User login
A Case of Bloom Syndrome With Uncommon Clinical Manifestations Confirmed on Genetic Testing
Bloom syndrome, also called congenital telangiectatic erythema and stunted growth, was first described by David Bloom in 1954.1 It is a rare autosomal-recessive disorder (Online Mendelian Inheritance in Man 210900) characterized by specific clinical manifestations including photosensitivity, telangiectatic facial erythema, proportionate growth deficiency, hypogonadism, immunodeficiency, and a tendency to develop various malignancies.2 Linkage analysis revealed that the Bloom syndrome gene locus resides on chromosome arm 15q26.1,3 and the BLM gene in this region has been identified as being responsible for the development of Bloom syndrome.4,5 We report the case of a 12-year-old Chinese girl with Bloom syndrome and detected BLM gene. The evaluation was approved by the Institutional Ethical Review Boards of Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China).
Case Report
We evaluated a Bloom syndrome family, which consisted of the patient and her parents. The patient was a 12-year-old Chinese girl who was apparently healthy until 3 months of age when her parents noticed an erythematous eruption with blisters on the face. Exacerbation after exposure to sunlight is usual, which results in the eruption becoming prominent in summer and fainter in winter.2 Gradually, the patient’s skin lesions became more progressive, extending to the forehead, nose, and ears, with oozing, crusting, atrophy, and telangiectases developing on the face despite treatment. In the last 3 years, no blisters were present on the patient’s face because of her efforts to avoid sun exposure. She had no history of recurrent infections.
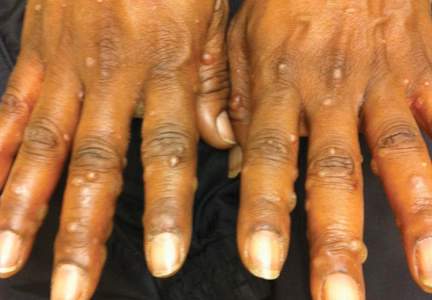
On physical examination, the patient was generally healthy with normal intelligence and short stature. She weighed 26 kg and was approximately 122-cm tall. Telangiectatic erythema and slight scaling were noted on the face, which simulated lupus erythematosus (Figures 1A and 1B). She had additional abnormalities including alopecia areata (Figure 1C), eyebrow hair loss, flat nose, reticular pigmentation on the forehead and trunk, and finger swelling. The distal phalanges on all 10 fingers became short and sharpened and the fingernails became wider than they were long (Figure 1D). Laboratory investigations, including a complete blood cell count, liver and kidney function tests, stool examination, serum complement, and albumin and globulin levels, were within reference range.

After informed consent was obtained, a mutation analysis of the BLM gene was performed in the patient and her parents. We used a genomic DNA purification kit to extract genomic DNA from peripheral blood according to the manufacturer’s protocol. Genomic DNA was used to amplify the exons of the BLM gene with intron flanking sequences by polymerase chain reaction with the primer described elsewhere.6 After the amplification, the polymerase chain reaction products were purified and the BLM gene was sequenced. Sequence comparisons and analysis were performed using Phred/Phrap/Consed version 12.0.
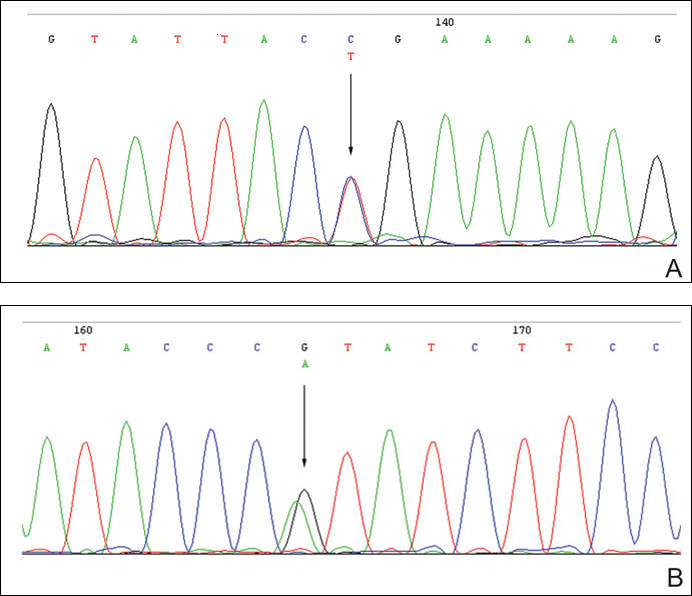
The patient was found to carry changes in 2 heterozygous nucleotide sites, including c.2603C>T in exon 13 and c.3961G>A in exon 21 of the BLM gene. The patient’s father was found to carry c.2603C>T and her mother carried c.3961G>A (Figure 2).
Comment
Patients with Bloom syndrome have a characteristic clinical appearance that typically includes photosensitivity, telangiectatic facial erythema, and growth deficiency. Telangiectatic erythema of the face develops during infancy or early childhood as red macules or plaques and may simulate lupus erythematosus. The lesions are described as a butterfly rash affecting the bridge of the nose and cheeks but also may involve the margins of the eyelids, forehead, ears, and sometimes the dorsa of the hands and forearms. Moderate and proportionate growth deficiencies develop both in utero and postnatally. Patients with Bloom syndrome characteristically have narrow, slender, distinct facial features with micrognathism and a relatively prominent nose. They usually may have mild microcephaly, meaning the head is longer and narrower than normal.2,7-10
German and Takebe11 reported 14 Japanese patients with Bloom syndrome. The phenotype differs somewhat from most cases recognized elsewhere in that dolichocephaly was a less constant feature, the facial skin was less prominent, and life-threatening infections were less common. Our patient had typical telangiectatic facial erythema without microcephaly, dolichocephaly, or any infections. She also had some uncommon manifestations such as alopecia areata, eyebrow hair loss, flat nose, reticular pigmentation, and short sharpened distal phalanges with fingernails that were wider than they were long. Although she had no recurrent infections and laboratory tests were within reference range, the alopecia areata and eyebrow hair loss may be associated with an abnormal immune response. The reasons for the short sharpened distal phalanges and the fingernail findings are unclear. The presence of reticular pigmentation also is unclear but may be associated with photosensitivity. Since the BLM gene was discovered to be the disease-causing gene of Bloom syndrome in 1995,4,5 approximately 70 mutations were reported. The BLM gene encodes for the Bloom syndrome protein, a DNA helicase of the highly conserved RecQ subfamily of helicases, a group of nuclear proteins important in the maintenance of genomic stability.12
Mutation analysis of the BLM gene in our patient showed changes in 2 heterozygous nucleotide sites, including c.2603C>T in exon 13 and c.3961G>A in exon 21 of the BLM gene, which altered proline residue with leucine residue at 868 and valine residue with isoleucine residue at 1321, respectively. According to GenBank,13,14 c.2603C>T and c.3961G>A are single nucleotide polymorphisms of the BLM gene. The genotypic distribution of International HapMap Project15 showed that C=602/602 and T=0/602 on c.2603 in 301 unrelated Chinese patients and G=585/602 and A=17/602 on c.3961 in 301 unrelated Chinese patients. Because of the low prevalence of genotypes c.2603T and c.3961A in China, the relationship between clinical features and c.2603C>T and c.3961G>A of the BLM gene in our patient requires further study.
In conclusion, we report a patient with Bloom syndrome with uncommon clinical manifestations. Our findings indicate that c.2603C>T and c.3961G>A of the BLM gene may be the pathogenic nature for Bloom syndrome in China.
Acknowledgments
The authors would like to thank the patient and her family for their participation in the study. The authors also thank Li Qi, BA, Beijing, China, for his contribution to the review of the data in the literature.
- Bloom D. Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs; probably a syndrome entity. AMA Am J Dis Child. 1954;88:754-758.
- German J. Bloom’s syndrome, I: genetical and clinical observations in the first twenty-seven patients. Am J Hum Genet. 1969;21:196-227.
- German J, Roe AM, Leppert MF, et al. Bloom syndrome: an analysis of consanguineous families assigns the locus mutated to chromosome band 15q26.1. Proc Natl Acad Sci U S A. 1994;91:6669-6673.
- Passarge E. A DNA helicase in full Bloom. Nat Genet. 1995;11:356-357.
- Ellis NA, Groden J, Ye TZ, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655-666.
- German J, Sanz MM, Ciocci S, et al. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum Mutat. 2007;28:743-753.
- Landau JW, Sasaki MS, Newcomer VD, et al. Bloom’s syndrome: the syndrome of telangiectatic erythema and growth retardation. Arch Dermatol. 1966;94:687-694.
- Gretzula JC, Hevia O, Weber PJ. Bloom’s syndrome. J Am Acad Dermatol. 1987;17:479-488.
- Passarge E. Bloom’s syndrome: the German experience. Ann Genet. 1991;34:179-197.
- German J. Bloom’s syndrome. Dermatol Clin. 1995;13:7-18.
- German J, Takebe H. Bloom’s syndrome, XIV: the disorder in Japan. Clin Genet. 1989;35:93-110.
- Bennett RJ, Keck JL. Structure and function of RecQ DNA helicases. Crit Rev Biochem Mol Biol. 2004;39:79-97.
- Reference SNP (refSNP) Cluster Report: rs2227935. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=2227935. Accessed February 3, 2016.
- Reference SNP (refSNP) Cluster Report: rs7167216. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=7167216. Accessed February 3, 2016.
- Homo sapiens:GRCh37.p13 (GCF_000001405.25)Chr 1 (NC_000001.10):1 - 249.3M. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/variationtools/1000genomes/?=%EF%BC%86=. Accessed February 3, 2016.
Bloom syndrome, also called congenital telangiectatic erythema and stunted growth, was first described by David Bloom in 1954.1 It is a rare autosomal-recessive disorder (Online Mendelian Inheritance in Man 210900) characterized by specific clinical manifestations including photosensitivity, telangiectatic facial erythema, proportionate growth deficiency, hypogonadism, immunodeficiency, and a tendency to develop various malignancies.2 Linkage analysis revealed that the Bloom syndrome gene locus resides on chromosome arm 15q26.1,3 and the BLM gene in this region has been identified as being responsible for the development of Bloom syndrome.4,5 We report the case of a 12-year-old Chinese girl with Bloom syndrome and detected BLM gene. The evaluation was approved by the Institutional Ethical Review Boards of Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China).
Case Report
We evaluated a Bloom syndrome family, which consisted of the patient and her parents. The patient was a 12-year-old Chinese girl who was apparently healthy until 3 months of age when her parents noticed an erythematous eruption with blisters on the face. Exacerbation after exposure to sunlight is usual, which results in the eruption becoming prominent in summer and fainter in winter.2 Gradually, the patient’s skin lesions became more progressive, extending to the forehead, nose, and ears, with oozing, crusting, atrophy, and telangiectases developing on the face despite treatment. In the last 3 years, no blisters were present on the patient’s face because of her efforts to avoid sun exposure. She had no history of recurrent infections.
On physical examination, the patient was generally healthy with normal intelligence and short stature. She weighed 26 kg and was approximately 122-cm tall. Telangiectatic erythema and slight scaling were noted on the face, which simulated lupus erythematosus (Figures 1A and 1B). She had additional abnormalities including alopecia areata (Figure 1C), eyebrow hair loss, flat nose, reticular pigmentation on the forehead and trunk, and finger swelling. The distal phalanges on all 10 fingers became short and sharpened and the fingernails became wider than they were long (Figure 1D). Laboratory investigations, including a complete blood cell count, liver and kidney function tests, stool examination, serum complement, and albumin and globulin levels, were within reference range.
After informed consent was obtained, a mutation analysis of the BLM gene was performed in the patient and her parents. We used a genomic DNA purification kit to extract genomic DNA from peripheral blood according to the manufacturer’s protocol. Genomic DNA was used to amplify the exons of the BLM gene with intron flanking sequences by polymerase chain reaction with the primer described elsewhere.6 After the amplification, the polymerase chain reaction products were purified and the BLM gene was sequenced. Sequence comparisons and analysis were performed using Phred/Phrap/Consed version 12.0.
The patient was found to carry changes in 2 heterozygous nucleotide sites, including c.2603C>T in exon 13 and c.3961G>A in exon 21 of the BLM gene. The patient’s father was found to carry c.2603C>T and her mother carried c.3961G>A (Figure 2).
Comment
Patients with Bloom syndrome have a characteristic clinical appearance that typically includes photosensitivity, telangiectatic facial erythema, and growth deficiency. Telangiectatic erythema of the face develops during infancy or early childhood as red macules or plaques and may simulate lupus erythematosus. The lesions are described as a butterfly rash affecting the bridge of the nose and cheeks but also may involve the margins of the eyelids, forehead, ears, and sometimes the dorsa of the hands and forearms. Moderate and proportionate growth deficiencies develop both in utero and postnatally. Patients with Bloom syndrome characteristically have narrow, slender, distinct facial features with micrognathism and a relatively prominent nose. They usually may have mild microcephaly, meaning the head is longer and narrower than normal.2,7-10
German and Takebe11 reported 14 Japanese patients with Bloom syndrome. The phenotype differs somewhat from most cases recognized elsewhere in that dolichocephaly was a less constant feature, the facial skin was less prominent, and life-threatening infections were less common. Our patient had typical telangiectatic facial erythema without microcephaly, dolichocephaly, or any infections. She also had some uncommon manifestations such as alopecia areata, eyebrow hair loss, flat nose, reticular pigmentation, and short sharpened distal phalanges with fingernails that were wider than they were long. Although she had no recurrent infections and laboratory tests were within reference range, the alopecia areata and eyebrow hair loss may be associated with an abnormal immune response. The reasons for the short sharpened distal phalanges and the fingernail findings are unclear. The presence of reticular pigmentation also is unclear but may be associated with photosensitivity. Since the BLM gene was discovered to be the disease-causing gene of Bloom syndrome in 1995,4,5 approximately 70 mutations were reported. The BLM gene encodes for the Bloom syndrome protein, a DNA helicase of the highly conserved RecQ subfamily of helicases, a group of nuclear proteins important in the maintenance of genomic stability.12
Mutation analysis of the BLM gene in our patient showed changes in 2 heterozygous nucleotide sites, including c.2603C>T in exon 13 and c.3961G>A in exon 21 of the BLM gene, which altered proline residue with leucine residue at 868 and valine residue with isoleucine residue at 1321, respectively. According to GenBank,13,14 c.2603C>T and c.3961G>A are single nucleotide polymorphisms of the BLM gene. The genotypic distribution of International HapMap Project15 showed that C=602/602 and T=0/602 on c.2603 in 301 unrelated Chinese patients and G=585/602 and A=17/602 on c.3961 in 301 unrelated Chinese patients. Because of the low prevalence of genotypes c.2603T and c.3961A in China, the relationship between clinical features and c.2603C>T and c.3961G>A of the BLM gene in our patient requires further study.
In conclusion, we report a patient with Bloom syndrome with uncommon clinical manifestations. Our findings indicate that c.2603C>T and c.3961G>A of the BLM gene may be the pathogenic nature for Bloom syndrome in China.
Acknowledgments
The authors would like to thank the patient and her family for their participation in the study. The authors also thank Li Qi, BA, Beijing, China, for his contribution to the review of the data in the literature.
Bloom syndrome, also called congenital telangiectatic erythema and stunted growth, was first described by David Bloom in 1954.1 It is a rare autosomal-recessive disorder (Online Mendelian Inheritance in Man 210900) characterized by specific clinical manifestations including photosensitivity, telangiectatic facial erythema, proportionate growth deficiency, hypogonadism, immunodeficiency, and a tendency to develop various malignancies.2 Linkage analysis revealed that the Bloom syndrome gene locus resides on chromosome arm 15q26.1,3 and the BLM gene in this region has been identified as being responsible for the development of Bloom syndrome.4,5 We report the case of a 12-year-old Chinese girl with Bloom syndrome and detected BLM gene. The evaluation was approved by the Institutional Ethical Review Boards of Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College (Beijing, China).
Case Report
We evaluated a Bloom syndrome family, which consisted of the patient and her parents. The patient was a 12-year-old Chinese girl who was apparently healthy until 3 months of age when her parents noticed an erythematous eruption with blisters on the face. Exacerbation after exposure to sunlight is usual, which results in the eruption becoming prominent in summer and fainter in winter.2 Gradually, the patient’s skin lesions became more progressive, extending to the forehead, nose, and ears, with oozing, crusting, atrophy, and telangiectases developing on the face despite treatment. In the last 3 years, no blisters were present on the patient’s face because of her efforts to avoid sun exposure. She had no history of recurrent infections.
On physical examination, the patient was generally healthy with normal intelligence and short stature. She weighed 26 kg and was approximately 122-cm tall. Telangiectatic erythema and slight scaling were noted on the face, which simulated lupus erythematosus (Figures 1A and 1B). She had additional abnormalities including alopecia areata (Figure 1C), eyebrow hair loss, flat nose, reticular pigmentation on the forehead and trunk, and finger swelling. The distal phalanges on all 10 fingers became short and sharpened and the fingernails became wider than they were long (Figure 1D). Laboratory investigations, including a complete blood cell count, liver and kidney function tests, stool examination, serum complement, and albumin and globulin levels, were within reference range.
After informed consent was obtained, a mutation analysis of the BLM gene was performed in the patient and her parents. We used a genomic DNA purification kit to extract genomic DNA from peripheral blood according to the manufacturer’s protocol. Genomic DNA was used to amplify the exons of the BLM gene with intron flanking sequences by polymerase chain reaction with the primer described elsewhere.6 After the amplification, the polymerase chain reaction products were purified and the BLM gene was sequenced. Sequence comparisons and analysis were performed using Phred/Phrap/Consed version 12.0.
The patient was found to carry changes in 2 heterozygous nucleotide sites, including c.2603C>T in exon 13 and c.3961G>A in exon 21 of the BLM gene. The patient’s father was found to carry c.2603C>T and her mother carried c.3961G>A (Figure 2).
Comment
Patients with Bloom syndrome have a characteristic clinical appearance that typically includes photosensitivity, telangiectatic facial erythema, and growth deficiency. Telangiectatic erythema of the face develops during infancy or early childhood as red macules or plaques and may simulate lupus erythematosus. The lesions are described as a butterfly rash affecting the bridge of the nose and cheeks but also may involve the margins of the eyelids, forehead, ears, and sometimes the dorsa of the hands and forearms. Moderate and proportionate growth deficiencies develop both in utero and postnatally. Patients with Bloom syndrome characteristically have narrow, slender, distinct facial features with micrognathism and a relatively prominent nose. They usually may have mild microcephaly, meaning the head is longer and narrower than normal.2,7-10
German and Takebe11 reported 14 Japanese patients with Bloom syndrome. The phenotype differs somewhat from most cases recognized elsewhere in that dolichocephaly was a less constant feature, the facial skin was less prominent, and life-threatening infections were less common. Our patient had typical telangiectatic facial erythema without microcephaly, dolichocephaly, or any infections. She also had some uncommon manifestations such as alopecia areata, eyebrow hair loss, flat nose, reticular pigmentation, and short sharpened distal phalanges with fingernails that were wider than they were long. Although she had no recurrent infections and laboratory tests were within reference range, the alopecia areata and eyebrow hair loss may be associated with an abnormal immune response. The reasons for the short sharpened distal phalanges and the fingernail findings are unclear. The presence of reticular pigmentation also is unclear but may be associated with photosensitivity. Since the BLM gene was discovered to be the disease-causing gene of Bloom syndrome in 1995,4,5 approximately 70 mutations were reported. The BLM gene encodes for the Bloom syndrome protein, a DNA helicase of the highly conserved RecQ subfamily of helicases, a group of nuclear proteins important in the maintenance of genomic stability.12
Mutation analysis of the BLM gene in our patient showed changes in 2 heterozygous nucleotide sites, including c.2603C>T in exon 13 and c.3961G>A in exon 21 of the BLM gene, which altered proline residue with leucine residue at 868 and valine residue with isoleucine residue at 1321, respectively. According to GenBank,13,14 c.2603C>T and c.3961G>A are single nucleotide polymorphisms of the BLM gene. The genotypic distribution of International HapMap Project15 showed that C=602/602 and T=0/602 on c.2603 in 301 unrelated Chinese patients and G=585/602 and A=17/602 on c.3961 in 301 unrelated Chinese patients. Because of the low prevalence of genotypes c.2603T and c.3961A in China, the relationship between clinical features and c.2603C>T and c.3961G>A of the BLM gene in our patient requires further study.
In conclusion, we report a patient with Bloom syndrome with uncommon clinical manifestations. Our findings indicate that c.2603C>T and c.3961G>A of the BLM gene may be the pathogenic nature for Bloom syndrome in China.
Acknowledgments
The authors would like to thank the patient and her family for their participation in the study. The authors also thank Li Qi, BA, Beijing, China, for his contribution to the review of the data in the literature.
- Bloom D. Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs; probably a syndrome entity. AMA Am J Dis Child. 1954;88:754-758.
- German J. Bloom’s syndrome, I: genetical and clinical observations in the first twenty-seven patients. Am J Hum Genet. 1969;21:196-227.
- German J, Roe AM, Leppert MF, et al. Bloom syndrome: an analysis of consanguineous families assigns the locus mutated to chromosome band 15q26.1. Proc Natl Acad Sci U S A. 1994;91:6669-6673.
- Passarge E. A DNA helicase in full Bloom. Nat Genet. 1995;11:356-357.
- Ellis NA, Groden J, Ye TZ, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655-666.
- German J, Sanz MM, Ciocci S, et al. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum Mutat. 2007;28:743-753.
- Landau JW, Sasaki MS, Newcomer VD, et al. Bloom’s syndrome: the syndrome of telangiectatic erythema and growth retardation. Arch Dermatol. 1966;94:687-694.
- Gretzula JC, Hevia O, Weber PJ. Bloom’s syndrome. J Am Acad Dermatol. 1987;17:479-488.
- Passarge E. Bloom’s syndrome: the German experience. Ann Genet. 1991;34:179-197.
- German J. Bloom’s syndrome. Dermatol Clin. 1995;13:7-18.
- German J, Takebe H. Bloom’s syndrome, XIV: the disorder in Japan. Clin Genet. 1989;35:93-110.
- Bennett RJ, Keck JL. Structure and function of RecQ DNA helicases. Crit Rev Biochem Mol Biol. 2004;39:79-97.
- Reference SNP (refSNP) Cluster Report: rs2227935. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=2227935. Accessed February 3, 2016.
- Reference SNP (refSNP) Cluster Report: rs7167216. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=7167216. Accessed February 3, 2016.
- Homo sapiens:GRCh37.p13 (GCF_000001405.25)Chr 1 (NC_000001.10):1 - 249.3M. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/variationtools/1000genomes/?=%EF%BC%86=. Accessed February 3, 2016.
- Bloom D. Congenital telangiectatic erythema resembling lupus erythematosus in dwarfs; probably a syndrome entity. AMA Am J Dis Child. 1954;88:754-758.
- German J. Bloom’s syndrome, I: genetical and clinical observations in the first twenty-seven patients. Am J Hum Genet. 1969;21:196-227.
- German J, Roe AM, Leppert MF, et al. Bloom syndrome: an analysis of consanguineous families assigns the locus mutated to chromosome band 15q26.1. Proc Natl Acad Sci U S A. 1994;91:6669-6673.
- Passarge E. A DNA helicase in full Bloom. Nat Genet. 1995;11:356-357.
- Ellis NA, Groden J, Ye TZ, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995;83:655-666.
- German J, Sanz MM, Ciocci S, et al. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum Mutat. 2007;28:743-753.
- Landau JW, Sasaki MS, Newcomer VD, et al. Bloom’s syndrome: the syndrome of telangiectatic erythema and growth retardation. Arch Dermatol. 1966;94:687-694.
- Gretzula JC, Hevia O, Weber PJ. Bloom’s syndrome. J Am Acad Dermatol. 1987;17:479-488.
- Passarge E. Bloom’s syndrome: the German experience. Ann Genet. 1991;34:179-197.
- German J. Bloom’s syndrome. Dermatol Clin. 1995;13:7-18.
- German J, Takebe H. Bloom’s syndrome, XIV: the disorder in Japan. Clin Genet. 1989;35:93-110.
- Bennett RJ, Keck JL. Structure and function of RecQ DNA helicases. Crit Rev Biochem Mol Biol. 2004;39:79-97.
- Reference SNP (refSNP) Cluster Report: rs2227935. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=2227935. Accessed February 3, 2016.
- Reference SNP (refSNP) Cluster Report: rs7167216. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=7167216. Accessed February 3, 2016.
- Homo sapiens:GRCh37.p13 (GCF_000001405.25)Chr 1 (NC_000001.10):1 - 249.3M. National Center for Biotechnology Information website. http://www.ncbi.nlm.nih.gov/variationtools/1000genomes/?=%EF%BC%86=. Accessed February 3, 2016.
Case Report: Hypertension in a Pediatric Patient With Repeat Aortic Coarctation Repair
Introduction
Coarctation of the aorta comprises approximately 5% to 8% of congenital heart defects and is often associated with valvular malformations.1 These defects are typically diagnosed early and are managed with surgical repair, balloon angioplasty, or endovascular stent placement. However, as the following case illustrates, complications can occur in this population despite early intervention.
Case
A 15-year-old male adolescent presented to the pediatric ED after repeated blood pressure (BP) checks by the school nurse revealed consistently elevated systolic and diastolic pressures. The patient’s hypertension was associated with symptoms of intermittent headache and light-headedness. His medical history was remarkable for a congenital aortic coarctation and a bicuspid aortic valve. The patient had undergone a subclavian flap repair prior to 1 month of age, followed by a balloon dilatation 1 year later for recurrent coarctation. The rest of the patient’s medical history was unremarkable, including normal renal function. He denied illicit drug or alcohol use, sexual activity, or trauma.
On evaluation, the patient’s cardiac examination revealed a regular rate and rhythm with normally split S2; there were no rubs, murmurs, or gallops on auscultation. He had normal and equal pulses of the upper and lower extremities bilaterally. The patient presented without cyanosis. He was alert and oriented with normal upper and lower extremity reflexes. The neurological examination, including cranial nerve, strength, and gait testing, was unremarkable. The gastrointestinal examination showed a soft, nondistended abdomen, with no pulsatile masses. There was no abnormal swelling of his extremities. Although the physical examination findings were unremarkable, the patient’s vital signs were concerning as BP in his right upper extremity was as high as 208/110 mm Hg, while BP in his right leg was 130/68 mm Hg.
The patient followed up with his cardiologist, who ordered cardiac magnetic resonance imaging (MRI). The MRI showed mild narrowing of the distal aortic arch with a minimal and clinically insignificant pressure gradient. Based on the MRI findings, the patient was referred to a pediatric nephrologist, who performed a 24-hour ambulatory BP evaluation. The results of this study showed the patient to have systolic hypertension at the 95th percentile for his age and height. Based on the patient’s athletic predilection, β-blockers were avoided, and he was instead started on the angiotensin-converting enzyme (ACE) inhibitor lisinopril, along with annual follow-up cardiac evaluation.
Discussion
The authors’ initial concern for this patient was the possibility of a recurrent coarctation causing a significant pressure gradient between the upper and lower extremities with associated symptoms. A review of the literature demonstrates such an occurrence is not uncommon in this patient population, especially in patients with a history of early intervention (ie, within the first year of life).2
Causes and Incidence
One of the factors believed to contribute to recurrent coarctation is insufficient growth versus retraction of the manipulated tissues over time. The rates of recurrence vary based on the initial technique used for repair. These recurrences have been found to be approximately 6% in patients who had subclavian flap repairs; 31% for those who had balloon angioplasty alone; and approximately 20% in patients who had aortic stenting.3-5 As seen in this case, balloon angioplasty is usually performed in patients requiring revascularization. However, up to 32% of these patients will require further intervention due to subsequent recurrence.6
Evaluation
Although emergency physicians (EPs) have numerous diagnostic modalities available to evaluate patients with suspected aortic coarctation, as long as the patient is in no acute distress, much of the work-up can be performed on an outpatient basis—in conjunction with the primary- and subspecialty-care team. Regarding appropriate imaging modalities, echocardiography with Doppler or 3D reconstruction of MR angiogram can be useful in detecting both anatomical abnormalities as well as the associated gradient dysfunction; computed tomography can be used for assessing the anatomy.7 All of these modalities can also be used to evaluate late-term complications of aortic coarctation pathology, including aortic aneurysms. To help ensure good outcome, the EP should always keep the possibility of recurrence in the differential when evaluating these patients, regardless of the number of previous interventions attempted.
Hypertension
As this case illustrates, patients with a history of coarctation repair often develop high BP. Unfortunately, up to 23% of these patients will go on to have BP above the 95th percentile.5 Moreover, a significant number of patients in this population will also suffer from exercise-induced hypertension, even when at-rest BP is controlled with antihypertensive medications.8
β-blockers, angiotensin-receptor blockers, and ACE inhibitors are considered the first-line medications for hypertension in adults and adult-sized patients with this condition.9
Since a high proportion of patients as young as age 7 years may develop high BP postrepair,10 the EP should discuss the initiation of an antihypertensive agent with the patient’s care team prior to discharge. It is also important to keep in mind that elevated BP is present to a significant degree even in patients without recurrent obstruction. The negative sequelae associated with uncontrolled hypertension is well known, and patients with congenital anatomical anomalies are at higher risk for such negative outcomes.
Conclusion
This case illustrates a common presentation of a teenaged patient with a chronic medical condition due to a corrected congenital cardiac defect. It also demonstrates the unique and early opportunity the EP has to evaluate and provide appropriate intervention for patients with potentially life-threatening diseases.
Patients with a history of corrective vascular surgery due to congenital heart malformations are an at-risk population. Therefore, during evaluation, the EP should always keep in mind that that these patients have a higher prevalence of related abnormalities at earlier ages than the general population. Steps initiated in the ED prior to discharge, in collaboration with the patient’s primary- and specialty-care team, can assist in expediting appropriate outpatient management of any sequelae. If a patient does not have a cardiologist, a referral to one should always be made prior to discharge.
Dr Smith is a postgraduate year 3 resident in the department of emergency medicine at Alpert Medical School of Brown University, Providence, Rhode Island. Dr Merritt is an assistant professor and pediatric emergency medicine attending in the department of emergency medicine, Brown Alpert Medical School, Providence, Rhode Island.
- Hypertension in a Pediatric Patient With Repeat Aortic Coarctation Repair
- Saxena A. Recurrent coarctation: interventional techniques and results. World J Pediatr Congenit Heart Surg. 2015;6(2):257-265.
- Uchytil B, Ceryny J, Nicovsky J, et al. Surgery for coarctation of the aorta: long-term post-operative results. Scripta Medica. 2003;76(6):347-356.
- Jahangiri M, Shinebourne EA, Zurakowski D, Rigby ML, Redington AN, Lincoln C. Subclavian flap angioplasty: does the arch look after itself? J Thorac Cardiovasc Surg. 2000;120(2):224-229.
- Rao PS, Thapar MK, Galal O, Wilson AD. Follow-up results of balloon angioplasty of native coarctation in neonates and infants. Am Heart J. 1990;120(6 Pt 1):1310-1304.
- Holzer R, Qureshi S, Ghasemi A, et al. Stenting of aortic coarctation: acute, intermediate, and long-term results of a prospective multi-institutional registry--Congenital Cardiovascular Interventional Study Consortium (CCISC). Catheter Cardiovasc Interv. 2010;76(4):553-563.
- Yetman AT, Nykanen D, McCrindle BW, et al. Balloon angioplasty of recurrent coarctation: a 12-year review. J Am Coll Cardiol. 1997;30(3):811-816.
- Bashore TM, Granger CB, Jackson KP, Patel MR. Heart disease. In: Current Medical Diagnosis and Treatment 2016. Papadakis MA, McPhee SJ. The McGraw-Hill Companies, Inc: New York; 2010:322,323
- Correia AS, Gonçalves A, Paiva M, et al. Long-term follow-up after aortic coarctation repair: the unsolved issue of exercise-induced hypertension. Rev Port Cardiol. 2013;32(11):879-883.
- Warnes CA, Williams RG, Bashore TM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease. Circulation. 2008;111(23):e766,e767. Available at: http://circ.ahajournals.org/content/118/23/e714.full.pdf. Accessed January 12, 2016.
- O’Sullivan JJ, Derrick G, Darnell R. Prevalence of hypertension in children after early repair of coarctation of the aorta: a cohort study using casual and 24 hour blood pressure measurement. Heart. 2002;88(2):163-166.
Introduction
Coarctation of the aorta comprises approximately 5% to 8% of congenital heart defects and is often associated with valvular malformations.1 These defects are typically diagnosed early and are managed with surgical repair, balloon angioplasty, or endovascular stent placement. However, as the following case illustrates, complications can occur in this population despite early intervention.
Case
A 15-year-old male adolescent presented to the pediatric ED after repeated blood pressure (BP) checks by the school nurse revealed consistently elevated systolic and diastolic pressures. The patient’s hypertension was associated with symptoms of intermittent headache and light-headedness. His medical history was remarkable for a congenital aortic coarctation and a bicuspid aortic valve. The patient had undergone a subclavian flap repair prior to 1 month of age, followed by a balloon dilatation 1 year later for recurrent coarctation. The rest of the patient’s medical history was unremarkable, including normal renal function. He denied illicit drug or alcohol use, sexual activity, or trauma.
On evaluation, the patient’s cardiac examination revealed a regular rate and rhythm with normally split S2; there were no rubs, murmurs, or gallops on auscultation. He had normal and equal pulses of the upper and lower extremities bilaterally. The patient presented without cyanosis. He was alert and oriented with normal upper and lower extremity reflexes. The neurological examination, including cranial nerve, strength, and gait testing, was unremarkable. The gastrointestinal examination showed a soft, nondistended abdomen, with no pulsatile masses. There was no abnormal swelling of his extremities. Although the physical examination findings were unremarkable, the patient’s vital signs were concerning as BP in his right upper extremity was as high as 208/110 mm Hg, while BP in his right leg was 130/68 mm Hg.
