User login
Erythema Multiforme–like Dermatitis Due to Isoniazid Hypersensitivity in a Patient With Psoriasis
To the Editor:
Psoriasis vulgaris is a chronic autoimmune inflammatory disease and biologic agents, such as anti–tumor necrosis factor α (TNF-α), are alternative drugs in case of resistance or adverse events to conventional ones.1 The limitation of these agents is immunosuppression that may cause infections such as tuberculosis (TB). Prophylaxis is indicated to latent TB diseases if the purified protein derivative (tuberculin) skin test is higher than 5 mm before starting these treatments. The challenge in TB treatment is adverse drug reactions (ADRs) that are reported in 4% to 6% of cases.2,3
Erythema multiforme–like dermatitis is a rare skin rash that develops due to isoniazid (INH). The clinical presentation includes erythematoedematous lesions in an acral distribution with no mucosal involvement and systemic exposure to INH. Skin biopsy and patch tests are the supportive diagnostic methods. Isoniazid-associated skin rashes rarely are reported and generally are not severe enough to terminate the drug. We present a patient with psoriasis who received TB prophylaxis before anti–TNF-α use. He presented with erythema multiforme–like dermatitis due to INH. Withdrawal of the drug and treatment of the lesions were the first steps of intolerance, followed by a patch test with the culprit drug after recovery. We discuss the diagnostic drug allergy evaluation and treatment approach.
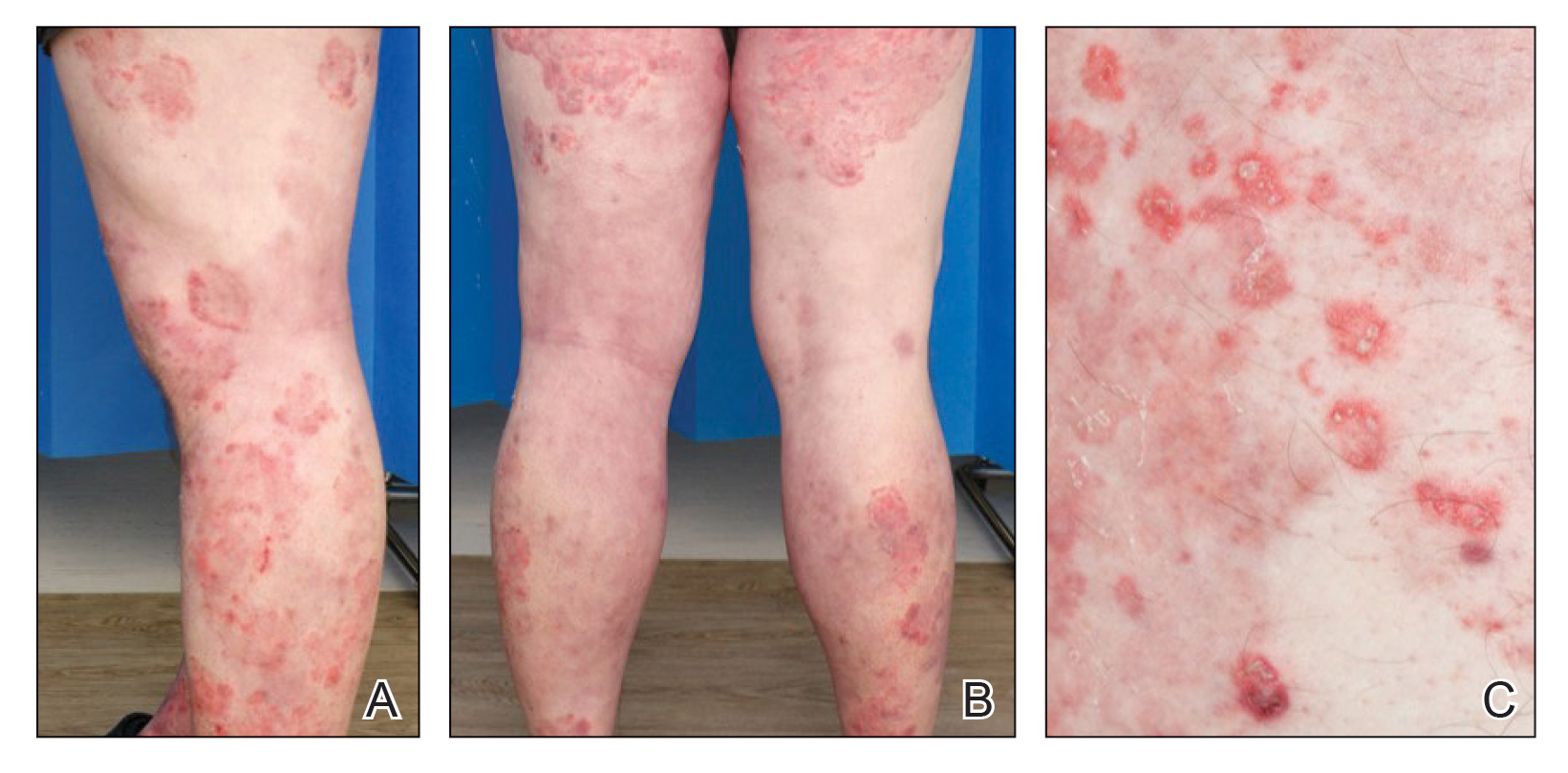
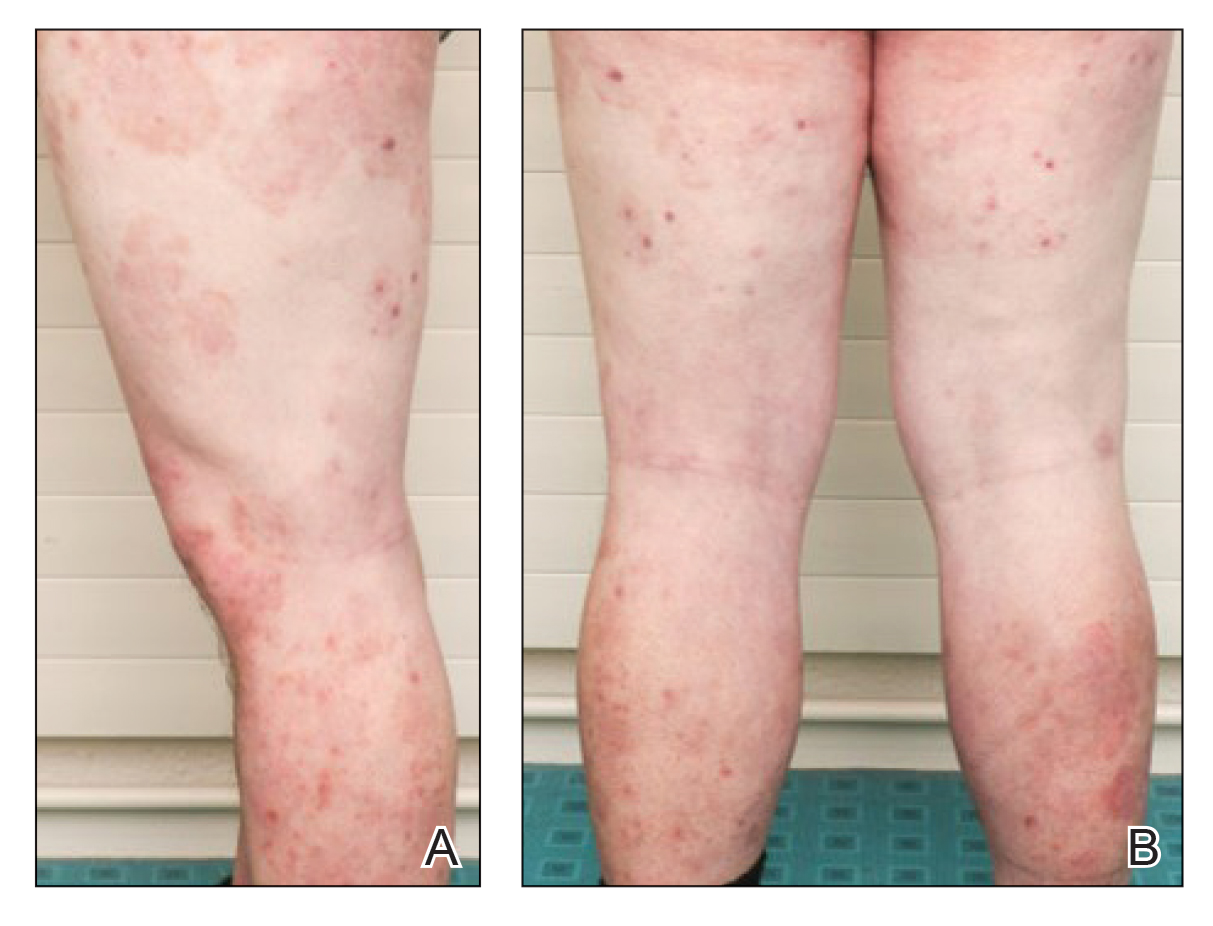
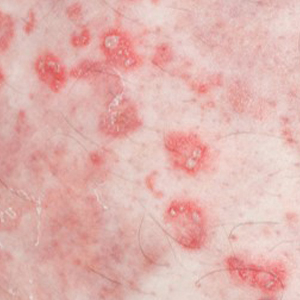
A 37-year-old man presented with a 15-year history of severe psoriasis with frequent flares. He was treated with various topical and systemic agents including acitretin and methotrexate at 4-year intervals. Despite the addition of phototherapy, he underwent a new treatment with anti–TNF-α, as the disease control with other treatments was insufficient. Before starting anti–TNF-α, preventive treatment against TB with INH (300 mg/d) was indicated with 20 mm of purified protein derivative. On approximately the 20th day of treatment, he developed pruritic erythema with desquamation and exfoliation localized to the hands and feet (Figure 1). Isoniazid was discontinued and a topical steroid was initiated. After 3 weeks, the skin lesions were completely improved and INH was reinitiated at the same dose with antihistamine prophylaxis (oral levocetirizine 5 mg/d). Seven days later, similar skin lesions presented that were more extensive on the arms and legs (Figure 2). Complete blood cell counts, renal and hepatic function tests, and hepatitis markers were within reference range in consultation with the allergy division. To distinguish the lesions from a psoriasis attack, a punch biopsy of the eruptive dermatitis showed erythema multiforme–like dermatitis including dermal edema and perivascular lymphocytic infiltration with no relation to psoriasis but consistent with a drug eruption. Isoniazid was discontinued, and the skin lesions resolved after 4 weeks of topical steroid and oral antihistamine use (Figure 3). There was no other drug use except INH, and a skin patch test with INH was positive at 72 hours (Figure 4). Skin tests with INH were done to 5 healthy lesions that were negative. Finally, TB prophylaxis was performed with rifampicin (10 mg/kg/d [600 mg/d]) for 4 months with no ADRs. The patient’s psoriasis lesions improved with anti–TNF-α that was initiated 1 month after starting TB prevention with rifampicin.
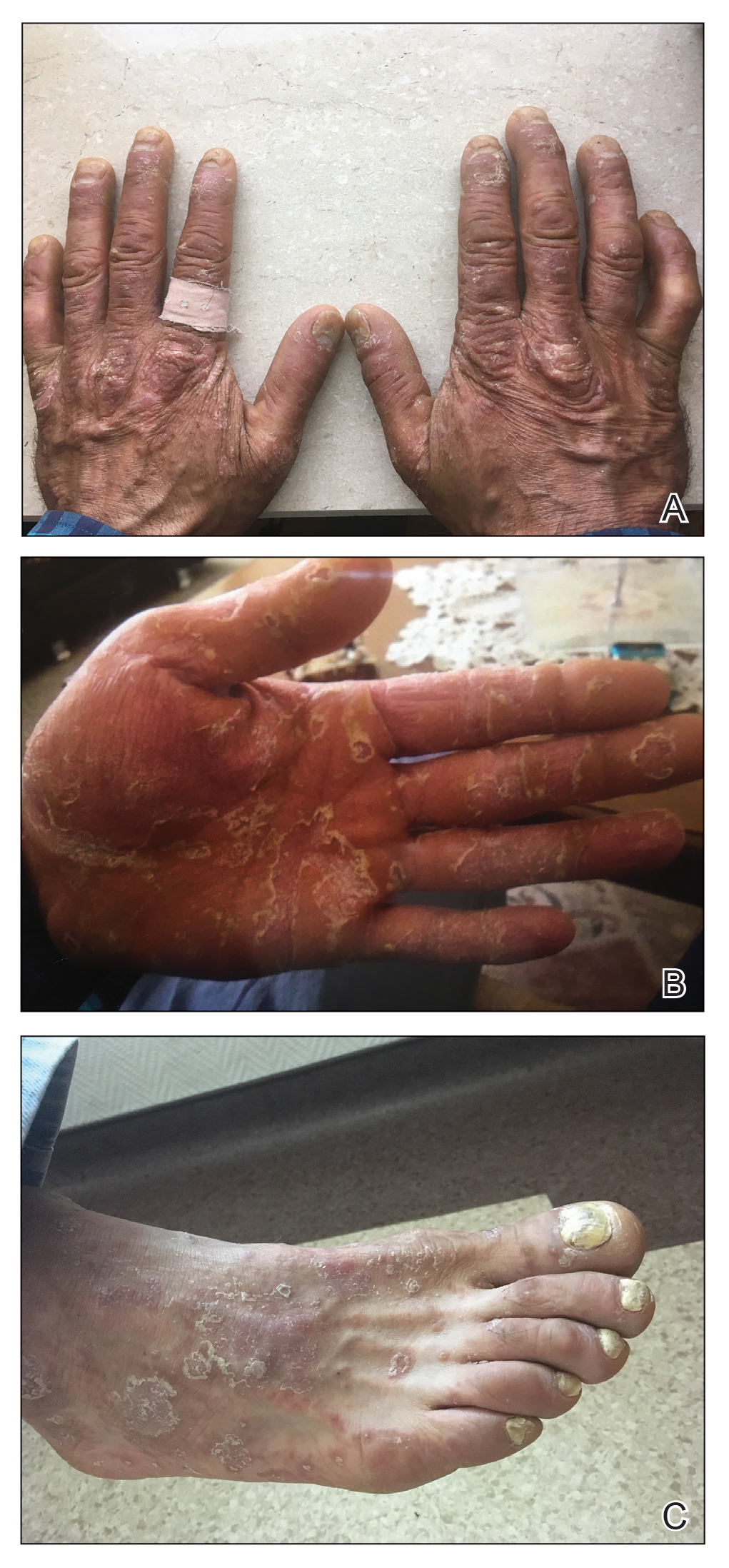
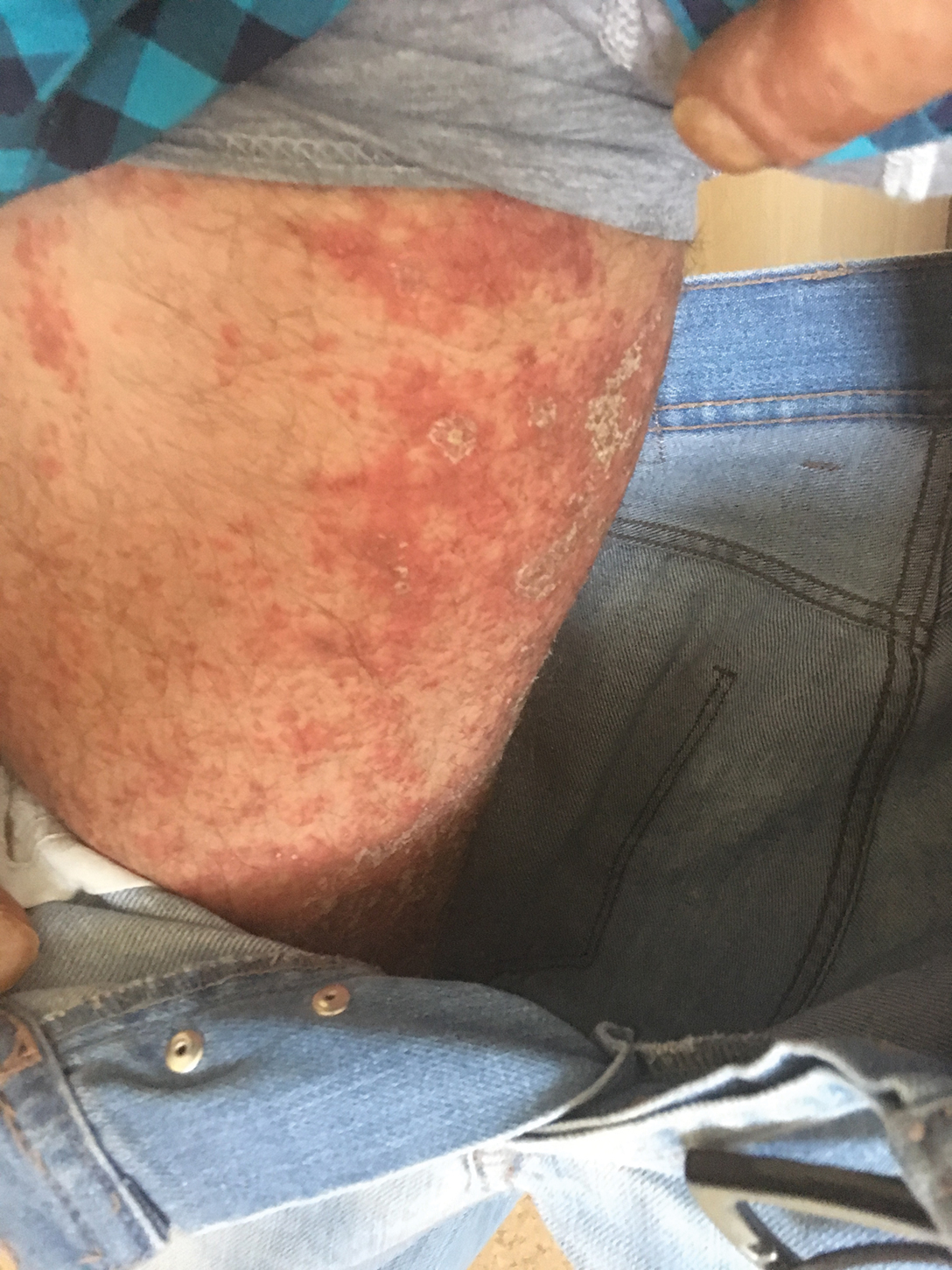
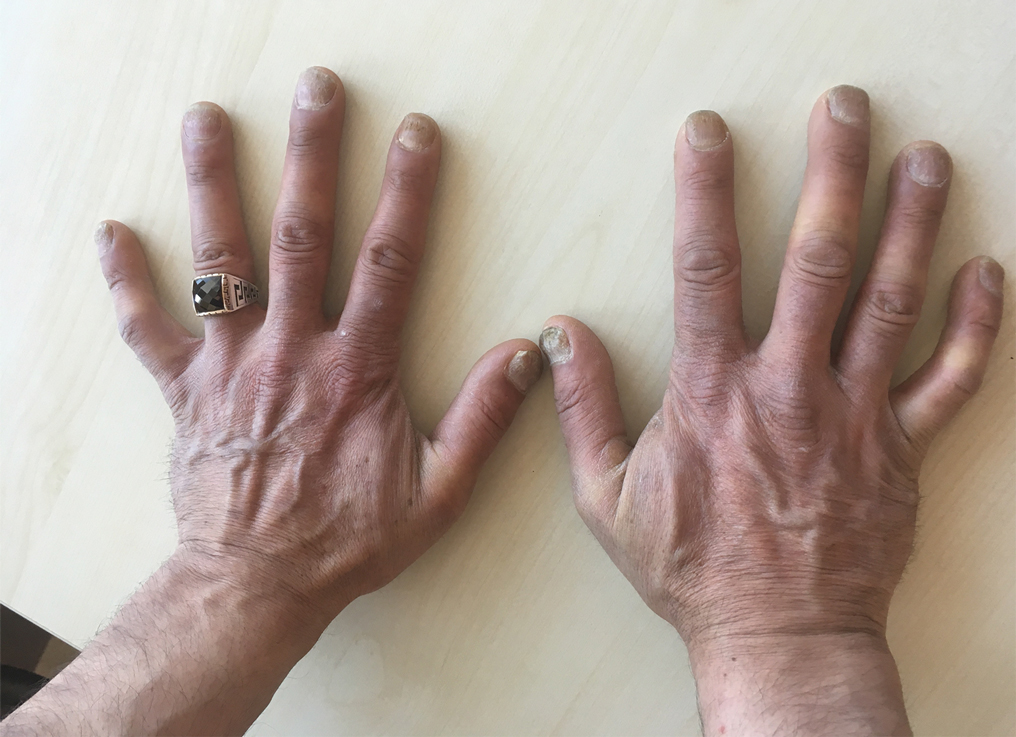
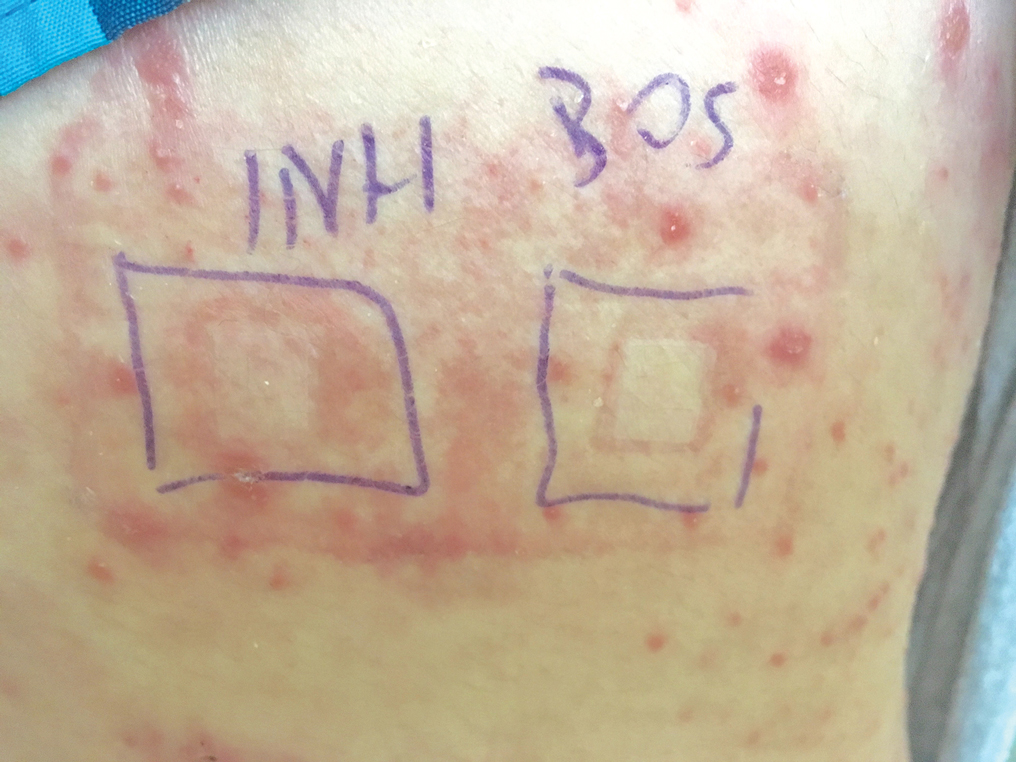
This case of erythema multiforme–like dermatitis was diagnosed with acral involvement, a positive patch test to INH, and lymphocytic inflammation in a skin biopsy. It was a drug-induced reaction, as skin lesions developed during INH intake and improved after drug withdrawal.
Isoniazid, also known as isonicotinylhydrazide, is an oral antibiotic used for the treatment of TB and other mycobacteria. Protective treatment against latent TB primarily is done with daily INH for 6 or 9 months; alternatively, INH may be taken weekly with rifapentine for 3 months or daily with rifampicin for 4 months. Daily rifampicin alone for 4 months also is an option. In general, these regimens have similar efficacy; however, in terms of safety, the rifampicin and rifapentine combination regimens have fewer hepatotoxicity events compared to the INH alone regimen, but there are more cutaneous and flulike reactions and gastrointestinal intolerance.4 Cutaneous ADRs to TB treatment such as mild itchiness and cutaneous eruptions usually are observed within 2 months of drug initiation. Pyrazinamide was reported as the most common drug associated with cutaneous ADRs, and INH was the rarest offending drug.5
The frequency of ADRs to INH is approximately 5.4%, and the most prevalent ADRs include asymptomatic elevation of serum liver enzyme concentrations, peripheral neuropathy, and hepatotoxicity, and skin lesions are less common.2 Our patient’s laboratory test results excluded vitamin B deficiency, hepatic and renal dysfunction, and neuropathy.
Previously reported skin reactions related to INH were late-type reactions such as maculopapular rash, dermatitis, erythema multiforme, drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, Stevens-Johnson syndrome, and toxic epidermal necrolysis.5,6 The concerning prediagnosis of psoriatic exacerbation in our patient was ruled out by the absence of typical skin lesions such as well-defined, erythematous plaques and pustules and atypical localization such as the dorsal hands and feet rather than the knees, elbows, lumbosacral region, scalp, and abdomen, which is typical of psoriasis. DRESS syndrome was unlikely with the absence of fever, lymphadenopathy, hypereosinophilia, leukocytosis, and renal and hepatic dysfunction.7 There were no widespread blisters, epidermal detachment, or mucosal involvement on the trunk or face typically associated with Stevens-Johnson syndrome and toxic epidermal necrolysis.7,8 A possible diagnosis of contact dermatitis was suspected with likely skin lesions as exfoliation and chapping, typical localization on the hands and feet, and positive patch test that supported sensitization to the drug. However, the patient’s skin lesions were not eczematous (characterized by erythema, vesiculation, exudation, or bullous edema in the acute phase), and were not localized to areas of irritant exposure.3 In our patient, erythematoedematous lesions in an acral distribution with no mucosal involvement and systemic exposure to INH was compatible with erythema multiforme, whereas the absence of target appearance, positive patch test, and late appearance were incompatible with erythema multiforme.8
Because the clinical picture did not fit contact dermatitis or erythema multiforme, a diagnosis of erythema multiforme–like noneczematous dermatitis was suggested. Noneczematous dermatitis has subtypes that include purpuric, lichenoid, pustular, lymphomatoid, dyshidrosiform, and pigmented, as well as erythema multiforme–like contact eruptions.9 These clinical entities are not associated with contact exposure, but are related to systemic exposure, as seen in our patient.10 The patch test positivity and skin biopsy report also supported the diagnosis of erythema multiforme–like dermatitis. Erythema multiforme–like dermatitis is thought to be caused by medications or infections inducing immunocomplexes and lymphocytic infiltration in the dermis and subepidermis. Nevertheless, the prognosis was self-limiting in both.8 The clinical polymorphism caused by INH in this patient was suggested to be related with individual susceptibility, variability of contact-activating modalities, and the targeted cutaneous structures. Furthermore, among the risk factors for cutaneous ADRs—HIV, polypharmacy, older age, and preexisting renal and liver impairment—the only notable factor in this patient was psoriasis as an autoimmune disorder.
Patients with skin diseases such as psoriasis should be followed up by closer monitoring during INH use. Withdrawal of the drug and symptomatic treatment of the lesions with corticosteroid and antihistamine are the first steps of drug intolerance. After complete recovery and termination of antiallergic drugs, diagnostic tests are recommended if the drug reaction was not life-threatening. Skin prick and intradermal tests are useful in early-type drug reactions, whereas patch testing and late evaluation of an intradermal test may be helpful in the diagnosis of delayed-type reactions. The full dose of INH is avoided in an intradermal test against irritation. A patch test with INH was performed by diluting a 100-mg tablet with 1 mL of distilled water, and used as 1/100, 1/10, and 1/1 dilutions.8 Patch testing with INH also was done in 5 healthy control patients to exclude the irritation effect in this case. The rechallenge of INH was done in a controlled manner in our patient to rule out psoriasis activation since it was a localized skin reaction with no serious ADR. An oral provocation test with the culprit drug is the gold standard of drug allergy diagnosis that should be done in a tertiary hospital with an intensive care unit.
This case of erythema multiforme–like dermatitis due to INH is interesting due to systemic intake of INH, which resulted in dermatitis with localized involvement similar to erythema multiforme but with no immunologic processes or prior sensitization. With the increasing use of anti–TNF-α treatment, INH use will be more prevalent than in the past for the treatment of latent TB. Even though the skin-restricted ADRs of INH are rare and minor, particular attention should be paid to patients with dermatologic diseases. In our case, diagnostic drug allergy evaluation was performed to optimize the second-line treatment of TB infection, in addition to early withdrawal of the culprit drug.
- Vide J, Magina S. Moderate to severe psoriasis treatment challenges through the era of biological drugs.An Bras Dermatol. 2017;92:668-674.
- Gülbay BE, Gürkan OU, Yildiz OA, et al. Side effects due to primary antituberculosis drugs during the initial phase of therapy in 1149 hospitalized patients for tuberculosis. Respir Med. 2006;100:1834-1842.
- Holdiness MR. Contact dermatitis to antituberculosis drugs. Contact Dermatitis. 1986;15:282-288.
- Getahun H, Matteelli A, Abubakar I, et al. Management of latent Mycobacterium tuberculosis infection: WHO guidelines for low tuberculosis burden countries. Eur Respir J. 2015;46:1563-1576.
- Tan WC, Ong CK, Kang SC, et al. Two years review of cutaneous adverse drug reaction from first line anti-tuberculous drugs. Med J Malaysia. 2007;62:143-146.
- Özkaya E.Eczematous-type multiple drug allergy from isoniazid and ethambutol with positive patch test results. Cutis. 2013;92:121-124.
- Fernando SL. Drug-reaction eosinophilia and systemic symptoms and drug-induced hypersensitivity syndrome. Australas J Dermatol. 2014;55:15-23.
- Rebollo S, Sanchez P, Vega JM, et al. Hypersensitivity syndrome from isoniazid with positive patch test. Contact Dermatitis. 2001;45:306.
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902.
- Bonamonte D, Foti C, Vestita M, et al. Nummular eczema and contact allergy: a retrospective study. Dermatitis. 2012;23:153-157.
To the Editor:
Psoriasis vulgaris is a chronic autoimmune inflammatory disease and biologic agents, such as anti–tumor necrosis factor α (TNF-α), are alternative drugs in case of resistance or adverse events to conventional ones.1 The limitation of these agents is immunosuppression that may cause infections such as tuberculosis (TB). Prophylaxis is indicated to latent TB diseases if the purified protein derivative (tuberculin) skin test is higher than 5 mm before starting these treatments. The challenge in TB treatment is adverse drug reactions (ADRs) that are reported in 4% to 6% of cases.2,3
Erythema multiforme–like dermatitis is a rare skin rash that develops due to isoniazid (INH). The clinical presentation includes erythematoedematous lesions in an acral distribution with no mucosal involvement and systemic exposure to INH. Skin biopsy and patch tests are the supportive diagnostic methods. Isoniazid-associated skin rashes rarely are reported and generally are not severe enough to terminate the drug. We present a patient with psoriasis who received TB prophylaxis before anti–TNF-α use. He presented with erythema multiforme–like dermatitis due to INH. Withdrawal of the drug and treatment of the lesions were the first steps of intolerance, followed by a patch test with the culprit drug after recovery. We discuss the diagnostic drug allergy evaluation and treatment approach.
A 37-year-old man presented with a 15-year history of severe psoriasis with frequent flares. He was treated with various topical and systemic agents including acitretin and methotrexate at 4-year intervals. Despite the addition of phototherapy, he underwent a new treatment with anti–TNF-α, as the disease control with other treatments was insufficient. Before starting anti–TNF-α, preventive treatment against TB with INH (300 mg/d) was indicated with 20 mm of purified protein derivative. On approximately the 20th day of treatment, he developed pruritic erythema with desquamation and exfoliation localized to the hands and feet (Figure 1). Isoniazid was discontinued and a topical steroid was initiated. After 3 weeks, the skin lesions were completely improved and INH was reinitiated at the same dose with antihistamine prophylaxis (oral levocetirizine 5 mg/d). Seven days later, similar skin lesions presented that were more extensive on the arms and legs (Figure 2). Complete blood cell counts, renal and hepatic function tests, and hepatitis markers were within reference range in consultation with the allergy division. To distinguish the lesions from a psoriasis attack, a punch biopsy of the eruptive dermatitis showed erythema multiforme–like dermatitis including dermal edema and perivascular lymphocytic infiltration with no relation to psoriasis but consistent with a drug eruption. Isoniazid was discontinued, and the skin lesions resolved after 4 weeks of topical steroid and oral antihistamine use (Figure 3). There was no other drug use except INH, and a skin patch test with INH was positive at 72 hours (Figure 4). Skin tests with INH were done to 5 healthy lesions that were negative. Finally, TB prophylaxis was performed with rifampicin (10 mg/kg/d [600 mg/d]) for 4 months with no ADRs. The patient’s psoriasis lesions improved with anti–TNF-α that was initiated 1 month after starting TB prevention with rifampicin.
