User login
New cystic fibrosis therapy raises hopes among specialists and patients
A newly approved triple-combination modulator to treat cystic fibrosis (CF) has raised expectations of a treatment turning point among patients and specialists. If the early results are sustained, elexacaftor/ivacaftor/tezacaftor (Trikafta) could prove to be the rare case of a much-touted new medicine that meets high expectations.
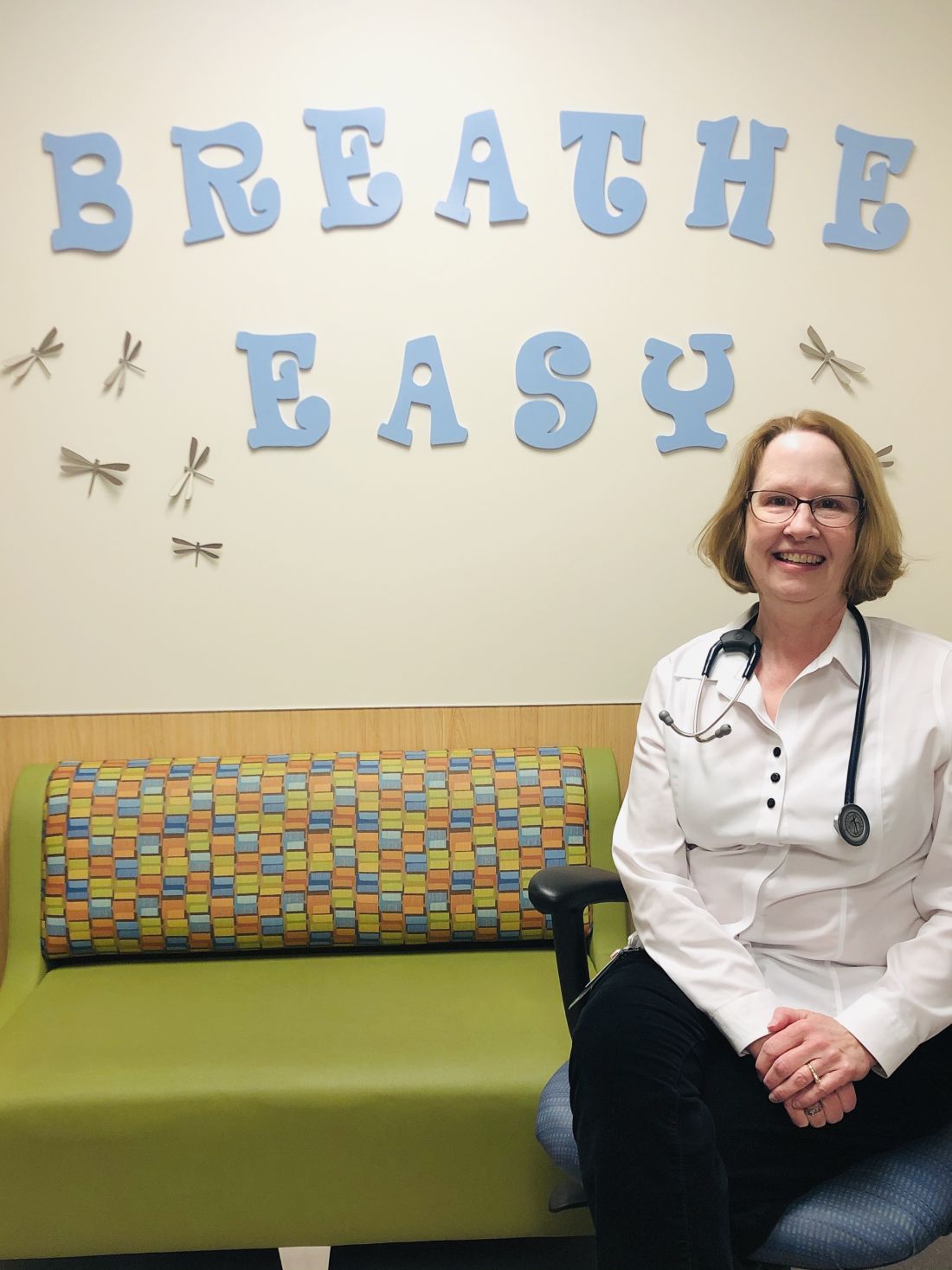
“CF even in infants causes inflammation, so we know that lung damage can start early and progress,” said Susan Millard, MD, FCCP, of Helen DeVos Children’s Hospital in Grand Rapids, Mich., and the local clinical research director for the pediatric pulmonary and sleep medicine section. “This oral drug therapy is actually treating the underlying problem, as opposed to many of the therapies we have that take hours to nebulize and only work locally in the airways.”
Dr. Millard is the recent past pediatric editor for Chest Physician and has been a local principal investigator at Helen DeVos Children’s Hospital for many Vertex-sponsored clinical studies.
The pivotal studies
The Food and Drug Administration approval of Trikafta rested on two pivotal phase 3, placebo-controlled studies, one in patients with two copies of the most common CF mutations, F508del, and the second in patients with one copy of F508del and a second mutation that was called a “minimal-function” mutation. The findings have ignited the hopes of many people with CF and their physicians. The drug was approved in October 2019 for patients aged 12 years and older who have at least one F508del mutation of the cystic fibrosis transmembrane conductance regulator gene. About 90% of patients in the United States have at least one copy of F508del. In the study looking at patients with one copy of F508del, the mean predicted forced expiratory volume in 1 second increased 13.8% in patients taking the drug versus placebo (N Engl J Med. 2019 Oct 31. doi: 10.1056/NEJMoa1908639). The number of pulmonary exacerbations decreased by 63% in the Trikafta group, compared with placebo. Pulmonary exacerbations were described as a change in specific symptoms that required treatment with a new oral, intravenous, or inhaled antibiotic. Serious adverse drug reactions that occurred more frequently in patients receiving Trikafta, compared with placebo, were rash and influenza events.
In the study that included patients with two copies of F508del, on average, the lung function increased 10% versus patients on ivacaftor/tezacaftor at 4 weeks. In addition, there was a 45.1 mmol/L on average decrease in the sweat chloride level in the Trikafta group, compared with ivacaftor/tezacaftor.
A hopeful start
Robert Giusti, MD, a pediatric pulmonologist at New York University Langone Health, is also hopeful. “This could be the kind of treatment that will make a revolution in terms of [cystic fibrosis] care if it can be started very early in life shortly after diagnosis. We anticipate that patients will be disease free for a longer period of time.”

The Cystic Fibrosis Foundation’s (CFF) “venture philanthropy” initiative played an important role in the development of the drug by Vertex Pharmaceuticals. The CFF has invested many millions of dollars in research by drug companies since the 1980s and was an early backer of Vertex. According to a statement on the CFF website, the Foundation sold its royalty rights for treatments developed by Vertex for $3.3 billion in 2014. The drug has a list price of about $311,000 a year. Payment issues may arise in the future, but for now, Vertex has stated that insurers and some Medicaid programs have begun paying claims for Trikafta
Specialists who treat CF now are watching to see how well patients tolerate this highly anticipated drug – and how well it meets expectations. The Therapeutic Development Network, the clinical research division of the CFF, is enrolling patients taking Trikafta in an observational study to follow for long-term follow-up.
Meeting expectations
“[Long-term efficacy is] something that we’re always concerned about. When the drug comes to market, is it going to be as effective as we thought it might be?” said Ryan Thomas, MD, director of the Cystic Fibrosis Center at Michigan State University, East Lansing. The MSU Cystic Fibrosis Center receives funding from the Cystic Fibrosis Foundation.

Almost one in five patients could not tolerate treatment with Orkambi, most often because of adverse breathing events, according to a French study published in the American Journal of Respiratory and Critical Care Medicine. The investigators wrote: “Among the 845 patients (292 adolescents, 553 adults) who initiated lumacaftor/ivacaftor, 18.2% (154 patients) discontinued treatment, often due to respiratory (48.1%, 74 patients) or nonrespiratory (27.9%, 43 patients) adverse events” and that the discontinuation rate was considerably higher than previously reported in clinical trials.
“We thought [Orkambi] was going to be something that could have a big effect,” Dr. Thomas said. “It turned out that it was harder for people to tolerate than we thought and the improvements weren’t as sustained as we thought they might be. I really don’t think this will end up being the case with Trikafta.”
Longer-term data are starting to emerge, which may ease some of the concerns inherent in working with a newer medicine. “These [data] suggest that this is going to be a game changer,” Dr. Thomas said. “If Trikafta is this efficacious, well, we’re talking about having people with CF who will live full lifespans without a lung transplant, and that is so rare.”
The decrease in hospitalizations, improved CT scans, and lower rates of lung function decline suggest it could be “the Holy Grail,” Dr. Thomas said.
A different disease
Trikafta is the latest in a series of improvements of CF treatment in recent decades, recalled Dr. Giusti, who has been in this field for about 3 decades. “It used to be that I attended many funerals for children with CF. Now with patients living longer and healthier lives I am invited to attend their weddings and even their children’s baptisms and bris ceremonies. It is a very different disease than it used to be.”
The promise of Trikafta leaves behind the minority of patients for whom the drug won’t work. This is for the 10% of patients that have rare mutations. That can lead to difficult conversations with parents about why this new option is not a choice for their child, Dr. Millard said. “It just crushes you, but the Cystic Fibrosis Foundation is committing a lot of new research in that direction. Their mantra is ‘until it is done.’ ”
Realistic expectations
William (Randy) Hunt, MD, FAAP, FACP, assistant professor of medicine in the Division of Pulmonary, Allergy, Critical Care and Sleep, Emory University School of Medicine, Atlanta, agrees that Trikafta is an exciting development in CF treatment. He noted, “Starting this medication early in life may very well significantly attenuate the disease, but it is not a cure. For individuals who already have significant disease, we may not see the same level of improvements in lung function as what we saw in the studies. The studies generally excluded individuals with ppFEV1 < 40%. Nevertheless, I remain optimistic and have been prescribing it to nearly everyone that qualifies after a discussion.”
Dr. Hunt added, “Patients are asking if they can stop their current chronic CF therapies once they start Trikafta. The answer is “no, at least not right now.” While all the relatively short-term data around Trikafta are very promising, we do not yet know how sustained the long-term benefits will be. Still, safely removing therapeutic burden from our patient population is a real interest. There are plans underway by the CFF and other institutions to systematically research whether discontinuing chronic CF therapies is safe in the setting of Trikafta.”