The patient followed up with his cardiologist, who ordered cardiac magnetic resonance imaging (MRI). The MRI showed mild narrowing of the distal aortic arch with a minimal and clinically insignificant pressure gradient. Based on the MRI findings, the patient was referred to a pediatric nephrologist, who performed a 24-hour ambulatory BP evaluation. The results of this study showed the patient to have systolic hypertension at the 95th percentile for his age and height. Based on the patient’s athletic predilection, β-blockers were avoided, and he was instead started on the angiotensin-converting enzyme (ACE) inhibitor lisinopril, along with annual follow-up cardiac evaluation.
Discussion
The authors’ initial concern for this patient was the possibility of a recurrent coarctation causing a significant pressure gradient between the upper and lower extremities with associated symptoms. A review of the literature demonstrates such an occurrence is not uncommon in this patient population, especially in patients with a history of early intervention (ie, within the first year of life).2
Causes and Incidence
One of the factors believed to contribute to recurrent coarctation is insufficient growth versus retraction of the manipulated tissues over time. The rates of recurrence vary based on the initial technique used for repair. These recurrences have been found to be approximately 6% in patients who had subclavian flap repairs; 31% for those who had balloon angioplasty alone; and approximately 20% in patients who had aortic stenting.3-5 As seen in this case, balloon angioplasty is usually performed in patients requiring revascularization. However, up to 32% of these patients will require further intervention due to subsequent recurrence.6
Evaluation
Although emergency physicians (EPs) have numerous diagnostic modalities available to evaluate patients with suspected aortic coarctation, as long as the patient is in no acute distress, much of the work-up can be performed on an outpatient basis—in conjunction with the primary- and subspecialty-care team. Regarding appropriate imaging modalities, echocardiography with Doppler or 3D reconstruction of MR angiogram can be useful in detecting both anatomical abnormalities as well as the associated gradient dysfunction; computed tomography can be used for assessing the anatomy.7 All of these modalities can also be used to evaluate late-term complications of aortic coarctation pathology, including aortic aneurysms. To help ensure good outcome, the EP should always keep the possibility of recurrence in the differential when evaluating these patients, regardless of the number of previous interventions attempted.
Hypertension
As this case illustrates, patients with a history of coarctation repair often develop high BP. Unfortunately, up to 23% of these patients will go on to have BP above the 95th percentile.5 Moreover, a significant number of patients in this population will also suffer from exercise-induced hypertension, even when at-rest BP is controlled with antihypertensive medications.8
β-blockers, angiotensin-receptor blockers, and ACE inhibitors are considered the first-line medications for hypertension in adults and adult-sized patients with this condition.9
Since a high proportion of patients as young as age 7 years may develop high BP postrepair,10 the EP should discuss the initiation of an antihypertensive agent with the patient’s care team prior to discharge. It is also important to keep in mind that elevated BP is present to a significant degree even in patients without recurrent obstruction. The negative sequelae associated with uncontrolled hypertension is well known, and patients with congenital anatomical anomalies are at higher risk for such negative outcomes.
Conclusion
This case illustrates a common presentation of a teenaged patient with a chronic medical condition due to a corrected congenital cardiac defect. It also demonstrates the unique and early opportunity the EP has to evaluate and provide appropriate intervention for patients with potentially life-threatening diseases.
Patients with a history of corrective vascular surgery due to congenital heart malformations are an at-risk population. Therefore, during evaluation, the EP should always keep in mind that that these patients have a higher prevalence of related abnormalities at earlier ages than the general population. Steps initiated in the ED prior to discharge, in collaboration with the patient’s primary- and specialty-care team, can assist in expediting appropriate outpatient management of any sequelae. If a patient does not have a cardiologist, a referral to one should always be made prior to discharge.
Dr Smith is a postgraduate year 3 resident in the department of emergency medicine at Alpert Medical School of Brown University, Providence, Rhode Island. Dr Merritt is an assistant professor and pediatric emergency medicine attending in the department of emergency medicine, Brown Alpert Medical School, Providence, Rhode Island.
Introduction
Coarctation of the aorta comprises approximately 5% to 8% of congenital heart defects and is often associated with valvular malformations.1 These defects are typically diagnosed early and are managed with surgical repair, balloon angioplasty, or endovascular stent placement. However, as the following case illustrates, complications can occur in this population despite early intervention.
Case
A 15-year-old male adolescent presented to the pediatric ED after repeated blood pressure (BP) checks by the school nurse revealed consistently elevated systolic and diastolic pressures. The patient’s hypertension was associated with symptoms of intermittent headache and light-headedness. His medical history was remarkable for a congenital aortic coarctation and a bicuspid aortic valve. The patient had undergone a subclavian flap repair prior to 1 month of age, followed by a balloon dilatation 1 year later for recurrent coarctation. The rest of the patient’s medical history was unremarkable, including normal renal function. He denied illicit drug or alcohol use, sexual activity, or trauma.
On evaluation, the patient’s cardiac examination revealed a regular rate and rhythm with normally split S2; there were no rubs, murmurs, or gallops on auscultation. He had normal and equal pulses of the upper and lower extremities bilaterally. The patient presented without cyanosis. He was alert and oriented with normal upper and lower extremity reflexes. The neurological examination, including cranial nerve, strength, and gait testing, was unremarkable. The gastrointestinal examination showed a soft, nondistended abdomen, with no pulsatile masses. There was no abnormal swelling of his extremities. Although the physical examination findings were unremarkable, the patient’s vital signs were concerning as BP in his right upper extremity was as high as 208/110 mm Hg, while BP in his right leg was 130/68 mm Hg.
The patient followed up with his cardiologist, who ordered cardiac magnetic resonance imaging (MRI). The MRI showed mild narrowing of the distal aortic arch with a minimal and clinically insignificant pressure gradient. Based on the MRI findings, the patient was referred to a pediatric nephrologist, who performed a 24-hour ambulatory BP evaluation. The results of this study showed the patient to have systolic hypertension at the 95th percentile for his age and height. Based on the patient’s athletic predilection, β-blockers were avoided, and he was instead started on the angiotensin-converting enzyme (ACE) inhibitor lisinopril, along with annual follow-up cardiac evaluation.
Discussion
The authors’ initial concern for this patient was the possibility of a recurrent coarctation causing a significant pressure gradient between the upper and lower extremities with associated symptoms. A review of the literature demonstrates such an occurrence is not uncommon in this patient population, especially in patients with a history of early intervention (ie, within the first year of life).2
Causes and Incidence
One of the factors believed to contribute to recurrent coarctation is insufficient growth versus retraction of the manipulated tissues over time. The rates of recurrence vary based on the initial technique used for repair. These recurrences have been found to be approximately 6% in patients who had subclavian flap repairs; 31% for those who had balloon angioplasty alone; and approximately 20% in patients who had aortic stenting.3-5 As seen in this case, balloon angioplasty is usually performed in patients requiring revascularization. However, up to 32% of these patients will require further intervention due to subsequent recurrence.6
Evaluation
Although emergency physicians (EPs) have numerous diagnostic modalities available to evaluate patients with suspected aortic coarctation, as long as the patient is in no acute distress, much of the work-up can be performed on an outpatient basis—in conjunction with the primary- and subspecialty-care team. Regarding appropriate imaging modalities, echocardiography with Doppler or 3D reconstruction of MR angiogram can be useful in detecting both anatomical abnormalities as well as the associated gradient dysfunction; computed tomography can be used for assessing the anatomy.7 All of these modalities can also be used to evaluate late-term complications of aortic coarctation pathology, including aortic aneurysms. To help ensure good outcome, the EP should always keep the possibility of recurrence in the differential when evaluating these patients, regardless of the number of previous interventions attempted.
Hypertension
As this case illustrates, patients with a history of coarctation repair often develop high BP. Unfortunately, up to 23% of these patients will go on to have BP above the 95th percentile.5 Moreover, a significant number of patients in this population will also suffer from exercise-induced hypertension, even when at-rest BP is controlled with antihypertensive medications.8
β-blockers, angiotensin-receptor blockers, and ACE inhibitors are considered the first-line medications for hypertension in adults and adult-sized patients with this condition.9
Since a high proportion of patients as young as age 7 years may develop high BP postrepair,10 the EP should discuss the initiation of an antihypertensive agent with the patient’s care team prior to discharge. It is also important to keep in mind that elevated BP is present to a significant degree even in patients without recurrent obstruction. The negative sequelae associated with uncontrolled hypertension is well known, and patients with congenital anatomical anomalies are at higher risk for such negative outcomes.
Conclusion
This case illustrates a common presentation of a teenaged patient with a chronic medical condition due to a corrected congenital cardiac defect. It also demonstrates the unique and early opportunity the EP has to evaluate and provide appropriate intervention for patients with potentially life-threatening diseases.
Patients with a history of corrective vascular surgery due to congenital heart malformations are an at-risk population. Therefore, during evaluation, the EP should always keep in mind that that these patients have a higher prevalence of related abnormalities at earlier ages than the general population. Steps initiated in the ED prior to discharge, in collaboration with the patient’s primary- and specialty-care team, can assist in expediting appropriate outpatient management of any sequelae. If a patient does not have a cardiologist, a referral to one should always be made prior to discharge.
Dr Smith is a postgraduate year 3 resident in the department of emergency medicine at Alpert Medical School of Brown University, Providence, Rhode Island. Dr Merritt is an assistant professor and pediatric emergency medicine attending in the department of emergency medicine, Brown Alpert Medical School, Providence, Rhode Island.
- Hypertension in a Pediatric Patient With Repeat Aortic Coarctation Repair
- Saxena A. Recurrent coarctation: interventional techniques and results. World J Pediatr Congenit Heart Surg. 2015;6(2):257-265.
- Uchytil B, Ceryny J, Nicovsky J, et al. Surgery for coarctation of the aorta: long-term post-operative results. Scripta Medica. 2003;76(6):347-356.
- Jahangiri M, Shinebourne EA, Zurakowski D, Rigby ML, Redington AN, Lincoln C. Subclavian flap angioplasty: does the arch look after itself? J Thorac Cardiovasc Surg. 2000;120(2):224-229.
- Rao PS, Thapar MK, Galal O, Wilson AD. Follow-up results of balloon angioplasty of native coarctation in neonates and infants. Am Heart J. 1990;120(6 Pt 1):1310-1304.
- Holzer R, Qureshi S, Ghasemi A, et al. Stenting of aortic coarctation: acute, intermediate, and long-term results of a prospective multi-institutional registry--Congenital Cardiovascular Interventional Study Consortium (CCISC). Catheter Cardiovasc Interv. 2010;76(4):553-563.
- Yetman AT, Nykanen D, McCrindle BW, et al. Balloon angioplasty of recurrent coarctation: a 12-year review. J Am Coll Cardiol. 1997;30(3):811-816.
- Bashore TM, Granger CB, Jackson KP, Patel MR. Heart disease. In: Current Medical Diagnosis and Treatment 2016. Papadakis MA, McPhee SJ. The McGraw-Hill Companies, Inc: New York; 2010:322,323
- Correia AS, Gonçalves A, Paiva M, et al. Long-term follow-up after aortic coarctation repair: the unsolved issue of exercise-induced hypertension. Rev Port Cardiol. 2013;32(11):879-883.
- Warnes CA, Williams RG, Bashore TM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease. Circulation. 2008;111(23):e766,e767. Available at: http://circ.ahajournals.org/content/118/23/e714.full.pdf. Accessed January 12, 2016.
- O’Sullivan JJ, Derrick G, Darnell R. Prevalence of hypertension in children after early repair of coarctation of the aorta: a cohort study using casual and 24 hour blood pressure measurement. Heart. 2002;88(2):163-166.
- Hypertension in a Pediatric Patient With Repeat Aortic Coarctation Repair
- Saxena A. Recurrent coarctation: interventional techniques and results. World J Pediatr Congenit Heart Surg. 2015;6(2):257-265.
- Uchytil B, Ceryny J, Nicovsky J, et al. Surgery for coarctation of the aorta: long-term post-operative results. Scripta Medica. 2003;76(6):347-356.
- Jahangiri M, Shinebourne EA, Zurakowski D, Rigby ML, Redington AN, Lincoln C. Subclavian flap angioplasty: does the arch look after itself? J Thorac Cardiovasc Surg. 2000;120(2):224-229.
- Rao PS, Thapar MK, Galal O, Wilson AD. Follow-up results of balloon angioplasty of native coarctation in neonates and infants. Am Heart J. 1990;120(6 Pt 1):1310-1304.
- Holzer R, Qureshi S, Ghasemi A, et al. Stenting of aortic coarctation: acute, intermediate, and long-term results of a prospective multi-institutional registry--Congenital Cardiovascular Interventional Study Consortium (CCISC). Catheter Cardiovasc Interv. 2010;76(4):553-563.
- Yetman AT, Nykanen D, McCrindle BW, et al. Balloon angioplasty of recurrent coarctation: a 12-year review. J Am Coll Cardiol. 1997;30(3):811-816.
- Bashore TM, Granger CB, Jackson KP, Patel MR. Heart disease. In: Current Medical Diagnosis and Treatment 2016. Papadakis MA, McPhee SJ. The McGraw-Hill Companies, Inc: New York; 2010:322,323
- Correia AS, Gonçalves A, Paiva M, et al. Long-term follow-up after aortic coarctation repair: the unsolved issue of exercise-induced hypertension. Rev Port Cardiol. 2013;32(11):879-883.
- Warnes CA, Williams RG, Bashore TM, et al; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. ACC/AHA 2008 guidelines for the management of adults with congenital heart disease. Circulation. 2008;111(23):e766,e767. Available at: http://circ.ahajournals.org/content/118/23/e714.full.pdf. Accessed January 12, 2016.
- O’Sullivan JJ, Derrick G, Darnell R. Prevalence of hypertension in children after early repair of coarctation of the aorta: a cohort study using casual and 24 hour blood pressure measurement. Heart. 2002;88(2):163-166.
Case Studies In Toxicology: Withdrawal: Another Danger of Diversion
Case
A 34-year-old man with a history of polysubstance abuse presented to the ED after he had a seizure during his regular methadone-treatment program meeting. While at the clinic, attendees witnessed the patient experience a loss of consciousness accompanied by generalized shaking movements of his extremities, which lasted for several minutes.
Upon arrival in the ED, the patient stated that he had a mild headache; he was otherwise asymptomatic. Initial vital signs were: blood pressure, 126/80 mm Hg; heart rate, 82 beats/minute; respiratory rate, 16 breaths/minute; and temperature, 97.3°F. Oxygen saturation was 98% on room air, and a finger-stick glucose test was 140 mg/dL.
Physical examination revealed a small right-sided parietal hematoma. The patient had no tremors and his neurological examination, including mental status, was normal. When reviewing the patient’s medical history and medications in the health record, it was noted that the patient had a prescription for alprazolam for an anxiety disorder. On further questioning, the patient admitted that he had sold his last alprazolam prescription and had not been taking the drug for the past week.
What characterizes the benzodiazepine withdrawal syndrome?

Although introduced into clinical practice in the 1960s, the potential for dependence and a withdrawal syndrome was not appreciated until the early 1980s. This clinical syndrome can manifest with a wide variety of findings, most commonly with what are termed “rebound effects” or “rebound hyperexcitability.” These effects include anxiety, insomnia or sleep disturbance, tremulousness, irritability, sweating, psychomotor agitation, difficulty in concentration, nausea, weight loss, palpitations, headache, muscular pain and stiffness, or generalized weakness.2 More severe manifestations include delirium, seizures, or psychosis. Often, these symptoms and signs may be confused with the very manifestations that prompted the initial use of the BZD, a reemergence of which can exacerbate the withdrawal syndrome.
When does benzodiazepine withdrawal occur?
The exact time course of BZD withdrawal can vary considerably and, unlike alcohol withdrawal (which occurs from a single compound, ethanol), can be difficult to characterize. The onset of withdrawal symptoms is dependent on a number of factors, including the half-life of the BZD involved. For example, delayed onset withdrawal symptoms of up to 3 weeks after cessation of the medication are described with long-acting BZDs such as chlordiazepoxide and diazepam. Conversely, symptoms may present as early as 24 to 48 hours after abrupt termination of BZDs with shorter half-lives, alprazolam and lorazepam. This variable time of onset differs considerably from other withdrawal syndromes, notably ethanol withdrawal. While both syndromes correlate to the individual patient’s severity of dependence, alcohol withdrawal follows a more predictable time course.
Some authors distinguish a rebound syndrome from a true withdrawal syndrome, the former of which is self-limited in nature and the result of cessation of treatment for the primary disease process. In this model, rebound symptoms begin 1 to 4 days after the abrupt cessation or dose reduction of the BZD, and are relatively short-lived, lasting 2 to 3 days.2
What is the appropriate treatment for benzodiazepine withdrawal?
The standard therapy for almost all withdrawal syndromes is reinstitution of the causal agent. A number of non-BZD-based treatment strategies have been investigated, and all have met with limited success. Of these, anticonvulsant drugs such as carbamazepine and valproic acid were initially considered promising based on case reports and small case series.4 These medications ultimately proved ineffective in randomized, placebo-controlled studies.5 β-Adrenergic antagonists, such as propranolol, have been studied as a method to normalize a patient’s vital signs but also proved nonbeneficial in managing withdrawal.5,6
The safest and most effective management approach for patients with BZD withdrawal is reinstitution of the BZD followed by a prolonged and gradual tapering until cessation, if that is desired.1,2,5,6 While all BZDs share structural and mechanistic similarities, there are subtle variations within this class that can affect their pharmacologic effects. These structural differences may result in incomplete cross-tolerance, which may lead to inadequate mitigation of the withdrawal syndrome. For example, previous reports suggest that alprazolam and clonazepam are structurally unique and bind to the BZD receptor with higher affinity than other BZDs. Therefore, while in general any BZD can be used to treat withdrawal from another BZD, it is recommended to treat withdrawal from these two agents with the implicated BZD.
There are, however, limitations to this approach. Namely, some BZDs are only available in oral formulations (eg, alprazolam and clonazepam) or the BZD of choice may not be readily available or on formulary within a given institution. In a patient with a severe withdrawal syndrome where it is not feasible or potentially harmful to administer an oral medication, it is reasonable to provide parenteral (preferably intravenous [IV]) BZD therapy. The optimal approach is to start with a small “standard” dose and titrate to effect while monitoring for adverse effects (eg, oversedation, ventilatory depression). Redosing should be triggered by symptoms or signs, and not performed in a timed or standing-order fashion. If this approach proves ineffective and withdrawal symptoms persist despite adequate BZD therapy, a direct GABA agonist such as propofol is a sensible alternative or adjuvant treatment. This may sound similar to the management of patients with ethanol withdrawal; indeed, this approach is essentially the same, with the exception of the more drawn-out time course.
Case Conclusion
After arrival in the ED, the patient received diazepam 10 mg IV and was subsequently admitted to the hospital for further evaluation. During his hospitalization, the patient was re-started on his usual dose of oral alprazolam. No further withdrawal syndrome was observed, and he was discharged on hospital day 2 with a plan to slowly taper his alprazolam dose with his outpatient psychiatrist.
Dr Repplinger is a senior medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Withdrawal: Another Danger of Diversion
- Marriott S, Tyrer P. Benzodiazepine dependence. Avoidance and withdrawal. Drug Saf. 1993;9(2):93-103.
- Pétursson H. The benzodiazepine withdrawal syndrome. Addiction. 1994;89(11):1455-1459.
- Authier N, Balayssac D, Sautereau M, et al. Benzodiazepine dependence: focus on withdrawal syndrome. Ann Pharm Fr. 2009;67(6):408-413.
- Pages KP, Ries RK. Use of anticonvulsants in benzodiazepine withdrawal. Am J Addict. 1998;7(3):198-204.
- Ashton H. The treatment of benzodiazepine dependence. Addiction. 1994;89(11):1535-1541.
- Parr JM, Kavanagh DJ, Cahill L, Mitchell G, McD Young R. Effectiveness of current treatment approaches for benzodiazepine discontinuation: a meta-analysis. Addiction. 2009;104(1):13-24.
Case
A 34-year-old man with a history of polysubstance abuse presented to the ED after he had a seizure during his regular methadone-treatment program meeting. While at the clinic, attendees witnessed the patient experience a loss of consciousness accompanied by generalized shaking movements of his extremities, which lasted for several minutes.
Upon arrival in the ED, the patient stated that he had a mild headache; he was otherwise asymptomatic. Initial vital signs were: blood pressure, 126/80 mm Hg; heart rate, 82 beats/minute; respiratory rate, 16 breaths/minute; and temperature, 97.3°F. Oxygen saturation was 98% on room air, and a finger-stick glucose test was 140 mg/dL.
Physical examination revealed a small right-sided parietal hematoma. The patient had no tremors and his neurological examination, including mental status, was normal. When reviewing the patient’s medical history and medications in the health record, it was noted that the patient had a prescription for alprazolam for an anxiety disorder. On further questioning, the patient admitted that he had sold his last alprazolam prescription and had not been taking the drug for the past week.
What characterizes the benzodiazepine withdrawal syndrome?
Although introduced into clinical practice in the 1960s, the potential for dependence and a withdrawal syndrome was not appreciated until the early 1980s. This clinical syndrome can manifest with a wide variety of findings, most commonly with what are termed “rebound effects” or “rebound hyperexcitability.” These effects include anxiety, insomnia or sleep disturbance, tremulousness, irritability, sweating, psychomotor agitation, difficulty in concentration, nausea, weight loss, palpitations, headache, muscular pain and stiffness, or generalized weakness.2 More severe manifestations include delirium, seizures, or psychosis. Often, these symptoms and signs may be confused with the very manifestations that prompted the initial use of the BZD, a reemergence of which can exacerbate the withdrawal syndrome.
When does benzodiazepine withdrawal occur?
The exact time course of BZD withdrawal can vary considerably and, unlike alcohol withdrawal (which occurs from a single compound, ethanol), can be difficult to characterize. The onset of withdrawal symptoms is dependent on a number of factors, including the half-life of the BZD involved. For example, delayed onset withdrawal symptoms of up to 3 weeks after cessation of the medication are described with long-acting BZDs such as chlordiazepoxide and diazepam. Conversely, symptoms may present as early as 24 to 48 hours after abrupt termination of BZDs with shorter half-lives, alprazolam and lorazepam. This variable time of onset differs considerably from other withdrawal syndromes, notably ethanol withdrawal. While both syndromes correlate to the individual patient’s severity of dependence, alcohol withdrawal follows a more predictable time course.
Some authors distinguish a rebound syndrome from a true withdrawal syndrome, the former of which is self-limited in nature and the result of cessation of treatment for the primary disease process. In this model, rebound symptoms begin 1 to 4 days after the abrupt cessation or dose reduction of the BZD, and are relatively short-lived, lasting 2 to 3 days.2
What is the appropriate treatment for benzodiazepine withdrawal?
The standard therapy for almost all withdrawal syndromes is reinstitution of the causal agent. A number of non-BZD-based treatment strategies have been investigated, and all have met with limited success. Of these, anticonvulsant drugs such as carbamazepine and valproic acid were initially considered promising based on case reports and small case series.4 These medications ultimately proved ineffective in randomized, placebo-controlled studies.5 β-Adrenergic antagonists, such as propranolol, have been studied as a method to normalize a patient’s vital signs but also proved nonbeneficial in managing withdrawal.5,6
The safest and most effective management approach for patients with BZD withdrawal is reinstitution of the BZD followed by a prolonged and gradual tapering until cessation, if that is desired.1,2,5,6 While all BZDs share structural and mechanistic similarities, there are subtle variations within this class that can affect their pharmacologic effects. These structural differences may result in incomplete cross-tolerance, which may lead to inadequate mitigation of the withdrawal syndrome. For example, previous reports suggest that alprazolam and clonazepam are structurally unique and bind to the BZD receptor with higher affinity than other BZDs. Therefore, while in general any BZD can be used to treat withdrawal from another BZD, it is recommended to treat withdrawal from these two agents with the implicated BZD.
There are, however, limitations to this approach. Namely, some BZDs are only available in oral formulations (eg, alprazolam and clonazepam) or the BZD of choice may not be readily available or on formulary within a given institution. In a patient with a severe withdrawal syndrome where it is not feasible or potentially harmful to administer an oral medication, it is reasonable to provide parenteral (preferably intravenous [IV]) BZD therapy. The optimal approach is to start with a small “standard” dose and titrate to effect while monitoring for adverse effects (eg, oversedation, ventilatory depression). Redosing should be triggered by symptoms or signs, and not performed in a timed or standing-order fashion. If this approach proves ineffective and withdrawal symptoms persist despite adequate BZD therapy, a direct GABA agonist such as propofol is a sensible alternative or adjuvant treatment. This may sound similar to the management of patients with ethanol withdrawal; indeed, this approach is essentially the same, with the exception of the more drawn-out time course.
Case Conclusion
After arrival in the ED, the patient received diazepam 10 mg IV and was subsequently admitted to the hospital for further evaluation. During his hospitalization, the patient was re-started on his usual dose of oral alprazolam. No further withdrawal syndrome was observed, and he was discharged on hospital day 2 with a plan to slowly taper his alprazolam dose with his outpatient psychiatrist.
Dr Repplinger is a senior medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 34-year-old man with a history of polysubstance abuse presented to the ED after he had a seizure during his regular methadone-treatment program meeting. While at the clinic, attendees witnessed the patient experience a loss of consciousness accompanied by generalized shaking movements of his extremities, which lasted for several minutes.
Upon arrival in the ED, the patient stated that he had a mild headache; he was otherwise asymptomatic. Initial vital signs were: blood pressure, 126/80 mm Hg; heart rate, 82 beats/minute; respiratory rate, 16 breaths/minute; and temperature, 97.3°F. Oxygen saturation was 98% on room air, and a finger-stick glucose test was 140 mg/dL.
Physical examination revealed a small right-sided parietal hematoma. The patient had no tremors and his neurological examination, including mental status, was normal. When reviewing the patient’s medical history and medications in the health record, it was noted that the patient had a prescription for alprazolam for an anxiety disorder. On further questioning, the patient admitted that he had sold his last alprazolam prescription and had not been taking the drug for the past week.
What characterizes the benzodiazepine withdrawal syndrome?
Although introduced into clinical practice in the 1960s, the potential for dependence and a withdrawal syndrome was not appreciated until the early 1980s. This clinical syndrome can manifest with a wide variety of findings, most commonly with what are termed “rebound effects” or “rebound hyperexcitability.” These effects include anxiety, insomnia or sleep disturbance, tremulousness, irritability, sweating, psychomotor agitation, difficulty in concentration, nausea, weight loss, palpitations, headache, muscular pain and stiffness, or generalized weakness.2 More severe manifestations include delirium, seizures, or psychosis. Often, these symptoms and signs may be confused with the very manifestations that prompted the initial use of the BZD, a reemergence of which can exacerbate the withdrawal syndrome.
When does benzodiazepine withdrawal occur?
The exact time course of BZD withdrawal can vary considerably and, unlike alcohol withdrawal (which occurs from a single compound, ethanol), can be difficult to characterize. The onset of withdrawal symptoms is dependent on a number of factors, including the half-life of the BZD involved. For example, delayed onset withdrawal symptoms of up to 3 weeks after cessation of the medication are described with long-acting BZDs such as chlordiazepoxide and diazepam. Conversely, symptoms may present as early as 24 to 48 hours after abrupt termination of BZDs with shorter half-lives, alprazolam and lorazepam. This variable time of onset differs considerably from other withdrawal syndromes, notably ethanol withdrawal. While both syndromes correlate to the individual patient’s severity of dependence, alcohol withdrawal follows a more predictable time course.
Some authors distinguish a rebound syndrome from a true withdrawal syndrome, the former of which is self-limited in nature and the result of cessation of treatment for the primary disease process. In this model, rebound symptoms begin 1 to 4 days after the abrupt cessation or dose reduction of the BZD, and are relatively short-lived, lasting 2 to 3 days.2
What is the appropriate treatment for benzodiazepine withdrawal?
The standard therapy for almost all withdrawal syndromes is reinstitution of the causal agent. A number of non-BZD-based treatment strategies have been investigated, and all have met with limited success. Of these, anticonvulsant drugs such as carbamazepine and valproic acid were initially considered promising based on case reports and small case series.4 These medications ultimately proved ineffective in randomized, placebo-controlled studies.5 β-Adrenergic antagonists, such as propranolol, have been studied as a method to normalize a patient’s vital signs but also proved nonbeneficial in managing withdrawal.5,6
The safest and most effective management approach for patients with BZD withdrawal is reinstitution of the BZD followed by a prolonged and gradual tapering until cessation, if that is desired.1,2,5,6 While all BZDs share structural and mechanistic similarities, there are subtle variations within this class that can affect their pharmacologic effects. These structural differences may result in incomplete cross-tolerance, which may lead to inadequate mitigation of the withdrawal syndrome. For example, previous reports suggest that alprazolam and clonazepam are structurally unique and bind to the BZD receptor with higher affinity than other BZDs. Therefore, while in general any BZD can be used to treat withdrawal from another BZD, it is recommended to treat withdrawal from these two agents with the implicated BZD.
There are, however, limitations to this approach. Namely, some BZDs are only available in oral formulations (eg, alprazolam and clonazepam) or the BZD of choice may not be readily available or on formulary within a given institution. In a patient with a severe withdrawal syndrome where it is not feasible or potentially harmful to administer an oral medication, it is reasonable to provide parenteral (preferably intravenous [IV]) BZD therapy. The optimal approach is to start with a small “standard” dose and titrate to effect while monitoring for adverse effects (eg, oversedation, ventilatory depression). Redosing should be triggered by symptoms or signs, and not performed in a timed or standing-order fashion. If this approach proves ineffective and withdrawal symptoms persist despite adequate BZD therapy, a direct GABA agonist such as propofol is a sensible alternative or adjuvant treatment. This may sound similar to the management of patients with ethanol withdrawal; indeed, this approach is essentially the same, with the exception of the more drawn-out time course.
Case Conclusion
After arrival in the ED, the patient received diazepam 10 mg IV and was subsequently admitted to the hospital for further evaluation. During his hospitalization, the patient was re-started on his usual dose of oral alprazolam. No further withdrawal syndrome was observed, and he was discharged on hospital day 2 with a plan to slowly taper his alprazolam dose with his outpatient psychiatrist.
Dr Repplinger is a senior medical toxicology fellow in the department of emergency medicine at New York University Langone Medical Center. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Withdrawal: Another Danger of Diversion
- Marriott S, Tyrer P. Benzodiazepine dependence. Avoidance and withdrawal. Drug Saf. 1993;9(2):93-103.
- Pétursson H. The benzodiazepine withdrawal syndrome. Addiction. 1994;89(11):1455-1459.
- Authier N, Balayssac D, Sautereau M, et al. Benzodiazepine dependence: focus on withdrawal syndrome. Ann Pharm Fr. 2009;67(6):408-413.
- Pages KP, Ries RK. Use of anticonvulsants in benzodiazepine withdrawal. Am J Addict. 1998;7(3):198-204.
- Ashton H. The treatment of benzodiazepine dependence. Addiction. 1994;89(11):1535-1541.
- Parr JM, Kavanagh DJ, Cahill L, Mitchell G, McD Young R. Effectiveness of current treatment approaches for benzodiazepine discontinuation: a meta-analysis. Addiction. 2009;104(1):13-24.
- Withdrawal: Another Danger of Diversion
- Marriott S, Tyrer P. Benzodiazepine dependence. Avoidance and withdrawal. Drug Saf. 1993;9(2):93-103.
- Pétursson H. The benzodiazepine withdrawal syndrome. Addiction. 1994;89(11):1455-1459.
- Authier N, Balayssac D, Sautereau M, et al. Benzodiazepine dependence: focus on withdrawal syndrome. Ann Pharm Fr. 2009;67(6):408-413.
- Pages KP, Ries RK. Use of anticonvulsants in benzodiazepine withdrawal. Am J Addict. 1998;7(3):198-204.
- Ashton H. The treatment of benzodiazepine dependence. Addiction. 1994;89(11):1535-1541.
- Parr JM, Kavanagh DJ, Cahill L, Mitchell G, McD Young R. Effectiveness of current treatment approaches for benzodiazepine discontinuation: a meta-analysis. Addiction. 2009;104(1):13-24.

Knee pain • no popping • no previous trauma • Dx?
THE CASE
A 36-year-old man sought care at our family medicine clinic for knee pain that he’d had for the past year. He denied any previous injury or trauma to the knee. The pain affected the posterolateral left knee and was aggravated by squatting and deep flexion. Daily activities did not bother him, but skiing, golfing, mountain biking, and lifting weights worsened the pain. His pain had gradually become more severe and frequent. He denied any mechanical symptoms such as catching, popping, or locking.
Examination of his left knee demonstrated range of motion from 0 to 120 degrees; further flexion caused significant pain. McMurray and Thessaly tests were positive for posterolateral pain, particularly with knee flexion >120 degrees. Physical examination was otherwise unremarkable. Standard x-rays of the left knee were normal. Our patient completed a month of physical therapy, but his symptoms did not improve.
THE DIAGNOSIS
After the patient completed physical therapy, magnetic resonance imaging (MRI) was performed. The MRI did not reveal any left knee effusion, and the menisci, collateral ligaments, and cartilage surfaces were normal. And, while the cruciate ligaments were intact, a large pericruciate ganglion cyst was noted (FIGURES 1 AND 2).
