User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
NORD Publishes Report on Spontaneous Intracranial Hypotension (SIH)
As part of its ongoing educational outreach, NORD has published a report on spontaneous intracranial hypotension (SIH) in its Rare Disease Database. The database covers approximately 1,300 rare diseases and is available to all on the NORD website. Reports may be read online or downloaded free of charge.
The report on SIH was developed in collaboration with Connie Deline, MD, co-founder of the Spinal CSF Leak Foundation, and Wouter I. Schievink, MD, Professor of Neurosurgery, Department of Neurosurgery, Cedars-Sinai Medical Center. The hope is that the new report will reduce diagnosis delay and also be helpful to newly diagnosed patients.
The new report may be accessed here. SIH is secondary to a cerebrospinal fluid leak at the level of the spine, and the resulting loss of CSF volume to support the brain and spinal cord. To promote awareness of the condition, February 26 to March 4 has been designated the first spinal CSF leak awareness week.
As part of its ongoing educational outreach, NORD has published a report on spontaneous intracranial hypotension (SIH) in its Rare Disease Database. The database covers approximately 1,300 rare diseases and is available to all on the NORD website. Reports may be read online or downloaded free of charge.
The report on SIH was developed in collaboration with Connie Deline, MD, co-founder of the Spinal CSF Leak Foundation, and Wouter I. Schievink, MD, Professor of Neurosurgery, Department of Neurosurgery, Cedars-Sinai Medical Center. The hope is that the new report will reduce diagnosis delay and also be helpful to newly diagnosed patients.
The new report may be accessed here. SIH is secondary to a cerebrospinal fluid leak at the level of the spine, and the resulting loss of CSF volume to support the brain and spinal cord. To promote awareness of the condition, February 26 to March 4 has been designated the first spinal CSF leak awareness week.
As part of its ongoing educational outreach, NORD has published a report on spontaneous intracranial hypotension (SIH) in its Rare Disease Database. The database covers approximately 1,300 rare diseases and is available to all on the NORD website. Reports may be read online or downloaded free of charge.
The report on SIH was developed in collaboration with Connie Deline, MD, co-founder of the Spinal CSF Leak Foundation, and Wouter I. Schievink, MD, Professor of Neurosurgery, Department of Neurosurgery, Cedars-Sinai Medical Center. The hope is that the new report will reduce diagnosis delay and also be helpful to newly diagnosed patients.
The new report may be accessed here. SIH is secondary to a cerebrospinal fluid leak at the level of the spine, and the resulting loss of CSF volume to support the brain and spinal cord. To promote awareness of the condition, February 26 to March 4 has been designated the first spinal CSF leak awareness week.
CORD Offers Consensus Framework for Ethical Collaboration
The Canadian Organization for Rare Disorders (CORD) has developed a Consensus Framework to encourage ethical collaboration among patient organizations, health care professionals, and the pharmaceutical industry.
The Canadian Organization for Rare Disorders (CORD) has developed a Consensus Framework to encourage ethical collaboration among patient organizations, health care professionals, and the pharmaceutical industry.
The Canadian Organization for Rare Disorders (CORD) has developed a Consensus Framework to encourage ethical collaboration among patient organizations, health care professionals, and the pharmaceutical industry.
Apply Now to Join NORD’s Charity Marathon Team
Running for Rare, NORD’s charity marathon team, will be running the Los Angeles Marathon on March 19 to raise funds for NORD’s patient assistance program for undiagnosed patients and to raise awareness of rare diseases. Each runner is paired with a community partner (a patient or caregiver) who provides inspiration to the runner.
Applications are open for runners and community partners. The LA Marathon is the 5th largest marathon in the US and the 11th largest worldwide. More than 25,000 people participate. Details.
Running for Rare, NORD’s charity marathon team, will be running the Los Angeles Marathon on March 19 to raise funds for NORD’s patient assistance program for undiagnosed patients and to raise awareness of rare diseases. Each runner is paired with a community partner (a patient or caregiver) who provides inspiration to the runner.
Applications are open for runners and community partners. The LA Marathon is the 5th largest marathon in the US and the 11th largest worldwide. More than 25,000 people participate. Details.
Running for Rare, NORD’s charity marathon team, will be running the Los Angeles Marathon on March 19 to raise funds for NORD’s patient assistance program for undiagnosed patients and to raise awareness of rare diseases. Each runner is paired with a community partner (a patient or caregiver) who provides inspiration to the runner.
Applications are open for runners and community partners. The LA Marathon is the 5th largest marathon in the US and the 11th largest worldwide. More than 25,000 people participate. Details.
NORD to Partner Again With Hole in the Wall Gang Camp
NORD is proud to once again partner with the Hole in the Wall Gang Camp on a rare disease summer family camp in Connecticut. The camp provides a special opportunity for children and families affected by rare diseases to join together for a weekend of pure fun—free of charge. The camp is open to 25 families who are located in the Northeast, and it will take place June 1 to 4 in Ashford, Connecticut. Apply here.
NORD is proud to once again partner with the Hole in the Wall Gang Camp on a rare disease summer family camp in Connecticut. The camp provides a special opportunity for children and families affected by rare diseases to join together for a weekend of pure fun—free of charge. The camp is open to 25 families who are located in the Northeast, and it will take place June 1 to 4 in Ashford, Connecticut. Apply here.
NORD is proud to once again partner with the Hole in the Wall Gang Camp on a rare disease summer family camp in Connecticut. The camp provides a special opportunity for children and families affected by rare diseases to join together for a weekend of pure fun—free of charge. The camp is open to 25 families who are located in the Northeast, and it will take place June 1 to 4 in Ashford, Connecticut. Apply here.
Rare Disease Day 2017 Will Highlight Research Theme
On February 28th, medical professionals, patients, and advocates will observe Rare Disease Day in more than 90 nations worldwide. This awareness day was established in 2008 to promote education regarding rare diseases. As the national sponsor of Rare Disease Day in the US, NORD hosts the official website at www.rarediseaseday.us.
The theme for 2017 is Research and, in particular, the importance of research on rare diseases. This theme will be observed in all participating nations.
NORD provides tools and resources for those hosting events at academic centers and hospitals. Download this presentation for other suggestions regarding how to get involved. NORD will be co-hosting a tweetchat with ABC News and Dr. Richard Besser on Rare Disease Day. Details on this and other activities will be available on the Rare Disease Day US website.
On February 28th, medical professionals, patients, and advocates will observe Rare Disease Day in more than 90 nations worldwide. This awareness day was established in 2008 to promote education regarding rare diseases. As the national sponsor of Rare Disease Day in the US, NORD hosts the official website at www.rarediseaseday.us.
The theme for 2017 is Research and, in particular, the importance of research on rare diseases. This theme will be observed in all participating nations.
NORD provides tools and resources for those hosting events at academic centers and hospitals. Download this presentation for other suggestions regarding how to get involved. NORD will be co-hosting a tweetchat with ABC News and Dr. Richard Besser on Rare Disease Day. Details on this and other activities will be available on the Rare Disease Day US website.
On February 28th, medical professionals, patients, and advocates will observe Rare Disease Day in more than 90 nations worldwide. This awareness day was established in 2008 to promote education regarding rare diseases. As the national sponsor of Rare Disease Day in the US, NORD hosts the official website at www.rarediseaseday.us.
The theme for 2017 is Research and, in particular, the importance of research on rare diseases. This theme will be observed in all participating nations.
NORD provides tools and resources for those hosting events at academic centers and hospitals. Download this presentation for other suggestions regarding how to get involved. NORD will be co-hosting a tweetchat with ABC News and Dr. Richard Besser on Rare Disease Day. Details on this and other activities will be available on the Rare Disease Day US website.
NORD Seeks Input Regarding Patient Protections and ACA
As the new administration and Congress consider replacing the Affordable Care Act with a new system, NORD is seeking input from patients and health care providers for its advocacy on behalf of rare disease patients. Patients or providers who have experiences or concerns to share regarding pre-existing conditions, annual or lifetime insurance caps, high-risk pools, or other topics may share their experiences with NORD.
As the new administration and Congress consider replacing the Affordable Care Act with a new system, NORD is seeking input from patients and health care providers for its advocacy on behalf of rare disease patients. Patients or providers who have experiences or concerns to share regarding pre-existing conditions, annual or lifetime insurance caps, high-risk pools, or other topics may share their experiences with NORD.
As the new administration and Congress consider replacing the Affordable Care Act with a new system, NORD is seeking input from patients and health care providers for its advocacy on behalf of rare disease patients. Patients or providers who have experiences or concerns to share regarding pre-existing conditions, annual or lifetime insurance caps, high-risk pools, or other topics may share their experiences with NORD.
Pediatric Nail Diseases: Clinical Pearls
Our dermatology department recently sponsored a pediatric dermatology lecture series for the pediatric residency program. Within this series, Antonella Tosti, MD, a professor at the University of Miami Health System, Florida, and a renowned expert in nail disorders and allergic contact dermatitis, presented her clinical expertise on the presentation and management of common pediatric nail diseases. This article highlights pearls from her unique and enlightening lecture.
Pearl: Hand-foot-and-mouth disease is a recognized trigger for onychomadesis
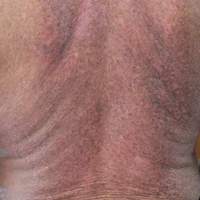
An arrest in nail matrix activity is responsible for onychomadesis, or shedding of the nail. Its presentation in children can be further divided based upon the degree of involvement. If a few nails are affected, trauma should be implicated. In contrast, if all nails are involved, a systemic etiology should be suspected. Hand-foot-and-mouth disease (HFMD) has been recognized as a trigger for onychomadesis in school-aged children. Onychomadesis presents with characteristic proximal nail detachment (Figure 1). The association of HFMD with onychomadesis and Beau lines was first reported in 2000. Five patients who resided within close proximity and shared a physician-diagnosed case of HFMD presented with representative nail findings 4 weeks after illness.1 Hypotheses for these changes include viral-induced nail pathology, inflammation from cutaneous lesions of HFMD, and systemic effects from the disease.2 Given the prevalence of HFMD and benign outcome, clinicians should be cognizant of this unique cutaneous manifestation.
Pearl: Management of pediatric melanonychia can take a wait-and-see approach
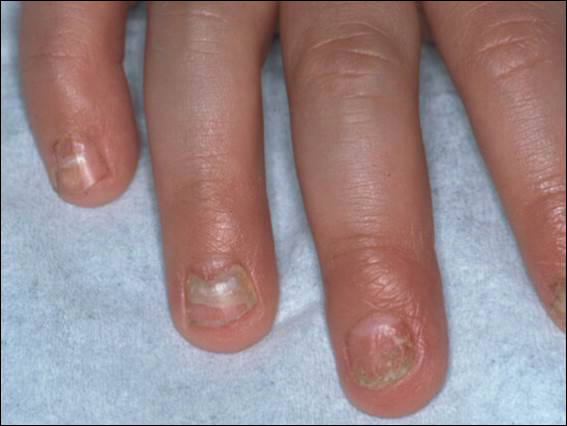
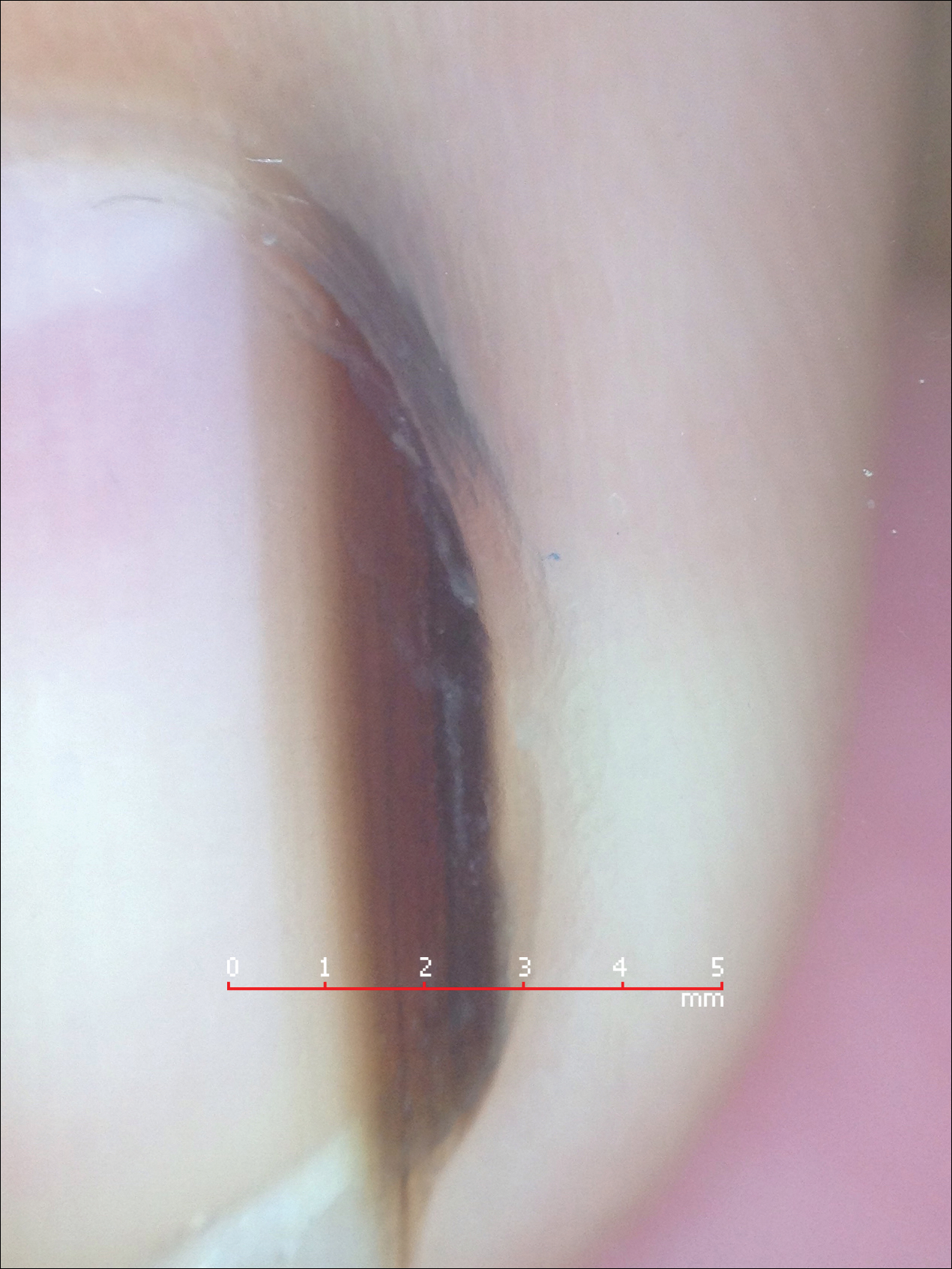
Melanonychia is the presence of a longitudinal brown-black band extending from the proximal nail fold. The cause of melanonychia can be due to either activation or hyperplasia. Activation is the less common etiology in children; however, if present, activation can be due to Laugier-Hunziker syndrome or trauma such as onychotillomania. Melanonychia in children usually is the result of hyperplasia of melanocytes and can manifest as a lentigo, nevus, or more rarely melanoma. Nail matrix nevi are typically exhibited on the fingernails, particularly the thumb, and frequently are junctional nevi (Figure 2). Spontaneous fading of nevi is expected with time due to decreased melanin production. Therapeutic options for melanonychia include regular clinical monitoring, biopsy, or excision. Dr. Tosti explained that one must be wary when pursuing a biopsy, as it can result in a false-negative finding due to missed pathology. If clinically indicated, a shave biopsy of the nail matrix can be performed to best analyze the lesion. She noted that if more than 3 mm of the matrix is removed, a resultant scar will ensue. Conservative management is recommended given the indolent clinical behavior of the majority of cases of melanonychia in children.3
Pearl: Congenital hypertrophy of the lateral nail folds can be treated with tape
Congenital hypertrophy of the lateral nail folds is relatively common in children and normally improves with age. Koilonychia may also occur simultaneously and can be viewed as a physiologic process in this age group. The etiology of the underlying disorder is due to anomalous periungual soft-tissue changes of the bilateral halluces; the resulting overgrowth can partially cover the nail plate. Although usually a self-limiting condition, the changes can cause inflammation and discomfort due to an ingrown nail.4 Dr. Tosti advised that by simply taping and retracting the bilateral overgrowth, the condition can be more readily resolved. This simple treatment can be demonstrated in the office and subsequently performed at home.
