User login
Zebrafish research solves mystery of HSC generation

A cure for a range of blood and immune disorders is in sight, according to researchers who say they’ve solved a mystery of hematopoietic stem cell (HSC) generation.
By studying zebrafish embryos, the investigators found that migratory cells from a region known as the endotome are essential for HSC formation.
The team also discovered some of the signals required for HSC generation and identified genes necessary for endotome formation.
The researchers believe these findings, published in Nature, bring us one step closer to creating viable HSCs in the lab.
“HSCs are one of the best therapeutic tools at our disposal because they can make any blood cell in the body,” said study author Peter Currie, PhD, of the Australian Regenerative Medicine Institute at Monash University in Victoria, Australia.
“Potentially, we could use these cells in many more ways than current transplantation strategies to treat serious blood disorders and diseases, but only if we can figure out how they are generated in the first place.”
In an attempt to do just that, Dr Currie and his colleagues studied developing zebrafish. They used high-resolution microscopy to film how HSCs form inside the embryo.
The investigators noted that, in vertebrate embryos, HSCs are initially generated within the dorsal aorta. And previous research showed that signaling relayed from adjacent somites coordinates HSC induction.
Dr Currie and his colleagues found that somite specification of HSCs occurs thanks to an endothelial precursor cell population. These cells arise in a sub-compartment of the zebrafish somite the researchers dubbed “the endotome.”
Endothelial cells from the endotome are specified thanks to activity of the homeobox gene meox1. Specified endotome cells then migrate to and colonize the dorsal aorta, where they induce HSC formation via chemokine signaling that’s activated during endotome formation.
“Endotome cells act like a comfy sofa for pre-HSCs to snuggle into, helping them progress to become fully fledged stem cells,” Dr Currie said. “Not only did we identify some of the cells and signals required for HSC formation, we also pinpointed the genes required for endotome formation in the first place.”
“The really exciting thing about these results is that if we can find the signals present in the endotome cells responsible for embryonic HSC formation, then we can use them in vitro to make different blood cells on demand for all sorts of blood-related disorders.”
For the next phase of this research, Dr Currie and his colleagues are attempting to identify more of the molecular cues that trigger HSC production. ![]()
A cure for a range of blood and immune disorders is in sight, according to researchers who say they’ve solved a mystery of hematopoietic stem cell (HSC) generation.
By studying zebrafish embryos, the investigators found that migratory cells from a region known as the endotome are essential for HSC formation.
The team also discovered some of the signals required for HSC generation and identified genes necessary for endotome formation.
The researchers believe these findings, published in Nature, bring us one step closer to creating viable HSCs in the lab.
“HSCs are one of the best therapeutic tools at our disposal because they can make any blood cell in the body,” said study author Peter Currie, PhD, of the Australian Regenerative Medicine Institute at Monash University in Victoria, Australia.
“Potentially, we could use these cells in many more ways than current transplantation strategies to treat serious blood disorders and diseases, but only if we can figure out how they are generated in the first place.”
In an attempt to do just that, Dr Currie and his colleagues studied developing zebrafish. They used high-resolution microscopy to film how HSCs form inside the embryo.
The investigators noted that, in vertebrate embryos, HSCs are initially generated within the dorsal aorta. And previous research showed that signaling relayed from adjacent somites coordinates HSC induction.
Dr Currie and his colleagues found that somite specification of HSCs occurs thanks to an endothelial precursor cell population. These cells arise in a sub-compartment of the zebrafish somite the researchers dubbed “the endotome.”
Endothelial cells from the endotome are specified thanks to activity of the homeobox gene meox1. Specified endotome cells then migrate to and colonize the dorsal aorta, where they induce HSC formation via chemokine signaling that’s activated during endotome formation.
“Endotome cells act like a comfy sofa for pre-HSCs to snuggle into, helping them progress to become fully fledged stem cells,” Dr Currie said. “Not only did we identify some of the cells and signals required for HSC formation, we also pinpointed the genes required for endotome formation in the first place.”
“The really exciting thing about these results is that if we can find the signals present in the endotome cells responsible for embryonic HSC formation, then we can use them in vitro to make different blood cells on demand for all sorts of blood-related disorders.”
For the next phase of this research, Dr Currie and his colleagues are attempting to identify more of the molecular cues that trigger HSC production. ![]()
A cure for a range of blood and immune disorders is in sight, according to researchers who say they’ve solved a mystery of hematopoietic stem cell (HSC) generation.
By studying zebrafish embryos, the investigators found that migratory cells from a region known as the endotome are essential for HSC formation.
The team also discovered some of the signals required for HSC generation and identified genes necessary for endotome formation.
The researchers believe these findings, published in Nature, bring us one step closer to creating viable HSCs in the lab.
“HSCs are one of the best therapeutic tools at our disposal because they can make any blood cell in the body,” said study author Peter Currie, PhD, of the Australian Regenerative Medicine Institute at Monash University in Victoria, Australia.
“Potentially, we could use these cells in many more ways than current transplantation strategies to treat serious blood disorders and diseases, but only if we can figure out how they are generated in the first place.”
In an attempt to do just that, Dr Currie and his colleagues studied developing zebrafish. They used high-resolution microscopy to film how HSCs form inside the embryo.
The investigators noted that, in vertebrate embryos, HSCs are initially generated within the dorsal aorta. And previous research showed that signaling relayed from adjacent somites coordinates HSC induction.
Dr Currie and his colleagues found that somite specification of HSCs occurs thanks to an endothelial precursor cell population. These cells arise in a sub-compartment of the zebrafish somite the researchers dubbed “the endotome.”
Endothelial cells from the endotome are specified thanks to activity of the homeobox gene meox1. Specified endotome cells then migrate to and colonize the dorsal aorta, where they induce HSC formation via chemokine signaling that’s activated during endotome formation.
“Endotome cells act like a comfy sofa for pre-HSCs to snuggle into, helping them progress to become fully fledged stem cells,” Dr Currie said. “Not only did we identify some of the cells and signals required for HSC formation, we also pinpointed the genes required for endotome formation in the first place.”
“The really exciting thing about these results is that if we can find the signals present in the endotome cells responsible for embryonic HSC formation, then we can use them in vitro to make different blood cells on demand for all sorts of blood-related disorders.”
For the next phase of this research, Dr Currie and his colleagues are attempting to identify more of the molecular cues that trigger HSC production. ![]()
Technique may predict progression in MM

Credit: Graham Colm
Scientists have found that a technique used in Earth science research may be able to help physicians predict the course of multiple myeloma (MM).
The team showed they could use calcium isotope analysis to predict whether MM patients are at risk for developing bone lesions.
The group believes this technique could be used to chart the progression or recurrence of MM and help tailor therapies to better protect patients’ bones.
“At present, there is no good way to track changes in bone balance, except retrospectively using X-ray methods,” said Ariel Anbar, PhD, of Arizona State University in Tempe. “By the time the X-rays show something, the damage has been done.”
“Right now, pain is usually the first indication that cancer is affecting the bones,” added Rafael Fonseca, MD, of the Mayo Clinic in Scottsdale, Arizona. “If we could detect it earlier by an analysis of urine or blood in high-risk patients, it could significantly improve their care.”
To explore this possibility, Drs Fonseca, Anbar, and their colleagues performed calcium isotope analysis using blood samples from MM patients. The team recounted their results in a letter to Leukemia.
As the name suggests, calcium isotope analysis measures calcium isotopes that are naturally present in blood. The technique makes use of a fact well known to Earth scientists but not normally used in biomedicine: different isotopes of a chemical element can react at slightly different rates.
An earlier study testing this technique in healthy subjects showed that when bones form, the lighter isotopes of calcium enter bone a little faster than the heavier isotopes. That difference, called isotope fractionation, is the key to the method.
In healthy, active humans, bone is forming at about the same rate as it resorbs. But if bone loss is occurring, the isotopic composition of blood becomes enriched in the lighter isotopes as bones resorb more quickly than they are formed.
The effect on calcium isotopes is very small, typically less than a 0.02% change in the isotope ratio. But even effects that small can be measured using precise mass spectrometry methods.
Using these methods, the researchers tested peripheral blood samples from 71 adult patients—55 with MM, 9 with smoldering MM, and 7 with monoclonal gammopathy of undetermined significance.
The researchers found an association between how active a patient’s disease was and the change in the isotope ratios. In fact, the isotope ratios predicted disease activity better than, and independent from, standard clinical variables.
Blood samples from patients with active disease had significantly lower mean calcium isotope compositions than samples from patients with non-active disease (mean δ44/42Ca=-0.8 vs -0.67, P=0.025), regardless of diagnosis.
For MM patients specifically, those with active disease had a significantly lower mean δ44/42Ca than patients with non-active disease (-0.79 vs -0.63, P=0.016), which was consistent with resorption of bone containing lighter 42Ca.
Dr Anbar noted that, although calcium isotope analysis has worked in this small set of patients, additional research is needed to verify these initial findings and improve the efficiency of analysis.
“If the method proves to be robust after more careful validation, it could provide earlier detection of bone involvement than presently possible,” he said, “and also provide the possibility to monitor the effectiveness of drugs to combat bone loss.” ![]()
Credit: Graham Colm
Scientists have found that a technique used in Earth science research may be able to help physicians predict the course of multiple myeloma (MM).
