User login
Drug reverses anticoagulation activity of rivaroxaban

Credit: Kevin MacKenzie
An intravenous (IV) bolus of the factor Xa inhibitor antidote andexanet alfa can significantly and immediately reverse the steady-state anticoagulation activity of rivaroxaban in healthy subjects, according to initial results of the phase 3 ANNEXA-R study.
Portola Pharmaceuticals, the company developing andexanet alfa, recently announced these results from the first part of the study.
The company expects to present the full data set on March 16 at the American College of Cardiology’s 64th Annual Scientific Session & Expo in San Diego.
The second part of the ANNEXA-R study, in which researchers are evaluating a bolus plus a continuous infusion of andexanet alfa to sustain reversal, is ongoing.
Portola is developing andexanet alfa as a universal antidote for patients treated with oral and injectable factor Xa inhibitors who are experiencing a major bleeding episode or who require emergency surgery.
Andexanet alfa acts as a factor Xa decoy that targets and sequesters both oral and injectable factor Xa inhibitors in the blood. Once bound, the factor Xa inhibitors are unable to bind to and inhibit native factor Xa, thus allowing for the restoration of normal hemostatic processes.
ANNEXA-R details
This randomized, double-blind, placebo-controlled study is an evaluation of andexanet alfa in reversing rivaroxaban-induced anticoagulation in healthy volunteers ages 50 to 75 years.
In the first part of the study, 41 subjects received rivaroxaban at 20 mg once daily for 4 days. Then, they were randomized in a 2:1 ratio to receive either andexanet alfa administered as an 800 mg IV bolus (n=27) or to placebo (n=14).
Results showed that andexanet alfa significantly and immediately reversed the anticoagulation activity of rivaroxaban. Furthermore, andexanet alfa appeared to be well tolerated.
For the second part of the ANNEXA-R study, researchers plan to enroll about 40 healthy volunteers and give them rivaroxaban at 20 mg once daily for 4 days.
Then, subjects will be randomized to receive either placebo or andexanet alfa administered as an 800 mg IV bolus, followed by a continuous infusion of 8 mg/min for 120 minutes. Data from this part of the study are expected in mid-2015.
Andexanet alfa development
Andexanet alfa is the only compound being studied as a reversal agent for factor Xa inhibitors that directly and specifically corrects anti-factor Xa activity.
Portola is evaluating andexanet alfa in randomized, placebo-controlled phase 3 ANNEXA registration studies using pharmacodynamic endpoints agreed to with the US Food and Drug Administration (FDA), such as anti-factor Xa inhibitor units, to demonstrate efficacy.
Researchers recently reported statistically significant results from the first part of the phase 3 ANNEXA-A study, in which researchers evaluated andexanet alfa administered as a single IV bolus dose with the direct factor Xa inhibitor apixaban.
The second part of the study is ongoing. It’s an evaluation of an IV bolus plus a continuous infusion of andexanet alfa to sustain the reversal of anticoagulation activity.
“The statistically significant phase 3 ANNEXA-R study data, together with results presented previously with apixaban, provide compelling evidence that this ground-breaking agent could serve as a universal antidote for factor Xa inhibitor anticoagulants,” said John T. Curnutte, MD, PhD, executive vice president, research and development for Portola.
“Andexanet alfa is unique among the other reversal agents in development in that it has been the only agent to immediately and significantly reverse all of the key pharmacodynamic measurements of coagulation that have been agreed to with the FDA for accelerated approval. These include anti-factor Xa levels, thrombin generation, and unbound anticoagulant (free-fraction). This has been demonstrated with all of the factor Xa inhibitors studied to date—apixaban, rivaroxaban, edoxaban, and enoxaparin.” ![]()
Credit: Kevin MacKenzie
An intravenous (IV) bolus of the factor Xa inhibitor antidote andexanet alfa can significantly and immediately reverse the steady-state anticoagulation activity of rivaroxaban in healthy subjects, according to initial results of the phase 3 ANNEXA-R study.
Portola Pharmaceuticals, the company developing andexanet alfa, recently announced these results from the first part of the study.
The company expects to present the full data set on March 16 at the American College of Cardiology’s 64th Annual Scientific Session & Expo in San Diego.
The second part of the ANNEXA-R study, in which researchers are evaluating a bolus plus a continuous infusion of andexanet alfa to sustain reversal, is ongoing.
Portola is developing andexanet alfa as a universal antidote for patients treated with oral and injectable factor Xa inhibitors who are experiencing a major bleeding episode or who require emergency surgery.
Andexanet alfa acts as a factor Xa decoy that targets and sequesters both oral and injectable factor Xa inhibitors in the blood. Once bound, the factor Xa inhibitors are unable to bind to and inhibit native factor Xa, thus allowing for the restoration of normal hemostatic processes.
ANNEXA-R details
This randomized, double-blind, placebo-controlled study is an evaluation of andexanet alfa in reversing rivaroxaban-induced anticoagulation in healthy volunteers ages 50 to 75 years.
In the first part of the study, 41 subjects received rivaroxaban at 20 mg once daily for 4 days. Then, they were randomized in a 2:1 ratio to receive either andexanet alfa administered as an 800 mg IV bolus (n=27) or to placebo (n=14).
Results showed that andexanet alfa significantly and immediately reversed the anticoagulation activity of rivaroxaban. Furthermore, andexanet alfa appeared to be well tolerated.
For the second part of the ANNEXA-R study, researchers plan to enroll about 40 healthy volunteers and give them rivaroxaban at 20 mg once daily for 4 days.
Then, subjects will be randomized to receive either placebo or andexanet alfa administered as an 800 mg IV bolus, followed by a continuous infusion of 8 mg/min for 120 minutes. Data from this part of the study are expected in mid-2015.
Andexanet alfa development
Andexanet alfa is the only compound being studied as a reversal agent for factor Xa inhibitors that directly and specifically corrects anti-factor Xa activity.
Portola is evaluating andexanet alfa in randomized, placebo-controlled phase 3 ANNEXA registration studies using pharmacodynamic endpoints agreed to with the US Food and Drug Administration (FDA), such as anti-factor Xa inhibitor units, to demonstrate efficacy.
Researchers recently reported statistically significant results from the first part of the phase 3 ANNEXA-A study, in which researchers evaluated andexanet alfa administered as a single IV bolus dose with the direct factor Xa inhibitor apixaban.
The second part of the study is ongoing. It’s an evaluation of an IV bolus plus a continuous infusion of andexanet alfa to sustain the reversal of anticoagulation activity.
“The statistically significant phase 3 ANNEXA-R study data, together with results presented previously with apixaban, provide compelling evidence that this ground-breaking agent could serve as a universal antidote for factor Xa inhibitor anticoagulants,” said John T. Curnutte, MD, PhD, executive vice president, research and development for Portola.
“Andexanet alfa is unique among the other reversal agents in development in that it has been the only agent to immediately and significantly reverse all of the key pharmacodynamic measurements of coagulation that have been agreed to with the FDA for accelerated approval. These include anti-factor Xa levels, thrombin generation, and unbound anticoagulant (free-fraction). This has been demonstrated with all of the factor Xa inhibitors studied to date—apixaban, rivaroxaban, edoxaban, and enoxaparin.” ![]()
Credit: Kevin MacKenzie
An intravenous (IV) bolus of the factor Xa inhibitor antidote andexanet alfa can significantly and immediately reverse the steady-state anticoagulation activity of rivaroxaban in healthy subjects, according to initial results of the phase 3 ANNEXA-R study.
Portola Pharmaceuticals, the company developing andexanet alfa, recently announced these results from the first part of the study.
The company expects to present the full data set on March 16 at the American College of Cardiology’s 64th Annual Scientific Session & Expo in San Diego.
The second part of the ANNEXA-R study, in which researchers are evaluating a bolus plus a continuous infusion of andexanet alfa to sustain reversal, is ongoing.
Portola is developing andexanet alfa as a universal antidote for patients treated with oral and injectable factor Xa inhibitors who are experiencing a major bleeding episode or who require emergency surgery.
Andexanet alfa acts as a factor Xa decoy that targets and sequesters both oral and injectable factor Xa inhibitors in the blood. Once bound, the factor Xa inhibitors are unable to bind to and inhibit native factor Xa, thus allowing for the restoration of normal hemostatic processes.
ANNEXA-R details
This randomized, double-blind, placebo-controlled study is an evaluation of andexanet alfa in reversing rivaroxaban-induced anticoagulation in healthy volunteers ages 50 to 75 years.
In the first part of the study, 41 subjects received rivaroxaban at 20 mg once daily for 4 days. Then, they were randomized in a 2:1 ratio to receive either andexanet alfa administered as an 800 mg IV bolus (n=27) or to placebo (n=14).
Results showed that andexanet alfa significantly and immediately reversed the anticoagulation activity of rivaroxaban. Furthermore, andexanet alfa appeared to be well tolerated.
For the second part of the ANNEXA-R study, researchers plan to enroll about 40 healthy volunteers and give them rivaroxaban at 20 mg once daily for 4 days.
Then, subjects will be randomized to receive either placebo or andexanet alfa administered as an 800 mg IV bolus, followed by a continuous infusion of 8 mg/min for 120 minutes. Data from this part of the study are expected in mid-2015.
Andexanet alfa development
Andexanet alfa is the only compound being studied as a reversal agent for factor Xa inhibitors that directly and specifically corrects anti-factor Xa activity.
Portola is evaluating andexanet alfa in randomized, placebo-controlled phase 3 ANNEXA registration studies using pharmacodynamic endpoints agreed to with the US Food and Drug Administration (FDA), such as anti-factor Xa inhibitor units, to demonstrate efficacy.
Researchers recently reported statistically significant results from the first part of the phase 3 ANNEXA-A study, in which researchers evaluated andexanet alfa administered as a single IV bolus dose with the direct factor Xa inhibitor apixaban.
The second part of the study is ongoing. It’s an evaluation of an IV bolus plus a continuous infusion of andexanet alfa to sustain the reversal of anticoagulation activity.
“The statistically significant phase 3 ANNEXA-R study data, together with results presented previously with apixaban, provide compelling evidence that this ground-breaking agent could serve as a universal antidote for factor Xa inhibitor anticoagulants,” said John T. Curnutte, MD, PhD, executive vice president, research and development for Portola.
“Andexanet alfa is unique among the other reversal agents in development in that it has been the only agent to immediately and significantly reverse all of the key pharmacodynamic measurements of coagulation that have been agreed to with the FDA for accelerated approval. These include anti-factor Xa levels, thrombin generation, and unbound anticoagulant (free-fraction). This has been demonstrated with all of the factor Xa inhibitors studied to date—apixaban, rivaroxaban, edoxaban, and enoxaparin.” ![]()
Localized Argyria With Pseudo-ochronosis
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
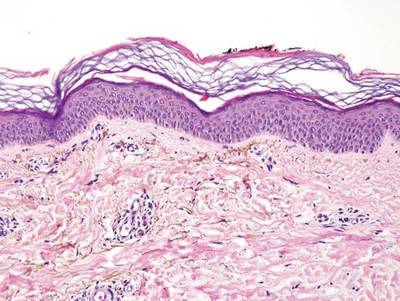
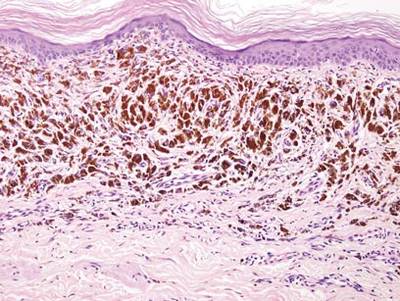
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.

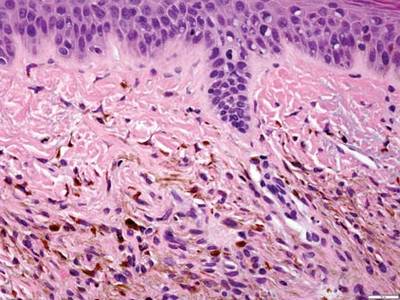
A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
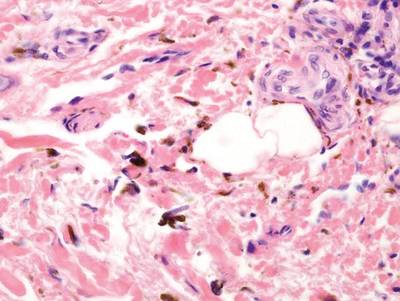
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
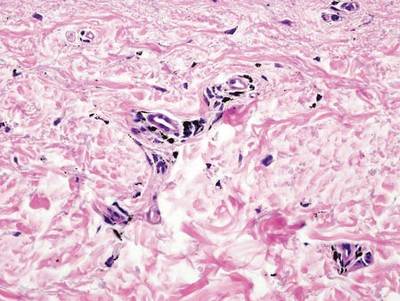
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.
A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
Localized cutaneous argyria often presents as asymptomatic black or blue-gray pigmented macules in areas of the skin exposed to silver-containing compounds.1 Silver may enter the skin by traumatic implantation or absorption via eccrine sweat glands.2 Our patient witnessed a gun fight several years ago while on a mission trip and sustained multiple shrapnel wounds.
As in our patient, hyperpigmentation may appear years following initial exposure. Over time, incident light reduces colorless silver salts and compounds to black elemental silver.3 It also has been suggested that metallic silver granules stimulate tyrosine kinase activity, leading to locally increased melanin production.4 Together, these processes result in the clinical appearance of a blue-black macule. Despite its long-standing association with silver, this appearance also has been noted with deposition of other metals.5 Histologically, metal deposits can be seen as black granules surrounding eccrine glands, blood vessels, and elastic fibers on higher magnification.6 Granules also may be found in sebaceous glands and arrector pili muscle fibers. These findings do not distinguish from generalized argyria due to increased serum silver levels; however, some cases of localized cutaneous argyria have demonstrated spheroid black globules with surrounding collagen necrosis,1 which have not been reported with generalized disease. Localized cutaneous argyria also may be associated with ocher pigmentation of thickened collagen fibers, resembling changes typically found in alkaptonuria, an inherited deficiency of homogentisic acid oxidase (an enzyme involved in tyrosine metabolism).7 The resulting buildup of metabolic intermediates leads to ochronosis, a deposition of ocher-pigmented intermediates in connective tissue throughout the body. In the skin, ocher pigmentation occurs in elastic fibers of the reticular dermis.1 Grossly, these changes result in a blue-gray discoloration of the skin due to a light-scattering phenomenon known as the Tyndall effect. Exogenous ochronosis also can occur, most commonly from the topical application of hydroquinone or other skin-lightening compounds.1,5 Ocher pigmentation occurring in the setting of localized cutaneous argyria is referred to as pseudo-ochronosis, a finding first described by Robinson-Bostom et al.1 The etiology of this condition is poorly understood, but Robinson-Bostom et al1 noted the appearance of dark metal granules surrounding collagen bundles and hypothesized that metal aggregates surrounding collagen bundles in pseudo-ochronosis cause a homogenized appearance under light microscopy. Yellow-brown, swollen, homogenized collagen bundles can be visualized in the reticular dermis with surrounding deposition of metal granules (Figures 1 and 2).1 Typical patterns of granule deposition in localized argyria also are present.
A blue nevus is a collection of proliferating dermal melanocytes. Many histologic subtypes exist and there may be extensive variability in the extent of sclerosis, cellular architecture, and tissue cellularity between each variant.8 Blue nevi commonly present as blue-black hyperpigmentation in the dermis and subcutaneous tissue.9 Histologically, they are characterized by slender, bipolar, dendritic melanocytes in a sclerotic stroma (Figure 3).8 Melanocytes are highly pigmented and contain small monomorphic nuclei. Lesions are relatively homogenous and typically are restricted to the dermis with epidermal sparing.9 Dark granules and ocher fibers are absent.
Long-term use of hydroxychloroquine or other antimalarials may cause a macular pattern of blue-gray hyperpigmentation.10 Biopsy specimens typically reveal coarse, yellow-brown pigment granules primarily affecting the superficial dermis (Figure 4). Granules are found both extracellularly and within macrophages. Fontana-Masson silver staining may identify melanin, as hydroxychloroquine-melanin binding may contribute to patterns of hyperpigmentation.10 Hemosiderin often is present in cases of hydroxychloroquine pigmentation. Preceding ecchymosis appears to favor the deposition of hydroxychloroquine in the skin.11 The absence of dark metal granules helps distinguish hydroxychloroquine pigmentation from argyria.
