User login
Hypertrophic Lichen Planus–like Eruption Following Pembrolizumab
To the Editor:
Pembrolizumab, a humanized monoclonal anti–programmed cell death protein 1 (PD-1) antibody, acts by blocking negative immune regulators such as PD-1.1 Since its approval by the US Food and Drug Administration in 2014, the use of PD-1 inhibitors such as pembrolizumab has dramatically increased, and they are now the standard of care for cancers such as melanoma, lung cancer, and renal cell carcinoma.2,3 With increased use comes a better understanding of the cutaneous adverse effects that may occur. To date, almost 50% of patients treated with PD-1 inhibitors will develop an adverse cutaneous reaction.4 Thus far, cases of patients developing vitiligo, bullous pemphigoid, psoriasis, granulomatous skin reactions, severe cutaneous reactions (ie, toxic epidermal necrolysis), lupus erythematosus, and lichenoid reactions have been described.3,5,6 There are fewer than 30 documented cases of lichenoid reactions due to anti–PD-1 treatment described in the literature, increasing the importance of case reports to demonstrate a full range of cutaneous findings.3 We present a case of a reaction to pembrolizumab with an eruption of lichenoid papules predominantly involving the hands and feet as well as nail changes.
A 60-year-old man with ocular melanoma metastatic to the right lung, transverse colon, and right axillary lymph nodes presented with a chief concern of growing skin lesions present for 6 weeks on the hands and feet. The lesions were tender to the touch and occasionally drained a clear fluid. He also reported nail fragility. Of note, the patient was being treated for metastatic melanoma with pembrolizumab infusions every 3 weeks, which started 6 weeks prior to the onset of the eruption.
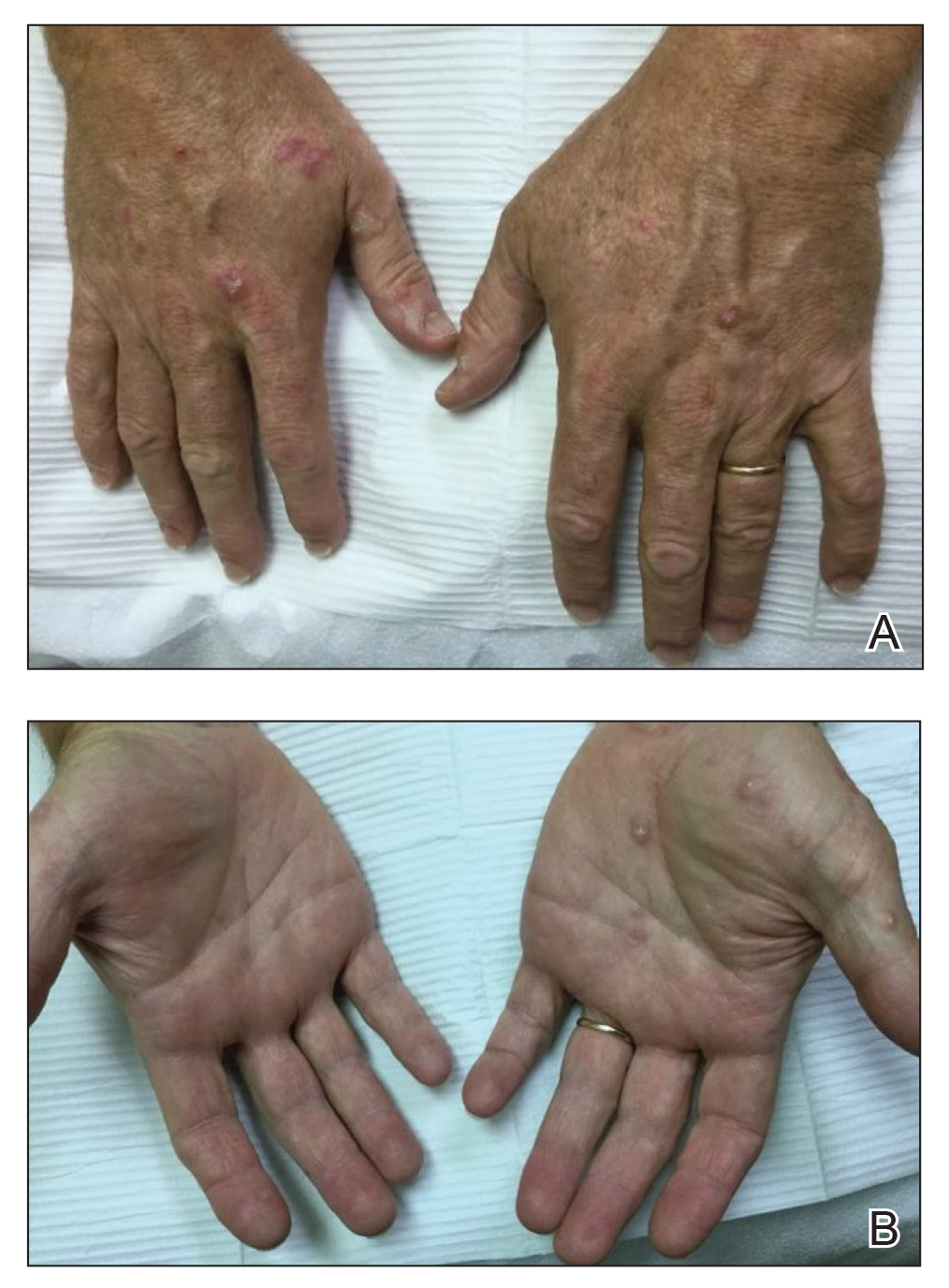
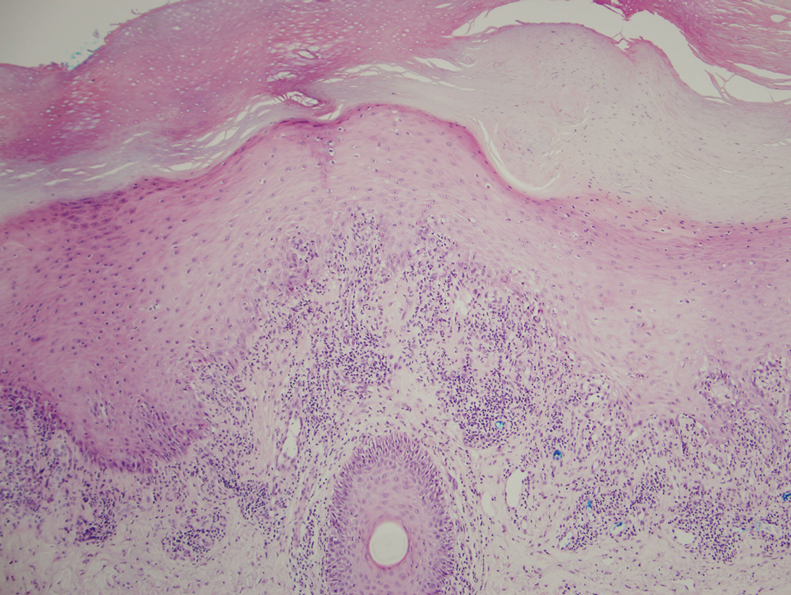
Physical examination demonstrated lichenoid papules on the dorsal and ventral aspects of the hands and feet (Figure 1), as well as longitudinal ridging on numerous fingernails and mild koilonychia. A punch biopsy revealed lichenoid interface dermatitis with irregular epidermal hyperplasia (Figure 2). A diagnosis of hypertrophic lichen planus–like drug eruption in response to pembrolizumab was made and clobetasol cream was prescribed.
At 1-month follow-up, the patient reported notable improvement with clobetasol, and he was transitioned to tacrolimus ointment 0.1%. He continued to improve until a month later when he reported new lesions arising a week after a pembrolizumab infusion. He continued to use clobetasol cream for flares and tacrolimus ointment for maintenance.
Almost 3 months after the initial visit, the patient presented with inflammation around his right third fingernail of 1 week’s duration, with more notable fragility than his other nails. No trauma was described, and the nail abnormality was attributed to pembrolizumab. Clobetasol cream and biotin 3 mg daily resulted in improvement, and no other nails were affected in a similar way.
Programmed cell death protein 1 blockers are associated with a variety of adverse events including hypothyroidism, gastrointestinal abnormalities, fatigue, and skin disorders.7 In one study (N=83), cutaneous adverse drug events were found to occur in 42% (35/83) of patients following pembrolizumab therapy, with the most common cutaneous lesions being maculopapular eruptions (29% [24/83]), pruritus (12% [10/83]), and hypopigmentation (8% [7/83]).5
A total of 29 cases of lichenoid dermatitis following anti–PD-1 therapy have been described in the literature.3 Cases range from an eruption of photodistributed hyperkeratotic papules and plaques to hypertrophic vesiculobullous lesions.3,6 Suggested pathophysiology involves blocking the interaction of programmed death ligand 1 on keratinocytes with PD-1 on T cells.3 Management typically includes topical or systemic steroids. Cyclosporine and acitretin also have been successful in a small number of patients. Most patients continue anti–PD-1 treatment with systemic therapy.3
Our patient represents a similar lichenoid eruption; however, the distribution on the dorsal and ventral aspects of the hands and feet as well as nail dystrophy make the presentation unique. Anticancer drugs that increase the T-cell immune response by altering the complex signaling among T cells, antigen-presenting cells, and tumor cells have been associated with cutaneous eruptions. Although the exact mechanism is still not fully understood, clinical suspicion of a pembrolizumab reaction should remain high on the differential in the setting of hyperkeratotic papules in association with anti–PD-1 therapy.
- Homet Moreno B, Ribas A. Anti-programmed cell death protein-1/ligand-1 therapy in different cancers. Br J Cancer. 2015;112:1421-1427.
- Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109-1117.
- Simonsen AB, Kaae J, Elleback E, et al. Cutaneous adverse reactions to anti-PD-1 treatment: a systematic review. J Am Acad Dermatol. 2020;83:1415-1424.
- Hwang SJ, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort. J Am Acad Dermatol. 2016;74:455-461.
- Sanlorenzo M, Vujic I, Daud A, et al. Pembrolizumab cutaneous adverse events and their association with disease progression. JAMA Dermatol. 2015;151:1206-1212.
- Joseph RW, Cappel M, Goedjen B, et al. Lichenoid dermatitis in three patients with metastatic melanoma treated with anti-PD-1 therapy. Cancer Immunol Res. 2015;3:18-22.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134-144.
To the Editor:
Pembrolizumab, a humanized monoclonal anti–programmed cell death protein 1 (PD-1) antibody, acts by blocking negative immune regulators such as PD-1.1 Since its approval by the US Food and Drug Administration in 2014, the use of PD-1 inhibitors such as pembrolizumab has dramatically increased, and they are now the standard of care for cancers such as melanoma, lung cancer, and renal cell carcinoma.2,3 With increased use comes a better understanding of the cutaneous adverse effects that may occur. To date, almost 50% of patients treated with PD-1 inhibitors will develop an adverse cutaneous reaction.4 Thus far, cases of patients developing vitiligo, bullous pemphigoid, psoriasis, granulomatous skin reactions, severe cutaneous reactions (ie, toxic epidermal necrolysis), lupus erythematosus, and lichenoid reactions have been described.3,5,6 There are fewer than 30 documented cases of lichenoid reactions due to anti–PD-1 treatment described in the literature, increasing the importance of case reports to demonstrate a full range of cutaneous findings.3 We present a case of a reaction to pembrolizumab with an eruption of lichenoid papules predominantly involving the hands and feet as well as nail changes.
A 60-year-old man with ocular melanoma metastatic to the right lung, transverse colon, and right axillary lymph nodes presented with a chief concern of growing skin lesions present for 6 weeks on the hands and feet. The lesions were tender to the touch and occasionally drained a clear fluid. He also reported nail fragility. Of note, the patient was being treated for metastatic melanoma with pembrolizumab infusions every 3 weeks, which started 6 weeks prior to the onset of the eruption.
Physical examination demonstrated lichenoid papules on the dorsal and ventral aspects of the hands and feet (Figure 1), as well as longitudinal ridging on numerous fingernails and mild koilonychia. A punch biopsy revealed lichenoid interface dermatitis with irregular epidermal hyperplasia (Figure 2). A diagnosis of hypertrophic lichen planus–like drug eruption in response to pembrolizumab was made and clobetasol cream was prescribed.
At 1-month follow-up, the patient reported notable improvement with clobetasol, and he was transitioned to tacrolimus ointment 0.1%. He continued to improve until a month later when he reported new lesions arising a week after a pembrolizumab infusion. He continued to use clobetasol cream for flares and tacrolimus ointment for maintenance.
Almost 3 months after the initial visit, the patient presented with inflammation around his right third fingernail of 1 week’s duration, with more notable fragility than his other nails. No trauma was described, and the nail abnormality was attributed to pembrolizumab. Clobetasol cream and biotin 3 mg daily resulted in improvement, and no other nails were affected in a similar way.
Programmed cell death protein 1 blockers are associated with a variety of adverse events including hypothyroidism, gastrointestinal abnormalities, fatigue, and skin disorders.7 In one study (N=83), cutaneous adverse drug events were found to occur in 42% (35/83) of patients following pembrolizumab therapy, with the most common cutaneous lesions being maculopapular eruptions (29% [24/83]), pruritus (12% [10/83]), and hypopigmentation (8% [7/83]).5
A total of 29 cases of lichenoid dermatitis following anti–PD-1 therapy have been described in the literature.3 Cases range from an eruption of photodistributed hyperkeratotic papules and plaques to hypertrophic vesiculobullous lesions.3,6 Suggested pathophysiology involves blocking the interaction of programmed death ligand 1 on keratinocytes with PD-1 on T cells.3 Management typically includes topical or systemic steroids. Cyclosporine and acitretin also have been successful in a small number of patients. Most patients continue anti–PD-1 treatment with systemic therapy.3
Our patient represents a similar lichenoid eruption; however, the distribution on the dorsal and ventral aspects of the hands and feet as well as nail dystrophy make the presentation unique. Anticancer drugs that increase the T-cell immune response by altering the complex signaling among T cells, antigen-presenting cells, and tumor cells have been associated with cutaneous eruptions. Although the exact mechanism is still not fully understood, clinical suspicion of a pembrolizumab reaction should remain high on the differential in the setting of hyperkeratotic papules in association with anti–PD-1 therapy.
To the Editor:
Pembrolizumab, a humanized monoclonal anti–programmed cell death protein 1 (PD-1) antibody, acts by blocking negative immune regulators such as PD-1.1 Since its approval by the US Food and Drug Administration in 2014, the use of PD-1 inhibitors such as pembrolizumab has dramatically increased, and they are now the standard of care for cancers such as melanoma, lung cancer, and renal cell carcinoma.2,3 With increased use comes a better understanding of the cutaneous adverse effects that may occur. To date, almost 50% of patients treated with PD-1 inhibitors will develop an adverse cutaneous reaction.4 Thus far, cases of patients developing vitiligo, bullous pemphigoid, psoriasis, granulomatous skin reactions, severe cutaneous reactions (ie, toxic epidermal necrolysis), lupus erythematosus, and lichenoid reactions have been described.3,5,6 There are fewer than 30 documented cases of lichenoid reactions due to anti–PD-1 treatment described in the literature, increasing the importance of case reports to demonstrate a full range of cutaneous findings.3 We present a case of a reaction to pembrolizumab with an eruption of lichenoid papules predominantly involving the hands and feet as well as nail changes.
A 60-year-old man with ocular melanoma metastatic to the right lung, transverse colon, and right axillary lymph nodes presented with a chief concern of growing skin lesions present for 6 weeks on the hands and feet. The lesions were tender to the touch and occasionally drained a clear fluid. He also reported nail fragility. Of note, the patient was being treated for metastatic melanoma with pembrolizumab infusions every 3 weeks, which started 6 weeks prior to the onset of the eruption.
Physical examination demonstrated lichenoid papules on the dorsal and ventral aspects of the hands and feet (Figure 1), as well as longitudinal ridging on numerous fingernails and mild koilonychia. A punch biopsy revealed lichenoid interface dermatitis with irregular epidermal hyperplasia (Figure 2). A diagnosis of hypertrophic lichen planus–like drug eruption in response to pembrolizumab was made and clobetasol cream was prescribed.
At 1-month follow-up, the patient reported notable improvement with clobetasol, and he was transitioned to tacrolimus ointment 0.1%. He continued to improve until a month later when he reported new lesions arising a week after a pembrolizumab infusion. He continued to use clobetasol cream for flares and tacrolimus ointment for maintenance.
Almost 3 months after the initial visit, the patient presented with inflammation around his right third fingernail of 1 week’s duration, with more notable fragility than his other nails. No trauma was described, and the nail abnormality was attributed to pembrolizumab. Clobetasol cream and biotin 3 mg daily resulted in improvement, and no other nails were affected in a similar way.
Programmed cell death protein 1 blockers are associated with a variety of adverse events including hypothyroidism, gastrointestinal abnormalities, fatigue, and skin disorders.7 In one study (N=83), cutaneous adverse drug events were found to occur in 42% (35/83) of patients following pembrolizumab therapy, with the most common cutaneous lesions being maculopapular eruptions (29% [24/83]), pruritus (12% [10/83]), and hypopigmentation (8% [7/83]).5
A total of 29 cases of lichenoid dermatitis following anti–PD-1 therapy have been described in the literature.3 Cases range from an eruption of photodistributed hyperkeratotic papules and plaques to hypertrophic vesiculobullous lesions.3,6 Suggested pathophysiology involves blocking the interaction of programmed death ligand 1 on keratinocytes with PD-1 on T cells.3 Management typically includes topical or systemic steroids. Cyclosporine and acitretin also have been successful in a small number of patients. Most patients continue anti–PD-1 treatment with systemic therapy.3
Our patient represents a similar lichenoid eruption; however, the distribution on the dorsal and ventral aspects of the hands and feet as well as nail dystrophy make the presentation unique. Anticancer drugs that increase the T-cell immune response by altering the complex signaling among T cells, antigen-presenting cells, and tumor cells have been associated with cutaneous eruptions. Although the exact mechanism is still not fully understood, clinical suspicion of a pembrolizumab reaction should remain high on the differential in the setting of hyperkeratotic papules in association with anti–PD-1 therapy.
- Homet Moreno B, Ribas A. Anti-programmed cell death protein-1/ligand-1 therapy in different cancers. Br J Cancer. 2015;112:1421-1427.
- Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109-1117.
- Simonsen AB, Kaae J, Elleback E, et al. Cutaneous adverse reactions to anti-PD-1 treatment: a systematic review. J Am Acad Dermatol. 2020;83:1415-1424.
- Hwang SJ, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort. J Am Acad Dermatol. 2016;74:455-461.
- Sanlorenzo M, Vujic I, Daud A, et al. Pembrolizumab cutaneous adverse events and their association with disease progression. JAMA Dermatol. 2015;151:1206-1212.
- Joseph RW, Cappel M, Goedjen B, et al. Lichenoid dermatitis in three patients with metastatic melanoma treated with anti-PD-1 therapy. Cancer Immunol Res. 2015;3:18-22.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134-144.
- Homet Moreno B, Ribas A. Anti-programmed cell death protein-1/ligand-1 therapy in different cancers. Br J Cancer. 2015;112:1421-1427.
- Robert C, Ribas A, Wolchok JD, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109-1117.
- Simonsen AB, Kaae J, Elleback E, et al. Cutaneous adverse reactions to anti-PD-1 treatment: a systematic review. J Am Acad Dermatol. 2020;83:1415-1424.
- Hwang SJ, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort. J Am Acad Dermatol. 2016;74:455-461.
- Sanlorenzo M, Vujic I, Daud A, et al. Pembrolizumab cutaneous adverse events and their association with disease progression. JAMA Dermatol. 2015;151:1206-1212.
- Joseph RW, Cappel M, Goedjen B, et al. Lichenoid dermatitis in three patients with metastatic melanoma treated with anti-PD-1 therapy. Cancer Immunol Res. 2015;3:18-22.
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134-144.
Practice Points
- With an increased use of immunotherapy medications such as pembrolizumab for various cancers, it is important that dermatologists are aware of the wide range of adverse cutaneous reactions that can occur, including lichenoid reactions.
- Hypertrophic lichen planus should be considered in the differential diagnosis of patients with cutaneous lesions in addition to nail findings developing after starting programmed cell death protein 1 inhibitor therapy.
Testosterone Pellet–Induced Generalized Drug Eruption
To the Editor:
Testosterone-replacement therapy (TRT) is indicated for hypogonadism. The benefits of TRT are well documented, with multiple options available for delivery. Testosterone pellet implantation (TPI) is an effective treatment option for hypogonadism with minimal adverse reactions. Availability of TRT is increasing, as facilities are offering off-label applications. Although TPI generally is well tolerated, cutaneous reactions have been documented. We present a patient with drug-induced dermatitis following TPI.
A 51-year-old man with hypogonadism presented with an extremely pruritic rash that began on the left buttock 3 days after receiving his fourth TPI. The patient had received subcutaneous insertions of 8 testosterone pellets (75 mg per pellet every 6 months) to the left buttock. He denied any history of a similar rash. His medical history was remarkable for hyperlipidemia, which was controlled with niacin and omega-3 fatty acids (fish oil). Other medications included glucosamine. Before presenting to our clinic, he was given a 40-mg intramuscular injection of triamcinolone acetonide and trimethoprim-sulfamethoxazole twice daily for 7 days, a methylprednisolone dose pack, and triamcinolone ointment 0.1% twice daily by his primary care physician, all without improvement of the rash.
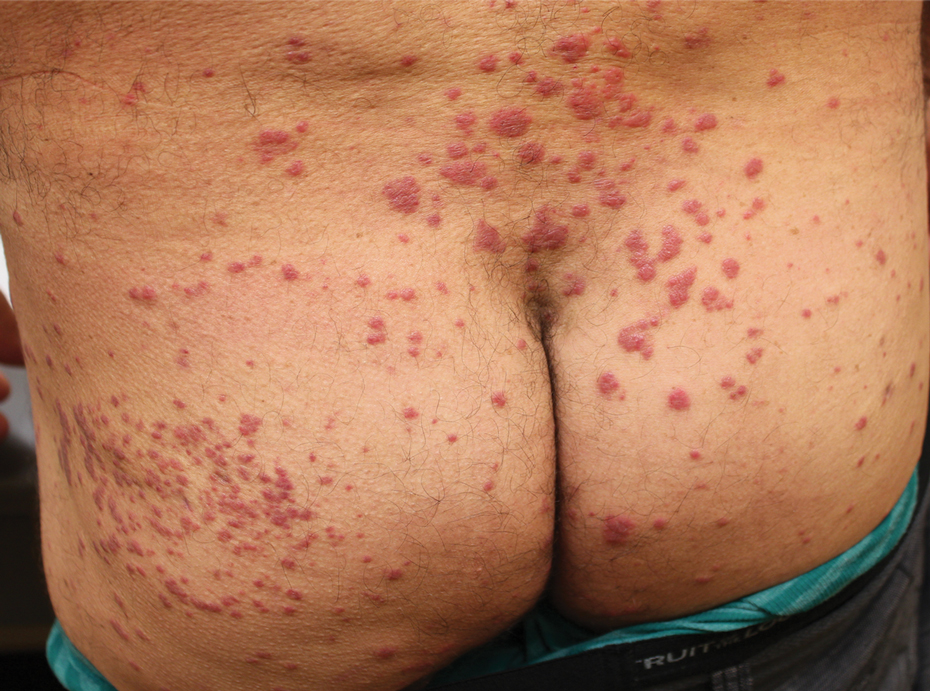
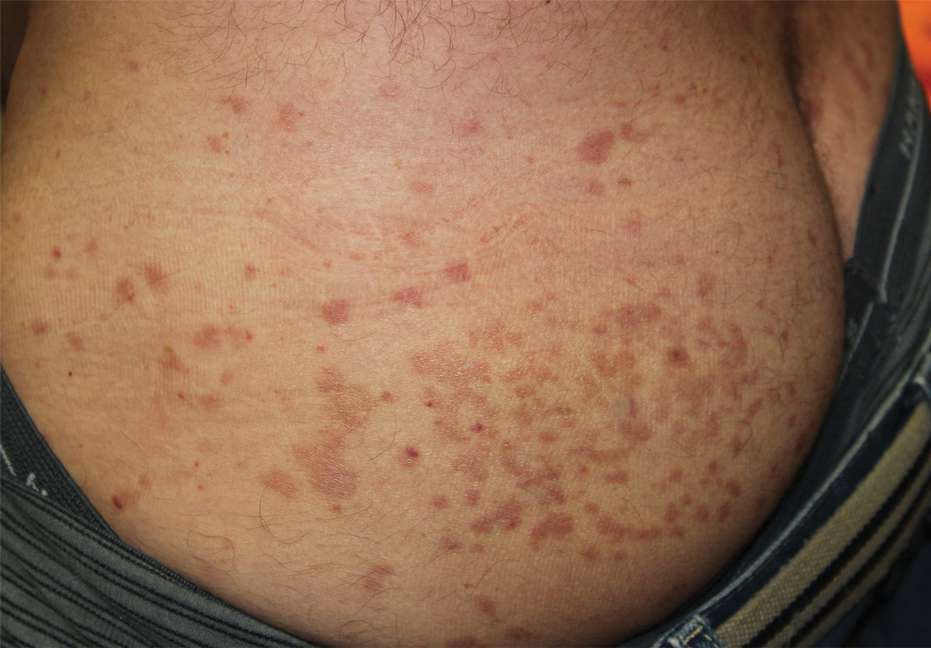
Physical examination revealed multiple well-circumscribed, coalescing clusters of darkly erythematous papules and dermal plaques of varying size on the buttocks with extension to the lower back, abdomen, and thighs (Figure 1). The differential diagnosis included lichenoid eruption, pseudolymphoma, sarcoidosis, and granuloma annulare.
