User login
FDA approves raltegravir for newborns at risk for HIV-1 infection
The Food and Drug Administration has approved the use of raltegravir (Isentress), in combination with other antiretrovirals, for HIV-1 exposed newborns who weigh at least 2 kg and are at high risk for acquiring HIV-1 infection from their mothers, according to a press release from Merck, the manufacturer of Isentress.
“With this FDA approval, Isentress becomes the only integrase inhibitor approved in the U.S. for the treatment of HIV-1, in combination with other antiretroviral agents, for neonates weighing at least 2 kg,” Eliav Barr, MD, senior vice president, global clinical development, infectious diseases and vaccines, Merck Research Laboratories, said in a statement.

The study, IMPAACT P1110, was composed of two cohorts receiving different doses of raltegravir at different time intervals. The first cohort, of 16 newborns, received two single doses of raltegravir: the first within 48 hours of birth and the second between 7 and 10 days of age. The second cohort of 26 newborns received daily doses based on weight for 6 weeks. After observation for 24 weeks, all infants were HIV-1 negative.
Raltegravir does not cure HIV-1 infection or AIDS and severe side effects have been reported, including hypersensitivity reaction and toxic epidermal necrolysis, according to the Merck release.
Raltegravir should not be used in newborns and infants weighing under 2 kg. If raltegravir has been administered to the mother 2-24 hours before delivery, the first dose for the newborn should be given between 24 and 48 hours after birth.
The Food and Drug Administration has approved the use of raltegravir (Isentress), in combination with other antiretrovirals, for HIV-1 exposed newborns who weigh at least 2 kg and are at high risk for acquiring HIV-1 infection from their mothers, according to a press release from Merck, the manufacturer of Isentress.
“With this FDA approval, Isentress becomes the only integrase inhibitor approved in the U.S. for the treatment of HIV-1, in combination with other antiretroviral agents, for neonates weighing at least 2 kg,” Eliav Barr, MD, senior vice president, global clinical development, infectious diseases and vaccines, Merck Research Laboratories, said in a statement.
The study, IMPAACT P1110, was composed of two cohorts receiving different doses of raltegravir at different time intervals. The first cohort, of 16 newborns, received two single doses of raltegravir: the first within 48 hours of birth and the second between 7 and 10 days of age. The second cohort of 26 newborns received daily doses based on weight for 6 weeks. After observation for 24 weeks, all infants were HIV-1 negative.
Raltegravir does not cure HIV-1 infection or AIDS and severe side effects have been reported, including hypersensitivity reaction and toxic epidermal necrolysis, according to the Merck release.
Raltegravir should not be used in newborns and infants weighing under 2 kg. If raltegravir has been administered to the mother 2-24 hours before delivery, the first dose for the newborn should be given between 24 and 48 hours after birth.
The Food and Drug Administration has approved the use of raltegravir (Isentress), in combination with other antiretrovirals, for HIV-1 exposed newborns who weigh at least 2 kg and are at high risk for acquiring HIV-1 infection from their mothers, according to a press release from Merck, the manufacturer of Isentress.
“With this FDA approval, Isentress becomes the only integrase inhibitor approved in the U.S. for the treatment of HIV-1, in combination with other antiretroviral agents, for neonates weighing at least 2 kg,” Eliav Barr, MD, senior vice president, global clinical development, infectious diseases and vaccines, Merck Research Laboratories, said in a statement.
The study, IMPAACT P1110, was composed of two cohorts receiving different doses of raltegravir at different time intervals. The first cohort, of 16 newborns, received two single doses of raltegravir: the first within 48 hours of birth and the second between 7 and 10 days of age. The second cohort of 26 newborns received daily doses based on weight for 6 weeks. After observation for 24 weeks, all infants were HIV-1 negative.
Raltegravir does not cure HIV-1 infection or AIDS and severe side effects have been reported, including hypersensitivity reaction and toxic epidermal necrolysis, according to the Merck release.
Raltegravir should not be used in newborns and infants weighing under 2 kg. If raltegravir has been administered to the mother 2-24 hours before delivery, the first dose for the newborn should be given between 24 and 48 hours after birth.
FDA axes asthma drugs’ boxed warning
The Food and Drug Administration has eliminated the boxed warning for risk of asthma-related death from the labels of products containing both an inhaled corticosteroid (ICS) and a long-acting beta agonist (LABA), the agency announced.
In 2011, the FDA required companies manufacturing fixed-dose LABA-ICS combination products to conduct 26-week clinical safety trials to evaluate the risks of serious adverse asthma-related events in patients treated with these drugs. Specifically, the companies had to compare the risks of taking a LABA in combination with an ICS with the risks of taking an ICS alone.
The removal of the boxed warning follows the FDA’s review of these trials, which found that treating asthma with LABAs in combination with ICS did not result in patients experiencing significantly more serious asthma-related side effects and asthma-related deaths, compared with those being treated with an ICS alone, according to the FDA announcement. “Results of subgroup analyses for gender, adolescents 12-18 years, and African Americans are consistent with the primary endpoint results,” the statement added.