He concluded that 10% of individuals with CF mutations still do not respond to the modulators currently available. “We will not leave that population behind, but treating these remaining mutations is going to take continued efforts and likely modulators that are therapeutically differently from the mechanism of actions of those that are currently available,” he said.
Therese Borden contributed to this article.
1/2/2020 - This story was updated.
A newly approved triple-combination modulator to treat cystic fibrosis (CF) has raised expectations of a treatment turning point among patients and specialists. If the early results are sustained, elexacaftor/ivacaftor/tezacaftor (Trikafta) could prove to be the rare case of a much-touted new medicine that meets high expectations.
“CF even in infants causes inflammation, so we know that lung damage can start early and progress,” said Susan Millard, MD, FCCP, of Helen DeVos Children’s Hospital in Grand Rapids, Mich., and the local clinical research director for the pediatric pulmonary and sleep medicine section. “This oral drug therapy is actually treating the underlying problem, as opposed to many of the therapies we have that take hours to nebulize and only work locally in the airways.”
Dr. Millard is the recent past pediatric editor for Chest Physician and has been a local principal investigator at Helen DeVos Children’s Hospital for many Vertex-sponsored clinical studies.
The pivotal studies
The Food and Drug Administration approval of Trikafta rested on two pivotal phase 3, placebo-controlled studies, one in patients with two copies of the most common CF mutations, F508del, and the second in patients with one copy of F508del and a second mutation that was called a “minimal-function” mutation. The findings have ignited the hopes of many people with CF and their physicians. The drug was approved in October 2019 for patients aged 12 years and older who have at least one F508del mutation of the cystic fibrosis transmembrane conductance regulator gene. About 90% of patients in the United States have at least one copy of F508del. In the study looking at patients with one copy of F508del, the mean predicted forced expiratory volume in 1 second increased 13.8% in patients taking the drug versus placebo (N Engl J Med. 2019 Oct 31. doi: 10.1056/NEJMoa1908639). The number of pulmonary exacerbations decreased by 63% in the Trikafta group, compared with placebo. Pulmonary exacerbations were described as a change in specific symptoms that required treatment with a new oral, intravenous, or inhaled antibiotic. Serious adverse drug reactions that occurred more frequently in patients receiving Trikafta, compared with placebo, were rash and influenza events.
In the study that included patients with two copies of F508del, on average, the lung function increased 10% versus patients on ivacaftor/tezacaftor at 4 weeks. In addition, there was a 45.1 mmol/L on average decrease in the sweat chloride level in the Trikafta group, compared with ivacaftor/tezacaftor.
A hopeful start
Robert Giusti, MD, a pediatric pulmonologist at New York University Langone Health, is also hopeful. “This could be the kind of treatment that will make a revolution in terms of [cystic fibrosis] care if it can be started very early in life shortly after diagnosis. We anticipate that patients will be disease free for a longer period of time.”
The Cystic Fibrosis Foundation’s (CFF) “venture philanthropy” initiative played an important role in the development of the drug by Vertex Pharmaceuticals. The CFF has invested many millions of dollars in research by drug companies since the 1980s and was an early backer of Vertex. According to a statement on the CFF website, the Foundation sold its royalty rights for treatments developed by Vertex for $3.3 billion in 2014. The drug has a list price of about $311,000 a year. Payment issues may arise in the future, but for now, Vertex has stated that insurers and some Medicaid programs have begun paying claims for Trikafta
Specialists who treat CF now are watching to see how well patients tolerate this highly anticipated drug – and how well it meets expectations. The Therapeutic Development Network, the clinical research division of the CFF, is enrolling patients taking Trikafta in an observational study to follow for long-term follow-up.
Meeting expectations
“[Long-term efficacy is] something that we’re always concerned about. When the drug comes to market, is it going to be as effective as we thought it might be?” said Ryan Thomas, MD, director of the Cystic Fibrosis Center at Michigan State University, East Lansing. The MSU Cystic Fibrosis Center receives funding from the Cystic Fibrosis Foundation.
Almost one in five patients could not tolerate treatment with Orkambi, most often because of adverse breathing events, according to a French study published in the American Journal of Respiratory and Critical Care Medicine. The investigators wrote: “Among the 845 patients (292 adolescents, 553 adults) who initiated lumacaftor/ivacaftor, 18.2% (154 patients) discontinued treatment, often due to respiratory (48.1%, 74 patients) or nonrespiratory (27.9%, 43 patients) adverse events” and that the discontinuation rate was considerably higher than previously reported in clinical trials.
“We thought [Orkambi] was going to be something that could have a big effect,” Dr. Thomas said. “It turned out that it was harder for people to tolerate than we thought and the improvements weren’t as sustained as we thought they might be. I really don’t think this will end up being the case with Trikafta.”
Longer-term data are starting to emerge, which may ease some of the concerns inherent in working with a newer medicine. “These [data] suggest that this is going to be a game changer,” Dr. Thomas said. “If Trikafta is this efficacious, well, we’re talking about having people with CF who will live full lifespans without a lung transplant, and that is so rare.”
The decrease in hospitalizations, improved CT scans, and lower rates of lung function decline suggest it could be “the Holy Grail,” Dr. Thomas said.
A different disease
Trikafta is the latest in a series of improvements of CF treatment in recent decades, recalled Dr. Giusti, who has been in this field for about 3 decades. “It used to be that I attended many funerals for children with CF. Now with patients living longer and healthier lives I am invited to attend their weddings and even their children’s baptisms and bris ceremonies. It is a very different disease than it used to be.”
The promise of Trikafta leaves behind the minority of patients for whom the drug won’t work. This is for the 10% of patients that have rare mutations. That can lead to difficult conversations with parents about why this new option is not a choice for their child, Dr. Millard said. “It just crushes you, but the Cystic Fibrosis Foundation is committing a lot of new research in that direction. Their mantra is ‘until it is done.’ ”
Realistic expectations
William (Randy) Hunt, MD, FAAP, FACP, assistant professor of medicine in the Division of Pulmonary, Allergy, Critical Care and Sleep, Emory University School of Medicine, Atlanta, agrees that Trikafta is an exciting development in CF treatment. He noted, “Starting this medication early in life may very well significantly attenuate the disease, but it is not a cure. For individuals who already have significant disease, we may not see the same level of improvements in lung function as what we saw in the studies. The studies generally excluded individuals with ppFEV1 < 40%. Nevertheless, I remain optimistic and have been prescribing it to nearly everyone that qualifies after a discussion.”
Dr. Hunt added, “Patients are asking if they can stop their current chronic CF therapies once they start Trikafta. The answer is “no, at least not right now.” While all the relatively short-term data around Trikafta are very promising, we do not yet know how sustained the long-term benefits will be. Still, safely removing therapeutic burden from our patient population is a real interest. There are plans underway by the CFF and other institutions to systematically research whether discontinuing chronic CF therapies is safe in the setting of Trikafta.”
He concluded that 10% of individuals with CF mutations still do not respond to the modulators currently available. “We will not leave that population behind, but treating these remaining mutations is going to take continued efforts and likely modulators that are therapeutically differently from the mechanism of actions of those that are currently available,” he said.
Therese Borden contributed to this article.
1/2/2020 - This story was updated.
A newly approved triple-combination modulator to treat cystic fibrosis (CF) has raised expectations of a treatment turning point among patients and specialists. If the early results are sustained, elexacaftor/ivacaftor/tezacaftor (Trikafta) could prove to be the rare case of a much-touted new medicine that meets high expectations.
“CF even in infants causes inflammation, so we know that lung damage can start early and progress,” said Susan Millard, MD, FCCP, of Helen DeVos Children’s Hospital in Grand Rapids, Mich., and the local clinical research director for the pediatric pulmonary and sleep medicine section. “This oral drug therapy is actually treating the underlying problem, as opposed to many of the therapies we have that take hours to nebulize and only work locally in the airways.”
Dr. Millard is the recent past pediatric editor for Chest Physician and has been a local principal investigator at Helen DeVos Children’s Hospital for many Vertex-sponsored clinical studies.
The pivotal studies
The Food and Drug Administration approval of Trikafta rested on two pivotal phase 3, placebo-controlled studies, one in patients with two copies of the most common CF mutations, F508del, and the second in patients with one copy of F508del and a second mutation that was called a “minimal-function” mutation. The findings have ignited the hopes of many people with CF and their physicians. The drug was approved in October 2019 for patients aged 12 years and older who have at least one F508del mutation of the cystic fibrosis transmembrane conductance regulator gene. About 90% of patients in the United States have at least one copy of F508del. In the study looking at patients with one copy of F508del, the mean predicted forced expiratory volume in 1 second increased 13.8% in patients taking the drug versus placebo (N Engl J Med. 2019 Oct 31. doi: 10.1056/NEJMoa1908639). The number of pulmonary exacerbations decreased by 63% in the Trikafta group, compared with placebo. Pulmonary exacerbations were described as a change in specific symptoms that required treatment with a new oral, intravenous, or inhaled antibiotic. Serious adverse drug reactions that occurred more frequently in patients receiving Trikafta, compared with placebo, were rash and influenza events.
In the study that included patients with two copies of F508del, on average, the lung function increased 10% versus patients on ivacaftor/tezacaftor at 4 weeks. In addition, there was a 45.1 mmol/L on average decrease in the sweat chloride level in the Trikafta group, compared with ivacaftor/tezacaftor.
A hopeful start
Robert Giusti, MD, a pediatric pulmonologist at New York University Langone Health, is also hopeful. “This could be the kind of treatment that will make a revolution in terms of [cystic fibrosis] care if it can be started very early in life shortly after diagnosis. We anticipate that patients will be disease free for a longer period of time.”
The Cystic Fibrosis Foundation’s (CFF) “venture philanthropy” initiative played an important role in the development of the drug by Vertex Pharmaceuticals. The CFF has invested many millions of dollars in research by drug companies since the 1980s and was an early backer of Vertex. According to a statement on the CFF website, the Foundation sold its royalty rights for treatments developed by Vertex for $3.3 billion in 2014. The drug has a list price of about $311,000 a year. Payment issues may arise in the future, but for now, Vertex has stated that insurers and some Medicaid programs have begun paying claims for Trikafta
Specialists who treat CF now are watching to see how well patients tolerate this highly anticipated drug – and how well it meets expectations. The Therapeutic Development Network, the clinical research division of the CFF, is enrolling patients taking Trikafta in an observational study to follow for long-term follow-up.