Pearl: Onychomycosis is uncommon in children
Onychomycosis occurs in less than 1% of children.5 Several factors are responsible for this decreased prevalence. More rapid nail growth and smaller nail surface area decreases the ability of the fungi to penetrate the nail plate.6 Furthermore, children have a diminished rate of tinea pedis, leading to less neighboring infection. When onychomycosis does affect this patient population, it commonly presents as distal subungual onychomycosis and favors the fingernails over the toenails. Treatment options usually parallel those of the adult population; however, all medications for children are considered off-label use by the US Food and Drug Administration. Dr. Tosti explained that oral granules of terbinafine can be sprinkled on food to help with pediatric ingestion. Topical therapies should also be considered; children usually respond better than their adult counterparts due to their thinner nails, which grant enhanced drug delivery and penetration.6
Pearl: Acute paronychia can be due to nail-biting and sucking
Acute paronychia is inflammation of the proximal nail fold. In children, it frequently is a result of mixed flora induced by nail-biting and sucking. Management involves culturing the affected lesions and is effectively treated with warm soaks alone. Dr. Tosti highlighted that Candida in the subungual space is a common colonizer and is typically self-limiting in nature if isolated. Candida can be cultured more readily in premature infants, immunosuppressed patients, and those with chronic mucocutaneous candidiasis. Patients with chronic mucocutaneous candidiasis can exhibit periungual inflammation involving several digits. The differential can include nail psoriasis, as both can demonstrate dystrophic changes. The differential for localized paronychia includes herpetic whitlow and can manifest as vesicles under the proximal nail fold.
Final Thoughts
These clinical pearls are shared to help deliver utmost care to our pediatric patients presenting with nail pathology. For example, a child exhibiting melanonychia can cause alarm due to the possibility of underlying melanoma; given the rarity of neoplasia in these patients, a conservative approach is favored to help avoid unnecessary biopsies and subsequent scarring. Similarly, it is important to be aware of the common colonizers of the subungual area, particularly Candida, to avoid unessential medications with potential side effects. The examples demonstrated help shed light on the management of pediatric nail diseases.
Acknowledgment
This article is possible thanks to the help of Antonella Tosti, MD (Miami, Florida), who contributed her time and expertise at the University of Miami Pediatric Grand Rounds to expand the foundation and knowledge of pediatric nail diseases.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Yuksel S, Evrengul H, Ozhan B, et al. Onychomadesis-a late complication of hand-foot-mouth disease [published online May 2, 2016]. J Pediatr. 2016;174:274.
- Cooper C, Arva NC, Lee C, et al. A clinical, histopathologic, and outcome study of melanonychia striata in childhood. J Am Acad Dermatol. 2015;72:773-779.
- Piraccini BM, Parente GL, Varotti E, et al. Congenital hypertrophy of the lateral nail folds of the hallux: clinical features and follow-up of seven cases. Pediatr Dermatol. 2000;17:348-351.
- Totri CR, Feldstein S, Admani S, et al. Epidemiologic analysis of onychomycosis in the San Diego pediatric population [published online October 4, 2016]. Pediatr Dermatol. 2017;34:46-49.
- Feldstein S, Totri C, Friedlander SF. Antifungal therapy for onychomycosis in children. Clin Dermatol. 2015;33:333-339.
Our dermatology department recently sponsored a pediatric dermatology lecture series for the pediatric residency program. Within this series, Antonella Tosti, MD, a professor at the University of Miami Health System, Florida, and a renowned expert in nail disorders and allergic contact dermatitis, presented her clinical expertise on the presentation and management of common pediatric nail diseases. This article highlights pearls from her unique and enlightening lecture.
Pearl: Hand-foot-and-mouth disease is a recognized trigger for onychomadesis
An arrest in nail matrix activity is responsible for onychomadesis, or shedding of the nail. Its presentation in children can be further divided based upon the degree of involvement. If a few nails are affected, trauma should be implicated. In contrast, if all nails are involved, a systemic etiology should be suspected. Hand-foot-and-mouth disease (HFMD) has been recognized as a trigger for onychomadesis in school-aged children. Onychomadesis presents with characteristic proximal nail detachment (Figure 1). The association of HFMD with onychomadesis and Beau lines was first reported in 2000. Five patients who resided within close proximity and shared a physician-diagnosed case of HFMD presented with representative nail findings 4 weeks after illness.1 Hypotheses for these changes include viral-induced nail pathology, inflammation from cutaneous lesions of HFMD, and systemic effects from the disease.2 Given the prevalence of HFMD and benign outcome, clinicians should be cognizant of this unique cutaneous manifestation.
Pearl: Management of pediatric melanonychia can take a wait-and-see approach
Melanonychia is the presence of a longitudinal brown-black band extending from the proximal nail fold. The cause of melanonychia can be due to either activation or hyperplasia. Activation is the less common etiology in children; however, if present, activation can be due to Laugier-Hunziker syndrome or trauma such as onychotillomania. Melanonychia in children usually is the result of hyperplasia of melanocytes and can manifest as a lentigo, nevus, or more rarely melanoma. Nail matrix nevi are typically exhibited on the fingernails, particularly the thumb, and frequently are junctional nevi (Figure 2). Spontaneous fading of nevi is expected with time due to decreased melanin production. Therapeutic options for melanonychia include regular clinical monitoring, biopsy, or excision. Dr. Tosti explained that one must be wary when pursuing a biopsy, as it can result in a false-negative finding due to missed pathology. If clinically indicated, a shave biopsy of the nail matrix can be performed to best analyze the lesion. She noted that if more than 3 mm of the matrix is removed, a resultant scar will ensue. Conservative management is recommended given the indolent clinical behavior of the majority of cases of melanonychia in children.3
Pearl: Congenital hypertrophy of the lateral nail folds can be treated with tape
Congenital hypertrophy of the lateral nail folds is relatively common in children and normally improves with age. Koilonychia may also occur simultaneously and can be viewed as a physiologic process in this age group. The etiology of the underlying disorder is due to anomalous periungual soft-tissue changes of the bilateral halluces; the resulting overgrowth can partially cover the nail plate. Although usually a self-limiting condition, the changes can cause inflammation and discomfort due to an ingrown nail.4 Dr. Tosti advised that by simply taping and retracting the bilateral overgrowth, the condition can be more readily resolved. This simple treatment can be demonstrated in the office and subsequently performed at home.
Pearl: Onychomycosis is uncommon in children
Onychomycosis occurs in less than 1% of children.5 Several factors are responsible for this decreased prevalence. More rapid nail growth and smaller nail surface area decreases the ability of the fungi to penetrate the nail plate.6 Furthermore, children have a diminished rate of tinea pedis, leading to less neighboring infection. When onychomycosis does affect this patient population, it commonly presents as distal subungual onychomycosis and favors the fingernails over the toenails. Treatment options usually parallel those of the adult population; however, all medications for children are considered off-label use by the US Food and Drug Administration. Dr. Tosti explained that oral granules of terbinafine can be sprinkled on food to help with pediatric ingestion. Topical therapies should also be considered; children usually respond better than their adult counterparts due to their thinner nails, which grant enhanced drug delivery and penetration.6
Pearl: Acute paronychia can be due to nail-biting and sucking
Acute paronychia is inflammation of the proximal nail fold. In children, it frequently is a result of mixed flora induced by nail-biting and sucking. Management involves culturing the affected lesions and is effectively treated with warm soaks alone. Dr. Tosti highlighted that Candida in the subungual space is a common colonizer and is typically self-limiting in nature if isolated. Candida can be cultured more readily in premature infants, immunosuppressed patients, and those with chronic mucocutaneous candidiasis. Patients with chronic mucocutaneous candidiasis can exhibit periungual inflammation involving several digits. The differential can include nail psoriasis, as both can demonstrate dystrophic changes. The differential for localized paronychia includes herpetic whitlow and can manifest as vesicles under the proximal nail fold.
Final Thoughts
These clinical pearls are shared to help deliver utmost care to our pediatric patients presenting with nail pathology. For example, a child exhibiting melanonychia can cause alarm due to the possibility of underlying melanoma; given the rarity of neoplasia in these patients, a conservative approach is favored to help avoid unnecessary biopsies and subsequent scarring. Similarly, it is important to be aware of the common colonizers of the subungual area, particularly Candida, to avoid unessential medications with potential side effects. The examples demonstrated help shed light on the management of pediatric nail diseases.
Acknowledgment
This article is possible thanks to the help of Antonella Tosti, MD (Miami, Florida), who contributed her time and expertise at the University of Miami Pediatric Grand Rounds to expand the foundation and knowledge of pediatric nail diseases.
Our dermatology department recently sponsored a pediatric dermatology lecture series for the pediatric residency program. Within this series, Antonella Tosti, MD, a professor at the University of Miami Health System, Florida, and a renowned expert in nail disorders and allergic contact dermatitis, presented her clinical expertise on the presentation and management of common pediatric nail diseases. This article highlights pearls from her unique and enlightening lecture.
Pearl: Hand-foot-and-mouth disease is a recognized trigger for onychomadesis
An arrest in nail matrix activity is responsible for onychomadesis, or shedding of the nail. Its presentation in children can be further divided based upon the degree of involvement. If a few nails are affected, trauma should be implicated. In contrast, if all nails are involved, a systemic etiology should be suspected. Hand-foot-and-mouth disease (HFMD) has been recognized as a trigger for onychomadesis in school-aged children. Onychomadesis presents with characteristic proximal nail detachment (Figure 1). The association of HFMD with onychomadesis and Beau lines was first reported in 2000. Five patients who resided within close proximity and shared a physician-diagnosed case of HFMD presented with representative nail findings 4 weeks after illness.1 Hypotheses for these changes include viral-induced nail pathology, inflammation from cutaneous lesions of HFMD, and systemic effects from the disease.2 Given the prevalence of HFMD and benign outcome, clinicians should be cognizant of this unique cutaneous manifestation.
Pearl: Management of pediatric melanonychia can take a wait-and-see approach
Melanonychia is the presence of a longitudinal brown-black band extending from the proximal nail fold. The cause of melanonychia can be due to either activation or hyperplasia. Activation is the less common etiology in children; however, if present, activation can be due to Laugier-Hunziker syndrome or trauma such as onychotillomania. Melanonychia in children usually is the result of hyperplasia of melanocytes and can manifest as a lentigo, nevus, or more rarely melanoma. Nail matrix nevi are typically exhibited on the fingernails, particularly the thumb, and frequently are junctional nevi (Figure 2). Spontaneous fading of nevi is expected with time due to decreased melanin production. Therapeutic options for melanonychia include regular clinical monitoring, biopsy, or excision. Dr. Tosti explained that one must be wary when pursuing a biopsy, as it can result in a false-negative finding due to missed pathology. If clinically indicated, a shave biopsy of the nail matrix can be performed to best analyze the lesion. She noted that if more than 3 mm of the matrix is removed, a resultant scar will ensue. Conservative management is recommended given the indolent clinical behavior of the majority of cases of melanonychia in children.3
Pearl: Congenital hypertrophy of the lateral nail folds can be treated with tape
Congenital hypertrophy of the lateral nail folds is relatively common in children and normally improves with age. Koilonychia may also occur simultaneously and can be viewed as a physiologic process in this age group. The etiology of the underlying disorder is due to anomalous periungual soft-tissue changes of the bilateral halluces; the resulting overgrowth can partially cover the nail plate. Although usually a self-limiting condition, the changes can cause inflammation and discomfort due to an ingrown nail.4 Dr. Tosti advised that by simply taping and retracting the bilateral overgrowth, the condition can be more readily resolved. This simple treatment can be demonstrated in the office and subsequently performed at home.
Pearl: Onychomycosis is uncommon in children
Onychomycosis occurs in less than 1% of children.5 Several factors are responsible for this decreased prevalence. More rapid nail growth and smaller nail surface area decreases the ability of the fungi to penetrate the nail plate.6 Furthermore, children have a diminished rate of tinea pedis, leading to less neighboring infection. When onychomycosis does affect this patient population, it commonly presents as distal subungual onychomycosis and favors the fingernails over the toenails. Treatment options usually parallel those of the adult population; however, all medications for children are considered off-label use by the US Food and Drug Administration. Dr. Tosti explained that oral granules of terbinafine can be sprinkled on food to help with pediatric ingestion. Topical therapies should also be considered; children usually respond better than their adult counterparts due to their thinner nails, which grant enhanced drug delivery and penetration.6
Pearl: Acute paronychia can be due to nail-biting and sucking
Acute paronychia is inflammation of the proximal nail fold. In children, it frequently is a result of mixed flora induced by nail-biting and sucking. Management involves culturing the affected lesions and is effectively treated with warm soaks alone. Dr. Tosti highlighted that Candida in the subungual space is a common colonizer and is typically self-limiting in nature if isolated. Candida can be cultured more readily in premature infants, immunosuppressed patients, and those with chronic mucocutaneous candidiasis. Patients with chronic mucocutaneous candidiasis can exhibit periungual inflammation involving several digits. The differential can include nail psoriasis, as both can demonstrate dystrophic changes. The differential for localized paronychia includes herpetic whitlow and can manifest as vesicles under the proximal nail fold.
Final Thoughts
These clinical pearls are shared to help deliver utmost care to our pediatric patients presenting with nail pathology. For example, a child exhibiting melanonychia can cause alarm due to the possibility of underlying melanoma; given the rarity of neoplasia in these patients, a conservative approach is favored to help avoid unnecessary biopsies and subsequent scarring. Similarly, it is important to be aware of the common colonizers of the subungual area, particularly Candida, to avoid unessential medications with potential side effects. The examples demonstrated help shed light on the management of pediatric nail diseases.
Acknowledgment
This article is possible thanks to the help of Antonella Tosti, MD (Miami, Florida), who contributed her time and expertise at the University of Miami Pediatric Grand Rounds to expand the foundation and knowledge of pediatric nail diseases.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Yuksel S, Evrengul H, Ozhan B, et al. Onychomadesis-a late complication of hand-foot-mouth disease [published online May 2, 2016]. J Pediatr. 2016;174:274.
- Cooper C, Arva NC, Lee C, et al. A clinical, histopathologic, and outcome study of melanonychia striata in childhood. J Am Acad Dermatol. 2015;72:773-779.
- Piraccini BM, Parente GL, Varotti E, et al. Congenital hypertrophy of the lateral nail folds of the hallux: clinical features and follow-up of seven cases. Pediatr Dermatol. 2000;17:348-351.
- Totri CR, Feldstein S, Admani S, et al. Epidemiologic analysis of onychomycosis in the San Diego pediatric population [published online October 4, 2016]. Pediatr Dermatol. 2017;34:46-49.
- Feldstein S, Totri C, Friedlander SF. Antifungal therapy for onychomycosis in children. Clin Dermatol. 2015;33:333-339.
- Clementz GC, Mancini AJ. Nail matrix arrest following hand-foot-mouth disease: a report of five children. Pediatr Dermatol. 2000;17:7-11.
- Yuksel S, Evrengul H, Ozhan B, et al. Onychomadesis-a late complication of hand-foot-mouth disease [published online May 2, 2016]. J Pediatr. 2016;174:274.
- Cooper C, Arva NC, Lee C, et al. A clinical, histopathologic, and outcome study of melanonychia striata in childhood. J Am Acad Dermatol. 2015;72:773-779.
- Piraccini BM, Parente GL, Varotti E, et al. Congenital hypertrophy of the lateral nail folds of the hallux: clinical features and follow-up of seven cases. Pediatr Dermatol. 2000;17:348-351.
- Totri CR, Feldstein S, Admani S, et al. Epidemiologic analysis of onychomycosis in the San Diego pediatric population [published online October 4, 2016]. Pediatr Dermatol. 2017;34:46-49.
- Feldstein S, Totri C, Friedlander SF. Antifungal therapy for onychomycosis in children. Clin Dermatol. 2015;33:333-339.

Actinomycetoma: An Update on Diagnosis and Treatment
Mycetoma is a subcutaneous disease that can be caused by aerobic bacteria (actinomycetoma) or fungi (eumycetoma). Diagnosis is based on clinical manifestations, including swelling and deformity of affected areas, as well as the presence of granulation tissue, scars, abscesses, sinus tracts, and a purulent exudate that contains the microorganisms.