This case of erythema multiforme–like dermatitis was diagnosed with acral involvement, a positive patch test to INH, and lymphocytic inflammation in a skin biopsy. It was a drug-induced reaction, as skin lesions developed during INH intake and improved after drug withdrawal.
Isoniazid, also known as isonicotinylhydrazide, is an oral antibiotic used for the treatment of TB and other mycobacteria. Protective treatment against latent TB primarily is done with daily INH for 6 or 9 months; alternatively, INH may be taken weekly with rifapentine for 3 months or daily with rifampicin for 4 months. Daily rifampicin alone for 4 months also is an option. In general, these regimens have similar efficacy; however, in terms of safety, the rifampicin and rifapentine combination regimens have fewer hepatotoxicity events compared to the INH alone regimen, but there are more cutaneous and flulike reactions and gastrointestinal intolerance.4 Cutaneous ADRs to TB treatment such as mild itchiness and cutaneous eruptions usually are observed within 2 months of drug initiation. Pyrazinamide was reported as the most common drug associated with cutaneous ADRs, and INH was the rarest offending drug.5
The frequency of ADRs to INH is approximately 5.4%, and the most prevalent ADRs include asymptomatic elevation of serum liver enzyme concentrations, peripheral neuropathy, and hepatotoxicity, and skin lesions are less common.2 Our patient’s laboratory test results excluded vitamin B deficiency, hepatic and renal dysfunction, and neuropathy.
Previously reported skin reactions related to INH were late-type reactions such as maculopapular rash, dermatitis, erythema multiforme, drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, Stevens-Johnson syndrome, and toxic epidermal necrolysis.5,6 The concerning prediagnosis of psoriatic exacerbation in our patient was ruled out by the absence of typical skin lesions such as well-defined, erythematous plaques and pustules and atypical localization such as the dorsal hands and feet rather than the knees, elbows, lumbosacral region, scalp, and abdomen, which is typical of psoriasis. DRESS syndrome was unlikely with the absence of fever, lymphadenopathy, hypereosinophilia, leukocytosis, and renal and hepatic dysfunction.7 There were no widespread blisters, epidermal detachment, or mucosal involvement on the trunk or face typically associated with Stevens-Johnson syndrome and toxic epidermal necrolysis.7,8 A possible diagnosis of contact dermatitis was suspected with likely skin lesions as exfoliation and chapping, typical localization on the hands and feet, and positive patch test that supported sensitization to the drug. However, the patient’s skin lesions were not eczematous (characterized by erythema, vesiculation, exudation, or bullous edema in the acute phase), and were not localized to areas of irritant exposure.3 In our patient, erythematoedematous lesions in an acral distribution with no mucosal involvement and systemic exposure to INH was compatible with erythema multiforme, whereas the absence of target appearance, positive patch test, and late appearance were incompatible with erythema multiforme.8
Because the clinical picture did not fit contact dermatitis or erythema multiforme, a diagnosis of erythema multiforme–like noneczematous dermatitis was suggested. Noneczematous dermatitis has subtypes that include purpuric, lichenoid, pustular, lymphomatoid, dyshidrosiform, and pigmented, as well as erythema multiforme–like contact eruptions.9 These clinical entities are not associated with contact exposure, but are related to systemic exposure, as seen in our patient.10 The patch test positivity and skin biopsy report also supported the diagnosis of erythema multiforme–like dermatitis. Erythema multiforme–like dermatitis is thought to be caused by medications or infections inducing immunocomplexes and lymphocytic infiltration in the dermis and subepidermis. Nevertheless, the prognosis was self-limiting in both.8 The clinical polymorphism caused by INH in this patient was suggested to be related with individual susceptibility, variability of contact-activating modalities, and the targeted cutaneous structures. Furthermore, among the risk factors for cutaneous ADRs—HIV, polypharmacy, older age, and preexisting renal and liver impairment—the only notable factor in this patient was psoriasis as an autoimmune disorder.
Patients with skin diseases such as psoriasis should be followed up by closer monitoring during INH use. Withdrawal of the drug and symptomatic treatment of the lesions with corticosteroid and antihistamine are the first steps of drug intolerance. After complete recovery and termination of antiallergic drugs, diagnostic tests are recommended if the drug reaction was not life-threatening. Skin prick and intradermal tests are useful in early-type drug reactions, whereas patch testing and late evaluation of an intradermal test may be helpful in the diagnosis of delayed-type reactions. The full dose of INH is avoided in an intradermal test against irritation. A patch test with INH was performed by diluting a 100-mg tablet with 1 mL of distilled water, and used as 1/100, 1/10, and 1/1 dilutions.8 Patch testing with INH also was done in 5 healthy control patients to exclude the irritation effect in this case. The rechallenge of INH was done in a controlled manner in our patient to rule out psoriasis activation since it was a localized skin reaction with no serious ADR. An oral provocation test with the culprit drug is the gold standard of drug allergy diagnosis that should be done in a tertiary hospital with an intensive care unit.
This case of erythema multiforme–like dermatitis due to INH is interesting due to systemic intake of INH, which resulted in dermatitis with localized involvement similar to erythema multiforme but with no immunologic processes or prior sensitization. With the increasing use of anti–TNF-α treatment, INH use will be more prevalent than in the past for the treatment of latent TB. Even though the skin-restricted ADRs of INH are rare and minor, particular attention should be paid to patients with dermatologic diseases. In our case, diagnostic drug allergy evaluation was performed to optimize the second-line treatment of TB infection, in addition to early withdrawal of the culprit drug.
To the Editor:
Psoriasis vulgaris is a chronic autoimmune inflammatory disease and biologic agents, such as anti–tumor necrosis factor α (TNF-α), are alternative drugs in case of resistance or adverse events to conventional ones.1 The limitation of these agents is immunosuppression that may cause infections such as tuberculosis (TB). Prophylaxis is indicated to latent TB diseases if the purified protein derivative (tuberculin) skin test is higher than 5 mm before starting these treatments. The challenge in TB treatment is adverse drug reactions (ADRs) that are reported in 4% to 6% of cases.2,3
Erythema multiforme–like dermatitis is a rare skin rash that develops due to isoniazid (INH). The clinical presentation includes erythematoedematous lesions in an acral distribution with no mucosal involvement and systemic exposure to INH. Skin biopsy and patch tests are the supportive diagnostic methods. Isoniazid-associated skin rashes rarely are reported and generally are not severe enough to terminate the drug. We present a patient with psoriasis who received TB prophylaxis before anti–TNF-α use. He presented with erythema multiforme–like dermatitis due to INH. Withdrawal of the drug and treatment of the lesions were the first steps of intolerance, followed by a patch test with the culprit drug after recovery. We discuss the diagnostic drug allergy evaluation and treatment approach.
A 37-year-old man presented with a 15-year history of severe psoriasis with frequent flares. He was treated with various topical and systemic agents including acitretin and methotrexate at 4-year intervals. Despite the addition of phototherapy, he underwent a new treatment with anti–TNF-α, as the disease control with other treatments was insufficient. Before starting anti–TNF-α, preventive treatment against TB with INH (300 mg/d) was indicated with 20 mm of purified protein derivative. On approximately the 20th day of treatment, he developed pruritic erythema with desquamation and exfoliation localized to the hands and feet (Figure 1). Isoniazid was discontinued and a topical steroid was initiated. After 3 weeks, the skin lesions were completely improved and INH was reinitiated at the same dose with antihistamine prophylaxis (oral levocetirizine 5 mg/d). Seven days later, similar skin lesions presented that were more extensive on the arms and legs (Figure 2). Complete blood cell counts, renal and hepatic function tests, and hepatitis markers were within reference range in consultation with the allergy division. To distinguish the lesions from a psoriasis attack, a punch biopsy of the eruptive dermatitis showed erythema multiforme–like dermatitis including dermal edema and perivascular lymphocytic infiltration with no relation to psoriasis but consistent with a drug eruption. Isoniazid was discontinued, and the skin lesions resolved after 4 weeks of topical steroid and oral antihistamine use (Figure 3). There was no other drug use except INH, and a skin patch test with INH was positive at 72 hours (Figure 4). Skin tests with INH were done to 5 healthy lesions that were negative. Finally, TB prophylaxis was performed with rifampicin (10 mg/kg/d [600 mg/d]) for 4 months with no ADRs. The patient’s psoriasis lesions improved with anti–TNF-α that was initiated 1 month after starting TB prevention with rifampicin.
This case of erythema multiforme–like dermatitis was diagnosed with acral involvement, a positive patch test to INH, and lymphocytic inflammation in a skin biopsy. It was a drug-induced reaction, as skin lesions developed during INH intake and improved after drug withdrawal.
Isoniazid, also known as isonicotinylhydrazide, is an oral antibiotic used for the treatment of TB and other mycobacteria. Protective treatment against latent TB primarily is done with daily INH for 6 or 9 months; alternatively, INH may be taken weekly with rifapentine for 3 months or daily with rifampicin for 4 months. Daily rifampicin alone for 4 months also is an option. In general, these regimens have similar efficacy; however, in terms of safety, the rifampicin and rifapentine combination regimens have fewer hepatotoxicity events compared to the INH alone regimen, but there are more cutaneous and flulike reactions and gastrointestinal intolerance.4 Cutaneous ADRs to TB treatment such as mild itchiness and cutaneous eruptions usually are observed within 2 months of drug initiation. Pyrazinamide was reported as the most common drug associated with cutaneous ADRs, and INH was the rarest offending drug.5
The frequency of ADRs to INH is approximately 5.4%, and the most prevalent ADRs include asymptomatic elevation of serum liver enzyme concentrations, peripheral neuropathy, and hepatotoxicity, and skin lesions are less common.2 Our patient’s laboratory test results excluded vitamin B deficiency, hepatic and renal dysfunction, and neuropathy.
Previously reported skin reactions related to INH were late-type reactions such as maculopapular rash, dermatitis, erythema multiforme, drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, Stevens-Johnson syndrome, and toxic epidermal necrolysis.5,6 The concerning prediagnosis of psoriatic exacerbation in our patient was ruled out by the absence of typical skin lesions such as well-defined, erythematous plaques and pustules and atypical localization such as the dorsal hands and feet rather than the knees, elbows, lumbosacral region, scalp, and abdomen, which is typical of psoriasis. DRESS syndrome was unlikely with the absence of fever, lymphadenopathy, hypereosinophilia, leukocytosis, and renal and hepatic dysfunction.7 There were no widespread blisters, epidermal detachment, or mucosal involvement on the trunk or face typically associated with Stevens-Johnson syndrome and toxic epidermal necrolysis.7,8 A possible diagnosis of contact dermatitis was suspected with likely skin lesions as exfoliation and chapping, typical localization on the hands and feet, and positive patch test that supported sensitization to the drug. However, the patient’s skin lesions were not eczematous (characterized by erythema, vesiculation, exudation, or bullous edema in the acute phase), and were not localized to areas of irritant exposure.3 In our patient, erythematoedematous lesions in an acral distribution with no mucosal involvement and systemic exposure to INH was compatible with erythema multiforme, whereas the absence of target appearance, positive patch test, and late appearance were incompatible with erythema multiforme.8
Because the clinical picture did not fit contact dermatitis or erythema multiforme, a diagnosis of erythema multiforme–like noneczematous dermatitis was suggested. Noneczematous dermatitis has subtypes that include purpuric, lichenoid, pustular, lymphomatoid, dyshidrosiform, and pigmented, as well as erythema multiforme–like contact eruptions.9 These clinical entities are not associated with contact exposure, but are related to systemic exposure, as seen in our patient.10 The patch test positivity and skin biopsy report also supported the diagnosis of erythema multiforme–like dermatitis. Erythema multiforme–like dermatitis is thought to be caused by medications or infections inducing immunocomplexes and lymphocytic infiltration in the dermis and subepidermis. Nevertheless, the prognosis was self-limiting in both.8 The clinical polymorphism caused by INH in this patient was suggested to be related with individual susceptibility, variability of contact-activating modalities, and the targeted cutaneous structures. Furthermore, among the risk factors for cutaneous ADRs—HIV, polypharmacy, older age, and preexisting renal and liver impairment—the only notable factor in this patient was psoriasis as an autoimmune disorder.
Patients with skin diseases such as psoriasis should be followed up by closer monitoring during INH use. Withdrawal of the drug and symptomatic treatment of the lesions with corticosteroid and antihistamine are the first steps of drug intolerance. After complete recovery and termination of antiallergic drugs, diagnostic tests are recommended if the drug reaction was not life-threatening. Skin prick and intradermal tests are useful in early-type drug reactions, whereas patch testing and late evaluation of an intradermal test may be helpful in the diagnosis of delayed-type reactions. The full dose of INH is avoided in an intradermal test against irritation. A patch test with INH was performed by diluting a 100-mg tablet with 1 mL of distilled water, and used as 1/100, 1/10, and 1/1 dilutions.8 Patch testing with INH also was done in 5 healthy control patients to exclude the irritation effect in this case. The rechallenge of INH was done in a controlled manner in our patient to rule out psoriasis activation since it was a localized skin reaction with no serious ADR. An oral provocation test with the culprit drug is the gold standard of drug allergy diagnosis that should be done in a tertiary hospital with an intensive care unit.
This case of erythema multiforme–like dermatitis due to INH is interesting due to systemic intake of INH, which resulted in dermatitis with localized involvement similar to erythema multiforme but with no immunologic processes or prior sensitization. With the increasing use of anti–TNF-α treatment, INH use will be more prevalent than in the past for the treatment of latent TB. Even though the skin-restricted ADRs of INH are rare and minor, particular attention should be paid to patients with dermatologic diseases. In our case, diagnostic drug allergy evaluation was performed to optimize the second-line treatment of TB infection, in addition to early withdrawal of the culprit drug.
- Vide J, Magina S. Moderate to severe psoriasis treatment challenges through the era of biological drugs.An Bras Dermatol. 2017;92:668-674.
- Gülbay BE, Gürkan OU, Yildiz OA, et al. Side effects due to primary antituberculosis drugs during the initial phase of therapy in 1149 hospitalized patients for tuberculosis. Respir Med. 2006;100:1834-1842.
- Holdiness MR. Contact dermatitis to antituberculosis drugs. Contact Dermatitis. 1986;15:282-288.
- Getahun H, Matteelli A, Abubakar I, et al. Management of latent Mycobacterium tuberculosis infection: WHO guidelines for low tuberculosis burden countries. Eur Respir J. 2015;46:1563-1576.
- Tan WC, Ong CK, Kang SC, et al. Two years review of cutaneous adverse drug reaction from first line anti-tuberculous drugs. Med J Malaysia. 2007;62:143-146.
- Özkaya E.Eczematous-type multiple drug allergy from isoniazid and ethambutol with positive patch test results. Cutis. 2013;92:121-124.
- Fernando SL. Drug-reaction eosinophilia and systemic symptoms and drug-induced hypersensitivity syndrome. Australas J Dermatol. 2014;55:15-23.
- Rebollo S, Sanchez P, Vega JM, et al. Hypersensitivity syndrome from isoniazid with positive patch test. Contact Dermatitis. 2001;45:306.
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902.
- Bonamonte D, Foti C, Vestita M, et al. Nummular eczema and contact allergy: a retrospective study. Dermatitis. 2012;23:153-157.
- Vide J, Magina S. Moderate to severe psoriasis treatment challenges through the era of biological drugs.An Bras Dermatol. 2017;92:668-674.
- Gülbay BE, Gürkan OU, Yildiz OA, et al. Side effects due to primary antituberculosis drugs during the initial phase of therapy in 1149 hospitalized patients for tuberculosis. Respir Med. 2006;100:1834-1842.
- Holdiness MR. Contact dermatitis to antituberculosis drugs. Contact Dermatitis. 1986;15:282-288.
- Getahun H, Matteelli A, Abubakar I, et al. Management of latent Mycobacterium tuberculosis infection: WHO guidelines for low tuberculosis burden countries. Eur Respir J. 2015;46:1563-1576.
- Tan WC, Ong CK, Kang SC, et al. Two years review of cutaneous adverse drug reaction from first line anti-tuberculous drugs. Med J Malaysia. 2007;62:143-146.
- Özkaya E.Eczematous-type multiple drug allergy from isoniazid and ethambutol with positive patch test results. Cutis. 2013;92:121-124.
- Fernando SL. Drug-reaction eosinophilia and systemic symptoms and drug-induced hypersensitivity syndrome. Australas J Dermatol. 2014;55:15-23.
- Rebollo S, Sanchez P, Vega JM, et al. Hypersensitivity syndrome from isoniazid with positive patch test. Contact Dermatitis. 2001;45:306.
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902.
- Bonamonte D, Foti C, Vestita M, et al. Nummular eczema and contact allergy: a retrospective study. Dermatitis. 2012;23:153-157.
Practice Points
- Hypersensitivity skin reactions to antituberculosis (TB) drugs are on the rise due to the increasing use of anti–tumor necrosis factor α. Isoniazid (INH) use will be more prevalent than in the past for the treatment of latent TB.
- Even though the skin-restricted adverse events to INH are rare and minor, particular attention should be paid to patients with dermatologic diseases such as psoriasis.
Tinea Incognito Mimicking Pustular Psoriasis in a Patient With Psoriasis and Cushing Syndrome
To the Editor:
The term tinea incognito was introduced by Ive and Marks1 in 1968 and refers to unusual clinical presentations of tinea due to the application of topical corticosteroids. Tinea incognito, which does not feature the classical clinical characteristics of tinea corporis such as well-defined, erythematous, scaly patches and elevated borders, is regularly misdiagnosed as inflammatory dermatosis.2 Immunosuppression caused by topical and/or systemic steroids predisposes patients to the development of tinea.3 Herein, a case of widespread pustular tinea incognito mimicking pustular psoriasis along with failure of tumor necrosis factor (TNF) inhibitor treatment is reported in a patient with chronic plaque psoriasis and steroid-induced Cushing syndrome.
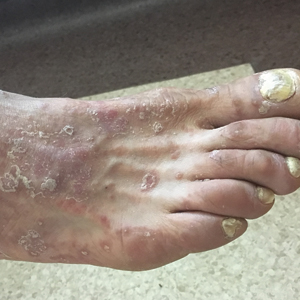
A 46-year-old man with a 25-year history of psoriasis was referred to the dermatologic outpatient clinic with a severe flare-up of chronic plaque psoriasis. Prior treatments included methotrexate and acitretin without response. Narrowband UVB treatment was discontinued due to claustrophobia. Topical treatment with calcipotriol 0.005%–betamethasone dipropionate 0.05% gel was reported to be ineffective. The patient was administered prednisone over several months in a primary care setting at a dosage of 35 mg daily when he presented to the dermatology clinic. Physical examination revealed widespread chronic plaque psoriasis of the trunk and extremities, and a psoriasis area and severity index score of 15 was calculated. The patient had onychodystrophy with subungual hyperkeratosis of all toenails. Signs of prednisone-induced Cushing syndrome, including central obesity, lipodystrophy, and red striae, were noted.
Treatment was started by dermatology with the TNF inhibitor adalimumab at an initial dose of 80 mg, followed by subsequent 40-mg doses every other week; prednisone was tapered off. Topical treatment with a 4-week course of clobetasol propionate cream 0.05% daily for psoriatic lesions was initiated.
Six weeks after the initial consultation, the patient presented to the hospital’s emergency department with worsening symptoms of itchy, burning, and painful skin after good initial improvement. The patient’s skin started to burn upon application of clobetasol and the rash worsened. The patient did not use emollients. At that point, the patient was on a daily dose of 15 mg of prednisone. On dermatologic review, multiple partially annular lesions with subtle scaling and multiple pustules on the arms and legs as well as the buttocks and groin were noticed. These lesions were confined to sites of prior psoriasis as marked by postinflammatory hyperpigmentation (Figure 1). Widespread tinea was assumed, and treatment with fluconazole 50 mg daily was administered for 4 weeks. Direct examination of skin scrapings from the patient’s thigh showed hyphae, and fungal culture was positive for Trichophyton rubrum. Scrapings from the patient’s hallux nail remained inconclusive due to bacterial overgrowth. At 4-week follow-up, the patient’s skin had cleared entirely and showed only postinflammatory changes (Figure 2). Healthy proximal nail growth was observed. Fluconazole was continued at a once-weekly dose of 150 mg together with adalimumab at a dose of 40 mg every 2 weeks and a prednisone tapering schedule.
This case describes pustular tinea incognito in a patient with chronic plaque psoriasis. As the name indicates, tinea incognito can mimic other skin conditions and classically is linked to topical application of corticosteroids.1 Tinea incognito can be a diagnostic challenge. Kim et al4 reported a diagnostic delay of 15 months and the frequent requirement for the involvement of a second physician or dermatologist. Treatment with topical or systemic corticosteroids is a risk factor for dermatophyte infections because of their immunosuppressive action.3,5 Although recommended by current guidelines, a large number of psoriatic patients are treated with systemic steroids, predominantly prescribed in primary care, that can lead to iatrogenic Cushing syndrome, as demonstrated in this patient.6
In addition to systemic and topical steroids, the reported patient was started on the TNF inhibitor adalimumab prior to the onset of the tinea. Cases of patients on TNF inhibitors with widespread tinea are scarce. Bardazzi et al7 reported 2 cases of widespread nonpustular tinea in patients with psoriasis on TNF inhibitor treatment without further immunomodulating treatment. They hypothesized that TNF-α could be an important cytokine in the defense against dermatophytes.7
Whether psoriasis itself is a risk factor for tinea is still under debate, but tinea pedum and onychomycosis seem to have higher prevalence among psoriatic patients.8,9 As in this patient, bacterial overgrowth of hyperkeratotic nail samples can confound the culture’s clinical significance, thereby hindering the diagnosis of onychomycosis in patients with psoriasis.10 Alteras et al8 hypothesized that autoinoculation from preexisting onychomycosis or tinea pedum was the underlying mechanism of tinea incognito.
This patient’s hyperkeratotic nails showed healthy regrowth after initiation of both fluconazole and adalimumab, though it remained unclear whether preexisting onychomycosis was a possible source of tinea incognito. The finding that the patient’s tinea was almost exclusively limited to the sites of prior psoriatic lesions argues for autoinoculation and spreading accelerated by application of topical steroids triggered by the immunosuppressive effects of both topical and systemic steroids. The TNF inhibitor treatment may have helped to unmask the dermatophyte infection rather than contributing to it, as it cleared the psoriatic plaques.
Apart from psoriasis, tinea incognito most commonly is mistaken for other inflammatory conditions such as eczema, folliculitis, rosacea, granuloma annulare, and discoid lupus erythematosus.2 Inflammatory tinea can present with pustules due to the increased occurrence of neutrophil invasion.11This patient’s symptoms worsened 4 weeks after the initiation of TNF inhibitor treatment, which suggested treatment failure. However, clearance of the preexisting psoriatic lesions with remnant hyperpigmentation only argued for good response to TNF inhibitor treatment. The main differential diagnosis of this case of tinea incognito was generalized pustular psoriasis. The patient also was being treated with systemic and topical steroids, both known for their potential to trigger pustular psoriasis.12,13 Furthermore, TNF inhibitors have been described as a trigger for predominantly palmoplantar pustulosis but also are additionally associated with generalized pustular psoriasis.14
This case aims to raise awareness that tinea incognito can imitate both pustular psoriasis and TNF inhibitor treatment failure. Furthermore, the presented findings highlight risks associated with the treatment of psoriasis with systemic steroids. Pustular tinea incognito should be considered in the differential diagnosis of pustular psoriasis, especially in the setting of immunosuppression. After initial improvement, worsening of symptoms such as itching and burning as well as extension of the lesions upon application of topical steroids are regularly described in tinea incognito and can be present in addition to the more typical annular presentation of lesions as a clue to the diagnosis.
- Ive FA, Marks R. Tinea incognito. Br Med J. 1968;3:149-152.
- Arenas R, Moreno-Coutiño G, Vera L, et al. Tinea incognito. Clin Dermatol. 2010;28:137-139.
- Rouzaud C, Chosidow O, Brocard A, et al. Severe dermatophytosis in solid organ transplant recipients: a French retrospective series and literature review [published online January 25, 2018]. Transpl Infect Dis. doi:10.1111/tid.12799
- Kim WJ, Kim TW, Mun JH, et al. Tinea incognito in Korea and its risk factors: nine-year multicenter survey. J Korean Med Sci. 2013;28:145-151.
- Ohta Y, Saitoh N, Tanuma H, et al. Local cytokine expression in steroid-modified tinea faciei. J Dermatol. 1998;25:362-366.
- Augustin M, Schäfer I, Reich K, et al. Systemic treatment with corticosteroids in psoriasis-health care provision far beyond the S3-guidelines. J Dtsch Dermatol Ges. 2011;9:833-838.
- Bardazzi F, Balestri R, Rech G, et al. Dermatophytosis during anti-TNF-α monoclonal antibody therapy. Mycoses. 2011;54:E619-E620.
- Alteras I, Ingberg A, Segal R, et al. The incidence of skin manifestations by dermatophytes in patients with psoriasis. Mycopathologia. 1986;95:37-39.
- Leibovici V, Ramot Y, Siam R, et al. Prevalence of tinea pedis in psoriasis, compared to atopic dermatitis and normal controls—a prospective study. Mycoses. 2014;57:754-758.
- Tsentemeidou A, Vyzantiadis TA, Kyriakou A, et al. Prevalence of onychomycosis amongst patients with nail psoriasis who are not receiving immunosuppressive agents: results of a pilot study. Mycoses. 2017;60:830-835.
- Hirschmann JV, Raugi GJ. Pustular tinea pedis. J Am Acad Dermatol. 2000;42:132-133.
- Brenner M, Molin S, Ruebsam K, et al. Generalized pustular psoriasis induced by systemic glucocorticosteroids: four cases and recommendations for treatment. Br J Dermatol. 2009;161:964-966.
- Boxley JD, Dawber RP, Summerly R. Generalized pustular psoriasis on withdrawal of clobetasol propionate ointment. Br Med J. 1975;2:255-256.
- Kucharekova M, Winnepenninckx V, Frank J, et al. Generalized pustulosis induced by adalimumab in a patient with rheumatoid arthritis—a therapeutic challenge. Int J Dermatol. 2008;47:25-28.
To the Editor:
The term tinea incognito was introduced by Ive and Marks1 in 1968 and refers to unusual clinical presentations of tinea due to the application of topical corticosteroids. Tinea incognito, which does not feature the classical clinical characteristics of tinea corporis such as well-defined, erythematous, scaly patches and elevated borders, is regularly misdiagnosed as inflammatory dermatosis.2 Immunosuppression caused by topical and/or systemic steroids predisposes patients to the development of tinea.3 Herein, a case of widespread pustular tinea incognito mimicking pustular psoriasis along with failure of tumor necrosis factor (TNF) inhibitor treatment is reported in a patient with chronic plaque psoriasis and steroid-induced Cushing syndrome.
A 46-year-old man with a 25-year history of psoriasis was referred to the dermatologic outpatient clinic with a severe flare-up of chronic plaque psoriasis. Prior treatments included methotrexate and acitretin without response. Narrowband UVB treatment was discontinued due to claustrophobia. Topical treatment with calcipotriol 0.005%–betamethasone dipropionate 0.05% gel was reported to be ineffective. The patient was administered prednisone over several months in a primary care setting at a dosage of 35 mg daily when he presented to the dermatology clinic. Physical examination revealed widespread chronic plaque psoriasis of the trunk and extremities, and a psoriasis area and severity index score of 15 was calculated. The patient had onychodystrophy with subungual hyperkeratosis of all toenails. Signs of prednisone-induced Cushing syndrome, including central obesity, lipodystrophy, and red striae, were noted.
Treatment was started by dermatology with the TNF inhibitor adalimumab at an initial dose of 80 mg, followed by subsequent 40-mg doses every other week; prednisone was tapered off. Topical treatment with a 4-week course of clobetasol propionate cream 0.05% daily for psoriatic lesions was initiated.
Six weeks after the initial consultation, the patient presented to the hospital’s emergency department with worsening symptoms of itchy, burning, and painful skin after good initial improvement. The patient’s skin started to burn upon application of clobetasol and the rash worsened. The patient did not use emollients. At that point, the patient was on a daily dose of 15 mg of prednisone. On dermatologic review, multiple partially annular lesions with subtle scaling and multiple pustules on the arms and legs as well as the buttocks and groin were noticed. These lesions were confined to sites of prior psoriasis as marked by postinflammatory hyperpigmentation (Figure 1). Widespread tinea was assumed, and treatment with fluconazole 50 mg daily was administered for 4 weeks. Direct examination of skin scrapings from the patient’s thigh showed hyphae, and fungal culture was positive for Trichophyton rubrum. Scrapings from the patient’s hallux nail remained inconclusive due to bacterial overgrowth. At 4-week follow-up, the patient’s skin had cleared entirely and showed only postinflammatory changes (Figure 2). Healthy proximal nail growth was observed. Fluconazole was continued at a once-weekly dose of 150 mg together with adalimumab at a dose of 40 mg every 2 weeks and a prednisone tapering schedule.
This case describes pustular tinea incognito in a patient with chronic plaque psoriasis. As the name indicates, tinea incognito can mimic other skin conditions and classically is linked to topical application of corticosteroids.1 Tinea incognito can be a diagnostic challenge. Kim et al4 reported a diagnostic delay of 15 months and the frequent requirement for the involvement of a second physician or dermatologist. Treatment with topical or systemic corticosteroids is a risk factor for dermatophyte infections because of their immunosuppressive action.3,5 Although recommended by current guidelines, a large number of psoriatic patients are treated with systemic steroids, predominantly prescribed in primary care, that can lead to iatrogenic Cushing syndrome, as demonstrated in this patient.6
In addition to systemic and topical steroids, the reported patient was started on the TNF inhibitor adalimumab prior to the onset of the tinea. Cases of patients on TNF inhibitors with widespread tinea are scarce. Bardazzi et al7 reported 2 cases of widespread nonpustular tinea in patients with psoriasis on TNF inhibitor treatment without further immunomodulating treatment. They hypothesized that TNF-α could be an important cytokine in the defense against dermatophytes.7
Whether psoriasis itself is a risk factor for tinea is still under debate, but tinea pedum and onychomycosis seem to have higher prevalence among psoriatic patients.8,9 As in this patient, bacterial overgrowth of hyperkeratotic nail samples can confound the culture’s clinical significance, thereby hindering the diagnosis of onychomycosis in patients with psoriasis.10 Alteras et al8 hypothesized that autoinoculation from preexisting onychomycosis or tinea pedum was the underlying mechanism of tinea incognito.
This patient’s hyperkeratotic nails showed healthy regrowth after initiation of both fluconazole and adalimumab, though it remained unclear whether preexisting onychomycosis was a possible source of tinea incognito. The finding that the patient’s tinea was almost exclusively limited to the sites of prior psoriatic lesions argues for autoinoculation and spreading accelerated by application of topical steroids triggered by the immunosuppressive effects of both topical and systemic steroids. The TNF inhibitor treatment may have helped to unmask the dermatophyte infection rather than contributing to it, as it cleared the psoriatic plaques.
Apart from psoriasis, tinea incognito most commonly is mistaken for other inflammatory conditions such as eczema, folliculitis, rosacea, granuloma annulare, and discoid lupus erythematosus.2 Inflammatory tinea can present with pustules due to the increased occurrence of neutrophil invasion.11This patient’s symptoms worsened 4 weeks after the initiation of TNF inhibitor treatment, which suggested treatment failure. However, clearance of the preexisting psoriatic lesions with remnant hyperpigmentation only argued for good response to TNF inhibitor treatment. The main differential diagnosis of this case of tinea incognito was generalized pustular psoriasis. The patient also was being treated with systemic and topical steroids, both known for their potential to trigger pustular psoriasis.12,13 Furthermore, TNF inhibitors have been described as a trigger for predominantly palmoplantar pustulosis but also are additionally associated with generalized pustular psoriasis.14
This case aims to raise awareness that tinea incognito can imitate both pustular psoriasis and TNF inhibitor treatment failure. Furthermore, the presented findings highlight risks associated with the treatment of psoriasis with systemic steroids. Pustular tinea incognito should be considered in the differential diagnosis of pustular psoriasis, especially in the setting of immunosuppression. After initial improvement, worsening of symptoms such as itching and burning as well as extension of the lesions upon application of topical steroids are regularly described in tinea incognito and can be present in addition to the more typical annular presentation of lesions as a clue to the diagnosis.
To the Editor:
The term tinea incognito was introduced by Ive and Marks1 in 1968 and refers to unusual clinical presentations of tinea due to the application of topical corticosteroids. Tinea incognito, which does not feature the classical clinical characteristics of tinea corporis such as well-defined, erythematous, scaly patches and elevated borders, is regularly misdiagnosed as inflammatory dermatosis.2 Immunosuppression caused by topical and/or systemic steroids predisposes patients to the development of tinea.3 Herein, a case of widespread pustular tinea incognito mimicking pustular psoriasis along with failure of tumor necrosis factor (TNF) inhibitor treatment is reported in a patient with chronic plaque psoriasis and steroid-induced Cushing syndrome.
