User login
Oxybutynin Treatment for Hyperhidrosis in Spinal Cord Injury Patients
Hyperhidrosis (HH) is sweating beyond that which is required for thermoregulation.1 Secondary HH, which is usually caused by an underlying medical condition or drug, may be seen in patients with spinal cord injury (SCI) and can negatively impact psychosocial well-being and quality of life (QOL) if not treated.1
Information on the current prevalence of HH is lacking. In 2004, one study projected the prevalence of HH in the U.S. to be 2.8%.2 A previous study found that about 27% of the 154 patients with SCI reported experiencing HH that was annoying, with 28 (14.6%) of those reporting no contributing somatic causes.3 Somatic causes of HH include autonomic dysreflexia, posttraumatic syringomyelia, or orthostatic hypotension.4
Autonomic dysreflexia is a syndrome that describes a dramatic increase in blood pressure (BP) in patients with spinal cord lesion at or above T6 and is characterized by exaggerated autonomic responses to stimuli that are innocuous to individuals without SCI.5,6 Noxious stimuli that may trigger autonomic dysreflexia include bowel or bladder distension or obstruction, urinary infection, catheter insertion, suprapubic palpation, or skin irritation.6 Syringomyelia, another somatic cause of HH, is a cystic lesion on the spinal cord that may develop secondary to congenital anomalies or SCI.6-8
The following case reports describe 2 patients with SCI with different diagnoses and presentations of HH. Both were treated with oxybutynin for HH.
Case 1 Presentation
Mr. J is a 49-year-old with C6-C7 SCI attributed to a motor vehicle accident 26 years before. He presented to the primary care clinic for a routine visit in a self-propelled wheelchair. His diagnosis included tetraplegia, muscle spasms, osteoporosis, chronic pain syndrome, benign prosthetic hypertrophy, neurogenic bladder, and neurogenic bowel. He was noted to have a bath towel around his neck to wipe sweat from his face and neck. He did not recall when this condition started; however, he reported a prior trial of diazepam 5 mg as needed in 2006 in the mornings and before transfers when sweating was usually worst. He continued to use diazepam because it helped with his muscle spasms but not with sweating. His other medications included oral baclofen, alendronate, ibuprofen, docusate sodium, tamsulosin, calcium/vitamin C supplement, and bisacodyl suppository.
The patient’s surgical history was significant for anterior discectomy with C6-C7 fusion, sphincterotomy, transurethral resection of the prostate, and right urethral stent placement for hydronephrosis in 2004. His cystoscopy and renal sonogram were within normal limits. On physical examination, his vital signs were within normal limits. However, his long-sleeved shirt was wet on the front, and his neck, chest, and arms also were moist from excessive sweating. During his transfer to and from his wheelchair, he was noted to have chattering of his teeth. The remainder of the physical examination was negative for any other acute findings.
Mr. J was prescribed a 30-day trial of oxybutynin 5 mg 1 tablet by mouth per day for HH. During a 3-week follow-up telephone call, Mr. J reported that the oxybutynin was working well; the sweating on his chest had improved and had resolved on his face. Except for mild dryness of mouth, he was tolerating the medication well. There were no changes in his neurogenic bladder, which was managed with an external urinary device.
Six months later, Mr. J reported that oxybutynin continued to work well, and he no longer had to travel with a towel. He was able to go to a football game, social activities were more enjoyable, and he was not embarrassed because of excessive sweating.
Case 2 Presentation
Mr. B is a 48-year-old with T12 paraplegia secondary to a motor vehicle accident in 1994. He called the primary clinic for a visit because he was concerned about cold clammy hands for the past 6 to 7 months when he woke up in the morning and sometimes throughout the day. He was preparing to start his first semester in college. His diagnosis included neurogenic bowel and bladder and muscle spasms. There was no history of posttraumatic syringomyelia, and his medications included baclofen, dantrolene, diazepam, and multivitamins.
Mr. B took tolterodine 4 mg/d for several years, and for unknown reasons, about 6 years previously the prescription was changed to oxybutynin 15-mg extended release for his neurogenic bladder. He continued to have urinary leakage between the every 4-hour intermittent catherization, and oxybutynin was increased to 10-mg tablet twice per day.
About 7 months prior to this appointment, Mr. B independently stopped the oxybutynin as he felt that it was not making a difference in the management of his neurogenic bladder. It was noted that his cold clammy hands started about the same time that he discontinued the oxybutynin. He could not recall whether he had this symptom prior to initiation of any medication. It was mutually decided to restart the oxybutynin at a lower dose, this time not for his bladder but for the HH. He was ordered a 30-day trial of a 5-mg tablet once per day. About 3 weeks later, he sent a secure message to report oxybutynin’s effectiveness and to request a refill.
Discussion
Sweating is a physiological process that involves the active secretion of water by specialized sweat glands in the skin.9-11 There are 2 types of sweat glands in the skin; apocrine and eccrine.9 Collectively the 3 million eccrine sweat glands of the average person approximately equal the mass of a kidney and exceed the secretory rate of exocrine glands.9 They function in evaporative cooling in response to thermal or physiologic stimuli and are widely distributed over the body, especially on the forehead, back, palms, and soles.10
Sympathetic cholinergic nerves are mainly responsible for sweat secretion by the release of acetylcholine to activate muscarinic receptors on the gland.11 Postganglionic fibers from sympathetic nerve cells innervate sweat glands that release cholinergics.6 Postganglionic cholinergic receptors that are activated by muscarinic drugs are termed muscarinic receptors and are readily accessible to antimuscarinic drugs.6,12 Anticholinergic/antimuscarinic agents antagonize muscarinic receptors and suppress premature detrusor contractions to enhance bladder storage.13 They include oxybutynin, tolterodine, trospium, solifenacin, darifenacin, and fesoterodine.13 Oxybutynin was used in both cases because it is on VA formulary. It was effective in treating HH, although the etiology is unclear and the presentations were different.
One retrospective study that analyzed 20 patients who received oxybutynin for primary HH at uncommon sites, such as the back and groin, found that QOL improved in 85% of the subjects after 6 weeks.14 Randomized placebo-controlled trials also have found oxybutynin effective for treatment of palmar and axillary HH and generalized HH.15,16
Syringomyelia was ruled out in both cases based on history and radiologic studies, specifically magnetic resonance imaging. Autonomic dysreflexia was ruled out as the HH was not an acute finding and BP was within normal limits. Orthostatic hypotension is a common finding in SCI, mainly in tetraplegic patients, and could be suspected in both cases. Sweating was usually worse in the mornings in both cases and during transfers, as noted in the first case.17 However, chronic autoregulation allows for chronic adaption to tissue hypoperfusion over time.16
Hyperhidrosis or other disorders of eccrine sweating can occur for various reasons, including changes in the spinal sympathetic preganglionic, ganglionic, or postganglionic neurons; dysfunction of the thermoregulatory centers in the brain’s central autonomic network; or changes in the muscarinic cholinergic synapse on sweat glands.18
Conclusion
Patients with SCI may have an acute or chronic presentation of HH. Removal of the inciting cause in the case of autonomic dysreflexia and/or the administration of a pharmaceutical agent is the usual treatment.
Regardless of the etiology of HH that persists, effective treatment should be a goal, especially in those patients whose QOL is affected by this condition. The outcome of treatment with oxybutynin in these case reports is consistent with the findings of the limited retrospective study and randomized placebo-controlled studies that show oxybutynin is effective for treating bothersome HH.14-16
The results of these case reports are not generalizable to patients with SCI and HH, nor are the results of the limited retrospective study and randomized placebo-controlled studies, as their sample sizes were small.14,16,17 However, information on the use of oxybutynin for the effective treatment of HH in the SCI population is promising. Research studies on the prevalence of HH and randomized placebo-controlled trials with a larger SCI population are considerations for future studies.
1. Strutton DR, Kowalski JW, Glaser DA, Stang PE. US prevalence of hyperhidrosis and impact on individuals with axillary hyperhidrosis: results from a national survey. J Am Acad Dermatol. 2004;51(2):241-248.
2. Walling HW. Clinical differentiation of primary from secondary hyperhidrosis. J Am Acad Dermatol. 2011;64(4):690-695.
3. Andersen LS, Biering-Sørensen F, Müller PG, Jensen IL, Aggerbeck B. The prevalence of hyperhidrosis in patients with spinal cord injuries and an evaluation of the effect of dextropropoxyphene hydrochloride in therapy. Paraplegia. 1992;(30):184-191.
4. Sato K, Kang WH, Saga K, Sato KT. Biology of sweat glands and their disorders. II. Disorders of sweat gland function. J Am Acad Dermatol. 1989;20(5, pt 1):713-726.
5. Kewalramani LS. Autonomic dysreflexia in traumatic myelopathy. Am J Phys Med. 1980;59(1):1-21.
6. Low PA, Engstrom JW. Disorders of the autonomic nervous system. In: Kasper D, Fauci A, Hauser S, Longo D, Jameson J, Loscalzo J, eds. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw-Hill; 2015.
7. Milhort TH. Classification of syringomyelia. Neurosurg Focus. 2000;8(3):E1.
8. National Institute of Neurological Disorders and Stroke. Syringomyelia fact sheet. https://www.ninds .nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Syringomyelia-Fact-Sheet. Accessed February 13, 2017.
9. Meschner AL. Junqueira’s Basic Histology. 14th ed. New York, NY: McGraw-Hill; 2016:371-392.
10. Mauro TM. Biology of eccrine and apocrine glands. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K. eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
11. Barrett KE, Barman SM, Boitano S, Brooks HL. Ganong’s Review of Medical Physiology. 25th ed. New York, NY: McGraw-Hill; 2016:261-272.
12. Kellogg DL Jr. Thermoregulation. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
13. Rovner ES, Wyman J, Lam S. Urinary Incontinence. In: DiPiro JT, Talbert RL, Yee GC, Matzke GR, Wells BG, Posey L. eds. Pharmacotherapy: A Pathophysiologic Approach. 9th ed. New York, NY: McGraw-Hill; 2014:1377-1396.
14. Teivelis MP, Wolosker N, Krutman M, Kauffman P, Campos JR, Puech-Leão P. Treatment of uncommon sites of focal primary hyperhidrosis: experience with pharmacological therapy using oxybutynin. Clinics (Sao Paulo). 2014;69(9):608-614.
15. Wolosker N, de Campos JR, Kauffman P, Puech-Leão P. A randomized-placebo-controlled trial of oxybutynin for the initial treatment of palmar and axillary hyperhidrosis. J Vasc Surg. 2012;55(6):1696-1700.
16. Schollhammer M, Brenaut E, Menard-Andivot N, et al. Oxybutynin as a treatment for generalized hyperhidrosis. Br J Dermatol. 2015;173(5):1163-1168.
17. Gonzalez F, Chang JY, Banovac K, Messina D, Martinez-Arizala A, Kelley RE. Autoregulation of cerebral blood flow in patients with orthostatic hypotension after spinal cord injury. Paraplegia. 1991;29(1):1-7.
18. Fealey RD, Hebert AA. Disorders of the eccrine sweat glands and sweating. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K, eds. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw-Hill; 2012:chap 84.
Hyperhidrosis (HH) is sweating beyond that which is required for thermoregulation.1 Secondary HH, which is usually caused by an underlying medical condition or drug, may be seen in patients with spinal cord injury (SCI) and can negatively impact psychosocial well-being and quality of life (QOL) if not treated.1
Information on the current prevalence of HH is lacking. In 2004, one study projected the prevalence of HH in the U.S. to be 2.8%.2 A previous study found that about 27% of the 154 patients with SCI reported experiencing HH that was annoying, with 28 (14.6%) of those reporting no contributing somatic causes.3 Somatic causes of HH include autonomic dysreflexia, posttraumatic syringomyelia, or orthostatic hypotension.4
Autonomic dysreflexia is a syndrome that describes a dramatic increase in blood pressure (BP) in patients with spinal cord lesion at or above T6 and is characterized by exaggerated autonomic responses to stimuli that are innocuous to individuals without SCI.5,6 Noxious stimuli that may trigger autonomic dysreflexia include bowel or bladder distension or obstruction, urinary infection, catheter insertion, suprapubic palpation, or skin irritation.6 Syringomyelia, another somatic cause of HH, is a cystic lesion on the spinal cord that may develop secondary to congenital anomalies or SCI.6-8
The following case reports describe 2 patients with SCI with different diagnoses and presentations of HH. Both were treated with oxybutynin for HH.
Case 1 Presentation
Mr. J is a 49-year-old with C6-C7 SCI attributed to a motor vehicle accident 26 years before. He presented to the primary care clinic for a routine visit in a self-propelled wheelchair. His diagnosis included tetraplegia, muscle spasms, osteoporosis, chronic pain syndrome, benign prosthetic hypertrophy, neurogenic bladder, and neurogenic bowel. He was noted to have a bath towel around his neck to wipe sweat from his face and neck. He did not recall when this condition started; however, he reported a prior trial of diazepam 5 mg as needed in 2006 in the mornings and before transfers when sweating was usually worst. He continued to use diazepam because it helped with his muscle spasms but not with sweating. His other medications included oral baclofen, alendronate, ibuprofen, docusate sodium, tamsulosin, calcium/vitamin C supplement, and bisacodyl suppository.
The patient’s surgical history was significant for anterior discectomy with C6-C7 fusion, sphincterotomy, transurethral resection of the prostate, and right urethral stent placement for hydronephrosis in 2004. His cystoscopy and renal sonogram were within normal limits. On physical examination, his vital signs were within normal limits. However, his long-sleeved shirt was wet on the front, and his neck, chest, and arms also were moist from excessive sweating. During his transfer to and from his wheelchair, he was noted to have chattering of his teeth. The remainder of the physical examination was negative for any other acute findings.
Mr. J was prescribed a 30-day trial of oxybutynin 5 mg 1 tablet by mouth per day for HH. During a 3-week follow-up telephone call, Mr. J reported that the oxybutynin was working well; the sweating on his chest had improved and had resolved on his face. Except for mild dryness of mouth, he was tolerating the medication well. There were no changes in his neurogenic bladder, which was managed with an external urinary device.
Six months later, Mr. J reported that oxybutynin continued to work well, and he no longer had to travel with a towel. He was able to go to a football game, social activities were more enjoyable, and he was not embarrassed because of excessive sweating.
Case 2 Presentation
Mr. B is a 48-year-old with T12 paraplegia secondary to a motor vehicle accident in 1994. He called the primary clinic for a visit because he was concerned about cold clammy hands for the past 6 to 7 months when he woke up in the morning and sometimes throughout the day. He was preparing to start his first semester in college. His diagnosis included neurogenic bowel and bladder and muscle spasms. There was no history of posttraumatic syringomyelia, and his medications included baclofen, dantrolene, diazepam, and multivitamins.
Mr. B took tolterodine 4 mg/d for several years, and for unknown reasons, about 6 years previously the prescription was changed to oxybutynin 15-mg extended release for his neurogenic bladder. He continued to have urinary leakage between the every 4-hour intermittent catherization, and oxybutynin was increased to 10-mg tablet twice per day.
About 7 months prior to this appointment, Mr. B independently stopped the oxybutynin as he felt that it was not making a difference in the management of his neurogenic bladder. It was noted that his cold clammy hands started about the same time that he discontinued the oxybutynin. He could not recall whether he had this symptom prior to initiation of any medication. It was mutually decided to restart the oxybutynin at a lower dose, this time not for his bladder but for the HH. He was ordered a 30-day trial of a 5-mg tablet once per day. About 3 weeks later, he sent a secure message to report oxybutynin’s effectiveness and to request a refill.
Discussion
Sweating is a physiological process that involves the active secretion of water by specialized sweat glands in the skin.9-11 There are 2 types of sweat glands in the skin; apocrine and eccrine.9 Collectively the 3 million eccrine sweat glands of the average person approximately equal the mass of a kidney and exceed the secretory rate of exocrine glands.9 They function in evaporative cooling in response to thermal or physiologic stimuli and are widely distributed over the body, especially on the forehead, back, palms, and soles.10
Sympathetic cholinergic nerves are mainly responsible for sweat secretion by the release of acetylcholine to activate muscarinic receptors on the gland.11 Postganglionic fibers from sympathetic nerve cells innervate sweat glands that release cholinergics.6 Postganglionic cholinergic receptors that are activated by muscarinic drugs are termed muscarinic receptors and are readily accessible to antimuscarinic drugs.6,12 Anticholinergic/antimuscarinic agents antagonize muscarinic receptors and suppress premature detrusor contractions to enhance bladder storage.13 They include oxybutynin, tolterodine, trospium, solifenacin, darifenacin, and fesoterodine.13 Oxybutynin was used in both cases because it is on VA formulary. It was effective in treating HH, although the etiology is unclear and the presentations were different.
One retrospective study that analyzed 20 patients who received oxybutynin for primary HH at uncommon sites, such as the back and groin, found that QOL improved in 85% of the subjects after 6 weeks.14 Randomized placebo-controlled trials also have found oxybutynin effective for treatment of palmar and axillary HH and generalized HH.15,16
Syringomyelia was ruled out in both cases based on history and radiologic studies, specifically magnetic resonance imaging. Autonomic dysreflexia was ruled out as the HH was not an acute finding and BP was within normal limits. Orthostatic hypotension is a common finding in SCI, mainly in tetraplegic patients, and could be suspected in both cases. Sweating was usually worse in the mornings in both cases and during transfers, as noted in the first case.17 However, chronic autoregulation allows for chronic adaption to tissue hypoperfusion over time.16
Hyperhidrosis or other disorders of eccrine sweating can occur for various reasons, including changes in the spinal sympathetic preganglionic, ganglionic, or postganglionic neurons; dysfunction of the thermoregulatory centers in the brain’s central autonomic network; or changes in the muscarinic cholinergic synapse on sweat glands.18
Conclusion
Patients with SCI may have an acute or chronic presentation of HH. Removal of the inciting cause in the case of autonomic dysreflexia and/or the administration of a pharmaceutical agent is the usual treatment.
Regardless of the etiology of HH that persists, effective treatment should be a goal, especially in those patients whose QOL is affected by this condition. The outcome of treatment with oxybutynin in these case reports is consistent with the findings of the limited retrospective study and randomized placebo-controlled studies that show oxybutynin is effective for treating bothersome HH.14-16
The results of these case reports are not generalizable to patients with SCI and HH, nor are the results of the limited retrospective study and randomized placebo-controlled studies, as their sample sizes were small.14,16,17 However, information on the use of oxybutynin for the effective treatment of HH in the SCI population is promising. Research studies on the prevalence of HH and randomized placebo-controlled trials with a larger SCI population are considerations for future studies.
Hyperhidrosis (HH) is sweating beyond that which is required for thermoregulation.1 Secondary HH, which is usually caused by an underlying medical condition or drug, may be seen in patients with spinal cord injury (SCI) and can negatively impact psychosocial well-being and quality of life (QOL) if not treated.1
Information on the current prevalence of HH is lacking. In 2004, one study projected the prevalence of HH in the U.S. to be 2.8%.2 A previous study found that about 27% of the 154 patients with SCI reported experiencing HH that was annoying, with 28 (14.6%) of those reporting no contributing somatic causes.3 Somatic causes of HH include autonomic dysreflexia, posttraumatic syringomyelia, or orthostatic hypotension.4
Autonomic dysreflexia is a syndrome that describes a dramatic increase in blood pressure (BP) in patients with spinal cord lesion at or above T6 and is characterized by exaggerated autonomic responses to stimuli that are innocuous to individuals without SCI.5,6 Noxious stimuli that may trigger autonomic dysreflexia include bowel or bladder distension or obstruction, urinary infection, catheter insertion, suprapubic palpation, or skin irritation.6 Syringomyelia, another somatic cause of HH, is a cystic lesion on the spinal cord that may develop secondary to congenital anomalies or SCI.6-8
The following case reports describe 2 patients with SCI with different diagnoses and presentations of HH. Both were treated with oxybutynin for HH.
Case 1 Presentation
Mr. J is a 49-year-old with C6-C7 SCI attributed to a motor vehicle accident 26 years before. He presented to the primary care clinic for a routine visit in a self-propelled wheelchair. His diagnosis included tetraplegia, muscle spasms, osteoporosis, chronic pain syndrome, benign prosthetic hypertrophy, neurogenic bladder, and neurogenic bowel. He was noted to have a bath towel around his neck to wipe sweat from his face and neck. He did not recall when this condition started; however, he reported a prior trial of diazepam 5 mg as needed in 2006 in the mornings and before transfers when sweating was usually worst. He continued to use diazepam because it helped with his muscle spasms but not with sweating. His other medications included oral baclofen, alendronate, ibuprofen, docusate sodium, tamsulosin, calcium/vitamin C supplement, and bisacodyl suppository.
The patient’s surgical history was significant for anterior discectomy with C6-C7 fusion, sphincterotomy, transurethral resection of the prostate, and right urethral stent placement for hydronephrosis in 2004. His cystoscopy and renal sonogram were within normal limits. On physical examination, his vital signs were within normal limits. However, his long-sleeved shirt was wet on the front, and his neck, chest, and arms also were moist from excessive sweating. During his transfer to and from his wheelchair, he was noted to have chattering of his teeth. The remainder of the physical examination was negative for any other acute findings.
Mr. J was prescribed a 30-day trial of oxybutynin 5 mg 1 tablet by mouth per day for HH. During a 3-week follow-up telephone call, Mr. J reported that the oxybutynin was working well; the sweating on his chest had improved and had resolved on his face. Except for mild dryness of mouth, he was tolerating the medication well. There were no changes in his neurogenic bladder, which was managed with an external urinary device.
Six months later, Mr. J reported that oxybutynin continued to work well, and he no longer had to travel with a towel. He was able to go to a football game, social activities were more enjoyable, and he was not embarrassed because of excessive sweating.
Case 2 Presentation
Mr. B is a 48-year-old with T12 paraplegia secondary to a motor vehicle accident in 1994. He called the primary clinic for a visit because he was concerned about cold clammy hands for the past 6 to 7 months when he woke up in the morning and sometimes throughout the day. He was preparing to start his first semester in college. His diagnosis included neurogenic bowel and bladder and muscle spasms. There was no history of posttraumatic syringomyelia, and his medications included baclofen, dantrolene, diazepam, and multivitamins.
Mr. B took tolterodine 4 mg/d for several years, and for unknown reasons, about 6 years previously the prescription was changed to oxybutynin 15-mg extended release for his neurogenic bladder. He continued to have urinary leakage between the every 4-hour intermittent catherization, and oxybutynin was increased to 10-mg tablet twice per day.
About 7 months prior to this appointment, Mr. B independently stopped the oxybutynin as he felt that it was not making a difference in the management of his neurogenic bladder. It was noted that his cold clammy hands started about the same time that he discontinued the oxybutynin. He could not recall whether he had this symptom prior to initiation of any medication. It was mutually decided to restart the oxybutynin at a lower dose, this time not for his bladder but for the HH. He was ordered a 30-day trial of a 5-mg tablet once per day. About 3 weeks later, he sent a secure message to report oxybutynin’s effectiveness and to request a refill.
Discussion
Sweating is a physiological process that involves the active secretion of water by specialized sweat glands in the skin.9-11 There are 2 types of sweat glands in the skin; apocrine and eccrine.9 Collectively the 3 million eccrine sweat glands of the average person approximately equal the mass of a kidney and exceed the secretory rate of exocrine glands.9 They function in evaporative cooling in response to thermal or physiologic stimuli and are widely distributed over the body, especially on the forehead, back, palms, and soles.10
Sympathetic cholinergic nerves are mainly responsible for sweat secretion by the release of acetylcholine to activate muscarinic receptors on the gland.11 Postganglionic fibers from sympathetic nerve cells innervate sweat glands that release cholinergics.6 Postganglionic cholinergic receptors that are activated by muscarinic drugs are termed muscarinic receptors and are readily accessible to antimuscarinic drugs.6,12 Anticholinergic/antimuscarinic agents antagonize muscarinic receptors and suppress premature detrusor contractions to enhance bladder storage.13 They include oxybutynin, tolterodine, trospium, solifenacin, darifenacin, and fesoterodine.13 Oxybutynin was used in both cases because it is on VA formulary. It was effective in treating HH, although the etiology is unclear and the presentations were different.
One retrospective study that analyzed 20 patients who received oxybutynin for primary HH at uncommon sites, such as the back and groin, found that QOL improved in 85% of the subjects after 6 weeks.14 Randomized placebo-controlled trials also have found oxybutynin effective for treatment of palmar and axillary HH and generalized HH.15,16
Syringomyelia was ruled out in both cases based on history and radiologic studies, specifically magnetic resonance imaging. Autonomic dysreflexia was ruled out as the HH was not an acute finding and BP was within normal limits. Orthostatic hypotension is a common finding in SCI, mainly in tetraplegic patients, and could be suspected in both cases. Sweating was usually worse in the mornings in both cases and during transfers, as noted in the first case.17 However, chronic autoregulation allows for chronic adaption to tissue hypoperfusion over time.16
Hyperhidrosis or other disorders of eccrine sweating can occur for various reasons, including changes in the spinal sympathetic preganglionic, ganglionic, or postganglionic neurons; dysfunction of the thermoregulatory centers in the brain’s central autonomic network; or changes in the muscarinic cholinergic synapse on sweat glands.18
Conclusion
Patients with SCI may have an acute or chronic presentation of HH. Removal of the inciting cause in the case of autonomic dysreflexia and/or the administration of a pharmaceutical agent is the usual treatment.
Regardless of the etiology of HH that persists, effective treatment should be a goal, especially in those patients whose QOL is affected by this condition. The outcome of treatment with oxybutynin in these case reports is consistent with the findings of the limited retrospective study and randomized placebo-controlled studies that show oxybutynin is effective for treating bothersome HH.14-16
The results of these case reports are not generalizable to patients with SCI and HH, nor are the results of the limited retrospective study and randomized placebo-controlled studies, as their sample sizes were small.14,16,17 However, information on the use of oxybutynin for the effective treatment of HH in the SCI population is promising. Research studies on the prevalence of HH and randomized placebo-controlled trials with a larger SCI population are considerations for future studies.
1. Strutton DR, Kowalski JW, Glaser DA, Stang PE. US prevalence of hyperhidrosis and impact on individuals with axillary hyperhidrosis: results from a national survey. J Am Acad Dermatol. 2004;51(2):241-248.
2. Walling HW. Clinical differentiation of primary from secondary hyperhidrosis. J Am Acad Dermatol. 2011;64(4):690-695.
3. Andersen LS, Biering-Sørensen F, Müller PG, Jensen IL, Aggerbeck B. The prevalence of hyperhidrosis in patients with spinal cord injuries and an evaluation of the effect of dextropropoxyphene hydrochloride in therapy. Paraplegia. 1992;(30):184-191.
4. Sato K, Kang WH, Saga K, Sato KT. Biology of sweat glands and their disorders. II. Disorders of sweat gland function. J Am Acad Dermatol. 1989;20(5, pt 1):713-726.
5. Kewalramani LS. Autonomic dysreflexia in traumatic myelopathy. Am J Phys Med. 1980;59(1):1-21.
6. Low PA, Engstrom JW. Disorders of the autonomic nervous system. In: Kasper D, Fauci A, Hauser S, Longo D, Jameson J, Loscalzo J, eds. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw-Hill; 2015.
7. Milhort TH. Classification of syringomyelia. Neurosurg Focus. 2000;8(3):E1.
8. National Institute of Neurological Disorders and Stroke. Syringomyelia fact sheet. https://www.ninds .nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Syringomyelia-Fact-Sheet. Accessed February 13, 2017.
9. Meschner AL. Junqueira’s Basic Histology. 14th ed. New York, NY: McGraw-Hill; 2016:371-392.
10. Mauro TM. Biology of eccrine and apocrine glands. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K. eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
11. Barrett KE, Barman SM, Boitano S, Brooks HL. Ganong’s Review of Medical Physiology. 25th ed. New York, NY: McGraw-Hill; 2016:261-272.
12. Kellogg DL Jr. Thermoregulation. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
13. Rovner ES, Wyman J, Lam S. Urinary Incontinence. In: DiPiro JT, Talbert RL, Yee GC, Matzke GR, Wells BG, Posey L. eds. Pharmacotherapy: A Pathophysiologic Approach. 9th ed. New York, NY: McGraw-Hill; 2014:1377-1396.
14. Teivelis MP, Wolosker N, Krutman M, Kauffman P, Campos JR, Puech-Leão P. Treatment of uncommon sites of focal primary hyperhidrosis: experience with pharmacological therapy using oxybutynin. Clinics (Sao Paulo). 2014;69(9):608-614.
15. Wolosker N, de Campos JR, Kauffman P, Puech-Leão P. A randomized-placebo-controlled trial of oxybutynin for the initial treatment of palmar and axillary hyperhidrosis. J Vasc Surg. 2012;55(6):1696-1700.
16. Schollhammer M, Brenaut E, Menard-Andivot N, et al. Oxybutynin as a treatment for generalized hyperhidrosis. Br J Dermatol. 2015;173(5):1163-1168.
17. Gonzalez F, Chang JY, Banovac K, Messina D, Martinez-Arizala A, Kelley RE. Autoregulation of cerebral blood flow in patients with orthostatic hypotension after spinal cord injury. Paraplegia. 1991;29(1):1-7.
18. Fealey RD, Hebert AA. Disorders of the eccrine sweat glands and sweating. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K, eds. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw-Hill; 2012:chap 84.
1. Strutton DR, Kowalski JW, Glaser DA, Stang PE. US prevalence of hyperhidrosis and impact on individuals with axillary hyperhidrosis: results from a national survey. J Am Acad Dermatol. 2004;51(2):241-248.
2. Walling HW. Clinical differentiation of primary from secondary hyperhidrosis. J Am Acad Dermatol. 2011;64(4):690-695.
3. Andersen LS, Biering-Sørensen F, Müller PG, Jensen IL, Aggerbeck B. The prevalence of hyperhidrosis in patients with spinal cord injuries and an evaluation of the effect of dextropropoxyphene hydrochloride in therapy. Paraplegia. 1992;(30):184-191.
4. Sato K, Kang WH, Saga K, Sato KT. Biology of sweat glands and their disorders. II. Disorders of sweat gland function. J Am Acad Dermatol. 1989;20(5, pt 1):713-726.
5. Kewalramani LS. Autonomic dysreflexia in traumatic myelopathy. Am J Phys Med. 1980;59(1):1-21.
6. Low PA, Engstrom JW. Disorders of the autonomic nervous system. In: Kasper D, Fauci A, Hauser S, Longo D, Jameson J, Loscalzo J, eds. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw-Hill; 2015.
7. Milhort TH. Classification of syringomyelia. Neurosurg Focus. 2000;8(3):E1.
8. National Institute of Neurological Disorders and Stroke. Syringomyelia fact sheet. https://www.ninds .nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Syringomyelia-Fact-Sheet. Accessed February 13, 2017.
9. Meschner AL. Junqueira’s Basic Histology. 14th ed. New York, NY: McGraw-Hill; 2016:371-392.
10. Mauro TM. Biology of eccrine and apocrine glands. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K. eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
11. Barrett KE, Barman SM, Boitano S, Brooks HL. Ganong’s Review of Medical Physiology. 25th ed. New York, NY: McGraw-Hill; 2016:261-272.
12. Kellogg DL Jr. Thermoregulation. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K, eds. Fitzpatrick’s Dermatology in General Medicine. 8th ed. New York, NY: McGraw-Hill; 2012.
13. Rovner ES, Wyman J, Lam S. Urinary Incontinence. In: DiPiro JT, Talbert RL, Yee GC, Matzke GR, Wells BG, Posey L. eds. Pharmacotherapy: A Pathophysiologic Approach. 9th ed. New York, NY: McGraw-Hill; 2014:1377-1396.
14. Teivelis MP, Wolosker N, Krutman M, Kauffman P, Campos JR, Puech-Leão P. Treatment of uncommon sites of focal primary hyperhidrosis: experience with pharmacological therapy using oxybutynin. Clinics (Sao Paulo). 2014;69(9):608-614.
15. Wolosker N, de Campos JR, Kauffman P, Puech-Leão P. A randomized-placebo-controlled trial of oxybutynin for the initial treatment of palmar and axillary hyperhidrosis. J Vasc Surg. 2012;55(6):1696-1700.
16. Schollhammer M, Brenaut E, Menard-Andivot N, et al. Oxybutynin as a treatment for generalized hyperhidrosis. Br J Dermatol. 2015;173(5):1163-1168.
17. Gonzalez F, Chang JY, Banovac K, Messina D, Martinez-Arizala A, Kelley RE. Autoregulation of cerebral blood flow in patients with orthostatic hypotension after spinal cord injury. Paraplegia. 1991;29(1):1-7.
18. Fealey RD, Hebert AA. Disorders of the eccrine sweat glands and sweating. In: Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K, eds. Fitzpatrick’s Dermatology in General Medicine. New York, NY: McGraw-Hill; 2012:chap 84.
Case Studies in Toxicology: Drink the Water, but Don’t Eat the Paint
Case
A 2-year-old boy and his mother were referred to the ED by the child’s pediatrician after a routine venous blood lead level (BLL) taken at the boy’s recent well visit revealed an elevated lead level of 52 mcg/dL (normal range, <5 mcg/dL). The child’s mother reported that although her son had always been a picky eater, he had recently been complaining of abdominal pain.
The patient’s well-child visits had been normal until his recent 2-year checkup, at which time his pediatrician noticed some speech delay. On further history taking, the emergency physician (EP) learned the patient and his mother resided in an older home (built in the 1950s) that was in disrepair. The mother asked the EP if the elevation in the child’s BLL could be due to the drinking water in their town.
What are the most likely sources of environmental lead exposure?
In 2016, the topic of lead poisoning grabbed national attention when a pediatrician in Flint, Michigan detected an abrupt doubling of the number of children with elevated lead levels in her practice.1 Upon further investigation, it was discovered that these kids had one thing in common: the source of their drinking water. The City of Flint had recently switched the source of its potable water from Lake Huron to the Flint River. The lower quality water, which was not properly treated with an anticorrosive agent such as orthophosphate, led to widespread pipe corrosion and lead contamination. This finding resulted in a cascade of water testing by other municipalities and school systems, many of which identified lead concentrations above the currently accepted drinking water standard of 15 parts per billion (ppb).
Thousands of children each year are identified to have elevated BLLs, based on the Centers for Disease Control and Prevention definition of a “level of concern” as more than 5 mcg/dL.2 The majority of these exposures stem from environmental exposure to lead paint dust in the home, but drinking water normally contributes as a low-level, constant, “basal” exposure. While lead-contaminated drinking water is not acceptable, it is unlikely to generate many ED visits. However, there are a variety of other lead sources that may prompt children to present to the ED with acute or subacute lead poisoning.
Lead is a heavy metal whose physical properties indicate its common uses. It provides durability and opacity to pigments, which is why it is found in oil paint, house paint used before 1976, and on paint for large outdoor structures, where it is still used. Lead is also found in the pigments used in cosmetics, stained glass, and painted pottery, and as an adulterant in highly colored foodstuffs such as imported turmeric.3
The physicochemical characteristics of lead make it an ideal component of solder. Many plumbing pipes in use today are not lead, but join one another using lead solder at the joints, sites that are vulnerable to corrosion. The heavy molecular weight of lead makes it a useful component of bullets and munitions.
Tetraethyl lead was used as an “anti-knock” agent to smooth out the combustion of heterogenous compounds in automotive fuel before it was removed in the mid-1970s.4 Prior to its removal, leaded gasoline was the largest source of air, soil, and groundwater contamination leading to environmental exposures.4 At present, the most common source of environmental lead exposure among young children is through peeling paint in deteriorating residential buildings. Hazardous occupational lead exposures arise from work involving munitions, reclamation and salvage, painting, welding, and numerous other settings—particularly sites where industrial hygiene is suboptimal. Lead from these sites can be inadvertently transported home on clothing or shoes, raising the exposure risk for children in the household.4
What are the health effects of lead exposure?
Like most heavy metals, lead is toxic to many organ systems in the body. The signs and symptoms of lead poisoning vary depending on the patient’s BLL and age (Table 1).5 The most common clinical effect of lead in the adult population is hypertension.6 Additional renal effects include a Fanconi-type syndrome with glycosuria and proteinuria. Lead can cause a peripheral neuropathy that is predominantly motor, classically causing foot or wrist drop. Abdominal pain from lead exposure is sometimes termed “lead colic” due to its intermittent and often severe nature. Abnormalities in urate metabolism cause a gouty arthritis referred to as “saturnine gout.” 6

The young pediatric central nervous system (CNS) is much more vulnerable to the effects of lead than the adult CNS. Even low-level lead exposure to the developing brain causes deficits in intelligence quotient, attention, impulse control, and other neurocognitive functions that are largely irreversible.7
Children with an elevated BLL may also develop constipation, anorexia, pallor, and pica.8 The development of geophagia (subtype of pica in which one craves and ingests nonfood clay or soil-like materials), represents a “chicken-or-egg” phenomena as it both causes and results from lead poisoning.
Lead impairs multiple steps of the heme synthesis pathway, causing microcytic anemia with basophilic stippling. Lead-induced anemia exacerbates pica as anemic patients are more likely to eat leaded paint chips and other lead-containing materials such as pottery.8 Of note, leaded white paint is reported to have a pleasant taste due to the sweet-tasting lead acetate used as a pigment.
The most dramatic and consequential manifestation of lead poisoning is lead encephalopathy. This can occur at any age, but manifests in children at much lower BLLs than in adults. Patients can be altered or obtunded, have convulsive activity, and may develop cerebral edema. Encephalopathy is a life-threatening emergency and must be recognized and treated immediately. Lead encephalopathy should be suspected in any young child with hand-to-mouth behavior who has any of the above environmental risk factors.4 The findings of anemia or the other diagnostic signs described below are too unreliable and take too long to be truly helpful in making the diagnosis.
