User login
Peristomal Pyoderma Gangrenosum at an Ileostomy Site
To the Editor:
Peristomal pyoderma gangrenosum (PPG) is a rare entity first described in 1984.1 Lesions usually begin as pustules that coalesce into an erythematous skin ulceration that contains purulent material. The lesion appears on the skin that surrounds an abdominal stoma. Peristomal pyoderma gangrenosum typically is associated with Crohn disease and ulcerative colitis, cancer, blood dyscrasia, diabetes mellitus, and hepatitis.2 We describe a case of PPG following an ileostomy in a patient with colon cancer and a related history of Crohn disease.
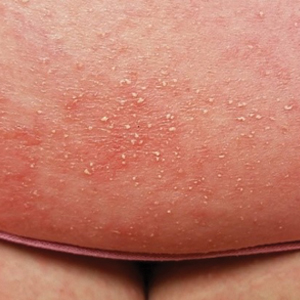
A 32-year-old woman presented to a dermatology office with a spontaneously painful, 3.2-cm ulceration that was extremely tender to palpation, located immediately adjacent to the site of an ileostomy (Figure). The patient had a history of refractory constipation that failed to respond to standard conservative measures 4 years prior. She underwent a colonoscopy, which revealed a 6.5-cm, irregularly shaped, exophytic mass in the rectosigmoid portion of the colon. Histopathologic examination of several biopsies confirmed the diagnosis of moderately well-differentiated adenocarcinoma, and additional evaluation determined the cancer to be stage IIB. She had a medical history of pancolonic Crohn disease since high school that was treated with periodic infusions of infliximab at the standard dose of 5 mg/kg. Colon cancer treatment consisted of preoperative radiotherapy, complete colectomy with ileoanal anastomosis, and creation of a J-pouch and formation of a temporary ileostomy, along with postoperative capecitabine chemotherapy.

The ileostomy eventually was reversed, and the patient did well for 3 years. When the patient developed severe abdominal pain, the J-pouch was examined and found to be remarkably involved with Crohn disease. However, during the colonoscopy, the J-pouch was inadvertently punctured, leading to the formation of a large pelvic abscess. The latter necessitated diversion of stool, and the patient had the original ileostomy recreated.
Prior to presentation to dermatology, various consultants suspected the ulceration was possibly a deep fungal infection, cutaneous Crohn disease, a factitious ulceration, or acute allergic contact dermatitis related to some element of ostomy care. However, dermatologic consultation suggested that the troublesome lesion was classic PPG and recommended administration of a tumor necrosis factor (TNF) α–blocking agent and concomitant intralesional injections of dilute triamcinolone acetonide.
The patient was treated with subcutaneous adalimumab 40 mg once weekly, and received near weekly subcutaneous injections of triamcinolone acetonide 10 mg/mL. After 2 months, the discomfort subsided, and the ulceration gradually resolved into a depressed scar. Eighteen months later, the scar was barely perceptible as a minimally erythematous depression. Adalimumab ultimately was discontinued, as the residual J-pouch was removed, and the biologic drug was associated with extensive alopecia areata–like hair loss. There has been no recurrence of PPG in the 40 months since clinical resolution.
Peristomal pyoderma gangrenosum is an uncommon subtype of pyoderma gangrenosum, which is characterized by chronic, persistent, or recurrent painful ulceration(s) close to an abdominal stoma. In total, fewer than 100 cases of PPG have been reported thus far in the readily available medical literature.3 Inflammatory bowel disease (IBD) is the most frequently diagnosed systemic condition associated with PPG, though other associated conditions include diverticular disease, abdominal malignancy, and neurologic dysfunction. Approximately 2% to 4.3% of all patients who have stoma creation surgery related to underlying IBD develop PPG. It is estimated that the yearly incidence rate of PPG in all abdominal stomas is quite low (approximately 0.6%).4
Peristomal pyoderma gangrenosum can occur at any age, but it tends to predominate in young to middle-aged adults, with a slight female predilection. The etiology and pathogenesis of PPG are largely unknown, though studies have shown that an abnormal immune response may be critical to its development. Risk factors for PPG are not well defined but potentially include autoimmune disorders, a high body mass index, and females or African Americans with IBD.4 Because PPG does not have characteristic histopathologic features, it is a diagnosis of exclusion that is based on the clinical examination and histologic findings that rule out other potential disorders.
There are 4 types of PPG based on the clinical and histopathologic characteristics: ulcerative, pustular, bullous, and vegetative. Peristomal pyoderma gangrenosum tends to be either ulcerative or vegetative, with ulcerative being by far the predominant type. The onset of PPG is quite variable, occurring a few weeks to several years after stoma formation.5 Ulcer size can range from less than 3 cm to 30 cm.4 Lesions begin as deep painful nodules or as superficial hemorrhagic pustules, either idiopathic or following ostensibly minimal trauma. Subsequently, they become necrotic and form an ulceration. The ulcers can be single or multiple lesions, typically with erythematous raised borders and purulent discharge. The ulcers are extremely painful and rapidly progressive. After the ulcers heal, they often leave a characteristic weblike atrophic scar that can break down further following any form of irritation or trauma.5
A prompt diagnosis of PPG is important. A diagnosis of PPG should be considered when dealing with a noninfectious ulcer surrounding a stoma in patients with IBD or other autoimmune conditions.6 Because PPG is a rare skin disorder, it is likely to be missed and lead to unnecessary diagnostic workup and a delay in proper therapy. In our patient, a diagnosis of PPG was overlooked for other infectious and autoimmune causes. The diagnostic evaluation of a patient with PPG is based on 3 principles: (1) ruling out other causes of a peristomal ulcer, such as an abscess, contact dermatitis, or wound infection; (2) determining whether there is an underlying intestinal bowel disease in the stoma; and (3) identifying associated systemic disorders such as vasculitis, erythema nodosum, or similar processes.4 The differential diagnosis depends on the type and stage of PPG and can include malignancy, vasculitis, extraintestinal IBD, infectious disease, and insect bites. A review of the history of the ulcer is helpful in ruling out other diseases, and a colonoscopy or ileoscopy can identify if patients have an underlying active IBD. Swabs for smear and both bacterial and fungal cultures should be taken from the exudate and directly from the ulcer base. Biopsy of the ulcer also helps to exclude alternative diagnoses.6
The primary goals of treating PPG include to reduce pain and the risk for secondary infection, increase pouch adherence, and decrease purulent exudate.7 Although there is not one well-defined optimal therapeutic intervention, there are a variety of effective approaches that may be considered and used. In mild cases, management methods such as dressings, topical agents, or intralesional steroids may be capable of controlling the disease. Daily wound care is important. Moisture-retentive dressings can control pain, induce collagen formation, promote angiogenesis, and prevent contamination. Cleaning the wound with sterile saline and applying an anti-infective agent also may be effective. Application of ultrapotent topical steroids and tacrolimus ointment 0.3% can be used in patients without concomitant secondary infection. In patients who are in remission, human platelet-derived growth factor may be used. Intralesional injections of dilute triamcinolone acetonide or cyclosporine solution also can be helpful. Cyclosporin A was used as a systemic monotherapy to treat a 48-year-old man and 50-year-old woman with the idiopathic form of PPG. After 3 months of treatment, PPG had completely resolved and there were no major side effects.8 Other potential topical therapies that control inflammation and promote wound healing include benzoyl peroxide, chlormethine (topical alkylating agent and nitrogen mustard that has anti-inflammatory properties), nicotine, and 5-aminosalicylic acid. If an ulcer becomes infected, empiric antibiotic therapy should be given immediately and adjusted based on culture and sensitivity results.4
Systemic therapy should be considered in patients who do not respond to topical or local interventions, have a rapid and severe course, or have an active underlying bowel disease. Oral prednisone (1 mg/kg/d) has proved to be one of the most successful drugs used to treat PPG. Treatment should be continued until complete lesion healing, and low-dose maintenance therapy should be administered in recurrent cases. Intravenous corticosteroid therapy—hydrocortisone 100 mg 4 times daily or pulse therapy with intravenous methylprednisolone 1 g/d)—can be used for up to 5 days and may be effective. Oral minocycline 100 mg twice daily may be helpful as an adjunctive therapy to corticosteroids. When corticosteroids fail, oral cyclosporine 3 to 5 mg/kg/d often is prescribed. Studies have shown that patients demonstrate clinical improvement within 3 weeks of cyclosporine initiation, and it has been shown further to be more effective than either azathioprine or methotrexate.4,8
Infliximab, a chimeric antibody that binds both circulating and tissue-bound TNF-α, has been shown to effectively treat PPG. A clinical trial conducted by Brooklyn et al9 found that 46% of patients (6/13) treated with infliximab responded compared with only 6% in a placebo control group (1/17). Although infliximab may result in sepsis, the benefits far outweigh the risks, especially for patients with steroid-refractory PPG.4 Adalimumab is a human monoclonal IgG1 antibody to TNF-α that neutralizes its function by blocking the interaction between the molecule and its receptor. Many clinical studies have shown that adalimumab induces and maintains a clinical response in patients with active Crohn disease. The biologic proved to be effective in our patient, but it is associated with potential side effects that should be monitored including injection-site reactions, pruritus, leukopenia, urticaria, and rare instances of alopecia.10 Etanercept is another potentially effective biologic agent.7 Plasma exchange, immunoglobulin infusion, and interferon-alfa therapy also can be used in refractory PPG cases, though data on these treatments are very limited.4
Unlike routine pyoderma gangrenosum—for which surgical intervention is contraindicated—surgical intervention may be appropriate for the peristomal variant. Surgical treatment options include stoma revision and/or relocation; however, both of these procedures are accompanied by failure rates ranging from 40% to 100%.5 Removal of a diseased intestinal segment, especially one with active IBD, may result in healing of the skin lesion. In our patient, removal of the residual and diseased J-pouch was part of the management plan. However,it generally is recommended that any surgical intervention be accompanied by medical therapy including oral metronidazole 500 mg/d and concomitant administration of an immunosuppressant.1,3
Because PPG tends to recur, long-term maintenance therapy should always be considered. Pain reduction, anemia correction, proper nutrition, and management of associated and underlying diseases should be performed. Meticulous care of the stoma and prevention of leaks also should be emphasized. Overall, if PPG is detected and diagnosed early as well as treated appropriately and aggressively, the patient likely will have a good prognosis.4
- Sheldon DG, Sawchuk LL, Kozarek RA, et al. Twenty cases of peristomal pyoderma gangrenosum: diagnostic implications and management. Arch Surg. 2000;135:564-569.
- Hughes AP, Jackson JM, Callen JP. Clinical features and treatment of peristomal pyoderma gangrenosum. JAMA. 2000;284:1546-1548.
- Afifi L, Sanchez IM, Wallace MM, et al. Diagnosis and management of peristomal pyoderma gangrenosum: a systematic review. J Am Acad Dermatol. 2018;78:1195-1204.
- Wu XR, Shen B. Diagnosis and management of parastomal pyoderma gangrenosum. Gastroenterol Rep (Oxf). 2013;1:1-8.
- Javed A, Pal S, Ahuja V, et al. Management of peristomal pyoderma gangrenosum: two different approaches for the same clinical problem. Trop Gastroenterol. 2011;32:153-156.
- Toh JW, Whiteley I. Devastating peristomal pyoderma gangrenosum: challenges in diagnosis and management. Clin Gastroenterol Hepatol. 2017;15:A19-A20.
- DeMartyn LE, Faller NA, Miller L. Treating peristomal pyoderma gangrenosum with topical crushed prednisone: a report of three cases. Ostomy Wound Manage. 2014;60:50-54.
- V’lckova-Laskoska MT, Laskoski DS, Caca-Biljanovska NG, et al. Pyoderma gangrenosum successfully treated with cyclosporin A.Adv Exp Med Biol. 1999;455:541-555.
- Brooklyn TN, Dunnill MGS, Shetty A, at al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Alkhouri N, Hupertz V, Mahajan L. Adalimumab treatment for peristomal pyoderma gangrenosum associated with Crohn’s disease. Inflamm Bowel Dis. 2009;15:803-806.
To the Editor:
Peristomal pyoderma gangrenosum (PPG) is a rare entity first described in 1984.1 Lesions usually begin as pustules that coalesce into an erythematous skin ulceration that contains purulent material. The lesion appears on the skin that surrounds an abdominal stoma. Peristomal pyoderma gangrenosum typically is associated with Crohn disease and ulcerative colitis, cancer, blood dyscrasia, diabetes mellitus, and hepatitis.2 We describe a case of PPG following an ileostomy in a patient with colon cancer and a related history of Crohn disease.
A 32-year-old woman presented to a dermatology office with a spontaneously painful, 3.2-cm ulceration that was extremely tender to palpation, located immediately adjacent to the site of an ileostomy (Figure). The patient had a history of refractory constipation that failed to respond to standard conservative measures 4 years prior. She underwent a colonoscopy, which revealed a 6.5-cm, irregularly shaped, exophytic mass in the rectosigmoid portion of the colon. Histopathologic examination of several biopsies confirmed the diagnosis of moderately well-differentiated adenocarcinoma, and additional evaluation determined the cancer to be stage IIB. She had a medical history of pancolonic Crohn disease since high school that was treated with periodic infusions of infliximab at the standard dose of 5 mg/kg. Colon cancer treatment consisted of preoperative radiotherapy, complete colectomy with ileoanal anastomosis, and creation of a J-pouch and formation of a temporary ileostomy, along with postoperative capecitabine chemotherapy.
The ileostomy eventually was reversed, and the patient did well for 3 years. When the patient developed severe abdominal pain, the J-pouch was examined and found to be remarkably involved with Crohn disease. However, during the colonoscopy, the J-pouch was inadvertently punctured, leading to the formation of a large pelvic abscess. The latter necessitated diversion of stool, and the patient had the original ileostomy recreated.
Prior to presentation to dermatology, various consultants suspected the ulceration was possibly a deep fungal infection, cutaneous Crohn disease, a factitious ulceration, or acute allergic contact dermatitis related to some element of ostomy care. However, dermatologic consultation suggested that the troublesome lesion was classic PPG and recommended administration of a tumor necrosis factor (TNF) α–blocking agent and concomitant intralesional injections of dilute triamcinolone acetonide.
The patient was treated with subcutaneous adalimumab 40 mg once weekly, and received near weekly subcutaneous injections of triamcinolone acetonide 10 mg/mL. After 2 months, the discomfort subsided, and the ulceration gradually resolved into a depressed scar. Eighteen months later, the scar was barely perceptible as a minimally erythematous depression. Adalimumab ultimately was discontinued, as the residual J-pouch was removed, and the biologic drug was associated with extensive alopecia areata–like hair loss. There has been no recurrence of PPG in the 40 months since clinical resolution.
Peristomal pyoderma gangrenosum is an uncommon subtype of pyoderma gangrenosum, which is characterized by chronic, persistent, or recurrent painful ulceration(s) close to an abdominal stoma. In total, fewer than 100 cases of PPG have been reported thus far in the readily available medical literature.3 Inflammatory bowel disease (IBD) is the most frequently diagnosed systemic condition associated with PPG, though other associated conditions include diverticular disease, abdominal malignancy, and neurologic dysfunction. Approximately 2% to 4.3% of all patients who have stoma creation surgery related to underlying IBD develop PPG. It is estimated that the yearly incidence rate of PPG in all abdominal stomas is quite low (approximately 0.6%).4
Peristomal pyoderma gangrenosum can occur at any age, but it tends to predominate in young to middle-aged adults, with a slight female predilection. The etiology and pathogenesis of PPG are largely unknown, though studies have shown that an abnormal immune response may be critical to its development. Risk factors for PPG are not well defined but potentially include autoimmune disorders, a high body mass index, and females or African Americans with IBD.4 Because PPG does not have characteristic histopathologic features, it is a diagnosis of exclusion that is based on the clinical examination and histologic findings that rule out other potential disorders.
There are 4 types of PPG based on the clinical and histopathologic characteristics: ulcerative, pustular, bullous, and vegetative. Peristomal pyoderma gangrenosum tends to be either ulcerative or vegetative, with ulcerative being by far the predominant type. The onset of PPG is quite variable, occurring a few weeks to several years after stoma formation.5 Ulcer size can range from less than 3 cm to 30 cm.4 Lesions begin as deep painful nodules or as superficial hemorrhagic pustules, either idiopathic or following ostensibly minimal trauma. Subsequently, they become necrotic and form an ulceration. The ulcers can be single or multiple lesions, typically with erythematous raised borders and purulent discharge. The ulcers are extremely painful and rapidly progressive. After the ulcers heal, they often leave a characteristic weblike atrophic scar that can break down further following any form of irritation or trauma.5
A prompt diagnosis of PPG is important. A diagnosis of PPG should be considered when dealing with a noninfectious ulcer surrounding a stoma in patients with IBD or other autoimmune conditions.6 Because PPG is a rare skin disorder, it is likely to be missed and lead to unnecessary diagnostic workup and a delay in proper therapy. In our patient, a diagnosis of PPG was overlooked for other infectious and autoimmune causes. The diagnostic evaluation of a patient with PPG is based on 3 principles: (1) ruling out other causes of a peristomal ulcer, such as an abscess, contact dermatitis, or wound infection; (2) determining whether there is an underlying intestinal bowel disease in the stoma; and (3) identifying associated systemic disorders such as vasculitis, erythema nodosum, or similar processes.4 The differential diagnosis depends on the type and stage of PPG and can include malignancy, vasculitis, extraintestinal IBD, infectious disease, and insect bites. A review of the history of the ulcer is helpful in ruling out other diseases, and a colonoscopy or ileoscopy can identify if patients have an underlying active IBD. Swabs for smear and both bacterial and fungal cultures should be taken from the exudate and directly from the ulcer base. Biopsy of the ulcer also helps to exclude alternative diagnoses.6
The primary goals of treating PPG include to reduce pain and the risk for secondary infection, increase pouch adherence, and decrease purulent exudate.7 Although there is not one well-defined optimal therapeutic intervention, there are a variety of effective approaches that may be considered and used. In mild cases, management methods such as dressings, topical agents, or intralesional steroids may be capable of controlling the disease. Daily wound care is important. Moisture-retentive dressings can control pain, induce collagen formation, promote angiogenesis, and prevent contamination. Cleaning the wound with sterile saline and applying an anti-infective agent also may be effective. Application of ultrapotent topical steroids and tacrolimus ointment 0.3% can be used in patients without concomitant secondary infection. In patients who are in remission, human platelet-derived growth factor may be used. Intralesional injections of dilute triamcinolone acetonide or cyclosporine solution also can be helpful. Cyclosporin A was used as a systemic monotherapy to treat a 48-year-old man and 50-year-old woman with the idiopathic form of PPG. After 3 months of treatment, PPG had completely resolved and there were no major side effects.8 Other potential topical therapies that control inflammation and promote wound healing include benzoyl peroxide, chlormethine (topical alkylating agent and nitrogen mustard that has anti-inflammatory properties), nicotine, and 5-aminosalicylic acid. If an ulcer becomes infected, empiric antibiotic therapy should be given immediately and adjusted based on culture and sensitivity results.4
Systemic therapy should be considered in patients who do not respond to topical or local interventions, have a rapid and severe course, or have an active underlying bowel disease. Oral prednisone (1 mg/kg/d) has proved to be one of the most successful drugs used to treat PPG. Treatment should be continued until complete lesion healing, and low-dose maintenance therapy should be administered in recurrent cases. Intravenous corticosteroid therapy—hydrocortisone 100 mg 4 times daily or pulse therapy with intravenous methylprednisolone 1 g/d)—can be used for up to 5 days and may be effective. Oral minocycline 100 mg twice daily may be helpful as an adjunctive therapy to corticosteroids. When corticosteroids fail, oral cyclosporine 3 to 5 mg/kg/d often is prescribed. Studies have shown that patients demonstrate clinical improvement within 3 weeks of cyclosporine initiation, and it has been shown further to be more effective than either azathioprine or methotrexate.4,8
Infliximab, a chimeric antibody that binds both circulating and tissue-bound TNF-α, has been shown to effectively treat PPG. A clinical trial conducted by Brooklyn et al9 found that 46% of patients (6/13) treated with infliximab responded compared with only 6% in a placebo control group (1/17). Although infliximab may result in sepsis, the benefits far outweigh the risks, especially for patients with steroid-refractory PPG.4 Adalimumab is a human monoclonal IgG1 antibody to TNF-α that neutralizes its function by blocking the interaction between the molecule and its receptor. Many clinical studies have shown that adalimumab induces and maintains a clinical response in patients with active Crohn disease. The biologic proved to be effective in our patient, but it is associated with potential side effects that should be monitored including injection-site reactions, pruritus, leukopenia, urticaria, and rare instances of alopecia.10 Etanercept is another potentially effective biologic agent.7 Plasma exchange, immunoglobulin infusion, and interferon-alfa therapy also can be used in refractory PPG cases, though data on these treatments are very limited.4
Unlike routine pyoderma gangrenosum—for which surgical intervention is contraindicated—surgical intervention may be appropriate for the peristomal variant. Surgical treatment options include stoma revision and/or relocation; however, both of these procedures are accompanied by failure rates ranging from 40% to 100%.5 Removal of a diseased intestinal segment, especially one with active IBD, may result in healing of the skin lesion. In our patient, removal of the residual and diseased J-pouch was part of the management plan. However,it generally is recommended that any surgical intervention be accompanied by medical therapy including oral metronidazole 500 mg/d and concomitant administration of an immunosuppressant.1,3
Because PPG tends to recur, long-term maintenance therapy should always be considered. Pain reduction, anemia correction, proper nutrition, and management of associated and underlying diseases should be performed. Meticulous care of the stoma and prevention of leaks also should be emphasized. Overall, if PPG is detected and diagnosed early as well as treated appropriately and aggressively, the patient likely will have a good prognosis.4
To the Editor:
Peristomal pyoderma gangrenosum (PPG) is a rare entity first described in 1984.1 Lesions usually begin as pustules that coalesce into an erythematous skin ulceration that contains purulent material. The lesion appears on the skin that surrounds an abdominal stoma. Peristomal pyoderma gangrenosum typically is associated with Crohn disease and ulcerative colitis, cancer, blood dyscrasia, diabetes mellitus, and hepatitis.2 We describe a case of PPG following an ileostomy in a patient with colon cancer and a related history of Crohn disease.
A 32-year-old woman presented to a dermatology office with a spontaneously painful, 3.2-cm ulceration that was extremely tender to palpation, located immediately adjacent to the site of an ileostomy (Figure). The patient had a history of refractory constipation that failed to respond to standard conservative measures 4 years prior. She underwent a colonoscopy, which revealed a 6.5-cm, irregularly shaped, exophytic mass in the rectosigmoid portion of the colon. Histopathologic examination of several biopsies confirmed the diagnosis of moderately well-differentiated adenocarcinoma, and additional evaluation determined the cancer to be stage IIB. She had a medical history of pancolonic Crohn disease since high school that was treated with periodic infusions of infliximab at the standard dose of 5 mg/kg. Colon cancer treatment consisted of preoperative radiotherapy, complete colectomy with ileoanal anastomosis, and creation of a J-pouch and formation of a temporary ileostomy, along with postoperative capecitabine chemotherapy.
The ileostomy eventually was reversed, and the patient did well for 3 years. When the patient developed severe abdominal pain, the J-pouch was examined and found to be remarkably involved with Crohn disease. However, during the colonoscopy, the J-pouch was inadvertently punctured, leading to the formation of a large pelvic abscess. The latter necessitated diversion of stool, and the patient had the original ileostomy recreated.
Prior to presentation to dermatology, various consultants suspected the ulceration was possibly a deep fungal infection, cutaneous Crohn disease, a factitious ulceration, or acute allergic contact dermatitis related to some element of ostomy care. However, dermatologic consultation suggested that the troublesome lesion was classic PPG and recommended administration of a tumor necrosis factor (TNF) α–blocking agent and concomitant intralesional injections of dilute triamcinolone acetonide.
The patient was treated with subcutaneous adalimumab 40 mg once weekly, and received near weekly subcutaneous injections of triamcinolone acetonide 10 mg/mL. After 2 months, the discomfort subsided, and the ulceration gradually resolved into a depressed scar. Eighteen months later, the scar was barely perceptible as a minimally erythematous depression. Adalimumab ultimately was discontinued, as the residual J-pouch was removed, and the biologic drug was associated with extensive alopecia areata–like hair loss. There has been no recurrence of PPG in the 40 months since clinical resolution.
Peristomal pyoderma gangrenosum is an uncommon subtype of pyoderma gangrenosum, which is characterized by chronic, persistent, or recurrent painful ulceration(s) close to an abdominal stoma. In total, fewer than 100 cases of PPG have been reported thus far in the readily available medical literature.3 Inflammatory bowel disease (IBD) is the most frequently diagnosed systemic condition associated with PPG, though other associated conditions include diverticular disease, abdominal malignancy, and neurologic dysfunction. Approximately 2% to 4.3% of all patients who have stoma creation surgery related to underlying IBD develop PPG. It is estimated that the yearly incidence rate of PPG in all abdominal stomas is quite low (approximately 0.6%).4
Peristomal pyoderma gangrenosum can occur at any age, but it tends to predominate in young to middle-aged adults, with a slight female predilection. The etiology and pathogenesis of PPG are largely unknown, though studies have shown that an abnormal immune response may be critical to its development. Risk factors for PPG are not well defined but potentially include autoimmune disorders, a high body mass index, and females or African Americans with IBD.4 Because PPG does not have characteristic histopathologic features, it is a diagnosis of exclusion that is based on the clinical examination and histologic findings that rule out other potential disorders.
There are 4 types of PPG based on the clinical and histopathologic characteristics: ulcerative, pustular, bullous, and vegetative. Peristomal pyoderma gangrenosum tends to be either ulcerative or vegetative, with ulcerative being by far the predominant type. The onset of PPG is quite variable, occurring a few weeks to several years after stoma formation.5 Ulcer size can range from less than 3 cm to 30 cm.4 Lesions begin as deep painful nodules or as superficial hemorrhagic pustules, either idiopathic or following ostensibly minimal trauma. Subsequently, they become necrotic and form an ulceration. The ulcers can be single or multiple lesions, typically with erythematous raised borders and purulent discharge. The ulcers are extremely painful and rapidly progressive. After the ulcers heal, they often leave a characteristic weblike atrophic scar that can break down further following any form of irritation or trauma.5
A prompt diagnosis of PPG is important. A diagnosis of PPG should be considered when dealing with a noninfectious ulcer surrounding a stoma in patients with IBD or other autoimmune conditions.6 Because PPG is a rare skin disorder, it is likely to be missed and lead to unnecessary diagnostic workup and a delay in proper therapy. In our patient, a diagnosis of PPG was overlooked for other infectious and autoimmune causes. The diagnostic evaluation of a patient with PPG is based on 3 principles: (1) ruling out other causes of a peristomal ulcer, such as an abscess, contact dermatitis, or wound infection; (2) determining whether there is an underlying intestinal bowel disease in the stoma; and (3) identifying associated systemic disorders such as vasculitis, erythema nodosum, or similar processes.4 The differential diagnosis depends on the type and stage of PPG and can include malignancy, vasculitis, extraintestinal IBD, infectious disease, and insect bites. A review of the history of the ulcer is helpful in ruling out other diseases, and a colonoscopy or ileoscopy can identify if patients have an underlying active IBD. Swabs for smear and both bacterial and fungal cultures should be taken from the exudate and directly from the ulcer base. Biopsy of the ulcer also helps to exclude alternative diagnoses.6
The primary goals of treating PPG include to reduce pain and the risk for secondary infection, increase pouch adherence, and decrease purulent exudate.7 Although there is not one well-defined optimal therapeutic intervention, there are a variety of effective approaches that may be considered and used. In mild cases, management methods such as dressings, topical agents, or intralesional steroids may be capable of controlling the disease. Daily wound care is important. Moisture-retentive dressings can control pain, induce collagen formation, promote angiogenesis, and prevent contamination. Cleaning the wound with sterile saline and applying an anti-infective agent also may be effective. Application of ultrapotent topical steroids and tacrolimus ointment 0.3% can be used in patients without concomitant secondary infection. In patients who are in remission, human platelet-derived growth factor may be used. Intralesional injections of dilute triamcinolone acetonide or cyclosporine solution also can be helpful. Cyclosporin A was used as a systemic monotherapy to treat a 48-year-old man and 50-year-old woman with the idiopathic form of PPG. After 3 months of treatment, PPG had completely resolved and there were no major side effects.8 Other potential topical therapies that control inflammation and promote wound healing include benzoyl peroxide, chlormethine (topical alkylating agent and nitrogen mustard that has anti-inflammatory properties), nicotine, and 5-aminosalicylic acid. If an ulcer becomes infected, empiric antibiotic therapy should be given immediately and adjusted based on culture and sensitivity results.4
Systemic therapy should be considered in patients who do not respond to topical or local interventions, have a rapid and severe course, or have an active underlying bowel disease. Oral prednisone (1 mg/kg/d) has proved to be one of the most successful drugs used to treat PPG. Treatment should be continued until complete lesion healing, and low-dose maintenance therapy should be administered in recurrent cases. Intravenous corticosteroid therapy—hydrocortisone 100 mg 4 times daily or pulse therapy with intravenous methylprednisolone 1 g/d)—can be used for up to 5 days and may be effective. Oral minocycline 100 mg twice daily may be helpful as an adjunctive therapy to corticosteroids. When corticosteroids fail, oral cyclosporine 3 to 5 mg/kg/d often is prescribed. Studies have shown that patients demonstrate clinical improvement within 3 weeks of cyclosporine initiation, and it has been shown further to be more effective than either azathioprine or methotrexate.4,8
Infliximab, a chimeric antibody that binds both circulating and tissue-bound TNF-α, has been shown to effectively treat PPG. A clinical trial conducted by Brooklyn et al9 found that 46% of patients (6/13) treated with infliximab responded compared with only 6% in a placebo control group (1/17). Although infliximab may result in sepsis, the benefits far outweigh the risks, especially for patients with steroid-refractory PPG.4 Adalimumab is a human monoclonal IgG1 antibody to TNF-α that neutralizes its function by blocking the interaction between the molecule and its receptor. Many clinical studies have shown that adalimumab induces and maintains a clinical response in patients with active Crohn disease. The biologic proved to be effective in our patient, but it is associated with potential side effects that should be monitored including injection-site reactions, pruritus, leukopenia, urticaria, and rare instances of alopecia.10 Etanercept is another potentially effective biologic agent.7 Plasma exchange, immunoglobulin infusion, and interferon-alfa therapy also can be used in refractory PPG cases, though data on these treatments are very limited.4
Unlike routine pyoderma gangrenosum—for which surgical intervention is contraindicated—surgical intervention may be appropriate for the peristomal variant. Surgical treatment options include stoma revision and/or relocation; however, both of these procedures are accompanied by failure rates ranging from 40% to 100%.5 Removal of a diseased intestinal segment, especially one with active IBD, may result in healing of the skin lesion. In our patient, removal of the residual and diseased J-pouch was part of the management plan. However,it generally is recommended that any surgical intervention be accompanied by medical therapy including oral metronidazole 500 mg/d and concomitant administration of an immunosuppressant.1,3
Because PPG tends to recur, long-term maintenance therapy should always be considered. Pain reduction, anemia correction, proper nutrition, and management of associated and underlying diseases should be performed. Meticulous care of the stoma and prevention of leaks also should be emphasized. Overall, if PPG is detected and diagnosed early as well as treated appropriately and aggressively, the patient likely will have a good prognosis.4
- Sheldon DG, Sawchuk LL, Kozarek RA, et al. Twenty cases of peristomal pyoderma gangrenosum: diagnostic implications and management. Arch Surg. 2000;135:564-569.
- Hughes AP, Jackson JM, Callen JP. Clinical features and treatment of peristomal pyoderma gangrenosum. JAMA. 2000;284:1546-1548.
- Afifi L, Sanchez IM, Wallace MM, et al. Diagnosis and management of peristomal pyoderma gangrenosum: a systematic review. J Am Acad Dermatol. 2018;78:1195-1204.
- Wu XR, Shen B. Diagnosis and management of parastomal pyoderma gangrenosum. Gastroenterol Rep (Oxf). 2013;1:1-8.
- Javed A, Pal S, Ahuja V, et al. Management of peristomal pyoderma gangrenosum: two different approaches for the same clinical problem. Trop Gastroenterol. 2011;32:153-156.
- Toh JW, Whiteley I. Devastating peristomal pyoderma gangrenosum: challenges in diagnosis and management. Clin Gastroenterol Hepatol. 2017;15:A19-A20.
- DeMartyn LE, Faller NA, Miller L. Treating peristomal pyoderma gangrenosum with topical crushed prednisone: a report of three cases. Ostomy Wound Manage. 2014;60:50-54.
- V’lckova-Laskoska MT, Laskoski DS, Caca-Biljanovska NG, et al. Pyoderma gangrenosum successfully treated with cyclosporin A.Adv Exp Med Biol. 1999;455:541-555.
- Brooklyn TN, Dunnill MGS, Shetty A, at al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Alkhouri N, Hupertz V, Mahajan L. Adalimumab treatment for peristomal pyoderma gangrenosum associated with Crohn’s disease. Inflamm Bowel Dis. 2009;15:803-806.
- Sheldon DG, Sawchuk LL, Kozarek RA, et al. Twenty cases of peristomal pyoderma gangrenosum: diagnostic implications and management. Arch Surg. 2000;135:564-569.
- Hughes AP, Jackson JM, Callen JP. Clinical features and treatment of peristomal pyoderma gangrenosum. JAMA. 2000;284:1546-1548.
- Afifi L, Sanchez IM, Wallace MM, et al. Diagnosis and management of peristomal pyoderma gangrenosum: a systematic review. J Am Acad Dermatol. 2018;78:1195-1204.
- Wu XR, Shen B. Diagnosis and management of parastomal pyoderma gangrenosum. Gastroenterol Rep (Oxf). 2013;1:1-8.
- Javed A, Pal S, Ahuja V, et al. Management of peristomal pyoderma gangrenosum: two different approaches for the same clinical problem. Trop Gastroenterol. 2011;32:153-156.
- Toh JW, Whiteley I. Devastating peristomal pyoderma gangrenosum: challenges in diagnosis and management. Clin Gastroenterol Hepatol. 2017;15:A19-A20.
- DeMartyn LE, Faller NA, Miller L. Treating peristomal pyoderma gangrenosum with topical crushed prednisone: a report of three cases. Ostomy Wound Manage. 2014;60:50-54.
- V’lckova-Laskoska MT, Laskoski DS, Caca-Biljanovska NG, et al. Pyoderma gangrenosum successfully treated with cyclosporin A.Adv Exp Med Biol. 1999;455:541-555.
- Brooklyn TN, Dunnill MGS, Shetty A, at al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55:505-509.
- Alkhouri N, Hupertz V, Mahajan L. Adalimumab treatment for peristomal pyoderma gangrenosum associated with Crohn’s disease. Inflamm Bowel Dis. 2009;15:803-806.
Practice Points
- A pyoderma gangrenosum subtype occurs in close proximity to an abdominal stoma.
- Peristomal pyoderma gangrenosum is a diagnosis of exclusion.
- Peristomal pyoderma gangrenosum typically responds best to tumor necrosis factor α blockers and corticosteroid therapy (intralesional and systemic).