A 46-year-old man with a 25-year history of psoriasis was referred to the dermatologic outpatient clinic with a severe flare-up of chronic plaque psoriasis. Prior treatments included methotrexate and acitretin without response. Narrowband UVB treatment was discontinued due to claustrophobia. Topical treatment with calcipotriol 0.005%–betamethasone dipropionate 0.05% gel was reported to be ineffective. The patient was administered prednisone over several months in a primary care setting at a dosage of 35 mg daily when he presented to the dermatology clinic. Physical examination revealed widespread chronic plaque psoriasis of the trunk and extremities, and a psoriasis area and severity index score of 15 was calculated. The patient had onychodystrophy with subungual hyperkeratosis of all toenails. Signs of prednisone-induced Cushing syndrome, including central obesity, lipodystrophy, and red striae, were noted.
Treatment was started by dermatology with the TNF inhibitor adalimumab at an initial dose of 80 mg, followed by subsequent 40-mg doses every other week; prednisone was tapered off. Topical treatment with a 4-week course of clobetasol propionate cream 0.05% daily for psoriatic lesions was initiated.
Six weeks after the initial consultation, the patient presented to the hospital’s emergency department with worsening symptoms of itchy, burning, and painful skin after good initial improvement. The patient’s skin started to burn upon application of clobetasol and the rash worsened. The patient did not use emollients. At that point, the patient was on a daily dose of 15 mg of prednisone. On dermatologic review, multiple partially annular lesions with subtle scaling and multiple pustules on the arms and legs as well as the buttocks and groin were noticed. These lesions were confined to sites of prior psoriasis as marked by postinflammatory hyperpigmentation (Figure 1). Widespread tinea was assumed, and treatment with fluconazole 50 mg daily was administered for 4 weeks. Direct examination of skin scrapings from the patient’s thigh showed hyphae, and fungal culture was positive for Trichophyton rubrum. Scrapings from the patient’s hallux nail remained inconclusive due to bacterial overgrowth. At 4-week follow-up, the patient’s skin had cleared entirely and showed only postinflammatory changes (Figure 2). Healthy proximal nail growth was observed. Fluconazole was continued at a once-weekly dose of 150 mg together with adalimumab at a dose of 40 mg every 2 weeks and a prednisone tapering schedule.
This case describes pustular tinea incognito in a patient with chronic plaque psoriasis. As the name indicates, tinea incognito can mimic other skin conditions and classically is linked to topical application of corticosteroids.1 Tinea incognito can be a diagnostic challenge. Kim et al4 reported a diagnostic delay of 15 months and the frequent requirement for the involvement of a second physician or dermatologist. Treatment with topical or systemic corticosteroids is a risk factor for dermatophyte infections because of their immunosuppressive action.3,5 Although recommended by current guidelines, a large number of psoriatic patients are treated with systemic steroids, predominantly prescribed in primary care, that can lead to iatrogenic Cushing syndrome, as demonstrated in this patient.6
In addition to systemic and topical steroids, the reported patient was started on the TNF inhibitor adalimumab prior to the onset of the tinea. Cases of patients on TNF inhibitors with widespread tinea are scarce. Bardazzi et al7 reported 2 cases of widespread nonpustular tinea in patients with psoriasis on TNF inhibitor treatment without further immunomodulating treatment. They hypothesized that TNF-α could be an important cytokine in the defense against dermatophytes.7
Whether psoriasis itself is a risk factor for tinea is still under debate, but tinea pedum and onychomycosis seem to have higher prevalence among psoriatic patients.8,9 As in this patient, bacterial overgrowth of hyperkeratotic nail samples can confound the culture’s clinical significance, thereby hindering the diagnosis of onychomycosis in patients with psoriasis.10 Alteras et al8 hypothesized that autoinoculation from preexisting onychomycosis or tinea pedum was the underlying mechanism of tinea incognito.
This patient’s hyperkeratotic nails showed healthy regrowth after initiation of both fluconazole and adalimumab, though it remained unclear whether preexisting onychomycosis was a possible source of tinea incognito. The finding that the patient’s tinea was almost exclusively limited to the sites of prior psoriatic lesions argues for autoinoculation and spreading accelerated by application of topical steroids triggered by the immunosuppressive effects of both topical and systemic steroids. The TNF inhibitor treatment may have helped to unmask the dermatophyte infection rather than contributing to it, as it cleared the psoriatic plaques.
Apart from psoriasis, tinea incognito most commonly is mistaken for other inflammatory conditions such as eczema, folliculitis, rosacea, granuloma annulare, and discoid lupus erythematosus.2 Inflammatory tinea can present with pustules due to the increased occurrence of neutrophil invasion.11This patient’s symptoms worsened 4 weeks after the initiation of TNF inhibitor treatment, which suggested treatment failure. However, clearance of the preexisting psoriatic lesions with remnant hyperpigmentation only argued for good response to TNF inhibitor treatment. The main differential diagnosis of this case of tinea incognito was generalized pustular psoriasis. The patient also was being treated with systemic and topical steroids, both known for their potential to trigger pustular psoriasis.12,13 Furthermore, TNF inhibitors have been described as a trigger for predominantly palmoplantar pustulosis but also are additionally associated with generalized pustular psoriasis.14
This case aims to raise awareness that tinea incognito can imitate both pustular psoriasis and TNF inhibitor treatment failure. Furthermore, the presented findings highlight risks associated with the treatment of psoriasis with systemic steroids. Pustular tinea incognito should be considered in the differential diagnosis of pustular psoriasis, especially in the setting of immunosuppression. After initial improvement, worsening of symptoms such as itching and burning as well as extension of the lesions upon application of topical steroids are regularly described in tinea incognito and can be present in addition to the more typical annular presentation of lesions as a clue to the diagnosis.
- Ive FA, Marks R. Tinea incognito. Br Med J. 1968;3:149-152.
- Arenas R, Moreno-Coutiño G, Vera L, et al. Tinea incognito. Clin Dermatol. 2010;28:137-139.
- Rouzaud C, Chosidow O, Brocard A, et al. Severe dermatophytosis in solid organ transplant recipients: a French retrospective series and literature review [published online January 25, 2018]. Transpl Infect Dis. doi:10.1111/tid.12799
- Kim WJ, Kim TW, Mun JH, et al. Tinea incognito in Korea and its risk factors: nine-year multicenter survey. J Korean Med Sci. 2013;28:145-151.
- Ohta Y, Saitoh N, Tanuma H, et al. Local cytokine expression in steroid-modified tinea faciei. J Dermatol. 1998;25:362-366.
- Augustin M, Schäfer I, Reich K, et al. Systemic treatment with corticosteroids in psoriasis-health care provision far beyond the S3-guidelines. J Dtsch Dermatol Ges. 2011;9:833-838.
- Bardazzi F, Balestri R, Rech G, et al. Dermatophytosis during anti-TNF-α monoclonal antibody therapy. Mycoses. 2011;54:E619-E620.
- Alteras I, Ingberg A, Segal R, et al. The incidence of skin manifestations by dermatophytes in patients with psoriasis. Mycopathologia. 1986;95:37-39.
- Leibovici V, Ramot Y, Siam R, et al. Prevalence of tinea pedis in psoriasis, compared to atopic dermatitis and normal controls—a prospective study. Mycoses. 2014;57:754-758.
- Tsentemeidou A, Vyzantiadis TA, Kyriakou A, et al. Prevalence of onychomycosis amongst patients with nail psoriasis who are not receiving immunosuppressive agents: results of a pilot study. Mycoses. 2017;60:830-835.
- Hirschmann JV, Raugi GJ. Pustular tinea pedis. J Am Acad Dermatol. 2000;42:132-133.
- Brenner M, Molin S, Ruebsam K, et al. Generalized pustular psoriasis induced by systemic glucocorticosteroids: four cases and recommendations for treatment. Br J Dermatol. 2009;161:964-966.
- Boxley JD, Dawber RP, Summerly R. Generalized pustular psoriasis on withdrawal of clobetasol propionate ointment. Br Med J. 1975;2:255-256.
- Kucharekova M, Winnepenninckx V, Frank J, et al. Generalized pustulosis induced by adalimumab in a patient with rheumatoid arthritis—a therapeutic challenge. Int J Dermatol. 2008;47:25-28.
- Ive FA, Marks R. Tinea incognito. Br Med J. 1968;3:149-152.
- Arenas R, Moreno-Coutiño G, Vera L, et al. Tinea incognito. Clin Dermatol. 2010;28:137-139.
- Rouzaud C, Chosidow O, Brocard A, et al. Severe dermatophytosis in solid organ transplant recipients: a French retrospective series and literature review [published online January 25, 2018]. Transpl Infect Dis. doi:10.1111/tid.12799
- Kim WJ, Kim TW, Mun JH, et al. Tinea incognito in Korea and its risk factors: nine-year multicenter survey. J Korean Med Sci. 2013;28:145-151.
- Ohta Y, Saitoh N, Tanuma H, et al. Local cytokine expression in steroid-modified tinea faciei. J Dermatol. 1998;25:362-366.
- Augustin M, Schäfer I, Reich K, et al. Systemic treatment with corticosteroids in psoriasis-health care provision far beyond the S3-guidelines. J Dtsch Dermatol Ges. 2011;9:833-838.
- Bardazzi F, Balestri R, Rech G, et al. Dermatophytosis during anti-TNF-α monoclonal antibody therapy. Mycoses. 2011;54:E619-E620.
- Alteras I, Ingberg A, Segal R, et al. The incidence of skin manifestations by dermatophytes in patients with psoriasis. Mycopathologia. 1986;95:37-39.
- Leibovici V, Ramot Y, Siam R, et al. Prevalence of tinea pedis in psoriasis, compared to atopic dermatitis and normal controls—a prospective study. Mycoses. 2014;57:754-758.
- Tsentemeidou A, Vyzantiadis TA, Kyriakou A, et al. Prevalence of onychomycosis amongst patients with nail psoriasis who are not receiving immunosuppressive agents: results of a pilot study. Mycoses. 2017;60:830-835.
- Hirschmann JV, Raugi GJ. Pustular tinea pedis. J Am Acad Dermatol. 2000;42:132-133.
- Brenner M, Molin S, Ruebsam K, et al. Generalized pustular psoriasis induced by systemic glucocorticosteroids: four cases and recommendations for treatment. Br J Dermatol. 2009;161:964-966.
- Boxley JD, Dawber RP, Summerly R. Generalized pustular psoriasis on withdrawal of clobetasol propionate ointment. Br Med J. 1975;2:255-256.
- Kucharekova M, Winnepenninckx V, Frank J, et al. Generalized pustulosis induced by adalimumab in a patient with rheumatoid arthritis—a therapeutic challenge. Int J Dermatol. 2008;47:25-28.
Practice Points
- Tinea incognito and its altered clinical presentation can provide clinical challenges and often is diagnosed with delay.
- Immunosuppression, such as iatrogenic Cushing syndrome, is a risk factor for tinea incognito.
- Pustular tinea incognito is a differential diagnosis of pustular psoriasis that can mimic tumor necrosis factor inhibitor treatment failure in patients with psoriasis.
Squamous Cell Carcinoma in Hidradenitis Suppurativa Lesions Following Tumor Necrosis Factor α Inhibitors
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition with high morbidity rates. Symptoms typically develop between puberty and the third decade of life, affecting twice as many females as males, with an overall disease prevalence of 1% to 4%.1 The pathogenesis is theorized to be related to an immune response to follicular occlusion and rupture in genetically susceptible individuals.
Among the complications associated with HS, the development of cutaneous squamous cell carcinoma (SCC) is 4.6-times more likely within HS lesions than in normal skin and typically is seen in the setting of long-standing disease, particularly in men with HS lesions located on the buttocks and genital region for more than 20 years.2 In 2015, the tumor necrosis factor (TNF) inhibitor adalimumab was approved by the US Food and Drug Administration for the treatment of HS. Tumor necrosis factor α inhibitors have been associated with an increased risk for skin cancer in other clinical settings.3,4 We present a case of locally advanced SCC that developed in a patient with HS who was treated with adalimumab and infliximab (both TNF-α inhibitors), ultimately leading to the patient’s death.
A 59-year-old man who smoked with a 40-year history of severe HS, who previously was lost to follow-up, presented to our dermatology clinic with lesions on the buttocks. Physical examination demonstrated confluent, indurated, boggy plaques; scattered sinus tracts with purulent drainage; scattered cystlike nodules; and tenderness to palpation consistent with Hurley stage III disease (Figure 1A). No involvement of the axillae or groin was noted. He was started on doxycycline and a prednisone taper with minimal improvement and subsequently was switched to adalimumab 3 months later. Adalimumab provided little relief and was discontinued; therapy was transitioned to infliximab 3 months later.
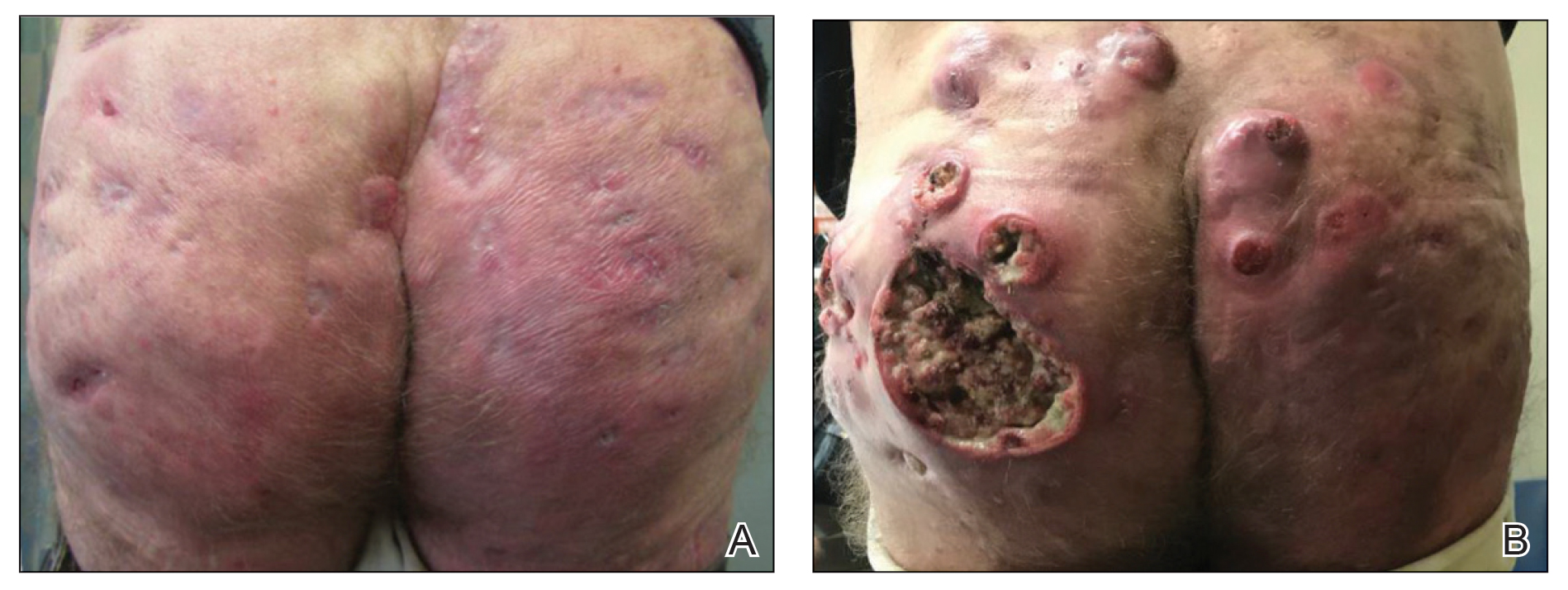
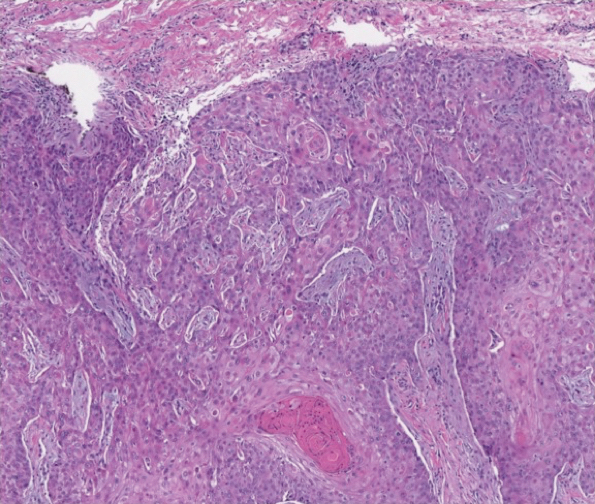
The patient returned to our clinic 3 months later with a severe flare and intractable pain after 4 infusions of infliximab. Physical examination showed a 7×5-cm deep malodorous ulcer with fibrinous exudate on the left buttock, several 2- to 3-cm shallow ulcers draining yellow exudate, and numerous fluctuant subcutaneous nodules on a background of scarring and sinus tracts. He was started again on doxycycline and a prednisone taper. At follow-up 2 weeks later, the largest ulcer had increased to 8 cm, and more indurated and tender subcutaneous nodules and scattered ulcerations developed (Figure 1B). Two punch biopsies of the left buttock revealed an invasive keratinizing carcinoma with no connection to the epidermis, consistent with SCC (Figure 2). Human papillomavirus (HPV) test results with probes for 37 HPV types—13 that were high risk (HPV-16, −18, −31, −33, −35, −39, −45, −51, −52, −56, −58, −59, −68)—were negative. Computerized tomography demonstrated diffuse thickening of the skin on the buttocks, inguinal adenopathy suspicious for nodal metastases, and no evidence of distant metastatic disease. Given the extent of the disease, surgical treatment was not an option, and he began receiving palliative radiotherapy. However, his health declined, and he developed aspiration pneumonia and hypotension requiring pressor support. He was transitioned to hospice care and died 3 months after presentation.
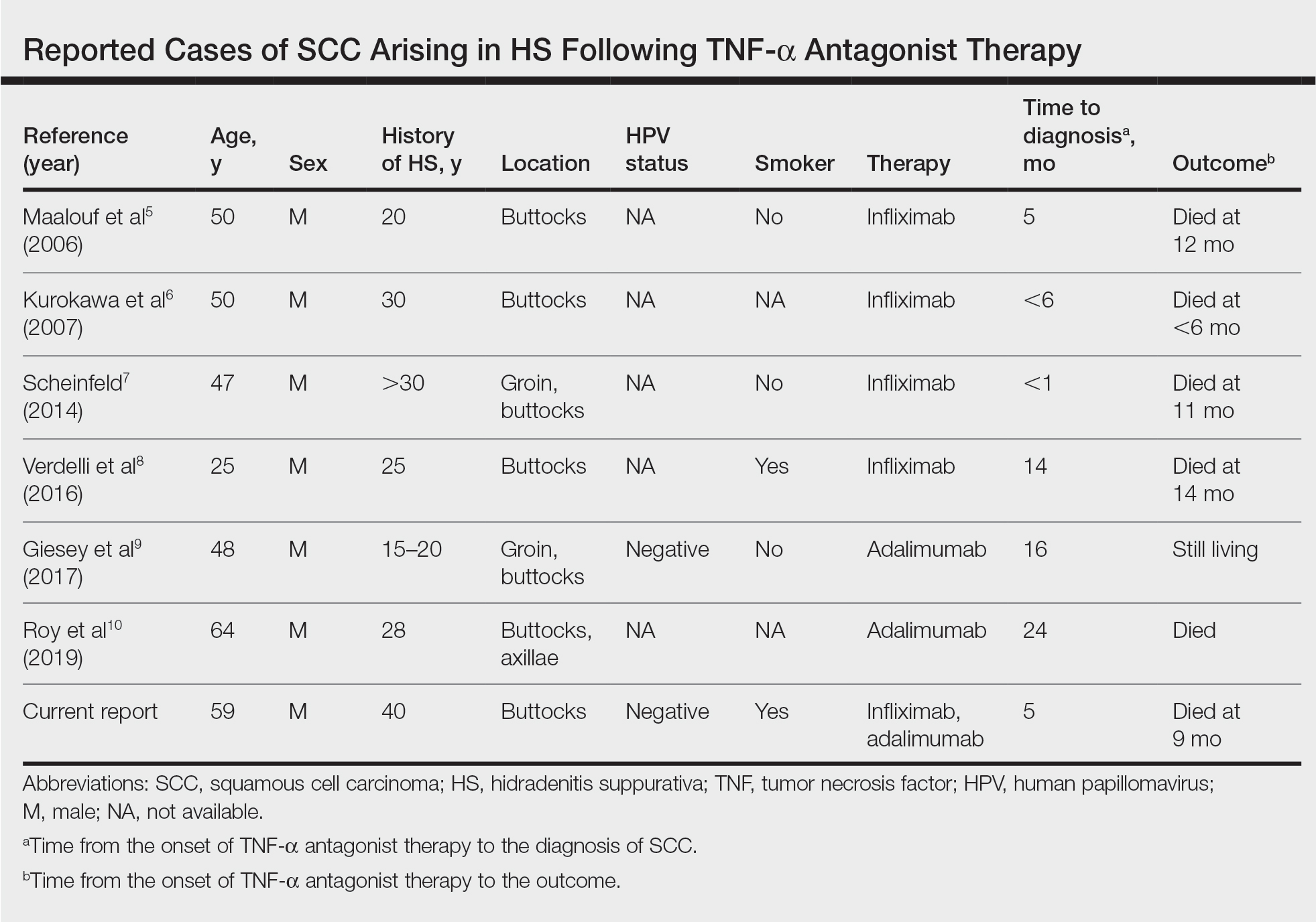
Tumor necrosis factor α antagonist treatment is being increasingly used to control HS but also may increase the risk for SCC development. We performed a search of PubMed articles indexed for MEDLINE as well as Web of Science using the terms hidradenitis suppurativa or acne inversa and one of the following—tumor necrosis factor inhibitor, infliximab, adalimumab, or etanercept—and squamous cell carcinoma or Marjolin ulcer. Seven cases of SCC arising in an HS patient treated with a TNF-α inhibitor have been reported (Table).5-10 Four cases were associated with infliximab use, 2 with adalimumab, and our case occurred after both adalimumab and infliximab treatment. All individuals were men with severe, long-standing disease of the anogenital region. In addition to smoking, HPV-16 positivity also has been reported as a risk factor for developing SCC in the setting of HS.11 In our patient, however, HPV testing did not cover all HPV strains, but several high-risk strains, including HPV-16, were negative.
Hidradenitis suppurativa is caused by an immune response to ruptured follicles and TNF-α antagonists are useful in suppressing this response; however, immunosuppression can lead to an increased susceptibility to malignancy, especially in SCC. It is unclear whether the use of infliximab or adalimumab is causal, additive, or a confounder in the development of SCC in patients with severe HS. It is possible that these agents increase the rapidity of the development of SCC in already-susceptible patients. Although TNF-α antagonists can be an effective therapeutic option for patients with moderate to severe HS, the potential risk for contributing to skin cancer development should raise provider suspicion in high-risk patients. Given the findings in this report, it may be suitable for providers to consider a biopsy prior to initiating TNF-α therapy in men older than 20 years with moderate to severe HS of the groin or buttocks, in addition to more frequent monitoring and a lower threshold to biopsy lesions with rapid growth or ulceration.
- Alikhan A, Lynch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-561; quiz 562-533.
- Lapins J, Ye W, Nyren O, et al. Incidence of cancer among patients with hidradenitis suppurativa. Arch Dermatol. 2001;137:730-734.
- Askling J, Fahrbach K, Nordstrom B, et al. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol Drug Saf. 2011;20:119-130.
- Mariette X, Matucci-Cerinic M, Pavelka K, et al. Malignancies associated with tumour necrosis factor inhibitors in registries and prospective observational studies: a systematic review and meta-analysis. Ann Rheum Dis. 2011;70:1895-1904.
- Maalouf E, Faye O, Poli F, et al. Fatal epidermoid carcinoma in hidradenitis suppurativa following treatment with infliximab. Ann Dermatol Venereol. 2006;133(5 pt 1):473-474.
- Kurokawa I, Nishimura K, Yamanaka K, et al. Cytokeratin expression in squamous cell carcinoma arising from hidradenitis suppurativa (acne inversa). J Cutan Pathol. 2007;34:675-678.
- Scheinfeld N. A case of a patient with stage III familial hidradenitis suppurativa treated with 3 courses of infliximab and died of metastatic squamous cell carcinoma. Dermatol Online J. 2014;20(3).
- Verdelli A, Antiga E, Bonciani D, et al. A fatal case of hidradenitis suppurativa associated with sepsis and squamous cell carcinoma. Int J Dermatol. 2016;55:E52-E53.
- Giesey R, Delost GR, Honaker J, et al. Metastatic squamous cell carcinoma in a patient treated with adalimumab for hidradenitis suppurativa. JAAD Case Rep. 2017;3:489-491.
- Roy C, Roy S, Ghazawi F, et al. Cutaneous squamous cell carcinoma arising in hidradenitis suppurativa: a case report. SAGE Open Med Case Rep. 2019;7:2050313X19847359.
- Lavogiez C, Delaporte E, Darras-Vercambre S, et al. Clinicopathological study of 13 cases of squamous cell carcinoma complicating hidradenitis suppurativa. Dermatology. 2010;220:147-153.
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition with high morbidity rates. Symptoms typically develop between puberty and the third decade of life, affecting twice as many females as males, with an overall disease prevalence of 1% to 4%.1 The pathogenesis is theorized to be related to an immune response to follicular occlusion and rupture in genetically susceptible individuals.
Among the complications associated with HS, the development of cutaneous squamous cell carcinoma (SCC) is 4.6-times more likely within HS lesions than in normal skin and typically is seen in the setting of long-standing disease, particularly in men with HS lesions located on the buttocks and genital region for more than 20 years.2 In 2015, the tumor necrosis factor (TNF) inhibitor adalimumab was approved by the US Food and Drug Administration for the treatment of HS. Tumor necrosis factor α inhibitors have been associated with an increased risk for skin cancer in other clinical settings.3,4 We present a case of locally advanced SCC that developed in a patient with HS who was treated with adalimumab and infliximab (both TNF-α inhibitors), ultimately leading to the patient’s death.
A 59-year-old man who smoked with a 40-year history of severe HS, who previously was lost to follow-up, presented to our dermatology clinic with lesions on the buttocks. Physical examination demonstrated confluent, indurated, boggy plaques; scattered sinus tracts with purulent drainage; scattered cystlike nodules; and tenderness to palpation consistent with Hurley stage III disease (Figure 1A). No involvement of the axillae or groin was noted. He was started on doxycycline and a prednisone taper with minimal improvement and subsequently was switched to adalimumab 3 months later. Adalimumab provided little relief and was discontinued; therapy was transitioned to infliximab 3 months later.
The patient returned to our clinic 3 months later with a severe flare and intractable pain after 4 infusions of infliximab. Physical examination showed a 7×5-cm deep malodorous ulcer with fibrinous exudate on the left buttock, several 2- to 3-cm shallow ulcers draining yellow exudate, and numerous fluctuant subcutaneous nodules on a background of scarring and sinus tracts. He was started again on doxycycline and a prednisone taper. At follow-up 2 weeks later, the largest ulcer had increased to 8 cm, and more indurated and tender subcutaneous nodules and scattered ulcerations developed (Figure 1B). Two punch biopsies of the left buttock revealed an invasive keratinizing carcinoma with no connection to the epidermis, consistent with SCC (Figure 2). Human papillomavirus (HPV) test results with probes for 37 HPV types—13 that were high risk (HPV-16, −18, −31, −33, −35, −39, −45, −51, −52, −56, −58, −59, −68)—were negative. Computerized tomography demonstrated diffuse thickening of the skin on the buttocks, inguinal adenopathy suspicious for nodal metastases, and no evidence of distant metastatic disease. Given the extent of the disease, surgical treatment was not an option, and he began receiving palliative radiotherapy. However, his health declined, and he developed aspiration pneumonia and hypotension requiring pressor support. He was transitioned to hospice care and died 3 months after presentation.
Tumor necrosis factor α antagonist treatment is being increasingly used to control HS but also may increase the risk for SCC development. We performed a search of PubMed articles indexed for MEDLINE as well as Web of Science using the terms hidradenitis suppurativa or acne inversa and one of the following—tumor necrosis factor inhibitor, infliximab, adalimumab, or etanercept—and squamous cell carcinoma or Marjolin ulcer. Seven cases of SCC arising in an HS patient treated with a TNF-α inhibitor have been reported (Table).5-10 Four cases were associated with infliximab use, 2 with adalimumab, and our case occurred after both adalimumab and infliximab treatment. All individuals were men with severe, long-standing disease of the anogenital region. In addition to smoking, HPV-16 positivity also has been reported as a risk factor for developing SCC in the setting of HS.11 In our patient, however, HPV testing did not cover all HPV strains, but several high-risk strains, including HPV-16, were negative.
Hidradenitis suppurativa is caused by an immune response to ruptured follicles and TNF-α antagonists are useful in suppressing this response; however, immunosuppression can lead to an increased susceptibility to malignancy, especially in SCC. It is unclear whether the use of infliximab or adalimumab is causal, additive, or a confounder in the development of SCC in patients with severe HS. It is possible that these agents increase the rapidity of the development of SCC in already-susceptible patients. Although TNF-α antagonists can be an effective therapeutic option for patients with moderate to severe HS, the potential risk for contributing to skin cancer development should raise provider suspicion in high-risk patients. Given the findings in this report, it may be suitable for providers to consider a biopsy prior to initiating TNF-α therapy in men older than 20 years with moderate to severe HS of the groin or buttocks, in addition to more frequent monitoring and a lower threshold to biopsy lesions with rapid growth or ulceration.
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition with high morbidity rates. Symptoms typically develop between puberty and the third decade of life, affecting twice as many females as males, with an overall disease prevalence of 1% to 4%.1 The pathogenesis is theorized to be related to an immune response to follicular occlusion and rupture in genetically susceptible individuals.
Among the complications associated with HS, the development of cutaneous squamous cell carcinoma (SCC) is 4.6-times more likely within HS lesions than in normal skin and typically is seen in the setting of long-standing disease, particularly in men with HS lesions located on the buttocks and genital region for more than 20 years.2 In 2015, the tumor necrosis factor (TNF) inhibitor adalimumab was approved by the US Food and Drug Administration for the treatment of HS. Tumor necrosis factor α inhibitors have been associated with an increased risk for skin cancer in other clinical settings.3,4 We present a case of locally advanced SCC that developed in a patient with HS who was treated with adalimumab and infliximab (both TNF-α inhibitors), ultimately leading to the patient’s death.
A 59-year-old man who smoked with a 40-year history of severe HS, who previously was lost to follow-up, presented to our dermatology clinic with lesions on the buttocks. Physical examination demonstrated confluent, indurated, boggy plaques; scattered sinus tracts with purulent drainage; scattered cystlike nodules; and tenderness to palpation consistent with Hurley stage III disease (Figure 1A). No involvement of the axillae or groin was noted. He was started on doxycycline and a prednisone taper with minimal improvement and subsequently was switched to adalimumab 3 months later. Adalimumab provided little relief and was discontinued; therapy was transitioned to infliximab 3 months later.
The patient returned to our clinic 3 months later with a severe flare and intractable pain after 4 infusions of infliximab. Physical examination showed a 7×5-cm deep malodorous ulcer with fibrinous exudate on the left buttock, several 2- to 3-cm shallow ulcers draining yellow exudate, and numerous fluctuant subcutaneous nodules on a background of scarring and sinus tracts. He was started again on doxycycline and a prednisone taper. At follow-up 2 weeks later, the largest ulcer had increased to 8 cm, and more indurated and tender subcutaneous nodules and scattered ulcerations developed (Figure 1B). Two punch biopsies of the left buttock revealed an invasive keratinizing carcinoma with no connection to the epidermis, consistent with SCC (Figure 2). Human papillomavirus (HPV) test results with probes for 37 HPV types—13 that were high risk (HPV-16, −18, −31, −33, −35, −39, −45, −51, −52, −56, −58, −59, −68)—were negative. Computerized tomography demonstrated diffuse thickening of the skin on the buttocks, inguinal adenopathy suspicious for nodal metastases, and no evidence of distant metastatic disease. Given the extent of the disease, surgical treatment was not an option, and he began receiving palliative radiotherapy. However, his health declined, and he developed aspiration pneumonia and hypotension requiring pressor support. He was transitioned to hospice care and died 3 months after presentation.
Tumor necrosis factor α antagonist treatment is being increasingly used to control HS but also may increase the risk for SCC development. We performed a search of PubMed articles indexed for MEDLINE as well as Web of Science using the terms hidradenitis suppurativa or acne inversa and one of the following—tumor necrosis factor inhibitor, infliximab, adalimumab, or etanercept—and squamous cell carcinoma or Marjolin ulcer. Seven cases of SCC arising in an HS patient treated with a TNF-α inhibitor have been reported (Table).5-10 Four cases were associated with infliximab use, 2 with adalimumab, and our case occurred after both adalimumab and infliximab treatment. All individuals were men with severe, long-standing disease of the anogenital region. In addition to smoking, HPV-16 positivity also has been reported as a risk factor for developing SCC in the setting of HS.11 In our patient, however, HPV testing did not cover all HPV strains, but several high-risk strains, including HPV-16, were negative.