Meeting expectations
“[Long-term efficacy is] something that we’re always concerned about. When the drug comes to market, is it going to be as effective as we thought it might be?” said Ryan Thomas, MD, director of the Cystic Fibrosis Center at Michigan State University, East Lansing. The MSU Cystic Fibrosis Center receives funding from the Cystic Fibrosis Foundation.
Almost one in five patients could not tolerate treatment with Orkambi, most often because of adverse breathing events, according to a French study published in the American Journal of Respiratory and Critical Care Medicine. The investigators wrote: “Among the 845 patients (292 adolescents, 553 adults) who initiated lumacaftor/ivacaftor, 18.2% (154 patients) discontinued treatment, often due to respiratory (48.1%, 74 patients) or nonrespiratory (27.9%, 43 patients) adverse events” and that the discontinuation rate was considerably higher than previously reported in clinical trials.
“We thought [Orkambi] was going to be something that could have a big effect,” Dr. Thomas said. “It turned out that it was harder for people to tolerate than we thought and the improvements weren’t as sustained as we thought they might be. I really don’t think this will end up being the case with Trikafta.”
Longer-term data are starting to emerge, which may ease some of the concerns inherent in working with a newer medicine. “These [data] suggest that this is going to be a game changer,” Dr. Thomas said. “If Trikafta is this efficacious, well, we’re talking about having people with CF who will live full lifespans without a lung transplant, and that is so rare.”
The decrease in hospitalizations, improved CT scans, and lower rates of lung function decline suggest it could be “the Holy Grail,” Dr. Thomas said.
A different disease
Trikafta is the latest in a series of improvements of CF treatment in recent decades, recalled Dr. Giusti, who has been in this field for about 3 decades. “It used to be that I attended many funerals for children with CF. Now with patients living longer and healthier lives I am invited to attend their weddings and even their children’s baptisms and bris ceremonies. It is a very different disease than it used to be.”
The promise of Trikafta leaves behind the minority of patients for whom the drug won’t work. This is for the 10% of patients that have rare mutations. That can lead to difficult conversations with parents about why this new option is not a choice for their child, Dr. Millard said. “It just crushes you, but the Cystic Fibrosis Foundation is committing a lot of new research in that direction. Their mantra is ‘until it is done.’ ”
Realistic expectations
William (Randy) Hunt, MD, FAAP, FACP, assistant professor of medicine in the Division of Pulmonary, Allergy, Critical Care and Sleep, Emory University School of Medicine, Atlanta, agrees that Trikafta is an exciting development in CF treatment. He noted, “Starting this medication early in life may very well significantly attenuate the disease, but it is not a cure. For individuals who already have significant disease, we may not see the same level of improvements in lung function as what we saw in the studies. The studies generally excluded individuals with ppFEV1 < 40%. Nevertheless, I remain optimistic and have been prescribing it to nearly everyone that qualifies after a discussion.”
Dr. Hunt added, “Patients are asking if they can stop their current chronic CF therapies once they start Trikafta. The answer is “no, at least not right now.” While all the relatively short-term data around Trikafta are very promising, we do not yet know how sustained the long-term benefits will be. Still, safely removing therapeutic burden from our patient population is a real interest. There are plans underway by the CFF and other institutions to systematically research whether discontinuing chronic CF therapies is safe in the setting of Trikafta.”
He concluded that 10% of individuals with CF mutations still do not respond to the modulators currently available. “We will not leave that population behind, but treating these remaining mutations is going to take continued efforts and likely modulators that are therapeutically differently from the mechanism of actions of those that are currently available,” he said.
Therese Borden contributed to this article.
1/2/2020 - This story was updated.
Rand analysis of proposed Medicare buy-in uncovers surprising findings
Early buy-in by people aged 50-64 might raise insurance premiums
Allowing people aged 50-64 to buy into Medicare might result in higher premiums for people who purchase their health insurance on the individual market, a finding that runs counter to many people’s expectations, said the authors of a newly released report.
In the report, Christine Eibner, PhD, of the RAND Corporation and colleagues estimated that the premium for so-called bronze market plans might increase by 2%-10%, depending on the design of a Medicare buy-in program. (The bronze plans are ones with fewer benefits sold on exchanges created through the implementation of the Affordable Care Act of 2010 [ACA].)
A perception has been that younger adults have been in effect subsidizing the cost of older ones in the marketplace plans. Instead, it appears that younger adults who enroll in the individual market tend to be relatively unhealthy and thus expensive to cover.
“When older adults leave the market, insurers are left with a smaller pool of younger, less healthy, and relatively expensive people given their age, leading to higher premiums,” Dr. Eibner and colleagues said in the report.
This result was unexpected, as there has been discussion of using a Medicare buy-in to reduce the cost of premiums for others in the marketplace, Dr. Eibner and colleagues said. But this result is consistent with other recent findings, including research presented by the consulting firm Milliman at a Society of Actuaries meeting in June. The Blue Cross Blue Shield Association estimates that losing a large group of customers could raise premiums by about 10% for the remaining pool of insured people, the New York Times has reported.
That might make it attractive to middle-aged Americans. Dr. Eibner and colleagues estimated the annual premium for a Medicare buy-in at $9,747 in 2022. For a 50-year-old, a bronze-level ACA plan would cost $9,208 and a gold-level one, $12,277. For a 60-year-old, the annual premium for a bronze-level plan might be $13,512 and $18,016 for a gold-level plan.
Total out-of-pocket health spending, including premiums, would fall, on average, by 16%-35% for those who moved from ACA-compliant individual market coverage to a buy-in. The lower spending reflects that buy-in enrollees would have access to Medicare payment rates, which are substantially lower than private rates, and lower administrative costs.
There may be growing interest in allowing people aged 50-64 to buy into Medicare if enthusiasm wanes for bids to create a giant national health program, Dr. Eibner said in an interview.
“If there is concern that Medicare for all is going too far, I think this option is something that could become more prominent,” she said.
Many Democratic lawmakers already are focused on a Medicare buy-in approach. Sen. Debbie Stabenow (D-Mich.) has 20 Democratic cosponsors for her bill, which would allow for a Medicare buy-in at age 50. Sen. Bernie Sanders (I-Vt.) has 14 Democratic cosponsors for the current version of his well-known “Medicare-for-all” bill. That’s two fewer than he had for the Medicare-for-all bill he offered in the 115th session of Congress (Jan. 3, 2017–Jan. 3, 2019).
Among the supporters of Sen. Stabenow’s bill are several 2020 presidential contenders: Sen. Cory Booker (D-N.J.), Sen. Amy Klobuchar (D-Minn.), and Sen. Kamala Harris (D-Calif). As of Saturday, Sen. Elizabeth Warren (D-Mass.) backed Sanders’ bill, but not Stabenow’s. But Sen. Warren also has spoken recently of a Medicare expansion for people at age 50 as a step on the path toward universal coverage. Former Vice President Joseph R. Biden and Mayor Pete Buttigieg of South Bend, Ind., have said they would like to offer Americans the option to buy into Medicare or a public plan.
The idea of lowering the Medicare age has been considered for many years by Democrats. It was seen as a way to help older Americans afford medical care before the enactment of the ACA. Before that law took effect, consumers were not guaranteed access to a health plan, causing many older Americans to go without coverage.
But Dr. Eibner and colleagues found that a Medicare buy-in would have little to no effect on total health insurance enrollment. A Medicare buy-in might increase enrollment by 400,000 to 1.6 million for those over age 50, while decreasing enrollment by 100,000 to 800,000 for those under age 50 because of rising premiums.
“It’s not doing a lot to get people covered,” Dr. Eibner said in the interview.
In the report, Dr. Eibner and colleagues estimated that between 2.8 million and 7.0 million people would choose to enroll, depending on the approach used to design a Medicare buy-in. They considered numerous potential options for the design of a Medicare expansion, including various levels of federal subsidy for people using the buy-in. Dr. Eibner and colleagues also considered whether insurers would respond by trying to selectively market to healthier individuals, increasing their chance of enrolling.
The envisioned Medicare buy-in would have no effect on the Medicare Trust Fund, which pools money available through previously collected dedicated taxes, Dr. Eibner and colleagues said. In creating their model, they drew upon data from the Survey of Income and Program Participation, the Medical Expenditure Panel Survey, and the Kaiser Family Foundation and Health Research and Educational Trust Employer Health Benefits Survey.
Dr. Eibner and colleagues noted “several important limitations” for their work. It does not look at how the buy-in might affect clinicians and hospitals. Lower Medicare payment rates might cause some physicians to turn away patients covered by a Medicare buy-in.
“On the other hand, even if some providers decided not to participate in the buy-in, the buy-in might offer enrollees a broader network than what is available on the individual market,” Dr. Eibner and colleagues wrote. “Furthermore, it is not clear that providers would be legally allowed to opt out of the buy-in while still accepting payment for current Medicare beneficiaries.”
The Rand report was developed with the support of funding by the AARP.
SOURCE: Eibner C et al. RAND Corporation. Research Report. 2019. doi: 10.7249/RR4246.
Early buy-in by people aged 50-64 might raise insurance premiums
Early buy-in by people aged 50-64 might raise insurance premiums
Allowing people aged 50-64 to buy into Medicare might result in higher premiums for people who purchase their health insurance on the individual market, a finding that runs counter to many people’s expectations, said the authors of a newly released report.
In the report, Christine Eibner, PhD, of the RAND Corporation and colleagues estimated that the premium for so-called bronze market plans might increase by 2%-10%, depending on the design of a Medicare buy-in program. (The bronze plans are ones with fewer benefits sold on exchanges created through the implementation of the Affordable Care Act of 2010 [ACA].)
A perception has been that younger adults have been in effect subsidizing the cost of older ones in the marketplace plans. Instead, it appears that younger adults who enroll in the individual market tend to be relatively unhealthy and thus expensive to cover.
“When older adults leave the market, insurers are left with a smaller pool of younger, less healthy, and relatively expensive people given their age, leading to higher premiums,” Dr. Eibner and colleagues said in the report.
