit vaccine, H4:IC31, which also reduced the rate of sustained QFT conversion, though the efficacy estimate did not reach statistical significance, investigators reported.
Neither H4:IC31 nor BCG revaccination prevented initial QFT conversion, the primary endpoint of the study; however, both vaccines were immunogenic, they said.
Moreover, the significantly reduced rate of sustained conversion with BCG revaccination provides a “promising signal,” study authors said in the New England Journal of Medicine.
“The durability of this important finding and potential public health significance for protection against tuberculosis disease warrants epidemiologic modeling and further clinical evaluation,” wrote Elisa Nemes, PhD, of the South African Tuberculosis Vaccine Initiative, which is part of the Institute of Infectious Disease and Molecular Medicine at the University of Cape Town (South Africa), and her coauthors.
Similarly, the nonsignificantly reduced rate of sustained QFT conversion seen with H4:IC31 suggested that subunit vaccines can have a biologic effect in this setting, which may inform development of new tuberculosis vaccines, Dr. Nemes and her colleagues added.
The phase 2 trial included 990 adolescents in South Africa who had undergone neonatal BCG vaccination. They were randomly assigned to receive BCG revaccination, H4:IC31 vaccine, or placebo.
Neither vaccine met the primary efficacy criterion based on initial QFT conversion rates, which were 13.1% for BCG revaccination, 14.3% for H4:IC31 vaccine, and 15.8% for placebo.
For the secondary endpoint of sustained QFT conversion, the efficacy of BCG revaccination was 45.4% (95% confidence interval, 6.4%-68.1%; P = .03), while the efficacy of H4:IC31 vaccine was 34.2% (95% CI, –10.4% to 60.7%; P = .11).
“These encouraging findings provide an impetus to reevaluate the use of BCG revaccination of populations that are free of M. tuberculosis infection for the prevention of disease,” Dr. Nemes and her coauthors wrote in their report.
Revaccination with BCG was associated with more adverse events, compared with the other groups, although adverse events in the trial were predominantly injection-site reactions that were mild to moderate in severity, investigators reported. There were no serious adverse events judged by investigators to be related to trial vaccine.
Taken together, these results raise important questions regarding the potential benefits of vaccine-mediated prevention of M. tuberculosis infection for control of tuberculosis disease, according to Dr. Nemes and her coauthors.
However, interpretation of the findings is limited because there is no definitive test for M. tuberculosis infection.
Recent infection diagnosed by tuberculin skin test or QFT conversion has been associated with higher risk of disease, compared with nonconversion, according to investigators, while reversion to a negative tuberculin skin test correlates with infection containment and lower risk of tuberculosis.
“Although the clinical significance of QFT reversion remains to be established, we propose that sustained QFT conversion more likely represents sustained M. tuberculosis infection and a higher risk of progression to disease than transient QFT conversion,” they wrote.
The study was supported by Aeras, Sanofi Pasteur, the Bill & Melinda Gates Foundation, the Government of the Netherlands Directorate-General for International Cooperation and Development, and the United Kingdom Department for International Development. Study authors reported disclosures related to GlaxoSmithKline, Sanofi Pasteur, and Aeras.
SOURCE: Nemes E et al. N Engl J Med. 2018;379:138-49.
Vaccination may have reduced the rate of sustained Mycobacterium tuberculosis infection in a recent randomized, placebo-controlled clinical trial conducted in a high-risk setting for tuberculosis transmission, despite not meeting the primary endpoint of the study.
In adolescents who had received the bacille Calmette-Guérin (BCG) vaccine in infancy, BCG revaccination reduced the rate of sustained conversion of QuantiFERON-TB Gold In-Tube assay (QFT), a test that is thought to reflect sustained M. tuberculosis infection.
The study also evaluated a candidate subunit vaccine, H4:IC31, which also reduced the rate of sustained QFT conversion, though the efficacy estimate did not reach statistical significance, investigators reported.
Neither H4:IC31 nor BCG revaccination prevented initial QFT conversion, the primary endpoint of the study; however, both vaccines were immunogenic, they said.
Moreover, the significantly reduced rate of sustained conversion with BCG revaccination provides a “promising signal,” study authors said in the New England Journal of Medicine.
“The durability of this important finding and potential public health significance for protection against tuberculosis disease warrants epidemiologic modeling and further clinical evaluation,” wrote Elisa Nemes, PhD, of the South African Tuberculosis Vaccine Initiative, which is part of the Institute of Infectious Disease and Molecular Medicine at the University of Cape Town (South Africa), and her coauthors.
Similarly, the nonsignificantly reduced rate of sustained QFT conversion seen with H4:IC31 suggested that subunit vaccines can have a biologic effect in this setting, which may inform development of new tuberculosis vaccines, Dr. Nemes and her colleagues added.
The phase 2 trial included 990 adolescents in South Africa who had undergone neonatal BCG vaccination. They were randomly assigned to receive BCG revaccination, H4:IC31 vaccine, or placebo.
Neither vaccine met the primary efficacy criterion based on initial QFT conversion rates, which were 13.1% for BCG revaccination, 14.3% for H4:IC31 vaccine, and 15.8% for placebo.
For the secondary endpoint of sustained QFT conversion, the efficacy of BCG revaccination was 45.4% (95% confidence interval, 6.4%-68.1%; P = .03), while the efficacy of H4:IC31 vaccine was 34.2% (95% CI, –10.4% to 60.7%; P = .11).
“These encouraging findings provide an impetus to reevaluate the use of BCG revaccination of populations that are free of M. tuberculosis infection for the prevention of disease,” Dr. Nemes and her coauthors wrote in their report.
Revaccination with BCG was associated with more adverse events, compared with the other groups, although adverse events in the trial were predominantly injection-site reactions that were mild to moderate in severity, investigators reported. There were no serious adverse events judged by investigators to be related to trial vaccine.
Taken together, these results raise important questions regarding the potential benefits of vaccine-mediated prevention of M. tuberculosis infection for control of tuberculosis disease, according to Dr. Nemes and her coauthors.
However, interpretation of the findings is limited because there is no definitive test for M. tuberculosis infection.
Recent infection diagnosed by tuberculin skin test or QFT conversion has been associated with higher risk of disease, compared with nonconversion, according to investigators, while reversion to a negative tuberculin skin test correlates with infection containment and lower risk of tuberculosis.
“Although the clinical significance of QFT reversion remains to be established, we propose that sustained QFT conversion more likely represents sustained M. tuberculosis infection and a higher risk of progression to disease than transient QFT conversion,” they wrote.
The study was supported by Aeras, Sanofi Pasteur, the Bill & Melinda Gates Foundation, the Government of the Netherlands Directorate-General for International Cooperation and Development, and the United Kingdom Department for International Development. Study authors reported disclosures related to GlaxoSmithKline, Sanofi Pasteur, and Aeras.
SOURCE: Nemes E et al. N Engl J Med. 2018;379:138-49.
Vaccination may have reduced the rate of sustained Mycobacterium tuberculosis infection in a recent randomized, placebo-controlled clinical trial conducted in a high-risk setting for tuberculosis transmission, despite not meeting the primary endpoint of the study.
In adolescents who had received the bacille Calmette-Guérin (BCG) vaccine in infancy, BCG revaccination reduced the rate of sustained conversion of QuantiFERON-TB Gold In-Tube assay (QFT), a test that is thought to reflect sustained M. tuberculosis infection.
The study also evaluated a candidate subunit vaccine, H4:IC31, which also reduced the rate of sustained QFT conversion, though the efficacy estimate did not reach statistical significance, investigators reported.
Neither H4:IC31 nor BCG revaccination prevented initial QFT conversion, the primary endpoint of the study; however, both vaccines were immunogenic, they said.
Moreover, the significantly reduced rate of sustained conversion with BCG revaccination provides a “promising signal,” study authors said in the New England Journal of Medicine.
“The durability of this important finding and potential public health significance for protection against tuberculosis disease warrants epidemiologic modeling and further clinical evaluation,” wrote Elisa Nemes, PhD, of the South African Tuberculosis Vaccine Initiative, which is part of the Institute of Infectious Disease and Molecular Medicine at the University of Cape Town (South Africa), and her coauthors.
Similarly, the nonsignificantly reduced rate of sustained QFT conversion seen with H4:IC31 suggested that subunit vaccines can have a biologic effect in this setting, which may inform development of new tuberculosis vaccines, Dr. Nemes and her colleagues added.
The phase 2 trial included 990 adolescents in South Africa who had undergone neonatal BCG vaccination. They were randomly assigned to receive BCG revaccination, H4:IC31 vaccine, or placebo.
Neither vaccine met the primary efficacy criterion based on initial QFT conversion rates, which were 13.1% for BCG revaccination, 14.3% for H4:IC31 vaccine, and 15.8% for placebo.
For the secondary endpoint of sustained QFT conversion, the efficacy of BCG revaccination was 45.4% (95% confidence interval, 6.4%-68.1%; P = .03), while the efficacy of H4:IC31 vaccine was 34.2% (95% CI, –10.4% to 60.7%; P = .11).