Regressed melanomas may appear clinically as gray macules. These lesions arise in cases of malignant melanoma that spontaneously regress without treatment. Spontaneous regression occurs in 10% to 35% of cases depending on tumor subtype.12 Lesions can have a variable appearance based on the degree of regression. Partial regression is demonstrated by mixed melanosis and fibrosis in the dermis (Figure 5).13,14 Melanin is housed within melanophages present in a variably expanded papillary dermis. Tumors in early stages of regression can be surrounded by an inflammatory infiltrate, which becomes diminished at later stages. However, a few exceptional cases have been noted with extensive inflammatory infiltrate and no residual tumor.14 Completely regressed lesions typically appear as a band of dermal melanophages in the absence of inflammation or melanocytic atypia.15 The finding of regressed melanoma should prompt further investigation including sentinel lymph node biopsy, as it may be associated with metastasis.
Tattooing occurs following traumatic penetration of the skin with impregnation of pigmented foreign material into deep dermal layers.16 Histologic examination usually reveals clumps of fine particulate material in the dermis (Figure 6). The color of the pigment depends on the agent used. For example, graphite appears as black particles that may be confused with localized cutaneous argyria. Distinction can be made using elemental identification techniques such as energy-dispersive X-ray spectroscopy.1 The intensity of the pigment in granules found in tattoos or localized cutaneous argyria will fail to diminish with the application of melanin bleach.6
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
- Robinson-Bostom L, Pomerantz D, Wilkel C, et al. Localized argyria with pseudo-ochronosis. J Am Acad Dermatol. 2002;46:222-227.
- Tajirian AL, Campbell RM, Robinson-Bostom L. Localized argyria after exposure to aerosolized solder. Cutis. 2006;78:305-308.
- Shelley WB, Shelley ED, Burmeister V. Argyria: the intradermal photograph, a manifestation of passive photosensitivity. J Am Acad Dermatol. 1987;16:211-217.
- Buckley WR, Terhaar CJ. The skin as an excretory organ in argyria. Trans St Johns Hosp Dermatol Soc. 1973;59:39-44.
- Shimizu I, Dill SW, McBean J, et al. Metal-induced granule deposition with pseudo-ochronosis. J Am Acad Dermatol. 2010;63:357-359.
- Rackoff EMJ, Benbenisty KM, Maize JC, et al. Localized cutaneous argyria from an acupuncture needle clini-cally concerning for metastatic melanoma. Cutis. 2007;80:423-426.
- Fernandez-Canon JM, Granadino B, Beltran-Valero de Bernabe D, et al. The molecular basis of alkaptonuria. Nat Genet. 1996;14:5-6.
- Busam KJ, Woodruff JM, Erlandson RA, et al. Large plaque-type blue nevus with subcutaneous cellular nodules. Am J Surg Pathol. 2000;24:92-99.
- Granter SR, McKee PH, Calonje E, et al. Melanoma associated with blue nevus and melanoma mimicking cellular blue nevus: a clinicopathologic study of 10 cases on the spectrum of so-called ‘malignant blue nevus.’ Am J Surg Pathol. 2001;25:316.
- Puri PK, Lountzis NI, Tyler W, et al. Hydroxychloroquine-induced hyperpigmentation: the staining pattern. J Cutan Pathol. 2008;35:1134-1137.
- Jallouli M, Francès C, Piette JC, et al. Hydroxychloroquine-induced pigmentation in patients with systemic lupus erythematosus: a case-control study. JAMA Dermatol. 2013;149:935-940.
- Blessing K, McLaren KM. Histological regression in primary cutaneous melanoma: recognition, prevalence and significance. Histopathology. 1992;20:315-322.
- LeBoit PE. Melanosis and its meanings. Am J Dermatopathol. 2002;24:369-372.
- Emanuel PO, Mannion M, Phelps RG. Complete regression of primary malignant melanoma. Am J Dermatopathol. 2008;30:178-181.
- Yang CH, Yeh JT, Shen SC, et al. Regressed subungual melanoma simulating cellular blue nevus: managed with sentinel lymph node biopsy. Dermatol Surg. 2006;32:577-581.
- Apfelberg DB, Manchester GH. Decorative and traumatic tattoo biophysics and removal. Clin Plast Surg. 1987;14:243-251.
Edoxaban approved for atrial fib, DVT, and PE indications
Edoxaban, a selective factor Xa-inhibitor, has been approved by the Food and Drug Administration for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, with a statement in the boxed warning that it should not be used in patients with normal renal function.
The warning reflects the results of a subgroup analysis in the pivotal trial, which found that the 60-mg dose was superior to warfarin in terms of reducing the stroke risk in mildly renally impaired patients, but was worse in patients with normal renal function. This was the main focus of a meeting of the FDA’s Cardiovascular and Renal Drugs Advisory Panel meeting in October, in which the panel voted 9-1 to recommend approval of edoxaban for this indication, but had mixed opinions on whether approval should be limited to patients with mild to moderate renal impairment.
The approved prescribing information recommends that a patient’s creatinine clearance should be checked before edoxaban is prescribed. “Patients with creatinine clearance greater than 95 mL/min have an increased risk of stroke, compared to similar patients given warfarin,” and should be treated with another anticoagulant, the FDA said in the Jan. 9 statement announcing the approval. The recommended dose for those with a creatinine clearance between 50 mL/min and 95 mL/min is 60 mg once a day; for those with a creatinine clearance of 15-50 mL/min, the recommended dose is 30 mg once a day, according to the prescribing information.
Edoxaban, the fourth novel oral anticoagulant drug approved by the FDA, will be marketed as Savaysa by Daiichi Sankyo. It was also approved to treat deep vein thrombosis and pulmonary embolism following 5-10 days of initial therapy with a parenteral anticoagulant. The recommended dose for this indication is 60 mg once a day. For patients with a creatinine clearance of 15-50 mL/min, or who weigh up to 60 kg (about 132 pounds), or who are taking “certain P-glycoprotein inhibitors,” the 30-mg/day dose is recommended.
Approval for the nonvalvular AF indication was based on ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation) study, comparing once-daily edoxaban (60 mg and 30 mg) to warfarin in 21,015 patients with nonvalvular AF, at a moderate to high risk of thromboembolic events (N. Engl. J. Med. 2013;369:2093-104). Over a median of almost 3 years, both doses were noninferior to warfarin in the primary efficacy endpoint, the occurrence of first stroke or of a systemic embolic event. Overall, major bleeding events were significantly lower among those on the 60-mg and 30-mg doses, compared with those on warfarin. However, the rate of ischemic stroke was higher relative to warfarin in patients with a creatinine clearance over 95 mL/min.
About half of the edoxaban dose is eliminated by the kidneys, and patients with a creatinine clearance above 95 mL/min have lower plasma edoxaban levels, according to a statement in the clinical trials section of the prescribing information, which adds: “Given the clear relationship of dose and blood levels to effectiveness in the ENGAGE AF-TIMI 48 study, it could be anticipated that patients with better renal function would show a smaller effect of Savaysa, compared to warfarin than would patients with mildly impaired renal function, and this was in fact observed.”
Approval of the DVT and PE indication was based on the Hokusai-VTE study of about 8,200 people comparing edoxaban to warfarin, which found that the edoxaban 60 mg once a day was noninferior to warfarin in the rate of symptomatic venous thromboembolism (3.2% vs. 3.5% in those on warfarin). The rate of major or clinically relevant nonmajor bleeding events was 8.5% among those on edoxaban vs. 10.3% in those on warfarin (N. Engl. J. Med. 2013;369:1406-15).
Bleeding and anemia were the most common adverse events among patients with nonvalvular atrial fibrillation in clinical trials, and “as with other FDA-approved anticlotting drugs, bleeding, including life-threatening bleeding, is the most serious risk with Savaysa,” the FDA statement said. Among those treated for DVT and PE, the most common adverse events were bleeding, rash, abnormal liver function tests, and anemia.
Savaysa is the fourth novel oral anticoagulant to be cleared by the FDA, after dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis).
Serious adverse events associated with edoxaban should be reported to the FDA’s MedWatch program or at 800-332-1088.
A precedent does not come readily to mind where one restricts the use of a drug to patients with mild to moderately impaired renal function to optimize the benefit-risk balance. Typically, one avoids the drug or reduces the dose in such subgroups. For this reason, I think the drug is likely going to be a “nonstarter” for many clinicians.
During the FDA Advisory Committee panel meeting on edoxaban, the emphasis was on excluding patients with a creatinine clearance greater than 80 mL/min (representing about 37% of patients in the pivotal trial). The FDA approval used a cut-off of 95 mL/min (representing about 22% of patients in the pivotal trial). Even though the latter increases the eligible pool of patients for edoxaban, the major challenge is going to be clinical acceptability and marketability, especially given no unique advantage of this drug over other approved novel anticoagulants.
The CrCl cut off of 95mL/min applies to nonvalvular atrial fibrillation indication, and not to the venous thromboembolism (DVT/PE) indication. However, the latter indication requires 5-10 days of parenteral anticoagulant therapy, which puts it at a disadvantage compared with rivaroxaban or apixaban.
It is interesting to note that active pathological bleeding qualifies as a contraindication, but use in patients with normal renal function does not. There is a twofold increased risk of overall stroke or systemic embolism (primary endpoint) and ischemic stroke in patients with normal renal function (CrCl > 95 mL/min) along with an attenuation of the hemorrhagic risk advantage in this subgroup. Even though adjudicated major bleeding is still favorable for edoxaban in this subgroup, in my opinion the ischemic stroke risk outweighs the major bleeding advantage.
Sanjay Kaul, M.D., is professor of medicine at the University of California, Los Angeles. He was a member of the FDA’s Cardiovascular and Renal Drugs Advisory Committee that reviewed edoxaban at a meeting on Oct. 30, 2014.
A precedent does not come readily to mind where one restricts the use of a drug to patients with mild to moderately impaired renal function to optimize the benefit-risk balance. Typically, one avoids the drug or reduces the dose in such subgroups. For this reason, I think the drug is likely going to be a “nonstarter” for many clinicians.
During the FDA Advisory Committee panel meeting on edoxaban, the emphasis was on excluding patients with a creatinine clearance greater than 80 mL/min (representing about 37% of patients in the pivotal trial). The FDA approval used a cut-off of 95 mL/min (representing about 22% of patients in the pivotal trial). Even though the latter increases the eligible pool of patients for edoxaban, the major challenge is going to be clinical acceptability and marketability, especially given no unique advantage of this drug over other approved novel anticoagulants.
The CrCl cut off of 95mL/min applies to nonvalvular atrial fibrillation indication, and not to the venous thromboembolism (DVT/PE) indication. However, the latter indication requires 5-10 days of parenteral anticoagulant therapy, which puts it at a disadvantage compared with rivaroxaban or apixaban.
It is interesting to note that active pathological bleeding qualifies as a contraindication, but use in patients with normal renal function does not. There is a twofold increased risk of overall stroke or systemic embolism (primary endpoint) and ischemic stroke in patients with normal renal function (CrCl > 95 mL/min) along with an attenuation of the hemorrhagic risk advantage in this subgroup. Even though adjudicated major bleeding is still favorable for edoxaban in this subgroup, in my opinion the ischemic stroke risk outweighs the major bleeding advantage.
Sanjay Kaul, M.D., is professor of medicine at the University of California, Los Angeles. He was a member of the FDA’s Cardiovascular and Renal Drugs Advisory Committee that reviewed edoxaban at a meeting on Oct. 30, 2014.
A precedent does not come readily to mind where one restricts the use of a drug to patients with mild to moderately impaired renal function to optimize the benefit-risk balance. Typically, one avoids the drug or reduces the dose in such subgroups. For this reason, I think the drug is likely going to be a “nonstarter” for many clinicians.
During the FDA Advisory Committee panel meeting on edoxaban, the emphasis was on excluding patients with a creatinine clearance greater than 80 mL/min (representing about 37% of patients in the pivotal trial). The FDA approval used a cut-off of 95 mL/min (representing about 22% of patients in the pivotal trial). Even though the latter increases the eligible pool of patients for edoxaban, the major challenge is going to be clinical acceptability and marketability, especially given no unique advantage of this drug over other approved novel anticoagulants.
The CrCl cut off of 95mL/min applies to nonvalvular atrial fibrillation indication, and not to the venous thromboembolism (DVT/PE) indication. However, the latter indication requires 5-10 days of parenteral anticoagulant therapy, which puts it at a disadvantage compared with rivaroxaban or apixaban.
It is interesting to note that active pathological bleeding qualifies as a contraindication, but use in patients with normal renal function does not. There is a twofold increased risk of overall stroke or systemic embolism (primary endpoint) and ischemic stroke in patients with normal renal function (CrCl > 95 mL/min) along with an attenuation of the hemorrhagic risk advantage in this subgroup. Even though adjudicated major bleeding is still favorable for edoxaban in this subgroup, in my opinion the ischemic stroke risk outweighs the major bleeding advantage.
Sanjay Kaul, M.D., is professor of medicine at the University of California, Los Angeles. He was a member of the FDA’s Cardiovascular and Renal Drugs Advisory Committee that reviewed edoxaban at a meeting on Oct. 30, 2014.
Edoxaban, a selective factor Xa-inhibitor, has been approved by the Food and Drug Administration for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, with a statement in the boxed warning that it should not be used in patients with normal renal function.
The warning reflects the results of a subgroup analysis in the pivotal trial, which found that the 60-mg dose was superior to warfarin in terms of reducing the stroke risk in mildly renally impaired patients, but was worse in patients with normal renal function. This was the main focus of a meeting of the FDA’s Cardiovascular and Renal Drugs Advisory Panel meeting in October, in which the panel voted 9-1 to recommend approval of edoxaban for this indication, but had mixed opinions on whether approval should be limited to patients with mild to moderate renal impairment.
The approved prescribing information recommends that a patient’s creatinine clearance should be checked before edoxaban is prescribed. “Patients with creatinine clearance greater than 95 mL/min have an increased risk of stroke, compared to similar patients given warfarin,” and should be treated with another anticoagulant, the FDA said in the Jan. 9 statement announcing the approval. The recommended dose for those with a creatinine clearance between 50 mL/min and 95 mL/min is 60 mg once a day; for those with a creatinine clearance of 15-50 mL/min, the recommended dose is 30 mg once a day, according to the prescribing information.
Edoxaban, the fourth novel oral anticoagulant drug approved by the FDA, will be marketed as Savaysa by Daiichi Sankyo. It was also approved to treat deep vein thrombosis and pulmonary embolism following 5-10 days of initial therapy with a parenteral anticoagulant. The recommended dose for this indication is 60 mg once a day. For patients with a creatinine clearance of 15-50 mL/min, or who weigh up to 60 kg (about 132 pounds), or who are taking “certain P-glycoprotein inhibitors,” the 30-mg/day dose is recommended.
Approval for the nonvalvular AF indication was based on ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation) study, comparing once-daily edoxaban (60 mg and 30 mg) to warfarin in 21,015 patients with nonvalvular AF, at a moderate to high risk of thromboembolic events (N. Engl. J. Med. 2013;369:2093-104). Over a median of almost 3 years, both doses were noninferior to warfarin in the primary efficacy endpoint, the occurrence of first stroke or of a systemic embolic event. Overall, major bleeding events were significantly lower among those on the 60-mg and 30-mg doses, compared with those on warfarin. However, the rate of ischemic stroke was higher relative to warfarin in patients with a creatinine clearance over 95 mL/min.
About half of the edoxaban dose is eliminated by the kidneys, and patients with a creatinine clearance above 95 mL/min have lower plasma edoxaban levels, according to a statement in the clinical trials section of the prescribing information, which adds: “Given the clear relationship of dose and blood levels to effectiveness in the ENGAGE AF-TIMI 48 study, it could be anticipated that patients with better renal function would show a smaller effect of Savaysa, compared to warfarin than would patients with mildly impaired renal function, and this was in fact observed.”
