User login
Wiping Away Cellulitis: A Case of Factitious Disorder
To the Editor:
Patients with psychocutaneous disorders present unique challenges to physicians. We illustrate the critical role that dermoscopy may play to illuminate exogenous skin pathology.
A 50-year-old woman with a reported medical history of systemic lupus erythematosus, chronic pain, and nonhealing leg ulcers presented to the emergency department with severe pain of the left lower leg and redness that was concerning for cellulitis. She sought treatment at an outside hospital for cellulitis 2 weeks prior but left against medical advice. Symptomatic review revealed chest pain, shortness of breath, nausea, vomiting, and diarrhea. The primary team started her on intravenous clindamycin and vancomycin for the presumed infection and scheduled narcotic medications due to concerns of intractable pain in the left leg. The dermatology department was consulted after failure to improve with 1 week of systemic antibiotics.
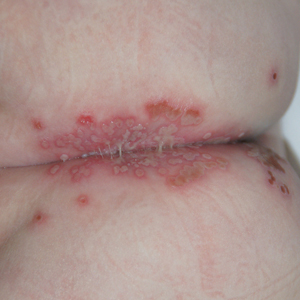
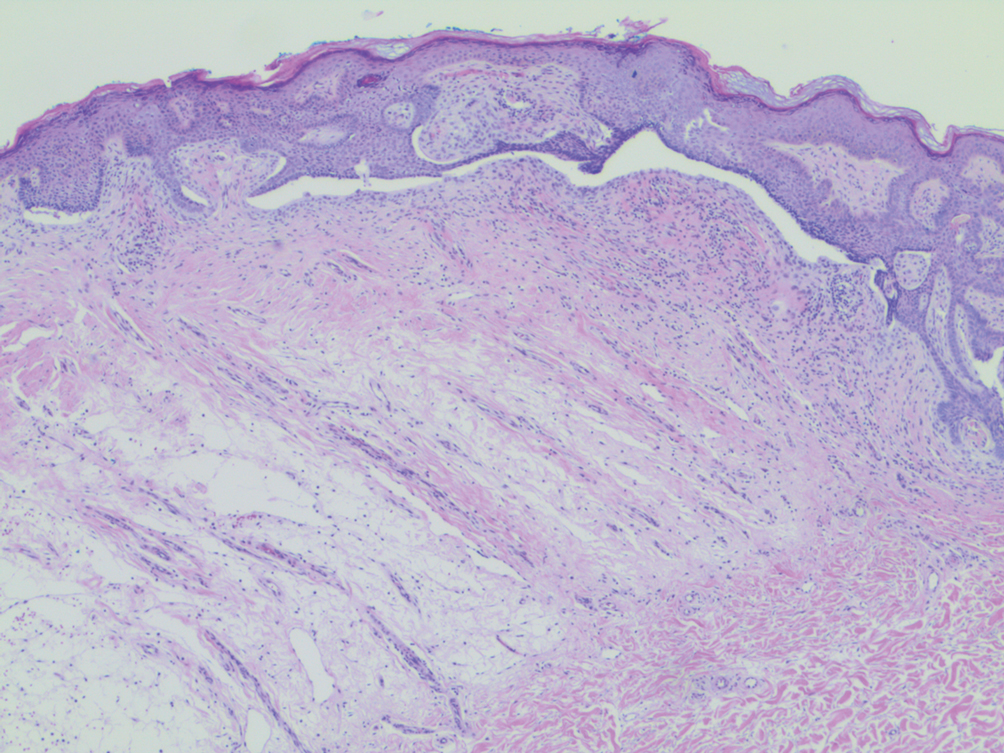
Physical examination revealed a geometric, atrophic, purple plaque on the left anterior shin from a prior leg ulcer as well as a diffuse red-pink patch extending from the knee to the ankle. Notably, the cellulitis spared the left posterior calf resting against the sheet and had a sharp line of demarcation at the distal shin. The leg was cool to the touch while the patient was distractible. She later reported that the leg was extremely tender to palpation. Dermoscopy revealed linear red pigments within skin furrows that accentuated skin lines (Figure). These findings raised suspicions of an external manipulation. The skin was wiped with an alcohol pad that removed a shimmering pink substance consistent in appearance to a cosmetic product. The skin beneath the cellulitis appeared normal.
On further review of the patient’s medical record, it was noted that she was admitted several months ago for ulcers of the left leg. She had been to multiple hospitals and had numerous rounds of antibiotics. Biopsy of an ulcer revealed dermal fibrosis consistent with scarring. Aerobic bacteria, atypical mycobacteria, and fungal cultures were all negative. The physicians suspected a self-induced etiology consistent with dermatitis artefacta. The patient emphasized multiple psychosocial stressors as well as having frequent lupus flares despite repeated negative workup. Given the exaggerated symptoms and unnecessary hospital visits, she was given the diagnosis of factitious disorder (malingering or Munchausen syndrome). After extensive discussion, the patient was amenable to outpatient mental health counseling.
Dermoscopy is not a standard method to diagnose cellulitis of the skin; however, when patients present with an atypical response to appropriate care, the presumed diagnosis must be challenged. This patient had dramatized symptoms, false medical history, and numerous hospitalizations that were suspicious for factitious disorder.1 Furthermore, the physical examination was inconsistent with the classic course of cellulitis. In this case, dermoscopy had advantages over biopsies because it was noninvasive, gave immediate feedback, and provided a macroscopic view of the morphology. Via dermoscopy, we had an objective lens to distinguish cellulitis from cosmetic product and to obtain the correct diagnosis.
- Harth W, Taube KM, Gieler U. Facticious disorders in dermatology. J Dtsch Dermatol Ges. 2010;8:361-372.
To the Editor:
Patients with psychocutaneous disorders present unique challenges to physicians. We illustrate the critical role that dermoscopy may play to illuminate exogenous skin pathology.
A 50-year-old woman with a reported medical history of systemic lupus erythematosus, chronic pain, and nonhealing leg ulcers presented to the emergency department with severe pain of the left lower leg and redness that was concerning for cellulitis. She sought treatment at an outside hospital for cellulitis 2 weeks prior but left against medical advice. Symptomatic review revealed chest pain, shortness of breath, nausea, vomiting, and diarrhea. The primary team started her on intravenous clindamycin and vancomycin for the presumed infection and scheduled narcotic medications due to concerns of intractable pain in the left leg. The dermatology department was consulted after failure to improve with 1 week of systemic antibiotics.
Physical examination revealed a geometric, atrophic, purple plaque on the left anterior shin from a prior leg ulcer as well as a diffuse red-pink patch extending from the knee to the ankle. Notably, the cellulitis spared the left posterior calf resting against the sheet and had a sharp line of demarcation at the distal shin. The leg was cool to the touch while the patient was distractible. She later reported that the leg was extremely tender to palpation. Dermoscopy revealed linear red pigments within skin furrows that accentuated skin lines (Figure). These findings raised suspicions of an external manipulation. The skin was wiped with an alcohol pad that removed a shimmering pink substance consistent in appearance to a cosmetic product. The skin beneath the cellulitis appeared normal.
On further review of the patient’s medical record, it was noted that she was admitted several months ago for ulcers of the left leg. She had been to multiple hospitals and had numerous rounds of antibiotics. Biopsy of an ulcer revealed dermal fibrosis consistent with scarring. Aerobic bacteria, atypical mycobacteria, and fungal cultures were all negative. The physicians suspected a self-induced etiology consistent with dermatitis artefacta. The patient emphasized multiple psychosocial stressors as well as having frequent lupus flares despite repeated negative workup. Given the exaggerated symptoms and unnecessary hospital visits, she was given the diagnosis of factitious disorder (malingering or Munchausen syndrome). After extensive discussion, the patient was amenable to outpatient mental health counseling.
Dermoscopy is not a standard method to diagnose cellulitis of the skin; however, when patients present with an atypical response to appropriate care, the presumed diagnosis must be challenged. This patient had dramatized symptoms, false medical history, and numerous hospitalizations that were suspicious for factitious disorder.1 Furthermore, the physical examination was inconsistent with the classic course of cellulitis. In this case, dermoscopy had advantages over biopsies because it was noninvasive, gave immediate feedback, and provided a macroscopic view of the morphology. Via dermoscopy, we had an objective lens to distinguish cellulitis from cosmetic product and to obtain the correct diagnosis.
To the Editor:
Patients with psychocutaneous disorders present unique challenges to physicians. We illustrate the critical role that dermoscopy may play to illuminate exogenous skin pathology.
A 50-year-old woman with a reported medical history of systemic lupus erythematosus, chronic pain, and nonhealing leg ulcers presented to the emergency department with severe pain of the left lower leg and redness that was concerning for cellulitis. She sought treatment at an outside hospital for cellulitis 2 weeks prior but left against medical advice. Symptomatic review revealed chest pain, shortness of breath, nausea, vomiting, and diarrhea. The primary team started her on intravenous clindamycin and vancomycin for the presumed infection and scheduled narcotic medications due to concerns of intractable pain in the left leg. The dermatology department was consulted after failure to improve with 1 week of systemic antibiotics.
Physical examination revealed a geometric, atrophic, purple plaque on the left anterior shin from a prior leg ulcer as well as a diffuse red-pink patch extending from the knee to the ankle. Notably, the cellulitis spared the left posterior calf resting against the sheet and had a sharp line of demarcation at the distal shin. The leg was cool to the touch while the patient was distractible. She later reported that the leg was extremely tender to palpation. Dermoscopy revealed linear red pigments within skin furrows that accentuated skin lines (Figure). These findings raised suspicions of an external manipulation. The skin was wiped with an alcohol pad that removed a shimmering pink substance consistent in appearance to a cosmetic product. The skin beneath the cellulitis appeared normal.
On further review of the patient’s medical record, it was noted that she was admitted several months ago for ulcers of the left leg. She had been to multiple hospitals and had numerous rounds of antibiotics. Biopsy of an ulcer revealed dermal fibrosis consistent with scarring. Aerobic bacteria, atypical mycobacteria, and fungal cultures were all negative. The physicians suspected a self-induced etiology consistent with dermatitis artefacta. The patient emphasized multiple psychosocial stressors as well as having frequent lupus flares despite repeated negative workup. Given the exaggerated symptoms and unnecessary hospital visits, she was given the diagnosis of factitious disorder (malingering or Munchausen syndrome). After extensive discussion, the patient was amenable to outpatient mental health counseling.
Dermoscopy is not a standard method to diagnose cellulitis of the skin; however, when patients present with an atypical response to appropriate care, the presumed diagnosis must be challenged. This patient had dramatized symptoms, false medical history, and numerous hospitalizations that were suspicious for factitious disorder.1 Furthermore, the physical examination was inconsistent with the classic course of cellulitis. In this case, dermoscopy had advantages over biopsies because it was noninvasive, gave immediate feedback, and provided a macroscopic view of the morphology. Via dermoscopy, we had an objective lens to distinguish cellulitis from cosmetic product and to obtain the correct diagnosis.
- Harth W, Taube KM, Gieler U. Facticious disorders in dermatology. J Dtsch Dermatol Ges. 2010;8:361-372.
- Harth W, Taube KM, Gieler U. Facticious disorders in dermatology. J Dtsch Dermatol Ges. 2010;8:361-372.
Practice Points
- Consider exogenous factors or alternative diagnoses when a patient does not respond to appropriate care.
- Although dermoscopy is not used to diagnose cellulitis, it could be helpful in distinguishing cosmetic products used in dermatitis artefacta.
Pigmented Basal Cell Carcinoma With Annular Leukoderma
To the Editor:
Annular leukoderma, or the halo phenomenon, is a circular reaction of hypopigmentation that most commonly is observed alongside congenital nevi, acquired melanocytic nevi, blue nevi, Spitz nevi, vitiligo, and rarely melanoma.1 There is limited literature on the mechanism of the halo phenomenon. Most of the literature proposes a T cell–mediated immune response to antigens, which causes not only surrounding pigment loss but also heralds the regression of central lesions.2 Others have suggested a vascular mechanism, with blood shunted away from the lesions.3 Because guidelines discourage biopsy of typical halo nevi, it becomes important to evaluate lesions for worrisome features such as ulceration or asymmetry, especially in older patients. We present a case of a pigmented basal cell carcinoma (BCC) that exhibited the halo phenomenon. Four other cases have been described in the literature.3-6
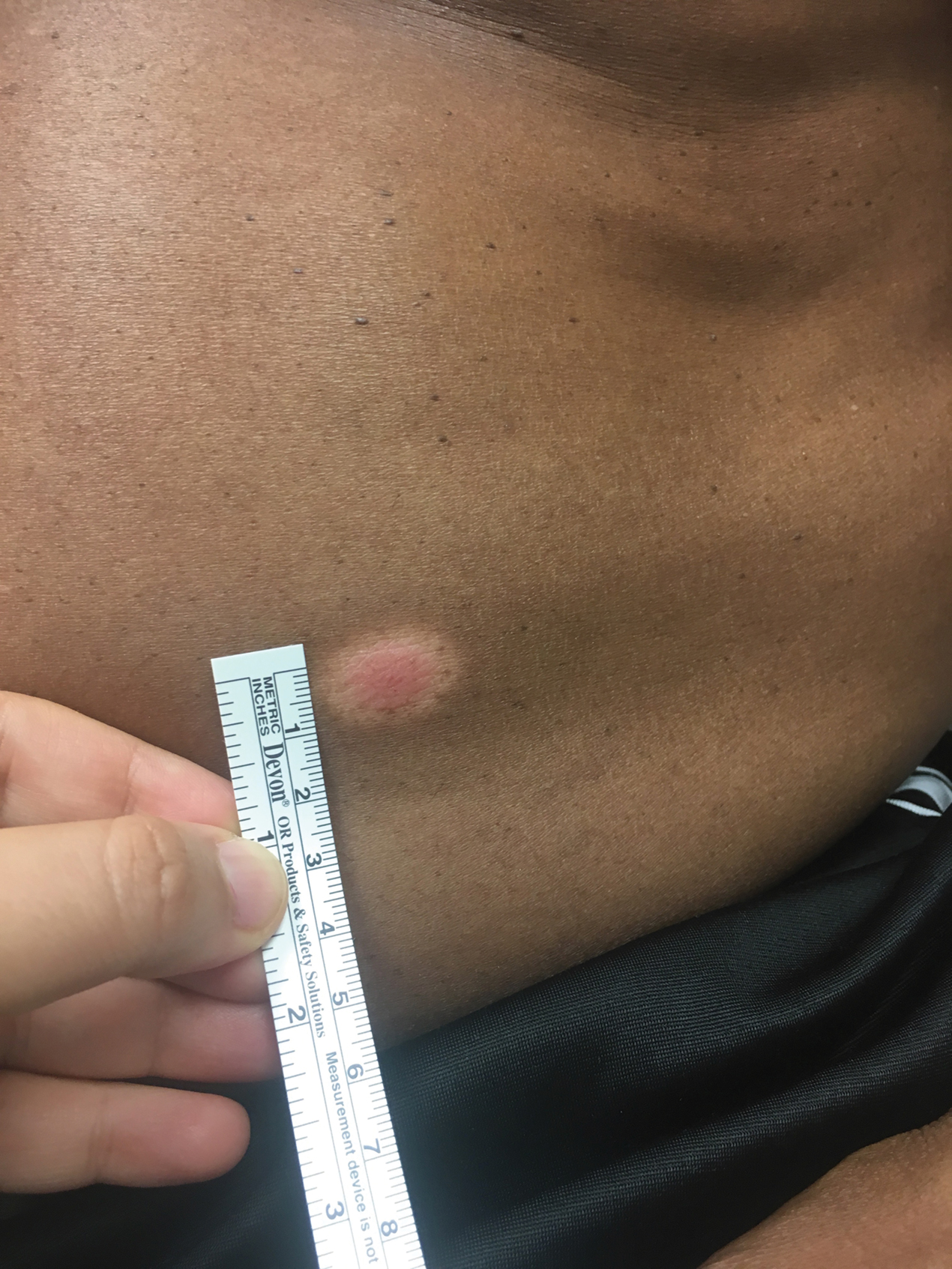
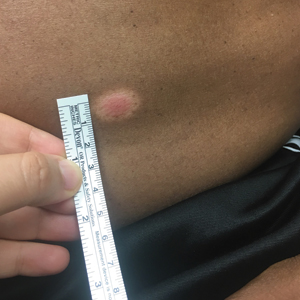
A 53-year-old man presented for evaluation of an asymptomatic lesion on the left side of the abdomen of approximately 8 months’ duration. He had no personal or family history of skin cancer. Physical examination revealed a central 1-cm, pink, verrucous papule surrounded by a 2×1.2-cm, depigmented, circular patch on the left side of the inferior abdomen (Figure 1). Upon questioning, the patient produced cell phone photographs of the trunk from 3 years prior, which did not show any lesions present. Full-body skin examination did not reveal any other concerning pigmented lesions. Excisional biopsy was performed due to concern for amelanotic melanoma, and histopathology revealed a superficial and pigmented BCC (Figure 2). Immunohistochemistry with Melan-A was negative for atypical melanocytes, with no uptake in the leukoderma areas.
The clinical presentation initially was concerning for amelanotic melanoma. All melanoma subtypes may appear as hypomelanotic lesions, though these most commonly are observed in the desmoplastic or nodular subtypes. Amelanotic melanomas may present as well-defined red or pink macules, plaques, or nodules, with some tumors presenting with light brown pigmentation.7
The differential diagnosis for lesions with the halo phenomenon is large. In adults, the halo phenomenon may be concerning for malignant or regressing melanoma. As an immunogenic tumor, melanoma’s immunogenic melanocytes may incite a cell-mediated immune response to antigens common to neoplastic and normal melanocytes, which can clinically manifest not only as local annular leukoderma but also as distant vitiligo or halo nevi.7 The halo phenomenon more commonly is associated with benign processes such as vitiligo and halo nevi in children. In most children, halo nevi occur as an isolated phenomenon but still warrant a complete skin examination for melanoma and vitiligo. The presence of halo nevi has been associated with distant vitiligo—possibly through shared immunologic mechanisms—especially if patients present with the Koebner phenomenon, multiple halo nevi, or a family history of vitiligo.8 A prospective study also found that the presence of halo nevi was an independent risk factor for the progression of segmental vitiligo to mixed vitiligo.9 Hormones also may play a role in the leukoderma acquisitum centrifugum, or halo, nevi. Halo nevi most commonly affect adolescents and pregnant women. It has been postulated that congenital nevi may be unique in their response to altered estrogen levels, increasing the rate not only of halo nevi but also of melanoma in pregnant women.10
Our patient’s final histologic diagnosis was pigmented BCC, which comprises only 6% of all BCCs.3 The proposed mechanism is that melanocytes colonize the tumor in the surrounding stroma and produce excess melanin. Basal cell carcinoma with halo phenomenon is a rare presentation. As in our case, 2 prior BCC reports also involved patients older than 50 years,3,5 with the 2 other cases describing women in their late twenties and early thirties.4,6 Additionally, 2 of 4 reports described patients with a history of multiple BCCs.3,5
In summary, the seemingly benign halo phenomenon may accompany malignant processes such as nonmelanoma skin cancer. Careful consideration of lesion time course and atypia is imperative for proper clinical suspicion in such cases.
- Mooney MA, Barr RJ, Buxton MG. Halo nevus or halo phenomenon? a study of 142 cases. J Cutan Pathol. 1995;22:342-348.
- Zeff RA, Freitag A, Grin CM, et al. The immune response in halo nevi. J Am Acad Dermatol. 1997;37:620-624.
- Johnson DB, Ceilley RI. Basal cell carcinoma with annular leukoderma mimicking leukoderma acquisitum centrifugum. Arch Dermatol. 1980;116:352-353.
- Basak PY, Meric G, Ciris M. Basal cell carcinoma with halo phenomenon in a young female: significance of dermatoscopy in early diagnosis. Indian J Dermatol. 2015;60:214.
- Pembroke AC, Liddell K. Basal cell epithelioma with a hypopigmented halo. Arch Dermatol. 1981;117:317.
- Rustemeyer J, Günther L, Deichert L. A rare association: basal cell carcinoma in a vitiliginous macula. Oral Maxillofac Surg. 2011;15:175-177.
- Naveh HP, Rao UN, Butterfield LH. Melanoma‐associated leukoderma—immunology in black and white? Pigment Cell Melanoma Res. 2013;26:796-804.
- Zhou H, Wu L-C, Chen M-K, et al. Factors associated with development of vitiligo in patients with halo nevus. Chinese Med J. 2017;130:2703.
- Ezzedine K, Diallo A, Léauté‐Labrèze C, et al. Halo naevi and leukotrichia are strong predictors of the passage to mixed vitiligo in a subgroup of segmental vitiligo. Br J Dermatol. 2012;166:539-544.
- Nading MA, Nanney LB, Ellis DL. Pregnancy and estrogen receptor β expression in a large congenital nevus. Arch Dermatol. 2009;145:691-694.
To the Editor:
Annular leukoderma, or the halo phenomenon, is a circular reaction of hypopigmentation that most commonly is observed alongside congenital nevi, acquired melanocytic nevi, blue nevi, Spitz nevi, vitiligo, and rarely melanoma.1 There is limited literature on the mechanism of the halo phenomenon. Most of the literature proposes a T cell–mediated immune response to antigens, which causes not only surrounding pigment loss but also heralds the regression of central lesions.2 Others have suggested a vascular mechanism, with blood shunted away from the lesions.3 Because guidelines discourage biopsy of typical halo nevi, it becomes important to evaluate lesions for worrisome features such as ulceration or asymmetry, especially in older patients. We present a case of a pigmented basal cell carcinoma (BCC) that exhibited the halo phenomenon. Four other cases have been described in the literature.3-6
A 53-year-old man presented for evaluation of an asymptomatic lesion on the left side of the abdomen of approximately 8 months’ duration. He had no personal or family history of skin cancer. Physical examination revealed a central 1-cm, pink, verrucous papule surrounded by a 2×1.2-cm, depigmented, circular patch on the left side of the inferior abdomen (Figure 1). Upon questioning, the patient produced cell phone photographs of the trunk from 3 years prior, which did not show any lesions present. Full-body skin examination did not reveal any other concerning pigmented lesions. Excisional biopsy was performed due to concern for amelanotic melanoma, and histopathology revealed a superficial and pigmented BCC (Figure 2). Immunohistochemistry with Melan-A was negative for atypical melanocytes, with no uptake in the leukoderma areas.
The clinical presentation initially was concerning for amelanotic melanoma. All melanoma subtypes may appear as hypomelanotic lesions, though these most commonly are observed in the desmoplastic or nodular subtypes. Amelanotic melanomas may present as well-defined red or pink macules, plaques, or nodules, with some tumors presenting with light brown pigmentation.7
The differential diagnosis for lesions with the halo phenomenon is large. In adults, the halo phenomenon may be concerning for malignant or regressing melanoma. As an immunogenic tumor, melanoma’s immunogenic melanocytes may incite a cell-mediated immune response to antigens common to neoplastic and normal melanocytes, which can clinically manifest not only as local annular leukoderma but also as distant vitiligo or halo nevi.7 The halo phenomenon more commonly is associated with benign processes such as vitiligo and halo nevi in children. In most children, halo nevi occur as an isolated phenomenon but still warrant a complete skin examination for melanoma and vitiligo. The presence of halo nevi has been associated with distant vitiligo—possibly through shared immunologic mechanisms—especially if patients present with the Koebner phenomenon, multiple halo nevi, or a family history of vitiligo.8 A prospective study also found that the presence of halo nevi was an independent risk factor for the progression of segmental vitiligo to mixed vitiligo.9 Hormones also may play a role in the leukoderma acquisitum centrifugum, or halo, nevi. Halo nevi most commonly affect adolescents and pregnant women. It has been postulated that congenital nevi may be unique in their response to altered estrogen levels, increasing the rate not only of halo nevi but also of melanoma in pregnant women.10
Our patient’s final histologic diagnosis was pigmented BCC, which comprises only 6% of all BCCs.3 The proposed mechanism is that melanocytes colonize the tumor in the surrounding stroma and produce excess melanin. Basal cell carcinoma with halo phenomenon is a rare presentation. As in our case, 2 prior BCC reports also involved patients older than 50 years,3,5 with the 2 other cases describing women in their late twenties and early thirties.4,6 Additionally, 2 of 4 reports described patients with a history of multiple BCCs.3,5
In summary, the seemingly benign halo phenomenon may accompany malignant processes such as nonmelanoma skin cancer. Careful consideration of lesion time course and atypia is imperative for proper clinical suspicion in such cases.
To the Editor:
Annular leukoderma, or the halo phenomenon, is a circular reaction of hypopigmentation that most commonly is observed alongside congenital nevi, acquired melanocytic nevi, blue nevi, Spitz nevi, vitiligo, and rarely melanoma.1 There is limited literature on the mechanism of the halo phenomenon. Most of the literature proposes a T cell–mediated immune response to antigens, which causes not only surrounding pigment loss but also heralds the regression of central lesions.2 Others have suggested a vascular mechanism, with blood shunted away from the lesions.3 Because guidelines discourage biopsy of typical halo nevi, it becomes important to evaluate lesions for worrisome features such as ulceration or asymmetry, especially in older patients. We present a case of a pigmented basal cell carcinoma (BCC) that exhibited the halo phenomenon. Four other cases have been described in the literature.3-6
A 53-year-old man presented for evaluation of an asymptomatic lesion on the left side of the abdomen of approximately 8 months’ duration. He had no personal or family history of skin cancer. Physical examination revealed a central 1-cm, pink, verrucous papule surrounded by a 2×1.2-cm, depigmented, circular patch on the left side of the inferior abdomen (Figure 1). Upon questioning, the patient produced cell phone photographs of the trunk from 3 years prior, which did not show any lesions present. Full-body skin examination did not reveal any other concerning pigmented lesions. Excisional biopsy was performed due to concern for amelanotic melanoma, and histopathology revealed a superficial and pigmented BCC (Figure 2). Immunohistochemistry with Melan-A was negative for atypical melanocytes, with no uptake in the leukoderma areas.