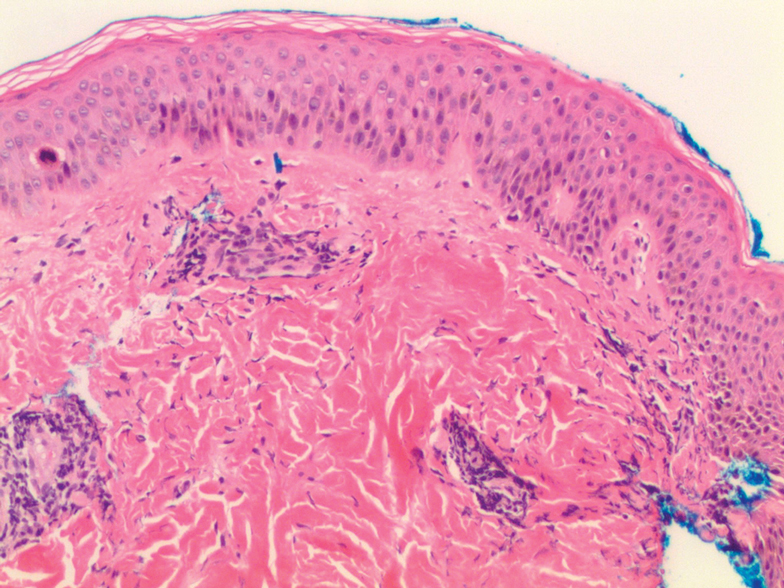
Histologic examination of a punch biopsy revealed an epidermis with a normal stratum corneum and subtle cell-poor vacuolar interface dermatitis with rare necrotic keratinocytes. There was a mild perivascular lymphocytic infiltrate with slight edema within the dermis without notable eosinophils or findings indicative of a vasculitic process (Figure 2).
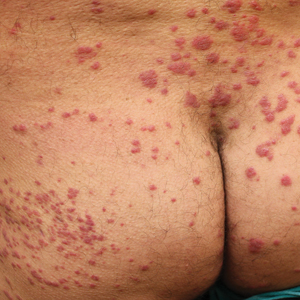
Oral prednisone 60 mg daily and betamethasone ointment 0.05% applied twice daily were started, with notable improvement of the rash in 1 week (Figure 3). Given the temporal relationship of the TPI, histologic findings suggestive of drug eruption, and resolution of symptoms shortly after treatment, a diagnosis of testosterone pellet–induced generalized dermatitis was established.
Testosterone-replacement therapy is the principal treatment of male pathologic hypoandrogenism, but off-label prescription frequently occurs for age-related hypogonadism and hypoactive sexual desire disorder.1 Testosterone-replacement therapy also can enhance sexual desire and function and improve mood in premenopausal and postmenopausal women with testosterone deficiency.2 Delivery options include topicals, intramuscular injections, oral formulations, transdermal patches and gels, and subcutaneous placement of testosterone pellets (TPI).Cutaneous reactions to TPI are rare. Hirsutism, male-pattern hair loss, and acne are possible cutaneous adverse reactions.3 In addition, a localized erythematous pruritic eruption at the implantation site and an immunologic foreign-body reaction to testosterone pellets have been reported.4
In one case report, a man developed recurrent ill-defined, erythematous, scaly plaques and patches over the buttocks and thighs, consistent with testosterone-induced eczematous dermatitis, subsequent to his second TPI. The patient presented with the eruption within 4 weeks after the most recent implantation, similar to our case, but differed temporally in initial presentation, presenting after the second implantation.5 Our case differed in morphologic presentation (dermal plaques as opposed to eczematous change) and refractoriness to triamcinolone injection.
Testosterone-replacement therapy is becoming more widely available. Lack of regulation of proper marketing by such facilities as medical spas that offer TPI for off-label applications has led to a rampant increase in TRT prescribing, possibly foreshadowing an increase in adverse cutaneous reactions to TRT.6
Our case of histologically consistent testosterone pellet–induced dermatitis highlights a rare cutaneous adverse reaction that can occur subsequent to TPI and illustrates the efficacy of high-dose oral steroids as a treatment option. With increased use of TRT, physicians should be cognizant of the potential adverse cutaneous effects related to this treatment and counsel patients appropriately prior to initiating treatment.
Acknowledgment
We thank the patient for granting permission to publish this case.
- Clayton AH, Kingsberg SA, Goldstein I. Evaluation and management of hypoactive sexual desire disorder. Sex Med. 2018;6:59-74.
- Glaser R, Dimitrakakis C. Testosterone therapy in women: myths and misconceptions. Maturitas. 2013;74:230-234.
- Testopel (testosterone pellet) [package insert]. Endo Pharmaceuticals, Inc; 2016. Accessed December 16, 2020. https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=a1741a0b-3d4c-42dc-880d-a06e96cce9ef&type=display
- Cavender RK, Fairall M. Subcutaneous testosterone pellet implant (Testopel) therapy for men with testosterone deficiency syndrome: a single-site retrospective safety analysis. J Sex Med. 2009;6:3177-3192.
- Heldt Manica LA, Cohen PR. Testosterone pellet associated dermatitis: report and review of Testopel-related cutaneous adverse effects. Cureus. 2017;9:e1560.
- Mintzes B. The marketing of testosterone treatments for age-related low testosterone or ‘Low T’. Curr Opin Endocrinol Diabetes Obes. 2018;25:224-230.
To the Editor:
Testosterone-replacement therapy (TRT) is indicated for hypogonadism. The benefits of TRT are well documented, with multiple options available for delivery. Testosterone pellet implantation (TPI) is an effective treatment option for hypogonadism with minimal adverse reactions. Availability of TRT is increasing, as facilities are offering off-label applications. Although TPI generally is well tolerated, cutaneous reactions have been documented. We present a patient with drug-induced dermatitis following TPI.
A 51-year-old man with hypogonadism presented with an extremely pruritic rash that began on the left buttock 3 days after receiving his fourth TPI. The patient had received subcutaneous insertions of 8 testosterone pellets (75 mg per pellet every 6 months) to the left buttock. He denied any history of a similar rash. His medical history was remarkable for hyperlipidemia, which was controlled with niacin and omega-3 fatty acids (fish oil). Other medications included glucosamine. Before presenting to our clinic, he was given a 40-mg intramuscular injection of triamcinolone acetonide and trimethoprim-sulfamethoxazole twice daily for 7 days, a methylprednisolone dose pack, and triamcinolone ointment 0.1% twice daily by his primary care physician, all without improvement of the rash.
Physical examination revealed multiple well-circumscribed, coalescing clusters of darkly erythematous papules and dermal plaques of varying size on the buttocks with extension to the lower back, abdomen, and thighs (Figure 1). The differential diagnosis included lichenoid eruption, pseudolymphoma, sarcoidosis, and granuloma annulare.
Histologic examination of a punch biopsy revealed an epidermis with a normal stratum corneum and subtle cell-poor vacuolar interface dermatitis with rare necrotic keratinocytes. There was a mild perivascular lymphocytic infiltrate with slight edema within the dermis without notable eosinophils or findings indicative of a vasculitic process (Figure 2).
Oral prednisone 60 mg daily and betamethasone ointment 0.05% applied twice daily were started, with notable improvement of the rash in 1 week (Figure 3). Given the temporal relationship of the TPI, histologic findings suggestive of drug eruption, and resolution of symptoms shortly after treatment, a diagnosis of testosterone pellet–induced generalized dermatitis was established.
Testosterone-replacement therapy is the principal treatment of male pathologic hypoandrogenism, but off-label prescription frequently occurs for age-related hypogonadism and hypoactive sexual desire disorder.1 Testosterone-replacement therapy also can enhance sexual desire and function and improve mood in premenopausal and postmenopausal women with testosterone deficiency.2 Delivery options include topicals, intramuscular injections, oral formulations, transdermal patches and gels, and subcutaneous placement of testosterone pellets (TPI).Cutaneous reactions to TPI are rare. Hirsutism, male-pattern hair loss, and acne are possible cutaneous adverse reactions.3 In addition, a localized erythematous pruritic eruption at the implantation site and an immunologic foreign-body reaction to testosterone pellets have been reported.4
In one case report, a man developed recurrent ill-defined, erythematous, scaly plaques and patches over the buttocks and thighs, consistent with testosterone-induced eczematous dermatitis, subsequent to his second TPI. The patient presented with the eruption within 4 weeks after the most recent implantation, similar to our case, but differed temporally in initial presentation, presenting after the second implantation.5 Our case differed in morphologic presentation (dermal plaques as opposed to eczematous change) and refractoriness to triamcinolone injection.
Testosterone-replacement therapy is becoming more widely available. Lack of regulation of proper marketing by such facilities as medical spas that offer TPI for off-label applications has led to a rampant increase in TRT prescribing, possibly foreshadowing an increase in adverse cutaneous reactions to TRT.6
Our case of histologically consistent testosterone pellet–induced dermatitis highlights a rare cutaneous adverse reaction that can occur subsequent to TPI and illustrates the efficacy of high-dose oral steroids as a treatment option. With increased use of TRT, physicians should be cognizant of the potential adverse cutaneous effects related to this treatment and counsel patients appropriately prior to initiating treatment.
Acknowledgment
We thank the patient for granting permission to publish this case.
To the Editor:
Testosterone-replacement therapy (TRT) is indicated for hypogonadism. The benefits of TRT are well documented, with multiple options available for delivery. Testosterone pellet implantation (TPI) is an effective treatment option for hypogonadism with minimal adverse reactions. Availability of TRT is increasing, as facilities are offering off-label applications. Although TPI generally is well tolerated, cutaneous reactions have been documented. We present a patient with drug-induced dermatitis following TPI.
A 51-year-old man with hypogonadism presented with an extremely pruritic rash that began on the left buttock 3 days after receiving his fourth TPI. The patient had received subcutaneous insertions of 8 testosterone pellets (75 mg per pellet every 6 months) to the left buttock. He denied any history of a similar rash. His medical history was remarkable for hyperlipidemia, which was controlled with niacin and omega-3 fatty acids (fish oil). Other medications included glucosamine. Before presenting to our clinic, he was given a 40-mg intramuscular injection of triamcinolone acetonide and trimethoprim-sulfamethoxazole twice daily for 7 days, a methylprednisolone dose pack, and triamcinolone ointment 0.1% twice daily by his primary care physician, all without improvement of the rash.
Physical examination revealed multiple well-circumscribed, coalescing clusters of darkly erythematous papules and dermal plaques of varying size on the buttocks with extension to the lower back, abdomen, and thighs (Figure 1). The differential diagnosis included lichenoid eruption, pseudolymphoma, sarcoidosis, and granuloma annulare.
Histologic examination of a punch biopsy revealed an epidermis with a normal stratum corneum and subtle cell-poor vacuolar interface dermatitis with rare necrotic keratinocytes. There was a mild perivascular lymphocytic infiltrate with slight edema within the dermis without notable eosinophils or findings indicative of a vasculitic process (Figure 2).
Oral prednisone 60 mg daily and betamethasone ointment 0.05% applied twice daily were started, with notable improvement of the rash in 1 week (Figure 3). Given the temporal relationship of the TPI, histologic findings suggestive of drug eruption, and resolution of symptoms shortly after treatment, a diagnosis of testosterone pellet–induced generalized dermatitis was established.
Testosterone-replacement therapy is the principal treatment of male pathologic hypoandrogenism, but off-label prescription frequently occurs for age-related hypogonadism and hypoactive sexual desire disorder.1 Testosterone-replacement therapy also can enhance sexual desire and function and improve mood in premenopausal and postmenopausal women with testosterone deficiency.2 Delivery options include topicals, intramuscular injections, oral formulations, transdermal patches and gels, and subcutaneous placement of testosterone pellets (TPI).Cutaneous reactions to TPI are rare. Hirsutism, male-pattern hair loss, and acne are possible cutaneous adverse reactions.3 In addition, a localized erythematous pruritic eruption at the implantation site and an immunologic foreign-body reaction to testosterone pellets have been reported.4
In one case report, a man developed recurrent ill-defined, erythematous, scaly plaques and patches over the buttocks and thighs, consistent with testosterone-induced eczematous dermatitis, subsequent to his second TPI. The patient presented with the eruption within 4 weeks after the most recent implantation, similar to our case, but differed temporally in initial presentation, presenting after the second implantation.5 Our case differed in morphologic presentation (dermal plaques as opposed to eczematous change) and refractoriness to triamcinolone injection.
Testosterone-replacement therapy is becoming more widely available. Lack of regulation of proper marketing by such facilities as medical spas that offer TPI for off-label applications has led to a rampant increase in TRT prescribing, possibly foreshadowing an increase in adverse cutaneous reactions to TRT.6
Our case of histologically consistent testosterone pellet–induced dermatitis highlights a rare cutaneous adverse reaction that can occur subsequent to TPI and illustrates the efficacy of high-dose oral steroids as a treatment option. With increased use of TRT, physicians should be cognizant of the potential adverse cutaneous effects related to this treatment and counsel patients appropriately prior to initiating treatment.
Acknowledgment
We thank the patient for granting permission to publish this case.
- Clayton AH, Kingsberg SA, Goldstein I. Evaluation and management of hypoactive sexual desire disorder. Sex Med. 2018;6:59-74.
- Glaser R, Dimitrakakis C. Testosterone therapy in women: myths and misconceptions. Maturitas. 2013;74:230-234.
- Testopel (testosterone pellet) [package insert]. Endo Pharmaceuticals, Inc; 2016. Accessed December 16, 2020. https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=a1741a0b-3d4c-42dc-880d-a06e96cce9ef&type=display
- Cavender RK, Fairall M. Subcutaneous testosterone pellet implant (Testopel) therapy for men with testosterone deficiency syndrome: a single-site retrospective safety analysis. J Sex Med. 2009;6:3177-3192.
- Heldt Manica LA, Cohen PR. Testosterone pellet associated dermatitis: report and review of Testopel-related cutaneous adverse effects. Cureus. 2017;9:e1560.
- Mintzes B. The marketing of testosterone treatments for age-related low testosterone or ‘Low T’. Curr Opin Endocrinol Diabetes Obes. 2018;25:224-230.
- Clayton AH, Kingsberg SA, Goldstein I. Evaluation and management of hypoactive sexual desire disorder. Sex Med. 2018;6:59-74.
- Glaser R, Dimitrakakis C. Testosterone therapy in women: myths and misconceptions. Maturitas. 2013;74:230-234.
- Testopel (testosterone pellet) [package insert]. Endo Pharmaceuticals, Inc; 2016. Accessed December 16, 2020. https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=a1741a0b-3d4c-42dc-880d-a06e96cce9ef&type=display
- Cavender RK, Fairall M. Subcutaneous testosterone pellet implant (Testopel) therapy for men with testosterone deficiency syndrome: a single-site retrospective safety analysis. J Sex Med. 2009;6:3177-3192.
- Heldt Manica LA, Cohen PR. Testosterone pellet associated dermatitis: report and review of Testopel-related cutaneous adverse effects. Cureus. 2017;9:e1560.
- Mintzes B. The marketing of testosterone treatments for age-related low testosterone or ‘Low T’. Curr Opin Endocrinol Diabetes Obes. 2018;25:224-230.
Practice Points
- Dermatologists should be aware that testosterone pellet implantation can cause dermatitis overlying the implantation site, which can generalize and differ in morphologic presentation.
- For patients presenting with a suspected case of testosterone pellet–induced dermatitis, a high-dose oral corticosteroid can be deployed as an effective therapy.
High-Grade Ovarian Serous Carcinoma Presenting as Androgenetic Alopecia
To the Editor:
Female pattern hair loss is common, and the literature suggests that up to 56% of women experience hair thinning in their lifetime, with increased prevalence in older women.1 Pathophysiology is incompletely understood and involves the nonscarring progressive miniaturization of hair follicles, causing decreased production of terminal hairs relative to more delicate vellus hairs. Because vellus hairs have a shorter anagen growth phase than terminal hairs, hair loss is expedited. Androgen excess, when present, hastens the process by inducing early transition of hair follicles from the anagen phase to the senescent telogen phase. Serum testosterone levels are within reference range in most female patients with hair loss, suggesting the presence of additional contributing factors.2
Given the high prevalence of female pattern hair loss and the harm of overlooking androgen excess and an androgen-secreting neoplasm, dermatologists must recognize indications for further evaluation. Additional signs of hyperandrogenism, such as menstrual irregularities, acne, hirsutism, anabolic appearance, voice deepening, and clitoromegaly, are reasons for concern.3 Elevated serum androgen levels also should raise suspicion of malignancy. Historically, a total testosterone level above 200 ng/dL or a dehydroepiandrosterone sulfate (DHEA-S) level greater than 700 µg/dL prompted evaluation for a tumor.4 More recent studies show that tumor-induced increases in serum androgen levels are highly variable, challenging the utility of these cutoffs.5
A 70-year-old woman presented with hair loss over the last 12 years with accentuated thinning on the frontal and vertex scalp. The patient’s primary care physician previously made a diagnosis of androgenetic alopecia and recommended topical minoxidil. Although the patient had a history of excess facial and body hair since young adulthood, she noted a progressive increase in the density of chest and back hair, prominent coarsening of the texture of the facial and body hair, and new facial acne in the last 3 years. Prior to these changes, the density and texture of the scalp and body hair had been stable for many years.
Although other postmenopausal females in the patient’s family displayed patterned hair loss, they did not possess coarse and dense hair on the face and trunk. Her family history was notable for ovarian cancer in her mother (in her 70s) and breast cancer in her maternal grandmother (in her 80s).
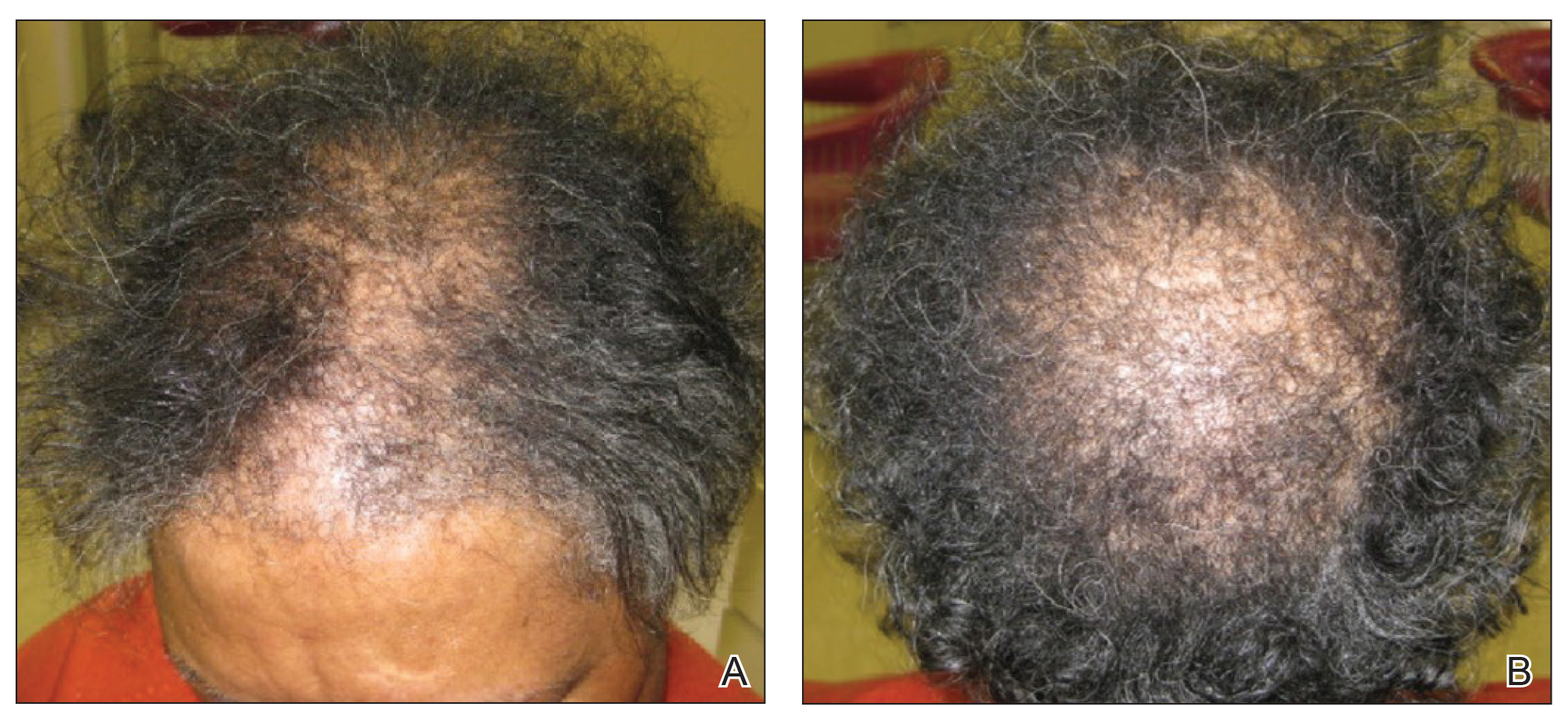
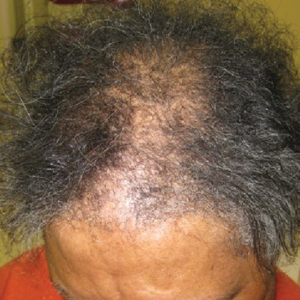
A review of systems was notable only for decreased energy. Physical examination revealed a well-appearing older woman with coarse terminal hair growth on the cheeks, submental chin, neck, chest, back, and forearms. Scalp examination indicated diffusely decreased hair density, most marked over the vertex, crown, and frontal scalp, without scale, erythema, or loss of follicular ostia (Figure 1).
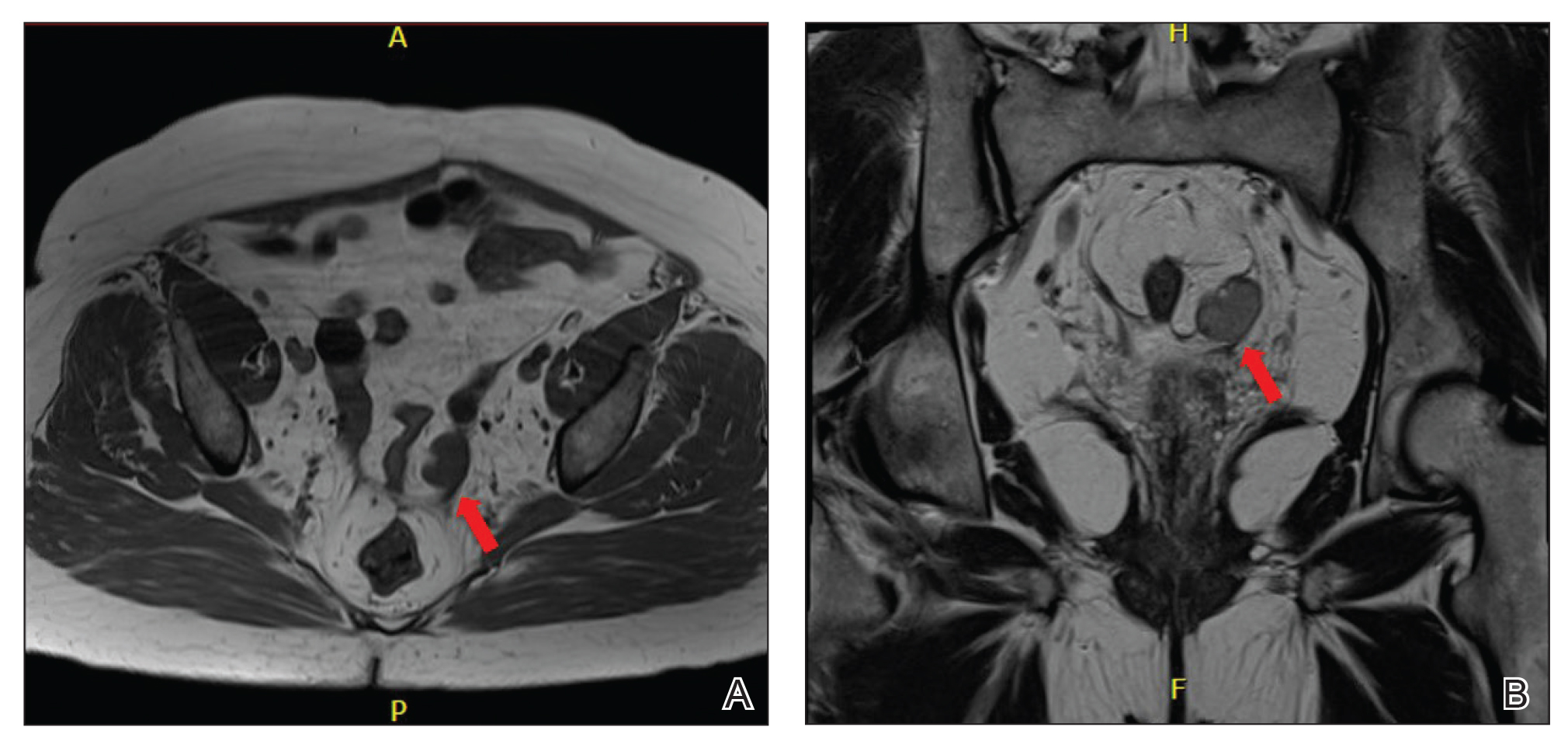
Laboratory evaluation revealed elevated levels of total testosterone (106 ng/dL [reference range, <40 ng/dL]) and free testosterone (32.9 pg/mL [reference range, 1.8–10.4 pg/mL]) but a DHEA-S level within reference range, suggesting an ovarian source of androgen excess. The CA-125 level was elevated (89 U/mL [reference range, <39 U/mL]).