The team showed they could use calcium isotope analysis to predict whether MM patients are at risk for developing bone lesions.
The group believes this technique could be used to chart the progression or recurrence of MM and help tailor therapies to better protect patients’ bones.
“At present, there is no good way to track changes in bone balance, except retrospectively using X-ray methods,” said Ariel Anbar, PhD, of Arizona State University in Tempe. “By the time the X-rays show something, the damage has been done.”
“Right now, pain is usually the first indication that cancer is affecting the bones,” added Rafael Fonseca, MD, of the Mayo Clinic in Scottsdale, Arizona. “If we could detect it earlier by an analysis of urine or blood in high-risk patients, it could significantly improve their care.”
To explore this possibility, Drs Fonseca, Anbar, and their colleagues performed calcium isotope analysis using blood samples from MM patients. The team recounted their results in a letter to Leukemia.
As the name suggests, calcium isotope analysis measures calcium isotopes that are naturally present in blood. The technique makes use of a fact well known to Earth scientists but not normally used in biomedicine: different isotopes of a chemical element can react at slightly different rates.
An earlier study testing this technique in healthy subjects showed that when bones form, the lighter isotopes of calcium enter bone a little faster than the heavier isotopes. That difference, called isotope fractionation, is the key to the method.
In healthy, active humans, bone is forming at about the same rate as it resorbs. But if bone loss is occurring, the isotopic composition of blood becomes enriched in the lighter isotopes as bones resorb more quickly than they are formed.
The effect on calcium isotopes is very small, typically less than a 0.02% change in the isotope ratio. But even effects that small can be measured using precise mass spectrometry methods.
Using these methods, the researchers tested peripheral blood samples from 71 adult patients—55 with MM, 9 with smoldering MM, and 7 with monoclonal gammopathy of undetermined significance.
The researchers found an association between how active a patient’s disease was and the change in the isotope ratios. In fact, the isotope ratios predicted disease activity better than, and independent from, standard clinical variables.
Blood samples from patients with active disease had significantly lower mean calcium isotope compositions than samples from patients with non-active disease (mean δ44/42Ca=-0.8 vs -0.67, P=0.025), regardless of diagnosis.
For MM patients specifically, those with active disease had a significantly lower mean δ44/42Ca than patients with non-active disease (-0.79 vs -0.63, P=0.016), which was consistent with resorption of bone containing lighter 42Ca.
Dr Anbar noted that, although calcium isotope analysis has worked in this small set of patients, additional research is needed to verify these initial findings and improve the efficiency of analysis.
“If the method proves to be robust after more careful validation, it could provide earlier detection of bone involvement than presently possible,” he said, “and also provide the possibility to monitor the effectiveness of drugs to combat bone loss.” ![]()
Credit: Graham Colm
Scientists have found that a technique used in Earth science research may be able to help physicians predict the course of multiple myeloma (MM).
The team showed they could use calcium isotope analysis to predict whether MM patients are at risk for developing bone lesions.
The group believes this technique could be used to chart the progression or recurrence of MM and help tailor therapies to better protect patients’ bones.
“At present, there is no good way to track changes in bone balance, except retrospectively using X-ray methods,” said Ariel Anbar, PhD, of Arizona State University in Tempe. “By the time the X-rays show something, the damage has been done.”
“Right now, pain is usually the first indication that cancer is affecting the bones,” added Rafael Fonseca, MD, of the Mayo Clinic in Scottsdale, Arizona. “If we could detect it earlier by an analysis of urine or blood in high-risk patients, it could significantly improve their care.”
To explore this possibility, Drs Fonseca, Anbar, and their colleagues performed calcium isotope analysis using blood samples from MM patients. The team recounted their results in a letter to Leukemia.
As the name suggests, calcium isotope analysis measures calcium isotopes that are naturally present in blood. The technique makes use of a fact well known to Earth scientists but not normally used in biomedicine: different isotopes of a chemical element can react at slightly different rates.
An earlier study testing this technique in healthy subjects showed that when bones form, the lighter isotopes of calcium enter bone a little faster than the heavier isotopes. That difference, called isotope fractionation, is the key to the method.
In healthy, active humans, bone is forming at about the same rate as it resorbs. But if bone loss is occurring, the isotopic composition of blood becomes enriched in the lighter isotopes as bones resorb more quickly than they are formed.
The effect on calcium isotopes is very small, typically less than a 0.02% change in the isotope ratio. But even effects that small can be measured using precise mass spectrometry methods.
Using these methods, the researchers tested peripheral blood samples from 71 adult patients—55 with MM, 9 with smoldering MM, and 7 with monoclonal gammopathy of undetermined significance.
The researchers found an association between how active a patient’s disease was and the change in the isotope ratios. In fact, the isotope ratios predicted disease activity better than, and independent from, standard clinical variables.
Blood samples from patients with active disease had significantly lower mean calcium isotope compositions than samples from patients with non-active disease (mean δ44/42Ca=-0.8 vs -0.67, P=0.025), regardless of diagnosis.
For MM patients specifically, those with active disease had a significantly lower mean δ44/42Ca than patients with non-active disease (-0.79 vs -0.63, P=0.016), which was consistent with resorption of bone containing lighter 42Ca.
Dr Anbar noted that, although calcium isotope analysis has worked in this small set of patients, additional research is needed to verify these initial findings and improve the efficiency of analysis.
“If the method proves to be robust after more careful validation, it could provide earlier detection of bone involvement than presently possible,” he said, “and also provide the possibility to monitor the effectiveness of drugs to combat bone loss.” ![]()
Cancer survivors aren’t living healthy, study shows

Credit: Bill Branson
Childhood cancer survivors are no more likely than their cancer-free peers to adhere to healthy living guidelines, according to a study published in the Journal of Cancer Survivorship.
Survivors were less likely to be smokers and had a lower average body mass index (BMI).
But there were no significant differences between survivors and cancer-free control subjects with regard to overall diet, physical activity, or alcohol consumption.
Chloe Berdan, of Promedica in Toledo, Ohio, and her colleagues uncovered these results by examining data from the Chicago Healthy Living Study.
The team assessed adherence to American Cancer Society Guidelines on Nutrition and Physical Activity via interviews with 431 childhood cancer survivors and 361 control subjects who never had cancer. The survivors, ages 18 to 59, were all diagnosed with a malignant cancer before their 21st birthdays.
There were no significant differences in sex or race between survivors and controls. Survivors were younger than controls (28.4±7.8 vs 29.6± 8.3 years, P=0.04) and had less education (14.0±2.0 vs 14.4±2.0 years, P=0.01).
Overall, there was no significant difference between survivors and control subjects in adhering to the American Cancer Society guidelines.
Survivors and controls also had similar scores for several individual measures, including alcohol consumption, overall physical activity, overall diet, the servings of fruits/vegetables consumed, and the consumption of red/processed meat.
However, survivors were significantly less likely than controls to be smokers—11.4% vs 17.5% (P=0.02). Survivors had, on average, a BMI of about 1.2 kg/m² lower than controls (P=0.01). And survivors consumed significantly less fiber than controls—9.2±3.5 vs 9.7±3.8 kcal (P=0.05).
Only about 1 in 10 survivors (10.2%) met fiber recommendations, 17.7% ate 5 fruits or vegetables per day, and 46.2% met the red/processed meat recommendation of less than 18 oz per week. On average, survivors scored under 50% for the quality of their diets.
Survivors were better at meeting the goal of at least 5 hours of moderate activity per week (60.5%) than to sticking to any of the other guidelines.
About 36% of survivors were within a healthy BMI range, 2.9% were underweight, 28.9% were overweight, and 32.4% were obese.
The 0.7% of survivors who adhered fully to the guidelines tended to be women, non-smokers, and people with a good view of their own health.
“There is still much room for improvement in educating and encouraging survivors to follow healthier diets and lifestyles,” Berdan said. “Adopting such behavior during early adulthood may have a lasting impact on their quality of life and overall survival.” ![]()
Credit: Bill Branson
Childhood cancer survivors are no more likely than their cancer-free peers to adhere to healthy living guidelines, according to a study published in the Journal of Cancer Survivorship.
Survivors were less likely to be smokers and had a lower average body mass index (BMI).
But there were no significant differences between survivors and cancer-free control subjects with regard to overall diet, physical activity, or alcohol consumption.
Chloe Berdan, of Promedica in Toledo, Ohio, and her colleagues uncovered these results by examining data from the Chicago Healthy Living Study.
The team assessed adherence to American Cancer Society Guidelines on Nutrition and Physical Activity via interviews with 431 childhood cancer survivors and 361 control subjects who never had cancer. The survivors, ages 18 to 59, were all diagnosed with a malignant cancer before their 21st birthdays.
There were no significant differences in sex or race between survivors and controls. Survivors were younger than controls (28.4±7.8 vs 29.6± 8.3 years, P=0.04) and had less education (14.0±2.0 vs 14.4±2.0 years, P=0.01).
Overall, there was no significant difference between survivors and control subjects in adhering to the American Cancer Society guidelines.
