User login
Resolution of Psoriatic Lesions on the Gingiva and Hard Palate Following Administration of Adalimumab for Cutaneous Psoriasis
Psoriasis is a chronic, relapsing, inflammatory systemic disorder of the skin with an incidence of 2% to 3% and is estimated to affect 125 million individuals worldwide.1 Environmental triggers of disease modulation may include cutaneous microbiota, smoking, alcohol use, drugs (ie, beta-blockers, lithium, antimalarials), stress, and trauma.2 Comorbidities associated with cutaneous lesions include psoriatic arthritis, Crohn disease, type 2 diabetes mellitus, metabolic syndrome, stroke, and cardiovascular disease.3 In some studies, patients with psoriasis also had a 24% to 27% increased propensity for periodontal bone loss versus 10% of controls.4,5
Oral psoriasis is rare and case reports have been preferentially published in dental journals, usually with regard to glossal lesions, leaving gingival and palatal psoriatic involvement infrequently reported in the dermatologic literature.6,7 In fact, oral assessments involving 535 psoriatic patients from a dermatology center only yielded cases of geographic and fissured tongue.8 Another study at a psoriasis clinic found 3.8% (21/547) of patients with geographic tongue, 3.1% (17/547) with buccal mucosal plaques, and only 0.4% (2/547) with palatal lesions.9 To extend the knowledge of oral psoriasis, we provide the clinical and histopathologic findings of a patient with synchronous oral and cutaneous psoriatic lesions that responded well to the administration of adalimumab for management of recurrent cutaneous disease.
Case Report
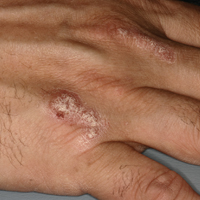
A 51-year-old man presented to the attending periodontist for comprehensive treatment of multiple quadrants of gingival recession. His medical history was remarkable for psoriasis; Prinzmetal angina, which led to myocardial infarction; and diverticulitis. The cutaneous psoriasis began approximately 18 years prior to the current presentation and was initially managed with various topical therapeutics. At an 11-year follow-up, the patient was experiencing poor lesional control as well as severe pruritus and was prescribed etanercept by a dermatologist. His inconsistent compliance with frequency and dosing failed to achieve satisfactory disease suppression and etanercept was discontinued after approximately 2.5 years. Two years later the patient was switched to adalimumab by a dermatologist, and around this time he had developed psoriatic arthritis of the hands and knees and pitting of the nail plates. The patient elected to discontinue adalimumab usage after 3 years due to successful management of the skin lesions, cost considerations, and his perception that the psoriasis could “remain in remission.” After a 6-month lapse, the patient resumed adalimumab due to cutaneous lesional recurrence (Figure 1A).
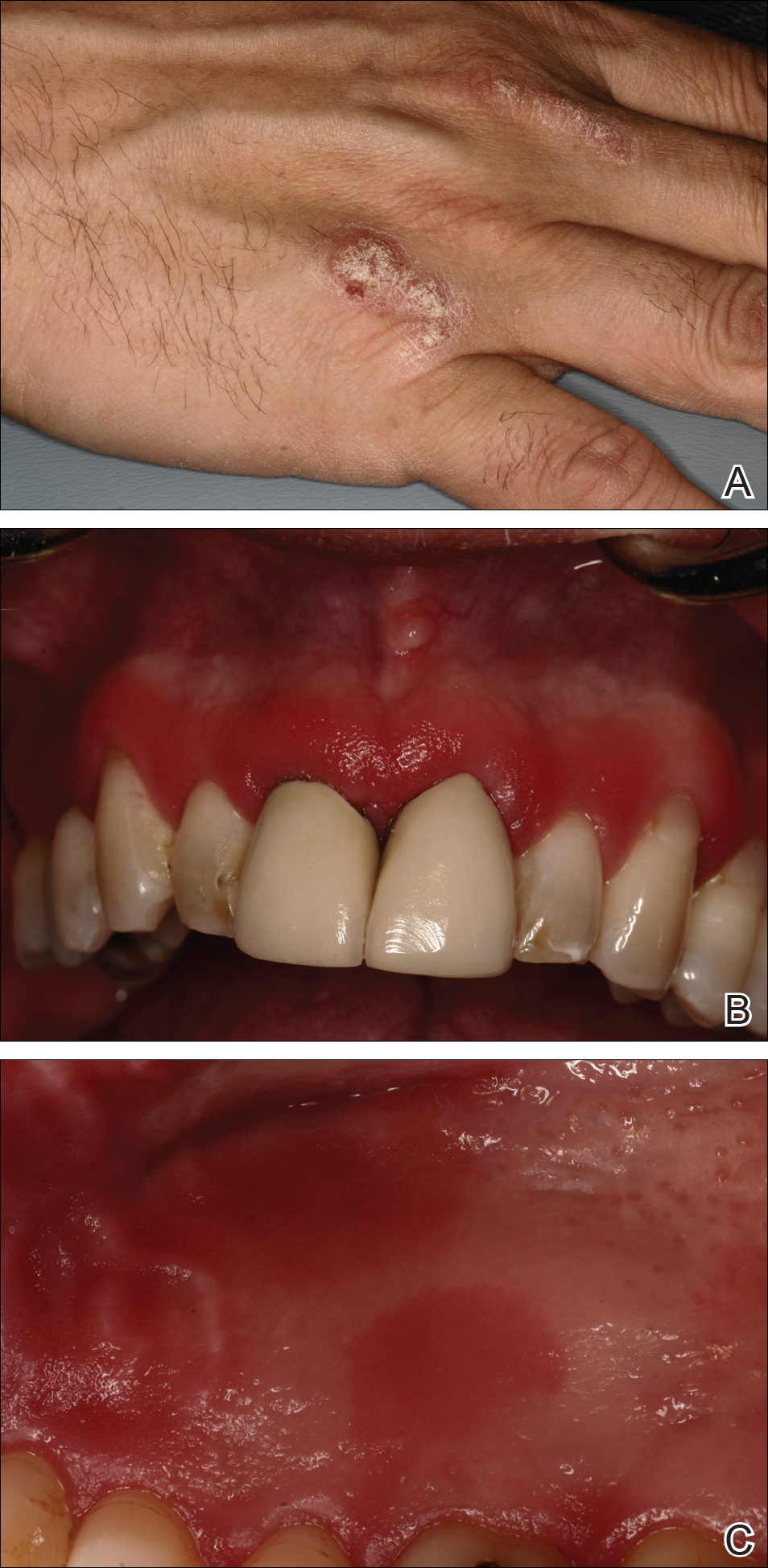
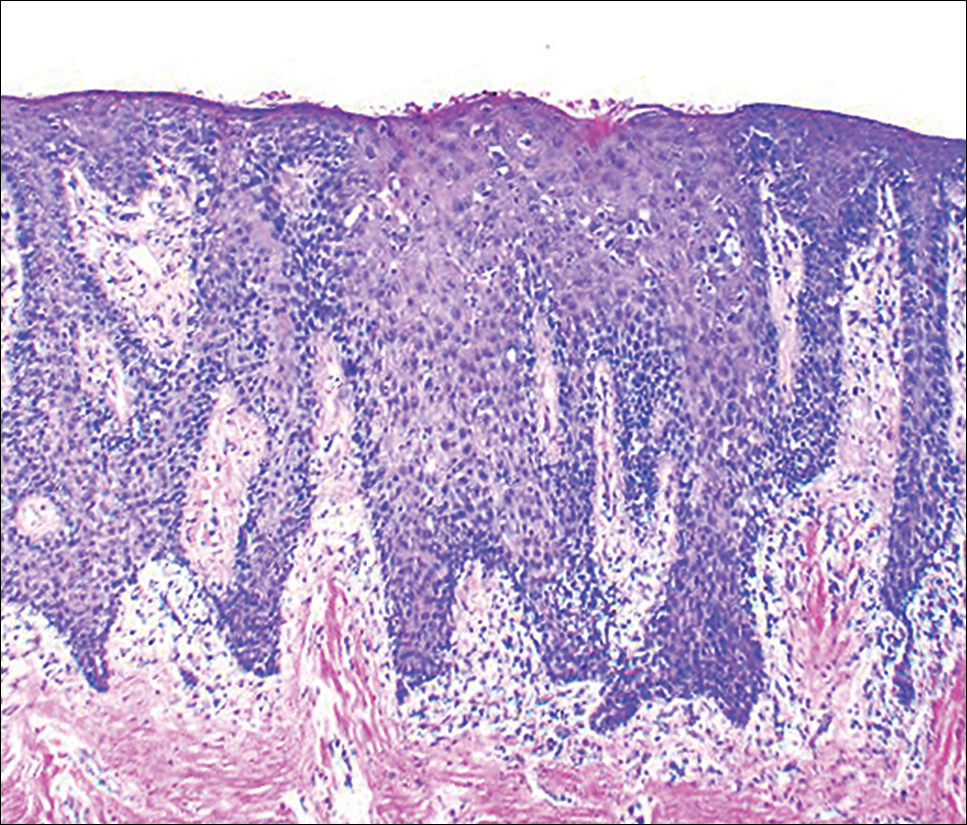
At the current presentation, an oral examination performed 2 days after the reinstitution of adalim-umab revealed generalized severe gingivitis with an atypical inflammatory response that extended from just beyond the mucogingival junction to the marginal gingiva. The gingiva also appeared edematous with a conspicuously granular surface (Figure 1B). The hard palate displayed multiple red macules of varying sizes (Figure 1C). A maxillary gingival biopsy demonstrated hyperkeratosis, parakeratosis, spongiosis, acanthosis, elongation of the rete ridges, numerous collections of neutrophils (Munro microabscesses), and abundant lymphocytes in the subjacent connective tissue (Figure 2). Periodic acid–Schiff staining was negative for fungal hyphae. These features were consistent with oral mucosal psoriasis.
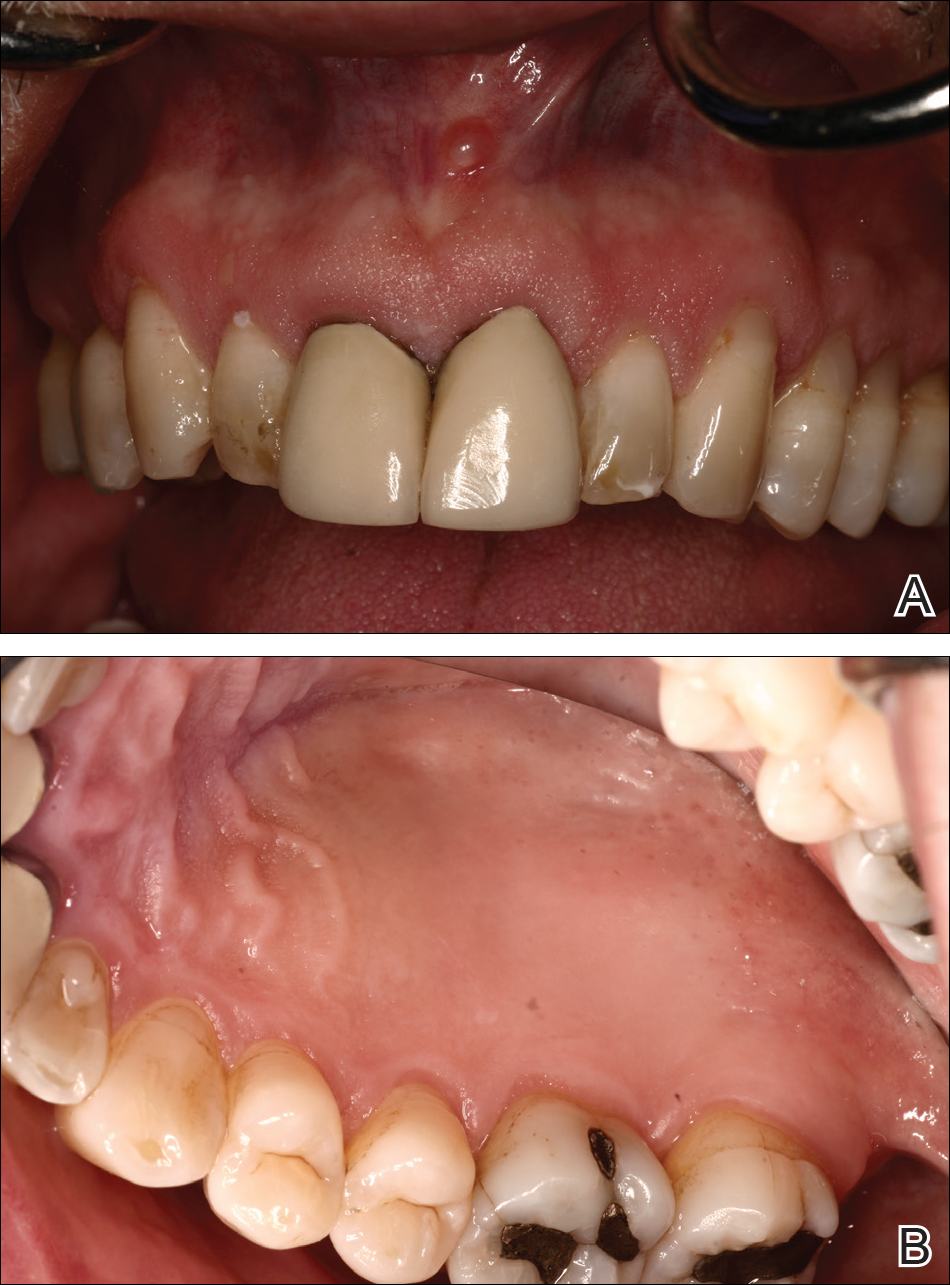
At a 2-month follow-up, the biopsy site had healed without incident and without loss of the gingival architecture. There was an almost-complete resolution of the gingival erythema (Figure 3A) and the patient has since noticed a lack of bleeding using floss. Additionally, the red macules on the palate were no longer present (Figure 3B). The cutaneous plaques were greatly reduced in size and the patient experienced a proportionate decline in pruritus. Based on the uneventful surgical biopsy procedure, the patient was advised to undergo gingival grafting and has not returned for periodontal care.
Comment
Psoriasis of the oral cavity is rare and typically occurs on the tongue and less frequently on the hard palate, lip, buccal mucosa, and gingiva.2,7 The lesions are almost always concordant with cutaneous psoriasis, and only sporadic examples exclusive to the oral mucosa have been recognized.7,10 Gingival psoriasis usually is described as intensely erythematous and occasionally laced with white scaly streaks involving the marginal gingiva that extend toward the mucogingival junction. In general, the erythematous presentation of gingival psoriasis may not be commensurate with the degree of inflammation induced by dental plaque-based periodontal disease. Doben11 documented gingival psoriasis as appearing “deeply stippled and grainy” and commented that the tissue was “friable” and incapable of maintaining a “clean incision line” during periodontal surgery. In our patient, the gingiva also had exhibited a granular surface. Patients with oral psoriasis often report soreness or a burning sensation of the gingiva, which may easily bleed on manipulation or brushing the teeth, whereas other patients are asymptomatic,12 as in our case. Psoriasis of the hard palate usually presents as multiple painless red macules. Unlike cutaneous psoriasis, oral lesions rarely evoke pruritus.10 Histopathologically, oral psoriasis bears a striking resemblance to its cutaneous counterpart. The epithelium has a pronounced parakeratinized surface with elongated rete ridges and aggregations of Munro microabscesses. The connective tissue often is composed of dilated capillaries that closely approximate the epithelium as well as infiltrations of lymphocytes. Specimens suspected for oral psoriasis should routinely be stained with periodic acid–Schiff to rule out candidiasis coinfection. The microscopic findings of our patient were congruent with prior reports of oral psoriasis.7,10-12 Some clinicians have questioned if psoriasis can actually occur in the oral cavity, but most authorities in the field have recognized its true existence, as evidenced by various shared HLA antigens, specifically HLA-Cw.13
Another group of oral lesions collectively referred to as psoriasiform mucositis, notably geographic tongue (benign migratory glossitis, erythema migrans) and its extraglossal variant geographic stomatitis,14,15 have histopathologic features and HLAs similar to those seen in cutaneous psoriasis.13 Interestingly, geographic tongue has been found in 3.8% to 9.1% of cohorts with cutaneous psoriasis,8,9 but in the extant population, the vast majority of patients with oral psoriasiform mucositis do not have cutaneous psoriasis. Other differential diagnoses for gingival psoriasis are lichen planus, human immunodeficiency virus–associated periodontitis, desquamative gingivitis, plasma cell gingivitis, erythematous candidiasis, mucous membrane pemphigoid, pemphigus vulgaris, leukemia, systemic lupus erythematosus, granulomatosis with polyangiitis, orofacial granulomatosis, localized juvenile spongiotic gingivitis hyperplasia, and primary gingivostomatitis.
Management of gingival psoriasis focuses on strategies to reduce inflammation and discomfort and measures to achieve meticulous oral plaque control. Judicious efforts should be exercised to avoid oral soft-tissue injury when performing periodontal scaling, although it has not been established whether gingival psoriasis is associated with the Köbner phenomenon, as seen with cutaneous lesions. Adjunctive measures employed for symptomatic patients have involved the use of corticosteroids (eg, lesional injection, oral rinse, systemic) and oral rinses with retinoic acid, chlorhexidine gluconate, and warm saline.7,10,16 Prolonged utilization of corticosteroids, however, may necessitate supplemental administration of antifungal agents.
This case report represents a rare documentation of a successful outcome of gingival and palatal psoriasis subsequent to the reinstitution of adalimumab solely for treatment of recurrent cutaneous disease. There likely is a pharmacologic basis for the amelioration of oral psoriasis in our patient. Adalimumab is a bivalent IgG monoclonal antibody that binds to activated dermal dendritic cell receptors of tumor necrosis factor α, thereby attenuating a cytokine-derived inflammatory response and apoptosis.17 In fact, patients with rheumatoid arthritis showed notable reductions in both gingival inflammation and bleeding following a 3-month regimen of adalimumab.18
Conclusion
Practitioners should be aware of the phenotypic overlap of cutaneous and oral psoriasis, particularly involving the gingiva and palate. It is recommended that psoriasis patients routinely receive a dental prophylaxis and engage in oral hygiene efforts to reduce the presence of oral microbiota. Furthermore, it is emphasized that psoriatic patients who maintain an atypical erythematous presentation on the oral mucosa undergo a biopsy for identification of the lesions and correlation with disease dissemination. Prospective studies are needed to characterize the clinical courses of oral psoriasis, ascertain their correlative behavior with cutaneous flares, and determine if lesional improvement can be achieved with the use of biologic agents or other therapeutic modalities.
- Gupta R, Debbaneh MG, Liao W. Genetic epidemiology of psoriasis. Curr Dermatol Rep. 2014;3:61-78.
- Younai FS, Phelan JA. Oral mucositis with features of psoriasis: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:61-67.
- Xu T, Zhang YH. Association of psoriasis with stroke and myocardial infarction: meta-analysis of cohort studies. Br J Dermatol. 2012;167:1345-1350.
- Lazaridou E, Tsikrikoni A, Fotiadou C, et al. Association of chronic plaque psoriasis and severe periodontitis: a hospital based case-control study. J Eur Acad Dermatol Venereol. 2013;27:967-972.
- Skudutyte-Rysstad R, Slevolden EM, Hansen BF, et al. Association between moderate to severe psoriasis and periodontitis in a Scandinavian population. BMC Oral Health. 2014;14:139.
- Zunt SL, Tomich CE. Erythema migrans—a psoriasiform lesion of the oral mucosa. J Dermatol Surg Oncol. 1989;15:1067-1070.
- Reis V, Artico G, Seo J, et al. Psoriasiform mucositis on the gingival and palatal mucosae treated with retinoic-acid mouthwash. Int J Dermatol. 2013;52:113-115.
- Germi L, De Giorgi V, Bergamo F, et al. Psoriasis and oral lesions: multicentric study of oral mucosa diseases Italian group (GIPMO). Dermatol Online J. 2012;18:11.
- Kaur I, Handa S, Kumar B. Oral lesions in psoriasis. Int J Dermatol. 1997;36:78-79.
- Brayshaw HA, Orban B. Psoriasis gingivae. J Periodontol. 1953;24:156-160.
- Doben DI. Psoriasis of the attached gingiva. J Periodontol. 1976;47:38-40.
- Mattsson U, Warfvinge G, Jontell M. Oral psoriasis—a diagnostic dilemma: a report of two cases and a review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;120:e183-e189.
- Dermatologic diseases. In: Neville BW, Damm DD, Allen CM, et al, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: Saunders/Elsevier; 2009:792-794.
- Brooks JK, Balciunas BA. Geographic stomatitis: review of the literature and report of five cases. J Am Dent Assoc. 1987;115:421-424.
- Brooks JK, Nikitakis NG. Multiple mucosal lesions. erythema migrans. Gen Dent. 2007;55:160, 163.
- Ulmansky M, Michelle R, Azaz B. Oral psoriasis: report of six new cases. J Oral Pathol Med. 1995;24:42-45.
- Lis K, Kuzawinska O, Bałkowiec-Iskra E. Tumor necrosis factor inhibitors—state of knowledge. Arch Med Sci. 2014;10:1175-1185.
- Kobayashi T, Yokoyama T, Ito S, et al. Periodontal and serum protein profiles in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitor adalimumab. J Periodontol. 2014;85:1480-1488.
Psoriasis is a chronic, relapsing, inflammatory systemic disorder of the skin with an incidence of 2% to 3% and is estimated to affect 125 million individuals worldwide.1 Environmental triggers of disease modulation may include cutaneous microbiota, smoking, alcohol use, drugs (ie, beta-blockers, lithium, antimalarials), stress, and trauma.2 Comorbidities associated with cutaneous lesions include psoriatic arthritis, Crohn disease, type 2 diabetes mellitus, metabolic syndrome, stroke, and cardiovascular disease.3 In some studies, patients with psoriasis also had a 24% to 27% increased propensity for periodontal bone loss versus 10% of controls.4,5
Oral psoriasis is rare and case reports have been preferentially published in dental journals, usually with regard to glossal lesions, leaving gingival and palatal psoriatic involvement infrequently reported in the dermatologic literature.6,7 In fact, oral assessments involving 535 psoriatic patients from a dermatology center only yielded cases of geographic and fissured tongue.8 Another study at a psoriasis clinic found 3.8% (21/547) of patients with geographic tongue, 3.1% (17/547) with buccal mucosal plaques, and only 0.4% (2/547) with palatal lesions.9 To extend the knowledge of oral psoriasis, we provide the clinical and histopathologic findings of a patient with synchronous oral and cutaneous psoriatic lesions that responded well to the administration of adalimumab for management of recurrent cutaneous disease.
Case Report
A 51-year-old man presented to the attending periodontist for comprehensive treatment of multiple quadrants of gingival recession. His medical history was remarkable for psoriasis; Prinzmetal angina, which led to myocardial infarction; and diverticulitis. The cutaneous psoriasis began approximately 18 years prior to the current presentation and was initially managed with various topical therapeutics. At an 11-year follow-up, the patient was experiencing poor lesional control as well as severe pruritus and was prescribed etanercept by a dermatologist. His inconsistent compliance with frequency and dosing failed to achieve satisfactory disease suppression and etanercept was discontinued after approximately 2.5 years. Two years later the patient was switched to adalimumab by a dermatologist, and around this time he had developed psoriatic arthritis of the hands and knees and pitting of the nail plates. The patient elected to discontinue adalimumab usage after 3 years due to successful management of the skin lesions, cost considerations, and his perception that the psoriasis could “remain in remission.” After a 6-month lapse, the patient resumed adalimumab due to cutaneous lesional recurrence (Figure 1A).
At the current presentation, an oral examination performed 2 days after the reinstitution of adalim-umab revealed generalized severe gingivitis with an atypical inflammatory response that extended from just beyond the mucogingival junction to the marginal gingiva. The gingiva also appeared edematous with a conspicuously granular surface (Figure 1B). The hard palate displayed multiple red macules of varying sizes (Figure 1C). A maxillary gingival biopsy demonstrated hyperkeratosis, parakeratosis, spongiosis, acanthosis, elongation of the rete ridges, numerous collections of neutrophils (Munro microabscesses), and abundant lymphocytes in the subjacent connective tissue (Figure 2). Periodic acid–Schiff staining was negative for fungal hyphae. These features were consistent with oral mucosal psoriasis.
At a 2-month follow-up, the biopsy site had healed without incident and without loss of the gingival architecture. There was an almost-complete resolution of the gingival erythema (Figure 3A) and the patient has since noticed a lack of bleeding using floss. Additionally, the red macules on the palate were no longer present (Figure 3B). The cutaneous plaques were greatly reduced in size and the patient experienced a proportionate decline in pruritus. Based on the uneventful surgical biopsy procedure, the patient was advised to undergo gingival grafting and has not returned for periodontal care.
Comment
Psoriasis of the oral cavity is rare and typically occurs on the tongue and less frequently on the hard palate, lip, buccal mucosa, and gingiva.2,7 The lesions are almost always concordant with cutaneous psoriasis, and only sporadic examples exclusive to the oral mucosa have been recognized.7,10 Gingival psoriasis usually is described as intensely erythematous and occasionally laced with white scaly streaks involving the marginal gingiva that extend toward the mucogingival junction. In general, the erythematous presentation of gingival psoriasis may not be commensurate with the degree of inflammation induced by dental plaque-based periodontal disease. Doben11 documented gingival psoriasis as appearing “deeply stippled and grainy” and commented that the tissue was “friable” and incapable of maintaining a “clean incision line” during periodontal surgery. In our patient, the gingiva also had exhibited a granular surface. Patients with oral psoriasis often report soreness or a burning sensation of the gingiva, which may easily bleed on manipulation or brushing the teeth, whereas other patients are asymptomatic,12 as in our case. Psoriasis of the hard palate usually presents as multiple painless red macules. Unlike cutaneous psoriasis, oral lesions rarely evoke pruritus.10 Histopathologically, oral psoriasis bears a striking resemblance to its cutaneous counterpart. The epithelium has a pronounced parakeratinized surface with elongated rete ridges and aggregations of Munro microabscesses. The connective tissue often is composed of dilated capillaries that closely approximate the epithelium as well as infiltrations of lymphocytes. Specimens suspected for oral psoriasis should routinely be stained with periodic acid–Schiff to rule out candidiasis coinfection. The microscopic findings of our patient were congruent with prior reports of oral psoriasis.7,10-12 Some clinicians have questioned if psoriasis can actually occur in the oral cavity, but most authorities in the field have recognized its true existence, as evidenced by various shared HLA antigens, specifically HLA-Cw.13
Another group of oral lesions collectively referred to as psoriasiform mucositis, notably geographic tongue (benign migratory glossitis, erythema migrans) and its extraglossal variant geographic stomatitis,14,15 have histopathologic features and HLAs similar to those seen in cutaneous psoriasis.13 Interestingly, geographic tongue has been found in 3.8% to 9.1% of cohorts with cutaneous psoriasis,8,9 but in the extant population, the vast majority of patients with oral psoriasiform mucositis do not have cutaneous psoriasis. Other differential diagnoses for gingival psoriasis are lichen planus, human immunodeficiency virus–associated periodontitis, desquamative gingivitis, plasma cell gingivitis, erythematous candidiasis, mucous membrane pemphigoid, pemphigus vulgaris, leukemia, systemic lupus erythematosus, granulomatosis with polyangiitis, orofacial granulomatosis, localized juvenile spongiotic gingivitis hyperplasia, and primary gingivostomatitis.
Management of gingival psoriasis focuses on strategies to reduce inflammation and discomfort and measures to achieve meticulous oral plaque control. Judicious efforts should be exercised to avoid oral soft-tissue injury when performing periodontal scaling, although it has not been established whether gingival psoriasis is associated with the Köbner phenomenon, as seen with cutaneous lesions. Adjunctive measures employed for symptomatic patients have involved the use of corticosteroids (eg, lesional injection, oral rinse, systemic) and oral rinses with retinoic acid, chlorhexidine gluconate, and warm saline.7,10,16 Prolonged utilization of corticosteroids, however, may necessitate supplemental administration of antifungal agents.
This case report represents a rare documentation of a successful outcome of gingival and palatal psoriasis subsequent to the reinstitution of adalimumab solely for treatment of recurrent cutaneous disease. There likely is a pharmacologic basis for the amelioration of oral psoriasis in our patient. Adalimumab is a bivalent IgG monoclonal antibody that binds to activated dermal dendritic cell receptors of tumor necrosis factor α, thereby attenuating a cytokine-derived inflammatory response and apoptosis.17 In fact, patients with rheumatoid arthritis showed notable reductions in both gingival inflammation and bleeding following a 3-month regimen of adalimumab.18
Conclusion
Practitioners should be aware of the phenotypic overlap of cutaneous and oral psoriasis, particularly involving the gingiva and palate. It is recommended that psoriasis patients routinely receive a dental prophylaxis and engage in oral hygiene efforts to reduce the presence of oral microbiota. Furthermore, it is emphasized that psoriatic patients who maintain an atypical erythematous presentation on the oral mucosa undergo a biopsy for identification of the lesions and correlation with disease dissemination. Prospective studies are needed to characterize the clinical courses of oral psoriasis, ascertain their correlative behavior with cutaneous flares, and determine if lesional improvement can be achieved with the use of biologic agents or other therapeutic modalities.
Psoriasis is a chronic, relapsing, inflammatory systemic disorder of the skin with an incidence of 2% to 3% and is estimated to affect 125 million individuals worldwide.1 Environmental triggers of disease modulation may include cutaneous microbiota, smoking, alcohol use, drugs (ie, beta-blockers, lithium, antimalarials), stress, and trauma.2 Comorbidities associated with cutaneous lesions include psoriatic arthritis, Crohn disease, type 2 diabetes mellitus, metabolic syndrome, stroke, and cardiovascular disease.3 In some studies, patients with psoriasis also had a 24% to 27% increased propensity for periodontal bone loss versus 10% of controls.4,5
Oral psoriasis is rare and case reports have been preferentially published in dental journals, usually with regard to glossal lesions, leaving gingival and palatal psoriatic involvement infrequently reported in the dermatologic literature.6,7 In fact, oral assessments involving 535 psoriatic patients from a dermatology center only yielded cases of geographic and fissured tongue.8 Another study at a psoriasis clinic found 3.8% (21/547) of patients with geographic tongue, 3.1% (17/547) with buccal mucosal plaques, and only 0.4% (2/547) with palatal lesions.9 To extend the knowledge of oral psoriasis, we provide the clinical and histopathologic findings of a patient with synchronous oral and cutaneous psoriatic lesions that responded well to the administration of adalimumab for management of recurrent cutaneous disease.
Case Report
A 51-year-old man presented to the attending periodontist for comprehensive treatment of multiple quadrants of gingival recession. His medical history was remarkable for psoriasis; Prinzmetal angina, which led to myocardial infarction; and diverticulitis. The cutaneous psoriasis began approximately 18 years prior to the current presentation and was initially managed with various topical therapeutics. At an 11-year follow-up, the patient was experiencing poor lesional control as well as severe pruritus and was prescribed etanercept by a dermatologist. His inconsistent compliance with frequency and dosing failed to achieve satisfactory disease suppression and etanercept was discontinued after approximately 2.5 years. Two years later the patient was switched to adalimumab by a dermatologist, and around this time he had developed psoriatic arthritis of the hands and knees and pitting of the nail plates. The patient elected to discontinue adalimumab usage after 3 years due to successful management of the skin lesions, cost considerations, and his perception that the psoriasis could “remain in remission.” After a 6-month lapse, the patient resumed adalimumab due to cutaneous lesional recurrence (Figure 1A).
At the current presentation, an oral examination performed 2 days after the reinstitution of adalim-umab revealed generalized severe gingivitis with an atypical inflammatory response that extended from just beyond the mucogingival junction to the marginal gingiva. The gingiva also appeared edematous with a conspicuously granular surface (Figure 1B). The hard palate displayed multiple red macules of varying sizes (Figure 1C). A maxillary gingival biopsy demonstrated hyperkeratosis, parakeratosis, spongiosis, acanthosis, elongation of the rete ridges, numerous collections of neutrophils (Munro microabscesses), and abundant lymphocytes in the subjacent connective tissue (Figure 2). Periodic acid–Schiff staining was negative for fungal hyphae. These features were consistent with oral mucosal psoriasis.
At a 2-month follow-up, the biopsy site had healed without incident and without loss of the gingival architecture. There was an almost-complete resolution of the gingival erythema (Figure 3A) and the patient has since noticed a lack of bleeding using floss. Additionally, the red macules on the palate were no longer present (Figure 3B). The cutaneous plaques were greatly reduced in size and the patient experienced a proportionate decline in pruritus. Based on the uneventful surgical biopsy procedure, the patient was advised to undergo gingival grafting and has not returned for periodontal care.
Comment
Psoriasis of the oral cavity is rare and typically occurs on the tongue and less frequently on the hard palate, lip, buccal mucosa, and gingiva.2,7 The lesions are almost always concordant with cutaneous psoriasis, and only sporadic examples exclusive to the oral mucosa have been recognized.7,10 Gingival psoriasis usually is described as intensely erythematous and occasionally laced with white scaly streaks involving the marginal gingiva that extend toward the mucogingival junction. In general, the erythematous presentation of gingival psoriasis may not be commensurate with the degree of inflammation induced by dental plaque-based periodontal disease. Doben11 documented gingival psoriasis as appearing “deeply stippled and grainy” and commented that the tissue was “friable” and incapable of maintaining a “clean incision line” during periodontal surgery. In our patient, the gingiva also had exhibited a granular surface. Patients with oral psoriasis often report soreness or a burning sensation of the gingiva, which may easily bleed on manipulation or brushing the teeth, whereas other patients are asymptomatic,12 as in our case. Psoriasis of the hard palate usually presents as multiple painless red macules. Unlike cutaneous psoriasis, oral lesions rarely evoke pruritus.10 Histopathologically, oral psoriasis bears a striking resemblance to its cutaneous counterpart. The epithelium has a pronounced parakeratinized surface with elongated rete ridges and aggregations of Munro microabscesses. The connective tissue often is composed of dilated capillaries that closely approximate the epithelium as well as infiltrations of lymphocytes. Specimens suspected for oral psoriasis should routinely be stained with periodic acid–Schiff to rule out candidiasis coinfection. The microscopic findings of our patient were congruent with prior reports of oral psoriasis.7,10-12 Some clinicians have questioned if psoriasis can actually occur in the oral cavity, but most authorities in the field have recognized its true existence, as evidenced by various shared HLA antigens, specifically HLA-Cw.13
Another group of oral lesions collectively referred to as psoriasiform mucositis, notably geographic tongue (benign migratory glossitis, erythema migrans) and its extraglossal variant geographic stomatitis,14,15 have histopathologic features and HLAs similar to those seen in cutaneous psoriasis.13 Interestingly, geographic tongue has been found in 3.8% to 9.1% of cohorts with cutaneous psoriasis,8,9 but in the extant population, the vast majority of patients with oral psoriasiform mucositis do not have cutaneous psoriasis. Other differential diagnoses for gingival psoriasis are lichen planus, human immunodeficiency virus–associated periodontitis, desquamative gingivitis, plasma cell gingivitis, erythematous candidiasis, mucous membrane pemphigoid, pemphigus vulgaris, leukemia, systemic lupus erythematosus, granulomatosis with polyangiitis, orofacial granulomatosis, localized juvenile spongiotic gingivitis hyperplasia, and primary gingivostomatitis.
Management of gingival psoriasis focuses on strategies to reduce inflammation and discomfort and measures to achieve meticulous oral plaque control. Judicious efforts should be exercised to avoid oral soft-tissue injury when performing periodontal scaling, although it has not been established whether gingival psoriasis is associated with the Köbner phenomenon, as seen with cutaneous lesions. Adjunctive measures employed for symptomatic patients have involved the use of corticosteroids (eg, lesional injection, oral rinse, systemic) and oral rinses with retinoic acid, chlorhexidine gluconate, and warm saline.7,10,16 Prolonged utilization of corticosteroids, however, may necessitate supplemental administration of antifungal agents.
