User login
Bleeding Factors
An 80‐year‐old man with coronary artery disease and chronic obstructive pulmonary disease (COPD) was admitted to an outside hospital after a mechanical fall. On presentation to the emergency room his systolic blood pressure was found to be 86/62 mm Hg. He complained of right flank, groin, and thigh pain. On physical exam, a hematoma extending from his right groin down to his right knee was found, as well as scattered ecchymoses involving his trunk and all 4 extremities. His hemoglobin was low, at 5.6 g/dL (14‐17 g/dL). A computed tomography (CT) scan revealed a right‐sided retroperitoneal bleed extending from the iliopsoas into his right thigh. The patient received 13 transfusions of packed red blood cells over the course of 9 days as he continued to bleed. Transfer to our facility for further workup and management ensued.
On serial testing at our institution his activated partial thromboplastin time (aPTT) was elevated at >160 seconds (normal range, 24‐36 seconds). Further coagulation parameters were found as follows: platelets 182.000/L; prothrombin time 17.6 seconds; international normalized ratio (INR) 1.4; thrombin 18 seconds; fibrinogen 778 mg/dL; and D‐dimer 3866 ng/mL. Of note, the patient had not received any medications known to potentially interfere with the measured aPTT. Because the source of his bleeding was not apparent at this point, disorders of primary hemostasis, including hereditary disease states (eg, von Willebrand disease), iatrogenic disorders (eg, drug‐induced), or acquired disorders, such as immune thrombocytopenia, were considered and ruled out. At this point the differential diagnoses had to be expanded, and secondary disorders of hemostasis were considered. A deficiency or decreased activity of coagulation factors was suspected. Whereas factor IX and XI were found to be normal, the factor VIII level was significantly decreased at 3% (50%‐150% being normal). This prompted an assay to check for the presence of a factor VIII inhibitor. It proved to be significantly elevated at 25.6 Bethesda Units (BU) (normal, 0.00‐0.04 BU). On that basis we arrived at the diagnosis of acquired factor VIII deficiency, but the etiology of such remained unclear to this point. Hence a search for the specific etiology of acquired factor IIII deficiency was launched, and connective tissue disease, as well as malignancy, was ruled out. While inflammatory bowel disease is a known potential cause for this condition, the clinical picture was not consistent with such and this diagnosis was not considered further. The patient received immunosuppressive therapy with prednisone 1 mg/kg orally per day. Rituximab and cyclophosphamide were considered, but due to bacteremia from bilateral parotitis, this was deferred. Of note, his bleeding abnormality was apparent prior to initiation of antibiotic therapy.
The bleeding stopped 2 days after initiation of treatment. At the time of discharge, 2 weeks after presentation, factor VIII inhibitor levels had decreased to 13 BU and his partial thromboplastin time (PTT) was 100 seconds.
Discussion
Acquired factor VIII inhibitor, also called acquired hemophilia A, is a rare, potentially life‐threatening bleeding disorder. It is caused by autoantibodies directed against coagulation factor VIII.1
The estimated incidence in the general population is 1 in 4 million/year. Risk factors include advanced age, pregnancy and the postpartum period, rheumatoid disease/connective tissue disease, inflammatory bowel disease, medications (especially antibiotics and psychiatric drugs), and malignancy. Both solid tumors as well as hematologic malignancies have been associated with acquired hemophilia A.2
Patients older than 85 years are more frequently affected. The annual incidence is 14.7 in 1 million in this age group. Hence, it is found rarely in young patients, but pregnancy and the postpartum period represent the exception.
Patients with acquired factor VIII inhibitor tend to bleed into the skin, soft tissue, muscle, brain, and mucous membranes. Most of the time, they present with epistaxis, retroperitoneal hematomas, or gastrointestinal bleeds, while patients with congenital factor VIII deficiency3 are more likely to bleed into the large joints. Acquired factor VIII inhibitor is associated with a high morbidity and mortality.
In the presence of an isolated elevated aPTT, once heparin has been ruled out, specific factor deficiencies and/or inhibitors need to be considered. The inhibitor assay helps to establish the diagnosis of acquired factor VIII deficiency and allows the quantification of factor VIII inhibitor. A search for specific etiologies of acquired factor VIII inhibitors should be undertaken; however, in 50% of cases no concomitant condition is found. The differential diagnoses should be expanded within the appropriate framework and tailored to the individual patient.
Control of bleeding might be achieved by factor VIII concentrate if the bleeding is mild. However, if the hemorrhage is life‐threatening, recombinant factor VII is frequently required to stop the bleeding.4 One has to be aware that recombinant factor VII may precipitate thromboembolic events and as such might pose a dilemma, as the degree of bleeding has to be balanced with the risk of unintended side effects. Therapy to eliminate factor VIII inhibitor is the combination of prednisone and cyclophosphamide, though monoclonal CD20 antibody (Rituximab) has become the first‐line agent in the appropriate setting.5 Risk and benefit of therapy have to be balanced with the severity of the bleed and potential unintended side effects of immunosuppression, especially in the presence of infection.
As hospitalists, we are challenged daily by a high degree of complexities in inpatient care. Hospitalists are well trained to manage a wide variety of conditions, and coagulopathies are no exception. They are so common in the inpatient setting that every hospitalist should be familiar with the basic principles of diagnosing and managing bleeding disorders. Because of the hospitalist's ability to promptly react, the consulting role of the hematologist can be reserved for the more unusual blood dyscrasias.
This article is intended to raise physician awareness for the discussed condition because early recognition and treatment are of paramount importance in patient outcome.
- ,.Acquired inhibitors.Bailleres Clin Haematol.1996:9:331–354.
- ,,,.Acquired hemophilia A: a concise review.Am J Hematol.2005;80:50–63.
- ,.Acquired hemophilia.Rev Clin Exp Hematol.2005;5:389–404.
- ,,,.Treatment of acquired haemophilia with recombinant activated VII: a critical appraisal.Haemophilia.2007;13:451–461.
- ,,,.Selective B‐cell depletion with rituximab for the treatment of patients with acquired hemophilia.Blood.2004:103:4424–4428.
An 80‐year‐old man with coronary artery disease and chronic obstructive pulmonary disease (COPD) was admitted to an outside hospital after a mechanical fall. On presentation to the emergency room his systolic blood pressure was found to be 86/62 mm Hg. He complained of right flank, groin, and thigh pain. On physical exam, a hematoma extending from his right groin down to his right knee was found, as well as scattered ecchymoses involving his trunk and all 4 extremities. His hemoglobin was low, at 5.6 g/dL (14‐17 g/dL). A computed tomography (CT) scan revealed a right‐sided retroperitoneal bleed extending from the iliopsoas into his right thigh. The patient received 13 transfusions of packed red blood cells over the course of 9 days as he continued to bleed. Transfer to our facility for further workup and management ensued.
