User login
Transcranial Direct Current Stimulation Enhances Cognitive Training in Parkinson’s Disease
PORTLAND, OR—Combining transcranial direct current stimulation (tDCS) and cognitive training resulted in an improvement in a greater number of cognitive outcomes than either intervention alone in a small, randomized, controlled trial of patients with Parkinson’s disease and mild cognitive impairment.
Researchers at Curtin University in Perth, Australia, conducted the trial comparing the effects of standard (ie, not individualized) cognitive training (SCT), tailored (ie, individualized) cognitive training (TCT), tDCS, and a combination of tDCS with either form of cognitive training on cognitive outcomes, activities of daily living, and quality of life. Previously, it was not known whether either form of cognitive training, tDCS, or a combination of the two would be most efficacious in improving cognition.
“Executive function, attention and working memory, memory, and language were the cognitive domains that improved for some groups, and we also found that activities of daily living and quality of life improved for the different groups as well,” PhD candidate Blake Lawrence said at the Fourth World Parkinson Congress.

Patients had cognitive deficits that did not interfere with functional independence and were responding to stable doses of antiparkinsonian medication. Forty-two eligible participants underwent neuropsychologic testing at baseline and were randomly and equally assigned to one of the following six groups: SCT, TCT, tDCS, SCT+tDCS, TCT+tDCS, or control.
Cognitive training consisted of three 45-minute sessions per week for four weeks using Smartbrain Pro software in participants’ homes. tDCS involved constant 1.5-mA stimulation for 20 minutes in one session per week for four weeks at the university, with the anode placed over area F3 to stimulate the left dorsal lateral prefrontal cortex. Neuropsychologic testing was conducted post intervention (ie, at five weeks). Follow-up evaluations were at 12 weeks.
The researchers administered tests to evaluate executive function (Stockings of Cambridge), attention and working memory (Stroop test), memory (paragraph recall), quality of life (Parkinson’s Disease Questionnaire), activities of daily living (Unified Parkinson’s Disease Rating Scale-II), and language (similarities test). In general, combining tDCS with either form of cognitive training resulted in significantly greater improvements in more outcomes than any of the modalities alone. SCT showed positive results when compared against the control group in memory improvement at follow-up (effect size, 1.30), as well as in quality of life and activities of daily living post intervention (effect sizes, 0.24 and 0.33, respectively). TCT showed benefits on quality of life at both time points (effect sizes, 0.26 and 0.12, respectively).
When combined with tDCS, SCT produced improvements in attention and working memory both post intervention and at 12-week follow-up (effect sizes, 0.60 and 0.24, respectively), and executive function at post intervention and follow-up (0.41 and 0.23, respectively). Improvement in activities of daily living and language were statistically significant only immediately post intervention.
Combining tDCS with TCT resulted in improvements post intervention and at follow-up on measures of memory (1.36 and 1.75, respectively) and executive function (0.19 and 0.92, respectively), as well as in language post intervention (1.06).
“The groups that completed both cognitive training and brain stimulation improved to a greater extent and in more outcomes than the groups that just completed the brain training or the stimulation individually,” Mr. Lawrence said. “The majority of the effects were shown immediately after the intervention, but some of the promising results ... actually maintained improvement at the 12-week follow-up, so that was after about eight weeks, when they did not complete any intervention whatsoever.”
The improvements are probably clinically meaningful to patients, since they themselves reported the outcomes on quality of life and activities of daily living scales, he said.
—Daniel M. Keller
PORTLAND, OR—Combining transcranial direct current stimulation (tDCS) and cognitive training resulted in an improvement in a greater number of cognitive outcomes than either intervention alone in a small, randomized, controlled trial of patients with Parkinson’s disease and mild cognitive impairment.
Researchers at Curtin University in Perth, Australia, conducted the trial comparing the effects of standard (ie, not individualized) cognitive training (SCT), tailored (ie, individualized) cognitive training (TCT), tDCS, and a combination of tDCS with either form of cognitive training on cognitive outcomes, activities of daily living, and quality of life. Previously, it was not known whether either form of cognitive training, tDCS, or a combination of the two would be most efficacious in improving cognition.
“Executive function, attention and working memory, memory, and language were the cognitive domains that improved for some groups, and we also found that activities of daily living and quality of life improved for the different groups as well,” PhD candidate Blake Lawrence said at the Fourth World Parkinson Congress.
Patients had cognitive deficits that did not interfere with functional independence and were responding to stable doses of antiparkinsonian medication. Forty-two eligible participants underwent neuropsychologic testing at baseline and were randomly and equally assigned to one of the following six groups: SCT, TCT, tDCS, SCT+tDCS, TCT+tDCS, or control.
Cognitive training consisted of three 45-minute sessions per week for four weeks using Smartbrain Pro software in participants’ homes. tDCS involved constant 1.5-mA stimulation for 20 minutes in one session per week for four weeks at the university, with the anode placed over area F3 to stimulate the left dorsal lateral prefrontal cortex. Neuropsychologic testing was conducted post intervention (ie, at five weeks). Follow-up evaluations were at 12 weeks.
The researchers administered tests to evaluate executive function (Stockings of Cambridge), attention and working memory (Stroop test), memory (paragraph recall), quality of life (Parkinson’s Disease Questionnaire), activities of daily living (Unified Parkinson’s Disease Rating Scale-II), and language (similarities test). In general, combining tDCS with either form of cognitive training resulted in significantly greater improvements in more outcomes than any of the modalities alone. SCT showed positive results when compared against the control group in memory improvement at follow-up (effect size, 1.30), as well as in quality of life and activities of daily living post intervention (effect sizes, 0.24 and 0.33, respectively). TCT showed benefits on quality of life at both time points (effect sizes, 0.26 and 0.12, respectively).
When combined with tDCS, SCT produced improvements in attention and working memory both post intervention and at 12-week follow-up (effect sizes, 0.60 and 0.24, respectively), and executive function at post intervention and follow-up (0.41 and 0.23, respectively). Improvement in activities of daily living and language were statistically significant only immediately post intervention.
Combining tDCS with TCT resulted in improvements post intervention and at follow-up on measures of memory (1.36 and 1.75, respectively) and executive function (0.19 and 0.92, respectively), as well as in language post intervention (1.06).
“The groups that completed both cognitive training and brain stimulation improved to a greater extent and in more outcomes than the groups that just completed the brain training or the stimulation individually,” Mr. Lawrence said. “The majority of the effects were shown immediately after the intervention, but some of the promising results ... actually maintained improvement at the 12-week follow-up, so that was after about eight weeks, when they did not complete any intervention whatsoever.”
The improvements are probably clinically meaningful to patients, since they themselves reported the outcomes on quality of life and activities of daily living scales, he said.
—Daniel M. Keller
PORTLAND, OR—Combining transcranial direct current stimulation (tDCS) and cognitive training resulted in an improvement in a greater number of cognitive outcomes than either intervention alone in a small, randomized, controlled trial of patients with Parkinson’s disease and mild cognitive impairment.
Researchers at Curtin University in Perth, Australia, conducted the trial comparing the effects of standard (ie, not individualized) cognitive training (SCT), tailored (ie, individualized) cognitive training (TCT), tDCS, and a combination of tDCS with either form of cognitive training on cognitive outcomes, activities of daily living, and quality of life. Previously, it was not known whether either form of cognitive training, tDCS, or a combination of the two would be most efficacious in improving cognition.
“Executive function, attention and working memory, memory, and language were the cognitive domains that improved for some groups, and we also found that activities of daily living and quality of life improved for the different groups as well,” PhD candidate Blake Lawrence said at the Fourth World Parkinson Congress.
Patients had cognitive deficits that did not interfere with functional independence and were responding to stable doses of antiparkinsonian medication. Forty-two eligible participants underwent neuropsychologic testing at baseline and were randomly and equally assigned to one of the following six groups: SCT, TCT, tDCS, SCT+tDCS, TCT+tDCS, or control.
Cognitive training consisted of three 45-minute sessions per week for four weeks using Smartbrain Pro software in participants’ homes. tDCS involved constant 1.5-mA stimulation for 20 minutes in one session per week for four weeks at the university, with the anode placed over area F3 to stimulate the left dorsal lateral prefrontal cortex. Neuropsychologic testing was conducted post intervention (ie, at five weeks). Follow-up evaluations were at 12 weeks.
The researchers administered tests to evaluate executive function (Stockings of Cambridge), attention and working memory (Stroop test), memory (paragraph recall), quality of life (Parkinson’s Disease Questionnaire), activities of daily living (Unified Parkinson’s Disease Rating Scale-II), and language (similarities test). In general, combining tDCS with either form of cognitive training resulted in significantly greater improvements in more outcomes than any of the modalities alone. SCT showed positive results when compared against the control group in memory improvement at follow-up (effect size, 1.30), as well as in quality of life and activities of daily living post intervention (effect sizes, 0.24 and 0.33, respectively). TCT showed benefits on quality of life at both time points (effect sizes, 0.26 and 0.12, respectively).
When combined with tDCS, SCT produced improvements in attention and working memory both post intervention and at 12-week follow-up (effect sizes, 0.60 and 0.24, respectively), and executive function at post intervention and follow-up (0.41 and 0.23, respectively). Improvement in activities of daily living and language were statistically significant only immediately post intervention.
Combining tDCS with TCT resulted in improvements post intervention and at follow-up on measures of memory (1.36 and 1.75, respectively) and executive function (0.19 and 0.92, respectively), as well as in language post intervention (1.06).
“The groups that completed both cognitive training and brain stimulation improved to a greater extent and in more outcomes than the groups that just completed the brain training or the stimulation individually,” Mr. Lawrence said. “The majority of the effects were shown immediately after the intervention, but some of the promising results ... actually maintained improvement at the 12-week follow-up, so that was after about eight weeks, when they did not complete any intervention whatsoever.”
The improvements are probably clinically meaningful to patients, since they themselves reported the outcomes on quality of life and activities of daily living scales, he said.
—Daniel M. Keller

ASCO: Patients with advanced cancer should receive palliative care within 8 weeks of diagnosis
Patients with advanced cancer should receive dedicated palliative care services early in the disease course, concurrently with active treatment, according to the American Society of Clinical Oncology’s new guidelines on the integration of palliative care into standard oncology care.