This result was unexpected, as there has been discussion of using a Medicare buy-in to reduce the cost of premiums for others in the marketplace, Dr. Eibner and colleagues said. But this result is consistent with other recent findings, including research presented by the consulting firm Milliman at a Society of Actuaries meeting in June. The Blue Cross Blue Shield Association estimates that losing a large group of customers could raise premiums by about 10% for the remaining pool of insured people, the New York Times has reported.
That might make it attractive to middle-aged Americans. Dr. Eibner and colleagues estimated the annual premium for a Medicare buy-in at $9,747 in 2022. For a 50-year-old, a bronze-level ACA plan would cost $9,208 and a gold-level one, $12,277. For a 60-year-old, the annual premium for a bronze-level plan might be $13,512 and $18,016 for a gold-level plan.
Total out-of-pocket health spending, including premiums, would fall, on average, by 16%-35% for those who moved from ACA-compliant individual market coverage to a buy-in. The lower spending reflects that buy-in enrollees would have access to Medicare payment rates, which are substantially lower than private rates, and lower administrative costs.
There may be growing interest in allowing people aged 50-64 to buy into Medicare if enthusiasm wanes for bids to create a giant national health program, Dr. Eibner said in an interview.
“If there is concern that Medicare for all is going too far, I think this option is something that could become more prominent,” she said.
Many Democratic lawmakers already are focused on a Medicare buy-in approach. Sen. Debbie Stabenow (D-Mich.) has 20 Democratic cosponsors for her bill, which would allow for a Medicare buy-in at age 50. Sen. Bernie Sanders (I-Vt.) has 14 Democratic cosponsors for the current version of his well-known “Medicare-for-all” bill. That’s two fewer than he had for the Medicare-for-all bill he offered in the 115th session of Congress (Jan. 3, 2017–Jan. 3, 2019).
Among the supporters of Sen. Stabenow’s bill are several 2020 presidential contenders: Sen. Cory Booker (D-N.J.), Sen. Amy Klobuchar (D-Minn.), and Sen. Kamala Harris (D-Calif). As of Saturday, Sen. Elizabeth Warren (D-Mass.) backed Sanders’ bill, but not Stabenow’s. But Sen. Warren also has spoken recently of a Medicare expansion for people at age 50 as a step on the path toward universal coverage. Former Vice President Joseph R. Biden and Mayor Pete Buttigieg of South Bend, Ind., have said they would like to offer Americans the option to buy into Medicare or a public plan.
The idea of lowering the Medicare age has been considered for many years by Democrats. It was seen as a way to help older Americans afford medical care before the enactment of the ACA. Before that law took effect, consumers were not guaranteed access to a health plan, causing many older Americans to go without coverage.
But Dr. Eibner and colleagues found that a Medicare buy-in would have little to no effect on total health insurance enrollment. A Medicare buy-in might increase enrollment by 400,000 to 1.6 million for those over age 50, while decreasing enrollment by 100,000 to 800,000 for those under age 50 because of rising premiums.
“It’s not doing a lot to get people covered,” Dr. Eibner said in the interview.
In the report, Dr. Eibner and colleagues estimated that between 2.8 million and 7.0 million people would choose to enroll, depending on the approach used to design a Medicare buy-in. They considered numerous potential options for the design of a Medicare expansion, including various levels of federal subsidy for people using the buy-in. Dr. Eibner and colleagues also considered whether insurers would respond by trying to selectively market to healthier individuals, increasing their chance of enrolling.
The envisioned Medicare buy-in would have no effect on the Medicare Trust Fund, which pools money available through previously collected dedicated taxes, Dr. Eibner and colleagues said. In creating their model, they drew upon data from the Survey of Income and Program Participation, the Medical Expenditure Panel Survey, and the Kaiser Family Foundation and Health Research and Educational Trust Employer Health Benefits Survey.
Dr. Eibner and colleagues noted “several important limitations” for their work. It does not look at how the buy-in might affect clinicians and hospitals. Lower Medicare payment rates might cause some physicians to turn away patients covered by a Medicare buy-in.
“On the other hand, even if some providers decided not to participate in the buy-in, the buy-in might offer enrollees a broader network than what is available on the individual market,” Dr. Eibner and colleagues wrote. “Furthermore, it is not clear that providers would be legally allowed to opt out of the buy-in while still accepting payment for current Medicare beneficiaries.”
The Rand report was developed with the support of funding by the AARP.
SOURCE: Eibner C et al. RAND Corporation. Research Report. 2019. doi: 10.7249/RR4246.
Allowing people aged 50-64 to buy into Medicare might result in higher premiums for people who purchase their health insurance on the individual market, a finding that runs counter to many people’s expectations, said the authors of a newly released report.
In the report, Christine Eibner, PhD, of the RAND Corporation and colleagues estimated that the premium for so-called bronze market plans might increase by 2%-10%, depending on the design of a Medicare buy-in program. (The bronze plans are ones with fewer benefits sold on exchanges created through the implementation of the Affordable Care Act of 2010 [ACA].)
A perception has been that younger adults have been in effect subsidizing the cost of older ones in the marketplace plans. Instead, it appears that younger adults who enroll in the individual market tend to be relatively unhealthy and thus expensive to cover.
“When older adults leave the market, insurers are left with a smaller pool of younger, less healthy, and relatively expensive people given their age, leading to higher premiums,” Dr. Eibner and colleagues said in the report.
This result was unexpected, as there has been discussion of using a Medicare buy-in to reduce the cost of premiums for others in the marketplace, Dr. Eibner and colleagues said. But this result is consistent with other recent findings, including research presented by the consulting firm Milliman at a Society of Actuaries meeting in June. The Blue Cross Blue Shield Association estimates that losing a large group of customers could raise premiums by about 10% for the remaining pool of insured people, the New York Times has reported.
That might make it attractive to middle-aged Americans. Dr. Eibner and colleagues estimated the annual premium for a Medicare buy-in at $9,747 in 2022. For a 50-year-old, a bronze-level ACA plan would cost $9,208 and a gold-level one, $12,277. For a 60-year-old, the annual premium for a bronze-level plan might be $13,512 and $18,016 for a gold-level plan.
Total out-of-pocket health spending, including premiums, would fall, on average, by 16%-35% for those who moved from ACA-compliant individual market coverage to a buy-in. The lower spending reflects that buy-in enrollees would have access to Medicare payment rates, which are substantially lower than private rates, and lower administrative costs.
There may be growing interest in allowing people aged 50-64 to buy into Medicare if enthusiasm wanes for bids to create a giant national health program, Dr. Eibner said in an interview.
“If there is concern that Medicare for all is going too far, I think this option is something that could become more prominent,” she said.
Many Democratic lawmakers already are focused on a Medicare buy-in approach. Sen. Debbie Stabenow (D-Mich.) has 20 Democratic cosponsors for her bill, which would allow for a Medicare buy-in at age 50. Sen. Bernie Sanders (I-Vt.) has 14 Democratic cosponsors for the current version of his well-known “Medicare-for-all” bill. That’s two fewer than he had for the Medicare-for-all bill he offered in the 115th session of Congress (Jan. 3, 2017–Jan. 3, 2019).
Among the supporters of Sen. Stabenow’s bill are several 2020 presidential contenders: Sen. Cory Booker (D-N.J.), Sen. Amy Klobuchar (D-Minn.), and Sen. Kamala Harris (D-Calif). As of Saturday, Sen. Elizabeth Warren (D-Mass.) backed Sanders’ bill, but not Stabenow’s. But Sen. Warren also has spoken recently of a Medicare expansion for people at age 50 as a step on the path toward universal coverage. Former Vice President Joseph R. Biden and Mayor Pete Buttigieg of South Bend, Ind., have said they would like to offer Americans the option to buy into Medicare or a public plan.
The idea of lowering the Medicare age has been considered for many years by Democrats. It was seen as a way to help older Americans afford medical care before the enactment of the ACA. Before that law took effect, consumers were not guaranteed access to a health plan, causing many older Americans to go without coverage.
But Dr. Eibner and colleagues found that a Medicare buy-in would have little to no effect on total health insurance enrollment. A Medicare buy-in might increase enrollment by 400,000 to 1.6 million for those over age 50, while decreasing enrollment by 100,000 to 800,000 for those under age 50 because of rising premiums.
“It’s not doing a lot to get people covered,” Dr. Eibner said in the interview.
In the report, Dr. Eibner and colleagues estimated that between 2.8 million and 7.0 million people would choose to enroll, depending on the approach used to design a Medicare buy-in. They considered numerous potential options for the design of a Medicare expansion, including various levels of federal subsidy for people using the buy-in. Dr. Eibner and colleagues also considered whether insurers would respond by trying to selectively market to healthier individuals, increasing their chance of enrolling.
The envisioned Medicare buy-in would have no effect on the Medicare Trust Fund, which pools money available through previously collected dedicated taxes, Dr. Eibner and colleagues said. In creating their model, they drew upon data from the Survey of Income and Program Participation, the Medical Expenditure Panel Survey, and the Kaiser Family Foundation and Health Research and Educational Trust Employer Health Benefits Survey.
Dr. Eibner and colleagues noted “several important limitations” for their work. It does not look at how the buy-in might affect clinicians and hospitals. Lower Medicare payment rates might cause some physicians to turn away patients covered by a Medicare buy-in.
“On the other hand, even if some providers decided not to participate in the buy-in, the buy-in might offer enrollees a broader network than what is available on the individual market,” Dr. Eibner and colleagues wrote. “Furthermore, it is not clear that providers would be legally allowed to opt out of the buy-in while still accepting payment for current Medicare beneficiaries.”
The Rand report was developed with the support of funding by the AARP.
SOURCE: Eibner C et al. RAND Corporation. Research Report. 2019. doi: 10.7249/RR4246.
FDA panel rejects new empagliflozin indication for type 1 diabetes
A Food and Drug Administration advisory panel voted 14-2 against recommending approval of a supplemental New Drug Application for empagliflozin (Jardiance) as an adjunct to insulin therapy to improve glycemic control in adults with type 1 diabetes. The drug is already is approved for people with type 2 diabetes.

Members of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee cited persisting concerns about the risk for diabetic ketoacidosis (DKA) seen with the drug, as well as the limited pool of evidence Boehringer Ingelheim presented. Patients with type 1 diabetes are at increased risk for DKA.
