User login
Patient Signout Is Not Uniformly Comprehensive and Often Lacks Critical Information
Clinical question: Do signouts vary in the quality and quantity of information, and what are the various factors affecting signout quality?
Background: Miscommunication during transfers of responsibility for hospitalized patients is common and can result in harm. Recommendations for safe and effective handoffs emphasize key content, clear communication, senior staff supervision, and adequate time for questions. Still, little is known about adherence to these recommendations in clinical practice.
Study design: Prospective, observational cohort.
Setting: Medical unit of an acute-care teaching hospital.
Synopsis: Oral signouts were audiotaped among IM house staff teams and the accompanying written signouts were collected for review of content. Signout sessions (n=88) included eight IM teams at one hospital and contained 503 patient signouts.
The median signout duration was 35 seconds (IQR 19-62) per patient. Key clinical information was present in just 62% of combined written or oral signouts. Most signouts included no questions from the recipient. Factors associated with higher rate of content inclusion included: familiarity with the patient, sense of responsibility (primary team vs. covering team), only one signout per day (as compared to sequential signout), presence of a senior resident, and comprehensive, written signouts.
Study limitations include the Hawthorne effect, as several participants mentioned that the presence of audiotape led to more comprehensive signouts than are typical. Also, the signout quality assessment in this study has not been validated with patient-safety outcomes.
Bottom line: Signouts among internal-medicine residents at this one hospital showed variability in terms of quantitative and qualitative information and often missed crucial information about patient care.
Citation: Horwitz LI, Moin T, Krumholz HM, Wang L, Bradley EH. What are covering doctors told about their patients? Analysis of sign-out among internal medicine house staff. Qual Saf Health Care. 2009;18(4):248-255.
Clinical question: Do signouts vary in the quality and quantity of information, and what are the various factors affecting signout quality?
Background: Miscommunication during transfers of responsibility for hospitalized patients is common and can result in harm. Recommendations for safe and effective handoffs emphasize key content, clear communication, senior staff supervision, and adequate time for questions. Still, little is known about adherence to these recommendations in clinical practice.
Study design: Prospective, observational cohort.
Setting: Medical unit of an acute-care teaching hospital.
Synopsis: Oral signouts were audiotaped among IM house staff teams and the accompanying written signouts were collected for review of content. Signout sessions (n=88) included eight IM teams at one hospital and contained 503 patient signouts.
The median signout duration was 35 seconds (IQR 19-62) per patient. Key clinical information was present in just 62% of combined written or oral signouts. Most signouts included no questions from the recipient. Factors associated with higher rate of content inclusion included: familiarity with the patient, sense of responsibility (primary team vs. covering team), only one signout per day (as compared to sequential signout), presence of a senior resident, and comprehensive, written signouts.
Study limitations include the Hawthorne effect, as several participants mentioned that the presence of audiotape led to more comprehensive signouts than are typical. Also, the signout quality assessment in this study has not been validated with patient-safety outcomes.
Bottom line: Signouts among internal-medicine residents at this one hospital showed variability in terms of quantitative and qualitative information and often missed crucial information about patient care.
Citation: Horwitz LI, Moin T, Krumholz HM, Wang L, Bradley EH. What are covering doctors told about their patients? Analysis of sign-out among internal medicine house staff. Qual Saf Health Care. 2009;18(4):248-255.
Clinical question: Do signouts vary in the quality and quantity of information, and what are the various factors affecting signout quality?
Background: Miscommunication during transfers of responsibility for hospitalized patients is common and can result in harm. Recommendations for safe and effective handoffs emphasize key content, clear communication, senior staff supervision, and adequate time for questions. Still, little is known about adherence to these recommendations in clinical practice.
Study design: Prospective, observational cohort.
Setting: Medical unit of an acute-care teaching hospital.
Synopsis: Oral signouts were audiotaped among IM house staff teams and the accompanying written signouts were collected for review of content. Signout sessions (n=88) included eight IM teams at one hospital and contained 503 patient signouts.
The median signout duration was 35 seconds (IQR 19-62) per patient. Key clinical information was present in just 62% of combined written or oral signouts. Most signouts included no questions from the recipient. Factors associated with higher rate of content inclusion included: familiarity with the patient, sense of responsibility (primary team vs. covering team), only one signout per day (as compared to sequential signout), presence of a senior resident, and comprehensive, written signouts.
Study limitations include the Hawthorne effect, as several participants mentioned that the presence of audiotape led to more comprehensive signouts than are typical. Also, the signout quality assessment in this study has not been validated with patient-safety outcomes.
Bottom line: Signouts among internal-medicine residents at this one hospital showed variability in terms of quantitative and qualitative information and often missed crucial information about patient care.
Citation: Horwitz LI, Moin T, Krumholz HM, Wang L, Bradley EH. What are covering doctors told about their patients? Analysis of sign-out among internal medicine house staff. Qual Saf Health Care. 2009;18(4):248-255.
Emergency Department Signout via Voicemail Yields Mixed Reviews
Clinical question: How does traditional, oral signout from emergency providers to inpatient medicine physicians compare to dictated, voicemail signout?
Background: Communication failures contribute to errors in care transition from ED to inpatient medicine units. Signout between ED providers and internal medicine (IM) physicians is typically oral (“synchronous communication”). It is not known how dictated signout to a voicemail system (“asynchronous communication”) affects the quality and safety of handoff communications.
Study design: Prospective, pre-post analysis.
Setting: A 944-bed urban academic medical center in Connecticut.
Synopsis: Surveys were administered to all IM and ED providers before and after the implementation of a voicemail signout system. In the new system, ED providers dictated signout for stable patients, rather than giving traditional synchronous telephone signout. It was the responsibility of the admitting IM physician to listen to the voicemail after receiving a text notification that a patient was being admitted.
ED providers recorded signouts in 89.5% of medicine admissions. However, voicemails were accessed only 58.5% of the time by receiving physicians. All ED providers and 56% of IM physicians believed signout was easier following the voicemail intervention. Overall, ED providers gave the quality, content, and accuracy of their signout communication higher ratings than IM physicians did; 69% of all providers felt the interaction among participants was worse following the intervention. There was no change in the rate of perceived adverse events or ICU transfers within 24 hours after admission.
This intervention was a QI initiative at a single center. Mixed results and small sample size limit generalizability of the study.
Bottom line: Asynchronous signout by voicemail increased efficiency, particularly among ED providers but decreased perceived quality of interaction between medical providers without obviously affecting patient safety.
Citation: Horwitz LI, Parwani V, Shah NR, et al. Evaluation of an asynchronous physician voicemail sign-out for emergency department admissions. Ann Emerg Med. 2009;54:368-378.
Clinical question: How does traditional, oral signout from emergency providers to inpatient medicine physicians compare to dictated, voicemail signout?
Background: Communication failures contribute to errors in care transition from ED to inpatient medicine units. Signout between ED providers and internal medicine (IM) physicians is typically oral (“synchronous communication”). It is not known how dictated signout to a voicemail system (“asynchronous communication”) affects the quality and safety of handoff communications.
Study design: Prospective, pre-post analysis.
Setting: A 944-bed urban academic medical center in Connecticut.
Synopsis: Surveys were administered to all IM and ED providers before and after the implementation of a voicemail signout system. In the new system, ED providers dictated signout for stable patients, rather than giving traditional synchronous telephone signout. It was the responsibility of the admitting IM physician to listen to the voicemail after receiving a text notification that a patient was being admitted.
ED providers recorded signouts in 89.5% of medicine admissions. However, voicemails were accessed only 58.5% of the time by receiving physicians. All ED providers and 56% of IM physicians believed signout was easier following the voicemail intervention. Overall, ED providers gave the quality, content, and accuracy of their signout communication higher ratings than IM physicians did; 69% of all providers felt the interaction among participants was worse following the intervention. There was no change in the rate of perceived adverse events or ICU transfers within 24 hours after admission.