This case report represents a rare documentation of a successful outcome of gingival and palatal psoriasis subsequent to the reinstitution of adalimumab solely for treatment of recurrent cutaneous disease. There likely is a pharmacologic basis for the amelioration of oral psoriasis in our patient. Adalimumab is a bivalent IgG monoclonal antibody that binds to activated dermal dendritic cell receptors of tumor necrosis factor α, thereby attenuating a cytokine-derived inflammatory response and apoptosis.17 In fact, patients with rheumatoid arthritis showed notable reductions in both gingival inflammation and bleeding following a 3-month regimen of adalimumab.18
Conclusion
Practitioners should be aware of the phenotypic overlap of cutaneous and oral psoriasis, particularly involving the gingiva and palate. It is recommended that psoriasis patients routinely receive a dental prophylaxis and engage in oral hygiene efforts to reduce the presence of oral microbiota. Furthermore, it is emphasized that psoriatic patients who maintain an atypical erythematous presentation on the oral mucosa undergo a biopsy for identification of the lesions and correlation with disease dissemination. Prospective studies are needed to characterize the clinical courses of oral psoriasis, ascertain their correlative behavior with cutaneous flares, and determine if lesional improvement can be achieved with the use of biologic agents or other therapeutic modalities.
- Gupta R, Debbaneh MG, Liao W. Genetic epidemiology of psoriasis. Curr Dermatol Rep. 2014;3:61-78.
- Younai FS, Phelan JA. Oral mucositis with features of psoriasis: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:61-67.
- Xu T, Zhang YH. Association of psoriasis with stroke and myocardial infarction: meta-analysis of cohort studies. Br J Dermatol. 2012;167:1345-1350.
- Lazaridou E, Tsikrikoni A, Fotiadou C, et al. Association of chronic plaque psoriasis and severe periodontitis: a hospital based case-control study. J Eur Acad Dermatol Venereol. 2013;27:967-972.
- Skudutyte-Rysstad R, Slevolden EM, Hansen BF, et al. Association between moderate to severe psoriasis and periodontitis in a Scandinavian population. BMC Oral Health. 2014;14:139.
- Zunt SL, Tomich CE. Erythema migrans—a psoriasiform lesion of the oral mucosa. J Dermatol Surg Oncol. 1989;15:1067-1070.
- Reis V, Artico G, Seo J, et al. Psoriasiform mucositis on the gingival and palatal mucosae treated with retinoic-acid mouthwash. Int J Dermatol. 2013;52:113-115.
- Germi L, De Giorgi V, Bergamo F, et al. Psoriasis and oral lesions: multicentric study of oral mucosa diseases Italian group (GIPMO). Dermatol Online J. 2012;18:11.
- Kaur I, Handa S, Kumar B. Oral lesions in psoriasis. Int J Dermatol. 1997;36:78-79.
- Brayshaw HA, Orban B. Psoriasis gingivae. J Periodontol. 1953;24:156-160.
- Doben DI. Psoriasis of the attached gingiva. J Periodontol. 1976;47:38-40.
- Mattsson U, Warfvinge G, Jontell M. Oral psoriasis—a diagnostic dilemma: a report of two cases and a review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;120:e183-e189.
- Dermatologic diseases. In: Neville BW, Damm DD, Allen CM, et al, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: Saunders/Elsevier; 2009:792-794.
- Brooks JK, Balciunas BA. Geographic stomatitis: review of the literature and report of five cases. J Am Dent Assoc. 1987;115:421-424.
- Brooks JK, Nikitakis NG. Multiple mucosal lesions. erythema migrans. Gen Dent. 2007;55:160, 163.
- Ulmansky M, Michelle R, Azaz B. Oral psoriasis: report of six new cases. J Oral Pathol Med. 1995;24:42-45.
- Lis K, Kuzawinska O, Bałkowiec-Iskra E. Tumor necrosis factor inhibitors—state of knowledge. Arch Med Sci. 2014;10:1175-1185.
- Kobayashi T, Yokoyama T, Ito S, et al. Periodontal and serum protein profiles in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitor adalimumab. J Periodontol. 2014;85:1480-1488.
- Gupta R, Debbaneh MG, Liao W. Genetic epidemiology of psoriasis. Curr Dermatol Rep. 2014;3:61-78.
- Younai FS, Phelan JA. Oral mucositis with features of psoriasis: report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:61-67.
- Xu T, Zhang YH. Association of psoriasis with stroke and myocardial infarction: meta-analysis of cohort studies. Br J Dermatol. 2012;167:1345-1350.
- Lazaridou E, Tsikrikoni A, Fotiadou C, et al. Association of chronic plaque psoriasis and severe periodontitis: a hospital based case-control study. J Eur Acad Dermatol Venereol. 2013;27:967-972.
- Skudutyte-Rysstad R, Slevolden EM, Hansen BF, et al. Association between moderate to severe psoriasis and periodontitis in a Scandinavian population. BMC Oral Health. 2014;14:139.
- Zunt SL, Tomich CE. Erythema migrans—a psoriasiform lesion of the oral mucosa. J Dermatol Surg Oncol. 1989;15:1067-1070.
- Reis V, Artico G, Seo J, et al. Psoriasiform mucositis on the gingival and palatal mucosae treated with retinoic-acid mouthwash. Int J Dermatol. 2013;52:113-115.
- Germi L, De Giorgi V, Bergamo F, et al. Psoriasis and oral lesions: multicentric study of oral mucosa diseases Italian group (GIPMO). Dermatol Online J. 2012;18:11.
- Kaur I, Handa S, Kumar B. Oral lesions in psoriasis. Int J Dermatol. 1997;36:78-79.
- Brayshaw HA, Orban B. Psoriasis gingivae. J Periodontol. 1953;24:156-160.
- Doben DI. Psoriasis of the attached gingiva. J Periodontol. 1976;47:38-40.
- Mattsson U, Warfvinge G, Jontell M. Oral psoriasis—a diagnostic dilemma: a report of two cases and a review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol. 2015;120:e183-e189.
- Dermatologic diseases. In: Neville BW, Damm DD, Allen CM, et al, eds. Oral and Maxillofacial Pathology. 3rd ed. St. Louis, MO: Saunders/Elsevier; 2009:792-794.
- Brooks JK, Balciunas BA. Geographic stomatitis: review of the literature and report of five cases. J Am Dent Assoc. 1987;115:421-424.
- Brooks JK, Nikitakis NG. Multiple mucosal lesions. erythema migrans. Gen Dent. 2007;55:160, 163.
- Ulmansky M, Michelle R, Azaz B. Oral psoriasis: report of six new cases. J Oral Pathol Med. 1995;24:42-45.
- Lis K, Kuzawinska O, Bałkowiec-Iskra E. Tumor necrosis factor inhibitors—state of knowledge. Arch Med Sci. 2014;10:1175-1185.
- Kobayashi T, Yokoyama T, Ito S, et al. Periodontal and serum protein profiles in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitor adalimumab. J Periodontol. 2014;85:1480-1488.
Practice Points
- A subset of patients with cutaneous psoriasis may be associated with oral psoriatic outbreaks.
- Oral psoriasis presents as an atypical inflammatory response, and histopathologic assessment is recommended for lesional identity.
- Use of adalimumab for management of cutaneous psoriasis may demonstrate efficacy for oral psoriasis.
Diagnosis of Severe Acute Lower Gastrointestinal Bleeding with CTA
Case
A 31-year-old white man presented to the ED with abdominal and rectal pain accompanied by multiple episodes of bloody diarrhea. He stated he had mild rectal pain the previous night but was pain-free and in his usual state of health the morning of his presentation. Approximately 2 hours before presenting to the ED, however, he began experiencing mild stomach pain, then bloody diarrhea which he described as bright red and “filling the toilet bowl with blood.” He had no history of inflammatory bowel disease or other gastrointestinal (GI) disorder, no recent travel, no complaints of nausea or vomiting, and no infectious symptoms. He described a remote history of external hemorrhoids, and review of his family history was significant for multiple paternal relatives with aortic aneurysms. He was not taking any medications and was a nonsmoker with a normal body mass index (24.3 kg/m2).
Upon arrival at the ED, the patient’s vital signs were: heart rate, 112 beats/min; and blood pressure, 139/102 mm Hg; respiratory rate and temperature were normal, as was the patient’s oxygen saturation on room air. Physical examination was notable for no subjective or objective findings of orthostatic hypotension; increased bowel sounds and diffuse mild abdominal tenderness; and no external hemorrhoids, fissures, or rectal tenderness. Laboratory evaluation was significant for hemoglobin (Hgb), 15.0 g/dL; blood urea nitrogen (BUN)-to-creatinine (Cr) ratio, 11.6; and anion gap, 17 mEq/L.
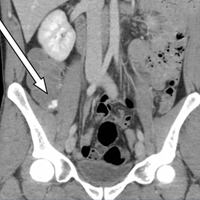
Upon initial presentation, there was some concern for an infection. However, as the patient continued to have bowel movements consisting almost entirely of frank blood and did not have any infectious signs, a vascular etiology was more strongly considered. Given the patient’s relatively stable vital signs, BUN-to-Cr ratio of less than 20, and lack of orthostatic hypotension, there was low concern for an upper GI etiology, and endoscopy was not obtained emergently. The patient instead underwent abdominal computed tomography angiography (CTA), which identified active extravasation and contrast pooling within the cecum and appendix (Figure 1).

Shortly after the patient returned from imaging, repeat laboratory studies were performed, demonstrating an Hgb drop from 15.0 g/dL to 12.3 g/dL, and surgical services was emergently consulted. The surgeon recommended that embolization first be attempted, with surgery as the option of last resort given the poor localization of the bleed on CTA and the long-term consequences of colonic resection in a young, otherwise healthy man.
Interventional radiology was consulted, and the patient was brought immediately to the angiography suite, where he was found to have “active extravasation arising from a distal descending branch off the right colic artery” (Figure 2). Coil embolization resulted in complete resolution of the hemorrhage.

Later that evening, the patient’s Hgb continued to drop, reaching nadir at 7.3 g/dL, and he continued to have severe hematochezia. His falling Hgb was thought to be indicative of the degree of hemorrhage he had sustained prior to embolization, and the clearance of such blood as the source of his ongoing hematochezia. Following transfusion of 2 U of packed red blood cells (PRBCs), the patient’s Hgb improved to 12.0 g/dL, and he did not experience any significant bleeding for the remainder of his hospital stay.
The following morning, the patient underwent an extensive colonoscopy (extending 25 cm into the terminal ileum), which was unable to detect any signs of arteriovenous malformations, angiodysplasia, or any other possible source of bleeding. After 24 hours with stable vital signs and Hgb levels, the patient was discharged home with close surgical and gastroenterological follow-up, with possible genetic testing for connective tissue diseases. The diagnosis at discharge was spontaneous mesenteric hemorrhage of unknown etiology.
Discussion
Acute lower GI bleeding has an estimated annual hospitalization rate of 36 patients per 100,000, or about half the rate for upper GI bleeding.1,2 The majority of patients (>80%) will have spontaneous resolution and can be worked up nonemergently.
Etiology and Work-Up
Assessment of the etiology of hematochezia begins with ruling out an upper GI source of the bleed; 10% to 15% of patients presenting with hematochezia without hematemesis are ultimately diagnosed with an upper GI etiology. 4,5
BUN-to-Cr Ratio. In a study of patients presenting with hematochezia but no hematemesis or renal failure, Srygley et al6 found a BUN-to-Cr ratio greater than 93% to be sensitive for an upper GI source, with a likelihood ratio of 7.5. The proposed etiology is some combination of absorption of digested blood products and prerenal azotemia due to hypovolemia.
Tachycardia and Orthostatic Hypotension. There have been discussions in the literature about other findings to rule in/out upper GI bleeding. While some studies have found statistically significant results between upper and lower GI bleeding for tachycardia and orthostatic hypotension (increased percentage of both in upper GI bleeding), there is disagreement about whether these findings are clinically significant.7-9
Nasogastric Lavage. Although nasogastric (NG) lavage is no longer the standard of care in the ED due to poor sensitivity and marked discomfort to the patient, most current gastroenterology guidelines still recommend its use; therefore, NG may be requested by the GI consultant.10-12
Diagnosis
Once an upper GI source has been ruled out, identification of the lesion is the next step. The differential diagnosis includes common sources such as diverticular disease, angiodysplasia, colitis, anorectal sources, and neoplasm.5 Less common, but associated with a high risk of mortality, is aortoenteric fistula (100% mortality without surgical intervention).5
Colonoscopy. Emergent colonoscopy can be used for both diagnosis and (potential) therapeutic intervention and is therefore the first option of choice.1,3,4,9 However, as seen in our case, some patients experience such profound hemorrhage that visualization of the colon may be difficult or impossible; patients may also be too unstable to await bowel preparation or undergo a procedure.
Computed Tomography Angiography. For patients in whom colonoscopy is contraindicated, CTA is the imaging modality of choice, and has a 91% to 92% sensitivity in identifying active bleeding (>0.35 mL/min).13-16
Computed tomography of the abdomen and pelvis with contrast alone, as opposed to CTA, is insufficient for detecting GI bleeding, as it is timed so that imaging is obtained when the contrast is in the portal venous capillary beds, rather than in the arteries or arterioles. By protocol, though, many institutions require abdominal and pelvic CTA to include both arterial phase and venous phase images, allowing for assessment of both active arterial bleeding and alternative lower GI sources of hematochezia (eg, mesenteric ischemia).
When ordering a CT study, an awareness of local practice is important in understanding the information that will be obtained from the study. Protocols for lower GI bleed that include CTA have reported accuracy and efficiency without worsening of renal function, despite the increased contrast load.17
Triphasic CT Enterography. Another CT modality to consider is triphasic CT enterography, which uses IV and oral contrast. In a preliminary trial, this modality achieved a specificity of 100% (sensitivity 42%) in detecting GI bleeding.18
Red Blood Cell Scintigraphy. An additional imaging modality that has been the subject of much debate in the GI literature is tagged RBC scintigraphy with Technetium-99m. Various studies have found bleeding-site confirmation in 24% to 97% of patients, and correct localization in 41% to 100% of patients. Given the extensive variability within the literature on selection criteria, localization, site confirmation, and other variables, as well as evidence from one prospective trial by Zink et al19 that found a significant disagreement between CTA and scintigraphy, RBC scintigraphy is not recommended as an alternative imaging modality for the rapid diagnosis of an acute lower GI bleed.
Conclusion
Severe hematochezia is a potential surgical emergency with a broad differential diagnosis. While emergent colonoscopy is an excellent first option, in patients with severe hematochezia, there may be too much blood in the colon to obtain adequate visual images; additionally, depending on practice setting, emergency colonoscopy may not be immediately available. In either case, CTA—a readily available, noninvasive, rapid, and repeatable diagnostic tool—should be considered as an alternate to colonoscopy, particularly in patients with brisk hematochezia.
If a patient with severe hematochezia presents to the ED, the emergency physician (EP) must recognize that the degree of hemorrhage may not correlate with the patient’s vital signs or initial laboratory values. For this reason, the EP must have a high index of suspicion, and consider CTA to allow for a rapid definitive diagnosis and prompt discussion between surgical, interventional radiology, and/or gastroenterology teams to improve clinical outcomes and decrease morbidity and mortality.20
1. Ghassemi K, Jensen D. Lower GI bleeding: epidemiology and management. Curr Gastroenterol Rep. 2013;15(7):333. doi:10.1007/s11894-013-0333-5.
2. Strate LL, Ayanian JZ, Kotler G, Syngal S. Risk factors for mortality in lower intestinal bleeding. Clin Gastroenterol Hepatol. 2008;6(9):1004-1010. doi:10.1016/j.cgh.2008.03.021.
3. Qayad E, Dagar G, Nanchal R. Lower gastrointestinal hemorrhage. Crit Care Clin. 2016;32(2):241-254. doi:10.1016/j.ccc.2015.12.004.
4. Strate LL. Lower GI bleeding: epidemiology and diagnosis. Gastroenterol Clin North Am. 2005;34(4):643-664.
5. Goralnick E, Meguerdichian D. Gastrointestinal bleeding. In: Marx J, Hockberger R, Walls R. (Eds.). Rosen’s Emergency Medicine, 8th Edition. Philadelphia, PA: Saunders, 2014;248-253.
6. Srygley FD, Gerando CJ, Tran T, Fisher DA. Does this patient have a severe upper gastrointestinal bleed? JAMA. 2012;307(10):1072-1079. doi:10.1001/jama.2012.253.
7. Whelen C, Chen C, Kaboli P, Siddique J, Prochaska M, Meltzer DO. Upper versus lower gastrointestinal bleeding: a direct comparison of clinical presentation, outcomes, and resource utilization. J Hosp Med. 2010;5(3):141-147. doi:10.1002/jhm.606.
8. Sittichanbunch Y, Senasu S, Thongkrau T, Keeratiksikorn C, Sawanyawisuth K. How to differentiate sites of gastrointestinal bleeding in patients with hematochezia by using clinical factors? Gastroenterol Res Pract. 2013;2013:265076. doi:10.1155/2013/265076.
9. Velayos F, Williamson A, Sousa KH, et al. Early predictors of severe lower gastrointestinal bleeding and adverse outcomes: a prospective study. Clin Gastroenterol Hepatol. 2004;2(6):485-490.
10. Palamidessi N, Sinert R, Falzon L, Zehtabchi S. Nasogastric aspiration and lavage in emergency department patients with hematochezia or melena without hematemesis. Acad Emerg Med. 2010;17(2):126-132. doi:10.1111/j.1553-2712.2009.00609.x.
11. Singer AJ, Richman PB, Kowalska A, Thode HC Jr. Comparison of patient and practitioner assessments of pain from commonly performed emergency department procedures. Ann Emerg Med. 1999;33(6):652-658.
12. Strate L, Gralnek I. ACG clinical guideline: management of patients with acute lower gastrointestinal bleeding. Am J Gastroenterol. 2016;111(4):459-474. doi:10.1038/ajg.2016.41.
13. Wu LM, Xu JR, Yin Y, Qu XH. Usefulness of CT angiography in diagnosing acute gastrointestinal bleeding: a meta-analysis. World J Gastroenterol. 2010;16(31):3957-3963.
14. Geffroy Y, Rodallec MH, Boulay-Coletta I, Julles MC, Ridereau-Zins C, Zins M. Multidetector CT angiography in acute gastrointestinal bleeding: why, when, and how. Radiographics. 2011;31(3):E35-E46.
15. Reis F, Cardia P, D’Ippolito G. Computed tomography angiography in patients with active gastrointestinal bleeding. Radiol Bras. 2015;48(6):381-390. doi:10.1590/0100-3984.2014.0014.
16. Chan V, Tse D, Dixon S, et al. Outcome following a negative CT angiogram for gastrointestinal hemorrhage. Cardiovasc Intervent Radiol. 2015;38(2):329-335. doi:10.1007/s00270-014-0928-8.
17. Jacovides T, Nadolski G, Allen S, et al. Arteriography for lower gastrointestinal hemorrhage: role of preceding abdominal computed tomographic angiogram in diagnosis and localization. JAMA Surgery. 2015;150(7):650-656. doi:10.1001/jamasurg.2015.97.
18. Hara AK, Walker FB, Silva AC, Leighton JA. Preliminary estimate of triphasic CT enterography performance in hemodynamically stable patients with suspected gastrointestinal bleeding. AJR Am J Roentgenol. 2009;193(5):1252-1260. doi:10.2214/AJR.08.1494.
19. Zink SI, Ohki SK, Stein B, et al. Noninvasive evaluation of active lower gastrointestinal bleeding: comparison between contrast-enhanced MDCT and 99mTc-labeled RBC scintigraphy. AJR Am J Roentgenol. 2008;91(4):1107-1114. doi:10.2214/AJR.07.3642.
20. Nable J, Graham A. Gastrointestinal bleeding. Emerg Med Clin N Am. 2016;34(2):309-325. doi:10.1016/j.emc.2015.12.001.
Case
A 31-year-old white man presented to the ED with abdominal and rectal pain accompanied by multiple episodes of bloody diarrhea. He stated he had mild rectal pain the previous night but was pain-free and in his usual state of health the morning of his presentation. Approximately 2 hours before presenting to the ED, however, he began experiencing mild stomach pain, then bloody diarrhea which he described as bright red and “filling the toilet bowl with blood.” He had no history of inflammatory bowel disease or other gastrointestinal (GI) disorder, no recent travel, no complaints of nausea or vomiting, and no infectious symptoms. He described a remote history of external hemorrhoids, and review of his family history was significant for multiple paternal relatives with aortic aneurysms. He was not taking any medications and was a nonsmoker with a normal body mass index (24.3 kg/m2).
Upon arrival at the ED, the patient’s vital signs were: heart rate, 112 beats/min; and blood pressure, 139/102 mm Hg; respiratory rate and temperature were normal, as was the patient’s oxygen saturation on room air. Physical examination was notable for no subjective or objective findings of orthostatic hypotension; increased bowel sounds and diffuse mild abdominal tenderness; and no external hemorrhoids, fissures, or rectal tenderness. Laboratory evaluation was significant for hemoglobin (Hgb), 15.0 g/dL; blood urea nitrogen (BUN)-to-creatinine (Cr) ratio, 11.6; and anion gap, 17 mEq/L.
Upon initial presentation, there was some concern for an infection. However, as the patient continued to have bowel movements consisting almost entirely of frank blood and did not have any infectious signs, a vascular etiology was more strongly considered. Given the patient’s relatively stable vital signs, BUN-to-Cr ratio of less than 20, and lack of orthostatic hypotension, there was low concern for an upper GI etiology, and endoscopy was not obtained emergently. The patient instead underwent abdominal computed tomography angiography (CTA), which identified active extravasation and contrast pooling within the cecum and appendix (Figure 1).
Shortly after the patient returned from imaging, repeat laboratory studies were performed, demonstrating an Hgb drop from 15.0 g/dL to 12.3 g/dL, and surgical services was emergently consulted. The surgeon recommended that embolization first be attempted, with surgery as the option of last resort given the poor localization of the bleed on CTA and the long-term consequences of colonic resection in a young, otherwise healthy man.
Interventional radiology was consulted, and the patient was brought immediately to the angiography suite, where he was found to have “active extravasation arising from a distal descending branch off the right colic artery” (Figure 2). Coil embolization resulted in complete resolution of the hemorrhage.
Later that evening, the patient’s Hgb continued to drop, reaching nadir at 7.3 g/dL, and he continued to have severe hematochezia. His falling Hgb was thought to be indicative of the degree of hemorrhage he had sustained prior to embolization, and the clearance of such blood as the source of his ongoing hematochezia. Following transfusion of 2 U of packed red blood cells (PRBCs), the patient’s Hgb improved to 12.0 g/dL, and he did not experience any significant bleeding for the remainder of his hospital stay.
The following morning, the patient underwent an extensive colonoscopy (extending 25 cm into the terminal ileum), which was unable to detect any signs of arteriovenous malformations, angiodysplasia, or any other possible source of bleeding. After 24 hours with stable vital signs and Hgb levels, the patient was discharged home with close surgical and gastroenterological follow-up, with possible genetic testing for connective tissue diseases. The diagnosis at discharge was spontaneous mesenteric hemorrhage of unknown etiology.
Discussion
Acute lower GI bleeding has an estimated annual hospitalization rate of 36 patients per 100,000, or about half the rate for upper GI bleeding.1,2 The majority of patients (>80%) will have spontaneous resolution and can be worked up nonemergently.
Etiology and Work-Up
Assessment of the etiology of hematochezia begins with ruling out an upper GI source of the bleed; 10% to 15% of patients presenting with hematochezia without hematemesis are ultimately diagnosed with an upper GI etiology. 4,5
BUN-to-Cr Ratio. In a study of patients presenting with hematochezia but no hematemesis or renal failure, Srygley et al6 found a BUN-to-Cr ratio greater than 93% to be sensitive for an upper GI source, with a likelihood ratio of 7.5. The proposed etiology is some combination of absorption of digested blood products and prerenal azotemia due to hypovolemia.
Tachycardia and Orthostatic Hypotension. There have been discussions in the literature about other findings to rule in/out upper GI bleeding. While some studies have found statistically significant results between upper and lower GI bleeding for tachycardia and orthostatic hypotension (increased percentage of both in upper GI bleeding), there is disagreement about whether these findings are clinically significant.7-9
Nasogastric Lavage. Although nasogastric (NG) lavage is no longer the standard of care in the ED due to poor sensitivity and marked discomfort to the patient, most current gastroenterology guidelines still recommend its use; therefore, NG may be requested by the GI consultant.10-12
Diagnosis
Once an upper GI source has been ruled out, identification of the lesion is the next step. The differential diagnosis includes common sources such as diverticular disease, angiodysplasia, colitis, anorectal sources, and neoplasm.5 Less common, but associated with a high risk of mortality, is aortoenteric fistula (100% mortality without surgical intervention).5
Colonoscopy. Emergent colonoscopy can be used for both diagnosis and (potential) therapeutic intervention and is therefore the first option of choice.1,3,4,9 However, as seen in our case, some patients experience such profound hemorrhage that visualization of the colon may be difficult or impossible; patients may also be too unstable to await bowel preparation or undergo a procedure.
Computed Tomography Angiography. For patients in whom colonoscopy is contraindicated, CTA is the imaging modality of choice, and has a 91% to 92% sensitivity in identifying active bleeding (>0.35 mL/min).13-16
Computed tomography of the abdomen and pelvis with contrast alone, as opposed to CTA, is insufficient for detecting GI bleeding, as it is timed so that imaging is obtained when the contrast is in the portal venous capillary beds, rather than in the arteries or arterioles. By protocol, though, many institutions require abdominal and pelvic CTA to include both arterial phase and venous phase images, allowing for assessment of both active arterial bleeding and alternative lower GI sources of hematochezia (eg, mesenteric ischemia).
When ordering a CT study, an awareness of local practice is important in understanding the information that will be obtained from the study. Protocols for lower GI bleed that include CTA have reported accuracy and efficiency without worsening of renal function, despite the increased contrast load.17
Triphasic CT Enterography. Another CT modality to consider is triphasic CT enterography, which uses IV and oral contrast. In a preliminary trial, this modality achieved a specificity of 100% (sensitivity 42%) in detecting GI bleeding.18
Red Blood Cell Scintigraphy. An additional imaging modality that has been the subject of much debate in the GI literature is tagged RBC scintigraphy with Technetium-99m. Various studies have found bleeding-site confirmation in 24% to 97% of patients, and correct localization in 41% to 100% of patients. Given the extensive variability within the literature on selection criteria, localization, site confirmation, and other variables, as well as evidence from one prospective trial by Zink et al19 that found a significant disagreement between CTA and scintigraphy, RBC scintigraphy is not recommended as an alternative imaging modality for the rapid diagnosis of an acute lower GI bleed.
Conclusion
Severe hematochezia is a potential surgical emergency with a broad differential diagnosis. While emergent colonoscopy is an excellent first option, in patients with severe hematochezia, there may be too much blood in the colon to obtain adequate visual images; additionally, depending on practice setting, emergency colonoscopy may not be immediately available. In either case, CTA—a readily available, noninvasive, rapid, and repeatable diagnostic tool—should be considered as an alternate to colonoscopy, particularly in patients with brisk hematochezia.
If a patient with severe hematochezia presents to the ED, the emergency physician (EP) must recognize that the degree of hemorrhage may not correlate with the patient’s vital signs or initial laboratory values. For this reason, the EP must have a high index of suspicion, and consider CTA to allow for a rapid definitive diagnosis and prompt discussion between surgical, interventional radiology, and/or gastroenterology teams to improve clinical outcomes and decrease morbidity and mortality.20
Case
A 31-year-old white man presented to the ED with abdominal and rectal pain accompanied by multiple episodes of bloody diarrhea. He stated he had mild rectal pain the previous night but was pain-free and in his usual state of health the morning of his presentation. Approximately 2 hours before presenting to the ED, however, he began experiencing mild stomach pain, then bloody diarrhea which he described as bright red and “filling the toilet bowl with blood.” He had no history of inflammatory bowel disease or other gastrointestinal (GI) disorder, no recent travel, no complaints of nausea or vomiting, and no infectious symptoms. He described a remote history of external hemorrhoids, and review of his family history was significant for multiple paternal relatives with aortic aneurysms. He was not taking any medications and was a nonsmoker with a normal body mass index (24.3 kg/m2).
Upon arrival at the ED, the patient’s vital signs were: heart rate, 112 beats/min; and blood pressure, 139/102 mm Hg; respiratory rate and temperature were normal, as was the patient’s oxygen saturation on room air. Physical examination was notable for no subjective or objective findings of orthostatic hypotension; increased bowel sounds and diffuse mild abdominal tenderness; and no external hemorrhoids, fissures, or rectal tenderness. Laboratory evaluation was significant for hemoglobin (Hgb), 15.0 g/dL; blood urea nitrogen (BUN)-to-creatinine (Cr) ratio, 11.6; and anion gap, 17 mEq/L.
Upon initial presentation, there was some concern for an infection. However, as the patient continued to have bowel movements consisting almost entirely of frank blood and did not have any infectious signs, a vascular etiology was more strongly considered. Given the patient’s relatively stable vital signs, BUN-to-Cr ratio of less than 20, and lack of orthostatic hypotension, there was low concern for an upper GI etiology, and endoscopy was not obtained emergently. The patient instead underwent abdominal computed tomography angiography (CTA), which identified active extravasation and contrast pooling within the cecum and appendix (Figure 1).
Shortly after the patient returned from imaging, repeat laboratory studies were performed, demonstrating an Hgb drop from 15.0 g/dL to 12.3 g/dL, and surgical services was emergently consulted. The surgeon recommended that embolization first be attempted, with surgery as the option of last resort given the poor localization of the bleed on CTA and the long-term consequences of colonic resection in a young, otherwise healthy man.
Interventional radiology was consulted, and the patient was brought immediately to the angiography suite, where he was found to have “active extravasation arising from a distal descending branch off the right colic artery” (Figure 2). Coil embolization resulted in complete resolution of the hemorrhage.
Later that evening, the patient’s Hgb continued to drop, reaching nadir at 7.3 g/dL, and he continued to have severe hematochezia. His falling Hgb was thought to be indicative of the degree of hemorrhage he had sustained prior to embolization, and the clearance of such blood as the source of his ongoing hematochezia. Following transfusion of 2 U of packed red blood cells (PRBCs), the patient’s Hgb improved to 12.0 g/dL, and he did not experience any significant bleeding for the remainder of his hospital stay.
The following morning, the patient underwent an extensive colonoscopy (extending 25 cm into the terminal ileum), which was unable to detect any signs of arteriovenous malformations, angiodysplasia, or any other possible source of bleeding. After 24 hours with stable vital signs and Hgb levels, the patient was discharged home with close surgical and gastroenterological follow-up, with possible genetic testing for connective tissue diseases. The diagnosis at discharge was spontaneous mesenteric hemorrhage of unknown etiology.
Discussion
Acute lower GI bleeding has an estimated annual hospitalization rate of 36 patients per 100,000, or about half the rate for upper GI bleeding.1,2 The majority of patients (>80%) will have spontaneous resolution and can be worked up nonemergently.
Etiology and Work-Up
Assessment of the etiology of hematochezia begins with ruling out an upper GI source of the bleed; 10% to 15% of patients presenting with hematochezia without hematemesis are ultimately diagnosed with an upper GI etiology. 4,5
BUN-to-Cr Ratio. In a study of patients presenting with hematochezia but no hematemesis or renal failure, Srygley et al6 found a BUN-to-Cr ratio greater than 93% to be sensitive for an upper GI source, with a likelihood ratio of 7.5. The proposed etiology is some combination of absorption of digested blood products and prerenal azotemia due to hypovolemia.
Tachycardia and Orthostatic Hypotension. There have been discussions in the literature about other findings to rule in/out upper GI bleeding. While some studies have found statistically significant results between upper and lower GI bleeding for tachycardia and orthostatic hypotension (increased percentage of both in upper GI bleeding), there is disagreement about whether these findings are clinically significant.7-9
Nasogastric Lavage. Although nasogastric (NG) lavage is no longer the standard of care in the ED due to poor sensitivity and marked discomfort to the patient, most current gastroenterology guidelines still recommend its use; therefore, NG may be requested by the GI consultant.10-12
Diagnosis
Once an upper GI source has been ruled out, identification of the lesion is the next step. The differential diagnosis includes common sources such as diverticular disease, angiodysplasia, colitis, anorectal sources, and neoplasm.5 Less common, but associated with a high risk of mortality, is aortoenteric fistula (100% mortality without surgical intervention).5
Colonoscopy. Emergent colonoscopy can be used for both diagnosis and (potential) therapeutic intervention and is therefore the first option of choice.1,3,4,9 However, as seen in our case, some patients experience such profound hemorrhage that visualization of the colon may be difficult or impossible; patients may also be too unstable to await bowel preparation or undergo a procedure.
Computed Tomography Angiography. For patients in whom colonoscopy is contraindicated, CTA is the imaging modality of choice, and has a 91% to 92% sensitivity in identifying active bleeding (>0.35 mL/min).13-16
Computed tomography of the abdomen and pelvis with contrast alone, as opposed to CTA, is insufficient for detecting GI bleeding, as it is timed so that imaging is obtained when the contrast is in the portal venous capillary beds, rather than in the arteries or arterioles. By protocol, though, many institutions require abdominal and pelvic CTA to include both arterial phase and venous phase images, allowing for assessment of both active arterial bleeding and alternative lower GI sources of hematochezia (eg, mesenteric ischemia).
When ordering a CT study, an awareness of local practice is important in understanding the information that will be obtained from the study. Protocols for lower GI bleed that include CTA have reported accuracy and efficiency without worsening of renal function, despite the increased contrast load.17
Triphasic CT Enterography. Another CT modality to consider is triphasic CT enterography, which uses IV and oral contrast. In a preliminary trial, this modality achieved a specificity of 100% (sensitivity 42%) in detecting GI bleeding.18
Red Blood Cell Scintigraphy. An additional imaging modality that has been the subject of much debate in the GI literature is tagged RBC scintigraphy with Technetium-99m. Various studies have found bleeding-site confirmation in 24% to 97% of patients, and correct localization in 41% to 100% of patients. Given the extensive variability within the literature on selection criteria, localization, site confirmation, and other variables, as well as evidence from one prospective trial by Zink et al19 that found a significant disagreement between CTA and scintigraphy, RBC scintigraphy is not recommended as an alternative imaging modality for the rapid diagnosis of an acute lower GI bleed.
Conclusion
Severe hematochezia is a potential surgical emergency with a broad differential diagnosis. While emergent colonoscopy is an excellent first option, in patients with severe hematochezia, there may be too much blood in the colon to obtain adequate visual images; additionally, depending on practice setting, emergency colonoscopy may not be immediately available. In either case, CTA—a readily available, noninvasive, rapid, and repeatable diagnostic tool—should be considered as an alternate to colonoscopy, particularly in patients with brisk hematochezia.
If a patient with severe hematochezia presents to the ED, the emergency physician (EP) must recognize that the degree of hemorrhage may not correlate with the patient’s vital signs or initial laboratory values. For this reason, the EP must have a high index of suspicion, and consider CTA to allow for a rapid definitive diagnosis and prompt discussion between surgical, interventional radiology, and/or gastroenterology teams to improve clinical outcomes and decrease morbidity and mortality.20
1. Ghassemi K, Jensen D. Lower GI bleeding: epidemiology and management. Curr Gastroenterol Rep. 2013;15(7):333. doi:10.1007/s11894-013-0333-5.
2. Strate LL, Ayanian JZ, Kotler G, Syngal S. Risk factors for mortality in lower intestinal bleeding. Clin Gastroenterol Hepatol. 2008;6(9):1004-1010. doi:10.1016/j.cgh.2008.03.021.
