User login
Individuals with DS are at significantly increased risk of developing Alzheimer’s disease (AD) because of the overexpression of the amyloid precursor protein (APP) gene on chromosome 21.
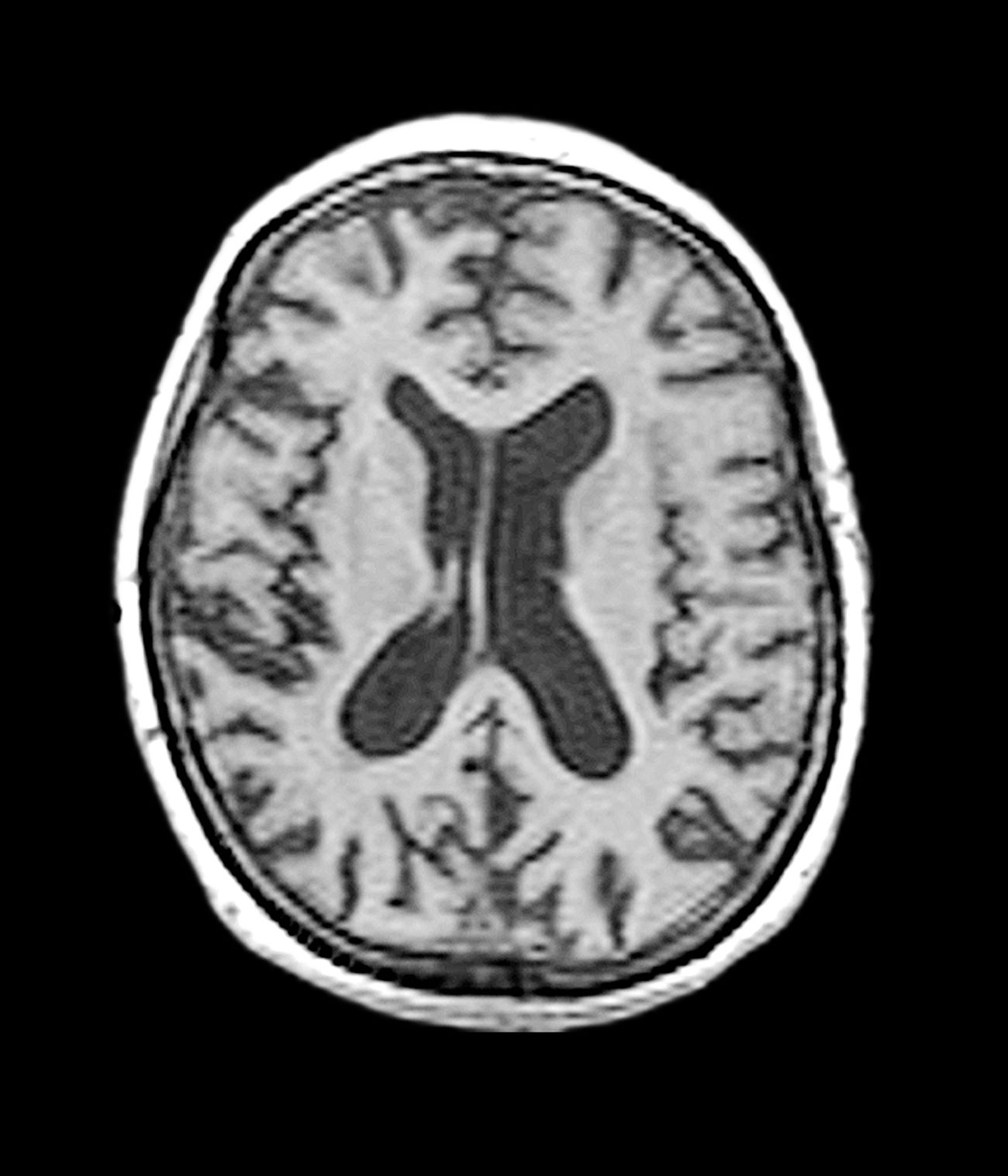
The patient exhibits hallmark symptoms of AD, including progressive memory loss, disorientation, difficulty performing daily tasks, and behavioral changes such as irritability and social withdrawal. The MRI findings of ventricular enlargement and cortical atrophy are consistent with brain changes commonly seen in AD, particularly in individuals with DS who often develop these changes earlier in life (typically in their thirties or forties).
Frontotemporal dementia primarily causes behavioral and language changes with relative memory sparing early on, making it inconsistent with this patient's prominent memory loss, disorientation, and generalized cortical atrophy — features more typical of AD.
