User login
Discovery could help halt malaria transmission

Credit: Swiss TPH
Malaria parasites exploit the epigenetic regulator HP1 to promote their survival and transmission between human hosts, a new study suggests.
It appears that Plasmodium falciparum uses HP1 to control the expression of surface antigens and escape the body’s immune responses. This prolongs the parasite’s survival and enables its transmission.
Researchers believe this discovery paves the way for new strategies to prevent malaria transmission.
Till Voss, PhD, of the Swiss Tropical and Public Health Institute in Basel, and his colleagues detailed the discovery in Cell Host & Microbe.
The team knew that HP1 induces heritable condensation of chromosomal regions. As a result, genes located within these regions are not expressed.
Since this conformation is reversible, HP1-controlled genes can become activated without requiring changes in the underlying DNA sequence.
With this in mind, the researchers engineered a mutant P falciparum parasite in which HP1 expression can be shut down. And the team observed that, in HP1-depleted parasites, all of the 60 var genes became highly active.
Each var gene encodes a distinct variant of the virulence factor PfEMP1, which is displayed on the surface of the parasite-infected red blood cell. PfEMP1 is a major target of the immune system in infected humans.
Individual parasites normally express only 1 of the 60 var/PfEMP1 proteins, while keeping all other members silenced. By switching to another var/PfEMP1 variant, the parasite is able to escape existing immune responses raised against previous variants.
Dr Voss and his colleagues found that HP1 protects the PfEMP1 antigenic repertoire from being exposed to the immune system all at once.
“This finding is a major step forward in understanding the complex mechanisms responsible for antigenic variation,” Dr Voss said. “Furthermore, the tools generated in our study may be relevant for future research on malaria vaccines and immunity.”
The researchers also found that parasites lacking HP1 fail to copy their genomes and are therefore unable to proliferate. Initially, this led the team to believe that all the parasites they had cultured were dead.
However, more than 50% of these parasites turned out to be fully viable and differentiated into gametocytes, the sexual form of the malaria parasite. Gametocytes are the only form of the parasite capable of infecting a mosquito and are a prerequisite to transmit malaria between humans.
“Such a high sexual conversion rate is unprecedented,” Dr Voss said. “Usually, only around 1% of parasites undergo this switch.”
Further experiments revealed that a master transcription factor triggering sexual differentiation—AP2-G—is expressed at much higher levels in parasites lacking HP1. Under normal conditions, HP1 silences the expression of AP2-G and therefore prevents sexual conversion in most parasites.
“The switch from parasite proliferation to gametocyte differentiation is controlled epigenetically by an HP1-dependent mechanism,” Dr Voss said.
“With this knowledge in hand, and with the identification of another epigenetic regulator involved in the same process [also published in Cell Host & Microbe], we are now able to specifically track the sexual conversion pathway in molecular detail.”
This may enable the development of new drugs to prevent sexual conversion and, consequently, malaria transmission. ![]()
Credit: Swiss TPH
Malaria parasites exploit the epigenetic regulator HP1 to promote their survival and transmission between human hosts, a new study suggests.
It appears that Plasmodium falciparum uses HP1 to control the expression of surface antigens and escape the body’s immune responses. This prolongs the parasite’s survival and enables its transmission.
Researchers believe this discovery paves the way for new strategies to prevent malaria transmission.
Till Voss, PhD, of the Swiss Tropical and Public Health Institute in Basel, and his colleagues detailed the discovery in Cell Host & Microbe.
The team knew that HP1 induces heritable condensation of chromosomal regions. As a result, genes located within these regions are not expressed.
Since this conformation is reversible, HP1-controlled genes can become activated without requiring changes in the underlying DNA sequence.
With this in mind, the researchers engineered a mutant P falciparum parasite in which HP1 expression can be shut down. And the team observed that, in HP1-depleted parasites, all of the 60 var genes became highly active.
Each var gene encodes a distinct variant of the virulence factor PfEMP1, which is displayed on the surface of the parasite-infected red blood cell. PfEMP1 is a major target of the immune system in infected humans.
Individual parasites normally express only 1 of the 60 var/PfEMP1 proteins, while keeping all other members silenced. By switching to another var/PfEMP1 variant, the parasite is able to escape existing immune responses raised against previous variants.
Dr Voss and his colleagues found that HP1 protects the PfEMP1 antigenic repertoire from being exposed to the immune system all at once.
“This finding is a major step forward in understanding the complex mechanisms responsible for antigenic variation,” Dr Voss said. “Furthermore, the tools generated in our study may be relevant for future research on malaria vaccines and immunity.”
The researchers also found that parasites lacking HP1 fail to copy their genomes and are therefore unable to proliferate. Initially, this led the team to believe that all the parasites they had cultured were dead.
However, more than 50% of these parasites turned out to be fully viable and differentiated into gametocytes, the sexual form of the malaria parasite. Gametocytes are the only form of the parasite capable of infecting a mosquito and are a prerequisite to transmit malaria between humans.
“Such a high sexual conversion rate is unprecedented,” Dr Voss said. “Usually, only around 1% of parasites undergo this switch.”
Further experiments revealed that a master transcription factor triggering sexual differentiation—AP2-G—is expressed at much higher levels in parasites lacking HP1. Under normal conditions, HP1 silences the expression of AP2-G and therefore prevents sexual conversion in most parasites.
“The switch from parasite proliferation to gametocyte differentiation is controlled epigenetically by an HP1-dependent mechanism,” Dr Voss said.
“With this knowledge in hand, and with the identification of another epigenetic regulator involved in the same process [also published in Cell Host & Microbe], we are now able to specifically track the sexual conversion pathway in molecular detail.”
This may enable the development of new drugs to prevent sexual conversion and, consequently, malaria transmission. ![]()
Credit: Swiss TPH
Malaria parasites exploit the epigenetic regulator HP1 to promote their survival and transmission between human hosts, a new study suggests.
It appears that Plasmodium falciparum uses HP1 to control the expression of surface antigens and escape the body’s immune responses. This prolongs the parasite’s survival and enables its transmission.
Researchers believe this discovery paves the way for new strategies to prevent malaria transmission.
Till Voss, PhD, of the Swiss Tropical and Public Health Institute in Basel, and his colleagues detailed the discovery in Cell Host & Microbe.
The team knew that HP1 induces heritable condensation of chromosomal regions. As a result, genes located within these regions are not expressed.
Since this conformation is reversible, HP1-controlled genes can become activated without requiring changes in the underlying DNA sequence.
With this in mind, the researchers engineered a mutant P falciparum parasite in which HP1 expression can be shut down. And the team observed that, in HP1-depleted parasites, all of the 60 var genes became highly active.
Each var gene encodes a distinct variant of the virulence factor PfEMP1, which is displayed on the surface of the parasite-infected red blood cell. PfEMP1 is a major target of the immune system in infected humans.
Individual parasites normally express only 1 of the 60 var/PfEMP1 proteins, while keeping all other members silenced. By switching to another var/PfEMP1 variant, the parasite is able to escape existing immune responses raised against previous variants.
Dr Voss and his colleagues found that HP1 protects the PfEMP1 antigenic repertoire from being exposed to the immune system all at once.
“This finding is a major step forward in understanding the complex mechanisms responsible for antigenic variation,” Dr Voss said. “Furthermore, the tools generated in our study may be relevant for future research on malaria vaccines and immunity.”
The researchers also found that parasites lacking HP1 fail to copy their genomes and are therefore unable to proliferate. Initially, this led the team to believe that all the parasites they had cultured were dead.
However, more than 50% of these parasites turned out to be fully viable and differentiated into gametocytes, the sexual form of the malaria parasite. Gametocytes are the only form of the parasite capable of infecting a mosquito and are a prerequisite to transmit malaria between humans.
“Such a high sexual conversion rate is unprecedented,” Dr Voss said. “Usually, only around 1% of parasites undergo this switch.”
Further experiments revealed that a master transcription factor triggering sexual differentiation—AP2-G—is expressed at much higher levels in parasites lacking HP1. Under normal conditions, HP1 silences the expression of AP2-G and therefore prevents sexual conversion in most parasites.
“The switch from parasite proliferation to gametocyte differentiation is controlled epigenetically by an HP1-dependent mechanism,” Dr Voss said.
“With this knowledge in hand, and with the identification of another epigenetic regulator involved in the same process [also published in Cell Host & Microbe], we are now able to specifically track the sexual conversion pathway in molecular detail.”