3. Qayad E, Dagar G, Nanchal R. Lower gastrointestinal hemorrhage. Crit Care Clin. 2016;32(2):241-254. doi:10.1016/j.ccc.2015.12.004.
4. Strate LL. Lower GI bleeding: epidemiology and diagnosis. Gastroenterol Clin North Am. 2005;34(4):643-664.
5. Goralnick E, Meguerdichian D. Gastrointestinal bleeding. In: Marx J, Hockberger R, Walls R. (Eds.). Rosen’s Emergency Medicine, 8th Edition. Philadelphia, PA: Saunders, 2014;248-253.
6. Srygley FD, Gerando CJ, Tran T, Fisher DA. Does this patient have a severe upper gastrointestinal bleed? JAMA. 2012;307(10):1072-1079. doi:10.1001/jama.2012.253.
7. Whelen C, Chen C, Kaboli P, Siddique J, Prochaska M, Meltzer DO. Upper versus lower gastrointestinal bleeding: a direct comparison of clinical presentation, outcomes, and resource utilization. J Hosp Med. 2010;5(3):141-147. doi:10.1002/jhm.606.
8. Sittichanbunch Y, Senasu S, Thongkrau T, Keeratiksikorn C, Sawanyawisuth K. How to differentiate sites of gastrointestinal bleeding in patients with hematochezia by using clinical factors? Gastroenterol Res Pract. 2013;2013:265076. doi:10.1155/2013/265076.
9. Velayos F, Williamson A, Sousa KH, et al. Early predictors of severe lower gastrointestinal bleeding and adverse outcomes: a prospective study. Clin Gastroenterol Hepatol. 2004;2(6):485-490.
10. Palamidessi N, Sinert R, Falzon L, Zehtabchi S. Nasogastric aspiration and lavage in emergency department patients with hematochezia or melena without hematemesis. Acad Emerg Med. 2010;17(2):126-132. doi:10.1111/j.1553-2712.2009.00609.x.
11. Singer AJ, Richman PB, Kowalska A, Thode HC Jr. Comparison of patient and practitioner assessments of pain from commonly performed emergency department procedures. Ann Emerg Med. 1999;33(6):652-658.
12. Strate L, Gralnek I. ACG clinical guideline: management of patients with acute lower gastrointestinal bleeding. Am J Gastroenterol. 2016;111(4):459-474. doi:10.1038/ajg.2016.41.
13. Wu LM, Xu JR, Yin Y, Qu XH. Usefulness of CT angiography in diagnosing acute gastrointestinal bleeding: a meta-analysis. World J Gastroenterol. 2010;16(31):3957-3963.
14. Geffroy Y, Rodallec MH, Boulay-Coletta I, Julles MC, Ridereau-Zins C, Zins M. Multidetector CT angiography in acute gastrointestinal bleeding: why, when, and how. Radiographics. 2011;31(3):E35-E46.
15. Reis F, Cardia P, D’Ippolito G. Computed tomography angiography in patients with active gastrointestinal bleeding. Radiol Bras. 2015;48(6):381-390. doi:10.1590/0100-3984.2014.0014.
16. Chan V, Tse D, Dixon S, et al. Outcome following a negative CT angiogram for gastrointestinal hemorrhage. Cardiovasc Intervent Radiol. 2015;38(2):329-335. doi:10.1007/s00270-014-0928-8.
17. Jacovides T, Nadolski G, Allen S, et al. Arteriography for lower gastrointestinal hemorrhage: role of preceding abdominal computed tomographic angiogram in diagnosis and localization. JAMA Surgery. 2015;150(7):650-656. doi:10.1001/jamasurg.2015.97.
18. Hara AK, Walker FB, Silva AC, Leighton JA. Preliminary estimate of triphasic CT enterography performance in hemodynamically stable patients with suspected gastrointestinal bleeding. AJR Am J Roentgenol. 2009;193(5):1252-1260. doi:10.2214/AJR.08.1494.
19. Zink SI, Ohki SK, Stein B, et al. Noninvasive evaluation of active lower gastrointestinal bleeding: comparison between contrast-enhanced MDCT and 99mTc-labeled RBC scintigraphy. AJR Am J Roentgenol. 2008;91(4):1107-1114. doi:10.2214/AJR.07.3642.
20. Nable J, Graham A. Gastrointestinal bleeding. Emerg Med Clin N Am. 2016;34(2):309-325. doi:10.1016/j.emc.2015.12.001.
1. Ghassemi K, Jensen D. Lower GI bleeding: epidemiology and management. Curr Gastroenterol Rep. 2013;15(7):333. doi:10.1007/s11894-013-0333-5.
2. Strate LL, Ayanian JZ, Kotler G, Syngal S. Risk factors for mortality in lower intestinal bleeding. Clin Gastroenterol Hepatol. 2008;6(9):1004-1010. doi:10.1016/j.cgh.2008.03.021.
3. Qayad E, Dagar G, Nanchal R. Lower gastrointestinal hemorrhage. Crit Care Clin. 2016;32(2):241-254. doi:10.1016/j.ccc.2015.12.004.
4. Strate LL. Lower GI bleeding: epidemiology and diagnosis. Gastroenterol Clin North Am. 2005;34(4):643-664.
5. Goralnick E, Meguerdichian D. Gastrointestinal bleeding. In: Marx J, Hockberger R, Walls R. (Eds.). Rosen’s Emergency Medicine, 8th Edition. Philadelphia, PA: Saunders, 2014;248-253.
6. Srygley FD, Gerando CJ, Tran T, Fisher DA. Does this patient have a severe upper gastrointestinal bleed? JAMA. 2012;307(10):1072-1079. doi:10.1001/jama.2012.253.
7. Whelen C, Chen C, Kaboli P, Siddique J, Prochaska M, Meltzer DO. Upper versus lower gastrointestinal bleeding: a direct comparison of clinical presentation, outcomes, and resource utilization. J Hosp Med. 2010;5(3):141-147. doi:10.1002/jhm.606.
8. Sittichanbunch Y, Senasu S, Thongkrau T, Keeratiksikorn C, Sawanyawisuth K. How to differentiate sites of gastrointestinal bleeding in patients with hematochezia by using clinical factors? Gastroenterol Res Pract. 2013;2013:265076. doi:10.1155/2013/265076.
9. Velayos F, Williamson A, Sousa KH, et al. Early predictors of severe lower gastrointestinal bleeding and adverse outcomes: a prospective study. Clin Gastroenterol Hepatol. 2004;2(6):485-490.
10. Palamidessi N, Sinert R, Falzon L, Zehtabchi S. Nasogastric aspiration and lavage in emergency department patients with hematochezia or melena without hematemesis. Acad Emerg Med. 2010;17(2):126-132. doi:10.1111/j.1553-2712.2009.00609.x.
11. Singer AJ, Richman PB, Kowalska A, Thode HC Jr. Comparison of patient and practitioner assessments of pain from commonly performed emergency department procedures. Ann Emerg Med. 1999;33(6):652-658.
12. Strate L, Gralnek I. ACG clinical guideline: management of patients with acute lower gastrointestinal bleeding. Am J Gastroenterol. 2016;111(4):459-474. doi:10.1038/ajg.2016.41.
13. Wu LM, Xu JR, Yin Y, Qu XH. Usefulness of CT angiography in diagnosing acute gastrointestinal bleeding: a meta-analysis. World J Gastroenterol. 2010;16(31):3957-3963.
14. Geffroy Y, Rodallec MH, Boulay-Coletta I, Julles MC, Ridereau-Zins C, Zins M. Multidetector CT angiography in acute gastrointestinal bleeding: why, when, and how. Radiographics. 2011;31(3):E35-E46.
15. Reis F, Cardia P, D’Ippolito G. Computed tomography angiography in patients with active gastrointestinal bleeding. Radiol Bras. 2015;48(6):381-390. doi:10.1590/0100-3984.2014.0014.
16. Chan V, Tse D, Dixon S, et al. Outcome following a negative CT angiogram for gastrointestinal hemorrhage. Cardiovasc Intervent Radiol. 2015;38(2):329-335. doi:10.1007/s00270-014-0928-8.
17. Jacovides T, Nadolski G, Allen S, et al. Arteriography for lower gastrointestinal hemorrhage: role of preceding abdominal computed tomographic angiogram in diagnosis and localization. JAMA Surgery. 2015;150(7):650-656. doi:10.1001/jamasurg.2015.97.
18. Hara AK, Walker FB, Silva AC, Leighton JA. Preliminary estimate of triphasic CT enterography performance in hemodynamically stable patients with suspected gastrointestinal bleeding. AJR Am J Roentgenol. 2009;193(5):1252-1260. doi:10.2214/AJR.08.1494.
19. Zink SI, Ohki SK, Stein B, et al. Noninvasive evaluation of active lower gastrointestinal bleeding: comparison between contrast-enhanced MDCT and 99mTc-labeled RBC scintigraphy. AJR Am J Roentgenol. 2008;91(4):1107-1114. doi:10.2214/AJR.07.3642.
20. Nable J, Graham A. Gastrointestinal bleeding. Emerg Med Clin N Am. 2016;34(2):309-325. doi:10.1016/j.emc.2015.12.001.
Idiopathic Intracranial Hypertension in a 24-Year-Old Woman
Case
A 24-year-old woman presented to the ED for evaluation of a 3-week history of worsening headache and a 5-day history of increasingly blurry vision. The patient stated that she had initially contacted her primary care physician, but instead presented to the ED because he had no open appointments until the following week and recommended that she go to the ED.
The patient described her headache as a pulsating and throbbing pain over her entire head, which only mildly improved after taking over-the-counter (OTC) ibuprofen. She further noted that her headache was somewhat worse when lying down, and reported the sensation of hearing her own pulsating heartbeat in her ears.
The patient had no personal or family history of migraines, tension headaches, aneurysms, clotting disorders, bleeding disorders, or renal disease, and stated that she had never experienced this type of headache before. She denied photophobia, phonophobia, neck stiffness, fever, vomiting, cough, numbness or weakness in her extremities, or pain anywhere else in her body.
Over the past 5 days, the patient noticed her vision had become increasingly blurry. She was not on any prescription medications, stating the only medication she used was occasional OTC ibuprofen. She had no known allergy to medications and denied smoking or recreational drug use; she admitted to occasional alcohol consumption.
The patient resided with her husband, who had no similar symptoms. Physical examination showed an obese woman (height, 5 ft 6 in; weight, 195 lb; body mass index, 32 kg/m2), lying supine in apparent discomfort. Vital signs at presentation were all normal, and oxygen saturation was normal on room air.
A bedside ocular examination showed 20/100 in both eyes while using glasses; no visual field cuts or obvious central scotoma was present. The patient was alert and oriented to time and place. The neurological examination showed intact cranial nerves, 5/5 strength in all extremities, intact sensation in all extremities, no pronator drift, negative Romberg test, and a normal gait. Fundoscopic examination revealed mildly blurred medial optic discs bilaterally. The rest of the physical examination was normal.
Discussion
Pseudotumor cerebri, more commonly referred to as idiopathic intracranial hypertension (IIH), is characterized by increased intracranial pressure (ICP) with no explanatory findings on imaging studies or in cerebrospinal fluid (CSF) analysis, and may be accompanied by symptoms of chronic headache, tinnitus, papilledema and progressive vision loss caused by optic nerve damage.1 Though historically IIH was referred to by several other names, including “benign intracranial hypertension,” the condition is not benign—when untreated, IIH can cause chronic disabling headaches and permanent vision loss.1
Clinical Course
The clinical course of IIH is unpredictable: In some patients, vision loss occurs gradually over the course of several weeks, while in others, loss occurs over a several month period. There are also patients with IIH who do not experience any alteration or loss of vision. Furthermore, some patients will experience permanent resolution of symptoms after a single lumbar puncture (LP); others have symptom recurrence after less than 24 hours; and some patients spontaneously remit on their own with no treatment whatsoever.1-4
Etiology
In the United States, IIH is a rare cause of headache, occurring in just 1 person per 100,000 annually.1 Though 90% of IIH cases occur in obese women of childbearing age, the etiology of IIH is unknown. Lumbar puncture usually alleviates the patient’s headache, but the CSF pressure typically returns to its pre-tap levels after a few hours.4,5 Neither CSF overproduction nor insufficient CSF resorption is responsible for causing IIH. One theory on the etiology of IIH proposes its cause to be due to a congenital malformation of the venous sinuses. This theory would explain why the symptoms so closely mimic those of venous sinus thrombosis, and why some IIH patients experience relief of symptoms after placement of a venous sinus stent.2
Symptoms
As noted previously, the most common symptom of IIH is headache, which patients usually describe as pressure-like and throbbing, and often involving retro-ocular pain. One feature in over half of patients is pulse-synchronous tinnitus (ie, hearing their own heartbeat in their ears). Eye pain, photophobia, blurry vision, and nausea/vomiting are all common symptoms in IIH, but these symptoms are also present in other causes of headache. The IIH headache might be relapsing and remitting, and can last from a few hours to weeks.2-4,6
Diagnosis
Imaging Studies. Noncontrast computed tomography (CT) imaging studies do not typically demonstrate any abnormal findings.1 Magnetic resonance imaging (MRI) studies show some inconsistent and subtle findings, such as flattening of the backs of the eyeballs, empty sella, or tortuous optic nerves.1
Lumbar Puncture. On LP, a very high opening pressure is a hallmark of IIH. An opening pressure <20 cm H2O is generally considered normal, 20 cm to 25 cm H2O is “equivocal,” and >25 cm H2O is abnormal.7 Patients presenting with IIH commonly have an opening pressure that exceeds 200 cm H2O.1-3 Extremely high pressures, however, are not required for the diagnosis, but some elevations in opening pressure will always be present.2,5 With the exception of a high opening pressure, the patient’s CSF analysis is normal.
Differential Diagnosis
Idiopathic intracranial hypertension is essentially a diagnosis of exclusion, one that is made after exclusion of all other potential causes of increased ICP (Table). Since contrast CT and MRI can identify subtle anatomical deformities and small lesions, their absence on these studies can help establish a diagnosis of IIH.

Venous Sinus Thrombosis. Venous sinus thrombosis is a rare but devastating condition that also cannot be diagnosed from a noncontrast CT but must always be considered in the differential diagnosis of IIH.8-10 Venous sinus thrombosis is characterized by a clot in one of the large venous sinuses that drain blood from the brain; the clot causes pressure to back up into the smaller cerebral vasculature, eventually inducing either a hemorrhagic stroke from a stressed vessel rupturing, or an ischemic stroke from lack of blood flow to the affected area of the brain. This condition is even more rare than IIH (0.5 cases per 100,000 population), but it can be devastating if missed, carrying a mortality rate as high as 15% in some studies.11
Risk Factors
Risk factors known to cause cerebral venous clots include genetic thrombophilias, pregnancy or recent pregnancy, oral contraceptive use, inflammatory bowel disease, severe dehydration, local infection/trauma, and substance abuse. Regardless of risk factors, the most recent guidelines of the American Heart Association/American Stroke Association recommend imaging studies of the cerebral venous sinuses for any patient presenting with new-onset symptoms suggestive of IIH (Class 1, Level of Evidence C).11 The two imaging options for evaluation of the cerebral venous sinuses are CT venography or MR venography. Since the 2013 American College of Radiology Appropriateness Criteria do not indicate a preference of one modality over the other, the choice of can be left to your radiologist.12
Patient Disposition
Patients with IIH typically do not require inpatient admission. Only about 3% of IIH patients will have a fulminant course of rapid-onset of vision loss, but even the most severe and acute cases will deteriorate over weeks, not hours or days.13 Nevertheless, close neurology follow-up is essential. If rapid and thorough outpatient neurological care is unavailable, admission is required.
Management
Not every patient with IIH experiences amelioration or resolution of symptoms following an LP; moreover, there is no clear way to differentiate patients who will experience therapeutic effects from LP from those who will not. Serial LPs as treatment for IIH have been discussed in the literature, but a ventriculoperitoneal shunt is a more practical approach in patients who do not respond to an initial LP.2,14
CSF Volume. The volume of CSF that can be removed safely may be 15 to 25 mL or more. A 1974 paper by Johnston and Paterson15 described five pseudotumor patients whose CSF was drained until their pressure had normalized; the amount removed varied from 15 to 25 mL, without adverse effects. A 1975 case series by Weisberg6 described safe removal of up to 30 mL of CSF in pseudotumor patients—the precise amount removed was determined by that which was necessary to lower the CSF pressure into the normal range. In 2007, a case report by Aly and Lawther16 of a pregnant woman with IIH describes twice weekly LP drainage of 30 mL.
There is nothing in the current literature to suggest that removing 10 to 30 mL of CSF instead of the 4 to 8 mL typically drawn in a diagnostic LP is going to pose any risk to the patient. The main complication associated with therapeutic LP is post-LP headache.5,17,18 There are currently no studies documenting outcomes after specific amounts of CSF removal.
Lifestyle Modifications: Weight Loss. No prospective, randomized controlled trials have proven weight loss to be effective in ameliorating the symptoms of IIH; however, several studies have found that rapid weight loss—whether through aggressive dieting or gastric bypass surgery—can improve symptoms dramatically within several months.19,20 One small study by Johnson et al has suggested that a 6% weight reduction is associated with marked improvement in papilledema.21Pharmacotherapy. The accepted first-line medication to alleviate symptoms of IIH is acetazolamide, and its use is supported by a recent randomized controlled trial conducted by the Neuro-Ophthalmology Research Disease Investigator Consortium (NORDIC).22 Most neurologists will administer a starting dose of acetazolamide 500 mg twice a day, and then increase the dose until symptoms are controlled or adverse effects appear (eg, fatigue, nausea/vomiting/diarrhea, electrolyte abnormalities, kidney stones) that contraindicate further dosage increases. In the NORDIC trial, patients were given up to 4 g of acetazolamide daily.22
Other medications, including loop diuretics and corticosteroids, should not be used except under the direct supervision of a neurologist.2,14
Refractory Cases
A patient who fails conservative treatment should be referred to a neurosurgeon for placement of a CSF shunt, optic nerve sheath fenestration, or placement of a venous sinus stent.23
Case Conclusion
After a noncontrast CT of the head was interpreted as completely normal, an LP was performed with the patient in the left lateral recumbent position. The opening CSF pressure exceeded 55 cm H2O (the upper limit of the manometer). The CSF was clear, and opening pressure was rechecked after each 5 mL draw. After 15 mL had been removed, the patient reported a sudden, dramatic disappearance of her headache and clearing of her vision. After 19 mL of CSF had been removed, the CSF pressure had dropped into the normal range (<20 cm H2O), and the procedure was ended.
To definitively rule out venous sinus thrombosis, a CT venogram was performed in the ED, and interpreted as normal. All other CSF results (cell count, protein, glucose, and gram stain) were normal. After complete resolution of the patient’s symptoms, she was discharged home with a prescription for acetazolamide 500 mg twice daily and instructions to follow-up with a neurologist within 48 hours. At discharge, the patient also received weight-loss counseling and was instructed to return immediately to the ED if her headache recurred or if she experienced any new neurological symptoms.
Summary
Idiopathic intracranial hypertension, also referred to as pseudotumor cerebri, is a rare but potentially vision-threatening cause of headache. Patients with signs and symptoms of IIH often initially present to the ED for evaluation and management. While the etiology of IIH is poorly understood, its clinical picture is unique: elevated ICP (sometimes markedly so) with no other significant findings on noncontrast head CT or CSF analysis. Venous sinus thrombosis, a life-threatening mimic of IIH, must always be included in the differential diagnosis.
Idiopathic intracranial hypertension is initially treated with rapid weight loss and acetazolamide. Many patients experience instant, though sometimes only transient, symptom relief from LP. No definitive studies to support any specific approach, including “therapeutic lumbar punctures.” The condition is rarely fulminant, and hospital admission is not typically required as long as urgent outpatient neurology follow-up is available.
1. Degnan AJ, Levy LM. Pseudotumor cerebri: brief review of clinical syndrome and imaging findings. AJNR Am J Neuroradiol. 2011;32(11):1986-1893. doi:10.3174/ajnr.A2404.
2. Biousse V, Bruce BB, Newman NJ. Update on the pathophysiology and management of idiopathic intracranial hypertension. J Neurol Neurosurg Psychiatry. 2012;83(5):488-494. doi:10.1136/jnnp-2011-302029. 3. Wall M, George D. Idiopathic intracranial hypertension: a prospective study of 50 patients. Brain. 1991;114(Pt 1A):155-180.
4. Wall M. Idiopathic intracranial hypertension. Neurol Clin. 2010;28(3):593-617. doi:10.1016/j.ncl.2010.03.003.
5. Friedman DI, Rausch EA. Headache diagnoses in patients with treated idiopathic intracranial hypertension. Neurology. 2002;58(10):1551-1553.
6. Weisberg LA. Benign intracranial hypertension. Medicine (Baltimore). 1975;54(3):197-207.
7. Whiteley W, Al-Shahi R, Warlow CP, Zeidler M, Lueck CJ. CSF opening pressure: reference interval and the effect of body mass index. Neurology. 2006;67(9):1690-1691.
8. Biousse V, Ameri A, Bousser MG. Isolated intracranial hypertension as the only sign of cerebral venous thrombosis. Neurology. 1999;53(7):1537-1542.
9. Leker RR, Steiner I. Features of dural sinus thrombosis simulating pseudotumor cerebri. Eur J Neurol. 1999;6(5):601-604.
10. Sylaja PN, Ahsan Moosa NV, Radhakrishnan K, Sankara Sarma P, Pradeep Kumar S. Differential diagnosis of patients with intracranial sinus venous thrombosis related isolated intracranial hypertension from those with idiopathic intracranial hypertension. J Neurol Sci. 2003;215(1-2):9-12.
11. Saposnik G, Barinagarrementeria F, Brown RD Jr, et al; American Heart Association Stroke Council and the Council on Epidemiology and Prevention. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(4):1158-1192. doi:10.1161/STR.0b013e31820a8364.
12. American College of Radiology ACR Appropriateness Criteria: Headache. https://acsearch.acr.org/docs/69482/Narrative/. Updated 2013. Accessed January 19, 2017.
13. Thambisetty M, Lavin PJ, Newman NJ, Biousse V. Fulminant idiopathic intracranial hypertension. Neurology. 2007;68(3):229-232.
14. Mollan SP, Markey KA, Benzimra JD, et al. A practical approach to, diagnosis, assessment and management of idiopathic intracranial hypertension. Pract Neurol. 2014;14(6):380-390. doi:10.1136/practneurol-2014-000821.
15. Johnston I, Paterson A. Benign intracranial hypertension. II. CSF pressure and circulation. Brain. 1974;97(2):301-312.
16. Aly EE, Lawther BK. Anaesthetic management of uncontrolled idiopathic intracranial hypertension during labour and delivery using an intrathecal catheter. Anesthesia. 2007;62(2):178-181.
17. Panikkath R, Welker J, Johnston R, Lado-Abeal J. Intracranial hypertension and intracranial hypotension causing headache in the same patient. Proc (Bayl Univ Med Cent). 2014;27(3):217-218.
18. Nafiu OO, Monterosso D, Walton SR, Bradin S. Post dural puncture headache in a pediatric patient with idiopathic intracranial hypertension. Paediatr Anaesth. 2005;15(9):778-781. doi:10.1111/j.1460-9592.2004.01529.x.
19. Sinclair AJ, Burdon MA, Nightingale PG, et al. Low energy diet and intracranial pressure in women with idiopathic intracranial hypertension: prospective cohort study. BMJ. 2010;341:c2701. doi:10.1136/bmj.c2701.
20. Kupersmith MJ, Gamell L, Turbin R, Peck V, Spiegel P, Wall M. Effects of weight loss on the course of idiopathic intracranial hypertension in women. Neurology. 1998;50(4):1094-1098.
21. Johnson LN, Krohel GB, Madsen RW, March GA Jr. The role of weight loss and acetazolamide in the treatment of idiopathic intracranial hypertension (pseudotumor cerebri) Ophthalmology. 1998;105(12):2313-2317. doi:10.1016/S0161-6420(98)91234-9.
22. NORDIC Idiopathic Intracranial Hypertension Study Group Writing Committee; Wall M, McDermott MP, Kieburtz KD, et al. Effect of acetazolamide on visual function in patients with idiopathic intracranial hypertension and mild visual loss: the idiopathic intracranial hypertension treatment trial. JAMA. 2014;311(16):1641-1651. doi:10.1001/jama.2014.3312.
23. Fonseca PL, Rigamonti D, Miller NR, Subramanian PS. Visual outcomes of surgical intervention for pseudotumour cerebri: optic nerve sheath fenestration versus cerebrospinal fluid diversion. Br J Ophthalmol. 2014;98(10):1360-1363. doi:10.1136/bjophthalmol-2014-304953.
Case
A 24-year-old woman presented to the ED for evaluation of a 3-week history of worsening headache and a 5-day history of increasingly blurry vision. The patient stated that she had initially contacted her primary care physician, but instead presented to the ED because he had no open appointments until the following week and recommended that she go to the ED.
The patient described her headache as a pulsating and throbbing pain over her entire head, which only mildly improved after taking over-the-counter (OTC) ibuprofen. She further noted that her headache was somewhat worse when lying down, and reported the sensation of hearing her own pulsating heartbeat in her ears.
The patient had no personal or family history of migraines, tension headaches, aneurysms, clotting disorders, bleeding disorders, or renal disease, and stated that she had never experienced this type of headache before. She denied photophobia, phonophobia, neck stiffness, fever, vomiting, cough, numbness or weakness in her extremities, or pain anywhere else in her body.
Over the past 5 days, the patient noticed her vision had become increasingly blurry. She was not on any prescription medications, stating the only medication she used was occasional OTC ibuprofen. She had no known allergy to medications and denied smoking or recreational drug use; she admitted to occasional alcohol consumption.
The patient resided with her husband, who had no similar symptoms. Physical examination showed an obese woman (height, 5 ft 6 in; weight, 195 lb; body mass index, 32 kg/m2), lying supine in apparent discomfort. Vital signs at presentation were all normal, and oxygen saturation was normal on room air.
A bedside ocular examination showed 20/100 in both eyes while using glasses; no visual field cuts or obvious central scotoma was present. The patient was alert and oriented to time and place. The neurological examination showed intact cranial nerves, 5/5 strength in all extremities, intact sensation in all extremities, no pronator drift, negative Romberg test, and a normal gait. Fundoscopic examination revealed mildly blurred medial optic discs bilaterally. The rest of the physical examination was normal.
Discussion
Pseudotumor cerebri, more commonly referred to as idiopathic intracranial hypertension (IIH), is characterized by increased intracranial pressure (ICP) with no explanatory findings on imaging studies or in cerebrospinal fluid (CSF) analysis, and may be accompanied by symptoms of chronic headache, tinnitus, papilledema and progressive vision loss caused by optic nerve damage.1 Though historically IIH was referred to by several other names, including “benign intracranial hypertension,” the condition is not benign—when untreated, IIH can cause chronic disabling headaches and permanent vision loss.1
Clinical Course
The clinical course of IIH is unpredictable: In some patients, vision loss occurs gradually over the course of several weeks, while in others, loss occurs over a several month period. There are also patients with IIH who do not experience any alteration or loss of vision. Furthermore, some patients will experience permanent resolution of symptoms after a single lumbar puncture (LP); others have symptom recurrence after less than 24 hours; and some patients spontaneously remit on their own with no treatment whatsoever.1-4
Etiology
In the United States, IIH is a rare cause of headache, occurring in just 1 person per 100,000 annually.1 Though 90% of IIH cases occur in obese women of childbearing age, the etiology of IIH is unknown. Lumbar puncture usually alleviates the patient’s headache, but the CSF pressure typically returns to its pre-tap levels after a few hours.4,5 Neither CSF overproduction nor insufficient CSF resorption is responsible for causing IIH. One theory on the etiology of IIH proposes its cause to be due to a congenital malformation of the venous sinuses. This theory would explain why the symptoms so closely mimic those of venous sinus thrombosis, and why some IIH patients experience relief of symptoms after placement of a venous sinus stent.2
Symptoms
As noted previously, the most common symptom of IIH is headache, which patients usually describe as pressure-like and throbbing, and often involving retro-ocular pain. One feature in over half of patients is pulse-synchronous tinnitus (ie, hearing their own heartbeat in their ears). Eye pain, photophobia, blurry vision, and nausea/vomiting are all common symptoms in IIH, but these symptoms are also present in other causes of headache. The IIH headache might be relapsing and remitting, and can last from a few hours to weeks.2-4,6
Diagnosis
Imaging Studies. Noncontrast computed tomography (CT) imaging studies do not typically demonstrate any abnormal findings.1 Magnetic resonance imaging (MRI) studies show some inconsistent and subtle findings, such as flattening of the backs of the eyeballs, empty sella, or tortuous optic nerves.1
Lumbar Puncture. On LP, a very high opening pressure is a hallmark of IIH. An opening pressure <20 cm H2O is generally considered normal, 20 cm to 25 cm H2O is “equivocal,” and >25 cm H2O is abnormal.7 Patients presenting with IIH commonly have an opening pressure that exceeds 200 cm H2O.1-3 Extremely high pressures, however, are not required for the diagnosis, but some elevations in opening pressure will always be present.2,5 With the exception of a high opening pressure, the patient’s CSF analysis is normal.
Differential Diagnosis
Idiopathic intracranial hypertension is essentially a diagnosis of exclusion, one that is made after exclusion of all other potential causes of increased ICP (Table). Since contrast CT and MRI can identify subtle anatomical deformities and small lesions, their absence on these studies can help establish a diagnosis of IIH.
Venous Sinus Thrombosis. Venous sinus thrombosis is a rare but devastating condition that also cannot be diagnosed from a noncontrast CT but must always be considered in the differential diagnosis of IIH.8-10 Venous sinus thrombosis is characterized by a clot in one of the large venous sinuses that drain blood from the brain; the clot causes pressure to back up into the smaller cerebral vasculature, eventually inducing either a hemorrhagic stroke from a stressed vessel rupturing, or an ischemic stroke from lack of blood flow to the affected area of the brain. This condition is even more rare than IIH (0.5 cases per 100,000 population), but it can be devastating if missed, carrying a mortality rate as high as 15% in some studies.11
Risk Factors
Risk factors known to cause cerebral venous clots include genetic thrombophilias, pregnancy or recent pregnancy, oral contraceptive use, inflammatory bowel disease, severe dehydration, local infection/trauma, and substance abuse. Regardless of risk factors, the most recent guidelines of the American Heart Association/American Stroke Association recommend imaging studies of the cerebral venous sinuses for any patient presenting with new-onset symptoms suggestive of IIH (Class 1, Level of Evidence C).11 The two imaging options for evaluation of the cerebral venous sinuses are CT venography or MR venography. Since the 2013 American College of Radiology Appropriateness Criteria do not indicate a preference of one modality over the other, the choice of can be left to your radiologist.12
Patient Disposition
Patients with IIH typically do not require inpatient admission. Only about 3% of IIH patients will have a fulminant course of rapid-onset of vision loss, but even the most severe and acute cases will deteriorate over weeks, not hours or days.13 Nevertheless, close neurology follow-up is essential. If rapid and thorough outpatient neurological care is unavailable, admission is required.
Management
Not every patient with IIH experiences amelioration or resolution of symptoms following an LP; moreover, there is no clear way to differentiate patients who will experience therapeutic effects from LP from those who will not. Serial LPs as treatment for IIH have been discussed in the literature, but a ventriculoperitoneal shunt is a more practical approach in patients who do not respond to an initial LP.2,14
CSF Volume. The volume of CSF that can be removed safely may be 15 to 25 mL or more. A 1974 paper by Johnston and Paterson15 described five pseudotumor patients whose CSF was drained until their pressure had normalized; the amount removed varied from 15 to 25 mL, without adverse effects. A 1975 case series by Weisberg6 described safe removal of up to 30 mL of CSF in pseudotumor patients—the precise amount removed was determined by that which was necessary to lower the CSF pressure into the normal range. In 2007, a case report by Aly and Lawther16 of a pregnant woman with IIH describes twice weekly LP drainage of 30 mL.
There is nothing in the current literature to suggest that removing 10 to 30 mL of CSF instead of the 4 to 8 mL typically drawn in a diagnostic LP is going to pose any risk to the patient. The main complication associated with therapeutic LP is post-LP headache.5,17,18 There are currently no studies documenting outcomes after specific amounts of CSF removal.
Lifestyle Modifications: Weight Loss. No prospective, randomized controlled trials have proven weight loss to be effective in ameliorating the symptoms of IIH; however, several studies have found that rapid weight loss—whether through aggressive dieting or gastric bypass surgery—can improve symptoms dramatically within several months.19,20 One small study by Johnson et al has suggested that a 6% weight reduction is associated with marked improvement in papilledema.21Pharmacotherapy. The accepted first-line medication to alleviate symptoms of IIH is acetazolamide, and its use is supported by a recent randomized controlled trial conducted by the Neuro-Ophthalmology Research Disease Investigator Consortium (NORDIC).22 Most neurologists will administer a starting dose of acetazolamide 500 mg twice a day, and then increase the dose until symptoms are controlled or adverse effects appear (eg, fatigue, nausea/vomiting/diarrhea, electrolyte abnormalities, kidney stones) that contraindicate further dosage increases. In the NORDIC trial, patients were given up to 4 g of acetazolamide daily.22
Other medications, including loop diuretics and corticosteroids, should not be used except under the direct supervision of a neurologist.2,14
Refractory Cases
A patient who fails conservative treatment should be referred to a neurosurgeon for placement of a CSF shunt, optic nerve sheath fenestration, or placement of a venous sinus stent.23
Case Conclusion
After a noncontrast CT of the head was interpreted as completely normal, an LP was performed with the patient in the left lateral recumbent position. The opening CSF pressure exceeded 55 cm H2O (the upper limit of the manometer). The CSF was clear, and opening pressure was rechecked after each 5 mL draw. After 15 mL had been removed, the patient reported a sudden, dramatic disappearance of her headache and clearing of her vision. After 19 mL of CSF had been removed, the CSF pressure had dropped into the normal range (<20 cm H2O), and the procedure was ended.
To definitively rule out venous sinus thrombosis, a CT venogram was performed in the ED, and interpreted as normal. All other CSF results (cell count, protein, glucose, and gram stain) were normal. After complete resolution of the patient’s symptoms, she was discharged home with a prescription for acetazolamide 500 mg twice daily and instructions to follow-up with a neurologist within 48 hours. At discharge, the patient also received weight-loss counseling and was instructed to return immediately to the ED if her headache recurred or if she experienced any new neurological symptoms.
Summary
Idiopathic intracranial hypertension, also referred to as pseudotumor cerebri, is a rare but potentially vision-threatening cause of headache. Patients with signs and symptoms of IIH often initially present to the ED for evaluation and management. While the etiology of IIH is poorly understood, its clinical picture is unique: elevated ICP (sometimes markedly so) with no other significant findings on noncontrast head CT or CSF analysis. Venous sinus thrombosis, a life-threatening mimic of IIH, must always be included in the differential diagnosis.
Idiopathic intracranial hypertension is initially treated with rapid weight loss and acetazolamide. Many patients experience instant, though sometimes only transient, symptom relief from LP. No definitive studies to support any specific approach, including “therapeutic lumbar punctures.” The condition is rarely fulminant, and hospital admission is not typically required as long as urgent outpatient neurology follow-up is available.
Case
A 24-year-old woman presented to the ED for evaluation of a 3-week history of worsening headache and a 5-day history of increasingly blurry vision. The patient stated that she had initially contacted her primary care physician, but instead presented to the ED because he had no open appointments until the following week and recommended that she go to the ED.
The patient described her headache as a pulsating and throbbing pain over her entire head, which only mildly improved after taking over-the-counter (OTC) ibuprofen. She further noted that her headache was somewhat worse when lying down, and reported the sensation of hearing her own pulsating heartbeat in her ears.
