User login
FDA clears first-ever neurostimulation device for opioid withdrawal symptoms
The Food and Drug Administration has cleared a medical device that applies electrical stimulation to cranial nerves in order to reduce opioid withdrawal symptoms.
The clearance was granted amid the opioid crisis, which is killing 175 Americans each day, according to the recent report by The President’s Commission on Combating Drug Addiction and the Opioid Crisis. Currently, opioid addiction is treatable by three approved medications, said FDA Commissioner Scott Gottlieb, MD, in a press statement announcing the agency’s decision to permit marketing of the NSS-2 Bridge device.
“While we continue to pursue better medicines for the treatment of opioid use disorder, we also need to look to devices that can assist in this therapy,” Dr. Gottlieb said.
The NSS-2 Bridge device was cleared as an acupuncture aid in 2014. The new use of the device to reduce the symptoms of opioid withdrawal required a new clearance, which was granted based on a single, one-arm clinical study. In the study, 73 patients who were experiencing physical withdrawal from opioids used the neurostimulation device. They reported an initial mean score of 20.1 on the Clinical Opiate Withdrawal Score (COWS). All patients using the device had a reduction of at least 31% on the COWS within 30 minutes of beginning use of the NSS-2 Bridge. A total of 88% of participating patients transitioned to medication-assisted treatment after 5 days of using the device. Additional medications used to treat specific symptoms, such as nausea and vomiting, were permitted during the trial.
The physician-placed battery-powered device sits behind the ear and uses three percutaneous electrode arrays and one single-point needle to provide neurostimulation. The electrode placement is assisted with a transillumination technique, and also is based on known neuroanatomic landmarks for branches of cranial nerves V, VII, and IX, along with branches of the occipital nerve (Clin Med Diagnostics. 2015;5[4]:70-9).
The single-use device is designed to be used for up to 5 days during acute opioid withdrawal and is contraindicated for patients with hemophilia, patients with cardiac pacemakers, or those diagnosed with psoriasis vulgaris. The NSS-2 Bridge device requires a prescription and was cleared through the de novo premarket review pathway. This, said the FDA in the press statement, is “a regulatory pathway for some low- to moderate-risk devices that are novel and for which there is no legally marketed predicate device to which the device can claim substantial equivalence.”
The NSS-2 Bridge device will be marketed by Innovative Health Solutions.
koakes@frontlinemedcom.com
On Twitter @karioakes
The Food and Drug Administration has cleared a medical device that applies electrical stimulation to cranial nerves in order to reduce opioid withdrawal symptoms.
The clearance was granted amid the opioid crisis, which is killing 175 Americans each day, according to the recent report by The President’s Commission on Combating Drug Addiction and the Opioid Crisis. Currently, opioid addiction is treatable by three approved medications, said FDA Commissioner Scott Gottlieb, MD, in a press statement announcing the agency’s decision to permit marketing of the NSS-2 Bridge device.
“While we continue to pursue better medicines for the treatment of opioid use disorder, we also need to look to devices that can assist in this therapy,” Dr. Gottlieb said.
The NSS-2 Bridge device was cleared as an acupuncture aid in 2014. The new use of the device to reduce the symptoms of opioid withdrawal required a new clearance, which was granted based on a single, one-arm clinical study. In the study, 73 patients who were experiencing physical withdrawal from opioids used the neurostimulation device. They reported an initial mean score of 20.1 on the Clinical Opiate Withdrawal Score (COWS). All patients using the device had a reduction of at least 31% on the COWS within 30 minutes of beginning use of the NSS-2 Bridge. A total of 88% of participating patients transitioned to medication-assisted treatment after 5 days of using the device. Additional medications used to treat specific symptoms, such as nausea and vomiting, were permitted during the trial.
The physician-placed battery-powered device sits behind the ear and uses three percutaneous electrode arrays and one single-point needle to provide neurostimulation. The electrode placement is assisted with a transillumination technique, and also is based on known neuroanatomic landmarks for branches of cranial nerves V, VII, and IX, along with branches of the occipital nerve (Clin Med Diagnostics. 2015;5[4]:70-9).
The single-use device is designed to be used for up to 5 days during acute opioid withdrawal and is contraindicated for patients with hemophilia, patients with cardiac pacemakers, or those diagnosed with psoriasis vulgaris. The NSS-2 Bridge device requires a prescription and was cleared through the de novo premarket review pathway. This, said the FDA in the press statement, is “a regulatory pathway for some low- to moderate-risk devices that are novel and for which there is no legally marketed predicate device to which the device can claim substantial equivalence.”
The NSS-2 Bridge device will be marketed by Innovative Health Solutions.
koakes@frontlinemedcom.com
On Twitter @karioakes
The Food and Drug Administration has cleared a medical device that applies electrical stimulation to cranial nerves in order to reduce opioid withdrawal symptoms.
The clearance was granted amid the opioid crisis, which is killing 175 Americans each day, according to the recent report by The President’s Commission on Combating Drug Addiction and the Opioid Crisis. Currently, opioid addiction is treatable by three approved medications, said FDA Commissioner Scott Gottlieb, MD, in a press statement announcing the agency’s decision to permit marketing of the NSS-2 Bridge device.
“While we continue to pursue better medicines for the treatment of opioid use disorder, we also need to look to devices that can assist in this therapy,” Dr. Gottlieb said.
The NSS-2 Bridge device was cleared as an acupuncture aid in 2014. The new use of the device to reduce the symptoms of opioid withdrawal required a new clearance, which was granted based on a single, one-arm clinical study. In the study, 73 patients who were experiencing physical withdrawal from opioids used the neurostimulation device. They reported an initial mean score of 20.1 on the Clinical Opiate Withdrawal Score (COWS). All patients using the device had a reduction of at least 31% on the COWS within 30 minutes of beginning use of the NSS-2 Bridge. A total of 88% of participating patients transitioned to medication-assisted treatment after 5 days of using the device. Additional medications used to treat specific symptoms, such as nausea and vomiting, were permitted during the trial.
The physician-placed battery-powered device sits behind the ear and uses three percutaneous electrode arrays and one single-point needle to provide neurostimulation. The electrode placement is assisted with a transillumination technique, and also is based on known neuroanatomic landmarks for branches of cranial nerves V, VII, and IX, along with branches of the occipital nerve (Clin Med Diagnostics. 2015;5[4]:70-9).