Focal Palmoplantar Keratoderma and Gingival Keratosis Caused by a KRT16 Mutation
To the Editor:
Focal palmoplantar keratoderma and gingival keratosis (FPGK)(Online Mendelian Inheritance in Man [OMIM] 148730) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma and gingival hyperkeratosis presenting as leukokeratosis. Focal palmoplantar keratoderma and gingival keratosis was first defined by Gorlin1 in 1976. Since then, only a few cases have been reported, but no causative mutations have been identified.2
Focal pressure-related palmoplantar keratoderma (PPK) and oral hyperkeratosis also are seen in pachyonychia congenita (PC)(OMIM 167200, 615726, 615728, 167210), a rare autosomal-dominant disorder of keratinization characterized by PPK and nail dystrophy. Patients with PC often present with plantar pain; more variable features include oral leukokeratosis, follicular hyperkeratosis, pilosebaceous and epidermal inclusion cysts, hoarseness, hyperhidrosis, and natal teeth. Pachyonychia congenita is caused by mutation in keratin genes KRT6A, KRT6B, KRT16, or KRT17.
Focal palmoplantar keratoderma and gingival keratosis as well as PC are distinct from other forms of PPK with gingival involvement such as
Despite the common features of FPGK and PC, they are considered distinct disorders due to absence of nail changes in FPGK and no prior evidence of a common genetic cause. We present a patient with familial FPGK found by whole exome sequencing to be caused by a mutation in KRT16.
 showing focal palmoplantar keratoderma and gingival keratosis in those heterozygous for KRT16 mutation p.R127H")
The proband was a 57-year-old man born to unrelated parents (Figure 1). He had no skin problems at birth, and his development was normal. He had painful focal keratoderma since childhood that were most prominent at pressure points on the soles and toes (Figure 2A), in addition to gingival hyperkeratosis and oral leukokeratosis (Figure 2B). He had no associated abnormalities of the skin, hair, or teeth and no nail findings (Figure 2C). He reported that his father and 2 of his 3 sisters were affected with similar symptoms. A punch biopsy of the right fifth toe was consistent with verrucous epidermal hyperplasia with perinuclear keratinization in the spinous layer (Figure 3A). A gingival biopsy showed perinuclear eosinophilic globules and basophilic stranding in the cytoplasm (Figure 3B). His older sister had more severe and painful focal keratoderma of the soles, punctate keratoderma of the palms, gingival hyperkeratosis, and leukokeratosis of the tongue.

Whole exome sequencing of the proband revealed a heterozygous missense mutation in KRT16 (c.380G>A, p.R127H, rs57424749). Sanger sequencing confirmed this mutation and showed that it was heterozygous in both of his affected sisters and absent in his unaffected niece (Figure 1). The patient was treated with topical and systemic retinoids, keratolytics, and mechanical removal to moderate effect, with noted improvement in the appearance and associated pain of the plantar keratoderma.

Phenotypic heterogeneity is common in PC, though PC due to KRT6A mutations demonstrates more severe nail disease with oral lesions, cysts, and follicular hyperkeratosis, while PC caused by KRT16 mutations generally presents with more extensive and painful PPK.4KRT16 mutations affecting p.R127 are frequent causes of PC, and genotype-phenotype correlations have been observed. Individuals with p.R127P mutations exhibit more severe disease with earlier age of onset, more extensive nail involvement and oral leukokeratosis, and greater impact on daily quality of life than in individuals with p.R127C mutations.5 Cases of PC with KRT16 p.R127S and p.R127G mutations also have been observed. The KRT16 c.380G>A, p.R127H mutation we documented has been reported in one kindred with PC who presented with PPK, oral leukokeratosis, toenail thickening, and pilosebaceous and follicular hyperkeratosis.6
Although patients with FPGK lack the thickening of fingernails and/or toenails considered a defining feature of PC, the disorders otherwise are phenotypically similar, suggesting the possibility of common pathogenesis. One linkage study of familial FPGK excluded genetic intervals containing type I and type II keratins but was limited to a single small kindred.2 This study and our data together suggest that, similar to PC, there are multiple genes in which mutations cause FPGK.
Murine Krt16 knockouts show distinct phenotypes depending on the mouse strain in which they are propagated, ranging from perinatal lethality to differences in the severity of oral and PPK lesions.7 These observations provide evidence that additional genetic variants contribute to Krt16 phenotypes in mice and suggest the same could be true for humans.
We propose that some cases of FPGK are due to mutations in KRT16 and thus share a genetic pathogenesis with PC, underscoring the utility of whole exome sequencing in providing genetic diagnoses for disorders that are genetically and clinically heterogeneous. Further biologic investigation of phenotypes caused by KRT16 mutation may reveal respective contributions of additional genetic variation and environmental effects to the variable clinical presentations.
- Gorlin RJ. Focal palmoplantar and marginal gingival hyperkeratosis—a syndrome. Birth Defects Orig Artic Ser. 1976;12:239-242.
- Kolde G, Hennies HC, Bethke G, et al. Focal palmoplantar and gingival keratosis: a distinct palmoplantar ectodermal dysplasia with epidermolytic alterations but lack of mutations in known keratins. J Am Acad Dermatol. 2005;52(3 pt 1):403-409.
- Duchatelet S, Hovnanian A. Olmsted syndrome: clinical, molecular and therapeutic aspects. Orphanet J Rare Dis. 2015;10:33.
- Spaunhurst KM, Hogendorf AM, Smith FJ, et al. Pachyonychia congenita patients with mutations in KRT6A have more extensive disease compared with patients who have mutations in KRT16. Br J Dermatol. 2012;166:875-878.
- Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachyonychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131:1025-1028.
- Wilson NJ, O’Toole EA, Milstone LM, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol. 2014;171:343-355.
- Zieman A, Coulombe PA. The keratin 16 null phenotype is modestly impacted by genetic strain background in mice. Exp Dermatol. 2018;27:672-674.
To the Editor:
Focal palmoplantar keratoderma and gingival keratosis (FPGK)(Online Mendelian Inheritance in Man [OMIM] 148730) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma and gingival hyperkeratosis presenting as leukokeratosis. Focal palmoplantar keratoderma and gingival keratosis was first defined by Gorlin1 in 1976. Since then, only a few cases have been reported, but no causative mutations have been identified.2
Focal pressure-related palmoplantar keratoderma (PPK) and oral hyperkeratosis also are seen in pachyonychia congenita (PC)(OMIM 167200, 615726, 615728, 167210), a rare autosomal-dominant disorder of keratinization characterized by PPK and nail dystrophy. Patients with PC often present with plantar pain; more variable features include oral leukokeratosis, follicular hyperkeratosis, pilosebaceous and epidermal inclusion cysts, hoarseness, hyperhidrosis, and natal teeth. Pachyonychia congenita is caused by mutation in keratin genes KRT6A, KRT6B, KRT16, or KRT17.
Focal palmoplantar keratoderma and gingival keratosis as well as PC are distinct from other forms of PPK with gingival involvement such as
Despite the common features of FPGK and PC, they are considered distinct disorders due to absence of nail changes in FPGK and no prior evidence of a common genetic cause. We present a patient with familial FPGK found by whole exome sequencing to be caused by a mutation in KRT16.
The proband was a 57-year-old man born to unrelated parents (Figure 1). He had no skin problems at birth, and his development was normal. He had painful focal keratoderma since childhood that were most prominent at pressure points on the soles and toes (Figure 2A), in addition to gingival hyperkeratosis and oral leukokeratosis (Figure 2B). He had no associated abnormalities of the skin, hair, or teeth and no nail findings (Figure 2C). He reported that his father and 2 of his 3 sisters were affected with similar symptoms. A punch biopsy of the right fifth toe was consistent with verrucous epidermal hyperplasia with perinuclear keratinization in the spinous layer (Figure 3A). A gingival biopsy showed perinuclear eosinophilic globules and basophilic stranding in the cytoplasm (Figure 3B). His older sister had more severe and painful focal keratoderma of the soles, punctate keratoderma of the palms, gingival hyperkeratosis, and leukokeratosis of the tongue.
Whole exome sequencing of the proband revealed a heterozygous missense mutation in KRT16 (c.380G>A, p.R127H, rs57424749). Sanger sequencing confirmed this mutation and showed that it was heterozygous in both of his affected sisters and absent in his unaffected niece (Figure 1). The patient was treated with topical and systemic retinoids, keratolytics, and mechanical removal to moderate effect, with noted improvement in the appearance and associated pain of the plantar keratoderma.
Phenotypic heterogeneity is common in PC, though PC due to KRT6A mutations demonstrates more severe nail disease with oral lesions, cysts, and follicular hyperkeratosis, while PC caused by KRT16 mutations generally presents with more extensive and painful PPK.4KRT16 mutations affecting p.R127 are frequent causes of PC, and genotype-phenotype correlations have been observed. Individuals with p.R127P mutations exhibit more severe disease with earlier age of onset, more extensive nail involvement and oral leukokeratosis, and greater impact on daily quality of life than in individuals with p.R127C mutations.5 Cases of PC with KRT16 p.R127S and p.R127G mutations also have been observed. The KRT16 c.380G>A, p.R127H mutation we documented has been reported in one kindred with PC who presented with PPK, oral leukokeratosis, toenail thickening, and pilosebaceous and follicular hyperkeratosis.6
Although patients with FPGK lack the thickening of fingernails and/or toenails considered a defining feature of PC, the disorders otherwise are phenotypically similar, suggesting the possibility of common pathogenesis. One linkage study of familial FPGK excluded genetic intervals containing type I and type II keratins but was limited to a single small kindred.2 This study and our data together suggest that, similar to PC, there are multiple genes in which mutations cause FPGK.
Murine Krt16 knockouts show distinct phenotypes depending on the mouse strain in which they are propagated, ranging from perinatal lethality to differences in the severity of oral and PPK lesions.7 These observations provide evidence that additional genetic variants contribute to Krt16 phenotypes in mice and suggest the same could be true for humans.
We propose that some cases of FPGK are due to mutations in KRT16 and thus share a genetic pathogenesis with PC, underscoring the utility of whole exome sequencing in providing genetic diagnoses for disorders that are genetically and clinically heterogeneous. Further biologic investigation of phenotypes caused by KRT16 mutation may reveal respective contributions of additional genetic variation and environmental effects to the variable clinical presentations.
To the Editor:
Focal palmoplantar keratoderma and gingival keratosis (FPGK)(Online Mendelian Inheritance in Man [OMIM] 148730) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma and gingival hyperkeratosis presenting as leukokeratosis. Focal palmoplantar keratoderma and gingival keratosis was first defined by Gorlin1 in 1976. Since then, only a few cases have been reported, but no causative mutations have been identified.2
Focal pressure-related palmoplantar keratoderma (PPK) and oral hyperkeratosis also are seen in pachyonychia congenita (PC)(OMIM 167200, 615726, 615728, 167210), a rare autosomal-dominant disorder of keratinization characterized by PPK and nail dystrophy. Patients with PC often present with plantar pain; more variable features include oral leukokeratosis, follicular hyperkeratosis, pilosebaceous and epidermal inclusion cysts, hoarseness, hyperhidrosis, and natal teeth. Pachyonychia congenita is caused by mutation in keratin genes KRT6A, KRT6B, KRT16, or KRT17.
Focal palmoplantar keratoderma and gingival keratosis as well as PC are distinct from other forms of PPK with gingival involvement such as
Despite the common features of FPGK and PC, they are considered distinct disorders due to absence of nail changes in FPGK and no prior evidence of a common genetic cause. We present a patient with familial FPGK found by whole exome sequencing to be caused by a mutation in KRT16.
The proband was a 57-year-old man born to unrelated parents (Figure 1). He had no skin problems at birth, and his development was normal. He had painful focal keratoderma since childhood that were most prominent at pressure points on the soles and toes (Figure 2A), in addition to gingival hyperkeratosis and oral leukokeratosis (Figure 2B). He had no associated abnormalities of the skin, hair, or teeth and no nail findings (Figure 2C). He reported that his father and 2 of his 3 sisters were affected with similar symptoms. A punch biopsy of the right fifth toe was consistent with verrucous epidermal hyperplasia with perinuclear keratinization in the spinous layer (Figure 3A). A gingival biopsy showed perinuclear eosinophilic globules and basophilic stranding in the cytoplasm (Figure 3B). His older sister had more severe and painful focal keratoderma of the soles, punctate keratoderma of the palms, gingival hyperkeratosis, and leukokeratosis of the tongue.
Whole exome sequencing of the proband revealed a heterozygous missense mutation in KRT16 (c.380G>A, p.R127H, rs57424749). Sanger sequencing confirmed this mutation and showed that it was heterozygous in both of his affected sisters and absent in his unaffected niece (Figure 1). The patient was treated with topical and systemic retinoids, keratolytics, and mechanical removal to moderate effect, with noted improvement in the appearance and associated pain of the plantar keratoderma.
Phenotypic heterogeneity is common in PC, though PC due to KRT6A mutations demonstrates more severe nail disease with oral lesions, cysts, and follicular hyperkeratosis, while PC caused by KRT16 mutations generally presents with more extensive and painful PPK.4KRT16 mutations affecting p.R127 are frequent causes of PC, and genotype-phenotype correlations have been observed. Individuals with p.R127P mutations exhibit more severe disease with earlier age of onset, more extensive nail involvement and oral leukokeratosis, and greater impact on daily quality of life than in individuals with p.R127C mutations.5 Cases of PC with KRT16 p.R127S and p.R127G mutations also have been observed. The KRT16 c.380G>A, p.R127H mutation we documented has been reported in one kindred with PC who presented with PPK, oral leukokeratosis, toenail thickening, and pilosebaceous and follicular hyperkeratosis.6
Although patients with FPGK lack the thickening of fingernails and/or toenails considered a defining feature of PC, the disorders otherwise are phenotypically similar, suggesting the possibility of common pathogenesis. One linkage study of familial FPGK excluded genetic intervals containing type I and type II keratins but was limited to a single small kindred.2 This study and our data together suggest that, similar to PC, there are multiple genes in which mutations cause FPGK.
Murine Krt16 knockouts show distinct phenotypes depending on the mouse strain in which they are propagated, ranging from perinatal lethality to differences in the severity of oral and PPK lesions.7 These observations provide evidence that additional genetic variants contribute to Krt16 phenotypes in mice and suggest the same could be true for humans.
We propose that some cases of FPGK are due to mutations in KRT16 and thus share a genetic pathogenesis with PC, underscoring the utility of whole exome sequencing in providing genetic diagnoses for disorders that are genetically and clinically heterogeneous. Further biologic investigation of phenotypes caused by KRT16 mutation may reveal respective contributions of additional genetic variation and environmental effects to the variable clinical presentations.
- Gorlin RJ. Focal palmoplantar and marginal gingival hyperkeratosis—a syndrome. Birth Defects Orig Artic Ser. 1976;12:239-242.
- Kolde G, Hennies HC, Bethke G, et al. Focal palmoplantar and gingival keratosis: a distinct palmoplantar ectodermal dysplasia with epidermolytic alterations but lack of mutations in known keratins. J Am Acad Dermatol. 2005;52(3 pt 1):403-409.
- Duchatelet S, Hovnanian A. Olmsted syndrome: clinical, molecular and therapeutic aspects. Orphanet J Rare Dis. 2015;10:33.
- Spaunhurst KM, Hogendorf AM, Smith FJ, et al. Pachyonychia congenita patients with mutations in KRT6A have more extensive disease compared with patients who have mutations in KRT16. Br J Dermatol. 2012;166:875-878.
- Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachyonychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131:1025-1028.
- Wilson NJ, O’Toole EA, Milstone LM, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol. 2014;171:343-355.
- Zieman A, Coulombe PA. The keratin 16 null phenotype is modestly impacted by genetic strain background in mice. Exp Dermatol. 2018;27:672-674.
- Gorlin RJ. Focal palmoplantar and marginal gingival hyperkeratosis—a syndrome. Birth Defects Orig Artic Ser. 1976;12:239-242.
- Kolde G, Hennies HC, Bethke G, et al. Focal palmoplantar and gingival keratosis: a distinct palmoplantar ectodermal dysplasia with epidermolytic alterations but lack of mutations in known keratins. J Am Acad Dermatol. 2005;52(3 pt 1):403-409.
- Duchatelet S, Hovnanian A. Olmsted syndrome: clinical, molecular and therapeutic aspects. Orphanet J Rare Dis. 2015;10:33.
- Spaunhurst KM, Hogendorf AM, Smith FJ, et al. Pachyonychia congenita patients with mutations in KRT6A have more extensive disease compared with patients who have mutations in KRT16. Br J Dermatol. 2012;166:875-878.
- Fu T, Leachman SA, Wilson NJ, et al. Genotype-phenotype correlations among pachyonychia congenita patients with K16 mutations. J Invest Dermatol. 2011;131:1025-1028.
- Wilson NJ, O’Toole EA, Milstone LM, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol. 2014;171:343-355.
- Zieman A, Coulombe PA. The keratin 16 null phenotype is modestly impacted by genetic strain background in mice. Exp Dermatol. 2018;27:672-674.
Practice Points
- Focal palmoplantar keratoderma and gingival keratosis (FPGK) is a rare autosomal-dominant syndrome featuring focal, pressure-related, painful palmoplantar keratoderma (PPK) and gingival hyperkeratosis presenting as leukokeratosis.
- Focal pressure-related PPK and oral hyperkeratosis also are seen in pachyonychia congenita (PC), which is caused by mutations in keratin genes and is distinguished from FPGK by characteristic nail changes.
- A shared causative gene suggests that FPGK should be considered part of the PC spectrum.