Hidradenitis suppurativa is caused by an immune response to ruptured follicles and TNF-α antagonists are useful in suppressing this response; however, immunosuppression can lead to an increased susceptibility to malignancy, especially in SCC. It is unclear whether the use of infliximab or adalimumab is causal, additive, or a confounder in the development of SCC in patients with severe HS. It is possible that these agents increase the rapidity of the development of SCC in already-susceptible patients. Although TNF-α antagonists can be an effective therapeutic option for patients with moderate to severe HS, the potential risk for contributing to skin cancer development should raise provider suspicion in high-risk patients. Given the findings in this report, it may be suitable for providers to consider a biopsy prior to initiating TNF-α therapy in men older than 20 years with moderate to severe HS of the groin or buttocks, in addition to more frequent monitoring and a lower threshold to biopsy lesions with rapid growth or ulceration.
- Alikhan A, Lynch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-561; quiz 562-533.
- Lapins J, Ye W, Nyren O, et al. Incidence of cancer among patients with hidradenitis suppurativa. Arch Dermatol. 2001;137:730-734.
- Askling J, Fahrbach K, Nordstrom B, et al. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol Drug Saf. 2011;20:119-130.
- Mariette X, Matucci-Cerinic M, Pavelka K, et al. Malignancies associated with tumour necrosis factor inhibitors in registries and prospective observational studies: a systematic review and meta-analysis. Ann Rheum Dis. 2011;70:1895-1904.
- Maalouf E, Faye O, Poli F, et al. Fatal epidermoid carcinoma in hidradenitis suppurativa following treatment with infliximab. Ann Dermatol Venereol. 2006;133(5 pt 1):473-474.
- Kurokawa I, Nishimura K, Yamanaka K, et al. Cytokeratin expression in squamous cell carcinoma arising from hidradenitis suppurativa (acne inversa). J Cutan Pathol. 2007;34:675-678.
- Scheinfeld N. A case of a patient with stage III familial hidradenitis suppurativa treated with 3 courses of infliximab and died of metastatic squamous cell carcinoma. Dermatol Online J. 2014;20(3).
- Verdelli A, Antiga E, Bonciani D, et al. A fatal case of hidradenitis suppurativa associated with sepsis and squamous cell carcinoma. Int J Dermatol. 2016;55:E52-E53.
- Giesey R, Delost GR, Honaker J, et al. Metastatic squamous cell carcinoma in a patient treated with adalimumab for hidradenitis suppurativa. JAAD Case Rep. 2017;3:489-491.
- Roy C, Roy S, Ghazawi F, et al. Cutaneous squamous cell carcinoma arising in hidradenitis suppurativa: a case report. SAGE Open Med Case Rep. 2019;7:2050313X19847359.
- Lavogiez C, Delaporte E, Darras-Vercambre S, et al. Clinicopathological study of 13 cases of squamous cell carcinoma complicating hidradenitis suppurativa. Dermatology. 2010;220:147-153.
- Alikhan A, Lynch PJ, Eisen DB. Hidradenitis suppurativa: a comprehensive review. J Am Acad Dermatol. 2009;60:539-561; quiz 562-533.
- Lapins J, Ye W, Nyren O, et al. Incidence of cancer among patients with hidradenitis suppurativa. Arch Dermatol. 2001;137:730-734.
- Askling J, Fahrbach K, Nordstrom B, et al. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol Drug Saf. 2011;20:119-130.
- Mariette X, Matucci-Cerinic M, Pavelka K, et al. Malignancies associated with tumour necrosis factor inhibitors in registries and prospective observational studies: a systematic review and meta-analysis. Ann Rheum Dis. 2011;70:1895-1904.
- Maalouf E, Faye O, Poli F, et al. Fatal epidermoid carcinoma in hidradenitis suppurativa following treatment with infliximab. Ann Dermatol Venereol. 2006;133(5 pt 1):473-474.
- Kurokawa I, Nishimura K, Yamanaka K, et al. Cytokeratin expression in squamous cell carcinoma arising from hidradenitis suppurativa (acne inversa). J Cutan Pathol. 2007;34:675-678.
- Scheinfeld N. A case of a patient with stage III familial hidradenitis suppurativa treated with 3 courses of infliximab and died of metastatic squamous cell carcinoma. Dermatol Online J. 2014;20(3).
- Verdelli A, Antiga E, Bonciani D, et al. A fatal case of hidradenitis suppurativa associated with sepsis and squamous cell carcinoma. Int J Dermatol. 2016;55:E52-E53.
- Giesey R, Delost GR, Honaker J, et al. Metastatic squamous cell carcinoma in a patient treated with adalimumab for hidradenitis suppurativa. JAAD Case Rep. 2017;3:489-491.
- Roy C, Roy S, Ghazawi F, et al. Cutaneous squamous cell carcinoma arising in hidradenitis suppurativa: a case report. SAGE Open Med Case Rep. 2019;7:2050313X19847359.
- Lavogiez C, Delaporte E, Darras-Vercambre S, et al. Clinicopathological study of 13 cases of squamous cell carcinoma complicating hidradenitis suppurativa. Dermatology. 2010;220:147-153.
Practice Points
- Consider biopsy of representative lesions in men older than 20 years with moderate to severe disease of the groin and/or buttocks prior to initiation of tumor necrosis factor inhibitors.
- Consider more frequent clinical monitoring with a decrease in threshold to perform biopsy of any new or ulcerating lesions.
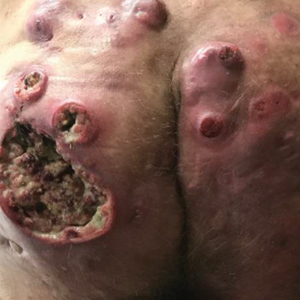
Dupilumab for the Treatment of Lichen Planus
To the Editor:
Lichen planus (LP) is an inflammatory mucocutaneous disorder that primarily affects adults aged 30 to 60 years.1 It can present across various regions such as the skin, scalp, oral cavity, genitalia, nails, and hair. It classically presents with pruritic, purple, polygonal papules or plaques. The proposed pathogenesis of this condition involves autoimmune destruction of epidermal basal keratinocytes.2 Management involves a stepwise approach, beginning with topical therapies such as corticosteroids and phototherapy and proceeding to systemic therapy including oral corticosteroids and retinoids. Additional medications with reported positive results include immunomodulators such as cyclosporine, tacrolimus, and mycophenolate mofetil.2-4 Dupilumab is a biologic immunomodulator and antagonist to the IL-4Rα on helper T cells (TH1). Although indicated for the treatment of moderate to severe atopic dermatitis, this medication’s immunomodulatory properties have been shown to aid various inflammatory cutaneous conditions, including prurigo nodularis.5-9 We present a case of dupilumab therapy for treatment-refractory LP.
A 52-year-old man presented with a new-onset progressive rash over the prior 6 months. He reported no history of atopic dermatitis. The patient described the rash as “severely pruritic” with a numeric rating scale itch intensity of 9/10 (0 being no itch; 10 being the worst itch imaginable). Physical examination revealed purple polygonal scaly papules on the arms, hands, legs, feet, chest, and back (Figure 1).
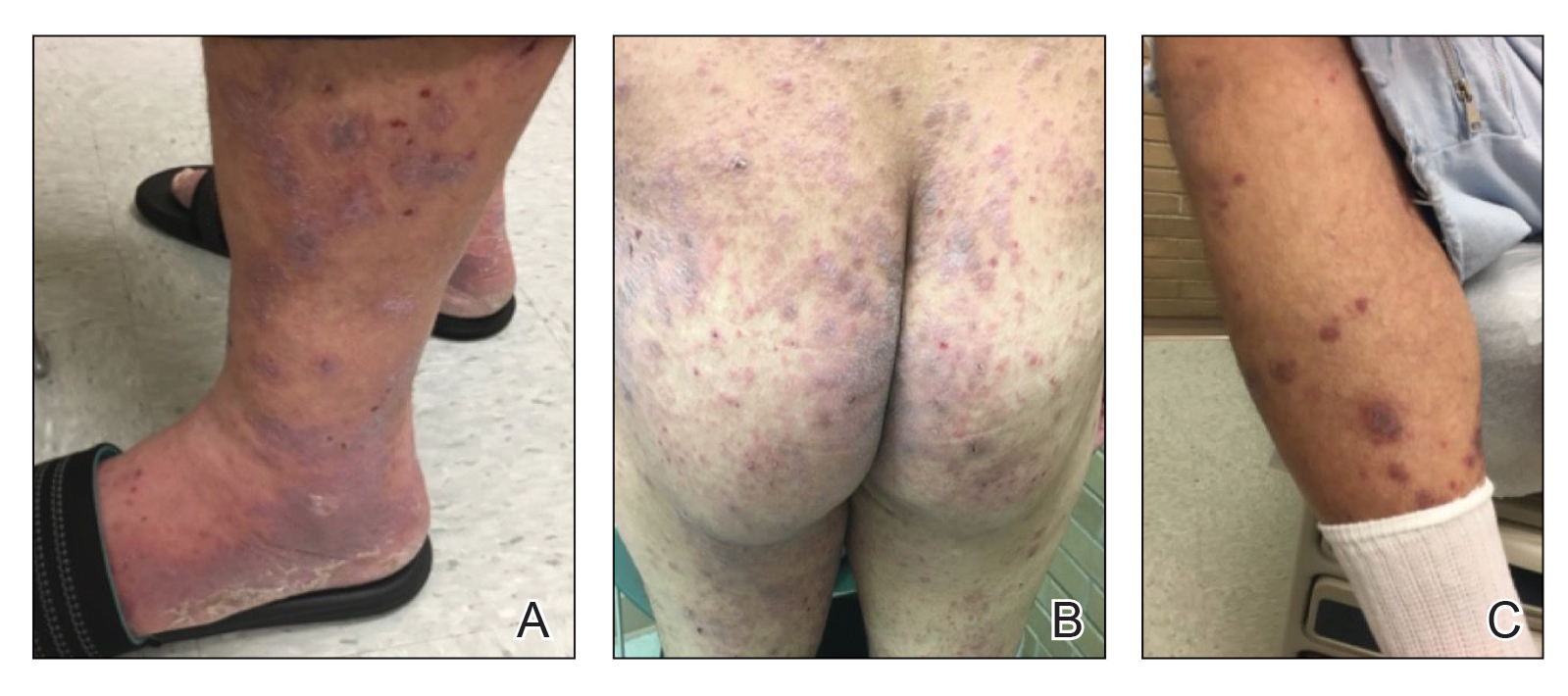
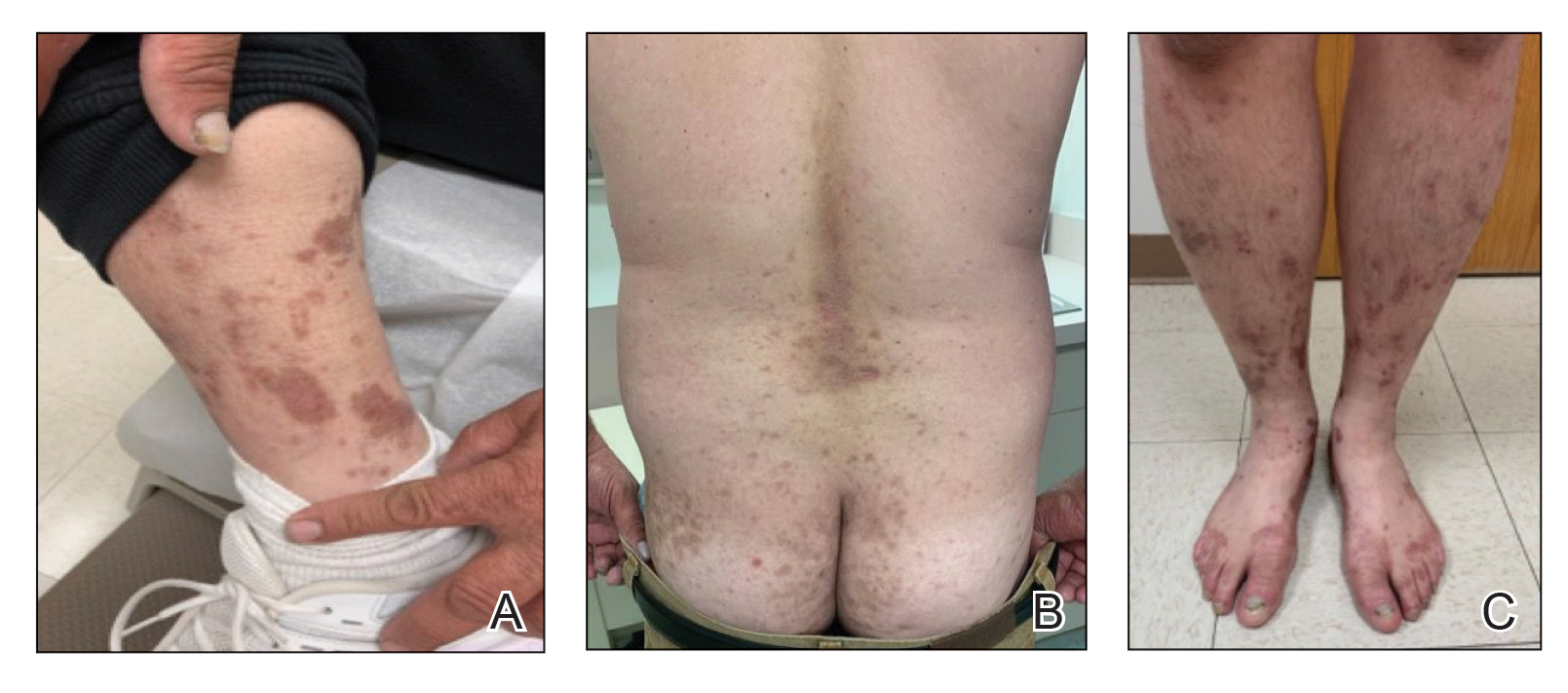
Three biopsies were taken, all indicative of lichenoid dermatitis consistent with LP. Rapid plasma reagin as well as HIV and hepatitis C virus serology tests were negative. Halobetasol ointment, tacrolimus ointment, and oral prednisone (28-day taper starting at 40 mg) all failed. Acitretin subsequently was initiated and failed to provide any benefit. The patient was unable to come to clinic 3 times a week for phototherapy due to his work schedule.
Due to the chronic, severe, and recalcitrant nature of his condition, as well as the lack of US Food and Drug Administration–approved treatments, the patient agreed to begin off-label treatment with dupilumab. Upon documentation, the patient’s primary diagnosis was listed as LP, clearly stating all commonly accepted treatments were attempted, except off-label therapy, and failed, and the plan was to treat him with dupilumab as if he had a severe form of atopic dermatitis. Dupilumab was approved with this documentation with a minimal co-pay, as the patient was on Medicaid. At 3-month follow-up (after 4 administrations of the medication), the patient showed remarkable improvement in appearance, and his numeric rating scale itch intensity score improved to 1/10.
Lichen planus is an immune-mediated, inflammatory condition that can affect the skin, hair, nails, and oral cavity. Although its etiology is not fully understood, research supports a primarily TH1 immunologic reaction.10 These T cells promote cytotoxic CD8 T-cell differentiation and migration, leading to subsequent destruction of epidermal basal keratinocytes. An important cytokine in this pathway—tumor necrosis factor α—stimulates a series of proinflammatory factors, including IL-1α, IL-8, and IL-6. IL-6 is of particular interest, as its elevation has been identified in the serum of patients with LP, with levels correlating to disease severity.11 This increase is thought to be multifactorial and a reliable predictor of disease activity.12,13 In addition to its proinflammatory role, IL-6 promotes the activity of IL-4, an essential cytokine in TH2 T-cell differentiation.
The TH2 pathway, enhanced by IL-6, increases the activity of downstream cytokines IL-4, IL-5, and IL-13. This pathway promotes IgE class switching and eosinophil maturation, pivotal factors in the development of atopic conditions such as allergic rhinitis, asthma, and atopic dermatitis. The role of IL-4 and TH2 cells in the pathogenesis of LP remains poorly understood.14 In prior basic laboratory studies, utilizing tissue sampling, RNA extraction, and real-time polymerase chain reaction assays, Yamauchi et al15 proposed that TH2-related chemokines played a pathogenic role in oral LP. Additional reports propose the pathogenic involvement of TH17, TH0, and TH2 T cells.16 These findings suggest that elevated IL-6 in those with LP may stimulate an increase in IL-4 and subsequent TH2 response. Dupilumab, a monoclonal antibody that targets IL-4Rα found on T cells, inhibits both IL-4 and IL-13 signaling, decreasing subsequent effector cell function.17,18 Several case reports have described dupilumab successfully treating various additional dermatoses, including prurigo nodularis, chronic pruritus, and bullous pemphigoid.5-9 Our case demonstrates an example of LP responsive to dupilumab. Our findings suggest that dupilumab interacts with the pathogenic cascade of LP, potentially implicating the role of TH2 in the pathophysiology of LP.
Treatment-refractory LP remains difficult to manage for both the patient and provider. Treatment regimens remain limited to small uncontrolled studies and case reports. Although primarily considered a TH1-mediated disease, the interplay of various alternative signaling pathways has been suggested. Our case of dupilumab-responsive LP suggests an underlying pathologic role of TH2-mediated activity. Dupilumab shows promise as an effective therapy for refractory LP, as evidenced by our patient’s remarkable response. Larger studies are warranted regarding the role of TH2-mediated inflammation and the use of dupilumab in LP.
- Cleach LL, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;266:723-732.
- Lehman, JS, Tollefson MM, Gibson LE. Lichen planus. Int J Dermatol. 2009;48:682-694.
- Frieling U, Bonsmann G, Schwarz T, et al. Treatment of severe lichen planus with mycophenolate mofetil. J Am Acad Dermatol. 2003;49:1063-1066.
- Cribier B, Frances C, Chosidow O. Treatment of lichen planus. an evidence-based medicine analysis of efficacy. Arch Dermatol. 1998;134:1521-1530.
- Calugareanu A, Jachiet C, Lepelletier C, et al. Dramatic improvement of generalized prurigo nodularis with dupilumab. J Eur Acad Dermatol Venereol. 2019;33:E303-E304.
- Kaye A, Gordon SC, Deverapalli SC, et al. Dupilumab for the treatment of recalcitrant bullous pemphigoid. JAMA Dermatol. 2018;154:1225-1226.
- Mollanazar NK, Qiu CC, Aldrich JL, et al. Use of dupilumab in HIV-positive patients: report of four cases. Br J Dermatol. 2019;181:1311-1312.
- Zhai LL, Savage KT, Qiu CC, et al. Chronic pruritus responding to dupilumab—a case series. Medicines (Basel). 2019;6:72.
- Mollanazar NK, Elgash M, Weaver L, et al. Reduced itch associated with dupilumab treatment in 4 patients with prurigo nodularis. JAMA Dermatol. 2019;155:121-122.
- Lodi G, Scully C, Carrozzo M, et al. Current controversies in oral lichen planus: report of an international consensus meeting. part 1. viral infections and etiopathogenesis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:40-51.
- Yin M, Li G, Song H, et al. Identifying the association between interleukin-6 and lichen planus: a meta-analysis. Biomed Rep. 2017;6:571-575.
- Sun A, Chia JS, Chang YF, et al. Serum interleukin-6 level is a useful marker in evaluating therapeutic effects of levamisole and Chinese medicinal herbs on patients with oral lichen planus. J Oral Pathol Med. 2002;31:196-203.
- Rhodus NL, Cheng B, Bowles W, et al. Proinflammatory cytokine levels in saliva before and after treatment of (erosive) oral lichen planus with dexamethasone. Oral Dis. 2006;12:112-116.
- Carrozzo M. Understanding the pathobiology of oral lichen planus. Curr Oral Health Rep. 2014;1:173-179.
- Yamauchi M, Moriyama M, Hayashida JN, et al. Myeloid dendritic cells stimulated by thymic stromal lymphopoietin promote Th2 immune responses and the pathogenesis of oral lichen planus. Plos One. 2017:12:e0173017.
- Piccinni M-P, Lombardell L, Logidice F, et al. Potential pathogenetic role of Th17, Th0, and Th2 cells in erosive and reticular oral lichen planus. Oral Dis. 2013:20:212-218.
- Kidd P. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev. 2003;8:223-246.
- Noda S, Kruefer JG, Guttum-Yassky E. The translational revolution and use of biologics in patients with inflammatory skin diseases. J Allergy Clin Immunol. 2015;135:324-336.
To the Editor:
Lichen planus (LP) is an inflammatory mucocutaneous disorder that primarily affects adults aged 30 to 60 years.1 It can present across various regions such as the skin, scalp, oral cavity, genitalia, nails, and hair. It classically presents with pruritic, purple, polygonal papules or plaques. The proposed pathogenesis of this condition involves autoimmune destruction of epidermal basal keratinocytes.2 Management involves a stepwise approach, beginning with topical therapies such as corticosteroids and phototherapy and proceeding to systemic therapy including oral corticosteroids and retinoids. Additional medications with reported positive results include immunomodulators such as cyclosporine, tacrolimus, and mycophenolate mofetil.2-4 Dupilumab is a biologic immunomodulator and antagonist to the IL-4Rα on helper T cells (TH1). Although indicated for the treatment of moderate to severe atopic dermatitis, this medication’s immunomodulatory properties have been shown to aid various inflammatory cutaneous conditions, including prurigo nodularis.5-9 We present a case of dupilumab therapy for treatment-refractory LP.
A 52-year-old man presented with a new-onset progressive rash over the prior 6 months. He reported no history of atopic dermatitis. The patient described the rash as “severely pruritic” with a numeric rating scale itch intensity of 9/10 (0 being no itch; 10 being the worst itch imaginable). Physical examination revealed purple polygonal scaly papules on the arms, hands, legs, feet, chest, and back (Figure 1).
Three biopsies were taken, all indicative of lichenoid dermatitis consistent with LP. Rapid plasma reagin as well as HIV and hepatitis C virus serology tests were negative. Halobetasol ointment, tacrolimus ointment, and oral prednisone (28-day taper starting at 40 mg) all failed. Acitretin subsequently was initiated and failed to provide any benefit. The patient was unable to come to clinic 3 times a week for phototherapy due to his work schedule.
Due to the chronic, severe, and recalcitrant nature of his condition, as well as the lack of US Food and Drug Administration–approved treatments, the patient agreed to begin off-label treatment with dupilumab. Upon documentation, the patient’s primary diagnosis was listed as LP, clearly stating all commonly accepted treatments were attempted, except off-label therapy, and failed, and the plan was to treat him with dupilumab as if he had a severe form of atopic dermatitis. Dupilumab was approved with this documentation with a minimal co-pay, as the patient was on Medicaid. At 3-month follow-up (after 4 administrations of the medication), the patient showed remarkable improvement in appearance, and his numeric rating scale itch intensity score improved to 1/10.
Lichen planus is an immune-mediated, inflammatory condition that can affect the skin, hair, nails, and oral cavity. Although its etiology is not fully understood, research supports a primarily TH1 immunologic reaction.10 These T cells promote cytotoxic CD8 T-cell differentiation and migration, leading to subsequent destruction of epidermal basal keratinocytes. An important cytokine in this pathway—tumor necrosis factor α—stimulates a series of proinflammatory factors, including IL-1α, IL-8, and IL-6. IL-6 is of particular interest, as its elevation has been identified in the serum of patients with LP, with levels correlating to disease severity.11 This increase is thought to be multifactorial and a reliable predictor of disease activity.12,13 In addition to its proinflammatory role, IL-6 promotes the activity of IL-4, an essential cytokine in TH2 T-cell differentiation.
The TH2 pathway, enhanced by IL-6, increases the activity of downstream cytokines IL-4, IL-5, and IL-13. This pathway promotes IgE class switching and eosinophil maturation, pivotal factors in the development of atopic conditions such as allergic rhinitis, asthma, and atopic dermatitis. The role of IL-4 and TH2 cells in the pathogenesis of LP remains poorly understood.14 In prior basic laboratory studies, utilizing tissue sampling, RNA extraction, and real-time polymerase chain reaction assays, Yamauchi et al15 proposed that TH2-related chemokines played a pathogenic role in oral LP. Additional reports propose the pathogenic involvement of TH17, TH0, and TH2 T cells.16 These findings suggest that elevated IL-6 in those with LP may stimulate an increase in IL-4 and subsequent TH2 response. Dupilumab, a monoclonal antibody that targets IL-4Rα found on T cells, inhibits both IL-4 and IL-13 signaling, decreasing subsequent effector cell function.17,18 Several case reports have described dupilumab successfully treating various additional dermatoses, including prurigo nodularis, chronic pruritus, and bullous pemphigoid.5-9 Our case demonstrates an example of LP responsive to dupilumab. Our findings suggest that dupilumab interacts with the pathogenic cascade of LP, potentially implicating the role of TH2 in the pathophysiology of LP.
Treatment-refractory LP remains difficult to manage for both the patient and provider. Treatment regimens remain limited to small uncontrolled studies and case reports. Although primarily considered a TH1-mediated disease, the interplay of various alternative signaling pathways has been suggested. Our case of dupilumab-responsive LP suggests an underlying pathologic role of TH2-mediated activity. Dupilumab shows promise as an effective therapy for refractory LP, as evidenced by our patient’s remarkable response. Larger studies are warranted regarding the role of TH2-mediated inflammation and the use of dupilumab in LP.
To the Editor:
Lichen planus (LP) is an inflammatory mucocutaneous disorder that primarily affects adults aged 30 to 60 years.1 It can present across various regions such as the skin, scalp, oral cavity, genitalia, nails, and hair. It classically presents with pruritic, purple, polygonal papules or plaques. The proposed pathogenesis of this condition involves autoimmune destruction of epidermal basal keratinocytes.2 Management involves a stepwise approach, beginning with topical therapies such as corticosteroids and phototherapy and proceeding to systemic therapy including oral corticosteroids and retinoids. Additional medications with reported positive results include immunomodulators such as cyclosporine, tacrolimus, and mycophenolate mofetil.2-4 Dupilumab is a biologic immunomodulator and antagonist to the IL-4Rα on helper T cells (TH1). Although indicated for the treatment of moderate to severe atopic dermatitis, this medication’s immunomodulatory properties have been shown to aid various inflammatory cutaneous conditions, including prurigo nodularis.5-9 We present a case of dupilumab therapy for treatment-refractory LP.
A 52-year-old man presented with a new-onset progressive rash over the prior 6 months. He reported no history of atopic dermatitis. The patient described the rash as “severely pruritic” with a numeric rating scale itch intensity of 9/10 (0 being no itch; 10 being the worst itch imaginable). Physical examination revealed purple polygonal scaly papules on the arms, hands, legs, feet, chest, and back (Figure 1).
Three biopsies were taken, all indicative of lichenoid dermatitis consistent with LP. Rapid plasma reagin as well as HIV and hepatitis C virus serology tests were negative. Halobetasol ointment, tacrolimus ointment, and oral prednisone (28-day taper starting at 40 mg) all failed. Acitretin subsequently was initiated and failed to provide any benefit. The patient was unable to come to clinic 3 times a week for phototherapy due to his work schedule.
Due to the chronic, severe, and recalcitrant nature of his condition, as well as the lack of US Food and Drug Administration–approved treatments, the patient agreed to begin off-label treatment with dupilumab. Upon documentation, the patient’s primary diagnosis was listed as LP, clearly stating all commonly accepted treatments were attempted, except off-label therapy, and failed, and the plan was to treat him with dupilumab as if he had a severe form of atopic dermatitis. Dupilumab was approved with this documentation with a minimal co-pay, as the patient was on Medicaid. At 3-month follow-up (after 4 administrations of the medication), the patient showed remarkable improvement in appearance, and his numeric rating scale itch intensity score improved to 1/10.
Lichen planus is an immune-mediated, inflammatory condition that can affect the skin, hair, nails, and oral cavity. Although its etiology is not fully understood, research supports a primarily TH1 immunologic reaction.10 These T cells promote cytotoxic CD8 T-cell differentiation and migration, leading to subsequent destruction of epidermal basal keratinocytes. An important cytokine in this pathway—tumor necrosis factor α—stimulates a series of proinflammatory factors, including IL-1α, IL-8, and IL-6. IL-6 is of particular interest, as its elevation has been identified in the serum of patients with LP, with levels correlating to disease severity.11 This increase is thought to be multifactorial and a reliable predictor of disease activity.12,13 In addition to its proinflammatory role, IL-6 promotes the activity of IL-4, an essential cytokine in TH2 T-cell differentiation.
The TH2 pathway, enhanced by IL-6, increases the activity of downstream cytokines IL-4, IL-5, and IL-13. This pathway promotes IgE class switching and eosinophil maturation, pivotal factors in the development of atopic conditions such as allergic rhinitis, asthma, and atopic dermatitis. The role of IL-4 and TH2 cells in the pathogenesis of LP remains poorly understood.14 In prior basic laboratory studies, utilizing tissue sampling, RNA extraction, and real-time polymerase chain reaction assays, Yamauchi et al15 proposed that TH2-related chemokines played a pathogenic role in oral LP. Additional reports propose the pathogenic involvement of TH17, TH0, and TH2 T cells.16 These findings suggest that elevated IL-6 in those with LP may stimulate an increase in IL-4 and subsequent TH2 response. Dupilumab, a monoclonal antibody that targets IL-4Rα found on T cells, inhibits both IL-4 and IL-13 signaling, decreasing subsequent effector cell function.17,18 Several case reports have described dupilumab successfully treating various additional dermatoses, including prurigo nodularis, chronic pruritus, and bullous pemphigoid.5-9 Our case demonstrates an example of LP responsive to dupilumab. Our findings suggest that dupilumab interacts with the pathogenic cascade of LP, potentially implicating the role of TH2 in the pathophysiology of LP.
Treatment-refractory LP remains difficult to manage for both the patient and provider. Treatment regimens remain limited to small uncontrolled studies and case reports. Although primarily considered a TH1-mediated disease, the interplay of various alternative signaling pathways has been suggested. Our case of dupilumab-responsive LP suggests an underlying pathologic role of TH2-mediated activity. Dupilumab shows promise as an effective therapy for refractory LP, as evidenced by our patient’s remarkable response. Larger studies are warranted regarding the role of TH2-mediated inflammation and the use of dupilumab in LP.
- Cleach LL, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;266:723-732.
- Lehman, JS, Tollefson MM, Gibson LE. Lichen planus. Int J Dermatol. 2009;48:682-694.
- Frieling U, Bonsmann G, Schwarz T, et al. Treatment of severe lichen planus with mycophenolate mofetil. J Am Acad Dermatol. 2003;49:1063-1066.
- Cribier B, Frances C, Chosidow O. Treatment of lichen planus. an evidence-based medicine analysis of efficacy. Arch Dermatol. 1998;134:1521-1530.
- Calugareanu A, Jachiet C, Lepelletier C, et al. Dramatic improvement of generalized prurigo nodularis with dupilumab. J Eur Acad Dermatol Venereol. 2019;33:E303-E304.
- Kaye A, Gordon SC, Deverapalli SC, et al. Dupilumab for the treatment of recalcitrant bullous pemphigoid. JAMA Dermatol. 2018;154:1225-1226.
- Mollanazar NK, Qiu CC, Aldrich JL, et al. Use of dupilumab in HIV-positive patients: report of four cases. Br J Dermatol. 2019;181:1311-1312.
- Zhai LL, Savage KT, Qiu CC, et al. Chronic pruritus responding to dupilumab—a case series. Medicines (Basel). 2019;6:72.
- Mollanazar NK, Elgash M, Weaver L, et al. Reduced itch associated with dupilumab treatment in 4 patients with prurigo nodularis. JAMA Dermatol. 2019;155:121-122.
- Lodi G, Scully C, Carrozzo M, et al. Current controversies in oral lichen planus: report of an international consensus meeting. part 1. viral infections and etiopathogenesis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:40-51.
- Yin M, Li G, Song H, et al. Identifying the association between interleukin-6 and lichen planus: a meta-analysis. Biomed Rep. 2017;6:571-575.
- Sun A, Chia JS, Chang YF, et al. Serum interleukin-6 level is a useful marker in evaluating therapeutic effects of levamisole and Chinese medicinal herbs on patients with oral lichen planus. J Oral Pathol Med. 2002;31:196-203.
- Rhodus NL, Cheng B, Bowles W, et al. Proinflammatory cytokine levels in saliva before and after treatment of (erosive) oral lichen planus with dexamethasone. Oral Dis. 2006;12:112-116.
- Carrozzo M. Understanding the pathobiology of oral lichen planus. Curr Oral Health Rep. 2014;1:173-179.
