User login
PD-1 inhibitor granted accelerated approval for cHL

Photo courtesy of Business Wire
The US Food and Drug Administration (FDA) has granted accelerated approval for the PD-1 inhibitor nivolumab (Opdivo) to treat classical Hodgkin lymphoma (cHL).
The drug is approved to treat patients with relapsed or refractory cHL who have received an autologous hematopoietic stem cell transplant (HSCT) and post-transplant brentuximab vedotin.
Nivolumab received accelerated approval because it has not yet shown a clinical benefit in these patients. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of nivolumab for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted nivolumab breakthrough therapy designation, priority review status, and orphan drug designation.
Dosing and precautions
The recommended dose and schedule of nivolumab for cHL patients is 3 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity.
The FDA added a new “Warning and Precaution” to the label for nivolumab, regarding complications of allogeneic HSCT after nivolumab.
Transplant-related deaths have occurred. So the FDA said healthcare professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions.
The FDA has required the manufacturer of nivolumab, Bristol-Myers Squibb, to further study the safety of allogeneic HSCT after nivolumab.
Full prescribing information for the drug is available here.
Trials of nivolumab
The FDA granted nivolumab accelerated approval in cHL patients based on the results of 2 single-arm, multicenter trials—the phase 1 Checkmate 039 trial (presented at ICML last year) and the phase 2 CheckMate 205 trial (to be presented at ASCO 2016).
Efficacy
Thus far, researchers have evaluated the efficacy of nivolumab in 95 cHL patients from both trials. All of these patients previously received an autologous HSCT and post-transplant brentuximab vedotin. They received a median of 5 prior systemic regimens (range, 3 to 15).
The patients received a median of 17 doses of nivolumab (range, 3 to 48). The overall response rate was 65%, and the complete response rate was 7%.
The median time to response was 2.1 months (range, 0.7 to 5.7), and the estimated median duration of response was 8.7 months (range, 0+ to 23.1+).
Safety
Researchers evaluated the safety of nivolumab in 263 patients with relapsed or refractory cHL. Ninety-eight percent of these patients had received an autologous HSCT. The patients received a median of 10 doses of nivolumab (range, 1 to 48) at the approved dose and schedule.
The most common (≥20%) adverse events (AEs) of any grade were fatigue, upper respiratory tract infection, cough, pyrexia, and diarrhea.
Additional common (≥10%) AEs included rash, pruritus, musculoskeletal pain, nausea, vomiting, abdominal pain, headache, peripheral neuropathy, arthralgia, dyspnea, infusion-related reactions, and hypothyroidism or thyroiditis.
Serious AEs were reported in 21% of patients. The most common, reported in 1% to 3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash. ![]()
Photo courtesy of Business Wire
The US Food and Drug Administration (FDA) has granted accelerated approval for the PD-1 inhibitor nivolumab (Opdivo) to treat classical Hodgkin lymphoma (cHL).
The drug is approved to treat patients with relapsed or refractory cHL who have received an autologous hematopoietic stem cell transplant (HSCT) and post-transplant brentuximab vedotin.
Nivolumab received accelerated approval because it has not yet shown a clinical benefit in these patients. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of nivolumab for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted nivolumab breakthrough therapy designation, priority review status, and orphan drug designation.
Dosing and precautions
The recommended dose and schedule of nivolumab for cHL patients is 3 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity.
The FDA added a new “Warning and Precaution” to the label for nivolumab, regarding complications of allogeneic HSCT after nivolumab.
Transplant-related deaths have occurred. So the FDA said healthcare professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions.
The FDA has required the manufacturer of nivolumab, Bristol-Myers Squibb, to further study the safety of allogeneic HSCT after nivolumab.
Full prescribing information for the drug is available here.
Trials of nivolumab
The FDA granted nivolumab accelerated approval in cHL patients based on the results of 2 single-arm, multicenter trials—the phase 1 Checkmate 039 trial (presented at ICML last year) and the phase 2 CheckMate 205 trial (to be presented at ASCO 2016).
Efficacy
Thus far, researchers have evaluated the efficacy of nivolumab in 95 cHL patients from both trials. All of these patients previously received an autologous HSCT and post-transplant brentuximab vedotin. They received a median of 5 prior systemic regimens (range, 3 to 15).
The patients received a median of 17 doses of nivolumab (range, 3 to 48). The overall response rate was 65%, and the complete response rate was 7%.
The median time to response was 2.1 months (range, 0.7 to 5.7), and the estimated median duration of response was 8.7 months (range, 0+ to 23.1+).
Safety
Researchers evaluated the safety of nivolumab in 263 patients with relapsed or refractory cHL. Ninety-eight percent of these patients had received an autologous HSCT. The patients received a median of 10 doses of nivolumab (range, 1 to 48) at the approved dose and schedule.
The most common (≥20%) adverse events (AEs) of any grade were fatigue, upper respiratory tract infection, cough, pyrexia, and diarrhea.
Additional common (≥10%) AEs included rash, pruritus, musculoskeletal pain, nausea, vomiting, abdominal pain, headache, peripheral neuropathy, arthralgia, dyspnea, infusion-related reactions, and hypothyroidism or thyroiditis.
Serious AEs were reported in 21% of patients. The most common, reported in 1% to 3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash. ![]()
Photo courtesy of Business Wire
The US Food and Drug Administration (FDA) has granted accelerated approval for the PD-1 inhibitor nivolumab (Opdivo) to treat classical Hodgkin lymphoma (cHL).
The drug is approved to treat patients with relapsed or refractory cHL who have received an autologous hematopoietic stem cell transplant (HSCT) and post-transplant brentuximab vedotin.
Nivolumab received accelerated approval because it has not yet shown a clinical benefit in these patients. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of nivolumab for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted nivolumab breakthrough therapy designation, priority review status, and orphan drug designation.
Dosing and precautions
The recommended dose and schedule of nivolumab for cHL patients is 3 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity.
The FDA added a new “Warning and Precaution” to the label for nivolumab, regarding complications of allogeneic HSCT after nivolumab.
Transplant-related deaths have occurred. So the FDA said healthcare professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions.
The FDA has required the manufacturer of nivolumab, Bristol-Myers Squibb, to further study the safety of allogeneic HSCT after nivolumab.
Full prescribing information for the drug is available here.
Trials of nivolumab
The FDA granted nivolumab accelerated approval in cHL patients based on the results of 2 single-arm, multicenter trials—the phase 1 Checkmate 039 trial (presented at ICML last year) and the phase 2 CheckMate 205 trial (to be presented at ASCO 2016).
Efficacy
Thus far, researchers have evaluated the efficacy of nivolumab in 95 cHL patients from both trials. All of these patients previously received an autologous HSCT and post-transplant brentuximab vedotin. They received a median of 5 prior systemic regimens (range, 3 to 15).
The patients received a median of 17 doses of nivolumab (range, 3 to 48). The overall response rate was 65%, and the complete response rate was 7%.
The median time to response was 2.1 months (range, 0.7 to 5.7), and the estimated median duration of response was 8.7 months (range, 0+ to 23.1+).
Safety
Researchers evaluated the safety of nivolumab in 263 patients with relapsed or refractory cHL. Ninety-eight percent of these patients had received an autologous HSCT. The patients received a median of 10 doses of nivolumab (range, 1 to 48) at the approved dose and schedule.
The most common (≥20%) adverse events (AEs) of any grade were fatigue, upper respiratory tract infection, cough, pyrexia, and diarrhea.
Additional common (≥10%) AEs included rash, pruritus, musculoskeletal pain, nausea, vomiting, abdominal pain, headache, peripheral neuropathy, arthralgia, dyspnea, infusion-related reactions, and hypothyroidism or thyroiditis.
Serious AEs were reported in 21% of patients. The most common, reported in 1% to 3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash. ![]()
Team describes new approach to cancer immunotherapy
Image by Kathryn T. Iacono
A new approach to cancer immunotherapy may avoid some of the shortcomings associated with other methods, according to researchers.
The group found that eliminating a key protein in regulatory T cells (Tregs) makes them so unstable that they become effector T cells (Teff) and begin to attack the cancer.
And this conversion from Treg to Teff occurs only in the inflammatory conditions that prevail within many tumors.
As a result, Tregs embedded in normal tissue throughout the body continue to have a restraining effect on their local Teffs, protecting healthy organs and tissues from attack.
The researchers said this raises the prospect of therapies that concentrate the immune system’s firepower on tumors without producing residual damage and harmful side effects.