The patient had no personal or family history of migraines, tension headaches, aneurysms, clotting disorders, bleeding disorders, or renal disease, and stated that she had never experienced this type of headache before. She denied photophobia, phonophobia, neck stiffness, fever, vomiting, cough, numbness or weakness in her extremities, or pain anywhere else in her body.
Over the past 5 days, the patient noticed her vision had become increasingly blurry. She was not on any prescription medications, stating the only medication she used was occasional OTC ibuprofen. She had no known allergy to medications and denied smoking or recreational drug use; she admitted to occasional alcohol consumption.
The patient resided with her husband, who had no similar symptoms. Physical examination showed an obese woman (height, 5 ft 6 in; weight, 195 lb; body mass index, 32 kg/m2), lying supine in apparent discomfort. Vital signs at presentation were all normal, and oxygen saturation was normal on room air.
A bedside ocular examination showed 20/100 in both eyes while using glasses; no visual field cuts or obvious central scotoma was present. The patient was alert and oriented to time and place. The neurological examination showed intact cranial nerves, 5/5 strength in all extremities, intact sensation in all extremities, no pronator drift, negative Romberg test, and a normal gait. Fundoscopic examination revealed mildly blurred medial optic discs bilaterally. The rest of the physical examination was normal.
Discussion
Pseudotumor cerebri, more commonly referred to as idiopathic intracranial hypertension (IIH), is characterized by increased intracranial pressure (ICP) with no explanatory findings on imaging studies or in cerebrospinal fluid (CSF) analysis, and may be accompanied by symptoms of chronic headache, tinnitus, papilledema and progressive vision loss caused by optic nerve damage.1 Though historically IIH was referred to by several other names, including “benign intracranial hypertension,” the condition is not benign—when untreated, IIH can cause chronic disabling headaches and permanent vision loss.1
Clinical Course
The clinical course of IIH is unpredictable: In some patients, vision loss occurs gradually over the course of several weeks, while in others, loss occurs over a several month period. There are also patients with IIH who do not experience any alteration or loss of vision. Furthermore, some patients will experience permanent resolution of symptoms after a single lumbar puncture (LP); others have symptom recurrence after less than 24 hours; and some patients spontaneously remit on their own with no treatment whatsoever.1-4
Etiology
In the United States, IIH is a rare cause of headache, occurring in just 1 person per 100,000 annually.1 Though 90% of IIH cases occur in obese women of childbearing age, the etiology of IIH is unknown. Lumbar puncture usually alleviates the patient’s headache, but the CSF pressure typically returns to its pre-tap levels after a few hours.4,5 Neither CSF overproduction nor insufficient CSF resorption is responsible for causing IIH. One theory on the etiology of IIH proposes its cause to be due to a congenital malformation of the venous sinuses. This theory would explain why the symptoms so closely mimic those of venous sinus thrombosis, and why some IIH patients experience relief of symptoms after placement of a venous sinus stent.2
Symptoms
As noted previously, the most common symptom of IIH is headache, which patients usually describe as pressure-like and throbbing, and often involving retro-ocular pain. One feature in over half of patients is pulse-synchronous tinnitus (ie, hearing their own heartbeat in their ears). Eye pain, photophobia, blurry vision, and nausea/vomiting are all common symptoms in IIH, but these symptoms are also present in other causes of headache. The IIH headache might be relapsing and remitting, and can last from a few hours to weeks.2-4,6
Diagnosis
Imaging Studies. Noncontrast computed tomography (CT) imaging studies do not typically demonstrate any abnormal findings.1 Magnetic resonance imaging (MRI) studies show some inconsistent and subtle findings, such as flattening of the backs of the eyeballs, empty sella, or tortuous optic nerves.1
Lumbar Puncture. On LP, a very high opening pressure is a hallmark of IIH. An opening pressure <20 cm H2O is generally considered normal, 20 cm to 25 cm H2O is “equivocal,” and >25 cm H2O is abnormal.7 Patients presenting with IIH commonly have an opening pressure that exceeds 200 cm H2O.1-3 Extremely high pressures, however, are not required for the diagnosis, but some elevations in opening pressure will always be present.2,5 With the exception of a high opening pressure, the patient’s CSF analysis is normal.
Differential Diagnosis
Idiopathic intracranial hypertension is essentially a diagnosis of exclusion, one that is made after exclusion of all other potential causes of increased ICP (Table). Since contrast CT and MRI can identify subtle anatomical deformities and small lesions, their absence on these studies can help establish a diagnosis of IIH.
Venous Sinus Thrombosis. Venous sinus thrombosis is a rare but devastating condition that also cannot be diagnosed from a noncontrast CT but must always be considered in the differential diagnosis of IIH.8-10 Venous sinus thrombosis is characterized by a clot in one of the large venous sinuses that drain blood from the brain; the clot causes pressure to back up into the smaller cerebral vasculature, eventually inducing either a hemorrhagic stroke from a stressed vessel rupturing, or an ischemic stroke from lack of blood flow to the affected area of the brain. This condition is even more rare than IIH (0.5 cases per 100,000 population), but it can be devastating if missed, carrying a mortality rate as high as 15% in some studies.11
Risk Factors
Risk factors known to cause cerebral venous clots include genetic thrombophilias, pregnancy or recent pregnancy, oral contraceptive use, inflammatory bowel disease, severe dehydration, local infection/trauma, and substance abuse. Regardless of risk factors, the most recent guidelines of the American Heart Association/American Stroke Association recommend imaging studies of the cerebral venous sinuses for any patient presenting with new-onset symptoms suggestive of IIH (Class 1, Level of Evidence C).11 The two imaging options for evaluation of the cerebral venous sinuses are CT venography or MR venography. Since the 2013 American College of Radiology Appropriateness Criteria do not indicate a preference of one modality over the other, the choice of can be left to your radiologist.12
Patient Disposition
Patients with IIH typically do not require inpatient admission. Only about 3% of IIH patients will have a fulminant course of rapid-onset of vision loss, but even the most severe and acute cases will deteriorate over weeks, not hours or days.13 Nevertheless, close neurology follow-up is essential. If rapid and thorough outpatient neurological care is unavailable, admission is required.
Management
Not every patient with IIH experiences amelioration or resolution of symptoms following an LP; moreover, there is no clear way to differentiate patients who will experience therapeutic effects from LP from those who will not. Serial LPs as treatment for IIH have been discussed in the literature, but a ventriculoperitoneal shunt is a more practical approach in patients who do not respond to an initial LP.2,14
CSF Volume. The volume of CSF that can be removed safely may be 15 to 25 mL or more. A 1974 paper by Johnston and Paterson15 described five pseudotumor patients whose CSF was drained until their pressure had normalized; the amount removed varied from 15 to 25 mL, without adverse effects. A 1975 case series by Weisberg6 described safe removal of up to 30 mL of CSF in pseudotumor patients—the precise amount removed was determined by that which was necessary to lower the CSF pressure into the normal range. In 2007, a case report by Aly and Lawther16 of a pregnant woman with IIH describes twice weekly LP drainage of 30 mL.
There is nothing in the current literature to suggest that removing 10 to 30 mL of CSF instead of the 4 to 8 mL typically drawn in a diagnostic LP is going to pose any risk to the patient. The main complication associated with therapeutic LP is post-LP headache.5,17,18 There are currently no studies documenting outcomes after specific amounts of CSF removal.
Lifestyle Modifications: Weight Loss. No prospective, randomized controlled trials have proven weight loss to be effective in ameliorating the symptoms of IIH; however, several studies have found that rapid weight loss—whether through aggressive dieting or gastric bypass surgery—can improve symptoms dramatically within several months.19,20 One small study by Johnson et al has suggested that a 6% weight reduction is associated with marked improvement in papilledema.21Pharmacotherapy. The accepted first-line medication to alleviate symptoms of IIH is acetazolamide, and its use is supported by a recent randomized controlled trial conducted by the Neuro-Ophthalmology Research Disease Investigator Consortium (NORDIC).22 Most neurologists will administer a starting dose of acetazolamide 500 mg twice a day, and then increase the dose until symptoms are controlled or adverse effects appear (eg, fatigue, nausea/vomiting/diarrhea, electrolyte abnormalities, kidney stones) that contraindicate further dosage increases. In the NORDIC trial, patients were given up to 4 g of acetazolamide daily.22
Other medications, including loop diuretics and corticosteroids, should not be used except under the direct supervision of a neurologist.2,14
Refractory Cases
A patient who fails conservative treatment should be referred to a neurosurgeon for placement of a CSF shunt, optic nerve sheath fenestration, or placement of a venous sinus stent.23
Case Conclusion
After a noncontrast CT of the head was interpreted as completely normal, an LP was performed with the patient in the left lateral recumbent position. The opening CSF pressure exceeded 55 cm H2O (the upper limit of the manometer). The CSF was clear, and opening pressure was rechecked after each 5 mL draw. After 15 mL had been removed, the patient reported a sudden, dramatic disappearance of her headache and clearing of her vision. After 19 mL of CSF had been removed, the CSF pressure had dropped into the normal range (<20 cm H2O), and the procedure was ended.
To definitively rule out venous sinus thrombosis, a CT venogram was performed in the ED, and interpreted as normal. All other CSF results (cell count, protein, glucose, and gram stain) were normal. After complete resolution of the patient’s symptoms, she was discharged home with a prescription for acetazolamide 500 mg twice daily and instructions to follow-up with a neurologist within 48 hours. At discharge, the patient also received weight-loss counseling and was instructed to return immediately to the ED if her headache recurred or if she experienced any new neurological symptoms.
Summary
Idiopathic intracranial hypertension, also referred to as pseudotumor cerebri, is a rare but potentially vision-threatening cause of headache. Patients with signs and symptoms of IIH often initially present to the ED for evaluation and management. While the etiology of IIH is poorly understood, its clinical picture is unique: elevated ICP (sometimes markedly so) with no other significant findings on noncontrast head CT or CSF analysis. Venous sinus thrombosis, a life-threatening mimic of IIH, must always be included in the differential diagnosis.
Idiopathic intracranial hypertension is initially treated with rapid weight loss and acetazolamide. Many patients experience instant, though sometimes only transient, symptom relief from LP. No definitive studies to support any specific approach, including “therapeutic lumbar punctures.” The condition is rarely fulminant, and hospital admission is not typically required as long as urgent outpatient neurology follow-up is available.
1. Degnan AJ, Levy LM. Pseudotumor cerebri: brief review of clinical syndrome and imaging findings. AJNR Am J Neuroradiol. 2011;32(11):1986-1893. doi:10.3174/ajnr.A2404.
2. Biousse V, Bruce BB, Newman NJ. Update on the pathophysiology and management of idiopathic intracranial hypertension. J Neurol Neurosurg Psychiatry. 2012;83(5):488-494. doi:10.1136/jnnp-2011-302029. 3. Wall M, George D. Idiopathic intracranial hypertension: a prospective study of 50 patients. Brain. 1991;114(Pt 1A):155-180.
4. Wall M. Idiopathic intracranial hypertension. Neurol Clin. 2010;28(3):593-617. doi:10.1016/j.ncl.2010.03.003.
5. Friedman DI, Rausch EA. Headache diagnoses in patients with treated idiopathic intracranial hypertension. Neurology. 2002;58(10):1551-1553.
6. Weisberg LA. Benign intracranial hypertension. Medicine (Baltimore). 1975;54(3):197-207.
7. Whiteley W, Al-Shahi R, Warlow CP, Zeidler M, Lueck CJ. CSF opening pressure: reference interval and the effect of body mass index. Neurology. 2006;67(9):1690-1691.
8. Biousse V, Ameri A, Bousser MG. Isolated intracranial hypertension as the only sign of cerebral venous thrombosis. Neurology. 1999;53(7):1537-1542.
9. Leker RR, Steiner I. Features of dural sinus thrombosis simulating pseudotumor cerebri. Eur J Neurol. 1999;6(5):601-604.
10. Sylaja PN, Ahsan Moosa NV, Radhakrishnan K, Sankara Sarma P, Pradeep Kumar S. Differential diagnosis of patients with intracranial sinus venous thrombosis related isolated intracranial hypertension from those with idiopathic intracranial hypertension. J Neurol Sci. 2003;215(1-2):9-12.
11. Saposnik G, Barinagarrementeria F, Brown RD Jr, et al; American Heart Association Stroke Council and the Council on Epidemiology and Prevention. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(4):1158-1192. doi:10.1161/STR.0b013e31820a8364.
12. American College of Radiology ACR Appropriateness Criteria: Headache. https://acsearch.acr.org/docs/69482/Narrative/. Updated 2013. Accessed January 19, 2017.
13. Thambisetty M, Lavin PJ, Newman NJ, Biousse V. Fulminant idiopathic intracranial hypertension. Neurology. 2007;68(3):229-232.
14. Mollan SP, Markey KA, Benzimra JD, et al. A practical approach to, diagnosis, assessment and management of idiopathic intracranial hypertension. Pract Neurol. 2014;14(6):380-390. doi:10.1136/practneurol-2014-000821.
15. Johnston I, Paterson A. Benign intracranial hypertension. II. CSF pressure and circulation. Brain. 1974;97(2):301-312.
16. Aly EE, Lawther BK. Anaesthetic management of uncontrolled idiopathic intracranial hypertension during labour and delivery using an intrathecal catheter. Anesthesia. 2007;62(2):178-181.
17. Panikkath R, Welker J, Johnston R, Lado-Abeal J. Intracranial hypertension and intracranial hypotension causing headache in the same patient. Proc (Bayl Univ Med Cent). 2014;27(3):217-218.
18. Nafiu OO, Monterosso D, Walton SR, Bradin S. Post dural puncture headache in a pediatric patient with idiopathic intracranial hypertension. Paediatr Anaesth. 2005;15(9):778-781. doi:10.1111/j.1460-9592.2004.01529.x.
19. Sinclair AJ, Burdon MA, Nightingale PG, et al. Low energy diet and intracranial pressure in women with idiopathic intracranial hypertension: prospective cohort study. BMJ. 2010;341:c2701. doi:10.1136/bmj.c2701.
20. Kupersmith MJ, Gamell L, Turbin R, Peck V, Spiegel P, Wall M. Effects of weight loss on the course of idiopathic intracranial hypertension in women. Neurology. 1998;50(4):1094-1098.
21. Johnson LN, Krohel GB, Madsen RW, March GA Jr. The role of weight loss and acetazolamide in the treatment of idiopathic intracranial hypertension (pseudotumor cerebri) Ophthalmology. 1998;105(12):2313-2317. doi:10.1016/S0161-6420(98)91234-9.
22. NORDIC Idiopathic Intracranial Hypertension Study Group Writing Committee; Wall M, McDermott MP, Kieburtz KD, et al. Effect of acetazolamide on visual function in patients with idiopathic intracranial hypertension and mild visual loss: the idiopathic intracranial hypertension treatment trial. JAMA. 2014;311(16):1641-1651. doi:10.1001/jama.2014.3312.
23. Fonseca PL, Rigamonti D, Miller NR, Subramanian PS. Visual outcomes of surgical intervention for pseudotumour cerebri: optic nerve sheath fenestration versus cerebrospinal fluid diversion. Br J Ophthalmol. 2014;98(10):1360-1363. doi:10.1136/bjophthalmol-2014-304953.
1. Degnan AJ, Levy LM. Pseudotumor cerebri: brief review of clinical syndrome and imaging findings. AJNR Am J Neuroradiol. 2011;32(11):1986-1893. doi:10.3174/ajnr.A2404.
2. Biousse V, Bruce BB, Newman NJ. Update on the pathophysiology and management of idiopathic intracranial hypertension. J Neurol Neurosurg Psychiatry. 2012;83(5):488-494. doi:10.1136/jnnp-2011-302029. 3. Wall M, George D. Idiopathic intracranial hypertension: a prospective study of 50 patients. Brain. 1991;114(Pt 1A):155-180.
4. Wall M. Idiopathic intracranial hypertension. Neurol Clin. 2010;28(3):593-617. doi:10.1016/j.ncl.2010.03.003.
5. Friedman DI, Rausch EA. Headache diagnoses in patients with treated idiopathic intracranial hypertension. Neurology. 2002;58(10):1551-1553.
6. Weisberg LA. Benign intracranial hypertension. Medicine (Baltimore). 1975;54(3):197-207.
7. Whiteley W, Al-Shahi R, Warlow CP, Zeidler M, Lueck CJ. CSF opening pressure: reference interval and the effect of body mass index. Neurology. 2006;67(9):1690-1691.
8. Biousse V, Ameri A, Bousser MG. Isolated intracranial hypertension as the only sign of cerebral venous thrombosis. Neurology. 1999;53(7):1537-1542.
9. Leker RR, Steiner I. Features of dural sinus thrombosis simulating pseudotumor cerebri. Eur J Neurol. 1999;6(5):601-604.
10. Sylaja PN, Ahsan Moosa NV, Radhakrishnan K, Sankara Sarma P, Pradeep Kumar S. Differential diagnosis of patients with intracranial sinus venous thrombosis related isolated intracranial hypertension from those with idiopathic intracranial hypertension. J Neurol Sci. 2003;215(1-2):9-12.
11. Saposnik G, Barinagarrementeria F, Brown RD Jr, et al; American Heart Association Stroke Council and the Council on Epidemiology and Prevention. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(4):1158-1192. doi:10.1161/STR.0b013e31820a8364.
12. American College of Radiology ACR Appropriateness Criteria: Headache. https://acsearch.acr.org/docs/69482/Narrative/. Updated 2013. Accessed January 19, 2017.
13. Thambisetty M, Lavin PJ, Newman NJ, Biousse V. Fulminant idiopathic intracranial hypertension. Neurology. 2007;68(3):229-232.
14. Mollan SP, Markey KA, Benzimra JD, et al. A practical approach to, diagnosis, assessment and management of idiopathic intracranial hypertension. Pract Neurol. 2014;14(6):380-390. doi:10.1136/practneurol-2014-000821.
15. Johnston I, Paterson A. Benign intracranial hypertension. II. CSF pressure and circulation. Brain. 1974;97(2):301-312.
16. Aly EE, Lawther BK. Anaesthetic management of uncontrolled idiopathic intracranial hypertension during labour and delivery using an intrathecal catheter. Anesthesia. 2007;62(2):178-181.
17. Panikkath R, Welker J, Johnston R, Lado-Abeal J. Intracranial hypertension and intracranial hypotension causing headache in the same patient. Proc (Bayl Univ Med Cent). 2014;27(3):217-218.
18. Nafiu OO, Monterosso D, Walton SR, Bradin S. Post dural puncture headache in a pediatric patient with idiopathic intracranial hypertension. Paediatr Anaesth. 2005;15(9):778-781. doi:10.1111/j.1460-9592.2004.01529.x.
19. Sinclair AJ, Burdon MA, Nightingale PG, et al. Low energy diet and intracranial pressure in women with idiopathic intracranial hypertension: prospective cohort study. BMJ. 2010;341:c2701. doi:10.1136/bmj.c2701.
20. Kupersmith MJ, Gamell L, Turbin R, Peck V, Spiegel P, Wall M. Effects of weight loss on the course of idiopathic intracranial hypertension in women. Neurology. 1998;50(4):1094-1098.
21. Johnson LN, Krohel GB, Madsen RW, March GA Jr. The role of weight loss and acetazolamide in the treatment of idiopathic intracranial hypertension (pseudotumor cerebri) Ophthalmology. 1998;105(12):2313-2317. doi:10.1016/S0161-6420(98)91234-9.
22. NORDIC Idiopathic Intracranial Hypertension Study Group Writing Committee; Wall M, McDermott MP, Kieburtz KD, et al. Effect of acetazolamide on visual function in patients with idiopathic intracranial hypertension and mild visual loss: the idiopathic intracranial hypertension treatment trial. JAMA. 2014;311(16):1641-1651. doi:10.1001/jama.2014.3312.
23. Fonseca PL, Rigamonti D, Miller NR, Subramanian PS. Visual outcomes of surgical intervention for pseudotumour cerebri: optic nerve sheath fenestration versus cerebrospinal fluid diversion. Br J Ophthalmol. 2014;98(10):1360-1363. doi:10.1136/bjophthalmol-2014-304953.

Emergency Imaging: Abdominal Pain 6 Months After Cesarean Delivery
A 45-year-old woman with a history of polycystic ovary syndrome presented to the ED for evaluation of acute abdominal pain. The patient’s surgical history was significant for a cesarean delivery 6 months prior to presentation. Abdominal examination revealed a well-healed suprapubic cesarean incision scar, which was tender upon palpation. A computed tomography (CT) scan of the abdomen and pelvis with contrast were ordered; representative images are shown above (Figure 1a-1d).

What is the diagnosis? What are the associated complications and preferred management for this entity?
Answer
The scout image from the CT scan shows multiple dilated loops of small bowel (white arrows, Figure 2a) and only a small amount of air within a decompressed colon (red arrow, Figure 2a). The multiplanar CT image confirmed multiple dilated small bowel loops (white arrows, Figure 2b) and the decompressed large bowel (red arrows, Figure 2b), indicating the presence of a small bowel obstruction. A distal small bowel loop (white arrows, Figure 2c and 2d) was identified in a hernia sac within the walls of the rectus abdominis muscle (red arrows, Figure 2c and 2d). Mesenteric stranding within the hernia sac was suggestive of incarceration (black arrow, Figure 2d). No signs of intestinal ischemia, such as pneumatosis or wall thickening, were present.

An exploratory laparotomy was emergently performed, which confirmed the presence of incarcerated small bowel within the posterior rectus sheath defect without evidence of strangulation. Reduction of small bowel and primary closure of the hernia defect was subsequently performed without complication.
Abdominal Wall Hernias
Abdominal wall hernias are common in the United States, with more than 1 million abdominal wall hernia repairs performed annually.1 A posterior rectus sheath hernia is a rare type of abdominal wall hernia; the majority are postsurgical (as seen in this patient) or posttraumatic, with only a few reported congenital cases.2
Anatomy
The rectus sheath encloses the rectus abdominis muscle and is composed of the aponeuroses of the transversus abdominis, external oblique, and internal oblique muscles. The aponeuroses form an anterior and posterior sheath, which together serve as a strong barrier against the herniation of abdominal contents, accounting for the rarity of a spontaneous rectus sheath hernia. However, inferior to the umbilicus (below the arcuate line), the posterior rectus sheath is composed primarily of transversalis fascia, which may make this region more susceptible to herniation.3 Additional predisposing factors to herniation include increased muscle weakness and elevated intra-abdominal pressure, such as that which occurs during pregnancy or from ascites.4
Clinical Presentation
Like other abdominal wall hernias, the clinical presentation of posterior rectus sheath hernias is nonspecific. Patients may be asymptomatic or may develop abdominal pain, distension, and vomiting as a result of acute complications that necessitate emergent surgery. During history-taking, inquiry into a patient’s surgical history is crucial because it may raise clinical suspicion for an abdominal wall hernia, as was the case in our patient, who recently had a cesarean delivery.
Diagnosis
Because prompt and accurate diagnosis of acute complications of abdominal wall hernias is essential, imaging studies are typically required for diagnosis. Computed tomography is the modality of choice based on its ability to provide superior anatomic detail of the abdominal wall, permitting identification of hernias and differentiating them from other abdominal masses, such as hematomas, abscesses, or tumors. Additionally, CT is able to detect early signs of hernia sac complications, including bowel obstruction, incarceration, and strangulation.5
Treatment
Treatment for a posterior rectus sheath hernia is surgical with primary closure being the preferred method. Prosthetic repair may also be performed, particularly when the hernia defect is large, but it has been shown to be associated with an increased risk of intestinal strangulation.3
1. Rutkow IM. Demographic and socioeconomic aspects of hernia repair in the United States in 2003. Surg Clin North Am. 2003;83(5):1045-1051, v-vi. doi:10.1016/S0039-6109(03)00132-4.
2. Lenobel S, Lenobel R, Yu J. Posterior rectus sheath hernia causing intermittent small bowel obstruction. J Radiol Case Rep J. 2014;8(9):25-29. doi:10.3941/jrcr.v8i9.2081.
3. Losanoff JE, Basson MD, Gruber SA. Spontaneous hernia through the posterior rectus abdominis sheath: case report and review of the published literature 1937-2008. Hernia. 2009;13(5):555-558. doi:10.1007/s10029-009-0481-6.
4. Bentzon N, Adamsen S. Hernia of the posterior rectus sheath: a new entity? Eur J Surg. 1995;161(3):215-216.
5. Aguirre DA, Santosa AC, Casola G, Sirlin CB. Abdominal wall hernias: imaging features, complications, and diagnostic pitfalls at mutli-detector row CT. Radiographics. 2005;25(6):1501-1520. doi:10.1148/rg.256055018.
A 45-year-old woman with a history of polycystic ovary syndrome presented to the ED for evaluation of acute abdominal pain. The patient’s surgical history was significant for a cesarean delivery 6 months prior to presentation. Abdominal examination revealed a well-healed suprapubic cesarean incision scar, which was tender upon palpation. A computed tomography (CT) scan of the abdomen and pelvis with contrast were ordered; representative images are shown above (Figure 1a-1d).
What is the diagnosis? What are the associated complications and preferred management for this entity?
Answer
The scout image from the CT scan shows multiple dilated loops of small bowel (white arrows, Figure 2a) and only a small amount of air within a decompressed colon (red arrow, Figure 2a). The multiplanar CT image confirmed multiple dilated small bowel loops (white arrows, Figure 2b) and the decompressed large bowel (red arrows, Figure 2b), indicating the presence of a small bowel obstruction. A distal small bowel loop (white arrows, Figure 2c and 2d) was identified in a hernia sac within the walls of the rectus abdominis muscle (red arrows, Figure 2c and 2d). Mesenteric stranding within the hernia sac was suggestive of incarceration (black arrow, Figure 2d). No signs of intestinal ischemia, such as pneumatosis or wall thickening, were present.
An exploratory laparotomy was emergently performed, which confirmed the presence of incarcerated small bowel within the posterior rectus sheath defect without evidence of strangulation. Reduction of small bowel and primary closure of the hernia defect was subsequently performed without complication.
Abdominal Wall Hernias
Abdominal wall hernias are common in the United States, with more than 1 million abdominal wall hernia repairs performed annually.1 A posterior rectus sheath hernia is a rare type of abdominal wall hernia; the majority are postsurgical (as seen in this patient) or posttraumatic, with only a few reported congenital cases.2
Anatomy
The rectus sheath encloses the rectus abdominis muscle and is composed of the aponeuroses of the transversus abdominis, external oblique, and internal oblique muscles. The aponeuroses form an anterior and posterior sheath, which together serve as a strong barrier against the herniation of abdominal contents, accounting for the rarity of a spontaneous rectus sheath hernia. However, inferior to the umbilicus (below the arcuate line), the posterior rectus sheath is composed primarily of transversalis fascia, which may make this region more susceptible to herniation.3 Additional predisposing factors to herniation include increased muscle weakness and elevated intra-abdominal pressure, such as that which occurs during pregnancy or from ascites.4
Clinical Presentation
Like other abdominal wall hernias, the clinical presentation of posterior rectus sheath hernias is nonspecific. Patients may be asymptomatic or may develop abdominal pain, distension, and vomiting as a result of acute complications that necessitate emergent surgery. During history-taking, inquiry into a patient’s surgical history is crucial because it may raise clinical suspicion for an abdominal wall hernia, as was the case in our patient, who recently had a cesarean delivery.
Diagnosis
Because prompt and accurate diagnosis of acute complications of abdominal wall hernias is essential, imaging studies are typically required for diagnosis. Computed tomography is the modality of choice based on its ability to provide superior anatomic detail of the abdominal wall, permitting identification of hernias and differentiating them from other abdominal masses, such as hematomas, abscesses, or tumors. Additionally, CT is able to detect early signs of hernia sac complications, including bowel obstruction, incarceration, and strangulation.5
Treatment
Treatment for a posterior rectus sheath hernia is surgical with primary closure being the preferred method. Prosthetic repair may also be performed, particularly when the hernia defect is large, but it has been shown to be associated with an increased risk of intestinal strangulation.3
A 45-year-old woman with a history of polycystic ovary syndrome presented to the ED for evaluation of acute abdominal pain. The patient’s surgical history was significant for a cesarean delivery 6 months prior to presentation. Abdominal examination revealed a well-healed suprapubic cesarean incision scar, which was tender upon palpation. A computed tomography (CT) scan of the abdomen and pelvis with contrast were ordered; representative images are shown above (Figure 1a-1d).
What is the diagnosis? What are the associated complications and preferred management for this entity?
Answer
The scout image from the CT scan shows multiple dilated loops of small bowel (white arrows, Figure 2a) and only a small amount of air within a decompressed colon (red arrow, Figure 2a). The multiplanar CT image confirmed multiple dilated small bowel loops (white arrows, Figure 2b) and the decompressed large bowel (red arrows, Figure 2b), indicating the presence of a small bowel obstruction. A distal small bowel loop (white arrows, Figure 2c and 2d) was identified in a hernia sac within the walls of the rectus abdominis muscle (red arrows, Figure 2c and 2d). Mesenteric stranding within the hernia sac was suggestive of incarceration (black arrow, Figure 2d). No signs of intestinal ischemia, such as pneumatosis or wall thickening, were present.
An exploratory laparotomy was emergently performed, which confirmed the presence of incarcerated small bowel within the posterior rectus sheath defect without evidence of strangulation. Reduction of small bowel and primary closure of the hernia defect was subsequently performed without complication.
Abdominal Wall Hernias
Abdominal wall hernias are common in the United States, with more than 1 million abdominal wall hernia repairs performed annually.1 A posterior rectus sheath hernia is a rare type of abdominal wall hernia; the majority are postsurgical (as seen in this patient) or posttraumatic, with only a few reported congenital cases.2
Anatomy
The rectus sheath encloses the rectus abdominis muscle and is composed of the aponeuroses of the transversus abdominis, external oblique, and internal oblique muscles. The aponeuroses form an anterior and posterior sheath, which together serve as a strong barrier against the herniation of abdominal contents, accounting for the rarity of a spontaneous rectus sheath hernia. However, inferior to the umbilicus (below the arcuate line), the posterior rectus sheath is composed primarily of transversalis fascia, which may make this region more susceptible to herniation.3 Additional predisposing factors to herniation include increased muscle weakness and elevated intra-abdominal pressure, such as that which occurs during pregnancy or from ascites.4
Clinical Presentation
Like other abdominal wall hernias, the clinical presentation of posterior rectus sheath hernias is nonspecific. Patients may be asymptomatic or may develop abdominal pain, distension, and vomiting as a result of acute complications that necessitate emergent surgery. During history-taking, inquiry into a patient’s surgical history is crucial because it may raise clinical suspicion for an abdominal wall hernia, as was the case in our patient, who recently had a cesarean delivery.
Diagnosis
Because prompt and accurate diagnosis of acute complications of abdominal wall hernias is essential, imaging studies are typically required for diagnosis. Computed tomography is the modality of choice based on its ability to provide superior anatomic detail of the abdominal wall, permitting identification of hernias and differentiating them from other abdominal masses, such as hematomas, abscesses, or tumors. Additionally, CT is able to detect early signs of hernia sac complications, including bowel obstruction, incarceration, and strangulation.5
Treatment
Treatment for a posterior rectus sheath hernia is surgical with primary closure being the preferred method. Prosthetic repair may also be performed, particularly when the hernia defect is large, but it has been shown to be associated with an increased risk of intestinal strangulation.3
1. Rutkow IM. Demographic and socioeconomic aspects of hernia repair in the United States in 2003. Surg Clin North Am. 2003;83(5):1045-1051, v-vi. doi:10.1016/S0039-6109(03)00132-4.
2. Lenobel S, Lenobel R, Yu J. Posterior rectus sheath hernia causing intermittent small bowel obstruction. J Radiol Case Rep J. 2014;8(9):25-29. doi:10.3941/jrcr.v8i9.2081.
3. Losanoff JE, Basson MD, Gruber SA. Spontaneous hernia through the posterior rectus abdominis sheath: case report and review of the published literature 1937-2008. Hernia. 2009;13(5):555-558. doi:10.1007/s10029-009-0481-6.
4. Bentzon N, Adamsen S. Hernia of the posterior rectus sheath: a new entity? Eur J Surg. 1995;161(3):215-216.
5. Aguirre DA, Santosa AC, Casola G, Sirlin CB. Abdominal wall hernias: imaging features, complications, and diagnostic pitfalls at mutli-detector row CT. Radiographics. 2005;25(6):1501-1520. doi:10.1148/rg.256055018.
1. Rutkow IM. Demographic and socioeconomic aspects of hernia repair in the United States in 2003. Surg Clin North Am. 2003;83(5):1045-1051, v-vi. doi:10.1016/S0039-6109(03)00132-4.
2. Lenobel S, Lenobel R, Yu J. Posterior rectus sheath hernia causing intermittent small bowel obstruction. J Radiol Case Rep J. 2014;8(9):25-29. doi:10.3941/jrcr.v8i9.2081.
3. Losanoff JE, Basson MD, Gruber SA. Spontaneous hernia through the posterior rectus abdominis sheath: case report and review of the published literature 1937-2008. Hernia. 2009;13(5):555-558. doi:10.1007/s10029-009-0481-6.
4. Bentzon N, Adamsen S. Hernia of the posterior rectus sheath: a new entity? Eur J Surg. 1995;161(3):215-216.
5. Aguirre DA, Santosa AC, Casola G, Sirlin CB. Abdominal wall hernias: imaging features, complications, and diagnostic pitfalls at mutli-detector row CT. Radiographics. 2005;25(6):1501-1520. doi:10.1148/rg.256055018.
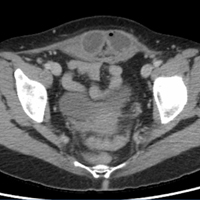
Nausea/vomiting • tachycardia • unintentional weight loss • Dx?
THE CASE
A 22-year-old woman presented to the emergency department (ED) with a 24-hour history of nausea, vomiting, diarrhea, generalized abdominal pain, and mild headache. She denied shortness of breath, chest pain, or anxiety, and didn’t have a history of cardiac problems. The physical examination revealed tachycardia (heart rate, 135 beats/min) and a respiratory rate of 24 breaths per minute. The patient was diagnosed with dehydration and was given 3 liters of intravenous (IV) fluids. After fluid administration, her heart rate decreased to 94 beats/min and she was discharged home.
The patient returned to the ED later that same day with recurrent nausea, vomiting, and a mild fever. This time she reported a several week history of palpitations, heat intolerance, agitation, mild cognitive impairment, and difficulty sleeping. Her mother accompanied her to this visit and added that the patient had unintentionally lost 13 pounds over the past 2 weeks. The patient denied pain or enlargement in her neck, obstructive symptoms, hives, pruritus, or changes in vision. Reexamination revealed tachycardia (132 beats/min) with no murmurs, rubs, or gallops; increased respiratory rate (26 breaths/min); and diffuse thyromegaly without distinct nodules. The thyroid was nontender to palpation. The patient was also found to have a fine resting tremor, hyperactive deep tendon reflexes, and clonus in her lower extremities. Bibasilar crackles were noted on lung exam.
THE DIAGNOSIS
An electrocardiogram (EKG) revealed sinus tachycardia with some sinus arrhythmia. A chest radiograph revealed prominent pulmonary vasculature and the presence of Kerley B lines consistent with marked pulmonary edema. Laboratory testing revealed an elevated N-terminal pro b-type natriuretic peptide level of 2420 pg/mL (normal range: <100 pg/mL). Evaluation of thyroid function revealed overt hyperthyroidism with an elevated free thyroxine of 4.6 ng/dL (normal range: 0.8-1.8 ng/dL), a total triiodothyronine of 199 ng/dL (normal range: 60-181 ng/dL), and a suppressed thyroid-stimulating hormone level of <0.02 mcU/mL (normal range: 0.35-5 mcU/mL). A subsequent thyroid ultrasound showed a diffusely enlarged thyroid gland with a thickened isthmus, but no nodules.