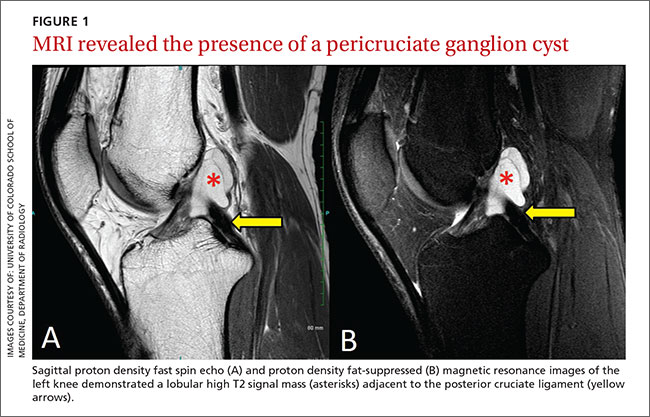
DISCUSSION
Ganglion cysts are dense, encapsulated structures filled with clear viscous fluid that often arise adjacent to tendon sheaths or joint capsules, most commonly over the dorsum of the hand.1 Intra-articular ganglia involving the cruciate ligaments of the knee are relatively uncommon.2 The estimated prevalence of cruciate ligament ganglion cysts at arthroscopy is 0.2% to 1.9%; similar rates have been demonstrated with MRI.3-6 There are more reported cases of these cysts involving the anterior cruciate ligament (ACL) compared to those affecting the posterior cruciate ligament (PCL).2,6
Classification of these cysts is based on relative location with respect to the ligaments. Type 1 cysts originate anterior to the ACL; type 2, between the ACL and PCL; and type 3, posterior to the PCL.6,7 Cruciate ligament ganglion cysts are more common in men, are typically discovered between age 20 and 40, and are usually incidental findings.8
The pathogenesis of ganglion cyst formation is unknown.1,6,7 The most widely accepted theory is that ganglion cysts result from mucinous degeneration of connective tissue in areas of repetitive stress.1,6,7 Other theories suggest hyaluronic acid production secondary to mesenchymal stem cell proliferation within the ligaments, synovial tissue herniation, or congenital translocation of synovial tissue as possible etiologies.2,6,7
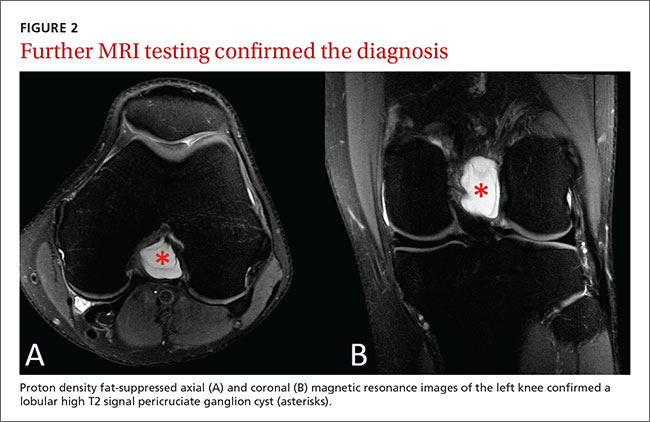
Concurrent pathologies such as meniscal tears or chondral lesions may also be present; however, there is some disagreement as to what role, if any, antecedent trauma has in the pathogenesis of cyst formation.1,6 Several investigators have suggested that prior knee trauma is a likely risk factor.2,8,9
In most patients, cruciate ligament ganglion cysts are asymptomatic.7 The most common presenting symptom is nonspecific pain that is exacerbated by activity, such as stair climbing, squatting, or other activities that require extreme flexion or extension of the knee.6,9 Other possible symptoms include limited range of motion (extension block with ACL involvement, limited flexion with PCL lesions), a catching or locking sensation, instability, or joint line tenderness.5,6 A palpable mass on physical exam is not usually present.6 Some investigators suggest that larger lesions and those closer to the femoral ligamentous attachments are more likely to cause symptoms.5
Cruciate ligament ganglion cysts can be an easily overlooked source of a patient’s symptoms because they often mimic more common pathologies.2 The differential diagnosis of cruciate ligament ganglion cysts and posterior knee pain includes any other intra-articular cysts (eg, meniscal cysts), posterior meniscal tear, popliteus tendinopathy, or neoplasms (eg, hemangioma and synovial sarcoma).2,6
MRI is the best method of diagnosis
Because the symptoms of cruciate ligament ganglion cysts are variable and nonspecific, the diagnosis is rarely made on clinical grounds alone.1 The best method of evaluating suspected intra-articular pathologies such as cruciate ligament ganglion cysts is MRI.5,10
Cruciate ligament ganglion cysts typically follow fluid signal on all sequences, with low signal intensity on T1-weighted images and high signal intensity on T2-weighted images.1,2,5,6 A pericruciate location with a multilocular appearance is usually sufficient evidence to make a diagnosis. However, solid or semi-solid pathologies (such as synovial cell sarcoma, synovial hemangioma, or synovial chondromatosis) can have similar signal intensity.
If necessary, intravenous contrast can be helpful; a lack of central contrast enhancement can differentiate ganglion cysts from other solid, enhancing, or partially enhancing lesions. Other diagnostic modalities, such as ultrasound, computed tomography (CT), and diagnostic arthroscopy, are less practical and have a wide range of sensitivity and specificity.5,6,10
Arthroscopic excision is the treatment of choice
Asymptomatic cruciate ligament ganglion cysts are usually managed with clinical follow-up. For patients with symptomatic cysts, ultrasound- or CT-guided percutaneous cyst aspiration may temporarily improve symptoms, but recurrence rates have not been well studied.2,6,9,10 Additionally, accessibility to cysts in this location via these approaches is limited. Arthroscopic excision of the cyst is the treatment of choice for symptomatic cases.1,2,5,6,10
Our patient underwent arthroscopic cyst resection, which resulted in complete resolution of his symptoms. In 3 months, he returned to his regular physical activities with no pain or discomfort. One year later, he remained asymptomatic.
THE TAKEAWAY
Cruciate ligament ganglion cysts are a rare cause of posterior knee pain. An MRI is the best diagnostic modality to evaluate and confirm the diagnosis, as well as rule out other pathologies. The treatment of choice for symptomatic cases is arthroscopic excision of the cyst.
1. Mao Y, Dong Q, Wang Y. Ganglion cysts of the cruciate ligaments: a series of 31 cases and review of the literature. BMC Musculoskelet Disord. 2012;13:137.
2. Krudwig WK, Schulte KK, Heinemann C. Intra-articular ganglion cysts of the knee joint: a report of 85 cases and review of the literature. Knee Surg Sports Traumatol Arthrosc. 2004;12:123-129.
3. Bergin D, Morrison WB, Carrino JA, et al. Anterior cruciate ligament ganglia and mucoid degeneration: coexistence and clinical correlation. AJR Am J Roentgenol. 2004;182:1283-1287.
4. Bui-Mansfield LT, Youngberg RA. Intraarticular ganglia of the knee: prevalence, presentation, etiology, and management. AJR Am J Roentgenol. 1997;168:123-127.
5. Lunhao B, Yu S, Jiashi W. Diagnosis and treatment of ganglion cysts of the cruciate ligaments. Arch Orthop Trauma Surg. 2011;131:1053-1057.
6. Stein D, Cantlon M, Mackay B, et al. Cysts about the knee: evaluation and management. J Am Acad Orthop Surg. 2013;21:469-479.
7. Zantop T, Rusch A, Hassenpflug J, et al. Intra-articular ganglion cysts of the cruciate ligaments: case report and review of the literature. Arch Orthop Trauma Surg. 2003;123:195-198.
8. Tsai TY, Yang YS, Tseng FJ, et al. Arthroscopic excision of ganglion cysts of the posterior cruciate ligaments using posterior trans-septal portal. Arthroscopy. 2012;28:95-99.
9. Huang GS, Lee CH, Chan WP, et al. Ganglion cysts of the cruciate ligaments. Acta Radiol. 2002;43:419-424.
10. Tyrrell PN, Cassar-Pullicino VN, McCall IW. Intra-articular ganglion cysts of the cruciate ligaments. Eur Radiol. 2000;10:1233-1238.
THE CASE
A 36-year-old man sought care at our family medicine clinic for knee pain that he’d had for the past year. He denied any previous injury or trauma to the knee. The pain affected the posterolateral left knee and was aggravated by squatting and deep flexion. Daily activities did not bother him, but skiing, golfing, mountain biking, and lifting weights worsened the pain. His pain had gradually become more severe and frequent. He denied any mechanical symptoms such as catching, popping, or locking.
Examination of his left knee demonstrated range of motion from 0 to 120 degrees; further flexion caused significant pain. McMurray and Thessaly tests were positive for posterolateral pain, particularly with knee flexion >120 degrees. Physical examination was otherwise unremarkable. Standard x-rays of the left knee were normal. Our patient completed a month of physical therapy, but his symptoms did not improve.
THE DIAGNOSIS
After the patient completed physical therapy, magnetic resonance imaging (MRI) was performed. The MRI did not reveal any left knee effusion, and the menisci, collateral ligaments, and cartilage surfaces were normal. And, while the cruciate ligaments were intact, a large pericruciate ganglion cyst was noted (FIGURES 1 AND 2).
DISCUSSION
Ganglion cysts are dense, encapsulated structures filled with clear viscous fluid that often arise adjacent to tendon sheaths or joint capsules, most commonly over the dorsum of the hand.1 Intra-articular ganglia involving the cruciate ligaments of the knee are relatively uncommon.2 The estimated prevalence of cruciate ligament ganglion cysts at arthroscopy is 0.2% to 1.9%; similar rates have been demonstrated with MRI.3-6 There are more reported cases of these cysts involving the anterior cruciate ligament (ACL) compared to those affecting the posterior cruciate ligament (PCL).2,6
Classification of these cysts is based on relative location with respect to the ligaments. Type 1 cysts originate anterior to the ACL; type 2, between the ACL and PCL; and type 3, posterior to the PCL.6,7 Cruciate ligament ganglion cysts are more common in men, are typically discovered between age 20 and 40, and are usually incidental findings.8
The pathogenesis of ganglion cyst formation is unknown.1,6,7 The most widely accepted theory is that ganglion cysts result from mucinous degeneration of connective tissue in areas of repetitive stress.1,6,7 Other theories suggest hyaluronic acid production secondary to mesenchymal stem cell proliferation within the ligaments, synovial tissue herniation, or congenital translocation of synovial tissue as possible etiologies.2,6,7
Concurrent pathologies such as meniscal tears or chondral lesions may also be present; however, there is some disagreement as to what role, if any, antecedent trauma has in the pathogenesis of cyst formation.1,6 Several investigators have suggested that prior knee trauma is a likely risk factor.2,8,9
In most patients, cruciate ligament ganglion cysts are asymptomatic.7 The most common presenting symptom is nonspecific pain that is exacerbated by activity, such as stair climbing, squatting, or other activities that require extreme flexion or extension of the knee.6,9 Other possible symptoms include limited range of motion (extension block with ACL involvement, limited flexion with PCL lesions), a catching or locking sensation, instability, or joint line tenderness.5,6 A palpable mass on physical exam is not usually present.6 Some investigators suggest that larger lesions and those closer to the femoral ligamentous attachments are more likely to cause symptoms.5
Cruciate ligament ganglion cysts can be an easily overlooked source of a patient’s symptoms because they often mimic more common pathologies.2 The differential diagnosis of cruciate ligament ganglion cysts and posterior knee pain includes any other intra-articular cysts (eg, meniscal cysts), posterior meniscal tear, popliteus tendinopathy, or neoplasms (eg, hemangioma and synovial sarcoma).2,6
MRI is the best method of diagnosis
Because the symptoms of cruciate ligament ganglion cysts are variable and nonspecific, the diagnosis is rarely made on clinical grounds alone.1 The best method of evaluating suspected intra-articular pathologies such as cruciate ligament ganglion cysts is MRI.5,10
Cruciate ligament ganglion cysts typically follow fluid signal on all sequences, with low signal intensity on T1-weighted images and high signal intensity on T2-weighted images.1,2,5,6 A pericruciate location with a multilocular appearance is usually sufficient evidence to make a diagnosis. However, solid or semi-solid pathologies (such as synovial cell sarcoma, synovial hemangioma, or synovial chondromatosis) can have similar signal intensity.
If necessary, intravenous contrast can be helpful; a lack of central contrast enhancement can differentiate ganglion cysts from other solid, enhancing, or partially enhancing lesions. Other diagnostic modalities, such as ultrasound, computed tomography (CT), and diagnostic arthroscopy, are less practical and have a wide range of sensitivity and specificity.5,6,10
Arthroscopic excision is the treatment of choice
Asymptomatic cruciate ligament ganglion cysts are usually managed with clinical follow-up. For patients with symptomatic cysts, ultrasound- or CT-guided percutaneous cyst aspiration may temporarily improve symptoms, but recurrence rates have not been well studied.2,6,9,10 Additionally, accessibility to cysts in this location via these approaches is limited. Arthroscopic excision of the cyst is the treatment of choice for symptomatic cases.1,2,5,6,10
Our patient underwent arthroscopic cyst resection, which resulted in complete resolution of his symptoms. In 3 months, he returned to his regular physical activities with no pain or discomfort. One year later, he remained asymptomatic.
THE TAKEAWAY
Cruciate ligament ganglion cysts are a rare cause of posterior knee pain. An MRI is the best diagnostic modality to evaluate and confirm the diagnosis, as well as rule out other pathologies. The treatment of choice for symptomatic cases is arthroscopic excision of the cyst.
THE CASE
A 36-year-old man sought care at our family medicine clinic for knee pain that he’d had for the past year. He denied any previous injury or trauma to the knee. The pain affected the posterolateral left knee and was aggravated by squatting and deep flexion. Daily activities did not bother him, but skiing, golfing, mountain biking, and lifting weights worsened the pain. His pain had gradually become more severe and frequent. He denied any mechanical symptoms such as catching, popping, or locking.
Examination of his left knee demonstrated range of motion from 0 to 120 degrees; further flexion caused significant pain. McMurray and Thessaly tests were positive for posterolateral pain, particularly with knee flexion >120 degrees. Physical examination was otherwise unremarkable. Standard x-rays of the left knee were normal. Our patient completed a month of physical therapy, but his symptoms did not improve.
THE DIAGNOSIS
After the patient completed physical therapy, magnetic resonance imaging (MRI) was performed. The MRI did not reveal any left knee effusion, and the menisci, collateral ligaments, and cartilage surfaces were normal. And, while the cruciate ligaments were intact, a large pericruciate ganglion cyst was noted (FIGURES 1 AND 2).
DISCUSSION
Ganglion cysts are dense, encapsulated structures filled with clear viscous fluid that often arise adjacent to tendon sheaths or joint capsules, most commonly over the dorsum of the hand.1 Intra-articular ganglia involving the cruciate ligaments of the knee are relatively uncommon.2 The estimated prevalence of cruciate ligament ganglion cysts at arthroscopy is 0.2% to 1.9%; similar rates have been demonstrated with MRI.3-6 There are more reported cases of these cysts involving the anterior cruciate ligament (ACL) compared to those affecting the posterior cruciate ligament (PCL).2,6
Classification of these cysts is based on relative location with respect to the ligaments. Type 1 cysts originate anterior to the ACL; type 2, between the ACL and PCL; and type 3, posterior to the PCL.6,7 Cruciate ligament ganglion cysts are more common in men, are typically discovered between age 20 and 40, and are usually incidental findings.8
The pathogenesis of ganglion cyst formation is unknown.1,6,7 The most widely accepted theory is that ganglion cysts result from mucinous degeneration of connective tissue in areas of repetitive stress.1,6,7 Other theories suggest hyaluronic acid production secondary to mesenchymal stem cell proliferation within the ligaments, synovial tissue herniation, or congenital translocation of synovial tissue as possible etiologies.2,6,7
Concurrent pathologies such as meniscal tears or chondral lesions may also be present; however, there is some disagreement as to what role, if any, antecedent trauma has in the pathogenesis of cyst formation.1,6 Several investigators have suggested that prior knee trauma is a likely risk factor.2,8,9
In most patients, cruciate ligament ganglion cysts are asymptomatic.7 The most common presenting symptom is nonspecific pain that is exacerbated by activity, such as stair climbing, squatting, or other activities that require extreme flexion or extension of the knee.6,9 Other possible symptoms include limited range of motion (extension block with ACL involvement, limited flexion with PCL lesions), a catching or locking sensation, instability, or joint line tenderness.5,6 A palpable mass on physical exam is not usually present.6 Some investigators suggest that larger lesions and those closer to the femoral ligamentous attachments are more likely to cause symptoms.5
Cruciate ligament ganglion cysts can be an easily overlooked source of a patient’s symptoms because they often mimic more common pathologies.2 The differential diagnosis of cruciate ligament ganglion cysts and posterior knee pain includes any other intra-articular cysts (eg, meniscal cysts), posterior meniscal tear, popliteus tendinopathy, or neoplasms (eg, hemangioma and synovial sarcoma).2,6
MRI is the best method of diagnosis
Because the symptoms of cruciate ligament ganglion cysts are variable and nonspecific, the diagnosis is rarely made on clinical grounds alone.1 The best method of evaluating suspected intra-articular pathologies such as cruciate ligament ganglion cysts is MRI.5,10
Cruciate ligament ganglion cysts typically follow fluid signal on all sequences, with low signal intensity on T1-weighted images and high signal intensity on T2-weighted images.1,2,5,6 A pericruciate location with a multilocular appearance is usually sufficient evidence to make a diagnosis. However, solid or semi-solid pathologies (such as synovial cell sarcoma, synovial hemangioma, or synovial chondromatosis) can have similar signal intensity.
If necessary, intravenous contrast can be helpful; a lack of central contrast enhancement can differentiate ganglion cysts from other solid, enhancing, or partially enhancing lesions. Other diagnostic modalities, such as ultrasound, computed tomography (CT), and diagnostic arthroscopy, are less practical and have a wide range of sensitivity and specificity.5,6,10
Arthroscopic excision is the treatment of choice
Asymptomatic cruciate ligament ganglion cysts are usually managed with clinical follow-up. For patients with symptomatic cysts, ultrasound- or CT-guided percutaneous cyst aspiration may temporarily improve symptoms, but recurrence rates have not been well studied.2,6,9,10 Additionally, accessibility to cysts in this location via these approaches is limited. Arthroscopic excision of the cyst is the treatment of choice for symptomatic cases.1,2,5,6,10
Our patient underwent arthroscopic cyst resection, which resulted in complete resolution of his symptoms. In 3 months, he returned to his regular physical activities with no pain or discomfort. One year later, he remained asymptomatic.
THE TAKEAWAY
Cruciate ligament ganglion cysts are a rare cause of posterior knee pain. An MRI is the best diagnostic modality to evaluate and confirm the diagnosis, as well as rule out other pathologies. The treatment of choice for symptomatic cases is arthroscopic excision of the cyst.
1. Mao Y, Dong Q, Wang Y. Ganglion cysts of the cruciate ligaments: a series of 31 cases and review of the literature. BMC Musculoskelet Disord. 2012;13:137.
2. Krudwig WK, Schulte KK, Heinemann C. Intra-articular ganglion cysts of the knee joint: a report of 85 cases and review of the literature. Knee Surg Sports Traumatol Arthrosc. 2004;12:123-129.
3. Bergin D, Morrison WB, Carrino JA, et al. Anterior cruciate ligament ganglia and mucoid degeneration: coexistence and clinical correlation. AJR Am J Roentgenol. 2004;182:1283-1287.
4. Bui-Mansfield LT, Youngberg RA. Intraarticular ganglia of the knee: prevalence, presentation, etiology, and management. AJR Am J Roentgenol. 1997;168:123-127.
5. Lunhao B, Yu S, Jiashi W. Diagnosis and treatment of ganglion cysts of the cruciate ligaments. Arch Orthop Trauma Surg. 2011;131:1053-1057.
6. Stein D, Cantlon M, Mackay B, et al. Cysts about the knee: evaluation and management. J Am Acad Orthop Surg. 2013;21:469-479.
7. Zantop T, Rusch A, Hassenpflug J, et al. Intra-articular ganglion cysts of the cruciate ligaments: case report and review of the literature. Arch Orthop Trauma Surg. 2003;123:195-198.
8. Tsai TY, Yang YS, Tseng FJ, et al. Arthroscopic excision of ganglion cysts of the posterior cruciate ligaments using posterior trans-septal portal. Arthroscopy. 2012;28:95-99.
9. Huang GS, Lee CH, Chan WP, et al. Ganglion cysts of the cruciate ligaments. Acta Radiol. 2002;43:419-424.
10. Tyrrell PN, Cassar-Pullicino VN, McCall IW. Intra-articular ganglion cysts of the cruciate ligaments. Eur Radiol. 2000;10:1233-1238.
1. Mao Y, Dong Q, Wang Y. Ganglion cysts of the cruciate ligaments: a series of 31 cases and review of the literature. BMC Musculoskelet Disord. 2012;13:137.
2. Krudwig WK, Schulte KK, Heinemann C. Intra-articular ganglion cysts of the knee joint: a report of 85 cases and review of the literature. Knee Surg Sports Traumatol Arthrosc. 2004;12:123-129.
3. Bergin D, Morrison WB, Carrino JA, et al. Anterior cruciate ligament ganglia and mucoid degeneration: coexistence and clinical correlation. AJR Am J Roentgenol. 2004;182:1283-1287.
4. Bui-Mansfield LT, Youngberg RA. Intraarticular ganglia of the knee: prevalence, presentation, etiology, and management. AJR Am J Roentgenol. 1997;168:123-127.
5. Lunhao B, Yu S, Jiashi W. Diagnosis and treatment of ganglion cysts of the cruciate ligaments. Arch Orthop Trauma Surg. 2011;131:1053-1057.
6. Stein D, Cantlon M, Mackay B, et al. Cysts about the knee: evaluation and management. J Am Acad Orthop Surg. 2013;21:469-479.
7. Zantop T, Rusch A, Hassenpflug J, et al. Intra-articular ganglion cysts of the cruciate ligaments: case report and review of the literature. Arch Orthop Trauma Surg. 2003;123:195-198.
8. Tsai TY, Yang YS, Tseng FJ, et al. Arthroscopic excision of ganglion cysts of the posterior cruciate ligaments using posterior trans-septal portal. Arthroscopy. 2012;28:95-99.
9. Huang GS, Lee CH, Chan WP, et al. Ganglion cysts of the cruciate ligaments. Acta Radiol. 2002;43:419-424.
10. Tyrrell PN, Cassar-Pullicino VN, McCall IW. Intra-articular ganglion cysts of the cruciate ligaments. Eur Radiol. 2000;10:1233-1238.

These umbilical lesions weren't granulomas after all
THE CASES
CASE 1 › A 15-month-old boy was brought to our center for plastic surgery after being referred by his general practitioner (GP). The patient had a non-healing lesion on his umbilicus that had been present since birth. It had remained the same size, but bled occasionally. The GP initially presumed the lesion was a granuloma and treated it with silver nitrate cautery, but this did not eradicate it.
After talking with the boy’s mother further, we learned that there had been a constant oozing from the area since birth and that the lesion protruded slightly from the abdomen when the child cried. The boy had congenital heart disease, but his bowel and genitourinary history were normal. A clinical examination revealed pink, moist tissue herniating from the umbilicus with surrounding abdominal fullness when the boy stood up (FIGURE 1A). An ultrasound showed a focal 19 x 7 mm complex area around the umbilicus with no definite track. The lesion was surgically removed. Histology revealed a completely excised vitellointestinal duct remnant.
CASE 2 › A 6-year-old boy with a history of attention-deficit/hyperactivity disorder was brought to our clinic with a non-healing umbilical lesion after being referred by his GP. The lesion had been present since birth and had failed to resolve despite several attempts to treat it with silver nitrate cautery. Clinically, the patient appeared to have a granulomatous umbilical polyp (FIGURE 1B). The patient underwent surgical excision of the lesion. Histological analysis revealed a completely excised vitellointestinal duct remnant (FIGURE 2).
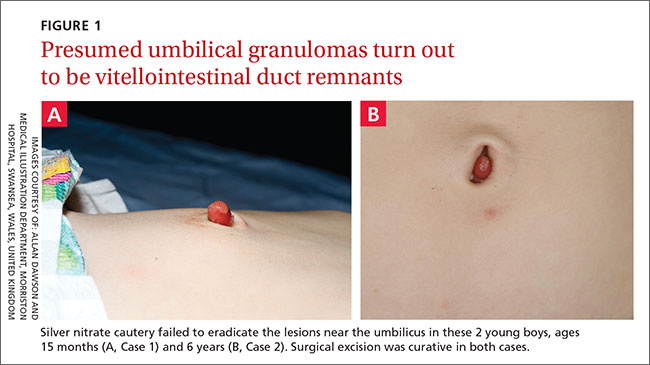
DISCUSSION
The vitellointestinal duct (VID), also called the omphalomesenteric duct (OMD), connects the alimentary canal and the yolk sac in early embryogenesis. Failure of involution of the duct results in abnormalities such as Meckel’s diverticulum, cysts, and polyps.
VID anomalies occur in approximately 2% of newborns; a small percentage of these have patent connections to the intestine.1 Parents are often the first to notice the abnormality and will typically see a reddish protrusion around the umbilicus or a persistent serous discharge around the umbilicus soon after birth.
VID remnants are similar in presentation to benign granulomas or granulation tissue, which are benign lesions that present in the first few weeks of life. Granulomas are reddish in color, bleed minimally when irritated by trauma, and respond well to silver nitrate cautery.2 When the lesion fails to respond to treatment, an alternative diagnosis should be investigated further.

Ultrasonography is the best way to evaluate a suspected VID remnant
A suspected VID remnant should first be assessed with ultrasonography to determine the extent of the remnant and guide surgical treatment. Ultrasonography can also delineate the relationship of these congenital remnants with the umbilicus and bladder.3
Potential complications that can arise from these lesions include an intestinal hernia, intussusception, volvulus, abdominal pain, or a persistent discharge that can lead to infection.3 Mortality following complications is significantly high.4
Although the etiology of patent VIDs and their remnants remains unknown, the presence of such ducts is associated with other congenital anomalies, including Down Syndrome, structural cardiac malformation, conduction abnormalities, and cleft lip and palate.5-7 Therefore, additional history taking and examinations may be required to identify these associated pathologies. In Case 1, the 15-month-old boy had congenital heart disease.
Surgical excision will prevent complications
A simple surgical excision should be performed for VID remnants. The prognosis is excellent when such procedures are performed in the non-acute setting. Some debate exists as to whether all remnants require formal abdominal exploration.8,9
Treatment of patent VIDs requires surgical excision of the duct, with or without a segment of the small bowel, to obliterate the connection.10 Reconstruction of the umbilicus is then performed, depending on the surgical technique used.
Our patients both made complete recoveries following their surgeries with resolution of their symptoms.
THE TAKEAWAY
Consider a VID remnant as part of the differential diagnosis for any patient who has what appears to be a granulomatous umbilical lesion. Order ultrasonography to evaluate a suspected VID, especially for lesions that fail to respond to 2 or 3 silver nitrate treatments. Surgical excision of a VID remnant is usually curative.
1. Vane DW, West KW, Grosfeld JL. Vitelline duct anomalies. Experience with 217 childhood cases. Arch Surg. 1987;122:542-547.
2. Piparsaliya S, Joshi M, Rajput N, et al. Patent vitellointestinal duct: A close differential diagnosis of umbilical granuloma: A case report and review of literature. Surgical Science. 2011;2:134-136.
3. Khati NJ, Enquist EG, Javitt MC. Imaging of the umbilicus and periumbilical region. Radiographics. 1998;18:413-431.
4. Yamada T, Seiki Y, Ueda M, et al. Patent omphalomesenteric duct: a case report and review of Japanese literature. Asia Oceania J Obstet Gynaecol. 1989;15:229-236.
5. Martin RH, Doublestein GL, Jarvis MR. Concurrent ectopic pregnancy, Meckel’s diverticulum with vitelline duct remnant, cecal volvulus, and congenital complete heart block: report of a case. J Am Osteopath Assoc. 1986;86:589-591.
6. Elebute EA, Ransome-Kuti O. Patent vitello-intestinal duct with ileal prolapse. Arch Surg. 1965;91:456-460.
7. Blair SP, Beasley SW. Intussusception of vitello-intestinal tract through an exomphalos in trisomy 13. Pediatric Surgery International. 1989;4:422-423.
8. Kutin ND, Allen JE, Jewett TC. The umbilical polyp. J Pediatr Surg. 1979;14:741-744.
9. Pacilli M, Sebire NJ, Maritsi D, et al. Umbilical polyp in infants and children. Eur J Pediatr Surg. 2007;17:397-399.
10. Storms P, Pexsters J, Vandekerkhof J. Small omphalocele with ileal prolapse through a patent omphalomesenteric duct. A case report and review of literature. Acta Chir Belg. 1988;88:392-394.
THE CASES
CASE 1 › A 15-month-old boy was brought to our center for plastic surgery after being referred by his general practitioner (GP). The patient had a non-healing lesion on his umbilicus that had been present since birth. It had remained the same size, but bled occasionally. The GP initially presumed the lesion was a granuloma and treated it with silver nitrate cautery, but this did not eradicate it.
After talking with the boy’s mother further, we learned that there had been a constant oozing from the area since birth and that the lesion protruded slightly from the abdomen when the child cried. The boy had congenital heart disease, but his bowel and genitourinary history were normal. A clinical examination revealed pink, moist tissue herniating from the umbilicus with surrounding abdominal fullness when the boy stood up (FIGURE 1A). An ultrasound showed a focal 19 x 7 mm complex area around the umbilicus with no definite track. The lesion was surgically removed. Histology revealed a completely excised vitellointestinal duct remnant.
CASE 2 › A 6-year-old boy with a history of attention-deficit/hyperactivity disorder was brought to our clinic with a non-healing umbilical lesion after being referred by his GP. The lesion had been present since birth and had failed to resolve despite several attempts to treat it with silver nitrate cautery. Clinically, the patient appeared to have a granulomatous umbilical polyp (FIGURE 1B). The patient underwent surgical excision of the lesion. Histological analysis revealed a completely excised vitellointestinal duct remnant (FIGURE 2).
DISCUSSION
The vitellointestinal duct (VID), also called the omphalomesenteric duct (OMD), connects the alimentary canal and the yolk sac in early embryogenesis. Failure of involution of the duct results in abnormalities such as Meckel’s diverticulum, cysts, and polyps.
VID anomalies occur in approximately 2% of newborns; a small percentage of these have patent connections to the intestine.1 Parents are often the first to notice the abnormality and will typically see a reddish protrusion around the umbilicus or a persistent serous discharge around the umbilicus soon after birth.
VID remnants are similar in presentation to benign granulomas or granulation tissue, which are benign lesions that present in the first few weeks of life. Granulomas are reddish in color, bleed minimally when irritated by trauma, and respond well to silver nitrate cautery.2 When the lesion fails to respond to treatment, an alternative diagnosis should be investigated further.
Ultrasonography is the best way to evaluate a suspected VID remnant
A suspected VID remnant should first be assessed with ultrasonography to determine the extent of the remnant and guide surgical treatment. Ultrasonography can also delineate the relationship of these congenital remnants with the umbilicus and bladder.3
Potential complications that can arise from these lesions include an intestinal hernia, intussusception, volvulus, abdominal pain, or a persistent discharge that can lead to infection.3 Mortality following complications is significantly high.4
Although the etiology of patent VIDs and their remnants remains unknown, the presence of such ducts is associated with other congenital anomalies, including Down Syndrome, structural cardiac malformation, conduction abnormalities, and cleft lip and palate.5-7 Therefore, additional history taking and examinations may be required to identify these associated pathologies. In Case 1, the 15-month-old boy had congenital heart disease.
Surgical excision will prevent complications
A simple surgical excision should be performed for VID remnants. The prognosis is excellent when such procedures are performed in the non-acute setting. Some debate exists as to whether all remnants require formal abdominal exploration.8,9
Treatment of patent VIDs requires surgical excision of the duct, with or without a segment of the small bowel, to obliterate the connection.10 Reconstruction of the umbilicus is then performed, depending on the surgical technique used.
Our patients both made complete recoveries following their surgeries with resolution of their symptoms.
THE TAKEAWAY
Consider a VID remnant as part of the differential diagnosis for any patient who has what appears to be a granulomatous umbilical lesion. Order ultrasonography to evaluate a suspected VID, especially for lesions that fail to respond to 2 or 3 silver nitrate treatments. Surgical excision of a VID remnant is usually curative.
THE CASES
CASE 1 › A 15-month-old boy was brought to our center for plastic surgery after being referred by his general practitioner (GP). The patient had a non-healing lesion on his umbilicus that had been present since birth. It had remained the same size, but bled occasionally. The GP initially presumed the lesion was a granuloma and treated it with silver nitrate cautery, but this did not eradicate it.
After talking with the boy’s mother further, we learned that there had been a constant oozing from the area since birth and that the lesion protruded slightly from the abdomen when the child cried. The boy had congenital heart disease, but his bowel and genitourinary history were normal. A clinical examination revealed pink, moist tissue herniating from the umbilicus with surrounding abdominal fullness when the boy stood up (FIGURE 1A). An ultrasound showed a focal 19 x 7 mm complex area around the umbilicus with no definite track. The lesion was surgically removed. Histology revealed a completely excised vitellointestinal duct remnant.