The single-use device is designed to be used for up to 5 days during acute opioid withdrawal and is contraindicated for patients with hemophilia, patients with cardiac pacemakers, or those diagnosed with psoriasis vulgaris. The NSS-2 Bridge device requires a prescription and was cleared through the de novo premarket review pathway. This, said the FDA in the press statement, is “a regulatory pathway for some low- to moderate-risk devices that are novel and for which there is no legally marketed predicate device to which the device can claim substantial equivalence.”
The NSS-2 Bridge device will be marketed by Innovative Health Solutions.
koakes@frontlinemedcom.com
On Twitter @karioakes
DTaP vaccination rate highest in Maryland
, according to the Centers for Disease Control and Prevention.
Maryland’s coverage of required DTaP vaccine doses came in at a national high of 99.6% for children entering kindergarten in 2016-2017, while the District of Columbia had the nation’s lowest rate at 82.2%, Ranee Seither, MPH, of the National Center for Immunization and Respiratory Disease, and associates at the CDC, Atlanta, reported (MMWR. 2017 Oct 13;66[40]:1073-80).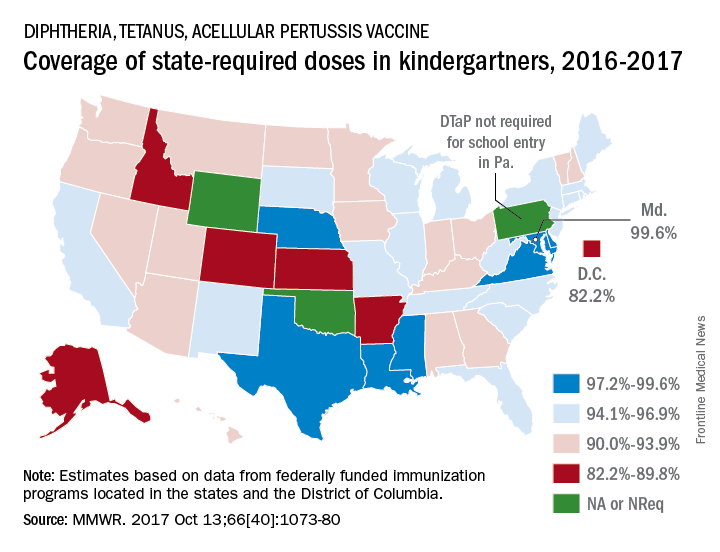
There is also variation among the states in the number of doses required for kindergarten entry: Most require five, but Illinois, Maryland, Virginia, and Wisconsin require four; Nebraska requires three; and Pennsylvania does not require pertussis vaccine. Oklahoma and Wyoming did not report vaccination coverage “because of widespread problems with the quality of data reported by schools,” they noted.
Nationally, median coverage for state-required doses of the DTaP vaccine was 94.5%, according to data from federally funded immunization programs in the 50 states and D.C., which included 3,973,172 kindergartners for the 2016-2017 school year.
, according to the Centers for Disease Control and Prevention.
Maryland’s coverage of required DTaP vaccine doses came in at a national high of 99.6% for children entering kindergarten in 2016-2017, while the District of Columbia had the nation’s lowest rate at 82.2%, Ranee Seither, MPH, of the National Center for Immunization and Respiratory Disease, and associates at the CDC, Atlanta, reported (MMWR. 2017 Oct 13;66[40]:1073-80).
There is also variation among the states in the number of doses required for kindergarten entry: Most require five, but Illinois, Maryland, Virginia, and Wisconsin require four; Nebraska requires three; and Pennsylvania does not require pertussis vaccine. Oklahoma and Wyoming did not report vaccination coverage “because of widespread problems with the quality of data reported by schools,” they noted.
Nationally, median coverage for state-required doses of the DTaP vaccine was 94.5%, according to data from federally funded immunization programs in the 50 states and D.C., which included 3,973,172 kindergartners for the 2016-2017 school year.
, according to the Centers for Disease Control and Prevention.
Maryland’s coverage of required DTaP vaccine doses came in at a national high of 99.6% for children entering kindergarten in 2016-2017, while the District of Columbia had the nation’s lowest rate at 82.2%, Ranee Seither, MPH, of the National Center for Immunization and Respiratory Disease, and associates at the CDC, Atlanta, reported (MMWR. 2017 Oct 13;66[40]:1073-80).
There is also variation among the states in the number of doses required for kindergarten entry: Most require five, but Illinois, Maryland, Virginia, and Wisconsin require four; Nebraska requires three; and Pennsylvania does not require pertussis vaccine. Oklahoma and Wyoming did not report vaccination coverage “because of widespread problems with the quality of data reported by schools,” they noted.
Nationally, median coverage for state-required doses of the DTaP vaccine was 94.5%, according to data from federally funded immunization programs in the 50 states and D.C., which included 3,973,172 kindergartners for the 2016-2017 school year.
FROM MMWR
Adherence boon, or Big Brother loom?
The Food and Drug Administration has approved the first drug in the United States with a digital ingestion tracking system. Abilify MyCite (aripiprazole tablets with sensor) has an ingestible sensor embedded in the pill that records that the medication was taken. The product is approved for the treatment of schizophrenia, acute treatment of manic and mixed episodes associated with bipolar I disorder, and for use as an add-on treatment for depression in adults.
The system works by sending a message from the pill’s sensor to a wearable patch, according to a statement issued by the FDA. The patch transmits the information to a mobile application so that patients can track the ingestion of the medication on their smartphones. Patients can also permit their caregivers and physician to access the information through a web-based portal.
[polldaddy:9874958]
The Food and Drug Administration has approved the first drug in the United States with a digital ingestion tracking system. Abilify MyCite (aripiprazole tablets with sensor) has an ingestible sensor embedded in the pill that records that the medication was taken. The product is approved for the treatment of schizophrenia, acute treatment of manic and mixed episodes associated with bipolar I disorder, and for use as an add-on treatment for depression in adults.
The system works by sending a message from the pill’s sensor to a wearable patch, according to a statement issued by the FDA. The patch transmits the information to a mobile application so that patients can track the ingestion of the medication on their smartphones. Patients can also permit their caregivers and physician to access the information through a web-based portal.