Vascular dementia often presents with stepwise decline and focal neurologic deficits; it is unlikely in this patient, given the absence of cerebrovascular events or risk factors like hypertension or diabetes.
While normal pressure hydrocephalus can cause ventricular enlargement, its classic triad of gait disturbance, urinary incontinence, and dementia is incomplete here, making this answer unlikely.
DS is the most common genetic cause of intellectual disability, occurring in approximately 1 in 700 live births worldwide. Nearly all adults with DS develop neuropathologic changes associated with AD by age 40, and the lifetime risk of developing dementia exceeds 90%; by the age of 55-60, at least 70% of individuals with DS exhibit clinical signs of dementia.
The link between DS and neurodegeneration is largely attributed to the triplication of chromosome 21, which includes the APP gene. Overexpression of APP leads to excessive production and accumulation of amyloid-β (Aβ), which forms the hallmark plaques seen in AD. In DS, amyloid plaques begin to form as early as the teenage years, and by the fourth decade, neurofibrillary tangles (tau protein aggregates) and widespread neurodegeneration are nearly universal.
Initial symptoms of AD often include memory impairment, particularly short-term memory deficits, and executive dysfunction, difficulty with planning and problem-solving, and visuospatial deficits. Behavioral and personality changes, such as irritability, withdrawal, or apathy, are also commonly reported. Compared with sporadic AD, individuals with DS often show earlier behavioral symptoms, including impulsivity and changes in social interactions, which may precede noticeable memory deficits. Additionally, late-onset myoclonic epilepsy is common in individuals with DS and dementia, further complicating diagnosis and care.
The diagnosis of dementia in DS is challenging because of baseline intellectual disability, which makes it difficult to assess cognitive decline using standard neuropsychological tests. However, a combination of clinical history, caregiver reports, neuroimaging, and biomarker analysis can aid in early detection. Structural MRI of the brain often reveals ventricular enlargement and cortical atrophy, while PET imaging can detect early amyloid and tau accumulation.
Because AD is now the leading cause of death in individuals with DS, the lack of effective disease-modifying treatments highlights an important need for clinical trials focused on this population. Current research aims to explore the role of anti-amyloid monoclonal antibodies, neuroprotective agents, and lifestyle interventions to delay or prevent neurodegeneration.
Shaheen E. Lakhan, MD, PhD, MS, MEd, Chief of Pain Management, Carilion Clinic and Virginia Tech Carilion School of Medicine, Roanoke, Virginia.
Disclosure: Shaheen E. Lakhan, MD, PhD, MS, MEd, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
Individuals with DS are at significantly increased risk of developing Alzheimer’s disease (AD) because of the overexpression of the amyloid precursor protein (APP) gene on chromosome 21.
The patient exhibits hallmark symptoms of AD, including progressive memory loss, disorientation, difficulty performing daily tasks, and behavioral changes such as irritability and social withdrawal. The MRI findings of ventricular enlargement and cortical atrophy are consistent with brain changes commonly seen in AD, particularly in individuals with DS who often develop these changes earlier in life (typically in their thirties or forties).
Frontotemporal dementia primarily causes behavioral and language changes with relative memory sparing early on, making it inconsistent with this patient's prominent memory loss, disorientation, and generalized cortical atrophy — features more typical of AD.
Vascular dementia often presents with stepwise decline and focal neurologic deficits; it is unlikely in this patient, given the absence of cerebrovascular events or risk factors like hypertension or diabetes.
While normal pressure hydrocephalus can cause ventricular enlargement, its classic triad of gait disturbance, urinary incontinence, and dementia is incomplete here, making this answer unlikely.
DS is the most common genetic cause of intellectual disability, occurring in approximately 1 in 700 live births worldwide. Nearly all adults with DS develop neuropathologic changes associated with AD by age 40, and the lifetime risk of developing dementia exceeds 90%; by the age of 55-60, at least 70% of individuals with DS exhibit clinical signs of dementia.
The link between DS and neurodegeneration is largely attributed to the triplication of chromosome 21, which includes the APP gene. Overexpression of APP leads to excessive production and accumulation of amyloid-β (Aβ), which forms the hallmark plaques seen in AD. In DS, amyloid plaques begin to form as early as the teenage years, and by the fourth decade, neurofibrillary tangles (tau protein aggregates) and widespread neurodegeneration are nearly universal.