“These trials showed that LABAs, when used with ICS, did not significantly increase the risk of asthma-related hospitalizations, the need to insert a breathing tube known as intubation, or asthma-related deaths, compared to ICS alone,” the FDA said in the statement.
The trials also demonstrated that using the combination reduced asthma exacerbations, compared with using ICS alone, and that most of the exacerbations “were those that required at least 3 days of systemic corticosteroids” – information that is being added the product labels, according to the FDA.
The products that will no longer carry this boxed warning in their labels include AstraZeneca’s budesonide/formoterol fumarate dihydrate (Symbicort) and GlaxoSmithKline’s fluticasone furoate/vilanterol (Breo Ellipta) and fluticasone propionate/salmeterol (Advair Diskus and Advair HFA).
The FDA also approved updates to the Warnings and Precautions section of labeling for the ICS/LABA class, which now includes a description of the four trials. Information on the efficacy of the drugs, found in the trials, has been added to the Clinical Studies section of the labels as well.
In a related safety announcement, the FDA stated the following: “Using LABAs alone to treat asthma without an ICS to treat lung inflammation is associated with an increased risk of asthma-related death. Therefore, the Boxed Warning stating this will remain in the labels of all single-ingredient LABA medicines, which are approved to treat asthma, chronic obstructive pulmonary disease (COPD), and wheezing caused by exercise. The labels of medicines that contain both an ICS and LABA also retain a Warning and Precaution related to the increased risk of asthma-related death when LABAs are used without an ICS to treat asthma.

The Food and Drug Administration has eliminated the boxed warning for risk of asthma-related death from the labels of products containing both an inhaled corticosteroid (ICS) and a long-acting beta agonist (LABA), the agency announced.
In 2011, the FDA required companies manufacturing fixed-dose LABA-ICS combination products to conduct 26-week clinical safety trials to evaluate the risks of serious adverse asthma-related events in patients treated with these drugs. Specifically, the companies had to compare the risks of taking a LABA in combination with an ICS with the risks of taking an ICS alone.
The removal of the boxed warning follows the FDA’s review of these trials, which found that treating asthma with LABAs in combination with ICS did not result in patients experiencing significantly more serious asthma-related side effects and asthma-related deaths, compared with those being treated with an ICS alone, according to the FDA announcement. “Results of subgroup analyses for gender, adolescents 12-18 years, and African Americans are consistent with the primary endpoint results,” the statement added.
“These trials showed that LABAs, when used with ICS, did not significantly increase the risk of asthma-related hospitalizations, the need to insert a breathing tube known as intubation, or asthma-related deaths, compared to ICS alone,” the FDA said in the statement.
The trials also demonstrated that using the combination reduced asthma exacerbations, compared with using ICS alone, and that most of the exacerbations “were those that required at least 3 days of systemic corticosteroids” – information that is being added the product labels, according to the FDA.
The products that will no longer carry this boxed warning in their labels include AstraZeneca’s budesonide/formoterol fumarate dihydrate (Symbicort) and GlaxoSmithKline’s fluticasone furoate/vilanterol (Breo Ellipta) and fluticasone propionate/salmeterol (Advair Diskus and Advair HFA).
The FDA also approved updates to the Warnings and Precautions section of labeling for the ICS/LABA class, which now includes a description of the four trials. Information on the efficacy of the drugs, found in the trials, has been added to the Clinical Studies section of the labels as well.
In a related safety announcement, the FDA stated the following: “Using LABAs alone to treat asthma without an ICS to treat lung inflammation is associated with an increased risk of asthma-related death. Therefore, the Boxed Warning stating this will remain in the labels of all single-ingredient LABA medicines, which are approved to treat asthma, chronic obstructive pulmonary disease (COPD), and wheezing caused by exercise. The labels of medicines that contain both an ICS and LABA also retain a Warning and Precaution related to the increased risk of asthma-related death when LABAs are used without an ICS to treat asthma.
The Food and Drug Administration has eliminated the boxed warning for risk of asthma-related death from the labels of products containing both an inhaled corticosteroid (ICS) and a long-acting beta agonist (LABA), the agency announced.
In 2011, the FDA required companies manufacturing fixed-dose LABA-ICS combination products to conduct 26-week clinical safety trials to evaluate the risks of serious adverse asthma-related events in patients treated with these drugs. Specifically, the companies had to compare the risks of taking a LABA in combination with an ICS with the risks of taking an ICS alone.
The removal of the boxed warning follows the FDA’s review of these trials, which found that treating asthma with LABAs in combination with ICS did not result in patients experiencing significantly more serious asthma-related side effects and asthma-related deaths, compared with those being treated with an ICS alone, according to the FDA announcement. “Results of subgroup analyses for gender, adolescents 12-18 years, and African Americans are consistent with the primary endpoint results,” the statement added.
“These trials showed that LABAs, when used with ICS, did not significantly increase the risk of asthma-related hospitalizations, the need to insert a breathing tube known as intubation, or asthma-related deaths, compared to ICS alone,” the FDA said in the statement.