- Yamauchi M, Moriyama M, Hayashida JN, et al. Myeloid dendritic cells stimulated by thymic stromal lymphopoietin promote Th2 immune responses and the pathogenesis of oral lichen planus. Plos One. 2017:12:e0173017.
- Piccinni M-P, Lombardell L, Logidice F, et al. Potential pathogenetic role of Th17, Th0, and Th2 cells in erosive and reticular oral lichen planus. Oral Dis. 2013:20:212-218.
- Kidd P. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev. 2003;8:223-246.
- Noda S, Kruefer JG, Guttum-Yassky E. The translational revolution and use of biologics in patients with inflammatory skin diseases. J Allergy Clin Immunol. 2015;135:324-336.
- Cleach LL, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;266:723-732.
- Lehman, JS, Tollefson MM, Gibson LE. Lichen planus. Int J Dermatol. 2009;48:682-694.
- Frieling U, Bonsmann G, Schwarz T, et al. Treatment of severe lichen planus with mycophenolate mofetil. J Am Acad Dermatol. 2003;49:1063-1066.
- Cribier B, Frances C, Chosidow O. Treatment of lichen planus. an evidence-based medicine analysis of efficacy. Arch Dermatol. 1998;134:1521-1530.
- Calugareanu A, Jachiet C, Lepelletier C, et al. Dramatic improvement of generalized prurigo nodularis with dupilumab. J Eur Acad Dermatol Venereol. 2019;33:E303-E304.
- Kaye A, Gordon SC, Deverapalli SC, et al. Dupilumab for the treatment of recalcitrant bullous pemphigoid. JAMA Dermatol. 2018;154:1225-1226.
- Mollanazar NK, Qiu CC, Aldrich JL, et al. Use of dupilumab in HIV-positive patients: report of four cases. Br J Dermatol. 2019;181:1311-1312.
- Zhai LL, Savage KT, Qiu CC, et al. Chronic pruritus responding to dupilumab—a case series. Medicines (Basel). 2019;6:72.
- Mollanazar NK, Elgash M, Weaver L, et al. Reduced itch associated with dupilumab treatment in 4 patients with prurigo nodularis. JAMA Dermatol. 2019;155:121-122.
- Lodi G, Scully C, Carrozzo M, et al. Current controversies in oral lichen planus: report of an international consensus meeting. part 1. viral infections and etiopathogenesis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:40-51.
- Yin M, Li G, Song H, et al. Identifying the association between interleukin-6 and lichen planus: a meta-analysis. Biomed Rep. 2017;6:571-575.
- Sun A, Chia JS, Chang YF, et al. Serum interleukin-6 level is a useful marker in evaluating therapeutic effects of levamisole and Chinese medicinal herbs on patients with oral lichen planus. J Oral Pathol Med. 2002;31:196-203.
- Rhodus NL, Cheng B, Bowles W, et al. Proinflammatory cytokine levels in saliva before and after treatment of (erosive) oral lichen planus with dexamethasone. Oral Dis. 2006;12:112-116.
- Carrozzo M. Understanding the pathobiology of oral lichen planus. Curr Oral Health Rep. 2014;1:173-179.
- Yamauchi M, Moriyama M, Hayashida JN, et al. Myeloid dendritic cells stimulated by thymic stromal lymphopoietin promote Th2 immune responses and the pathogenesis of oral lichen planus. Plos One. 2017:12:e0173017.
- Piccinni M-P, Lombardell L, Logidice F, et al. Potential pathogenetic role of Th17, Th0, and Th2 cells in erosive and reticular oral lichen planus. Oral Dis. 2013:20:212-218.
- Kidd P. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev. 2003;8:223-246.
- Noda S, Kruefer JG, Guttum-Yassky E. The translational revolution and use of biologics in patients with inflammatory skin diseases. J Allergy Clin Immunol. 2015;135:324-336.
Practice Points
- Lichen planus (LP) is an inflammatory mucocutaneous disorder that can present across various regions of the body with pruritic, purple, polygonal papules or plaques.
- The proposed pathogenesis of LP involves autoimmune destruction of epidermal basal keratinocytes.
- The immunomodulatory properties of dupilumab have been shown to aid various inflammatory cutaneous conditions.
Isolated Perianal Erosive Lichen Planus: A Diagnostic Challenge
To the Editor:
Erosive lichen planus (LP) often is painful, debilitating, and resistant to topical therapy making it both a diagnostic and therapeutic challenge. We report the case of an elderly woman with isolated perianal erosive LP, a rare clinical manifestation. We also review cases of erosive perianal LP reported in the literature.
A 72-year-old woman was referred to our dermatology clinic for evaluation of multiple pruritic and painful perianal lesions of 1 year’s duration. The lesions had remained stable since onset, with no other reported lesions elsewhere on body, including the mucosae. Her medical history was notable for rheumatoid arthritis, osteoporosis, hypercholesterolemia, and hypertension. She was taking methotrexate, folic acid, abatacept, alendronate, atorvastatin, and lisinopril. The patient reported she had been using abatacept for 3 years and lisinopril for 2 years. Her primary care physician initially treated the lesions as hemorrhoids but referred her to a gastroenterologist when they failed to improve. Gastroenterology evaluated the patient, and a colonoscopy was performed with unremarkable results. Thus, she was referred to dermatology for further evaluation.
Physical examination revealed 2 tender, sharply defined, angulated erosions with irregular violaceous borders involving the perianal skin (Figure 1). A biopsy of one of the lesions was taken. Histopathologic examination revealed acanthosis of the epidermis with slight compact hyperkeratosis, scattered dyskeratotic keratinocytes, and a dense bandlike lymphohistiocytic infiltrate that obliterated the dermoepidermal junction (Figure 2). A diagnosis of perianal erosive LP was made. The patient was prescribed mometasone ointment 0.1% daily with notable improvement after 2 months.
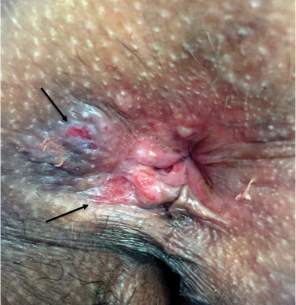
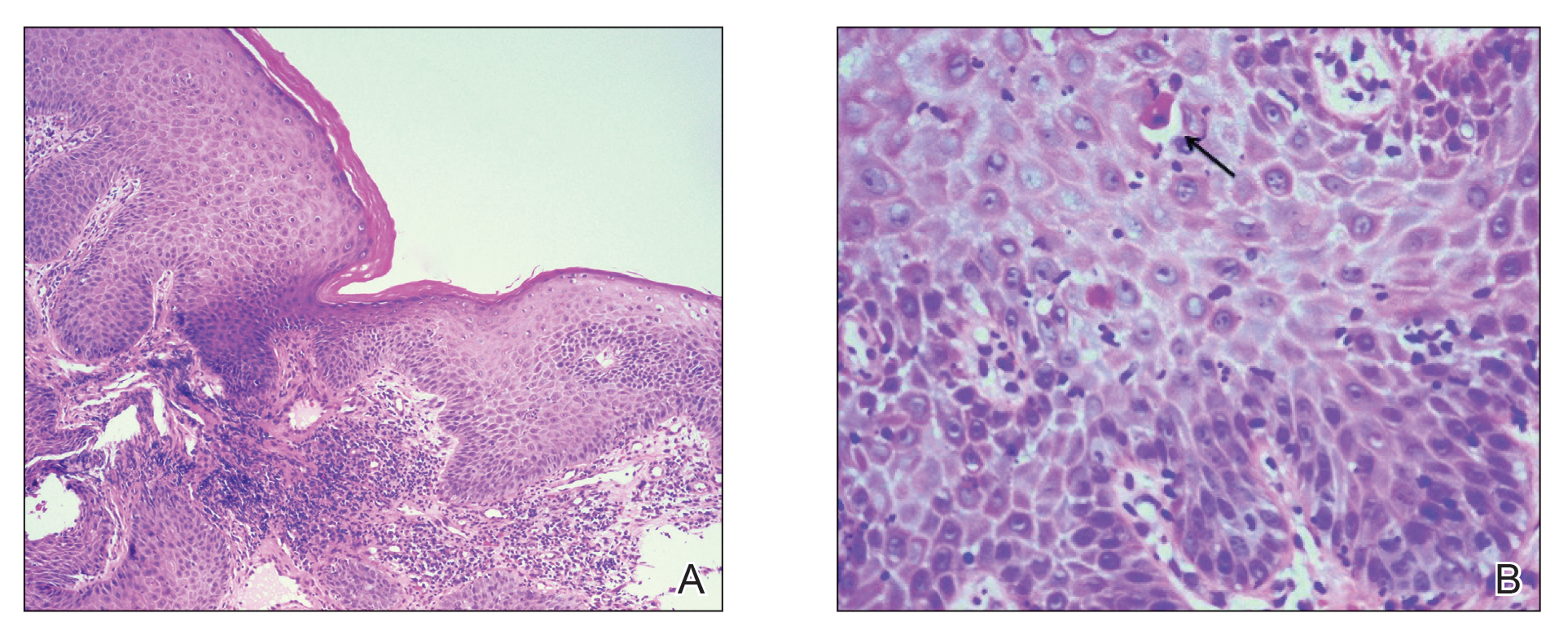
Erosive LP is an extremely rare variant of LP.1 It typically manifests as chronic painful erosions that often can progress to scarring, ulceration, and tissue destruction. Although erosive LP most commonly involves the mucosal surfaces of the genitalia and oral mucosa, it also has been reported in the palmoplantar skin, lacrimal duct, external auditory meatus, and esophagus.2-7 However, isolated perianal involvement is extremely rare. A PubMed search of articles indexed for MEDLINE using the terms erosive or ulcerative and lichen planus and perianal revealed 10 cases of perianal erosive LP, and weak data exist regarding therapy (Table).8-12 Of these cases, only 3 reported isolated perianal involvement.8-10 In most reported cases, perianal involvement manifested as extremely painful and occasionally pruritic, sharply angulated erosions and ulcers arising 0.5 to 3 cm from the anus with macerated, whitish, and violaceous borders. Most of the lesions occurred unilaterally, with only 1 case of bilateral perianal involvement.10
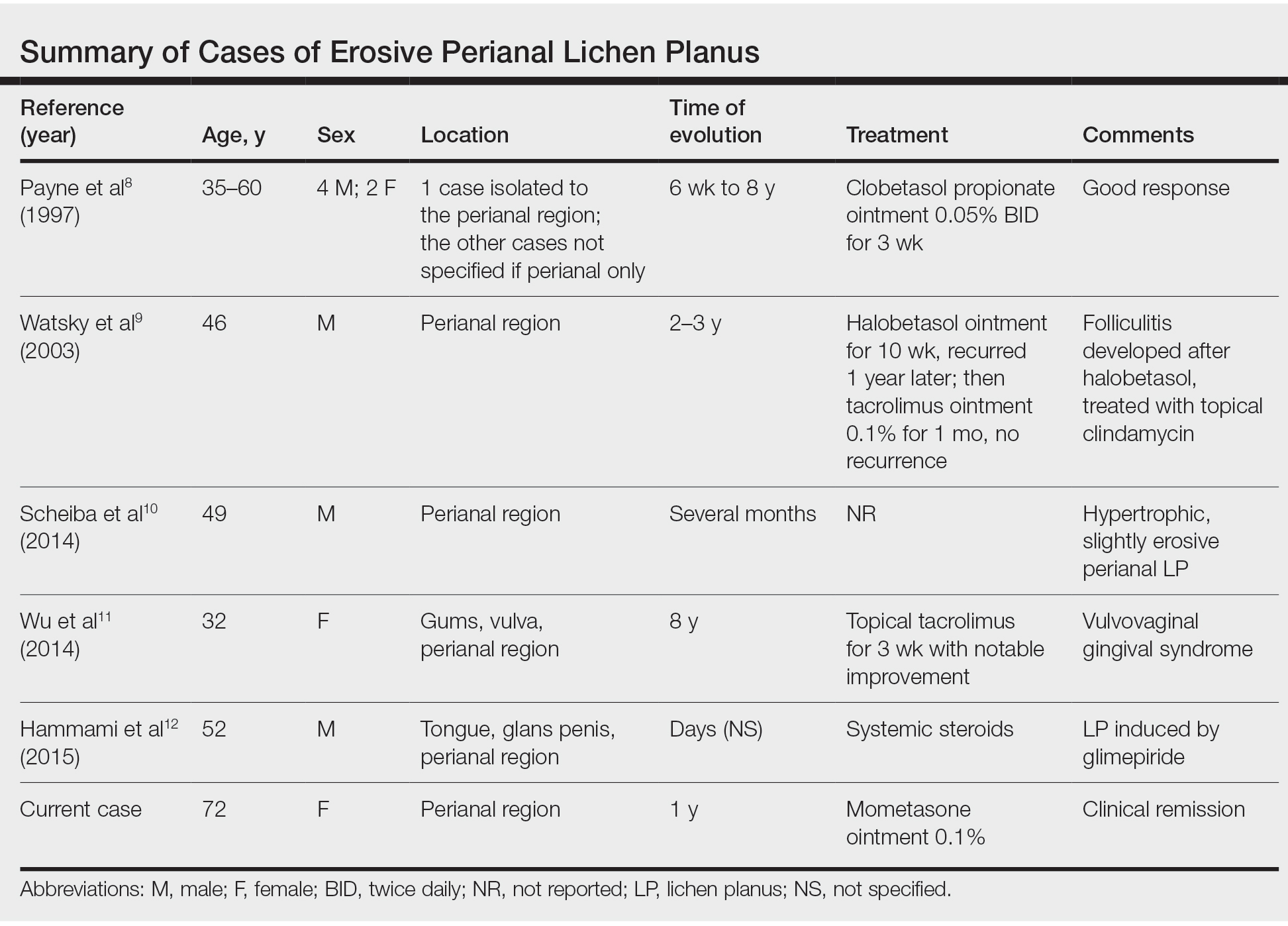
The differential diagnosis of perianal erosions is extensive and includes cutaneous Crohn disease, extramammary Paget disease, cutaneous malignancy, herpes simplex virus, cytomegalovirus, external hemorrhoids, lichen sclerosus, Behçet disease, lichen simplex chronicus, and drug-induced lichenoid reaction, among others. It is worth emphasizing infectious processes and cutaneous malignancies in light of our patient’s immunosuppression. Perianal cytomegalovirus has been reported in the literature in association with HIV, and it is a clinically challenging diagnosis.13 Cutaneous malignancy associated with the use of methotrexate also was considered in the differential diagnosis for our patient, given the increased risk for nonmelanoma skin cancer with the use of immunosuppresants.14
Along with a thorough patient history and physical examination, skin biopsy and clinicopathologic correlation are key to determine the exact etiology. Histologically, LP is characterized by a lichenoid interface dermatitis with a dense bandlike lymphohistiocytic infiltrate at the dermoepidermal junction. Other distinguishing factors include irregular acanthosis, hyperkeratosis, basal cell vacuolar degeneration, and Civatte bodies. Drug-induced LP is a possibility, but it is unclear if abatacept or lisinopril may have played a role in our patient. However, absence of eosinophils and parakeratosis suggested an idiopathic rather than drug-induced etiology. In 2016, Day et al2 published a clinicopathologic review of 60 cases of perianal lichenoid dermatoses in which only 17% of lesions were LP. Of note, 90% of perianal LP lesions were of the hypertrophic variant, and none were of the erosive variant, further supporting that our case represents a rare clinical manifestation of perianal LP.
Treatment of LP varies depending on the location and subtype of the lesions and is primarily aimed at improving symptoms. Topical corticosteroids are the standard treatment of LP; however, there is limited evidence regarding their efficacy for mucosal LP. Although randomized controlled trials assessing the efficacy of different interventions on oral erosive LP are available in the literature,15 there is a paucity of studies addressing this topic for genital or perianal LP. A review of the literature regarding perianal erosive LP suggests good response to high-potency topical steroids and calcineurin inhibitors with resolution of lesions within 3 to 4 weeks.11,15-18
Erosive LP is a painful variant that can cause erosions, ulcerations, and scarring. It rarely is seen in the perianal region alone and presents a diagnostic challenge. Treatment with high-potency topical steroid therapy seems to be effective in the few cases that have been reported as well as in our case. More comprehensive data from randomized controlled trials would be needed to evaluate their efficacy compared to other therapies.
- Rebora A. Erosive lichen planus: what is this? Dermatology. 2002;205:226-228; discussion 227.
- Day T, Bohl TG, Scurry J. Perianal lichen dermatoses: a review of 60 cases. Australas J Dermatol. 2016;57:210-215.
- Fox LP, Lightdale CJ, Grossman ME. Lichen planus of the esophagus: what dermatologists need to know. J Am Acad Dermatol. 2011;65:175-883.
- Holmstrup P, Thorn JJ, Rindum J, et al. Malignant development of lichen planus-affected oral mucosa. J Oral Pathol. 1988;17:219-225.
- Lewi, FM, Bogliatto F. Erosive vulval lichen planus—a diagnosis not to be missed: a clinical review. Eur J Obstet Gynecol Reprod Biol. 2013;171:214-219.
- Webber NK, Setterfield JF, Lewis FM, et al. Lacrimal canalicular duct scarring in patients with lichen planus. Arch Dermatol. 2012;148:224-227.
- Martin L, Moriniere S, Machet MC, et al. Bilateral conductive deafness related to erosive lichen planus. J Laryngol Otol. 1998;112:365-366.
- Payne CM, McPartlin JF, Hawley PR. Ulcerative perianal lichen planus. Br J Dermatol. 1997;136:479.
- Watsky KL. Erosive perianal lichen planus responsive to tacrolimus. Int J Dermatol. 2003;42:217-218.
- Scheiba N, Toberer F, Lenhard BH, et al. Erythema and erosions of the perianal region in a 49-year-old man. J Dtsch Dermatol Ges. 2014;12:162-165.
- Wu Y, Qiao J, Fang H. Syndrome in question. An Bras Dermatol. 2014;89:843-844.
- Hammami S, Ksouda K, Affes H, et al. Mucosal lichenoid drug reaction associated with glimepiride: a case report. Eur Rev Med Pharmacol Sci. 2015;19:2301-2302.
- Meyerle JH, Turiansky GW. Perianal ulcer in a patient with AIDS. Arch Dermatol. 2004;140:877-882.
- Scott FI, Mamtani R, Brensinger CM, et al. Risk of nonmelanoma skin cancer associated with the use of immunosuppressant and biologic agents in patients with a history of autoimmune disease and nonmelanoma skin cancer. JAMA Dermatol. 2016;152:164-172.
- Cheng S, Kirtschig G, Cooper S, et al. Interventions for erosive lichen planus affecting mucosal sites. Cochrane Database Syst Rev. 2012:Cd008092.
- Gunther S. Effect of retinoic acid in lichen planus of the genitalia and perianal region. Br J Vener Dis. 1973;49:553-554.
- Vente C, Reich K, Neumann C. Erosive mucosal lichen planus: response to topical treatment with tacrolimus. Br J Dermatol. 1999;140:338-342.
- Lonsdale-Eccles AA, Velangi S. Topical pimecrolimus in the treatment of genital lichen planus: a prospective case series. Br J Dermatol. 2005;153:390-394.
To the Editor:
Erosive lichen planus (LP) often is painful, debilitating, and resistant to topical therapy making it both a diagnostic and therapeutic challenge. We report the case of an elderly woman with isolated perianal erosive LP, a rare clinical manifestation. We also review cases of erosive perianal LP reported in the literature.
A 72-year-old woman was referred to our dermatology clinic for evaluation of multiple pruritic and painful perianal lesions of 1 year’s duration. The lesions had remained stable since onset, with no other reported lesions elsewhere on body, including the mucosae. Her medical history was notable for rheumatoid arthritis, osteoporosis, hypercholesterolemia, and hypertension. She was taking methotrexate, folic acid, abatacept, alendronate, atorvastatin, and lisinopril. The patient reported she had been using abatacept for 3 years and lisinopril for 2 years. Her primary care physician initially treated the lesions as hemorrhoids but referred her to a gastroenterologist when they failed to improve. Gastroenterology evaluated the patient, and a colonoscopy was performed with unremarkable results. Thus, she was referred to dermatology for further evaluation.
Physical examination revealed 2 tender, sharply defined, angulated erosions with irregular violaceous borders involving the perianal skin (Figure 1). A biopsy of one of the lesions was taken. Histopathologic examination revealed acanthosis of the epidermis with slight compact hyperkeratosis, scattered dyskeratotic keratinocytes, and a dense bandlike lymphohistiocytic infiltrate that obliterated the dermoepidermal junction (Figure 2). A diagnosis of perianal erosive LP was made. The patient was prescribed mometasone ointment 0.1% daily with notable improvement after 2 months.
Erosive LP is an extremely rare variant of LP.1 It typically manifests as chronic painful erosions that often can progress to scarring, ulceration, and tissue destruction. Although erosive LP most commonly involves the mucosal surfaces of the genitalia and oral mucosa, it also has been reported in the palmoplantar skin, lacrimal duct, external auditory meatus, and esophagus.2-7 However, isolated perianal involvement is extremely rare. A PubMed search of articles indexed for MEDLINE using the terms erosive or ulcerative and lichen planus and perianal revealed 10 cases of perianal erosive LP, and weak data exist regarding therapy (Table).8-12 Of these cases, only 3 reported isolated perianal involvement.8-10 In most reported cases, perianal involvement manifested as extremely painful and occasionally pruritic, sharply angulated erosions and ulcers arising 0.5 to 3 cm from the anus with macerated, whitish, and violaceous borders. Most of the lesions occurred unilaterally, with only 1 case of bilateral perianal involvement.10
The differential diagnosis of perianal erosions is extensive and includes cutaneous Crohn disease, extramammary Paget disease, cutaneous malignancy, herpes simplex virus, cytomegalovirus, external hemorrhoids, lichen sclerosus, Behçet disease, lichen simplex chronicus, and drug-induced lichenoid reaction, among others. It is worth emphasizing infectious processes and cutaneous malignancies in light of our patient’s immunosuppression. Perianal cytomegalovirus has been reported in the literature in association with HIV, and it is a clinically challenging diagnosis.13 Cutaneous malignancy associated with the use of methotrexate also was considered in the differential diagnosis for our patient, given the increased risk for nonmelanoma skin cancer with the use of immunosuppresants.14
Along with a thorough patient history and physical examination, skin biopsy and clinicopathologic correlation are key to determine the exact etiology. Histologically, LP is characterized by a lichenoid interface dermatitis with a dense bandlike lymphohistiocytic infiltrate at the dermoepidermal junction. Other distinguishing factors include irregular acanthosis, hyperkeratosis, basal cell vacuolar degeneration, and Civatte bodies. Drug-induced LP is a possibility, but it is unclear if abatacept or lisinopril may have played a role in our patient. However, absence of eosinophils and parakeratosis suggested an idiopathic rather than drug-induced etiology. In 2016, Day et al2 published a clinicopathologic review of 60 cases of perianal lichenoid dermatoses in which only 17% of lesions were LP. Of note, 90% of perianal LP lesions were of the hypertrophic variant, and none were of the erosive variant, further supporting that our case represents a rare clinical manifestation of perianal LP.
Treatment of LP varies depending on the location and subtype of the lesions and is primarily aimed at improving symptoms. Topical corticosteroids are the standard treatment of LP; however, there is limited evidence regarding their efficacy for mucosal LP. Although randomized controlled trials assessing the efficacy of different interventions on oral erosive LP are available in the literature,15 there is a paucity of studies addressing this topic for genital or perianal LP. A review of the literature regarding perianal erosive LP suggests good response to high-potency topical steroids and calcineurin inhibitors with resolution of lesions within 3 to 4 weeks.11,15-18
Erosive LP is a painful variant that can cause erosions, ulcerations, and scarring. It rarely is seen in the perianal region alone and presents a diagnostic challenge. Treatment with high-potency topical steroid therapy seems to be effective in the few cases that have been reported as well as in our case. More comprehensive data from randomized controlled trials would be needed to evaluate their efficacy compared to other therapies.
To the Editor:
Erosive lichen planus (LP) often is painful, debilitating, and resistant to topical therapy making it both a diagnostic and therapeutic challenge. We report the case of an elderly woman with isolated perianal erosive LP, a rare clinical manifestation. We also review cases of erosive perianal LP reported in the literature.
A 72-year-old woman was referred to our dermatology clinic for evaluation of multiple pruritic and painful perianal lesions of 1 year’s duration. The lesions had remained stable since onset, with no other reported lesions elsewhere on body, including the mucosae. Her medical history was notable for rheumatoid arthritis, osteoporosis, hypercholesterolemia, and hypertension. She was taking methotrexate, folic acid, abatacept, alendronate, atorvastatin, and lisinopril. The patient reported she had been using abatacept for 3 years and lisinopril for 2 years. Her primary care physician initially treated the lesions as hemorrhoids but referred her to a gastroenterologist when they failed to improve. Gastroenterology evaluated the patient, and a colonoscopy was performed with unremarkable results. Thus, she was referred to dermatology for further evaluation.
Physical examination revealed 2 tender, sharply defined, angulated erosions with irregular violaceous borders involving the perianal skin (Figure 1). A biopsy of one of the lesions was taken. Histopathologic examination revealed acanthosis of the epidermis with slight compact hyperkeratosis, scattered dyskeratotic keratinocytes, and a dense bandlike lymphohistiocytic infiltrate that obliterated the dermoepidermal junction (Figure 2). A diagnosis of perianal erosive LP was made. The patient was prescribed mometasone ointment 0.1% daily with notable improvement after 2 months.
Erosive LP is an extremely rare variant of LP.1 It typically manifests as chronic painful erosions that often can progress to scarring, ulceration, and tissue destruction. Although erosive LP most commonly involves the mucosal surfaces of the genitalia and oral mucosa, it also has been reported in the palmoplantar skin, lacrimal duct, external auditory meatus, and esophagus.2-7 However, isolated perianal involvement is extremely rare. A PubMed search of articles indexed for MEDLINE using the terms erosive or ulcerative and lichen planus and perianal revealed 10 cases of perianal erosive LP, and weak data exist regarding therapy (Table).8-12 Of these cases, only 3 reported isolated perianal involvement.8-10 In most reported cases, perianal involvement manifested as extremely painful and occasionally pruritic, sharply angulated erosions and ulcers arising 0.5 to 3 cm from the anus with macerated, whitish, and violaceous borders. Most of the lesions occurred unilaterally, with only 1 case of bilateral perianal involvement.10
The differential diagnosis of perianal erosions is extensive and includes cutaneous Crohn disease, extramammary Paget disease, cutaneous malignancy, herpes simplex virus, cytomegalovirus, external hemorrhoids, lichen sclerosus, Behçet disease, lichen simplex chronicus, and drug-induced lichenoid reaction, among others. It is worth emphasizing infectious processes and cutaneous malignancies in light of our patient’s immunosuppression. Perianal cytomegalovirus has been reported in the literature in association with HIV, and it is a clinically challenging diagnosis.13 Cutaneous malignancy associated with the use of methotrexate also was considered in the differential diagnosis for our patient, given the increased risk for nonmelanoma skin cancer with the use of immunosuppresants.14
Along with a thorough patient history and physical examination, skin biopsy and clinicopathologic correlation are key to determine the exact etiology. Histologically, LP is characterized by a lichenoid interface dermatitis with a dense bandlike lymphohistiocytic infiltrate at the dermoepidermal junction. Other distinguishing factors include irregular acanthosis, hyperkeratosis, basal cell vacuolar degeneration, and Civatte bodies. Drug-induced LP is a possibility, but it is unclear if abatacept or lisinopril may have played a role in our patient. However, absence of eosinophils and parakeratosis suggested an idiopathic rather than drug-induced etiology. In 2016, Day et al2 published a clinicopathologic review of 60 cases of perianal lichenoid dermatoses in which only 17% of lesions were LP. Of note, 90% of perianal LP lesions were of the hypertrophic variant, and none were of the erosive variant, further supporting that our case represents a rare clinical manifestation of perianal LP.
Treatment of LP varies depending on the location and subtype of the lesions and is primarily aimed at improving symptoms. Topical corticosteroids are the standard treatment of LP; however, there is limited evidence regarding their efficacy for mucosal LP. Although randomized controlled trials assessing the efficacy of different interventions on oral erosive LP are available in the literature,15 there is a paucity of studies addressing this topic for genital or perianal LP. A review of the literature regarding perianal erosive LP suggests good response to high-potency topical steroids and calcineurin inhibitors with resolution of lesions within 3 to 4 weeks.11,15-18
Erosive LP is a painful variant that can cause erosions, ulcerations, and scarring. It rarely is seen in the perianal region alone and presents a diagnostic challenge. Treatment with high-potency topical steroid therapy seems to be effective in the few cases that have been reported as well as in our case. More comprehensive data from randomized controlled trials would be needed to evaluate their efficacy compared to other therapies.
- Rebora A. Erosive lichen planus: what is this? Dermatology. 2002;205:226-228; discussion 227.
- Day T, Bohl TG, Scurry J. Perianal lichen dermatoses: a review of 60 cases. Australas J Dermatol. 2016;57:210-215.
- Fox LP, Lightdale CJ, Grossman ME. Lichen planus of the esophagus: what dermatologists need to know. J Am Acad Dermatol. 2011;65:175-883.
- Holmstrup P, Thorn JJ, Rindum J, et al. Malignant development of lichen planus-affected oral mucosa. J Oral Pathol. 1988;17:219-225.
- Lewi, FM, Bogliatto F. Erosive vulval lichen planus—a diagnosis not to be missed: a clinical review. Eur J Obstet Gynecol Reprod Biol. 2013;171:214-219.
- Webber NK, Setterfield JF, Lewis FM, et al. Lacrimal canalicular duct scarring in patients with lichen planus. Arch Dermatol. 2012;148:224-227.
- Martin L, Moriniere S, Machet MC, et al. Bilateral conductive deafness related to erosive lichen planus. J Laryngol Otol. 1998;112:365-366.
- Payne CM, McPartlin JF, Hawley PR. Ulcerative perianal lichen planus. Br J Dermatol. 1997;136:479.
- Watsky KL. Erosive perianal lichen planus responsive to tacrolimus. Int J Dermatol. 2003;42:217-218.
- Scheiba N, Toberer F, Lenhard BH, et al. Erythema and erosions of the perianal region in a 49-year-old man. J Dtsch Dermatol Ges. 2014;12:162-165.
- Wu Y, Qiao J, Fang H. Syndrome in question. An Bras Dermatol. 2014;89:843-844.
- Hammami S, Ksouda K, Affes H, et al. Mucosal lichenoid drug reaction associated with glimepiride: a case report. Eur Rev Med Pharmacol Sci. 2015;19:2301-2302.
- Meyerle JH, Turiansky GW. Perianal ulcer in a patient with AIDS. Arch Dermatol. 2004;140:877-882.
- Scott FI, Mamtani R, Brensinger CM, et al. Risk of nonmelanoma skin cancer associated with the use of immunosuppressant and biologic agents in patients with a history of autoimmune disease and nonmelanoma skin cancer. JAMA Dermatol. 2016;152:164-172.
- Cheng S, Kirtschig G, Cooper S, et al. Interventions for erosive lichen planus affecting mucosal sites. Cochrane Database Syst Rev. 2012:Cd008092.
- Gunther S. Effect of retinoic acid in lichen planus of the genitalia and perianal region. Br J Vener Dis. 1973;49:553-554.
- Vente C, Reich K, Neumann C. Erosive mucosal lichen planus: response to topical treatment with tacrolimus. Br J Dermatol. 1999;140:338-342.
- Lonsdale-Eccles AA, Velangi S. Topical pimecrolimus in the treatment of genital lichen planus: a prospective case series. Br J Dermatol. 2005;153:390-394.
- Rebora A. Erosive lichen planus: what is this? Dermatology. 2002;205:226-228; discussion 227.
- Day T, Bohl TG, Scurry J. Perianal lichen dermatoses: a review of 60 cases. Australas J Dermatol. 2016;57:210-215.
- Fox LP, Lightdale CJ, Grossman ME. Lichen planus of the esophagus: what dermatologists need to know. J Am Acad Dermatol. 2011;65:175-883.
- Holmstrup P, Thorn JJ, Rindum J, et al. Malignant development of lichen planus-affected oral mucosa. J Oral Pathol. 1988;17:219-225.
- Lewi, FM, Bogliatto F. Erosive vulval lichen planus—a diagnosis not to be missed: a clinical review. Eur J Obstet Gynecol Reprod Biol. 2013;171:214-219.
- Webber NK, Setterfield JF, Lewis FM, et al. Lacrimal canalicular duct scarring in patients with lichen planus. Arch Dermatol. 2012;148:224-227.
- Martin L, Moriniere S, Machet MC, et al. Bilateral conductive deafness related to erosive lichen planus. J Laryngol Otol. 1998;112:365-366.
- Payne CM, McPartlin JF, Hawley PR. Ulcerative perianal lichen planus. Br J Dermatol. 1997;136:479.
