User login
Giving flu and COVID-19 shots at same time appears safe, effective: Study
Overall, the NVX-CoV2373 vaccine (Novavax) is showing 89.8% efficacy in an ongoing, placebo-controlled phase 3 study. When the researchers gave a smaller group of 431 volunteers from the same study an influenza shot at the same time, efficacy dropped slightly to 87.5%.
“These results demonstrate the promising opportunity for concomitant vaccination, which may lead to higher vaccination rates and further protection against both viruses,” said study coauthor Raja Rajaram, MD, medical affairs lead, Europe, Middle East, and Africa at Seqirus, the company that supplied the influenza vaccines for the research.
The research was published online June 13 as a medRxiv preprint.
“With these COVID-19 vaccines, there are essentially no concurrent use studies,” Paul A. Offit, MD, told this news organization when asked to comment.
Traditionally, how a new vaccine might interact with existing vaccines is studied before the product is cleared for use. That was not the case, however, with the COVID-19 vaccines made available through expedited emergency use authorization.
The researchers found no major safety concerns associated with concomitant vaccination, Dr. Rajaram said. In addition to safety, the aim of the current study was to determine whether either vaccine changes the immunogenicity or effectiveness of the other.
“It’s a small study, but it’s certainly encouraging to know that there didn’t seem to be a big decrease in immunogenicity either way and the safety profile was similar. Not identical, but similar,” added Dr. Offit, director of the Vaccine Education Center at Children’s Hospital of Philadelphia.
Some adverse events were more common in the co-administration group. For example, injection-site tenderness was reported by 70%, versus 58% for those who got the COVID-19 shot alone. The same was true for pain at the injection site, 40% versus 29%; fatigue, 28% versus 19%; and muscle pain, 28% versus 21%.
Rates of unsolicited adverse events, adverse events that required medical attention, and serious adverse events were low and well balanced between groups.
Fewer antibodies important?
Although co-administering the two vaccines did not change the immune response for the influenza vaccine, the spike protein antibody response to the COVID-19 vaccine was less robust.
Antibody titer levels at day 35 were 46,678 among people in the Novavax vaccine alone group, compared with 31,236 titers in the participants who received both vaccines.
“This impact did not seem to be clinically meaningful as vaccine efficacy appeared to be preserved,” the researchers noted.
Gregory A. Poland, MD, an internist and part of the Vaccine Research Group at Mayo Clinic in Rochester, Minn., agreed. “I highly doubt that is significant,” he said in an interview.
Dr. Rajaram said the antibody findings are “slightly surprising but not completely unexpected” because the same observation has been made in other combination vaccine studies. He added that the antibody levels “remain very high, although we do not yet know what antibody levels are required to achieve protection against COVID-19.”
The decrease could become more concerning if people start with fewer antibodies and they drop over time with normal waning of protection, Dr. Poland said. This group could include people over age 65 or people who are immunocompromised. More data would be needed to confirm this, he added.
A boost for booster vaccines?
The research could carry implications for future COVID-19 booster shots, Dr. Poland said.
“Overall, the study results are reassuring and of potential practical importance if we have to give booster doses. It will make it easier to give them both in one visit,” said Dr. Poland, who was not affiliated with the research.
Although Novavax could be positioning itself as a logical choice for a COVID-19 booster based on the findings, Dr. Offit believes it is more important to focus on having more COVID-19 vaccine options available.
“There may be, as we say at the track, ‘courses for horses,’ ” he said, meaning that different vaccines may be better suited for different situations.
“It’s likely we’re going to find these vaccines have different safety profiles, they may have different populations for whom they work best, and they may have differences in terms of their long-term durability,” he added. Also, some may prove more effective against certain variants of concern.
The Novavax vaccine would add a new class of COVID-19 vaccine to the mRNA and adenovirus vaccines. NVX-CoV2373 is a recombinant spike protein vaccine.
“I think the more vaccines that are available here, the better,” Dr. Offit said.
Study limitations
Dr. Poland shared some caveats. The study was primarily conducted in adults aged 18-64 years, so there is less certainty on what could happen in people over 65. Furthermore, co-administration was evaluated after the first dose of the Novavax vaccine. “The reason I bring that up is most of the COVID-19 vaccine reactogenicity occurs with dose two, not dose one.
“All in all, it’s an important first step – but it’s only a first step,” Dr. Poland said. “We need more data, including in elderly people who are primarily at risk for morbidity and mortality from the flu.”
He suggested expanding the research to study co-administration of COVID-19 vaccines with different formulations of influenza vaccines.
The study was supported by Novavax. Dr. Offit had no relevant financial disclosures. Dr. Poland serves as a consultant to all of the COVID-19 vaccine companies.
A version of this article first appeared on Medscape.com.
Overall, the NVX-CoV2373 vaccine (Novavax) is showing 89.8% efficacy in an ongoing, placebo-controlled phase 3 study. When the researchers gave a smaller group of 431 volunteers from the same study an influenza shot at the same time, efficacy dropped slightly to 87.5%.
“These results demonstrate the promising opportunity for concomitant vaccination, which may lead to higher vaccination rates and further protection against both viruses,” said study coauthor Raja Rajaram, MD, medical affairs lead, Europe, Middle East, and Africa at Seqirus, the company that supplied the influenza vaccines for the research.
The research was published online June 13 as a medRxiv preprint.
“With these COVID-19 vaccines, there are essentially no concurrent use studies,” Paul A. Offit, MD, told this news organization when asked to comment.
Traditionally, how a new vaccine might interact with existing vaccines is studied before the product is cleared for use. That was not the case, however, with the COVID-19 vaccines made available through expedited emergency use authorization.
The researchers found no major safety concerns associated with concomitant vaccination, Dr. Rajaram said. In addition to safety, the aim of the current study was to determine whether either vaccine changes the immunogenicity or effectiveness of the other.
“It’s a small study, but it’s certainly encouraging to know that there didn’t seem to be a big decrease in immunogenicity either way and the safety profile was similar. Not identical, but similar,” added Dr. Offit, director of the Vaccine Education Center at Children’s Hospital of Philadelphia.
Some adverse events were more common in the co-administration group. For example, injection-site tenderness was reported by 70%, versus 58% for those who got the COVID-19 shot alone. The same was true for pain at the injection site, 40% versus 29%; fatigue, 28% versus 19%; and muscle pain, 28% versus 21%.
Rates of unsolicited adverse events, adverse events that required medical attention, and serious adverse events were low and well balanced between groups.
Fewer antibodies important?
Although co-administering the two vaccines did not change the immune response for the influenza vaccine, the spike protein antibody response to the COVID-19 vaccine was less robust.
Antibody titer levels at day 35 were 46,678 among people in the Novavax vaccine alone group, compared with 31,236 titers in the participants who received both vaccines.
“This impact did not seem to be clinically meaningful as vaccine efficacy appeared to be preserved,” the researchers noted.
Gregory A. Poland, MD, an internist and part of the Vaccine Research Group at Mayo Clinic in Rochester, Minn., agreed. “I highly doubt that is significant,” he said in an interview.
Dr. Rajaram said the antibody findings are “slightly surprising but not completely unexpected” because the same observation has been made in other combination vaccine studies. He added that the antibody levels “remain very high, although we do not yet know what antibody levels are required to achieve protection against COVID-19.”
The decrease could become more concerning if people start with fewer antibodies and they drop over time with normal waning of protection, Dr. Poland said. This group could include people over age 65 or people who are immunocompromised. More data would be needed to confirm this, he added.
A boost for booster vaccines?
The research could carry implications for future COVID-19 booster shots, Dr. Poland said.
“Overall, the study results are reassuring and of potential practical importance if we have to give booster doses. It will make it easier to give them both in one visit,” said Dr. Poland, who was not affiliated with the research.
Although Novavax could be positioning itself as a logical choice for a COVID-19 booster based on the findings, Dr. Offit believes it is more important to focus on having more COVID-19 vaccine options available.
“There may be, as we say at the track, ‘courses for horses,’ ” he said, meaning that different vaccines may be better suited for different situations.
“It’s likely we’re going to find these vaccines have different safety profiles, they may have different populations for whom they work best, and they may have differences in terms of their long-term durability,” he added. Also, some may prove more effective against certain variants of concern.
The Novavax vaccine would add a new class of COVID-19 vaccine to the mRNA and adenovirus vaccines. NVX-CoV2373 is a recombinant spike protein vaccine.
“I think the more vaccines that are available here, the better,” Dr. Offit said.
Study limitations
Dr. Poland shared some caveats. The study was primarily conducted in adults aged 18-64 years, so there is less certainty on what could happen in people over 65. Furthermore, co-administration was evaluated after the first dose of the Novavax vaccine. “The reason I bring that up is most of the COVID-19 vaccine reactogenicity occurs with dose two, not dose one.
“All in all, it’s an important first step – but it’s only a first step,” Dr. Poland said. “We need more data, including in elderly people who are primarily at risk for morbidity and mortality from the flu.”
He suggested expanding the research to study co-administration of COVID-19 vaccines with different formulations of influenza vaccines.
The study was supported by Novavax. Dr. Offit had no relevant financial disclosures. Dr. Poland serves as a consultant to all of the COVID-19 vaccine companies.
A version of this article first appeared on Medscape.com.
Overall, the NVX-CoV2373 vaccine (Novavax) is showing 89.8% efficacy in an ongoing, placebo-controlled phase 3 study. When the researchers gave a smaller group of 431 volunteers from the same study an influenza shot at the same time, efficacy dropped slightly to 87.5%.
“These results demonstrate the promising opportunity for concomitant vaccination, which may lead to higher vaccination rates and further protection against both viruses,” said study coauthor Raja Rajaram, MD, medical affairs lead, Europe, Middle East, and Africa at Seqirus, the company that supplied the influenza vaccines for the research.
The research was published online June 13 as a medRxiv preprint.
“With these COVID-19 vaccines, there are essentially no concurrent use studies,” Paul A. Offit, MD, told this news organization when asked to comment.
Traditionally, how a new vaccine might interact with existing vaccines is studied before the product is cleared for use. That was not the case, however, with the COVID-19 vaccines made available through expedited emergency use authorization.
The researchers found no major safety concerns associated with concomitant vaccination, Dr. Rajaram said. In addition to safety, the aim of the current study was to determine whether either vaccine changes the immunogenicity or effectiveness of the other.
“It’s a small study, but it’s certainly encouraging to know that there didn’t seem to be a big decrease in immunogenicity either way and the safety profile was similar. Not identical, but similar,” added Dr. Offit, director of the Vaccine Education Center at Children’s Hospital of Philadelphia.
Some adverse events were more common in the co-administration group. For example, injection-site tenderness was reported by 70%, versus 58% for those who got the COVID-19 shot alone. The same was true for pain at the injection site, 40% versus 29%; fatigue, 28% versus 19%; and muscle pain, 28% versus 21%.
Rates of unsolicited adverse events, adverse events that required medical attention, and serious adverse events were low and well balanced between groups.
Fewer antibodies important?
Although co-administering the two vaccines did not change the immune response for the influenza vaccine, the spike protein antibody response to the COVID-19 vaccine was less robust.
Antibody titer levels at day 35 were 46,678 among people in the Novavax vaccine alone group, compared with 31,236 titers in the participants who received both vaccines.
“This impact did not seem to be clinically meaningful as vaccine efficacy appeared to be preserved,” the researchers noted.
Gregory A. Poland, MD, an internist and part of the Vaccine Research Group at Mayo Clinic in Rochester, Minn., agreed. “I highly doubt that is significant,” he said in an interview.
Dr. Rajaram said the antibody findings are “slightly surprising but not completely unexpected” because the same observation has been made in other combination vaccine studies. He added that the antibody levels “remain very high, although we do not yet know what antibody levels are required to achieve protection against COVID-19.”
The decrease could become more concerning if people start with fewer antibodies and they drop over time with normal waning of protection, Dr. Poland said. This group could include people over age 65 or people who are immunocompromised. More data would be needed to confirm this, he added.
A boost for booster vaccines?
The research could carry implications for future COVID-19 booster shots, Dr. Poland said.
“Overall, the study results are reassuring and of potential practical importance if we have to give booster doses. It will make it easier to give them both in one visit,” said Dr. Poland, who was not affiliated with the research.
Although Novavax could be positioning itself as a logical choice for a COVID-19 booster based on the findings, Dr. Offit believes it is more important to focus on having more COVID-19 vaccine options available.
“There may be, as we say at the track, ‘courses for horses,’ ” he said, meaning that different vaccines may be better suited for different situations.
“It’s likely we’re going to find these vaccines have different safety profiles, they may have different populations for whom they work best, and they may have differences in terms of their long-term durability,” he added. Also, some may prove more effective against certain variants of concern.
The Novavax vaccine would add a new class of COVID-19 vaccine to the mRNA and adenovirus vaccines. NVX-CoV2373 is a recombinant spike protein vaccine.
“I think the more vaccines that are available here, the better,” Dr. Offit said.
Study limitations
Dr. Poland shared some caveats. The study was primarily conducted in adults aged 18-64 years, so there is less certainty on what could happen in people over 65. Furthermore, co-administration was evaluated after the first dose of the Novavax vaccine. “The reason I bring that up is most of the COVID-19 vaccine reactogenicity occurs with dose two, not dose one.
“All in all, it’s an important first step – but it’s only a first step,” Dr. Poland said. “We need more data, including in elderly people who are primarily at risk for morbidity and mortality from the flu.”
He suggested expanding the research to study co-administration of COVID-19 vaccines with different formulations of influenza vaccines.
The study was supported by Novavax. Dr. Offit had no relevant financial disclosures. Dr. Poland serves as a consultant to all of the COVID-19 vaccine companies.
A version of this article first appeared on Medscape.com.
AHA: Don’t delay COVID shot while CDC reviews myocarditis cases
While the investigation into cases of myocarditis possibly associated with COVID vaccines proceeds, the American Heart Association/American Stroke Association (ASA) continue to urge everyone who is eligible for the vaccine to get it without delay.
“We remain confident that the benefits of vaccination far exceed the very unusual risks,” the leadership of the AHA/ASA said in a statement issued June 12.
“The risks of COVID-19 infection include its potentially fatal consequences and the potential long-term health effects that are still revealing themselves, including lingering consequences affecting the heart, brain, vascular system, and other organs after infection,” they point out.
Late last week, the Centers for Disease Control and Prevention alerted health care providers that the COVID-19 Vaccine Safety Technical Work Group (VaST) of the Advisory Committee on Immunization Practices (ACIP) will meet June 18 to review cases of myocarditis reported in adolescents and young adults after they received a COVID-19 vaccine manufactured by Pfizer-BioNTech or Moderna.
The CDC is monitoring the Vaccine Adverse Events Reporting System (VAERS) and the Vaccine Safety Datalink (VSD) for cases of myocarditis that have been associated with the mRNA vaccines against SARS-CoV-2 from Pfizer and Moderna.
These cases may occur more often in males than females and more frequently after the second dose than the first dose of either mRNA vaccine. Symptoms typically occur in the 3 days after administration.
“The CDC’s ongoing investigation into cases of suspected myocarditis reflects a strong and steadfast commitment to transparency and the importance of scientific rigor on all fronts. We applaud the CDC’s unwavering efforts to lead our nation’s scientific and public health efforts, including ensuring the continued safety of the COVID-19 vaccines,” the AHA/ASA states.
They emphasize that vaccinations should continue, and say it’s important to consider the details of the suspected myocarditis cases being investigated by the CDC.
As of June 11, more than 306 million doses of COVID-19 vaccines have been administered in the United States (since Dec. 14, 2020) and nearly 43% of Americans – more than 142 million people – are now fully vaccinated.
According to the June 10 CDC VAERS report detailing adverse events through May 31:
- 789 cases of suspected myocarditis have been reported, with 475 involving people younger than 30 years; 79 cases reported were in patients 16 or 17 years old.
- The vast majority (81%) of the 270 patients younger than 30 years who were discharged from care after suspected myocarditis related to COVID-19 vaccination have recovered fully; the remaining 19% of patients report ongoing symptoms or complete data are missing.
- 196 cases of suspected myocarditis after a COVID-19 vaccine were reported in young adults 18 to 24 years of age, which is higher than expected for this age group.
As of May 31, only about 9% of the COVID-19 vaccine doses administered were to people 16 to 24 years of age, which is why this “higher-than-normal rate of possible myocarditis cases” warrants investigation, the AHA/ASA says.
They note that these suspected myocarditis cases were reported to VAERS because of their proximity to COVID-19 vaccine administration.
It remains to be determined which cases meet the clinical criteria for a diagnosis of myocarditis and whether they have any direct connection to the COVID-19 vaccine, the AHA/ASA says.
They urge all health care professionals to be aware of “very rare” adverse events that could be related to a COVID-19 vaccine, including myocarditis, blood clots, low platelets, and symptoms of severe inflammation.
They advise asking patients who present with symptoms related to these conditions about the timing of recent COVID vaccinations, as needed, to confirm the diagnosis and provide appropriate treatment quickly.
The AHA will be at the CDC’s June 18 meeting to review the latest evidence on cases of suspected myocarditis after the COVID-19 vaccine, the statement adds.
The statement notes that it reflects the views of the AHA/ASA and its scientific leadership, including current president Mitchel S.V. Elkind, MD, PhD; immediate past-president Robert A. Harrington, MD; president-elect Donald M. Lloyd-Jones, MD; AHA/ASA chief science and medical officer Mariell Jessup, MD; and chief medical officer for prevention Eduardo Sanchez, MD, MPH.
A version of this article first appeared on Medscape.com.
While the investigation into cases of myocarditis possibly associated with COVID vaccines proceeds, the American Heart Association/American Stroke Association (ASA) continue to urge everyone who is eligible for the vaccine to get it without delay.
“We remain confident that the benefits of vaccination far exceed the very unusual risks,” the leadership of the AHA/ASA said in a statement issued June 12.
“The risks of COVID-19 infection include its potentially fatal consequences and the potential long-term health effects that are still revealing themselves, including lingering consequences affecting the heart, brain, vascular system, and other organs after infection,” they point out.
Late last week, the Centers for Disease Control and Prevention alerted health care providers that the COVID-19 Vaccine Safety Technical Work Group (VaST) of the Advisory Committee on Immunization Practices (ACIP) will meet June 18 to review cases of myocarditis reported in adolescents and young adults after they received a COVID-19 vaccine manufactured by Pfizer-BioNTech or Moderna.
The CDC is monitoring the Vaccine Adverse Events Reporting System (VAERS) and the Vaccine Safety Datalink (VSD) for cases of myocarditis that have been associated with the mRNA vaccines against SARS-CoV-2 from Pfizer and Moderna.
These cases may occur more often in males than females and more frequently after the second dose than the first dose of either mRNA vaccine. Symptoms typically occur in the 3 days after administration.
“The CDC’s ongoing investigation into cases of suspected myocarditis reflects a strong and steadfast commitment to transparency and the importance of scientific rigor on all fronts. We applaud the CDC’s unwavering efforts to lead our nation’s scientific and public health efforts, including ensuring the continued safety of the COVID-19 vaccines,” the AHA/ASA states.
They emphasize that vaccinations should continue, and say it’s important to consider the details of the suspected myocarditis cases being investigated by the CDC.