This may enable the development of new drugs to prevent sexual conversion and, consequently, malaria transmission. ![]()
Protein appears essential for NK cell survival

Credit: Walter and Eliza Hall
Institute of Medical Research
New research suggests the Mcl-1 protein is crucial for the survival of natural killer (NK) cells and, therefore, innate immune responses.
Researchers deleted Mcl-1 from NK cells in mice and observed a loss of the cells from all tissues.
This made the mice more receptive to allogeneic hematopoietic stem cell transplants and resistant to toxic shock following a sepsis challenge, but it also made the mice susceptible to melanoma metastases.
The researchers believe their findings, published in Nature Communications, will help to determine how NK cells can be manipulated to treat a range of disorders.
They said Mcl-1 could be a target for boosting or depleting NK cell populations when necessary.
The researchers first discovered that Mcl-1 is highly expressed in NK cells. And Mcl-1 is regulated by IL-15 in a dose-dependent manner via STAT5 phosphorylation and subsequent binding to the 3′-UTR of Mcl-1.
“We showed Mcl-1 levels inside the cell increase in response to [IL-15],” said study author Nick Huntington, PhD, of the Walter and Eliza Hall Institute of Medical Research in Victoria, Australia.
“We previously knew IL-15 boosted production and survival of natural killer cells, and we have shown that IL-15 does this by initiating a cascade of signals that tell the natural killer cell to produce Mcl-1 to keep it alive.”
To further explore this phenomenon, the researchers deleted Mcl-1 from NK cells in mice and observed depletion of the cells in all tissues. The team said this was the result of a failure to antagonize pro-apoptotic proteins in the outer mitochondrial membrane.
Additional experiments showed that the mice needed the NK cells to fight off invading melanoma cells that had spread past the original cancer site.
“Without natural killer cells, the body was unable to destroy melanoma metastases that had spread throughout the body, and the cancers overwhelmed the lungs,” Dr Huntington said.
However, the loss of NK cells also made mice more receptive to allogeneic stem cell transplants and resistant to toxic shock after polymicrobial sepsis challenge.
“Natural killer cells led the response that caused rejection of donor stem cells in bone marrow transplantations,” Dr Huntington said. “They also produced inflammatory signals that can result in toxic shock syndrome, a potentially fatal illness caused by bacterial toxins that causes a whole-body inflammatory reaction.”
The researchers said these results clearly show a non-redundant pathway linking IL-15 to Mcl-1 in the maintenance of NK cells and innate immune responses.
Dr Huntington said the discovery provides a solid lead to look for ways of boosting or depleting NK cells when necessary.
“Now that we know the critical importance of Mcl-1 in the survival of natural killer cells,” he said, “we are investigating how we might manipulate this protein, or other proteins in the pathway, to treat disease.” ![]()
Credit: Walter and Eliza Hall
Institute of Medical Research
New research suggests the Mcl-1 protein is crucial for the survival of natural killer (NK) cells and, therefore, innate immune responses.
Researchers deleted Mcl-1 from NK cells in mice and observed a loss of the cells from all tissues.
This made the mice more receptive to allogeneic hematopoietic stem cell transplants and resistant to toxic shock following a sepsis challenge, but it also made the mice susceptible to melanoma metastases.
The researchers believe their findings, published in Nature Communications, will help to determine how NK cells can be manipulated to treat a range of disorders.
They said Mcl-1 could be a target for boosting or depleting NK cell populations when necessary.
The researchers first discovered that Mcl-1 is highly expressed in NK cells. And Mcl-1 is regulated by IL-15 in a dose-dependent manner via STAT5 phosphorylation and subsequent binding to the 3′-UTR of Mcl-1.
“We showed Mcl-1 levels inside the cell increase in response to [IL-15],” said study author Nick Huntington, PhD, of the Walter and Eliza Hall Institute of Medical Research in Victoria, Australia.
“We previously knew IL-15 boosted production and survival of natural killer cells, and we have shown that IL-15 does this by initiating a cascade of signals that tell the natural killer cell to produce Mcl-1 to keep it alive.”
To further explore this phenomenon, the researchers deleted Mcl-1 from NK cells in mice and observed depletion of the cells in all tissues. The team said this was the result of a failure to antagonize pro-apoptotic proteins in the outer mitochondrial membrane.
Additional experiments showed that the mice needed the NK cells to fight off invading melanoma cells that had spread past the original cancer site.
“Without natural killer cells, the body was unable to destroy melanoma metastases that had spread throughout the body, and the cancers overwhelmed the lungs,” Dr Huntington said.
However, the loss of NK cells also made mice more receptive to allogeneic stem cell transplants and resistant to toxic shock after polymicrobial sepsis challenge.
“Natural killer cells led the response that caused rejection of donor stem cells in bone marrow transplantations,” Dr Huntington said. “They also produced inflammatory signals that can result in toxic shock syndrome, a potentially fatal illness caused by bacterial toxins that causes a whole-body inflammatory reaction.”
The researchers said these results clearly show a non-redundant pathway linking IL-15 to Mcl-1 in the maintenance of NK cells and innate immune responses.
Dr Huntington said the discovery provides a solid lead to look for ways of boosting or depleting NK cells when necessary.
“Now that we know the critical importance of Mcl-1 in the survival of natural killer cells,” he said, “we are investigating how we might manipulate this protein, or other proteins in the pathway, to treat disease.” ![]()
Credit: Walter and Eliza Hall
Institute of Medical Research
New research suggests the Mcl-1 protein is crucial for the survival of natural killer (NK) cells and, therefore, innate immune responses.
Researchers deleted Mcl-1 from NK cells in mice and observed a loss of the cells from all tissues.
This made the mice more receptive to allogeneic hematopoietic stem cell transplants and resistant to toxic shock following a sepsis challenge, but it also made the mice susceptible to melanoma metastases.
The researchers believe their findings, published in Nature Communications, will help to determine how NK cells can be manipulated to treat a range of disorders.
They said Mcl-1 could be a target for boosting or depleting NK cell populations when necessary.
The researchers first discovered that Mcl-1 is highly expressed in NK cells. And Mcl-1 is regulated by IL-15 in a dose-dependent manner via STAT5 phosphorylation and subsequent binding to the 3′-UTR of Mcl-1.
“We showed Mcl-1 levels inside the cell increase in response to [IL-15],” said study author Nick Huntington, PhD, of the Walter and Eliza Hall Institute of Medical Research in Victoria, Australia.
“We previously knew IL-15 boosted production and survival of natural killer cells, and we have shown that IL-15 does this by initiating a cascade of signals that tell the natural killer cell to produce Mcl-1 to keep it alive.”
To further explore this phenomenon, the researchers deleted Mcl-1 from NK cells in mice and observed depletion of the cells in all tissues. The team said this was the result of a failure to antagonize pro-apoptotic proteins in the outer mitochondrial membrane.
Additional experiments showed that the mice needed the NK cells to fight off invading melanoma cells that had spread past the original cancer site.
“Without natural killer cells, the body was unable to destroy melanoma metastases that had spread throughout the body, and the cancers overwhelmed the lungs,” Dr Huntington said.
However, the loss of NK cells also made mice more receptive to allogeneic stem cell transplants and resistant to toxic shock after polymicrobial sepsis challenge.
“Natural killer cells led the response that caused rejection of donor stem cells in bone marrow transplantations,” Dr Huntington said. “They also produced inflammatory signals that can result in toxic shock syndrome, a potentially fatal illness caused by bacterial toxins that causes a whole-body inflammatory reaction.”
The researchers said these results clearly show a non-redundant pathway linking IL-15 to Mcl-1 in the maintenance of NK cells and innate immune responses.
Dr Huntington said the discovery provides a solid lead to look for ways of boosting or depleting NK cells when necessary.
“Now that we know the critical importance of Mcl-1 in the survival of natural killer cells,” he said, “we are investigating how we might manipulate this protein, or other proteins in the pathway, to treat disease.” ![]()
Method may help treat SCD, other disorders
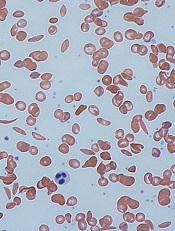
Credit: Graham Beards
Scientists have discovered that manipulating gene regulation can cause red blood cells to produce fetal hemoglobin, a finding that may have implications for sickle cell disease (SCD) and other hemoglobinopathies.
The researchers used protein-engineering techniques to force chromatin fiber into looped structures that contact DNA at specific sites to preferentially activate genes that regulate hemoglobin.
The team described the work in Cell. The research was previously presented at the 2013 ASH Annual Meeting.
Key to the researcher’s method is a developmental transition that normally occurs in the blood of newborns. A biological switch regulates a changeover from fetal hemoglobin to adult hemoglobin as it begins to silence the genes that produce fetal hemoglobin.