On serial testing at our institution his activated partial thromboplastin time (aPTT) was elevated at >160 seconds (normal range, 24‐36 seconds). Further coagulation parameters were found as follows: platelets 182.000/L; prothrombin time 17.6 seconds; international normalized ratio (INR) 1.4; thrombin 18 seconds; fibrinogen 778 mg/dL; and D‐dimer 3866 ng/mL. Of note, the patient had not received any medications known to potentially interfere with the measured aPTT. Because the source of his bleeding was not apparent at this point, disorders of primary hemostasis, including hereditary disease states (eg, von Willebrand disease), iatrogenic disorders (eg, drug‐induced), or acquired disorders, such as immune thrombocytopenia, were considered and ruled out. At this point the differential diagnoses had to be expanded, and secondary disorders of hemostasis were considered. A deficiency or decreased activity of coagulation factors was suspected. Whereas factor IX and XI were found to be normal, the factor VIII level was significantly decreased at 3% (50%‐150% being normal). This prompted an assay to check for the presence of a factor VIII inhibitor. It proved to be significantly elevated at 25.6 Bethesda Units (BU) (normal, 0.00‐0.04 BU). On that basis we arrived at the diagnosis of acquired factor VIII deficiency, but the etiology of such remained unclear to this point. Hence a search for the specific etiology of acquired factor IIII deficiency was launched, and connective tissue disease, as well as malignancy, was ruled out. While inflammatory bowel disease is a known potential cause for this condition, the clinical picture was not consistent with such and this diagnosis was not considered further. The patient received immunosuppressive therapy with prednisone 1 mg/kg orally per day. Rituximab and cyclophosphamide were considered, but due to bacteremia from bilateral parotitis, this was deferred. Of note, his bleeding abnormality was apparent prior to initiation of antibiotic therapy.
The bleeding stopped 2 days after initiation of treatment. At the time of discharge, 2 weeks after presentation, factor VIII inhibitor levels had decreased to 13 BU and his partial thromboplastin time (PTT) was 100 seconds.
Discussion
Acquired factor VIII inhibitor, also called acquired hemophilia A, is a rare, potentially life‐threatening bleeding disorder. It is caused by autoantibodies directed against coagulation factor VIII.1
The estimated incidence in the general population is 1 in 4 million/year. Risk factors include advanced age, pregnancy and the postpartum period, rheumatoid disease/connective tissue disease, inflammatory bowel disease, medications (especially antibiotics and psychiatric drugs), and malignancy. Both solid tumors as well as hematologic malignancies have been associated with acquired hemophilia A.2
Patients older than 85 years are more frequently affected. The annual incidence is 14.7 in 1 million in this age group. Hence, it is found rarely in young patients, but pregnancy and the postpartum period represent the exception.
Patients with acquired factor VIII inhibitor tend to bleed into the skin, soft tissue, muscle, brain, and mucous membranes. Most of the time, they present with epistaxis, retroperitoneal hematomas, or gastrointestinal bleeds, while patients with congenital factor VIII deficiency3 are more likely to bleed into the large joints. Acquired factor VIII inhibitor is associated with a high morbidity and mortality.
In the presence of an isolated elevated aPTT, once heparin has been ruled out, specific factor deficiencies and/or inhibitors need to be considered. The inhibitor assay helps to establish the diagnosis of acquired factor VIII deficiency and allows the quantification of factor VIII inhibitor. A search for specific etiologies of acquired factor VIII inhibitors should be undertaken; however, in 50% of cases no concomitant condition is found. The differential diagnoses should be expanded within the appropriate framework and tailored to the individual patient.
Control of bleeding might be achieved by factor VIII concentrate if the bleeding is mild. However, if the hemorrhage is life‐threatening, recombinant factor VII is frequently required to stop the bleeding.4 One has to be aware that recombinant factor VII may precipitate thromboembolic events and as such might pose a dilemma, as the degree of bleeding has to be balanced with the risk of unintended side effects. Therapy to eliminate factor VIII inhibitor is the combination of prednisone and cyclophosphamide, though monoclonal CD20 antibody (Rituximab) has become the first‐line agent in the appropriate setting.5 Risk and benefit of therapy have to be balanced with the severity of the bleed and potential unintended side effects of immunosuppression, especially in the presence of infection.
As hospitalists, we are challenged daily by a high degree of complexities in inpatient care. Hospitalists are well trained to manage a wide variety of conditions, and coagulopathies are no exception. They are so common in the inpatient setting that every hospitalist should be familiar with the basic principles of diagnosing and managing bleeding disorders. Because of the hospitalist's ability to promptly react, the consulting role of the hematologist can be reserved for the more unusual blood dyscrasias.
This article is intended to raise physician awareness for the discussed condition because early recognition and treatment are of paramount importance in patient outcome.
An 80‐year‐old man with coronary artery disease and chronic obstructive pulmonary disease (COPD) was admitted to an outside hospital after a mechanical fall. On presentation to the emergency room his systolic blood pressure was found to be 86/62 mm Hg. He complained of right flank, groin, and thigh pain. On physical exam, a hematoma extending from his right groin down to his right knee was found, as well as scattered ecchymoses involving his trunk and all 4 extremities. His hemoglobin was low, at 5.6 g/dL (14‐17 g/dL). A computed tomography (CT) scan revealed a right‐sided retroperitoneal bleed extending from the iliopsoas into his right thigh. The patient received 13 transfusions of packed red blood cells over the course of 9 days as he continued to bleed. Transfer to our facility for further workup and management ensued.
On serial testing at our institution his activated partial thromboplastin time (aPTT) was elevated at >160 seconds (normal range, 24‐36 seconds). Further coagulation parameters were found as follows: platelets 182.000/L; prothrombin time 17.6 seconds; international normalized ratio (INR) 1.4; thrombin 18 seconds; fibrinogen 778 mg/dL; and D‐dimer 3866 ng/mL. Of note, the patient had not received any medications known to potentially interfere with the measured aPTT. Because the source of his bleeding was not apparent at this point, disorders of primary hemostasis, including hereditary disease states (eg, von Willebrand disease), iatrogenic disorders (eg, drug‐induced), or acquired disorders, such as immune thrombocytopenia, were considered and ruled out. At this point the differential diagnoses had to be expanded, and secondary disorders of hemostasis were considered. A deficiency or decreased activity of coagulation factors was suspected. Whereas factor IX and XI were found to be normal, the factor VIII level was significantly decreased at 3% (50%‐150% being normal). This prompted an assay to check for the presence of a factor VIII inhibitor. It proved to be significantly elevated at 25.6 Bethesda Units (BU) (normal, 0.00‐0.04 BU). On that basis we arrived at the diagnosis of acquired factor VIII deficiency, but the etiology of such remained unclear to this point. Hence a search for the specific etiology of acquired factor IIII deficiency was launched, and connective tissue disease, as well as malignancy, was ruled out. While inflammatory bowel disease is a known potential cause for this condition, the clinical picture was not consistent with such and this diagnosis was not considered further. The patient received immunosuppressive therapy with prednisone 1 mg/kg orally per day. Rituximab and cyclophosphamide were considered, but due to bacteremia from bilateral parotitis, this was deferred. Of note, his bleeding abnormality was apparent prior to initiation of antibiotic therapy.
The bleeding stopped 2 days after initiation of treatment. At the time of discharge, 2 weeks after presentation, factor VIII inhibitor levels had decreased to 13 BU and his partial thromboplastin time (PTT) was 100 seconds.
Discussion
Acquired factor VIII inhibitor, also called acquired hemophilia A, is a rare, potentially life‐threatening bleeding disorder. It is caused by autoantibodies directed against coagulation factor VIII.1
The estimated incidence in the general population is 1 in 4 million/year. Risk factors include advanced age, pregnancy and the postpartum period, rheumatoid disease/connective tissue disease, inflammatory bowel disease, medications (especially antibiotics and psychiatric drugs), and malignancy. Both solid tumors as well as hematologic malignancies have been associated with acquired hemophilia A.2
Patients older than 85 years are more frequently affected. The annual incidence is 14.7 in 1 million in this age group. Hence, it is found rarely in young patients, but pregnancy and the postpartum period represent the exception.
Patients with acquired factor VIII inhibitor tend to bleed into the skin, soft tissue, muscle, brain, and mucous membranes. Most of the time, they present with epistaxis, retroperitoneal hematomas, or gastrointestinal bleeds, while patients with congenital factor VIII deficiency3 are more likely to bleed into the large joints. Acquired factor VIII inhibitor is associated with a high morbidity and mortality.