How is the diagnosis of lead poisoning made?
The gold standard for the diagnosis of lead poisoning is the measurement of BLL. However, the turnaround time for this test is usually at least 24 hours, but may take up to several days. As such, adjunctive testing can accelerate obtaining a diagnosis. A complete blood count (CBC) to evaluate for microcytic anemia may demonstrate a characteristic pattern of basophilic stippling.9 A protoporphyrin level—either a free erythrocyte protoporphyrin (FEP) or a zinc protoporphyrin level—will be elevated, a result of heme synthesis disruption.9 Urinalysis may demonstrate glycosuria or proteinuria.6 Hypertension is often present, even in pediatric patients.
An abdominal radiograph is essential in children to determine whether a lead foreign body, such as a paint chip, is present in the intestinal lumen. Long bone films may demonstrate “lead lines” at the metaphysis, which in fact do not reflect lead itself but abnormal calcium deposition in growing bone due to lead’s interference with bone remodeling. A computed tomography (CT) scan of the brain in patients with encephalopathy will often demonstrate cerebral edema.6
Of note, capillary BLLs taken via finger-stick can be falsely elevated due contamination during collection (eg, the presence of lead dust on the skin). However, this screening method is often used by clinicians in the pediatric primary care setting because of its feasibility. Elevated BLLs from capillary testing should always be followed by a BLL obtained by venipuncture.2
Case Continuation
The patient’s mother was counseled on sources of lead contamination. She was informed that although drinking water may contribute some amount to an elevated BLL, the most likely source of contamination is still lead paint found in older homes such as the one in which she and her son resided.
Diagnostic studies to support the diagnosis of lead poisoning were performed. A CBC revealed a hemoglobin of 9.8 g/dL with a mean corpuscular volume of 68 fL. A microscopic smear of blood demonstrated basophilic stippling of red blood cells. An FEP level was 386 mcg/dL. An abdominal radiograph demonstrated small radiopacities throughout the large intestine, without obstruction, which was suggestive of ingested lead paint chips.
What is the best management approach to patients with suspected lead poisoning?
The first-line treatment for patients with lead poisoning is removal from the exposure source, which first and foremost requires identification of the hazard through careful history taking and scene investigation by the local health department. This will avoid recurrent visits following successful chelation for repeat exposure to an unidentified source. Relocation to another dwelling will often be required for patients with presumed exposure until the hazard can be identified and abated.
Patients who have ingested or have embedded leaded foreign bodies will require removal via whole bowel irrigation or surgical means.
Following decontamination, chelation is required for children with a BLL more than 45 mcg/dL, and adults with CNS symptomatology and a BLL more than 70 mcg/dL. Table 2 provides guidelines for chelation therapy based on BLL.5

There are three chelating agents commonly used to reduce the body lead burden (Table 2).5 The most common, owing largely to it being the only agent used orally, is succimer (or dimercaptosuccinic acid, DMSA). The second agent is calcium disodium edetate (CaNa2EDTA), which is given intravenously. In patients with encephalopathy, EDTA should be given after the first dose of the third agent, British anti-Lewisite (BAL; 2,3-dimercaptopropanol), in order to prevent redistribution of lead from the peripheral compartment into the CNS.10 However, BAL is the most difficult of the three agents to administer as it is suspended in peanut oil and is given via intramuscular injection every 4 hours.
Unfortunately, while chelation therapy is highly beneficial for patients with severe lead poisoning, it has not been demonstrated to positively impact children who already have developed neurocognitive sequelae associated with lower level lead exposure.11 This highlights the importance of prevention.
Work-up and Management in the ED
The patient with lead poisoning, while an unusual presentation in the ED, requires specialized management to minimize sequelae of exposure. Careful attention to history is vital. When in doubt, the EP should consult with her or his regional poison control center (800-222-1222) or with a medical toxicologist or other expert.
There are several scenarios in which a patient may present to the ED with lead toxicity. The following scenarios, along with their respective clinical approach strategies, represent three of the most common presentations.
Scenario 1: The Pediatric Patient With Elevated Venous Blood Lead Levels
The EP should employ the following clinical approach when evaluating and managing the pediatric patient with normal mental status whose routine screening reveals a BLL sufficiently elevated to warrant evaluation or admission—perhaps to discontinue exposure or initiate chelation therapy.
- Obtain a history, including possible lead sources; perform a complete physical examination; and obtain a repeat BLL, CBC with microscopic examination, and renal function test.
- Obtain an abdominal film to look for radiopacities, including paint chips or larger ingested foreign bodies.
- If radiopaque foreign bodies are present on abdominal radiograph, whole bowel irrigation with polyethylene glycol solution given via a nasogastric tube at 250 to 500 cc/h for a pediatric patient (1 to 2 L/h for adult patients) should be given until no residual foreign bodies remain.
- Obtain a radiograph of the long bone, which may demonstrate metaphyseal enhancement in the pediatric patient, suggesting long-term exposure.
- Ensure local or state health departments are contacted to arrange for environmental inspection of the home. This is essential prior to discharge to the home environment.
- Begin chelation therapy according to the BLL (Table 2).
Scenario 2: Adult Patients Presenting With Signs and Symptoms of Lead Toxicity
The adult patient who presents to the ED with complaints suggestive of lead poisoning and whose history is indicative of lead exposure should be evaluated and managed as follows:
- Obtain a thorough history, including the occupation and hobbies of the patient and all family members.
- Obtain vital signs to evaluate for hypertension; repeat BLL, CBC with smear, and serum creatinine test. Perform a physical examination to evaluate for lead lines.
- Obtain radiographic images, which may demonstrate a leaded foreign body, such as in the patient with prior history of gunshot wounds.
- If the BLL is sufficiently elevated or clinical findings are sufficiently severe, admit for chelation.
Scenario 3: The Pediatric or Adult Patient Presenting With Altered Mental Status
The patient presenting with altered mental status of unclear etiology—regardless of age—and in whom lead encephalopathy is a possible cause, should be worked-up and managed as follows:
- Obtain BLL, CBC, FEP levels. Consider radiographic imaging to assess for ingested or embedded foreign bodies.
- If abnormalities in the above laboratory studies are consistent with lead poisoning, initiate chelation immediately—prior to receiving repeat BLL result.
- Obtain a CT scan of the head to assess for cerebral edema.
- Provide supportive care for encephalopathy, including airway control and management of increased intracranial pressure.
Case Conclusion
The patient was admitted to the hospital for whole bowel irrigation and chelation therapy with succimer. The local health department conducted an investigation of the home and found multiple areas of peeling lead paint and lead dust, and ordered remediation of the property before it could be re-occupied by the family. A test of the home’s drinking water found no elevation above the 15 ppb standard.
The patient was discharged from the hospital in the care of his mother. They were relocated to a lead-free home, with follow-up by the pediatrician for ongoing monitoring of the BLL and developmental milestones.
1. Hanna-Attisha M, LaChance J, Sadler RC, Champney Schnepp A. Elevated blood lead levels in children associated with the flint drinking water crisis: A spatial analysis of risk and public health response. Am J Public Health. 2016;106(2):283-290. doi:0.2105/AJPH.2015.303003.
2. Centers for Disease Control and Prevention Advisory Committee on Childhood Lead Poisoning Prevention. Low level lead exposure harms children: a renewed call for primary prevention. January 4, 2012. Available at https://www.cdc.gov/nceh/lead/acclpp/final_document_030712.pdf. Accessed February 27, 2017.
3. Food and Drug Administration. Spices USA Inc. issues alert on elevated levels of lead in ground turmeric. http://www.fda.gov/safety/recalls/ucm523561.htm, September 26, 2016. Accessed February 27, 2017.
4. US Department of Health and Human Services - Agency for Toxic Substances & Disease Registry. Toxic substances portal: lead. US Department of Health and Human Services Web site. Available at https://www.atsdr.cdc.gov/ToxProfiles/TP.asp?id=96&tid=22. Updated January 21, 2015. Accessed February 27, 2017.
5. Calello DP, Henretig FM. Lead. In: Goldfrank LG, Flomenbaum NE, Lewin NA, Howland MA, Hoffman RS, Nelson LS (eds.). Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2014:1219-1234.
6. US Department of Health and Human Services - Agency for Toxic Substances & Disease Registry. Environmental health and medicine education: lead toxicity. https://www.atsdr.cdc.gov/csem/csem.asp?csem=7&po=10. Updated August 26, 2016. Accessed February 27, 2017.
7. Canfield RL, Henderson Jr CR, Cory-Slechta DA, Cox C, Jusko TA, Lanphear BP. Intellectual impairment in children with blood lead concentrations below 10 microg per deciliter. New Engl J Med. 2003;348:1517-1526.
8. Kathuria P, Rowden AK. Lead toxicity. Medscape Web site. Available at http://emedicine.medscape.com/article/1174752-clinical. Updated January 31, 2017. Accessed February 27, 2017.
9. US Department of Health and Human Services - Agency for Toxic Substances & Disease Registry. Environmental health and medicine education. Lead toxicity: what tests can assist with diagnosis of lead toxicity? https://www.atsdr.cdc.gov/csem/csem.asp?csem=7&po=12. Updated August 25, 2016. Accessed February 27, 2017.
10. Chisholm JJ Jr. The use of chelating agents in the treatment of acute and chronic lead intoxication in childhood. J Pediatr. 1968;73(1):1-38.
11. Rogan WJ, Dietrich KN, Ware JH, et al; Treatment of Lead-Exposed Children Trial Group. The effect of chelation therapy with succimer on neuropsychological development in children exposed to lead. N Engl J Med. 2001;344(19):1421-1426.
Case
A 2-year-old boy and his mother were referred to the ED by the child’s pediatrician after a routine venous blood lead level (BLL) taken at the boy’s recent well visit revealed an elevated lead level of 52 mcg/dL (normal range, <5 mcg/dL). The child’s mother reported that although her son had always been a picky eater, he had recently been complaining of abdominal pain.
The patient’s well-child visits had been normal until his recent 2-year checkup, at which time his pediatrician noticed some speech delay. On further history taking, the emergency physician (EP) learned the patient and his mother resided in an older home (built in the 1950s) that was in disrepair. The mother asked the EP if the elevation in the child’s BLL could be due to the drinking water in their town.
What are the most likely sources of environmental lead exposure?
In 2016, the topic of lead poisoning grabbed national attention when a pediatrician in Flint, Michigan detected an abrupt doubling of the number of children with elevated lead levels in her practice.1 Upon further investigation, it was discovered that these kids had one thing in common: the source of their drinking water. The City of Flint had recently switched the source of its potable water from Lake Huron to the Flint River. The lower quality water, which was not properly treated with an anticorrosive agent such as orthophosphate, led to widespread pipe corrosion and lead contamination. This finding resulted in a cascade of water testing by other municipalities and school systems, many of which identified lead concentrations above the currently accepted drinking water standard of 15 parts per billion (ppb).
Thousands of children each year are identified to have elevated BLLs, based on the Centers for Disease Control and Prevention definition of a “level of concern” as more than 5 mcg/dL.2 The majority of these exposures stem from environmental exposure to lead paint dust in the home, but drinking water normally contributes as a low-level, constant, “basal” exposure. While lead-contaminated drinking water is not acceptable, it is unlikely to generate many ED visits. However, there are a variety of other lead sources that may prompt children to present to the ED with acute or subacute lead poisoning.
Lead is a heavy metal whose physical properties indicate its common uses. It provides durability and opacity to pigments, which is why it is found in oil paint, house paint used before 1976, and on paint for large outdoor structures, where it is still used. Lead is also found in the pigments used in cosmetics, stained glass, and painted pottery, and as an adulterant in highly colored foodstuffs such as imported turmeric.3
The physicochemical characteristics of lead make it an ideal component of solder. Many plumbing pipes in use today are not lead, but join one another using lead solder at the joints, sites that are vulnerable to corrosion. The heavy molecular weight of lead makes it a useful component of bullets and munitions.
Tetraethyl lead was used as an “anti-knock” agent to smooth out the combustion of heterogenous compounds in automotive fuel before it was removed in the mid-1970s.4 Prior to its removal, leaded gasoline was the largest source of air, soil, and groundwater contamination leading to environmental exposures.4 At present, the most common source of environmental lead exposure among young children is through peeling paint in deteriorating residential buildings. Hazardous occupational lead exposures arise from work involving munitions, reclamation and salvage, painting, welding, and numerous other settings—particularly sites where industrial hygiene is suboptimal. Lead from these sites can be inadvertently transported home on clothing or shoes, raising the exposure risk for children in the household.4
What are the health effects of lead exposure?
Like most heavy metals, lead is toxic to many organ systems in the body. The signs and symptoms of lead poisoning vary depending on the patient’s BLL and age (Table 1).5 The most common clinical effect of lead in the adult population is hypertension.6 Additional renal effects include a Fanconi-type syndrome with glycosuria and proteinuria. Lead can cause a peripheral neuropathy that is predominantly motor, classically causing foot or wrist drop. Abdominal pain from lead exposure is sometimes termed “lead colic” due to its intermittent and often severe nature. Abnormalities in urate metabolism cause a gouty arthritis referred to as “saturnine gout.” 6
The young pediatric central nervous system (CNS) is much more vulnerable to the effects of lead than the adult CNS. Even low-level lead exposure to the developing brain causes deficits in intelligence quotient, attention, impulse control, and other neurocognitive functions that are largely irreversible.7
Children with an elevated BLL may also develop constipation, anorexia, pallor, and pica.8 The development of geophagia (subtype of pica in which one craves and ingests nonfood clay or soil-like materials), represents a “chicken-or-egg” phenomena as it both causes and results from lead poisoning.
Lead impairs multiple steps of the heme synthesis pathway, causing microcytic anemia with basophilic stippling. Lead-induced anemia exacerbates pica as anemic patients are more likely to eat leaded paint chips and other lead-containing materials such as pottery.8 Of note, leaded white paint is reported to have a pleasant taste due to the sweet-tasting lead acetate used as a pigment.
The most dramatic and consequential manifestation of lead poisoning is lead encephalopathy. This can occur at any age, but manifests in children at much lower BLLs than in adults. Patients can be altered or obtunded, have convulsive activity, and may develop cerebral edema. Encephalopathy is a life-threatening emergency and must be recognized and treated immediately. Lead encephalopathy should be suspected in any young child with hand-to-mouth behavior who has any of the above environmental risk factors.4 The findings of anemia or the other diagnostic signs described below are too unreliable and take too long to be truly helpful in making the diagnosis.
How is the diagnosis of lead poisoning made?
The gold standard for the diagnosis of lead poisoning is the measurement of BLL. However, the turnaround time for this test is usually at least 24 hours, but may take up to several days. As such, adjunctive testing can accelerate obtaining a diagnosis. A complete blood count (CBC) to evaluate for microcytic anemia may demonstrate a characteristic pattern of basophilic stippling.9 A protoporphyrin level—either a free erythrocyte protoporphyrin (FEP) or a zinc protoporphyrin level—will be elevated, a result of heme synthesis disruption.9 Urinalysis may demonstrate glycosuria or proteinuria.6 Hypertension is often present, even in pediatric patients.
An abdominal radiograph is essential in children to determine whether a lead foreign body, such as a paint chip, is present in the intestinal lumen. Long bone films may demonstrate “lead lines” at the metaphysis, which in fact do not reflect lead itself but abnormal calcium deposition in growing bone due to lead’s interference with bone remodeling. A computed tomography (CT) scan of the brain in patients with encephalopathy will often demonstrate cerebral edema.6
Of note, capillary BLLs taken via finger-stick can be falsely elevated due contamination during collection (eg, the presence of lead dust on the skin). However, this screening method is often used by clinicians in the pediatric primary care setting because of its feasibility. Elevated BLLs from capillary testing should always be followed by a BLL obtained by venipuncture.2
Case Continuation
The patient’s mother was counseled on sources of lead contamination. She was informed that although drinking water may contribute some amount to an elevated BLL, the most likely source of contamination is still lead paint found in older homes such as the one in which she and her son resided.
Diagnostic studies to support the diagnosis of lead poisoning were performed. A CBC revealed a hemoglobin of 9.8 g/dL with a mean corpuscular volume of 68 fL. A microscopic smear of blood demonstrated basophilic stippling of red blood cells. An FEP level was 386 mcg/dL. An abdominal radiograph demonstrated small radiopacities throughout the large intestine, without obstruction, which was suggestive of ingested lead paint chips.
What is the best management approach to patients with suspected lead poisoning?
The first-line treatment for patients with lead poisoning is removal from the exposure source, which first and foremost requires identification of the hazard through careful history taking and scene investigation by the local health department. This will avoid recurrent visits following successful chelation for repeat exposure to an unidentified source. Relocation to another dwelling will often be required for patients with presumed exposure until the hazard can be identified and abated.
Patients who have ingested or have embedded leaded foreign bodies will require removal via whole bowel irrigation or surgical means.
Following decontamination, chelation is required for children with a BLL more than 45 mcg/dL, and adults with CNS symptomatology and a BLL more than 70 mcg/dL. Table 2 provides guidelines for chelation therapy based on BLL.5
There are three chelating agents commonly used to reduce the body lead burden (Table 2).5 The most common, owing largely to it being the only agent used orally, is succimer (or dimercaptosuccinic acid, DMSA). The second agent is calcium disodium edetate (CaNa2EDTA), which is given intravenously. In patients with encephalopathy, EDTA should be given after the first dose of the third agent, British anti-Lewisite (BAL; 2,3-dimercaptopropanol), in order to prevent redistribution of lead from the peripheral compartment into the CNS.10 However, BAL is the most difficult of the three agents to administer as it is suspended in peanut oil and is given via intramuscular injection every 4 hours.
Unfortunately, while chelation therapy is highly beneficial for patients with severe lead poisoning, it has not been demonstrated to positively impact children who already have developed neurocognitive sequelae associated with lower level lead exposure.11 This highlights the importance of prevention.
Work-up and Management in the ED
The patient with lead poisoning, while an unusual presentation in the ED, requires specialized management to minimize sequelae of exposure. Careful attention to history is vital. When in doubt, the EP should consult with her or his regional poison control center (800-222-1222) or with a medical toxicologist or other expert.
There are several scenarios in which a patient may present to the ED with lead toxicity. The following scenarios, along with their respective clinical approach strategies, represent three of the most common presentations.
Scenario 1: The Pediatric Patient With Elevated Venous Blood Lead Levels
The EP should employ the following clinical approach when evaluating and managing the pediatric patient with normal mental status whose routine screening reveals a BLL sufficiently elevated to warrant evaluation or admission—perhaps to discontinue exposure or initiate chelation therapy.
- Obtain a history, including possible lead sources; perform a complete physical examination; and obtain a repeat BLL, CBC with microscopic examination, and renal function test.
- Obtain an abdominal film to look for radiopacities, including paint chips or larger ingested foreign bodies.
- If radiopaque foreign bodies are present on abdominal radiograph, whole bowel irrigation with polyethylene glycol solution given via a nasogastric tube at 250 to 500 cc/h for a pediatric patient (1 to 2 L/h for adult patients) should be given until no residual foreign bodies remain.
- Obtain a radiograph of the long bone, which may demonstrate metaphyseal enhancement in the pediatric patient, suggesting long-term exposure.
- Ensure local or state health departments are contacted to arrange for environmental inspection of the home. This is essential prior to discharge to the home environment.
- Begin chelation therapy according to the BLL (Table 2).
Scenario 2: Adult Patients Presenting With Signs and Symptoms of Lead Toxicity
The adult patient who presents to the ED with complaints suggestive of lead poisoning and whose history is indicative of lead exposure should be evaluated and managed as follows:
- Obtain a thorough history, including the occupation and hobbies of the patient and all family members.
- Obtain vital signs to evaluate for hypertension; repeat BLL, CBC with smear, and serum creatinine test. Perform a physical examination to evaluate for lead lines.
- Obtain radiographic images, which may demonstrate a leaded foreign body, such as in the patient with prior history of gunshot wounds.
- If the BLL is sufficiently elevated or clinical findings are sufficiently severe, admit for chelation.
Scenario 3: The Pediatric or Adult Patient Presenting With Altered Mental Status
The patient presenting with altered mental status of unclear etiology—regardless of age—and in whom lead encephalopathy is a possible cause, should be worked-up and managed as follows:
- Obtain BLL, CBC, FEP levels. Consider radiographic imaging to assess for ingested or embedded foreign bodies.
- If abnormalities in the above laboratory studies are consistent with lead poisoning, initiate chelation immediately—prior to receiving repeat BLL result.
- Obtain a CT scan of the head to assess for cerebral edema.
- Provide supportive care for encephalopathy, including airway control and management of increased intracranial pressure.
Case Conclusion
The patient was admitted to the hospital for whole bowel irrigation and chelation therapy with succimer. The local health department conducted an investigation of the home and found multiple areas of peeling lead paint and lead dust, and ordered remediation of the property before it could be re-occupied by the family. A test of the home’s drinking water found no elevation above the 15 ppb standard.
The patient was discharged from the hospital in the care of his mother. They were relocated to a lead-free home, with follow-up by the pediatrician for ongoing monitoring of the BLL and developmental milestones.
Case
A 2-year-old boy and his mother were referred to the ED by the child’s pediatrician after a routine venous blood lead level (BLL) taken at the boy’s recent well visit revealed an elevated lead level of 52 mcg/dL (normal range, <5 mcg/dL). The child’s mother reported that although her son had always been a picky eater, he had recently been complaining of abdominal pain.
The patient’s well-child visits had been normal until his recent 2-year checkup, at which time his pediatrician noticed some speech delay. On further history taking, the emergency physician (EP) learned the patient and his mother resided in an older home (built in the 1950s) that was in disrepair. The mother asked the EP if the elevation in the child’s BLL could be due to the drinking water in their town.
What are the most likely sources of environmental lead exposure?
In 2016, the topic of lead poisoning grabbed national attention when a pediatrician in Flint, Michigan detected an abrupt doubling of the number of children with elevated lead levels in her practice.1 Upon further investigation, it was discovered that these kids had one thing in common: the source of their drinking water. The City of Flint had recently switched the source of its potable water from Lake Huron to the Flint River. The lower quality water, which was not properly treated with an anticorrosive agent such as orthophosphate, led to widespread pipe corrosion and lead contamination. This finding resulted in a cascade of water testing by other municipalities and school systems, many of which identified lead concentrations above the currently accepted drinking water standard of 15 parts per billion (ppb).
Thousands of children each year are identified to have elevated BLLs, based on the Centers for Disease Control and Prevention definition of a “level of concern” as more than 5 mcg/dL.2 The majority of these exposures stem from environmental exposure to lead paint dust in the home, but drinking water normally contributes as a low-level, constant, “basal” exposure. While lead-contaminated drinking water is not acceptable, it is unlikely to generate many ED visits. However, there are a variety of other lead sources that may prompt children to present to the ED with acute or subacute lead poisoning.
Lead is a heavy metal whose physical properties indicate its common uses. It provides durability and opacity to pigments, which is why it is found in oil paint, house paint used before 1976, and on paint for large outdoor structures, where it is still used. Lead is also found in the pigments used in cosmetics, stained glass, and painted pottery, and as an adulterant in highly colored foodstuffs such as imported turmeric.3
The physicochemical characteristics of lead make it an ideal component of solder. Many plumbing pipes in use today are not lead, but join one another using lead solder at the joints, sites that are vulnerable to corrosion. The heavy molecular weight of lead makes it a useful component of bullets and munitions.
Tetraethyl lead was used as an “anti-knock” agent to smooth out the combustion of heterogenous compounds in automotive fuel before it was removed in the mid-1970s.4 Prior to its removal, leaded gasoline was the largest source of air, soil, and groundwater contamination leading to environmental exposures.4 At present, the most common source of environmental lead exposure among young children is through peeling paint in deteriorating residential buildings. Hazardous occupational lead exposures arise from work involving munitions, reclamation and salvage, painting, welding, and numerous other settings—particularly sites where industrial hygiene is suboptimal. Lead from these sites can be inadvertently transported home on clothing or shoes, raising the exposure risk for children in the household.4
What are the health effects of lead exposure?
Like most heavy metals, lead is toxic to many organ systems in the body. The signs and symptoms of lead poisoning vary depending on the patient’s BLL and age (Table 1).5 The most common clinical effect of lead in the adult population is hypertension.6 Additional renal effects include a Fanconi-type syndrome with glycosuria and proteinuria. Lead can cause a peripheral neuropathy that is predominantly motor, classically causing foot or wrist drop. Abdominal pain from lead exposure is sometimes termed “lead colic” due to its intermittent and often severe nature. Abnormalities in urate metabolism cause a gouty arthritis referred to as “saturnine gout.” 6
The young pediatric central nervous system (CNS) is much more vulnerable to the effects of lead than the adult CNS. Even low-level lead exposure to the developing brain causes deficits in intelligence quotient, attention, impulse control, and other neurocognitive functions that are largely irreversible.7
Children with an elevated BLL may also develop constipation, anorexia, pallor, and pica.8 The development of geophagia (subtype of pica in which one craves and ingests nonfood clay or soil-like materials), represents a “chicken-or-egg” phenomena as it both causes and results from lead poisoning.
Lead impairs multiple steps of the heme synthesis pathway, causing microcytic anemia with basophilic stippling. Lead-induced anemia exacerbates pica as anemic patients are more likely to eat leaded paint chips and other lead-containing materials such as pottery.8 Of note, leaded white paint is reported to have a pleasant taste due to the sweet-tasting lead acetate used as a pigment.
The most dramatic and consequential manifestation of lead poisoning is lead encephalopathy. This can occur at any age, but manifests in children at much lower BLLs than in adults. Patients can be altered or obtunded, have convulsive activity, and may develop cerebral edema. Encephalopathy is a life-threatening emergency and must be recognized and treated immediately. Lead encephalopathy should be suspected in any young child with hand-to-mouth behavior who has any of the above environmental risk factors.4 The findings of anemia or the other diagnostic signs described below are too unreliable and take too long to be truly helpful in making the diagnosis.
How is the diagnosis of lead poisoning made?
The gold standard for the diagnosis of lead poisoning is the measurement of BLL. However, the turnaround time for this test is usually at least 24 hours, but may take up to several days. As such, adjunctive testing can accelerate obtaining a diagnosis. A complete blood count (CBC) to evaluate for microcytic anemia may demonstrate a characteristic pattern of basophilic stippling.9 A protoporphyrin level—either a free erythrocyte protoporphyrin (FEP) or a zinc protoporphyrin level—will be elevated, a result of heme synthesis disruption.9 Urinalysis may demonstrate glycosuria or proteinuria.6 Hypertension is often present, even in pediatric patients.
An abdominal radiograph is essential in children to determine whether a lead foreign body, such as a paint chip, is present in the intestinal lumen. Long bone films may demonstrate “lead lines” at the metaphysis, which in fact do not reflect lead itself but abnormal calcium deposition in growing bone due to lead’s interference with bone remodeling. A computed tomography (CT) scan of the brain in patients with encephalopathy will often demonstrate cerebral edema.6
Of note, capillary BLLs taken via finger-stick can be falsely elevated due contamination during collection (eg, the presence of lead dust on the skin). However, this screening method is often used by clinicians in the pediatric primary care setting because of its feasibility. Elevated BLLs from capillary testing should always be followed by a BLL obtained by venipuncture.2
Case Continuation
The patient’s mother was counseled on sources of lead contamination. She was informed that although drinking water may contribute some amount to an elevated BLL, the most likely source of contamination is still lead paint found in older homes such as the one in which she and her son resided.
Diagnostic studies to support the diagnosis of lead poisoning were performed. A CBC revealed a hemoglobin of 9.8 g/dL with a mean corpuscular volume of 68 fL. A microscopic smear of blood demonstrated basophilic stippling of red blood cells. An FEP level was 386 mcg/dL. An abdominal radiograph demonstrated small radiopacities throughout the large intestine, without obstruction, which was suggestive of ingested lead paint chips.
What is the best management approach to patients with suspected lead poisoning?
The first-line treatment for patients with lead poisoning is removal from the exposure source, which first and foremost requires identification of the hazard through careful history taking and scene investigation by the local health department. This will avoid recurrent visits following successful chelation for repeat exposure to an unidentified source. Relocation to another dwelling will often be required for patients with presumed exposure until the hazard can be identified and abated.
Patients who have ingested or have embedded leaded foreign bodies will require removal via whole bowel irrigation or surgical means.
Following decontamination, chelation is required for children with a BLL more than 45 mcg/dL, and adults with CNS symptomatology and a BLL more than 70 mcg/dL. Table 2 provides guidelines for chelation therapy based on BLL.5
There are three chelating agents commonly used to reduce the body lead burden (Table 2).5 The most common, owing largely to it being the only agent used orally, is succimer (or dimercaptosuccinic acid, DMSA). The second agent is calcium disodium edetate (CaNa2EDTA), which is given intravenously. In patients with encephalopathy, EDTA should be given after the first dose of the third agent, British anti-Lewisite (BAL; 2,3-dimercaptopropanol), in order to prevent redistribution of lead from the peripheral compartment into the CNS.10 However, BAL is the most difficult of the three agents to administer as it is suspended in peanut oil and is given via intramuscular injection every 4 hours.
Unfortunately, while chelation therapy is highly beneficial for patients with severe lead poisoning, it has not been demonstrated to positively impact children who already have developed neurocognitive sequelae associated with lower level lead exposure.11 This highlights the importance of prevention.
Work-up and Management in the ED
The patient with lead poisoning, while an unusual presentation in the ED, requires specialized management to minimize sequelae of exposure. Careful attention to history is vital. When in doubt, the EP should consult with her or his regional poison control center (800-222-1222) or with a medical toxicologist or other expert.
There are several scenarios in which a patient may present to the ED with lead toxicity. The following scenarios, along with their respective clinical approach strategies, represent three of the most common presentations.
Scenario 1: The Pediatric Patient With Elevated Venous Blood Lead Levels
The EP should employ the following clinical approach when evaluating and managing the pediatric patient with normal mental status whose routine screening reveals a BLL sufficiently elevated to warrant evaluation or admission—perhaps to discontinue exposure or initiate chelation therapy.
- Obtain a history, including possible lead sources; perform a complete physical examination; and obtain a repeat BLL, CBC with microscopic examination, and renal function test.
- Obtain an abdominal film to look for radiopacities, including paint chips or larger ingested foreign bodies.
- If radiopaque foreign bodies are present on abdominal radiograph, whole bowel irrigation with polyethylene glycol solution given via a nasogastric tube at 250 to 500 cc/h for a pediatric patient (1 to 2 L/h for adult patients) should be given until no residual foreign bodies remain.
- Obtain a radiograph of the long bone, which may demonstrate metaphyseal enhancement in the pediatric patient, suggesting long-term exposure.
- Ensure local or state health departments are contacted to arrange for environmental inspection of the home. This is essential prior to discharge to the home environment.
- Begin chelation therapy according to the BLL (Table 2).
Scenario 2: Adult Patients Presenting With Signs and Symptoms of Lead Toxicity
The adult patient who presents to the ED with complaints suggestive of lead poisoning and whose history is indicative of lead exposure should be evaluated and managed as follows:
- Obtain a thorough history, including the occupation and hobbies of the patient and all family members.
- Obtain vital signs to evaluate for hypertension; repeat BLL, CBC with smear, and serum creatinine test. Perform a physical examination to evaluate for lead lines.
- Obtain radiographic images, which may demonstrate a leaded foreign body, such as in the patient with prior history of gunshot wounds.
- If the BLL is sufficiently elevated or clinical findings are sufficiently severe, admit for chelation.
Scenario 3: The Pediatric or Adult Patient Presenting With Altered Mental Status
The patient presenting with altered mental status of unclear etiology—regardless of age—and in whom lead encephalopathy is a possible cause, should be worked-up and managed as follows:
- Obtain BLL, CBC, FEP levels. Consider radiographic imaging to assess for ingested or embedded foreign bodies.
- If abnormalities in the above laboratory studies are consistent with lead poisoning, initiate chelation immediately—prior to receiving repeat BLL result.
- Obtain a CT scan of the head to assess for cerebral edema.
- Provide supportive care for encephalopathy, including airway control and management of increased intracranial pressure.
Case Conclusion
The patient was admitted to the hospital for whole bowel irrigation and chelation therapy with succimer. The local health department conducted an investigation of the home and found multiple areas of peeling lead paint and lead dust, and ordered remediation of the property before it could be re-occupied by the family. A test of the home’s drinking water found no elevation above the 15 ppb standard.
The patient was discharged from the hospital in the care of his mother. They were relocated to a lead-free home, with follow-up by the pediatrician for ongoing monitoring of the BLL and developmental milestones.
1. Hanna-Attisha M, LaChance J, Sadler RC, Champney Schnepp A. Elevated blood lead levels in children associated with the flint drinking water crisis: A spatial analysis of risk and public health response. Am J Public Health. 2016;106(2):283-290. doi:0.2105/AJPH.2015.303003.
2. Centers for Disease Control and Prevention Advisory Committee on Childhood Lead Poisoning Prevention. Low level lead exposure harms children: a renewed call for primary prevention. January 4, 2012. Available at https://www.cdc.gov/nceh/lead/acclpp/final_document_030712.pdf. Accessed February 27, 2017.
3. Food and Drug Administration. Spices USA Inc. issues alert on elevated levels of lead in ground turmeric. http://www.fda.gov/safety/recalls/ucm523561.htm, September 26, 2016. Accessed February 27, 2017.
4. US Department of Health and Human Services - Agency for Toxic Substances & Disease Registry. Toxic substances portal: lead. US Department of Health and Human Services Web site. Available at https://www.atsdr.cdc.gov/ToxProfiles/TP.asp?id=96&tid=22. Updated January 21, 2015. Accessed February 27, 2017.
5. Calello DP, Henretig FM. Lead. In: Goldfrank LG, Flomenbaum NE, Lewin NA, Howland MA, Hoffman RS, Nelson LS (eds.). Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2014:1219-1234.
6. US Department of Health and Human Services - Agency for Toxic Substances & Disease Registry. Environmental health and medicine education: lead toxicity. https://www.atsdr.cdc.gov/csem/csem.asp?csem=7&po=10. Updated August 26, 2016. Accessed February 27, 2017.
7. Canfield RL, Henderson Jr CR, Cory-Slechta DA, Cox C, Jusko TA, Lanphear BP. Intellectual impairment in children with blood lead concentrations below 10 microg per deciliter. New Engl J Med. 2003;348:1517-1526.
8. Kathuria P, Rowden AK. Lead toxicity. Medscape Web site. Available at http://emedicine.medscape.com/article/1174752-clinical. Updated January 31, 2017. Accessed February 27, 2017.
9. US Department of Health and Human Services - Agency for Toxic Substances & Disease Registry. Environmental health and medicine education. Lead toxicity: what tests can assist with diagnosis of lead toxicity? https://www.atsdr.cdc.gov/csem/csem.asp?csem=7&po=12. Updated August 25, 2016. Accessed February 27, 2017.
10. Chisholm JJ Jr. The use of chelating agents in the treatment of acute and chronic lead intoxication in childhood. J Pediatr. 1968;73(1):1-38.
11. Rogan WJ, Dietrich KN, Ware JH, et al; Treatment of Lead-Exposed Children Trial Group. The effect of chelation therapy with succimer on neuropsychological development in children exposed to lead. N Engl J Med. 2001;344(19):1421-1426.
1. Hanna-Attisha M, LaChance J, Sadler RC, Champney Schnepp A. Elevated blood lead levels in children associated with the flint drinking water crisis: A spatial analysis of risk and public health response. Am J Public Health. 2016;106(2):283-290. doi:0.2105/AJPH.2015.303003.
2. Centers for Disease Control and Prevention Advisory Committee on Childhood Lead Poisoning Prevention. Low level lead exposure harms children: a renewed call for primary prevention. January 4, 2012. Available at https://www.cdc.gov/nceh/lead/acclpp/final_document_030712.pdf. Accessed February 27, 2017.
3. Food and Drug Administration. Spices USA Inc. issues alert on elevated levels of lead in ground turmeric. http://www.fda.gov/safety/recalls/ucm523561.htm, September 26, 2016. Accessed February 27, 2017.
4. US Department of Health and Human Services - Agency for Toxic Substances & Disease Registry. Toxic substances portal: lead. US Department of Health and Human Services Web site. Available at https://www.atsdr.cdc.gov/ToxProfiles/TP.asp?id=96&tid=22. Updated January 21, 2015. Accessed February 27, 2017.
5. Calello DP, Henretig FM. Lead. In: Goldfrank LG, Flomenbaum NE, Lewin NA, Howland MA, Hoffman RS, Nelson LS (eds.). Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill; 2014:1219-1234.
6. US Department of Health and Human Services - Agency for Toxic Substances & Disease Registry. Environmental health and medicine education: lead toxicity. https://www.atsdr.cdc.gov/csem/csem.asp?csem=7&po=10. Updated August 26, 2016. Accessed February 27, 2017.
7. Canfield RL, Henderson Jr CR, Cory-Slechta DA, Cox C, Jusko TA, Lanphear BP. Intellectual impairment in children with blood lead concentrations below 10 microg per deciliter. New Engl J Med. 2003;348:1517-1526.
8. Kathuria P, Rowden AK. Lead toxicity. Medscape Web site. Available at http://emedicine.medscape.com/article/1174752-clinical. Updated January 31, 2017. Accessed February 27, 2017.
9. US Department of Health and Human Services - Agency for Toxic Substances & Disease Registry. Environmental health and medicine education. Lead toxicity: what tests can assist with diagnosis of lead toxicity? https://www.atsdr.cdc.gov/csem/csem.asp?csem=7&po=12. Updated August 25, 2016. Accessed February 27, 2017.
10. Chisholm JJ Jr. The use of chelating agents in the treatment of acute and chronic lead intoxication in childhood. J Pediatr. 1968;73(1):1-38.