Survivors and controls also had similar scores for several individual measures, including alcohol consumption, overall physical activity, overall diet, the servings of fruits/vegetables consumed, and the consumption of red/processed meat.
However, survivors were significantly less likely than controls to be smokers—11.4% vs 17.5% (P=0.02). Survivors had, on average, a BMI of about 1.2 kg/m² lower than controls (P=0.01). And survivors consumed significantly less fiber than controls—9.2±3.5 vs 9.7±3.8 kcal (P=0.05).
Only about 1 in 10 survivors (10.2%) met fiber recommendations, 17.7% ate 5 fruits or vegetables per day, and 46.2% met the red/processed meat recommendation of less than 18 oz per week. On average, survivors scored under 50% for the quality of their diets.
Survivors were better at meeting the goal of at least 5 hours of moderate activity per week (60.5%) than to sticking to any of the other guidelines.
About 36% of survivors were within a healthy BMI range, 2.9% were underweight, 28.9% were overweight, and 32.4% were obese.
The 0.7% of survivors who adhered fully to the guidelines tended to be women, non-smokers, and people with a good view of their own health.
“There is still much room for improvement in educating and encouraging survivors to follow healthier diets and lifestyles,” Berdan said. “Adopting such behavior during early adulthood may have a lasting impact on their quality of life and overall survival.” ![]()
Credit: Bill Branson
Childhood cancer survivors are no more likely than their cancer-free peers to adhere to healthy living guidelines, according to a study published in the Journal of Cancer Survivorship.
Survivors were less likely to be smokers and had a lower average body mass index (BMI).
But there were no significant differences between survivors and cancer-free control subjects with regard to overall diet, physical activity, or alcohol consumption.
Chloe Berdan, of Promedica in Toledo, Ohio, and her colleagues uncovered these results by examining data from the Chicago Healthy Living Study.
The team assessed adherence to American Cancer Society Guidelines on Nutrition and Physical Activity via interviews with 431 childhood cancer survivors and 361 control subjects who never had cancer. The survivors, ages 18 to 59, were all diagnosed with a malignant cancer before their 21st birthdays.
There were no significant differences in sex or race between survivors and controls. Survivors were younger than controls (28.4±7.8 vs 29.6± 8.3 years, P=0.04) and had less education (14.0±2.0 vs 14.4±2.0 years, P=0.01).
Overall, there was no significant difference between survivors and control subjects in adhering to the American Cancer Society guidelines.
Survivors and controls also had similar scores for several individual measures, including alcohol consumption, overall physical activity, overall diet, the servings of fruits/vegetables consumed, and the consumption of red/processed meat.
However, survivors were significantly less likely than controls to be smokers—11.4% vs 17.5% (P=0.02). Survivors had, on average, a BMI of about 1.2 kg/m² lower than controls (P=0.01). And survivors consumed significantly less fiber than controls—9.2±3.5 vs 9.7±3.8 kcal (P=0.05).
Only about 1 in 10 survivors (10.2%) met fiber recommendations, 17.7% ate 5 fruits or vegetables per day, and 46.2% met the red/processed meat recommendation of less than 18 oz per week. On average, survivors scored under 50% for the quality of their diets.
Survivors were better at meeting the goal of at least 5 hours of moderate activity per week (60.5%) than to sticking to any of the other guidelines.
About 36% of survivors were within a healthy BMI range, 2.9% were underweight, 28.9% were overweight, and 32.4% were obese.
The 0.7% of survivors who adhered fully to the guidelines tended to be women, non-smokers, and people with a good view of their own health.
“There is still much room for improvement in educating and encouraging survivors to follow healthier diets and lifestyles,” Berdan said. “Adopting such behavior during early adulthood may have a lasting impact on their quality of life and overall survival.” ![]()
A quicker way to manipulate malaria genes

(left) and Jeffrey Wagner
Credit: Bryce Vickmark
The gene-editing technique CRISPR can disrupt a single gene from the malaria parasite Plasmodium falciparum in a matter of weeks, a new study suggests.
Although CRISPR’s success rate ranged from 50% to 100%, the researchers believe the technique shows promise and could greatly speed up gene analysis.
At present, it can take up to a year to determine the function of a single gene in P falciparum, which can hinder efforts to develop drugs and vaccines.
“Even though we’ve sequenced the entire genome of Plasmodium falciparum, half of it still remains functionally uncharacterized,” said Jacquin Niles, MD, PhD, of the Massachusetts Institute of Technology in Cambridge.
“That’s about 2500 genes that, if only we knew what they did, we could think about novel therapeutics, whether it’s drugs or vaccines.”
Dr Niles and his colleagues described their use of CRISPR in P falciparum in Nature Methods.
The team noted that, in P falciparum, gene editing can take up to a year because it relies on homologous recombination, a type of genetic swapping that cells use to repair broken DNA strands and that occurs very rarely in the genome of the malaria parasite.
“You have to rely on this really inefficient process that occurs only if you have spontaneous DNA strand breaks that happen to fall within your region of interest,” Dr Niles said.
More recently, researchers have successfully used zinc finger nucleases to cut out specific genes, but this approach is costly because it requires a new nuclease to be designed for each gene target.
CRISPR exploits a set of bacterial proteins that protect microbes from viral infection. The system includes a DNA-cutting enzyme, Cas9, bound to a short RNA guide strand that is programmed to bind to a specific genome sequence, telling Cas9 where to make its cut. This approach allows scientists to target and delete any gene by simply changing the RNA guide strand sequence.
As soon as researchers proved this system could work in cells other than bacteria, Dr Niles started to think about using it to manipulate P falciparum.
To test this approach, he and his colleagues tried using CRISPR to disrupt 2 genes, kahrp and eba-175, that had previously been knocked out in malaria using traditional approaches.
The kahrp gene produces a protein that causes red blood cells to develop a knobby appearance when infected with malaria. Dr Niles’s team was able to disrupt this gene in 100% of parasites treated with the CRISPR system. The red blood cells infected by parasites remained smooth.
The other gene, eba-175, codes for a protein that binds to red blood cell receptors and helps the malaria parasite enter cells. The researchers were only able to disrupt this gene in 50% to 80% of parasites manipulated with CRISPR.
“We consider this to be a win,” Dr Niles said. “Compared to the efficiency with which P falciparum genetics have been done in the past, even 50% is pretty substantial.”
Now that CRISPR technology has been validated in P falciparum, Dr Niles expects many scientists will adopt it for genetic studies of the parasite. Such efforts could reveal more about how the parasite invades red blood cells and replicates inside cells, which could generate new drug and vaccine targets.
“I think the impact could be quite huge,” he said. “It lowers the barrier to really being more imaginative in terms of how we do experiments and the kinds of questions that we can ask.” ![]()
(left) and Jeffrey Wagner
Credit: Bryce Vickmark
The gene-editing technique CRISPR can disrupt a single gene from the malaria parasite Plasmodium falciparum in a matter of weeks, a new study suggests.
Although CRISPR’s success rate ranged from 50% to 100%, the researchers believe the technique shows promise and could greatly speed up gene analysis.
At present, it can take up to a year to determine the function of a single gene in P falciparum, which can hinder efforts to develop drugs and vaccines.
“Even though we’ve sequenced the entire genome of Plasmodium falciparum, half of it still remains functionally uncharacterized,” said Jacquin Niles, MD, PhD, of the Massachusetts Institute of Technology in Cambridge.
“That’s about 2500 genes that, if only we knew what they did, we could think about novel therapeutics, whether it’s drugs or vaccines.”
Dr Niles and his colleagues described their use of CRISPR in P falciparum in Nature Methods.
The team noted that, in P falciparum, gene editing can take up to a year because it relies on homologous recombination, a type of genetic swapping that cells use to repair broken DNA strands and that occurs very rarely in the genome of the malaria parasite.
“You have to rely on this really inefficient process that occurs only if you have spontaneous DNA strand breaks that happen to fall within your region of interest,” Dr Niles said.
More recently, researchers have successfully used zinc finger nucleases to cut out specific genes, but this approach is costly because it requires a new nuclease to be designed for each gene target.
CRISPR exploits a set of bacterial proteins that protect microbes from viral infection. The system includes a DNA-cutting enzyme, Cas9, bound to a short RNA guide strand that is programmed to bind to a specific genome sequence, telling Cas9 where to make its cut. This approach allows scientists to target and delete any gene by simply changing the RNA guide strand sequence.
As soon as researchers proved this system could work in cells other than bacteria, Dr Niles started to think about using it to manipulate P falciparum.
To test this approach, he and his colleagues tried using CRISPR to disrupt 2 genes, kahrp and eba-175, that had previously been knocked out in malaria using traditional approaches.
The kahrp gene produces a protein that causes red blood cells to develop a knobby appearance when infected with malaria. Dr Niles’s team was able to disrupt this gene in 100% of parasites treated with the CRISPR system. The red blood cells infected by parasites remained smooth.
The other gene, eba-175, codes for a protein that binds to red blood cell receptors and helps the malaria parasite enter cells. The researchers were only able to disrupt this gene in 50% to 80% of parasites manipulated with CRISPR.
“We consider this to be a win,” Dr Niles said. “Compared to the efficiency with which P falciparum genetics have been done in the past, even 50% is pretty substantial.”