The patient’s results were discussed with the on-call endocrinology provider at the time of her revisit to the ED. The patient was started on antithyroid medications (methimazole 20 mg/d) and a beta-blocker (atenolol 25 mg/d). Arrangements were made for an outpatient endocrine consultation within 3 days of her visit to the ED.
Upon evaluation in the outpatient endocrinology clinic, a thyrotropin receptor antibody test was positive, confirming Graves’ disease. The patient was given a diagnosis of thyrotoxicosis secondary to hyperthyroidism due to Graves’ disease. Her marked pulmonary edema was secondary to thyrotoxicosis and aggressive hydration with IV fluids.
DISCUSSION
Hyperthyroidism is a common metabolic disorder with prominent cardiovascular manifestations.1 Classically, patients with hyperthyroidism develop irritability, heat intolerance, emotional lability, muscle weakness, menstrual abnormalities, and weight loss (despite an increased appetite). Cardiovascular manifestations include palpitations in up to 85% of patients, and dyspnea on exertion and fatigue in approximately 50% of patients.2 Hyperthyroidism has also been shown to produce changes in cardiac contractility, myocardial oxygen consumption, cardiac output, blood pressure, and systemic vascular resistance.3,4 Hyperthyroidism may complicate preexisting cardiac disease or may cause cardiac complications in individuals without structural abnormalities. (Our patient had no known structural abnormalities.)
In a small subset of patients with severe hyperthyroidism and exaggerated sinus tachycardia or atrial fibrillation, rate-related left ventricular dysfunction may cause heart failure.5 The assessment of thyrotoxic manifestations, especially potential cardiovascular complications, is essential to formulating an appropriate treatment plan.6 Cardiac evaluation may require an echocardiogram, EKG, Holter monitor, or myocardial perfusion studies.
Beta-blockers, diuretics among treatment options
Treatment with beta-blockers to reduce heart rate should be first-line therapy.7 In patients with overt heart failure involving pulmonary congestion, the use of diuretics may be appropriate.8
Our patient continued to take the medications prescribed during her ED visit: methimazole 20 mg/d and atenolol 25 mg/d for her Graves’ disease. A
THE TAKEAWAY
The cardiovascular manifestations of hyperthyroidism remain some of the most common signs and symptoms of thyroid disease. Pulmonary edema and congestive heart failure, however, are uncommon. Physicians need to be aware of this rare—but important—clinical presentation of a common condition.
1. Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001;344:501-509.
2. Fadel BM, Ellahham S, Ringel MD, et al. Hyperthyroid heart disease. Clin Cardiol. 2000;23:402-408.
3. Biondi B, Palmieri EA, Lombardi G, et al. Effects of thyroid hormone on cardiac function: the relative importance of heart rate, loading conditions, and myocardial contractility in the regulation of cardiac performance in human hyperthyroidism. J Clin Endocrinol Metab. 2002;87:968-974.
4. Kahaly GJ, Dillmann WH. Thyroid hormone action in the heart. Endocr Rev. 2005;26:704-728.
5. Klein I, Danzi S. Thyroid disease and the heart. Circulation. 2007;116:1725-1735.
6. Bahn Chair RS, Burch HB, Cooper DS, et al; American Thyroid Association; American Association of Clinical Endocrinologists. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Thyroid. 2011;21:593-646.
7. Klein I, Becker DV, Levey GS. Treatment of hyperthyroid disease. Ann Intern Med. 1994;121:281-288.
8. Danzi S, Klein I. Thyroid hormone and blood pressure regulation. Curr Hypertens Rep. 2003;5:513-520.
THE CASE
A 22-year-old woman presented to the emergency department (ED) with a 24-hour history of nausea, vomiting, diarrhea, generalized abdominal pain, and mild headache. She denied shortness of breath, chest pain, or anxiety, and didn’t have a history of cardiac problems. The physical examination revealed tachycardia (heart rate, 135 beats/min) and a respiratory rate of 24 breaths per minute. The patient was diagnosed with dehydration and was given 3 liters of intravenous (IV) fluids. After fluid administration, her heart rate decreased to 94 beats/min and she was discharged home.
The patient returned to the ED later that same day with recurrent nausea, vomiting, and a mild fever. This time she reported a several week history of palpitations, heat intolerance, agitation, mild cognitive impairment, and difficulty sleeping. Her mother accompanied her to this visit and added that the patient had unintentionally lost 13 pounds over the past 2 weeks. The patient denied pain or enlargement in her neck, obstructive symptoms, hives, pruritus, or changes in vision. Reexamination revealed tachycardia (132 beats/min) with no murmurs, rubs, or gallops; increased respiratory rate (26 breaths/min); and diffuse thyromegaly without distinct nodules. The thyroid was nontender to palpation. The patient was also found to have a fine resting tremor, hyperactive deep tendon reflexes, and clonus in her lower extremities. Bibasilar crackles were noted on lung exam.
THE DIAGNOSIS
An electrocardiogram (EKG) revealed sinus tachycardia with some sinus arrhythmia. A chest radiograph revealed prominent pulmonary vasculature and the presence of Kerley B lines consistent with marked pulmonary edema. Laboratory testing revealed an elevated N-terminal pro b-type natriuretic peptide level of 2420 pg/mL (normal range: <100 pg/mL). Evaluation of thyroid function revealed overt hyperthyroidism with an elevated free thyroxine of 4.6 ng/dL (normal range: 0.8-1.8 ng/dL), a total triiodothyronine of 199 ng/dL (normal range: 60-181 ng/dL), and a suppressed thyroid-stimulating hormone level of <0.02 mcU/mL (normal range: 0.35-5 mcU/mL). A subsequent thyroid ultrasound showed a diffusely enlarged thyroid gland with a thickened isthmus, but no nodules.
The patient’s results were discussed with the on-call endocrinology provider at the time of her revisit to the ED. The patient was started on antithyroid medications (methimazole 20 mg/d) and a beta-blocker (atenolol 25 mg/d). Arrangements were made for an outpatient endocrine consultation within 3 days of her visit to the ED.
Upon evaluation in the outpatient endocrinology clinic, a thyrotropin receptor antibody test was positive, confirming Graves’ disease. The patient was given a diagnosis of thyrotoxicosis secondary to hyperthyroidism due to Graves’ disease. Her marked pulmonary edema was secondary to thyrotoxicosis and aggressive hydration with IV fluids.
DISCUSSION
Hyperthyroidism is a common metabolic disorder with prominent cardiovascular manifestations.1 Classically, patients with hyperthyroidism develop irritability, heat intolerance, emotional lability, muscle weakness, menstrual abnormalities, and weight loss (despite an increased appetite). Cardiovascular manifestations include palpitations in up to 85% of patients, and dyspnea on exertion and fatigue in approximately 50% of patients.2 Hyperthyroidism has also been shown to produce changes in cardiac contractility, myocardial oxygen consumption, cardiac output, blood pressure, and systemic vascular resistance.3,4 Hyperthyroidism may complicate preexisting cardiac disease or may cause cardiac complications in individuals without structural abnormalities. (Our patient had no known structural abnormalities.)
In a small subset of patients with severe hyperthyroidism and exaggerated sinus tachycardia or atrial fibrillation, rate-related left ventricular dysfunction may cause heart failure.5 The assessment of thyrotoxic manifestations, especially potential cardiovascular complications, is essential to formulating an appropriate treatment plan.6 Cardiac evaluation may require an echocardiogram, EKG, Holter monitor, or myocardial perfusion studies.
Beta-blockers, diuretics among treatment options
Treatment with beta-blockers to reduce heart rate should be first-line therapy.7 In patients with overt heart failure involving pulmonary congestion, the use of diuretics may be appropriate.8
Our patient continued to take the medications prescribed during her ED visit: methimazole 20 mg/d and atenolol 25 mg/d for her Graves’ disease. A
THE TAKEAWAY
The cardiovascular manifestations of hyperthyroidism remain some of the most common signs and symptoms of thyroid disease. Pulmonary edema and congestive heart failure, however, are uncommon. Physicians need to be aware of this rare—but important—clinical presentation of a common condition.
THE CASE
A 22-year-old woman presented to the emergency department (ED) with a 24-hour history of nausea, vomiting, diarrhea, generalized abdominal pain, and mild headache. She denied shortness of breath, chest pain, or anxiety, and didn’t have a history of cardiac problems. The physical examination revealed tachycardia (heart rate, 135 beats/min) and a respiratory rate of 24 breaths per minute. The patient was diagnosed with dehydration and was given 3 liters of intravenous (IV) fluids. After fluid administration, her heart rate decreased to 94 beats/min and she was discharged home.
The patient returned to the ED later that same day with recurrent nausea, vomiting, and a mild fever. This time she reported a several week history of palpitations, heat intolerance, agitation, mild cognitive impairment, and difficulty sleeping. Her mother accompanied her to this visit and added that the patient had unintentionally lost 13 pounds over the past 2 weeks. The patient denied pain or enlargement in her neck, obstructive symptoms, hives, pruritus, or changes in vision. Reexamination revealed tachycardia (132 beats/min) with no murmurs, rubs, or gallops; increased respiratory rate (26 breaths/min); and diffuse thyromegaly without distinct nodules. The thyroid was nontender to palpation. The patient was also found to have a fine resting tremor, hyperactive deep tendon reflexes, and clonus in her lower extremities. Bibasilar crackles were noted on lung exam.
THE DIAGNOSIS
An electrocardiogram (EKG) revealed sinus tachycardia with some sinus arrhythmia. A chest radiograph revealed prominent pulmonary vasculature and the presence of Kerley B lines consistent with marked pulmonary edema. Laboratory testing revealed an elevated N-terminal pro b-type natriuretic peptide level of 2420 pg/mL (normal range: <100 pg/mL). Evaluation of thyroid function revealed overt hyperthyroidism with an elevated free thyroxine of 4.6 ng/dL (normal range: 0.8-1.8 ng/dL), a total triiodothyronine of 199 ng/dL (normal range: 60-181 ng/dL), and a suppressed thyroid-stimulating hormone level of <0.02 mcU/mL (normal range: 0.35-5 mcU/mL). A subsequent thyroid ultrasound showed a diffusely enlarged thyroid gland with a thickened isthmus, but no nodules.
The patient’s results were discussed with the on-call endocrinology provider at the time of her revisit to the ED. The patient was started on antithyroid medications (methimazole 20 mg/d) and a beta-blocker (atenolol 25 mg/d). Arrangements were made for an outpatient endocrine consultation within 3 days of her visit to the ED.
Upon evaluation in the outpatient endocrinology clinic, a thyrotropin receptor antibody test was positive, confirming Graves’ disease. The patient was given a diagnosis of thyrotoxicosis secondary to hyperthyroidism due to Graves’ disease. Her marked pulmonary edema was secondary to thyrotoxicosis and aggressive hydration with IV fluids.
DISCUSSION
Hyperthyroidism is a common metabolic disorder with prominent cardiovascular manifestations.1 Classically, patients with hyperthyroidism develop irritability, heat intolerance, emotional lability, muscle weakness, menstrual abnormalities, and weight loss (despite an increased appetite). Cardiovascular manifestations include palpitations in up to 85% of patients, and dyspnea on exertion and fatigue in approximately 50% of patients.2 Hyperthyroidism has also been shown to produce changes in cardiac contractility, myocardial oxygen consumption, cardiac output, blood pressure, and systemic vascular resistance.3,4 Hyperthyroidism may complicate preexisting cardiac disease or may cause cardiac complications in individuals without structural abnormalities. (Our patient had no known structural abnormalities.)
In a small subset of patients with severe hyperthyroidism and exaggerated sinus tachycardia or atrial fibrillation, rate-related left ventricular dysfunction may cause heart failure.5 The assessment of thyrotoxic manifestations, especially potential cardiovascular complications, is essential to formulating an appropriate treatment plan.6 Cardiac evaluation may require an echocardiogram, EKG, Holter monitor, or myocardial perfusion studies.
Beta-blockers, diuretics among treatment options
Treatment with beta-blockers to reduce heart rate should be first-line therapy.7 In patients with overt heart failure involving pulmonary congestion, the use of diuretics may be appropriate.8
Our patient continued to take the medications prescribed during her ED visit: methimazole 20 mg/d and atenolol 25 mg/d for her Graves’ disease. A
THE TAKEAWAY
The cardiovascular manifestations of hyperthyroidism remain some of the most common signs and symptoms of thyroid disease. Pulmonary edema and congestive heart failure, however, are uncommon. Physicians need to be aware of this rare—but important—clinical presentation of a common condition.
1. Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001;344:501-509.
2. Fadel BM, Ellahham S, Ringel MD, et al. Hyperthyroid heart disease. Clin Cardiol. 2000;23:402-408.
3. Biondi B, Palmieri EA, Lombardi G, et al. Effects of thyroid hormone on cardiac function: the relative importance of heart rate, loading conditions, and myocardial contractility in the regulation of cardiac performance in human hyperthyroidism. J Clin Endocrinol Metab. 2002;87:968-974.
4. Kahaly GJ, Dillmann WH. Thyroid hormone action in the heart. Endocr Rev. 2005;26:704-728.
5. Klein I, Danzi S. Thyroid disease and the heart. Circulation. 2007;116:1725-1735.
6. Bahn Chair RS, Burch HB, Cooper DS, et al; American Thyroid Association; American Association of Clinical Endocrinologists. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Thyroid. 2011;21:593-646.
7. Klein I, Becker DV, Levey GS. Treatment of hyperthyroid disease. Ann Intern Med. 1994;121:281-288.
8. Danzi S, Klein I. Thyroid hormone and blood pressure regulation. Curr Hypertens Rep. 2003;5:513-520.
1. Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001;344:501-509.
2. Fadel BM, Ellahham S, Ringel MD, et al. Hyperthyroid heart disease. Clin Cardiol. 2000;23:402-408.
3. Biondi B, Palmieri EA, Lombardi G, et al. Effects of thyroid hormone on cardiac function: the relative importance of heart rate, loading conditions, and myocardial contractility in the regulation of cardiac performance in human hyperthyroidism. J Clin Endocrinol Metab. 2002;87:968-974.
4. Kahaly GJ, Dillmann WH. Thyroid hormone action in the heart. Endocr Rev. 2005;26:704-728.
5. Klein I, Danzi S. Thyroid disease and the heart. Circulation. 2007;116:1725-1735.
6. Bahn Chair RS, Burch HB, Cooper DS, et al; American Thyroid Association; American Association of Clinical Endocrinologists. Hyperthyroidism and other causes of thyrotoxicosis: management guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Thyroid. 2011;21:593-646.
7. Klein I, Becker DV, Levey GS. Treatment of hyperthyroid disease. Ann Intern Med. 1994;121:281-288.
8. Danzi S, Klein I. Thyroid hormone and blood pressure regulation. Curr Hypertens Rep. 2003;5:513-520.

Muscle cramps/pain • weakness • muscle twitching • Dx?
THE CASE
A 39-year-old man who worked in construction presented to our clinic with complaints of muscle cramps and muscle pain that had been bothering him for several months. The cramps and pain started in both of his arms and subsequently became diffuse and generalized. He also reported an unintentional 15-pound weight loss.
His exam at that time was unremarkable. He was diagnosed with dehydration and cramping due to overexertion at work. A basic metabolic panel, hemogram, lipid panel, and thyroid stimulating hormone level were ordered. The patient’s triglyceride level, which was 227 mg/dL, was the only significant result (normal level: <150 mg/dL).
The patient’s symptoms continued to worsen until he returned to the clinic 6 months later, again complaining of muscle cramps and pain throughout his body. At that second visit, he also reported profound overall weakness and the development of diffuse muscle twitching, which his wife had observed while he was sleeping. As a result of these worrisome symptoms, he had become anxious and depressed.
A review of his medical record revealed a weight loss of about 20 pounds over the previous year. On exam, he had diffuse fasciculations in all the major muscle groups, including his tongue. The patient’s strength was 4/5 in all muscle groups. His deep tendon reflexes were 3+. He had a negative Babinski reflex (ie, he had downward facing toes with plantar stimulation), and cranial nerves II to XII were all intact. His rapid alternating movements and gait were slow.
THE DIAGNOSIS
Based on the exam, the primary diagnostic consideration for the patient was amyotrophic lateral sclerosis (ALS). Lab tests were ordered and revealed normal calcium and electrolyte levels, a normal erythrocyte sedimentation rate, a normal C-reactive protein level, and a negative test for acetylcholine receptor antibodies. However, the patient had an elevated creatine kinase level of 664 U/L (normal: 30-200 U/L). The patient was sent to a neuromuscular specialist, who identified signs of upper and lower motor neuron disease in all 4 of the patient’s extremities (he had foot drop that had not been present previously) and a very brisk jaw jerk. Along with the tongue fasciculations, the results of the specialist’s physical exam suggested ALS. Four-limb electromyography (EMG) showed widespread fasciculations and some large motor unit potentials and recruitment abnormalities, which were also consistent with ALS. It appeared that the patient’s weight loss was due to both muscle atrophy and the amount of calories burned from his constant twitching.
Extensive testing was done to rule out other potential causes of the patient’s symptoms, including magnetic resonance imaging (MRI) of the spine and brain (which was normal). In addition, the patient’s aldolase level and antineutrophil cytoplasmic antibodies were normal. The patient tested negative for human immunodeficiency virus and antibodies to double-stranded DNA. After serial neurologic exams, the final diagnosis of ALS was made.
DISCUSSION
ALS, also known as Lou Gehrig’s disease, is a degenerative motor neuron disease.1-3 The incidence in North America is 1.5 to 2.7 per 100,000 per year, and the prevalence is 2.7 to 7.4 per 100,000.4 The incidence of ALS increases with each decade of life, especially after age 40, and peaks at 74 years of age.4 The male to female ratio is 1:1.5-2.4 ALS affects upper and lower motor neurons and is progressive; however, the rate of progression and phenotype vary greatly between individuals.2 Most patients with ALS die within 2 to 5 years of onset.5
There is no specific test for ALS; the diagnosis is made clinically based on the revised El Escorial World Federation of Neurology criteria, also known as the Airlie House criteria.2,6,7 These criteria include evidence of lower motor neuron degeneration by clinical, electrophysiologic, or neuropathologic exam; evidence of upper motor neuron disease by clinical exam; progressive spread of symptoms or signs within a region or to other regions (by history or exam); and the absence of electrophysiologic, neuroimaging, or pathologic evidence of other disease processes that could explain the symptoms. If patients have evidence of upper and lower motor neuron disease, they should be reevaluated in 4 weeks to see if symptoms are improving or progressing.
Like our patient, many patients will have an elevated creatine kinase level (some with levels as high as 1000 U/L), and calcium may also be elevated because, rarely, ALS is associated with primary hyperparathyroidism.8 Electrophysiologic studies can be helpful in identifying active denervation of lower motor neurons.4,6,7
The differential diagnosis for ALS includes myasthenia gravis, inclusion-body myositis, multifocal motor neuropathy, benign fasciculations, hereditary spastic paraplegia, primary lateral sclerosis, post-polio progressive muscle atrophy, cervical spondylosis, and multiple sclerosis. A negative acetylcholine receptor antibody test will rule out myasthenia gravis, imaging of the spine can rule out cervical spondylosis, and electrophysiologic testing helps eliminate the other conditions (TABLE 14).

Treatment in specialty clinics can prolong survival
The mainstays of treatment are symptom management, multidisciplinary care (by physicians, physical/occupational/speech therapists, nutritionists, psychologists, psychotherapists, and genetic counselors), palliative care, and counseling about end-of-life issues for patients and family.1,5 Utilization of an ALS specialty clinic can provide access to all of these services and should be considered, as there is evidence that treatment in such clinics can prolong survival.5 The location of ALS specialty clinics can be found on the ALS Association’s Web site at http://www.alsa.org/community/.
Despite treatment, however, ALS is a progressive disease. The prognosis is poor, with a median survival of 2 to 5 years after diagnosis.9
The El Escorial World Federation of Neurology criteria for the diagnosis of ALS address how to treat the most common symptoms of ALS that occur as the disease progresses. These symptoms include dyspnea, muscle spasms, spasticity, sialorrhea, and pseudobulbar affect (TABLE 21,5).

Our patient was started on baclofen 10 mg 3 times per day (titrated up as needed) for muscle spasms and cramps, which resulted in some improvement of his cramps, but no improvement in the spasms. He was also started on sertraline 50 mg for anxiety and depression. His overall weakness continued to progress, and we recommended that the patient get ankle-foot orthosis braces to help with the mobility impairment caused by foot drop.
We then referred him to an ALS specialty clinic recommended by the neuromuscular specialist. The patient is now enrolled in a clinical trial designed to test a cerebrospinal fluid marker for diagnosis and for a new drug aimed at symptom management.
THE TAKEAWAY
Muscle cramps and pain are early signs of ALS. Although ALS is uncommon, patients who present with muscle cramps and muscle pain should have a creatine kinase test ordered (which, if elevated, should prompt further investigation into ALS as the possible cause). Patients should also undergo a neurologic examination to seek evidence of upper and lower motor neuron disease. They should then be reevaluated in 4 weeks to see if symptoms are improving or progressing. If no improvement is seen and symptoms are progressive, a work-up for ALS should be considered.
The mainstay of treatment for patients with ALS is multidisciplinary symptom management and palliative care. Utilization of an ALS specialty clinic should also be recommended, as it can improve survival.5
1. Miller RG, Gelinas D, O’Connor P. Amyotrophic Lateral Sclerosis: American Academy of Neurology Press Quality of Life Guide Series. Demos Medical Publishing; 2004.
2. Simon NG, Turner MR, Vucic S, et al. Quantifying disease progression in amyotrophic lateral sclerosis. Ann Neurol. 2014;76:643-657.
3. Worms PM. The epidemiology of motor neuron diseases: a review of recent studies. J Neurol Sci. 2001;191:3-9.
4. Shaw PJ. ALS and other motor neuron diseases. In: Goldman L, Schafer AI, eds. Goldman’s Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier Saunders; 2015:chap 418.
5. Miller RG, Jackson CE, Kasarskis EJ, et al. Practice Parameter update: The Care of the Patient with Amyotrophic Lateral Sclerosis: Multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1227-1233.
6. Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. 1994;124:96-107.
7. Brooks BR, Miller RG, Swash M, et al; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293-299.
8. Jackson CE, Amato AA, Bryan WW, et al. Primary hyperparathyroidism and ALS: is there a relation? Neurology. 1998;50:1795-1799.
9. Jablecki CK, Berry C, Leach J. Survival prediction in amyotrophic lateral sclerosis. Muscle Nerve. 1989;12:833-841.
THE CASE
A 39-year-old man who worked in construction presented to our clinic with complaints of muscle cramps and muscle pain that had been bothering him for several months. The cramps and pain started in both of his arms and subsequently became diffuse and generalized. He also reported an unintentional 15-pound weight loss.
His exam at that time was unremarkable. He was diagnosed with dehydration and cramping due to overexertion at work. A basic metabolic panel, hemogram, lipid panel, and thyroid stimulating hormone level were ordered. The patient’s triglyceride level, which was 227 mg/dL, was the only significant result (normal level: <150 mg/dL).
The patient’s symptoms continued to worsen until he returned to the clinic 6 months later, again complaining of muscle cramps and pain throughout his body. At that second visit, he also reported profound overall weakness and the development of diffuse muscle twitching, which his wife had observed while he was sleeping. As a result of these worrisome symptoms, he had become anxious and depressed.
A review of his medical record revealed a weight loss of about 20 pounds over the previous year. On exam, he had diffuse fasciculations in all the major muscle groups, including his tongue. The patient’s strength was 4/5 in all muscle groups. His deep tendon reflexes were 3+. He had a negative Babinski reflex (ie, he had downward facing toes with plantar stimulation), and cranial nerves II to XII were all intact. His rapid alternating movements and gait were slow.
THE DIAGNOSIS
Based on the exam, the primary diagnostic consideration for the patient was amyotrophic lateral sclerosis (ALS). Lab tests were ordered and revealed normal calcium and electrolyte levels, a normal erythrocyte sedimentation rate, a normal C-reactive protein level, and a negative test for acetylcholine receptor antibodies. However, the patient had an elevated creatine kinase level of 664 U/L (normal: 30-200 U/L). The patient was sent to a neuromuscular specialist, who identified signs of upper and lower motor neuron disease in all 4 of the patient’s extremities (he had foot drop that had not been present previously) and a very brisk jaw jerk. Along with the tongue fasciculations, the results of the specialist’s physical exam suggested ALS. Four-limb electromyography (EMG) showed widespread fasciculations and some large motor unit potentials and recruitment abnormalities, which were also consistent with ALS. It appeared that the patient’s weight loss was due to both muscle atrophy and the amount of calories burned from his constant twitching.
Extensive testing was done to rule out other potential causes of the patient’s symptoms, including magnetic resonance imaging (MRI) of the spine and brain (which was normal). In addition, the patient’s aldolase level and antineutrophil cytoplasmic antibodies were normal. The patient tested negative for human immunodeficiency virus and antibodies to double-stranded DNA. After serial neurologic exams, the final diagnosis of ALS was made.
DISCUSSION
ALS, also known as Lou Gehrig’s disease, is a degenerative motor neuron disease.1-3 The incidence in North America is 1.5 to 2.7 per 100,000 per year, and the prevalence is 2.7 to 7.4 per 100,000.4 The incidence of ALS increases with each decade of life, especially after age 40, and peaks at 74 years of age.4 The male to female ratio is 1:1.5-2.4 ALS affects upper and lower motor neurons and is progressive; however, the rate of progression and phenotype vary greatly between individuals.2 Most patients with ALS die within 2 to 5 years of onset.5
There is no specific test for ALS; the diagnosis is made clinically based on the revised El Escorial World Federation of Neurology criteria, also known as the Airlie House criteria.2,6,7 These criteria include evidence of lower motor neuron degeneration by clinical, electrophysiologic, or neuropathologic exam; evidence of upper motor neuron disease by clinical exam; progressive spread of symptoms or signs within a region or to other regions (by history or exam); and the absence of electrophysiologic, neuroimaging, or pathologic evidence of other disease processes that could explain the symptoms. If patients have evidence of upper and lower motor neuron disease, they should be reevaluated in 4 weeks to see if symptoms are improving or progressing.
Like our patient, many patients will have an elevated creatine kinase level (some with levels as high as 1000 U/L), and calcium may also be elevated because, rarely, ALS is associated with primary hyperparathyroidism.8 Electrophysiologic studies can be helpful in identifying active denervation of lower motor neurons.4,6,7
The differential diagnosis for ALS includes myasthenia gravis, inclusion-body myositis, multifocal motor neuropathy, benign fasciculations, hereditary spastic paraplegia, primary lateral sclerosis, post-polio progressive muscle atrophy, cervical spondylosis, and multiple sclerosis. A negative acetylcholine receptor antibody test will rule out myasthenia gravis, imaging of the spine can rule out cervical spondylosis, and electrophysiologic testing helps eliminate the other conditions (TABLE 14).
Treatment in specialty clinics can prolong survival
The mainstays of treatment are symptom management, multidisciplinary care (by physicians, physical/occupational/speech therapists, nutritionists, psychologists, psychotherapists, and genetic counselors), palliative care, and counseling about end-of-life issues for patients and family.1,5 Utilization of an ALS specialty clinic can provide access to all of these services and should be considered, as there is evidence that treatment in such clinics can prolong survival.5 The location of ALS specialty clinics can be found on the ALS Association’s Web site at http://www.alsa.org/community/.
Despite treatment, however, ALS is a progressive disease. The prognosis is poor, with a median survival of 2 to 5 years after diagnosis.9
The El Escorial World Federation of Neurology criteria for the diagnosis of ALS address how to treat the most common symptoms of ALS that occur as the disease progresses. These symptoms include dyspnea, muscle spasms, spasticity, sialorrhea, and pseudobulbar affect (TABLE 21,5).
Our patient was started on baclofen 10 mg 3 times per day (titrated up as needed) for muscle spasms and cramps, which resulted in some improvement of his cramps, but no improvement in the spasms. He was also started on sertraline 50 mg for anxiety and depression. His overall weakness continued to progress, and we recommended that the patient get ankle-foot orthosis braces to help with the mobility impairment caused by foot drop.
We then referred him to an ALS specialty clinic recommended by the neuromuscular specialist. The patient is now enrolled in a clinical trial designed to test a cerebrospinal fluid marker for diagnosis and for a new drug aimed at symptom management.
THE TAKEAWAY
Muscle cramps and pain are early signs of ALS. Although ALS is uncommon, patients who present with muscle cramps and muscle pain should have a creatine kinase test ordered (which, if elevated, should prompt further investigation into ALS as the possible cause). Patients should also undergo a neurologic examination to seek evidence of upper and lower motor neuron disease. They should then be reevaluated in 4 weeks to see if symptoms are improving or progressing. If no improvement is seen and symptoms are progressive, a work-up for ALS should be considered.
The mainstay of treatment for patients with ALS is multidisciplinary symptom management and palliative care. Utilization of an ALS specialty clinic should also be recommended, as it can improve survival.5
THE CASE
A 39-year-old man who worked in construction presented to our clinic with complaints of muscle cramps and muscle pain that had been bothering him for several months. The cramps and pain started in both of his arms and subsequently became diffuse and generalized. He also reported an unintentional 15-pound weight loss.
His exam at that time was unremarkable. He was diagnosed with dehydration and cramping due to overexertion at work. A basic metabolic panel, hemogram, lipid panel, and thyroid stimulating hormone level were ordered. The patient’s triglyceride level, which was 227 mg/dL, was the only significant result (normal level: <150 mg/dL).
The patient’s symptoms continued to worsen until he returned to the clinic 6 months later, again complaining of muscle cramps and pain throughout his body. At that second visit, he also reported profound overall weakness and the development of diffuse muscle twitching, which his wife had observed while he was sleeping. As a result of these worrisome symptoms, he had become anxious and depressed.
A review of his medical record revealed a weight loss of about 20 pounds over the previous year. On exam, he had diffuse fasciculations in all the major muscle groups, including his tongue. The patient’s strength was 4/5 in all muscle groups. His deep tendon reflexes were 3+. He had a negative Babinski reflex (ie, he had downward facing toes with plantar stimulation), and cranial nerves II to XII were all intact. His rapid alternating movements and gait were slow.
THE DIAGNOSIS
Based on the exam, the primary diagnostic consideration for the patient was amyotrophic lateral sclerosis (ALS). Lab tests were ordered and revealed normal calcium and electrolyte levels, a normal erythrocyte sedimentation rate, a normal C-reactive protein level, and a negative test for acetylcholine receptor antibodies. However, the patient had an elevated creatine kinase level of 664 U/L (normal: 30-200 U/L). The patient was sent to a neuromuscular specialist, who identified signs of upper and lower motor neuron disease in all 4 of the patient’s extremities (he had foot drop that had not been present previously) and a very brisk jaw jerk. Along with the tongue fasciculations, the results of the specialist’s physical exam suggested ALS. Four-limb electromyography (EMG) showed widespread fasciculations and some large motor unit potentials and recruitment abnormalities, which were also consistent with ALS. It appeared that the patient’s weight loss was due to both muscle atrophy and the amount of calories burned from his constant twitching.
Extensive testing was done to rule out other potential causes of the patient’s symptoms, including magnetic resonance imaging (MRI) of the spine and brain (which was normal). In addition, the patient’s aldolase level and antineutrophil cytoplasmic antibodies were normal. The patient tested negative for human immunodeficiency virus and antibodies to double-stranded DNA. After serial neurologic exams, the final diagnosis of ALS was made.
DISCUSSION
ALS, also known as Lou Gehrig’s disease, is a degenerative motor neuron disease.1-3 The incidence in North America is 1.5 to 2.7 per 100,000 per year, and the prevalence is 2.7 to 7.4 per 100,000.4 The incidence of ALS increases with each decade of life, especially after age 40, and peaks at 74 years of age.4 The male to female ratio is 1:1.5-2.4 ALS affects upper and lower motor neurons and is progressive; however, the rate of progression and phenotype vary greatly between individuals.2 Most patients with ALS die within 2 to 5 years of onset.5
There is no specific test for ALS; the diagnosis is made clinically based on the revised El Escorial World Federation of Neurology criteria, also known as the Airlie House criteria.2,6,7 These criteria include evidence of lower motor neuron degeneration by clinical, electrophysiologic, or neuropathologic exam; evidence of upper motor neuron disease by clinical exam; progressive spread of symptoms or signs within a region or to other regions (by history or exam); and the absence of electrophysiologic, neuroimaging, or pathologic evidence of other disease processes that could explain the symptoms. If patients have evidence of upper and lower motor neuron disease, they should be reevaluated in 4 weeks to see if symptoms are improving or progressing.
Like our patient, many patients will have an elevated creatine kinase level (some with levels as high as 1000 U/L), and calcium may also be elevated because, rarely, ALS is associated with primary hyperparathyroidism.8 Electrophysiologic studies can be helpful in identifying active denervation of lower motor neurons.4,6,7
The differential diagnosis for ALS includes myasthenia gravis, inclusion-body myositis, multifocal motor neuropathy, benign fasciculations, hereditary spastic paraplegia, primary lateral sclerosis, post-polio progressive muscle atrophy, cervical spondylosis, and multiple sclerosis. A negative acetylcholine receptor antibody test will rule out myasthenia gravis, imaging of the spine can rule out cervical spondylosis, and electrophysiologic testing helps eliminate the other conditions (TABLE 14).
Treatment in specialty clinics can prolong survival
The mainstays of treatment are symptom management, multidisciplinary care (by physicians, physical/occupational/speech therapists, nutritionists, psychologists, psychotherapists, and genetic counselors), palliative care, and counseling about end-of-life issues for patients and family.1,5 Utilization of an ALS specialty clinic can provide access to all of these services and should be considered, as there is evidence that treatment in such clinics can prolong survival.5 The location of ALS specialty clinics can be found on the ALS Association’s Web site at http://www.alsa.org/community/.
Despite treatment, however, ALS is a progressive disease. The prognosis is poor, with a median survival of 2 to 5 years after diagnosis.9
The El Escorial World Federation of Neurology criteria for the diagnosis of ALS address how to treat the most common symptoms of ALS that occur as the disease progresses. These symptoms include dyspnea, muscle spasms, spasticity, sialorrhea, and pseudobulbar affect (TABLE 21,5).
Our patient was started on baclofen 10 mg 3 times per day (titrated up as needed) for muscle spasms and cramps, which resulted in some improvement of his cramps, but no improvement in the spasms. He was also started on sertraline 50 mg for anxiety and depression. His overall weakness continued to progress, and we recommended that the patient get ankle-foot orthosis braces to help with the mobility impairment caused by foot drop.
We then referred him to an ALS specialty clinic recommended by the neuromuscular specialist. The patient is now enrolled in a clinical trial designed to test a cerebrospinal fluid marker for diagnosis and for a new drug aimed at symptom management.
THE TAKEAWAY
Muscle cramps and pain are early signs of ALS. Although ALS is uncommon, patients who present with muscle cramps and muscle pain should have a creatine kinase test ordered (which, if elevated, should prompt further investigation into ALS as the possible cause). Patients should also undergo a neurologic examination to seek evidence of upper and lower motor neuron disease. They should then be reevaluated in 4 weeks to see if symptoms are improving or progressing. If no improvement is seen and symptoms are progressive, a work-up for ALS should be considered.
The mainstay of treatment for patients with ALS is multidisciplinary symptom management and palliative care. Utilization of an ALS specialty clinic should also be recommended, as it can improve survival.5
1. Miller RG, Gelinas D, O’Connor P. Amyotrophic Lateral Sclerosis: American Academy of Neurology Press Quality of Life Guide Series. Demos Medical Publishing; 2004.
2. Simon NG, Turner MR, Vucic S, et al. Quantifying disease progression in amyotrophic lateral sclerosis. Ann Neurol. 2014;76:643-657.