11. Rogan WJ, Dietrich KN, Ware JH, et al; Treatment of Lead-Exposed Children Trial Group. The effect of chelation therapy with succimer on neuropsychological development in children exposed to lead. N Engl J Med. 2001;344(19):1421-1426.

Acute Submandibular Sialadenitis
Case
A 21-year-old woman presented to the ED with pain and swelling on the right side of her neck. She stated the pain started earlier that morning and worsened when she ate or swallowed. The patient denied a recent or remote history of drooling, voice changes, or neck swelling. She reported no fevers, chills, or any other complaints, and had no pertinent medical history—specifically, no history of recent dental work. Her surgical history included tonsillectomy and cholecystectomy. There was no family history of diabetes, thyroid disease, autoimmune disease, or any other diseases. The patient stated that she was not on any prescription or over-the-counter medications. Regarding her social history, she denied past or cu

Vital signs at presentation were: blood pressure, 124/63 mm Hg (sitting); heart rate, 73 beats/min; respiratory rate, 15 breaths/min; and temperature, 98°F. Oxygen saturation was 99% on room air. On clinical examination, pain was noted in the patient’s right submandibular area and was tender to palpation. The swelling extended to the angle of the mandible posteriorly (Figure 1a and 1b). There was no erythema or increased surface temperature to suggest overlying cellulitis. The oral examination showed no evidence of dental infection, angioedema, or Ludwig angina. The pharynx was normal in appearance. The otological examination was unremarkable, and there was no evidence of mastoiditis.
Laboratory evaluation included a complete blood count (CBC), basic metabolic profile (BMP), and rapid streptococcal test (RST). The results of the patient’s CBC revealed a white blood cell count (WBC) of 11.1 x 109/L; the BMP was unremarkable; and the RST was negative.
A soft tissue neck computed tomography (CT) scan with contrast was obtained, which revealed mild right submandibular gland enlargement with abnormal enhancement (Figure 2). Stranding was also noted in the right submandibular space along with thickening of the right platysma muscle, and few surrounding lymph nodes were prominent (Figure 3). The findings were consistent with acute submandibular sialadenitis.

The patient received intravenous (IV) normal saline for hydration and IV ketorolac for analgesia, as well as an initial dose of oral amoxicillin/clavulanate 875/125 mg. At discharge, she was given a 10-day course of oral amoxicillin/clavulanate 875/125 mg with instructions to follow-up with her primary care physician and otolaryngologist within 2 days. The patient did well on follow-up, and her symptoms resolved within a few days of discharge.

Discussion
Comparatively little has been published on acute submandibular sialadenitis over the past three decades, and much of that which is cited in the literature comes from a rather small pool of case reports.1 In a literature review, Raad et al1 noted, “Pertinent literature on [this] subject includes case reports but no studies describing the microbial and clinical characteristics of this disease.” Further, many of the published case reports describe neonatal presentations of submandibular sialadenitis, the incidence of which is rare in this patient population.2-4
Submandibular and Parotid Glands
The submandibular gland is the second largest salivary gland, the parotid gland being the largest. The duct of the salivary gland, the Wharton’s duct, opens under the tongue in the area of the lingual frenulum. Ductal obstruction is more frequently seen with the submandibular gland than with the parotid gland.1 The reason for this is unclear, but may be related to several factors. One factor may be that, unlike the Stenson’s duct of the parotid gland, the Wharton’s duct does not pass through a muscle; thus, there is no muscular massage supporting the movement of secretion, as there is with buccinator muscle massage of Stenson’s duct. In addition, submandibular saliva is more viscous than parotid saliva due to its higher protein content and higher concentration of calcium phosphate.1
Etiology
Submandibular gland obstruction can occur in the absence of infection. Noninfectious cases typically present with pain upon eating and swallowing. A bacterial infectious etiology is associated with odynophagia, but also includes persistent pain and tenderness. This presents as pain associated with eating. Bacterial infection of the submandibular gland adds the element of persistent pain, associated with such features as tenderness. In addition, purulent discharge from the Wharton’s duct may be present in infectious cases, and accompanied by fever, chills, and an elevated WBC.1
Several bacteria have been isolated in infectious submandibular sialadenitis, the most common pathogens being Staphylococcus aureus. However, streptococci, Pseudomonas aeruginosa, Moraxella catarrhalis, and Escherichia coli bacteria have also been identified in cases of infectious submandibular sialadenitis.5
Viral etiologies of sialadenitis, such as mumps, are generally bilateral and nonsuppurative. The human immunodeficiency virus can also cause bilateral nonsuppurative salivary gland infections.6
Imaging Studies
As illustrated in our case, CT imaging can assist in confirming the diagnosis of acute submandibular sialadenitis by defining the anatomic involvement and identifying the presence of an abscess. Ultrasound can also be used and has been described as a first-line imaging procedure.7,8
Treatment
Surgical Intervention. Abscesses may require surgical intervention. However, most cases without abscess formation respond to outpatient treatment with antibiotics.5 If ductal obstruction is identified, removal of the calculus may be needed. This may involve ductal dilation, sialolithectomy, or even ductoplasty if a stricture is identified.1
Antibiotic Therapy. With respect to antibiotic selection, Chandak et al5 recommend oral amoxicillin-clavulanic acid. Other antistaphylococcal coverage recommendations have been made in the literature. Gland massage may be helpful after the tenderness has resolved,5 and sialogogues (eg, lemon drops, vitamin C lozenges) can also provide some relief.6 In addition, to avoid disease recurrence and prevent dental complications, Chandak et al5 emphasize the crucial role of hydration and excellent oral hygiene.
Conclusion
We suspected acute submandibular sialadenitis in our patient based on clinical findings, which were confirmed on CT imaging. Patients with acute submandibular sialadenitis may present with submandibular gland obstruction in the absence of bacterial infection. Noninfectious obstruction typically presents as pain associated with eating and swallowing, whereas infectious cases include constant pain and tenderness in the affected area. In addition, patients with infectious etiology may also have purulent discharge from Wharton’s duct, fever, chills, and an elevated WBC. Several bacteria have been isolated, the most common being S aureus. However, streptococci, P aeruginosa, M catarrhalis and E coli have also been identified. Computed tomography studies are helpful in confirming the diagnosis, defining anatomical involvement, and in identifying abscess formation.
Abscesses may require surgical intervention. However, most cases without abscess formation respond to outpatient treatment with antibiotics. Antibiotic selection involves antistaphylococcal coverage, such as amoxicillin-clavulanic acid. Glandular massage may be helpful after the tenderness has resolved. In addition, the literature emphasizes the crucial role of hydration and excellent oral hygiene in disease recurrence and to prevent dental complications.
1. Raad II, Sabbagh MF, Caranasos GJ. Acute bacterial sialadenitis: a study of 29 cases and review. Rev Infect Dis. 1990;12(4):591-601.
2. Banks WW, Handler SD, Glade GB, Turner HD. Neonatal submandibular sialadenitis. Am J Otolaryngol. 1980;1(3):261-263.
3. Wells DH. Suppuration of the submandibular salivary glands in the neonate. Am J Dis Child. 1975;129(5):628-630.
4. Ryan RF, Padmakumar B. Neonatal suppurative sialadenitis: an important clinical diagnosis. BMJ Case Rep. 2015;2015. pii:bcr2014208535. doi:10.1136/bcr-2014-208535.
5. Chandak R, Degwekar S, Chandak M, Rawlani S. Acute submandibular sialadenitis—a case report. Case Rep Dent. 2012;2012:615375. doi:10.1155/2012/615375.
6. Wilson KF, Meier JD, Ward PD. Salivary gland disorders. Am Fam Physician. 2014;89(11):882-888.
7. Alyas F, Lewis K, Williams M, et al. Diseases of the submandibular gland as demonstrated using high resolution ultrasound. Br J Radiol. 2005;78(928):362-369. doi:10.1259/bjr/93120352.
8. Howlett DC, Alyas F, Wong KT, et al. Sonographic assessment of the submandibular space. Clin Radiol. 2004;59(12):1070-1078. doi:10.1016/j.crad.2004.06.025.
Case
A 21-year-old woman presented to the ED with pain and swelling on the right side of her neck. She stated the pain started earlier that morning and worsened when she ate or swallowed. The patient denied a recent or remote history of drooling, voice changes, or neck swelling. She reported no fevers, chills, or any other complaints, and had no pertinent medical history—specifically, no history of recent dental work. Her surgical history included tonsillectomy and cholecystectomy. There was no family history of diabetes, thyroid disease, autoimmune disease, or any other diseases. The patient stated that she was not on any prescription or over-the-counter medications. Regarding her social history, she denied past or cu
Vital signs at presentation were: blood pressure, 124/63 mm Hg (sitting); heart rate, 73 beats/min; respiratory rate, 15 breaths/min; and temperature, 98°F. Oxygen saturation was 99% on room air. On clinical examination, pain was noted in the patient’s right submandibular area and was tender to palpation. The swelling extended to the angle of the mandible posteriorly (Figure 1a and 1b). There was no erythema or increased surface temperature to suggest overlying cellulitis. The oral examination showed no evidence of dental infection, angioedema, or Ludwig angina. The pharynx was normal in appearance. The otological examination was unremarkable, and there was no evidence of mastoiditis.
Laboratory evaluation included a complete blood count (CBC), basic metabolic profile (BMP), and rapid streptococcal test (RST). The results of the patient’s CBC revealed a white blood cell count (WBC) of 11.1 x 109/L; the BMP was unremarkable; and the RST was negative.
A soft tissue neck computed tomography (CT) scan with contrast was obtained, which revealed mild right submandibular gland enlargement with abnormal enhancement (Figure 2). Stranding was also noted in the right submandibular space along with thickening of the right platysma muscle, and few surrounding lymph nodes were prominent (Figure 3). The findings were consistent with acute submandibular sialadenitis.
The patient received intravenous (IV) normal saline for hydration and IV ketorolac for analgesia, as well as an initial dose of oral amoxicillin/clavulanate 875/125 mg. At discharge, she was given a 10-day course of oral amoxicillin/clavulanate 875/125 mg with instructions to follow-up with her primary care physician and otolaryngologist within 2 days. The patient did well on follow-up, and her symptoms resolved within a few days of discharge.
Discussion
Comparatively little has been published on acute submandibular sialadenitis over the past three decades, and much of that which is cited in the literature comes from a rather small pool of case reports.1 In a literature review, Raad et al1 noted, “Pertinent literature on [this] subject includes case reports but no studies describing the microbial and clinical characteristics of this disease.” Further, many of the published case reports describe neonatal presentations of submandibular sialadenitis, the incidence of which is rare in this patient population.2-4
Submandibular and Parotid Glands
The submandibular gland is the second largest salivary gland, the parotid gland being the largest. The duct of the salivary gland, the Wharton’s duct, opens under the tongue in the area of the lingual frenulum. Ductal obstruction is more frequently seen with the submandibular gland than with the parotid gland.1 The reason for this is unclear, but may be related to several factors. One factor may be that, unlike the Stenson’s duct of the parotid gland, the Wharton’s duct does not pass through a muscle; thus, there is no muscular massage supporting the movement of secretion, as there is with buccinator muscle massage of Stenson’s duct. In addition, submandibular saliva is more viscous than parotid saliva due to its higher protein content and higher concentration of calcium phosphate.1
Etiology
Submandibular gland obstruction can occur in the absence of infection. Noninfectious cases typically present with pain upon eating and swallowing. A bacterial infectious etiology is associated with odynophagia, but also includes persistent pain and tenderness. This presents as pain associated with eating. Bacterial infection of the submandibular gland adds the element of persistent pain, associated with such features as tenderness. In addition, purulent discharge from the Wharton’s duct may be present in infectious cases, and accompanied by fever, chills, and an elevated WBC.1
Several bacteria have been isolated in infectious submandibular sialadenitis, the most common pathogens being Staphylococcus aureus. However, streptococci, Pseudomonas aeruginosa, Moraxella catarrhalis, and Escherichia coli bacteria have also been identified in cases of infectious submandibular sialadenitis.5
Viral etiologies of sialadenitis, such as mumps, are generally bilateral and nonsuppurative. The human immunodeficiency virus can also cause bilateral nonsuppurative salivary gland infections.6
Imaging Studies
As illustrated in our case, CT imaging can assist in confirming the diagnosis of acute submandibular sialadenitis by defining the anatomic involvement and identifying the presence of an abscess. Ultrasound can also be used and has been described as a first-line imaging procedure.7,8
Treatment
Surgical Intervention. Abscesses may require surgical intervention. However, most cases without abscess formation respond to outpatient treatment with antibiotics.5 If ductal obstruction is identified, removal of the calculus may be needed. This may involve ductal dilation, sialolithectomy, or even ductoplasty if a stricture is identified.1
Antibiotic Therapy. With respect to antibiotic selection, Chandak et al5 recommend oral amoxicillin-clavulanic acid. Other antistaphylococcal coverage recommendations have been made in the literature. Gland massage may be helpful after the tenderness has resolved,5 and sialogogues (eg, lemon drops, vitamin C lozenges) can also provide some relief.6 In addition, to avoid disease recurrence and prevent dental complications, Chandak et al5 emphasize the crucial role of hydration and excellent oral hygiene.
Conclusion
We suspected acute submandibular sialadenitis in our patient based on clinical findings, which were confirmed on CT imaging. Patients with acute submandibular sialadenitis may present with submandibular gland obstruction in the absence of bacterial infection. Noninfectious obstruction typically presents as pain associated with eating and swallowing, whereas infectious cases include constant pain and tenderness in the affected area. In addition, patients with infectious etiology may also have purulent discharge from Wharton’s duct, fever, chills, and an elevated WBC. Several bacteria have been isolated, the most common being S aureus. However, streptococci, P aeruginosa, M catarrhalis and E coli have also been identified. Computed tomography studies are helpful in confirming the diagnosis, defining anatomical involvement, and in identifying abscess formation.
Abscesses may require surgical intervention. However, most cases without abscess formation respond to outpatient treatment with antibiotics. Antibiotic selection involves antistaphylococcal coverage, such as amoxicillin-clavulanic acid. Glandular massage may be helpful after the tenderness has resolved. In addition, the literature emphasizes the crucial role of hydration and excellent oral hygiene in disease recurrence and to prevent dental complications.
Case
A 21-year-old woman presented to the ED with pain and swelling on the right side of her neck. She stated the pain started earlier that morning and worsened when she ate or swallowed. The patient denied a recent or remote history of drooling, voice changes, or neck swelling. She reported no fevers, chills, or any other complaints, and had no pertinent medical history—specifically, no history of recent dental work. Her surgical history included tonsillectomy and cholecystectomy. There was no family history of diabetes, thyroid disease, autoimmune disease, or any other diseases. The patient stated that she was not on any prescription or over-the-counter medications. Regarding her social history, she denied past or cu
Vital signs at presentation were: blood pressure, 124/63 mm Hg (sitting); heart rate, 73 beats/min; respiratory rate, 15 breaths/min; and temperature, 98°F. Oxygen saturation was 99% on room air. On clinical examination, pain was noted in the patient’s right submandibular area and was tender to palpation. The swelling extended to the angle of the mandible posteriorly (Figure 1a and 1b). There was no erythema or increased surface temperature to suggest overlying cellulitis. The oral examination showed no evidence of dental infection, angioedema, or Ludwig angina. The pharynx was normal in appearance. The otological examination was unremarkable, and there was no evidence of mastoiditis.
Laboratory evaluation included a complete blood count (CBC), basic metabolic profile (BMP), and rapid streptococcal test (RST). The results of the patient’s CBC revealed a white blood cell count (WBC) of 11.1 x 109/L; the BMP was unremarkable; and the RST was negative.
A soft tissue neck computed tomography (CT) scan with contrast was obtained, which revealed mild right submandibular gland enlargement with abnormal enhancement (Figure 2). Stranding was also noted in the right submandibular space along with thickening of the right platysma muscle, and few surrounding lymph nodes were prominent (Figure 3). The findings were consistent with acute submandibular sialadenitis.
The patient received intravenous (IV) normal saline for hydration and IV ketorolac for analgesia, as well as an initial dose of oral amoxicillin/clavulanate 875/125 mg. At discharge, she was given a 10-day course of oral amoxicillin/clavulanate 875/125 mg with instructions to follow-up with her primary care physician and otolaryngologist within 2 days. The patient did well on follow-up, and her symptoms resolved within a few days of discharge.
Discussion
Comparatively little has been published on acute submandibular sialadenitis over the past three decades, and much of that which is cited in the literature comes from a rather small pool of case reports.1 In a literature review, Raad et al1 noted, “Pertinent literature on [this] subject includes case reports but no studies describing the microbial and clinical characteristics of this disease.” Further, many of the published case reports describe neonatal presentations of submandibular sialadenitis, the incidence of which is rare in this patient population.2-4
Submandibular and Parotid Glands
The submandibular gland is the second largest salivary gland, the parotid gland being the largest. The duct of the salivary gland, the Wharton’s duct, opens under the tongue in the area of the lingual frenulum. Ductal obstruction is more frequently seen with the submandibular gland than with the parotid gland.1 The reason for this is unclear, but may be related to several factors. One factor may be that, unlike the Stenson’s duct of the parotid gland, the Wharton’s duct does not pass through a muscle; thus, there is no muscular massage supporting the movement of secretion, as there is with buccinator muscle massage of Stenson’s duct. In addition, submandibular saliva is more viscous than parotid saliva due to its higher protein content and higher concentration of calcium phosphate.1
Etiology
Submandibular gland obstruction can occur in the absence of infection. Noninfectious cases typically present with pain upon eating and swallowing. A bacterial infectious etiology is associated with odynophagia, but also includes persistent pain and tenderness. This presents as pain associated with eating. Bacterial infection of the submandibular gland adds the element of persistent pain, associated with such features as tenderness. In addition, purulent discharge from the Wharton’s duct may be present in infectious cases, and accompanied by fever, chills, and an elevated WBC.1
Several bacteria have been isolated in infectious submandibular sialadenitis, the most common pathogens being Staphylococcus aureus. However, streptococci, Pseudomonas aeruginosa, Moraxella catarrhalis, and Escherichia coli bacteria have also been identified in cases of infectious submandibular sialadenitis.5
Viral etiologies of sialadenitis, such as mumps, are generally bilateral and nonsuppurative. The human immunodeficiency virus can also cause bilateral nonsuppurative salivary gland infections.6
Imaging Studies
As illustrated in our case, CT imaging can assist in confirming the diagnosis of acute submandibular sialadenitis by defining the anatomic involvement and identifying the presence of an abscess. Ultrasound can also be used and has been described as a first-line imaging procedure.7,8
Treatment
Surgical Intervention. Abscesses may require surgical intervention. However, most cases without abscess formation respond to outpatient treatment with antibiotics.5 If ductal obstruction is identified, removal of the calculus may be needed. This may involve ductal dilation, sialolithectomy, or even ductoplasty if a stricture is identified.1
Antibiotic Therapy. With respect to antibiotic selection, Chandak et al5 recommend oral amoxicillin-clavulanic acid. Other antistaphylococcal coverage recommendations have been made in the literature. Gland massage may be helpful after the tenderness has resolved,5 and sialogogues (eg, lemon drops, vitamin C lozenges) can also provide some relief.6 In addition, to avoid disease recurrence and prevent dental complications, Chandak et al5 emphasize the crucial role of hydration and excellent oral hygiene.
Conclusion
We suspected acute submandibular sialadenitis in our patient based on clinical findings, which were confirmed on CT imaging. Patients with acute submandibular sialadenitis may present with submandibular gland obstruction in the absence of bacterial infection. Noninfectious obstruction typically presents as pain associated with eating and swallowing, whereas infectious cases include constant pain and tenderness in the affected area. In addition, patients with infectious etiology may also have purulent discharge from Wharton’s duct, fever, chills, and an elevated WBC. Several bacteria have been isolated, the most common being S aureus. However, streptococci, P aeruginosa, M catarrhalis and E coli have also been identified. Computed tomography studies are helpful in confirming the diagnosis, defining anatomical involvement, and in identifying abscess formation.
Abscesses may require surgical intervention. However, most cases without abscess formation respond to outpatient treatment with antibiotics. Antibiotic selection involves antistaphylococcal coverage, such as amoxicillin-clavulanic acid. Glandular massage may be helpful after the tenderness has resolved. In addition, the literature emphasizes the crucial role of hydration and excellent oral hygiene in disease recurrence and to prevent dental complications.
1. Raad II, Sabbagh MF, Caranasos GJ. Acute bacterial sialadenitis: a study of 29 cases and review. Rev Infect Dis. 1990;12(4):591-601.
2. Banks WW, Handler SD, Glade GB, Turner HD. Neonatal submandibular sialadenitis. Am J Otolaryngol. 1980;1(3):261-263.
3. Wells DH. Suppuration of the submandibular salivary glands in the neonate. Am J Dis Child. 1975;129(5):628-630.
4. Ryan RF, Padmakumar B. Neonatal suppurative sialadenitis: an important clinical diagnosis. BMJ Case Rep. 2015;2015. pii:bcr2014208535. doi:10.1136/bcr-2014-208535.
5. Chandak R, Degwekar S, Chandak M, Rawlani S. Acute submandibular sialadenitis—a case report. Case Rep Dent. 2012;2012:615375. doi:10.1155/2012/615375.
6. Wilson KF, Meier JD, Ward PD. Salivary gland disorders. Am Fam Physician. 2014;89(11):882-888.
7. Alyas F, Lewis K, Williams M, et al. Diseases of the submandibular gland as demonstrated using high resolution ultrasound. Br J Radiol. 2005;78(928):362-369. doi:10.1259/bjr/93120352.
8. Howlett DC, Alyas F, Wong KT, et al. Sonographic assessment of the submandibular space. Clin Radiol. 2004;59(12):1070-1078. doi:10.1016/j.crad.2004.06.025.
1. Raad II, Sabbagh MF, Caranasos GJ. Acute bacterial sialadenitis: a study of 29 cases and review. Rev Infect Dis. 1990;12(4):591-601.
2. Banks WW, Handler SD, Glade GB, Turner HD. Neonatal submandibular sialadenitis. Am J Otolaryngol. 1980;1(3):261-263.
3. Wells DH. Suppuration of the submandibular salivary glands in the neonate. Am J Dis Child. 1975;129(5):628-630.
4. Ryan RF, Padmakumar B. Neonatal suppurative sialadenitis: an important clinical diagnosis. BMJ Case Rep. 2015;2015. pii:bcr2014208535. doi:10.1136/bcr-2014-208535.
5. Chandak R, Degwekar S, Chandak M, Rawlani S. Acute submandibular sialadenitis—a case report. Case Rep Dent. 2012;2012:615375. doi:10.1155/2012/615375.
6. Wilson KF, Meier JD, Ward PD. Salivary gland disorders. Am Fam Physician. 2014;89(11):882-888.
7. Alyas F, Lewis K, Williams M, et al. Diseases of the submandibular gland as demonstrated using high resolution ultrasound. Br J Radiol. 2005;78(928):362-369. doi:10.1259/bjr/93120352.
8. Howlett DC, Alyas F, Wong KT, et al. Sonographic assessment of the submandibular space. Clin Radiol. 2004;59(12):1070-1078. doi:10.1016/j.crad.2004.06.025.
Misdiagnosed Crusted Scabies in an AIDS Patient Leads to Hyperinfestation
Case Report
A recently incarcerated 34-year-old man with an 11-year history of multidrug-resistant human immunodeficiency virus/AIDS (CD4 count, 121 cells/mm3; viral load, 49,625 particles/mm3 one week prior to presentation) was admitted to the hospital for an intensely pruritic, hyperkeratotic, scaly rash involving the entire body. The rash first appeared on the feet approximately 1 year prior to admission. At that time the patient was given oral fluconazole and a steroid cream with near resolution of the rash. He was then transferred multiple times to different units with subsequent discontinuation of the medications. The rash flared and progressed to involve the knees. He was restarted on the fluconazole and steroid cream and placed in isolation by medical personnel at the prison 6 months prior to presentation. The rash continued to spread, and he was given a working diagnosis of plaque-type psoriasis by several providers after several months of nonresponse to treatment. Additional attempts at treatment at outside facilities included oral fluconazole, trimethoprim-sulfamethoxazole, and other antibiotics. He was referred to dermatology at our institution but missed the appointment and was admitted to the hospital before the appointment could be rescheduled.
On admission to the hospital, he denied similar lesions in close contacts. On review of systems he had subjective fevers and chills, decreased appetite, nausea without vomiting, dysphagia to solids, epigastric pain, and 70-lb weight loss over the last 6 months. Facial involvement of the rash impaired the ability to open the mouth, speak, and eat. He had no known drug allergies. His only medications at the time of admission were nortriptyline, trimethoprim-sulfamethoxazole, and oral combination elvitegravir-cobicistat-emtricitabine-tenofovir for hu-man immunodeficiency virus treatment.
On physical examination he was cachectic, shivering, and foul smelling. He was afebrile, slightly tachycardic (112 beats per minute), and hypertensive (144/83 mm Hg) with a respiratory rate of 18 breaths per minute. His height was 1.83 m (6 ft) and weight was 48.5 kg (107 lb) with a body mass index of 14.5. Extensive erythematous, hyperkeratotic, crusted, and fissured plaques covered the entire body including the face, hands, and feet. The tongue was covered with bilateral white-colored plaques, and he had patches of alopecia, excoriations, and scales on the scalp. The elbows were fixed in a flexed position and he had decreased range of motion in the wrists and fingers due to the severe hyperkeratosis (Figure 1A). Hyperkeratosis also was prominent on the knees and feet with associated burrows (Figure 2A). He had foot drop on the left.
The differential diagnosis included a drug eruption; fungal or parasite infestation, such as crusted scabies; psoriasis; or cutaneous lymphoma. Laboratory studies were difficult to obtain, as there were limited areas suitable for vascular access. Blood work showed leukocytosis (18.9×109 cells/L [reference range, 4.8–10.8×109 cells/L) with 13.3% eosinophils (reference range, 1%–6%). This eosinophilia narrowed the likely diagnoses to a drug eruption or parasite infection.
The dermatology service was consulted. A mineral oil preparation was performed and showed numerous mites and feces consistent with a diagnosis of crusted scabies (Figure 3). The patient was started on a regimen of permethrin cream 5% applied to the entire body, except the face, which was left on overnight and washed off. This regimen was repeated daily for 1 week, then twice weekly until the rash resolved after a total of 3 weeks. Due to the severity of his condition, immunocompromised status, and concern for superinfection, oral ivermectin 200 μg/kg once daily was added on days 1, 2, 8, 9, 15, 22, and 29.1
Our patient’s hospital course was further complicated by symptomatic hypoglycemia, altered mental status, and superimposed methicillin-resistant Staphylococcus aureus bacteremia, as well as Pseudomonas aeruginosa bacteremia, pneumonia, and coffee ground emesis. He was transferred to the intensive care unit but fortunately did not require intubation. His overall condition, mental status, and rash gradually improved. Three weeks after admission he only had a few residual lesions on the feet with clearing elsewhere (Figures 1B and 2B). He was discharged with a skin moisturizer and was referred for physical and occupational therapy. On follow-up clinic visits at 3 and 6 months, he had recovered well with general improvement in his condition.
Comment
Classic (noncrusted) scabies is common worldwide, with an estimated 300 million cases per year. It is caused by the mite Sarcoptes scabiei var hominis, and transmission occurs by direct skin-to-skin contact or less commonly by fomites (eg, linens, bedsheets) and therefore is common in overcrowded environments.2 Crusted scabies is a severe, highly contagious form of the disease in which the host’s immune system is overwhelmed and unable to defend against mites on the skin, resulting in hyperinfestation of the host. The mites use secretions to dissolve the epidermis and burrow through the skin, leaving feces in their tracks.3 Interestingly, the native aboriginal populations of Australia have a high incidence of crusted scabies even though they show no signs of immunosuppression. The reason remains unclear but may be due to a skewed T-cell response.4 Various mechanisms have been described for the symptoms of scabies, and it is believed that there is a hypersensitivity reaction to the mites and the feces. Increased IL-17 production by skin T cells may be responsible.5
Clinical Features
Crusted scabies is characterized by severe hyperkeratosis and plaques with desquamation and erythroderma that is worse in the acral regions and large joints, such as the elbows and the knees, as seen in our patient. Because of the deep burrows, patients are predisposed to secondary superinfections by bacteria. In our case, the patient had methicillin-resistant S aureus bacteremia, which persisted for some time despite treatment with intravenous antibiotics.
Diagnosis
Because scabies can imitate different conditions, it can be difficult to diagnose. Misdiagnosis of psoriasis in our patient led to ineffective treatment and subsequent worsening of his condition. Burrows are pathognomonic for scabies, though in severe cases, the burrows may be concealed by extreme hyperkeratosis. Diagnosis is confirmed by mineral oil preparation from the plaques showing numerous scabies mites and feces.
Treatment
It is important to control the spread of scabies, as it is highly contagious, and if the living environment is not properly cleaned, the patient can be reinfected. All clothing, bedsheets, and linens in the household must be washed in hot water and dried in a hot dryer, and nonwashable items should be placed in a closed plastic bag for 72 hours. All contacts also should be treated with 1 application of permethrin cream to the entire body including the head and neck, left on overnight, and washed off with warm water.1 The washing also helps remove some of the skin crusts. Patients should be educated that pruritus and burning may initially worsen with permethrin treatment due to the body’s reaction to the parasite.1,2 In addition, keratolytic agents such as topical urea or salicylic acid can be used as an adjuvant therapy to improve the efficacy of permethrin.
Permethrin is effective against both mites and eggs and works by inhibiting sodium channels, resulting in nerve signal conduction block and subsequent paralysis. Ivermectin is thought to act on glutamate-gated chloride channels, which are present in invertebrates but absent in vertebrates, causing hyperpolarization and paralysis of the adult mite.1,6
Conclusion
Crusted scabies is a highly contagious and intensely pruritic condition. Scabies can mimic other conditions, such as psoriasis or severe dermatitis, so it is important to keep this diagnosis in mind, especially in immunocompromised patients or populations in overcrowded areas (eg, those who are incarcerated or in nursing homes). Treatment consists of isolating the patient, starting topical permethrin and oral ivermectin (in severe cases), washing all linens, and prophylactically treating contacts. A delay in diagnosis can lead to severe debilitating disease, as seen in the extreme case of our patient. However, our patient made a full recovery with appropriate treatment and care.
- Currie BJ, McCarthy JS. Permethrin and ivermectin for scabies. N Engl J Med. 2010;362:717-725.
- World Health Organization. Water-related diseases: scabies. http://www.who.int/water_sanitation_health/diseases-risks/diseases/scabies/en/. Accessed February 23, 2017.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381.
- Liu X, Walton SF, Murray HC, et al. Crusted scabies is associated with increased IL-17 secretion by skin T cells. Parasite Immunol. 2014;36:594-604.
- Geary TG. Ivermectin 20 years on: maturation of a wonder drug [published online August 26, 2005]. Trends Parasitol. 2005;21:530-532.
Case Report
A recently incarcerated 34-year-old man with an 11-year history of multidrug-resistant human immunodeficiency virus/AIDS (CD4 count, 121 cells/mm3; viral load, 49,625 particles/mm3 one week prior to presentation) was admitted to the hospital for an intensely pruritic, hyperkeratotic, scaly rash involving the entire body. The rash first appeared on the feet approximately 1 year prior to admission. At that time the patient was given oral fluconazole and a steroid cream with near resolution of the rash. He was then transferred multiple times to different units with subsequent discontinuation of the medications. The rash flared and progressed to involve the knees. He was restarted on the fluconazole and steroid cream and placed in isolation by medical personnel at the prison 6 months prior to presentation. The rash continued to spread, and he was given a working diagnosis of plaque-type psoriasis by several providers after several months of nonresponse to treatment. Additional attempts at treatment at outside facilities included oral fluconazole, trimethoprim-sulfamethoxazole, and other antibiotics. He was referred to dermatology at our institution but missed the appointment and was admitted to the hospital before the appointment could be rescheduled.
On admission to the hospital, he denied similar lesions in close contacts. On review of systems he had subjective fevers and chills, decreased appetite, nausea without vomiting, dysphagia to solids, epigastric pain, and 70-lb weight loss over the last 6 months. Facial involvement of the rash impaired the ability to open the mouth, speak, and eat. He had no known drug allergies. His only medications at the time of admission were nortriptyline, trimethoprim-sulfamethoxazole, and oral combination elvitegravir-cobicistat-emtricitabine-tenofovir for hu-man immunodeficiency virus treatment.
On physical examination he was cachectic, shivering, and foul smelling. He was afebrile, slightly tachycardic (112 beats per minute), and hypertensive (144/83 mm Hg) with a respiratory rate of 18 breaths per minute. His height was 1.83 m (6 ft) and weight was 48.5 kg (107 lb) with a body mass index of 14.5. Extensive erythematous, hyperkeratotic, crusted, and fissured plaques covered the entire body including the face, hands, and feet. The tongue was covered with bilateral white-colored plaques, and he had patches of alopecia, excoriations, and scales on the scalp. The elbows were fixed in a flexed position and he had decreased range of motion in the wrists and fingers due to the severe hyperkeratosis (Figure 1A). Hyperkeratosis also was prominent on the knees and feet with associated burrows (Figure 2A). He had foot drop on the left.
The differential diagnosis included a drug eruption; fungal or parasite infestation, such as crusted scabies; psoriasis; or cutaneous lymphoma. Laboratory studies were difficult to obtain, as there were limited areas suitable for vascular access. Blood work showed leukocytosis (18.9×109 cells/L [reference range, 4.8–10.8×109 cells/L) with 13.3% eosinophils (reference range, 1%–6%). This eosinophilia narrowed the likely diagnoses to a drug eruption or parasite infection.
The dermatology service was consulted. A mineral oil preparation was performed and showed numerous mites and feces consistent with a diagnosis of crusted scabies (Figure 3). The patient was started on a regimen of permethrin cream 5% applied to the entire body, except the face, which was left on overnight and washed off. This regimen was repeated daily for 1 week, then twice weekly until the rash resolved after a total of 3 weeks. Due to the severity of his condition, immunocompromised status, and concern for superinfection, oral ivermectin 200 μg/kg once daily was added on days 1, 2, 8, 9, 15, 22, and 29.1
Our patient’s hospital course was further complicated by symptomatic hypoglycemia, altered mental status, and superimposed methicillin-resistant Staphylococcus aureus bacteremia, as well as Pseudomonas aeruginosa bacteremia, pneumonia, and coffee ground emesis. He was transferred to the intensive care unit but fortunately did not require intubation. His overall condition, mental status, and rash gradually improved. Three weeks after admission he only had a few residual lesions on the feet with clearing elsewhere (Figures 1B and 2B). He was discharged with a skin moisturizer and was referred for physical and occupational therapy. On follow-up clinic visits at 3 and 6 months, he had recovered well with general improvement in his condition.
Comment
Classic (noncrusted) scabies is common worldwide, with an estimated 300 million cases per year. It is caused by the mite Sarcoptes scabiei var hominis, and transmission occurs by direct skin-to-skin contact or less commonly by fomites (eg, linens, bedsheets) and therefore is common in overcrowded environments.2 Crusted scabies is a severe, highly contagious form of the disease in which the host’s immune system is overwhelmed and unable to defend against mites on the skin, resulting in hyperinfestation of the host. The mites use secretions to dissolve the epidermis and burrow through the skin, leaving feces in their tracks.3 Interestingly, the native aboriginal populations of Australia have a high incidence of crusted scabies even though they show no signs of immunosuppression. The reason remains unclear but may be due to a skewed T-cell response.4 Various mechanisms have been described for the symptoms of scabies, and it is believed that there is a hypersensitivity reaction to the mites and the feces. Increased IL-17 production by skin T cells may be responsible.5
Clinical Features
Crusted scabies is characterized by severe hyperkeratosis and plaques with desquamation and erythroderma that is worse in the acral regions and large joints, such as the elbows and the knees, as seen in our patient. Because of the deep burrows, patients are predisposed to secondary superinfections by bacteria. In our case, the patient had methicillin-resistant S aureus bacteremia, which persisted for some time despite treatment with intravenous antibiotics.
Diagnosis
Because scabies can imitate different conditions, it can be difficult to diagnose. Misdiagnosis of psoriasis in our patient led to ineffective treatment and subsequent worsening of his condition. Burrows are pathognomonic for scabies, though in severe cases, the burrows may be concealed by extreme hyperkeratosis. Diagnosis is confirmed by mineral oil preparation from the plaques showing numerous scabies mites and feces.
Treatment
It is important to control the spread of scabies, as it is highly contagious, and if the living environment is not properly cleaned, the patient can be reinfected. All clothing, bedsheets, and linens in the household must be washed in hot water and dried in a hot dryer, and nonwashable items should be placed in a closed plastic bag for 72 hours. All contacts also should be treated with 1 application of permethrin cream to the entire body including the head and neck, left on overnight, and washed off with warm water.1 The washing also helps remove some of the skin crusts. Patients should be educated that pruritus and burning may initially worsen with permethrin treatment due to the body’s reaction to the parasite.1,2 In addition, keratolytic agents such as topical urea or salicylic acid can be used as an adjuvant therapy to improve the efficacy of permethrin.
Permethrin is effective against both mites and eggs and works by inhibiting sodium channels, resulting in nerve signal conduction block and subsequent paralysis. Ivermectin is thought to act on glutamate-gated chloride channels, which are present in invertebrates but absent in vertebrates, causing hyperpolarization and paralysis of the adult mite.1,6
Conclusion
Crusted scabies is a highly contagious and intensely pruritic condition. Scabies can mimic other conditions, such as psoriasis or severe dermatitis, so it is important to keep this diagnosis in mind, especially in immunocompromised patients or populations in overcrowded areas (eg, those who are incarcerated or in nursing homes). Treatment consists of isolating the patient, starting topical permethrin and oral ivermectin (in severe cases), washing all linens, and prophylactically treating contacts. A delay in diagnosis can lead to severe debilitating disease, as seen in the extreme case of our patient. However, our patient made a full recovery with appropriate treatment and care.