Acute Generalized Exanthematous Pustulosis Induced by the Second-Generation Antipsychotic Cariprazine
To the Editor:
A 57-year-old woman presented to an outpatient clinic with severe pruritus and burning of the skin as well as subjective fevers and chills. She had been discharged from a psychiatric hospital for attempted suicide 1 day prior. There were no recent changes in the medication regimen, which consisted of linaclotide, fluoxetine, lorazepam, and gabapentin. While admitted, the patient was started on the atypical antipsychotic cariprazine. Within 24 hours of the first dose, she developed severe facial erythema that progressed to diffuse erythema over more than 60% of the body surface area. The attending psychiatrist promptly discontinued cariprazine. During the next 24 hours, there were no reports of fever, leukocytosis, or signs of systemic organ involvement. Given the patient’s mental and medical stability, she was discharged with instructions to follow up with the outpatient dermatology clinic.
At the current presentation, physical examination revealed innumerable 1- to 4-mm pustules coalescing to lakes of pus on an erythematous base over more than 60% of the body surface area (Figure 1). The mucous membranes were clear of lesions, the Nikolsky sign was negative, and the patient’s temperature was 99.6 °F in the office. Complete blood cell count and complete metabolic panel results were within reference range.

A 4-mm abdominal punch biopsy showed subcorneal neutrophilic pustules, papillary dermal edema, and superficial dermal lymphohistiocytic inflammation with numerous neutrophils, eosinophils, and extravasated red blood cells, consistent with acute generalized exanthematous pustulosis (AGEP)(Figure 2). The patient was started on wet wraps with triamcinolone cream 0.1%.

Two days later, physical examination revealed the erythema noted on initial examination had notably decreased, and the patient no longer reported burning or pruritus. One week after initial presentation to the clinic, the patient’s rash had resolved, and only a few small areas of desquamation remained.
Acute generalized exanthematous pustulosis is a severe cutaneous adverse reaction characterized by the development of numerous nonfollicular sterile pustules on an edematous and erythematous base. In almost 90% of reported cases, the cause is related to use of antibiotics, antifungals, antimalarials, or diltiazem (a calcium channel blocker). This rare cutaneous reaction occurs in 1 to 5 patients per million per year1; it carries a 1% to 2% mortality rate with proper supportive treatment.
The clinical symptoms of AGEP typically present 24 to 48 hours after drug initiation with the rapid development of dozens to thousands of 1- to 4-mm pustules, typically localized to the flexor surfaces and face. In the setting of AGEP, acute onset of fever and leukocytosis typically occur at the time of the cutaneous eruption. These features were absent in this patient. The eruption usually starts on the face and then migrates to the trunk and extremities, sparing the palms and soles. Systemic involvement most commonly presents as hepatic, renal, or pulmonary insufficiency, which has been seen in 20% of cases.2
The immunologic response associated with the reaction has been studied in vitro. Drug-specific CD8 T cells use perforin/granzyme B and Fas ligand mechanisms to induce apoptosis of the keratinocytes within the epidermis, leading to vesicle formation.3 During the very first stages of formation, vesicles mainly comprise CD8 T cells and keratinocytes. These cells then begin producing CXC-18, a potent neutrophil chemokine, leading to extensive chemotaxis of neutrophils into vesicles, which then rapidly transform to pustules.3 This rapid transformation leads to the lakes of pustules, a description often associated with AGEP.
Treatment of AGEP is mainly supportive and consists of discontinuing use of the causative agent. Topical corticosteroids can be used during the pustular phase for symptom management. There is no evidence that systemic steroids reduce the duration of the disease.2 Other supportive measures such as application of wet wraps can be used to provide comfort.
Cutaneous adverse drug reactions commonly are associated with psychiatric pharmacotherapy, but first-and second-generation antipsychotics rarely are associated with these types of reactions. In this patient, the causative agent of the AGEP was cariprazine, an atypical antipsychotic that had no reported association with AGEP or cutaneous adverse drug reactions prior to this presentation.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:1214.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73:843-848.
To the Editor:
A 57-year-old woman presented to an outpatient clinic with severe pruritus and burning of the skin as well as subjective fevers and chills. She had been discharged from a psychiatric hospital for attempted suicide 1 day prior. There were no recent changes in the medication regimen, which consisted of linaclotide, fluoxetine, lorazepam, and gabapentin. While admitted, the patient was started on the atypical antipsychotic cariprazine. Within 24 hours of the first dose, she developed severe facial erythema that progressed to diffuse erythema over more than 60% of the body surface area. The attending psychiatrist promptly discontinued cariprazine. During the next 24 hours, there were no reports of fever, leukocytosis, or signs of systemic organ involvement. Given the patient’s mental and medical stability, she was discharged with instructions to follow up with the outpatient dermatology clinic.
At the current presentation, physical examination revealed innumerable 1- to 4-mm pustules coalescing to lakes of pus on an erythematous base over more than 60% of the body surface area (Figure 1). The mucous membranes were clear of lesions, the Nikolsky sign was negative, and the patient’s temperature was 99.6 °F in the office. Complete blood cell count and complete metabolic panel results were within reference range.
A 4-mm abdominal punch biopsy showed subcorneal neutrophilic pustules, papillary dermal edema, and superficial dermal lymphohistiocytic inflammation with numerous neutrophils, eosinophils, and extravasated red blood cells, consistent with acute generalized exanthematous pustulosis (AGEP)(Figure 2). The patient was started on wet wraps with triamcinolone cream 0.1%.
Two days later, physical examination revealed the erythema noted on initial examination had notably decreased, and the patient no longer reported burning or pruritus. One week after initial presentation to the clinic, the patient’s rash had resolved, and only a few small areas of desquamation remained.
Acute generalized exanthematous pustulosis is a severe cutaneous adverse reaction characterized by the development of numerous nonfollicular sterile pustules on an edematous and erythematous base. In almost 90% of reported cases, the cause is related to use of antibiotics, antifungals, antimalarials, or diltiazem (a calcium channel blocker). This rare cutaneous reaction occurs in 1 to 5 patients per million per year1; it carries a 1% to 2% mortality rate with proper supportive treatment.
The clinical symptoms of AGEP typically present 24 to 48 hours after drug initiation with the rapid development of dozens to thousands of 1- to 4-mm pustules, typically localized to the flexor surfaces and face. In the setting of AGEP, acute onset of fever and leukocytosis typically occur at the time of the cutaneous eruption. These features were absent in this patient. The eruption usually starts on the face and then migrates to the trunk and extremities, sparing the palms and soles. Systemic involvement most commonly presents as hepatic, renal, or pulmonary insufficiency, which has been seen in 20% of cases.2
The immunologic response associated with the reaction has been studied in vitro. Drug-specific CD8 T cells use perforin/granzyme B and Fas ligand mechanisms to induce apoptosis of the keratinocytes within the epidermis, leading to vesicle formation.3 During the very first stages of formation, vesicles mainly comprise CD8 T cells and keratinocytes. These cells then begin producing CXC-18, a potent neutrophil chemokine, leading to extensive chemotaxis of neutrophils into vesicles, which then rapidly transform to pustules.3 This rapid transformation leads to the lakes of pustules, a description often associated with AGEP.
Treatment of AGEP is mainly supportive and consists of discontinuing use of the causative agent. Topical corticosteroids can be used during the pustular phase for symptom management. There is no evidence that systemic steroids reduce the duration of the disease.2 Other supportive measures such as application of wet wraps can be used to provide comfort.
Cutaneous adverse drug reactions commonly are associated with psychiatric pharmacotherapy, but first-and second-generation antipsychotics rarely are associated with these types of reactions. In this patient, the causative agent of the AGEP was cariprazine, an atypical antipsychotic that had no reported association with AGEP or cutaneous adverse drug reactions prior to this presentation.
To the Editor:
A 57-year-old woman presented to an outpatient clinic with severe pruritus and burning of the skin as well as subjective fevers and chills. She had been discharged from a psychiatric hospital for attempted suicide 1 day prior. There were no recent changes in the medication regimen, which consisted of linaclotide, fluoxetine, lorazepam, and gabapentin. While admitted, the patient was started on the atypical antipsychotic cariprazine. Within 24 hours of the first dose, she developed severe facial erythema that progressed to diffuse erythema over more than 60% of the body surface area. The attending psychiatrist promptly discontinued cariprazine. During the next 24 hours, there were no reports of fever, leukocytosis, or signs of systemic organ involvement. Given the patient’s mental and medical stability, she was discharged with instructions to follow up with the outpatient dermatology clinic.
At the current presentation, physical examination revealed innumerable 1- to 4-mm pustules coalescing to lakes of pus on an erythematous base over more than 60% of the body surface area (Figure 1). The mucous membranes were clear of lesions, the Nikolsky sign was negative, and the patient’s temperature was 99.6 °F in the office. Complete blood cell count and complete metabolic panel results were within reference range.
A 4-mm abdominal punch biopsy showed subcorneal neutrophilic pustules, papillary dermal edema, and superficial dermal lymphohistiocytic inflammation with numerous neutrophils, eosinophils, and extravasated red blood cells, consistent with acute generalized exanthematous pustulosis (AGEP)(Figure 2). The patient was started on wet wraps with triamcinolone cream 0.1%.
Two days later, physical examination revealed the erythema noted on initial examination had notably decreased, and the patient no longer reported burning or pruritus. One week after initial presentation to the clinic, the patient’s rash had resolved, and only a few small areas of desquamation remained.
Acute generalized exanthematous pustulosis is a severe cutaneous adverse reaction characterized by the development of numerous nonfollicular sterile pustules on an edematous and erythematous base. In almost 90% of reported cases, the cause is related to use of antibiotics, antifungals, antimalarials, or diltiazem (a calcium channel blocker). This rare cutaneous reaction occurs in 1 to 5 patients per million per year1; it carries a 1% to 2% mortality rate with proper supportive treatment.
The clinical symptoms of AGEP typically present 24 to 48 hours after drug initiation with the rapid development of dozens to thousands of 1- to 4-mm pustules, typically localized to the flexor surfaces and face. In the setting of AGEP, acute onset of fever and leukocytosis typically occur at the time of the cutaneous eruption. These features were absent in this patient. The eruption usually starts on the face and then migrates to the trunk and extremities, sparing the palms and soles. Systemic involvement most commonly presents as hepatic, renal, or pulmonary insufficiency, which has been seen in 20% of cases.2
The immunologic response associated with the reaction has been studied in vitro. Drug-specific CD8 T cells use perforin/granzyme B and Fas ligand mechanisms to induce apoptosis of the keratinocytes within the epidermis, leading to vesicle formation.3 During the very first stages of formation, vesicles mainly comprise CD8 T cells and keratinocytes. These cells then begin producing CXC-18, a potent neutrophil chemokine, leading to extensive chemotaxis of neutrophils into vesicles, which then rapidly transform to pustules.3 This rapid transformation leads to the lakes of pustules, a description often associated with AGEP.
Treatment of AGEP is mainly supportive and consists of discontinuing use of the causative agent. Topical corticosteroids can be used during the pustular phase for symptom management. There is no evidence that systemic steroids reduce the duration of the disease.2 Other supportive measures such as application of wet wraps can be used to provide comfort.
Cutaneous adverse drug reactions commonly are associated with psychiatric pharmacotherapy, but first-and second-generation antipsychotics rarely are associated with these types of reactions. In this patient, the causative agent of the AGEP was cariprazine, an atypical antipsychotic that had no reported association with AGEP or cutaneous adverse drug reactions prior to this presentation.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:1214.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73:843-848.
- Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
- Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis: pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:1214.
- Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP): a review and update. J Am Acad Dermatol. 2015;73:843-848.
Practice Points
- The second-generation antipsychotic cariprazine has been shown to be a potential causative agent in acute generalized exanthematous pustulosis (AGEP).
- Treatment of AGEP is mainly supportive and consists of discontinuation of the causative agent as well as symptom control using cold compresses and topical corticosteroids.
Nevus Lipomatosis Deemed Suspicious by Airport Security
To the Editor:
A 47-year-old man presented at the dermatology clinic with a growing lesion on the left medial thigh.
Physical examination revealed a 5-cm, pedunculated, fatty nodule on the left medial thigh that was clinically consistent with nevus lipomatosis (NL)(Figure). Although benign, trouble traveling through airport security prompted the patient to request shave removal, which subsequently was performed. Histology showed a large pedunculated nodule with prominent adipose tissue, consistent with NL. At 3-month follow-up, the patient reported getting through airport security multiple times without incident.

Nevus lipomatosis is a benign fatty lesion most commonly found on the medial thighs or trunk of adults. The lesion usually is asymptomatic but can become irritated by rubbing or catching on clothing. Our patient had symptomatic NL that caused delays getting through airport security; he experienced full resolution after simple shave removal. In rare instances, both benign and malignant skin conditions have been seen on airport scanning devices since the introduction of increased security measures following September 11, 2001. In 2016, Heymann1 reported a man with a 1.5-cm epidermal inclusion cyst detected by airport security scanners, prompting the traveler to request and carry a medically explanatory letter used to get through security. In 2015 Mayer and Adams2 described a case of nodular melanoma that was detected 20 times over a period of 2 months by airport scanners, and in 2016, Caine et al3 reported a case of desmoplastic melanoma that was detected by airport security, but after its removal was not identified by security for the next 40 flights. Noncutaneous pathology also can be detected by airport scanners. In 2013, Naraynsingh et al4 reported a man with a large left reducible inguinal hernia who was stopped by airport security and subjected to an invasive physical examination of the area. These instances demonstrate the breadth of conditions that can be cumbersome when individuals are traveling by airplane in our current security climate.
Our patient had to go through the trouble of having the benign NL lesion removed to avoid the hassle of repeatedly being stopped by airport security. The patient had the lesion removed and is doing well, but the procedure could have been avoided if systems existed to help patients with dermatologic and medical conditions at airport security. Our patient likely will never be stopped again for the suspicious lump on the left inner thigh, but many others will be stopped for similar reasons.
- Heymann WR. A cyst misinterpreted on airport scan as security threat. JAMA Dermatol. 2016;152:1388. doi:10.1001/jamadermatol.2016.3329
- Mayer JE, Adams BB. Nodular melanoma serendipitously detected by airport full body scanners. Dermatology. 2015;230:16-17. doi:10.1159/000368045
- Caine P, Javed MU, Karoo ROS. A desmoplastic melanoma detected by an airport security scanner. J Plast Reconstr Aesthet Surg. 2016;69:874-876. doi:10.1016/j.bjps.2016.02.022
- Naraynsingh V, Cawich SO, Maharaj R, et al. Inguinal hernia and airport scanners: an emerging indication for repair? 2013;2013:952835. Case Rep Med. doi:10.1155/2013/952835
To the Editor:
A 47-year-old man presented at the dermatology clinic with a growing lesion on the left medial thigh.
Physical examination revealed a 5-cm, pedunculated, fatty nodule on the left medial thigh that was clinically consistent with nevus lipomatosis (NL)(Figure). Although benign, trouble traveling through airport security prompted the patient to request shave removal, which subsequently was performed. Histology showed a large pedunculated nodule with prominent adipose tissue, consistent with NL. At 3-month follow-up, the patient reported getting through airport security multiple times without incident.
Nevus lipomatosis is a benign fatty lesion most commonly found on the medial thighs or trunk of adults. The lesion usually is asymptomatic but can become irritated by rubbing or catching on clothing. Our patient had symptomatic NL that caused delays getting through airport security; he experienced full resolution after simple shave removal. In rare instances, both benign and malignant skin conditions have been seen on airport scanning devices since the introduction of increased security measures following September 11, 2001. In 2016, Heymann1 reported a man with a 1.5-cm epidermal inclusion cyst detected by airport security scanners, prompting the traveler to request and carry a medically explanatory letter used to get through security. In 2015 Mayer and Adams2 described a case of nodular melanoma that was detected 20 times over a period of 2 months by airport scanners, and in 2016, Caine et al3 reported a case of desmoplastic melanoma that was detected by airport security, but after its removal was not identified by security for the next 40 flights. Noncutaneous pathology also can be detected by airport scanners. In 2013, Naraynsingh et al4 reported a man with a large left reducible inguinal hernia who was stopped by airport security and subjected to an invasive physical examination of the area. These instances demonstrate the breadth of conditions that can be cumbersome when individuals are traveling by airplane in our current security climate.
Our patient had to go through the trouble of having the benign NL lesion removed to avoid the hassle of repeatedly being stopped by airport security. The patient had the lesion removed and is doing well, but the procedure could have been avoided if systems existed to help patients with dermatologic and medical conditions at airport security. Our patient likely will never be stopped again for the suspicious lump on the left inner thigh, but many others will be stopped for similar reasons.
To the Editor:
A 47-year-old man presented at the dermatology clinic with a growing lesion on the left medial thigh.
Physical examination revealed a 5-cm, pedunculated, fatty nodule on the left medial thigh that was clinically consistent with nevus lipomatosis (NL)(Figure). Although benign, trouble traveling through airport security prompted the patient to request shave removal, which subsequently was performed. Histology showed a large pedunculated nodule with prominent adipose tissue, consistent with NL. At 3-month follow-up, the patient reported getting through airport security multiple times without incident.
Nevus lipomatosis is a benign fatty lesion most commonly found on the medial thighs or trunk of adults. The lesion usually is asymptomatic but can become irritated by rubbing or catching on clothing. Our patient had symptomatic NL that caused delays getting through airport security; he experienced full resolution after simple shave removal. In rare instances, both benign and malignant skin conditions have been seen on airport scanning devices since the introduction of increased security measures following September 11, 2001. In 2016, Heymann1 reported a man with a 1.5-cm epidermal inclusion cyst detected by airport security scanners, prompting the traveler to request and carry a medically explanatory letter used to get through security. In 2015 Mayer and Adams2 described a case of nodular melanoma that was detected 20 times over a period of 2 months by airport scanners, and in 2016, Caine et al3 reported a case of desmoplastic melanoma that was detected by airport security, but after its removal was not identified by security for the next 40 flights. Noncutaneous pathology also can be detected by airport scanners. In 2013, Naraynsingh et al4 reported a man with a large left reducible inguinal hernia who was stopped by airport security and subjected to an invasive physical examination of the area. These instances demonstrate the breadth of conditions that can be cumbersome when individuals are traveling by airplane in our current security climate.
Our patient had to go through the trouble of having the benign NL lesion removed to avoid the hassle of repeatedly being stopped by airport security. The patient had the lesion removed and is doing well, but the procedure could have been avoided if systems existed to help patients with dermatologic and medical conditions at airport security. Our patient likely will never be stopped again for the suspicious lump on the left inner thigh, but many others will be stopped for similar reasons.
- Heymann WR. A cyst misinterpreted on airport scan as security threat. JAMA Dermatol. 2016;152:1388. doi:10.1001/jamadermatol.2016.3329
- Mayer JE, Adams BB. Nodular melanoma serendipitously detected by airport full body scanners. Dermatology. 2015;230:16-17. doi:10.1159/000368045
- Caine P, Javed MU, Karoo ROS. A desmoplastic melanoma detected by an airport security scanner. J Plast Reconstr Aesthet Surg. 2016;69:874-876. doi:10.1016/j.bjps.2016.02.022
- Naraynsingh V, Cawich SO, Maharaj R, et al. Inguinal hernia and airport scanners: an emerging indication for repair? 2013;2013:952835. Case Rep Med. doi:10.1155/2013/952835
- Heymann WR. A cyst misinterpreted on airport scan as security threat. JAMA Dermatol. 2016;152:1388. doi:10.1001/jamadermatol.2016.3329
- Mayer JE, Adams BB. Nodular melanoma serendipitously detected by airport full body scanners. Dermatology. 2015;230:16-17. doi:10.1159/000368045
- Caine P, Javed MU, Karoo ROS. A desmoplastic melanoma detected by an airport security scanner. J Plast Reconstr Aesthet Surg. 2016;69:874-876. doi:10.1016/j.bjps.2016.02.022
- Naraynsingh V, Cawich SO, Maharaj R, et al. Inguinal hernia and airport scanners: an emerging indication for repair? 2013;2013:952835. Case Rep Med. doi:10.1155/2013/952835
Practice Points
- Nevus lipomatosis is a benign fatty lesion that most commonly is found on the medial thighs or trunk of adults.
- Both benign and malignant skin conditions have been detected on airport scanning devices.
- At times, patients must go through the hassle of having the benign lesions removed to avoid repeated problems at airport security.

Chronic Retiform Purpura of the Abdomen and Thighs: A Fatal Case of Intravascular Large Cell Lymphoma
To the Editor:
Intravascular large cell lymphoma (ILCL) is a rare B-cell lymphoma that is defined by the presence of large neoplastic B cells in the lumen of blood vessels.1 At least 3 variants of ILCL have been described based on case reports and a small case series: classic, cutaneous, and hemophagocytic. The classic variant presents in elderly patients as nonspecific constitutional symptoms (fever or pain, or less frequently weight loss) or as signs of multiorgan failure (most commonly of the central nervous system). Skin involvement, which is present in nearly half of these patients, can take on multiple morphologies, including retiform purpura, ulcerated nodules, or pseudocellulitis. The cutaneous variant typically presents in middle-aged women with normal hematologic studies. Systemic involvement is less common in this variant of disease than the classic variant, which may partly explain why overall survival is superior in this variant. The hemophagocytic variant manifests as intravascular lymphoma accompanied by hemophagocytic syndrome (fever, hepatosplenomegaly, thrombocytopenia, and bone marrow involvement).

