User login
Chimerism and the use of fludarabine associated with secondary graft failure in aplastic anemia
Inferior overall survival was observed in patients with aplastic anemia who had mixed or complete recipient-type chimerism or complete donor chimerism after hematopoietic stem cell transplantation (HSCT), according to the results of a registry database study in Japan.
Researchers examined four groups of patients with AA age greater than 15 years who underwent a first allogeneic bone marrow or peripheral blood stem cell transplantation and achieved engraftment.
Group 1 consisted of patients with mixed chimerism (MC) that did not require either granulocyte-colony stimulating factor (G-CSF) or transfusion support; group 2 consisted of MC (with no secondary graft failure (SGF) that required G-CSF and/or transfusion support ; group 3 consisted of patients with SGF with MC or complete recipient-type chimerism; and group 4 consisted of SGF with complete donor-type chimerism.
The overall median follow-up of survivors was 1,727 days. The overall survival (OS) was 90.4% at 1 year and 83.5% at 5 years in patients without MC or SGF (n = 340), which was not different from the OS in groups 1 and 2. However, inferior OS was observed in group 3 (1 year, 52.1%; 5 years, 52.1%) and group 4 (1 year, 82.4%; 5 years, 56.3%). In addition, multivariate analyses showed that the use of fludarabine (Flu) and the absence of irradiation in conditioning were associated with the development of SGF with MC or complete recipient-type chimerism. The use of Flu in conditioning also was associated with SGF with complete donor-type chimerism.
“The occurrence of SGF with both MC/recipient-type and donor-type chimerism after HSCT for AA was associated with inferior OS, and the conditioning regimens influenced the occurrence of SGF,” the researchers concluded.
The Japan Agency for Medical Research and Development funded the research. Two of the authors reported receiving honoraria or research funding from Sanofi KK and one from Shionogi.
SOURCE: Kako S et al. Biol Blood Marrow Transplant 2020. 26:445-50.
Inferior overall survival was observed in patients with aplastic anemia who had mixed or complete recipient-type chimerism or complete donor chimerism after hematopoietic stem cell transplantation (HSCT), according to the results of a registry database study in Japan.
Researchers examined four groups of patients with AA age greater than 15 years who underwent a first allogeneic bone marrow or peripheral blood stem cell transplantation and achieved engraftment.
Group 1 consisted of patients with mixed chimerism (MC) that did not require either granulocyte-colony stimulating factor (G-CSF) or transfusion support; group 2 consisted of MC (with no secondary graft failure (SGF) that required G-CSF and/or transfusion support ; group 3 consisted of patients with SGF with MC or complete recipient-type chimerism; and group 4 consisted of SGF with complete donor-type chimerism.
The overall median follow-up of survivors was 1,727 days. The overall survival (OS) was 90.4% at 1 year and 83.5% at 5 years in patients without MC or SGF (n = 340), which was not different from the OS in groups 1 and 2. However, inferior OS was observed in group 3 (1 year, 52.1%; 5 years, 52.1%) and group 4 (1 year, 82.4%; 5 years, 56.3%). In addition, multivariate analyses showed that the use of fludarabine (Flu) and the absence of irradiation in conditioning were associated with the development of SGF with MC or complete recipient-type chimerism. The use of Flu in conditioning also was associated with SGF with complete donor-type chimerism.
“The occurrence of SGF with both MC/recipient-type and donor-type chimerism after HSCT for AA was associated with inferior OS, and the conditioning regimens influenced the occurrence of SGF,” the researchers concluded.
The Japan Agency for Medical Research and Development funded the research. Two of the authors reported receiving honoraria or research funding from Sanofi KK and one from Shionogi.
SOURCE: Kako S et al. Biol Blood Marrow Transplant 2020. 26:445-50.
Inferior overall survival was observed in patients with aplastic anemia who had mixed or complete recipient-type chimerism or complete donor chimerism after hematopoietic stem cell transplantation (HSCT), according to the results of a registry database study in Japan.
Researchers examined four groups of patients with AA age greater than 15 years who underwent a first allogeneic bone marrow or peripheral blood stem cell transplantation and achieved engraftment.
Group 1 consisted of patients with mixed chimerism (MC) that did not require either granulocyte-colony stimulating factor (G-CSF) or transfusion support; group 2 consisted of MC (with no secondary graft failure (SGF) that required G-CSF and/or transfusion support ; group 3 consisted of patients with SGF with MC or complete recipient-type chimerism; and group 4 consisted of SGF with complete donor-type chimerism.
The overall median follow-up of survivors was 1,727 days. The overall survival (OS) was 90.4% at 1 year and 83.5% at 5 years in patients without MC or SGF (n = 340), which was not different from the OS in groups 1 and 2. However, inferior OS was observed in group 3 (1 year, 52.1%; 5 years, 52.1%) and group 4 (1 year, 82.4%; 5 years, 56.3%). In addition, multivariate analyses showed that the use of fludarabine (Flu) and the absence of irradiation in conditioning were associated with the development of SGF with MC or complete recipient-type chimerism. The use of Flu in conditioning also was associated with SGF with complete donor-type chimerism.
“The occurrence of SGF with both MC/recipient-type and donor-type chimerism after HSCT for AA was associated with inferior OS, and the conditioning regimens influenced the occurrence of SGF,” the researchers concluded.
The Japan Agency for Medical Research and Development funded the research. Two of the authors reported receiving honoraria or research funding from Sanofi KK and one from Shionogi.
SOURCE: Kako S et al. Biol Blood Marrow Transplant 2020. 26:445-50.
FROM BIOLOGY OF BLOOD AND MARROW TRANSPLANTATION
SARS epidemiology provides clues to potential treatment for COVID-19
A team of researchers has discovered important commonalities between SARS-CoV-2 and SARS-CoV infection that could lead to a potential targets for antiviral intervention.
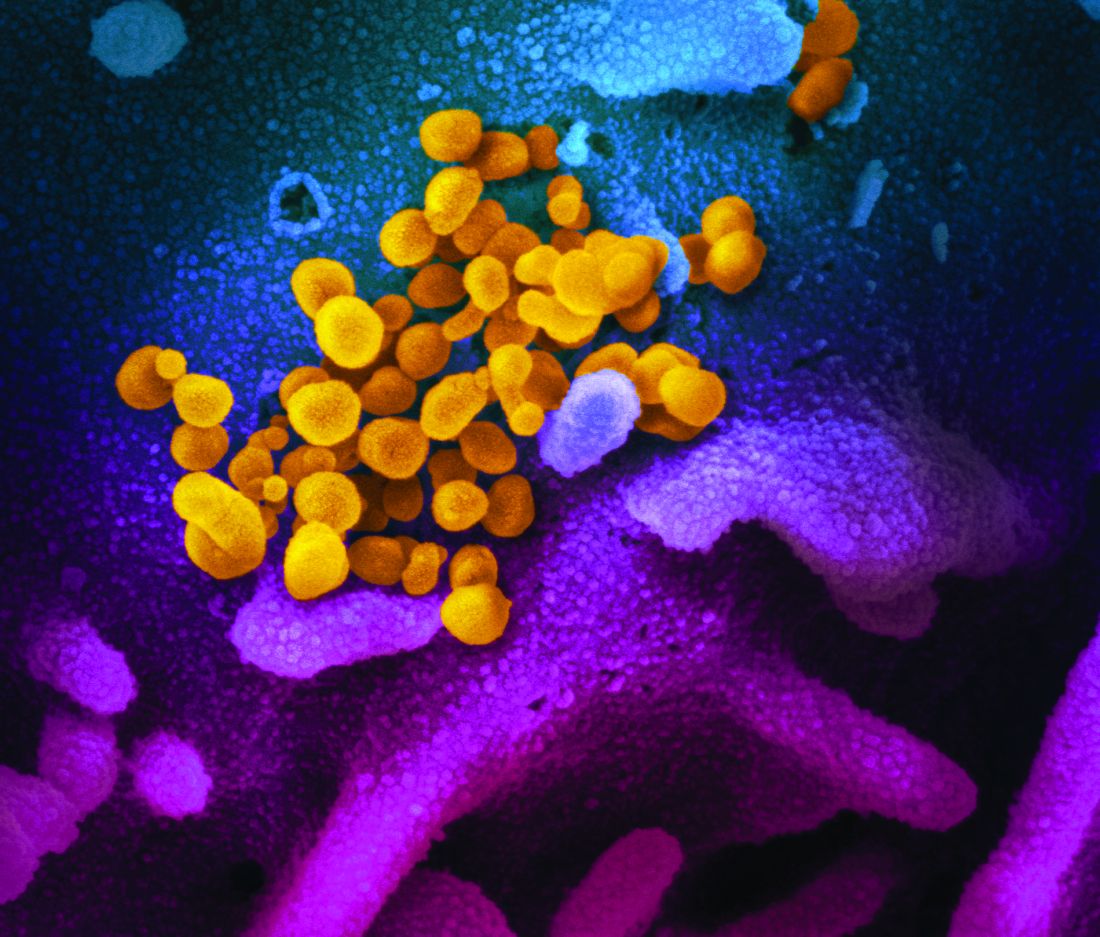
Markus Hoffmann, of the Leibniz Institute for Primate Research, Göttingen, Germany, and a team of investigators also found that antibody responses raised against SARS-S during infection or vaccination might offer some level of protection against SARS-CoV-2 infection. Their findings were published in Cell.
In order for coronaviruses to enter a cell, they must first bind their viral spike (S) proteins to cellular receptors and depend on S protein priming by host cell proteases. The study found that the SARS-CoV-2, causal agent for COVID-19, uses the same SARS-CoV receptor, ACE2, for entry and uses the serine protease TMPRSS2 for S protein priming as the original SARS-CoV-1 (SARS). Importantly, the researchers also found that the cellular serine protease TMPRSS2 primes SARS-CoV-2-S for entry and that a serine protease inhibitor blocks SARS-CoV-2 infection of lung cells, providing opportunities for potential therapeutic intervention.
The researchers performed a sequence analysis that showed SARS-CoV-2 clusters with SARS-CoV–related viruses from bats, of which some – but not all – can use ACE2 for host cell entry. Further analysis of the receptor binding motif known to make contact with ACE2 showed that most amino acid residues essential for ACE2 binding by SARS-S were conserved in SARS-2-S but were absent from S proteins of those SARS-related coronaviruses previously found not to use ACE2.
In addition, the researchers found that SARS-CoV-2–infected BHK-21 cells transfected to express ACE2 with high efficiency, but not the parental BHK-21 cells indicating that SARS-CoV-2-S, like the original SARS virus S protein, uses ACE2 for cellular entry.
Using cultured cells, the researchers found that the protease inhibitor, camostat mesylate, inhibited SARS-S and SARS-2-S entry into primary human lung cells, demonstrating that SARS-CoV-2 can use TMPRSS2 for S protein priming and that camostat mesylate can block SARS-CoV-2 infection of lung cells. Camostat mesylate has been used as a therapy for some forms of cancer and other viral infections.
In addition to their research on the protease inhibitor, the researchers also found that sera from convalescent SARS patients cross-neutralized SARS-2-S–driven entry. They found that four sera obtained from three convalescent SARS patients inhibited SARS-S entry into cell lines in a concentration dependent fashion.
“We demonstrate that SARS-CoV-2 uses the SARS55 CoV receptor, ACE2, for entry and the serine protease TMPRSS2 for S protein priming. A TMPRSS2 inhibitor approved for clinical use blocked entry and might constitute a treatment option. Finally, we show that the sera from convalescent SARS patients cross-neutralized SARS-2-S–driven entry,” the authors concluded.
The study was supported by BMBF (RAPID Consortium) and German Research Foundation (DFG). The authors reported that they had no conflicts.
SOURCE: Hoffmann M et al. Cell 2020. doi: 10.1016/j.cell.2020.02.052.
A team of researchers has discovered important commonalities between SARS-CoV-2 and SARS-CoV infection that could lead to a potential targets for antiviral intervention.
Markus Hoffmann, of the Leibniz Institute for Primate Research, Göttingen, Germany, and a team of investigators also found that antibody responses raised against SARS-S during infection or vaccination might offer some level of protection against SARS-CoV-2 infection. Their findings were published in Cell.
In order for coronaviruses to enter a cell, they must first bind their viral spike (S) proteins to cellular receptors and depend on S protein priming by host cell proteases. The study found that the SARS-CoV-2, causal agent for COVID-19, uses the same SARS-CoV receptor, ACE2, for entry and uses the serine protease TMPRSS2 for S protein priming as the original SARS-CoV-1 (SARS). Importantly, the researchers also found that the cellular serine protease TMPRSS2 primes SARS-CoV-2-S for entry and that a serine protease inhibitor blocks SARS-CoV-2 infection of lung cells, providing opportunities for potential therapeutic intervention.
The researchers performed a sequence analysis that showed SARS-CoV-2 clusters with SARS-CoV–related viruses from bats, of which some – but not all – can use ACE2 for host cell entry. Further analysis of the receptor binding motif known to make contact with ACE2 showed that most amino acid residues essential for ACE2 binding by SARS-S were conserved in SARS-2-S but were absent from S proteins of those SARS-related coronaviruses previously found not to use ACE2.
In addition, the researchers found that SARS-CoV-2–infected BHK-21 cells transfected to express ACE2 with high efficiency, but not the parental BHK-21 cells indicating that SARS-CoV-2-S, like the original SARS virus S protein, uses ACE2 for cellular entry.
Using cultured cells, the researchers found that the protease inhibitor, camostat mesylate, inhibited SARS-S and SARS-2-S entry into primary human lung cells, demonstrating that SARS-CoV-2 can use TMPRSS2 for S protein priming and that camostat mesylate can block SARS-CoV-2 infection of lung cells. Camostat mesylate has been used as a therapy for some forms of cancer and other viral infections.