As of June 11, more than 306 million doses of COVID-19 vaccines have been administered in the United States (since Dec. 14, 2020) and nearly 43% of Americans – more than 142 million people – are now fully vaccinated.
According to the June 10 CDC VAERS report detailing adverse events through May 31:
- 789 cases of suspected myocarditis have been reported, with 475 involving people younger than 30 years; 79 cases reported were in patients 16 or 17 years old.
- The vast majority (81%) of the 270 patients younger than 30 years who were discharged from care after suspected myocarditis related to COVID-19 vaccination have recovered fully; the remaining 19% of patients report ongoing symptoms or complete data are missing.
- 196 cases of suspected myocarditis after a COVID-19 vaccine were reported in young adults 18 to 24 years of age, which is higher than expected for this age group.
As of May 31, only about 9% of the COVID-19 vaccine doses administered were to people 16 to 24 years of age, which is why this “higher-than-normal rate of possible myocarditis cases” warrants investigation, the AHA/ASA says.
They note that these suspected myocarditis cases were reported to VAERS because of their proximity to COVID-19 vaccine administration.
It remains to be determined which cases meet the clinical criteria for a diagnosis of myocarditis and whether they have any direct connection to the COVID-19 vaccine, the AHA/ASA says.
They urge all health care professionals to be aware of “very rare” adverse events that could be related to a COVID-19 vaccine, including myocarditis, blood clots, low platelets, and symptoms of severe inflammation.
They advise asking patients who present with symptoms related to these conditions about the timing of recent COVID vaccinations, as needed, to confirm the diagnosis and provide appropriate treatment quickly.
The AHA will be at the CDC’s June 18 meeting to review the latest evidence on cases of suspected myocarditis after the COVID-19 vaccine, the statement adds.
The statement notes that it reflects the views of the AHA/ASA and its scientific leadership, including current president Mitchel S.V. Elkind, MD, PhD; immediate past-president Robert A. Harrington, MD; president-elect Donald M. Lloyd-Jones, MD; AHA/ASA chief science and medical officer Mariell Jessup, MD; and chief medical officer for prevention Eduardo Sanchez, MD, MPH.
A version of this article first appeared on Medscape.com.
While the investigation into cases of myocarditis possibly associated with COVID vaccines proceeds, the American Heart Association/American Stroke Association (ASA) continue to urge everyone who is eligible for the vaccine to get it without delay.
“We remain confident that the benefits of vaccination far exceed the very unusual risks,” the leadership of the AHA/ASA said in a statement issued June 12.
“The risks of COVID-19 infection include its potentially fatal consequences and the potential long-term health effects that are still revealing themselves, including lingering consequences affecting the heart, brain, vascular system, and other organs after infection,” they point out.
Late last week, the Centers for Disease Control and Prevention alerted health care providers that the COVID-19 Vaccine Safety Technical Work Group (VaST) of the Advisory Committee on Immunization Practices (ACIP) will meet June 18 to review cases of myocarditis reported in adolescents and young adults after they received a COVID-19 vaccine manufactured by Pfizer-BioNTech or Moderna.
The CDC is monitoring the Vaccine Adverse Events Reporting System (VAERS) and the Vaccine Safety Datalink (VSD) for cases of myocarditis that have been associated with the mRNA vaccines against SARS-CoV-2 from Pfizer and Moderna.
These cases may occur more often in males than females and more frequently after the second dose than the first dose of either mRNA vaccine. Symptoms typically occur in the 3 days after administration.
“The CDC’s ongoing investigation into cases of suspected myocarditis reflects a strong and steadfast commitment to transparency and the importance of scientific rigor on all fronts. We applaud the CDC’s unwavering efforts to lead our nation’s scientific and public health efforts, including ensuring the continued safety of the COVID-19 vaccines,” the AHA/ASA states.
They emphasize that vaccinations should continue, and say it’s important to consider the details of the suspected myocarditis cases being investigated by the CDC.
As of June 11, more than 306 million doses of COVID-19 vaccines have been administered in the United States (since Dec. 14, 2020) and nearly 43% of Americans – more than 142 million people – are now fully vaccinated.
According to the June 10 CDC VAERS report detailing adverse events through May 31:
- 789 cases of suspected myocarditis have been reported, with 475 involving people younger than 30 years; 79 cases reported were in patients 16 or 17 years old.
- The vast majority (81%) of the 270 patients younger than 30 years who were discharged from care after suspected myocarditis related to COVID-19 vaccination have recovered fully; the remaining 19% of patients report ongoing symptoms or complete data are missing.
- 196 cases of suspected myocarditis after a COVID-19 vaccine were reported in young adults 18 to 24 years of age, which is higher than expected for this age group.
As of May 31, only about 9% of the COVID-19 vaccine doses administered were to people 16 to 24 years of age, which is why this “higher-than-normal rate of possible myocarditis cases” warrants investigation, the AHA/ASA says.
They note that these suspected myocarditis cases were reported to VAERS because of their proximity to COVID-19 vaccine administration.
It remains to be determined which cases meet the clinical criteria for a diagnosis of myocarditis and whether they have any direct connection to the COVID-19 vaccine, the AHA/ASA says.
They urge all health care professionals to be aware of “very rare” adverse events that could be related to a COVID-19 vaccine, including myocarditis, blood clots, low platelets, and symptoms of severe inflammation.
They advise asking patients who present with symptoms related to these conditions about the timing of recent COVID vaccinations, as needed, to confirm the diagnosis and provide appropriate treatment quickly.
The AHA will be at the CDC’s June 18 meeting to review the latest evidence on cases of suspected myocarditis after the COVID-19 vaccine, the statement adds.
The statement notes that it reflects the views of the AHA/ASA and its scientific leadership, including current president Mitchel S.V. Elkind, MD, PhD; immediate past-president Robert A. Harrington, MD; president-elect Donald M. Lloyd-Jones, MD; AHA/ASA chief science and medical officer Mariell Jessup, MD; and chief medical officer for prevention Eduardo Sanchez, MD, MPH.
A version of this article first appeared on Medscape.com.
Watchdog group demands removal of FDA leaders after aducanumab approval
In a letter to the U.S. Department of Health and Human Services Secretary Xavier Becerra, Michael A. Carome, MD, director of Public Citizen’s Health Research Group, said: “The FDA’s decision to approve aducanumab for anyone with Alzheimer’s disease, regardless of severity, showed a stunning disregard for science, eviscerated the agency’s standards for approving new drugs, and ranks as one of the most irresponsible and egregious decisions in the history of the agency.”
Public Citizen urged Mr. Becerra to seek the resignations or the removal of the three FDA officials it said were most responsible for the approval – Acting FDA Commissioner Janet Woodcock, MD; Center for Drug Evaluation and Research (CDER) Director Patrizia Cavazzoni, MD; and CDER’s Office of Neuroscience Director Billy Dunn, MD.
“This decision is a disastrous blow to the agency’s credibility, public health, and the financial sustainability of the Medicare program,” writes Dr. Carome, noting that Biogen said it would charge $56,000 annually for the infusion.
Aaron Kesselheim, MD, one of three FDA Peripheral and Central Nervous System Drugs advisory committee members who resigned in the wake of the approval, agreed with Public Citizen that the agency’s credibility is suffering.
“The aducanumab decision is the worst example yet of the FDA’s movement away from its high standards,” Dr. Kesselheim, a professor of medicine at Harvard Medical School, Boston, and Harvard colleague Jerry Avorn, MD, wrote in the New York Times on June 15.
“As physicians, we know well that Alzheimer’s disease is a terrible condition,” they wrote. However, they added, “approving a drug that has such poor evidence that it works and causes such worrisome side effects is not the solution.”
In his resignation letter, Dr. Kesselheim said he had also been dismayed by the agency’s 2016 approval of eteplirsen (Exondys 51, Sarepta Therapeutics) for Duchenne muscular dystrophy. In both the eteplirsen and aducanumab approvals, the agency went against its advisers’ recommendations, Dr. Kesselheim said.
Advocates who backed approval decry cost
Aducanumab had a rocky road to approval but had unwavering backing from the Alzheimer’s Association and at least one other organization, UsAgainstAlzheimer’s.
The Alzheimer’s Association was particularly outspoken in its support and, in March, was accused of potential conflict of interest by Public Citizen and several neurologists because the association accepted at least $1.4 million from Biogen and its partner Eisai since fiscal year 2018.
The association applauded the FDA approval but, a few days later, expressed outrage over the $56,000-a-year price tag.
“This price is simply unacceptable,” the Alzheimer’s Association said in the statement. “For many, this price will pose an insurmountable barrier to access, it complicates and jeopardizes sustainable access to this treatment, and may further deepen issues of health equity,” the association said, adding, “We call on Biogen to change this price.”
UsAgainstAlzheimer’s also expressed concerns about access, even before it knew aducanumab’s price.
“Shockingly, Medicare does not reimburse patients for the expensive PET scans important to determine whether someone is appropriate for this drug,” noted George Vradenburg, chairman and cofounder of the group, in a June 7 statement. “We intend to work with Biogen and Medicare to make access to this drug affordable for every American who needs it,” Mr. Vradenburg said.
Dr. Carome said the advocates’ complaints were hard to fathom.
“This should not have come as a surprise to anyone,” Dr. Carome said, adding that “it’s essentially the ballpark figure the company threw out weeks ago.”
“Fifty-six-thousand-dollars is particularly egregiously overpriced for a drug that doesn’t work,” Dr. Carome said. “If the [Alzheimer’s Association] truly finds this objectionable, hopefully they’ll stop accepting money from Biogen and its partner Eisai,” he added.
“The Alzheimer’s Association is recognizing that the genie is out of the bottle and that they are going to have trouble reining in the inevitable run-away costs,” said Mike Greicius, MD, MPH, associate professor of neurology at Stanford University’s Wu Tsai Neurosciences Institute, Stanford, California.
“In addition to the eye-popping annual cost that Biogen has invented, I hope the Alzheimer’s Association is also concerned about the dangerously loose and broad FDA labeling which does not require screening for amyloid-positivity and does not restrict use to the milder forms of disease studied in the Phase 3 trials,” Dr. Greicius said.
Another advocacy group, Patients For Affordable Drugs, commended the Alzheimer’s Association. Its statement “was nothing short of courageous, especially in light of the Alzheimer’s Association’s reliance on funding from drug corporations, including Biogen,” said David Mitchell, a cancer patient and founder of Patients For Affordable Drugs, in a statement.
Mr. Mitchell said his members “stand with the Alzheimer’s Association in its denunciation of the price set by Biogen” and called for a new law that would allow Medicare to negotiate drug prices.
A version of this article first appeared on Medscape.com.
In a letter to the U.S. Department of Health and Human Services Secretary Xavier Becerra, Michael A. Carome, MD, director of Public Citizen’s Health Research Group, said: “The FDA’s decision to approve aducanumab for anyone with Alzheimer’s disease, regardless of severity, showed a stunning disregard for science, eviscerated the agency’s standards for approving new drugs, and ranks as one of the most irresponsible and egregious decisions in the history of the agency.”
Public Citizen urged Mr. Becerra to seek the resignations or the removal of the three FDA officials it said were most responsible for the approval – Acting FDA Commissioner Janet Woodcock, MD; Center for Drug Evaluation and Research (CDER) Director Patrizia Cavazzoni, MD; and CDER’s Office of Neuroscience Director Billy Dunn, MD.
“This decision is a disastrous blow to the agency’s credibility, public health, and the financial sustainability of the Medicare program,” writes Dr. Carome, noting that Biogen said it would charge $56,000 annually for the infusion.
Aaron Kesselheim, MD, one of three FDA Peripheral and Central Nervous System Drugs advisory committee members who resigned in the wake of the approval, agreed with Public Citizen that the agency’s credibility is suffering.
“The aducanumab decision is the worst example yet of the FDA’s movement away from its high standards,” Dr. Kesselheim, a professor of medicine at Harvard Medical School, Boston, and Harvard colleague Jerry Avorn, MD, wrote in the New York Times on June 15.
“As physicians, we know well that Alzheimer’s disease is a terrible condition,” they wrote. However, they added, “approving a drug that has such poor evidence that it works and causes such worrisome side effects is not the solution.”
In his resignation letter, Dr. Kesselheim said he had also been dismayed by the agency’s 2016 approval of eteplirsen (Exondys 51, Sarepta Therapeutics) for Duchenne muscular dystrophy. In both the eteplirsen and aducanumab approvals, the agency went against its advisers’ recommendations, Dr. Kesselheim said.
Advocates who backed approval decry cost
Aducanumab had a rocky road to approval but had unwavering backing from the Alzheimer’s Association and at least one other organization, UsAgainstAlzheimer’s.
The Alzheimer’s Association was particularly outspoken in its support and, in March, was accused of potential conflict of interest by Public Citizen and several neurologists because the association accepted at least $1.4 million from Biogen and its partner Eisai since fiscal year 2018.
The association applauded the FDA approval but, a few days later, expressed outrage over the $56,000-a-year price tag.
“This price is simply unacceptable,” the Alzheimer’s Association said in the statement. “For many, this price will pose an insurmountable barrier to access, it complicates and jeopardizes sustainable access to this treatment, and may further deepen issues of health equity,” the association said, adding, “We call on Biogen to change this price.”
UsAgainstAlzheimer’s also expressed concerns about access, even before it knew aducanumab’s price.
“Shockingly, Medicare does not reimburse patients for the expensive PET scans important to determine whether someone is appropriate for this drug,” noted George Vradenburg, chairman and cofounder of the group, in a June 7 statement. “We intend to work with Biogen and Medicare to make access to this drug affordable for every American who needs it,” Mr. Vradenburg said.
Dr. Carome said the advocates’ complaints were hard to fathom.
“This should not have come as a surprise to anyone,” Dr. Carome said, adding that “it’s essentially the ballpark figure the company threw out weeks ago.”
“Fifty-six-thousand-dollars is particularly egregiously overpriced for a drug that doesn’t work,” Dr. Carome said. “If the [Alzheimer’s Association] truly finds this objectionable, hopefully they’ll stop accepting money from Biogen and its partner Eisai,” he added.
“The Alzheimer’s Association is recognizing that the genie is out of the bottle and that they are going to have trouble reining in the inevitable run-away costs,” said Mike Greicius, MD, MPH, associate professor of neurology at Stanford University’s Wu Tsai Neurosciences Institute, Stanford, California.
“In addition to the eye-popping annual cost that Biogen has invented, I hope the Alzheimer’s Association is also concerned about the dangerously loose and broad FDA labeling which does not require screening for amyloid-positivity and does not restrict use to the milder forms of disease studied in the Phase 3 trials,” Dr. Greicius said.
Another advocacy group, Patients For Affordable Drugs, commended the Alzheimer’s Association. Its statement “was nothing short of courageous, especially in light of the Alzheimer’s Association’s reliance on funding from drug corporations, including Biogen,” said David Mitchell, a cancer patient and founder of Patients For Affordable Drugs, in a statement.
Mr. Mitchell said his members “stand with the Alzheimer’s Association in its denunciation of the price set by Biogen” and called for a new law that would allow Medicare to negotiate drug prices.
A version of this article first appeared on Medscape.com.
In a letter to the U.S. Department of Health and Human Services Secretary Xavier Becerra, Michael A. Carome, MD, director of Public Citizen’s Health Research Group, said: “The FDA’s decision to approve aducanumab for anyone with Alzheimer’s disease, regardless of severity, showed a stunning disregard for science, eviscerated the agency’s standards for approving new drugs, and ranks as one of the most irresponsible and egregious decisions in the history of the agency.”
Public Citizen urged Mr. Becerra to seek the resignations or the removal of the three FDA officials it said were most responsible for the approval – Acting FDA Commissioner Janet Woodcock, MD; Center for Drug Evaluation and Research (CDER) Director Patrizia Cavazzoni, MD; and CDER’s Office of Neuroscience Director Billy Dunn, MD.
“This decision is a disastrous blow to the agency’s credibility, public health, and the financial sustainability of the Medicare program,” writes Dr. Carome, noting that Biogen said it would charge $56,000 annually for the infusion.
Aaron Kesselheim, MD, one of three FDA Peripheral and Central Nervous System Drugs advisory committee members who resigned in the wake of the approval, agreed with Public Citizen that the agency’s credibility is suffering.
“The aducanumab decision is the worst example yet of the FDA’s movement away from its high standards,” Dr. Kesselheim, a professor of medicine at Harvard Medical School, Boston, and Harvard colleague Jerry Avorn, MD, wrote in the New York Times on June 15.
“As physicians, we know well that Alzheimer’s disease is a terrible condition,” they wrote. However, they added, “approving a drug that has such poor evidence that it works and causes such worrisome side effects is not the solution.”
In his resignation letter, Dr. Kesselheim said he had also been dismayed by the agency’s 2016 approval of eteplirsen (Exondys 51, Sarepta Therapeutics) for Duchenne muscular dystrophy. In both the eteplirsen and aducanumab approvals, the agency went against its advisers’ recommendations, Dr. Kesselheim said.
Advocates who backed approval decry cost
Aducanumab had a rocky road to approval but had unwavering backing from the Alzheimer’s Association and at least one other organization, UsAgainstAlzheimer’s.
The Alzheimer’s Association was particularly outspoken in its support and, in March, was accused of potential conflict of interest by Public Citizen and several neurologists because the association accepted at least $1.4 million from Biogen and its partner Eisai since fiscal year 2018.
The association applauded the FDA approval but, a few days later, expressed outrage over the $56,000-a-year price tag.
“This price is simply unacceptable,” the Alzheimer’s Association said in the statement. “For many, this price will pose an insurmountable barrier to access, it complicates and jeopardizes sustainable access to this treatment, and may further deepen issues of health equity,” the association said, adding, “We call on Biogen to change this price.”
UsAgainstAlzheimer’s also expressed concerns about access, even before it knew aducanumab’s price.
“Shockingly, Medicare does not reimburse patients for the expensive PET scans important to determine whether someone is appropriate for this drug,” noted George Vradenburg, chairman and cofounder of the group, in a June 7 statement. “We intend to work with Biogen and Medicare to make access to this drug affordable for every American who needs it,” Mr. Vradenburg said.
Dr. Carome said the advocates’ complaints were hard to fathom.
“This should not have come as a surprise to anyone,” Dr. Carome said, adding that “it’s essentially the ballpark figure the company threw out weeks ago.”
“Fifty-six-thousand-dollars is particularly egregiously overpriced for a drug that doesn’t work,” Dr. Carome said. “If the [Alzheimer’s Association] truly finds this objectionable, hopefully they’ll stop accepting money from Biogen and its partner Eisai,” he added.
“The Alzheimer’s Association is recognizing that the genie is out of the bottle and that they are going to have trouble reining in the inevitable run-away costs,” said Mike Greicius, MD, MPH, associate professor of neurology at Stanford University’s Wu Tsai Neurosciences Institute, Stanford, California.
“In addition to the eye-popping annual cost that Biogen has invented, I hope the Alzheimer’s Association is also concerned about the dangerously loose and broad FDA labeling which does not require screening for amyloid-positivity and does not restrict use to the milder forms of disease studied in the Phase 3 trials,” Dr. Greicius said.