“Many current approaches to immunotherapy involve depleting or blocking Tregs in order to shift the balance toward Teff cells,” said Harvey Cantor, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“This, however, runs the risk of triggering an autoimmune response in which the Teff cells attack normal as well as malignant tissue. The key to our approach is that it singles out the Tregs inside a tumor for conversion, leaving Tregs elsewhere in the body unchanged.”
Dr Cantor and his colleagues described the approach in PNAS.
The study builds on research published last year in Science. That study showed that Tregs maintain their immune-suppressive properties under inflammatory conditions as long as they have high enough levels of a protein called Helios. Depriving Tregs of sufficient Helios caused them to lose that stability and turn into Teff cells.
The new study explored whether this convertibility could be harnessed for therapeutic purposes in cancers.
The first set of experiments involved mice engineered to lack Helios in their Tregs. When the animals were injected with melanoma or colon cancer cells, they developed tumors far more slowly than animals with normal Tregs.
“Inspection of the animals’ tumor tissue showed an unstable set of T regulatory cells, many of which had converted into Teffs,” said Hye-Jung Kim, PhD, also of the Dana-Farber Cancer Institute.
The researchers then explored whether stanching Helios production in tumor-dwelling Tregs could have the same effect. They tested several antibodies that bind to key receptors on Tregs and cause a downturn in Helios production.
The team chose an antibody that worked well, DTA-1, and tested it in mice with Treg-laden tumors. When they analyzed the tumor tissue, it was clear that DTA-1 had triggered conversion of Tregs to Teffs.
“This represents a next stage in cancer immunotherapy,” Dr Cantor said. “We now have a very specific, targeted way of inducing a T-effector-cell attack on cancer while lowering the risk of adverse effects on healthy tissue. The next step will be to organize a clinical trial using this approach in patients.” ![]()
Image by Kathryn T. Iacono
A new approach to cancer immunotherapy may avoid some of the shortcomings associated with other methods, according to researchers.
The group found that eliminating a key protein in regulatory T cells (Tregs) makes them so unstable that they become effector T cells (Teff) and begin to attack the cancer.
And this conversion from Treg to Teff occurs only in the inflammatory conditions that prevail within many tumors.
As a result, Tregs embedded in normal tissue throughout the body continue to have a restraining effect on their local Teffs, protecting healthy organs and tissues from attack.
The researchers said this raises the prospect of therapies that concentrate the immune system’s firepower on tumors without producing residual damage and harmful side effects.
“Many current approaches to immunotherapy involve depleting or blocking Tregs in order to shift the balance toward Teff cells,” said Harvey Cantor, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“This, however, runs the risk of triggering an autoimmune response in which the Teff cells attack normal as well as malignant tissue. The key to our approach is that it singles out the Tregs inside a tumor for conversion, leaving Tregs elsewhere in the body unchanged.”
Dr Cantor and his colleagues described the approach in PNAS.
The study builds on research published last year in Science. That study showed that Tregs maintain their immune-suppressive properties under inflammatory conditions as long as they have high enough levels of a protein called Helios. Depriving Tregs of sufficient Helios caused them to lose that stability and turn into Teff cells.
The new study explored whether this convertibility could be harnessed for therapeutic purposes in cancers.
The first set of experiments involved mice engineered to lack Helios in their Tregs. When the animals were injected with melanoma or colon cancer cells, they developed tumors far more slowly than animals with normal Tregs.
“Inspection of the animals’ tumor tissue showed an unstable set of T regulatory cells, many of which had converted into Teffs,” said Hye-Jung Kim, PhD, also of the Dana-Farber Cancer Institute.
The researchers then explored whether stanching Helios production in tumor-dwelling Tregs could have the same effect. They tested several antibodies that bind to key receptors on Tregs and cause a downturn in Helios production.
The team chose an antibody that worked well, DTA-1, and tested it in mice with Treg-laden tumors. When they analyzed the tumor tissue, it was clear that DTA-1 had triggered conversion of Tregs to Teffs.
“This represents a next stage in cancer immunotherapy,” Dr Cantor said. “We now have a very specific, targeted way of inducing a T-effector-cell attack on cancer while lowering the risk of adverse effects on healthy tissue. The next step will be to organize a clinical trial using this approach in patients.” ![]()
Image by Kathryn T. Iacono
A new approach to cancer immunotherapy may avoid some of the shortcomings associated with other methods, according to researchers.
The group found that eliminating a key protein in regulatory T cells (Tregs) makes them so unstable that they become effector T cells (Teff) and begin to attack the cancer.
And this conversion from Treg to Teff occurs only in the inflammatory conditions that prevail within many tumors.
As a result, Tregs embedded in normal tissue throughout the body continue to have a restraining effect on their local Teffs, protecting healthy organs and tissues from attack.
The researchers said this raises the prospect of therapies that concentrate the immune system’s firepower on tumors without producing residual damage and harmful side effects.
“Many current approaches to immunotherapy involve depleting or blocking Tregs in order to shift the balance toward Teff cells,” said Harvey Cantor, MD, of the Dana-Farber Cancer Institute in Boston, Massachusetts.
“This, however, runs the risk of triggering an autoimmune response in which the Teff cells attack normal as well as malignant tissue. The key to our approach is that it singles out the Tregs inside a tumor for conversion, leaving Tregs elsewhere in the body unchanged.”
Dr Cantor and his colleagues described the approach in PNAS.
The study builds on research published last year in Science. That study showed that Tregs maintain their immune-suppressive properties under inflammatory conditions as long as they have high enough levels of a protein called Helios. Depriving Tregs of sufficient Helios caused them to lose that stability and turn into Teff cells.
The new study explored whether this convertibility could be harnessed for therapeutic purposes in cancers.
The first set of experiments involved mice engineered to lack Helios in their Tregs. When the animals were injected with melanoma or colon cancer cells, they developed tumors far more slowly than animals with normal Tregs.
“Inspection of the animals’ tumor tissue showed an unstable set of T regulatory cells, many of which had converted into Teffs,” said Hye-Jung Kim, PhD, also of the Dana-Farber Cancer Institute.
The researchers then explored whether stanching Helios production in tumor-dwelling Tregs could have the same effect. They tested several antibodies that bind to key receptors on Tregs and cause a downturn in Helios production.
The team chose an antibody that worked well, DTA-1, and tested it in mice with Treg-laden tumors. When they analyzed the tumor tissue, it was clear that DTA-1 had triggered conversion of Tregs to Teffs.
“This represents a next stage in cancer immunotherapy,” Dr Cantor said. “We now have a very specific, targeted way of inducing a T-effector-cell attack on cancer while lowering the risk of adverse effects on healthy tissue. The next step will be to organize a clinical trial using this approach in patients.” ![]()
Physical activity may lower risk of some cancers

Photo by K. Johansson
Being physically active during leisure time may lower a person’s risk of certain cancers, according to a new study.
A high level of physical activity was associated with a 20% lower risk of myeloid leukemia, a 17% lower risk of myeloma, a 9% lower risk of non-Hodgkin lymphoma, and a 7% lower risk of cancer in general.
On the other hand, a high level of physical activity was also associated with a higher risk of malignant melanoma and prostate cancer.
Steven C. Moore, PhD, of the National Cancer Institute in Bethesda, Maryland, and his colleagues reported these findings in JAMA Internal Medicine.
The researchers pooled data from 12 US and European study cohorts with self-reported physical activity (1987-2004). And they analyzed associations between physical activity and 26 types of cancer.
The study included 1.4 million participants, and 186,932 cancers were identified during a median of 11 years of follow-up.
Compared with the lowest level of leisure-time physical activity (10th percentile), the highest level of activity (90th percentile) had strong inverse associations (a 20% or greater reduction in risk) for 7 cancer types:
- Myeloid leukemia (hazard ratio [HR]=0.80 [95% CI, 0.70-0.92])
- Esophageal adenocarcinoma (HR=0.58 [95% CI, 0.37-0.89])
- Liver cancer (HR=0.73 [95% CI, 0.55-0.98])
- Lung cancer (HR=0.74 [95% CI, 0.71-0.77])
- Kidney cancer (HR=0.77 [95% CI, 0.70-0.85])
- Gastric cardia (HR=0.78 [95% CI, 0.64-0.95])
- Endometrial cancer (HR=0.79 [95% CI, 0.68-0.92]).
There were moderate inverse associations (a 10% to 20% reduction in risk) between the highest level of activity and 6 cancers:
- Myeloma (HR=0.83 [95% CI, 0.72-0.95])
- Colon cancer (HR=0.84 [95% CI, 0.77-0.91])
- Head and neck cancer (HR=0.85 [95% CI, 0.78-0.93])
- Rectal cancer (HR=0.87 [95% CI, 0.80-0.95])
- Bladder cancer (HR=0.87 [95% CI, 0.82-0.92])
- Breast cancer (HR=0.90 [95% CI, 0.87-0.93]).