CASE 2 › A 6-year-old boy with a history of attention-deficit/hyperactivity disorder was brought to our clinic with a non-healing umbilical lesion after being referred by his GP. The lesion had been present since birth and had failed to resolve despite several attempts to treat it with silver nitrate cautery. Clinically, the patient appeared to have a granulomatous umbilical polyp (FIGURE 1B). The patient underwent surgical excision of the lesion. Histological analysis revealed a completely excised vitellointestinal duct remnant (FIGURE 2).
DISCUSSION
The vitellointestinal duct (VID), also called the omphalomesenteric duct (OMD), connects the alimentary canal and the yolk sac in early embryogenesis. Failure of involution of the duct results in abnormalities such as Meckel’s diverticulum, cysts, and polyps.
VID anomalies occur in approximately 2% of newborns; a small percentage of these have patent connections to the intestine.1 Parents are often the first to notice the abnormality and will typically see a reddish protrusion around the umbilicus or a persistent serous discharge around the umbilicus soon after birth.
VID remnants are similar in presentation to benign granulomas or granulation tissue, which are benign lesions that present in the first few weeks of life. Granulomas are reddish in color, bleed minimally when irritated by trauma, and respond well to silver nitrate cautery.2 When the lesion fails to respond to treatment, an alternative diagnosis should be investigated further.
Ultrasonography is the best way to evaluate a suspected VID remnant
A suspected VID remnant should first be assessed with ultrasonography to determine the extent of the remnant and guide surgical treatment. Ultrasonography can also delineate the relationship of these congenital remnants with the umbilicus and bladder.3
Potential complications that can arise from these lesions include an intestinal hernia, intussusception, volvulus, abdominal pain, or a persistent discharge that can lead to infection.3 Mortality following complications is significantly high.4
Although the etiology of patent VIDs and their remnants remains unknown, the presence of such ducts is associated with other congenital anomalies, including Down Syndrome, structural cardiac malformation, conduction abnormalities, and cleft lip and palate.5-7 Therefore, additional history taking and examinations may be required to identify these associated pathologies. In Case 1, the 15-month-old boy had congenital heart disease.
Surgical excision will prevent complications
A simple surgical excision should be performed for VID remnants. The prognosis is excellent when such procedures are performed in the non-acute setting. Some debate exists as to whether all remnants require formal abdominal exploration.8,9
Treatment of patent VIDs requires surgical excision of the duct, with or without a segment of the small bowel, to obliterate the connection.10 Reconstruction of the umbilicus is then performed, depending on the surgical technique used.
Our patients both made complete recoveries following their surgeries with resolution of their symptoms.
THE TAKEAWAY
Consider a VID remnant as part of the differential diagnosis for any patient who has what appears to be a granulomatous umbilical lesion. Order ultrasonography to evaluate a suspected VID, especially for lesions that fail to respond to 2 or 3 silver nitrate treatments. Surgical excision of a VID remnant is usually curative.
1. Vane DW, West KW, Grosfeld JL. Vitelline duct anomalies. Experience with 217 childhood cases. Arch Surg. 1987;122:542-547.
2. Piparsaliya S, Joshi M, Rajput N, et al. Patent vitellointestinal duct: A close differential diagnosis of umbilical granuloma: A case report and review of literature. Surgical Science. 2011;2:134-136.
3. Khati NJ, Enquist EG, Javitt MC. Imaging of the umbilicus and periumbilical region. Radiographics. 1998;18:413-431.
4. Yamada T, Seiki Y, Ueda M, et al. Patent omphalomesenteric duct: a case report and review of Japanese literature. Asia Oceania J Obstet Gynaecol. 1989;15:229-236.
5. Martin RH, Doublestein GL, Jarvis MR. Concurrent ectopic pregnancy, Meckel’s diverticulum with vitelline duct remnant, cecal volvulus, and congenital complete heart block: report of a case. J Am Osteopath Assoc. 1986;86:589-591.
6. Elebute EA, Ransome-Kuti O. Patent vitello-intestinal duct with ileal prolapse. Arch Surg. 1965;91:456-460.
7. Blair SP, Beasley SW. Intussusception of vitello-intestinal tract through an exomphalos in trisomy 13. Pediatric Surgery International. 1989;4:422-423.
8. Kutin ND, Allen JE, Jewett TC. The umbilical polyp. J Pediatr Surg. 1979;14:741-744.
9. Pacilli M, Sebire NJ, Maritsi D, et al. Umbilical polyp in infants and children. Eur J Pediatr Surg. 2007;17:397-399.
10. Storms P, Pexsters J, Vandekerkhof J. Small omphalocele with ileal prolapse through a patent omphalomesenteric duct. A case report and review of literature. Acta Chir Belg. 1988;88:392-394.
1. Vane DW, West KW, Grosfeld JL. Vitelline duct anomalies. Experience with 217 childhood cases. Arch Surg. 1987;122:542-547.
2. Piparsaliya S, Joshi M, Rajput N, et al. Patent vitellointestinal duct: A close differential diagnosis of umbilical granuloma: A case report and review of literature. Surgical Science. 2011;2:134-136.
3. Khati NJ, Enquist EG, Javitt MC. Imaging of the umbilicus and periumbilical region. Radiographics. 1998;18:413-431.
4. Yamada T, Seiki Y, Ueda M, et al. Patent omphalomesenteric duct: a case report and review of Japanese literature. Asia Oceania J Obstet Gynaecol. 1989;15:229-236.
5. Martin RH, Doublestein GL, Jarvis MR. Concurrent ectopic pregnancy, Meckel’s diverticulum with vitelline duct remnant, cecal volvulus, and congenital complete heart block: report of a case. J Am Osteopath Assoc. 1986;86:589-591.
6. Elebute EA, Ransome-Kuti O. Patent vitello-intestinal duct with ileal prolapse. Arch Surg. 1965;91:456-460.
7. Blair SP, Beasley SW. Intussusception of vitello-intestinal tract through an exomphalos in trisomy 13. Pediatric Surgery International. 1989;4:422-423.
8. Kutin ND, Allen JE, Jewett TC. The umbilical polyp. J Pediatr Surg. 1979;14:741-744.
9. Pacilli M, Sebire NJ, Maritsi D, et al. Umbilical polyp in infants and children. Eur J Pediatr Surg. 2007;17:397-399.
10. Storms P, Pexsters J, Vandekerkhof J. Small omphalocele with ileal prolapse through a patent omphalomesenteric duct. A case report and review of literature. Acta Chir Belg. 1988;88:392-394.

Ureter and Nerve Root Compression Secondary to Expansile Fibrous Dysplasia of the Transverse Process
Fibrous dysplasia is a developmental abnormality caused by excessive proliferation of immature spindle-cell fibrous tissues in bones. It is characterized by benign bony growths, which can lead to local swelling, bony deformities, and lytic conversion, predisposing the bone to pathologic fractures. Although this process can occur in cortical bone, it primarily affects the trabecular bone, leading to enlargement and expansion from within the medullary space. Malignant transformation to osteosarcoma or fibrosarcoma can occur, although this is exceedingly rare (<0.5%).1,2
This case report describes a patient who presented with an expansile lytic mass in a lumbar transverse process that was postoperatively identified on pathology as monostotic fibrous dysplasia. Such lesions that involve the transverse processes are rare and have been associated with pain and significant discomfort.3-5 This is the first reported case of a transverse process fibrous dysplasia causing urinary retention and neurologic symptoms simultaneously. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
History
A 52-year-old black man presented to us with 6 to 8 months of increasing right flank pain, difficulty with urination, and right lower extremity pain in the area of his anterior thigh. He also complained of “buckling” of his thigh with ambulation. On review of systems, the patient denied any fevers, chills, headache, changes in weight or vision, or hearing problems. He had no systemic symptoms except for 6 months of frequent urinary tract infections and difficulty emptying his bladder, which resulted in urinary retention. He denied any significant medical history and denied any use of alcohol or tobacco.
Physical Examination
On physical examination, the patient was a well-appearing 52-year-old man in no apparent distress. No signs of gross deformity, erythema, ecchymosis, or infection were noted upon examination of his lower extremities. His motor examination was within normal limits from L2 to S1. However, both fine and gross sensation were decreased in the L3 distribution. Sensation was intact to the remaining nerve-root distributions. The Babinski sign was negative for both lower extremities, and clonus was within physiologic limits. Examination of his gait was notable for quadriceps buckling with ambulation.
Radiographic Examination
The patient initially presented to his primary care physician, who evaluated his symptoms with a computed tomography scan of his abdomen and pelvis. This showed a mass of the right L3 transverse process (Figure 1). The patient was referred to us for further management of this lesion. Dedicated magnetic resonance imaging of his lumbar spine was performed, showing an expansile, lytic, homogeneous mass in the patient’s right L3 transverse process. The mass showed a significant mass effect, compressing the exiting nerve roots and, presumably, his right ureter (Figure 2). A bone scan showed monostotic disease. The patient had failed conservative management, including physical therapy and anti-inflammatory medications. His right-sided radiculopathy was worsening, and he complained that the pain was affecting his quality of life and limiting his performance of his daily activities. A pain management specialist was requested to better manage his pain. Considering progression of his condition, surgical management was discussed, leading to a planned biopsy and resection of the mass.
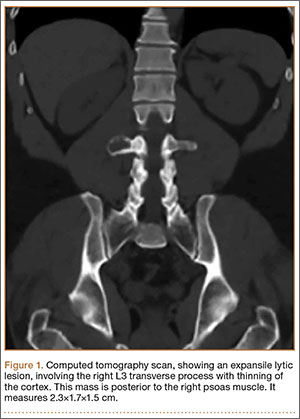
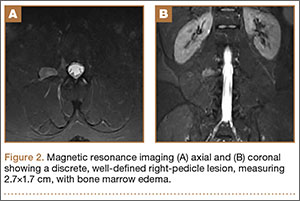
Surgical Procedure
The patient was taken into the operating room and positioned prone on a Jackson table with a Wilson frame. Fluoroscopy was used to localize the right L3 transverse process. An incision was made over the right L3 transverse process and a Wiltse intramuscular approach was performed. After the right L3 transverse process was identified, the soft tissue from the transverse process was retracted in all directions, including medially up to the pedicle. The intertransverse ligament was detached from both the cephalad and caudal edges of the transverse process. We used a Woodson elevator to perform subperiosteal dissection to remove the soft tissue circumferentially. After dissection, we placed a Cobb elevator to protect the rostral and caudal soft tissue and used a high-speed burr to amputate the lytic transverse process at its base. The transverse process was removed en bloc (Figure 3) and sent for frozen pathologic evaluation (Figure 4). After the diagnosis of a benign lesion, the wound was closed in layers.

Complete resolution of both urinary and neurologic symptoms were immediately noted and up to 1 month postoperatively.
Discussion
Primary bone tumors of the spine are rare, with a reported incidence of 2.5 to 8.5 per 100,000 people per year.6 The estimated incidence of benign primary tumors involving the spine accounts for about 1% of all primary skeletal tumors and nearly 5% for malignant tumors.7-9 In contrast, secondary tumors involving the bony spinal column are relatively common. Postmortem studies indicate up to 70% of cancer patients demonstrate axial skeletal involvement.10,11 The most commonly encountered benign tumors affecting the spine include giant cell tumors, osteoid osteomas, osteoblastomas, and hemangiomas. Chordomas are frequently reported as the most common malignant primary spine neoplasms. Of all primary benign bone lesions, fibrous dysplasia accounts for approximately 1.4%.8
Primary and secondary malignant osseous tumors have a predilection for the anterior column, and primary benign lesions usually affect the posterior column.8,12-14 Because of the greater blood supply and more direct communication with the viscera via the Batson plexus, the anterior column is most likely to be seeded by metastatic disease. Similarly, hemangiomas and multiple myeloma are typically located in the anterior column, most likely because of the more abundant blood supply there. Chordomas are also found in this cancellous anterior column. Osteoid osteoma, osteoblastoma, and aneurysmal bone cysts are found within the more cortical architecture of posterior elements. The location of this patient’s lesion within the transverse process elevates confidence in the diagnosis of a benign lesion.
The conventional, isolated form of fibrous dysplasia was originally described in 1942 by Lichtenstein and Jaffe.2 They described 15 cases of benign “nonosteogenic fibromas” near the ends of long bones in young patients. Monostotic fibrous dysplasia constitutes the majority of these cases, approximately 80%.1,2,8,15 Fibrous dysplasia may also present as part of McCune-Albright syndrome, in which case it is associated with precocious puberty and café au lait spots. Less commonly, they are associated with intramuscular myxomas, as in Mazabraud syndrome. The lesions in these syndromes are typically polyostotic. In all forms, fibrous dysplasia develops from an activating mutation in the gene that encodes the alpha subunit of the G protein on chromosome 20q13, activating cyclic adenylate cyclase and slowing the differentiation of osteoblasts.3,8
With regard to presentation, fibrous dysplasia is usually asymptomatic and discovered incidentally. The literature reports that the most common presenting symptom for patients with monostotic fibrous dysplasia of the spine is back pain localized to the lesion.15 Meredith and Healey2 completed a comprehensive review of 54 cases of monostotic fibrous dysplasia involving the spine in which the majority of symptoms included back pain, neck pain, sacral region pain, pathologic fracture, painful torticollis, progressive myelopathy, paresthesias of the foot, and only 1 case of radiculopathy involving thoracic vertebra. In normal anatomy, the ureter lies within retroperitoneal fat anterior to the psoas muscle and L2-L5 transverse processes and is normally mobile.16-18 This becomes clinically significant in lean patients as the ureter becomes closer to the spine. There are several reports of iatrogenic ureter injury in lumbar disc surgery.16-18 Normal variants, including medialization towards the spine, may predispose the ureters to injury, iatrogenic, or otherwise. In fact, medialization of the ureters occurs commonly in black men and usually involves the right side, which may have occurred in this black patient.19
Fibrous dysplasia is most often diagnosed by its radiographic appearance or biopsy. However, recent data suggest that deoxyribonucleic acid (DNA) analysis may soon be able to diagnose this process.20 Imaging typically reveals expansile, central lytic lesions within the medullary cavity. Pathology shows dense fibroblasts around immature woven bone, commonly referred to as “Chinese lettering.” The treatment varies from observation to en bloc surgical resection. Clinical observation is warranted for asymptomatic or incidental findings of monostotic fibrous dysplasia, as long as the risk for pathologic fracture is low.11 Bisphosphonate therapy, both oral and intravenous, offers promising outcomes for the treatment of fibrous dysplasia, with improvement in pain and function as well as in the radiographic findings.11,21 Management of monostotic fibrous dysplasia presenting as an isolated expansile mass of the transverse process in lumbar spine has rarely been described.3-5 Troop and Herring5 reported a case of monostotic fibrous dysplasia in the lumbar spine, with involvement of the vertebral body and the posterior elements. Chow and coauthors3 and Harris and colleagues4 described the involvement of the transverse process of L4. Chow and coauthors’3 treatment consisted of excision that resulted in an asymptomatic patient at 8-year follow-up, while Harris and colleagues4 chose observation. In the latter study, the patient’s lower back pain persisted at 4-year follow-up.
Progressive enlargement, recurrence, and malignant transformation have all been described. Meredith and Healey2 reported the reappearance of monostotic fibrous dysplasia affecting C2, extending through the fusion mass to involve a previously unaffected vertebra 20 years after the original C2 posterior elements excision via posterior spinal fusion from C1 to C3. In the literature, the incidence of malignant transformation ranges from 0.4% to 4%.8 One case of malignant transformation in thoracic spine was reported by Fu and colleagues.22 Therefore, complete removal of all affected bone is recommended.1,2,4,5,15,22,23
Conclusion
We present an unusual condition with complete resolution of symptoms after surgical resection. Several points may be considered from this report. Fibrous dysplasia lesions have been found in all bones of the body, including the skull, face, and extremities; however, monostotic fibrous dysplasia involving the spine is rare.11,23,24 Furthermore, there are no other reports of these lesions causing simultaneous nerve compression and urologic symptoms. Considering anatomy, clinicians may consider lesions of the lumbar transverse process in patients presenting to orthopedic surgeons with urinary symptoms, especially when combined with neurologic symptoms. In these lesions, fibrous dysplasia should be within the differential diagnosis. Clinicians should also recognize that complete resolution of symptoms has been reported with wide resection of these lesions.
1. Leet AI, Magur E, Lee JS, Weintroub S, Robey PG, Collins MT. Fibrous dysplasia in the spine: prevalence of lesions and association with scoliosis. J Bone Joint Surg Am. 2004;86(3):531-537.
2. Meredith DS, Healey JH. Twenty-year follow-up of monostotic fibrous dysplasia of the second cervical vertebra: a case report and review of the literature. J Bone Joint Surg Am. 2011;93(13):e74.
3. Chow LT, Griffith J, Chow WH, Kumta SM. Monostotic fibrous dysplasia of the spine: report of a case involving the lumbar transverse process and review of the literature. Arch Orthop Trauma Surg. 2000;120(7-8):460-464.
4. Harris WH, Dudley HR Jr, Barry RJ. The natural history of fibrous dysplasia. An orthopaedic, pathologic, and roentgenographic study. J Bone Joint Surg Am. 1962;44(2):207-233.
5. Troop JK, Herring JA. Monostotic fibrous dysplasia of the lumbar spine: case report and review of the literature. J Pediatr Orthop. 1988;8(5):599-601.
6. Dreghorn CR, Newman RJ, Hardy GJ, Dickson RA. Primary tumors of the axial skeleton. Experience of the Leeds Regional Bone Tumor Registry. Spine. 1990;15(2):137-140.
7. Schuster JM, Grady MS. Medical management and adjuvant therapies in spinal metastatic disease. Neurosurg Focus. 2001;11(6):e3.
8. Unni K. Introduction and scope. In: Unni K, ed. Dahlin’s Bone Tumors—General Aspects and Data on 11,087 Cases. Philadelphia, PA: Lippincott-Raven; 1996:1-9.
9. Wong DA, Fornasier VL, MacNab I. Spinal metastases: the obvious, the occult, and the impostors. Spine. 1990;15(1):1-4.
10. Dagi TF, Schmidek HH. Vascular tumors of the spine. In: Sundaresan N, Schmidek HH, Schiller AL, eds. Tumors of the Spine: Diagnosis and Clinical Management. Philadelphia, PA: W.B. Saunders Co; 1990:181-191.
11. DiCaprio M, Enneking W. Fibrous dysplasia. Pathophysiology, evaluation, and treatment. J Bone Joint Surg Am. 2005;87(8):1848-1864.
12. Gasbarrini A, Cappuccio M, Mirabile L, et al. Spinal metastases: treatment evaluation algorithm. Eur Rev Med Pharmacol Sci. 2004;8(6):265-274.
13. Loblaw DA, Laperriere NJ, Mackillop WJ. A population-based study of malignant spinal cord compression in Ontario. Clin Oncol. 2003;15(4):211-217.
14. Ortiz Gómez JA. The incidence of vertebral body metastases. Int Orthop. 1995;19(5):309-311.
15. Avimadje AM, Goupille P, Zerkak D, Begnard G, Brunais-Besse J, Valat JP. Monostotic fibrous dysplasia of the lumbar spine. Joint Bone Spine. 2000;67(1):65-70.
16. Isiklar ZU, Lindsey RW, Coburn M. Ureteral injury after anterior lumbar interbody fusion. A case report. Spine. 1996;21(20):2379-2382.
17. Krone A, Heller V, Osterhage HR. Ureteral injury in lumbar disc surgery. Acta Neurochir (Wien). 1985;78(3-4):108–112.
18. Cho KT, Im SH, Hong SK. Ureteral injury after inadvertent violation of the intertransverse space during posterior lumbar diskectomy: a case report. Surg Neurol. 2008;69(2):135-137.
19. Adam EJ, Desai SC, Lawton G. Racial variations in normal ureteric course. Clin Radiol. 1985;36(4):373-375.
20. Stathopoulos IP, Balanika AP, Baltas CS, et al. Fibrous dysplasia; confirmation of clinical diagnosis by DNA tests instead of biopsy. J Musculoskelet Neuronal Interact. 2013;13(1):120-123.
21. Lane JM, Khan SN, O’Connor WJ, et al. Bisphosphonate therapy in fibrous dysplasia. Clin Orthop Relat Res. 2001;382:6-12.
22. Fu CJ, Hsu CY, Shih TT, Wu MZ. Monostotic fibrous dysplasia of the thoracic spine with malignant transformation. J Formos Med Assoc. 2004;103(9):711-714.
23. McCarthy EF. Fibro-osseous lesions of the maxillofacial bones. Head Neck Pathol. 2013;7(1):5-10.
24. Manjila S, Zender CA, Weaver J, Rodgers M, Cohen AR. Aneurysmal bone cyst within fibrous dysplasia of the anterior skull base: continued intracranial extension after endoscopic resections requiring craniofacial approach with free tissue transfer reconstruction [published online ahead of print February 26, 2013]. Childs Nerv Syst. 2013;29(7).
Fibrous dysplasia is a developmental abnormality caused by excessive proliferation of immature spindle-cell fibrous tissues in bones. It is characterized by benign bony growths, which can lead to local swelling, bony deformities, and lytic conversion, predisposing the bone to pathologic fractures. Although this process can occur in cortical bone, it primarily affects the trabecular bone, leading to enlargement and expansion from within the medullary space. Malignant transformation to osteosarcoma or fibrosarcoma can occur, although this is exceedingly rare (<0.5%).1,2
This case report describes a patient who presented with an expansile lytic mass in a lumbar transverse process that was postoperatively identified on pathology as monostotic fibrous dysplasia. Such lesions that involve the transverse processes are rare and have been associated with pain and significant discomfort.3-5 This is the first reported case of a transverse process fibrous dysplasia causing urinary retention and neurologic symptoms simultaneously. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
History
A 52-year-old black man presented to us with 6 to 8 months of increasing right flank pain, difficulty with urination, and right lower extremity pain in the area of his anterior thigh. He also complained of “buckling” of his thigh with ambulation. On review of systems, the patient denied any fevers, chills, headache, changes in weight or vision, or hearing problems. He had no systemic symptoms except for 6 months of frequent urinary tract infections and difficulty emptying his bladder, which resulted in urinary retention. He denied any significant medical history and denied any use of alcohol or tobacco.
Physical Examination
On physical examination, the patient was a well-appearing 52-year-old man in no apparent distress. No signs of gross deformity, erythema, ecchymosis, or infection were noted upon examination of his lower extremities. His motor examination was within normal limits from L2 to S1. However, both fine and gross sensation were decreased in the L3 distribution. Sensation was intact to the remaining nerve-root distributions. The Babinski sign was negative for both lower extremities, and clonus was within physiologic limits. Examination of his gait was notable for quadriceps buckling with ambulation.
Radiographic Examination
The patient initially presented to his primary care physician, who evaluated his symptoms with a computed tomography scan of his abdomen and pelvis. This showed a mass of the right L3 transverse process (Figure 1). The patient was referred to us for further management of this lesion. Dedicated magnetic resonance imaging of his lumbar spine was performed, showing an expansile, lytic, homogeneous mass in the patient’s right L3 transverse process. The mass showed a significant mass effect, compressing the exiting nerve roots and, presumably, his right ureter (Figure 2). A bone scan showed monostotic disease. The patient had failed conservative management, including physical therapy and anti-inflammatory medications. His right-sided radiculopathy was worsening, and he complained that the pain was affecting his quality of life and limiting his performance of his daily activities. A pain management specialist was requested to better manage his pain. Considering progression of his condition, surgical management was discussed, leading to a planned biopsy and resection of the mass.
Surgical Procedure
The patient was taken into the operating room and positioned prone on a Jackson table with a Wilson frame. Fluoroscopy was used to localize the right L3 transverse process. An incision was made over the right L3 transverse process and a Wiltse intramuscular approach was performed. After the right L3 transverse process was identified, the soft tissue from the transverse process was retracted in all directions, including medially up to the pedicle. The intertransverse ligament was detached from both the cephalad and caudal edges of the transverse process. We used a Woodson elevator to perform subperiosteal dissection to remove the soft tissue circumferentially. After dissection, we placed a Cobb elevator to protect the rostral and caudal soft tissue and used a high-speed burr to amputate the lytic transverse process at its base. The transverse process was removed en bloc (Figure 3) and sent for frozen pathologic evaluation (Figure 4). After the diagnosis of a benign lesion, the wound was closed in layers.
Complete resolution of both urinary and neurologic symptoms were immediately noted and up to 1 month postoperatively.
Discussion
Primary bone tumors of the spine are rare, with a reported incidence of 2.5 to 8.5 per 100,000 people per year.6 The estimated incidence of benign primary tumors involving the spine accounts for about 1% of all primary skeletal tumors and nearly 5% for malignant tumors.7-9 In contrast, secondary tumors involving the bony spinal column are relatively common. Postmortem studies indicate up to 70% of cancer patients demonstrate axial skeletal involvement.10,11 The most commonly encountered benign tumors affecting the spine include giant cell tumors, osteoid osteomas, osteoblastomas, and hemangiomas. Chordomas are frequently reported as the most common malignant primary spine neoplasms. Of all primary benign bone lesions, fibrous dysplasia accounts for approximately 1.4%.8
Primary and secondary malignant osseous tumors have a predilection for the anterior column, and primary benign lesions usually affect the posterior column.8,12-14 Because of the greater blood supply and more direct communication with the viscera via the Batson plexus, the anterior column is most likely to be seeded by metastatic disease. Similarly, hemangiomas and multiple myeloma are typically located in the anterior column, most likely because of the more abundant blood supply there. Chordomas are also found in this cancellous anterior column. Osteoid osteoma, osteoblastoma, and aneurysmal bone cysts are found within the more cortical architecture of posterior elements. The location of this patient’s lesion within the transverse process elevates confidence in the diagnosis of a benign lesion.
The conventional, isolated form of fibrous dysplasia was originally described in 1942 by Lichtenstein and Jaffe.2 They described 15 cases of benign “nonosteogenic fibromas” near the ends of long bones in young patients. Monostotic fibrous dysplasia constitutes the majority of these cases, approximately 80%.1,2,8,15 Fibrous dysplasia may also present as part of McCune-Albright syndrome, in which case it is associated with precocious puberty and café au lait spots. Less commonly, they are associated with intramuscular myxomas, as in Mazabraud syndrome. The lesions in these syndromes are typically polyostotic. In all forms, fibrous dysplasia develops from an activating mutation in the gene that encodes the alpha subunit of the G protein on chromosome 20q13, activating cyclic adenylate cyclase and slowing the differentiation of osteoblasts.3,8
With regard to presentation, fibrous dysplasia is usually asymptomatic and discovered incidentally. The literature reports that the most common presenting symptom for patients with monostotic fibrous dysplasia of the spine is back pain localized to the lesion.15 Meredith and Healey2 completed a comprehensive review of 54 cases of monostotic fibrous dysplasia involving the spine in which the majority of symptoms included back pain, neck pain, sacral region pain, pathologic fracture, painful torticollis, progressive myelopathy, paresthesias of the foot, and only 1 case of radiculopathy involving thoracic vertebra. In normal anatomy, the ureter lies within retroperitoneal fat anterior to the psoas muscle and L2-L5 transverse processes and is normally mobile.16-18 This becomes clinically significant in lean patients as the ureter becomes closer to the spine. There are several reports of iatrogenic ureter injury in lumbar disc surgery.16-18 Normal variants, including medialization towards the spine, may predispose the ureters to injury, iatrogenic, or otherwise. In fact, medialization of the ureters occurs commonly in black men and usually involves the right side, which may have occurred in this black patient.19
Fibrous dysplasia is most often diagnosed by its radiographic appearance or biopsy. However, recent data suggest that deoxyribonucleic acid (DNA) analysis may soon be able to diagnose this process.20 Imaging typically reveals expansile, central lytic lesions within the medullary cavity. Pathology shows dense fibroblasts around immature woven bone, commonly referred to as “Chinese lettering.” The treatment varies from observation to en bloc surgical resection. Clinical observation is warranted for asymptomatic or incidental findings of monostotic fibrous dysplasia, as long as the risk for pathologic fracture is low.11 Bisphosphonate therapy, both oral and intravenous, offers promising outcomes for the treatment of fibrous dysplasia, with improvement in pain and function as well as in the radiographic findings.11,21 Management of monostotic fibrous dysplasia presenting as an isolated expansile mass of the transverse process in lumbar spine has rarely been described.3-5 Troop and Herring5 reported a case of monostotic fibrous dysplasia in the lumbar spine, with involvement of the vertebral body and the posterior elements. Chow and coauthors3 and Harris and colleagues4 described the involvement of the transverse process of L4. Chow and coauthors’3 treatment consisted of excision that resulted in an asymptomatic patient at 8-year follow-up, while Harris and colleagues4 chose observation. In the latter study, the patient’s lower back pain persisted at 4-year follow-up.
Progressive enlargement, recurrence, and malignant transformation have all been described. Meredith and Healey2 reported the reappearance of monostotic fibrous dysplasia affecting C2, extending through the fusion mass to involve a previously unaffected vertebra 20 years after the original C2 posterior elements excision via posterior spinal fusion from C1 to C3. In the literature, the incidence of malignant transformation ranges from 0.4% to 4%.8 One case of malignant transformation in thoracic spine was reported by Fu and colleagues.22 Therefore, complete removal of all affected bone is recommended.1,2,4,5,15,22,23
Conclusion
We present an unusual condition with complete resolution of symptoms after surgical resection. Several points may be considered from this report. Fibrous dysplasia lesions have been found in all bones of the body, including the skull, face, and extremities; however, monostotic fibrous dysplasia involving the spine is rare.11,23,24 Furthermore, there are no other reports of these lesions causing simultaneous nerve compression and urologic symptoms. Considering anatomy, clinicians may consider lesions of the lumbar transverse process in patients presenting to orthopedic surgeons with urinary symptoms, especially when combined with neurologic symptoms. In these lesions, fibrous dysplasia should be within the differential diagnosis. Clinicians should also recognize that complete resolution of symptoms has been reported with wide resection of these lesions.
Fibrous dysplasia is a developmental abnormality caused by excessive proliferation of immature spindle-cell fibrous tissues in bones. It is characterized by benign bony growths, which can lead to local swelling, bony deformities, and lytic conversion, predisposing the bone to pathologic fractures. Although this process can occur in cortical bone, it primarily affects the trabecular bone, leading to enlargement and expansion from within the medullary space. Malignant transformation to osteosarcoma or fibrosarcoma can occur, although this is exceedingly rare (<0.5%).1,2
This case report describes a patient who presented with an expansile lytic mass in a lumbar transverse process that was postoperatively identified on pathology as monostotic fibrous dysplasia. Such lesions that involve the transverse processes are rare and have been associated with pain and significant discomfort.3-5 This is the first reported case of a transverse process fibrous dysplasia causing urinary retention and neurologic symptoms simultaneously. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
History
A 52-year-old black man presented to us with 6 to 8 months of increasing right flank pain, difficulty with urination, and right lower extremity pain in the area of his anterior thigh. He also complained of “buckling” of his thigh with ambulation. On review of systems, the patient denied any fevers, chills, headache, changes in weight or vision, or hearing problems. He had no systemic symptoms except for 6 months of frequent urinary tract infections and difficulty emptying his bladder, which resulted in urinary retention. He denied any significant medical history and denied any use of alcohol or tobacco.
Physical Examination
On physical examination, the patient was a well-appearing 52-year-old man in no apparent distress. No signs of gross deformity, erythema, ecchymosis, or infection were noted upon examination of his lower extremities. His motor examination was within normal limits from L2 to S1. However, both fine and gross sensation were decreased in the L3 distribution. Sensation was intact to the remaining nerve-root distributions. The Babinski sign was negative for both lower extremities, and clonus was within physiologic limits. Examination of his gait was notable for quadriceps buckling with ambulation.
Radiographic Examination
The patient initially presented to his primary care physician, who evaluated his symptoms with a computed tomography scan of his abdomen and pelvis. This showed a mass of the right L3 transverse process (Figure 1). The patient was referred to us for further management of this lesion. Dedicated magnetic resonance imaging of his lumbar spine was performed, showing an expansile, lytic, homogeneous mass in the patient’s right L3 transverse process. The mass showed a significant mass effect, compressing the exiting nerve roots and, presumably, his right ureter (Figure 2). A bone scan showed monostotic disease. The patient had failed conservative management, including physical therapy and anti-inflammatory medications. His right-sided radiculopathy was worsening, and he complained that the pain was affecting his quality of life and limiting his performance of his daily activities. A pain management specialist was requested to better manage his pain. Considering progression of his condition, surgical management was discussed, leading to a planned biopsy and resection of the mass.
Surgical Procedure
The patient was taken into the operating room and positioned prone on a Jackson table with a Wilson frame. Fluoroscopy was used to localize the right L3 transverse process. An incision was made over the right L3 transverse process and a Wiltse intramuscular approach was performed. After the right L3 transverse process was identified, the soft tissue from the transverse process was retracted in all directions, including medially up to the pedicle. The intertransverse ligament was detached from both the cephalad and caudal edges of the transverse process. We used a Woodson elevator to perform subperiosteal dissection to remove the soft tissue circumferentially. After dissection, we placed a Cobb elevator to protect the rostral and caudal soft tissue and used a high-speed burr to amputate the lytic transverse process at its base. The transverse process was removed en bloc (Figure 3) and sent for frozen pathologic evaluation (Figure 4). After the diagnosis of a benign lesion, the wound was closed in layers.
Complete resolution of both urinary and neurologic symptoms were immediately noted and up to 1 month postoperatively.