A 69-year-old man presented to the emergency department for failure to thrive and nonhealing wounds of 1 year’s duration. His medical history was notable for poorly controlled diabetes mellitus, progressive multifocal ischemic and hemorrhagic cerebral infarcts, and bilateral deep venous thromboses. Physical examination revealed large purpuric to brown plaques in a retiform configuration with central necrotic eschars on the thighs and abdomen (Figure 1). There was no palpable lymphadenopathy. Laboratory tests revealed normocytic anemia with a hemoglobin level of 10.5 g/dL (reference range, 12–18 g/dL), elevated lactate dehydrogenase level of 525 U/L (reference range, 118–242 U/L), elevated erythrocyte sedimentation rate of 73 mm/h (reference range, <20 mm/h), antinuclear antibody (ANA) titer of 1:2560 (reference range, <1:80), and polyclonal hypergammaglobulinemia. The patient’s white blood cell and platelet counts, creatinine level, and liver function tests were within reference range. Cryoglobulins, coagulation studies, and cardiolipin antibodies were negative. Chest and abdominal imaging also were negative. An incisional skin biopsy and skin punch biopsy showed thrombotic coagulopathy and dilated vessels. A bone marrow biopsy revealed a hypercellular marrow but no plasma cell neoplasm. A repeat incisional skin biopsy demonstrated large CD20+ and CD45+ atypical lymphocytes within the small capillaries of the deep dermis and subcutaneous fat (Figure 2), which confirmed ILCL. Too deconditioned to tolerate chemotherapy, the patient opted for palliative care and died 18 months after initial presentation.
. B, CD20 immunohistochemical staining highlighted atypical B cells")
The diagnosis of ILCL often is delayed for several reasons.2 Patients can present with a variety of signs and symptoms related to small vessel occlusion that can be misattributed to other conditions.3,4 In our case, the patient’s recurrent infarcts were thought to be due to his poorly controlled diabetes mellitus, which was diagnosed a few weeks prior, and a positive ANA, even though the workup for antiphospholipid syndrome was negative. Interestingly, a positive ANA (without signs or symptoms of lupus or other autoimmune conditions) has been reported in patients with lymphoma.3 A positive antineutrophil cytoplasmic antibody level (without symptoms or other signs of vasculitis) has been reported in patients with ILCL.4,5 Therefore, distractors are common.
Multiple incisional skin biopsies in the absence of clinical findings (ie, random skin biopsy) are moderately sensitive (77.8%) for the diagnosis of ILCL.2 In a study by Matsue et al,2 111 suspected cases of ILCL underwent 3 incisional biopsies of fat-containing areas of the skin, such as the thigh, abdomen, and upper arm. Intravascular large cell lymphoma was confirmed in 26 cases. Seven additional cases were diagnosed as ILCL, 2 by additional skin biopsies (1 by a second round and 1 by a third round) and 5 by internal organ biopsy (4 bone marrow and 1 adrenal gland). The remaining cases ultimately were found to be a diagnostic mimicker of ILCL, including non-ILCL.2 Although random skin biopsies are reasonably sensitive for ILCL, multiple biopsies are needed, and in some cases, sampling of an internal organ may be required to establish the diagnosis of ILCL.
The prognosis of ILCL is poor; the 3-year overall survival rate for classic and cutaneous variants is 22% and 56%, respectively.6 Anthracycline-based chemotherapy, such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), is considered first-line treatment, and the addition of rituximab to the CHOP regimen may improve remission rates and survival.7
- Ponzoni M, Campo E, Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks [published online August 15, 2018]. Blood. 2018;132:1561-1567. doi:10.1182/blood-2017-04-737445
- Matsue K, Abe Y, Kitadate A, et al. Sensitivity and specificity of incisional random skin biopsy for diagnosis of intravascular large B-cell lymphoma. Blood. 2019;133:1257-1259.
- Altintas A, Cil T, Pasa S, et al. Clinical significance of elevated antinuclear antibody test in patients with Hodgkin’s and non-Hodgkin’s lymphoma. Minerva Med. 2008;99:7-14.
- Shinkawa Y, Hatachi S, Yagita M. Intravascular large B-cell lymphoma with a high titer of proteinase-3-anti-neutrophil cytoplasmic antibody mimicking granulomatosis with polyangiitis. Mod Rheumatol. 2019;29:195-197.
- Sugiyama A, Kobayashi M, Daizo A, et al. Diffuse cerebral vasoconstriction in a intravascular lymphoma patient with a high serum MPO-ANCA level. Intern Med. 2017;56:1715-1718.
- Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant.’ Br J Haematol. 2004;127:173-183.
- Ferreri AJM, Dognini GP, Bairey O, et al; International Extranodal Lyphoma Study Group. The addition of rituximab to anthracycline-based chemotherapy significantly improves outcome in ‘Western’ patients with intravascular large B-cell lymphoma [published online August 10, 2008]. Br J Haematol. 2008;143:253-257. doi:10.1111/j.1365-2141.2008.07338.x
To the Editor:
Intravascular large cell lymphoma (ILCL) is a rare B-cell lymphoma that is defined by the presence of large neoplastic B cells in the lumen of blood vessels.1 At least 3 variants of ILCL have been described based on case reports and a small case series: classic, cutaneous, and hemophagocytic. The classic variant presents in elderly patients as nonspecific constitutional symptoms (fever or pain, or less frequently weight loss) or as signs of multiorgan failure (most commonly of the central nervous system). Skin involvement, which is present in nearly half of these patients, can take on multiple morphologies, including retiform purpura, ulcerated nodules, or pseudocellulitis. The cutaneous variant typically presents in middle-aged women with normal hematologic studies. Systemic involvement is less common in this variant of disease than the classic variant, which may partly explain why overall survival is superior in this variant. The hemophagocytic variant manifests as intravascular lymphoma accompanied by hemophagocytic syndrome (fever, hepatosplenomegaly, thrombocytopenia, and bone marrow involvement).
A 69-year-old man presented to the emergency department for failure to thrive and nonhealing wounds of 1 year’s duration. His medical history was notable for poorly controlled diabetes mellitus, progressive multifocal ischemic and hemorrhagic cerebral infarcts, and bilateral deep venous thromboses. Physical examination revealed large purpuric to brown plaques in a retiform configuration with central necrotic eschars on the thighs and abdomen (Figure 1). There was no palpable lymphadenopathy. Laboratory tests revealed normocytic anemia with a hemoglobin level of 10.5 g/dL (reference range, 12–18 g/dL), elevated lactate dehydrogenase level of 525 U/L (reference range, 118–242 U/L), elevated erythrocyte sedimentation rate of 73 mm/h (reference range, <20 mm/h), antinuclear antibody (ANA) titer of 1:2560 (reference range, <1:80), and polyclonal hypergammaglobulinemia. The patient’s white blood cell and platelet counts, creatinine level, and liver function tests were within reference range. Cryoglobulins, coagulation studies, and cardiolipin antibodies were negative. Chest and abdominal imaging also were negative. An incisional skin biopsy and skin punch biopsy showed thrombotic coagulopathy and dilated vessels. A bone marrow biopsy revealed a hypercellular marrow but no plasma cell neoplasm. A repeat incisional skin biopsy demonstrated large CD20+ and CD45+ atypical lymphocytes within the small capillaries of the deep dermis and subcutaneous fat (Figure 2), which confirmed ILCL. Too deconditioned to tolerate chemotherapy, the patient opted for palliative care and died 18 months after initial presentation.
The diagnosis of ILCL often is delayed for several reasons.2 Patients can present with a variety of signs and symptoms related to small vessel occlusion that can be misattributed to other conditions.3,4 In our case, the patient’s recurrent infarcts were thought to be due to his poorly controlled diabetes mellitus, which was diagnosed a few weeks prior, and a positive ANA, even though the workup for antiphospholipid syndrome was negative. Interestingly, a positive ANA (without signs or symptoms of lupus or other autoimmune conditions) has been reported in patients with lymphoma.3 A positive antineutrophil cytoplasmic antibody level (without symptoms or other signs of vasculitis) has been reported in patients with ILCL.4,5 Therefore, distractors are common.
Multiple incisional skin biopsies in the absence of clinical findings (ie, random skin biopsy) are moderately sensitive (77.8%) for the diagnosis of ILCL.2 In a study by Matsue et al,2 111 suspected cases of ILCL underwent 3 incisional biopsies of fat-containing areas of the skin, such as the thigh, abdomen, and upper arm. Intravascular large cell lymphoma was confirmed in 26 cases. Seven additional cases were diagnosed as ILCL, 2 by additional skin biopsies (1 by a second round and 1 by a third round) and 5 by internal organ biopsy (4 bone marrow and 1 adrenal gland). The remaining cases ultimately were found to be a diagnostic mimicker of ILCL, including non-ILCL.2 Although random skin biopsies are reasonably sensitive for ILCL, multiple biopsies are needed, and in some cases, sampling of an internal organ may be required to establish the diagnosis of ILCL.
The prognosis of ILCL is poor; the 3-year overall survival rate for classic and cutaneous variants is 22% and 56%, respectively.6 Anthracycline-based chemotherapy, such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), is considered first-line treatment, and the addition of rituximab to the CHOP regimen may improve remission rates and survival.7
To the Editor:
Intravascular large cell lymphoma (ILCL) is a rare B-cell lymphoma that is defined by the presence of large neoplastic B cells in the lumen of blood vessels.1 At least 3 variants of ILCL have been described based on case reports and a small case series: classic, cutaneous, and hemophagocytic. The classic variant presents in elderly patients as nonspecific constitutional symptoms (fever or pain, or less frequently weight loss) or as signs of multiorgan failure (most commonly of the central nervous system). Skin involvement, which is present in nearly half of these patients, can take on multiple morphologies, including retiform purpura, ulcerated nodules, or pseudocellulitis. The cutaneous variant typically presents in middle-aged women with normal hematologic studies. Systemic involvement is less common in this variant of disease than the classic variant, which may partly explain why overall survival is superior in this variant. The hemophagocytic variant manifests as intravascular lymphoma accompanied by hemophagocytic syndrome (fever, hepatosplenomegaly, thrombocytopenia, and bone marrow involvement).
A 69-year-old man presented to the emergency department for failure to thrive and nonhealing wounds of 1 year’s duration. His medical history was notable for poorly controlled diabetes mellitus, progressive multifocal ischemic and hemorrhagic cerebral infarcts, and bilateral deep venous thromboses. Physical examination revealed large purpuric to brown plaques in a retiform configuration with central necrotic eschars on the thighs and abdomen (Figure 1). There was no palpable lymphadenopathy. Laboratory tests revealed normocytic anemia with a hemoglobin level of 10.5 g/dL (reference range, 12–18 g/dL), elevated lactate dehydrogenase level of 525 U/L (reference range, 118–242 U/L), elevated erythrocyte sedimentation rate of 73 mm/h (reference range, <20 mm/h), antinuclear antibody (ANA) titer of 1:2560 (reference range, <1:80), and polyclonal hypergammaglobulinemia. The patient’s white blood cell and platelet counts, creatinine level, and liver function tests were within reference range. Cryoglobulins, coagulation studies, and cardiolipin antibodies were negative. Chest and abdominal imaging also were negative. An incisional skin biopsy and skin punch biopsy showed thrombotic coagulopathy and dilated vessels. A bone marrow biopsy revealed a hypercellular marrow but no plasma cell neoplasm. A repeat incisional skin biopsy demonstrated large CD20+ and CD45+ atypical lymphocytes within the small capillaries of the deep dermis and subcutaneous fat (Figure 2), which confirmed ILCL. Too deconditioned to tolerate chemotherapy, the patient opted for palliative care and died 18 months after initial presentation.
The diagnosis of ILCL often is delayed for several reasons.2 Patients can present with a variety of signs and symptoms related to small vessel occlusion that can be misattributed to other conditions.3,4 In our case, the patient’s recurrent infarcts were thought to be due to his poorly controlled diabetes mellitus, which was diagnosed a few weeks prior, and a positive ANA, even though the workup for antiphospholipid syndrome was negative. Interestingly, a positive ANA (without signs or symptoms of lupus or other autoimmune conditions) has been reported in patients with lymphoma.3 A positive antineutrophil cytoplasmic antibody level (without symptoms or other signs of vasculitis) has been reported in patients with ILCL.4,5 Therefore, distractors are common.
Multiple incisional skin biopsies in the absence of clinical findings (ie, random skin biopsy) are moderately sensitive (77.8%) for the diagnosis of ILCL.2 In a study by Matsue et al,2 111 suspected cases of ILCL underwent 3 incisional biopsies of fat-containing areas of the skin, such as the thigh, abdomen, and upper arm. Intravascular large cell lymphoma was confirmed in 26 cases. Seven additional cases were diagnosed as ILCL, 2 by additional skin biopsies (1 by a second round and 1 by a third round) and 5 by internal organ biopsy (4 bone marrow and 1 adrenal gland). The remaining cases ultimately were found to be a diagnostic mimicker of ILCL, including non-ILCL.2 Although random skin biopsies are reasonably sensitive for ILCL, multiple biopsies are needed, and in some cases, sampling of an internal organ may be required to establish the diagnosis of ILCL.
The prognosis of ILCL is poor; the 3-year overall survival rate for classic and cutaneous variants is 22% and 56%, respectively.6 Anthracycline-based chemotherapy, such as CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone), is considered first-line treatment, and the addition of rituximab to the CHOP regimen may improve remission rates and survival.7
- Ponzoni M, Campo E, Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks [published online August 15, 2018]. Blood. 2018;132:1561-1567. doi:10.1182/blood-2017-04-737445
- Matsue K, Abe Y, Kitadate A, et al. Sensitivity and specificity of incisional random skin biopsy for diagnosis of intravascular large B-cell lymphoma. Blood. 2019;133:1257-1259.
- Altintas A, Cil T, Pasa S, et al. Clinical significance of elevated antinuclear antibody test in patients with Hodgkin’s and non-Hodgkin’s lymphoma. Minerva Med. 2008;99:7-14.
- Shinkawa Y, Hatachi S, Yagita M. Intravascular large B-cell lymphoma with a high titer of proteinase-3-anti-neutrophil cytoplasmic antibody mimicking granulomatosis with polyangiitis. Mod Rheumatol. 2019;29:195-197.
- Sugiyama A, Kobayashi M, Daizo A, et al. Diffuse cerebral vasoconstriction in a intravascular lymphoma patient with a high serum MPO-ANCA level. Intern Med. 2017;56:1715-1718.
- Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant.’ Br J Haematol. 2004;127:173-183.
- Ferreri AJM, Dognini GP, Bairey O, et al; International Extranodal Lyphoma Study Group. The addition of rituximab to anthracycline-based chemotherapy significantly improves outcome in ‘Western’ patients with intravascular large B-cell lymphoma [published online August 10, 2008]. Br J Haematol. 2008;143:253-257. doi:10.1111/j.1365-2141.2008.07338.x
- Ponzoni M, Campo E, Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks [published online August 15, 2018]. Blood. 2018;132:1561-1567. doi:10.1182/blood-2017-04-737445
- Matsue K, Abe Y, Kitadate A, et al. Sensitivity and specificity of incisional random skin biopsy for diagnosis of intravascular large B-cell lymphoma. Blood. 2019;133:1257-1259.
- Altintas A, Cil T, Pasa S, et al. Clinical significance of elevated antinuclear antibody test in patients with Hodgkin’s and non-Hodgkin’s lymphoma. Minerva Med. 2008;99:7-14.
- Shinkawa Y, Hatachi S, Yagita M. Intravascular large B-cell lymphoma with a high titer of proteinase-3-anti-neutrophil cytoplasmic antibody mimicking granulomatosis with polyangiitis. Mod Rheumatol. 2019;29:195-197.
- Sugiyama A, Kobayashi M, Daizo A, et al. Diffuse cerebral vasoconstriction in a intravascular lymphoma patient with a high serum MPO-ANCA level. Intern Med. 2017;56:1715-1718.
- Ferreri AJ, Campo E, Seymour JF, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant.’ Br J Haematol. 2004;127:173-183.
- Ferreri AJM, Dognini GP, Bairey O, et al; International Extranodal Lyphoma Study Group. The addition of rituximab to anthracycline-based chemotherapy significantly improves outcome in ‘Western’ patients with intravascular large B-cell lymphoma [published online August 10, 2008]. Br J Haematol. 2008;143:253-257. doi:10.1111/j.1365-2141.2008.07338.x
Practice Points
- Intravascular large cell lymphoma (ILCL) is a life-threatening malignancy that can present with retiform purpura and other symptoms of vascular occlusion.
- The diagnosis of ILCL can be challenging because of the presence of distractors, and multiple biopsies may be required to establish pathology.

Tumor Necrosis Factor α Inhibitor–Induced Lupuslike Syndrome in a Patient Prescribed Certolizumab Pegol
To the Editor:
Tumor necrosis factor α (TNF-α) inhibitor–induced lupuslike syndrome (TAILS) is a newly described entity that refers to the onset of subacute cutaneous lupus erythematosus (SCLE) during drug therapy with TNF-α antagonists. The condition is unique because it is thought to occur via a separate pathophysiologic mechanism than all other agents implicated in the development of drug-induced lupus erythematosus (DILE). Infliximab and etanercept are the 2 most common TNF-α antagonists associated with TAILS. Although rare, adalimumab, golimumab, and certolizumab pegol have been reported to induce this state of autoimmunity. We report an uncommon presentation of TAILS in a patient taking certolizumab pegol with a brief discussion of the pathogenesis underlying TAILS.

A 71-year-old woman presented to the dermatology clinic with a rash located on the arms, face, and trunk that she reported as having been present for months. She had a medical history of rheumatoid arthritis and currently was receiving certolizumab pegol injections. Physical examination revealed erythematous patches and plaques with overlying scaling and evidence of atrophic scarring on sun-exposed areas of the body. The lesions predominantly were in a symmetrical distribution across the extensor surfaces of both outer arms as well as the posterior superior thoracic region extending anteriorly along the bilateral supraclavicular area (Figures 1 and 2). A 4-mm punch biopsy was obtained and sent for histologic analysis, along with a sample of the patient’s serum for antinuclear antibody (ANA) testing.
.")
Hematoxylin and eosin–stained tissue sections of the right superior thoracic lesions revealed epidermal atrophy, hyperkeratosis, and vacuolar alteration of the basal layer with apoptosis, consistent with a lichenoid tissue reaction. In addition, both superficial and deep perivascular and periadnexal lymphocytic infiltrates were observed as well as increased dermal mucin. Serologic testing was performed with a comprehensive ANA panel of the patient’s serum (Table). Of note, there was a speckled ANA pattern (1:1280), with elevated anti–double-stranded DNA (anti-dsDNA) and anti–Sjögren syndrome–related antigen A (anti-SSA)(also called anti-Ro antibodies) levels. The patient’s rheumatologist was consulted; certolizumab pegol was removed from the current drug regimen and switched to a daily regimen of hydroxychloroquine and prednisone. Seven weeks after discontinuation of certolizumab pegol, the patient was symptom free and without any cutaneous involvement. Based on the histologic analysis, presence of anti-SSA (Ro) autoantibodies, and the resolution of symptoms following withdrawal of anti–TNF-α therapy, a diagnosis of TAILS was made.