Ideally, patients should be referred to interdisciplinary palliative care teams within 8 weeks of cancer diagnosis, and palliative care should be available in both the inpatient and outpatient setting, recommended ASCO.
The guidelines, which updated and expanded the 2012 ASCO provisional clinical opinion, were developed by a multidisciplinary expert panel that systematically reviewed phase III randomized controlled trials, secondary analyses of those trials, and meta-analyses that were published between March 2010 and January 2016.
According to the panel, essential components of palliative care include:
• Rapport and relationship building with patient and family caregivers.
• Symptom, distress, and functional status management.
• Exploration of understanding and education about illness and prognosis.
• Clarification of treatment goals.
• Assessment and support of coping needs.
• Assistance with medical decision making.

• Provision of referrals to other care providers as indicated.
The panel makes the case that not only does palliative care improve care for patients and families, it also likely reduces the total cost of care, often substantially. However, “race, poverty and low socioeconomic and/or immigration status are determinants of barriers to palliative care,” wrote the expert panel, which was cochaired by Betty Ferrell, PhD, of the City of Hope Medical Center, Duarte, Calif., and Thomas Smith, MD, of the Sidney Kimmel Comprehensive Cancer Center in Baltimore.

Read the full guidelines here.
jcraig@frontlinemedcom.com
On Twitter @jessnicolecraig
Patients with advanced cancer should receive dedicated palliative care services early in the disease course, concurrently with active treatment, according to the American Society of Clinical Oncology’s new guidelines on the integration of palliative care into standard oncology care.
Ideally, patients should be referred to interdisciplinary palliative care teams within 8 weeks of cancer diagnosis, and palliative care should be available in both the inpatient and outpatient setting, recommended ASCO.
The guidelines, which updated and expanded the 2012 ASCO provisional clinical opinion, were developed by a multidisciplinary expert panel that systematically reviewed phase III randomized controlled trials, secondary analyses of those trials, and meta-analyses that were published between March 2010 and January 2016.
According to the panel, essential components of palliative care include:
• Rapport and relationship building with patient and family caregivers.
• Symptom, distress, and functional status management.
• Exploration of understanding and education about illness and prognosis.
• Clarification of treatment goals.
• Assessment and support of coping needs.
• Assistance with medical decision making.
• Provision of referrals to other care providers as indicated.
The panel makes the case that not only does palliative care improve care for patients and families, it also likely reduces the total cost of care, often substantially. However, “race, poverty and low socioeconomic and/or immigration status are determinants of barriers to palliative care,” wrote the expert panel, which was cochaired by Betty Ferrell, PhD, of the City of Hope Medical Center, Duarte, Calif., and Thomas Smith, MD, of the Sidney Kimmel Comprehensive Cancer Center in Baltimore.
Read the full guidelines here.
jcraig@frontlinemedcom.com
On Twitter @jessnicolecraig
Patients with advanced cancer should receive dedicated palliative care services early in the disease course, concurrently with active treatment, according to the American Society of Clinical Oncology’s new guidelines on the integration of palliative care into standard oncology care.
Ideally, patients should be referred to interdisciplinary palliative care teams within 8 weeks of cancer diagnosis, and palliative care should be available in both the inpatient and outpatient setting, recommended ASCO.
The guidelines, which updated and expanded the 2012 ASCO provisional clinical opinion, were developed by a multidisciplinary expert panel that systematically reviewed phase III randomized controlled trials, secondary analyses of those trials, and meta-analyses that were published between March 2010 and January 2016.
According to the panel, essential components of palliative care include:
• Rapport and relationship building with patient and family caregivers.
• Symptom, distress, and functional status management.
• Exploration of understanding and education about illness and prognosis.
• Clarification of treatment goals.
• Assessment and support of coping needs.
• Assistance with medical decision making.
• Provision of referrals to other care providers as indicated.
The panel makes the case that not only does palliative care improve care for patients and families, it also likely reduces the total cost of care, often substantially. However, “race, poverty and low socioeconomic and/or immigration status are determinants of barriers to palliative care,” wrote the expert panel, which was cochaired by Betty Ferrell, PhD, of the City of Hope Medical Center, Duarte, Calif., and Thomas Smith, MD, of the Sidney Kimmel Comprehensive Cancer Center in Baltimore.
Read the full guidelines here.
jcraig@frontlinemedcom.com
On Twitter @jessnicolecraig
FROM THE JOURNAL OF CLINICAL ONCOLOGY
The march of technology
Each year the American Academy of Pediatrics National Conference and Exhibition fills a huge convention hall with the latest products that can improve health and generate practice revenue.
Some products are solutions to the minor annoyances of everyday practice. For instance, there are ear curettes equipped with their own LED light and a magnifying lens. There are countless creams to treat rashes. There are new automated devices for testing hearing, vision, and attention. And at the far extreme, there are products with the potential to revolutionize clinical care or to bankrupt it. The latest technology in that category is whole exome sequencing.

A couple weeks earlier I had listened to a national meeting of pediatric ethicists discuss this technology. Some proponents discussed the possibility of doing whole exome sequencing (WES) for every newborn. Alas, many ethicists can’t do math. Even if the cost goes below $1,000 per test, at 4 million babies per year in the United States, that is $4 billion per year. That sounds like a small sum, compared with the current federal deficit, but the original budget for the entire, 10-year-long Human Genome Project (HGP) was $4.5 billion. There were complaints in that era that diverting such an enormous amount of money into the HGP would cut the funding of a lot of other very good research at the National Institutes of Health. Conversely, Medicare spends $4.5 billion on hepatitis C treatment.
Viewed differently, the yearly per capita payment to general pediatricians, excluding vaccine costs, is around $1,000. Perhaps I’m biased, but I think I provide much more value than a genetic sequence.
Precision medicine has a lot of potential. So far, it is mostly potential. One colleague related that, in the past year, he has done WES on three patients, at about $4,000 charge for each, and gotten positive results in two cases. He figures soon he will be ordering it on every child with symptoms of autism, developmental delay, or failure to thrive. Is that a wise idea? That, it seems, is the area in which there is the least illuminating research.
Dr. Powell is a pediatric hospitalist and clinical ethics consultant living in St. Louis.
Each year the American Academy of Pediatrics National Conference and Exhibition fills a huge convention hall with the latest products that can improve health and generate practice revenue.
Some products are solutions to the minor annoyances of everyday practice. For instance, there are ear curettes equipped with their own LED light and a magnifying lens. There are countless creams to treat rashes. There are new automated devices for testing hearing, vision, and attention. And at the far extreme, there are products with the potential to revolutionize clinical care or to bankrupt it. The latest technology in that category is whole exome sequencing.
A couple weeks earlier I had listened to a national meeting of pediatric ethicists discuss this technology. Some proponents discussed the possibility of doing whole exome sequencing (WES) for every newborn. Alas, many ethicists can’t do math. Even if the cost goes below $1,000 per test, at 4 million babies per year in the United States, that is $4 billion per year. That sounds like a small sum, compared with the current federal deficit, but the original budget for the entire, 10-year-long Human Genome Project (HGP) was $4.5 billion. There were complaints in that era that diverting such an enormous amount of money into the HGP would cut the funding of a lot of other very good research at the National Institutes of Health. Conversely, Medicare spends $4.5 billion on hepatitis C treatment.
Viewed differently, the yearly per capita payment to general pediatricians, excluding vaccine costs, is around $1,000. Perhaps I’m biased, but I think I provide much more value than a genetic sequence.
Precision medicine has a lot of potential. So far, it is mostly potential. One colleague related that, in the past year, he has done WES on three patients, at about $4,000 charge for each, and gotten positive results in two cases. He figures soon he will be ordering it on every child with symptoms of autism, developmental delay, or failure to thrive. Is that a wise idea? That, it seems, is the area in which there is the least illuminating research.
Dr. Powell is a pediatric hospitalist and clinical ethics consultant living in St. Louis.
Each year the American Academy of Pediatrics National Conference and Exhibition fills a huge convention hall with the latest products that can improve health and generate practice revenue.
Some products are solutions to the minor annoyances of everyday practice. For instance, there are ear curettes equipped with their own LED light and a magnifying lens. There are countless creams to treat rashes. There are new automated devices for testing hearing, vision, and attention. And at the far extreme, there are products with the potential to revolutionize clinical care or to bankrupt it. The latest technology in that category is whole exome sequencing.
A couple weeks earlier I had listened to a national meeting of pediatric ethicists discuss this technology. Some proponents discussed the possibility of doing whole exome sequencing (WES) for every newborn. Alas, many ethicists can’t do math. Even if the cost goes below $1,000 per test, at 4 million babies per year in the United States, that is $4 billion per year. That sounds like a small sum, compared with the current federal deficit, but the original budget for the entire, 10-year-long Human Genome Project (HGP) was $4.5 billion. There were complaints in that era that diverting such an enormous amount of money into the HGP would cut the funding of a lot of other very good research at the National Institutes of Health. Conversely, Medicare spends $4.5 billion on hepatitis C treatment.
Viewed differently, the yearly per capita payment to general pediatricians, excluding vaccine costs, is around $1,000. Perhaps I’m biased, but I think I provide much more value than a genetic sequence.
Precision medicine has a lot of potential. So far, it is mostly potential. One colleague related that, in the past year, he has done WES on three patients, at about $4,000 charge for each, and gotten positive results in two cases. He figures soon he will be ordering it on every child with symptoms of autism, developmental delay, or failure to thrive. Is that a wise idea? That, it seems, is the area in which there is the least illuminating research.
Dr. Powell is a pediatric hospitalist and clinical ethics consultant living in St. Louis.
Clinical Challenges - November 2016
What’s your diagnosis?