Discussion
Primary bone tumors of the spine are rare, with a reported incidence of 2.5 to 8.5 per 100,000 people per year.6 The estimated incidence of benign primary tumors involving the spine accounts for about 1% of all primary skeletal tumors and nearly 5% for malignant tumors.7-9 In contrast, secondary tumors involving the bony spinal column are relatively common. Postmortem studies indicate up to 70% of cancer patients demonstrate axial skeletal involvement.10,11 The most commonly encountered benign tumors affecting the spine include giant cell tumors, osteoid osteomas, osteoblastomas, and hemangiomas. Chordomas are frequently reported as the most common malignant primary spine neoplasms. Of all primary benign bone lesions, fibrous dysplasia accounts for approximately 1.4%.8
Primary and secondary malignant osseous tumors have a predilection for the anterior column, and primary benign lesions usually affect the posterior column.8,12-14 Because of the greater blood supply and more direct communication with the viscera via the Batson plexus, the anterior column is most likely to be seeded by metastatic disease. Similarly, hemangiomas and multiple myeloma are typically located in the anterior column, most likely because of the more abundant blood supply there. Chordomas are also found in this cancellous anterior column. Osteoid osteoma, osteoblastoma, and aneurysmal bone cysts are found within the more cortical architecture of posterior elements. The location of this patient’s lesion within the transverse process elevates confidence in the diagnosis of a benign lesion.
The conventional, isolated form of fibrous dysplasia was originally described in 1942 by Lichtenstein and Jaffe.2 They described 15 cases of benign “nonosteogenic fibromas” near the ends of long bones in young patients. Monostotic fibrous dysplasia constitutes the majority of these cases, approximately 80%.1,2,8,15 Fibrous dysplasia may also present as part of McCune-Albright syndrome, in which case it is associated with precocious puberty and café au lait spots. Less commonly, they are associated with intramuscular myxomas, as in Mazabraud syndrome. The lesions in these syndromes are typically polyostotic. In all forms, fibrous dysplasia develops from an activating mutation in the gene that encodes the alpha subunit of the G protein on chromosome 20q13, activating cyclic adenylate cyclase and slowing the differentiation of osteoblasts.3,8
With regard to presentation, fibrous dysplasia is usually asymptomatic and discovered incidentally. The literature reports that the most common presenting symptom for patients with monostotic fibrous dysplasia of the spine is back pain localized to the lesion.15 Meredith and Healey2 completed a comprehensive review of 54 cases of monostotic fibrous dysplasia involving the spine in which the majority of symptoms included back pain, neck pain, sacral region pain, pathologic fracture, painful torticollis, progressive myelopathy, paresthesias of the foot, and only 1 case of radiculopathy involving thoracic vertebra. In normal anatomy, the ureter lies within retroperitoneal fat anterior to the psoas muscle and L2-L5 transverse processes and is normally mobile.16-18 This becomes clinically significant in lean patients as the ureter becomes closer to the spine. There are several reports of iatrogenic ureter injury in lumbar disc surgery.16-18 Normal variants, including medialization towards the spine, may predispose the ureters to injury, iatrogenic, or otherwise. In fact, medialization of the ureters occurs commonly in black men and usually involves the right side, which may have occurred in this black patient.19
Fibrous dysplasia is most often diagnosed by its radiographic appearance or biopsy. However, recent data suggest that deoxyribonucleic acid (DNA) analysis may soon be able to diagnose this process.20 Imaging typically reveals expansile, central lytic lesions within the medullary cavity. Pathology shows dense fibroblasts around immature woven bone, commonly referred to as “Chinese lettering.” The treatment varies from observation to en bloc surgical resection. Clinical observation is warranted for asymptomatic or incidental findings of monostotic fibrous dysplasia, as long as the risk for pathologic fracture is low.11 Bisphosphonate therapy, both oral and intravenous, offers promising outcomes for the treatment of fibrous dysplasia, with improvement in pain and function as well as in the radiographic findings.11,21 Management of monostotic fibrous dysplasia presenting as an isolated expansile mass of the transverse process in lumbar spine has rarely been described.3-5 Troop and Herring5 reported a case of monostotic fibrous dysplasia in the lumbar spine, with involvement of the vertebral body and the posterior elements. Chow and coauthors3 and Harris and colleagues4 described the involvement of the transverse process of L4. Chow and coauthors’3 treatment consisted of excision that resulted in an asymptomatic patient at 8-year follow-up, while Harris and colleagues4 chose observation. In the latter study, the patient’s lower back pain persisted at 4-year follow-up.
Progressive enlargement, recurrence, and malignant transformation have all been described. Meredith and Healey2 reported the reappearance of monostotic fibrous dysplasia affecting C2, extending through the fusion mass to involve a previously unaffected vertebra 20 years after the original C2 posterior elements excision via posterior spinal fusion from C1 to C3. In the literature, the incidence of malignant transformation ranges from 0.4% to 4%.8 One case of malignant transformation in thoracic spine was reported by Fu and colleagues.22 Therefore, complete removal of all affected bone is recommended.1,2,4,5,15,22,23
Conclusion
We present an unusual condition with complete resolution of symptoms after surgical resection. Several points may be considered from this report. Fibrous dysplasia lesions have been found in all bones of the body, including the skull, face, and extremities; however, monostotic fibrous dysplasia involving the spine is rare.11,23,24 Furthermore, there are no other reports of these lesions causing simultaneous nerve compression and urologic symptoms. Considering anatomy, clinicians may consider lesions of the lumbar transverse process in patients presenting to orthopedic surgeons with urinary symptoms, especially when combined with neurologic symptoms. In these lesions, fibrous dysplasia should be within the differential diagnosis. Clinicians should also recognize that complete resolution of symptoms has been reported with wide resection of these lesions.
1. Leet AI, Magur E, Lee JS, Weintroub S, Robey PG, Collins MT. Fibrous dysplasia in the spine: prevalence of lesions and association with scoliosis. J Bone Joint Surg Am. 2004;86(3):531-537.
2. Meredith DS, Healey JH. Twenty-year follow-up of monostotic fibrous dysplasia of the second cervical vertebra: a case report and review of the literature. J Bone Joint Surg Am. 2011;93(13):e74.
3. Chow LT, Griffith J, Chow WH, Kumta SM. Monostotic fibrous dysplasia of the spine: report of a case involving the lumbar transverse process and review of the literature. Arch Orthop Trauma Surg. 2000;120(7-8):460-464.
4. Harris WH, Dudley HR Jr, Barry RJ. The natural history of fibrous dysplasia. An orthopaedic, pathologic, and roentgenographic study. J Bone Joint Surg Am. 1962;44(2):207-233.
5. Troop JK, Herring JA. Monostotic fibrous dysplasia of the lumbar spine: case report and review of the literature. J Pediatr Orthop. 1988;8(5):599-601.
6. Dreghorn CR, Newman RJ, Hardy GJ, Dickson RA. Primary tumors of the axial skeleton. Experience of the Leeds Regional Bone Tumor Registry. Spine. 1990;15(2):137-140.
7. Schuster JM, Grady MS. Medical management and adjuvant therapies in spinal metastatic disease. Neurosurg Focus. 2001;11(6):e3.
8. Unni K. Introduction and scope. In: Unni K, ed. Dahlin’s Bone Tumors—General Aspects and Data on 11,087 Cases. Philadelphia, PA: Lippincott-Raven; 1996:1-9.
9. Wong DA, Fornasier VL, MacNab I. Spinal metastases: the obvious, the occult, and the impostors. Spine. 1990;15(1):1-4.
10. Dagi TF, Schmidek HH. Vascular tumors of the spine. In: Sundaresan N, Schmidek HH, Schiller AL, eds. Tumors of the Spine: Diagnosis and Clinical Management. Philadelphia, PA: W.B. Saunders Co; 1990:181-191.
11. DiCaprio M, Enneking W. Fibrous dysplasia. Pathophysiology, evaluation, and treatment. J Bone Joint Surg Am. 2005;87(8):1848-1864.
12. Gasbarrini A, Cappuccio M, Mirabile L, et al. Spinal metastases: treatment evaluation algorithm. Eur Rev Med Pharmacol Sci. 2004;8(6):265-274.
13. Loblaw DA, Laperriere NJ, Mackillop WJ. A population-based study of malignant spinal cord compression in Ontario. Clin Oncol. 2003;15(4):211-217.
14. Ortiz Gómez JA. The incidence of vertebral body metastases. Int Orthop. 1995;19(5):309-311.
15. Avimadje AM, Goupille P, Zerkak D, Begnard G, Brunais-Besse J, Valat JP. Monostotic fibrous dysplasia of the lumbar spine. Joint Bone Spine. 2000;67(1):65-70.
16. Isiklar ZU, Lindsey RW, Coburn M. Ureteral injury after anterior lumbar interbody fusion. A case report. Spine. 1996;21(20):2379-2382.
17. Krone A, Heller V, Osterhage HR. Ureteral injury in lumbar disc surgery. Acta Neurochir (Wien). 1985;78(3-4):108–112.
18. Cho KT, Im SH, Hong SK. Ureteral injury after inadvertent violation of the intertransverse space during posterior lumbar diskectomy: a case report. Surg Neurol. 2008;69(2):135-137.
19. Adam EJ, Desai SC, Lawton G. Racial variations in normal ureteric course. Clin Radiol. 1985;36(4):373-375.
20. Stathopoulos IP, Balanika AP, Baltas CS, et al. Fibrous dysplasia; confirmation of clinical diagnosis by DNA tests instead of biopsy. J Musculoskelet Neuronal Interact. 2013;13(1):120-123.
21. Lane JM, Khan SN, O’Connor WJ, et al. Bisphosphonate therapy in fibrous dysplasia. Clin Orthop Relat Res. 2001;382:6-12.
22. Fu CJ, Hsu CY, Shih TT, Wu MZ. Monostotic fibrous dysplasia of the thoracic spine with malignant transformation. J Formos Med Assoc. 2004;103(9):711-714.
23. McCarthy EF. Fibro-osseous lesions of the maxillofacial bones. Head Neck Pathol. 2013;7(1):5-10.
24. Manjila S, Zender CA, Weaver J, Rodgers M, Cohen AR. Aneurysmal bone cyst within fibrous dysplasia of the anterior skull base: continued intracranial extension after endoscopic resections requiring craniofacial approach with free tissue transfer reconstruction [published online ahead of print February 26, 2013]. Childs Nerv Syst. 2013;29(7).
1. Leet AI, Magur E, Lee JS, Weintroub S, Robey PG, Collins MT. Fibrous dysplasia in the spine: prevalence of lesions and association with scoliosis. J Bone Joint Surg Am. 2004;86(3):531-537.
2. Meredith DS, Healey JH. Twenty-year follow-up of monostotic fibrous dysplasia of the second cervical vertebra: a case report and review of the literature. J Bone Joint Surg Am. 2011;93(13):e74.
3. Chow LT, Griffith J, Chow WH, Kumta SM. Monostotic fibrous dysplasia of the spine: report of a case involving the lumbar transverse process and review of the literature. Arch Orthop Trauma Surg. 2000;120(7-8):460-464.
4. Harris WH, Dudley HR Jr, Barry RJ. The natural history of fibrous dysplasia. An orthopaedic, pathologic, and roentgenographic study. J Bone Joint Surg Am. 1962;44(2):207-233.
5. Troop JK, Herring JA. Monostotic fibrous dysplasia of the lumbar spine: case report and review of the literature. J Pediatr Orthop. 1988;8(5):599-601.
6. Dreghorn CR, Newman RJ, Hardy GJ, Dickson RA. Primary tumors of the axial skeleton. Experience of the Leeds Regional Bone Tumor Registry. Spine. 1990;15(2):137-140.
7. Schuster JM, Grady MS. Medical management and adjuvant therapies in spinal metastatic disease. Neurosurg Focus. 2001;11(6):e3.
8. Unni K. Introduction and scope. In: Unni K, ed. Dahlin’s Bone Tumors—General Aspects and Data on 11,087 Cases. Philadelphia, PA: Lippincott-Raven; 1996:1-9.
9. Wong DA, Fornasier VL, MacNab I. Spinal metastases: the obvious, the occult, and the impostors. Spine. 1990;15(1):1-4.
10. Dagi TF, Schmidek HH. Vascular tumors of the spine. In: Sundaresan N, Schmidek HH, Schiller AL, eds. Tumors of the Spine: Diagnosis and Clinical Management. Philadelphia, PA: W.B. Saunders Co; 1990:181-191.
11. DiCaprio M, Enneking W. Fibrous dysplasia. Pathophysiology, evaluation, and treatment. J Bone Joint Surg Am. 2005;87(8):1848-1864.
12. Gasbarrini A, Cappuccio M, Mirabile L, et al. Spinal metastases: treatment evaluation algorithm. Eur Rev Med Pharmacol Sci. 2004;8(6):265-274.
13. Loblaw DA, Laperriere NJ, Mackillop WJ. A population-based study of malignant spinal cord compression in Ontario. Clin Oncol. 2003;15(4):211-217.
14. Ortiz Gómez JA. The incidence of vertebral body metastases. Int Orthop. 1995;19(5):309-311.
15. Avimadje AM, Goupille P, Zerkak D, Begnard G, Brunais-Besse J, Valat JP. Monostotic fibrous dysplasia of the lumbar spine. Joint Bone Spine. 2000;67(1):65-70.
16. Isiklar ZU, Lindsey RW, Coburn M. Ureteral injury after anterior lumbar interbody fusion. A case report. Spine. 1996;21(20):2379-2382.
17. Krone A, Heller V, Osterhage HR. Ureteral injury in lumbar disc surgery. Acta Neurochir (Wien). 1985;78(3-4):108–112.
18. Cho KT, Im SH, Hong SK. Ureteral injury after inadvertent violation of the intertransverse space during posterior lumbar diskectomy: a case report. Surg Neurol. 2008;69(2):135-137.
19. Adam EJ, Desai SC, Lawton G. Racial variations in normal ureteric course. Clin Radiol. 1985;36(4):373-375.
20. Stathopoulos IP, Balanika AP, Baltas CS, et al. Fibrous dysplasia; confirmation of clinical diagnosis by DNA tests instead of biopsy. J Musculoskelet Neuronal Interact. 2013;13(1):120-123.
21. Lane JM, Khan SN, O’Connor WJ, et al. Bisphosphonate therapy in fibrous dysplasia. Clin Orthop Relat Res. 2001;382:6-12.
22. Fu CJ, Hsu CY, Shih TT, Wu MZ. Monostotic fibrous dysplasia of the thoracic spine with malignant transformation. J Formos Med Assoc. 2004;103(9):711-714.
23. McCarthy EF. Fibro-osseous lesions of the maxillofacial bones. Head Neck Pathol. 2013;7(1):5-10.
24. Manjila S, Zender CA, Weaver J, Rodgers M, Cohen AR. Aneurysmal bone cyst within fibrous dysplasia of the anterior skull base: continued intracranial extension after endoscopic resections requiring craniofacial approach with free tissue transfer reconstruction [published online ahead of print February 26, 2013]. Childs Nerv Syst. 2013;29(7).
Thyroid nodule: not as clear-cut as it seems
Benign etiologies and primary thyroid cancers are the most common causes of incidental thyroid nodules. Clinically evident metastases to the thyroid gland are not common and account for 2%-3% of thyroid cancers, though the incidence of thyroid metastases reaches 24% in autopsy studies.1 The most common clinically detected thyroid metastases originate from renal cell carcinoma (RCC; 48.1%).2 We report here a rare case of a man with clear-cell RCC with late recurrence in the thyroid gland as a solitary metastasis, 13 years after the primary diagnosis.
Click on the PDF icon at the top of this introduction to read the full article.
Benign etiologies and primary thyroid cancers are the most common causes of incidental thyroid nodules. Clinically evident metastases to the thyroid gland are not common and account for 2%-3% of thyroid cancers, though the incidence of thyroid metastases reaches 24% in autopsy studies.1 The most common clinically detected thyroid metastases originate from renal cell carcinoma (RCC; 48.1%).2 We report here a rare case of a man with clear-cell RCC with late recurrence in the thyroid gland as a solitary metastasis, 13 years after the primary diagnosis.
Click on the PDF icon at the top of this introduction to read the full article.
Benign etiologies and primary thyroid cancers are the most common causes of incidental thyroid nodules. Clinically evident metastases to the thyroid gland are not common and account for 2%-3% of thyroid cancers, though the incidence of thyroid metastases reaches 24% in autopsy studies.1 The most common clinically detected thyroid metastases originate from renal cell carcinoma (RCC; 48.1%).2 We report here a rare case of a man with clear-cell RCC with late recurrence in the thyroid gland as a solitary metastasis, 13 years after the primary diagnosis.
Click on the PDF icon at the top of this introduction to read the full article.
Distal Ulna Fracture With Delayed Ulnar Nerve Palsy in a Baseball Player
Ulnar nerve injury leads to clawing of the ulnar digits and loss of digital abduction and adduction because of paralysis of the ulnar innervated extrinsic and intrinsic muscles. Isolated motor paralysis without sensory deficit can occur from compression within the Guyon canal.1 Cubital tunnel at the elbow is the most common site for ulnar nerve compression.2 Compression at both levels can be encountered in sports-related activities. Nerve compression in the Guyon canal can occur with bicycling and is known as cyclist’s palsy,3-6 but it can also develop from canoeing.7 Cubital tunnel syndrome is the most common neuropathy of the elbow among throwing athletes, especially in baseball pitchers and can result from nerve traction and compression within the fibro-osseous tunnel or subluxation out of the tunnel.2 Both compression syndromes can develop from repetitive stress and/or pressure to the nerve in the retrocondylar groove.
Ulnar nerve palsy may be associated with forearm fractures, which is usually caused by simultaneous ulna and radius fractures, especially in children.8-12 To our knowledge, there are no reports in the literature of an ulnar nerve palsy associated with an isolated ulnar shaft fracture in an adult. We report a case of delayed ulnar nerve palsy after an ulnar shaft fracture in a baseball player. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 19-year-old, right hand–dominant college baseball player was batting right-handed in an intrasquad scrimmage when a high and inside pitched ball from a right-handed pitcher struck the volar-ulnar aspect of his right forearm. Examination in the training room and emergency department revealed moderate swelling and ecchymosis over the distal third of the ulna. He had a normal neurovascular examination, including normal sensation to light touch and normal finger abduction/adduction and wrist flexion/extension. He was otherwise healthy. Radiographs of the right forearm showed a minimally displaced transverse fracture of the distal third of the ulna (Figures 1A, 1B).
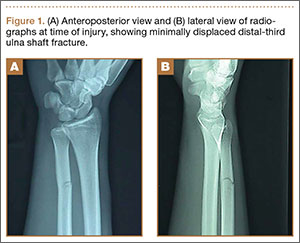
The patient was initially treated with a well-padded, removable, long-arm posterior splint for 2 weeks with serial examinations each day in the training room. At 2-week follow-up, he reported less pain and swelling but stated that his hand had “felt funny” the past several days. Examination revealed clawing of the ulnar digits with paresthesias in the ulnar nerve distribution (Figures 2A, 2B). His extrinsic muscle function was normal. Radiographs showed stable fracture alignment. Ulnar neuropathy was diagnosed, and treatment was observation with a plan for electromyography (EMG) at 6 weeks after injury if there were no signs of nerve recovery. Physical therapy was instituted and focused on improving intrinsic muscle and proprioceptive functions with the goal of an expeditious, but safe, return to playing baseball. Three weeks after his injury, the patient had decreased tenderness at his fracture site and was given a forearm pad and sleeve for light, noncontact baseball activity (Figure 3). A long velcro wrist splint was used during conditioning and when not playing baseball. Forearm supination and pronation were limited initially because of patient discomfort and to prevent torsional fracture displacement or delayed healing. Six weeks after his injury, the patient returned to hitting and was showing early signs of improved sensation and intrinsic hand strength. He had progressed to a light throwing program and reported difficult hand coordination, poor ball control, and overall difficulty in accurately throwing over the next 3 to 4 months. Because of his difficulty with ball control, the patient began a progressive return to full-game activity over 6 weeks, which initially included a return to batting only, then playing in the outfield, and, eventually, a return to his normal position in the infield. Serial radiographs continued to show good fracture alignment with appropriate new bone formation (Figures 4A, 4B). Normal motor strength was noted at 3 months after injury and normal sensation at 4 months after injury.
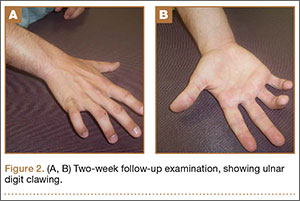
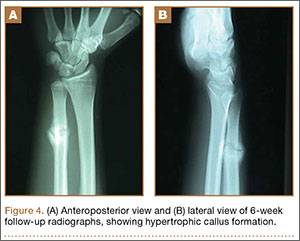
By the end of his summer league, 6 months after his injury, the patient was named Most Valuable Player and had a batting average over .400. He reported near-normal hand function. One year after injury, his examination revealed normal hand function (Figure 5), including normal sensation to light touch, 5/5 intrinsic hand function, and symmetric grip strength. Radiographs showed a healed fracture (Figures 6A, 6B). The patient has gone on to play more than 9 years of professional baseball.
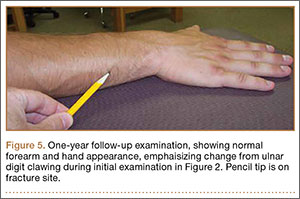
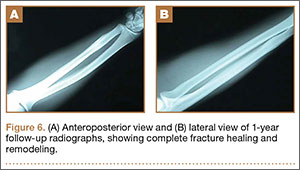
Discussion
The ulnar nerve has a course that runs down the volar compartment of the distal forearm. The flexor carpi ulnaris provides coverage to the nerve in this area. Proximal to the wrist, the nerve emerges from under the flexor carpi ulnaris tendon and passes deep to the flexor retinaculum, which is the distal extension of the antebrachial fascia and blends distally into the palmar carpal ligament.13 In our patient, the most likely cause of this presentation of ulnar neuropathy was the direct blow to the nerve from the high-intensity impact of a thrown baseball to this superficial and exposed area of the forearm. Since the patient presented with delayed paresthesias and ulnar clawing 2 weeks after injury, possible contributing causes could be evolving pressure or nerve damage from a perineural hematoma and/or intraneural hematoma or increased local pressure from intramuscular hemorrhage.14 There are both acute and chronic cases of ulnar nerve entrapment by bone or scar tissue that resolved by surgical decompression.8-12 Surgical exploration was not deemed necessary in our case because the fracture was minimally displaced, and the patient regained sensation and motor function over the course of 3 to 4 months.
Nerve injuries can be classified as neurapraxia, axonotmesis, or neurotmesis. Neurapraxia is the mildest form of nerve injury and neurotmesis the most severe. Neurapraxia may be associated with a temporary block to conduction or nerve demyelination without axonal disruption. Spontaneous recovery takes 2 weeks to 2 months. Axonotmesis involves an actual loss of axonal continuity; however, connective tissue supporting structures remain intact and allow axonal regeneration. Finally, neurotmesis is transection of the peripheral nerve, and spontaneous regeneration is not possible. The mechanism of injury in our patient suggests that the pathology was neurapraxia.1,15
Management of these injuries should proceed according to basic extremity injury–care practices. Initial care should include thorough neurovascular and radiographic evaluations. If nerve deficits are present with a closed injury and minimal fracture displacement, treatment can include observation and serial examinations with a baseline EMG, or waiting until 4 to 6 weeks after injury to obtain an EMG if there are no signs of nerve recovery. Early EMG testing and surgical exploration may be warranted if there is a concern for nerve disruption or entrapment, such as marked fracture displacement or an open injury. Additional early-care measures should include swelling control modalities and immobilization based on the type of fracture. Ultrasound was not readily available at the time of our patient’s injury, but it may be a helpful adjunct in guiding decision-making regarding whether to perform early surgical exploration for hematoma evacuation or nerve injury.16-18 Our case report was intended to provide an awareness of the unusual association between an isolated ulnar shaft fracture and a delayed ulnar nerve palsy in an athlete. Nerve injuries may be unrecognized in some patients in a trauma situation, since the focus is usually on the fracture and the typical patient does not have to return to high-demand, coordinated athletic activity, such as throwing a ball. Because of the possible delayed presentation of these nerve injuries, close observation of nerve function after ulna fractures from blunt trauma is warranted.
1. Dhillon MS, Chu ML, Posner MA. Demyelinating focal motor neuropathy of the ulnar nerve masquerading as compression in Guyon’s canal: a case report. J Hand Surg Am. 2003;28(1):48-51.
2. Hariri S, McAdams TR. Nerve injuries about the elbow. Clin Sports Med. 2010;29(4):655-675.
3. Akuthota V, Plastaras C, Lindberg K, Tobey J, Press J, Garvan C. The effect of long-distance bicycling on ulnar and median nerves: an electrophysiologic evaluation of cyclist palsy. Am J Sports Med. 2005;33(8):1224-1230.
4. Capitani D, Beer S. Handlebar palsy--a compression syndrome of the deep terminal (motor) branch of the ulnar nerve in biking. J Neurol. 2002;249(10):1441-1445.
5. Patterson JM, Jaggars MM, Boyer MI. Ulnar and median nerve palsy in long-distance cyclists. A prospective study. Am J Sports Med. 2003;31(4):585-589.
6. Slane J, Timmerman M, Ploeg HL, Thelen DG. The influence of glove and hand position on pressure over the ulnar nerve during cycling. Clin Biomech (Bristol, Avon). 2011;26(6):642-648.
7. Paul F, Diesta FJ, Ratzlaff T, Vogel HP, Zipp F. Combined ulnar nerve palsy in Guyon’s canal and distal median nerve irritation following excessive canoeing. Clinical Neurophysiology. 2007;118(4):e81-e82.
8. Hirasawa H, Sakai A, Toba N, Kamiuttanai M, Nakamura T, Tanaka K. Bony entrapment of ulnar nerve after closed forearm fracture: a case report. J Orthop Surg (Hong Kong). 2004;12(1):122-125.
9. Dahlin LB, Düppe H. Injuries to the nerves associated with fractured forearms in children. Scand J Plast Reconstr Surg Hand Surg. 2007;41(4):207-210.
10. Neiman R, Maiocco B, Deeney VF. Ulnar nerve injury after closed forearm fractures in children. J Pediatr Orthop. 1998;18(5):683-685.
11. Pai VS. Injury of the ulnar nerve associated with fracture of the ulna: A case report. J Orthop Surgery. 1999;7(2):73.
12. Suganuma S, Tada K, Hayashi H, Segawa T, Tsuchiya H. Ulnar nerve palsy associated with closed midshaft forearm fractures. Orthopedics. 2012;35(11):e1680-e1683.
13. Ombaba J, Kuo M, Rayan G. Anatomy of the ulnar tunnel and the influence of wrist motion on its morphology. J Hand Surg Am. 2010;35A:760-768.
14. Vijayakumar R, Nesathurai S, Abbott KM, Eustace S. Ulnar neuropathy resulting from diffuse intramuscular hemorrhage: a case report. Arch Phys Med Rehabil. 2000;81(8):1127-1130.
15. Browner, Bruce. Skeletal Trauma: Basic Science, Management, and Reconstruction [eBook]. 4th ed. Philadelphia, PA: WB Saunders Company; 2009:1487.
16. Koenig RW, Pedro MT, Heinen CP, et al. High-resolution ultrasonography in evaluating peripheral nerve entrapment and trauma. Neurosurg Focus. 2009;26(2):E13.
17. Zhu J, Liu F, Li D, Shao J, Hu B. Preliminary study of the types of traumatic peripheral nerve injuries by ultrasound. Eur Radiol. 2011;21(5):1097-1101.
18. Lee FC, Singh H, Nazarian LN, Ratliff JK. High-resolution ultrasonography in the diagnosis and intra-operative management of peripheral nerve lesions. J Neurosurg. 2011;114(1):206-221.
Ulnar nerve injury leads to clawing of the ulnar digits and loss of digital abduction and adduction because of paralysis of the ulnar innervated extrinsic and intrinsic muscles. Isolated motor paralysis without sensory deficit can occur from compression within the Guyon canal.1 Cubital tunnel at the elbow is the most common site for ulnar nerve compression.2 Compression at both levels can be encountered in sports-related activities. Nerve compression in the Guyon canal can occur with bicycling and is known as cyclist’s palsy,3-6 but it can also develop from canoeing.7 Cubital tunnel syndrome is the most common neuropathy of the elbow among throwing athletes, especially in baseball pitchers and can result from nerve traction and compression within the fibro-osseous tunnel or subluxation out of the tunnel.2 Both compression syndromes can develop from repetitive stress and/or pressure to the nerve in the retrocondylar groove.
Ulnar nerve palsy may be associated with forearm fractures, which is usually caused by simultaneous ulna and radius fractures, especially in children.8-12 To our knowledge, there are no reports in the literature of an ulnar nerve palsy associated with an isolated ulnar shaft fracture in an adult. We report a case of delayed ulnar nerve palsy after an ulnar shaft fracture in a baseball player. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 19-year-old, right hand–dominant college baseball player was batting right-handed in an intrasquad scrimmage when a high and inside pitched ball from a right-handed pitcher struck the volar-ulnar aspect of his right forearm. Examination in the training room and emergency department revealed moderate swelling and ecchymosis over the distal third of the ulna. He had a normal neurovascular examination, including normal sensation to light touch and normal finger abduction/adduction and wrist flexion/extension. He was otherwise healthy. Radiographs of the right forearm showed a minimally displaced transverse fracture of the distal third of the ulna (Figures 1A, 1B).
The patient was initially treated with a well-padded, removable, long-arm posterior splint for 2 weeks with serial examinations each day in the training room. At 2-week follow-up, he reported less pain and swelling but stated that his hand had “felt funny” the past several days. Examination revealed clawing of the ulnar digits with paresthesias in the ulnar nerve distribution (Figures 2A, 2B). His extrinsic muscle function was normal. Radiographs showed stable fracture alignment. Ulnar neuropathy was diagnosed, and treatment was observation with a plan for electromyography (EMG) at 6 weeks after injury if there were no signs of nerve recovery. Physical therapy was instituted and focused on improving intrinsic muscle and proprioceptive functions with the goal of an expeditious, but safe, return to playing baseball. Three weeks after his injury, the patient had decreased tenderness at his fracture site and was given a forearm pad and sleeve for light, noncontact baseball activity (Figure 3). A long velcro wrist splint was used during conditioning and when not playing baseball. Forearm supination and pronation were limited initially because of patient discomfort and to prevent torsional fracture displacement or delayed healing. Six weeks after his injury, the patient returned to hitting and was showing early signs of improved sensation and intrinsic hand strength. He had progressed to a light throwing program and reported difficult hand coordination, poor ball control, and overall difficulty in accurately throwing over the next 3 to 4 months. Because of his difficulty with ball control, the patient began a progressive return to full-game activity over 6 weeks, which initially included a return to batting only, then playing in the outfield, and, eventually, a return to his normal position in the infield. Serial radiographs continued to show good fracture alignment with appropriate new bone formation (Figures 4A, 4B). Normal motor strength was noted at 3 months after injury and normal sensation at 4 months after injury.
By the end of his summer league, 6 months after his injury, the patient was named Most Valuable Player and had a batting average over .400. He reported near-normal hand function. One year after injury, his examination revealed normal hand function (Figure 5), including normal sensation to light touch, 5/5 intrinsic hand function, and symmetric grip strength. Radiographs showed a healed fracture (Figures 6A, 6B). The patient has gone on to play more than 9 years of professional baseball.
Discussion
The ulnar nerve has a course that runs down the volar compartment of the distal forearm. The flexor carpi ulnaris provides coverage to the nerve in this area. Proximal to the wrist, the nerve emerges from under the flexor carpi ulnaris tendon and passes deep to the flexor retinaculum, which is the distal extension of the antebrachial fascia and blends distally into the palmar carpal ligament.13 In our patient, the most likely cause of this presentation of ulnar neuropathy was the direct blow to the nerve from the high-intensity impact of a thrown baseball to this superficial and exposed area of the forearm. Since the patient presented with delayed paresthesias and ulnar clawing 2 weeks after injury, possible contributing causes could be evolving pressure or nerve damage from a perineural hematoma and/or intraneural hematoma or increased local pressure from intramuscular hemorrhage.14 There are both acute and chronic cases of ulnar nerve entrapment by bone or scar tissue that resolved by surgical decompression.8-12 Surgical exploration was not deemed necessary in our case because the fracture was minimally displaced, and the patient regained sensation and motor function over the course of 3 to 4 months.
Nerve injuries can be classified as neurapraxia, axonotmesis, or neurotmesis. Neurapraxia is the mildest form of nerve injury and neurotmesis the most severe. Neurapraxia may be associated with a temporary block to conduction or nerve demyelination without axonal disruption. Spontaneous recovery takes 2 weeks to 2 months. Axonotmesis involves an actual loss of axonal continuity; however, connective tissue supporting structures remain intact and allow axonal regeneration. Finally, neurotmesis is transection of the peripheral nerve, and spontaneous regeneration is not possible. The mechanism of injury in our patient suggests that the pathology was neurapraxia.1,15
Management of these injuries should proceed according to basic extremity injury–care practices. Initial care should include thorough neurovascular and radiographic evaluations. If nerve deficits are present with a closed injury and minimal fracture displacement, treatment can include observation and serial examinations with a baseline EMG, or waiting until 4 to 6 weeks after injury to obtain an EMG if there are no signs of nerve recovery. Early EMG testing and surgical exploration may be warranted if there is a concern for nerve disruption or entrapment, such as marked fracture displacement or an open injury. Additional early-care measures should include swelling control modalities and immobilization based on the type of fracture. Ultrasound was not readily available at the time of our patient’s injury, but it may be a helpful adjunct in guiding decision-making regarding whether to perform early surgical exploration for hematoma evacuation or nerve injury.16-18 Our case report was intended to provide an awareness of the unusual association between an isolated ulnar shaft fracture and a delayed ulnar nerve palsy in an athlete. Nerve injuries may be unrecognized in some patients in a trauma situation, since the focus is usually on the fracture and the typical patient does not have to return to high-demand, coordinated athletic activity, such as throwing a ball. Because of the possible delayed presentation of these nerve injuries, close observation of nerve function after ulna fractures from blunt trauma is warranted.