The agency said it typically gets two major studies to support applications for drug approvals, but the application reviewed Nov. 13 rested largely on a single phase 3 trial, in which 241 people with type 1 diabetes took a low dose (2.5 mg) of empagliflozin for about 6 months. Panelists repeatedly objected to the paucity of data they had to consider this expanded approval.
“We owe it to patients with type 1 diabetes to do this right,” said Brendan M. Everett, MD, MPH, of Harvard Medical School, Boston, who served as a panelist. “It’s out of respect for them that I voted no.”
Boehringer Ingelheim, and members of the public argued that people with type 1 diabetes want access to new medicines such as empagliflozin that already are available for people with type 2 diabetes. The agency approved the drug in 2014 at doses of 10 mg and 25 mg as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. In 2016, it approved a new indication for empagliflozin for reducing risk of cardiovascular death in people with type 2 disease.
The panelists said they were sympathetic to that point of view, but stressed the need for further larger studies of empagliflozin in people with type 1 diabetes.
“I do think this is promising,” said panelist Michael Blaha, MD, MPH, of Johns Hopkins University, Baltimore. “But I’m not sure we are here to evaluate promising things. We’re here to evaluate proven things.”
The FDA advisers also said there was a need for greater clarity about reporting of adverse events in testing of this drug. The FDA reviewers reported disagreements with Boehringer about some cases classified as “unlikely ketoacidosis but ketosis” in the study.
“Some events adjudicated as ‘unlikely ketoacidosis but ketosis’ were clinically significant serious events requiring hospitalizations and prompt intervention, such as discontinuation of study drug,” wrote Mahtab Niyyati, MD, an FDA clinical reviewer, in her slides for the meeting.
The FDA is not obliged to accept the suggestions of its advisory panels, but it often does.
The agency did not ask the panel to weigh in directly on whether to approve the drug. Instead, the question put to the panel for a vote was whether the available data suggest that the benefits outweigh the risks of empagliflozin and support approval of a 2.5-mg dose as an adjunct to insulin for people with type 1 diabetes.
Empagliflozin is part of the sodium-glucose cotransporter 2 (SGLT2) inhibitor class of medicines, already known to have a risk for DKA, the agency noted in its briefing document for the meeting. DKA occurs as a result of insulin deficiency and subsequent ketogenesis.
There are no SGLT2 inhibitors approved for type 1 diabetes, the staff said in the review. The agency has rebuffed recent bids by makers of other SGLT2 drugs for people with type 1 diabetes. In July, AstraZeneca said the FDA had not approved its application for use of dapagliflozin (Farxiga) as an adjunct treatment to insulin to improve glycemic control in adult patients with type1 diabetes, when insulin alone does not provide adequate glycemic control. AstraZeneca said it was working with the agency on issues raised in the response letter it received.
In March, the FDA blocked a bid by Sanofi for approval of its investigational SGLT1/2 inhibitor, sotagliflozin (Zynquista) for use in people with type 1 diabetes. A panel had voted 8-8 in January on a question about the additional approval for this drug.
At the Nov. 13 meeting, panel members offered comments about the potential design of a new test for empagliflozin in type 1 diabetes, including a suggestion for a 2-year trial.
Anna McCollister-Slipp, the consumer representative on the FDA panel, cast one of the two votes in favor of use of the drug for people with type 1 diabetes. She said the agency needed to press for more research in this field but also argued that patients can manage the risks of treatments they find valuable. She cited, as an example, how she has stuck with an insulin pump to manage her own type 1 diabetes, despite having setbacks with the device that sent her to the emergency department.
The other vote in support of the empagliflozin application came from panelist Kashif M. Munir, MD, medical director of the University of Maryland Center for Diabetes and Endrocrinology, Baltimore. In explaining his vote, Dr. Munir noted that he and other physicians already are prescribing medications such as empagliflozin for people with type 1 diabetes, even though it is an off-label use. Boehringer’s strategy of using a lower dose of the drug for this group of patients would mean a reduction in effectiveness but also would lower the risk for side effects.
“Some of us do use existing medications” and have patients take partial doses, Dr. Munir said, adding that the current off-label use of the drug persuaded him to vote in favor of expanded approval, despite his concerns about the data.
Empagliflozin given at 2.5 mg resulted in a statistically significant difference of 0.26% in change in hemoglobin HbA1c at week 26, compared with placebo, said Roberto Crackel, PhD, an FDA mathematical statistician, during the presentation. There was a numerically small benefit in body weight and systolic blood pressure, but no benefit in reducing hypoglycemic events, he said.
In concluding, the agency’s presentation, Dr. Niyyati presented a slide depicting potential risk and benefit for empagliflozin with 6 months and then 6.5 years of follow-up. It showed that in terms of the benefit of HbA1c control, there is a potential but undemonstrated reduction in the risk of microvascular complications at 6 months and an estimated 2.8% reduction in microvascular complications after 6.5 years.
In terms of risk of DKA, there are limited data with unstable estimates, ranging to perhaps as many as 468 additional patients-with-events per 10,000 patients at the 6-month point. By the 6.5-year mark, treating 10,000 patients could result in 1,494 additional events.
In a statement issued after the panel’s vote, Boehringer and its partner on empagliflozin, Eli Lilly, stressed the benefit seen with the drug, a statistically significant reduction in HbA1c (0.28%), compared with insulin given with a matched placebo in adults with type 1 diabetes. Secondary endpoints of the trial demonstrated reductions in weight (1.8 kg) and systolic blood pressure (2.1 mm Hg), compared with insulin plus placebo, the companies said.
“We continue to believe the totality of data from the EASE [Empagliflozin as Adjunctive to Insulin Therapy] program indicates a favorable benefit-risk profile for empagliflozin 2.5 mg in adults with type 1 diabetes and look forward to continuing to work with the FDA in this review process,” said Mohamed Eid, MD, MPH, vice president, clinical development & medical affairs, cardiometabolism & respiratory medicine, Boehringer Ingelheim, in a statement.
Speaking as a member of the public, Kelly L. Close, founder of the diaTribe Foundation, urged the FDA to “think creatively” about approval of the drug.” Many people already are using empagliflozin off label, she said. An FDA approval would help physicians and their patients manage the risks of this medicine. Without such help, patients may be needlessly exposed to harm, she argued.
“That’s what happens with popular unregulated drugs that payers cover, and we know that many, many payers are covering this drug for people with type 1,” she said.
Sidney Wolfe, MD, founder and senior adviser to Public Citizen’s health research group, made the opposing argument during the public session. The trial, in which 241 took the low dose of the drug and 241 on placebo, was “underpowered” in Dr. Wolfe’s view. He also stressed the issue that the FDA had raised about adjudication of the cases of side effects.
An FDA approval of the 2.5-mg dose would send “a dangerous false green signal to those doctors who are already prescribing off-label” drugs in SGLT2 inhibitor class, he said.
That would foster a misleading perception “that we have found the sweet spot” balancing safety and risk, he added. “I can’t see how the FDA or the advisory committee would suggest approval” of empagliflozin for type 1 diabetes.”
A Food and Drug Administration advisory panel voted 14-2 against recommending approval of a supplemental New Drug Application for empagliflozin (Jardiance) as an adjunct to insulin therapy to improve glycemic control in adults with type 1 diabetes. The drug is already is approved for people with type 2 diabetes.
Members of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee cited persisting concerns about the risk for diabetic ketoacidosis (DKA) seen with the drug, as well as the limited pool of evidence Boehringer Ingelheim presented. Patients with type 1 diabetes are at increased risk for DKA.
The agency said it typically gets two major studies to support applications for drug approvals, but the application reviewed Nov. 13 rested largely on a single phase 3 trial, in which 241 people with type 1 diabetes took a low dose (2.5 mg) of empagliflozin for about 6 months. Panelists repeatedly objected to the paucity of data they had to consider this expanded approval.
“We owe it to patients with type 1 diabetes to do this right,” said Brendan M. Everett, MD, MPH, of Harvard Medical School, Boston, who served as a panelist. “It’s out of respect for them that I voted no.”
Boehringer Ingelheim, and members of the public argued that people with type 1 diabetes want access to new medicines such as empagliflozin that already are available for people with type 2 diabetes. The agency approved the drug in 2014 at doses of 10 mg and 25 mg as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. In 2016, it approved a new indication for empagliflozin for reducing risk of cardiovascular death in people with type 2 disease.
The panelists said they were sympathetic to that point of view, but stressed the need for further larger studies of empagliflozin in people with type 1 diabetes.
“I do think this is promising,” said panelist Michael Blaha, MD, MPH, of Johns Hopkins University, Baltimore. “But I’m not sure we are here to evaluate promising things. We’re here to evaluate proven things.”
The FDA advisers also said there was a need for greater clarity about reporting of adverse events in testing of this drug. The FDA reviewers reported disagreements with Boehringer about some cases classified as “unlikely ketoacidosis but ketosis” in the study.
“Some events adjudicated as ‘unlikely ketoacidosis but ketosis’ were clinically significant serious events requiring hospitalizations and prompt intervention, such as discontinuation of study drug,” wrote Mahtab Niyyati, MD, an FDA clinical reviewer, in her slides for the meeting.
The FDA is not obliged to accept the suggestions of its advisory panels, but it often does.
The agency did not ask the panel to weigh in directly on whether to approve the drug. Instead, the question put to the panel for a vote was whether the available data suggest that the benefits outweigh the risks of empagliflozin and support approval of a 2.5-mg dose as an adjunct to insulin for people with type 1 diabetes.
Empagliflozin is part of the sodium-glucose cotransporter 2 (SGLT2) inhibitor class of medicines, already known to have a risk for DKA, the agency noted in its briefing document for the meeting. DKA occurs as a result of insulin deficiency and subsequent ketogenesis.
There are no SGLT2 inhibitors approved for type 1 diabetes, the staff said in the review. The agency has rebuffed recent bids by makers of other SGLT2 drugs for people with type 1 diabetes. In July, AstraZeneca said the FDA had not approved its application for use of dapagliflozin (Farxiga) as an adjunct treatment to insulin to improve glycemic control in adult patients with type1 diabetes, when insulin alone does not provide adequate glycemic control. AstraZeneca said it was working with the agency on issues raised in the response letter it received.