Approval of the DVT and PE indication was based on the Hokusai-VTE study of about 8,200 people comparing edoxaban to warfarin, which found that the edoxaban 60 mg once a day was noninferior to warfarin in the rate of symptomatic venous thromboembolism (3.2% vs. 3.5% in those on warfarin). The rate of major or clinically relevant nonmajor bleeding events was 8.5% among those on edoxaban vs. 10.3% in those on warfarin (N. Engl. J. Med. 2013;369:1406-15).
Bleeding and anemia were the most common adverse events among patients with nonvalvular atrial fibrillation in clinical trials, and “as with other FDA-approved anticlotting drugs, bleeding, including life-threatening bleeding, is the most serious risk with Savaysa,” the FDA statement said. Among those treated for DVT and PE, the most common adverse events were bleeding, rash, abnormal liver function tests, and anemia.
Savaysa is the fourth novel oral anticoagulant to be cleared by the FDA, after dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis).
Serious adverse events associated with edoxaban should be reported to the FDA’s MedWatch program or at 800-332-1088.
Edoxaban, a selective factor Xa-inhibitor, has been approved by the Food and Drug Administration for reducing the risk of stroke and systemic embolism in patients with nonvalvular atrial fibrillation, with a statement in the boxed warning that it should not be used in patients with normal renal function.
The warning reflects the results of a subgroup analysis in the pivotal trial, which found that the 60-mg dose was superior to warfarin in terms of reducing the stroke risk in mildly renally impaired patients, but was worse in patients with normal renal function. This was the main focus of a meeting of the FDA’s Cardiovascular and Renal Drugs Advisory Panel meeting in October, in which the panel voted 9-1 to recommend approval of edoxaban for this indication, but had mixed opinions on whether approval should be limited to patients with mild to moderate renal impairment.
The approved prescribing information recommends that a patient’s creatinine clearance should be checked before edoxaban is prescribed. “Patients with creatinine clearance greater than 95 mL/min have an increased risk of stroke, compared to similar patients given warfarin,” and should be treated with another anticoagulant, the FDA said in the Jan. 9 statement announcing the approval. The recommended dose for those with a creatinine clearance between 50 mL/min and 95 mL/min is 60 mg once a day; for those with a creatinine clearance of 15-50 mL/min, the recommended dose is 30 mg once a day, according to the prescribing information.
Edoxaban, the fourth novel oral anticoagulant drug approved by the FDA, will be marketed as Savaysa by Daiichi Sankyo. It was also approved to treat deep vein thrombosis and pulmonary embolism following 5-10 days of initial therapy with a parenteral anticoagulant. The recommended dose for this indication is 60 mg once a day. For patients with a creatinine clearance of 15-50 mL/min, or who weigh up to 60 kg (about 132 pounds), or who are taking “certain P-glycoprotein inhibitors,” the 30-mg/day dose is recommended.
Approval for the nonvalvular AF indication was based on ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation) study, comparing once-daily edoxaban (60 mg and 30 mg) to warfarin in 21,015 patients with nonvalvular AF, at a moderate to high risk of thromboembolic events (N. Engl. J. Med. 2013;369:2093-104). Over a median of almost 3 years, both doses were noninferior to warfarin in the primary efficacy endpoint, the occurrence of first stroke or of a systemic embolic event. Overall, major bleeding events were significantly lower among those on the 60-mg and 30-mg doses, compared with those on warfarin. However, the rate of ischemic stroke was higher relative to warfarin in patients with a creatinine clearance over 95 mL/min.
About half of the edoxaban dose is eliminated by the kidneys, and patients with a creatinine clearance above 95 mL/min have lower plasma edoxaban levels, according to a statement in the clinical trials section of the prescribing information, which adds: “Given the clear relationship of dose and blood levels to effectiveness in the ENGAGE AF-TIMI 48 study, it could be anticipated that patients with better renal function would show a smaller effect of Savaysa, compared to warfarin than would patients with mildly impaired renal function, and this was in fact observed.”
Approval of the DVT and PE indication was based on the Hokusai-VTE study of about 8,200 people comparing edoxaban to warfarin, which found that the edoxaban 60 mg once a day was noninferior to warfarin in the rate of symptomatic venous thromboembolism (3.2% vs. 3.5% in those on warfarin). The rate of major or clinically relevant nonmajor bleeding events was 8.5% among those on edoxaban vs. 10.3% in those on warfarin (N. Engl. J. Med. 2013;369:1406-15).
Bleeding and anemia were the most common adverse events among patients with nonvalvular atrial fibrillation in clinical trials, and “as with other FDA-approved anticlotting drugs, bleeding, including life-threatening bleeding, is the most serious risk with Savaysa,” the FDA statement said. Among those treated for DVT and PE, the most common adverse events were bleeding, rash, abnormal liver function tests, and anemia.
Savaysa is the fourth novel oral anticoagulant to be cleared by the FDA, after dabigatran (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis).
Serious adverse events associated with edoxaban should be reported to the FDA’s MedWatch program or at 800-332-1088.
Taking a look at neurologist burnout
There’s a lot in the news these days about doctor burnout. More specifically, neurologist burnout.
In a 2012 survey study, about 53% of neurologists reported burnout, which was third among all specialties surveyed, behind emergency medicine physicians and general internists. Neurologists also reported the fourth lowest job satisfaction with work-life balance, with about 41% satisfied that work leaves enough time for personal or family life. Neurology was the only one out of five specialties with the highest rates of burnout that was also among the five specialties with the lowest work-life balance.

Granted, the term “burnout” can mean a lot, but these days seems to refer to the fall of the American physician: Overworked, with rising costs, and falling reimbursements, sandwiched between patients who want to be cured immediately and those who want to sue us, and even on a good day facing a litany of terrible diseases.
Heck, I’d be burned out, too. Maybe I am.
Some say this is from the worries of solo practice, since we’re usually more pressed for time and money. I disagree, as I’ve seen it on both sides.
Recently, I saw my own internist. Six months ago she closed her own solo practice to join a large, hospital-owned group. She looked exhausted, worse than I’d ever seen her. She told me that she now gets a secure paycheck, but her stress level is worse. The hospital sets her schedule, tells her how much time she can spend with each patient, gives her quotas she has to meet, and has supplied an electronic health record (EHR) system that’s less than user friendly. (Personally, all of the ones I’ve tried are terrible.) When she goes home, she told me that now after dinner she still has to log on and do 2-3 more hours of charting just to catch up.
The grass is always greener. In her, I see a doctor who doesn’t have to watch each penny and worry about whether she’ll get a paycheck next week. In me, she looks at someone who’s free to pick their vacation days and isn’t chained to a quota system and a burdensome EHR.
Who’s right? I suppose it depends on what your life preferences are. Are we both burned out? We probably are, but in different ways.
But why the high rate of burnout for neurologists? Likely because of the issues I mentioned above. For myself, I’ve seen my salary drop 50% since its highest point in 2005. We’re faced with rising costs (like many other businesses). Unlike other professions, however, we don’t have much control over our reimbursement. Peculiar to medicine is the simple fact that what we charge has no bearing on what we get paid. Those rates are set by factors over which we have no control. Worse, they’re often set by politicians and insurance executives, who see us as the enemy.
There’s also the way reimbursements are set-up: they still favor docs who do a lot of procedures. While neurologists have a few, most of our job is thinking. And that’s not compensated nearly as well as jabbing needles and scalpels in people.
Then you get beyond financial issues. Many of us go through the day feeling like we have a target on our backs, in fear of patients becoming plaintiffs. What else? The nature of our field is such that we deal with diseases that are often challenging to diagnose and sometimes difficult, if not impossible, to treat. Yet, we still have to put on our best show and attitude for those afflicted. Part of why they come to us is to have questions answered and be given any glimmer of hope we can find.
In spite of this, the majority of us go on. Even burned out, we came here to help others. It’s part of what makes us tick and drives us to look in the mirror and head to the office. I wouldn’t trade what I do for anything. But I wish I could do it in a less adversarial world where I’m forced to choose between freedom and a (even temporary) sense of security.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
There’s a lot in the news these days about doctor burnout. More specifically, neurologist burnout.
In a 2012 survey study, about 53% of neurologists reported burnout, which was third among all specialties surveyed, behind emergency medicine physicians and general internists. Neurologists also reported the fourth lowest job satisfaction with work-life balance, with about 41% satisfied that work leaves enough time for personal or family life. Neurology was the only one out of five specialties with the highest rates of burnout that was also among the five specialties with the lowest work-life balance.
Granted, the term “burnout” can mean a lot, but these days seems to refer to the fall of the American physician: Overworked, with rising costs, and falling reimbursements, sandwiched between patients who want to be cured immediately and those who want to sue us, and even on a good day facing a litany of terrible diseases.
Heck, I’d be burned out, too. Maybe I am.
Some say this is from the worries of solo practice, since we’re usually more pressed for time and money. I disagree, as I’ve seen it on both sides.
Recently, I saw my own internist. Six months ago she closed her own solo practice to join a large, hospital-owned group. She looked exhausted, worse than I’d ever seen her. She told me that she now gets a secure paycheck, but her stress level is worse. The hospital sets her schedule, tells her how much time she can spend with each patient, gives her quotas she has to meet, and has supplied an electronic health record (EHR) system that’s less than user friendly. (Personally, all of the ones I’ve tried are terrible.) When she goes home, she told me that now after dinner she still has to log on and do 2-3 more hours of charting just to catch up.
The grass is always greener. In her, I see a doctor who doesn’t have to watch each penny and worry about whether she’ll get a paycheck next week. In me, she looks at someone who’s free to pick their vacation days and isn’t chained to a quota system and a burdensome EHR.
Who’s right? I suppose it depends on what your life preferences are. Are we both burned out? We probably are, but in different ways.
But why the high rate of burnout for neurologists? Likely because of the issues I mentioned above. For myself, I’ve seen my salary drop 50% since its highest point in 2005. We’re faced with rising costs (like many other businesses). Unlike other professions, however, we don’t have much control over our reimbursement. Peculiar to medicine is the simple fact that what we charge has no bearing on what we get paid. Those rates are set by factors over which we have no control. Worse, they’re often set by politicians and insurance executives, who see us as the enemy.
There’s also the way reimbursements are set-up: they still favor docs who do a lot of procedures. While neurologists have a few, most of our job is thinking. And that’s not compensated nearly as well as jabbing needles and scalpels in people.
Then you get beyond financial issues. Many of us go through the day feeling like we have a target on our backs, in fear of patients becoming plaintiffs. What else? The nature of our field is such that we deal with diseases that are often challenging to diagnose and sometimes difficult, if not impossible, to treat. Yet, we still have to put on our best show and attitude for those afflicted. Part of why they come to us is to have questions answered and be given any glimmer of hope we can find.
In spite of this, the majority of us go on. Even burned out, we came here to help others. It’s part of what makes us tick and drives us to look in the mirror and head to the office. I wouldn’t trade what I do for anything. But I wish I could do it in a less adversarial world where I’m forced to choose between freedom and a (even temporary) sense of security.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
There’s a lot in the news these days about doctor burnout. More specifically, neurologist burnout.
In a 2012 survey study, about 53% of neurologists reported burnout, which was third among all specialties surveyed, behind emergency medicine physicians and general internists. Neurologists also reported the fourth lowest job satisfaction with work-life balance, with about 41% satisfied that work leaves enough time for personal or family life. Neurology was the only one out of five specialties with the highest rates of burnout that was also among the five specialties with the lowest work-life balance.
Granted, the term “burnout” can mean a lot, but these days seems to refer to the fall of the American physician: Overworked, with rising costs, and falling reimbursements, sandwiched between patients who want to be cured immediately and those who want to sue us, and even on a good day facing a litany of terrible diseases.
Heck, I’d be burned out, too. Maybe I am.
Some say this is from the worries of solo practice, since we’re usually more pressed for time and money. I disagree, as I’ve seen it on both sides.
Recently, I saw my own internist. Six months ago she closed her own solo practice to join a large, hospital-owned group. She looked exhausted, worse than I’d ever seen her. She told me that she now gets a secure paycheck, but her stress level is worse. The hospital sets her schedule, tells her how much time she can spend with each patient, gives her quotas she has to meet, and has supplied an electronic health record (EHR) system that’s less than user friendly. (Personally, all of the ones I’ve tried are terrible.) When she goes home, she told me that now after dinner she still has to log on and do 2-3 more hours of charting just to catch up.
The grass is always greener. In her, I see a doctor who doesn’t have to watch each penny and worry about whether she’ll get a paycheck next week. In me, she looks at someone who’s free to pick their vacation days and isn’t chained to a quota system and a burdensome EHR.
Who’s right? I suppose it depends on what your life preferences are. Are we both burned out? We probably are, but in different ways.
But why the high rate of burnout for neurologists? Likely because of the issues I mentioned above. For myself, I’ve seen my salary drop 50% since its highest point in 2005. We’re faced with rising costs (like many other businesses). Unlike other professions, however, we don’t have much control over our reimbursement. Peculiar to medicine is the simple fact that what we charge has no bearing on what we get paid. Those rates are set by factors over which we have no control. Worse, they’re often set by politicians and insurance executives, who see us as the enemy.
There’s also the way reimbursements are set-up: they still favor docs who do a lot of procedures. While neurologists have a few, most of our job is thinking. And that’s not compensated nearly as well as jabbing needles and scalpels in people.
Then you get beyond financial issues. Many of us go through the day feeling like we have a target on our backs, in fear of patients becoming plaintiffs. What else? The nature of our field is such that we deal with diseases that are often challenging to diagnose and sometimes difficult, if not impossible, to treat. Yet, we still have to put on our best show and attitude for those afflicted. Part of why they come to us is to have questions answered and be given any glimmer of hope we can find.
In spite of this, the majority of us go on. Even burned out, we came here to help others. It’s part of what makes us tick and drives us to look in the mirror and head to the office. I wouldn’t trade what I do for anything. But I wish I could do it in a less adversarial world where I’m forced to choose between freedom and a (even temporary) sense of security.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.

Cutaneous Side Effects of Chemotherapy in Pediatric Oncology Patients
Pediatric oncology patients can present with various skin lesions related to both their primary disease and immunosuppressive treatments. In the majority of cases, cutaneous findings are associated with the use of chemotherapeutic agents. The toxic effects of chemotherapeutic agents, which generally are associated with treatment of solid organ malignancies (eg, liver, kidneys), can be detected by oncologists using clinical signs and laboratory tests.1-3 However, it also is important for dermatologists to recognize and evaluate cutaneous side effects associated with chemotherapeutic agents. Reports in the literature of cutaneous side effects of chemotherapy in pediatric patients generally are limited to case studies. This study aimed to evaluate the characteristics of cutaneous side effects of chemotherapy in pediatric oncology patients.
Materials and Methods
The study was performed through the collaboration of the departments of dermatology and venereology and pediatric oncology in the Faculty of Medicine at Ege University, Izmir, Turkey. Sixty-five pediatric oncology patients who were scheduled to undergo chemotherapy from May 2011 to May 2013 were included in the study. Clinical examination of dermatologic findings was conducted at baseline (prior to beginning chemotherapy) and at months 1, 3, and 6 of treatment. Patients were examined a total of 4 times during the study. Patients with a history of skin disease prior to diagnosis of their malignancy were excluded, as the study aimed to evaluate cutaneous side effects of chemotherapy. Patients who developed cutaneous side effects during the study period were photographed. Skin biopsy was performed to confirm clinical diagnosis. Patients were split into 5 groups according to oncological diagnoses, including hematological malignancies, solid organ tumors, bone and soft tissue tumors, central nervous system tumors, and Langerhans cell histiocytosis. Data regarding age, gender, treatments administered (ie, chemotherapeutics, antibiotics, antifungals, antivirals), and dermatologic signs were recorded. Mucocutaneous findings were classified as infectious (viral, bacterial, fungal) lesions, bullous lesions, inflammatory dermatoses (eg, diaper dermatitis, asteatotic eczema, contact dermatitis, seborrheic dermatitis), xeroderma, petechiae/ecchymoses, nail signs, alopecia, mucositis, cheilitis, oral aphthae, drug reactions confirmed by histopathology, cushingoid signs (eg, striae, acneform eruption, hypertrichosis), and cutaneous hyperpigmentation.
Statistical analysis was performed using SPSS version 15.0 and χ2 test was applied to the analysis.