Ulnar nerve injury leads to clawing of the ulnar digits and loss of digital abduction and adduction because of paralysis of the ulnar innervated extrinsic and intrinsic muscles. Isolated motor paralysis without sensory deficit can occur from compression within the Guyon canal.1 Cubital tunnel at the elbow is the most common site for ulnar nerve compression.2 Compression at both levels can be encountered in sports-related activities. Nerve compression in the Guyon canal can occur with bicycling and is known as cyclist’s palsy,3-6 but it can also develop from canoeing.7 Cubital tunnel syndrome is the most common neuropathy of the elbow among throwing athletes, especially in baseball pitchers and can result from nerve traction and compression within the fibro-osseous tunnel or subluxation out of the tunnel.2 Both compression syndromes can develop from repetitive stress and/or pressure to the nerve in the retrocondylar groove.
Ulnar nerve palsy may be associated with forearm fractures, which is usually caused by simultaneous ulna and radius fractures, especially in children.8-12 To our knowledge, there are no reports in the literature of an ulnar nerve palsy associated with an isolated ulnar shaft fracture in an adult. We report a case of delayed ulnar nerve palsy after an ulnar shaft fracture in a baseball player. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A 19-year-old, right hand–dominant college baseball player was batting right-handed in an intrasquad scrimmage when a high and inside pitched ball from a right-handed pitcher struck the volar-ulnar aspect of his right forearm. Examination in the training room and emergency department revealed moderate swelling and ecchymosis over the distal third of the ulna. He had a normal neurovascular examination, including normal sensation to light touch and normal finger abduction/adduction and wrist flexion/extension. He was otherwise healthy. Radiographs of the right forearm showed a minimally displaced transverse fracture of the distal third of the ulna (Figures 1A, 1B).
The patient was initially treated with a well-padded, removable, long-arm posterior splint for 2 weeks with serial examinations each day in the training room. At 2-week follow-up, he reported less pain and swelling but stated that his hand had “felt funny” the past several days. Examination revealed clawing of the ulnar digits with paresthesias in the ulnar nerve distribution (Figures 2A, 2B). His extrinsic muscle function was normal. Radiographs showed stable fracture alignment. Ulnar neuropathy was diagnosed, and treatment was observation with a plan for electromyography (EMG) at 6 weeks after injury if there were no signs of nerve recovery. Physical therapy was instituted and focused on improving intrinsic muscle and proprioceptive functions with the goal of an expeditious, but safe, return to playing baseball. Three weeks after his injury, the patient had decreased tenderness at his fracture site and was given a forearm pad and sleeve for light, noncontact baseball activity (Figure 3). A long velcro wrist splint was used during conditioning and when not playing baseball. Forearm supination and pronation were limited initially because of patient discomfort and to prevent torsional fracture displacement or delayed healing. Six weeks after his injury, the patient returned to hitting and was showing early signs of improved sensation and intrinsic hand strength. He had progressed to a light throwing program and reported difficult hand coordination, poor ball control, and overall difficulty in accurately throwing over the next 3 to 4 months. Because of his difficulty with ball control, the patient began a progressive return to full-game activity over 6 weeks, which initially included a return to batting only, then playing in the outfield, and, eventually, a return to his normal position in the infield. Serial radiographs continued to show good fracture alignment with appropriate new bone formation (Figures 4A, 4B). Normal motor strength was noted at 3 months after injury and normal sensation at 4 months after injury.
By the end of his summer league, 6 months after his injury, the patient was named Most Valuable Player and had a batting average over .400. He reported near-normal hand function. One year after injury, his examination revealed normal hand function (Figure 5), including normal sensation to light touch, 5/5 intrinsic hand function, and symmetric grip strength. Radiographs showed a healed fracture (Figures 6A, 6B). The patient has gone on to play more than 9 years of professional baseball.
Discussion
The ulnar nerve has a course that runs down the volar compartment of the distal forearm. The flexor carpi ulnaris provides coverage to the nerve in this area. Proximal to the wrist, the nerve emerges from under the flexor carpi ulnaris tendon and passes deep to the flexor retinaculum, which is the distal extension of the antebrachial fascia and blends distally into the palmar carpal ligament.13 In our patient, the most likely cause of this presentation of ulnar neuropathy was the direct blow to the nerve from the high-intensity impact of a thrown baseball to this superficial and exposed area of the forearm. Since the patient presented with delayed paresthesias and ulnar clawing 2 weeks after injury, possible contributing causes could be evolving pressure or nerve damage from a perineural hematoma and/or intraneural hematoma or increased local pressure from intramuscular hemorrhage.14 There are both acute and chronic cases of ulnar nerve entrapment by bone or scar tissue that resolved by surgical decompression.8-12 Surgical exploration was not deemed necessary in our case because the fracture was minimally displaced, and the patient regained sensation and motor function over the course of 3 to 4 months.
Nerve injuries can be classified as neurapraxia, axonotmesis, or neurotmesis. Neurapraxia is the mildest form of nerve injury and neurotmesis the most severe. Neurapraxia may be associated with a temporary block to conduction or nerve demyelination without axonal disruption. Spontaneous recovery takes 2 weeks to 2 months. Axonotmesis involves an actual loss of axonal continuity; however, connective tissue supporting structures remain intact and allow axonal regeneration. Finally, neurotmesis is transection of the peripheral nerve, and spontaneous regeneration is not possible. The mechanism of injury in our patient suggests that the pathology was neurapraxia.1,15
Management of these injuries should proceed according to basic extremity injury–care practices. Initial care should include thorough neurovascular and radiographic evaluations. If nerve deficits are present with a closed injury and minimal fracture displacement, treatment can include observation and serial examinations with a baseline EMG, or waiting until 4 to 6 weeks after injury to obtain an EMG if there are no signs of nerve recovery. Early EMG testing and surgical exploration may be warranted if there is a concern for nerve disruption or entrapment, such as marked fracture displacement or an open injury. Additional early-care measures should include swelling control modalities and immobilization based on the type of fracture. Ultrasound was not readily available at the time of our patient’s injury, but it may be a helpful adjunct in guiding decision-making regarding whether to perform early surgical exploration for hematoma evacuation or nerve injury.16-18 Our case report was intended to provide an awareness of the unusual association between an isolated ulnar shaft fracture and a delayed ulnar nerve palsy in an athlete. Nerve injuries may be unrecognized in some patients in a trauma situation, since the focus is usually on the fracture and the typical patient does not have to return to high-demand, coordinated athletic activity, such as throwing a ball. Because of the possible delayed presentation of these nerve injuries, close observation of nerve function after ulna fractures from blunt trauma is warranted.
1. Dhillon MS, Chu ML, Posner MA. Demyelinating focal motor neuropathy of the ulnar nerve masquerading as compression in Guyon’s canal: a case report. J Hand Surg Am. 2003;28(1):48-51.
2. Hariri S, McAdams TR. Nerve injuries about the elbow. Clin Sports Med. 2010;29(4):655-675.
3. Akuthota V, Plastaras C, Lindberg K, Tobey J, Press J, Garvan C. The effect of long-distance bicycling on ulnar and median nerves: an electrophysiologic evaluation of cyclist palsy. Am J Sports Med. 2005;33(8):1224-1230.
4. Capitani D, Beer S. Handlebar palsy--a compression syndrome of the deep terminal (motor) branch of the ulnar nerve in biking. J Neurol. 2002;249(10):1441-1445.
5. Patterson JM, Jaggars MM, Boyer MI. Ulnar and median nerve palsy in long-distance cyclists. A prospective study. Am J Sports Med. 2003;31(4):585-589.
6. Slane J, Timmerman M, Ploeg HL, Thelen DG. The influence of glove and hand position on pressure over the ulnar nerve during cycling. Clin Biomech (Bristol, Avon). 2011;26(6):642-648.
7. Paul F, Diesta FJ, Ratzlaff T, Vogel HP, Zipp F. Combined ulnar nerve palsy in Guyon’s canal and distal median nerve irritation following excessive canoeing. Clinical Neurophysiology. 2007;118(4):e81-e82.
8. Hirasawa H, Sakai A, Toba N, Kamiuttanai M, Nakamura T, Tanaka K. Bony entrapment of ulnar nerve after closed forearm fracture: a case report. J Orthop Surg (Hong Kong). 2004;12(1):122-125.
9. Dahlin LB, Düppe H. Injuries to the nerves associated with fractured forearms in children. Scand J Plast Reconstr Surg Hand Surg. 2007;41(4):207-210.
10. Neiman R, Maiocco B, Deeney VF. Ulnar nerve injury after closed forearm fractures in children. J Pediatr Orthop. 1998;18(5):683-685.
11. Pai VS. Injury of the ulnar nerve associated with fracture of the ulna: A case report. J Orthop Surgery. 1999;7(2):73.
12. Suganuma S, Tada K, Hayashi H, Segawa T, Tsuchiya H. Ulnar nerve palsy associated with closed midshaft forearm fractures. Orthopedics. 2012;35(11):e1680-e1683.
13. Ombaba J, Kuo M, Rayan G. Anatomy of the ulnar tunnel and the influence of wrist motion on its morphology. J Hand Surg Am. 2010;35A:760-768.
14. Vijayakumar R, Nesathurai S, Abbott KM, Eustace S. Ulnar neuropathy resulting from diffuse intramuscular hemorrhage: a case report. Arch Phys Med Rehabil. 2000;81(8):1127-1130.
15. Browner, Bruce. Skeletal Trauma: Basic Science, Management, and Reconstruction [eBook]. 4th ed. Philadelphia, PA: WB Saunders Company; 2009:1487.
16. Koenig RW, Pedro MT, Heinen CP, et al. High-resolution ultrasonography in evaluating peripheral nerve entrapment and trauma. Neurosurg Focus. 2009;26(2):E13.
17. Zhu J, Liu F, Li D, Shao J, Hu B. Preliminary study of the types of traumatic peripheral nerve injuries by ultrasound. Eur Radiol. 2011;21(5):1097-1101.
18. Lee FC, Singh H, Nazarian LN, Ratliff JK. High-resolution ultrasonography in the diagnosis and intra-operative management of peripheral nerve lesions. J Neurosurg. 2011;114(1):206-221.
1. Dhillon MS, Chu ML, Posner MA. Demyelinating focal motor neuropathy of the ulnar nerve masquerading as compression in Guyon’s canal: a case report. J Hand Surg Am. 2003;28(1):48-51.
2. Hariri S, McAdams TR. Nerve injuries about the elbow. Clin Sports Med. 2010;29(4):655-675.
3. Akuthota V, Plastaras C, Lindberg K, Tobey J, Press J, Garvan C. The effect of long-distance bicycling on ulnar and median nerves: an electrophysiologic evaluation of cyclist palsy. Am J Sports Med. 2005;33(8):1224-1230.
4. Capitani D, Beer S. Handlebar palsy--a compression syndrome of the deep terminal (motor) branch of the ulnar nerve in biking. J Neurol. 2002;249(10):1441-1445.
5. Patterson JM, Jaggars MM, Boyer MI. Ulnar and median nerve palsy in long-distance cyclists. A prospective study. Am J Sports Med. 2003;31(4):585-589.
6. Slane J, Timmerman M, Ploeg HL, Thelen DG. The influence of glove and hand position on pressure over the ulnar nerve during cycling. Clin Biomech (Bristol, Avon). 2011;26(6):642-648.
7. Paul F, Diesta FJ, Ratzlaff T, Vogel HP, Zipp F. Combined ulnar nerve palsy in Guyon’s canal and distal median nerve irritation following excessive canoeing. Clinical Neurophysiology. 2007;118(4):e81-e82.
8. Hirasawa H, Sakai A, Toba N, Kamiuttanai M, Nakamura T, Tanaka K. Bony entrapment of ulnar nerve after closed forearm fracture: a case report. J Orthop Surg (Hong Kong). 2004;12(1):122-125.
9. Dahlin LB, Düppe H. Injuries to the nerves associated with fractured forearms in children. Scand J Plast Reconstr Surg Hand Surg. 2007;41(4):207-210.
10. Neiman R, Maiocco B, Deeney VF. Ulnar nerve injury after closed forearm fractures in children. J Pediatr Orthop. 1998;18(5):683-685.
11. Pai VS. Injury of the ulnar nerve associated with fracture of the ulna: A case report. J Orthop Surgery. 1999;7(2):73.
12. Suganuma S, Tada K, Hayashi H, Segawa T, Tsuchiya H. Ulnar nerve palsy associated with closed midshaft forearm fractures. Orthopedics. 2012;35(11):e1680-e1683.
13. Ombaba J, Kuo M, Rayan G. Anatomy of the ulnar tunnel and the influence of wrist motion on its morphology. J Hand Surg Am. 2010;35A:760-768.
14. Vijayakumar R, Nesathurai S, Abbott KM, Eustace S. Ulnar neuropathy resulting from diffuse intramuscular hemorrhage: a case report. Arch Phys Med Rehabil. 2000;81(8):1127-1130.
15. Browner, Bruce. Skeletal Trauma: Basic Science, Management, and Reconstruction [eBook]. 4th ed. Philadelphia, PA: WB Saunders Company; 2009:1487.
16. Koenig RW, Pedro MT, Heinen CP, et al. High-resolution ultrasonography in evaluating peripheral nerve entrapment and trauma. Neurosurg Focus. 2009;26(2):E13.
17. Zhu J, Liu F, Li D, Shao J, Hu B. Preliminary study of the types of traumatic peripheral nerve injuries by ultrasound. Eur Radiol. 2011;21(5):1097-1101.
18. Lee FC, Singh H, Nazarian LN, Ratliff JK. High-resolution ultrasonography in the diagnosis and intra-operative management of peripheral nerve lesions. J Neurosurg. 2011;114(1):206-221.
Diagnosis and Management of Cold Urticaria
Cold urticaria is a rare condition characterized by a localized or systemic eruption of papules upon exposure of the skin to cold air, liquids, and/or objects. In some cases, angioedema and anaphylaxis can occur. The wheal-and-flare reaction results from a localized or systemic release of histamine, leukotrienes, and various other proinflammatory mast cell mediators. Cold urticaria can be acquired or follow an autosomal-dominant familial transmission pattern. Acquired cold urticaria often presents in young adulthood with a mean duration of 4 to 5 years and remission or improvement of symptoms after 5 years in 50% of cases.1 The familial variant most commonly presents in early childhood and endures throughout the patient’s life.2 Cold urticaria generally is classified as acute or chronic if symptoms persist for more than 6 weeks. Pharmacologic therapies with prophylactic effects that may reduce the intensity of symptoms or inhibit their development include antihistamines, leuko-triene receptor antagonists, biologics, and glucocorticoids. We present the case of a 23-year-old man with cold urticaria that was refractory to initial treatment with H1 antihistamines along with a review of the literature.
Case Report
A 23-year-old man presented to the dermatology clinic for evaluation of recurrent burning, itching, and sometimes development of a painful rash on the face, neck, and arms of 2 years’ duration that typically occurred following exposure to cold, wind, and rain. He also developed symptoms in warm weather when exposed to wind while sweating. His medical history was remarkable for asthma, which was not active. He was not taking any medications and had no known drug or environmental allergies. No other members of his household developed similar symptoms. His only successful means of prevention was to stay indoors, which thereby limited his activities.
Physical examination of the dorsal hands following an ice cube test revealed numerous 3- to 5-mm urticarial papules with surrounding erythema (Figure).
Following the initial evaluation, the patient was treated unsuccessfully with a mix of first- and second-generation antihistamines in gradually increasing doses to a maximum dose of loratadine 20 mg once daily, cetirizine 20 mg once daily, and hydroxyzine 20 mg once daily. A course of montelukast 10 mg once daily was started in addition to the antihistamines and led to a reduction in the severity of the lesions but not the frequency and did not relieve the burning sensation; the patient subsequently discontinued therapy. Next, a trial of cyclosporine was attempted, but the patient reported that it caused emesis and subsequently discontinued treatment. The patient also did not tolerate prednisone. He eventually decided to treat his symptoms with lifestyle choices only, such as making sure to be well covered in cold temperatures.
Comment
Cold urticaria is a physical urticaria resulting from mast cell degranulation and the subsequent release of histamine and proinflammatory cytokines upon exposure of the skin to cold air, liquid, and/or objects. Symtpoms usually are limited to localized exposed areas of the skin but also can be generalized. Cold urticaria typically manifests as erythematous, pruritic papules and also may be accompanied by deep tissue involvement resulting in angioedema and/or anaphylaxis. Symptoms usually occur within minutes of cold exposure; however, in delayed-type cold urticaria, symptoms may develop 24 to 72 hours later.3 Prevalence is relatively equal in both sexes and is highest among young adults (ie, 18–27 years old), with a greater incidence associated with cold climates.4 In one study, the overall incidence of acquired cold urticaria in Central Europe was estimated to be 0.05%.1
Systemic involvement may occur with extensive cold contact, ranging in severity from generalized urticaria to anaphylaxis and involvement of the cardiovascular, respiratory, and/or gastrointestinal systems.5 Patients who exhibit systemic responses to cold exposure should avoid swimming in cold water, as this may induce anaphylaxis and result in injury or death. In a 2004 study that included 30 children with cold urticaria at a tertiary center in Boston,6 11 (36.7%) participants who underwent cold stimulation testing developed systemic symptoms; 5 (45.5%) participants experienced respiratory distress and 8 (72.7%) experienced a decrease in level of consciousness (eg, faintness, dizziness, hypotension). Aquatic activity was the trigger in all 11 participants except for 1 (9.0%), who experienced systemic symptoms on exposure to cold air. In the same study, 14 (46.7%) participants were diagnosed with asthma and 15 (50%) were diagnosed with allergic rhinitis. Of the 28 participants whose family histories were available for review, 25 (89.3%) had a family history of atopic disease.6 A 2008 Greek study4 of 62 adults with acquired cold urticaria found that 18 (29%) participants had at least 1 serious systemic response resulting in generalized urticaria or angioedema associated with hypotension (eg, dizziness, fainting, disorientation, shock). In both of these studies, a majority of the serious systemic reactions were associated with cold water activities.
Cold urticaria is primarily an idiopathic phenomenon but can be classified as acquired or familial. Acquired cold urticaria may result from primary or secondary causes, which can include cryoglobulinemia, human immunodeficiency virus, syphilis, mononucleosis, rubeola, toxoplasmosis, varicella, hepatitis, and various drugs (eg, penicillin, angiotensin-converting enzyme inhibitors, oral contraceptives).7 Familial causes include cryopyrin-associated periodic syndrome, phospholipase Cγ2 gene–associated antibody deficiency and immune dysregulation, Muckle-Wells syndrome, and neonatal-onset multisystem inflammatory disease.
Typically, cold urticaria is diagnosed using cold stimulation tests such as the ice cube test, in which an ice cube is applied directly to the patient’s skin for 3 to 5 minutes and a response is measured 10 minutes after its removal.8 This test has been shown to have a sensitivity of 83% to 90% and a specificity of 100%.9 Alternatively, cold urticaria may be diagnosed through the use of a Peltier element-based cold-provocation device, which exposes the patient to a variety of temperatures in order for clinicians to determine the threshold upon which there is an observable reaction. With a sensitivity of 93% and specificity of 100%, the accuracy of this test is similar to that of the ice cube test.10 If a patient has a history of serious systemic involvement, any testing that exposes the patient to extensive cold exposure should be used with caution.
Patients should be counseled about potential serious systemic symptoms and the importance of wearing appropriate cold-weather clothing. Avoidance of cold water activities and overexposure to cold weather also should be emphasized. Pharmacologic therapy for prophylaxis typically includes a second-generation H1 antihistamine (eg, cetirizine, loratadine, desloratadine). Since these drugs have been shown to be less sedating than first-generation antihistamines, they are considered a better choice for chronic treatment. At high doses, however, these medications may have a sedative effect; therefore, nighttime use is preferable if possible. The standard dosage is 5 mg to 10 mg daily for oral cetirizine, 10 mg daily for oral loratadine, and 5 mg daily for oral desloratadine; however, up to 4 times the standard dosage of these medications may be required for effective treatment of cold urticaria.11 Given the associated risk of anaphylaxis, patients should be prescribed an epinephrine pen and educated about its appropriate use, including the importance of keeping the pen accessible at all times.
In refractory cases of cold urticaria, an H2 antihistamine (eg, ranitidine) can be used in conjunction with H1 antihistamines.12 Omalizumab, an IgE-mediated treatment, also has been shown to be safe and effective in patients with recalcitrant physical urticaria, including cold urticaria.13,14 One report described the case of a 69-year-old woman with cold urticaria who was unable to leave the house without developing a widespread eruption on the face, trunk, and limbs.15 After undergoing a series of unsuccessful treatments, the patient was started on cyclosporine 125 mg twice daily, which was reduced to 100 mg twice daily after 4 weeks of therapy and then reduced to 75 mg twice daily after 4 months of treatment. One week after therapy was initiated the patient reported that she was able to leave the house, and after 4 weeks of treatment the lesions only developed on the hands and feet. The patient remained in remission with a low-dose therapy of cyclosporine 75 mg twice daily with lesions only occurring on the hands and feet. The low-dose maintenance therapy was associated with minimal adverse effects.15 To our knowledge, there are no known large studies on the efficacy of cyclosporine in the treatment of cold urticaria.
Leukotriene receptor antagonists (eg, montelukast, zafirlukast, zileuton) have been used to treat chronic urticaria. In one report, montelukast was used in a 29-year-old woman with cold urticaria who had initially been treated with cetirizine 30 mg daily, cyproheptadine 4 mg daily, and doxycycline 200 mg daily with minimal to no relief. After treatment with montelukast, she experienced notable and stable improvements in symptoms.16 Hydroxychloroquine also has been shown to be safe and to substantially improve quality of life in patients with idiopathic chronic urticaria.17 Methotrexate (with close patient monitoring for adverse effects) has been reported to benefit some patients whose chronic urticaria was unresponsive to standard treatment.18 Treatment regimens for chronic urticaria have shown variable success in the treatment of cold urticaria and may be considered in cases refractory to treatment with high-dose second-generation H1 antihistamines.
Topical application of capsaicin for 4 to 7 days has been shown to deplete the neuropeptides in sensory fibers that may be involved in cold reactions, although skin irritation may prevent usage.19
Prednisone therapy was used in a small study of 6 patients with acquired cold urticaria.20 Three patients were treated for periods of 3 to 5 days with prednisone 20 mg each morning. Three other patients were given a single dose of prednisone 20 mg or 25 mg in the morning, depending on body weight. Following prednisone therapy, complete or partial pruritus was subjectively improved in all 6 patients. Additionally, significant reductions in venous histamine concentrations at 5 and 10 minutes following cold immersion were noted (P<.05 and P<.025, respectively); however, no significant improvement in either erythema or edema was noted posttreatment following cold immersion.20 Despite these findings, prednisone has not been shown to consistently prevent histamine release. Another report noted the case of a 47-year-old man with cold urticaria who required hypothermic cardiopulmonary bypass. Pretreatment with prednisone 20 mg daily and preoperative hydrocortisone 100 mg intravenously did not prevent histamine release.21
Cold desensitization (ie, exposing progressively larger areas of the patient’s skin to increasingly colder water) may induce tolerance to cold and decrease the temperature threshold at which symptoms develop; however, patients with known serious systemic reactions should be tested with extreme caution and only under the supervision of a clinician.22,23 Tolerance may wane when cold desensitization therapy is stopped.
The prognosis for patients with acquired cold urticaria generally is good. Improvement of symptoms or full remission occurs within 5 to 6 years in 50% of patients.24 Once remission has occurred, patients generally remain symptom free. For other familial variants, symptoms may last a lifetime.
Conclusion
This case report and review of the literature highlights the limitations of cold urticaria and the importance of effective management in improving quality of life in affected patients. Symptoms may limit patients’ ability to work in certain environments, inhibit them from engaging in daily activities, and even prevent them from leaving their homes in colder temperatures. In addition to behavioral modifications, pharmacologic management may provide symptomatic relief. Antihistamines are the first line of treatment in cold urticaria. Second-generation antihistamines, which are more selective for H1 receptors and less sedating, are generally recommended. Up to 4 times the standard dosage of these medications may be required for effective treatment.5 The primary goal of therapy in mild to moderate cases is improvement in quality of life.
- Siebenhaar F, Weller K, Mlynek A, et al. Acquired cold urticaria: clinical picture and update on diagnosis and treatment. Clin Exp Dermatol. 2007;32:241-245.
- Gandhi C, Healy C, Wanderer AA, et al. Familial atypical cold urticaria: description of a new hereditary disease. J Allergy Clin Immunol. 2009;124:1245-1250.
- Bäck O, Larsen A. Delayed cold urticaria. Acta Derm Venereol. 1978;58:369-371.
- Katsarou-Katsari A, Makris M, Lagogianni E, et al. Clinical features and natural history of acquired cold urticaria in a tertiary referral hospital: a 10-year prospective study. J Eur Acad Dermatol Venereol. 2008;22:1405-1411.
- Wanderer AA, Grandel KE, Wasserman SI, et al. Clinical characteristics of cold-induced systemic reactions in acquired cold urticaria syndromes: recommendations for prevention of this complication and a proposal for a diagnostic classification of cold urticaria. J Allergy Clin Immunol. 1986;78(3 Pt 1):417-423.
- Alangari AA, Twarog FJ, Shih MC, et al. Clinical features and anaphylaxis in children with cold urticaria. Pediatrics. 2004;113:e313-e317.
- Wanderer AA, Hoffman HM. The spectrum of acquired and familial cold-induced urticaria/urticaria-like syndromes. Immunol Allergy Clin North Am. 2004;24:259-286.
- Visitsuntorn N, Tuchinda M, Arunyanark N, et al. Ice cube test in children with cold urticaria. Asian Pac J Allergy Immunol. 1992;10:111-115.
- Neittaanmäki H. Cold urticaria. clinical findings in 220 patients. J Am Acad Dermatol. 1985;13:636-644.
- Siebenhaar F, Staubach P, Metz M, et al. Peltier effect-based temperature challenge: an improved method for diagnosing cold urticaria. J Allergy Clin Immunol. 2004;114:1224-1225.
- Siebenhaar F, Degener F, Zuberbier T, et al. High-dose desloratadine decreases wheal volume and improves cold provocation thresholds compared with standard-dose treatment in patients with acquired cold urticaria: a randomized, placebo-controlled, crossover study. J Allergy Clin Immunol. 2009;123:672-679.
- Duc J, Pécoud A. Successful treatment of idiopathic cold urticaria with the association of H1 and H2 antagonists: a case report. Ann Allergy. 1986;56:355-357.
- Metz M, Altrichter S, Ardelean E, et al. Anti-immunoglobulin E treatment of patients with recalcitrant physical urticaria. Int Arch Allergy Immunol. 2011;154:177-180.
- Boyce JA. Successful treatment of cold-induced urticaria/anaphylaxis with anti-IgE. J Allergy Clin Immunol. 2006;117:1415-1418.
- Marsland AM, Beck MH. Cold urticaria responding to systemic cyclosporine. Br J Dermatol. 2003;149:214-215.
- Hani N, Hartmann K, Casper C, et al. Improvement of cold urticaria by treatment with the leukotriene receptor antagonist montelukast. Acta Derm Venereol. 2000;80:229.
- Reeves GE, Boyle MJ, Bonfield J, et al. Impact of hydroxychloroquine therapy on chronic urticaria: chronic autoimmune urticaria study and evaluation. Intern Med J. 2004;34:182-186.
- Perez A, Woods A, Grattan CE. Methotrexate: a useful steroid-sparing agent in recalcitrant chronic urticaria. Br J Dermatol. 2010;162:191-194.
- Tóth-Kása I, Jancsó G, Obál F Jr, et al. Involvement of sensory nerve endings in cold and heat urticaria. J Invest Dermatol. 1983;80:34-36.
- Black AK, Keahey TM, Eady RA, et al. Dissociation of histamine release and clinical improvement following treatment of acquired cold urticaria by prednisone. Br J Clin Pharmacol. 1981;12:327-331.
- Johnston WE, Moss J, Philbin DM, et al. Management of cold urticaria during hypothermic cardiopulmonary bypass. N Engl J Med. 1982;306:219-221.
- Krause K, Zuberbier T, Maurer, M. Modern Approaches to the diagnosis and treatment of cold contact urticaria. Curr Allergy Asthma Rep. 2010;10:273-279.
- von Mackensen YA, Sticherling M. Cold urticaria: tolerance induction with cold baths. Br J Dermatol. 2007;157:835-836.
- Möller A, Henning M, Zuberbier T, et al. Epidemiology and clinical aspects of cold urticaria [article in German]. Hautarzt. 1996;47:510-514.
Cold urticaria is a rare condition characterized by a localized or systemic eruption of papules upon exposure of the skin to cold air, liquids, and/or objects. In some cases, angioedema and anaphylaxis can occur. The wheal-and-flare reaction results from a localized or systemic release of histamine, leukotrienes, and various other proinflammatory mast cell mediators. Cold urticaria can be acquired or follow an autosomal-dominant familial transmission pattern. Acquired cold urticaria often presents in young adulthood with a mean duration of 4 to 5 years and remission or improvement of symptoms after 5 years in 50% of cases.1 The familial variant most commonly presents in early childhood and endures throughout the patient’s life.2 Cold urticaria generally is classified as acute or chronic if symptoms persist for more than 6 weeks. Pharmacologic therapies with prophylactic effects that may reduce the intensity of symptoms or inhibit their development include antihistamines, leuko-triene receptor antagonists, biologics, and glucocorticoids. We present the case of a 23-year-old man with cold urticaria that was refractory to initial treatment with H1 antihistamines along with a review of the literature.
Case Report
A 23-year-old man presented to the dermatology clinic for evaluation of recurrent burning, itching, and sometimes development of a painful rash on the face, neck, and arms of 2 years’ duration that typically occurred following exposure to cold, wind, and rain. He also developed symptoms in warm weather when exposed to wind while sweating. His medical history was remarkable for asthma, which was not active. He was not taking any medications and had no known drug or environmental allergies. No other members of his household developed similar symptoms. His only successful means of prevention was to stay indoors, which thereby limited his activities.
Physical examination of the dorsal hands following an ice cube test revealed numerous 3- to 5-mm urticarial papules with surrounding erythema (Figure).
Following the initial evaluation, the patient was treated unsuccessfully with a mix of first- and second-generation antihistamines in gradually increasing doses to a maximum dose of loratadine 20 mg once daily, cetirizine 20 mg once daily, and hydroxyzine 20 mg once daily. A course of montelukast 10 mg once daily was started in addition to the antihistamines and led to a reduction in the severity of the lesions but not the frequency and did not relieve the burning sensation; the patient subsequently discontinued therapy. Next, a trial of cyclosporine was attempted, but the patient reported that it caused emesis and subsequently discontinued treatment. The patient also did not tolerate prednisone. He eventually decided to treat his symptoms with lifestyle choices only, such as making sure to be well covered in cold temperatures.
Comment
Cold urticaria is a physical urticaria resulting from mast cell degranulation and the subsequent release of histamine and proinflammatory cytokines upon exposure of the skin to cold air, liquid, and/or objects. Symtpoms usually are limited to localized exposed areas of the skin but also can be generalized. Cold urticaria typically manifests as erythematous, pruritic papules and also may be accompanied by deep tissue involvement resulting in angioedema and/or anaphylaxis. Symptoms usually occur within minutes of cold exposure; however, in delayed-type cold urticaria, symptoms may develop 24 to 72 hours later.3 Prevalence is relatively equal in both sexes and is highest among young adults (ie, 18–27 years old), with a greater incidence associated with cold climates.4 In one study, the overall incidence of acquired cold urticaria in Central Europe was estimated to be 0.05%.1
Systemic involvement may occur with extensive cold contact, ranging in severity from generalized urticaria to anaphylaxis and involvement of the cardiovascular, respiratory, and/or gastrointestinal systems.5 Patients who exhibit systemic responses to cold exposure should avoid swimming in cold water, as this may induce anaphylaxis and result in injury or death. In a 2004 study that included 30 children with cold urticaria at a tertiary center in Boston,6 11 (36.7%) participants who underwent cold stimulation testing developed systemic symptoms; 5 (45.5%) participants experienced respiratory distress and 8 (72.7%) experienced a decrease in level of consciousness (eg, faintness, dizziness, hypotension). Aquatic activity was the trigger in all 11 participants except for 1 (9.0%), who experienced systemic symptoms on exposure to cold air. In the same study, 14 (46.7%) participants were diagnosed with asthma and 15 (50%) were diagnosed with allergic rhinitis. Of the 28 participants whose family histories were available for review, 25 (89.3%) had a family history of atopic disease.6 A 2008 Greek study4 of 62 adults with acquired cold urticaria found that 18 (29%) participants had at least 1 serious systemic response resulting in generalized urticaria or angioedema associated with hypotension (eg, dizziness, fainting, disorientation, shock). In both of these studies, a majority of the serious systemic reactions were associated with cold water activities.
Cold urticaria is primarily an idiopathic phenomenon but can be classified as acquired or familial. Acquired cold urticaria may result from primary or secondary causes, which can include cryoglobulinemia, human immunodeficiency virus, syphilis, mononucleosis, rubeola, toxoplasmosis, varicella, hepatitis, and various drugs (eg, penicillin, angiotensin-converting enzyme inhibitors, oral contraceptives).7 Familial causes include cryopyrin-associated periodic syndrome, phospholipase Cγ2 gene–associated antibody deficiency and immune dysregulation, Muckle-Wells syndrome, and neonatal-onset multisystem inflammatory disease.