Case Report
A recently incarcerated 34-year-old man with an 11-year history of multidrug-resistant human immunodeficiency virus/AIDS (CD4 count, 121 cells/mm3; viral load, 49,625 particles/mm3 one week prior to presentation) was admitted to the hospital for an intensely pruritic, hyperkeratotic, scaly rash involving the entire body. The rash first appeared on the feet approximately 1 year prior to admission. At that time the patient was given oral fluconazole and a steroid cream with near resolution of the rash. He was then transferred multiple times to different units with subsequent discontinuation of the medications. The rash flared and progressed to involve the knees. He was restarted on the fluconazole and steroid cream and placed in isolation by medical personnel at the prison 6 months prior to presentation. The rash continued to spread, and he was given a working diagnosis of plaque-type psoriasis by several providers after several months of nonresponse to treatment. Additional attempts at treatment at outside facilities included oral fluconazole, trimethoprim-sulfamethoxazole, and other antibiotics. He was referred to dermatology at our institution but missed the appointment and was admitted to the hospital before the appointment could be rescheduled.
On admission to the hospital, he denied similar lesions in close contacts. On review of systems he had subjective fevers and chills, decreased appetite, nausea without vomiting, dysphagia to solids, epigastric pain, and 70-lb weight loss over the last 6 months. Facial involvement of the rash impaired the ability to open the mouth, speak, and eat. He had no known drug allergies. His only medications at the time of admission were nortriptyline, trimethoprim-sulfamethoxazole, and oral combination elvitegravir-cobicistat-emtricitabine-tenofovir for hu-man immunodeficiency virus treatment.
On physical examination he was cachectic, shivering, and foul smelling. He was afebrile, slightly tachycardic (112 beats per minute), and hypertensive (144/83 mm Hg) with a respiratory rate of 18 breaths per minute. His height was 1.83 m (6 ft) and weight was 48.5 kg (107 lb) with a body mass index of 14.5. Extensive erythematous, hyperkeratotic, crusted, and fissured plaques covered the entire body including the face, hands, and feet. The tongue was covered with bilateral white-colored plaques, and he had patches of alopecia, excoriations, and scales on the scalp. The elbows were fixed in a flexed position and he had decreased range of motion in the wrists and fingers due to the severe hyperkeratosis (Figure 1A). Hyperkeratosis also was prominent on the knees and feet with associated burrows (Figure 2A). He had foot drop on the left.
The differential diagnosis included a drug eruption; fungal or parasite infestation, such as crusted scabies; psoriasis; or cutaneous lymphoma. Laboratory studies were difficult to obtain, as there were limited areas suitable for vascular access. Blood work showed leukocytosis (18.9×109 cells/L [reference range, 4.8–10.8×109 cells/L) with 13.3% eosinophils (reference range, 1%–6%). This eosinophilia narrowed the likely diagnoses to a drug eruption or parasite infection.
The dermatology service was consulted. A mineral oil preparation was performed and showed numerous mites and feces consistent with a diagnosis of crusted scabies (Figure 3). The patient was started on a regimen of permethrin cream 5% applied to the entire body, except the face, which was left on overnight and washed off. This regimen was repeated daily for 1 week, then twice weekly until the rash resolved after a total of 3 weeks. Due to the severity of his condition, immunocompromised status, and concern for superinfection, oral ivermectin 200 μg/kg once daily was added on days 1, 2, 8, 9, 15, 22, and 29.1
Our patient’s hospital course was further complicated by symptomatic hypoglycemia, altered mental status, and superimposed methicillin-resistant Staphylococcus aureus bacteremia, as well as Pseudomonas aeruginosa bacteremia, pneumonia, and coffee ground emesis. He was transferred to the intensive care unit but fortunately did not require intubation. His overall condition, mental status, and rash gradually improved. Three weeks after admission he only had a few residual lesions on the feet with clearing elsewhere (Figures 1B and 2B). He was discharged with a skin moisturizer and was referred for physical and occupational therapy. On follow-up clinic visits at 3 and 6 months, he had recovered well with general improvement in his condition.
Comment
Classic (noncrusted) scabies is common worldwide, with an estimated 300 million cases per year. It is caused by the mite Sarcoptes scabiei var hominis, and transmission occurs by direct skin-to-skin contact or less commonly by fomites (eg, linens, bedsheets) and therefore is common in overcrowded environments.2 Crusted scabies is a severe, highly contagious form of the disease in which the host’s immune system is overwhelmed and unable to defend against mites on the skin, resulting in hyperinfestation of the host. The mites use secretions to dissolve the epidermis and burrow through the skin, leaving feces in their tracks.3 Interestingly, the native aboriginal populations of Australia have a high incidence of crusted scabies even though they show no signs of immunosuppression. The reason remains unclear but may be due to a skewed T-cell response.4 Various mechanisms have been described for the symptoms of scabies, and it is believed that there is a hypersensitivity reaction to the mites and the feces. Increased IL-17 production by skin T cells may be responsible.5
Clinical Features
Crusted scabies is characterized by severe hyperkeratosis and plaques with desquamation and erythroderma that is worse in the acral regions and large joints, such as the elbows and the knees, as seen in our patient. Because of the deep burrows, patients are predisposed to secondary superinfections by bacteria. In our case, the patient had methicillin-resistant S aureus bacteremia, which persisted for some time despite treatment with intravenous antibiotics.
Diagnosis
Because scabies can imitate different conditions, it can be difficult to diagnose. Misdiagnosis of psoriasis in our patient led to ineffective treatment and subsequent worsening of his condition. Burrows are pathognomonic for scabies, though in severe cases, the burrows may be concealed by extreme hyperkeratosis. Diagnosis is confirmed by mineral oil preparation from the plaques showing numerous scabies mites and feces.
Treatment
It is important to control the spread of scabies, as it is highly contagious, and if the living environment is not properly cleaned, the patient can be reinfected. All clothing, bedsheets, and linens in the household must be washed in hot water and dried in a hot dryer, and nonwashable items should be placed in a closed plastic bag for 72 hours. All contacts also should be treated with 1 application of permethrin cream to the entire body including the head and neck, left on overnight, and washed off with warm water.1 The washing also helps remove some of the skin crusts. Patients should be educated that pruritus and burning may initially worsen with permethrin treatment due to the body’s reaction to the parasite.1,2 In addition, keratolytic agents such as topical urea or salicylic acid can be used as an adjuvant therapy to improve the efficacy of permethrin.
Permethrin is effective against both mites and eggs and works by inhibiting sodium channels, resulting in nerve signal conduction block and subsequent paralysis. Ivermectin is thought to act on glutamate-gated chloride channels, which are present in invertebrates but absent in vertebrates, causing hyperpolarization and paralysis of the adult mite.1,6
Conclusion
Crusted scabies is a highly contagious and intensely pruritic condition. Scabies can mimic other conditions, such as psoriasis or severe dermatitis, so it is important to keep this diagnosis in mind, especially in immunocompromised patients or populations in overcrowded areas (eg, those who are incarcerated or in nursing homes). Treatment consists of isolating the patient, starting topical permethrin and oral ivermectin (in severe cases), washing all linens, and prophylactically treating contacts. A delay in diagnosis can lead to severe debilitating disease, as seen in the extreme case of our patient. However, our patient made a full recovery with appropriate treatment and care.
- Currie BJ, McCarthy JS. Permethrin and ivermectin for scabies. N Engl J Med. 2010;362:717-725.
- World Health Organization. Water-related diseases: scabies. http://www.who.int/water_sanitation_health/diseases-risks/diseases/scabies/en/. Accessed February 23, 2017.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381.
- Liu X, Walton SF, Murray HC, et al. Crusted scabies is associated with increased IL-17 secretion by skin T cells. Parasite Immunol. 2014;36:594-604.
- Geary TG. Ivermectin 20 years on: maturation of a wonder drug [published online August 26, 2005]. Trends Parasitol. 2005;21:530-532.
- Currie BJ, McCarthy JS. Permethrin and ivermectin for scabies. N Engl J Med. 2010;362:717-725.
- World Health Organization. Water-related diseases: scabies. http://www.who.int/water_sanitation_health/diseases-risks/diseases/scabies/en/. Accessed February 23, 2017.
- Chosidow O. Scabies and pediculosis. Lancet. 2000;355:819-826.
- Roberts LJ, Huffam SE, Walton SF, et al. Crusted scabies: clinical and immunological findings in seventy-eight patients and a review of the literature. J Infect. 2005;50:375-381.
- Liu X, Walton SF, Murray HC, et al. Crusted scabies is associated with increased IL-17 secretion by skin T cells. Parasite Immunol. 2014;36:594-604.
- Geary TG. Ivermectin 20 years on: maturation of a wonder drug [published online August 26, 2005]. Trends Parasitol. 2005;21:530-532.
Practice Points
- Keep scabies in mind, especially in immunocompromised patients or populations in overcrowded areas.
- Treatment consists of isolating the patient, starting topical permethrin and oral ivermectin (in severe cases), washing all linens, and prophylactically treating contacts.
Imatinib Mesylate–Induced Lichenoid Drug Eruption
Imatinib mesylate is a tyrosine kinase inhibitor initially approved by the US Food and Drug Administration in 2001 for chronic myeloid leukemia (CML). The indications for imatinib have expanded since its initial approval. It is increasingly important that dermatologists recognize adverse cutaneous manifestations associated with imatinib and are aware of their management and outcomes to avoid unnecessarily discontinuing a potentially lifesaving medication.
Adverse cutaneous manifestations in response to imatinib are not infrequent, accounting for 7% to 21% of all side effects.1 The most frequent cutaneous manifestations of imatinib are dry skin, alopecia, facial edema, and photosensitivity rash, respectively.1 Other less common manifestations include exfoliative dermatitis, nail disorders, psoriasis, folliculitis, hypotrichosis, urticaria, petechiae, Stevens-Johnson syndrome, erythema multiforme, Sweet syndrome, and leukocytoclastic vasculitis.
We report a case of imatinib-induced lichenoid drug eruption (LDE), a rare cutaneous side effect of imatinib use, along with a review of the literature.
Case Report
An 86-year-old man with a history of gastrointestinal stromal tumors (GISTs) and myelodysplastic syndrome presented with diffuse hyperpigmented skin lesions on the trunk, arms, legs, and lower lip of 2 weeks’ duration. He had been taking imatinib 400 mg once daily for 5 months for GIST. Although the oncologist stopped the medication 2 weeks prior, the lesions were persistent and gradually expanded to involve the trunk, arms, legs, and lower lip. He denied any pain or pruritus. Physical examination revealed multiple ill-defined, brown to violaceous, slightly scaly macules and patches on the trunk (Figures 1A and 1B), arms, and legs (Figure 1C), as well as violaceous to erythematous patches on the mucosal aspect of the lower lip (Figure 2). Two 4-mm punch biopsies were performed from the chest and back, which revealed an atrophic epidermis, lichenoid infiltration, and multiple melanophages in the upper dermis consistent with LDE (Figure 3). Direct immunofluorescence was negative. Therefore, based on the clinicopathologic correlation, the diagnosis of imatinib-induced LDE was made. He was treated with clobetasol ointment twice daily for 3 weeks with some improvement. His GIST was stable on follow-up computed tomography 3 months after presentation, and imatinib was resumed 1 month later with continued rash that was stable with topical corticosteroid treatment.
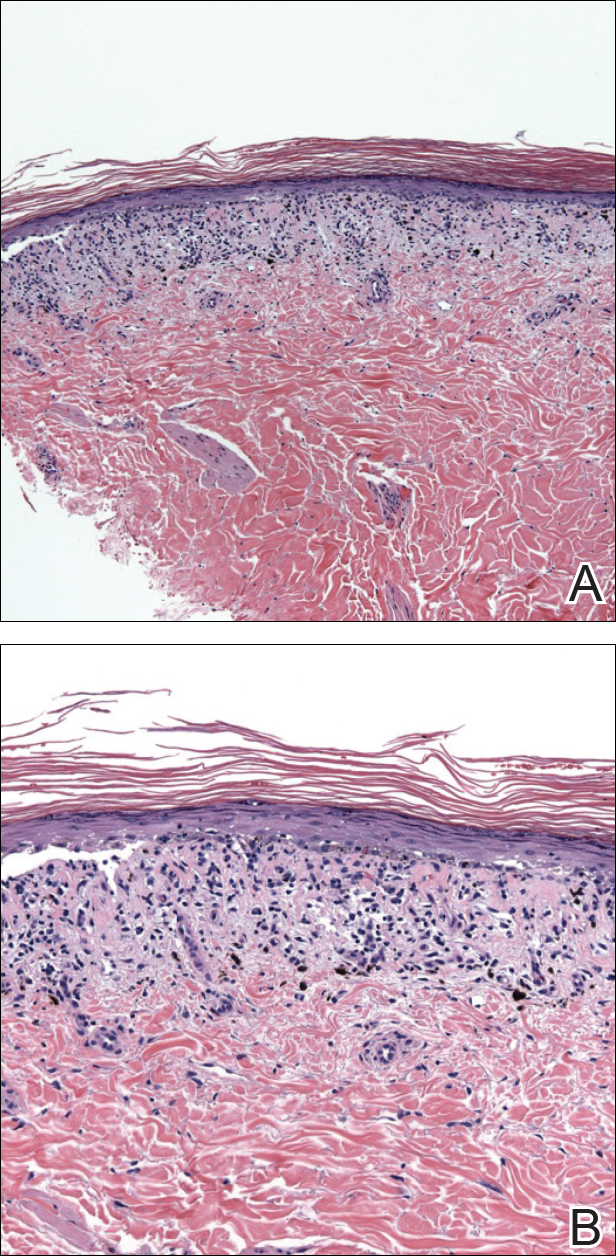
Comment
In addition to CML, imatinib has been approved for acute lymphoblastic leukemia, myelodysplastic syndromes, aggressive systemic mastocytosis, hypereosinophilic syndrome, chronic eosinophilic leukemia, dermatofibrosarcoma protuberans, and GIST. Moreover, off-label use of imatinib for various other tyrosine kinase–positive cancers and rheumatologic conditions have been documented.2,3 With the expanding use of imatinib, there will be more occasions for dermatologists to encounter cutaneous manifestations associated with its use.
According to a PubMed search of articles indexed for MEDLINE using the terms imatinib mesylate lichenoid drug, there have been few case reports of LDE associated with imatinib in the literature (eTable).4-24 Compared to classic LDE, imatinib-induced LDE has a few characteristic findings. Classic LDE frequently spares the oral mucosa and genitalia, but imatinib-induced LDE with manifestations on the oral mucosa and genitalia as well as cutaneous eruptions have been reported.4-9 In fact, the first known case of imatinib-induced LDE was an oral eruption in a patient with CML.4 In patients with oral involvement, lesions have been described as lacy reticular macules and violaceous papules, erosions, and ulcers.4,5,12 Interestingly, of those cases manifesting as concomitant oral and cutaneous LDE, the oral eruptions recurred more frequently, with 3 of 12 patients having recurrence of oral lesions after the cutaneous manifestations resolved.8,16 Genital manifestations of imatinib-induced LDE were much less common.9,11
To date, subsequent reports of imatinib-induced LDE have documented skin manifestations consistent with classic LDE occurring in a diffuse, bilateral, photodistributed pattern.10,15,16 One case presented with diffuse hyperpigmentation associated with LDE in a Japanese patient.20 The authors suggested this finding may be more prominent in patients with skin of color,20 which is consistent with the current case. Nail findings such as subungual hyperkeratosis and longitudinal ridging also have been reported.9,11
The latency period between initiation of imat-inib and onset of LDE generally ranges from 1 to 12 months, with onset most commonly occurring between 2 to 5 months or with dosage increase (eTable). Imatinib-induced LDE primarily has been documented with a 400-mg dose, with 1 case of a 600-mg dose and 1 case of an 800-mg dose, which suggests dose dependency. Furthermore, reports exist of several patients responding well to dose reduction with subsequent recurrence on dose reescalation.13,15
Historically, LDE resolves with discontinuation of the drug after a few weeks to months. When discontinuation of imatinib is unfavorable or patients report symptoms including severe pruritus or pain, treatment should be considered. Topical or oral corticosteroids can be used to treat imatinib-induced LDE, similar to lichen planus. When oral corticosteroids are contraindicated (eg, due to poor patient tolerance), oral acitretin at 25 to 35 mg once daily for 6 to 12 weeks has been reported as an alternative treatment.25
In the majority of cases of imatinib-induced LDE, it was undesirable to stop imatinib (eTable). Notably, in half the reported cases, imatinib was able to be continued and patients were treated symptomatically with either oral and/or topical steroids and/or acitretin with complete remission or tolerable recurrences. Dalmau et al9 reported 3 patients who responded poorly to topical and oral steroids and were subsequently treated with acitretin 25 mg once daily; 2 of 3 patients responded favorably to treatment and imatinib was able to be continued. In the current case imatinib initially helped, but because his rash was relatively asymptomatic, imatinib was restarted with control of rash with topical steroids. He developed some pancytopenia, which required intermittent stoppage of the imatinib.
Conclusion
We present a case of imatinib-induced cutaneous and oral LDE in a patient with GIST. Topical corticosteroids, oral acitretin, and oral steroids all may be reasonable treatment options if discontinuing imatinib is not possible in a symptomatic patient. If these therapies fail and the eruption is extensive or intolerable, dosage adjustment is another option to consider before discontinuation of imatinib.
- Scheinfeld N. Imatinib mesylate and dermatology part 2: a review of the cutaneous side effects of imatinib mesylate. J Drugs Dermatol. 2006;5:228-231.
- Kim H, Kim NH, Kang HJ, et al. Successful long-term use of imatinib mesylate in pediatric patients with sclerodermatous chronic GVHD. Pediatr Transplant. 2012;16:910-912.
- Prey S, Ezzedine K, Doussau A, et al. Imatinib mesylate in scleroderma-associated diffuse skin fibrosis: a phase II multicentre randomized double-blinded controlled trial. Br J Dermatol. 2012;167:1138-1144.
- Lim DS, Muir J. Oral lichenoid reaction to imatinib (STI 571, gleevec). Dermatology. 2002;205:169-171.
- Ena P, Chiarolini F, Siddi GM, et al. Oral lichenoid eruption secondary to imatinib (glivec). J Dermatolog Treat. 2004;15:253-255.
- Roux C, Boisseau-Garsaud AM, Saint-Cyr I, et al. Lichenoid cutaneous reaction to imatinib. Ann Dermatol Venereol. 2004;131:571-573.
- Prabhash K, Doval DC. Lichenoid eruption due to imat-inib. Indian J Dermatol Venereol Leprol. 2005;71:287-288.
- Pascual JC, Matarredona J, Miralles J, et al. Oral and cutaneous lichenoid reaction secondary to imatinib: report of two cases. Int J Dermatol. 2006;45:1471-1473.
- Dalmau J, Peramiquel L, Puig L, et al. Imatinib-associated lichenoid eruption: acitretin treatment allows maintained antineoplastic effect. Br J Dermatol. 2006;154:1213-1216.
- Chan CY, Browning J, Smith-Zagone MJ, et al. Cutaneous lichenoid dermatitis associated with imatinib mesylate. Dermatol Online J. 2007;13:29.
- Wahiduzzaman M, Pubalan M. Oral and cutaneous lichenoid reaction with nail changes secondary to imatinib: report of a case and literature review. Dermatol Online J. 2008;14:14.
- Basso FG, Boer CC, Correa ME, et al. Skin and oral lesions associated to imatinib mesylate therapy. Support Care Cancer. 2009;17:465-468.
- Kawakami T, Kawanabe T, Soma Y. Cutaneous lichenoid eruption caused by imatinib mesylate in a Japanese patient with chronic myeloid leukaemia. Acta Derm Venereol. 2009;89:325-326.
- Sendagorta E, Herranz P, Feito M, et al. Lichenoid drug eruption related to imatinib: report of a new case and review of the literature. Clin Exp Dermatol. 2009;34:E315-E316.
- Kuraishi N, Nagai Y, Hasegawa M, et al. Lichenoid drug eruption with palmoplantar hyperkeratosis due to imatinib mesylate: a case report and a review of the literature. Acta Derm Venereol. 2010;90:73-76.
- Brazzelli V, Muzio F, Manna G, et al. Photo-induced dermatitis and oral lichenoid reaction in a chronic myeloid leukemia patient treated with imatinib mesylate. Photodermatol Photoimmunol Photomed. 2012;28:2-5.
- Ghosh SK. Generalized lichenoid drug eruption associated with imatinib mesylate therapy. Indian J Dermatol. 2013;58:388-392.
- Lee J, Chung J, Jung M, et al. Lichenoid drug eruption after low-dose imatinib mesylate therapy. Ann Dermatol. 2013;25:500-502.
- Machaczka M, Gossart M. Multiple skin lesions caused by imatinib mesylate treatment of chronic myeloid leukemia. Pol Arch Med Wewn. 2013;123:251-252.
- Kagimoto Y, Mizuashi M, Kikuchi K, et al. Lichenoid drug eruption with hyperpigmentation caused by imatinib mesylate [published online June 20, 2013]. Int J Dermatol. 2014;53:E161-E162.
- Arshdeep, De D, Malhotra P, et al. Imatinib mesylate-induced severe lichenoid rash. Indian J Dermatol Venereol Leprol. 2014;80:93-95.
- Lau YM, Lam YK, Leung KH, et al. Trachyonychia in a patient with chronic myeloid leukaemia after imatinib mesylate. Hong Kong Med J. 2014;20:464.e2.
- Bhatia A, Kanish B, Chaudhary P. Lichenoid drug eruption due to imatinib mesylate. Int J Appl Basic Med Res. 2015;5:68-69.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Laurberg G, Geiger JM, Hjorth N, et al. Treatment of lichen planus with acitretin. a double-blind, placebo-controlled study in 65 patients. J Am Acad Dermatol. 1991;24:434-437.
Imatinib mesylate is a tyrosine kinase inhibitor initially approved by the US Food and Drug Administration in 2001 for chronic myeloid leukemia (CML). The indications for imatinib have expanded since its initial approval. It is increasingly important that dermatologists recognize adverse cutaneous manifestations associated with imatinib and are aware of their management and outcomes to avoid unnecessarily discontinuing a potentially lifesaving medication.
Adverse cutaneous manifestations in response to imatinib are not infrequent, accounting for 7% to 21% of all side effects.1 The most frequent cutaneous manifestations of imatinib are dry skin, alopecia, facial edema, and photosensitivity rash, respectively.1 Other less common manifestations include exfoliative dermatitis, nail disorders, psoriasis, folliculitis, hypotrichosis, urticaria, petechiae, Stevens-Johnson syndrome, erythema multiforme, Sweet syndrome, and leukocytoclastic vasculitis.
We report a case of imatinib-induced lichenoid drug eruption (LDE), a rare cutaneous side effect of imatinib use, along with a review of the literature.
Case Report
An 86-year-old man with a history of gastrointestinal stromal tumors (GISTs) and myelodysplastic syndrome presented with diffuse hyperpigmented skin lesions on the trunk, arms, legs, and lower lip of 2 weeks’ duration. He had been taking imatinib 400 mg once daily for 5 months for GIST. Although the oncologist stopped the medication 2 weeks prior, the lesions were persistent and gradually expanded to involve the trunk, arms, legs, and lower lip. He denied any pain or pruritus. Physical examination revealed multiple ill-defined, brown to violaceous, slightly scaly macules and patches on the trunk (Figures 1A and 1B), arms, and legs (Figure 1C), as well as violaceous to erythematous patches on the mucosal aspect of the lower lip (Figure 2). Two 4-mm punch biopsies were performed from the chest and back, which revealed an atrophic epidermis, lichenoid infiltration, and multiple melanophages in the upper dermis consistent with LDE (Figure 3). Direct immunofluorescence was negative. Therefore, based on the clinicopathologic correlation, the diagnosis of imatinib-induced LDE was made. He was treated with clobetasol ointment twice daily for 3 weeks with some improvement. His GIST was stable on follow-up computed tomography 3 months after presentation, and imatinib was resumed 1 month later with continued rash that was stable with topical corticosteroid treatment.
Comment
In addition to CML, imatinib has been approved for acute lymphoblastic leukemia, myelodysplastic syndromes, aggressive systemic mastocytosis, hypereosinophilic syndrome, chronic eosinophilic leukemia, dermatofibrosarcoma protuberans, and GIST. Moreover, off-label use of imatinib for various other tyrosine kinase–positive cancers and rheumatologic conditions have been documented.2,3 With the expanding use of imatinib, there will be more occasions for dermatologists to encounter cutaneous manifestations associated with its use.
According to a PubMed search of articles indexed for MEDLINE using the terms imatinib mesylate lichenoid drug, there have been few case reports of LDE associated with imatinib in the literature (eTable).4-24 Compared to classic LDE, imatinib-induced LDE has a few characteristic findings. Classic LDE frequently spares the oral mucosa and genitalia, but imatinib-induced LDE with manifestations on the oral mucosa and genitalia as well as cutaneous eruptions have been reported.4-9 In fact, the first known case of imatinib-induced LDE was an oral eruption in a patient with CML.4 In patients with oral involvement, lesions have been described as lacy reticular macules and violaceous papules, erosions, and ulcers.4,5,12 Interestingly, of those cases manifesting as concomitant oral and cutaneous LDE, the oral eruptions recurred more frequently, with 3 of 12 patients having recurrence of oral lesions after the cutaneous manifestations resolved.8,16 Genital manifestations of imatinib-induced LDE were much less common.9,11
To date, subsequent reports of imatinib-induced LDE have documented skin manifestations consistent with classic LDE occurring in a diffuse, bilateral, photodistributed pattern.10,15,16 One case presented with diffuse hyperpigmentation associated with LDE in a Japanese patient.20 The authors suggested this finding may be more prominent in patients with skin of color,20 which is consistent with the current case. Nail findings such as subungual hyperkeratosis and longitudinal ridging also have been reported.9,11
The latency period between initiation of imat-inib and onset of LDE generally ranges from 1 to 12 months, with onset most commonly occurring between 2 to 5 months or with dosage increase (eTable). Imatinib-induced LDE primarily has been documented with a 400-mg dose, with 1 case of a 600-mg dose and 1 case of an 800-mg dose, which suggests dose dependency. Furthermore, reports exist of several patients responding well to dose reduction with subsequent recurrence on dose reescalation.13,15
Historically, LDE resolves with discontinuation of the drug after a few weeks to months. When discontinuation of imatinib is unfavorable or patients report symptoms including severe pruritus or pain, treatment should be considered. Topical or oral corticosteroids can be used to treat imatinib-induced LDE, similar to lichen planus. When oral corticosteroids are contraindicated (eg, due to poor patient tolerance), oral acitretin at 25 to 35 mg once daily for 6 to 12 weeks has been reported as an alternative treatment.25
In the majority of cases of imatinib-induced LDE, it was undesirable to stop imatinib (eTable). Notably, in half the reported cases, imatinib was able to be continued and patients were treated symptomatically with either oral and/or topical steroids and/or acitretin with complete remission or tolerable recurrences. Dalmau et al9 reported 3 patients who responded poorly to topical and oral steroids and were subsequently treated with acitretin 25 mg once daily; 2 of 3 patients responded favorably to treatment and imatinib was able to be continued. In the current case imatinib initially helped, but because his rash was relatively asymptomatic, imatinib was restarted with control of rash with topical steroids. He developed some pancytopenia, which required intermittent stoppage of the imatinib.
Conclusion
We present a case of imatinib-induced cutaneous and oral LDE in a patient with GIST. Topical corticosteroids, oral acitretin, and oral steroids all may be reasonable treatment options if discontinuing imatinib is not possible in a symptomatic patient. If these therapies fail and the eruption is extensive or intolerable, dosage adjustment is another option to consider before discontinuation of imatinib.
Imatinib mesylate is a tyrosine kinase inhibitor initially approved by the US Food and Drug Administration in 2001 for chronic myeloid leukemia (CML). The indications for imatinib have expanded since its initial approval. It is increasingly important that dermatologists recognize adverse cutaneous manifestations associated with imatinib and are aware of their management and outcomes to avoid unnecessarily discontinuing a potentially lifesaving medication.
Adverse cutaneous manifestations in response to imatinib are not infrequent, accounting for 7% to 21% of all side effects.1 The most frequent cutaneous manifestations of imatinib are dry skin, alopecia, facial edema, and photosensitivity rash, respectively.1 Other less common manifestations include exfoliative dermatitis, nail disorders, psoriasis, folliculitis, hypotrichosis, urticaria, petechiae, Stevens-Johnson syndrome, erythema multiforme, Sweet syndrome, and leukocytoclastic vasculitis.
We report a case of imatinib-induced lichenoid drug eruption (LDE), a rare cutaneous side effect of imatinib use, along with a review of the literature.
Case Report
An 86-year-old man with a history of gastrointestinal stromal tumors (GISTs) and myelodysplastic syndrome presented with diffuse hyperpigmented skin lesions on the trunk, arms, legs, and lower lip of 2 weeks’ duration. He had been taking imatinib 400 mg once daily for 5 months for GIST. Although the oncologist stopped the medication 2 weeks prior, the lesions were persistent and gradually expanded to involve the trunk, arms, legs, and lower lip. He denied any pain or pruritus. Physical examination revealed multiple ill-defined, brown to violaceous, slightly scaly macules and patches on the trunk (Figures 1A and 1B), arms, and legs (Figure 1C), as well as violaceous to erythematous patches on the mucosal aspect of the lower lip (Figure 2). Two 4-mm punch biopsies were performed from the chest and back, which revealed an atrophic epidermis, lichenoid infiltration, and multiple melanophages in the upper dermis consistent with LDE (Figure 3). Direct immunofluorescence was negative. Therefore, based on the clinicopathologic correlation, the diagnosis of imatinib-induced LDE was made. He was treated with clobetasol ointment twice daily for 3 weeks with some improvement. His GIST was stable on follow-up computed tomography 3 months after presentation, and imatinib was resumed 1 month later with continued rash that was stable with topical corticosteroid treatment.
Comment
In addition to CML, imatinib has been approved for acute lymphoblastic leukemia, myelodysplastic syndromes, aggressive systemic mastocytosis, hypereosinophilic syndrome, chronic eosinophilic leukemia, dermatofibrosarcoma protuberans, and GIST. Moreover, off-label use of imatinib for various other tyrosine kinase–positive cancers and rheumatologic conditions have been documented.2,3 With the expanding use of imatinib, there will be more occasions for dermatologists to encounter cutaneous manifestations associated with its use.
According to a PubMed search of articles indexed for MEDLINE using the terms imatinib mesylate lichenoid drug, there have been few case reports of LDE associated with imatinib in the literature (eTable).4-24 Compared to classic LDE, imatinib-induced LDE has a few characteristic findings. Classic LDE frequently spares the oral mucosa and genitalia, but imatinib-induced LDE with manifestations on the oral mucosa and genitalia as well as cutaneous eruptions have been reported.4-9 In fact, the first known case of imatinib-induced LDE was an oral eruption in a patient with CML.4 In patients with oral involvement, lesions have been described as lacy reticular macules and violaceous papules, erosions, and ulcers.4,5,12 Interestingly, of those cases manifesting as concomitant oral and cutaneous LDE, the oral eruptions recurred more frequently, with 3 of 12 patients having recurrence of oral lesions after the cutaneous manifestations resolved.8,16 Genital manifestations of imatinib-induced LDE were much less common.9,11
To date, subsequent reports of imatinib-induced LDE have documented skin manifestations consistent with classic LDE occurring in a diffuse, bilateral, photodistributed pattern.10,15,16 One case presented with diffuse hyperpigmentation associated with LDE in a Japanese patient.20 The authors suggested this finding may be more prominent in patients with skin of color,20 which is consistent with the current case. Nail findings such as subungual hyperkeratosis and longitudinal ridging also have been reported.9,11
The latency period between initiation of imat-inib and onset of LDE generally ranges from 1 to 12 months, with onset most commonly occurring between 2 to 5 months or with dosage increase (eTable). Imatinib-induced LDE primarily has been documented with a 400-mg dose, with 1 case of a 600-mg dose and 1 case of an 800-mg dose, which suggests dose dependency. Furthermore, reports exist of several patients responding well to dose reduction with subsequent recurrence on dose reescalation.13,15
Historically, LDE resolves with discontinuation of the drug after a few weeks to months. When discontinuation of imatinib is unfavorable or patients report symptoms including severe pruritus or pain, treatment should be considered. Topical or oral corticosteroids can be used to treat imatinib-induced LDE, similar to lichen planus. When oral corticosteroids are contraindicated (eg, due to poor patient tolerance), oral acitretin at 25 to 35 mg once daily for 6 to 12 weeks has been reported as an alternative treatment.25
In the majority of cases of imatinib-induced LDE, it was undesirable to stop imatinib (eTable). Notably, in half the reported cases, imatinib was able to be continued and patients were treated symptomatically with either oral and/or topical steroids and/or acitretin with complete remission or tolerable recurrences. Dalmau et al9 reported 3 patients who responded poorly to topical and oral steroids and were subsequently treated with acitretin 25 mg once daily; 2 of 3 patients responded favorably to treatment and imatinib was able to be continued. In the current case imatinib initially helped, but because his rash was relatively asymptomatic, imatinib was restarted with control of rash with topical steroids. He developed some pancytopenia, which required intermittent stoppage of the imatinib.
Conclusion
We present a case of imatinib-induced cutaneous and oral LDE in a patient with GIST. Topical corticosteroids, oral acitretin, and oral steroids all may be reasonable treatment options if discontinuing imatinib is not possible in a symptomatic patient. If these therapies fail and the eruption is extensive or intolerable, dosage adjustment is another option to consider before discontinuation of imatinib.
- Scheinfeld N. Imatinib mesylate and dermatology part 2: a review of the cutaneous side effects of imatinib mesylate. J Drugs Dermatol. 2006;5:228-231.
- Kim H, Kim NH, Kang HJ, et al. Successful long-term use of imatinib mesylate in pediatric patients with sclerodermatous chronic GVHD. Pediatr Transplant. 2012;16:910-912.
- Prey S, Ezzedine K, Doussau A, et al. Imatinib mesylate in scleroderma-associated diffuse skin fibrosis: a phase II multicentre randomized double-blinded controlled trial. Br J Dermatol. 2012;167:1138-1144.
- Lim DS, Muir J. Oral lichenoid reaction to imatinib (STI 571, gleevec). Dermatology. 2002;205:169-171.
- Ena P, Chiarolini F, Siddi GM, et al. Oral lichenoid eruption secondary to imatinib (glivec). J Dermatolog Treat. 2004;15:253-255.
- Roux C, Boisseau-Garsaud AM, Saint-Cyr I, et al. Lichenoid cutaneous reaction to imatinib. Ann Dermatol Venereol. 2004;131:571-573.
- Prabhash K, Doval DC. Lichenoid eruption due to imat-inib. Indian J Dermatol Venereol Leprol. 2005;71:287-288.
- Pascual JC, Matarredona J, Miralles J, et al. Oral and cutaneous lichenoid reaction secondary to imatinib: report of two cases. Int J Dermatol. 2006;45:1471-1473.
- Dalmau J, Peramiquel L, Puig L, et al. Imatinib-associated lichenoid eruption: acitretin treatment allows maintained antineoplastic effect. Br J Dermatol. 2006;154:1213-1216.
- Chan CY, Browning J, Smith-Zagone MJ, et al. Cutaneous lichenoid dermatitis associated with imatinib mesylate. Dermatol Online J. 2007;13:29.
- Wahiduzzaman M, Pubalan M. Oral and cutaneous lichenoid reaction with nail changes secondary to imatinib: report of a case and literature review. Dermatol Online J. 2008;14:14.
- Basso FG, Boer CC, Correa ME, et al. Skin and oral lesions associated to imatinib mesylate therapy. Support Care Cancer. 2009;17:465-468.
- Kawakami T, Kawanabe T, Soma Y. Cutaneous lichenoid eruption caused by imatinib mesylate in a Japanese patient with chronic myeloid leukaemia. Acta Derm Venereol. 2009;89:325-326.
- Sendagorta E, Herranz P, Feito M, et al. Lichenoid drug eruption related to imatinib: report of a new case and review of the literature. Clin Exp Dermatol. 2009;34:E315-E316.
- Kuraishi N, Nagai Y, Hasegawa M, et al. Lichenoid drug eruption with palmoplantar hyperkeratosis due to imatinib mesylate: a case report and a review of the literature. Acta Derm Venereol. 2010;90:73-76.
- Brazzelli V, Muzio F, Manna G, et al. Photo-induced dermatitis and oral lichenoid reaction in a chronic myeloid leukemia patient treated with imatinib mesylate. Photodermatol Photoimmunol Photomed. 2012;28:2-5.
- Ghosh SK. Generalized lichenoid drug eruption associated with imatinib mesylate therapy. Indian J Dermatol. 2013;58:388-392.
- Lee J, Chung J, Jung M, et al. Lichenoid drug eruption after low-dose imatinib mesylate therapy. Ann Dermatol. 2013;25:500-502.
- Machaczka M, Gossart M. Multiple skin lesions caused by imatinib mesylate treatment of chronic myeloid leukemia. Pol Arch Med Wewn. 2013;123:251-252.
- Kagimoto Y, Mizuashi M, Kikuchi K, et al. Lichenoid drug eruption with hyperpigmentation caused by imatinib mesylate [published online June 20, 2013]. Int J Dermatol. 2014;53:E161-E162.
- Arshdeep, De D, Malhotra P, et al. Imatinib mesylate-induced severe lichenoid rash. Indian J Dermatol Venereol Leprol. 2014;80:93-95.