Pelvic ultrasonography was suspicious for an ovarian pathology. Follow-up pelvic magnetic resonance imaging (MRI) demonstrated a 2.5-cm mass abutting the left ovary (Figure 2). The patient was given a diagnosis of stage IIIA high-grade ovarian serous carcinoma with lymph node involvement. Other notable findings from the workup included a BRCA2 mutation and concurrent renal cell carcinoma. After bilateral salpingo-oophorectomy, partial nephrectomy, and chemotherapy with carboplatin and paclitaxel, the testosterone level returned to within reference range and remained stable for the next 2 years of follow-up.
Female pattern hair loss is common in postmenopausal women and is a frequent concern in patients presenting to dermatology. Although most cases of androgenetic alopecia are isolated or secondary to benign conditions, such as polycystic ovary syndrome or nonclassic congenital adrenal hyperplasia, a small minority(<1% of women presenting with signs of hyperandrogenism) have an androgen-secreting tumor.6
Rapid onset or worsening of clinical hyperandrogenism, as seen in our patient, should raise concern for pathology; serum total testosterone and DHEA-S levels should be evaluated. Abnormally elevated serum androgens are associated with malignancy; however, there is variability in the recommended cutoff levels to prompt suspicion for an androgen-producing tumor and further workup in postmenopausal women.7 In the case of testosterone elevation, classic teaching designates a testosterone level greater than 200 ng/dL as the appropriate threshold for concern, but this level is now debated. In a series of women with hyperandrogenism referred to a center for suspicion of an androgen-secreting tumor, those with a tumor had, on average, a significantly higher (260 ng/dL) testosterone level than women who had other causes (90 ng/dL)(P<.05).6 The authors of that study proposed a cutoff of 1.4 ng/mL because women in their series who had a tumor were 8.4 times more likely to have a testosterone level of 1.4 ng/mL or higher than women without a tumor. However, this cutoff was only 92% sensitive and 70% specific.6 The degree of androgen elevation is highly variable in both tumorous and benign pathologies with notable overlap, challenging the notion of a clear cutoff.
Imaging is indicated for a patient presenting with both clinical and biochemical hyperandrogenism. Patients with an isolated testosterone level elevation can be evaluated with transvaginal ultrasonography; however, detection and characterization of malignancies is highly dependent on the skill of the examiner.8,9 The higher sensitivity and specificity of pelvic MRI reduces the likelihood of missing a malignancy and unnecessary surgery. Tumors too small to be visualized by MRI rarely are malignant.10
Sex cord-stromal cell tumors, despite representing fewer than 10% of ovarian tumors, are responsible for the majority of androgen-secreting malignancies. Our patient presented with clinical hyperandrogenism with an elevated testosterone level in the setting of a serous ovarian carcinoma, which is an epithelial neoplasm. Epithelial tumors are the most common type of ovarian tumor and typically are nonfunctional, though they have been reported to cause hyperandrogenism through indirect mechanisms. It is thought that both benign and malignant epithelial tumors can induce stromal hyperplasia or luteinization, leading to an increase in androgen levels.6
Due to the high prevalence of androgenetic alopecia and hirsutism in aging women, identification of androgen-secreting neoplasms by clinical presentation is challenging. A wide range of serum testosterone levels is possible at presentation, which complicates diagnosis. This case highlights the importance of correlating clinical and biochemical hyperandrogenism in raising suspicion of malignancy in older women presenting with hair loss.
- Carmina E, Azziz R, Bergfeld W, et al. Female pattern hair loss and androgen excess: a report from the multidisciplinary androgen excess and PCOS committee. J Clin Endocrinol Metab. 2019;104:2875-2891.
- Herskovitz I, Tosti A. Female pattern hair loss. Int J Endocrinol Metab. 2013;11:e9860.
- Rothman MS, Wierman ME. How should postmenopausal androgen excess be evaluated? Clin Endocrinol (Oxf). 2011;75:160-164.
- Derksen J, Nagesser SK, Meinders AE, et al. Identification of virilizing adrenal tumors in hirsute women. N Engl J Med. 1994;331:968-973.
- Kaltsas GA, Isidori AM, Kola BP, et al. The value of the low-dose dexamethasone suppression test in the differential diagnosis of hyperandrogenism in women. J Clin Endocrinol Metab. 2003;88:2634-2643.
- Sarfati J, Bachelot A, Coussieu C, et al; Study Group Hyperandrogenism in Postmenopausal Women. Impact of clinical, hormonal, radiological, immunohistochemical studies on the diagnosis of postmenopausal hyperandrogenism. Eur J Endocrinol. 2011;165:779-788.
- Glintborg D, Altinok ML, Petersen KR, et al. Total testosterone levels are often more than three times elevated in patients with androgen-secreting tumours. BMJ Case Rep. 2015;2015:bcr2014204797.
- Iyer VR, Lee SI. MRI, CT, and PET/CT for ovarian cancer detection and adnexal lesion characterization. AJR Am J Roentgenol. 2010;194:311-321.
- Rauh-Hain JA, Krivak TC, Del Carmen MG, et al. Ovarian cancer screening and early detection in the general population. Rev Obstet Gynecol. 2011;4:15-21.
- Horta M, Cunha TM. Sex cord-stromal tumors of the ovary: a comprehensive review and update for radiologists. Diagn Interv Radiol. 2015;21:277-286.
To the Editor:
Female pattern hair loss is common, and the literature suggests that up to 56% of women experience hair thinning in their lifetime, with increased prevalence in older women.1 Pathophysiology is incompletely understood and involves the nonscarring progressive miniaturization of hair follicles, causing decreased production of terminal hairs relative to more delicate vellus hairs. Because vellus hairs have a shorter anagen growth phase than terminal hairs, hair loss is expedited. Androgen excess, when present, hastens the process by inducing early transition of hair follicles from the anagen phase to the senescent telogen phase. Serum testosterone levels are within reference range in most female patients with hair loss, suggesting the presence of additional contributing factors.2
Given the high prevalence of female pattern hair loss and the harm of overlooking androgen excess and an androgen-secreting neoplasm, dermatologists must recognize indications for further evaluation. Additional signs of hyperandrogenism, such as menstrual irregularities, acne, hirsutism, anabolic appearance, voice deepening, and clitoromegaly, are reasons for concern.3 Elevated serum androgen levels also should raise suspicion of malignancy. Historically, a total testosterone level above 200 ng/dL or a dehydroepiandrosterone sulfate (DHEA-S) level greater than 700 µg/dL prompted evaluation for a tumor.4 More recent studies show that tumor-induced increases in serum androgen levels are highly variable, challenging the utility of these cutoffs.5
A 70-year-old woman presented with hair loss over the last 12 years with accentuated thinning on the frontal and vertex scalp. The patient’s primary care physician previously made a diagnosis of androgenetic alopecia and recommended topical minoxidil. Although the patient had a history of excess facial and body hair since young adulthood, she noted a progressive increase in the density of chest and back hair, prominent coarsening of the texture of the facial and body hair, and new facial acne in the last 3 years. Prior to these changes, the density and texture of the scalp and body hair had been stable for many years.
Although other postmenopausal females in the patient’s family displayed patterned hair loss, they did not possess coarse and dense hair on the face and trunk. Her family history was notable for ovarian cancer in her mother (in her 70s) and breast cancer in her maternal grandmother (in her 80s).
A review of systems was notable only for decreased energy. Physical examination revealed a well-appearing older woman with coarse terminal hair growth on the cheeks, submental chin, neck, chest, back, and forearms. Scalp examination indicated diffusely decreased hair density, most marked over the vertex, crown, and frontal scalp, without scale, erythema, or loss of follicular ostia (Figure 1).
Laboratory evaluation revealed elevated levels of total testosterone (106 ng/dL [reference range, <40 ng/dL]) and free testosterone (32.9 pg/mL [reference range, 1.8–10.4 pg/mL]) but a DHEA-S level within reference range, suggesting an ovarian source of androgen excess. The CA-125 level was elevated (89 U/mL [reference range, <39 U/mL]).
Pelvic ultrasonography was suspicious for an ovarian pathology. Follow-up pelvic magnetic resonance imaging (MRI) demonstrated a 2.5-cm mass abutting the left ovary (Figure 2). The patient was given a diagnosis of stage IIIA high-grade ovarian serous carcinoma with lymph node involvement. Other notable findings from the workup included a BRCA2 mutation and concurrent renal cell carcinoma. After bilateral salpingo-oophorectomy, partial nephrectomy, and chemotherapy with carboplatin and paclitaxel, the testosterone level returned to within reference range and remained stable for the next 2 years of follow-up.
Female pattern hair loss is common in postmenopausal women and is a frequent concern in patients presenting to dermatology. Although most cases of androgenetic alopecia are isolated or secondary to benign conditions, such as polycystic ovary syndrome or nonclassic congenital adrenal hyperplasia, a small minority(<1% of women presenting with signs of hyperandrogenism) have an androgen-secreting tumor.6
Rapid onset or worsening of clinical hyperandrogenism, as seen in our patient, should raise concern for pathology; serum total testosterone and DHEA-S levels should be evaluated. Abnormally elevated serum androgens are associated with malignancy; however, there is variability in the recommended cutoff levels to prompt suspicion for an androgen-producing tumor and further workup in postmenopausal women.7 In the case of testosterone elevation, classic teaching designates a testosterone level greater than 200 ng/dL as the appropriate threshold for concern, but this level is now debated. In a series of women with hyperandrogenism referred to a center for suspicion of an androgen-secreting tumor, those with a tumor had, on average, a significantly higher (260 ng/dL) testosterone level than women who had other causes (90 ng/dL)(P<.05).6 The authors of that study proposed a cutoff of 1.4 ng/mL because women in their series who had a tumor were 8.4 times more likely to have a testosterone level of 1.4 ng/mL or higher than women without a tumor. However, this cutoff was only 92% sensitive and 70% specific.6 The degree of androgen elevation is highly variable in both tumorous and benign pathologies with notable overlap, challenging the notion of a clear cutoff.
Imaging is indicated for a patient presenting with both clinical and biochemical hyperandrogenism. Patients with an isolated testosterone level elevation can be evaluated with transvaginal ultrasonography; however, detection and characterization of malignancies is highly dependent on the skill of the examiner.8,9 The higher sensitivity and specificity of pelvic MRI reduces the likelihood of missing a malignancy and unnecessary surgery. Tumors too small to be visualized by MRI rarely are malignant.10
Sex cord-stromal cell tumors, despite representing fewer than 10% of ovarian tumors, are responsible for the majority of androgen-secreting malignancies. Our patient presented with clinical hyperandrogenism with an elevated testosterone level in the setting of a serous ovarian carcinoma, which is an epithelial neoplasm. Epithelial tumors are the most common type of ovarian tumor and typically are nonfunctional, though they have been reported to cause hyperandrogenism through indirect mechanisms. It is thought that both benign and malignant epithelial tumors can induce stromal hyperplasia or luteinization, leading to an increase in androgen levels.6
Due to the high prevalence of androgenetic alopecia and hirsutism in aging women, identification of androgen-secreting neoplasms by clinical presentation is challenging. A wide range of serum testosterone levels is possible at presentation, which complicates diagnosis. This case highlights the importance of correlating clinical and biochemical hyperandrogenism in raising suspicion of malignancy in older women presenting with hair loss.
To the Editor:
Female pattern hair loss is common, and the literature suggests that up to 56% of women experience hair thinning in their lifetime, with increased prevalence in older women.1 Pathophysiology is incompletely understood and involves the nonscarring progressive miniaturization of hair follicles, causing decreased production of terminal hairs relative to more delicate vellus hairs. Because vellus hairs have a shorter anagen growth phase than terminal hairs, hair loss is expedited. Androgen excess, when present, hastens the process by inducing early transition of hair follicles from the anagen phase to the senescent telogen phase. Serum testosterone levels are within reference range in most female patients with hair loss, suggesting the presence of additional contributing factors.2
Given the high prevalence of female pattern hair loss and the harm of overlooking androgen excess and an androgen-secreting neoplasm, dermatologists must recognize indications for further evaluation. Additional signs of hyperandrogenism, such as menstrual irregularities, acne, hirsutism, anabolic appearance, voice deepening, and clitoromegaly, are reasons for concern.3 Elevated serum androgen levels also should raise suspicion of malignancy. Historically, a total testosterone level above 200 ng/dL or a dehydroepiandrosterone sulfate (DHEA-S) level greater than 700 µg/dL prompted evaluation for a tumor.4 More recent studies show that tumor-induced increases in serum androgen levels are highly variable, challenging the utility of these cutoffs.5
A 70-year-old woman presented with hair loss over the last 12 years with accentuated thinning on the frontal and vertex scalp. The patient’s primary care physician previously made a diagnosis of androgenetic alopecia and recommended topical minoxidil. Although the patient had a history of excess facial and body hair since young adulthood, she noted a progressive increase in the density of chest and back hair, prominent coarsening of the texture of the facial and body hair, and new facial acne in the last 3 years. Prior to these changes, the density and texture of the scalp and body hair had been stable for many years.
Although other postmenopausal females in the patient’s family displayed patterned hair loss, they did not possess coarse and dense hair on the face and trunk. Her family history was notable for ovarian cancer in her mother (in her 70s) and breast cancer in her maternal grandmother (in her 80s).
A review of systems was notable only for decreased energy. Physical examination revealed a well-appearing older woman with coarse terminal hair growth on the cheeks, submental chin, neck, chest, back, and forearms. Scalp examination indicated diffusely decreased hair density, most marked over the vertex, crown, and frontal scalp, without scale, erythema, or loss of follicular ostia (Figure 1).
Laboratory evaluation revealed elevated levels of total testosterone (106 ng/dL [reference range, <40 ng/dL]) and free testosterone (32.9 pg/mL [reference range, 1.8–10.4 pg/mL]) but a DHEA-S level within reference range, suggesting an ovarian source of androgen excess. The CA-125 level was elevated (89 U/mL [reference range, <39 U/mL]).
Pelvic ultrasonography was suspicious for an ovarian pathology. Follow-up pelvic magnetic resonance imaging (MRI) demonstrated a 2.5-cm mass abutting the left ovary (Figure 2). The patient was given a diagnosis of stage IIIA high-grade ovarian serous carcinoma with lymph node involvement. Other notable findings from the workup included a BRCA2 mutation and concurrent renal cell carcinoma. After bilateral salpingo-oophorectomy, partial nephrectomy, and chemotherapy with carboplatin and paclitaxel, the testosterone level returned to within reference range and remained stable for the next 2 years of follow-up.
Female pattern hair loss is common in postmenopausal women and is a frequent concern in patients presenting to dermatology. Although most cases of androgenetic alopecia are isolated or secondary to benign conditions, such as polycystic ovary syndrome or nonclassic congenital adrenal hyperplasia, a small minority(<1% of women presenting with signs of hyperandrogenism) have an androgen-secreting tumor.6
Rapid onset or worsening of clinical hyperandrogenism, as seen in our patient, should raise concern for pathology; serum total testosterone and DHEA-S levels should be evaluated. Abnormally elevated serum androgens are associated with malignancy; however, there is variability in the recommended cutoff levels to prompt suspicion for an androgen-producing tumor and further workup in postmenopausal women.7 In the case of testosterone elevation, classic teaching designates a testosterone level greater than 200 ng/dL as the appropriate threshold for concern, but this level is now debated. In a series of women with hyperandrogenism referred to a center for suspicion of an androgen-secreting tumor, those with a tumor had, on average, a significantly higher (260 ng/dL) testosterone level than women who had other causes (90 ng/dL)(P<.05).6 The authors of that study proposed a cutoff of 1.4 ng/mL because women in their series who had a tumor were 8.4 times more likely to have a testosterone level of 1.4 ng/mL or higher than women without a tumor. However, this cutoff was only 92% sensitive and 70% specific.6 The degree of androgen elevation is highly variable in both tumorous and benign pathologies with notable overlap, challenging the notion of a clear cutoff.
Imaging is indicated for a patient presenting with both clinical and biochemical hyperandrogenism. Patients with an isolated testosterone level elevation can be evaluated with transvaginal ultrasonography; however, detection and characterization of malignancies is highly dependent on the skill of the examiner.8,9 The higher sensitivity and specificity of pelvic MRI reduces the likelihood of missing a malignancy and unnecessary surgery. Tumors too small to be visualized by MRI rarely are malignant.10
Sex cord-stromal cell tumors, despite representing fewer than 10% of ovarian tumors, are responsible for the majority of androgen-secreting malignancies. Our patient presented with clinical hyperandrogenism with an elevated testosterone level in the setting of a serous ovarian carcinoma, which is an epithelial neoplasm. Epithelial tumors are the most common type of ovarian tumor and typically are nonfunctional, though they have been reported to cause hyperandrogenism through indirect mechanisms. It is thought that both benign and malignant epithelial tumors can induce stromal hyperplasia or luteinization, leading to an increase in androgen levels.6
Due to the high prevalence of androgenetic alopecia and hirsutism in aging women, identification of androgen-secreting neoplasms by clinical presentation is challenging. A wide range of serum testosterone levels is possible at presentation, which complicates diagnosis. This case highlights the importance of correlating clinical and biochemical hyperandrogenism in raising suspicion of malignancy in older women presenting with hair loss.
- Carmina E, Azziz R, Bergfeld W, et al. Female pattern hair loss and androgen excess: a report from the multidisciplinary androgen excess and PCOS committee. J Clin Endocrinol Metab. 2019;104:2875-2891.
- Herskovitz I, Tosti A. Female pattern hair loss. Int J Endocrinol Metab. 2013;11:e9860.
- Rothman MS, Wierman ME. How should postmenopausal androgen excess be evaluated? Clin Endocrinol (Oxf). 2011;75:160-164.
- Derksen J, Nagesser SK, Meinders AE, et al. Identification of virilizing adrenal tumors in hirsute women. N Engl J Med. 1994;331:968-973.
- Kaltsas GA, Isidori AM, Kola BP, et al. The value of the low-dose dexamethasone suppression test in the differential diagnosis of hyperandrogenism in women. J Clin Endocrinol Metab. 2003;88:2634-2643.
- Sarfati J, Bachelot A, Coussieu C, et al; Study Group Hyperandrogenism in Postmenopausal Women. Impact of clinical, hormonal, radiological, immunohistochemical studies on the diagnosis of postmenopausal hyperandrogenism. Eur J Endocrinol. 2011;165:779-788.
- Glintborg D, Altinok ML, Petersen KR, et al. Total testosterone levels are often more than three times elevated in patients with androgen-secreting tumours. BMJ Case Rep. 2015;2015:bcr2014204797.
- Iyer VR, Lee SI. MRI, CT, and PET/CT for ovarian cancer detection and adnexal lesion characterization. AJR Am J Roentgenol. 2010;194:311-321.
- Rauh-Hain JA, Krivak TC, Del Carmen MG, et al. Ovarian cancer screening and early detection in the general population. Rev Obstet Gynecol. 2011;4:15-21.
- Horta M, Cunha TM. Sex cord-stromal tumors of the ovary: a comprehensive review and update for radiologists. Diagn Interv Radiol. 2015;21:277-286.
- Carmina E, Azziz R, Bergfeld W, et al. Female pattern hair loss and androgen excess: a report from the multidisciplinary androgen excess and PCOS committee. J Clin Endocrinol Metab. 2019;104:2875-2891.
- Herskovitz I, Tosti A. Female pattern hair loss. Int J Endocrinol Metab. 2013;11:e9860.
- Rothman MS, Wierman ME. How should postmenopausal androgen excess be evaluated? Clin Endocrinol (Oxf). 2011;75:160-164.
- Derksen J, Nagesser SK, Meinders AE, et al. Identification of virilizing adrenal tumors in hirsute women. N Engl J Med. 1994;331:968-973.
- Kaltsas GA, Isidori AM, Kola BP, et al. The value of the low-dose dexamethasone suppression test in the differential diagnosis of hyperandrogenism in women. J Clin Endocrinol Metab. 2003;88:2634-2643.
- Sarfati J, Bachelot A, Coussieu C, et al; Study Group Hyperandrogenism in Postmenopausal Women. Impact of clinical, hormonal, radiological, immunohistochemical studies on the diagnosis of postmenopausal hyperandrogenism. Eur J Endocrinol. 2011;165:779-788.
- Glintborg D, Altinok ML, Petersen KR, et al. Total testosterone levels are often more than three times elevated in patients with androgen-secreting tumours. BMJ Case Rep. 2015;2015:bcr2014204797.
- Iyer VR, Lee SI. MRI, CT, and PET/CT for ovarian cancer detection and adnexal lesion characterization. AJR Am J Roentgenol. 2010;194:311-321.
- Rauh-Hain JA, Krivak TC, Del Carmen MG, et al. Ovarian cancer screening and early detection in the general population. Rev Obstet Gynecol. 2011;4:15-21.
- Horta M, Cunha TM. Sex cord-stromal tumors of the ovary: a comprehensive review and update for radiologists. Diagn Interv Radiol. 2015;21:277-286.
Practice Points
- Laboratory assessment for possible androgen excess should be performed in patients with female pattern hair loss and include baseline serum total testosterone and dehydroepiandrosterone sulfate.
- Rapid onset or worsening of clinical hyperandrogenism should raise suspicion of malignancy.
- Transvaginal ultrasonography and possible pelvic magnetic resonance imaging are indicated for patients with clinical hyperandrogenism and an isolated testosterone level elevation.
Symmetric Drug-Related Intertriginous and Flexural Exanthema
To the Editor:
Symmetric drug-related intertriginous and flexural exanthema (SDRIFE) is a curious disorder that has undergone many clinical transformations since first being described by Andersen et al1 in 1984 using the term baboon syndrome. Initially described as a mercury hypersensitivity reaction resulting in an eruption resembling the red-bottomed baboon, this exanthema has expanded in definition with inciting agents, clinical features, and diagnostic criteria. Its prognosis, however, has remained stable and favorable throughout the decades. The condition is almost universally benign and self-limited.1-3 As new cases are reported in the literature and the paradigm of SDRIFE continues to shift, its prognosis also may warrant reconsideration and respect as a potentially destructive reaction.
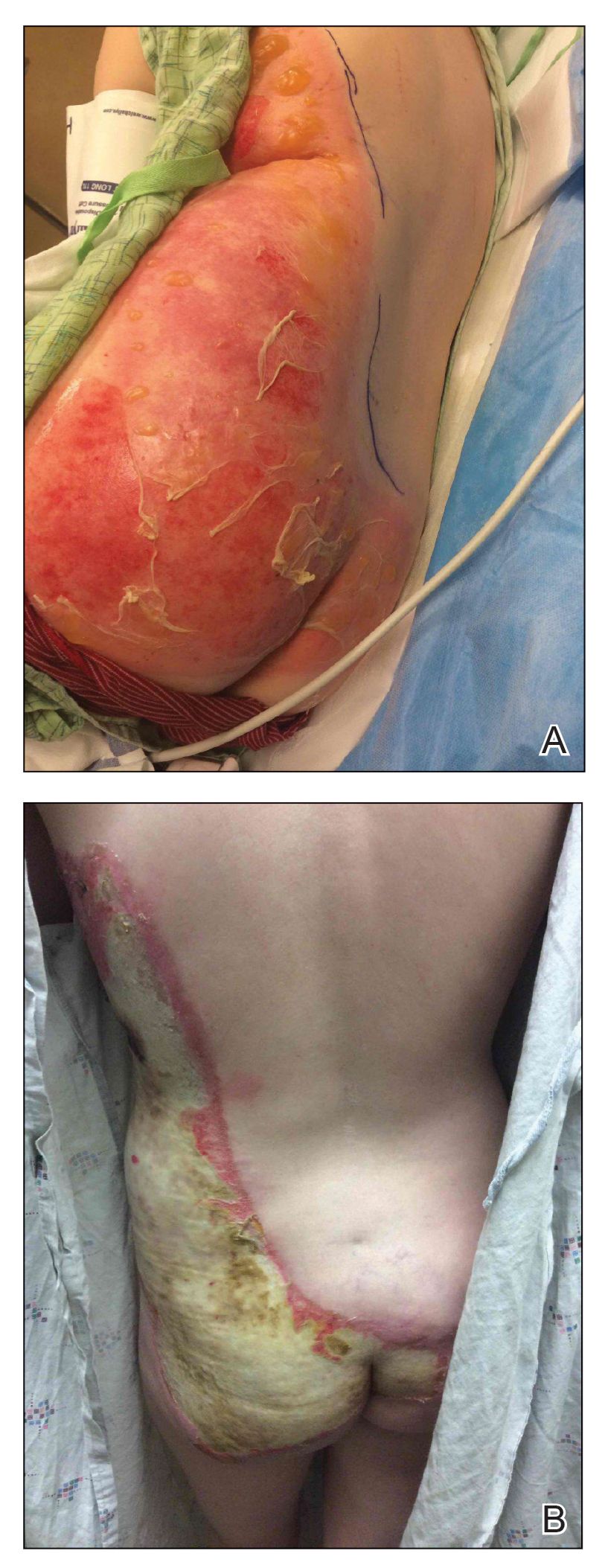
A 39-year-old woman who was otherwise healthy presented to the emergency department after developing a rapidly evolving and blistering rash on the left flank. Hours later, the rash had progressed to a sharply demarcated, confluent, erythematous plaque with central ulceration and large flaccid bullae peripherally, encompassing 18% of total body surface area and extending from the gluteal cleft to the tip of the scapula along the left flank (Figure 1) with no vaginal or mucosal involvement. The patient recently had completed a 10-day course of amoxicillin–clavulanic acid 2 days prior for a cat bite on the right dorsal wrist. Additional history confirmed the absence of prodromal fever, fatigue, or chills. Inciting trauma, including chemical and thermal burns, was denied. Potential underlying psychosocial cofounders were explored and were unrevealing.