3. Worms PM. The epidemiology of motor neuron diseases: a review of recent studies. J Neurol Sci. 2001;191:3-9.
4. Shaw PJ. ALS and other motor neuron diseases. In: Goldman L, Schafer AI, eds. Goldman’s Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier Saunders; 2015:chap 418.
5. Miller RG, Jackson CE, Kasarskis EJ, et al. Practice Parameter update: The Care of the Patient with Amyotrophic Lateral Sclerosis: Multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1227-1233.
6. Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. 1994;124:96-107.
7. Brooks BR, Miller RG, Swash M, et al; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293-299.
8. Jackson CE, Amato AA, Bryan WW, et al. Primary hyperparathyroidism and ALS: is there a relation? Neurology. 1998;50:1795-1799.
9. Jablecki CK, Berry C, Leach J. Survival prediction in amyotrophic lateral sclerosis. Muscle Nerve. 1989;12:833-841.
1. Miller RG, Gelinas D, O’Connor P. Amyotrophic Lateral Sclerosis: American Academy of Neurology Press Quality of Life Guide Series. Demos Medical Publishing; 2004.
2. Simon NG, Turner MR, Vucic S, et al. Quantifying disease progression in amyotrophic lateral sclerosis. Ann Neurol. 2014;76:643-657.
3. Worms PM. The epidemiology of motor neuron diseases: a review of recent studies. J Neurol Sci. 2001;191:3-9.
4. Shaw PJ. ALS and other motor neuron diseases. In: Goldman L, Schafer AI, eds. Goldman’s Cecil Medicine. 24th ed. Philadelphia, PA: Elsevier Saunders; 2015:chap 418.
5. Miller RG, Jackson CE, Kasarskis EJ, et al. Practice Parameter update: The Care of the Patient with Amyotrophic Lateral Sclerosis: Multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1227-1233.
6. Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. 1994;124:96-107.
7. Brooks BR, Miller RG, Swash M, et al; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293-299.
8. Jackson CE, Amato AA, Bryan WW, et al. Primary hyperparathyroidism and ALS: is there a relation? Neurology. 1998;50:1795-1799.
9. Jablecki CK, Berry C, Leach J. Survival prediction in amyotrophic lateral sclerosis. Muscle Nerve. 1989;12:833-841.

Torsades de Pointes in Severe Alcohol Withdrawal and Cirrhosis: Implications for Risk Stratification and Management
Torsades de pointes (TdP) is a life-threatening ventricular arrhythmia that is associated with both congenital and acquired QT interval prolongation. QT interval prolongation is commonly observed in acute alcohol withdrawal and cirrhotic cardiomyopathy.1-3 In both conditions, there is a positive correlation between the degree of QT interval prolongation and disease severity.4,5 The precise mechanisms of QT interval prolongation in these conditions are not well understood. One hypothesis is that autonomic hyperexcitability results in altered ventricular repolarization and QT interval prolongation. This mechanism of QT prolongation has been found in acute alcohol withdrawal independent of electrolyte abnormalities, use of QT-prolonging medications, and cirrhosis.1,2,6
The authors report the case of a veteran who was hospitalized for acute alcohol withdrawal and decompensated cirrhosis and was found to have a newly prolonged QT interval. On hospital day 3, the patient developed TdP, which required external defibrillation. Despite correction of electrolyte abnormalities, abstinence from alcohol, avoidance of QT-prolonging medications, and exclusion of cardiac ischemia, there was significant and persistent prolongation of the QT interval—ultimately attributed to cirrhotic cardiomyopathy. Acquired QT interval prolongation is common in both acute alcohol withdrawal and cirrhosis.This case highlights the importance of close monitoring of the QT interval and TdP susceptibility in patients being treated for acute alcohol withdrawal, particularly those with cirrhosis.
Case Report
A 66-year-old male veteran with a 35-year history of alcohol dependence presented for alcohol detoxification. He reported having drunk at least 32 ounces of vodka every day of the preceding 5 years and reported having unsuccessfully attempted self-detoxification several times. Prior detoxification efforts were unsuccessful because of intractable nausea and tremulousness. Additional presenting symptoms included lethargy, anorexia, and a fall with transient right-side hemiparesis (findings on magnetic resonance imaging of the head had been normal).
His medical history included type 2 diabetes, tobacco dependence, and macular degeneration. The only medication being taken was glargine 25 units daily. On admission, the patient was afebrile (98.1°F), normotensive (103/77 mm Hg), and oriented to person, place, and time. Examination also revealed a protuberant abdomen with caput medusae, and no shifting dullness or lower extremity edema. The neurologic examination was nonfocal.
Laboratory test results on admission were significant for elevated serum alcohol level (243.8 mg/dL); elevated levels of aspartate aminotransferase (144 units/L) alanine aminotransferase (25 units/L), and total bilirubin (4.2 mg/L); hypoalbuminemia; normocalemia; hypomagnesemia; normal corrected calcium level; and normal renal function (0.84 mg/dL)(Table). The patient’s admission Child-Pugh score of 10 indicated class C liver disease. Admission electrocardiogram (EKG) revealed normal sinus rhythm, first-degree atrioventricular block, and prolongation of the QTc interval (519 ms). Six years earlier, the patient’s QTc interval had been 409 ms (Figures 1 and 2). As QT interval depends on heart rate, it is most commonly expressed as corrected QT, or QTc, where QTc = QT/(√RR). 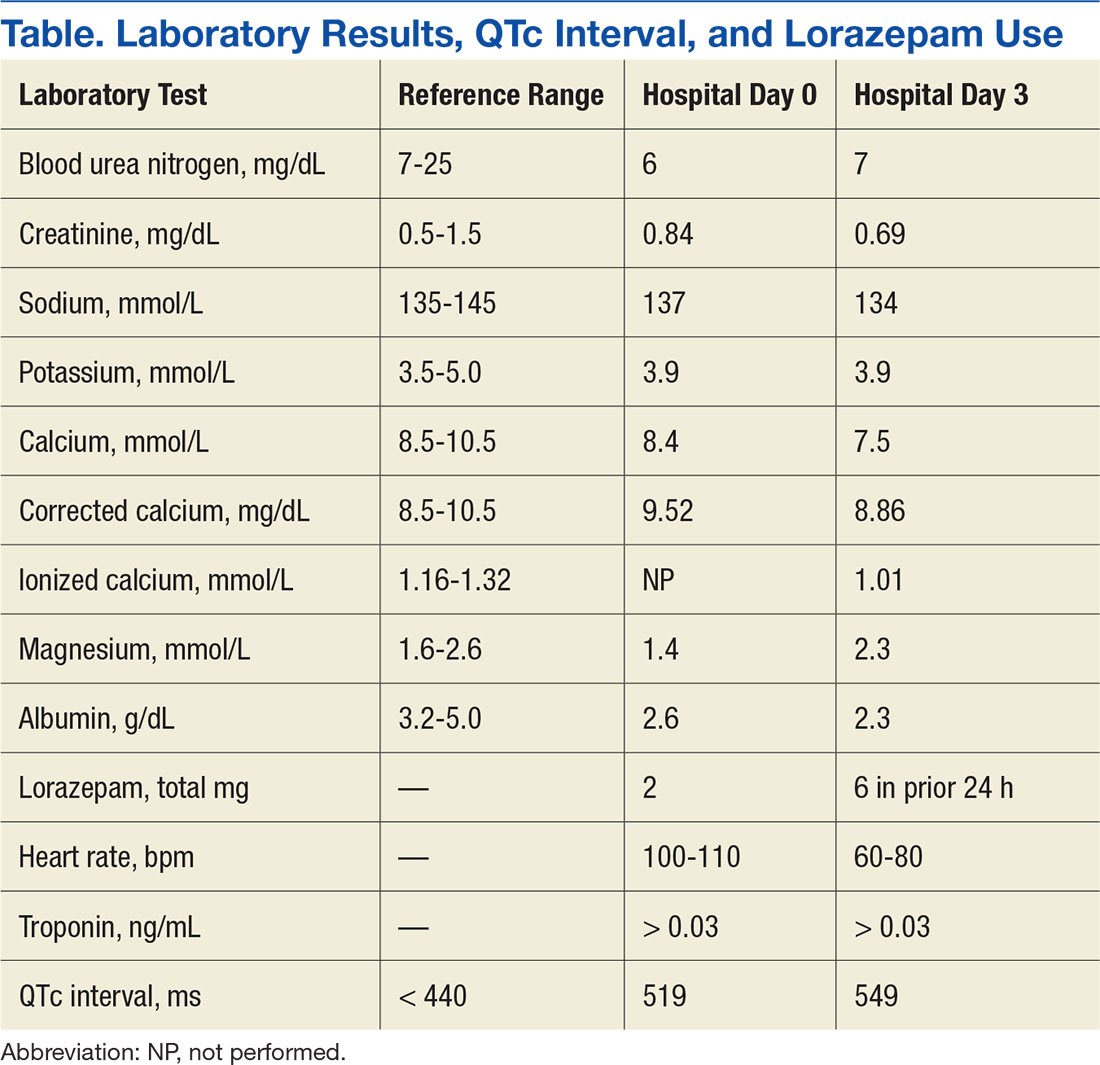
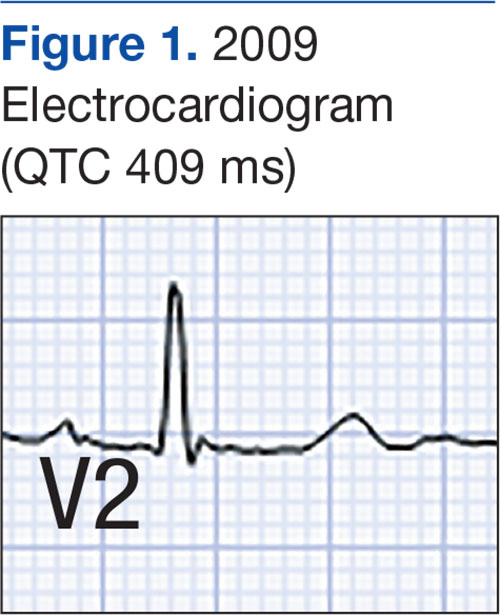
Symptom-triggered therapy for alcohol withdrawal was instituted, and the patient’s electrolyte abnormalities were corrected. Telemetry monitoring demonstrated polymorphic ventricular ectopy, including a 6.8-s run of polymorphic ventricular tachycardia and several shorter runs (4-10 beats) of nonsustained ventricular tachycardia, prompting initiation of a low-dose beta blocker. Based on elevated scores on the symptom-triggered scale for alcohol withdrawal, the Clinical Institute Withdrawal Assessement for Alcohol Withdrawal (CIWA), several doses of oral lorazepam were given for withdrawal symptoms.
The patient became increasingly confused, and new-onset nystagmus was noted. These findings raised concern for Wernicke encephalopathy, so the patient was empirically started on IV high-dose thiamine supplementation. The CIWA scores remained high, and there were frequent episodes of ventricular ectopy during the first 2 hospital days. Interval EKG revealed further prolongation of the QTc interval (549 ms) without evidence of cardiac ischemia (Figure 3). Cardiac enzymes were negative, and electrolyte levels were within normal limits.
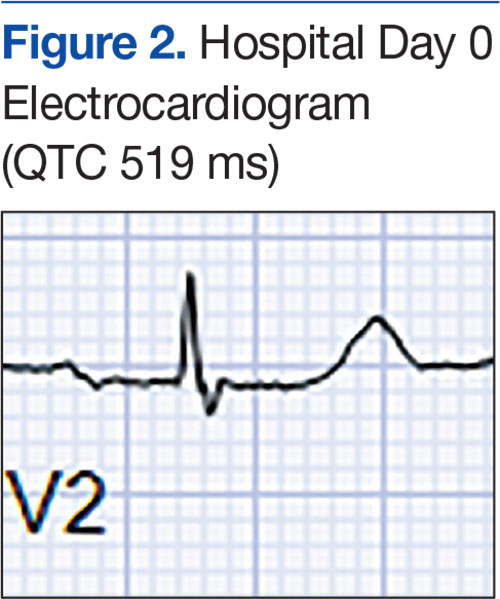
The month-long hospitalization was notable for development of significant ascites, continual electrolyte repletion in the setting of diuresis, formal diagnosis of alcoholic cirrhoisis, cognitive and physical rehabilitation. During the hospitalization, QTc interval remained prolonged (range, 460-500 ms), despite electrolyte repletion, and he was discharged with a wearable cardioverter defibrillator. A month
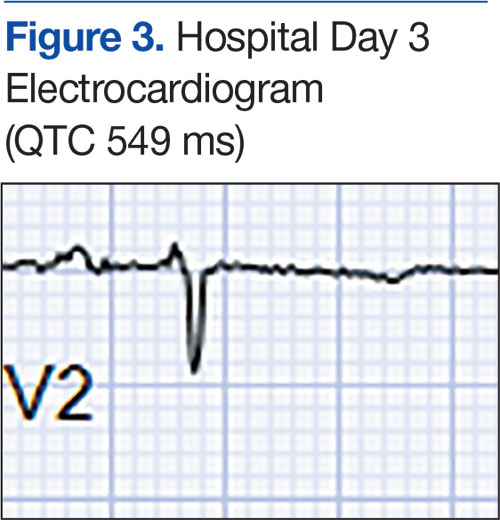
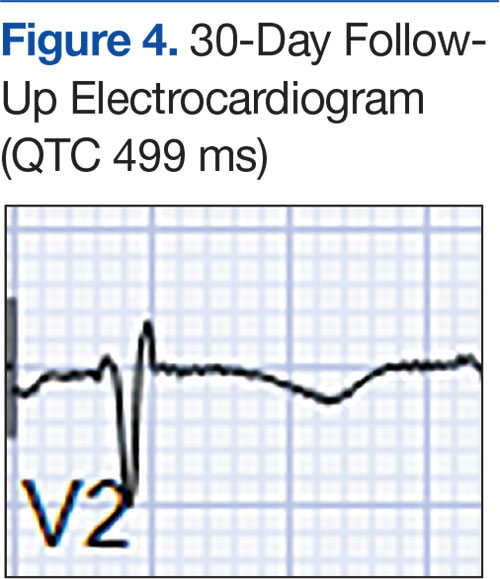
Discussion
Alcohol dependence is a common chronic and relapsing disease that often requires controlled detoxification. Investigators have found a high incidence of QT interval prolongation in alcohol withdrawal and hepatic disease.3,6 Common causes of QT interval prolongation in this setting are poor nutrition, electrolyte abnormalities (particularly hypocalcemia and hypomagnesemia), and use of certain medications.1-3,7,8 In addition, alcohol is directly toxic to the renal tubules, resulting in renal wasting of divalent cations, which may persist up to 30 days after the most recent alcohol exposure.9,10
The patient in this case report was initially thought to have hypomagnesemia-induced long QT syndrome (leading to TdP cardiac arrest), but the authors’ review of laboratory test results revealed the QT interval remained markedly prolonged, despite adequate correction of hypomagnesemia implicating the hyperadrenergic state of acute alcohol withdrawal in QT interval prolongation and TdP cardiac arrest. Interestingly, the QT interval remained prolonged 2 months after TdP arrest, despite sustained normalization of electrolyte levels and the absence of active ischemia or use of QT-prolonging medications.
Given the exclusion of other causes of QT interval prolongation, the authors hypothesized that autonomic hyperactivity of acute alcohol withdrawal and resultant QT interval prolongation were potentiated by underlying cirrhotic cardiomyopathy, a well described cause of QT interval prolongation. Cirrhotic cardiomyopathy is thought to cause QT interval prolongation by delayed repolarization of cardiomyocytes and promotion of sympatho-adrenergic hyperactivity.6 In other case series, TdP development has been associated with severe withdrawal symptoms, particularly delirium tremens.2 In cirrhosis, QT interval prolongation often is described as an early manifestation of cirrhotic cardiomyopathy, irrespective of the underlying etiology, and precedes systolic and diastolic dysfunction.6
The magnitude of QT prolongation has been associated with severity of liver disease as expressed by Child-Pugh score, with reports of QT normalization after liver transplantation.4,5 Patients with higher Child-Pugh scores should be considered to be at elevated risk for malignant ventricular arrhythmias. The authors recommend checking an EKG on admission of any patient who has liver disease or has presented for alcohol withdrawal. Patients with a prolonged QTc interval should be monitored on telemetry. The authors also recommend aggressive repletion of electrolytes, particularly potassium and magnesium, in patients who present with cirrhosis and alcohol withdrawal.
Avoidance of QT-prolonging medications is advisable for all patients with a long QT interval. Beta blockers shorten the QT interval in cirrhotic patients, but the role of beta blockers in preventing malignant arrhythmias in this group of patients is not yet clear.11 The present patient’s QT interval had been normal before he developed cirrhotic liver disease. His presentation was suggestive of acquired long QT syndrome, likely caused by cirrhotic cardiomyopathy given the exhaustive exclusion of other causes of QT interval prolongation.
Conclusion
This case highlights the importance of close monitoring of the QT interval in patients being treated for acute alcohol withdrawal, particularly those with cirrhosis, and suggests that timely and aggressive management of withdrawal, repletion of electrolytes, and telemetry monitoring may prevent life-threatening arrhythmia.
1. Otero-Antón E, González-Quintela A, Saborido J, Torre JA, Virgós A, Barrio E. Prolongation of the QTc interval during alcohol withdrawal syndrome. Acta Cardiol. 1997;52(3):285-294.
2. Cuculi F, Kobza R, Ehmann T, Erne P. ECG changes amongst patients with alcohol withdrawal seizures and delirium tremens. Swiss Med Wkly. 2006;136(13-14):223-227.
3. Mimidis K, Thomopoulos K, Tziakas D, et al. Prolongation of the QTc interval in patients with cirrhosis. Ann Gastroenterol. 2003;16(2):155-158.
4. Bernardi M, Calandra S, Colantoni A, et al. Q-T interval prolongation in cirrhosis: prevalence, relationship with severity, and etiology of the disease and possible pathogenetic factors. Hepatology. 1998;27(1):28-34.
5. Bal JS, Thuluvath PJ. Prolongation of QTc interval: relationship with etiology and severity of liver disease, mortality and liver transplantation. Liver Int. 2003;23(4):243-248.
6. Zardi EM, Abbate A, Zardi DM, et al. Cirrhotic cardiomyopathy. J Am Coll Cardiol. 2010;56(7):539-549.
7. Faigel DO, Metz DC, Kochman ML. Torsade de pointes complicating the treatment of bleeding esophageal varices: association with neuroleptics, vasopressin, and electrolyte imbalance. Am J Gastroenterol. 1995;90(5):822-824.
8. Kotsia AP, Dimitriadis G, Baltogiannis GG, Kolettis TM. Torsade de pointes and persistent QTc prolongation after intravenous amiodarone. Case Rep Med. 2012;2012:673019.
9. Denison H, Jern S, Jagenburg R, Wendestam C, Wallerstedt S. Influence of increased adrenergic activity and magnesium depletion on cardiac rhythm in alcohol withdrawal. Br Heart J. 1994;72(6):554-560.
10. Plaza de los Reyes M, Orozco R, Rosemblitt M, Rendic Y, Espinace M. Renal secretion of magnesium and other electrolytes under the influence of acute ingestion of alcohol, in normal subjects [in Spanish]. Rev Med Chil. 1968;96(3):138-141.
11. Bernardi M, Maggioli C, Dibra V, Zaccherini G. QT interval prolongation in liver cirrhosis: innocent bystander or serious threat? Expert Rev Gastroenterol Hepatol. 2012;6(1):57-66.
Torsades de pointes (TdP) is a life-threatening ventricular arrhythmia that is associated with both congenital and acquired QT interval prolongation. QT interval prolongation is commonly observed in acute alcohol withdrawal and cirrhotic cardiomyopathy.1-3 In both conditions, there is a positive correlation between the degree of QT interval prolongation and disease severity.4,5 The precise mechanisms of QT interval prolongation in these conditions are not well understood. One hypothesis is that autonomic hyperexcitability results in altered ventricular repolarization and QT interval prolongation. This mechanism of QT prolongation has been found in acute alcohol withdrawal independent of electrolyte abnormalities, use of QT-prolonging medications, and cirrhosis.1,2,6
The authors report the case of a veteran who was hospitalized for acute alcohol withdrawal and decompensated cirrhosis and was found to have a newly prolonged QT interval. On hospital day 3, the patient developed TdP, which required external defibrillation. Despite correction of electrolyte abnormalities, abstinence from alcohol, avoidance of QT-prolonging medications, and exclusion of cardiac ischemia, there was significant and persistent prolongation of the QT interval—ultimately attributed to cirrhotic cardiomyopathy. Acquired QT interval prolongation is common in both acute alcohol withdrawal and cirrhosis.This case highlights the importance of close monitoring of the QT interval and TdP susceptibility in patients being treated for acute alcohol withdrawal, particularly those with cirrhosis.
Case Report
A 66-year-old male veteran with a 35-year history of alcohol dependence presented for alcohol detoxification. He reported having drunk at least 32 ounces of vodka every day of the preceding 5 years and reported having unsuccessfully attempted self-detoxification several times. Prior detoxification efforts were unsuccessful because of intractable nausea and tremulousness. Additional presenting symptoms included lethargy, anorexia, and a fall with transient right-side hemiparesis (findings on magnetic resonance imaging of the head had been normal).
His medical history included type 2 diabetes, tobacco dependence, and macular degeneration. The only medication being taken was glargine 25 units daily. On admission, the patient was afebrile (98.1°F), normotensive (103/77 mm Hg), and oriented to person, place, and time. Examination also revealed a protuberant abdomen with caput medusae, and no shifting dullness or lower extremity edema. The neurologic examination was nonfocal.
Laboratory test results on admission were significant for elevated serum alcohol level (243.8 mg/dL); elevated levels of aspartate aminotransferase (144 units/L) alanine aminotransferase (25 units/L), and total bilirubin (4.2 mg/L); hypoalbuminemia; normocalemia; hypomagnesemia; normal corrected calcium level; and normal renal function (0.84 mg/dL)(Table). The patient’s admission Child-Pugh score of 10 indicated class C liver disease. Admission electrocardiogram (EKG) revealed normal sinus rhythm, first-degree atrioventricular block, and prolongation of the QTc interval (519 ms). Six years earlier, the patient’s QTc interval had been 409 ms (Figures 1 and 2). As QT interval depends on heart rate, it is most commonly expressed as corrected QT, or QTc, where QTc = QT/(√RR).
Symptom-triggered therapy for alcohol withdrawal was instituted, and the patient’s electrolyte abnormalities were corrected. Telemetry monitoring demonstrated polymorphic ventricular ectopy, including a 6.8-s run of polymorphic ventricular tachycardia and several shorter runs (4-10 beats) of nonsustained ventricular tachycardia, prompting initiation of a low-dose beta blocker. Based on elevated scores on the symptom-triggered scale for alcohol withdrawal, the Clinical Institute Withdrawal Assessement for Alcohol Withdrawal (CIWA), several doses of oral lorazepam were given for withdrawal symptoms.
The patient became increasingly confused, and new-onset nystagmus was noted. These findings raised concern for Wernicke encephalopathy, so the patient was empirically started on IV high-dose thiamine supplementation. The CIWA scores remained high, and there were frequent episodes of ventricular ectopy during the first 2 hospital days. Interval EKG revealed further prolongation of the QTc interval (549 ms) without evidence of cardiac ischemia (Figure 3). Cardiac enzymes were negative, and electrolyte levels were within normal limits.
The month-long hospitalization was notable for development of significant ascites, continual electrolyte repletion in the setting of diuresis, formal diagnosis of alcoholic cirrhoisis, cognitive and physical rehabilitation. During the hospitalization, QTc interval remained prolonged (range, 460-500 ms), despite electrolyte repletion, and he was discharged with a wearable cardioverter defibrillator. A month
Discussion
Alcohol dependence is a common chronic and relapsing disease that often requires controlled detoxification. Investigators have found a high incidence of QT interval prolongation in alcohol withdrawal and hepatic disease.3,6 Common causes of QT interval prolongation in this setting are poor nutrition, electrolyte abnormalities (particularly hypocalcemia and hypomagnesemia), and use of certain medications.1-3,7,8 In addition, alcohol is directly toxic to the renal tubules, resulting in renal wasting of divalent cations, which may persist up to 30 days after the most recent alcohol exposure.9,10
The patient in this case report was initially thought to have hypomagnesemia-induced long QT syndrome (leading to TdP cardiac arrest), but the authors’ review of laboratory test results revealed the QT interval remained markedly prolonged, despite adequate correction of hypomagnesemia implicating the hyperadrenergic state of acute alcohol withdrawal in QT interval prolongation and TdP cardiac arrest. Interestingly, the QT interval remained prolonged 2 months after TdP arrest, despite sustained normalization of electrolyte levels and the absence of active ischemia or use of QT-prolonging medications.
Given the exclusion of other causes of QT interval prolongation, the authors hypothesized that autonomic hyperactivity of acute alcohol withdrawal and resultant QT interval prolongation were potentiated by underlying cirrhotic cardiomyopathy, a well described cause of QT interval prolongation. Cirrhotic cardiomyopathy is thought to cause QT interval prolongation by delayed repolarization of cardiomyocytes and promotion of sympatho-adrenergic hyperactivity.6 In other case series, TdP development has been associated with severe withdrawal symptoms, particularly delirium tremens.2 In cirrhosis, QT interval prolongation often is described as an early manifestation of cirrhotic cardiomyopathy, irrespective of the underlying etiology, and precedes systolic and diastolic dysfunction.6
The magnitude of QT prolongation has been associated with severity of liver disease as expressed by Child-Pugh score, with reports of QT normalization after liver transplantation.4,5 Patients with higher Child-Pugh scores should be considered to be at elevated risk for malignant ventricular arrhythmias. The authors recommend checking an EKG on admission of any patient who has liver disease or has presented for alcohol withdrawal. Patients with a prolonged QTc interval should be monitored on telemetry. The authors also recommend aggressive repletion of electrolytes, particularly potassium and magnesium, in patients who present with cirrhosis and alcohol withdrawal.
Avoidance of QT-prolonging medications is advisable for all patients with a long QT interval. Beta blockers shorten the QT interval in cirrhotic patients, but the role of beta blockers in preventing malignant arrhythmias in this group of patients is not yet clear.11 The present patient’s QT interval had been normal before he developed cirrhotic liver disease. His presentation was suggestive of acquired long QT syndrome, likely caused by cirrhotic cardiomyopathy given the exhaustive exclusion of other causes of QT interval prolongation.
Conclusion
This case highlights the importance of close monitoring of the QT interval in patients being treated for acute alcohol withdrawal, particularly those with cirrhosis, and suggests that timely and aggressive management of withdrawal, repletion of electrolytes, and telemetry monitoring may prevent life-threatening arrhythmia.
Torsades de pointes (TdP) is a life-threatening ventricular arrhythmia that is associated with both congenital and acquired QT interval prolongation. QT interval prolongation is commonly observed in acute alcohol withdrawal and cirrhotic cardiomyopathy.1-3 In both conditions, there is a positive correlation between the degree of QT interval prolongation and disease severity.4,5 The precise mechanisms of QT interval prolongation in these conditions are not well understood. One hypothesis is that autonomic hyperexcitability results in altered ventricular repolarization and QT interval prolongation. This mechanism of QT prolongation has been found in acute alcohol withdrawal independent of electrolyte abnormalities, use of QT-prolonging medications, and cirrhosis.1,2,6
The authors report the case of a veteran who was hospitalized for acute alcohol withdrawal and decompensated cirrhosis and was found to have a newly prolonged QT interval. On hospital day 3, the patient developed TdP, which required external defibrillation. Despite correction of electrolyte abnormalities, abstinence from alcohol, avoidance of QT-prolonging medications, and exclusion of cardiac ischemia, there was significant and persistent prolongation of the QT interval—ultimately attributed to cirrhotic cardiomyopathy. Acquired QT interval prolongation is common in both acute alcohol withdrawal and cirrhosis.This case highlights the importance of close monitoring of the QT interval and TdP susceptibility in patients being treated for acute alcohol withdrawal, particularly those with cirrhosis.
Case Report
A 66-year-old male veteran with a 35-year history of alcohol dependence presented for alcohol detoxification. He reported having drunk at least 32 ounces of vodka every day of the preceding 5 years and reported having unsuccessfully attempted self-detoxification several times. Prior detoxification efforts were unsuccessful because of intractable nausea and tremulousness. Additional presenting symptoms included lethargy, anorexia, and a fall with transient right-side hemiparesis (findings on magnetic resonance imaging of the head had been normal).
His medical history included type 2 diabetes, tobacco dependence, and macular degeneration. The only medication being taken was glargine 25 units daily. On admission, the patient was afebrile (98.1°F), normotensive (103/77 mm Hg), and oriented to person, place, and time. Examination also revealed a protuberant abdomen with caput medusae, and no shifting dullness or lower extremity edema. The neurologic examination was nonfocal.
Laboratory test results on admission were significant for elevated serum alcohol level (243.8 mg/dL); elevated levels of aspartate aminotransferase (144 units/L) alanine aminotransferase (25 units/L), and total bilirubin (4.2 mg/L); hypoalbuminemia; normocalemia; hypomagnesemia; normal corrected calcium level; and normal renal function (0.84 mg/dL)(Table). The patient’s admission Child-Pugh score of 10 indicated class C liver disease. Admission electrocardiogram (EKG) revealed normal sinus rhythm, first-degree atrioventricular block, and prolongation of the QTc interval (519 ms). Six years earlier, the patient’s QTc interval had been 409 ms (Figures 1 and 2). As QT interval depends on heart rate, it is most commonly expressed as corrected QT, or QTc, where QTc = QT/(√RR).
Symptom-triggered therapy for alcohol withdrawal was instituted, and the patient’s electrolyte abnormalities were corrected. Telemetry monitoring demonstrated polymorphic ventricular ectopy, including a 6.8-s run of polymorphic ventricular tachycardia and several shorter runs (4-10 beats) of nonsustained ventricular tachycardia, prompting initiation of a low-dose beta blocker. Based on elevated scores on the symptom-triggered scale for alcohol withdrawal, the Clinical Institute Withdrawal Assessement for Alcohol Withdrawal (CIWA), several doses of oral lorazepam were given for withdrawal symptoms.
The patient became increasingly confused, and new-onset nystagmus was noted. These findings raised concern for Wernicke encephalopathy, so the patient was empirically started on IV high-dose thiamine supplementation. The CIWA scores remained high, and there were frequent episodes of ventricular ectopy during the first 2 hospital days. Interval EKG revealed further prolongation of the QTc interval (549 ms) without evidence of cardiac ischemia (Figure 3). Cardiac enzymes were negative, and electrolyte levels were within normal limits.
The month-long hospitalization was notable for development of significant ascites, continual electrolyte repletion in the setting of diuresis, formal diagnosis of alcoholic cirrhoisis, cognitive and physical rehabilitation. During the hospitalization, QTc interval remained prolonged (range, 460-500 ms), despite electrolyte repletion, and he was discharged with a wearable cardioverter defibrillator. A month
Discussion
Alcohol dependence is a common chronic and relapsing disease that often requires controlled detoxification. Investigators have found a high incidence of QT interval prolongation in alcohol withdrawal and hepatic disease.3,6 Common causes of QT interval prolongation in this setting are poor nutrition, electrolyte abnormalities (particularly hypocalcemia and hypomagnesemia), and use of certain medications.1-3,7,8 In addition, alcohol is directly toxic to the renal tubules, resulting in renal wasting of divalent cations, which may persist up to 30 days after the most recent alcohol exposure.9,10
The patient in this case report was initially thought to have hypomagnesemia-induced long QT syndrome (leading to TdP cardiac arrest), but the authors’ review of laboratory test results revealed the QT interval remained markedly prolonged, despite adequate correction of hypomagnesemia implicating the hyperadrenergic state of acute alcohol withdrawal in QT interval prolongation and TdP cardiac arrest. Interestingly, the QT interval remained prolonged 2 months after TdP arrest, despite sustained normalization of electrolyte levels and the absence of active ischemia or use of QT-prolonging medications.
Given the exclusion of other causes of QT interval prolongation, the authors hypothesized that autonomic hyperactivity of acute alcohol withdrawal and resultant QT interval prolongation were potentiated by underlying cirrhotic cardiomyopathy, a well described cause of QT interval prolongation. Cirrhotic cardiomyopathy is thought to cause QT interval prolongation by delayed repolarization of cardiomyocytes and promotion of sympatho-adrenergic hyperactivity.6 In other case series, TdP development has been associated with severe withdrawal symptoms, particularly delirium tremens.2 In cirrhosis, QT interval prolongation often is described as an early manifestation of cirrhotic cardiomyopathy, irrespective of the underlying etiology, and precedes systolic and diastolic dysfunction.6
The magnitude of QT prolongation has been associated with severity of liver disease as expressed by Child-Pugh score, with reports of QT normalization after liver transplantation.4,5 Patients with higher Child-Pugh scores should be considered to be at elevated risk for malignant ventricular arrhythmias. The authors recommend checking an EKG on admission of any patient who has liver disease or has presented for alcohol withdrawal. Patients with a prolonged QTc interval should be monitored on telemetry. The authors also recommend aggressive repletion of electrolytes, particularly potassium and magnesium, in patients who present with cirrhosis and alcohol withdrawal.
Avoidance of QT-prolonging medications is advisable for all patients with a long QT interval. Beta blockers shorten the QT interval in cirrhotic patients, but the role of beta blockers in preventing malignant arrhythmias in this group of patients is not yet clear.11 The present patient’s QT interval had been normal before he developed cirrhotic liver disease. His presentation was suggestive of acquired long QT syndrome, likely caused by cirrhotic cardiomyopathy given the exhaustive exclusion of other causes of QT interval prolongation.
Conclusion
This case highlights the importance of close monitoring of the QT interval in patients being treated for acute alcohol withdrawal, particularly those with cirrhosis, and suggests that timely and aggressive management of withdrawal, repletion of electrolytes, and telemetry monitoring may prevent life-threatening arrhythmia.
1. Otero-Antón E, González-Quintela A, Saborido J, Torre JA, Virgós A, Barrio E. Prolongation of the QTc interval during alcohol withdrawal syndrome. Acta Cardiol. 1997;52(3):285-294.
2. Cuculi F, Kobza R, Ehmann T, Erne P. ECG changes amongst patients with alcohol withdrawal seizures and delirium tremens. Swiss Med Wkly. 2006;136(13-14):223-227.
3. Mimidis K, Thomopoulos K, Tziakas D, et al. Prolongation of the QTc interval in patients with cirrhosis. Ann Gastroenterol. 2003;16(2):155-158.
4. Bernardi M, Calandra S, Colantoni A, et al. Q-T interval prolongation in cirrhosis: prevalence, relationship with severity, and etiology of the disease and possible pathogenetic factors. Hepatology. 1998;27(1):28-34.
5. Bal JS, Thuluvath PJ. Prolongation of QTc interval: relationship with etiology and severity of liver disease, mortality and liver transplantation. Liver Int. 2003;23(4):243-248.
6. Zardi EM, Abbate A, Zardi DM, et al. Cirrhotic cardiomyopathy. J Am Coll Cardiol. 2010;56(7):539-549.
7. Faigel DO, Metz DC, Kochman ML. Torsade de pointes complicating the treatment of bleeding esophageal varices: association with neuroleptics, vasopressin, and electrolyte imbalance. Am J Gastroenterol. 1995;90(5):822-824.
8. Kotsia AP, Dimitriadis G, Baltogiannis GG, Kolettis TM. Torsade de pointes and persistent QTc prolongation after intravenous amiodarone. Case Rep Med. 2012;2012:673019.
9. Denison H, Jern S, Jagenburg R, Wendestam C, Wallerstedt S. Influence of increased adrenergic activity and magnesium depletion on cardiac rhythm in alcohol withdrawal. Br Heart J. 1994;72(6):554-560.