Another advocacy group, Patients For Affordable Drugs, commended the Alzheimer’s Association. Its statement “was nothing short of courageous, especially in light of the Alzheimer’s Association’s reliance on funding from drug corporations, including Biogen,” said David Mitchell, a cancer patient and founder of Patients For Affordable Drugs, in a statement.
Mr. Mitchell said his members “stand with the Alzheimer’s Association in its denunciation of the price set by Biogen” and called for a new law that would allow Medicare to negotiate drug prices.
A version of this article first appeared on Medscape.com.
AMA acknowledges medical education racism of past, vows better future
The report received overwhelming support at the House of Delegates, the AMA’s legislative policy making body, during an online meeting held June 13.
The Council on Medical Education’s report recommends that the AMA acknowledge the harm caused by the Flexner Report, which was issued in 1910 and has since shaped medical education. The Flexner Report caused harm not only to historically Black medical schools, but also to physician workforce diversity and to the clinical outcomes of minority and marginalized patients, according to the medical education advisory body.
The council also recommended conducting a study on medical education with a focus on health equity and racial justice, improving diversity among healthcare workers, and fixing inequitable outcomes from minorities and marginalized patient populations.
The report comes on the heels of the resignation of JAMA editor-in-chief Howard Bauchner, MD, and another high-ranking editor following a February podcast on systemic racism in medicine. The AMA has since released a strategic plan addressing racism and health inequity that has divided membership.
Flexner Report’s effect on physician diversity
The Council on Medical Education’s report observed that as a result of the Flexner Report’s recommendations, 89 medical schools, including 5 of the 7 existing medical schools training Black physicians, were closed because they didn’t meet the report’s standards. In addition, the report created a limited role for Black physicians while “hint[ing] that Black physicians possessed less potential and ability than their White counterparts,” read the Council’s report.
In addition to consigning the role of the Black physician to “educating the [Black] race to know and to practice fundamental hygienic principles,” the Flexner Report also observed that “a well-taught negro sanitarian will be immensely useful,” per the Council’s report.
The impact of the closure of medical schools training Black physicians was dramatic. According to the Council’s report, in 1964, 93% of medical students in the United States were men and 97% of those students were non-Hispanic White.
Today, 56% of physicians identify as White, 17% as Asian, 6% as Hispanic, and 5% as Black or African American, per the Association of American Medical Colleges; nearly 14% of active physicians didn’t report their race in the survey. By means of contrast, the U.S. population in 2019 was 60% White, 19% Latino/Hispanic, 13% Black or African American, and 6% Asian American, according to the Brookings Institute.
Abraham Flexner, who wrote the Flexner Report, is often referred to as the “father of modern medical education,” according to the AAMC. In November, the AAMC observed that the Flexner Report contained racist and sexist ideas and that his work contributed to the closure of historically Black medical schools. Both statements were included in AAMC’s announcement about the removal of Flexner’s name from its most prestigious award. As of January, the award is now called the AAMC Award for Excellence in Medical Education.
Pathway programs can increase diversity
Pathway programs, which leverage targeted milestones along the journey to becoming a physician in order to increase diversity, were an area of focus in the council’s report. These programs “can exert a meaningful, positive effect on student outcomes and increase diversity across various levels of educational settings,” according to its report.
Centers of Excellence, which provides grants for mentorship and training programs, is one of many pathway programs. During the 2018-2019 academic year, Centers of Excellence supported more than 1,300 trainees – 99% of them were underrepresented minorities and 64% came from financially or educationally disadvantaged backgrounds. In 2006, federal funding was cut to these programs and the number of Centers of Excellence fell.
Still, the report cites the passage of federal funding in 2020 of $50 million for public institutions of higher education that train physicians; educational institutions in states with a projected primary care shortage in 2025 are given priority in the grant-funding process.
AMA council’s report garners support from delegates
Delegates voiced overwhelming support of the council’s report during the June 13 meeting. Lou Edje, MD, a Perrysburgh, Ohio–based family physician, voiced strong support for the council’s report, in particular its recommendations that recognize the harm caused by the Flexner Report. Dr. Edje observed that the Flexner Report, with its elimination of five of seven Black medical schools, “[set] back admissions of Black students into medicine by 50 years.”
“Empathy is what we are called to have as physicians. I implore you to simply substitute your ethnicity into these quotes to help understand the historic need for health equity in medicine today. This CME report is part of the antidote to Flexner. We support [it] fully,” concluded Dr. Edje, who spoke for the Great Lakes States Coalition of the AMA.
Rohan Khazanchi, a medical student at the University of Nebraska, Omaha, and a member of the council, said, “Our broad attempt with this report was twofold: to fill gaps in AMA policy with evidence-based recommendations which could improve diversity in our health workforce and, second, to enhance our organization’s vision for truth, reconciliation, and healing to redress the historic marginalization of minoritized physicians in medicine.”
According to an AMA spokesperson, the House of Delegates will vote on this and other policies this week, after which the policies are considered final.
A version of this article first appeared on Medscape.com.
The report received overwhelming support at the House of Delegates, the AMA’s legislative policy making body, during an online meeting held June 13.
The Council on Medical Education’s report recommends that the AMA acknowledge the harm caused by the Flexner Report, which was issued in 1910 and has since shaped medical education. The Flexner Report caused harm not only to historically Black medical schools, but also to physician workforce diversity and to the clinical outcomes of minority and marginalized patients, according to the medical education advisory body.
The council also recommended conducting a study on medical education with a focus on health equity and racial justice, improving diversity among healthcare workers, and fixing inequitable outcomes from minorities and marginalized patient populations.
The report comes on the heels of the resignation of JAMA editor-in-chief Howard Bauchner, MD, and another high-ranking editor following a February podcast on systemic racism in medicine. The AMA has since released a strategic plan addressing racism and health inequity that has divided membership.
Flexner Report’s effect on physician diversity
The Council on Medical Education’s report observed that as a result of the Flexner Report’s recommendations, 89 medical schools, including 5 of the 7 existing medical schools training Black physicians, were closed because they didn’t meet the report’s standards. In addition, the report created a limited role for Black physicians while “hint[ing] that Black physicians possessed less potential and ability than their White counterparts,” read the Council’s report.
In addition to consigning the role of the Black physician to “educating the [Black] race to know and to practice fundamental hygienic principles,” the Flexner Report also observed that “a well-taught negro sanitarian will be immensely useful,” per the Council’s report.
The impact of the closure of medical schools training Black physicians was dramatic. According to the Council’s report, in 1964, 93% of medical students in the United States were men and 97% of those students were non-Hispanic White.
Today, 56% of physicians identify as White, 17% as Asian, 6% as Hispanic, and 5% as Black or African American, per the Association of American Medical Colleges; nearly 14% of active physicians didn’t report their race in the survey. By means of contrast, the U.S. population in 2019 was 60% White, 19% Latino/Hispanic, 13% Black or African American, and 6% Asian American, according to the Brookings Institute.
Abraham Flexner, who wrote the Flexner Report, is often referred to as the “father of modern medical education,” according to the AAMC. In November, the AAMC observed that the Flexner Report contained racist and sexist ideas and that his work contributed to the closure of historically Black medical schools. Both statements were included in AAMC’s announcement about the removal of Flexner’s name from its most prestigious award. As of January, the award is now called the AAMC Award for Excellence in Medical Education.
Pathway programs can increase diversity
Pathway programs, which leverage targeted milestones along the journey to becoming a physician in order to increase diversity, were an area of focus in the council’s report. These programs “can exert a meaningful, positive effect on student outcomes and increase diversity across various levels of educational settings,” according to its report.
Centers of Excellence, which provides grants for mentorship and training programs, is one of many pathway programs. During the 2018-2019 academic year, Centers of Excellence supported more than 1,300 trainees – 99% of them were underrepresented minorities and 64% came from financially or educationally disadvantaged backgrounds. In 2006, federal funding was cut to these programs and the number of Centers of Excellence fell.
Still, the report cites the passage of federal funding in 2020 of $50 million for public institutions of higher education that train physicians; educational institutions in states with a projected primary care shortage in 2025 are given priority in the grant-funding process.
AMA council’s report garners support from delegates
Delegates voiced overwhelming support of the council’s report during the June 13 meeting. Lou Edje, MD, a Perrysburgh, Ohio–based family physician, voiced strong support for the council’s report, in particular its recommendations that recognize the harm caused by the Flexner Report. Dr. Edje observed that the Flexner Report, with its elimination of five of seven Black medical schools, “[set] back admissions of Black students into medicine by 50 years.”
“Empathy is what we are called to have as physicians. I implore you to simply substitute your ethnicity into these quotes to help understand the historic need for health equity in medicine today. This CME report is part of the antidote to Flexner. We support [it] fully,” concluded Dr. Edje, who spoke for the Great Lakes States Coalition of the AMA.
Rohan Khazanchi, a medical student at the University of Nebraska, Omaha, and a member of the council, said, “Our broad attempt with this report was twofold: to fill gaps in AMA policy with evidence-based recommendations which could improve diversity in our health workforce and, second, to enhance our organization’s vision for truth, reconciliation, and healing to redress the historic marginalization of minoritized physicians in medicine.”
According to an AMA spokesperson, the House of Delegates will vote on this and other policies this week, after which the policies are considered final.
A version of this article first appeared on Medscape.com.
The report received overwhelming support at the House of Delegates, the AMA’s legislative policy making body, during an online meeting held June 13.
The Council on Medical Education’s report recommends that the AMA acknowledge the harm caused by the Flexner Report, which was issued in 1910 and has since shaped medical education. The Flexner Report caused harm not only to historically Black medical schools, but also to physician workforce diversity and to the clinical outcomes of minority and marginalized patients, according to the medical education advisory body.
The council also recommended conducting a study on medical education with a focus on health equity and racial justice, improving diversity among healthcare workers, and fixing inequitable outcomes from minorities and marginalized patient populations.
The report comes on the heels of the resignation of JAMA editor-in-chief Howard Bauchner, MD, and another high-ranking editor following a February podcast on systemic racism in medicine. The AMA has since released a strategic plan addressing racism and health inequity that has divided membership.
Flexner Report’s effect on physician diversity
The Council on Medical Education’s report observed that as a result of the Flexner Report’s recommendations, 89 medical schools, including 5 of the 7 existing medical schools training Black physicians, were closed because they didn’t meet the report’s standards. In addition, the report created a limited role for Black physicians while “hint[ing] that Black physicians possessed less potential and ability than their White counterparts,” read the Council’s report.
In addition to consigning the role of the Black physician to “educating the [Black] race to know and to practice fundamental hygienic principles,” the Flexner Report also observed that “a well-taught negro sanitarian will be immensely useful,” per the Council’s report.
The impact of the closure of medical schools training Black physicians was dramatic. According to the Council’s report, in 1964, 93% of medical students in the United States were men and 97% of those students were non-Hispanic White.
Today, 56% of physicians identify as White, 17% as Asian, 6% as Hispanic, and 5% as Black or African American, per the Association of American Medical Colleges; nearly 14% of active physicians didn’t report their race in the survey. By means of contrast, the U.S. population in 2019 was 60% White, 19% Latino/Hispanic, 13% Black or African American, and 6% Asian American, according to the Brookings Institute.
Abraham Flexner, who wrote the Flexner Report, is often referred to as the “father of modern medical education,” according to the AAMC. In November, the AAMC observed that the Flexner Report contained racist and sexist ideas and that his work contributed to the closure of historically Black medical schools. Both statements were included in AAMC’s announcement about the removal of Flexner’s name from its most prestigious award. As of January, the award is now called the AAMC Award for Excellence in Medical Education.
Pathway programs can increase diversity
Pathway programs, which leverage targeted milestones along the journey to becoming a physician in order to increase diversity, were an area of focus in the council’s report. These programs “can exert a meaningful, positive effect on student outcomes and increase diversity across various levels of educational settings,” according to its report.
Centers of Excellence, which provides grants for mentorship and training programs, is one of many pathway programs. During the 2018-2019 academic year, Centers of Excellence supported more than 1,300 trainees – 99% of them were underrepresented minorities and 64% came from financially or educationally disadvantaged backgrounds. In 2006, federal funding was cut to these programs and the number of Centers of Excellence fell.
Still, the report cites the passage of federal funding in 2020 of $50 million for public institutions of higher education that train physicians; educational institutions in states with a projected primary care shortage in 2025 are given priority in the grant-funding process.
AMA council’s report garners support from delegates
Delegates voiced overwhelming support of the council’s report during the June 13 meeting. Lou Edje, MD, a Perrysburgh, Ohio–based family physician, voiced strong support for the council’s report, in particular its recommendations that recognize the harm caused by the Flexner Report. Dr. Edje observed that the Flexner Report, with its elimination of five of seven Black medical schools, “[set] back admissions of Black students into medicine by 50 years.”
“Empathy is what we are called to have as physicians. I implore you to simply substitute your ethnicity into these quotes to help understand the historic need for health equity in medicine today. This CME report is part of the antidote to Flexner. We support [it] fully,” concluded Dr. Edje, who spoke for the Great Lakes States Coalition of the AMA.
Rohan Khazanchi, a medical student at the University of Nebraska, Omaha, and a member of the council, said, “Our broad attempt with this report was twofold: to fill gaps in AMA policy with evidence-based recommendations which could improve diversity in our health workforce and, second, to enhance our organization’s vision for truth, reconciliation, and healing to redress the historic marginalization of minoritized physicians in medicine.”
According to an AMA spokesperson, the House of Delegates will vote on this and other policies this week, after which the policies are considered final.
A version of this article first appeared on Medscape.com.
New biomarkers may predict interstitial lung disease progression in patients with systemic sclerosis
Quantitative assessment of the extent of interstitial lung disease in patients with systemic sclerosis and levels of certain proteins in bronchoalveolar lavage samples have potential for predicting mortality and disease progression, according to two analyses of data from the Scleroderma Lung Study I and II.

The analyses, presented at the annual European Congress of Rheumatology, aim to improve current prognostic abilities in patients with systemic sclerosis–interstitial lung disease (SSc-ILD). Although forced vital capacity is commonly used as a biomarker for survival in many SSc-ILD trials, other factors can affect FVC, such as respiratory muscle weakness and skin fibrosis. Further, FVC correlates poorly with patient-reported outcomes, explained first author Elizabeth Volkmann, MD, director of the scleroderma program at the University of California, Los Angeles, and the founder and codirector of the UCLA connective tissue disease–related interstitial lung disease program.
Dr. Volkmann presented two studies that investigated the potential of radiographic and protein biomarkers for predicting mortality and identifying patients at risk for ILD progression. The biomarkers may also help to identify patients who would benefit most from immunosuppressive therapy.
The first study found that tracking the quantitative extent of ILD (QILD) over time with high-resolution CT (HRCT) predicted poorer outcomes and could therefore act as a surrogate endpoint for mortality among patients with SSc-ILD. The other study identified associations between specific proteins from bronchoalveolar lavage (BAL) and the likelihood of ILD progression, although some associations were treatment dependent.

Jacob M. van Laar, MD, PhD, professor of rheumatology at the University Medical Center Utrecht (the Netherlands), who was not involved in the study, found the results intriguing and noted the importance of further validation in research before these biomarkers are considered for clinical use.
“It would be wonderful if we can tailor therapy based on BAL biomarkers in the future, as clinicians often struggle to decide on selection, timing, and duration of immunosuppressive treatment,” Dr. van Laar told this news organization. “This has become even more relevant with the introduction of new drugs such as nintedanib.”
Extent of ILD progression as a surrogate for mortality
Scleroderma Lung Study I involved 158 patients with SSc-ILD who were randomly assigned to receive either cyclophosphamide or placebo for 12 months. Scleroderma Lung Study II included 142 patients with SSc-ILD who were randomly assigned to receive either mycophenolate for 24 months or cyclophosphamide for 12 months followed by placebo for 12 months.
The researchers calculated QILD in the whole lung at baseline, at 12 months in the first trial, and at 24 months in the second trial. However, only 82 participants from the first trial and 90 participants from the second trial underwent HRCT. Demographic and disease characteristics were similar between the two groups on follow-up scans.
Follow-up continued for 12 years for patients in the first trial and 8 years in the second. The researchers compared survival rates between the 41% of participants from the first study and 31% of participants from the second study who had poorer QILD scores (at least a 2% increase) with the participants who had stable or improved scores (less than 2% increase).
Participants from both trials had significantly poorer long-term survival if their QILD scores had increased by at least 2% at follow-up (P = .01 for I; P = .019 for II). The association was no longer significant after adjustment for baseline FVC, age, and modified Rodnan skin score in the first trial (hazard ratio, 1.98; P = .089), but it remained significant for participants of the second trial (HR, 3.86; P = .014).
“Data from two independent trial cohorts demonstrated that radiographic progression of SSc-ILD at 1 and 2 years is associated with worse long-term survival,” Dr. Volkmann told attendees.
However, FVC did not significantly predict risk of mortality in either trial.
“To me, the most striking finding from the first study was that change in QILD performed better as a predictor of survival than change in FVC,” Dr. van Laar said in an interview. “This indicates QILD is fit for purpose and worth including in future clinical trials.”
Limitations of the study included lack of HRCT for all participants in the trials and the difference in timing (1 year and 2 years) of HRCT assessment between the two trials. The greater hazard ratio for worsened QILD in the second trial may suggest that assessment at 2 years provides more reliable data as a biomarker, Dr. Volkmann said.
“QILD may represent a better proxy for how a patient feels, functions, and survives than FVC,” she said.
Treatment-dependent biomarkers for worsening lung fibrosis
In the second study, the researchers looked for any associations between changes in the radiographic extent of SSc-ILD and 68 proteins from BAL.
“Being able to risk-stratify patients with interstitial lung disease at the time of diagnosis and predict which patients are likely to have a stable versus progressive disease course is critical for making important treatment decisions for these patients,” Dr. Volkmann told attendees.
The second study she presented involved Scleroderma Lung Study I. Of the 158 participants, 144 underwent a bronchoscopy, yielding BAL protein samples from 103 participants. The researchers determined the extent of radiographic fibrosis in the whole lung with quantitative imaging analysis of HRCT of the chest at baseline and 12 months.
Although the researchers identified several statistically significant associations between certain proteins and changes in radiographic fibrosis, “baseline protein levels were differentially associated with the course of ILD based on treatment status,” she told attendees.
For example, increased levels of the following proteins were linked to poor radiographic fibrosis scores for patients who received placebo:
- Granulocyte-macrophage colony-stimulating factor
- Interleukin-1
- Monocyte chemoattractant protein–3
- Chemokine ligand–5
- Transforming growth factor–beta
- Hepatocyte growth factor
- Stem cell factor
- IL-4
- TGF-alpha
Yet increases in these proteins predicted improvement in radiographic fibrosis in patients who had taken cyclophosphamide.
Independently of treatment, the researchers also identified an association between higher levels of fractalkine and poorer radiographic fibrosis scores and between higher IL-7 levels and improved radiographic fibrosis scores.
After adjusting for treatment arm and baseline severity of ILD, significant associations remained between change in radiographic fibrosis score and IL-1, MCP-3, surfactant protein C, IL-7 and CCL-5 levels.
“Biomarker discovery is really central to our ability to risk stratify patients with SSc-ILD,” Dr. Volkmann told attendees. “Understanding how biomarkers predict outcomes in treated and untreated patients may improve personalized medicine to patients with SSc-ILD and could also reveal novel treatment targets.”