And there were suggestive inverse associations between the highest level of activity and 3 cancers:
- Non-Hodgkin lymphoma (HR=0.91 [95% CI, 0.83-1.00])
- Gallbladder cancer (HR=0.72 [95% CI, 0.51-1.01])
- Small intestine cancer (HR=0.78 [95% CI, 0.60-1.00]).
However, the highest level of activity was also associated with an increased risk of prostate cancer (HR=1.05 [95% CI, 1.03-1.08]) and malignant melanoma (HR=1.27 [95% CI, 1.16-1.40]).
The researchers said the main limitation of this study is that they cannot fully exclude the possibility that diet, smoking, and other factors may have affected these results. Also, the study used self-reported physical activity, which can mean errors in recall.
Still, the team said these findings support promoting physical activity as a key component of population-wide cancer prevention and control efforts. ![]()
Photo by K. Johansson
Being physically active during leisure time may lower a person’s risk of certain cancers, according to a new study.
A high level of physical activity was associated with a 20% lower risk of myeloid leukemia, a 17% lower risk of myeloma, a 9% lower risk of non-Hodgkin lymphoma, and a 7% lower risk of cancer in general.
On the other hand, a high level of physical activity was also associated with a higher risk of malignant melanoma and prostate cancer.
Steven C. Moore, PhD, of the National Cancer Institute in Bethesda, Maryland, and his colleagues reported these findings in JAMA Internal Medicine.
The researchers pooled data from 12 US and European study cohorts with self-reported physical activity (1987-2004). And they analyzed associations between physical activity and 26 types of cancer.
The study included 1.4 million participants, and 186,932 cancers were identified during a median of 11 years of follow-up.
Compared with the lowest level of leisure-time physical activity (10th percentile), the highest level of activity (90th percentile) had strong inverse associations (a 20% or greater reduction in risk) for 7 cancer types:
- Myeloid leukemia (hazard ratio [HR]=0.80 [95% CI, 0.70-0.92])
- Esophageal adenocarcinoma (HR=0.58 [95% CI, 0.37-0.89])
- Liver cancer (HR=0.73 [95% CI, 0.55-0.98])
- Lung cancer (HR=0.74 [95% CI, 0.71-0.77])
- Kidney cancer (HR=0.77 [95% CI, 0.70-0.85])
- Gastric cardia (HR=0.78 [95% CI, 0.64-0.95])
- Endometrial cancer (HR=0.79 [95% CI, 0.68-0.92]).
There were moderate inverse associations (a 10% to 20% reduction in risk) between the highest level of activity and 6 cancers:
- Myeloma (HR=0.83 [95% CI, 0.72-0.95])
- Colon cancer (HR=0.84 [95% CI, 0.77-0.91])
- Head and neck cancer (HR=0.85 [95% CI, 0.78-0.93])
- Rectal cancer (HR=0.87 [95% CI, 0.80-0.95])
- Bladder cancer (HR=0.87 [95% CI, 0.82-0.92])
- Breast cancer (HR=0.90 [95% CI, 0.87-0.93]).
And there were suggestive inverse associations between the highest level of activity and 3 cancers:
- Non-Hodgkin lymphoma (HR=0.91 [95% CI, 0.83-1.00])
- Gallbladder cancer (HR=0.72 [95% CI, 0.51-1.01])
- Small intestine cancer (HR=0.78 [95% CI, 0.60-1.00]).
However, the highest level of activity was also associated with an increased risk of prostate cancer (HR=1.05 [95% CI, 1.03-1.08]) and malignant melanoma (HR=1.27 [95% CI, 1.16-1.40]).
The researchers said the main limitation of this study is that they cannot fully exclude the possibility that diet, smoking, and other factors may have affected these results. Also, the study used self-reported physical activity, which can mean errors in recall.
Still, the team said these findings support promoting physical activity as a key component of population-wide cancer prevention and control efforts. ![]()
Photo by K. Johansson
Being physically active during leisure time may lower a person’s risk of certain cancers, according to a new study.
A high level of physical activity was associated with a 20% lower risk of myeloid leukemia, a 17% lower risk of myeloma, a 9% lower risk of non-Hodgkin lymphoma, and a 7% lower risk of cancer in general.
On the other hand, a high level of physical activity was also associated with a higher risk of malignant melanoma and prostate cancer.
Steven C. Moore, PhD, of the National Cancer Institute in Bethesda, Maryland, and his colleagues reported these findings in JAMA Internal Medicine.
The researchers pooled data from 12 US and European study cohorts with self-reported physical activity (1987-2004). And they analyzed associations between physical activity and 26 types of cancer.
The study included 1.4 million participants, and 186,932 cancers were identified during a median of 11 years of follow-up.
Compared with the lowest level of leisure-time physical activity (10th percentile), the highest level of activity (90th percentile) had strong inverse associations (a 20% or greater reduction in risk) for 7 cancer types:
- Myeloid leukemia (hazard ratio [HR]=0.80 [95% CI, 0.70-0.92])
- Esophageal adenocarcinoma (HR=0.58 [95% CI, 0.37-0.89])
- Liver cancer (HR=0.73 [95% CI, 0.55-0.98])
- Lung cancer (HR=0.74 [95% CI, 0.71-0.77])
- Kidney cancer (HR=0.77 [95% CI, 0.70-0.85])
- Gastric cardia (HR=0.78 [95% CI, 0.64-0.95])
- Endometrial cancer (HR=0.79 [95% CI, 0.68-0.92]).
There were moderate inverse associations (a 10% to 20% reduction in risk) between the highest level of activity and 6 cancers:
- Myeloma (HR=0.83 [95% CI, 0.72-0.95])
- Colon cancer (HR=0.84 [95% CI, 0.77-0.91])
- Head and neck cancer (HR=0.85 [95% CI, 0.78-0.93])
- Rectal cancer (HR=0.87 [95% CI, 0.80-0.95])
- Bladder cancer (HR=0.87 [95% CI, 0.82-0.92])
- Breast cancer (HR=0.90 [95% CI, 0.87-0.93]).
And there were suggestive inverse associations between the highest level of activity and 3 cancers:
- Non-Hodgkin lymphoma (HR=0.91 [95% CI, 0.83-1.00])
- Gallbladder cancer (HR=0.72 [95% CI, 0.51-1.01])
- Small intestine cancer (HR=0.78 [95% CI, 0.60-1.00]).
However, the highest level of activity was also associated with an increased risk of prostate cancer (HR=1.05 [95% CI, 1.03-1.08]) and malignant melanoma (HR=1.27 [95% CI, 1.16-1.40]).
The researchers said the main limitation of this study is that they cannot fully exclude the possibility that diet, smoking, and other factors may have affected these results. Also, the study used self-reported physical activity, which can mean errors in recall.
Still, the team said these findings support promoting physical activity as a key component of population-wide cancer prevention and control efforts. ![]()
Reversal agent granted conditional approval in Canada

to prevent thrombosis after
knee replacement surgery
© Boehringer Ingelheim
Health Canada has granted conditional approval for idarucizumab (Praxbind), a humanized antibody fragment designed to reverse the anticoagulant effects of dabigatran etexilate (Pradaxa) in cases of emergency surgery/urgent procedures or in situations of life-threatening or uncontrolled bleeding.
The conditional approval of idarucizumab reflects the promising nature of the available clinical evidence.
For the drug to gain full approval, Boehringer Ingelheim—the company that markets both idarucizumab and dabigatran—must provide Health Canada with data confirming that idarucizumab provides a clinical benefit.
To date, study results have demonstrated that 5g of idarucizumab provides immediate, complete, and sustained reversal of the anticoagulant effects of dabigatran in most patients.
In the ongoing phase 3 RE-VERSE AD trial, researchers are evaluating idarucizumab in emergency settings.
Interim results from this trial showed that idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of dabigatran-treated patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
Researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events. ![]()
to prevent thrombosis after
knee replacement surgery
© Boehringer Ingelheim
Health Canada has granted conditional approval for idarucizumab (Praxbind), a humanized antibody fragment designed to reverse the anticoagulant effects of dabigatran etexilate (Pradaxa) in cases of emergency surgery/urgent procedures or in situations of life-threatening or uncontrolled bleeding.
The conditional approval of idarucizumab reflects the promising nature of the available clinical evidence.
For the drug to gain full approval, Boehringer Ingelheim—the company that markets both idarucizumab and dabigatran—must provide Health Canada with data confirming that idarucizumab provides a clinical benefit.
To date, study results have demonstrated that 5g of idarucizumab provides immediate, complete, and sustained reversal of the anticoagulant effects of dabigatran in most patients.