In the presence of an isolated elevated aPTT, once heparin has been ruled out, specific factor deficiencies and/or inhibitors need to be considered. The inhibitor assay helps to establish the diagnosis of acquired factor VIII deficiency and allows the quantification of factor VIII inhibitor. A search for specific etiologies of acquired factor VIII inhibitors should be undertaken; however, in 50% of cases no concomitant condition is found. The differential diagnoses should be expanded within the appropriate framework and tailored to the individual patient.
Control of bleeding might be achieved by factor VIII concentrate if the bleeding is mild. However, if the hemorrhage is life‐threatening, recombinant factor VII is frequently required to stop the bleeding.4 One has to be aware that recombinant factor VII may precipitate thromboembolic events and as such might pose a dilemma, as the degree of bleeding has to be balanced with the risk of unintended side effects. Therapy to eliminate factor VIII inhibitor is the combination of prednisone and cyclophosphamide, though monoclonal CD20 antibody (Rituximab) has become the first‐line agent in the appropriate setting.5 Risk and benefit of therapy have to be balanced with the severity of the bleed and potential unintended side effects of immunosuppression, especially in the presence of infection.
As hospitalists, we are challenged daily by a high degree of complexities in inpatient care. Hospitalists are well trained to manage a wide variety of conditions, and coagulopathies are no exception. They are so common in the inpatient setting that every hospitalist should be familiar with the basic principles of diagnosing and managing bleeding disorders. Because of the hospitalist's ability to promptly react, the consulting role of the hematologist can be reserved for the more unusual blood dyscrasias.
This article is intended to raise physician awareness for the discussed condition because early recognition and treatment are of paramount importance in patient outcome.
- ,.Acquired inhibitors.Bailleres Clin Haematol.1996:9:331–354.
- ,,,.Acquired hemophilia A: a concise review.Am J Hematol.2005;80:50–63.
- ,.Acquired hemophilia.Rev Clin Exp Hematol.2005;5:389–404.
- ,,,.Treatment of acquired haemophilia with recombinant activated VII: a critical appraisal.Haemophilia.2007;13:451–461.
- ,,,.Selective B‐cell depletion with rituximab for the treatment of patients with acquired hemophilia.Blood.2004:103:4424–4428.
- ,.Acquired inhibitors.Bailleres Clin Haematol.1996:9:331–354.
- ,,,.Acquired hemophilia A: a concise review.Am J Hematol.2005;80:50–63.
- ,.Acquired hemophilia.Rev Clin Exp Hematol.2005;5:389–404.
- ,,,.Treatment of acquired haemophilia with recombinant activated VII: a critical appraisal.Haemophilia.2007;13:451–461.
- ,,,.Selective B‐cell depletion with rituximab for the treatment of patients with acquired hemophilia.Blood.2004:103:4424–4428.
Benign Pneumatosis Intestinalis
A 9‐year‐old male, with a history of immune thrombocytopenic purpura (ITP) and hypoplastic left heart syndrome (HLHS) repaired by total cavopulmonary shunt, presented to the Emergency Department with a 4‐day history of crampy abdominal pain with defecation and a 4‐day history of nonbloody diarrhea with intermittent nonbilious vomiting. The abdominal pain was diffuse and nonspecific without any radiation. He had a history of encopresis and constipation over the past 3 months. No anorexia was noted and the pain did not keep him from his activities of daily living. Review of all other systems was noncontributory.
His past medical history consisted of HLHS repaired by total cavopulmonary shunt with excellent results. He was diagnosed with ITP about 6 weeks prior to this presentation and had been treated with intravenous immunoglobulin and oral prednisone. His home medications included prednisone (1.5 mg/kg/day), lansoprazole, digoxin, enalapril, furosemide, and warfarin.
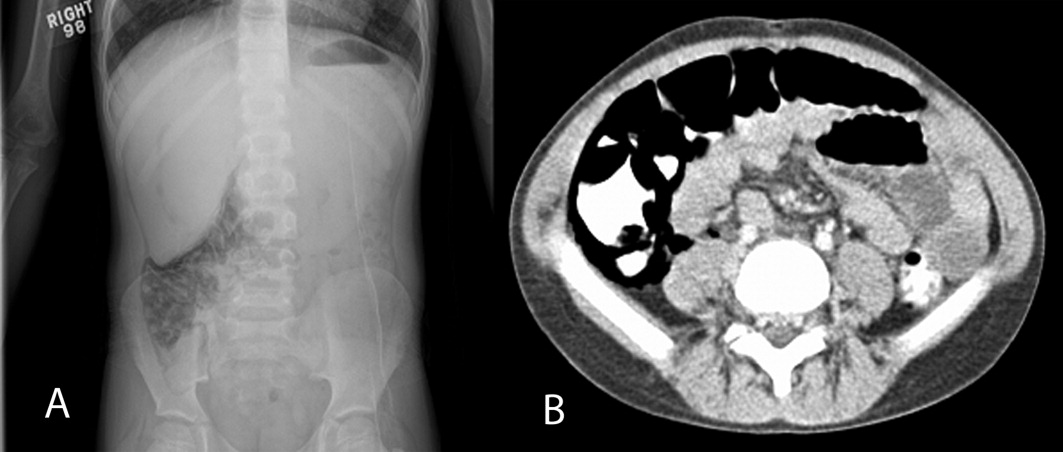
On physical examination, his temperature was 37C, pulse rate 97, respiratory rate 16, unlabored blood pressure was 112/74 mm Hg, oxygen saturation 91% on room air, and weight was 22.8 kg. He had a grade 4/6 holosystolic murmur across the precordium and multiple healed surgical incisions. His abdomen was soft without tenderness or distention with normoactive bowel sounds. The rest of his physical exam was unremarkable. An acute abdominal series was obtained, which showed pneumatosis intestinalis (PI) of the right colon, pneumoperitoneum, and possible portal venous gas (Figure 1A). Initial laboratory evaluation including a complete blood count and a comprehensive metabolic panel; amylase, lipase, lactate, and venous blood gas were all within normal limits, with the exception of a platelet level of 75,000/L (normal, 150,000450,000 cells/mL). Stool was negative for Rotavirus antigen, Clostridium difficile toxin, Helicobacter pylori antigen, and Shiga toxin 1 and 2. Bacterial cultures and trichrome stain for ova and parasites were both negative. Stool analysis for occult blood was negative on admission and became positive during his hospital course. A contrast computed tomography (CT) of the abdomen and pelvis confirmed the findings of pneumatosis intestinalis in the cecum and ascending colon with intraperitoneal and retroperitoneal air, but did not reveal any portal venous gas (Figure 1B).

The patient was admitted to the Children's Hospital and made nil per os (NPO; ie, nothing by mouth), placed on ampicillin/sulbactam and metronidazole prophylaxis, and observed with serial abdominal examinations. Total parenteral nutrition (TPN) was begun on hospital day 2 and continued for 10 days. Surgical intervention was not required at the initial presentation secondary to his clinical and hemodynamic stability. Since immunosuppression from chronic steroid therapy is a known risk factor for the development of PI,1 a slow steroid taper with intravenous methylprednisolone was initiated and was transitioned to oral prednisone after he resumed oral nutrition. He remained NPO for 10 days until the PI radiographically resolved. Oral feeds were reintroduced slowly without complications or recurrence of PI.