In March, the FDA blocked a bid by Sanofi for approval of its investigational SGLT1/2 inhibitor, sotagliflozin (Zynquista) for use in people with type 1 diabetes. A panel had voted 8-8 in January on a question about the additional approval for this drug.
At the Nov. 13 meeting, panel members offered comments about the potential design of a new test for empagliflozin in type 1 diabetes, including a suggestion for a 2-year trial.
Anna McCollister-Slipp, the consumer representative on the FDA panel, cast one of the two votes in favor of use of the drug for people with type 1 diabetes. She said the agency needed to press for more research in this field but also argued that patients can manage the risks of treatments they find valuable. She cited, as an example, how she has stuck with an insulin pump to manage her own type 1 diabetes, despite having setbacks with the device that sent her to the emergency department.
The other vote in support of the empagliflozin application came from panelist Kashif M. Munir, MD, medical director of the University of Maryland Center for Diabetes and Endrocrinology, Baltimore. In explaining his vote, Dr. Munir noted that he and other physicians already are prescribing medications such as empagliflozin for people with type 1 diabetes, even though it is an off-label use. Boehringer’s strategy of using a lower dose of the drug for this group of patients would mean a reduction in effectiveness but also would lower the risk for side effects.
“Some of us do use existing medications” and have patients take partial doses, Dr. Munir said, adding that the current off-label use of the drug persuaded him to vote in favor of expanded approval, despite his concerns about the data.
Empagliflozin given at 2.5 mg resulted in a statistically significant difference of 0.26% in change in hemoglobin HbA1c at week 26, compared with placebo, said Roberto Crackel, PhD, an FDA mathematical statistician, during the presentation. There was a numerically small benefit in body weight and systolic blood pressure, but no benefit in reducing hypoglycemic events, he said.
In concluding, the agency’s presentation, Dr. Niyyati presented a slide depicting potential risk and benefit for empagliflozin with 6 months and then 6.5 years of follow-up. It showed that in terms of the benefit of HbA1c control, there is a potential but undemonstrated reduction in the risk of microvascular complications at 6 months and an estimated 2.8% reduction in microvascular complications after 6.5 years.
In terms of risk of DKA, there are limited data with unstable estimates, ranging to perhaps as many as 468 additional patients-with-events per 10,000 patients at the 6-month point. By the 6.5-year mark, treating 10,000 patients could result in 1,494 additional events.
In a statement issued after the panel’s vote, Boehringer and its partner on empagliflozin, Eli Lilly, stressed the benefit seen with the drug, a statistically significant reduction in HbA1c (0.28%), compared with insulin given with a matched placebo in adults with type 1 diabetes. Secondary endpoints of the trial demonstrated reductions in weight (1.8 kg) and systolic blood pressure (2.1 mm Hg), compared with insulin plus placebo, the companies said.
“We continue to believe the totality of data from the EASE [Empagliflozin as Adjunctive to Insulin Therapy] program indicates a favorable benefit-risk profile for empagliflozin 2.5 mg in adults with type 1 diabetes and look forward to continuing to work with the FDA in this review process,” said Mohamed Eid, MD, MPH, vice president, clinical development & medical affairs, cardiometabolism & respiratory medicine, Boehringer Ingelheim, in a statement.
Speaking as a member of the public, Kelly L. Close, founder of the diaTribe Foundation, urged the FDA to “think creatively” about approval of the drug.” Many people already are using empagliflozin off label, she said. An FDA approval would help physicians and their patients manage the risks of this medicine. Without such help, patients may be needlessly exposed to harm, she argued.
“That’s what happens with popular unregulated drugs that payers cover, and we know that many, many payers are covering this drug for people with type 1,” she said.
Sidney Wolfe, MD, founder and senior adviser to Public Citizen’s health research group, made the opposing argument during the public session. The trial, in which 241 took the low dose of the drug and 241 on placebo, was “underpowered” in Dr. Wolfe’s view. He also stressed the issue that the FDA had raised about adjudication of the cases of side effects.
An FDA approval of the 2.5-mg dose would send “a dangerous false green signal to those doctors who are already prescribing off-label” drugs in SGLT2 inhibitor class, he said.
That would foster a misleading perception “that we have found the sweet spot” balancing safety and risk, he added. “I can’t see how the FDA or the advisory committee would suggest approval” of empagliflozin for type 1 diabetes.”
A Food and Drug Administration advisory panel voted 14-2 against recommending approval of a supplemental New Drug Application for empagliflozin (Jardiance) as an adjunct to insulin therapy to improve glycemic control in adults with type 1 diabetes. The drug is already is approved for people with type 2 diabetes.
Members of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee cited persisting concerns about the risk for diabetic ketoacidosis (DKA) seen with the drug, as well as the limited pool of evidence Boehringer Ingelheim presented. Patients with type 1 diabetes are at increased risk for DKA.
The agency said it typically gets two major studies to support applications for drug approvals, but the application reviewed Nov. 13 rested largely on a single phase 3 trial, in which 241 people with type 1 diabetes took a low dose (2.5 mg) of empagliflozin for about 6 months. Panelists repeatedly objected to the paucity of data they had to consider this expanded approval.
“We owe it to patients with type 1 diabetes to do this right,” said Brendan M. Everett, MD, MPH, of Harvard Medical School, Boston, who served as a panelist. “It’s out of respect for them that I voted no.”
Boehringer Ingelheim, and members of the public argued that people with type 1 diabetes want access to new medicines such as empagliflozin that already are available for people with type 2 diabetes. The agency approved the drug in 2014 at doses of 10 mg and 25 mg as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. In 2016, it approved a new indication for empagliflozin for reducing risk of cardiovascular death in people with type 2 disease.
The panelists said they were sympathetic to that point of view, but stressed the need for further larger studies of empagliflozin in people with type 1 diabetes.
“I do think this is promising,” said panelist Michael Blaha, MD, MPH, of Johns Hopkins University, Baltimore. “But I’m not sure we are here to evaluate promising things. We’re here to evaluate proven things.”
The FDA advisers also said there was a need for greater clarity about reporting of adverse events in testing of this drug. The FDA reviewers reported disagreements with Boehringer about some cases classified as “unlikely ketoacidosis but ketosis” in the study.
“Some events adjudicated as ‘unlikely ketoacidosis but ketosis’ were clinically significant serious events requiring hospitalizations and prompt intervention, such as discontinuation of study drug,” wrote Mahtab Niyyati, MD, an FDA clinical reviewer, in her slides for the meeting.
The FDA is not obliged to accept the suggestions of its advisory panels, but it often does.
The agency did not ask the panel to weigh in directly on whether to approve the drug. Instead, the question put to the panel for a vote was whether the available data suggest that the benefits outweigh the risks of empagliflozin and support approval of a 2.5-mg dose as an adjunct to insulin for people with type 1 diabetes.
Empagliflozin is part of the sodium-glucose cotransporter 2 (SGLT2) inhibitor class of medicines, already known to have a risk for DKA, the agency noted in its briefing document for the meeting. DKA occurs as a result of insulin deficiency and subsequent ketogenesis.
There are no SGLT2 inhibitors approved for type 1 diabetes, the staff said in the review. The agency has rebuffed recent bids by makers of other SGLT2 drugs for people with type 1 diabetes. In July, AstraZeneca said the FDA had not approved its application for use of dapagliflozin (Farxiga) as an adjunct treatment to insulin to improve glycemic control in adult patients with type1 diabetes, when insulin alone does not provide adequate glycemic control. AstraZeneca said it was working with the agency on issues raised in the response letter it received.
In March, the FDA blocked a bid by Sanofi for approval of its investigational SGLT1/2 inhibitor, sotagliflozin (Zynquista) for use in people with type 1 diabetes. A panel had voted 8-8 in January on a question about the additional approval for this drug.
At the Nov. 13 meeting, panel members offered comments about the potential design of a new test for empagliflozin in type 1 diabetes, including a suggestion for a 2-year trial.
Anna McCollister-Slipp, the consumer representative on the FDA panel, cast one of the two votes in favor of use of the drug for people with type 1 diabetes. She said the agency needed to press for more research in this field but also argued that patients can manage the risks of treatments they find valuable. She cited, as an example, how she has stuck with an insulin pump to manage her own type 1 diabetes, despite having setbacks with the device that sent her to the emergency department.
The other vote in support of the empagliflozin application came from panelist Kashif M. Munir, MD, medical director of the University of Maryland Center for Diabetes and Endrocrinology, Baltimore. In explaining his vote, Dr. Munir noted that he and other physicians already are prescribing medications such as empagliflozin for people with type 1 diabetes, even though it is an off-label use. Boehringer’s strategy of using a lower dose of the drug for this group of patients would mean a reduction in effectiveness but also would lower the risk for side effects.
“Some of us do use existing medications” and have patients take partial doses, Dr. Munir said, adding that the current off-label use of the drug persuaded him to vote in favor of expanded approval, despite his concerns about the data.
Empagliflozin given at 2.5 mg resulted in a statistically significant difference of 0.26% in change in hemoglobin HbA1c at week 26, compared with placebo, said Roberto Crackel, PhD, an FDA mathematical statistician, during the presentation. There was a numerically small benefit in body weight and systolic blood pressure, but no benefit in reducing hypoglycemic events, he said.
In concluding, the agency’s presentation, Dr. Niyyati presented a slide depicting potential risk and benefit for empagliflozin with 6 months and then 6.5 years of follow-up. It showed that in terms of the benefit of HbA1c control, there is a potential but undemonstrated reduction in the risk of microvascular complications at 6 months and an estimated 2.8% reduction in microvascular complications after 6.5 years.
In terms of risk of DKA, there are limited data with unstable estimates, ranging to perhaps as many as 468 additional patients-with-events per 10,000 patients at the 6-month point. By the 6.5-year mark, treating 10,000 patients could result in 1,494 additional events.
In a statement issued after the panel’s vote, Boehringer and its partner on empagliflozin, Eli Lilly, stressed the benefit seen with the drug, a statistically significant reduction in HbA1c (0.28%), compared with insulin given with a matched placebo in adults with type 1 diabetes. Secondary endpoints of the trial demonstrated reductions in weight (1.8 kg) and systolic blood pressure (2.1 mm Hg), compared with insulin plus placebo, the companies said.