The clinical presentation initially was concerning for amelanotic melanoma. All melanoma subtypes may appear as hypomelanotic lesions, though these most commonly are observed in the desmoplastic or nodular subtypes. Amelanotic melanomas may present as well-defined red or pink macules, plaques, or nodules, with some tumors presenting with light brown pigmentation.7
The differential diagnosis for lesions with the halo phenomenon is large. In adults, the halo phenomenon may be concerning for malignant or regressing melanoma. As an immunogenic tumor, melanoma’s immunogenic melanocytes may incite a cell-mediated immune response to antigens common to neoplastic and normal melanocytes, which can clinically manifest not only as local annular leukoderma but also as distant vitiligo or halo nevi.7 The halo phenomenon more commonly is associated with benign processes such as vitiligo and halo nevi in children. In most children, halo nevi occur as an isolated phenomenon but still warrant a complete skin examination for melanoma and vitiligo. The presence of halo nevi has been associated with distant vitiligo—possibly through shared immunologic mechanisms—especially if patients present with the Koebner phenomenon, multiple halo nevi, or a family history of vitiligo.8 A prospective study also found that the presence of halo nevi was an independent risk factor for the progression of segmental vitiligo to mixed vitiligo.9 Hormones also may play a role in the leukoderma acquisitum centrifugum, or halo, nevi. Halo nevi most commonly affect adolescents and pregnant women. It has been postulated that congenital nevi may be unique in their response to altered estrogen levels, increasing the rate not only of halo nevi but also of melanoma in pregnant women.10
Our patient’s final histologic diagnosis was pigmented BCC, which comprises only 6% of all BCCs.3 The proposed mechanism is that melanocytes colonize the tumor in the surrounding stroma and produce excess melanin. Basal cell carcinoma with halo phenomenon is a rare presentation. As in our case, 2 prior BCC reports also involved patients older than 50 years,3,5 with the 2 other cases describing women in their late twenties and early thirties.4,6 Additionally, 2 of 4 reports described patients with a history of multiple BCCs.3,5
In summary, the seemingly benign halo phenomenon may accompany malignant processes such as nonmelanoma skin cancer. Careful consideration of lesion time course and atypia is imperative for proper clinical suspicion in such cases.
- Mooney MA, Barr RJ, Buxton MG. Halo nevus or halo phenomenon? a study of 142 cases. J Cutan Pathol. 1995;22:342-348.
- Zeff RA, Freitag A, Grin CM, et al. The immune response in halo nevi. J Am Acad Dermatol. 1997;37:620-624.
- Johnson DB, Ceilley RI. Basal cell carcinoma with annular leukoderma mimicking leukoderma acquisitum centrifugum. Arch Dermatol. 1980;116:352-353.
- Basak PY, Meric G, Ciris M. Basal cell carcinoma with halo phenomenon in a young female: significance of dermatoscopy in early diagnosis. Indian J Dermatol. 2015;60:214.
- Pembroke AC, Liddell K. Basal cell epithelioma with a hypopigmented halo. Arch Dermatol. 1981;117:317.
- Rustemeyer J, Günther L, Deichert L. A rare association: basal cell carcinoma in a vitiliginous macula. Oral Maxillofac Surg. 2011;15:175-177.
- Naveh HP, Rao UN, Butterfield LH. Melanoma‐associated leukoderma—immunology in black and white? Pigment Cell Melanoma Res. 2013;26:796-804.
- Zhou H, Wu L-C, Chen M-K, et al. Factors associated with development of vitiligo in patients with halo nevus. Chinese Med J. 2017;130:2703.
- Ezzedine K, Diallo A, Léauté‐Labrèze C, et al. Halo naevi and leukotrichia are strong predictors of the passage to mixed vitiligo in a subgroup of segmental vitiligo. Br J Dermatol. 2012;166:539-544.
- Nading MA, Nanney LB, Ellis DL. Pregnancy and estrogen receptor β expression in a large congenital nevus. Arch Dermatol. 2009;145:691-694.
- Mooney MA, Barr RJ, Buxton MG. Halo nevus or halo phenomenon? a study of 142 cases. J Cutan Pathol. 1995;22:342-348.
- Zeff RA, Freitag A, Grin CM, et al. The immune response in halo nevi. J Am Acad Dermatol. 1997;37:620-624.
- Johnson DB, Ceilley RI. Basal cell carcinoma with annular leukoderma mimicking leukoderma acquisitum centrifugum. Arch Dermatol. 1980;116:352-353.
- Basak PY, Meric G, Ciris M. Basal cell carcinoma with halo phenomenon in a young female: significance of dermatoscopy in early diagnosis. Indian J Dermatol. 2015;60:214.
- Pembroke AC, Liddell K. Basal cell epithelioma with a hypopigmented halo. Arch Dermatol. 1981;117:317.
- Rustemeyer J, Günther L, Deichert L. A rare association: basal cell carcinoma in a vitiliginous macula. Oral Maxillofac Surg. 2011;15:175-177.
- Naveh HP, Rao UN, Butterfield LH. Melanoma‐associated leukoderma—immunology in black and white? Pigment Cell Melanoma Res. 2013;26:796-804.
- Zhou H, Wu L-C, Chen M-K, et al. Factors associated with development of vitiligo in patients with halo nevus. Chinese Med J. 2017;130:2703.
- Ezzedine K, Diallo A, Léauté‐Labrèze C, et al. Halo naevi and leukotrichia are strong predictors of the passage to mixed vitiligo in a subgroup of segmental vitiligo. Br J Dermatol. 2012;166:539-544.
- Nading MA, Nanney LB, Ellis DL. Pregnancy and estrogen receptor β expression in a large congenital nevus. Arch Dermatol. 2009;145:691-694.
Practice Points
- Annular leukoderma, or the halo phenomenon, is a circular reaction of hypopigmentation that more commonly is associated with benign processes such as halo nevi.
- The halo phenomenon may accompany malignant processes, such as nonmelanoma skin cancer. Careful consideration of lesion time course and atypia is imperative for proper clinical suspicion in such cases.
Argyria From a Topical Home Remedy
To the Editor:
Argyria is a rare disease caused by chronic exposure to products with high silver content (eg, oral ingestion, inhalation, percutaneous absorption). With time, the blood levels of silver surpass the body’s renal and hepatic excretory capacities that lead to silver granules being deposited in the skin and internal organs, including the liver, spleen, adrenal glands, and bone marrow.1 The cutaneous deposition results in a blue or blue-gray pigmentation of the skin, mucous membranes, and nails. Intervals of exposure that span from 8 months to 5 years prior to symptom onset have been described in the literature.2 The discoloration that results often is permanent, with no established way of effectively removing silver deposits from the tissue.3
A 22-year-old autistic man, who was completely dependent on his mother’s care, presented to the emergency department with a primary concern of abdominal pain. The mother reported that he was indicating abdominal pain by motioning to his stomach for the last 5 days. The mother also reported he did not have a bowel movement during this time, and she noticed his hands were shaking. Prior to presentation, the mother had given him 2 enemas and had him on a 3-day strict liquid fast consisting of water, lemon juice, cayenne pepper, honey, and orange juice. Notably, the mother had a strong history of using naturopathic remedies for treatment of her son’s ailments.
On admission, the patient was stable. There was a 2-point decrease in the patient’s body mass index over the last month. Initial serum electrolytes were highly abnormal with a serum sodium level of 124 mEq/L (reference range, 135–145 mEq/L), blood urea nitrogen of 3 mg/dL (reference range, 7–20 mg/dL), creatinine of 0.77 mg/dL (reference range, 0.74–1.35 mg/dL), and lactic acid of 2.1 mEq/L (reference range, 0.5–1 mEq/L). Serum osmolality was 272 mOsm/kg (reference range, 275–295 mOsm/kg). Urine osmolality was 114 mOsm/kg (reference range, 500–850 mOsm/kg) with a low-normal urine sodium level of 41 mmol/24 hr (reference range, 40–220 mmol/24 hr). Abnormalities were felt to be secondary to malnutrition from the strict liquid diet (blood urea nitrogen and creatinine ratio of 3:1 suggestive of notable protein calorie malnutrition). The patient was given 1 L of normal saline in the emergency department, with further fluids held so as not to increase serum sodium level too rapidly. A regular diet was started.
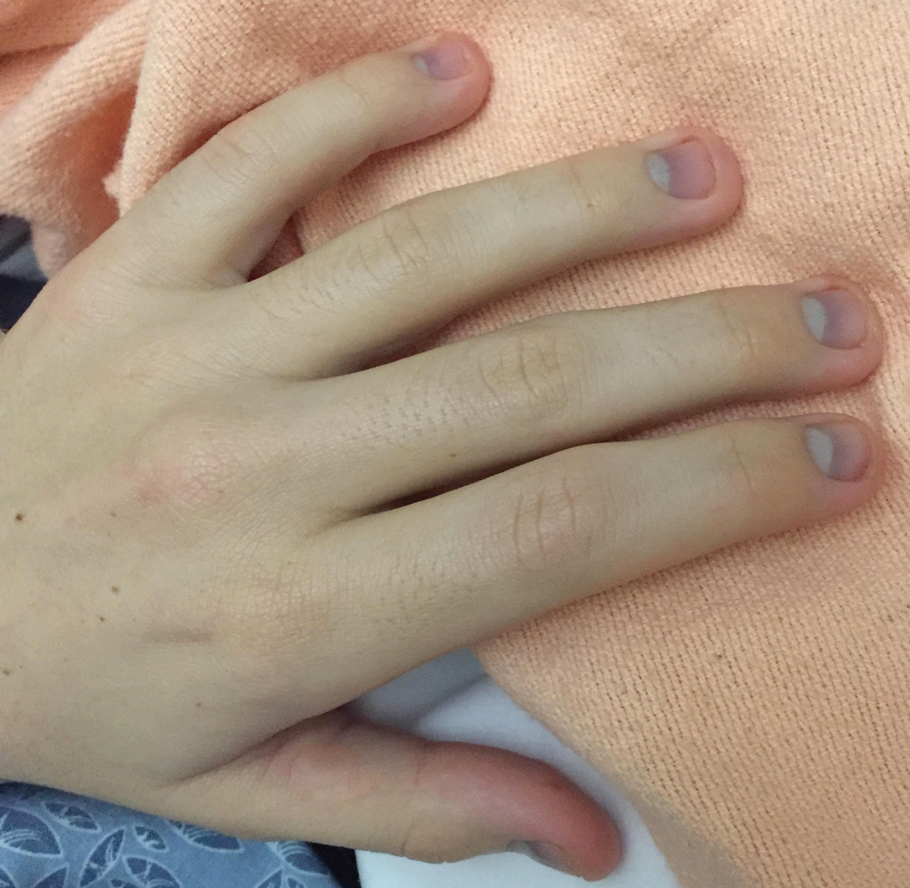
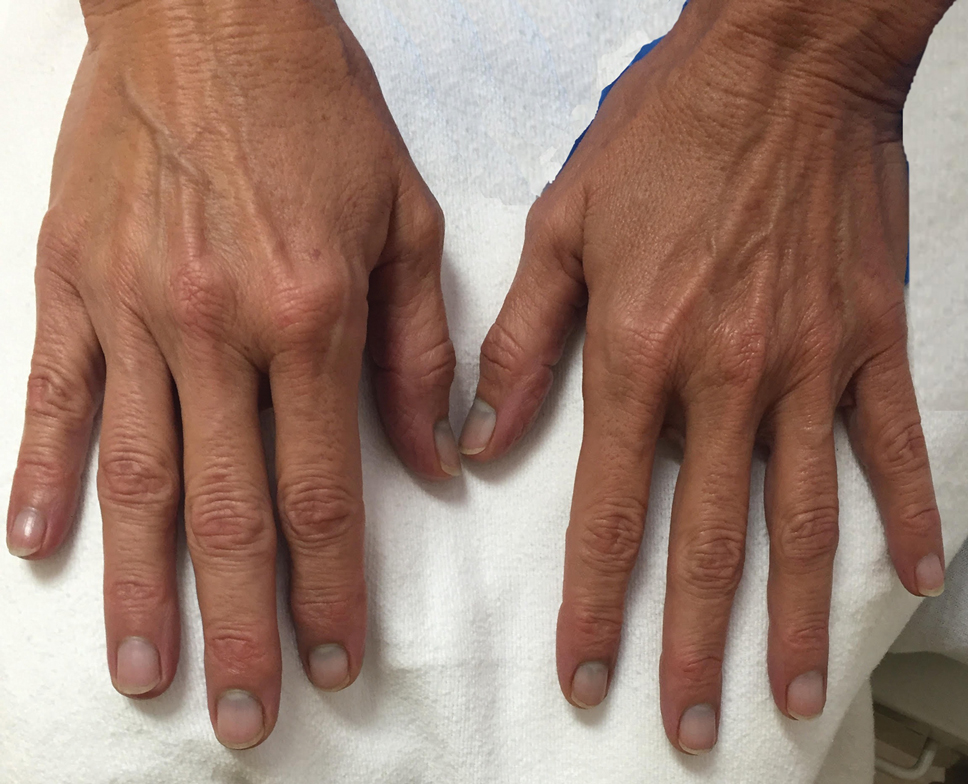
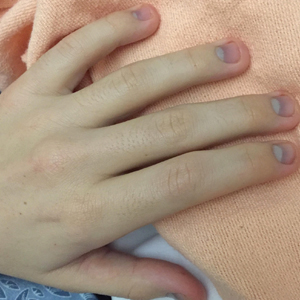
Physical examination revealed dry mucosal membranes but otherwise was unremarkable. Active bowel sounds were noted, as well as a soft, nontender, and nondistended abdomen; however, when examining the patient’s hands for reported shaking, a distinct abnormality of the nails was noticed. The patient had slate blue discoloration of the lunula, along with hyperpigmented violaceous discoloration of the proximal nail bed on all 10 fingernails (Figure 1). No abnormalities were seen on the toenails. The mother had a distinct bluish gray discoloration of the face as well as similar nail findings (Figure 2), strongly suggestive of colloidal silver use. An urgent serum silver level was ordered on the patient as well as a heavy metal panel. The mother was found applying numerous “natural remedies” to the patient’s skin while in the hospital, including a liquid spray and lotion, both in unmarked bottles. At that time, the mother was informed that no external supplements should be applied to her son. The serum silver level was elevated substantially at 94.3 ng/mL (reference range, <1.0 ng/mL). When the mother was confronted, she initially denied use of silver but later admitted to notable silver content in the cream she was applying to her son’s skin. The mother reported that she read online that colloidal silver had been historically used to cure numerous ailments and she was ordering products from an online company. She was counseled on the dangers of both topical application and ingestion of silver, and all supplements were removed from the home.
Argyria is a rare condition caused by chronic exposure to silver and is characterized by a blue-gray pigmentation in the skin and appendages, mucous membranes, and internal organs.4 Clinically, argyria is classified as generalized or localized. Generalized argyria results from ingestion or inhalation of silver compounds, where granules deposit preferentially in sun-exposed areas of skin as well as internal organs, with the highest concentration in the liver, spleen, and adrenal glands; discoloration often is permanent.5 On the contrary, localized argyria results from direct external contact with silver and granules deposited in the hands, eyes, and mucosa.5 Although the exact mechanism of penetration from topical silver remains unknown, it is thought to enter via the eccrine sweat ducts, as histopathology reveals silver granules found in highest concentration surrounding sweat glands in the dermis.6
Initial differential diagnoses for altered nail pigmentation include drug-induced causes, systemic diseases, cyanosis, and exposure to metals.7 The most commonly indicated medications resulting in blue nail pigment changes include antimalarials, minocycline, zidovudine, and phenothiazine. Systemic diseases that may cause blue nail color change include Wilson disease, hemochromatosis, Addison disease, methemoglobinemia, and alkaptonuria.7 Metals include gold, mercury, arsenic, bismuth, lead, and silver.4 After a thorough review of the patient’s medications and lack of support for any underlying disease process, contact with metals, particularly silver, was ranked highly on our differential list. In support of this theory, the mother’s bluish gray facial skin led to high clinical suspicion that she was ingesting colloidal silver and also was exposing her son to silver.
Treatment of argyria is challenging but first and foremost involves discontinuation of the source of chronic silver exposure. Unfortunately, the discoloration of generalized argyria often is permanent. Sunscreen can be used to help prevent any further darkening of pigment. The pigment in localized argyria has been reported to slowly fade with time, and there also have been reports of successful treatment using a low-fluence Q-switched 1064-nm Nd:YAG laser.8
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Lencastre A, Lobo M, João A. Argyria—case report. An Bras Dermatol. 2013;88:413-416.
- Park S-W, Kim J-H, Shin H-T, et al. An effective modality for argyria treatment: Q-switched 1,064-nm Nd:YAG laser. Ann Dermatol. 2013;25:511-512.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Garcias-Ladaria J, Hernandez-Bel P, Torregrosa-Calatayud JL, et al. Localized cutaneous argyria: a report of 2 cases. Actas Dermosifiliogr. 2013;104:253-254.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-192.
- Kubba A, Kubba R, Batrani M, Pal T. Argyria an unrecognized cause of cutaneous pigmentation in Indian patients: a case series and review of the literature. Indian J Dermatol Venereol Leprol. 2013;79:805-811.
- Han TY, Chang HS, Lee HK, et al. Successful treatment of argyria using a low-fluence Q-switched 1064-nm Nd:YAG laser. Int J Dermatol. 2011;50:751-753.
To the Editor:
Argyria is a rare disease caused by chronic exposure to products with high silver content (eg, oral ingestion, inhalation, percutaneous absorption). With time, the blood levels of silver surpass the body’s renal and hepatic excretory capacities that lead to silver granules being deposited in the skin and internal organs, including the liver, spleen, adrenal glands, and bone marrow.1 The cutaneous deposition results in a blue or blue-gray pigmentation of the skin, mucous membranes, and nails. Intervals of exposure that span from 8 months to 5 years prior to symptom onset have been described in the literature.2 The discoloration that results often is permanent, with no established way of effectively removing silver deposits from the tissue.3
A 22-year-old autistic man, who was completely dependent on his mother’s care, presented to the emergency department with a primary concern of abdominal pain. The mother reported that he was indicating abdominal pain by motioning to his stomach for the last 5 days. The mother also reported he did not have a bowel movement during this time, and she noticed his hands were shaking. Prior to presentation, the mother had given him 2 enemas and had him on a 3-day strict liquid fast consisting of water, lemon juice, cayenne pepper, honey, and orange juice. Notably, the mother had a strong history of using naturopathic remedies for treatment of her son’s ailments.
On admission, the patient was stable. There was a 2-point decrease in the patient’s body mass index over the last month. Initial serum electrolytes were highly abnormal with a serum sodium level of 124 mEq/L (reference range, 135–145 mEq/L), blood urea nitrogen of 3 mg/dL (reference range, 7–20 mg/dL), creatinine of 0.77 mg/dL (reference range, 0.74–1.35 mg/dL), and lactic acid of 2.1 mEq/L (reference range, 0.5–1 mEq/L). Serum osmolality was 272 mOsm/kg (reference range, 275–295 mOsm/kg). Urine osmolality was 114 mOsm/kg (reference range, 500–850 mOsm/kg) with a low-normal urine sodium level of 41 mmol/24 hr (reference range, 40–220 mmol/24 hr). Abnormalities were felt to be secondary to malnutrition from the strict liquid diet (blood urea nitrogen and creatinine ratio of 3:1 suggestive of notable protein calorie malnutrition). The patient was given 1 L of normal saline in the emergency department, with further fluids held so as not to increase serum sodium level too rapidly. A regular diet was started.
Physical examination revealed dry mucosal membranes but otherwise was unremarkable. Active bowel sounds were noted, as well as a soft, nontender, and nondistended abdomen; however, when examining the patient’s hands for reported shaking, a distinct abnormality of the nails was noticed. The patient had slate blue discoloration of the lunula, along with hyperpigmented violaceous discoloration of the proximal nail bed on all 10 fingernails (Figure 1). No abnormalities were seen on the toenails. The mother had a distinct bluish gray discoloration of the face as well as similar nail findings (Figure 2), strongly suggestive of colloidal silver use. An urgent serum silver level was ordered on the patient as well as a heavy metal panel. The mother was found applying numerous “natural remedies” to the patient’s skin while in the hospital, including a liquid spray and lotion, both in unmarked bottles. At that time, the mother was informed that no external supplements should be applied to her son. The serum silver level was elevated substantially at 94.3 ng/mL (reference range, <1.0 ng/mL). When the mother was confronted, she initially denied use of silver but later admitted to notable silver content in the cream she was applying to her son’s skin. The mother reported that she read online that colloidal silver had been historically used to cure numerous ailments and she was ordering products from an online company. She was counseled on the dangers of both topical application and ingestion of silver, and all supplements were removed from the home.
Argyria is a rare condition caused by chronic exposure to silver and is characterized by a blue-gray pigmentation in the skin and appendages, mucous membranes, and internal organs.4 Clinically, argyria is classified as generalized or localized. Generalized argyria results from ingestion or inhalation of silver compounds, where granules deposit preferentially in sun-exposed areas of skin as well as internal organs, with the highest concentration in the liver, spleen, and adrenal glands; discoloration often is permanent.5 On the contrary, localized argyria results from direct external contact with silver and granules deposited in the hands, eyes, and mucosa.5 Although the exact mechanism of penetration from topical silver remains unknown, it is thought to enter via the eccrine sweat ducts, as histopathology reveals silver granules found in highest concentration surrounding sweat glands in the dermis.6
Initial differential diagnoses for altered nail pigmentation include drug-induced causes, systemic diseases, cyanosis, and exposure to metals.7 The most commonly indicated medications resulting in blue nail pigment changes include antimalarials, minocycline, zidovudine, and phenothiazine. Systemic diseases that may cause blue nail color change include Wilson disease, hemochromatosis, Addison disease, methemoglobinemia, and alkaptonuria.7 Metals include gold, mercury, arsenic, bismuth, lead, and silver.4 After a thorough review of the patient’s medications and lack of support for any underlying disease process, contact with metals, particularly silver, was ranked highly on our differential list. In support of this theory, the mother’s bluish gray facial skin led to high clinical suspicion that she was ingesting colloidal silver and also was exposing her son to silver.
Treatment of argyria is challenging but first and foremost involves discontinuation of the source of chronic silver exposure. Unfortunately, the discoloration of generalized argyria often is permanent. Sunscreen can be used to help prevent any further darkening of pigment. The pigment in localized argyria has been reported to slowly fade with time, and there also have been reports of successful treatment using a low-fluence Q-switched 1064-nm Nd:YAG laser.8
To the Editor:
Argyria is a rare disease caused by chronic exposure to products with high silver content (eg, oral ingestion, inhalation, percutaneous absorption). With time, the blood levels of silver surpass the body’s renal and hepatic excretory capacities that lead to silver granules being deposited in the skin and internal organs, including the liver, spleen, adrenal glands, and bone marrow.1 The cutaneous deposition results in a blue or blue-gray pigmentation of the skin, mucous membranes, and nails. Intervals of exposure that span from 8 months to 5 years prior to symptom onset have been described in the literature.2 The discoloration that results often is permanent, with no established way of effectively removing silver deposits from the tissue.3
A 22-year-old autistic man, who was completely dependent on his mother’s care, presented to the emergency department with a primary concern of abdominal pain. The mother reported that he was indicating abdominal pain by motioning to his stomach for the last 5 days. The mother also reported he did not have a bowel movement during this time, and she noticed his hands were shaking. Prior to presentation, the mother had given him 2 enemas and had him on a 3-day strict liquid fast consisting of water, lemon juice, cayenne pepper, honey, and orange juice. Notably, the mother had a strong history of using naturopathic remedies for treatment of her son’s ailments.
On admission, the patient was stable. There was a 2-point decrease in the patient’s body mass index over the last month. Initial serum electrolytes were highly abnormal with a serum sodium level of 124 mEq/L (reference range, 135–145 mEq/L), blood urea nitrogen of 3 mg/dL (reference range, 7–20 mg/dL), creatinine of 0.77 mg/dL (reference range, 0.74–1.35 mg/dL), and lactic acid of 2.1 mEq/L (reference range, 0.5–1 mEq/L). Serum osmolality was 272 mOsm/kg (reference range, 275–295 mOsm/kg). Urine osmolality was 114 mOsm/kg (reference range, 500–850 mOsm/kg) with a low-normal urine sodium level of 41 mmol/24 hr (reference range, 40–220 mmol/24 hr). Abnormalities were felt to be secondary to malnutrition from the strict liquid diet (blood urea nitrogen and creatinine ratio of 3:1 suggestive of notable protein calorie malnutrition). The patient was given 1 L of normal saline in the emergency department, with further fluids held so as not to increase serum sodium level too rapidly. A regular diet was started.
