User login
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.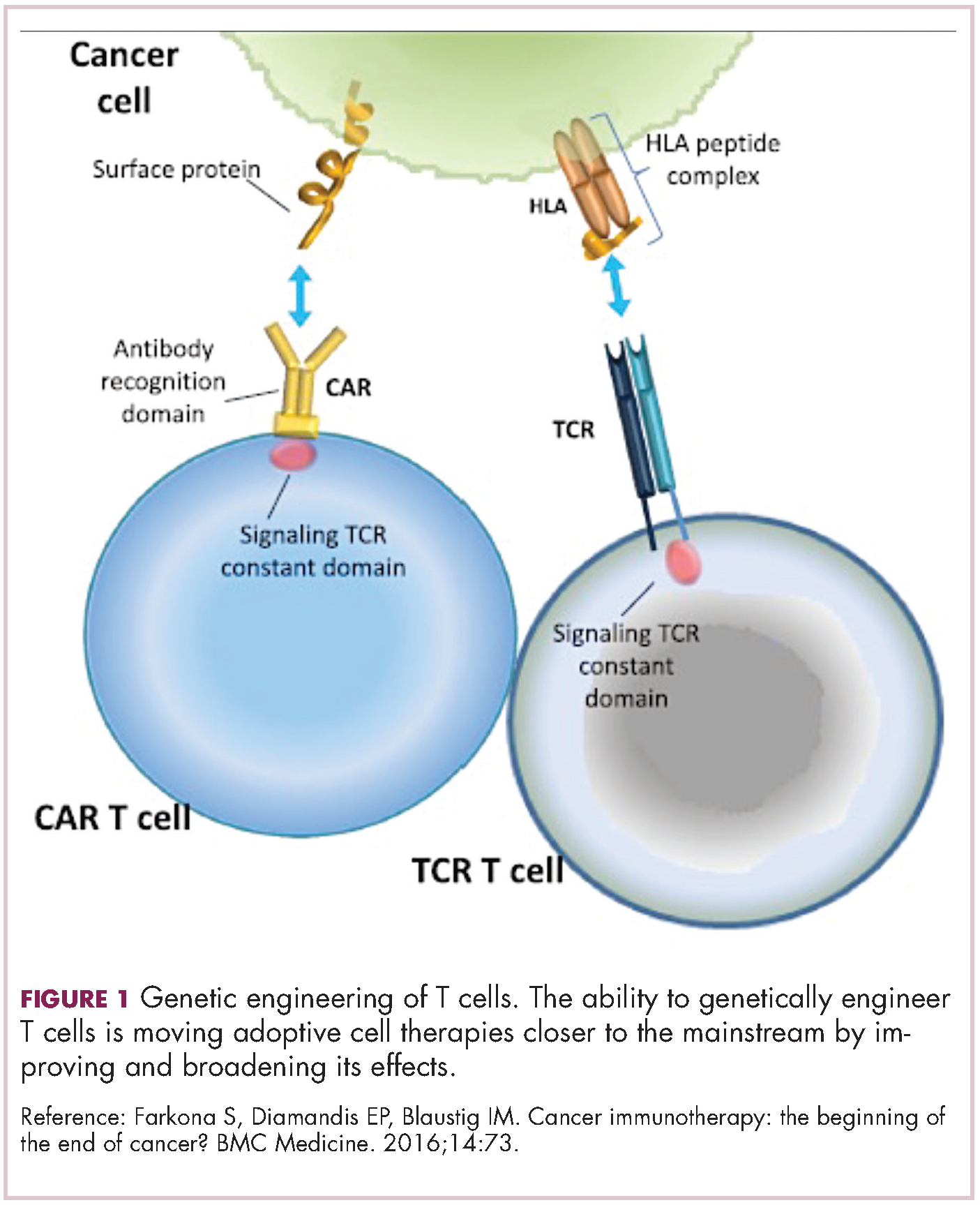
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17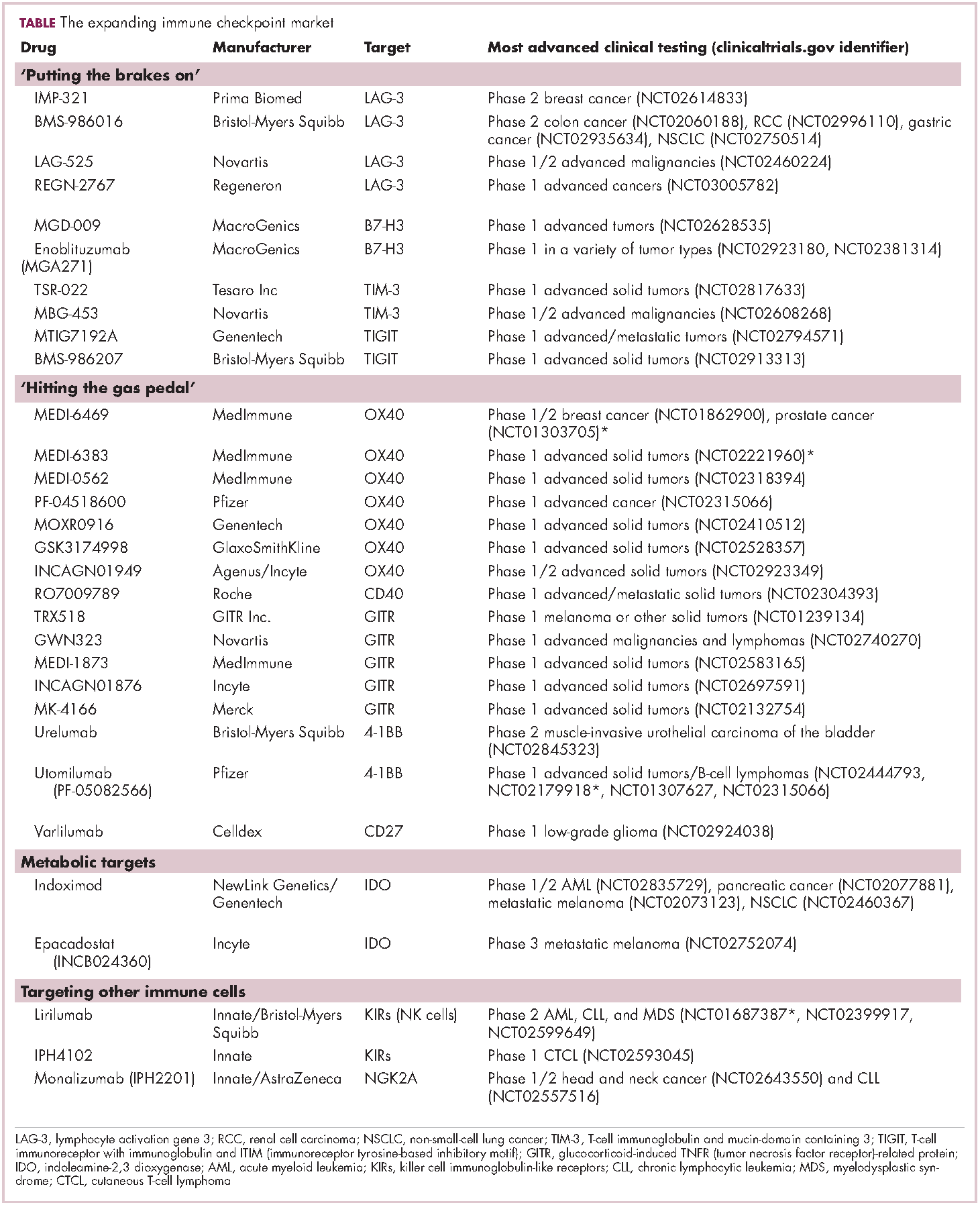
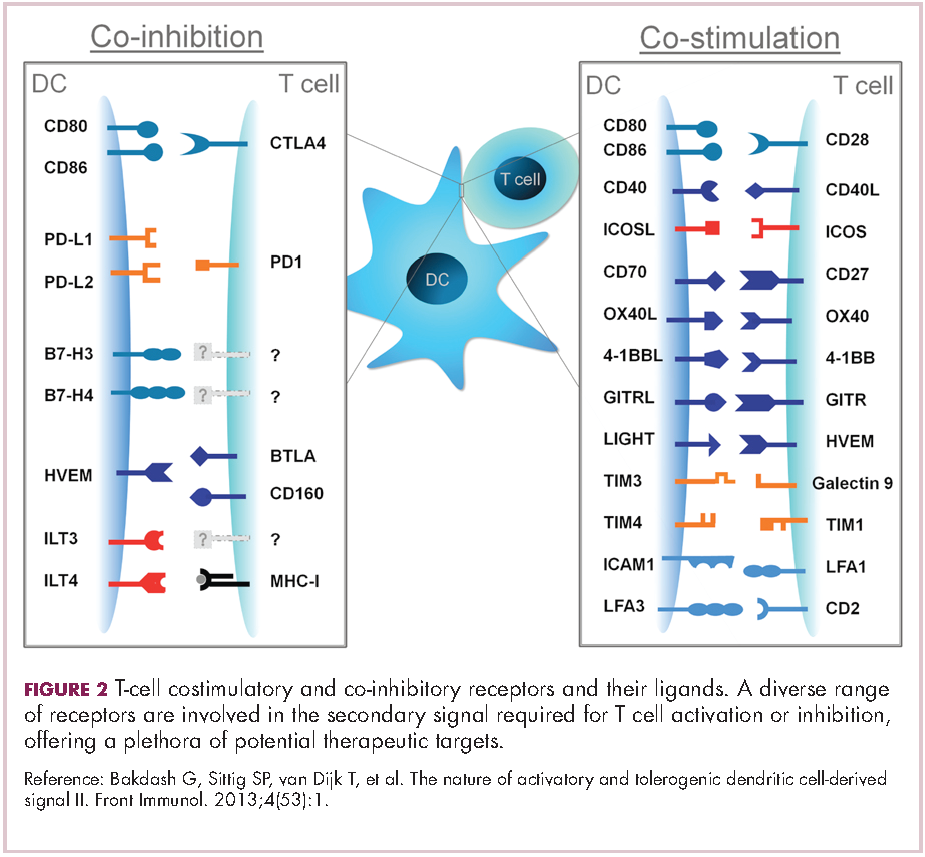
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).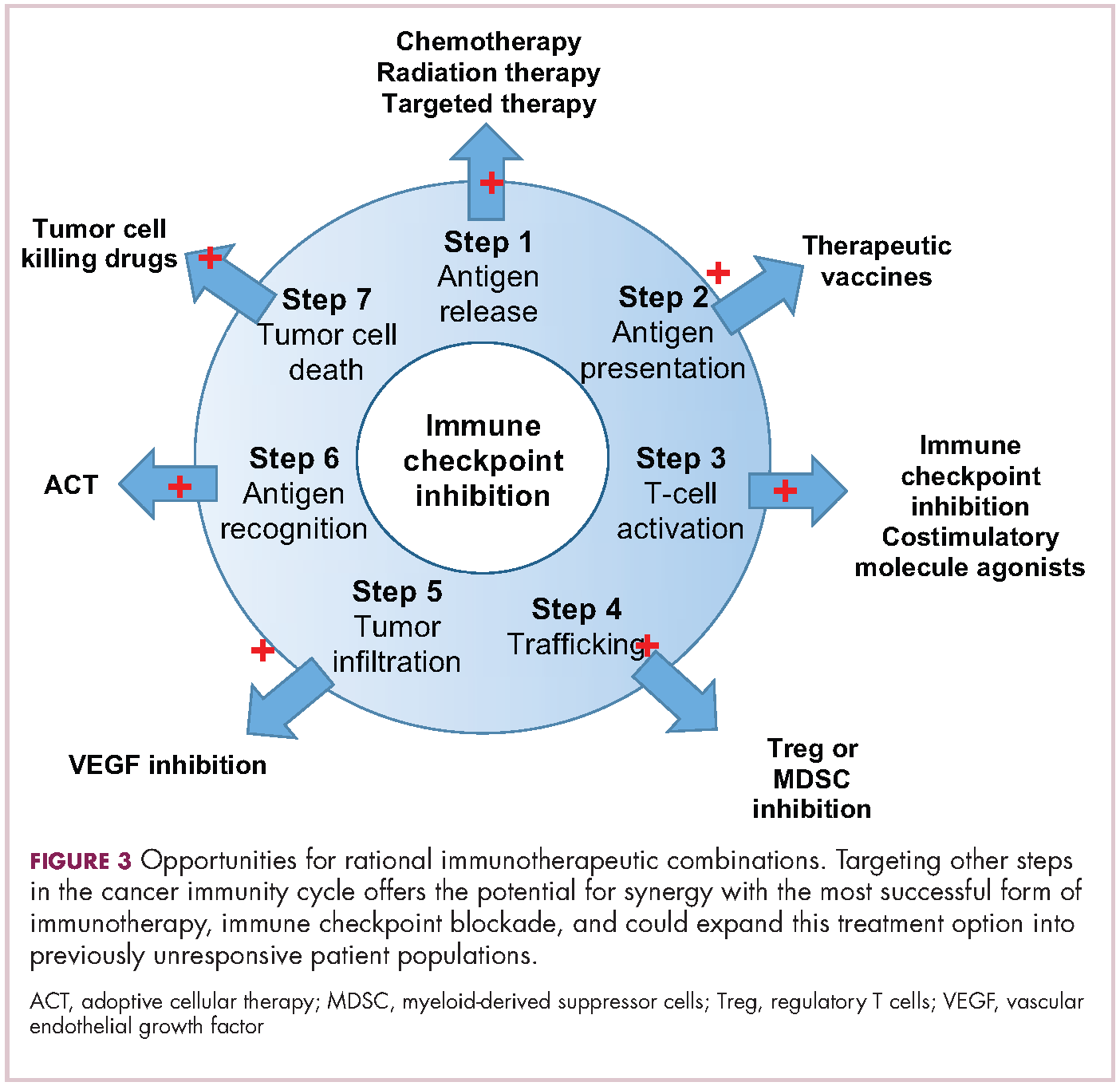
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
The relationship between the immune system and tumors is complex and dynamic, and for immunotherapy to reach its full potential it will likely need to attack on multiple fronts. Here, we discuss some of the latest and most promising developments in the immuno-oncology field designed to build on the successes and address limitations.
The anti-tumor immune response
Cancer is a disease of genomic instability, whereby genetic alterations ranging from a single nucleotide to the whole chromosome level frequently occur. Although cancers derive from a patient’s own tissues, these genetic differences can mark the cancer cell as non-self, triggering an immune response to eliminate these cells.