In addition to their research on the protease inhibitor, the researchers also found that sera from convalescent SARS patients cross-neutralized SARS-2-S–driven entry. They found that four sera obtained from three convalescent SARS patients inhibited SARS-S entry into cell lines in a concentration dependent fashion.
“We demonstrate that SARS-CoV-2 uses the SARS55 CoV receptor, ACE2, for entry and the serine protease TMPRSS2 for S protein priming. A TMPRSS2 inhibitor approved for clinical use blocked entry and might constitute a treatment option. Finally, we show that the sera from convalescent SARS patients cross-neutralized SARS-2-S–driven entry,” the authors concluded.
The study was supported by BMBF (RAPID Consortium) and German Research Foundation (DFG). The authors reported that they had no conflicts.
SOURCE: Hoffmann M et al. Cell 2020. doi: 10.1016/j.cell.2020.02.052.
A team of researchers has discovered important commonalities between SARS-CoV-2 and SARS-CoV infection that could lead to a potential targets for antiviral intervention.
Markus Hoffmann, of the Leibniz Institute for Primate Research, Göttingen, Germany, and a team of investigators also found that antibody responses raised against SARS-S during infection or vaccination might offer some level of protection against SARS-CoV-2 infection. Their findings were published in Cell.
In order for coronaviruses to enter a cell, they must first bind their viral spike (S) proteins to cellular receptors and depend on S protein priming by host cell proteases. The study found that the SARS-CoV-2, causal agent for COVID-19, uses the same SARS-CoV receptor, ACE2, for entry and uses the serine protease TMPRSS2 for S protein priming as the original SARS-CoV-1 (SARS). Importantly, the researchers also found that the cellular serine protease TMPRSS2 primes SARS-CoV-2-S for entry and that a serine protease inhibitor blocks SARS-CoV-2 infection of lung cells, providing opportunities for potential therapeutic intervention.
The researchers performed a sequence analysis that showed SARS-CoV-2 clusters with SARS-CoV–related viruses from bats, of which some – but not all – can use ACE2 for host cell entry. Further analysis of the receptor binding motif known to make contact with ACE2 showed that most amino acid residues essential for ACE2 binding by SARS-S were conserved in SARS-2-S but were absent from S proteins of those SARS-related coronaviruses previously found not to use ACE2.
In addition, the researchers found that SARS-CoV-2–infected BHK-21 cells transfected to express ACE2 with high efficiency, but not the parental BHK-21 cells indicating that SARS-CoV-2-S, like the original SARS virus S protein, uses ACE2 for cellular entry.
Using cultured cells, the researchers found that the protease inhibitor, camostat mesylate, inhibited SARS-S and SARS-2-S entry into primary human lung cells, demonstrating that SARS-CoV-2 can use TMPRSS2 for S protein priming and that camostat mesylate can block SARS-CoV-2 infection of lung cells. Camostat mesylate has been used as a therapy for some forms of cancer and other viral infections.
In addition to their research on the protease inhibitor, the researchers also found that sera from convalescent SARS patients cross-neutralized SARS-2-S–driven entry. They found that four sera obtained from three convalescent SARS patients inhibited SARS-S entry into cell lines in a concentration dependent fashion.
“We demonstrate that SARS-CoV-2 uses the SARS55 CoV receptor, ACE2, for entry and the serine protease TMPRSS2 for S protein priming. A TMPRSS2 inhibitor approved for clinical use blocked entry and might constitute a treatment option. Finally, we show that the sera from convalescent SARS patients cross-neutralized SARS-2-S–driven entry,” the authors concluded.
The study was supported by BMBF (RAPID Consortium) and German Research Foundation (DFG). The authors reported that they had no conflicts.
SOURCE: Hoffmann M et al. Cell 2020. doi: 10.1016/j.cell.2020.02.052.
FROM CELL
Monthly injection therapy for HIV found noninferior to daily oral dosing
Two international phase 3 randomized trials of according to reports published in the New England Journal of Medicine.
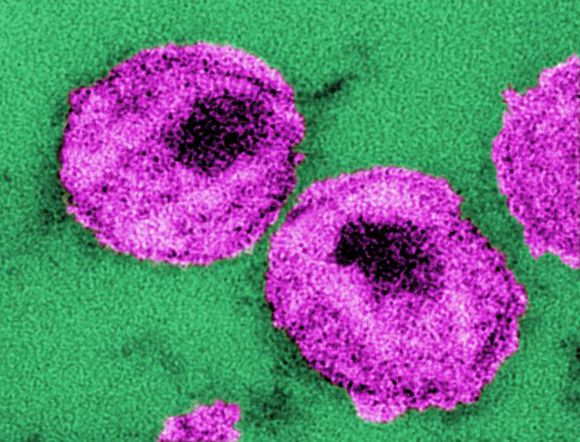
The Long-Acting Cabotegravir and Rilpivirine after Oral Induction for HIV-1 Infection (FLAIR) study and the Long-Acting Cabotegravir and Rilpivirine for Maintenance of HIV-1 Suppression (ATLAS) study looked at a separate facet of the use of a monthly therapeutic injection as a replacement for daily oral HIV therapy.
The FLAIR trial (ClinicalTrials.gov number, NCT02938520) was a phase 3, randomized, open-label study in which adults with HIV-1 infection who had not previously received antiretroviral therapy were given 20 weeks of daily oral induction therapy with dolutegravir–abacavir–lamivudine. Those patients with an HIV-1 RNA level of less than 50 copies per milliliter after 16 weeks were then randomly assigned (1:1) to continue their current oral therapy or switch to oral cabotegravir plus rilpivirine for 1 month followed by monthly intramuscular injections of long-acting cabotegravir, an HIV-1 integrase strand-transfer inhibitor, and rilpivirine, a nonnucleoside reverse-transcriptase inhibitor.
At week 48, an HIV-1 RNA level of 50 copies per milliliter or higher was found in 6 of 283 patients (2.1%) who received the long-acting therapy and in 7 of 283 (2.5%) who received oral therapy, which met the criterion for noninferiority for the primary endpoint. An HIV-1 RNA level of less than 50 copies per milliliter at week 48 was found in 93.6% of patients who received long-acting therapy and in 93.3% who received oral therapy, which also met the criterion for noninferiority, according to the study published in the New England Journal of Medicine.
Injection site reactions were reported in 86% of the long-acting therapy patients, 4 of whom withdrew from the trial for injection-related reasons. Grade 3 or higher adverse events and events that met liver-related stopping criteria occurred in 11% and 2%, respectively, of those who received long-acting therapy and in 4% and 1% of those who received oral therapy.
An assessment of treatment satisfaction at 48 weeks showed that 91% of the patients who switched to long-acting therapy preferred it to their daily oral therapy.
The ATLAS trial (ClinicalTrials.gov number, NCT02951052) was a phase 3, open-label, multicenter, noninferiority trial involving patients who had plasma HIV-1 RNA levels of less than 50 copies per milliliter for at least 6 months while taking standard oral antiretroviral therapy. These patients were randomized (308 in each group) to the long-acting cabotegravir plus rilpivirine injection therapy or daily oral therapy.
At 48 weeks, HIV-1 RNA levels of 50 copies per milliliter or higher were found in five participants (1.6%) receiving long-acting therapy and in three (1.0%) receiving oral therapy, which met the criterion for noninferiority for the primary endpoint, according to a study reported in the New England Journal of Medicine.
HIV-1 RNA levels of less than 50 copies per milliliter at week 48 occurred in 92.5% of patients on long-acting therapy and in 95.5% of those receiving oral therapy, which also met the criterion for noninferiority for this endpoint. Three patients in the long-acting therapy group had virologic failure, compared with four participants who received oral therapy.
Adverse events were more common in the long-acting–therapy group and included injection-site pain, which occurred in 231 recipients (75%) of long-acting therapy. This was mild or moderate in most cases, according to the authors. However, 1% of the participants in this group withdrew because of it. Overall, serious adverse events were reported in no more than 5% of participants in each group.
Together, the ATLAS and the FLAIR trials show that long-acting intramuscular injection therapy is noninferior to oral therapy as both an early regimen for HIV treatment, as well as for later, maintenance dosing. The use of long-acting therapy may improve patient adherence to treatment, according to both sets of study authors.
The ATLAS and FLAIR trials were funded by ViiV Healthcare and Janssen. The authors of both studies reported ties to pharmaceutical associations, and some authors are employees of the two funding sources.
SOURCE: Orkin C et al. N Engl J Med. 2020 Mar 4. doi: 10.1056/NEJMoa1909512 and Swindells S et al. N Engl J Med. 2020 Mar 4. doi: 10.1056/NEJMoa1904398.
Two international phase 3 randomized trials of according to reports published in the New England Journal of Medicine.
The Long-Acting Cabotegravir and Rilpivirine after Oral Induction for HIV-1 Infection (FLAIR) study and the Long-Acting Cabotegravir and Rilpivirine for Maintenance of HIV-1 Suppression (ATLAS) study looked at a separate facet of the use of a monthly therapeutic injection as a replacement for daily oral HIV therapy.
The FLAIR trial (ClinicalTrials.gov number, NCT02938520) was a phase 3, randomized, open-label study in which adults with HIV-1 infection who had not previously received antiretroviral therapy were given 20 weeks of daily oral induction therapy with dolutegravir–abacavir–lamivudine. Those patients with an HIV-1 RNA level of less than 50 copies per milliliter after 16 weeks were then randomly assigned (1:1) to continue their current oral therapy or switch to oral cabotegravir plus rilpivirine for 1 month followed by monthly intramuscular injections of long-acting cabotegravir, an HIV-1 integrase strand-transfer inhibitor, and rilpivirine, a nonnucleoside reverse-transcriptase inhibitor.
At week 48, an HIV-1 RNA level of 50 copies per milliliter or higher was found in 6 of 283 patients (2.1%) who received the long-acting therapy and in 7 of 283 (2.5%) who received oral therapy, which met the criterion for noninferiority for the primary endpoint. An HIV-1 RNA level of less than 50 copies per milliliter at week 48 was found in 93.6% of patients who received long-acting therapy and in 93.3% who received oral therapy, which also met the criterion for noninferiority, according to the study published in the New England Journal of Medicine.
Injection site reactions were reported in 86% of the long-acting therapy patients, 4 of whom withdrew from the trial for injection-related reasons. Grade 3 or higher adverse events and events that met liver-related stopping criteria occurred in 11% and 2%, respectively, of those who received long-acting therapy and in 4% and 1% of those who received oral therapy.
An assessment of treatment satisfaction at 48 weeks showed that 91% of the patients who switched to long-acting therapy preferred it to their daily oral therapy.
The ATLAS trial (ClinicalTrials.gov number, NCT02951052) was a phase 3, open-label, multicenter, noninferiority trial involving patients who had plasma HIV-1 RNA levels of less than 50 copies per milliliter for at least 6 months while taking standard oral antiretroviral therapy. These patients were randomized (308 in each group) to the long-acting cabotegravir plus rilpivirine injection therapy or daily oral therapy.
At 48 weeks, HIV-1 RNA levels of 50 copies per milliliter or higher were found in five participants (1.6%) receiving long-acting therapy and in three (1.0%) receiving oral therapy, which met the criterion for noninferiority for the primary endpoint, according to a study reported in the New England Journal of Medicine.
HIV-1 RNA levels of less than 50 copies per milliliter at week 48 occurred in 92.5% of patients on long-acting therapy and in 95.5% of those receiving oral therapy, which also met the criterion for noninferiority for this endpoint. Three patients in the long-acting therapy group had virologic failure, compared with four participants who received oral therapy.
Adverse events were more common in the long-acting–therapy group and included injection-site pain, which occurred in 231 recipients (75%) of long-acting therapy. This was mild or moderate in most cases, according to the authors. However, 1% of the participants in this group withdrew because of it. Overall, serious adverse events were reported in no more than 5% of participants in each group.
Together, the ATLAS and the FLAIR trials show that long-acting intramuscular injection therapy is noninferior to oral therapy as both an early regimen for HIV treatment, as well as for later, maintenance dosing. The use of long-acting therapy may improve patient adherence to treatment, according to both sets of study authors.
The ATLAS and FLAIR trials were funded by ViiV Healthcare and Janssen. The authors of both studies reported ties to pharmaceutical associations, and some authors are employees of the two funding sources.
SOURCE: Orkin C et al. N Engl J Med. 2020 Mar 4. doi: 10.1056/NEJMoa1909512 and Swindells S et al. N Engl J Med. 2020 Mar 4. doi: 10.1056/NEJMoa1904398.
Two international phase 3 randomized trials of according to reports published in the New England Journal of Medicine.
The Long-Acting Cabotegravir and Rilpivirine after Oral Induction for HIV-1 Infection (FLAIR) study and the Long-Acting Cabotegravir and Rilpivirine for Maintenance of HIV-1 Suppression (ATLAS) study looked at a separate facet of the use of a monthly therapeutic injection as a replacement for daily oral HIV therapy.
The FLAIR trial (ClinicalTrials.gov number, NCT02938520) was a phase 3, randomized, open-label study in which adults with HIV-1 infection who had not previously received antiretroviral therapy were given 20 weeks of daily oral induction therapy with dolutegravir–abacavir–lamivudine. Those patients with an HIV-1 RNA level of less than 50 copies per milliliter after 16 weeks were then randomly assigned (1:1) to continue their current oral therapy or switch to oral cabotegravir plus rilpivirine for 1 month followed by monthly intramuscular injections of long-acting cabotegravir, an HIV-1 integrase strand-transfer inhibitor, and rilpivirine, a nonnucleoside reverse-transcriptase inhibitor.