[polldaddy:9874958]
The Food and Drug Administration has approved the first drug in the United States with a digital ingestion tracking system. Abilify MyCite (aripiprazole tablets with sensor) has an ingestible sensor embedded in the pill that records that the medication was taken. The product is approved for the treatment of schizophrenia, acute treatment of manic and mixed episodes associated with bipolar I disorder, and for use as an add-on treatment for depression in adults.
The system works by sending a message from the pill’s sensor to a wearable patch, according to a statement issued by the FDA. The patch transmits the information to a mobile application so that patients can track the ingestion of the medication on their smartphones. Patients can also permit their caregivers and physician to access the information through a web-based portal.
[polldaddy:9874958]
FDA approves cariprazine for schizophrenia maintenance treatment
The Food and Drug Administration has approved a supplemental new drug application for cariprazine (Vraylar) for maintenance treatment of adults with schizophrenia, the drug’s licensor, Allergan, announced Nov. 13. The drug was approved in 2015 for the acute treatment of schizophrenia or for manic or mixed episodes of bipolar I disorder in adults.
The efficacy of the atypical antipsychotic for maintenance treatment of schizophrenia was demonstrated by a 72-week multinational, double-blind, randomized study of a stabilized cariprazine dose of 3, 6, or 9 mg daily, compared with placebo. The daily dose had a significant effect on the study’s primary endpoint – time to relapse. Nearly twice as many placebo-treated patients as cariprazine-treated patients relapsed (49.5% vs. 29.7%).
“The goal of clinicians is to minimize relapses, which can cause significant personal distress and can often have serious implications for a patient’s health,” said Herbert Y. Meltzer, MD, professor of psychiatry and behavioral sciences, pharmacology, and physiology, at Northwestern University, Chicago, in the release. “The approval of Vraylar for the maintenance treatment of schizophrenia provides an important therapy for patients and physicians who are in need of long-term treatment options.”
Cariprazine may cause rash, pruritus, urticaria, and events suggestive of angioedema and is not approved for patients with dementia-related psychosis, as it has an increased mortality risk for elderly patients with dementia. In approved schizophrenia patients, it carries a risk of extrapyramidal symptoms and akathisia.
The Food and Drug Administration has approved a supplemental new drug application for cariprazine (Vraylar) for maintenance treatment of adults with schizophrenia, the drug’s licensor, Allergan, announced Nov. 13. The drug was approved in 2015 for the acute treatment of schizophrenia or for manic or mixed episodes of bipolar I disorder in adults.
The efficacy of the atypical antipsychotic for maintenance treatment of schizophrenia was demonstrated by a 72-week multinational, double-blind, randomized study of a stabilized cariprazine dose of 3, 6, or 9 mg daily, compared with placebo. The daily dose had a significant effect on the study’s primary endpoint – time to relapse. Nearly twice as many placebo-treated patients as cariprazine-treated patients relapsed (49.5% vs. 29.7%).
“The goal of clinicians is to minimize relapses, which can cause significant personal distress and can often have serious implications for a patient’s health,” said Herbert Y. Meltzer, MD, professor of psychiatry and behavioral sciences, pharmacology, and physiology, at Northwestern University, Chicago, in the release. “The approval of Vraylar for the maintenance treatment of schizophrenia provides an important therapy for patients and physicians who are in need of long-term treatment options.”
Cariprazine may cause rash, pruritus, urticaria, and events suggestive of angioedema and is not approved for patients with dementia-related psychosis, as it has an increased mortality risk for elderly patients with dementia. In approved schizophrenia patients, it carries a risk of extrapyramidal symptoms and akathisia.
The Food and Drug Administration has approved a supplemental new drug application for cariprazine (Vraylar) for maintenance treatment of adults with schizophrenia, the drug’s licensor, Allergan, announced Nov. 13. The drug was approved in 2015 for the acute treatment of schizophrenia or for manic or mixed episodes of bipolar I disorder in adults.
The efficacy of the atypical antipsychotic for maintenance treatment of schizophrenia was demonstrated by a 72-week multinational, double-blind, randomized study of a stabilized cariprazine dose of 3, 6, or 9 mg daily, compared with placebo. The daily dose had a significant effect on the study’s primary endpoint – time to relapse. Nearly twice as many placebo-treated patients as cariprazine-treated patients relapsed (49.5% vs. 29.7%).
“The goal of clinicians is to minimize relapses, which can cause significant personal distress and can often have serious implications for a patient’s health,” said Herbert Y. Meltzer, MD, professor of psychiatry and behavioral sciences, pharmacology, and physiology, at Northwestern University, Chicago, in the release. “The approval of Vraylar for the maintenance treatment of schizophrenia provides an important therapy for patients and physicians who are in need of long-term treatment options.”
Cariprazine may cause rash, pruritus, urticaria, and events suggestive of angioedema and is not approved for patients with dementia-related psychosis, as it has an increased mortality risk for elderly patients with dementia. In approved schizophrenia patients, it carries a risk of extrapyramidal symptoms and akathisia.
AML candidate drug back in the pipeline
The Food and Drug Administration has given the biopharmaceutical company Cellectis permission to resume phase 1 trials of UCART123, a gene-edited T-cell investigational drug that targets CD123, as a potential treatment for acute myeloid leukemia (AML) and blastic plasmacytoid dendritic cell neoplasm (BPDCN), according to a press release from the company.
UCART123 is the first allogeneic, “off-the-shelf” gene-edited chimeric antigen receptor (CAR) T-cell product candidate that the FDA has approved for clinical trials. The agency had placed a clinical hold on phase 1 trials of the gene-edited CAR T-cell drug on Sept. 4, following a patient death in the BPDCN clinical study. In order to proceed with the trials, Cellectis agreed to several changes in the study protocols.
The changes include decreasing the dose of the UCART123 therapy to 6.25x104 cells/kg and lowering the dose of the lympho-depleting regimen of cyclophosphamide to 750 mg/m2 per day over 3 days with a maximum daily dose of 1.33 g. Additionally, there can be no uncontrolled infection after receipt of the lympho-depleting preconditioning regimen. Patients must be afebrile at the start of treatment, off all but a replacement dose of corticosteroids, and have no organ dysfunction. Plus, the next three patients treated in each study must be under age 65.
There’s also a condition that patient enrollments be staggered by at least 28 days.
The drug sponsor is working with investigators and each clinical site to obtain the Institutional Review Board’s approval of the revised protocols.