This intervention was a QI initiative at a single center. Mixed results and small sample size limit generalizability of the study.
Bottom line: Asynchronous signout by voicemail increased efficiency, particularly among ED providers but decreased perceived quality of interaction between medical providers without obviously affecting patient safety.
Citation: Horwitz LI, Parwani V, Shah NR, et al. Evaluation of an asynchronous physician voicemail sign-out for emergency department admissions. Ann Emerg Med. 2009;54:368-378.
Clinical question: How does traditional, oral signout from emergency providers to inpatient medicine physicians compare to dictated, voicemail signout?
Background: Communication failures contribute to errors in care transition from ED to inpatient medicine units. Signout between ED providers and internal medicine (IM) physicians is typically oral (“synchronous communication”). It is not known how dictated signout to a voicemail system (“asynchronous communication”) affects the quality and safety of handoff communications.
Study design: Prospective, pre-post analysis.
Setting: A 944-bed urban academic medical center in Connecticut.
Synopsis: Surveys were administered to all IM and ED providers before and after the implementation of a voicemail signout system. In the new system, ED providers dictated signout for stable patients, rather than giving traditional synchronous telephone signout. It was the responsibility of the admitting IM physician to listen to the voicemail after receiving a text notification that a patient was being admitted.
ED providers recorded signouts in 89.5% of medicine admissions. However, voicemails were accessed only 58.5% of the time by receiving physicians. All ED providers and 56% of IM physicians believed signout was easier following the voicemail intervention. Overall, ED providers gave the quality, content, and accuracy of their signout communication higher ratings than IM physicians did; 69% of all providers felt the interaction among participants was worse following the intervention. There was no change in the rate of perceived adverse events or ICU transfers within 24 hours after admission.
This intervention was a QI initiative at a single center. Mixed results and small sample size limit generalizability of the study.
Bottom line: Asynchronous signout by voicemail increased efficiency, particularly among ED providers but decreased perceived quality of interaction between medical providers without obviously affecting patient safety.
Citation: Horwitz LI, Parwani V, Shah NR, et al. Evaluation of an asynchronous physician voicemail sign-out for emergency department admissions. Ann Emerg Med. 2009;54:368-378.
Emergency Department “Boarding” Results in Undesirable Events
Clinical question: What is the frequency and nature of undesirable events experienced by patients who “board” in the ED?
Background: Hospital crowding results in patients spending extended amounts of time—also known as “boarding”—in the ED as they wait for an inpatient bed. Prior studies have shown that longer ED boarding times are associated with adverse outcomes. Few studies have examined the nature and frequency of undesirable events that patients experience while boarding.
Study design: Retrospective chart review.
Setting: Urban academic medical center.
Synopsis: In this pilot study, authors reviewed the charts of patients who were treated in the ED and subsequently admitted to the hospital on three different days during the study period (n=151). More than a quarter (27.8%) of patients experienced an undesirable event, such as missing a scheduled medication, while they were boarding. Older patients, those with comorbid illnesses, and those who endured prolonged boarding times (greater than six hours) were more likely to experience an undesirable event. In addition, 3.3% of patients experienced such adverse events as suboptimal blood pressure control, hypotension, hypoxia, or arrhythmia.
This study was performed at a single center and lacks a comparison group (i.e., nonboarded patients). It is intended to serve as an exploratory study for future analysis of adverse events in boarded patients.
Bottom line: Undesirable events are common among boarded patients, although it is unknown whether they are more common than in nonboarded patients.
Citation: Liu SW, Thomas SH, Gordon JA, Hamedani AG, Weissman JS. A pilot study examining undesirable events among emergency-department boarded patients awaiting inpatient beds. Ann Emerg Med. 2009;54(3):381-385.
Clinical question: What is the frequency and nature of undesirable events experienced by patients who “board” in the ED?
Background: Hospital crowding results in patients spending extended amounts of time—also known as “boarding”—in the ED as they wait for an inpatient bed. Prior studies have shown that longer ED boarding times are associated with adverse outcomes. Few studies have examined the nature and frequency of undesirable events that patients experience while boarding.
Study design: Retrospective chart review.
Setting: Urban academic medical center.
Synopsis: In this pilot study, authors reviewed the charts of patients who were treated in the ED and subsequently admitted to the hospital on three different days during the study period (n=151). More than a quarter (27.8%) of patients experienced an undesirable event, such as missing a scheduled medication, while they were boarding. Older patients, those with comorbid illnesses, and those who endured prolonged boarding times (greater than six hours) were more likely to experience an undesirable event. In addition, 3.3% of patients experienced such adverse events as suboptimal blood pressure control, hypotension, hypoxia, or arrhythmia.
This study was performed at a single center and lacks a comparison group (i.e., nonboarded patients). It is intended to serve as an exploratory study for future analysis of adverse events in boarded patients.
Bottom line: Undesirable events are common among boarded patients, although it is unknown whether they are more common than in nonboarded patients.
Citation: Liu SW, Thomas SH, Gordon JA, Hamedani AG, Weissman JS. A pilot study examining undesirable events among emergency-department boarded patients awaiting inpatient beds. Ann Emerg Med. 2009;54(3):381-385.
Clinical question: What is the frequency and nature of undesirable events experienced by patients who “board” in the ED?
Background: Hospital crowding results in patients spending extended amounts of time—also known as “boarding”—in the ED as they wait for an inpatient bed. Prior studies have shown that longer ED boarding times are associated with adverse outcomes. Few studies have examined the nature and frequency of undesirable events that patients experience while boarding.
Study design: Retrospective chart review.
Setting: Urban academic medical center.
Synopsis: In this pilot study, authors reviewed the charts of patients who were treated in the ED and subsequently admitted to the hospital on three different days during the study period (n=151). More than a quarter (27.8%) of patients experienced an undesirable event, such as missing a scheduled medication, while they were boarding. Older patients, those with comorbid illnesses, and those who endured prolonged boarding times (greater than six hours) were more likely to experience an undesirable event. In addition, 3.3% of patients experienced such adverse events as suboptimal blood pressure control, hypotension, hypoxia, or arrhythmia.
This study was performed at a single center and lacks a comparison group (i.e., nonboarded patients). It is intended to serve as an exploratory study for future analysis of adverse events in boarded patients.
Bottom line: Undesirable events are common among boarded patients, although it is unknown whether they are more common than in nonboarded patients.
Citation: Liu SW, Thomas SH, Gordon JA, Hamedani AG, Weissman JS. A pilot study examining undesirable events among emergency-department boarded patients awaiting inpatient beds. Ann Emerg Med. 2009;54(3):381-385.
Decreased ICU Duty Hours Does Not Affect Patient Mortality
Clinical question: Does the reduction in work hours for residents affect mortality in medical and surgical ICUs?
Background: A reduction in work hours for residents was enforced in July 2003. Several prior studies using administrative or claims data did not show an association of the reduced work hours for residents with mortality in teaching hospitals when compared with nonteaching hospitals.
Study design: Observational retrospective registry cohort.
Setting: Twelve academic, 12 community, and 16 nonteaching hospitals in the U.S.
Synopsis: Data from 230,151 patients were extracted as post-hoc analysis from a voluntary clinical registry that uses a well-validated severity-of-illness scoring system. The exposure was defined as date of admission to ICU within two years before and after the reform. Hospitals were categorized as academic, community with residents, or nonteaching. Sophisticated statistical analyses were performed, including interaction terms for teaching status and time. To test the effect the reduced work hours had on mortality, the mortality trends of academic hospitals and community hospitals with residents were compared with the baseline trend of nonteaching hospitals. After risk adjustments, all hospitals had improved in-hospital and ICU mortality after the reform. None of the statistical improvements were significantly different.
Study limitations include the selection bias, as only highly motivated hospitals participating in the registry were included, and misclassification bias, as not all hospitals implemented the reform at the same time. Nevertheless, this study supports the consistent literature on the topic and adds a more robust assessment of severity of illness.