The worldwide proportion of mycetomas is 60% actinomycetomas and 40% eumycetomas.1 The disease is endemic in tropical, subtropical, and temperate regions, predominating between latitudes 30°N and 15°S. Most cases occur in Africa, especially Sudan, Mauritania, and Senegal; India; Yemen; and Pakistan. In the Americas, the countries with the most reported cases are Mexico and Venezuela.1
Although mycetoma is rare in developed countries, migration of patients from endemic areas makes knowledge of this condition crucial for dermatologists worldwide. We present a review of the current concepts in the epidemiology, clinical presentation, diagnosis, and treatment of actinomycetoma.
Epidemiology
Actinomycetoma is more common in Latin America, with Mexico having the highest incidence. At last count, there were 2631 cases reported in Mexico.2 The majority of cases of mycetoma in Mexico are actinomycetoma (98%), including Nocardia (86%) and Actinomadura madurae (10%). Eumycetoma is rare in Mexico, constituting only 2% of cases.2 Worldwide, men are affected more commonly than women, which is thought to be related to a higher occupational risk during agricultural labor.
Clinical Features
Mycetoma can affect the skin, subcutaneous tissue, bones, and occasionally the internal organs. It is characterized by swelling, deformation of the affected area, and fistulae that drain serosanguineous or purulent exudates.
In Mexico, 60% of cases of mycetoma affect the lower extremities; the feet are the most commonly affected area, followed by the trunk (back and chest), arms, forearms, legs, knees, and thighs.1 Other sites include the hands, shoulders, and abdominal wall. The head and neck area are seldom affected.3 Mycetoma lesions grow and disseminate locally. Bone lesions are possible depending on the osteophilic affinity of the etiological agent and on the interactions between the fungus and the host’s immune system. In severe advanced cases of mycetoma, the lesions may involve tendons and nerves. Dissemination via blood or lymphatics is extremely rare.4
Diagnosis
Diagnosis of actinomycetoma is suspected based on clinical features and confirmed by direct examination of exudates with Lugol iodine or saline solution. On direct microscopy, actinomycetes are recognized by the production of filaments with a width of 0.5 to 1 μm. On hematoxylin and eosin stain, the small grains of Nocardia appear eosinophilic with a blue center and pink filaments. On Gram stain, actinomycetoma grains show positive branching filaments. Culture of grains recovered from aspirated material or biopsy specimens provides specific etiologic diagnosis. Cultures should be held for at least 4 weeks. Additionally, there are some enzymatic, molecular, and serologic tests available for diagnosis.5-7 Serologic diagnosis is available in a few centers in Mexico and can be helpful in some cases for diagnosis or follow-up during treatment. Antibodies can be determined via enzyme-linked immunosorbent assay, Western blot analysis, immunodiffusion, or counterimmunoelectrophoresis.8
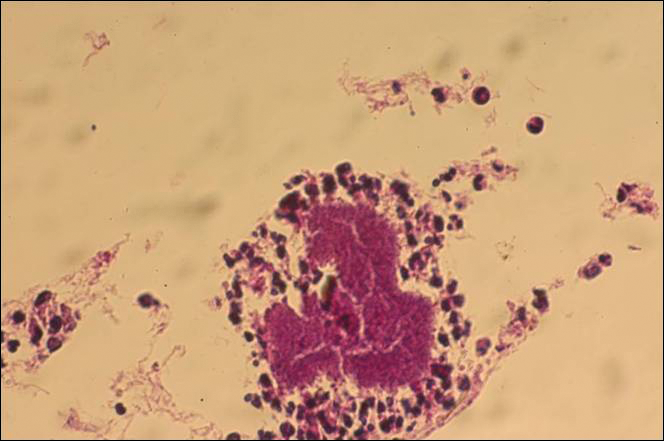
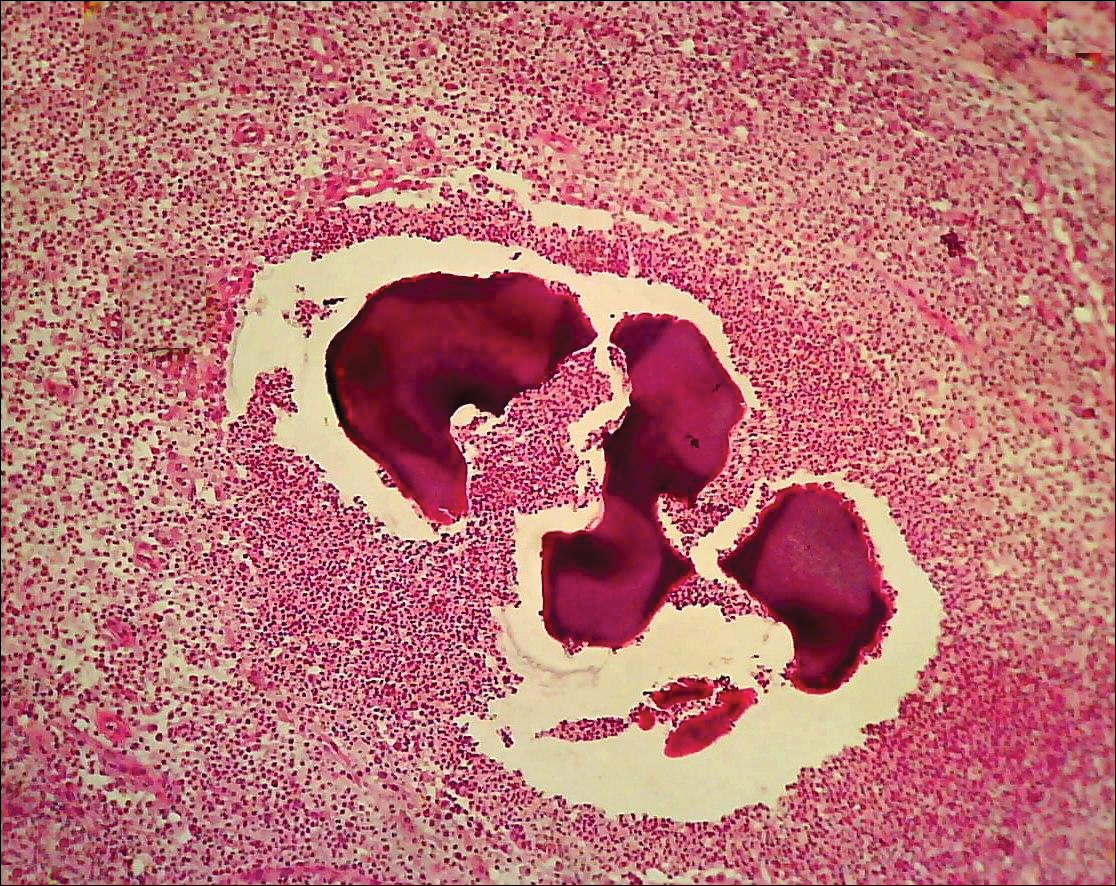
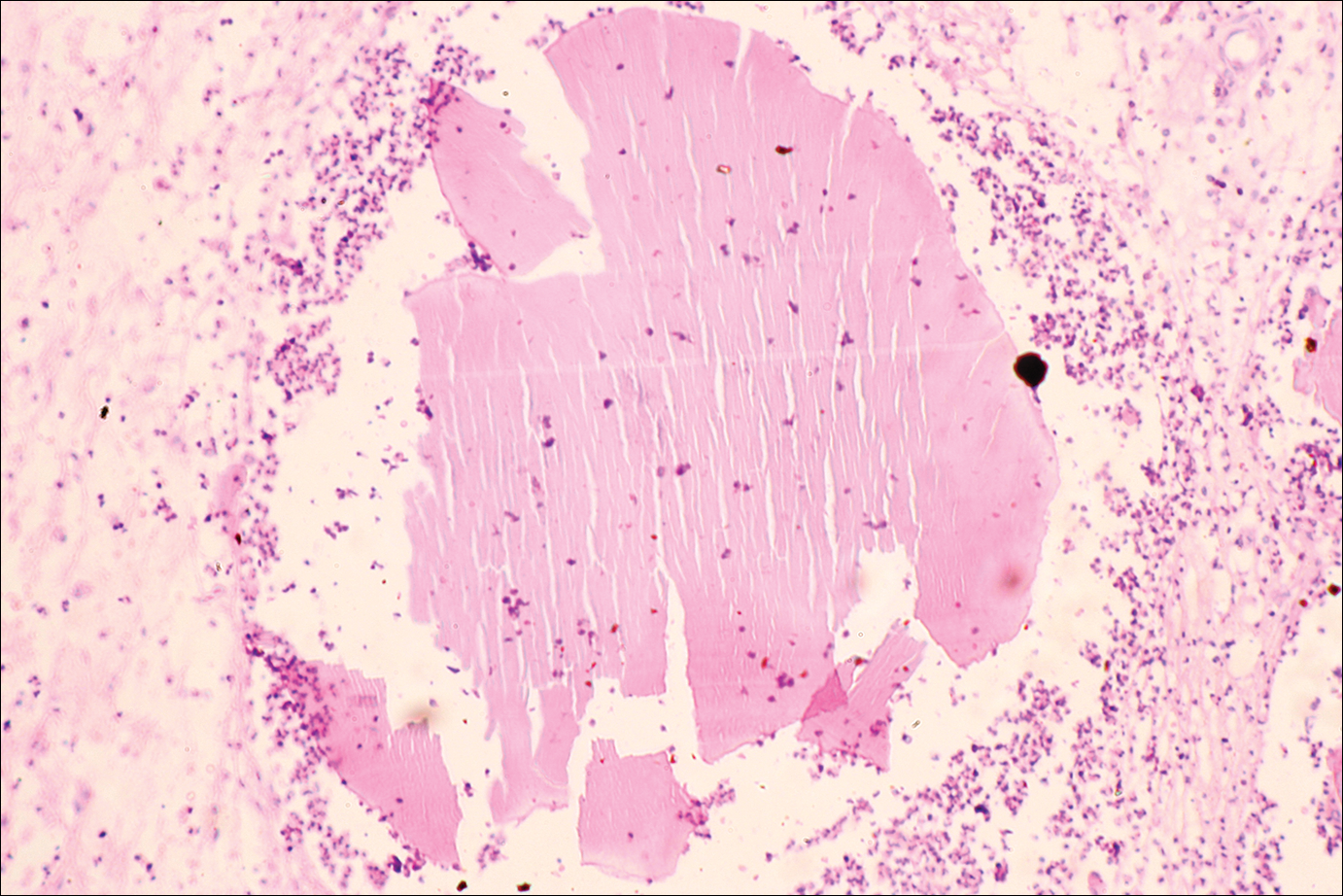
The causative agents of actinomycetoma can be isolated in Sabouraud dextrose agar. Deep wedge biopsies (or puncture aspiration) are useful in observing the diagnostic grains, which can be identified adequately with Gram stain. Grains usually are surrounded and/or infiltrated by neutrophils. The size, form, and color of grains can identify the causative agent.1 The granules of Nocardia are small (80–130 mm) and reniform or wormlike, with club structures in their periphery (Figure 1). Actinomadura madurae is characterized by large, white-yellow granules that can be seen with the naked eye (1–3 mm). On microscopic examination with hematoxylin and eosin stain, these grains are purple and exhibit peripheral pink pseudofilaments (Figure 2).2 The grains of Actinomadura pelletieri are large (1–3 mm) and red or violaceous. They fragment or break easily, giving the appearance of a broken dish (Figure 3). Streptomyces somaliensis forms round grains approximately 0.5 to 1 cm in diameter. These grains stain poorly and are extremely hard. Cutting the grains during processing results in striation, giving them the appearance of a potato chip (Figure 4).2

Treatment of Actinomycetoma
Precise identification of the etiologic agent is essential to provide effective treatment of actinomycetoma. Without treatment, or in resistant cases, progressive osseous and visceral involvement is inevitable.9 Actinomycetoma without osseous involvement usually responds well to medical treatment.
The treatment of choice for actinomycetoma involving Nocardia brasiliensis is a combination of dapsone 100 to 200 mg once daily and trimethoprim-sulfamethoxazole (TMP-SMX) 80/400 to 160/800 mg once daily for 2 to 3 years.10 Other treatments have included the following: (1) amikacin 15 mg/kg or 500 mg intramuscularly twice daily for 3 weeks plus dapsone 100 to 200 mg once daily plus TMP-SMX 80/400 to 160/800 mg daily for 2 to 3 years (amikacin, however, is expensive and potentially toxic [nephrotoxicity and ototoxicity] and therefore is used only in resistant cases); (2) dapsone 100 to 200 mg once daily or TMP-SMX 80/400 to 160/800 mg daily for 2 to 3 years plus intramuscular kanamycin 15 mg/kg once daily for 2 weeks at the beginning of treatment, alternating with rest periods to reduce the risk for nephrotoxicity and ototoxicity10; (3) dapsone 1.5 mg/kg orally twice daily plus phosphomycin 500 mg once daily; (4) dapsone 1.5 mg/kg orally twice daily plus streptomycin 1 g once daily (14 mg/kg/d) for 1 month, then the same dose every other day for 1 to 2 months monitoring for ototoxicity; and (5) TMP-SMX 80/400 to 160/800 mg once daily for 2 to 3 years or rifampicin (15–20 mg/kg/d) plus streptomycin 1 g once daily (14 mg/kg/d) for 1 month at the beginning of treatment, then the same dose every other day for 2 to 3 months until a total dose of 60 g is administered, monitoring for ototoxicity.11 Audiometric tests and creatinine levels must be performed every 5 weeks during the treatment to monitor toxicity.10
The best results for infections with A pelletieri, A madurae, and S somaliensis have been with streptomycin (1 g once daily in adults; 20 mg/kg once daily in children) until a total dose of 50 g is reached in combination with TMP-SMX or dapsone12 (Figure 5). Alternatives for A madurae infections include streptomycin plus oral clofazimine (100 mg once daily), oral rifampicin (300 mg twice daily), oral tetracycline (1 g once daily), oral isoniazid (300–600 mg once daily), or oral minocycline (100 mg twice daily; also effective for A pelletieri).

More recently, other drugs have been used such as carbapenems (eg, imipenem, meropenem), which have wide-spectrum efficacy and are resistant to β-lactamases. Patients should be hospitalized to receive intravenous therapy with imipenem.2 Carbapenems are effective against gram-positive and gram-negative as well as Nocardia species.13,14 Mycetoma that is resistant, severe, or has visceral involvement can be treated with a combination of amikacin and imipenem.15,16 Meropenem is a similar drug that is available as an oral formulation. Both imipenem and meropenem are recommended in cases with bone involvement.17,18 Alternatives for resistant cases include amoxicillin–clavulanic acid 500/125 mg orally 3 times daily for 3 to 6 months or intravenous cefotaxime 1 g every 8 hours plus intramuscular amikacin 500 mg twice daily plus oral levamisole 300 mg once weekly for 4 weeks.19-23
For resistant cases associated with Nocardia species, clindamycin plus quinolones (eg, ciprofloxacin, moxifloxacin, garenoxacin) at a dose of 25 mg/kg once daily for at least 3 months has been suggested in in vivo studies.23
Overall, the cure rate for actinomycetoma treated with any of the prior therapies ranges from 60% to 90%. Treatment must be modified or stopped if there is clinical or laboratory evidence of drug toxicity.13,24 Surgical treatment of actinomycetoma is contraindicated, as it may cause hematogenous dissemination.
Prognosis
Actinomycetomas of a few months’ duration and without bone involvement respond well to therapy. If no therapy is provided or if there is resistance, the functional and cosmetic prognosis is poor, mainly for the feet. There is a risk for spine involvement with mycetoma on the back and posterior head. Thoracic lesions may penetrate into the lungs. The muscular fascia impedes the penetration of abdominal lesions, but the inguinal canals can offer a path for intra-abdominal dissemination.4 Advanced cases lead to a poor general condition of patients, difficulty in using affected extremities, and in extreme cases even death.
The criteria used to guide the discontinuation of initial therapy for any mycetoma include a decrease in the volume of the lesion, closure of fistulae, 3 consecutive negative monthly cultures, imaging studies showing bone regeneration, lack of echoes and cavities on echography, and absence of grains on examination of fine-needle aspirates.11 After the initial treatment protocol is finished, most experts recommend continuing treatment with dapsone 100 to 300 mg once daily for several years to prevent recurrence.12
Prevention
Mycetoma is a disease associated with poverty. It could be prevented by improving living conditions and by regular use of shoes in rural populations.2
Conclusion
Mycetoma is a chronic infection that develops after traumatic inoculation of the skin with either true fungi or aerobic actinomycetes. The resultant infections are known as eumycetoma or actinomycetoma, respectively. The etiologic agents can be found in the so-called grains. Black grains suggest a fungal infection, minute white grains suggest Nocardia, and red grains are due to A pelletieri. Larger white grains or yellow-white grains may be fungal or actinomycotic in origin.