“We continue to believe the totality of data from the EASE [Empagliflozin as Adjunctive to Insulin Therapy] program indicates a favorable benefit-risk profile for empagliflozin 2.5 mg in adults with type 1 diabetes and look forward to continuing to work with the FDA in this review process,” said Mohamed Eid, MD, MPH, vice president, clinical development & medical affairs, cardiometabolism & respiratory medicine, Boehringer Ingelheim, in a statement.
Speaking as a member of the public, Kelly L. Close, founder of the diaTribe Foundation, urged the FDA to “think creatively” about approval of the drug.” Many people already are using empagliflozin off label, she said. An FDA approval would help physicians and their patients manage the risks of this medicine. Without such help, patients may be needlessly exposed to harm, she argued.
“That’s what happens with popular unregulated drugs that payers cover, and we know that many, many payers are covering this drug for people with type 1,” she said.
Sidney Wolfe, MD, founder and senior adviser to Public Citizen’s health research group, made the opposing argument during the public session. The trial, in which 241 took the low dose of the drug and 241 on placebo, was “underpowered” in Dr. Wolfe’s view. He also stressed the issue that the FDA had raised about adjudication of the cases of side effects.
An FDA approval of the 2.5-mg dose would send “a dangerous false green signal to those doctors who are already prescribing off-label” drugs in SGLT2 inhibitor class, he said.
That would foster a misleading perception “that we have found the sweet spot” balancing safety and risk, he added. “I can’t see how the FDA or the advisory committee would suggest approval” of empagliflozin for type 1 diabetes.”
REPORTING FROM AN FDA ADVISORY COMMITTEE MEETING
NAM offers recommendations to fight clinician burnout
WASHINGTON – a condition now estimated to affect a third to a half of clinicians in the United States, according to a report from an influential federal panel.
The National Academy of Medicine (NAM) on Oct. 23 released a report, “Taking Action Against Clinician Burnout: A Systems Approach to Professional Well-Being.” The report calls for a broad and unified approach to tackling the root causes of burnout.
There must be a concerted effort by leaders of many fields of health care to create less stressful workplaces for clinicians, Pascale Carayon, PhD, cochair of the NAM committee that produced the report, said during the NAM press event.
“This is not an easy process,” said Dr. Carayon, a researcher into patient safety issues at the University of Wisconsin–Madison. “There is no single solution.”
The NAM report assigns specific tasks to many different participants in health care through a six-goal approach, as described below.
–Create positive workplaces. Leaders of health care systems should consider how their business and management decisions will affect clinicians’ jobs, taking into account the potential to add to their levels of burnout. Executives need to continuously monitor and evaluate the extent of burnout in their organizations, and report on this at least annually.
–Address burnout in training and in clinicians’ early years. Medical, nursing, and pharmacy schools should consider steps such as monitoring workload, implementing pass-fail grading, improving access to scholarships and affordable loans, and creating new loan repayment systems.
–Reduce administrative burden. Federal and state bodies and organizations such as the National Quality Forum should reconsider how their regulations and recommendations contribute to burnout. Organizations should seek to eliminate tasks that do not improve the care of patients.
–Improve usability and relevance of health information technology (IT). Medical organizations should develop and buy systems that are as user-friendly and easy to operate as possible. They also should look to use IT to reduce documentation demands and automate nonessential tasks.
–Reduce stigma and improve burnout recovery services. State officials and legislative bodies should make it easier for clinicians to use employee assistance programs, peer support programs, and mental health providers without the information being admissible in malpractice litigation. The report notes the recommendations from the Federation of State Medical Boards, American Medical Association, and the American Psychiatric Association on limiting inquiries in licensing applications about a clinician’s mental health. Questions should focus on current impairment rather than reach well into a clinician’s past.
–Create a national research agenda on clinician well-being. By the end of 2020, federal agencies – including the Agency for Healthcare Research and Quality, the National Institute for Occupational Safety and Health, the Health Resources and Services Administration, and the U.S. Department of Veterans Affairs – should develop a coordinated research agenda on clinician burnout, the report said.
In casting a wide net and assigning specific tasks, the NAM report seeks to establish efforts to address clinician burnout as a broad and shared responsibility. It would be too easy for different medical organizations to depict addressing burnout as being outside of their responsibilities, Christine K. Cassel, MD, the cochair of the NAM committee that produced the report, said during the press event.

“Nothing could be farther from the truth. Everyone is necessary to solve this problem,” said Dr. Cassel, who is a former chief executive officer of the National Quality Forum.
Darrell G. Kirch, MD, chief executive of the Association of American Medical Colleges, described the report as a “call to action” at the press event.
Previously published research has found between 35% and 54% of nurses and physicians in the United States have substantial symptoms of burnout, with the prevalence of burnout ranging between 45% and 60% for medical students and residents, the NAM report said.
Leaders of health organizations must consider how the policies they set will add stress for clinicians and make them less effective in caring for patients, said Vindell Washington, MD, chief medical officer of Blue Cross Blue Shield of Louisiana and a member of the NAM committee that wrote the report.

“Those linkages should be incentives and motivations for boards and leaders more broadly to act on the problem,” Dr. Washington said at the NAM event.
Dr. Kirch said he experienced burnout as a first-year medical student. He said a “brilliant aspect” of the NAM report is its emphasis on burnout as a response to the conditions under which medicine is practiced. In the past, burnout has been viewed as being the fault of the physician or nurse experiencing it, with the response then being to try to “fix” this individual, Dr. Kirch said at the event.
The NAM report instead defines burnout as a “work-related phenomenon studied since at least the 1970s,” in which an individual may experience exhaustion and detachment. Depression and other mental health issues such as anxiety disorders and addiction can follow burnout, he said. “That involves a real human toll.”
Joe Rotella, MD, MBA, chief medical officer at American Academy of Hospice and Palliative Medicine, said in an interview that this NAM paper has the potential to spark the kind of transformation that its earlier research did for the quality of care. Then called the Institute of Medicine(IOM), NAM in 1999 issued a report, “To Err Is Human,” which is broadly seen as a key catalyst in efforts in the ensuing decades to improve the quality of care. IOM then followed up with a 2001 report, “Crossing the Quality Chasm.”
“Those papers over a period of time really did change the way we do health care,” said Dr. Rotella, who was not involved with the NAM report.
In Dr. Rotella’s view, the NAM report provides a solid framework for what remains a daunting task, addressing the many factors involved in burnout.
“The most exciting thing about this is that they don’t have 500 recommendations. They had six and that’s something people can organize around,” he said. “They are not small goals. I’m not saying they are simple.”
The NAM report delves into the factors that contribute to burnout. These include a maze of government and commercial insurance plans that create “a confusing and onerous environment for clinicians,” with many of them juggling “multiple payment systems with complex rules, processes, metrics, and incentives that may frequently change.”
Clinicians face a growing field of measurements intended to judge the quality of their performance. While some of these are useful, others are duplicative and some are not relevant to patient care, the NAM report said.
The report also noted that many clinicians describe electronic health records (EHRs) as taking a toll on their work and private lives. Previously published research has found that for every hour spent with a patient, physicians spend an additional 1-2 hours on the EHR at work, with additional time needed to complete this data entry at home after work hours, the report said.
In an interview, Cynda Rushton, RN, PhD, a Johns Hopkins University researcher and a member of the NAM committee that produced the report, said this new publication will support efforts to overhaul many aspects of current medical practice. She said she hopes it will be a “catalyst for bold and fundamental reform.
“It’s taking a deep dive into the evidence to see how we can begin to dismantle the system’s contributions to burnout,” she said. “No longer can we put Band-Aids on a gaping wound.”
WASHINGTON – a condition now estimated to affect a third to a half of clinicians in the United States, according to a report from an influential federal panel.
The National Academy of Medicine (NAM) on Oct. 23 released a report, “Taking Action Against Clinician Burnout: A Systems Approach to Professional Well-Being.” The report calls for a broad and unified approach to tackling the root causes of burnout.
There must be a concerted effort by leaders of many fields of health care to create less stressful workplaces for clinicians, Pascale Carayon, PhD, cochair of the NAM committee that produced the report, said during the NAM press event.
“This is not an easy process,” said Dr. Carayon, a researcher into patient safety issues at the University of Wisconsin–Madison. “There is no single solution.”
The NAM report assigns specific tasks to many different participants in health care through a six-goal approach, as described below.
–Create positive workplaces. Leaders of health care systems should consider how their business and management decisions will affect clinicians’ jobs, taking into account the potential to add to their levels of burnout. Executives need to continuously monitor and evaluate the extent of burnout in their organizations, and report on this at least annually.
–Address burnout in training and in clinicians’ early years. Medical, nursing, and pharmacy schools should consider steps such as monitoring workload, implementing pass-fail grading, improving access to scholarships and affordable loans, and creating new loan repayment systems.
–Reduce administrative burden. Federal and state bodies and organizations such as the National Quality Forum should reconsider how their regulations and recommendations contribute to burnout. Organizations should seek to eliminate tasks that do not improve the care of patients.
–Improve usability and relevance of health information technology (IT). Medical organizations should develop and buy systems that are as user-friendly and easy to operate as possible. They also should look to use IT to reduce documentation demands and automate nonessential tasks.
–Reduce stigma and improve burnout recovery services. State officials and legislative bodies should make it easier for clinicians to use employee assistance programs, peer support programs, and mental health providers without the information being admissible in malpractice litigation. The report notes the recommendations from the Federation of State Medical Boards, American Medical Association, and the American Psychiatric Association on limiting inquiries in licensing applications about a clinician’s mental health. Questions should focus on current impairment rather than reach well into a clinician’s past.
–Create a national research agenda on clinician well-being. By the end of 2020, federal agencies – including the Agency for Healthcare Research and Quality, the National Institute for Occupational Safety and Health, the Health Resources and Services Administration, and the U.S. Department of Veterans Affairs – should develop a coordinated research agenda on clinician burnout, the report said.
In casting a wide net and assigning specific tasks, the NAM report seeks to establish efforts to address clinician burnout as a broad and shared responsibility. It would be too easy for different medical organizations to depict addressing burnout as being outside of their responsibilities, Christine K. Cassel, MD, the cochair of the NAM committee that produced the report, said during the press event.