Bottom line: The restriction on resident duty hours does not appear to affect patient mortality.
Citation: Prasad M, Iwashyna TJ, Christie JD, et al. Effect of work-hours regulations on intensive care unit mortality in United States teaching hospitals. Crit Care Med. 2009;37(9):2564-2569.
Clinical question: Does the reduction in work hours for residents affect mortality in medical and surgical ICUs?
Background: A reduction in work hours for residents was enforced in July 2003. Several prior studies using administrative or claims data did not show an association of the reduced work hours for residents with mortality in teaching hospitals when compared with nonteaching hospitals.
Study design: Observational retrospective registry cohort.
Setting: Twelve academic, 12 community, and 16 nonteaching hospitals in the U.S.
Synopsis: Data from 230,151 patients were extracted as post-hoc analysis from a voluntary clinical registry that uses a well-validated severity-of-illness scoring system. The exposure was defined as date of admission to ICU within two years before and after the reform. Hospitals were categorized as academic, community with residents, or nonteaching. Sophisticated statistical analyses were performed, including interaction terms for teaching status and time. To test the effect the reduced work hours had on mortality, the mortality trends of academic hospitals and community hospitals with residents were compared with the baseline trend of nonteaching hospitals. After risk adjustments, all hospitals had improved in-hospital and ICU mortality after the reform. None of the statistical improvements were significantly different.
Study limitations include the selection bias, as only highly motivated hospitals participating in the registry were included, and misclassification bias, as not all hospitals implemented the reform at the same time. Nevertheless, this study supports the consistent literature on the topic and adds a more robust assessment of severity of illness.
Bottom line: The restriction on resident duty hours does not appear to affect patient mortality.
Citation: Prasad M, Iwashyna TJ, Christie JD, et al. Effect of work-hours regulations on intensive care unit mortality in United States teaching hospitals. Crit Care Med. 2009;37(9):2564-2569.
Clinical question: Does the reduction in work hours for residents affect mortality in medical and surgical ICUs?
Background: A reduction in work hours for residents was enforced in July 2003. Several prior studies using administrative or claims data did not show an association of the reduced work hours for residents with mortality in teaching hospitals when compared with nonteaching hospitals.
Study design: Observational retrospective registry cohort.
Setting: Twelve academic, 12 community, and 16 nonteaching hospitals in the U.S.
Synopsis: Data from 230,151 patients were extracted as post-hoc analysis from a voluntary clinical registry that uses a well-validated severity-of-illness scoring system. The exposure was defined as date of admission to ICU within two years before and after the reform. Hospitals were categorized as academic, community with residents, or nonteaching. Sophisticated statistical analyses were performed, including interaction terms for teaching status and time. To test the effect the reduced work hours had on mortality, the mortality trends of academic hospitals and community hospitals with residents were compared with the baseline trend of nonteaching hospitals. After risk adjustments, all hospitals had improved in-hospital and ICU mortality after the reform. None of the statistical improvements were significantly different.
Study limitations include the selection bias, as only highly motivated hospitals participating in the registry were included, and misclassification bias, as not all hospitals implemented the reform at the same time. Nevertheless, this study supports the consistent literature on the topic and adds a more robust assessment of severity of illness.
Bottom line: The restriction on resident duty hours does not appear to affect patient mortality.
Citation: Prasad M, Iwashyna TJ, Christie JD, et al. Effect of work-hours regulations on intensive care unit mortality in United States teaching hospitals. Crit Care Med. 2009;37(9):2564-2569.
Initiatives Aim at Improving HIV and Mental Health Services
Two new HHS initiatives will expand health services for people with HIV and for people with mental health and substance use disorders.
A new 3-year multi-agency project, Partnerships for Care: Health Departments and Health Centers Collaborating to Improve HIV Health Outcomes, is putting $11 million toward integrating high-quality HIV services into primary care. Run jointly by the CDC and the Health Resources and Services Administration (HRSA), and funded through the Affordable Care Act and HHS Minority AIDS Initiative Fund, the program will develop innovative partnerships between health centers and state health departments in Florida, Massachusetts, Maryland, and New York. The HRSA-funded health centers will work with CDC-funded state health departments to expand the provision of HIV prevention, testing, care, and treatment services, especially among racial and ethnic minorities.
In June 2014, CDC awarded cooperative agreements to the 4 state health departments to begin putting the program into practice in communities most affected by HIV. Those health departments identified 22 health centers as their partners; the health centers are eligible to apply for funding to support workforce development, infrastructure development, HIV service delivery, partnership building, and quality improvement activities.
The HHS also announced $54.6 million in funding to support 221 health centers in 47 states and Puerto Rico to establish or expand behavioral health services for over 450,000 patients. The funds will be used for hiring new mental health professionals, adding mental health and substance use disorder health services, and employing integrated models of primary care.
In 2013, more than 1.2 million patients visited health centers for behavioral health services. The new grant funding, said HHS Secretary Sylvia M. Burwell, will “further reduce the barriers that too often prevent people from getting the help they need for mental health problems. Health centers with these awards are on the front lines of better integrating mental health into primary care and improving access to care through the Affordable Care Act.”
For more information on the projects and their funding, visit http://www.hrsa.gov/grants/apply/assistance/bphchiv and http://www.hrsa.gov/about/news/2014tables/behavioralhealth.
Two new HHS initiatives will expand health services for people with HIV and for people with mental health and substance use disorders.
A new 3-year multi-agency project, Partnerships for Care: Health Departments and Health Centers Collaborating to Improve HIV Health Outcomes, is putting $11 million toward integrating high-quality HIV services into primary care. Run jointly by the CDC and the Health Resources and Services Administration (HRSA), and funded through the Affordable Care Act and HHS Minority AIDS Initiative Fund, the program will develop innovative partnerships between health centers and state health departments in Florida, Massachusetts, Maryland, and New York. The HRSA-funded health centers will work with CDC-funded state health departments to expand the provision of HIV prevention, testing, care, and treatment services, especially among racial and ethnic minorities.
In June 2014, CDC awarded cooperative agreements to the 4 state health departments to begin putting the program into practice in communities most affected by HIV. Those health departments identified 22 health centers as their partners; the health centers are eligible to apply for funding to support workforce development, infrastructure development, HIV service delivery, partnership building, and quality improvement activities.
The HHS also announced $54.6 million in funding to support 221 health centers in 47 states and Puerto Rico to establish or expand behavioral health services for over 450,000 patients. The funds will be used for hiring new mental health professionals, adding mental health and substance use disorder health services, and employing integrated models of primary care.
In 2013, more than 1.2 million patients visited health centers for behavioral health services. The new grant funding, said HHS Secretary Sylvia M. Burwell, will “further reduce the barriers that too often prevent people from getting the help they need for mental health problems. Health centers with these awards are on the front lines of better integrating mental health into primary care and improving access to care through the Affordable Care Act.”
For more information on the projects and their funding, visit http://www.hrsa.gov/grants/apply/assistance/bphchiv and http://www.hrsa.gov/about/news/2014tables/behavioralhealth.
Two new HHS initiatives will expand health services for people with HIV and for people with mental health and substance use disorders.
A new 3-year multi-agency project, Partnerships for Care: Health Departments and Health Centers Collaborating to Improve HIV Health Outcomes, is putting $11 million toward integrating high-quality HIV services into primary care. Run jointly by the CDC and the Health Resources and Services Administration (HRSA), and funded through the Affordable Care Act and HHS Minority AIDS Initiative Fund, the program will develop innovative partnerships between health centers and state health departments in Florida, Massachusetts, Maryland, and New York. The HRSA-funded health centers will work with CDC-funded state health departments to expand the provision of HIV prevention, testing, care, and treatment services, especially among racial and ethnic minorities.