Physical examination revealed dry mucosal membranes but otherwise was unremarkable. Active bowel sounds were noted, as well as a soft, nontender, and nondistended abdomen; however, when examining the patient’s hands for reported shaking, a distinct abnormality of the nails was noticed. The patient had slate blue discoloration of the lunula, along with hyperpigmented violaceous discoloration of the proximal nail bed on all 10 fingernails (Figure 1). No abnormalities were seen on the toenails. The mother had a distinct bluish gray discoloration of the face as well as similar nail findings (Figure 2), strongly suggestive of colloidal silver use. An urgent serum silver level was ordered on the patient as well as a heavy metal panel. The mother was found applying numerous “natural remedies” to the patient’s skin while in the hospital, including a liquid spray and lotion, both in unmarked bottles. At that time, the mother was informed that no external supplements should be applied to her son. The serum silver level was elevated substantially at 94.3 ng/mL (reference range, <1.0 ng/mL). When the mother was confronted, she initially denied use of silver but later admitted to notable silver content in the cream she was applying to her son’s skin. The mother reported that she read online that colloidal silver had been historically used to cure numerous ailments and she was ordering products from an online company. She was counseled on the dangers of both topical application and ingestion of silver, and all supplements were removed from the home.
Argyria is a rare condition caused by chronic exposure to silver and is characterized by a blue-gray pigmentation in the skin and appendages, mucous membranes, and internal organs.4 Clinically, argyria is classified as generalized or localized. Generalized argyria results from ingestion or inhalation of silver compounds, where granules deposit preferentially in sun-exposed areas of skin as well as internal organs, with the highest concentration in the liver, spleen, and adrenal glands; discoloration often is permanent.5 On the contrary, localized argyria results from direct external contact with silver and granules deposited in the hands, eyes, and mucosa.5 Although the exact mechanism of penetration from topical silver remains unknown, it is thought to enter via the eccrine sweat ducts, as histopathology reveals silver granules found in highest concentration surrounding sweat glands in the dermis.6
Initial differential diagnoses for altered nail pigmentation include drug-induced causes, systemic diseases, cyanosis, and exposure to metals.7 The most commonly indicated medications resulting in blue nail pigment changes include antimalarials, minocycline, zidovudine, and phenothiazine. Systemic diseases that may cause blue nail color change include Wilson disease, hemochromatosis, Addison disease, methemoglobinemia, and alkaptonuria.7 Metals include gold, mercury, arsenic, bismuth, lead, and silver.4 After a thorough review of the patient’s medications and lack of support for any underlying disease process, contact with metals, particularly silver, was ranked highly on our differential list. In support of this theory, the mother’s bluish gray facial skin led to high clinical suspicion that she was ingesting colloidal silver and also was exposing her son to silver.
Treatment of argyria is challenging but first and foremost involves discontinuation of the source of chronic silver exposure. Unfortunately, the discoloration of generalized argyria often is permanent. Sunscreen can be used to help prevent any further darkening of pigment. The pigment in localized argyria has been reported to slowly fade with time, and there also have been reports of successful treatment using a low-fluence Q-switched 1064-nm Nd:YAG laser.8
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Lencastre A, Lobo M, João A. Argyria—case report. An Bras Dermatol. 2013;88:413-416.
- Park S-W, Kim J-H, Shin H-T, et al. An effective modality for argyria treatment: Q-switched 1,064-nm Nd:YAG laser. Ann Dermatol. 2013;25:511-512.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Garcias-Ladaria J, Hernandez-Bel P, Torregrosa-Calatayud JL, et al. Localized cutaneous argyria: a report of 2 cases. Actas Dermosifiliogr. 2013;104:253-254.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-192.
- Kubba A, Kubba R, Batrani M, Pal T. Argyria an unrecognized cause of cutaneous pigmentation in Indian patients: a case series and review of the literature. Indian J Dermatol Venereol Leprol. 2013;79:805-811.
- Han TY, Chang HS, Lee HK, et al. Successful treatment of argyria using a low-fluence Q-switched 1064-nm Nd:YAG laser. Int J Dermatol. 2011;50:751-753.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Lencastre A, Lobo M, João A. Argyria—case report. An Bras Dermatol. 2013;88:413-416.
- Park S-W, Kim J-H, Shin H-T, et al. An effective modality for argyria treatment: Q-switched 1,064-nm Nd:YAG laser. Ann Dermatol. 2013;25:511-512.
- Molina-Hernandez AI, Diaz-Gonzalez JM, Saeb-Lima M, et al. Argyria after silver nitrate intake: case report and brief review of literature. Indian J Dermatol. 2015;60:520.
- Garcias-Ladaria J, Hernandez-Bel P, Torregrosa-Calatayud JL, et al. Localized cutaneous argyria: a report of 2 cases. Actas Dermosifiliogr. 2013;104:253-254.
- Kapur N, Landon G, Yu RC. Localized argyria in an antique restorer. Br J Dermatol. 2001;144:191-192.
- Kubba A, Kubba R, Batrani M, Pal T. Argyria an unrecognized cause of cutaneous pigmentation in Indian patients: a case series and review of the literature. Indian J Dermatol Venereol Leprol. 2013;79:805-811.
- Han TY, Chang HS, Lee HK, et al. Successful treatment of argyria using a low-fluence Q-switched 1064-nm Nd:YAG laser. Int J Dermatol. 2011;50:751-753.
Practice Points
- Argyria results from chronic exposure to products with a high silver content and may result in abnormalities of the skin and internal organs.
- Examination of the fingernails can provide important clues to underlying systemic conditions or external exposures.
Phacomatosis Pigmentokeratotica Associated With Raynaud Phenomenon, Segmental Nevi, Hyperhidrosis, and Scoliosis
To the Editor:
Phacomatosis pigmentokeratotica (PPK) is a rare epidermal nevus syndrome complicated by multiple extracutaneous anomalies, including skeletal defects and neurologic anomalies. Less common associations include lateral curvature of the spine and hyperhidrosis. We present a patient with PPK and unilateral Raynaud phenomenon in addition to a segmental distribution of melanocytic nevi, hyperhidrosis, and scoliosis.
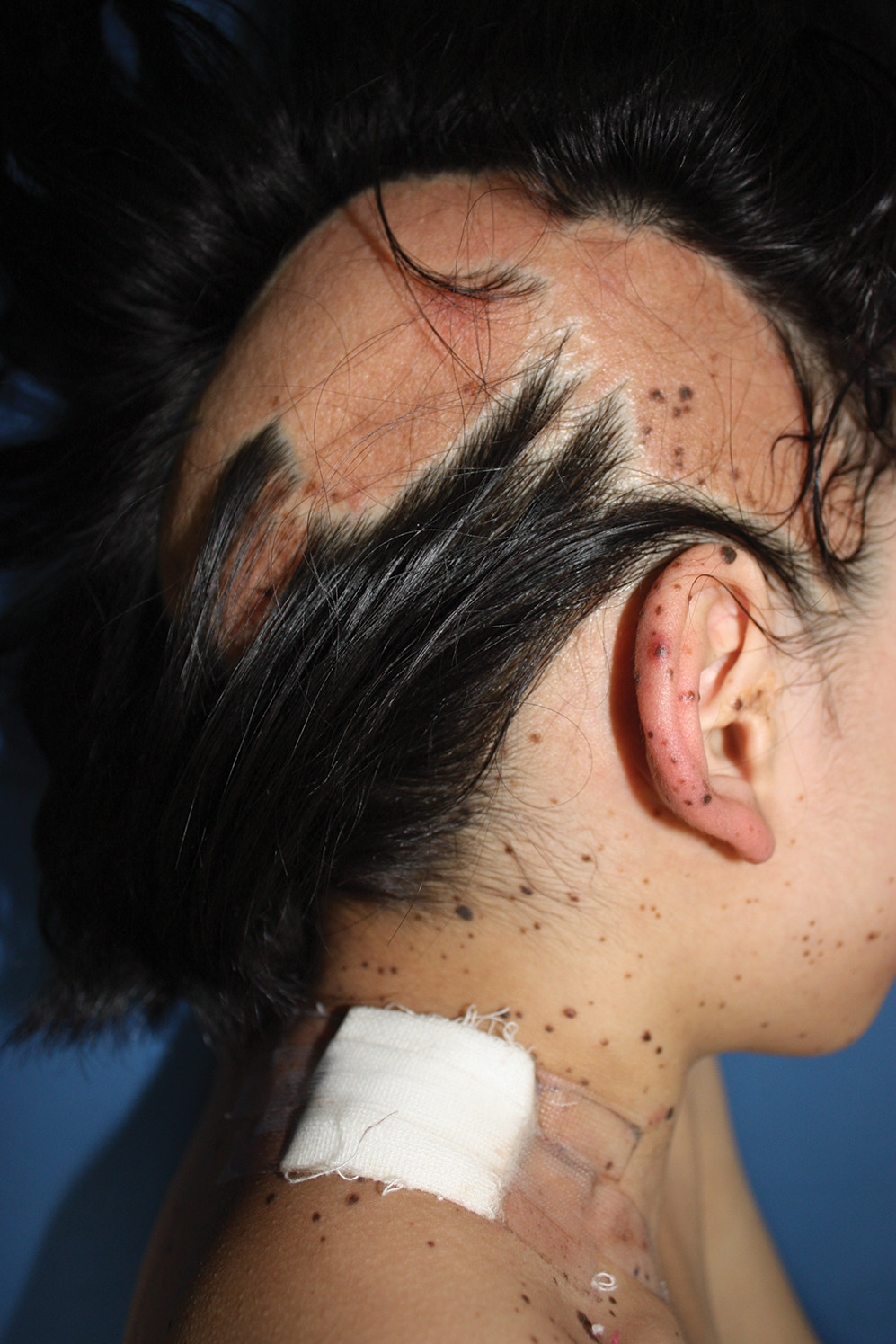
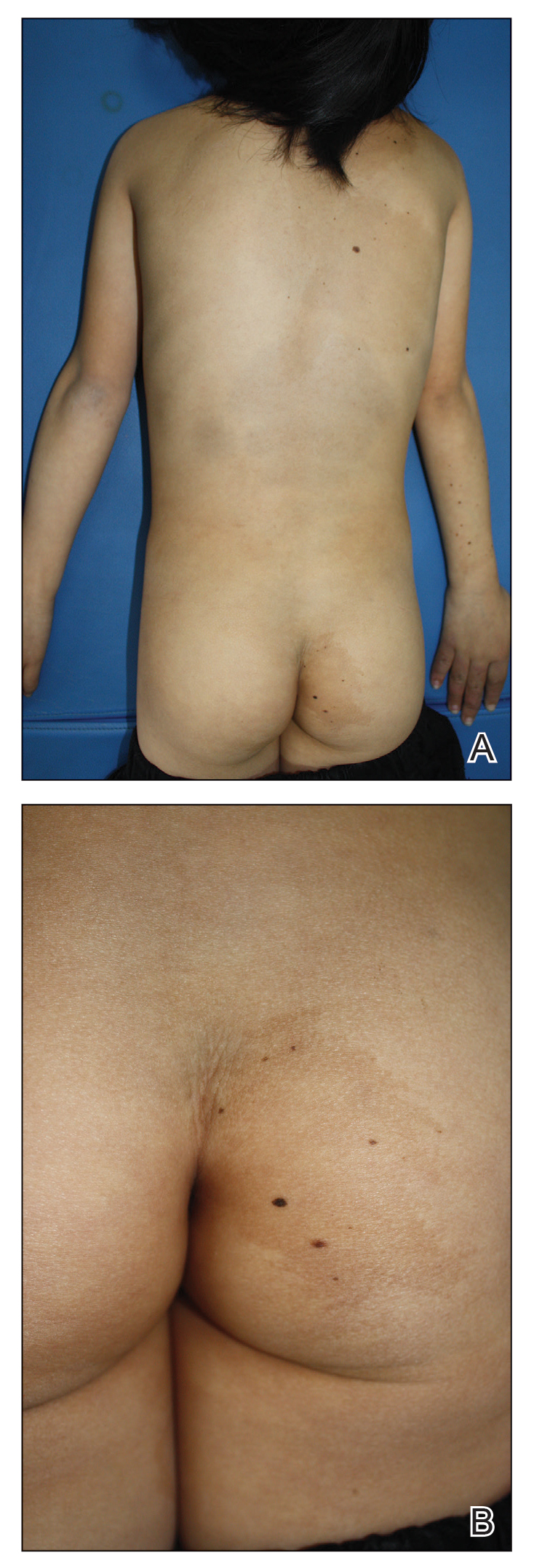
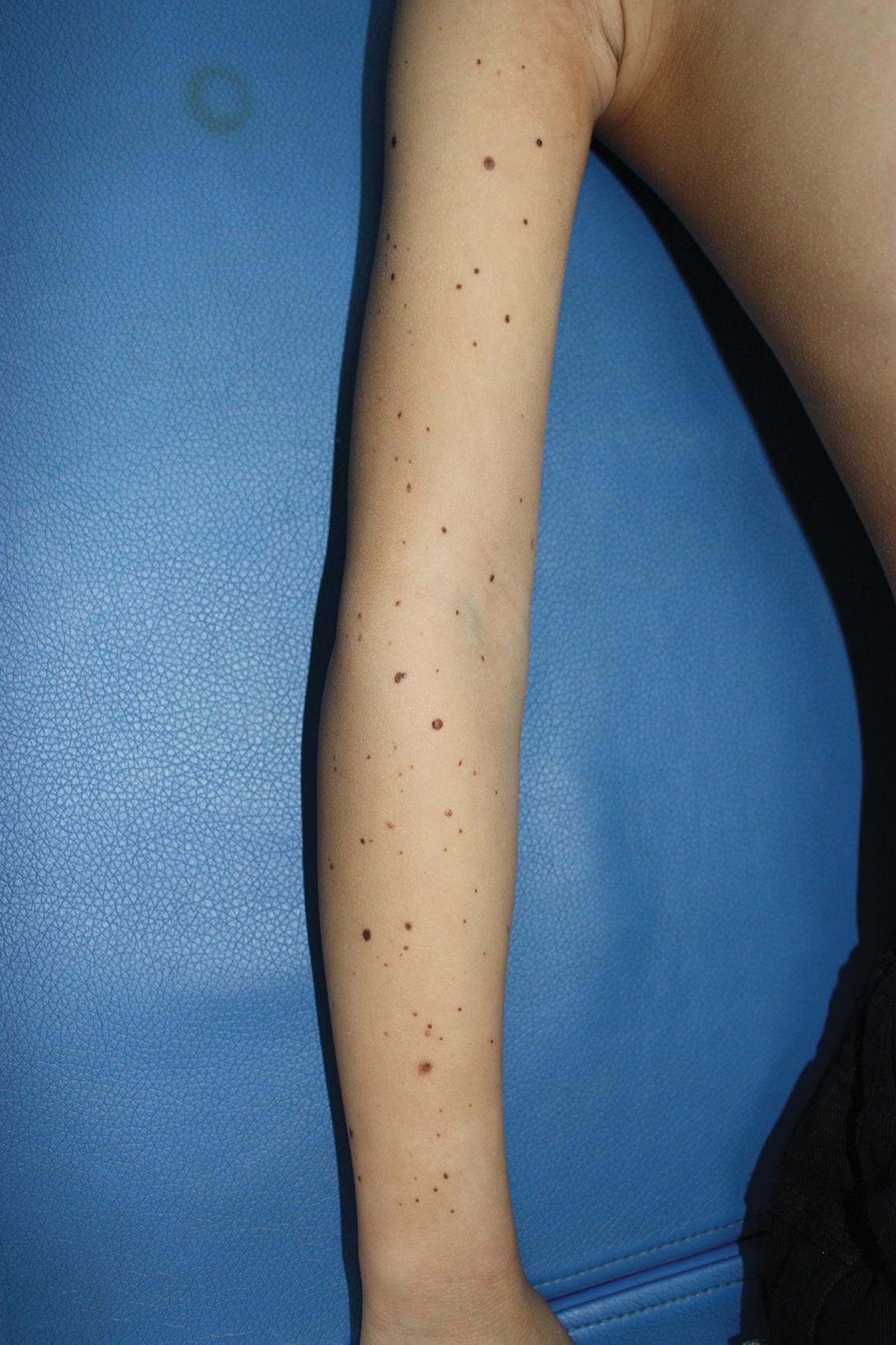
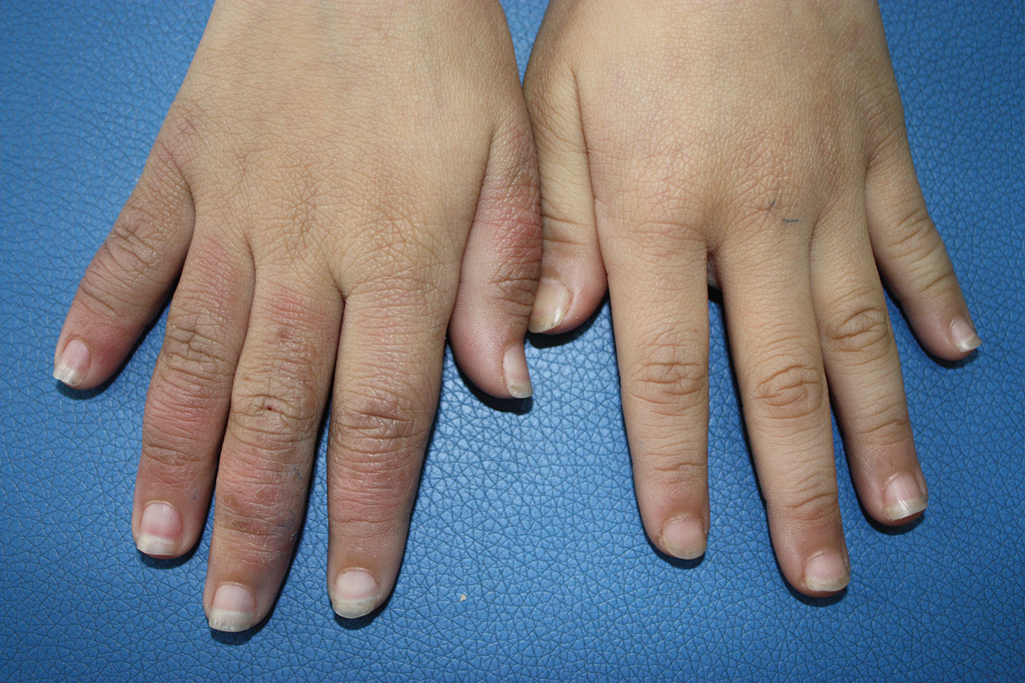
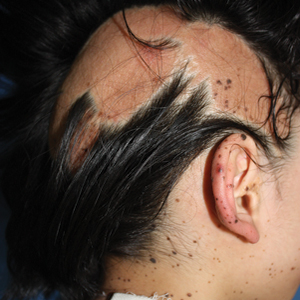
A 9-year-old girl was born with a yellow-orange alopecic plaque on the right side of the scalp (Figure 1). There also were 2 large, irregularly pigmented patches localized on the right side of the upper back and buttock. Over 3 years, numerous papular nevi developed within these pigmented patches and were diagnosed as speckled lentiginous nevi (Figure 2). In addition, numerous nevi of various sizes affected the right face, right shoulder, right arm (Figure 3), and right neck and were clearly demarcated along the midline. Several nevi also were noted within the nevus sebaceous on the right scalp. These skin lesions expanded progressively with age. At 6 years of age, she was diagnosed with hyperhidrosis of the right half of the body, which was most pronounced on the face. Raynaud phenomenon restricted to the right hand also was noted (Figure 4). Upon cold exposure, the digits become pale white, cold, and numb; then blue; and finally red. She lacked other features of connective tissue disease, and autoantibody testing was negative. She also was noted to have an abnormal lateral curvature of the spine (scoliosis). Auditory, ocular, and neurologic examinations were normal. Cranial and cerebral magnetic resonance imaging showed no central nervous system abnormalities. Her family history was negative for nevus spilus, nevus sebaceous, and neurofibromatosis. The clinical findings in our patient led to the diagnosis of PPK.
Phacomatosis pigmentokeratotica is a distinctive epidermal nevus syndrome characterized by the coexistence of a speckled lentiginous nevus, also known as a nevus spilus, and a nevus sebaceous1; PPK frequently is complicated by skeletal, ophthalmic, or neurologic abnormalities.2 Most cases reported are sporadic, and a postzygotic mosaic HRas proto-oncogene, GTPase, HRAS, mutation has been demonstrated in some patients and may contribute to the phenotype of PPK.3,4
Other anomalies have included ichthyosislike diffuse hyperkeratosis, laxity of the hands, pelvic hypoplasia, glaucoma, psychomotor retardation, and hypophosphatemic rickets. These patients also should be monitored for the development of malignant neoplasms within the nevus sebaceous.5 Segmental hyperhidrosis may be seen in association with the nevus spilus component.2
Raynaud phenomenon involving only the right hand was a unique finding in our patient. In 3 years of follow-up, our patient developed no evidence of connective tissue disease or other systemic illness. We speculate that Raynaud phenomenon of the right hand along with hyperhidrosis of the right side of the body could be a result of dysfunction of the autonomic nervous system. We propose that Raynaud phenomenon represents an unusual manifestation of PPK and may broaden the spectrum of extracutaneous anomalies associated with the disease. The finding of segmental nevi outside of the confines of the nevus spilus was another unusual manifestation of mosaicism.
- Happle R, Hoffmann R, Restano L, et al. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet. 1996;65:363-365.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22, 23-24.
- Groesser L, Herschberger E, Sagrera A, et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol. 2013;133:1998-2003.
- Martin RJ, Arefi M, Splitt M, et al. Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br J Dermatol. 2018;178:289-291.
- Chu GY, Wu CY. Phacomatosis pigmentokeratotica: a follow-up report with fatal outcome. Acta Derm Venereol. 2014;94:467-468.
To the Editor:
Phacomatosis pigmentokeratotica (PPK) is a rare epidermal nevus syndrome complicated by multiple extracutaneous anomalies, including skeletal defects and neurologic anomalies. Less common associations include lateral curvature of the spine and hyperhidrosis. We present a patient with PPK and unilateral Raynaud phenomenon in addition to a segmental distribution of melanocytic nevi, hyperhidrosis, and scoliosis.
A 9-year-old girl was born with a yellow-orange alopecic plaque on the right side of the scalp (Figure 1). There also were 2 large, irregularly pigmented patches localized on the right side of the upper back and buttock. Over 3 years, numerous papular nevi developed within these pigmented patches and were diagnosed as speckled lentiginous nevi (Figure 2). In addition, numerous nevi of various sizes affected the right face, right shoulder, right arm (Figure 3), and right neck and were clearly demarcated along the midline. Several nevi also were noted within the nevus sebaceous on the right scalp. These skin lesions expanded progressively with age. At 6 years of age, she was diagnosed with hyperhidrosis of the right half of the body, which was most pronounced on the face. Raynaud phenomenon restricted to the right hand also was noted (Figure 4). Upon cold exposure, the digits become pale white, cold, and numb; then blue; and finally red. She lacked other features of connective tissue disease, and autoantibody testing was negative. She also was noted to have an abnormal lateral curvature of the spine (scoliosis). Auditory, ocular, and neurologic examinations were normal. Cranial and cerebral magnetic resonance imaging showed no central nervous system abnormalities. Her family history was negative for nevus spilus, nevus sebaceous, and neurofibromatosis. The clinical findings in our patient led to the diagnosis of PPK.
Phacomatosis pigmentokeratotica is a distinctive epidermal nevus syndrome characterized by the coexistence of a speckled lentiginous nevus, also known as a nevus spilus, and a nevus sebaceous1; PPK frequently is complicated by skeletal, ophthalmic, or neurologic abnormalities.2 Most cases reported are sporadic, and a postzygotic mosaic HRas proto-oncogene, GTPase, HRAS, mutation has been demonstrated in some patients and may contribute to the phenotype of PPK.3,4
Other anomalies have included ichthyosislike diffuse hyperkeratosis, laxity of the hands, pelvic hypoplasia, glaucoma, psychomotor retardation, and hypophosphatemic rickets. These patients also should be monitored for the development of malignant neoplasms within the nevus sebaceous.5 Segmental hyperhidrosis may be seen in association with the nevus spilus component.2
Raynaud phenomenon involving only the right hand was a unique finding in our patient. In 3 years of follow-up, our patient developed no evidence of connective tissue disease or other systemic illness. We speculate that Raynaud phenomenon of the right hand along with hyperhidrosis of the right side of the body could be a result of dysfunction of the autonomic nervous system. We propose that Raynaud phenomenon represents an unusual manifestation of PPK and may broaden the spectrum of extracutaneous anomalies associated with the disease. The finding of segmental nevi outside of the confines of the nevus spilus was another unusual manifestation of mosaicism.
To the Editor:
Phacomatosis pigmentokeratotica (PPK) is a rare epidermal nevus syndrome complicated by multiple extracutaneous anomalies, including skeletal defects and neurologic anomalies. Less common associations include lateral curvature of the spine and hyperhidrosis. We present a patient with PPK and unilateral Raynaud phenomenon in addition to a segmental distribution of melanocytic nevi, hyperhidrosis, and scoliosis.
A 9-year-old girl was born with a yellow-orange alopecic plaque on the right side of the scalp (Figure 1). There also were 2 large, irregularly pigmented patches localized on the right side of the upper back and buttock. Over 3 years, numerous papular nevi developed within these pigmented patches and were diagnosed as speckled lentiginous nevi (Figure 2). In addition, numerous nevi of various sizes affected the right face, right shoulder, right arm (Figure 3), and right neck and were clearly demarcated along the midline. Several nevi also were noted within the nevus sebaceous on the right scalp. These skin lesions expanded progressively with age. At 6 years of age, she was diagnosed with hyperhidrosis of the right half of the body, which was most pronounced on the face. Raynaud phenomenon restricted to the right hand also was noted (Figure 4). Upon cold exposure, the digits become pale white, cold, and numb; then blue; and finally red. She lacked other features of connective tissue disease, and autoantibody testing was negative. She also was noted to have an abnormal lateral curvature of the spine (scoliosis). Auditory, ocular, and neurologic examinations were normal. Cranial and cerebral magnetic resonance imaging showed no central nervous system abnormalities. Her family history was negative for nevus spilus, nevus sebaceous, and neurofibromatosis. The clinical findings in our patient led to the diagnosis of PPK.