Subacute cutaneous lupus erythematosus, the most common subset of DILE, typically presents with annular polycyclic or papulosquamous skin eruptions on the legs; patients often test positive for anti-SSA/Ro and/or anti–Sjögren syndrome–related antigen B (also called anti-La) antibodies. Pharmaceutical agents linked to the development of SCLE are calcium channel blockers, angiotensin-converting enzyme inhibitors, thiazide diuretics, terbinafine, the chemotherapeutic agent gemcitabine, and TNF-α antagonists.1,2 Tumor necrosis factor α antagonists are biologic agents that commonly are used in the management of systemic inflammatory diseases such as ulcerative colitis, Crohn disease, seronegative spondyloarthropathies, and rheumatoid arthritis. Among this family of therapeutics includes adalimumab (humanized monoclonal antibody), infliximab (chimeric monoclonal TNF-α antagonist), etanercept (soluble receptor fusion protein), certolizumab pegol (Fab fraction of a human IgG monoclonal antibody), and golimumab (humanized monoclonal antibody).
Tumor necrosis factor α inhibitor–induced lupuslike syndrome most commonly occurs in women in the fifth decade of life, and it is seen more often in those using infliximab or entanercept.3 Although reports do exist, TAILS rarely complicates treatment with adalimumab, golimumab, or certolizumab.4,5 Due to the lack of reports, there are no diagnostic criteria nor an acceptable theory regarding the pathogenesis. In one study in France, the estimated incidence was thought to be 0.19% for infliximab and 0.18% for etanercept.6 Tumor necrosis factor α inhibitor–induced lupuslike syndrome is unique in that it is thought to occur by a different mechanism than that of other known offending agents in the development of DILE. Molecular mimicry, direct cytotoxicity, altered T-cell gene expression, and disruption of central immune tolerance have all been hypothesized to cause drug-induced systemic lupus erythematosus, SCLE, and chronic cutaneous lupus erythematosus. Tumor necrosis factor α inhibitors, are postulated to cause the induction of SCLE via an independent route separate from not only other drugs that cause SCLE but also all forms of DILE as a whole, making it a distinctive player within the realm of agents known to cause a lupuslike syndrome. The following hypotheses may explain this occurrence:
1. Increased humoral autoimmunity: Under normal circumstances, TNF-α activation leads to upregulation in the production of cytotoxic CD8+ T lymphocytes. The upregulation of CD8+ T lymphocytes concurrently leads to a simultaneous suppression of B lymphocytes. Inhibiting the effects of TNF-α on the other hand promotes cytotoxic T-lymphocyte suppression, leading to an increased synthesis of B cells and subsequently a state of increased humoral autoimmunity.7
2. Infection: The immunosuppressive effects of TNF-α inhibitors are well known, and the propensity to develop microbial infections, such as tuberculosis, is markedly increased on the use of these agents. Infections brought on by TNF-α inhibitor usage are hypothesized to induce a widespread activation of polyclonal B lymphocytes, eventually leading to the formation of antibodies against these polyclonal B lymphocytes and subsequently SCLE.8
3. Helper T cell (TH2) response: The inhibition of TH1 CD4+ lymphocytes by TNF-α inversely leads to an increased production of TH2 CD4+ lymphocytes. This increase in the levels of circulating TH2 CD4+ lymphocytes brought on by the action of anti–TNF-α agents is thought to promote the development of SCLE.9,10
4. Apoptosis theory: Molecules of TNF-α inhibitors are capable of binding to TNF-α receptors on the cell surface. In doing so, cellular apoptosis is triggered, resulting in the release of nucleosomal autoantigens from the apoptotic cells. In susceptible individuals, autoantibodies then begin to form against the nucleosomal autoantigens, leading to an autoimmune reaction that is characterized by SCLE.11,12
Major histone compatibility (MHC) antigen testing performed by Sontheimer et al12 established the presence of the HLA class I, HLA-B8, and/or HLA-DR3 haplotypes in patients with SCLE.13,14 Furthermore, there is a well-known association between the antinuclear profile of known SCLE patients and the presence of anti-SSA (Ro) antibodies.13 Therefore, we propose that in susceptible individuals, such as those with the HLA class I, HLA-B8, or HLA-DR3 haplotypes, the initiation of a TNF-α inhibitor causes cellular apoptosis with the subsequent release of nucleosomal and cytoplasmic components (namely that of the Ro autoantigens), inducing a state of autoimmunity. An ensuing immunogenic response is then initiated in predisposed individuals for which anti-SSA (Ro) autoantibodies are produced against these previously mentioned autoantigens.
Drug-induced SCLE is most common in females (71%), with a median age of 58 years. The most common site of cutaneous manifestations is the legs.15 Although our patient was in the eighth decade of life with predominant cutaneous involvement of the upper extremity, the erythematous plaques with a symmetric, annular, polycyclic appearance in photosensitive regions raised a heightened suspicion for lupus erythematosus. Histology classically involves an interface dermatitis with vacuolar or hydropic change and lymphocytic infiltrates,16 consistent with the analysis of tissue sections from our patient. Moreover, the speckled ANA profile with positive anti-dsDNA and anti-SSA (Ro) antibodies in the absence of a negative rheumatoid factor and anticyclic citrullinated peptide antibodies strongly favored the diagnosis of SCLE over alternative diagnoses.2
The supraclavicular rash in our patient raises clinical suspicion for the shawl sign of dermatomyositis, which also is associated with musculoskeletal pain and photosensitivity. In addition, skin biopsy revealed vacuolar alteration of the basement membrane zoneand dermal mucin in both lupus erythematosus and dermatomyositis; therefore, skin biopsy is of little use in distinguishing the 2 conditions, and antibody testing must be performed. Although anti-SSA (Ro) antibodies commonly are associated with SCLE, there are reports involving positivity for the extractable nuclear antigen in cases of dermatomyositis.17 Based on our patient’s current drug regimen, including that of a known offending agent for SCLE, a presumptive diagnosis of TAILS was made. Following withdrawal of certolizumab pegol injections and subsequent resolution of the skin lesions, our patient was given a definitive diagnosis of TAILS based on clinical and pathological assessments.
The clinical diagnosis of TAILS should be made according to the triad of at least 1 serologic and 1 nonserologic American College of Rheumatology criteria, such as anti-SSA (Ro) antibodies and a photosensitive rash, respectively, as well as a relationship between the onset of symptoms and TNF-α inhibitor therapy.18 Both the definitive diagnosis and the treatment of TAILS can be made via withdrawal of the TNF-α inhibitor, which was true in our case whereby chronologically the onset of use with a TNF-α inhibitor was associated with disease onset. Furthermore, withdrawal led to complete improvement of all signs and symptoms, collectively supporting a diagnosis of TAILS. Notably, switching to a different TNF-α inhibitor has been shown to be safe and effective.19
- Marzano AV, Vezzoli P, Crosti C. Drug-induced lupus: an update on its dermatological aspects. Lupus. 2009;18:935-940.
- Wiznia LE, Subtil A, Choi JN. Subacute cutaneous lupus erythematosus induced by chemotherapy: gemcitabine as a causative agent. JAMA Dermatol. 2013;149:1071-1075.
- Williams VL, Cohen PR. TNF alpha antagonist-induced lupus-like syndrome: report and review of the literature with implications for treatment with alternative TNF alpha antagonists. Int J Dermatol. 2011;50:619-625.
- Pasut G. Pegylation of biological molecules and potential benefits: pharmacological properties of certolizumab pegol. Bio Drugs. 2014;28(suppl 1):15-23.
- Mudduluru BM, Shah S, Shamah S. et al. TNF-alpha antagonist induced lupus on three different agents. Postgrad Med. 2017;129:304-306.
- De Bandt M. Anti-TNF-alpha-induced lupus. Arthritis Res Ther. 2019;21:235.
- Costa MF, Said NR, Zimmermann B. Drug-induced lupus due to anti-tumor necrosis factor alfa agents. Semin Arthritis Rheum. 2008;37:381-387.
- Caramaschi P, Biasi D, Colombatti M. Anti-TNF alpha therapy in rheumatoid arthritis and autoimmunity. Rheumatol Int. 2006;26:209-214.
- Yung RL, Quddus J, Chrisp CE, et al. Mechanism of drug-induced lupus. I. cloned Th2 cells modified with DNA methylation inhibitors in vitro cause autoimmunity in vivo. J Immunol. 1995;154:3025-3035.
- Yung R, Powers D, Johnson K, et al. Mechanisms of drug-induced lupus. II. T cells overexpressing lymphocyte function-associated antigen 1 become autoreactive and cause a lupuslike disease in syngeneic mice. J Clin Invest. 1996;97:2866-2871.
- Sontheimer RD, Stastny P, Gilliam JN. Human histocompatibility antigen associations in subacute cutaneous lupus erythematosus. J Clin Invest. 1981;67:312-316.
- Sontheimer RD, Maddison PJ, Reichlin M, et al. Serologic and HLA associations in subacute cutaneous lupus erythematosus, a clinical subset of lupus erythematosus. Ann Intern Med. 1982;97:664-671.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
- Deutscher SL, Harley JB, Keene JD. Molecular analysis of the 60-kDa human Ro ribonucleoprotein. Proc Natl Acad Sci. 1988;85:9479-9483.
- DalleVedove C, Simon JC, Girolomoni G. Drug-induced lupus erythematosus with emphasis on skin manifestations and the role of anti-TNFα agents [article in German]. J Dtsch Dermatol Ges. 2012;10:889-897.
- Okon LG, Werth VP. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol. 2013;27:391-404.
- Schulte-Pelkum J, Fritzler M, Mahler M. Latest update on the Ro/SS-A autoantibody system. Autoimmun Rev. 2009;8:632-637.
- De Bandt M, Sibilia J, Le Loët X, et al. Systemic lupus erythematosus induced by anti-tumour necrosis factor alpha therapy: a French national survey. Arthritis Res Ther. 2005;7:R545-R551.
- Lupu A, Tieranu C, Constantinescu CL, et al. TNFα inhibitor induced lupus-like syndrome (TAILS) in a patient with IBD. Current Health Sci J. 2014;40:285-288.
To the Editor:
Tumor necrosis factor α (TNF-α) inhibitor–induced lupuslike syndrome (TAILS) is a newly described entity that refers to the onset of subacute cutaneous lupus erythematosus (SCLE) during drug therapy with TNF-α antagonists. The condition is unique because it is thought to occur via a separate pathophysiologic mechanism than all other agents implicated in the development of drug-induced lupus erythematosus (DILE). Infliximab and etanercept are the 2 most common TNF-α antagonists associated with TAILS. Although rare, adalimumab, golimumab, and certolizumab pegol have been reported to induce this state of autoimmunity. We report an uncommon presentation of TAILS in a patient taking certolizumab pegol with a brief discussion of the pathogenesis underlying TAILS.
A 71-year-old woman presented to the dermatology clinic with a rash located on the arms, face, and trunk that she reported as having been present for months. She had a medical history of rheumatoid arthritis and currently was receiving certolizumab pegol injections. Physical examination revealed erythematous patches and plaques with overlying scaling and evidence of atrophic scarring on sun-exposed areas of the body. The lesions predominantly were in a symmetrical distribution across the extensor surfaces of both outer arms as well as the posterior superior thoracic region extending anteriorly along the bilateral supraclavicular area (Figures 1 and 2). A 4-mm punch biopsy was obtained and sent for histologic analysis, along with a sample of the patient’s serum for antinuclear antibody (ANA) testing.
Hematoxylin and eosin–stained tissue sections of the right superior thoracic lesions revealed epidermal atrophy, hyperkeratosis, and vacuolar alteration of the basal layer with apoptosis, consistent with a lichenoid tissue reaction. In addition, both superficial and deep perivascular and periadnexal lymphocytic infiltrates were observed as well as increased dermal mucin. Serologic testing was performed with a comprehensive ANA panel of the patient’s serum (Table). Of note, there was a speckled ANA pattern (1:1280), with elevated anti–double-stranded DNA (anti-dsDNA) and anti–Sjögren syndrome–related antigen A (anti-SSA)(also called anti-Ro antibodies) levels. The patient’s rheumatologist was consulted; certolizumab pegol was removed from the current drug regimen and switched to a daily regimen of hydroxychloroquine and prednisone. Seven weeks after discontinuation of certolizumab pegol, the patient was symptom free and without any cutaneous involvement. Based on the histologic analysis, presence of anti-SSA (Ro) autoantibodies, and the resolution of symptoms following withdrawal of anti–TNF-α therapy, a diagnosis of TAILS was made.
Subacute cutaneous lupus erythematosus, the most common subset of DILE, typically presents with annular polycyclic or papulosquamous skin eruptions on the legs; patients often test positive for anti-SSA/Ro and/or anti–Sjögren syndrome–related antigen B (also called anti-La) antibodies. Pharmaceutical agents linked to the development of SCLE are calcium channel blockers, angiotensin-converting enzyme inhibitors, thiazide diuretics, terbinafine, the chemotherapeutic agent gemcitabine, and TNF-α antagonists.1,2 Tumor necrosis factor α antagonists are biologic agents that commonly are used in the management of systemic inflammatory diseases such as ulcerative colitis, Crohn disease, seronegative spondyloarthropathies, and rheumatoid arthritis. Among this family of therapeutics includes adalimumab (humanized monoclonal antibody), infliximab (chimeric monoclonal TNF-α antagonist), etanercept (soluble receptor fusion protein), certolizumab pegol (Fab fraction of a human IgG monoclonal antibody), and golimumab (humanized monoclonal antibody).
Tumor necrosis factor α inhibitor–induced lupuslike syndrome most commonly occurs in women in the fifth decade of life, and it is seen more often in those using infliximab or entanercept.3 Although reports do exist, TAILS rarely complicates treatment with adalimumab, golimumab, or certolizumab.4,5 Due to the lack of reports, there are no diagnostic criteria nor an acceptable theory regarding the pathogenesis. In one study in France, the estimated incidence was thought to be 0.19% for infliximab and 0.18% for etanercept.6 Tumor necrosis factor α inhibitor–induced lupuslike syndrome is unique in that it is thought to occur by a different mechanism than that of other known offending agents in the development of DILE. Molecular mimicry, direct cytotoxicity, altered T-cell gene expression, and disruption of central immune tolerance have all been hypothesized to cause drug-induced systemic lupus erythematosus, SCLE, and chronic cutaneous lupus erythematosus. Tumor necrosis factor α inhibitors, are postulated to cause the induction of SCLE via an independent route separate from not only other drugs that cause SCLE but also all forms of DILE as a whole, making it a distinctive player within the realm of agents known to cause a lupuslike syndrome. The following hypotheses may explain this occurrence:
1. Increased humoral autoimmunity: Under normal circumstances, TNF-α activation leads to upregulation in the production of cytotoxic CD8+ T lymphocytes. The upregulation of CD8+ T lymphocytes concurrently leads to a simultaneous suppression of B lymphocytes. Inhibiting the effects of TNF-α on the other hand promotes cytotoxic T-lymphocyte suppression, leading to an increased synthesis of B cells and subsequently a state of increased humoral autoimmunity.7
2. Infection: The immunosuppressive effects of TNF-α inhibitors are well known, and the propensity to develop microbial infections, such as tuberculosis, is markedly increased on the use of these agents. Infections brought on by TNF-α inhibitor usage are hypothesized to induce a widespread activation of polyclonal B lymphocytes, eventually leading to the formation of antibodies against these polyclonal B lymphocytes and subsequently SCLE.8
3. Helper T cell (TH2) response: The inhibition of TH1 CD4+ lymphocytes by TNF-α inversely leads to an increased production of TH2 CD4+ lymphocytes. This increase in the levels of circulating TH2 CD4+ lymphocytes brought on by the action of anti–TNF-α agents is thought to promote the development of SCLE.9,10
4. Apoptosis theory: Molecules of TNF-α inhibitors are capable of binding to TNF-α receptors on the cell surface. In doing so, cellular apoptosis is triggered, resulting in the release of nucleosomal autoantigens from the apoptotic cells. In susceptible individuals, autoantibodies then begin to form against the nucleosomal autoantigens, leading to an autoimmune reaction that is characterized by SCLE.11,12
Major histone compatibility (MHC) antigen testing performed by Sontheimer et al12 established the presence of the HLA class I, HLA-B8, and/or HLA-DR3 haplotypes in patients with SCLE.13,14 Furthermore, there is a well-known association between the antinuclear profile of known SCLE patients and the presence of anti-SSA (Ro) antibodies.13 Therefore, we propose that in susceptible individuals, such as those with the HLA class I, HLA-B8, or HLA-DR3 haplotypes, the initiation of a TNF-α inhibitor causes cellular apoptosis with the subsequent release of nucleosomal and cytoplasmic components (namely that of the Ro autoantigens), inducing a state of autoimmunity. An ensuing immunogenic response is then initiated in predisposed individuals for which anti-SSA (Ro) autoantibodies are produced against these previously mentioned autoantigens.
Drug-induced SCLE is most common in females (71%), with a median age of 58 years. The most common site of cutaneous manifestations is the legs.15 Although our patient was in the eighth decade of life with predominant cutaneous involvement of the upper extremity, the erythematous plaques with a symmetric, annular, polycyclic appearance in photosensitive regions raised a heightened suspicion for lupus erythematosus. Histology classically involves an interface dermatitis with vacuolar or hydropic change and lymphocytic infiltrates,16 consistent with the analysis of tissue sections from our patient. Moreover, the speckled ANA profile with positive anti-dsDNA and anti-SSA (Ro) antibodies in the absence of a negative rheumatoid factor and anticyclic citrullinated peptide antibodies strongly favored the diagnosis of SCLE over alternative diagnoses.2
The supraclavicular rash in our patient raises clinical suspicion for the shawl sign of dermatomyositis, which also is associated with musculoskeletal pain and photosensitivity. In addition, skin biopsy revealed vacuolar alteration of the basement membrane zoneand dermal mucin in both lupus erythematosus and dermatomyositis; therefore, skin biopsy is of little use in distinguishing the 2 conditions, and antibody testing must be performed. Although anti-SSA (Ro) antibodies commonly are associated with SCLE, there are reports involving positivity for the extractable nuclear antigen in cases of dermatomyositis.17 Based on our patient’s current drug regimen, including that of a known offending agent for SCLE, a presumptive diagnosis of TAILS was made. Following withdrawal of certolizumab pegol injections and subsequent resolution of the skin lesions, our patient was given a definitive diagnosis of TAILS based on clinical and pathological assessments.
The clinical diagnosis of TAILS should be made according to the triad of at least 1 serologic and 1 nonserologic American College of Rheumatology criteria, such as anti-SSA (Ro) antibodies and a photosensitive rash, respectively, as well as a relationship between the onset of symptoms and TNF-α inhibitor therapy.18 Both the definitive diagnosis and the treatment of TAILS can be made via withdrawal of the TNF-α inhibitor, which was true in our case whereby chronologically the onset of use with a TNF-α inhibitor was associated with disease onset. Furthermore, withdrawal led to complete improvement of all signs and symptoms, collectively supporting a diagnosis of TAILS. Notably, switching to a different TNF-α inhibitor has been shown to be safe and effective.19
To the Editor:
Tumor necrosis factor α (TNF-α) inhibitor–induced lupuslike syndrome (TAILS) is a newly described entity that refers to the onset of subacute cutaneous lupus erythematosus (SCLE) during drug therapy with TNF-α antagonists. The condition is unique because it is thought to occur via a separate pathophysiologic mechanism than all other agents implicated in the development of drug-induced lupus erythematosus (DILE). Infliximab and etanercept are the 2 most common TNF-α antagonists associated with TAILS. Although rare, adalimumab, golimumab, and certolizumab pegol have been reported to induce this state of autoimmunity. We report an uncommon presentation of TAILS in a patient taking certolizumab pegol with a brief discussion of the pathogenesis underlying TAILS.
A 71-year-old woman presented to the dermatology clinic with a rash located on the arms, face, and trunk that she reported as having been present for months. She had a medical history of rheumatoid arthritis and currently was receiving certolizumab pegol injections. Physical examination revealed erythematous patches and plaques with overlying scaling and evidence of atrophic scarring on sun-exposed areas of the body. The lesions predominantly were in a symmetrical distribution across the extensor surfaces of both outer arms as well as the posterior superior thoracic region extending anteriorly along the bilateral supraclavicular area (Figures 1 and 2). A 4-mm punch biopsy was obtained and sent for histologic analysis, along with a sample of the patient’s serum for antinuclear antibody (ANA) testing.
Hematoxylin and eosin–stained tissue sections of the right superior thoracic lesions revealed epidermal atrophy, hyperkeratosis, and vacuolar alteration of the basal layer with apoptosis, consistent with a lichenoid tissue reaction. In addition, both superficial and deep perivascular and periadnexal lymphocytic infiltrates were observed as well as increased dermal mucin. Serologic testing was performed with a comprehensive ANA panel of the patient’s serum (Table). Of note, there was a speckled ANA pattern (1:1280), with elevated anti–double-stranded DNA (anti-dsDNA) and anti–Sjögren syndrome–related antigen A (anti-SSA)(also called anti-Ro antibodies) levels. The patient’s rheumatologist was consulted; certolizumab pegol was removed from the current drug regimen and switched to a daily regimen of hydroxychloroquine and prednisone. Seven weeks after discontinuation of certolizumab pegol, the patient was symptom free and without any cutaneous involvement. Based on the histologic analysis, presence of anti-SSA (Ro) autoantibodies, and the resolution of symptoms following withdrawal of anti–TNF-α therapy, a diagnosis of TAILS was made.
Subacute cutaneous lupus erythematosus, the most common subset of DILE, typically presents with annular polycyclic or papulosquamous skin eruptions on the legs; patients often test positive for anti-SSA/Ro and/or anti–Sjögren syndrome–related antigen B (also called anti-La) antibodies. Pharmaceutical agents linked to the development of SCLE are calcium channel blockers, angiotensin-converting enzyme inhibitors, thiazide diuretics, terbinafine, the chemotherapeutic agent gemcitabine, and TNF-α antagonists.1,2 Tumor necrosis factor α antagonists are biologic agents that commonly are used in the management of systemic inflammatory diseases such as ulcerative colitis, Crohn disease, seronegative spondyloarthropathies, and rheumatoid arthritis. Among this family of therapeutics includes adalimumab (humanized monoclonal antibody), infliximab (chimeric monoclonal TNF-α antagonist), etanercept (soluble receptor fusion protein), certolizumab pegol (Fab fraction of a human IgG monoclonal antibody), and golimumab (humanized monoclonal antibody).
Tumor necrosis factor α inhibitor–induced lupuslike syndrome most commonly occurs in women in the fifth decade of life, and it is seen more often in those using infliximab or entanercept.3 Although reports do exist, TAILS rarely complicates treatment with adalimumab, golimumab, or certolizumab.4,5 Due to the lack of reports, there are no diagnostic criteria nor an acceptable theory regarding the pathogenesis. In one study in France, the estimated incidence was thought to be 0.19% for infliximab and 0.18% for etanercept.6 Tumor necrosis factor α inhibitor–induced lupuslike syndrome is unique in that it is thought to occur by a different mechanism than that of other known offending agents in the development of DILE. Molecular mimicry, direct cytotoxicity, altered T-cell gene expression, and disruption of central immune tolerance have all been hypothesized to cause drug-induced systemic lupus erythematosus, SCLE, and chronic cutaneous lupus erythematosus. Tumor necrosis factor α inhibitors, are postulated to cause the induction of SCLE via an independent route separate from not only other drugs that cause SCLE but also all forms of DILE as a whole, making it a distinctive player within the realm of agents known to cause a lupuslike syndrome. The following hypotheses may explain this occurrence:
1. Increased humoral autoimmunity: Under normal circumstances, TNF-α activation leads to upregulation in the production of cytotoxic CD8+ T lymphocytes. The upregulation of CD8+ T lymphocytes concurrently leads to a simultaneous suppression of B lymphocytes. Inhibiting the effects of TNF-α on the other hand promotes cytotoxic T-lymphocyte suppression, leading to an increased synthesis of B cells and subsequently a state of increased humoral autoimmunity.7
2. Infection: The immunosuppressive effects of TNF-α inhibitors are well known, and the propensity to develop microbial infections, such as tuberculosis, is markedly increased on the use of these agents. Infections brought on by TNF-α inhibitor usage are hypothesized to induce a widespread activation of polyclonal B lymphocytes, eventually leading to the formation of antibodies against these polyclonal B lymphocytes and subsequently SCLE.8
3. Helper T cell (TH2) response: The inhibition of TH1 CD4+ lymphocytes by TNF-α inversely leads to an increased production of TH2 CD4+ lymphocytes. This increase in the levels of circulating TH2 CD4+ lymphocytes brought on by the action of anti–TNF-α agents is thought to promote the development of SCLE.9,10
4. Apoptosis theory: Molecules of TNF-α inhibitors are capable of binding to TNF-α receptors on the cell surface. In doing so, cellular apoptosis is triggered, resulting in the release of nucleosomal autoantigens from the apoptotic cells. In susceptible individuals, autoantibodies then begin to form against the nucleosomal autoantigens, leading to an autoimmune reaction that is characterized by SCLE.11,12
Major histone compatibility (MHC) antigen testing performed by Sontheimer et al12 established the presence of the HLA class I, HLA-B8, and/or HLA-DR3 haplotypes in patients with SCLE.13,14 Furthermore, there is a well-known association between the antinuclear profile of known SCLE patients and the presence of anti-SSA (Ro) antibodies.13 Therefore, we propose that in susceptible individuals, such as those with the HLA class I, HLA-B8, or HLA-DR3 haplotypes, the initiation of a TNF-α inhibitor causes cellular apoptosis with the subsequent release of nucleosomal and cytoplasmic components (namely that of the Ro autoantigens), inducing a state of autoimmunity. An ensuing immunogenic response is then initiated in predisposed individuals for which anti-SSA (Ro) autoantibodies are produced against these previously mentioned autoantigens.
Drug-induced SCLE is most common in females (71%), with a median age of 58 years. The most common site of cutaneous manifestations is the legs.15 Although our patient was in the eighth decade of life with predominant cutaneous involvement of the upper extremity, the erythematous plaques with a symmetric, annular, polycyclic appearance in photosensitive regions raised a heightened suspicion for lupus erythematosus. Histology classically involves an interface dermatitis with vacuolar or hydropic change and lymphocytic infiltrates,16 consistent with the analysis of tissue sections from our patient. Moreover, the speckled ANA profile with positive anti-dsDNA and anti-SSA (Ro) antibodies in the absence of a negative rheumatoid factor and anticyclic citrullinated peptide antibodies strongly favored the diagnosis of SCLE over alternative diagnoses.2
The supraclavicular rash in our patient raises clinical suspicion for the shawl sign of dermatomyositis, which also is associated with musculoskeletal pain and photosensitivity. In addition, skin biopsy revealed vacuolar alteration of the basement membrane zoneand dermal mucin in both lupus erythematosus and dermatomyositis; therefore, skin biopsy is of little use in distinguishing the 2 conditions, and antibody testing must be performed. Although anti-SSA (Ro) antibodies commonly are associated with SCLE, there are reports involving positivity for the extractable nuclear antigen in cases of dermatomyositis.17 Based on our patient’s current drug regimen, including that of a known offending agent for SCLE, a presumptive diagnosis of TAILS was made. Following withdrawal of certolizumab pegol injections and subsequent resolution of the skin lesions, our patient was given a definitive diagnosis of TAILS based on clinical and pathological assessments.
The clinical diagnosis of TAILS should be made according to the triad of at least 1 serologic and 1 nonserologic American College of Rheumatology criteria, such as anti-SSA (Ro) antibodies and a photosensitive rash, respectively, as well as a relationship between the onset of symptoms and TNF-α inhibitor therapy.18 Both the definitive diagnosis and the treatment of TAILS can be made via withdrawal of the TNF-α inhibitor, which was true in our case whereby chronologically the onset of use with a TNF-α inhibitor was associated with disease onset. Furthermore, withdrawal led to complete improvement of all signs and symptoms, collectively supporting a diagnosis of TAILS. Notably, switching to a different TNF-α inhibitor has been shown to be safe and effective.19
- Marzano AV, Vezzoli P, Crosti C. Drug-induced lupus: an update on its dermatological aspects. Lupus. 2009;18:935-940.
- Wiznia LE, Subtil A, Choi JN. Subacute cutaneous lupus erythematosus induced by chemotherapy: gemcitabine as a causative agent. JAMA Dermatol. 2013;149:1071-1075.
- Williams VL, Cohen PR. TNF alpha antagonist-induced lupus-like syndrome: report and review of the literature with implications for treatment with alternative TNF alpha antagonists. Int J Dermatol. 2011;50:619-625.
- Pasut G. Pegylation of biological molecules and potential benefits: pharmacological properties of certolizumab pegol. Bio Drugs. 2014;28(suppl 1):15-23.
- Mudduluru BM, Shah S, Shamah S. et al. TNF-alpha antagonist induced lupus on three different agents. Postgrad Med. 2017;129:304-306.
- De Bandt M. Anti-TNF-alpha-induced lupus. Arthritis Res Ther. 2019;21:235.
- Costa MF, Said NR, Zimmermann B. Drug-induced lupus due to anti-tumor necrosis factor alfa agents. Semin Arthritis Rheum. 2008;37:381-387.
- Caramaschi P, Biasi D, Colombatti M. Anti-TNF alpha therapy in rheumatoid arthritis and autoimmunity. Rheumatol Int. 2006;26:209-214.
- Yung RL, Quddus J, Chrisp CE, et al. Mechanism of drug-induced lupus. I. cloned Th2 cells modified with DNA methylation inhibitors in vitro cause autoimmunity in vivo. J Immunol. 1995;154:3025-3035.
- Yung R, Powers D, Johnson K, et al. Mechanisms of drug-induced lupus. II. T cells overexpressing lymphocyte function-associated antigen 1 become autoreactive and cause a lupuslike disease in syngeneic mice. J Clin Invest. 1996;97:2866-2871.
- Sontheimer RD, Stastny P, Gilliam JN. Human histocompatibility antigen associations in subacute cutaneous lupus erythematosus. J Clin Invest. 1981;67:312-316.
- Sontheimer RD, Maddison PJ, Reichlin M, et al. Serologic and HLA associations in subacute cutaneous lupus erythematosus, a clinical subset of lupus erythematosus. Ann Intern Med. 1982;97:664-671.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
- Deutscher SL, Harley JB, Keene JD. Molecular analysis of the 60-kDa human Ro ribonucleoprotein. Proc Natl Acad Sci. 1988;85:9479-9483.
- DalleVedove C, Simon JC, Girolomoni G. Drug-induced lupus erythematosus with emphasis on skin manifestations and the role of anti-TNFα agents [article in German]. J Dtsch Dermatol Ges. 2012;10:889-897.
- Okon LG, Werth VP. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol. 2013;27:391-404.
- Schulte-Pelkum J, Fritzler M, Mahler M. Latest update on the Ro/SS-A autoantibody system. Autoimmun Rev. 2009;8:632-637.
- De Bandt M, Sibilia J, Le Loët X, et al. Systemic lupus erythematosus induced by anti-tumour necrosis factor alpha therapy: a French national survey. Arthritis Res Ther. 2005;7:R545-R551.
- Lupu A, Tieranu C, Constantinescu CL, et al. TNFα inhibitor induced lupus-like syndrome (TAILS) in a patient with IBD. Current Health Sci J. 2014;40:285-288.
- Marzano AV, Vezzoli P, Crosti C. Drug-induced lupus: an update on its dermatological aspects. Lupus. 2009;18:935-940.
- Wiznia LE, Subtil A, Choi JN. Subacute cutaneous lupus erythematosus induced by chemotherapy: gemcitabine as a causative agent. JAMA Dermatol. 2013;149:1071-1075.
- Williams VL, Cohen PR. TNF alpha antagonist-induced lupus-like syndrome: report and review of the literature with implications for treatment with alternative TNF alpha antagonists. Int J Dermatol. 2011;50:619-625.
- Pasut G. Pegylation of biological molecules and potential benefits: pharmacological properties of certolizumab pegol. Bio Drugs. 2014;28(suppl 1):15-23.
- Mudduluru BM, Shah S, Shamah S. et al. TNF-alpha antagonist induced lupus on three different agents. Postgrad Med. 2017;129:304-306.
- De Bandt M. Anti-TNF-alpha-induced lupus. Arthritis Res Ther. 2019;21:235.
- Costa MF, Said NR, Zimmermann B. Drug-induced lupus due to anti-tumor necrosis factor alfa agents. Semin Arthritis Rheum. 2008;37:381-387.
- Caramaschi P, Biasi D, Colombatti M. Anti-TNF alpha therapy in rheumatoid arthritis and autoimmunity. Rheumatol Int. 2006;26:209-214.
- Yung RL, Quddus J, Chrisp CE, et al. Mechanism of drug-induced lupus. I. cloned Th2 cells modified with DNA methylation inhibitors in vitro cause autoimmunity in vivo. J Immunol. 1995;154:3025-3035.
- Yung R, Powers D, Johnson K, et al. Mechanisms of drug-induced lupus. II. T cells overexpressing lymphocyte function-associated antigen 1 become autoreactive and cause a lupuslike disease in syngeneic mice. J Clin Invest. 1996;97:2866-2871.
- Sontheimer RD, Stastny P, Gilliam JN. Human histocompatibility antigen associations in subacute cutaneous lupus erythematosus. J Clin Invest. 1981;67:312-316.
- Sontheimer RD, Maddison PJ, Reichlin M, et al. Serologic and HLA associations in subacute cutaneous lupus erythematosus, a clinical subset of lupus erythematosus. Ann Intern Med. 1982;97:664-671.
- Lee LA, Roberts CM, Frank MB, et al. The autoantibody response to Ro/SSA in cutaneous lupus erythematosus. Arch Dermatol. 1994;130:1262-1268.
- Deutscher SL, Harley JB, Keene JD. Molecular analysis of the 60-kDa human Ro ribonucleoprotein. Proc Natl Acad Sci. 1988;85:9479-9483.
- DalleVedove C, Simon JC, Girolomoni G. Drug-induced lupus erythematosus with emphasis on skin manifestations and the role of anti-TNFα agents [article in German]. J Dtsch Dermatol Ges. 2012;10:889-897.
- Okon LG, Werth VP. Cutaneous lupus erythematosus: diagnosis and treatment. Best Pract Res Clin Rheumatol. 2013;27:391-404.
- Schulte-Pelkum J, Fritzler M, Mahler M. Latest update on the Ro/SS-A autoantibody system. Autoimmun Rev. 2009;8:632-637.
- De Bandt M, Sibilia J, Le Loët X, et al. Systemic lupus erythematosus induced by anti-tumour necrosis factor alpha therapy: a French national survey. Arthritis Res Ther. 2005;7:R545-R551.
- Lupu A, Tieranu C, Constantinescu CL, et al. TNFα inhibitor induced lupus-like syndrome (TAILS) in a patient with IBD. Current Health Sci J. 2014;40:285-288.
Practice Points
- Tumor necrosis factor α (TNF-α) inhibitor–induced lupuslike syndrome (TAILS) is a form of drug-induced lupus specific to patients on anti–TNF-α therapy.
- The underlying mechanism of disease development is unique compared to other types of drug-induced lupus.
- TAILS most commonly is associated with the use of infliximab and etanercept but also has been reported with adalimumab, golimumab, and certolizumab pegol.