At week 48, an HIV-1 RNA level of 50 copies per milliliter or higher was found in 6 of 283 patients (2.1%) who received the long-acting therapy and in 7 of 283 (2.5%) who received oral therapy, which met the criterion for noninferiority for the primary endpoint. An HIV-1 RNA level of less than 50 copies per milliliter at week 48 was found in 93.6% of patients who received long-acting therapy and in 93.3% who received oral therapy, which also met the criterion for noninferiority, according to the study published in the New England Journal of Medicine.
Injection site reactions were reported in 86% of the long-acting therapy patients, 4 of whom withdrew from the trial for injection-related reasons. Grade 3 or higher adverse events and events that met liver-related stopping criteria occurred in 11% and 2%, respectively, of those who received long-acting therapy and in 4% and 1% of those who received oral therapy.
An assessment of treatment satisfaction at 48 weeks showed that 91% of the patients who switched to long-acting therapy preferred it to their daily oral therapy.
The ATLAS trial (ClinicalTrials.gov number, NCT02951052) was a phase 3, open-label, multicenter, noninferiority trial involving patients who had plasma HIV-1 RNA levels of less than 50 copies per milliliter for at least 6 months while taking standard oral antiretroviral therapy. These patients were randomized (308 in each group) to the long-acting cabotegravir plus rilpivirine injection therapy or daily oral therapy.
At 48 weeks, HIV-1 RNA levels of 50 copies per milliliter or higher were found in five participants (1.6%) receiving long-acting therapy and in three (1.0%) receiving oral therapy, which met the criterion for noninferiority for the primary endpoint, according to a study reported in the New England Journal of Medicine.
HIV-1 RNA levels of less than 50 copies per milliliter at week 48 occurred in 92.5% of patients on long-acting therapy and in 95.5% of those receiving oral therapy, which also met the criterion for noninferiority for this endpoint. Three patients in the long-acting therapy group had virologic failure, compared with four participants who received oral therapy.
Adverse events were more common in the long-acting–therapy group and included injection-site pain, which occurred in 231 recipients (75%) of long-acting therapy. This was mild or moderate in most cases, according to the authors. However, 1% of the participants in this group withdrew because of it. Overall, serious adverse events were reported in no more than 5% of participants in each group.
Together, the ATLAS and the FLAIR trials show that long-acting intramuscular injection therapy is noninferior to oral therapy as both an early regimen for HIV treatment, as well as for later, maintenance dosing. The use of long-acting therapy may improve patient adherence to treatment, according to both sets of study authors.
The ATLAS and FLAIR trials were funded by ViiV Healthcare and Janssen. The authors of both studies reported ties to pharmaceutical associations, and some authors are employees of the two funding sources.
SOURCE: Orkin C et al. N Engl J Med. 2020 Mar 4. doi: 10.1056/NEJMoa1909512 and Swindells S et al. N Engl J Med. 2020 Mar 4. doi: 10.1056/NEJMoa1904398.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
New CAR T-cell therapy eliminates MM and tumor propagating cells without fratricide in lab study
These cells proved to be active in vitro and in vivo against MM plasma cells, memory B cells, and MM-propagating cells, according to a report in Nature Communications.
This research is important because most MM patients eventually succumb to the disease and previously developed CAR T cells targeting B-cell maturation antigen (BCMA) on MM cells have shown high-response rates but limited durability.
Previous research showed that CD229/LY9 is a potential target for CAR T-cell therapy in MM because of its strong and homogeneous expression on the bulk of tumor cells, as well as chemotherapy-resistant myeloma progenitors; its absence from most normal cells; and dependence of MM cells on CD229 for their survival, according to Sabarinath V. Radhakrishnan, MD, of the University of Utah, Salt Lake City, and colleagues.
Using primary CD138+ tumor cells from three patients with plasma cell leukemia, a highly aggressive form of MM, which all showed high expression of CD229, the researchers found that CD229 CAR T cells exhibited high cytotoxic activity against these cells. In addition, when assessing two MM cell lines, U-266 and RPMI-8226, expressing different levels of CD229, they found that CD229 CAR T cells efficiently killed both cell lines in vitro.
“We do not observe fratricide during CD229 CAR T-cell production, as CD229 is downregulated in T cells during activation. In addition, while CD229 CAR T cells target normal CD229high T cells, they spare functional CD229neg/low T cells. These findings indicate that CD229 CAR T cells may be an effective treatment for patients with MM,” the authors concluded.
The study was funded by several nongovernmental organizations and the National Cancer Institute. Three of the authors are inventors on PCT application US2017/42840 “Antibodies and CAR T Cells for the Treatment of Multiple Myeloma” describing the therapeutic use of CD229 CAR T cells.
SOURCE: Radhakrishnan SV et al. Nat Commun. 2020 Feb 7;11(1):798. doi: 10.1038/s41467-020-14619-z.
These cells proved to be active in vitro and in vivo against MM plasma cells, memory B cells, and MM-propagating cells, according to a report in Nature Communications.
This research is important because most MM patients eventually succumb to the disease and previously developed CAR T cells targeting B-cell maturation antigen (BCMA) on MM cells have shown high-response rates but limited durability.
Previous research showed that CD229/LY9 is a potential target for CAR T-cell therapy in MM because of its strong and homogeneous expression on the bulk of tumor cells, as well as chemotherapy-resistant myeloma progenitors; its absence from most normal cells; and dependence of MM cells on CD229 for their survival, according to Sabarinath V. Radhakrishnan, MD, of the University of Utah, Salt Lake City, and colleagues.
Using primary CD138+ tumor cells from three patients with plasma cell leukemia, a highly aggressive form of MM, which all showed high expression of CD229, the researchers found that CD229 CAR T cells exhibited high cytotoxic activity against these cells. In addition, when assessing two MM cell lines, U-266 and RPMI-8226, expressing different levels of CD229, they found that CD229 CAR T cells efficiently killed both cell lines in vitro.
“We do not observe fratricide during CD229 CAR T-cell production, as CD229 is downregulated in T cells during activation. In addition, while CD229 CAR T cells target normal CD229high T cells, they spare functional CD229neg/low T cells. These findings indicate that CD229 CAR T cells may be an effective treatment for patients with MM,” the authors concluded.
The study was funded by several nongovernmental organizations and the National Cancer Institute. Three of the authors are inventors on PCT application US2017/42840 “Antibodies and CAR T Cells for the Treatment of Multiple Myeloma” describing the therapeutic use of CD229 CAR T cells.
SOURCE: Radhakrishnan SV et al. Nat Commun. 2020 Feb 7;11(1):798. doi: 10.1038/s41467-020-14619-z.
These cells proved to be active in vitro and in vivo against MM plasma cells, memory B cells, and MM-propagating cells, according to a report in Nature Communications.
This research is important because most MM patients eventually succumb to the disease and previously developed CAR T cells targeting B-cell maturation antigen (BCMA) on MM cells have shown high-response rates but limited durability.
Previous research showed that CD229/LY9 is a potential target for CAR T-cell therapy in MM because of its strong and homogeneous expression on the bulk of tumor cells, as well as chemotherapy-resistant myeloma progenitors; its absence from most normal cells; and dependence of MM cells on CD229 for their survival, according to Sabarinath V. Radhakrishnan, MD, of the University of Utah, Salt Lake City, and colleagues.
Using primary CD138+ tumor cells from three patients with plasma cell leukemia, a highly aggressive form of MM, which all showed high expression of CD229, the researchers found that CD229 CAR T cells exhibited high cytotoxic activity against these cells. In addition, when assessing two MM cell lines, U-266 and RPMI-8226, expressing different levels of CD229, they found that CD229 CAR T cells efficiently killed both cell lines in vitro.
“We do not observe fratricide during CD229 CAR T-cell production, as CD229 is downregulated in T cells during activation. In addition, while CD229 CAR T cells target normal CD229high T cells, they spare functional CD229neg/low T cells. These findings indicate that CD229 CAR T cells may be an effective treatment for patients with MM,” the authors concluded.
The study was funded by several nongovernmental organizations and the National Cancer Institute. Three of the authors are inventors on PCT application US2017/42840 “Antibodies and CAR T Cells for the Treatment of Multiple Myeloma” describing the therapeutic use of CD229 CAR T cells.
SOURCE: Radhakrishnan SV et al. Nat Commun. 2020 Feb 7;11(1):798. doi: 10.1038/s41467-020-14619-z.
FROM NATURE COMMUNICATIONS
ID Blog: SARS-CoV-2 – What’s in a name?
Coming up with a moniker for the new coronavirus shows the perils of naming names.
There is no Baby Book of Names or hurricane alphabet to readily name diseases and their causal entities. Throughout history and even in the modern era, a host of considerations have intruded on the decision as to what to call these blights upon humanity. Names have varied from inflammatory to misleading, from colloquial to scientific. And when it concerns a new epidemiological entity such as the latest coronavirus outbreak originating in China, health organizations, media, politicians, scientific taxonomy commissions, and the public at large all have a stake in the naming.
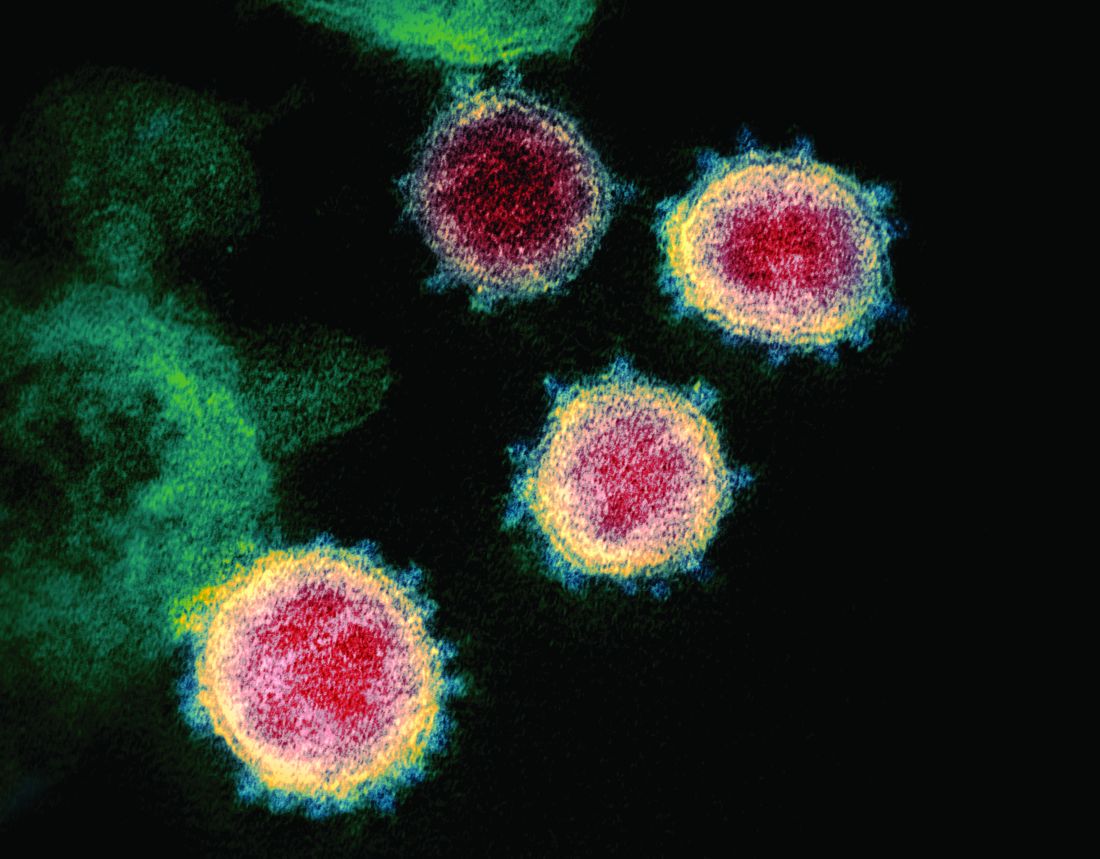
From “Wuhan virus” to “novel coronavirus-2019” to “COVID-19 virus,” the name of the new coronavirus that first appeared in China has been evolving to its now official designation: SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2). But where did the final name come from, how does such a name become official, and who makes it so?
Virus taxonomy
The Coronavirus Study Group (CSG) of the International Committee on Taxonomy of Viruses (ICTV) named the new coronavirus SARS-CoV-2 based upon its genetic relationship to the original SARS-CoV that caused an outbreak of disease in 2002–2003.
According to the ICTV website, the first internationally organized attempts to introduce order into the bewildering variety of viruses took place at the International Congress of Microbiology held in Moscow in 1966 where a committee was created that later became the ICTV and was given the task of developing a single, universal taxonomic scheme for all the viruses infecting animals, plants, fungi, bacteria, and archaea. The ICTV was created as a committee of the virology division of the International Union of Microbiological Societies and is governed by statutes approved by the virology division. Virus classification and nomenclature are subject to rules set out in an International Code.
These designate that: “The universal virus classification system shall employ the hierarchical levels of realm, subrealm, kingdom, subkingdom, phylum, subphylum, class, subclass, order, suborder, family, subfamily, genus, subgenus and species.”
Many of the topmost areas of classification are based on whether the viruses are DNA or RNA, single or double stranded, and have a simple protein shell or a complex lipoprotein envelope. Other levels of classification include host species, type of replication, and type of diseases they cause, the later exemplified in the SARS designation for this virus.
There are 98 international study groups (SGs) covering all major virus orders, families, and genera that are part of the ICTV, and it was the one dedicated to the single-stranded RNA coronaviruses, the CSG, that came up with the SARS-CoV-2 name and first referenced it in their Feb 11 publication in the Cold Springs Harbor preprint journal bioRxiv.