10. Plaza de los Reyes M, Orozco R, Rosemblitt M, Rendic Y, Espinace M. Renal secretion of magnesium and other electrolytes under the influence of acute ingestion of alcohol, in normal subjects [in Spanish]. Rev Med Chil. 1968;96(3):138-141.
11. Bernardi M, Maggioli C, Dibra V, Zaccherini G. QT interval prolongation in liver cirrhosis: innocent bystander or serious threat? Expert Rev Gastroenterol Hepatol. 2012;6(1):57-66.
1. Otero-Antón E, González-Quintela A, Saborido J, Torre JA, Virgós A, Barrio E. Prolongation of the QTc interval during alcohol withdrawal syndrome. Acta Cardiol. 1997;52(3):285-294.
2. Cuculi F, Kobza R, Ehmann T, Erne P. ECG changes amongst patients with alcohol withdrawal seizures and delirium tremens. Swiss Med Wkly. 2006;136(13-14):223-227.
3. Mimidis K, Thomopoulos K, Tziakas D, et al. Prolongation of the QTc interval in patients with cirrhosis. Ann Gastroenterol. 2003;16(2):155-158.
4. Bernardi M, Calandra S, Colantoni A, et al. Q-T interval prolongation in cirrhosis: prevalence, relationship with severity, and etiology of the disease and possible pathogenetic factors. Hepatology. 1998;27(1):28-34.
5. Bal JS, Thuluvath PJ. Prolongation of QTc interval: relationship with etiology and severity of liver disease, mortality and liver transplantation. Liver Int. 2003;23(4):243-248.
6. Zardi EM, Abbate A, Zardi DM, et al. Cirrhotic cardiomyopathy. J Am Coll Cardiol. 2010;56(7):539-549.
7. Faigel DO, Metz DC, Kochman ML. Torsade de pointes complicating the treatment of bleeding esophageal varices: association with neuroleptics, vasopressin, and electrolyte imbalance. Am J Gastroenterol. 1995;90(5):822-824.
8. Kotsia AP, Dimitriadis G, Baltogiannis GG, Kolettis TM. Torsade de pointes and persistent QTc prolongation after intravenous amiodarone. Case Rep Med. 2012;2012:673019.
9. Denison H, Jern S, Jagenburg R, Wendestam C, Wallerstedt S. Influence of increased adrenergic activity and magnesium depletion on cardiac rhythm in alcohol withdrawal. Br Heart J. 1994;72(6):554-560.
10. Plaza de los Reyes M, Orozco R, Rosemblitt M, Rendic Y, Espinace M. Renal secretion of magnesium and other electrolytes under the influence of acute ingestion of alcohol, in normal subjects [in Spanish]. Rev Med Chil. 1968;96(3):138-141.
11. Bernardi M, Maggioli C, Dibra V, Zaccherini G. QT interval prolongation in liver cirrhosis: innocent bystander or serious threat? Expert Rev Gastroenterol Hepatol. 2012;6(1):57-66.
Questioning the Specificity and Sensitivity of ELISA for Bullous Pemphigoid Diagnosis
Bullous pemphigoid (BP) is the most common autoimmune blistering disease. The classic presentation of BP is a generalized, pruritic, bullous eruption in elderly patients, which is occasionally preceded by an urticarial prodrome. Immunopathologically, BP is characterized by IgG and sometimes IgE autoantibodies that target basement membrane zone proteins BP180 and BP230 of the epidermis.1
The diagnosis of BP should be suspected when an elderly patient presents with tense blisters and can be confirmed via diagnostic testing, including tissue histology and direct immunofluorescence (DIF) as the gold standard, as well as indirect immunofluorescence (IIF), enzyme-linked immunosorbent assay (ELISA), and most recently biochip technology as supportive tests.2 Since its advent, ELISA has gained popularity as a trustworthy diagnostic test for BP. The specificity of ELISA for BP diagnosis is reported to be 98% to 100%, which leads clinicians to believe that a positive ELISA equals certain diagnosis of BP; however, misdiagnosis of BP based on a positive ELISA result can occur.3-13 The treatment of BP often involves lifelong immunosuppressive therapy. Complications of immunosuppressive therapy contribute to morbidity and mortality in these patients, thus an accurate diagnosis is paramount before introducing therapy.14
We present the case of a 74-year-old man with a history of a pruritic nonbullous eruption who was diagnosed with BP and treated for 3 years based on positive ELISA results in the absence of confirmatory histology or DIF.
Case Report
A 74-year-old man with diabetes mellitus, hypertension, hyperlipidemia, benign prostatic hypertrophy, and obstructive sleep apnea presented for further evaluation and confirmation of a prior diagnosis of BP by an outside dermatologist. He reported a pruritic rash on the trunk, back, and extremities of 3 years’ duration. He denied occurrence of blisters at any time.
On presentation to an outside dermatologist 3 years ago, a biopsy was performed along with serologic studies due to the patient’s age and the possibility of an urticarial prodrome in BP. The biopsy revealed epidermal acanthosis, subepidermal separation, and a perivascular and interstitial infiltrate of lymphocytes and eosinophils in the papillary dermis. Direct immunofluorescence was nondiagnostic with a weak discontinuous pattern of IgG and IgA linearly along the basement membrane zone as well as few scattered and clumped cytoid bodies of IgM and IgA. Indirect immunofluoresence revealed a positive IgG titer of 1:40 on monkey esophagus substrate and a positive epidermal pattern on human split-skin substrate with a titer of 1:80. An ELISA for IgG autoantibodies against BP180 and BP230 yielded 15 U and 6 U, respectively (cut off value, 9 U). Based on the positive ELISA for IgG against BP180, a diagnosis of BP was made.
Over the following 3 years, the treatment included prednisone, tetracycline, nicotinamide, doxycycline, and dapsone. Therapy was suboptimal due to the patient’s comorbidities and socioeconomic status. Poorly controlled diabetes mellitus precluded consistent use of prednisone as recommended for BP. Tetracycline and nicotinamide were transiently effective in controlling the patient’s symptoms but were discontinued due to changes in his health insurance. Doxycycline and dapsone were ineffective. Throughout this 3-year period, the patient remained blister free, but the pruritic eruption was persistent.
The patient presented to our clinic due to his frustration with the lack of improvement and doubts about the BP diagnosis given the persistent absence of bullous lesions. Physical examination revealed numerous eroded, scaly, crusted papules on erythematous edematous plaques on all extremities, trunk, and back (Figure 1). The head, neck, face, and oral mucosa were spared. His history and clinical findings were atypical for BP and skin biopsies were performed. Histology revealed epidermal erosion with parakeratosis, spongiosis, and superficial perivascular lymphocytic inflammation with rare eosinophils without subepidermal split (Figure 2). Direct immunofluorescence was negative for IgG, IgA, IgM, C3, and C1q. Additionally, further review of the initial histology by another dermatopathologist revealed that the subepidermal separation reported was more likely artifactual clefts. These findings were not consistent with BP.
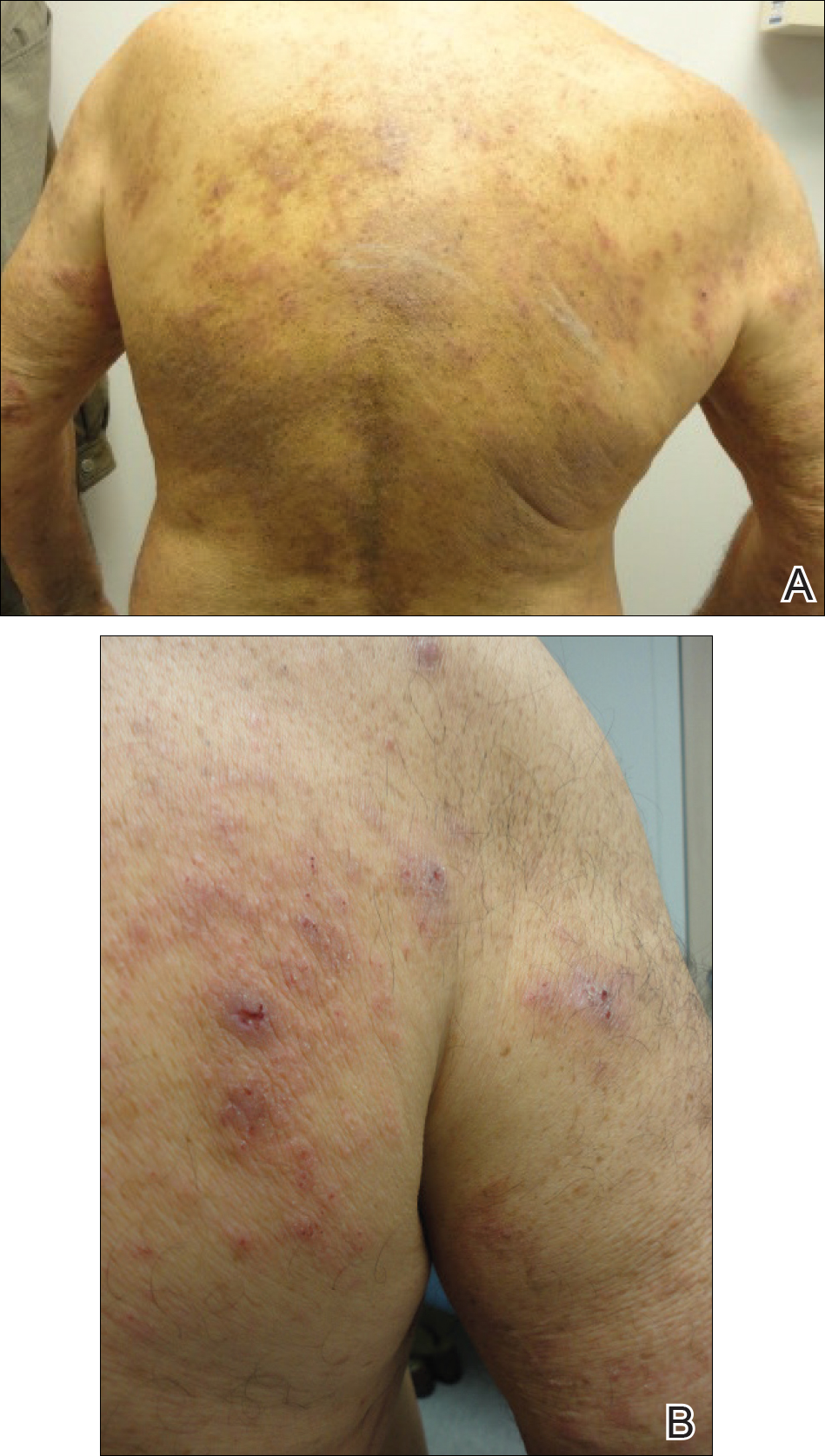

Given the patient’s clinical history, lack of bullae, and twice-negative DIF, the diagnosis was determined to be more consistent with eczematous spongiotic dermatitis. He refused a referral for phototherapy due to scheduling inconvenience. The patient was started on cyclosporine 0.5 mg/kg twice daily. After 10 days of treatment, he returned for follow-up and reported notable improvement in the pruritus. On physical examination, his dermatitis was improved with decreased erythema and inflammation.
The patient is being continued on extensive dry skin care with thick moisturizers and additional topical corticosteroid application on an as-needed basis.
Comment
Chronic immunosuppression contributes to morbidity and mortality in patients with BP; therefore, accurate diagnosis of BP is of utmost importance.14 A meta-analysis described ELISA as a test with high sensitivity and specificity (87% and 98%–100%, respectively) for diagnosis of BP.3 Nevertheless, there are opportunities for misdiagnosis using ELISA, as demonstrated in our case. To determine if the reported sensitivity and specificity of ELISA is accurate and reliable for clinical use, individual studies from the meta-analysis were reviewed.4,5,7-10,13,15 Issues identified in our review included dissimilar diagnostic procedures and patient populations among individual studies, several reports of positive ELISA in patients without BP, and a lack of explanation for these false-positive results.
There are notable differences in diagnostic procedures and patient populations among reports that establish the sensitivity and specificity of ELISA for BP diagnosis.3-13 Studies have detected IgG that targets the NC16A domain of the BP180 kD antigen, the C-terminal of the BP180 kD antigen, or the entire ectodomain of the BP180 kD antigen. Study patient populations varied in disease activity, stage, and treatment. Control patients included healthy patients as well as those with many dermatoses, including pemphigus vulgaris, systemic scleroderma, systemic lupus erythematosus, rheumatoid arthritis, lichen planus, and discoid lupus erythematosus.3-13 Due to these differences between individual studies, we believe the results that determine the overall sensitivity and specificity of ELISA for BP diagnosis must be interpreted with caution. For ELISA statistics to be clinically applicable to a specific patient, he/she should be similar to the patients studied. Therefore, we believe each study must be evaluated individually for applicability, given the differences that exist between them.
Furthermore, there have been several reports of false-positive ELISA results in patients with other dermatologic disorders, specifically in elderly patients with pruritus who do not fulfill clinical criteria for diagnosis with BP.16-18 In a population of elderly patients with pruritus for which no specific dermatological or systemic cause was identified, Hofmann et al18 found that 12% (3/25) of patients showed IgG reactivity to BP180 despite having negative DIF results. In another study of elderly patients with pruritic dermatoses, Feliciani et al17 found that 33% (5/15) of patients had IgG reactivity against BP230 or BP180, though they did not fulfill BP criteria based on clinical presentation and showed negative DIF and IIF results. These findings suggest that IgG reactivity against BP autoantibodies as determined by ELISA is not uncommon in pruritic diseases of the elderly.
Explanations for false-positive ELISA results were rare. A few authors suggested that false-positives could be attributed to an excessively low cutoff value,7-9 which was consistent with reports that the titer of autoantibodies to BP180 correlates with disease severity, suggesting that the higher titer of antibodies correlates with more severe disease and likely more accurate diagnosis.10,19,20 It is important to consider that patients who have low titers of BP180 autoantibodies with inconsistent clinical characteristics and DIF results may not truly have BP. Furthermore, to determine the clinical value of ELISA in identifying patients in the initial phase of BP, sera of BP patients should be compared with sera of elderly patients with pruritic skin disorders because they comprise the patient population that often requires diagnosis.18
Given the issues identified in our review of the literature, the published sensitivity and specificity of ELISA for BP diagnosis are likely overstated. In conclusion, ELISA should not be relied on as a single criterion adequate for diagnosis of BP.12,21 Rather, the diagnosis of BP can be obtained with a positive predictive value of 95% when a patient meets 3 of 4 clinical criteria (ie, absence of atrophic scars, absence of head and neck involvement, absence of mucosal involvement, and older than 70 years) and demonstrates linear deposits of predominantly IgG and/or C3 along the basement membrane zone of a perilesional biopsy on DIF.15 The gold standard for diagnosis of BP remains clinical presentation along with DIF, which can be supported by histology, IIF, and ELISA.22
- Delaporte E, Dubost-Brama A, Ghohestani R, et al. IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol. 1996;157:3642-3647.
- Schmidt E, Zillikens D. Diagnosis and clinical severity markers of bullous pemphigoid. F1000 Med Rep. 2009;1:15.
- Tampoia M, Giavarina D, Di Giorgio C, et al. Diagnostic accuracy of enzyme-linked immunosorbent assays (ELISA) to detect anti-skin autoantibodies in autoimmune blistering diseases: a systematic review and meta-analysis. Autoimmun Rev. 2012;12:121-126.
- Zillikens D, Mascaro JM, Rose PA, et al. A highly sensitive enzyme-linked immunosorbent assay for the detection of circulating anti-BP180 autoantibodies in patients with bullous pemphigoid. J Invest Dermatol. 1997;109:679-683.
- Sitaru C, Dahnrich C, Probst C, et al. Enzyme-linked immunosorbent assay using multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and specific detection of pemphigoid autoantibodies. Exp Dermatol. 2007;16:770-777.
- Yang B, Wang C, Chen S, et al. Evaluation of the combination of BP180-NC16a enzyme-linked immunosorbent assay and BP230 enzyme-linked immunosorbent assay in the diagnosis of bullous pemphigoid. Indian J Dermatol Venereol Leprol. 2012;78:722-727.
- Sakuma-Oyama Y, Powell AM, Oyama N, et al. Evaluation of a BP180-NC16a enzyme-linked immunosorbent assay in the initial diagnosis of bullous pemphigoid. Br J Dermatol. 2004;151:126-131.
- Tampoia M, Lattanzi V, Zucano A, et al. Evaluation of a new ELISA assay for detection of BP230 autoantibodies in bullous pemphigoid. Ann N Y Acad Sci. 2009;1173:15-20.
- Feng S, Lin L, Jin P, et al. Role of BP180NC16a-enzyme-linked immunosorbent assay (ELISA) in the diagnosis of bullous pemphigoid in China. Int J Dermatol. 2008;47:24-28.
- Kobayashi M, Amagai M, Kuroda-Kinoshita K, et al. BP180 ELISA using bacterial recombinant NC16a protein as a diagnostic and monitoring tool for bullous pemphigoid. J Dermatol Sci. 2002;30:224-232.
- Roussel A, Benichou J, Arivelo Randriamanantany Z, et al. Enzyme-linked immunosorbent assay for the combination of bullous pemphigoid antigens 1 and 2 in the diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:293-298.
- Chan, Lawrence S. ELISA instead of indirect IF in patients with BP. Arch Dermatol. 2011;147:291-292.
- Barnadas MA, Rubiales V, González J, et al. Enzyme-linked immunosorbent assay (ELISA) and indirect immunofluorescence testing in a bullous pemphigoid and pemphigoid gestationis. Int J Dermatol. 2008;47:1245-1249.
- Borradori L, Bernard P. Pemphigoid group. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. New York, NY: Mosby; 2003:469.
- Vaillant L, Bernard P, Joly P, et al. Evaluation of clinical criteria for diagnosis of bullous pemphigoid. Arch Dermatol. 1998;134:1075-1080.
- Fania L, Caldarola G, Muller R, et al. IgE recognition of bullous pemphigoid (BP)180 and BP230 in BP patients and elderly individuals with pruritic dermatoses. Clin Immunol. 2012;143:236-245.
- Feliciani C, Caldarola G, Kneisel A, et al. IgG autoantibody reactivity against bullous pemphigoid (BP) 180 and BP230 in elderly patients with pruritic dermatoses. Br J Dermatol. 2009;61:306-312.
- Hofmann SC, Tamm K, Hertl M, et al. Diagnostic value of an enzyme-linked immunosorbent assay using BP180 recombinant proteins in elderly patients with pruritic skin disorders. Br J Dermatol. 2003;149:910-911.
- Schmidt E, Obe K, Brocker EB, et al. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Arch Dermatol. 2000;136:174-178.
- Feng S, Wu Q, Jin P, et al. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Int J Dermatol. 2008;47:225-228.
- Di Zenzo G, Joly P, Zambruno G, et al. Sensitivity of immunofluorescence studies vs enzyme-linked immunosorbent assay for diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:1454-1456.
- Schmidt E, Zillikens D. Modern diagnosis of autoimmune blistering skin diseases. Autoimmun Rev. 2010;10:84-89.
Bullous pemphigoid (BP) is the most common autoimmune blistering disease. The classic presentation of BP is a generalized, pruritic, bullous eruption in elderly patients, which is occasionally preceded by an urticarial prodrome. Immunopathologically, BP is characterized by IgG and sometimes IgE autoantibodies that target basement membrane zone proteins BP180 and BP230 of the epidermis.1
The diagnosis of BP should be suspected when an elderly patient presents with tense blisters and can be confirmed via diagnostic testing, including tissue histology and direct immunofluorescence (DIF) as the gold standard, as well as indirect immunofluorescence (IIF), enzyme-linked immunosorbent assay (ELISA), and most recently biochip technology as supportive tests.2 Since its advent, ELISA has gained popularity as a trustworthy diagnostic test for BP. The specificity of ELISA for BP diagnosis is reported to be 98% to 100%, which leads clinicians to believe that a positive ELISA equals certain diagnosis of BP; however, misdiagnosis of BP based on a positive ELISA result can occur.3-13 The treatment of BP often involves lifelong immunosuppressive therapy. Complications of immunosuppressive therapy contribute to morbidity and mortality in these patients, thus an accurate diagnosis is paramount before introducing therapy.14
We present the case of a 74-year-old man with a history of a pruritic nonbullous eruption who was diagnosed with BP and treated for 3 years based on positive ELISA results in the absence of confirmatory histology or DIF.
Case Report
A 74-year-old man with diabetes mellitus, hypertension, hyperlipidemia, benign prostatic hypertrophy, and obstructive sleep apnea presented for further evaluation and confirmation of a prior diagnosis of BP by an outside dermatologist. He reported a pruritic rash on the trunk, back, and extremities of 3 years’ duration. He denied occurrence of blisters at any time.
On presentation to an outside dermatologist 3 years ago, a biopsy was performed along with serologic studies due to the patient’s age and the possibility of an urticarial prodrome in BP. The biopsy revealed epidermal acanthosis, subepidermal separation, and a perivascular and interstitial infiltrate of lymphocytes and eosinophils in the papillary dermis. Direct immunofluorescence was nondiagnostic with a weak discontinuous pattern of IgG and IgA linearly along the basement membrane zone as well as few scattered and clumped cytoid bodies of IgM and IgA. Indirect immunofluoresence revealed a positive IgG titer of 1:40 on monkey esophagus substrate and a positive epidermal pattern on human split-skin substrate with a titer of 1:80. An ELISA for IgG autoantibodies against BP180 and BP230 yielded 15 U and 6 U, respectively (cut off value, 9 U). Based on the positive ELISA for IgG against BP180, a diagnosis of BP was made.
Over the following 3 years, the treatment included prednisone, tetracycline, nicotinamide, doxycycline, and dapsone. Therapy was suboptimal due to the patient’s comorbidities and socioeconomic status. Poorly controlled diabetes mellitus precluded consistent use of prednisone as recommended for BP. Tetracycline and nicotinamide were transiently effective in controlling the patient’s symptoms but were discontinued due to changes in his health insurance. Doxycycline and dapsone were ineffective. Throughout this 3-year period, the patient remained blister free, but the pruritic eruption was persistent.
The patient presented to our clinic due to his frustration with the lack of improvement and doubts about the BP diagnosis given the persistent absence of bullous lesions. Physical examination revealed numerous eroded, scaly, crusted papules on erythematous edematous plaques on all extremities, trunk, and back (Figure 1). The head, neck, face, and oral mucosa were spared. His history and clinical findings were atypical for BP and skin biopsies were performed. Histology revealed epidermal erosion with parakeratosis, spongiosis, and superficial perivascular lymphocytic inflammation with rare eosinophils without subepidermal split (Figure 2). Direct immunofluorescence was negative for IgG, IgA, IgM, C3, and C1q. Additionally, further review of the initial histology by another dermatopathologist revealed that the subepidermal separation reported was more likely artifactual clefts. These findings were not consistent with BP.
Given the patient’s clinical history, lack of bullae, and twice-negative DIF, the diagnosis was determined to be more consistent with eczematous spongiotic dermatitis. He refused a referral for phototherapy due to scheduling inconvenience. The patient was started on cyclosporine 0.5 mg/kg twice daily. After 10 days of treatment, he returned for follow-up and reported notable improvement in the pruritus. On physical examination, his dermatitis was improved with decreased erythema and inflammation.
The patient is being continued on extensive dry skin care with thick moisturizers and additional topical corticosteroid application on an as-needed basis.
Comment
Chronic immunosuppression contributes to morbidity and mortality in patients with BP; therefore, accurate diagnosis of BP is of utmost importance.14 A meta-analysis described ELISA as a test with high sensitivity and specificity (87% and 98%–100%, respectively) for diagnosis of BP.3 Nevertheless, there are opportunities for misdiagnosis using ELISA, as demonstrated in our case. To determine if the reported sensitivity and specificity of ELISA is accurate and reliable for clinical use, individual studies from the meta-analysis were reviewed.4,5,7-10,13,15 Issues identified in our review included dissimilar diagnostic procedures and patient populations among individual studies, several reports of positive ELISA in patients without BP, and a lack of explanation for these false-positive results.
There are notable differences in diagnostic procedures and patient populations among reports that establish the sensitivity and specificity of ELISA for BP diagnosis.3-13 Studies have detected IgG that targets the NC16A domain of the BP180 kD antigen, the C-terminal of the BP180 kD antigen, or the entire ectodomain of the BP180 kD antigen. Study patient populations varied in disease activity, stage, and treatment. Control patients included healthy patients as well as those with many dermatoses, including pemphigus vulgaris, systemic scleroderma, systemic lupus erythematosus, rheumatoid arthritis, lichen planus, and discoid lupus erythematosus.3-13 Due to these differences between individual studies, we believe the results that determine the overall sensitivity and specificity of ELISA for BP diagnosis must be interpreted with caution. For ELISA statistics to be clinically applicable to a specific patient, he/she should be similar to the patients studied. Therefore, we believe each study must be evaluated individually for applicability, given the differences that exist between them.
Furthermore, there have been several reports of false-positive ELISA results in patients with other dermatologic disorders, specifically in elderly patients with pruritus who do not fulfill clinical criteria for diagnosis with BP.16-18 In a population of elderly patients with pruritus for which no specific dermatological or systemic cause was identified, Hofmann et al18 found that 12% (3/25) of patients showed IgG reactivity to BP180 despite having negative DIF results. In another study of elderly patients with pruritic dermatoses, Feliciani et al17 found that 33% (5/15) of patients had IgG reactivity against BP230 or BP180, though they did not fulfill BP criteria based on clinical presentation and showed negative DIF and IIF results. These findings suggest that IgG reactivity against BP autoantibodies as determined by ELISA is not uncommon in pruritic diseases of the elderly.
Explanations for false-positive ELISA results were rare. A few authors suggested that false-positives could be attributed to an excessively low cutoff value,7-9 which was consistent with reports that the titer of autoantibodies to BP180 correlates with disease severity, suggesting that the higher titer of antibodies correlates with more severe disease and likely more accurate diagnosis.10,19,20 It is important to consider that patients who have low titers of BP180 autoantibodies with inconsistent clinical characteristics and DIF results may not truly have BP. Furthermore, to determine the clinical value of ELISA in identifying patients in the initial phase of BP, sera of BP patients should be compared with sera of elderly patients with pruritic skin disorders because they comprise the patient population that often requires diagnosis.18
Given the issues identified in our review of the literature, the published sensitivity and specificity of ELISA for BP diagnosis are likely overstated. In conclusion, ELISA should not be relied on as a single criterion adequate for diagnosis of BP.12,21 Rather, the diagnosis of BP can be obtained with a positive predictive value of 95% when a patient meets 3 of 4 clinical criteria (ie, absence of atrophic scars, absence of head and neck involvement, absence of mucosal involvement, and older than 70 years) and demonstrates linear deposits of predominantly IgG and/or C3 along the basement membrane zone of a perilesional biopsy on DIF.15 The gold standard for diagnosis of BP remains clinical presentation along with DIF, which can be supported by histology, IIF, and ELISA.22
Bullous pemphigoid (BP) is the most common autoimmune blistering disease. The classic presentation of BP is a generalized, pruritic, bullous eruption in elderly patients, which is occasionally preceded by an urticarial prodrome. Immunopathologically, BP is characterized by IgG and sometimes IgE autoantibodies that target basement membrane zone proteins BP180 and BP230 of the epidermis.1
The diagnosis of BP should be suspected when an elderly patient presents with tense blisters and can be confirmed via diagnostic testing, including tissue histology and direct immunofluorescence (DIF) as the gold standard, as well as indirect immunofluorescence (IIF), enzyme-linked immunosorbent assay (ELISA), and most recently biochip technology as supportive tests.2 Since its advent, ELISA has gained popularity as a trustworthy diagnostic test for BP. The specificity of ELISA for BP diagnosis is reported to be 98% to 100%, which leads clinicians to believe that a positive ELISA equals certain diagnosis of BP; however, misdiagnosis of BP based on a positive ELISA result can occur.3-13 The treatment of BP often involves lifelong immunosuppressive therapy. Complications of immunosuppressive therapy contribute to morbidity and mortality in these patients, thus an accurate diagnosis is paramount before introducing therapy.14
We present the case of a 74-year-old man with a history of a pruritic nonbullous eruption who was diagnosed with BP and treated for 3 years based on positive ELISA results in the absence of confirmatory histology or DIF.
Case Report
A 74-year-old man with diabetes mellitus, hypertension, hyperlipidemia, benign prostatic hypertrophy, and obstructive sleep apnea presented for further evaluation and confirmation of a prior diagnosis of BP by an outside dermatologist. He reported a pruritic rash on the trunk, back, and extremities of 3 years’ duration. He denied occurrence of blisters at any time.
On presentation to an outside dermatologist 3 years ago, a biopsy was performed along with serologic studies due to the patient’s age and the possibility of an urticarial prodrome in BP. The biopsy revealed epidermal acanthosis, subepidermal separation, and a perivascular and interstitial infiltrate of lymphocytes and eosinophils in the papillary dermis. Direct immunofluorescence was nondiagnostic with a weak discontinuous pattern of IgG and IgA linearly along the basement membrane zone as well as few scattered and clumped cytoid bodies of IgM and IgA. Indirect immunofluoresence revealed a positive IgG titer of 1:40 on monkey esophagus substrate and a positive epidermal pattern on human split-skin substrate with a titer of 1:80. An ELISA for IgG autoantibodies against BP180 and BP230 yielded 15 U and 6 U, respectively (cut off value, 9 U). Based on the positive ELISA for IgG against BP180, a diagnosis of BP was made.
Over the following 3 years, the treatment included prednisone, tetracycline, nicotinamide, doxycycline, and dapsone. Therapy was suboptimal due to the patient’s comorbidities and socioeconomic status. Poorly controlled diabetes mellitus precluded consistent use of prednisone as recommended for BP. Tetracycline and nicotinamide were transiently effective in controlling the patient’s symptoms but were discontinued due to changes in his health insurance. Doxycycline and dapsone were ineffective. Throughout this 3-year period, the patient remained blister free, but the pruritic eruption was persistent.
The patient presented to our clinic due to his frustration with the lack of improvement and doubts about the BP diagnosis given the persistent absence of bullous lesions. Physical examination revealed numerous eroded, scaly, crusted papules on erythematous edematous plaques on all extremities, trunk, and back (Figure 1). The head, neck, face, and oral mucosa were spared. His history and clinical findings were atypical for BP and skin biopsies were performed. Histology revealed epidermal erosion with parakeratosis, spongiosis, and superficial perivascular lymphocytic inflammation with rare eosinophils without subepidermal split (Figure 2). Direct immunofluorescence was negative for IgG, IgA, IgM, C3, and C1q. Additionally, further review of the initial histology by another dermatopathologist revealed that the subepidermal separation reported was more likely artifactual clefts. These findings were not consistent with BP.
Given the patient’s clinical history, lack of bullae, and twice-negative DIF, the diagnosis was determined to be more consistent with eczematous spongiotic dermatitis. He refused a referral for phototherapy due to scheduling inconvenience. The patient was started on cyclosporine 0.5 mg/kg twice daily. After 10 days of treatment, he returned for follow-up and reported notable improvement in the pruritus. On physical examination, his dermatitis was improved with decreased erythema and inflammation.
The patient is being continued on extensive dry skin care with thick moisturizers and additional topical corticosteroid application on an as-needed basis.
Comment
Chronic immunosuppression contributes to morbidity and mortality in patients with BP; therefore, accurate diagnosis of BP is of utmost importance.14 A meta-analysis described ELISA as a test with high sensitivity and specificity (87% and 98%–100%, respectively) for diagnosis of BP.3 Nevertheless, there are opportunities for misdiagnosis using ELISA, as demonstrated in our case. To determine if the reported sensitivity and specificity of ELISA is accurate and reliable for clinical use, individual studies from the meta-analysis were reviewed.4,5,7-10,13,15 Issues identified in our review included dissimilar diagnostic procedures and patient populations among individual studies, several reports of positive ELISA in patients without BP, and a lack of explanation for these false-positive results.
There are notable differences in diagnostic procedures and patient populations among reports that establish the sensitivity and specificity of ELISA for BP diagnosis.3-13 Studies have detected IgG that targets the NC16A domain of the BP180 kD antigen, the C-terminal of the BP180 kD antigen, or the entire ectodomain of the BP180 kD antigen. Study patient populations varied in disease activity, stage, and treatment. Control patients included healthy patients as well as those with many dermatoses, including pemphigus vulgaris, systemic scleroderma, systemic lupus erythematosus, rheumatoid arthritis, lichen planus, and discoid lupus erythematosus.3-13 Due to these differences between individual studies, we believe the results that determine the overall sensitivity and specificity of ELISA for BP diagnosis must be interpreted with caution. For ELISA statistics to be clinically applicable to a specific patient, he/she should be similar to the patients studied. Therefore, we believe each study must be evaluated individually for applicability, given the differences that exist between them.
Furthermore, there have been several reports of false-positive ELISA results in patients with other dermatologic disorders, specifically in elderly patients with pruritus who do not fulfill clinical criteria for diagnosis with BP.16-18 In a population of elderly patients with pruritus for which no specific dermatological or systemic cause was identified, Hofmann et al18 found that 12% (3/25) of patients showed IgG reactivity to BP180 despite having negative DIF results. In another study of elderly patients with pruritic dermatoses, Feliciani et al17 found that 33% (5/15) of patients had IgG reactivity against BP230 or BP180, though they did not fulfill BP criteria based on clinical presentation and showed negative DIF and IIF results. These findings suggest that IgG reactivity against BP autoantibodies as determined by ELISA is not uncommon in pruritic diseases of the elderly.
Explanations for false-positive ELISA results were rare. A few authors suggested that false-positives could be attributed to an excessively low cutoff value,7-9 which was consistent with reports that the titer of autoantibodies to BP180 correlates with disease severity, suggesting that the higher titer of antibodies correlates with more severe disease and likely more accurate diagnosis.10,19,20 It is important to consider that patients who have low titers of BP180 autoantibodies with inconsistent clinical characteristics and DIF results may not truly have BP. Furthermore, to determine the clinical value of ELISA in identifying patients in the initial phase of BP, sera of BP patients should be compared with sera of elderly patients with pruritic skin disorders because they comprise the patient population that often requires diagnosis.18
Given the issues identified in our review of the literature, the published sensitivity and specificity of ELISA for BP diagnosis are likely overstated. In conclusion, ELISA should not be relied on as a single criterion adequate for diagnosis of BP.12,21 Rather, the diagnosis of BP can be obtained with a positive predictive value of 95% when a patient meets 3 of 4 clinical criteria (ie, absence of atrophic scars, absence of head and neck involvement, absence of mucosal involvement, and older than 70 years) and demonstrates linear deposits of predominantly IgG and/or C3 along the basement membrane zone of a perilesional biopsy on DIF.15 The gold standard for diagnosis of BP remains clinical presentation along with DIF, which can be supported by histology, IIF, and ELISA.22
- Delaporte E, Dubost-Brama A, Ghohestani R, et al. IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol. 1996;157:3642-3647.
- Schmidt E, Zillikens D. Diagnosis and clinical severity markers of bullous pemphigoid. F1000 Med Rep. 2009;1:15.
- Tampoia M, Giavarina D, Di Giorgio C, et al. Diagnostic accuracy of enzyme-linked immunosorbent assays (ELISA) to detect anti-skin autoantibodies in autoimmune blistering diseases: a systematic review and meta-analysis. Autoimmun Rev. 2012;12:121-126.
- Zillikens D, Mascaro JM, Rose PA, et al. A highly sensitive enzyme-linked immunosorbent assay for the detection of circulating anti-BP180 autoantibodies in patients with bullous pemphigoid. J Invest Dermatol. 1997;109:679-683.
- Sitaru C, Dahnrich C, Probst C, et al. Enzyme-linked immunosorbent assay using multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and specific detection of pemphigoid autoantibodies. Exp Dermatol. 2007;16:770-777.