Discussion
Three major hypotheses for the origin of bowel wall gas have been proposed: intraluminal gastrointestinal (GI) gas; bacterial production of gas; and pulmonary gas.1 The intralumenal GI gas hypothesis states that intralumenal gas translocates to the bowel wall due to increased intralumenal pressure, mucosal injury from direct trauma, reduction is size of Peyer's patches from immunosuppressive medications, or a combination of factors.1, 2 The bacterial theory proposes direct invasion of the bowel wall by gas‐producing bacteria; this hypothesis is not adequately supported by bacteriologic data.2 The pulmonary gas hypothesis states that alveolar rupture could result in dissection of air through the mediastinum to the retroperitoneum and eventually along vascular channels to the gut.2 Increased intralumenal gut pressure due to coughing also drives this dissection of gas into the bowel wall.2
Chronic immunosuppression with steroids and congenital heart disease are both known risk factors for the development of PI.3 Our patient presented with common complaints of abdominal pain, encopresis, and vomiting, with a benign exam. However, he had a radiographic finding of PI. With bowel rest, antibiotics, and TPN, our patient made a full recovery without requiring surgical intervention. With patients at higher risk for PI, there needs to be a higher index of suspicion for PI in the setting of GI complaints and hemodynamic instability. It has been reported in the literature that patients at higher risk can include those with inflammatory bowel disease, chronic pulmonary disease, immunosuppressive states such as leukemia or acquired immunodeficiency syndrome, short gut syndrome, and malignancies.1, 3 Kurbegov and Sondheimer4 published a series of 32 nonneonatal cases of PI, looking for characteristics that predicted higher risk of poor outcome. Their findings showed low serum bicarbonate and PI with free air and portal venous gas as significant predictors of poor outcome.4 A recent study by Morris et al.5 showed similar results: lactic acidosis is a predictor of poor patient outcome. The clinical examination of this patient, both initially and longitudinally, and the lack of laboratory abnormalities were the key factors in the disposition of this patient in the setting of alarming abdominal radiographs.
- ,,.The spectrum of pneumatosis intestinalis.Arch Surg.2003;138(1):68–75.
- ,,, et al.Pneumatosis intestinalis with pneumoperitoneum mimicking intestinal perforation in a patient with myelodysplastic syndrome after hematopoietic stem cell transplantation.Korean J Intern Med.2007;22(1):40–44.
- ,.Benign pneumatosis in children.Pediatr Radiol.2000;30(11):786–793.
- ,.Pneumatosis intestinalis in non‐neonatal pediatric patients.Pediatrics.2001;108(2):402–406.
- ,,, et al.Management and outcome of pneumatosis intestinalis.Am J Surg.2008;195(5):679–682; discussion 682–683.
A 9‐year‐old male, with a history of immune thrombocytopenic purpura (ITP) and hypoplastic left heart syndrome (HLHS) repaired by total cavopulmonary shunt, presented to the Emergency Department with a 4‐day history of crampy abdominal pain with defecation and a 4‐day history of nonbloody diarrhea with intermittent nonbilious vomiting. The abdominal pain was diffuse and nonspecific without any radiation. He had a history of encopresis and constipation over the past 3 months. No anorexia was noted and the pain did not keep him from his activities of daily living. Review of all other systems was noncontributory.
His past medical history consisted of HLHS repaired by total cavopulmonary shunt with excellent results. He was diagnosed with ITP about 6 weeks prior to this presentation and had been treated with intravenous immunoglobulin and oral prednisone. His home medications included prednisone (1.5 mg/kg/day), lansoprazole, digoxin, enalapril, furosemide, and warfarin.
On physical examination, his temperature was 37C, pulse rate 97, respiratory rate 16, unlabored blood pressure was 112/74 mm Hg, oxygen saturation 91% on room air, and weight was 22.8 kg. He had a grade 4/6 holosystolic murmur across the precordium and multiple healed surgical incisions. His abdomen was soft without tenderness or distention with normoactive bowel sounds. The rest of his physical exam was unremarkable. An acute abdominal series was obtained, which showed pneumatosis intestinalis (PI) of the right colon, pneumoperitoneum, and possible portal venous gas (Figure 1A). Initial laboratory evaluation including a complete blood count and a comprehensive metabolic panel; amylase, lipase, lactate, and venous blood gas were all within normal limits, with the exception of a platelet level of 75,000/L (normal, 150,000450,000 cells/mL). Stool was negative for Rotavirus antigen, Clostridium difficile toxin, Helicobacter pylori antigen, and Shiga toxin 1 and 2. Bacterial cultures and trichrome stain for ova and parasites were both negative. Stool analysis for occult blood was negative on admission and became positive during his hospital course. A contrast computed tomography (CT) of the abdomen and pelvis confirmed the findings of pneumatosis intestinalis in the cecum and ascending colon with intraperitoneal and retroperitoneal air, but did not reveal any portal venous gas (Figure 1B).
The patient was admitted to the Children's Hospital and made nil per os (NPO; ie, nothing by mouth), placed on ampicillin/sulbactam and metronidazole prophylaxis, and observed with serial abdominal examinations. Total parenteral nutrition (TPN) was begun on hospital day 2 and continued for 10 days. Surgical intervention was not required at the initial presentation secondary to his clinical and hemodynamic stability. Since immunosuppression from chronic steroid therapy is a known risk factor for the development of PI,1 a slow steroid taper with intravenous methylprednisolone was initiated and was transitioned to oral prednisone after he resumed oral nutrition. He remained NPO for 10 days until the PI radiographically resolved. Oral feeds were reintroduced slowly without complications or recurrence of PI.
Discussion
Three major hypotheses for the origin of bowel wall gas have been proposed: intraluminal gastrointestinal (GI) gas; bacterial production of gas; and pulmonary gas.1 The intralumenal GI gas hypothesis states that intralumenal gas translocates to the bowel wall due to increased intralumenal pressure, mucosal injury from direct trauma, reduction is size of Peyer's patches from immunosuppressive medications, or a combination of factors.1, 2 The bacterial theory proposes direct invasion of the bowel wall by gas‐producing bacteria; this hypothesis is not adequately supported by bacteriologic data.2 The pulmonary gas hypothesis states that alveolar rupture could result in dissection of air through the mediastinum to the retroperitoneum and eventually along vascular channels to the gut.2 Increased intralumenal gut pressure due to coughing also drives this dissection of gas into the bowel wall.2
Chronic immunosuppression with steroids and congenital heart disease are both known risk factors for the development of PI.3 Our patient presented with common complaints of abdominal pain, encopresis, and vomiting, with a benign exam. However, he had a radiographic finding of PI. With bowel rest, antibiotics, and TPN, our patient made a full recovery without requiring surgical intervention. With patients at higher risk for PI, there needs to be a higher index of suspicion for PI in the setting of GI complaints and hemodynamic instability. It has been reported in the literature that patients at higher risk can include those with inflammatory bowel disease, chronic pulmonary disease, immunosuppressive states such as leukemia or acquired immunodeficiency syndrome, short gut syndrome, and malignancies.1, 3 Kurbegov and Sondheimer4 published a series of 32 nonneonatal cases of PI, looking for characteristics that predicted higher risk of poor outcome. Their findings showed low serum bicarbonate and PI with free air and portal venous gas as significant predictors of poor outcome.4 A recent study by Morris et al.5 showed similar results: lactic acidosis is a predictor of poor patient outcome. The clinical examination of this patient, both initially and longitudinally, and the lack of laboratory abnormalities were the key factors in the disposition of this patient in the setting of alarming abdominal radiographs.