Phacomatosis pigmentokeratotica is a distinctive epidermal nevus syndrome characterized by the coexistence of a speckled lentiginous nevus, also known as a nevus spilus, and a nevus sebaceous1; PPK frequently is complicated by skeletal, ophthalmic, or neurologic abnormalities.2 Most cases reported are sporadic, and a postzygotic mosaic HRas proto-oncogene, GTPase, HRAS, mutation has been demonstrated in some patients and may contribute to the phenotype of PPK.3,4
Other anomalies have included ichthyosislike diffuse hyperkeratosis, laxity of the hands, pelvic hypoplasia, glaucoma, psychomotor retardation, and hypophosphatemic rickets. These patients also should be monitored for the development of malignant neoplasms within the nevus sebaceous.5 Segmental hyperhidrosis may be seen in association with the nevus spilus component.2
Raynaud phenomenon involving only the right hand was a unique finding in our patient. In 3 years of follow-up, our patient developed no evidence of connective tissue disease or other systemic illness. We speculate that Raynaud phenomenon of the right hand along with hyperhidrosis of the right side of the body could be a result of dysfunction of the autonomic nervous system. We propose that Raynaud phenomenon represents an unusual manifestation of PPK and may broaden the spectrum of extracutaneous anomalies associated with the disease. The finding of segmental nevi outside of the confines of the nevus spilus was another unusual manifestation of mosaicism.
- Happle R, Hoffmann R, Restano L, et al. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet. 1996;65:363-365.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22, 23-24.
- Groesser L, Herschberger E, Sagrera A, et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol. 2013;133:1998-2003.
- Martin RJ, Arefi M, Splitt M, et al. Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br J Dermatol. 2018;178:289-291.
- Chu GY, Wu CY. Phacomatosis pigmentokeratotica: a follow-up report with fatal outcome. Acta Derm Venereol. 2014;94:467-468.
- Happle R, Hoffmann R, Restano L, et al. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet. 1996;65:363-365.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22, 23-24.
- Groesser L, Herschberger E, Sagrera A, et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol. 2013;133:1998-2003.
- Martin RJ, Arefi M, Splitt M, et al. Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br J Dermatol. 2018;178:289-291.
- Chu GY, Wu CY. Phacomatosis pigmentokeratotica: a follow-up report with fatal outcome. Acta Derm Venereol. 2014;94:467-468.
Practice Points
- Phacomatosis pigmentokeratotica (PPK) is characterized by the coexistence of speckled lentiginous nevus and nevus sebaceous.
- Raynaud phenomenon may be an unreported association with PPK.
Urticarial Vasculitis Successfully Treated With Omalizumab
To the Editor:
Urticarial vasculitis (UV) is a clinicopathologic entity. It manifests as an eruption of erythematous wheals that clinically resemble urticaria, but the lesions of UV last longer, may leave residual hyperpigmentation, and may or may not be pruritic.1 Therapies most often employed include oral antihistamines and systemic immunosuppressant drugs such as corticosteroids, dapsone, colchicine, or hydroxychloroquine.2 We present a woman with UV who successfully was treated with omalizumab.
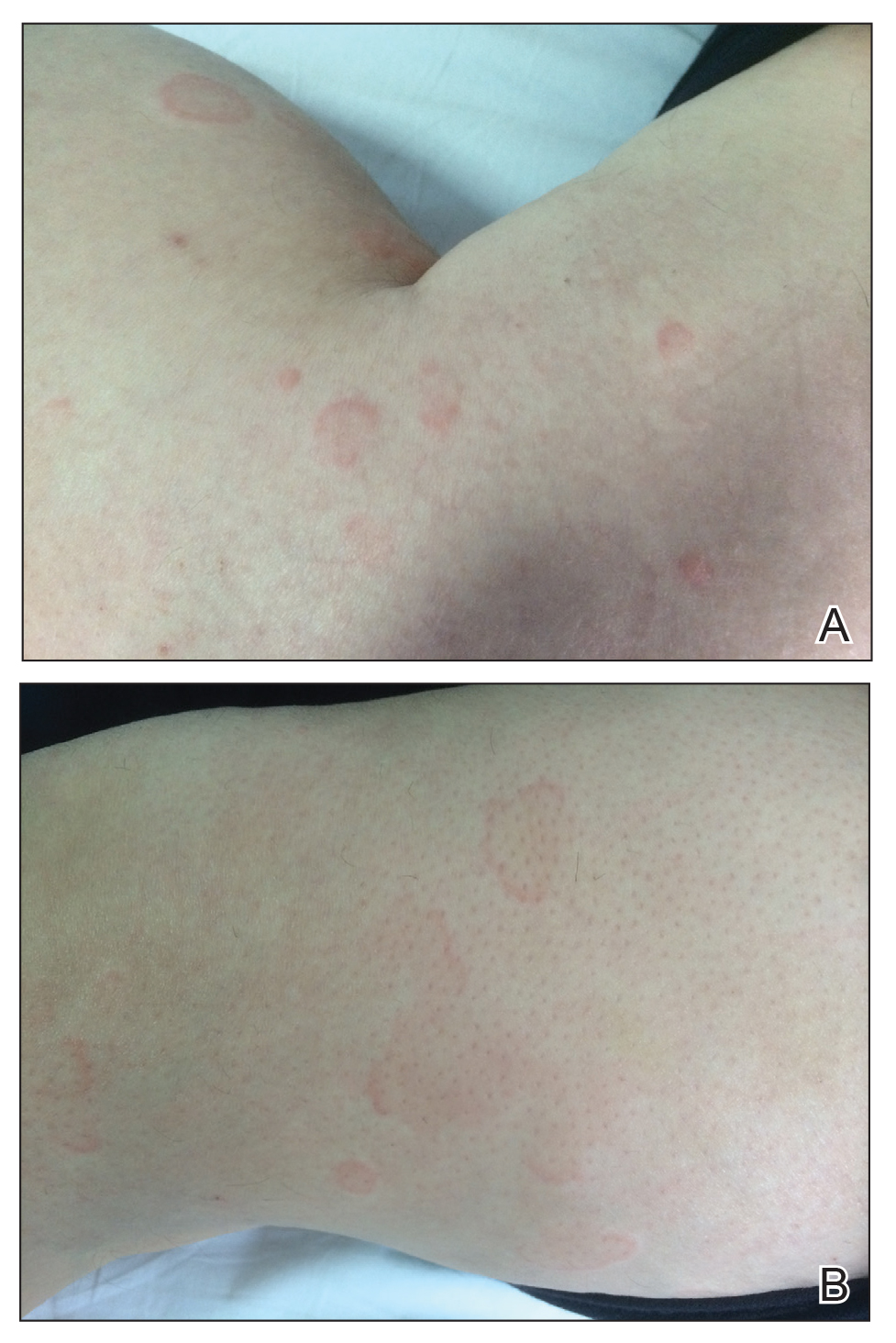
A 49-year-old woman presented to our outpatient clinic with generalized pruritic skin rashes of 2 years’ duration. She also described swelling on the upper eyelids 2 times monthly. She used several antihistamines (up to 4 times daily) and was taking systemic corticosteroids and antidepressants. Physical examination revealed generalized erythematous and edematous papules and plaques on the trunk and extremities (Figure 1). At follow-up a few days later, we observed that the lesions were lasting for more than 24 hours, but there was no residual pigmentation. According to clinical concerns and the association with angioedema, we initially thought the diagnosis was chronic urticaria and angioedema. The patient had no extracutaneous manifestations such as fever, arthralgia, or lymphadenopathy. Routine laboratory examinations including antinuclear antibodies were within reference range. She had normal C3 and C4 levels and an elevated total IgE level (344 IU/mL [reference range, 0–170 IU/mL]). Because the IgE level was elevated and she had no response to the highest dosages of antihistamines, we decided to start omalizumab therapy. Prior to starting omalizumab, we performed a skin biopsy for histopathologic and direct immunofluorescence examinations for UV, as the duration of the lesions was more than 24 hours. Histopathologic examination revealed lymphocytes within the vessel wall and perivascular lymphocytic infiltration with eosinophils (Figure 2). On direct immunofluorescence, perivascular IgA deposition was observed (Figure 3). Histopathologic findings were associated with lymphocytic vasculitis. Systemic involvement was not detected on detailed laboratory and radiologic examinations.


After the first application of omalizumab, the lesions disappeared within a few days. She was treated with subcutaneous omalizumab 300 mg every 4 weeks for 6 months, and we did not observe any adverse effects related to the drug. There was no relapse after therapy cessation.
Omalizumab is a recombinant humanized anti-IgE monoclonal antibody that is approved by the US Food and Drug Administration for treatment of chronic idiopathic urticaria.3-5 Studies have suggested that omalizumab might play an important role in the treatment of other potentially IgE-mediated disease processes including allergic asthma, atopic dermatitis, allergic rhinitis, nasal polyposis, and severe ocular allergies.6 The proposed mechanism of action of omalizumab includes reduction of free IgE through the reversible formation of tiny, biologically inert complexes; targeting IgE-expressing B cells; and inhibiting production of IgE. Because it reduces free IgE, omalizumab has been used in normal IgE or hyper-IgE situations. Omalizumab also induces eosinophil apoptosis; increases IL-2, IL-3, tumor necrosis factor α, and IFN-γ; and reduces IL-4.7 A number of off-label uses have been described such as atopic dermatitis, bullous pemphigoid, hyper-IgE syndrome, cutaneous mastocytosis, toxic epidermal necrolysis, and eosinophilic granulomatosis with polyangitis.8 There are no clinical studies of omalizumab for UV, and only a few case reports have shown that omalizumab also might be beneficial for this condition.2-4 Diez et al4 reported 3 cases of women aged 28, 51, and 54 years with spontaneous chronic urticaria with autoimmune and pressure components as well as vasculitis whose symptoms completely improved after starting omalizumab. Kai et al3 successfully treated a patient with normocomplementemic UV with omalizumab and suggested that omalizumab markedly improved the patient’s quality of life with chronic urticaria and UV. Ghazanfar and Thomsen2 reported the case of a 68-year-old man diagnosed with histopathologically confirmed leukocytoclastic vasculitis. He had used systemic corticosteroid therapy and dapsone without notable improvement. The patient was switched to subcutaneous omalizumab 300 mg once every 4 weeks; after 1 month, he observed complete remission of the UV and symptoms.2
Our case suggests that omalizumab has a beneficial effect on patients with UV. Omalizumab may be effective in UV through its reduction of IgE, as in chronic urticaria, and through downstream effects on cellular activation mechanisms (possibly a reduction in chemotaxis or immune complex formation). However, the mechanism of action of omalizumab for UV remains, in part, unresolved. It is not known whether omalizumab is efficacious against both normocomplementemic and hypocomplementemic UV. Further studies with a greater number of patients are needed to confirm the effects of omalizumab for vasculitic patients.
- Chang S, Carr W. Urticarial vasculitis. Allergy Asthma Proc. 2007;28:97-100.
- Ghazanfar MN, Thomsen SF. Omalizumab for urticarial vasculitis: case report and review of the literature. Case Rep Dermatol Med. 2015:576893.
- Kai AC, Flohr C, Grattan CE. Improvement in quality of life impairment followed by relapse with 6-monthly periodic administration of omalizumab for severe treatment-refractory chronic urticaria and urticarial vasculitis. Clin Exp Dermatol. 2014;39:651-652.
- Diez LS, Tamayo LM, Cardona R. Omalizumab: therapeutic option in chronic spontaneous urticaria difficult to control with associated vasculitis, report of three cases. Biomedica. 2013;33:503-512.
- Maurer M, Rosen K, Hsieh HJ. Omalizumab for chronic urticaria. N Engl J Med. 2013;368:2530.
- Ben Shoshan M. Omalizumab: not only for asthma. Recent Pat Inflamm Allergy Drug Discov. 2008;2:191-201.
- Fueyo-Casado A, Campos-Munoz L, Gonzalez-Guerra E, et al. Effectiveness of omalizumab in a case of urticarial vasculitis. Clin Exp Dermatol. Published March 1, 2017. doi:10.1111/ced.13076
- Chia JC, Mydlarski PR. Dermatologic uses of omalizumab. J Dermatol Treat. Published November 7, 2016. doi:10.1080/09546634.2016.1249819
To the Editor:
Urticarial vasculitis (UV) is a clinicopathologic entity. It manifests as an eruption of erythematous wheals that clinically resemble urticaria, but the lesions of UV last longer, may leave residual hyperpigmentation, and may or may not be pruritic.1 Therapies most often employed include oral antihistamines and systemic immunosuppressant drugs such as corticosteroids, dapsone, colchicine, or hydroxychloroquine.2 We present a woman with UV who successfully was treated with omalizumab.
A 49-year-old woman presented to our outpatient clinic with generalized pruritic skin rashes of 2 years’ duration. She also described swelling on the upper eyelids 2 times monthly. She used several antihistamines (up to 4 times daily) and was taking systemic corticosteroids and antidepressants. Physical examination revealed generalized erythematous and edematous papules and plaques on the trunk and extremities (Figure 1). At follow-up a few days later, we observed that the lesions were lasting for more than 24 hours, but there was no residual pigmentation. According to clinical concerns and the association with angioedema, we initially thought the diagnosis was chronic urticaria and angioedema. The patient had no extracutaneous manifestations such as fever, arthralgia, or lymphadenopathy. Routine laboratory examinations including antinuclear antibodies were within reference range. She had normal C3 and C4 levels and an elevated total IgE level (344 IU/mL [reference range, 0–170 IU/mL]). Because the IgE level was elevated and she had no response to the highest dosages of antihistamines, we decided to start omalizumab therapy. Prior to starting omalizumab, we performed a skin biopsy for histopathologic and direct immunofluorescence examinations for UV, as the duration of the lesions was more than 24 hours. Histopathologic examination revealed lymphocytes within the vessel wall and perivascular lymphocytic infiltration with eosinophils (Figure 2). On direct immunofluorescence, perivascular IgA deposition was observed (Figure 3). Histopathologic findings were associated with lymphocytic vasculitis. Systemic involvement was not detected on detailed laboratory and radiologic examinations.
After the first application of omalizumab, the lesions disappeared within a few days. She was treated with subcutaneous omalizumab 300 mg every 4 weeks for 6 months, and we did not observe any adverse effects related to the drug. There was no relapse after therapy cessation.
Omalizumab is a recombinant humanized anti-IgE monoclonal antibody that is approved by the US Food and Drug Administration for treatment of chronic idiopathic urticaria.3-5 Studies have suggested that omalizumab might play an important role in the treatment of other potentially IgE-mediated disease processes including allergic asthma, atopic dermatitis, allergic rhinitis, nasal polyposis, and severe ocular allergies.6 The proposed mechanism of action of omalizumab includes reduction of free IgE through the reversible formation of tiny, biologically inert complexes; targeting IgE-expressing B cells; and inhibiting production of IgE. Because it reduces free IgE, omalizumab has been used in normal IgE or hyper-IgE situations. Omalizumab also induces eosinophil apoptosis; increases IL-2, IL-3, tumor necrosis factor α, and IFN-γ; and reduces IL-4.7 A number of off-label uses have been described such as atopic dermatitis, bullous pemphigoid, hyper-IgE syndrome, cutaneous mastocytosis, toxic epidermal necrolysis, and eosinophilic granulomatosis with polyangitis.8 There are no clinical studies of omalizumab for UV, and only a few case reports have shown that omalizumab also might be beneficial for this condition.2-4 Diez et al4 reported 3 cases of women aged 28, 51, and 54 years with spontaneous chronic urticaria with autoimmune and pressure components as well as vasculitis whose symptoms completely improved after starting omalizumab. Kai et al3 successfully treated a patient with normocomplementemic UV with omalizumab and suggested that omalizumab markedly improved the patient’s quality of life with chronic urticaria and UV. Ghazanfar and Thomsen2 reported the case of a 68-year-old man diagnosed with histopathologically confirmed leukocytoclastic vasculitis. He had used systemic corticosteroid therapy and dapsone without notable improvement. The patient was switched to subcutaneous omalizumab 300 mg once every 4 weeks; after 1 month, he observed complete remission of the UV and symptoms.2
Our case suggests that omalizumab has a beneficial effect on patients with UV. Omalizumab may be effective in UV through its reduction of IgE, as in chronic urticaria, and through downstream effects on cellular activation mechanisms (possibly a reduction in chemotaxis or immune complex formation). However, the mechanism of action of omalizumab for UV remains, in part, unresolved. It is not known whether omalizumab is efficacious against both normocomplementemic and hypocomplementemic UV. Further studies with a greater number of patients are needed to confirm the effects of omalizumab for vasculitic patients.
To the Editor:
Urticarial vasculitis (UV) is a clinicopathologic entity. It manifests as an eruption of erythematous wheals that clinically resemble urticaria, but the lesions of UV last longer, may leave residual hyperpigmentation, and may or may not be pruritic.1 Therapies most often employed include oral antihistamines and systemic immunosuppressant drugs such as corticosteroids, dapsone, colchicine, or hydroxychloroquine.2 We present a woman with UV who successfully was treated with omalizumab.
A 49-year-old woman presented to our outpatient clinic with generalized pruritic skin rashes of 2 years’ duration. She also described swelling on the upper eyelids 2 times monthly. She used several antihistamines (up to 4 times daily) and was taking systemic corticosteroids and antidepressants. Physical examination revealed generalized erythematous and edematous papules and plaques on the trunk and extremities (Figure 1). At follow-up a few days later, we observed that the lesions were lasting for more than 24 hours, but there was no residual pigmentation. According to clinical concerns and the association with angioedema, we initially thought the diagnosis was chronic urticaria and angioedema. The patient had no extracutaneous manifestations such as fever, arthralgia, or lymphadenopathy. Routine laboratory examinations including antinuclear antibodies were within reference range. She had normal C3 and C4 levels and an elevated total IgE level (344 IU/mL [reference range, 0–170 IU/mL]). Because the IgE level was elevated and she had no response to the highest dosages of antihistamines, we decided to start omalizumab therapy. Prior to starting omalizumab, we performed a skin biopsy for histopathologic and direct immunofluorescence examinations for UV, as the duration of the lesions was more than 24 hours. Histopathologic examination revealed lymphocytes within the vessel wall and perivascular lymphocytic infiltration with eosinophils (Figure 2). On direct immunofluorescence, perivascular IgA deposition was observed (Figure 3). Histopathologic findings were associated with lymphocytic vasculitis. Systemic involvement was not detected on detailed laboratory and radiologic examinations.
After the first application of omalizumab, the lesions disappeared within a few days. She was treated with subcutaneous omalizumab 300 mg every 4 weeks for 6 months, and we did not observe any adverse effects related to the drug. There was no relapse after therapy cessation.
Omalizumab is a recombinant humanized anti-IgE monoclonal antibody that is approved by the US Food and Drug Administration for treatment of chronic idiopathic urticaria.3-5 Studies have suggested that omalizumab might play an important role in the treatment of other potentially IgE-mediated disease processes including allergic asthma, atopic dermatitis, allergic rhinitis, nasal polyposis, and severe ocular allergies.6 The proposed mechanism of action of omalizumab includes reduction of free IgE through the reversible formation of tiny, biologically inert complexes; targeting IgE-expressing B cells; and inhibiting production of IgE. Because it reduces free IgE, omalizumab has been used in normal IgE or hyper-IgE situations. Omalizumab also induces eosinophil apoptosis; increases IL-2, IL-3, tumor necrosis factor α, and IFN-γ; and reduces IL-4.7 A number of off-label uses have been described such as atopic dermatitis, bullous pemphigoid, hyper-IgE syndrome, cutaneous mastocytosis, toxic epidermal necrolysis, and eosinophilic granulomatosis with polyangitis.8 There are no clinical studies of omalizumab for UV, and only a few case reports have shown that omalizumab also might be beneficial for this condition.2-4 Diez et al4 reported 3 cases of women aged 28, 51, and 54 years with spontaneous chronic urticaria with autoimmune and pressure components as well as vasculitis whose symptoms completely improved after starting omalizumab. Kai et al3 successfully treated a patient with normocomplementemic UV with omalizumab and suggested that omalizumab markedly improved the patient’s quality of life with chronic urticaria and UV. Ghazanfar and Thomsen2 reported the case of a 68-year-old man diagnosed with histopathologically confirmed leukocytoclastic vasculitis. He had used systemic corticosteroid therapy and dapsone without notable improvement. The patient was switched to subcutaneous omalizumab 300 mg once every 4 weeks; after 1 month, he observed complete remission of the UV and symptoms.2
Our case suggests that omalizumab has a beneficial effect on patients with UV. Omalizumab may be effective in UV through its reduction of IgE, as in chronic urticaria, and through downstream effects on cellular activation mechanisms (possibly a reduction in chemotaxis or immune complex formation). However, the mechanism of action of omalizumab for UV remains, in part, unresolved. It is not known whether omalizumab is efficacious against both normocomplementemic and hypocomplementemic UV. Further studies with a greater number of patients are needed to confirm the effects of omalizumab for vasculitic patients.
- Chang S, Carr W. Urticarial vasculitis. Allergy Asthma Proc. 2007;28:97-100.
- Ghazanfar MN, Thomsen SF. Omalizumab for urticarial vasculitis: case report and review of the literature. Case Rep Dermatol Med. 2015:576893.
- Kai AC, Flohr C, Grattan CE. Improvement in quality of life impairment followed by relapse with 6-monthly periodic administration of omalizumab for severe treatment-refractory chronic urticaria and urticarial vasculitis. Clin Exp Dermatol. 2014;39:651-652.
- Diez LS, Tamayo LM, Cardona R. Omalizumab: therapeutic option in chronic spontaneous urticaria difficult to control with associated vasculitis, report of three cases. Biomedica. 2013;33:503-512.
- Maurer M, Rosen K, Hsieh HJ. Omalizumab for chronic urticaria. N Engl J Med. 2013;368:2530.
- Ben Shoshan M. Omalizumab: not only for asthma. Recent Pat Inflamm Allergy Drug Discov. 2008;2:191-201.
- Fueyo-Casado A, Campos-Munoz L, Gonzalez-Guerra E, et al. Effectiveness of omalizumab in a case of urticarial vasculitis. Clin Exp Dermatol. Published March 1, 2017. doi:10.1111/ced.13076
- Chia JC, Mydlarski PR. Dermatologic uses of omalizumab. J Dermatol Treat. Published November 7, 2016. doi:10.1080/09546634.2016.1249819
- Chang S, Carr W. Urticarial vasculitis. Allergy Asthma Proc. 2007;28:97-100.
- Ghazanfar MN, Thomsen SF. Omalizumab for urticarial vasculitis: case report and review of the literature. Case Rep Dermatol Med. 2015:576893.
- Kai AC, Flohr C, Grattan CE. Improvement in quality of life impairment followed by relapse with 6-monthly periodic administration of omalizumab for severe treatment-refractory chronic urticaria and urticarial vasculitis. Clin Exp Dermatol. 2014;39:651-652.
- Diez LS, Tamayo LM, Cardona R. Omalizumab: therapeutic option in chronic spontaneous urticaria difficult to control with associated vasculitis, report of three cases. Biomedica. 2013;33:503-512.
- Maurer M, Rosen K, Hsieh HJ. Omalizumab for chronic urticaria. N Engl J Med. 2013;368:2530.
- Ben Shoshan M. Omalizumab: not only for asthma. Recent Pat Inflamm Allergy Drug Discov. 2008;2:191-201.
- Fueyo-Casado A, Campos-Munoz L, Gonzalez-Guerra E, et al. Effectiveness of omalizumab in a case of urticarial vasculitis. Clin Exp Dermatol. Published March 1, 2017. doi:10.1111/ced.13076
- Chia JC, Mydlarski PR. Dermatologic uses of omalizumab. J Dermatol Treat. Published November 7, 2016. doi:10.1080/09546634.2016.1249819
Practice Points
- The differential diagnosis of urticaria and urticarial vasculitis may be complicated.
- Omalizumab is an effective urticaria treatment and also can be an alternative treatment choice in resistant urticarial vasculitis.
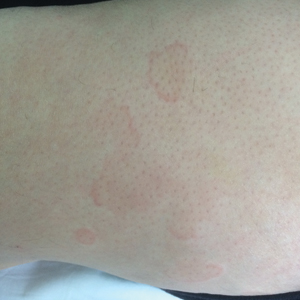
Bullous Pemphigoid Triggered by Liraglutide
To the Editor:
Bullous pemphigoid (BP) is an autoimmune blistering disease that typically affects the elderly, with an incidence of approximately 7 new cases per million.1 The pathogenesis of BP involves autoantibodies to BP antigens 180 and 230 at the dermoepidermal junction. Bullous pemphigoid has been associated with the use of multiple medications; vaccines; and physical damage to the skin, including trauma, radiation, and surgery.2
Several classes of medications may cause BP; one study described an association of BP with loop diuretics,3 while others found higher incidences of BP in patients taking aldosterone antagonists and neuroleptics.4 We describe a case of drug-triggered BP to liraglutide, a glucagonlike peptide 1 (GLP-1) receptor agonist.