Typically, cold urticaria is diagnosed using cold stimulation tests such as the ice cube test, in which an ice cube is applied directly to the patient’s skin for 3 to 5 minutes and a response is measured 10 minutes after its removal.8 This test has been shown to have a sensitivity of 83% to 90% and a specificity of 100%.9 Alternatively, cold urticaria may be diagnosed through the use of a Peltier element-based cold-provocation device, which exposes the patient to a variety of temperatures in order for clinicians to determine the threshold upon which there is an observable reaction. With a sensitivity of 93% and specificity of 100%, the accuracy of this test is similar to that of the ice cube test.10 If a patient has a history of serious systemic involvement, any testing that exposes the patient to extensive cold exposure should be used with caution.
Patients should be counseled about potential serious systemic symptoms and the importance of wearing appropriate cold-weather clothing. Avoidance of cold water activities and overexposure to cold weather also should be emphasized. Pharmacologic therapy for prophylaxis typically includes a second-generation H1 antihistamine (eg, cetirizine, loratadine, desloratadine). Since these drugs have been shown to be less sedating than first-generation antihistamines, they are considered a better choice for chronic treatment. At high doses, however, these medications may have a sedative effect; therefore, nighttime use is preferable if possible. The standard dosage is 5 mg to 10 mg daily for oral cetirizine, 10 mg daily for oral loratadine, and 5 mg daily for oral desloratadine; however, up to 4 times the standard dosage of these medications may be required for effective treatment of cold urticaria.11 Given the associated risk of anaphylaxis, patients should be prescribed an epinephrine pen and educated about its appropriate use, including the importance of keeping the pen accessible at all times.
In refractory cases of cold urticaria, an H2 antihistamine (eg, ranitidine) can be used in conjunction with H1 antihistamines.12 Omalizumab, an IgE-mediated treatment, also has been shown to be safe and effective in patients with recalcitrant physical urticaria, including cold urticaria.13,14 One report described the case of a 69-year-old woman with cold urticaria who was unable to leave the house without developing a widespread eruption on the face, trunk, and limbs.15 After undergoing a series of unsuccessful treatments, the patient was started on cyclosporine 125 mg twice daily, which was reduced to 100 mg twice daily after 4 weeks of therapy and then reduced to 75 mg twice daily after 4 months of treatment. One week after therapy was initiated the patient reported that she was able to leave the house, and after 4 weeks of treatment the lesions only developed on the hands and feet. The patient remained in remission with a low-dose therapy of cyclosporine 75 mg twice daily with lesions only occurring on the hands and feet. The low-dose maintenance therapy was associated with minimal adverse effects.15 To our knowledge, there are no known large studies on the efficacy of cyclosporine in the treatment of cold urticaria.
Leukotriene receptor antagonists (eg, montelukast, zafirlukast, zileuton) have been used to treat chronic urticaria. In one report, montelukast was used in a 29-year-old woman with cold urticaria who had initially been treated with cetirizine 30 mg daily, cyproheptadine 4 mg daily, and doxycycline 200 mg daily with minimal to no relief. After treatment with montelukast, she experienced notable and stable improvements in symptoms.16 Hydroxychloroquine also has been shown to be safe and to substantially improve quality of life in patients with idiopathic chronic urticaria.17 Methotrexate (with close patient monitoring for adverse effects) has been reported to benefit some patients whose chronic urticaria was unresponsive to standard treatment.18 Treatment regimens for chronic urticaria have shown variable success in the treatment of cold urticaria and may be considered in cases refractory to treatment with high-dose second-generation H1 antihistamines.
Topical application of capsaicin for 4 to 7 days has been shown to deplete the neuropeptides in sensory fibers that may be involved in cold reactions, although skin irritation may prevent usage.19
Prednisone therapy was used in a small study of 6 patients with acquired cold urticaria.20 Three patients were treated for periods of 3 to 5 days with prednisone 20 mg each morning. Three other patients were given a single dose of prednisone 20 mg or 25 mg in the morning, depending on body weight. Following prednisone therapy, complete or partial pruritus was subjectively improved in all 6 patients. Additionally, significant reductions in venous histamine concentrations at 5 and 10 minutes following cold immersion were noted (P<.05 and P<.025, respectively); however, no significant improvement in either erythema or edema was noted posttreatment following cold immersion.20 Despite these findings, prednisone has not been shown to consistently prevent histamine release. Another report noted the case of a 47-year-old man with cold urticaria who required hypothermic cardiopulmonary bypass. Pretreatment with prednisone 20 mg daily and preoperative hydrocortisone 100 mg intravenously did not prevent histamine release.21
Cold desensitization (ie, exposing progressively larger areas of the patient’s skin to increasingly colder water) may induce tolerance to cold and decrease the temperature threshold at which symptoms develop; however, patients with known serious systemic reactions should be tested with extreme caution and only under the supervision of a clinician.22,23 Tolerance may wane when cold desensitization therapy is stopped.
The prognosis for patients with acquired cold urticaria generally is good. Improvement of symptoms or full remission occurs within 5 to 6 years in 50% of patients.24 Once remission has occurred, patients generally remain symptom free. For other familial variants, symptoms may last a lifetime.
Conclusion
This case report and review of the literature highlights the limitations of cold urticaria and the importance of effective management in improving quality of life in affected patients. Symptoms may limit patients’ ability to work in certain environments, inhibit them from engaging in daily activities, and even prevent them from leaving their homes in colder temperatures. In addition to behavioral modifications, pharmacologic management may provide symptomatic relief. Antihistamines are the first line of treatment in cold urticaria. Second-generation antihistamines, which are more selective for H1 receptors and less sedating, are generally recommended. Up to 4 times the standard dosage of these medications may be required for effective treatment.5 The primary goal of therapy in mild to moderate cases is improvement in quality of life.
Cold urticaria is a rare condition characterized by a localized or systemic eruption of papules upon exposure of the skin to cold air, liquids, and/or objects. In some cases, angioedema and anaphylaxis can occur. The wheal-and-flare reaction results from a localized or systemic release of histamine, leukotrienes, and various other proinflammatory mast cell mediators. Cold urticaria can be acquired or follow an autosomal-dominant familial transmission pattern. Acquired cold urticaria often presents in young adulthood with a mean duration of 4 to 5 years and remission or improvement of symptoms after 5 years in 50% of cases.1 The familial variant most commonly presents in early childhood and endures throughout the patient’s life.2 Cold urticaria generally is classified as acute or chronic if symptoms persist for more than 6 weeks. Pharmacologic therapies with prophylactic effects that may reduce the intensity of symptoms or inhibit their development include antihistamines, leuko-triene receptor antagonists, biologics, and glucocorticoids. We present the case of a 23-year-old man with cold urticaria that was refractory to initial treatment with H1 antihistamines along with a review of the literature.
Case Report
A 23-year-old man presented to the dermatology clinic for evaluation of recurrent burning, itching, and sometimes development of a painful rash on the face, neck, and arms of 2 years’ duration that typically occurred following exposure to cold, wind, and rain. He also developed symptoms in warm weather when exposed to wind while sweating. His medical history was remarkable for asthma, which was not active. He was not taking any medications and had no known drug or environmental allergies. No other members of his household developed similar symptoms. His only successful means of prevention was to stay indoors, which thereby limited his activities.
Physical examination of the dorsal hands following an ice cube test revealed numerous 3- to 5-mm urticarial papules with surrounding erythema (Figure).
Following the initial evaluation, the patient was treated unsuccessfully with a mix of first- and second-generation antihistamines in gradually increasing doses to a maximum dose of loratadine 20 mg once daily, cetirizine 20 mg once daily, and hydroxyzine 20 mg once daily. A course of montelukast 10 mg once daily was started in addition to the antihistamines and led to a reduction in the severity of the lesions but not the frequency and did not relieve the burning sensation; the patient subsequently discontinued therapy. Next, a trial of cyclosporine was attempted, but the patient reported that it caused emesis and subsequently discontinued treatment. The patient also did not tolerate prednisone. He eventually decided to treat his symptoms with lifestyle choices only, such as making sure to be well covered in cold temperatures.
Comment
Cold urticaria is a physical urticaria resulting from mast cell degranulation and the subsequent release of histamine and proinflammatory cytokines upon exposure of the skin to cold air, liquid, and/or objects. Symtpoms usually are limited to localized exposed areas of the skin but also can be generalized. Cold urticaria typically manifests as erythematous, pruritic papules and also may be accompanied by deep tissue involvement resulting in angioedema and/or anaphylaxis. Symptoms usually occur within minutes of cold exposure; however, in delayed-type cold urticaria, symptoms may develop 24 to 72 hours later.3 Prevalence is relatively equal in both sexes and is highest among young adults (ie, 18–27 years old), with a greater incidence associated with cold climates.4 In one study, the overall incidence of acquired cold urticaria in Central Europe was estimated to be 0.05%.1
Systemic involvement may occur with extensive cold contact, ranging in severity from generalized urticaria to anaphylaxis and involvement of the cardiovascular, respiratory, and/or gastrointestinal systems.5 Patients who exhibit systemic responses to cold exposure should avoid swimming in cold water, as this may induce anaphylaxis and result in injury or death. In a 2004 study that included 30 children with cold urticaria at a tertiary center in Boston,6 11 (36.7%) participants who underwent cold stimulation testing developed systemic symptoms; 5 (45.5%) participants experienced respiratory distress and 8 (72.7%) experienced a decrease in level of consciousness (eg, faintness, dizziness, hypotension). Aquatic activity was the trigger in all 11 participants except for 1 (9.0%), who experienced systemic symptoms on exposure to cold air. In the same study, 14 (46.7%) participants were diagnosed with asthma and 15 (50%) were diagnosed with allergic rhinitis. Of the 28 participants whose family histories were available for review, 25 (89.3%) had a family history of atopic disease.6 A 2008 Greek study4 of 62 adults with acquired cold urticaria found that 18 (29%) participants had at least 1 serious systemic response resulting in generalized urticaria or angioedema associated with hypotension (eg, dizziness, fainting, disorientation, shock). In both of these studies, a majority of the serious systemic reactions were associated with cold water activities.
Cold urticaria is primarily an idiopathic phenomenon but can be classified as acquired or familial. Acquired cold urticaria may result from primary or secondary causes, which can include cryoglobulinemia, human immunodeficiency virus, syphilis, mononucleosis, rubeola, toxoplasmosis, varicella, hepatitis, and various drugs (eg, penicillin, angiotensin-converting enzyme inhibitors, oral contraceptives).7 Familial causes include cryopyrin-associated periodic syndrome, phospholipase Cγ2 gene–associated antibody deficiency and immune dysregulation, Muckle-Wells syndrome, and neonatal-onset multisystem inflammatory disease.
Typically, cold urticaria is diagnosed using cold stimulation tests such as the ice cube test, in which an ice cube is applied directly to the patient’s skin for 3 to 5 minutes and a response is measured 10 minutes after its removal.8 This test has been shown to have a sensitivity of 83% to 90% and a specificity of 100%.9 Alternatively, cold urticaria may be diagnosed through the use of a Peltier element-based cold-provocation device, which exposes the patient to a variety of temperatures in order for clinicians to determine the threshold upon which there is an observable reaction. With a sensitivity of 93% and specificity of 100%, the accuracy of this test is similar to that of the ice cube test.10 If a patient has a history of serious systemic involvement, any testing that exposes the patient to extensive cold exposure should be used with caution.
Patients should be counseled about potential serious systemic symptoms and the importance of wearing appropriate cold-weather clothing. Avoidance of cold water activities and overexposure to cold weather also should be emphasized. Pharmacologic therapy for prophylaxis typically includes a second-generation H1 antihistamine (eg, cetirizine, loratadine, desloratadine). Since these drugs have been shown to be less sedating than first-generation antihistamines, they are considered a better choice for chronic treatment. At high doses, however, these medications may have a sedative effect; therefore, nighttime use is preferable if possible. The standard dosage is 5 mg to 10 mg daily for oral cetirizine, 10 mg daily for oral loratadine, and 5 mg daily for oral desloratadine; however, up to 4 times the standard dosage of these medications may be required for effective treatment of cold urticaria.11 Given the associated risk of anaphylaxis, patients should be prescribed an epinephrine pen and educated about its appropriate use, including the importance of keeping the pen accessible at all times.
In refractory cases of cold urticaria, an H2 antihistamine (eg, ranitidine) can be used in conjunction with H1 antihistamines.12 Omalizumab, an IgE-mediated treatment, also has been shown to be safe and effective in patients with recalcitrant physical urticaria, including cold urticaria.13,14 One report described the case of a 69-year-old woman with cold urticaria who was unable to leave the house without developing a widespread eruption on the face, trunk, and limbs.15 After undergoing a series of unsuccessful treatments, the patient was started on cyclosporine 125 mg twice daily, which was reduced to 100 mg twice daily after 4 weeks of therapy and then reduced to 75 mg twice daily after 4 months of treatment. One week after therapy was initiated the patient reported that she was able to leave the house, and after 4 weeks of treatment the lesions only developed on the hands and feet. The patient remained in remission with a low-dose therapy of cyclosporine 75 mg twice daily with lesions only occurring on the hands and feet. The low-dose maintenance therapy was associated with minimal adverse effects.15 To our knowledge, there are no known large studies on the efficacy of cyclosporine in the treatment of cold urticaria.
Leukotriene receptor antagonists (eg, montelukast, zafirlukast, zileuton) have been used to treat chronic urticaria. In one report, montelukast was used in a 29-year-old woman with cold urticaria who had initially been treated with cetirizine 30 mg daily, cyproheptadine 4 mg daily, and doxycycline 200 mg daily with minimal to no relief. After treatment with montelukast, she experienced notable and stable improvements in symptoms.16 Hydroxychloroquine also has been shown to be safe and to substantially improve quality of life in patients with idiopathic chronic urticaria.17 Methotrexate (with close patient monitoring for adverse effects) has been reported to benefit some patients whose chronic urticaria was unresponsive to standard treatment.18 Treatment regimens for chronic urticaria have shown variable success in the treatment of cold urticaria and may be considered in cases refractory to treatment with high-dose second-generation H1 antihistamines.
Topical application of capsaicin for 4 to 7 days has been shown to deplete the neuropeptides in sensory fibers that may be involved in cold reactions, although skin irritation may prevent usage.19
Prednisone therapy was used in a small study of 6 patients with acquired cold urticaria.20 Three patients were treated for periods of 3 to 5 days with prednisone 20 mg each morning. Three other patients were given a single dose of prednisone 20 mg or 25 mg in the morning, depending on body weight. Following prednisone therapy, complete or partial pruritus was subjectively improved in all 6 patients. Additionally, significant reductions in venous histamine concentrations at 5 and 10 minutes following cold immersion were noted (P<.05 and P<.025, respectively); however, no significant improvement in either erythema or edema was noted posttreatment following cold immersion.20 Despite these findings, prednisone has not been shown to consistently prevent histamine release. Another report noted the case of a 47-year-old man with cold urticaria who required hypothermic cardiopulmonary bypass. Pretreatment with prednisone 20 mg daily and preoperative hydrocortisone 100 mg intravenously did not prevent histamine release.21
Cold desensitization (ie, exposing progressively larger areas of the patient’s skin to increasingly colder water) may induce tolerance to cold and decrease the temperature threshold at which symptoms develop; however, patients with known serious systemic reactions should be tested with extreme caution and only under the supervision of a clinician.22,23 Tolerance may wane when cold desensitization therapy is stopped.
The prognosis for patients with acquired cold urticaria generally is good. Improvement of symptoms or full remission occurs within 5 to 6 years in 50% of patients.24 Once remission has occurred, patients generally remain symptom free. For other familial variants, symptoms may last a lifetime.
Conclusion
This case report and review of the literature highlights the limitations of cold urticaria and the importance of effective management in improving quality of life in affected patients. Symptoms may limit patients’ ability to work in certain environments, inhibit them from engaging in daily activities, and even prevent them from leaving their homes in colder temperatures. In addition to behavioral modifications, pharmacologic management may provide symptomatic relief. Antihistamines are the first line of treatment in cold urticaria. Second-generation antihistamines, which are more selective for H1 receptors and less sedating, are generally recommended. Up to 4 times the standard dosage of these medications may be required for effective treatment.5 The primary goal of therapy in mild to moderate cases is improvement in quality of life.
- Siebenhaar F, Weller K, Mlynek A, et al. Acquired cold urticaria: clinical picture and update on diagnosis and treatment. Clin Exp Dermatol. 2007;32:241-245.
- Gandhi C, Healy C, Wanderer AA, et al. Familial atypical cold urticaria: description of a new hereditary disease. J Allergy Clin Immunol. 2009;124:1245-1250.
- Bäck O, Larsen A. Delayed cold urticaria. Acta Derm Venereol. 1978;58:369-371.
- Katsarou-Katsari A, Makris M, Lagogianni E, et al. Clinical features and natural history of acquired cold urticaria in a tertiary referral hospital: a 10-year prospective study. J Eur Acad Dermatol Venereol. 2008;22:1405-1411.
- Wanderer AA, Grandel KE, Wasserman SI, et al. Clinical characteristics of cold-induced systemic reactions in acquired cold urticaria syndromes: recommendations for prevention of this complication and a proposal for a diagnostic classification of cold urticaria. J Allergy Clin Immunol. 1986;78(3 Pt 1):417-423.
- Alangari AA, Twarog FJ, Shih MC, et al. Clinical features and anaphylaxis in children with cold urticaria. Pediatrics. 2004;113:e313-e317.
- Wanderer AA, Hoffman HM. The spectrum of acquired and familial cold-induced urticaria/urticaria-like syndromes. Immunol Allergy Clin North Am. 2004;24:259-286.
- Visitsuntorn N, Tuchinda M, Arunyanark N, et al. Ice cube test in children with cold urticaria. Asian Pac J Allergy Immunol. 1992;10:111-115.
- Neittaanmäki H. Cold urticaria. clinical findings in 220 patients. J Am Acad Dermatol. 1985;13:636-644.
- Siebenhaar F, Staubach P, Metz M, et al. Peltier effect-based temperature challenge: an improved method for diagnosing cold urticaria. J Allergy Clin Immunol. 2004;114:1224-1225.
- Siebenhaar F, Degener F, Zuberbier T, et al. High-dose desloratadine decreases wheal volume and improves cold provocation thresholds compared with standard-dose treatment in patients with acquired cold urticaria: a randomized, placebo-controlled, crossover study. J Allergy Clin Immunol. 2009;123:672-679.
- Duc J, Pécoud A. Successful treatment of idiopathic cold urticaria with the association of H1 and H2 antagonists: a case report. Ann Allergy. 1986;56:355-357.
- Metz M, Altrichter S, Ardelean E, et al. Anti-immunoglobulin E treatment of patients with recalcitrant physical urticaria. Int Arch Allergy Immunol. 2011;154:177-180.
- Boyce JA. Successful treatment of cold-induced urticaria/anaphylaxis with anti-IgE. J Allergy Clin Immunol. 2006;117:1415-1418.
- Marsland AM, Beck MH. Cold urticaria responding to systemic cyclosporine. Br J Dermatol. 2003;149:214-215.
- Hani N, Hartmann K, Casper C, et al. Improvement of cold urticaria by treatment with the leukotriene receptor antagonist montelukast. Acta Derm Venereol. 2000;80:229.
- Reeves GE, Boyle MJ, Bonfield J, et al. Impact of hydroxychloroquine therapy on chronic urticaria: chronic autoimmune urticaria study and evaluation. Intern Med J. 2004;34:182-186.
- Perez A, Woods A, Grattan CE. Methotrexate: a useful steroid-sparing agent in recalcitrant chronic urticaria. Br J Dermatol. 2010;162:191-194.
- Tóth-Kása I, Jancsó G, Obál F Jr, et al. Involvement of sensory nerve endings in cold and heat urticaria. J Invest Dermatol. 1983;80:34-36.
- Black AK, Keahey TM, Eady RA, et al. Dissociation of histamine release and clinical improvement following treatment of acquired cold urticaria by prednisone. Br J Clin Pharmacol. 1981;12:327-331.
- Johnston WE, Moss J, Philbin DM, et al. Management of cold urticaria during hypothermic cardiopulmonary bypass. N Engl J Med. 1982;306:219-221.
- Krause K, Zuberbier T, Maurer, M. Modern Approaches to the diagnosis and treatment of cold contact urticaria. Curr Allergy Asthma Rep. 2010;10:273-279.
- von Mackensen YA, Sticherling M. Cold urticaria: tolerance induction with cold baths. Br J Dermatol. 2007;157:835-836.
- Möller A, Henning M, Zuberbier T, et al. Epidemiology and clinical aspects of cold urticaria [article in German]. Hautarzt. 1996;47:510-514.
- Siebenhaar F, Weller K, Mlynek A, et al. Acquired cold urticaria: clinical picture and update on diagnosis and treatment. Clin Exp Dermatol. 2007;32:241-245.
- Gandhi C, Healy C, Wanderer AA, et al. Familial atypical cold urticaria: description of a new hereditary disease. J Allergy Clin Immunol. 2009;124:1245-1250.
- Bäck O, Larsen A. Delayed cold urticaria. Acta Derm Venereol. 1978;58:369-371.
- Katsarou-Katsari A, Makris M, Lagogianni E, et al. Clinical features and natural history of acquired cold urticaria in a tertiary referral hospital: a 10-year prospective study. J Eur Acad Dermatol Venereol. 2008;22:1405-1411.
- Wanderer AA, Grandel KE, Wasserman SI, et al. Clinical characteristics of cold-induced systemic reactions in acquired cold urticaria syndromes: recommendations for prevention of this complication and a proposal for a diagnostic classification of cold urticaria. J Allergy Clin Immunol. 1986;78(3 Pt 1):417-423.
- Alangari AA, Twarog FJ, Shih MC, et al. Clinical features and anaphylaxis in children with cold urticaria. Pediatrics. 2004;113:e313-e317.
- Wanderer AA, Hoffman HM. The spectrum of acquired and familial cold-induced urticaria/urticaria-like syndromes. Immunol Allergy Clin North Am. 2004;24:259-286.
- Visitsuntorn N, Tuchinda M, Arunyanark N, et al. Ice cube test in children with cold urticaria. Asian Pac J Allergy Immunol. 1992;10:111-115.
- Neittaanmäki H. Cold urticaria. clinical findings in 220 patients. J Am Acad Dermatol. 1985;13:636-644.
- Siebenhaar F, Staubach P, Metz M, et al. Peltier effect-based temperature challenge: an improved method for diagnosing cold urticaria. J Allergy Clin Immunol. 2004;114:1224-1225.
- Siebenhaar F, Degener F, Zuberbier T, et al. High-dose desloratadine decreases wheal volume and improves cold provocation thresholds compared with standard-dose treatment in patients with acquired cold urticaria: a randomized, placebo-controlled, crossover study. J Allergy Clin Immunol. 2009;123:672-679.
- Duc J, Pécoud A. Successful treatment of idiopathic cold urticaria with the association of H1 and H2 antagonists: a case report. Ann Allergy. 1986;56:355-357.
- Metz M, Altrichter S, Ardelean E, et al. Anti-immunoglobulin E treatment of patients with recalcitrant physical urticaria. Int Arch Allergy Immunol. 2011;154:177-180.
- Boyce JA. Successful treatment of cold-induced urticaria/anaphylaxis with anti-IgE. J Allergy Clin Immunol. 2006;117:1415-1418.
- Marsland AM, Beck MH. Cold urticaria responding to systemic cyclosporine. Br J Dermatol. 2003;149:214-215.
- Hani N, Hartmann K, Casper C, et al. Improvement of cold urticaria by treatment with the leukotriene receptor antagonist montelukast. Acta Derm Venereol. 2000;80:229.
- Reeves GE, Boyle MJ, Bonfield J, et al. Impact of hydroxychloroquine therapy on chronic urticaria: chronic autoimmune urticaria study and evaluation. Intern Med J. 2004;34:182-186.
- Perez A, Woods A, Grattan CE. Methotrexate: a useful steroid-sparing agent in recalcitrant chronic urticaria. Br J Dermatol. 2010;162:191-194.
- Tóth-Kása I, Jancsó G, Obál F Jr, et al. Involvement of sensory nerve endings in cold and heat urticaria. J Invest Dermatol. 1983;80:34-36.
- Black AK, Keahey TM, Eady RA, et al. Dissociation of histamine release and clinical improvement following treatment of acquired cold urticaria by prednisone. Br J Clin Pharmacol. 1981;12:327-331.
- Johnston WE, Moss J, Philbin DM, et al. Management of cold urticaria during hypothermic cardiopulmonary bypass. N Engl J Med. 1982;306:219-221.
- Krause K, Zuberbier T, Maurer, M. Modern Approaches to the diagnosis and treatment of cold contact urticaria. Curr Allergy Asthma Rep. 2010;10:273-279.
- von Mackensen YA, Sticherling M. Cold urticaria: tolerance induction with cold baths. Br J Dermatol. 2007;157:835-836.
- Möller A, Henning M, Zuberbier T, et al. Epidemiology and clinical aspects of cold urticaria [article in German]. Hautarzt. 1996;47:510-514.
Practice Points
- Cold urticaria is a physical urticaria characterized by a localized or systemic eruption of papules upon exposure of the skin to cold air, liquids, and/or objects.
- Symptoms of cold urticaria, which range from erythema, pruritus, and hives to angioedema and sometimes anaphylaxis, may be debilitating for patients; therefore, effective treatment is required to improve quality of life.
- First-line treatment for cold urticaria includes second-generation H1 antihistamines at up to 4 times the standard dosage.
Recurrent Varicella in an Immunocompetent Woman
Case Report
A 52-year-old black woman presented to our dermatology clinic for evaluation of a generalized pruritic rash of 5 days’ duration. The eruption had started on the trunk and subsequently spread to the face, legs, and arms, including the dorsal surfaces of the hands (Figure 1). The patient reported that she had developed a similar rash 4 years prior. She recalled no sick contacts but had occupational exposure to many people as a food service worker. Two days prior, the referring physician had initiated treatment with oral acyclovir 400 mg every 6 hours. The patient was in otherwise good health and reported no fever, chills, diaphoresis, or fatigue. She did not recall any recent insect bites, and a review of systems was negative.
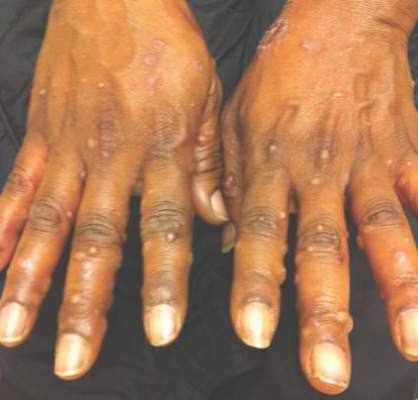
The patient’s medical history was remarkable for 2 cases of varicella: the first, which occurred at 5 years of age, was diagnosed by a pediatrician and manifested as diffuse papules, vesicles, and crusts with concurrent mild fever. The infection followed a typical clinical course and resolved without complications after 1 week. The second case of varicella was diagnosed clinically at our dermatology clinic approximately 4 years prior to the current presentation and manifested as widespread pruritic lesions that were too numerous to count. Given her history of varicella in childhood, a punch biopsy specimen was taken from a lesion on the left trunk and a dermatopathologist confirmed the diagnosis of a herpesvirus infection. The second infection also resolved without sequelae after 12 days. Her medical history was otherwise unremarkable, revealing no exceptional sinopulmonary or gastrointestinal infections. The patient was not currently taking any medications or supplements and reported no known drug allergies.
Physical examination at the current presentation revealed a well-nourished, afebrile woman with vesicles and papules on the hands, arms, and legs along with vesicular and crusted papules in various stages of healing distributed on the chest, abdomen, and back. Lesions on the legs and feet were present but scant. The eruption was not confined to a single dermatome. No lesions were noted on the palms, soles, or oral mucosa and no epitrochlear, axillary, or supraclavicular lymphadenopathy was noted.
Initial laboratory values were obtained. A complete blood count demonstrated a normal leukocyte number of 5700 cells/μL (reference range, 4500–11,000 cells/μL) and mild anemia with a hemoglobin level of 10.3 g/dL (reference range, 14.0–17.5 g/dL). Monocytes were mildly elevated at 11% (reference range, 1%–9%). Serologic tests showed positive titers for varicella-zoster virus (VZV) IgM at 1.64 (negative, <0.91) and VZV IgG at 1.72 (negative, <0.91), indicating current and past VZV infection, respectively. Antibodies against herpes simplex virus (HSV) types 1 and 2 were negative for IgM and positive for IgG at >5.00 (negative, <0.90), indicating a remote HSV infection. Furthermore, results from a culture of a lesion on the left hand were negative for HSV.
After consultation with the Department of Infectious Diseases, further laboratory studies were performed. The absolute lymphocyte number was within normal range at 1600 cells/μL (reference range, 850–3900 cells/μL). Likewise, CD4+ T lymphocytes were normal at 618 cells/μL (reference range, 490–1740 cells/μL) or 39% of total lymphocytes (reference range, 30%–61%). Screening results were negative for human immunodeficiency virus types 1 and 2. Immunoglobulin subtype analysis revealed slightly elevated IgG at 1709 mg/dL (reference range, 723–1685 mg/dL), elevated IgA at 487 mg/dL (reference range, 65–382 mg/dL), and normal IgM at 238 mg/dL (reference range, 63–277 mg/dL).
Consistent with the clinical presentation and serologic studies, recurrent varicella was accepted as the most plausible diagnosis. Over the next 2 weeks, the eruption resolved with postinflammatory hyperpigmentation (Figure 2). The patient returned to work without further incident.
Comment
As denoted by its hyphenated name, VZV infection can cause 2 distinct disease processes.1,2 Varicella, the generalized initial exanthem known as chickenpox, appears predominantly in childhood. With resolution of this primary infection, the virus lies dormant in sensory ganglia, persisting in neurons. Stress, advanced age, and/or compromised immunity may reactivate latent VZV. This secondary expression is known as herpes zoster (shingles), a unilateral eruption of lesions localized to a single dermatome.
In most cases, morphology of the varicella eruption confirms the diagnosis. Lesions evolve through stages from macules and papules to vesicles and pustules and then to crusts. This evolution typically takes 24 to 48 hours.2 The varicella eruption contains an admixture of elements from each stage simultaneously. Crusts usually resolve over an average of 14 days. Serologically, IgM is measurable as early as 1 to 2 days after appearance of the eruption.3,4 Next to appear are IgG antibodies, which generally remain detectable for life. With more than 90% of the US population being seropositive for VZV,5 diagnosis and management of varicella and herpes zoster usually are straightforward; however, there have been unusual variations on this classic sequence of pathogenesis.
In disseminated zoster, the clinical presentation includes more than 20 lesions outside the dermatome primarily affected.6 Another permutation of VZV infection is zoster sine herpete, which causes the characteristic dermatomal pain of herpes zoster but without the rash.7 Occasionally, 2 cases of chickenpox occur in the same person, usually indicating an underlying immune deficiency. Recurrent varicella in those with intact immunity is purportedly rare. A PubMed search of articles indexed for MEDLINE using the search terms recurrent varicella, chickenpox reinfection, and immunocompetent revealed 41 cases of recurrent varicella in immunocompetent patients in the English language literature occurring among children,1,8-11 adults,8 the elderly,12 health care workers,13-15 and pregnant women16 (Table).
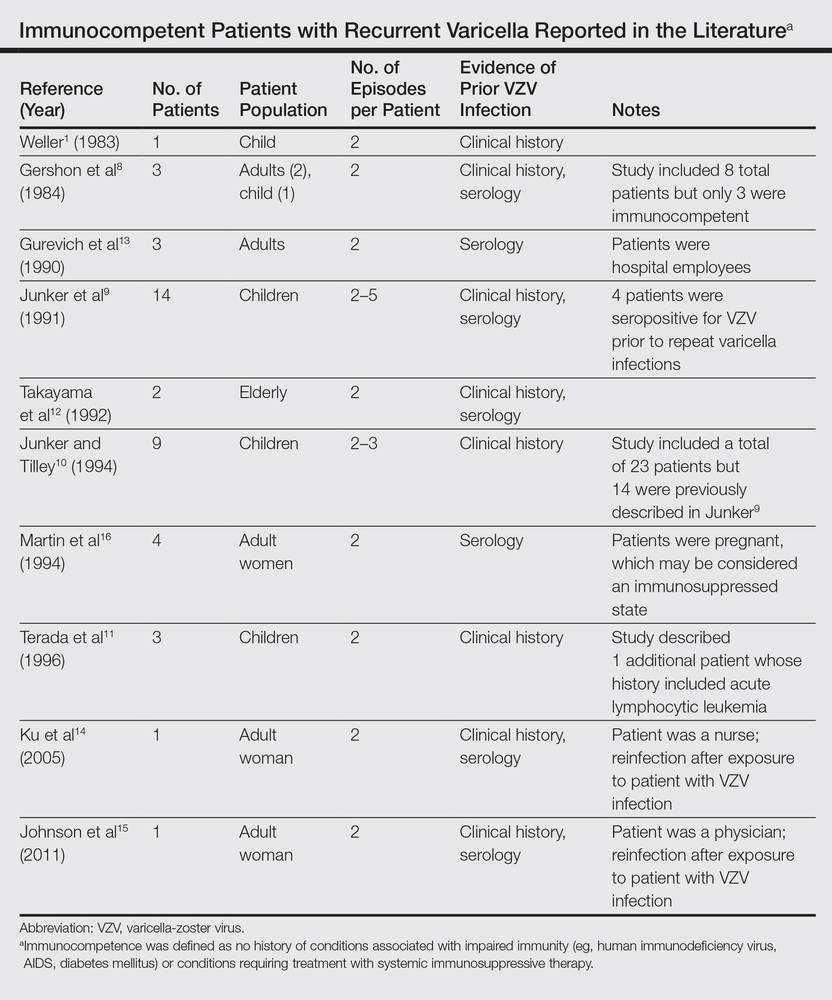
|
Surveillance studies, however, have challenged the apparent rarity of recurrent varicella, asserting that varicella may recur more frequently than is generally recognized.17,18 Hall et al17 described 9947 cases of varicella, with nearly 6.9% reporting prior varicella infection. Another surveillance report by Marin et al18 evaluating data from 1047 adults with varicella noted that 21% of participants reported prior VZV infections. Both of these studies defined varicella by clinical parameters as a condition with acute onset of generalized maculopapulovesicular rash without other known cause. Although laboratory confirmation of VZV infection was not documented in either study, a history of varicella is considered a reliable indicator of immunity. In fact, studies show that a history of varicella is associated with serologic evidence of immunity 97% to 100% of the time.19,20
Immunity against VZV in humans is not well understood. Although both humoral and cellular factors play a role, cell-mediated immunity may be more important in suppressing primary infection and defending against reinfection. Varicella is more likely to disseminate in lymphopenic patients,21,22 while its course is uninfluenced by hypogammaglobulinemia.1,23 One study of simian varicella virus, which demonstrates 75% genetic homology with VZV, noted that simian varicella virus–infected rhesus macaques without CD4+ T lymphocyte response experienced higher viral loads, prolonged viremia, and disseminated varicella.24 The loss of CD20+ B lymphocytes did not intensify the severity of varicella in the primate model. It is accepted, however, that waning humoral immunity and lower antibody levels correlate with varicella recurrence.25 Ethnicity may impact immunoglobulin persistence. One investigation postulated that individuals with darker skin types experience reduced viral shedding and therefore less antigenic boosting from secondary VZV infections, as they may less readily maintain protective levels of VZV-specific immunoglobulins.25 This phenomenon may have contributed to the 3 episodes of varicella in our patient.