In the ongoing phase 3 RE-VERSE AD trial, researchers are evaluating idarucizumab in emergency settings.
Interim results from this trial showed that idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of dabigatran-treated patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
Researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events. ![]()
to prevent thrombosis after
knee replacement surgery
© Boehringer Ingelheim
Health Canada has granted conditional approval for idarucizumab (Praxbind), a humanized antibody fragment designed to reverse the anticoagulant effects of dabigatran etexilate (Pradaxa) in cases of emergency surgery/urgent procedures or in situations of life-threatening or uncontrolled bleeding.
The conditional approval of idarucizumab reflects the promising nature of the available clinical evidence.
For the drug to gain full approval, Boehringer Ingelheim—the company that markets both idarucizumab and dabigatran—must provide Health Canada with data confirming that idarucizumab provides a clinical benefit.
To date, study results have demonstrated that 5g of idarucizumab provides immediate, complete, and sustained reversal of the anticoagulant effects of dabigatran in most patients.
In the ongoing phase 3 RE-VERSE AD trial, researchers are evaluating idarucizumab in emergency settings.
Interim results from this trial showed that idarucizumab normalized diluted thrombin time and ecarin clotting time in a majority of dabigatran-treated patients with uncontrolled or life-threatening bleeding complications and most patients who required emergency surgery or an invasive procedure.
Researchers said there were no safety concerns related to idarucizumab. However, 23% of patients in this trial experienced serious adverse events, 20% of patients died, and several patients had thrombotic or bleeding events. ![]()
Improving NK cell therapy

Image by Joshua Stokes
New findings published in PNAS may help scientists improve the efficacy of natural killer (NK) cell therapy for patients with leukemia.
The preclinical research revealed a tolerance mechanism that restrains the activity of NK cells, as well as a potential way to overcome this problem.
Investigators found that a transcription factor, Kruppel-like factor 2 (KFL2), is critical for NK cell expansion and survival.
Specifically, KLF2 limits immature NK cell proliferation and instructs mature NK cells to home to niches rich in interleukin 15 (IL-15), which is necessary for their continued survival.
“This is the same process likely used by cancer cells to avoid destruction by NK cells,” said study author Eric Sebzda, PhD, of Vanderbilt University Medical Center in Nashville, Tennessee.
In particular, tumors may avoid immune clearance by promoting KLF2 destruction within the NK cell population, thereby starving these cells of IL-15.
Dr Sebzda and his colleagues noted that increased expression of IL-15 can improve immune responses against tumors. Unfortunately, it’s not easy to introduce the cytokine only within a tumor microenvironment, and high systemic levels of IL-15 can be toxic.
Recruiting cells that transpresent IL-15 to the tumor microenvironment may overcome this barrier and therefore improve NK cell-mediated cancer therapy, the investigators said. However, the methodology hasn’t been worked out yet.
“Our paper should encourage this line of inquiry,” Dr Sebzda concluded. ![]()
Image by Joshua Stokes
New findings published in PNAS may help scientists improve the efficacy of natural killer (NK) cell therapy for patients with leukemia.
The preclinical research revealed a tolerance mechanism that restrains the activity of NK cells, as well as a potential way to overcome this problem.
Investigators found that a transcription factor, Kruppel-like factor 2 (KFL2), is critical for NK cell expansion and survival.
Specifically, KLF2 limits immature NK cell proliferation and instructs mature NK cells to home to niches rich in interleukin 15 (IL-15), which is necessary for their continued survival.
“This is the same process likely used by cancer cells to avoid destruction by NK cells,” said study author Eric Sebzda, PhD, of Vanderbilt University Medical Center in Nashville, Tennessee.
In particular, tumors may avoid immune clearance by promoting KLF2 destruction within the NK cell population, thereby starving these cells of IL-15.
Dr Sebzda and his colleagues noted that increased expression of IL-15 can improve immune responses against tumors. Unfortunately, it’s not easy to introduce the cytokine only within a tumor microenvironment, and high systemic levels of IL-15 can be toxic.
Recruiting cells that transpresent IL-15 to the tumor microenvironment may overcome this barrier and therefore improve NK cell-mediated cancer therapy, the investigators said. However, the methodology hasn’t been worked out yet.
“Our paper should encourage this line of inquiry,” Dr Sebzda concluded. ![]()
Image by Joshua Stokes
New findings published in PNAS may help scientists improve the efficacy of natural killer (NK) cell therapy for patients with leukemia.
The preclinical research revealed a tolerance mechanism that restrains the activity of NK cells, as well as a potential way to overcome this problem.
Investigators found that a transcription factor, Kruppel-like factor 2 (KFL2), is critical for NK cell expansion and survival.
Specifically, KLF2 limits immature NK cell proliferation and instructs mature NK cells to home to niches rich in interleukin 15 (IL-15), which is necessary for their continued survival.
“This is the same process likely used by cancer cells to avoid destruction by NK cells,” said study author Eric Sebzda, PhD, of Vanderbilt University Medical Center in Nashville, Tennessee.
In particular, tumors may avoid immune clearance by promoting KLF2 destruction within the NK cell population, thereby starving these cells of IL-15.
Dr Sebzda and his colleagues noted that increased expression of IL-15 can improve immune responses against tumors. Unfortunately, it’s not easy to introduce the cytokine only within a tumor microenvironment, and high systemic levels of IL-15 can be toxic.
Recruiting cells that transpresent IL-15 to the tumor microenvironment may overcome this barrier and therefore improve NK cell-mediated cancer therapy, the investigators said. However, the methodology hasn’t been worked out yet.
“Our paper should encourage this line of inquiry,” Dr Sebzda concluded. ![]()
CDC announces availability of funds to fight Zika

Photo courtesy of
Muhammad Mahdi Karim
The US Centers for Disease Control and Prevention (CDC) has announced that US states and territories can now apply for funds to fight Zika locally.
The agency said more than $85 million in redirected funds identified by the Department of Health and Human Services is being made available to support efforts to protect Americans from Zika infection and associated adverse health outcomes.
“These funds will allow states and territories to continue implementation of their Zika preparedness plans but are not enough to support a comprehensive Zika response and can only temporarily address what is needed,” said Stephen C. Redd, MD, director of the CDC’s Office of Public Health Preparedness and Response.
“Without the full amount of requested emergency supplemental funding, many activities that need to start now are being delayed or may have to be stopped within months.”
The more than $85 million in funds includes the more $60 million reported earlier this year (Funding opportunity number CDC-RFA-CK14-1401CONTPPHF16) as well as another $25 million that was just announced (Funding opportunity number CDC-RFA-TP16-1602).
Earlier this year, states and cities participating in the Epidemiology and Lab Capacity program became eligible for more than $60 million in funds to:
- Build laboratory capacity
- Enhance epidemiological surveillance and investigation
- Improve mosquito control and monitoring
- Keep blood supplies safe
- Contribute data to the US Zika Pregnancy registry.
Applications for these funds are due May 27, 2016, and will be disbursed during the summer.
Under the latest announcement, $25 million in FY 2016 preparedness and response funding will go to 53 states, cities, and territories at risk for outbreaks of Zika virus infection.
Recipients will receive funds based on the geographic locations of the two mosquitoes known to transmit Zika virus (Aedes aegypti and Aedes albopictus), history of mosquito-borne disease outbreaks, and size of population.
The funds are intended to strengthen incident management and emergency operations coordination, information management and sharing, and community recovery and resilience.
State, local, and territorial health officials can use the funds to identify and investigate a possible outbreak of Zika virus disease in their communities, coordinate a comprehensive response across all levels of government and non-governmental partners (including the healthcare sector), and identify and connect to community services families affected by Zika virus disease.
Applications for the funds are due June 13, 2016. Funds will be disbursed during the summer and remain available through July 2017.
Zika virus disease is caused by the Zika virus, which is spread to people primarily through the bite of infected Aedes aegypti and Aedes albopictus mosquitoes, though Aedes aegypti are more likely to spread Zika. Sexual transmission and transmission via blood transfusion have been documented as well.
There is currently no vaccine or treatment for Zika. The most common symptoms of Zika are fever, rash, joint pain, and conjunctivitis. In previous outbreaks, the illness has typically been mild, with symptoms lasting for several days to a week after a person is bitten by an infected mosquito.
Zika virus infection in pregnant women is a cause of microcephaly and other severe fetal brain defects. Zika has also been linked to Guillain-Barré syndrome. ![]()
Photo courtesy of
Muhammad Mahdi Karim
The US Centers for Disease Control and Prevention (CDC) has announced that US states and territories can now apply for funds to fight Zika locally.