Results
Of 65 patients, 62 completed the study and were included in the analysis. Three patients were excluded from the results, as 2 patients died during treatment and 1 patient withdrew from the study prior to completion. Twenty-seven (43.5%) patients were female and 35 (56.5%) were male ranging in age from 1 to 17 years (mean age, 8.14 years; median age [standard deviation], 7.25 [5.42] years). There were 31 (50%) patients in the hematological malignancies group, 11 (17.7%) in the solid organ tumors group, 10 (16.1%) in the bone and soft tissue tumors group, and 9 (14.5%) in the central nervous system tumors group; Langerhans cell histiocytosis was diagnosed in 1 (1.6%) patient. Hodgkin lymphoma made up 29.0% (n=9) of hematological malignancies. Other hematological malignancies included acute myeloblastic leukemia (n=7 [22.5%]), acute lymphoblastic leukemia (n=7 [22.5%]), T-cell lymphoma (n=5 [16.1%]), non-Hodgkin lym-phoma (n=1 [3.2%]), anaplastic giant cell lymphoma (n=1 [3.2%]), and diffuse giant cell lymphoma (n=1 [3.2%]).
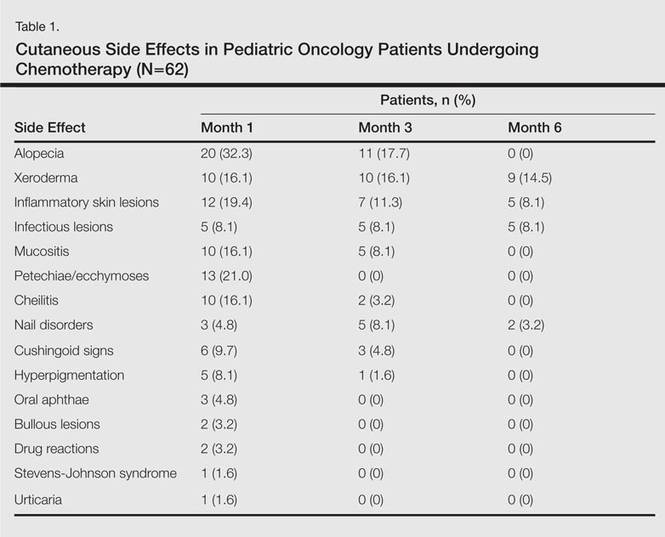
In addition to chemotherapeutic agents, 7 (11.3%) patients in this study also received antibiotics and 3 (4.8%) received antivirals. The most frequently employed chemotherapeutic agents were vincristine, methotrexate, cytarabine, etoposide, and dexamethasone. Cyclophosphamide, doxorubicin, ifosfamide, asparaginase, carboplatin, procarbazine, daunorubicin, actinomycin D, vinblastine, cisplatin, bleomycin, idarubicin, 6-mercaptopurine, temozolamide, and cyclosporine also were administered. The most commonly encountered dermatological side effects were alopecia, xeroderma, inflammatory skin lesions, infectious lesions, and mucositis, respectively (Table 1). Cutaneous side effects were frequently seen at months 1 and 3 of treatment.
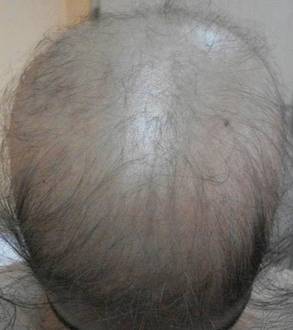
The most commonly encountered dermatologic side effect was alopecia (31/62 [50%]). Anagen effluvium (Figure 1) was detected in half of the cases, while complete scalp hair loss was noted in the rest. Alopecia was encountered more commonly in cases with central nervous system tumors (5/9 [55.6%]) and hematological malignancies (16/31 [51.6%])(Table 2).
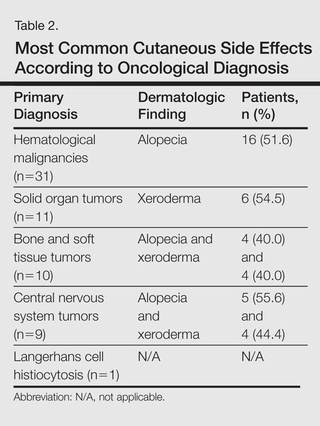
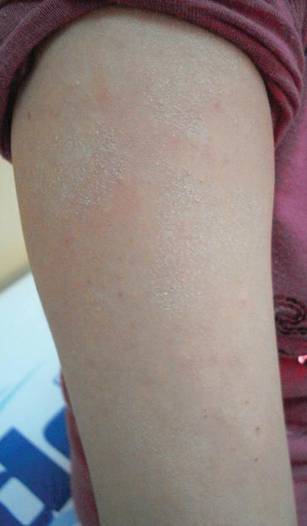
The second most commonly encountered side effect was xeroderma (29/62 [46.8%])(Figure 2). This side effect was most commonly encountered in patients with solid organ tumors (6/11 [54.5%]) and central nervous system tumors (4/9 [44.4%]), and occurred less frequently with bone and soft tissue tumors (4/10 [40.0%]).
Findings of eczema accounted for the majority of inflammatory lesions, which were the third most commonly encountered side effects. Among 24 cases of inflammatory skin lesions, 8 patients (33.3%) had diaper dermatitis, 7 (29.2%) had asteatotic eczema, 6 (25.0%) had contact dermatitis, and 3 (12.5%) had seborrheic dermatitis. Although inflammatory skin lesions were commonly encountered in patients with hematological malignancies (14/31 [45.2%]), the difference was not statistically significant.
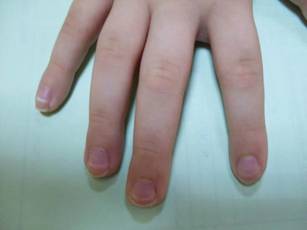
Mucositis and oral aphthous lesions were observed in 15 (24.2%) and 3 (4.8%) patients, respectively. Nail signs were noted in 10 (16.1%) patients; 4 patients had transverse streaks on the nail plates, 3 had linear streaks, 2 had nail plate fragility, and 1 had increased pigmentation at the nail bed and periungual area. Figure 3 shows linear streaks on the nail plate. These side effects were most commonly encountered in patients with solid organ tumors (5/11 [45.5%]); however, the difference was not statistically significant when compared with the other diagnostic groups.
Dermatologic signs with infectious origins were detected in 15 (24.2%) patients; 2 patients had herpes labialis, 2 had verruca vulgaris, 3 had bacterial folliculitis, 1 had acute paronychia, 1 had soft tissue infection, 2 had tinea versicolor, and 4 had mucocutaneous candidiasis. Dermatologic side effects due to infectious causes were more commonly encountered in patients with bone and soft tissue tumors (4/11 [36.4%]), and the difference was statistically significant when compared with the other diagnostic groups (P=.04).
Petechiae and ecchymotic lesions were present in 13 (21.0%) patients. These side effects occurred mainly in the first month of chemotherapy, namely when patients were in the pancytopenic phase.
Comment
Variability among the oncological diagnosis and drugs used in treatment as well as increased numbers of chemotherapeutic agents available have led to many side effects and complications in pediatric oncology patients undergoing chemotherapy.1,2 Comprehensive studies regarding the cutaneous side effects of chemotherapeuticagents in cancer treatment have been conducted in adult patients. Side effects in pediatric patients have only been documented in case reports in the literature. In our study of pediatric oncology patients undergoing treatment with chemotherapy, the most commonly observed dermatologic side effect was alopecia, followed by xeroderma, inflammatory lesions, infectious lesions, mucositis, petechiae/ecchymoses, cheilitis, nail disorders, cushingoid signs, oral aphthae, bullous lesions, and drug reactions confirmed histopathologically (Table 1).
Because the common effects of chemotherapeutic agents used in cancer treatment are greatest in areas of rapidly dividing cells, the skin and skin appendages frequently are affected by these drugs.1-3 Cutaneous signs are frequently observed, especially in regions with increased mitotic activity such as the hair, mucosa, and nails.
Kamil et al1 reported that the incidence of alopecia was 64.3% (74/115) in a study of adult cancer patients who underwent chemotherapy. Chemotherapeutic agents that have commonly caused alopecia are vincristine, daunorubicin, doxorubicin, cyclophosphamide, etoposide, cytarabine, and carboplatin.1,2 In our study, alopecia was noted in 31 (50.0%) patients, especially with the use of vincristine (7/31 [22.6%]), daunorubicin (8/31 [25.8%]), doxorubicin (6/31 [19.4%]), and cyclophosphamide (10/31 [32.3%]).
Darkening of the skin and paleness accompanied the majority of cases of xeroderma in our study. Skin dryness was in an ichthyosiform appearance and was severe in 1 patient who was diagnosed with osteosarcoma. Asteatotic eczema and cheilitis were related to skin dryness. It has been reported that acquired paraneoplastic ichthyosis can develop in hematological malignancies, primarily in patients with Hodgkin lymphoma.4
The incidence of mucositis has been related to the doses of chemotherapeutic agents. Although it is a commonly encountered side effect, there is no standard treatment of mucositis; therefore, preventive care in patients undergoing chemotherapy is important. It has been reported that practicing good oral hygiene before the treatment period can decrease the incidence of mucositis.5-9 The lower incidence of mucositis in our study compared to the literature (55.6%)5 can be attributed to the lower doses of chemotherapy drugs administered to children due to their weights; they also had active oral mucosa care during chemotherapy.
Another common complication observed in our study was nail disorders. Transverse streaks commonly are encountered due to damage in the nail matrix. Other signs are increased linear streaks, longitudinal melanonychia, nail plate fragility, and onycholysis.10
Cancer patients acquire infections more frequently because of immunosuppression from chemotherapy and malignancy.11,12 In our study, cutaneous side effects with infectious causes were noted in 15 patients. Steroids, which are included in the majority of chemotherapeutic protocols, can cause cushingoid changes. Striae from rapid weight gain, acneform eruptions, hypertrichosis, and atrophy of the skin also have been observed among secondary changes to chemotherapy.1,11
Other skin signs observed in the study were acute urticaria in 1 patient (1.6%) following administration of intrathecal methotrexate; Stevens-Johnson syndrome related to voriconazole was noted in 1 (1.6%) patient.
Hyperpigmentation is a common side effect observed in oncology patients.13-15 It can be observed locally in the skin as well as the mucosa, teeth, hair, and nails, and it generally develops secondary to alkylating agents.16 Moreover, hyperpigmentation may develop in regions with occlusions (eg, electrocardiogram pads, adhesion sites of plasters), and commonly is associated with ifosfamide, etoposide, carboplatin, and cyclosporine. Although the development mechanism of hyperpigmentation related to chemotherapy drugs is not clearly known, it is thought to be due to direct toxicity, melanocyte stimulation, or postinflammatory changes.1,6,17 In our study, xeroderma was noted in some patients with hyperpigmentation; all of them had received cyclosporine and systemic steroid treatments. The other chemotherapeutics were defined as etoposide, cytarabine, dacarbazine, and ifosfamide.1 Our patients with hyperpigmentation were not taking these therapies.
Increased skin malignancies have been reported in adult cases with hematological malignancies.18 None of the patients in our study had a secondary skin malignancy, likely because we evaluated a pediatric population and the follow-up period (6 months) was too short for the development of a secondary malignancy.
Conclusion
A wide range of cutaneous side effects can be observed in pediatric oncology patients undergoing chemotherapy based on oncological diagnosis and treatment protocol. Although these side effects are not fatal, they may negatively affect morbidity and can lead to emotional distress. Knowing the possible cutaneous side effects of chemotherapy in pediatric patients and their causes is important for early diagnosis and minimal treatment.
- Kamil N, Kamil S, Ahmed SP, et al. Toxic effects of multiple anticancer drugs on skin. Pak J Pharm Sci. 2010;23:7-14.
- Alley E, Green R, Schuchter L. Cutaneous toxicities of cancer therapy. Curr Opin Oncol. 2002;14:212-216.
- Ozkan A, Apak H, Celkan T, et al. Toxic epidermal necrolysis after the use of high-dose cytosine arabinoside. Pediatr Dermatol. 2001;18:38-40.
- Rizos E, Milionis HJ, Pavlidis N, et al. Acquired ichthyosis: a paraneoplastic skin manifestation of Hodgkin’s disease. Lancet Oncol. 2002;3:727.
- Otmani N, Alami R, Hessissen L, et al. Determinants of severe oral mucositis in pediatric cancer patients: a prospective study. Int J Pediatr Dent. 2011;21:210-216.
- Mateus C, Robert C. New drugs in oncology and skin toxicity [in French]. Rev Med Interne. 2009;30:401-410.
- Manji A, Tomlinson D, Ethier MC, et al. Psychometric properties of the Oral Mucositis Daily Questionnaire for child self-report and importance of mucositis in children treated with chemotherapy. Support Care Cancer. 2012;20:1251-1258.
- Keefe DM. Mucositis management in patients with cancer. Support Cancer Ther. 2006;3:154-157.
- Raber-Durlacher JE, Elad S, Barasch A. Oral mucositis. Oral Oncol. 2010;46:452-456.
- Utas S, Kulluk P. A case of hydroxyurea-induced longitudinal melanonychia. Int J Dermatol. 2010;49:466-474.
- Ott H, Höger PH. Dermatologic manifestations of infections in pediatric cancer patients [in German]. Klin Padiatr. 2005;217(suppl 1):110-119.
- Ramphal R, Grant RM, Dzolganovski B, et al. Herpes simplex virus in febrile neutropenic children undergoing chemotherapy for cancer: a prospective cohort study. Pediatr Infect Dis J. 2007;26:700-704.
- Yaris N, Cakir M, Kalyoncu M, et al. Bleomycin induced hyperpigmentation with yolk sac tumor. Indian J Pediatr. 2007;74:505-506.
- Kleynberg RL, Sofi AA, Chaudhary RT. Hand-foot hyperpigmentation skin lesions associated with combination gemcitabine-carboplatin (GemCarbo) therapy. Am J Ther. 2011;18:261-263.
- Blaya M, Saba N. Chemotherapy-induced hyperpigmentation of the tongue. N Engl J Med. 2011;365:e20.
- Anandajeya WV, Corrêa ZM, Augsburger JJ. Primary acquired melanosis with atypia treated with mitomycin C. Int Ophthalmol. 2009;29:285-288.
- Torres C, Wong L, Welsh O, et al. Skin manifestations associated with chemotherapy in children with hematologic malignancies. Pediatr Dermatol. 2011;2:123-147.
- Mays SR, Cohen PR. Emerging dermatologic issues in the oncology patient. Semin Cutan Med Surg. 2006;25:179-189.
Pediatric oncology patients can present with various skin lesions related to both their primary disease and immunosuppressive treatments. In the majority of cases, cutaneous findings are associated with the use of chemotherapeutic agents. The toxic effects of chemotherapeutic agents, which generally are associated with treatment of solid organ malignancies (eg, liver, kidneys), can be detected by oncologists using clinical signs and laboratory tests.1-3 However, it also is important for dermatologists to recognize and evaluate cutaneous side effects associated with chemotherapeutic agents. Reports in the literature of cutaneous side effects of chemotherapy in pediatric patients generally are limited to case studies. This study aimed to evaluate the characteristics of cutaneous side effects of chemotherapy in pediatric oncology patients.
Materials and Methods
The study was performed through the collaboration of the departments of dermatology and venereology and pediatric oncology in the Faculty of Medicine at Ege University, Izmir, Turkey. Sixty-five pediatric oncology patients who were scheduled to undergo chemotherapy from May 2011 to May 2013 were included in the study. Clinical examination of dermatologic findings was conducted at baseline (prior to beginning chemotherapy) and at months 1, 3, and 6 of treatment. Patients were examined a total of 4 times during the study. Patients with a history of skin disease prior to diagnosis of their malignancy were excluded, as the study aimed to evaluate cutaneous side effects of chemotherapy. Patients who developed cutaneous side effects during the study period were photographed. Skin biopsy was performed to confirm clinical diagnosis. Patients were split into 5 groups according to oncological diagnoses, including hematological malignancies, solid organ tumors, bone and soft tissue tumors, central nervous system tumors, and Langerhans cell histiocytosis. Data regarding age, gender, treatments administered (ie, chemotherapeutics, antibiotics, antifungals, antivirals), and dermatologic signs were recorded. Mucocutaneous findings were classified as infectious (viral, bacterial, fungal) lesions, bullous lesions, inflammatory dermatoses (eg, diaper dermatitis, asteatotic eczema, contact dermatitis, seborrheic dermatitis), xeroderma, petechiae/ecchymoses, nail signs, alopecia, mucositis, cheilitis, oral aphthae, drug reactions confirmed by histopathology, cushingoid signs (eg, striae, acneform eruption, hypertrichosis), and cutaneous hyperpigmentation.