- Watsky KL. Erosive perianal lichen planus responsive to tacrolimus. Int J Dermatol. 2003;42:217-218.
- Scheiba N, Toberer F, Lenhard BH, et al. Erythema and erosions of the perianal region in a 49-year-old man. J Dtsch Dermatol Ges. 2014;12:162-165.
- Wu Y, Qiao J, Fang H. Syndrome in question. An Bras Dermatol. 2014;89:843-844.
- Hammami S, Ksouda K, Affes H, et al. Mucosal lichenoid drug reaction associated with glimepiride: a case report. Eur Rev Med Pharmacol Sci. 2015;19:2301-2302.
- Meyerle JH, Turiansky GW. Perianal ulcer in a patient with AIDS. Arch Dermatol. 2004;140:877-882.
- Scott FI, Mamtani R, Brensinger CM, et al. Risk of nonmelanoma skin cancer associated with the use of immunosuppressant and biologic agents in patients with a history of autoimmune disease and nonmelanoma skin cancer. JAMA Dermatol. 2016;152:164-172.
- Cheng S, Kirtschig G, Cooper S, et al. Interventions for erosive lichen planus affecting mucosal sites. Cochrane Database Syst Rev. 2012:Cd008092.
- Gunther S. Effect of retinoic acid in lichen planus of the genitalia and perianal region. Br J Vener Dis. 1973;49:553-554.
- Vente C, Reich K, Neumann C. Erosive mucosal lichen planus: response to topical treatment with tacrolimus. Br J Dermatol. 1999;140:338-342.
- Lonsdale-Eccles AA, Velangi S. Topical pimecrolimus in the treatment of genital lichen planus: a prospective case series. Br J Dermatol. 2005;153:390-394.
Practice Points
- Erosive lichen planus (LP) is an underrecognized variant of LP presenting with painful erosions, ulcerations, and scarring.
- Although rare, perianal erosive LP should be included in the differential diagnosis of perianal erosions.
- Treatment with high-potency steroids is an effective therapeutic option resulting in notable improvement.
Incontinentia Pigmenti: Initial Presentation of Encephalopathy and Seizures
To the Editor:
A 7-day-old full-term infant presented to the neonatal intensive care unit with poor feeding and altered consciousness. She was born at 39 weeks and 3 days to a gravida 1 mother with a pregnancy history complicated by maternal chorioamnionitis and gestational diabetes. During labor, nonreassuring fetal heart tones and arrest of labor prompted an uncomplicated cesarean delivery with normal Apgar scores at birth. The infant’s family history revealed only beta thalassemia minor in her father. At 5 to 7 days of life, the mother noted difficulty with feeding and poor latch along with lethargy and depressed consciousness in the infant.
Upon arrival to the neonatal intensive care unit, the infant was noted to have rhythmic lip-smacking behavior, intermittent nystagmus, mild hypotonia, and clonic movements of the left upper extremity. An electroencephalogram was markedly abnormal, capturing multiple seizures in the bilateral cortical hemispheres. She was loaded with phenobarbital with no further seizure activity. Brain magnetic resonance imaging revealed innumerable punctate foci of restricted diffusion with corresponding punctate hemorrhage within the frontal and parietal white matter, as well as cortical diffusion restriction within the occipital lobe, inferior temporal lobe, bilateral thalami, and corpus callosum (Figure 1). An exhaustive infectious workup also was completed and was unremarkable, though she was treated with broad-spectrum antimicrobials, including intravenous acyclovir.
Five days after being hospitalized (day 10 of life), a vesicular rash was noted on the arms and legs (Figure 2). Discussion with the patient’s mother revealed that the first signs of unusual skin lesions occurred as early as several days prior. There were no oral mucosal lesions or gross ocular abnormalities. No nail changes were appreciated. A bedside Tzanck preparation was negative for viral cytopathic changes. A skin biopsy was performed that demonstrated eosinophilic spongiosis with necrotic keratinocytes, typical of the vesicular stage of incontinentia pigmenti (IP)(Figure 3). An ophthalmology examination showed an arteriovenous malformation of the right eye with subtle neovascularization at the infratemporal periphery, consistent with known ocular manifestations of IP. The infant’s mother reported no history of notable dental abnormalities, hair loss, skin rashes, or nail changes. Genetic testing demonstrated the common IKBKG (inhibitor of κ light polypeptide gene enhancer in B cells, kinase gamma [formerly known as NEMO]) gene deletion on the X chromosome, consistent with IP.
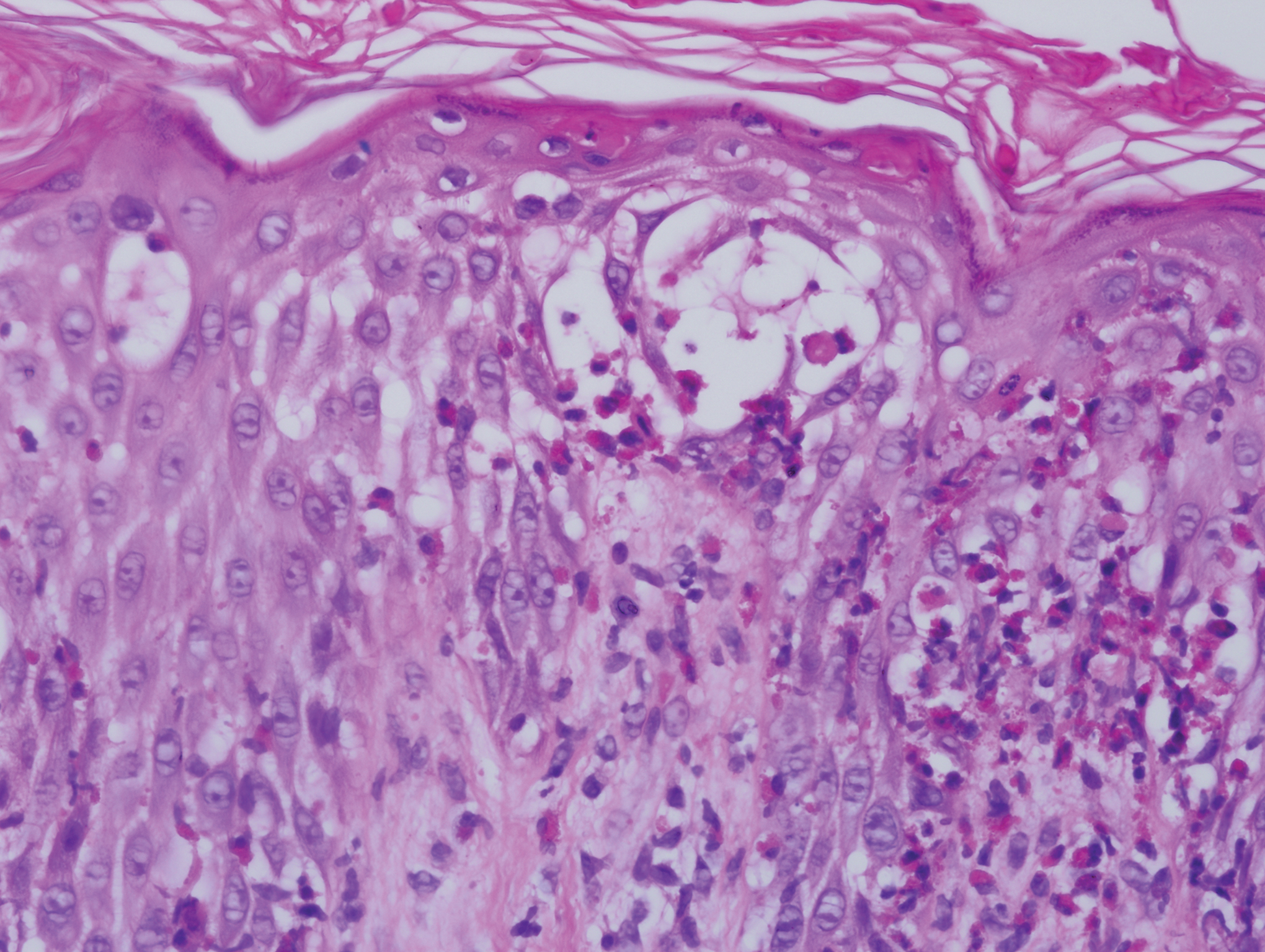
She successfully underwent retinal laser ablative therapy for the ocular manifestations without further evidence of neovascularization. She developed a mild cataract that was not visually significant and required no intervention. Her brain abnormalities were thought to represent foci of necrosis with superimposed hemorrhagic transformation due to spontaneous degeneration of brain cells in which the mutated X chromosome was activated. No further treatment was indicated beyond suppression of the consequent seizures. There was no notable cortical edema or other medical indication for systemic glucocorticoid therapy. Phenobarbital was continued without further seizure events.
Several months after the initial presentation, a follow-up electroencephalogram was normal. Phenobarbital was slowly weaned and finally discontinued approximately 6 months after the initial event with no other reported seizures. She currently is achieving normal developmental milestones with the exception of slight motor delay and expected residual hypotonia.
Incontinentia pigmenti, also known as Bloch-Sulzberger syndrome, is a rare multisystem neuroectodermal disorder, primarily affecting the skin, central nervous system (CNS), and retinas. The disorder can be inherited in an X-linked dominant fashion and appears almost exclusively in women with typical in utero lethality seen in males. Most affected individuals have a sporadic, or de novo, mutation, which was likely the case in our patient given that her mother demonstrated no signs or symptoms.1 The pathogenesis of disease is a defect at chromosome Xq28 that is a region encoding the nuclear factor–κB essential modulator, IKBKG. Absence or mutation of IKBKG in IP results in failure to activate nuclear factor–κB and leaves cells vulnerable to cytokine-mediated apoptosis, especially after exposure to tumor necrosis factor α.2
Clinical manifestations of IP are present at or soon after birth. The cutaneous findings of this disorder are classically described as a step-wise progression through 4 distinct stages: (1) a linear and/or whorled vesicular eruption predominantly on the extremities at birth or within the first few weeks of life; (2) thickened linear or whorled verrucous plaques; (3) hyperpigmented streaks and whorls that may or may not correspond with prior affected areas that may resolve by adolescence; and (4) hypopigmented, possibly atrophic plaques on the extremities that may persist lifelong. Importantly, not every patient will experience each of these stages. Overlap can occur, and the time course of each stage is highly variable. Other ectodermal manifestations include dental abnormalities such as small, misshaped, or missing teeth; alopecia; and nail abnormalities. Ocular abnormalities associated with IP primarily occur in the retina, including vascular occlusion, neovascularization, hemorrhages, foveal abnormalities, as well as exudative and tractional detachments.3,4
It is crucial to recognize CNS anomalies in association with the cutaneous findings of IP, as CNS pathology can be severe with profound developmental implications. Central nervous system findings have been noted to correlate with the appearance of the vesicular stage of IP. A high index of suspicion is needed, as the disease can demonstrate progression within a short time.5-8 The most frequent anomalies include seizures, motor impairment, intellectual disability, and microcephaly.9,10 Some of the most commonly identified CNS lesions on imaging include necrosis or brain infarcts, atrophy, and lesions of the corpus callosum.7
The pathogenesis of observed CNS changes in IP is not well understood. There have been numerous proposals of a vascular mechanism, and a microangiopathic process appears to be most plausible. Mutations in IKBKG may result in interruption of signaling via vascular endothelial growth factor receptor 3 with a consequent impact on angiogenesis, supporting a vascular mechanism. Additionally, mutations in IKBKG lead to activation of eotaxin, an eosinophil-selective chemokine.9 Eotaxin activation results in eosinophilic degranulation that mediates the classic eosinophilic infiltrate seen in the classic skin histology of IP. Additionally, it has been shown that eotaxin is strongly expressed by endothelial cells in IP, and more abundant eosinophil degranulation may play a role in mediating vaso-occlusion.7 Other studies have found that the highest expression level of the IKBKG gene is in the CNS, potentially explaining the extensive imaging findings of hemorrhage and diffusion restriction in our patient. These features likely are attributable to apoptosis of cells possessing the mutated IKBKG gene.9-11
- Ehrenreich M, Tarlow MM, Godlewska-Janusz E, et al. Incontinentia pigmenti (Bloch-Sulzberger syndrome): a systemic disorder. Cutis. 2007;79:355-362.
- Smahi A, Courtois G, Rabia SH, et al. The NF-kappaB signaling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet. 2002;11:2371-2375.
- O’Doherty M, McCreery K, Green AJ, et al. Incontinentia pigmenti—ophthalmological observation of a series of cases and review of the literature. Br J Ophthalmol. 2011;95:11-16.
- Swinney CC, Han DP, Karth PA. Incontinentia pigmenti: a comprehensive review and update. Ophthalmic Surg Lasers Imaging Retina. 2015;46:650-657.
- Hennel SJ, Ekert PG, Volpe JJ, et al. Insights into the pathogenesis of cerebral lesions in incontinentia pigmenti. Pediatr Neurol. 2003;29:148-150.
- Maingay-de Groof F, Lequin MH, Roofthooft DW, et al. Extensive cerebral infarction in the newborn due to incontinentia pigmenti. Eur J Paediatr Neurol. 2008;12:284-289.
- Minic´ S, Trpinac D, Obradovic´ M. Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet J Rare Dis. 2013;8:25-35.
- Wolf NI, Kramer N, Harting I, et al. Diffuse cortical necrosis in a neonate with incontinentia pigmenti and an encephalitis-like presentation. AJNR Am J Neuroradiol. 2005;26:1580-1582.
- Phan TA, Wargon O, Turner AM. Incontinentia pigmenti case series: clinical spectrum of incontinentia pigmenti in 53 female patients and their relatives. Clin Exp Dermatol. 2005;30:474-480.
- Volpe J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50:553-562.
- Pascual-Castroviejo I, Pascual-Pascual SI, Velazquez-Fragua R, et al. Incontinentia pigmenti: clinical and neuroimaging findings in a series of 12 patients. Neurologia. 2006;21:239-248.
To the Editor:
A 7-day-old full-term infant presented to the neonatal intensive care unit with poor feeding and altered consciousness. She was born at 39 weeks and 3 days to a gravida 1 mother with a pregnancy history complicated by maternal chorioamnionitis and gestational diabetes. During labor, nonreassuring fetal heart tones and arrest of labor prompted an uncomplicated cesarean delivery with normal Apgar scores at birth. The infant’s family history revealed only beta thalassemia minor in her father. At 5 to 7 days of life, the mother noted difficulty with feeding and poor latch along with lethargy and depressed consciousness in the infant.
Upon arrival to the neonatal intensive care unit, the infant was noted to have rhythmic lip-smacking behavior, intermittent nystagmus, mild hypotonia, and clonic movements of the left upper extremity. An electroencephalogram was markedly abnormal, capturing multiple seizures in the bilateral cortical hemispheres. She was loaded with phenobarbital with no further seizure activity. Brain magnetic resonance imaging revealed innumerable punctate foci of restricted diffusion with corresponding punctate hemorrhage within the frontal and parietal white matter, as well as cortical diffusion restriction within the occipital lobe, inferior temporal lobe, bilateral thalami, and corpus callosum (Figure 1). An exhaustive infectious workup also was completed and was unremarkable, though she was treated with broad-spectrum antimicrobials, including intravenous acyclovir.
Five days after being hospitalized (day 10 of life), a vesicular rash was noted on the arms and legs (Figure 2). Discussion with the patient’s mother revealed that the first signs of unusual skin lesions occurred as early as several days prior. There were no oral mucosal lesions or gross ocular abnormalities. No nail changes were appreciated. A bedside Tzanck preparation was negative for viral cytopathic changes. A skin biopsy was performed that demonstrated eosinophilic spongiosis with necrotic keratinocytes, typical of the vesicular stage of incontinentia pigmenti (IP)(Figure 3). An ophthalmology examination showed an arteriovenous malformation of the right eye with subtle neovascularization at the infratemporal periphery, consistent with known ocular manifestations of IP. The infant’s mother reported no history of notable dental abnormalities, hair loss, skin rashes, or nail changes. Genetic testing demonstrated the common IKBKG (inhibitor of κ light polypeptide gene enhancer in B cells, kinase gamma [formerly known as NEMO]) gene deletion on the X chromosome, consistent with IP.
She successfully underwent retinal laser ablative therapy for the ocular manifestations without further evidence of neovascularization. She developed a mild cataract that was not visually significant and required no intervention. Her brain abnormalities were thought to represent foci of necrosis with superimposed hemorrhagic transformation due to spontaneous degeneration of brain cells in which the mutated X chromosome was activated. No further treatment was indicated beyond suppression of the consequent seizures. There was no notable cortical edema or other medical indication for systemic glucocorticoid therapy. Phenobarbital was continued without further seizure events.
Several months after the initial presentation, a follow-up electroencephalogram was normal. Phenobarbital was slowly weaned and finally discontinued approximately 6 months after the initial event with no other reported seizures. She currently is achieving normal developmental milestones with the exception of slight motor delay and expected residual hypotonia.
Incontinentia pigmenti, also known as Bloch-Sulzberger syndrome, is a rare multisystem neuroectodermal disorder, primarily affecting the skin, central nervous system (CNS), and retinas. The disorder can be inherited in an X-linked dominant fashion and appears almost exclusively in women with typical in utero lethality seen in males. Most affected individuals have a sporadic, or de novo, mutation, which was likely the case in our patient given that her mother demonstrated no signs or symptoms.1 The pathogenesis of disease is a defect at chromosome Xq28 that is a region encoding the nuclear factor–κB essential modulator, IKBKG. Absence or mutation of IKBKG in IP results in failure to activate nuclear factor–κB and leaves cells vulnerable to cytokine-mediated apoptosis, especially after exposure to tumor necrosis factor α.2
Clinical manifestations of IP are present at or soon after birth. The cutaneous findings of this disorder are classically described as a step-wise progression through 4 distinct stages: (1) a linear and/or whorled vesicular eruption predominantly on the extremities at birth or within the first few weeks of life; (2) thickened linear or whorled verrucous plaques; (3) hyperpigmented streaks and whorls that may or may not correspond with prior affected areas that may resolve by adolescence; and (4) hypopigmented, possibly atrophic plaques on the extremities that may persist lifelong. Importantly, not every patient will experience each of these stages. Overlap can occur, and the time course of each stage is highly variable. Other ectodermal manifestations include dental abnormalities such as small, misshaped, or missing teeth; alopecia; and nail abnormalities. Ocular abnormalities associated with IP primarily occur in the retina, including vascular occlusion, neovascularization, hemorrhages, foveal abnormalities, as well as exudative and tractional detachments.3,4
It is crucial to recognize CNS anomalies in association with the cutaneous findings of IP, as CNS pathology can be severe with profound developmental implications. Central nervous system findings have been noted to correlate with the appearance of the vesicular stage of IP. A high index of suspicion is needed, as the disease can demonstrate progression within a short time.5-8 The most frequent anomalies include seizures, motor impairment, intellectual disability, and microcephaly.9,10 Some of the most commonly identified CNS lesions on imaging include necrosis or brain infarcts, atrophy, and lesions of the corpus callosum.7
The pathogenesis of observed CNS changes in IP is not well understood. There have been numerous proposals of a vascular mechanism, and a microangiopathic process appears to be most plausible. Mutations in IKBKG may result in interruption of signaling via vascular endothelial growth factor receptor 3 with a consequent impact on angiogenesis, supporting a vascular mechanism. Additionally, mutations in IKBKG lead to activation of eotaxin, an eosinophil-selective chemokine.9 Eotaxin activation results in eosinophilic degranulation that mediates the classic eosinophilic infiltrate seen in the classic skin histology of IP. Additionally, it has been shown that eotaxin is strongly expressed by endothelial cells in IP, and more abundant eosinophil degranulation may play a role in mediating vaso-occlusion.7 Other studies have found that the highest expression level of the IKBKG gene is in the CNS, potentially explaining the extensive imaging findings of hemorrhage and diffusion restriction in our patient. These features likely are attributable to apoptosis of cells possessing the mutated IKBKG gene.9-11
To the Editor:
A 7-day-old full-term infant presented to the neonatal intensive care unit with poor feeding and altered consciousness. She was born at 39 weeks and 3 days to a gravida 1 mother with a pregnancy history complicated by maternal chorioamnionitis and gestational diabetes. During labor, nonreassuring fetal heart tones and arrest of labor prompted an uncomplicated cesarean delivery with normal Apgar scores at birth. The infant’s family history revealed only beta thalassemia minor in her father. At 5 to 7 days of life, the mother noted difficulty with feeding and poor latch along with lethargy and depressed consciousness in the infant.
Upon arrival to the neonatal intensive care unit, the infant was noted to have rhythmic lip-smacking behavior, intermittent nystagmus, mild hypotonia, and clonic movements of the left upper extremity. An electroencephalogram was markedly abnormal, capturing multiple seizures in the bilateral cortical hemispheres. She was loaded with phenobarbital with no further seizure activity. Brain magnetic resonance imaging revealed innumerable punctate foci of restricted diffusion with corresponding punctate hemorrhage within the frontal and parietal white matter, as well as cortical diffusion restriction within the occipital lobe, inferior temporal lobe, bilateral thalami, and corpus callosum (Figure 1). An exhaustive infectious workup also was completed and was unremarkable, though she was treated with broad-spectrum antimicrobials, including intravenous acyclovir.
Five days after being hospitalized (day 10 of life), a vesicular rash was noted on the arms and legs (Figure 2). Discussion with the patient’s mother revealed that the first signs of unusual skin lesions occurred as early as several days prior. There were no oral mucosal lesions or gross ocular abnormalities. No nail changes were appreciated. A bedside Tzanck preparation was negative for viral cytopathic changes. A skin biopsy was performed that demonstrated eosinophilic spongiosis with necrotic keratinocytes, typical of the vesicular stage of incontinentia pigmenti (IP)(Figure 3). An ophthalmology examination showed an arteriovenous malformation of the right eye with subtle neovascularization at the infratemporal periphery, consistent with known ocular manifestations of IP. The infant’s mother reported no history of notable dental abnormalities, hair loss, skin rashes, or nail changes. Genetic testing demonstrated the common IKBKG (inhibitor of κ light polypeptide gene enhancer in B cells, kinase gamma [formerly known as NEMO]) gene deletion on the X chromosome, consistent with IP.
She successfully underwent retinal laser ablative therapy for the ocular manifestations without further evidence of neovascularization. She developed a mild cataract that was not visually significant and required no intervention. Her brain abnormalities were thought to represent foci of necrosis with superimposed hemorrhagic transformation due to spontaneous degeneration of brain cells in which the mutated X chromosome was activated. No further treatment was indicated beyond suppression of the consequent seizures. There was no notable cortical edema or other medical indication for systemic glucocorticoid therapy. Phenobarbital was continued without further seizure events.
Several months after the initial presentation, a follow-up electroencephalogram was normal. Phenobarbital was slowly weaned and finally discontinued approximately 6 months after the initial event with no other reported seizures. She currently is achieving normal developmental milestones with the exception of slight motor delay and expected residual hypotonia.
Incontinentia pigmenti, also known as Bloch-Sulzberger syndrome, is a rare multisystem neuroectodermal disorder, primarily affecting the skin, central nervous system (CNS), and retinas. The disorder can be inherited in an X-linked dominant fashion and appears almost exclusively in women with typical in utero lethality seen in males. Most affected individuals have a sporadic, or de novo, mutation, which was likely the case in our patient given that her mother demonstrated no signs or symptoms.1 The pathogenesis of disease is a defect at chromosome Xq28 that is a region encoding the nuclear factor–κB essential modulator, IKBKG. Absence or mutation of IKBKG in IP results in failure to activate nuclear factor–κB and leaves cells vulnerable to cytokine-mediated apoptosis, especially after exposure to tumor necrosis factor α.2
Clinical manifestations of IP are present at or soon after birth. The cutaneous findings of this disorder are classically described as a step-wise progression through 4 distinct stages: (1) a linear and/or whorled vesicular eruption predominantly on the extremities at birth or within the first few weeks of life; (2) thickened linear or whorled verrucous plaques; (3) hyperpigmented streaks and whorls that may or may not correspond with prior affected areas that may resolve by adolescence; and (4) hypopigmented, possibly atrophic plaques on the extremities that may persist lifelong. Importantly, not every patient will experience each of these stages. Overlap can occur, and the time course of each stage is highly variable. Other ectodermal manifestations include dental abnormalities such as small, misshaped, or missing teeth; alopecia; and nail abnormalities. Ocular abnormalities associated with IP primarily occur in the retina, including vascular occlusion, neovascularization, hemorrhages, foveal abnormalities, as well as exudative and tractional detachments.3,4
It is crucial to recognize CNS anomalies in association with the cutaneous findings of IP, as CNS pathology can be severe with profound developmental implications. Central nervous system findings have been noted to correlate with the appearance of the vesicular stage of IP. A high index of suspicion is needed, as the disease can demonstrate progression within a short time.5-8 The most frequent anomalies include seizures, motor impairment, intellectual disability, and microcephaly.9,10 Some of the most commonly identified CNS lesions on imaging include necrosis or brain infarcts, atrophy, and lesions of the corpus callosum.7
The pathogenesis of observed CNS changes in IP is not well understood. There have been numerous proposals of a vascular mechanism, and a microangiopathic process appears to be most plausible. Mutations in IKBKG may result in interruption of signaling via vascular endothelial growth factor receptor 3 with a consequent impact on angiogenesis, supporting a vascular mechanism. Additionally, mutations in IKBKG lead to activation of eotaxin, an eosinophil-selective chemokine.9 Eotaxin activation results in eosinophilic degranulation that mediates the classic eosinophilic infiltrate seen in the classic skin histology of IP. Additionally, it has been shown that eotaxin is strongly expressed by endothelial cells in IP, and more abundant eosinophil degranulation may play a role in mediating vaso-occlusion.7 Other studies have found that the highest expression level of the IKBKG gene is in the CNS, potentially explaining the extensive imaging findings of hemorrhage and diffusion restriction in our patient. These features likely are attributable to apoptosis of cells possessing the mutated IKBKG gene.9-11
- Ehrenreich M, Tarlow MM, Godlewska-Janusz E, et al. Incontinentia pigmenti (Bloch-Sulzberger syndrome): a systemic disorder. Cutis. 2007;79:355-362.
- Smahi A, Courtois G, Rabia SH, et al. The NF-kappaB signaling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet. 2002;11:2371-2375.
- O’Doherty M, McCreery K, Green AJ, et al. Incontinentia pigmenti—ophthalmological observation of a series of cases and review of the literature. Br J Ophthalmol. 2011;95:11-16.
- Swinney CC, Han DP, Karth PA. Incontinentia pigmenti: a comprehensive review and update. Ophthalmic Surg Lasers Imaging Retina. 2015;46:650-657.
- Hennel SJ, Ekert PG, Volpe JJ, et al. Insights into the pathogenesis of cerebral lesions in incontinentia pigmenti. Pediatr Neurol. 2003;29:148-150.
- Maingay-de Groof F, Lequin MH, Roofthooft DW, et al. Extensive cerebral infarction in the newborn due to incontinentia pigmenti. Eur J Paediatr Neurol. 2008;12:284-289.
- Minic´ S, Trpinac D, Obradovic´ M. Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet J Rare Dis. 2013;8:25-35.
- Wolf NI, Kramer N, Harting I, et al. Diffuse cortical necrosis in a neonate with incontinentia pigmenti and an encephalitis-like presentation. AJNR Am J Neuroradiol. 2005;26:1580-1582.
- Phan TA, Wargon O, Turner AM. Incontinentia pigmenti case series: clinical spectrum of incontinentia pigmenti in 53 female patients and their relatives. Clin Exp Dermatol. 2005;30:474-480.
- Volpe J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50:553-562.
- Pascual-Castroviejo I, Pascual-Pascual SI, Velazquez-Fragua R, et al. Incontinentia pigmenti: clinical and neuroimaging findings in a series of 12 patients. Neurologia. 2006;21:239-248.
- Ehrenreich M, Tarlow MM, Godlewska-Janusz E, et al. Incontinentia pigmenti (Bloch-Sulzberger syndrome): a systemic disorder. Cutis. 2007;79:355-362.
- Smahi A, Courtois G, Rabia SH, et al. The NF-kappaB signaling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum Mol Genet. 2002;11:2371-2375.
- O’Doherty M, McCreery K, Green AJ, et al. Incontinentia pigmenti—ophthalmological observation of a series of cases and review of the literature. Br J Ophthalmol. 2011;95:11-16.
- Swinney CC, Han DP, Karth PA. Incontinentia pigmenti: a comprehensive review and update. Ophthalmic Surg Lasers Imaging Retina. 2015;46:650-657.
- Hennel SJ, Ekert PG, Volpe JJ, et al. Insights into the pathogenesis of cerebral lesions in incontinentia pigmenti. Pediatr Neurol. 2003;29:148-150.
- Maingay-de Groof F, Lequin MH, Roofthooft DW, et al. Extensive cerebral infarction in the newborn due to incontinentia pigmenti. Eur J Paediatr Neurol. 2008;12:284-289.
- Minic´ S, Trpinac D, Obradovic´ M. Systematic review of central nervous system anomalies in incontinentia pigmenti. Orphanet J Rare Dis. 2013;8:25-35.
- Wolf NI, Kramer N, Harting I, et al. Diffuse cortical necrosis in a neonate with incontinentia pigmenti and an encephalitis-like presentation. AJNR Am J Neuroradiol. 2005;26:1580-1582.
- Phan TA, Wargon O, Turner AM. Incontinentia pigmenti case series: clinical spectrum of incontinentia pigmenti in 53 female patients and their relatives. Clin Exp Dermatol. 2005;30:474-480.
- Volpe J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50:553-562.
- Pascual-Castroviejo I, Pascual-Pascual SI, Velazquez-Fragua R, et al. Incontinentia pigmenti: clinical and neuroimaging findings in a series of 12 patients. Neurologia. 2006;21:239-248.
Practice Points
- Central nervous system involvement in incontinentia pigmenti (IP) may be profound and can present prior to the classic cutaneous findings.
- A high index of suspicion for IP should be maintained in neonatal vesicular eruptions of unclear etiology, especially in the setting of unexplained seizures and/or abnormal brain imaging.
Enfuvirtide-Induced Cutaneous Amyloidosis
To the Editor:
Cutaneous amyloidosis can be secondary to many causes. We describe a case of amyloidosis that was secondary to the deposition of an antiretroviral drug enfuvirtide and clinically presented as bullae over the anterior abdominal wall.
A 65-year-old man with HIV presented with pink vesicles and flaccid bullae on the anterolateral aspect of the lower abdomen (Figure 1) in areas of self-administered subcutaneous injections of enfuvirtide. He reported tissue swelling with a yellow discoloration immediately after injections that would spontaneously subside after a few minutes.
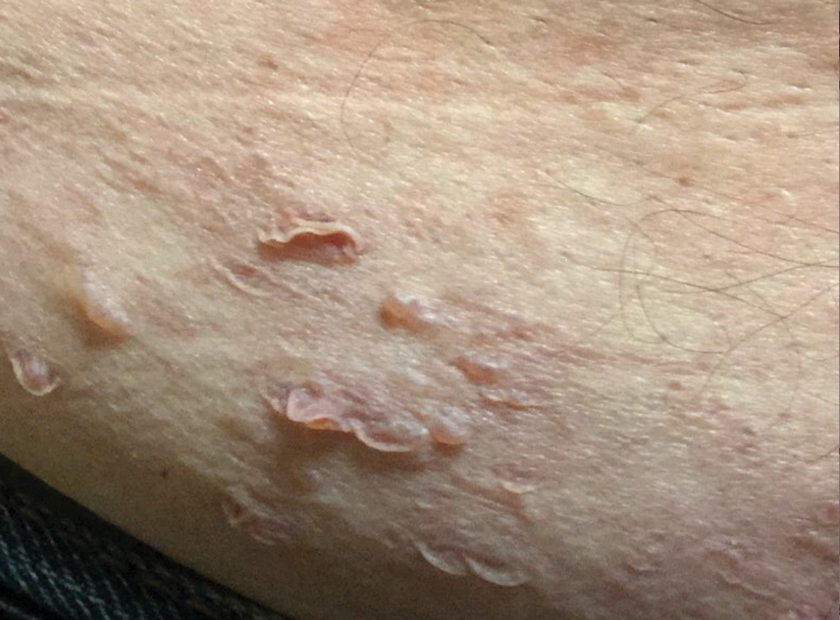
A biopsy from the left lateral abdomen revealed dilated vessels concentrically encompassed by pink globular material and nodular collections of the pink amorphous substance in the upper dermis (Figure 2), which was accompanied by a sparse, perivascular, lymphohistiocytic inflammatory infiltrate; scattered plasma cells; and rare eosinophils in a background of dermal edema. Although Congo red stain was negative, crystal violet revealed metachromatic staining of the globular material that was highlighted as dark violet against a blue background. Given these clinical and histopathologic findings, a diagnosis of drug-induced amyloidosis was made.