Hematologists have long known that SCD patients with elevated levels of fetal hemoglobin have a milder form of the disease.
“This observation has been a major driver in the field to understand the molecular basis of the mechanisms that control the biological switch, with the ultimate goal to reverse it,” said study author Gerd A. Blobel, MD, PhD, of The Children’s Hospital of Philadelphia in Pennsylvania.
In previous research, his team used bioengineering techniques to adapt zinc-finger proteins to latch onto specific DNA sites far apart on a chromosome. The chromatin loop that results transmits regulatory signals for specific genes.
In their current work, the researchers custom-designed zinc fingers to flip the biological switch in hematopoietic stem cells, reactivating the genes expressing fetal hemoglobin at the expense of the genes expressing adult hemoglobin.
The team achieved these results in cultured blood cells from adult mice and adult humans.
The researchers are now planning to test the approach in animal models of SCD. If this strategy corrects the disease in animals, it may set the stage to move to human trials.
Dr Blobel also noted that, in principle, the forced chromatin looping approach could be applied to other hemoglobin-related disorders, such as certain forms of thalassemia. ![]()
Credit: Graham Beards
Scientists have discovered that manipulating gene regulation can cause red blood cells to produce fetal hemoglobin, a finding that may have implications for sickle cell disease (SCD) and other hemoglobinopathies.
The researchers used protein-engineering techniques to force chromatin fiber into looped structures that contact DNA at specific sites to preferentially activate genes that regulate hemoglobin.
The team described the work in Cell. The research was previously presented at the 2013 ASH Annual Meeting.
Key to the researcher’s method is a developmental transition that normally occurs in the blood of newborns. A biological switch regulates a changeover from fetal hemoglobin to adult hemoglobin as it begins to silence the genes that produce fetal hemoglobin.
Hematologists have long known that SCD patients with elevated levels of fetal hemoglobin have a milder form of the disease.
“This observation has been a major driver in the field to understand the molecular basis of the mechanisms that control the biological switch, with the ultimate goal to reverse it,” said study author Gerd A. Blobel, MD, PhD, of The Children’s Hospital of Philadelphia in Pennsylvania.
In previous research, his team used bioengineering techniques to adapt zinc-finger proteins to latch onto specific DNA sites far apart on a chromosome. The chromatin loop that results transmits regulatory signals for specific genes.
In their current work, the researchers custom-designed zinc fingers to flip the biological switch in hematopoietic stem cells, reactivating the genes expressing fetal hemoglobin at the expense of the genes expressing adult hemoglobin.
The team achieved these results in cultured blood cells from adult mice and adult humans.
The researchers are now planning to test the approach in animal models of SCD. If this strategy corrects the disease in animals, it may set the stage to move to human trials.
Dr Blobel also noted that, in principle, the forced chromatin looping approach could be applied to other hemoglobin-related disorders, such as certain forms of thalassemia. ![]()
Credit: Graham Beards
Scientists have discovered that manipulating gene regulation can cause red blood cells to produce fetal hemoglobin, a finding that may have implications for sickle cell disease (SCD) and other hemoglobinopathies.
The researchers used protein-engineering techniques to force chromatin fiber into looped structures that contact DNA at specific sites to preferentially activate genes that regulate hemoglobin.
The team described the work in Cell. The research was previously presented at the 2013 ASH Annual Meeting.
Key to the researcher’s method is a developmental transition that normally occurs in the blood of newborns. A biological switch regulates a changeover from fetal hemoglobin to adult hemoglobin as it begins to silence the genes that produce fetal hemoglobin.
Hematologists have long known that SCD patients with elevated levels of fetal hemoglobin have a milder form of the disease.
“This observation has been a major driver in the field to understand the molecular basis of the mechanisms that control the biological switch, with the ultimate goal to reverse it,” said study author Gerd A. Blobel, MD, PhD, of The Children’s Hospital of Philadelphia in Pennsylvania.
In previous research, his team used bioengineering techniques to adapt zinc-finger proteins to latch onto specific DNA sites far apart on a chromosome. The chromatin loop that results transmits regulatory signals for specific genes.
In their current work, the researchers custom-designed zinc fingers to flip the biological switch in hematopoietic stem cells, reactivating the genes expressing fetal hemoglobin at the expense of the genes expressing adult hemoglobin.
The team achieved these results in cultured blood cells from adult mice and adult humans.
The researchers are now planning to test the approach in animal models of SCD. If this strategy corrects the disease in animals, it may set the stage to move to human trials.
Dr Blobel also noted that, in principle, the forced chromatin looping approach could be applied to other hemoglobin-related disorders, such as certain forms of thalassemia. ![]()
MM drug disappoints in phase 3 trial
The proteasome inhibitor carfilzomib (Kyprolis) did not meet its primary endpoint in the phase 3 FOCUS trial, according to the drug’s developers.
Single-agent carfilzomib did not improve overall survival compared to an active control regimen of corticosteroids plus optional cyclophosphamide in patients with relapsed and refractory multiple myeloma (MM).
This result raises questions about carfilzomib’s chances for regulatory approval around the world.
However, the companies developing the drug, Amgen and its subsidiary Onyx Pharmaceuticals, Inc., said results of the phase 3 ASPIRE trial should be sufficient to support carfilzomib’s approval.
At present, carfilzomib has accelerated approval from the US Food and Drug Administration for the treatment of MM patients who have received at least 2 prior therapies, including bortezomib and an immunomodulatory agent, and have demonstrated disease progression on or within 60 days of completing their last therapy.
That approval was based on response rates observed with carfilzomib. For the drug to gain full approval, it must demonstrate a clinical benefit.
The FOCUS trial
For the FOCUS trial, researchers enrolled 315 patients with relapsed and advanced refractory MM.
Patients were randomized to receive carfilzomib or an active control regimen consisting of low-dose dexamethasone, or equivalent corticosteroids, plus optional cyclophosphamide.
Nearly all patients in the control arm received cyclophosphamide. Patients were heavily pretreated and had received a median of 5 treatment regimens prior to study entry.
The trial’s primary endpoint was overall survival, and there was no significant difference between the 2 treatment arms. The hazard ratio was 0.975.
Treatment discontinuation due to adverse events and on-study deaths were comparable between the treatment arms. The rate of cardiac events in the carfilzomib arm was consistent with the current US label.
However, there was an increase in the incidence of renal adverse events of all grades observed in the carfilzomib arm compared to the active control arm and the label.
Full prescribing information for carfilzomib is available at www.kyprolis.com.
Amgen and Onyx said detailed results from the FOCUS trial will be submitted for presentation at an upcoming scientific meeting. ![]()
The proteasome inhibitor carfilzomib (Kyprolis) did not meet its primary endpoint in the phase 3 FOCUS trial, according to the drug’s developers.
Single-agent carfilzomib did not improve overall survival compared to an active control regimen of corticosteroids plus optional cyclophosphamide in patients with relapsed and refractory multiple myeloma (MM).
This result raises questions about carfilzomib’s chances for regulatory approval around the world.
However, the companies developing the drug, Amgen and its subsidiary Onyx Pharmaceuticals, Inc., said results of the phase 3 ASPIRE trial should be sufficient to support carfilzomib’s approval.
At present, carfilzomib has accelerated approval from the US Food and Drug Administration for the treatment of MM patients who have received at least 2 prior therapies, including bortezomib and an immunomodulatory agent, and have demonstrated disease progression on or within 60 days of completing their last therapy.
That approval was based on response rates observed with carfilzomib. For the drug to gain full approval, it must demonstrate a clinical benefit.
The FOCUS trial
For the FOCUS trial, researchers enrolled 315 patients with relapsed and advanced refractory MM.
Patients were randomized to receive carfilzomib or an active control regimen consisting of low-dose dexamethasone, or equivalent corticosteroids, plus optional cyclophosphamide.
Nearly all patients in the control arm received cyclophosphamide. Patients were heavily pretreated and had received a median of 5 treatment regimens prior to study entry.
The trial’s primary endpoint was overall survival, and there was no significant difference between the 2 treatment arms. The hazard ratio was 0.975.
Treatment discontinuation due to adverse events and on-study deaths were comparable between the treatment arms. The rate of cardiac events in the carfilzomib arm was consistent with the current US label.
However, there was an increase in the incidence of renal adverse events of all grades observed in the carfilzomib arm compared to the active control arm and the label.
Full prescribing information for carfilzomib is available at www.kyprolis.com.