Answer to “What’s your diagnosis?” on page X: Collagenous gastritis and collagenous sprue
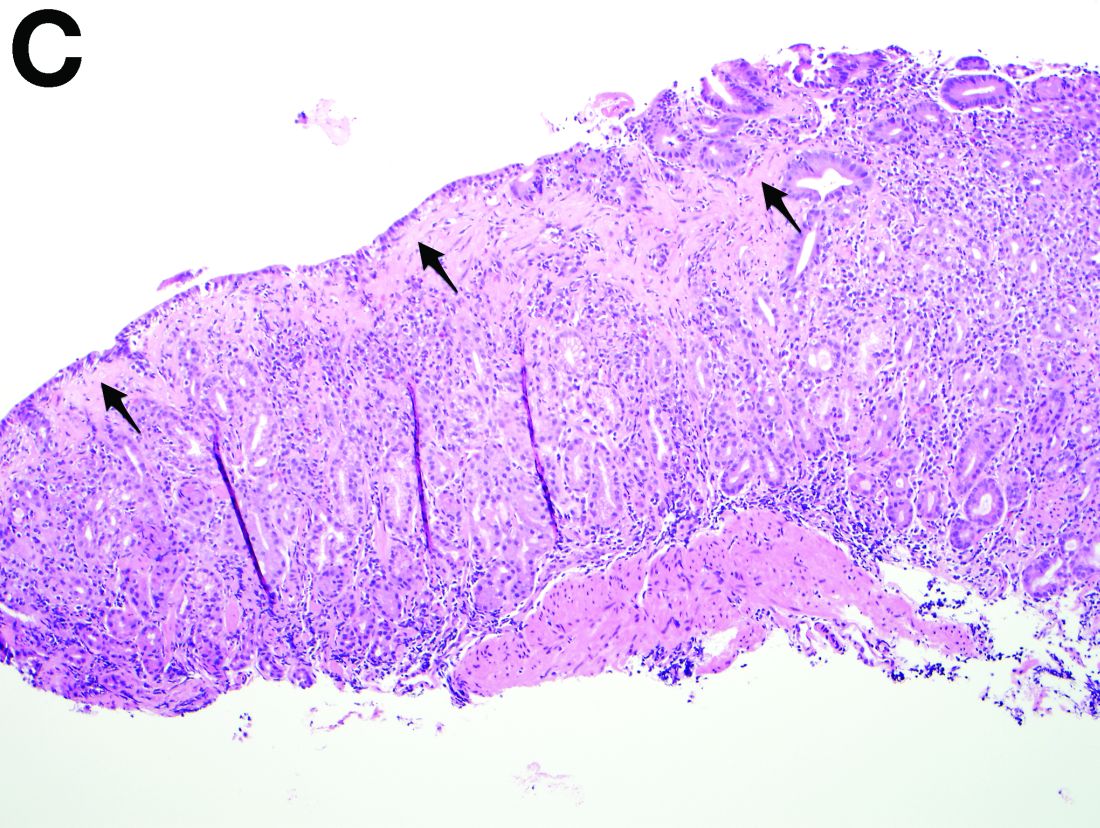
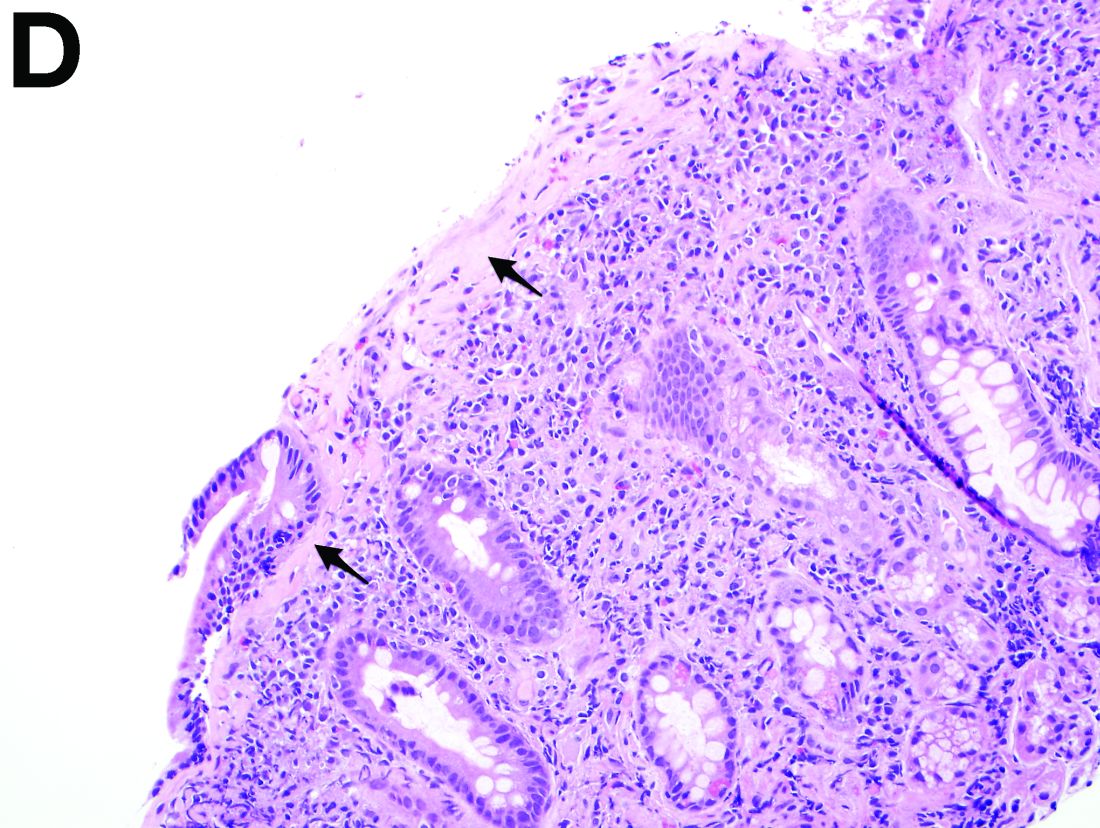
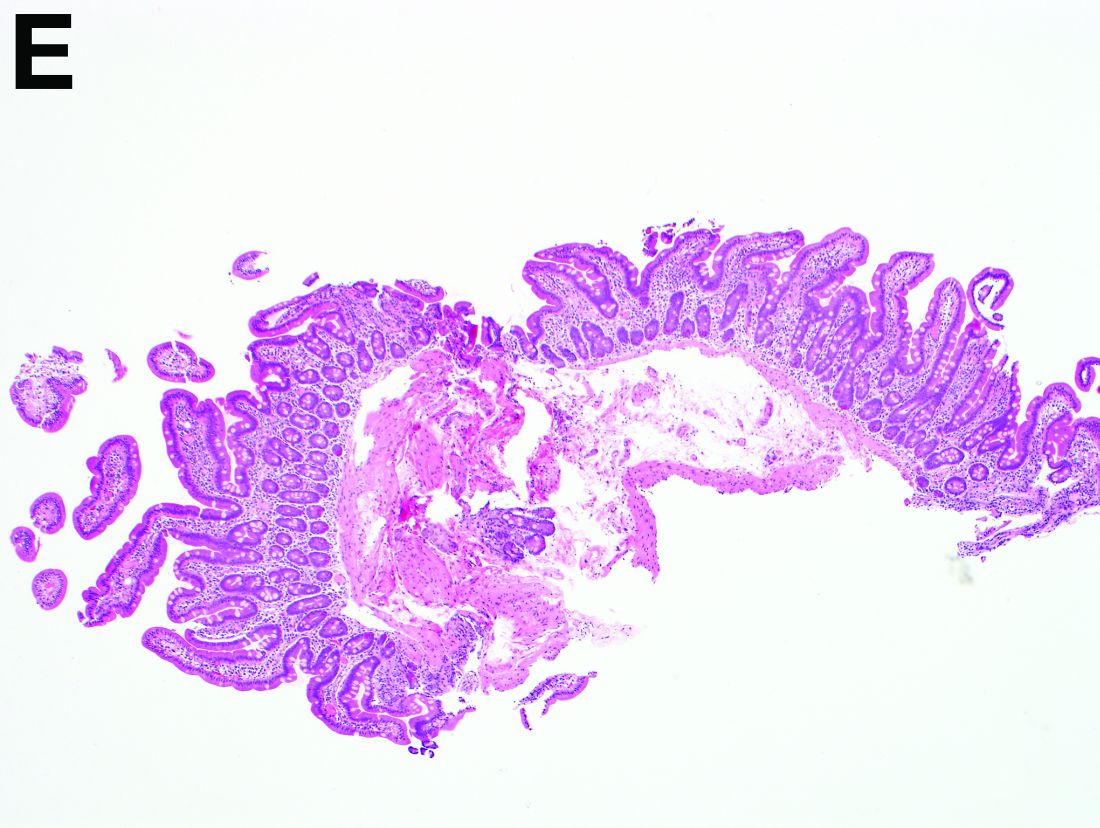
Recent studies have also shown the importance of obtaining at least 1 biopsy from the duodenal bulb to avoid missing the diagnosis of celiac disease. In 126 patients with newly established celiac disease and 85 patients with a previous diagnosis on a gluten-free diet presenting for reevaluation, villous atrophy was limited to the duodenal bulb in 9% and 14% of cases, respectively.3
References
1. Brain, O., Rajaguru, C., Warren, B. et al. Collagenous gastritis: reports and systematic review. Eur J Gastroenterol Hepatol. 2009;21:1419-24.
2. Gopal, P., McKenna, B.J. The collagenous gastroenteritides: similarities and differences. Arch Pathol Lab Med. 2010;134:1485-9.
3. Evans, K.E., Aziz, I., Cross, S.S. et al. A prospective study of duodenal bulb biopsy in newly diagnosed and established adult celiac disease. Am J Gastroenterol. 2011;106:1837-742.
Answer to “What’s your diagnosis?” on page X: Collagenous gastritis and collagenous sprue
Recent studies have also shown the importance of obtaining at least 1 biopsy from the duodenal bulb to avoid missing the diagnosis of celiac disease. In 126 patients with newly established celiac disease and 85 patients with a previous diagnosis on a gluten-free diet presenting for reevaluation, villous atrophy was limited to the duodenal bulb in 9% and 14% of cases, respectively.3
References
1. Brain, O., Rajaguru, C., Warren, B. et al. Collagenous gastritis: reports and systematic review. Eur J Gastroenterol Hepatol. 2009;21:1419-24.