A 75-year-old man presented to dermatology for evaluation of a vesicular eruption on the head, neck, trunk, and arms of 6 months’ duration. The eruption developed 2 weeks after starting liraglutide 1.2 mg subcutaneously daily for diabetes mellitus. The patient had a medical history of type 2 diabetes mellitus, hypertension, stroke, and prostate cancer treated with prostatectomy, and he also was taking insulin. Liraglutide was discontinued shortly after the onset of the eruption.
Physical examination revealed annular plaques on the head, neck, trunk, and arms with central hypopigmentation and hyperpigmented borders (Figure 1). Two tense bullae were evident on the left flank (Figure 2). Histopathology revealed a subepidermal blister, mixed perivascular infiltrate with numerous eosinophils, and pigment incontinence (Figure 3). Direct immunofluorescence showed linear deposition of IgG and C3 along the basement membrane zone that was localized to the roof of the blister on salt-split analysis. No microorganisms were identified on periodic acid–Schiff, Grocott-Gomori methenamine-silver, acid-fast bacilli, and Fite stains. The patient initially was treated with clobetasol ointment 0.05%, leading to marginal improvement. He declined treatment with prednisone or dapsone, and he was started on doxycycline. Seven months after stopping liraglutide and starting doxycycline, the patient had no blisters, but residual pigmentary changes remained.


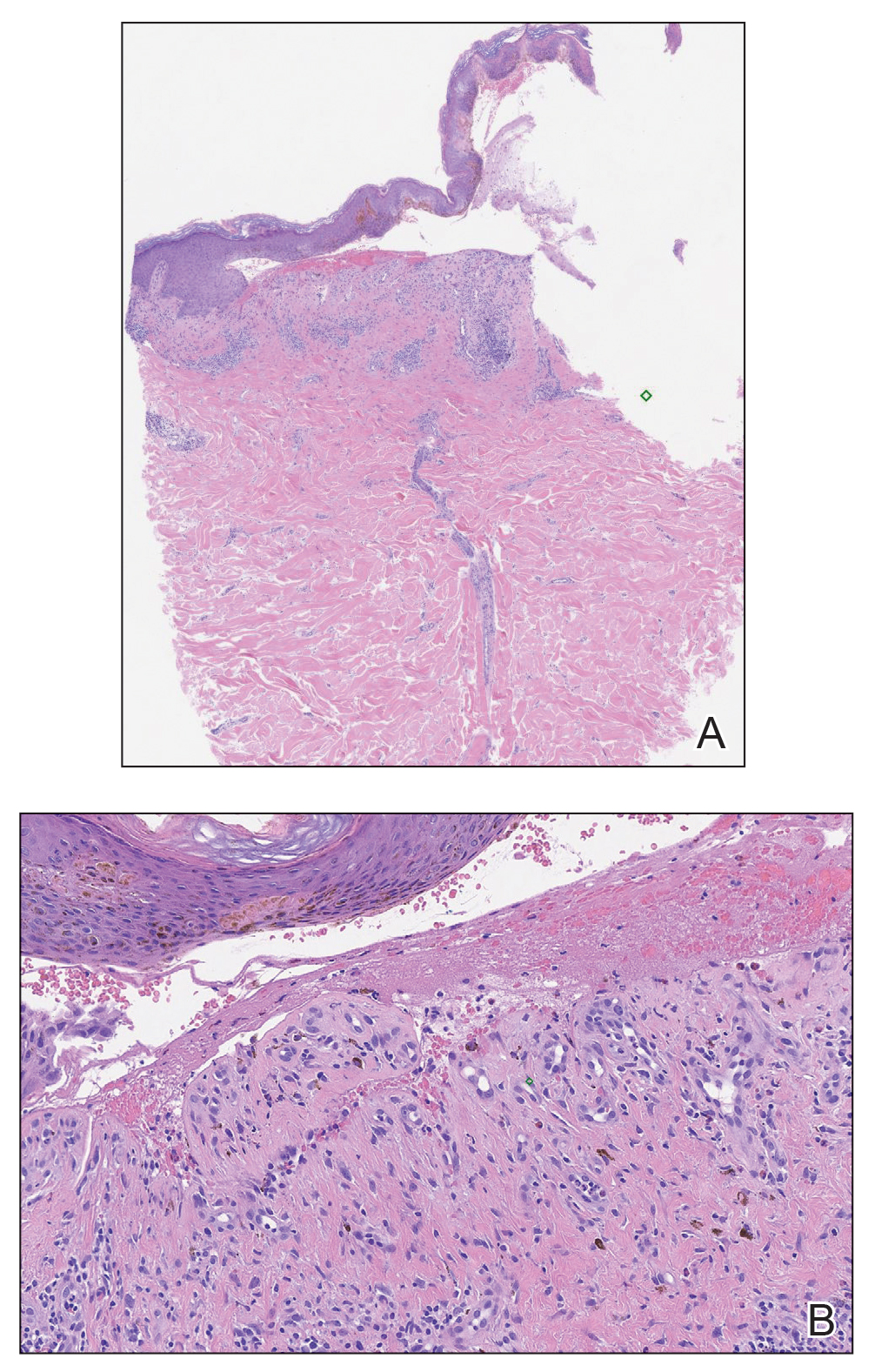
Two types of BP have been described in response to medications: drug-induced BP and drug-triggered BP. Drug-induced BP presents as an acute, self-limited eruption that typically resolves after withdrawal of the offending agent. It tends to involve a younger population and may present with mucosal involvement and target lesions on the palms and soles. Direct immunofluorescence shows linear IgG and C3 deposition at the basement membrane zone. Patients tend to respond quickly to systemic corticosteroids and have low recurrence rates. Drug-triggered BP is a chronic form of BP that is caused by a medication and is not resolved with removal of the offending agent.5 Therefore, drug-triggered BP is more difficult to detect, especially in patients taking multiple medications.
Our patient represents a case of drug-triggered BP to liraglutide. Liraglutide is a GLP-1 receptor agonist that is US Food and Drug Administration approved for the treatment of type 2 diabetes mellitus. Glucagonlike peptide 1 is an incretin hormone that is secreted by the intestine during digestion. It binds to the GLP-1 receptor leading to an increase in glucose-dependent insulin secretion and a decrease in glucagon secretion.6 Glucagonlike peptide 1 agonists also affect the immune system; liraglutide has been shown to modestly improve psoriasis, reduce the number of dermal gamma delta T cells, and decrease IL-17 expression.7 Glucagonlike peptide 1 agonists also produce anti-inflammatory effects on multiple organs including the liver, brain, vasculature, kidney, and skin.8
Dipeptidyl peptidase 4 (DPP-4) inhibitors that function to inhibit the degradation of GLP-1 and other peptides also have been reported to cause BP. In several patients, the DPP-4 inhibitors vildagliptin and sitagliptin caused drug-induced BP that resolved with discontinuation of the medication.9 Dipeptidyl peptidase 4 is expressed in various organ systems including the skin, and inhibition of DPP-4 enhances eosinophil mobilization in the blood and recruitment to the skin in animal models.10
Although the pathogenesis of BP involves autoantibodies to BP antigens 180 and 230, these antibodies are not sufficient to cause disease, as antibasement antibodies have been detected in patients without clinically evident BP. These patients, however, may be more susceptible to developing medication-induced BP. Several hypotheses regarding the pathogenesis of medication-induced BP have been proposed, including immune dysregulation, molecular mimicry, and cross-reactivity to a prior sensitizing agent.5 Liraglutide and the DPP-4 inhibitors affect the immune system, supporting the hypothesis of immune dysregulation; however, the exact mechanism of how immune modulating medications such as GLP-1 agonists and DPP-4 inhibitors cause BP remains unclear.
The effects of liraglutide and the DPP-4 inhibitors on the immune system may play a role in the pathogenesis of drug-triggered BP and drug-induced BP, respectively. Additional studies of the immunomodulatory effects of GLP-1 agonists and DPP-4 inhibitors may help elucidate the pathogenesis of drug-triggered or drug-induced BP.
- Serwin AB, Musialkowska E, Piascik M. Incidence and mortality of bullous pemphigoid in north-east Poland (Podlaskie Province), 1999-2012: a retrospective bicentric cohort study. Int J Dermatol. 2014;53:E432-E437.
- Danescu S, Chiorean R, Macovei V, et al. Role of physical factors in the pathogenesis of bullous pemphigoid: case report series and a comprehensive review of the published work. J Dermatol. 2016;43:134-130.
- Lloyd-Lavery A, Chi CC, Wojnarowska F, et al. The associations between bullous pemphigoid and drug use: a UK case-control study. JAMA Dermatol. 2013;149:58-62.
- Bastuji-Garin S, Joly P, Picard-Dahan C, et al. Drugs associated with bullous pemphigoid. a case-control study. Arch Dermatol. 1996;132:272-276.
- Stavropoulos PG, Soura E, Antoniou C. Drug-induced pemphigoid: a review of the literature. J Eur Acad Dermatol Venereol. 2014;28:1133-1140.
- Triplitt C, Solis-Herrera C. GLP-1 receptor agonists: practical considerations for clinical practice. Diabetes Educ. 2015;41(suppl 1):32S-46S.
- Buysschaert M, Baeck M, Preumont V, et al. Improvement of psoriasis during glucagon-like peptide-1 analogue therapy in type 2 diabetes is associated with decreasing dermal gammadelta T-cell number: a prospective case-series study. Br J Dermatol. 2014;171:155-161.
- Lee YS, Jun HS. Anti-inflammatory effects of GLP-1-based therapies beyond glucose control. Mediators Inflamm. 2016;2016:3094642.
- Skandalis K, Spirova M, Gaitanis G, et al Drug-induced bullous pemphigoid in diabetes mellitus patients receiving dipeptidyl peptidase-IV inhibitors plus metformin. J Eur Acad Dermatol Venereol. 2012;26:249-253.
- Forssmann U, Stoetzer C, Stephan M, et al. Inhibition of CD26/dipeptidyl peptidase IV enhances CCL11/eotaxin-mediated recruitment of eosinophils in vivo. J Immunol. 2008;181:1120-1127.
To the Editor:
Bullous pemphigoid (BP) is an autoimmune blistering disease that typically affects the elderly, with an incidence of approximately 7 new cases per million.1 The pathogenesis of BP involves autoantibodies to BP antigens 180 and 230 at the dermoepidermal junction. Bullous pemphigoid has been associated with the use of multiple medications; vaccines; and physical damage to the skin, including trauma, radiation, and surgery.2
Several classes of medications may cause BP; one study described an association of BP with loop diuretics,3 while others found higher incidences of BP in patients taking aldosterone antagonists and neuroleptics.4 We describe a case of drug-triggered BP to liraglutide, a glucagonlike peptide 1 (GLP-1) receptor agonist.
A 75-year-old man presented to dermatology for evaluation of a vesicular eruption on the head, neck, trunk, and arms of 6 months’ duration. The eruption developed 2 weeks after starting liraglutide 1.2 mg subcutaneously daily for diabetes mellitus. The patient had a medical history of type 2 diabetes mellitus, hypertension, stroke, and prostate cancer treated with prostatectomy, and he also was taking insulin. Liraglutide was discontinued shortly after the onset of the eruption.
Physical examination revealed annular plaques on the head, neck, trunk, and arms with central hypopigmentation and hyperpigmented borders (Figure 1). Two tense bullae were evident on the left flank (Figure 2). Histopathology revealed a subepidermal blister, mixed perivascular infiltrate with numerous eosinophils, and pigment incontinence (Figure 3). Direct immunofluorescence showed linear deposition of IgG and C3 along the basement membrane zone that was localized to the roof of the blister on salt-split analysis. No microorganisms were identified on periodic acid–Schiff, Grocott-Gomori methenamine-silver, acid-fast bacilli, and Fite stains. The patient initially was treated with clobetasol ointment 0.05%, leading to marginal improvement. He declined treatment with prednisone or dapsone, and he was started on doxycycline. Seven months after stopping liraglutide and starting doxycycline, the patient had no blisters, but residual pigmentary changes remained.
Two types of BP have been described in response to medications: drug-induced BP and drug-triggered BP. Drug-induced BP presents as an acute, self-limited eruption that typically resolves after withdrawal of the offending agent. It tends to involve a younger population and may present with mucosal involvement and target lesions on the palms and soles. Direct immunofluorescence shows linear IgG and C3 deposition at the basement membrane zone. Patients tend to respond quickly to systemic corticosteroids and have low recurrence rates. Drug-triggered BP is a chronic form of BP that is caused by a medication and is not resolved with removal of the offending agent.5 Therefore, drug-triggered BP is more difficult to detect, especially in patients taking multiple medications.
Our patient represents a case of drug-triggered BP to liraglutide. Liraglutide is a GLP-1 receptor agonist that is US Food and Drug Administration approved for the treatment of type 2 diabetes mellitus. Glucagonlike peptide 1 is an incretin hormone that is secreted by the intestine during digestion. It binds to the GLP-1 receptor leading to an increase in glucose-dependent insulin secretion and a decrease in glucagon secretion.6 Glucagonlike peptide 1 agonists also affect the immune system; liraglutide has been shown to modestly improve psoriasis, reduce the number of dermal gamma delta T cells, and decrease IL-17 expression.7 Glucagonlike peptide 1 agonists also produce anti-inflammatory effects on multiple organs including the liver, brain, vasculature, kidney, and skin.8
Dipeptidyl peptidase 4 (DPP-4) inhibitors that function to inhibit the degradation of GLP-1 and other peptides also have been reported to cause BP. In several patients, the DPP-4 inhibitors vildagliptin and sitagliptin caused drug-induced BP that resolved with discontinuation of the medication.9 Dipeptidyl peptidase 4 is expressed in various organ systems including the skin, and inhibition of DPP-4 enhances eosinophil mobilization in the blood and recruitment to the skin in animal models.10
Although the pathogenesis of BP involves autoantibodies to BP antigens 180 and 230, these antibodies are not sufficient to cause disease, as antibasement antibodies have been detected in patients without clinically evident BP. These patients, however, may be more susceptible to developing medication-induced BP. Several hypotheses regarding the pathogenesis of medication-induced BP have been proposed, including immune dysregulation, molecular mimicry, and cross-reactivity to a prior sensitizing agent.5 Liraglutide and the DPP-4 inhibitors affect the immune system, supporting the hypothesis of immune dysregulation; however, the exact mechanism of how immune modulating medications such as GLP-1 agonists and DPP-4 inhibitors cause BP remains unclear.
The effects of liraglutide and the DPP-4 inhibitors on the immune system may play a role in the pathogenesis of drug-triggered BP and drug-induced BP, respectively. Additional studies of the immunomodulatory effects of GLP-1 agonists and DPP-4 inhibitors may help elucidate the pathogenesis of drug-triggered or drug-induced BP.
To the Editor:
Bullous pemphigoid (BP) is an autoimmune blistering disease that typically affects the elderly, with an incidence of approximately 7 new cases per million.1 The pathogenesis of BP involves autoantibodies to BP antigens 180 and 230 at the dermoepidermal junction. Bullous pemphigoid has been associated with the use of multiple medications; vaccines; and physical damage to the skin, including trauma, radiation, and surgery.2
Several classes of medications may cause BP; one study described an association of BP with loop diuretics,3 while others found higher incidences of BP in patients taking aldosterone antagonists and neuroleptics.4 We describe a case of drug-triggered BP to liraglutide, a glucagonlike peptide 1 (GLP-1) receptor agonist.
A 75-year-old man presented to dermatology for evaluation of a vesicular eruption on the head, neck, trunk, and arms of 6 months’ duration. The eruption developed 2 weeks after starting liraglutide 1.2 mg subcutaneously daily for diabetes mellitus. The patient had a medical history of type 2 diabetes mellitus, hypertension, stroke, and prostate cancer treated with prostatectomy, and he also was taking insulin. Liraglutide was discontinued shortly after the onset of the eruption.
Physical examination revealed annular plaques on the head, neck, trunk, and arms with central hypopigmentation and hyperpigmented borders (Figure 1). Two tense bullae were evident on the left flank (Figure 2). Histopathology revealed a subepidermal blister, mixed perivascular infiltrate with numerous eosinophils, and pigment incontinence (Figure 3). Direct immunofluorescence showed linear deposition of IgG and C3 along the basement membrane zone that was localized to the roof of the blister on salt-split analysis. No microorganisms were identified on periodic acid–Schiff, Grocott-Gomori methenamine-silver, acid-fast bacilli, and Fite stains. The patient initially was treated with clobetasol ointment 0.05%, leading to marginal improvement. He declined treatment with prednisone or dapsone, and he was started on doxycycline. Seven months after stopping liraglutide and starting doxycycline, the patient had no blisters, but residual pigmentary changes remained.
Two types of BP have been described in response to medications: drug-induced BP and drug-triggered BP. Drug-induced BP presents as an acute, self-limited eruption that typically resolves after withdrawal of the offending agent. It tends to involve a younger population and may present with mucosal involvement and target lesions on the palms and soles. Direct immunofluorescence shows linear IgG and C3 deposition at the basement membrane zone. Patients tend to respond quickly to systemic corticosteroids and have low recurrence rates. Drug-triggered BP is a chronic form of BP that is caused by a medication and is not resolved with removal of the offending agent.5 Therefore, drug-triggered BP is more difficult to detect, especially in patients taking multiple medications.
Our patient represents a case of drug-triggered BP to liraglutide. Liraglutide is a GLP-1 receptor agonist that is US Food and Drug Administration approved for the treatment of type 2 diabetes mellitus. Glucagonlike peptide 1 is an incretin hormone that is secreted by the intestine during digestion. It binds to the GLP-1 receptor leading to an increase in glucose-dependent insulin secretion and a decrease in glucagon secretion.6 Glucagonlike peptide 1 agonists also affect the immune system; liraglutide has been shown to modestly improve psoriasis, reduce the number of dermal gamma delta T cells, and decrease IL-17 expression.7 Glucagonlike peptide 1 agonists also produce anti-inflammatory effects on multiple organs including the liver, brain, vasculature, kidney, and skin.8
Dipeptidyl peptidase 4 (DPP-4) inhibitors that function to inhibit the degradation of GLP-1 and other peptides also have been reported to cause BP. In several patients, the DPP-4 inhibitors vildagliptin and sitagliptin caused drug-induced BP that resolved with discontinuation of the medication.9 Dipeptidyl peptidase 4 is expressed in various organ systems including the skin, and inhibition of DPP-4 enhances eosinophil mobilization in the blood and recruitment to the skin in animal models.10
Although the pathogenesis of BP involves autoantibodies to BP antigens 180 and 230, these antibodies are not sufficient to cause disease, as antibasement antibodies have been detected in patients without clinically evident BP. These patients, however, may be more susceptible to developing medication-induced BP. Several hypotheses regarding the pathogenesis of medication-induced BP have been proposed, including immune dysregulation, molecular mimicry, and cross-reactivity to a prior sensitizing agent.5 Liraglutide and the DPP-4 inhibitors affect the immune system, supporting the hypothesis of immune dysregulation; however, the exact mechanism of how immune modulating medications such as GLP-1 agonists and DPP-4 inhibitors cause BP remains unclear.
The effects of liraglutide and the DPP-4 inhibitors on the immune system may play a role in the pathogenesis of drug-triggered BP and drug-induced BP, respectively. Additional studies of the immunomodulatory effects of GLP-1 agonists and DPP-4 inhibitors may help elucidate the pathogenesis of drug-triggered or drug-induced BP.
- Serwin AB, Musialkowska E, Piascik M. Incidence and mortality of bullous pemphigoid in north-east Poland (Podlaskie Province), 1999-2012: a retrospective bicentric cohort study. Int J Dermatol. 2014;53:E432-E437.
- Danescu S, Chiorean R, Macovei V, et al. Role of physical factors in the pathogenesis of bullous pemphigoid: case report series and a comprehensive review of the published work. J Dermatol. 2016;43:134-130.
- Lloyd-Lavery A, Chi CC, Wojnarowska F, et al. The associations between bullous pemphigoid and drug use: a UK case-control study. JAMA Dermatol. 2013;149:58-62.
- Bastuji-Garin S, Joly P, Picard-Dahan C, et al. Drugs associated with bullous pemphigoid. a case-control study. Arch Dermatol. 1996;132:272-276.
- Stavropoulos PG, Soura E, Antoniou C. Drug-induced pemphigoid: a review of the literature. J Eur Acad Dermatol Venereol. 2014;28:1133-1140.
- Triplitt C, Solis-Herrera C. GLP-1 receptor agonists: practical considerations for clinical practice. Diabetes Educ. 2015;41(suppl 1):32S-46S.
- Buysschaert M, Baeck M, Preumont V, et al. Improvement of psoriasis during glucagon-like peptide-1 analogue therapy in type 2 diabetes is associated with decreasing dermal gammadelta T-cell number: a prospective case-series study. Br J Dermatol. 2014;171:155-161.
- Lee YS, Jun HS. Anti-inflammatory effects of GLP-1-based therapies beyond glucose control. Mediators Inflamm. 2016;2016:3094642.
- Skandalis K, Spirova M, Gaitanis G, et al Drug-induced bullous pemphigoid in diabetes mellitus patients receiving dipeptidyl peptidase-IV inhibitors plus metformin. J Eur Acad Dermatol Venereol. 2012;26:249-253.
- Forssmann U, Stoetzer C, Stephan M, et al. Inhibition of CD26/dipeptidyl peptidase IV enhances CCL11/eotaxin-mediated recruitment of eosinophils in vivo. J Immunol. 2008;181:1120-1127.
- Serwin AB, Musialkowska E, Piascik M. Incidence and mortality of bullous pemphigoid in north-east Poland (Podlaskie Province), 1999-2012: a retrospective bicentric cohort study. Int J Dermatol. 2014;53:E432-E437.
- Danescu S, Chiorean R, Macovei V, et al. Role of physical factors in the pathogenesis of bullous pemphigoid: case report series and a comprehensive review of the published work. J Dermatol. 2016;43:134-130.
- Lloyd-Lavery A, Chi CC, Wojnarowska F, et al. The associations between bullous pemphigoid and drug use: a UK case-control study. JAMA Dermatol. 2013;149:58-62.
- Bastuji-Garin S, Joly P, Picard-Dahan C, et al. Drugs associated with bullous pemphigoid. a case-control study. Arch Dermatol. 1996;132:272-276.
- Stavropoulos PG, Soura E, Antoniou C. Drug-induced pemphigoid: a review of the literature. J Eur Acad Dermatol Venereol. 2014;28:1133-1140.
- Triplitt C, Solis-Herrera C. GLP-1 receptor agonists: practical considerations for clinical practice. Diabetes Educ. 2015;41(suppl 1):32S-46S.
- Buysschaert M, Baeck M, Preumont V, et al. Improvement of psoriasis during glucagon-like peptide-1 analogue therapy in type 2 diabetes is associated with decreasing dermal gammadelta T-cell number: a prospective case-series study. Br J Dermatol. 2014;171:155-161.
- Lee YS, Jun HS. Anti-inflammatory effects of GLP-1-based therapies beyond glucose control. Mediators Inflamm. 2016;2016:3094642.
- Skandalis K, Spirova M, Gaitanis G, et al Drug-induced bullous pemphigoid in diabetes mellitus patients receiving dipeptidyl peptidase-IV inhibitors plus metformin. J Eur Acad Dermatol Venereol. 2012;26:249-253.
- Forssmann U, Stoetzer C, Stephan M, et al. Inhibition of CD26/dipeptidyl peptidase IV enhances CCL11/eotaxin-mediated recruitment of eosinophils in vivo. J Immunol. 2008;181:1120-1127.
Practice Points
- Liraglutide and dipeptidyl peptidase 4 inhibitors, medications used in the treatment of diabetes mellitus, may be linked to the development of bullous pemphigoid (BP).
- Further study of the mechanism of action of these medications may lead to improved understanding of the pathogenesis of BP.