Amgen and Onyx said detailed results from the FOCUS trial will be submitted for presentation at an upcoming scientific meeting. ![]()
The proteasome inhibitor carfilzomib (Kyprolis) did not meet its primary endpoint in the phase 3 FOCUS trial, according to the drug’s developers.
Single-agent carfilzomib did not improve overall survival compared to an active control regimen of corticosteroids plus optional cyclophosphamide in patients with relapsed and refractory multiple myeloma (MM).
This result raises questions about carfilzomib’s chances for regulatory approval around the world.
However, the companies developing the drug, Amgen and its subsidiary Onyx Pharmaceuticals, Inc., said results of the phase 3 ASPIRE trial should be sufficient to support carfilzomib’s approval.
At present, carfilzomib has accelerated approval from the US Food and Drug Administration for the treatment of MM patients who have received at least 2 prior therapies, including bortezomib and an immunomodulatory agent, and have demonstrated disease progression on or within 60 days of completing their last therapy.
That approval was based on response rates observed with carfilzomib. For the drug to gain full approval, it must demonstrate a clinical benefit.
The FOCUS trial
For the FOCUS trial, researchers enrolled 315 patients with relapsed and advanced refractory MM.
Patients were randomized to receive carfilzomib or an active control regimen consisting of low-dose dexamethasone, or equivalent corticosteroids, plus optional cyclophosphamide.
Nearly all patients in the control arm received cyclophosphamide. Patients were heavily pretreated and had received a median of 5 treatment regimens prior to study entry.
The trial’s primary endpoint was overall survival, and there was no significant difference between the 2 treatment arms. The hazard ratio was 0.975.
Treatment discontinuation due to adverse events and on-study deaths were comparable between the treatment arms. The rate of cardiac events in the carfilzomib arm was consistent with the current US label.
However, there was an increase in the incidence of renal adverse events of all grades observed in the carfilzomib arm compared to the active control arm and the label.
Full prescribing information for carfilzomib is available at www.kyprolis.com.
Amgen and Onyx said detailed results from the FOCUS trial will be submitted for presentation at an upcoming scientific meeting. ![]()
Discovery could change management of ALL
Two genes appear to play a key role in acute lymphoblastic leukemia (ALL), particularly for patients who also have Down syndrome.
Researchers found evidence suggesting that 2 in 3 cases of ALL among patients with Down syndrome may be caused by mutations in RAS or JAK2, but the mutations tend not to occur together.
The group therefore believes that if we can begin to identify which of the genes is mutated in ALL patients, we can tailor therapy accordingly.
“We believe our findings are a breakthrough in understanding the underlying causes of leukemia, and, eventually, we hope to design more tailored and effective treatment for this cancer, with less toxic drugs and less side effects,” said study author Dean Nizetic, MD, PhD, a professor at both Queen Mary University of London in the UK and Lee Kong Chian School of Medicine in Singapore.
He and his colleagues reported their findings in Nature Communications.
The researchers conducted full-exome or targeted sequencing in 42 samples from 39 patients with ALL and Down syndrome. The team found similar recurrence rates for driver mutations in RAS (15/42), JAK2 mutations (12/42), and P2RY8-CRLF2 fusions (14/42).
Together, RAS and JAK2 mutations drove two-thirds of the ALL cases, though the mutations were almost completely mutually exclusive (P=0.016).
The researchers also noted that, in two-thirds of patients with relapsed ALL, there was a switch from a primary JAK2- or PTPN11-mutated subclone to a RAS-mutated subclone in relapse.
Moving forward, the team plans to conduct more studies to determine how else RAS and JAK2 might affect children who have ALL, with or without Down syndrome. ![]()
Two genes appear to play a key role in acute lymphoblastic leukemia (ALL), particularly for patients who also have Down syndrome.
Researchers found evidence suggesting that 2 in 3 cases of ALL among patients with Down syndrome may be caused by mutations in RAS or JAK2, but the mutations tend not to occur together.
The group therefore believes that if we can begin to identify which of the genes is mutated in ALL patients, we can tailor therapy accordingly.
“We believe our findings are a breakthrough in understanding the underlying causes of leukemia, and, eventually, we hope to design more tailored and effective treatment for this cancer, with less toxic drugs and less side effects,” said study author Dean Nizetic, MD, PhD, a professor at both Queen Mary University of London in the UK and Lee Kong Chian School of Medicine in Singapore.
He and his colleagues reported their findings in Nature Communications.
The researchers conducted full-exome or targeted sequencing in 42 samples from 39 patients with ALL and Down syndrome. The team found similar recurrence rates for driver mutations in RAS (15/42), JAK2 mutations (12/42), and P2RY8-CRLF2 fusions (14/42).
Together, RAS and JAK2 mutations drove two-thirds of the ALL cases, though the mutations were almost completely mutually exclusive (P=0.016).
The researchers also noted that, in two-thirds of patients with relapsed ALL, there was a switch from a primary JAK2- or PTPN11-mutated subclone to a RAS-mutated subclone in relapse.
Moving forward, the team plans to conduct more studies to determine how else RAS and JAK2 might affect children who have ALL, with or without Down syndrome. ![]()
Two genes appear to play a key role in acute lymphoblastic leukemia (ALL), particularly for patients who also have Down syndrome.
Researchers found evidence suggesting that 2 in 3 cases of ALL among patients with Down syndrome may be caused by mutations in RAS or JAK2, but the mutations tend not to occur together.
The group therefore believes that if we can begin to identify which of the genes is mutated in ALL patients, we can tailor therapy accordingly.
“We believe our findings are a breakthrough in understanding the underlying causes of leukemia, and, eventually, we hope to design more tailored and effective treatment for this cancer, with less toxic drugs and less side effects,” said study author Dean Nizetic, MD, PhD, a professor at both Queen Mary University of London in the UK and Lee Kong Chian School of Medicine in Singapore.
He and his colleagues reported their findings in Nature Communications.
The researchers conducted full-exome or targeted sequencing in 42 samples from 39 patients with ALL and Down syndrome. The team found similar recurrence rates for driver mutations in RAS (15/42), JAK2 mutations (12/42), and P2RY8-CRLF2 fusions (14/42).
Together, RAS and JAK2 mutations drove two-thirds of the ALL cases, though the mutations were almost completely mutually exclusive (P=0.016).
The researchers also noted that, in two-thirds of patients with relapsed ALL, there was a switch from a primary JAK2- or PTPN11-mutated subclone to a RAS-mutated subclone in relapse.
Moving forward, the team plans to conduct more studies to determine how else RAS and JAK2 might affect children who have ALL, with or without Down syndrome. ![]()
Drug on the fast track to treat AML

Credit: Esther Dyson
The US Food and Drug Administration (FDA) has granted fast track designation for AG-221 to treat acute myelogenous leukemia (AML) patients with mutated isocitrate dehydrogenase-2 (IDH2).
AG-221 is an IDH2 inhibitor under evaluation in a phase 1 trial of patients with advanced hematologic malignancies.
Results from this trial were presented at the 19th Congress of the European Hematology Association, which took place in Milan in June.
The FDA’s fast track drug development program is designed to expedite clinical development and submission of a new drug application (NDA) for drugs with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates meetings between the FDA and the company developing a drug to discuss all aspects of development to support approval. It also affords the developer the opportunity to submit sections of an NDA on a rolling basis as data become available, so the FDA does not have wait for the entire NDA submission before beginning its review.
AG-221 also recently received orphan designation as a treatment for AML. The FDA grants orphan status to support the development of drugs for underserved patient populations or rare disorders that affect fewer than 200,000 people in the US.
Orphan designation affords the drug’s developer certain benefits, including market exclusivity upon regulatory approval, exemption of FDA application fees, and tax credits for qualified clinical trials.
Phase 1 trial results
Thus far in the phase 1 study, AG-221 has proven active and well-tolerated in patients with AML, myelodysplastic syndromes (MDS), and chronic myelomonocytic leukemia (CMML).
The trial included 35 patients with a median age of 68 years (range, 48-81).
Twenty-seven patients had relapsed/refractory AML, 4 had relapsed/refractory MDS, 2 had untreated AML, 1 had CMML, and 1 had granulocytic sarcoma. Thirty-one patients had R140Q IDH2 mutations, and 4 had R172K IDH2 mutations.
The patients received AG-221 at doses ranging from 30 mg BID to 150 mg QD. Patients completed a median of 1 cycle of treatment (range, <1-5+) and a mean of 2 cycles.
The drug was generally well-tolerated, largely prompting grade 1 or 2 adverse events. Grade 3 or higher events included thrombocytopenia (n=3), anemia (n=1), febrile neutropenia (n=3), sepsis (n=3), diarrhea (n=1), fatigue (n=1), leukocytosis (n=2), neutropenia (n=1), and rash (n=1).