“These encouraging findings provide an impetus to reevaluate the use of BCG revaccination of populations that are free of M. tuberculosis infection for the prevention of disease,” Dr. Nemes and her coauthors wrote in their report.
Revaccination with BCG was associated with more adverse events, compared with the other groups, although adverse events in the trial were predominantly injection-site reactions that were mild to moderate in severity, investigators reported. There were no serious adverse events judged by investigators to be related to trial vaccine.
Taken together, these results raise important questions regarding the potential benefits of vaccine-mediated prevention of M. tuberculosis infection for control of tuberculosis disease, according to Dr. Nemes and her coauthors.
However, interpretation of the findings is limited because there is no definitive test for M. tuberculosis infection.
Recent infection diagnosed by tuberculin skin test or QFT conversion has been associated with higher risk of disease, compared with nonconversion, according to investigators, while reversion to a negative tuberculin skin test correlates with infection containment and lower risk of tuberculosis.
“Although the clinical significance of QFT reversion remains to be established, we propose that sustained QFT conversion more likely represents sustained M. tuberculosis infection and a higher risk of progression to disease than transient QFT conversion,” they wrote.
The study was supported by Aeras, Sanofi Pasteur, the Bill & Melinda Gates Foundation, the Government of the Netherlands Directorate-General for International Cooperation and Development, and the United Kingdom Department for International Development. Study authors reported disclosures related to GlaxoSmithKline, Sanofi Pasteur, and Aeras.
SOURCE: Nemes E et al. N Engl J Med. 2018;379:138-49.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Neither H4:IC31 nor BCG revaccination prevented initial QFT conversion, the primary endpoint; however, both vaccines were immunogenic.
Major finding: For the secondary endpoint of sustained QuantiFERON-TB Gold In-Tube Assay (QFT) conversion, efficacy was 45.4% (P = .03) for BCG revaccination and 34.2% (P = .11) for H4:IC31, a candidate subunit vaccine.
Study details: A phase 2, randomized, placebo-controlled trial including 990 adolescents in South Africa who had received BCG vaccine in infancy.
Disclosures: The study was supported by Aeras, Sanofi Pasteur, the Bill & Melinda Gates Foundation, the Government of the Netherlands Directorate-General for International Cooperation and Development, and the United Kingdom Department for International Development. Study authors reported disclosures related to GlaxoSmithKline, Sanofi Pasteur, and Aeras.
Source: Nemes E et al. N Engl J Med. 2018;379:138-49.
Transplanting HCV-infected kidneys in HCV-infected patients showed positive outcomes, costs
Transplanting a kidney infected with hepatitis C into individuals infected with HCV, followed by treatment, was more effective and less costly than transplanting an uninfected kidney, preceded by HCV treatment, according to Mark H. Eckman, MD, of the University of Cincinnati and his colleagues.
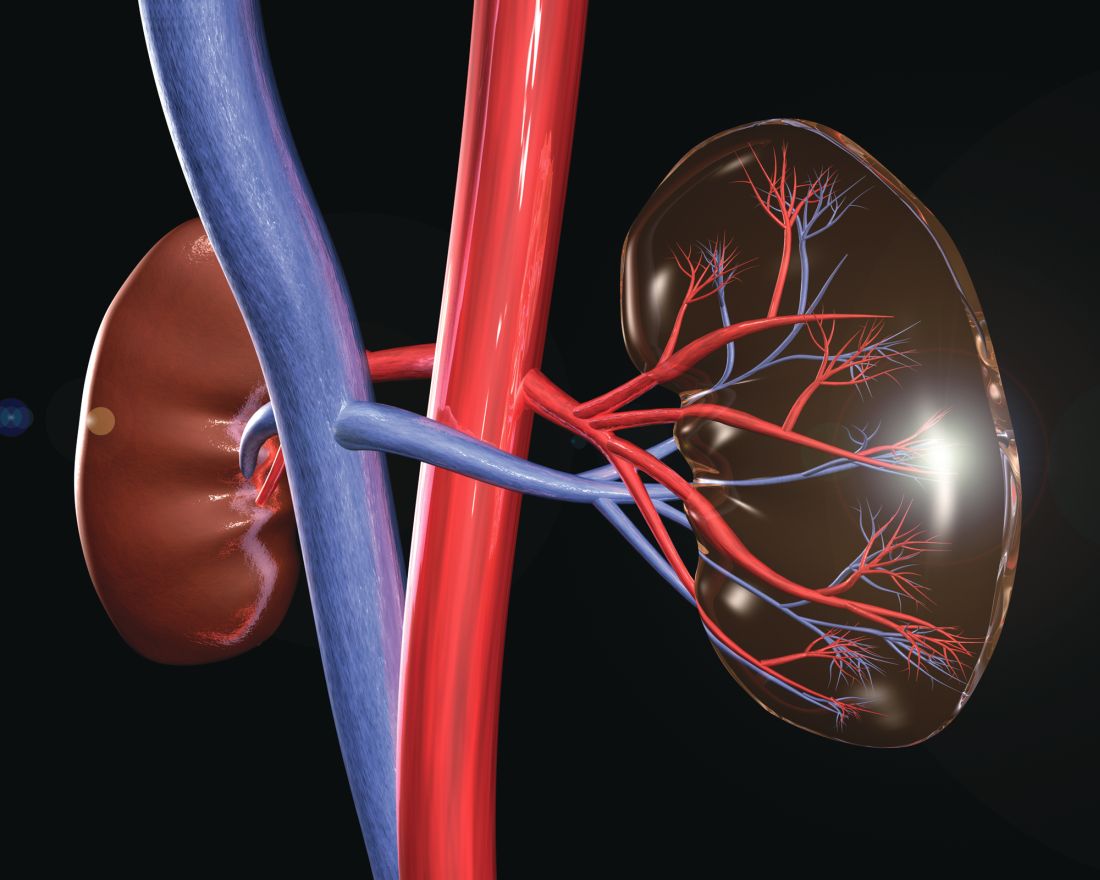
Largely because of the longer wait times for uninfected kidneys, a typical patient aged 58 years on hemodialysis would gain an average of 0.5 quality-adjusted life-years at a lifetime cost savings of $41,591 dollars, according to the model.
“In an era of increasing success for kidney transplants and demand that far outstrips supply, deferring antiviral therapy until after transplant of HCV-infected kidneys, when available, should be both cost saving and effective,” the researchers wrote.
The study was funded by grants from Merck Sharpe & Dohme and the National Center for Advancing Translational Science. Several of the authors reported having grants from Merck and grants and personal fees from a variety of other pharmaceutical companies.
SOURCE: Eckman MH et al. Ann Intern Med. 2018 Jul 10. doi: 10.7326/M17-3088.
Transplanting a kidney infected with hepatitis C into individuals infected with HCV, followed by treatment, was more effective and less costly than transplanting an uninfected kidney, preceded by HCV treatment, according to Mark H. Eckman, MD, of the University of Cincinnati and his colleagues.
Largely because of the longer wait times for uninfected kidneys, a typical patient aged 58 years on hemodialysis would gain an average of 0.5 quality-adjusted life-years at a lifetime cost savings of $41,591 dollars, according to the model.
“In an era of increasing success for kidney transplants and demand that far outstrips supply, deferring antiviral therapy until after transplant of HCV-infected kidneys, when available, should be both cost saving and effective,” the researchers wrote.
The study was funded by grants from Merck Sharpe & Dohme and the National Center for Advancing Translational Science. Several of the authors reported having grants from Merck and grants and personal fees from a variety of other pharmaceutical companies.
SOURCE: Eckman MH et al. Ann Intern Med. 2018 Jul 10. doi: 10.7326/M17-3088.
Transplanting a kidney infected with hepatitis C into individuals infected with HCV, followed by treatment, was more effective and less costly than transplanting an uninfected kidney, preceded by HCV treatment, according to Mark H. Eckman, MD, of the University of Cincinnati and his colleagues.
Largely because of the longer wait times for uninfected kidneys, a typical patient aged 58 years on hemodialysis would gain an average of 0.5 quality-adjusted life-years at a lifetime cost savings of $41,591 dollars, according to the model.
“In an era of increasing success for kidney transplants and demand that far outstrips supply, deferring antiviral therapy until after transplant of HCV-infected kidneys, when available, should be both cost saving and effective,” the researchers wrote.
The study was funded by grants from Merck Sharpe & Dohme and the National Center for Advancing Translational Science. Several of the authors reported having grants from Merck and grants and personal fees from a variety of other pharmaceutical companies.
SOURCE: Eckman MH et al. Ann Intern Med. 2018 Jul 10. doi: 10.7326/M17-3088.
FROM ANNALS OF INTERNAL MEDICINE
Fluoroquinolones can cause fatal hypoglycemia, FDA warns
Fluoroquinolones have caused at least 67 cases of life-threatening hypoglycemic coma, including 13 deaths and 9 permanent and disabling injuries, according to an internal safety review by the Food and Drug Administration. Most cases (44) were associated with levofloxacin.

The review also found new neuropsychiatric side effects associated with fluoroquinolones, including disturbances in attention, memory impairment, and delirium.