A 9‐year‐old male, with a history of immune thrombocytopenic purpura (ITP) and hypoplastic left heart syndrome (HLHS) repaired by total cavopulmonary shunt, presented to the Emergency Department with a 4‐day history of crampy abdominal pain with defecation and a 4‐day history of nonbloody diarrhea with intermittent nonbilious vomiting. The abdominal pain was diffuse and nonspecific without any radiation. He had a history of encopresis and constipation over the past 3 months. No anorexia was noted and the pain did not keep him from his activities of daily living. Review of all other systems was noncontributory.
His past medical history consisted of HLHS repaired by total cavopulmonary shunt with excellent results. He was diagnosed with ITP about 6 weeks prior to this presentation and had been treated with intravenous immunoglobulin and oral prednisone. His home medications included prednisone (1.5 mg/kg/day), lansoprazole, digoxin, enalapril, furosemide, and warfarin.
On physical examination, his temperature was 37C, pulse rate 97, respiratory rate 16, unlabored blood pressure was 112/74 mm Hg, oxygen saturation 91% on room air, and weight was 22.8 kg. He had a grade 4/6 holosystolic murmur across the precordium and multiple healed surgical incisions. His abdomen was soft without tenderness or distention with normoactive bowel sounds. The rest of his physical exam was unremarkable. An acute abdominal series was obtained, which showed pneumatosis intestinalis (PI) of the right colon, pneumoperitoneum, and possible portal venous gas (Figure 1A). Initial laboratory evaluation including a complete blood count and a comprehensive metabolic panel; amylase, lipase, lactate, and venous blood gas were all within normal limits, with the exception of a platelet level of 75,000/L (normal, 150,000450,000 cells/mL). Stool was negative for Rotavirus antigen, Clostridium difficile toxin, Helicobacter pylori antigen, and Shiga toxin 1 and 2. Bacterial cultures and trichrome stain for ova and parasites were both negative. Stool analysis for occult blood was negative on admission and became positive during his hospital course. A contrast computed tomography (CT) of the abdomen and pelvis confirmed the findings of pneumatosis intestinalis in the cecum and ascending colon with intraperitoneal and retroperitoneal air, but did not reveal any portal venous gas (Figure 1B).
The patient was admitted to the Children's Hospital and made nil per os (NPO; ie, nothing by mouth), placed on ampicillin/sulbactam and metronidazole prophylaxis, and observed with serial abdominal examinations. Total parenteral nutrition (TPN) was begun on hospital day 2 and continued for 10 days. Surgical intervention was not required at the initial presentation secondary to his clinical and hemodynamic stability. Since immunosuppression from chronic steroid therapy is a known risk factor for the development of PI,1 a slow steroid taper with intravenous methylprednisolone was initiated and was transitioned to oral prednisone after he resumed oral nutrition. He remained NPO for 10 days until the PI radiographically resolved. Oral feeds were reintroduced slowly without complications or recurrence of PI.
Discussion
Three major hypotheses for the origin of bowel wall gas have been proposed: intraluminal gastrointestinal (GI) gas; bacterial production of gas; and pulmonary gas.1 The intralumenal GI gas hypothesis states that intralumenal gas translocates to the bowel wall due to increased intralumenal pressure, mucosal injury from direct trauma, reduction is size of Peyer's patches from immunosuppressive medications, or a combination of factors.1, 2 The bacterial theory proposes direct invasion of the bowel wall by gas‐producing bacteria; this hypothesis is not adequately supported by bacteriologic data.2 The pulmonary gas hypothesis states that alveolar rupture could result in dissection of air through the mediastinum to the retroperitoneum and eventually along vascular channels to the gut.2 Increased intralumenal gut pressure due to coughing also drives this dissection of gas into the bowel wall.2
Chronic immunosuppression with steroids and congenital heart disease are both known risk factors for the development of PI.3 Our patient presented with common complaints of abdominal pain, encopresis, and vomiting, with a benign exam. However, he had a radiographic finding of PI. With bowel rest, antibiotics, and TPN, our patient made a full recovery without requiring surgical intervention. With patients at higher risk for PI, there needs to be a higher index of suspicion for PI in the setting of GI complaints and hemodynamic instability. It has been reported in the literature that patients at higher risk can include those with inflammatory bowel disease, chronic pulmonary disease, immunosuppressive states such as leukemia or acquired immunodeficiency syndrome, short gut syndrome, and malignancies.1, 3 Kurbegov and Sondheimer4 published a series of 32 nonneonatal cases of PI, looking for characteristics that predicted higher risk of poor outcome. Their findings showed low serum bicarbonate and PI with free air and portal venous gas as significant predictors of poor outcome.4 A recent study by Morris et al.5 showed similar results: lactic acidosis is a predictor of poor patient outcome. The clinical examination of this patient, both initially and longitudinally, and the lack of laboratory abnormalities were the key factors in the disposition of this patient in the setting of alarming abdominal radiographs.
- ,,.The spectrum of pneumatosis intestinalis.Arch Surg.2003;138(1):68–75.
- ,,, et al.Pneumatosis intestinalis with pneumoperitoneum mimicking intestinal perforation in a patient with myelodysplastic syndrome after hematopoietic stem cell transplantation.Korean J Intern Med.2007;22(1):40–44.
- ,.Benign pneumatosis in children.Pediatr Radiol.2000;30(11):786–793.
- ,.Pneumatosis intestinalis in non‐neonatal pediatric patients.Pediatrics.2001;108(2):402–406.
- ,,, et al.Management and outcome of pneumatosis intestinalis.Am J Surg.2008;195(5):679–682; discussion 682–683.
- ,,.The spectrum of pneumatosis intestinalis.Arch Surg.2003;138(1):68–75.
- ,,, et al.Pneumatosis intestinalis with pneumoperitoneum mimicking intestinal perforation in a patient with myelodysplastic syndrome after hematopoietic stem cell transplantation.Korean J Intern Med.2007;22(1):40–44.
- ,.Benign pneumatosis in children.Pediatr Radiol.2000;30(11):786–793.
- ,.Pneumatosis intestinalis in non‐neonatal pediatric patients.Pediatrics.2001;108(2):402–406.
- ,,, et al.Management and outcome of pneumatosis intestinalis.Am J Surg.2008;195(5):679–682; discussion 682–683.
Pleural Effusion with IFNα for HCV
Case Report
A 52‐year‐old woman with chronic hepatitis C was admitted with complaints of dry cough, shortness of breath, and fever. Four days prior to admission, she had successfully finished a 44‐week course of pegylated interferon (IFN) alpha and ribavirin with undetectable viral load on completion of treatment. At 30 weeks, she had developed a dry cough, which she initially ignored. Three weeks later, as a result of a violent coughing episode, she sustained a spontaneous uncomplicated fracture of the left sixth rib. Chest x‐ray at that time did not show an infiltrate or opacity. She continued treatment, and over the next 6 weeks developed progressive dyspnea on exertion. Five days prior to admission, she had developed fever of 101F. Repeat chest x‐ray revealed a left lingular infiltrate and she was prescribed levofloxacin. Her symptoms failed to improve and she was admitted to the hospital.