In June 2014, CDC awarded cooperative agreements to the 4 state health departments to begin putting the program into practice in communities most affected by HIV. Those health departments identified 22 health centers as their partners; the health centers are eligible to apply for funding to support workforce development, infrastructure development, HIV service delivery, partnership building, and quality improvement activities.
The HHS also announced $54.6 million in funding to support 221 health centers in 47 states and Puerto Rico to establish or expand behavioral health services for over 450,000 patients. The funds will be used for hiring new mental health professionals, adding mental health and substance use disorder health services, and employing integrated models of primary care.
In 2013, more than 1.2 million patients visited health centers for behavioral health services. The new grant funding, said HHS Secretary Sylvia M. Burwell, will “further reduce the barriers that too often prevent people from getting the help they need for mental health problems. Health centers with these awards are on the front lines of better integrating mental health into primary care and improving access to care through the Affordable Care Act.”
For more information on the projects and their funding, visit http://www.hrsa.gov/grants/apply/assistance/bphchiv and http://www.hrsa.gov/about/news/2014tables/behavioralhealth.
Bortezomib can treat chronic GVHD, study shows
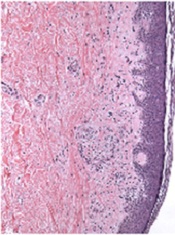
Credit: PLOS ONE
The proteasome inhibitor bortezomib can treat chronic graft-vs-host disease (GVHD), according to research published in Blood.
The study showed that bortezomib provides better outcomes than existing treatments and does not impair the graft-vs-tumor effect.
“Bortezomib helped a group of patients who desperately needed a treatment, having failed multiple different therapies,” said study author Mehrdad Abedi, MD, of the University of California, Davis.
“The drug fights chronic graft-vs-host disease, and, unlike other GVHD therapies such as steroid, cyclosporine, or mycophenolate, it treats chronic GVHD without dampening the graft-vs-tumor effect, which can be critically important to help patients avoid relapse. In fact, because bortezomib is an anticancer drug, it potentially attacks cancer cells in its own right.”
The researchers first studied bortezomib in mice and found the drug suppresses the donor immune cells that cause GVHD.
“We then tested this concept in patients with chronic GVHD . . . ,” Dr Abedi said. “Almost all the patients who tolerated and remained on the treatment responded. In some cases, individual responses were quite dramatic. We were able to stop their other immunosuppressive medications and keep the patients under control with just weekly injections of bortezomib.”
Dr Abedi added that one patient had severe hemolytic anemia that did not respond to several lines of therapy.
“After receiving bortezomib, the patient’s symptoms improved, and we were able to take her completely off steroid and other immunosuppressive medications,” he said. “Another person had multiple ulcers, which completely healed. These were patients who had been on all different kinds of medications and had no response.”
This research is ongoing. Dr Abedi and his colleagues are now looking at a potential oral version of the drug and a similar agent that would alleviate the need for weekly injections and could have fewer side effects.
This research was funded by the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, and Millennium Pharmaceuticals, makers of bortezomib. ![]()
Credit: PLOS ONE
The proteasome inhibitor bortezomib can treat chronic graft-vs-host disease (GVHD), according to research published in Blood.
The study showed that bortezomib provides better outcomes than existing treatments and does not impair the graft-vs-tumor effect.
“Bortezomib helped a group of patients who desperately needed a treatment, having failed multiple different therapies,” said study author Mehrdad Abedi, MD, of the University of California, Davis.
“The drug fights chronic graft-vs-host disease, and, unlike other GVHD therapies such as steroid, cyclosporine, or mycophenolate, it treats chronic GVHD without dampening the graft-vs-tumor effect, which can be critically important to help patients avoid relapse. In fact, because bortezomib is an anticancer drug, it potentially attacks cancer cells in its own right.”
The researchers first studied bortezomib in mice and found the drug suppresses the donor immune cells that cause GVHD.
“We then tested this concept in patients with chronic GVHD . . . ,” Dr Abedi said. “Almost all the patients who tolerated and remained on the treatment responded. In some cases, individual responses were quite dramatic. We were able to stop their other immunosuppressive medications and keep the patients under control with just weekly injections of bortezomib.”
Dr Abedi added that one patient had severe hemolytic anemia that did not respond to several lines of therapy.
“After receiving bortezomib, the patient’s symptoms improved, and we were able to take her completely off steroid and other immunosuppressive medications,” he said. “Another person had multiple ulcers, which completely healed. These were patients who had been on all different kinds of medications and had no response.”
This research is ongoing. Dr Abedi and his colleagues are now looking at a potential oral version of the drug and a similar agent that would alleviate the need for weekly injections and could have fewer side effects.
This research was funded by the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, and Millennium Pharmaceuticals, makers of bortezomib. ![]()
Credit: PLOS ONE
The proteasome inhibitor bortezomib can treat chronic graft-vs-host disease (GVHD), according to research published in Blood.
The study showed that bortezomib provides better outcomes than existing treatments and does not impair the graft-vs-tumor effect.
“Bortezomib helped a group of patients who desperately needed a treatment, having failed multiple different therapies,” said study author Mehrdad Abedi, MD, of the University of California, Davis.
“The drug fights chronic graft-vs-host disease, and, unlike other GVHD therapies such as steroid, cyclosporine, or mycophenolate, it treats chronic GVHD without dampening the graft-vs-tumor effect, which can be critically important to help patients avoid relapse. In fact, because bortezomib is an anticancer drug, it potentially attacks cancer cells in its own right.”
The researchers first studied bortezomib in mice and found the drug suppresses the donor immune cells that cause GVHD.
“We then tested this concept in patients with chronic GVHD . . . ,” Dr Abedi said. “Almost all the patients who tolerated and remained on the treatment responded. In some cases, individual responses were quite dramatic. We were able to stop their other immunosuppressive medications and keep the patients under control with just weekly injections of bortezomib.”
Dr Abedi added that one patient had severe hemolytic anemia that did not respond to several lines of therapy.
“After receiving bortezomib, the patient’s symptoms improved, and we were able to take her completely off steroid and other immunosuppressive medications,” he said. “Another person had multiple ulcers, which completely healed. These were patients who had been on all different kinds of medications and had no response.”
This research is ongoing. Dr Abedi and his colleagues are now looking at a potential oral version of the drug and a similar agent that would alleviate the need for weekly injections and could have fewer side effects.
This research was funded by the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, and Millennium Pharmaceuticals, makers of bortezomib. ![]()
Team creates functional vascular grafts in a week
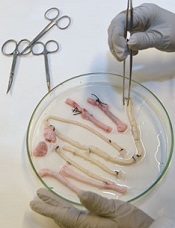
Credit: Robert Emilsson
Researchers have found they can engineer vascular grafts using autologous peripheral blood and successfully transplant them into patients with portal vein thrombosis.
The team said this research provides early evidence for generating personalized blood vessels using stem cells from a blood sample rather than the bone marrow.
Suchitra Sumitran-Holgersson, PhD, of Sahlgrenska University Hospital in Sweden, and her colleagues described the work in EBioMedicine.
The researchers tested their new method on two young children with portal vein thrombosis.
The team used the patients’ own stem cells to grow new blood vessels, a procedure that had been performed at Sahlgrenska University Hospital once before. The novel finding was that they could do this without extracting the cells from the bone marrow.
The researchers took decellularized allogeneic vascular scaffolds and repopulated them with peripheral whole blood in a bioreactor.
The team found they could use 25 mL of blood, the minimum quantity needed to obtain enough stem cells. And the extraction procedure worked the first time.
“Not only that, but the blood itself accelerated growth of the new vein,” Dr Sumitran-Holgersson said. “The entire process took only a week, as opposed to a month in the first case. The blood contains substances that naturally promote growth.”
Dr Sumitran-Holgersson and her colleagues have treated three patients thus far. Two of those patients are still doing well and have veins that are functioning as they should. In the third case, the child is under medical surveillance, and the outcome is more uncertain.