Considering these findings, the agency will strengthen warning labels on all fluoroquinolones, which already warn that the antibiotics may cause hypoglycemia and mental health issues, especially in older people, the FDA said in a press statement.
“Health care professionals should be aware of the potential risk of hypoglycemia, sometimes resulting in coma, occurring more frequently in the elderly and those with diabetes taking an oral hypoglycemic medicine or insulin,” the statement said. “Alert patients of the symptoms of hypoglycemia and carefully monitor blood glucose levels in these patients and discuss with them how to treat themselves if they have symptoms of hypoglycemia. Inform patients about the risk of psychiatric adverse reactions that can occur after just one dose. Stop fluoroquinolone treatment immediately if a patient reports any central nervous system side effects, including psychiatric adverse reactions, or blood glucose disturbances and switch to a non–fluoroquinolone antibiotic if possible. Stop fluoroquinolone treatment immediately if a patient reports serious side effects involving the tendons, muscles, joints, or nerves, and switch to a non–fluoroquinolone antibiotic to complete the patient’s treatment course.”
The statement also warned not to prescribe fluoroquinolones to patients who have other treatment options for acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections because the risks outweigh the benefits in these patients.
The FDA conducted the postmarketing review on all five of the fluoroquinolones (ciprofloxacin, gemifloxacin, levofloxacin, moxifloxacin, and ofloxacin). The newest fluoroquinolone, delafloxacin, approved a year ago, was not included in the class review. However, the agency expects that similar adverse events will be associated with delafloxacin and labeling on that drug will include the new warnings.
The agency reviewed cases in the FDA Adverse Event Reporting System, and in published medical literature, during 1987-2017. Most of the incidents (56) were in the system; 11 additional cases were published. Levofloxacin caused most of the incidents (44), followed by ciprofloxacin (12), moxifloxacin (9), and ofloxacin (2). Four of the fluoroquinolones have a labeled drug interaction with sulfonylurea agents, which can cause hypoglycemia.
Some of those who died were getting the antibiotics for complicated infections, including urinary tract and upper respiratory tract infections, and postoperative antibiotic prophylaxis. Others had renal insufficiency – a risk factor for hypoglycemia.
Of the 54 patients who survived, 9 never fully recovered and had permanent disabilities. Four patients remained in a coma for at least 1 month, despite blood sugar normalization. Five experienced some type of neurologic injury.
The new label changes will also fortify the existing warning about mental health side effects, after the review found new reactions that are not listed in the current warning, including the new reports of disturbance in attention, memory impairment, and delirium.
The FDA statement did not include the number of cases found or the associated drugs. Again, the safety review was based on reports in the FAERS database and published medical literature.
“We found that psychiatric adverse reactions were not consistent in the drug labels. The labels of fluoroquinolones currently include many psychiatric adverse reactions in the Warnings and Precautions section, for example, hallucination, psychoses, confusion, depression, anxiety, and paranoia. In an effort to harmonize the psychiatric adverse reactions described in the drug labels across the class of fluoroquinolones, we are requiring that all fluoroquinolones include six psychiatric adverse reactions (disturbance in attention, memory impairment, delirium, nervousness, agitation, and disorientation) in the Central Nervous System Effects of the Warnings and Precautions section of the labels. Disturbance in attention, memory impairment, and delirium are new adverse reactions to be added to the labels of the entire class of fluoroquinolones. Nervousness, agitation, and disorientation had been previously listed in the fluoroquinolone drug labels and will now be added to the Warnings and Precautions section of each drug label to harmonize labels across the fluoroquinolone drug class. The new label changes will make the psychiatric adverse reactions more prominent and more consistent.”
The FDA has previously warned about other adverse events associated with fluoroquinolones in May 2016, restricting use for certain uncomplicated infections; July 2016, for disabling side effects; August 2013, for peripheral neuropathy, and July 2008, for tendinitis and tendon rupture.
Fluoroquinolones have caused at least 67 cases of life-threatening hypoglycemic coma, including 13 deaths and 9 permanent and disabling injuries, according to an internal safety review by the Food and Drug Administration. Most cases (44) were associated with levofloxacin.
The review also found new neuropsychiatric side effects associated with fluoroquinolones, including disturbances in attention, memory impairment, and delirium.
Considering these findings, the agency will strengthen warning labels on all fluoroquinolones, which already warn that the antibiotics may cause hypoglycemia and mental health issues, especially in older people, the FDA said in a press statement.
“Health care professionals should be aware of the potential risk of hypoglycemia, sometimes resulting in coma, occurring more frequently in the elderly and those with diabetes taking an oral hypoglycemic medicine or insulin,” the statement said. “Alert patients of the symptoms of hypoglycemia and carefully monitor blood glucose levels in these patients and discuss with them how to treat themselves if they have symptoms of hypoglycemia. Inform patients about the risk of psychiatric adverse reactions that can occur after just one dose. Stop fluoroquinolone treatment immediately if a patient reports any central nervous system side effects, including psychiatric adverse reactions, or blood glucose disturbances and switch to a non–fluoroquinolone antibiotic if possible. Stop fluoroquinolone treatment immediately if a patient reports serious side effects involving the tendons, muscles, joints, or nerves, and switch to a non–fluoroquinolone antibiotic to complete the patient’s treatment course.”
The statement also warned not to prescribe fluoroquinolones to patients who have other treatment options for acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections because the risks outweigh the benefits in these patients.
The FDA conducted the postmarketing review on all five of the fluoroquinolones (ciprofloxacin, gemifloxacin, levofloxacin, moxifloxacin, and ofloxacin). The newest fluoroquinolone, delafloxacin, approved a year ago, was not included in the class review. However, the agency expects that similar adverse events will be associated with delafloxacin and labeling on that drug will include the new warnings.
The agency reviewed cases in the FDA Adverse Event Reporting System, and in published medical literature, during 1987-2017. Most of the incidents (56) were in the system; 11 additional cases were published. Levofloxacin caused most of the incidents (44), followed by ciprofloxacin (12), moxifloxacin (9), and ofloxacin (2). Four of the fluoroquinolones have a labeled drug interaction with sulfonylurea agents, which can cause hypoglycemia.
Some of those who died were getting the antibiotics for complicated infections, including urinary tract and upper respiratory tract infections, and postoperative antibiotic prophylaxis. Others had renal insufficiency – a risk factor for hypoglycemia.
Of the 54 patients who survived, 9 never fully recovered and had permanent disabilities. Four patients remained in a coma for at least 1 month, despite blood sugar normalization. Five experienced some type of neurologic injury.
The new label changes will also fortify the existing warning about mental health side effects, after the review found new reactions that are not listed in the current warning, including the new reports of disturbance in attention, memory impairment, and delirium.
The FDA statement did not include the number of cases found or the associated drugs. Again, the safety review was based on reports in the FAERS database and published medical literature.
“We found that psychiatric adverse reactions were not consistent in the drug labels. The labels of fluoroquinolones currently include many psychiatric adverse reactions in the Warnings and Precautions section, for example, hallucination, psychoses, confusion, depression, anxiety, and paranoia. In an effort to harmonize the psychiatric adverse reactions described in the drug labels across the class of fluoroquinolones, we are requiring that all fluoroquinolones include six psychiatric adverse reactions (disturbance in attention, memory impairment, delirium, nervousness, agitation, and disorientation) in the Central Nervous System Effects of the Warnings and Precautions section of the labels. Disturbance in attention, memory impairment, and delirium are new adverse reactions to be added to the labels of the entire class of fluoroquinolones. Nervousness, agitation, and disorientation had been previously listed in the fluoroquinolone drug labels and will now be added to the Warnings and Precautions section of each drug label to harmonize labels across the fluoroquinolone drug class. The new label changes will make the psychiatric adverse reactions more prominent and more consistent.”
The FDA has previously warned about other adverse events associated with fluoroquinolones in May 2016, restricting use for certain uncomplicated infections; July 2016, for disabling side effects; August 2013, for peripheral neuropathy, and July 2008, for tendinitis and tendon rupture.
Fluoroquinolones have caused at least 67 cases of life-threatening hypoglycemic coma, including 13 deaths and 9 permanent and disabling injuries, according to an internal safety review by the Food and Drug Administration. Most cases (44) were associated with levofloxacin.
The review also found new neuropsychiatric side effects associated with fluoroquinolones, including disturbances in attention, memory impairment, and delirium.
Considering these findings, the agency will strengthen warning labels on all fluoroquinolones, which already warn that the antibiotics may cause hypoglycemia and mental health issues, especially in older people, the FDA said in a press statement.
“Health care professionals should be aware of the potential risk of hypoglycemia, sometimes resulting in coma, occurring more frequently in the elderly and those with diabetes taking an oral hypoglycemic medicine or insulin,” the statement said. “Alert patients of the symptoms of hypoglycemia and carefully monitor blood glucose levels in these patients and discuss with them how to treat themselves if they have symptoms of hypoglycemia. Inform patients about the risk of psychiatric adverse reactions that can occur after just one dose. Stop fluoroquinolone treatment immediately if a patient reports any central nervous system side effects, including psychiatric adverse reactions, or blood glucose disturbances and switch to a non–fluoroquinolone antibiotic if possible. Stop fluoroquinolone treatment immediately if a patient reports serious side effects involving the tendons, muscles, joints, or nerves, and switch to a non–fluoroquinolone antibiotic to complete the patient’s treatment course.”