The trials also demonstrated that using the combination reduced asthma exacerbations, compared with using ICS alone, and that most of the exacerbations “were those that required at least 3 days of systemic corticosteroids” – information that is being added the product labels, according to the FDA.
The products that will no longer carry this boxed warning in their labels include AstraZeneca’s budesonide/formoterol fumarate dihydrate (Symbicort) and GlaxoSmithKline’s fluticasone furoate/vilanterol (Breo Ellipta) and fluticasone propionate/salmeterol (Advair Diskus and Advair HFA).
The FDA also approved updates to the Warnings and Precautions section of labeling for the ICS/LABA class, which now includes a description of the four trials. Information on the efficacy of the drugs, found in the trials, has been added to the Clinical Studies section of the labels as well.
In a related safety announcement, the FDA stated the following: “Using LABAs alone to treat asthma without an ICS to treat lung inflammation is associated with an increased risk of asthma-related death. Therefore, the Boxed Warning stating this will remain in the labels of all single-ingredient LABA medicines, which are approved to treat asthma, chronic obstructive pulmonary disease (COPD), and wheezing caused by exercise. The labels of medicines that contain both an ICS and LABA also retain a Warning and Precaution related to the increased risk of asthma-related death when LABAs are used without an ICS to treat asthma.
FDA: Gadolinium retention prompts new GBCA class warning, safety measures
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.

Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
sworcester@frontlinemedcom.com
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
sworcester@frontlinemedcom.com
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
sworcester@frontlinemedcom.com
FDA: Gadolinium retention prompts new GBCA class warning, safety measures
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
Gadolinium-based contrast agents (GBCAs) used for MRI will now carry a warning regarding their potential retention in the bodies and brains of treated patients, according to the Food and Drug Administration.
The FDA is requiring the new class warning, along with other safety measures, based on evidence showing that trace amounts of gadolinium can be retained in the body for months to years after treatment.
Specifically, the agency will require that patients receiving GBCAs first receive a Medication Guide and that GBCA manufacturers conduct human and animal studies to further assess GBCA safety. At this time, the only known adverse health effect of gadolinium retention is nephrogenic systemic fibrosis, which affects a small subgroup of patients with pre-existing kidney failure. No causal association has been established between gadolinium retention and reported adverse events in those with normal kidney function.
The FDA recommended that health care professionals consider the retention characteristics of GBCAs for patients who may be at higher risk for retention, including those requiring multiple lifetime doses, pregnant women, children, and patients with inflammatory conditions, but stressed that, although repeated GBCA imaging studies should be minimized when possible, they should not be avoided or deferred when they are necessary. In the safety alert, the FDA noted that administration of the GBCAs Dotarem (gadoterate meglumine), Gadavist (gadobutrol), and ProHance (gadoteridol) produce the lowest gadolinium levels in the body, and the three agents leave similar gadolinium levels in the body.
The agency encourages reports of adverse events or side effects related to the use of GBCAs to its MedWatch Safety information and Adverse Event Reporting Program. Reports can be submitted online at www.fda.gov/MedWatch/report or by calling 1-800-332-1088 to request a preaddressed form that can be mailed or faxed to 1-800-FDA-0178.
FDA: Laparoscopic power morcellators can spread malignant cells when used in women with occult uterine cancers*
Laparoscopic power morcellation appears capable of spreading fulminant uterine malignancies when used to remove uterine fibroids from women who have unsuspected uterine cancers.
A new Food and Drug Administration literature review of 23 studies found consistent evidence that women who undergo surgery using laparoscopic power morcellators (LPMs) for fibroids that were assumed to be benign may have an occult uterine sarcoma or leiomyosarcoma. In the FDA’s literature review of 12 studies from 2014 to 2016, women who received power morcellation were at a significantly increased risk of death, compared with those whose fibroids were removed by other methods.
The findings reaffirm the agency’s 2014 warnings about LPMs:
• LPMs are contraindicated in gynecologic surgery in which the tissue to be morcellated is known or suspected to contain malignancy.
• LPMs are contraindicated for removal of uterine tissue containing suspected fibroids in patients who are peri- or postmenopausal, or in candidates for en bloc tissue removal.
• Boxed warning: Uterine tissue may contain unsuspected cancer. The use of LPMs during fibroid surgery may spread cancer and decrease long-term survival of patients. This information should be shared with patients when considering surgery with the use of these devices.
“We recognize that some health organizations have reported a lower estimate of risk and that some groups continue to request that we scale back our recommendations. However, after looking at all the relevant data, we believe our estimates remain accurate, and our recommendation against the use of this device to remove fibroids in the vast majority of women is appropriate and critical to better protecting these women. We are committed to continuing to review new relevant data to assure patient safety.”
The review determined that 1 in 350 women undergoing hysterectomy or myomectomy for fibroids may have an occult uterine sarcoma, and that 1 in about 500 have an unsuspected leiomyosarcoma. The literature review clearly identified increased risk of uterine cancers and decreased survival when these women are treated with an LPM, as opposed to manual morcellation or en bloc removal.