2. Gopal, P., McKenna, B.J. The collagenous gastroenteritides: similarities and differences. Arch Pathol Lab Med. 2010;134:1485-9.
3. Evans, K.E., Aziz, I., Cross, S.S. et al. A prospective study of duodenal bulb biopsy in newly diagnosed and established adult celiac disease. Am J Gastroenterol. 2011;106:1837-742.
Answer to “What’s your diagnosis?” on page X: Collagenous gastritis and collagenous sprue
Recent studies have also shown the importance of obtaining at least 1 biopsy from the duodenal bulb to avoid missing the diagnosis of celiac disease. In 126 patients with newly established celiac disease and 85 patients with a previous diagnosis on a gluten-free diet presenting for reevaluation, villous atrophy was limited to the duodenal bulb in 9% and 14% of cases, respectively.3
References
1. Brain, O., Rajaguru, C., Warren, B. et al. Collagenous gastritis: reports and systematic review. Eur J Gastroenterol Hepatol. 2009;21:1419-24.
2. Gopal, P., McKenna, B.J. The collagenous gastroenteritides: similarities and differences. Arch Pathol Lab Med. 2010;134:1485-9.
3. Evans, K.E., Aziz, I., Cross, S.S. et al. A prospective study of duodenal bulb biopsy in newly diagnosed and established adult celiac disease. Am J Gastroenterol. 2011;106:1837-742.
What’s your diagnosis?
What’s your diagnosis?
What’s your diagnosis?
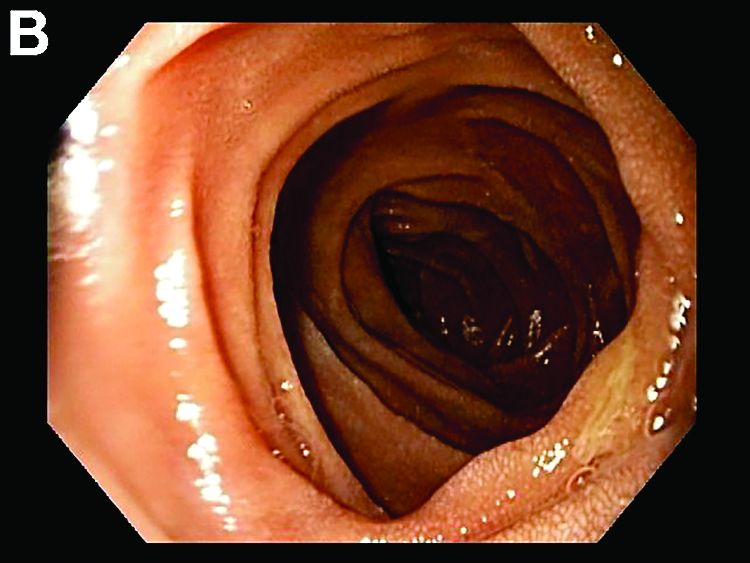
By Benjamin Kloesel, MD, Vishal S. Chandan, MD, and Glenn L. Alexander, MD. Published previously in Gastroenterology (2012;143:1439, 1692).
A 30-year-old woman with a past medical history of hypothyroidism presents for evaluation of epigastric discomfort, nausea without emesis, abdominal bloating, and watery, nonbloody diarrhea for 5 months. This was associated with a 15-pound weight loss. Complete blood count, liver function tests, thyroid-stimulating hormone, immunoglobulin (Ig) levels, and IgG/IgA tissue transglutaminase (tTG) were within normal limits. Stool studies for bacterial pathogens, Giardia, Clostridium difficile toxin, and ova/parasites were negative. 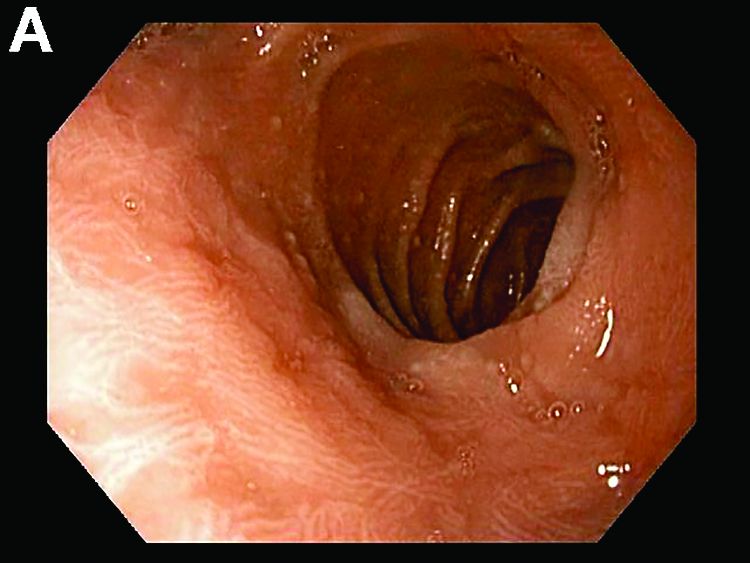
Results puzzling for embolic protection during TAVR
The largest randomized clinical trial to assess the safety and efficacy of cerebral embolic protection systems during transcatheter aortic valve replacement yielded puzzling and somewhat contradictory results, according to a report presented at the Transcatheter Cardiovascular Therapeutics annual meeting and published simultaneously in the Journal of the American College of Cardiology.
Virtually every device in this industry-sponsored study involving 363 elderly patients (mean age, 83.4 years) with severe aortic stenosis trapped particulate debris as intended, the mean volume of new lesions in the protected areas of the brain was reduced by 42%, and the number and volume of new lesions correlated with neurocognitive outcomes at 30 days.
However, the reduction in lesion volume did not achieve statistical significance, and the improvement in neurocognitive function also did not reach statistical significance.
In addition, “the sample size was clearly too low to assess clinical outcomes, and in retrospect, was also too low to evaluate follow-up MRI findings or neurocognitive outcomes.” Nevertheless, the trial “provides reassuring evidence of device safety,” said Samir R. Kapadia, MD, of the Cleveland Clinic (J Am Coll Cardiol. 2016 Nov 1. doi: 10.1016/j.jacc.2016.10.023).
In this prospective study, the investigators assessed patients at 17 medical centers in the United States and 2 in Germany. In addition to being elderly, the study patients were at high risk because of frequent comorbidities, including atrial fibrillation (31.7%) and prior stroke (5.8%).

The remaining 123 patients underwent TAVR but not MRI in a safety arm of the trial.
The protection devices were placed “without safety concerns” in most patients. The rate of major adverse events with the device was 7.3%, markedly less than the 18.3% prespecified performance goal for this outcome. Total procedure time was lengthened by only 13 minutes when the device was used, and total fluoroscopy time was increased by only 3 minutes. These findings demonstrate the overall safety of using the device, Dr. Kapadia said.
Debris including thrombus with tissue elements, artery wall particles, calcifications, valve tissue, and foreign materials was retrieved from the filters in 99% of patients.
The mean volume of new cerebral lesions in areas of the brain protected by the device was reduced by 42%, compared with that in patients who underwent TAVR without the protection device. However, this reduction was not statistically significant, so the primary efficacy endpoint of the study was not met.
Similarly, neurocognitive testing at 30 days showed that the volume of new lesions correlated with poorer outcomes. However, the difference in neurocognitive function between the intervention group and the control group did not reach statistical significance.
Several limitations likely contributed to this lack of statistical significance, Dr. Kapadia said.
First, the 5-day “window” for MRI assessment was too long. Both the number and the volume of new lesions rapidly changed over time, which led to marked variance in MRI findings depending on when the images were taken.
In addition, only one TAVR device was available at the time the trial was designed, so the study wasn’t stratified by type of valve device. But several new devices became available during the study, and the study investigators were permitted to use any of them. Both pre- and postimplantation techniques differ among these TAVR devices, but these differences could not be accounted for, given the study design.
Also, certain risk factors for stroke, especially certain findings on baseline MRI, were not understood when the trial was designed, and those factors also were not accounted for, Dr. Kapadia said.
Claret Medical funded the study. Dr. Kapadia reported having no relevant financial disclosures; his associates reported numerous ties to industry sources. The meeting was sponsored by the Cardiovascular Research Foundation.
From a logical standpoint, a device that collects cerebral embolic material in 99% of cases should prevent ischemic brain injury, yet the findings from this randomized trial don’t appear to support the routine use of such devices. But it would be inappropriate and unfair to close the book on cerebral protection after this chapter.
The authors acknowledge that an MRI “window” of 5 days creates too much heterogeneity in the data, that multiple TAVR devices requiring different implantation techniques further muddy the picture, and that in retrospect the sample size was inadequate and the study was underpowered. In addition, rigorous neurocognitive assessment can be challenging in elderly, recovering patients, and results can depend on the time of day and the patient’s alertness.
Despite the negative findings regarding both primary and secondary endpoints, the data do show the overall safety of embolic protection devices. We are dealing with a potential benefit that cannot be ignored as TAVR shifts to younger and lower-risk patients.
Azeem Latib, MD, is in the interventional cardiology unit at San Raffaele Scientific Institute in Milan. Matteo Pagnesi, MD, is in the interventional cardiology unit at EMO-GVM Centro Cuore Columbus in Milan. San Raffaele Scientific Institute has been involved in clinical studies of embolic protection devices made by Claret Medical, Innovative Cardiovascular Solutions, and Keystone Heart. Dr. Latib and Dr. Pagnesi reported having no other relevant financial disclosures. They made these remarks in an editorial accompanying Dr. Kapadia’s report (J Am Coll Cardiol. 2016 Nov 1. doi: 10.1016/j.jacc.2016.10.036).
From a logical standpoint, a device that collects cerebral embolic material in 99% of cases should prevent ischemic brain injury, yet the findings from this randomized trial don’t appear to support the routine use of such devices. But it would be inappropriate and unfair to close the book on cerebral protection after this chapter.
The authors acknowledge that an MRI “window” of 5 days creates too much heterogeneity in the data, that multiple TAVR devices requiring different implantation techniques further muddy the picture, and that in retrospect the sample size was inadequate and the study was underpowered. In addition, rigorous neurocognitive assessment can be challenging in elderly, recovering patients, and results can depend on the time of day and the patient’s alertness.