Specific diagnosis requires direct examination, culture, and biopsy. The treatment of choice for actinomycetoma by N brasiliensis is a combination of dapsone 100 to 200 mg once daily and TMP-SMX 80/400 to 160/800 mg once daily for 2 to 3 years. Other effective treatments include aminoglycosides (eg, amikacine, streptomycin) and quinolones. More recently, some other agents have been used such as carbapenems and natural products of Streptomyces cattleya (imipenem), which have wide-spectrum efficacy and are resistant to β-lactamases.
- Welsh O, Vera-Cabrera L, Welsh E, et al. Actinomycetoma and advances in its treatment. Clin Dermatol. 2012;30:372-381.
- Arenas R. Micología Medica Ilustrada. 4th ed. Mexico City, Mexico: McGraw-Hill Interamericana; 2011:125-146.
- McGinnis MR. Mycetoma. Dermatol Clin. 1996;14:97-104.
- Fahal AH. Mycetoma: Clinico-pathological Monograph. Khartoum, Sudan: University of Khartoum Press; 2006:20-23, 81-82.
- Estrada-Chavez GE, Vega-Memije ME, Arenas R, et al. Eumycotic mycetoma caused by Madurella mycetomatis successfully treated with antifungals, surgery, and topical negative pressure therapy. Int J Dermatol. 2009;48:401-403.
- Chávez G, Arenas R, Pérez-Polito A, et al. Eumycetic mycetoma due to Madurella mycetomatis. report of six cases. Rev Iberoam Micol. 1998;15:90-93.
- Vasquez del Mercado E, Arenas R, Moreno G. Sequelae and long-term consequences of systemic and subcutaneous mycoses. In: Fratamico PM, Smith JL, Brogden KA, eds. Sequelae and Long-term Consequences of Infectious Diseases. Washington, DC: ASM Press; 2009:415-420.
- Mancini N, Ossi CM, Perotti M, et al. Molecular mycological diagnosis and correct antimycotic treatments. J Clin Microbiol. 2005;43:3584-3585.
- Arenas R, Lavalle P. Micetoma (madura foot). In: Arenas R, Estrada R, eds. Tropical Dermatology. Austin, TX: Landes Bioscience; 2001:51-61.
- Welsh O, Sauceda E, González J, et al. Amikacin alone and in combination with trimethoprim-sulfamethoxazole in the treatment of actinomycotic mycetoma. J Am Acad Dermatol. 1987;17:443-448.
- Fahal AH. Mycetoma: clinico-pathological monograph. In: Fahal AH. Evidence Based Guidelines for the Management of Mycetoma Patients. Khartoum, Sudan: University of Khartoum Press; 2002:5-15.
- Welsh O, Salinas MC, Rodríguez MA. Treatment of eumycetoma and actinomycetoma. Curr Top Med Mycol. 1995;6:47-71.
- Valle ACF, Welsh O, Vera-Cabrera L. Subcutaneous mycoses—mycetoma. In: Tyring SK, Lupi O, Hengge UR, eds. Tropical Dermatology. Philadelphia, PA: Elsevier Churchill Livingstone; 2006:197-200.
- Fuentes A, Arenas R, Reyes M, et al. Actinomicetoma por Nocardia sp. Informe de cinco casos tratados con imipenem solo o combinado con amikacina. Gac Med Mex. 2006;142:247-252.
- Gombert ME, Aulicino TM, DuBouchet L, et al. Therapy of experimental cerebral nocardiosis with imipenem, amikacin, trimethoprim-sulfamethoxazole, and minocylina. Antimicrob Agents Chemother. 1986;30:270-273.
- Calandra GB, Ricci FM, Wang C, et al. Safety and tolerance comparison of imipenem-cilastatin to cephalotin and cefazolin. J Antimicrob Chemother. 1983;12:125-131.
- Ameen M, Arenas R, Vasquez del Mercado E, et al. Efficacy of imipenem therapy for Nocardia actinomycetomas refractory to sulfonamides. J Am Acad Dermatol. 2010;62:239-246.
- Ameen M, Vargas F, Vasquez del Mercado E, et al. Successful treatment of Nocardia actinomycetoma with meropenem and amikacin combination therapy. Int J Dermatol. 2011;50:443-445.
- Ameen M, Arenas R. Emerging therapeutic regimes for the management of mycetomas. Expert Opin Pharmacother. 2008;9:2077-2085.
- Vera-Cabrera L, Daw-Garza A, Said-Fernández S, et al. Therapeutic effect of a novel oxazolidinone, DA-7867 in BALB/c mice infected with Nocardia brasiliensis. PloS Negl Trop Dis. 2008;2:e289.
- Gómez A, Saúl A, Bonifaz A. Amoxicillin and clavulanic acid in the treatment of actinomicetoma. Int J Dermatol. 1993;32:218-220.
- Méndez-Tovar L, Serrano-Jaen L, Almeida-Arvizu VM. Cefotaxima mas amikacina asociadas a inmunomodulación en el tratamiento de actinomicetoma resistente a tratamiento convencional. Gac Med Mex. 1999;135:517-521.
- Chacon-Moreno BE, Welsh O, Cavazos-Rocha N, et al. Efficacy of ciprofloxacin and moxifloxacin against Nocardia brasiliensis in vitro in an experimental model of actinomycetoma in BALB/c mice. Antimicrob Agents Chemother. 2009;53:295-297.
- Welsh O. Treatment of actinomycetoma. Arch Med Res. 1993;24:413-415.
Mycetoma is a subcutaneous disease that can be caused by aerobic bacteria (actinomycetoma) or fungi (eumycetoma). Diagnosis is based on clinical manifestations, including swelling and deformity of affected areas, as well as the presence of granulation tissue, scars, abscesses, sinus tracts, and a purulent exudate that contains the microorganisms.
The worldwide proportion of mycetomas is 60% actinomycetomas and 40% eumycetomas.1 The disease is endemic in tropical, subtropical, and temperate regions, predominating between latitudes 30°N and 15°S. Most cases occur in Africa, especially Sudan, Mauritania, and Senegal; India; Yemen; and Pakistan. In the Americas, the countries with the most reported cases are Mexico and Venezuela.1
Although mycetoma is rare in developed countries, migration of patients from endemic areas makes knowledge of this condition crucial for dermatologists worldwide. We present a review of the current concepts in the epidemiology, clinical presentation, diagnosis, and treatment of actinomycetoma.
Epidemiology
Actinomycetoma is more common in Latin America, with Mexico having the highest incidence. At last count, there were 2631 cases reported in Mexico.2 The majority of cases of mycetoma in Mexico are actinomycetoma (98%), including Nocardia (86%) and Actinomadura madurae (10%). Eumycetoma is rare in Mexico, constituting only 2% of cases.2 Worldwide, men are affected more commonly than women, which is thought to be related to a higher occupational risk during agricultural labor.
Clinical Features
Mycetoma can affect the skin, subcutaneous tissue, bones, and occasionally the internal organs. It is characterized by swelling, deformation of the affected area, and fistulae that drain serosanguineous or purulent exudates.
In Mexico, 60% of cases of mycetoma affect the lower extremities; the feet are the most commonly affected area, followed by the trunk (back and chest), arms, forearms, legs, knees, and thighs.1 Other sites include the hands, shoulders, and abdominal wall. The head and neck area are seldom affected.3 Mycetoma lesions grow and disseminate locally. Bone lesions are possible depending on the osteophilic affinity of the etiological agent and on the interactions between the fungus and the host’s immune system. In severe advanced cases of mycetoma, the lesions may involve tendons and nerves. Dissemination via blood or lymphatics is extremely rare.4
Diagnosis
Diagnosis of actinomycetoma is suspected based on clinical features and confirmed by direct examination of exudates with Lugol iodine or saline solution. On direct microscopy, actinomycetes are recognized by the production of filaments with a width of 0.5 to 1 μm. On hematoxylin and eosin stain, the small grains of Nocardia appear eosinophilic with a blue center and pink filaments. On Gram stain, actinomycetoma grains show positive branching filaments. Culture of grains recovered from aspirated material or biopsy specimens provides specific etiologic diagnosis. Cultures should be held for at least 4 weeks. Additionally, there are some enzymatic, molecular, and serologic tests available for diagnosis.5-7 Serologic diagnosis is available in a few centers in Mexico and can be helpful in some cases for diagnosis or follow-up during treatment. Antibodies can be determined via enzyme-linked immunosorbent assay, Western blot analysis, immunodiffusion, or counterimmunoelectrophoresis.8
The causative agents of actinomycetoma can be isolated in Sabouraud dextrose agar. Deep wedge biopsies (or puncture aspiration) are useful in observing the diagnostic grains, which can be identified adequately with Gram stain. Grains usually are surrounded and/or infiltrated by neutrophils. The size, form, and color of grains can identify the causative agent.1 The granules of Nocardia are small (80–130 mm) and reniform or wormlike, with club structures in their periphery (Figure 1). Actinomadura madurae is characterized by large, white-yellow granules that can be seen with the naked eye (1–3 mm). On microscopic examination with hematoxylin and eosin stain, these grains are purple and exhibit peripheral pink pseudofilaments (Figure 2).2 The grains of Actinomadura pelletieri are large (1–3 mm) and red or violaceous. They fragment or break easily, giving the appearance of a broken dish (Figure 3). Streptomyces somaliensis forms round grains approximately 0.5 to 1 cm in diameter. These grains stain poorly and are extremely hard. Cutting the grains during processing results in striation, giving them the appearance of a potato chip (Figure 4).2
Treatment of Actinomycetoma
Precise identification of the etiologic agent is essential to provide effective treatment of actinomycetoma. Without treatment, or in resistant cases, progressive osseous and visceral involvement is inevitable.9 Actinomycetoma without osseous involvement usually responds well to medical treatment.
The treatment of choice for actinomycetoma involving Nocardia brasiliensis is a combination of dapsone 100 to 200 mg once daily and trimethoprim-sulfamethoxazole (TMP-SMX) 80/400 to 160/800 mg once daily for 2 to 3 years.10 Other treatments have included the following: (1) amikacin 15 mg/kg or 500 mg intramuscularly twice daily for 3 weeks plus dapsone 100 to 200 mg once daily plus TMP-SMX 80/400 to 160/800 mg daily for 2 to 3 years (amikacin, however, is expensive and potentially toxic [nephrotoxicity and ototoxicity] and therefore is used only in resistant cases); (2) dapsone 100 to 200 mg once daily or TMP-SMX 80/400 to 160/800 mg daily for 2 to 3 years plus intramuscular kanamycin 15 mg/kg once daily for 2 weeks at the beginning of treatment, alternating with rest periods to reduce the risk for nephrotoxicity and ototoxicity10; (3) dapsone 1.5 mg/kg orally twice daily plus phosphomycin 500 mg once daily; (4) dapsone 1.5 mg/kg orally twice daily plus streptomycin 1 g once daily (14 mg/kg/d) for 1 month, then the same dose every other day for 1 to 2 months monitoring for ototoxicity; and (5) TMP-SMX 80/400 to 160/800 mg once daily for 2 to 3 years or rifampicin (15–20 mg/kg/d) plus streptomycin 1 g once daily (14 mg/kg/d) for 1 month at the beginning of treatment, then the same dose every other day for 2 to 3 months until a total dose of 60 g is administered, monitoring for ototoxicity.11 Audiometric tests and creatinine levels must be performed every 5 weeks during the treatment to monitor toxicity.10
The best results for infections with A pelletieri, A madurae, and S somaliensis have been with streptomycin (1 g once daily in adults; 20 mg/kg once daily in children) until a total dose of 50 g is reached in combination with TMP-SMX or dapsone12 (Figure 5). Alternatives for A madurae infections include streptomycin plus oral clofazimine (100 mg once daily), oral rifampicin (300 mg twice daily), oral tetracycline (1 g once daily), oral isoniazid (300–600 mg once daily), or oral minocycline (100 mg twice daily; also effective for A pelletieri).
More recently, other drugs have been used such as carbapenems (eg, imipenem, meropenem), which have wide-spectrum efficacy and are resistant to β-lactamases. Patients should be hospitalized to receive intravenous therapy with imipenem.2 Carbapenems are effective against gram-positive and gram-negative as well as Nocardia species.13,14 Mycetoma that is resistant, severe, or has visceral involvement can be treated with a combination of amikacin and imipenem.15,16 Meropenem is a similar drug that is available as an oral formulation. Both imipenem and meropenem are recommended in cases with bone involvement.17,18 Alternatives for resistant cases include amoxicillin–clavulanic acid 500/125 mg orally 3 times daily for 3 to 6 months or intravenous cefotaxime 1 g every 8 hours plus intramuscular amikacin 500 mg twice daily plus oral levamisole 300 mg once weekly for 4 weeks.19-23
For resistant cases associated with Nocardia species, clindamycin plus quinolones (eg, ciprofloxacin, moxifloxacin, garenoxacin) at a dose of 25 mg/kg once daily for at least 3 months has been suggested in in vivo studies.23
Overall, the cure rate for actinomycetoma treated with any of the prior therapies ranges from 60% to 90%. Treatment must be modified or stopped if there is clinical or laboratory evidence of drug toxicity.13,24 Surgical treatment of actinomycetoma is contraindicated, as it may cause hematogenous dissemination.
Prognosis
Actinomycetomas of a few months’ duration and without bone involvement respond well to therapy. If no therapy is provided or if there is resistance, the functional and cosmetic prognosis is poor, mainly for the feet. There is a risk for spine involvement with mycetoma on the back and posterior head. Thoracic lesions may penetrate into the lungs. The muscular fascia impedes the penetration of abdominal lesions, but the inguinal canals can offer a path for intra-abdominal dissemination.4 Advanced cases lead to a poor general condition of patients, difficulty in using affected extremities, and in extreme cases even death.
The criteria used to guide the discontinuation of initial therapy for any mycetoma include a decrease in the volume of the lesion, closure of fistulae, 3 consecutive negative monthly cultures, imaging studies showing bone regeneration, lack of echoes and cavities on echography, and absence of grains on examination of fine-needle aspirates.11 After the initial treatment protocol is finished, most experts recommend continuing treatment with dapsone 100 to 300 mg once daily for several years to prevent recurrence.12
Prevention
Mycetoma is a disease associated with poverty. It could be prevented by improving living conditions and by regular use of shoes in rural populations.2
Conclusion
Mycetoma is a chronic infection that develops after traumatic inoculation of the skin with either true fungi or aerobic actinomycetes. The resultant infections are known as eumycetoma or actinomycetoma, respectively. The etiologic agents can be found in the so-called grains. Black grains suggest a fungal infection, minute white grains suggest Nocardia, and red grains are due to A pelletieri. Larger white grains or yellow-white grains may be fungal or actinomycotic in origin.
Specific diagnosis requires direct examination, culture, and biopsy. The treatment of choice for actinomycetoma by N brasiliensis is a combination of dapsone 100 to 200 mg once daily and TMP-SMX 80/400 to 160/800 mg once daily for 2 to 3 years. Other effective treatments include aminoglycosides (eg, amikacine, streptomycin) and quinolones. More recently, some other agents have been used such as carbapenems and natural products of Streptomyces cattleya (imipenem), which have wide-spectrum efficacy and are resistant to β-lactamases.
Mycetoma is a subcutaneous disease that can be caused by aerobic bacteria (actinomycetoma) or fungi (eumycetoma). Diagnosis is based on clinical manifestations, including swelling and deformity of affected areas, as well as the presence of granulation tissue, scars, abscesses, sinus tracts, and a purulent exudate that contains the microorganisms.
The worldwide proportion of mycetomas is 60% actinomycetomas and 40% eumycetomas.1 The disease is endemic in tropical, subtropical, and temperate regions, predominating between latitudes 30°N and 15°S. Most cases occur in Africa, especially Sudan, Mauritania, and Senegal; India; Yemen; and Pakistan. In the Americas, the countries with the most reported cases are Mexico and Venezuela.1
Although mycetoma is rare in developed countries, migration of patients from endemic areas makes knowledge of this condition crucial for dermatologists worldwide. We present a review of the current concepts in the epidemiology, clinical presentation, diagnosis, and treatment of actinomycetoma.