Exuberant Lymphomatoid Papulosis of the Head and Upper Trunk
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
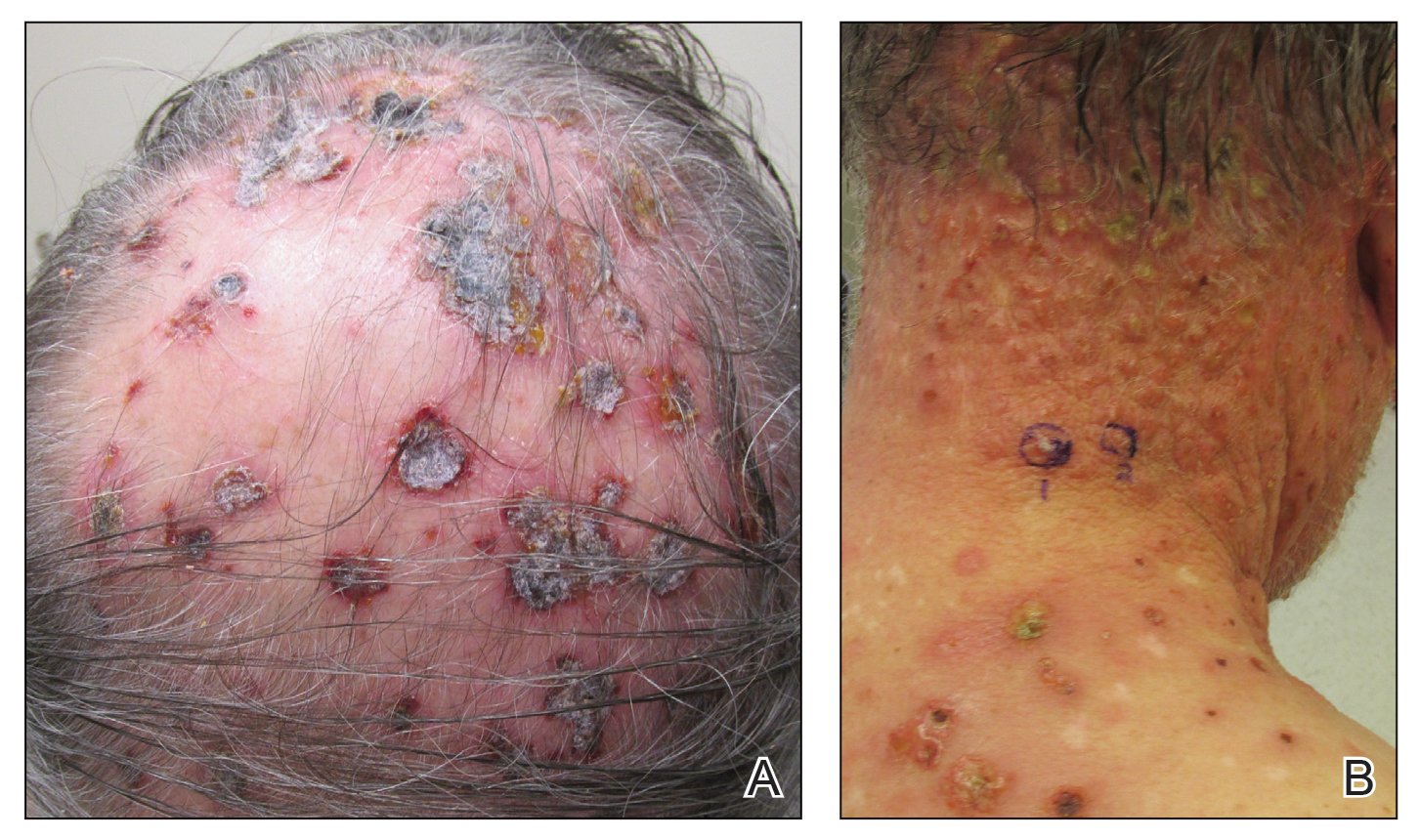
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
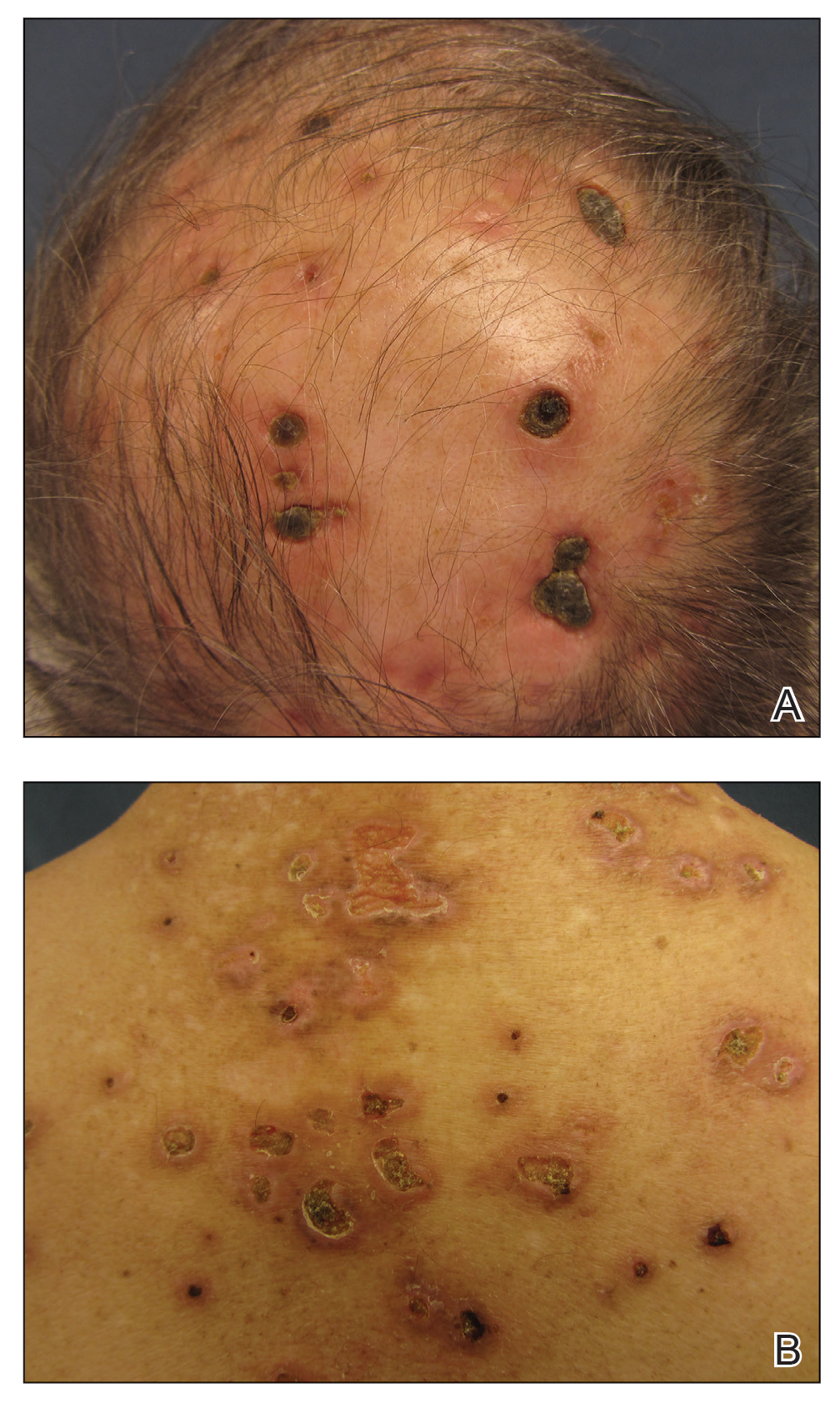
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
To the Editor:
Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder. This disease affects patients of all ages but most commonly presents in the fifth decade with a slight male predominance.1 The estimated worldwide incidence is 1.2 to 1.9 cases per 1,000,000 individuals, and the 10-year survival rate is close to 100%.1 Clinically, LyP presents as a few to more than 100 red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution. They most commonly are distributed on the trunk and extremities; however, the face, scalp, and oral mucosa rarely may be involved. Each lesion may last on average 3 to 8 weeks, with residual hyperpigmentation or hypopigmentation of the skin or superficial varioliform scars. The clinical characteristic of spontaneous regression is crucial for distinguishing LyP from other forms of cutaneous lymphoma.2 The disease course is variable, lasting anywhere from a few months to decades. Histopathologically, LyP consists of a frequently CD30+ lymphocytic proliferation in multiple described patterns.1 We report a case of LyP in a patient who initially presented with pink edematous papules and vesicles that progressed to crusted ulcerations, nodules, and deep necrotic eschars on the scalp, neck, and upper trunk. Multiple biopsies and T-cell gene rearrangement studies were necessary to make the diagnosis.
A 73-year-old man presented with edematous crusted papules and nodules as well as scarring with serous drainage on the scalp and upper trunk of several months’ duration. He also reported pain and pruritus. He had a medical history of B-cell CD20− chronic lymphocytic leukemia (CLL) that was treated with fludarabine, cyclophosphamide, rituximab, and intravenous immunoglobulin approximately one year prior and currently was in remission; prostate cancer treated with prostatectomy; hypertension; and type 2 diabetes mellitus. His medications included metoprolol, valsartan, and glipizide.
Histopathology revealed a hypersensitivity reaction, and the clinicopathologic correlation was believed to represent an exuberant arthropod bite reaction in the setting of CLL. The eruption responded well to oral prednisone and topical corticosteroids but recurred when the medications were withdrawn. A repeat biopsy resulted in a diagnosis of atypical eosinophil-predominant Sweet syndrome. The condition resolved.
Three years later he developed multiple honey-crusted, superficial ulcers as well as serous, fluid-filled vesiculobullae on the head. A tissue culture revealed Proteus mirabilis, Staphylococcus aureus, and Enterococcus faecalis, and was negative for acid-fast bacteria and fungus. Biopsy of these lesions revealed dermal ulceration with a mixed inflammatory infiltrate and numerous eosinophils as well as a few clustered CD30+ cells; direct immunofluorescence was negative. An extensive laboratory workup including bullous pemphigoid antigens, C-reactive protein, antinuclear antibodies comprehensive profile, antineutrophil cytoplasmic antibodies, rheumatoid factor, anticyclic citrullinated peptide antibodies, serum protein electrophoresis, lactate dehydrogenase, complete blood cell count with differential, complete metabolic profile, thyroid-stimulating hormone, uric acid, C3, C4, immunoglobulin profile, angiotensin-converting enzyme level, and urinalysis was unremarkable. He improved with courses of minocycline, prednisone, and topical clobetasol, but he had periodic and progressive flares over several months with punched-out crusted ulcerations developing on the scalp (Figure 1A) and neck (Figure 1B). The oral and ocular mucosae were uninvolved, but the nasal mucosa had some involvement.
A repeat biopsy demonstrated an atypical CD30+ lymphoid infiltrate favoring LyP. T-cell clonality performed on this specimen and the prior biopsy demonstrated identical T-cell receptor β and γ clones. CD3, CD5, CD7, and CD4 immunostains highlighted the perivascular, perifollicular, and folliculotropic lymphocytic infiltrate. CD8 highlighted occasional background small T cells with only a few folliculotropic forms. A CD30 study revealed several scattered enlarged lymphocytes, and CD20 displayed a few dispersed B cells. A repeat perilesional direct immunofluorescence study was again negative. With treatment, he later formed multiple dry punched-out ulcers with dark eschars on the scalp, posterior neck, and upper back. There were multiple scars on the head, chest, and back, and no vesicles or bullae were present (Figure 2). The patient was presented at a meeting of the Philadelphia Dermatological Society and a consensus diagnosis of LyP was reached. The patient has continued to improve with oral minocycline 100 mg twice daily, topical clobetasol, and topical mupirocin.
Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy, with some disagreement in the literature concerning the exact percentage.3 In some studies, lymphoma has been estimated to occur in less than 20% of cases.4,5 Wieser et al1 reported a retrospective analysis of 180 patients with LyP that revealed a secondary malignancy in 52% of patients. They also reported that the number of lesions and the symptom severity were not associated with lymphoma development.1 Similarly, Cordel et al6 reported a diagnosis of lymphoma in 41% of 106 patients. These analyses reveal that the association with lymphoma may be higher than previously thought, but referral bias may be a confounding factor in these numbers.1,5,6 Associated malignancies may occur prior to, concomitantly, or years after the diagnosis of LyP. The most frequently reported malignancies include mycosis fungoides, Hodgkin lymphoma, and primary cutaneous anaplastic large cell lymphoma.1,4
Nicolaou et al3 indicated that head involvement was more likely associated with lymphoma. Our patient had a history of CLL prior to the development of LyP, and it continues to be in remission. The incidence of CLL in patients with LyP is reported to be 0.8%.4 Our patient had an exuberant case of LyP predominantly involving the head, neck, and upper torso, which is an unusual distribution. Vesiculobullous lesions also are uncharacteristic of LyP and may have represented concomitant bullous impetigo, but bullous variants of LyP also have been reported.7 Due to the unique distribution and characteristic scarring, Brunsting-Perry cicatricial pemphigoid also was considered in the clinical differential diagnosis.
The pathogenesis of LyP associated with malignancy is not definitively known. Theories propose that progression to a malignant clonal T-cell population may come from cytogenetic events, inadequate host response, or persistent antigenic or viral stimulation.4 Studies have demonstrated overlapping T-cell receptor gene rearrangement clones in lesions in patients with both LyP and mycosis fungoides, suggesting a common origin between the diseases.8 Other theories suggest that LyP may arise from an early, reactive, polyclonal lymphoid expansion that evolves into a clonal neoplastic process.4 Interestingly, LyP is a clonal T-cell disorder, while Hodgkin lymphoma and CLL are B-cell disorders. Thus, reports of CLL occurring with LyP, as in our patient, may support the theory that LyP arises from an early stem-cell or precursor-cell defect.4
There is no cure for LyP and data regarding the potential of aggressive therapy on the prevention of secondary lymphomas is lacking. Wieser et al1 reported that treatment did not prevent the progression to lymphoma in their retrospective analysis of 180 patients. The number of lesions, frequency of outbreaks, and extent of the scarring can dictate the treatment approach for LyP. Conservative topical therapies include corticosteroids, bexarotene, and imiquimod. Mupirocin may help to prevent infection of ulcerated lesions.1,2 Low-dose methotrexate has been shown to be the most efficacious treatment in reducing the number of lesions, particularly for scarring or cosmetically sensitive areas. Oral methotrexate at a dosage of 10 mg to 25 mg weekly tapered to the lowest effective dose may suppress outbreaks of LyP lesions.1,2 Other therapies include psoralen plus UVA, UVB, interferon alfa-2a, oral bexarotene, oral acyclovir or valacyclovir, etretinate, mycophenolic acid, photodynamic therapy, oral antibiotics, excision, and radiotherapy.1,2 Systemic chemotherapy and total-skin electron beam therapy have shown efficacy in clearing the lesions; however, the disease recurs after discontinuation of therapy.2 Systemic chemotherapy is not recommended for the treatment of LyP, as risks outweigh the benefits and it does not reduce the risk for developing lymphoma.1 The prognosis generally is good, though long-term follow-up is imperative to monitor for the development of other lymphomas.
Our patient presented with LyP a few months after completing chemotherapy for his CLL. It is unknown if he developed LyP just before the time of presentation, or if he may have developed it at the same time as his CLL by a common inciting event. In the latter case, it is speculative that the LyP may have been controlled by chemotherapy for his CLL, only to become clinically apparent after discontinuation, then naturally remit for a longer period. Case reports such as ours with unusual clinical presentations, B-cell lymphoma associations, and unique timing of lymphoma onset may help to provide insight into the pathogenesis of this disease.
We highlighted an unusual case of LyP that presented clinically with crusted ulcerations as well as vesiculobullous and edematous papules that progressed into deep punched-out ulcers with eschars, nodules, and scarring on the head and upper trunk. Lymphomatoid papulosis can be difficult to diagnose histopathologically at the early stages, and multiple repeat biopsies may be necessary to confirm the diagnosis. T-cell gene rearrangement and immunohistochemistry studies are helpful along with clinical correlation to establish a diagnosis in these cases. We recommend that physicians keep LyP on the differential diagnosis for patients with similar clinical presentations and remain vigilant in monitoring for the development of secondary lymphoma.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
- Wieser I, Oh C, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Duvic M. CD30+ neoplasms of the skin. Curr Hematol Malig Rep. 2011;6:245-250.
- Nicolaou V, Papadavid E, Ekonomise A, et al. Association of clinicopathological characteristics with secondary neoplastic lymphoproliferative disorders in patients with lymphomatoid papulosis. Leuk Lymphoma. 2015;56:1303-1307.
- Ahn C, Orscheln C, Huang W. Lymphomatoid papulosis as a harbinger of chronic lymphocytic leukemia. Ann Hematol. 2014;93:1923-1925.
- Kunishige J, McDonald H, Alvarez G, et al. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009;34:576-5781.
- Cordelet al. Frequency and risk factors for associated lymphomas in patients with lymphomatoid papulosis. Oncologist. 2016;21:76-83.
- Sureda N, Thomas L, Bathelier E, et al. Bullous lymphomatoid papulosis. Clin Exp Dermatol. 2011;36:800-801.
- de la Garza Bravo M, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin. Hum Pathol. 2015;46:558-569.
Practice Points
- Lymphomatoid papulosis (LyP) is a chronic, recurring, self-healing, primary cutaneous lymphoproliferative disorder characterized by red-brown papules or nodules, some with hemorrhagic crust or central necrosis, often occurring in crops and in various stages of evolution.
- Histopathologically, LyP consists of a frequently CD30Mathematical Pi LT Std+ lymphocytic proliferation in multiple described patterns.
- Lymphomatoid papulosis is an indolent cutaneous lymphoma; however, it is associated with the potential development of a second hematologic malignancy.
Home Treatment of Presumed Melanocytic Nevus With Frankincense
To the Editor:
Melanocytic nevi are ubiquitous, and although they are benign, patients often desire to have them removed. We report a patient who presented to our clinic after attempting home removal of a concerning mole on the back with frankincense, a remedy that she found online.
A 43-year-old woman presented with a worrisome mole on the back. She had no personal history of skin cancer, but her father had a history of melanoma in situ in his 60s. The patient reported that she had the mole for years, but approximately 1 month prior to her visit she noticed that it began to bleed and crust, causing concern for melanoma. She read online that the lesion could be removed with topical application of the essential oil frankincense; she applied it directly to the lesion on the back. Within hours she developed a burn where it was applied with associated blistering.
Clinically, the lesion appeared as a darkly pigmented, well-circumscribed papule with hemorrhagic crust overlying a well-demarcated pink plaque (Figure 1). Dermatoscopically, the lesion lacked a pigment network and demonstrated 2 distinct pink papules with peripheral telangiectasia and a pink background with white streaks (Figure 2). A shave biopsy of the lesion demonstrated a nodular basal cell carcinoma extending to the base and margin.
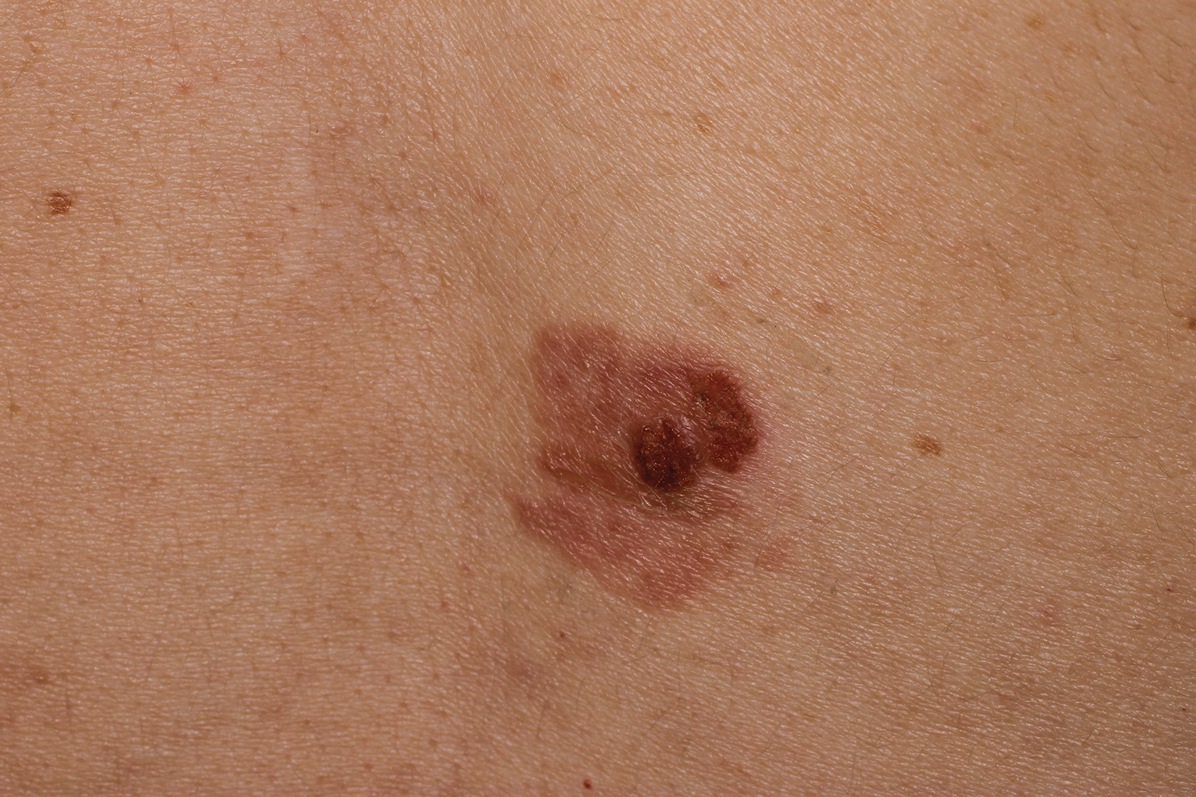
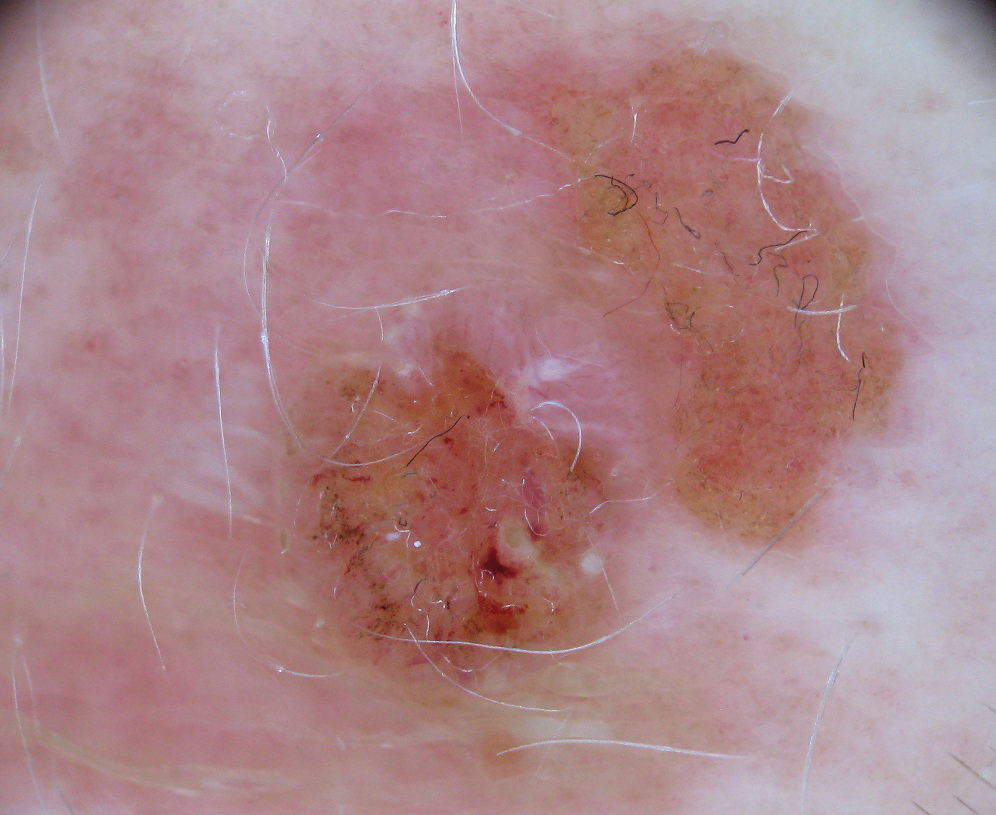
Frankincense is the common name given to oleo-gum-resins of Boswellia species.1 It has been studied extensively for anti-inflammatory and antitumoral properties. It has been demonstrated that high concentrations of its active component, boswellic acid, can have a cytotoxic or cytostatic effect on certain malignant cell lines, such as melanoma, in vitro.2,3 It also has been shown to be antitumoral in mouse models.4 There are limited in vivo studies in the literature assessing the effects of boswellic acid or frankincense on cutaneous melanocytic lesions or other cutaneous malignancies, such as basal cell carcinoma.
A Google search of home remedy mole removal yielded more than 1,000,000 results. At the time of submission, the top 5 results all listed frankincense as a potential treatment along with garlic, iodine, castor oil, onion juice, pineapple juice, banana peels, honey, and aloe vera. None of the results cited evidence for their treatments. Although all recommended dilution of the frankincense prior to application, none warned of potential risks or side effects of its use.
Natural methods of home mole removal have long been sought after. Escharotics are most commonly utilized, including bloodroot (Sanguinaria canadensis), zinc chloride, Chelidonium majus, and Solanum sodomaeum. Many formulations are commercially available online, despite the fact that they can be mutilating and potentially dangerous when used without appropriate supervision.5 This case and an online search demonstrated that these agents are not only potentially harmful home remedies but also are currently falsely advertised as effective therapeutic management for melanocytic nevi.
Approximately 6 million individuals in the United States search the internet for health information daily, and as many as 41% of those do so to learn about alternative medicine.5,6 Although information gleaned from search engines can be useful, it is unregulated and often can be inaccurate. Clinicians generally are unaware of the erroneous material presented online and, therefore, cannot appropriately combat patient misinformation. Our case demonstrates the need to maintain an awareness of common online fallacies to better answer patient questions and guide them to more accurate sources of dermatologic information and appropriate treatment.
- Du Z, Liu Z, Ning Z, et al. Prospects of boswellic acids as potential pharmaceutics. Planta Med. 2015;81:259-271.
- Eichhorn T, Greten HJ, Efferth T. Molecular determinants of the response of tumor cells to boswellic acids. Pharmaceuticals (Basel). 2011;4:1171-1182.
- Zhao W, Entschladen F, Liu H, et al. Boswellic acid acetate induces differentiation and apoptosis in highly metastatic melanoma and fibrosarcoma cell. Cancer Detect Prev. 2003;27:67-75.
- Huang MT, Badmaev V, Ding Y, et al. Anti-tumor and anti-carcinogenic activities of triterpenoid, beta-boswellic acid. Biofactors. 2000;13:225-230.
- Adler BL, Friedman AJ. Safety & efficacy of agents used for home mole removal and skin cancer treatment in the internet age, and analysis of cases. J Drugs Dermatol. 2013;12:1058-1063.