Statistical analysis was performed using SPSS version 15.0 and χ2 test was applied to the analysis.
Results
Of 65 patients, 62 completed the study and were included in the analysis. Three patients were excluded from the results, as 2 patients died during treatment and 1 patient withdrew from the study prior to completion. Twenty-seven (43.5%) patients were female and 35 (56.5%) were male ranging in age from 1 to 17 years (mean age, 8.14 years; median age [standard deviation], 7.25 [5.42] years). There were 31 (50%) patients in the hematological malignancies group, 11 (17.7%) in the solid organ tumors group, 10 (16.1%) in the bone and soft tissue tumors group, and 9 (14.5%) in the central nervous system tumors group; Langerhans cell histiocytosis was diagnosed in 1 (1.6%) patient. Hodgkin lymphoma made up 29.0% (n=9) of hematological malignancies. Other hematological malignancies included acute myeloblastic leukemia (n=7 [22.5%]), acute lymphoblastic leukemia (n=7 [22.5%]), T-cell lymphoma (n=5 [16.1%]), non-Hodgkin lym-phoma (n=1 [3.2%]), anaplastic giant cell lymphoma (n=1 [3.2%]), and diffuse giant cell lymphoma (n=1 [3.2%]).
In addition to chemotherapeutic agents, 7 (11.3%) patients in this study also received antibiotics and 3 (4.8%) received antivirals. The most frequently employed chemotherapeutic agents were vincristine, methotrexate, cytarabine, etoposide, and dexamethasone. Cyclophosphamide, doxorubicin, ifosfamide, asparaginase, carboplatin, procarbazine, daunorubicin, actinomycin D, vinblastine, cisplatin, bleomycin, idarubicin, 6-mercaptopurine, temozolamide, and cyclosporine also were administered. The most commonly encountered dermatological side effects were alopecia, xeroderma, inflammatory skin lesions, infectious lesions, and mucositis, respectively (Table 1). Cutaneous side effects were frequently seen at months 1 and 3 of treatment.
The most commonly encountered dermatologic side effect was alopecia (31/62 [50%]). Anagen effluvium (Figure 1) was detected in half of the cases, while complete scalp hair loss was noted in the rest. Alopecia was encountered more commonly in cases with central nervous system tumors (5/9 [55.6%]) and hematological malignancies (16/31 [51.6%])(Table 2).
The second most commonly encountered side effect was xeroderma (29/62 [46.8%])(Figure 2). This side effect was most commonly encountered in patients with solid organ tumors (6/11 [54.5%]) and central nervous system tumors (4/9 [44.4%]), and occurred less frequently with bone and soft tissue tumors (4/10 [40.0%]).
Findings of eczema accounted for the majority of inflammatory lesions, which were the third most commonly encountered side effects. Among 24 cases of inflammatory skin lesions, 8 patients (33.3%) had diaper dermatitis, 7 (29.2%) had asteatotic eczema, 6 (25.0%) had contact dermatitis, and 3 (12.5%) had seborrheic dermatitis. Although inflammatory skin lesions were commonly encountered in patients with hematological malignancies (14/31 [45.2%]), the difference was not statistically significant.
Mucositis and oral aphthous lesions were observed in 15 (24.2%) and 3 (4.8%) patients, respectively. Nail signs were noted in 10 (16.1%) patients; 4 patients had transverse streaks on the nail plates, 3 had linear streaks, 2 had nail plate fragility, and 1 had increased pigmentation at the nail bed and periungual area. Figure 3 shows linear streaks on the nail plate. These side effects were most commonly encountered in patients with solid organ tumors (5/11 [45.5%]); however, the difference was not statistically significant when compared with the other diagnostic groups.
Dermatologic signs with infectious origins were detected in 15 (24.2%) patients; 2 patients had herpes labialis, 2 had verruca vulgaris, 3 had bacterial folliculitis, 1 had acute paronychia, 1 had soft tissue infection, 2 had tinea versicolor, and 4 had mucocutaneous candidiasis. Dermatologic side effects due to infectious causes were more commonly encountered in patients with bone and soft tissue tumors (4/11 [36.4%]), and the difference was statistically significant when compared with the other diagnostic groups (P=.04).
Petechiae and ecchymotic lesions were present in 13 (21.0%) patients. These side effects occurred mainly in the first month of chemotherapy, namely when patients were in the pancytopenic phase.
Comment
Variability among the oncological diagnosis and drugs used in treatment as well as increased numbers of chemotherapeutic agents available have led to many side effects and complications in pediatric oncology patients undergoing chemotherapy.1,2 Comprehensive studies regarding the cutaneous side effects of chemotherapeuticagents in cancer treatment have been conducted in adult patients. Side effects in pediatric patients have only been documented in case reports in the literature. In our study of pediatric oncology patients undergoing treatment with chemotherapy, the most commonly observed dermatologic side effect was alopecia, followed by xeroderma, inflammatory lesions, infectious lesions, mucositis, petechiae/ecchymoses, cheilitis, nail disorders, cushingoid signs, oral aphthae, bullous lesions, and drug reactions confirmed histopathologically (Table 1).
Because the common effects of chemotherapeutic agents used in cancer treatment are greatest in areas of rapidly dividing cells, the skin and skin appendages frequently are affected by these drugs.1-3 Cutaneous signs are frequently observed, especially in regions with increased mitotic activity such as the hair, mucosa, and nails.
Kamil et al1 reported that the incidence of alopecia was 64.3% (74/115) in a study of adult cancer patients who underwent chemotherapy. Chemotherapeutic agents that have commonly caused alopecia are vincristine, daunorubicin, doxorubicin, cyclophosphamide, etoposide, cytarabine, and carboplatin.1,2 In our study, alopecia was noted in 31 (50.0%) patients, especially with the use of vincristine (7/31 [22.6%]), daunorubicin (8/31 [25.8%]), doxorubicin (6/31 [19.4%]), and cyclophosphamide (10/31 [32.3%]).
Darkening of the skin and paleness accompanied the majority of cases of xeroderma in our study. Skin dryness was in an ichthyosiform appearance and was severe in 1 patient who was diagnosed with osteosarcoma. Asteatotic eczema and cheilitis were related to skin dryness. It has been reported that acquired paraneoplastic ichthyosis can develop in hematological malignancies, primarily in patients with Hodgkin lymphoma.4
The incidence of mucositis has been related to the doses of chemotherapeutic agents. Although it is a commonly encountered side effect, there is no standard treatment of mucositis; therefore, preventive care in patients undergoing chemotherapy is important. It has been reported that practicing good oral hygiene before the treatment period can decrease the incidence of mucositis.5-9 The lower incidence of mucositis in our study compared to the literature (55.6%)5 can be attributed to the lower doses of chemotherapy drugs administered to children due to their weights; they also had active oral mucosa care during chemotherapy.
Another common complication observed in our study was nail disorders. Transverse streaks commonly are encountered due to damage in the nail matrix. Other signs are increased linear streaks, longitudinal melanonychia, nail plate fragility, and onycholysis.10
Cancer patients acquire infections more frequently because of immunosuppression from chemotherapy and malignancy.11,12 In our study, cutaneous side effects with infectious causes were noted in 15 patients. Steroids, which are included in the majority of chemotherapeutic protocols, can cause cushingoid changes. Striae from rapid weight gain, acneform eruptions, hypertrichosis, and atrophy of the skin also have been observed among secondary changes to chemotherapy.1,11
Other skin signs observed in the study were acute urticaria in 1 patient (1.6%) following administration of intrathecal methotrexate; Stevens-Johnson syndrome related to voriconazole was noted in 1 (1.6%) patient.
Hyperpigmentation is a common side effect observed in oncology patients.13-15 It can be observed locally in the skin as well as the mucosa, teeth, hair, and nails, and it generally develops secondary to alkylating agents.16 Moreover, hyperpigmentation may develop in regions with occlusions (eg, electrocardiogram pads, adhesion sites of plasters), and commonly is associated with ifosfamide, etoposide, carboplatin, and cyclosporine. Although the development mechanism of hyperpigmentation related to chemotherapy drugs is not clearly known, it is thought to be due to direct toxicity, melanocyte stimulation, or postinflammatory changes.1,6,17 In our study, xeroderma was noted in some patients with hyperpigmentation; all of them had received cyclosporine and systemic steroid treatments. The other chemotherapeutics were defined as etoposide, cytarabine, dacarbazine, and ifosfamide.1 Our patients with hyperpigmentation were not taking these therapies.
Increased skin malignancies have been reported in adult cases with hematological malignancies.18 None of the patients in our study had a secondary skin malignancy, likely because we evaluated a pediatric population and the follow-up period (6 months) was too short for the development of a secondary malignancy.
Conclusion
A wide range of cutaneous side effects can be observed in pediatric oncology patients undergoing chemotherapy based on oncological diagnosis and treatment protocol. Although these side effects are not fatal, they may negatively affect morbidity and can lead to emotional distress. Knowing the possible cutaneous side effects of chemotherapy in pediatric patients and their causes is important for early diagnosis and minimal treatment.
Pediatric oncology patients can present with various skin lesions related to both their primary disease and immunosuppressive treatments. In the majority of cases, cutaneous findings are associated with the use of chemotherapeutic agents. The toxic effects of chemotherapeutic agents, which generally are associated with treatment of solid organ malignancies (eg, liver, kidneys), can be detected by oncologists using clinical signs and laboratory tests.1-3 However, it also is important for dermatologists to recognize and evaluate cutaneous side effects associated with chemotherapeutic agents. Reports in the literature of cutaneous side effects of chemotherapy in pediatric patients generally are limited to case studies. This study aimed to evaluate the characteristics of cutaneous side effects of chemotherapy in pediatric oncology patients.
Materials and Methods
The study was performed through the collaboration of the departments of dermatology and venereology and pediatric oncology in the Faculty of Medicine at Ege University, Izmir, Turkey. Sixty-five pediatric oncology patients who were scheduled to undergo chemotherapy from May 2011 to May 2013 were included in the study. Clinical examination of dermatologic findings was conducted at baseline (prior to beginning chemotherapy) and at months 1, 3, and 6 of treatment. Patients were examined a total of 4 times during the study. Patients with a history of skin disease prior to diagnosis of their malignancy were excluded, as the study aimed to evaluate cutaneous side effects of chemotherapy. Patients who developed cutaneous side effects during the study period were photographed. Skin biopsy was performed to confirm clinical diagnosis. Patients were split into 5 groups according to oncological diagnoses, including hematological malignancies, solid organ tumors, bone and soft tissue tumors, central nervous system tumors, and Langerhans cell histiocytosis. Data regarding age, gender, treatments administered (ie, chemotherapeutics, antibiotics, antifungals, antivirals), and dermatologic signs were recorded. Mucocutaneous findings were classified as infectious (viral, bacterial, fungal) lesions, bullous lesions, inflammatory dermatoses (eg, diaper dermatitis, asteatotic eczema, contact dermatitis, seborrheic dermatitis), xeroderma, petechiae/ecchymoses, nail signs, alopecia, mucositis, cheilitis, oral aphthae, drug reactions confirmed by histopathology, cushingoid signs (eg, striae, acneform eruption, hypertrichosis), and cutaneous hyperpigmentation.
Statistical analysis was performed using SPSS version 15.0 and χ2 test was applied to the analysis.
Results
Of 65 patients, 62 completed the study and were included in the analysis. Three patients were excluded from the results, as 2 patients died during treatment and 1 patient withdrew from the study prior to completion. Twenty-seven (43.5%) patients were female and 35 (56.5%) were male ranging in age from 1 to 17 years (mean age, 8.14 years; median age [standard deviation], 7.25 [5.42] years). There were 31 (50%) patients in the hematological malignancies group, 11 (17.7%) in the solid organ tumors group, 10 (16.1%) in the bone and soft tissue tumors group, and 9 (14.5%) in the central nervous system tumors group; Langerhans cell histiocytosis was diagnosed in 1 (1.6%) patient. Hodgkin lymphoma made up 29.0% (n=9) of hematological malignancies. Other hematological malignancies included acute myeloblastic leukemia (n=7 [22.5%]), acute lymphoblastic leukemia (n=7 [22.5%]), T-cell lymphoma (n=5 [16.1%]), non-Hodgkin lym-phoma (n=1 [3.2%]), anaplastic giant cell lymphoma (n=1 [3.2%]), and diffuse giant cell lymphoma (n=1 [3.2%]).
In addition to chemotherapeutic agents, 7 (11.3%) patients in this study also received antibiotics and 3 (4.8%) received antivirals. The most frequently employed chemotherapeutic agents were vincristine, methotrexate, cytarabine, etoposide, and dexamethasone. Cyclophosphamide, doxorubicin, ifosfamide, asparaginase, carboplatin, procarbazine, daunorubicin, actinomycin D, vinblastine, cisplatin, bleomycin, idarubicin, 6-mercaptopurine, temozolamide, and cyclosporine also were administered. The most commonly encountered dermatological side effects were alopecia, xeroderma, inflammatory skin lesions, infectious lesions, and mucositis, respectively (Table 1). Cutaneous side effects were frequently seen at months 1 and 3 of treatment.
The most commonly encountered dermatologic side effect was alopecia (31/62 [50%]). Anagen effluvium (Figure 1) was detected in half of the cases, while complete scalp hair loss was noted in the rest. Alopecia was encountered more commonly in cases with central nervous system tumors (5/9 [55.6%]) and hematological malignancies (16/31 [51.6%])(Table 2).
The second most commonly encountered side effect was xeroderma (29/62 [46.8%])(Figure 2). This side effect was most commonly encountered in patients with solid organ tumors (6/11 [54.5%]) and central nervous system tumors (4/9 [44.4%]), and occurred less frequently with bone and soft tissue tumors (4/10 [40.0%]).
Findings of eczema accounted for the majority of inflammatory lesions, which were the third most commonly encountered side effects. Among 24 cases of inflammatory skin lesions, 8 patients (33.3%) had diaper dermatitis, 7 (29.2%) had asteatotic eczema, 6 (25.0%) had contact dermatitis, and 3 (12.5%) had seborrheic dermatitis. Although inflammatory skin lesions were commonly encountered in patients with hematological malignancies (14/31 [45.2%]), the difference was not statistically significant.
Mucositis and oral aphthous lesions were observed in 15 (24.2%) and 3 (4.8%) patients, respectively. Nail signs were noted in 10 (16.1%) patients; 4 patients had transverse streaks on the nail plates, 3 had linear streaks, 2 had nail plate fragility, and 1 had increased pigmentation at the nail bed and periungual area. Figure 3 shows linear streaks on the nail plate. These side effects were most commonly encountered in patients with solid organ tumors (5/11 [45.5%]); however, the difference was not statistically significant when compared with the other diagnostic groups.
Dermatologic signs with infectious origins were detected in 15 (24.2%) patients; 2 patients had herpes labialis, 2 had verruca vulgaris, 3 had bacterial folliculitis, 1 had acute paronychia, 1 had soft tissue infection, 2 had tinea versicolor, and 4 had mucocutaneous candidiasis. Dermatologic side effects due to infectious causes were more commonly encountered in patients with bone and soft tissue tumors (4/11 [36.4%]), and the difference was statistically significant when compared with the other diagnostic groups (P=.04).
Petechiae and ecchymotic lesions were present in 13 (21.0%) patients. These side effects occurred mainly in the first month of chemotherapy, namely when patients were in the pancytopenic phase.