On admission, she denied expectoration, sore throat, night sweats, or rashes. She also denied tobacco use, pets at home, or recent travel outside the Midwest. Examination revealed a temperature of 99.4F and decreased breath sounds over the left lower chest. Chest x‐ray revealed left‐sided pleural effusion. D‐dimer was negative. Computed tomography (CT) scan of the chest showed a left lingular infiltrate, right lower lobe ground‐glass opacity, and a moderately‐sized left pleural effusion. Azithromycin, piperacillin/tazobactam, and vancomycin were empirically started. Over the next 36 hours, she became increasingly tachypneic and short of breath. A diagnostic and therapeutic thoracentesis with aspiration of 800 mL of light‐yellow‐colored fluid brought symptomatic relief. Pleural fluid analysis revealed an exudative effusion with 3.8 gm/dL of protein (serum protein = 6.2 gm/dL), lactic dehydrogenase (LDH) of 998 IU/L (serum LDH = 293 IU/L), and normal adenosine deaminase. The cell count was 362 per mm3 with 37% lymphocytes, 32% macrophages, 26% neutrophils, and 1% eosinophils. There were no atypical or malignant cells. Bacterial, fungal, viral, acid‐fast stains and cultures, and polymerase chain reaction (PCR) for Mycobacterium tuberculosis were all negative. An echocardiogram and plasma B‐type natriuretic peptide were normal.
Serum antinuclear and antineutrophilic cytoplasmic antibodies, Bordetella pertussis PCR, serologies for Mycoplasma, Chlamydia, Coxiella, and urinary antigens for Legionella and Blastomyces were all negative. Bronchoscopy with bronchoalveolar lavage (BAL) was performed on hospital day 5. BAL stains and cultures for bacteria, fungi, acid‐fast organisms, Cytomegalovirus, Herpes simplex virus, Legionella, and Pneumocystis were negative. Cytology revealed mild acute inflammation with macrophage predominance and no malignant cells.
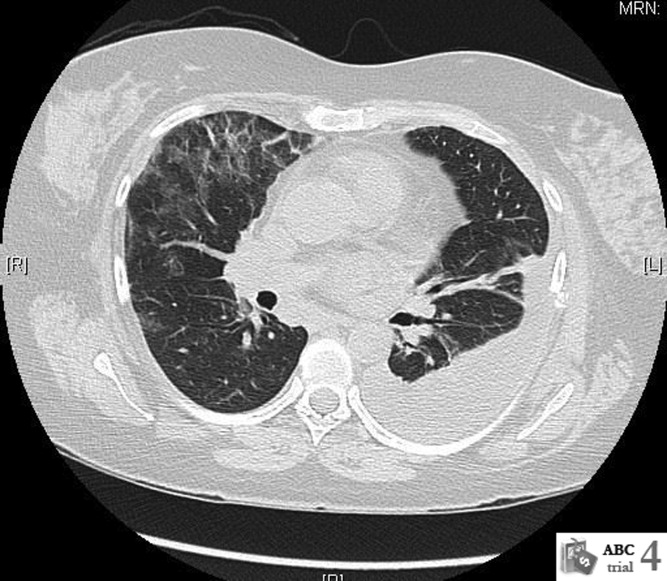
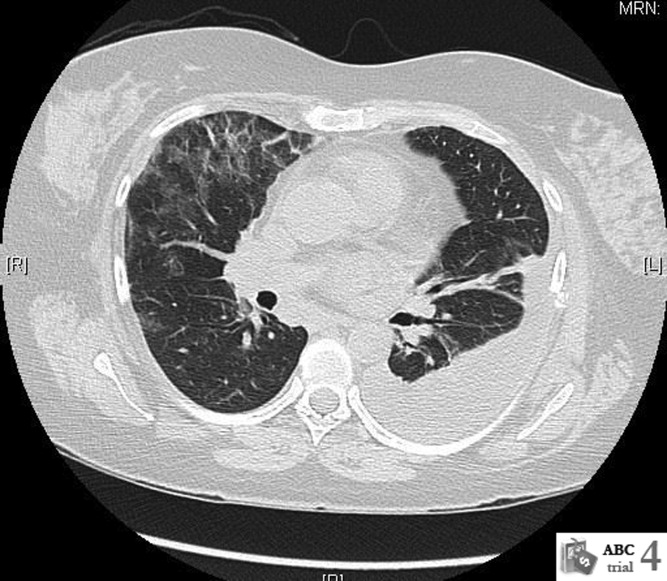
Repeat CT scan of the chest on day 6 showed bilateral ground‐glass infiltrates and persistent left pleural effusion (Figure 1). In the absence of an identifiable cause, the patient was diagnosed with interstitial pneumonitis and pleural effusion secondary to pegylated IFN alpha and ribavirin. Treatment with steroids was considered, but was not used due to recent successful suppression of hepatitis C. She was discharged with continued close follow‐up. Her fever gradually subsided over the next 2 weeks and her cough continued to improve over the next 6 weeks. Follow‐up CT scan of the chest 3 months after discharge showed complete resolution of the left pleural effusion and near‐resolution of the bilateral basal infiltrates.
Discussion
Use of IFN alpha has been associated with multiple forms of lung toxicity, of which interstitial pneumonitis and granulomatous inflammation resembling sarcoidosis are the most common. Unusual forms include isolated nonproductive cough, exacerbation of asthma, organizing pneumonia, pleural effusion, adult respiratory distress syndrome, and exacerbation of vasculitis.1 Reports of adverse pulmonary effects of ribavirin are sparse, and it has not been implicated as a sole etiologic agent in causing lung toxicity. It is therefore likely that pulmonary toxicity observed in patients with hepatitis C virus (HCV) infection undergoing IFN alpha and ribavirin therapy is due to the IFN.
Pleural effusion may accompany the IFN‐induced capillary leak syndrome.2
There have been only 2 other cases of pleural effusion during treatment with IFN alpha described to date.3, 4 Takeda et al.3 described a 54‐year‐old male who was accidentally detected to have a moderate‐sized right pleural effusion on magnetic resonance imaging (MRI) of the abdomen, 14 days after therapy with recombinant IFN alpha was initiated. The pleural fluid was a lymphocyte‐predominant exudate and resolved approximately 4 months after discontinuation of IFN treatment. Tsushima et al.4 reported bilateral pleural effusions and ground‐glass opacities in a patient treated with IFN for metastatic renal cell cancer that resolved following a course of steroids.
IFN‐related pulmonary toxicity has been reported to typically develop between 2 and 16 weeks of treatment. Our patient had a delayed onset of symptoms at 30 weeks and progressed on to develop left pleural effusion and pulmonary infiltrates by the time she finished 44 weeks of treatment. We ruled out infectious, malignant, cardiac, and autoimmune causes, which often present in a similar fashion.
BAL fluid cytology in our patient revealed predominant macrophages. Yamaguchi et al., in their analysis of BAL fluid in patients with hepatitis C, demonstrated increased macrophages (76% and 77.5%) and lymphocytes (19.8% and 18.8%) before and after treatment with IFN alpha, respectively.
The cornerstone of management of lung toxicity due to IFN is to diminish or stop use of the offending agent. Our patient demonstrated complete recovery of symptoms and radiological resolution within 3 months of completion of IFN therapy, without corticosteroid therapy. Although corticosteroid regimes of 6 to 12 months have been used to manage IFN related lung toxicity, most patients recover without them.6 Moreover, corticosteroids have been implicated in the recurrence of hepatitis C.
We believe that our patient's pathology is most consistent with lung and pleural toxicity temporally related to IFN treatment. Through our case report, we bring to attention this infrequent complication, and emphasize its self‐limited course upon withdrawal of the offending agent.
Acknowledgements
The authors thank Dr. Philippe Camus, Hpital Le Bocage, Dijon, France, for his invaluable suggestions and for reviewing this case report prior to submission.
- ,.Groupe d'Etudes de la Pathologie Pulmonaire Iatrogène (GEPPI). Pneumotox Online. The drug‐induced lung diseases. Available at: http://www.pneumotox.com. Accessed February 2009.
- ,,, et al.Fatality and interferon alpha for malignant melanoma.Lancet.1998;352(9138):1443–1444.