Twelve studies of women with uterine cancers examined this outcome in comparison groups. LPMs were associated with a 2- to 3-times increased risk of disease recurrence, compared with manual morcellation or other fibroid removal methods. In some studies, disease-free survival was significantly shorter among those who had undergone an LPM procedure; others found a higher risk of death – including, in one study, almost a quintupling of mortality risk by 1 year (crude risk ratio, 4.75).
Device-related malignancies began appearing in 2013; since then, 262 cases have been reported. But after the 2014 warning, use of LPMs in this application has decreased sharply. Two studies examined this, one finding that LPM use in fibroid surgery dropped from 14% to 3% and the other, that it dropped from 11% to 0.02%.
“The agency also continues to recommend that the advantages and risks of using LPMs during fibroid surgery be thoroughly discussed between the patient and physician before surgery,” the paper concluded. “FDA continues to actively encourage and engage in research to evaluate outcomes for a range of treatment options for fibroids and support the development of safer alternatives for providing a minimally invasive approach.”
msullivan@frontlinemedcom.com
Laparoscopic power morcellation appears capable of spreading fulminant uterine malignancies when used to remove uterine fibroids from women who have unsuspected uterine cancers.
A new Food and Drug Administration literature review of 23 studies found consistent evidence that women who undergo surgery using laparoscopic power morcellators (LPMs) for fibroids that were assumed to be benign may have an occult uterine sarcoma or leiomyosarcoma. In the FDA’s literature review of 12 studies from 2014 to 2016, women who received power morcellation were at a significantly increased risk of death, compared with those whose fibroids were removed by other methods.
The findings reaffirm the agency’s 2014 warnings about LPMs:
• LPMs are contraindicated in gynecologic surgery in which the tissue to be morcellated is known or suspected to contain malignancy.
• LPMs are contraindicated for removal of uterine tissue containing suspected fibroids in patients who are peri- or postmenopausal, or in candidates for en bloc tissue removal.
• Boxed warning: Uterine tissue may contain unsuspected cancer. The use of LPMs during fibroid surgery may spread cancer and decrease long-term survival of patients. This information should be shared with patients when considering surgery with the use of these devices.
“We recognize that some health organizations have reported a lower estimate of risk and that some groups continue to request that we scale back our recommendations. However, after looking at all the relevant data, we believe our estimates remain accurate, and our recommendation against the use of this device to remove fibroids in the vast majority of women is appropriate and critical to better protecting these women. We are committed to continuing to review new relevant data to assure patient safety.”
The review determined that 1 in 350 women undergoing hysterectomy or myomectomy for fibroids may have an occult uterine sarcoma, and that 1 in about 500 have an unsuspected leiomyosarcoma. The literature review clearly identified increased risk of uterine cancers and decreased survival when these women are treated with an LPM, as opposed to manual morcellation or en bloc removal.
Twelve studies of women with uterine cancers examined this outcome in comparison groups. LPMs were associated with a 2- to 3-times increased risk of disease recurrence, compared with manual morcellation or other fibroid removal methods. In some studies, disease-free survival was significantly shorter among those who had undergone an LPM procedure; others found a higher risk of death – including, in one study, almost a quintupling of mortality risk by 1 year (crude risk ratio, 4.75).
Device-related malignancies began appearing in 2013; since then, 262 cases have been reported. But after the 2014 warning, use of LPMs in this application has decreased sharply. Two studies examined this, one finding that LPM use in fibroid surgery dropped from 14% to 3% and the other, that it dropped from 11% to 0.02%.
“The agency also continues to recommend that the advantages and risks of using LPMs during fibroid surgery be thoroughly discussed between the patient and physician before surgery,” the paper concluded. “FDA continues to actively encourage and engage in research to evaluate outcomes for a range of treatment options for fibroids and support the development of safer alternatives for providing a minimally invasive approach.”
msullivan@frontlinemedcom.com
Laparoscopic power morcellation appears capable of spreading fulminant uterine malignancies when used to remove uterine fibroids from women who have unsuspected uterine cancers.
A new Food and Drug Administration literature review of 23 studies found consistent evidence that women who undergo surgery using laparoscopic power morcellators (LPMs) for fibroids that were assumed to be benign may have an occult uterine sarcoma or leiomyosarcoma. In the FDA’s literature review of 12 studies from 2014 to 2016, women who received power morcellation were at a significantly increased risk of death, compared with those whose fibroids were removed by other methods.
The findings reaffirm the agency’s 2014 warnings about LPMs:
• LPMs are contraindicated in gynecologic surgery in which the tissue to be morcellated is known or suspected to contain malignancy.
• LPMs are contraindicated for removal of uterine tissue containing suspected fibroids in patients who are peri- or postmenopausal, or in candidates for en bloc tissue removal.
• Boxed warning: Uterine tissue may contain unsuspected cancer. The use of LPMs during fibroid surgery may spread cancer and decrease long-term survival of patients. This information should be shared with patients when considering surgery with the use of these devices.