- Lau YM, Lam YK, Leung KH, et al. Trachyonychia in a patient with chronic myeloid leukaemia after imatinib mesylate. Hong Kong Med J. 2014;20:464.e2.
- Bhatia A, Kanish B, Chaudhary P. Lichenoid drug eruption due to imatinib mesylate. Int J Appl Basic Med Res. 2015;5:68-69.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Laurberg G, Geiger JM, Hjorth N, et al. Treatment of lichen planus with acitretin. a double-blind, placebo-controlled study in 65 patients. J Am Acad Dermatol. 1991;24:434-437.
- Scheinfeld N. Imatinib mesylate and dermatology part 2: a review of the cutaneous side effects of imatinib mesylate. J Drugs Dermatol. 2006;5:228-231.
- Kim H, Kim NH, Kang HJ, et al. Successful long-term use of imatinib mesylate in pediatric patients with sclerodermatous chronic GVHD. Pediatr Transplant. 2012;16:910-912.
- Prey S, Ezzedine K, Doussau A, et al. Imatinib mesylate in scleroderma-associated diffuse skin fibrosis: a phase II multicentre randomized double-blinded controlled trial. Br J Dermatol. 2012;167:1138-1144.
- Lim DS, Muir J. Oral lichenoid reaction to imatinib (STI 571, gleevec). Dermatology. 2002;205:169-171.
- Ena P, Chiarolini F, Siddi GM, et al. Oral lichenoid eruption secondary to imatinib (glivec). J Dermatolog Treat. 2004;15:253-255.
- Roux C, Boisseau-Garsaud AM, Saint-Cyr I, et al. Lichenoid cutaneous reaction to imatinib. Ann Dermatol Venereol. 2004;131:571-573.
- Prabhash K, Doval DC. Lichenoid eruption due to imat-inib. Indian J Dermatol Venereol Leprol. 2005;71:287-288.
- Pascual JC, Matarredona J, Miralles J, et al. Oral and cutaneous lichenoid reaction secondary to imatinib: report of two cases. Int J Dermatol. 2006;45:1471-1473.
- Dalmau J, Peramiquel L, Puig L, et al. Imatinib-associated lichenoid eruption: acitretin treatment allows maintained antineoplastic effect. Br J Dermatol. 2006;154:1213-1216.
- Chan CY, Browning J, Smith-Zagone MJ, et al. Cutaneous lichenoid dermatitis associated with imatinib mesylate. Dermatol Online J. 2007;13:29.
- Wahiduzzaman M, Pubalan M. Oral and cutaneous lichenoid reaction with nail changes secondary to imatinib: report of a case and literature review. Dermatol Online J. 2008;14:14.
- Basso FG, Boer CC, Correa ME, et al. Skin and oral lesions associated to imatinib mesylate therapy. Support Care Cancer. 2009;17:465-468.
- Kawakami T, Kawanabe T, Soma Y. Cutaneous lichenoid eruption caused by imatinib mesylate in a Japanese patient with chronic myeloid leukaemia. Acta Derm Venereol. 2009;89:325-326.
- Sendagorta E, Herranz P, Feito M, et al. Lichenoid drug eruption related to imatinib: report of a new case and review of the literature. Clin Exp Dermatol. 2009;34:E315-E316.
- Kuraishi N, Nagai Y, Hasegawa M, et al. Lichenoid drug eruption with palmoplantar hyperkeratosis due to imatinib mesylate: a case report and a review of the literature. Acta Derm Venereol. 2010;90:73-76.
- Brazzelli V, Muzio F, Manna G, et al. Photo-induced dermatitis and oral lichenoid reaction in a chronic myeloid leukemia patient treated with imatinib mesylate. Photodermatol Photoimmunol Photomed. 2012;28:2-5.
- Ghosh SK. Generalized lichenoid drug eruption associated with imatinib mesylate therapy. Indian J Dermatol. 2013;58:388-392.
- Lee J, Chung J, Jung M, et al. Lichenoid drug eruption after low-dose imatinib mesylate therapy. Ann Dermatol. 2013;25:500-502.
- Machaczka M, Gossart M. Multiple skin lesions caused by imatinib mesylate treatment of chronic myeloid leukemia. Pol Arch Med Wewn. 2013;123:251-252.
- Kagimoto Y, Mizuashi M, Kikuchi K, et al. Lichenoid drug eruption with hyperpigmentation caused by imatinib mesylate [published online June 20, 2013]. Int J Dermatol. 2014;53:E161-E162.
- Arshdeep, De D, Malhotra P, et al. Imatinib mesylate-induced severe lichenoid rash. Indian J Dermatol Venereol Leprol. 2014;80:93-95.
- Lau YM, Lam YK, Leung KH, et al. Trachyonychia in a patient with chronic myeloid leukaemia after imatinib mesylate. Hong Kong Med J. 2014;20:464.e2.
- Bhatia A, Kanish B, Chaudhary P. Lichenoid drug eruption due to imatinib mesylate. Int J Appl Basic Med Res. 2015;5:68-69.
- Luo JR, Xiang XJ, Xiong JP. Lichenoid drug eruption caused by imatinib mesylate in a Chinese patient with gastrointestinal stromal tumor. Int J Clin Pharmacol Ther. 2016;54:719-722.
- Laurberg G, Geiger JM, Hjorth N, et al. Treatment of lichen planus with acitretin. a double-blind, placebo-controlled study in 65 patients. J Am Acad Dermatol. 1991;24:434-437.
Practice Points
- Imatinib mesylate can cause cutaneous adverse reactions including dry skin, alopecia, facial edema, photosensitivity rash, and lichenoid drug eruption (LDE).
- Topical corticosteroids, oral acitretin, and oral steroids may be reasonable treatment options for imatinib-induced LDE if discontinuing imatinib is not possible in a symptomatic patient.
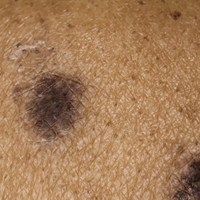
Weakness and pain in arms and legs • dark urine • history of vertebral osteomyelitis • Dx?
THE CASE
A 76-year-old Caucasian woman presented to the emergency department with a 7-day history of weakness and pain in her arms and legs. She had a history of Candida albicans vertebral osteomyelitis that had been treated for 3 months with fluconazole; non-Hodgkin lymphoma that had been in remission for 6 months; diabetes mellitus; hyperlipidemia; and hypothyroidism. The woman had dark urine, but denied chills, fever, respiratory symptoms, bowel or bladder leakage, falls/trauma, or grapefruit juice intake.
Her current medications included oral fluconazole 400 mg/d, simvastatin 20 mg/d, levothyroxine 88 mcg/d, pregabalin 75 mg/d, metformin 1000 mg twice daily, 6 units of subcutaneous insulin glargine at bedtime, and 2 units of insulin lispro with each meal. During the examination, we noted marked proximal muscle weakness, significant tenderness in all extremities, and diminished deep tendon reflexes. The patient had no saddle anesthesia, impaired rectal tone, or sensory abnormalities.
THE DIAGNOSIS
Magnetic resonance imaging of the patient’s spine confirmed multilevel discitis and osteomyelitis (T7-T9, L5-S1) with no cord compression. Laboratory data included a creatinine level of 1.42 mg/dL (the patient’s baseline was 0.8 mg/dL); a creatine kinase (CK) level of 8876 U/L (normal range, 0-220 U/L); a thyroid-stimulating hormone (TSH) level of 9.35 mIU/L (normal range, 0.4-5.5 mIU/L); and an erythrocyte sedimentation rate of 27 mm/hr (normal range, 0-31 mm/hr).
The patient received aggressive fluid hydration, orally and intravenously. On Day 2, the patient’s serum myoglobin level was 14,301 ng/mL (normal range, 30-90 ng/mL) and her aldolase level was 87.6 U/L (normal range, 1.5-8.5 U/L).
Zeroing in on the cause. There were no signs of drug abuse or use of other non-statin culprit medications that could have caused the patient’s rhabdomyolysis. She also did not describe any triggers of rhabdomyolysis, such as trauma, viral infection, metabolic disturbances, or temperature dysregulation. We believed the most likely cause of our patient’s signs and symptoms was statin-induced rhabdomyolysis, likely due to an interaction between simvastatin and fluconazole. We considered hypothyroidism-induced rhabdomyolysis, but thought it was unlikely because the patient had a mildly increased TSH level on admission, and one would expect to see levels higher than 100 mIU/L.1-3
We also considered viral myositis in the differential, but it was an unlikely culprit because the patient lacked any history of fever or respiratory or gastrointestinal symptoms. And while paraneoplastic polymyositis could have caused the patient’s weakness, the marked muscle pain and acute kidney injury were far more suggestive of rhabdomyolysis.
DISCUSSION
Rhabdomyolysis is a serious complication of statin treatment. Both higher statin doses and pharmacokinetic factors can raise statin levels, leading to this serious muscle-related syndrome.4,5 Co-administration of statins with drugs that are strong inhibitors of cytochrome P450 (CYP) 3A4 (the main cytochrome P450 isoform that metabolizes most statins) can increase statin levels several fold.6,7 The trigger for our patient’s statin-induced rhabdomyolysis was fluconazole, a known moderate inhibitor of CYP3A4, which is comparatively weaker than certain potent azoles like itraconazole or ketoconazole.7-10 Doses of fluconazole generally ≥200 mg/d are needed to produce clinical interactions with CYP3A4 substrates.7 There are only 3 reported cases of fluconazole-simvastatin–induced rhabdomyolysis (TABLE 1).11-13
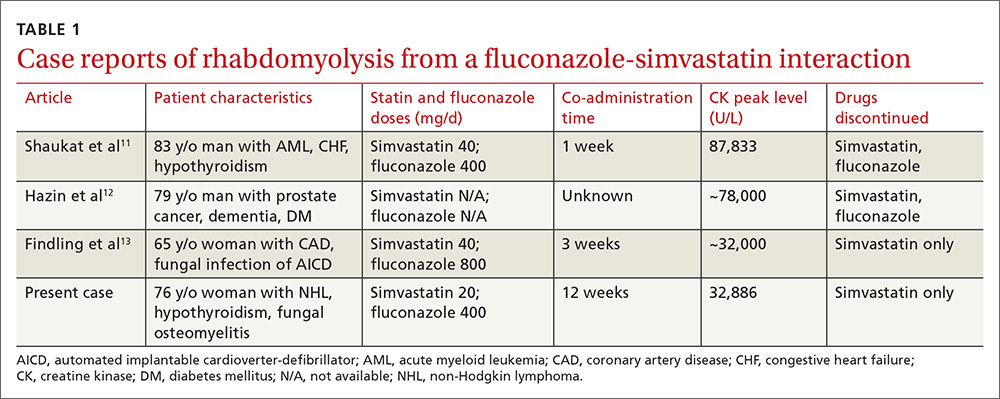
The Food and Drug Administration advises against simvastatin co-prescription with itraconazole and ketoconazole, but doesn’t mention fluconazole in its Drug Safety communication on simvastatin.14
Lexicomp places the simvastatin-fluconazole drug interaction into category C, which means that the agents can interact in a clinically significant manner (and a monitoring plan should be implemented), but that the benefits of concomitant use usually outweigh the risks.15
How our patient’s case differs from previous cases
Several features distinguish our patient’s scenario from previous cases. First, unlike other cases in which both drugs were stopped, only simvastatin was discontinued in our patient. Simvastatin and fluconazole have a half-life of 3 hours6 and 32 hours,7 respectively, suggesting that when simvastatin has fully cleared, fluconazole’s concentration will not even have halved. Thus, fluconazole was safely continued to treat the patient’s osteomyelitis.
Second, compared to previous case reports, our patient was taking a lower dose of simvastatin (20 mg). A 20-mg dose can make the drug interaction easier to miss; pharmacists are more likely to inform the physician of a potential drug interaction when the dose of a statin is ≥40 mg compared to when it is <40 mg (odds ratio=1.89; 95% confidence interval, 0.98-3.63).16
Researchers involved in the British randomized trial SEARCH (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine) sought to evaluate any added benefit to a higher dose of simvastatin in post-myocardial infarction patients. Among approximately 12,000 patients in the trial, there were 7 cases of rhabdomyolysis for the 80-mg simvastatin group and none for the 20-mg group.5 Another large case-control study showed that a 40-mg simvastatin dose was 5 times more likely to cause rhabdomyolysis than a 20-mg dose.17 Yet, based on our patient’s case, even 20 mg/d simvastatin should not decrease physician suspicion for rhabdomyolysis if patients are also taking a CYP3A4 inhibitor.
Third, the simvastatin-fluconazole co-administration time in our patient was 12 weeks, which is longer than previously reported (TABLE 111-13). Azole inhibition of CYP450 occurs relatively rapidly, but that does not mean that rhabdomyolysis will always occur immediately. For example, in cases of statin monotherapy, rhabdomyolysis secondary to statin biochemical toxicity can occur up to 1050 (mean=348) days after the drug’s initiation.18
Avoiding a drug-drug interaction in your patient
Physicians can use pharmacokinetic profiles to choose among different statins and azoles to help avoid a drug interaction (TABLE 26,7,10,19). Pravastatin’s serum concentration, for example, is not influenced by CYP3A4 inhibitors such as itraconazole11 because pravastatin is metabolized by sulfation6 and not by the CYP450 system. Rosuvastatin and pitavastatin are minimally metabolized by the CYP450 system.19,20

Among approximately 2700 statin-treated outpatients,4 the prevalence of potentially harmful statin interactions with other drugs (including CYP3A4 inhibitors), was significantly higher among patients treated with simvastatin or atorvastatin (CYP3A4-metabolized statins), than among patients treated with fluvastatin (CYP2C9-metabolized statin) or pravastatin (metabolized by sulfation). Apart from drug-drug interactions, other risk factors for statin-induced rhabdomyolysis include use of lipophilic statins, advanced age, and female gender.21
We discontinued our patient’s simvastatin on the day she was admitted to the hospital, but continued with the fluconazole throughout her hospitalization. Her CK level continued to rise, peaked on hospital Day 3 at 32,886 U/L, and then progressively decreased. The patient’s weakness and pain improved and her acute kidney injury resolved with hydration. She was discharged on hospital Day 7 on oral fluconazole, but no statin, and her muscle symptoms have since resolved.
THE TAKEAWAY
When hyperlipidemic patients have to take an azole for an extended period (eg, cancer prophylaxis or chronic osteomyelitis) and the azole is a strong CYP450 inhibitor (eg, itraconazole), switching to a statin that is not primarily metabolized by the CYP450 system (eg, pravastatin, pitavastatin) is wise. If the azole is a moderate CYP450 inhibitor (eg, fluconazole), we suggest that therapy should be closely monitored. In the case of short-term azole treatment (eg, such as for oral candidiasis), the statin should be stopped or the dose reduced by at least 50% (eg, from 40 or 20 mg to 10 mg).6
Prescriber knowledge is sometimes a limiting factor in identifying clinically significant interactions.22 This is especially pertinent in a case like this one, where a lower statin dose may result in a lower chance of the pharmacist alerting the prescribing physician16 and when an azole is used that is a comparatively weaker CYP450 inhibitor than other azoles such as itraconazole. Even in the era of electronic medical records, approximately 90% of drug interaction alerts are overridden by physicians, and alert fatigue is pronounced.23
The intricacies and pharmacokinetic principles of this case should contribute to greater provider familiarity with even low-dose simvastatin-fluconazole interactions and help prevent iatrogenic complications such as rhabdomyolysis.
1. Kisakol G, Tunc R, Kaya A. Rhabdomyolysis in a patient with hypothyroidism. Endocr J. 2003;50:221-223.
2. Scott KR, Simmons Z, Boyer PJ. Hypothyroid myopathy with a strikingly elevated serum creatine kinase level. Muscle Nerve. 2002;26:141-144.
3. Barahona MJ, Mauri A, Sucunza N, et al. Hypothyroidism as a cause of rhabdomyolysis. Endocr J. 2002;49:621-623.
4. Rätz Bravo AE, Tchambaz L, Krähenbühl-Melcher A, et al. Prevalence of potentially severe drug-drug interactions in ambulatory patients with dyslipidaemia receiving HMG-CoA reductase inhibitor therapy. Drug Saf. 2005;28:263-275.
5. Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group, Armitage J, Bowman L, Wallendszus K, et al. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376:1658-1669.
6. Chong PH, Seeger JD, Franklin C. Clinically relevant differences between the statins: implications for therapeutic selection. Am J Med. 2001;111:390-400.
7. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism: clinical relevance. Clin Pharmacokinet. 2000;38:111-180.
8. Malhotra B, Dickins M, Alvey C, et al. Effects of the moderate CYP3A4 inhibitor, fluconazole, on the pharmacokinetics of fesoterodine in healthy subjects. Br J Clin Pharmacol. 2011;72:263-269.
9. US Food and Drug Administration. Drug development and drug interactions: Table of substrates, inhibitors and inducers. Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm. Accessed February 9, 2017.
10. Niwa T, Shiraga T, Takagi A. Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol Pharm Bull. 2005;28:1805-1808.
11. Shaukat A, Benekli M, Vladutiu GD, et al. Simvastatin-fluconazole causing rhabdomyolysis. Ann Pharmacother. 2003;37:1032-1035.
12. Hazin R, Abuzetun JY, Suker M, et al. Rhabdomyolysis induced by simvastatin-fluconazole combination. J Natl Med Assoc. 2008;100:444-446.
13. Findling O, Meier N, Sellner J, et al. Clinical reasoning: rhabdomyolysis after combined treatment with simvastatin and fluconazole. Neurology. 2008;71:e34-e37.
14. US Food and Drug Administration. FDA Drug Safety Communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. June 8, 2011. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm256581.htm. Accessed February 1, 2017.
15. Wolters Kluwer. Lexicomp online. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed February 9, 2017.
16. Molden E, Skovlund E, Braathen P. Risk management of simvastatin or atorvastatin interactions with CYP3A4 inhibitors. Drug Saf. 2008;31:587-596.
17. Parkin L, Paul C, Herbison GP. Simvastatin dose and risk of rhabdomyolysis: nested case-control study based on national health and drug dispensing data. Int J Cardiol. 2014;174:83-89.
18. Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292:2585-2590.
19. Saito Y. Pitavastatin: an overview. Atheroscler Suppl. 2011;12:271-276.
20. Olsson AG, McTaggart F, Raza A. Rosuvastatin: a highly effective new HMG-CoA reductase inhibitor. Cardiovasc Drug Rev. 2002;20:303-328.
21. Magni P, Macchi C, Morlotti B, et al. Risk identification and possible countermeasures for muscle adverse effects during statin therapy. Eur J Intern Med. 2015;26:82-88.
22. Ko Y, Malone DC, Skrepnek GH, et al. Prescribers’ knowledge of and sources of information for potential drug-drug interactions: a postal survey of US prescribers. Drug Saf. 2008;31:525-536.
23. Phansalkar S, van der Sijs H, Tucker AD, et al. Drug-drug interactions that should be non-interruptive in order to reduce alert fatigue in electronic health records. J Am Med Inform Assoc. 2013;20:489-493.
THE CASE
A 76-year-old Caucasian woman presented to the emergency department with a 7-day history of weakness and pain in her arms and legs. She had a history of Candida albicans vertebral osteomyelitis that had been treated for 3 months with fluconazole; non-Hodgkin lymphoma that had been in remission for 6 months; diabetes mellitus; hyperlipidemia; and hypothyroidism. The woman had dark urine, but denied chills, fever, respiratory symptoms, bowel or bladder leakage, falls/trauma, or grapefruit juice intake.
Her current medications included oral fluconazole 400 mg/d, simvastatin 20 mg/d, levothyroxine 88 mcg/d, pregabalin 75 mg/d, metformin 1000 mg twice daily, 6 units of subcutaneous insulin glargine at bedtime, and 2 units of insulin lispro with each meal. During the examination, we noted marked proximal muscle weakness, significant tenderness in all extremities, and diminished deep tendon reflexes. The patient had no saddle anesthesia, impaired rectal tone, or sensory abnormalities.
THE DIAGNOSIS
Magnetic resonance imaging of the patient’s spine confirmed multilevel discitis and osteomyelitis (T7-T9, L5-S1) with no cord compression. Laboratory data included a creatinine level of 1.42 mg/dL (the patient’s baseline was 0.8 mg/dL); a creatine kinase (CK) level of 8876 U/L (normal range, 0-220 U/L); a thyroid-stimulating hormone (TSH) level of 9.35 mIU/L (normal range, 0.4-5.5 mIU/L); and an erythrocyte sedimentation rate of 27 mm/hr (normal range, 0-31 mm/hr).
The patient received aggressive fluid hydration, orally and intravenously. On Day 2, the patient’s serum myoglobin level was 14,301 ng/mL (normal range, 30-90 ng/mL) and her aldolase level was 87.6 U/L (normal range, 1.5-8.5 U/L).
Zeroing in on the cause. There were no signs of drug abuse or use of other non-statin culprit medications that could have caused the patient’s rhabdomyolysis. She also did not describe any triggers of rhabdomyolysis, such as trauma, viral infection, metabolic disturbances, or temperature dysregulation. We believed the most likely cause of our patient’s signs and symptoms was statin-induced rhabdomyolysis, likely due to an interaction between simvastatin and fluconazole. We considered hypothyroidism-induced rhabdomyolysis, but thought it was unlikely because the patient had a mildly increased TSH level on admission, and one would expect to see levels higher than 100 mIU/L.1-3
We also considered viral myositis in the differential, but it was an unlikely culprit because the patient lacked any history of fever or respiratory or gastrointestinal symptoms. And while paraneoplastic polymyositis could have caused the patient’s weakness, the marked muscle pain and acute kidney injury were far more suggestive of rhabdomyolysis.
DISCUSSION
Rhabdomyolysis is a serious complication of statin treatment. Both higher statin doses and pharmacokinetic factors can raise statin levels, leading to this serious muscle-related syndrome.4,5 Co-administration of statins with drugs that are strong inhibitors of cytochrome P450 (CYP) 3A4 (the main cytochrome P450 isoform that metabolizes most statins) can increase statin levels several fold.6,7 The trigger for our patient’s statin-induced rhabdomyolysis was fluconazole, a known moderate inhibitor of CYP3A4, which is comparatively weaker than certain potent azoles like itraconazole or ketoconazole.7-10 Doses of fluconazole generally ≥200 mg/d are needed to produce clinical interactions with CYP3A4 substrates.7 There are only 3 reported cases of fluconazole-simvastatin–induced rhabdomyolysis (TABLE 1).11-13
The Food and Drug Administration advises against simvastatin co-prescription with itraconazole and ketoconazole, but doesn’t mention fluconazole in its Drug Safety communication on simvastatin.14
Lexicomp places the simvastatin-fluconazole drug interaction into category C, which means that the agents can interact in a clinically significant manner (and a monitoring plan should be implemented), but that the benefits of concomitant use usually outweigh the risks.15
How our patient’s case differs from previous cases
Several features distinguish our patient’s scenario from previous cases. First, unlike other cases in which both drugs were stopped, only simvastatin was discontinued in our patient. Simvastatin and fluconazole have a half-life of 3 hours6 and 32 hours,7 respectively, suggesting that when simvastatin has fully cleared, fluconazole’s concentration will not even have halved. Thus, fluconazole was safely continued to treat the patient’s osteomyelitis.
Second, compared to previous case reports, our patient was taking a lower dose of simvastatin (20 mg). A 20-mg dose can make the drug interaction easier to miss; pharmacists are more likely to inform the physician of a potential drug interaction when the dose of a statin is ≥40 mg compared to when it is <40 mg (odds ratio=1.89; 95% confidence interval, 0.98-3.63).16
Researchers involved in the British randomized trial SEARCH (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine) sought to evaluate any added benefit to a higher dose of simvastatin in post-myocardial infarction patients. Among approximately 12,000 patients in the trial, there were 7 cases of rhabdomyolysis for the 80-mg simvastatin group and none for the 20-mg group.5 Another large case-control study showed that a 40-mg simvastatin dose was 5 times more likely to cause rhabdomyolysis than a 20-mg dose.17 Yet, based on our patient’s case, even 20 mg/d simvastatin should not decrease physician suspicion for rhabdomyolysis if patients are also taking a CYP3A4 inhibitor.
Third, the simvastatin-fluconazole co-administration time in our patient was 12 weeks, which is longer than previously reported (TABLE 111-13). Azole inhibition of CYP450 occurs relatively rapidly, but that does not mean that rhabdomyolysis will always occur immediately. For example, in cases of statin monotherapy, rhabdomyolysis secondary to statin biochemical toxicity can occur up to 1050 (mean=348) days after the drug’s initiation.18
Avoiding a drug-drug interaction in your patient
Physicians can use pharmacokinetic profiles to choose among different statins and azoles to help avoid a drug interaction (TABLE 26,7,10,19). Pravastatin’s serum concentration, for example, is not influenced by CYP3A4 inhibitors such as itraconazole11 because pravastatin is metabolized by sulfation6 and not by the CYP450 system. Rosuvastatin and pitavastatin are minimally metabolized by the CYP450 system.19,20
Among approximately 2700 statin-treated outpatients,4 the prevalence of potentially harmful statin interactions with other drugs (including CYP3A4 inhibitors), was significantly higher among patients treated with simvastatin or atorvastatin (CYP3A4-metabolized statins), than among patients treated with fluvastatin (CYP2C9-metabolized statin) or pravastatin (metabolized by sulfation). Apart from drug-drug interactions, other risk factors for statin-induced rhabdomyolysis include use of lipophilic statins, advanced age, and female gender.21
We discontinued our patient’s simvastatin on the day she was admitted to the hospital, but continued with the fluconazole throughout her hospitalization. Her CK level continued to rise, peaked on hospital Day 3 at 32,886 U/L, and then progressively decreased. The patient’s weakness and pain improved and her acute kidney injury resolved with hydration. She was discharged on hospital Day 7 on oral fluconazole, but no statin, and her muscle symptoms have since resolved.
THE TAKEAWAY
When hyperlipidemic patients have to take an azole for an extended period (eg, cancer prophylaxis or chronic osteomyelitis) and the azole is a strong CYP450 inhibitor (eg, itraconazole), switching to a statin that is not primarily metabolized by the CYP450 system (eg, pravastatin, pitavastatin) is wise. If the azole is a moderate CYP450 inhibitor (eg, fluconazole), we suggest that therapy should be closely monitored. In the case of short-term azole treatment (eg, such as for oral candidiasis), the statin should be stopped or the dose reduced by at least 50% (eg, from 40 or 20 mg to 10 mg).6
Prescriber knowledge is sometimes a limiting factor in identifying clinically significant interactions.22 This is especially pertinent in a case like this one, where a lower statin dose may result in a lower chance of the pharmacist alerting the prescribing physician16 and when an azole is used that is a comparatively weaker CYP450 inhibitor than other azoles such as itraconazole. Even in the era of electronic medical records, approximately 90% of drug interaction alerts are overridden by physicians, and alert fatigue is pronounced.23
The intricacies and pharmacokinetic principles of this case should contribute to greater provider familiarity with even low-dose simvastatin-fluconazole interactions and help prevent iatrogenic complications such as rhabdomyolysis.
THE CASE
A 76-year-old Caucasian woman presented to the emergency department with a 7-day history of weakness and pain in her arms and legs. She had a history of Candida albicans vertebral osteomyelitis that had been treated for 3 months with fluconazole; non-Hodgkin lymphoma that had been in remission for 6 months; diabetes mellitus; hyperlipidemia; and hypothyroidism. The woman had dark urine, but denied chills, fever, respiratory symptoms, bowel or bladder leakage, falls/trauma, or grapefruit juice intake.
Her current medications included oral fluconazole 400 mg/d, simvastatin 20 mg/d, levothyroxine 88 mcg/d, pregabalin 75 mg/d, metformin 1000 mg twice daily, 6 units of subcutaneous insulin glargine at bedtime, and 2 units of insulin lispro with each meal. During the examination, we noted marked proximal muscle weakness, significant tenderness in all extremities, and diminished deep tendon reflexes. The patient had no saddle anesthesia, impaired rectal tone, or sensory abnormalities.
THE DIAGNOSIS
Magnetic resonance imaging of the patient’s spine confirmed multilevel discitis and osteomyelitis (T7-T9, L5-S1) with no cord compression. Laboratory data included a creatinine level of 1.42 mg/dL (the patient’s baseline was 0.8 mg/dL); a creatine kinase (CK) level of 8876 U/L (normal range, 0-220 U/L); a thyroid-stimulating hormone (TSH) level of 9.35 mIU/L (normal range, 0.4-5.5 mIU/L); and an erythrocyte sedimentation rate of 27 mm/hr (normal range, 0-31 mm/hr).
The patient received aggressive fluid hydration, orally and intravenously. On Day 2, the patient’s serum myoglobin level was 14,301 ng/mL (normal range, 30-90 ng/mL) and her aldolase level was 87.6 U/L (normal range, 1.5-8.5 U/L).
Zeroing in on the cause. There were no signs of drug abuse or use of other non-statin culprit medications that could have caused the patient’s rhabdomyolysis. She also did not describe any triggers of rhabdomyolysis, such as trauma, viral infection, metabolic disturbances, or temperature dysregulation. We believed the most likely cause of our patient’s signs and symptoms was statin-induced rhabdomyolysis, likely due to an interaction between simvastatin and fluconazole. We considered hypothyroidism-induced rhabdomyolysis, but thought it was unlikely because the patient had a mildly increased TSH level on admission, and one would expect to see levels higher than 100 mIU/L.1-3
We also considered viral myositis in the differential, but it was an unlikely culprit because the patient lacked any history of fever or respiratory or gastrointestinal symptoms. And while paraneoplastic polymyositis could have caused the patient’s weakness, the marked muscle pain and acute kidney injury were far more suggestive of rhabdomyolysis.
DISCUSSION
Rhabdomyolysis is a serious complication of statin treatment. Both higher statin doses and pharmacokinetic factors can raise statin levels, leading to this serious muscle-related syndrome.4,5 Co-administration of statins with drugs that are strong inhibitors of cytochrome P450 (CYP) 3A4 (the main cytochrome P450 isoform that metabolizes most statins) can increase statin levels several fold.6,7 The trigger for our patient’s statin-induced rhabdomyolysis was fluconazole, a known moderate inhibitor of CYP3A4, which is comparatively weaker than certain potent azoles like itraconazole or ketoconazole.7-10 Doses of fluconazole generally ≥200 mg/d are needed to produce clinical interactions with CYP3A4 substrates.7 There are only 3 reported cases of fluconazole-simvastatin–induced rhabdomyolysis (TABLE 1).11-13
The Food and Drug Administration advises against simvastatin co-prescription with itraconazole and ketoconazole, but doesn’t mention fluconazole in its Drug Safety communication on simvastatin.14
Lexicomp places the simvastatin-fluconazole drug interaction into category C, which means that the agents can interact in a clinically significant manner (and a monitoring plan should be implemented), but that the benefits of concomitant use usually outweigh the risks.15
How our patient’s case differs from previous cases
Several features distinguish our patient’s scenario from previous cases. First, unlike other cases in which both drugs were stopped, only simvastatin was discontinued in our patient. Simvastatin and fluconazole have a half-life of 3 hours6 and 32 hours,7 respectively, suggesting that when simvastatin has fully cleared, fluconazole’s concentration will not even have halved. Thus, fluconazole was safely continued to treat the patient’s osteomyelitis.
Second, compared to previous case reports, our patient was taking a lower dose of simvastatin (20 mg). A 20-mg dose can make the drug interaction easier to miss; pharmacists are more likely to inform the physician of a potential drug interaction when the dose of a statin is ≥40 mg compared to when it is <40 mg (odds ratio=1.89; 95% confidence interval, 0.98-3.63).16
Researchers involved in the British randomized trial SEARCH (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine) sought to evaluate any added benefit to a higher dose of simvastatin in post-myocardial infarction patients. Among approximately 12,000 patients in the trial, there were 7 cases of rhabdomyolysis for the 80-mg simvastatin group and none for the 20-mg group.5 Another large case-control study showed that a 40-mg simvastatin dose was 5 times more likely to cause rhabdomyolysis than a 20-mg dose.17 Yet, based on our patient’s case, even 20 mg/d simvastatin should not decrease physician suspicion for rhabdomyolysis if patients are also taking a CYP3A4 inhibitor.
Third, the simvastatin-fluconazole co-administration time in our patient was 12 weeks, which is longer than previously reported (TABLE 111-13). Azole inhibition of CYP450 occurs relatively rapidly, but that does not mean that rhabdomyolysis will always occur immediately. For example, in cases of statin monotherapy, rhabdomyolysis secondary to statin biochemical toxicity can occur up to 1050 (mean=348) days after the drug’s initiation.18
Avoiding a drug-drug interaction in your patient
Physicians can use pharmacokinetic profiles to choose among different statins and azoles to help avoid a drug interaction (TABLE 26,7,10,19). Pravastatin’s serum concentration, for example, is not influenced by CYP3A4 inhibitors such as itraconazole11 because pravastatin is metabolized by sulfation6 and not by the CYP450 system. Rosuvastatin and pitavastatin are minimally metabolized by the CYP450 system.19,20
Among approximately 2700 statin-treated outpatients,4 the prevalence of potentially harmful statin interactions with other drugs (including CYP3A4 inhibitors), was significantly higher among patients treated with simvastatin or atorvastatin (CYP3A4-metabolized statins), than among patients treated with fluvastatin (CYP2C9-metabolized statin) or pravastatin (metabolized by sulfation). Apart from drug-drug interactions, other risk factors for statin-induced rhabdomyolysis include use of lipophilic statins, advanced age, and female gender.21
We discontinued our patient’s simvastatin on the day she was admitted to the hospital, but continued with the fluconazole throughout her hospitalization. Her CK level continued to rise, peaked on hospital Day 3 at 32,886 U/L, and then progressively decreased. The patient’s weakness and pain improved and her acute kidney injury resolved with hydration. She was discharged on hospital Day 7 on oral fluconazole, but no statin, and her muscle symptoms have since resolved.
THE TAKEAWAY
When hyperlipidemic patients have to take an azole for an extended period (eg, cancer prophylaxis or chronic osteomyelitis) and the azole is a strong CYP450 inhibitor (eg, itraconazole), switching to a statin that is not primarily metabolized by the CYP450 system (eg, pravastatin, pitavastatin) is wise. If the azole is a moderate CYP450 inhibitor (eg, fluconazole), we suggest that therapy should be closely monitored. In the case of short-term azole treatment (eg, such as for oral candidiasis), the statin should be stopped or the dose reduced by at least 50% (eg, from 40 or 20 mg to 10 mg).6
Prescriber knowledge is sometimes a limiting factor in identifying clinically significant interactions.22 This is especially pertinent in a case like this one, where a lower statin dose may result in a lower chance of the pharmacist alerting the prescribing physician16 and when an azole is used that is a comparatively weaker CYP450 inhibitor than other azoles such as itraconazole. Even in the era of electronic medical records, approximately 90% of drug interaction alerts are overridden by physicians, and alert fatigue is pronounced.23
The intricacies and pharmacokinetic principles of this case should contribute to greater provider familiarity with even low-dose simvastatin-fluconazole interactions and help prevent iatrogenic complications such as rhabdomyolysis.
1. Kisakol G, Tunc R, Kaya A. Rhabdomyolysis in a patient with hypothyroidism. Endocr J. 2003;50:221-223.
2. Scott KR, Simmons Z, Boyer PJ. Hypothyroid myopathy with a strikingly elevated serum creatine kinase level. Muscle Nerve. 2002;26:141-144.
3. Barahona MJ, Mauri A, Sucunza N, et al. Hypothyroidism as a cause of rhabdomyolysis. Endocr J. 2002;49:621-623.
4. Rätz Bravo AE, Tchambaz L, Krähenbühl-Melcher A, et al. Prevalence of potentially severe drug-drug interactions in ambulatory patients with dyslipidaemia receiving HMG-CoA reductase inhibitor therapy. Drug Saf. 2005;28:263-275.
5. Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group, Armitage J, Bowman L, Wallendszus K, et al. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376:1658-1669.
6. Chong PH, Seeger JD, Franklin C. Clinically relevant differences between the statins: implications for therapeutic selection. Am J Med. 2001;111:390-400.
7. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism: clinical relevance. Clin Pharmacokinet. 2000;38:111-180.
8. Malhotra B, Dickins M, Alvey C, et al. Effects of the moderate CYP3A4 inhibitor, fluconazole, on the pharmacokinetics of fesoterodine in healthy subjects. Br J Clin Pharmacol. 2011;72:263-269.
9. US Food and Drug Administration. Drug development and drug interactions: Table of substrates, inhibitors and inducers. Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm. Accessed February 9, 2017.
10. Niwa T, Shiraga T, Takagi A. Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol Pharm Bull. 2005;28:1805-1808.
11. Shaukat A, Benekli M, Vladutiu GD, et al. Simvastatin-fluconazole causing rhabdomyolysis. Ann Pharmacother. 2003;37:1032-1035.
12. Hazin R, Abuzetun JY, Suker M, et al. Rhabdomyolysis induced by simvastatin-fluconazole combination. J Natl Med Assoc. 2008;100:444-446.
13. Findling O, Meier N, Sellner J, et al. Clinical reasoning: rhabdomyolysis after combined treatment with simvastatin and fluconazole. Neurology. 2008;71:e34-e37.
14. US Food and Drug Administration. FDA Drug Safety Communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. June 8, 2011. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm256581.htm. Accessed February 1, 2017.
15. Wolters Kluwer. Lexicomp online. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed February 9, 2017.
16. Molden E, Skovlund E, Braathen P. Risk management of simvastatin or atorvastatin interactions with CYP3A4 inhibitors. Drug Saf. 2008;31:587-596.
17. Parkin L, Paul C, Herbison GP. Simvastatin dose and risk of rhabdomyolysis: nested case-control study based on national health and drug dispensing data. Int J Cardiol. 2014;174:83-89.
18. Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292:2585-2590.
19. Saito Y. Pitavastatin: an overview. Atheroscler Suppl. 2011;12:271-276.
20. Olsson AG, McTaggart F, Raza A. Rosuvastatin: a highly effective new HMG-CoA reductase inhibitor. Cardiovasc Drug Rev. 2002;20:303-328.
21. Magni P, Macchi C, Morlotti B, et al. Risk identification and possible countermeasures for muscle adverse effects during statin therapy. Eur J Intern Med. 2015;26:82-88.
22. Ko Y, Malone DC, Skrepnek GH, et al. Prescribers’ knowledge of and sources of information for potential drug-drug interactions: a postal survey of US prescribers. Drug Saf. 2008;31:525-536.
23. Phansalkar S, van der Sijs H, Tucker AD, et al. Drug-drug interactions that should be non-interruptive in order to reduce alert fatigue in electronic health records. J Am Med Inform Assoc. 2013;20:489-493.
1. Kisakol G, Tunc R, Kaya A. Rhabdomyolysis in a patient with hypothyroidism. Endocr J. 2003;50:221-223.
2. Scott KR, Simmons Z, Boyer PJ. Hypothyroid myopathy with a strikingly elevated serum creatine kinase level. Muscle Nerve. 2002;26:141-144.
3. Barahona MJ, Mauri A, Sucunza N, et al. Hypothyroidism as a cause of rhabdomyolysis. Endocr J. 2002;49:621-623.
4. Rätz Bravo AE, Tchambaz L, Krähenbühl-Melcher A, et al. Prevalence of potentially severe drug-drug interactions in ambulatory patients with dyslipidaemia receiving HMG-CoA reductase inhibitor therapy. Drug Saf. 2005;28:263-275.
5. Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine (SEARCH) Collaborative Group, Armitage J, Bowman L, Wallendszus K, et al. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376:1658-1669.
6. Chong PH, Seeger JD, Franklin C. Clinically relevant differences between the statins: implications for therapeutic selection. Am J Med. 2001;111:390-400.
7. Venkatakrishnan K, von Moltke LL, Greenblatt DJ. Effects of the antifungal agents on oxidative drug metabolism: clinical relevance. Clin Pharmacokinet. 2000;38:111-180.
8. Malhotra B, Dickins M, Alvey C, et al. Effects of the moderate CYP3A4 inhibitor, fluconazole, on the pharmacokinetics of fesoterodine in healthy subjects. Br J Clin Pharmacol. 2011;72:263-269.
9. US Food and Drug Administration. Drug development and drug interactions: Table of substrates, inhibitors and inducers. Available at: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm. Accessed February 9, 2017.
10. Niwa T, Shiraga T, Takagi A. Effect of antifungal drugs on cytochrome P450 (CYP) 2C9, CYP2C19, and CYP3A4 activities in human liver microsomes. Biol Pharm Bull. 2005;28:1805-1808.
11. Shaukat A, Benekli M, Vladutiu GD, et al. Simvastatin-fluconazole causing rhabdomyolysis. Ann Pharmacother. 2003;37:1032-1035.
12. Hazin R, Abuzetun JY, Suker M, et al. Rhabdomyolysis induced by simvastatin-fluconazole combination. J Natl Med Assoc. 2008;100:444-446.
13. Findling O, Meier N, Sellner J, et al. Clinical reasoning: rhabdomyolysis after combined treatment with simvastatin and fluconazole. Neurology. 2008;71:e34-e37.
14. US Food and Drug Administration. FDA Drug Safety Communication: New restrictions, contraindications, and dose limitations for Zocor (simvastatin) to reduce the risk of muscle injury. June 8, 2011. Available at: http://www.fda.gov/Drugs/DrugSafety/ucm256581.htm. Accessed February 1, 2017.
15. Wolters Kluwer. Lexicomp online. Available at: http://www.wolterskluwercdi.com/lexicomp-online/. Accessed February 9, 2017.
16. Molden E, Skovlund E, Braathen P. Risk management of simvastatin or atorvastatin interactions with CYP3A4 inhibitors. Drug Saf. 2008;31:587-596.
17. Parkin L, Paul C, Herbison GP. Simvastatin dose and risk of rhabdomyolysis: nested case-control study based on national health and drug dispensing data. Int J Cardiol. 2014;174:83-89.
18. Graham DJ, Staffa JA, Shatin D, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA. 2004;292:2585-2590.
19. Saito Y. Pitavastatin: an overview. Atheroscler Suppl. 2011;12:271-276.
20. Olsson AG, McTaggart F, Raza A. Rosuvastatin: a highly effective new HMG-CoA reductase inhibitor. Cardiovasc Drug Rev. 2002;20:303-328.
21. Magni P, Macchi C, Morlotti B, et al. Risk identification and possible countermeasures for muscle adverse effects during statin therapy. Eur J Intern Med. 2015;26:82-88.
22. Ko Y, Malone DC, Skrepnek GH, et al. Prescribers’ knowledge of and sources of information for potential drug-drug interactions: a postal survey of US prescribers. Drug Saf. 2008;31:525-536.
23. Phansalkar S, van der Sijs H, Tucker AD, et al. Drug-drug interactions that should be non-interruptive in order to reduce alert fatigue in electronic health records. J Am Med Inform Assoc. 2013;20:489-493.
Allergic Reaction to Phenylephrine
Phenylephrine, a sympathomimetic drug, is commonly used in eye exams to dilate the pupil of the eye and to differentiate scleritis from episcleritis. Common adverse effects (AEs) of phenylephrine include subjective burning, stinging with lacrimation, rebound hyperemia, and liberation of iris pigment into the anterior chamber. Less common, systemic AEs include tachycardia and elevation of systemic blood pressure. Although instances of allergic reactions are rare, phenylephrine has been reported to cause contact dermatitis, blepharoconjunctivitis, and as in this case, keratoconjunctivitis.
Case Report
An 83-year-old white male presented for a red eye evaluation 2 days after having undergone a comprehensive eye exam with dilation at the Malcom Randall VAMC clinic in Gainesville, Florida. The patient reported onset of blurred vision, which he described as looking through a fog. He further compared the feeling to pins sticking in his eyes. The patient noted he had experienced similar symptoms on a few other occasions following eye exams. At the most recent eye exam, proparacaine and fluorescein had been used for tonometry, and phenylephrine 2.5% and tropicamide 0.5% had been used for pupillary dilation.
The patient’s best-corrected visual acuity was counting fingers at 2 feet in the right eye (OD) and left eye (OS). The best-corrected visual acuity 2 days prior had been 20/20 OD and OS. Pupils and extraocular motilities were unremarkable. Intraocular pressures were not obtained due to concern for a possible adverse reaction to proparacaine.
Slit-lamp evaluation revealed the lids to be lax, erythematous, and edematous in both eyes (Figure 1).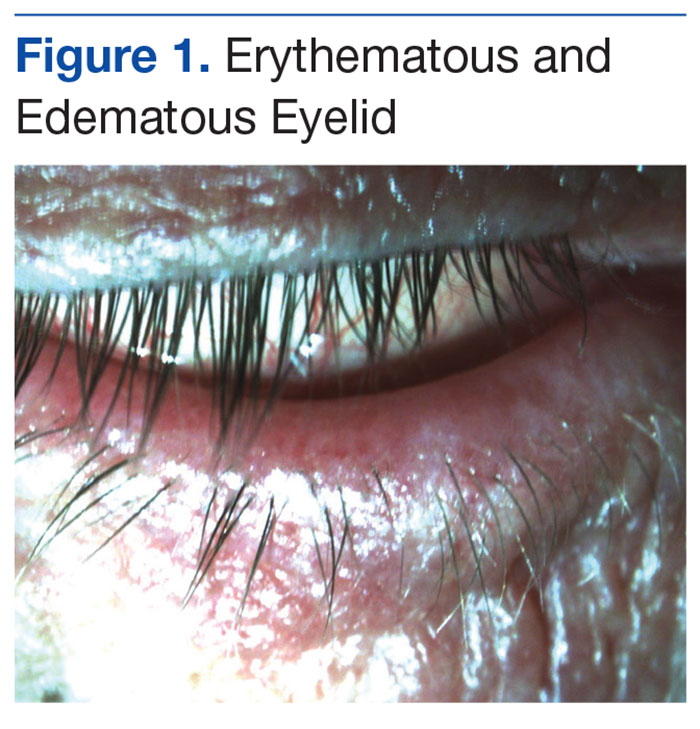

The initial diagnosis was acute chemical conjunctivitis most likely due to an AE to proparacaine. The plan was to start the patient on antibiotic eye drops qid OU, prednisolone qid OU, and artificial tears every hour OU. The patient was scheduled to return to clinic 4 days later for an anterior segment follow-up.
At the follow-up visit, the patient reported significant visual improvement. His best-corrected visual acuity was 20/40-2 without improvement on pinhole OD and 20/50-2 with improvement to 20/30+ on pinhole OS. Slit-lamp evaluation revealed 1+ bulbar conjunctival injection OU, intact corneal epithelium OU, and no cells or flare in the anterior chambers OU. Due to improving punctate epitheliopathy, the frequency of the antibiotic drops, the prednisolone, and the artificial tears was reduced to bid. After 3 days, he was instructed to discontinue them. The patient was scheduled to return in 2 weeks for an anterior segment follow-up.
At the next follow-up visit, the patient reported that his vision had returned to normal, and he had no further ocular AEs. His best-corrected visual acuity was 20/20-2 OD and 20/20 OS. Slit-lamp evaluation revealed mild blepharitis OU, trace bulbar conjunctival injection OU, and complete resolution of the keratitis OU. The assessment was acute allergic conjunctivitis thought to be secondary to an AE to proparacaine OU, yet the need to rule out hypersensitivity to tropicamide and/or phenylephrine remained. The plan was to educate the patient of the possibility of allergic reaction on future visits and to recommend continued use of artificial tears as needed.
Through a careful and extensive chart review of all past visits, it was suspected that phenylephrine might be to blame rather than proparacaine. At the subsequent visit, the patient agreed to undergo testing to determine the culprit via instillation of proparacaine in one eye and tropicamide in the other. The patient had no reaction to either drop (checked 45 minutes after instillation and the following day). By process of elimination, phenylephrine was determined to be the offending agent.
Discussion
Following a thorough review of the patient’s chart, it was found that on other occasions he had presented with suspected allergic reactions following routine eye examinations. The patient reported he had experienced a reaction in 2007 but could not recall what drops were instilled in his eyes at the time. In addition, there was no documentation in his medical record of the subsequent reaction following that visit. Another reaction occurred in July 2010 with instillation of tropicamide 1%, phenylephrine 2.5%, and Fluress (fluorescein sodium and benoxinate hydrochloride ophthalmic solution USP). In October 2013, when tropicamide 0.5%, proparacaine, and fluorescein strips were instilled, there was no reaction. The next reaction occurred in October 2014, when tropicamide 0.5%, phenylephrine 2.5%, proparacaine, and fluorescein strips were instilled.
This careful review of past exam notes revealed that phenylephrine and Fluress were the only drops that had not been instilled at the October 2013 visit when no AE was reported. However, Fluress was an unlikely culprit since it was not instilled in October 2014, and the patient still experienced an AE. Therefore, the agent most likely responsible for the allergic reaction in the patient, as confirmed by a review of the past notes and by the aforementioned pharmacologic test, was deemed to be phenylephrine (Table).
Adverse reactions to topical ocular medications and specifically to diagnostic eye drops have long been recognized. Mathias, Camarasa, Barber, Ducombs, and Monsálvezhave reported on variations of conjunctivitis and periorbital erythema with positive patch testing to phenylephrine.1-5 Geyer and colleagues reported on a study of 21 patients who had blepharoconjunctivitis after instillation of phenylephrine.6 In this case study patient, severe keratoconjunctivitis was the clinical manifestation observed.
Villarreal and colleagues studied 31 patients who had a previous reaction to mydriatic drops. The study found that phenylephrine was the drug that most frequently caused an AE (93.5%).7 One patient reacted to the preservative thimerosal, and 1 patient reacted to benoxiprocaine. Tropicamide was demonstrated to be very well tolerated as none of the patients tested positive on either the patch test or the pharmacologic test.
Tropicamide is a nonselective muscarinic antagonist commonly used for mydriasis due to its fast onset and short duration.8 Adverse reactions to tropicamide are rare. Three studies reported on patients who had a positive patch test to tropicamide.9-11 However, the reaction was not provoked by direct instillation of tropicamide into the eye.
Common in-office topical anesthetics, proparacaine, tetracaine, benoxinate, and lidocaine also can cause AEs. Corneal toxicity is a well-known complication with topical anesthetic abuse, whereas allergic reactions are considered rare. The most common symptoms include stingingand discomfort upon instillation. Common signs include punctate corneal epithelial erosionsresulting indirectly from a decrease in reflex tearing, infrequent blinking, and increased tear evaporation.12 Topical anesthetics also inhibit the migration of corneal epithelial cells and cause direct damage to the cells that are present, leading to impaired healing and epithelial defects.13
Manifestations of allergic reaction to topical anesthetics can include conjunctival hyperemia and edema, edematous eyelids, and lacrimation. One published case described a 60-year-old woman who developed eczematous dermatitis of the eyelids after ophthalmic anesthetic drops were instilled prior to laser surgery. Patch testing showed a positive response to benzocaine 5%, proparacaine, and tetracaine 0.5%.14
Preservatives, in general, can cause an allergic reaction. Benzalkonium chloride’s (BAK) cytotoxic sequelae include possible trabecular cell death in glaucoma patients, disruption of tear film stability (even at low concentrations), and immune-allergenic properties. One article reported BAK as one of the 30 most frequent allergens causing allergic periorbital dermatitis.15 Benzalkonium chloride is used in most brands of phenylephrine. However, preservatives in this patient’s case were ruled out as instigating agents since both phenylephrine and tropicamide contain the same preservative, BAK 0.01%, yet this patient did not develop a reaction to tropicamide when used without phenylephrine. Expired medications also were not considered to be a factor as none of the medications used on the patient were indeed expired (the Malcom Randall VAMC clinic maintains a strict policy of discarding medications 28 days after being opened).
Although uncommon, phenylephrine sometimes has been found to cause a type 4 hypersensitivity reaction, also known as cell-mediated or delayed-type hypersensitivity.16 First, helper T cells secrete cytokines. Activation of cytokines recruits and activates cytotoxic T cells, monocytes, and macrophages, leading to inflammation of the surrounding tissue. Examples of cell-mediated hypersensitivity include reactions to the tuberculin skin test and to poison ivy.
Type 1 hypersensitivity reactions, also known as immediate or anaphylactic hypersensitivity reactions, are not triggered by phenylephrine. In this type of reaction, IgE binds to the mast cell on initial exposure to an allergen. On second exposure, the allergen binds to the IgE, causing the mast cell to release mediators of inflammation, triggering physiologic responses. Examples of this type of hypersensitivity include those seen with penicillin, bee stings, hay fever, bronchial asthma, and food allergies, for example, to shellfish.
A toxic reaction’s mechanism differs from that of a type 4 hypersensitivity reaction. Toxic reactions occur due to direct cytotoxicity of a drug caused by a low or high pH and either hyper- or hypo-osmolarity. Toxicity can lead to corneal and conjunctival cell necrosis or induce apoptosis, stimulating inflammatory reactions. Clinically, toxic reactions will present with follicles, whereas allergic reactions will present with papillae.
The definitive diagnostic methods used to determine the allergic agent causing ocular or periocular AEs are patch testing and conjunctival challenge.7 Mathias, Camarasa, Barber, Ducombs,and Monsálvezused patch testing to confirm phenylephrine as the allergic agent in their series of cases. Patch testing entails the application of a small amount of an allergic agent that is taped onto the skin. The allergic agent is confirmed if the patient has a dermal reaction, wherein the area patched will become erythematous. When patch testing is negative or inconclusive, a conjunctival challenge is performed by instillation of the suspected allergic agent into the eye with subsequent observation to determine whether a reaction occurs. The sequelae found in Villarreal’s study included itching, lacrimation, edema, erythema, and sometimes blepharitis.7
A direct conjunctival challenge with the suspected culprit was not pursued in this patient’s case due to the known severity of the potential resulting reaction. The authors instead chose an indirect method of determining the implicating agent and used the process of elimination to whittle down the most likely suspect. A challenge with the medications suspected not to be likely offenders was undertaken. This spared the patient a likely repeat of the AE he had just recovered from.
Management
Allergic reactions can resolve without medical intervention. The first step is to remove the allergen. For delayed hypersensitivity reactions, treatments may include topical decongestants, cool compresses, and corticosteroids.8 The treatment for immediate hypersensitivity reaction differs from that of delayed hypersensitivity reaction in that antihistamines are used.17,18
This patient reported receiving no treatment for his ocular symptoms following eye examinations in the past, yet he experienced complete resolution after each AE. In this case, both a steroid and a prophylactic antibiotic to facilitate a more rapid improvement were used.
Conclusion
Although uncommon, cases of allergic reaction to phenylephrine can occur. The incidence of phenylephrine allergy is 0.6%.6 The case study patient presented with a severe keratoconjunctivitis following routine eye examination with an accompanying history of adverse ocular signs and symptoms following multiple past exams.
It is important for all eye care clinicians to realize that AEs to diagnostic eye drops are possible and can occur following the most routine of visits. Such reactions can be caused by dilating agents, anesthetics, or preservatives, and these may be allergic or toxic. Clinicians should take special care to identify the instigating agent, and if possible, to avoid using such agents on patients during future exams. Clinicians also should understand how best to manage iatrogenic AEs when they encounter them in order to restore a patient’s visual function as quickly as possible.
1. Mathias CG, Maibach HI, Irvine A, Adler W. Allergic contact dermatitis to echothiophate iodide and phenylephrine. Arch Ophthalmol. 1979;97(2):286-287.
2. Camarasa JG. Contact dermatitis to phenylephrine. Contact Dermatitis. 1984;10(3):182.
3. Barber K. Allergic contact eczema to phenylephrine. Contact Dermatitis. 1983;9(4):274-277.
4. Ducombs G, de Casamayor J, Verin P, Maleville J. Allergic contact dermatitis to phenylephrine. Contact Dermatitis. 1986;15(2):107-108.
5. Monsálvez V, Fuertes L, García-Cano I, Vanaclocha F, Ortez de Frutos J. Blepharoconjunctivitis due to phenylephrine [in Spanish]. Actas Dermosifiliogr. 2010;101(5):466-467.
6. Geyer O, Yust I, Lazar M. Allergic blepharoconjunctivitis due to phenylephrine. J Ocul Pharmacol. 1988;4(2):123-126.
7. Villarreal O. Reliability of diagnostic tests for contact allergy to mydriatic eyedrops. Contact Dermatitis. 1998;38(3):150-154.
8. Frazier M, Jaanus SD. Cycloplegics. In: Bartlett JD, Jaanus SD. Clinical Ocular Pharmacology. 5th ed. St. Louis, MO: Butterworth-Heinemann; 2009:125-138.
9. Decraene T, Goossens A. Contact allergy to atropine and other mydriatic agents in eye drops. Contact Dermatitis. 2001;45(5):309-310.
10. Boukhman MP, Maibach HI. Allergic contact dermatitis from tropicamide ophthalmic solution. Contact Dermatitis. 1999;41(1):47-48.
11. Yoshikawa K, Kawahara S. Contact allergy to atropine and other mydriatic agents. Contact Dermatitis. 1985;12(1):56-57.
12. Mcgee HT, Fraunfelder FW. Toxicities of topical ophthalmic anesthetics. Expert Opin Drug Saf. 2007;6(6):637-640.
13. Dass BA, Soong HK, Lee B. Effects of proparacaine of actin cytoskeleton of corneal epithelium. J Ocul Pharmacol. 1988;4(3):187-194.
14. Dannaker CJ, Maibach HI, Austin E. Allergic contact dermatitis to proparacaine with subsequent cross-sensitization to tetracaine from ophthalmic preparations. Am J Contact Dermat. 2001;12(3):177-179.
15. Hong J, Bielory L. Allergy to ophthalmic preservatives. Curr Opin Allergy Clin Immunol. 2009;9(5):447-453.
16. Gonzalo-Garijo MA, Pérez-Calderón R, de Argila D, Rodríguez-Nevado I. Erythrodermia to pseudoephedrine in a patient with contact allergy to phenylephrine. Allergol Immunopathol (Madr). 2002;30(4):239-242.
17. Platts-Mills TAE. Immediate hypersensitivity (Type I). In: Male D, Brostoff J, Roth DB, Roitt I. Immunology. 7th ed. Canada: Elsevier Limited; 2006:423-446.
18. Britton W. Type IV hypersensitivity. In: Male D, Brostoff J, Roth DB, Roitt I. Immunology. 7th ed. Canada: Elsevier Limited; 2006:477-491.
Phenylephrine, a sympathomimetic drug, is commonly used in eye exams to dilate the pupil of the eye and to differentiate scleritis from episcleritis. Common adverse effects (AEs) of phenylephrine include subjective burning, stinging with lacrimation, rebound hyperemia, and liberation of iris pigment into the anterior chamber. Less common, systemic AEs include tachycardia and elevation of systemic blood pressure. Although instances of allergic reactions are rare, phenylephrine has been reported to cause contact dermatitis, blepharoconjunctivitis, and as in this case, keratoconjunctivitis.
Case Report
An 83-year-old white male presented for a red eye evaluation 2 days after having undergone a comprehensive eye exam with dilation at the Malcom Randall VAMC clinic in Gainesville, Florida. The patient reported onset of blurred vision, which he described as looking through a fog. He further compared the feeling to pins sticking in his eyes. The patient noted he had experienced similar symptoms on a few other occasions following eye exams. At the most recent eye exam, proparacaine and fluorescein had been used for tonometry, and phenylephrine 2.5% and tropicamide 0.5% had been used for pupillary dilation.
The patient’s best-corrected visual acuity was counting fingers at 2 feet in the right eye (OD) and left eye (OS). The best-corrected visual acuity 2 days prior had been 20/20 OD and OS. Pupils and extraocular motilities were unremarkable. Intraocular pressures were not obtained due to concern for a possible adverse reaction to proparacaine.
Slit-lamp evaluation revealed the lids to be lax, erythematous, and edematous in both eyes (Figure 1).
The initial diagnosis was acute chemical conjunctivitis most likely due to an AE to proparacaine. The plan was to start the patient on antibiotic eye drops qid OU, prednisolone qid OU, and artificial tears every hour OU. The patient was scheduled to return to clinic 4 days later for an anterior segment follow-up.
At the follow-up visit, the patient reported significant visual improvement. His best-corrected visual acuity was 20/40-2 without improvement on pinhole OD and 20/50-2 with improvement to 20/30+ on pinhole OS. Slit-lamp evaluation revealed 1+ bulbar conjunctival injection OU, intact corneal epithelium OU, and no cells or flare in the anterior chambers OU. Due to improving punctate epitheliopathy, the frequency of the antibiotic drops, the prednisolone, and the artificial tears was reduced to bid. After 3 days, he was instructed to discontinue them. The patient was scheduled to return in 2 weeks for an anterior segment follow-up.
At the next follow-up visit, the patient reported that his vision had returned to normal, and he had no further ocular AEs. His best-corrected visual acuity was 20/20-2 OD and 20/20 OS. Slit-lamp evaluation revealed mild blepharitis OU, trace bulbar conjunctival injection OU, and complete resolution of the keratitis OU. The assessment was acute allergic conjunctivitis thought to be secondary to an AE to proparacaine OU, yet the need to rule out hypersensitivity to tropicamide and/or phenylephrine remained. The plan was to educate the patient of the possibility of allergic reaction on future visits and to recommend continued use of artificial tears as needed.
Through a careful and extensive chart review of all past visits, it was suspected that phenylephrine might be to blame rather than proparacaine. At the subsequent visit, the patient agreed to undergo testing to determine the culprit via instillation of proparacaine in one eye and tropicamide in the other. The patient had no reaction to either drop (checked 45 minutes after instillation and the following day). By process of elimination, phenylephrine was determined to be the offending agent.
Discussion
Following a thorough review of the patient’s chart, it was found that on other occasions he had presented with suspected allergic reactions following routine eye examinations. The patient reported he had experienced a reaction in 2007 but could not recall what drops were instilled in his eyes at the time. In addition, there was no documentation in his medical record of the subsequent reaction following that visit. Another reaction occurred in July 2010 with instillation of tropicamide 1%, phenylephrine 2.5%, and Fluress (fluorescein sodium and benoxinate hydrochloride ophthalmic solution USP). In October 2013, when tropicamide 0.5%, proparacaine, and fluorescein strips were instilled, there was no reaction. The next reaction occurred in October 2014, when tropicamide 0.5%, phenylephrine 2.5%, proparacaine, and fluorescein strips were instilled.
This careful review of past exam notes revealed that phenylephrine and Fluress were the only drops that had not been instilled at the October 2013 visit when no AE was reported. However, Fluress was an unlikely culprit since it was not instilled in October 2014, and the patient still experienced an AE. Therefore, the agent most likely responsible for the allergic reaction in the patient, as confirmed by a review of the past notes and by the aforementioned pharmacologic test, was deemed to be phenylephrine (Table).
Adverse reactions to topical ocular medications and specifically to diagnostic eye drops have long been recognized. Mathias, Camarasa, Barber, Ducombs, and Monsálvezhave reported on variations of conjunctivitis and periorbital erythema with positive patch testing to phenylephrine.1-5 Geyer and colleagues reported on a study of 21 patients who had blepharoconjunctivitis after instillation of phenylephrine.6 In this case study patient, severe keratoconjunctivitis was the clinical manifestation observed.
Villarreal and colleagues studied 31 patients who had a previous reaction to mydriatic drops. The study found that phenylephrine was the drug that most frequently caused an AE (93.5%).7 One patient reacted to the preservative thimerosal, and 1 patient reacted to benoxiprocaine. Tropicamide was demonstrated to be very well tolerated as none of the patients tested positive on either the patch test or the pharmacologic test.
Tropicamide is a nonselective muscarinic antagonist commonly used for mydriasis due to its fast onset and short duration.8 Adverse reactions to tropicamide are rare. Three studies reported on patients who had a positive patch test to tropicamide.9-11 However, the reaction was not provoked by direct instillation of tropicamide into the eye.
Common in-office topical anesthetics, proparacaine, tetracaine, benoxinate, and lidocaine also can cause AEs. Corneal toxicity is a well-known complication with topical anesthetic abuse, whereas allergic reactions are considered rare. The most common symptoms include stingingand discomfort upon instillation. Common signs include punctate corneal epithelial erosionsresulting indirectly from a decrease in reflex tearing, infrequent blinking, and increased tear evaporation.12 Topical anesthetics also inhibit the migration of corneal epithelial cells and cause direct damage to the cells that are present, leading to impaired healing and epithelial defects.13
Manifestations of allergic reaction to topical anesthetics can include conjunctival hyperemia and edema, edematous eyelids, and lacrimation. One published case described a 60-year-old woman who developed eczematous dermatitis of the eyelids after ophthalmic anesthetic drops were instilled prior to laser surgery. Patch testing showed a positive response to benzocaine 5%, proparacaine, and tetracaine 0.5%.14
Preservatives, in general, can cause an allergic reaction. Benzalkonium chloride’s (BAK) cytotoxic sequelae include possible trabecular cell death in glaucoma patients, disruption of tear film stability (even at low concentrations), and immune-allergenic properties. One article reported BAK as one of the 30 most frequent allergens causing allergic periorbital dermatitis.15 Benzalkonium chloride is used in most brands of phenylephrine. However, preservatives in this patient’s case were ruled out as instigating agents since both phenylephrine and tropicamide contain the same preservative, BAK 0.01%, yet this patient did not develop a reaction to tropicamide when used without phenylephrine. Expired medications also were not considered to be a factor as none of the medications used on the patient were indeed expired (the Malcom Randall VAMC clinic maintains a strict policy of discarding medications 28 days after being opened).
Although uncommon, phenylephrine sometimes has been found to cause a type 4 hypersensitivity reaction, also known as cell-mediated or delayed-type hypersensitivity.16 First, helper T cells secrete cytokines. Activation of cytokines recruits and activates cytotoxic T cells, monocytes, and macrophages, leading to inflammation of the surrounding tissue. Examples of cell-mediated hypersensitivity include reactions to the tuberculin skin test and to poison ivy.
Type 1 hypersensitivity reactions, also known as immediate or anaphylactic hypersensitivity reactions, are not triggered by phenylephrine. In this type of reaction, IgE binds to the mast cell on initial exposure to an allergen. On second exposure, the allergen binds to the IgE, causing the mast cell to release mediators of inflammation, triggering physiologic responses. Examples of this type of hypersensitivity include those seen with penicillin, bee stings, hay fever, bronchial asthma, and food allergies, for example, to shellfish.
A toxic reaction’s mechanism differs from that of a type 4 hypersensitivity reaction. Toxic reactions occur due to direct cytotoxicity of a drug caused by a low or high pH and either hyper- or hypo-osmolarity. Toxicity can lead to corneal and conjunctival cell necrosis or induce apoptosis, stimulating inflammatory reactions. Clinically, toxic reactions will present with follicles, whereas allergic reactions will present with papillae.
The definitive diagnostic methods used to determine the allergic agent causing ocular or periocular AEs are patch testing and conjunctival challenge.7 Mathias, Camarasa, Barber, Ducombs,and Monsálvezused patch testing to confirm phenylephrine as the allergic agent in their series of cases. Patch testing entails the application of a small amount of an allergic agent that is taped onto the skin. The allergic agent is confirmed if the patient has a dermal reaction, wherein the area patched will become erythematous. When patch testing is negative or inconclusive, a conjunctival challenge is performed by instillation of the suspected allergic agent into the eye with subsequent observation to determine whether a reaction occurs. The sequelae found in Villarreal’s study included itching, lacrimation, edema, erythema, and sometimes blepharitis.7
A direct conjunctival challenge with the suspected culprit was not pursued in this patient’s case due to the known severity of the potential resulting reaction. The authors instead chose an indirect method of determining the implicating agent and used the process of elimination to whittle down the most likely suspect. A challenge with the medications suspected not to be likely offenders was undertaken. This spared the patient a likely repeat of the AE he had just recovered from.
Management
Allergic reactions can resolve without medical intervention. The first step is to remove the allergen. For delayed hypersensitivity reactions, treatments may include topical decongestants, cool compresses, and corticosteroids.8 The treatment for immediate hypersensitivity reaction differs from that of delayed hypersensitivity reaction in that antihistamines are used.17,18
This patient reported receiving no treatment for his ocular symptoms following eye examinations in the past, yet he experienced complete resolution after each AE. In this case, both a steroid and a prophylactic antibiotic to facilitate a more rapid improvement were used.
Conclusion
Although uncommon, cases of allergic reaction to phenylephrine can occur. The incidence of phenylephrine allergy is 0.6%.6 The case study patient presented with a severe keratoconjunctivitis following routine eye examination with an accompanying history of adverse ocular signs and symptoms following multiple past exams.
It is important for all eye care clinicians to realize that AEs to diagnostic eye drops are possible and can occur following the most routine of visits. Such reactions can be caused by dilating agents, anesthetics, or preservatives, and these may be allergic or toxic. Clinicians should take special care to identify the instigating agent, and if possible, to avoid using such agents on patients during future exams. Clinicians also should understand how best to manage iatrogenic AEs when they encounter them in order to restore a patient’s visual function as quickly as possible.
Phenylephrine, a sympathomimetic drug, is commonly used in eye exams to dilate the pupil of the eye and to differentiate scleritis from episcleritis. Common adverse effects (AEs) of phenylephrine include subjective burning, stinging with lacrimation, rebound hyperemia, and liberation of iris pigment into the anterior chamber. Less common, systemic AEs include tachycardia and elevation of systemic blood pressure. Although instances of allergic reactions are rare, phenylephrine has been reported to cause contact dermatitis, blepharoconjunctivitis, and as in this case, keratoconjunctivitis.
Case Report
An 83-year-old white male presented for a red eye evaluation 2 days after having undergone a comprehensive eye exam with dilation at the Malcom Randall VAMC clinic in Gainesville, Florida. The patient reported onset of blurred vision, which he described as looking through a fog. He further compared the feeling to pins sticking in his eyes. The patient noted he had experienced similar symptoms on a few other occasions following eye exams. At the most recent eye exam, proparacaine and fluorescein had been used for tonometry, and phenylephrine 2.5% and tropicamide 0.5% had been used for pupillary dilation.
The patient’s best-corrected visual acuity was counting fingers at 2 feet in the right eye (OD) and left eye (OS). The best-corrected visual acuity 2 days prior had been 20/20 OD and OS. Pupils and extraocular motilities were unremarkable. Intraocular pressures were not obtained due to concern for a possible adverse reaction to proparacaine.
Slit-lamp evaluation revealed the lids to be lax, erythematous, and edematous in both eyes (Figure 1).
The initial diagnosis was acute chemical conjunctivitis most likely due to an AE to proparacaine. The plan was to start the patient on antibiotic eye drops qid OU, prednisolone qid OU, and artificial tears every hour OU. The patient was scheduled to return to clinic 4 days later for an anterior segment follow-up.
At the follow-up visit, the patient reported significant visual improvement. His best-corrected visual acuity was 20/40-2 without improvement on pinhole OD and 20/50-2 with improvement to 20/30+ on pinhole OS. Slit-lamp evaluation revealed 1+ bulbar conjunctival injection OU, intact corneal epithelium OU, and no cells or flare in the anterior chambers OU. Due to improving punctate epitheliopathy, the frequency of the antibiotic drops, the prednisolone, and the artificial tears was reduced to bid. After 3 days, he was instructed to discontinue them. The patient was scheduled to return in 2 weeks for an anterior segment follow-up.
At the next follow-up visit, the patient reported that his vision had returned to normal, and he had no further ocular AEs. His best-corrected visual acuity was 20/20-2 OD and 20/20 OS. Slit-lamp evaluation revealed mild blepharitis OU, trace bulbar conjunctival injection OU, and complete resolution of the keratitis OU. The assessment was acute allergic conjunctivitis thought to be secondary to an AE to proparacaine OU, yet the need to rule out hypersensitivity to tropicamide and/or phenylephrine remained. The plan was to educate the patient of the possibility of allergic reaction on future visits and to recommend continued use of artificial tears as needed.
Through a careful and extensive chart review of all past visits, it was suspected that phenylephrine might be to blame rather than proparacaine. At the subsequent visit, the patient agreed to undergo testing to determine the culprit via instillation of proparacaine in one eye and tropicamide in the other. The patient had no reaction to either drop (checked 45 minutes after instillation and the following day). By process of elimination, phenylephrine was determined to be the offending agent.
Discussion
Following a thorough review of the patient’s chart, it was found that on other occasions he had presented with suspected allergic reactions following routine eye examinations. The patient reported he had experienced a reaction in 2007 but could not recall what drops were instilled in his eyes at the time. In addition, there was no documentation in his medical record of the subsequent reaction following that visit. Another reaction occurred in July 2010 with instillation of tropicamide 1%, phenylephrine 2.5%, and Fluress (fluorescein sodium and benoxinate hydrochloride ophthalmic solution USP). In October 2013, when tropicamide 0.5%, proparacaine, and fluorescein strips were instilled, there was no reaction. The next reaction occurred in October 2014, when tropicamide 0.5%, phenylephrine 2.5%, proparacaine, and fluorescein strips were instilled.
This careful review of past exam notes revealed that phenylephrine and Fluress were the only drops that had not been instilled at the October 2013 visit when no AE was reported. However, Fluress was an unlikely culprit since it was not instilled in October 2014, and the patient still experienced an AE. Therefore, the agent most likely responsible for the allergic reaction in the patient, as confirmed by a review of the past notes and by the aforementioned pharmacologic test, was deemed to be phenylephrine (Table).
Adverse reactions to topical ocular medications and specifically to diagnostic eye drops have long been recognized. Mathias, Camarasa, Barber, Ducombs, and Monsálvezhave reported on variations of conjunctivitis and periorbital erythema with positive patch testing to phenylephrine.1-5 Geyer and colleagues reported on a study of 21 patients who had blepharoconjunctivitis after instillation of phenylephrine.6 In this case study patient, severe keratoconjunctivitis was the clinical manifestation observed.
Villarreal and colleagues studied 31 patients who had a previous reaction to mydriatic drops. The study found that phenylephrine was the drug that most frequently caused an AE (93.5%).7 One patient reacted to the preservative thimerosal, and 1 patient reacted to benoxiprocaine. Tropicamide was demonstrated to be very well tolerated as none of the patients tested positive on either the patch test or the pharmacologic test.
Tropicamide is a nonselective muscarinic antagonist commonly used for mydriasis due to its fast onset and short duration.8 Adverse reactions to tropicamide are rare. Three studies reported on patients who had a positive patch test to tropicamide.9-11 However, the reaction was not provoked by direct instillation of tropicamide into the eye.
Common in-office topical anesthetics, proparacaine, tetracaine, benoxinate, and lidocaine also can cause AEs. Corneal toxicity is a well-known complication with topical anesthetic abuse, whereas allergic reactions are considered rare. The most common symptoms include stingingand discomfort upon instillation. Common signs include punctate corneal epithelial erosionsresulting indirectly from a decrease in reflex tearing, infrequent blinking, and increased tear evaporation.12 Topical anesthetics also inhibit the migration of corneal epithelial cells and cause direct damage to the cells that are present, leading to impaired healing and epithelial defects.13
Manifestations of allergic reaction to topical anesthetics can include conjunctival hyperemia and edema, edematous eyelids, and lacrimation. One published case described a 60-year-old woman who developed eczematous dermatitis of the eyelids after ophthalmic anesthetic drops were instilled prior to laser surgery. Patch testing showed a positive response to benzocaine 5%, proparacaine, and tetracaine 0.5%.14
Preservatives, in general, can cause an allergic reaction. Benzalkonium chloride’s (BAK) cytotoxic sequelae include possible trabecular cell death in glaucoma patients, disruption of tear film stability (even at low concentrations), and immune-allergenic properties. One article reported BAK as one of the 30 most frequent allergens causing allergic periorbital dermatitis.15 Benzalkonium chloride is used in most brands of phenylephrine. However, preservatives in this patient’s case were ruled out as instigating agents since both phenylephrine and tropicamide contain the same preservative, BAK 0.01%, yet this patient did not develop a reaction to tropicamide when used without phenylephrine. Expired medications also were not considered to be a factor as none of the medications used on the patient were indeed expired (the Malcom Randall VAMC clinic maintains a strict policy of discarding medications 28 days after being opened).