The hold followed the death of a 78-year-old man with relapsed/refractory BPDCN with 30% blasts in his bone marrow and cutaneous lesions. The first dose of UCART123 at 6.25x105 cells/kg was administered without complication, but at day 5 the patient began experiencing side effects, including cytokine release syndrome and a lung infection. At day 8, the cytokine release syndrome had worsened and the patient had also developed capillary leak syndrome. He died on day 9 of the study.
In the AML phase 1 study, a 58-year-old woman with AML and 84% blasts in her bone marrow received the same dose of UCART123. She also developed cytokine release syndrome and capillary leak syndrome but both resolved with treatment.
Both patients also received the same preconditioning treatment: 30 mg/m2 per day fludarabine for 4 days and 1g/m2 per day cyclophosphamide for 3 days.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
The Food and Drug Administration has given the biopharmaceutical company Cellectis permission to resume phase 1 trials of UCART123, a gene-edited T-cell investigational drug that targets CD123, as a potential treatment for acute myeloid leukemia (AML) and blastic plasmacytoid dendritic cell neoplasm (BPDCN), according to a press release from the company.
UCART123 is the first allogeneic, “off-the-shelf” gene-edited chimeric antigen receptor (CAR) T-cell product candidate that the FDA has approved for clinical trials. The agency had placed a clinical hold on phase 1 trials of the gene-edited CAR T-cell drug on Sept. 4, following a patient death in the BPDCN clinical study. In order to proceed with the trials, Cellectis agreed to several changes in the study protocols.
The changes include decreasing the dose of the UCART123 therapy to 6.25x104 cells/kg and lowering the dose of the lympho-depleting regimen of cyclophosphamide to 750 mg/m2 per day over 3 days with a maximum daily dose of 1.33 g. Additionally, there can be no uncontrolled infection after receipt of the lympho-depleting preconditioning regimen. Patients must be afebrile at the start of treatment, off all but a replacement dose of corticosteroids, and have no organ dysfunction. Plus, the next three patients treated in each study must be under age 65.
There’s also a condition that patient enrollments be staggered by at least 28 days.
The drug sponsor is working with investigators and each clinical site to obtain the Institutional Review Board’s approval of the revised protocols.
The hold followed the death of a 78-year-old man with relapsed/refractory BPDCN with 30% blasts in his bone marrow and cutaneous lesions. The first dose of UCART123 at 6.25x105 cells/kg was administered without complication, but at day 5 the patient began experiencing side effects, including cytokine release syndrome and a lung infection. At day 8, the cytokine release syndrome had worsened and the patient had also developed capillary leak syndrome. He died on day 9 of the study.
In the AML phase 1 study, a 58-year-old woman with AML and 84% blasts in her bone marrow received the same dose of UCART123. She also developed cytokine release syndrome and capillary leak syndrome but both resolved with treatment.
Both patients also received the same preconditioning treatment: 30 mg/m2 per day fludarabine for 4 days and 1g/m2 per day cyclophosphamide for 3 days.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
The Food and Drug Administration has given the biopharmaceutical company Cellectis permission to resume phase 1 trials of UCART123, a gene-edited T-cell investigational drug that targets CD123, as a potential treatment for acute myeloid leukemia (AML) and blastic plasmacytoid dendritic cell neoplasm (BPDCN), according to a press release from the company.
UCART123 is the first allogeneic, “off-the-shelf” gene-edited chimeric antigen receptor (CAR) T-cell product candidate that the FDA has approved for clinical trials. The agency had placed a clinical hold on phase 1 trials of the gene-edited CAR T-cell drug on Sept. 4, following a patient death in the BPDCN clinical study. In order to proceed with the trials, Cellectis agreed to several changes in the study protocols.
The changes include decreasing the dose of the UCART123 therapy to 6.25x104 cells/kg and lowering the dose of the lympho-depleting regimen of cyclophosphamide to 750 mg/m2 per day over 3 days with a maximum daily dose of 1.33 g. Additionally, there can be no uncontrolled infection after receipt of the lympho-depleting preconditioning regimen. Patients must be afebrile at the start of treatment, off all but a replacement dose of corticosteroids, and have no organ dysfunction. Plus, the next three patients treated in each study must be under age 65.
There’s also a condition that patient enrollments be staggered by at least 28 days.
The drug sponsor is working with investigators and each clinical site to obtain the Institutional Review Board’s approval of the revised protocols.
The hold followed the death of a 78-year-old man with relapsed/refractory BPDCN with 30% blasts in his bone marrow and cutaneous lesions. The first dose of UCART123 at 6.25x105 cells/kg was administered without complication, but at day 5 the patient began experiencing side effects, including cytokine release syndrome and a lung infection. At day 8, the cytokine release syndrome had worsened and the patient had also developed capillary leak syndrome. He died on day 9 of the study.
In the AML phase 1 study, a 58-year-old woman with AML and 84% blasts in her bone marrow received the same dose of UCART123. She also developed cytokine release syndrome and capillary leak syndrome but both resolved with treatment.
Both patients also received the same preconditioning treatment: 30 mg/m2 per day fludarabine for 4 days and 1g/m2 per day cyclophosphamide for 3 days.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
FDA asked to approve add-on drug for eosinophilic COPD
GlaxoSmithKline asked the Food and Drug Administration to approve an interleuklin-5 antagonist as an add-on maintenance therapy for patients with eosinophilic chronic obstructive pulmonary disease (COPD).
The pharmaceutical and health care company is seeking approval of mepolizumab to be used specifically to treat COPD patients with an eosinophilic phenotype. The drug currently is indicated to treat patients aged 12 years or older with severe asthma and asthma with an eosinophilic phenotype and is sold under the name Nucala, according to a GlaxoSmithKline statement issued November 7.![]()
Headache, injection site reaction, back pain, and fatigue are the most common adverse reactions seen in patients who took mepolizumab during clinical trials.
Mepolizumab is not approved for the treatment of COPD anywhere in the world, and GlaxoSmithKline intends to also ask other countries’ regulatory authorities to allow this drug to be sold as a therapy for COPD.
GlaxoSmithKline asked the Food and Drug Administration to approve an interleuklin-5 antagonist as an add-on maintenance therapy for patients with eosinophilic chronic obstructive pulmonary disease (COPD).