The statement also warned not to prescribe fluoroquinolones to patients who have other treatment options for acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections because the risks outweigh the benefits in these patients.
The FDA conducted the postmarketing review on all five of the fluoroquinolones (ciprofloxacin, gemifloxacin, levofloxacin, moxifloxacin, and ofloxacin). The newest fluoroquinolone, delafloxacin, approved a year ago, was not included in the class review. However, the agency expects that similar adverse events will be associated with delafloxacin and labeling on that drug will include the new warnings.
The agency reviewed cases in the FDA Adverse Event Reporting System, and in published medical literature, during 1987-2017. Most of the incidents (56) were in the system; 11 additional cases were published. Levofloxacin caused most of the incidents (44), followed by ciprofloxacin (12), moxifloxacin (9), and ofloxacin (2). Four of the fluoroquinolones have a labeled drug interaction with sulfonylurea agents, which can cause hypoglycemia.
Some of those who died were getting the antibiotics for complicated infections, including urinary tract and upper respiratory tract infections, and postoperative antibiotic prophylaxis. Others had renal insufficiency – a risk factor for hypoglycemia.
Of the 54 patients who survived, 9 never fully recovered and had permanent disabilities. Four patients remained in a coma for at least 1 month, despite blood sugar normalization. Five experienced some type of neurologic injury.
The new label changes will also fortify the existing warning about mental health side effects, after the review found new reactions that are not listed in the current warning, including the new reports of disturbance in attention, memory impairment, and delirium.
The FDA statement did not include the number of cases found or the associated drugs. Again, the safety review was based on reports in the FAERS database and published medical literature.
“We found that psychiatric adverse reactions were not consistent in the drug labels. The labels of fluoroquinolones currently include many psychiatric adverse reactions in the Warnings and Precautions section, for example, hallucination, psychoses, confusion, depression, anxiety, and paranoia. In an effort to harmonize the psychiatric adverse reactions described in the drug labels across the class of fluoroquinolones, we are requiring that all fluoroquinolones include six psychiatric adverse reactions (disturbance in attention, memory impairment, delirium, nervousness, agitation, and disorientation) in the Central Nervous System Effects of the Warnings and Precautions section of the labels. Disturbance in attention, memory impairment, and delirium are new adverse reactions to be added to the labels of the entire class of fluoroquinolones. Nervousness, agitation, and disorientation had been previously listed in the fluoroquinolone drug labels and will now be added to the Warnings and Precautions section of each drug label to harmonize labels across the fluoroquinolone drug class. The new label changes will make the psychiatric adverse reactions more prominent and more consistent.”
The FDA has previously warned about other adverse events associated with fluoroquinolones in May 2016, restricting use for certain uncomplicated infections; July 2016, for disabling side effects; August 2013, for peripheral neuropathy, and July 2008, for tendinitis and tendon rupture.
FDA Efforts to Advance Development of Gene Therapies
FDA Commissioner Scott Gottlieb, MD, releases a statement on the agency’s efforts to advance development of gene therapies.
Once just a theory, gene therapies are now a therapeutic reality for some patients. These platforms may have the potential to treat and cure some of our most intractable and vexing diseases. The policy framework we construct for how these products should be developed, reviewed by regulators, and reimbursed, will help set the stage for the continued advancement of this new market. Last year, we announced our comprehensive policy framework for regenerative medicine, including a draft guidance that describes the expedited programs, such as the breakthrough therapy designation, and the regenerative medicine advanced therapy (RMAT) designation, that may be available to sponsors of these therapies. Today, we’re unveiling a complementary framework for the development, review and approval of gene therapies.

In the past 12 months, we’ve seen three separate gene therapy products approved by the FDA. This reflects the rapid advancements in this field. An inflection point was reached with the development of vectors that could reliably deliver gene cassettes in vivo, into cells and human tissue. In the future, we expect this field to continue to expand, with the potential approval of new treatments for many debilitating diseases. These therapies hold great promise. Our new steps are aimed at fostering developments in this innovative field.
Gene therapies are being studied in many areas, including genetic disorders, autoimmune diseases, heart disease, cancer and HIV/AIDS. We look forward to working with the academic and research communities to make safe and effective products a reality for more patients. But we know that we still have much to learn about how these products work, how to administer them safely, and whether they will continue to work properly in the body without causing adverse side effects over long periods of time. In contrast to traditional drug review, some of the more challenging questions when it comes to gene therapy relate to product manufacturing and quality, or questions about the durability of response, which often can’t be fully answered in any reasonably sized pre-market trial. For some of these products, we may need to accept some level of uncertainty around these questions at the time of approval. For example, in some cases the long-term durability of the effect won’t be fully understood at the time of approval. Effective tools for reliable post-market follow up, such as required post-market clinical trials, are going to be one key to advancing this field and helping to ensure that our approach fosters safe and innovative treatments.
Even when there may be uncertainty about some questions, we need to make certain we assure patient safety and adequately characterize the potential risks and demonstrated benefits of these products. In part because of the added questions that often surround a new technology like gene therapy, these products are initially being aimed at devastating diseases, many of which lack available therapies, including some diseases that are fatal. In such cases of devastating diseases without available therapies, we’ve traditionally been willing to accept more uncertainty to facilitate timely access to promising therapies. In such cases, drug sponsors are generally required to conduct post-marketing clinical trials, known as phase 4 confirmatory trials, to confirm clinical benefit of the drug. This is the direction Congress gave the FDA by creating vehicles like the accelerated approval pathway.
When it comes to novel technologies like gene therapy, the FDA is steadfastly committed to a regulatory path that maintains the agency’s gold standard for assuring safety and efficacy. As we develop this evidence-based framework, we’re going to have to modernize how we approach certain aspects of these products in order to make sure our approach is tailored to the unique challenges created by these new platforms.
Today, we’re taking a step toward shaping this modern structure for the regulation of gene therapy. The agency is issuing a suite of six scientific guidance documents intended to serve as the building blocks of a modern, comprehensive framework for how we’ll help advance the field of gene therapy while making sure new products meet the FDA’s gold standard for safety and effectiveness.
These policies are part of our efforts to communicate the steps we’re taking to provide clear recommendations to sponsors and researchers, so that we can better support innovation. The documents are being issued in draft form so that we can solicit public input on these new policies. As with all draft guidances, all of the comments we receive will be carefully considered prior to finalizing these documents. We’re committed to working with stakeholders to bring novel treatments to the market while ensuring the safety of patients.
Disease-Specific Gene Therapy Guidances
The FDA has issuing three new draft guidance documents on the development of gene therapy products for specific disease categories. These are the first three disease-specific guidances that the agency is issuing for gene therapy products. Our new commitment to develop disease-specific guidance documents reflects the increasing activity in this field, and its growing importance to advancing public health.
Human Gene Therapy for Hemophilia: Gene therapy products for hemophilia are now being developed as single-dose treatments that may enable long-term production of the missing or abnormal coagulation factor in patients. This may reduce or eliminate the need for coagulation factor replacement. To define the proper development pathway for such products, we’re issuing a new draft guidance on gene therapy products that are targeted to the treatment of hemophilia. Once finalized, this new guidance will provide recommendations on the FDA’s current thinking on clinical trial design and preclinical considerations to support the development of these gene therapy products. Among other elements, the draft guidance provides recommendations regarding surrogate endpoints that could be used by sponsors pursuing accelerated approval of gene therapy products that are intended for treatment of hemophilia.
Human Gene Therapy for Retinal Disorders: Another area of fast-paced activity is gene therapy products targeted to the treatment of retinal disorders. The Human Gene Therapy for Retinal Disorders guidance, once finalized, will assist those developing gene therapy products for a wide variety of retinal disorders affecting both adult and pediatric patients. Gene therapy products currently undergoing clinical trials in the United States for retinal disorders are commonly delivered by intravitreal injections (into the fluid portion of the eye), or by subretinal injections (beneath the retina). In some cases, the gene therapy products are encapsulated in a device to be implanted within the eye. This new guidance document will focus on issues that are specific to gene therapies for retinal disorders. The document provides recommendations related to product development, preclinical testing, and clinical trial design for such products.
Human Gene Therapy for Rare Diseases: Rare diseases are those that affect fewer than 200,000 people in the United States. The National Institutes of Health reports that nearly 7,000 rare diseases affect more than 25 million Americans. About 80 percent of rare diseases are caused by a single-gene defect, and about half of all rare diseases affect children. Since most rare diseases have no approved therapies, there is a significant unmet need. The Human Gene Therapy for Rare Diseases guidance, once finalized, will provide recommendations on preclinical, manufacturing and clinical trial design for all phases of the clinical development program for these types of gene therapies. The information is intended to assist sponsors in the design of clinical development programs, where there may be limited study population size, potential feasibility and safety issues, as well as issues relating to the interpretation of effectiveness.