- ,,, et al.Pleural effusion during interferon treatment for chronic hepatitis C.Hepatogastroenterology.2000;47(35):1431–1435.
- ,,, et al.A case of renal cell carcinoma complicated with interstitial pneumonitis, complete A‐V block and pleural effusion during interferon‐alpha therapy.Nihon Kokyuki Gakkai Zasshi.2001;39:893–898.
- ,,, et al.Analysis of bronchoalveolar lavage fluid of patients with chronic hepatitis c before and after treatment with interferon alpha.Thorax.1997;52:33–37.
- ,,, et al.Spectrum of pulmonary toxicity associated with the use of interferon therapy for hepatitis C: case report and review of the literature.Clin Infect Dis.2004;39:1724–1729.
Case Report
A 52‐year‐old woman with chronic hepatitis C was admitted with complaints of dry cough, shortness of breath, and fever. Four days prior to admission, she had successfully finished a 44‐week course of pegylated interferon (IFN) alpha and ribavirin with undetectable viral load on completion of treatment. At 30 weeks, she had developed a dry cough, which she initially ignored. Three weeks later, as a result of a violent coughing episode, she sustained a spontaneous uncomplicated fracture of the left sixth rib. Chest x‐ray at that time did not show an infiltrate or opacity. She continued treatment, and over the next 6 weeks developed progressive dyspnea on exertion. Five days prior to admission, she had developed fever of 101F. Repeat chest x‐ray revealed a left lingular infiltrate and she was prescribed levofloxacin. Her symptoms failed to improve and she was admitted to the hospital.
On admission, she denied expectoration, sore throat, night sweats, or rashes. She also denied tobacco use, pets at home, or recent travel outside the Midwest. Examination revealed a temperature of 99.4F and decreased breath sounds over the left lower chest. Chest x‐ray revealed left‐sided pleural effusion. D‐dimer was negative. Computed tomography (CT) scan of the chest showed a left lingular infiltrate, right lower lobe ground‐glass opacity, and a moderately‐sized left pleural effusion. Azithromycin, piperacillin/tazobactam, and vancomycin were empirically started. Over the next 36 hours, she became increasingly tachypneic and short of breath. A diagnostic and therapeutic thoracentesis with aspiration of 800 mL of light‐yellow‐colored fluid brought symptomatic relief. Pleural fluid analysis revealed an exudative effusion with 3.8 gm/dL of protein (serum protein = 6.2 gm/dL), lactic dehydrogenase (LDH) of 998 IU/L (serum LDH = 293 IU/L), and normal adenosine deaminase. The cell count was 362 per mm3 with 37% lymphocytes, 32% macrophages, 26% neutrophils, and 1% eosinophils. There were no atypical or malignant cells. Bacterial, fungal, viral, acid‐fast stains and cultures, and polymerase chain reaction (PCR) for Mycobacterium tuberculosis were all negative. An echocardiogram and plasma B‐type natriuretic peptide were normal.
Serum antinuclear and antineutrophilic cytoplasmic antibodies, Bordetella pertussis PCR, serologies for Mycoplasma, Chlamydia, Coxiella, and urinary antigens for Legionella and Blastomyces were all negative. Bronchoscopy with bronchoalveolar lavage (BAL) was performed on hospital day 5. BAL stains and cultures for bacteria, fungi, acid‐fast organisms, Cytomegalovirus, Herpes simplex virus, Legionella, and Pneumocystis were negative. Cytology revealed mild acute inflammation with macrophage predominance and no malignant cells.
Repeat CT scan of the chest on day 6 showed bilateral ground‐glass infiltrates and persistent left pleural effusion (Figure 1). In the absence of an identifiable cause, the patient was diagnosed with interstitial pneumonitis and pleural effusion secondary to pegylated IFN alpha and ribavirin. Treatment with steroids was considered, but was not used due to recent successful suppression of hepatitis C. She was discharged with continued close follow‐up. Her fever gradually subsided over the next 2 weeks and her cough continued to improve over the next 6 weeks. Follow‐up CT scan of the chest 3 months after discharge showed complete resolution of the left pleural effusion and near‐resolution of the bilateral basal infiltrates.
Discussion
Use of IFN alpha has been associated with multiple forms of lung toxicity, of which interstitial pneumonitis and granulomatous inflammation resembling sarcoidosis are the most common. Unusual forms include isolated nonproductive cough, exacerbation of asthma, organizing pneumonia, pleural effusion, adult respiratory distress syndrome, and exacerbation of vasculitis.1 Reports of adverse pulmonary effects of ribavirin are sparse, and it has not been implicated as a sole etiologic agent in causing lung toxicity. It is therefore likely that pulmonary toxicity observed in patients with hepatitis C virus (HCV) infection undergoing IFN alpha and ribavirin therapy is due to the IFN.
Pleural effusion may accompany the IFN‐induced capillary leak syndrome.2
There have been only 2 other cases of pleural effusion during treatment with IFN alpha described to date.3, 4 Takeda et al.3 described a 54‐year‐old male who was accidentally detected to have a moderate‐sized right pleural effusion on magnetic resonance imaging (MRI) of the abdomen, 14 days after therapy with recombinant IFN alpha was initiated. The pleural fluid was a lymphocyte‐predominant exudate and resolved approximately 4 months after discontinuation of IFN treatment. Tsushima et al.4 reported bilateral pleural effusions and ground‐glass opacities in a patient treated with IFN for metastatic renal cell cancer that resolved following a course of steroids.
IFN‐related pulmonary toxicity has been reported to typically develop between 2 and 16 weeks of treatment. Our patient had a delayed onset of symptoms at 30 weeks and progressed on to develop left pleural effusion and pulmonary infiltrates by the time she finished 44 weeks of treatment. We ruled out infectious, malignant, cardiac, and autoimmune causes, which often present in a similar fashion.
BAL fluid cytology in our patient revealed predominant macrophages. Yamaguchi et al., in their analysis of BAL fluid in patients with hepatitis C, demonstrated increased macrophages (76% and 77.5%) and lymphocytes (19.8% and 18.8%) before and after treatment with IFN alpha, respectively.
The cornerstone of management of lung toxicity due to IFN is to diminish or stop use of the offending agent. Our patient demonstrated complete recovery of symptoms and radiological resolution within 3 months of completion of IFN therapy, without corticosteroid therapy. Although corticosteroid regimes of 6 to 12 months have been used to manage IFN related lung toxicity, most patients recover without them.6 Moreover, corticosteroids have been implicated in the recurrence of hepatitis C.
We believe that our patient's pathology is most consistent with lung and pleural toxicity temporally related to IFN treatment. Through our case report, we bring to attention this infrequent complication, and emphasize its self‐limited course upon withdrawal of the offending agent.
Acknowledgements
The authors thank Dr. Philippe Camus, Hpital Le Bocage, Dijon, France, for his invaluable suggestions and for reviewing this case report prior to submission.
Case Report
A 52‐year‐old woman with chronic hepatitis C was admitted with complaints of dry cough, shortness of breath, and fever. Four days prior to admission, she had successfully finished a 44‐week course of pegylated interferon (IFN) alpha and ribavirin with undetectable viral load on completion of treatment. At 30 weeks, she had developed a dry cough, which she initially ignored. Three weeks later, as a result of a violent coughing episode, she sustained a spontaneous uncomplicated fracture of the left sixth rib. Chest x‐ray at that time did not show an infiltrate or opacity. She continued treatment, and over the next 6 weeks developed progressive dyspnea on exertion. Five days prior to admission, she had developed fever of 101F. Repeat chest x‐ray revealed a left lingular infiltrate and she was prescribed levofloxacin. Her symptoms failed to improve and she was admitted to the hospital.