“We recognize that some health organizations have reported a lower estimate of risk and that some groups continue to request that we scale back our recommendations. However, after looking at all the relevant data, we believe our estimates remain accurate, and our recommendation against the use of this device to remove fibroids in the vast majority of women is appropriate and critical to better protecting these women. We are committed to continuing to review new relevant data to assure patient safety.”
The review determined that 1 in 350 women undergoing hysterectomy or myomectomy for fibroids may have an occult uterine sarcoma, and that 1 in about 500 have an unsuspected leiomyosarcoma. The literature review clearly identified increased risk of uterine cancers and decreased survival when these women are treated with an LPM, as opposed to manual morcellation or en bloc removal.
Twelve studies of women with uterine cancers examined this outcome in comparison groups. LPMs were associated with a 2- to 3-times increased risk of disease recurrence, compared with manual morcellation or other fibroid removal methods. In some studies, disease-free survival was significantly shorter among those who had undergone an LPM procedure; others found a higher risk of death – including, in one study, almost a quintupling of mortality risk by 1 year (crude risk ratio, 4.75).
Device-related malignancies began appearing in 2013; since then, 262 cases have been reported. But after the 2014 warning, use of LPMs in this application has decreased sharply. Two studies examined this, one finding that LPM use in fibroid surgery dropped from 14% to 3% and the other, that it dropped from 11% to 0.02%.
“The agency also continues to recommend that the advantages and risks of using LPMs during fibroid surgery be thoroughly discussed between the patient and physician before surgery,” the paper concluded. “FDA continues to actively encourage and engage in research to evaluate outcomes for a range of treatment options for fibroids and support the development of safer alternatives for providing a minimally invasive approach.”
msullivan@frontlinemedcom.com
FDA approves topical antibiotic for impetigo infections
The Food and Drug Administration has approved in patients aged 2 months or older.
This is the first topical treatment for impetigo to be approved in more than 10 years, according to the press release from the manufacturer, Medimetriks Pharmaceuticals.
Ozenoxacin is a quinolone antimicrobial. The prescribing information is available on the FDA website.
The Food and Drug Administration has approved in patients aged 2 months or older.
This is the first topical treatment for impetigo to be approved in more than 10 years, according to the press release from the manufacturer, Medimetriks Pharmaceuticals.
Ozenoxacin is a quinolone antimicrobial. The prescribing information is available on the FDA website.
The Food and Drug Administration has approved in patients aged 2 months or older.
This is the first topical treatment for impetigo to be approved in more than 10 years, according to the press release from the manufacturer, Medimetriks Pharmaceuticals.
Ozenoxacin is a quinolone antimicrobial. The prescribing information is available on the FDA website.
New DBS device gains approval for Parkinson’s disease
The approval is based on as-yet unpublished results of the INTREPID trial, which successfully met its primary endpoint of mean change in waking hours with good symptom control, according to an announcement from the device’s manufacturer, Boston Scientific. The INTREPID study is the first multicenter, prospective, double-blind, randomized, sham-controlled study of DBS for Parkinson’s disease in the United States and enrolled in 292 patients at 23 sites.
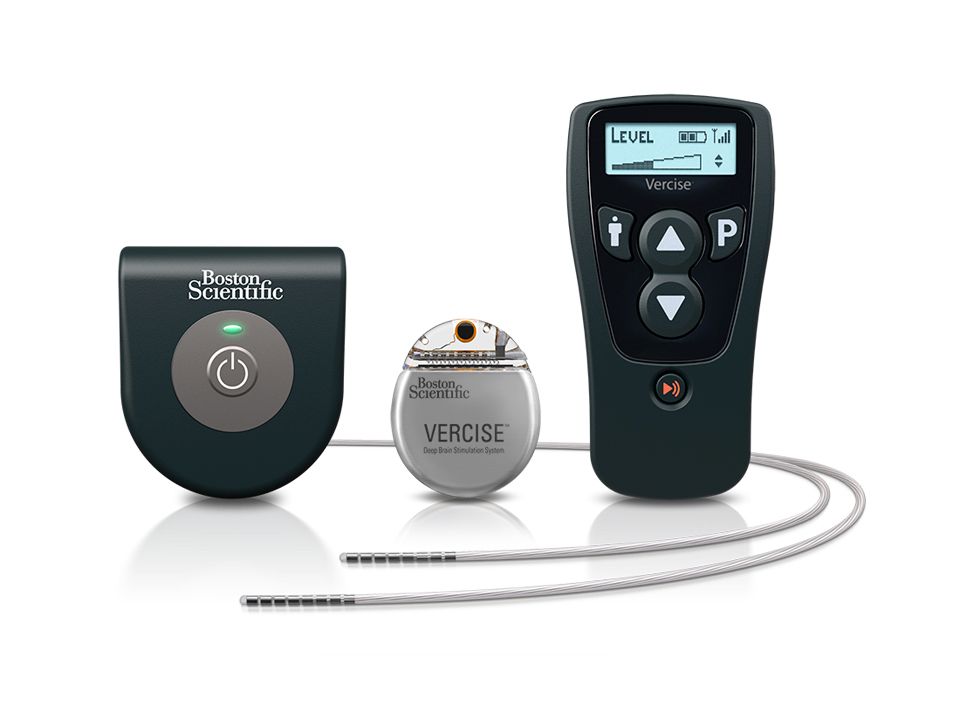
The implantable pulse generator of the device is “the smallest, rechargeable DBS device available in the U.S.,” according to Boston Scientific, and can independently control the amount of current delivered by each of the electrodes on the implanted leads, allowing them “to work together to address common challenges in DBS therapy such as fluctuations in symptoms and the progressive nature of the condition by offering more adaptable delivery of stimulation.”