Epidemiology
Actinomycetoma is more common in Latin America, with Mexico having the highest incidence. At last count, there were 2631 cases reported in Mexico.2 The majority of cases of mycetoma in Mexico are actinomycetoma (98%), including Nocardia (86%) and Actinomadura madurae (10%). Eumycetoma is rare in Mexico, constituting only 2% of cases.2 Worldwide, men are affected more commonly than women, which is thought to be related to a higher occupational risk during agricultural labor.
Clinical Features
Mycetoma can affect the skin, subcutaneous tissue, bones, and occasionally the internal organs. It is characterized by swelling, deformation of the affected area, and fistulae that drain serosanguineous or purulent exudates.
In Mexico, 60% of cases of mycetoma affect the lower extremities; the feet are the most commonly affected area, followed by the trunk (back and chest), arms, forearms, legs, knees, and thighs.1 Other sites include the hands, shoulders, and abdominal wall. The head and neck area are seldom affected.3 Mycetoma lesions grow and disseminate locally. Bone lesions are possible depending on the osteophilic affinity of the etiological agent and on the interactions between the fungus and the host’s immune system. In severe advanced cases of mycetoma, the lesions may involve tendons and nerves. Dissemination via blood or lymphatics is extremely rare.4
Diagnosis
Diagnosis of actinomycetoma is suspected based on clinical features and confirmed by direct examination of exudates with Lugol iodine or saline solution. On direct microscopy, actinomycetes are recognized by the production of filaments with a width of 0.5 to 1 μm. On hematoxylin and eosin stain, the small grains of Nocardia appear eosinophilic with a blue center and pink filaments. On Gram stain, actinomycetoma grains show positive branching filaments. Culture of grains recovered from aspirated material or biopsy specimens provides specific etiologic diagnosis. Cultures should be held for at least 4 weeks. Additionally, there are some enzymatic, molecular, and serologic tests available for diagnosis.5-7 Serologic diagnosis is available in a few centers in Mexico and can be helpful in some cases for diagnosis or follow-up during treatment. Antibodies can be determined via enzyme-linked immunosorbent assay, Western blot analysis, immunodiffusion, or counterimmunoelectrophoresis.8
The causative agents of actinomycetoma can be isolated in Sabouraud dextrose agar. Deep wedge biopsies (or puncture aspiration) are useful in observing the diagnostic grains, which can be identified adequately with Gram stain. Grains usually are surrounded and/or infiltrated by neutrophils. The size, form, and color of grains can identify the causative agent.1 The granules of Nocardia are small (80–130 mm) and reniform or wormlike, with club structures in their periphery (Figure 1). Actinomadura madurae is characterized by large, white-yellow granules that can be seen with the naked eye (1–3 mm). On microscopic examination with hematoxylin and eosin stain, these grains are purple and exhibit peripheral pink pseudofilaments (Figure 2).2 The grains of Actinomadura pelletieri are large (1–3 mm) and red or violaceous. They fragment or break easily, giving the appearance of a broken dish (Figure 3). Streptomyces somaliensis forms round grains approximately 0.5 to 1 cm in diameter. These grains stain poorly and are extremely hard. Cutting the grains during processing results in striation, giving them the appearance of a potato chip (Figure 4).2
Treatment of Actinomycetoma
Precise identification of the etiologic agent is essential to provide effective treatment of actinomycetoma. Without treatment, or in resistant cases, progressive osseous and visceral involvement is inevitable.9 Actinomycetoma without osseous involvement usually responds well to medical treatment.
The treatment of choice for actinomycetoma involving Nocardia brasiliensis is a combination of dapsone 100 to 200 mg once daily and trimethoprim-sulfamethoxazole (TMP-SMX) 80/400 to 160/800 mg once daily for 2 to 3 years.10 Other treatments have included the following: (1) amikacin 15 mg/kg or 500 mg intramuscularly twice daily for 3 weeks plus dapsone 100 to 200 mg once daily plus TMP-SMX 80/400 to 160/800 mg daily for 2 to 3 years (amikacin, however, is expensive and potentially toxic [nephrotoxicity and ototoxicity] and therefore is used only in resistant cases); (2) dapsone 100 to 200 mg once daily or TMP-SMX 80/400 to 160/800 mg daily for 2 to 3 years plus intramuscular kanamycin 15 mg/kg once daily for 2 weeks at the beginning of treatment, alternating with rest periods to reduce the risk for nephrotoxicity and ototoxicity10; (3) dapsone 1.5 mg/kg orally twice daily plus phosphomycin 500 mg once daily; (4) dapsone 1.5 mg/kg orally twice daily plus streptomycin 1 g once daily (14 mg/kg/d) for 1 month, then the same dose every other day for 1 to 2 months monitoring for ototoxicity; and (5) TMP-SMX 80/400 to 160/800 mg once daily for 2 to 3 years or rifampicin (15–20 mg/kg/d) plus streptomycin 1 g once daily (14 mg/kg/d) for 1 month at the beginning of treatment, then the same dose every other day for 2 to 3 months until a total dose of 60 g is administered, monitoring for ototoxicity.11 Audiometric tests and creatinine levels must be performed every 5 weeks during the treatment to monitor toxicity.10
The best results for infections with A pelletieri, A madurae, and S somaliensis have been with streptomycin (1 g once daily in adults; 20 mg/kg once daily in children) until a total dose of 50 g is reached in combination with TMP-SMX or dapsone12 (Figure 5). Alternatives for A madurae infections include streptomycin plus oral clofazimine (100 mg once daily), oral rifampicin (300 mg twice daily), oral tetracycline (1 g once daily), oral isoniazid (300–600 mg once daily), or oral minocycline (100 mg twice daily; also effective for A pelletieri).
More recently, other drugs have been used such as carbapenems (eg, imipenem, meropenem), which have wide-spectrum efficacy and are resistant to β-lactamases. Patients should be hospitalized to receive intravenous therapy with imipenem.2 Carbapenems are effective against gram-positive and gram-negative as well as Nocardia species.13,14 Mycetoma that is resistant, severe, or has visceral involvement can be treated with a combination of amikacin and imipenem.15,16 Meropenem is a similar drug that is available as an oral formulation. Both imipenem and meropenem are recommended in cases with bone involvement.17,18 Alternatives for resistant cases include amoxicillin–clavulanic acid 500/125 mg orally 3 times daily for 3 to 6 months or intravenous cefotaxime 1 g every 8 hours plus intramuscular amikacin 500 mg twice daily plus oral levamisole 300 mg once weekly for 4 weeks.19-23
For resistant cases associated with Nocardia species, clindamycin plus quinolones (eg, ciprofloxacin, moxifloxacin, garenoxacin) at a dose of 25 mg/kg once daily for at least 3 months has been suggested in in vivo studies.23
Overall, the cure rate for actinomycetoma treated with any of the prior therapies ranges from 60% to 90%. Treatment must be modified or stopped if there is clinical or laboratory evidence of drug toxicity.13,24 Surgical treatment of actinomycetoma is contraindicated, as it may cause hematogenous dissemination.
Prognosis
Actinomycetomas of a few months’ duration and without bone involvement respond well to therapy. If no therapy is provided or if there is resistance, the functional and cosmetic prognosis is poor, mainly for the feet. There is a risk for spine involvement with mycetoma on the back and posterior head. Thoracic lesions may penetrate into the lungs. The muscular fascia impedes the penetration of abdominal lesions, but the inguinal canals can offer a path for intra-abdominal dissemination.4 Advanced cases lead to a poor general condition of patients, difficulty in using affected extremities, and in extreme cases even death.
The criteria used to guide the discontinuation of initial therapy for any mycetoma include a decrease in the volume of the lesion, closure of fistulae, 3 consecutive negative monthly cultures, imaging studies showing bone regeneration, lack of echoes and cavities on echography, and absence of grains on examination of fine-needle aspirates.11 After the initial treatment protocol is finished, most experts recommend continuing treatment with dapsone 100 to 300 mg once daily for several years to prevent recurrence.12
Prevention
Mycetoma is a disease associated with poverty. It could be prevented by improving living conditions and by regular use of shoes in rural populations.2
Conclusion
Mycetoma is a chronic infection that develops after traumatic inoculation of the skin with either true fungi or aerobic actinomycetes. The resultant infections are known as eumycetoma or actinomycetoma, respectively. The etiologic agents can be found in the so-called grains. Black grains suggest a fungal infection, minute white grains suggest Nocardia, and red grains are due to A pelletieri. Larger white grains or yellow-white grains may be fungal or actinomycotic in origin.
Specific diagnosis requires direct examination, culture, and biopsy. The treatment of choice for actinomycetoma by N brasiliensis is a combination of dapsone 100 to 200 mg once daily and TMP-SMX 80/400 to 160/800 mg once daily for 2 to 3 years. Other effective treatments include aminoglycosides (eg, amikacine, streptomycin) and quinolones. More recently, some other agents have been used such as carbapenems and natural products of Streptomyces cattleya (imipenem), which have wide-spectrum efficacy and are resistant to β-lactamases.
- Welsh O, Vera-Cabrera L, Welsh E, et al. Actinomycetoma and advances in its treatment. Clin Dermatol. 2012;30:372-381.
- Arenas R. Micología Medica Ilustrada. 4th ed. Mexico City, Mexico: McGraw-Hill Interamericana; 2011:125-146.
- McGinnis MR. Mycetoma. Dermatol Clin. 1996;14:97-104.
- Fahal AH. Mycetoma: Clinico-pathological Monograph. Khartoum, Sudan: University of Khartoum Press; 2006:20-23, 81-82.
- Estrada-Chavez GE, Vega-Memije ME, Arenas R, et al. Eumycotic mycetoma caused by Madurella mycetomatis successfully treated with antifungals, surgery, and topical negative pressure therapy. Int J Dermatol. 2009;48:401-403.
- Chávez G, Arenas R, Pérez-Polito A, et al. Eumycetic mycetoma due to Madurella mycetomatis. report of six cases. Rev Iberoam Micol. 1998;15:90-93.
- Vasquez del Mercado E, Arenas R, Moreno G. Sequelae and long-term consequences of systemic and subcutaneous mycoses. In: Fratamico PM, Smith JL, Brogden KA, eds. Sequelae and Long-term Consequences of Infectious Diseases. Washington, DC: ASM Press; 2009:415-420.
- Mancini N, Ossi CM, Perotti M, et al. Molecular mycological diagnosis and correct antimycotic treatments. J Clin Microbiol. 2005;43:3584-3585.
- Arenas R, Lavalle P. Micetoma (madura foot). In: Arenas R, Estrada R, eds. Tropical Dermatology. Austin, TX: Landes Bioscience; 2001:51-61.
- Welsh O, Sauceda E, González J, et al. Amikacin alone and in combination with trimethoprim-sulfamethoxazole in the treatment of actinomycotic mycetoma. J Am Acad Dermatol. 1987;17:443-448.
- Fahal AH. Mycetoma: clinico-pathological monograph. In: Fahal AH. Evidence Based Guidelines for the Management of Mycetoma Patients. Khartoum, Sudan: University of Khartoum Press; 2002:5-15.
- Welsh O, Salinas MC, Rodríguez MA. Treatment of eumycetoma and actinomycetoma. Curr Top Med Mycol. 1995;6:47-71.
- Valle ACF, Welsh O, Vera-Cabrera L. Subcutaneous mycoses—mycetoma. In: Tyring SK, Lupi O, Hengge UR, eds. Tropical Dermatology. Philadelphia, PA: Elsevier Churchill Livingstone; 2006:197-200.
- Fuentes A, Arenas R, Reyes M, et al. Actinomicetoma por Nocardia sp. Informe de cinco casos tratados con imipenem solo o combinado con amikacina. Gac Med Mex. 2006;142:247-252.
- Gombert ME, Aulicino TM, DuBouchet L, et al. Therapy of experimental cerebral nocardiosis with imipenem, amikacin, trimethoprim-sulfamethoxazole, and minocylina. Antimicrob Agents Chemother. 1986;30:270-273.
- Calandra GB, Ricci FM, Wang C, et al. Safety and tolerance comparison of imipenem-cilastatin to cephalotin and cefazolin. J Antimicrob Chemother. 1983;12:125-131.
- Ameen M, Arenas R, Vasquez del Mercado E, et al. Efficacy of imipenem therapy for Nocardia actinomycetomas refractory to sulfonamides. J Am Acad Dermatol. 2010;62:239-246.
- Ameen M, Vargas F, Vasquez del Mercado E, et al. Successful treatment of Nocardia actinomycetoma with meropenem and amikacin combination therapy. Int J Dermatol. 2011;50:443-445.
- Ameen M, Arenas R. Emerging therapeutic regimes for the management of mycetomas. Expert Opin Pharmacother. 2008;9:2077-2085.
- Vera-Cabrera L, Daw-Garza A, Said-Fernández S, et al. Therapeutic effect of a novel oxazolidinone, DA-7867 in BALB/c mice infected with Nocardia brasiliensis. PloS Negl Trop Dis. 2008;2:e289.
- Gómez A, Saúl A, Bonifaz A. Amoxicillin and clavulanic acid in the treatment of actinomicetoma. Int J Dermatol. 1993;32:218-220.
- Méndez-Tovar L, Serrano-Jaen L, Almeida-Arvizu VM. Cefotaxima mas amikacina asociadas a inmunomodulación en el tratamiento de actinomicetoma resistente a tratamiento convencional. Gac Med Mex. 1999;135:517-521.
- Chacon-Moreno BE, Welsh O, Cavazos-Rocha N, et al. Efficacy of ciprofloxacin and moxifloxacin against Nocardia brasiliensis in vitro in an experimental model of actinomycetoma in BALB/c mice. Antimicrob Agents Chemother. 2009;53:295-297.
- Welsh O. Treatment of actinomycetoma. Arch Med Res. 1993;24:413-415.
- Welsh O, Vera-Cabrera L, Welsh E, et al. Actinomycetoma and advances in its treatment. Clin Dermatol. 2012;30:372-381.
- Arenas R. Micología Medica Ilustrada. 4th ed. Mexico City, Mexico: McGraw-Hill Interamericana; 2011:125-146.
- McGinnis MR. Mycetoma. Dermatol Clin. 1996;14:97-104.
- Fahal AH. Mycetoma: Clinico-pathological Monograph. Khartoum, Sudan: University of Khartoum Press; 2006:20-23, 81-82.
- Estrada-Chavez GE, Vega-Memije ME, Arenas R, et al. Eumycotic mycetoma caused by Madurella mycetomatis successfully treated with antifungals, surgery, and topical negative pressure therapy. Int J Dermatol. 2009;48:401-403.
- Chávez G, Arenas R, Pérez-Polito A, et al. Eumycetic mycetoma due to Madurella mycetomatis. report of six cases. Rev Iberoam Micol. 1998;15:90-93.
- Vasquez del Mercado E, Arenas R, Moreno G. Sequelae and long-term consequences of systemic and subcutaneous mycoses. In: Fratamico PM, Smith JL, Brogden KA, eds. Sequelae and Long-term Consequences of Infectious Diseases. Washington, DC: ASM Press; 2009:415-420.
- Mancini N, Ossi CM, Perotti M, et al. Molecular mycological diagnosis and correct antimycotic treatments. J Clin Microbiol. 2005;43:3584-3585.
- Arenas R, Lavalle P. Micetoma (madura foot). In: Arenas R, Estrada R, eds. Tropical Dermatology. Austin, TX: Landes Bioscience; 2001:51-61.
- Welsh O, Sauceda E, González J, et al. Amikacin alone and in combination with trimethoprim-sulfamethoxazole in the treatment of actinomycotic mycetoma. J Am Acad Dermatol. 1987;17:443-448.
- Fahal AH. Mycetoma: clinico-pathological monograph. In: Fahal AH. Evidence Based Guidelines for the Management of Mycetoma Patients. Khartoum, Sudan: University of Khartoum Press; 2002:5-15.
- Welsh O, Salinas MC, Rodríguez MA. Treatment of eumycetoma and actinomycetoma. Curr Top Med Mycol. 1995;6:47-71.
- Valle ACF, Welsh O, Vera-Cabrera L. Subcutaneous mycoses—mycetoma. In: Tyring SK, Lupi O, Hengge UR, eds. Tropical Dermatology. Philadelphia, PA: Elsevier Churchill Livingstone; 2006:197-200.
- Fuentes A, Arenas R, Reyes M, et al. Actinomicetoma por Nocardia sp. Informe de cinco casos tratados con imipenem solo o combinado con amikacina. Gac Med Mex. 2006;142:247-252.
- Gombert ME, Aulicino TM, DuBouchet L, et al. Therapy of experimental cerebral nocardiosis with imipenem, amikacin, trimethoprim-sulfamethoxazole, and minocylina. Antimicrob Agents Chemother. 1986;30:270-273.