Virulence factors that are intrinsic to VZV may also prompt reinfection. Although taxonomy is still in flux, 3 to 5 major genotypes of VZV have been recognized to date, categorized into European (Dumas), Japanese (Oka), and mosaic clades.26-28 In one study population, approximately 80% of the VZV strains isolated in the United States were of the European variety.26 It is unclear whether infection with one strain of VZV affords immunoprotection against the other strains. Interestingly, one report documented recurrent herpes zoster caused by 2 distinct VZV strains in the same individual.29 Since subtypes of VZV vary geographically, it is possible that increasing global travel may correlate with increased incidence and reporting of varicella reinfection, particularly in cosmopolitan centers. In patients with recurrent varicella, a careful investigation of their international travel history may be necessary.
1. Weller T. Varicella and herpes zoster: changing concepts of the natural history, control, and importance of a not-so-benign virus. N Engl J Med. 1983;309:1362-1368.
2. Heininger U, Seward JF. Varicella. Lancet. 2006;368:1365-1376.
3. Krah DL. Assays for antibodies to varicella-zoster virus. Infect Dis Clin North Am. 1996;10:507-527.
4. Oladepo DK, Klapper PE, Percival D, et al. Serological diagnosis of varicella-zoster virus in sera with antibody-capture enzyme-linked immunosorbent assay of IgM. J Virol Meth. 2000;84:169-173.
5. Kilgore PE, Kruszon-Moran D, Seward JF, et al. Varicella in Americans from NHANES III: implications for control through routine immunizations. J Med Virol. 2003;70:S111-S118.
6. Gupta S, Jain A, Gardiner C, et al. A rare case of disseminated cutaneous zoster in an immunocompetent patient. BMC Fam Pract. 2005;6:50.
7. Lewis GW. Zoster sine herpete. Br Med J. 1958;8:418-421.
8. Gershon AA, Steinberg SP, Gelb L. Clinical reinfection with varicella-zoster virus. J Infect Dis. 1984;149:137-142.
9. Junker AK, Angus E, Thomas EE. Recurrent varicella-zoster virus infections in apparently immunocompetent children. Pediatr Infect Dis J. 1991;10:569-575.
10. Junker AK, Tilley P. Varicella-zoster virus antibody avidity and IgG-subclass patterns in children with recurrent chickenpox. J Med Virol. 1994;43:119-124.
11. Terada K, Kawano S, Shimada Y, et al. Recurrent chickenpox after natural infection. Ped Infect Dis J. 1996;15:179-181.
12. Takayama N, Takayama M, Negishi M. Clinical varicella-zoster virus reinfection observed in two advanced-age persons [article in Japanese]. Kansenshogaku Zasshi. 1992;66:1373-1377.
13. Gurevich I, Jensen L, Kalter R, et al. Chickenpox in apparently “immune” hospital workers. Infect Control Hosp Epidemiol. 1990;11:510, 512.
14. Ku C, Liu Y, Christiani DC. Case report: occupationally related recurrent varicella (chickenpox) in a hospital nurse. Environ Health Perspect. 2005;113:1373-1375.
15. Johnson JA, Bloch KC, Dang BN. Varicella reinfection in a seropositive physician following occupational exposure to localized zoster. Clin Infect Dis. 2011;52:907-909.
16. Martin KA, Junker AK, Thomas EE, et al. Occurrence of chickenpox during pregnancy in women seropositive for varicella-zoster virus. J Infect Dis. 1994;170:991-995.
17. Hall S, Maupin T, Seward J, et al. Second varicella infections: are they more common than previously thought? Pediatrics. 2002;109:1068-1073.
18. Marin M, Watson TL, Chaves SS, et al. Varicella among adults: data from an active surveillance project, 1995-2005. J Infect Dis. 2008;197(Suppl 2):S94-S100.
19. Perella DM, Fiks A, Spain CV. Validity of reported varicella history as a marker for varicella-zoster virus immunity. Paper presented at: Pediatric Academic Societies Annual Meeting; 2005; Washington, DC.
20. Ferson MJ, Bell SM, Robertson PW. Determination and importance of varicella immune status of nursing staff in a children’s hospital. J Hosp Infect. 1990;15:347-351.
21. Arvin AM, Pollard RB, Rasmussen LE, et al. Selective impairment of lymphocyte reactivity to varicella-zoster virus antigen among untreated patients with lymphoma. J Infect Dis. 1978;137:531-540.
22. Feldman S, Hughes WT, Daniel CB. Varicella in children with cancer: seventy-seven cases. Pediatrics. 1975;56:388-397.
23. Arvin AM. Varicella-zoster virus. Clin Microbiol Rev. 1996;9:361-381.
24. Haberthur K, Engelmann F, Park B, et al. CD4 T cell immunity is critical for the control of simian varicella virus infection in nonhuman primate model of VZV infection. PLoS Pathog. 2011;7:e1002367.
25. Ayres KL, Talukder Y, Breuer J. Humoral immunity following chickenpox is influenced by geography and ethnicity. J Infect. 2010;61:244-251.
26. Loparev VN, Gonzalez A, Deleon-Carnes M, et al. Global identification of three major genotypes of varicella-zoster virus: longitudinal clustering and strategies for genotyping. J Virol. 2004;78:8349-8358.
27. Parker SP, Breuer J, Taha Y, et al. Genotyping of varicella-zoster virus and the discrimination of Oka vaccine strains by TaqMan real-time PCR. J Clin Microbiol. 2006;44:3911-3914.
28. Loparev VN, Rubtcova EN, Bostik V, et al. Identification of five major and two minor genotypes of varicella-zoster virus strains: a practical two-amplicon approach used to genotype clinical isolates in Australia and New Zealand. J Virol. 2007;81:12758-12765.
29. Taha Y, Scott FT, Parker SP, et al. Reactivation of 2 genetically distinct varicella-zoster viruses in the same individual. Clin Infect Dis. 2006;43:1301-1303.
Case Report
A 52-year-old black woman presented to our dermatology clinic for evaluation of a generalized pruritic rash of 5 days’ duration. The eruption had started on the trunk and subsequently spread to the face, legs, and arms, including the dorsal surfaces of the hands (Figure 1). The patient reported that she had developed a similar rash 4 years prior. She recalled no sick contacts but had occupational exposure to many people as a food service worker. Two days prior, the referring physician had initiated treatment with oral acyclovir 400 mg every 6 hours. The patient was in otherwise good health and reported no fever, chills, diaphoresis, or fatigue. She did not recall any recent insect bites, and a review of systems was negative.
The patient’s medical history was remarkable for 2 cases of varicella: the first, which occurred at 5 years of age, was diagnosed by a pediatrician and manifested as diffuse papules, vesicles, and crusts with concurrent mild fever. The infection followed a typical clinical course and resolved without complications after 1 week. The second case of varicella was diagnosed clinically at our dermatology clinic approximately 4 years prior to the current presentation and manifested as widespread pruritic lesions that were too numerous to count. Given her history of varicella in childhood, a punch biopsy specimen was taken from a lesion on the left trunk and a dermatopathologist confirmed the diagnosis of a herpesvirus infection. The second infection also resolved without sequelae after 12 days. Her medical history was otherwise unremarkable, revealing no exceptional sinopulmonary or gastrointestinal infections. The patient was not currently taking any medications or supplements and reported no known drug allergies.
Physical examination at the current presentation revealed a well-nourished, afebrile woman with vesicles and papules on the hands, arms, and legs along with vesicular and crusted papules in various stages of healing distributed on the chest, abdomen, and back. Lesions on the legs and feet were present but scant. The eruption was not confined to a single dermatome. No lesions were noted on the palms, soles, or oral mucosa and no epitrochlear, axillary, or supraclavicular lymphadenopathy was noted.
Initial laboratory values were obtained. A complete blood count demonstrated a normal leukocyte number of 5700 cells/μL (reference range, 4500–11,000 cells/μL) and mild anemia with a hemoglobin level of 10.3 g/dL (reference range, 14.0–17.5 g/dL). Monocytes were mildly elevated at 11% (reference range, 1%–9%). Serologic tests showed positive titers for varicella-zoster virus (VZV) IgM at 1.64 (negative, <0.91) and VZV IgG at 1.72 (negative, <0.91), indicating current and past VZV infection, respectively. Antibodies against herpes simplex virus (HSV) types 1 and 2 were negative for IgM and positive for IgG at >5.00 (negative, <0.90), indicating a remote HSV infection. Furthermore, results from a culture of a lesion on the left hand were negative for HSV.
After consultation with the Department of Infectious Diseases, further laboratory studies were performed. The absolute lymphocyte number was within normal range at 1600 cells/μL (reference range, 850–3900 cells/μL). Likewise, CD4+ T lymphocytes were normal at 618 cells/μL (reference range, 490–1740 cells/μL) or 39% of total lymphocytes (reference range, 30%–61%). Screening results were negative for human immunodeficiency virus types 1 and 2. Immunoglobulin subtype analysis revealed slightly elevated IgG at 1709 mg/dL (reference range, 723–1685 mg/dL), elevated IgA at 487 mg/dL (reference range, 65–382 mg/dL), and normal IgM at 238 mg/dL (reference range, 63–277 mg/dL).
Consistent with the clinical presentation and serologic studies, recurrent varicella was accepted as the most plausible diagnosis. Over the next 2 weeks, the eruption resolved with postinflammatory hyperpigmentation (Figure 2). The patient returned to work without further incident.
Comment
As denoted by its hyphenated name, VZV infection can cause 2 distinct disease processes.1,2 Varicella, the generalized initial exanthem known as chickenpox, appears predominantly in childhood. With resolution of this primary infection, the virus lies dormant in sensory ganglia, persisting in neurons. Stress, advanced age, and/or compromised immunity may reactivate latent VZV. This secondary expression is known as herpes zoster (shingles), a unilateral eruption of lesions localized to a single dermatome.
In most cases, morphology of the varicella eruption confirms the diagnosis. Lesions evolve through stages from macules and papules to vesicles and pustules and then to crusts. This evolution typically takes 24 to 48 hours.2 The varicella eruption contains an admixture of elements from each stage simultaneously. Crusts usually resolve over an average of 14 days. Serologically, IgM is measurable as early as 1 to 2 days after appearance of the eruption.3,4 Next to appear are IgG antibodies, which generally remain detectable for life. With more than 90% of the US population being seropositive for VZV,5 diagnosis and management of varicella and herpes zoster usually are straightforward; however, there have been unusual variations on this classic sequence of pathogenesis.
In disseminated zoster, the clinical presentation includes more than 20 lesions outside the dermatome primarily affected.6 Another permutation of VZV infection is zoster sine herpete, which causes the characteristic dermatomal pain of herpes zoster but without the rash.7 Occasionally, 2 cases of chickenpox occur in the same person, usually indicating an underlying immune deficiency. Recurrent varicella in those with intact immunity is purportedly rare. A PubMed search of articles indexed for MEDLINE using the search terms recurrent varicella, chickenpox reinfection, and immunocompetent revealed 41 cases of recurrent varicella in immunocompetent patients in the English language literature occurring among children,1,8-11 adults,8 the elderly,12 health care workers,13-15 and pregnant women16 (Table).
|
|
Surveillance studies, however, have challenged the apparent rarity of recurrent varicella, asserting that varicella may recur more frequently than is generally recognized.17,18 Hall et al17 described 9947 cases of varicella, with nearly 6.9% reporting prior varicella infection. Another surveillance report by Marin et al18 evaluating data from 1047 adults with varicella noted that 21% of participants reported prior VZV infections. Both of these studies defined varicella by clinical parameters as a condition with acute onset of generalized maculopapulovesicular rash without other known cause. Although laboratory confirmation of VZV infection was not documented in either study, a history of varicella is considered a reliable indicator of immunity. In fact, studies show that a history of varicella is associated with serologic evidence of immunity 97% to 100% of the time.19,20
Immunity against VZV in humans is not well understood. Although both humoral and cellular factors play a role, cell-mediated immunity may be more important in suppressing primary infection and defending against reinfection. Varicella is more likely to disseminate in lymphopenic patients,21,22 while its course is uninfluenced by hypogammaglobulinemia.1,23 One study of simian varicella virus, which demonstrates 75% genetic homology with VZV, noted that simian varicella virus–infected rhesus macaques without CD4+ T lymphocyte response experienced higher viral loads, prolonged viremia, and disseminated varicella.24 The loss of CD20+ B lymphocytes did not intensify the severity of varicella in the primate model. It is accepted, however, that waning humoral immunity and lower antibody levels correlate with varicella recurrence.25 Ethnicity may impact immunoglobulin persistence. One investigation postulated that individuals with darker skin types experience reduced viral shedding and therefore less antigenic boosting from secondary VZV infections, as they may less readily maintain protective levels of VZV-specific immunoglobulins.25 This phenomenon may have contributed to the 3 episodes of varicella in our patient.
Virulence factors that are intrinsic to VZV may also prompt reinfection. Although taxonomy is still in flux, 3 to 5 major genotypes of VZV have been recognized to date, categorized into European (Dumas), Japanese (Oka), and mosaic clades.26-28 In one study population, approximately 80% of the VZV strains isolated in the United States were of the European variety.26 It is unclear whether infection with one strain of VZV affords immunoprotection against the other strains. Interestingly, one report documented recurrent herpes zoster caused by 2 distinct VZV strains in the same individual.29 Since subtypes of VZV vary geographically, it is possible that increasing global travel may correlate with increased incidence and reporting of varicella reinfection, particularly in cosmopolitan centers. In patients with recurrent varicella, a careful investigation of their international travel history may be necessary.
Case Report
A 52-year-old black woman presented to our dermatology clinic for evaluation of a generalized pruritic rash of 5 days’ duration. The eruption had started on the trunk and subsequently spread to the face, legs, and arms, including the dorsal surfaces of the hands (Figure 1). The patient reported that she had developed a similar rash 4 years prior. She recalled no sick contacts but had occupational exposure to many people as a food service worker. Two days prior, the referring physician had initiated treatment with oral acyclovir 400 mg every 6 hours. The patient was in otherwise good health and reported no fever, chills, diaphoresis, or fatigue. She did not recall any recent insect bites, and a review of systems was negative.
The patient’s medical history was remarkable for 2 cases of varicella: the first, which occurred at 5 years of age, was diagnosed by a pediatrician and manifested as diffuse papules, vesicles, and crusts with concurrent mild fever. The infection followed a typical clinical course and resolved without complications after 1 week. The second case of varicella was diagnosed clinically at our dermatology clinic approximately 4 years prior to the current presentation and manifested as widespread pruritic lesions that were too numerous to count. Given her history of varicella in childhood, a punch biopsy specimen was taken from a lesion on the left trunk and a dermatopathologist confirmed the diagnosis of a herpesvirus infection. The second infection also resolved without sequelae after 12 days. Her medical history was otherwise unremarkable, revealing no exceptional sinopulmonary or gastrointestinal infections. The patient was not currently taking any medications or supplements and reported no known drug allergies.
Physical examination at the current presentation revealed a well-nourished, afebrile woman with vesicles and papules on the hands, arms, and legs along with vesicular and crusted papules in various stages of healing distributed on the chest, abdomen, and back. Lesions on the legs and feet were present but scant. The eruption was not confined to a single dermatome. No lesions were noted on the palms, soles, or oral mucosa and no epitrochlear, axillary, or supraclavicular lymphadenopathy was noted.
Initial laboratory values were obtained. A complete blood count demonstrated a normal leukocyte number of 5700 cells/μL (reference range, 4500–11,000 cells/μL) and mild anemia with a hemoglobin level of 10.3 g/dL (reference range, 14.0–17.5 g/dL). Monocytes were mildly elevated at 11% (reference range, 1%–9%). Serologic tests showed positive titers for varicella-zoster virus (VZV) IgM at 1.64 (negative, <0.91) and VZV IgG at 1.72 (negative, <0.91), indicating current and past VZV infection, respectively. Antibodies against herpes simplex virus (HSV) types 1 and 2 were negative for IgM and positive for IgG at >5.00 (negative, <0.90), indicating a remote HSV infection. Furthermore, results from a culture of a lesion on the left hand were negative for HSV.
After consultation with the Department of Infectious Diseases, further laboratory studies were performed. The absolute lymphocyte number was within normal range at 1600 cells/μL (reference range, 850–3900 cells/μL). Likewise, CD4+ T lymphocytes were normal at 618 cells/μL (reference range, 490–1740 cells/μL) or 39% of total lymphocytes (reference range, 30%–61%). Screening results were negative for human immunodeficiency virus types 1 and 2. Immunoglobulin subtype analysis revealed slightly elevated IgG at 1709 mg/dL (reference range, 723–1685 mg/dL), elevated IgA at 487 mg/dL (reference range, 65–382 mg/dL), and normal IgM at 238 mg/dL (reference range, 63–277 mg/dL).
Consistent with the clinical presentation and serologic studies, recurrent varicella was accepted as the most plausible diagnosis. Over the next 2 weeks, the eruption resolved with postinflammatory hyperpigmentation (Figure 2). The patient returned to work without further incident.
Comment
As denoted by its hyphenated name, VZV infection can cause 2 distinct disease processes.1,2 Varicella, the generalized initial exanthem known as chickenpox, appears predominantly in childhood. With resolution of this primary infection, the virus lies dormant in sensory ganglia, persisting in neurons. Stress, advanced age, and/or compromised immunity may reactivate latent VZV. This secondary expression is known as herpes zoster (shingles), a unilateral eruption of lesions localized to a single dermatome.
In most cases, morphology of the varicella eruption confirms the diagnosis. Lesions evolve through stages from macules and papules to vesicles and pustules and then to crusts. This evolution typically takes 24 to 48 hours.2 The varicella eruption contains an admixture of elements from each stage simultaneously. Crusts usually resolve over an average of 14 days. Serologically, IgM is measurable as early as 1 to 2 days after appearance of the eruption.3,4 Next to appear are IgG antibodies, which generally remain detectable for life. With more than 90% of the US population being seropositive for VZV,5 diagnosis and management of varicella and herpes zoster usually are straightforward; however, there have been unusual variations on this classic sequence of pathogenesis.
In disseminated zoster, the clinical presentation includes more than 20 lesions outside the dermatome primarily affected.6 Another permutation of VZV infection is zoster sine herpete, which causes the characteristic dermatomal pain of herpes zoster but without the rash.7 Occasionally, 2 cases of chickenpox occur in the same person, usually indicating an underlying immune deficiency. Recurrent varicella in those with intact immunity is purportedly rare. A PubMed search of articles indexed for MEDLINE using the search terms recurrent varicella, chickenpox reinfection, and immunocompetent revealed 41 cases of recurrent varicella in immunocompetent patients in the English language literature occurring among children,1,8-11 adults,8 the elderly,12 health care workers,13-15 and pregnant women16 (Table).
|
|
Surveillance studies, however, have challenged the apparent rarity of recurrent varicella, asserting that varicella may recur more frequently than is generally recognized.17,18 Hall et al17 described 9947 cases of varicella, with nearly 6.9% reporting prior varicella infection. Another surveillance report by Marin et al18 evaluating data from 1047 adults with varicella noted that 21% of participants reported prior VZV infections. Both of these studies defined varicella by clinical parameters as a condition with acute onset of generalized maculopapulovesicular rash without other known cause. Although laboratory confirmation of VZV infection was not documented in either study, a history of varicella is considered a reliable indicator of immunity. In fact, studies show that a history of varicella is associated with serologic evidence of immunity 97% to 100% of the time.19,20
Immunity against VZV in humans is not well understood. Although both humoral and cellular factors play a role, cell-mediated immunity may be more important in suppressing primary infection and defending against reinfection. Varicella is more likely to disseminate in lymphopenic patients,21,22 while its course is uninfluenced by hypogammaglobulinemia.1,23 One study of simian varicella virus, which demonstrates 75% genetic homology with VZV, noted that simian varicella virus–infected rhesus macaques without CD4+ T lymphocyte response experienced higher viral loads, prolonged viremia, and disseminated varicella.24 The loss of CD20+ B lymphocytes did not intensify the severity of varicella in the primate model. It is accepted, however, that waning humoral immunity and lower antibody levels correlate with varicella recurrence.25 Ethnicity may impact immunoglobulin persistence. One investigation postulated that individuals with darker skin types experience reduced viral shedding and therefore less antigenic boosting from secondary VZV infections, as they may less readily maintain protective levels of VZV-specific immunoglobulins.25 This phenomenon may have contributed to the 3 episodes of varicella in our patient.
Virulence factors that are intrinsic to VZV may also prompt reinfection. Although taxonomy is still in flux, 3 to 5 major genotypes of VZV have been recognized to date, categorized into European (Dumas), Japanese (Oka), and mosaic clades.26-28 In one study population, approximately 80% of the VZV strains isolated in the United States were of the European variety.26 It is unclear whether infection with one strain of VZV affords immunoprotection against the other strains. Interestingly, one report documented recurrent herpes zoster caused by 2 distinct VZV strains in the same individual.29 Since subtypes of VZV vary geographically, it is possible that increasing global travel may correlate with increased incidence and reporting of varicella reinfection, particularly in cosmopolitan centers. In patients with recurrent varicella, a careful investigation of their international travel history may be necessary.
1. Weller T. Varicella and herpes zoster: changing concepts of the natural history, control, and importance of a not-so-benign virus. N Engl J Med. 1983;309:1362-1368.
2. Heininger U, Seward JF. Varicella. Lancet. 2006;368:1365-1376.
3. Krah DL. Assays for antibodies to varicella-zoster virus. Infect Dis Clin North Am. 1996;10:507-527.
4. Oladepo DK, Klapper PE, Percival D, et al. Serological diagnosis of varicella-zoster virus in sera with antibody-capture enzyme-linked immunosorbent assay of IgM. J Virol Meth. 2000;84:169-173.
5. Kilgore PE, Kruszon-Moran D, Seward JF, et al. Varicella in Americans from NHANES III: implications for control through routine immunizations. J Med Virol. 2003;70:S111-S118.
6. Gupta S, Jain A, Gardiner C, et al. A rare case of disseminated cutaneous zoster in an immunocompetent patient. BMC Fam Pract. 2005;6:50.
7. Lewis GW. Zoster sine herpete. Br Med J. 1958;8:418-421.
8. Gershon AA, Steinberg SP, Gelb L. Clinical reinfection with varicella-zoster virus. J Infect Dis. 1984;149:137-142.
9. Junker AK, Angus E, Thomas EE. Recurrent varicella-zoster virus infections in apparently immunocompetent children. Pediatr Infect Dis J. 1991;10:569-575.
10. Junker AK, Tilley P. Varicella-zoster virus antibody avidity and IgG-subclass patterns in children with recurrent chickenpox. J Med Virol. 1994;43:119-124.
11. Terada K, Kawano S, Shimada Y, et al. Recurrent chickenpox after natural infection. Ped Infect Dis J. 1996;15:179-181.
12. Takayama N, Takayama M, Negishi M. Clinical varicella-zoster virus reinfection observed in two advanced-age persons [article in Japanese]. Kansenshogaku Zasshi. 1992;66:1373-1377.
13. Gurevich I, Jensen L, Kalter R, et al. Chickenpox in apparently “immune” hospital workers. Infect Control Hosp Epidemiol. 1990;11:510, 512.
14. Ku C, Liu Y, Christiani DC. Case report: occupationally related recurrent varicella (chickenpox) in a hospital nurse. Environ Health Perspect. 2005;113:1373-1375.
15. Johnson JA, Bloch KC, Dang BN. Varicella reinfection in a seropositive physician following occupational exposure to localized zoster. Clin Infect Dis. 2011;52:907-909.
16. Martin KA, Junker AK, Thomas EE, et al. Occurrence of chickenpox during pregnancy in women seropositive for varicella-zoster virus. J Infect Dis. 1994;170:991-995.
17. Hall S, Maupin T, Seward J, et al. Second varicella infections: are they more common than previously thought? Pediatrics. 2002;109:1068-1073.
18. Marin M, Watson TL, Chaves SS, et al. Varicella among adults: data from an active surveillance project, 1995-2005. J Infect Dis. 2008;197(Suppl 2):S94-S100.
19. Perella DM, Fiks A, Spain CV. Validity of reported varicella history as a marker for varicella-zoster virus immunity. Paper presented at: Pediatric Academic Societies Annual Meeting; 2005; Washington, DC.
20. Ferson MJ, Bell SM, Robertson PW. Determination and importance of varicella immune status of nursing staff in a children’s hospital. J Hosp Infect. 1990;15:347-351.
21. Arvin AM, Pollard RB, Rasmussen LE, et al. Selective impairment of lymphocyte reactivity to varicella-zoster virus antigen among untreated patients with lymphoma. J Infect Dis. 1978;137:531-540.
22. Feldman S, Hughes WT, Daniel CB. Varicella in children with cancer: seventy-seven cases. Pediatrics. 1975;56:388-397.
23. Arvin AM. Varicella-zoster virus. Clin Microbiol Rev. 1996;9:361-381.
24. Haberthur K, Engelmann F, Park B, et al. CD4 T cell immunity is critical for the control of simian varicella virus infection in nonhuman primate model of VZV infection. PLoS Pathog. 2011;7:e1002367.
25. Ayres KL, Talukder Y, Breuer J. Humoral immunity following chickenpox is influenced by geography and ethnicity. J Infect. 2010;61:244-251.
26. Loparev VN, Gonzalez A, Deleon-Carnes M, et al. Global identification of three major genotypes of varicella-zoster virus: longitudinal clustering and strategies for genotyping. J Virol. 2004;78:8349-8358.
27. Parker SP, Breuer J, Taha Y, et al. Genotyping of varicella-zoster virus and the discrimination of Oka vaccine strains by TaqMan real-time PCR. J Clin Microbiol. 2006;44:3911-3914.
28. Loparev VN, Rubtcova EN, Bostik V, et al. Identification of five major and two minor genotypes of varicella-zoster virus strains: a practical two-amplicon approach used to genotype clinical isolates in Australia and New Zealand. J Virol. 2007;81:12758-12765.
29. Taha Y, Scott FT, Parker SP, et al. Reactivation of 2 genetically distinct varicella-zoster viruses in the same individual. Clin Infect Dis. 2006;43:1301-1303.
1. Weller T. Varicella and herpes zoster: changing concepts of the natural history, control, and importance of a not-so-benign virus. N Engl J Med. 1983;309:1362-1368.
2. Heininger U, Seward JF. Varicella. Lancet. 2006;368:1365-1376.
3. Krah DL. Assays for antibodies to varicella-zoster virus. Infect Dis Clin North Am. 1996;10:507-527.
4. Oladepo DK, Klapper PE, Percival D, et al. Serological diagnosis of varicella-zoster virus in sera with antibody-capture enzyme-linked immunosorbent assay of IgM. J Virol Meth. 2000;84:169-173.
5. Kilgore PE, Kruszon-Moran D, Seward JF, et al. Varicella in Americans from NHANES III: implications for control through routine immunizations. J Med Virol. 2003;70:S111-S118.
6. Gupta S, Jain A, Gardiner C, et al. A rare case of disseminated cutaneous zoster in an immunocompetent patient. BMC Fam Pract. 2005;6:50.
7. Lewis GW. Zoster sine herpete. Br Med J. 1958;8:418-421.
8. Gershon AA, Steinberg SP, Gelb L. Clinical reinfection with varicella-zoster virus. J Infect Dis. 1984;149:137-142.
9. Junker AK, Angus E, Thomas EE. Recurrent varicella-zoster virus infections in apparently immunocompetent children. Pediatr Infect Dis J. 1991;10:569-575.
10. Junker AK, Tilley P. Varicella-zoster virus antibody avidity and IgG-subclass patterns in children with recurrent chickenpox. J Med Virol. 1994;43:119-124.
11. Terada K, Kawano S, Shimada Y, et al. Recurrent chickenpox after natural infection. Ped Infect Dis J. 1996;15:179-181.
12. Takayama N, Takayama M, Negishi M. Clinical varicella-zoster virus reinfection observed in two advanced-age persons [article in Japanese]. Kansenshogaku Zasshi. 1992;66:1373-1377.
13. Gurevich I, Jensen L, Kalter R, et al. Chickenpox in apparently “immune” hospital workers. Infect Control Hosp Epidemiol. 1990;11:510, 512.
14. Ku C, Liu Y, Christiani DC. Case report: occupationally related recurrent varicella (chickenpox) in a hospital nurse. Environ Health Perspect. 2005;113:1373-1375.
15. Johnson JA, Bloch KC, Dang BN. Varicella reinfection in a seropositive physician following occupational exposure to localized zoster. Clin Infect Dis. 2011;52:907-909.
16. Martin KA, Junker AK, Thomas EE, et al. Occurrence of chickenpox during pregnancy in women seropositive for varicella-zoster virus. J Infect Dis. 1994;170:991-995.
17. Hall S, Maupin T, Seward J, et al. Second varicella infections: are they more common than previously thought? Pediatrics. 2002;109:1068-1073.
18. Marin M, Watson TL, Chaves SS, et al. Varicella among adults: data from an active surveillance project, 1995-2005. J Infect Dis. 2008;197(Suppl 2):S94-S100.
19. Perella DM, Fiks A, Spain CV. Validity of reported varicella history as a marker for varicella-zoster virus immunity. Paper presented at: Pediatric Academic Societies Annual Meeting; 2005; Washington, DC.
20. Ferson MJ, Bell SM, Robertson PW. Determination and importance of varicella immune status of nursing staff in a children’s hospital. J Hosp Infect. 1990;15:347-351.
21. Arvin AM, Pollard RB, Rasmussen LE, et al. Selective impairment of lymphocyte reactivity to varicella-zoster virus antigen among untreated patients with lymphoma. J Infect Dis. 1978;137:531-540.
22. Feldman S, Hughes WT, Daniel CB. Varicella in children with cancer: seventy-seven cases. Pediatrics. 1975;56:388-397.
23. Arvin AM. Varicella-zoster virus. Clin Microbiol Rev. 1996;9:361-381.
24. Haberthur K, Engelmann F, Park B, et al. CD4 T cell immunity is critical for the control of simian varicella virus infection in nonhuman primate model of VZV infection. PLoS Pathog. 2011;7:e1002367.
25. Ayres KL, Talukder Y, Breuer J. Humoral immunity following chickenpox is influenced by geography and ethnicity. J Infect. 2010;61:244-251.
26. Loparev VN, Gonzalez A, Deleon-Carnes M, et al. Global identification of three major genotypes of varicella-zoster virus: longitudinal clustering and strategies for genotyping. J Virol. 2004;78:8349-8358.
27. Parker SP, Breuer J, Taha Y, et al. Genotyping of varicella-zoster virus and the discrimination of Oka vaccine strains by TaqMan real-time PCR. J Clin Microbiol. 2006;44:3911-3914.
28. Loparev VN, Rubtcova EN, Bostik V, et al. Identification of five major and two minor genotypes of varicella-zoster virus strains: a practical two-amplicon approach used to genotype clinical isolates in Australia and New Zealand. J Virol. 2007;81:12758-12765.
29. Taha Y, Scott FT, Parker SP, et al. Reactivation of 2 genetically distinct varicella-zoster viruses in the same individual. Clin Infect Dis. 2006;43:1301-1303.
Practice Points
- Varicella is a viral exanthem that manifests as generalized and pruritic papules, vesicles, and crusted lesions and is the initial expression of infection with the varicella-zoster virus (VZV).
- Secondary expression of VZV infection is typified by herpes zoster, where painful papules and vesicles are confined within a single dermatome.
- Although contradictory to dogma and public perception, varicella may occur more than once; therefore, patients with a generalized pruritic exanthem should be screened for varicella, even if they report a history of varicella.