The pharmaceutical and health care company is seeking approval of mepolizumab to be used specifically to treat COPD patients with an eosinophilic phenotype. The drug currently is indicated to treat patients aged 12 years or older with severe asthma and asthma with an eosinophilic phenotype and is sold under the name Nucala, according to a GlaxoSmithKline statement issued November 7.![]()
Headache, injection site reaction, back pain, and fatigue are the most common adverse reactions seen in patients who took mepolizumab during clinical trials.
Mepolizumab is not approved for the treatment of COPD anywhere in the world, and GlaxoSmithKline intends to also ask other countries’ regulatory authorities to allow this drug to be sold as a therapy for COPD.
GlaxoSmithKline asked the Food and Drug Administration to approve an interleuklin-5 antagonist as an add-on maintenance therapy for patients with eosinophilic chronic obstructive pulmonary disease (COPD).
The pharmaceutical and health care company is seeking approval of mepolizumab to be used specifically to treat COPD patients with an eosinophilic phenotype. The drug currently is indicated to treat patients aged 12 years or older with severe asthma and asthma with an eosinophilic phenotype and is sold under the name Nucala, according to a GlaxoSmithKline statement issued November 7.![]()
Headache, injection site reaction, back pain, and fatigue are the most common adverse reactions seen in patients who took mepolizumab during clinical trials.
Mepolizumab is not approved for the treatment of COPD anywhere in the world, and GlaxoSmithKline intends to also ask other countries’ regulatory authorities to allow this drug to be sold as a therapy for COPD.
Public health hazard: Bring your flu to work day
Slightly more than 41% of health care personnel who had the flu during the 2014-2015 influenza season went to work while they were ill, according to an annual survey.
Physicians, however, were well above this average, with 63% reporting they had worked with an influenza-like illness (ILI); they were not quite as far above average as pharmacists, though, who had a 67% rate of “presenteeism” – the highest among all of the health care occupations included in the survey, said Sophia Chiu, MD, MPH, of the Centers for Disease Control and Prevention’s National Institute for Occupational Safety and Health, and her associates.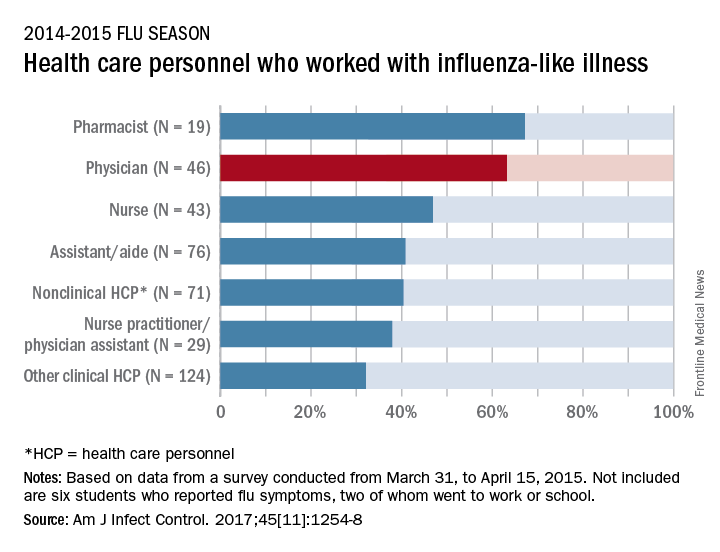
“The statistics are alarming. At least one earlier study has shown that patients who are exposed to a health care worker who is sick are five times more likely to get a health care–associated infection,” Dr. Chiu said in a separate written statement.
For the study, ILI was defined as “fever (without a specified temperature cutoff) and sore throat or cough.” The “nonclinical personnel” category included managers, food service workers, and janitors, while the “other clinical personnel” category included technicians and technologists. The annual Internet panel survey was conducted from March 31, 2015, to April 15, 2015, and 414 of its 1,914 respondents self-reported having an ILI, of whom 183 said that they worked during their illness, Dr. Chiu and her associates said.
The investigators are all CDC employees. The respondents were recruited from Internet panels operated by Survey Sampling International through a contract with Abt Associates.
Slightly more than 41% of health care personnel who had the flu during the 2014-2015 influenza season went to work while they were ill, according to an annual survey.
Physicians, however, were well above this average, with 63% reporting they had worked with an influenza-like illness (ILI); they were not quite as far above average as pharmacists, though, who had a 67% rate of “presenteeism” – the highest among all of the health care occupations included in the survey, said Sophia Chiu, MD, MPH, of the Centers for Disease Control and Prevention’s National Institute for Occupational Safety and Health, and her associates.
“The statistics are alarming. At least one earlier study has shown that patients who are exposed to a health care worker who is sick are five times more likely to get a health care–associated infection,” Dr. Chiu said in a separate written statement.
For the study, ILI was defined as “fever (without a specified temperature cutoff) and sore throat or cough.” The “nonclinical personnel” category included managers, food service workers, and janitors, while the “other clinical personnel” category included technicians and technologists. The annual Internet panel survey was conducted from March 31, 2015, to April 15, 2015, and 414 of its 1,914 respondents self-reported having an ILI, of whom 183 said that they worked during their illness, Dr. Chiu and her associates said.
The investigators are all CDC employees. The respondents were recruited from Internet panels operated by Survey Sampling International through a contract with Abt Associates.
Slightly more than 41% of health care personnel who had the flu during the 2014-2015 influenza season went to work while they were ill, according to an annual survey.
Physicians, however, were well above this average, with 63% reporting they had worked with an influenza-like illness (ILI); they were not quite as far above average as pharmacists, though, who had a 67% rate of “presenteeism” – the highest among all of the health care occupations included in the survey, said Sophia Chiu, MD, MPH, of the Centers for Disease Control and Prevention’s National Institute for Occupational Safety and Health, and her associates.
“The statistics are alarming. At least one earlier study has shown that patients who are exposed to a health care worker who is sick are five times more likely to get a health care–associated infection,” Dr. Chiu said in a separate written statement.
For the study, ILI was defined as “fever (without a specified temperature cutoff) and sore throat or cough.” The “nonclinical personnel” category included managers, food service workers, and janitors, while the “other clinical personnel” category included technicians and technologists. The annual Internet panel survey was conducted from March 31, 2015, to April 15, 2015, and 414 of its 1,914 respondents self-reported having an ILI, of whom 183 said that they worked during their illness, Dr. Chiu and her associates said.