Initial symptoms of AD often include memory impairment, particularly short-term memory deficits, and executive dysfunction, difficulty with planning and problem-solving, and visuospatial deficits. Behavioral and personality changes, such as irritability, withdrawal, or apathy, are also commonly reported. Compared with sporadic AD, individuals with DS often show earlier behavioral symptoms, including impulsivity and changes in social interactions, which may precede noticeable memory deficits. Additionally, late-onset myoclonic epilepsy is common in individuals with DS and dementia, further complicating diagnosis and care.
The diagnosis of dementia in DS is challenging because of baseline intellectual disability, which makes it difficult to assess cognitive decline using standard neuropsychological tests. However, a combination of clinical history, caregiver reports, neuroimaging, and biomarker analysis can aid in early detection. Structural MRI of the brain often reveals ventricular enlargement and cortical atrophy, while PET imaging can detect early amyloid and tau accumulation.
Because AD is now the leading cause of death in individuals with DS, the lack of effective disease-modifying treatments highlights an important need for clinical trials focused on this population. Current research aims to explore the role of anti-amyloid monoclonal antibodies, neuroprotective agents, and lifestyle interventions to delay or prevent neurodegeneration.
Shaheen E. Lakhan, MD, PhD, MS, MEd, Chief of Pain Management, Carilion Clinic and Virginia Tech Carilion School of Medicine, Roanoke, Virginia.
Disclosure: Shaheen E. Lakhan, MD, PhD, MS, MEd, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
Individuals with DS are at significantly increased risk of developing Alzheimer’s disease (AD) because of the overexpression of the amyloid precursor protein (APP) gene on chromosome 21.
The patient exhibits hallmark symptoms of AD, including progressive memory loss, disorientation, difficulty performing daily tasks, and behavioral changes such as irritability and social withdrawal. The MRI findings of ventricular enlargement and cortical atrophy are consistent with brain changes commonly seen in AD, particularly in individuals with DS who often develop these changes earlier in life (typically in their thirties or forties).
Frontotemporal dementia primarily causes behavioral and language changes with relative memory sparing early on, making it inconsistent with this patient's prominent memory loss, disorientation, and generalized cortical atrophy — features more typical of AD.
Vascular dementia often presents with stepwise decline and focal neurologic deficits; it is unlikely in this patient, given the absence of cerebrovascular events or risk factors like hypertension or diabetes.
While normal pressure hydrocephalus can cause ventricular enlargement, its classic triad of gait disturbance, urinary incontinence, and dementia is incomplete here, making this answer unlikely.
DS is the most common genetic cause of intellectual disability, occurring in approximately 1 in 700 live births worldwide. Nearly all adults with DS develop neuropathologic changes associated with AD by age 40, and the lifetime risk of developing dementia exceeds 90%; by the age of 55-60, at least 70% of individuals with DS exhibit clinical signs of dementia.
The link between DS and neurodegeneration is largely attributed to the triplication of chromosome 21, which includes the APP gene. Overexpression of APP leads to excessive production and accumulation of amyloid-β (Aβ), which forms the hallmark plaques seen in AD. In DS, amyloid plaques begin to form as early as the teenage years, and by the fourth decade, neurofibrillary tangles (tau protein aggregates) and widespread neurodegeneration are nearly universal.
Initial symptoms of AD often include memory impairment, particularly short-term memory deficits, and executive dysfunction, difficulty with planning and problem-solving, and visuospatial deficits. Behavioral and personality changes, such as irritability, withdrawal, or apathy, are also commonly reported. Compared with sporadic AD, individuals with DS often show earlier behavioral symptoms, including impulsivity and changes in social interactions, which may precede noticeable memory deficits. Additionally, late-onset myoclonic epilepsy is common in individuals with DS and dementia, further complicating diagnosis and care.
The diagnosis of dementia in DS is challenging because of baseline intellectual disability, which makes it difficult to assess cognitive decline using standard neuropsychological tests. However, a combination of clinical history, caregiver reports, neuroimaging, and biomarker analysis can aid in early detection. Structural MRI of the brain often reveals ventricular enlargement and cortical atrophy, while PET imaging can detect early amyloid and tau accumulation.
Because AD is now the leading cause of death in individuals with DS, the lack of effective disease-modifying treatments highlights an important need for clinical trials focused on this population. Current research aims to explore the role of anti-amyloid monoclonal antibodies, neuroprotective agents, and lifestyle interventions to delay or prevent neurodegeneration.
Shaheen E. Lakhan, MD, PhD, MS, MEd, Chief of Pain Management, Carilion Clinic and Virginia Tech Carilion School of Medicine, Roanoke, Virginia.