The first hints of this anti-tumor immunity date back more than a century and a half and sparked the concept of mobilizing the immune system to treat patients.1-3 Although early pioneers achieved little progress in this regard, their efforts provided invaluable insights into the complex and dynamic relationship between a tumor and the immune system that are now translating into real clinical successes.
We now understand that the immune system has a dual role in both restraining and promoting cancer development and have translated this understanding into the theory of cancer immunoediting. Immunoediting has three stages: elimination, wherein the tumor is seemingly destroyed by the innate and adaptive immune response; equilibrium, in which cancer cells that were able to escape elimination are selected for growth; and escape, whereby these resistant cancer cells overwhelm the immune system and develop into a symptomatic lesion.4,5
Immuno-oncologists have also described the cancer immunity cycle to capture the steps that are required for an effective anti-tumor immune response and defects in this cycle form the basis of the most common mechanisms used by cancer cells to subvert the anti-tumor immune response. Much like the cancer hallmarks did for molecularly targeted cancer drugs, the cancer immunity cycle serves as the intellectual framework for cancer immunotherapy.6,7
Exploiting nature’s weapon of mass destruction
Initially, attempts at immunotherapy focused on boosting the immune response using adjuvants and cytokines. The characterization of subtle differences between tumor cells and normal cells led to the development of vaccines and cell-based therapies that exploited these tumor-associated antigens (TAAs).1-6
Despite the approval of a therapeutic vaccine, sipuleucel-T, in 2010 for the treatment of metastatic prostate cancer, in general the success of vaccines has been limited. Marketing authorization for sipuleucel-T was recently withdrawn in Europe, and although it is still available in the United States, it is not widely used because of issues with production and administration. Other vaccines, such as GVAX, which looked particularly promising in early-stage clinical trials, failed to show clinical efficacy in subsequent testing.8,9
Cell-based therapies, such as adoptive cellular therapy (ACT), in which immune cells are removed from the host, primed to attack cancer cells, and then reinfused back into the patient, have focused on T cells because they are the major effectors of the adaptive immune response. Clinical success with the most common approach, tumor-infiltrating lymphocyte (TIL)
Two key techniques have been developed (Figure 1). T-cell receptor (TCR) therapy involves genetically modifying the receptor on the surface of T cells that is responsible for recognizing antigens bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs). The TCR can be altered to recognize a specific TAA or modified to improve its antigen recognition and binding capabilities. This type of therapy is limited by the fact that the TCRs need to be genetically matched to the patient’s immune type.