The investigators are all CDC employees. The respondents were recruited from Internet panels operated by Survey Sampling International through a contract with Abt Associates.
FROM THE AMERICAN JOURNAL OF INFECTION CONTROL
FDA grants 510k clearance for glucose monitoring system
OptiScan Biomedical Corporation announced Oct. 18 that the Food and Drug Administration has granted 510(k) clearance for the OptiScanner 5000 Glucose Monitoring System.
The clearance allows the device to be used for monitoring plasma glucose levels and determining dysglycemia in surgical intensive care unit (SICU) patients. It is a bedside glucose monitoring system that provides physicians with critical trending and tracking information to manage patient glucose levels in the ICU.
It is estimated that roughly 20% of ICU patients have pre-existing diabetes and an additional 40- to- 60% of ICU patients suffer from “stress hyperglycemia” or a temporary elevation of glucose levels, with all of these patients requiring accurate glucose monitoring to maintain glycemic control.
“There is a broad consensus in the medical community regarding the need for automated, continuous and highly accurate glucose monitoring in the ICU and my experience with the OptiScanner 5000 indicates that this device will play a critical role in delivering this enhanced level of care. I look forward to implementing this technology as soon as possible,” said Grant V. Bochicchio, MD, MPH, FACS, chief of acute and critical care surgery, and Harry Edison Professor of Surgery, Washington University School of Medicine, in a press release.
Read the full press release here.
OptiScan Biomedical Corporation announced Oct. 18 that the Food and Drug Administration has granted 510(k) clearance for the OptiScanner 5000 Glucose Monitoring System.
The clearance allows the device to be used for monitoring plasma glucose levels and determining dysglycemia in surgical intensive care unit (SICU) patients. It is a bedside glucose monitoring system that provides physicians with critical trending and tracking information to manage patient glucose levels in the ICU.
It is estimated that roughly 20% of ICU patients have pre-existing diabetes and an additional 40- to- 60% of ICU patients suffer from “stress hyperglycemia” or a temporary elevation of glucose levels, with all of these patients requiring accurate glucose monitoring to maintain glycemic control.
“There is a broad consensus in the medical community regarding the need for automated, continuous and highly accurate glucose monitoring in the ICU and my experience with the OptiScanner 5000 indicates that this device will play a critical role in delivering this enhanced level of care. I look forward to implementing this technology as soon as possible,” said Grant V. Bochicchio, MD, MPH, FACS, chief of acute and critical care surgery, and Harry Edison Professor of Surgery, Washington University School of Medicine, in a press release.
Read the full press release here.
OptiScan Biomedical Corporation announced Oct. 18 that the Food and Drug Administration has granted 510(k) clearance for the OptiScanner 5000 Glucose Monitoring System.
The clearance allows the device to be used for monitoring plasma glucose levels and determining dysglycemia in surgical intensive care unit (SICU) patients. It is a bedside glucose monitoring system that provides physicians with critical trending and tracking information to manage patient glucose levels in the ICU.
It is estimated that roughly 20% of ICU patients have pre-existing diabetes and an additional 40- to- 60% of ICU patients suffer from “stress hyperglycemia” or a temporary elevation of glucose levels, with all of these patients requiring accurate glucose monitoring to maintain glycemic control.
“There is a broad consensus in the medical community regarding the need for automated, continuous and highly accurate glucose monitoring in the ICU and my experience with the OptiScanner 5000 indicates that this device will play a critical role in delivering this enhanced level of care. I look forward to implementing this technology as soon as possible,” said Grant V. Bochicchio, MD, MPH, FACS, chief of acute and critical care surgery, and Harry Edison Professor of Surgery, Washington University School of Medicine, in a press release.
Read the full press release here.
FDA panels support two NDAs for buprenorphine subcutaneous injections
SILVER SPRING, MD. – Two Food and Drug Administration advisory panels have recommended approval of two new drug applications (NDA) for buprenorphine subcutaneous injections for the treatment of opioid dependence.
On Nov. 1, panelists recommended approval of some of the doses proposed in the NDA submitted by Braeburn Pharmaceuticals at the joint meeting of the Psychopharmacologic Drugs Advisory and the Drug Safety and Risk Management committees. The formulation, currently known as CAM2038, is intended to be used as part of a treatment plan that can include counseling and psychosocial support. The subcutaneous depot is available weekly, in 8-, 16-, 24-, and 32-mg injections, and monthly, in 64-, 96-, 128-, and 160-mg injections.
On the previous day, Oct. 31, the panelists voted 18-1 with no abstentions to recommend an NDA submitted by Indivior. This formulation is known as RBP-6000.
Both formulations must be administered by a health care provider using a prefilled syringe with a predetermined dosage. The injection forms a biodegradable subcutaneous depot that, as it degrades, releases buprenorphine at a steady and controlled pace over the course of treatment – increasing the success of treatment for opioid use disorder.
Braeburn’s NDA was based on results of a double-blind, randomized, within-subject, inpatient laboratory study of 47 patients over 14 days. Patients were randomized into two groups: 22 patients in the 24-mg group and 25 patients in the 32-mg group. Patients were administered an initial dose on day 0 and a follow-up dose on day 7. The results of the study found a complete blockade of opioids after the first injection that was sustained over the 1-week interdosing interval.
The committees said that of most of the doses should be approved, but a majority of committee members were uncomfortable with the higher doses.
Voting on Indivior’s NDA was based, in part, on the results of a randomized, double-blind, placebo-controlled, multicenter phase 3 study. The study lasted 24 weeks and randomly assigned 504 patients into one of three groups based on monthly dosing regimen of buprenorphine: 300 mg/300 mg, 300 mg/100 mg, and placebo. After randomization, the 300 mg/300 mg group had 201 patients, the 300 mg/100 mg group had 203 patients, and the placebo group had 100 patients. The study found that the primary and secondary endpoints were met, and significantly higher percentage of abstinence with subcutaneous buprenorphine were observed. Patients in both the 300 mg/300 mg and 300 mg/100 mg groups had very similar distributions of percentage of weeks patients abstained from opioid use with more than 20% of patients achieving 80%-100% abstinence from opioids during the course of the study, a significant improvement over the placebo group.