Disclosure: Shaheen E. Lakhan, MD, PhD, MS, MEd, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
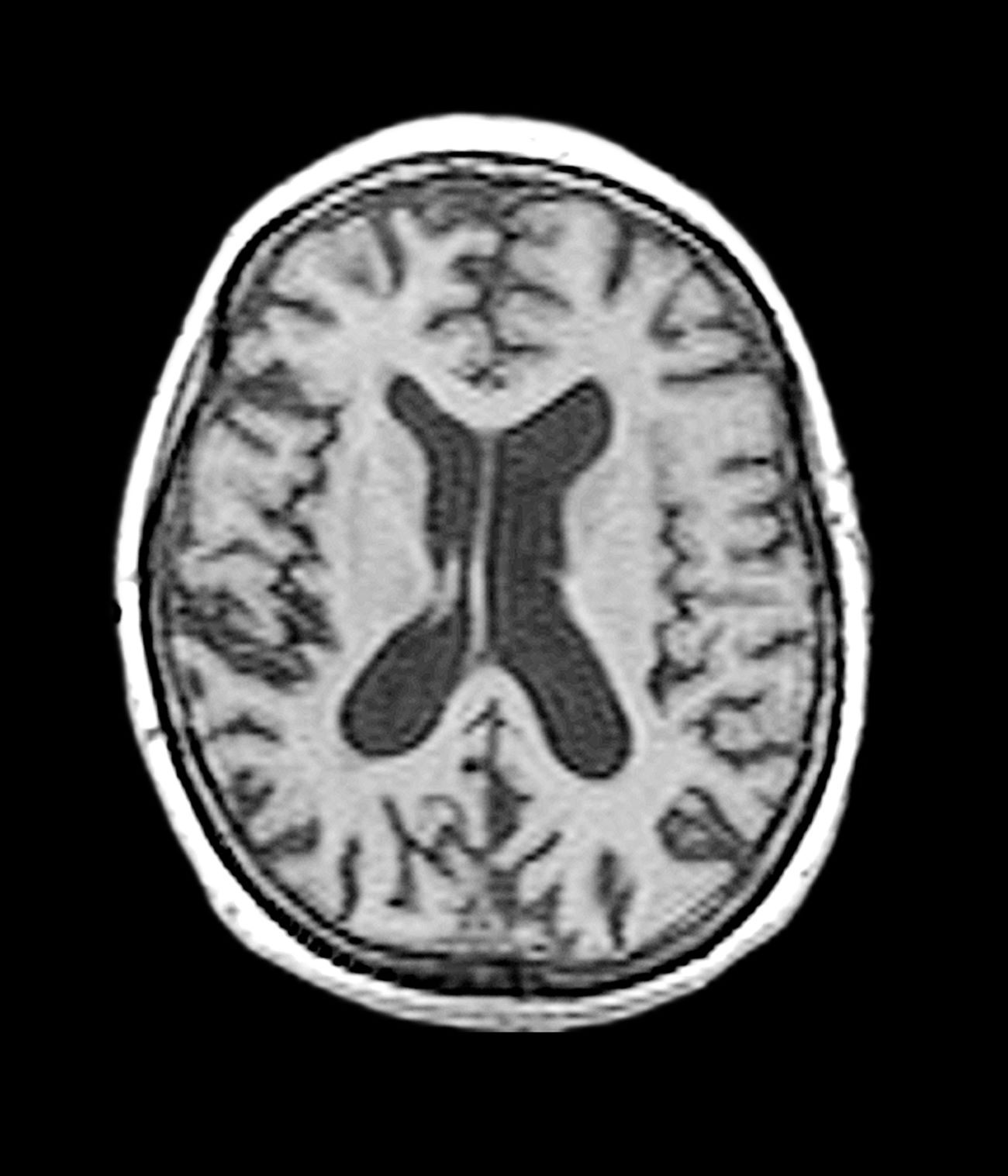
A 40-year-old man with Down syndrome (DS) presented with progressive cognitive decline over 2 years, characterized by memory impairment, difficulty performing familiar tasks, and increasing disorientation. His caregivers noted mood changes, including irritability and withdrawal, and occasional episodes of agitation. Clinical history revealed congenital heart disease (surgically repaired in childhood) and hypothyroidism, which was well controlled with levothyroxine. Physical examination showed no focal neurologic deficits, but he exhibited mild hypotonia, a characteristic feature of DS, and a shuffling gait. Routine blood tests revealed normal thyroid function, no evidence of vitamin B12 deficiency, and unremarkable metabolic panels. An MRI scan (as shown in the image) demonstrated marked ventricular enlargement and generalized cortical atrophy.