Four patients had serious events possibly related to treatment. One patient had grade 3 confusion and grade 5 respiratory failure. One patient had grade 3 leukocytosis, grade 3 anorexia, and grade 1 nausea. One patient had grade 3 diarrhea. And 1 patient had grade 3 leukocytosis.
Twenty-five patients were evaluable for response. There were 6 complete responses (CRs), 2 CRs with incomplete platelet recovery, 1 CR with incomplete hematologic recovery, and 5 partial responses. Five patients had stable disease, and 6 had progressive disease.
The most responses occurred among patients who received AG-221 at 50 mg BID, and most responses occurred in cycle 1.
Twelve of the 14 responses are ongoing. Of the 8 patients who achieved a CR or CR with incomplete platelet recovery, 5 have lasted more than 2.5 months (range, 1-4+ months). And the 5 patients with stable disease remain on study.
This study is sponsored by Agios Pharmaceuticals Inc., the company developing AG-221 in collaboration with Celgene. ![]()
Credit: Esther Dyson
The US Food and Drug Administration (FDA) has granted fast track designation for AG-221 to treat acute myelogenous leukemia (AML) patients with mutated isocitrate dehydrogenase-2 (IDH2).
AG-221 is an IDH2 inhibitor under evaluation in a phase 1 trial of patients with advanced hematologic malignancies.
Results from this trial were presented at the 19th Congress of the European Hematology Association, which took place in Milan in June.
The FDA’s fast track drug development program is designed to expedite clinical development and submission of a new drug application (NDA) for drugs with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates meetings between the FDA and the company developing a drug to discuss all aspects of development to support approval. It also affords the developer the opportunity to submit sections of an NDA on a rolling basis as data become available, so the FDA does not have wait for the entire NDA submission before beginning its review.
AG-221 also recently received orphan designation as a treatment for AML. The FDA grants orphan status to support the development of drugs for underserved patient populations or rare disorders that affect fewer than 200,000 people in the US.
Orphan designation affords the drug’s developer certain benefits, including market exclusivity upon regulatory approval, exemption of FDA application fees, and tax credits for qualified clinical trials.
Phase 1 trial results
Thus far in the phase 1 study, AG-221 has proven active and well-tolerated in patients with AML, myelodysplastic syndromes (MDS), and chronic myelomonocytic leukemia (CMML).
The trial included 35 patients with a median age of 68 years (range, 48-81).
Twenty-seven patients had relapsed/refractory AML, 4 had relapsed/refractory MDS, 2 had untreated AML, 1 had CMML, and 1 had granulocytic sarcoma. Thirty-one patients had R140Q IDH2 mutations, and 4 had R172K IDH2 mutations.
The patients received AG-221 at doses ranging from 30 mg BID to 150 mg QD. Patients completed a median of 1 cycle of treatment (range, <1-5+) and a mean of 2 cycles.
The drug was generally well-tolerated, largely prompting grade 1 or 2 adverse events. Grade 3 or higher events included thrombocytopenia (n=3), anemia (n=1), febrile neutropenia (n=3), sepsis (n=3), diarrhea (n=1), fatigue (n=1), leukocytosis (n=2), neutropenia (n=1), and rash (n=1).
Four patients had serious events possibly related to treatment. One patient had grade 3 confusion and grade 5 respiratory failure. One patient had grade 3 leukocytosis, grade 3 anorexia, and grade 1 nausea. One patient had grade 3 diarrhea. And 1 patient had grade 3 leukocytosis.
Twenty-five patients were evaluable for response. There were 6 complete responses (CRs), 2 CRs with incomplete platelet recovery, 1 CR with incomplete hematologic recovery, and 5 partial responses. Five patients had stable disease, and 6 had progressive disease.
The most responses occurred among patients who received AG-221 at 50 mg BID, and most responses occurred in cycle 1.
Twelve of the 14 responses are ongoing. Of the 8 patients who achieved a CR or CR with incomplete platelet recovery, 5 have lasted more than 2.5 months (range, 1-4+ months). And the 5 patients with stable disease remain on study.
This study is sponsored by Agios Pharmaceuticals Inc., the company developing AG-221 in collaboration with Celgene. ![]()
Credit: Esther Dyson
The US Food and Drug Administration (FDA) has granted fast track designation for AG-221 to treat acute myelogenous leukemia (AML) patients with mutated isocitrate dehydrogenase-2 (IDH2).
AG-221 is an IDH2 inhibitor under evaluation in a phase 1 trial of patients with advanced hematologic malignancies.
Results from this trial were presented at the 19th Congress of the European Hematology Association, which took place in Milan in June.
The FDA’s fast track drug development program is designed to expedite clinical development and submission of a new drug application (NDA) for drugs with the potential to treat serious or life-threatening conditions and address unmet medical needs.
Fast track designation facilitates meetings between the FDA and the company developing a drug to discuss all aspects of development to support approval. It also affords the developer the opportunity to submit sections of an NDA on a rolling basis as data become available, so the FDA does not have wait for the entire NDA submission before beginning its review.
AG-221 also recently received orphan designation as a treatment for AML. The FDA grants orphan status to support the development of drugs for underserved patient populations or rare disorders that affect fewer than 200,000 people in the US.
Orphan designation affords the drug’s developer certain benefits, including market exclusivity upon regulatory approval, exemption of FDA application fees, and tax credits for qualified clinical trials.
Phase 1 trial results
Thus far in the phase 1 study, AG-221 has proven active and well-tolerated in patients with AML, myelodysplastic syndromes (MDS), and chronic myelomonocytic leukemia (CMML).
The trial included 35 patients with a median age of 68 years (range, 48-81).
Twenty-seven patients had relapsed/refractory AML, 4 had relapsed/refractory MDS, 2 had untreated AML, 1 had CMML, and 1 had granulocytic sarcoma. Thirty-one patients had R140Q IDH2 mutations, and 4 had R172K IDH2 mutations.
The patients received AG-221 at doses ranging from 30 mg BID to 150 mg QD. Patients completed a median of 1 cycle of treatment (range, <1-5+) and a mean of 2 cycles.
The drug was generally well-tolerated, largely prompting grade 1 or 2 adverse events. Grade 3 or higher events included thrombocytopenia (n=3), anemia (n=1), febrile neutropenia (n=3), sepsis (n=3), diarrhea (n=1), fatigue (n=1), leukocytosis (n=2), neutropenia (n=1), and rash (n=1).
Four patients had serious events possibly related to treatment. One patient had grade 3 confusion and grade 5 respiratory failure. One patient had grade 3 leukocytosis, grade 3 anorexia, and grade 1 nausea. One patient had grade 3 diarrhea. And 1 patient had grade 3 leukocytosis.
Twenty-five patients were evaluable for response. There were 6 complete responses (CRs), 2 CRs with incomplete platelet recovery, 1 CR with incomplete hematologic recovery, and 5 partial responses. Five patients had stable disease, and 6 had progressive disease.
The most responses occurred among patients who received AG-221 at 50 mg BID, and most responses occurred in cycle 1.
Twelve of the 14 responses are ongoing. Of the 8 patients who achieved a CR or CR with incomplete platelet recovery, 5 have lasted more than 2.5 months (range, 1-4+ months). And the 5 patients with stable disease remain on study.
This study is sponsored by Agios Pharmaceuticals Inc., the company developing AG-221 in collaboration with Celgene. ![]()
Method could overcome chemoresistance in lymphoma

Credit: Rhoda Baer
Indirectly targeting the prosurvival protein Mcl-1 can reverse chemoresistance in lymphoma and other cancer cells, investigators have reported in The Journal of Biological Chemistry.
The team found that targeting an enzyme known as protein phosphatase 2A (PP2A) inhibited Mcl-1 dephosphorylation, which prompted the loss of Mcl-1 in chemoresistant Burkitt lymphoma cells.
“These findings may lead to a new target for chemoresistant cancer cells,” said Ruth W. Craig, PhD, of Geisel School of Medicine at Dartmouth in Hanover, New Hampshire.
“These cells are resistant to multiple types of standard chemotherapeutic agents because of [Mcl-1] overexpression. However, Mcl-1 expression plummets when we inhibit [PP2A], and then cancer cells subsequently die.”
Dr Craig and her colleagues found that PP2A can be inhibited to stop the removal of phosphate groups from a regulatory motif in Mcl-1 referred to as the PEST region (enriched with amino acids proline, glutamic acid, serine, and threonine).