“We believe that this technological progress can lead to dissemination of the method for the benefit of additional groups of patients, such as those with varicose veins or myocardial infarction, who need new blood vessels,” Dr Sumitran-Holgersson said. “Our dream is to be able to grow complete organs as a way of overcoming the current shortage from donors.” ![]()
Credit: Robert Emilsson
Researchers have found they can engineer vascular grafts using autologous peripheral blood and successfully transplant them into patients with portal vein thrombosis.
The team said this research provides early evidence for generating personalized blood vessels using stem cells from a blood sample rather than the bone marrow.
Suchitra Sumitran-Holgersson, PhD, of Sahlgrenska University Hospital in Sweden, and her colleagues described the work in EBioMedicine.
The researchers tested their new method on two young children with portal vein thrombosis.
The team used the patients’ own stem cells to grow new blood vessels, a procedure that had been performed at Sahlgrenska University Hospital once before. The novel finding was that they could do this without extracting the cells from the bone marrow.
The researchers took decellularized allogeneic vascular scaffolds and repopulated them with peripheral whole blood in a bioreactor.
The team found they could use 25 mL of blood, the minimum quantity needed to obtain enough stem cells. And the extraction procedure worked the first time.
“Not only that, but the blood itself accelerated growth of the new vein,” Dr Sumitran-Holgersson said. “The entire process took only a week, as opposed to a month in the first case. The blood contains substances that naturally promote growth.”
Dr Sumitran-Holgersson and her colleagues have treated three patients thus far. Two of those patients are still doing well and have veins that are functioning as they should. In the third case, the child is under medical surveillance, and the outcome is more uncertain.
“We believe that this technological progress can lead to dissemination of the method for the benefit of additional groups of patients, such as those with varicose veins or myocardial infarction, who need new blood vessels,” Dr Sumitran-Holgersson said. “Our dream is to be able to grow complete organs as a way of overcoming the current shortage from donors.” ![]()
Credit: Robert Emilsson
Researchers have found they can engineer vascular grafts using autologous peripheral blood and successfully transplant them into patients with portal vein thrombosis.
The team said this research provides early evidence for generating personalized blood vessels using stem cells from a blood sample rather than the bone marrow.
Suchitra Sumitran-Holgersson, PhD, of Sahlgrenska University Hospital in Sweden, and her colleagues described the work in EBioMedicine.
The researchers tested their new method on two young children with portal vein thrombosis.
The team used the patients’ own stem cells to grow new blood vessels, a procedure that had been performed at Sahlgrenska University Hospital once before. The novel finding was that they could do this without extracting the cells from the bone marrow.
The researchers took decellularized allogeneic vascular scaffolds and repopulated them with peripheral whole blood in a bioreactor.
The team found they could use 25 mL of blood, the minimum quantity needed to obtain enough stem cells. And the extraction procedure worked the first time.
“Not only that, but the blood itself accelerated growth of the new vein,” Dr Sumitran-Holgersson said. “The entire process took only a week, as opposed to a month in the first case. The blood contains substances that naturally promote growth.”
Dr Sumitran-Holgersson and her colleagues have treated three patients thus far. Two of those patients are still doing well and have veins that are functioning as they should. In the third case, the child is under medical surveillance, and the outcome is more uncertain.
“We believe that this technological progress can lead to dissemination of the method for the benefit of additional groups of patients, such as those with varicose veins or myocardial infarction, who need new blood vessels,” Dr Sumitran-Holgersson said. “Our dream is to be able to grow complete organs as a way of overcoming the current shortage from donors.” ![]()
Guidelines for children’s bronchiolitis treatment issued by AAP
The main treatment for bronchiolitis in young children should be support and observation, according to new clinical practice guidelines for diagnosing, managing, and preventing bronchiolitis.
The guidelines apply to children aged 1-23 months and emphasize clinical diagnosis and no medications except nebulized hypertonic saline for infants hospitalized with bronchiolitis, wrote Dr. Shawn L. Ralston, Dr. Allan S. Lieberthal, and their associates (Pediatrics 2014 October 27 [doi:10.1542/peds.2014-2742]). These guidelines update and replace the ones issued by the American Academy of Pediatrics in 2006 (Pediatrics 2006 118:1774-93). The findings are based on a review of the evidence in the Cochrane Library, Medline, and the Cumulative Index of Nursing and Allied Health Literature (CINAHL) from 2004 through May 2014.

The most notable change to these updated guidelines, according to Dr. Lieberthal, is the preventive recommendation for palivizumab, which is now not indicated for children born at 29 weeks’ gestation or older unless they have hemodynamically significant heart disease or chronic lung disease of prematurity (those born at less than 32 weeks’ gestation who needed at least 21% oxygen for their first month). Infants who qualify for prophylactic palivizumab should receive five monthly doses during respiratory syncytial virus season.
Dr. Lieberthal noted in an interview that several other recommendations state that certain treatments should not be used at all rather than simply not being routinely used. These include albuterol, epinephrine, corticosteroids, chest physiotherapy, and antibiotics.
“Bronchiolitis is a self-limited viral illness,” he said. Because it is diagnosed by signs and symptoms, no lab tests, oximetry, imaging, or other tests are needed, and treatment involves only support and observation. “None of the treatments that have been tested have been shown to affect the outcome of the illness,” said Dr. Lieberthal, who practices general pediatrics and clinical pediatric pulmonology at Kaiser-Permanente in Panorama City, Calif.
Dr. Ralston noted in an interview that a new recommendation exists for using hypertonic saline to children who are hospitalized for bronchiolitis (although not in the emergency department), but the evidence for it is weak and its therapeutic value limited.
“This medication appears to have a slow onset and to provide a favorable response only in settings where patients are hospitalized for longer than is typical in most U.S. hospitals, as most of the studies were performed outside the U.S.,” said Dr. Ralston, a pediatrician at Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
The guidelines also note that clinicians “may choose not to administer supplemental oxygen if the oxyhemoglobin saturation exceeds 90%” in children, although the evidence for this recommendation is also weak. Children should receive nasogastric or intravenous fluids if they cannot maintain oral hydration.
Parents should be advised that children who avoid secondhand tobacco smoke and are exclusively breastfed for at least 6 months have a reduced risk of bronchiolitis. Further, anyone caring for a child with bronchiolitis should disinfect their hands using an alcohol-based rub or soap and water after direct contact with the child and the child’s immediate environment.
Dr. Ralston said that important points stressed in both this recommendation and in the previous one include clinical diagnosis and avoiding exposure to tobacco smoke to reduce children’s risk of bronchiolitis.
“This guideline is mostly about what you shouldn’t do for the disease since because of the high volume of disease bronchiolitis represents a major area of unnecessary medical intervention in children,” she said. “We know that the vast majority of children will suffer only side effects from the medications or testing typically used in bronchiolitis care.”
Funding was provided by the American Academy of Pediatrics with travel support from the American Academy of Family Physicians, the American College of Chest Physicians, the American Thoracic Society, and the American College of Emergency Physicians for their representatives.
These guidelines, written with clarity, give incredibly direct and helpful direction on the diagnosis and treatment of bronchiolitis. It is great that they are coming out now, prior to RSV season. Bronchiolitis is a clinical diagnosis and these guidelines reaffirm that there is not usually any need for x-ray or laboratory confirmation of the diagnosis. The guidelines are primarily important for clarifying, based on the evidence, that many commonly used treatments, including albuterol, epinephrine, and steroids are not recommended for treatment of bronchiolitis as they are simply not helpful.
The guidance on administration of palivizumab is also important. It should not be administered in infants with a gestational age of > 29 weeks, and it should be reserved for infants in the first year of life who had a gestational age < 32 weeks and who had hemodynamically significant heart disease or chronic lung disease of prematurity.
Neil Skolnik, M.D., is the associate director of the family medicine program at Abington (Pa.) Memorial Hospital and professor of family and community medicine at Temple University in Philadelphia.