The panels’ recommendations come against the backdrop of the opioid epidemic in the United States, which President Trump has deemed a public health emergency. Many of the panel members and speakers at both meetings expressed support for the NDAs in that context and emphasized that, unlike sublingual administration of buprenorphine, these treatments do not require daily intervention. In addition, sublingual tablets are easier to abuse or more likely to lead to overdose because the patient must self-administer the medication. Expanding the toolkit of physicians who treat opioid use disorder might help stem the tide of the epidemic, some speakers said.
Usually, the FDA follows its advisory panels’ recommendations, which are not binding.
SILVER SPRING, MD. – Two Food and Drug Administration advisory panels have recommended approval of two new drug applications (NDA) for buprenorphine subcutaneous injections for the treatment of opioid dependence.
On Nov. 1, panelists recommended approval of some of the doses proposed in the NDA submitted by Braeburn Pharmaceuticals at the joint meeting of the Psychopharmacologic Drugs Advisory and the Drug Safety and Risk Management committees. The formulation, currently known as CAM2038, is intended to be used as part of a treatment plan that can include counseling and psychosocial support. The subcutaneous depot is available weekly, in 8-, 16-, 24-, and 32-mg injections, and monthly, in 64-, 96-, 128-, and 160-mg injections.
On the previous day, Oct. 31, the panelists voted 18-1 with no abstentions to recommend an NDA submitted by Indivior. This formulation is known as RBP-6000.
Both formulations must be administered by a health care provider using a prefilled syringe with a predetermined dosage. The injection forms a biodegradable subcutaneous depot that, as it degrades, releases buprenorphine at a steady and controlled pace over the course of treatment – increasing the success of treatment for opioid use disorder.
Braeburn’s NDA was based on results of a double-blind, randomized, within-subject, inpatient laboratory study of 47 patients over 14 days. Patients were randomized into two groups: 22 patients in the 24-mg group and 25 patients in the 32-mg group. Patients were administered an initial dose on day 0 and a follow-up dose on day 7. The results of the study found a complete blockade of opioids after the first injection that was sustained over the 1-week interdosing interval.
The committees said that of most of the doses should be approved, but a majority of committee members were uncomfortable with the higher doses.
Voting on Indivior’s NDA was based, in part, on the results of a randomized, double-blind, placebo-controlled, multicenter phase 3 study. The study lasted 24 weeks and randomly assigned 504 patients into one of three groups based on monthly dosing regimen of buprenorphine: 300 mg/300 mg, 300 mg/100 mg, and placebo. After randomization, the 300 mg/300 mg group had 201 patients, the 300 mg/100 mg group had 203 patients, and the placebo group had 100 patients. The study found that the primary and secondary endpoints were met, and significantly higher percentage of abstinence with subcutaneous buprenorphine were observed. Patients in both the 300 mg/300 mg and 300 mg/100 mg groups had very similar distributions of percentage of weeks patients abstained from opioid use with more than 20% of patients achieving 80%-100% abstinence from opioids during the course of the study, a significant improvement over the placebo group.
The panels’ recommendations come against the backdrop of the opioid epidemic in the United States, which President Trump has deemed a public health emergency. Many of the panel members and speakers at both meetings expressed support for the NDAs in that context and emphasized that, unlike sublingual administration of buprenorphine, these treatments do not require daily intervention. In addition, sublingual tablets are easier to abuse or more likely to lead to overdose because the patient must self-administer the medication. Expanding the toolkit of physicians who treat opioid use disorder might help stem the tide of the epidemic, some speakers said.
Usually, the FDA follows its advisory panels’ recommendations, which are not binding.
SILVER SPRING, MD. – Two Food and Drug Administration advisory panels have recommended approval of two new drug applications (NDA) for buprenorphine subcutaneous injections for the treatment of opioid dependence.
On Nov. 1, panelists recommended approval of some of the doses proposed in the NDA submitted by Braeburn Pharmaceuticals at the joint meeting of the Psychopharmacologic Drugs Advisory and the Drug Safety and Risk Management committees. The formulation, currently known as CAM2038, is intended to be used as part of a treatment plan that can include counseling and psychosocial support. The subcutaneous depot is available weekly, in 8-, 16-, 24-, and 32-mg injections, and monthly, in 64-, 96-, 128-, and 160-mg injections.
On the previous day, Oct. 31, the panelists voted 18-1 with no abstentions to recommend an NDA submitted by Indivior. This formulation is known as RBP-6000.
Both formulations must be administered by a health care provider using a prefilled syringe with a predetermined dosage. The injection forms a biodegradable subcutaneous depot that, as it degrades, releases buprenorphine at a steady and controlled pace over the course of treatment – increasing the success of treatment for opioid use disorder.
Braeburn’s NDA was based on results of a double-blind, randomized, within-subject, inpatient laboratory study of 47 patients over 14 days. Patients were randomized into two groups: 22 patients in the 24-mg group and 25 patients in the 32-mg group. Patients were administered an initial dose on day 0 and a follow-up dose on day 7. The results of the study found a complete blockade of opioids after the first injection that was sustained over the 1-week interdosing interval.
The committees said that of most of the doses should be approved, but a majority of committee members were uncomfortable with the higher doses.
Voting on Indivior’s NDA was based, in part, on the results of a randomized, double-blind, placebo-controlled, multicenter phase 3 study. The study lasted 24 weeks and randomly assigned 504 patients into one of three groups based on monthly dosing regimen of buprenorphine: 300 mg/300 mg, 300 mg/100 mg, and placebo. After randomization, the 300 mg/300 mg group had 201 patients, the 300 mg/100 mg group had 203 patients, and the placebo group had 100 patients. The study found that the primary and secondary endpoints were met, and significantly higher percentage of abstinence with subcutaneous buprenorphine were observed. Patients in both the 300 mg/300 mg and 300 mg/100 mg groups had very similar distributions of percentage of weeks patients abstained from opioid use with more than 20% of patients achieving 80%-100% abstinence from opioids during the course of the study, a significant improvement over the placebo group.
The panels’ recommendations come against the backdrop of the opioid epidemic in the United States, which President Trump has deemed a public health emergency. Many of the panel members and speakers at both meetings expressed support for the NDAs in that context and emphasized that, unlike sublingual administration of buprenorphine, these treatments do not require daily intervention. In addition, sublingual tablets are easier to abuse or more likely to lead to overdose because the patient must self-administer the medication. Expanding the toolkit of physicians who treat opioid use disorder might help stem the tide of the epidemic, some speakers said.
Usually, the FDA follows its advisory panels’ recommendations, which are not binding.