- Calandra GB, Ricci FM, Wang C, et al. Safety and tolerance comparison of imipenem-cilastatin to cephalotin and cefazolin. J Antimicrob Chemother. 1983;12:125-131.
- Ameen M, Arenas R, Vasquez del Mercado E, et al. Efficacy of imipenem therapy for Nocardia actinomycetomas refractory to sulfonamides. J Am Acad Dermatol. 2010;62:239-246.
- Ameen M, Vargas F, Vasquez del Mercado E, et al. Successful treatment of Nocardia actinomycetoma with meropenem and amikacin combination therapy. Int J Dermatol. 2011;50:443-445.
- Ameen M, Arenas R. Emerging therapeutic regimes for the management of mycetomas. Expert Opin Pharmacother. 2008;9:2077-2085.
- Vera-Cabrera L, Daw-Garza A, Said-Fernández S, et al. Therapeutic effect of a novel oxazolidinone, DA-7867 in BALB/c mice infected with Nocardia brasiliensis. PloS Negl Trop Dis. 2008;2:e289.
- Gómez A, Saúl A, Bonifaz A. Amoxicillin and clavulanic acid in the treatment of actinomicetoma. Int J Dermatol. 1993;32:218-220.
- Méndez-Tovar L, Serrano-Jaen L, Almeida-Arvizu VM. Cefotaxima mas amikacina asociadas a inmunomodulación en el tratamiento de actinomicetoma resistente a tratamiento convencional. Gac Med Mex. 1999;135:517-521.
- Chacon-Moreno BE, Welsh O, Cavazos-Rocha N, et al. Efficacy of ciprofloxacin and moxifloxacin against Nocardia brasiliensis in vitro in an experimental model of actinomycetoma in BALB/c mice. Antimicrob Agents Chemother. 2009;53:295-297.
- Welsh O. Treatment of actinomycetoma. Arch Med Res. 1993;24:413-415.
Practice Points
- Diagnosis of actinomycetoma is based on clinical manifestations including increased swelling and deformity of affected areas, presence of granulation tissue, scars, abscesses, sinus tracts, and a purulent exudate containing microorganisms.
- The feet are the most commonly affected location, followed by the trunk (back and chest), arms, forearms, legs, knees, and thighs.
- Specific diagnosis of actinomycetoma requires clinical examination as well as direct examination of culture and biopsy results.
- Overall, the cure rate for actinomycetoma ranges from 60% to 90%.
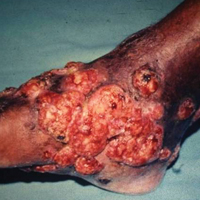
Verrucous Carcinoma of the Buccal Mucosa With Extension to the Cheek
To the Editor:
Verrucous carcinoma is an uncommon type of squamous cell carcinoma (SCC) and was first described by Ackerman1 in 1948. Rock and Fisher2 called this condition oral florid papillomatosis. The distinctive features of this tumor are low-grade malignancy, slow growth, local invasiveness, and rarely intraoral and extraoral metastasis. Extraorally, it can occur in any part of the body,3 a common site being the anogenital region. Depending on the area of occurrence, the condition also is known as Buschke-Lowenstein tumor4 or giant condyloma acuminatum (anogenital region) and carcinoma cuniculatum5 (plantar region). The exact etiology of the condition is unknown, though it is associated with human papillomavirus infection, traumatic scars, chronic infection, tobacco, and chemical carcinogens.3 We report a rare case of verrucous carcinoma originating from the buccal mucosa that subsequently spread to involve the lip and cheek as a large cauliflowerlike growth, which is an unusual presentation.
A 65-year-old man presented to the dermatology department with a painless growth inside the left side of the oral cavity that had developed 5 years prior as a growth on the left buccal mucosa. The lesion gradually increased in size to involve the left oral commissure including the upper and lower lips and the skin of the left cheek; it extended beyond the nasolabial fold in a cauliflowerlike pattern. The lesion was insidious in onset and was not associated with pain, itching, or bleeding. The patient chewed tobacco for the last 40 years, with no similar lesions on any part of the body. On physical examination a warty papilliform lesion was seen on the left buccal mucosa with extension to 2 cm of the upper and lower lip on the left side including the left oral commissure and the skin of the left cheek beyond the nasolabial fold where it appeared as a cauliflowerlike growth measuring 4×5 cm in size (Figure 1). No notable lymphadenopathy was present.
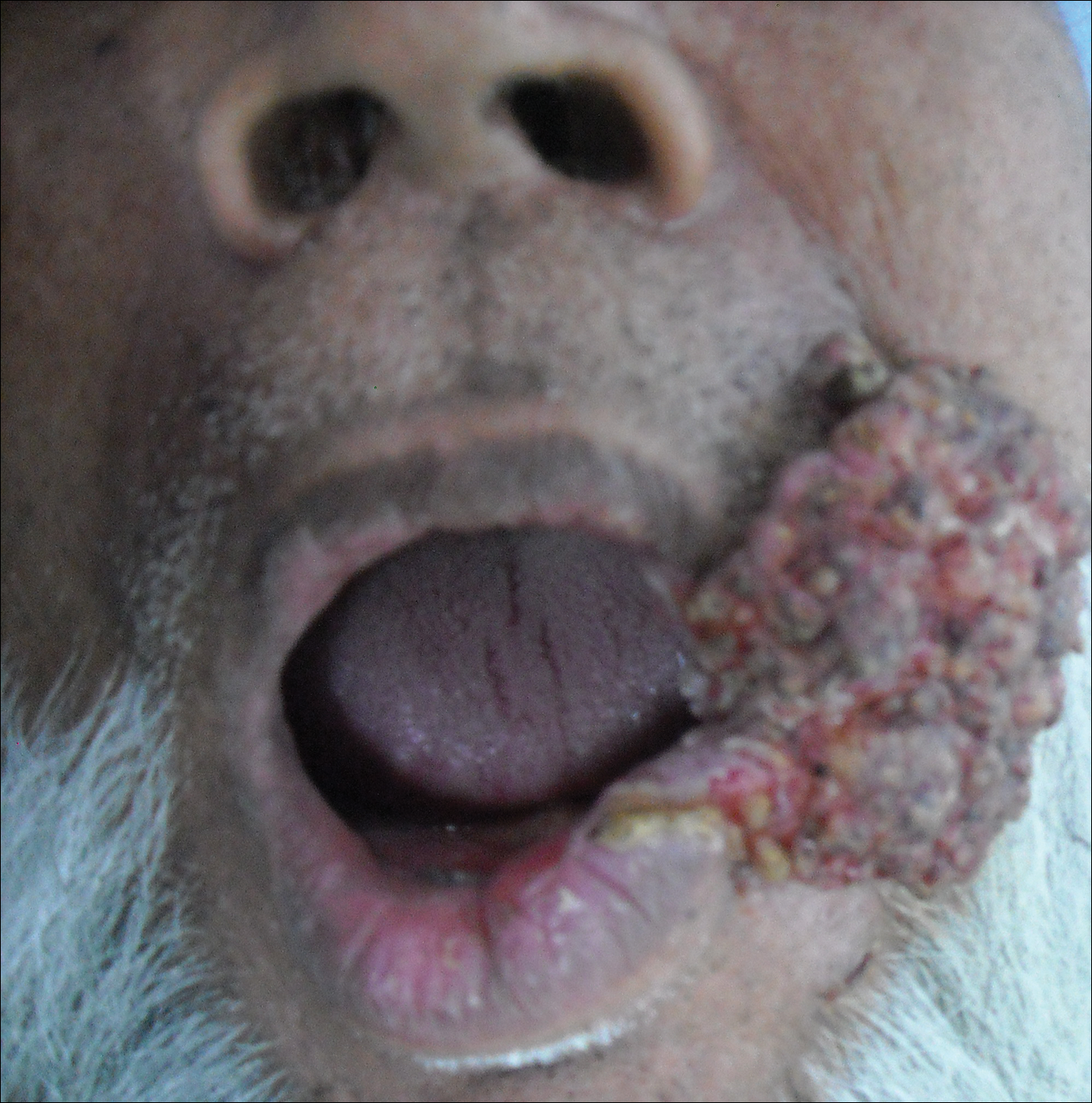
Digital radiographs of the skull (posteroanterior oblique view)(Figure 2) and mandible (left oblique view) showed a lobulated soft-tissue density lesion overlying the left half of the mandible (near the mandibular angle) with involvement of both the upper and lower lips on the left side. However, no obvious underlying bony erosion was noted.
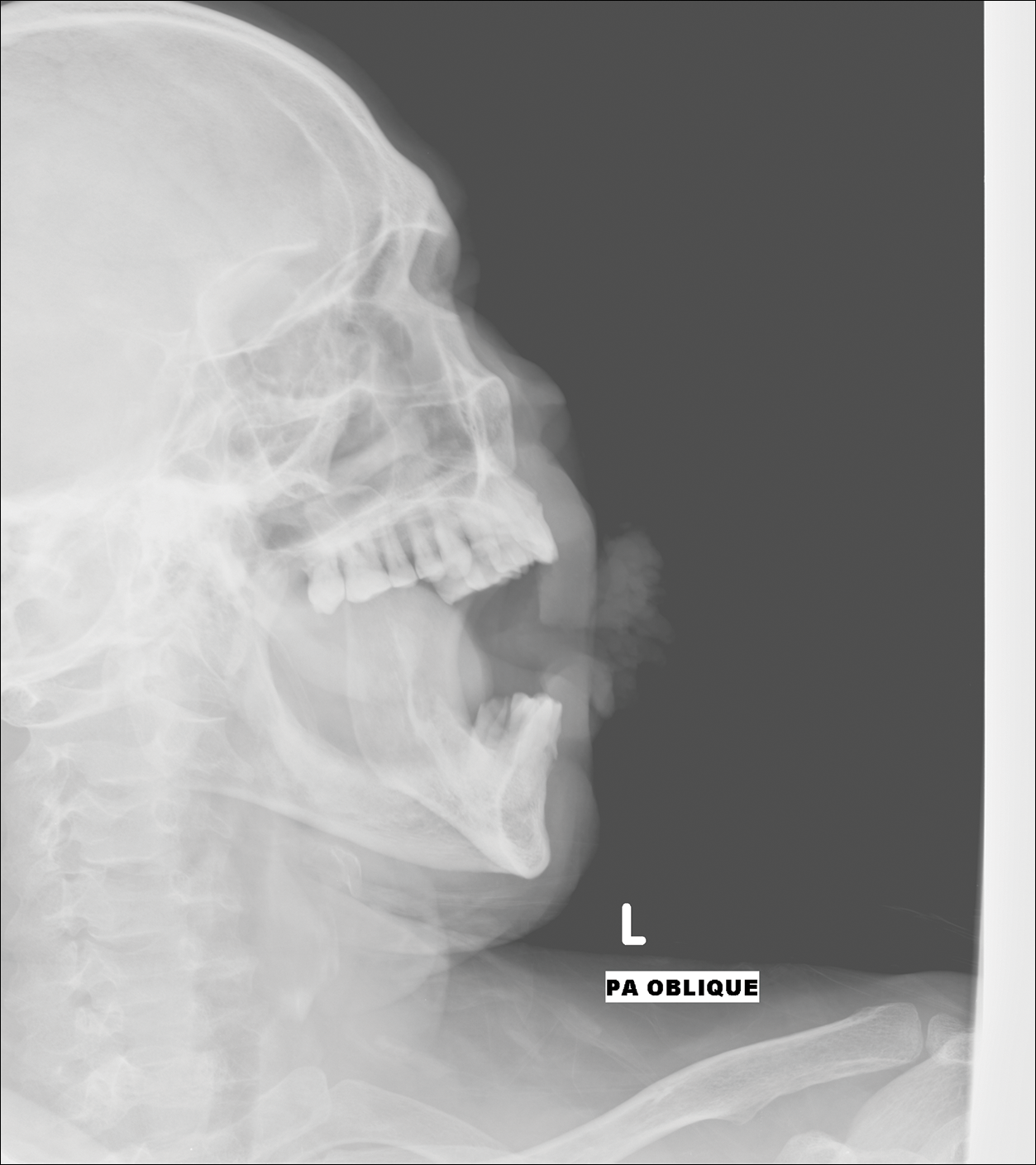
Computed tomography revealed a large soft-tissue mass (41.3×35.3 mm)(Figure 3A) involving the left buccal mucosa with extension into overlying muscle, subcutaneous tissue, and skin. Externally, the lesion was exophytic, irregular, and polypoidal with surface ulceration. Medially, the lesion involved the left oral commissure and parts of the adjoining upper and lower lips. No underlying bony erosion was seen. An enlarged lymph node measuring 20×15 mm was noted in the left upper deep cervical group in the submandibular region (Figure 3B).
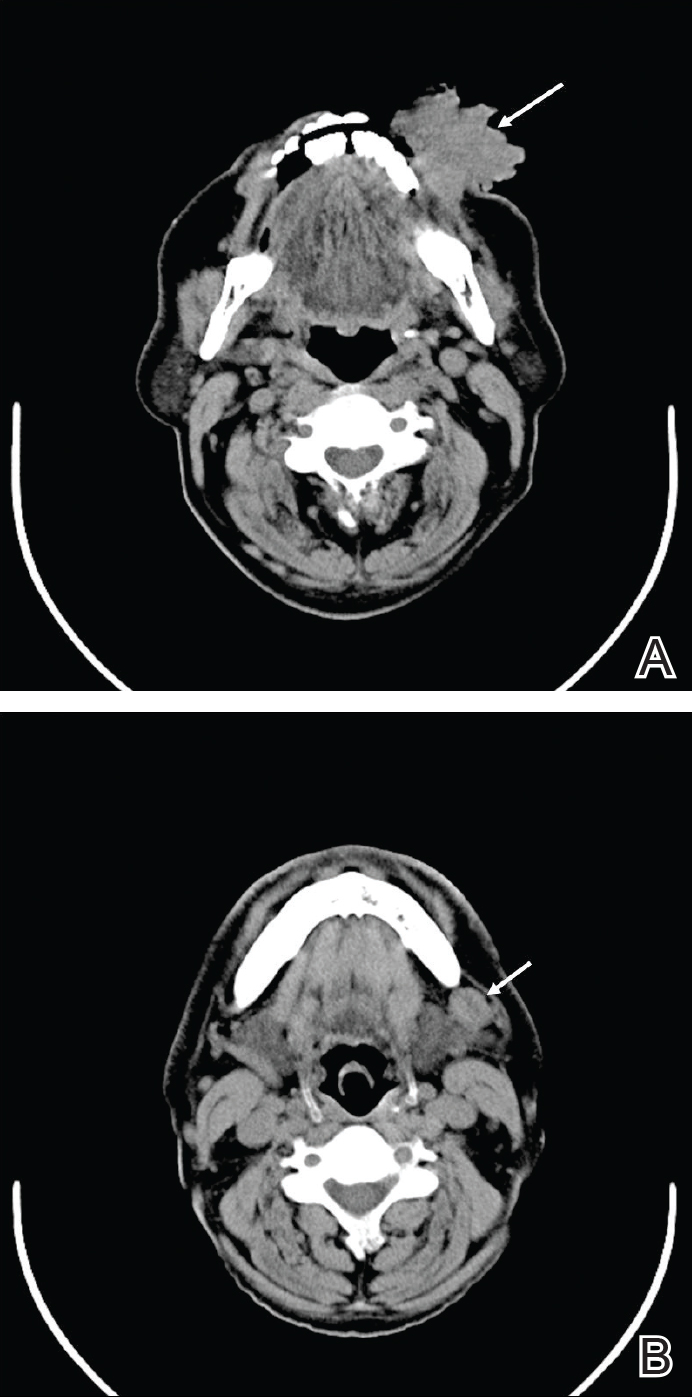
Our clinical differential diagnosis included verrucous carcinoma and hypertrophic variety of lupus vulgaris. A 1×2-cm diagnostic incisional biopsy was performed from the cauliflowerlike growth and ultrasound-guided fine-needle aspiration was done from the lymph node. Histopathology revealed a hyperplastic stratified squamous epithelium with upward extension of verrucous projections, which was largely superficial to the adjacent epithelium (Figure 4A). In addition to the surface verrucous projections, there was lesion extension into the subepithelial zone in the form of round club-shaped protrusions (Figure 4B). There was no loss of polarity in these downward proliferations. No horn pearl formation was present. Fine-needle aspiration revealed reactive lymphadenitis.