These guidelines, written with clarity, give incredibly direct and helpful direction on the diagnosis and treatment of bronchiolitis. It is great that they are coming out now, prior to RSV season. Bronchiolitis is a clinical diagnosis and these guidelines reaffirm that there is not usually any need for x-ray or laboratory confirmation of the diagnosis. The guidelines are primarily important for clarifying, based on the evidence, that many commonly used treatments, including albuterol, epinephrine, and steroids are not recommended for treatment of bronchiolitis as they are simply not helpful.
The guidance on administration of palivizumab is also important. It should not be administered in infants with a gestational age of > 29 weeks, and it should be reserved for infants in the first year of life who had a gestational age < 32 weeks and who had hemodynamically significant heart disease or chronic lung disease of prematurity.
Neil Skolnik, M.D., is the associate director of the family medicine program at Abington (Pa.) Memorial Hospital and professor of family and community medicine at Temple University in Philadelphia.
These guidelines, written with clarity, give incredibly direct and helpful direction on the diagnosis and treatment of bronchiolitis. It is great that they are coming out now, prior to RSV season. Bronchiolitis is a clinical diagnosis and these guidelines reaffirm that there is not usually any need for x-ray or laboratory confirmation of the diagnosis. The guidelines are primarily important for clarifying, based on the evidence, that many commonly used treatments, including albuterol, epinephrine, and steroids are not recommended for treatment of bronchiolitis as they are simply not helpful.
The guidance on administration of palivizumab is also important. It should not be administered in infants with a gestational age of > 29 weeks, and it should be reserved for infants in the first year of life who had a gestational age < 32 weeks and who had hemodynamically significant heart disease or chronic lung disease of prematurity.
Neil Skolnik, M.D., is the associate director of the family medicine program at Abington (Pa.) Memorial Hospital and professor of family and community medicine at Temple University in Philadelphia.
The main treatment for bronchiolitis in young children should be support and observation, according to new clinical practice guidelines for diagnosing, managing, and preventing bronchiolitis.
The guidelines apply to children aged 1-23 months and emphasize clinical diagnosis and no medications except nebulized hypertonic saline for infants hospitalized with bronchiolitis, wrote Dr. Shawn L. Ralston, Dr. Allan S. Lieberthal, and their associates (Pediatrics 2014 October 27 [doi:10.1542/peds.2014-2742]). These guidelines update and replace the ones issued by the American Academy of Pediatrics in 2006 (Pediatrics 2006 118:1774-93). The findings are based on a review of the evidence in the Cochrane Library, Medline, and the Cumulative Index of Nursing and Allied Health Literature (CINAHL) from 2004 through May 2014.
The most notable change to these updated guidelines, according to Dr. Lieberthal, is the preventive recommendation for palivizumab, which is now not indicated for children born at 29 weeks’ gestation or older unless they have hemodynamically significant heart disease or chronic lung disease of prematurity (those born at less than 32 weeks’ gestation who needed at least 21% oxygen for their first month). Infants who qualify for prophylactic palivizumab should receive five monthly doses during respiratory syncytial virus season.
Dr. Lieberthal noted in an interview that several other recommendations state that certain treatments should not be used at all rather than simply not being routinely used. These include albuterol, epinephrine, corticosteroids, chest physiotherapy, and antibiotics.
“Bronchiolitis is a self-limited viral illness,” he said. Because it is diagnosed by signs and symptoms, no lab tests, oximetry, imaging, or other tests are needed, and treatment involves only support and observation. “None of the treatments that have been tested have been shown to affect the outcome of the illness,” said Dr. Lieberthal, who practices general pediatrics and clinical pediatric pulmonology at Kaiser-Permanente in Panorama City, Calif.
Dr. Ralston noted in an interview that a new recommendation exists for using hypertonic saline to children who are hospitalized for bronchiolitis (although not in the emergency department), but the evidence for it is weak and its therapeutic value limited.
“This medication appears to have a slow onset and to provide a favorable response only in settings where patients are hospitalized for longer than is typical in most U.S. hospitals, as most of the studies were performed outside the U.S.,” said Dr. Ralston, a pediatrician at Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
The guidelines also note that clinicians “may choose not to administer supplemental oxygen if the oxyhemoglobin saturation exceeds 90%” in children, although the evidence for this recommendation is also weak. Children should receive nasogastric or intravenous fluids if they cannot maintain oral hydration.
Parents should be advised that children who avoid secondhand tobacco smoke and are exclusively breastfed for at least 6 months have a reduced risk of bronchiolitis. Further, anyone caring for a child with bronchiolitis should disinfect their hands using an alcohol-based rub or soap and water after direct contact with the child and the child’s immediate environment.
Dr. Ralston said that important points stressed in both this recommendation and in the previous one include clinical diagnosis and avoiding exposure to tobacco smoke to reduce children’s risk of bronchiolitis.
“This guideline is mostly about what you shouldn’t do for the disease since because of the high volume of disease bronchiolitis represents a major area of unnecessary medical intervention in children,” she said. “We know that the vast majority of children will suffer only side effects from the medications or testing typically used in bronchiolitis care.”
Funding was provided by the American Academy of Pediatrics with travel support from the American Academy of Family Physicians, the American College of Chest Physicians, the American Thoracic Society, and the American College of Emergency Physicians for their representatives.
The main treatment for bronchiolitis in young children should be support and observation, according to new clinical practice guidelines for diagnosing, managing, and preventing bronchiolitis.
The guidelines apply to children aged 1-23 months and emphasize clinical diagnosis and no medications except nebulized hypertonic saline for infants hospitalized with bronchiolitis, wrote Dr. Shawn L. Ralston, Dr. Allan S. Lieberthal, and their associates (Pediatrics 2014 October 27 [doi:10.1542/peds.2014-2742]). These guidelines update and replace the ones issued by the American Academy of Pediatrics in 2006 (Pediatrics 2006 118:1774-93). The findings are based on a review of the evidence in the Cochrane Library, Medline, and the Cumulative Index of Nursing and Allied Health Literature (CINAHL) from 2004 through May 2014.
The most notable change to these updated guidelines, according to Dr. Lieberthal, is the preventive recommendation for palivizumab, which is now not indicated for children born at 29 weeks’ gestation or older unless they have hemodynamically significant heart disease or chronic lung disease of prematurity (those born at less than 32 weeks’ gestation who needed at least 21% oxygen for their first month). Infants who qualify for prophylactic palivizumab should receive five monthly doses during respiratory syncytial virus season.
Dr. Lieberthal noted in an interview that several other recommendations state that certain treatments should not be used at all rather than simply not being routinely used. These include albuterol, epinephrine, corticosteroids, chest physiotherapy, and antibiotics.
“Bronchiolitis is a self-limited viral illness,” he said. Because it is diagnosed by signs and symptoms, no lab tests, oximetry, imaging, or other tests are needed, and treatment involves only support and observation. “None of the treatments that have been tested have been shown to affect the outcome of the illness,” said Dr. Lieberthal, who practices general pediatrics and clinical pediatric pulmonology at Kaiser-Permanente in Panorama City, Calif.
Dr. Ralston noted in an interview that a new recommendation exists for using hypertonic saline to children who are hospitalized for bronchiolitis (although not in the emergency department), but the evidence for it is weak and its therapeutic value limited.
“This medication appears to have a slow onset and to provide a favorable response only in settings where patients are hospitalized for longer than is typical in most U.S. hospitals, as most of the studies were performed outside the U.S.,” said Dr. Ralston, a pediatrician at Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
The guidelines also note that clinicians “may choose not to administer supplemental oxygen if the oxyhemoglobin saturation exceeds 90%” in children, although the evidence for this recommendation is also weak. Children should receive nasogastric or intravenous fluids if they cannot maintain oral hydration.