Nodules on the Anterior Neck Following Poly-L-lactic Acid Injection
Poly-L-lactic acid (PLLA) is a synthetic biologic polymer that is suspended in solution and can be injected for soft-tissue augmentation. The stimulatory molecule functions to increase collagen synthesis as a by-product of its degradation.1 Poly-L-lactic acid measures 40 to 63 μm and is irregularly shaped, which inhibits product mobility and allows for precise tissue augmentation.2 Clinical trials of injectable PLLA have proven its safety with no reported cases of infection, allergies, or serious adverse reactions.3-5 The most common patient concerns generally are transient in nature, such as swelling, tenderness, pain, bruising, and bleeding. Persistent adverse events of PLLA primarily are papule and nodule formation.6 Clinical trials showed a variable incidence of papule/nodule formation between 6% and 44%.2 Nodule formation remains a major challenge to achieving optimal results from injectable PLLA. We present a case in which a hyperdiluted formulation of PLLA produced a relatively acute (3-week) onset of multiple nodule formations dispersed on the anterior neck. The nodules were resistant to less-invasive treatment modalities and were further requested to be surgically excised.
Case Report
A 38-year-old woman presented for soft-tissue augmentation of the anterior neck using PLLA to achieve correction of skin laxity and static rhytides. She had a history of successful PLLA injections in the temples, knees, chest, and buttocks over a 5-year period. Forty-eight hours prior to injection, 1 PLLA vial was hydrated with 7 cc bacteriostatic water by using a continuous rotation suspension method over the 48 hours. On the day of injection, the PLLA was further hyperdiluted with 2 cc of 2% lidocaine and an additional 7 cc of bacteriostatic water, for a total of 16 cc diluent. The product was injected using a cannula in the anterior and lateral neck. According to the patient, 3 weeks after the procedure she noticed that some nodules began to form at the cannula insertion sites, while others formed distant from those sites; a total of 10 nodules had formed on the anterior neck (Figure 1).

The bacteriostatic water, lidocaine, and PLLA vial were all confirmed not to be expired. The manufacturer was contacted, and no other adverse reactions have been reported with this particular lot number of PLLA. The nodules initially were treated with injections of large boluses of bacteriostatic saline, which was ineffective. Treatment was then attempted using injections of a solution containing 1.0 mL of 5-fluorouracil (5-FU) 50 mg/mL, 0.4 mL of dexamethasone 4 mg/mL, 0.1 mL of triamcinolone 10 mg/mL, and 0.3 mL hyaluronidase. A series of 4 injections was performed in 2- to 4-week intervals. Two of the nodules resolved completely with this treatment. The remaining 8 nodules subjectively improved in size and softened to palpation but did not resolve completely. At 2 of the injection sites, treatment was complicated with steroid atrophy of the overlying skin. At the patient’s request, the remaining nodules were surgically excised (Figure 2). Histopathology revealed exogenous foreign material consistent with dermal filler (Figure 3).

Comment
Causes of Nodule Formation—Two factors that could contribute to nodule formation are inadequate dispersion of molecules and an insufficient volume of dilution. One study demonstrated that hydration for at least 24 hours is required for adequate PLLA dispersion. Furthermore, sonification for 5 minutes after a 2-hour hydration disperses molecules similarly to the 48-hour hydration.7 The PLLA in the current case was hydrated for 48 hours using a continuous rotation suspension method. Therefore, this likely did not play a role in our patient’s nodule formation. The volume of dilution has been shown to impact the incidence of nodule formation.8 At present, most injectors (60.4%) reconstitute each vial of PLLA with 9 to 10 mL of diluent.9 The PLLA in our patient was reconstituted with 16 mL; therefore, we believe that the anatomic location was the main contributor of nodule formation.
.")
Fillers should be injected in the subcutaneous or deep dermal plane of tissue.10 The platysma is a superficial muscle that is intimately involved with the overlying skin of the anterior neck, and injections in this area could inadvertently be intramuscular. Intramuscular injections have a higher incidence of nodule formation.1 Our patient had prior PLLA injections without adverse reactions in numerous other sites, supporting the claim that the anterior neck is prone to nodule formation from PLLA injections.
Management of Noninflammatory Nodules—Initial treatment of nodules with injections of saline was ineffective. This treatment can be used in an attempt to disperse the product. Treatment was then attempted with injections of a solution containing 5-FU, dexamethasone, triamcinolone, and hyaluronidase. Combination steroid therapy may be superior to monotherapy.11 Dexamethasone may exhibit a cytoprotective effect on cells such as fibroblasts when used in combination with triamcinolone; monotherapy steroid use with triamcinolone alone induced fibroblast apoptosis at a much higher level.12 Hyaluronidase works by breaking cross-links in hyaluronic acid, a glycosaminoglycan polysaccharide prevalent in the skin and connective tissue, which increases tissue permeability and aids in delivery of the other injected fluids.13 5-Fluorouracil is an antimetabolite that may aid in treating nodules by discouraging additional fibroblast activity and fibrosis.14
The combination of 5-FU, dexamethasone, and triamcinolone has been shown to be successful in treating noninflammatory nodules in as few as 1 treatment.14 In our patient, hyaluronidase also was used in an attempt to aid delivery of the other injected fluids. If nodules do not resolve with 1 injection, it is recommended to wait at least 8 weeks before repeating the injection to prevent steroid atrophy of the overlying skin. In our patient, the intramuscular placement of the filler contributed to the nodules being resistant to this treatment. During excision, the nodules were tightly embedded in the underlying tissue, which may have prevented the solution from being delivered to the nodule (Figure 2).
Conclusion
Injectable PLLA is approved by the US Food and Drug Administration for soft-tissue augmentation of deep nasolabial folds and facial wrinkles. Off-label use of this product may cause higher incidence of nodule formation. Injectors should be cautious of injecting into the anterior neck. If nodules do form, treatment can be attempted with injections of saline. If that treatment fails, another treatment option is injection(s) of a mixture of 5-FU, dexamethasone, triamcinolone, and hyaluronidase separated by 8-week intervals. Finally, surgical excision is a viable treatment option, as presented in our case.
- Bartus C, William HC, Daro-Kaftan E. A decade of experience with injectable poly-L-lactic acid: a focus on safety. Dermatol Surg. 2013;39:698-705.
- Engelhard P, Humble G, Mest D. Safety of Sculptra: a review of clinical trial data. J Cosmet Laser Ther. 2005;7:201-205.
- Mest DR, Humble G. Safety and efficacy of poly-L-lactic acid injections in persons with HIV-associated lipoatrophy: the US experience. Dermatol Surg. 2006;32:1336-1345.
- Burgess CM, Quiroga RM. Assessment of the safety and efficacy of poly-L-lactic acid for the treatment of HIV associated facial lipoatrophy. J Am Acad Dermatol. 2005;52:233-239.
- Cattelan AM, Bauer U, Trevenzoli M, et al. Use of polylactic acid implants to correct facial lipoatrophy in human immunodeficiency virus 1-positive individuals receiving combination antiretroviral therapy. Arch Dermatol. 2006;142:329-334.
- Sculptra. Package insert. sanofi-aventis U.S. LLC; 2009.
- Li CN, Wang CC, Huang CC, et al. A novel, optimized method to accelerate the preparation of injectable poly-L-lactic acid by sonication. J Drugs Dermatol. 2018;17:894-898.
- Rossner F, Rossner M, Hartmann V, et al. Decrease of reported adverse events to injectable polylactic acid after recommending an increased dilution: 8-year results from the Injectable Filler Safety study. J Cosmet Dermatol. 2009;8:14-18.
- Lin MJ, Dubin DP, Goldberg DJ, et al. Practices in the usage and reconstitution of poly-L-lactic acid. J Drugs Dermatol. 2019;18:880-886.
- Sieber DA, Scheuer JF 3rd, Villanueva NL, et al. Review of 3-dimensional facial anatomy: injecting fillers and neuromodulators. Plast Reconstr Surg Glob Open. 2016;4(12 suppl Anatomy and Safety in Cosmetic Medicine: Cosmetic Bootcamp):E1166.
- Syed F, Singh S, Bayat A. Superior effect of combination vs. single steroid therapy in keloid disease: a comparative in vitro analysis of glucocorticoids. Wound Repair Regen. 2013;21:88-102.
- Brody HJ. Use of hyaluronidase in the treatment of granulomatous hyaluronic acid reactions or unwanted hyaluronic acid misplacement. Dermatol Surg. 2005;31:893-897.
- Funt D, Pavicic T. Dermal fillers in aesthetics: an overview of adverse events and treatment approaches. Clin Cosm Investig Dermatol. 2013;6:295-316.
- Aguilera SB, Aristizabal M, Reed A. Successful treatment of calcium hydroxylapatite nodules with intralesional 5-fluorouracil, dexamethasone, and triamcinolone. J Drugs Dermatol. 2016;15:1142-1143.
Poly-L-lactic acid (PLLA) is a synthetic biologic polymer that is suspended in solution and can be injected for soft-tissue augmentation. The stimulatory molecule functions to increase collagen synthesis as a by-product of its degradation.1 Poly-L-lactic acid measures 40 to 63 μm and is irregularly shaped, which inhibits product mobility and allows for precise tissue augmentation.2 Clinical trials of injectable PLLA have proven its safety with no reported cases of infection, allergies, or serious adverse reactions.3-5 The most common patient concerns generally are transient in nature, such as swelling, tenderness, pain, bruising, and bleeding. Persistent adverse events of PLLA primarily are papule and nodule formation.6 Clinical trials showed a variable incidence of papule/nodule formation between 6% and 44%.2 Nodule formation remains a major challenge to achieving optimal results from injectable PLLA. We present a case in which a hyperdiluted formulation of PLLA produced a relatively acute (3-week) onset of multiple nodule formations dispersed on the anterior neck. The nodules were resistant to less-invasive treatment modalities and were further requested to be surgically excised.
Case Report
A 38-year-old woman presented for soft-tissue augmentation of the anterior neck using PLLA to achieve correction of skin laxity and static rhytides. She had a history of successful PLLA injections in the temples, knees, chest, and buttocks over a 5-year period. Forty-eight hours prior to injection, 1 PLLA vial was hydrated with 7 cc bacteriostatic water by using a continuous rotation suspension method over the 48 hours. On the day of injection, the PLLA was further hyperdiluted with 2 cc of 2% lidocaine and an additional 7 cc of bacteriostatic water, for a total of 16 cc diluent. The product was injected using a cannula in the anterior and lateral neck. According to the patient, 3 weeks after the procedure she noticed that some nodules began to form at the cannula insertion sites, while others formed distant from those sites; a total of 10 nodules had formed on the anterior neck (Figure 1).
The bacteriostatic water, lidocaine, and PLLA vial were all confirmed not to be expired. The manufacturer was contacted, and no other adverse reactions have been reported with this particular lot number of PLLA. The nodules initially were treated with injections of large boluses of bacteriostatic saline, which was ineffective. Treatment was then attempted using injections of a solution containing 1.0 mL of 5-fluorouracil (5-FU) 50 mg/mL, 0.4 mL of dexamethasone 4 mg/mL, 0.1 mL of triamcinolone 10 mg/mL, and 0.3 mL hyaluronidase. A series of 4 injections was performed in 2- to 4-week intervals. Two of the nodules resolved completely with this treatment. The remaining 8 nodules subjectively improved in size and softened to palpation but did not resolve completely. At 2 of the injection sites, treatment was complicated with steroid atrophy of the overlying skin. At the patient’s request, the remaining nodules were surgically excised (Figure 2). Histopathology revealed exogenous foreign material consistent with dermal filler (Figure 3).
Comment
Causes of Nodule Formation—Two factors that could contribute to nodule formation are inadequate dispersion of molecules and an insufficient volume of dilution. One study demonstrated that hydration for at least 24 hours is required for adequate PLLA dispersion. Furthermore, sonification for 5 minutes after a 2-hour hydration disperses molecules similarly to the 48-hour hydration.7 The PLLA in the current case was hydrated for 48 hours using a continuous rotation suspension method. Therefore, this likely did not play a role in our patient’s nodule formation. The volume of dilution has been shown to impact the incidence of nodule formation.8 At present, most injectors (60.4%) reconstitute each vial of PLLA with 9 to 10 mL of diluent.9 The PLLA in our patient was reconstituted with 16 mL; therefore, we believe that the anatomic location was the main contributor of nodule formation.
Fillers should be injected in the subcutaneous or deep dermal plane of tissue.10 The platysma is a superficial muscle that is intimately involved with the overlying skin of the anterior neck, and injections in this area could inadvertently be intramuscular. Intramuscular injections have a higher incidence of nodule formation.1 Our patient had prior PLLA injections without adverse reactions in numerous other sites, supporting the claim that the anterior neck is prone to nodule formation from PLLA injections.
Management of Noninflammatory Nodules—Initial treatment of nodules with injections of saline was ineffective. This treatment can be used in an attempt to disperse the product. Treatment was then attempted with injections of a solution containing 5-FU, dexamethasone, triamcinolone, and hyaluronidase. Combination steroid therapy may be superior to monotherapy.11 Dexamethasone may exhibit a cytoprotective effect on cells such as fibroblasts when used in combination with triamcinolone; monotherapy steroid use with triamcinolone alone induced fibroblast apoptosis at a much higher level.12 Hyaluronidase works by breaking cross-links in hyaluronic acid, a glycosaminoglycan polysaccharide prevalent in the skin and connective tissue, which increases tissue permeability and aids in delivery of the other injected fluids.13 5-Fluorouracil is an antimetabolite that may aid in treating nodules by discouraging additional fibroblast activity and fibrosis.14
The combination of 5-FU, dexamethasone, and triamcinolone has been shown to be successful in treating noninflammatory nodules in as few as 1 treatment.14 In our patient, hyaluronidase also was used in an attempt to aid delivery of the other injected fluids. If nodules do not resolve with 1 injection, it is recommended to wait at least 8 weeks before repeating the injection to prevent steroid atrophy of the overlying skin. In our patient, the intramuscular placement of the filler contributed to the nodules being resistant to this treatment. During excision, the nodules were tightly embedded in the underlying tissue, which may have prevented the solution from being delivered to the nodule (Figure 2).
Conclusion
Injectable PLLA is approved by the US Food and Drug Administration for soft-tissue augmentation of deep nasolabial folds and facial wrinkles. Off-label use of this product may cause higher incidence of nodule formation. Injectors should be cautious of injecting into the anterior neck. If nodules do form, treatment can be attempted with injections of saline. If that treatment fails, another treatment option is injection(s) of a mixture of 5-FU, dexamethasone, triamcinolone, and hyaluronidase separated by 8-week intervals. Finally, surgical excision is a viable treatment option, as presented in our case.
Poly-L-lactic acid (PLLA) is a synthetic biologic polymer that is suspended in solution and can be injected for soft-tissue augmentation. The stimulatory molecule functions to increase collagen synthesis as a by-product of its degradation.1 Poly-L-lactic acid measures 40 to 63 μm and is irregularly shaped, which inhibits product mobility and allows for precise tissue augmentation.2 Clinical trials of injectable PLLA have proven its safety with no reported cases of infection, allergies, or serious adverse reactions.3-5 The most common patient concerns generally are transient in nature, such as swelling, tenderness, pain, bruising, and bleeding. Persistent adverse events of PLLA primarily are papule and nodule formation.6 Clinical trials showed a variable incidence of papule/nodule formation between 6% and 44%.2 Nodule formation remains a major challenge to achieving optimal results from injectable PLLA. We present a case in which a hyperdiluted formulation of PLLA produced a relatively acute (3-week) onset of multiple nodule formations dispersed on the anterior neck. The nodules were resistant to less-invasive treatment modalities and were further requested to be surgically excised.
Case Report
A 38-year-old woman presented for soft-tissue augmentation of the anterior neck using PLLA to achieve correction of skin laxity and static rhytides. She had a history of successful PLLA injections in the temples, knees, chest, and buttocks over a 5-year period. Forty-eight hours prior to injection, 1 PLLA vial was hydrated with 7 cc bacteriostatic water by using a continuous rotation suspension method over the 48 hours. On the day of injection, the PLLA was further hyperdiluted with 2 cc of 2% lidocaine and an additional 7 cc of bacteriostatic water, for a total of 16 cc diluent. The product was injected using a cannula in the anterior and lateral neck. According to the patient, 3 weeks after the procedure she noticed that some nodules began to form at the cannula insertion sites, while others formed distant from those sites; a total of 10 nodules had formed on the anterior neck (Figure 1).
The bacteriostatic water, lidocaine, and PLLA vial were all confirmed not to be expired. The manufacturer was contacted, and no other adverse reactions have been reported with this particular lot number of PLLA. The nodules initially were treated with injections of large boluses of bacteriostatic saline, which was ineffective. Treatment was then attempted using injections of a solution containing 1.0 mL of 5-fluorouracil (5-FU) 50 mg/mL, 0.4 mL of dexamethasone 4 mg/mL, 0.1 mL of triamcinolone 10 mg/mL, and 0.3 mL hyaluronidase. A series of 4 injections was performed in 2- to 4-week intervals. Two of the nodules resolved completely with this treatment. The remaining 8 nodules subjectively improved in size and softened to palpation but did not resolve completely. At 2 of the injection sites, treatment was complicated with steroid atrophy of the overlying skin. At the patient’s request, the remaining nodules were surgically excised (Figure 2). Histopathology revealed exogenous foreign material consistent with dermal filler (Figure 3).
Comment
Causes of Nodule Formation—Two factors that could contribute to nodule formation are inadequate dispersion of molecules and an insufficient volume of dilution. One study demonstrated that hydration for at least 24 hours is required for adequate PLLA dispersion. Furthermore, sonification for 5 minutes after a 2-hour hydration disperses molecules similarly to the 48-hour hydration.7 The PLLA in the current case was hydrated for 48 hours using a continuous rotation suspension method. Therefore, this likely did not play a role in our patient’s nodule formation. The volume of dilution has been shown to impact the incidence of nodule formation.8 At present, most injectors (60.4%) reconstitute each vial of PLLA with 9 to 10 mL of diluent.9 The PLLA in our patient was reconstituted with 16 mL; therefore, we believe that the anatomic location was the main contributor of nodule formation.
Fillers should be injected in the subcutaneous or deep dermal plane of tissue.10 The platysma is a superficial muscle that is intimately involved with the overlying skin of the anterior neck, and injections in this area could inadvertently be intramuscular. Intramuscular injections have a higher incidence of nodule formation.1 Our patient had prior PLLA injections without adverse reactions in numerous other sites, supporting the claim that the anterior neck is prone to nodule formation from PLLA injections.
Management of Noninflammatory Nodules—Initial treatment of nodules with injections of saline was ineffective. This treatment can be used in an attempt to disperse the product. Treatment was then attempted with injections of a solution containing 5-FU, dexamethasone, triamcinolone, and hyaluronidase. Combination steroid therapy may be superior to monotherapy.11 Dexamethasone may exhibit a cytoprotective effect on cells such as fibroblasts when used in combination with triamcinolone; monotherapy steroid use with triamcinolone alone induced fibroblast apoptosis at a much higher level.12 Hyaluronidase works by breaking cross-links in hyaluronic acid, a glycosaminoglycan polysaccharide prevalent in the skin and connective tissue, which increases tissue permeability and aids in delivery of the other injected fluids.13 5-Fluorouracil is an antimetabolite that may aid in treating nodules by discouraging additional fibroblast activity and fibrosis.14
The combination of 5-FU, dexamethasone, and triamcinolone has been shown to be successful in treating noninflammatory nodules in as few as 1 treatment.14 In our patient, hyaluronidase also was used in an attempt to aid delivery of the other injected fluids. If nodules do not resolve with 1 injection, it is recommended to wait at least 8 weeks before repeating the injection to prevent steroid atrophy of the overlying skin. In our patient, the intramuscular placement of the filler contributed to the nodules being resistant to this treatment. During excision, the nodules were tightly embedded in the underlying tissue, which may have prevented the solution from being delivered to the nodule (Figure 2).
Conclusion
Injectable PLLA is approved by the US Food and Drug Administration for soft-tissue augmentation of deep nasolabial folds and facial wrinkles. Off-label use of this product may cause higher incidence of nodule formation. Injectors should be cautious of injecting into the anterior neck. If nodules do form, treatment can be attempted with injections of saline. If that treatment fails, another treatment option is injection(s) of a mixture of 5-FU, dexamethasone, triamcinolone, and hyaluronidase separated by 8-week intervals. Finally, surgical excision is a viable treatment option, as presented in our case.
- Bartus C, William HC, Daro-Kaftan E. A decade of experience with injectable poly-L-lactic acid: a focus on safety. Dermatol Surg. 2013;39:698-705.
- Engelhard P, Humble G, Mest D. Safety of Sculptra: a review of clinical trial data. J Cosmet Laser Ther. 2005;7:201-205.
- Mest DR, Humble G. Safety and efficacy of poly-L-lactic acid injections in persons with HIV-associated lipoatrophy: the US experience. Dermatol Surg. 2006;32:1336-1345.
- Burgess CM, Quiroga RM. Assessment of the safety and efficacy of poly-L-lactic acid for the treatment of HIV associated facial lipoatrophy. J Am Acad Dermatol. 2005;52:233-239.
- Cattelan AM, Bauer U, Trevenzoli M, et al. Use of polylactic acid implants to correct facial lipoatrophy in human immunodeficiency virus 1-positive individuals receiving combination antiretroviral therapy. Arch Dermatol. 2006;142:329-334.
- Sculptra. Package insert. sanofi-aventis U.S. LLC; 2009.
- Li CN, Wang CC, Huang CC, et al. A novel, optimized method to accelerate the preparation of injectable poly-L-lactic acid by sonication. J Drugs Dermatol. 2018;17:894-898.
- Rossner F, Rossner M, Hartmann V, et al. Decrease of reported adverse events to injectable polylactic acid after recommending an increased dilution: 8-year results from the Injectable Filler Safety study. J Cosmet Dermatol. 2009;8:14-18.
- Lin MJ, Dubin DP, Goldberg DJ, et al. Practices in the usage and reconstitution of poly-L-lactic acid. J Drugs Dermatol. 2019;18:880-886.
- Sieber DA, Scheuer JF 3rd, Villanueva NL, et al. Review of 3-dimensional facial anatomy: injecting fillers and neuromodulators. Plast Reconstr Surg Glob Open. 2016;4(12 suppl Anatomy and Safety in Cosmetic Medicine: Cosmetic Bootcamp):E1166.
- Syed F, Singh S, Bayat A. Superior effect of combination vs. single steroid therapy in keloid disease: a comparative in vitro analysis of glucocorticoids. Wound Repair Regen. 2013;21:88-102.
- Brody HJ. Use of hyaluronidase in the treatment of granulomatous hyaluronic acid reactions or unwanted hyaluronic acid misplacement. Dermatol Surg. 2005;31:893-897.
- Funt D, Pavicic T. Dermal fillers in aesthetics: an overview of adverse events and treatment approaches. Clin Cosm Investig Dermatol. 2013;6:295-316.
- Aguilera SB, Aristizabal M, Reed A. Successful treatment of calcium hydroxylapatite nodules with intralesional 5-fluorouracil, dexamethasone, and triamcinolone. J Drugs Dermatol. 2016;15:1142-1143.
- Bartus C, William HC, Daro-Kaftan E. A decade of experience with injectable poly-L-lactic acid: a focus on safety. Dermatol Surg. 2013;39:698-705.
- Engelhard P, Humble G, Mest D. Safety of Sculptra: a review of clinical trial data. J Cosmet Laser Ther. 2005;7:201-205.
- Mest DR, Humble G. Safety and efficacy of poly-L-lactic acid injections in persons with HIV-associated lipoatrophy: the US experience. Dermatol Surg. 2006;32:1336-1345.
- Burgess CM, Quiroga RM. Assessment of the safety and efficacy of poly-L-lactic acid for the treatment of HIV associated facial lipoatrophy. J Am Acad Dermatol. 2005;52:233-239.
- Cattelan AM, Bauer U, Trevenzoli M, et al. Use of polylactic acid implants to correct facial lipoatrophy in human immunodeficiency virus 1-positive individuals receiving combination antiretroviral therapy. Arch Dermatol. 2006;142:329-334.
- Sculptra. Package insert. sanofi-aventis U.S. LLC; 2009.
- Li CN, Wang CC, Huang CC, et al. A novel, optimized method to accelerate the preparation of injectable poly-L-lactic acid by sonication. J Drugs Dermatol. 2018;17:894-898.
- Rossner F, Rossner M, Hartmann V, et al. Decrease of reported adverse events to injectable polylactic acid after recommending an increased dilution: 8-year results from the Injectable Filler Safety study. J Cosmet Dermatol. 2009;8:14-18.
- Lin MJ, Dubin DP, Goldberg DJ, et al. Practices in the usage and reconstitution of poly-L-lactic acid. J Drugs Dermatol. 2019;18:880-886.
- Sieber DA, Scheuer JF 3rd, Villanueva NL, et al. Review of 3-dimensional facial anatomy: injecting fillers and neuromodulators. Plast Reconstr Surg Glob Open. 2016;4(12 suppl Anatomy and Safety in Cosmetic Medicine: Cosmetic Bootcamp):E1166.
- Syed F, Singh S, Bayat A. Superior effect of combination vs. single steroid therapy in keloid disease: a comparative in vitro analysis of glucocorticoids. Wound Repair Regen. 2013;21:88-102.
- Brody HJ. Use of hyaluronidase in the treatment of granulomatous hyaluronic acid reactions or unwanted hyaluronic acid misplacement. Dermatol Surg. 2005;31:893-897.
- Funt D, Pavicic T. Dermal fillers in aesthetics: an overview of adverse events and treatment approaches. Clin Cosm Investig Dermatol. 2013;6:295-316.
- Aguilera SB, Aristizabal M, Reed A. Successful treatment of calcium hydroxylapatite nodules with intralesional 5-fluorouracil, dexamethasone, and triamcinolone. J Drugs Dermatol. 2016;15:1142-1143.
Practice Points
- Injecting poly-L-lactic acid (PLLA) into the anterior neck is an off-label procedure and may cause a higher incidence of nodule formation.
- Most nodules from PLLA can be treated with injections of 5-fluorouracil, dexamethasone, triamcinolone, and hyaluronidase separated by 8-week intervals.
- Treatment-resistant nodules may require surgical excision.