On admission, she denied expectoration, sore throat, night sweats, or rashes. She also denied tobacco use, pets at home, or recent travel outside the Midwest. Examination revealed a temperature of 99.4F and decreased breath sounds over the left lower chest. Chest x‐ray revealed left‐sided pleural effusion. D‐dimer was negative. Computed tomography (CT) scan of the chest showed a left lingular infiltrate, right lower lobe ground‐glass opacity, and a moderately‐sized left pleural effusion. Azithromycin, piperacillin/tazobactam, and vancomycin were empirically started. Over the next 36 hours, she became increasingly tachypneic and short of breath. A diagnostic and therapeutic thoracentesis with aspiration of 800 mL of light‐yellow‐colored fluid brought symptomatic relief. Pleural fluid analysis revealed an exudative effusion with 3.8 gm/dL of protein (serum protein = 6.2 gm/dL), lactic dehydrogenase (LDH) of 998 IU/L (serum LDH = 293 IU/L), and normal adenosine deaminase. The cell count was 362 per mm3 with 37% lymphocytes, 32% macrophages, 26% neutrophils, and 1% eosinophils. There were no atypical or malignant cells. Bacterial, fungal, viral, acid‐fast stains and cultures, and polymerase chain reaction (PCR) for Mycobacterium tuberculosis were all negative. An echocardiogram and plasma B‐type natriuretic peptide were normal.
Serum antinuclear and antineutrophilic cytoplasmic antibodies, Bordetella pertussis PCR, serologies for Mycoplasma, Chlamydia, Coxiella, and urinary antigens for Legionella and Blastomyces were all negative. Bronchoscopy with bronchoalveolar lavage (BAL) was performed on hospital day 5. BAL stains and cultures for bacteria, fungi, acid‐fast organisms, Cytomegalovirus, Herpes simplex virus, Legionella, and Pneumocystis were negative. Cytology revealed mild acute inflammation with macrophage predominance and no malignant cells.
Repeat CT scan of the chest on day 6 showed bilateral ground‐glass infiltrates and persistent left pleural effusion (Figure 1). In the absence of an identifiable cause, the patient was diagnosed with interstitial pneumonitis and pleural effusion secondary to pegylated IFN alpha and ribavirin. Treatment with steroids was considered, but was not used due to recent successful suppression of hepatitis C. She was discharged with continued close follow‐up. Her fever gradually subsided over the next 2 weeks and her cough continued to improve over the next 6 weeks. Follow‐up CT scan of the chest 3 months after discharge showed complete resolution of the left pleural effusion and near‐resolution of the bilateral basal infiltrates.
Discussion
Use of IFN alpha has been associated with multiple forms of lung toxicity, of which interstitial pneumonitis and granulomatous inflammation resembling sarcoidosis are the most common. Unusual forms include isolated nonproductive cough, exacerbation of asthma, organizing pneumonia, pleural effusion, adult respiratory distress syndrome, and exacerbation of vasculitis.1 Reports of adverse pulmonary effects of ribavirin are sparse, and it has not been implicated as a sole etiologic agent in causing lung toxicity. It is therefore likely that pulmonary toxicity observed in patients with hepatitis C virus (HCV) infection undergoing IFN alpha and ribavirin therapy is due to the IFN.
Pleural effusion may accompany the IFN‐induced capillary leak syndrome.2
There have been only 2 other cases of pleural effusion during treatment with IFN alpha described to date.3, 4 Takeda et al.3 described a 54‐year‐old male who was accidentally detected to have a moderate‐sized right pleural effusion on magnetic resonance imaging (MRI) of the abdomen, 14 days after therapy with recombinant IFN alpha was initiated. The pleural fluid was a lymphocyte‐predominant exudate and resolved approximately 4 months after discontinuation of IFN treatment. Tsushima et al.4 reported bilateral pleural effusions and ground‐glass opacities in a patient treated with IFN for metastatic renal cell cancer that resolved following a course of steroids.
IFN‐related pulmonary toxicity has been reported to typically develop between 2 and 16 weeks of treatment. Our patient had a delayed onset of symptoms at 30 weeks and progressed on to develop left pleural effusion and pulmonary infiltrates by the time she finished 44 weeks of treatment. We ruled out infectious, malignant, cardiac, and autoimmune causes, which often present in a similar fashion.
BAL fluid cytology in our patient revealed predominant macrophages. Yamaguchi et al., in their analysis of BAL fluid in patients with hepatitis C, demonstrated increased macrophages (76% and 77.5%) and lymphocytes (19.8% and 18.8%) before and after treatment with IFN alpha, respectively.
The cornerstone of management of lung toxicity due to IFN is to diminish or stop use of the offending agent. Our patient demonstrated complete recovery of symptoms and radiological resolution within 3 months of completion of IFN therapy, without corticosteroid therapy. Although corticosteroid regimes of 6 to 12 months have been used to manage IFN related lung toxicity, most patients recover without them.6 Moreover, corticosteroids have been implicated in the recurrence of hepatitis C.
We believe that our patient's pathology is most consistent with lung and pleural toxicity temporally related to IFN treatment. Through our case report, we bring to attention this infrequent complication, and emphasize its self‐limited course upon withdrawal of the offending agent.
Acknowledgements
The authors thank Dr. Philippe Camus, Hpital Le Bocage, Dijon, France, for his invaluable suggestions and for reviewing this case report prior to submission.
- ,.Groupe d'Etudes de la Pathologie Pulmonaire Iatrogène (GEPPI). Pneumotox Online. The drug‐induced lung diseases. Available at: http://www.pneumotox.com. Accessed February 2009.
- ,,, et al.Fatality and interferon alpha for malignant melanoma.Lancet.1998;352(9138):1443–1444.
- ,,, et al.Pleural effusion during interferon treatment for chronic hepatitis C.Hepatogastroenterology.2000;47(35):1431–1435.
- ,,, et al.A case of renal cell carcinoma complicated with interstitial pneumonitis, complete A‐V block and pleural effusion during interferon‐alpha therapy.Nihon Kokyuki Gakkai Zasshi.2001;39:893–898.
- ,,, et al.Analysis of bronchoalveolar lavage fluid of patients with chronic hepatitis c before and after treatment with interferon alpha.Thorax.1997;52:33–37.
- ,,, et al.Spectrum of pulmonary toxicity associated with the use of interferon therapy for hepatitis C: case report and review of the literature.Clin Infect Dis.2004;39:1724–1729.
- ,.Groupe d'Etudes de la Pathologie Pulmonaire Iatrogène (GEPPI). Pneumotox Online. The drug‐induced lung diseases. Available at: http://www.pneumotox.com. Accessed February 2009.
- ,,, et al.Fatality and interferon alpha for malignant melanoma.Lancet.1998;352(9138):1443–1444.
- ,,, et al.Pleural effusion during interferon treatment for chronic hepatitis C.Hepatogastroenterology.2000;47(35):1431–1435.
- ,,, et al.A case of renal cell carcinoma complicated with interstitial pneumonitis, complete A‐V block and pleural effusion during interferon‐alpha therapy.Nihon Kokyuki Gakkai Zasshi.2001;39:893–898.
- ,,, et al.Analysis of bronchoalveolar lavage fluid of patients with chronic hepatitis c before and after treatment with interferon alpha.Thorax.1997;52:33–37.
- ,,, et al.Spectrum of pulmonary toxicity associated with the use of interferon therapy for hepatitis C: case report and review of the literature.Clin Infect Dis.2004;39:1724–1729.