Dr. van Laar said in an interview that this study’s biggest strength lay in its large sample size and in the comprehensiveness of the biomarkers studied.
“The findings are interesting from a research perspective and potentially relevant for clinical practice, but the utility of measuring biomarkers in BAL should be further studied for predictive value on clinical endpoints,” Dr. van Laar said. “BAL is an invasive procedure [that] is not routinely done.”
The research was funded by the National Institutes of Health. Dr. Volkmann has consulted for Boehringer Ingelheim and received grant funding from Corbus, Forbius, and Kadmon. Dr. van Laar has received grant funding or personal fees from Arthrogen, Arxx Therapeutics, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Gesynta, Leadiant, Merck Sharp & Dohme, Roche, Sanofi, and Thermofisher.
A version of this article first appeared on Medscape.com.
Quantitative assessment of the extent of interstitial lung disease in patients with systemic sclerosis and levels of certain proteins in bronchoalveolar lavage samples have potential for predicting mortality and disease progression, according to two analyses of data from the Scleroderma Lung Study I and II.
The analyses, presented at the annual European Congress of Rheumatology, aim to improve current prognostic abilities in patients with systemic sclerosis–interstitial lung disease (SSc-ILD). Although forced vital capacity is commonly used as a biomarker for survival in many SSc-ILD trials, other factors can affect FVC, such as respiratory muscle weakness and skin fibrosis. Further, FVC correlates poorly with patient-reported outcomes, explained first author Elizabeth Volkmann, MD, director of the scleroderma program at the University of California, Los Angeles, and the founder and codirector of the UCLA connective tissue disease–related interstitial lung disease program.
Dr. Volkmann presented two studies that investigated the potential of radiographic and protein biomarkers for predicting mortality and identifying patients at risk for ILD progression. The biomarkers may also help to identify patients who would benefit most from immunosuppressive therapy.
The first study found that tracking the quantitative extent of ILD (QILD) over time with high-resolution CT (HRCT) predicted poorer outcomes and could therefore act as a surrogate endpoint for mortality among patients with SSc-ILD. The other study identified associations between specific proteins from bronchoalveolar lavage (BAL) and the likelihood of ILD progression, although some associations were treatment dependent.
Jacob M. van Laar, MD, PhD, professor of rheumatology at the University Medical Center Utrecht (the Netherlands), who was not involved in the study, found the results intriguing and noted the importance of further validation in research before these biomarkers are considered for clinical use.
“It would be wonderful if we can tailor therapy based on BAL biomarkers in the future, as clinicians often struggle to decide on selection, timing, and duration of immunosuppressive treatment,” Dr. van Laar told this news organization. “This has become even more relevant with the introduction of new drugs such as nintedanib.”
Extent of ILD progression as a surrogate for mortality
Scleroderma Lung Study I involved 158 patients with SSc-ILD who were randomly assigned to receive either cyclophosphamide or placebo for 12 months. Scleroderma Lung Study II included 142 patients with SSc-ILD who were randomly assigned to receive either mycophenolate for 24 months or cyclophosphamide for 12 months followed by placebo for 12 months.
The researchers calculated QILD in the whole lung at baseline, at 12 months in the first trial, and at 24 months in the second trial. However, only 82 participants from the first trial and 90 participants from the second trial underwent HRCT. Demographic and disease characteristics were similar between the two groups on follow-up scans.
Follow-up continued for 12 years for patients in the first trial and 8 years in the second. The researchers compared survival rates between the 41% of participants from the first study and 31% of participants from the second study who had poorer QILD scores (at least a 2% increase) with the participants who had stable or improved scores (less than 2% increase).
Participants from both trials had significantly poorer long-term survival if their QILD scores had increased by at least 2% at follow-up (P = .01 for I; P = .019 for II). The association was no longer significant after adjustment for baseline FVC, age, and modified Rodnan skin score in the first trial (hazard ratio, 1.98; P = .089), but it remained significant for participants of the second trial (HR, 3.86; P = .014).
“Data from two independent trial cohorts demonstrated that radiographic progression of SSc-ILD at 1 and 2 years is associated with worse long-term survival,” Dr. Volkmann told attendees.
However, FVC did not significantly predict risk of mortality in either trial.
“To me, the most striking finding from the first study was that change in QILD performed better as a predictor of survival than change in FVC,” Dr. van Laar said in an interview. “This indicates QILD is fit for purpose and worth including in future clinical trials.”
Limitations of the study included lack of HRCT for all participants in the trials and the difference in timing (1 year and 2 years) of HRCT assessment between the two trials. The greater hazard ratio for worsened QILD in the second trial may suggest that assessment at 2 years provides more reliable data as a biomarker, Dr. Volkmann said.
“QILD may represent a better proxy for how a patient feels, functions, and survives than FVC,” she said.
Treatment-dependent biomarkers for worsening lung fibrosis
In the second study, the researchers looked for any associations between changes in the radiographic extent of SSc-ILD and 68 proteins from BAL.
“Being able to risk-stratify patients with interstitial lung disease at the time of diagnosis and predict which patients are likely to have a stable versus progressive disease course is critical for making important treatment decisions for these patients,” Dr. Volkmann told attendees.
The second study she presented involved Scleroderma Lung Study I. Of the 158 participants, 144 underwent a bronchoscopy, yielding BAL protein samples from 103 participants. The researchers determined the extent of radiographic fibrosis in the whole lung with quantitative imaging analysis of HRCT of the chest at baseline and 12 months.
Although the researchers identified several statistically significant associations between certain proteins and changes in radiographic fibrosis, “baseline protein levels were differentially associated with the course of ILD based on treatment status,” she told attendees.
For example, increased levels of the following proteins were linked to poor radiographic fibrosis scores for patients who received placebo:
- Granulocyte-macrophage colony-stimulating factor
- Interleukin-1
- Monocyte chemoattractant protein–3
- Chemokine ligand–5
- Transforming growth factor–beta
- Hepatocyte growth factor
- Stem cell factor
- IL-4
- TGF-alpha
Yet increases in these proteins predicted improvement in radiographic fibrosis in patients who had taken cyclophosphamide.
Independently of treatment, the researchers also identified an association between higher levels of fractalkine and poorer radiographic fibrosis scores and between higher IL-7 levels and improved radiographic fibrosis scores.
After adjusting for treatment arm and baseline severity of ILD, significant associations remained between change in radiographic fibrosis score and IL-1, MCP-3, surfactant protein C, IL-7 and CCL-5 levels.
“Biomarker discovery is really central to our ability to risk stratify patients with SSc-ILD,” Dr. Volkmann told attendees. “Understanding how biomarkers predict outcomes in treated and untreated patients may improve personalized medicine to patients with SSc-ILD and could also reveal novel treatment targets.”
Dr. van Laar said in an interview that this study’s biggest strength lay in its large sample size and in the comprehensiveness of the biomarkers studied.
“The findings are interesting from a research perspective and potentially relevant for clinical practice, but the utility of measuring biomarkers in BAL should be further studied for predictive value on clinical endpoints,” Dr. van Laar said. “BAL is an invasive procedure [that] is not routinely done.”
The research was funded by the National Institutes of Health. Dr. Volkmann has consulted for Boehringer Ingelheim and received grant funding from Corbus, Forbius, and Kadmon. Dr. van Laar has received grant funding or personal fees from Arthrogen, Arxx Therapeutics, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Gesynta, Leadiant, Merck Sharp & Dohme, Roche, Sanofi, and Thermofisher.
A version of this article first appeared on Medscape.com.
Quantitative assessment of the extent of interstitial lung disease in patients with systemic sclerosis and levels of certain proteins in bronchoalveolar lavage samples have potential for predicting mortality and disease progression, according to two analyses of data from the Scleroderma Lung Study I and II.
The analyses, presented at the annual European Congress of Rheumatology, aim to improve current prognostic abilities in patients with systemic sclerosis–interstitial lung disease (SSc-ILD). Although forced vital capacity is commonly used as a biomarker for survival in many SSc-ILD trials, other factors can affect FVC, such as respiratory muscle weakness and skin fibrosis. Further, FVC correlates poorly with patient-reported outcomes, explained first author Elizabeth Volkmann, MD, director of the scleroderma program at the University of California, Los Angeles, and the founder and codirector of the UCLA connective tissue disease–related interstitial lung disease program.
Dr. Volkmann presented two studies that investigated the potential of radiographic and protein biomarkers for predicting mortality and identifying patients at risk for ILD progression. The biomarkers may also help to identify patients who would benefit most from immunosuppressive therapy.
The first study found that tracking the quantitative extent of ILD (QILD) over time with high-resolution CT (HRCT) predicted poorer outcomes and could therefore act as a surrogate endpoint for mortality among patients with SSc-ILD. The other study identified associations between specific proteins from bronchoalveolar lavage (BAL) and the likelihood of ILD progression, although some associations were treatment dependent.
Jacob M. van Laar, MD, PhD, professor of rheumatology at the University Medical Center Utrecht (the Netherlands), who was not involved in the study, found the results intriguing and noted the importance of further validation in research before these biomarkers are considered for clinical use.
“It would be wonderful if we can tailor therapy based on BAL biomarkers in the future, as clinicians often struggle to decide on selection, timing, and duration of immunosuppressive treatment,” Dr. van Laar told this news organization. “This has become even more relevant with the introduction of new drugs such as nintedanib.”
Extent of ILD progression as a surrogate for mortality
Scleroderma Lung Study I involved 158 patients with SSc-ILD who were randomly assigned to receive either cyclophosphamide or placebo for 12 months. Scleroderma Lung Study II included 142 patients with SSc-ILD who were randomly assigned to receive either mycophenolate for 24 months or cyclophosphamide for 12 months followed by placebo for 12 months.
The researchers calculated QILD in the whole lung at baseline, at 12 months in the first trial, and at 24 months in the second trial. However, only 82 participants from the first trial and 90 participants from the second trial underwent HRCT. Demographic and disease characteristics were similar between the two groups on follow-up scans.
Follow-up continued for 12 years for patients in the first trial and 8 years in the second. The researchers compared survival rates between the 41% of participants from the first study and 31% of participants from the second study who had poorer QILD scores (at least a 2% increase) with the participants who had stable or improved scores (less than 2% increase).
Participants from both trials had significantly poorer long-term survival if their QILD scores had increased by at least 2% at follow-up (P = .01 for I; P = .019 for II). The association was no longer significant after adjustment for baseline FVC, age, and modified Rodnan skin score in the first trial (hazard ratio, 1.98; P = .089), but it remained significant for participants of the second trial (HR, 3.86; P = .014).
“Data from two independent trial cohorts demonstrated that radiographic progression of SSc-ILD at 1 and 2 years is associated with worse long-term survival,” Dr. Volkmann told attendees.
However, FVC did not significantly predict risk of mortality in either trial.
“To me, the most striking finding from the first study was that change in QILD performed better as a predictor of survival than change in FVC,” Dr. van Laar said in an interview. “This indicates QILD is fit for purpose and worth including in future clinical trials.”
Limitations of the study included lack of HRCT for all participants in the trials and the difference in timing (1 year and 2 years) of HRCT assessment between the two trials. The greater hazard ratio for worsened QILD in the second trial may suggest that assessment at 2 years provides more reliable data as a biomarker, Dr. Volkmann said.
“QILD may represent a better proxy for how a patient feels, functions, and survives than FVC,” she said.
Treatment-dependent biomarkers for worsening lung fibrosis
In the second study, the researchers looked for any associations between changes in the radiographic extent of SSc-ILD and 68 proteins from BAL.
“Being able to risk-stratify patients with interstitial lung disease at the time of diagnosis and predict which patients are likely to have a stable versus progressive disease course is critical for making important treatment decisions for these patients,” Dr. Volkmann told attendees.
The second study she presented involved Scleroderma Lung Study I. Of the 158 participants, 144 underwent a bronchoscopy, yielding BAL protein samples from 103 participants. The researchers determined the extent of radiographic fibrosis in the whole lung with quantitative imaging analysis of HRCT of the chest at baseline and 12 months.
Although the researchers identified several statistically significant associations between certain proteins and changes in radiographic fibrosis, “baseline protein levels were differentially associated with the course of ILD based on treatment status,” she told attendees.
For example, increased levels of the following proteins were linked to poor radiographic fibrosis scores for patients who received placebo:
- Granulocyte-macrophage colony-stimulating factor
- Interleukin-1
- Monocyte chemoattractant protein–3
- Chemokine ligand–5
- Transforming growth factor–beta
- Hepatocyte growth factor
- Stem cell factor
- IL-4
- TGF-alpha
Yet increases in these proteins predicted improvement in radiographic fibrosis in patients who had taken cyclophosphamide.
Independently of treatment, the researchers also identified an association between higher levels of fractalkine and poorer radiographic fibrosis scores and between higher IL-7 levels and improved radiographic fibrosis scores.
After adjusting for treatment arm and baseline severity of ILD, significant associations remained between change in radiographic fibrosis score and IL-1, MCP-3, surfactant protein C, IL-7 and CCL-5 levels.
“Biomarker discovery is really central to our ability to risk stratify patients with SSc-ILD,” Dr. Volkmann told attendees. “Understanding how biomarkers predict outcomes in treated and untreated patients may improve personalized medicine to patients with SSc-ILD and could also reveal novel treatment targets.”
Dr. van Laar said in an interview that this study’s biggest strength lay in its large sample size and in the comprehensiveness of the biomarkers studied.
“The findings are interesting from a research perspective and potentially relevant for clinical practice, but the utility of measuring biomarkers in BAL should be further studied for predictive value on clinical endpoints,” Dr. van Laar said. “BAL is an invasive procedure [that] is not routinely done.”
The research was funded by the National Institutes of Health. Dr. Volkmann has consulted for Boehringer Ingelheim and received grant funding from Corbus, Forbius, and Kadmon. Dr. van Laar has received grant funding or personal fees from Arthrogen, Arxx Therapeutics, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Gesynta, Leadiant, Merck Sharp & Dohme, Roche, Sanofi, and Thermofisher.
A version of this article first appeared on Medscape.com.
The aducanumab revolution
The approval was hailed by advocacy groups and some practitioners as a victory for patients and families, as the drug – the first anti-Alzheimer’s agent to reach the market in 18 years – is a potentially disease-modifying therapy, which acts to clear amyloid plaques from the brain.
But several prominent Alzheimer’s researchers lambasted the agency’s decision, citing unclear evidence of benefit, trials that did not meet their primary endpoints, and reliance on a post hoc analysis of a high-dose subgroup of patients in a halted trial to argue that aducanumab (Aduhelm, Biogen, and Eisai), slowed cognitive and functional decline by 22% on one measure. In November 2020, 10 of 11 members of an independent FDA advisory committee voted against aducanumab’s approval, citing holes in the data and concerns about the quality of the evidence. After the agency went on to approve anyway, three members of that committee resigned in protest.
The FDA decision on aducanumab was made using the agency’s accelerated approval pathway, which allows for the use of a surrogate endpoint – in this case imaging that showed amyloid clearance from the brain – to predict clinical benefit. But amyloid clearance, which a number of experimental antiamyloid antibodies have been shown capable of, has not been definitively linked to clinical benefit. Aducanumab, which is delivered by monthly intravenous infusion, will be marketed pending results from a phase 4 clinical trial, which the manufacturer has nearly a decade to complete. The drug’s price was announced at $56,000 per year, underscoring concern over its modest-at-best benefits.
Clinicians prescribing aducanumab must obtain magnetic resonance imaging at baseline and repeatedly during the course of treatment to detect brain edema and microhemorrhages, which occurred in a third of high-dose patients in clinical trials. Beyond this, there are few restrictions. The FDA label allows for its use in any patient deemed to have Alzheimer’s disease, without stipulations as to disease stage or evidence of brain amyloid. Payers, of course, are likely to restrict use to certain patient groups, and to require evidence of amyloid positivity. The FDA offered no guidance on when treatment should be ceased, leaving payers to make that call as well. Whatever aducanumab’s value and role turns out to be, the first-in-class treatment for Alzheimer’s disease is likely to have a major impact on how patients are assessed and treated in the coming years, and embolden manufactures of similar agents to seek FDA approval.
This news organization reached out to researchers, advocates, and specialists in the community to learn how they see this change playing out.
Fielding broad interest
Maria C. Carrillo, PhD, chief science officer of the Alzheimer’s Association, which was a strong proponent of aducanumab’s approval, acknowledged in an interview that the months to come are likely to be confusing for practitioners and families alike as the drug makes its way into community practices.

“We understand that off the bat millions of Americans will not have access to this tomorrow, but over time that will build. And the physician community, the specialists most likely to be prescribing this, over the next few years will even expand further,” Dr. Carrillo said.
For now, those specialists are mostly just struggling to respond responsibly to a deluge of inquiries from patients and their families.
“I’ve gotten like 20 calls in the just the past 2 days,” said neurologist Philip R. Delio, MD, who practices in Santa Barbara, Calif. “This is a longstanding issue that physicians have with patients’ access to information. Patients are getting information about a drug which isn’t available yet. They don’t know that it’s not ready to be sold. They don’t necessarily realize that a biopharma company won’t go into production until the FDA approves the drug.”

Many patients, Dr. Delio said, are aware of the controversy surrounding aducanumab and eager to hear their neurologist’s opinion. “I have tried to let them know that I want to see the trial data and to better understand the FDA’s rationale in approving it. I always caution patients that the devil will be in the details.”
While aducanumab’s label gives physicians remarkably wide latitude in whom to treat, clinicians say that until payers weigh in, the label is all but meaningless. Neurologist Douglas Scharre, MD, of the Ohio State University Wexner Medical Center, and a site investigator on a trial of aducanumab, said that he and his colleagues at the university’s memory center have tried to anticipate who might be deemed eligible by triaging calls.

Dr. Scharre and colleagues have been working under the assumption that payers will support aducanumab only for patients like those who seemed to benefit in the trials – people with mild cognitive impairment (MCI) or in the earliest stages of dementia with evidence of brain amyloid.
“I don’t want to fill up our new patient slots with people who are not even appropriate for this drug,” Dr. Scharre said. “We have a call center, and we have a few triage questions. After that a nurse practitioner collects some more data, and there’s a review process. Only then do we decide whether that person could be a candidate. If we deem that they are, we will want them in and to order an amyloid PET” – a type of brain scan that is seldom used outside research settings and not reimbursed by Medicare.
Dr. Scharre predicts that regardless of payer limitations, “there will be people hounding for the drug who are not appropriate for the drug. There will be very wealthy people who will want to pay for tests and get it no matter what.” Another concern, he said, was that having poorly selected patients on the drug could make definitive trial results even more elusive.
“The label the way it’s written is not going to help the drug in phase 4 trials,” he said. “It’s good to have real-world patient data, but if you have all these people in your cohort who are too early or too late, you won’t have good results.”
The challenge of delivery
Intravenous infusions are new to Alzheimer’s disease and pose all sorts of logistical hurdles. The Alzheimer’s Association’s Dr. Carrillo described the situation as “manageable,” noting that infusions are standard of care for many diseases, and that neurologists now have more than 15 years’ experience with them for multiple sclerosis.