And inhibiting the removal of phosphate groups, such as at threonine-163 and serine-159, targets the Mcl-1 protein for rapid destruction.
To reach this conclusion, the investigators studied BL41-3 cells, a Burkitt lymphoma cell line that overexpresses Mcl-1 and has proven resistant to multiple treatments.
The team exposed BL41-3 cells to 2 different PP2A inhibitors, okadaic acid and calyculin A. Both drugs prompted an increase in phosphorylation at threonine-163 and serine-159, as well as a decrease in Mcl-1 expression.
Further investigation confirmed that PP2A interacts with Mcl-1. And, as with therapeutic targeting, shRNA knockdown of PP2A/Aα increased phosphorylation while decreasing Mcl-1 expression.
Finally, the investigators showed the increase in Mcl-1 phosphorylation and decrease in its expression occurred well before markers of cell death appeared—about 2 to 3 hours before.
“PP2A is a complex multi-subunit enzyme, and we hope to identify more specifically which form of PP2A is involved in dephosphorylating Mcl-1,” Dr Craig said. “This could [provide us with] a more specific way of causing Mcl-1 destruction.” ![]()
Credit: Rhoda Baer
Indirectly targeting the prosurvival protein Mcl-1 can reverse chemoresistance in lymphoma and other cancer cells, investigators have reported in The Journal of Biological Chemistry.
The team found that targeting an enzyme known as protein phosphatase 2A (PP2A) inhibited Mcl-1 dephosphorylation, which prompted the loss of Mcl-1 in chemoresistant Burkitt lymphoma cells.
“These findings may lead to a new target for chemoresistant cancer cells,” said Ruth W. Craig, PhD, of Geisel School of Medicine at Dartmouth in Hanover, New Hampshire.
“These cells are resistant to multiple types of standard chemotherapeutic agents because of [Mcl-1] overexpression. However, Mcl-1 expression plummets when we inhibit [PP2A], and then cancer cells subsequently die.”
Dr Craig and her colleagues found that PP2A can be inhibited to stop the removal of phosphate groups from a regulatory motif in Mcl-1 referred to as the PEST region (enriched with amino acids proline, glutamic acid, serine, and threonine).
And inhibiting the removal of phosphate groups, such as at threonine-163 and serine-159, targets the Mcl-1 protein for rapid destruction.
To reach this conclusion, the investigators studied BL41-3 cells, a Burkitt lymphoma cell line that overexpresses Mcl-1 and has proven resistant to multiple treatments.
The team exposed BL41-3 cells to 2 different PP2A inhibitors, okadaic acid and calyculin A. Both drugs prompted an increase in phosphorylation at threonine-163 and serine-159, as well as a decrease in Mcl-1 expression.
Further investigation confirmed that PP2A interacts with Mcl-1. And, as with therapeutic targeting, shRNA knockdown of PP2A/Aα increased phosphorylation while decreasing Mcl-1 expression.
Finally, the investigators showed the increase in Mcl-1 phosphorylation and decrease in its expression occurred well before markers of cell death appeared—about 2 to 3 hours before.
“PP2A is a complex multi-subunit enzyme, and we hope to identify more specifically which form of PP2A is involved in dephosphorylating Mcl-1,” Dr Craig said. “This could [provide us with] a more specific way of causing Mcl-1 destruction.” ![]()
Credit: Rhoda Baer
Indirectly targeting the prosurvival protein Mcl-1 can reverse chemoresistance in lymphoma and other cancer cells, investigators have reported in The Journal of Biological Chemistry.
The team found that targeting an enzyme known as protein phosphatase 2A (PP2A) inhibited Mcl-1 dephosphorylation, which prompted the loss of Mcl-1 in chemoresistant Burkitt lymphoma cells.
“These findings may lead to a new target for chemoresistant cancer cells,” said Ruth W. Craig, PhD, of Geisel School of Medicine at Dartmouth in Hanover, New Hampshire.
“These cells are resistant to multiple types of standard chemotherapeutic agents because of [Mcl-1] overexpression. However, Mcl-1 expression plummets when we inhibit [PP2A], and then cancer cells subsequently die.”
Dr Craig and her colleagues found that PP2A can be inhibited to stop the removal of phosphate groups from a regulatory motif in Mcl-1 referred to as the PEST region (enriched with amino acids proline, glutamic acid, serine, and threonine).
And inhibiting the removal of phosphate groups, such as at threonine-163 and serine-159, targets the Mcl-1 protein for rapid destruction.
To reach this conclusion, the investigators studied BL41-3 cells, a Burkitt lymphoma cell line that overexpresses Mcl-1 and has proven resistant to multiple treatments.
The team exposed BL41-3 cells to 2 different PP2A inhibitors, okadaic acid and calyculin A. Both drugs prompted an increase in phosphorylation at threonine-163 and serine-159, as well as a decrease in Mcl-1 expression.
Further investigation confirmed that PP2A interacts with Mcl-1. And, as with therapeutic targeting, shRNA knockdown of PP2A/Aα increased phosphorylation while decreasing Mcl-1 expression.
Finally, the investigators showed the increase in Mcl-1 phosphorylation and decrease in its expression occurred well before markers of cell death appeared—about 2 to 3 hours before.
“PP2A is a complex multi-subunit enzyme, and we hope to identify more specifically which form of PP2A is involved in dephosphorylating Mcl-1,” Dr Craig said. “This could [provide us with] a more specific way of causing Mcl-1 destruction.”
ESA recalled due to particulates

Amgen is recalling lots of the erythropoiesis-stimulating agent Aranesp (darbepoetin alfa) that were distributed in several countries outside the US.
The recall applies to 9 lots of Aranesp 500 mcg prefilled syringes from non-US distributors, wholesalers, and hospital pharmacies.
A routine quality examination revealed cellulose and/or polyester particles in a small number of syringes, so Amgen is recalling the 9 lots as a precautionary measure.
To date, there have been no complaints or adverse events that can be attributed to the presence of these particles.
The presence of particulate matter could elicit inflammatory and allergic responses, both chronic and acute, and may be life-threatening.
However, health implications may vary depending on the route of drug administration, the amount of particulate matter injected into the patient, the size of the particles, the patient’s underlying medical condition, and the presence of a right-to-left cardiac shunt.
The products impacted by the recall are Aranesp 500 mcg prefilled syringes. A single lot of Aranesp was packaged for different countries into 9 lots: 1042847, 1044141A, 1044141C, 1044141D, 1046891A, 1046891B, 1047394A, 1047622A, and 1047996A.
The impacted syringes were distributed in Belgium, Denmark, Finland, France, Ireland, Italy, Kuwait, Luxemburg, Russia, Saudi Arabia, Slovenia, Sweden, Switzerland, and the UK.
Consumers in the US who have questions regarding this recall can contact Amgen at 1-800-77-AMGEN (open 24 hours per day, 7 days per week).
Adverse events or quality problems associated with Aranesp can be reported to the US Food and Drug Administration’s MedWatch Adverse Event Reporting Program.
In the US, Aranesp is indicated for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, or in patients with non-myeloid malignancies where anemia results from concomitantly administered chemotherapy.
Amgen is recalling lots of the erythropoiesis-stimulating agent Aranesp (darbepoetin alfa) that were distributed in several countries outside the US.
The recall applies to 9 lots of Aranesp 500 mcg prefilled syringes from non-US distributors, wholesalers, and hospital pharmacies.
A routine quality examination revealed cellulose and/or polyester particles in a small number of syringes, so Amgen is recalling the 9 lots as a precautionary measure.
To date, there have been no complaints or adverse events that can be attributed to the presence of these particles.
The presence of particulate matter could elicit inflammatory and allergic responses, both chronic and acute, and may be life-threatening.
However, health implications may vary depending on the route of drug administration, the amount of particulate matter injected into the patient, the size of the particles, the patient’s underlying medical condition, and the presence of a right-to-left cardiac shunt.
The products impacted by the recall are Aranesp 500 mcg prefilled syringes. A single lot of Aranesp was packaged for different countries into 9 lots: 1042847, 1044141A, 1044141C, 1044141D, 1046891A, 1046891B, 1047394A, 1047622A, and 1047996A.
The impacted syringes were distributed in Belgium, Denmark, Finland, France, Ireland, Italy, Kuwait, Luxemburg, Russia, Saudi Arabia, Slovenia, Sweden, Switzerland, and the UK.
Consumers in the US who have questions regarding this recall can contact Amgen at 1-800-77-AMGEN (open 24 hours per day, 7 days per week).
Adverse events or quality problems associated with Aranesp can be reported to the US Food and Drug Administration’s MedWatch Adverse Event Reporting Program.