The agency said more than $85 million in redirected funds identified by the Department of Health and Human Services is being made available to support efforts to protect Americans from Zika infection and associated adverse health outcomes.
“These funds will allow states and territories to continue implementation of their Zika preparedness plans but are not enough to support a comprehensive Zika response and can only temporarily address what is needed,” said Stephen C. Redd, MD, director of the CDC’s Office of Public Health Preparedness and Response.
“Without the full amount of requested emergency supplemental funding, many activities that need to start now are being delayed or may have to be stopped within months.”
The more than $85 million in funds includes the more $60 million reported earlier this year (Funding opportunity number CDC-RFA-CK14-1401CONTPPHF16) as well as another $25 million that was just announced (Funding opportunity number CDC-RFA-TP16-1602).
Earlier this year, states and cities participating in the Epidemiology and Lab Capacity program became eligible for more than $60 million in funds to:
- Build laboratory capacity
- Enhance epidemiological surveillance and investigation
- Improve mosquito control and monitoring
- Keep blood supplies safe
- Contribute data to the US Zika Pregnancy registry.
Applications for these funds are due May 27, 2016, and will be disbursed during the summer.
Under the latest announcement, $25 million in FY 2016 preparedness and response funding will go to 53 states, cities, and territories at risk for outbreaks of Zika virus infection.
Recipients will receive funds based on the geographic locations of the two mosquitoes known to transmit Zika virus (Aedes aegypti and Aedes albopictus), history of mosquito-borne disease outbreaks, and size of population.
The funds are intended to strengthen incident management and emergency operations coordination, information management and sharing, and community recovery and resilience.
State, local, and territorial health officials can use the funds to identify and investigate a possible outbreak of Zika virus disease in their communities, coordinate a comprehensive response across all levels of government and non-governmental partners (including the healthcare sector), and identify and connect to community services families affected by Zika virus disease.
Applications for the funds are due June 13, 2016. Funds will be disbursed during the summer and remain available through July 2017.
Zika virus disease is caused by the Zika virus, which is spread to people primarily through the bite of infected Aedes aegypti and Aedes albopictus mosquitoes, though Aedes aegypti are more likely to spread Zika. Sexual transmission and transmission via blood transfusion have been documented as well.
There is currently no vaccine or treatment for Zika. The most common symptoms of Zika are fever, rash, joint pain, and conjunctivitis. In previous outbreaks, the illness has typically been mild, with symptoms lasting for several days to a week after a person is bitten by an infected mosquito.
Zika virus infection in pregnant women is a cause of microcephaly and other severe fetal brain defects. Zika has also been linked to Guillain-Barré syndrome. ![]()
Photo courtesy of
Muhammad Mahdi Karim
The US Centers for Disease Control and Prevention (CDC) has announced that US states and territories can now apply for funds to fight Zika locally.
The agency said more than $85 million in redirected funds identified by the Department of Health and Human Services is being made available to support efforts to protect Americans from Zika infection and associated adverse health outcomes.
“These funds will allow states and territories to continue implementation of their Zika preparedness plans but are not enough to support a comprehensive Zika response and can only temporarily address what is needed,” said Stephen C. Redd, MD, director of the CDC’s Office of Public Health Preparedness and Response.
“Without the full amount of requested emergency supplemental funding, many activities that need to start now are being delayed or may have to be stopped within months.”
The more than $85 million in funds includes the more $60 million reported earlier this year (Funding opportunity number CDC-RFA-CK14-1401CONTPPHF16) as well as another $25 million that was just announced (Funding opportunity number CDC-RFA-TP16-1602).
Earlier this year, states and cities participating in the Epidemiology and Lab Capacity program became eligible for more than $60 million in funds to:
- Build laboratory capacity
- Enhance epidemiological surveillance and investigation
- Improve mosquito control and monitoring
- Keep blood supplies safe
- Contribute data to the US Zika Pregnancy registry.
Applications for these funds are due May 27, 2016, and will be disbursed during the summer.
Under the latest announcement, $25 million in FY 2016 preparedness and response funding will go to 53 states, cities, and territories at risk for outbreaks of Zika virus infection.
Recipients will receive funds based on the geographic locations of the two mosquitoes known to transmit Zika virus (Aedes aegypti and Aedes albopictus), history of mosquito-borne disease outbreaks, and size of population.
The funds are intended to strengthen incident management and emergency operations coordination, information management and sharing, and community recovery and resilience.
State, local, and territorial health officials can use the funds to identify and investigate a possible outbreak of Zika virus disease in their communities, coordinate a comprehensive response across all levels of government and non-governmental partners (including the healthcare sector), and identify and connect to community services families affected by Zika virus disease.
Applications for the funds are due June 13, 2016. Funds will be disbursed during the summer and remain available through July 2017.
Zika virus disease is caused by the Zika virus, which is spread to people primarily through the bite of infected Aedes aegypti and Aedes albopictus mosquitoes, though Aedes aegypti are more likely to spread Zika. Sexual transmission and transmission via blood transfusion have been documented as well.
There is currently no vaccine or treatment for Zika. The most common symptoms of Zika are fever, rash, joint pain, and conjunctivitis. In previous outbreaks, the illness has typically been mild, with symptoms lasting for several days to a week after a person is bitten by an infected mosquito.
Zika virus infection in pregnant women is a cause of microcephaly and other severe fetal brain defects. Zika has also been linked to Guillain-Barré syndrome. ![]()
PRAC: Products may not increase risk of inhibitors

Results of a meta-analysis suggest a pair of recombinant factor VIII (FVIII) products may not increase the risk of FVIII inhibitors in previously untreated patients with severe hemophilia A.
The European Medicines Agency’s Pharmacovigilance Risk Assessment Committee (PRAC) analyzed data from 3 observational studies and found that patients who received Kogenate or Helixate had a higher incidence of inhibitors than patients who received other recombinant FVIII products.
However, the difference was not significant, and the PRAC concluded that, overall, the evidence does not confirm an increased risk of inhibitors with Kogenate or Helixate.
This conclusion is consistent with the PRAC’s previous conclusions from a review carried out in 2013.
For the current analysis, the PRAC reviewed data from 3 studies, which were conducted by the RODIN study group, the UK Haemophilia Centre Doctors’ Organisation (UKHCDO), and the FranceCoag Network.
The analysis included 1102 previously untreated patients—481 in the RODIN study, 293 in the FranceCoag study, and 328 in the UKHCDO study—who received octocog alfa (Advate, Helixate, or Kogenate), moroctocog alfa (Refacto or Refacto AF), or other recombinant FVIII products.
Results suggested a trend toward an increase of high-titer inhibitor development and all inhibitor development with Kogenate or Helixate compared to Advate.
Overall, 37% of patients treated with Kogenate or Helixate (147/400) developed inhibitory antibodies, 22% of whom (n=88) had high-titer inhibitors. Twenty-six percent of patients who received Advate (100/385) developed inhibitors, 15% of whom (n=57) had high-titer inhibitors.
The PRAC said a similar trend was observed for other recombinant FVIII products, but the results were less pronounced due to sample size constraints.
The committee pointed out that there were several limitations with this meta-analysis, including the possibility of residual confounding. The group also said a number of parameters may have had an impact on the incidence of inhibitors in these previously untreated patients, and adjusting for all of these factors may not be possible.
In addition, the PRAC noted that there has been no signal for a similar trend of increases in inhibitor development with Kogenate in previously treated patients in other studies.
Finally, the committee recommended that companies marketing recombinant FVIII products monitor published studies on inhibitor development with the aim of keeping the product information up to date. ![]()
Results of a meta-analysis suggest a pair of recombinant factor VIII (FVIII) products may not increase the risk of FVIII inhibitors in previously untreated patients with severe hemophilia A.
The European Medicines Agency’s Pharmacovigilance Risk Assessment Committee (PRAC) analyzed data from 3 observational studies and found that patients who received Kogenate or Helixate had a higher incidence of inhibitors than patients who received other recombinant FVIII products.
However, the difference was not significant, and the PRAC concluded that, overall, the evidence does not confirm an increased risk of inhibitors with Kogenate or Helixate.
This conclusion is consistent with the PRAC’s previous conclusions from a review carried out in 2013.
For the current analysis, the PRAC reviewed data from 3 studies, which were conducted by the RODIN study group, the UK Haemophilia Centre Doctors’ Organisation (UKHCDO), and the FranceCoag Network.
The analysis included 1102 previously untreated patients—481 in the RODIN study, 293 in the FranceCoag study, and 328 in the UKHCDO study—who received octocog alfa (Advate, Helixate, or Kogenate), moroctocog alfa (Refacto or Refacto AF), or other recombinant FVIII products.