Releasing the brakes
To ensure that it is only activated at the appropriate time and not in response to the antigens expressed on the surface of the host’s own tissues or harmless materials, the immune system has developed numerous mechanisms for immunological tolerance. Cancer cells are able to exploit these mechanisms to allow them to evade the anti-tumor immune response. One of the main ways in which they do this is by manipulating the signaling pathways involved in T-cell activation, which play a vital role in tolerance.12
To become fully activated, T cells require a primary signal generated by an interaction between the TCR and the antigen-MHC complex on the surface of an APC, followed by secondary costimulatory signals generated by a range of different receptors present on the T-cell surface binding to their ligands on the APC.
If the second signal is inhibitory rather than stimulatory, then the T cell is deactivated instead of becoming activated. Two key coinhibitory receptors are programmed cell death 1 (PD-1) and cytotoxic T-lymphocyte antigen 4 (CTLA-4) and tumor cells are able to overcome the anti-tumor immune response in part by expressing the ligands that bind these receptors to dampen the activity of tumor-infiltrating T cells and induce tolerance.13
The development of inhibitors of CTLA-4 and PD-1 and their respective ligands has driven some of the most dramatic successes with cancer immunotherapy, particularly with PD-1-targeting drugs which have fewer side effects. Targeting of this pathway has resulted in durable responses, revolutionizing the treatment of metastatic melanoma, with recently published long-term survival data for pembrolizumab showing that 40% of patients were alive 3 years after initiating treatment and, in a separate study, 34% of nivolumab-treated patients were still alive after 5 years.14,15 More recently, PD-1 inhibitors have been slowly expanding into a range of other cancer types and 4 immune checkpoint inhibitors are now approved by the United States Food and Drug Administration (FDA): ipilimumab (Yervoy), nivolumab (Opdivo), pembrolizumab (Keytruda) and atezolizumab (Tecentriq).
Six years on from the first approval in this drug class and an extensive network of coinhibitory receptors has been uncovered – so-called immune checkpoints – many of which are now also serving as therapeutic targets (Table, Figure 2).16 Lymphocyte activation gene 3 (LAG-3) is a member of the immunoglobulin superfamily of receptors that is expressed on a number of different types of immune cell. In addition to negatively regulating cytotoxic T-cell activation like PD-1 and CTLA-4, it is also thought to regulate the immunosuppressive functions of regulatory T cells and the maturation and activation of dendritic cells. T-cell immunoglobulin and mucin domain-containing 3 (TIM-3) is found on the surface of helper and cytotoxic T cells and regulates T-cell inhibition as well as macrophage activation. Inhibitors of both proteins have been developed that are being evaluated in phase 1 or 2 clinical trials in a variety of tumor types.17
Indeed, although T cells have commanded the most attention, there is growing appreciation of the potential for targeting other types of immune cell that play a role in the anti-tumor immune response or in fostering an immunosuppressive microenvironment. NK cells have been a particular focus, since they represent the body’s first line of immune defense and they appear to have analogous inhibitory and activating receptors expressed on their surface that regulate their cytotoxic activity.