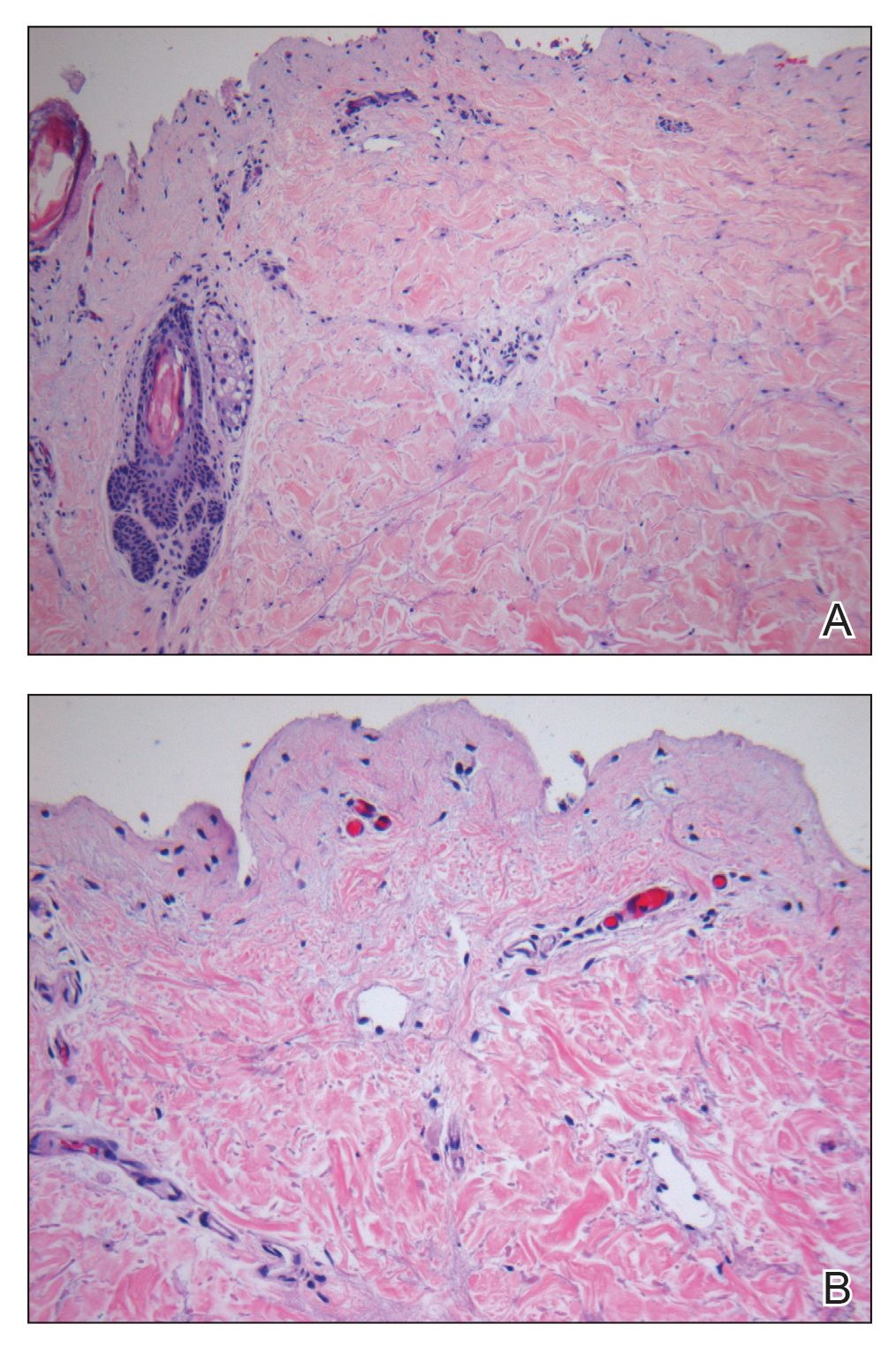
Laboratory test results, including complete blood cell count and metabolic panel as well as vital signs were unremarkable, except for slight leukocytosis at 14,000/µL (reference range 4500–11,000/µL). A punch biopsy was taken from the patient’s left upper back at the time of admission, which revealed a sparse, superficial, perivascular infiltrate of lymphocytes and rare neutrophils with largely absent epidermis and an occasional focal necrosis of adnexal epithelium (Figure 2). Immunofluorescence was negative for specific deposition of IgG, IgA, IgM, C3, or fibrinogen. Wound culture also returned negative, and the Naranjo adverse drug reaction probability scale score was calculated to be 4 out of 12, indicating possible adverse drug reaction.4
Given the extent and distribution of the rash as well as the full-thickness dermal involvement, the patient was transferred to the burn unit for subsequent care. At 8-month follow-up, she experienced severe, symptomatic, hypertrophic scarring and was awaiting intralesional triamcinolone acetonide injections. The patient subsequently was lost to follow up.
The clinical picture of SDRIFE has remained obscure over the last 30 years, likely owing to its rarity and unclear pathogenesis. Diagnostic criteria for SDRIFE were first proposed by Häusermann et al2 in 2004 and contained 5 elements: (1) occurrence after (re)exposure to systemic drugs, (2) sharply demarcated erythema of the gluteal region or V-shaped erythema of the inguinal area, (3) involvement of at least 1 other intertriginous location, (4) symmetry of affected areas, and (5) absence of systemic symptoms and signs. Based on these clinical criteria, our patients fulfilled 3 of 5 elements, with deductions for symmetry of affected areas and involvement of other intertriginous locations. Histopathologic findings in SDRIFE predominantly are nonspecific with superficial perivascular mononuclear infiltrates; however, prior reports have confirmed the potential for vacuolar changes and hydropic degeneration in the basal cell layer with subepidermal bullae formation.5,6 Similarly, although the presence of bullae are somewhat atypical in SDRIFE, it has been described.3 Taken together, we speculate that these findings may support a diagnosis of SDRIFE with atypical presentation, though an alternative diagnosis of bullous fixed drug eruption (FDE) cannot be ruled out.
Historically, SDRIFE has been associated with a benign course. The condition typically arises within a few hours to days following administration of the offending agent, most commonly amoxicillin or another β-lactam antibiotic.1 Most cases spontaneously resolve via desquamation within 1 to 2 weeks. We present an unusual case of amoxicillin-induced full-thickness epidermal necrosis resulting in symptomatic sequelae, which exhibits findings of SDRIFE, bullous FDE, or Stevens-Johnson syndrome/toxic epidermal necrolysis, suggesting the possibility for a common pathway underlying the pathogenesis of these conditions.
The diagnostic uncertainty that commonly accompanies these various toxic drug reactions may in part relate to their underlying immunopathogenesis. Although the exact mechanism by which SDRIFE results in its characteristic skin lesions has not been fully elucidated, prior work through patch testing, lymphocyte transformation assays, and immunohistochemical staining of biopsies suggests a type IV delayed hypersensitivity (DTH) reaction.7-10 Specifically, SDRIFE appears to share features of both DTH type IVa—involving CD4+ helper T cells (TH1), monocytes, and IFN-γ signaling—and DTH type IVc—involving cytotoxic CD4 and CD8 cells, granzyme B action, and FasL signaling.11,12 A similar inflammatory milieu has been implicated in numerous toxic drug eruptions, including Stevens-Johnson syndrome/toxic epidermal necrolysis and FDE.11,13 This mechanistic overlap may explain the overlap seen clinically among such conditions.
In the undifferentiated patient, categorization of the clinical syndrome proves helpful in prognostication and therapeutic approach. The complexities and commonalities intrinsic to these syndromes, however, may simultaneously preclude certain cases from neatly following the predefined rules. These atypical presentations, while diagnostically challenging, can in turn offer a unique opportunity to reexamine the current state of disease understanding to better allow for appropriate classification.
Despite its rarity, SDRIFE should be considered in the differential of undiagnosed drug eruptions, particularly as new clinical presentations emerge. Careful documentation and timely declaration of future cases will prove invaluable for diagnostic and therapeutic advancements should this once-benign condition develop a more destructive potential.
- Andersen KE, Hjorth N, Menné T. The baboon syndrome: systemically-induced allergic contact dermatitis. Contact Dermatitis. 1984;10:97-100.
- Häusermann P, Harr TH, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Tan SC, Tan JW. Symmetrical drug-related intertriginous and flexural exanthema. Curr Opin Allergy Clin Immunol. 2011;11:313-318.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Wolf R, Orion E, Matz H. The baboon syndrome or intertriginous drug eruption: a report of eleven cases and a second look at its pathomechanism. Dermatol Online J. 2003;9:2.
- Elmariah SB, Cheung W, Wang N, et al. Systemic drug-related intertriginous and flexural exanthema (SDRIFE). Dermatol Online J. 2009;15:3.
- Hembold P, Hegemann B, Dickert C, et al. Symptomatic psychotropic and nonpigmenting fixed drug eruption due to cimetidine (so-called baboon syndrome). Dermatology. 1998;197:402-403.
- Barbaud A, Trechot P, Granel F, et al. A baboon syndrome induced by intravenous human immunoglobulins: a report of a case and immunological analysis. Dermatology. 1999;199:258-260.
- Miyahara A, Kawashima H, Okubo Y, et al. A new proposal for a clinical-oriented subclassification of baboon syndrome and review of baboon syndrome. Asian Pac J Allergy Immunol. 2011;29:150-160.
- Goossens C, Sass U, Song M. Baboon syndrome. Dermatology. 1997;194:421-422.
- Pichler WJ. Delayed drug hypersensitivity reactions. Ann Intern Med. 2003;139:123-129.
- Ozkaya E. Current understanding of baboon syndrome. Expert Rev Dermatol. 2009;4:163-175.
- Ozakaya E. Fixed drug eruption: state of the art. J Dtsch Dermatol Ges. 2008;6:181-188.
To the Editor:
Symmetric drug-related intertriginous and flexural exanthema (SDRIFE) is a curious disorder that has undergone many clinical transformations since first being described by Andersen et al1 in 1984 using the term baboon syndrome. Initially described as a mercury hypersensitivity reaction resulting in an eruption resembling the red-bottomed baboon, this exanthema has expanded in definition with inciting agents, clinical features, and diagnostic criteria. Its prognosis, however, has remained stable and favorable throughout the decades. The condition is almost universally benign and self-limited.1-3 As new cases are reported in the literature and the paradigm of SDRIFE continues to shift, its prognosis also may warrant reconsideration and respect as a potentially destructive reaction.
A 39-year-old woman who was otherwise healthy presented to the emergency department after developing a rapidly evolving and blistering rash on the left flank. Hours later, the rash had progressed to a sharply demarcated, confluent, erythematous plaque with central ulceration and large flaccid bullae peripherally, encompassing 18% of total body surface area and extending from the gluteal cleft to the tip of the scapula along the left flank (Figure 1) with no vaginal or mucosal involvement. The patient recently had completed a 10-day course of amoxicillin–clavulanic acid 2 days prior for a cat bite on the right dorsal wrist. Additional history confirmed the absence of prodromal fever, fatigue, or chills. Inciting trauma, including chemical and thermal burns, was denied. Potential underlying psychosocial cofounders were explored and were unrevealing.
Laboratory test results, including complete blood cell count and metabolic panel as well as vital signs were unremarkable, except for slight leukocytosis at 14,000/µL (reference range 4500–11,000/µL). A punch biopsy was taken from the patient’s left upper back at the time of admission, which revealed a sparse, superficial, perivascular infiltrate of lymphocytes and rare neutrophils with largely absent epidermis and an occasional focal necrosis of adnexal epithelium (Figure 2). Immunofluorescence was negative for specific deposition of IgG, IgA, IgM, C3, or fibrinogen. Wound culture also returned negative, and the Naranjo adverse drug reaction probability scale score was calculated to be 4 out of 12, indicating possible adverse drug reaction.4
Given the extent and distribution of the rash as well as the full-thickness dermal involvement, the patient was transferred to the burn unit for subsequent care. At 8-month follow-up, she experienced severe, symptomatic, hypertrophic scarring and was awaiting intralesional triamcinolone acetonide injections. The patient subsequently was lost to follow up.
The clinical picture of SDRIFE has remained obscure over the last 30 years, likely owing to its rarity and unclear pathogenesis. Diagnostic criteria for SDRIFE were first proposed by Häusermann et al2 in 2004 and contained 5 elements: (1) occurrence after (re)exposure to systemic drugs, (2) sharply demarcated erythema of the gluteal region or V-shaped erythema of the inguinal area, (3) involvement of at least 1 other intertriginous location, (4) symmetry of affected areas, and (5) absence of systemic symptoms and signs. Based on these clinical criteria, our patients fulfilled 3 of 5 elements, with deductions for symmetry of affected areas and involvement of other intertriginous locations. Histopathologic findings in SDRIFE predominantly are nonspecific with superficial perivascular mononuclear infiltrates; however, prior reports have confirmed the potential for vacuolar changes and hydropic degeneration in the basal cell layer with subepidermal bullae formation.5,6 Similarly, although the presence of bullae are somewhat atypical in SDRIFE, it has been described.3 Taken together, we speculate that these findings may support a diagnosis of SDRIFE with atypical presentation, though an alternative diagnosis of bullous fixed drug eruption (FDE) cannot be ruled out.
Historically, SDRIFE has been associated with a benign course. The condition typically arises within a few hours to days following administration of the offending agent, most commonly amoxicillin or another β-lactam antibiotic.1 Most cases spontaneously resolve via desquamation within 1 to 2 weeks. We present an unusual case of amoxicillin-induced full-thickness epidermal necrosis resulting in symptomatic sequelae, which exhibits findings of SDRIFE, bullous FDE, or Stevens-Johnson syndrome/toxic epidermal necrolysis, suggesting the possibility for a common pathway underlying the pathogenesis of these conditions.
The diagnostic uncertainty that commonly accompanies these various toxic drug reactions may in part relate to their underlying immunopathogenesis. Although the exact mechanism by which SDRIFE results in its characteristic skin lesions has not been fully elucidated, prior work through patch testing, lymphocyte transformation assays, and immunohistochemical staining of biopsies suggests a type IV delayed hypersensitivity (DTH) reaction.7-10 Specifically, SDRIFE appears to share features of both DTH type IVa—involving CD4+ helper T cells (TH1), monocytes, and IFN-γ signaling—and DTH type IVc—involving cytotoxic CD4 and CD8 cells, granzyme B action, and FasL signaling.11,12 A similar inflammatory milieu has been implicated in numerous toxic drug eruptions, including Stevens-Johnson syndrome/toxic epidermal necrolysis and FDE.11,13 This mechanistic overlap may explain the overlap seen clinically among such conditions.
In the undifferentiated patient, categorization of the clinical syndrome proves helpful in prognostication and therapeutic approach. The complexities and commonalities intrinsic to these syndromes, however, may simultaneously preclude certain cases from neatly following the predefined rules. These atypical presentations, while diagnostically challenging, can in turn offer a unique opportunity to reexamine the current state of disease understanding to better allow for appropriate classification.
Despite its rarity, SDRIFE should be considered in the differential of undiagnosed drug eruptions, particularly as new clinical presentations emerge. Careful documentation and timely declaration of future cases will prove invaluable for diagnostic and therapeutic advancements should this once-benign condition develop a more destructive potential.
To the Editor:
Symmetric drug-related intertriginous and flexural exanthema (SDRIFE) is a curious disorder that has undergone many clinical transformations since first being described by Andersen et al1 in 1984 using the term baboon syndrome. Initially described as a mercury hypersensitivity reaction resulting in an eruption resembling the red-bottomed baboon, this exanthema has expanded in definition with inciting agents, clinical features, and diagnostic criteria. Its prognosis, however, has remained stable and favorable throughout the decades. The condition is almost universally benign and self-limited.1-3 As new cases are reported in the literature and the paradigm of SDRIFE continues to shift, its prognosis also may warrant reconsideration and respect as a potentially destructive reaction.
A 39-year-old woman who was otherwise healthy presented to the emergency department after developing a rapidly evolving and blistering rash on the left flank. Hours later, the rash had progressed to a sharply demarcated, confluent, erythematous plaque with central ulceration and large flaccid bullae peripherally, encompassing 18% of total body surface area and extending from the gluteal cleft to the tip of the scapula along the left flank (Figure 1) with no vaginal or mucosal involvement. The patient recently had completed a 10-day course of amoxicillin–clavulanic acid 2 days prior for a cat bite on the right dorsal wrist. Additional history confirmed the absence of prodromal fever, fatigue, or chills. Inciting trauma, including chemical and thermal burns, was denied. Potential underlying psychosocial cofounders were explored and were unrevealing.
Laboratory test results, including complete blood cell count and metabolic panel as well as vital signs were unremarkable, except for slight leukocytosis at 14,000/µL (reference range 4500–11,000/µL). A punch biopsy was taken from the patient’s left upper back at the time of admission, which revealed a sparse, superficial, perivascular infiltrate of lymphocytes and rare neutrophils with largely absent epidermis and an occasional focal necrosis of adnexal epithelium (Figure 2). Immunofluorescence was negative for specific deposition of IgG, IgA, IgM, C3, or fibrinogen. Wound culture also returned negative, and the Naranjo adverse drug reaction probability scale score was calculated to be 4 out of 12, indicating possible adverse drug reaction.4
Given the extent and distribution of the rash as well as the full-thickness dermal involvement, the patient was transferred to the burn unit for subsequent care. At 8-month follow-up, she experienced severe, symptomatic, hypertrophic scarring and was awaiting intralesional triamcinolone acetonide injections. The patient subsequently was lost to follow up.
The clinical picture of SDRIFE has remained obscure over the last 30 years, likely owing to its rarity and unclear pathogenesis. Diagnostic criteria for SDRIFE were first proposed by Häusermann et al2 in 2004 and contained 5 elements: (1) occurrence after (re)exposure to systemic drugs, (2) sharply demarcated erythema of the gluteal region or V-shaped erythema of the inguinal area, (3) involvement of at least 1 other intertriginous location, (4) symmetry of affected areas, and (5) absence of systemic symptoms and signs. Based on these clinical criteria, our patients fulfilled 3 of 5 elements, with deductions for symmetry of affected areas and involvement of other intertriginous locations. Histopathologic findings in SDRIFE predominantly are nonspecific with superficial perivascular mononuclear infiltrates; however, prior reports have confirmed the potential for vacuolar changes and hydropic degeneration in the basal cell layer with subepidermal bullae formation.5,6 Similarly, although the presence of bullae are somewhat atypical in SDRIFE, it has been described.3 Taken together, we speculate that these findings may support a diagnosis of SDRIFE with atypical presentation, though an alternative diagnosis of bullous fixed drug eruption (FDE) cannot be ruled out.
Historically, SDRIFE has been associated with a benign course. The condition typically arises within a few hours to days following administration of the offending agent, most commonly amoxicillin or another β-lactam antibiotic.1 Most cases spontaneously resolve via desquamation within 1 to 2 weeks. We present an unusual case of amoxicillin-induced full-thickness epidermal necrosis resulting in symptomatic sequelae, which exhibits findings of SDRIFE, bullous FDE, or Stevens-Johnson syndrome/toxic epidermal necrolysis, suggesting the possibility for a common pathway underlying the pathogenesis of these conditions.
The diagnostic uncertainty that commonly accompanies these various toxic drug reactions may in part relate to their underlying immunopathogenesis. Although the exact mechanism by which SDRIFE results in its characteristic skin lesions has not been fully elucidated, prior work through patch testing, lymphocyte transformation assays, and immunohistochemical staining of biopsies suggests a type IV delayed hypersensitivity (DTH) reaction.7-10 Specifically, SDRIFE appears to share features of both DTH type IVa—involving CD4+ helper T cells (TH1), monocytes, and IFN-γ signaling—and DTH type IVc—involving cytotoxic CD4 and CD8 cells, granzyme B action, and FasL signaling.11,12 A similar inflammatory milieu has been implicated in numerous toxic drug eruptions, including Stevens-Johnson syndrome/toxic epidermal necrolysis and FDE.11,13 This mechanistic overlap may explain the overlap seen clinically among such conditions.
In the undifferentiated patient, categorization of the clinical syndrome proves helpful in prognostication and therapeutic approach. The complexities and commonalities intrinsic to these syndromes, however, may simultaneously preclude certain cases from neatly following the predefined rules. These atypical presentations, while diagnostically challenging, can in turn offer a unique opportunity to reexamine the current state of disease understanding to better allow for appropriate classification.
Despite its rarity, SDRIFE should be considered in the differential of undiagnosed drug eruptions, particularly as new clinical presentations emerge. Careful documentation and timely declaration of future cases will prove invaluable for diagnostic and therapeutic advancements should this once-benign condition develop a more destructive potential.
- Andersen KE, Hjorth N, Menné T. The baboon syndrome: systemically-induced allergic contact dermatitis. Contact Dermatitis. 1984;10:97-100.
- Häusermann P, Harr TH, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Tan SC, Tan JW. Symmetrical drug-related intertriginous and flexural exanthema. Curr Opin Allergy Clin Immunol. 2011;11:313-318.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Wolf R, Orion E, Matz H. The baboon syndrome or intertriginous drug eruption: a report of eleven cases and a second look at its pathomechanism. Dermatol Online J. 2003;9:2.
- Elmariah SB, Cheung W, Wang N, et al. Systemic drug-related intertriginous and flexural exanthema (SDRIFE). Dermatol Online J. 2009;15:3.
- Hembold P, Hegemann B, Dickert C, et al. Symptomatic psychotropic and nonpigmenting fixed drug eruption due to cimetidine (so-called baboon syndrome). Dermatology. 1998;197:402-403.
- Barbaud A, Trechot P, Granel F, et al. A baboon syndrome induced by intravenous human immunoglobulins: a report of a case and immunological analysis. Dermatology. 1999;199:258-260.
- Miyahara A, Kawashima H, Okubo Y, et al. A new proposal for a clinical-oriented subclassification of baboon syndrome and review of baboon syndrome. Asian Pac J Allergy Immunol. 2011;29:150-160.
- Goossens C, Sass U, Song M. Baboon syndrome. Dermatology. 1997;194:421-422.
- Pichler WJ. Delayed drug hypersensitivity reactions. Ann Intern Med. 2003;139:123-129.
- Ozkaya E. Current understanding of baboon syndrome. Expert Rev Dermatol. 2009;4:163-175.
- Ozakaya E. Fixed drug eruption: state of the art. J Dtsch Dermatol Ges. 2008;6:181-188.
- Andersen KE, Hjorth N, Menné T. The baboon syndrome: systemically-induced allergic contact dermatitis. Contact Dermatitis. 1984;10:97-100.
- Häusermann P, Harr TH, Bircher AJ. Baboon syndrome resulting from systemic drugs: is there strife between SDRIFE and allergic contact dermatitis syndrome? Contact Dermatitis. 2004;51:297-310.
- Tan SC, Tan JW. Symmetrical drug-related intertriginous and flexural exanthema. Curr Opin Allergy Clin Immunol. 2011;11:313-318.
- Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239-245.
- Wolf R, Orion E, Matz H. The baboon syndrome or intertriginous drug eruption: a report of eleven cases and a second look at its pathomechanism. Dermatol Online J. 2003;9:2.
- Elmariah SB, Cheung W, Wang N, et al. Systemic drug-related intertriginous and flexural exanthema (SDRIFE). Dermatol Online J. 2009;15:3.
- Hembold P, Hegemann B, Dickert C, et al. Symptomatic psychotropic and nonpigmenting fixed drug eruption due to cimetidine (so-called baboon syndrome). Dermatology. 1998;197:402-403.
- Barbaud A, Trechot P, Granel F, et al. A baboon syndrome induced by intravenous human immunoglobulins: a report of a case and immunological analysis. Dermatology. 1999;199:258-260.
- Miyahara A, Kawashima H, Okubo Y, et al. A new proposal for a clinical-oriented subclassification of baboon syndrome and review of baboon syndrome. Asian Pac J Allergy Immunol. 2011;29:150-160.
- Goossens C, Sass U, Song M. Baboon syndrome. Dermatology. 1997;194:421-422.
- Pichler WJ. Delayed drug hypersensitivity reactions. Ann Intern Med. 2003;139:123-129.