Results suggested a trend toward an increase of high-titer inhibitor development and all inhibitor development with Kogenate or Helixate compared to Advate.
Overall, 37% of patients treated with Kogenate or Helixate (147/400) developed inhibitory antibodies, 22% of whom (n=88) had high-titer inhibitors. Twenty-six percent of patients who received Advate (100/385) developed inhibitors, 15% of whom (n=57) had high-titer inhibitors.
The PRAC said a similar trend was observed for other recombinant FVIII products, but the results were less pronounced due to sample size constraints.
The committee pointed out that there were several limitations with this meta-analysis, including the possibility of residual confounding. The group also said a number of parameters may have had an impact on the incidence of inhibitors in these previously untreated patients, and adjusting for all of these factors may not be possible.
In addition, the PRAC noted that there has been no signal for a similar trend of increases in inhibitor development with Kogenate in previously treated patients in other studies.
Finally, the committee recommended that companies marketing recombinant FVIII products monitor published studies on inhibitor development with the aim of keeping the product information up to date. ![]()
Results of a meta-analysis suggest a pair of recombinant factor VIII (FVIII) products may not increase the risk of FVIII inhibitors in previously untreated patients with severe hemophilia A.
The European Medicines Agency’s Pharmacovigilance Risk Assessment Committee (PRAC) analyzed data from 3 observational studies and found that patients who received Kogenate or Helixate had a higher incidence of inhibitors than patients who received other recombinant FVIII products.
However, the difference was not significant, and the PRAC concluded that, overall, the evidence does not confirm an increased risk of inhibitors with Kogenate or Helixate.
This conclusion is consistent with the PRAC’s previous conclusions from a review carried out in 2013.
For the current analysis, the PRAC reviewed data from 3 studies, which were conducted by the RODIN study group, the UK Haemophilia Centre Doctors’ Organisation (UKHCDO), and the FranceCoag Network.
The analysis included 1102 previously untreated patients—481 in the RODIN study, 293 in the FranceCoag study, and 328 in the UKHCDO study—who received octocog alfa (Advate, Helixate, or Kogenate), moroctocog alfa (Refacto or Refacto AF), or other recombinant FVIII products.
Results suggested a trend toward an increase of high-titer inhibitor development and all inhibitor development with Kogenate or Helixate compared to Advate.
Overall, 37% of patients treated with Kogenate or Helixate (147/400) developed inhibitory antibodies, 22% of whom (n=88) had high-titer inhibitors. Twenty-six percent of patients who received Advate (100/385) developed inhibitors, 15% of whom (n=57) had high-titer inhibitors.
The PRAC said a similar trend was observed for other recombinant FVIII products, but the results were less pronounced due to sample size constraints.
The committee pointed out that there were several limitations with this meta-analysis, including the possibility of residual confounding. The group also said a number of parameters may have had an impact on the incidence of inhibitors in these previously untreated patients, and adjusting for all of these factors may not be possible.
In addition, the PRAC noted that there has been no signal for a similar trend of increases in inhibitor development with Kogenate in previously treated patients in other studies.
Finally, the committee recommended that companies marketing recombinant FVIII products monitor published studies on inhibitor development with the aim of keeping the product information up to date.
FDA warns of counterfeit BiCNU
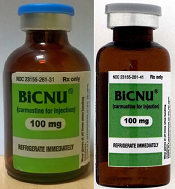
shown on the left and a
counterfeit vial on the right
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) is warning healthcare professionals that a counterfeit version of BiCNU (carmustine for injection) 100 mg has been detected in some foreign countries.
The agency said there is no indication that counterfeit BiCNU has entered the legitimate US drug supply chain and no indication that any US patients have received counterfeit BiCNU.
Still, the FDA is advising that healthcare professionals inspect BiCNU vials as an added precaution to ensure the product administered to patients is authentic.
BiCNU is approved to treat brain cancers, multiple myeloma, and lymphoma. It is manufactured by Emcure Pharmaceuticals Ltd. and distributed in the US by Heritage Pharmaceuticals Inc.
Heritage previously announced that counterfeit BiCNU had been found in India, Ireland, and Israel.
How to identify counterfeit BiCNU
BiCNU is available as a vial of BiCNU and dehydrated alcohol co-packaged together.
While the NDC on the outer package of the authentic and counterfeit versions might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging. The authentic product has a blue flip top, while the counterfeit product may have a gray flip top.
The product may also be counterfeit if the vial displays the following lot numbers, batch numbers, manufacturing dates, and expiration dates.
| Product | Expiration
date |
Manufacturing
date |
Lot number | Batch number |
| BiCNU | 01/18 | 2/16 | BCEM771322 | EM/BC20161990 |
| Diluent | 01/18 | 2/16 | SBCDA224736 | EM/BCD2220 |
| BiCNU | 12/17 | 1/16 | BCEM771318 | EM/BC20151896 |
| Diluent | 12/17 | 1/16 | SBCDA224732 | EM/BCD2216 |
| BiCNU | 10/17 | 11/15 | BCEM771317 | EM/BC20151895 |
| Diluent | 10/17 | 11/15 | SBCDA224731 | EM/BCD2215 |
The FDA urges healthcare professionals to purchase drug products only from legitimate suppliers.
Healthcare professionals are encouraged to report sales solicitation of suspect drug products by calling the FDA’s Office of Criminal Investigations (OCI) at 800-551-3989, reporting via OCI’s website, or emailing DrugSupplyChainIntegrity@fda.hhs.gov.
Healthcare professionals and patients should report adverse events related to the use of any suspect medications to the FDA’s MedWatch Adverse Event Reporting Program.
shown on the left and a
counterfeit vial on the right
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) is warning healthcare professionals that a counterfeit version of BiCNU (carmustine for injection) 100 mg has been detected in some foreign countries.
The agency said there is no indication that counterfeit BiCNU has entered the legitimate US drug supply chain and no indication that any US patients have received counterfeit BiCNU.
Still, the FDA is advising that healthcare professionals inspect BiCNU vials as an added precaution to ensure the product administered to patients is authentic.
BiCNU is approved to treat brain cancers, multiple myeloma, and lymphoma. It is manufactured by Emcure Pharmaceuticals Ltd. and distributed in the US by Heritage Pharmaceuticals Inc.
Heritage previously announced that counterfeit BiCNU had been found in India, Ireland, and Israel.
How to identify counterfeit BiCNU
BiCNU is available as a vial of BiCNU and dehydrated alcohol co-packaged together.
While the NDC on the outer package of the authentic and counterfeit versions might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging. The authentic product has a blue flip top, while the counterfeit product may have a gray flip top.
The product may also be counterfeit if the vial displays the following lot numbers, batch numbers, manufacturing dates, and expiration dates.
| Product | Expiration
date |
Manufacturing
date |
Lot number | Batch number |
| BiCNU | 01/18 | 2/16 | BCEM771322 | EM/BC20161990 |
| Diluent | 01/18 | 2/16 | SBCDA224736 | EM/BCD2220 |
| BiCNU | 12/17 | 1/16 | BCEM771318 | EM/BC20151896 |
| Diluent | 12/17 | 1/16 | SBCDA224732 | EM/BCD2216 |
| BiCNU | 10/17 | 11/15 | BCEM771317 | EM/BC20151895 |
| Diluent | 10/17 | 11/15 | SBCDA224731 | EM/BCD2215 |
The FDA urges healthcare professionals to purchase drug products only from legitimate suppliers.
Healthcare professionals are encouraged to report sales solicitation of suspect drug products by calling the FDA’s Office of Criminal Investigations (OCI) at 800-551-3989, reporting via OCI’s website, or emailing DrugSupplyChainIntegrity@fda.hhs.gov.
Healthcare professionals and patients should report adverse events related to the use of any suspect medications to the FDA’s MedWatch Adverse Event Reporting Program.
shown on the left and a
counterfeit vial on the right
Photo courtesy of the FDA
The US Food and Drug Administration (FDA) is warning healthcare professionals that a counterfeit version of BiCNU (carmustine for injection) 100 mg has been detected in some foreign countries.
The agency said there is no indication that counterfeit BiCNU has entered the legitimate US drug supply chain and no indication that any US patients have received counterfeit BiCNU.
Still, the FDA is advising that healthcare professionals inspect BiCNU vials as an added precaution to ensure the product administered to patients is authentic.
BiCNU is approved to treat brain cancers, multiple myeloma, and lymphoma. It is manufactured by Emcure Pharmaceuticals Ltd. and distributed in the US by Heritage Pharmaceuticals Inc.
Heritage previously announced that counterfeit BiCNU had been found in India, Ireland, and Israel.