Now that CRISPR technology has been validated in P falciparum, Dr Niles expects many scientists will adopt it for genetic studies of the parasite. Such efforts could reveal more about how the parasite invades red blood cells and replicates inside cells, which could generate new drug and vaccine targets.
“I think the impact could be quite huge,” he said. “It lowers the barrier to really being more imaginative in terms of how we do experiments and the kinds of questions that we can ask.” ![]()
(left) and Jeffrey Wagner
Credit: Bryce Vickmark
The gene-editing technique CRISPR can disrupt a single gene from the malaria parasite Plasmodium falciparum in a matter of weeks, a new study suggests.
Although CRISPR’s success rate ranged from 50% to 100%, the researchers believe the technique shows promise and could greatly speed up gene analysis.
At present, it can take up to a year to determine the function of a single gene in P falciparum, which can hinder efforts to develop drugs and vaccines.
“Even though we’ve sequenced the entire genome of Plasmodium falciparum, half of it still remains functionally uncharacterized,” said Jacquin Niles, MD, PhD, of the Massachusetts Institute of Technology in Cambridge.
“That’s about 2500 genes that, if only we knew what they did, we could think about novel therapeutics, whether it’s drugs or vaccines.”
Dr Niles and his colleagues described their use of CRISPR in P falciparum in Nature Methods.
The team noted that, in P falciparum, gene editing can take up to a year because it relies on homologous recombination, a type of genetic swapping that cells use to repair broken DNA strands and that occurs very rarely in the genome of the malaria parasite.
“You have to rely on this really inefficient process that occurs only if you have spontaneous DNA strand breaks that happen to fall within your region of interest,” Dr Niles said.
More recently, researchers have successfully used zinc finger nucleases to cut out specific genes, but this approach is costly because it requires a new nuclease to be designed for each gene target.
CRISPR exploits a set of bacterial proteins that protect microbes from viral infection. The system includes a DNA-cutting enzyme, Cas9, bound to a short RNA guide strand that is programmed to bind to a specific genome sequence, telling Cas9 where to make its cut. This approach allows scientists to target and delete any gene by simply changing the RNA guide strand sequence.
As soon as researchers proved this system could work in cells other than bacteria, Dr Niles started to think about using it to manipulate P falciparum.
To test this approach, he and his colleagues tried using CRISPR to disrupt 2 genes, kahrp and eba-175, that had previously been knocked out in malaria using traditional approaches.
The kahrp gene produces a protein that causes red blood cells to develop a knobby appearance when infected with malaria. Dr Niles’s team was able to disrupt this gene in 100% of parasites treated with the CRISPR system. The red blood cells infected by parasites remained smooth.
The other gene, eba-175, codes for a protein that binds to red blood cell receptors and helps the malaria parasite enter cells. The researchers were only able to disrupt this gene in 50% to 80% of parasites manipulated with CRISPR.
“We consider this to be a win,” Dr Niles said. “Compared to the efficiency with which P falciparum genetics have been done in the past, even 50% is pretty substantial.”
Now that CRISPR technology has been validated in P falciparum, Dr Niles expects many scientists will adopt it for genetic studies of the parasite. Such efforts could reveal more about how the parasite invades red blood cells and replicates inside cells, which could generate new drug and vaccine targets.
“I think the impact could be quite huge,” he said. “It lowers the barrier to really being more imaginative in terms of how we do experiments and the kinds of questions that we can ask.” ![]()
Drug decreases need for blood transfusions
![]()
Credit: UAB Hospital
Results of a retrospective study suggest an antifibrinolytic agent can significantly reduce the need for blood transfusions after surgery, without increasing the risk of complications.
The agent, tranexamic acid, has been shown to reduce blood loss during or shortly after major joint surgery, but safety concerns remain because large-scale effectiveness studies are lacking.
So researchers set out to evaluate tranexamic acid in a large sample of surgical patients.
The team recounted their efforts in BMJ.
Stavros Memtsoudis, MD, PhD, of the Hospital for Special Surgery in New York, New York, and his colleagues analyzed data from 872,416 patients who underwent total hip or knee replacement procedures at 510 US hospitals between 2006 and 2012.
The researchers compared patients who received tranexamic acid (at 1000 mg,
2000 mg, or 3000 mg) on the day of surgery to patients who did not. The team adjusted their analysis for factors such as patient age and sex, hospital size and location, the type of procedure, and the anesthesia used.
Results showed that use of tranexamic acid was associated with an up to 69% reduction in the need for blood transfusions. Overall, the rate of allogeneic or autologous transfusion was 7.7% among patients who received tranexamic acid and 20.1% among those who did not (P<0.01).
Tranexamic acid use was also linked to a decreased risk of all complications (1.9% vs 2.6%, P<0.001), thromboembolic events (0.6% vs 0.8%, P=0.0057), the need for mechanical ventilation (0.1% vs 0.2%, P=0.0003), and admission to an intensive care unit (3.1% vs 7.5%, P<0.001).
The median length of hospital stay was the same for treated and untreated patients—3 days. But the median cost of hospital stay was lower among tranexamic acid-treated patients—$14,890 vs $15,110 (P<0.001).
A tranexamic acid dose of 2000 mg appeared to have the best effectiveness and safety profile. But the researchers said additional studies are needed to establish optimal dosing schemes and assess subgroup-specific effectiveness and safety. ![]()
![]()
Credit: UAB Hospital
Results of a retrospective study suggest an antifibrinolytic agent can significantly reduce the need for blood transfusions after surgery, without increasing the risk of complications.
The agent, tranexamic acid, has been shown to reduce blood loss during or shortly after major joint surgery, but safety concerns remain because large-scale effectiveness studies are lacking.
So researchers set out to evaluate tranexamic acid in a large sample of surgical patients.
The team recounted their efforts in BMJ.
Stavros Memtsoudis, MD, PhD, of the Hospital for Special Surgery in New York, New York, and his colleagues analyzed data from 872,416 patients who underwent total hip or knee replacement procedures at 510 US hospitals between 2006 and 2012.
The researchers compared patients who received tranexamic acid (at 1000 mg,
2000 mg, or 3000 mg) on the day of surgery to patients who did not. The team adjusted their analysis for factors such as patient age and sex, hospital size and location, the type of procedure, and the anesthesia used.
Results showed that use of tranexamic acid was associated with an up to 69% reduction in the need for blood transfusions. Overall, the rate of allogeneic or autologous transfusion was 7.7% among patients who received tranexamic acid and 20.1% among those who did not (P<0.01).
Tranexamic acid use was also linked to a decreased risk of all complications (1.9% vs 2.6%, P<0.001), thromboembolic events (0.6% vs 0.8%, P=0.0057), the need for mechanical ventilation (0.1% vs 0.2%, P=0.0003), and admission to an intensive care unit (3.1% vs 7.5%, P<0.001).
The median length of hospital stay was the same for treated and untreated patients—3 days. But the median cost of hospital stay was lower among tranexamic acid-treated patients—$14,890 vs $15,110 (P<0.001).
A tranexamic acid dose of 2000 mg appeared to have the best effectiveness and safety profile. But the researchers said additional studies are needed to establish optimal dosing schemes and assess subgroup-specific effectiveness and safety. ![]()
![]()
Credit: UAB Hospital
Results of a retrospective study suggest an antifibrinolytic agent can significantly reduce the need for blood transfusions after surgery, without increasing the risk of complications.
The agent, tranexamic acid, has been shown to reduce blood loss during or shortly after major joint surgery, but safety concerns remain because large-scale effectiveness studies are lacking.
So researchers set out to evaluate tranexamic acid in a large sample of surgical patients.
The team recounted their efforts in BMJ.
Stavros Memtsoudis, MD, PhD, of the Hospital for Special Surgery in New York, New York, and his colleagues analyzed data from 872,416 patients who underwent total hip or knee replacement procedures at 510 US hospitals between 2006 and 2012.
The researchers compared patients who received tranexamic acid (at 1000 mg,
2000 mg, or 3000 mg) on the day of surgery to patients who did not. The team adjusted their analysis for factors such as patient age and sex, hospital size and location, the type of procedure, and the anesthesia used.
Results showed that use of tranexamic acid was associated with an up to 69% reduction in the need for blood transfusions. Overall, the rate of allogeneic or autologous transfusion was 7.7% among patients who received tranexamic acid and 20.1% among those who did not (P<0.01).
Tranexamic acid use was also linked to a decreased risk of all complications (1.9% vs 2.6%, P<0.001), thromboembolic events (0.6% vs 0.8%, P=0.0057), the need for mechanical ventilation (0.1% vs 0.2%, P=0.0003), and admission to an intensive care unit (3.1% vs 7.5%, P<0.001).
The median length of hospital stay was the same for treated and untreated patients—3 days. But the median cost of hospital stay was lower among tranexamic acid-treated patients—$14,890 vs $15,110 (P<0.001).
A tranexamic acid dose of 2000 mg appeared to have the best effectiveness and safety profile. But the researchers said additional studies are needed to establish optimal dosing schemes and assess subgroup-specific effectiveness and safety. ![]()
Molecule is active against MYC-driven malignancies

Credit: Ed Uthman
A small molecule can disrupt the interactions between MYC and its binding partner MAX in MYC-driven cancers, according to research published in PNAS.