Still, most clinicians treating Alzheimer’s disease in the community – neurologists, geriatricians, psychiatrists, and primary care physicians – do not have infusion centers in their practices. Virtually none have experience with or access to PET-amyloid, or with screening for amyloid-related imaging abnormalities–edema (ARIA-e) on MRI, as required by the FDA.
“I contacted the hospital infusion center we use and said I could end up sending five or six patients a week, can you handle this? They only have so many chairs,” Dr. Delio said. “I am one neurologist in a local community, and I might have 50 candidates for this drug. That’s a lot for them.” Patients with cognitive impairment are also difficult to infuse and may need to be treated at home, he noted.
“MRIs are easy enough to do,” Dr. Delio said. “But do we know what ARIA-e looks like on imaging? You’d have to talk to the radiologists – this is another element of uncertainty. Do we even know what we’re looking for with these scans? Will we recognize this?”
Neurologist Jeffrey L. Cummings, MD, ScD, of the University of Nevada, Las Vegas, a vocal proponent of aducanumab and lead author of a May 2021 paper defending the evidence for it, acknowledged that the field was unprepared for a wide-scale adoption of infusions in dementia treatment, pointing to a Rand Corporation study from 2017 that warned that screening, diagnosis, and availability of infusion chairs would have to be drastically scaled up to meet demand.

“There are few clinicians who know how to identify MCI, too few imaging centers, too few radiologists who know how to identify ARIA-e on MRI, so all of these things will be required to be put into place. The label doesn’t specify any of this, but good clinical practice will require that, and getting this up and running will take 18 to 24 months,” Dr. Cummings said.
Neurologist David S. Knopman, MD, of the Mayo Clinic in Rochester, Minn., a leading critic of the evidence for aducanumab who recently resigned his position on the independent committee that advises the FDA on neurology drugs, said that for large research institutions like his that have served as trial sites, the transition to offering PET-amyloid, MRI, and infusions in clinical practice will be easier.

“We have all this because this is what we do every day. And we have a very extensive understanding of MCI and mild dementia staging,” Dr. Knopman said. “But the amount of infrastructure that is implied by this, and all the extra steps it would take, would be a real challenge for people in general neurology practice.”
In addition to routine use of PET-amyloid and MRI screening for ARIA-e, Dr. Knopman said, clinicians will have to provide genetic screening and counseling before administering aducanumab, as clinical trials showed that treated patients have a higher risk of developing ARIA-e if they have APOE4, a risk variant for Alzheimer’s disease. “And that has real implications for the families and the children of patients,” he said.
Uncertainty over costs
Aducanumab’s true costs, to patients and to taxpayers, remain unknown. The $56,000 per year currently cited by its manufacturer “doesn’t count the PET scans and MRIs,” Dr. Knopman noted. “We’re probably pushing $100,00 a year for the first year of treatment.”
Most of that expense will likely be borne by Medicare, he said, and if not, “that will exacerbate existing health care disparities. People who can pay out of pocket are a pretty limited group.”
Dr. Scharre agreed that the costs of treatment were concerning, and that “at least you should be able to narrow it down and hopefully just use health care dollars for people who might stand to benefit,” he said – namely patients in an earlier stage of disease.
The Alzheimer’s Association’s Dr. Carrillo declined to address the high price of aducanumab or its implications, saying only that the association is “very invested in all aspects of access including covering costs associated with the drug and the rest of treatment.”
Access also means “infrastructure, access to physicians to diagnose, access to diagnostics,” Dr. Carrillo said.
Dr. Cummings said aducanumab’s price would likely come down through negotiations with the Centers for Medicare & Medicaid Services, copayments, and bulk purchases.
The FDA has offered no guidance on how long treatment with aducanumab should last, or what should prompt withdrawal of treatment, meaning that patients could, in theory, stay on it to the end of their lives – raising costs further.
Critics have also noted that a built-in financial incentive under Medicare Part B, which covers infusion drugs, could result in overprescription of aducanumab. Under Medicare Part B, prescribing physicians are reimbursed 6% of a drug’s average sales price.
Geriatricians wary
On social media and in the lay press, geriatricians have been among the most outspoken opponents of the FDA decision and the Alzheimer’s Association’s advocacy of aducanumab.
Eric Widera, MD, a geriatrician at the University of California, San Francisco, said that the specialty might be less likely than others to embrace aducanumab. “I think part of the reasons geriatricians don’t make a lot of money is they have strong commitment to their values,” Dr. Widera said.

The American Geriatrics Society opposed the drug’s approval, citing concerns about evidence, side effects, and cost. “Additional considerations are the unintended consequences of overstressing Medicare’s limited financial reserves, and of challenging health care systems … to divert precious resources to an expensive treatment of uncertain value,” the society’s president, Peter Hollmann, MD, and chief executive officer, Nancy E. Lundebjerg, wrote in a June 2 letter to the FDA.
Dr. Widera said the approval was likely to undermine confidence in the FDA and in the Alzheimer’s Association, which receives significant funding from drug manufacturers, including Biogen and Eisai. “There’s a lot of reasons that the Geriatrics Society could have done what the Alzheimer’s Association did, and yet they came out against it, which I applaud.”
Dr. Widera pointed to a study showing that dementia patients were less likely to be on an antidementia drug if they were treated by a geriatrician, compared with a psychiatrist or a neurologist. But whether the specialty will prove as cautious with aducanumab remains to be seen. Some geriatricians will be tempted to open lucrative infusion centers, he predicted.
What is especially worrisome, Dr. Widera said, is that aducanumab’s label offers no guidance as to when to withdraw treatment. “We’ll probably see something similar to what happened with the cholinesterase inhibitors” – the class of marginally effective antidementia drugs that includes donepezil (Aricept, Pfizer) and rivastigmine (Exelon, Novartis). “No one thinks about deprescribing them. People are prescribed them even in their last months of life. There is no reason to think these infusions won’t be continued for a very long time, well beyond how long people were dosed in the trials.”
“Taking care of someone with dementia is hard enough,” Dr. Widera added. “We can’t even get normal support in the home for someone with dementia. But we are more than happy to throw money to Biogen for a drug they have not yet showed benefit for. Hopefully in 5 years we’ll have a drug that actually works,” Dr. Widera said. “After 5 years of giving this to people at $50,000 a year.”
A fractured research community
Ever since October 2019, when Biogen and Eisai announced that despite two trials halted for futility, they would go ahead and seek FDA approval for aducanumab, the Alzheimer’s research community has been bitterly divided over the drug and the FDA’s accelerated approval process.
Top researchers published critical editorials in journals, with some eventually taking their case to major newspapers as well. The Alzheimer’s Association’s position on the drug has clashed with that of many researchers whose work it supports.
“The Alzheimer’s community has been wonderfully collegial – we all have a common purpose,” Dr. Cummings said. “Now we have people taking extreme positions and I’m hoping this will not result in a permanent fracturing of the community.”
Chief among the critics’ concerns is that the FDA decision ratified the use of antiamyloid therapies based on biomarker evidence, opening the door for makers of similar drugs – those still under development or even those whose development has been halted – to seek approval on weak evidence of clinical benefit.
Whether the approval will chill research into drugs targeting pathways other than amyloid is uncertain.
Dr. Cummings said he felt that while the aducanumab decision would spur other manufacturers of antiamyloid drugs to seek accelerated approval, other classes of Alzheimer’s therapies in development also stand to get a boost. Many Alzheimer’s experts believe that a combination of drugs targeting different elements of the disease pathway – not just amyloid – will be needed in the long run.
Dr. Scharre said that the buzz over aducanumab’s approval will have at least one concrete benefit: people getting into doctors’ offices sooner.
“The people who come into our memory centers represent only a fraction of people walking around with MCI – there are people out there who may have heard that it’s normal aging; they have decreased insight; there’s denial, there’s embarrassment – there’s hundreds of reasons people avoid getting seen,” he said.
“Perhaps they come in and learn that they don’t have any degenerative process but their thyroid is out of whack, or there’s something else causing cognitive impairment. And if they do have a degenerative process, they’ll have time to start [aducanumab], and hopefully get to see a reduction in the decline.”
Dr. Knopman was a site investigator for the Biogen aducanumab trials and has consulted for Samus Therapeutics, Third Rock, Roche, and Alzeca Biosciences. A former member of the FDA’s Peripheral and Central Nervous System Drugs Advisory Committee, he was recused from the Nov. 6, 2020, meeting that voted against aducanumab. Dr. Cummings has consulted for Biogen, Eisai, and other manufacturers. Dr. Scharre reports financial relationships with Biogen, Brain Test, Acadia, and Vascular Scientific. Dr. Widera has no disclosures. Dr. Delio is a speaker for Gore Medical, Allergan, and Biohaven Pharmaceuticals.
The approval was hailed by advocacy groups and some practitioners as a victory for patients and families, as the drug – the first anti-Alzheimer’s agent to reach the market in 18 years – is a potentially disease-modifying therapy, which acts to clear amyloid plaques from the brain.
But several prominent Alzheimer’s researchers lambasted the agency’s decision, citing unclear evidence of benefit, trials that did not meet their primary endpoints, and reliance on a post hoc analysis of a high-dose subgroup of patients in a halted trial to argue that aducanumab (Aduhelm, Biogen, and Eisai), slowed cognitive and functional decline by 22% on one measure. In November 2020, 10 of 11 members of an independent FDA advisory committee voted against aducanumab’s approval, citing holes in the data and concerns about the quality of the evidence. After the agency went on to approve anyway, three members of that committee resigned in protest.
The FDA decision on aducanumab was made using the agency’s accelerated approval pathway, which allows for the use of a surrogate endpoint – in this case imaging that showed amyloid clearance from the brain – to predict clinical benefit. But amyloid clearance, which a number of experimental antiamyloid antibodies have been shown capable of, has not been definitively linked to clinical benefit. Aducanumab, which is delivered by monthly intravenous infusion, will be marketed pending results from a phase 4 clinical trial, which the manufacturer has nearly a decade to complete. The drug’s price was announced at $56,000 per year, underscoring concern over its modest-at-best benefits.
Clinicians prescribing aducanumab must obtain magnetic resonance imaging at baseline and repeatedly during the course of treatment to detect brain edema and microhemorrhages, which occurred in a third of high-dose patients in clinical trials. Beyond this, there are few restrictions. The FDA label allows for its use in any patient deemed to have Alzheimer’s disease, without stipulations as to disease stage or evidence of brain amyloid. Payers, of course, are likely to restrict use to certain patient groups, and to require evidence of amyloid positivity. The FDA offered no guidance on when treatment should be ceased, leaving payers to make that call as well. Whatever aducanumab’s value and role turns out to be, the first-in-class treatment for Alzheimer’s disease is likely to have a major impact on how patients are assessed and treated in the coming years, and embolden manufactures of similar agents to seek FDA approval.
This news organization reached out to researchers, advocates, and specialists in the community to learn how they see this change playing out.
Fielding broad interest
Maria C. Carrillo, PhD, chief science officer of the Alzheimer’s Association, which was a strong proponent of aducanumab’s approval, acknowledged in an interview that the months to come are likely to be confusing for practitioners and families alike as the drug makes its way into community practices.
“We understand that off the bat millions of Americans will not have access to this tomorrow, but over time that will build. And the physician community, the specialists most likely to be prescribing this, over the next few years will even expand further,” Dr. Carrillo said.
For now, those specialists are mostly just struggling to respond responsibly to a deluge of inquiries from patients and their families.
“I’ve gotten like 20 calls in the just the past 2 days,” said neurologist Philip R. Delio, MD, who practices in Santa Barbara, Calif. “This is a longstanding issue that physicians have with patients’ access to information. Patients are getting information about a drug which isn’t available yet. They don’t know that it’s not ready to be sold. They don’t necessarily realize that a biopharma company won’t go into production until the FDA approves the drug.”
Many patients, Dr. Delio said, are aware of the controversy surrounding aducanumab and eager to hear their neurologist’s opinion. “I have tried to let them know that I want to see the trial data and to better understand the FDA’s rationale in approving it. I always caution patients that the devil will be in the details.”
While aducanumab’s label gives physicians remarkably wide latitude in whom to treat, clinicians say that until payers weigh in, the label is all but meaningless. Neurologist Douglas Scharre, MD, of the Ohio State University Wexner Medical Center, and a site investigator on a trial of aducanumab, said that he and his colleagues at the university’s memory center have tried to anticipate who might be deemed eligible by triaging calls.
Dr. Scharre and colleagues have been working under the assumption that payers will support aducanumab only for patients like those who seemed to benefit in the trials – people with mild cognitive impairment (MCI) or in the earliest stages of dementia with evidence of brain amyloid.
“I don’t want to fill up our new patient slots with people who are not even appropriate for this drug,” Dr. Scharre said. “We have a call center, and we have a few triage questions. After that a nurse practitioner collects some more data, and there’s a review process. Only then do we decide whether that person could be a candidate. If we deem that they are, we will want them in and to order an amyloid PET” – a type of brain scan that is seldom used outside research settings and not reimbursed by Medicare.
Dr. Scharre predicts that regardless of payer limitations, “there will be people hounding for the drug who are not appropriate for the drug. There will be very wealthy people who will want to pay for tests and get it no matter what.” Another concern, he said, was that having poorly selected patients on the drug could make definitive trial results even more elusive.
“The label the way it’s written is not going to help the drug in phase 4 trials,” he said. “It’s good to have real-world patient data, but if you have all these people in your cohort who are too early or too late, you won’t have good results.”
The challenge of delivery
Intravenous infusions are new to Alzheimer’s disease and pose all sorts of logistical hurdles. The Alzheimer’s Association’s Dr. Carrillo described the situation as “manageable,” noting that infusions are standard of care for many diseases, and that neurologists now have more than 15 years’ experience with them for multiple sclerosis.
Still, most clinicians treating Alzheimer’s disease in the community – neurologists, geriatricians, psychiatrists, and primary care physicians – do not have infusion centers in their practices. Virtually none have experience with or access to PET-amyloid, or with screening for amyloid-related imaging abnormalities–edema (ARIA-e) on MRI, as required by the FDA.
“I contacted the hospital infusion center we use and said I could end up sending five or six patients a week, can you handle this? They only have so many chairs,” Dr. Delio said. “I am one neurologist in a local community, and I might have 50 candidates for this drug. That’s a lot for them.” Patients with cognitive impairment are also difficult to infuse and may need to be treated at home, he noted.
“MRIs are easy enough to do,” Dr. Delio said. “But do we know what ARIA-e looks like on imaging? You’d have to talk to the radiologists – this is another element of uncertainty. Do we even know what we’re looking for with these scans? Will we recognize this?”
Neurologist Jeffrey L. Cummings, MD, ScD, of the University of Nevada, Las Vegas, a vocal proponent of aducanumab and lead author of a May 2021 paper defending the evidence for it, acknowledged that the field was unprepared for a wide-scale adoption of infusions in dementia treatment, pointing to a Rand Corporation study from 2017 that warned that screening, diagnosis, and availability of infusion chairs would have to be drastically scaled up to meet demand.
“There are few clinicians who know how to identify MCI, too few imaging centers, too few radiologists who know how to identify ARIA-e on MRI, so all of these things will be required to be put into place. The label doesn’t specify any of this, but good clinical practice will require that, and getting this up and running will take 18 to 24 months,” Dr. Cummings said.
Neurologist David S. Knopman, MD, of the Mayo Clinic in Rochester, Minn., a leading critic of the evidence for aducanumab who recently resigned his position on the independent committee that advises the FDA on neurology drugs, said that for large research institutions like his that have served as trial sites, the transition to offering PET-amyloid, MRI, and infusions in clinical practice will be easier.
“We have all this because this is what we do every day. And we have a very extensive understanding of MCI and mild dementia staging,” Dr. Knopman said. “But the amount of infrastructure that is implied by this, and all the extra steps it would take, would be a real challenge for people in general neurology practice.”
In addition to routine use of PET-amyloid and MRI screening for ARIA-e, Dr. Knopman said, clinicians will have to provide genetic screening and counseling before administering aducanumab, as clinical trials showed that treated patients have a higher risk of developing ARIA-e if they have APOE4, a risk variant for Alzheimer’s disease. “And that has real implications for the families and the children of patients,” he said.
Uncertainty over costs
Aducanumab’s true costs, to patients and to taxpayers, remain unknown. The $56,000 per year currently cited by its manufacturer “doesn’t count the PET scans and MRIs,” Dr. Knopman noted. “We’re probably pushing $100,00 a year for the first year of treatment.”
Most of that expense will likely be borne by Medicare, he said, and if not, “that will exacerbate existing health care disparities. People who can pay out of pocket are a pretty limited group.”
Dr. Scharre agreed that the costs of treatment were concerning, and that “at least you should be able to narrow it down and hopefully just use health care dollars for people who might stand to benefit,” he said – namely patients in an earlier stage of disease.
The Alzheimer’s Association’s Dr. Carrillo declined to address the high price of aducanumab or its implications, saying only that the association is “very invested in all aspects of access including covering costs associated with the drug and the rest of treatment.”
Access also means “infrastructure, access to physicians to diagnose, access to diagnostics,” Dr. Carrillo said.
Dr. Cummings said aducanumab’s price would likely come down through negotiations with the Centers for Medicare & Medicaid Services, copayments, and bulk purchases.
The FDA has offered no guidance on how long treatment with aducanumab should last, or what should prompt withdrawal of treatment, meaning that patients could, in theory, stay on it to the end of their lives – raising costs further.
Critics have also noted that a built-in financial incentive under Medicare Part B, which covers infusion drugs, could result in overprescription of aducanumab. Under Medicare Part B, prescribing physicians are reimbursed 6% of a drug’s average sales price.
Geriatricians wary
On social media and in the lay press, geriatricians have been among the most outspoken opponents of the FDA decision and the Alzheimer’s Association’s advocacy of aducanumab.
Eric Widera, MD, a geriatrician at the University of California, San Francisco, said that the specialty might be less likely than others to embrace aducanumab. “I think part of the reasons geriatricians don’t make a lot of money is they have strong commitment to their values,” Dr. Widera said.
The American Geriatrics Society opposed the drug’s approval, citing concerns about evidence, side effects, and cost. “Additional considerations are the unintended consequences of overstressing Medicare’s limited financial reserves, and of challenging health care systems … to divert precious resources to an expensive treatment of uncertain value,” the society’s president, Peter Hollmann, MD, and chief executive officer, Nancy E. Lundebjerg, wrote in a June 2 letter to the FDA.
Dr. Widera said the approval was likely to undermine confidence in the FDA and in the Alzheimer’s Association, which receives significant funding from drug manufacturers, including Biogen and Eisai. “There’s a lot of reasons that the Geriatrics Society could have done what the Alzheimer’s Association did, and yet they came out against it, which I applaud.”
Dr. Widera pointed to a study showing that dementia patients were less likely to be on an antidementia drug if they were treated by a geriatrician, compared with a psychiatrist or a neurologist. But whether the specialty will prove as cautious with aducanumab remains to be seen. Some geriatricians will be tempted to open lucrative infusion centers, he predicted.
What is especially worrisome, Dr. Widera said, is that aducanumab’s label offers no guidance as to when to withdraw treatment. “We’ll probably see something similar to what happened with the cholinesterase inhibitors” – the class of marginally effective antidementia drugs that includes donepezil (Aricept, Pfizer) and rivastigmine (Exelon, Novartis). “No one thinks about deprescribing them. People are prescribed them even in their last months of life. There is no reason to think these infusions won’t be continued for a very long time, well beyond how long people were dosed in the trials.”