How to identify counterfeit BiCNU
BiCNU is available as a vial of BiCNU and dehydrated alcohol co-packaged together.
While the NDC on the outer package of the authentic and counterfeit versions might match, the best way to distinguish a counterfeit is to look at the BiCNU vial inside the packaging. The authentic product has a blue flip top, while the counterfeit product may have a gray flip top.
The product may also be counterfeit if the vial displays the following lot numbers, batch numbers, manufacturing dates, and expiration dates.
| Product | Expiration
date |
Manufacturing
date |
Lot number | Batch number |
| BiCNU | 01/18 | 2/16 | BCEM771322 | EM/BC20161990 |
| Diluent | 01/18 | 2/16 | SBCDA224736 | EM/BCD2220 |
| BiCNU | 12/17 | 1/16 | BCEM771318 | EM/BC20151896 |
| Diluent | 12/17 | 1/16 | SBCDA224732 | EM/BCD2216 |
| BiCNU | 10/17 | 11/15 | BCEM771317 | EM/BC20151895 |
| Diluent | 10/17 | 11/15 | SBCDA224731 | EM/BCD2215 |
The FDA urges healthcare professionals to purchase drug products only from legitimate suppliers.
Healthcare professionals are encouraged to report sales solicitation of suspect drug products by calling the FDA’s Office of Criminal Investigations (OCI) at 800-551-3989, reporting via OCI’s website, or emailing DrugSupplyChainIntegrity@fda.hhs.gov.
Healthcare professionals and patients should report adverse events related to the use of any suspect medications to the FDA’s MedWatch Adverse Event Reporting Program.
EC approves FC fusion protein for hemophilia B

Photo courtesy of Biogen
The European Commission (EC) has approved eftrenonacog alfa (Alprolix) to treat hemophilia B.
This recombinant factor IX Fc fusion protein therapy is indicated for both on-demand and prophylactic treatment in patients of all ages.
Eftrenonacog alfa is developed by fusing factor IX to the Fc portion of immunoglobulin G subclass 1. This enables eftrenonacog alfa to use a naturally occurring pathway to prolong the time the therapy remains in the body.
Prophylactically, eftrenonacog alfa can be administered with an initial dose every 7 days or every 10 days, and the dosing interval can be adjusted based on individual response.
Sobi and Biogen are collaborators in the development and commercialization of eftrenonacog alfa for hemophilia B.
Sobi said it is working to make the drug available in Europe as quickly as possible.
The EC’s decision to approve eftrenonacog alfa was based on results from two phase 3 trials: the B-LONG study and the Kids B-LONG study.
B-LONG study
The B-LONG study included 123 male subjects with severe hemophilia B who were 12 years of age or older. They had no current or previous factor IX inhibitors and a history of 100 or more documented prior exposure days to factor IX products.
Patients received eftrenonacog alfa in 1 of 4 treatment arms:
- Weekly prophylaxis starting at 50 IU/kg, with pharmacokinetic (PK)-driven dose adjustments (n=63)
- Individualized interval prophylaxis starting at 100 IU/kg every 10 days, with PK-driven interval adjustments (n=29)
- On-demand treatment at 20 IU/kg to 100 IU/kg (n=27)
- Perioperative management (n=12, including 8 from arms 1-3).
Researchers assessed control of bleeding in all patients who experienced a bleeding episode while on study. In total, 90.4% of bleeding episodes were controlled by a single injection of eftrenonacog alfa.
The overall median annualized bleeding rates (ABRs)—including spontaneous and traumatic bleeds—were 2.95 in the weekly prophylaxis arm, 1.38 in the individualized interval prophylaxis arm, and 17.69 in the episodic treatment arm.
The perioperative management arm consisted of 12 patients undergoing 14 major surgical procedures. The treating physicians rated the hemostatic efficacy of eftrenonacog alfa as “excellent” or “good” in all surgeries.
Eftrenonacog alfa was considered generally well-tolerated. None of the patients developed inhibitors, and none reported anaphylaxis.
The most common adverse events—with an incidence of 5% or greater—occurring outside of the perioperative management arm were nasopharyngitis, influenza, arthralgia, upper respiratory infection, hypertension, and headache.
One serious adverse event may have been drug-related. The patient experienced obstructive uropathy in the setting of hematuria. However, he continued to receive eftrenonacog alfa, and the event resolved with medical management.
Kids B-LONG
In Kids B-LONG, researchers tested eftrenonacog alfa in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Children who received eftrenonacog alfa prophylactically had an overall median ABR of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of eftrenonacog alfa.
None of the patients developed inhibitors. Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events.
None of the patients discontinued the study due to an adverse event. One adverse event—decreased appetite occurring in 1 patient—was considered related to eftrenonacog alfa treatment.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the B-LONG study.
Photo courtesy of Biogen
The European Commission (EC) has approved eftrenonacog alfa (Alprolix) to treat hemophilia B.
This recombinant factor IX Fc fusion protein therapy is indicated for both on-demand and prophylactic treatment in patients of all ages.
Eftrenonacog alfa is developed by fusing factor IX to the Fc portion of immunoglobulin G subclass 1. This enables eftrenonacog alfa to use a naturally occurring pathway to prolong the time the therapy remains in the body.
Prophylactically, eftrenonacog alfa can be administered with an initial dose every 7 days or every 10 days, and the dosing interval can be adjusted based on individual response.
Sobi and Biogen are collaborators in the development and commercialization of eftrenonacog alfa for hemophilia B.
Sobi said it is working to make the drug available in Europe as quickly as possible.
The EC’s decision to approve eftrenonacog alfa was based on results from two phase 3 trials: the B-LONG study and the Kids B-LONG study.
B-LONG study
The B-LONG study included 123 male subjects with severe hemophilia B who were 12 years of age or older. They had no current or previous factor IX inhibitors and a history of 100 or more documented prior exposure days to factor IX products.
Patients received eftrenonacog alfa in 1 of 4 treatment arms:
- Weekly prophylaxis starting at 50 IU/kg, with pharmacokinetic (PK)-driven dose adjustments (n=63)
- Individualized interval prophylaxis starting at 100 IU/kg every 10 days, with PK-driven interval adjustments (n=29)
- On-demand treatment at 20 IU/kg to 100 IU/kg (n=27)
- Perioperative management (n=12, including 8 from arms 1-3).
Researchers assessed control of bleeding in all patients who experienced a bleeding episode while on study. In total, 90.4% of bleeding episodes were controlled by a single injection of eftrenonacog alfa.
The overall median annualized bleeding rates (ABRs)—including spontaneous and traumatic bleeds—were 2.95 in the weekly prophylaxis arm, 1.38 in the individualized interval prophylaxis arm, and 17.69 in the episodic treatment arm.
The perioperative management arm consisted of 12 patients undergoing 14 major surgical procedures. The treating physicians rated the hemostatic efficacy of eftrenonacog alfa as “excellent” or “good” in all surgeries.
Eftrenonacog alfa was considered generally well-tolerated. None of the patients developed inhibitors, and none reported anaphylaxis.
The most common adverse events—with an incidence of 5% or greater—occurring outside of the perioperative management arm were nasopharyngitis, influenza, arthralgia, upper respiratory infection, hypertension, and headache.
One serious adverse event may have been drug-related. The patient experienced obstructive uropathy in the setting of hematuria. However, he continued to receive eftrenonacog alfa, and the event resolved with medical management.
Kids B-LONG
In Kids B-LONG, researchers tested eftrenonacog alfa in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Children who received eftrenonacog alfa prophylactically had an overall median ABR of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of eftrenonacog alfa.
None of the patients developed inhibitors. Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events.
None of the patients discontinued the study due to an adverse event. One adverse event—decreased appetite occurring in 1 patient—was considered related to eftrenonacog alfa treatment.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the B-LONG study.
Photo courtesy of Biogen
The European Commission (EC) has approved eftrenonacog alfa (Alprolix) to treat hemophilia B.
This recombinant factor IX Fc fusion protein therapy is indicated for both on-demand and prophylactic treatment in patients of all ages.
Eftrenonacog alfa is developed by fusing factor IX to the Fc portion of immunoglobulin G subclass 1. This enables eftrenonacog alfa to use a naturally occurring pathway to prolong the time the therapy remains in the body.
Prophylactically, eftrenonacog alfa can be administered with an initial dose every 7 days or every 10 days, and the dosing interval can be adjusted based on individual response.
Sobi and Biogen are collaborators in the development and commercialization of eftrenonacog alfa for hemophilia B.
Sobi said it is working to make the drug available in Europe as quickly as possible.
The EC’s decision to approve eftrenonacog alfa was based on results from two phase 3 trials: the B-LONG study and the Kids B-LONG study.