Guidances on Manufacturing Gene Therapies
The FDA is also providing new and comprehensive updates to three existing guidances that address manufacturing issues related to gene therapy. These updates reflect input from many stakeholders. We encourage additional feedback on these documents during the comment period.
The first draft guidance, Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs), provides sponsors with recommendations on how to provide sufficient CMC information to assure safety, identity, quality, purity and strength/potency of investigational gene therapy products. The guidance applies to human gene therapies and to combination products that contain a human gene therapy in combination with a drug or device. In addition, this guidance is organized to follow the structure of the FDA guidance on the Common Technical Document.
The second draft guidance, Testing of Retroviral Vector-Based Gene Therapy Products for Replication Competent Retrovirus (RCR) during Product Manufacture and Patient Follow-up, provides additional recommendations regarding the proper testing for RCR during the manufacture of retroviral vector-based gene therapy products, as well as during the follow-up monitoring of patients who’ve received retroviral vector-based gene therapy products. Specifically, the draft guidance recommends the identification and amount of material to be tested. The guidance also provides advice on general testing methods.
The third draft guidance, Long Term Follow-Up After Administration of Human Gene Therapy Products, provides recommendations regarding the design of long-term follow-up (LTFU) observational studies for the collection of data on delayed adverse events following administration of a gene therapy product. Because of some of the additional uncertainty intrinsic to a novel platform like gene therapy -- including questions related to the durability of the treatment effects as well as the theoretical potential for off-target effects if the genes do not insert correctly -- there’s an increased need for robust long-term follow-up of patients in the post-market period. This guidance describes product characteristics, patient-related factors, and the preclinical and clinical data that should be considered when assessing the need for LTFU observations and describes the features related to effective post-market follow up.
Once finalized, these draft guidances will replace previous guidances issued by the FDA in April 2008 (CMC) and November 2006 (RCR and LTFU).
The field of gene therapy has progressed rapidly since these guidances were first issued. Therefore, the FDA is updating these guidances to provide sponsors with the agency’s most up-to-date thinking.
Our goal is to help promote safe and effective product development in this field. We’ll continue to work with the product sponsors to help make the development and approval of these innovative gene therapies more efficient, while putting in place the regulatory controls needed to ensure that the resulting therapies are both safe and effective. We’ll also make full use of our expedited programs such as breakthrough therapy designation and regenerative medicine advanced therapy designation whenever possible.
Gene therapy represents one of the most promising opportunities for developing highly effective and even curative treatments for many vexing disorders. Some of these products are almost certainly going to change the contours of medical practice, and the destiny of patients with some debilitating diseases.
The FDA’s goal is to help these innovations advance in a framework that assures the safety and effectiveness of these resulting treatments, and continues to build peoples’ confidence in this novel area of medicine.
–Scott Gottlieb, MD
FDA Commissioner Scott Gottlieb, MD, releases a statement on the agency’s efforts to advance development of gene therapies.
FDA Commissioner Scott Gottlieb, MD, releases a statement on the agency’s efforts to advance development of gene therapies.
Once just a theory, gene therapies are now a therapeutic reality for some patients. These platforms may have the potential to treat and cure some of our most intractable and vexing diseases. The policy framework we construct for how these products should be developed, reviewed by regulators, and reimbursed, will help set the stage for the continued advancement of this new market. Last year, we announced our comprehensive policy framework for regenerative medicine, including a draft guidance that describes the expedited programs, such as the breakthrough therapy designation, and the regenerative medicine advanced therapy (RMAT) designation, that may be available to sponsors of these therapies. Today, we’re unveiling a complementary framework for the development, review and approval of gene therapies.
In the past 12 months, we’ve seen three separate gene therapy products approved by the FDA. This reflects the rapid advancements in this field. An inflection point was reached with the development of vectors that could reliably deliver gene cassettes in vivo, into cells and human tissue. In the future, we expect this field to continue to expand, with the potential approval of new treatments for many debilitating diseases. These therapies hold great promise. Our new steps are aimed at fostering developments in this innovative field.
Gene therapies are being studied in many areas, including genetic disorders, autoimmune diseases, heart disease, cancer and HIV/AIDS. We look forward to working with the academic and research communities to make safe and effective products a reality for more patients. But we know that we still have much to learn about how these products work, how to administer them safely, and whether they will continue to work properly in the body without causing adverse side effects over long periods of time. In contrast to traditional drug review, some of the more challenging questions when it comes to gene therapy relate to product manufacturing and quality, or questions about the durability of response, which often can’t be fully answered in any reasonably sized pre-market trial. For some of these products, we may need to accept some level of uncertainty around these questions at the time of approval. For example, in some cases the long-term durability of the effect won’t be fully understood at the time of approval. Effective tools for reliable post-market follow up, such as required post-market clinical trials, are going to be one key to advancing this field and helping to ensure that our approach fosters safe and innovative treatments.
Even when there may be uncertainty about some questions, we need to make certain we assure patient safety and adequately characterize the potential risks and demonstrated benefits of these products. In part because of the added questions that often surround a new technology like gene therapy, these products are initially being aimed at devastating diseases, many of which lack available therapies, including some diseases that are fatal. In such cases of devastating diseases without available therapies, we’ve traditionally been willing to accept more uncertainty to facilitate timely access to promising therapies. In such cases, drug sponsors are generally required to conduct post-marketing clinical trials, known as phase 4 confirmatory trials, to confirm clinical benefit of the drug. This is the direction Congress gave the FDA by creating vehicles like the accelerated approval pathway.
When it comes to novel technologies like gene therapy, the FDA is steadfastly committed to a regulatory path that maintains the agency’s gold standard for assuring safety and efficacy. As we develop this evidence-based framework, we’re going to have to modernize how we approach certain aspects of these products in order to make sure our approach is tailored to the unique challenges created by these new platforms.
Today, we’re taking a step toward shaping this modern structure for the regulation of gene therapy. The agency is issuing a suite of six scientific guidance documents intended to serve as the building blocks of a modern, comprehensive framework for how we’ll help advance the field of gene therapy while making sure new products meet the FDA’s gold standard for safety and effectiveness.
These policies are part of our efforts to communicate the steps we’re taking to provide clear recommendations to sponsors and researchers, so that we can better support innovation. The documents are being issued in draft form so that we can solicit public input on these new policies. As with all draft guidances, all of the comments we receive will be carefully considered prior to finalizing these documents. We’re committed to working with stakeholders to bring novel treatments to the market while ensuring the safety of patients.
Disease-Specific Gene Therapy Guidances
The FDA has issuing three new draft guidance documents on the development of gene therapy products for specific disease categories. These are the first three disease-specific guidances that the agency is issuing for gene therapy products. Our new commitment to develop disease-specific guidance documents reflects the increasing activity in this field, and its growing importance to advancing public health.
Human Gene Therapy for Hemophilia: Gene therapy products for hemophilia are now being developed as single-dose treatments that may enable long-term production of the missing or abnormal coagulation factor in patients. This may reduce or eliminate the need for coagulation factor replacement. To define the proper development pathway for such products, we’re issuing a new draft guidance on gene therapy products that are targeted to the treatment of hemophilia. Once finalized, this new guidance will provide recommendations on the FDA’s current thinking on clinical trial design and preclinical considerations to support the development of these gene therapy products. Among other elements, the draft guidance provides recommendations regarding surrogate endpoints that could be used by sponsors pursuing accelerated approval of gene therapy products that are intended for treatment of hemophilia.
Human Gene Therapy for Retinal Disorders: Another area of fast-paced activity is gene therapy products targeted to the treatment of retinal disorders. The Human Gene Therapy for Retinal Disorders guidance, once finalized, will assist those developing gene therapy products for a wide variety of retinal disorders affecting both adult and pediatric patients. Gene therapy products currently undergoing clinical trials in the United States for retinal disorders are commonly delivered by intravitreal injections (into the fluid portion of the eye), or by subretinal injections (beneath the retina). In some cases, the gene therapy products are encapsulated in a device to be implanted within the eye. This new guidance document will focus on issues that are specific to gene therapies for retinal disorders. The document provides recommendations related to product development, preclinical testing, and clinical trial design for such products.
Human Gene Therapy for Rare Diseases: Rare diseases are those that affect fewer than 200,000 people in the United States. The National Institutes of Health reports that nearly 7,000 rare diseases affect more than 25 million Americans. About 80 percent of rare diseases are caused by a single-gene defect, and about half of all rare diseases affect children. Since most rare diseases have no approved therapies, there is a significant unmet need. The Human Gene Therapy for Rare Diseases guidance, once finalized, will provide recommendations on preclinical, manufacturing and clinical trial design for all phases of the clinical development program for these types of gene therapies. The information is intended to assist sponsors in the design of clinical development programs, where there may be limited study population size, potential feasibility and safety issues, as well as issues relating to the interpretation of effectiveness.
Guidances on Manufacturing Gene Therapies
The FDA is also providing new and comprehensive updates to three existing guidances that address manufacturing issues related to gene therapy. These updates reflect input from many stakeholders. We encourage additional feedback on these documents during the comment period.