Although uncommon, phenylephrine sometimes has been found to cause a type 4 hypersensitivity reaction, also known as cell-mediated or delayed-type hypersensitivity.16 First, helper T cells secrete cytokines. Activation of cytokines recruits and activates cytotoxic T cells, monocytes, and macrophages, leading to inflammation of the surrounding tissue. Examples of cell-mediated hypersensitivity include reactions to the tuberculin skin test and to poison ivy.
Type 1 hypersensitivity reactions, also known as immediate or anaphylactic hypersensitivity reactions, are not triggered by phenylephrine. In this type of reaction, IgE binds to the mast cell on initial exposure to an allergen. On second exposure, the allergen binds to the IgE, causing the mast cell to release mediators of inflammation, triggering physiologic responses. Examples of this type of hypersensitivity include those seen with penicillin, bee stings, hay fever, bronchial asthma, and food allergies, for example, to shellfish.
A toxic reaction’s mechanism differs from that of a type 4 hypersensitivity reaction. Toxic reactions occur due to direct cytotoxicity of a drug caused by a low or high pH and either hyper- or hypo-osmolarity. Toxicity can lead to corneal and conjunctival cell necrosis or induce apoptosis, stimulating inflammatory reactions. Clinically, toxic reactions will present with follicles, whereas allergic reactions will present with papillae.
The definitive diagnostic methods used to determine the allergic agent causing ocular or periocular AEs are patch testing and conjunctival challenge.7 Mathias, Camarasa, Barber, Ducombs,and Monsálvezused patch testing to confirm phenylephrine as the allergic agent in their series of cases. Patch testing entails the application of a small amount of an allergic agent that is taped onto the skin. The allergic agent is confirmed if the patient has a dermal reaction, wherein the area patched will become erythematous. When patch testing is negative or inconclusive, a conjunctival challenge is performed by instillation of the suspected allergic agent into the eye with subsequent observation to determine whether a reaction occurs. The sequelae found in Villarreal’s study included itching, lacrimation, edema, erythema, and sometimes blepharitis.7
A direct conjunctival challenge with the suspected culprit was not pursued in this patient’s case due to the known severity of the potential resulting reaction. The authors instead chose an indirect method of determining the implicating agent and used the process of elimination to whittle down the most likely suspect. A challenge with the medications suspected not to be likely offenders was undertaken. This spared the patient a likely repeat of the AE he had just recovered from.
Management
Allergic reactions can resolve without medical intervention. The first step is to remove the allergen. For delayed hypersensitivity reactions, treatments may include topical decongestants, cool compresses, and corticosteroids.8 The treatment for immediate hypersensitivity reaction differs from that of delayed hypersensitivity reaction in that antihistamines are used.17,18
This patient reported receiving no treatment for his ocular symptoms following eye examinations in the past, yet he experienced complete resolution after each AE. In this case, both a steroid and a prophylactic antibiotic to facilitate a more rapid improvement were used.
Conclusion
Although uncommon, cases of allergic reaction to phenylephrine can occur. The incidence of phenylephrine allergy is 0.6%.6 The case study patient presented with a severe keratoconjunctivitis following routine eye examination with an accompanying history of adverse ocular signs and symptoms following multiple past exams.
It is important for all eye care clinicians to realize that AEs to diagnostic eye drops are possible and can occur following the most routine of visits. Such reactions can be caused by dilating agents, anesthetics, or preservatives, and these may be allergic or toxic. Clinicians should take special care to identify the instigating agent, and if possible, to avoid using such agents on patients during future exams. Clinicians also should understand how best to manage iatrogenic AEs when they encounter them in order to restore a patient’s visual function as quickly as possible.
1. Mathias CG, Maibach HI, Irvine A, Adler W. Allergic contact dermatitis to echothiophate iodide and phenylephrine. Arch Ophthalmol. 1979;97(2):286-287.
2. Camarasa JG. Contact dermatitis to phenylephrine. Contact Dermatitis. 1984;10(3):182.
3. Barber K. Allergic contact eczema to phenylephrine. Contact Dermatitis. 1983;9(4):274-277.
4. Ducombs G, de Casamayor J, Verin P, Maleville J. Allergic contact dermatitis to phenylephrine. Contact Dermatitis. 1986;15(2):107-108.
5. Monsálvez V, Fuertes L, García-Cano I, Vanaclocha F, Ortez de Frutos J. Blepharoconjunctivitis due to phenylephrine [in Spanish]. Actas Dermosifiliogr. 2010;101(5):466-467.
6. Geyer O, Yust I, Lazar M. Allergic blepharoconjunctivitis due to phenylephrine. J Ocul Pharmacol. 1988;4(2):123-126.
7. Villarreal O. Reliability of diagnostic tests for contact allergy to mydriatic eyedrops. Contact Dermatitis. 1998;38(3):150-154.
8. Frazier M, Jaanus SD. Cycloplegics. In: Bartlett JD, Jaanus SD. Clinical Ocular Pharmacology. 5th ed. St. Louis, MO: Butterworth-Heinemann; 2009:125-138.
9. Decraene T, Goossens A. Contact allergy to atropine and other mydriatic agents in eye drops. Contact Dermatitis. 2001;45(5):309-310.
10. Boukhman MP, Maibach HI. Allergic contact dermatitis from tropicamide ophthalmic solution. Contact Dermatitis. 1999;41(1):47-48.
11. Yoshikawa K, Kawahara S. Contact allergy to atropine and other mydriatic agents. Contact Dermatitis. 1985;12(1):56-57.
12. Mcgee HT, Fraunfelder FW. Toxicities of topical ophthalmic anesthetics. Expert Opin Drug Saf. 2007;6(6):637-640.
13. Dass BA, Soong HK, Lee B. Effects of proparacaine of actin cytoskeleton of corneal epithelium. J Ocul Pharmacol. 1988;4(3):187-194.
14. Dannaker CJ, Maibach HI, Austin E. Allergic contact dermatitis to proparacaine with subsequent cross-sensitization to tetracaine from ophthalmic preparations. Am J Contact Dermat. 2001;12(3):177-179.
15. Hong J, Bielory L. Allergy to ophthalmic preservatives. Curr Opin Allergy Clin Immunol. 2009;9(5):447-453.
16. Gonzalo-Garijo MA, Pérez-Calderón R, de Argila D, Rodríguez-Nevado I. Erythrodermia to pseudoephedrine in a patient with contact allergy to phenylephrine. Allergol Immunopathol (Madr). 2002;30(4):239-242.
17. Platts-Mills TAE. Immediate hypersensitivity (Type I). In: Male D, Brostoff J, Roth DB, Roitt I. Immunology. 7th ed. Canada: Elsevier Limited; 2006:423-446.
18. Britton W. Type IV hypersensitivity. In: Male D, Brostoff J, Roth DB, Roitt I. Immunology. 7th ed. Canada: Elsevier Limited; 2006:477-491.
1. Mathias CG, Maibach HI, Irvine A, Adler W. Allergic contact dermatitis to echothiophate iodide and phenylephrine. Arch Ophthalmol. 1979;97(2):286-287.
2. Camarasa JG. Contact dermatitis to phenylephrine. Contact Dermatitis. 1984;10(3):182.
3. Barber K. Allergic contact eczema to phenylephrine. Contact Dermatitis. 1983;9(4):274-277.
4. Ducombs G, de Casamayor J, Verin P, Maleville J. Allergic contact dermatitis to phenylephrine. Contact Dermatitis. 1986;15(2):107-108.
5. Monsálvez V, Fuertes L, García-Cano I, Vanaclocha F, Ortez de Frutos J. Blepharoconjunctivitis due to phenylephrine [in Spanish]. Actas Dermosifiliogr. 2010;101(5):466-467.
6. Geyer O, Yust I, Lazar M. Allergic blepharoconjunctivitis due to phenylephrine. J Ocul Pharmacol. 1988;4(2):123-126.
7. Villarreal O. Reliability of diagnostic tests for contact allergy to mydriatic eyedrops. Contact Dermatitis. 1998;38(3):150-154.
8. Frazier M, Jaanus SD. Cycloplegics. In: Bartlett JD, Jaanus SD. Clinical Ocular Pharmacology. 5th ed. St. Louis, MO: Butterworth-Heinemann; 2009:125-138.
9. Decraene T, Goossens A. Contact allergy to atropine and other mydriatic agents in eye drops. Contact Dermatitis. 2001;45(5):309-310.
10. Boukhman MP, Maibach HI. Allergic contact dermatitis from tropicamide ophthalmic solution. Contact Dermatitis. 1999;41(1):47-48.
11. Yoshikawa K, Kawahara S. Contact allergy to atropine and other mydriatic agents. Contact Dermatitis. 1985;12(1):56-57.
12. Mcgee HT, Fraunfelder FW. Toxicities of topical ophthalmic anesthetics. Expert Opin Drug Saf. 2007;6(6):637-640.
13. Dass BA, Soong HK, Lee B. Effects of proparacaine of actin cytoskeleton of corneal epithelium. J Ocul Pharmacol. 1988;4(3):187-194.
14. Dannaker CJ, Maibach HI, Austin E. Allergic contact dermatitis to proparacaine with subsequent cross-sensitization to tetracaine from ophthalmic preparations. Am J Contact Dermat. 2001;12(3):177-179.
15. Hong J, Bielory L. Allergy to ophthalmic preservatives. Curr Opin Allergy Clin Immunol. 2009;9(5):447-453.
16. Gonzalo-Garijo MA, Pérez-Calderón R, de Argila D, Rodríguez-Nevado I. Erythrodermia to pseudoephedrine in a patient with contact allergy to phenylephrine. Allergol Immunopathol (Madr). 2002;30(4):239-242.
17. Platts-Mills TAE. Immediate hypersensitivity (Type I). In: Male D, Brostoff J, Roth DB, Roitt I. Immunology. 7th ed. Canada: Elsevier Limited; 2006:423-446.
18. Britton W. Type IV hypersensitivity. In: Male D, Brostoff J, Roth DB, Roitt I. Immunology. 7th ed. Canada: Elsevier Limited; 2006:477-491.
Late-Onset Bexarotene-Induced CD4 Lymphopenia in a Cutaneous T-cell Lymphoma Patient
Infections, autoimmune disease, bone marrow failure, medications, and total-body irradiation may induce CD4 lymphopenia, defined as a CD4 T-cell count below 300 cells/mL or less than 20% of total lymphocytes.1 Human immunodeficiency virus (HIV) is the most common cause of CD4 lymphopenia, with sepsis (bacterial and fungal) and postoperative states the most common causes in hospital settings.2 No underlying factors are found in 0.02% of CD4 lymphopenia cases, which are considered to be idiopathic.3,4 We report a patient with cutaneous T-cell lymphoma (CTCL) who developed profound CD4 lymphopenia in the setting of long-term bexarotene therapy.
Case Report
A 63-year-old man with hypertension presented to our dermatology clinic with pruritic scaly plaques on the scalp of 4 months’ duration that had progressed to full-body exfoliative erythroderma (Figure 1). He had diffuse palmoplantar keratoderma and lymphadenopathy. His only long-term medications were terazosin for benign prostatic hyperplasia and atenolol for hypertension; he reported no new medications. Laboratory evaluation revealed normal liver and kidney function. A complete blood cell count (CBC) revealed a white blood cell (WBC) count within reference range (8000/µL [reference range, 4500–11,000/µL]) but with increased eosinophils (12.9% [reference range, 2.7%]) and monocytes (11.8% [reference range, 4%]) and reduced lymphocytes (16.8% [reference range, 34%]). Flow cytometry showed a CD4:CD8 ratio of 1.18 to 1 (reference range, 0.8–4.2)(absolute CD4+ cells, 764/µL [reference range, 297–1551/µL]; absolute CD8+ cells, 654/µL [reference range, 100–1047/µL]). Skin biopsy revealed subacute spongiotic dermatitis with numerous eosinophils, exocytosis including folliculotropism, and rare atypical lymphocytes (Figure 2). Molecular studies showed T-cell receptor γ gene rearrangement. The patient did not have any other underlying conditions that would predispose him to lymphopenia. Based on these findings, a diagnosis of CTCL stage IIIA was made and agreed on by experts at the University of California, San Diego Dermatology Grand Rounds.

The patient was subsequently started on acitretin, topical corticosteroids, and hydroxyzine. However, the erythroderma progressed and he developed fever, chills, and malaise, and he was hospitalized 2 months later for intensive therapy and to rule out infection. He improved on daily wet wraps, topical steroids, oral antibiotics, and initiation of narrowband UVB therapy. He was discharged 1 week later. Acitretin was switched to bexarotene 3 months later due to peeling and cracking of the palmoplantar skin. The initial dose was 225 mg once daily, which was steadily increased over the next 4 months to a therapeutic dose of 600 mg once daily, which was much lower than the maximum dose of 400 mg/m2 daily (calculated at 750 mg/d in our patient). The patient achieved clinical remission 1 year after initiation of bexarotene in conjunction with narrowband UVB therapy. Serum eosinophils also normalized. Because there were no intolerable side effects, this dose was continued for 2 more years before it was slowly tapered to 375 mg once daily over a 1-year period. The new dose was maintained thereafter. Secondary hypertriglyceridemia and hypothyroidism, known side effects of bexarotene, developed 1 and 5 months after initiating therapy, respectively, and were treated with levothyroxine and fenofibrate. Blood counts were checked every 3 months and remained within reference range. Within the first few months of therapy, lymphocytes did trend down to 16.8%, but segmented neutrophils were normal at 59.4%. For the next 5 years the total WBC count and differential remained within reference range. T-cell subsets and flow cytometry data were not measured. No new medications were started during this period, and none of his existing medications had lymphopenia as a known side effect.
Five years after the initial diagnosis, the patient was still on bexarotene and was suspected to have pneumonia that was treated by his primary care provider with cefuroxime and azithromycin for 2 weeks with no improvement. He was then admitted to the hospital with shortness of breath, productive cough, night sweats, and dyspnea of 1 month’s duration. There was no associated weight loss or fever. Notably, the skin was clear. He was further treated for community-acquired pneumonia, first with vancomycin and ceftazidime, then with ciprofloxacin and sulfamethoxazole-trimethoprim, with no improvement. A CBC with differential was obtained on the patient’s first admission and revealed a WBC count of 3600/µL with decreased lymphocytes (8.6%), no eosinophilia, and anemia (hemoglobin, 10.5 g/dL [reference range, 33–37 g/dL]). T-cell subset studies revealed a CD4:CD8 ratio of 0.06 to 1 (absolute CD4+ cells, 6/µL; absolute CD8+ cells, 107/µL). The patient also had an elevated lactate dehydrogenase level of 1015 U/L (reference range, 100–200 U/L) and a normal comprehensive metabolic panel. A comprehensive workup, including urine and blood cultures, serum Cryptococcus and coccidioidomycosis IgG/IgM, histoplasmosis urine antigen, legionella, HIV, purified protein derivative (tuberculin), and aspergillosis galactomannan antigen panel, was negative. Blood tests for HIV and human T-lymphotropic virus also were negative. Bronchoscopy with cytology and sputum cultures for fungi, acid-fast bacteria, and viruses identified Pneumocystis jiroveci in the bronchial wash. Pneumocystis pneumonia was treated with intravenous clindamycin, primaquine, and leucovorin. The patient’s WBC count continued to drop over the next 2 weeks to a nadir of 1.7% with few lymphocytes noted on the differential. At that point, the bexarotene was stopped and was considered causative in inducing CD4 lymphopenia, resulting in opportunistic infection. The patient steadily improved and was discharged on sulfamethoxazole-trimethoprim prophyla
His CD4 count slowly improved over the next 18 months; however, his skin disease recurred and progressed to exfoliative erythroderma with marked scarring alopecia (Figure 3), facial swelling, extreme pruritus, and notable eosinophilia. Repeat computed tomography was negative for extracutaneous involvement. A repeat skin biopsy showed recurrent mycosis fungoides similar to the original biopsy (Figure 4). Topical steroids and narrowband UVB therapy were restarted. A bone marrow biopsy revealed no definitive lymphoma, but the peripheral blood showed occasional CD8+ “flower cells” and no CD4+ Sézary cells. Two repeat molecular studies failed to show the T-cell receptor gene rearrangement. Localized electron beam radiation therapy, lenalidomide, and clobetasol were tried without benefit. The patient was hospitalized 3 months later and was started on wet wraps as well as weekly infusions of the histone deacetylase inhibitor romidepsin (14 mg/m2 over a 4-hour period) on days 1, 8, and 15 of a 28-day cycle with rapid improvement. He experienced transient slight neutropenia with the first several treatments that quickly resolved. His skin was clear while on a regimen of triamcinolone, wet wraps, and intravenous romidepsin. He demonstrated visible improvement after 3 weekly infusions of romedepsin (Figure 5). His skin disease cleared after 9 infusions of romidepsin, and he currently remains in remission; however, he developed presumed bronchopneumonia after approximately 3 to 4 infusions. He then presented with severe headaches after his ninth infusion and was found to have cryptococcal meningitis. Romedepsin was stopped and he was treated with systemic antifungal therapy. His CTCL never recurred despite not restarting romidepsin.
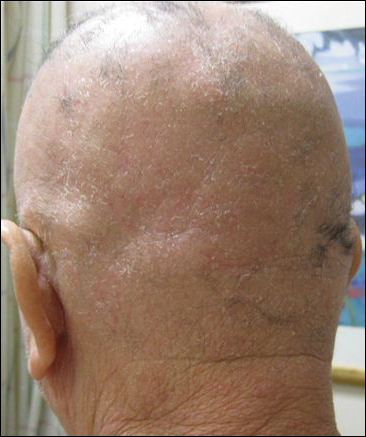
Comment
The retinoids are chemically related to vitamin A. They regulate epithelial cell growth and are beneficial in inflammatory skin disorders and in patients with increased cell turnover as well as in skin cancer and precancer prevention/treatment.5 The first- and second-generation retinoids, isotretinoin and acitretin, respectively, cause anemia or leukopenia in less than 10% of patients; adverse effects are noted more commonly in doses greater than 1 mg/kg daily.6-8
Bexarotene is a third-generation retinoid drug that is more selective for retinoid X receptors. It was approved in 1991 for treatment of advanced CTCL (stages IIB–IVB) in adult patients who have failed at least 1 prior systemic therapy. Bexarotene is noted to promote cell cycle arrest and apoptosis in CTCL cell lines.9 However, one study suggested that for bexarotene, inhibition of proliferation is more important than causing apoptosis in CTCL cells, and this effect is achieved through triggering the p53/p73-dependent cell cycle inhibition pathway.10 Studies in patients with Sézary syndrome have shown that bexarotene changes the chemokine receptor expression in circulating malignant T cells, making them less likely to traffic to the skin (lower chemokine receptor type 4 expression),11 which may explain why some CTCL cases have shown improvement of skin disease on bexarotene despite progression of extradermal disease.12
Common side effects of bexarotene include hyperlipidemia and central hypothyroidism.13 In addition, dose-related myelosuppression with isolated leukopenia, particularly neutropenia, also has been reported (18% of patients at a dosage of 300 mg/m2/d and 43% of patients with a dosage greater than 300 mg/m2/d). Leukopenia generally occurs within the first 4 to 8 weeks of treatment, is relatively mild (WBC, 1000–2999/µL), and generally is reversible.13-15 One review of 66 mycosis fungoides patients treated with bexarotene described a patient who developed leukopenia 15 months after initiating bexarotene therapy.14 The manufacturer recommends that treatment with bexarotene be continued as long as the patient is receiving benefit from the treatment. One trial of 70 mycosis fungoides patient treated with bexarotene reported response rates of 48% on bexarotene monotherapy (n=54) and 69% on bexarotene plus an additional agent (n=16).15 The authors noted higher response rates in patients on 2 lipid-lowering agents. They concluded that bexarotene was a safe and effective agent for treatment of cutaneous T-cell lymphoma and recommended continued treatment with a lowered dose of bexarotene in those achieving complete responses for a period of 2 years. Although the recommended initial dose is 300 mg/m2/d, bexarotene can be increased to 400 mg/m2/d after 8 weeks if no response to treatment is appreciated.16 Our patient was on a maximum bexarotene dose of 600 mg once daily (280 mg/m2/d) for the first 2 years, and a maintenance dose of 300 mg once daily for the next 3 years. He was not on any medicines known to induce leukopenia and he was not given any known cytochrome P450 3A4 inhibitors that could increase the toxicity of bexarotene.
The patient’s CBC was checked routinely every 2 to 3 months after he was started on bexarotene. For 5 years, the CBC and differential remained within reference range; however, his CD4 counts were not followed during those 5 years. We attribute his CD4 lymphopenia and subsequent pneumocystis pneumonia to bexarotene. After our patient’s CD4 lymphopenia was discovered, he developed a precipitous drop in his WBC and lymphocyte counts while hospitalized that worsened over a 2-week period. At this point, the bexarotene was discontinued and his WBC count slowly recovered. We believe that one of the initial antibiotics prescribed by the patient’s primary care physician at initial onset of pneumonia symptoms as an outpatient could have acted synergistically with bexarotene to worsen lymphopenia. Specifically, ceftazidime, vancomycin, and ciprofloxacin have all been reported to cause leukopenia; however, it was neutropenia in these cases, not lymphopenia.17,18 Notwithstanding, the opportunistic pneumonia and therefore CD4 lymphopenia was present prior to any antibiotic use.
The CD4 lymphopenia was unlikely due to underlying infection(s) because an extensive workup was negative, except for the pneumocystis, which likely resulted from the lymphopenia. The CD4 lymphopenia also could be idiopathic, as it has been reported in 3 patients with mycosis fungoides.19 All 3 patients were erythrodermic at presentation and were noted to have numerous CD4+ lymphocytes in the cutaneous lesions but few circulating CD4+ T lymphocytes in the blood. The authors attributed the CD4 lymphopenia to cutaneous sequestration of CD4+ T lymphocytes.19 These cases contrast with our patient who was in clinical remission at the time of CD4 lymphopenia, which improved and normalized following discontinuation of bexarotene.
Conclusion
This case emphasizes the importance of monitoring for leukopenia, specifically CD4 lymphopenia, in patients on long-term bexarotene therapy. Routine CBC as well as T-cell subset counts should be performed during treatment. Rotation off bexarotene after several years of therapy should be considered, even in patients with continuous benefit from this systemic therapy.
- Smith DK, Neal JJ, Holmberg SD. Unexplained opportunistic infections and CD4+ T-lymphocytopenia without HIV infection. an investigation of cases in the United States. The Centers for Disease Control Idiopathic CD4+ T-lymphocytopenia Task Force. N Engl J Med. 1993;328:373-379.
- Castelino DJ, McNair P, Kay TW. Lymphocytopenia in a hospital population: what does it signify? Aust N Z J Med. 1997;27:170-174.
- Zonios DI, Falloon J, Bennett JE, et al. Idiopathic CD4+ lymphocytopenia: natural history and prognostic factors. Blood. 2008;112:287-294.
- Duncan RA, von Reyn CF, Alliegro GM, et al. Idiopathic CD4+ T-lymphocytopenia: four patients with opportunistic infections and no evidence of HIV infection. N Engl J Med. 1993;328:393-398.
- Bruno NP, Beacham BE, Burnett JW. Adverse effects of isotretinoin therapy. Cutis. 1984;33:484-486, 489.
- Strauss JS, Rapini RP, Shalita AR, et al. Isotretinoin therapy for acne: results of a multicenter dose-response study. J Am Acad Dermatol. 1984;10:490-496.
- Windhorst DB, Nigra T. General clinical toxicology of oral retinoids. J Am Acad Dermatol.1982;6:675-682.
- Glinnick SE. Leucopenia from accutane: in ten percent? Schoch Let. 1985;35:9.
- Wilcox RA. Cutaneous T-cell lymphoma: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86:928-948.
- Nieto-Rementería N, Pérez-Yarza G, Boyano MD, et al. Bexarotene activates the p53/p73 pathway in human cutaneous T-cell lymphoma. Br J Dermatol. 2009;160:519-526.
- Richardson SK, Newton SB, Bach TL, et al. Bexarotene blunts malignant T-cell chemotaxis in Sézary syndrome: reduction of chemokine receptor 4-positive lymphocytes and decreased chemotaxis to thymus and activation-regulated chemokine. Am J Hematol. 2007;82:792-797.
- Bouwhuis SA, Davis MD, el-Azhary RA, et al. Bexarotene treatment of late-stage mycosis fungoides and Sézary syndrome: development of extracutaneous lymphoma in 6 patients. J Am Acad Dermatol. 2005;52:991-996.
- Targretin [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals International, Inc; 2015.
- , , , et al. Bexarotene therapy for mycosis fungoides and Sézary syndrome. Br J Dermatol. 2009;160:1299-1307.
- , , et al. Optimizing bexarotene therapy for cutaneous T-cell lymphoma. J Am Acad Dermatol. 2002;47:672-684.
- Scarisbrick JJ, Morris S, Azurdia R, et al. U.K. consensus statement on safe clinical prescribing of bexarotene for patients with cutaneous T-cell lymphoma. Br J Dermatol. 2013;168:192-200.
- Black E, Lau TT, Ensom MH. Vancomycin-induced neutropenia: is it dose-or duration related? Ann Pharmacother. 2011;45:629-638.
- Choo PW, Gantz NM. Reversible leukopenia related to ciprofloxacin therapy. South Med J. 1990;83:597-598.
- Stevens SR, Griffiths TW, Cooper KD. Idiopathic CD4+ T lymphocytopenia in a patient with mycosis fungoides. J Am Acad Dermatol. 1995;32:1063-1064.
Infections, autoimmune disease, bone marrow failure, medications, and total-body irradiation may induce CD4 lymphopenia, defined as a CD4 T-cell count below 300 cells/mL or less than 20% of total lymphocytes.1 Human immunodeficiency virus (HIV) is the most common cause of CD4 lymphopenia, with sepsis (bacterial and fungal) and postoperative states the most common causes in hospital settings.2 No underlying factors are found in 0.02% of CD4 lymphopenia cases, which are considered to be idiopathic.3,4 We report a patient with cutaneous T-cell lymphoma (CTCL) who developed profound CD4 lymphopenia in the setting of long-term bexarotene therapy.
Case Report
A 63-year-old man with hypertension presented to our dermatology clinic with pruritic scaly plaques on the scalp of 4 months’ duration that had progressed to full-body exfoliative erythroderma (Figure 1). He had diffuse palmoplantar keratoderma and lymphadenopathy. His only long-term medications were terazosin for benign prostatic hyperplasia and atenolol for hypertension; he reported no new medications. Laboratory evaluation revealed normal liver and kidney function. A complete blood cell count (CBC) revealed a white blood cell (WBC) count within reference range (8000/µL [reference range, 4500–11,000/µL]) but with increased eosinophils (12.9% [reference range, 2.7%]) and monocytes (11.8% [reference range, 4%]) and reduced lymphocytes (16.8% [reference range, 34%]). Flow cytometry showed a CD4:CD8 ratio of 1.18 to 1 (reference range, 0.8–4.2)(absolute CD4+ cells, 764/µL [reference range, 297–1551/µL]; absolute CD8+ cells, 654/µL [reference range, 100–1047/µL]). Skin biopsy revealed subacute spongiotic dermatitis with numerous eosinophils, exocytosis including folliculotropism, and rare atypical lymphocytes (Figure 2). Molecular studies showed T-cell receptor γ gene rearrangement. The patient did not have any other underlying conditions that would predispose him to lymphopenia. Based on these findings, a diagnosis of CTCL stage IIIA was made and agreed on by experts at the University of California, San Diego Dermatology Grand Rounds.
The patient was subsequently started on acitretin, topical corticosteroids, and hydroxyzine. However, the erythroderma progressed and he developed fever, chills, and malaise, and he was hospitalized 2 months later for intensive therapy and to rule out infection. He improved on daily wet wraps, topical steroids, oral antibiotics, and initiation of narrowband UVB therapy. He was discharged 1 week later. Acitretin was switched to bexarotene 3 months later due to peeling and cracking of the palmoplantar skin. The initial dose was 225 mg once daily, which was steadily increased over the next 4 months to a therapeutic dose of 600 mg once daily, which was much lower than the maximum dose of 400 mg/m2 daily (calculated at 750 mg/d in our patient). The patient achieved clinical remission 1 year after initiation of bexarotene in conjunction with narrowband UVB therapy. Serum eosinophils also normalized. Because there were no intolerable side effects, this dose was continued for 2 more years before it was slowly tapered to 375 mg once daily over a 1-year period. The new dose was maintained thereafter. Secondary hypertriglyceridemia and hypothyroidism, known side effects of bexarotene, developed 1 and 5 months after initiating therapy, respectively, and were treated with levothyroxine and fenofibrate. Blood counts were checked every 3 months and remained within reference range. Within the first few months of therapy, lymphocytes did trend down to 16.8%, but segmented neutrophils were normal at 59.4%. For the next 5 years the total WBC count and differential remained within reference range. T-cell subsets and flow cytometry data were not measured. No new medications were started during this period, and none of his existing medications had lymphopenia as a known side effect.
Five years after the initial diagnosis, the patient was still on bexarotene and was suspected to have pneumonia that was treated by his primary care provider with cefuroxime and azithromycin for 2 weeks with no improvement. He was then admitted to the hospital with shortness of breath, productive cough, night sweats, and dyspnea of 1 month’s duration. There was no associated weight loss or fever. Notably, the skin was clear. He was further treated for community-acquired pneumonia, first with vancomycin and ceftazidime, then with ciprofloxacin and sulfamethoxazole-trimethoprim, with no improvement. A CBC with differential was obtained on the patient’s first admission and revealed a WBC count of 3600/µL with decreased lymphocytes (8.6%), no eosinophilia, and anemia (hemoglobin, 10.5 g/dL [reference range, 33–37 g/dL]). T-cell subset studies revealed a CD4:CD8 ratio of 0.06 to 1 (absolute CD4+ cells, 6/µL; absolute CD8+ cells, 107/µL). The patient also had an elevated lactate dehydrogenase level of 1015 U/L (reference range, 100–200 U/L) and a normal comprehensive metabolic panel. A comprehensive workup, including urine and blood cultures, serum Cryptococcus and coccidioidomycosis IgG/IgM, histoplasmosis urine antigen, legionella, HIV, purified protein derivative (tuberculin), and aspergillosis galactomannan antigen panel, was negative. Blood tests for HIV and human T-lymphotropic virus also were negative. Bronchoscopy with cytology and sputum cultures for fungi, acid-fast bacteria, and viruses identified Pneumocystis jiroveci in the bronchial wash. Pneumocystis pneumonia was treated with intravenous clindamycin, primaquine, and leucovorin. The patient’s WBC count continued to drop over the next 2 weeks to a nadir of 1.7% with few lymphocytes noted on the differential. At that point, the bexarotene was stopped and was considered causative in inducing CD4 lymphopenia, resulting in opportunistic infection. The patient steadily improved and was discharged on sulfamethoxazole-trimethoprim prophyla
His CD4 count slowly improved over the next 18 months; however, his skin disease recurred and progressed to exfoliative erythroderma with marked scarring alopecia (Figure 3), facial swelling, extreme pruritus, and notable eosinophilia. Repeat computed tomography was negative for extracutaneous involvement. A repeat skin biopsy showed recurrent mycosis fungoides similar to the original biopsy (Figure 4). Topical steroids and narrowband UVB therapy were restarted. A bone marrow biopsy revealed no definitive lymphoma, but the peripheral blood showed occasional CD8+ “flower cells” and no CD4+ Sézary cells. Two repeat molecular studies failed to show the T-cell receptor gene rearrangement. Localized electron beam radiation therapy, lenalidomide, and clobetasol were tried without benefit. The patient was hospitalized 3 months later and was started on wet wraps as well as weekly infusions of the histone deacetylase inhibitor romidepsin (14 mg/m2 over a 4-hour period) on days 1, 8, and 15 of a 28-day cycle with rapid improvement. He experienced transient slight neutropenia with the first several treatments that quickly resolved. His skin was clear while on a regimen of triamcinolone, wet wraps, and intravenous romidepsin. He demonstrated visible improvement after 3 weekly infusions of romedepsin (Figure 5). His skin disease cleared after 9 infusions of romidepsin, and he currently remains in remission; however, he developed presumed bronchopneumonia after approximately 3 to 4 infusions. He then presented with severe headaches after his ninth infusion and was found to have cryptococcal meningitis. Romedepsin was stopped and he was treated with systemic antifungal therapy. His CTCL never recurred despite not restarting romidepsin.
Comment
The retinoids are chemically related to vitamin A. They regulate epithelial cell growth and are beneficial in inflammatory skin disorders and in patients with increased cell turnover as well as in skin cancer and precancer prevention/treatment.5 The first- and second-generation retinoids, isotretinoin and acitretin, respectively, cause anemia or leukopenia in less than 10% of patients; adverse effects are noted more commonly in doses greater than 1 mg/kg daily.6-8
Bexarotene is a third-generation retinoid drug that is more selective for retinoid X receptors. It was approved in 1991 for treatment of advanced CTCL (stages IIB–IVB) in adult patients who have failed at least 1 prior systemic therapy. Bexarotene is noted to promote cell cycle arrest and apoptosis in CTCL cell lines.9 However, one study suggested that for bexarotene, inhibition of proliferation is more important than causing apoptosis in CTCL cells, and this effect is achieved through triggering the p53/p73-dependent cell cycle inhibition pathway.10 Studies in patients with Sézary syndrome have shown that bexarotene changes the chemokine receptor expression in circulating malignant T cells, making them less likely to traffic to the skin (lower chemokine receptor type 4 expression),11 which may explain why some CTCL cases have shown improvement of skin disease on bexarotene despite progression of extradermal disease.12
Common side effects of bexarotene include hyperlipidemia and central hypothyroidism.13 In addition, dose-related myelosuppression with isolated leukopenia, particularly neutropenia, also has been reported (18% of patients at a dosage of 300 mg/m2/d and 43% of patients with a dosage greater than 300 mg/m2/d). Leukopenia generally occurs within the first 4 to 8 weeks of treatment, is relatively mild (WBC, 1000–2999/µL), and generally is reversible.13-15 One review of 66 mycosis fungoides patients treated with bexarotene described a patient who developed leukopenia 15 months after initiating bexarotene therapy.14 The manufacturer recommends that treatment with bexarotene be continued as long as the patient is receiving benefit from the treatment. One trial of 70 mycosis fungoides patient treated with bexarotene reported response rates of 48% on bexarotene monotherapy (n=54) and 69% on bexarotene plus an additional agent (n=16).15 The authors noted higher response rates in patients on 2 lipid-lowering agents. They concluded that bexarotene was a safe and effective agent for treatment of cutaneous T-cell lymphoma and recommended continued treatment with a lowered dose of bexarotene in those achieving complete responses for a period of 2 years. Although the recommended initial dose is 300 mg/m2/d, bexarotene can be increased to 400 mg/m2/d after 8 weeks if no response to treatment is appreciated.16 Our patient was on a maximum bexarotene dose of 600 mg once daily (280 mg/m2/d) for the first 2 years, and a maintenance dose of 300 mg once daily for the next 3 years. He was not on any medicines known to induce leukopenia and he was not given any known cytochrome P450 3A4 inhibitors that could increase the toxicity of bexarotene.
The patient’s CBC was checked routinely every 2 to 3 months after he was started on bexarotene. For 5 years, the CBC and differential remained within reference range; however, his CD4 counts were not followed during those 5 years. We attribute his CD4 lymphopenia and subsequent pneumocystis pneumonia to bexarotene. After our patient’s CD4 lymphopenia was discovered, he developed a precipitous drop in his WBC and lymphocyte counts while hospitalized that worsened over a 2-week period. At this point, the bexarotene was discontinued and his WBC count slowly recovered. We believe that one of the initial antibiotics prescribed by the patient’s primary care physician at initial onset of pneumonia symptoms as an outpatient could have acted synergistically with bexarotene to worsen lymphopenia. Specifically, ceftazidime, vancomycin, and ciprofloxacin have all been reported to cause leukopenia; however, it was neutropenia in these cases, not lymphopenia.17,18 Notwithstanding, the opportunistic pneumonia and therefore CD4 lymphopenia was present prior to any antibiotic use.
The CD4 lymphopenia was unlikely due to underlying infection(s) because an extensive workup was negative, except for the pneumocystis, which likely resulted from the lymphopenia. The CD4 lymphopenia also could be idiopathic, as it has been reported in 3 patients with mycosis fungoides.19 All 3 patients were erythrodermic at presentation and were noted to have numerous CD4+ lymphocytes in the cutaneous lesions but few circulating CD4+ T lymphocytes in the blood. The authors attributed the CD4 lymphopenia to cutaneous sequestration of CD4+ T lymphocytes.19 These cases contrast with our patient who was in clinical remission at the time of CD4 lymphopenia, which improved and normalized following discontinuation of bexarotene.
Conclusion
This case emphasizes the importance of monitoring for leukopenia, specifically CD4 lymphopenia, in patients on long-term bexarotene therapy. Routine CBC as well as T-cell subset counts should be performed during treatment. Rotation off bexarotene after several years of therapy should be considered, even in patients with continuous benefit from this systemic therapy.