- Ozkaya E. Current understanding of baboon syndrome. Expert Rev Dermatol. 2009;4:163-175.
- Ozakaya E. Fixed drug eruption: state of the art. J Dtsch Dermatol Ges. 2008;6:181-188.
Practice Points
- Symmetric drug-related intertriginous and flexural exanthema (SDRIFE) appears in the absence of systemic signs and symptoms such as fever, which may help differentiate it from infectious causes.
- β-Lactam antibiotics, particularly amoxicillin, are common offenders in the pathogenesis of SDRIFE, but new drug relationships frequently are being described.
- Symmetric drug-related intertriginous and flexural exanthema commonly follows a benign course but warrants respect, as it may have devastating potential.
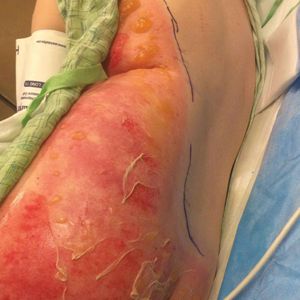
Multiple Glomangiomas in a Patient With a History of Metastatic Melanoma
To the Editor:
A 32-year-old man presented to the dermatology clinic with multiple asymptomatic blue lesions on the arms and upper torso of 15 years’ duration. His medical history was notable for a recent diagnosis of malignant melanoma following excision of a mole on the upper back 4 months prior. He reported that the mole had been present since childhood, but his sister noticed that it increased in size and changed in color over the course of a year. Physical examination showed multiple blue subcutaneous nodules on the bilateral arms and lower back. The nodules were soft and nontender, and some had telangiectasia on the overlying skin.
Given the atypical distribution of nodules and the patient’s recent history of melanoma, there was concern for cutaneous metastases. A punch biopsy of one of the nodules on the right upper arm was performed. Microscopic examination of the biopsy specimen revealed a proliferation of multiple cavernous vessels surrounded by several rows of monotonous round cells with moderate eosinophilic cytoplasm and monomorphic nuclei, which was consistent with a diagnosis of glomangioma (Figure 1). Immunohistochemical analysis showed diffuse positive staining for smooth muscle actin (Figure 2); CD34 immunostain was positive in endothelial cells and negative in tumor cells (Figure 3).



Two weeks after the first punch biopsy, the patient returned for follow-up. He noted a new soft, painless, nontender mass in the left axillary region. Positron emission tomography–computed tomography and a lymphoscintigram were performed to assess for lymphadenopathy, but they were not contributory. Subsequently, the patient underwent bilateral axillary sentinel lymph node dissection, which revealed the presence of metastatic melanoma in one lymph node in the left axilla. No metastatic disease was identified in the right axillary sentinel lymph nodes. A second skin biopsy was performed on another blue nodule to confirm the diagnosis and to exclude the possibility of sampling error. The histopathologic examination again revealed glomangioma, which established the diagnosis of multiple glomangiomas.
Glomus tumors arise from modified smooth muscle cells located in glomus bodies. The glomus body is a component of the dermis involved in regulation of body temperature that is composed of an afferent arteriole and an efferent venule. The arterial end of this apparatus, known as the Sucquet-Hoyer canal, is surrounded by glomus cells that have a contractile capability similar to smooth muscle cells. Glomus tumors usually present as painful masses on the fingers with a typical subungual location and almost always are solitary.1 Glomangiomas, sometimes known as glomuvenous malformations, tend to be larger and usually are painless. They mostly are found on the trunk and extremities and can appear in groups.2,3 Histopathologically, glomus tumors are circumscribed lesions that show a predominance of glomus cells surrounding inconspicuous blood vessels. Glomangiomas are less well-circumscribed and show a more vascular architecture with prominent dilated vessels and a smaller number of glomus cells.4
We present a case of a patient with multiple glomangiomas. There are few reports of multiple glomangiomas in the literature. This case is particularly interesting in that our patient had a history of malignant melanoma, and there was a concern for skin metastases. Despite the patient’s personal history of blue lesions that predated the diagnosis of melanoma for many years, we could not exclude the possibility of cutaneous metastases without performing biopsies.
Tumors of glomus cell origin usually are benign. It has been suggested to replace the term glomangioma with glomuvenous malformations to emphasize the hamartomatous nature of these lesions.5 Glomuvenous malformations, or glomangiomas, can occur sporadically or can be inherited as a familial disorder. Inheritable glomangioma has been linked to the chromosome 1p21-22 locus and mutations in the glomulin gene, GLMN, with variable penetrance.6 Our patient did not report a family history of such lesions.
Glomangiomas typically are solitary but rarely can present as multiple lesions in fewer than 10% of cases.7 Multiple glomangiomas are classified into 3 subtypes: localized, disseminated, and congenital plaque type. Localized multiple glomangiomas present as blue nodules confined to 1 anatomic location such as the hand or arm. Disseminated glomangiomas are more widely distributed and involve more than 1 anatomic location.8 Plaque-type glomangiomas consist of numerous confluent lesions occurring either as solitary or multiple plaques.2 Clinically, glomangiomas manifest as painless to mildly painful cutaneous nodules. Compared to venous malformations, glomangiomas are less compressible under external pressure.
Histopathologically, glomangiomas appear as nonencapsulated tumors with large, irregular, prominent vessels lined by glomus cells. Glomus cells may be so sparse that the distinction from venous malformations and hemangiomas becomes difficult. Immunohistochemistry can play an important role in diagnosis. As modified smooth muscle cells, glomus cells stain positive with a-smooth muscle actin, while CD34 highlights the vascular endothelium.1The clinical differential diagnosis of multiple blue or violaceous subcutaneous nodules includes blue rubber bleb nevus syndrome, Maffucci syndrome, glomus tumor, pyogenic granuloma, hemangioma, spiradenoma, angiolipoma, leiomyoma, or hemangiopericytoma.9-12
Different treatment modalities are available for solitary glomangiomas, including surgical excision, sclerotherapy, and laser application. Treatment of multiple glomangiomas may not be feasible, and excision of isolated symptomatic lesions may be the only option; however, it is crucial to reach the correct diagnosis in these patients to avoid improper treatments and interventions.
- Patterson JW. Weedon’s Skin Pathology. 4th ed. Edinburgh, Scotland: Churchill Livingstone Elsevier; 2016.
- Mallory SB, Enjolras O, Boon LM, et al. Congenital plaque-type glomuvenous malformations presenting in childhood. Arch Dermatol. 2006;142:892-896.
- Boon L, Mulliken JB, Enjolras O, et al. Glomuvenous malformation (glomangioma) and venous malformation distinct clinicopathologic and genetic entities. Arch Dermatol. 2004;140:971-976.
- Gombos Z, Zhang PJ. Glomus tumor. Arch Pathol Lab Med. 2008;132:1448-1452.
- Brouillard P, Boon LM, Mulliken JB, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am J Hum Genet. 2002;70:866-874.
- Brouillard P, Ghassibé M, Penington A, et al. Four common glomulin mutations cause two thirds of glomuvenous malformations (“familial glomangiomas”): evidence for a founder effect. J Med Genet. 2005;42:E13.
- Goodman TF, Abele DC. Multiple glomus tumors. a clinical and electron microscopic study. Arch Dermatol. 1971;103:11-23.
- Miyamoto H, Wada H. Localized multiple glomangiomas on the foot. J Dermatol. 2009;36:604-607.
- Borovaya A, Kunte C, Flaig MJ, et al. Disseminated cutaneousglomangiomas in an adolescent boy. Acta Derm Venereol. 2012;92:324-325.
- Leger M, Patel U, Mandal R, et al. Glomangioma. Dermatol Online J. 2010;16:11.
- Ertem D, Acar Y, Kotiloglu E, et al. Blue rubber bleb nevus syndrome. Pediatrics. 2001;107:418-420.
- Faik A, Allali F, El Hassani S, et al. Maffucci’s syndrome: a case report. Clin Rheumatol. 2006;25:88-91.
To the Editor:
A 32-year-old man presented to the dermatology clinic with multiple asymptomatic blue lesions on the arms and upper torso of 15 years’ duration. His medical history was notable for a recent diagnosis of malignant melanoma following excision of a mole on the upper back 4 months prior. He reported that the mole had been present since childhood, but his sister noticed that it increased in size and changed in color over the course of a year. Physical examination showed multiple blue subcutaneous nodules on the bilateral arms and lower back. The nodules were soft and nontender, and some had telangiectasia on the overlying skin.
Given the atypical distribution of nodules and the patient’s recent history of melanoma, there was concern for cutaneous metastases. A punch biopsy of one of the nodules on the right upper arm was performed. Microscopic examination of the biopsy specimen revealed a proliferation of multiple cavernous vessels surrounded by several rows of monotonous round cells with moderate eosinophilic cytoplasm and monomorphic nuclei, which was consistent with a diagnosis of glomangioma (Figure 1). Immunohistochemical analysis showed diffuse positive staining for smooth muscle actin (Figure 2); CD34 immunostain was positive in endothelial cells and negative in tumor cells (Figure 3).
Two weeks after the first punch biopsy, the patient returned for follow-up. He noted a new soft, painless, nontender mass in the left axillary region. Positron emission tomography–computed tomography and a lymphoscintigram were performed to assess for lymphadenopathy, but they were not contributory. Subsequently, the patient underwent bilateral axillary sentinel lymph node dissection, which revealed the presence of metastatic melanoma in one lymph node in the left axilla. No metastatic disease was identified in the right axillary sentinel lymph nodes. A second skin biopsy was performed on another blue nodule to confirm the diagnosis and to exclude the possibility of sampling error. The histopathologic examination again revealed glomangioma, which established the diagnosis of multiple glomangiomas.
Glomus tumors arise from modified smooth muscle cells located in glomus bodies. The glomus body is a component of the dermis involved in regulation of body temperature that is composed of an afferent arteriole and an efferent venule. The arterial end of this apparatus, known as the Sucquet-Hoyer canal, is surrounded by glomus cells that have a contractile capability similar to smooth muscle cells. Glomus tumors usually present as painful masses on the fingers with a typical subungual location and almost always are solitary.1 Glomangiomas, sometimes known as glomuvenous malformations, tend to be larger and usually are painless. They mostly are found on the trunk and extremities and can appear in groups.2,3 Histopathologically, glomus tumors are circumscribed lesions that show a predominance of glomus cells surrounding inconspicuous blood vessels. Glomangiomas are less well-circumscribed and show a more vascular architecture with prominent dilated vessels and a smaller number of glomus cells.4
We present a case of a patient with multiple glomangiomas. There are few reports of multiple glomangiomas in the literature. This case is particularly interesting in that our patient had a history of malignant melanoma, and there was a concern for skin metastases. Despite the patient’s personal history of blue lesions that predated the diagnosis of melanoma for many years, we could not exclude the possibility of cutaneous metastases without performing biopsies.
Tumors of glomus cell origin usually are benign. It has been suggested to replace the term glomangioma with glomuvenous malformations to emphasize the hamartomatous nature of these lesions.5 Glomuvenous malformations, or glomangiomas, can occur sporadically or can be inherited as a familial disorder. Inheritable glomangioma has been linked to the chromosome 1p21-22 locus and mutations in the glomulin gene, GLMN, with variable penetrance.6 Our patient did not report a family history of such lesions.
Glomangiomas typically are solitary but rarely can present as multiple lesions in fewer than 10% of cases.7 Multiple glomangiomas are classified into 3 subtypes: localized, disseminated, and congenital plaque type. Localized multiple glomangiomas present as blue nodules confined to 1 anatomic location such as the hand or arm. Disseminated glomangiomas are more widely distributed and involve more than 1 anatomic location.8 Plaque-type glomangiomas consist of numerous confluent lesions occurring either as solitary or multiple plaques.2 Clinically, glomangiomas manifest as painless to mildly painful cutaneous nodules. Compared to venous malformations, glomangiomas are less compressible under external pressure.
Histopathologically, glomangiomas appear as nonencapsulated tumors with large, irregular, prominent vessels lined by glomus cells. Glomus cells may be so sparse that the distinction from venous malformations and hemangiomas becomes difficult. Immunohistochemistry can play an important role in diagnosis. As modified smooth muscle cells, glomus cells stain positive with a-smooth muscle actin, while CD34 highlights the vascular endothelium.1The clinical differential diagnosis of multiple blue or violaceous subcutaneous nodules includes blue rubber bleb nevus syndrome, Maffucci syndrome, glomus tumor, pyogenic granuloma, hemangioma, spiradenoma, angiolipoma, leiomyoma, or hemangiopericytoma.9-12
Different treatment modalities are available for solitary glomangiomas, including surgical excision, sclerotherapy, and laser application. Treatment of multiple glomangiomas may not be feasible, and excision of isolated symptomatic lesions may be the only option; however, it is crucial to reach the correct diagnosis in these patients to avoid improper treatments and interventions.
To the Editor:
A 32-year-old man presented to the dermatology clinic with multiple asymptomatic blue lesions on the arms and upper torso of 15 years’ duration. His medical history was notable for a recent diagnosis of malignant melanoma following excision of a mole on the upper back 4 months prior. He reported that the mole had been present since childhood, but his sister noticed that it increased in size and changed in color over the course of a year. Physical examination showed multiple blue subcutaneous nodules on the bilateral arms and lower back. The nodules were soft and nontender, and some had telangiectasia on the overlying skin.
Given the atypical distribution of nodules and the patient’s recent history of melanoma, there was concern for cutaneous metastases. A punch biopsy of one of the nodules on the right upper arm was performed. Microscopic examination of the biopsy specimen revealed a proliferation of multiple cavernous vessels surrounded by several rows of monotonous round cells with moderate eosinophilic cytoplasm and monomorphic nuclei, which was consistent with a diagnosis of glomangioma (Figure 1). Immunohistochemical analysis showed diffuse positive staining for smooth muscle actin (Figure 2); CD34 immunostain was positive in endothelial cells and negative in tumor cells (Figure 3).
Two weeks after the first punch biopsy, the patient returned for follow-up. He noted a new soft, painless, nontender mass in the left axillary region. Positron emission tomography–computed tomography and a lymphoscintigram were performed to assess for lymphadenopathy, but they were not contributory. Subsequently, the patient underwent bilateral axillary sentinel lymph node dissection, which revealed the presence of metastatic melanoma in one lymph node in the left axilla. No metastatic disease was identified in the right axillary sentinel lymph nodes. A second skin biopsy was performed on another blue nodule to confirm the diagnosis and to exclude the possibility of sampling error. The histopathologic examination again revealed glomangioma, which established the diagnosis of multiple glomangiomas.
Glomus tumors arise from modified smooth muscle cells located in glomus bodies. The glomus body is a component of the dermis involved in regulation of body temperature that is composed of an afferent arteriole and an efferent venule. The arterial end of this apparatus, known as the Sucquet-Hoyer canal, is surrounded by glomus cells that have a contractile capability similar to smooth muscle cells. Glomus tumors usually present as painful masses on the fingers with a typical subungual location and almost always are solitary.1 Glomangiomas, sometimes known as glomuvenous malformations, tend to be larger and usually are painless. They mostly are found on the trunk and extremities and can appear in groups.2,3 Histopathologically, glomus tumors are circumscribed lesions that show a predominance of glomus cells surrounding inconspicuous blood vessels. Glomangiomas are less well-circumscribed and show a more vascular architecture with prominent dilated vessels and a smaller number of glomus cells.4
We present a case of a patient with multiple glomangiomas. There are few reports of multiple glomangiomas in the literature. This case is particularly interesting in that our patient had a history of malignant melanoma, and there was a concern for skin metastases. Despite the patient’s personal history of blue lesions that predated the diagnosis of melanoma for many years, we could not exclude the possibility of cutaneous metastases without performing biopsies.
Tumors of glomus cell origin usually are benign. It has been suggested to replace the term glomangioma with glomuvenous malformations to emphasize the hamartomatous nature of these lesions.5 Glomuvenous malformations, or glomangiomas, can occur sporadically or can be inherited as a familial disorder. Inheritable glomangioma has been linked to the chromosome 1p21-22 locus and mutations in the glomulin gene, GLMN, with variable penetrance.6 Our patient did not report a family history of such lesions.
Glomangiomas typically are solitary but rarely can present as multiple lesions in fewer than 10% of cases.7 Multiple glomangiomas are classified into 3 subtypes: localized, disseminated, and congenital plaque type. Localized multiple glomangiomas present as blue nodules confined to 1 anatomic location such as the hand or arm. Disseminated glomangiomas are more widely distributed and involve more than 1 anatomic location.8 Plaque-type glomangiomas consist of numerous confluent lesions occurring either as solitary or multiple plaques.2 Clinically, glomangiomas manifest as painless to mildly painful cutaneous nodules. Compared to venous malformations, glomangiomas are less compressible under external pressure.
Histopathologically, glomangiomas appear as nonencapsulated tumors with large, irregular, prominent vessels lined by glomus cells. Glomus cells may be so sparse that the distinction from venous malformations and hemangiomas becomes difficult. Immunohistochemistry can play an important role in diagnosis. As modified smooth muscle cells, glomus cells stain positive with a-smooth muscle actin, while CD34 highlights the vascular endothelium.1The clinical differential diagnosis of multiple blue or violaceous subcutaneous nodules includes blue rubber bleb nevus syndrome, Maffucci syndrome, glomus tumor, pyogenic granuloma, hemangioma, spiradenoma, angiolipoma, leiomyoma, or hemangiopericytoma.9-12
Different treatment modalities are available for solitary glomangiomas, including surgical excision, sclerotherapy, and laser application. Treatment of multiple glomangiomas may not be feasible, and excision of isolated symptomatic lesions may be the only option; however, it is crucial to reach the correct diagnosis in these patients to avoid improper treatments and interventions.
- Patterson JW. Weedon’s Skin Pathology. 4th ed. Edinburgh, Scotland: Churchill Livingstone Elsevier; 2016.
- Mallory SB, Enjolras O, Boon LM, et al. Congenital plaque-type glomuvenous malformations presenting in childhood. Arch Dermatol. 2006;142:892-896.
- Boon L, Mulliken JB, Enjolras O, et al. Glomuvenous malformation (glomangioma) and venous malformation distinct clinicopathologic and genetic entities. Arch Dermatol. 2004;140:971-976.
- Gombos Z, Zhang PJ. Glomus tumor. Arch Pathol Lab Med. 2008;132:1448-1452.
- Brouillard P, Boon LM, Mulliken JB, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am J Hum Genet. 2002;70:866-874.
- Brouillard P, Ghassibé M, Penington A, et al. Four common glomulin mutations cause two thirds of glomuvenous malformations (“familial glomangiomas”): evidence for a founder effect. J Med Genet. 2005;42:E13.
- Goodman TF, Abele DC. Multiple glomus tumors. a clinical and electron microscopic study. Arch Dermatol. 1971;103:11-23.
- Miyamoto H, Wada H. Localized multiple glomangiomas on the foot. J Dermatol. 2009;36:604-607.
- Borovaya A, Kunte C, Flaig MJ, et al. Disseminated cutaneousglomangiomas in an adolescent boy. Acta Derm Venereol. 2012;92:324-325.
- Leger M, Patel U, Mandal R, et al. Glomangioma. Dermatol Online J. 2010;16:11.
- Ertem D, Acar Y, Kotiloglu E, et al. Blue rubber bleb nevus syndrome. Pediatrics. 2001;107:418-420.
- Faik A, Allali F, El Hassani S, et al. Maffucci’s syndrome: a case report. Clin Rheumatol. 2006;25:88-91.
- Patterson JW. Weedon’s Skin Pathology. 4th ed. Edinburgh, Scotland: Churchill Livingstone Elsevier; 2016.
- Mallory SB, Enjolras O, Boon LM, et al. Congenital plaque-type glomuvenous malformations presenting in childhood. Arch Dermatol. 2006;142:892-896.
- Boon L, Mulliken JB, Enjolras O, et al. Glomuvenous malformation (glomangioma) and venous malformation distinct clinicopathologic and genetic entities. Arch Dermatol. 2004;140:971-976.
- Gombos Z, Zhang PJ. Glomus tumor. Arch Pathol Lab Med. 2008;132:1448-1452.
- Brouillard P, Boon LM, Mulliken JB, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am J Hum Genet. 2002;70:866-874.
- Brouillard P, Ghassibé M, Penington A, et al. Four common glomulin mutations cause two thirds of glomuvenous malformations (“familial glomangiomas”): evidence for a founder effect. J Med Genet. 2005;42:E13.
- Goodman TF, Abele DC. Multiple glomus tumors. a clinical and electron microscopic study. Arch Dermatol. 1971;103:11-23.
- Miyamoto H, Wada H. Localized multiple glomangiomas on the foot. J Dermatol. 2009;36:604-607.
- Borovaya A, Kunte C, Flaig MJ, et al. Disseminated cutaneousglomangiomas in an adolescent boy. Acta Derm Venereol. 2012;92:324-325.
- Leger M, Patel U, Mandal R, et al. Glomangioma. Dermatol Online J. 2010;16:11.
- Ertem D, Acar Y, Kotiloglu E, et al. Blue rubber bleb nevus syndrome. Pediatrics. 2001;107:418-420.
- Faik A, Allali F, El Hassani S, et al. Maffucci’s syndrome: a case report. Clin Rheumatol. 2006;25:88-91.
Practice Points
- The diagnosis of glomus tumor and glomangioma is easily suspected when the lesions are in the digital or subungual region.
- Multiple glomangiomas are rare and can clinically pose a diagnostic challenge to dermatologists.
- In patients with a recent history of malignancy, multiple glomangiomas may mimic cutaneous metastases. Therefore, multiple biopsies and histologic examination may be necessary.

Foreign-Body Reaction to Orthopedic Hardware a Decade After Implantation
To the Editor:
Cutaneous reactions to implantable devices, such as dental implants, intracoronary stents, prosthetic valves, endovascular prostheses, gynecologic devices, and spinal cord stimulator devices, occur with varying frequency and include infectious, hypersensitivity, allergic, and foreign-body reactions. Manifestations have included contact dermatitis; urticarial, vasculitic, and bullous eruptions; extrusion; and granuloma formation.1,2 Immune complex reactions around implants causing pain, inflammation, and loosening of hardwarealso have been reported.3,4 Most reported cutaneous reactions typically occur within the first weeks or months after implantation; a reaction rarely presents several years after implantation. We report a cutaneous reaction to an orthopedic appliance almost 10 years after implantation.
A 67-year-old man presented with 2 painful nodules on the right clavicle that were present for several months. The patient denied fever, chills, weight loss, enlarged lymph nodes, or night sweats. Approximately 10 years prior to the appearance of the nodules, the patient fractured the right clavicle and underwent placement of a metal plate. His medical history included resection of the right tonsil and soft-palate carcinoma with radical neck dissection and postoperative radiation, which was completed approximately 4 years prior to placement of the metal plate. The patient recently completed 4 to 6 weeks of fluorouracil for shave biopsy–proven actinic keratosis overlying the entire irradiated area.
Physical examination revealed 2 pink friable nodules measuring 1.5 to 2.5 cm in diameter and leaking serous fluid within the irradiated area (Figure 1). The differential diagnosis included pyogenic granuloma, cutaneous recurrent metastasis, and atypical basal cell carcinoma. A skin biopsy specimen showed hemorrhagic ulcerated skin with acute and chronic inflammation and abscess.
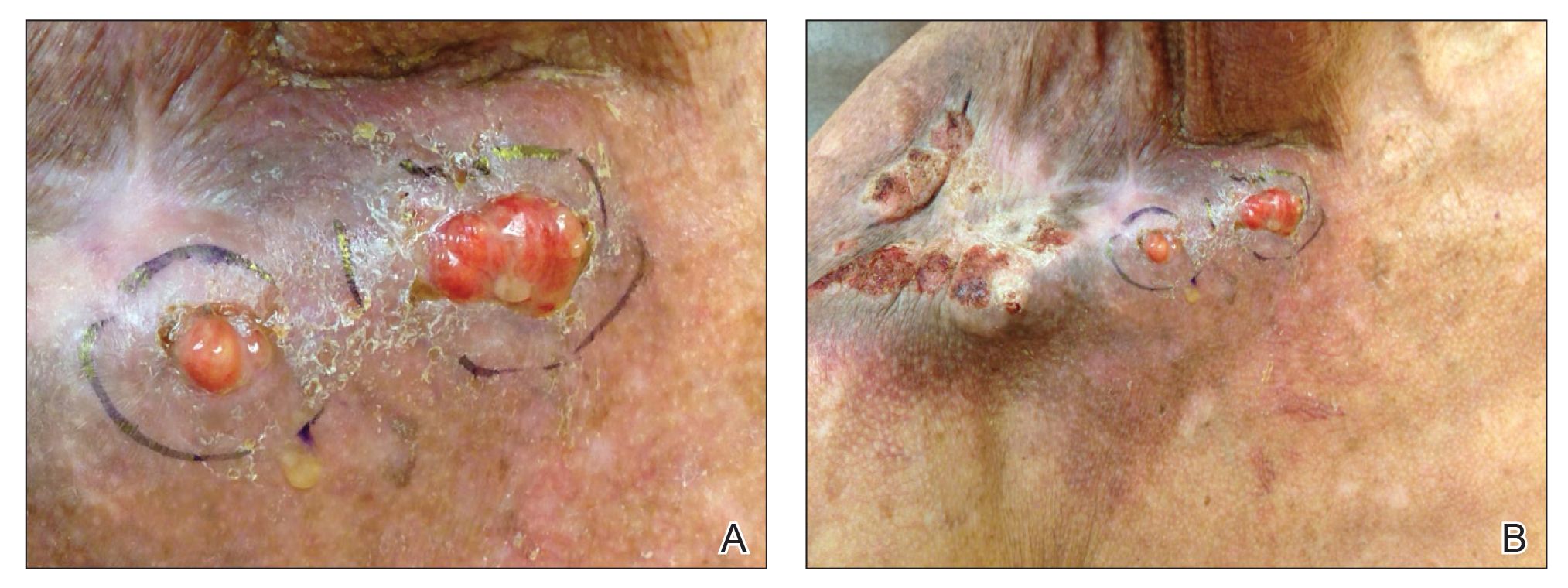
The patient presented for excisional biopsy of these areas on the right medial clavicle 1 week later. Physical examination revealed the 2 nodules had decreased in diameter; now, however, the patient had 4 discrete lesions measuring 4 to 7 mm in diameter, which were similar in appearance to the earlier nodules (Figure 2). He reported a low-grade fever, erythema, and increased tenderness of the area.