B-LONG study
The B-LONG study included 123 male subjects with severe hemophilia B who were 12 years of age or older. They had no current or previous factor IX inhibitors and a history of 100 or more documented prior exposure days to factor IX products.
Patients received eftrenonacog alfa in 1 of 4 treatment arms:
- Weekly prophylaxis starting at 50 IU/kg, with pharmacokinetic (PK)-driven dose adjustments (n=63)
- Individualized interval prophylaxis starting at 100 IU/kg every 10 days, with PK-driven interval adjustments (n=29)
- On-demand treatment at 20 IU/kg to 100 IU/kg (n=27)
- Perioperative management (n=12, including 8 from arms 1-3).
Researchers assessed control of bleeding in all patients who experienced a bleeding episode while on study. In total, 90.4% of bleeding episodes were controlled by a single injection of eftrenonacog alfa.
The overall median annualized bleeding rates (ABRs)—including spontaneous and traumatic bleeds—were 2.95 in the weekly prophylaxis arm, 1.38 in the individualized interval prophylaxis arm, and 17.69 in the episodic treatment arm.
The perioperative management arm consisted of 12 patients undergoing 14 major surgical procedures. The treating physicians rated the hemostatic efficacy of eftrenonacog alfa as “excellent” or “good” in all surgeries.
Eftrenonacog alfa was considered generally well-tolerated. None of the patients developed inhibitors, and none reported anaphylaxis.
The most common adverse events—with an incidence of 5% or greater—occurring outside of the perioperative management arm were nasopharyngitis, influenza, arthralgia, upper respiratory infection, hypertension, and headache.
One serious adverse event may have been drug-related. The patient experienced obstructive uropathy in the setting of hematuria. However, he continued to receive eftrenonacog alfa, and the event resolved with medical management.
Kids B-LONG
In Kids B-LONG, researchers tested eftrenonacog alfa in 30 previously treated children younger than 12 who had severe hemophilia B. Patients had at least 50 prior exposure days to factor IX therapies.
Children who received eftrenonacog alfa prophylactically had an overall median ABR of 1.97. The median ABR for spontaneous joint bleeds was 0.
Approximately 33% of patients did not experience any bleeding episodes. About 92% of bleeding episodes were controlled by 1 or 2 injections of eftrenonacog alfa.
None of the patients developed inhibitors. Researchers said there were no treatment-related serious adverse events and no cases of serious allergic reactions or vascular thrombotic events.
None of the patients discontinued the study due to an adverse event. One adverse event—decreased appetite occurring in 1 patient—was considered related to eftrenonacog alfa treatment.
The pattern of treatment-emergent adverse events in this study was generally consistent with results seen in adolescents and adults in the B-LONG study.
Test can monitor blood coagulation at home, team says

CINCINNATI—Researchers say they have developed a lateral flow assay device that patients can use at home to monitor their blood coagulation.
The device consists of nanofiber membranes inside a paper-based, porous test strip that is housed in a plastic cassette.
The researchers said this device can quickly reveal the level of the blood’s ability to clot using just a drop of blood from a finger prick.
“We have developed a blood-screening device for patients on medications like Coumadin (warfarin) or other blood thinners who need to monitor their blood-clotting levels on a regular basis,” said Andrew Steckl, PhD, of the University of Cincinnati in Ohio.
“Patients can soon monitor their blood coagulation characteristics from home quickly and painlessly before making needless trips to the lab or hospital.”
Hua Li, a student researcher in Dr Steckl’s lab, presented details on this device at the 8th International Conference on Porous Media and Annual Meeting of the International Society for Porous Media (abstract 1371).
Dr Steckl noted that slight changes in the level of coagulation properties will occur normally, but a major change in levels immediately shows up with his team’s device.
The researchers found the device was able to detect coagulation ability in rabbit blood and in blood from patients receiving warfarin.
“This simple test is not intended to replace the very careful and accurate measurements that get accomplished in a laboratory facility, but, at a relatively minimal cost, a patient can do this on their own between scheduled visits or when in doubt,” Dr Steckl said. “And it shouldn’t require a caregiver, as most patients can perform this test quickly on their own.”
Furthermore, the researchers said this technology can be calibrated to a specific patient’s condition. For example, a patient whose normal blood coagulation rate is significantly different from the general population because of a genetic disorder can use a tailor-made test kit that includes a different porous membrane.
The technology may also help patients who have a known inherited blood clotting disorder detect concerning levels early.
“By identifying potential blood-clotting problems early enough, we hope to prevent potential injury or death and the exorbitant associated costs,” Dr Steckl said. “By shifting from what were once mandatory expensive laboratory tests to using more in-home screening tests, patients can take more control of their lives, reduce healthcare costs, and, ultimately, save more lives.”
CINCINNATI—Researchers say they have developed a lateral flow assay device that patients can use at home to monitor their blood coagulation.
The device consists of nanofiber membranes inside a paper-based, porous test strip that is housed in a plastic cassette.
The researchers said this device can quickly reveal the level of the blood’s ability to clot using just a drop of blood from a finger prick.
“We have developed a blood-screening device for patients on medications like Coumadin (warfarin) or other blood thinners who need to monitor their blood-clotting levels on a regular basis,” said Andrew Steckl, PhD, of the University of Cincinnati in Ohio.
“Patients can soon monitor their blood coagulation characteristics from home quickly and painlessly before making needless trips to the lab or hospital.”
Hua Li, a student researcher in Dr Steckl’s lab, presented details on this device at the 8th International Conference on Porous Media and Annual Meeting of the International Society for Porous Media (abstract 1371).
Dr Steckl noted that slight changes in the level of coagulation properties will occur normally, but a major change in levels immediately shows up with his team’s device.
The researchers found the device was able to detect coagulation ability in rabbit blood and in blood from patients receiving warfarin.
“This simple test is not intended to replace the very careful and accurate measurements that get accomplished in a laboratory facility, but, at a relatively minimal cost, a patient can do this on their own between scheduled visits or when in doubt,” Dr Steckl said. “And it shouldn’t require a caregiver, as most patients can perform this test quickly on their own.”
Furthermore, the researchers said this technology can be calibrated to a specific patient’s condition. For example, a patient whose normal blood coagulation rate is significantly different from the general population because of a genetic disorder can use a tailor-made test kit that includes a different porous membrane.
The technology may also help patients who have a known inherited blood clotting disorder detect concerning levels early.
“By identifying potential blood-clotting problems early enough, we hope to prevent potential injury or death and the exorbitant associated costs,” Dr Steckl said. “By shifting from what were once mandatory expensive laboratory tests to using more in-home screening tests, patients can take more control of their lives, reduce healthcare costs, and, ultimately, save more lives.”
CINCINNATI—Researchers say they have developed a lateral flow assay device that patients can use at home to monitor their blood coagulation.
The device consists of nanofiber membranes inside a paper-based, porous test strip that is housed in a plastic cassette.
The researchers said this device can quickly reveal the level of the blood’s ability to clot using just a drop of blood from a finger prick.
“We have developed a blood-screening device for patients on medications like Coumadin (warfarin) or other blood thinners who need to monitor their blood-clotting levels on a regular basis,” said Andrew Steckl, PhD, of the University of Cincinnati in Ohio.
“Patients can soon monitor their blood coagulation characteristics from home quickly and painlessly before making needless trips to the lab or hospital.”
Hua Li, a student researcher in Dr Steckl’s lab, presented details on this device at the 8th International Conference on Porous Media and Annual Meeting of the International Society for Porous Media (abstract 1371).
Dr Steckl noted that slight changes in the level of coagulation properties will occur normally, but a major change in levels immediately shows up with his team’s device.
The researchers found the device was able to detect coagulation ability in rabbit blood and in blood from patients receiving warfarin.
“This simple test is not intended to replace the very careful and accurate measurements that get accomplished in a laboratory facility, but, at a relatively minimal cost, a patient can do this on their own between scheduled visits or when in doubt,” Dr Steckl said. “And it shouldn’t require a caregiver, as most patients can perform this test quickly on their own.”
Furthermore, the researchers said this technology can be calibrated to a specific patient’s condition. For example, a patient whose normal blood coagulation rate is significantly different from the general population because of a genetic disorder can use a tailor-made test kit that includes a different porous membrane.
The technology may also help patients who have a known inherited blood clotting disorder detect concerning levels early.
“By identifying potential blood-clotting problems early enough, we hope to prevent potential injury or death and the exorbitant associated costs,” Dr Steckl said. “By shifting from what were once mandatory expensive laboratory tests to using more in-home screening tests, patients can take more control of their lives, reduce healthcare costs, and, ultimately, save more lives.”