Infections, autoimmune disease, bone marrow failure, medications, and total-body irradiation may induce CD4 lymphopenia, defined as a CD4 T-cell count below 300 cells/mL or less than 20% of total lymphocytes.1 Human immunodeficiency virus (HIV) is the most common cause of CD4 lymphopenia, with sepsis (bacterial and fungal) and postoperative states the most common causes in hospital settings.2 No underlying factors are found in 0.02% of CD4 lymphopenia cases, which are considered to be idiopathic.3,4 We report a patient with cutaneous T-cell lymphoma (CTCL) who developed profound CD4 lymphopenia in the setting of long-term bexarotene therapy.
Case Report
A 63-year-old man with hypertension presented to our dermatology clinic with pruritic scaly plaques on the scalp of 4 months’ duration that had progressed to full-body exfoliative erythroderma (Figure 1). He had diffuse palmoplantar keratoderma and lymphadenopathy. His only long-term medications were terazosin for benign prostatic hyperplasia and atenolol for hypertension; he reported no new medications. Laboratory evaluation revealed normal liver and kidney function. A complete blood cell count (CBC) revealed a white blood cell (WBC) count within reference range (8000/µL [reference range, 4500–11,000/µL]) but with increased eosinophils (12.9% [reference range, 2.7%]) and monocytes (11.8% [reference range, 4%]) and reduced lymphocytes (16.8% [reference range, 34%]). Flow cytometry showed a CD4:CD8 ratio of 1.18 to 1 (reference range, 0.8–4.2)(absolute CD4+ cells, 764/µL [reference range, 297–1551/µL]; absolute CD8+ cells, 654/µL [reference range, 100–1047/µL]). Skin biopsy revealed subacute spongiotic dermatitis with numerous eosinophils, exocytosis including folliculotropism, and rare atypical lymphocytes (Figure 2). Molecular studies showed T-cell receptor γ gene rearrangement. The patient did not have any other underlying conditions that would predispose him to lymphopenia. Based on these findings, a diagnosis of CTCL stage IIIA was made and agreed on by experts at the University of California, San Diego Dermatology Grand Rounds.
The patient was subsequently started on acitretin, topical corticosteroids, and hydroxyzine. However, the erythroderma progressed and he developed fever, chills, and malaise, and he was hospitalized 2 months later for intensive therapy and to rule out infection. He improved on daily wet wraps, topical steroids, oral antibiotics, and initiation of narrowband UVB therapy. He was discharged 1 week later. Acitretin was switched to bexarotene 3 months later due to peeling and cracking of the palmoplantar skin. The initial dose was 225 mg once daily, which was steadily increased over the next 4 months to a therapeutic dose of 600 mg once daily, which was much lower than the maximum dose of 400 mg/m2 daily (calculated at 750 mg/d in our patient). The patient achieved clinical remission 1 year after initiation of bexarotene in conjunction with narrowband UVB therapy. Serum eosinophils also normalized. Because there were no intolerable side effects, this dose was continued for 2 more years before it was slowly tapered to 375 mg once daily over a 1-year period. The new dose was maintained thereafter. Secondary hypertriglyceridemia and hypothyroidism, known side effects of bexarotene, developed 1 and 5 months after initiating therapy, respectively, and were treated with levothyroxine and fenofibrate. Blood counts were checked every 3 months and remained within reference range. Within the first few months of therapy, lymphocytes did trend down to 16.8%, but segmented neutrophils were normal at 59.4%. For the next 5 years the total WBC count and differential remained within reference range. T-cell subsets and flow cytometry data were not measured. No new medications were started during this period, and none of his existing medications had lymphopenia as a known side effect.
Five years after the initial diagnosis, the patient was still on bexarotene and was suspected to have pneumonia that was treated by his primary care provider with cefuroxime and azithromycin for 2 weeks with no improvement. He was then admitted to the hospital with shortness of breath, productive cough, night sweats, and dyspnea of 1 month’s duration. There was no associated weight loss or fever. Notably, the skin was clear. He was further treated for community-acquired pneumonia, first with vancomycin and ceftazidime, then with ciprofloxacin and sulfamethoxazole-trimethoprim, with no improvement. A CBC with differential was obtained on the patient’s first admission and revealed a WBC count of 3600/µL with decreased lymphocytes (8.6%), no eosinophilia, and anemia (hemoglobin, 10.5 g/dL [reference range, 33–37 g/dL]). T-cell subset studies revealed a CD4:CD8 ratio of 0.06 to 1 (absolute CD4+ cells, 6/µL; absolute CD8+ cells, 107/µL). The patient also had an elevated lactate dehydrogenase level of 1015 U/L (reference range, 100–200 U/L) and a normal comprehensive metabolic panel. A comprehensive workup, including urine and blood cultures, serum Cryptococcus and coccidioidomycosis IgG/IgM, histoplasmosis urine antigen, legionella, HIV, purified protein derivative (tuberculin), and aspergillosis galactomannan antigen panel, was negative. Blood tests for HIV and human T-lymphotropic virus also were negative. Bronchoscopy with cytology and sputum cultures for fungi, acid-fast bacteria, and viruses identified Pneumocystis jiroveci in the bronchial wash. Pneumocystis pneumonia was treated with intravenous clindamycin, primaquine, and leucovorin. The patient’s WBC count continued to drop over the next 2 weeks to a nadir of 1.7% with few lymphocytes noted on the differential. At that point, the bexarotene was stopped and was considered causative in inducing CD4 lymphopenia, resulting in opportunistic infection. The patient steadily improved and was discharged on sulfamethoxazole-trimethoprim prophyla
His CD4 count slowly improved over the next 18 months; however, his skin disease recurred and progressed to exfoliative erythroderma with marked scarring alopecia (Figure 3), facial swelling, extreme pruritus, and notable eosinophilia. Repeat computed tomography was negative for extracutaneous involvement. A repeat skin biopsy showed recurrent mycosis fungoides similar to the original biopsy (Figure 4). Topical steroids and narrowband UVB therapy were restarted. A bone marrow biopsy revealed no definitive lymphoma, but the peripheral blood showed occasional CD8+ “flower cells” and no CD4+ Sézary cells. Two repeat molecular studies failed to show the T-cell receptor gene rearrangement. Localized electron beam radiation therapy, lenalidomide, and clobetasol were tried without benefit. The patient was hospitalized 3 months later and was started on wet wraps as well as weekly infusions of the histone deacetylase inhibitor romidepsin (14 mg/m2 over a 4-hour period) on days 1, 8, and 15 of a 28-day cycle with rapid improvement. He experienced transient slight neutropenia with the first several treatments that quickly resolved. His skin was clear while on a regimen of triamcinolone, wet wraps, and intravenous romidepsin. He demonstrated visible improvement after 3 weekly infusions of romedepsin (Figure 5). His skin disease cleared after 9 infusions of romidepsin, and he currently remains in remission; however, he developed presumed bronchopneumonia after approximately 3 to 4 infusions. He then presented with severe headaches after his ninth infusion and was found to have cryptococcal meningitis. Romedepsin was stopped and he was treated with systemic antifungal therapy. His CTCL never recurred despite not restarting romidepsin.
Comment
The retinoids are chemically related to vitamin A. They regulate epithelial cell growth and are beneficial in inflammatory skin disorders and in patients with increased cell turnover as well as in skin cancer and precancer prevention/treatment.5 The first- and second-generation retinoids, isotretinoin and acitretin, respectively, cause anemia or leukopenia in less than 10% of patients; adverse effects are noted more commonly in doses greater than 1 mg/kg daily.6-8
Bexarotene is a third-generation retinoid drug that is more selective for retinoid X receptors. It was approved in 1991 for treatment of advanced CTCL (stages IIB–IVB) in adult patients who have failed at least 1 prior systemic therapy. Bexarotene is noted to promote cell cycle arrest and apoptosis in CTCL cell lines.9 However, one study suggested that for bexarotene, inhibition of proliferation is more important than causing apoptosis in CTCL cells, and this effect is achieved through triggering the p53/p73-dependent cell cycle inhibition pathway.10 Studies in patients with Sézary syndrome have shown that bexarotene changes the chemokine receptor expression in circulating malignant T cells, making them less likely to traffic to the skin (lower chemokine receptor type 4 expression),11 which may explain why some CTCL cases have shown improvement of skin disease on bexarotene despite progression of extradermal disease.12
Common side effects of bexarotene include hyperlipidemia and central hypothyroidism.13 In addition, dose-related myelosuppression with isolated leukopenia, particularly neutropenia, also has been reported (18% of patients at a dosage of 300 mg/m2/d and 43% of patients with a dosage greater than 300 mg/m2/d). Leukopenia generally occurs within the first 4 to 8 weeks of treatment, is relatively mild (WBC, 1000–2999/µL), and generally is reversible.13-15 One review of 66 mycosis fungoides patients treated with bexarotene described a patient who developed leukopenia 15 months after initiating bexarotene therapy.14 The manufacturer recommends that treatment with bexarotene be continued as long as the patient is receiving benefit from the treatment. One trial of 70 mycosis fungoides patient treated with bexarotene reported response rates of 48% on bexarotene monotherapy (n=54) and 69% on bexarotene plus an additional agent (n=16).15 The authors noted higher response rates in patients on 2 lipid-lowering agents. They concluded that bexarotene was a safe and effective agent for treatment of cutaneous T-cell lymphoma and recommended continued treatment with a lowered dose of bexarotene in those achieving complete responses for a period of 2 years. Although the recommended initial dose is 300 mg/m2/d, bexarotene can be increased to 400 mg/m2/d after 8 weeks if no response to treatment is appreciated.16 Our patient was on a maximum bexarotene dose of 600 mg once daily (280 mg/m2/d) for the first 2 years, and a maintenance dose of 300 mg once daily for the next 3 years. He was not on any medicines known to induce leukopenia and he was not given any known cytochrome P450 3A4 inhibitors that could increase the toxicity of bexarotene.
The patient’s CBC was checked routinely every 2 to 3 months after he was started on bexarotene. For 5 years, the CBC and differential remained within reference range; however, his CD4 counts were not followed during those 5 years. We attribute his CD4 lymphopenia and subsequent pneumocystis pneumonia to bexarotene. After our patient’s CD4 lymphopenia was discovered, he developed a precipitous drop in his WBC and lymphocyte counts while hospitalized that worsened over a 2-week period. At this point, the bexarotene was discontinued and his WBC count slowly recovered. We believe that one of the initial antibiotics prescribed by the patient’s primary care physician at initial onset of pneumonia symptoms as an outpatient could have acted synergistically with bexarotene to worsen lymphopenia. Specifically, ceftazidime, vancomycin, and ciprofloxacin have all been reported to cause leukopenia; however, it was neutropenia in these cases, not lymphopenia.17,18 Notwithstanding, the opportunistic pneumonia and therefore CD4 lymphopenia was present prior to any antibiotic use.
The CD4 lymphopenia was unlikely due to underlying infection(s) because an extensive workup was negative, except for the pneumocystis, which likely resulted from the lymphopenia. The CD4 lymphopenia also could be idiopathic, as it has been reported in 3 patients with mycosis fungoides.19 All 3 patients were erythrodermic at presentation and were noted to have numerous CD4+ lymphocytes in the cutaneous lesions but few circulating CD4+ T lymphocytes in the blood. The authors attributed the CD4 lymphopenia to cutaneous sequestration of CD4+ T lymphocytes.19 These cases contrast with our patient who was in clinical remission at the time of CD4 lymphopenia, which improved and normalized following discontinuation of bexarotene.
Conclusion
This case emphasizes the importance of monitoring for leukopenia, specifically CD4 lymphopenia, in patients on long-term bexarotene therapy. Routine CBC as well as T-cell subset counts should be performed during treatment. Rotation off bexarotene after several years of therapy should be considered, even in patients with continuous benefit from this systemic therapy.
- Smith DK, Neal JJ, Holmberg SD. Unexplained opportunistic infections and CD4+ T-lymphocytopenia without HIV infection. an investigation of cases in the United States. The Centers for Disease Control Idiopathic CD4+ T-lymphocytopenia Task Force. N Engl J Med. 1993;328:373-379.
- Castelino DJ, McNair P, Kay TW. Lymphocytopenia in a hospital population: what does it signify? Aust N Z J Med. 1997;27:170-174.
- Zonios DI, Falloon J, Bennett JE, et al. Idiopathic CD4+ lymphocytopenia: natural history and prognostic factors. Blood. 2008;112:287-294.
- Duncan RA, von Reyn CF, Alliegro GM, et al. Idiopathic CD4+ T-lymphocytopenia: four patients with opportunistic infections and no evidence of HIV infection. N Engl J Med. 1993;328:393-398.
- Bruno NP, Beacham BE, Burnett JW. Adverse effects of isotretinoin therapy. Cutis. 1984;33:484-486, 489.
- Strauss JS, Rapini RP, Shalita AR, et al. Isotretinoin therapy for acne: results of a multicenter dose-response study. J Am Acad Dermatol. 1984;10:490-496.
- Windhorst DB, Nigra T. General clinical toxicology of oral retinoids. J Am Acad Dermatol.1982;6:675-682.
- Glinnick SE. Leucopenia from accutane: in ten percent? Schoch Let. 1985;35:9.
- Wilcox RA. Cutaneous T-cell lymphoma: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86:928-948.
- Nieto-Rementería N, Pérez-Yarza G, Boyano MD, et al. Bexarotene activates the p53/p73 pathway in human cutaneous T-cell lymphoma. Br J Dermatol. 2009;160:519-526.
- Richardson SK, Newton SB, Bach TL, et al. Bexarotene blunts malignant T-cell chemotaxis in Sézary syndrome: reduction of chemokine receptor 4-positive lymphocytes and decreased chemotaxis to thymus and activation-regulated chemokine. Am J Hematol. 2007;82:792-797.
- Bouwhuis SA, Davis MD, el-Azhary RA, et al. Bexarotene treatment of late-stage mycosis fungoides and Sézary syndrome: development of extracutaneous lymphoma in 6 patients. J Am Acad Dermatol. 2005;52:991-996.
- Targretin [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals International, Inc; 2015.
- , , , et al. Bexarotene therapy for mycosis fungoides and Sézary syndrome. Br J Dermatol. 2009;160:1299-1307.
- , , et al. Optimizing bexarotene therapy for cutaneous T-cell lymphoma. J Am Acad Dermatol. 2002;47:672-684.
- Scarisbrick JJ, Morris S, Azurdia R, et al. U.K. consensus statement on safe clinical prescribing of bexarotene for patients with cutaneous T-cell lymphoma. Br J Dermatol. 2013;168:192-200.
- Black E, Lau TT, Ensom MH. Vancomycin-induced neutropenia: is it dose-or duration related? Ann Pharmacother. 2011;45:629-638.
- Choo PW, Gantz NM. Reversible leukopenia related to ciprofloxacin therapy. South Med J. 1990;83:597-598.
- Stevens SR, Griffiths TW, Cooper KD. Idiopathic CD4+ T lymphocytopenia in a patient with mycosis fungoides. J Am Acad Dermatol. 1995;32:1063-1064.
- Smith DK, Neal JJ, Holmberg SD. Unexplained opportunistic infections and CD4+ T-lymphocytopenia without HIV infection. an investigation of cases in the United States. The Centers for Disease Control Idiopathic CD4+ T-lymphocytopenia Task Force. N Engl J Med. 1993;328:373-379.
- Castelino DJ, McNair P, Kay TW. Lymphocytopenia in a hospital population: what does it signify? Aust N Z J Med. 1997;27:170-174.
- Zonios DI, Falloon J, Bennett JE, et al. Idiopathic CD4+ lymphocytopenia: natural history and prognostic factors. Blood. 2008;112:287-294.
- Duncan RA, von Reyn CF, Alliegro GM, et al. Idiopathic CD4+ T-lymphocytopenia: four patients with opportunistic infections and no evidence of HIV infection. N Engl J Med. 1993;328:393-398.
- Bruno NP, Beacham BE, Burnett JW. Adverse effects of isotretinoin therapy. Cutis. 1984;33:484-486, 489.
- Strauss JS, Rapini RP, Shalita AR, et al. Isotretinoin therapy for acne: results of a multicenter dose-response study. J Am Acad Dermatol. 1984;10:490-496.
- Windhorst DB, Nigra T. General clinical toxicology of oral retinoids. J Am Acad Dermatol.1982;6:675-682.
- Glinnick SE. Leucopenia from accutane: in ten percent? Schoch Let. 1985;35:9.
- Wilcox RA. Cutaneous T-cell lymphoma: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86:928-948.
- Nieto-Rementería N, Pérez-Yarza G, Boyano MD, et al. Bexarotene activates the p53/p73 pathway in human cutaneous T-cell lymphoma. Br J Dermatol. 2009;160:519-526.
- Richardson SK, Newton SB, Bach TL, et al. Bexarotene blunts malignant T-cell chemotaxis in Sézary syndrome: reduction of chemokine receptor 4-positive lymphocytes and decreased chemotaxis to thymus and activation-regulated chemokine. Am J Hematol. 2007;82:792-797.
- Bouwhuis SA, Davis MD, el-Azhary RA, et al. Bexarotene treatment of late-stage mycosis fungoides and Sézary syndrome: development of extracutaneous lymphoma in 6 patients. J Am Acad Dermatol. 2005;52:991-996.
- Targretin [package insert]. Bridgewater, NJ: Valeant Pharmaceuticals International, Inc; 2015.
- , , , et al. Bexarotene therapy for mycosis fungoides and Sézary syndrome. Br J Dermatol. 2009;160:1299-1307.
- , , et al. Optimizing bexarotene therapy for cutaneous T-cell lymphoma. J Am Acad Dermatol. 2002;47:672-684.
- Scarisbrick JJ, Morris S, Azurdia R, et al. U.K. consensus statement on safe clinical prescribing of bexarotene for patients with cutaneous T-cell lymphoma. Br J Dermatol. 2013;168:192-200.
- Black E, Lau TT, Ensom MH. Vancomycin-induced neutropenia: is it dose-or duration related? Ann Pharmacother. 2011;45:629-638.
- Choo PW, Gantz NM. Reversible leukopenia related to ciprofloxacin therapy. South Med J. 1990;83:597-598.
- Stevens SR, Griffiths TW, Cooper KD. Idiopathic CD4+ T lymphocytopenia in a patient with mycosis fungoides. J Am Acad Dermatol. 1995;32:1063-1064.
Practice Points
- Most adverse effects of bexarotene (eg, hypothyroidism, hyperlipidemia, leukopenia) occur within the first several months of therapy.
- Delayed-onset leukopenia, including CD4 lymphopenia, may occur several years after initiating bexarotene therapy, resulting in opportunistic infections.
- Long-term periodic monitoring of T lymphocyte counts at least twice yearly in addition to standard quarterly complete blood cell count with differential are recommended.
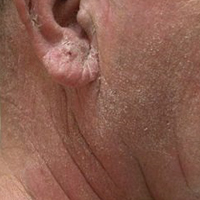
Lupus Erythematosus Tumidus of the Scalp Masquerading as Alopecia Areata
Lupus erythematosus tumidus (LET) is a relatively rare condition but may simply be underdiagnosed in the literature. It presents as urticarialike papules and plaques in sun-exposed areas, characterized by induration and erythema. Lesions occur on the face, neck, upper extremities, and trunk and heal without scarring.1,2 Rarely, lesions can show fine scaling and associated pruritus, but most often the lesions are asymptomatic.3
Case Report
A 45-year-old woman presented with 2 asymptomatic self-described bald spots on the top of the head of 2 months’ duration. The patient denied prior treatment of the lesions and noted one patch was resolving. She reported no involvement of the eyebrows, eyelashes, and axillary and pubic hair. A review of systems was negative. The patient denied personal or family history of lupus, thyroid disease, or vitiligo.
Clinical examination revealed a 1.1-cm round patch of nonscarring alopecia on the right vertex scalp and a 0.9-cm round patch of nonscarring alopecia with moderate hair regrowth on the left vertex scalp. There was no erythema, scaling, or induration. The rest of the scalp was normal in appearance and the eyebrows and eyelashes were uninvolved. The patient was diagnosed with alopecia areata and was treated with 10 mg/mL of intralesional triamcinolone once monthly for 4 months.
The patient initially showed improvement with moderate hair regrowth. After 4 months of treatment, she developed 3 new 1- to 1.5-cm erythematous alopecic patches on the vertex scalp and had worsening in the initial patches (Figure 1). Given the resistance to standard therapy and the onset of multiple new areas with evidence of inflammatory involvement, a punch biopsy was performed. Histopathologic examination revealed a fairly unremarkable epidermis and a dense dermal inflammatory infiltrate that was present both in the superficial and deep dermis (Figure 2). The inflammatory cells, which appeared to be predominantly comprised of lymphocytes, had a predilection for the vasculature but also were observed within the interstitial dermis. Additionally, mucin appeared to be slightly increased in the deep dermis. The lymphocytic phenotype was confirmed by immunohistochemical studies for CD20 and CD3. The most likely possibilities for this reaction pattern were LET, Jessner lymphocytic infiltrate of the skin (JLIS), gyrate erythema, and lymphoma; however, the immunohistochemical studies effectively ruled out lymphoma. Additionally, there was pronounced dermal mucin noted in the specimen. The patient was diagnosed with LET of the scalp based on the constellation of findings.
Comment
The classification of LET as a single unique entity or disease process sui generis has been in flux in the last decade. Its similarities to JLIS and other forms of chronic cutaneous lupus erythematosus (CCLE) have brought debate.4-6 In 1930, Gougerot and Burnier7 documented the first case of LET in the literature, describing smooth, infiltrated, erythematous lesions with no desquamation or other superficial changes seen in 5 patients.
In 2000, interest in LET and other forms of CCLE was increasing, and reports in the literature paralleled. That year, Kuhn et al4 reported 40 cases of LET, characterizing the clinical and histological features of each case to demonstrate that LET should be separate from other forms of CCLE. Until then, it is likely that many lesions that should have been classified as LET were instead classified as various forms of CCLE. The investigators maintained that LET also should be distinct from JLIS because it is associated with UV exposure.4 Kuhn et al8 reviewed phototesting in 60 patients with LET in 2001 and confirmed this subset was the most photosensitive type of lupus erythematosus.
In general, the histopathologic and immunohistochemical studies in LET and JLIS can be quite similar. Relatively distinguishing histopathologic findings in JLIS include no evidence of epidermal atrophy, basal vacuolar change, or follicular plugging, as well as negative immunofluorescence studies. Both entities show a predominantly T-cell population with a smaller component of B cells and thus a distinction cannot be made based on relative proportions of T and B cells in lesions.2
In 2003, Alexiades-Armenakas et al6 determined immunohistochemical criteria for LET, finding a predominance of T cells and more CD4 lymphocytes than CD8 lymphocytes with a mean ratio of roughly 3 to 1. Their study results maintained LET should be classified as a form of CCLE due to the chronicity of the lesions, the serologic profile with negative anti–double-stranded DNA, anticentromere, anti-Smith, anti-Ro/Sjögren syndrome antigen A, anti-La/Sjögren syndrome antigen B, and anti-nuclear ribonucleoprotein antibodies and the rare association with systemic disease.6 This conclusion was further solidified by a review published that same year citing unique histopathological features when compared to subacute cutaneous LE and discoid lupus erythematosus.5
This case illustrates the importance of histologic evaluation in determining the correct diagnosis in a patient with alopecia areata recalcitrant to treatment. Including LET in the differential of alopecic patches on the scalp could prove beneficial for patients, as LET responds well to antimalarial drugs and photoprotection.9 This patient had a normal antinuclear antibody panel and no signs or symptoms of systemic lupus. It was recommended that she avoid sun exposure and begin treatment with hydroxychloroquine but she declined. At a follow-up visit 6 months later she reported the lesions had improved, but a permanent wig had been sewn over the area, so it could not be examined.
- Lee L, Werth V. Rheumatologic disease. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 3rd ed. Mosby Elsevier; 2008:615-629.
- Weedon D. The lichenoid reaction pattern. In: Weedon D. Skin Pathology. 2nd ed. Edinburgh, Scotland: Churchill Livingstone; 2002:35-70.
- Dekle CL, Mannes KD, Davis LS, et al. Lupus tumidus. J Am Acad Dermatol. 1999;41:250-253.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Kuhn A, Sonntag M, Ruzicka T, et al. Histopathologic findings in lupus erythematosus tumidus: review of 80 patients. J Am Acad Dermatol. 2003;48:901-908.
- Alexiades-Armenakas MR, Baldassano M, Bince B, et al. Tumid lupus erythematosus: criteria for classification with immunohistochemical analysis. Arthritis Rheum. 2003;49:494-500.
- Gougerot H, Burnier R. Lupuse rythe mateux “tumidus.” Bull Soc Fr Dermatol Syph. 1930;37:1291-1292.
- Kuhn A, Sonntag M, Richter-Hintz D, et al. Phototesting in lupus erythematosus tumidus—review of 60 patients. Photochem Photobiol. 2001;73:532-536.
- Cozzani E, Christana K, Rongioletti F, et al. Lupus erythematosus tumidus: clinical, histopathological and serological aspects and therapy response of 21 patients. Eur J Dermatol. 2010;20:797-801.
Lupus erythematosus tumidus (LET) is a relatively rare condition but may simply be underdiagnosed in the literature. It presents as urticarialike papules and plaques in sun-exposed areas, characterized by induration and erythema. Lesions occur on the face, neck, upper extremities, and trunk and heal without scarring.1,2 Rarely, lesions can show fine scaling and associated pruritus, but most often the lesions are asymptomatic.3
Case Report
A 45-year-old woman presented with 2 asymptomatic self-described bald spots on the top of the head of 2 months’ duration. The patient denied prior treatment of the lesions and noted one patch was resolving. She reported no involvement of the eyebrows, eyelashes, and axillary and pubic hair. A review of systems was negative. The patient denied personal or family history of lupus, thyroid disease, or vitiligo.
Clinical examination revealed a 1.1-cm round patch of nonscarring alopecia on the right vertex scalp and a 0.9-cm round patch of nonscarring alopecia with moderate hair regrowth on the left vertex scalp. There was no erythema, scaling, or induration. The rest of the scalp was normal in appearance and the eyebrows and eyelashes were uninvolved. The patient was diagnosed with alopecia areata and was treated with 10 mg/mL of intralesional triamcinolone once monthly for 4 months.
The patient initially showed improvement with moderate hair regrowth. After 4 months of treatment, she developed 3 new 1- to 1.5-cm erythematous alopecic patches on the vertex scalp and had worsening in the initial patches (Figure 1). Given the resistance to standard therapy and the onset of multiple new areas with evidence of inflammatory involvement, a punch biopsy was performed. Histopathologic examination revealed a fairly unremarkable epidermis and a dense dermal inflammatory infiltrate that was present both in the superficial and deep dermis (Figure 2). The inflammatory cells, which appeared to be predominantly comprised of lymphocytes, had a predilection for the vasculature but also were observed within the interstitial dermis. Additionally, mucin appeared to be slightly increased in the deep dermis. The lymphocytic phenotype was confirmed by immunohistochemical studies for CD20 and CD3. The most likely possibilities for this reaction pattern were LET, Jessner lymphocytic infiltrate of the skin (JLIS), gyrate erythema, and lymphoma; however, the immunohistochemical studies effectively ruled out lymphoma. Additionally, there was pronounced dermal mucin noted in the specimen. The patient was diagnosed with LET of the scalp based on the constellation of findings.
Comment
The classification of LET as a single unique entity or disease process sui generis has been in flux in the last decade. Its similarities to JLIS and other forms of chronic cutaneous lupus erythematosus (CCLE) have brought debate.4-6 In 1930, Gougerot and Burnier7 documented the first case of LET in the literature, describing smooth, infiltrated, erythematous lesions with no desquamation or other superficial changes seen in 5 patients.
In 2000, interest in LET and other forms of CCLE was increasing, and reports in the literature paralleled. That year, Kuhn et al4 reported 40 cases of LET, characterizing the clinical and histological features of each case to demonstrate that LET should be separate from other forms of CCLE. Until then, it is likely that many lesions that should have been classified as LET were instead classified as various forms of CCLE. The investigators maintained that LET also should be distinct from JLIS because it is associated with UV exposure.4 Kuhn et al8 reviewed phototesting in 60 patients with LET in 2001 and confirmed this subset was the most photosensitive type of lupus erythematosus.
In general, the histopathologic and immunohistochemical studies in LET and JLIS can be quite similar. Relatively distinguishing histopathologic findings in JLIS include no evidence of epidermal atrophy, basal vacuolar change, or follicular plugging, as well as negative immunofluorescence studies. Both entities show a predominantly T-cell population with a smaller component of B cells and thus a distinction cannot be made based on relative proportions of T and B cells in lesions.2
In 2003, Alexiades-Armenakas et al6 determined immunohistochemical criteria for LET, finding a predominance of T cells and more CD4 lymphocytes than CD8 lymphocytes with a mean ratio of roughly 3 to 1. Their study results maintained LET should be classified as a form of CCLE due to the chronicity of the lesions, the serologic profile with negative anti–double-stranded DNA, anticentromere, anti-Smith, anti-Ro/Sjögren syndrome antigen A, anti-La/Sjögren syndrome antigen B, and anti-nuclear ribonucleoprotein antibodies and the rare association with systemic disease.6 This conclusion was further solidified by a review published that same year citing unique histopathological features when compared to subacute cutaneous LE and discoid lupus erythematosus.5
This case illustrates the importance of histologic evaluation in determining the correct diagnosis in a patient with alopecia areata recalcitrant to treatment. Including LET in the differential of alopecic patches on the scalp could prove beneficial for patients, as LET responds well to antimalarial drugs and photoprotection.9 This patient had a normal antinuclear antibody panel and no signs or symptoms of systemic lupus. It was recommended that she avoid sun exposure and begin treatment with hydroxychloroquine but she declined. At a follow-up visit 6 months later she reported the lesions had improved, but a permanent wig had been sewn over the area, so it could not be examined.
Lupus erythematosus tumidus (LET) is a relatively rare condition but may simply be underdiagnosed in the literature. It presents as urticarialike papules and plaques in sun-exposed areas, characterized by induration and erythema. Lesions occur on the face, neck, upper extremities, and trunk and heal without scarring.1,2 Rarely, lesions can show fine scaling and associated pruritus, but most often the lesions are asymptomatic.3
Case Report
A 45-year-old woman presented with 2 asymptomatic self-described bald spots on the top of the head of 2 months’ duration. The patient denied prior treatment of the lesions and noted one patch was resolving. She reported no involvement of the eyebrows, eyelashes, and axillary and pubic hair. A review of systems was negative. The patient denied personal or family history of lupus, thyroid disease, or vitiligo.
Clinical examination revealed a 1.1-cm round patch of nonscarring alopecia on the right vertex scalp and a 0.9-cm round patch of nonscarring alopecia with moderate hair regrowth on the left vertex scalp. There was no erythema, scaling, or induration. The rest of the scalp was normal in appearance and the eyebrows and eyelashes were uninvolved. The patient was diagnosed with alopecia areata and was treated with 10 mg/mL of intralesional triamcinolone once monthly for 4 months.
The patient initially showed improvement with moderate hair regrowth. After 4 months of treatment, she developed 3 new 1- to 1.5-cm erythematous alopecic patches on the vertex scalp and had worsening in the initial patches (Figure 1). Given the resistance to standard therapy and the onset of multiple new areas with evidence of inflammatory involvement, a punch biopsy was performed. Histopathologic examination revealed a fairly unremarkable epidermis and a dense dermal inflammatory infiltrate that was present both in the superficial and deep dermis (Figure 2). The inflammatory cells, which appeared to be predominantly comprised of lymphocytes, had a predilection for the vasculature but also were observed within the interstitial dermis. Additionally, mucin appeared to be slightly increased in the deep dermis. The lymphocytic phenotype was confirmed by immunohistochemical studies for CD20 and CD3. The most likely possibilities for this reaction pattern were LET, Jessner lymphocytic infiltrate of the skin (JLIS), gyrate erythema, and lymphoma; however, the immunohistochemical studies effectively ruled out lymphoma. Additionally, there was pronounced dermal mucin noted in the specimen. The patient was diagnosed with LET of the scalp based on the constellation of findings.
Comment
The classification of LET as a single unique entity or disease process sui generis has been in flux in the last decade. Its similarities to JLIS and other forms of chronic cutaneous lupus erythematosus (CCLE) have brought debate.4-6 In 1930, Gougerot and Burnier7 documented the first case of LET in the literature, describing smooth, infiltrated, erythematous lesions with no desquamation or other superficial changes seen in 5 patients.
In 2000, interest in LET and other forms of CCLE was increasing, and reports in the literature paralleled. That year, Kuhn et al4 reported 40 cases of LET, characterizing the clinical and histological features of each case to demonstrate that LET should be separate from other forms of CCLE. Until then, it is likely that many lesions that should have been classified as LET were instead classified as various forms of CCLE. The investigators maintained that LET also should be distinct from JLIS because it is associated with UV exposure.4 Kuhn et al8 reviewed phototesting in 60 patients with LET in 2001 and confirmed this subset was the most photosensitive type of lupus erythematosus.
In general, the histopathologic and immunohistochemical studies in LET and JLIS can be quite similar. Relatively distinguishing histopathologic findings in JLIS include no evidence of epidermal atrophy, basal vacuolar change, or follicular plugging, as well as negative immunofluorescence studies. Both entities show a predominantly T-cell population with a smaller component of B cells and thus a distinction cannot be made based on relative proportions of T and B cells in lesions.2
In 2003, Alexiades-Armenakas et al6 determined immunohistochemical criteria for LET, finding a predominance of T cells and more CD4 lymphocytes than CD8 lymphocytes with a mean ratio of roughly 3 to 1. Their study results maintained LET should be classified as a form of CCLE due to the chronicity of the lesions, the serologic profile with negative anti–double-stranded DNA, anticentromere, anti-Smith, anti-Ro/Sjögren syndrome antigen A, anti-La/Sjögren syndrome antigen B, and anti-nuclear ribonucleoprotein antibodies and the rare association with systemic disease.6 This conclusion was further solidified by a review published that same year citing unique histopathological features when compared to subacute cutaneous LE and discoid lupus erythematosus.5
This case illustrates the importance of histologic evaluation in determining the correct diagnosis in a patient with alopecia areata recalcitrant to treatment. Including LET in the differential of alopecic patches on the scalp could prove beneficial for patients, as LET responds well to antimalarial drugs and photoprotection.9 This patient had a normal antinuclear antibody panel and no signs or symptoms of systemic lupus. It was recommended that she avoid sun exposure and begin treatment with hydroxychloroquine but she declined. At a follow-up visit 6 months later she reported the lesions had improved, but a permanent wig had been sewn over the area, so it could not be examined.
- Lee L, Werth V. Rheumatologic disease. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 3rd ed. Mosby Elsevier; 2008:615-629.
- Weedon D. The lichenoid reaction pattern. In: Weedon D. Skin Pathology. 2nd ed. Edinburgh, Scotland: Churchill Livingstone; 2002:35-70.
- Dekle CL, Mannes KD, Davis LS, et al. Lupus tumidus. J Am Acad Dermatol. 1999;41:250-253.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Kuhn A, Sonntag M, Ruzicka T, et al. Histopathologic findings in lupus erythematosus tumidus: review of 80 patients. J Am Acad Dermatol. 2003;48:901-908.
- Alexiades-Armenakas MR, Baldassano M, Bince B, et al. Tumid lupus erythematosus: criteria for classification with immunohistochemical analysis. Arthritis Rheum. 2003;49:494-500.
- Gougerot H, Burnier R. Lupuse rythe mateux “tumidus.” Bull Soc Fr Dermatol Syph. 1930;37:1291-1292.
- Kuhn A, Sonntag M, Richter-Hintz D, et al. Phototesting in lupus erythematosus tumidus—review of 60 patients. Photochem Photobiol. 2001;73:532-536.
- Cozzani E, Christana K, Rongioletti F, et al. Lupus erythematosus tumidus: clinical, histopathological and serological aspects and therapy response of 21 patients. Eur J Dermatol. 2010;20:797-801.
- Lee L, Werth V. Rheumatologic disease. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. Vol 2. 3rd ed. Mosby Elsevier; 2008:615-629.
- Weedon D. The lichenoid reaction pattern. In: Weedon D. Skin Pathology. 2nd ed. Edinburgh, Scotland: Churchill Livingstone; 2002:35-70.
- Dekle CL, Mannes KD, Davis LS, et al. Lupus tumidus. J Am Acad Dermatol. 1999;41:250-253.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus—a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Kuhn A, Sonntag M, Ruzicka T, et al. Histopathologic findings in lupus erythematosus tumidus: review of 80 patients. J Am Acad Dermatol. 2003;48:901-908.
- Alexiades-Armenakas MR, Baldassano M, Bince B, et al. Tumid lupus erythematosus: criteria for classification with immunohistochemical analysis. Arthritis Rheum. 2003;49:494-500.
- Gougerot H, Burnier R. Lupuse rythe mateux “tumidus.” Bull Soc Fr Dermatol Syph. 1930;37:1291-1292.
- Kuhn A, Sonntag M, Richter-Hintz D, et al. Phototesting in lupus erythematosus tumidus—review of 60 patients. Photochem Photobiol. 2001;73:532-536.
- Cozzani E, Christana K, Rongioletti F, et al. Lupus erythematosus tumidus: clinical, histopathological and serological aspects and therapy response of 21 patients. Eur J Dermatol. 2010;20:797-801.
Practice Points
- Lupus erythematosus tumidus (LET) of the scalp can mimic alopecia areata on clinical presentation.
- A unique variant of chronic cutaneous lupus erythematosus, LET presents in sun-exposed areas without any corresponding systemic signs.
- Lupus erythematosus tumidus may respond well to antimalarial drugs.