Comment
Variability among the oncological diagnosis and drugs used in treatment as well as increased numbers of chemotherapeutic agents available have led to many side effects and complications in pediatric oncology patients undergoing chemotherapy.1,2 Comprehensive studies regarding the cutaneous side effects of chemotherapeuticagents in cancer treatment have been conducted in adult patients. Side effects in pediatric patients have only been documented in case reports in the literature. In our study of pediatric oncology patients undergoing treatment with chemotherapy, the most commonly observed dermatologic side effect was alopecia, followed by xeroderma, inflammatory lesions, infectious lesions, mucositis, petechiae/ecchymoses, cheilitis, nail disorders, cushingoid signs, oral aphthae, bullous lesions, and drug reactions confirmed histopathologically (Table 1).
Because the common effects of chemotherapeutic agents used in cancer treatment are greatest in areas of rapidly dividing cells, the skin and skin appendages frequently are affected by these drugs.1-3 Cutaneous signs are frequently observed, especially in regions with increased mitotic activity such as the hair, mucosa, and nails.
Kamil et al1 reported that the incidence of alopecia was 64.3% (74/115) in a study of adult cancer patients who underwent chemotherapy. Chemotherapeutic agents that have commonly caused alopecia are vincristine, daunorubicin, doxorubicin, cyclophosphamide, etoposide, cytarabine, and carboplatin.1,2 In our study, alopecia was noted in 31 (50.0%) patients, especially with the use of vincristine (7/31 [22.6%]), daunorubicin (8/31 [25.8%]), doxorubicin (6/31 [19.4%]), and cyclophosphamide (10/31 [32.3%]).
Darkening of the skin and paleness accompanied the majority of cases of xeroderma in our study. Skin dryness was in an ichthyosiform appearance and was severe in 1 patient who was diagnosed with osteosarcoma. Asteatotic eczema and cheilitis were related to skin dryness. It has been reported that acquired paraneoplastic ichthyosis can develop in hematological malignancies, primarily in patients with Hodgkin lymphoma.4
The incidence of mucositis has been related to the doses of chemotherapeutic agents. Although it is a commonly encountered side effect, there is no standard treatment of mucositis; therefore, preventive care in patients undergoing chemotherapy is important. It has been reported that practicing good oral hygiene before the treatment period can decrease the incidence of mucositis.5-9 The lower incidence of mucositis in our study compared to the literature (55.6%)5 can be attributed to the lower doses of chemotherapy drugs administered to children due to their weights; they also had active oral mucosa care during chemotherapy.
Another common complication observed in our study was nail disorders. Transverse streaks commonly are encountered due to damage in the nail matrix. Other signs are increased linear streaks, longitudinal melanonychia, nail plate fragility, and onycholysis.10
Cancer patients acquire infections more frequently because of immunosuppression from chemotherapy and malignancy.11,12 In our study, cutaneous side effects with infectious causes were noted in 15 patients. Steroids, which are included in the majority of chemotherapeutic protocols, can cause cushingoid changes. Striae from rapid weight gain, acneform eruptions, hypertrichosis, and atrophy of the skin also have been observed among secondary changes to chemotherapy.1,11
Other skin signs observed in the study were acute urticaria in 1 patient (1.6%) following administration of intrathecal methotrexate; Stevens-Johnson syndrome related to voriconazole was noted in 1 (1.6%) patient.
Hyperpigmentation is a common side effect observed in oncology patients.13-15 It can be observed locally in the skin as well as the mucosa, teeth, hair, and nails, and it generally develops secondary to alkylating agents.16 Moreover, hyperpigmentation may develop in regions with occlusions (eg, electrocardiogram pads, adhesion sites of plasters), and commonly is associated with ifosfamide, etoposide, carboplatin, and cyclosporine. Although the development mechanism of hyperpigmentation related to chemotherapy drugs is not clearly known, it is thought to be due to direct toxicity, melanocyte stimulation, or postinflammatory changes.1,6,17 In our study, xeroderma was noted in some patients with hyperpigmentation; all of them had received cyclosporine and systemic steroid treatments. The other chemotherapeutics were defined as etoposide, cytarabine, dacarbazine, and ifosfamide.1 Our patients with hyperpigmentation were not taking these therapies.
Increased skin malignancies have been reported in adult cases with hematological malignancies.18 None of the patients in our study had a secondary skin malignancy, likely because we evaluated a pediatric population and the follow-up period (6 months) was too short for the development of a secondary malignancy.
Conclusion
A wide range of cutaneous side effects can be observed in pediatric oncology patients undergoing chemotherapy based on oncological diagnosis and treatment protocol. Although these side effects are not fatal, they may negatively affect morbidity and can lead to emotional distress. Knowing the possible cutaneous side effects of chemotherapy in pediatric patients and their causes is important for early diagnosis and minimal treatment.
- Kamil N, Kamil S, Ahmed SP, et al. Toxic effects of multiple anticancer drugs on skin. Pak J Pharm Sci. 2010;23:7-14.
- Alley E, Green R, Schuchter L. Cutaneous toxicities of cancer therapy. Curr Opin Oncol. 2002;14:212-216.
- Ozkan A, Apak H, Celkan T, et al. Toxic epidermal necrolysis after the use of high-dose cytosine arabinoside. Pediatr Dermatol. 2001;18:38-40.
- Rizos E, Milionis HJ, Pavlidis N, et al. Acquired ichthyosis: a paraneoplastic skin manifestation of Hodgkin’s disease. Lancet Oncol. 2002;3:727.
- Otmani N, Alami R, Hessissen L, et al. Determinants of severe oral mucositis in pediatric cancer patients: a prospective study. Int J Pediatr Dent. 2011;21:210-216.
- Mateus C, Robert C. New drugs in oncology and skin toxicity [in French]. Rev Med Interne. 2009;30:401-410.
- Manji A, Tomlinson D, Ethier MC, et al. Psychometric properties of the Oral Mucositis Daily Questionnaire for child self-report and importance of mucositis in children treated with chemotherapy. Support Care Cancer. 2012;20:1251-1258.
- Keefe DM. Mucositis management in patients with cancer. Support Cancer Ther. 2006;3:154-157.
- Raber-Durlacher JE, Elad S, Barasch A. Oral mucositis. Oral Oncol. 2010;46:452-456.
- Utas S, Kulluk P. A case of hydroxyurea-induced longitudinal melanonychia. Int J Dermatol. 2010;49:466-474.
- Ott H, Höger PH. Dermatologic manifestations of infections in pediatric cancer patients [in German]. Klin Padiatr. 2005;217(suppl 1):110-119.
- Ramphal R, Grant RM, Dzolganovski B, et al. Herpes simplex virus in febrile neutropenic children undergoing chemotherapy for cancer: a prospective cohort study. Pediatr Infect Dis J. 2007;26:700-704.
- Yaris N, Cakir M, Kalyoncu M, et al. Bleomycin induced hyperpigmentation with yolk sac tumor. Indian J Pediatr. 2007;74:505-506.
- Kleynberg RL, Sofi AA, Chaudhary RT. Hand-foot hyperpigmentation skin lesions associated with combination gemcitabine-carboplatin (GemCarbo) therapy. Am J Ther. 2011;18:261-263.
- Blaya M, Saba N. Chemotherapy-induced hyperpigmentation of the tongue. N Engl J Med. 2011;365:e20.
- Anandajeya WV, Corrêa ZM, Augsburger JJ. Primary acquired melanosis with atypia treated with mitomycin C. Int Ophthalmol. 2009;29:285-288.
- Torres C, Wong L, Welsh O, et al. Skin manifestations associated with chemotherapy in children with hematologic malignancies. Pediatr Dermatol. 2011;2:123-147.
- Mays SR, Cohen PR. Emerging dermatologic issues in the oncology patient. Semin Cutan Med Surg. 2006;25:179-189.
- Kamil N, Kamil S, Ahmed SP, et al. Toxic effects of multiple anticancer drugs on skin. Pak J Pharm Sci. 2010;23:7-14.
- Alley E, Green R, Schuchter L. Cutaneous toxicities of cancer therapy. Curr Opin Oncol. 2002;14:212-216.
- Ozkan A, Apak H, Celkan T, et al. Toxic epidermal necrolysis after the use of high-dose cytosine arabinoside. Pediatr Dermatol. 2001;18:38-40.
- Rizos E, Milionis HJ, Pavlidis N, et al. Acquired ichthyosis: a paraneoplastic skin manifestation of Hodgkin’s disease. Lancet Oncol. 2002;3:727.
- Otmani N, Alami R, Hessissen L, et al. Determinants of severe oral mucositis in pediatric cancer patients: a prospective study. Int J Pediatr Dent. 2011;21:210-216.
- Mateus C, Robert C. New drugs in oncology and skin toxicity [in French]. Rev Med Interne. 2009;30:401-410.
- Manji A, Tomlinson D, Ethier MC, et al. Psychometric properties of the Oral Mucositis Daily Questionnaire for child self-report and importance of mucositis in children treated with chemotherapy. Support Care Cancer. 2012;20:1251-1258.
- Keefe DM. Mucositis management in patients with cancer. Support Cancer Ther. 2006;3:154-157.
- Raber-Durlacher JE, Elad S, Barasch A. Oral mucositis. Oral Oncol. 2010;46:452-456.
- Utas S, Kulluk P. A case of hydroxyurea-induced longitudinal melanonychia. Int J Dermatol. 2010;49:466-474.
- Ott H, Höger PH. Dermatologic manifestations of infections in pediatric cancer patients [in German]. Klin Padiatr. 2005;217(suppl 1):110-119.
- Ramphal R, Grant RM, Dzolganovski B, et al. Herpes simplex virus in febrile neutropenic children undergoing chemotherapy for cancer: a prospective cohort study. Pediatr Infect Dis J. 2007;26:700-704.
- Yaris N, Cakir M, Kalyoncu M, et al. Bleomycin induced hyperpigmentation with yolk sac tumor. Indian J Pediatr. 2007;74:505-506.
- Kleynberg RL, Sofi AA, Chaudhary RT. Hand-foot hyperpigmentation skin lesions associated with combination gemcitabine-carboplatin (GemCarbo) therapy. Am J Ther. 2011;18:261-263.
- Blaya M, Saba N. Chemotherapy-induced hyperpigmentation of the tongue. N Engl J Med. 2011;365:e20.
- Anandajeya WV, Corrêa ZM, Augsburger JJ. Primary acquired melanosis with atypia treated with mitomycin C. Int Ophthalmol. 2009;29:285-288.
- Torres C, Wong L, Welsh O, et al. Skin manifestations associated with chemotherapy in children with hematologic malignancies. Pediatr Dermatol. 2011;2:123-147.
- Mays SR, Cohen PR. Emerging dermatologic issues in the oncology patient. Semin Cutan Med Surg. 2006;25:179-189.
Practice Points
- Chemotherapeutic agents can cause a variety of cutaneous side effects.
- Pediatric oncology patients should be examined regularly for cutaneous side effects of chemotherapeutics.
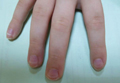
FDA approves oral anticoagulant for NVAF, VTE

Credit: FDA
The US Food and Drug Administration (FDA) has approved the oral, direct factor Xa inhibitor edoxaban (Savaysa) for use in two patient populations.
The anticoagulant is now approved to reduce the risk of stroke and systemic embolism in patients with non-valvular atrial fibrillation (NVAF) and to treat venous thromboembolism (VTE) in patients who have already received parenteral anticoagulation for 5 to 10 days.
The drug has been approved with a Boxed Warning.
The warning states that edoxaban is less effective in NVAF patients with a creatinine clearance greater than 95 mL/min. Patients with creatinine clearance above this limit have an increased risk of stroke if they receive edoxaban (compared to the risk with warfarin), so these patients should not receive edoxaban.
The warning also states that premature discontinuation of edoxaban increases the risk of stroke. Furthermore, spinal or epidural hematomas may occur in patients on edoxaban who are receiving anesthesia injected around the spine or undergoing spinal puncture.
Edoxaban for VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with deep vein thrombosis and 3319 with pulmonary embolism. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
Edoxaban proved superior when it came to the primary safety outcome. Clinically relevant bleeding occurred in 8.5% of edoxaban-treated patients and 10.3% of warfarin-treated patients (P=0.004 for superiority).
In the edoxaban arm, there were 2 fatal bleeds and 13 non-fatal bleeds in a critical site. With warfarin, there were 10 fatal bleeds and 25 non-fatal bleeds in a critical site.
Edoxaban in NVAF
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin for the prevention of stroke or systemic embolic events (SEE) in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or SEE was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
Annualized rates for the secondary composite endpoint of stroke, SEE, and cardiovascular death were 4.43% with warfarin, 3.85% with edoxaban at 60 mg (P=0.005), and 4.23% with edoxaban at 30 mg (P=0.32).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001).
Edoxaban is under development by Daiichi Sankyo Co., Ltd. ![]()
Credit: FDA
The US Food and Drug Administration (FDA) has approved the oral, direct factor Xa inhibitor edoxaban (Savaysa) for use in two patient populations.
The anticoagulant is now approved to reduce the risk of stroke and systemic embolism in patients with non-valvular atrial fibrillation (NVAF) and to treat venous thromboembolism (VTE) in patients who have already received parenteral anticoagulation for 5 to 10 days.
The drug has been approved with a Boxed Warning.
The warning states that edoxaban is less effective in NVAF patients with a creatinine clearance greater than 95 mL/min. Patients with creatinine clearance above this limit have an increased risk of stroke if they receive edoxaban (compared to the risk with warfarin), so these patients should not receive edoxaban.
The warning also states that premature discontinuation of edoxaban increases the risk of stroke. Furthermore, spinal or epidural hematomas may occur in patients on edoxaban who are receiving anesthesia injected around the spine or undergoing spinal puncture.
Edoxaban for VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with deep vein thrombosis and 3319 with pulmonary embolism. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
Edoxaban proved superior when it came to the primary safety outcome. Clinically relevant bleeding occurred in 8.5% of edoxaban-treated patients and 10.3% of warfarin-treated patients (P=0.004 for superiority).
In the edoxaban arm, there were 2 fatal bleeds and 13 non-fatal bleeds in a critical site. With warfarin, there were 10 fatal bleeds and 25 non-fatal bleeds in a critical site.
Edoxaban in NVAF
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin for the prevention of stroke or systemic embolic events (SEE) in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or SEE was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
Annualized rates for the secondary composite endpoint of stroke, SEE, and cardiovascular death were 4.43% with warfarin, 3.85% with edoxaban at 60 mg (P=0.005), and 4.23% with edoxaban at 30 mg (P=0.32).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001).
Edoxaban is under development by Daiichi Sankyo Co., Ltd. ![]()
Credit: FDA
The US Food and Drug Administration (FDA) has approved the oral, direct factor Xa inhibitor edoxaban (Savaysa) for use in two patient populations.
The anticoagulant is now approved to reduce the risk of stroke and systemic embolism in patients with non-valvular atrial fibrillation (NVAF) and to treat venous thromboembolism (VTE) in patients who have already received parenteral anticoagulation for 5 to 10 days.
The drug has been approved with a Boxed Warning.
The warning states that edoxaban is less effective in NVAF patients with a creatinine clearance greater than 95 mL/min. Patients with creatinine clearance above this limit have an increased risk of stroke if they receive edoxaban (compared to the risk with warfarin), so these patients should not receive edoxaban.
The warning also states that premature discontinuation of edoxaban increases the risk of stroke. Furthermore, spinal or epidural hematomas may occur in patients on edoxaban who are receiving anesthesia injected around the spine or undergoing spinal puncture.
Edoxaban for VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with deep vein thrombosis and 3319 with pulmonary embolism. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
Edoxaban proved superior when it came to the primary safety outcome. Clinically relevant bleeding occurred in 8.5% of edoxaban-treated patients and 10.3% of warfarin-treated patients (P=0.004 for superiority).
In the edoxaban arm, there were 2 fatal bleeds and 13 non-fatal bleeds in a critical site. With warfarin, there were 10 fatal bleeds and 25 non-fatal bleeds in a critical site.
Edoxaban in NVAF
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin for the prevention of stroke or systemic embolic events (SEE) in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or SEE was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
Annualized rates for the secondary composite endpoint of stroke, SEE, and cardiovascular death were 4.43% with warfarin, 3.85% with edoxaban at 60 mg (P=0.005), and 4.23% with edoxaban at 30 mg (P=0.32).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001).