The first draft guidance, Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs), provides sponsors with recommendations on how to provide sufficient CMC information to assure safety, identity, quality, purity and strength/potency of investigational gene therapy products. The guidance applies to human gene therapies and to combination products that contain a human gene therapy in combination with a drug or device. In addition, this guidance is organized to follow the structure of the FDA guidance on the Common Technical Document.
The second draft guidance, Testing of Retroviral Vector-Based Gene Therapy Products for Replication Competent Retrovirus (RCR) during Product Manufacture and Patient Follow-up, provides additional recommendations regarding the proper testing for RCR during the manufacture of retroviral vector-based gene therapy products, as well as during the follow-up monitoring of patients who’ve received retroviral vector-based gene therapy products. Specifically, the draft guidance recommends the identification and amount of material to be tested. The guidance also provides advice on general testing methods.
The third draft guidance, Long Term Follow-Up After Administration of Human Gene Therapy Products, provides recommendations regarding the design of long-term follow-up (LTFU) observational studies for the collection of data on delayed adverse events following administration of a gene therapy product. Because of some of the additional uncertainty intrinsic to a novel platform like gene therapy -- including questions related to the durability of the treatment effects as well as the theoretical potential for off-target effects if the genes do not insert correctly -- there’s an increased need for robust long-term follow-up of patients in the post-market period. This guidance describes product characteristics, patient-related factors, and the preclinical and clinical data that should be considered when assessing the need for LTFU observations and describes the features related to effective post-market follow up.
Once finalized, these draft guidances will replace previous guidances issued by the FDA in April 2008 (CMC) and November 2006 (RCR and LTFU).
The field of gene therapy has progressed rapidly since these guidances were first issued. Therefore, the FDA is updating these guidances to provide sponsors with the agency’s most up-to-date thinking.
Our goal is to help promote safe and effective product development in this field. We’ll continue to work with the product sponsors to help make the development and approval of these innovative gene therapies more efficient, while putting in place the regulatory controls needed to ensure that the resulting therapies are both safe and effective. We’ll also make full use of our expedited programs such as breakthrough therapy designation and regenerative medicine advanced therapy designation whenever possible.
Gene therapy represents one of the most promising opportunities for developing highly effective and even curative treatments for many vexing disorders. Some of these products are almost certainly going to change the contours of medical practice, and the destiny of patients with some debilitating diseases.
The FDA’s goal is to help these innovations advance in a framework that assures the safety and effectiveness of these resulting treatments, and continues to build peoples’ confidence in this novel area of medicine.
–Scott Gottlieb, MD
Once just a theory, gene therapies are now a therapeutic reality for some patients. These platforms may have the potential to treat and cure some of our most intractable and vexing diseases. The policy framework we construct for how these products should be developed, reviewed by regulators, and reimbursed, will help set the stage for the continued advancement of this new market. Last year, we announced our comprehensive policy framework for regenerative medicine, including a draft guidance that describes the expedited programs, such as the breakthrough therapy designation, and the regenerative medicine advanced therapy (RMAT) designation, that may be available to sponsors of these therapies. Today, we’re unveiling a complementary framework for the development, review and approval of gene therapies.
In the past 12 months, we’ve seen three separate gene therapy products approved by the FDA. This reflects the rapid advancements in this field. An inflection point was reached with the development of vectors that could reliably deliver gene cassettes in vivo, into cells and human tissue. In the future, we expect this field to continue to expand, with the potential approval of new treatments for many debilitating diseases. These therapies hold great promise. Our new steps are aimed at fostering developments in this innovative field.
Gene therapies are being studied in many areas, including genetic disorders, autoimmune diseases, heart disease, cancer and HIV/AIDS. We look forward to working with the academic and research communities to make safe and effective products a reality for more patients. But we know that we still have much to learn about how these products work, how to administer them safely, and whether they will continue to work properly in the body without causing adverse side effects over long periods of time. In contrast to traditional drug review, some of the more challenging questions when it comes to gene therapy relate to product manufacturing and quality, or questions about the durability of response, which often can’t be fully answered in any reasonably sized pre-market trial. For some of these products, we may need to accept some level of uncertainty around these questions at the time of approval. For example, in some cases the long-term durability of the effect won’t be fully understood at the time of approval. Effective tools for reliable post-market follow up, such as required post-market clinical trials, are going to be one key to advancing this field and helping to ensure that our approach fosters safe and innovative treatments.
Even when there may be uncertainty about some questions, we need to make certain we assure patient safety and adequately characterize the potential risks and demonstrated benefits of these products. In part because of the added questions that often surround a new technology like gene therapy, these products are initially being aimed at devastating diseases, many of which lack available therapies, including some diseases that are fatal. In such cases of devastating diseases without available therapies, we’ve traditionally been willing to accept more uncertainty to facilitate timely access to promising therapies. In such cases, drug sponsors are generally required to conduct post-marketing clinical trials, known as phase 4 confirmatory trials, to confirm clinical benefit of the drug. This is the direction Congress gave the FDA by creating vehicles like the accelerated approval pathway.
When it comes to novel technologies like gene therapy, the FDA is steadfastly committed to a regulatory path that maintains the agency’s gold standard for assuring safety and efficacy. As we develop this evidence-based framework, we’re going to have to modernize how we approach certain aspects of these products in order to make sure our approach is tailored to the unique challenges created by these new platforms.
Today, we’re taking a step toward shaping this modern structure for the regulation of gene therapy. The agency is issuing a suite of six scientific guidance documents intended to serve as the building blocks of a modern, comprehensive framework for how we’ll help advance the field of gene therapy while making sure new products meet the FDA’s gold standard for safety and effectiveness.
These policies are part of our efforts to communicate the steps we’re taking to provide clear recommendations to sponsors and researchers, so that we can better support innovation. The documents are being issued in draft form so that we can solicit public input on these new policies. As with all draft guidances, all of the comments we receive will be carefully considered prior to finalizing these documents. We’re committed to working with stakeholders to bring novel treatments to the market while ensuring the safety of patients.
Disease-Specific Gene Therapy Guidances
The FDA has issuing three new draft guidance documents on the development of gene therapy products for specific disease categories. These are the first three disease-specific guidances that the agency is issuing for gene therapy products. Our new commitment to develop disease-specific guidance documents reflects the increasing activity in this field, and its growing importance to advancing public health.
Human Gene Therapy for Hemophilia: Gene therapy products for hemophilia are now being developed as single-dose treatments that may enable long-term production of the missing or abnormal coagulation factor in patients. This may reduce or eliminate the need for coagulation factor replacement. To define the proper development pathway for such products, we’re issuing a new draft guidance on gene therapy products that are targeted to the treatment of hemophilia. Once finalized, this new guidance will provide recommendations on the FDA’s current thinking on clinical trial design and preclinical considerations to support the development of these gene therapy products. Among other elements, the draft guidance provides recommendations regarding surrogate endpoints that could be used by sponsors pursuing accelerated approval of gene therapy products that are intended for treatment of hemophilia.
Human Gene Therapy for Retinal Disorders: Another area of fast-paced activity is gene therapy products targeted to the treatment of retinal disorders. The Human Gene Therapy for Retinal Disorders guidance, once finalized, will assist those developing gene therapy products for a wide variety of retinal disorders affecting both adult and pediatric patients. Gene therapy products currently undergoing clinical trials in the United States for retinal disorders are commonly delivered by intravitreal injections (into the fluid portion of the eye), or by subretinal injections (beneath the retina). In some cases, the gene therapy products are encapsulated in a device to be implanted within the eye. This new guidance document will focus on issues that are specific to gene therapies for retinal disorders. The document provides recommendations related to product development, preclinical testing, and clinical trial design for such products.
Human Gene Therapy for Rare Diseases: Rare diseases are those that affect fewer than 200,000 people in the United States. The National Institutes of Health reports that nearly 7,000 rare diseases affect more than 25 million Americans. About 80 percent of rare diseases are caused by a single-gene defect, and about half of all rare diseases affect children. Since most rare diseases have no approved therapies, there is a significant unmet need. The Human Gene Therapy for Rare Diseases guidance, once finalized, will provide recommendations on preclinical, manufacturing and clinical trial design for all phases of the clinical development program for these types of gene therapies. The information is intended to assist sponsors in the design of clinical development programs, where there may be limited study population size, potential feasibility and safety issues, as well as issues relating to the interpretation of effectiveness.
Guidances on Manufacturing Gene Therapies
The FDA is also providing new and comprehensive updates to three existing guidances that address manufacturing issues related to gene therapy. These updates reflect input from many stakeholders. We encourage additional feedback on these documents during the comment period.
The first draft guidance, Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs), provides sponsors with recommendations on how to provide sufficient CMC information to assure safety, identity, quality, purity and strength/potency of investigational gene therapy products. The guidance applies to human gene therapies and to combination products that contain a human gene therapy in combination with a drug or device. In addition, this guidance is organized to follow the structure of the FDA guidance on the Common Technical Document.
The second draft guidance, Testing of Retroviral Vector-Based Gene Therapy Products for Replication Competent Retrovirus (RCR) during Product Manufacture and Patient Follow-up, provides additional recommendations regarding the proper testing for RCR during the manufacture of retroviral vector-based gene therapy products, as well as during the follow-up monitoring of patients who’ve received retroviral vector-based gene therapy products. Specifically, the draft guidance recommends the identification and amount of material to be tested. The guidance also provides advice on general testing methods.