- Kanthawala S, Vermeesch A, Given B, et al. Answers to health questions: internet search results versus online health community responses. J Med Internet Res. 2016;18:E95.
To the Editor:
Melanocytic nevi are ubiquitous, and although they are benign, patients often desire to have them removed. We report a patient who presented to our clinic after attempting home removal of a concerning mole on the back with frankincense, a remedy that she found online.
A 43-year-old woman presented with a worrisome mole on the back. She had no personal history of skin cancer, but her father had a history of melanoma in situ in his 60s. The patient reported that she had the mole for years, but approximately 1 month prior to her visit she noticed that it began to bleed and crust, causing concern for melanoma. She read online that the lesion could be removed with topical application of the essential oil frankincense; she applied it directly to the lesion on the back. Within hours she developed a burn where it was applied with associated blistering.
Clinically, the lesion appeared as a darkly pigmented, well-circumscribed papule with hemorrhagic crust overlying a well-demarcated pink plaque (Figure 1). Dermatoscopically, the lesion lacked a pigment network and demonstrated 2 distinct pink papules with peripheral telangiectasia and a pink background with white streaks (Figure 2). A shave biopsy of the lesion demonstrated a nodular basal cell carcinoma extending to the base and margin.
Frankincense is the common name given to oleo-gum-resins of Boswellia species.1 It has been studied extensively for anti-inflammatory and antitumoral properties. It has been demonstrated that high concentrations of its active component, boswellic acid, can have a cytotoxic or cytostatic effect on certain malignant cell lines, such as melanoma, in vitro.2,3 It also has been shown to be antitumoral in mouse models.4 There are limited in vivo studies in the literature assessing the effects of boswellic acid or frankincense on cutaneous melanocytic lesions or other cutaneous malignancies, such as basal cell carcinoma.
A Google search of home remedy mole removal yielded more than 1,000,000 results. At the time of submission, the top 5 results all listed frankincense as a potential treatment along with garlic, iodine, castor oil, onion juice, pineapple juice, banana peels, honey, and aloe vera. None of the results cited evidence for their treatments. Although all recommended dilution of the frankincense prior to application, none warned of potential risks or side effects of its use.
Natural methods of home mole removal have long been sought after. Escharotics are most commonly utilized, including bloodroot (Sanguinaria canadensis), zinc chloride, Chelidonium majus, and Solanum sodomaeum. Many formulations are commercially available online, despite the fact that they can be mutilating and potentially dangerous when used without appropriate supervision.5 This case and an online search demonstrated that these agents are not only potentially harmful home remedies but also are currently falsely advertised as effective therapeutic management for melanocytic nevi.
Approximately 6 million individuals in the United States search the internet for health information daily, and as many as 41% of those do so to learn about alternative medicine.5,6 Although information gleaned from search engines can be useful, it is unregulated and often can be inaccurate. Clinicians generally are unaware of the erroneous material presented online and, therefore, cannot appropriately combat patient misinformation. Our case demonstrates the need to maintain an awareness of common online fallacies to better answer patient questions and guide them to more accurate sources of dermatologic information and appropriate treatment.
To the Editor:
Melanocytic nevi are ubiquitous, and although they are benign, patients often desire to have them removed. We report a patient who presented to our clinic after attempting home removal of a concerning mole on the back with frankincense, a remedy that she found online.
A 43-year-old woman presented with a worrisome mole on the back. She had no personal history of skin cancer, but her father had a history of melanoma in situ in his 60s. The patient reported that she had the mole for years, but approximately 1 month prior to her visit she noticed that it began to bleed and crust, causing concern for melanoma. She read online that the lesion could be removed with topical application of the essential oil frankincense; she applied it directly to the lesion on the back. Within hours she developed a burn where it was applied with associated blistering.
Clinically, the lesion appeared as a darkly pigmented, well-circumscribed papule with hemorrhagic crust overlying a well-demarcated pink plaque (Figure 1). Dermatoscopically, the lesion lacked a pigment network and demonstrated 2 distinct pink papules with peripheral telangiectasia and a pink background with white streaks (Figure 2). A shave biopsy of the lesion demonstrated a nodular basal cell carcinoma extending to the base and margin.
Frankincense is the common name given to oleo-gum-resins of Boswellia species.1 It has been studied extensively for anti-inflammatory and antitumoral properties. It has been demonstrated that high concentrations of its active component, boswellic acid, can have a cytotoxic or cytostatic effect on certain malignant cell lines, such as melanoma, in vitro.2,3 It also has been shown to be antitumoral in mouse models.4 There are limited in vivo studies in the literature assessing the effects of boswellic acid or frankincense on cutaneous melanocytic lesions or other cutaneous malignancies, such as basal cell carcinoma.
A Google search of home remedy mole removal yielded more than 1,000,000 results. At the time of submission, the top 5 results all listed frankincense as a potential treatment along with garlic, iodine, castor oil, onion juice, pineapple juice, banana peels, honey, and aloe vera. None of the results cited evidence for their treatments. Although all recommended dilution of the frankincense prior to application, none warned of potential risks or side effects of its use.
Natural methods of home mole removal have long been sought after. Escharotics are most commonly utilized, including bloodroot (Sanguinaria canadensis), zinc chloride, Chelidonium majus, and Solanum sodomaeum. Many formulations are commercially available online, despite the fact that they can be mutilating and potentially dangerous when used without appropriate supervision.5 This case and an online search demonstrated that these agents are not only potentially harmful home remedies but also are currently falsely advertised as effective therapeutic management for melanocytic nevi.
Approximately 6 million individuals in the United States search the internet for health information daily, and as many as 41% of those do so to learn about alternative medicine.5,6 Although information gleaned from search engines can be useful, it is unregulated and often can be inaccurate. Clinicians generally are unaware of the erroneous material presented online and, therefore, cannot appropriately combat patient misinformation. Our case demonstrates the need to maintain an awareness of common online fallacies to better answer patient questions and guide them to more accurate sources of dermatologic information and appropriate treatment.
- Du Z, Liu Z, Ning Z, et al. Prospects of boswellic acids as potential pharmaceutics. Planta Med. 2015;81:259-271.
- Eichhorn T, Greten HJ, Efferth T. Molecular determinants of the response of tumor cells to boswellic acids. Pharmaceuticals (Basel). 2011;4:1171-1182.
- Zhao W, Entschladen F, Liu H, et al. Boswellic acid acetate induces differentiation and apoptosis in highly metastatic melanoma and fibrosarcoma cell. Cancer Detect Prev. 2003;27:67-75.
- Huang MT, Badmaev V, Ding Y, et al. Anti-tumor and anti-carcinogenic activities of triterpenoid, beta-boswellic acid. Biofactors. 2000;13:225-230.
- Adler BL, Friedman AJ. Safety & efficacy of agents used for home mole removal and skin cancer treatment in the internet age, and analysis of cases. J Drugs Dermatol. 2013;12:1058-1063.
- Kanthawala S, Vermeesch A, Given B, et al. Answers to health questions: internet search results versus online health community responses. J Med Internet Res. 2016;18:E95.
- Du Z, Liu Z, Ning Z, et al. Prospects of boswellic acids as potential pharmaceutics. Planta Med. 2015;81:259-271.
- Eichhorn T, Greten HJ, Efferth T. Molecular determinants of the response of tumor cells to boswellic acids. Pharmaceuticals (Basel). 2011;4:1171-1182.
- Zhao W, Entschladen F, Liu H, et al. Boswellic acid acetate induces differentiation and apoptosis in highly metastatic melanoma and fibrosarcoma cell. Cancer Detect Prev. 2003;27:67-75.
- Huang MT, Badmaev V, Ding Y, et al. Anti-tumor and anti-carcinogenic activities of triterpenoid, beta-boswellic acid. Biofactors. 2000;13:225-230.
- Adler BL, Friedman AJ. Safety & efficacy of agents used for home mole removal and skin cancer treatment in the internet age, and analysis of cases. J Drugs Dermatol. 2013;12:1058-1063.
- Kanthawala S, Vermeesch A, Given B, et al. Answers to health questions: internet search results versus online health community responses. J Med Internet Res. 2016;18:E95.
Practice Points
- Many patients seek natural methods of home mole removal online, including topical application of essential oils such as frankincense.
- These agents often are unregulated and can be potentially harmful when used without appropriate supervision.
- Dermatologists should be aware of common online fallacies to better answer patient questions and guide them to more accurate sources of dermatologic information and appropriate treatment.
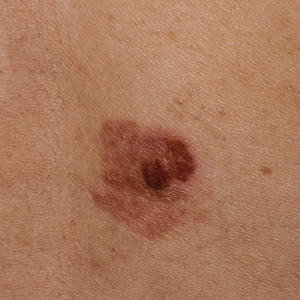
Fatal Case of Levamisole-Induced Vasculopathy in a Cocaine User
To the Editor:
Levamisole is a veterinary anthelmintic drug with immunomodulating properties that was once approved by the US Food and Drug Administration for the treatment of various conditions, including autoimmune diseases, cancer, pediatric kidney disease, and chronic infections.1-4 Levamisole was banned in 2000 after reports of associated agranulocytosis and a characteristic painful purpuric vasculitis.4,5 Despite the ban, its use persists due to its increasing incorporation as an adulterant in cocaine, presumably for its dopaminergic properties that potentiate psychotropic effects.6 In 2009, the Drug Enforcement Administration reported that 69% of seized cocaine in the United States contains this chemical, with an average concentration of 10%.5 Levamisole-induced vasculopathy (LIV) typically resolves following the cessation of cocaine without further treatment necessary. We present a fatal case of LIV to emphasize that early recognition and discontinuation of the offending agent could be lifesaving.
A 40-year-old woman with a history of cocaine abuse was admitted with tender, reticular, purpuric, and erythematous patches and plaques on the lower extremities with areas of necrosis (Figure 1). The lesions had been present intermittently for 6 months. She tried topical mupirocin and oral amoxicillin clavulanate without improvement. She also described polyarthralgia in the hands, but the remainder of the review of symptoms and physical examination was negative.
Coagulation studies and white blood cell counts were within reference range. A urine toxicology screen was positive for cocaine; however, urine testing for levamisole was not performed given the short half-life of levamisole in vivo. A biopsy of one of the skin lesions on the right thigh showed pauci-inflammatory superficial and deep vein thrombosis with recanalization (Figure 2). A rheumatology workup revealed an elevated C-reactive protein level, low C3, positive antinuclear antibody, positive anti–double-stranded DNA, positive anticardiolipin antibody, positive lupus anticoagulant, and positive perinuclear antineutrophil cytoplasmic antibody (ANCA). Tests for HIV, hepatitis B and C, cryoglobulinemia, and cytomegalovirus were negative. Given the clinical picture and laboratory findings, levamisole-induced vasculitis was deemed likely. The patient was treated with appropriate skin and wound care. She was discharged with a prednisone taper and oral cephalexin and was counseled on cocaine cessation.

Five months later, the patient was readmitted for lower extremity edema and worsening painful lesions that had progressed to involve the legs, thighs, buttocks, flanks, and the tip of her nose. A deep vein thrombosis workup was negative. She admitted to ongoing cocaine use that was confirmed with urine toxicology. Coagulation studies and white blood cell counts remained within reference range. Repeat skin biopsy was consistent with prior findings, demonstrating thrombosis of superficial and deep vessels with recanalization. In addition, it showed focal epidermal necrosis and a perivascular infiltrate of lymphocytes, histiocytes, and rare neutrophils. She was placed on high-dose methylprednisolone. Over the course of the next month, her urine continued to test positive for cocaine, and she developed necrotizing fasciitis necessitating lower extremity amputation, abdominal washout, and debridement. She quickly deteriorated, developing multiorgan failure with sepsis, leading to death. Of note, the patient was never found to have neutropenia or agranulocytosis throughout the disease course.
Because levamisole is no longer in clinical use, reports of its adverse effects come exclusively from users of cocaine, whether via smoking or snorting. Levamisole-induced vasculopathy typically is painful and purpuric, with or without necrosis, in a retiform or stellate pattern and commonly involves the extremities, trunk, face, and external ears.7 The average age of presentation is 43 years and it more commonly is seen in women.8
Levamisole-induced vasculopathy remains a diagnosis of exclusion, so it is important to rule out other treatable causes. The differential diagnosis for purpura associated with vasculitis also includes other antineutrophilic cytoplasmic–associated vasculitides (eg, granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis), infectious purpura fulminans, antiphospholipid syndrome, cryoglobulinemia, and disseminated intravascular coagulation.9 In LIV patients, perinuclear ANCAs are present in up to 90% of cases, and cytoplasmic ANCAs in 19% to 59% of cases.10,11 Although leukopenia and neutropenia complicate approximately 60% of LIV cases, they are not required to make the diagnosis.11,12 Elevated erythrocyte sedimentation rate, normal coagulation studies, and positive antineutrophil antibodies and lupus anticoagulant further aid in the diagnosis.8 Urine should be tested for cocaine in suspected patients. Urine also can be tested for levamisole, which is challenging because of the short half-life of 5.6 hours. Only 2% to 5% of levamisole is excreted unchanged in the urine, and testing requires gas chromatography and mass spectrometry that was not readily available to perform on our patient.7 In addition to laboratory and urine studies, hair strand testing,10 skin biopsy, and histologic findings also can be used to support the diagnosis.
The pathogenesis of LIV is not completely understood, but it is thought to be an immune complex–mediated process based on immunofluorescence studies in the skin.13,14 Classic pathologic findings include multiple fibrin thrombi within small vessels in the superficial and deep dermis, leukocytoclastic vasculitis of small vessels consisting of fibrinoid necrosis of the vessel wall, extravasated erythrocytes, karyorrhectic debris, and angiocentric inflammation.14 Direct immunofluorescence is not routinely performed but most commonly demonstrates deposition of IgA, IgM, and C3.14,15
Levamisole-induced vasculopathy usually resolves upon cessation of cocaine use without long-term sequelae. Steroids have been used as treatment of prominent vasculitis with variable success; however, immunosuppressive effects should be closely monitored, especially with inpatients with concurrent granulocytopenia. Broad-spectrum antibiotics have been used in cases with fever and agranulocytosis. Cutaneous lesions typically disappear within 2 to 3 weeks, and serologic markers resolve within 2 to 10 months. Recurrent use of cocaine generally results in recurrent neutropenia and skin eruptions, supporting the causal role. Our patient’s recurrent prolonged cocaine use with vasculopathy was assumed to be the source of the necrotizing fasciitis that led to a cascade of sepsis, rapidly progressing multiorgan failure, and ultimate demise.
Presentation of a purpuric vasculopathy, with or without associated neutropenia and positive autoantibodies, should prompt the consideration of levamisole-contaminated cocaine use in the clinician’s differential. Although the patient may initially deny cocaine use, it is important to keep this diagnosis in mind when the clinical picture fits, and urine toxicology screen should be ordered when there is question. Physicians and patients should be wary of potential complications, even death. Early recognition and discontinuation of the offending agent could be lifesaving.
- Menni S, Pistritto G, Gianotti R, et al. Ear lobe necrosis by levamisole-induced occlusive vasculitis in a pediatric patient. Pediatr Dermatol. 1997;14:477-479.
- Symoens J, Veys E, Mielants M, et al. Adverse reactions to levamisole. Cancer Treat Rep. 1978;62:1721-1730.
- Vogel CL, Silverman MA, Mansell PW, et al. Mechanism of levamisole-induced granulocytopenia in breast cancer patients. Am J Hematol. 1980;9:171-183.
- Rongioletti F, Ghio L, Ginevri F, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long-term treatment with levamisole in children. Br J Dermatol. 1999;140:948-951.
- Centers for Disease Control and Prevention (CDC). Agranulocytosis associated with cocaine use—four states, March 2008–November 2009. MMWR Morb Mortal Wkly Rep. 2009;58:1381-1385.
- Zhu NY, Legatt DF, Turner AR. Agranulocytosis after consumption of cocaine adulterated with levamisole. Ann Intern Med. 2009;150:287-289.
- Gross RL, Brucker J, Bahce-Altuntas A, et al. A novel cutaneous vasculitis syndrome induced by levamisole-contaminated cocaine. Clin Rheumatol. 2011;30:1385-1392.
- Trehy ML, Brown DJ, Woodruff JT, et al. Determination of levamisole in urine by gas chromatography-mass spectrometry. J Anal Toxicol. 2011;35:545-550.
- Waller JM, Feramisco JD, Alberta-Wszolek L, et al. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit? J Am Acad Dermatol. 2010;63:530-535.
- Pearson T, Bremmer M, Cohen J, et al. Vasculopathy related to cocaine adulterated with levamisole: a review of the literature. Dermatol Online J. 2012;18:1.
- Arora NP. Cutaneous vasculopathy and neutropenia associated with levamisole-adulterated cocaine. Am J Med Sci. 2013;345:45-51.
- Chai PR, Bastan W, Machan J, et al. Levamisole exposure and hematologic indices in cocaine users. Acad Emerg Med. 2011;18:1141-1147.
- Lazareth H, Peytavin G, Polivka L, et al. The hairy-print for levamisole-induced vasculitis. BMJ Case Rep. 2012;2012:bcr2012006602.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia—a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2011;65:722-725.
- Jenkins J, Babu K, Hsu-Hung E, et al. ANCA-positive necrotizing vasculitis and thrombotic vasculopathy induced by levamisole-adulterated cocaine: a distinctive clinicopathologic presentation. J Am Acad Dermatol. 2011;65:E14-E16.
To the Editor:
Levamisole is a veterinary anthelmintic drug with immunomodulating properties that was once approved by the US Food and Drug Administration for the treatment of various conditions, including autoimmune diseases, cancer, pediatric kidney disease, and chronic infections.1-4 Levamisole was banned in 2000 after reports of associated agranulocytosis and a characteristic painful purpuric vasculitis.4,5 Despite the ban, its use persists due to its increasing incorporation as an adulterant in cocaine, presumably for its dopaminergic properties that potentiate psychotropic effects.6 In 2009, the Drug Enforcement Administration reported that 69% of seized cocaine in the United States contains this chemical, with an average concentration of 10%.5 Levamisole-induced vasculopathy (LIV) typically resolves following the cessation of cocaine without further treatment necessary. We present a fatal case of LIV to emphasize that early recognition and discontinuation of the offending agent could be lifesaving.
A 40-year-old woman with a history of cocaine abuse was admitted with tender, reticular, purpuric, and erythematous patches and plaques on the lower extremities with areas of necrosis (Figure 1). The lesions had been present intermittently for 6 months. She tried topical mupirocin and oral amoxicillin clavulanate without improvement. She also described polyarthralgia in the hands, but the remainder of the review of symptoms and physical examination was negative.
Coagulation studies and white blood cell counts were within reference range. A urine toxicology screen was positive for cocaine; however, urine testing for levamisole was not performed given the short half-life of levamisole in vivo. A biopsy of one of the skin lesions on the right thigh showed pauci-inflammatory superficial and deep vein thrombosis with recanalization (Figure 2). A rheumatology workup revealed an elevated C-reactive protein level, low C3, positive antinuclear antibody, positive anti–double-stranded DNA, positive anticardiolipin antibody, positive lupus anticoagulant, and positive perinuclear antineutrophil cytoplasmic antibody (ANCA). Tests for HIV, hepatitis B and C, cryoglobulinemia, and cytomegalovirus were negative. Given the clinical picture and laboratory findings, levamisole-induced vasculitis was deemed likely. The patient was treated with appropriate skin and wound care. She was discharged with a prednisone taper and oral cephalexin and was counseled on cocaine cessation.
Five months later, the patient was readmitted for lower extremity edema and worsening painful lesions that had progressed to involve the legs, thighs, buttocks, flanks, and the tip of her nose. A deep vein thrombosis workup was negative. She admitted to ongoing cocaine use that was confirmed with urine toxicology. Coagulation studies and white blood cell counts remained within reference range. Repeat skin biopsy was consistent with prior findings, demonstrating thrombosis of superficial and deep vessels with recanalization. In addition, it showed focal epidermal necrosis and a perivascular infiltrate of lymphocytes, histiocytes, and rare neutrophils. She was placed on high-dose methylprednisolone. Over the course of the next month, her urine continued to test positive for cocaine, and she developed necrotizing fasciitis necessitating lower extremity amputation, abdominal washout, and debridement. She quickly deteriorated, developing multiorgan failure with sepsis, leading to death. Of note, the patient was never found to have neutropenia or agranulocytosis throughout the disease course.
Because levamisole is no longer in clinical use, reports of its adverse effects come exclusively from users of cocaine, whether via smoking or snorting. Levamisole-induced vasculopathy typically is painful and purpuric, with or without necrosis, in a retiform or stellate pattern and commonly involves the extremities, trunk, face, and external ears.7 The average age of presentation is 43 years and it more commonly is seen in women.8
Levamisole-induced vasculopathy remains a diagnosis of exclusion, so it is important to rule out other treatable causes. The differential diagnosis for purpura associated with vasculitis also includes other antineutrophilic cytoplasmic–associated vasculitides (eg, granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis), infectious purpura fulminans, antiphospholipid syndrome, cryoglobulinemia, and disseminated intravascular coagulation.9 In LIV patients, perinuclear ANCAs are present in up to 90% of cases, and cytoplasmic ANCAs in 19% to 59% of cases.10,11 Although leukopenia and neutropenia complicate approximately 60% of LIV cases, they are not required to make the diagnosis.11,12 Elevated erythrocyte sedimentation rate, normal coagulation studies, and positive antineutrophil antibodies and lupus anticoagulant further aid in the diagnosis.8 Urine should be tested for cocaine in suspected patients. Urine also can be tested for levamisole, which is challenging because of the short half-life of 5.6 hours. Only 2% to 5% of levamisole is excreted unchanged in the urine, and testing requires gas chromatography and mass spectrometry that was not readily available to perform on our patient.7 In addition to laboratory and urine studies, hair strand testing,10 skin biopsy, and histologic findings also can be used to support the diagnosis.
The pathogenesis of LIV is not completely understood, but it is thought to be an immune complex–mediated process based on immunofluorescence studies in the skin.13,14 Classic pathologic findings include multiple fibrin thrombi within small vessels in the superficial and deep dermis, leukocytoclastic vasculitis of small vessels consisting of fibrinoid necrosis of the vessel wall, extravasated erythrocytes, karyorrhectic debris, and angiocentric inflammation.14 Direct immunofluorescence is not routinely performed but most commonly demonstrates deposition of IgA, IgM, and C3.14,15
Levamisole-induced vasculopathy usually resolves upon cessation of cocaine use without long-term sequelae. Steroids have been used as treatment of prominent vasculitis with variable success; however, immunosuppressive effects should be closely monitored, especially with inpatients with concurrent granulocytopenia. Broad-spectrum antibiotics have been used in cases with fever and agranulocytosis. Cutaneous lesions typically disappear within 2 to 3 weeks, and serologic markers resolve within 2 to 10 months. Recurrent use of cocaine generally results in recurrent neutropenia and skin eruptions, supporting the causal role. Our patient’s recurrent prolonged cocaine use with vasculopathy was assumed to be the source of the necrotizing fasciitis that led to a cascade of sepsis, rapidly progressing multiorgan failure, and ultimate demise.
Presentation of a purpuric vasculopathy, with or without associated neutropenia and positive autoantibodies, should prompt the consideration of levamisole-contaminated cocaine use in the clinician’s differential. Although the patient may initially deny cocaine use, it is important to keep this diagnosis in mind when the clinical picture fits, and urine toxicology screen should be ordered when there is question. Physicians and patients should be wary of potential complications, even death. Early recognition and discontinuation of the offending agent could be lifesaving.
To the Editor:
Levamisole is a veterinary anthelmintic drug with immunomodulating properties that was once approved by the US Food and Drug Administration for the treatment of various conditions, including autoimmune diseases, cancer, pediatric kidney disease, and chronic infections.1-4 Levamisole was banned in 2000 after reports of associated agranulocytosis and a characteristic painful purpuric vasculitis.4,5 Despite the ban, its use persists due to its increasing incorporation as an adulterant in cocaine, presumably for its dopaminergic properties that potentiate psychotropic effects.6 In 2009, the Drug Enforcement Administration reported that 69% of seized cocaine in the United States contains this chemical, with an average concentration of 10%.5 Levamisole-induced vasculopathy (LIV) typically resolves following the cessation of cocaine without further treatment necessary. We present a fatal case of LIV to emphasize that early recognition and discontinuation of the offending agent could be lifesaving.
A 40-year-old woman with a history of cocaine abuse was admitted with tender, reticular, purpuric, and erythematous patches and plaques on the lower extremities with areas of necrosis (Figure 1). The lesions had been present intermittently for 6 months. She tried topical mupirocin and oral amoxicillin clavulanate without improvement. She also described polyarthralgia in the hands, but the remainder of the review of symptoms and physical examination was negative.
Coagulation studies and white blood cell counts were within reference range. A urine toxicology screen was positive for cocaine; however, urine testing for levamisole was not performed given the short half-life of levamisole in vivo. A biopsy of one of the skin lesions on the right thigh showed pauci-inflammatory superficial and deep vein thrombosis with recanalization (Figure 2). A rheumatology workup revealed an elevated C-reactive protein level, low C3, positive antinuclear antibody, positive anti–double-stranded DNA, positive anticardiolipin antibody, positive lupus anticoagulant, and positive perinuclear antineutrophil cytoplasmic antibody (ANCA). Tests for HIV, hepatitis B and C, cryoglobulinemia, and cytomegalovirus were negative. Given the clinical picture and laboratory findings, levamisole-induced vasculitis was deemed likely. The patient was treated with appropriate skin and wound care. She was discharged with a prednisone taper and oral cephalexin and was counseled on cocaine cessation.