The molecule, KJ-Pyr-9, inhibited MYC-induced oncogenic transformation in cell culture but had little to no effect on the oncogenic activity of several unrelated oncoproteins.
KJ-Pyr-9 preferentially interfered with proliferation in a range of cells that overexpressed MYC, including leukemia and lymphoma cells.
In vivo, the molecule inhibited the growth of MYC-amplified human cancer cells.
“We finally hit a home run with this—maybe a grand slam,” said study author Kim Janda, PhD, of The Scripps Research Institute in La Jolla, California.
For years, MYC has challenged researchers seeking to disrupt its activity in cancer cells.
“At room temperature or body temperature, MYC without any binding partners is random and constantly shifting,” said study author Jonathan Ross Hart, PhD, also of The Scripps Research Institute. “It’s like a piece of spaghetti.”
So instead of designing a compound to target the structure of MYC, the researchers tested a range of compounds from a library to see if any could disrupt the interactions between MYC and other proteins important in cell proliferation. One did—the small molecule KJ-Pyr-9.
To further investigate, the researchers ran tests in a variety of cell lines, including chronic myeloid leukemia, acute lymphoblastic leukemia, acute myeloid leukemia, Burkitt lymphoma, and solid tumors. And they tested KJ-Pyr-9 in mouse models of breast cancer.
The experiments showed that MYC-dependent cells die if treated with KJ-Pyr-9. In fact, a dose of KJ-Pyr-9 made it seem as if MYC was not present at all.
When mice with MYC-dependent tumors received KJ-Pyr-9, the tumors showed no growth after 31 days, compared with significant tumor growth in untreated mice.
Dr Janda said he hopes further research will reveal exactly how KJ-Pyr-9 interacts with MYC and how the compound can more effectively reach tumor cells. ![]()
Credit: Ed Uthman
A small molecule can disrupt the interactions between MYC and its binding partner MAX in MYC-driven cancers, according to research published in PNAS.
The molecule, KJ-Pyr-9, inhibited MYC-induced oncogenic transformation in cell culture but had little to no effect on the oncogenic activity of several unrelated oncoproteins.
KJ-Pyr-9 preferentially interfered with proliferation in a range of cells that overexpressed MYC, including leukemia and lymphoma cells.
In vivo, the molecule inhibited the growth of MYC-amplified human cancer cells.
“We finally hit a home run with this—maybe a grand slam,” said study author Kim Janda, PhD, of The Scripps Research Institute in La Jolla, California.
For years, MYC has challenged researchers seeking to disrupt its activity in cancer cells.
“At room temperature or body temperature, MYC without any binding partners is random and constantly shifting,” said study author Jonathan Ross Hart, PhD, also of The Scripps Research Institute. “It’s like a piece of spaghetti.”
So instead of designing a compound to target the structure of MYC, the researchers tested a range of compounds from a library to see if any could disrupt the interactions between MYC and other proteins important in cell proliferation. One did—the small molecule KJ-Pyr-9.
To further investigate, the researchers ran tests in a variety of cell lines, including chronic myeloid leukemia, acute lymphoblastic leukemia, acute myeloid leukemia, Burkitt lymphoma, and solid tumors. And they tested KJ-Pyr-9 in mouse models of breast cancer.
The experiments showed that MYC-dependent cells die if treated with KJ-Pyr-9. In fact, a dose of KJ-Pyr-9 made it seem as if MYC was not present at all.
When mice with MYC-dependent tumors received KJ-Pyr-9, the tumors showed no growth after 31 days, compared with significant tumor growth in untreated mice.
Dr Janda said he hopes further research will reveal exactly how KJ-Pyr-9 interacts with MYC and how the compound can more effectively reach tumor cells. ![]()
Credit: Ed Uthman
A small molecule can disrupt the interactions between MYC and its binding partner MAX in MYC-driven cancers, according to research published in PNAS.
The molecule, KJ-Pyr-9, inhibited MYC-induced oncogenic transformation in cell culture but had little to no effect on the oncogenic activity of several unrelated oncoproteins.
KJ-Pyr-9 preferentially interfered with proliferation in a range of cells that overexpressed MYC, including leukemia and lymphoma cells.
In vivo, the molecule inhibited the growth of MYC-amplified human cancer cells.
“We finally hit a home run with this—maybe a grand slam,” said study author Kim Janda, PhD, of The Scripps Research Institute in La Jolla, California.
For years, MYC has challenged researchers seeking to disrupt its activity in cancer cells.
“At room temperature or body temperature, MYC without any binding partners is random and constantly shifting,” said study author Jonathan Ross Hart, PhD, also of The Scripps Research Institute. “It’s like a piece of spaghetti.”
So instead of designing a compound to target the structure of MYC, the researchers tested a range of compounds from a library to see if any could disrupt the interactions between MYC and other proteins important in cell proliferation. One did—the small molecule KJ-Pyr-9.
To further investigate, the researchers ran tests in a variety of cell lines, including chronic myeloid leukemia, acute lymphoblastic leukemia, acute myeloid leukemia, Burkitt lymphoma, and solid tumors. And they tested KJ-Pyr-9 in mouse models of breast cancer.
The experiments showed that MYC-dependent cells die if treated with KJ-Pyr-9. In fact, a dose of KJ-Pyr-9 made it seem as if MYC was not present at all.
When mice with MYC-dependent tumors received KJ-Pyr-9, the tumors showed no growth after 31 days, compared with significant tumor growth in untreated mice.
Dr Janda said he hopes further research will reveal exactly how KJ-Pyr-9 interacts with MYC and how the compound can more effectively reach tumor cells. ![]()
Viruses can protect mice from malaria
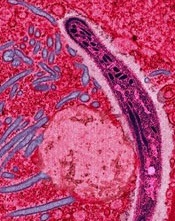
Credit: Ute Frevert
and Margaret Shear
In a new study, genetically altered viruses produced long-lasting antimalaria antibodies in mice and protected many of them from the disease.
The approach, known as vector immunoprophylaxis (VIP), produced antibodies against the Plasmodium falciparum circumsporozoite protein (CSP) and prevented malaria infection in 10% to 100% of mice, depending on the dose and type of viral vector used.
Researchers recounted these results in PNAS.
“We need better ways to fight malaria,” said study author Gary Ketner, PhD, of the Johns Hopkins Bloomberg School of Public Health in Baltimore, Maryland. “And our research suggests [VIP] could be a promising approach.”
To test the approach, Dr Ketner and his colleagues constructed adeno-associated virus (AAV) vectors encoding human immunoglobulin G (hIgG) specific for the P falciparum CSP central repeat by inserting the variable regions of mouse monoclonal antibodies (mAbs) 2A10 and 2C11 into the hIgG framework of the VIP expression plasmid.
The team then injected mice with 1 x 1011 genome copies (GC) of 2A10-AAV, 2C11-AAV, b12-AAV (which protects against HIV), or with buffer.
Within a week, the AAV-transduced mice expressed hIgG at 50 μg/mL to 500 μg/mL in serum. The expression increased until about the 4-week mark, when it reached 1000 μg/mL in some mice.
The mice that received 2A10-AAV or 2C11-AAV expressed antibodies that bound recombinant CSP and recognized whole P falciparum sporozoites. The b12-AAV-transduced mice and buffer-transduced mice did not.
In all AAV-transduced mice, serum antibody concentrations plateaued at 4 to 8 weeks and remained at that level through the end of the study, which was 52 weeks after transduction.
At the 8-week mark, the researchers tested the efficacy of VIP. They introduced—either intravenously or through a mosquito-bite challenge—transgenic Plasmodium berghei rodent sporozoites that incorporate the P falciparum target of the antibody in their CSP.
In the intravenously challenged group, 70% of 2A10-AAV-transduced mice were protected from malaria. In the mosquito-bite-challenged group, 60% of 2A10-AAV-transduced mice and 30% of 2C11-AAV-transduced mice were protected from malaria.
Role of dose and antibody level
To examine the effects of vector dose on mAb production and malaria protection, the researchers compared varying doses of 2A10-AAV to b12-AAV. They tested mice transduced with 3 x 1011 GC of b12-AAV or doses of 2A10-AAV ranging from 3 x 109 GC to 3 x 1011 GC.
The team conducted a mosquito-bite challenge at 11 weeks after transduction. And they found that 70% of the mice that received the highest AAV dose (1 x 1011 GC) were protected, as were 40% of mice that received 3 x 1010 GC and 10% of mice that received 1 x 1010 GC.
All mice transduced with 3 x 109 GC were parasitemic by day 7, and all b12-AAV mice were parasitemic by day 6. There was a signficant correlation between 2A10 antibody concentration and day to parasitemia.
In a subset of mice that produced higher levels of antibodies, the antimalaria protection was 100%. These mice expressed CSP-specific mAb 2A10 at 1 mg/mL or more.
So it seems the protection from malaria is dose-dependent, said study author Cailin Deal, PhD, of the Ragon Institute of MGH, MIT and Harvard in Cambridge, Massachusetts.