The final diagnosis of verrucous carcinoma was made and the patient was referred to the oncosurgery department for further management.
Verrucous carcinoma is a rare, low-grade, well-differentiated SCC of the skin or mucosa presenting with a verrucoid or cauliflowerlike appearance. It shows locally aggressive behavior and has low metastatic potential,6 a low degree of dysplasia, and a good prognosis. Because it is a tumor with predominantly horizontal growth, it tends to erode more than infiltrate. It does not present with remote metastasis.7 It has been known by several different names, usually related to anatomic sites (eg, Ackerman tumor, oral florid papillomatosis, carcinoma cuniculatum).
In the oral cavity, verrucous carcinoma constitutes 2% to 4.5% of all forms of SCC seen mainly in men older than 50 years and also is associated with a high incidence (37.7%) of a second primary tumor mainly in the oral mucosa (eg, tongue, lips, palate, salivary gland).8 Indudharan et al9 reported a case of verrucous carcinoma of the maxillary antrum in a young male patient, which also was a rare entity. Verrucous carcinoma is thought to predominantly affect elderly men. Walvekar et al10 reported a male to female ratio of 3.6 to 1 in patients with verrucous carcinoma, with a mean age of 53.9 years. According to Varshney et al,11 patients may range in age from the fourth to eighth decades of life, with a mean age of 60 years; 80% are male. The etiopathogenesis of verrucous carcinoma is related to the following carcinogens: biologic (eg, human papillomavirus), chemical (eg, smoking), and physical (eg, constant trauma).
Verrucous carcinoma should be considered in the differential diagnosis of slow-growing, locally spreading tumors. Oral tumors, especially in tobacco chewers, should raise suspicion of verrucous carcinoma, which will enable prompt management of the tumor.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Rock JA, Fisher ER. Florid papillomatosis of the oral cavity and larynx. Arch Otolaryngol. 1960;72:593-598.
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2007;56:506-507.
- Buschke A, Lowenstein L. Uber carcinomahnliche condylomata acuminata despenis. Klin Wochenschr. 1925;4:1726-1728.
- Aird I, Johnson HD, Lennox B, et al. Epithelioma cuniculatum: a variety of squamous carcinoma peculiar to the foot. Br J Surg. 1954;42:245-250.
- Schwartz RA. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-21.
- Zanini M, Wulkan C, Paschoal FM, et al. Verrucous carcinoma: a clinical histopathologic variant of squamous cell carcinoma. An Bras Dermatol. 2004;79:619-621.
- Kalsotra P, Manhas M, Sood R. Verrucous carcinoma of hard palate. JK Science. 2000;2:52-54.
- Indudharan R, Das PK, Thida T. Verrucous carcinoma of maxillary antrum. Singapore Med J. 1996;37:559-561.
- Walvekar RR, Chaukar DA, Deshpande MS, et al. Verrucous carcinoma of the oral cavity: a clinical and pathological study of 101 cases. Oral Oncol. 2009;45:47-51.
- Varshney S, Singh J, Saxena RK, et al. Verrucous carcinoma of larynx. Indian J Otolaryngol Head Neck Surg. 2004;56:54-56.
To the Editor:
Verrucous carcinoma is an uncommon type of squamous cell carcinoma (SCC) and was first described by Ackerman1 in 1948. Rock and Fisher2 called this condition oral florid papillomatosis. The distinctive features of this tumor are low-grade malignancy, slow growth, local invasiveness, and rarely intraoral and extraoral metastasis. Extraorally, it can occur in any part of the body,3 a common site being the anogenital region. Depending on the area of occurrence, the condition also is known as Buschke-Lowenstein tumor4 or giant condyloma acuminatum (anogenital region) and carcinoma cuniculatum5 (plantar region). The exact etiology of the condition is unknown, though it is associated with human papillomavirus infection, traumatic scars, chronic infection, tobacco, and chemical carcinogens.3 We report a rare case of verrucous carcinoma originating from the buccal mucosa that subsequently spread to involve the lip and cheek as a large cauliflowerlike growth, which is an unusual presentation.
A 65-year-old man presented to the dermatology department with a painless growth inside the left side of the oral cavity that had developed 5 years prior as a growth on the left buccal mucosa. The lesion gradually increased in size to involve the left oral commissure including the upper and lower lips and the skin of the left cheek; it extended beyond the nasolabial fold in a cauliflowerlike pattern. The lesion was insidious in onset and was not associated with pain, itching, or bleeding. The patient chewed tobacco for the last 40 years, with no similar lesions on any part of the body. On physical examination a warty papilliform lesion was seen on the left buccal mucosa with extension to 2 cm of the upper and lower lip on the left side including the left oral commissure and the skin of the left cheek beyond the nasolabial fold where it appeared as a cauliflowerlike growth measuring 4×5 cm in size (Figure 1). No notable lymphadenopathy was present.
Digital radiographs of the skull (posteroanterior oblique view)(Figure 2) and mandible (left oblique view) showed a lobulated soft-tissue density lesion overlying the left half of the mandible (near the mandibular angle) with involvement of both the upper and lower lips on the left side. However, no obvious underlying bony erosion was noted.
Computed tomography revealed a large soft-tissue mass (41.3×35.3 mm)(Figure 3A) involving the left buccal mucosa with extension into overlying muscle, subcutaneous tissue, and skin. Externally, the lesion was exophytic, irregular, and polypoidal with surface ulceration. Medially, the lesion involved the left oral commissure and parts of the adjoining upper and lower lips. No underlying bony erosion was seen. An enlarged lymph node measuring 20×15 mm was noted in the left upper deep cervical group in the submandibular region (Figure 3B).
Our clinical differential diagnosis included verrucous carcinoma and hypertrophic variety of lupus vulgaris. A 1×2-cm diagnostic incisional biopsy was performed from the cauliflowerlike growth and ultrasound-guided fine-needle aspiration was done from the lymph node. Histopathology revealed a hyperplastic stratified squamous epithelium with upward extension of verrucous projections, which was largely superficial to the adjacent epithelium (Figure 4A). In addition to the surface verrucous projections, there was lesion extension into the subepithelial zone in the form of round club-shaped protrusions (Figure 4B). There was no loss of polarity in these downward proliferations. No horn pearl formation was present. Fine-needle aspiration revealed reactive lymphadenitis.
The final diagnosis of verrucous carcinoma was made and the patient was referred to the oncosurgery department for further management.
Verrucous carcinoma is a rare, low-grade, well-differentiated SCC of the skin or mucosa presenting with a verrucoid or cauliflowerlike appearance. It shows locally aggressive behavior and has low metastatic potential,6 a low degree of dysplasia, and a good prognosis. Because it is a tumor with predominantly horizontal growth, it tends to erode more than infiltrate. It does not present with remote metastasis.7 It has been known by several different names, usually related to anatomic sites (eg, Ackerman tumor, oral florid papillomatosis, carcinoma cuniculatum).
In the oral cavity, verrucous carcinoma constitutes 2% to 4.5% of all forms of SCC seen mainly in men older than 50 years and also is associated with a high incidence (37.7%) of a second primary tumor mainly in the oral mucosa (eg, tongue, lips, palate, salivary gland).8 Indudharan et al9 reported a case of verrucous carcinoma of the maxillary antrum in a young male patient, which also was a rare entity. Verrucous carcinoma is thought to predominantly affect elderly men. Walvekar et al10 reported a male to female ratio of 3.6 to 1 in patients with verrucous carcinoma, with a mean age of 53.9 years. According to Varshney et al,11 patients may range in age from the fourth to eighth decades of life, with a mean age of 60 years; 80% are male. The etiopathogenesis of verrucous carcinoma is related to the following carcinogens: biologic (eg, human papillomavirus), chemical (eg, smoking), and physical (eg, constant trauma).
Verrucous carcinoma should be considered in the differential diagnosis of slow-growing, locally spreading tumors. Oral tumors, especially in tobacco chewers, should raise suspicion of verrucous carcinoma, which will enable prompt management of the tumor.
To the Editor:
Verrucous carcinoma is an uncommon type of squamous cell carcinoma (SCC) and was first described by Ackerman1 in 1948. Rock and Fisher2 called this condition oral florid papillomatosis. The distinctive features of this tumor are low-grade malignancy, slow growth, local invasiveness, and rarely intraoral and extraoral metastasis. Extraorally, it can occur in any part of the body,3 a common site being the anogenital region. Depending on the area of occurrence, the condition also is known as Buschke-Lowenstein tumor4 or giant condyloma acuminatum (anogenital region) and carcinoma cuniculatum5 (plantar region). The exact etiology of the condition is unknown, though it is associated with human papillomavirus infection, traumatic scars, chronic infection, tobacco, and chemical carcinogens.3 We report a rare case of verrucous carcinoma originating from the buccal mucosa that subsequently spread to involve the lip and cheek as a large cauliflowerlike growth, which is an unusual presentation.
A 65-year-old man presented to the dermatology department with a painless growth inside the left side of the oral cavity that had developed 5 years prior as a growth on the left buccal mucosa. The lesion gradually increased in size to involve the left oral commissure including the upper and lower lips and the skin of the left cheek; it extended beyond the nasolabial fold in a cauliflowerlike pattern. The lesion was insidious in onset and was not associated with pain, itching, or bleeding. The patient chewed tobacco for the last 40 years, with no similar lesions on any part of the body. On physical examination a warty papilliform lesion was seen on the left buccal mucosa with extension to 2 cm of the upper and lower lip on the left side including the left oral commissure and the skin of the left cheek beyond the nasolabial fold where it appeared as a cauliflowerlike growth measuring 4×5 cm in size (Figure 1). No notable lymphadenopathy was present.
Digital radiographs of the skull (posteroanterior oblique view)(Figure 2) and mandible (left oblique view) showed a lobulated soft-tissue density lesion overlying the left half of the mandible (near the mandibular angle) with involvement of both the upper and lower lips on the left side. However, no obvious underlying bony erosion was noted.
Computed tomography revealed a large soft-tissue mass (41.3×35.3 mm)(Figure 3A) involving the left buccal mucosa with extension into overlying muscle, subcutaneous tissue, and skin. Externally, the lesion was exophytic, irregular, and polypoidal with surface ulceration. Medially, the lesion involved the left oral commissure and parts of the adjoining upper and lower lips. No underlying bony erosion was seen. An enlarged lymph node measuring 20×15 mm was noted in the left upper deep cervical group in the submandibular region (Figure 3B).
Our clinical differential diagnosis included verrucous carcinoma and hypertrophic variety of lupus vulgaris. A 1×2-cm diagnostic incisional biopsy was performed from the cauliflowerlike growth and ultrasound-guided fine-needle aspiration was done from the lymph node. Histopathology revealed a hyperplastic stratified squamous epithelium with upward extension of verrucous projections, which was largely superficial to the adjacent epithelium (Figure 4A). In addition to the surface verrucous projections, there was lesion extension into the subepithelial zone in the form of round club-shaped protrusions (Figure 4B). There was no loss of polarity in these downward proliferations. No horn pearl formation was present. Fine-needle aspiration revealed reactive lymphadenitis.
The final diagnosis of verrucous carcinoma was made and the patient was referred to the oncosurgery department for further management.
Verrucous carcinoma is a rare, low-grade, well-differentiated SCC of the skin or mucosa presenting with a verrucoid or cauliflowerlike appearance. It shows locally aggressive behavior and has low metastatic potential,6 a low degree of dysplasia, and a good prognosis. Because it is a tumor with predominantly horizontal growth, it tends to erode more than infiltrate. It does not present with remote metastasis.7 It has been known by several different names, usually related to anatomic sites (eg, Ackerman tumor, oral florid papillomatosis, carcinoma cuniculatum).
In the oral cavity, verrucous carcinoma constitutes 2% to 4.5% of all forms of SCC seen mainly in men older than 50 years and also is associated with a high incidence (37.7%) of a second primary tumor mainly in the oral mucosa (eg, tongue, lips, palate, salivary gland).8 Indudharan et al9 reported a case of verrucous carcinoma of the maxillary antrum in a young male patient, which also was a rare entity. Verrucous carcinoma is thought to predominantly affect elderly men. Walvekar et al10 reported a male to female ratio of 3.6 to 1 in patients with verrucous carcinoma, with a mean age of 53.9 years. According to Varshney et al,11 patients may range in age from the fourth to eighth decades of life, with a mean age of 60 years; 80% are male. The etiopathogenesis of verrucous carcinoma is related to the following carcinogens: biologic (eg, human papillomavirus), chemical (eg, smoking), and physical (eg, constant trauma).
Verrucous carcinoma should be considered in the differential diagnosis of slow-growing, locally spreading tumors. Oral tumors, especially in tobacco chewers, should raise suspicion of verrucous carcinoma, which will enable prompt management of the tumor.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Rock JA, Fisher ER. Florid papillomatosis of the oral cavity and larynx. Arch Otolaryngol. 1960;72:593-598.
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2007;56:506-507.
- Buschke A, Lowenstein L. Uber carcinomahnliche condylomata acuminata despenis. Klin Wochenschr. 1925;4:1726-1728.
- Aird I, Johnson HD, Lennox B, et al. Epithelioma cuniculatum: a variety of squamous carcinoma peculiar to the foot. Br J Surg. 1954;42:245-250.
- Schwartz RA. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-21.
- Zanini M, Wulkan C, Paschoal FM, et al. Verrucous carcinoma: a clinical histopathologic variant of squamous cell carcinoma. An Bras Dermatol. 2004;79:619-621.
- Kalsotra P, Manhas M, Sood R. Verrucous carcinoma of hard palate. JK Science. 2000;2:52-54.
- Indudharan R, Das PK, Thida T. Verrucous carcinoma of maxillary antrum. Singapore Med J. 1996;37:559-561.
- Walvekar RR, Chaukar DA, Deshpande MS, et al. Verrucous carcinoma of the oral cavity: a clinical and pathological study of 101 cases. Oral Oncol. 2009;45:47-51.
- Varshney S, Singh J, Saxena RK, et al. Verrucous carcinoma of larynx. Indian J Otolaryngol Head Neck Surg. 2004;56:54-56.
- Ackerman LV. Verrucous carcinoma of the oral cavity. Surgery. 1948;23:670-678.
- Rock JA, Fisher ER. Florid papillomatosis of the oral cavity and larynx. Arch Otolaryngol. 1960;72:593-598.
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2007;56:506-507.
- Buschke A, Lowenstein L. Uber carcinomahnliche condylomata acuminata despenis. Klin Wochenschr. 1925;4:1726-1728.
- Aird I, Johnson HD, Lennox B, et al. Epithelioma cuniculatum: a variety of squamous carcinoma peculiar to the foot. Br J Surg. 1954;42:245-250.
- Schwartz RA. Verrucous carcinoma of the skin and mucosa. J Am Acad Dermatol. 1995;32:1-21.
- Zanini M, Wulkan C, Paschoal FM, et al. Verrucous carcinoma: a clinical histopathologic variant of squamous cell carcinoma. An Bras Dermatol. 2004;79:619-621.
- Kalsotra P, Manhas M, Sood R. Verrucous carcinoma of hard palate. JK Science. 2000;2:52-54.
- Indudharan R, Das PK, Thida T. Verrucous carcinoma of maxillary antrum. Singapore Med J. 1996;37:559-561.
- Walvekar RR, Chaukar DA, Deshpande MS, et al. Verrucous carcinoma of the oral cavity: a clinical and pathological study of 101 cases. Oral Oncol. 2009;45:47-51.
- Varshney S, Singh J, Saxena RK, et al. Verrucous carcinoma of larynx. Indian J Otolaryngol Head Neck Surg. 2004;56:54-56.
Practice Points
- Verrucous carcinoma is a slow-growing tumor that often presents in advanced clinical stages because it is poorly understood and underrecognized, especially in developing countries.
- Good clinicopathological correlation is required in cases of verrucous carcinoma to avoid misdiagnosis and provide appropriate treatment.
- Case-specific management should be considered, as presentation of verrucous carcinoma varies.
- Radiography should be considered to assess for lymph node involvement.
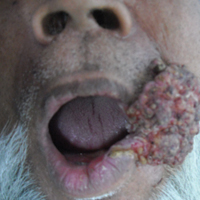
Widespread Poikilodermatous Dermatomyositis Associated With Chronic Lymphocytic Leukemia
To the Editor:
Dermatomyositis represents a rare idiopathic inflammatory process presenting with cutaneous lesions and muscular weakness. It often represents a paraneoplastic syndrome. We report the case of a 62-year-old man with a history of total-body poikiloderma and a recent diagnosis of chronic lymphocytic leukemia (CLL). Despite lacking typical features of the disease, a diagnosis of dermatomyositis was made. Our patient may represent a distinct poikilodermatous variant of dermatomyositis, sharing the generalized distribution of the erythrodermic subtype.