Despite the negative findings regarding both primary and secondary endpoints, the data do show the overall safety of embolic protection devices. We are dealing with a potential benefit that cannot be ignored as TAVR shifts to younger and lower-risk patients.
Azeem Latib, MD, is in the interventional cardiology unit at San Raffaele Scientific Institute in Milan. Matteo Pagnesi, MD, is in the interventional cardiology unit at EMO-GVM Centro Cuore Columbus in Milan. San Raffaele Scientific Institute has been involved in clinical studies of embolic protection devices made by Claret Medical, Innovative Cardiovascular Solutions, and Keystone Heart. Dr. Latib and Dr. Pagnesi reported having no other relevant financial disclosures. They made these remarks in an editorial accompanying Dr. Kapadia’s report (J Am Coll Cardiol. 2016 Nov 1. doi: 10.1016/j.jacc.2016.10.036).
From a logical standpoint, a device that collects cerebral embolic material in 99% of cases should prevent ischemic brain injury, yet the findings from this randomized trial don’t appear to support the routine use of such devices. But it would be inappropriate and unfair to close the book on cerebral protection after this chapter.
The authors acknowledge that an MRI “window” of 5 days creates too much heterogeneity in the data, that multiple TAVR devices requiring different implantation techniques further muddy the picture, and that in retrospect the sample size was inadequate and the study was underpowered. In addition, rigorous neurocognitive assessment can be challenging in elderly, recovering patients, and results can depend on the time of day and the patient’s alertness.
Despite the negative findings regarding both primary and secondary endpoints, the data do show the overall safety of embolic protection devices. We are dealing with a potential benefit that cannot be ignored as TAVR shifts to younger and lower-risk patients.
Azeem Latib, MD, is in the interventional cardiology unit at San Raffaele Scientific Institute in Milan. Matteo Pagnesi, MD, is in the interventional cardiology unit at EMO-GVM Centro Cuore Columbus in Milan. San Raffaele Scientific Institute has been involved in clinical studies of embolic protection devices made by Claret Medical, Innovative Cardiovascular Solutions, and Keystone Heart. Dr. Latib and Dr. Pagnesi reported having no other relevant financial disclosures. They made these remarks in an editorial accompanying Dr. Kapadia’s report (J Am Coll Cardiol. 2016 Nov 1. doi: 10.1016/j.jacc.2016.10.036).
The largest randomized clinical trial to assess the safety and efficacy of cerebral embolic protection systems during transcatheter aortic valve replacement yielded puzzling and somewhat contradictory results, according to a report presented at the Transcatheter Cardiovascular Therapeutics annual meeting and published simultaneously in the Journal of the American College of Cardiology.
Virtually every device in this industry-sponsored study involving 363 elderly patients (mean age, 83.4 years) with severe aortic stenosis trapped particulate debris as intended, the mean volume of new lesions in the protected areas of the brain was reduced by 42%, and the number and volume of new lesions correlated with neurocognitive outcomes at 30 days.
However, the reduction in lesion volume did not achieve statistical significance, and the improvement in neurocognitive function also did not reach statistical significance.
In addition, “the sample size was clearly too low to assess clinical outcomes, and in retrospect, was also too low to evaluate follow-up MRI findings or neurocognitive outcomes.” Nevertheless, the trial “provides reassuring evidence of device safety,” said Samir R. Kapadia, MD, of the Cleveland Clinic (J Am Coll Cardiol. 2016 Nov 1. doi: 10.1016/j.jacc.2016.10.023).
In this prospective study, the investigators assessed patients at 17 medical centers in the United States and 2 in Germany. In addition to being elderly, the study patients were at high risk because of frequent comorbidities, including atrial fibrillation (31.7%) and prior stroke (5.8%).
The remaining 123 patients underwent TAVR but not MRI in a safety arm of the trial.
The protection devices were placed “without safety concerns” in most patients. The rate of major adverse events with the device was 7.3%, markedly less than the 18.3% prespecified performance goal for this outcome. Total procedure time was lengthened by only 13 minutes when the device was used, and total fluoroscopy time was increased by only 3 minutes. These findings demonstrate the overall safety of using the device, Dr. Kapadia said.
Debris including thrombus with tissue elements, artery wall particles, calcifications, valve tissue, and foreign materials was retrieved from the filters in 99% of patients.
The mean volume of new cerebral lesions in areas of the brain protected by the device was reduced by 42%, compared with that in patients who underwent TAVR without the protection device. However, this reduction was not statistically significant, so the primary efficacy endpoint of the study was not met.
Similarly, neurocognitive testing at 30 days showed that the volume of new lesions correlated with poorer outcomes. However, the difference in neurocognitive function between the intervention group and the control group did not reach statistical significance.
Several limitations likely contributed to this lack of statistical significance, Dr. Kapadia said.
First, the 5-day “window” for MRI assessment was too long. Both the number and the volume of new lesions rapidly changed over time, which led to marked variance in MRI findings depending on when the images were taken.
In addition, only one TAVR device was available at the time the trial was designed, so the study wasn’t stratified by type of valve device. But several new devices became available during the study, and the study investigators were permitted to use any of them. Both pre- and postimplantation techniques differ among these TAVR devices, but these differences could not be accounted for, given the study design.
Also, certain risk factors for stroke, especially certain findings on baseline MRI, were not understood when the trial was designed, and those factors also were not accounted for, Dr. Kapadia said.
Claret Medical funded the study. Dr. Kapadia reported having no relevant financial disclosures; his associates reported numerous ties to industry sources. The meeting was sponsored by the Cardiovascular Research Foundation.
The largest randomized clinical trial to assess the safety and efficacy of cerebral embolic protection systems during transcatheter aortic valve replacement yielded puzzling and somewhat contradictory results, according to a report presented at the Transcatheter Cardiovascular Therapeutics annual meeting and published simultaneously in the Journal of the American College of Cardiology.
Virtually every device in this industry-sponsored study involving 363 elderly patients (mean age, 83.4 years) with severe aortic stenosis trapped particulate debris as intended, the mean volume of new lesions in the protected areas of the brain was reduced by 42%, and the number and volume of new lesions correlated with neurocognitive outcomes at 30 days.
However, the reduction in lesion volume did not achieve statistical significance, and the improvement in neurocognitive function also did not reach statistical significance.
In addition, “the sample size was clearly too low to assess clinical outcomes, and in retrospect, was also too low to evaluate follow-up MRI findings or neurocognitive outcomes.” Nevertheless, the trial “provides reassuring evidence of device safety,” said Samir R. Kapadia, MD, of the Cleveland Clinic (J Am Coll Cardiol. 2016 Nov 1. doi: 10.1016/j.jacc.2016.10.023).
In this prospective study, the investigators assessed patients at 17 medical centers in the United States and 2 in Germany. In addition to being elderly, the study patients were at high risk because of frequent comorbidities, including atrial fibrillation (31.7%) and prior stroke (5.8%).
The remaining 123 patients underwent TAVR but not MRI in a safety arm of the trial.
The protection devices were placed “without safety concerns” in most patients. The rate of major adverse events with the device was 7.3%, markedly less than the 18.3% prespecified performance goal for this outcome. Total procedure time was lengthened by only 13 minutes when the device was used, and total fluoroscopy time was increased by only 3 minutes. These findings demonstrate the overall safety of using the device, Dr. Kapadia said.
Debris including thrombus with tissue elements, artery wall particles, calcifications, valve tissue, and foreign materials was retrieved from the filters in 99% of patients.
The mean volume of new cerebral lesions in areas of the brain protected by the device was reduced by 42%, compared with that in patients who underwent TAVR without the protection device. However, this reduction was not statistically significant, so the primary efficacy endpoint of the study was not met.
Similarly, neurocognitive testing at 30 days showed that the volume of new lesions correlated with poorer outcomes. However, the difference in neurocognitive function between the intervention group and the control group did not reach statistical significance.
Several limitations likely contributed to this lack of statistical significance, Dr. Kapadia said.
First, the 5-day “window” for MRI assessment was too long. Both the number and the volume of new lesions rapidly changed over time, which led to marked variance in MRI findings depending on when the images were taken.
In addition, only one TAVR device was available at the time the trial was designed, so the study wasn’t stratified by type of valve device. But several new devices became available during the study, and the study investigators were permitted to use any of them. Both pre- and postimplantation techniques differ among these TAVR devices, but these differences could not be accounted for, given the study design.
Also, certain risk factors for stroke, especially certain findings on baseline MRI, were not understood when the trial was designed, and those factors also were not accounted for, Dr. Kapadia said.
Claret Medical funded the study. Dr. Kapadia reported having no relevant financial disclosures; his associates reported numerous ties to industry sources. The meeting was sponsored by the Cardiovascular Research Foundation.
Key clinical point: The largest randomized clinical trial to assess the safety and efficacy of cerebral embolic protection systems during TAVR yielded puzzling and contradictory results.
Major finding: Debris including thrombus with tissue elements, artery wall particles, calcifications, valve tissue, and foreign materials was retrieved from the cerebral protection filters in 99% of patients.
Data source: A prospective, international, randomized trial involving 363 elderly patients undergoing TAVR for severe aortic stenosis.
Disclosures: Claret Medical funded the study. Dr. Kapadia reported having no relevant financial disclosures; his associates reported numerous ties to industry sources.
Does Transdermal Nicotine Benefit Patients With Parkinson’s Disease?
PORTLAND, OR—High doses of transdermal nicotine failed to improve off motor symptoms in patients with Parkinson’s disease, according to trial results presented at the Fourth World Parkinson Congress. Nicotine may have provided benefit on secondary outcome measures, researchers said.
Gabriel Villafane, MD, a neurologist at Henri Mondor University Hospital in Créteil, France, and colleagues conducted the Nicopark2 Study, a single-blind, controlled, randomized trial to evaluate the effect of high doses of transdermal nicotine (approximately 90 mg per day) on motor symptoms in Parkinson’s disease. Cigarette smoking is associated with a dose-dependent reduction in risk of Parkinson’s disease. Nicotine’s effect on motor symptoms in Parkinson’s disease is controversial. Seven of eight open-label studies suggested that nicotine improves motor symptoms, but four placebo-controlled studies were negative.