Acute Alopecia Associated With Albendazole Toxicosis
To the Editor:
Albendazole is a commonly prescribed anthelmintic that typically is well tolerated. Its broadest application is in developing countries that have a high rate of endemic nematode infection.1,2 Albendazole belongs to the benzimidazole class of anthelmintic chemotherapeutic agents that function by inhibiting microtubule dynamics, resulting in cytotoxic antimitotic effects.3 Benzimidazoles (eg, albendazole, mebendazole) have a binding affinity for helminthic β-tubulin that is 25- to 400-times greater than their binding affinity for the mammalian counterpart.4 Consequently, benzimidazoles generally are afforded a very broad therapeutic index for helminthic infection.
A 53-year-old man presented to the emergency department (ED) after an episode of syncope and sudden hair loss. At presentation he had a fever (temperature, 103 °F [39.4 °C]), a heart rate of 120 bpm, and pancytopenia (white blood cell count, 0.4×103/μL [reference range, 4.0–10.0×103/μL]; hemoglobin, 7.0 g/dL [reference range, 11.2–15.7 g/dL]; platelet count, 100
The patient reported severe gastrointestinal (GI) distress and diarrhea for the last year as well as a 25-lb weight loss. He discussed his belief that his GI symptoms were due to a parasite he had acquired the year prior; however, he reported that an exhaustive outpatient GI workup had been negative. Two weeks before presentation to our ED, the patient presented to another ED with stomach upset and was given a dose of albendazole. Perceiving alleviation of his symptoms, he purchased 2 bottles of veterinary albendazole online and consumed 113,000 mg—approximately 300 times the standard dose of 400 mg.
A dermatologic examination in our ED demonstrated reticulated violaceous patches on the face and severe alopecia with preferential sparing of the occipital scalp (Figure 1). Photographs taken by the patient on his phone from a week prior to presentation showed no facial dyschromia or signs of hair loss. A punch biopsy of the chin demonstrated perivascular and perifollicular dermatitis with eosinophils, most consistent with a drug reaction.

The patient received broad-spectrum antibiotics and supportive care. Blood count parameters normalized, and his hair began to regrow within 2 weeks after albendazole discontinuation (Figure 2).

Our patient exhibited symptoms of tachycardia, pancytopenia, and acute massive hair loss with preferential sparing of the occipital and posterior hair line; this pattern of hair loss is classic in men with chemotherapy-induced anagen effluvium.5 Conventional chemotherapeutics include taxanes and Vinca alkaloids, both of which bind mammalian β-tubulin and commonly induce anagen effluvium.
Our patient’s toxicosis syndrome was strikingly similar to common adverse effects in patients treated with conventional chemotherapeutics, including aplastic anemia with severe neutropenia and anagen effluvium.6,7 This adverse effect profile suggests that albendazole exerts an effect on mammalian β-tubulin that is similar to conventional chemotherapy when albendazole is ingested in a massive quantity.
Other reports of albendazole-induced alopecia describe an idiosyncratic, dose-dependent telogen effluvium.8-10 Conventional chemotherapy uncommonly might induce telogen effluvium when given below a threshold necessary to induce anagen effluvium. In those cases, follicular matrix keratinocytes are disrupted without complete follicular fracture and attempt to repair the damaged elongating follicle before entering the telogen phase.7 This observed phenomenon and the inherent susceptibility of matrix keratinocytes to antimicrotubule agents might explain why a therapeutic dose of albendazole has been associated with telogen effluvium in certain individuals.
Our case of albendazole-related toxicosis of this magnitude is unique. Ghias et al11 reported a case of abendazole-induced anagen effluvium. Future reports might clarify whether this toxicosis syndrome is typical or atypical in massive albendazole overdose.
- Keiser J, Utzinger J. Efficacy of current drugs against soil-transmitted helminth infections: systematic review and meta-analysis. JAMA. 2008;299:1937-1948. doi:10.1001/jama.299.16.1937
- Bethony J, Brooker S, Albonico M, et al. Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. Lancet. 2006;367:1521-1532. doi:10.1016/S0140-6736(06)68653-4
- Lanusse CE, Prichard RK. Clinical pharmacokinetics and metabolism of benzimidazole anthelmintics in ruminants. Drug Metab Rev. 1993;25:235-279. doi:10.3109/03602539308993977
- Page SW. Antiparasitic drugs. In: Maddison JE, Church DB, Page SW, eds. Small Animal Clinical Pharmacology. 2nd ed. W.B. Saunders; 2008:198-260.
- Yun SJ, Kim S-J. Hair loss pattern due to chemotherapy-induced anagen effluvium: a cross-sectional observation. Dermatology. 2007;215:36-40. doi:10.1159/000102031
- de Weger VA, Beijnen JH, Schellens JHM. Cellular and clinical pharmacology of the taxanes docetaxel and paclitaxel—a review. Anticancer Drugs. 2014;25:488-494. doi:10.1097/CAD.0000000000000093
- Paus R, Haslam IS, Sharov AA, et al. Pathobiology of chemotherapy-induced hair loss. Lancet Oncol. 2013;14:E50-E59. doi:10.1016/S1470-2045(12)70553-3
- Imamkuliev KD, Alekseev VG, Dovgalev AS, et al. A case of alopecia in a patient with hydatid disease treated with Nemozole (albendazole)[in Russian]. Med Parazitol (Mosk). 2013:48-50.
- Tas A, Köklü S, Celik H. Loss of body hair as a side effect of albendazole. Wien Klin Wochenschr. 2012;124:220. doi:10.1007/s00508-011-0112-y
- Pilar García-Muret M, Sitjas D, Tuneu L, et al. Telogen effluvium associated with albendazole therapy. Int J Dermatol. 1990;29:669-670. doi:10.1111/j.1365-4362.1990.tb02597.x
- Ghias M, Amin B, Kutner A. Albendazole-induced anagen effluvium. JAAD Case Rep. 2020;6:54-56.
To the Editor:
Albendazole is a commonly prescribed anthelmintic that typically is well tolerated. Its broadest application is in developing countries that have a high rate of endemic nematode infection.1,2 Albendazole belongs to the benzimidazole class of anthelmintic chemotherapeutic agents that function by inhibiting microtubule dynamics, resulting in cytotoxic antimitotic effects.3 Benzimidazoles (eg, albendazole, mebendazole) have a binding affinity for helminthic β-tubulin that is 25- to 400-times greater than their binding affinity for the mammalian counterpart.4 Consequently, benzimidazoles generally are afforded a very broad therapeutic index for helminthic infection.
A 53-year-old man presented to the emergency department (ED) after an episode of syncope and sudden hair loss. At presentation he had a fever (temperature, 103 °F [39.4 °C]), a heart rate of 120 bpm, and pancytopenia (white blood cell count, 0.4×103/μL [reference range, 4.0–10.0×103/μL]; hemoglobin, 7.0 g/dL [reference range, 11.2–15.7 g/dL]; platelet count, 100
The patient reported severe gastrointestinal (GI) distress and diarrhea for the last year as well as a 25-lb weight loss. He discussed his belief that his GI symptoms were due to a parasite he had acquired the year prior; however, he reported that an exhaustive outpatient GI workup had been negative. Two weeks before presentation to our ED, the patient presented to another ED with stomach upset and was given a dose of albendazole. Perceiving alleviation of his symptoms, he purchased 2 bottles of veterinary albendazole online and consumed 113,000 mg—approximately 300 times the standard dose of 400 mg.
A dermatologic examination in our ED demonstrated reticulated violaceous patches on the face and severe alopecia with preferential sparing of the occipital scalp (Figure 1). Photographs taken by the patient on his phone from a week prior to presentation showed no facial dyschromia or signs of hair loss. A punch biopsy of the chin demonstrated perivascular and perifollicular dermatitis with eosinophils, most consistent with a drug reaction.
The patient received broad-spectrum antibiotics and supportive care. Blood count parameters normalized, and his hair began to regrow within 2 weeks after albendazole discontinuation (Figure 2).
Our patient exhibited symptoms of tachycardia, pancytopenia, and acute massive hair loss with preferential sparing of the occipital and posterior hair line; this pattern of hair loss is classic in men with chemotherapy-induced anagen effluvium.5 Conventional chemotherapeutics include taxanes and Vinca alkaloids, both of which bind mammalian β-tubulin and commonly induce anagen effluvium.
Our patient’s toxicosis syndrome was strikingly similar to common adverse effects in patients treated with conventional chemotherapeutics, including aplastic anemia with severe neutropenia and anagen effluvium.6,7 This adverse effect profile suggests that albendazole exerts an effect on mammalian β-tubulin that is similar to conventional chemotherapy when albendazole is ingested in a massive quantity.
Other reports of albendazole-induced alopecia describe an idiosyncratic, dose-dependent telogen effluvium.8-10 Conventional chemotherapy uncommonly might induce telogen effluvium when given below a threshold necessary to induce anagen effluvium. In those cases, follicular matrix keratinocytes are disrupted without complete follicular fracture and attempt to repair the damaged elongating follicle before entering the telogen phase.7 This observed phenomenon and the inherent susceptibility of matrix keratinocytes to antimicrotubule agents might explain why a therapeutic dose of albendazole has been associated with telogen effluvium in certain individuals.
Our case of albendazole-related toxicosis of this magnitude is unique. Ghias et al11 reported a case of abendazole-induced anagen effluvium. Future reports might clarify whether this toxicosis syndrome is typical or atypical in massive albendazole overdose.
To the Editor:
Albendazole is a commonly prescribed anthelmintic that typically is well tolerated. Its broadest application is in developing countries that have a high rate of endemic nematode infection.1,2 Albendazole belongs to the benzimidazole class of anthelmintic chemotherapeutic agents that function by inhibiting microtubule dynamics, resulting in cytotoxic antimitotic effects.3 Benzimidazoles (eg, albendazole, mebendazole) have a binding affinity for helminthic β-tubulin that is 25- to 400-times greater than their binding affinity for the mammalian counterpart.4 Consequently, benzimidazoles generally are afforded a very broad therapeutic index for helminthic infection.
A 53-year-old man presented to the emergency department (ED) after an episode of syncope and sudden hair loss. At presentation he had a fever (temperature, 103 °F [39.4 °C]), a heart rate of 120 bpm, and pancytopenia (white blood cell count, 0.4×103/μL [reference range, 4.0–10.0×103/μL]; hemoglobin, 7.0 g/dL [reference range, 11.2–15.7 g/dL]; platelet count, 100
The patient reported severe gastrointestinal (GI) distress and diarrhea for the last year as well as a 25-lb weight loss. He discussed his belief that his GI symptoms were due to a parasite he had acquired the year prior; however, he reported that an exhaustive outpatient GI workup had been negative. Two weeks before presentation to our ED, the patient presented to another ED with stomach upset and was given a dose of albendazole. Perceiving alleviation of his symptoms, he purchased 2 bottles of veterinary albendazole online and consumed 113,000 mg—approximately 300 times the standard dose of 400 mg.
A dermatologic examination in our ED demonstrated reticulated violaceous patches on the face and severe alopecia with preferential sparing of the occipital scalp (Figure 1). Photographs taken by the patient on his phone from a week prior to presentation showed no facial dyschromia or signs of hair loss. A punch biopsy of the chin demonstrated perivascular and perifollicular dermatitis with eosinophils, most consistent with a drug reaction.
The patient received broad-spectrum antibiotics and supportive care. Blood count parameters normalized, and his hair began to regrow within 2 weeks after albendazole discontinuation (Figure 2).
Our patient exhibited symptoms of tachycardia, pancytopenia, and acute massive hair loss with preferential sparing of the occipital and posterior hair line; this pattern of hair loss is classic in men with chemotherapy-induced anagen effluvium.5 Conventional chemotherapeutics include taxanes and Vinca alkaloids, both of which bind mammalian β-tubulin and commonly induce anagen effluvium.
Our patient’s toxicosis syndrome was strikingly similar to common adverse effects in patients treated with conventional chemotherapeutics, including aplastic anemia with severe neutropenia and anagen effluvium.6,7 This adverse effect profile suggests that albendazole exerts an effect on mammalian β-tubulin that is similar to conventional chemotherapy when albendazole is ingested in a massive quantity.
Other reports of albendazole-induced alopecia describe an idiosyncratic, dose-dependent telogen effluvium.8-10 Conventional chemotherapy uncommonly might induce telogen effluvium when given below a threshold necessary to induce anagen effluvium. In those cases, follicular matrix keratinocytes are disrupted without complete follicular fracture and attempt to repair the damaged elongating follicle before entering the telogen phase.7 This observed phenomenon and the inherent susceptibility of matrix keratinocytes to antimicrotubule agents might explain why a therapeutic dose of albendazole has been associated with telogen effluvium in certain individuals.
Our case of albendazole-related toxicosis of this magnitude is unique. Ghias et al11 reported a case of abendazole-induced anagen effluvium. Future reports might clarify whether this toxicosis syndrome is typical or atypical in massive albendazole overdose.
- Keiser J, Utzinger J. Efficacy of current drugs against soil-transmitted helminth infections: systematic review and meta-analysis. JAMA. 2008;299:1937-1948. doi:10.1001/jama.299.16.1937
- Bethony J, Brooker S, Albonico M, et al. Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. Lancet. 2006;367:1521-1532. doi:10.1016/S0140-6736(06)68653-4
- Lanusse CE, Prichard RK. Clinical pharmacokinetics and metabolism of benzimidazole anthelmintics in ruminants. Drug Metab Rev. 1993;25:235-279. doi:10.3109/03602539308993977
- Page SW. Antiparasitic drugs. In: Maddison JE, Church DB, Page SW, eds. Small Animal Clinical Pharmacology. 2nd ed. W.B. Saunders; 2008:198-260.
- Yun SJ, Kim S-J. Hair loss pattern due to chemotherapy-induced anagen effluvium: a cross-sectional observation. Dermatology. 2007;215:36-40. doi:10.1159/000102031
- de Weger VA, Beijnen JH, Schellens JHM. Cellular and clinical pharmacology of the taxanes docetaxel and paclitaxel—a review. Anticancer Drugs. 2014;25:488-494. doi:10.1097/CAD.0000000000000093
- Paus R, Haslam IS, Sharov AA, et al. Pathobiology of chemotherapy-induced hair loss. Lancet Oncol. 2013;14:E50-E59. doi:10.1016/S1470-2045(12)70553-3
- Imamkuliev KD, Alekseev VG, Dovgalev AS, et al. A case of alopecia in a patient with hydatid disease treated with Nemozole (albendazole)[in Russian]. Med Parazitol (Mosk). 2013:48-50.
- Tas A, Köklü S, Celik H. Loss of body hair as a side effect of albendazole. Wien Klin Wochenschr. 2012;124:220. doi:10.1007/s00508-011-0112-y
- Pilar García-Muret M, Sitjas D, Tuneu L, et al. Telogen effluvium associated with albendazole therapy. Int J Dermatol. 1990;29:669-670. doi:10.1111/j.1365-4362.1990.tb02597.x
- Ghias M, Amin B, Kutner A. Albendazole-induced anagen effluvium. JAAD Case Rep. 2020;6:54-56.
- Keiser J, Utzinger J. Efficacy of current drugs against soil-transmitted helminth infections: systematic review and meta-analysis. JAMA. 2008;299:1937-1948. doi:10.1001/jama.299.16.1937
- Bethony J, Brooker S, Albonico M, et al. Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. Lancet. 2006;367:1521-1532. doi:10.1016/S0140-6736(06)68653-4
- Lanusse CE, Prichard RK. Clinical pharmacokinetics and metabolism of benzimidazole anthelmintics in ruminants. Drug Metab Rev. 1993;25:235-279. doi:10.3109/03602539308993977
- Page SW. Antiparasitic drugs. In: Maddison JE, Church DB, Page SW, eds. Small Animal Clinical Pharmacology. 2nd ed. W.B. Saunders; 2008:198-260.
- Yun SJ, Kim S-J. Hair loss pattern due to chemotherapy-induced anagen effluvium: a cross-sectional observation. Dermatology. 2007;215:36-40. doi:10.1159/000102031
- de Weger VA, Beijnen JH, Schellens JHM. Cellular and clinical pharmacology of the taxanes docetaxel and paclitaxel—a review. Anticancer Drugs. 2014;25:488-494. doi:10.1097/CAD.0000000000000093
- Paus R, Haslam IS, Sharov AA, et al. Pathobiology of chemotherapy-induced hair loss. Lancet Oncol. 2013;14:E50-E59. doi:10.1016/S1470-2045(12)70553-3
- Imamkuliev KD, Alekseev VG, Dovgalev AS, et al. A case of alopecia in a patient with hydatid disease treated with Nemozole (albendazole)[in Russian]. Med Parazitol (Mosk). 2013:48-50.
- Tas A, Köklü S, Celik H. Loss of body hair as a side effect of albendazole. Wien Klin Wochenschr. 2012;124:220. doi:10.1007/s00508-011-0112-y
- Pilar García-Muret M, Sitjas D, Tuneu L, et al. Telogen effluvium associated with albendazole therapy. Int J Dermatol. 1990;29:669-670. doi:10.1111/j.1365-4362.1990.tb02597.x
- Ghias M, Amin B, Kutner A. Albendazole-induced anagen effluvium. JAAD Case Rep. 2020;6:54-56.
PRACTICE POINTS
- Albendazole functions by inhibiting microtubule dynamics and has a remarkably greater binding affinity for helminthic β-tubulin than for its mammalian counterpart.
- An uncommon adverse effect of albendazole at therapeutic dosing is a dose-dependent telogen effluvium in susceptible persons, likely caused by the inherent susceptibility of follicular matrix keratinocytes to antimicrotubule agents.
- Massive albendazole overdose can cause anagen effluvium and myelosuppression similar to the effects of conventional chemotherapy.

Sweet Syndrome With Pulmonary Involvement Preceding the Development of Myelodysplastic Syndrome
To the Editor:
A 59-year-old man was referred to our clinic for a rash, fever, and night sweats following treatment for metastatic seminoma with cisplatin and etoposide. Physical examination revealed indurated erythematous papules and plaques on the trunk and upper and lower extremities, some with annular or arcuate configuration with trailing scale (Figure, A). A skin biopsy demonstrated mild papillary dermal edema with a mixed infiltrate of mononuclear cells, neutrophils, eosinophils, mast cells, lymphocytes, and karyorrhectic debris without evidence of leukocytoclastic vasculitis. The histopathologic differential diagnosis included a histiocytoid variant of Sweet syndrome (SS), and our patient’s rapid clinical response to corticosteroids supported this diagnosis.