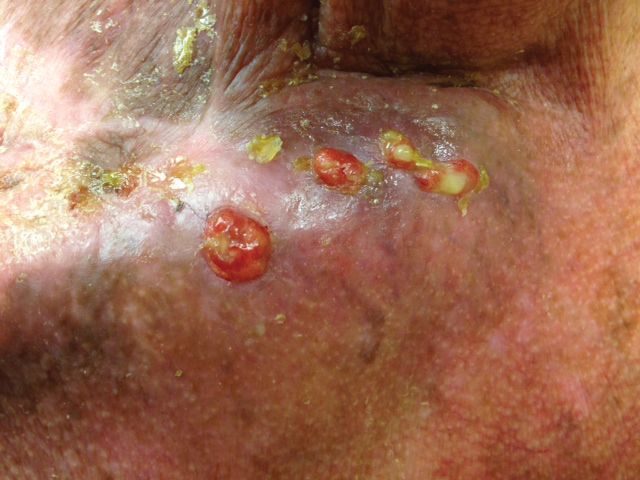
Underlying loosened orthopedic hardware screws were revealed upon punch biopsies of the involved areas (Figure 3). Wound cultures showed abundant Staphylococcus aureus and moderate group B Streptococcus; cultures for Mycobacterium were negative. The C-reactive protein level was elevated (5.47 mg/dL [reference range, ≤0.7 mg/dL]), and the erythrocyte sedimentation rate was increased (68 mm/h [reference range, 0–15 mm/h]). A complete blood cell count was within reference range, except for a mildly elevated eosinophil count (6.7% [reference range, 0%–5%]). The patient was admitted to the hospital, and antibiotics were started. Two days later, the orthopedic surgery service removed the hardware. At 3-week follow-up, physical examination revealed near closure of the wounds.
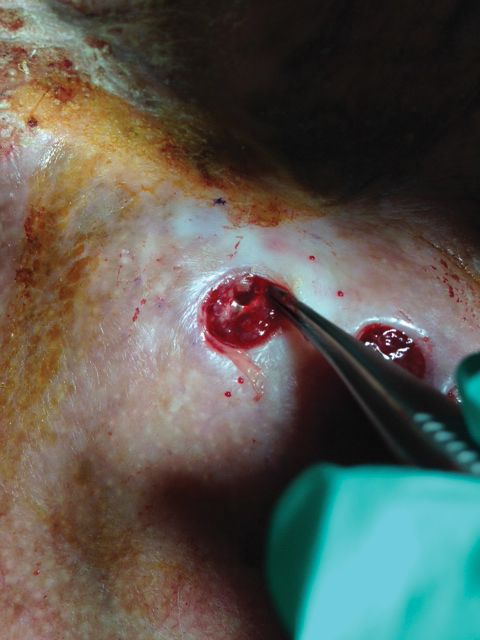
Cutaneous reactions to orthopedic implants include dermatitis, as well as urticarial, vasculitic, and bullous eruptions. Immune complex reactions can develop around implants, causing pain, inflammation, and loosening of hardware.1,3 Most inflammatory reactions take place within several months after implantation.3 Our patient’s reaction to hardware 10 years after implantation highlights the importance of taking a detailedand thorough history that includes queries about distant surgery.
- Basko-Plluska JL, Thyssen JP, Schalock PC. Cutaneous and systemic hypersensitivity reactions to metallic implants. Dermatitis. 2011;22:65-79.
- Chaudhry ZA, Najib U, Bajwa ZH, et al. Detailed analysis of allergic cutaneous reactions to spinal cord stimulator devices. J Pain Res. 2013;6:617-623.
- Huber M, Reinisch G, Trettenhahn G, et al. Presence of corrosion products and hypersensitivity-associated reactions in periprosthetic tissue after aseptic loosening of total hip replacements with metal bearing surfaces. Acta Biomater. 2009;5:172-180.
- Poncet-Wallet C, Ormezzano Y, Ernst E, et al. Study of a case of cochlear implant with recurrent cutaneous extrusion. Ann Otolaryngol Chir Cervicofac. 2009;126:264-268.
To the Editor:
Cutaneous reactions to implantable devices, such as dental implants, intracoronary stents, prosthetic valves, endovascular prostheses, gynecologic devices, and spinal cord stimulator devices, occur with varying frequency and include infectious, hypersensitivity, allergic, and foreign-body reactions. Manifestations have included contact dermatitis; urticarial, vasculitic, and bullous eruptions; extrusion; and granuloma formation.1,2 Immune complex reactions around implants causing pain, inflammation, and loosening of hardwarealso have been reported.3,4 Most reported cutaneous reactions typically occur within the first weeks or months after implantation; a reaction rarely presents several years after implantation. We report a cutaneous reaction to an orthopedic appliance almost 10 years after implantation.
A 67-year-old man presented with 2 painful nodules on the right clavicle that were present for several months. The patient denied fever, chills, weight loss, enlarged lymph nodes, or night sweats. Approximately 10 years prior to the appearance of the nodules, the patient fractured the right clavicle and underwent placement of a metal plate. His medical history included resection of the right tonsil and soft-palate carcinoma with radical neck dissection and postoperative radiation, which was completed approximately 4 years prior to placement of the metal plate. The patient recently completed 4 to 6 weeks of fluorouracil for shave biopsy–proven actinic keratosis overlying the entire irradiated area.
Physical examination revealed 2 pink friable nodules measuring 1.5 to 2.5 cm in diameter and leaking serous fluid within the irradiated area (Figure 1). The differential diagnosis included pyogenic granuloma, cutaneous recurrent metastasis, and atypical basal cell carcinoma. A skin biopsy specimen showed hemorrhagic ulcerated skin with acute and chronic inflammation and abscess.
The patient presented for excisional biopsy of these areas on the right medial clavicle 1 week later. Physical examination revealed the 2 nodules had decreased in diameter; now, however, the patient had 4 discrete lesions measuring 4 to 7 mm in diameter, which were similar in appearance to the earlier nodules (Figure 2). He reported a low-grade fever, erythema, and increased tenderness of the area.
Underlying loosened orthopedic hardware screws were revealed upon punch biopsies of the involved areas (Figure 3). Wound cultures showed abundant Staphylococcus aureus and moderate group B Streptococcus; cultures for Mycobacterium were negative. The C-reactive protein level was elevated (5.47 mg/dL [reference range, ≤0.7 mg/dL]), and the erythrocyte sedimentation rate was increased (68 mm/h [reference range, 0–15 mm/h]). A complete blood cell count was within reference range, except for a mildly elevated eosinophil count (6.7% [reference range, 0%–5%]). The patient was admitted to the hospital, and antibiotics were started. Two days later, the orthopedic surgery service removed the hardware. At 3-week follow-up, physical examination revealed near closure of the wounds.
Cutaneous reactions to orthopedic implants include dermatitis, as well as urticarial, vasculitic, and bullous eruptions. Immune complex reactions can develop around implants, causing pain, inflammation, and loosening of hardware.1,3 Most inflammatory reactions take place within several months after implantation.3 Our patient’s reaction to hardware 10 years after implantation highlights the importance of taking a detailedand thorough history that includes queries about distant surgery.
To the Editor:
Cutaneous reactions to implantable devices, such as dental implants, intracoronary stents, prosthetic valves, endovascular prostheses, gynecologic devices, and spinal cord stimulator devices, occur with varying frequency and include infectious, hypersensitivity, allergic, and foreign-body reactions. Manifestations have included contact dermatitis; urticarial, vasculitic, and bullous eruptions; extrusion; and granuloma formation.1,2 Immune complex reactions around implants causing pain, inflammation, and loosening of hardwarealso have been reported.3,4 Most reported cutaneous reactions typically occur within the first weeks or months after implantation; a reaction rarely presents several years after implantation. We report a cutaneous reaction to an orthopedic appliance almost 10 years after implantation.
A 67-year-old man presented with 2 painful nodules on the right clavicle that were present for several months. The patient denied fever, chills, weight loss, enlarged lymph nodes, or night sweats. Approximately 10 years prior to the appearance of the nodules, the patient fractured the right clavicle and underwent placement of a metal plate. His medical history included resection of the right tonsil and soft-palate carcinoma with radical neck dissection and postoperative radiation, which was completed approximately 4 years prior to placement of the metal plate. The patient recently completed 4 to 6 weeks of fluorouracil for shave biopsy–proven actinic keratosis overlying the entire irradiated area.
Physical examination revealed 2 pink friable nodules measuring 1.5 to 2.5 cm in diameter and leaking serous fluid within the irradiated area (Figure 1). The differential diagnosis included pyogenic granuloma, cutaneous recurrent metastasis, and atypical basal cell carcinoma. A skin biopsy specimen showed hemorrhagic ulcerated skin with acute and chronic inflammation and abscess.
The patient presented for excisional biopsy of these areas on the right medial clavicle 1 week later. Physical examination revealed the 2 nodules had decreased in diameter; now, however, the patient had 4 discrete lesions measuring 4 to 7 mm in diameter, which were similar in appearance to the earlier nodules (Figure 2). He reported a low-grade fever, erythema, and increased tenderness of the area.
Underlying loosened orthopedic hardware screws were revealed upon punch biopsies of the involved areas (Figure 3). Wound cultures showed abundant Staphylococcus aureus and moderate group B Streptococcus; cultures for Mycobacterium were negative. The C-reactive protein level was elevated (5.47 mg/dL [reference range, ≤0.7 mg/dL]), and the erythrocyte sedimentation rate was increased (68 mm/h [reference range, 0–15 mm/h]). A complete blood cell count was within reference range, except for a mildly elevated eosinophil count (6.7% [reference range, 0%–5%]). The patient was admitted to the hospital, and antibiotics were started. Two days later, the orthopedic surgery service removed the hardware. At 3-week follow-up, physical examination revealed near closure of the wounds.
Cutaneous reactions to orthopedic implants include dermatitis, as well as urticarial, vasculitic, and bullous eruptions. Immune complex reactions can develop around implants, causing pain, inflammation, and loosening of hardware.1,3 Most inflammatory reactions take place within several months after implantation.3 Our patient’s reaction to hardware 10 years after implantation highlights the importance of taking a detailedand thorough history that includes queries about distant surgery.
- Basko-Plluska JL, Thyssen JP, Schalock PC. Cutaneous and systemic hypersensitivity reactions to metallic implants. Dermatitis. 2011;22:65-79.
- Chaudhry ZA, Najib U, Bajwa ZH, et al. Detailed analysis of allergic cutaneous reactions to spinal cord stimulator devices. J Pain Res. 2013;6:617-623.
- Huber M, Reinisch G, Trettenhahn G, et al. Presence of corrosion products and hypersensitivity-associated reactions in periprosthetic tissue after aseptic loosening of total hip replacements with metal bearing surfaces. Acta Biomater. 2009;5:172-180.
- Poncet-Wallet C, Ormezzano Y, Ernst E, et al. Study of a case of cochlear implant with recurrent cutaneous extrusion. Ann Otolaryngol Chir Cervicofac. 2009;126:264-268.
- Basko-Plluska JL, Thyssen JP, Schalock PC. Cutaneous and systemic hypersensitivity reactions to metallic implants. Dermatitis. 2011;22:65-79.
- Chaudhry ZA, Najib U, Bajwa ZH, et al. Detailed analysis of allergic cutaneous reactions to spinal cord stimulator devices. J Pain Res. 2013;6:617-623.
- Huber M, Reinisch G, Trettenhahn G, et al. Presence of corrosion products and hypersensitivity-associated reactions in periprosthetic tissue after aseptic loosening of total hip replacements with metal bearing surfaces. Acta Biomater. 2009;5:172-180.
- Poncet-Wallet C, Ormezzano Y, Ernst E, et al. Study of a case of cochlear implant with recurrent cutaneous extrusion. Ann Otolaryngol Chir Cervicofac. 2009;126:264-268.
Practice Points
- Cutaneous reactions to implantable devices occur with varying frequency and include infectious, hypersensitivity, allergic, and foreign-body reactions.
- Most reactions typically occur within the first weeks or months after implantation; however, a reaction rarely may present several years after implantation.
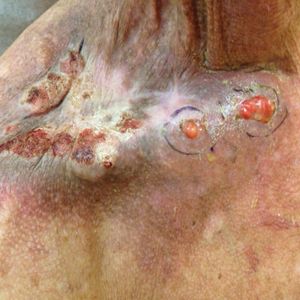
Recurrent Cutaneous Exophiala Phaeohyphomycosis in an Immunosuppressed Patient
To the Editor:
A 73-year-old man presented with a 2.5-cm, recurrent, fluctuant, multiloculated nodule on the left forearm. The lesion was nontender with occasional chalky, white to yellow discharge from multiple sinus tracts. He was otherwise well appearing without signs of systemic infection. He reported similar lesions in slightly different anatomic locations on the left forearm both 7 and 4 years prior to the current presentation. In both instances, the nodules were excised at an outside hospital without any additional treatment. Histopathology of the excised tissue from both prior occasions demonstrated brown septate hyphae surrounded by suppurative and granulomatous inflammation consistent with dematiaceous fungal infection of the dermis (Figures 1 and 2); the organisms were highlighted with periodic acid–Schiff stain.

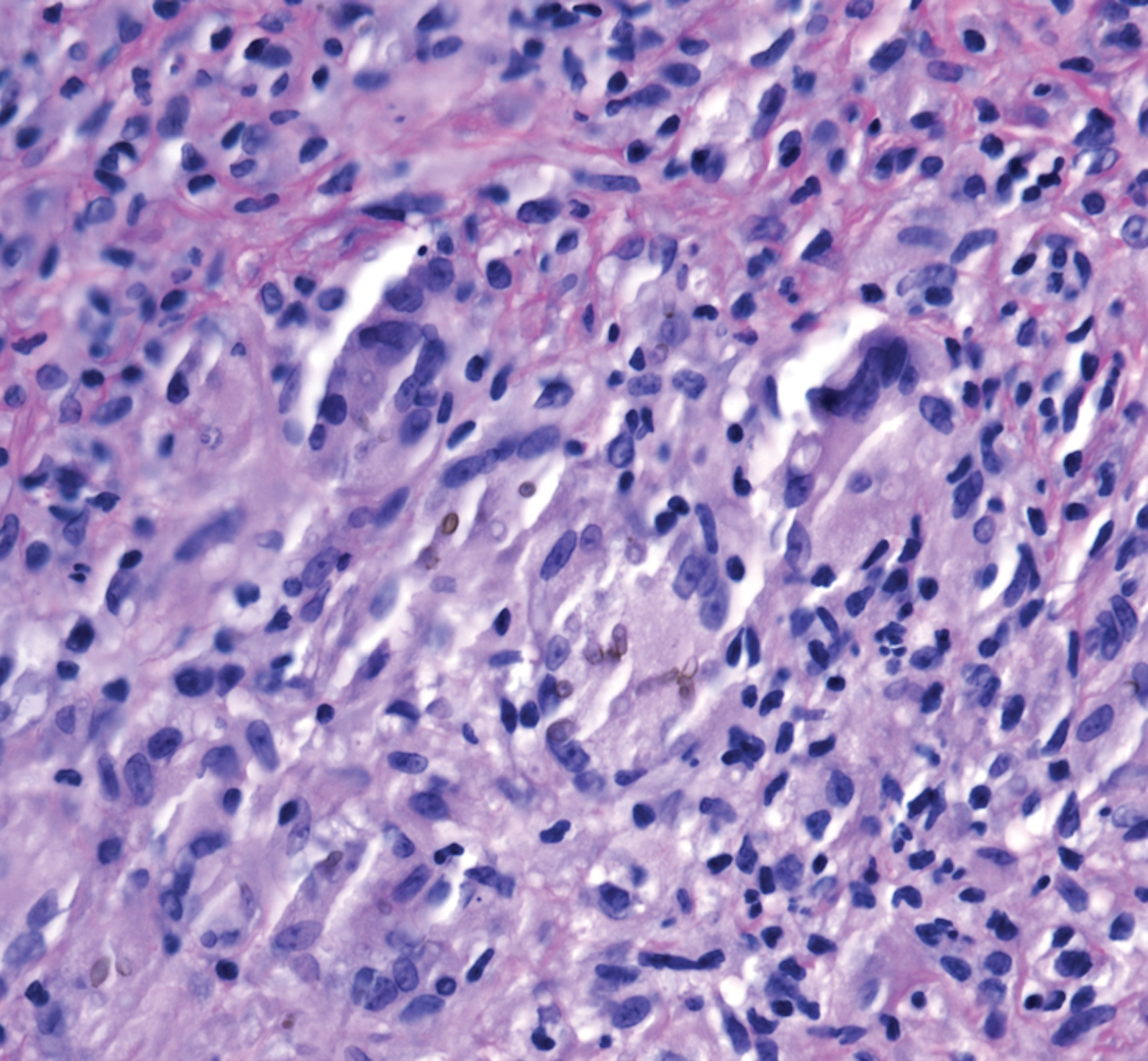
The patient’s medical history was notable for advanced heart failure with an ejection fraction of 25% and autosomal-dominant polycystic kidney disease. He received an orthotopic kidney transplant 17 years prior to the current presentation. Medications included tacrolimus, mycophenolate mofetil, and prednisone. He denied any trauma or notable exposures to vegetation, and his travel history was unremarkable. A review of systems was negative.
At the current presentation, a sterile fungal culture was performed and found positive for Exophiala species, while bacterial and mycobacterial cultures were negative. A diagnosis of phaeohyphomycosis was made, and he was scheduled for re-excision. Out of concern for interactions with his immunosuppressive regimen, he chose to forgo any systemic antifungal therapy. He died from hospital-acquired pneumonia and volume overload unresponsive to diuretics or dialysis.
Phaeohyphomycosis is a rare fungal infection caused by several genera of dematiaceous fungi that are characterized by the presence of melaninlike cell wall pigments thought to locally hinder immune clearance by scavenging phagocyte-derived free radicals. These fungi are ubiquitous in soil and vegetation and usually penetrate the skin at sites of minor trauma.1 Phaeohyphomycosis typically affects immunosuppressed hosts, and its incidence among organ transplant recipients currently is 9%.2 The incidence in this population has been rising, however, as recent advances in immunosuppressive therapies have increased posttransplant survival.3
Subcutaneous phaeohyphomycosis can present with nodules, cysts, tumors, and/or verrucous plaques, and the diagnosis almost always requires clinicopathologic correlation.3 Rapid diagnosis can be made when septate brown hyphae and/or yeast forms are observed on hematoxylin and eosin stain. Rarely, patients present with disseminated infection, characterized by fungemia; central nervous system involvement; and/or infection of multiple deep structures including the eyes, lungs, bones, and sinuses.4 The risk for dissemination from the skin likely is related to the culprit organism’s genus; Lomentospora, Cladophialophora, and Verruconis often are associated with dissemination, while Alternaria, Exophiala, and Fonsecaea typically remain confined to the skin and subcutis.5 Due to this difference and its potential to impact management, obtaining a tissue fungal culture is advisable when phaeohyphomycosis is suspected.
There is no standard treatment of phaeohyphomycosis. Regimens typically consist of excision and prolonged courses of azole therapy, though excision alone with close follow-up may be a reasonable alternative.6 The latter is a particularly important consideration when managing phaeohyphomycosis in organ transplant recipients, as azoles are known cytochrome P450 3A4 inhibitors that can affect serum levels of common immunosuppressive medications including calcineurin inhibitors and mammalian target of rapamycin inhibitors.3 Local recurrence is common regardless of whether azole therapy is pursued,7 and dissemination of localized Exophiala infections is exceedingly rare.8 There is a strong argument to be made for our patient’s decision to forgo antifungal therapy.
This case underscores the difficulty inherent to eradicating local subcutaneous Exophiala phaeohyphomycosis while providing reassurance that with treatment, the risk of life-threatening complications is low. Obtaining tissue for both hematoxylin and eosin stain and sterile culture is crucial to ensuring prompt diagnosis and tailoring the optimal treatment and surveillance strategy to the culprit organism. To avoid delays in diagnosis and treatment, it is important for clinicians to consider phaeohyphomycosis in the differential diagnosis for recurrent nodulocystic lesions in immunosuppressed patients and to recognize that presentations may span many years.
- Bhardwaj S, Capoor MR, Kolte S, et al. Phaeohyphomycosis due to Exophiala jeanselmei: an emerging pathogen in India—case report and review. Mycopathologia. 2016;181:279-284.
- Isa-Isa R, Garcia C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Tirico MCCP, Neto CF, Cruz LL, et al. Clinical spectrum of phaeohyphomycosis in solid organ transplant recipients. JAAD Case Rep. 2016;2:465-469.
- Revankar SG, Patterson JE, Sutton DA, et al. Disseminated phaeohyphomycosis: review of an emerging mycosis. Clin Infect Dis. 2002;34:467-476.
- Revankar SG, Baddley JW, Chen SC-A, et al. A mycoses study group international prospective study of phaeohyphomycosis: an analysis of 99 proven/probable cases. Open Forum Infect Dis. 2017;4:ofx200.
- Oberlin KE, Nichols AJ, Rosa R, et al. Phaeohyphomycosis due to Exophiala infections in solid organ transplant recipients: case report and literature review [published online June 26, 2017]. Transpl Infect Dis. 2017;19. doi:10.1111/tid.12723.
- Shirbur S, Telkar S, Goudar B, et al. Recurrent phaeohyphomycosis: a case report. J Clin Diagn Res. 2013;7:2015-2016.
- Li D-M, Li R-Y, de Hoog GS, et al. Fatal Exophiala infections in China, with a report of seven cases. Mycoses. 2011;54:E136-E142.
To the Editor:
A 73-year-old man presented with a 2.5-cm, recurrent, fluctuant, multiloculated nodule on the left forearm. The lesion was nontender with occasional chalky, white to yellow discharge from multiple sinus tracts. He was otherwise well appearing without signs of systemic infection. He reported similar lesions in slightly different anatomic locations on the left forearm both 7 and 4 years prior to the current presentation. In both instances, the nodules were excised at an outside hospital without any additional treatment. Histopathology of the excised tissue from both prior occasions demonstrated brown septate hyphae surrounded by suppurative and granulomatous inflammation consistent with dematiaceous fungal infection of the dermis (Figures 1 and 2); the organisms were highlighted with periodic acid–Schiff stain.
The patient’s medical history was notable for advanced heart failure with an ejection fraction of 25% and autosomal-dominant polycystic kidney disease. He received an orthotopic kidney transplant 17 years prior to the current presentation. Medications included tacrolimus, mycophenolate mofetil, and prednisone. He denied any trauma or notable exposures to vegetation, and his travel history was unremarkable. A review of systems was negative.
At the current presentation, a sterile fungal culture was performed and found positive for Exophiala species, while bacterial and mycobacterial cultures were negative. A diagnosis of phaeohyphomycosis was made, and he was scheduled for re-excision. Out of concern for interactions with his immunosuppressive regimen, he chose to forgo any systemic antifungal therapy. He died from hospital-acquired pneumonia and volume overload unresponsive to diuretics or dialysis.
Phaeohyphomycosis is a rare fungal infection caused by several genera of dematiaceous fungi that are characterized by the presence of melaninlike cell wall pigments thought to locally hinder immune clearance by scavenging phagocyte-derived free radicals. These fungi are ubiquitous in soil and vegetation and usually penetrate the skin at sites of minor trauma.1 Phaeohyphomycosis typically affects immunosuppressed hosts, and its incidence among organ transplant recipients currently is 9%.2 The incidence in this population has been rising, however, as recent advances in immunosuppressive therapies have increased posttransplant survival.3
Subcutaneous phaeohyphomycosis can present with nodules, cysts, tumors, and/or verrucous plaques, and the diagnosis almost always requires clinicopathologic correlation.3 Rapid diagnosis can be made when septate brown hyphae and/or yeast forms are observed on hematoxylin and eosin stain. Rarely, patients present with disseminated infection, characterized by fungemia; central nervous system involvement; and/or infection of multiple deep structures including the eyes, lungs, bones, and sinuses.4 The risk for dissemination from the skin likely is related to the culprit organism’s genus; Lomentospora, Cladophialophora, and Verruconis often are associated with dissemination, while Alternaria, Exophiala, and Fonsecaea typically remain confined to the skin and subcutis.5 Due to this difference and its potential to impact management, obtaining a tissue fungal culture is advisable when phaeohyphomycosis is suspected.