“Based on phylogeny, taxonomy and established practice, the CSG formally recognizes this virus as a sister to severe acute respiratory syndrome coronaviruses (SARS-CoVs) of the species severe acute respiratory syndrome–related coronavirus and designates it as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2),” they wrote.
According to the National Center for Biotechnology Information Taxonomy Browser, with respect to the original SARS CoV virus, of which this is a relative, the full taxonomic designation is: Viruses, Riboviria, Nidovirales, Cornidovirineae, Coronaviridae, Orthocoronavirinae, Betacoronavirus, Sarbecovirus.
The problem with naming names
The World Health Organization currently is not using the official scientific name of the virus, but rather is merely labeling it with regard to the disease: COVID-19, which simply refers to coronavirus disease 2019.
They are following a modern standard by which disease names avoid inflammatory connotations with people and places. Too often in the past from syphilis as the “French pox,” the 1918 influenza as the “Spanish flu,” AIDS as the “gay plague,” Middle East Respiratory Syndrome (MERS), and the currently named “WuFlu,” which made an appearance early in the new outbreak and which is symbolic of a sudden wave of anti-Asian, and specifically Chinese, prejudice.
Chinatown districts even in the United States are being affected economically through unwarranted fear associated with the virus. And there have been equivalently virulent outbreaks of hate speech against Asian individuals in places untouched by the new virus.
However, although SARS-CoV-2 as a name avoids such problems, different considerations led the WHO to reject it in its discussions, determining that its use ties it to tightly to the much more deadly SARS-CoV-1 virus in the public mind, risking greater fear and panic, especially in Asia, where SARS-CoV-1 had the biggest impact.
Back in 1896, William Sykes, MD, writing in the first flush of the triumph of germ theory in modern medicine, attempted to give some guidance to how medical science should best come up with new names of diseases by merging the demands of common parlance with those of taxonomic legitimacy. His “On the Origin and History of Disease-Names,” published in the Lancet, had clearcut advice: “It is vain to attempt to replace a folk name or one widely adopted by the people by a new one deliberately coined by scholars, and this for the following reasons: first, whatever names may be accepted by medical men must be translated by them into the vernacular of their patients, and by a resulting reaction the vernacular name comes to be the commoner one with themselves; and, secondly, there is no continuity or unchangeableness in the terms invented by savants, which are amended, improved upon, and displaced by the next writer on the subject, or, even more absurdly still, by the very inventors themselves in a subsequent publication.”
This is the reason that virus taxonomy provides names based upon unchangeable scientific descriptors of the actual disease causing entity, as illustrated by the decisions of the ICTV. In addition, the genomic sequences being provided by the scientific community are all being organized under the SARS-CoV-2 name and thus are cementing that moniker as the only acceptable scientific one.
Whether the rest of the world universally adopts SARS-CoV-2 as a name is still in question. If the outbreak spreads significantly beyond its current limits, fear and confusion – and simply the need for a more familiar-sounding label – may lead the general public to adopt more colloquial designations than those that science attempts to impose, as Dr. Sykes suggested back in 1896. That remains to be seen.
mlesney@mdedge.com
Mark Lesney is the managing editor of MDedge.com/IDPractioner. He has a PhD in plant virology and a PhD in the history of science, with a focus on the history of biotechnology and medicine. He has served as an adjunct assistant professor of the department of biochemistry and molecular & cellular biology at Georgetown University, Washington.
Coming up with a moniker for the new coronavirus shows the perils of naming names.
Coming up with a moniker for the new coronavirus shows the perils of naming names.
There is no Baby Book of Names or hurricane alphabet to readily name diseases and their causal entities. Throughout history and even in the modern era, a host of considerations have intruded on the decision as to what to call these blights upon humanity. Names have varied from inflammatory to misleading, from colloquial to scientific. And when it concerns a new epidemiological entity such as the latest coronavirus outbreak originating in China, health organizations, media, politicians, scientific taxonomy commissions, and the public at large all have a stake in the naming.
From “Wuhan virus” to “novel coronavirus-2019” to “COVID-19 virus,” the name of the new coronavirus that first appeared in China has been evolving to its now official designation: SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2). But where did the final name come from, how does such a name become official, and who makes it so?
Virus taxonomy
The Coronavirus Study Group (CSG) of the International Committee on Taxonomy of Viruses (ICTV) named the new coronavirus SARS-CoV-2 based upon its genetic relationship to the original SARS-CoV that caused an outbreak of disease in 2002–2003.
According to the ICTV website, the first internationally organized attempts to introduce order into the bewildering variety of viruses took place at the International Congress of Microbiology held in Moscow in 1966 where a committee was created that later became the ICTV and was given the task of developing a single, universal taxonomic scheme for all the viruses infecting animals, plants, fungi, bacteria, and archaea. The ICTV was created as a committee of the virology division of the International Union of Microbiological Societies and is governed by statutes approved by the virology division. Virus classification and nomenclature are subject to rules set out in an International Code.
These designate that: “The universal virus classification system shall employ the hierarchical levels of realm, subrealm, kingdom, subkingdom, phylum, subphylum, class, subclass, order, suborder, family, subfamily, genus, subgenus and species.”
Many of the topmost areas of classification are based on whether the viruses are DNA or RNA, single or double stranded, and have a simple protein shell or a complex lipoprotein envelope. Other levels of classification include host species, type of replication, and type of diseases they cause, the later exemplified in the SARS designation for this virus.
There are 98 international study groups (SGs) covering all major virus orders, families, and genera that are part of the ICTV, and it was the one dedicated to the single-stranded RNA coronaviruses, the CSG, that came up with the SARS-CoV-2 name and first referenced it in their Feb 11 publication in the Cold Springs Harbor preprint journal bioRxiv.
“Based on phylogeny, taxonomy and established practice, the CSG formally recognizes this virus as a sister to severe acute respiratory syndrome coronaviruses (SARS-CoVs) of the species severe acute respiratory syndrome–related coronavirus and designates it as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2),” they wrote.
According to the National Center for Biotechnology Information Taxonomy Browser, with respect to the original SARS CoV virus, of which this is a relative, the full taxonomic designation is: Viruses, Riboviria, Nidovirales, Cornidovirineae, Coronaviridae, Orthocoronavirinae, Betacoronavirus, Sarbecovirus.
The problem with naming names
The World Health Organization currently is not using the official scientific name of the virus, but rather is merely labeling it with regard to the disease: COVID-19, which simply refers to coronavirus disease 2019.
They are following a modern standard by which disease names avoid inflammatory connotations with people and places. Too often in the past from syphilis as the “French pox,” the 1918 influenza as the “Spanish flu,” AIDS as the “gay plague,” Middle East Respiratory Syndrome (MERS), and the currently named “WuFlu,” which made an appearance early in the new outbreak and which is symbolic of a sudden wave of anti-Asian, and specifically Chinese, prejudice.
Chinatown districts even in the United States are being affected economically through unwarranted fear associated with the virus. And there have been equivalently virulent outbreaks of hate speech against Asian individuals in places untouched by the new virus.
However, although SARS-CoV-2 as a name avoids such problems, different considerations led the WHO to reject it in its discussions, determining that its use ties it to tightly to the much more deadly SARS-CoV-1 virus in the public mind, risking greater fear and panic, especially in Asia, where SARS-CoV-1 had the biggest impact.
Back in 1896, William Sykes, MD, writing in the first flush of the triumph of germ theory in modern medicine, attempted to give some guidance to how medical science should best come up with new names of diseases by merging the demands of common parlance with those of taxonomic legitimacy. His “On the Origin and History of Disease-Names,” published in the Lancet, had clearcut advice: “It is vain to attempt to replace a folk name or one widely adopted by the people by a new one deliberately coined by scholars, and this for the following reasons: first, whatever names may be accepted by medical men must be translated by them into the vernacular of their patients, and by a resulting reaction the vernacular name comes to be the commoner one with themselves; and, secondly, there is no continuity or unchangeableness in the terms invented by savants, which are amended, improved upon, and displaced by the next writer on the subject, or, even more absurdly still, by the very inventors themselves in a subsequent publication.”
This is the reason that virus taxonomy provides names based upon unchangeable scientific descriptors of the actual disease causing entity, as illustrated by the decisions of the ICTV. In addition, the genomic sequences being provided by the scientific community are all being organized under the SARS-CoV-2 name and thus are cementing that moniker as the only acceptable scientific one.
Whether the rest of the world universally adopts SARS-CoV-2 as a name is still in question. If the outbreak spreads significantly beyond its current limits, fear and confusion – and simply the need for a more familiar-sounding label – may lead the general public to adopt more colloquial designations than those that science attempts to impose, as Dr. Sykes suggested back in 1896. That remains to be seen.
mlesney@mdedge.com
Mark Lesney is the managing editor of MDedge.com/IDPractioner. He has a PhD in plant virology and a PhD in the history of science, with a focus on the history of biotechnology and medicine. He has served as an adjunct assistant professor of the department of biochemistry and molecular & cellular biology at Georgetown University, Washington.
There is no Baby Book of Names or hurricane alphabet to readily name diseases and their causal entities. Throughout history and even in the modern era, a host of considerations have intruded on the decision as to what to call these blights upon humanity. Names have varied from inflammatory to misleading, from colloquial to scientific. And when it concerns a new epidemiological entity such as the latest coronavirus outbreak originating in China, health organizations, media, politicians, scientific taxonomy commissions, and the public at large all have a stake in the naming.
From “Wuhan virus” to “novel coronavirus-2019” to “COVID-19 virus,” the name of the new coronavirus that first appeared in China has been evolving to its now official designation: SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2). But where did the final name come from, how does such a name become official, and who makes it so?
Virus taxonomy
The Coronavirus Study Group (CSG) of the International Committee on Taxonomy of Viruses (ICTV) named the new coronavirus SARS-CoV-2 based upon its genetic relationship to the original SARS-CoV that caused an outbreak of disease in 2002–2003.
According to the ICTV website, the first internationally organized attempts to introduce order into the bewildering variety of viruses took place at the International Congress of Microbiology held in Moscow in 1966 where a committee was created that later became the ICTV and was given the task of developing a single, universal taxonomic scheme for all the viruses infecting animals, plants, fungi, bacteria, and archaea. The ICTV was created as a committee of the virology division of the International Union of Microbiological Societies and is governed by statutes approved by the virology division. Virus classification and nomenclature are subject to rules set out in an International Code.
These designate that: “The universal virus classification system shall employ the hierarchical levels of realm, subrealm, kingdom, subkingdom, phylum, subphylum, class, subclass, order, suborder, family, subfamily, genus, subgenus and species.”
Many of the topmost areas of classification are based on whether the viruses are DNA or RNA, single or double stranded, and have a simple protein shell or a complex lipoprotein envelope. Other levels of classification include host species, type of replication, and type of diseases they cause, the later exemplified in the SARS designation for this virus.
There are 98 international study groups (SGs) covering all major virus orders, families, and genera that are part of the ICTV, and it was the one dedicated to the single-stranded RNA coronaviruses, the CSG, that came up with the SARS-CoV-2 name and first referenced it in their Feb 11 publication in the Cold Springs Harbor preprint journal bioRxiv.
“Based on phylogeny, taxonomy and established practice, the CSG formally recognizes this virus as a sister to severe acute respiratory syndrome coronaviruses (SARS-CoVs) of the species severe acute respiratory syndrome–related coronavirus and designates it as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2),” they wrote.
According to the National Center for Biotechnology Information Taxonomy Browser, with respect to the original SARS CoV virus, of which this is a relative, the full taxonomic designation is: Viruses, Riboviria, Nidovirales, Cornidovirineae, Coronaviridae, Orthocoronavirinae, Betacoronavirus, Sarbecovirus.
The problem with naming names
The World Health Organization currently is not using the official scientific name of the virus, but rather is merely labeling it with regard to the disease: COVID-19, which simply refers to coronavirus disease 2019.
They are following a modern standard by which disease names avoid inflammatory connotations with people and places. Too often in the past from syphilis as the “French pox,” the 1918 influenza as the “Spanish flu,” AIDS as the “gay plague,” Middle East Respiratory Syndrome (MERS), and the currently named “WuFlu,” which made an appearance early in the new outbreak and which is symbolic of a sudden wave of anti-Asian, and specifically Chinese, prejudice.
Chinatown districts even in the United States are being affected economically through unwarranted fear associated with the virus. And there have been equivalently virulent outbreaks of hate speech against Asian individuals in places untouched by the new virus.
However, although SARS-CoV-2 as a name avoids such problems, different considerations led the WHO to reject it in its discussions, determining that its use ties it to tightly to the much more deadly SARS-CoV-1 virus in the public mind, risking greater fear and panic, especially in Asia, where SARS-CoV-1 had the biggest impact.
Back in 1896, William Sykes, MD, writing in the first flush of the triumph of germ theory in modern medicine, attempted to give some guidance to how medical science should best come up with new names of diseases by merging the demands of common parlance with those of taxonomic legitimacy. His “On the Origin and History of Disease-Names,” published in the Lancet, had clearcut advice: “It is vain to attempt to replace a folk name or one widely adopted by the people by a new one deliberately coined by scholars, and this for the following reasons: first, whatever names may be accepted by medical men must be translated by them into the vernacular of their patients, and by a resulting reaction the vernacular name comes to be the commoner one with themselves; and, secondly, there is no continuity or unchangeableness in the terms invented by savants, which are amended, improved upon, and displaced by the next writer on the subject, or, even more absurdly still, by the very inventors themselves in a subsequent publication.”
This is the reason that virus taxonomy provides names based upon unchangeable scientific descriptors of the actual disease causing entity, as illustrated by the decisions of the ICTV. In addition, the genomic sequences being provided by the scientific community are all being organized under the SARS-CoV-2 name and thus are cementing that moniker as the only acceptable scientific one.