“The Vercise DBS System changes the landscape of what physicians can do to help improve the quality of life for people living with Parkinson’s disease,” Jerry Vitek, MD, PhD, professor and chair of the department of neurology at the University of Minnesota, Minneapolis, and coordinating principal investigator for the INTREPID study said in the Boston Scientific announcement. “This system provides an ability to sculpt the current field in the DBS target using novel technology that offers flexibility in programming. This flexibility allows us to target different regions of the subthalamic nucleus, which we believe will improve outcomes while reducing side effects.”
The approval is based on as-yet unpublished results of the INTREPID trial, which successfully met its primary endpoint of mean change in waking hours with good symptom control, according to an announcement from the device’s manufacturer, Boston Scientific. The INTREPID study is the first multicenter, prospective, double-blind, randomized, sham-controlled study of DBS for Parkinson’s disease in the United States and enrolled in 292 patients at 23 sites.
The implantable pulse generator of the device is “the smallest, rechargeable DBS device available in the U.S.,” according to Boston Scientific, and can independently control the amount of current delivered by each of the electrodes on the implanted leads, allowing them “to work together to address common challenges in DBS therapy such as fluctuations in symptoms and the progressive nature of the condition by offering more adaptable delivery of stimulation.”
“The Vercise DBS System changes the landscape of what physicians can do to help improve the quality of life for people living with Parkinson’s disease,” Jerry Vitek, MD, PhD, professor and chair of the department of neurology at the University of Minnesota, Minneapolis, and coordinating principal investigator for the INTREPID study said in the Boston Scientific announcement. “This system provides an ability to sculpt the current field in the DBS target using novel technology that offers flexibility in programming. This flexibility allows us to target different regions of the subthalamic nucleus, which we believe will improve outcomes while reducing side effects.”
The approval is based on as-yet unpublished results of the INTREPID trial, which successfully met its primary endpoint of mean change in waking hours with good symptom control, according to an announcement from the device’s manufacturer, Boston Scientific. The INTREPID study is the first multicenter, prospective, double-blind, randomized, sham-controlled study of DBS for Parkinson’s disease in the United States and enrolled in 292 patients at 23 sites.
The implantable pulse generator of the device is “the smallest, rechargeable DBS device available in the U.S.,” according to Boston Scientific, and can independently control the amount of current delivered by each of the electrodes on the implanted leads, allowing them “to work together to address common challenges in DBS therapy such as fluctuations in symptoms and the progressive nature of the condition by offering more adaptable delivery of stimulation.”
“The Vercise DBS System changes the landscape of what physicians can do to help improve the quality of life for people living with Parkinson’s disease,” Jerry Vitek, MD, PhD, professor and chair of the department of neurology at the University of Minnesota, Minneapolis, and coordinating principal investigator for the INTREPID study said in the Boston Scientific announcement. “This system provides an ability to sculpt the current field in the DBS target using novel technology that offers flexibility in programming. This flexibility allows us to target different regions of the subthalamic nucleus, which we believe will improve outcomes while reducing side effects.”
FDA approves premixed, low-volume colon-cleansing solution
in adults preparing to undergo colonoscopy, according to Ferring Pharmaceuticals.
The sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution is a relatively low-volume, premixed, cranberry-flavored solution, making it easier to use and more palatable for patients.
Sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution is approved with two dosing options: split dose, one dose the evening prior and one dose the morning of the procedure, or the day before dose, which involves taking both doses the day prior to the procedure. Day before dosing is an alternative and should be used when split dosing is not appropriate. After each dose of sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution, clear liquids should be consumed based on the dosing recommendation. The American College of Gastroenterology recommends the split-dose regimen because of its improved cleansing quality of the colon and better tolerability of the liquid volume by patients.
Patients with impaired renal function should exercise caution if using sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution as it may effect renal function. A more comprehensive list of safety information is available at www.clenpiq.com.
To help your patients understand and prepare for a colonoscopy, use AGA’s patient education materials at http://www.gastro.org/patient-care/procedures/colonoscopy.
in adults preparing to undergo colonoscopy, according to Ferring Pharmaceuticals.
The sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution is a relatively low-volume, premixed, cranberry-flavored solution, making it easier to use and more palatable for patients.
Sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution is approved with two dosing options: split dose, one dose the evening prior and one dose the morning of the procedure, or the day before dose, which involves taking both doses the day prior to the procedure. Day before dosing is an alternative and should be used when split dosing is not appropriate. After each dose of sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution, clear liquids should be consumed based on the dosing recommendation. The American College of Gastroenterology recommends the split-dose regimen because of its improved cleansing quality of the colon and better tolerability of the liquid volume by patients.
Patients with impaired renal function should exercise caution if using sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution as it may effect renal function. A more comprehensive list of safety information is available at www.clenpiq.com.
To help your patients understand and prepare for a colonoscopy, use AGA’s patient education materials at http://www.gastro.org/patient-care/procedures/colonoscopy.
in adults preparing to undergo colonoscopy, according to Ferring Pharmaceuticals.
The sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution is a relatively low-volume, premixed, cranberry-flavored solution, making it easier to use and more palatable for patients.
Sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution is approved with two dosing options: split dose, one dose the evening prior and one dose the morning of the procedure, or the day before dose, which involves taking both doses the day prior to the procedure. Day before dosing is an alternative and should be used when split dosing is not appropriate. After each dose of sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution, clear liquids should be consumed based on the dosing recommendation. The American College of Gastroenterology recommends the split-dose regimen because of its improved cleansing quality of the colon and better tolerability of the liquid volume by patients.
Patients with impaired renal function should exercise caution if using sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution as it may effect renal function. A more comprehensive list of safety information is available at www.clenpiq.com.
To help your patients understand and prepare for a colonoscopy, use AGA’s patient education materials at http://www.gastro.org/patient-care/procedures/colonoscopy.
Nebulized LAMA for COPD approved
The Food and Drug Administration has given the nod to the first nebulized long-acting muscarinic antagonist (LAMA) treatment for chronic obstructive pulmonary disease (COPD) in the United States.
Glycopyrrolate (Lonhala Magnair) utilizes the eFlow technology system, developed by Pari Pharma. This nebulizing system is portable, virtually silent, and delivers the drug in 2-3 minutes, according to a statement from Sunovion Pharmaceuticals.![]()
The approval of glycopyrrolate is based on the results of the GOLDEN (Glycopyrrolate for Obstructive Lung Disease via Electronic Nebulizer) trials. The GOLDEN program comprised the GOLDEN-3 and GOLDEN-4 trials, both of which were phase 3, 12-week, randomized, double-blind, placebo-controlled, parallel-group, multicenter safety and efficacy trials, which compared adult glycopyrrolate patients to a placebo group with moderate to severe COPD. At 12 weeks, patients receiving treatment with glycopyrrolate showed clinical and statistically significant improvements in their baseline forced expiratory volume second (FEV1), compared with placebo.
GOLDEN-5, an additional study, followed the same criteria as previous studies, but increased its length to 48 weeks to evaluate the long-term safety and patient tolerability of glycopyrrolate. It also compared treatment of COPD with glycopyrrolate to treatment of COPD with the previously approved LAMA Spiriva (tiotropium bromide), delivered by the Handihaler device. Glycopyrrolate was well tolerated, and the overall treatment emergence of adverse events for glycopyrrolate and tiotropium bromide were similar.
Sunovion expects glycopyrrolate to be available in U.S. pharmacies in early 2018, according to the statement.
The Food and Drug Administration has given the nod to the first nebulized long-acting muscarinic antagonist (LAMA) treatment for chronic obstructive pulmonary disease (COPD) in the United States.
Glycopyrrolate (Lonhala Magnair) utilizes the eFlow technology system, developed by Pari Pharma. This nebulizing system is portable, virtually silent, and delivers the drug in 2-3 minutes, according to a statement from Sunovion Pharmaceuticals.![]()
The approval of glycopyrrolate is based on the results of the GOLDEN (Glycopyrrolate for Obstructive Lung Disease via Electronic Nebulizer) trials. The GOLDEN program comprised the GOLDEN-3 and GOLDEN-4 trials, both of which were phase 3, 12-week, randomized, double-blind, placebo-controlled, parallel-group, multicenter safety and efficacy trials, which compared adult glycopyrrolate patients to a placebo group with moderate to severe COPD. At 12 weeks, patients receiving treatment with glycopyrrolate showed clinical and statistically significant improvements in their baseline forced expiratory volume second (FEV1), compared with placebo.
GOLDEN-5, an additional study, followed the same criteria as previous studies, but increased its length to 48 weeks to evaluate the long-term safety and patient tolerability of glycopyrrolate. It also compared treatment of COPD with glycopyrrolate to treatment of COPD with the previously approved LAMA Spiriva (tiotropium bromide), delivered by the Handihaler device. Glycopyrrolate was well tolerated, and the overall treatment emergence of adverse events for glycopyrrolate and tiotropium bromide were similar.
Sunovion expects glycopyrrolate to be available in U.S. pharmacies in early 2018, according to the statement.
The Food and Drug Administration has given the nod to the first nebulized long-acting muscarinic antagonist (LAMA) treatment for chronic obstructive pulmonary disease (COPD) in the United States.