The investigators enrolled 40 patients with Parkinson’s disease in the study. Eligible patients were nonsmokers age 35 to 70 with a Hoehn and Yahr off stage of four or less and a Hoehn and Yahr on stage of three or less. Patients had received levodopa treatment for at least three years. Exclusion criteria included neurosurgery, psychiatric disease, and symptomatic orthostatic hypotension.
The primary outcome was mean difference between groups in change of Unified Parkinson’s Disease Rating Scale off motor score from baseline to week 39 on blinded video rating.
Twenty patients were randomized to receive transdermal nicotine, and 20 patients were randomized to a control group. The change in motor score between groups was not statistically significant. Change in quality of life assessed by the Parkinson’s Disease Questionnaire was not statistically significant. At 39 weeks, a reduction in levodopa doses, a reduction in dyskinesias, and an improvement in activities of daily living were observed in the nicotine group, the researchers said.Adverse events occurred in more patients in the nicotine group than in the control group. They included worsening of parkinsonism (30% vs 5%), cutaneous reactions (35% vs 5%), gastrointestinal complaints (65% vs 15%), hypotension (25% vs 5%), insomnia (25% vs 5%), and nervousness and anxiety (20% vs 0%). Ten patients in the nicotine group had a serious adverse event, compared with three patients in the control group.
—Jake Remaly
PORTLAND, OR—High doses of transdermal nicotine failed to improve off motor symptoms in patients with Parkinson’s disease, according to trial results presented at the Fourth World Parkinson Congress. Nicotine may have provided benefit on secondary outcome measures, researchers said.
Gabriel Villafane, MD, a neurologist at Henri Mondor University Hospital in Créteil, France, and colleagues conducted the Nicopark2 Study, a single-blind, controlled, randomized trial to evaluate the effect of high doses of transdermal nicotine (approximately 90 mg per day) on motor symptoms in Parkinson’s disease. Cigarette smoking is associated with a dose-dependent reduction in risk of Parkinson’s disease. Nicotine’s effect on motor symptoms in Parkinson’s disease is controversial. Seven of eight open-label studies suggested that nicotine improves motor symptoms, but four placebo-controlled studies were negative.
The investigators enrolled 40 patients with Parkinson’s disease in the study. Eligible patients were nonsmokers age 35 to 70 with a Hoehn and Yahr off stage of four or less and a Hoehn and Yahr on stage of three or less. Patients had received levodopa treatment for at least three years. Exclusion criteria included neurosurgery, psychiatric disease, and symptomatic orthostatic hypotension.
The primary outcome was mean difference between groups in change of Unified Parkinson’s Disease Rating Scale off motor score from baseline to week 39 on blinded video rating.
Twenty patients were randomized to receive transdermal nicotine, and 20 patients were randomized to a control group. The change in motor score between groups was not statistically significant. Change in quality of life assessed by the Parkinson’s Disease Questionnaire was not statistically significant. At 39 weeks, a reduction in levodopa doses, a reduction in dyskinesias, and an improvement in activities of daily living were observed in the nicotine group, the researchers said.Adverse events occurred in more patients in the nicotine group than in the control group. They included worsening of parkinsonism (30% vs 5%), cutaneous reactions (35% vs 5%), gastrointestinal complaints (65% vs 15%), hypotension (25% vs 5%), insomnia (25% vs 5%), and nervousness and anxiety (20% vs 0%). Ten patients in the nicotine group had a serious adverse event, compared with three patients in the control group.
—Jake Remaly
PORTLAND, OR—High doses of transdermal nicotine failed to improve off motor symptoms in patients with Parkinson’s disease, according to trial results presented at the Fourth World Parkinson Congress. Nicotine may have provided benefit on secondary outcome measures, researchers said.
Gabriel Villafane, MD, a neurologist at Henri Mondor University Hospital in Créteil, France, and colleagues conducted the Nicopark2 Study, a single-blind, controlled, randomized trial to evaluate the effect of high doses of transdermal nicotine (approximately 90 mg per day) on motor symptoms in Parkinson’s disease. Cigarette smoking is associated with a dose-dependent reduction in risk of Parkinson’s disease. Nicotine’s effect on motor symptoms in Parkinson’s disease is controversial. Seven of eight open-label studies suggested that nicotine improves motor symptoms, but four placebo-controlled studies were negative.
The investigators enrolled 40 patients with Parkinson’s disease in the study. Eligible patients were nonsmokers age 35 to 70 with a Hoehn and Yahr off stage of four or less and a Hoehn and Yahr on stage of three or less. Patients had received levodopa treatment for at least three years. Exclusion criteria included neurosurgery, psychiatric disease, and symptomatic orthostatic hypotension.
The primary outcome was mean difference between groups in change of Unified Parkinson’s Disease Rating Scale off motor score from baseline to week 39 on blinded video rating.
Twenty patients were randomized to receive transdermal nicotine, and 20 patients were randomized to a control group. The change in motor score between groups was not statistically significant. Change in quality of life assessed by the Parkinson’s Disease Questionnaire was not statistically significant. At 39 weeks, a reduction in levodopa doses, a reduction in dyskinesias, and an improvement in activities of daily living were observed in the nicotine group, the researchers said.Adverse events occurred in more patients in the nicotine group than in the control group. They included worsening of parkinsonism (30% vs 5%), cutaneous reactions (35% vs 5%), gastrointestinal complaints (65% vs 15%), hypotension (25% vs 5%), insomnia (25% vs 5%), and nervousness and anxiety (20% vs 0%). Ten patients in the nicotine group had a serious adverse event, compared with three patients in the control group.
—Jake Remaly

Hospitalizations for opioid poisoning tripled in preschool children
From 1997 to 2012, the annual number of hospitalizations for opioid poisoning rose 178% among children aged 1-19 years, according to data from over 13,000 discharge records.
In 2012, there were 2,918 hospitalizations for opioid poisoning among children aged 1-19, compared with 1,049 in 1997, reported Julie R. Gaither, PhD, MPH, RN, and her associates at Yale University in New Haven, Conn. (JAMA Pediatr. 2016 Oct 31. doi: 10.1001/jamapediatrics.2016.2154).
The greatest change occurred among the youngest children, as the number of those aged 1-4 years rose from 133 in 1997 to 421 in 2012 – an increase of 217%. For those aged 15-19 years, the annual number of hospitalizations went from 715 to 2,171 (204%) over that time period, which included a slight drop from 2009 to 2012, according to the investigators, who used data from 13,052 discharges in the Agency for Healthcare Research and Quality’s Kids’ Inpatient Database.
The increase in hospitalizations for prescription opioid poisoning in children aged 10-14 years was 58% from 1997 to 2012 (rising from 171 to 272), while estimates for 5- to 9-year-olds did not meet the criteria for statistical reliability and were not included in the analysis, Dr. Gaither and her associates said.
The study was supported by grants from the National Institute on Drug Abuse. The investigators did not report any conflicts of interest.
From 1997 to 2012, the annual number of hospitalizations for opioid poisoning rose 178% among children aged 1-19 years, according to data from over 13,000 discharge records.
In 2012, there were 2,918 hospitalizations for opioid poisoning among children aged 1-19, compared with 1,049 in 1997, reported Julie R. Gaither, PhD, MPH, RN, and her associates at Yale University in New Haven, Conn. (JAMA Pediatr. 2016 Oct 31. doi: 10.1001/jamapediatrics.2016.2154).
The greatest change occurred among the youngest children, as the number of those aged 1-4 years rose from 133 in 1997 to 421 in 2012 – an increase of 217%. For those aged 15-19 years, the annual number of hospitalizations went from 715 to 2,171 (204%) over that time period, which included a slight drop from 2009 to 2012, according to the investigators, who used data from 13,052 discharges in the Agency for Healthcare Research and Quality’s Kids’ Inpatient Database.
The increase in hospitalizations for prescription opioid poisoning in children aged 10-14 years was 58% from 1997 to 2012 (rising from 171 to 272), while estimates for 5- to 9-year-olds did not meet the criteria for statistical reliability and were not included in the analysis, Dr. Gaither and her associates said.
The study was supported by grants from the National Institute on Drug Abuse. The investigators did not report any conflicts of interest.
From 1997 to 2012, the annual number of hospitalizations for opioid poisoning rose 178% among children aged 1-19 years, according to data from over 13,000 discharge records.
In 2012, there were 2,918 hospitalizations for opioid poisoning among children aged 1-19, compared with 1,049 in 1997, reported Julie R. Gaither, PhD, MPH, RN, and her associates at Yale University in New Haven, Conn. (JAMA Pediatr. 2016 Oct 31. doi: 10.1001/jamapediatrics.2016.2154).
The greatest change occurred among the youngest children, as the number of those aged 1-4 years rose from 133 in 1997 to 421 in 2012 – an increase of 217%. For those aged 15-19 years, the annual number of hospitalizations went from 715 to 2,171 (204%) over that time period, which included a slight drop from 2009 to 2012, according to the investigators, who used data from 13,052 discharges in the Agency for Healthcare Research and Quality’s Kids’ Inpatient Database.
The increase in hospitalizations for prescription opioid poisoning in children aged 10-14 years was 58% from 1997 to 2012 (rising from 171 to 272), while estimates for 5- to 9-year-olds did not meet the criteria for statistical reliability and were not included in the analysis, Dr. Gaither and her associates said.
The study was supported by grants from the National Institute on Drug Abuse. The investigators did not report any conflicts of interest.
FROM JAMA PEDIATRICS
From 1997 to 2012, the annual number of hospitalizations for opioid poisoning rose 178% among children aged 1-19 years, according to data from over 13,000 discharge records.
ECMO patients need less sedation, pain meds than previously reported
Patients on extracorporeal membrane oxygenation (ECMO) received relatively low doses of sedatives and analgesics while at a light level of sedation in a single-center prospective study of 32 patients.
In addition, patients rarely required neuromuscular blockade, investigators reported online in the Journal of Critical Care.