Whether the rest of the world universally adopts SARS-CoV-2 as a name is still in question. If the outbreak spreads significantly beyond its current limits, fear and confusion – and simply the need for a more familiar-sounding label – may lead the general public to adopt more colloquial designations than those that science attempts to impose, as Dr. Sykes suggested back in 1896. That remains to be seen.
mlesney@mdedge.com
Mark Lesney is the managing editor of MDedge.com/IDPractioner. He has a PhD in plant virology and a PhD in the history of science, with a focus on the history of biotechnology and medicine. He has served as an adjunct assistant professor of the department of biochemistry and molecular & cellular biology at Georgetown University, Washington.
Initial FLAG-Ida outperforms 7+3 for high-risk AML
A treatment commonly used as a salvage regimen for relapsed/refractory acute myelogenous leukemia (AML) showed better results than did standard treatment when used as initial induction therapy.
Researchers found that the use of fludarabine, high-dose cytarabine, with granulocyte-colony stimulating factor (FLAG) – with or without idarubicin (Ida) – resulted in higher remission rates for nonfavorable risk AML patients after one course of induction, compared with standard 7+3 (anthracycline plus cytarabine) treatment.
The use of FLAG+/-Ida also resulted in a shorter time to complete remission (CR) and a shorter time to transplant, compared with 7+3. Additionally, postremission overall survival (OS) and disease-free survival (DFS) were better after achieving CR from FLAG-Ida, compared with 7+3, Melhem Solh, MD, of the Blood & Marrow Transplant Program at Northside Hospital, Atlanta, and colleagues reported in Leukemia Research.
The researchers retrospectively analyzed 304 consecutive AML patients, with nonfavorable National Comprehensive Cancer Network (NCCN) risk who received initial treatment at their center with either 7+3 (86 patients) or FLAG+/-Ida (218 patients). They found that patients in the FLAG+/-Ida group were more likely to achieve remission after one course of induction (74% vs. 62%; P less than .001) and had a faster time to achieve CR (30 days vs. 37.5 days; P less than .001), compared with patients receiving 7+3.
The time from diagnosis to allogeneic hematopoietic cell transplantation was shorter among CR patients after FLAG+/-Ida, compared with 7+3 (115 days vs. 151 days; P less than .003).
Additionally, the 3-year postremission OS and DFS were significantly better for patients receiving FLAG-Ida at 54% and 49%, compared with 39% and 32% for 7+3, respectively (P = .01).
Dr. Solh and colleagues found that the factors associated with postremission survival included age at first CR, NCCN risk, induction regimen (FLAG+/-Ida vs. 7+3; hazard ratio, 0.62; P = .01), and receipt of allogeneic hematopoietic cell transplantation.
“FLAG-Ida is a more efficacious regimen when used for initial induction compared to 3+7. It appears to produce better survival and improved postremission survival for AML patients with intermediate and poor risk AML without increasing toxicity,” Dr. Solh and colleagues wrote. “Further validation of these results in a well-designed randomized prospective trial will help define the best induction approaches for AML.”
There was no outside funding for this research and the authors reported that they had no relevant financial conflicts.
SOURCE: Solh MM et al. Leuk Res. 2020 Feb 14. doi: 10.1016/j.leukres.2020.106318.
A treatment commonly used as a salvage regimen for relapsed/refractory acute myelogenous leukemia (AML) showed better results than did standard treatment when used as initial induction therapy.
Researchers found that the use of fludarabine, high-dose cytarabine, with granulocyte-colony stimulating factor (FLAG) – with or without idarubicin (Ida) – resulted in higher remission rates for nonfavorable risk AML patients after one course of induction, compared with standard 7+3 (anthracycline plus cytarabine) treatment.
The use of FLAG+/-Ida also resulted in a shorter time to complete remission (CR) and a shorter time to transplant, compared with 7+3. Additionally, postremission overall survival (OS) and disease-free survival (DFS) were better after achieving CR from FLAG-Ida, compared with 7+3, Melhem Solh, MD, of the Blood & Marrow Transplant Program at Northside Hospital, Atlanta, and colleagues reported in Leukemia Research.
The researchers retrospectively analyzed 304 consecutive AML patients, with nonfavorable National Comprehensive Cancer Network (NCCN) risk who received initial treatment at their center with either 7+3 (86 patients) or FLAG+/-Ida (218 patients). They found that patients in the FLAG+/-Ida group were more likely to achieve remission after one course of induction (74% vs. 62%; P less than .001) and had a faster time to achieve CR (30 days vs. 37.5 days; P less than .001), compared with patients receiving 7+3.
The time from diagnosis to allogeneic hematopoietic cell transplantation was shorter among CR patients after FLAG+/-Ida, compared with 7+3 (115 days vs. 151 days; P less than .003).
Additionally, the 3-year postremission OS and DFS were significantly better for patients receiving FLAG-Ida at 54% and 49%, compared with 39% and 32% for 7+3, respectively (P = .01).
Dr. Solh and colleagues found that the factors associated with postremission survival included age at first CR, NCCN risk, induction regimen (FLAG+/-Ida vs. 7+3; hazard ratio, 0.62; P = .01), and receipt of allogeneic hematopoietic cell transplantation.
“FLAG-Ida is a more efficacious regimen when used for initial induction compared to 3+7. It appears to produce better survival and improved postremission survival for AML patients with intermediate and poor risk AML without increasing toxicity,” Dr. Solh and colleagues wrote. “Further validation of these results in a well-designed randomized prospective trial will help define the best induction approaches for AML.”
There was no outside funding for this research and the authors reported that they had no relevant financial conflicts.
SOURCE: Solh MM et al. Leuk Res. 2020 Feb 14. doi: 10.1016/j.leukres.2020.106318.
A treatment commonly used as a salvage regimen for relapsed/refractory acute myelogenous leukemia (AML) showed better results than did standard treatment when used as initial induction therapy.
Researchers found that the use of fludarabine, high-dose cytarabine, with granulocyte-colony stimulating factor (FLAG) – with or without idarubicin (Ida) – resulted in higher remission rates for nonfavorable risk AML patients after one course of induction, compared with standard 7+3 (anthracycline plus cytarabine) treatment.
The use of FLAG+/-Ida also resulted in a shorter time to complete remission (CR) and a shorter time to transplant, compared with 7+3. Additionally, postremission overall survival (OS) and disease-free survival (DFS) were better after achieving CR from FLAG-Ida, compared with 7+3, Melhem Solh, MD, of the Blood & Marrow Transplant Program at Northside Hospital, Atlanta, and colleagues reported in Leukemia Research.
The researchers retrospectively analyzed 304 consecutive AML patients, with nonfavorable National Comprehensive Cancer Network (NCCN) risk who received initial treatment at their center with either 7+3 (86 patients) or FLAG+/-Ida (218 patients). They found that patients in the FLAG+/-Ida group were more likely to achieve remission after one course of induction (74% vs. 62%; P less than .001) and had a faster time to achieve CR (30 days vs. 37.5 days; P less than .001), compared with patients receiving 7+3.
The time from diagnosis to allogeneic hematopoietic cell transplantation was shorter among CR patients after FLAG+/-Ida, compared with 7+3 (115 days vs. 151 days; P less than .003).
Additionally, the 3-year postremission OS and DFS were significantly better for patients receiving FLAG-Ida at 54% and 49%, compared with 39% and 32% for 7+3, respectively (P = .01).
Dr. Solh and colleagues found that the factors associated with postremission survival included age at first CR, NCCN risk, induction regimen (FLAG+/-Ida vs. 7+3; hazard ratio, 0.62; P = .01), and receipt of allogeneic hematopoietic cell transplantation.
“FLAG-Ida is a more efficacious regimen when used for initial induction compared to 3+7. It appears to produce better survival and improved postremission survival for AML patients with intermediate and poor risk AML without increasing toxicity,” Dr. Solh and colleagues wrote. “Further validation of these results in a well-designed randomized prospective trial will help define the best induction approaches for AML.”
There was no outside funding for this research and the authors reported that they had no relevant financial conflicts.
SOURCE: Solh MM et al. Leuk Res. 2020 Feb 14. doi: 10.1016/j.leukres.2020.106318.
FROM LEUKEMIA RESEARCH
WHO urges that ‘window of opportunity’ on containing novel coronavirus not be missed
As of 6 a.m. Geneva time, Feb. 21, China reported 75,567 cases of COVID-19 and 2,239 deaths, including a total of 892 new confirmed cases that were reported in China in the past 24 hours, with 118 deaths, stated Tedros Adhanom Ghebreyesus, MD, World Health Organization Director-General, in a Feb. 21 news conference on the COVID-19 outbreak.

What he described as “the significant decline in newly reported cases” is partly because of a change in reporting in which China switched from including “clinically diagnosed” cases to reporting only “suspected” and “laboratory confirmed cases.” The reporting procedure changed because the medical facilities in Wuhan regained the capability of checking all suspected cases with laboratory tests. As a result, some cases that were clinically confirmed were subtracted from the total because they tested negative, said Dr. Ghebreyesus.
Although the number of cases in Hubuei province continues to decline, the WHO is concerned about an increase seen in Shandong province and they are seeking more information. Outside China, there are now 1,152 cases in 26 countries and eight deaths. “Although the number of cases outside of China remains small, we are concerned about the cases with no clear epidemiological link, such as travel history to China, or contact with a confirmed case,” said Dr. Ghebreyesus. “Apart from the Diamond Princess cruise ship, the Republic of Korea now has the most cases outside China, and we are working closely with that government to understand the transmission dynamics that led to this increase.”
“We are also concerned about the increase of cases in the Islamic Republic of Iran, where there are now 18 cases and four deaths in just the past 2 days.”
“Our particular concern is for COVID-19 to spread in countries with weaker health systems,” he said, adding that tomorrow, he will address an emergency meeting of African health ministers held jointly by the African Union and the African Centres for Disease Control and Prevention on dealing with COVID-19.
Dr. Ghebreyesus also announced that today the WHO has designated six special envoys on COVID-19 to provide strategic advice and high-level political advocacy and engagement in different parts of the world.
In his remarks, Dr. Ghebreyesus particularly stressed that: “The measures that China and other countries have taken have given us a fighting chance of containing the spread of the virus. We call on all countries to continue their commitment for containment measures, while preparing for community transmission if it occurs. We must not look back and regret that we failed to take advantage of the window of opportunity that we have now.”
In the question and answer period, Dr. Ghebreyesus specifically addressed the issue of misinformation and conspiracy theories being promulgated by certain individuals and on social media about the source of the virus, especially those people who believe that it was designed in a Chinese virus laboratory. Scientists play an important role in refuting such particular misinformation, he said, and research must continue to track down the actual source in nature.
To that regard, a paper published online in the Lancet on Feb. 19, provided a consensus statement by more than 25 health scientists outside of China stating: “We stand together to strongly condemn conspiracy theories suggesting that COVID-19 does not have a natural origin. Scientists from multiple countries have published and analyzed genomes of the causative agent, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and they overwhelmingly conclude that this coronavirus originated in wildlife, as have so many other emerging pathogens.”
The WHO issues daily coronavirus disease (COVID-2019) situation reports on its website and provides these telebriefing updates daily.
As of 6 a.m. Geneva time, Feb. 21, China reported 75,567 cases of COVID-19 and 2,239 deaths, including a total of 892 new confirmed cases that were reported in China in the past 24 hours, with 118 deaths, stated Tedros Adhanom Ghebreyesus, MD, World Health Organization Director-General, in a Feb. 21 news conference on the COVID-19 outbreak.
What he described as “the significant decline in newly reported cases” is partly because of a change in reporting in which China switched from including “clinically diagnosed” cases to reporting only “suspected” and “laboratory confirmed cases.” The reporting procedure changed because the medical facilities in Wuhan regained the capability of checking all suspected cases with laboratory tests. As a result, some cases that were clinically confirmed were subtracted from the total because they tested negative, said Dr. Ghebreyesus.
Although the number of cases in Hubuei province continues to decline, the WHO is concerned about an increase seen in Shandong province and they are seeking more information. Outside China, there are now 1,152 cases in 26 countries and eight deaths. “Although the number of cases outside of China remains small, we are concerned about the cases with no clear epidemiological link, such as travel history to China, or contact with a confirmed case,” said Dr. Ghebreyesus. “Apart from the Diamond Princess cruise ship, the Republic of Korea now has the most cases outside China, and we are working closely with that government to understand the transmission dynamics that led to this increase.”
“We are also concerned about the increase of cases in the Islamic Republic of Iran, where there are now 18 cases and four deaths in just the past 2 days.”
“Our particular concern is for COVID-19 to spread in countries with weaker health systems,” he said, adding that tomorrow, he will address an emergency meeting of African health ministers held jointly by the African Union and the African Centres for Disease Control and Prevention on dealing with COVID-19.
Dr. Ghebreyesus also announced that today the WHO has designated six special envoys on COVID-19 to provide strategic advice and high-level political advocacy and engagement in different parts of the world.
In his remarks, Dr. Ghebreyesus particularly stressed that: “The measures that China and other countries have taken have given us a fighting chance of containing the spread of the virus. We call on all countries to continue their commitment for containment measures, while preparing for community transmission if it occurs. We must not look back and regret that we failed to take advantage of the window of opportunity that we have now.”
In the question and answer period, Dr. Ghebreyesus specifically addressed the issue of misinformation and conspiracy theories being promulgated by certain individuals and on social media about the source of the virus, especially those people who believe that it was designed in a Chinese virus laboratory. Scientists play an important role in refuting such particular misinformation, he said, and research must continue to track down the actual source in nature.
To that regard, a paper published online in the Lancet on Feb. 19, provided a consensus statement by more than 25 health scientists outside of China stating: “We stand together to strongly condemn conspiracy theories suggesting that COVID-19 does not have a natural origin. Scientists from multiple countries have published and analyzed genomes of the causative agent, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and they overwhelmingly conclude that this coronavirus originated in wildlife, as have so many other emerging pathogens.”