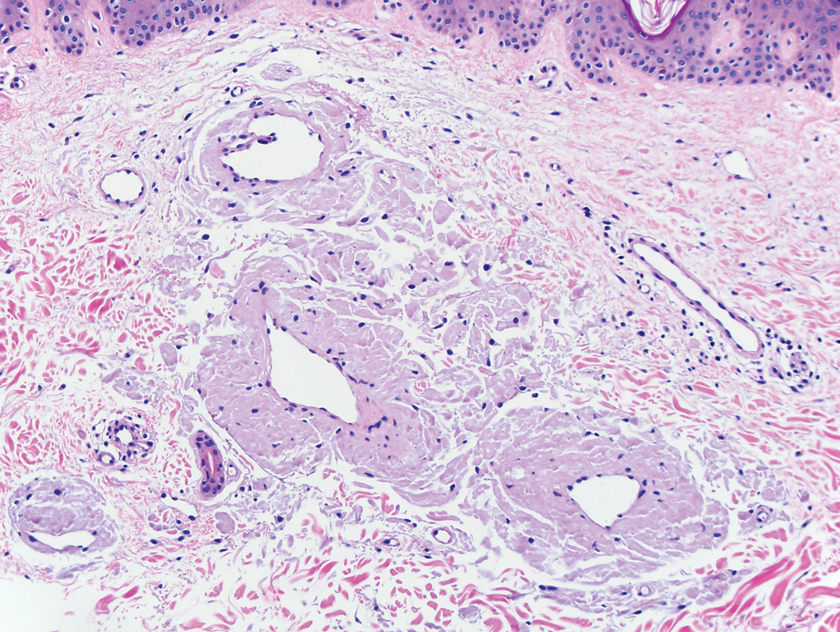
Amyloidosis refers to a group of disorders that result from misfolding of proteins in the characteristic beta-pleated sheet structure that can accumulate in various tissues. There are different subtypes of amyloidosis based on the type of protein deposited: immunoglobulin light chain protein (AL); serum amyloid A (AA), an acute-phase reactant accumulating in those with long-standing inflammatory conditions; beta-2 microglobulin (Ab2M) in patients with renal failure; keratin in macular and lichen amyloidosis; pharmaceutical-derived amyloid (eg, enfuvirtide, injectable insulin); and mutated proteins in hereditary amyloidosis such as transthyretin.1 Other familial forms include genetic variants of apolipolipoprotein AII (AApoAI, AApoAII), fibrinogen A alpha chain (AFib), lysozyme (ALys), cystatin C (ACys), and gelsolin (AGel).2
Cutaneous amyloidosis can stem from a systemic disease or arise as a localized phenomenon. Primary cutaneous amyloidosis can present as either macular, lichen, or nodular forms. The pathogenesis of cutaneous nodular amyloidosis differs from that of lichen and macular types and results from deposition of light chain–derived amyloid protein. In contrast, lichen and macular subtypes have keratin-derived amyloid deposits in the papillary dermis and stain positive for keratin antibodies, especially cytokeratins 5 and 6. Primary nodular amyloidosis has a 7% to 50% risk for developing systemic amyloidosis and a 9% risk for local recurrence, hence the necessity to assess for monoclonal gammopathy with urine light chains and serum immunoelectrophoresis.3
Drug-induced amyloidosis is a distinct type of cutaneous amyloidosis that histopathologically resembles nodular amyloidosis. Multiple drugs have been reported in this setting: insulin,4,5 enfuvirtide injections, and liraglutide.6 Enfuvirtide belongs to a class of antiretroviral agents and is a synthetic peptide composed of 36 amino acids. It inhibits the fusion of HIV with the host helper T cell by binding to glycoprotein 41.7 Enfuvirtide-related amyloidosis was described in 3 case reports, 2 that confirmed enfuvirtide as the amyloid constituent by protein analysis.8-10 One study analyzed the amyloid proteome in 50 cases of insulin-derived amyloidosis and 2 cases of enfuvirtide-derived amyloidosis. Laser microdissection–tandem microscopy revealed that the amyloid in such cases was composed of the drug enfuvirtide itself along with deposits of apolipoproteins (E, A-I, A-IV) and serum amyloid P component.4 Additional complications can occur at the site of enfuvirtide injections. A retrospective review of 7 patients with injection-site reactions to enfuvirtide described erythema, induration, and nodules, with histopathologic findings including hypersensitivity reactions and palisaded granulomas resembling granuloma annulare. Amorphous material was noted within histiocytes and in the surrounding connective tissue that was confirmed as enfuvirtide by immunoperoxidase staining.11
In summary, several types of cutaneous amyloidosis occur, including secondary cutaneous involvement by systemic amyloidosis and drug-induced amyloidosis, and notable histopathologic overlap exists between these types. Given the differing treatment requirements depending on the type of cutaneous amyloidosis, obtaining an appropriate clinical history, including the patient’s medication list, is important to ensure the correct diagnosis is reached. Protein analysis with mass spectrometry can be used if the nature of the amyloid remains indeterminate.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583-596.
- Ferri FF. Amyloidosis. In: Ferri F. Ferri’s Clinical Advisor 2016: 5 Books in 1. Elsevier; 2016.
- Kaltoft B, Schmidt G, Lauritzen AF, et al. Primary localised cutaneous amyloidosis—a systematic review. Dan Med J. 2013;60:A4727.
- D’Souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Sie MP, van der Wiel HE, Smedts FM, et al. Human recombinant insulin and amyloidosis: an unexpected association. Neth J Med. 2010;68:138-140.
- Martins CO, Lezcano C, Yi SS, et al. Novel iatrogenic amyloidosis caused by peptide drug liraglutide: a clinical mimic of AL amyloidosis. Haematologica. 2018;103:E610-E612.
- Lazzarin A, Clotet B, Cooper D, et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N Engl J Med. 2003;348:2186-2195.
- Naujokas A, Vidal CI, Mercer SE, et al. A novel form of amyloid deposited at the site of enfuvirtide injection. J Cutan Pathol. 2012;39:220-221; quiz 219.
- Mercer S, Whang T, Vidal C, et al. Massive amyloidosis at the site of enfuvirtide (Fuzeon) injection. J Cutan Pathol. 2011;38:93.
- Morilla ME, Kocher J, Harmaty M. Localized amyloidosis at the site of enfuvirtide injection. Ann Intern Med. 2009;151:515-516.
- Ball RA, Kinchelow T; ISR Substudy Group. Injection site reactions with the HIV-1 fusion inhibitor enfuvirtide. J Am Acad Dermatol. 2003;49:826-831.
To the Editor:
Cutaneous amyloidosis can be secondary to many causes. We describe a case of amyloidosis that was secondary to the deposition of an antiretroviral drug enfuvirtide and clinically presented as bullae over the anterior abdominal wall.
A 65-year-old man with HIV presented with pink vesicles and flaccid bullae on the anterolateral aspect of the lower abdomen (Figure 1) in areas of self-administered subcutaneous injections of enfuvirtide. He reported tissue swelling with a yellow discoloration immediately after injections that would spontaneously subside after a few minutes.
A biopsy from the left lateral abdomen revealed dilated vessels concentrically encompassed by pink globular material and nodular collections of the pink amorphous substance in the upper dermis (Figure 2), which was accompanied by a sparse, perivascular, lymphohistiocytic inflammatory infiltrate; scattered plasma cells; and rare eosinophils in a background of dermal edema. Although Congo red stain was negative, crystal violet revealed metachromatic staining of the globular material that was highlighted as dark violet against a blue background. Given these clinical and histopathologic findings, a diagnosis of drug-induced amyloidosis was made.
Amyloidosis refers to a group of disorders that result from misfolding of proteins in the characteristic beta-pleated sheet structure that can accumulate in various tissues. There are different subtypes of amyloidosis based on the type of protein deposited: immunoglobulin light chain protein (AL); serum amyloid A (AA), an acute-phase reactant accumulating in those with long-standing inflammatory conditions; beta-2 microglobulin (Ab2M) in patients with renal failure; keratin in macular and lichen amyloidosis; pharmaceutical-derived amyloid (eg, enfuvirtide, injectable insulin); and mutated proteins in hereditary amyloidosis such as transthyretin.1 Other familial forms include genetic variants of apolipolipoprotein AII (AApoAI, AApoAII), fibrinogen A alpha chain (AFib), lysozyme (ALys), cystatin C (ACys), and gelsolin (AGel).2
Cutaneous amyloidosis can stem from a systemic disease or arise as a localized phenomenon. Primary cutaneous amyloidosis can present as either macular, lichen, or nodular forms. The pathogenesis of cutaneous nodular amyloidosis differs from that of lichen and macular types and results from deposition of light chain–derived amyloid protein. In contrast, lichen and macular subtypes have keratin-derived amyloid deposits in the papillary dermis and stain positive for keratin antibodies, especially cytokeratins 5 and 6. Primary nodular amyloidosis has a 7% to 50% risk for developing systemic amyloidosis and a 9% risk for local recurrence, hence the necessity to assess for monoclonal gammopathy with urine light chains and serum immunoelectrophoresis.3
Drug-induced amyloidosis is a distinct type of cutaneous amyloidosis that histopathologically resembles nodular amyloidosis. Multiple drugs have been reported in this setting: insulin,4,5 enfuvirtide injections, and liraglutide.6 Enfuvirtide belongs to a class of antiretroviral agents and is a synthetic peptide composed of 36 amino acids. It inhibits the fusion of HIV with the host helper T cell by binding to glycoprotein 41.7 Enfuvirtide-related amyloidosis was described in 3 case reports, 2 that confirmed enfuvirtide as the amyloid constituent by protein analysis.8-10 One study analyzed the amyloid proteome in 50 cases of insulin-derived amyloidosis and 2 cases of enfuvirtide-derived amyloidosis. Laser microdissection–tandem microscopy revealed that the amyloid in such cases was composed of the drug enfuvirtide itself along with deposits of apolipoproteins (E, A-I, A-IV) and serum amyloid P component.4 Additional complications can occur at the site of enfuvirtide injections. A retrospective review of 7 patients with injection-site reactions to enfuvirtide described erythema, induration, and nodules, with histopathologic findings including hypersensitivity reactions and palisaded granulomas resembling granuloma annulare. Amorphous material was noted within histiocytes and in the surrounding connective tissue that was confirmed as enfuvirtide by immunoperoxidase staining.11
In summary, several types of cutaneous amyloidosis occur, including secondary cutaneous involvement by systemic amyloidosis and drug-induced amyloidosis, and notable histopathologic overlap exists between these types. Given the differing treatment requirements depending on the type of cutaneous amyloidosis, obtaining an appropriate clinical history, including the patient’s medication list, is important to ensure the correct diagnosis is reached. Protein analysis with mass spectrometry can be used if the nature of the amyloid remains indeterminate.
To the Editor:
Cutaneous amyloidosis can be secondary to many causes. We describe a case of amyloidosis that was secondary to the deposition of an antiretroviral drug enfuvirtide and clinically presented as bullae over the anterior abdominal wall.
A 65-year-old man with HIV presented with pink vesicles and flaccid bullae on the anterolateral aspect of the lower abdomen (Figure 1) in areas of self-administered subcutaneous injections of enfuvirtide. He reported tissue swelling with a yellow discoloration immediately after injections that would spontaneously subside after a few minutes.
A biopsy from the left lateral abdomen revealed dilated vessels concentrically encompassed by pink globular material and nodular collections of the pink amorphous substance in the upper dermis (Figure 2), which was accompanied by a sparse, perivascular, lymphohistiocytic inflammatory infiltrate; scattered plasma cells; and rare eosinophils in a background of dermal edema. Although Congo red stain was negative, crystal violet revealed metachromatic staining of the globular material that was highlighted as dark violet against a blue background. Given these clinical and histopathologic findings, a diagnosis of drug-induced amyloidosis was made.
Amyloidosis refers to a group of disorders that result from misfolding of proteins in the characteristic beta-pleated sheet structure that can accumulate in various tissues. There are different subtypes of amyloidosis based on the type of protein deposited: immunoglobulin light chain protein (AL); serum amyloid A (AA), an acute-phase reactant accumulating in those with long-standing inflammatory conditions; beta-2 microglobulin (Ab2M) in patients with renal failure; keratin in macular and lichen amyloidosis; pharmaceutical-derived amyloid (eg, enfuvirtide, injectable insulin); and mutated proteins in hereditary amyloidosis such as transthyretin.1 Other familial forms include genetic variants of apolipolipoprotein AII (AApoAI, AApoAII), fibrinogen A alpha chain (AFib), lysozyme (ALys), cystatin C (ACys), and gelsolin (AGel).2
Cutaneous amyloidosis can stem from a systemic disease or arise as a localized phenomenon. Primary cutaneous amyloidosis can present as either macular, lichen, or nodular forms. The pathogenesis of cutaneous nodular amyloidosis differs from that of lichen and macular types and results from deposition of light chain–derived amyloid protein. In contrast, lichen and macular subtypes have keratin-derived amyloid deposits in the papillary dermis and stain positive for keratin antibodies, especially cytokeratins 5 and 6. Primary nodular amyloidosis has a 7% to 50% risk for developing systemic amyloidosis and a 9% risk for local recurrence, hence the necessity to assess for monoclonal gammopathy with urine light chains and serum immunoelectrophoresis.3
Drug-induced amyloidosis is a distinct type of cutaneous amyloidosis that histopathologically resembles nodular amyloidosis. Multiple drugs have been reported in this setting: insulin,4,5 enfuvirtide injections, and liraglutide.6 Enfuvirtide belongs to a class of antiretroviral agents and is a synthetic peptide composed of 36 amino acids. It inhibits the fusion of HIV with the host helper T cell by binding to glycoprotein 41.7 Enfuvirtide-related amyloidosis was described in 3 case reports, 2 that confirmed enfuvirtide as the amyloid constituent by protein analysis.8-10 One study analyzed the amyloid proteome in 50 cases of insulin-derived amyloidosis and 2 cases of enfuvirtide-derived amyloidosis. Laser microdissection–tandem microscopy revealed that the amyloid in such cases was composed of the drug enfuvirtide itself along with deposits of apolipoproteins (E, A-I, A-IV) and serum amyloid P component.4 Additional complications can occur at the site of enfuvirtide injections. A retrospective review of 7 patients with injection-site reactions to enfuvirtide described erythema, induration, and nodules, with histopathologic findings including hypersensitivity reactions and palisaded granulomas resembling granuloma annulare. Amorphous material was noted within histiocytes and in the surrounding connective tissue that was confirmed as enfuvirtide by immunoperoxidase staining.11
In summary, several types of cutaneous amyloidosis occur, including secondary cutaneous involvement by systemic amyloidosis and drug-induced amyloidosis, and notable histopathologic overlap exists between these types. Given the differing treatment requirements depending on the type of cutaneous amyloidosis, obtaining an appropriate clinical history, including the patient’s medication list, is important to ensure the correct diagnosis is reached. Protein analysis with mass spectrometry can be used if the nature of the amyloid remains indeterminate.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583-596.
- Ferri FF. Amyloidosis. In: Ferri F. Ferri’s Clinical Advisor 2016: 5 Books in 1. Elsevier; 2016.
- Kaltoft B, Schmidt G, Lauritzen AF, et al. Primary localised cutaneous amyloidosis—a systematic review. Dan Med J. 2013;60:A4727.
- D’Souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Sie MP, van der Wiel HE, Smedts FM, et al. Human recombinant insulin and amyloidosis: an unexpected association. Neth J Med. 2010;68:138-140.
- Martins CO, Lezcano C, Yi SS, et al. Novel iatrogenic amyloidosis caused by peptide drug liraglutide: a clinical mimic of AL amyloidosis. Haematologica. 2018;103:E610-E612.
- Lazzarin A, Clotet B, Cooper D, et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N Engl J Med. 2003;348:2186-2195.
- Naujokas A, Vidal CI, Mercer SE, et al. A novel form of amyloid deposited at the site of enfuvirtide injection. J Cutan Pathol. 2012;39:220-221; quiz 219.
- Mercer S, Whang T, Vidal C, et al. Massive amyloidosis at the site of enfuvirtide (Fuzeon) injection. J Cutan Pathol. 2011;38:93.
- Morilla ME, Kocher J, Harmaty M. Localized amyloidosis at the site of enfuvirtide injection. Ann Intern Med. 2009;151:515-516.
- Ball RA, Kinchelow T; ISR Substudy Group. Injection site reactions with the HIV-1 fusion inhibitor enfuvirtide. J Am Acad Dermatol. 2003;49:826-831.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583-596.
- Ferri FF. Amyloidosis. In: Ferri F. Ferri’s Clinical Advisor 2016: 5 Books in 1. Elsevier; 2016.
- Kaltoft B, Schmidt G, Lauritzen AF, et al. Primary localised cutaneous amyloidosis—a systematic review. Dan Med J. 2013;60:A4727.
- D’Souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Sie MP, van der Wiel HE, Smedts FM, et al. Human recombinant insulin and amyloidosis: an unexpected association. Neth J Med. 2010;68:138-140.
- Martins CO, Lezcano C, Yi SS, et al. Novel iatrogenic amyloidosis caused by peptide drug liraglutide: a clinical mimic of AL amyloidosis. Haematologica. 2018;103:E610-E612.
- Lazzarin A, Clotet B, Cooper D, et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N Engl J Med. 2003;348:2186-2195.
- Naujokas A, Vidal CI, Mercer SE, et al. A novel form of amyloid deposited at the site of enfuvirtide injection. J Cutan Pathol. 2012;39:220-221; quiz 219.
- Mercer S, Whang T, Vidal C, et al. Massive amyloidosis at the site of enfuvirtide (Fuzeon) injection. J Cutan Pathol. 2011;38:93.
- Morilla ME, Kocher J, Harmaty M. Localized amyloidosis at the site of enfuvirtide injection. Ann Intern Med. 2009;151:515-516.
- Ball RA, Kinchelow T; ISR Substudy Group. Injection site reactions with the HIV-1 fusion inhibitor enfuvirtide. J Am Acad Dermatol. 2003;49:826-831.
Practice Points
- There are multiple types of cutaneous amyloidosis, and proper diagnosis is essential to direct treatment and follow-up care.
- Medication-associated amyloidosis is a rare type of amyloidosis that is not associated with systemic amyloidosis and is treated by switching to alternative medicines.
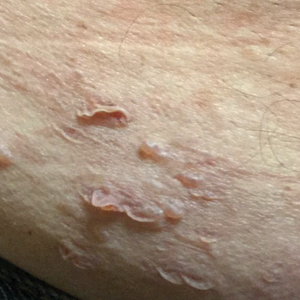
Wax Stripping and Isotretinoin Treatment: A Warning Not to Be Missed
To the Editor:
Oral isotretinoin is a widely used treatment modality in dermatologic practice that is highly effective for severe and recalcitrant acne vulgaris in addition to other conditions. Its use is accompanied by a variety of side effects that are mainly mucocutaneous. These dose-dependent side effects are experienced by almost all patients treated with this medication.1
A generally healthy 14-year-old adolescent girl presented with severe widespread erosions located in a linear pattern corresponding to areas of wax depilation on the shins and thighs (Figure). Approximately 5 months prior, the patient started oral isotretinoin 40 mg daily for severe and recalcitrant acne vulgaris. She was not taking other medications. After 4 months of treatment, during which the acne lesions improved and the patient experienced only mild xerosis and cheilitis, the dosage was increased to 60 mg daily. Three weeks later, the patient underwent wax depilation, which resulted in the erosions.
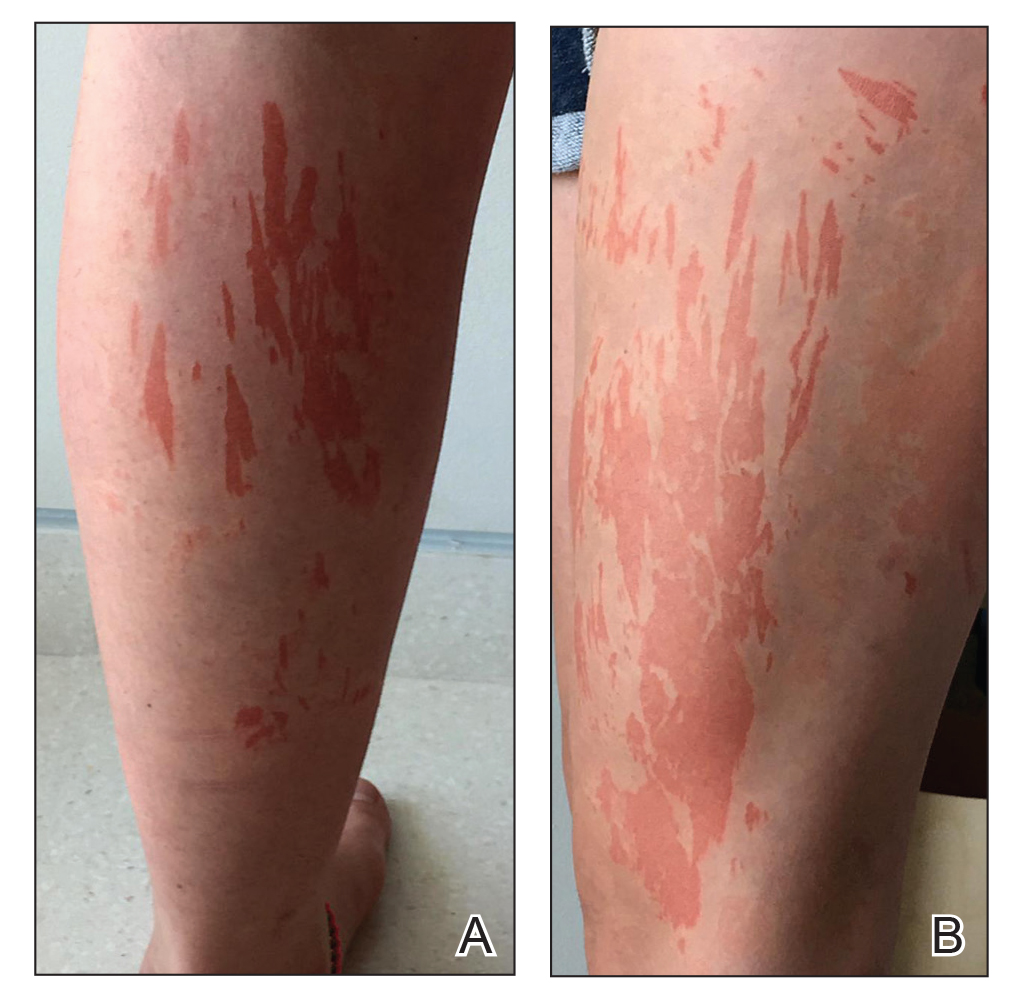
Oral isotretinoin treatment leads to structural and functional changes to the skin, related to epidermal dyscohesion and sebo-suppression. Although these changes may not be clinically evident in all patients, they still make the skin much more sensitive to external mechanical stimuli.1 Wax depilation commonly is used for treating excess hair on the body. Because it exerts remarkable mechanical stress on the epidermis, it may lead to epidermal stripping in patients taking isotretinoin, manifesting as widespread erosions and resulting in notable patient distress.
Dermatologists typically advise patients to avoid wax epilation while being treated with isotretinoin; however, some patients do not adhere to this recommendation. Also, there are dermatologists who are not aware of this potential side effect. In one survey (N=54), only 4% of consulting dermatologists were aware of this complication.2 A PubMed search of articles indexed for MEDLINE using the terms isotretinoin and wax revealed that this severe side effect with isotretinoin has been reported only 4 times in the medical literature.2-5 The fact that wax epilation should be avoided during isotretinoin treatment previously was not included in the prescribing information. It currently is included in the isotretinoin prescribing information6 with an indication not to perform wax depilation for 6 months after stopping treatment. This case should serve as a reminder to avoid wax depilation during isotretinoin treatment.
- Del Rosso JQ. Clinical relevance of skin barrier changes associated with the use of oral isotretinoin: the importance of barrier repair therapy in patient management. J Drugs Dermatol. 2013;12:626-631.
Woollons A, Price ML. Roaccutane and wax epilation: a cautionary tale. Br J Dermatol. 1997;137:839-840. - Egido Romo M. Isotretinoin and wax epilation. Br J Dermatol. 1991;124:393.
- Holmes SC, Thomson J. Isotretinoin and skin fragility. Br J Dermatol. 1995;132:165.
- Turel-Ermertcan A, Sahin MT, Yurtman D, et al. Inappropriate treatments at beauty centers: a case report of burns caused by hot wax stripping. J Dermatol. 2004;31:854-855.
- Accutane. Package insert. Roche; 2008.
To the Editor:
Oral isotretinoin is a widely used treatment modality in dermatologic practice that is highly effective for severe and recalcitrant acne vulgaris in addition to other conditions. Its use is accompanied by a variety of side effects that are mainly mucocutaneous. These dose-dependent side effects are experienced by almost all patients treated with this medication.1
A generally healthy 14-year-old adolescent girl presented with severe widespread erosions located in a linear pattern corresponding to areas of wax depilation on the shins and thighs (Figure). Approximately 5 months prior, the patient started oral isotretinoin 40 mg daily for severe and recalcitrant acne vulgaris. She was not taking other medications. After 4 months of treatment, during which the acne lesions improved and the patient experienced only mild xerosis and cheilitis, the dosage was increased to 60 mg daily. Three weeks later, the patient underwent wax depilation, which resulted in the erosions.
Oral isotretinoin treatment leads to structural and functional changes to the skin, related to epidermal dyscohesion and sebo-suppression. Although these changes may not be clinically evident in all patients, they still make the skin much more sensitive to external mechanical stimuli.1 Wax depilation commonly is used for treating excess hair on the body. Because it exerts remarkable mechanical stress on the epidermis, it may lead to epidermal stripping in patients taking isotretinoin, manifesting as widespread erosions and resulting in notable patient distress.
Dermatologists typically advise patients to avoid wax epilation while being treated with isotretinoin; however, some patients do not adhere to this recommendation. Also, there are dermatologists who are not aware of this potential side effect. In one survey (N=54), only 4% of consulting dermatologists were aware of this complication.2 A PubMed search of articles indexed for MEDLINE using the terms isotretinoin and wax revealed that this severe side effect with isotretinoin has been reported only 4 times in the medical literature.2-5 The fact that wax epilation should be avoided during isotretinoin treatment previously was not included in the prescribing information. It currently is included in the isotretinoin prescribing information6 with an indication not to perform wax depilation for 6 months after stopping treatment. This case should serve as a reminder to avoid wax depilation during isotretinoin treatment.
To the Editor:
Oral isotretinoin is a widely used treatment modality in dermatologic practice that is highly effective for severe and recalcitrant acne vulgaris in addition to other conditions. Its use is accompanied by a variety of side effects that are mainly mucocutaneous. These dose-dependent side effects are experienced by almost all patients treated with this medication.1
A generally healthy 14-year-old adolescent girl presented with severe widespread erosions located in a linear pattern corresponding to areas of wax depilation on the shins and thighs (Figure). Approximately 5 months prior, the patient started oral isotretinoin 40 mg daily for severe and recalcitrant acne vulgaris. She was not taking other medications. After 4 months of treatment, during which the acne lesions improved and the patient experienced only mild xerosis and cheilitis, the dosage was increased to 60 mg daily. Three weeks later, the patient underwent wax depilation, which resulted in the erosions.
Oral isotretinoin treatment leads to structural and functional changes to the skin, related to epidermal dyscohesion and sebo-suppression. Although these changes may not be clinically evident in all patients, they still make the skin much more sensitive to external mechanical stimuli.1 Wax depilation commonly is used for treating excess hair on the body. Because it exerts remarkable mechanical stress on the epidermis, it may lead to epidermal stripping in patients taking isotretinoin, manifesting as widespread erosions and resulting in notable patient distress.
Dermatologists typically advise patients to avoid wax epilation while being treated with isotretinoin; however, some patients do not adhere to this recommendation. Also, there are dermatologists who are not aware of this potential side effect. In one survey (N=54), only 4% of consulting dermatologists were aware of this complication.2 A PubMed search of articles indexed for MEDLINE using the terms isotretinoin and wax revealed that this severe side effect with isotretinoin has been reported only 4 times in the medical literature.2-5 The fact that wax epilation should be avoided during isotretinoin treatment previously was not included in the prescribing information. It currently is included in the isotretinoin prescribing information6 with an indication not to perform wax depilation for 6 months after stopping treatment. This case should serve as a reminder to avoid wax depilation during isotretinoin treatment.
- Del Rosso JQ. Clinical relevance of skin barrier changes associated with the use of oral isotretinoin: the importance of barrier repair therapy in patient management. J Drugs Dermatol. 2013;12:626-631.
Woollons A, Price ML. Roaccutane and wax epilation: a cautionary tale. Br J Dermatol. 1997;137:839-840. - Egido Romo M. Isotretinoin and wax epilation. Br J Dermatol. 1991;124:393.
- Holmes SC, Thomson J. Isotretinoin and skin fragility. Br J Dermatol. 1995;132:165.
- Turel-Ermertcan A, Sahin MT, Yurtman D, et al. Inappropriate treatments at beauty centers: a case report of burns caused by hot wax stripping. J Dermatol. 2004;31:854-855.
- Accutane. Package insert. Roche; 2008.
- Del Rosso JQ. Clinical relevance of skin barrier changes associated with the use of oral isotretinoin: the importance of barrier repair therapy in patient management. J Drugs Dermatol. 2013;12:626-631.
Woollons A, Price ML. Roaccutane and wax epilation: a cautionary tale. Br J Dermatol. 1997;137:839-840. - Egido Romo M. Isotretinoin and wax epilation. Br J Dermatol. 1991;124:393.
- Holmes SC, Thomson J. Isotretinoin and skin fragility. Br J Dermatol. 1995;132:165.
- Turel-Ermertcan A, Sahin MT, Yurtman D, et al. Inappropriate treatments at beauty centers: a case report of burns caused by hot wax stripping. J Dermatol. 2004;31:854-855.
- Accutane. Package insert. Roche; 2008.
Practice Points
- Oral isotretinoin treatment leads to structural and functional changes to the skin, making it much more sensitive to external mechanical stimuli.
- Wax depilation may lead to epidermal stripping in patients taking isotretinoin and therefore should be avoided in these patients.
Cutaneous Cholesterol Embolization to the Lower Trunk: An Underrecognized Presentation
To the Editor:
A 65-year-old man with severe atherosclerotic disease developed multiple painful eschars on the lower abdomen, thighs, sacrum, and perineum. He initially presented with myocardial ischemia and claudication and underwent 3 cardiac catheterizations as well as stenting of the superficial femoral artery. Within 2 weeks, he developed exquisitely tender nodules on the lower abdomen, clinically presumed to be sites of enoxaparin injections. These lesions gradually expanded and ulcerated to involve the sacrum, buttock, perineum, and upper thighs (Figure 1). Two punch biopsies from ulcerated skin taken 10 days apart demonstrated necrosis of skin and subcutaneous fat without evidence of vasculitis, vasculopathy, emboli, or notable inflammation despite examination of multiple levels of all submitted tissue. A definitive cause for the ulcerations remained elusive with development of new lesions. A third incisional biopsy of a newly developed, nonulcerated, subcutaneous nodule performed 8 weeks after presentation revealed multiple cholesterol emboli (Figure 2). He was treated with warfarin and clopidogrel bisulfate as well as local wound care. The lesions slowly resolved over the next 4 to 6 months.