With a relapsing and remitting course over 3 years, the rash eventually evolved into more edematous papules and plaques (Figure, B), and a repeat biopsy 3 years later was consistent with classic SS. Although the patient's condition improved with prednisone, attempts to taper prednisone invariably resulted in relapse. Multiple steroid-sparing agents were trialed over the course of 3 years including dapsone and mycophenolate mofetil, both of which resulted in hypersensitivity drug eruptions. Colchicine and methotrexate were ineffective. Thalidomide strongly was considered but ultimately was avoided due to substantial existing neuropathy associated with his prior chemotherapy for metastatic seminoma.
Four years after the initial diagnosis of SS, our patient presented with dyspnea and weight loss. Computed tomography revealed a nearly confluent miliary pattern of nodularity in the lungs. A wedge biopsy demonstrated pneumonitis with intra-alveolar fibrin and neutrophils with a notable absence of granulomatous inflammation. Fungal and acid-fast bacilli staining as well as tissue cultures were negative. He had a history of Mycobacterium kansasii pulmonary infection treated 18 months prior; however, in this instance, the histopathology, negative microbial cultures, and rapid steroid responsiveness were consistent with pulmonary involvement of SS. Over the ensuing 2 years, the patient developed worsening of his chronic anemia. He was diagnosed with myelodysplastic syndrome (MDS) by bone marrow biopsy, despite having a normal bone marrow biopsy more than 3 years prior to evaluate his anemia. At this time, thalidomide was initiated at 50 mg daily leading to notable improvement in his SS symptoms; however, he developed worsening neuropathy resulting in the discontinuation of this treatment 2 months later. An investigational combination of vosaroxin and azacytidine was used to treat his MDS, resulting in normalization of blood counts and remission from SS.
Sweet syndrome may occur in the setting of undiagnosed cancer or may signal the return of a previously treated malignancy. The first description of SS associated with solid tumors was in a patient with testicular cancer,1 which prompted continuous surveillance for recurrent seminoma in our patient, though none was found. Hematologic malignancies, as well as MDS, often are associated with SS.2 In our patient, multiple atypical features linked the development of SS to the ultimate presentation of MDS. The initial finding of a histiocytoid variant has been described in a case series of 9 patients with chronic relapsing SS who eventually developed MDS with latency of up to 7 years. The histopathology in these cases evolved over time to that of classic neutrophilic SS.3 Pulmonary involvement of SS is another interesting aspect of our case. In one analysis, 18 of 34 (53%) cases with pulmonary involvement featured hematologic pathology, including myelodysplasia and acute leukemia.4
In our patient, SS preceded the clinical manifestation of MDS by 6 years. A similar phenomenon has been described in a patient with SS that preceded myelodysplasia by 30 months and was recalcitrant to numerous steroid-sparing therapies except thalidomide, despite the persistence of myelodysplasia. Tapering thalidomide, however, resulted in recurrence of SS lesions in that patient.5 In another case, resolution of myelodysplasia from azacytidine treatment was associated with remission from SS.6
Our case represents a confluence of atypical features that seem to define myelodysplasia-associated SS, including the initial presentation with a clinically atypical histiocytoid variant, chronic relapsing and remitting course, and extracutaneous involvement of the lungs. These findings should prompt surveillance for hematologic malignancy or myelodysplasia. Serial bone marrow biopsies were required to evaluate persistent anemia before the histopathologic findings of MDS became apparent in our patient. Thalidomide was an effective treatment for the cutaneous manifestations in our patient and should be considered as a steroid-sparing agent in the treatment of recalcitrant SS. Despite the discontinuation of thalidomide therapy, effective control of our patient’s myelodysplasia with chemotherapy has kept him in remission from SS for more than 7 years of follow-up, suggesting a causal relationship between these disorders.
- Shapiro L, Baraf CS, Richheimer LL. Sweet’s syndrome (acute febrile neutrophilic dermatosis): report of a case. Arch Dermatol. 1971;103:81-84.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Vignon-Pennamen MD, Juillard C, Rybojad M, et al. Chronic recurrent lymphocytic Sweet syndrome as a predictive marker of myelodysplasia. Arch Dermatol. 2006;142:1170-1176.
- Fernandez-Bussy S, Labarca G, Cabello F, et al. Sweet’s syndrome with pulmonary involvement: case report and literature review. Respir Med Case Rep. 2012;6:16-19.
- Browning CE, Dixon DE, Malone JC, et al. Thalidomide in the treatment of recalcitrant Sweet’s syndrome associated with myelodysplasia. J Am Acad Dermatol. 2005;53(2 suppl 1):S135-S138.
- Martinelli S, Rigolin GM, Leo G, et al. Complete remission Sweet’s syndrome after azacytidine treatment for concomitant myelodysplastic syndrome. Int J Hematol. 2014;99:663-667.
To the Editor:
A 59-year-old man was referred to our clinic for a rash, fever, and night sweats following treatment for metastatic seminoma with cisplatin and etoposide. Physical examination revealed indurated erythematous papules and plaques on the trunk and upper and lower extremities, some with annular or arcuate configuration with trailing scale (Figure, A). A skin biopsy demonstrated mild papillary dermal edema with a mixed infiltrate of mononuclear cells, neutrophils, eosinophils, mast cells, lymphocytes, and karyorrhectic debris without evidence of leukocytoclastic vasculitis. The histopathologic differential diagnosis included a histiocytoid variant of Sweet syndrome (SS), and our patient’s rapid clinical response to corticosteroids supported this diagnosis.
With a relapsing and remitting course over 3 years, the rash eventually evolved into more edematous papules and plaques (Figure, B), and a repeat biopsy 3 years later was consistent with classic SS. Although the patient's condition improved with prednisone, attempts to taper prednisone invariably resulted in relapse. Multiple steroid-sparing agents were trialed over the course of 3 years including dapsone and mycophenolate mofetil, both of which resulted in hypersensitivity drug eruptions. Colchicine and methotrexate were ineffective. Thalidomide strongly was considered but ultimately was avoided due to substantial existing neuropathy associated with his prior chemotherapy for metastatic seminoma.
Four years after the initial diagnosis of SS, our patient presented with dyspnea and weight loss. Computed tomography revealed a nearly confluent miliary pattern of nodularity in the lungs. A wedge biopsy demonstrated pneumonitis with intra-alveolar fibrin and neutrophils with a notable absence of granulomatous inflammation. Fungal and acid-fast bacilli staining as well as tissue cultures were negative. He had a history of Mycobacterium kansasii pulmonary infection treated 18 months prior; however, in this instance, the histopathology, negative microbial cultures, and rapid steroid responsiveness were consistent with pulmonary involvement of SS. Over the ensuing 2 years, the patient developed worsening of his chronic anemia. He was diagnosed with myelodysplastic syndrome (MDS) by bone marrow biopsy, despite having a normal bone marrow biopsy more than 3 years prior to evaluate his anemia. At this time, thalidomide was initiated at 50 mg daily leading to notable improvement in his SS symptoms; however, he developed worsening neuropathy resulting in the discontinuation of this treatment 2 months later. An investigational combination of vosaroxin and azacytidine was used to treat his MDS, resulting in normalization of blood counts and remission from SS.
Sweet syndrome may occur in the setting of undiagnosed cancer or may signal the return of a previously treated malignancy. The first description of SS associated with solid tumors was in a patient with testicular cancer,1 which prompted continuous surveillance for recurrent seminoma in our patient, though none was found. Hematologic malignancies, as well as MDS, often are associated with SS.2 In our patient, multiple atypical features linked the development of SS to the ultimate presentation of MDS. The initial finding of a histiocytoid variant has been described in a case series of 9 patients with chronic relapsing SS who eventually developed MDS with latency of up to 7 years. The histopathology in these cases evolved over time to that of classic neutrophilic SS.3 Pulmonary involvement of SS is another interesting aspect of our case. In one analysis, 18 of 34 (53%) cases with pulmonary involvement featured hematologic pathology, including myelodysplasia and acute leukemia.4
In our patient, SS preceded the clinical manifestation of MDS by 6 years. A similar phenomenon has been described in a patient with SS that preceded myelodysplasia by 30 months and was recalcitrant to numerous steroid-sparing therapies except thalidomide, despite the persistence of myelodysplasia. Tapering thalidomide, however, resulted in recurrence of SS lesions in that patient.5 In another case, resolution of myelodysplasia from azacytidine treatment was associated with remission from SS.6
Our case represents a confluence of atypical features that seem to define myelodysplasia-associated SS, including the initial presentation with a clinically atypical histiocytoid variant, chronic relapsing and remitting course, and extracutaneous involvement of the lungs. These findings should prompt surveillance for hematologic malignancy or myelodysplasia. Serial bone marrow biopsies were required to evaluate persistent anemia before the histopathologic findings of MDS became apparent in our patient. Thalidomide was an effective treatment for the cutaneous manifestations in our patient and should be considered as a steroid-sparing agent in the treatment of recalcitrant SS. Despite the discontinuation of thalidomide therapy, effective control of our patient’s myelodysplasia with chemotherapy has kept him in remission from SS for more than 7 years of follow-up, suggesting a causal relationship between these disorders.
To the Editor:
A 59-year-old man was referred to our clinic for a rash, fever, and night sweats following treatment for metastatic seminoma with cisplatin and etoposide. Physical examination revealed indurated erythematous papules and plaques on the trunk and upper and lower extremities, some with annular or arcuate configuration with trailing scale (Figure, A). A skin biopsy demonstrated mild papillary dermal edema with a mixed infiltrate of mononuclear cells, neutrophils, eosinophils, mast cells, lymphocytes, and karyorrhectic debris without evidence of leukocytoclastic vasculitis. The histopathologic differential diagnosis included a histiocytoid variant of Sweet syndrome (SS), and our patient’s rapid clinical response to corticosteroids supported this diagnosis.
With a relapsing and remitting course over 3 years, the rash eventually evolved into more edematous papules and plaques (Figure, B), and a repeat biopsy 3 years later was consistent with classic SS. Although the patient's condition improved with prednisone, attempts to taper prednisone invariably resulted in relapse. Multiple steroid-sparing agents were trialed over the course of 3 years including dapsone and mycophenolate mofetil, both of which resulted in hypersensitivity drug eruptions. Colchicine and methotrexate were ineffective. Thalidomide strongly was considered but ultimately was avoided due to substantial existing neuropathy associated with his prior chemotherapy for metastatic seminoma.
Four years after the initial diagnosis of SS, our patient presented with dyspnea and weight loss. Computed tomography revealed a nearly confluent miliary pattern of nodularity in the lungs. A wedge biopsy demonstrated pneumonitis with intra-alveolar fibrin and neutrophils with a notable absence of granulomatous inflammation. Fungal and acid-fast bacilli staining as well as tissue cultures were negative. He had a history of Mycobacterium kansasii pulmonary infection treated 18 months prior; however, in this instance, the histopathology, negative microbial cultures, and rapid steroid responsiveness were consistent with pulmonary involvement of SS. Over the ensuing 2 years, the patient developed worsening of his chronic anemia. He was diagnosed with myelodysplastic syndrome (MDS) by bone marrow biopsy, despite having a normal bone marrow biopsy more than 3 years prior to evaluate his anemia. At this time, thalidomide was initiated at 50 mg daily leading to notable improvement in his SS symptoms; however, he developed worsening neuropathy resulting in the discontinuation of this treatment 2 months later. An investigational combination of vosaroxin and azacytidine was used to treat his MDS, resulting in normalization of blood counts and remission from SS.
Sweet syndrome may occur in the setting of undiagnosed cancer or may signal the return of a previously treated malignancy. The first description of SS associated with solid tumors was in a patient with testicular cancer,1 which prompted continuous surveillance for recurrent seminoma in our patient, though none was found. Hematologic malignancies, as well as MDS, often are associated with SS.2 In our patient, multiple atypical features linked the development of SS to the ultimate presentation of MDS. The initial finding of a histiocytoid variant has been described in a case series of 9 patients with chronic relapsing SS who eventually developed MDS with latency of up to 7 years. The histopathology in these cases evolved over time to that of classic neutrophilic SS.3 Pulmonary involvement of SS is another interesting aspect of our case. In one analysis, 18 of 34 (53%) cases with pulmonary involvement featured hematologic pathology, including myelodysplasia and acute leukemia.4
In our patient, SS preceded the clinical manifestation of MDS by 6 years. A similar phenomenon has been described in a patient with SS that preceded myelodysplasia by 30 months and was recalcitrant to numerous steroid-sparing therapies except thalidomide, despite the persistence of myelodysplasia. Tapering thalidomide, however, resulted in recurrence of SS lesions in that patient.5 In another case, resolution of myelodysplasia from azacytidine treatment was associated with remission from SS.6
Our case represents a confluence of atypical features that seem to define myelodysplasia-associated SS, including the initial presentation with a clinically atypical histiocytoid variant, chronic relapsing and remitting course, and extracutaneous involvement of the lungs. These findings should prompt surveillance for hematologic malignancy or myelodysplasia. Serial bone marrow biopsies were required to evaluate persistent anemia before the histopathologic findings of MDS became apparent in our patient. Thalidomide was an effective treatment for the cutaneous manifestations in our patient and should be considered as a steroid-sparing agent in the treatment of recalcitrant SS. Despite the discontinuation of thalidomide therapy, effective control of our patient’s myelodysplasia with chemotherapy has kept him in remission from SS for more than 7 years of follow-up, suggesting a causal relationship between these disorders.
- Shapiro L, Baraf CS, Richheimer LL. Sweet’s syndrome (acute febrile neutrophilic dermatosis): report of a case. Arch Dermatol. 1971;103:81-84.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Vignon-Pennamen MD, Juillard C, Rybojad M, et al. Chronic recurrent lymphocytic Sweet syndrome as a predictive marker of myelodysplasia. Arch Dermatol. 2006;142:1170-1176.
- Fernandez-Bussy S, Labarca G, Cabello F, et al. Sweet’s syndrome with pulmonary involvement: case report and literature review. Respir Med Case Rep. 2012;6:16-19.
- Browning CE, Dixon DE, Malone JC, et al. Thalidomide in the treatment of recalcitrant Sweet’s syndrome associated with myelodysplasia. J Am Acad Dermatol. 2005;53(2 suppl 1):S135-S138.
- Martinelli S, Rigolin GM, Leo G, et al. Complete remission Sweet’s syndrome after azacytidine treatment for concomitant myelodysplastic syndrome. Int J Hematol. 2014;99:663-667.
- Shapiro L, Baraf CS, Richheimer LL. Sweet’s syndrome (acute febrile neutrophilic dermatosis): report of a case. Arch Dermatol. 1971;103:81-84.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Vignon-Pennamen MD, Juillard C, Rybojad M, et al. Chronic recurrent lymphocytic Sweet syndrome as a predictive marker of myelodysplasia. Arch Dermatol. 2006;142:1170-1176.
- Fernandez-Bussy S, Labarca G, Cabello F, et al. Sweet’s syndrome with pulmonary involvement: case report and literature review. Respir Med Case Rep. 2012;6:16-19.
- Browning CE, Dixon DE, Malone JC, et al. Thalidomide in the treatment of recalcitrant Sweet’s syndrome associated with myelodysplasia. J Am Acad Dermatol. 2005;53(2 suppl 1):S135-S138.
- Martinelli S, Rigolin GM, Leo G, et al. Complete remission Sweet’s syndrome after azacytidine treatment for concomitant myelodysplastic syndrome. Int J Hematol. 2014;99:663-667.
Practice Points
- Sweet syndrome is characterized by the clinical constellation of fever, a skin eruption of tender erythematous papules or plaques, and response to corticosteroids.
- Skin biopsy characteristically demonstrates marked papillary dermal edema with a dense infiltrate of mature neutrophils without leukocytoclasia.
- Sweet syndrome often is idiopathic, though it has been associated with infection, autoimmunity, medication, and malignancy.

Calcified Urachal Remnant in a Young Adult: An Unusual Case
To the Editor:
An otherwise healthy 26-year-old man presented to our outpatient clinic with a 15- to 20-mm, shiny, friable-appearing, red umbilical nodule with clear malodorous discharge (Figure 1). The lesion developed 2 weeks prior and gradually increased in size and discomfort. The patient reported mild associated abdominal pain. He had no fever, changes in urination or bowel movements, or prior history of umbilical growths or drainage. The abdomen was tender to palpation.

Differential diagnoses included pyogenic granuloma, umbilical hernia, epidermoid cyst or abscess, and malignancy (low suspicion). A biopsy was not performed due to concern for bleeding or communication with the bowel. A complete blood cell count, comprehensive metabolic panel, and urinalysis were unremarkable except for mild leukocytosis and elevated C-reactive protein. Ultrasonography revealed a 1.4×1.3-cm inflammatory umbilical mass with no communication with the bowel. The patient was referred to the emergency department (ED) for further evaluation. Computed tomography (CT) revealed periumbilical inflammation and an associated 1-cm calcification that appeared to be connected to a potential tract from the bladder, suggestive of a urachal remnant calcification (Figure 2). The patient was diagnosed with a persistent urachal remnant, discharged home with ciprofloxacin, and scheduled for a follow-up with urology.

The patient returned to the ED 3 days later with painful umbilical bleeding (Figure 3). While there, the patient extracted a 1-cm stone from the lesion, consistent with the calcification visualized on CT scan. Computed tomographic virtual cystoscopy showed no connection between the bladder and umbilicus. He was diagnosed with an umbilical-urachal sinus. Complete surgical excision was recommended and performed by urology without complication.

We report an unusual presentation of a symptomatic urachal remnant in an adult. During embryogenesis, the urachus connects the umbilicus to the developing bladder and normally involutes during development. Incomplete regression can cause rare pathological urachal anomalies. The clinical presentation is nonspecific and differs between children and adults, with most cases presenting during infancy or childhood.1 Pediatric urachal abnormalities often present with umbilical drainage, abdominal pain, a palpable mass, an abnormal appearance of the umbilicus, or urinary tract infections.2,3 In adults, the most common symptoms include hematuria, pain, or dysuria. Alternatively, they may be asympomatic3 or present with periumbilical dermatitis4 or abscess. Rodrigues and Gandhi5 reported another case of a symptomatic calculus formed within a urachal remnant. Calcifications in urachal remnants are rare and usually are reported as incidental radiologic findings.
Overall, visible umbilical masses occur infrequently. In addition to urachal anomalies, the differential diagnosis includes several benign and malignant pathologies. Benign causes include epidermoid cysts, foreign body granulomas, pyogenic granulomas, abscesses, hamartomas, nevi, hemangiomas, dermatofibromas, neurofibromas, lipomas, granular cell tumors, desmoid tumors, keloid scars, omphaliths, hernias, or omphalomesenteric duct remnants.6 Primary malignancies (eg, skin cancers, urachal adenocarcinoma, mesenchymal tumors) or metastasis (ie, Sister Mary Joseph nodule) also can present as umbilical nodules.
The wide range of clinical presentations of urachal anomalies combined with the rarity make diagnosis difficult. Thus, it is essential to have a high index of suspicion and awareness of how they can present. Ultrasonography and CT scan are useful tools in making the diagnosis. Urachal anomalies are prone to infection or can be associated with malignancy; therefore, timely and correct diagnosis is critical. Although surgical removal is the primary treatment for urachal anomalies, it may not be the primary treatment of the other entities included in the differential diagnosis of umbilical nodules. For example, the Sister Mary Joseph nodule can be associated with various primary malignancies, which should be treated accordingly.
- Berman SM, Tolia BM, Laor E, et al. Urachal remnants in adults. Urology. 1988;31:17-21.
- Gleason JM, Bowlin PR, Bagli DJ, et al. A comprehensive review of pediatric urachal anomalies and predictive analysis for adult urachal adenocarcinoma. J Urol. 2015;193:632-636.
- Naiditch JA, Radhakrishnan J, Chin AC. Current diagnosis and management of urachal remnants. J Pediatr Surg. 2013;48:2148-2152.
- Cox GA, Chan I, Lloyd J, et al. Urachal sinus presenting as periumbilical dermatitis. Br J Dermatol. 2007;157:419-420.
- Rodrigues JCL, Gandhi S. Don’t get caught out! a rare case of a calcified urachal remnant mimicking a bladder calculus. J Radiol Case Rep. 2013;7:34-38.
- Ramoutar A, El Sheikh S, Aslam A. A persistent umbilical nodule. Clin Exp Dermatol. 2017;42:814-816.
To the Editor:
An otherwise healthy 26-year-old man presented to our outpatient clinic with a 15- to 20-mm, shiny, friable-appearing, red umbilical nodule with clear malodorous discharge (Figure 1). The lesion developed 2 weeks prior and gradually increased in size and discomfort. The patient reported mild associated abdominal pain. He had no fever, changes in urination or bowel movements, or prior history of umbilical growths or drainage. The abdomen was tender to palpation.
Differential diagnoses included pyogenic granuloma, umbilical hernia, epidermoid cyst or abscess, and malignancy (low suspicion). A biopsy was not performed due to concern for bleeding or communication with the bowel. A complete blood cell count, comprehensive metabolic panel, and urinalysis were unremarkable except for mild leukocytosis and elevated C-reactive protein. Ultrasonography revealed a 1.4×1.3-cm inflammatory umbilical mass with no communication with the bowel. The patient was referred to the emergency department (ED) for further evaluation. Computed tomography (CT) revealed periumbilical inflammation and an associated 1-cm calcification that appeared to be connected to a potential tract from the bladder, suggestive of a urachal remnant calcification (Figure 2). The patient was diagnosed with a persistent urachal remnant, discharged home with ciprofloxacin, and scheduled for a follow-up with urology.
The patient returned to the ED 3 days later with painful umbilical bleeding (Figure 3). While there, the patient extracted a 1-cm stone from the lesion, consistent with the calcification visualized on CT scan. Computed tomographic virtual cystoscopy showed no connection between the bladder and umbilicus. He was diagnosed with an umbilical-urachal sinus. Complete surgical excision was recommended and performed by urology without complication.
We report an unusual presentation of a symptomatic urachal remnant in an adult. During embryogenesis, the urachus connects the umbilicus to the developing bladder and normally involutes during development. Incomplete regression can cause rare pathological urachal anomalies. The clinical presentation is nonspecific and differs between children and adults, with most cases presenting during infancy or childhood.1 Pediatric urachal abnormalities often present with umbilical drainage, abdominal pain, a palpable mass, an abnormal appearance of the umbilicus, or urinary tract infections.2,3 In adults, the most common symptoms include hematuria, pain, or dysuria. Alternatively, they may be asympomatic3 or present with periumbilical dermatitis4 or abscess. Rodrigues and Gandhi5 reported another case of a symptomatic calculus formed within a urachal remnant. Calcifications in urachal remnants are rare and usually are reported as incidental radiologic findings.
Overall, visible umbilical masses occur infrequently. In addition to urachal anomalies, the differential diagnosis includes several benign and malignant pathologies. Benign causes include epidermoid cysts, foreign body granulomas, pyogenic granulomas, abscesses, hamartomas, nevi, hemangiomas, dermatofibromas, neurofibromas, lipomas, granular cell tumors, desmoid tumors, keloid scars, omphaliths, hernias, or omphalomesenteric duct remnants.6 Primary malignancies (eg, skin cancers, urachal adenocarcinoma, mesenchymal tumors) or metastasis (ie, Sister Mary Joseph nodule) also can present as umbilical nodules.
The wide range of clinical presentations of urachal anomalies combined with the rarity make diagnosis difficult. Thus, it is essential to have a high index of suspicion and awareness of how they can present. Ultrasonography and CT scan are useful tools in making the diagnosis. Urachal anomalies are prone to infection or can be associated with malignancy; therefore, timely and correct diagnosis is critical. Although surgical removal is the primary treatment for urachal anomalies, it may not be the primary treatment of the other entities included in the differential diagnosis of umbilical nodules. For example, the Sister Mary Joseph nodule can be associated with various primary malignancies, which should be treated accordingly.
To the Editor:
An otherwise healthy 26-year-old man presented to our outpatient clinic with a 15- to 20-mm, shiny, friable-appearing, red umbilical nodule with clear malodorous discharge (Figure 1). The lesion developed 2 weeks prior and gradually increased in size and discomfort. The patient reported mild associated abdominal pain. He had no fever, changes in urination or bowel movements, or prior history of umbilical growths or drainage. The abdomen was tender to palpation.
Differential diagnoses included pyogenic granuloma, umbilical hernia, epidermoid cyst or abscess, and malignancy (low suspicion). A biopsy was not performed due to concern for bleeding or communication with the bowel. A complete blood cell count, comprehensive metabolic panel, and urinalysis were unremarkable except for mild leukocytosis and elevated C-reactive protein. Ultrasonography revealed a 1.4×1.3-cm inflammatory umbilical mass with no communication with the bowel. The patient was referred to the emergency department (ED) for further evaluation. Computed tomography (CT) revealed periumbilical inflammation and an associated 1-cm calcification that appeared to be connected to a potential tract from the bladder, suggestive of a urachal remnant calcification (Figure 2). The patient was diagnosed with a persistent urachal remnant, discharged home with ciprofloxacin, and scheduled for a follow-up with urology.
The patient returned to the ED 3 days later with painful umbilical bleeding (Figure 3). While there, the patient extracted a 1-cm stone from the lesion, consistent with the calcification visualized on CT scan. Computed tomographic virtual cystoscopy showed no connection between the bladder and umbilicus. He was diagnosed with an umbilical-urachal sinus. Complete surgical excision was recommended and performed by urology without complication.
We report an unusual presentation of a symptomatic urachal remnant in an adult. During embryogenesis, the urachus connects the umbilicus to the developing bladder and normally involutes during development. Incomplete regression can cause rare pathological urachal anomalies. The clinical presentation is nonspecific and differs between children and adults, with most cases presenting during infancy or childhood.1 Pediatric urachal abnormalities often present with umbilical drainage, abdominal pain, a palpable mass, an abnormal appearance of the umbilicus, or urinary tract infections.2,3 In adults, the most common symptoms include hematuria, pain, or dysuria. Alternatively, they may be asympomatic3 or present with periumbilical dermatitis4 or abscess. Rodrigues and Gandhi5 reported another case of a symptomatic calculus formed within a urachal remnant. Calcifications in urachal remnants are rare and usually are reported as incidental radiologic findings.
Overall, visible umbilical masses occur infrequently. In addition to urachal anomalies, the differential diagnosis includes several benign and malignant pathologies. Benign causes include epidermoid cysts, foreign body granulomas, pyogenic granulomas, abscesses, hamartomas, nevi, hemangiomas, dermatofibromas, neurofibromas, lipomas, granular cell tumors, desmoid tumors, keloid scars, omphaliths, hernias, or omphalomesenteric duct remnants.6 Primary malignancies (eg, skin cancers, urachal adenocarcinoma, mesenchymal tumors) or metastasis (ie, Sister Mary Joseph nodule) also can present as umbilical nodules.
The wide range of clinical presentations of urachal anomalies combined with the rarity make diagnosis difficult. Thus, it is essential to have a high index of suspicion and awareness of how they can present. Ultrasonography and CT scan are useful tools in making the diagnosis. Urachal anomalies are prone to infection or can be associated with malignancy; therefore, timely and correct diagnosis is critical. Although surgical removal is the primary treatment for urachal anomalies, it may not be the primary treatment of the other entities included in the differential diagnosis of umbilical nodules. For example, the Sister Mary Joseph nodule can be associated with various primary malignancies, which should be treated accordingly.
- Berman SM, Tolia BM, Laor E, et al. Urachal remnants in adults. Urology. 1988;31:17-21.
- Gleason JM, Bowlin PR, Bagli DJ, et al. A comprehensive review of pediatric urachal anomalies and predictive analysis for adult urachal adenocarcinoma. J Urol. 2015;193:632-636.
- Naiditch JA, Radhakrishnan J, Chin AC. Current diagnosis and management of urachal remnants. J Pediatr Surg. 2013;48:2148-2152.
- Cox GA, Chan I, Lloyd J, et al. Urachal sinus presenting as periumbilical dermatitis. Br J Dermatol. 2007;157:419-420.
- Rodrigues JCL, Gandhi S. Don’t get caught out! a rare case of a calcified urachal remnant mimicking a bladder calculus. J Radiol Case Rep. 2013;7:34-38.
- Ramoutar A, El Sheikh S, Aslam A. A persistent umbilical nodule. Clin Exp Dermatol. 2017;42:814-816.
- Berman SM, Tolia BM, Laor E, et al. Urachal remnants in adults. Urology. 1988;31:17-21.
- Gleason JM, Bowlin PR, Bagli DJ, et al. A comprehensive review of pediatric urachal anomalies and predictive analysis for adult urachal adenocarcinoma. J Urol. 2015;193:632-636.
- Naiditch JA, Radhakrishnan J, Chin AC. Current diagnosis and management of urachal remnants. J Pediatr Surg. 2013;48:2148-2152.
- Cox GA, Chan I, Lloyd J, et al. Urachal sinus presenting as periumbilical dermatitis. Br J Dermatol. 2007;157:419-420.
- Rodrigues JCL, Gandhi S. Don’t get caught out! a rare case of a calcified urachal remnant mimicking a bladder calculus. J Radiol Case Rep. 2013;7:34-38.
- Ramoutar A, El Sheikh S, Aslam A. A persistent umbilical nodule. Clin Exp Dermatol. 2017;42:814-816.
Practice Points
- Visible umbilical nodules occur infrequently; the differential diagnosis is broad and consists of various benign and malignant pathologies.
- Disruption of the involution of the urachus during development can lead to various rare anomalies.
- Urachal anomalies are important to diagnose given the potential for secondary infection or malignancy.