Edoxaban is under development by Daiichi Sankyo Co., Ltd. ![]()
Product gets fast track designation for CTCL

mycosis fungoides
The US Food and Drug Administration (FDA) has granted fast track designation to SGX301 as a first-line treatment for cutaneous T-cell lymphoma (CTCL).
SGX301 is a photodynamic therapy utilizing safe, visible light for activation. The active ingredient in SGX301 is synthetic hypericin, a photosensitizer that is applied to skin lesions and then activated by fluorescent light 16 to 24 hours later.
Combined with photoactivation, hypericin has demonstrated significant antiproliferative effects on activated, normal human lymphoid cells and inhibited the growth of malignant T cells isolated from CTCL patients. Topical hypericin has also proven safe in a phase 1 study of healthy volunteers.
In a phase 2 trial of patients with CTCL (mycosis fungoides only) or psoriasis, topical hypericin conferred a significant improvement over placebo. Among CTCL patients, the treatment prompted a response rate of 58.3%, compared to an 8.3% response rate for placebo (P≤0.04).
Topical hypericin was also well tolerated in this trial. There were no deaths or serious adverse events related to the treatment. However, there were reports of mild to moderate burning, itching, erythema, and pruritus at the application site.
A phase 3 trial of SGX301 is set to begin in the first half of this year. In addition to its new fast track status, SGX301 also has orphan designation from the FDA.
About fast track designation
The FDA grants fast track designation to a drug that is intended to treat a serious or life-threatening condition and that demonstrates the potential to address an unmet medical need for the condition.
Fast track designation is designed to facilitate the development and expedite the review of new drugs. For instance, Soligenix, Inc., the company developing SGX301, is eligible to submit a new drug application (NDA) for SGX301 on a rolling basis, allowing the FDA to review sections of the NDA prior to receiving the complete submission.
Additionally, NDAs for fast track development programs ordinarily will be eligible for priority review, which imparts an abbreviated review time of approximately 6 months. ![]()
mycosis fungoides
The US Food and Drug Administration (FDA) has granted fast track designation to SGX301 as a first-line treatment for cutaneous T-cell lymphoma (CTCL).
SGX301 is a photodynamic therapy utilizing safe, visible light for activation. The active ingredient in SGX301 is synthetic hypericin, a photosensitizer that is applied to skin lesions and then activated by fluorescent light 16 to 24 hours later.
Combined with photoactivation, hypericin has demonstrated significant antiproliferative effects on activated, normal human lymphoid cells and inhibited the growth of malignant T cells isolated from CTCL patients. Topical hypericin has also proven safe in a phase 1 study of healthy volunteers.
In a phase 2 trial of patients with CTCL (mycosis fungoides only) or psoriasis, topical hypericin conferred a significant improvement over placebo. Among CTCL patients, the treatment prompted a response rate of 58.3%, compared to an 8.3% response rate for placebo (P≤0.04).
Topical hypericin was also well tolerated in this trial. There were no deaths or serious adverse events related to the treatment. However, there were reports of mild to moderate burning, itching, erythema, and pruritus at the application site.
A phase 3 trial of SGX301 is set to begin in the first half of this year. In addition to its new fast track status, SGX301 also has orphan designation from the FDA.
About fast track designation
The FDA grants fast track designation to a drug that is intended to treat a serious or life-threatening condition and that demonstrates the potential to address an unmet medical need for the condition.
Fast track designation is designed to facilitate the development and expedite the review of new drugs. For instance, Soligenix, Inc., the company developing SGX301, is eligible to submit a new drug application (NDA) for SGX301 on a rolling basis, allowing the FDA to review sections of the NDA prior to receiving the complete submission.
Additionally, NDAs for fast track development programs ordinarily will be eligible for priority review, which imparts an abbreviated review time of approximately 6 months. ![]()
mycosis fungoides
The US Food and Drug Administration (FDA) has granted fast track designation to SGX301 as a first-line treatment for cutaneous T-cell lymphoma (CTCL).
SGX301 is a photodynamic therapy utilizing safe, visible light for activation. The active ingredient in SGX301 is synthetic hypericin, a photosensitizer that is applied to skin lesions and then activated by fluorescent light 16 to 24 hours later.
Combined with photoactivation, hypericin has demonstrated significant antiproliferative effects on activated, normal human lymphoid cells and inhibited the growth of malignant T cells isolated from CTCL patients. Topical hypericin has also proven safe in a phase 1 study of healthy volunteers.
In a phase 2 trial of patients with CTCL (mycosis fungoides only) or psoriasis, topical hypericin conferred a significant improvement over placebo. Among CTCL patients, the treatment prompted a response rate of 58.3%, compared to an 8.3% response rate for placebo (P≤0.04).
Topical hypericin was also well tolerated in this trial. There were no deaths or serious adverse events related to the treatment. However, there were reports of mild to moderate burning, itching, erythema, and pruritus at the application site.
A phase 3 trial of SGX301 is set to begin in the first half of this year. In addition to its new fast track status, SGX301 also has orphan designation from the FDA.
About fast track designation
The FDA grants fast track designation to a drug that is intended to treat a serious or life-threatening condition and that demonstrates the potential to address an unmet medical need for the condition.
Fast track designation is designed to facilitate the development and expedite the review of new drugs. For instance, Soligenix, Inc., the company developing SGX301, is eligible to submit a new drug application (NDA) for SGX301 on a rolling basis, allowing the FDA to review sections of the NDA prior to receiving the complete submission.
Additionally, NDAs for fast track development programs ordinarily will be eligible for priority review, which imparts an abbreviated review time of approximately 6 months. ![]()
Drug granted orphan designation for MM

The US Food and Drug Administration (FDA) has granted selinexor (KPT-330) orphan drug designation to treat multiple myeloma (MM).
Selinexor already has orphan designation from the FDA to treat acute myeloid leukemia (AML) and diffuse large B-cell lymphoma (DLBCL).
The drug has also received orphan designation from the European Medicines Agency (EMA) to treat MM, AML, DLBCL, and chronic lymphocytic leukemia/small lymphocytic lymphoma, including Richter’s transformation.
“Orphan drug designation by the FDA for multiple myeloma is another significant milestone in the selinexor development program,” said Sharon Shacham, PhD, President and Chief Scientific Officer of Karyopharm Therapeutics, Inc., the company developing selinexor.
In the US, orphan designation qualifies a company for certain benefits, including an accelerated approval process, 7 years of market exclusivity following the drug’s approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
About selinexor
Selinexor (KPT-330) is a first-in-class, oral, selective inhibitor of nuclear export compound. The drug functions by inhibiting the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which subsequently reinitiates and amplifies their tumor suppressor function. This is thought to prompt apoptosis in cancer cells while largely sparing normal cells.
Selinexor combos in MM
In a poster presented at the 2014 ASH Annual Meeting (4773), researchers reported results observed with selinexor plus dexamethasone in preclinical models and in patients with heavily pretreated, refractory MM.
The study included 9 evaluable patients who received selinexor at 45 mg/m2 twice weekly and dexamethasone at 20 mg twice weekly. The combination prompted an overall response rate of 67%, with one stringent complete response (11%) and 5 partial responses (56%), as well as a clinical benefit rate of 89%.
The combination demonstrated a reduction in nausea grades and very little weight loss compared with selinexor alone. The most common grade 1/2 adverse events were nausea, fatigue, anorexia, and vomiting.
The combination was also associated with an increase in time on study relative to selinexor alone. Sixty-six percent of patients remained on study for at least 16 weeks, including one patient for 28 weeks and one for 43 weeks as of December 1, 2014.
During the dose-evaluation part of the study, the 60 mg/m2 selinexor dose was deemed intolerable in this heavily pretreated patient population. So 45 mg/m2 is the recommended future study dose.
In another poster presented at the 2014 ASH Annual Meeting (3443), researchers described the activity of selinexor in combination with carfilzomib. This preclinical study revealed a novel, intracellular, membrane-embedded mechanism of caspase activation.
The results suggested a model of synergy wherein the selinexor-carfilzomib combination promotes caspase activation, likely by induced proximity, cleavage of other caspases, and subsequent apoptosis as well as autophagy. ![]()
The US Food and Drug Administration (FDA) has granted selinexor (KPT-330) orphan drug designation to treat multiple myeloma (MM).
Selinexor already has orphan designation from the FDA to treat acute myeloid leukemia (AML) and diffuse large B-cell lymphoma (DLBCL).
The drug has also received orphan designation from the European Medicines Agency (EMA) to treat MM, AML, DLBCL, and chronic lymphocytic leukemia/small lymphocytic lymphoma, including Richter’s transformation.
“Orphan drug designation by the FDA for multiple myeloma is another significant milestone in the selinexor development program,” said Sharon Shacham, PhD, President and Chief Scientific Officer of Karyopharm Therapeutics, Inc., the company developing selinexor.
In the US, orphan designation qualifies a company for certain benefits, including an accelerated approval process, 7 years of market exclusivity following the drug’s approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
About selinexor
Selinexor (KPT-330) is a first-in-class, oral, selective inhibitor of nuclear export compound. The drug functions by inhibiting the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which subsequently reinitiates and amplifies their tumor suppressor function. This is thought to prompt apoptosis in cancer cells while largely sparing normal cells.
Selinexor combos in MM
In a poster presented at the 2014 ASH Annual Meeting (4773), researchers reported results observed with selinexor plus dexamethasone in preclinical models and in patients with heavily pretreated, refractory MM.
The study included 9 evaluable patients who received selinexor at 45 mg/m2 twice weekly and dexamethasone at 20 mg twice weekly. The combination prompted an overall response rate of 67%, with one stringent complete response (11%) and 5 partial responses (56%), as well as a clinical benefit rate of 89%.
The combination demonstrated a reduction in nausea grades and very little weight loss compared with selinexor alone. The most common grade 1/2 adverse events were nausea, fatigue, anorexia, and vomiting.
The combination was also associated with an increase in time on study relative to selinexor alone. Sixty-six percent of patients remained on study for at least 16 weeks, including one patient for 28 weeks and one for 43 weeks as of December 1, 2014.
During the dose-evaluation part of the study, the 60 mg/m2 selinexor dose was deemed intolerable in this heavily pretreated patient population. So 45 mg/m2 is the recommended future study dose.
In another poster presented at the 2014 ASH Annual Meeting (3443), researchers described the activity of selinexor in combination with carfilzomib. This preclinical study revealed a novel, intracellular, membrane-embedded mechanism of caspase activation.
The results suggested a model of synergy wherein the selinexor-carfilzomib combination promotes caspase activation, likely by induced proximity, cleavage of other caspases, and subsequent apoptosis as well as autophagy. ![]()
The US Food and Drug Administration (FDA) has granted selinexor (KPT-330) orphan drug designation to treat multiple myeloma (MM).
Selinexor already has orphan designation from the FDA to treat acute myeloid leukemia (AML) and diffuse large B-cell lymphoma (DLBCL).
The drug has also received orphan designation from the European Medicines Agency (EMA) to treat MM, AML, DLBCL, and chronic lymphocytic leukemia/small lymphocytic lymphoma, including Richter’s transformation.
“Orphan drug designation by the FDA for multiple myeloma is another significant milestone in the selinexor development program,” said Sharon Shacham, PhD, President and Chief Scientific Officer of Karyopharm Therapeutics, Inc., the company developing selinexor.
In the US, orphan designation qualifies a company for certain benefits, including an accelerated approval process, 7 years of market exclusivity following the drug’s approval, tax credits on US clinical trials, eligibility for orphan drug grants, and a waiver of certain administrative fees.
About selinexor
Selinexor (KPT-330) is a first-in-class, oral, selective inhibitor of nuclear export compound. The drug functions by inhibiting the nuclear export protein XPO1 (also called CRM1).
This leads to the accumulation of tumor suppressor proteins in the cell nucleus, which subsequently reinitiates and amplifies their tumor suppressor function. This is thought to prompt apoptosis in cancer cells while largely sparing normal cells.
Selinexor combos in MM
In a poster presented at the 2014 ASH Annual Meeting (4773), researchers reported results observed with selinexor plus dexamethasone in preclinical models and in patients with heavily pretreated, refractory MM.
The study included 9 evaluable patients who received selinexor at 45 mg/m2 twice weekly and dexamethasone at 20 mg twice weekly. The combination prompted an overall response rate of 67%, with one stringent complete response (11%) and 5 partial responses (56%), as well as a clinical benefit rate of 89%.
The combination demonstrated a reduction in nausea grades and very little weight loss compared with selinexor alone. The most common grade 1/2 adverse events were nausea, fatigue, anorexia, and vomiting.
The combination was also associated with an increase in time on study relative to selinexor alone. Sixty-six percent of patients remained on study for at least 16 weeks, including one patient for 28 weeks and one for 43 weeks as of December 1, 2014.
During the dose-evaluation part of the study, the 60 mg/m2 selinexor dose was deemed intolerable in this heavily pretreated patient population. So 45 mg/m2 is the recommended future study dose.
In another poster presented at the 2014 ASH Annual Meeting (3443), researchers described the activity of selinexor in combination with carfilzomib. This preclinical study revealed a novel, intracellular, membrane-embedded mechanism of caspase activation.
The results suggested a model of synergy wherein the selinexor-carfilzomib combination promotes caspase activation, likely by induced proximity, cleavage of other caspases, and subsequent apoptosis as well as autophagy. ![]()
Study raises questions about exchange transfusion

Credit: Vera Kratochvil
Results of a new study indicate that current guidelines for exchange transfusions in infants can successfully prevent kernicterus, a rare and life-threatening type of cerebral palsy triggered by escalating bilirubin that injures the brain.
However, the research also showed that only infants whose levels of bilirubin were well above the level for exchange transfusion actually developed kernicterus. And those infants all had additional risk factors for brain damage.
This suggests perhaps the threshold for exchange transfusion could safely be raised for infants with high bilirubin levels who have no other risk factors for brain injury, according to Yvonne W. Wu, MD, of the University of California, San Francisco (UCSF).
Dr Wu and her colleagues evaluated the health records of two groups of infants selected from 525,409 births. The children had been born at 15 hospitals within the Kaiser Permanente Northern California region from 1995 through 2011.
One group comprised 1833 newborns with levels of bilirubin above the level at which the American Academy of Pediatrics (AAP) recommends exchange transfusions.
The second group was made up of 104,716 randomly sampled newborns, born at at least 35 weeks’ gestation with lower levels of bilirubin. The two groups were followed for an average of 7 and 6 years, respectively.
The researchers confirmed 3 cases of kernicterus based on the brain MRIs of children with cerebral palsy. All 3 cases had occurred in newborns with the highest levels of bilirubin. But further study revealed that each child had 2 or more risk factors for brain damage.
”We found that cerebral palsy consistent with kernicterus did not occur in a single infant with high bilirubin without the presence of additional risk factors for neurotoxicity, such as prematurity, sepsis, and the hereditary blood disorder G6PD deficiency,” said Michael W. Kuzniewicz, MD, of UCSF. “This was the case even in infants with very high bilirubin.”
In 2004, the AAP published a guideline for treating infants whose bilirubin remained high despite phototherapy. It recommended exchange transfusions based on the level of bilirubin, the age of the infant, and other risk factors for brain damage.
“Our study was the first to evaluate how well the exchange transfusion guidelines predicted risk of cerebral palsy and kernicterus in babies with jaundice,” said Thomas B. Newman, MD, of UCSF.
“It was reassuring that brain injury due to high bilirubin was rare and that only those infants whose levels were well above exchange transfusion guidelines developed kernicterus.”
“Based on our study, the current guidelines for when to perform exchange transfusions have been quite successful in preventing kernicterus,” Dr Wu added. “However, our study also raises the question whether the threshold for exchange transfusion could be higher for infants with high bilirubin levels who are otherwise healthy and who have no other risk factors for brain injury.”
This is especially important, she noted, because exchange transfusions pose risks such as blood clot formation, blood pressure instability, bleeding, and changes in blood chemistry.
Dr Wu and her colleagues described this research in JAMA Pediatrics. ![]()
Credit: Vera Kratochvil
Results of a new study indicate that current guidelines for exchange transfusions in infants can successfully prevent kernicterus, a rare and life-threatening type of cerebral palsy triggered by escalating bilirubin that injures the brain.
However, the research also showed that only infants whose levels of bilirubin were well above the level for exchange transfusion actually developed kernicterus. And those infants all had additional risk factors for brain damage.