Parents should be advised that children who avoid secondhand tobacco smoke and are exclusively breastfed for at least 6 months have a reduced risk of bronchiolitis. Further, anyone caring for a child with bronchiolitis should disinfect their hands using an alcohol-based rub or soap and water after direct contact with the child and the child’s immediate environment.
Dr. Ralston said that important points stressed in both this recommendation and in the previous one include clinical diagnosis and avoiding exposure to tobacco smoke to reduce children’s risk of bronchiolitis.
“This guideline is mostly about what you shouldn’t do for the disease since because of the high volume of disease bronchiolitis represents a major area of unnecessary medical intervention in children,” she said. “We know that the vast majority of children will suffer only side effects from the medications or testing typically used in bronchiolitis care.”
Funding was provided by the American Academy of Pediatrics with travel support from the American Academy of Family Physicians, the American College of Chest Physicians, the American Thoracic Society, and the American College of Emergency Physicians for their representatives.
FROM PEDIATRICS
Key clinical point: Bronchiolitis should be diagnosed clinically and treated with support.
Major finding: Most treatments should not be administered because outcomes are not improved.
Data source: The findings are based on a review of the evidence in the Cochrane Library, Medline, and CINAHL from 2004 through May 2014.
Disclosures: Funding was provided by the American Academy of Pediatrics with travel support from the American Academy of Family Physicians, the American College of Chest Physicians, the American Thoracic Society, and the American College of Emergency Physicians for their representatives.

CHMP says ponatinib’s benefits outweigh risks

Credit: CDC
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) has adopted its final opinion on ponatinib (Iclusig), saying the drug’s benefits outweigh its risks.
The CHMP recommends that ponatinib continue to be used in accordance with its approved indications.
However, the drug’s product information should be updated with strengthened warnings, particularly about the risk of arterial and venous thrombotic events.
Ponatinib is approved in the European Union (EU) to treat adults with chronic phase, accelerated phase, or blast phase chronic myeloid leukemia (CML) who are resistant to dasatinib or nilotinib, who are intolerant to dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
The drug is also approved to treat adults with Philadelphia-chromosome positive acute lymphoblastic leukemia who are resistant to dasatinib, who cannot tolerate dasatinib and subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
Roughly a year ago, follow-up data revealed that ponatinib-treated patients had a higher incidence of arterial and venous thrombotic events than was observed when the drug first gained approval. So one ponatinib trial was discontinued, and the rest were placed on partial clinical hold.
Then, ponatinib was pulled from the US market. The drug ultimately returned to the marketplace with new recommendations designed to decrease the risk of thrombotic events. The EMA also revised its recommendations for ponatinib but kept the drug on the market.
PRAC review and recommendations
Because of these risks, the EMA’s Pharmacovigilance Risk Assessment Committee (PRAC) conducted an 11-month review of the available data on ponatinib and consulted with a scientific advisory group.
The PRAC assessed the available data on the nature, frequency, and severity of arterial and venous thrombotic events. And the committee concluded that the benefits of ponatinib outweigh its risks.
The PRAC said the risk of thrombotic events is likely dose-related, but there are insufficient data to formally recommend using lower doses of ponatinib. And there is a risk that lower doses might not be as effective in all patients and in long-term treatment.
The PRAC therefore concluded that the recommended starting dose of ponatinib should remain 45 mg once a day.
However, the committee also recommended updates to the product information to provide healthcare professionals with the latest evidence, in case they want to consider reducing the dose in patients with chronic phase CML who are responding well to treatment and who might be at particular risk of thrombotic events.
In addition, PRAC recommended that healthcare professionals stop ponatinib if there has been no response after 3 months of treatment and monitor patients for high blood pressure or signs of heart problems.
The CHMP concurred with these recommendations and is forwarding them to the European Commission. The commission is expected to issue a final, legally binding decision on ponatinib in December 2014, which will be valid throughout the EU.
A new study on the safety and benefits of ponatinib is in the works to help clarify if lower doses of the drug carry a lower risk of thrombotic events while still having a beneficial effect in patients with chronic phase CML. ![]()
Credit: CDC
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) has adopted its final opinion on ponatinib (Iclusig), saying the drug’s benefits outweigh its risks.
The CHMP recommends that ponatinib continue to be used in accordance with its approved indications.
However, the drug’s product information should be updated with strengthened warnings, particularly about the risk of arterial and venous thrombotic events.
Ponatinib is approved in the European Union (EU) to treat adults with chronic phase, accelerated phase, or blast phase chronic myeloid leukemia (CML) who are resistant to dasatinib or nilotinib, who are intolerant to dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
The drug is also approved to treat adults with Philadelphia-chromosome positive acute lymphoblastic leukemia who are resistant to dasatinib, who cannot tolerate dasatinib and subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
Roughly a year ago, follow-up data revealed that ponatinib-treated patients had a higher incidence of arterial and venous thrombotic events than was observed when the drug first gained approval. So one ponatinib trial was discontinued, and the rest were placed on partial clinical hold.
Then, ponatinib was pulled from the US market. The drug ultimately returned to the marketplace with new recommendations designed to decrease the risk of thrombotic events. The EMA also revised its recommendations for ponatinib but kept the drug on the market.
PRAC review and recommendations
Because of these risks, the EMA’s Pharmacovigilance Risk Assessment Committee (PRAC) conducted an 11-month review of the available data on ponatinib and consulted with a scientific advisory group.
The PRAC assessed the available data on the nature, frequency, and severity of arterial and venous thrombotic events. And the committee concluded that the benefits of ponatinib outweigh its risks.
The PRAC said the risk of thrombotic events is likely dose-related, but there are insufficient data to formally recommend using lower doses of ponatinib. And there is a risk that lower doses might not be as effective in all patients and in long-term treatment.
The PRAC therefore concluded that the recommended starting dose of ponatinib should remain 45 mg once a day.
However, the committee also recommended updates to the product information to provide healthcare professionals with the latest evidence, in case they want to consider reducing the dose in patients with chronic phase CML who are responding well to treatment and who might be at particular risk of thrombotic events.
In addition, PRAC recommended that healthcare professionals stop ponatinib if there has been no response after 3 months of treatment and monitor patients for high blood pressure or signs of heart problems.
The CHMP concurred with these recommendations and is forwarding them to the European Commission. The commission is expected to issue a final, legally binding decision on ponatinib in December 2014, which will be valid throughout the EU.
A new study on the safety and benefits of ponatinib is in the works to help clarify if lower doses of the drug carry a lower risk of thrombotic events while still having a beneficial effect in patients with chronic phase CML. ![]()
Credit: CDC
The European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) has adopted its final opinion on ponatinib (Iclusig), saying the drug’s benefits outweigh its risks.
The CHMP recommends that ponatinib continue to be used in accordance with its approved indications.
However, the drug’s product information should be updated with strengthened warnings, particularly about the risk of arterial and venous thrombotic events.
Ponatinib is approved in the European Union (EU) to treat adults with chronic phase, accelerated phase, or blast phase chronic myeloid leukemia (CML) who are resistant to dasatinib or nilotinib, who are intolerant to dasatinib or nilotinib and for whom subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
The drug is also approved to treat adults with Philadelphia-chromosome positive acute lymphoblastic leukemia who are resistant to dasatinib, who cannot tolerate dasatinib and subsequent treatment with imatinib is not clinically appropriate, or who have the T315I mutation.
Roughly a year ago, follow-up data revealed that ponatinib-treated patients had a higher incidence of arterial and venous thrombotic events than was observed when the drug first gained approval. So one ponatinib trial was discontinued, and the rest were placed on partial clinical hold.
Then, ponatinib was pulled from the US market. The drug ultimately returned to the marketplace with new recommendations designed to decrease the risk of thrombotic events. The EMA also revised its recommendations for ponatinib but kept the drug on the market.
PRAC review and recommendations
Because of these risks, the EMA’s Pharmacovigilance Risk Assessment Committee (PRAC) conducted an 11-month review of the available data on ponatinib and consulted with a scientific advisory group.