Glycopyrrolate (Lonhala Magnair) utilizes the eFlow technology system, developed by Pari Pharma. This nebulizing system is portable, virtually silent, and delivers the drug in 2-3 minutes, according to a statement from Sunovion Pharmaceuticals.![]()
The approval of glycopyrrolate is based on the results of the GOLDEN (Glycopyrrolate for Obstructive Lung Disease via Electronic Nebulizer) trials. The GOLDEN program comprised the GOLDEN-3 and GOLDEN-4 trials, both of which were phase 3, 12-week, randomized, double-blind, placebo-controlled, parallel-group, multicenter safety and efficacy trials, which compared adult glycopyrrolate patients to a placebo group with moderate to severe COPD. At 12 weeks, patients receiving treatment with glycopyrrolate showed clinical and statistically significant improvements in their baseline forced expiratory volume second (FEV1), compared with placebo.
GOLDEN-5, an additional study, followed the same criteria as previous studies, but increased its length to 48 weeks to evaluate the long-term safety and patient tolerability of glycopyrrolate. It also compared treatment of COPD with glycopyrrolate to treatment of COPD with the previously approved LAMA Spiriva (tiotropium bromide), delivered by the Handihaler device. Glycopyrrolate was well tolerated, and the overall treatment emergence of adverse events for glycopyrrolate and tiotropium bromide were similar.
Sunovion expects glycopyrrolate to be available in U.S. pharmacies in early 2018, according to the statement.
Developmental disabilities up significantly since 2014
In 2016, the prevalence of any diagnosed developmental disability in children aged 3-17 years was 6.99% – a statistically significant increase of 21% over the 5.76% recorded in 2014, the NCHS said in a recent Data Brief.
Autism spectrum disorder was up by a similar amount: 23% from 2014, when prevalence was 2.24%, to 2016, when the prevalence was 2.76% among children aged 3-17 years. Intellectual disability rose in 2015 but dropped in 2016, so the overall increase in prevalence was just 3.6%. The prevalence of other developmental delays, on the other hand, held steady from 2014 to 2015 and then took a big jump, 27.5%, in 2016, the NCHS investigators reported.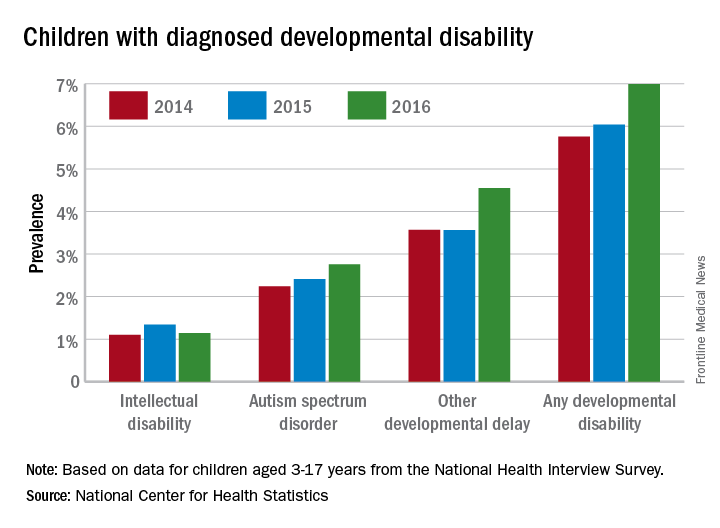
The estimates are based on reports by parents or guardians of ever receiving a diagnosis of each developmental disability from a physician or other medical professional.
In 2016, the prevalence of any diagnosed developmental disability in children aged 3-17 years was 6.99% – a statistically significant increase of 21% over the 5.76% recorded in 2014, the NCHS said in a recent Data Brief.
Autism spectrum disorder was up by a similar amount: 23% from 2014, when prevalence was 2.24%, to 2016, when the prevalence was 2.76% among children aged 3-17 years. Intellectual disability rose in 2015 but dropped in 2016, so the overall increase in prevalence was just 3.6%. The prevalence of other developmental delays, on the other hand, held steady from 2014 to 2015 and then took a big jump, 27.5%, in 2016, the NCHS investigators reported.
The estimates are based on reports by parents or guardians of ever receiving a diagnosis of each developmental disability from a physician or other medical professional.
In 2016, the prevalence of any diagnosed developmental disability in children aged 3-17 years was 6.99% – a statistically significant increase of 21% over the 5.76% recorded in 2014, the NCHS said in a recent Data Brief.
Autism spectrum disorder was up by a similar amount: 23% from 2014, when prevalence was 2.24%, to 2016, when the prevalence was 2.76% among children aged 3-17 years. Intellectual disability rose in 2015 but dropped in 2016, so the overall increase in prevalence was just 3.6%. The prevalence of other developmental delays, on the other hand, held steady from 2014 to 2015 and then took a big jump, 27.5%, in 2016, the NCHS investigators reported.
The estimates are based on reports by parents or guardians of ever receiving a diagnosis of each developmental disability from a physician or other medical professional.