“Taking care of someone with dementia is hard enough,” Dr. Widera added. “We can’t even get normal support in the home for someone with dementia. But we are more than happy to throw money to Biogen for a drug they have not yet showed benefit for. Hopefully in 5 years we’ll have a drug that actually works,” Dr. Widera said. “After 5 years of giving this to people at $50,000 a year.”
A fractured research community
Ever since October 2019, when Biogen and Eisai announced that despite two trials halted for futility, they would go ahead and seek FDA approval for aducanumab, the Alzheimer’s research community has been bitterly divided over the drug and the FDA’s accelerated approval process.
Top researchers published critical editorials in journals, with some eventually taking their case to major newspapers as well. The Alzheimer’s Association’s position on the drug has clashed with that of many researchers whose work it supports.
“The Alzheimer’s community has been wonderfully collegial – we all have a common purpose,” Dr. Cummings said. “Now we have people taking extreme positions and I’m hoping this will not result in a permanent fracturing of the community.”
Chief among the critics’ concerns is that the FDA decision ratified the use of antiamyloid therapies based on biomarker evidence, opening the door for makers of similar drugs – those still under development or even those whose development has been halted – to seek approval on weak evidence of clinical benefit.
Whether the approval will chill research into drugs targeting pathways other than amyloid is uncertain.
Dr. Cummings said he felt that while the aducanumab decision would spur other manufacturers of antiamyloid drugs to seek accelerated approval, other classes of Alzheimer’s therapies in development also stand to get a boost. Many Alzheimer’s experts believe that a combination of drugs targeting different elements of the disease pathway – not just amyloid – will be needed in the long run.
Dr. Scharre said that the buzz over aducanumab’s approval will have at least one concrete benefit: people getting into doctors’ offices sooner.
“The people who come into our memory centers represent only a fraction of people walking around with MCI – there are people out there who may have heard that it’s normal aging; they have decreased insight; there’s denial, there’s embarrassment – there’s hundreds of reasons people avoid getting seen,” he said.
“Perhaps they come in and learn that they don’t have any degenerative process but their thyroid is out of whack, or there’s something else causing cognitive impairment. And if they do have a degenerative process, they’ll have time to start [aducanumab], and hopefully get to see a reduction in the decline.”
Dr. Knopman was a site investigator for the Biogen aducanumab trials and has consulted for Samus Therapeutics, Third Rock, Roche, and Alzeca Biosciences. A former member of the FDA’s Peripheral and Central Nervous System Drugs Advisory Committee, he was recused from the Nov. 6, 2020, meeting that voted against aducanumab. Dr. Cummings has consulted for Biogen, Eisai, and other manufacturers. Dr. Scharre reports financial relationships with Biogen, Brain Test, Acadia, and Vascular Scientific. Dr. Widera has no disclosures. Dr. Delio is a speaker for Gore Medical, Allergan, and Biohaven Pharmaceuticals.
The approval was hailed by advocacy groups and some practitioners as a victory for patients and families, as the drug – the first anti-Alzheimer’s agent to reach the market in 18 years – is a potentially disease-modifying therapy, which acts to clear amyloid plaques from the brain.
But several prominent Alzheimer’s researchers lambasted the agency’s decision, citing unclear evidence of benefit, trials that did not meet their primary endpoints, and reliance on a post hoc analysis of a high-dose subgroup of patients in a halted trial to argue that aducanumab (Aduhelm, Biogen, and Eisai), slowed cognitive and functional decline by 22% on one measure. In November 2020, 10 of 11 members of an independent FDA advisory committee voted against aducanumab’s approval, citing holes in the data and concerns about the quality of the evidence. After the agency went on to approve anyway, three members of that committee resigned in protest.
The FDA decision on aducanumab was made using the agency’s accelerated approval pathway, which allows for the use of a surrogate endpoint – in this case imaging that showed amyloid clearance from the brain – to predict clinical benefit. But amyloid clearance, which a number of experimental antiamyloid antibodies have been shown capable of, has not been definitively linked to clinical benefit. Aducanumab, which is delivered by monthly intravenous infusion, will be marketed pending results from a phase 4 clinical trial, which the manufacturer has nearly a decade to complete. The drug’s price was announced at $56,000 per year, underscoring concern over its modest-at-best benefits.
Clinicians prescribing aducanumab must obtain magnetic resonance imaging at baseline and repeatedly during the course of treatment to detect brain edema and microhemorrhages, which occurred in a third of high-dose patients in clinical trials. Beyond this, there are few restrictions. The FDA label allows for its use in any patient deemed to have Alzheimer’s disease, without stipulations as to disease stage or evidence of brain amyloid. Payers, of course, are likely to restrict use to certain patient groups, and to require evidence of amyloid positivity. The FDA offered no guidance on when treatment should be ceased, leaving payers to make that call as well. Whatever aducanumab’s value and role turns out to be, the first-in-class treatment for Alzheimer’s disease is likely to have a major impact on how patients are assessed and treated in the coming years, and embolden manufactures of similar agents to seek FDA approval.
This news organization reached out to researchers, advocates, and specialists in the community to learn how they see this change playing out.
Fielding broad interest
Maria C. Carrillo, PhD, chief science officer of the Alzheimer’s Association, which was a strong proponent of aducanumab’s approval, acknowledged in an interview that the months to come are likely to be confusing for practitioners and families alike as the drug makes its way into community practices.
“We understand that off the bat millions of Americans will not have access to this tomorrow, but over time that will build. And the physician community, the specialists most likely to be prescribing this, over the next few years will even expand further,” Dr. Carrillo said.
For now, those specialists are mostly just struggling to respond responsibly to a deluge of inquiries from patients and their families.
“I’ve gotten like 20 calls in the just the past 2 days,” said neurologist Philip R. Delio, MD, who practices in Santa Barbara, Calif. “This is a longstanding issue that physicians have with patients’ access to information. Patients are getting information about a drug which isn’t available yet. They don’t know that it’s not ready to be sold. They don’t necessarily realize that a biopharma company won’t go into production until the FDA approves the drug.”
Many patients, Dr. Delio said, are aware of the controversy surrounding aducanumab and eager to hear their neurologist’s opinion. “I have tried to let them know that I want to see the trial data and to better understand the FDA’s rationale in approving it. I always caution patients that the devil will be in the details.”
While aducanumab’s label gives physicians remarkably wide latitude in whom to treat, clinicians say that until payers weigh in, the label is all but meaningless. Neurologist Douglas Scharre, MD, of the Ohio State University Wexner Medical Center, and a site investigator on a trial of aducanumab, said that he and his colleagues at the university’s memory center have tried to anticipate who might be deemed eligible by triaging calls.
Dr. Scharre and colleagues have been working under the assumption that payers will support aducanumab only for patients like those who seemed to benefit in the trials – people with mild cognitive impairment (MCI) or in the earliest stages of dementia with evidence of brain amyloid.
“I don’t want to fill up our new patient slots with people who are not even appropriate for this drug,” Dr. Scharre said. “We have a call center, and we have a few triage questions. After that a nurse practitioner collects some more data, and there’s a review process. Only then do we decide whether that person could be a candidate. If we deem that they are, we will want them in and to order an amyloid PET” – a type of brain scan that is seldom used outside research settings and not reimbursed by Medicare.
Dr. Scharre predicts that regardless of payer limitations, “there will be people hounding for the drug who are not appropriate for the drug. There will be very wealthy people who will want to pay for tests and get it no matter what.” Another concern, he said, was that having poorly selected patients on the drug could make definitive trial results even more elusive.
“The label the way it’s written is not going to help the drug in phase 4 trials,” he said. “It’s good to have real-world patient data, but if you have all these people in your cohort who are too early or too late, you won’t have good results.”
The challenge of delivery
Intravenous infusions are new to Alzheimer’s disease and pose all sorts of logistical hurdles. The Alzheimer’s Association’s Dr. Carrillo described the situation as “manageable,” noting that infusions are standard of care for many diseases, and that neurologists now have more than 15 years’ experience with them for multiple sclerosis.
Still, most clinicians treating Alzheimer’s disease in the community – neurologists, geriatricians, psychiatrists, and primary care physicians – do not have infusion centers in their practices. Virtually none have experience with or access to PET-amyloid, or with screening for amyloid-related imaging abnormalities–edema (ARIA-e) on MRI, as required by the FDA.
“I contacted the hospital infusion center we use and said I could end up sending five or six patients a week, can you handle this? They only have so many chairs,” Dr. Delio said. “I am one neurologist in a local community, and I might have 50 candidates for this drug. That’s a lot for them.” Patients with cognitive impairment are also difficult to infuse and may need to be treated at home, he noted.
“MRIs are easy enough to do,” Dr. Delio said. “But do we know what ARIA-e looks like on imaging? You’d have to talk to the radiologists – this is another element of uncertainty. Do we even know what we’re looking for with these scans? Will we recognize this?”
Neurologist Jeffrey L. Cummings, MD, ScD, of the University of Nevada, Las Vegas, a vocal proponent of aducanumab and lead author of a May 2021 paper defending the evidence for it, acknowledged that the field was unprepared for a wide-scale adoption of infusions in dementia treatment, pointing to a Rand Corporation study from 2017 that warned that screening, diagnosis, and availability of infusion chairs would have to be drastically scaled up to meet demand.
“There are few clinicians who know how to identify MCI, too few imaging centers, too few radiologists who know how to identify ARIA-e on MRI, so all of these things will be required to be put into place. The label doesn’t specify any of this, but good clinical practice will require that, and getting this up and running will take 18 to 24 months,” Dr. Cummings said.
Neurologist David S. Knopman, MD, of the Mayo Clinic in Rochester, Minn., a leading critic of the evidence for aducanumab who recently resigned his position on the independent committee that advises the FDA on neurology drugs, said that for large research institutions like his that have served as trial sites, the transition to offering PET-amyloid, MRI, and infusions in clinical practice will be easier.
“We have all this because this is what we do every day. And we have a very extensive understanding of MCI and mild dementia staging,” Dr. Knopman said. “But the amount of infrastructure that is implied by this, and all the extra steps it would take, would be a real challenge for people in general neurology practice.”
In addition to routine use of PET-amyloid and MRI screening for ARIA-e, Dr. Knopman said, clinicians will have to provide genetic screening and counseling before administering aducanumab, as clinical trials showed that treated patients have a higher risk of developing ARIA-e if they have APOE4, a risk variant for Alzheimer’s disease. “And that has real implications for the families and the children of patients,” he said.
Uncertainty over costs
Aducanumab’s true costs, to patients and to taxpayers, remain unknown. The $56,000 per year currently cited by its manufacturer “doesn’t count the PET scans and MRIs,” Dr. Knopman noted. “We’re probably pushing $100,00 a year for the first year of treatment.”
Most of that expense will likely be borne by Medicare, he said, and if not, “that will exacerbate existing health care disparities. People who can pay out of pocket are a pretty limited group.”
Dr. Scharre agreed that the costs of treatment were concerning, and that “at least you should be able to narrow it down and hopefully just use health care dollars for people who might stand to benefit,” he said – namely patients in an earlier stage of disease.
The Alzheimer’s Association’s Dr. Carrillo declined to address the high price of aducanumab or its implications, saying only that the association is “very invested in all aspects of access including covering costs associated with the drug and the rest of treatment.”
Access also means “infrastructure, access to physicians to diagnose, access to diagnostics,” Dr. Carrillo said.
Dr. Cummings said aducanumab’s price would likely come down through negotiations with the Centers for Medicare & Medicaid Services, copayments, and bulk purchases.
The FDA has offered no guidance on how long treatment with aducanumab should last, or what should prompt withdrawal of treatment, meaning that patients could, in theory, stay on it to the end of their lives – raising costs further.
Critics have also noted that a built-in financial incentive under Medicare Part B, which covers infusion drugs, could result in overprescription of aducanumab. Under Medicare Part B, prescribing physicians are reimbursed 6% of a drug’s average sales price.
Geriatricians wary
On social media and in the lay press, geriatricians have been among the most outspoken opponents of the FDA decision and the Alzheimer’s Association’s advocacy of aducanumab.
Eric Widera, MD, a geriatrician at the University of California, San Francisco, said that the specialty might be less likely than others to embrace aducanumab. “I think part of the reasons geriatricians don’t make a lot of money is they have strong commitment to their values,” Dr. Widera said.
The American Geriatrics Society opposed the drug’s approval, citing concerns about evidence, side effects, and cost. “Additional considerations are the unintended consequences of overstressing Medicare’s limited financial reserves, and of challenging health care systems … to divert precious resources to an expensive treatment of uncertain value,” the society’s president, Peter Hollmann, MD, and chief executive officer, Nancy E. Lundebjerg, wrote in a June 2 letter to the FDA.
Dr. Widera said the approval was likely to undermine confidence in the FDA and in the Alzheimer’s Association, which receives significant funding from drug manufacturers, including Biogen and Eisai. “There’s a lot of reasons that the Geriatrics Society could have done what the Alzheimer’s Association did, and yet they came out against it, which I applaud.”
Dr. Widera pointed to a study showing that dementia patients were less likely to be on an antidementia drug if they were treated by a geriatrician, compared with a psychiatrist or a neurologist. But whether the specialty will prove as cautious with aducanumab remains to be seen. Some geriatricians will be tempted to open lucrative infusion centers, he predicted.
What is especially worrisome, Dr. Widera said, is that aducanumab’s label offers no guidance as to when to withdraw treatment. “We’ll probably see something similar to what happened with the cholinesterase inhibitors” – the class of marginally effective antidementia drugs that includes donepezil (Aricept, Pfizer) and rivastigmine (Exelon, Novartis). “No one thinks about deprescribing them. People are prescribed them even in their last months of life. There is no reason to think these infusions won’t be continued for a very long time, well beyond how long people were dosed in the trials.”
“Taking care of someone with dementia is hard enough,” Dr. Widera added. “We can’t even get normal support in the home for someone with dementia. But we are more than happy to throw money to Biogen for a drug they have not yet showed benefit for. Hopefully in 5 years we’ll have a drug that actually works,” Dr. Widera said. “After 5 years of giving this to people at $50,000 a year.”
A fractured research community
Ever since October 2019, when Biogen and Eisai announced that despite two trials halted for futility, they would go ahead and seek FDA approval for aducanumab, the Alzheimer’s research community has been bitterly divided over the drug and the FDA’s accelerated approval process.
Top researchers published critical editorials in journals, with some eventually taking their case to major newspapers as well. The Alzheimer’s Association’s position on the drug has clashed with that of many researchers whose work it supports.
“The Alzheimer’s community has been wonderfully collegial – we all have a common purpose,” Dr. Cummings said. “Now we have people taking extreme positions and I’m hoping this will not result in a permanent fracturing of the community.”
Chief among the critics’ concerns is that the FDA decision ratified the use of antiamyloid therapies based on biomarker evidence, opening the door for makers of similar drugs – those still under development or even those whose development has been halted – to seek approval on weak evidence of clinical benefit.
Whether the approval will chill research into drugs targeting pathways other than amyloid is uncertain.
Dr. Cummings said he felt that while the aducanumab decision would spur other manufacturers of antiamyloid drugs to seek accelerated approval, other classes of Alzheimer’s therapies in development also stand to get a boost. Many Alzheimer’s experts believe that a combination of drugs targeting different elements of the disease pathway – not just amyloid – will be needed in the long run.
Dr. Scharre said that the buzz over aducanumab’s approval will have at least one concrete benefit: people getting into doctors’ offices sooner.
“The people who come into our memory centers represent only a fraction of people walking around with MCI – there are people out there who may have heard that it’s normal aging; they have decreased insight; there’s denial, there’s embarrassment – there’s hundreds of reasons people avoid getting seen,” he said.
“Perhaps they come in and learn that they don’t have any degenerative process but their thyroid is out of whack, or there’s something else causing cognitive impairment. And if they do have a degenerative process, they’ll have time to start [aducanumab], and hopefully get to see a reduction in the decline.”
Dr. Knopman was a site investigator for the Biogen aducanumab trials and has consulted for Samus Therapeutics, Third Rock, Roche, and Alzeca Biosciences. A former member of the FDA’s Peripheral and Central Nervous System Drugs Advisory Committee, he was recused from the Nov. 6, 2020, meeting that voted against aducanumab. Dr. Cummings has consulted for Biogen, Eisai, and other manufacturers. Dr. Scharre reports financial relationships with Biogen, Brain Test, Acadia, and Vascular Scientific. Dr. Widera has no disclosures. Dr. Delio is a speaker for Gore Medical, Allergan, and Biohaven Pharmaceuticals.
‘Remarkable’ results for targeted therapy of rare CNS tumors
The results from three small studies of targeted therapy for rare brain tumors were “remarkable,” according to Jaishri Blakeley, MD, a neurology professor at Johns Hopkins Medicine, Baltimore, who discussed the studies after they were presented at the American Society of Clinical Oncology meeting.
Although most patients don’t have targetable mutations, molecular testing “is well worth the effort,” for those that do. “I think it’s fair to say that precision medicine” – well established in other tumor types – “is finally here in full force for neuro-oncology,” Dr. Blakeley said.
A promising start
Fifteen of 16 patients (94%) in one study had newly diagnosed and untreated papillary craniopharyngiomas (PCPs) that harbored BRAF V600E mutations, a common finding in PCPs, which have no effective medical treatment.
Tumors shrunk 68%-99% in 14 patients (93%) after treatment with the BRAF inhibitor vemurafenib plus the MEK inhibitor cobimetinib, which was included to stave off resistance to vemurafenib. The 24-month progression free survival was 93%.
The combination resulted in significant response in all patients who received at least one cycle of therapy, with a median 91% volume reduction. “Our study indicates that BRAF/MEK inhibitors could be a powerful tool in the treatment of previously untreated PCP, with the potential to avoid the morbidity associated with radiation and surgery,” concluded lead investigator and presenter Priscilla K. Brastianos, MD, associate professor of medicine at Mass General Cancer Center, Boston.
Thirty-three people in the second study had a mix of high and low grade gliomas or other CNS tumors positive for TRK gene fusions, a known oncogenic driver; the majority were children. They were treated with the TRK inhibitor larotrectinib after progressing on other systemic therapies.
The objective response rate was 30%, and the disease control rate was 73% at 24 weeks, with a median time to best response of 1.9 months. Tumors shrank in 82% of evaluable patients. Median progression-free survival was 18.3 months, and overall survival was not reached.
“These results support testing for TRK gene fusions for all patients with CNS tumors, especially if there is no known driver and especially in infants,” concluded lead investigator and presenter Sebastien Perreault, MD, a clinical assistant neurosciences professor at the University of Montreal.
The third study tested ALK inhibitors such as crizotinib in seven patients with adult-onset neuroblastoma, a rare and almost invariably fatal tumor known to be enriched for ALK mutations; the subjects were positive for them.
Their disease remained stable anywhere from 3.4 to 37.4 months. Median time to progression was 15.5 months, and median overall survival was 46.5 months.
ALK inhibitors “can be a well-tolerated options for treatment, improving time to progression. Development of resistance to one agent does not preclude use of other agents in the same drug class. ALK inhibitors should be considered when treating patients with this diagnosis,” said lead investigator and presenter Jessica Stiefel, MD, a pediatric hematology oncology fellow at Memorial Sloan Kettering Cancer Center, New York.