“Of course, we don’t know what the human dosage would be,” she added, “but it’s conceivable that the right dosage could completely protect against malaria.” ![]()
Credit: Ute Frevert
and Margaret Shear
In a new study, genetically altered viruses produced long-lasting antimalaria antibodies in mice and protected many of them from the disease.
The approach, known as vector immunoprophylaxis (VIP), produced antibodies against the Plasmodium falciparum circumsporozoite protein (CSP) and prevented malaria infection in 10% to 100% of mice, depending on the dose and type of viral vector used.
Researchers recounted these results in PNAS.
“We need better ways to fight malaria,” said study author Gary Ketner, PhD, of the Johns Hopkins Bloomberg School of Public Health in Baltimore, Maryland. “And our research suggests [VIP] could be a promising approach.”
To test the approach, Dr Ketner and his colleagues constructed adeno-associated virus (AAV) vectors encoding human immunoglobulin G (hIgG) specific for the P falciparum CSP central repeat by inserting the variable regions of mouse monoclonal antibodies (mAbs) 2A10 and 2C11 into the hIgG framework of the VIP expression plasmid.
The team then injected mice with 1 x 1011 genome copies (GC) of 2A10-AAV, 2C11-AAV, b12-AAV (which protects against HIV), or with buffer.
Within a week, the AAV-transduced mice expressed hIgG at 50 μg/mL to 500 μg/mL in serum. The expression increased until about the 4-week mark, when it reached 1000 μg/mL in some mice.
The mice that received 2A10-AAV or 2C11-AAV expressed antibodies that bound recombinant CSP and recognized whole P falciparum sporozoites. The b12-AAV-transduced mice and buffer-transduced mice did not.
In all AAV-transduced mice, serum antibody concentrations plateaued at 4 to 8 weeks and remained at that level through the end of the study, which was 52 weeks after transduction.
At the 8-week mark, the researchers tested the efficacy of VIP. They introduced—either intravenously or through a mosquito-bite challenge—transgenic Plasmodium berghei rodent sporozoites that incorporate the P falciparum target of the antibody in their CSP.
In the intravenously challenged group, 70% of 2A10-AAV-transduced mice were protected from malaria. In the mosquito-bite-challenged group, 60% of 2A10-AAV-transduced mice and 30% of 2C11-AAV-transduced mice were protected from malaria.
Role of dose and antibody level
To examine the effects of vector dose on mAb production and malaria protection, the researchers compared varying doses of 2A10-AAV to b12-AAV. They tested mice transduced with 3 x 1011 GC of b12-AAV or doses of 2A10-AAV ranging from 3 x 109 GC to 3 x 1011 GC.
The team conducted a mosquito-bite challenge at 11 weeks after transduction. And they found that 70% of the mice that received the highest AAV dose (1 x 1011 GC) were protected, as were 40% of mice that received 3 x 1010 GC and 10% of mice that received 1 x 1010 GC.
All mice transduced with 3 x 109 GC were parasitemic by day 7, and all b12-AAV mice were parasitemic by day 6. There was a signficant correlation between 2A10 antibody concentration and day to parasitemia.
In a subset of mice that produced higher levels of antibodies, the antimalaria protection was 100%. These mice expressed CSP-specific mAb 2A10 at 1 mg/mL or more.
So it seems the protection from malaria is dose-dependent, said study author Cailin Deal, PhD, of the Ragon Institute of MGH, MIT and Harvard in Cambridge, Massachusetts.
“Of course, we don’t know what the human dosage would be,” she added, “but it’s conceivable that the right dosage could completely protect against malaria.” ![]()
Credit: Ute Frevert
and Margaret Shear
In a new study, genetically altered viruses produced long-lasting antimalaria antibodies in mice and protected many of them from the disease.
The approach, known as vector immunoprophylaxis (VIP), produced antibodies against the Plasmodium falciparum circumsporozoite protein (CSP) and prevented malaria infection in 10% to 100% of mice, depending on the dose and type of viral vector used.
Researchers recounted these results in PNAS.
“We need better ways to fight malaria,” said study author Gary Ketner, PhD, of the Johns Hopkins Bloomberg School of Public Health in Baltimore, Maryland. “And our research suggests [VIP] could be a promising approach.”
To test the approach, Dr Ketner and his colleagues constructed adeno-associated virus (AAV) vectors encoding human immunoglobulin G (hIgG) specific for the P falciparum CSP central repeat by inserting the variable regions of mouse monoclonal antibodies (mAbs) 2A10 and 2C11 into the hIgG framework of the VIP expression plasmid.
The team then injected mice with 1 x 1011 genome copies (GC) of 2A10-AAV, 2C11-AAV, b12-AAV (which protects against HIV), or with buffer.
Within a week, the AAV-transduced mice expressed hIgG at 50 μg/mL to 500 μg/mL in serum. The expression increased until about the 4-week mark, when it reached 1000 μg/mL in some mice.
The mice that received 2A10-AAV or 2C11-AAV expressed antibodies that bound recombinant CSP and recognized whole P falciparum sporozoites. The b12-AAV-transduced mice and buffer-transduced mice did not.
In all AAV-transduced mice, serum antibody concentrations plateaued at 4 to 8 weeks and remained at that level through the end of the study, which was 52 weeks after transduction.
At the 8-week mark, the researchers tested the efficacy of VIP. They introduced—either intravenously or through a mosquito-bite challenge—transgenic Plasmodium berghei rodent sporozoites that incorporate the P falciparum target of the antibody in their CSP.
In the intravenously challenged group, 70% of 2A10-AAV-transduced mice were protected from malaria. In the mosquito-bite-challenged group, 60% of 2A10-AAV-transduced mice and 30% of 2C11-AAV-transduced mice were protected from malaria.
Role of dose and antibody level
To examine the effects of vector dose on mAb production and malaria protection, the researchers compared varying doses of 2A10-AAV to b12-AAV. They tested mice transduced with 3 x 1011 GC of b12-AAV or doses of 2A10-AAV ranging from 3 x 109 GC to 3 x 1011 GC.
The team conducted a mosquito-bite challenge at 11 weeks after transduction. And they found that 70% of the mice that received the highest AAV dose (1 x 1011 GC) were protected, as were 40% of mice that received 3 x 1010 GC and 10% of mice that received 1 x 1010 GC.
All mice transduced with 3 x 109 GC were parasitemic by day 7, and all b12-AAV mice were parasitemic by day 6. There was a signficant correlation between 2A10 antibody concentration and day to parasitemia.
In a subset of mice that produced higher levels of antibodies, the antimalaria protection was 100%. These mice expressed CSP-specific mAb 2A10 at 1 mg/mL or more.
So it seems the protection from malaria is dose-dependent, said study author Cailin Deal, PhD, of the Ragon Institute of MGH, MIT and Harvard in Cambridge, Massachusetts.
“Of course, we don’t know what the human dosage would be,” she added, “but it’s conceivable that the right dosage could completely protect against malaria.”
HDAC inhibitor gets orphan status for DLBCL

The US Food and Drug Administration (FDA) has granted orphan designation for the histone deacetylase (HDAC) inhibitor mocetinostat to treat diffuse large B-cell lymphoma (DLBCL). The drug already had orphan designation as a treatment for myelodysplastic syndrome (MDS).
The FDA grants orphan status to support the development of drugs for underserved patient populations or rare disorders affecting fewer than 200,000 people in the US.
Orphan designation provides the drug’s developer, Mirati Therapeutics, Inc., with certain benefits, including market exclusivity upon regulatory approval, exemption of FDA application fees, and tax credits for qualified clinical trials.
Mocetinostat works by reversing aberrant acetylation resulting from mutations in histone acetyltransferases (HATs).
The drug is being developed as a single-agent treatment for patients with DLBCL or bladder cancer characterized by HAT mutations that Mirati believes are critical in the pathogenesis and progression of these cancers.
“We have identified genetic alterations in histone acetylation pathways (CREBBP and EP300) in approximately one-third of DLBCL and bladder tumors,” said Charles Baum, MD, PhD, president and CEO of Mirati.
He added that nonclinical tumor models with these mutations have proven responsive to mocetinostat, so Mirati predicts the HDAC inhibitor will halt tumor progression and reduce tumor burden in patients.
Mocetinostat is also under investigation in phase 2 studies in combination with azacitidine (Vidaza) as a treatment for intermediate- and high-risk MDS.
Mocetinostat previously demonstrated activity, as well as toxicity, in patients with Hodgkin lymphoma.
The US Food and Drug Administration (FDA) has granted orphan designation for the histone deacetylase (HDAC) inhibitor mocetinostat to treat diffuse large B-cell lymphoma (DLBCL). The drug already had orphan designation as a treatment for myelodysplastic syndrome (MDS).
The FDA grants orphan status to support the development of drugs for underserved patient populations or rare disorders affecting fewer than 200,000 people in the US.
Orphan designation provides the drug’s developer, Mirati Therapeutics, Inc., with certain benefits, including market exclusivity upon regulatory approval, exemption of FDA application fees, and tax credits for qualified clinical trials.
Mocetinostat works by reversing aberrant acetylation resulting from mutations in histone acetyltransferases (HATs).