The PRAC assessed the available data on the nature, frequency, and severity of arterial and venous thrombotic events. And the committee concluded that the benefits of ponatinib outweigh its risks.
The PRAC said the risk of thrombotic events is likely dose-related, but there are insufficient data to formally recommend using lower doses of ponatinib. And there is a risk that lower doses might not be as effective in all patients and in long-term treatment.
The PRAC therefore concluded that the recommended starting dose of ponatinib should remain 45 mg once a day.
However, the committee also recommended updates to the product information to provide healthcare professionals with the latest evidence, in case they want to consider reducing the dose in patients with chronic phase CML who are responding well to treatment and who might be at particular risk of thrombotic events.
In addition, PRAC recommended that healthcare professionals stop ponatinib if there has been no response after 3 months of treatment and monitor patients for high blood pressure or signs of heart problems.
The CHMP concurred with these recommendations and is forwarding them to the European Commission. The commission is expected to issue a final, legally binding decision on ponatinib in December 2014, which will be valid throughout the EU.
A new study on the safety and benefits of ponatinib is in the works to help clarify if lower doses of the drug carry a lower risk of thrombotic events while still having a beneficial effect in patients with chronic phase CML. ![]()
FDA approves treatment for acquired hemophilia A

The US Food and Drug Administration (FDA) has approved a recombinant porcine factor VIII (FVIII) product known as Obizur to treat bleeding episodes in adults with acquired hemophilia A.
Obizur replaces the inhibited human FVIII with a recombinant porcine sequence FVIII based on the rationale that porcine FVIII is less susceptible to inactivation by circulating human FVIII antibodies.
Physicians can manage Obizur’s efficacy and safety by measuring FVIII activity levels.
The ability to measure FVIII levels gives physicians an objective marker of hemostasis that can guide dosing and prevent overdosing, said Rebecca Kruse-Jarres, MD, Director of the Hemophilia Care Program at Puget Sound Blood Center in Seattle and principal investigator of a phase 2/3 trial of Obizur.
Trial results
The FDA approved Obizur based on results of a prospective, multicenter, phase 2/3 trial in which adults with acquired hemophilia A received the product to treat serious bleeding episodes.
Twenty-nine patients were enrolled and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or greater.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse reaction most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur. Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
About acquired hemophilia A and Obizur
Acquired hemophilia A is a rare but potentially life-threatening bleeding disorder caused by the development of antibodies directed against the body’s own FVIII.
Acquired hemophilia A development has been related to other medical conditions or health states, such as pregnancy, cancer, or the use of certain medications. However, in about half of acquired hemophilia A cases, no underlying cause can be found.
Diagnosis of this condition can be difficult, and the severity of bleeding can make treatment challenging.
“The approval of [Obizur] provides an important therapeutic option for use in the care of patients with this rare disease,” said Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The FDA previously granted Obizur orphan drug designation because it is intended for use in the treatment of a rare disease or condition. The product is manufactured by Baxter Healthcare Corporation.
Baxter said Obizur will be commercially available in the US in the coming months and is currently under regulatory review in Europe and Canada.
For more details on Obizur, see the full prescribing information. ![]()
The US Food and Drug Administration (FDA) has approved a recombinant porcine factor VIII (FVIII) product known as Obizur to treat bleeding episodes in adults with acquired hemophilia A.
Obizur replaces the inhibited human FVIII with a recombinant porcine sequence FVIII based on the rationale that porcine FVIII is less susceptible to inactivation by circulating human FVIII antibodies.
Physicians can manage Obizur’s efficacy and safety by measuring FVIII activity levels.
The ability to measure FVIII levels gives physicians an objective marker of hemostasis that can guide dosing and prevent overdosing, said Rebecca Kruse-Jarres, MD, Director of the Hemophilia Care Program at Puget Sound Blood Center in Seattle and principal investigator of a phase 2/3 trial of Obizur.
Trial results
The FDA approved Obizur based on results of a prospective, multicenter, phase 2/3 trial in which adults with acquired hemophilia A received the product to treat serious bleeding episodes.
Twenty-nine patients were enrolled and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or greater.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse reaction most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur. Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
About acquired hemophilia A and Obizur
Acquired hemophilia A is a rare but potentially life-threatening bleeding disorder caused by the development of antibodies directed against the body’s own FVIII.
Acquired hemophilia A development has been related to other medical conditions or health states, such as pregnancy, cancer, or the use of certain medications. However, in about half of acquired hemophilia A cases, no underlying cause can be found.
Diagnosis of this condition can be difficult, and the severity of bleeding can make treatment challenging.
“The approval of [Obizur] provides an important therapeutic option for use in the care of patients with this rare disease,” said Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The FDA previously granted Obizur orphan drug designation because it is intended for use in the treatment of a rare disease or condition. The product is manufactured by Baxter Healthcare Corporation.
Baxter said Obizur will be commercially available in the US in the coming months and is currently under regulatory review in Europe and Canada.
For more details on Obizur, see the full prescribing information. ![]()
The US Food and Drug Administration (FDA) has approved a recombinant porcine factor VIII (FVIII) product known as Obizur to treat bleeding episodes in adults with acquired hemophilia A.
Obizur replaces the inhibited human FVIII with a recombinant porcine sequence FVIII based on the rationale that porcine FVIII is less susceptible to inactivation by circulating human FVIII antibodies.
Physicians can manage Obizur’s efficacy and safety by measuring FVIII activity levels.
The ability to measure FVIII levels gives physicians an objective marker of hemostasis that can guide dosing and prevent overdosing, said Rebecca Kruse-Jarres, MD, Director of the Hemophilia Care Program at Puget Sound Blood Center in Seattle and principal investigator of a phase 2/3 trial of Obizur.
Trial results
The FDA approved Obizur based on results of a prospective, multicenter, phase 2/3 trial in which adults with acquired hemophilia A received the product to treat serious bleeding episodes.
Twenty-nine patients were enrolled and evaluated for safety. Researchers determined that one of the patients did not actually have acquired hemophilia A, so this patient could not be evaluated for efficacy.
At 24 hours after the initial infusion, all 28 patients in the efficacy analysis had a positive response to Obizur. This meant that bleeding stopped or decreased, the patients experienced clinical stabilization or improvement, and FVIII levels were 20% or greater.
Eighty-six percent of patients (24/28) had successful treatment of their initial bleeding episode. The overall treatment success was determined by the investigator based on the ability to discontinue or reduce the dose and/or dosing frequency of Obizur.
The adverse reaction most frequently reported in the 29 patients in the safety analysis was the development of inhibitors to porcine FVIII.
Nineteen patients were negative for anti-porcine FVIII antibodies at baseline, and 5 of these patients (26%) developed anti-porcine FVIII antibodies following exposure to Obizur. Of the 10 patients with detectable anti-porcine FVIII antibodies at baseline, 2 (20%) experienced an increase in titer, and 8 (80%) decreased to a non-detectable titer.
About acquired hemophilia A and Obizur
Acquired hemophilia A is a rare but potentially life-threatening bleeding disorder caused by the development of antibodies directed against the body’s own FVIII.
Acquired hemophilia A development has been related to other medical conditions or health states, such as pregnancy, cancer, or the use of certain medications. However, in about half of acquired hemophilia A cases, no underlying cause can be found.
Diagnosis of this condition can be difficult, and the severity of bleeding can make treatment challenging.
“The approval of [Obizur] provides an important therapeutic option for use in the care of patients with this rare disease,” said Karen Midthun, MD, director of the FDA’s Center for Biologics Evaluation and Research.
The FDA previously granted Obizur orphan drug designation because it is intended for use in the treatment of a rare disease or condition. The product is manufactured by Baxter Healthcare Corporation.
Baxter said Obizur will be commercially available in the US in the coming months and is currently under regulatory review in Europe and Canada.
For more details on Obizur, see the full prescribing information. ![]()