A ‘strong’ recommendation
The data “are great news” across the board. Targeted therapy applied to the right CNS tumor can have “dramatic” benefit for tumor control, Dr. Blakeley said.
But organizing molecular testing is not straightforward and requires strategies to balance “the use of precious resources, such as time money, and tissue,” with the potential benefit. Interpretation of testing results isn’t straightforward either, and is best handled by a molecular tumor board. Clinical pharmacists are also key to accessing expensive medications off label for CNS tumors.
Adverse events are also a consideration. Most of the subjects in the PCP study had grade 3/4 toxicity. Three patients in the ALK inhibitor study had to stop because of adverse events. Almost 40% on larotrectinib had grade 3 or 4 toxicity; nobody came off treatment, but a third had to skip doses.
Once an actionable mutation is identified, Dr. Blakeley’s “strong recommendation” is to enroll patients in a clinical trial that targets it, to take advantage the structure already in place to secure treatment, managed patients, and assess outcomes.
The National Cancer Institute’s MATCH trial is one of several options.
The BRAF/MEK inhibitor study was funded by Genentech and the National Institutes of Health. Dr. Brastianos had ties to numerous companies, including Pfizer, Lilly, and Merck. The TRK inhibitor study was funded by Bayer/Lilly. Dr. Perreault is a speaker and researcher for the company and has other ties. Dr. Blakeley is an adviser and/or researcher for a number of companies, including AbbVie, Astellas, BMS, and Exelixis. Dr. Stiefel didn’t have any disclosures, and didn’t report outside funding.
The results from three small studies of targeted therapy for rare brain tumors were “remarkable,” according to Jaishri Blakeley, MD, a neurology professor at Johns Hopkins Medicine, Baltimore, who discussed the studies after they were presented at the American Society of Clinical Oncology meeting.
Although most patients don’t have targetable mutations, molecular testing “is well worth the effort,” for those that do. “I think it’s fair to say that precision medicine” – well established in other tumor types – “is finally here in full force for neuro-oncology,” Dr. Blakeley said.
A promising start
Fifteen of 16 patients (94%) in one study had newly diagnosed and untreated papillary craniopharyngiomas (PCPs) that harbored BRAF V600E mutations, a common finding in PCPs, which have no effective medical treatment.
Tumors shrunk 68%-99% in 14 patients (93%) after treatment with the BRAF inhibitor vemurafenib plus the MEK inhibitor cobimetinib, which was included to stave off resistance to vemurafenib. The 24-month progression free survival was 93%.
The combination resulted in significant response in all patients who received at least one cycle of therapy, with a median 91% volume reduction. “Our study indicates that BRAF/MEK inhibitors could be a powerful tool in the treatment of previously untreated PCP, with the potential to avoid the morbidity associated with radiation and surgery,” concluded lead investigator and presenter Priscilla K. Brastianos, MD, associate professor of medicine at Mass General Cancer Center, Boston.
Thirty-three people in the second study had a mix of high and low grade gliomas or other CNS tumors positive for TRK gene fusions, a known oncogenic driver; the majority were children. They were treated with the TRK inhibitor larotrectinib after progressing on other systemic therapies.
The objective response rate was 30%, and the disease control rate was 73% at 24 weeks, with a median time to best response of 1.9 months. Tumors shrank in 82% of evaluable patients. Median progression-free survival was 18.3 months, and overall survival was not reached.
“These results support testing for TRK gene fusions for all patients with CNS tumors, especially if there is no known driver and especially in infants,” concluded lead investigator and presenter Sebastien Perreault, MD, a clinical assistant neurosciences professor at the University of Montreal.
The third study tested ALK inhibitors such as crizotinib in seven patients with adult-onset neuroblastoma, a rare and almost invariably fatal tumor known to be enriched for ALK mutations; the subjects were positive for them.
Their disease remained stable anywhere from 3.4 to 37.4 months. Median time to progression was 15.5 months, and median overall survival was 46.5 months.
ALK inhibitors “can be a well-tolerated options for treatment, improving time to progression. Development of resistance to one agent does not preclude use of other agents in the same drug class. ALK inhibitors should be considered when treating patients with this diagnosis,” said lead investigator and presenter Jessica Stiefel, MD, a pediatric hematology oncology fellow at Memorial Sloan Kettering Cancer Center, New York.
A ‘strong’ recommendation
The data “are great news” across the board. Targeted therapy applied to the right CNS tumor can have “dramatic” benefit for tumor control, Dr. Blakeley said.
But organizing molecular testing is not straightforward and requires strategies to balance “the use of precious resources, such as time money, and tissue,” with the potential benefit. Interpretation of testing results isn’t straightforward either, and is best handled by a molecular tumor board. Clinical pharmacists are also key to accessing expensive medications off label for CNS tumors.
Adverse events are also a consideration. Most of the subjects in the PCP study had grade 3/4 toxicity. Three patients in the ALK inhibitor study had to stop because of adverse events. Almost 40% on larotrectinib had grade 3 or 4 toxicity; nobody came off treatment, but a third had to skip doses.
Once an actionable mutation is identified, Dr. Blakeley’s “strong recommendation” is to enroll patients in a clinical trial that targets it, to take advantage the structure already in place to secure treatment, managed patients, and assess outcomes.
The National Cancer Institute’s MATCH trial is one of several options.
The BRAF/MEK inhibitor study was funded by Genentech and the National Institutes of Health. Dr. Brastianos had ties to numerous companies, including Pfizer, Lilly, and Merck. The TRK inhibitor study was funded by Bayer/Lilly. Dr. Perreault is a speaker and researcher for the company and has other ties. Dr. Blakeley is an adviser and/or researcher for a number of companies, including AbbVie, Astellas, BMS, and Exelixis. Dr. Stiefel didn’t have any disclosures, and didn’t report outside funding.
The results from three small studies of targeted therapy for rare brain tumors were “remarkable,” according to Jaishri Blakeley, MD, a neurology professor at Johns Hopkins Medicine, Baltimore, who discussed the studies after they were presented at the American Society of Clinical Oncology meeting.
Although most patients don’t have targetable mutations, molecular testing “is well worth the effort,” for those that do. “I think it’s fair to say that precision medicine” – well established in other tumor types – “is finally here in full force for neuro-oncology,” Dr. Blakeley said.
A promising start
Fifteen of 16 patients (94%) in one study had newly diagnosed and untreated papillary craniopharyngiomas (PCPs) that harbored BRAF V600E mutations, a common finding in PCPs, which have no effective medical treatment.
Tumors shrunk 68%-99% in 14 patients (93%) after treatment with the BRAF inhibitor vemurafenib plus the MEK inhibitor cobimetinib, which was included to stave off resistance to vemurafenib. The 24-month progression free survival was 93%.
The combination resulted in significant response in all patients who received at least one cycle of therapy, with a median 91% volume reduction. “Our study indicates that BRAF/MEK inhibitors could be a powerful tool in the treatment of previously untreated PCP, with the potential to avoid the morbidity associated with radiation and surgery,” concluded lead investigator and presenter Priscilla K. Brastianos, MD, associate professor of medicine at Mass General Cancer Center, Boston.
Thirty-three people in the second study had a mix of high and low grade gliomas or other CNS tumors positive for TRK gene fusions, a known oncogenic driver; the majority were children. They were treated with the TRK inhibitor larotrectinib after progressing on other systemic therapies.
The objective response rate was 30%, and the disease control rate was 73% at 24 weeks, with a median time to best response of 1.9 months. Tumors shrank in 82% of evaluable patients. Median progression-free survival was 18.3 months, and overall survival was not reached.
“These results support testing for TRK gene fusions for all patients with CNS tumors, especially if there is no known driver and especially in infants,” concluded lead investigator and presenter Sebastien Perreault, MD, a clinical assistant neurosciences professor at the University of Montreal.
The third study tested ALK inhibitors such as crizotinib in seven patients with adult-onset neuroblastoma, a rare and almost invariably fatal tumor known to be enriched for ALK mutations; the subjects were positive for them.
Their disease remained stable anywhere from 3.4 to 37.4 months. Median time to progression was 15.5 months, and median overall survival was 46.5 months.
ALK inhibitors “can be a well-tolerated options for treatment, improving time to progression. Development of resistance to one agent does not preclude use of other agents in the same drug class. ALK inhibitors should be considered when treating patients with this diagnosis,” said lead investigator and presenter Jessica Stiefel, MD, a pediatric hematology oncology fellow at Memorial Sloan Kettering Cancer Center, New York.
A ‘strong’ recommendation
The data “are great news” across the board. Targeted therapy applied to the right CNS tumor can have “dramatic” benefit for tumor control, Dr. Blakeley said.
But organizing molecular testing is not straightforward and requires strategies to balance “the use of precious resources, such as time money, and tissue,” with the potential benefit. Interpretation of testing results isn’t straightforward either, and is best handled by a molecular tumor board. Clinical pharmacists are also key to accessing expensive medications off label for CNS tumors.
Adverse events are also a consideration. Most of the subjects in the PCP study had grade 3/4 toxicity. Three patients in the ALK inhibitor study had to stop because of adverse events. Almost 40% on larotrectinib had grade 3 or 4 toxicity; nobody came off treatment, but a third had to skip doses.
Once an actionable mutation is identified, Dr. Blakeley’s “strong recommendation” is to enroll patients in a clinical trial that targets it, to take advantage the structure already in place to secure treatment, managed patients, and assess outcomes.
The National Cancer Institute’s MATCH trial is one of several options.
The BRAF/MEK inhibitor study was funded by Genentech and the National Institutes of Health. Dr. Brastianos had ties to numerous companies, including Pfizer, Lilly, and Merck. The TRK inhibitor study was funded by Bayer/Lilly. Dr. Perreault is a speaker and researcher for the company and has other ties. Dr. Blakeley is an adviser and/or researcher for a number of companies, including AbbVie, Astellas, BMS, and Exelixis. Dr. Stiefel didn’t have any disclosures, and didn’t report outside funding.
FROM ASCO 2021
Experimental antibody-drug conjugate shown active against r/r DLBCL
Patients with relapsed or refractory B-cell non-Hodgkin lymphomas who are not candidates for hematopoietic stem cell transplant have a generally poor prognosis and few treatment options, but an experimental combination of the antibody-drug conjugate naratuximab with rituximab showed promising efficacy and acceptable safety in these patients in a phase 2 trial.
Among patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) the combination was associated with a 44.7% overall response rate, including 31.6% complete responses, and two-thirds of patients had responses lasting more than 12 months, reported Moshe Yair Levy, MD, from Texas Oncology–Baylor Charles A Sammons Cancer Center in Dallas.
“This is, in my viewpoint, very exciting therapy,” he said in a question-and-answer session following his presentation of the data in a late-breaking abstract session during the European Hematology Association annual congress. (Abstract LB1903).
Naratuximab emtansine is an investigational antibody-drug conjugate (ADC) consisting of a humanized monoclonal antibody against CD37, a surface marker on B lymphocytes that is highly expressed in non-Hodgkin lymphoma (NHL), conjugated to a cytotoxic derivative of maitansine.
CD37 is also an internalizable cell-surface antigen, making it an attractive candidate for an ADC approach.
In a phase 1 trial, naratuximab monotherapy showed a good safety profile and a 22% overall response rate, Dr. Levy noted.
“What they found is that, if you coadminister this ADC with rituximab, you’re actually going to get more internalization of the CD37 monoclonal, therefore more payload delivered to your target cells,” he said.
He reported results of a multicenter, adaptive phase 2 study of the combination in patients with DLBCL and other relapsed/refractory NHL.
DLBCL and others
The trial was divided into two parts, with the first consisting of a safety run-in phase with expansion in patients with confirmed diagnoses of relapsed/refractory NHL, including DLBCL, follicular lymphoma, mantle cell lymphoma, and marginal zone lymphoma.
Patients with double- or triple-hit disease (with translocations in MYC plus either BCL2 and/or BCL6), bulky disease, or transformed lymphoma were eligible.
The second part consisted of two cohorts of patients with DLBCL treated with naratuximab and rituximab either weekly or every 3 weeks.
All patients in the study had received one to six prior lines of therapy, and had Eastern Cooperative Oncology Group performance status of 0-2. Patients with CNS lymphomas or prior anti-CD37 targeting therapy were excluded.
The safety population included 50 patients with DLBCL assigned to therapy every 3 weeks, 30 assigned to weekly therapy, and 20 patients with other NHL.
DLBCL efficacy
A total of 76 patients with DLBCL were evaluable for efficacy.
The ORR was 44% for patients in both the weekly and every 3 week cohorts, with 31.6% having complete responses.
Among 61 patients with nonbulky disease (longest diameter 7.5 cm or less), the ORR was 50.8%, and among 28 patients who had three or more prior lines of therapy the ORR was 46.4%, with 32.1% having a complete response.
Among responders followed for a median of 15 months, the median duration of response was not reached, and 66% had responses lasting beyond 12 months.
In the weekly dosing DLBCL cohort, 53.3% of patients discontinued treatment of both study drugs because of disease progression, as did 58% of those in the every 3 week cohort, and 30% of patients with other lymphomas. Only eight patients discontinued the combination because of treatment-emergent adverse events. Six patients had treatment-emergent adverse events leading to naratuximab dose reduction.
The most common grade 3 or 4 adverse events were neutropenia, leukopenia, lymphopenia and thrombocytopenias. Dr. Levy commented that the use of granulocyte colony-stimulating factor, which was not mandatory in the study, would likely have lowered the incidence of cytopenias.
There were 10 deaths during the study, 2 of which were considered to be treatment related, occurring in 1 patient each in the DLBCL dosing cohorts; 1 of the patients died from pneumonitis, and the other from left ventricular heart failure.
Other patients deaths were attributed to non–treatment-related cardiac arrest, acute renal failure, exacerbation of chronic heart failure, respiratory failure, multiorgan failure, lung infection, or colon adenocarcinoma.
Q 3 weeks suffices
In the question-and-answer session following the presentation, Kenny Lei, MD, from the Chinese University of Hong Kong asked Dr. Levy what the half-life of naratuximab is, and what was the investigator’s rationale for testing a weekly dosing schedule.
“I think the reason they checked the two different regimens, the Q week and the Q 3-week group, is that they noted that [naratuximab] was cleared relatively quickly, and they wanted to see whether or not, by giving Q weekly, when you get a continuous CD37 site occupancy if they would have a better outcome. But as you saw, in the groups there was really no clinically relevant difference in outcome,” Dr. Levy said.
Andrew Davies, MD, PhD, from the University of Southampton (England), asked whether the neutropenia seen in the study was related to myeloid expression of the target of from the off-target deconjugated payload.
“I don’t know that I necessarily have the answer to that,” Dr. Levy replied. “Remember there is the CD20 monoclonal rituximab which we know can cause neutropenia, as well as the CD37 and the target payload. I don’t know if we have enough information to attribute it to one specific component of the therapy,” he said.
The study was funded by Debiopharm International. Dr. Levy disclosed speaker activities for multiple companies, not including Debiopharm. Dr. Lei and Dr. Davies had no disclosures relevant to the study.
Patients with relapsed or refractory B-cell non-Hodgkin lymphomas who are not candidates for hematopoietic stem cell transplant have a generally poor prognosis and few treatment options, but an experimental combination of the antibody-drug conjugate naratuximab with rituximab showed promising efficacy and acceptable safety in these patients in a phase 2 trial.
Among patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) the combination was associated with a 44.7% overall response rate, including 31.6% complete responses, and two-thirds of patients had responses lasting more than 12 months, reported Moshe Yair Levy, MD, from Texas Oncology–Baylor Charles A Sammons Cancer Center in Dallas.
“This is, in my viewpoint, very exciting therapy,” he said in a question-and-answer session following his presentation of the data in a late-breaking abstract session during the European Hematology Association annual congress. (Abstract LB1903).
Naratuximab emtansine is an investigational antibody-drug conjugate (ADC) consisting of a humanized monoclonal antibody against CD37, a surface marker on B lymphocytes that is highly expressed in non-Hodgkin lymphoma (NHL), conjugated to a cytotoxic derivative of maitansine.
CD37 is also an internalizable cell-surface antigen, making it an attractive candidate for an ADC approach.
In a phase 1 trial, naratuximab monotherapy showed a good safety profile and a 22% overall response rate, Dr. Levy noted.
“What they found is that, if you coadminister this ADC with rituximab, you’re actually going to get more internalization of the CD37 monoclonal, therefore more payload delivered to your target cells,” he said.
He reported results of a multicenter, adaptive phase 2 study of the combination in patients with DLBCL and other relapsed/refractory NHL.
DLBCL and others
The trial was divided into two parts, with the first consisting of a safety run-in phase with expansion in patients with confirmed diagnoses of relapsed/refractory NHL, including DLBCL, follicular lymphoma, mantle cell lymphoma, and marginal zone lymphoma.
Patients with double- or triple-hit disease (with translocations in MYC plus either BCL2 and/or BCL6), bulky disease, or transformed lymphoma were eligible.
The second part consisted of two cohorts of patients with DLBCL treated with naratuximab and rituximab either weekly or every 3 weeks.
All patients in the study had received one to six prior lines of therapy, and had Eastern Cooperative Oncology Group performance status of 0-2. Patients with CNS lymphomas or prior anti-CD37 targeting therapy were excluded.
The safety population included 50 patients with DLBCL assigned to therapy every 3 weeks, 30 assigned to weekly therapy, and 20 patients with other NHL.
DLBCL efficacy
A total of 76 patients with DLBCL were evaluable for efficacy.
The ORR was 44% for patients in both the weekly and every 3 week cohorts, with 31.6% having complete responses.
Among 61 patients with nonbulky disease (longest diameter 7.5 cm or less), the ORR was 50.8%, and among 28 patients who had three or more prior lines of therapy the ORR was 46.4%, with 32.1% having a complete response.
Among responders followed for a median of 15 months, the median duration of response was not reached, and 66% had responses lasting beyond 12 months.
In the weekly dosing DLBCL cohort, 53.3% of patients discontinued treatment of both study drugs because of disease progression, as did 58% of those in the every 3 week cohort, and 30% of patients with other lymphomas. Only eight patients discontinued the combination because of treatment-emergent adverse events. Six patients had treatment-emergent adverse events leading to naratuximab dose reduction.
The most common grade 3 or 4 adverse events were neutropenia, leukopenia, lymphopenia and thrombocytopenias. Dr. Levy commented that the use of granulocyte colony-stimulating factor, which was not mandatory in the study, would likely have lowered the incidence of cytopenias.
There were 10 deaths during the study, 2 of which were considered to be treatment related, occurring in 1 patient each in the DLBCL dosing cohorts; 1 of the patients died from pneumonitis, and the other from left ventricular heart failure.
Other patients deaths were attributed to non–treatment-related cardiac arrest, acute renal failure, exacerbation of chronic heart failure, respiratory failure, multiorgan failure, lung infection, or colon adenocarcinoma.
Q 3 weeks suffices
In the question-and-answer session following the presentation, Kenny Lei, MD, from the Chinese University of Hong Kong asked Dr. Levy what the half-life of naratuximab is, and what was the investigator’s rationale for testing a weekly dosing schedule.