- Yang B, Wang C, Chen S, et al. Evaluation of the combination of BP180-NC16a enzyme-linked immunosorbent assay and BP230 enzyme-linked immunosorbent assay in the diagnosis of bullous pemphigoid. Indian J Dermatol Venereol Leprol. 2012;78:722-727.
- Sakuma-Oyama Y, Powell AM, Oyama N, et al. Evaluation of a BP180-NC16a enzyme-linked immunosorbent assay in the initial diagnosis of bullous pemphigoid. Br J Dermatol. 2004;151:126-131.
- Tampoia M, Lattanzi V, Zucano A, et al. Evaluation of a new ELISA assay for detection of BP230 autoantibodies in bullous pemphigoid. Ann N Y Acad Sci. 2009;1173:15-20.
- Feng S, Lin L, Jin P, et al. Role of BP180NC16a-enzyme-linked immunosorbent assay (ELISA) in the diagnosis of bullous pemphigoid in China. Int J Dermatol. 2008;47:24-28.
- Kobayashi M, Amagai M, Kuroda-Kinoshita K, et al. BP180 ELISA using bacterial recombinant NC16a protein as a diagnostic and monitoring tool for bullous pemphigoid. J Dermatol Sci. 2002;30:224-232.
- Roussel A, Benichou J, Arivelo Randriamanantany Z, et al. Enzyme-linked immunosorbent assay for the combination of bullous pemphigoid antigens 1 and 2 in the diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:293-298.
- Chan, Lawrence S. ELISA instead of indirect IF in patients with BP. Arch Dermatol. 2011;147:291-292.
- Barnadas MA, Rubiales V, González J, et al. Enzyme-linked immunosorbent assay (ELISA) and indirect immunofluorescence testing in a bullous pemphigoid and pemphigoid gestationis. Int J Dermatol. 2008;47:1245-1249.
- Borradori L, Bernard P. Pemphigoid group. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. New York, NY: Mosby; 2003:469.
- Vaillant L, Bernard P, Joly P, et al. Evaluation of clinical criteria for diagnosis of bullous pemphigoid. Arch Dermatol. 1998;134:1075-1080.
- Fania L, Caldarola G, Muller R, et al. IgE recognition of bullous pemphigoid (BP)180 and BP230 in BP patients and elderly individuals with pruritic dermatoses. Clin Immunol. 2012;143:236-245.
- Feliciani C, Caldarola G, Kneisel A, et al. IgG autoantibody reactivity against bullous pemphigoid (BP) 180 and BP230 in elderly patients with pruritic dermatoses. Br J Dermatol. 2009;61:306-312.
- Hofmann SC, Tamm K, Hertl M, et al. Diagnostic value of an enzyme-linked immunosorbent assay using BP180 recombinant proteins in elderly patients with pruritic skin disorders. Br J Dermatol. 2003;149:910-911.
- Schmidt E, Obe K, Brocker EB, et al. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Arch Dermatol. 2000;136:174-178.
- Feng S, Wu Q, Jin P, et al. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Int J Dermatol. 2008;47:225-228.
- Di Zenzo G, Joly P, Zambruno G, et al. Sensitivity of immunofluorescence studies vs enzyme-linked immunosorbent assay for diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:1454-1456.
- Schmidt E, Zillikens D. Modern diagnosis of autoimmune blistering skin diseases. Autoimmun Rev. 2010;10:84-89.
- Delaporte E, Dubost-Brama A, Ghohestani R, et al. IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol. 1996;157:3642-3647.
- Schmidt E, Zillikens D. Diagnosis and clinical severity markers of bullous pemphigoid. F1000 Med Rep. 2009;1:15.
- Tampoia M, Giavarina D, Di Giorgio C, et al. Diagnostic accuracy of enzyme-linked immunosorbent assays (ELISA) to detect anti-skin autoantibodies in autoimmune blistering diseases: a systematic review and meta-analysis. Autoimmun Rev. 2012;12:121-126.
- Zillikens D, Mascaro JM, Rose PA, et al. A highly sensitive enzyme-linked immunosorbent assay for the detection of circulating anti-BP180 autoantibodies in patients with bullous pemphigoid. J Invest Dermatol. 1997;109:679-683.
- Sitaru C, Dahnrich C, Probst C, et al. Enzyme-linked immunosorbent assay using multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and specific detection of pemphigoid autoantibodies. Exp Dermatol. 2007;16:770-777.
- Yang B, Wang C, Chen S, et al. Evaluation of the combination of BP180-NC16a enzyme-linked immunosorbent assay and BP230 enzyme-linked immunosorbent assay in the diagnosis of bullous pemphigoid. Indian J Dermatol Venereol Leprol. 2012;78:722-727.
- Sakuma-Oyama Y, Powell AM, Oyama N, et al. Evaluation of a BP180-NC16a enzyme-linked immunosorbent assay in the initial diagnosis of bullous pemphigoid. Br J Dermatol. 2004;151:126-131.
- Tampoia M, Lattanzi V, Zucano A, et al. Evaluation of a new ELISA assay for detection of BP230 autoantibodies in bullous pemphigoid. Ann N Y Acad Sci. 2009;1173:15-20.
- Feng S, Lin L, Jin P, et al. Role of BP180NC16a-enzyme-linked immunosorbent assay (ELISA) in the diagnosis of bullous pemphigoid in China. Int J Dermatol. 2008;47:24-28.
- Kobayashi M, Amagai M, Kuroda-Kinoshita K, et al. BP180 ELISA using bacterial recombinant NC16a protein as a diagnostic and monitoring tool for bullous pemphigoid. J Dermatol Sci. 2002;30:224-232.
- Roussel A, Benichou J, Arivelo Randriamanantany Z, et al. Enzyme-linked immunosorbent assay for the combination of bullous pemphigoid antigens 1 and 2 in the diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:293-298.
- Chan, Lawrence S. ELISA instead of indirect IF in patients with BP. Arch Dermatol. 2011;147:291-292.
- Barnadas MA, Rubiales V, González J, et al. Enzyme-linked immunosorbent assay (ELISA) and indirect immunofluorescence testing in a bullous pemphigoid and pemphigoid gestationis. Int J Dermatol. 2008;47:1245-1249.
- Borradori L, Bernard P. Pemphigoid group. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. New York, NY: Mosby; 2003:469.
- Vaillant L, Bernard P, Joly P, et al. Evaluation of clinical criteria for diagnosis of bullous pemphigoid. Arch Dermatol. 1998;134:1075-1080.
- Fania L, Caldarola G, Muller R, et al. IgE recognition of bullous pemphigoid (BP)180 and BP230 in BP patients and elderly individuals with pruritic dermatoses. Clin Immunol. 2012;143:236-245.
- Feliciani C, Caldarola G, Kneisel A, et al. IgG autoantibody reactivity against bullous pemphigoid (BP) 180 and BP230 in elderly patients with pruritic dermatoses. Br J Dermatol. 2009;61:306-312.
- Hofmann SC, Tamm K, Hertl M, et al. Diagnostic value of an enzyme-linked immunosorbent assay using BP180 recombinant proteins in elderly patients with pruritic skin disorders. Br J Dermatol. 2003;149:910-911.
- Schmidt E, Obe K, Brocker EB, et al. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Arch Dermatol. 2000;136:174-178.
- Feng S, Wu Q, Jin P, et al. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Int J Dermatol. 2008;47:225-228.
- Di Zenzo G, Joly P, Zambruno G, et al. Sensitivity of immunofluorescence studies vs enzyme-linked immunosorbent assay for diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:1454-1456.
- Schmidt E, Zillikens D. Modern diagnosis of autoimmune blistering skin diseases. Autoimmun Rev. 2010;10:84-89.
Practice Points
- A low serum level of autoantibodies to BP180 should be interpreted with caution because it is more likely to represent a false-positive than a high serum level.
- Rely on the gold standard for diagnosis of bullous pemphigoid: clinical presentation along with direct immunofluorescence, which can be supported by histology, indirect immunofluorescence, and enzyme-linked immunosorbent assay (ELISA) rather than ELISA alone.
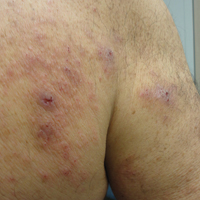
Plesiomonas shigelloides Periprosthetic Knee Infection After Consumption of Raw Oysters
Take-Home Points
- History and physical examination are key in identifying possible etiologies of orthopedic infections.
- If identified in the acute setting, periprosthetic infections can successfully be treated with irrigation, débridement, and polyethylene liner exchange.
- Discussion with an interdisciplinary medical team, including infectious disease specialists, can aide in improved diagnosis and treatment of periprosthetic infections.
Periprosthetic infection is a leading cause of morbidity after total joint arthroplasty.1 Despite advances in modern surgical practices, infection rates continue to range from 1% to 3% among all arthroplasty procedures performed in the United States.2-5 The most common causes of periprosthetic infection include Staphylococcus aureus, streptococcus, enterococcus, Escherichia coli, and Pseudomonas aeruginosa.6 However, many other pathogens that cause periprosthetic infection should be considered in the clinical setting. In this case report, periprosthetic knee infection with P shigelloides occurred after consumption of raw oysters.
P shigelloides is a gram-negative facultative anaerobic organism in the Vibrionaceae family,7 which also includes Vibrio vulnificus and Vibrio parahaemolyticus. P shigelloides is most well-known for causing diarrhea and septicemia in people who have consumed raw oysters or shellfish in the United States.8,9 Although P shigelloides infection is rare, there have been clinically significant outbreaks from contaminated water in Japan,10 consumption of freshwater fish in the Democratic Republic of the Congo,11 and consumption of raw oysters in the United States.8,9 Children and immunosuppressed people are most susceptible to the disease, which most commonly manifests as self-limiting watery diarrhea, with septicemia only in advanced cases.12There are very few reports of P shigelloides in the orthopedic population. In the medical literature, we found only 1 case of septic arthritis in a native knee; disease progression resulted in the patient’s death.13In this article, we report a case of P shigelloides septicemia that caused periprosthetic knee infection in a chemically and biologically immunosuppressed patient. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
Out of concern about a periprosthetic knee infection, a 66-year-old man was transferred from a regional medical center to our tertiary referral center. The patient reported a 3-day history of significant knee pain, swelling, and erythema that started the day after he consumed raw oysters at a seafood bar. He was unable to bear weight on the right knee and remained at home 1 day before presenting to the regional medical center.
The patient had undergone elective right total knee arthroplasty 18 months earlier, without previous issue (Figures A, B), and had a medical history of type 2 diabetes mellitus, psoriatic arthritis, hypertension, hyperlipidemia, hypothyroidism, and benign prostatic hypertrophy.
On presentation to our facility, the patient described pain in the right knee. Physical examination revealed swelling and erythema of the knee. Vital signs were within normal limits, with a temperature of 98.5°F. Laboratory work-up revealed white blood cell count of 17,700 with 79% neutrophils and 9% lymphocytes, serum C-reactive protein level of 270 mg/L, and erythrocyte sedimentation rate of 46 mm/h. Aspiration of the knee yielded about 100 mL of thick, brownish synovial fluid. Gram stain of the knee aspirate revealed gram-negative rods and many white blood cells. Nucleated cell count of the aspirate was 22,400 with 88% neutrophils. Blood cultures were obtained, and broad-spectrum antibiotics (vancomycin and ceftriaxone) were started in preparation for surgery.
Within 24 hours, the patient was taken for irrigation and débridement with polyethylene exchange of the right knee. Surgical exploration revealed brownish purulent fluid in the knee. The polyethylene insert was removed, and a complete synovectomy was performed for knee débridement. Nine liters of triple antibiotic (utilized bacitracin, polymyxin, and gentamicin) saline were used to copiously clean the metal surfaces of the implant, and a new polyethylene liner was inserted. Absorbable calcium sulfate antimicrobial beads, stimulant beads with 1 gram of vancomycin and 1.2 grams of tobramycin, were implanted both inside and over the knee capsule during closure.
Blood cultures, knee aspirate, and surgical cultures were all positive for P shigelloides. Of note, the patient did not describe having diarrhea, a symptom common in P shigelloides infection. After final cultures were received, the patient was placed on intravenous ceftriaxone and oral levofloxacin for 6 weeks. Three months later, he reported full return to activity and clearance of the infection.
Discussion
This case is a reminder that periprosthetic knee infection can occur from a variety of pathologic organisms and that obtaining a complete history is an important part of any diagnostic work-up. Although P shigelloides infection is rare, our patient had important historical findings that led to suspicion of Vibrionaceae infection: recent consumption of raw oysters, immunosuppression with etanercept and prednisone for psoriatic arthritis, and diabetes with hemoglobin A1c of 9.9% and presenting blood sugar of 338 mg/dL. His positive blood cultures represented P shigelloides septicemia, which seeded the knee prosthesis and led to acute periprosthetic infection. To our knowledge, this is the first report of P shigelloides periprosthetic infection in the orthopedic literature. The only other reported case of P shigelloides septicemia leading to septic arthritis in a native knee occurred in a 68-year-old Australian man who had end-stage liver disease and eventually died from complications of the P shigelloides infection.13
Although P shigelloides infection is rare, outbreaks have occurred around the world.7-11,14 Infections are most commonly associated with consumption of raw shellfish or freshwater fish or with water contamination.12 In the United States, the only described vector for disease has been consumption of raw oysters and shellfish—in particular, those harvested from the warm waters of the Gulf Coast.8,9P shigelloides usually causes a self-limiting watery diarrhea. However, in children and immunosuppressed patients, P shigelloides can lead to life-threatening septicemia.12 In the United States, P shigelloides cases often occur in the summer, likely related to the easy growth of the bacteria from shellfish in the Gulf Coast’s warm water and mud.8 This predilection for summer infections has been documented around the world.15Our patient reported eating raw oysters imported to the US Southwest from an unknown location. He likely was susceptible to P shigelloides infection, as he was immunosuppressed with etanercept and prednisone. However, there were no traditional diarrheal symptoms. Case reports have described nondiarrheal symptoms in children and other immunosuppressed people.12There is much to learn from this case report. Most important, it highlights the need to obtain a complete history and perform a thorough physical examination. Our patient’s 2 key historical findings, immunosuppressive medication use and raw oyster consumption, point strongly toward Vibrionaceae infection. Although a majority of periprosthetic infections are caused by common organisms, such as Staphylococcus and Streptococcus species, orthopedic clinicians should continue to expand their knowledge of periprosthetic infections, as many other pathogens can cause disease.
Am J Orthop. 2017;46(1):E32-E34. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Parvizi J, Adeli B, Zmistowski B, Restrepo C, Greenwald AS. Management of periprosthetic joint infection: the current knowledge: AAOS exhibit selection. J Bone Joint Surg Am. 2012;94(14):e104.
2. Fehring TK, Odum S, Griffin WL, Mason JB, Nadaud M. Early failures in total knee arthroplasty. Clin Orthop. 2001;(392):315-318.
3. Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty. 2008;23(7):984-991.
4. Clohisy JC, Calvert G, Tull F, McDonald D, Maloney WJ. Reasons for revision hip surgery: a retrospective review. Clin Orthop. 2004;(429):188-192.
5. Vessely MB, Whaley AL, Harmsen WS, Schleck CD, Berry DJ. The Chitranjan Ranawat Award: long-term survivorship and failure modes of 1000 cemented condylar total knee arthroplasties. Clin Orthop. 2006;(452):28-34.
6. Peel TN, Cheng AC, Buising KL, Choong PF. Microbiological aetiology, epidemiology, and clinical profile of prosthetic joint infections: are current antibiotic prophylaxis guidelines effective? Antimicrob Agents Chemother. 2012;56(5):2386-2391.
7. Wong TY, Tsui HY, So MK, et al. Plesiomonas shigelloides infection in Hong Kong: retrospective study of 167 laboratory-confirmed cases. Hong Kong Med J. 2000;6(4):375-380.
8. Holmberg SD, Wachsmuth IK, Hickman-Brenner FW, Blake PA, Farmer JJ 3rd. Plesiomonas enteric infections in the United States. Ann Intern Med. 1986;105(5):690-694.
9. Rutala WA, Sarubi FA Jr, Finch CS, McCormack JN, Steinkraus GE. Oyster-associated outbreak of diarrhoeal disease possibly caused by Plesiomonas shigelloides. Lancet. 1982;1(8274):739.
10. Tsukamoto T, Kinoshita Y, Shimada T, Sakazaki R. Two epidemics of diarrhoeal disease possibly caused by Plesiomonas shigelloides. J Hyg (Lond). 1978;80(2):275-280.
11. Van Damme LR, Vandepitte J. Frequent isolation of Edwardsiella tarda and Plesiomonas shigelloides from healthy Zairese freshwater fish: a possible source of sporadic diarrhea in the tropics. Appl Environ Microbiol. 1980;39(3):475-479.
12. Brenden RA, Miller MA, Janda JM. Clinical disease spectrum and pathogenic factors associated with Plesiomonas shigelloides infections in humans. Rev Infect Dis. 1988;10(2):303-316.
13. Gordon DL, Philpot CR, McGuire C. Plesiomonas shigelloides septic arthritis complicating rheumatoid arthritis. Aust N Z J Med. 1983;13(3):275-276.
14. Medema G, Schets C. Occurrence of Plesiomonas shigelloides in surface water: relationship with faecal pollution and trophic state. Zentralbl Hyg Umweltmed. 1993;194(4):398-404.
15. Huq MI, Islam MR. Microbiological & clinical studies in diarrhoea due to Plesiomonas shigelloides. Indian J Med Res. 1983;77:793-797.
Take-Home Points
- History and physical examination are key in identifying possible etiologies of orthopedic infections.
- If identified in the acute setting, periprosthetic infections can successfully be treated with irrigation, débridement, and polyethylene liner exchange.
- Discussion with an interdisciplinary medical team, including infectious disease specialists, can aide in improved diagnosis and treatment of periprosthetic infections.
Periprosthetic infection is a leading cause of morbidity after total joint arthroplasty.1 Despite advances in modern surgical practices, infection rates continue to range from 1% to 3% among all arthroplasty procedures performed in the United States.2-5 The most common causes of periprosthetic infection include Staphylococcus aureus, streptococcus, enterococcus, Escherichia coli, and Pseudomonas aeruginosa.6 However, many other pathogens that cause periprosthetic infection should be considered in the clinical setting. In this case report, periprosthetic knee infection with P shigelloides occurred after consumption of raw oysters.
P shigelloides is a gram-negative facultative anaerobic organism in the Vibrionaceae family,7 which also includes Vibrio vulnificus and Vibrio parahaemolyticus. P shigelloides is most well-known for causing diarrhea and septicemia in people who have consumed raw oysters or shellfish in the United States.8,9 Although P shigelloides infection is rare, there have been clinically significant outbreaks from contaminated water in Japan,10 consumption of freshwater fish in the Democratic Republic of the Congo,11 and consumption of raw oysters in the United States.8,9 Children and immunosuppressed people are most susceptible to the disease, which most commonly manifests as self-limiting watery diarrhea, with septicemia only in advanced cases.12There are very few reports of P shigelloides in the orthopedic population. In the medical literature, we found only 1 case of septic arthritis in a native knee; disease progression resulted in the patient’s death.13In this article, we report a case of P shigelloides septicemia that caused periprosthetic knee infection in a chemically and biologically immunosuppressed patient. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
Out of concern about a periprosthetic knee infection, a 66-year-old man was transferred from a regional medical center to our tertiary referral center. The patient reported a 3-day history of significant knee pain, swelling, and erythema that started the day after he consumed raw oysters at a seafood bar. He was unable to bear weight on the right knee and remained at home 1 day before presenting to the regional medical center.
The patient had undergone elective right total knee arthroplasty 18 months earlier, without previous issue (Figures A, B), and had a medical history of type 2 diabetes mellitus, psoriatic arthritis, hypertension, hyperlipidemia, hypothyroidism, and benign prostatic hypertrophy.
On presentation to our facility, the patient described pain in the right knee. Physical examination revealed swelling and erythema of the knee. Vital signs were within normal limits, with a temperature of 98.5°F. Laboratory work-up revealed white blood cell count of 17,700 with 79% neutrophils and 9% lymphocytes, serum C-reactive protein level of 270 mg/L, and erythrocyte sedimentation rate of 46 mm/h. Aspiration of the knee yielded about 100 mL of thick, brownish synovial fluid. Gram stain of the knee aspirate revealed gram-negative rods and many white blood cells. Nucleated cell count of the aspirate was 22,400 with 88% neutrophils. Blood cultures were obtained, and broad-spectrum antibiotics (vancomycin and ceftriaxone) were started in preparation for surgery.
Within 24 hours, the patient was taken for irrigation and débridement with polyethylene exchange of the right knee. Surgical exploration revealed brownish purulent fluid in the knee. The polyethylene insert was removed, and a complete synovectomy was performed for knee débridement. Nine liters of triple antibiotic (utilized bacitracin, polymyxin, and gentamicin) saline were used to copiously clean the metal surfaces of the implant, and a new polyethylene liner was inserted. Absorbable calcium sulfate antimicrobial beads, stimulant beads with 1 gram of vancomycin and 1.2 grams of tobramycin, were implanted both inside and over the knee capsule during closure.
Blood cultures, knee aspirate, and surgical cultures were all positive for P shigelloides. Of note, the patient did not describe having diarrhea, a symptom common in P shigelloides infection. After final cultures were received, the patient was placed on intravenous ceftriaxone and oral levofloxacin for 6 weeks. Three months later, he reported full return to activity and clearance of the infection.
Discussion
This case is a reminder that periprosthetic knee infection can occur from a variety of pathologic organisms and that obtaining a complete history is an important part of any diagnostic work-up. Although P shigelloides infection is rare, our patient had important historical findings that led to suspicion of Vibrionaceae infection: recent consumption of raw oysters, immunosuppression with etanercept and prednisone for psoriatic arthritis, and diabetes with hemoglobin A1c of 9.9% and presenting blood sugar of 338 mg/dL. His positive blood cultures represented P shigelloides septicemia, which seeded the knee prosthesis and led to acute periprosthetic infection. To our knowledge, this is the first report of P shigelloides periprosthetic infection in the orthopedic literature. The only other reported case of P shigelloides septicemia leading to septic arthritis in a native knee occurred in a 68-year-old Australian man who had end-stage liver disease and eventually died from complications of the P shigelloides infection.13
Although P shigelloides infection is rare, outbreaks have occurred around the world.7-11,14 Infections are most commonly associated with consumption of raw shellfish or freshwater fish or with water contamination.12 In the United States, the only described vector for disease has been consumption of raw oysters and shellfish—in particular, those harvested from the warm waters of the Gulf Coast.8,9P shigelloides usually causes a self-limiting watery diarrhea. However, in children and immunosuppressed patients, P shigelloides can lead to life-threatening septicemia.12 In the United States, P shigelloides cases often occur in the summer, likely related to the easy growth of the bacteria from shellfish in the Gulf Coast’s warm water and mud.8 This predilection for summer infections has been documented around the world.15Our patient reported eating raw oysters imported to the US Southwest from an unknown location. He likely was susceptible to P shigelloides infection, as he was immunosuppressed with etanercept and prednisone. However, there were no traditional diarrheal symptoms. Case reports have described nondiarrheal symptoms in children and other immunosuppressed people.12There is much to learn from this case report. Most important, it highlights the need to obtain a complete history and perform a thorough physical examination. Our patient’s 2 key historical findings, immunosuppressive medication use and raw oyster consumption, point strongly toward Vibrionaceae infection. Although a majority of periprosthetic infections are caused by common organisms, such as Staphylococcus and Streptococcus species, orthopedic clinicians should continue to expand their knowledge of periprosthetic infections, as many other pathogens can cause disease.
Am J Orthop. 2017;46(1):E32-E34. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
Take-Home Points
- History and physical examination are key in identifying possible etiologies of orthopedic infections.
- If identified in the acute setting, periprosthetic infections can successfully be treated with irrigation, débridement, and polyethylene liner exchange.
- Discussion with an interdisciplinary medical team, including infectious disease specialists, can aide in improved diagnosis and treatment of periprosthetic infections.
Periprosthetic infection is a leading cause of morbidity after total joint arthroplasty.1 Despite advances in modern surgical practices, infection rates continue to range from 1% to 3% among all arthroplasty procedures performed in the United States.2-5 The most common causes of periprosthetic infection include Staphylococcus aureus, streptococcus, enterococcus, Escherichia coli, and Pseudomonas aeruginosa.6 However, many other pathogens that cause periprosthetic infection should be considered in the clinical setting. In this case report, periprosthetic knee infection with P shigelloides occurred after consumption of raw oysters.
P shigelloides is a gram-negative facultative anaerobic organism in the Vibrionaceae family,7 which also includes Vibrio vulnificus and Vibrio parahaemolyticus. P shigelloides is most well-known for causing diarrhea and septicemia in people who have consumed raw oysters or shellfish in the United States.8,9 Although P shigelloides infection is rare, there have been clinically significant outbreaks from contaminated water in Japan,10 consumption of freshwater fish in the Democratic Republic of the Congo,11 and consumption of raw oysters in the United States.8,9 Children and immunosuppressed people are most susceptible to the disease, which most commonly manifests as self-limiting watery diarrhea, with septicemia only in advanced cases.12There are very few reports of P shigelloides in the orthopedic population. In the medical literature, we found only 1 case of septic arthritis in a native knee; disease progression resulted in the patient’s death.13In this article, we report a case of P shigelloides septicemia that caused periprosthetic knee infection in a chemically and biologically immunosuppressed patient. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
Out of concern about a periprosthetic knee infection, a 66-year-old man was transferred from a regional medical center to our tertiary referral center. The patient reported a 3-day history of significant knee pain, swelling, and erythema that started the day after he consumed raw oysters at a seafood bar. He was unable to bear weight on the right knee and remained at home 1 day before presenting to the regional medical center.
The patient had undergone elective right total knee arthroplasty 18 months earlier, without previous issue (Figures A, B), and had a medical history of type 2 diabetes mellitus, psoriatic arthritis, hypertension, hyperlipidemia, hypothyroidism, and benign prostatic hypertrophy.
On presentation to our facility, the patient described pain in the right knee. Physical examination revealed swelling and erythema of the knee. Vital signs were within normal limits, with a temperature of 98.5°F. Laboratory work-up revealed white blood cell count of 17,700 with 79% neutrophils and 9% lymphocytes, serum C-reactive protein level of 270 mg/L, and erythrocyte sedimentation rate of 46 mm/h. Aspiration of the knee yielded about 100 mL of thick, brownish synovial fluid. Gram stain of the knee aspirate revealed gram-negative rods and many white blood cells. Nucleated cell count of the aspirate was 22,400 with 88% neutrophils. Blood cultures were obtained, and broad-spectrum antibiotics (vancomycin and ceftriaxone) were started in preparation for surgery.
Within 24 hours, the patient was taken for irrigation and débridement with polyethylene exchange of the right knee. Surgical exploration revealed brownish purulent fluid in the knee. The polyethylene insert was removed, and a complete synovectomy was performed for knee débridement. Nine liters of triple antibiotic (utilized bacitracin, polymyxin, and gentamicin) saline were used to copiously clean the metal surfaces of the implant, and a new polyethylene liner was inserted. Absorbable calcium sulfate antimicrobial beads, stimulant beads with 1 gram of vancomycin and 1.2 grams of tobramycin, were implanted both inside and over the knee capsule during closure.
Blood cultures, knee aspirate, and surgical cultures were all positive for P shigelloides. Of note, the patient did not describe having diarrhea, a symptom common in P shigelloides infection. After final cultures were received, the patient was placed on intravenous ceftriaxone and oral levofloxacin for 6 weeks. Three months later, he reported full return to activity and clearance of the infection.
Discussion
This case is a reminder that periprosthetic knee infection can occur from a variety of pathologic organisms and that obtaining a complete history is an important part of any diagnostic work-up. Although P shigelloides infection is rare, our patient had important historical findings that led to suspicion of Vibrionaceae infection: recent consumption of raw oysters, immunosuppression with etanercept and prednisone for psoriatic arthritis, and diabetes with hemoglobin A1c of 9.9% and presenting blood sugar of 338 mg/dL. His positive blood cultures represented P shigelloides septicemia, which seeded the knee prosthesis and led to acute periprosthetic infection. To our knowledge, this is the first report of P shigelloides periprosthetic infection in the orthopedic literature. The only other reported case of P shigelloides septicemia leading to septic arthritis in a native knee occurred in a 68-year-old Australian man who had end-stage liver disease and eventually died from complications of the P shigelloides infection.13
Although P shigelloides infection is rare, outbreaks have occurred around the world.7-11,14 Infections are most commonly associated with consumption of raw shellfish or freshwater fish or with water contamination.12 In the United States, the only described vector for disease has been consumption of raw oysters and shellfish—in particular, those harvested from the warm waters of the Gulf Coast.8,9P shigelloides usually causes a self-limiting watery diarrhea. However, in children and immunosuppressed patients, P shigelloides can lead to life-threatening septicemia.12 In the United States, P shigelloides cases often occur in the summer, likely related to the easy growth of the bacteria from shellfish in the Gulf Coast’s warm water and mud.8 This predilection for summer infections has been documented around the world.15Our patient reported eating raw oysters imported to the US Southwest from an unknown location. He likely was susceptible to P shigelloides infection, as he was immunosuppressed with etanercept and prednisone. However, there were no traditional diarrheal symptoms. Case reports have described nondiarrheal symptoms in children and other immunosuppressed people.12There is much to learn from this case report. Most important, it highlights the need to obtain a complete history and perform a thorough physical examination. Our patient’s 2 key historical findings, immunosuppressive medication use and raw oyster consumption, point strongly toward Vibrionaceae infection. Although a majority of periprosthetic infections are caused by common organisms, such as Staphylococcus and Streptococcus species, orthopedic clinicians should continue to expand their knowledge of periprosthetic infections, as many other pathogens can cause disease.
Am J Orthop. 2017;46(1):E32-E34. Copyright Frontline Medical Communications Inc. 2017. All rights reserved.
1. Parvizi J, Adeli B, Zmistowski B, Restrepo C, Greenwald AS. Management of periprosthetic joint infection: the current knowledge: AAOS exhibit selection. J Bone Joint Surg Am. 2012;94(14):e104.
2. Fehring TK, Odum S, Griffin WL, Mason JB, Nadaud M. Early failures in total knee arthroplasty. Clin Orthop. 2001;(392):315-318.
3. Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty. 2008;23(7):984-991.
4. Clohisy JC, Calvert G, Tull F, McDonald D, Maloney WJ. Reasons for revision hip surgery: a retrospective review. Clin Orthop. 2004;(429):188-192.
5. Vessely MB, Whaley AL, Harmsen WS, Schleck CD, Berry DJ. The Chitranjan Ranawat Award: long-term survivorship and failure modes of 1000 cemented condylar total knee arthroplasties. Clin Orthop. 2006;(452):28-34.
6. Peel TN, Cheng AC, Buising KL, Choong PF. Microbiological aetiology, epidemiology, and clinical profile of prosthetic joint infections: are current antibiotic prophylaxis guidelines effective? Antimicrob Agents Chemother. 2012;56(5):2386-2391.
7. Wong TY, Tsui HY, So MK, et al. Plesiomonas shigelloides infection in Hong Kong: retrospective study of 167 laboratory-confirmed cases. Hong Kong Med J. 2000;6(4):375-380.
8. Holmberg SD, Wachsmuth IK, Hickman-Brenner FW, Blake PA, Farmer JJ 3rd. Plesiomonas enteric infections in the United States. Ann Intern Med. 1986;105(5):690-694.
9. Rutala WA, Sarubi FA Jr, Finch CS, McCormack JN, Steinkraus GE. Oyster-associated outbreak of diarrhoeal disease possibly caused by Plesiomonas shigelloides. Lancet. 1982;1(8274):739.
10. Tsukamoto T, Kinoshita Y, Shimada T, Sakazaki R. Two epidemics of diarrhoeal disease possibly caused by Plesiomonas shigelloides. J Hyg (Lond). 1978;80(2):275-280.
11. Van Damme LR, Vandepitte J. Frequent isolation of Edwardsiella tarda and Plesiomonas shigelloides from healthy Zairese freshwater fish: a possible source of sporadic diarrhea in the tropics. Appl Environ Microbiol. 1980;39(3):475-479.
12. Brenden RA, Miller MA, Janda JM. Clinical disease spectrum and pathogenic factors associated with Plesiomonas shigelloides infections in humans. Rev Infect Dis. 1988;10(2):303-316.
13. Gordon DL, Philpot CR, McGuire C. Plesiomonas shigelloides septic arthritis complicating rheumatoid arthritis. Aust N Z J Med. 1983;13(3):275-276.
14. Medema G, Schets C. Occurrence of Plesiomonas shigelloides in surface water: relationship with faecal pollution and trophic state. Zentralbl Hyg Umweltmed. 1993;194(4):398-404.
15. Huq MI, Islam MR. Microbiological & clinical studies in diarrhoea due to Plesiomonas shigelloides. Indian J Med Res. 1983;77:793-797.
1. Parvizi J, Adeli B, Zmistowski B, Restrepo C, Greenwald AS. Management of periprosthetic joint infection: the current knowledge: AAOS exhibit selection. J Bone Joint Surg Am. 2012;94(14):e104.
2. Fehring TK, Odum S, Griffin WL, Mason JB, Nadaud M. Early failures in total knee arthroplasty. Clin Orthop. 2001;(392):315-318.
3. Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty. 2008;23(7):984-991.
4. Clohisy JC, Calvert G, Tull F, McDonald D, Maloney WJ. Reasons for revision hip surgery: a retrospective review. Clin Orthop. 2004;(429):188-192.
5. Vessely MB, Whaley AL, Harmsen WS, Schleck CD, Berry DJ. The Chitranjan Ranawat Award: long-term survivorship and failure modes of 1000 cemented condylar total knee arthroplasties. Clin Orthop. 2006;(452):28-34.
6. Peel TN, Cheng AC, Buising KL, Choong PF. Microbiological aetiology, epidemiology, and clinical profile of prosthetic joint infections: are current antibiotic prophylaxis guidelines effective? Antimicrob Agents Chemother. 2012;56(5):2386-2391.
7. Wong TY, Tsui HY, So MK, et al. Plesiomonas shigelloides infection in Hong Kong: retrospective study of 167 laboratory-confirmed cases. Hong Kong Med J. 2000;6(4):375-380.
8. Holmberg SD, Wachsmuth IK, Hickman-Brenner FW, Blake PA, Farmer JJ 3rd. Plesiomonas enteric infections in the United States. Ann Intern Med. 1986;105(5):690-694.
9. Rutala WA, Sarubi FA Jr, Finch CS, McCormack JN, Steinkraus GE. Oyster-associated outbreak of diarrhoeal disease possibly caused by Plesiomonas shigelloides. Lancet. 1982;1(8274):739.
10. Tsukamoto T, Kinoshita Y, Shimada T, Sakazaki R. Two epidemics of diarrhoeal disease possibly caused by Plesiomonas shigelloides. J Hyg (Lond). 1978;80(2):275-280.
11. Van Damme LR, Vandepitte J. Frequent isolation of Edwardsiella tarda and Plesiomonas shigelloides from healthy Zairese freshwater fish: a possible source of sporadic diarrhea in the tropics. Appl Environ Microbiol. 1980;39(3):475-479.
12. Brenden RA, Miller MA, Janda JM. Clinical disease spectrum and pathogenic factors associated with Plesiomonas shigelloides infections in humans. Rev Infect Dis. 1988;10(2):303-316.
13. Gordon DL, Philpot CR, McGuire C. Plesiomonas shigelloides septic arthritis complicating rheumatoid arthritis. Aust N Z J Med. 1983;13(3):275-276.
14. Medema G, Schets C. Occurrence of Plesiomonas shigelloides in surface water: relationship with faecal pollution and trophic state. Zentralbl Hyg Umweltmed. 1993;194(4):398-404.
15. Huq MI, Islam MR. Microbiological & clinical studies in diarrhoea due to Plesiomonas shigelloides. Indian J Med Res. 1983;77:793-797.