The drug is being developed as a single-agent treatment for patients with DLBCL or bladder cancer characterized by HAT mutations that Mirati believes are critical in the pathogenesis and progression of these cancers.
“We have identified genetic alterations in histone acetylation pathways (CREBBP and EP300) in approximately one-third of DLBCL and bladder tumors,” said Charles Baum, MD, PhD, president and CEO of Mirati.
He added that nonclinical tumor models with these mutations have proven responsive to mocetinostat, so Mirati predicts the HDAC inhibitor will halt tumor progression and reduce tumor burden in patients.
Mocetinostat is also under investigation in phase 2 studies in combination with azacitidine (Vidaza) as a treatment for intermediate- and high-risk MDS.
Mocetinostat previously demonstrated activity, as well as toxicity, in patients with Hodgkin lymphoma.
The US Food and Drug Administration (FDA) has granted orphan designation for the histone deacetylase (HDAC) inhibitor mocetinostat to treat diffuse large B-cell lymphoma (DLBCL). The drug already had orphan designation as a treatment for myelodysplastic syndrome (MDS).
The FDA grants orphan status to support the development of drugs for underserved patient populations or rare disorders affecting fewer than 200,000 people in the US.
Orphan designation provides the drug’s developer, Mirati Therapeutics, Inc., with certain benefits, including market exclusivity upon regulatory approval, exemption of FDA application fees, and tax credits for qualified clinical trials.
Mocetinostat works by reversing aberrant acetylation resulting from mutations in histone acetyltransferases (HATs).
The drug is being developed as a single-agent treatment for patients with DLBCL or bladder cancer characterized by HAT mutations that Mirati believes are critical in the pathogenesis and progression of these cancers.
“We have identified genetic alterations in histone acetylation pathways (CREBBP and EP300) in approximately one-third of DLBCL and bladder tumors,” said Charles Baum, MD, PhD, president and CEO of Mirati.
He added that nonclinical tumor models with these mutations have proven responsive to mocetinostat, so Mirati predicts the HDAC inhibitor will halt tumor progression and reduce tumor burden in patients.
Mocetinostat is also under investigation in phase 2 studies in combination with azacitidine (Vidaza) as a treatment for intermediate- and high-risk MDS.
Mocetinostat previously demonstrated activity, as well as toxicity, in patients with Hodgkin lymphoma.
Enzyme plays key role in MM, group finds

Credit: Darren Baker
Researchers have identified an enzyme that appears key to prognosis and progression in multiple myeloma (MM).
The group found the enzyme, ST3GAL6, is overexpressed in MM cell lines and patients with disease.
This overexpression increases glycosylation, which escalates the interaction between MM cells and selectins.
And this encourages the circulation and spread of MM cells, as well as their retention in the bone marrow.
The researchers recounted this series of events in Blood.
“[W]e focused on alterations in [glycosylation] because of its role in cell-cell interactions and the spread of cancer cells in the blood,” said study author Michael O’Dwyer, MD, of National University of Ireland, Galway.
Using gene set enrichment analysis, he and his colleagues confirmed the overexpression of glycosylation-related signatures in MM. They also discovered the sialyltransferase ST3GAL6 was “one of the most significantly increased genes” in MM patients, when compared to healthy donors.
The team observed increased ST3GAL6 levels in both relapsed and newly diagnosed MM. And they found that higher ST3GAL6 levels were associated with decreased survival.
To expand upon these findings, the researchers went on to test 5 MM cell lines—MM1S, MM1R, U266, RPMI-8226, and H929. They found significantly higher levels of ST3GAL6 mRNA and protein in the cell lines compared to healthy CD138+ cells.
Knocking down ST3GAL6 in 2 of the cell lines—MM1S and RPMI-8226—significantly reduced the amount of alpha 2,3 sialic acid at the cell surface, which suggests that ST3GAL6 contributes to the synthesis of this glycan.
In addition, knocking down ST3GAL6 significantly reduced MM cells’ adhesion to bone marrow stem cells, human umbilical vein endothelial cells, and fibronectin. It also reduced MM cells’ transendothelial migration, attenuated Src activation in MM cells, and reduced the cells’ ability to roll on p-selectin.
Likewise, in mouse models, knockdown of ST3GAL6 reduced MM cell homing and engraftment. It also significantly decreased tumor burden and increased survival in the mice.
The researchers concluded that these findings highlight the importance of altered glycosylation, particularly sialylation, in MM cell adhesion and migration.
“Our research is crucial because it sheds new light on the biology of [MM], which could lead to new strategies to overcome resistance to treatment,” Dr O’Dwyer said. “Our aim now is to prevent these interactions that cause the spread [of MM cells] using specific enzyme and selectin inhibitors.”
Credit: Darren Baker
Researchers have identified an enzyme that appears key to prognosis and progression in multiple myeloma (MM).
The group found the enzyme, ST3GAL6, is overexpressed in MM cell lines and patients with disease.
This overexpression increases glycosylation, which escalates the interaction between MM cells and selectins.
And this encourages the circulation and spread of MM cells, as well as their retention in the bone marrow.
The researchers recounted this series of events in Blood.
“[W]e focused on alterations in [glycosylation] because of its role in cell-cell interactions and the spread of cancer cells in the blood,” said study author Michael O’Dwyer, MD, of National University of Ireland, Galway.
Using gene set enrichment analysis, he and his colleagues confirmed the overexpression of glycosylation-related signatures in MM. They also discovered the sialyltransferase ST3GAL6 was “one of the most significantly increased genes” in MM patients, when compared to healthy donors.
The team observed increased ST3GAL6 levels in both relapsed and newly diagnosed MM. And they found that higher ST3GAL6 levels were associated with decreased survival.
To expand upon these findings, the researchers went on to test 5 MM cell lines—MM1S, MM1R, U266, RPMI-8226, and H929. They found significantly higher levels of ST3GAL6 mRNA and protein in the cell lines compared to healthy CD138+ cells.
Knocking down ST3GAL6 in 2 of the cell lines—MM1S and RPMI-8226—significantly reduced the amount of alpha 2,3 sialic acid at the cell surface, which suggests that ST3GAL6 contributes to the synthesis of this glycan.
In addition, knocking down ST3GAL6 significantly reduced MM cells’ adhesion to bone marrow stem cells, human umbilical vein endothelial cells, and fibronectin. It also reduced MM cells’ transendothelial migration, attenuated Src activation in MM cells, and reduced the cells’ ability to roll on p-selectin.
Likewise, in mouse models, knockdown of ST3GAL6 reduced MM cell homing and engraftment. It also significantly decreased tumor burden and increased survival in the mice.
The researchers concluded that these findings highlight the importance of altered glycosylation, particularly sialylation, in MM cell adhesion and migration.
“Our research is crucial because it sheds new light on the biology of [MM], which could lead to new strategies to overcome resistance to treatment,” Dr O’Dwyer said. “Our aim now is to prevent these interactions that cause the spread [of MM cells] using specific enzyme and selectin inhibitors.”
Credit: Darren Baker
Researchers have identified an enzyme that appears key to prognosis and progression in multiple myeloma (MM).
The group found the enzyme, ST3GAL6, is overexpressed in MM cell lines and patients with disease.
This overexpression increases glycosylation, which escalates the interaction between MM cells and selectins.
And this encourages the circulation and spread of MM cells, as well as their retention in the bone marrow.
The researchers recounted this series of events in Blood.
“[W]e focused on alterations in [glycosylation] because of its role in cell-cell interactions and the spread of cancer cells in the blood,” said study author Michael O’Dwyer, MD, of National University of Ireland, Galway.
Using gene set enrichment analysis, he and his colleagues confirmed the overexpression of glycosylation-related signatures in MM. They also discovered the sialyltransferase ST3GAL6 was “one of the most significantly increased genes” in MM patients, when compared to healthy donors.
The team observed increased ST3GAL6 levels in both relapsed and newly diagnosed MM. And they found that higher ST3GAL6 levels were associated with decreased survival.
To expand upon these findings, the researchers went on to test 5 MM cell lines—MM1S, MM1R, U266, RPMI-8226, and H929. They found significantly higher levels of ST3GAL6 mRNA and protein in the cell lines compared to healthy CD138+ cells.
Knocking down ST3GAL6 in 2 of the cell lines—MM1S and RPMI-8226—significantly reduced the amount of alpha 2,3 sialic acid at the cell surface, which suggests that ST3GAL6 contributes to the synthesis of this glycan.
In addition, knocking down ST3GAL6 significantly reduced MM cells’ adhesion to bone marrow stem cells, human umbilical vein endothelial cells, and fibronectin. It also reduced MM cells’ transendothelial migration, attenuated Src activation in MM cells, and reduced the cells’ ability to roll on p-selectin.
Likewise, in mouse models, knockdown of ST3GAL6 reduced MM cell homing and engraftment. It also significantly decreased tumor burden and increased survival in the mice.
The researchers concluded that these findings highlight the importance of altered glycosylation, particularly sialylation, in MM cell adhesion and migration.
“Our research is crucial because it sheds new light on the biology of [MM], which could lead to new strategies to overcome resistance to treatment,” Dr O’Dwyer said. “Our aim now is to prevent these interactions that cause the spread [of MM cells] using specific enzyme and selectin inhibitors.”