In the US, Aranesp is indicated for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, or in patients with non-myeloid malignancies where anemia results from concomitantly administered chemotherapy.
Amgen is recalling lots of the erythropoiesis-stimulating agent Aranesp (darbepoetin alfa) that were distributed in several countries outside the US.
The recall applies to 9 lots of Aranesp 500 mcg prefilled syringes from non-US distributors, wholesalers, and hospital pharmacies.
A routine quality examination revealed cellulose and/or polyester particles in a small number of syringes, so Amgen is recalling the 9 lots as a precautionary measure.
To date, there have been no complaints or adverse events that can be attributed to the presence of these particles.
The presence of particulate matter could elicit inflammatory and allergic responses, both chronic and acute, and may be life-threatening.
However, health implications may vary depending on the route of drug administration, the amount of particulate matter injected into the patient, the size of the particles, the patient’s underlying medical condition, and the presence of a right-to-left cardiac shunt.
The products impacted by the recall are Aranesp 500 mcg prefilled syringes. A single lot of Aranesp was packaged for different countries into 9 lots: 1042847, 1044141A, 1044141C, 1044141D, 1046891A, 1046891B, 1047394A, 1047622A, and 1047996A.
The impacted syringes were distributed in Belgium, Denmark, Finland, France, Ireland, Italy, Kuwait, Luxemburg, Russia, Saudi Arabia, Slovenia, Sweden, Switzerland, and the UK.
Consumers in the US who have questions regarding this recall can contact Amgen at 1-800-77-AMGEN (open 24 hours per day, 7 days per week).
Adverse events or quality problems associated with Aranesp can be reported to the US Food and Drug Administration’s MedWatch Adverse Event Reporting Program.
In the US, Aranesp is indicated for the treatment of anemia associated with chronic renal failure, including patients on dialysis and patients not on dialysis, or in patients with non-myeloid malignancies where anemia results from concomitantly administered chemotherapy.
Higher BMI increases risk of leukemia, other cancers

A higher body mass index (BMI) increases the risk of developing 10 common cancers, according to a study of more than 5 million adults in the UK.
The research revealed an association between BMI and leukemia, as well as certain solid tumor malignancies, but no association for multiple myeloma or non-Hodgkin lymphoma.
Krishnan Bhaskaran, PhD, of the London School of Hygiene & Tropical Medicine in the UK, and his colleagues reported these findings in The Lancet.
“The number of people who are overweight or obese is rapidly increasing, both in the UK and worldwide,” Dr Bhaskaran said. “It is well recognized that this is likely to cause more diabetes and cardiovascular disease. Our results show that, if these trends continue, we can also expect to see substantially more cancers as a result.”
Using data from general practitioner records in the UK’s Clinical Practice Research Datalink, Dr Bhaskaran and his colleagues identified 5.24 million individuals aged 16 and older who were cancer-free and had been followed for an average of 7.5 years.
The researchers assessed the subjects’ risk of developing 22 of the most common cancers, which represent 90% of the cancers diagnosed in the UK. The team evaluated cancer risk according to BMI after adjusting for individual factors such as age, sex, smoking status, and socioeconomic status.
A total of 166,955 people developed 1 of the 22 cancers studied during the follow-up period.
BMI was associated with 17 of the cancer types—leukemia and melanoma, as well as cancers of the oral cavity, esophagus, stomach, colon, liver, gallbladder, pancreas, lung, breast, cervix, uterus, ovaries, prostate, kidney, and thyroid.
BMI was not associated with non-Hodgkin lymphoma, multiple myeloma, or cancers of the rectum, bladder, or brain/central nervous system.
Each 5 kg/m² increase in BMI was clearly linked with a higher risk of cancers of the uterus (62% increase), gallbladder (31%), kidney (25%), cervix (10%), thyroid (9%), and leukemia (9%).
Higher BMI also increased the overall risk of liver (19% increase), colon (10%), ovarian (9%), and breast cancers (5%), but the effects on these cancers varied by underlying BMI and by factors such as sex and menopausal status.
Even within normal BMI ranges, a higher BMI was associated with an increased risk of some cancers.
On the other hand, there was some evidence that subjects with a high BMI were at a slightly reduced risk of prostate cancer and premenopausal breast cancer.
“There was a lot of variation in the effects of BMI on different cancers, ” Dr Bhaskaran explained. “For example, risk of cancer of the uterus increased substantially at higher body mass index. For other cancers, we saw more modest increases in risk, or no effect at all.”
“For some cancers, like breast cancer occurring in younger women before the menopause, there even seemed to be a lower risk at higher BMI. This variation tells us that BMI must affect cancer risk through a number of different processes, depending on the cancer type.”
In a linked comment article, Peter Campbell, PhD, of the American Cancer Society in Atlanta, Georgia, said this research underscores the need for policy changes to reduce the incidence of obesity and being overweight in the UK and the rest of the world.
A higher body mass index (BMI) increases the risk of developing 10 common cancers, according to a study of more than 5 million adults in the UK.
The research revealed an association between BMI and leukemia, as well as certain solid tumor malignancies, but no association for multiple myeloma or non-Hodgkin lymphoma.
Krishnan Bhaskaran, PhD, of the London School of Hygiene & Tropical Medicine in the UK, and his colleagues reported these findings in The Lancet.
“The number of people who are overweight or obese is rapidly increasing, both in the UK and worldwide,” Dr Bhaskaran said. “It is well recognized that this is likely to cause more diabetes and cardiovascular disease. Our results show that, if these trends continue, we can also expect to see substantially more cancers as a result.”
Using data from general practitioner records in the UK’s Clinical Practice Research Datalink, Dr Bhaskaran and his colleagues identified 5.24 million individuals aged 16 and older who were cancer-free and had been followed for an average of 7.5 years.
The researchers assessed the subjects’ risk of developing 22 of the most common cancers, which represent 90% of the cancers diagnosed in the UK. The team evaluated cancer risk according to BMI after adjusting for individual factors such as age, sex, smoking status, and socioeconomic status.
A total of 166,955 people developed 1 of the 22 cancers studied during the follow-up period.
BMI was associated with 17 of the cancer types—leukemia and melanoma, as well as cancers of the oral cavity, esophagus, stomach, colon, liver, gallbladder, pancreas, lung, breast, cervix, uterus, ovaries, prostate, kidney, and thyroid.
BMI was not associated with non-Hodgkin lymphoma, multiple myeloma, or cancers of the rectum, bladder, or brain/central nervous system.
Each 5 kg/m² increase in BMI was clearly linked with a higher risk of cancers of the uterus (62% increase), gallbladder (31%), kidney (25%), cervix (10%), thyroid (9%), and leukemia (9%).
Higher BMI also increased the overall risk of liver (19% increase), colon (10%), ovarian (9%), and breast cancers (5%), but the effects on these cancers varied by underlying BMI and by factors such as sex and menopausal status.
Even within normal BMI ranges, a higher BMI was associated with an increased risk of some cancers.
On the other hand, there was some evidence that subjects with a high BMI were at a slightly reduced risk of prostate cancer and premenopausal breast cancer.
“There was a lot of variation in the effects of BMI on different cancers, ” Dr Bhaskaran explained. “For example, risk of cancer of the uterus increased substantially at higher body mass index. For other cancers, we saw more modest increases in risk, or no effect at all.”
“For some cancers, like breast cancer occurring in younger women before the menopause, there even seemed to be a lower risk at higher BMI. This variation tells us that BMI must affect cancer risk through a number of different processes, depending on the cancer type.”
In a linked comment article, Peter Campbell, PhD, of the American Cancer Society in Atlanta, Georgia, said this research underscores the need for policy changes to reduce the incidence of obesity and being overweight in the UK and the rest of the world.
A higher body mass index (BMI) increases the risk of developing 10 common cancers, according to a study of more than 5 million adults in the UK.
The research revealed an association between BMI and leukemia, as well as certain solid tumor malignancies, but no association for multiple myeloma or non-Hodgkin lymphoma.
Krishnan Bhaskaran, PhD, of the London School of Hygiene & Tropical Medicine in the UK, and his colleagues reported these findings in The Lancet.
“The number of people who are overweight or obese is rapidly increasing, both in the UK and worldwide,” Dr Bhaskaran said. “It is well recognized that this is likely to cause more diabetes and cardiovascular disease. Our results show that, if these trends continue, we can also expect to see substantially more cancers as a result.”
Using data from general practitioner records in the UK’s Clinical Practice Research Datalink, Dr Bhaskaran and his colleagues identified 5.24 million individuals aged 16 and older who were cancer-free and had been followed for an average of 7.5 years.