This suggests perhaps the threshold for exchange transfusion could safely be raised for infants with high bilirubin levels who have no other risk factors for brain injury, according to Yvonne W. Wu, MD, of the University of California, San Francisco (UCSF).
Dr Wu and her colleagues evaluated the health records of two groups of infants selected from 525,409 births. The children had been born at 15 hospitals within the Kaiser Permanente Northern California region from 1995 through 2011.
One group comprised 1833 newborns with levels of bilirubin above the level at which the American Academy of Pediatrics (AAP) recommends exchange transfusions.
The second group was made up of 104,716 randomly sampled newborns, born at at least 35 weeks’ gestation with lower levels of bilirubin. The two groups were followed for an average of 7 and 6 years, respectively.
The researchers confirmed 3 cases of kernicterus based on the brain MRIs of children with cerebral palsy. All 3 cases had occurred in newborns with the highest levels of bilirubin. But further study revealed that each child had 2 or more risk factors for brain damage.
”We found that cerebral palsy consistent with kernicterus did not occur in a single infant with high bilirubin without the presence of additional risk factors for neurotoxicity, such as prematurity, sepsis, and the hereditary blood disorder G6PD deficiency,” said Michael W. Kuzniewicz, MD, of UCSF. “This was the case even in infants with very high bilirubin.”
In 2004, the AAP published a guideline for treating infants whose bilirubin remained high despite phototherapy. It recommended exchange transfusions based on the level of bilirubin, the age of the infant, and other risk factors for brain damage.
“Our study was the first to evaluate how well the exchange transfusion guidelines predicted risk of cerebral palsy and kernicterus in babies with jaundice,” said Thomas B. Newman, MD, of UCSF.
“It was reassuring that brain injury due to high bilirubin was rare and that only those infants whose levels were well above exchange transfusion guidelines developed kernicterus.”
“Based on our study, the current guidelines for when to perform exchange transfusions have been quite successful in preventing kernicterus,” Dr Wu added. “However, our study also raises the question whether the threshold for exchange transfusion could be higher for infants with high bilirubin levels who are otherwise healthy and who have no other risk factors for brain injury.”
This is especially important, she noted, because exchange transfusions pose risks such as blood clot formation, blood pressure instability, bleeding, and changes in blood chemistry.
Dr Wu and her colleagues described this research in JAMA Pediatrics. ![]()
Credit: Vera Kratochvil
Results of a new study indicate that current guidelines for exchange transfusions in infants can successfully prevent kernicterus, a rare and life-threatening type of cerebral palsy triggered by escalating bilirubin that injures the brain.
However, the research also showed that only infants whose levels of bilirubin were well above the level for exchange transfusion actually developed kernicterus. And those infants all had additional risk factors for brain damage.
This suggests perhaps the threshold for exchange transfusion could safely be raised for infants with high bilirubin levels who have no other risk factors for brain injury, according to Yvonne W. Wu, MD, of the University of California, San Francisco (UCSF).
Dr Wu and her colleagues evaluated the health records of two groups of infants selected from 525,409 births. The children had been born at 15 hospitals within the Kaiser Permanente Northern California region from 1995 through 2011.
One group comprised 1833 newborns with levels of bilirubin above the level at which the American Academy of Pediatrics (AAP) recommends exchange transfusions.
The second group was made up of 104,716 randomly sampled newborns, born at at least 35 weeks’ gestation with lower levels of bilirubin. The two groups were followed for an average of 7 and 6 years, respectively.
The researchers confirmed 3 cases of kernicterus based on the brain MRIs of children with cerebral palsy. All 3 cases had occurred in newborns with the highest levels of bilirubin. But further study revealed that each child had 2 or more risk factors for brain damage.
”We found that cerebral palsy consistent with kernicterus did not occur in a single infant with high bilirubin without the presence of additional risk factors for neurotoxicity, such as prematurity, sepsis, and the hereditary blood disorder G6PD deficiency,” said Michael W. Kuzniewicz, MD, of UCSF. “This was the case even in infants with very high bilirubin.”
In 2004, the AAP published a guideline for treating infants whose bilirubin remained high despite phototherapy. It recommended exchange transfusions based on the level of bilirubin, the age of the infant, and other risk factors for brain damage.
“Our study was the first to evaluate how well the exchange transfusion guidelines predicted risk of cerebral palsy and kernicterus in babies with jaundice,” said Thomas B. Newman, MD, of UCSF.
“It was reassuring that brain injury due to high bilirubin was rare and that only those infants whose levels were well above exchange transfusion guidelines developed kernicterus.”
“Based on our study, the current guidelines for when to perform exchange transfusions have been quite successful in preventing kernicterus,” Dr Wu added. “However, our study also raises the question whether the threshold for exchange transfusion could be higher for infants with high bilirubin levels who are otherwise healthy and who have no other risk factors for brain injury.”
This is especially important, she noted, because exchange transfusions pose risks such as blood clot formation, blood pressure instability, bleeding, and changes in blood chemistry.
Dr Wu and her colleagues described this research in JAMA Pediatrics. ![]()
Excessive Sun Damage and a “Fungal Infection”?
A scaly lesion manifested on this 55-year-old man’s back several years ago. Over time, the lesion has grown, despite the application of several different OTC creams, including hydrocortisone and various antifungal creams. Previously asymptomatic, the lesion is now beginning to itch and occasionally bleed.
Additional history-taking reveals that for the first 10 years of his working life, the patient was a concrete finisher, working 12-hour shifts in the sun without a shirt, six days a week for months at a time. Aside from a 40-pack-year history of smoking, his health is “decent.”
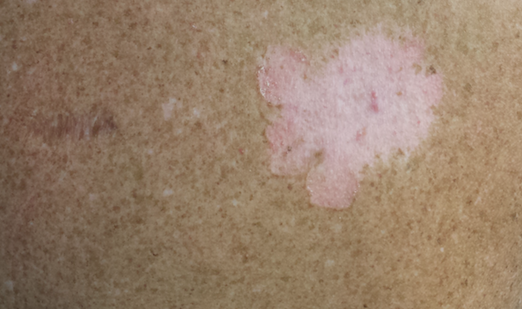
EXAMINATION
The lesion is an 8 x 10–cm pink, scaly plaque with thready, well-defined scaly margins. Tiny focal erosions are seen in the peripheral leading edge. Closer inspection of the lesion’s center reveals multiple telangiectasias and a complete lack of hairs or hair follicles. Elsewhere on sun-exposed areas of the patient’s truncal skin, there are thousands of solar lentigines. Multiple small actinic keratoses are noted on his face as well.
What is the diagnosis?
DISCUSSION
Prior to consulting dermatology, this patient had talked extensively with his primary care provider (PCP) about the large lesion on his back. The PCP was quite sure the cause was a fungal organism and assured the patient that the prescribed antifungal cream would almost certainly cure it. This is a scenario in which almost every PCP will participate at some point early in his/her career. When treatment fails, the patient is then referred to dermatology—where a totally unexpected diagnosis is made.
This case represents the natural history of superficial basal cell carcinoma (SBCC), the appearance of which is at odds with any other kind of skin cancer, including the other types of basal cell carcinoma. SBCC is especially common on the shoulders and trunks of patients with a history of excessive sun exposure. The mid-back location on this patient is quite typical, as is the annular, scaly morphology.
The cancer—which originates in a row of cells at the base of the epidermis—effectively destroys surface structures such as skin lines and hair follicles as it spreads laterally. Left in place long enough, SBCCs can eventually become focally invasive and can even spread into regional lymph nodes. Metastasis from them is extremely rare but has been reported.
Treatment is therefore necessary and can range from surgical excision of smaller lesions to electrodessication and curettage to nonsurgical options, such as the application of imiquimod creams or 5-fluorouracil cream, or even radiation therapy. This man was treated with thrice-weekly application of imiquimod 3.75% cream for at least two months, which will cause irritation and scant drainage but will likely result in a cure.
One of the main benefits of his presentation to dermatology was the opportunity to encourage and facilitate yearly skin check-ups—important, given his risk for other sun-caused skin cancers (especially melanoma).
A major learning point to be gained from this case: Just because a lesion is annular and scaly doesn’t mean it’s fungal. The fungi have to have a source (animal, child, soil), and it helps a lot if the affected area is damp and warm (eg, groin or under a bulletproof vest). Immunosuppression (from corticosteroid use, especially) helps as well.
Besides fungal infection, the differential for this type of lesion includes Bowen’s disease (intraepidermal squamous cell carcinoma), psoriasis, and eczema. The fact that the lesion was fixed (did not come and go), grew steadily, and was on the directly sun-exposed skin of a person with a history of heavy sun exposure all pointed to the diagnosis.
TAKE-HOME LEARNING POINTS
• Superficial BCC (SBCC) has also been called “field fire” skin cancer because of the prominent, well-defined scaly border that expands peripherally and leaves a “scorched earth” look behind.
• SBCCs are common on skin directly exposed to the sun, especially backs and arms.
• SBCCs do not resemble other types of BCC at all and are therefore often mistaken for “fungal infection.”
• Pay as much attention to the patient as to the lesion. There was abundant evidence of excessive sun damage on this patient’s trunk, making him an ideal patient for skin cancer.
A scaly lesion manifested on this 55-year-old man’s back several years ago. Over time, the lesion has grown, despite the application of several different OTC creams, including hydrocortisone and various antifungal creams. Previously asymptomatic, the lesion is now beginning to itch and occasionally bleed.
Additional history-taking reveals that for the first 10 years of his working life, the patient was a concrete finisher, working 12-hour shifts in the sun without a shirt, six days a week for months at a time. Aside from a 40-pack-year history of smoking, his health is “decent.”
EXAMINATION
The lesion is an 8 x 10–cm pink, scaly plaque with thready, well-defined scaly margins. Tiny focal erosions are seen in the peripheral leading edge. Closer inspection of the lesion’s center reveals multiple telangiectasias and a complete lack of hairs or hair follicles. Elsewhere on sun-exposed areas of the patient’s truncal skin, there are thousands of solar lentigines. Multiple small actinic keratoses are noted on his face as well.
What is the diagnosis?
DISCUSSION
Prior to consulting dermatology, this patient had talked extensively with his primary care provider (PCP) about the large lesion on his back. The PCP was quite sure the cause was a fungal organism and assured the patient that the prescribed antifungal cream would almost certainly cure it. This is a scenario in which almost every PCP will participate at some point early in his/her career. When treatment fails, the patient is then referred to dermatology—where a totally unexpected diagnosis is made.
This case represents the natural history of superficial basal cell carcinoma (SBCC), the appearance of which is at odds with any other kind of skin cancer, including the other types of basal cell carcinoma. SBCC is especially common on the shoulders and trunks of patients with a history of excessive sun exposure. The mid-back location on this patient is quite typical, as is the annular, scaly morphology.
The cancer—which originates in a row of cells at the base of the epidermis—effectively destroys surface structures such as skin lines and hair follicles as it spreads laterally. Left in place long enough, SBCCs can eventually become focally invasive and can even spread into regional lymph nodes. Metastasis from them is extremely rare but has been reported.
Treatment is therefore necessary and can range from surgical excision of smaller lesions to electrodessication and curettage to nonsurgical options, such as the application of imiquimod creams or 5-fluorouracil cream, or even radiation therapy. This man was treated with thrice-weekly application of imiquimod 3.75% cream for at least two months, which will cause irritation and scant drainage but will likely result in a cure.
One of the main benefits of his presentation to dermatology was the opportunity to encourage and facilitate yearly skin check-ups—important, given his risk for other sun-caused skin cancers (especially melanoma).
A major learning point to be gained from this case: Just because a lesion is annular and scaly doesn’t mean it’s fungal. The fungi have to have a source (animal, child, soil), and it helps a lot if the affected area is damp and warm (eg, groin or under a bulletproof vest). Immunosuppression (from corticosteroid use, especially) helps as well.
Besides fungal infection, the differential for this type of lesion includes Bowen’s disease (intraepidermal squamous cell carcinoma), psoriasis, and eczema. The fact that the lesion was fixed (did not come and go), grew steadily, and was on the directly sun-exposed skin of a person with a history of heavy sun exposure all pointed to the diagnosis.
TAKE-HOME LEARNING POINTS
• Superficial BCC (SBCC) has also been called “field fire” skin cancer because of the prominent, well-defined scaly border that expands peripherally and leaves a “scorched earth” look behind.
• SBCCs are common on skin directly exposed to the sun, especially backs and arms.
• SBCCs do not resemble other types of BCC at all and are therefore often mistaken for “fungal infection.”
• Pay as much attention to the patient as to the lesion. There was abundant evidence of excessive sun damage on this patient’s trunk, making him an ideal patient for skin cancer.
A scaly lesion manifested on this 55-year-old man’s back several years ago. Over time, the lesion has grown, despite the application of several different OTC creams, including hydrocortisone and various antifungal creams. Previously asymptomatic, the lesion is now beginning to itch and occasionally bleed.
Additional history-taking reveals that for the first 10 years of his working life, the patient was a concrete finisher, working 12-hour shifts in the sun without a shirt, six days a week for months at a time. Aside from a 40-pack-year history of smoking, his health is “decent.”
EXAMINATION
The lesion is an 8 x 10–cm pink, scaly plaque with thready, well-defined scaly margins. Tiny focal erosions are seen in the peripheral leading edge. Closer inspection of the lesion’s center reveals multiple telangiectasias and a complete lack of hairs or hair follicles. Elsewhere on sun-exposed areas of the patient’s truncal skin, there are thousands of solar lentigines. Multiple small actinic keratoses are noted on his face as well.
What is the diagnosis?
DISCUSSION
Prior to consulting dermatology, this patient had talked extensively with his primary care provider (PCP) about the large lesion on his back. The PCP was quite sure the cause was a fungal organism and assured the patient that the prescribed antifungal cream would almost certainly cure it. This is a scenario in which almost every PCP will participate at some point early in his/her career. When treatment fails, the patient is then referred to dermatology—where a totally unexpected diagnosis is made.
This case represents the natural history of superficial basal cell carcinoma (SBCC), the appearance of which is at odds with any other kind of skin cancer, including the other types of basal cell carcinoma. SBCC is especially common on the shoulders and trunks of patients with a history of excessive sun exposure. The mid-back location on this patient is quite typical, as is the annular, scaly morphology.
The cancer—which originates in a row of cells at the base of the epidermis—effectively destroys surface structures such as skin lines and hair follicles as it spreads laterally. Left in place long enough, SBCCs can eventually become focally invasive and can even spread into regional lymph nodes. Metastasis from them is extremely rare but has been reported.
Treatment is therefore necessary and can range from surgical excision of smaller lesions to electrodessication and curettage to nonsurgical options, such as the application of imiquimod creams or 5-fluorouracil cream, or even radiation therapy. This man was treated with thrice-weekly application of imiquimod 3.75% cream for at least two months, which will cause irritation and scant drainage but will likely result in a cure.
One of the main benefits of his presentation to dermatology was the opportunity to encourage and facilitate yearly skin check-ups—important, given his risk for other sun-caused skin cancers (especially melanoma).
A major learning point to be gained from this case: Just because a lesion is annular and scaly doesn’t mean it’s fungal. The fungi have to have a source (animal, child, soil), and it helps a lot if the affected area is damp and warm (eg, groin or under a bulletproof vest). Immunosuppression (from corticosteroid use, especially) helps as well.
Besides fungal infection, the differential for this type of lesion includes Bowen’s disease (intraepidermal squamous cell carcinoma), psoriasis, and eczema. The fact that the lesion was fixed (did not come and go), grew steadily, and was on the directly sun-exposed skin of a person with a history of heavy sun exposure all pointed to the diagnosis.
TAKE-HOME LEARNING POINTS
• Superficial BCC (SBCC) has also been called “field fire” skin cancer because of the prominent, well-defined scaly border that expands peripherally and leaves a “scorched earth” look behind.
• SBCCs are common on skin directly exposed to the sun, especially backs and arms.
• SBCCs do not resemble other types of BCC at all and are therefore often mistaken for “fungal infection.”
• Pay as much attention to the patient as to the lesion. There was abundant evidence of excessive sun damage on this patient’s trunk, making him an ideal patient for skin cancer.