This finding contrasts with current guidelines on the management of pain, agitation, and delirium in patients on ECMO. The guidelines are based upon previous research that indicated the need for significant increases in sedative and analgesic doses, as well as the need for neuromuscular blockade, wrote Jeremy R. DeGrado, PharmD, of the department of pharmacy at Brigham and Women’s Hospital, Boston, and his colleagues (J Crit Care. 2016 Aug 10;37:1-6. doi: 10.1016/j.jcrc.2016.07.020).
“Patients required significantly lower doses of opioids and sedatives than previously reported in the literature and did not demonstrate a need for increasing doses throughout the study period,” the investigators said. “Continuous infusions of opioids were utilized on most ECMO days, but continuous infusions of benzodiazepines were used on less than half of all ECMO days.”
Their 2-year, prospective, observational study assessed 32 adult intensive care unit patients on ECMO support for more than 48 hours. A total of 15 patients received VA (venoarterial) ECMO and 17 received VV (venovenous) ECMO. Patients received a median daily dose of benzodiazepines (midazolam equivalents) of 24 mg and a median daily dose of opioids (fentanyl equivalents) of 3,875 mcg.
The primary indication for VA ECMO was cardiogenic shock, while VV ECMO was mainly used as a bridge to lung transplant or in patients with severe acute respiratory distress syndrome. The researchers evaluated a total of 475 ECMO days: 110 VA ECMO and 365 VV ECMO.
On average, patients were sedated to Richmond Agitation Sedation Scale scores between 0 and −1. Across all 475 ECMO days, patients were treated with continuous infusions of opioids (on 85% of ECMO days), benzodiazepines (42%), propofol (20%), dexmedetomidine (7%), and neuromuscular blocking agents (13%).
In total, patients who received VV ECMO had a higher median dose of opioids and trended toward a lower dose of benzodiazepines than those who received VA ECMO, Dr. DeGrado and his associates reported.
In total, patients in the VA arm, compared with those in the VV arm, more frequently received a continuous infusion opioid (96% vs. 82% of days) and a benzodiazepine (58% vs. 37% of days). These differences were statistically significant.
Adjunctive therapies, including antipsychotics and clonidine, were administered frequently, according to the report.
“We did not observe an increase in dose requirement over time during ECMO support, possibly due to a multi-modal pharmacologic approach. Overall, patients were not deeply sedated and rarely required neuromuscular blockade. The hypothesis that patients on ECMO require high doses of sedatives and analgesics should be further investigated,” the researchers concluded.
The authors reported that they had no disclosures.
Patients on extracorporeal membrane oxygenation (ECMO) received relatively low doses of sedatives and analgesics while at a light level of sedation in a single-center prospective study of 32 patients.
In addition, patients rarely required neuromuscular blockade, investigators reported online in the Journal of Critical Care.
This finding contrasts with current guidelines on the management of pain, agitation, and delirium in patients on ECMO. The guidelines are based upon previous research that indicated the need for significant increases in sedative and analgesic doses, as well as the need for neuromuscular blockade, wrote Jeremy R. DeGrado, PharmD, of the department of pharmacy at Brigham and Women’s Hospital, Boston, and his colleagues (J Crit Care. 2016 Aug 10;37:1-6. doi: 10.1016/j.jcrc.2016.07.020).
“Patients required significantly lower doses of opioids and sedatives than previously reported in the literature and did not demonstrate a need for increasing doses throughout the study period,” the investigators said. “Continuous infusions of opioids were utilized on most ECMO days, but continuous infusions of benzodiazepines were used on less than half of all ECMO days.”
Their 2-year, prospective, observational study assessed 32 adult intensive care unit patients on ECMO support for more than 48 hours. A total of 15 patients received VA (venoarterial) ECMO and 17 received VV (venovenous) ECMO. Patients received a median daily dose of benzodiazepines (midazolam equivalents) of 24 mg and a median daily dose of opioids (fentanyl equivalents) of 3,875 mcg.
The primary indication for VA ECMO was cardiogenic shock, while VV ECMO was mainly used as a bridge to lung transplant or in patients with severe acute respiratory distress syndrome. The researchers evaluated a total of 475 ECMO days: 110 VA ECMO and 365 VV ECMO.
On average, patients were sedated to Richmond Agitation Sedation Scale scores between 0 and −1. Across all 475 ECMO days, patients were treated with continuous infusions of opioids (on 85% of ECMO days), benzodiazepines (42%), propofol (20%), dexmedetomidine (7%), and neuromuscular blocking agents (13%).
In total, patients who received VV ECMO had a higher median dose of opioids and trended toward a lower dose of benzodiazepines than those who received VA ECMO, Dr. DeGrado and his associates reported.
In total, patients in the VA arm, compared with those in the VV arm, more frequently received a continuous infusion opioid (96% vs. 82% of days) and a benzodiazepine (58% vs. 37% of days). These differences were statistically significant.
Adjunctive therapies, including antipsychotics and clonidine, were administered frequently, according to the report.
“We did not observe an increase in dose requirement over time during ECMO support, possibly due to a multi-modal pharmacologic approach. Overall, patients were not deeply sedated and rarely required neuromuscular blockade. The hypothesis that patients on ECMO require high doses of sedatives and analgesics should be further investigated,” the researchers concluded.
The authors reported that they had no disclosures.
Patients on extracorporeal membrane oxygenation (ECMO) received relatively low doses of sedatives and analgesics while at a light level of sedation in a single-center prospective study of 32 patients.
In addition, patients rarely required neuromuscular blockade, investigators reported online in the Journal of Critical Care.
This finding contrasts with current guidelines on the management of pain, agitation, and delirium in patients on ECMO. The guidelines are based upon previous research that indicated the need for significant increases in sedative and analgesic doses, as well as the need for neuromuscular blockade, wrote Jeremy R. DeGrado, PharmD, of the department of pharmacy at Brigham and Women’s Hospital, Boston, and his colleagues (J Crit Care. 2016 Aug 10;37:1-6. doi: 10.1016/j.jcrc.2016.07.020).
“Patients required significantly lower doses of opioids and sedatives than previously reported in the literature and did not demonstrate a need for increasing doses throughout the study period,” the investigators said. “Continuous infusions of opioids were utilized on most ECMO days, but continuous infusions of benzodiazepines were used on less than half of all ECMO days.”
Their 2-year, prospective, observational study assessed 32 adult intensive care unit patients on ECMO support for more than 48 hours. A total of 15 patients received VA (venoarterial) ECMO and 17 received VV (venovenous) ECMO. Patients received a median daily dose of benzodiazepines (midazolam equivalents) of 24 mg and a median daily dose of opioids (fentanyl equivalents) of 3,875 mcg.
The primary indication for VA ECMO was cardiogenic shock, while VV ECMO was mainly used as a bridge to lung transplant or in patients with severe acute respiratory distress syndrome. The researchers evaluated a total of 475 ECMO days: 110 VA ECMO and 365 VV ECMO.
On average, patients were sedated to Richmond Agitation Sedation Scale scores between 0 and −1. Across all 475 ECMO days, patients were treated with continuous infusions of opioids (on 85% of ECMO days), benzodiazepines (42%), propofol (20%), dexmedetomidine (7%), and neuromuscular blocking agents (13%).
In total, patients who received VV ECMO had a higher median dose of opioids and trended toward a lower dose of benzodiazepines than those who received VA ECMO, Dr. DeGrado and his associates reported.
In total, patients in the VA arm, compared with those in the VV arm, more frequently received a continuous infusion opioid (96% vs. 82% of days) and a benzodiazepine (58% vs. 37% of days). These differences were statistically significant.
Adjunctive therapies, including antipsychotics and clonidine, were administered frequently, according to the report.
“We did not observe an increase in dose requirement over time during ECMO support, possibly due to a multi-modal pharmacologic approach. Overall, patients were not deeply sedated and rarely required neuromuscular blockade. The hypothesis that patients on ECMO require high doses of sedatives and analgesics should be further investigated,” the researchers concluded.
The authors reported that they had no disclosures.
FROM JOURNAL OF CRITICAL CARE
Key clinical point:
Major finding: Patients required lower doses of opioids and sedatives than previously reported and did not need increasing doses.
Data source: A single-institution, prospective study of 32 patients on extracorporeal membrane oxygenation.
Disclosures: Dr. DeGrado reported having no financial disclosures.
NRS awards grants for rosacea studies
The National Rosacea Society (NRS) is awarding funding to three new studies and continuing funding for two ongoing studies on topics that include the impact of epigenetics on rosacea, the society announced.
The first study, by Dr. Luis Garza at John Hopkins University, Baltimore, will examine epigenetic lesions in rosacea. Epigenetics, the study of how DNA can be modified to act in certain ways, “may be responsible for why rosacea persists even though keratinocytes … slough off and are replaced every 2 months,” the NRS said in a written statement.
The second study, by Dr. Wenqing Li of Brown University, Providence, R.I., will use data from the Nurses’ Health Study II to examine how hormone use and hormone levels during menopause and pregnancy affect the risk of developing rosacea.
The third study, by Dr. Anna Di Nardo of the University of California, San Diego, and her associates, is looking at “whether the release of cathelicidin antimicrobial peptides, key players in the body’s normal innate immune system response, is central to the connection between the nervous system and skin inflammation through the activation of mast cells in rosacea,” the NRS statement noted.
Ongoing studies that also are receiving funding include work by Dr. Gideon Smith of Massachusetts General Hospital, Boston, and his associates, who are examining the risk of vascular disorders in people with rosacea, and a study by Dr. Lori Lee Stohl of Cornell University, New York, who is researching how stress-related biochemicals can increase mast-cell count.
“Research supported by the NRS has led to important insights into the physiology of the disorder, providing an essential foundation for developing new and better treatments. In addition, our growing knowledge is now pointing toward potentially meaningful connections between rosacea and other systemic illnesses,” Dr. Martin Steinhoff, chairman of dermatology and director of the Charles Institute of Dermatology, University College, Dublin, and a member of the NRS Medical Advisory Board, said in the statement.
Find the full NRS statement on the society’s website.