Fentanyl analogues an increasing factor in opioid deaths
Fentanyl analogues were involved in 14% of opioid overdose deaths in the second half of 2016, according to an analysis of 10 states reporting to the Enhanced State Opioid Overdose Surveillance program.
“Illicitly manufactured fentanyl is a key factor driving opioid overdose deaths and … fentanyl analogues [such as carfentanil, furanylfentanyl, and acetylfentanyl] are increasingly contributing to a complex illicit opioid market with significant public health implications,” investigators said in a report from the Centers for Disease Control and Prevention (MMWR 2017 Oct 27;66[early release]:1-6).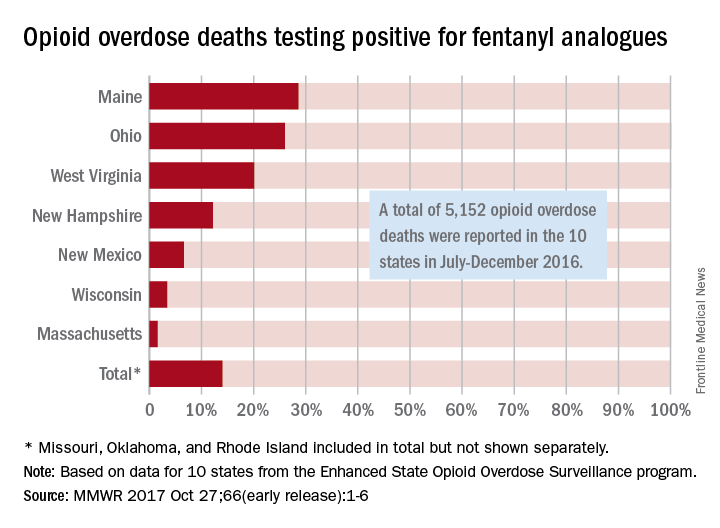
The overall rate of 14% represents 720 of the 5,152 total opioid deaths occurring in 10 states over the 6-month study period, they noted.
Of the 10 states in the analysis, Maine had the largest proportion (28.6%) of opioid overdose deaths involving fentanyl analogues, with Ohio second at 26% and West Virginia third at 20.1%. Ohio had the largest overall number of analogue-involved overdose deaths, however, at 531 during July-December 2016. At 1.6%, Massachusetts had the lowest rate of fentanyl analogue–involved deaths among the seven states for which separate figures were given, the published data show. (Three states – Missouri [22 counties], Oklahoma, and Rhode Island – were grouped together and had a combined rate of 1%.)
More than half of the overdose deaths involving fentanyl or a fentanyl analogue also involved heroin, cocaine, or methamphetamine, which means that almost half of the deaths “did not test positive for other illicit opioids, suggesting that fentanyl and fentanyl analogues might be emerging as unique illicit products,” the investigators wrote, adding that the fentanyl analogue situation “might mirror the rapidly rising trajectory of fentanyl overdose deaths that began in 2013 and become a major factor in opioid overdose deaths.”
Fentanyl analogues were involved in 14% of opioid overdose deaths in the second half of 2016, according to an analysis of 10 states reporting to the Enhanced State Opioid Overdose Surveillance program.
“Illicitly manufactured fentanyl is a key factor driving opioid overdose deaths and … fentanyl analogues [such as carfentanil, furanylfentanyl, and acetylfentanyl] are increasingly contributing to a complex illicit opioid market with significant public health implications,” investigators said in a report from the Centers for Disease Control and Prevention (MMWR 2017 Oct 27;66[early release]:1-6).
The overall rate of 14% represents 720 of the 5,152 total opioid deaths occurring in 10 states over the 6-month study period, they noted.
Of the 10 states in the analysis, Maine had the largest proportion (28.6%) of opioid overdose deaths involving fentanyl analogues, with Ohio second at 26% and West Virginia third at 20.1%. Ohio had the largest overall number of analogue-involved overdose deaths, however, at 531 during July-December 2016. At 1.6%, Massachusetts had the lowest rate of fentanyl analogue–involved deaths among the seven states for which separate figures were given, the published data show. (Three states – Missouri [22 counties], Oklahoma, and Rhode Island – were grouped together and had a combined rate of 1%.)
More than half of the overdose deaths involving fentanyl or a fentanyl analogue also involved heroin, cocaine, or methamphetamine, which means that almost half of the deaths “did not test positive for other illicit opioids, suggesting that fentanyl and fentanyl analogues might be emerging as unique illicit products,” the investigators wrote, adding that the fentanyl analogue situation “might mirror the rapidly rising trajectory of fentanyl overdose deaths that began in 2013 and become a major factor in opioid overdose deaths.”
Fentanyl analogues were involved in 14% of opioid overdose deaths in the second half of 2016, according to an analysis of 10 states reporting to the Enhanced State Opioid Overdose Surveillance program.
“Illicitly manufactured fentanyl is a key factor driving opioid overdose deaths and … fentanyl analogues [such as carfentanil, furanylfentanyl, and acetylfentanyl] are increasingly contributing to a complex illicit opioid market with significant public health implications,” investigators said in a report from the Centers for Disease Control and Prevention (MMWR 2017 Oct 27;66[early release]:1-6).
The overall rate of 14% represents 720 of the 5,152 total opioid deaths occurring in 10 states over the 6-month study period, they noted.
Of the 10 states in the analysis, Maine had the largest proportion (28.6%) of opioid overdose deaths involving fentanyl analogues, with Ohio second at 26% and West Virginia third at 20.1%. Ohio had the largest overall number of analogue-involved overdose deaths, however, at 531 during July-December 2016. At 1.6%, Massachusetts had the lowest rate of fentanyl analogue–involved deaths among the seven states for which separate figures were given, the published data show. (Three states – Missouri [22 counties], Oklahoma, and Rhode Island – were grouped together and had a combined rate of 1%.)
More than half of the overdose deaths involving fentanyl or a fentanyl analogue also involved heroin, cocaine, or methamphetamine, which means that almost half of the deaths “did not test positive for other illicit opioids, suggesting that fentanyl and fentanyl analogues might be emerging as unique illicit products,” the investigators wrote, adding that the fentanyl analogue situation “might mirror the rapidly rising trajectory of fentanyl overdose deaths that began in 2013 and become a major factor in opioid overdose deaths.”
FROM MMWR