Study reveals potential targets for MYC-dependent cancers

Credit: Juha Klefstrom
New research suggests the MYC protein drives cell growth by inhibiting a handful of genes involved in DNA packaging and cell death.
The study showed that MYC works through a microRNA to suppress the genes’ expression.
This marks the first time that a subset of MYC-controlled genes have been identified as critical players in the protein’s cancer-causing function, and it points to new therapeutic targets for MYC-dependent cancers.
“This is a different way of thinking about the roles of microRNA and chromatin packaging in cancer,” said Dean Felsher, MD, PhD, of the Stanford University School of Medicine in California.
“We were very surprised to learn that the overexpression of one microRNA can mimic the cancerous effect of MYC.”
Dr Felsher and his colleagues reported this discovery in Cancer Cell.
The team noted that MYC overexpression has been known to prompt an increase in the levels of a microRNA family called miR-17-92.
“People have known for several years that MYC regulates the expression of microRNAs,” Dr Felsher said. “But it wasn’t clear how this was related to MYC’s oncogenic function.”
To gain some insight, Dr Felsher and his colleagues analyzed MYC-dependent cancer cells in vitro and in vivo.
The cells in which miR-17-92 expression was locked in the “on” position kept dividing even when MYC expression was blocked. This suggested that MYC works through the microRNA family to exert its cancer-causing effects.
The researchers then looked for an overlap among genes affected by MYC overexpression and those affected by miR-17-92. There were about 401 genes whose expression was either increased or suppressed by both MYC and miR-17-92.
The team chose to focus on genes that were suppressed because these genes exhibited, on average, many more binding sites for the microRNAs. They further narrowed their panel down to 15 genes regulated by more than one miR-17-92 binding site.
Of these genes, 5 stood out. Four of them—Sin3b, Hbp1, Suv420h1, and Btg1—encode proteins known to regulate chromatin packaging.
These 4 proteins affect cell proliferation and senescence by regulating gene accessibility within the chromatin. They had never before been identified as MYC or miR-17-92 targets.
The fifth gene encodes the apoptotic protein Bim. Previous research suggested that Bim expression is affected by miR-17-92.
All 5 of the proteins are known to affect either cellular proliferation, entry into senescence, or apoptosis, in part by granting or prohibiting access to genes in tightly packaged stretches of DNA in the chromatin.
“MYC is still a general amplifier of gene transcription and expression,” Dr Felsher said. “But our study shows that the maintenance of the cancerous state relies on a more focused mechanism.”
Lastly, the researchers showed that suppressing the expression of the 5 target genes, effectively mimicking MYC overexpression, partially mitigates the effect of MYC deactivation.
Up to 30% of MYC-dependent cancer cells in culture continued to grow—compared to 11% of control cells—in the absence of MYC expression. And tumors in mice either failed to regress or recurred within a few weeks.
“One of the biggest unanswered questions in oncology is how oncogenes cause cancer, and whether you can replace an oncogene with another gene product,” Dr Felsher said.
“These experiments begin to reveal how MYC affects the self-renewal decisions of cells. They may also help us target those aspects of MYC overexpression that contribute to the cancer phenotype.”
Credit: Juha Klefstrom
New research suggests the MYC protein drives cell growth by inhibiting a handful of genes involved in DNA packaging and cell death.
The study showed that MYC works through a microRNA to suppress the genes’ expression.
This marks the first time that a subset of MYC-controlled genes have been identified as critical players in the protein’s cancer-causing function, and it points to new therapeutic targets for MYC-dependent cancers.
“This is a different way of thinking about the roles of microRNA and chromatin packaging in cancer,” said Dean Felsher, MD, PhD, of the Stanford University School of Medicine in California.
“We were very surprised to learn that the overexpression of one microRNA can mimic the cancerous effect of MYC.”
Dr Felsher and his colleagues reported this discovery in Cancer Cell.
The team noted that MYC overexpression has been known to prompt an increase in the levels of a microRNA family called miR-17-92.
“People have known for several years that MYC regulates the expression of microRNAs,” Dr Felsher said. “But it wasn’t clear how this was related to MYC’s oncogenic function.”
To gain some insight, Dr Felsher and his colleagues analyzed MYC-dependent cancer cells in vitro and in vivo.
The cells in which miR-17-92 expression was locked in the “on” position kept dividing even when MYC expression was blocked. This suggested that MYC works through the microRNA family to exert its cancer-causing effects.
The researchers then looked for an overlap among genes affected by MYC overexpression and those affected by miR-17-92. There were about 401 genes whose expression was either increased or suppressed by both MYC and miR-17-92.
The team chose to focus on genes that were suppressed because these genes exhibited, on average, many more binding sites for the microRNAs. They further narrowed their panel down to 15 genes regulated by more than one miR-17-92 binding site.
Of these genes, 5 stood out. Four of them—Sin3b, Hbp1, Suv420h1, and Btg1—encode proteins known to regulate chromatin packaging.
These 4 proteins affect cell proliferation and senescence by regulating gene accessibility within the chromatin. They had never before been identified as MYC or miR-17-92 targets.
The fifth gene encodes the apoptotic protein Bim. Previous research suggested that Bim expression is affected by miR-17-92.
All 5 of the proteins are known to affect either cellular proliferation, entry into senescence, or apoptosis, in part by granting or prohibiting access to genes in tightly packaged stretches of DNA in the chromatin.
“MYC is still a general amplifier of gene transcription and expression,” Dr Felsher said. “But our study shows that the maintenance of the cancerous state relies on a more focused mechanism.”
Lastly, the researchers showed that suppressing the expression of the 5 target genes, effectively mimicking MYC overexpression, partially mitigates the effect of MYC deactivation.
Up to 30% of MYC-dependent cancer cells in culture continued to grow—compared to 11% of control cells—in the absence of MYC expression. And tumors in mice either failed to regress or recurred within a few weeks.
“One of the biggest unanswered questions in oncology is how oncogenes cause cancer, and whether you can replace an oncogene with another gene product,” Dr Felsher said.
“These experiments begin to reveal how MYC affects the self-renewal decisions of cells. They may also help us target those aspects of MYC overexpression that contribute to the cancer phenotype.”
Credit: Juha Klefstrom
New research suggests the MYC protein drives cell growth by inhibiting a handful of genes involved in DNA packaging and cell death.
The study showed that MYC works through a microRNA to suppress the genes’ expression.
This marks the first time that a subset of MYC-controlled genes have been identified as critical players in the protein’s cancer-causing function, and it points to new therapeutic targets for MYC-dependent cancers.
“This is a different way of thinking about the roles of microRNA and chromatin packaging in cancer,” said Dean Felsher, MD, PhD, of the Stanford University School of Medicine in California.
“We were very surprised to learn that the overexpression of one microRNA can mimic the cancerous effect of MYC.”
Dr Felsher and his colleagues reported this discovery in Cancer Cell.
The team noted that MYC overexpression has been known to prompt an increase in the levels of a microRNA family called miR-17-92.
“People have known for several years that MYC regulates the expression of microRNAs,” Dr Felsher said. “But it wasn’t clear how this was related to MYC’s oncogenic function.”
To gain some insight, Dr Felsher and his colleagues analyzed MYC-dependent cancer cells in vitro and in vivo.
The cells in which miR-17-92 expression was locked in the “on” position kept dividing even when MYC expression was blocked. This suggested that MYC works through the microRNA family to exert its cancer-causing effects.
The researchers then looked for an overlap among genes affected by MYC overexpression and those affected by miR-17-92. There were about 401 genes whose expression was either increased or suppressed by both MYC and miR-17-92.
The team chose to focus on genes that were suppressed because these genes exhibited, on average, many more binding sites for the microRNAs. They further narrowed their panel down to 15 genes regulated by more than one miR-17-92 binding site.
Of these genes, 5 stood out. Four of them—Sin3b, Hbp1, Suv420h1, and Btg1—encode proteins known to regulate chromatin packaging.
These 4 proteins affect cell proliferation and senescence by regulating gene accessibility within the chromatin. They had never before been identified as MYC or miR-17-92 targets.
The fifth gene encodes the apoptotic protein Bim. Previous research suggested that Bim expression is affected by miR-17-92.
All 5 of the proteins are known to affect either cellular proliferation, entry into senescence, or apoptosis, in part by granting or prohibiting access to genes in tightly packaged stretches of DNA in the chromatin.
“MYC is still a general amplifier of gene transcription and expression,” Dr Felsher said. “But our study shows that the maintenance of the cancerous state relies on a more focused mechanism.”
Lastly, the researchers showed that suppressing the expression of the 5 target genes, effectively mimicking MYC overexpression, partially mitigates the effect of MYC deactivation.
Up to 30% of MYC-dependent cancer cells in culture continued to grow—compared to 11% of control cells—in the absence of MYC expression. And tumors in mice either failed to regress or recurred within a few weeks.
“One of the biggest unanswered questions in oncology is how oncogenes cause cancer, and whether you can replace an oncogene with another gene product,” Dr Felsher said.
“These experiments begin to reveal how MYC affects the self-renewal decisions of cells. They may also help us target those aspects of MYC overexpression that contribute to the cancer phenotype.”