The WHO issues daily coronavirus disease (COVID-2019) situation reports on its website and provides these telebriefing updates daily.
As of 6 a.m. Geneva time, Feb. 21, China reported 75,567 cases of COVID-19 and 2,239 deaths, including a total of 892 new confirmed cases that were reported in China in the past 24 hours, with 118 deaths, stated Tedros Adhanom Ghebreyesus, MD, World Health Organization Director-General, in a Feb. 21 news conference on the COVID-19 outbreak.
What he described as “the significant decline in newly reported cases” is partly because of a change in reporting in which China switched from including “clinically diagnosed” cases to reporting only “suspected” and “laboratory confirmed cases.” The reporting procedure changed because the medical facilities in Wuhan regained the capability of checking all suspected cases with laboratory tests. As a result, some cases that were clinically confirmed were subtracted from the total because they tested negative, said Dr. Ghebreyesus.
Although the number of cases in Hubuei province continues to decline, the WHO is concerned about an increase seen in Shandong province and they are seeking more information. Outside China, there are now 1,152 cases in 26 countries and eight deaths. “Although the number of cases outside of China remains small, we are concerned about the cases with no clear epidemiological link, such as travel history to China, or contact with a confirmed case,” said Dr. Ghebreyesus. “Apart from the Diamond Princess cruise ship, the Republic of Korea now has the most cases outside China, and we are working closely with that government to understand the transmission dynamics that led to this increase.”
“We are also concerned about the increase of cases in the Islamic Republic of Iran, where there are now 18 cases and four deaths in just the past 2 days.”
“Our particular concern is for COVID-19 to spread in countries with weaker health systems,” he said, adding that tomorrow, he will address an emergency meeting of African health ministers held jointly by the African Union and the African Centres for Disease Control and Prevention on dealing with COVID-19.
Dr. Ghebreyesus also announced that today the WHO has designated six special envoys on COVID-19 to provide strategic advice and high-level political advocacy and engagement in different parts of the world.
In his remarks, Dr. Ghebreyesus particularly stressed that: “The measures that China and other countries have taken have given us a fighting chance of containing the spread of the virus. We call on all countries to continue their commitment for containment measures, while preparing for community transmission if it occurs. We must not look back and regret that we failed to take advantage of the window of opportunity that we have now.”
In the question and answer period, Dr. Ghebreyesus specifically addressed the issue of misinformation and conspiracy theories being promulgated by certain individuals and on social media about the source of the virus, especially those people who believe that it was designed in a Chinese virus laboratory. Scientists play an important role in refuting such particular misinformation, he said, and research must continue to track down the actual source in nature.
To that regard, a paper published online in the Lancet on Feb. 19, provided a consensus statement by more than 25 health scientists outside of China stating: “We stand together to strongly condemn conspiracy theories suggesting that COVID-19 does not have a natural origin. Scientists from multiple countries have published and analyzed genomes of the causative agent, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), and they overwhelmingly conclude that this coronavirus originated in wildlife, as have so many other emerging pathogens.”
The WHO issues daily coronavirus disease (COVID-2019) situation reports on its website and provides these telebriefing updates daily.
REPORTING FROM A WHO TELEBRIEFING

2019-nCoV: Structure, characteristics of key potential therapy target determined
Researchers have identified the structure of a protein that could turn out to be a potential vaccine target for the 2019-nCoV.
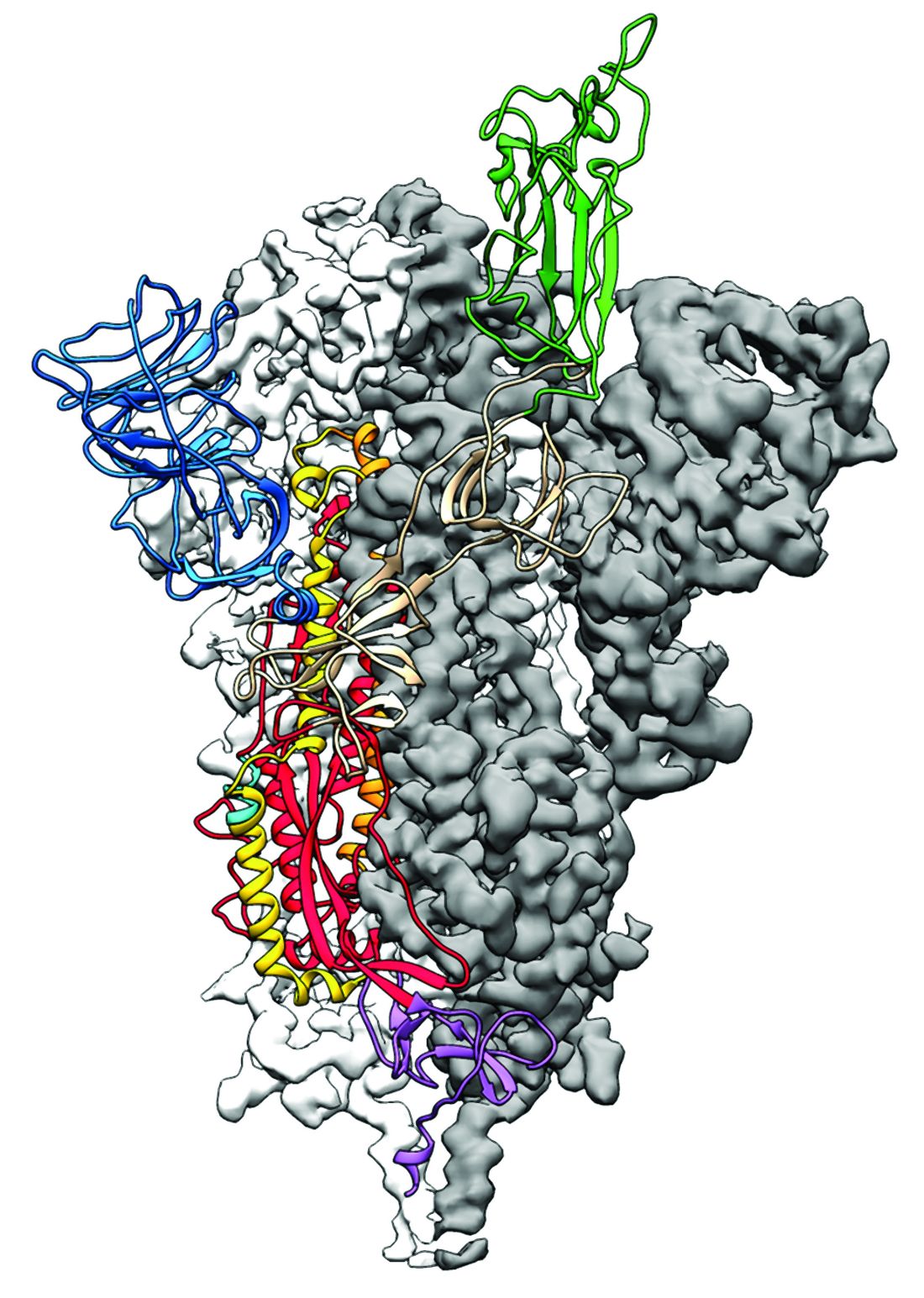
As is typical of other coronaviruses, 2019-nCoV makes use of a densely glycosylated spike protein to gain entry into host cells. The spike protein is a trimeric class I fusion protein that exists in a metastable prefusion conformation that undergoes a dramatic structural rearrangement to fuse the viral membrane with the host-cell membrane, according to Daniel Wrapp of the University of Texas at Austin and colleagues.
The researchers performed a study to synthesize and determine the 3-D structure of the spike protein because it is a logical target for vaccine development and for the development of targeted therapeutics for COVID-19, the disease caused by the virus.
“As soon as we knew this was a coronavirus, we felt we had to jump at it,” senior author Jason S. McLellan, PhD, associate professor of molecular science, said in a press release from the University, “because we could be one of the first ones to get this structure. We knew exactly what mutations to put into this because we’ve already shown these mutations work for a bunch of other coronaviruses.”
Because recent reports by other researchers demonstrated that 2019-nCoV and SARS-CoV spike proteins share the same functional host-cell receptor–angiotensin-converting enzyme 2 (ACE2), Dr. McLellan and his colleagues examined the relation between the two viruses. They found biophysical and structural evidence that the 2019-nCoV spike protein binds ACE2 with higher affinity than the closely related SARS-CoV spike protein. “The high affinity of 2019-nCoV S for human ACE2 may contribute to the apparent ease with which 2019-nCoV can spread from human-to-human; however, additional studies are needed to investigate this possibility,” the researchers wrote.
Focusing their attention on the receptor-binding domain (RBD) of the 2019-nCoV spike protein, they tested several published SARS-CoV RBD-specific monoclonal antibodies against it and found that these antibodies showed no appreciable binding to 2019-nCoV spike protein, which suggests limited antibody cross-reactivity. For this reason, they suggested that future antibody isolation and therapeutic design efforts will benefit from specifically using 2019-nCoV spike proteins as probes.
“This information will support precision vaccine design and discovery of anti-viral therapeutics, accelerating medical countermeasure development,” they concluded.
The research was supported in part by an National Institutes of Health/National Institute of Allergy and Infectious Diseases grant and by intramural funding from the National Institute of Allergy and Infectious Diseases. Four authors are inventors on US patent application No. 62/412,703 (Prefusion Coronavirus Spike Proteins and Their Use) and all are inventors on US patent application No. 62/972,886 (2019-nCoV Vaccine).
SOURCE: Wrapp D et al. Science. 2020 Feb 19. doi: 10.1126/science.abb2507.
Researchers have identified the structure of a protein that could turn out to be a potential vaccine target for the 2019-nCoV.
As is typical of other coronaviruses, 2019-nCoV makes use of a densely glycosylated spike protein to gain entry into host cells. The spike protein is a trimeric class I fusion protein that exists in a metastable prefusion conformation that undergoes a dramatic structural rearrangement to fuse the viral membrane with the host-cell membrane, according to Daniel Wrapp of the University of Texas at Austin and colleagues.
The researchers performed a study to synthesize and determine the 3-D structure of the spike protein because it is a logical target for vaccine development and for the development of targeted therapeutics for COVID-19, the disease caused by the virus.
“As soon as we knew this was a coronavirus, we felt we had to jump at it,” senior author Jason S. McLellan, PhD, associate professor of molecular science, said in a press release from the University, “because we could be one of the first ones to get this structure. We knew exactly what mutations to put into this because we’ve already shown these mutations work for a bunch of other coronaviruses.”
Because recent reports by other researchers demonstrated that 2019-nCoV and SARS-CoV spike proteins share the same functional host-cell receptor–angiotensin-converting enzyme 2 (ACE2), Dr. McLellan and his colleagues examined the relation between the two viruses. They found biophysical and structural evidence that the 2019-nCoV spike protein binds ACE2 with higher affinity than the closely related SARS-CoV spike protein. “The high affinity of 2019-nCoV S for human ACE2 may contribute to the apparent ease with which 2019-nCoV can spread from human-to-human; however, additional studies are needed to investigate this possibility,” the researchers wrote.
Focusing their attention on the receptor-binding domain (RBD) of the 2019-nCoV spike protein, they tested several published SARS-CoV RBD-specific monoclonal antibodies against it and found that these antibodies showed no appreciable binding to 2019-nCoV spike protein, which suggests limited antibody cross-reactivity. For this reason, they suggested that future antibody isolation and therapeutic design efforts will benefit from specifically using 2019-nCoV spike proteins as probes.
“This information will support precision vaccine design and discovery of anti-viral therapeutics, accelerating medical countermeasure development,” they concluded.
The research was supported in part by an National Institutes of Health/National Institute of Allergy and Infectious Diseases grant and by intramural funding from the National Institute of Allergy and Infectious Diseases. Four authors are inventors on US patent application No. 62/412,703 (Prefusion Coronavirus Spike Proteins and Their Use) and all are inventors on US patent application No. 62/972,886 (2019-nCoV Vaccine).
SOURCE: Wrapp D et al. Science. 2020 Feb 19. doi: 10.1126/science.abb2507.
Researchers have identified the structure of a protein that could turn out to be a potential vaccine target for the 2019-nCoV.
As is typical of other coronaviruses, 2019-nCoV makes use of a densely glycosylated spike protein to gain entry into host cells. The spike protein is a trimeric class I fusion protein that exists in a metastable prefusion conformation that undergoes a dramatic structural rearrangement to fuse the viral membrane with the host-cell membrane, according to Daniel Wrapp of the University of Texas at Austin and colleagues.
The researchers performed a study to synthesize and determine the 3-D structure of the spike protein because it is a logical target for vaccine development and for the development of targeted therapeutics for COVID-19, the disease caused by the virus.
“As soon as we knew this was a coronavirus, we felt we had to jump at it,” senior author Jason S. McLellan, PhD, associate professor of molecular science, said in a press release from the University, “because we could be one of the first ones to get this structure. We knew exactly what mutations to put into this because we’ve already shown these mutations work for a bunch of other coronaviruses.”
Because recent reports by other researchers demonstrated that 2019-nCoV and SARS-CoV spike proteins share the same functional host-cell receptor–angiotensin-converting enzyme 2 (ACE2), Dr. McLellan and his colleagues examined the relation between the two viruses. They found biophysical and structural evidence that the 2019-nCoV spike protein binds ACE2 with higher affinity than the closely related SARS-CoV spike protein. “The high affinity of 2019-nCoV S for human ACE2 may contribute to the apparent ease with which 2019-nCoV can spread from human-to-human; however, additional studies are needed to investigate this possibility,” the researchers wrote.