The third draft guidance, Long Term Follow-Up After Administration of Human Gene Therapy Products, provides recommendations regarding the design of long-term follow-up (LTFU) observational studies for the collection of data on delayed adverse events following administration of a gene therapy product. Because of some of the additional uncertainty intrinsic to a novel platform like gene therapy -- including questions related to the durability of the treatment effects as well as the theoretical potential for off-target effects if the genes do not insert correctly -- there’s an increased need for robust long-term follow-up of patients in the post-market period. This guidance describes product characteristics, patient-related factors, and the preclinical and clinical data that should be considered when assessing the need for LTFU observations and describes the features related to effective post-market follow up.
Once finalized, these draft guidances will replace previous guidances issued by the FDA in April 2008 (CMC) and November 2006 (RCR and LTFU).
The field of gene therapy has progressed rapidly since these guidances were first issued. Therefore, the FDA is updating these guidances to provide sponsors with the agency’s most up-to-date thinking.
Our goal is to help promote safe and effective product development in this field. We’ll continue to work with the product sponsors to help make the development and approval of these innovative gene therapies more efficient, while putting in place the regulatory controls needed to ensure that the resulting therapies are both safe and effective. We’ll also make full use of our expedited programs such as breakthrough therapy designation and regenerative medicine advanced therapy designation whenever possible.
Gene therapy represents one of the most promising opportunities for developing highly effective and even curative treatments for many vexing disorders. Some of these products are almost certainly going to change the contours of medical practice, and the destiny of patients with some debilitating diseases.
The FDA’s goal is to help these innovations advance in a framework that assures the safety and effectiveness of these resulting treatments, and continues to build peoples’ confidence in this novel area of medicine.
–Scott Gottlieb, MD
Does Combination Treatment Prevent Stroke?
Combining clopidogrel and aspirin following a small stroke or minor stroke symptoms reduces the risk of a new stroke, heart attack, or other ischemic event within 90 days, say researchers from the National Institute of Neurological Disorders and Stroke.
In POINT (Platelet-Oriented Inhibition in New TIA and minor ischemic stroke), an international clinical trial, 5% of the combination therapy group and 6.5% of the aspirin-only group had an ischemic event within 90 days. The benefit of the combination was concentrated in the first 2 weeks, while the risk of bleeding was constant over 90 days, says Walter Koroshetz, MD, director of NINDS, thus the treatment may be most valuable in acute management of a minor ischemic stroke or TIA.
The study was stopped early not only because the combination therapy was more effective than aspirin alone in preventing severe strokes, but also due to the risk of severe hemorrhage. The combination therapy was associated with an increase in major bleeding, although many of the episodes were not fatal and occurred outside the brain: 0.9% of the combination group had a major hemorrhage, compared with 0.4% of the aspirin-only group.
“Overall, the risk of severe bleeding was very small,” says lead investigator S. Claiborne Johnston, MD, PhD, “but it was not zero.”
Combining clopidogrel and aspirin following a small stroke or minor stroke symptoms reduces the risk of a new stroke, heart attack, or other ischemic event within 90 days, say researchers from the National Institute of Neurological Disorders and Stroke.
In POINT (Platelet-Oriented Inhibition in New TIA and minor ischemic stroke), an international clinical trial, 5% of the combination therapy group and 6.5% of the aspirin-only group had an ischemic event within 90 days. The benefit of the combination was concentrated in the first 2 weeks, while the risk of bleeding was constant over 90 days, says Walter Koroshetz, MD, director of NINDS, thus the treatment may be most valuable in acute management of a minor ischemic stroke or TIA.
The study was stopped early not only because the combination therapy was more effective than aspirin alone in preventing severe strokes, but also due to the risk of severe hemorrhage. The combination therapy was associated with an increase in major bleeding, although many of the episodes were not fatal and occurred outside the brain: 0.9% of the combination group had a major hemorrhage, compared with 0.4% of the aspirin-only group.
“Overall, the risk of severe bleeding was very small,” says lead investigator S. Claiborne Johnston, MD, PhD, “but it was not zero.”
Combining clopidogrel and aspirin following a small stroke or minor stroke symptoms reduces the risk of a new stroke, heart attack, or other ischemic event within 90 days, say researchers from the National Institute of Neurological Disorders and Stroke.
In POINT (Platelet-Oriented Inhibition in New TIA and minor ischemic stroke), an international clinical trial, 5% of the combination therapy group and 6.5% of the aspirin-only group had an ischemic event within 90 days. The benefit of the combination was concentrated in the first 2 weeks, while the risk of bleeding was constant over 90 days, says Walter Koroshetz, MD, director of NINDS, thus the treatment may be most valuable in acute management of a minor ischemic stroke or TIA.
The study was stopped early not only because the combination therapy was more effective than aspirin alone in preventing severe strokes, but also due to the risk of severe hemorrhage. The combination therapy was associated with an increase in major bleeding, although many of the episodes were not fatal and occurred outside the brain: 0.9% of the combination group had a major hemorrhage, compared with 0.4% of the aspirin-only group.
“Overall, the risk of severe bleeding was very small,” says lead investigator S. Claiborne Johnston, MD, PhD, “but it was not zero.”
Are PTSD Responses Inherited or Acquired?
Neuroimaging studies have consistently reported reduced activation of the medial prefrontal cortex (mPFC) in patients with posttraumatic stress disorder (PTSD) while they recall and imagine stressful personal events. During script-driven imagery (SDI) sessions, patients with PTSD exhibit increased psychophysiologic (eg, heart rate, skin conductance, and facial electromyographic) responses to trauma-related memories. However, the origin of the responses remained unclear. Are they familial, acquired, or resulting from trauma exposure?
Researchers from Harvard University, University of California Los Angeles, and University of New England conducted a study of 26 male identical twin pairs to help find the answer. The participants were divided into 4 groups: combat-exposed with PTSD (ExP+), their combat-unexposed twins without PTSD, combat-exposed participants without PTSD, and their combat-unexposed twins without PTSD. They engaged in SDI during functional magnetic resonance (fMRI) imaging and concurrent skin conductance measurement.
The results of the fMRI tests showed diminished activation in the medial prefrontal cortex of the patients with PTSD compared with the other groups. The SC response scores did not correlate significantly with PTSD symptom severity.
Contrary to the researchers’ predictions, mPFC activation was not inversely correlated with PTSD symptom severity. However, they say their finding of reduced mPFC activation in the ExP+ group provides evidence that the abnormality is an acquired characteristic. If those findings are replicated, such objectively measured biologic characteristics could potentially aid in diagnosing PTSD or assessing treatment response.
Source:
Dahlgren MK, Laifer LM, VanElzakker MB, et al. Psychol Med. 2018;48(7):1128-1138.
doi: 10.1017/S003329171700263X.
Neuroimaging studies have consistently reported reduced activation of the medial prefrontal cortex (mPFC) in patients with posttraumatic stress disorder (PTSD) while they recall and imagine stressful personal events. During script-driven imagery (SDI) sessions, patients with PTSD exhibit increased psychophysiologic (eg, heart rate, skin conductance, and facial electromyographic) responses to trauma-related memories. However, the origin of the responses remained unclear. Are they familial, acquired, or resulting from trauma exposure?
Researchers from Harvard University, University of California Los Angeles, and University of New England conducted a study of 26 male identical twin pairs to help find the answer. The participants were divided into 4 groups: combat-exposed with PTSD (ExP+), their combat-unexposed twins without PTSD, combat-exposed participants without PTSD, and their combat-unexposed twins without PTSD. They engaged in SDI during functional magnetic resonance (fMRI) imaging and concurrent skin conductance measurement.
The results of the fMRI tests showed diminished activation in the medial prefrontal cortex of the patients with PTSD compared with the other groups. The SC response scores did not correlate significantly with PTSD symptom severity.
Contrary to the researchers’ predictions, mPFC activation was not inversely correlated with PTSD symptom severity. However, they say their finding of reduced mPFC activation in the ExP+ group provides evidence that the abnormality is an acquired characteristic. If those findings are replicated, such objectively measured biologic characteristics could potentially aid in diagnosing PTSD or assessing treatment response.
Source:
Dahlgren MK, Laifer LM, VanElzakker MB, et al. Psychol Med. 2018;48(7):1128-1138.
doi: 10.1017/S003329171700263X.
Neuroimaging studies have consistently reported reduced activation of the medial prefrontal cortex (mPFC) in patients with posttraumatic stress disorder (PTSD) while they recall and imagine stressful personal events. During script-driven imagery (SDI) sessions, patients with PTSD exhibit increased psychophysiologic (eg, heart rate, skin conductance, and facial electromyographic) responses to trauma-related memories. However, the origin of the responses remained unclear. Are they familial, acquired, or resulting from trauma exposure?