There is no standard treatment of phaeohyphomycosis. Regimens typically consist of excision and prolonged courses of azole therapy, though excision alone with close follow-up may be a reasonable alternative.6 The latter is a particularly important consideration when managing phaeohyphomycosis in organ transplant recipients, as azoles are known cytochrome P450 3A4 inhibitors that can affect serum levels of common immunosuppressive medications including calcineurin inhibitors and mammalian target of rapamycin inhibitors.3 Local recurrence is common regardless of whether azole therapy is pursued,7 and dissemination of localized Exophiala infections is exceedingly rare.8 There is a strong argument to be made for our patient’s decision to forgo antifungal therapy.
This case underscores the difficulty inherent to eradicating local subcutaneous Exophiala phaeohyphomycosis while providing reassurance that with treatment, the risk of life-threatening complications is low. Obtaining tissue for both hematoxylin and eosin stain and sterile culture is crucial to ensuring prompt diagnosis and tailoring the optimal treatment and surveillance strategy to the culprit organism. To avoid delays in diagnosis and treatment, it is important for clinicians to consider phaeohyphomycosis in the differential diagnosis for recurrent nodulocystic lesions in immunosuppressed patients and to recognize that presentations may span many years.
To the Editor:
A 73-year-old man presented with a 2.5-cm, recurrent, fluctuant, multiloculated nodule on the left forearm. The lesion was nontender with occasional chalky, white to yellow discharge from multiple sinus tracts. He was otherwise well appearing without signs of systemic infection. He reported similar lesions in slightly different anatomic locations on the left forearm both 7 and 4 years prior to the current presentation. In both instances, the nodules were excised at an outside hospital without any additional treatment. Histopathology of the excised tissue from both prior occasions demonstrated brown septate hyphae surrounded by suppurative and granulomatous inflammation consistent with dematiaceous fungal infection of the dermis (Figures 1 and 2); the organisms were highlighted with periodic acid–Schiff stain.
The patient’s medical history was notable for advanced heart failure with an ejection fraction of 25% and autosomal-dominant polycystic kidney disease. He received an orthotopic kidney transplant 17 years prior to the current presentation. Medications included tacrolimus, mycophenolate mofetil, and prednisone. He denied any trauma or notable exposures to vegetation, and his travel history was unremarkable. A review of systems was negative.
At the current presentation, a sterile fungal culture was performed and found positive for Exophiala species, while bacterial and mycobacterial cultures were negative. A diagnosis of phaeohyphomycosis was made, and he was scheduled for re-excision. Out of concern for interactions with his immunosuppressive regimen, he chose to forgo any systemic antifungal therapy. He died from hospital-acquired pneumonia and volume overload unresponsive to diuretics or dialysis.
Phaeohyphomycosis is a rare fungal infection caused by several genera of dematiaceous fungi that are characterized by the presence of melaninlike cell wall pigments thought to locally hinder immune clearance by scavenging phagocyte-derived free radicals. These fungi are ubiquitous in soil and vegetation and usually penetrate the skin at sites of minor trauma.1 Phaeohyphomycosis typically affects immunosuppressed hosts, and its incidence among organ transplant recipients currently is 9%.2 The incidence in this population has been rising, however, as recent advances in immunosuppressive therapies have increased posttransplant survival.3
Subcutaneous phaeohyphomycosis can present with nodules, cysts, tumors, and/or verrucous plaques, and the diagnosis almost always requires clinicopathologic correlation.3 Rapid diagnosis can be made when septate brown hyphae and/or yeast forms are observed on hematoxylin and eosin stain. Rarely, patients present with disseminated infection, characterized by fungemia; central nervous system involvement; and/or infection of multiple deep structures including the eyes, lungs, bones, and sinuses.4 The risk for dissemination from the skin likely is related to the culprit organism’s genus; Lomentospora, Cladophialophora, and Verruconis often are associated with dissemination, while Alternaria, Exophiala, and Fonsecaea typically remain confined to the skin and subcutis.5 Due to this difference and its potential to impact management, obtaining a tissue fungal culture is advisable when phaeohyphomycosis is suspected.
There is no standard treatment of phaeohyphomycosis. Regimens typically consist of excision and prolonged courses of azole therapy, though excision alone with close follow-up may be a reasonable alternative.6 The latter is a particularly important consideration when managing phaeohyphomycosis in organ transplant recipients, as azoles are known cytochrome P450 3A4 inhibitors that can affect serum levels of common immunosuppressive medications including calcineurin inhibitors and mammalian target of rapamycin inhibitors.3 Local recurrence is common regardless of whether azole therapy is pursued,7 and dissemination of localized Exophiala infections is exceedingly rare.8 There is a strong argument to be made for our patient’s decision to forgo antifungal therapy.
This case underscores the difficulty inherent to eradicating local subcutaneous Exophiala phaeohyphomycosis while providing reassurance that with treatment, the risk of life-threatening complications is low. Obtaining tissue for both hematoxylin and eosin stain and sterile culture is crucial to ensuring prompt diagnosis and tailoring the optimal treatment and surveillance strategy to the culprit organism. To avoid delays in diagnosis and treatment, it is important for clinicians to consider phaeohyphomycosis in the differential diagnosis for recurrent nodulocystic lesions in immunosuppressed patients and to recognize that presentations may span many years.
- Bhardwaj S, Capoor MR, Kolte S, et al. Phaeohyphomycosis due to Exophiala jeanselmei: an emerging pathogen in India—case report and review. Mycopathologia. 2016;181:279-284.
- Isa-Isa R, Garcia C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Tirico MCCP, Neto CF, Cruz LL, et al. Clinical spectrum of phaeohyphomycosis in solid organ transplant recipients. JAAD Case Rep. 2016;2:465-469.
- Revankar SG, Patterson JE, Sutton DA, et al. Disseminated phaeohyphomycosis: review of an emerging mycosis. Clin Infect Dis. 2002;34:467-476.
- Revankar SG, Baddley JW, Chen SC-A, et al. A mycoses study group international prospective study of phaeohyphomycosis: an analysis of 99 proven/probable cases. Open Forum Infect Dis. 2017;4:ofx200.
- Oberlin KE, Nichols AJ, Rosa R, et al. Phaeohyphomycosis due to Exophiala infections in solid organ transplant recipients: case report and literature review [published online June 26, 2017]. Transpl Infect Dis. 2017;19. doi:10.1111/tid.12723.
- Shirbur S, Telkar S, Goudar B, et al. Recurrent phaeohyphomycosis: a case report. J Clin Diagn Res. 2013;7:2015-2016.
- Li D-M, Li R-Y, de Hoog GS, et al. Fatal Exophiala infections in China, with a report of seven cases. Mycoses. 2011;54:E136-E142.
- Bhardwaj S, Capoor MR, Kolte S, et al. Phaeohyphomycosis due to Exophiala jeanselmei: an emerging pathogen in India—case report and review. Mycopathologia. 2016;181:279-284.
- Isa-Isa R, Garcia C, Isa M, et al. Subcutaneous phaeohyphomycosis (mycotic cyst). Clin Dermatol. 2012;30:425-431.
- Tirico MCCP, Neto CF, Cruz LL, et al. Clinical spectrum of phaeohyphomycosis in solid organ transplant recipients. JAAD Case Rep. 2016;2:465-469.
- Revankar SG, Patterson JE, Sutton DA, et al. Disseminated phaeohyphomycosis: review of an emerging mycosis. Clin Infect Dis. 2002;34:467-476.
- Revankar SG, Baddley JW, Chen SC-A, et al. A mycoses study group international prospective study of phaeohyphomycosis: an analysis of 99 proven/probable cases. Open Forum Infect Dis. 2017;4:ofx200.
- Oberlin KE, Nichols AJ, Rosa R, et al. Phaeohyphomycosis due to Exophiala infections in solid organ transplant recipients: case report and literature review [published online June 26, 2017]. Transpl Infect Dis. 2017;19. doi:10.1111/tid.12723.
- Shirbur S, Telkar S, Goudar B, et al. Recurrent phaeohyphomycosis: a case report. J Clin Diagn Res. 2013;7:2015-2016.
- Li D-M, Li R-Y, de Hoog GS, et al. Fatal Exophiala infections in China, with a report of seven cases. Mycoses. 2011;54:E136-E142.
Practice Points
- Phaeohyphomycosis is an infection with dematiaceous fungi that most commonly affects immunosuppressed patients.
- Subcutaneous phaeohyphomycosis may present with nodulocystic lesions that recur over the course of years.
- Tissue fungal culture should be obtained when the diagnosis is suspected, as the risk for dissemination is related to the culprit organism.
- Surgical excision with close follow-up may be an appropriate management strategy for patients on immunosuppressive medications to avoid interactions with azole therapy.
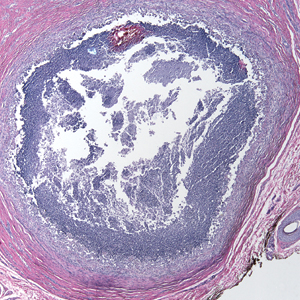
Scalp Arteriovenous Fistula With Intracranial Communication
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
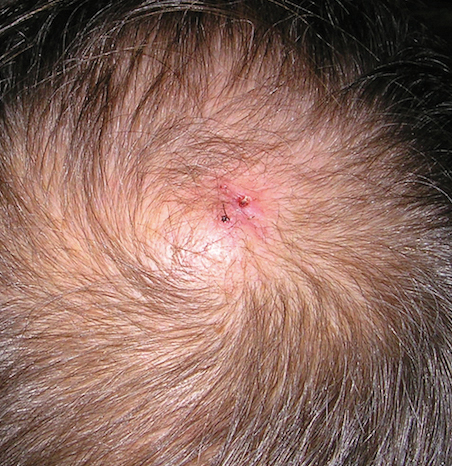
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
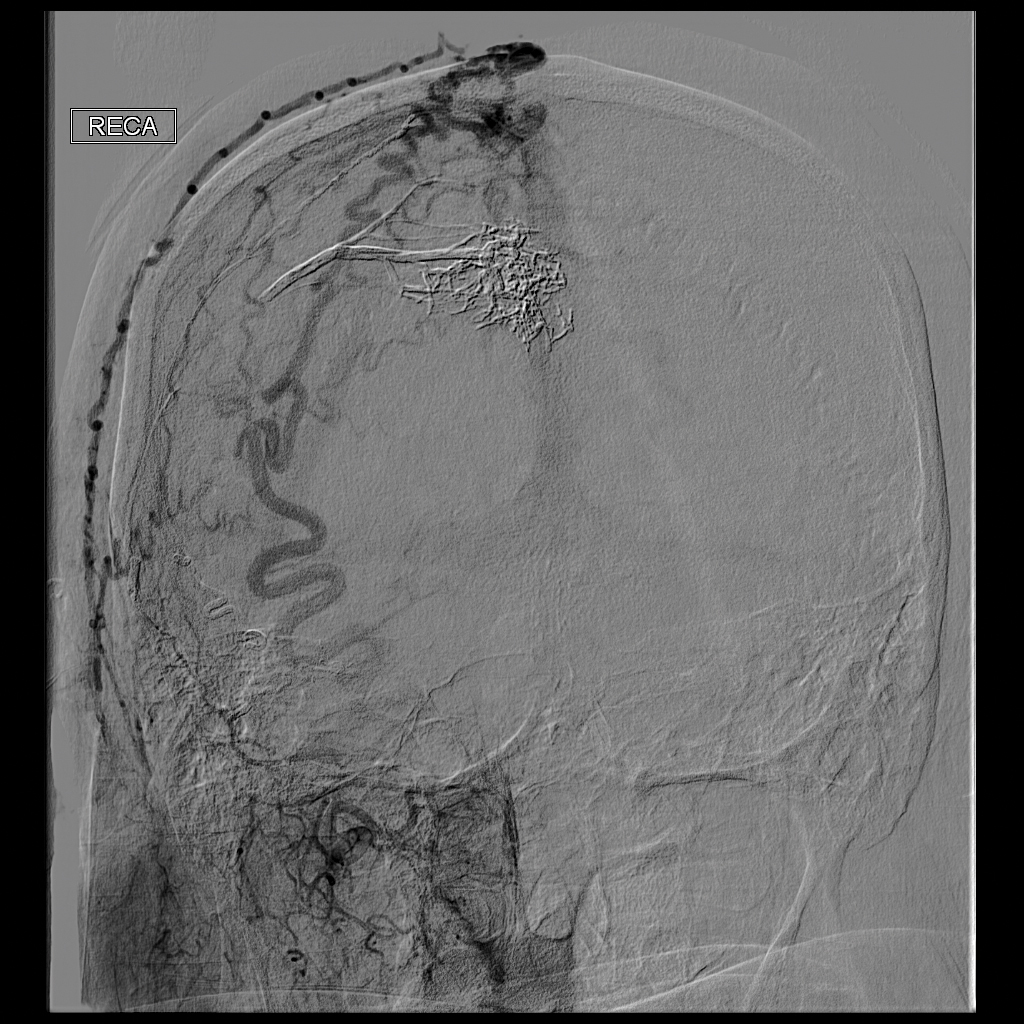
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
To the Editor:
A 71-year-old man presented with a nodule on the vertex of the scalp of 1 year’s duration. The lesion had become soft and tender during the week prior to presentation. He noted that he was experiencing headaches and a buzzing sound in his head. He denied all other neurologic symptoms. The patient was given amoxicillin from a primary care physician and was referred to our institution for evaluation of a presumed inflamed cyst.
The patient’s medical history included an intracranial arteriovenous fistula (AVF) treated with endovascular embolization 1 year prior to presentation, 2 substantial falls in childhood with head trauma and loss of consciousness, essential hypertension, and an aortic aneurysm. His medications included amlodipine, lisinopril, amoxicillin, a multivitamin, and grape seed extract.
Physical examination revealed a 2-cm, pink, somewhat rubbery, subcutaneous, nonmobile nodule on the vertex of the scalp (Figure 1). The lesion was not consistent with a common pilar cyst, and an excisional biopsy was performed to exclude malignancy. Upon superficial incision, the lesion bled moderately, and the procedure was immediately discontinued. Hemostasis was obtained, and the patient was sent for ultrasonography of the lesion.
Ultrasonography demonstrated a small hypoechoic nodule measuring up to 0.5 cm containing a tangle of vessels in the subcutaneous soft tissue corresponding to the palpable abnormality. A cerebral angiogram demonstrated a dural AVF of the superior sagittal sinus with multifocal supply that connected with this scalp nodule (Figure 2). The patient was treated by interventional neuroradiology with endovascular embolization, which resulted in complete resolution of the scalp nodule.
Scalp arteriovenous fistulas (S-AVFs) are characterized by abnormal connections between supplying arteries and draining veins in the subcutaneous plane of the scalp.1,2 The veins of an S-AVF undergo progressive aneurysmal dilatation from abnormal hemodynamics.1-3 Scalp arteriovenous fistulas are rare and may present as either an innocuous-looking scalp nodule or a progressively enlarging pulsatile mass on the scalp.2-4 Associated symptoms often include headache, local pain, bruits, tinnitus, and thrill.1,3,4 Recurrent hemorrhage, scalp necrosis, congestive heart failure, epilepsy, mental retardation, and intracranial ischemia also may occur.4
Scalp AVFs may occur with or without intracranial communication.4 Spontaneous S-AVFs with intracranial communication are uncommon, and their etiology is unclear. They may form as congenital malformations or may be idiopathic. Factors increasing circulation through the S-AVF such as trauma, pregnancy, hormonal changes, and inflammation prompt the development of symptoms.4 Scalp AVFs also may be caused by trauma.3 Scalp AVFs without intracranial communication have been reported following hair transplantation.1 Scalp AVFs with intracranial communication have been reported months to years after skull fracture or craniotomy.2 True spontaneous S-AVFs are difficult to differentiate from traumatic S-AVFs other than by history alone.2
Increased venous pressure has been shown to generate AVFs in rats.5 It has been suggested that S-AVFs can become enlarged by capturing subcutaneous or intracranial feeder vessels and that the consequent hemodynamic stress may induce de novo aneurysms in S-AVFs. Additionally, intracranial AVFs may alter the intracranial hemodynamics, leading to increased venous pressure in the superior sagittal sinus and the formation of communicating S-AVFs.5 Interestingly, our patient had an intracranial AVF treated with endovascular embolization 1 year prior to the formation of the S-AVF. An angiogram at the time of this embolization procedure did not demonstrate any S-AVFs. Furthermore, our patient has a history of 2 substantial falls in childhood with head trauma and loss of consciousness. Perhaps these traumas initiated a channel through the cranium where an S-AVF with intracranial communication was able to form and may have only become clinically or radiographically detectable once it enlarged due to the altered hemodynamics caused by the intracranial AVF 1 year prior.
The diagnosis of an S-AVF is confirmed with imaging studies. Doppler ultrasonography initially will help to detect that a lesion is vascular in nature. Intra-arterial digital subtraction angiography is the gold-standard imaging technique and is necessary to delineate the feeding arteries and the draining channels as well as possible communication with intracranial vasculature.1,2 There is controversy regarding the appropriate treatment of S-AVFs.2 Each S-AVF possesses unique anatomic features that dictate appropriate management. The prognosis for an S-AVF is extremely variable, and the decision to treat is based on the patient’s symptoms and risk for exsanguinating hemorrhage.2,4 Neurosurgical approaches include ligation of the feeding arteries, surgical resection, electrothrombosis, direct intralesional injection of sclerosing agents, and endovascular embolization. Endovascular intervention increasingly is utilized as a primary treatment or as a preoperative adjunct to surgery.2,4 Large S-AVFs have a high risk for recurrence after treatment with endovascular embolization alone. In cases with intracranial communication, the intracranial component is treated first.2
This case emphasizes the importance of including S-AVFs on the dermatologic differential diagnosis of a scalp nodule, especially in patients with any history of intracranial AVF. A thorough history, detailed intake of potential signs and symptoms of AVF, and palpation for bruits is recommended as part of the surgical evaluation of a scalp nodule. Imaging of scalp nodules also should be considered for patients with any history of intracranial AVF; S-AVFs should be referred to neurosurgery or interventional neuroradiology for evaluation and possible treatment.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
- Bernstein J, Podnos S, Leavitt M. Arteriovenous fistula following hair transplantation. Dermatol Surg. 2011;37:873-875.
- Kumar R, Sharma G, Sharma BS. Management of scalp arterio-venous malformation: case series and review of literature. Br J Neurosurg. 2012;26:371-377.
- Gurkanlar D, Gonul M, Solmaz I, et al. Cirsoid aneurysms of the scalp. Neurosurg Rev. 2006;29:208-212.
- Senoglu M, Yasim A, Gokce M, et al. Nontraumatic scalp arteriovenous fistula in an adult: technical report on an illustrative case. Surg Neurol. 2008;70:194-197.
- Lanzino G, Passacantilli E, Lemole G, et al. Scalp arteriovenous malformation draining into the superior sagittal sinus associated with an intracranial arteriovenous malformation: just a coincidence? case report. Neurosurgery. 2003;52:440-443.
Practice Points
- Scalp arteriovenous fistulas may be traumatic or spontaneous and present as either an innocuous-looking scalp nodule or as a progressively enlarging pulsatile mass on the scalp.
- Clinical detection followed by appropriate imaging and referral to neurosurgery or interventional neuroradiology is vital to patient safety.
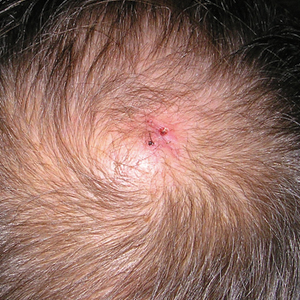
Oral Hairy Leukoplakia Associated With the Use of Adalimumab
To the Editor:
Oral hairy leukoplakia (OHL) is an Epstein-Barr virus (EBV)–mediated mucocutaneous disease that often involves the lingual epithelium. The lateral portions of the tongue are the most commonly affected sites. The lesions often are described as asymptomatic, white, corrugated patches or plaques that are unable to be scraped off.1 Oral hairy leukoplakia was first identified in 1984 and was considered to be associated with AIDS.2 An association between the presence of OHL and the degree of immunosuppression as well as the severity of human immunodeficiency virus (HIV) has been reported.3 Although OHL initially was considered to be pathognomonic for HIV, it has since been described in multiple other immunosuppressive conditions.4 Numerous medical conditions and combinations of immunosuppressive medications have been associated with OHL in patients who were HIV negative.5
Adalimumab is an injectable human IgG1 recombinant antibody to tumor necrosis factor α (TNF-α).6 It currently is approved by the US Food and Drug Administration for the treatment of rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult and pediatric Crohn disease, ulcerative colitis, noninfectious uveitis, hidradenitis suppurativa, and plaque psoriasis.7 We report a case of OHL associated with the use of adalimumab.
A 47-year-old woman initially presented with chronic plaque-type psoriasis. Her medical history was notable for bipolar disorder, migraines, hypertension, and tobacco use. The patient’s psoriasis initially was well controlled on a regimen of topical steroids and methotrexate; however, methotrexate was stopped after 2.5 years due to a mildly elevated alanine aminotransferase level, as well as an abnormal liver biopsy showing mildly active (grade 1 of 3) steatohepatitis with portal chronic inflammation, pericellular fibrosis, and portal and focal periportal fibrosis (stage 1-2 of 4). The patient and her dermatologist were uncomfortable continuing methotrexate with these findings. After baseline screening including a negative purified protein derivative skin test, adalimumab was initiated. A loading dose of 80 mg subcutaneously (SQ) was given, followed by adalimumab 40 mg SQ 1 week later and 40 mg every other week as maintenance.
The patient’s psoriasis was well controlled with adalimumab for 22 months, but she then developed a thin white plaque on the right lateral tongue (Figure 1). An incisional biopsy of the tongue performed by an oral surgeon revealed hyperkeratosis with Candida colonization and viral cytopathic effect (Figure 2). An EBV DNA in situ hybridization stain revealed focal positivity within these cells (Figure 3), leading to a diagnosis of OHL. Laboratory evaluation demonstrated a normal complete blood cell count with differential and liver panel as well as a negative HIV test. The patient otherwise felt well and denied fevers, lymphadenopathy, and weight loss.
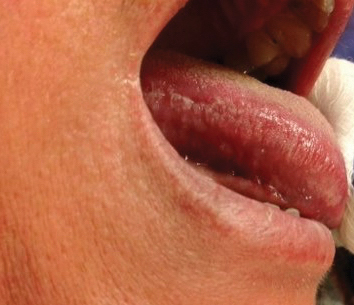
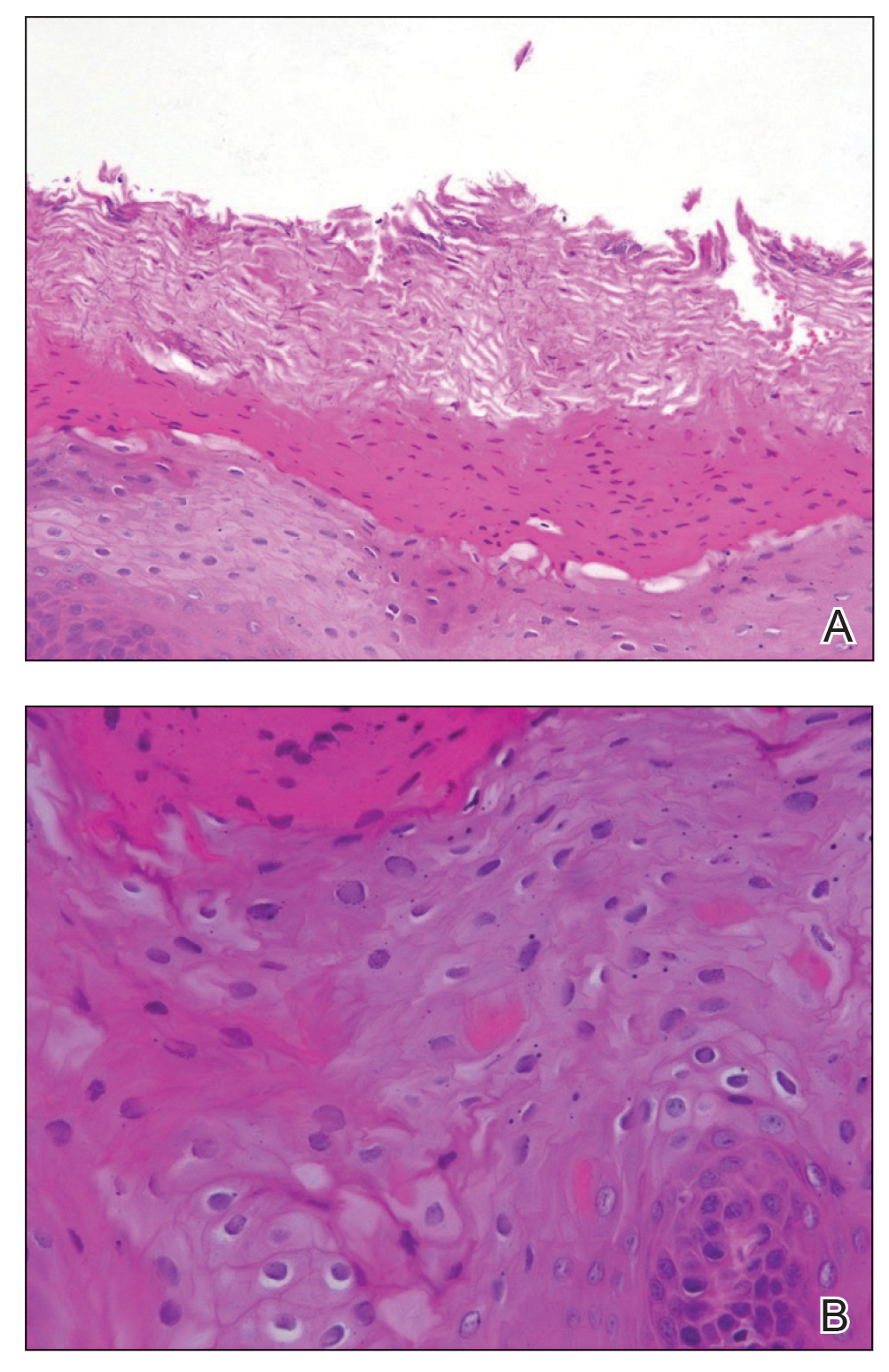
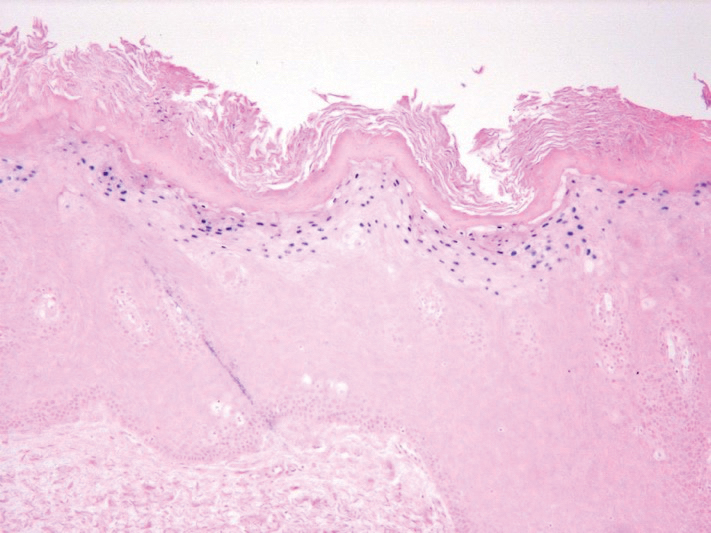
We consulted with an infectious disease and immunodeficiency specialist regarding the patient’s case. Before conducting further evaluation beyond HIV screening for immunodeficiency states, adalimumab was discontinued to see if the OHL would spontaneously resolve. Three months after discontinuation of adalimumab, the white plaque on the right lateral tongue was notably improved. The OHL continued to disappear and was completely resolved 1 year after discontinuation of adalimumab. The patient’s psoriasis had subsequently remained well controlled with diet and weight loss, smoking cessation, topical steroids, and apremilast without any recurrence of the OHL.