Focusing their attention on the receptor-binding domain (RBD) of the 2019-nCoV spike protein, they tested several published SARS-CoV RBD-specific monoclonal antibodies against it and found that these antibodies showed no appreciable binding to 2019-nCoV spike protein, which suggests limited antibody cross-reactivity. For this reason, they suggested that future antibody isolation and therapeutic design efforts will benefit from specifically using 2019-nCoV spike proteins as probes.
“This information will support precision vaccine design and discovery of anti-viral therapeutics, accelerating medical countermeasure development,” they concluded.
The research was supported in part by an National Institutes of Health/National Institute of Allergy and Infectious Diseases grant and by intramural funding from the National Institute of Allergy and Infectious Diseases. Four authors are inventors on US patent application No. 62/412,703 (Prefusion Coronavirus Spike Proteins and Their Use) and all are inventors on US patent application No. 62/972,886 (2019-nCoV Vaccine).
SOURCE: Wrapp D et al. Science. 2020 Feb 19. doi: 10.1126/science.abb2507.
FROM SCIENCE
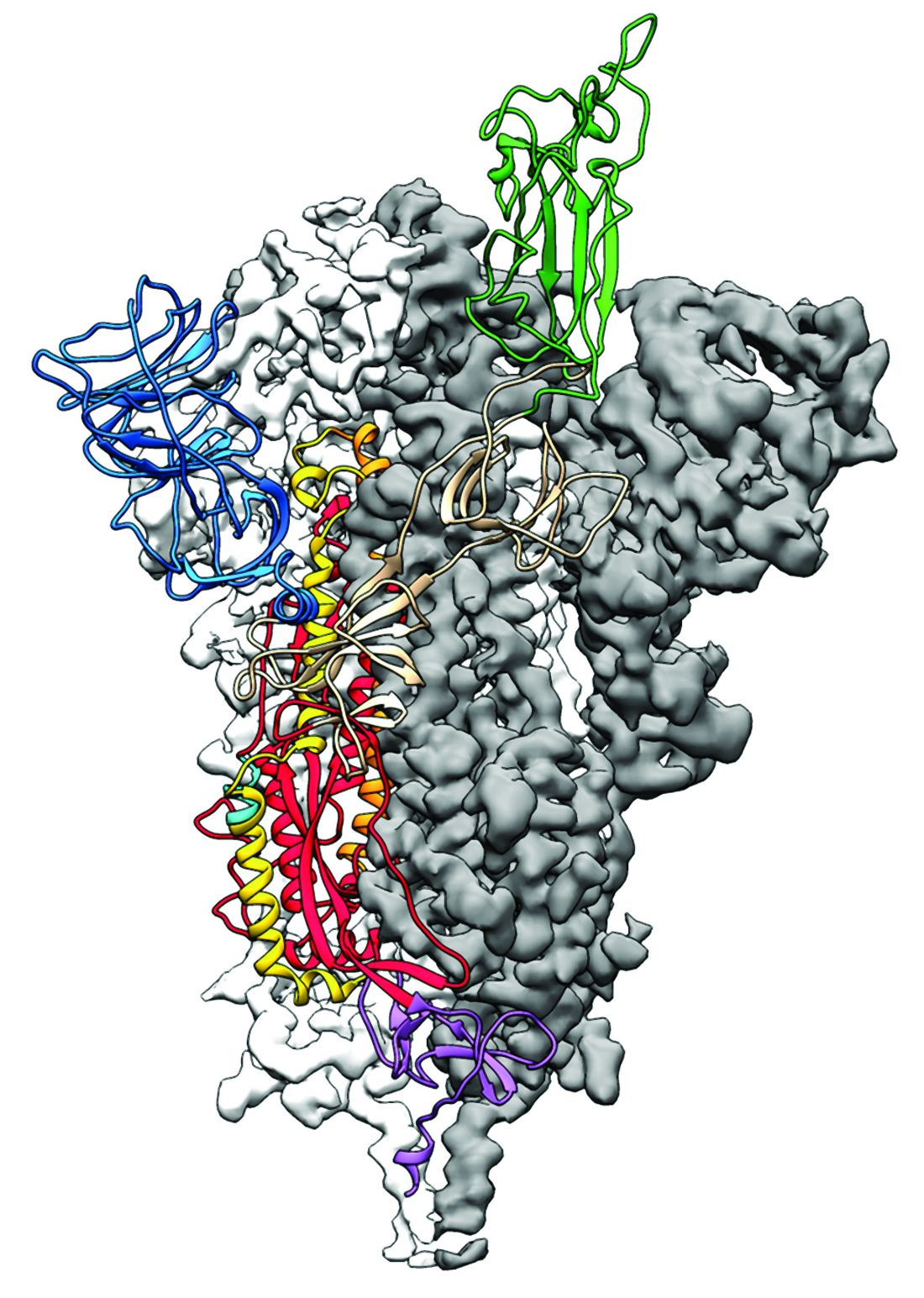
Screen pregnant women with suspected 2019-nCoV infection
It is too early yet to explicitly determine the effects of the Novel Coronavirus (2019-nCoV) on pregnant women and their fetuses. This is a critical concern, because members of the coronavirus family, which have been responsible for previous outbreaks of severe acute respiratory syndrome (SARS-CoV) and Middle East respiratory syndrome (MERS-CoV), have demonstrated their ability to cause severe complications during pregnancy, according to researchers.
The SARS virus outbreak and the more recent MERS virus outbreak provide the best available models with which to examine the potential impact of 2019-nCoV on pregnancy, according to a letter published online in the Lancet.
Twelve pregnant women were infected with SARS-CoV during the 2002-2003 pandemic. Three (25%) of these women died during pregnancy. Overall, four of seven women had a miscarriage in the first trimester. In the second or third trimester, two out of five women had fetal growth restriction, and four of the five had preterm birth (one case was spontaneous and three were induced because of the maternal condition), according to corresponding author David Baud, MD, PhD, of the maternal-fetal and obstetrics research unit at Lausanne (Switzerland) University Hospital, and colleagues.
A review of 11 pregnant women infected with the virus showed that 10 women (91%) presented with adverse outcomes. Six (55%) neonates were admitted to the ICU; three (27%) died. Two neonates were delivered prematurely because their mothers developed severe respiratory failure.
Because 2019-nCov has a potential for similar behavior, “we recommend systematic screening of any suspected 2019-nCoV infection during pregnancy. If 2019-nCoV infection during pregnancy is confirmed, extended follow-up should be recommended for mothers and their fetuses,” concluded Dr. Baud and colleagues.
Dr. Baud and associates are known for their previous research on the impacts of the Zika virus on pregnancy. They reported having no competing interests.
SOURCE: Baud D et al. Lancet. 2020 Feb 6. doi: 10.1016/S0140-6736(20)30311-1.
The coronavirus has been spreading rapidly in China, and recently, international cases have been identified, including within the United States. As the article by Locher et al. suggests, mechanical, physiological, and immune adaptations in pregnancy leave pregnant women at risk of severe complications from respiratory illnesses.
Obstetricians need to be prepared to screen, test, and promptly treat pregnant women with any severe respiratory illness to reduce maternal and perinatal morbidity. At this time, the Centers for Disease Control and Prevention advises that any patient with fever and signs of a lower respiratory infection, as well as an epidemiologic risk factor (such as recent travel to China), should be considered at risk for the coronavirus. Samples are collected and sent to the CDC as testing can be done only at the CDC at this time. Please refer to the CDC website for up-to-date guidance for health care professionals.
Unfortunately, there is no specific treatment for coronavirus. Clinical management includes prompt implementation of recommended infection prevention and control measures. Supportive management of complications, including fever reduction and advanced organ support, should be provided as necessary.
While coronavirus is a terrifying potential threat, it’s worth mentioning that, for most pregnant women, a much more likely threat is influenza. Pregnant women with influenza virus infection are at increased risk for progression to pneumonia, ICU admission, preterm delivery, and maternal death. The influenza vaccine can help reduce these risks, and we should continue to encourage vaccination for all pregnant women. Prompt treatment is important! Treatment within 48 hours of the onset of symptoms is ideal, but treatment should not be withheld if the ideal window is missed.
Finally, don’t forget to remind your pregnant patients to avoid close contact with sick family members and friends, wash hands frequently, and call the doctor’s office with any sign of a flu-like illness!
Angela Martin, MD, is an assistant professor of gynecology and obstetrics in the division of maternal-fetal medicine at the University of Kansas Medical Center in Kansas City. She is a member of the Ob.Gyn. News editorial advisory board.
The coronavirus has been spreading rapidly in China, and recently, international cases have been identified, including within the United States. As the article by Locher et al. suggests, mechanical, physiological, and immune adaptations in pregnancy leave pregnant women at risk of severe complications from respiratory illnesses.
Obstetricians need to be prepared to screen, test, and promptly treat pregnant women with any severe respiratory illness to reduce maternal and perinatal morbidity. At this time, the Centers for Disease Control and Prevention advises that any patient with fever and signs of a lower respiratory infection, as well as an epidemiologic risk factor (such as recent travel to China), should be considered at risk for the coronavirus. Samples are collected and sent to the CDC as testing can be done only at the CDC at this time. Please refer to the CDC website for up-to-date guidance for health care professionals.
Unfortunately, there is no specific treatment for coronavirus. Clinical management includes prompt implementation of recommended infection prevention and control measures. Supportive management of complications, including fever reduction and advanced organ support, should be provided as necessary.
While coronavirus is a terrifying potential threat, it’s worth mentioning that, for most pregnant women, a much more likely threat is influenza. Pregnant women with influenza virus infection are at increased risk for progression to pneumonia, ICU admission, preterm delivery, and maternal death. The influenza vaccine can help reduce these risks, and we should continue to encourage vaccination for all pregnant women. Prompt treatment is important! Treatment within 48 hours of the onset of symptoms is ideal, but treatment should not be withheld if the ideal window is missed.
Finally, don’t forget to remind your pregnant patients to avoid close contact with sick family members and friends, wash hands frequently, and call the doctor’s office with any sign of a flu-like illness!
Angela Martin, MD, is an assistant professor of gynecology and obstetrics in the division of maternal-fetal medicine at the University of Kansas Medical Center in Kansas City. She is a member of the Ob.Gyn. News editorial advisory board.
The coronavirus has been spreading rapidly in China, and recently, international cases have been identified, including within the United States. As the article by Locher et al. suggests, mechanical, physiological, and immune adaptations in pregnancy leave pregnant women at risk of severe complications from respiratory illnesses.
Obstetricians need to be prepared to screen, test, and promptly treat pregnant women with any severe respiratory illness to reduce maternal and perinatal morbidity. At this time, the Centers for Disease Control and Prevention advises that any patient with fever and signs of a lower respiratory infection, as well as an epidemiologic risk factor (such as recent travel to China), should be considered at risk for the coronavirus. Samples are collected and sent to the CDC as testing can be done only at the CDC at this time. Please refer to the CDC website for up-to-date guidance for health care professionals.
Unfortunately, there is no specific treatment for coronavirus. Clinical management includes prompt implementation of recommended infection prevention and control measures. Supportive management of complications, including fever reduction and advanced organ support, should be provided as necessary.
While coronavirus is a terrifying potential threat, it’s worth mentioning that, for most pregnant women, a much more likely threat is influenza. Pregnant women with influenza virus infection are at increased risk for progression to pneumonia, ICU admission, preterm delivery, and maternal death. The influenza vaccine can help reduce these risks, and we should continue to encourage vaccination for all pregnant women. Prompt treatment is important! Treatment within 48 hours of the onset of symptoms is ideal, but treatment should not be withheld if the ideal window is missed.
Finally, don’t forget to remind your pregnant patients to avoid close contact with sick family members and friends, wash hands frequently, and call the doctor’s office with any sign of a flu-like illness!
Angela Martin, MD, is an assistant professor of gynecology and obstetrics in the division of maternal-fetal medicine at the University of Kansas Medical Center in Kansas City. She is a member of the Ob.Gyn. News editorial advisory board.
It is too early yet to explicitly determine the effects of the Novel Coronavirus (2019-nCoV) on pregnant women and their fetuses. This is a critical concern, because members of the coronavirus family, which have been responsible for previous outbreaks of severe acute respiratory syndrome (SARS-CoV) and Middle East respiratory syndrome (MERS-CoV), have demonstrated their ability to cause severe complications during pregnancy, according to researchers.
The SARS virus outbreak and the more recent MERS virus outbreak provide the best available models with which to examine the potential impact of 2019-nCoV on pregnancy, according to a letter published online in the Lancet.
Twelve pregnant women were infected with SARS-CoV during the 2002-2003 pandemic. Three (25%) of these women died during pregnancy. Overall, four of seven women had a miscarriage in the first trimester. In the second or third trimester, two out of five women had fetal growth restriction, and four of the five had preterm birth (one case was spontaneous and three were induced because of the maternal condition), according to corresponding author David Baud, MD, PhD, of the maternal-fetal and obstetrics research unit at Lausanne (Switzerland) University Hospital, and colleagues.
A review of 11 pregnant women infected with the virus showed that 10 women (91%) presented with adverse outcomes. Six (55%) neonates were admitted to the ICU; three (27%) died. Two neonates were delivered prematurely because their mothers developed severe respiratory failure.
Because 2019-nCov has a potential for similar behavior, “we recommend systematic screening of any suspected 2019-nCoV infection during pregnancy. If 2019-nCoV infection during pregnancy is confirmed, extended follow-up should be recommended for mothers and their fetuses,” concluded Dr. Baud and colleagues.
Dr. Baud and associates are known for their previous research on the impacts of the Zika virus on pregnancy. They reported having no competing interests.
SOURCE: Baud D et al. Lancet. 2020 Feb 6. doi: 10.1016/S0140-6736(20)30311-1.