Researchers from Harvard University, University of California Los Angeles, and University of New England conducted a study of 26 male identical twin pairs to help find the answer. The participants were divided into 4 groups: combat-exposed with PTSD (ExP+), their combat-unexposed twins without PTSD, combat-exposed participants without PTSD, and their combat-unexposed twins without PTSD. They engaged in SDI during functional magnetic resonance (fMRI) imaging and concurrent skin conductance measurement.
The results of the fMRI tests showed diminished activation in the medial prefrontal cortex of the patients with PTSD compared with the other groups. The SC response scores did not correlate significantly with PTSD symptom severity.
Contrary to the researchers’ predictions, mPFC activation was not inversely correlated with PTSD symptom severity. However, they say their finding of reduced mPFC activation in the ExP+ group provides evidence that the abnormality is an acquired characteristic. If those findings are replicated, such objectively measured biologic characteristics could potentially aid in diagnosing PTSD or assessing treatment response.
Source:
Dahlgren MK, Laifer LM, VanElzakker MB, et al. Psychol Med. 2018;48(7):1128-1138.
doi: 10.1017/S003329171700263X.
Federal Health Care Data Trends: Gulf War & Post-9/11 Veterans
Currently, the largest growing percentage of veterans are those who served during the Gulf War and Post-9/11 era. Over the next few years, nearly every state in the US can expect growth in this veteran population greater than 20%. Although this group of 4.1 million veterans tend to use their VA health benefits less than do the veterans of previous eras, these veterans also exhibit some unique qualities not previously seen in VA patient populations. These veterans already have been identified to be at an increased risk of acquiring several diseases, which include arthritis, chronic fatigue syndrome, and sleep apnea. But perhaps the biggest shift, and the one that could require the most change in the way the Veterans Health Administration provides care, is along gender lines.
Click here to continue reading.
Currently, the largest growing percentage of veterans are those who served during the Gulf War and Post-9/11 era. Over the next few years, nearly every state in the US can expect growth in this veteran population greater than 20%. Although this group of 4.1 million veterans tend to use their VA health benefits less than do the veterans of previous eras, these veterans also exhibit some unique qualities not previously seen in VA patient populations. These veterans already have been identified to be at an increased risk of acquiring several diseases, which include arthritis, chronic fatigue syndrome, and sleep apnea. But perhaps the biggest shift, and the one that could require the most change in the way the Veterans Health Administration provides care, is along gender lines.
Click here to continue reading.
Currently, the largest growing percentage of veterans are those who served during the Gulf War and Post-9/11 era. Over the next few years, nearly every state in the US can expect growth in this veteran population greater than 20%. Although this group of 4.1 million veterans tend to use their VA health benefits less than do the veterans of previous eras, these veterans also exhibit some unique qualities not previously seen in VA patient populations. These veterans already have been identified to be at an increased risk of acquiring several diseases, which include arthritis, chronic fatigue syndrome, and sleep apnea. But perhaps the biggest shift, and the one that could require the most change in the way the Veterans Health Administration provides care, is along gender lines.
Click here to continue reading.
Diabetes Mellitus Federal Health Data Trends (FULL)
Diabetes mellitus (DM) is the seventh leading cause of death in the U.S., but after decades of rapid growth, both the incidence and prevalence appear to have leveled off. The VA spends $1.5 billion annually to treat patients with DM. Veterans are 2.5 times more likely than nonveterans to have diabetes, and many have comorbid conditions, including obesity, hypoglycemia, hypertension, dyslipidemia, cardiovascular disease, stroke, blindness, kidney disease, and amputations. Obesity and DM remain closely related, and more than two-thirds of women veterans with DM also have obesity.
A number of factors seem to increase the risk of type 2 DM for veterans. For example, DM is associated with exposure to herbicides, such as Agent Orange; past physical strain with chronic pain, and degenerative joint damage. Certain factors also increase risk of obesity, such as advanced age and low income, as well as limited access to healthy and high-quality foods. High-risk ethnic groups for diabetes include African Americans, Hispanics, Native Americans, Asians, and Pacific Islanders. Veterans with prediabetes, hypertension, low highdensity lipoprotein cholesterol levels, high triglyceride levels, and insufficient physical activity also are at increased risk.


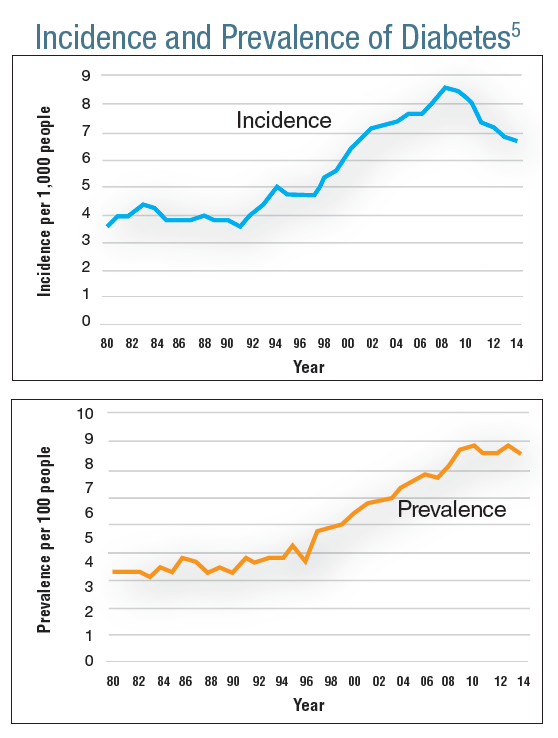
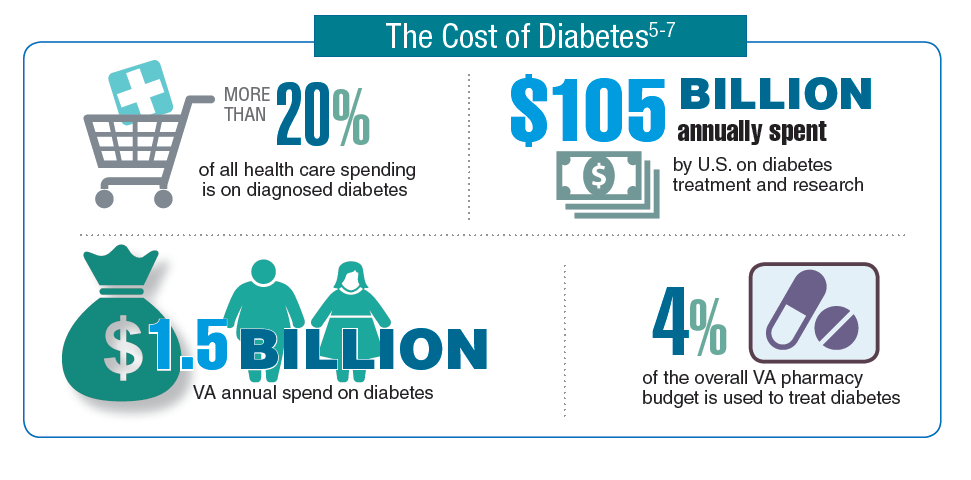
Click here to read the digital edition.
Diabetes mellitus (DM) is the seventh leading cause of death in the U.S., but after decades of rapid growth, both the incidence and prevalence appear to have leveled off. The VA spends $1.5 billion annually to treat patients with DM. Veterans are 2.5 times more likely than nonveterans to have diabetes, and many have comorbid conditions, including obesity, hypoglycemia, hypertension, dyslipidemia, cardiovascular disease, stroke, blindness, kidney disease, and amputations. Obesity and DM remain closely related, and more than two-thirds of women veterans with DM also have obesity.
A number of factors seem to increase the risk of type 2 DM for veterans. For example, DM is associated with exposure to herbicides, such as Agent Orange; past physical strain with chronic pain, and degenerative joint damage. Certain factors also increase risk of obesity, such as advanced age and low income, as well as limited access to healthy and high-quality foods. High-risk ethnic groups for diabetes include African Americans, Hispanics, Native Americans, Asians, and Pacific Islanders. Veterans with prediabetes, hypertension, low highdensity lipoprotein cholesterol levels, high triglyceride levels, and insufficient physical activity also are at increased risk.
Click here to read the digital edition.
Diabetes mellitus (DM) is the seventh leading cause of death in the U.S., but after decades of rapid growth, both the incidence and prevalence appear to have leveled off. The VA spends $1.5 billion annually to treat patients with DM. Veterans are 2.5 times more likely than nonveterans to have diabetes, and many have comorbid conditions, including obesity, hypoglycemia, hypertension, dyslipidemia, cardiovascular disease, stroke, blindness, kidney disease, and amputations. Obesity and DM remain closely related, and more than two-thirds of women veterans with DM also have obesity.
A number of factors seem to increase the risk of type 2 DM for veterans. For example, DM is associated with exposure to herbicides, such as Agent Orange; past physical strain with chronic pain, and degenerative joint damage. Certain factors also increase risk of obesity, such as advanced age and low income, as well as limited access to healthy and high-quality foods. High-risk ethnic groups for diabetes include African Americans, Hispanics, Native Americans, Asians, and Pacific Islanders. Veterans with prediabetes, hypertension, low highdensity lipoprotein cholesterol levels, high triglyceride levels, and insufficient physical activity also are at increased risk.