The National Rosacea Society (NRS) is awarding funding to three new studies and continuing funding for two ongoing studies on topics that include the impact of epigenetics on rosacea, the society announced.
The first study, by Dr. Luis Garza at John Hopkins University, Baltimore, will examine epigenetic lesions in rosacea. Epigenetics, the study of how DNA can be modified to act in certain ways, “may be responsible for why rosacea persists even though keratinocytes … slough off and are replaced every 2 months,” the NRS said in a written statement.
The second study, by Dr. Wenqing Li of Brown University, Providence, R.I., will use data from the Nurses’ Health Study II to examine how hormone use and hormone levels during menopause and pregnancy affect the risk of developing rosacea.
The third study, by Dr. Anna Di Nardo of the University of California, San Diego, and her associates, is looking at “whether the release of cathelicidin antimicrobial peptides, key players in the body’s normal innate immune system response, is central to the connection between the nervous system and skin inflammation through the activation of mast cells in rosacea,” the NRS statement noted.
Ongoing studies that also are receiving funding include work by Dr. Gideon Smith of Massachusetts General Hospital, Boston, and his associates, who are examining the risk of vascular disorders in people with rosacea, and a study by Dr. Lori Lee Stohl of Cornell University, New York, who is researching how stress-related biochemicals can increase mast-cell count.
“Research supported by the NRS has led to important insights into the physiology of the disorder, providing an essential foundation for developing new and better treatments. In addition, our growing knowledge is now pointing toward potentially meaningful connections between rosacea and other systemic illnesses,” Dr. Martin Steinhoff, chairman of dermatology and director of the Charles Institute of Dermatology, University College, Dublin, and a member of the NRS Medical Advisory Board, said in the statement.
Find the full NRS statement on the society’s website.
The National Rosacea Society (NRS) is awarding funding to three new studies and continuing funding for two ongoing studies on topics that include the impact of epigenetics on rosacea, the society announced.
The first study, by Dr. Luis Garza at John Hopkins University, Baltimore, will examine epigenetic lesions in rosacea. Epigenetics, the study of how DNA can be modified to act in certain ways, “may be responsible for why rosacea persists even though keratinocytes … slough off and are replaced every 2 months,” the NRS said in a written statement.
The second study, by Dr. Wenqing Li of Brown University, Providence, R.I., will use data from the Nurses’ Health Study II to examine how hormone use and hormone levels during menopause and pregnancy affect the risk of developing rosacea.
The third study, by Dr. Anna Di Nardo of the University of California, San Diego, and her associates, is looking at “whether the release of cathelicidin antimicrobial peptides, key players in the body’s normal innate immune system response, is central to the connection between the nervous system and skin inflammation through the activation of mast cells in rosacea,” the NRS statement noted.
Ongoing studies that also are receiving funding include work by Dr. Gideon Smith of Massachusetts General Hospital, Boston, and his associates, who are examining the risk of vascular disorders in people with rosacea, and a study by Dr. Lori Lee Stohl of Cornell University, New York, who is researching how stress-related biochemicals can increase mast-cell count.
“Research supported by the NRS has led to important insights into the physiology of the disorder, providing an essential foundation for developing new and better treatments. In addition, our growing knowledge is now pointing toward potentially meaningful connections between rosacea and other systemic illnesses,” Dr. Martin Steinhoff, chairman of dermatology and director of the Charles Institute of Dermatology, University College, Dublin, and a member of the NRS Medical Advisory Board, said in the statement.
Find the full NRS statement on the society’s website.
Blood pressure changes and persistent hypertension elevate dementia risk
BALTIMORE – The impact of blood pressure on the risk of dementia in late life follows a U-shaped curve in which elevated mid-life and late-life blood pressure, as well as late-life decline in blood pressure, all independently raise the risk of dementia, according to findings from a longitudinal study of the Framingham Offspring cohort of the Framingham Heart Study.
The findings highlight the benefits of establishing and maintaining lower blood pressure in midlife in preventing or lowering the risk of dementia, Emer McGrath, MD, said at the annual meeting of the American Neurological Association.

The researchers explored the issue using data from the Framingham Offspring cohort of the Framingham Heart Study. The cohort comprises 5,124 children, and spouses of children, of the original Framingham cohort. They have been examined clinically at regular intervals since 1971. The present study focused on 1,440 individuals who had five consecutive examinations during a 15-year period anytime between 1983 and 2001 and who had not been diagnosed with dementia at the time of the final blood pressure determination.
The 1,440 participants had a mean age of 69 years at their fifth examination. Just over half were female, 20% had been diagnosed with cardiovascular disease, and slightly less than 20% had been diagnosed with diabetes. Half were using antihypertensive medications.
During a mean 8-year follow-up period, 107 individuals developed dementia. Dementia was independently associated with midlife hypertension (140/90 mm Hg or higher), with a stronger association for systolic hypertension (hazard ratio, 1.57; 95% confidence interval, 1.05-2.35). Persistence of hypertension into late life, particularly systolic hypertension (HR, 1.96; 95% CI, 1.25-3.09), was another independent risk factor for dementia.
Among individuals who did not have hypertension at midlife, a decline in systolic blood pressure to less than 100/70 mm Hg in the ensuing years increased the risk of dementia (HR, 1.63; 95% CI, 1.08-2.46).
According to the researchers, the findings support the hypothesis of the U-shaped relationship between blood pressure and dementia. “Our data also highlight the potential sustained cognitive benefits of lower midlife blood pressure,” Dr. McGrath said.
The study was funded by the Framingham Heart Study, the National Institute on Aging, and the National Institute for Neurological Diseases and Stroke. Dr. McGrath had no disclosures.
BALTIMORE – The impact of blood pressure on the risk of dementia in late life follows a U-shaped curve in which elevated mid-life and late-life blood pressure, as well as late-life decline in blood pressure, all independently raise the risk of dementia, according to findings from a longitudinal study of the Framingham Offspring cohort of the Framingham Heart Study.
The findings highlight the benefits of establishing and maintaining lower blood pressure in midlife in preventing or lowering the risk of dementia, Emer McGrath, MD, said at the annual meeting of the American Neurological Association.
The researchers explored the issue using data from the Framingham Offspring cohort of the Framingham Heart Study. The cohort comprises 5,124 children, and spouses of children, of the original Framingham cohort. They have been examined clinically at regular intervals since 1971. The present study focused on 1,440 individuals who had five consecutive examinations during a 15-year period anytime between 1983 and 2001 and who had not been diagnosed with dementia at the time of the final blood pressure determination.
The 1,440 participants had a mean age of 69 years at their fifth examination. Just over half were female, 20% had been diagnosed with cardiovascular disease, and slightly less than 20% had been diagnosed with diabetes. Half were using antihypertensive medications.
During a mean 8-year follow-up period, 107 individuals developed dementia. Dementia was independently associated with midlife hypertension (140/90 mm Hg or higher), with a stronger association for systolic hypertension (hazard ratio, 1.57; 95% confidence interval, 1.05-2.35). Persistence of hypertension into late life, particularly systolic hypertension (HR, 1.96; 95% CI, 1.25-3.09), was another independent risk factor for dementia.
Among individuals who did not have hypertension at midlife, a decline in systolic blood pressure to less than 100/70 mm Hg in the ensuing years increased the risk of dementia (HR, 1.63; 95% CI, 1.08-2.46).
According to the researchers, the findings support the hypothesis of the U-shaped relationship between blood pressure and dementia. “Our data also highlight the potential sustained cognitive benefits of lower midlife blood pressure,” Dr. McGrath said.
The study was funded by the Framingham Heart Study, the National Institute on Aging, and the National Institute for Neurological Diseases and Stroke. Dr. McGrath had no disclosures.
BALTIMORE – The impact of blood pressure on the risk of dementia in late life follows a U-shaped curve in which elevated mid-life and late-life blood pressure, as well as late-life decline in blood pressure, all independently raise the risk of dementia, according to findings from a longitudinal study of the Framingham Offspring cohort of the Framingham Heart Study.
The findings highlight the benefits of establishing and maintaining lower blood pressure in midlife in preventing or lowering the risk of dementia, Emer McGrath, MD, said at the annual meeting of the American Neurological Association.
The researchers explored the issue using data from the Framingham Offspring cohort of the Framingham Heart Study. The cohort comprises 5,124 children, and spouses of children, of the original Framingham cohort. They have been examined clinically at regular intervals since 1971. The present study focused on 1,440 individuals who had five consecutive examinations during a 15-year period anytime between 1983 and 2001 and who had not been diagnosed with dementia at the time of the final blood pressure determination.
The 1,440 participants had a mean age of 69 years at their fifth examination. Just over half were female, 20% had been diagnosed with cardiovascular disease, and slightly less than 20% had been diagnosed with diabetes. Half were using antihypertensive medications.
During a mean 8-year follow-up period, 107 individuals developed dementia. Dementia was independently associated with midlife hypertension (140/90 mm Hg or higher), with a stronger association for systolic hypertension (hazard ratio, 1.57; 95% confidence interval, 1.05-2.35). Persistence of hypertension into late life, particularly systolic hypertension (HR, 1.96; 95% CI, 1.25-3.09), was another independent risk factor for dementia.
Among individuals who did not have hypertension at midlife, a decline in systolic blood pressure to less than 100/70 mm Hg in the ensuing years increased the risk of dementia (HR, 1.63; 95% CI, 1.08-2.46).
According to the researchers, the findings support the hypothesis of the U-shaped relationship between blood pressure and dementia. “Our data also highlight the potential sustained cognitive benefits of lower midlife blood pressure,” Dr. McGrath said.
The study was funded by the Framingham Heart Study, the National Institute on Aging, and the National Institute for Neurological Diseases and Stroke. Dr. McGrath had no disclosures.
AT ANA 2016
Key clinical point:
Major finding: Persistence of hypertension into late life, particularly systolic hypertension (HR, 1.96; 95% CI, 1.25-3.09), was another independent risk factor for dementia.
Data source: Offspring cohort of the Framingham Heart Study.
Disclosures: The study was funded by the Framingham Heart Study, the National Institute on Aging, and the National Institute for Neurological Diseases and Stroke. Dr. McGrath had no disclosures.