It is too early yet to explicitly determine the effects of the Novel Coronavirus (2019-nCoV) on pregnant women and their fetuses. This is a critical concern, because members of the coronavirus family, which have been responsible for previous outbreaks of severe acute respiratory syndrome (SARS-CoV) and Middle East respiratory syndrome (MERS-CoV), have demonstrated their ability to cause severe complications during pregnancy, according to researchers.
The SARS virus outbreak and the more recent MERS virus outbreak provide the best available models with which to examine the potential impact of 2019-nCoV on pregnancy, according to a letter published online in the Lancet.
Twelve pregnant women were infected with SARS-CoV during the 2002-2003 pandemic. Three (25%) of these women died during pregnancy. Overall, four of seven women had a miscarriage in the first trimester. In the second or third trimester, two out of five women had fetal growth restriction, and four of the five had preterm birth (one case was spontaneous and three were induced because of the maternal condition), according to corresponding author David Baud, MD, PhD, of the maternal-fetal and obstetrics research unit at Lausanne (Switzerland) University Hospital, and colleagues.
A review of 11 pregnant women infected with the virus showed that 10 women (91%) presented with adverse outcomes. Six (55%) neonates were admitted to the ICU; three (27%) died. Two neonates were delivered prematurely because their mothers developed severe respiratory failure.
Because 2019-nCov has a potential for similar behavior, “we recommend systematic screening of any suspected 2019-nCoV infection during pregnancy. If 2019-nCoV infection during pregnancy is confirmed, extended follow-up should be recommended for mothers and their fetuses,” concluded Dr. Baud and colleagues.
Dr. Baud and associates are known for their previous research on the impacts of the Zika virus on pregnancy. They reported having no competing interests.
SOURCE: Baud D et al. Lancet. 2020 Feb 6. doi: 10.1016/S0140-6736(20)30311-1.
FROM THE LANCET
Next-generation sequencing can expedite surveillance/discovery of new bat coronaviruses
Enrichment next-generation sequencing (NGS) provides a more cost-efficient and sensitive method for detecting and sequencing novel coronaviruses from wild bat populations, according to a study reported in mSphere, an open-access journal from the American Society for Microbiology.

With the appearance of the new zoonotic Wuhan coronavirus, the importance of monitoring the likelihood of new virus risks in wildlife reservoirs has been heightened. Bats in particular have been found to be the most common reservoir of coronaviruses, including being a probable source or mixing vessel for two previous modern epidemic coronaviruses: SARS (severe acute respiratory syndrome) and MERS (Middle East respiratory syndrome).
“We should be alert and vigilant with the knowledge that bat CoVs [coronaviruses] are likely to cause another disease outbreak, not only because of their prevalence but also because the high frequency of recombination between viruses may lead to the generation of viruses with changes in virulence,” according to Bei Li, MD, of the Wuhan (China) Institute of Virology, and colleagues.
“We previously provided serological evidence that [HKU8-related] CoV had jumped over from bats to camels and recombined with MERS-CoV, alerting other researchers that the CoV species could be dangerous. ... Genome-level comparison is needed to monitor the risk of alterations in species tropism and pathogenesis,” according to study authors. They performed a study to develop a more effective and cost efficient method for detecting and sequencing novel coronaviruses in the bat population.
The taxonomy of coronaviruses is particularly complex and may be too narrowly defined, given the high level of genetic plasticity found. There are four genera (Alpha-, Beta-, Gamma-, and Deltacoronavirus) consisting of 38 unique species in the CoV subfamily Orthocoronavirinae, and the number is increasing. Viral taxomists rely on the open reading frame 1b (ORF1b) gene for classification, but viruses in the same species may show great diversity in regions outside ORF1b, confounding the species designation. In particular, bat CoVs classed as the same species can differ significantly in terms of receptor usage or virus-host interaction, as observed in bat SARS-related CoVs, according to the researchers.
The researchers obtained RNA from previous bat CoV surveillance projects, which used bat rectal swabs. Libraries for NGS were constructed from total RNA and processed to generate RNA fragments larger than 300 nucleotides. Following first- and second-strand cDNA synthesis, double-stranded cDNA was purified and the library was amplified by polymerase chain reaction (PCR) technology.
Targeted CoV genome enrichment was achieved using 4,303 customized biotinylated 120-mer baits. These baits were designed from 90 representative CoV genomes, and in silico analysis determined that these baits should target the known CoV species tested. These baits were added and hybridized to the libraries. To capture virus-specific library fragments, streptavidin magnetic beads (which bind to biotin) were added to the hybridization reaction mixture. The beads were then washed to remove unbound DNA. The postcapture virus-specific library fragments were then amplified using a subsequent round of PCR.
The enrichment NGS were retrospectively complemented with unbiased NGS and/or additional Sanger sequencing to obtain full-length genomes. The study showed that enrichment NGS not only decreased the amount of data requiring analysis but produced full-length genome coverage in both laboratory and clinical samples.
Using this technology, the researchers “effectively reduced sequencing costs by increasing the sensitivity of detection. We discovered nine full genomes of bat CoVs in this study and revealed great genetic diversity for eight of them.” In addition, they noted that using standard targeted PCR, which is common practice for many surveillance studies, would not have discovered this diversity.
“We should be alert and vigilant with the knowledge that bat CoVs are likely to cause another disease outbreak, not only because of their prevalence but also because the high frequency of recombination between viruses may lead to the generation of viruses with changes in virulence,” according to the researchers.
“We have provided a cost-effective methodology for bat CoV surveillance. The high genetic diversity observed in our newly sequenced samples suggests further work is needed to characterize these bat CoVs prior to or in the early stages of spillover to humans,” the authors concluded.
This study was supported by the Chinese government. The authors reported that they had no conflicts.
Viral genome data for new CoVs from this study are available in GenBank under accession numbers MN611517 to MN611525.
SOURCE: Li B et al. mSphere 2020 Jan 29;5:e00807-19.
Enrichment next-generation sequencing (NGS) provides a more cost-efficient and sensitive method for detecting and sequencing novel coronaviruses from wild bat populations, according to a study reported in mSphere, an open-access journal from the American Society for Microbiology.
With the appearance of the new zoonotic Wuhan coronavirus, the importance of monitoring the likelihood of new virus risks in wildlife reservoirs has been heightened. Bats in particular have been found to be the most common reservoir of coronaviruses, including being a probable source or mixing vessel for two previous modern epidemic coronaviruses: SARS (severe acute respiratory syndrome) and MERS (Middle East respiratory syndrome).
“We should be alert and vigilant with the knowledge that bat CoVs [coronaviruses] are likely to cause another disease outbreak, not only because of their prevalence but also because the high frequency of recombination between viruses may lead to the generation of viruses with changes in virulence,” according to Bei Li, MD, of the Wuhan (China) Institute of Virology, and colleagues.
“We previously provided serological evidence that [HKU8-related] CoV had jumped over from bats to camels and recombined with MERS-CoV, alerting other researchers that the CoV species could be dangerous. ... Genome-level comparison is needed to monitor the risk of alterations in species tropism and pathogenesis,” according to study authors. They performed a study to develop a more effective and cost efficient method for detecting and sequencing novel coronaviruses in the bat population.
The taxonomy of coronaviruses is particularly complex and may be too narrowly defined, given the high level of genetic plasticity found. There are four genera (Alpha-, Beta-, Gamma-, and Deltacoronavirus) consisting of 38 unique species in the CoV subfamily Orthocoronavirinae, and the number is increasing. Viral taxomists rely on the open reading frame 1b (ORF1b) gene for classification, but viruses in the same species may show great diversity in regions outside ORF1b, confounding the species designation. In particular, bat CoVs classed as the same species can differ significantly in terms of receptor usage or virus-host interaction, as observed in bat SARS-related CoVs, according to the researchers.
The researchers obtained RNA from previous bat CoV surveillance projects, which used bat rectal swabs. Libraries for NGS were constructed from total RNA and processed to generate RNA fragments larger than 300 nucleotides. Following first- and second-strand cDNA synthesis, double-stranded cDNA was purified and the library was amplified by polymerase chain reaction (PCR) technology.
Targeted CoV genome enrichment was achieved using 4,303 customized biotinylated 120-mer baits. These baits were designed from 90 representative CoV genomes, and in silico analysis determined that these baits should target the known CoV species tested. These baits were added and hybridized to the libraries. To capture virus-specific library fragments, streptavidin magnetic beads (which bind to biotin) were added to the hybridization reaction mixture. The beads were then washed to remove unbound DNA. The postcapture virus-specific library fragments were then amplified using a subsequent round of PCR.
The enrichment NGS were retrospectively complemented with unbiased NGS and/or additional Sanger sequencing to obtain full-length genomes. The study showed that enrichment NGS not only decreased the amount of data requiring analysis but produced full-length genome coverage in both laboratory and clinical samples.
Using this technology, the researchers “effectively reduced sequencing costs by increasing the sensitivity of detection. We discovered nine full genomes of bat CoVs in this study and revealed great genetic diversity for eight of them.” In addition, they noted that using standard targeted PCR, which is common practice for many surveillance studies, would not have discovered this diversity.
“We should be alert and vigilant with the knowledge that bat CoVs are likely to cause another disease outbreak, not only because of their prevalence but also because the high frequency of recombination between viruses may lead to the generation of viruses with changes in virulence,” according to the researchers.
“We have provided a cost-effective methodology for bat CoV surveillance. The high genetic diversity observed in our newly sequenced samples suggests further work is needed to characterize these bat CoVs prior to or in the early stages of spillover to humans,” the authors concluded.
This study was supported by the Chinese government. The authors reported that they had no conflicts.
Viral genome data for new CoVs from this study are available in GenBank under accession numbers MN611517 to MN611525.
SOURCE: Li B et al. mSphere 2020 Jan 29;5:e00807-19.
Enrichment next-generation sequencing (NGS) provides a more cost-efficient and sensitive method for detecting and sequencing novel coronaviruses from wild bat populations, according to a study reported in mSphere, an open-access journal from the American Society for Microbiology.
With the appearance of the new zoonotic Wuhan coronavirus, the importance of monitoring the likelihood of new virus risks in wildlife reservoirs has been heightened. Bats in particular have been found to be the most common reservoir of coronaviruses, including being a probable source or mixing vessel for two previous modern epidemic coronaviruses: SARS (severe acute respiratory syndrome) and MERS (Middle East respiratory syndrome).
“We should be alert and vigilant with the knowledge that bat CoVs [coronaviruses] are likely to cause another disease outbreak, not only because of their prevalence but also because the high frequency of recombination between viruses may lead to the generation of viruses with changes in virulence,” according to Bei Li, MD, of the Wuhan (China) Institute of Virology, and colleagues.
“We previously provided serological evidence that [HKU8-related] CoV had jumped over from bats to camels and recombined with MERS-CoV, alerting other researchers that the CoV species could be dangerous. ... Genome-level comparison is needed to monitor the risk of alterations in species tropism and pathogenesis,” according to study authors. They performed a study to develop a more effective and cost efficient method for detecting and sequencing novel coronaviruses in the bat population.
The taxonomy of coronaviruses is particularly complex and may be too narrowly defined, given the high level of genetic plasticity found. There are four genera (Alpha-, Beta-, Gamma-, and Deltacoronavirus) consisting of 38 unique species in the CoV subfamily Orthocoronavirinae, and the number is increasing. Viral taxomists rely on the open reading frame 1b (ORF1b) gene for classification, but viruses in the same species may show great diversity in regions outside ORF1b, confounding the species designation. In particular, bat CoVs classed as the same species can differ significantly in terms of receptor usage or virus-host interaction, as observed in bat SARS-related CoVs, according to the researchers.
The researchers obtained RNA from previous bat CoV surveillance projects, which used bat rectal swabs. Libraries for NGS were constructed from total RNA and processed to generate RNA fragments larger than 300 nucleotides. Following first- and second-strand cDNA synthesis, double-stranded cDNA was purified and the library was amplified by polymerase chain reaction (PCR) technology.
Targeted CoV genome enrichment was achieved using 4,303 customized biotinylated 120-mer baits. These baits were designed from 90 representative CoV genomes, and in silico analysis determined that these baits should target the known CoV species tested. These baits were added and hybridized to the libraries. To capture virus-specific library fragments, streptavidin magnetic beads (which bind to biotin) were added to the hybridization reaction mixture. The beads were then washed to remove unbound DNA. The postcapture virus-specific library fragments were then amplified using a subsequent round of PCR.
The enrichment NGS were retrospectively complemented with unbiased NGS and/or additional Sanger sequencing to obtain full-length genomes. The study showed that enrichment NGS not only decreased the amount of data requiring analysis but produced full-length genome coverage in both laboratory and clinical samples.
Using this technology, the researchers “effectively reduced sequencing costs by increasing the sensitivity of detection. We discovered nine full genomes of bat CoVs in this study and revealed great genetic diversity for eight of them.” In addition, they noted that using standard targeted PCR, which is common practice for many surveillance studies, would not have discovered this diversity.
“We should be alert and vigilant with the knowledge that bat CoVs are likely to cause another disease outbreak, not only because of their prevalence but also because the high frequency of recombination between viruses may lead to the generation of viruses with changes in virulence,” according to the researchers.
“We have provided a cost-effective methodology for bat CoV surveillance. The high genetic diversity observed in our newly sequenced samples suggests further work is needed to characterize these bat CoVs prior to or in the early stages of spillover to humans,” the authors concluded.
This study was supported by the Chinese government. The authors reported that they had no conflicts.
Viral genome data for new CoVs from this study are available in GenBank under accession numbers MN611517 to MN611525.
SOURCE: Li B et al. mSphere 2020 Jan 29;5:e00807-19.
FROM MSPHERE