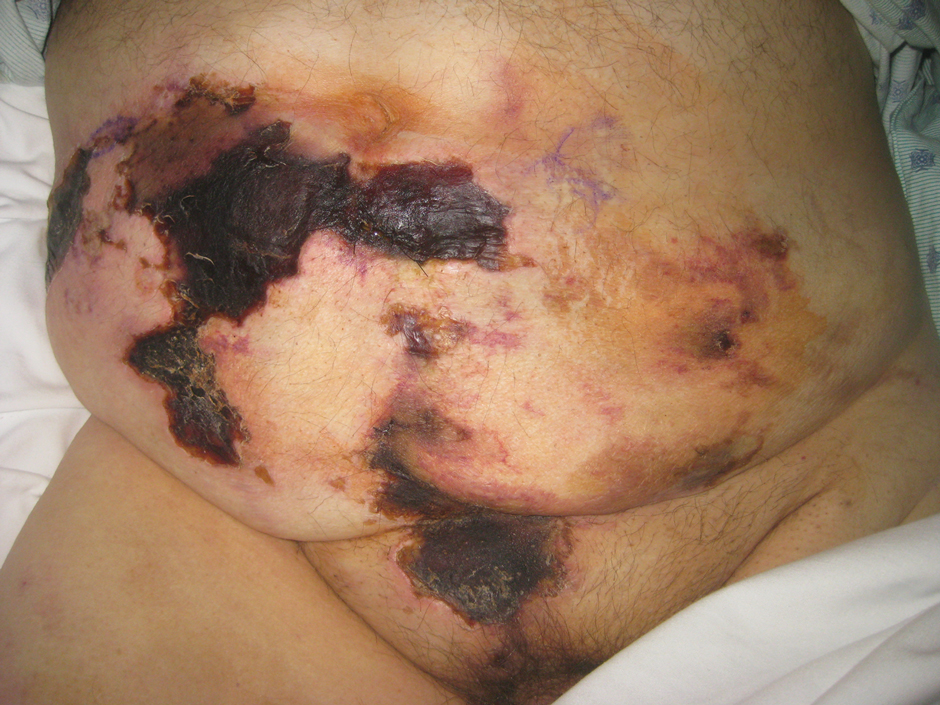
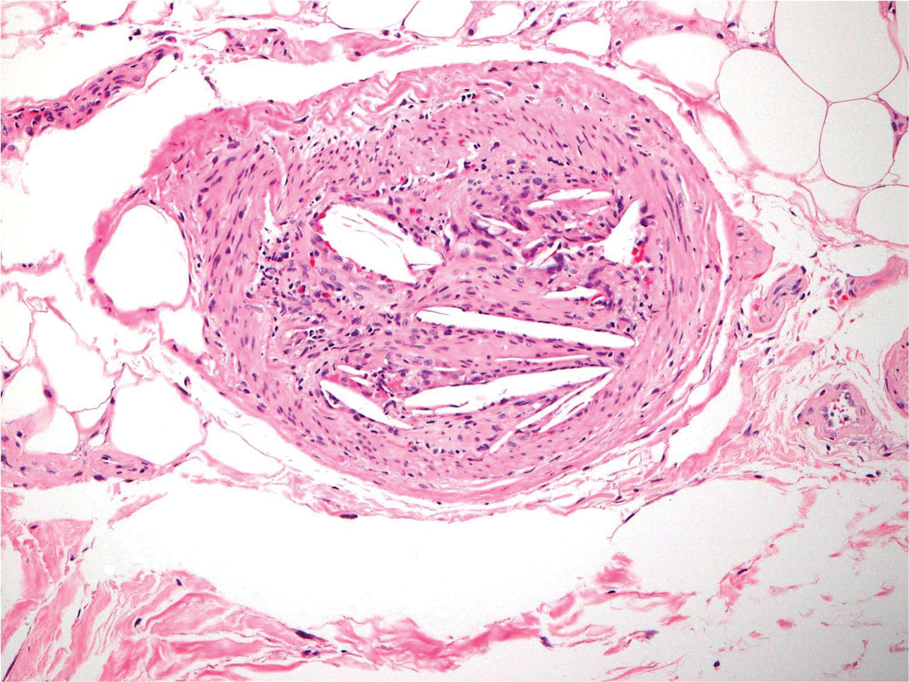
Cholesterol embolization syndrome occurs when disrupted atherosclerotic plaques embolize from large proximal arteries to more distal arterioles, resulting in ischemic damage to 1 or more organ systems.1 It can occur spontaneously but often is a consequence of thrombolytic therapy, anticoagulation, and angioinvasive procedures.2,3 Cutaneous manifestations include livedo reticularis, retiform purpura, nodules, and gangrene. Although livedo reticularis may extend from the legs to the trunk, gangrenous lesions predominantly involve the distal digits.
This case illustrates the challenge in diagnosis of cholesterol emboli, both clinically and histologically. Cutaneous lesions are morphologically variable and often occur with systemic manifestations, mimicking numerous conditions.1 Lower extremity involvement is a well-known occurrence in cholesterol embolization (ie, blue toe syndrome); however, periumbilical and lumbosacral lesions have not been emphasized in the dermatologic or peripheral vascular literature. Our patient’s initial diagnosis was enoxaparin necrosis at abdominal injection sites; however, this unusual distribution of lesions was ultimately determined to be the consequence of cholesterol embolization from the inferior epigastric and superficial external pudendal arteries at the time of stenting of the superficial femoral artery. Proximal truncal involvement should be recognized as an atypical but important cutaneous manifestation to facilitate timely diagnosis and treatment.4,5
Our patient’s course also highlights the potential need for multiple biopsies. Although the gold standard for diagnosis is histologic confirmation, a negative biopsy does not always exclude cholesterol emboli, and one should have a low threshold to perform additional biopsies in the appropriate clinical setting.
- Fine MJ, Kapoor W, Falanga V. Cholesterol crystal embolization: a review of 221 cases in the English literature. Angiology. 1987;38:769-784.
- Fukumoto Y, Tsutsui H, Tsuchihashi M, et al. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol. 2003;42:211-216.
- Karalis DG, Chandrasekaran K, Victor MF, et al. Recognition and embolic potential of intraaortic atherosclerotic debris. J Am Coll Cardiol. 1991;17:73.
- Zaytsev P, Miller K, Pellettiere EV. Cutaneous cholesterol emboli with infarction clinically mimicking heparin necrosis—a case report. Angiology. 1986;37:471-476.
- Erdim M, Tezel E, Biskin N. A case of skin necrosis as a result of cholesterol crystal embolisation. J Plast Reconstr Aesthet Surg. 2006;59:429-432.
To the Editor:
A 65-year-old man with severe atherosclerotic disease developed multiple painful eschars on the lower abdomen, thighs, sacrum, and perineum. He initially presented with myocardial ischemia and claudication and underwent 3 cardiac catheterizations as well as stenting of the superficial femoral artery. Within 2 weeks, he developed exquisitely tender nodules on the lower abdomen, clinically presumed to be sites of enoxaparin injections. These lesions gradually expanded and ulcerated to involve the sacrum, buttock, perineum, and upper thighs (Figure 1). Two punch biopsies from ulcerated skin taken 10 days apart demonstrated necrosis of skin and subcutaneous fat without evidence of vasculitis, vasculopathy, emboli, or notable inflammation despite examination of multiple levels of all submitted tissue. A definitive cause for the ulcerations remained elusive with development of new lesions. A third incisional biopsy of a newly developed, nonulcerated, subcutaneous nodule performed 8 weeks after presentation revealed multiple cholesterol emboli (Figure 2). He was treated with warfarin and clopidogrel bisulfate as well as local wound care. The lesions slowly resolved over the next 4 to 6 months.
Cholesterol embolization syndrome occurs when disrupted atherosclerotic plaques embolize from large proximal arteries to more distal arterioles, resulting in ischemic damage to 1 or more organ systems.1 It can occur spontaneously but often is a consequence of thrombolytic therapy, anticoagulation, and angioinvasive procedures.2,3 Cutaneous manifestations include livedo reticularis, retiform purpura, nodules, and gangrene. Although livedo reticularis may extend from the legs to the trunk, gangrenous lesions predominantly involve the distal digits.
This case illustrates the challenge in diagnosis of cholesterol emboli, both clinically and histologically. Cutaneous lesions are morphologically variable and often occur with systemic manifestations, mimicking numerous conditions.1 Lower extremity involvement is a well-known occurrence in cholesterol embolization (ie, blue toe syndrome); however, periumbilical and lumbosacral lesions have not been emphasized in the dermatologic or peripheral vascular literature. Our patient’s initial diagnosis was enoxaparin necrosis at abdominal injection sites; however, this unusual distribution of lesions was ultimately determined to be the consequence of cholesterol embolization from the inferior epigastric and superficial external pudendal arteries at the time of stenting of the superficial femoral artery. Proximal truncal involvement should be recognized as an atypical but important cutaneous manifestation to facilitate timely diagnosis and treatment.4,5
Our patient’s course also highlights the potential need for multiple biopsies. Although the gold standard for diagnosis is histologic confirmation, a negative biopsy does not always exclude cholesterol emboli, and one should have a low threshold to perform additional biopsies in the appropriate clinical setting.
To the Editor:
A 65-year-old man with severe atherosclerotic disease developed multiple painful eschars on the lower abdomen, thighs, sacrum, and perineum. He initially presented with myocardial ischemia and claudication and underwent 3 cardiac catheterizations as well as stenting of the superficial femoral artery. Within 2 weeks, he developed exquisitely tender nodules on the lower abdomen, clinically presumed to be sites of enoxaparin injections. These lesions gradually expanded and ulcerated to involve the sacrum, buttock, perineum, and upper thighs (Figure 1). Two punch biopsies from ulcerated skin taken 10 days apart demonstrated necrosis of skin and subcutaneous fat without evidence of vasculitis, vasculopathy, emboli, or notable inflammation despite examination of multiple levels of all submitted tissue. A definitive cause for the ulcerations remained elusive with development of new lesions. A third incisional biopsy of a newly developed, nonulcerated, subcutaneous nodule performed 8 weeks after presentation revealed multiple cholesterol emboli (Figure 2). He was treated with warfarin and clopidogrel bisulfate as well as local wound care. The lesions slowly resolved over the next 4 to 6 months.
Cholesterol embolization syndrome occurs when disrupted atherosclerotic plaques embolize from large proximal arteries to more distal arterioles, resulting in ischemic damage to 1 or more organ systems.1 It can occur spontaneously but often is a consequence of thrombolytic therapy, anticoagulation, and angioinvasive procedures.2,3 Cutaneous manifestations include livedo reticularis, retiform purpura, nodules, and gangrene. Although livedo reticularis may extend from the legs to the trunk, gangrenous lesions predominantly involve the distal digits.
This case illustrates the challenge in diagnosis of cholesterol emboli, both clinically and histologically. Cutaneous lesions are morphologically variable and often occur with systemic manifestations, mimicking numerous conditions.1 Lower extremity involvement is a well-known occurrence in cholesterol embolization (ie, blue toe syndrome); however, periumbilical and lumbosacral lesions have not been emphasized in the dermatologic or peripheral vascular literature. Our patient’s initial diagnosis was enoxaparin necrosis at abdominal injection sites; however, this unusual distribution of lesions was ultimately determined to be the consequence of cholesterol embolization from the inferior epigastric and superficial external pudendal arteries at the time of stenting of the superficial femoral artery. Proximal truncal involvement should be recognized as an atypical but important cutaneous manifestation to facilitate timely diagnosis and treatment.4,5
Our patient’s course also highlights the potential need for multiple biopsies. Although the gold standard for diagnosis is histologic confirmation, a negative biopsy does not always exclude cholesterol emboli, and one should have a low threshold to perform additional biopsies in the appropriate clinical setting.
- Fine MJ, Kapoor W, Falanga V. Cholesterol crystal embolization: a review of 221 cases in the English literature. Angiology. 1987;38:769-784.
- Fukumoto Y, Tsutsui H, Tsuchihashi M, et al. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol. 2003;42:211-216.
- Karalis DG, Chandrasekaran K, Victor MF, et al. Recognition and embolic potential of intraaortic atherosclerotic debris. J Am Coll Cardiol. 1991;17:73.
- Zaytsev P, Miller K, Pellettiere EV. Cutaneous cholesterol emboli with infarction clinically mimicking heparin necrosis—a case report. Angiology. 1986;37:471-476.
- Erdim M, Tezel E, Biskin N. A case of skin necrosis as a result of cholesterol crystal embolisation. J Plast Reconstr Aesthet Surg. 2006;59:429-432.
- Fine MJ, Kapoor W, Falanga V. Cholesterol crystal embolization: a review of 221 cases in the English literature. Angiology. 1987;38:769-784.
- Fukumoto Y, Tsutsui H, Tsuchihashi M, et al. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol. 2003;42:211-216.
- Karalis DG, Chandrasekaran K, Victor MF, et al. Recognition and embolic potential of intraaortic atherosclerotic debris. J Am Coll Cardiol. 1991;17:73.
- Zaytsev P, Miller K, Pellettiere EV. Cutaneous cholesterol emboli with infarction clinically mimicking heparin necrosis—a case report. Angiology. 1986;37:471-476.
- Erdim M, Tezel E, Biskin N. A case of skin necrosis as a result of cholesterol crystal embolisation. J Plast Reconstr Aesthet Surg. 2006;59:429-432.
Practice Points
- Cholesterol embolization may occur in proximal locations, and index of suspicion should be high in patients who are at risk.
- Several biopsies may be necessary to make a diagnosis of cholesterol emboli.

Candida Esophagitis Associated With Adalimumab for Hidradenitis Suppurativa
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory disease characterized by the development of painful abscesses, fistulous tracts, and scars. It most commonly affects the apocrine gland–bearing areas of the body such as the axillary, inguinal, and anogenital regions. With a prevalence of approximately 1%, HS can lead to notable morbidity.1 The pathogenesis is thought to be due to occlusion of terminal hair follicles that subsequently stimulates release of proinflammatory cytokines from nearby keratinocytes. The mechanism of initial occlusion is not well understood but may be due to friction or trauma. An inflammatory mechanism of disease also has been hypothesized; however, the exact cytokine profile is not known. Treatment of HS consists of several different modalities, including oral retinoids, antibiotics, antiandrogenic therapy, and surgery.1,2 Adalimumab is a well-known biologic that has been approved by the US Food and Drug Administration for the treatment of HS.
Adalimumab is a human monoclonal antibody against tumor necrosis factor (TNF) α and is thought to improve HS by several mechanisms. Inhibition of TNF-α and other proinflammatory cytokines found in inflammatory lesions and apocrine glands directly decreases the severity of lesion size and the frequency of recurrence.3 Adalimumab also is thought to downregulate expression of keratin 6 and prevent the hyperkeratinization seen in HS.4 Additionally, TNF-α inhibition decreases production of IL-1, which has been shown to cause hypercornification of follicles and perpetuate HS pathogenesis.5
A 41-year-old woman with a history of endometriosis, adenomyosis, polycystic ovary syndrome, interstitial cystitis, asthma, fibromyalgia, depression, and Hashimoto thyroiditis presented to our dermatology clinic with active draining lesions and sinus tracts in the perivaginal area that were consistent with HS, which initially was treated with doxycycline 100 mg twice daily. She experienced minimal improvement of the HS lesions at 2-month follow-up.
Due to disease severity, adalimumab was started. The patient received a loading dose of 4 injections totaling 160 mg and 80 mg on day 15, followed by a maintenance dose of 40 mg/0.4 mL weekly. The patient reported substantial improvement of pain, and complete resolution of active lesions was noted on physical examination after 4 weeks of treatment with adalimumab.
Six weeks after adalimumab was started, the patient developed severe dysphagia. She was evaluated by a gastroenterologist and underwent endoscopy (Figure), which led to a diagnosis of esophageal candidiasis. Adalimumab was discontinued immediately thereafter. The patient started treatment with nystatin oral rinse 4 times daily and oral fluconazole 200 mg daily. The candidiasis resolved within 2 weeks; however, she experienced recurrence of HS with draining lesions in the perivaginal area approximately 8 weeks after discontinuation of adalimumab. The patient requested to restart adalimumab treatment despite the recent history of esophagitis. Adalimumab 40 mg/0.4 mL weekly was restarted along with oral fluconazole 200 mg twice weekly and nystatin oral rinse 4 times daily. This regimen resulted in complete resolution of HS symptoms within 6 weeks with no recurrence of esophageal candidiasis during 6 months of follow-up.
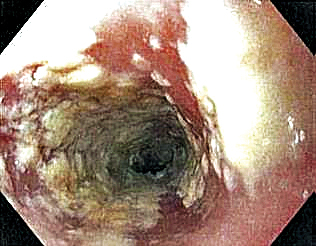
Although the side effect of Candida esophagitis associated with adalimumab treatment in our patient may be logical given the medication’s mechanism of action and side-effect profile, this case warrants additional attention. An increase in fungal infections occurs from treatment with adalimumab because TNF-α is involved in many immune regulatory steps that counteract infection. Candida typically activates the innate immune system through macrophages via pathogen-associated molecular pattern stimulation, subsequently stimulating the release of inflammatory cytokines such as TNF-α. The cellular immune system also is activated. Helper T cells (TH1) release TNF-α along with other proinflammatory cytokines to increase phagocytosis in polymorphonuclear cells and macrophages.6 Thus, inhibition of TNF-α compromises innate and cellular immunity, thereby increasing susceptibility to fungal organisms.
A PubMed search of articles indexed for MEDLINE using the terms Candida, candidiasis, esophageal, adalimumab, anti-TNF, and TNF revealed no reports of esophageal candidiasis in patients receiving adalimumab or any of the TNF inhibitors. Candida laryngitis was reported in a patient receiving adalimumab for treatment of rheumatoid arthritis.7 Other studies have demonstrated an incidence of mucocutaneous candidiasis, most notably oropharyngeal and vaginal candidiasis.8-10 One study found that anti-TNF medications were associated with an increased risk for candidiasis by a hazard ratio of 2.7 in patients with Crohn disease.8 Other studies have shown that the highest incidence of fungal infection is seen with the use of infliximab, while adalimumab is associated with lower rates of fungal infection.9,10 Although it is known that anti-TNF therapy predisposes patients to fungal infection, the dose of medication known to preclude the highest risk has not been studied. Furthermore, most studies assess rates of Candida infection in individuals receiving anti-TNF therapy in addition to several other immunosuppressant agents (ie, corticosteroids), which confounds the interpretation of results. Additional studies assessing rates of Candida and other opportunistic infections associated with use of adalimumab alone are needed to better guide clinical practices in dermatology.
Patients receiving adalimumab for dermatologic or other conditions should be closely monitored for opportunistic infections. Although immunomodulatory medications offer promising therapeutic benefits in patients with HS, larger studies regarding treatment with anti-TNF agents in HS are warranted to prevent complications from treatment and promote long-term efficacy and safety.
- Kurayev A, Ashkar H, Saraiya A, et al. Hidradenitis suppurativa: review of the pathogenesis and treatment. J Drugs Dermatol. 2016;15:1107-1022.
- Rambhatla PV, Lim HW, Hamzavi I. A systematic review of treatments for hidradenitis suppurativa. Arch Dermatol. 2012;148:439-446.
- van der Zee HH, de Ruiter L, van den Broecke DG, et al. Elevated levels of tumour necrosis factor (TNF)-alpha, interleukin (IL)-1beta and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-alpha and IL-1beta. Br J Dermatol. 2011;164:1292-1298.
- Shuja F, Chan CS, Rosen T. Biologic drugs for the treatment of hidradenitis suppurativa: an evidence-based review. Dermatol Clin. 2010;28:511-521, 523-514.
- Kutsch CL, Norris DA, Arend WP. Tumor necrosis factor-alpha induces interleukin-1 alpha and interleukin-1 receptor antagonist production by cultured human keratinocytes. J Invest Dermatol. 1993;101:79-85.
- Senet JM. Risk factors and physiopathology of candidiasis. Rev Iberoam Micol. 1997;14:6-13.
- Kobak S, Yilmaz H, Guclu O, et al. Severe candida laryngitis in a patient with rheumatoid arthritis treated with adalimumab. Eur J Rheumatol. 2014;1:167-169.
- Marehbian J, Arrighi HM, Hass S, et al. Adverse events associated with common therapy regimens for moderate-to-severe Crohn’s disease. Am J Gastroenterol. 2009;104:2524-2533.
- Tsiodras S, Samonis G, Boumpas DT, et al. Fungal infections complicating tumor necrosis factor alpha blockade therapy. Mayo Clin Proc. 2008;83:181-194.
- Aikawa NE, Rosa DT, Del Negro GM, et al. Systemic and localized infection by Candida species in patients with rheumatic diseases receiving anti-TNF therapy [in Portuguese]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.03.010
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory disease characterized by the development of painful abscesses, fistulous tracts, and scars. It most commonly affects the apocrine gland–bearing areas of the body such as the axillary, inguinal, and anogenital regions. With a prevalence of approximately 1%, HS can lead to notable morbidity.1 The pathogenesis is thought to be due to occlusion of terminal hair follicles that subsequently stimulates release of proinflammatory cytokines from nearby keratinocytes. The mechanism of initial occlusion is not well understood but may be due to friction or trauma. An inflammatory mechanism of disease also has been hypothesized; however, the exact cytokine profile is not known. Treatment of HS consists of several different modalities, including oral retinoids, antibiotics, antiandrogenic therapy, and surgery.1,2 Adalimumab is a well-known biologic that has been approved by the US Food and Drug Administration for the treatment of HS.
Adalimumab is a human monoclonal antibody against tumor necrosis factor (TNF) α and is thought to improve HS by several mechanisms. Inhibition of TNF-α and other proinflammatory cytokines found in inflammatory lesions and apocrine glands directly decreases the severity of lesion size and the frequency of recurrence.3 Adalimumab also is thought to downregulate expression of keratin 6 and prevent the hyperkeratinization seen in HS.4 Additionally, TNF-α inhibition decreases production of IL-1, which has been shown to cause hypercornification of follicles and perpetuate HS pathogenesis.5
A 41-year-old woman with a history of endometriosis, adenomyosis, polycystic ovary syndrome, interstitial cystitis, asthma, fibromyalgia, depression, and Hashimoto thyroiditis presented to our dermatology clinic with active draining lesions and sinus tracts in the perivaginal area that were consistent with HS, which initially was treated with doxycycline 100 mg twice daily. She experienced minimal improvement of the HS lesions at 2-month follow-up.
Due to disease severity, adalimumab was started. The patient received a loading dose of 4 injections totaling 160 mg and 80 mg on day 15, followed by a maintenance dose of 40 mg/0.4 mL weekly. The patient reported substantial improvement of pain, and complete resolution of active lesions was noted on physical examination after 4 weeks of treatment with adalimumab.
Six weeks after adalimumab was started, the patient developed severe dysphagia. She was evaluated by a gastroenterologist and underwent endoscopy (Figure), which led to a diagnosis of esophageal candidiasis. Adalimumab was discontinued immediately thereafter. The patient started treatment with nystatin oral rinse 4 times daily and oral fluconazole 200 mg daily. The candidiasis resolved within 2 weeks; however, she experienced recurrence of HS with draining lesions in the perivaginal area approximately 8 weeks after discontinuation of adalimumab. The patient requested to restart adalimumab treatment despite the recent history of esophagitis. Adalimumab 40 mg/0.4 mL weekly was restarted along with oral fluconazole 200 mg twice weekly and nystatin oral rinse 4 times daily. This regimen resulted in complete resolution of HS symptoms within 6 weeks with no recurrence of esophageal candidiasis during 6 months of follow-up.
Although the side effect of Candida esophagitis associated with adalimumab treatment in our patient may be logical given the medication’s mechanism of action and side-effect profile, this case warrants additional attention. An increase in fungal infections occurs from treatment with adalimumab because TNF-α is involved in many immune regulatory steps that counteract infection. Candida typically activates the innate immune system through macrophages via pathogen-associated molecular pattern stimulation, subsequently stimulating the release of inflammatory cytokines such as TNF-α. The cellular immune system also is activated. Helper T cells (TH1) release TNF-α along with other proinflammatory cytokines to increase phagocytosis in polymorphonuclear cells and macrophages.6 Thus, inhibition of TNF-α compromises innate and cellular immunity, thereby increasing susceptibility to fungal organisms.
A PubMed search of articles indexed for MEDLINE using the terms Candida, candidiasis, esophageal, adalimumab, anti-TNF, and TNF revealed no reports of esophageal candidiasis in patients receiving adalimumab or any of the TNF inhibitors. Candida laryngitis was reported in a patient receiving adalimumab for treatment of rheumatoid arthritis.7 Other studies have demonstrated an incidence of mucocutaneous candidiasis, most notably oropharyngeal and vaginal candidiasis.8-10 One study found that anti-TNF medications were associated with an increased risk for candidiasis by a hazard ratio of 2.7 in patients with Crohn disease.8 Other studies have shown that the highest incidence of fungal infection is seen with the use of infliximab, while adalimumab is associated with lower rates of fungal infection.9,10 Although it is known that anti-TNF therapy predisposes patients to fungal infection, the dose of medication known to preclude the highest risk has not been studied. Furthermore, most studies assess rates of Candida infection in individuals receiving anti-TNF therapy in addition to several other immunosuppressant agents (ie, corticosteroids), which confounds the interpretation of results. Additional studies assessing rates of Candida and other opportunistic infections associated with use of adalimumab alone are needed to better guide clinical practices in dermatology.
Patients receiving adalimumab for dermatologic or other conditions should be closely monitored for opportunistic infections. Although immunomodulatory medications offer promising therapeutic benefits in patients with HS, larger studies regarding treatment with anti-TNF agents in HS are warranted to prevent complications from treatment and promote long-term efficacy and safety.
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory disease characterized by the development of painful abscesses, fistulous tracts, and scars. It most commonly affects the apocrine gland–bearing areas of the body such as the axillary, inguinal, and anogenital regions. With a prevalence of approximately 1%, HS can lead to notable morbidity.1 The pathogenesis is thought to be due to occlusion of terminal hair follicles that subsequently stimulates release of proinflammatory cytokines from nearby keratinocytes. The mechanism of initial occlusion is not well understood but may be due to friction or trauma. An inflammatory mechanism of disease also has been hypothesized; however, the exact cytokine profile is not known. Treatment of HS consists of several different modalities, including oral retinoids, antibiotics, antiandrogenic therapy, and surgery.1,2 Adalimumab is a well-known biologic that has been approved by the US Food and Drug Administration for the treatment of HS.
Adalimumab is a human monoclonal antibody against tumor necrosis factor (TNF) α and is thought to improve HS by several mechanisms. Inhibition of TNF-α and other proinflammatory cytokines found in inflammatory lesions and apocrine glands directly decreases the severity of lesion size and the frequency of recurrence.3 Adalimumab also is thought to downregulate expression of keratin 6 and prevent the hyperkeratinization seen in HS.4 Additionally, TNF-α inhibition decreases production of IL-1, which has been shown to cause hypercornification of follicles and perpetuate HS pathogenesis.5
A 41-year-old woman with a history of endometriosis, adenomyosis, polycystic ovary syndrome, interstitial cystitis, asthma, fibromyalgia, depression, and Hashimoto thyroiditis presented to our dermatology clinic with active draining lesions and sinus tracts in the perivaginal area that were consistent with HS, which initially was treated with doxycycline 100 mg twice daily. She experienced minimal improvement of the HS lesions at 2-month follow-up.
Due to disease severity, adalimumab was started. The patient received a loading dose of 4 injections totaling 160 mg and 80 mg on day 15, followed by a maintenance dose of 40 mg/0.4 mL weekly. The patient reported substantial improvement of pain, and complete resolution of active lesions was noted on physical examination after 4 weeks of treatment with adalimumab.
Six weeks after adalimumab was started, the patient developed severe dysphagia. She was evaluated by a gastroenterologist and underwent endoscopy (Figure), which led to a diagnosis of esophageal candidiasis. Adalimumab was discontinued immediately thereafter. The patient started treatment with nystatin oral rinse 4 times daily and oral fluconazole 200 mg daily. The candidiasis resolved within 2 weeks; however, she experienced recurrence of HS with draining lesions in the perivaginal area approximately 8 weeks after discontinuation of adalimumab. The patient requested to restart adalimumab treatment despite the recent history of esophagitis. Adalimumab 40 mg/0.4 mL weekly was restarted along with oral fluconazole 200 mg twice weekly and nystatin oral rinse 4 times daily. This regimen resulted in complete resolution of HS symptoms within 6 weeks with no recurrence of esophageal candidiasis during 6 months of follow-up.
Although the side effect of Candida esophagitis associated with adalimumab treatment in our patient may be logical given the medication’s mechanism of action and side-effect profile, this case warrants additional attention. An increase in fungal infections occurs from treatment with adalimumab because TNF-α is involved in many immune regulatory steps that counteract infection. Candida typically activates the innate immune system through macrophages via pathogen-associated molecular pattern stimulation, subsequently stimulating the release of inflammatory cytokines such as TNF-α. The cellular immune system also is activated. Helper T cells (TH1) release TNF-α along with other proinflammatory cytokines to increase phagocytosis in polymorphonuclear cells and macrophages.6 Thus, inhibition of TNF-α compromises innate and cellular immunity, thereby increasing susceptibility to fungal organisms.
A PubMed search of articles indexed for MEDLINE using the terms Candida, candidiasis, esophageal, adalimumab, anti-TNF, and TNF revealed no reports of esophageal candidiasis in patients receiving adalimumab or any of the TNF inhibitors. Candida laryngitis was reported in a patient receiving adalimumab for treatment of rheumatoid arthritis.7 Other studies have demonstrated an incidence of mucocutaneous candidiasis, most notably oropharyngeal and vaginal candidiasis.8-10 One study found that anti-TNF medications were associated with an increased risk for candidiasis by a hazard ratio of 2.7 in patients with Crohn disease.8 Other studies have shown that the highest incidence of fungal infection is seen with the use of infliximab, while adalimumab is associated with lower rates of fungal infection.9,10 Although it is known that anti-TNF therapy predisposes patients to fungal infection, the dose of medication known to preclude the highest risk has not been studied. Furthermore, most studies assess rates of Candida infection in individuals receiving anti-TNF therapy in addition to several other immunosuppressant agents (ie, corticosteroids), which confounds the interpretation of results. Additional studies assessing rates of Candida and other opportunistic infections associated with use of adalimumab alone are needed to better guide clinical practices in dermatology.
Patients receiving adalimumab for dermatologic or other conditions should be closely monitored for opportunistic infections. Although immunomodulatory medications offer promising therapeutic benefits in patients with HS, larger studies regarding treatment with anti-TNF agents in HS are warranted to prevent complications from treatment and promote long-term efficacy and safety.
- Kurayev A, Ashkar H, Saraiya A, et al. Hidradenitis suppurativa: review of the pathogenesis and treatment. J Drugs Dermatol. 2016;15:1107-1022.
- Rambhatla PV, Lim HW, Hamzavi I. A systematic review of treatments for hidradenitis suppurativa. Arch Dermatol. 2012;148:439-446.
- van der Zee HH, de Ruiter L, van den Broecke DG, et al. Elevated levels of tumour necrosis factor (TNF)-alpha, interleukin (IL)-1beta and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-alpha and IL-1beta. Br J Dermatol. 2011;164:1292-1298.
- Shuja F, Chan CS, Rosen T. Biologic drugs for the treatment of hidradenitis suppurativa: an evidence-based review. Dermatol Clin. 2010;28:511-521, 523-514.
- Kutsch CL, Norris DA, Arend WP. Tumor necrosis factor-alpha induces interleukin-1 alpha and interleukin-1 receptor antagonist production by cultured human keratinocytes. J Invest Dermatol. 1993;101:79-85.
- Senet JM. Risk factors and physiopathology of candidiasis. Rev Iberoam Micol. 1997;14:6-13.
- Kobak S, Yilmaz H, Guclu O, et al. Severe candida laryngitis in a patient with rheumatoid arthritis treated with adalimumab. Eur J Rheumatol. 2014;1:167-169.
- Marehbian J, Arrighi HM, Hass S, et al. Adverse events associated with common therapy regimens for moderate-to-severe Crohn’s disease. Am J Gastroenterol. 2009;104:2524-2533.
- Tsiodras S, Samonis G, Boumpas DT, et al. Fungal infections complicating tumor necrosis factor alpha blockade therapy. Mayo Clin Proc. 2008;83:181-194.
- Aikawa NE, Rosa DT, Del Negro GM, et al. Systemic and localized infection by Candida species in patients with rheumatic diseases receiving anti-TNF therapy [in Portuguese]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.03.010
- Kurayev A, Ashkar H, Saraiya A, et al. Hidradenitis suppurativa: review of the pathogenesis and treatment. J Drugs Dermatol. 2016;15:1107-1022.
- Rambhatla PV, Lim HW, Hamzavi I. A systematic review of treatments for hidradenitis suppurativa. Arch Dermatol. 2012;148:439-446.
- van der Zee HH, de Ruiter L, van den Broecke DG, et al. Elevated levels of tumour necrosis factor (TNF)-alpha, interleukin (IL)-1beta and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-alpha and IL-1beta. Br J Dermatol. 2011;164:1292-1298.
- Shuja F, Chan CS, Rosen T. Biologic drugs for the treatment of hidradenitis suppurativa: an evidence-based review. Dermatol Clin. 2010;28:511-521, 523-514.
- Kutsch CL, Norris DA, Arend WP. Tumor necrosis factor-alpha induces interleukin-1 alpha and interleukin-1 receptor antagonist production by cultured human keratinocytes. J Invest Dermatol. 1993;101:79-85.
- Senet JM. Risk factors and physiopathology of candidiasis. Rev Iberoam Micol. 1997;14:6-13.
- Kobak S, Yilmaz H, Guclu O, et al. Severe candida laryngitis in a patient with rheumatoid arthritis treated with adalimumab. Eur J Rheumatol. 2014;1:167-169.
- Marehbian J, Arrighi HM, Hass S, et al. Adverse events associated with common therapy regimens for moderate-to-severe Crohn’s disease. Am J Gastroenterol. 2009;104:2524-2533.
- Tsiodras S, Samonis G, Boumpas DT, et al. Fungal infections complicating tumor necrosis factor alpha blockade therapy. Mayo Clin Proc. 2008;83:181-194.
- Aikawa NE, Rosa DT, Del Negro GM, et al. Systemic and localized infection by Candida species in patients with rheumatic diseases receiving anti-TNF therapy [in Portuguese]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.03.010
Practice Points
- Adalimumab is an effective treatment for patients with hidradenitis suppurativa.
- There is risk for opportunistic infections with adalimumab, and patients should be monitored closely.