The best-defined NK cell receptors are the killer cell immunoglobulin-like receptors (KIRs) that bind to the MHC class I proteins found on the surface of all cells that distinguish them as ‘self’ or ‘non-self’. KIRs can be either activating or inhibitory, depending upon their structure and the ligands to which they bind.19 To date, 2 antibodies targeting inhibitory KIRs have been developed. Though there has been some disappointment with these drugs, most recently a phase 2 trial of lirilumab in elderly patients with acute myeloid leukemia, which missed its primary endpoint, they continue to be evaluated in clinical trials.20
The inhibitory immune checkpoint field has also expanded to include molecules that regulate T-cell activity in other ways. Most prominently, this includes enzymes like indoleamine-2,3 dioxygenase (IDO), which is involved in the metabolism of the essential amino acid tryptophan. IDO-induced depletion of tryptophan and generation of tryptophan metabolites is toxic to cytotoxic T cells, and IDO is also thought to directly activate regulatory T cells, thus the net effect of IDO is immunosuppression. Two IDO inhibitors are currently being developed.21
Stepping on the gas
Despite their unprecedented success, immune checkpoint inhibitors are not effective in all patients or in all tumor types. Their efficacy is limited in large part by the requirement for a pre-existing anti-tumor immune response. If there are no T cells within the tumor microenvironment then releasing the brakes on the immune system won’t help.
More recently, researchers have returned to the idea of stimulating an anti-tumor immune response, this time by targeting the other side of the immune checkpoint coin, the costimulatory molecules. These drugs could prove more effective as they aren’t reliant on a pre-existing anti-tumor immune response. A number of agonist antibodies designed to target these receptors have now been developed and are undergoing clinical evaluation.22
Furthest along in development are those targeting OX40, a costimulatory molecule that is upregulated on the surface of T cells once they have been fully activated by the TCR signal and an initial costimulatory signal. OX40 is thought to be involved in a more long-term immune response and in the formation of a memory response. A mouse monoclonal antibody had a potent immune-stimulating effect accompanied by the regression of at least 1 metastatic lesion in 30% of patients treated in a phase 1 clinical trial, but was limited by the generation of anti-mouse antibodies. 7 OX40 agonists are now in clinical development, 6 fully human monoclonal antibodies and 1 OX40 ligand-Fc fusion protein, MEDI-6383.23
Combinations are key
Many researchers are now reaching the conclusion that combination therapy is likely to be key in expanding the scope of immunotherapy into currently unresponsive patient populations. Investigating rational combinations is already becoming a burgeoning area of the immuno-oncology field, with a variety of different strategies being tested.
Now the question becomes what are the optimal combinations and the timing and sequencing of combination therapy is likely to be a paramount consideration. Developing combinations that have distinct mechanisms of action or target multiple steps in the cancer immunity cycle offers the greatest potential for therapeutic synergy since this is most likely to address potential mechanisms of resistance by blocking other paths to immune evasion for cancer cells (Figure 3).
Given the expanding network of immune-checkpoint inhibitors and agonists, the focal point of combination therapy has been combining immune checkpoint-targeting drugs with different mechanisms of action, including those that would simultaneously release the brakes and step on the gas pedal. The vast majority of ongoing clinical trials of approved checkpoint inhibitors and the drugs in development listed in the table are combination trials.
These efforts yielded the first FDA-approved combination immunotherapy regimen in 2015; nivolumab and ipilimumab for the treatment of metastatic melanoma. Approval was based on the demonstration of improved ORR, prolonged response duration, and improved progression-free survival among 142 patients treated with the combination, compared to either drug alone.24
The results of a phase 1/2 trial evaluating the combination of a 4-1BB receptor agonist urelumab with nivolumab in hematologic malignancies and solid tumors found the combination to be safe and particularly effective in patients with advanced/metastatic melanoma, with an ORR of 50%.25 Nivolumab was also combined with the CD27 agonist varlilumab in a phase 1/2 clinical trial of patients with solid tumors, for which data was also recently released. Among 46 patients enrolled, primarily those with colorectal and ovarian cancer the combination had an acceptable safety profile and favorable changes in intratumoral immune biomarkers were observed. The phase 2 portion of the trial is ongoing.26
Meanwhile, Incyte’s IDO inhibitor epacadostat has recently been making waves in combination with pembrolizumab in patients with advanced solid tumors. It demonstrated particularly promising clinical activity in patients with metastatic melanoma, with an overall response rate (ORR) of 57%, including 2 complete responses (CRs), prompting initiation of a phase 3 trial of this combination (NCT02752074).27
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.