Five months later, the patient was readmitted for lower extremity edema and worsening painful lesions that had progressed to involve the legs, thighs, buttocks, flanks, and the tip of her nose. A deep vein thrombosis workup was negative. She admitted to ongoing cocaine use that was confirmed with urine toxicology. Coagulation studies and white blood cell counts remained within reference range. Repeat skin biopsy was consistent with prior findings, demonstrating thrombosis of superficial and deep vessels with recanalization. In addition, it showed focal epidermal necrosis and a perivascular infiltrate of lymphocytes, histiocytes, and rare neutrophils. She was placed on high-dose methylprednisolone. Over the course of the next month, her urine continued to test positive for cocaine, and she developed necrotizing fasciitis necessitating lower extremity amputation, abdominal washout, and debridement. She quickly deteriorated, developing multiorgan failure with sepsis, leading to death. Of note, the patient was never found to have neutropenia or agranulocytosis throughout the disease course.
Because levamisole is no longer in clinical use, reports of its adverse effects come exclusively from users of cocaine, whether via smoking or snorting. Levamisole-induced vasculopathy typically is painful and purpuric, with or without necrosis, in a retiform or stellate pattern and commonly involves the extremities, trunk, face, and external ears.7 The average age of presentation is 43 years and it more commonly is seen in women.8
Levamisole-induced vasculopathy remains a diagnosis of exclusion, so it is important to rule out other treatable causes. The differential diagnosis for purpura associated with vasculitis also includes other antineutrophilic cytoplasmic–associated vasculitides (eg, granulomatosis with polyangiitis, eosinophilic granulomatosis with polyangiitis), infectious purpura fulminans, antiphospholipid syndrome, cryoglobulinemia, and disseminated intravascular coagulation.9 In LIV patients, perinuclear ANCAs are present in up to 90% of cases, and cytoplasmic ANCAs in 19% to 59% of cases.10,11 Although leukopenia and neutropenia complicate approximately 60% of LIV cases, they are not required to make the diagnosis.11,12 Elevated erythrocyte sedimentation rate, normal coagulation studies, and positive antineutrophil antibodies and lupus anticoagulant further aid in the diagnosis.8 Urine should be tested for cocaine in suspected patients. Urine also can be tested for levamisole, which is challenging because of the short half-life of 5.6 hours. Only 2% to 5% of levamisole is excreted unchanged in the urine, and testing requires gas chromatography and mass spectrometry that was not readily available to perform on our patient.7 In addition to laboratory and urine studies, hair strand testing,10 skin biopsy, and histologic findings also can be used to support the diagnosis.
The pathogenesis of LIV is not completely understood, but it is thought to be an immune complex–mediated process based on immunofluorescence studies in the skin.13,14 Classic pathologic findings include multiple fibrin thrombi within small vessels in the superficial and deep dermis, leukocytoclastic vasculitis of small vessels consisting of fibrinoid necrosis of the vessel wall, extravasated erythrocytes, karyorrhectic debris, and angiocentric inflammation.14 Direct immunofluorescence is not routinely performed but most commonly demonstrates deposition of IgA, IgM, and C3.14,15
Levamisole-induced vasculopathy usually resolves upon cessation of cocaine use without long-term sequelae. Steroids have been used as treatment of prominent vasculitis with variable success; however, immunosuppressive effects should be closely monitored, especially with inpatients with concurrent granulocytopenia. Broad-spectrum antibiotics have been used in cases with fever and agranulocytosis. Cutaneous lesions typically disappear within 2 to 3 weeks, and serologic markers resolve within 2 to 10 months. Recurrent use of cocaine generally results in recurrent neutropenia and skin eruptions, supporting the causal role. Our patient’s recurrent prolonged cocaine use with vasculopathy was assumed to be the source of the necrotizing fasciitis that led to a cascade of sepsis, rapidly progressing multiorgan failure, and ultimate demise.
Presentation of a purpuric vasculopathy, with or without associated neutropenia and positive autoantibodies, should prompt the consideration of levamisole-contaminated cocaine use in the clinician’s differential. Although the patient may initially deny cocaine use, it is important to keep this diagnosis in mind when the clinical picture fits, and urine toxicology screen should be ordered when there is question. Physicians and patients should be wary of potential complications, even death. Early recognition and discontinuation of the offending agent could be lifesaving.
- Menni S, Pistritto G, Gianotti R, et al. Ear lobe necrosis by levamisole-induced occlusive vasculitis in a pediatric patient. Pediatr Dermatol. 1997;14:477-479.
- Symoens J, Veys E, Mielants M, et al. Adverse reactions to levamisole. Cancer Treat Rep. 1978;62:1721-1730.
- Vogel CL, Silverman MA, Mansell PW, et al. Mechanism of levamisole-induced granulocytopenia in breast cancer patients. Am J Hematol. 1980;9:171-183.
- Rongioletti F, Ghio L, Ginevri F, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long-term treatment with levamisole in children. Br J Dermatol. 1999;140:948-951.
- Centers for Disease Control and Prevention (CDC). Agranulocytosis associated with cocaine use—four states, March 2008–November 2009. MMWR Morb Mortal Wkly Rep. 2009;58:1381-1385.
- Zhu NY, Legatt DF, Turner AR. Agranulocytosis after consumption of cocaine adulterated with levamisole. Ann Intern Med. 2009;150:287-289.
- Gross RL, Brucker J, Bahce-Altuntas A, et al. A novel cutaneous vasculitis syndrome induced by levamisole-contaminated cocaine. Clin Rheumatol. 2011;30:1385-1392.
- Trehy ML, Brown DJ, Woodruff JT, et al. Determination of levamisole in urine by gas chromatography-mass spectrometry. J Anal Toxicol. 2011;35:545-550.
- Waller JM, Feramisco JD, Alberta-Wszolek L, et al. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit? J Am Acad Dermatol. 2010;63:530-535.
- Pearson T, Bremmer M, Cohen J, et al. Vasculopathy related to cocaine adulterated with levamisole: a review of the literature. Dermatol Online J. 2012;18:1.
- Arora NP. Cutaneous vasculopathy and neutropenia associated with levamisole-adulterated cocaine. Am J Med Sci. 2013;345:45-51.
- Chai PR, Bastan W, Machan J, et al. Levamisole exposure and hematologic indices in cocaine users. Acad Emerg Med. 2011;18:1141-1147.
- Lazareth H, Peytavin G, Polivka L, et al. The hairy-print for levamisole-induced vasculitis. BMJ Case Rep. 2012;2012:bcr2012006602.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia—a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2011;65:722-725.
- Jenkins J, Babu K, Hsu-Hung E, et al. ANCA-positive necrotizing vasculitis and thrombotic vasculopathy induced by levamisole-adulterated cocaine: a distinctive clinicopathologic presentation. J Am Acad Dermatol. 2011;65:E14-E16.
- Menni S, Pistritto G, Gianotti R, et al. Ear lobe necrosis by levamisole-induced occlusive vasculitis in a pediatric patient. Pediatr Dermatol. 1997;14:477-479.
- Symoens J, Veys E, Mielants M, et al. Adverse reactions to levamisole. Cancer Treat Rep. 1978;62:1721-1730.
- Vogel CL, Silverman MA, Mansell PW, et al. Mechanism of levamisole-induced granulocytopenia in breast cancer patients. Am J Hematol. 1980;9:171-183.
- Rongioletti F, Ghio L, Ginevri F, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating long-term treatment with levamisole in children. Br J Dermatol. 1999;140:948-951.
- Centers for Disease Control and Prevention (CDC). Agranulocytosis associated with cocaine use—four states, March 2008–November 2009. MMWR Morb Mortal Wkly Rep. 2009;58:1381-1385.
- Zhu NY, Legatt DF, Turner AR. Agranulocytosis after consumption of cocaine adulterated with levamisole. Ann Intern Med. 2009;150:287-289.
- Gross RL, Brucker J, Bahce-Altuntas A, et al. A novel cutaneous vasculitis syndrome induced by levamisole-contaminated cocaine. Clin Rheumatol. 2011;30:1385-1392.
- Trehy ML, Brown DJ, Woodruff JT, et al. Determination of levamisole in urine by gas chromatography-mass spectrometry. J Anal Toxicol. 2011;35:545-550.
- Waller JM, Feramisco JD, Alberta-Wszolek L, et al. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit? J Am Acad Dermatol. 2010;63:530-535.
- Pearson T, Bremmer M, Cohen J, et al. Vasculopathy related to cocaine adulterated with levamisole: a review of the literature. Dermatol Online J. 2012;18:1.
- Arora NP. Cutaneous vasculopathy and neutropenia associated with levamisole-adulterated cocaine. Am J Med Sci. 2013;345:45-51.
- Chai PR, Bastan W, Machan J, et al. Levamisole exposure and hematologic indices in cocaine users. Acad Emerg Med. 2011;18:1141-1147.
- Lazareth H, Peytavin G, Polivka L, et al. The hairy-print for levamisole-induced vasculitis. BMJ Case Rep. 2012;2012:bcr2012006602.
- Chung C, Tumeh PC, Birnbaum R, et al. Characteristic purpura of the ears, vasculitis, and neutropenia—a potential public health epidemic associated with levamisole-adulterated cocaine. J Am Acad Dermatol. 2011;65:722-725.
- Jenkins J, Babu K, Hsu-Hung E, et al. ANCA-positive necrotizing vasculitis and thrombotic vasculopathy induced by levamisole-adulterated cocaine: a distinctive clinicopathologic presentation. J Am Acad Dermatol. 2011;65:E14-E16.
Practice Points
- Levamisole-induced vasculopathy usually resolves upon cessation of cocaine use without long-term sequelae.
- Presentation of a purpuric vasculitis, with or without associated neutropenia and positive autoantibodies, should prompt the consideration of levamisole-contaminated cocaine use in the clinician’s differential. Early recognition and discontinuation of the offending agent could be lifesaving.
Genital Primary Herpetic Infection With Concurrent Hepatitis in an Infant
To the Editor:
Cutaneous herpes simplex virus (HSV) infection generally involves mucocutaneous junctions, but virtually any area of the skin can be affected.1 When the genital area of adult patients is affected, the disease usually is sexually transmitted and mainly caused by HSV-2. In infants, genital primary herpetic infection is rare and more commonly is caused by HSV-1 than by HSV-2. We report a rare case of genital primary herpetic infection with concurrent hepatitis in an infant.
An 8-month-old infant with no underlying medical problems, including atopic dermatitis, was referred for erythematous grouped vesicles with erosions on the perianal area of 4 days’ duration (Figure). The skin color appeared normal, not icterus. She also had a fever (temperature, 37.9 °C), and her urination pattern had changed from normal to frequent leakage, possibly owing to pain related to the eroded lesions. Physical examination did not reveal palpable inguinal lymph nodes. The oral mucosa was not involved. The patient’s father had a history of recurrent herpetic infection on both the perioral and perianal areas.
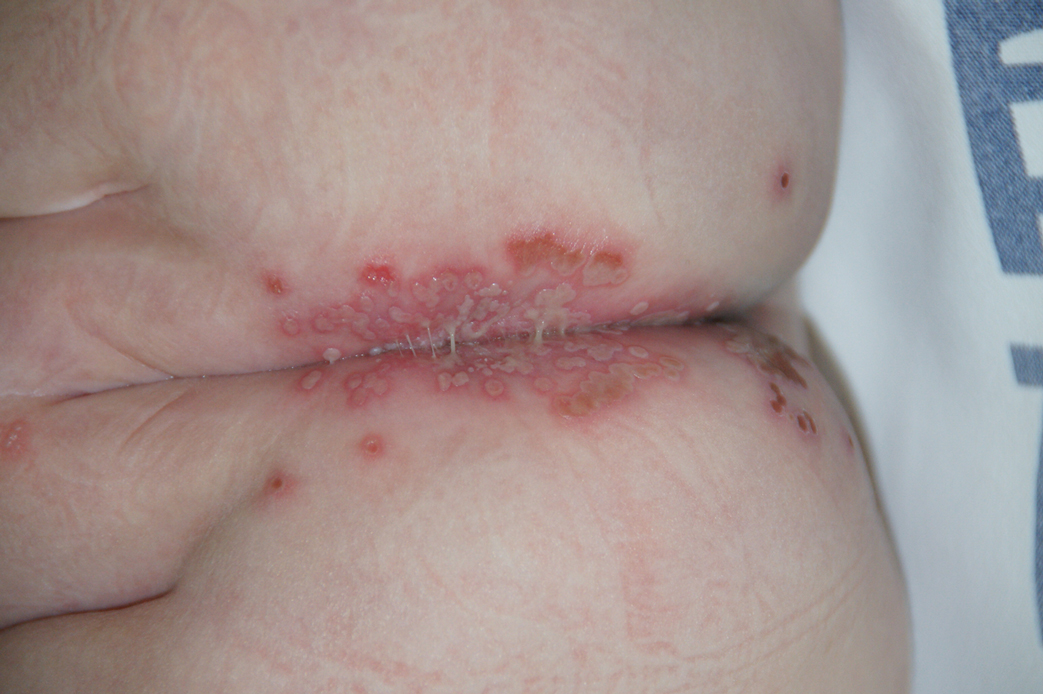
A Tzanck smear revealed giant multinucleated cells with multiple inflammatory cells. Laboratory tests revealed marked leukocytosis, elevated liver enzymes (aspartate aminotransferase, 141 IU/L [reference range, 15 IU/L–60 IU/L]; alanine aminotransferase, 422 IU/L [reference range, 13 IU/L–45 IU/L]), and was positive for herpes simplex viral IgM but negative for herpes simplex viral IgG. A viral culture also demonstrated the growth of HSV. An abdominal ultrasound was normal. Based on the cutaneous and laboratory findings, genital primary herpetic infection with concurrent hepatitis was diagnosed. Intravenous acyclovir 50 mg was administered 3 times daily for 7 days, and a wet dressing with topical mupirocin was employed daily until the skin lesions healed. The fever subsided soon after starting treatment. The liver enzyme counts decreased gradually in serial follow-up (aspartate aminotransferase, 75 IU/L; alanine aminotransferase, 70 IU/L).
Primary herpetic infection usually is asymptomatic, but when symptoms do occur, it is characterized by the sudden onset of painful vesicle clusters over erythematous edematous skin. Lesions can be associated with fever and malaise and may involve the perineum. Urinary symptoms may occur. The average age of onset ranges from 6 months to 4 years. The virus commonly is transmitted by asymptomatic carriers. Autoinoculation from concomitant oral primary herpetic infection or individuals with active herpetic infection is one possible route of transmission. In our patient, we assumed that she acquired the virus from her father during close contact. A diagnosis can be made clinically using direct methods including culture, Tzanck smear, or polymerase chain reaction, or indirect methods such as serologic tests.2
Hepatitis secondary to HSV infection is rare, especially in immunocompetent patients. It occurs during primary infection and rarely during recurrent infection with or without concomitant skin lesions.3 Symptoms include fever, anorexia, nausea, vomiting, abdominal pain, leukopenia, coagulopathy, and marked elevation of serum transaminase levels without jaundice. Based on our patient’s elevated liver enzyme levels and virological evidence of acute primary HSV infection, a lack of evidence of other hepatic viral infections, and the presence of herpes simplex viremia, we concluded that this infant had viral hepatitis as a part of the clinical presentation of primary HSV infection. We did not perform a direct liver biopsy considering her age and accompanying risks.4
Primary herpetic infection usually has a benign course and a short duration. In children, the prognosis depends on underlying immunologic status, not a particular type of HSV. In children with atopic dermatitis, primary herpetic infection tends to occur earlier and is more severe. Early treatment with acyclovir is effective; intravenous treatment is not required unless local complications or systemic involvement are present. Long-term follow-up is recommended because of the possibility of recurrence.
Although the possibility of systemic involvement including hepatitis due to HSV infection is low, awareness among dermatologists about primary herpetic infection and its possible complications would be helpful in the diagnosis and treatment, especially for atypical or extensive cases.
- Jenson HB, Shapiro ED. Primary herpes simplex virus infection of a diaper rash. Pediatr Infect Dis J. 1987;6:1136-1138.
- Batalla A, Flórez A, Dávila P, et al. Genital primary herpes simplexinfection in a 5-month-old infant. Dermatol Online J. 2011;17:8.
- Norvell JP, Blei AT, Jovanovic BD, et al. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl. 2007;13:1428-1434.
- Chen CK, Wu SH, Huang YC. Herpetic gingivostomatitis with severe hepatitis in a previously healthy child. J Microbiol Immunol Infect. 2012;45:324-325.
To the Editor:
Cutaneous herpes simplex virus (HSV) infection generally involves mucocutaneous junctions, but virtually any area of the skin can be affected.1 When the genital area of adult patients is affected, the disease usually is sexually transmitted and mainly caused by HSV-2. In infants, genital primary herpetic infection is rare and more commonly is caused by HSV-1 than by HSV-2. We report a rare case of genital primary herpetic infection with concurrent hepatitis in an infant.
An 8-month-old infant with no underlying medical problems, including atopic dermatitis, was referred for erythematous grouped vesicles with erosions on the perianal area of 4 days’ duration (Figure). The skin color appeared normal, not icterus. She also had a fever (temperature, 37.9 °C), and her urination pattern had changed from normal to frequent leakage, possibly owing to pain related to the eroded lesions. Physical examination did not reveal palpable inguinal lymph nodes. The oral mucosa was not involved. The patient’s father had a history of recurrent herpetic infection on both the perioral and perianal areas.
A Tzanck smear revealed giant multinucleated cells with multiple inflammatory cells. Laboratory tests revealed marked leukocytosis, elevated liver enzymes (aspartate aminotransferase, 141 IU/L [reference range, 15 IU/L–60 IU/L]; alanine aminotransferase, 422 IU/L [reference range, 13 IU/L–45 IU/L]), and was positive for herpes simplex viral IgM but negative for herpes simplex viral IgG. A viral culture also demonstrated the growth of HSV. An abdominal ultrasound was normal. Based on the cutaneous and laboratory findings, genital primary herpetic infection with concurrent hepatitis was diagnosed. Intravenous acyclovir 50 mg was administered 3 times daily for 7 days, and a wet dressing with topical mupirocin was employed daily until the skin lesions healed. The fever subsided soon after starting treatment. The liver enzyme counts decreased gradually in serial follow-up (aspartate aminotransferase, 75 IU/L; alanine aminotransferase, 70 IU/L).
Primary herpetic infection usually is asymptomatic, but when symptoms do occur, it is characterized by the sudden onset of painful vesicle clusters over erythematous edematous skin. Lesions can be associated with fever and malaise and may involve the perineum. Urinary symptoms may occur. The average age of onset ranges from 6 months to 4 years. The virus commonly is transmitted by asymptomatic carriers. Autoinoculation from concomitant oral primary herpetic infection or individuals with active herpetic infection is one possible route of transmission. In our patient, we assumed that she acquired the virus from her father during close contact. A diagnosis can be made clinically using direct methods including culture, Tzanck smear, or polymerase chain reaction, or indirect methods such as serologic tests.2
Hepatitis secondary to HSV infection is rare, especially in immunocompetent patients. It occurs during primary infection and rarely during recurrent infection with or without concomitant skin lesions.3 Symptoms include fever, anorexia, nausea, vomiting, abdominal pain, leukopenia, coagulopathy, and marked elevation of serum transaminase levels without jaundice. Based on our patient’s elevated liver enzyme levels and virological evidence of acute primary HSV infection, a lack of evidence of other hepatic viral infections, and the presence of herpes simplex viremia, we concluded that this infant had viral hepatitis as a part of the clinical presentation of primary HSV infection. We did not perform a direct liver biopsy considering her age and accompanying risks.4
Primary herpetic infection usually has a benign course and a short duration. In children, the prognosis depends on underlying immunologic status, not a particular type of HSV. In children with atopic dermatitis, primary herpetic infection tends to occur earlier and is more severe. Early treatment with acyclovir is effective; intravenous treatment is not required unless local complications or systemic involvement are present. Long-term follow-up is recommended because of the possibility of recurrence.
Although the possibility of systemic involvement including hepatitis due to HSV infection is low, awareness among dermatologists about primary herpetic infection and its possible complications would be helpful in the diagnosis and treatment, especially for atypical or extensive cases.
To the Editor:
Cutaneous herpes simplex virus (HSV) infection generally involves mucocutaneous junctions, but virtually any area of the skin can be affected.1 When the genital area of adult patients is affected, the disease usually is sexually transmitted and mainly caused by HSV-2. In infants, genital primary herpetic infection is rare and more commonly is caused by HSV-1 than by HSV-2. We report a rare case of genital primary herpetic infection with concurrent hepatitis in an infant.
An 8-month-old infant with no underlying medical problems, including atopic dermatitis, was referred for erythematous grouped vesicles with erosions on the perianal area of 4 days’ duration (Figure). The skin color appeared normal, not icterus. She also had a fever (temperature, 37.9 °C), and her urination pattern had changed from normal to frequent leakage, possibly owing to pain related to the eroded lesions. Physical examination did not reveal palpable inguinal lymph nodes. The oral mucosa was not involved. The patient’s father had a history of recurrent herpetic infection on both the perioral and perianal areas.
A Tzanck smear revealed giant multinucleated cells with multiple inflammatory cells. Laboratory tests revealed marked leukocytosis, elevated liver enzymes (aspartate aminotransferase, 141 IU/L [reference range, 15 IU/L–60 IU/L]; alanine aminotransferase, 422 IU/L [reference range, 13 IU/L–45 IU/L]), and was positive for herpes simplex viral IgM but negative for herpes simplex viral IgG. A viral culture also demonstrated the growth of HSV. An abdominal ultrasound was normal. Based on the cutaneous and laboratory findings, genital primary herpetic infection with concurrent hepatitis was diagnosed. Intravenous acyclovir 50 mg was administered 3 times daily for 7 days, and a wet dressing with topical mupirocin was employed daily until the skin lesions healed. The fever subsided soon after starting treatment. The liver enzyme counts decreased gradually in serial follow-up (aspartate aminotransferase, 75 IU/L; alanine aminotransferase, 70 IU/L).
Primary herpetic infection usually is asymptomatic, but when symptoms do occur, it is characterized by the sudden onset of painful vesicle clusters over erythematous edematous skin. Lesions can be associated with fever and malaise and may involve the perineum. Urinary symptoms may occur. The average age of onset ranges from 6 months to 4 years. The virus commonly is transmitted by asymptomatic carriers. Autoinoculation from concomitant oral primary herpetic infection or individuals with active herpetic infection is one possible route of transmission. In our patient, we assumed that she acquired the virus from her father during close contact. A diagnosis can be made clinically using direct methods including culture, Tzanck smear, or polymerase chain reaction, or indirect methods such as serologic tests.2
Hepatitis secondary to HSV infection is rare, especially in immunocompetent patients. It occurs during primary infection and rarely during recurrent infection with or without concomitant skin lesions.3 Symptoms include fever, anorexia, nausea, vomiting, abdominal pain, leukopenia, coagulopathy, and marked elevation of serum transaminase levels without jaundice. Based on our patient’s elevated liver enzyme levels and virological evidence of acute primary HSV infection, a lack of evidence of other hepatic viral infections, and the presence of herpes simplex viremia, we concluded that this infant had viral hepatitis as a part of the clinical presentation of primary HSV infection. We did not perform a direct liver biopsy considering her age and accompanying risks.4
Primary herpetic infection usually has a benign course and a short duration. In children, the prognosis depends on underlying immunologic status, not a particular type of HSV. In children with atopic dermatitis, primary herpetic infection tends to occur earlier and is more severe. Early treatment with acyclovir is effective; intravenous treatment is not required unless local complications or systemic involvement are present. Long-term follow-up is recommended because of the possibility of recurrence.
Although the possibility of systemic involvement including hepatitis due to HSV infection is low, awareness among dermatologists about primary herpetic infection and its possible complications would be helpful in the diagnosis and treatment, especially for atypical or extensive cases.
- Jenson HB, Shapiro ED. Primary herpes simplex virus infection of a diaper rash. Pediatr Infect Dis J. 1987;6:1136-1138.
- Batalla A, Flórez A, Dávila P, et al. Genital primary herpes simplexinfection in a 5-month-old infant. Dermatol Online J. 2011;17:8.
- Norvell JP, Blei AT, Jovanovic BD, et al. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl. 2007;13:1428-1434.
- Chen CK, Wu SH, Huang YC. Herpetic gingivostomatitis with severe hepatitis in a previously healthy child. J Microbiol Immunol Infect. 2012;45:324-325.
- Jenson HB, Shapiro ED. Primary herpes simplex virus infection of a diaper rash. Pediatr Infect Dis J. 1987;6:1136-1138.
- Batalla A, Flórez A, Dávila P, et al. Genital primary herpes simplexinfection in a 5-month-old infant. Dermatol Online J. 2011;17:8.
- Norvell JP, Blei AT, Jovanovic BD, et al. Herpes simplex virus hepatitis: an analysis of the published literature and institutional cases. Liver Transpl. 2007;13:1428-1434.
- Chen CK, Wu SH, Huang YC. Herpetic gingivostomatitis with severe hepatitis in a previously healthy child. J Microbiol Immunol Infect. 2012;45:324-325.
Practice Points
- Parents with a history of herpes simplex virus (HSV) need to be educated before the baby is born to be careful about direct skin contact with the child to prevent the spread of HSV infection.
- Although systemic involvement is not typical, additional tests to rule out internal organ involvement may be required, especially in children.