“I think the reason they checked the two different regimens, the Q week and the Q 3-week group, is that they noted that [naratuximab] was cleared relatively quickly, and they wanted to see whether or not, by giving Q weekly, when you get a continuous CD37 site occupancy if they would have a better outcome. But as you saw, in the groups there was really no clinically relevant difference in outcome,” Dr. Levy said.
Andrew Davies, MD, PhD, from the University of Southampton (England), asked whether the neutropenia seen in the study was related to myeloid expression of the target of from the off-target deconjugated payload.
“I don’t know that I necessarily have the answer to that,” Dr. Levy replied. “Remember there is the CD20 monoclonal rituximab which we know can cause neutropenia, as well as the CD37 and the target payload. I don’t know if we have enough information to attribute it to one specific component of the therapy,” he said.
The study was funded by Debiopharm International. Dr. Levy disclosed speaker activities for multiple companies, not including Debiopharm. Dr. Lei and Dr. Davies had no disclosures relevant to the study.
Patients with relapsed or refractory B-cell non-Hodgkin lymphomas who are not candidates for hematopoietic stem cell transplant have a generally poor prognosis and few treatment options, but an experimental combination of the antibody-drug conjugate naratuximab with rituximab showed promising efficacy and acceptable safety in these patients in a phase 2 trial.
Among patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL) the combination was associated with a 44.7% overall response rate, including 31.6% complete responses, and two-thirds of patients had responses lasting more than 12 months, reported Moshe Yair Levy, MD, from Texas Oncology–Baylor Charles A Sammons Cancer Center in Dallas.
“This is, in my viewpoint, very exciting therapy,” he said in a question-and-answer session following his presentation of the data in a late-breaking abstract session during the European Hematology Association annual congress. (Abstract LB1903).
Naratuximab emtansine is an investigational antibody-drug conjugate (ADC) consisting of a humanized monoclonal antibody against CD37, a surface marker on B lymphocytes that is highly expressed in non-Hodgkin lymphoma (NHL), conjugated to a cytotoxic derivative of maitansine.
CD37 is also an internalizable cell-surface antigen, making it an attractive candidate for an ADC approach.
In a phase 1 trial, naratuximab monotherapy showed a good safety profile and a 22% overall response rate, Dr. Levy noted.
“What they found is that, if you coadminister this ADC with rituximab, you’re actually going to get more internalization of the CD37 monoclonal, therefore more payload delivered to your target cells,” he said.
He reported results of a multicenter, adaptive phase 2 study of the combination in patients with DLBCL and other relapsed/refractory NHL.
DLBCL and others
The trial was divided into two parts, with the first consisting of a safety run-in phase with expansion in patients with confirmed diagnoses of relapsed/refractory NHL, including DLBCL, follicular lymphoma, mantle cell lymphoma, and marginal zone lymphoma.
Patients with double- or triple-hit disease (with translocations in MYC plus either BCL2 and/or BCL6), bulky disease, or transformed lymphoma were eligible.
The second part consisted of two cohorts of patients with DLBCL treated with naratuximab and rituximab either weekly or every 3 weeks.
All patients in the study had received one to six prior lines of therapy, and had Eastern Cooperative Oncology Group performance status of 0-2. Patients with CNS lymphomas or prior anti-CD37 targeting therapy were excluded.
The safety population included 50 patients with DLBCL assigned to therapy every 3 weeks, 30 assigned to weekly therapy, and 20 patients with other NHL.
DLBCL efficacy
A total of 76 patients with DLBCL were evaluable for efficacy.
The ORR was 44% for patients in both the weekly and every 3 week cohorts, with 31.6% having complete responses.
Among 61 patients with nonbulky disease (longest diameter 7.5 cm or less), the ORR was 50.8%, and among 28 patients who had three or more prior lines of therapy the ORR was 46.4%, with 32.1% having a complete response.
Among responders followed for a median of 15 months, the median duration of response was not reached, and 66% had responses lasting beyond 12 months.
In the weekly dosing DLBCL cohort, 53.3% of patients discontinued treatment of both study drugs because of disease progression, as did 58% of those in the every 3 week cohort, and 30% of patients with other lymphomas. Only eight patients discontinued the combination because of treatment-emergent adverse events. Six patients had treatment-emergent adverse events leading to naratuximab dose reduction.
The most common grade 3 or 4 adverse events were neutropenia, leukopenia, lymphopenia and thrombocytopenias. Dr. Levy commented that the use of granulocyte colony-stimulating factor, which was not mandatory in the study, would likely have lowered the incidence of cytopenias.
There were 10 deaths during the study, 2 of which were considered to be treatment related, occurring in 1 patient each in the DLBCL dosing cohorts; 1 of the patients died from pneumonitis, and the other from left ventricular heart failure.
Other patients deaths were attributed to non–treatment-related cardiac arrest, acute renal failure, exacerbation of chronic heart failure, respiratory failure, multiorgan failure, lung infection, or colon adenocarcinoma.
Q 3 weeks suffices
In the question-and-answer session following the presentation, Kenny Lei, MD, from the Chinese University of Hong Kong asked Dr. Levy what the half-life of naratuximab is, and what was the investigator’s rationale for testing a weekly dosing schedule.
“I think the reason they checked the two different regimens, the Q week and the Q 3-week group, is that they noted that [naratuximab] was cleared relatively quickly, and they wanted to see whether or not, by giving Q weekly, when you get a continuous CD37 site occupancy if they would have a better outcome. But as you saw, in the groups there was really no clinically relevant difference in outcome,” Dr. Levy said.
Andrew Davies, MD, PhD, from the University of Southampton (England), asked whether the neutropenia seen in the study was related to myeloid expression of the target of from the off-target deconjugated payload.
“I don’t know that I necessarily have the answer to that,” Dr. Levy replied. “Remember there is the CD20 monoclonal rituximab which we know can cause neutropenia, as well as the CD37 and the target payload. I don’t know if we have enough information to attribute it to one specific component of the therapy,” he said.
The study was funded by Debiopharm International. Dr. Levy disclosed speaker activities for multiple companies, not including Debiopharm. Dr. Lei and Dr. Davies had no disclosures relevant to the study.
FROM EHA 2021
As new cases fall, U.S. passes 4 million children with COVID-19
Even as the number of new COVID-19 cases continues to drop, the United States reached the 4-million mark for infected children, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
. That weekly total, the lowest since June of 2020, comes from 49 states (excluding N.Y.), the District of Columbia, New York City, Puerto Rico, and Guam, the AAP and CHA said in their weekly COVID-19 report.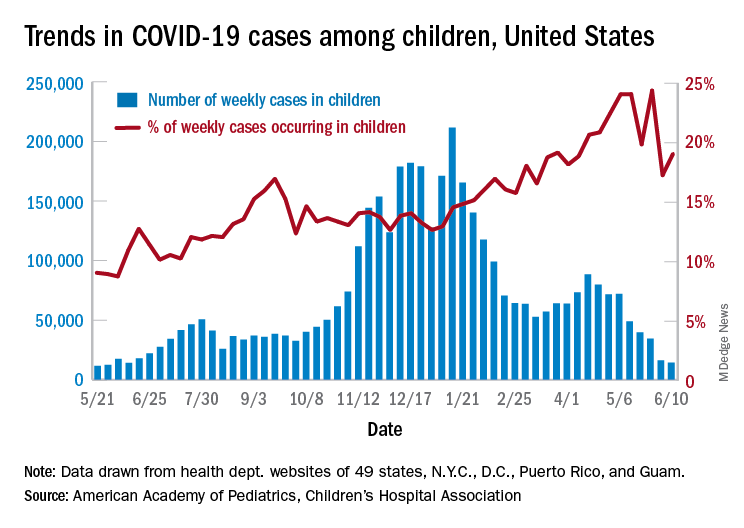
Children represent 14.1% of all COVID-19 cases since the beginning of the pandemic, while the corresponding figure for the week ending June 10 was 19.0%. That weekly proportion of cases among children had been rising pretty steadily through the winter and early spring, but the situation has become much more volatile over the last month, the AAP/CHA data show.
Use of the Pfizer-BioNTech vaccine in children aged 16-17 years, of course, didn’t begin until April, and the vaccine wasn’t authorized for children aged 12-15 years until mid-May. The Moderna and Johnson & Johnson vaccines have not received such authorization yet, but Moderna is in the process of seeking an emergency-use recommendation from the Food and Drug Administration.
In the younger group of children who are currently eligible, completion of the vaccine regimen took a big jump in the week ending June 14, according to the Centers for Disease Control and Prevention. The cumulative share of those aged 12-15 years who had received a second dose jumped from 4.1% on June 7 to 11.4% on June 14, with comparable numbers for 16- and 17-year-olds coming in at 26.4% and 29.1%.
Activity over just the last 14 days, however, shows a slight decrease in children aged 12-15 getting a first dose: For just the 2 weeks ending June 7, 17.9% of all children in the age group initiated a first dose, but for the 14 days ending June 14, only 17.1% of the age group did so, the CDC said on its COVID Data Tracker site.
For children aged 16-17 years – of whom less than 30% have reached full vaccination – activity seems to have stagnated: 4.8% of all 16- to 17-year-olds initiated a first vaccination during the 14 days ending June 7, compared with 4.7% who did so during the 14 days ending June 14, the CDC reported.
Older age groups with higher completion rates are still producing greater vaccine initiation. As of June 14, those aged 25-39 years had a completion rate of 41.9% and 24.0% of the age group had received a first dose in the previous 2 weeks, while 61.4% of those aged 50-64 were fully vaccinated, and 18.0% had gotten their first dose, the CDC data indicate.
Even as the number of new COVID-19 cases continues to drop, the United States reached the 4-million mark for infected children, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
. That weekly total, the lowest since June of 2020, comes from 49 states (excluding N.Y.), the District of Columbia, New York City, Puerto Rico, and Guam, the AAP and CHA said in their weekly COVID-19 report.
Children represent 14.1% of all COVID-19 cases since the beginning of the pandemic, while the corresponding figure for the week ending June 10 was 19.0%. That weekly proportion of cases among children had been rising pretty steadily through the winter and early spring, but the situation has become much more volatile over the last month, the AAP/CHA data show.
Use of the Pfizer-BioNTech vaccine in children aged 16-17 years, of course, didn’t begin until April, and the vaccine wasn’t authorized for children aged 12-15 years until mid-May. The Moderna and Johnson & Johnson vaccines have not received such authorization yet, but Moderna is in the process of seeking an emergency-use recommendation from the Food and Drug Administration.
In the younger group of children who are currently eligible, completion of the vaccine regimen took a big jump in the week ending June 14, according to the Centers for Disease Control and Prevention. The cumulative share of those aged 12-15 years who had received a second dose jumped from 4.1% on June 7 to 11.4% on June 14, with comparable numbers for 16- and 17-year-olds coming in at 26.4% and 29.1%.
Activity over just the last 14 days, however, shows a slight decrease in children aged 12-15 getting a first dose: For just the 2 weeks ending June 7, 17.9% of all children in the age group initiated a first dose, but for the 14 days ending June 14, only 17.1% of the age group did so, the CDC said on its COVID Data Tracker site.
For children aged 16-17 years – of whom less than 30% have reached full vaccination – activity seems to have stagnated: 4.8% of all 16- to 17-year-olds initiated a first vaccination during the 14 days ending June 7, compared with 4.7% who did so during the 14 days ending June 14, the CDC reported.
Older age groups with higher completion rates are still producing greater vaccine initiation. As of June 14, those aged 25-39 years had a completion rate of 41.9% and 24.0% of the age group had received a first dose in the previous 2 weeks, while 61.4% of those aged 50-64 were fully vaccinated, and 18.0% had gotten their first dose, the CDC data indicate.
Even as the number of new COVID-19 cases continues to drop, the United States reached the 4-million mark for infected children, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
. That weekly total, the lowest since June of 2020, comes from 49 states (excluding N.Y.), the District of Columbia, New York City, Puerto Rico, and Guam, the AAP and CHA said in their weekly COVID-19 report.
Children represent 14.1% of all COVID-19 cases since the beginning of the pandemic, while the corresponding figure for the week ending June 10 was 19.0%. That weekly proportion of cases among children had been rising pretty steadily through the winter and early spring, but the situation has become much more volatile over the last month, the AAP/CHA data show.
Use of the Pfizer-BioNTech vaccine in children aged 16-17 years, of course, didn’t begin until April, and the vaccine wasn’t authorized for children aged 12-15 years until mid-May. The Moderna and Johnson & Johnson vaccines have not received such authorization yet, but Moderna is in the process of seeking an emergency-use recommendation from the Food and Drug Administration.
In the younger group of children who are currently eligible, completion of the vaccine regimen took a big jump in the week ending June 14, according to the Centers for Disease Control and Prevention. The cumulative share of those aged 12-15 years who had received a second dose jumped from 4.1% on June 7 to 11.4% on June 14, with comparable numbers for 16- and 17-year-olds coming in at 26.4% and 29.1%.
Activity over just the last 14 days, however, shows a slight decrease in children aged 12-15 getting a first dose: For just the 2 weeks ending June 7, 17.9% of all children in the age group initiated a first dose, but for the 14 days ending June 14, only 17.1% of the age group did so, the CDC said on its COVID Data Tracker site.
For children aged 16-17 years – of whom less than 30% have reached full vaccination – activity seems to have stagnated: 4.8% of all 16- to 17-year-olds initiated a first vaccination during the 14 days ending June 7, compared with 4.7% who did so during the 14 days ending June 14, the CDC reported.
Older age groups with higher completion rates are still producing greater vaccine initiation. As of June 14, those aged 25-39 years had a completion rate of 41.9% and 24.0% of the age group had received a first dose in the previous 2 weeks, while 61.4% of those aged 50-64 were fully vaccinated, and 18.0% had gotten their first dose, the CDC data indicate.
Minnesota named best place to practice in 2021
For physicians who are just starting out or thinking about moving, the “Land of 10,000 Lakes” could be the land of opportunity, according to a recent Medscape analysis.
In a ranking of the 50 states, Minnesota “claimed top marks for livability, low incidence of adverse actions against doctors, and the performance of its health system,” Shelly Reese wrote in Medscape’s “Best & Worst Places to Practice 2021.”
Minnesota is below average where it’s good to be below average – share of physicians reporting burnout and/or depression – but above average in the share of physicians who say they’re “very happy” outside of work, Medscape said in the annual report.
and adverse actions and a high level of livability. Third place went to Washington (called the most livable state in the country by U.S. News and World Report), fourth to Colorado (physicians happy at and outside of work, high retention rate for residents), and fifth to Utah (low crime rate, high quality of life), Medscape said.
At the bottom of the list for 2021 is West Virginia, where physicians “may confront a bevy of challenges” in the form of low livability, a high rate of adverse actions, and relatively high malpractice payouts, Ms. Reese noted in the report.
State number 49 is Louisiana, where livability is low, malpractice payouts are high, and more than half of physicians say that they’re burned out and/or depressed. New Mexico is 48th (very high rate of adverse actions, poor resident retention), Nevada is 47th (low marks for avoidable hospital use and disparity in care), and Rhode Island is 46th (high malpractice payouts, low physician compensation), Medscape said.
Continuing with the group-of-five theme, America’s three most populous states finished in the top half of the ranking – California 16th, Texas 11th, and Florida 21st – but New York and Pennsylvania, numbers four and five by population size, did not.
The rankings are based on states’ performance in 10 different measures, three of which were sourced from Medscape surveys – happiness at work, happiness outside of work, and burnout/depression – and seven from other organizations: adverse actions against physicians, malpractice payouts, compensation (adjusted for cost of living), overall health, health system performance, overall livability, resident retention.
For physicians who are just starting out or thinking about moving, the “Land of 10,000 Lakes” could be the land of opportunity, according to a recent Medscape analysis.
In a ranking of the 50 states, Minnesota “claimed top marks for livability, low incidence of adverse actions against doctors, and the performance of its health system,” Shelly Reese wrote in Medscape’s “Best & Worst Places to Practice 2021.”
Minnesota is below average where it’s good to be below average – share of physicians reporting burnout and/or depression – but above average in the share of physicians who say they’re “very happy” outside of work, Medscape said in the annual report.
and adverse actions and a high level of livability. Third place went to Washington (called the most livable state in the country by U.S. News and World Report), fourth to Colorado (physicians happy at and outside of work, high retention rate for residents), and fifth to Utah (low crime rate, high quality of life), Medscape said.
At the bottom of the list for 2021 is West Virginia, where physicians “may confront a bevy of challenges” in the form of low livability, a high rate of adverse actions, and relatively high malpractice payouts, Ms. Reese noted in the report.
State number 49 is Louisiana, where livability is low, malpractice payouts are high, and more than half of physicians say that they’re burned out and/or depressed. New Mexico is 48th (very high rate of adverse actions, poor resident retention), Nevada is 47th (low marks for avoidable hospital use and disparity in care), and Rhode Island is 46th (high malpractice payouts, low physician compensation), Medscape said.
Continuing with the group-of-five theme, America’s three most populous states finished in the top half of the ranking – California 16th, Texas 11th, and Florida 21st – but New York and Pennsylvania, numbers four and five by population size, did not.
The rankings are based on states’ performance in 10 different measures, three of which were sourced from Medscape surveys – happiness at work, happiness outside of work, and burnout/depression – and seven from other organizations: adverse actions against physicians, malpractice payouts, compensation (adjusted for cost of living), overall health, health system performance, overall livability, resident retention.
For physicians who are just starting out or thinking about moving, the “Land of 10,000 Lakes” could be the land of opportunity, according to a recent Medscape analysis.
In a ranking of the 50 states, Minnesota “claimed top marks for livability, low incidence of adverse actions against doctors, and the performance of its health system,” Shelly Reese wrote in Medscape’s “Best & Worst Places to Practice 2021.”
Minnesota is below average where it’s good to be below average – share of physicians reporting burnout and/or depression – but above average in the share of physicians who say they’re “very happy” outside of work, Medscape said in the annual report.
and adverse actions and a high level of livability. Third place went to Washington (called the most livable state in the country by U.S. News and World Report), fourth to Colorado (physicians happy at and outside of work, high retention rate for residents), and fifth to Utah (low crime rate, high quality of life), Medscape said.
At the bottom of the list for 2021 is West Virginia, where physicians “may confront a bevy of challenges” in the form of low livability, a high rate of adverse actions, and relatively high malpractice payouts, Ms. Reese noted in the report.
State number 49 is Louisiana, where livability is low, malpractice payouts are high, and more than half of physicians say that they’re burned out and/or depressed. New Mexico is 48th (very high rate of adverse actions, poor resident retention), Nevada is 47th (low marks for avoidable hospital use and disparity in care), and Rhode Island is 46th (high malpractice payouts, low physician compensation), Medscape said.
Continuing with the group-of-five theme, America’s three most populous states finished in the top half of the ranking – California 16th, Texas 11th, and Florida 21st – but New York and Pennsylvania, numbers four and five by population size, did not.
The rankings are based on states’ performance in 10 different measures, three of which were sourced from Medscape surveys – happiness at work, happiness outside of work, and burnout/depression – and seven from other organizations: adverse actions against physicians, malpractice payouts, compensation (adjusted for cost of living), overall health, health system performance, overall livability, resident retention.