Oral hairy leukoplakia is associated with upregulated EBV replication and EBV-encoded proteins such as latent membrane protein 1.2 It often presents as white or gray patches on the lateral lingual margins with prominent folds and/or projections, giving a shaggy appearance. Oral hairy leukoplakia often is specific for HIV infection and rarely is associated with other immunodeficiencies.2 Prasad and Bilodeau5 performed a literature review of medical conditions and immunosuppressive medications associated with OHL in patients without HIV. Allogeneic transplant was associated with the highest incidence of OHL in HIV-negative patients (59.2% [45/76]).5 Various combinations of immunosuppressive medications (eg, prednisone, cyclosporine, azathioprine) also may be implicated in cases of HIV-negative patients with OHL. A case of OHL also has been reported with long-standing use of inhaled corticosteroids in an immunocompetent, HIV-negative patient.6 Another case was reported with long-term use of the aromatic antiepileptic lamotrigine, which resolved once stopping the medication.8 Although EBV is an oncovirus and has been associated with lymphoproliferative disorders and nasopharyngeal carcinoma, OHL is not considered to be a premalignant lesion.7 Despite the strong association between OHL and HIV, our patient was HIV negative. The only immunocompromising factor in our patient was the use of adalimumab to treat psoriasis. We did not conduct further testing for immunodeficiency states because the OHL spontaneously resolved when the adalimumab was discontinued.
PubMed and Ovid searches of articles indexed for MEDLINE using the terms adalimumab and oral hairy leukoplakia as well as TNF-alpha inhibitor and oral hairy leukoplakia with humans and English language as limitations revealed that no cases have been reported in the literature demonstrating an association between OHL and adalimumab or any other TNF-α inhibitor. However, Cetkovska et al9 reported a case of EBV hepatitis and subsequently chronic hepatitis as a complication of infliximab used for the treatment of chronic psoriasis. Because TNF-α and IFN-γ play an important role in controlling viral infections, there is an increased risk for reactivating a viral illness when depleting TNF through pharmacologic measures (ie, adalimumab, infliximab).8 Another case of EBV-associated plasmablastic lymphoma was reported after 1 year of adalimumab use in a patient with Crohn disease. The plasmablastic lymphoma resolved after 4 rounds of chemotherapy.10
The only contraindication for the use of adalimumab is a known hypersensitivity to the drug. Relative contraindications for use of adalimumab include active tuberculosis, demyelinating disease, hematologic diseases (ie, thrombocytopenia, pancytopenia), lymphoma, hepatitis C, and hepatitis B.11 The most common adverse effect of adalimumab is an injection-site reaction. Additional reported adverse effects of TNF-α inhibitors as a class are lymphoma, melanoma, nonmelanoma skin cancer, reactivation of latent tuberculosis, congestive heart failure, autoimmunity, and hematologic toxicity.11
This case demonstrates an association between adalimumab and OHL in an HIV-negative patient. Although the mechanism behind OHL and immunosuppression remains to be elucidated, this association is important to keep in mind when using adalimumab or other TNF-α inhibitors for the treatment of psoriasis or other medical conditions.
- Triantos D, Porter SR, Scully C, et al. Oral hairy leukoplakia: clinicopathologic features, pathogenesis, diagnosis, and clinical significance. Clin Infect Dis. 1997;25:1392-1396.
- Greenspan D, Greenspan JS, Conant M, et al. Oral “hairy” leucoplakia in male homosexuals: evidence of association with both papillomavirus and a herpes-group virus. Lancet. 1984;2:831-834.
- Glick M, Muzyka BC, Lurie D, et al. Oral manifestations associated with HIV-related disease as marks for immune suppression and AIDS. Oral Surg Oral Med Oral Pathol. 1994;77:344-349.
- Chambers AE, Conn B, Pemberton M, et al. Twenty-first-century oral hair leukoplakia—a non-HIV-associated entity. Oral Surg Oral Med Oral Patho Oral Radiol. 2015;119:326-332.
- Prasad JL, Bilodeau EA. Oral hairy leukoplakia in patients without HIV: presentation of 2 new cases. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:E151-E160.
- Moffat M, Jauhar S, Jones ME, et al. Oral hairy leukoplakia in an HIV-negative, immunocompetent patient. Oral Biosci Med. 2005;2:282-284.
- Greenspan JS, Greenspan D. Oral hairy leukoplakia: diagnosis and management. Oral Surg Oral Med Oral Pathol. 1989;67:396-403.
- Gordins P, Sloan P, Spickett GP, et al. Oral hairy leukoplakia in a patient on long-term anticonvulsant treatment with lamotrigine. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:E17-E23.
- Cetkovska P, Lomicova I, Mukensnabl P, et al. Anti-tumour necrosis factor treatment of severe psoriasis complicated by Epstein-Barr virus hepatitis and subsequently by chronic hepatitis. Dermatol Ther. 2015;28:369-372.
- Liu L, Charabaty A, Ozdemirli M. EBV-associated plasmablastic lymphoma in a patient with Crohn’s disease after adalimumab treatment. J Crohns Colitis. 2013;7:E118-E119.
- Humira [package insert]. North Chicago, IL: AbbVie Inc; 2018.
To the Editor:
Oral hairy leukoplakia (OHL) is an Epstein-Barr virus (EBV)–mediated mucocutaneous disease that often involves the lingual epithelium. The lateral portions of the tongue are the most commonly affected sites. The lesions often are described as asymptomatic, white, corrugated patches or plaques that are unable to be scraped off.1 Oral hairy leukoplakia was first identified in 1984 and was considered to be associated with AIDS.2 An association between the presence of OHL and the degree of immunosuppression as well as the severity of human immunodeficiency virus (HIV) has been reported.3 Although OHL initially was considered to be pathognomonic for HIV, it has since been described in multiple other immunosuppressive conditions.4 Numerous medical conditions and combinations of immunosuppressive medications have been associated with OHL in patients who were HIV negative.5
Adalimumab is an injectable human IgG1 recombinant antibody to tumor necrosis factor α (TNF-α).6 It currently is approved by the US Food and Drug Administration for the treatment of rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult and pediatric Crohn disease, ulcerative colitis, noninfectious uveitis, hidradenitis suppurativa, and plaque psoriasis.7 We report a case of OHL associated with the use of adalimumab.
A 47-year-old woman initially presented with chronic plaque-type psoriasis. Her medical history was notable for bipolar disorder, migraines, hypertension, and tobacco use. The patient’s psoriasis initially was well controlled on a regimen of topical steroids and methotrexate; however, methotrexate was stopped after 2.5 years due to a mildly elevated alanine aminotransferase level, as well as an abnormal liver biopsy showing mildly active (grade 1 of 3) steatohepatitis with portal chronic inflammation, pericellular fibrosis, and portal and focal periportal fibrosis (stage 1-2 of 4). The patient and her dermatologist were uncomfortable continuing methotrexate with these findings. After baseline screening including a negative purified protein derivative skin test, adalimumab was initiated. A loading dose of 80 mg subcutaneously (SQ) was given, followed by adalimumab 40 mg SQ 1 week later and 40 mg every other week as maintenance.
The patient’s psoriasis was well controlled with adalimumab for 22 months, but she then developed a thin white plaque on the right lateral tongue (Figure 1). An incisional biopsy of the tongue performed by an oral surgeon revealed hyperkeratosis with Candida colonization and viral cytopathic effect (Figure 2). An EBV DNA in situ hybridization stain revealed focal positivity within these cells (Figure 3), leading to a diagnosis of OHL. Laboratory evaluation demonstrated a normal complete blood cell count with differential and liver panel as well as a negative HIV test. The patient otherwise felt well and denied fevers, lymphadenopathy, and weight loss.
We consulted with an infectious disease and immunodeficiency specialist regarding the patient’s case. Before conducting further evaluation beyond HIV screening for immunodeficiency states, adalimumab was discontinued to see if the OHL would spontaneously resolve. Three months after discontinuation of adalimumab, the white plaque on the right lateral tongue was notably improved. The OHL continued to disappear and was completely resolved 1 year after discontinuation of adalimumab. The patient’s psoriasis had subsequently remained well controlled with diet and weight loss, smoking cessation, topical steroids, and apremilast without any recurrence of the OHL.
Oral hairy leukoplakia is associated with upregulated EBV replication and EBV-encoded proteins such as latent membrane protein 1.2 It often presents as white or gray patches on the lateral lingual margins with prominent folds and/or projections, giving a shaggy appearance. Oral hairy leukoplakia often is specific for HIV infection and rarely is associated with other immunodeficiencies.2 Prasad and Bilodeau5 performed a literature review of medical conditions and immunosuppressive medications associated with OHL in patients without HIV. Allogeneic transplant was associated with the highest incidence of OHL in HIV-negative patients (59.2% [45/76]).5 Various combinations of immunosuppressive medications (eg, prednisone, cyclosporine, azathioprine) also may be implicated in cases of HIV-negative patients with OHL. A case of OHL also has been reported with long-standing use of inhaled corticosteroids in an immunocompetent, HIV-negative patient.6 Another case was reported with long-term use of the aromatic antiepileptic lamotrigine, which resolved once stopping the medication.8 Although EBV is an oncovirus and has been associated with lymphoproliferative disorders and nasopharyngeal carcinoma, OHL is not considered to be a premalignant lesion.7 Despite the strong association between OHL and HIV, our patient was HIV negative. The only immunocompromising factor in our patient was the use of adalimumab to treat psoriasis. We did not conduct further testing for immunodeficiency states because the OHL spontaneously resolved when the adalimumab was discontinued.
PubMed and Ovid searches of articles indexed for MEDLINE using the terms adalimumab and oral hairy leukoplakia as well as TNF-alpha inhibitor and oral hairy leukoplakia with humans and English language as limitations revealed that no cases have been reported in the literature demonstrating an association between OHL and adalimumab or any other TNF-α inhibitor. However, Cetkovska et al9 reported a case of EBV hepatitis and subsequently chronic hepatitis as a complication of infliximab used for the treatment of chronic psoriasis. Because TNF-α and IFN-γ play an important role in controlling viral infections, there is an increased risk for reactivating a viral illness when depleting TNF through pharmacologic measures (ie, adalimumab, infliximab).8 Another case of EBV-associated plasmablastic lymphoma was reported after 1 year of adalimumab use in a patient with Crohn disease. The plasmablastic lymphoma resolved after 4 rounds of chemotherapy.10
The only contraindication for the use of adalimumab is a known hypersensitivity to the drug. Relative contraindications for use of adalimumab include active tuberculosis, demyelinating disease, hematologic diseases (ie, thrombocytopenia, pancytopenia), lymphoma, hepatitis C, and hepatitis B.11 The most common adverse effect of adalimumab is an injection-site reaction. Additional reported adverse effects of TNF-α inhibitors as a class are lymphoma, melanoma, nonmelanoma skin cancer, reactivation of latent tuberculosis, congestive heart failure, autoimmunity, and hematologic toxicity.11
This case demonstrates an association between adalimumab and OHL in an HIV-negative patient. Although the mechanism behind OHL and immunosuppression remains to be elucidated, this association is important to keep in mind when using adalimumab or other TNF-α inhibitors for the treatment of psoriasis or other medical conditions.
To the Editor:
Oral hairy leukoplakia (OHL) is an Epstein-Barr virus (EBV)–mediated mucocutaneous disease that often involves the lingual epithelium. The lateral portions of the tongue are the most commonly affected sites. The lesions often are described as asymptomatic, white, corrugated patches or plaques that are unable to be scraped off.1 Oral hairy leukoplakia was first identified in 1984 and was considered to be associated with AIDS.2 An association between the presence of OHL and the degree of immunosuppression as well as the severity of human immunodeficiency virus (HIV) has been reported.3 Although OHL initially was considered to be pathognomonic for HIV, it has since been described in multiple other immunosuppressive conditions.4 Numerous medical conditions and combinations of immunosuppressive medications have been associated with OHL in patients who were HIV negative.5
Adalimumab is an injectable human IgG1 recombinant antibody to tumor necrosis factor α (TNF-α).6 It currently is approved by the US Food and Drug Administration for the treatment of rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, adult and pediatric Crohn disease, ulcerative colitis, noninfectious uveitis, hidradenitis suppurativa, and plaque psoriasis.7 We report a case of OHL associated with the use of adalimumab.
A 47-year-old woman initially presented with chronic plaque-type psoriasis. Her medical history was notable for bipolar disorder, migraines, hypertension, and tobacco use. The patient’s psoriasis initially was well controlled on a regimen of topical steroids and methotrexate; however, methotrexate was stopped after 2.5 years due to a mildly elevated alanine aminotransferase level, as well as an abnormal liver biopsy showing mildly active (grade 1 of 3) steatohepatitis with portal chronic inflammation, pericellular fibrosis, and portal and focal periportal fibrosis (stage 1-2 of 4). The patient and her dermatologist were uncomfortable continuing methotrexate with these findings. After baseline screening including a negative purified protein derivative skin test, adalimumab was initiated. A loading dose of 80 mg subcutaneously (SQ) was given, followed by adalimumab 40 mg SQ 1 week later and 40 mg every other week as maintenance.
The patient’s psoriasis was well controlled with adalimumab for 22 months, but she then developed a thin white plaque on the right lateral tongue (Figure 1). An incisional biopsy of the tongue performed by an oral surgeon revealed hyperkeratosis with Candida colonization and viral cytopathic effect (Figure 2). An EBV DNA in situ hybridization stain revealed focal positivity within these cells (Figure 3), leading to a diagnosis of OHL. Laboratory evaluation demonstrated a normal complete blood cell count with differential and liver panel as well as a negative HIV test. The patient otherwise felt well and denied fevers, lymphadenopathy, and weight loss.
We consulted with an infectious disease and immunodeficiency specialist regarding the patient’s case. Before conducting further evaluation beyond HIV screening for immunodeficiency states, adalimumab was discontinued to see if the OHL would spontaneously resolve. Three months after discontinuation of adalimumab, the white plaque on the right lateral tongue was notably improved. The OHL continued to disappear and was completely resolved 1 year after discontinuation of adalimumab. The patient’s psoriasis had subsequently remained well controlled with diet and weight loss, smoking cessation, topical steroids, and apremilast without any recurrence of the OHL.
Oral hairy leukoplakia is associated with upregulated EBV replication and EBV-encoded proteins such as latent membrane protein 1.2 It often presents as white or gray patches on the lateral lingual margins with prominent folds and/or projections, giving a shaggy appearance. Oral hairy leukoplakia often is specific for HIV infection and rarely is associated with other immunodeficiencies.2 Prasad and Bilodeau5 performed a literature review of medical conditions and immunosuppressive medications associated with OHL in patients without HIV. Allogeneic transplant was associated with the highest incidence of OHL in HIV-negative patients (59.2% [45/76]).5 Various combinations of immunosuppressive medications (eg, prednisone, cyclosporine, azathioprine) also may be implicated in cases of HIV-negative patients with OHL. A case of OHL also has been reported with long-standing use of inhaled corticosteroids in an immunocompetent, HIV-negative patient.6 Another case was reported with long-term use of the aromatic antiepileptic lamotrigine, which resolved once stopping the medication.8 Although EBV is an oncovirus and has been associated with lymphoproliferative disorders and nasopharyngeal carcinoma, OHL is not considered to be a premalignant lesion.7 Despite the strong association between OHL and HIV, our patient was HIV negative. The only immunocompromising factor in our patient was the use of adalimumab to treat psoriasis. We did not conduct further testing for immunodeficiency states because the OHL spontaneously resolved when the adalimumab was discontinued.
PubMed and Ovid searches of articles indexed for MEDLINE using the terms adalimumab and oral hairy leukoplakia as well as TNF-alpha inhibitor and oral hairy leukoplakia with humans and English language as limitations revealed that no cases have been reported in the literature demonstrating an association between OHL and adalimumab or any other TNF-α inhibitor. However, Cetkovska et al9 reported a case of EBV hepatitis and subsequently chronic hepatitis as a complication of infliximab used for the treatment of chronic psoriasis. Because TNF-α and IFN-γ play an important role in controlling viral infections, there is an increased risk for reactivating a viral illness when depleting TNF through pharmacologic measures (ie, adalimumab, infliximab).8 Another case of EBV-associated plasmablastic lymphoma was reported after 1 year of adalimumab use in a patient with Crohn disease. The plasmablastic lymphoma resolved after 4 rounds of chemotherapy.10
The only contraindication for the use of adalimumab is a known hypersensitivity to the drug. Relative contraindications for use of adalimumab include active tuberculosis, demyelinating disease, hematologic diseases (ie, thrombocytopenia, pancytopenia), lymphoma, hepatitis C, and hepatitis B.11 The most common adverse effect of adalimumab is an injection-site reaction. Additional reported adverse effects of TNF-α inhibitors as a class are lymphoma, melanoma, nonmelanoma skin cancer, reactivation of latent tuberculosis, congestive heart failure, autoimmunity, and hematologic toxicity.11
This case demonstrates an association between adalimumab and OHL in an HIV-negative patient. Although the mechanism behind OHL and immunosuppression remains to be elucidated, this association is important to keep in mind when using adalimumab or other TNF-α inhibitors for the treatment of psoriasis or other medical conditions.
- Triantos D, Porter SR, Scully C, et al. Oral hairy leukoplakia: clinicopathologic features, pathogenesis, diagnosis, and clinical significance. Clin Infect Dis. 1997;25:1392-1396.
- Greenspan D, Greenspan JS, Conant M, et al. Oral “hairy” leucoplakia in male homosexuals: evidence of association with both papillomavirus and a herpes-group virus. Lancet. 1984;2:831-834.
- Glick M, Muzyka BC, Lurie D, et al. Oral manifestations associated with HIV-related disease as marks for immune suppression and AIDS. Oral Surg Oral Med Oral Pathol. 1994;77:344-349.
- Chambers AE, Conn B, Pemberton M, et al. Twenty-first-century oral hair leukoplakia—a non-HIV-associated entity. Oral Surg Oral Med Oral Patho Oral Radiol. 2015;119:326-332.
- Prasad JL, Bilodeau EA. Oral hairy leukoplakia in patients without HIV: presentation of 2 new cases. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:E151-E160.
- Moffat M, Jauhar S, Jones ME, et al. Oral hairy leukoplakia in an HIV-negative, immunocompetent patient. Oral Biosci Med. 2005;2:282-284.
- Greenspan JS, Greenspan D. Oral hairy leukoplakia: diagnosis and management. Oral Surg Oral Med Oral Pathol. 1989;67:396-403.
- Gordins P, Sloan P, Spickett GP, et al. Oral hairy leukoplakia in a patient on long-term anticonvulsant treatment with lamotrigine. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:E17-E23.
- Cetkovska P, Lomicova I, Mukensnabl P, et al. Anti-tumour necrosis factor treatment of severe psoriasis complicated by Epstein-Barr virus hepatitis and subsequently by chronic hepatitis. Dermatol Ther. 2015;28:369-372.
- Liu L, Charabaty A, Ozdemirli M. EBV-associated plasmablastic lymphoma in a patient with Crohn’s disease after adalimumab treatment. J Crohns Colitis. 2013;7:E118-E119.
- Humira [package insert]. North Chicago, IL: AbbVie Inc; 2018.
- Triantos D, Porter SR, Scully C, et al. Oral hairy leukoplakia: clinicopathologic features, pathogenesis, diagnosis, and clinical significance. Clin Infect Dis. 1997;25:1392-1396.
- Greenspan D, Greenspan JS, Conant M, et al. Oral “hairy” leucoplakia in male homosexuals: evidence of association with both papillomavirus and a herpes-group virus. Lancet. 1984;2:831-834.
- Glick M, Muzyka BC, Lurie D, et al. Oral manifestations associated with HIV-related disease as marks for immune suppression and AIDS. Oral Surg Oral Med Oral Pathol. 1994;77:344-349.
- Chambers AE, Conn B, Pemberton M, et al. Twenty-first-century oral hair leukoplakia—a non-HIV-associated entity. Oral Surg Oral Med Oral Patho Oral Radiol. 2015;119:326-332.
- Prasad JL, Bilodeau EA. Oral hairy leukoplakia in patients without HIV: presentation of 2 new cases. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:E151-E160.
- Moffat M, Jauhar S, Jones ME, et al. Oral hairy leukoplakia in an HIV-negative, immunocompetent patient. Oral Biosci Med. 2005;2:282-284.
- Greenspan JS, Greenspan D. Oral hairy leukoplakia: diagnosis and management. Oral Surg Oral Med Oral Pathol. 1989;67:396-403.
- Gordins P, Sloan P, Spickett GP, et al. Oral hairy leukoplakia in a patient on long-term anticonvulsant treatment with lamotrigine. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111:E17-E23.
- Cetkovska P, Lomicova I, Mukensnabl P, et al. Anti-tumour necrosis factor treatment of severe psoriasis complicated by Epstein-Barr virus hepatitis and subsequently by chronic hepatitis. Dermatol Ther. 2015;28:369-372.
- Liu L, Charabaty A, Ozdemirli M. EBV-associated plasmablastic lymphoma in a patient with Crohn’s disease after adalimumab treatment. J Crohns Colitis. 2013;7:E118-E119.
- Humira [package insert]. North Chicago, IL: AbbVie Inc; 2018.
Practice Points
- Workup for new-onset oral hairy leukoplakia should include a comprehensive medication history.
- Oral hairy leukoplakia is an uncommon side effect of adalimumab.
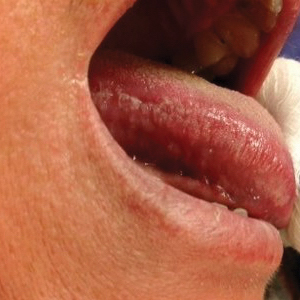
Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis Overlap Syndrome in a Patient With Relapsing Polychondritis
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.

Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.
Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.
Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
- Watkins S, Magill JM Jr, Ramos-Caro FA. Annular eruption preceding relapsing polychondritis: case report and review of the literature. Int J Dermatol. 2009;48:356-362.
- Damiani JM, Levine HL. Relapsing polychondritis—report of ten cases. Laryngoscope. 1979;89:929-46.
- Puéchal X, Terrier B, Mouthon L, et al. Relapsing polychondritis. Joint Bone Spine. 2014;81:118-24.
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis. Autoimmun Rev. 2014;13:90-95.
- Harr T, French L. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;16;5:39.
Practice Points
- The clinical presentation of relapsing polychondritis (RP) may demonstrate cutaneous manifestations other than the typical inflammation of cartilage-rich structures.
- Approximately 30% of patients with RP will have another autoimmune disease.