1. Adams JL, Smothers J, Srinivasan R, et al. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Disc. 2015;14:603-622.
2. D’Errico G, Machado HL, Sainz Jr B. A current perspective on cancer immune therapy: step-by-step approach to constructing the magic bullet. Clin Trans Med. 2017;6:3.
3. Farkona S, Diamandis EP, Blaustig IM. Cancer immunotherapy: the beginning of the end of cancer? BMC Med. 2016;14:73.
4. Meiliana A, Dewi NM, Wijaya A. Cancer immunotherapy: a review. Indones Biomed J. 2016;8(1):1-20.
5. Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumor microenvironment. Nat Rev Clin Oncol. 2016;13:143-158.
6. de Charette M, Marabelle A, Houot R. Turning tumor cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur J Cancer 2016;68:134-147.
7. Chen DS and Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013;39:1-10.
8. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature 2011;480:480-489.
9. Le DT, Wang-Gillam A, Picozzi V Jr, et al. A phase 2, randomized trial of GVAX Pancreas and CRS-207 immunotherapy versus GVAX alone in patients with metastatic pancreatic adenocarcinoma: Updated results. Presented at: the ASCO Gastrointestinal Cancers Symposium; January 16-18, 2014; San Francisco, CA. Abstract 177.
10. Sharpe M and Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech. 2015;8(4):337-350.
11. Perica K, Varela JC, Oelke M, et al. Adoptive T Cell Immunotherapy for Cancer. Ram Mai Med J. 2015;6(1):e0004.
12. Xing Y and Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol. 2012;4:a006957.
13. Buchbinder EI and Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98-106.
14. Robert C, Ribas A, Hamid O, et al. 3-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol. 2016(suppl;abstr 9503).
15. Hodi SF, Kluger HM, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Presented at the 2016 AACR Annual Meeting; April 16-20; New Orleans, LA. Abstract CT001.
16. Bakdash G, Sittig SP, van Dijk T, et al. The nature of activatory and tolerogenic dendritic cell-derived signal II. Front Immunol. 2013;4(53):1-18.
17. Sheridan C. Immuno-oncology moves beyond PD-1. Nat Biotechnol. 2015;33(7):673-675.
18. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res. 2016;22(21):5183-5188.
19. Carotta S. Targeting NK cells for anticancer immunotherapy: clinical and preclinical approaches. Front Immunol. 2016;7:152.
20. Innate Pharma Web site. Innate Pharma Announces Top-Line Results from EFFIKIR Trial Evaluating the Efficacy of Lirilumab as a Single Agent in Elderly Patients with Acute Myeloid Leukemia. http://www.innate-pharma.com/en/news-events/press-releases/innate-pharma-announces-top-line-results-effikir-trial-evaluating-efficacy-lirilumab-single-agent-elderly-patients-acute-myeloid-leukemia. Last updated February 6, 2017. Accessed online February 22, 2017.
21. Sheridan C. IDO inhibitors move center stage in immuno-oncology. Nat Biotechnol. 2015;33(4):321-322.
22. Sanmamed MF, Pastor F, Rodriguez A, et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol. 2015;42(4):640-655.
23. Linch SN, McNamara MJ, Redmond WL. OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front Oncol. 2015;5:34.
24. U.S. Food and Drug Administration Web site. Nivolumab in combination with ipilimumab. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm465274.htm. Last updated October 1, 2015. Accessed online February 22, 2017.
25. Massarelli E. Clinical safety and efficacy assessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. Presented at the 31st Annual Meeting of the Society for the Immunotherapy of Cancer; November 9-13, 2016; National Harbor, MD. Abstract 239.
26. Sanborn RE, Pishvain MJ, Callahan MK, et al. Phase I results from the combination of an immune-activating anti-CD27 antibody (varlilumab) in combination with PD-1 blockade (nivolumab): activation across multiple immune pathways without untoward immune-related adverse events. Clin Cancer Res. 2016;76(14):suppl. Abstract CT023.
27. Gangadhar T, Hamid O, Smith D.C, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol. 2016;27(6):379-400.