A 62-year-old man presented with pruritic poikiloderma involving the neck, arms, legs, abdomen, chest, and back of 2 years’ duration (Figure). He also experienced dysphagia and weakness of the legs. The rash was previously treated by other dermatologists with a combination of high-potency topical steroids and topical tacrolimus 0.1% without success. His history was notable for CLL, which had been diagnosed by a dermatologist 6 months prior to the current presentation. Prior to his visit to the dermatologist, the patient had received 6 chemotherapeutic sessions with a combination of rituximab and cyclophosphamide for the treatment of CLL. The rash did not improve with chemotherapy.

Repeat biopsies of affected regions only demonstrated features of mild interface dermatitis. Direct immunofluorescence studies showed scattered colloid body fluorescence for IgM. Because of bilateral weakness of the legs, a muscle biopsy was taken, which demonstrated severe atrophy and interstitial fibrosis, with neurogenic abnormalities detected in areas of lesser atrophy via abnormal muscle fiber–type grouping. Metabolic panel showed elevated muscle enzymes in the blood: creatine kinase, 243 U/L (reference range, 10–225 U/L); serum aldolase, 16 U/L (reference range, ≤8.1 U/L); lactate dehydrogenase, 314 U/L (reference range, 60–200 U/L). An autoimmune panel was negative for Jo-1, Scl-70, U1 ribonucleoprotein, DNA, desmoglein 1 and 3, and antiacetylcholine receptor antibodies. An elevated erythrocyte sedimentation rate was measured at 16 mm/h (reference range, 0–10 mm/h). Given these findings, the lesions were confirmed as a widespread poikilodermatous variant of dermatomyositis.
The patient was placed on a daily 50-mg dose of prednisone, which produced rapid improvement in scaling and erythema. Creatine kinase and serum aldolase levels normalized and motor strength increased. After 1 week the prednisone dosage was reduced to a daily 30-mg dose, and then 20 mg a week later. The skin lesions completely resolved within 4 to 5 months and the patient is currently on a prednisone dose of 5 mg, alternating with 2.5 mg of prednisone and rituximab infusion every 2 months.
Dermatomyositis is a rare entity with an incidence of approximately 0.5 to 1 per 100,000 individuals.1 It presents with a characteristic rash composed of Gottron papules; pathognomonic flat violaceous papules on the dorsal interphalangeal joints, elbows, or knees; and a heliotrope rash, a violaceous erythema involving the eyelids. Poikiloderma frequently is reported to present in a shawl-like distribution, encompassing the shoulders, arms, and upper back.1,2 Dermatomyositis of the poikilodermatous type can present in nonphotoexposed areas and photoexposed areas. The unusual feature is the total-body involvement, which is analogous to erythroderma.3
Our case may represent a distinct poikilodermatous manifestation sharing the distribution of the erythrodermic subtype. We believe that the skin lesions may have represented a paraneoplastic event presenting prior to diagnosis with CLL. Dermatomyositis has a strong association with cancer, with patients 3 times more likely to develop internal malignancy.4 Association is strongest for non-Hodgkin lymphoma, as well as ovarian, lung, colorectal, pancreatic, and gastric cancer. When associated with malignancy, symptoms of dermatomyositis or myositis typically precede the discovery of malignancy by an average of 1.9 years.5 Dermatomyositis has been previously reported to present as a paraneoplastic manifestation of CLL.6 One case has been reported of a patient with CLL who developed leukemia cutis presenting with poikiloderma in the characteristic dermatomyositis shawl-like distribution.7 The lack of dermal infiltration with leukemic cells in our patient, however, makes a paraneoplastic etiology much more likely.
Our patient’s rash did not initially improve with treatment of CLL, but dermatomyositis associated with hematological malignancy may precede, occur simultaneously, or follow the diagnosis of malignancy.8 Additionally, symptoms of dermatomyositis do not always parallel the course of hematological malignancy outcome. However, rituximab has been used as a treatment of dermatomyositis and may have contributed some synergistic effect in combination with prednisone in our patient.9
- Dourmishev LA, Dourmishev AL, Schwartz RA. Dermatomyositis: cutaneous manifestations of its variants. Int J Dermatol. 2002;41:625-630.
- Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-920; quiz 921-992.
- Liu ZH, Wang XD. Acute-onset adult dermatomyositis presenting with erythroderma and diplopia. Clin Exp Dermatol. 2007;32:751-752.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Bohan A, Peter JB, Bowman RL, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
- Ishida T, Aikawa K, Tamura T, et al. Chronic lymphocytic leukemia associated with nephrotic syndrome and dermatomyositis. Intern Med. 1995;34:15-17.
- Nousari HC, Kimyai-Asadi A, Huang CH, et al. T-cell chronic lymphocytic leukemia mimicking dermatomyositis. Int J Dermatol. 2000;39:144-146.
- Marie I, Guillevin L, Menard JF, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012;11:615-620.
- Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601-607.
To the Editor:
Dermatomyositis represents a rare idiopathic inflammatory process presenting with cutaneous lesions and muscular weakness. It often represents a paraneoplastic syndrome. We report the case of a 62-year-old man with a history of total-body poikiloderma and a recent diagnosis of chronic lymphocytic leukemia (CLL). Despite lacking typical features of the disease, a diagnosis of dermatomyositis was made. Our patient may represent a distinct poikilodermatous variant of dermatomyositis, sharing the generalized distribution of the erythrodermic subtype.
A 62-year-old man presented with pruritic poikiloderma involving the neck, arms, legs, abdomen, chest, and back of 2 years’ duration (Figure). He also experienced dysphagia and weakness of the legs. The rash was previously treated by other dermatologists with a combination of high-potency topical steroids and topical tacrolimus 0.1% without success. His history was notable for CLL, which had been diagnosed by a dermatologist 6 months prior to the current presentation. Prior to his visit to the dermatologist, the patient had received 6 chemotherapeutic sessions with a combination of rituximab and cyclophosphamide for the treatment of CLL. The rash did not improve with chemotherapy.
Repeat biopsies of affected regions only demonstrated features of mild interface dermatitis. Direct immunofluorescence studies showed scattered colloid body fluorescence for IgM. Because of bilateral weakness of the legs, a muscle biopsy was taken, which demonstrated severe atrophy and interstitial fibrosis, with neurogenic abnormalities detected in areas of lesser atrophy via abnormal muscle fiber–type grouping. Metabolic panel showed elevated muscle enzymes in the blood: creatine kinase, 243 U/L (reference range, 10–225 U/L); serum aldolase, 16 U/L (reference range, ≤8.1 U/L); lactate dehydrogenase, 314 U/L (reference range, 60–200 U/L). An autoimmune panel was negative for Jo-1, Scl-70, U1 ribonucleoprotein, DNA, desmoglein 1 and 3, and antiacetylcholine receptor antibodies. An elevated erythrocyte sedimentation rate was measured at 16 mm/h (reference range, 0–10 mm/h). Given these findings, the lesions were confirmed as a widespread poikilodermatous variant of dermatomyositis.
The patient was placed on a daily 50-mg dose of prednisone, which produced rapid improvement in scaling and erythema. Creatine kinase and serum aldolase levels normalized and motor strength increased. After 1 week the prednisone dosage was reduced to a daily 30-mg dose, and then 20 mg a week later. The skin lesions completely resolved within 4 to 5 months and the patient is currently on a prednisone dose of 5 mg, alternating with 2.5 mg of prednisone and rituximab infusion every 2 months.
Dermatomyositis is a rare entity with an incidence of approximately 0.5 to 1 per 100,000 individuals.1 It presents with a characteristic rash composed of Gottron papules; pathognomonic flat violaceous papules on the dorsal interphalangeal joints, elbows, or knees; and a heliotrope rash, a violaceous erythema involving the eyelids. Poikiloderma frequently is reported to present in a shawl-like distribution, encompassing the shoulders, arms, and upper back.1,2 Dermatomyositis of the poikilodermatous type can present in nonphotoexposed areas and photoexposed areas. The unusual feature is the total-body involvement, which is analogous to erythroderma.3
Our case may represent a distinct poikilodermatous manifestation sharing the distribution of the erythrodermic subtype. We believe that the skin lesions may have represented a paraneoplastic event presenting prior to diagnosis with CLL. Dermatomyositis has a strong association with cancer, with patients 3 times more likely to develop internal malignancy.4 Association is strongest for non-Hodgkin lymphoma, as well as ovarian, lung, colorectal, pancreatic, and gastric cancer. When associated with malignancy, symptoms of dermatomyositis or myositis typically precede the discovery of malignancy by an average of 1.9 years.5 Dermatomyositis has been previously reported to present as a paraneoplastic manifestation of CLL.6 One case has been reported of a patient with CLL who developed leukemia cutis presenting with poikiloderma in the characteristic dermatomyositis shawl-like distribution.7 The lack of dermal infiltration with leukemic cells in our patient, however, makes a paraneoplastic etiology much more likely.
Our patient’s rash did not initially improve with treatment of CLL, but dermatomyositis associated with hematological malignancy may precede, occur simultaneously, or follow the diagnosis of malignancy.8 Additionally, symptoms of dermatomyositis do not always parallel the course of hematological malignancy outcome. However, rituximab has been used as a treatment of dermatomyositis and may have contributed some synergistic effect in combination with prednisone in our patient.9
To the Editor:
Dermatomyositis represents a rare idiopathic inflammatory process presenting with cutaneous lesions and muscular weakness. It often represents a paraneoplastic syndrome. We report the case of a 62-year-old man with a history of total-body poikiloderma and a recent diagnosis of chronic lymphocytic leukemia (CLL). Despite lacking typical features of the disease, a diagnosis of dermatomyositis was made. Our patient may represent a distinct poikilodermatous variant of dermatomyositis, sharing the generalized distribution of the erythrodermic subtype.
A 62-year-old man presented with pruritic poikiloderma involving the neck, arms, legs, abdomen, chest, and back of 2 years’ duration (Figure). He also experienced dysphagia and weakness of the legs. The rash was previously treated by other dermatologists with a combination of high-potency topical steroids and topical tacrolimus 0.1% without success. His history was notable for CLL, which had been diagnosed by a dermatologist 6 months prior to the current presentation. Prior to his visit to the dermatologist, the patient had received 6 chemotherapeutic sessions with a combination of rituximab and cyclophosphamide for the treatment of CLL. The rash did not improve with chemotherapy.
Repeat biopsies of affected regions only demonstrated features of mild interface dermatitis. Direct immunofluorescence studies showed scattered colloid body fluorescence for IgM. Because of bilateral weakness of the legs, a muscle biopsy was taken, which demonstrated severe atrophy and interstitial fibrosis, with neurogenic abnormalities detected in areas of lesser atrophy via abnormal muscle fiber–type grouping. Metabolic panel showed elevated muscle enzymes in the blood: creatine kinase, 243 U/L (reference range, 10–225 U/L); serum aldolase, 16 U/L (reference range, ≤8.1 U/L); lactate dehydrogenase, 314 U/L (reference range, 60–200 U/L). An autoimmune panel was negative for Jo-1, Scl-70, U1 ribonucleoprotein, DNA, desmoglein 1 and 3, and antiacetylcholine receptor antibodies. An elevated erythrocyte sedimentation rate was measured at 16 mm/h (reference range, 0–10 mm/h). Given these findings, the lesions were confirmed as a widespread poikilodermatous variant of dermatomyositis.
The patient was placed on a daily 50-mg dose of prednisone, which produced rapid improvement in scaling and erythema. Creatine kinase and serum aldolase levels normalized and motor strength increased. After 1 week the prednisone dosage was reduced to a daily 30-mg dose, and then 20 mg a week later. The skin lesions completely resolved within 4 to 5 months and the patient is currently on a prednisone dose of 5 mg, alternating with 2.5 mg of prednisone and rituximab infusion every 2 months.
Dermatomyositis is a rare entity with an incidence of approximately 0.5 to 1 per 100,000 individuals.1 It presents with a characteristic rash composed of Gottron papules; pathognomonic flat violaceous papules on the dorsal interphalangeal joints, elbows, or knees; and a heliotrope rash, a violaceous erythema involving the eyelids. Poikiloderma frequently is reported to present in a shawl-like distribution, encompassing the shoulders, arms, and upper back.1,2 Dermatomyositis of the poikilodermatous type can present in nonphotoexposed areas and photoexposed areas. The unusual feature is the total-body involvement, which is analogous to erythroderma.3
Our case may represent a distinct poikilodermatous manifestation sharing the distribution of the erythrodermic subtype. We believe that the skin lesions may have represented a paraneoplastic event presenting prior to diagnosis with CLL. Dermatomyositis has a strong association with cancer, with patients 3 times more likely to develop internal malignancy.4 Association is strongest for non-Hodgkin lymphoma, as well as ovarian, lung, colorectal, pancreatic, and gastric cancer. When associated with malignancy, symptoms of dermatomyositis or myositis typically precede the discovery of malignancy by an average of 1.9 years.5 Dermatomyositis has been previously reported to present as a paraneoplastic manifestation of CLL.6 One case has been reported of a patient with CLL who developed leukemia cutis presenting with poikiloderma in the characteristic dermatomyositis shawl-like distribution.7 The lack of dermal infiltration with leukemic cells in our patient, however, makes a paraneoplastic etiology much more likely.
Our patient’s rash did not initially improve with treatment of CLL, but dermatomyositis associated with hematological malignancy may precede, occur simultaneously, or follow the diagnosis of malignancy.8 Additionally, symptoms of dermatomyositis do not always parallel the course of hematological malignancy outcome. However, rituximab has been used as a treatment of dermatomyositis and may have contributed some synergistic effect in combination with prednisone in our patient.9
- Dourmishev LA, Dourmishev AL, Schwartz RA. Dermatomyositis: cutaneous manifestations of its variants. Int J Dermatol. 2002;41:625-630.
- Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-920; quiz 921-992.
- Liu ZH, Wang XD. Acute-onset adult dermatomyositis presenting with erythroderma and diplopia. Clin Exp Dermatol. 2007;32:751-752.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Bohan A, Peter JB, Bowman RL, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
- Ishida T, Aikawa K, Tamura T, et al. Chronic lymphocytic leukemia associated with nephrotic syndrome and dermatomyositis. Intern Med. 1995;34:15-17.
- Nousari HC, Kimyai-Asadi A, Huang CH, et al. T-cell chronic lymphocytic leukemia mimicking dermatomyositis. Int J Dermatol. 2000;39:144-146.
- Marie I, Guillevin L, Menard JF, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012;11:615-620.
- Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601-607.
- Dourmishev LA, Dourmishev AL, Schwartz RA. Dermatomyositis: cutaneous manifestations of its variants. Int J Dermatol. 2002;41:625-630.
- Kovacs SO, Kovacs SC. Dermatomyositis. J Am Acad Dermatol. 1998;39:899-920; quiz 921-992.
- Liu ZH, Wang XD. Acute-onset adult dermatomyositis presenting with erythroderma and diplopia. Clin Exp Dermatol. 2007;32:751-752.
- Hill CL, Zhang Y, Sigurgeirsson B, et al. Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet. 2001;357:96-100.
- Bohan A, Peter JB, Bowman RL, et al. Computer-assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore). 1977;56:255-286.
- Ishida T, Aikawa K, Tamura T, et al. Chronic lymphocytic leukemia associated with nephrotic syndrome and dermatomyositis. Intern Med. 1995;34:15-17.
- Nousari HC, Kimyai-Asadi A, Huang CH, et al. T-cell chronic lymphocytic leukemia mimicking dermatomyositis. Int J Dermatol. 2000;39:144-146.
- Marie I, Guillevin L, Menard JF, et al. Hematological malignancy associated with polymyositis and dermatomyositis. Autoimmun Rev. 2012;11:615-620.
- Levine TD. Rituximab in the treatment of dermatomyositis: an open-label pilot study. Arthritis Rheum. 2005;52:601-607.
Practice Points
- Poikiloderma, even with an unusual clinical presentation, can be a useful clinical clue for the diagnosis of dermatomyositis or other collagen vascular disease.
- Dermatomyositis can be paraneoplastic and though often associated with epithelial malignancies and solid tumors can also be associated with leukemias.