The researchers assessed the subjects’ risk of developing 22 of the most common cancers, which represent 90% of the cancers diagnosed in the UK. The team evaluated cancer risk according to BMI after adjusting for individual factors such as age, sex, smoking status, and socioeconomic status.
A total of 166,955 people developed 1 of the 22 cancers studied during the follow-up period.
BMI was associated with 17 of the cancer types—leukemia and melanoma, as well as cancers of the oral cavity, esophagus, stomach, colon, liver, gallbladder, pancreas, lung, breast, cervix, uterus, ovaries, prostate, kidney, and thyroid.
BMI was not associated with non-Hodgkin lymphoma, multiple myeloma, or cancers of the rectum, bladder, or brain/central nervous system.
Each 5 kg/m² increase in BMI was clearly linked with a higher risk of cancers of the uterus (62% increase), gallbladder (31%), kidney (25%), cervix (10%), thyroid (9%), and leukemia (9%).
Higher BMI also increased the overall risk of liver (19% increase), colon (10%), ovarian (9%), and breast cancers (5%), but the effects on these cancers varied by underlying BMI and by factors such as sex and menopausal status.
Even within normal BMI ranges, a higher BMI was associated with an increased risk of some cancers.
On the other hand, there was some evidence that subjects with a high BMI were at a slightly reduced risk of prostate cancer and premenopausal breast cancer.
“There was a lot of variation in the effects of BMI on different cancers, ” Dr Bhaskaran explained. “For example, risk of cancer of the uterus increased substantially at higher body mass index. For other cancers, we saw more modest increases in risk, or no effect at all.”
“For some cancers, like breast cancer occurring in younger women before the menopause, there even seemed to be a lower risk at higher BMI. This variation tells us that BMI must affect cancer risk through a number of different processes, depending on the cancer type.”
In a linked comment article, Peter Campbell, PhD, of the American Cancer Society in Atlanta, Georgia, said this research underscores the need for policy changes to reduce the incidence of obesity and being overweight in the UK and the rest of the world.
More new insight into HSC generation
Credit: John Perry
Researchers say they have uncovered early clues that better explain how hematopoietic stem cells (HSCs) are generated.
Their research, conducted in zebrafish, provides new insight regarding the role of JAM proteins and the Notch signaling pathway, which was already known to be critical for HSC generation.
The new findings may help clear the way to producing HSCs from human pluripotent precursors, according to the researchers.
David Traver, PhD, of the University of California, San Diego School of Medicine, and his colleagues recounted these findings in a letter to Nature. A similar study, which revealed the role of endotome cells in HSC generation, was also recently published as a letter to Nature.
“Notch signaling between emitting and receiving cells is key to establishing HSC fate during development,” Dr Traver said. “What has not been known is where, when, and how Notch signal transduction is mediated.”
Through experiments in zebrafish models, Dr Traver and his colleagues found the Notch signal is transduced into HSC precursor cells from signal-emitting cells in the somite much earlier in the process than previously anticipated.
Specifically, the team found that JAM proteins, best known for helping maintain tight junctions between endothelial cells to prevent vascular leakage, were key mediators of Notch signaling.
When the researchers caused JAM proteins to lose function, Notch signaling and HSCs were also lost. When they enforced Notch signaling, the team rescued HSC development.
“To date, it has not been possible to generate HSCs de novo from human pluripotent precursors, like induced pluripotent stem cells,” Dr Traver said. “This has been due, in part, to a lack of understanding of the complete set of factors that the embryo uses to make HSCs in vivo. It has also likely been due to not knowing in what order each required factor is needed.”
“Our studies demonstrate that Notch signaling is required much earlier than previously thought. In fact, it may be one of the earliest determinants of HSC fate. This finding strongly suggests that in vitro approaches to instruct HSC fate from induced pluripotent stem cells must focus on the Notch pathway at early time-points in the process.”
“Our findings have also shown that JAM proteins serve as a sort of co-receptor for Notch signaling, in that they are required to maintain close contact between signal-emitting and signal-receiving cells to permit strong activation of Notch in the precursors of HSCs.”
Dr Traver and his colleagues believe the findings may have far-reaching implications for the eventual development of HSC-based therapies for diseases such as leukemia and congenital blood disorders.
Credit: John Perry
Researchers say they have uncovered early clues that better explain how hematopoietic stem cells (HSCs) are generated.
Their research, conducted in zebrafish, provides new insight regarding the role of JAM proteins and the Notch signaling pathway, which was already known to be critical for HSC generation.
The new findings may help clear the way to producing HSCs from human pluripotent precursors, according to the researchers.
David Traver, PhD, of the University of California, San Diego School of Medicine, and his colleagues recounted these findings in a letter to Nature. A similar study, which revealed the role of endotome cells in HSC generation, was also recently published as a letter to Nature.
“Notch signaling between emitting and receiving cells is key to establishing HSC fate during development,” Dr Traver said. “What has not been known is where, when, and how Notch signal transduction is mediated.”
Through experiments in zebrafish models, Dr Traver and his colleagues found the Notch signal is transduced into HSC precursor cells from signal-emitting cells in the somite much earlier in the process than previously anticipated.
Specifically, the team found that JAM proteins, best known for helping maintain tight junctions between endothelial cells to prevent vascular leakage, were key mediators of Notch signaling.
When the researchers caused JAM proteins to lose function, Notch signaling and HSCs were also lost. When they enforced Notch signaling, the team rescued HSC development.
“To date, it has not been possible to generate HSCs de novo from human pluripotent precursors, like induced pluripotent stem cells,” Dr Traver said. “This has been due, in part, to a lack of understanding of the complete set of factors that the embryo uses to make HSCs in vivo. It has also likely been due to not knowing in what order each required factor is needed.”
“Our studies demonstrate that Notch signaling is required much earlier than previously thought. In fact, it may be one of the earliest determinants of HSC fate. This finding strongly suggests that in vitro approaches to instruct HSC fate from induced pluripotent stem cells must focus on the Notch pathway at early time-points in the process.”
“Our findings have also shown that JAM proteins serve as a sort of co-receptor for Notch signaling, in that they are required to maintain close contact between signal-emitting and signal-receiving cells to permit strong activation of Notch in the precursors of HSCs.”
Dr Traver and his colleagues believe the findings may have far-reaching implications for the eventual development of HSC-based therapies for diseases such as leukemia and congenital blood disorders.
Credit: John Perry
Researchers say they have uncovered early clues that better explain how hematopoietic stem cells (HSCs) are generated.
Their research, conducted in zebrafish, provides new insight regarding the role of JAM proteins and the Notch signaling pathway, which was already known to be critical for HSC generation.
The new findings may help clear the way to producing HSCs from human pluripotent precursors, according to the researchers.
David Traver, PhD, of the University of California, San Diego School of Medicine, and his colleagues recounted these findings in a letter to Nature. A similar study, which revealed the role of endotome cells in HSC generation, was also recently published as a letter to Nature.
“Notch signaling between emitting and receiving cells is key to establishing HSC fate during development,” Dr Traver said. “What has not been known is where, when, and how Notch signal transduction is mediated.”
Through experiments in zebrafish models, Dr Traver and his colleagues found the Notch signal is transduced into HSC precursor cells from signal-emitting cells in the somite much earlier in the process than previously anticipated.
Specifically, the team found that JAM proteins, best known for helping maintain tight junctions between endothelial cells to prevent vascular leakage, were key mediators of Notch signaling.
When the researchers caused JAM proteins to lose function, Notch signaling and HSCs were also lost. When they enforced Notch signaling, the team rescued HSC development.
“To date, it has not been possible to generate HSCs de novo from human pluripotent precursors, like induced pluripotent stem cells,” Dr Traver said. “This has been due, in part, to a lack of understanding of the complete set of factors that the embryo uses to make HSCs in vivo. It has also likely been due to not knowing in what order each required factor is needed.”
“Our studies demonstrate that Notch signaling is required much earlier than previously thought. In fact, it may be one of the earliest determinants of HSC fate. This finding strongly suggests that in vitro approaches to instruct HSC fate from induced pluripotent stem cells must focus on the Notch pathway at early time-points in the process.”
“Our findings have also shown that JAM proteins serve as a sort of co-receptor for Notch signaling, in that they are required to maintain close contact between signal-emitting and signal-receiving cells to permit strong activation of Notch in the precursors of HSCs.”
Dr Traver and his colleagues believe the findings may have far-reaching implications for the eventual development of HSC-based therapies for diseases such as leukemia and congenital blood disorders.