“Nothing could be farther from the truth. Everyone is necessary to solve this problem,” said Dr. Cassel, who is a former chief executive officer of the National Quality Forum.
Darrell G. Kirch, MD, chief executive of the Association of American Medical Colleges, described the report as a “call to action” at the press event.
Previously published research has found between 35% and 54% of nurses and physicians in the United States have substantial symptoms of burnout, with the prevalence of burnout ranging between 45% and 60% for medical students and residents, the NAM report said.
Leaders of health organizations must consider how the policies they set will add stress for clinicians and make them less effective in caring for patients, said Vindell Washington, MD, chief medical officer of Blue Cross Blue Shield of Louisiana and a member of the NAM committee that wrote the report.
“Those linkages should be incentives and motivations for boards and leaders more broadly to act on the problem,” Dr. Washington said at the NAM event.
Dr. Kirch said he experienced burnout as a first-year medical student. He said a “brilliant aspect” of the NAM report is its emphasis on burnout as a response to the conditions under which medicine is practiced. In the past, burnout has been viewed as being the fault of the physician or nurse experiencing it, with the response then being to try to “fix” this individual, Dr. Kirch said at the event.
The NAM report instead defines burnout as a “work-related phenomenon studied since at least the 1970s,” in which an individual may experience exhaustion and detachment. Depression and other mental health issues such as anxiety disorders and addiction can follow burnout, he said. “That involves a real human toll.”
Joe Rotella, MD, MBA, chief medical officer at American Academy of Hospice and Palliative Medicine, said in an interview that this NAM paper has the potential to spark the kind of transformation that its earlier research did for the quality of care. Then called the Institute of Medicine(IOM), NAM in 1999 issued a report, “To Err Is Human,” which is broadly seen as a key catalyst in efforts in the ensuing decades to improve the quality of care. IOM then followed up with a 2001 report, “Crossing the Quality Chasm.”
“Those papers over a period of time really did change the way we do health care,” said Dr. Rotella, who was not involved with the NAM report.
In Dr. Rotella’s view, the NAM report provides a solid framework for what remains a daunting task, addressing the many factors involved in burnout.
“The most exciting thing about this is that they don’t have 500 recommendations. They had six and that’s something people can organize around,” he said. “They are not small goals. I’m not saying they are simple.”
The NAM report delves into the factors that contribute to burnout. These include a maze of government and commercial insurance plans that create “a confusing and onerous environment for clinicians,” with many of them juggling “multiple payment systems with complex rules, processes, metrics, and incentives that may frequently change.”
Clinicians face a growing field of measurements intended to judge the quality of their performance. While some of these are useful, others are duplicative and some are not relevant to patient care, the NAM report said.
The report also noted that many clinicians describe electronic health records (EHRs) as taking a toll on their work and private lives. Previously published research has found that for every hour spent with a patient, physicians spend an additional 1-2 hours on the EHR at work, with additional time needed to complete this data entry at home after work hours, the report said.
In an interview, Cynda Rushton, RN, PhD, a Johns Hopkins University researcher and a member of the NAM committee that produced the report, said this new publication will support efforts to overhaul many aspects of current medical practice. She said she hopes it will be a “catalyst for bold and fundamental reform.
“It’s taking a deep dive into the evidence to see how we can begin to dismantle the system’s contributions to burnout,” she said. “No longer can we put Band-Aids on a gaping wound.”
WASHINGTON – a condition now estimated to affect a third to a half of clinicians in the United States, according to a report from an influential federal panel.
The National Academy of Medicine (NAM) on Oct. 23 released a report, “Taking Action Against Clinician Burnout: A Systems Approach to Professional Well-Being.” The report calls for a broad and unified approach to tackling the root causes of burnout.
There must be a concerted effort by leaders of many fields of health care to create less stressful workplaces for clinicians, Pascale Carayon, PhD, cochair of the NAM committee that produced the report, said during the NAM press event.
“This is not an easy process,” said Dr. Carayon, a researcher into patient safety issues at the University of Wisconsin–Madison. “There is no single solution.”
The NAM report assigns specific tasks to many different participants in health care through a six-goal approach, as described below.
–Create positive workplaces. Leaders of health care systems should consider how their business and management decisions will affect clinicians’ jobs, taking into account the potential to add to their levels of burnout. Executives need to continuously monitor and evaluate the extent of burnout in their organizations, and report on this at least annually.
–Address burnout in training and in clinicians’ early years. Medical, nursing, and pharmacy schools should consider steps such as monitoring workload, implementing pass-fail grading, improving access to scholarships and affordable loans, and creating new loan repayment systems.
–Reduce administrative burden. Federal and state bodies and organizations such as the National Quality Forum should reconsider how their regulations and recommendations contribute to burnout. Organizations should seek to eliminate tasks that do not improve the care of patients.
–Improve usability and relevance of health information technology (IT). Medical organizations should develop and buy systems that are as user-friendly and easy to operate as possible. They also should look to use IT to reduce documentation demands and automate nonessential tasks.
–Reduce stigma and improve burnout recovery services. State officials and legislative bodies should make it easier for clinicians to use employee assistance programs, peer support programs, and mental health providers without the information being admissible in malpractice litigation. The report notes the recommendations from the Federation of State Medical Boards, American Medical Association, and the American Psychiatric Association on limiting inquiries in licensing applications about a clinician’s mental health. Questions should focus on current impairment rather than reach well into a clinician’s past.
–Create a national research agenda on clinician well-being. By the end of 2020, federal agencies – including the Agency for Healthcare Research and Quality, the National Institute for Occupational Safety and Health, the Health Resources and Services Administration, and the U.S. Department of Veterans Affairs – should develop a coordinated research agenda on clinician burnout, the report said.
In casting a wide net and assigning specific tasks, the NAM report seeks to establish efforts to address clinician burnout as a broad and shared responsibility. It would be too easy for different medical organizations to depict addressing burnout as being outside of their responsibilities, Christine K. Cassel, MD, the cochair of the NAM committee that produced the report, said during the press event.
“Nothing could be farther from the truth. Everyone is necessary to solve this problem,” said Dr. Cassel, who is a former chief executive officer of the National Quality Forum.
Darrell G. Kirch, MD, chief executive of the Association of American Medical Colleges, described the report as a “call to action” at the press event.
Previously published research has found between 35% and 54% of nurses and physicians in the United States have substantial symptoms of burnout, with the prevalence of burnout ranging between 45% and 60% for medical students and residents, the NAM report said.
Leaders of health organizations must consider how the policies they set will add stress for clinicians and make them less effective in caring for patients, said Vindell Washington, MD, chief medical officer of Blue Cross Blue Shield of Louisiana and a member of the NAM committee that wrote the report.
“Those linkages should be incentives and motivations for boards and leaders more broadly to act on the problem,” Dr. Washington said at the NAM event.
Dr. Kirch said he experienced burnout as a first-year medical student. He said a “brilliant aspect” of the NAM report is its emphasis on burnout as a response to the conditions under which medicine is practiced. In the past, burnout has been viewed as being the fault of the physician or nurse experiencing it, with the response then being to try to “fix” this individual, Dr. Kirch said at the event.
The NAM report instead defines burnout as a “work-related phenomenon studied since at least the 1970s,” in which an individual may experience exhaustion and detachment. Depression and other mental health issues such as anxiety disorders and addiction can follow burnout, he said. “That involves a real human toll.”
Joe Rotella, MD, MBA, chief medical officer at American Academy of Hospice and Palliative Medicine, said in an interview that this NAM paper has the potential to spark the kind of transformation that its earlier research did for the quality of care. Then called the Institute of Medicine(IOM), NAM in 1999 issued a report, “To Err Is Human,” which is broadly seen as a key catalyst in efforts in the ensuing decades to improve the quality of care. IOM then followed up with a 2001 report, “Crossing the Quality Chasm.”
“Those papers over a period of time really did change the way we do health care,” said Dr. Rotella, who was not involved with the NAM report.
In Dr. Rotella’s view, the NAM report provides a solid framework for what remains a daunting task, addressing the many factors involved in burnout.
“The most exciting thing about this is that they don’t have 500 recommendations. They had six and that’s something people can organize around,” he said. “They are not small goals. I’m not saying they are simple.”
The NAM report delves into the factors that contribute to burnout. These include a maze of government and commercial insurance plans that create “a confusing and onerous environment for clinicians,” with many of them juggling “multiple payment systems with complex rules, processes, metrics, and incentives that may frequently change.”
Clinicians face a growing field of measurements intended to judge the quality of their performance. While some of these are useful, others are duplicative and some are not relevant to patient care, the NAM report said.
The report also noted that many clinicians describe electronic health records (EHRs) as taking a toll on their work and private lives. Previously published research has found that for every hour spent with a patient, physicians spend an additional 1-2 hours on the EHR at work, with additional time needed to complete this data entry at home after work hours, the report said.
In an interview, Cynda Rushton, RN, PhD, a Johns Hopkins University researcher and a member of the NAM committee that produced the report, said this new publication will support efforts to overhaul many aspects of current medical practice. She said she hopes it will be a “catalyst for bold and fundamental reform.
“It’s taking a deep dive into the evidence to see how we can begin to dismantle the system’s contributions to burnout,” she said. “No longer can we put Band-Aids on a gaping wound.”
Medical boards change or consider amending mental health-related licensing questions
Delicia M. Haynes, MD, wants the Florida Board of Medicine to take another look at how its licensing applications query physicians about their mental health history. Her state’s board is one of several nationwide that has, in recent years, mulled whether the phrasing of its questions poses an unintended hurdle for physicians who need help for conditions such as depression.

The Federation of State Medical Boards (FSMB) recommends limiting such queries, if they must be asked, to questions about potential current impairment. But the Florida license application takes a more sweeping approach that may discourage physicians from seeking treatment, Dr. Haynes, founder and CEO of Family First Health Center in Daytona Beach, Fla., said in a video. She describes herself as a direct primary care physician.
In the video, which was posted on YouTube in January, 2019, Dr. Haynes discussed her own experience with having been treated for depression and then needing to report that to the state board. Florida’s license application asks if physicians have been treated within the past 5 years for a mental disorder that has impaired the ability to practice medicine. Those who have had such a condition may need to give the board an explanation providing the names of the physicians, therapists, and counselors they have seen, as well as details and dates for the institutions where they received treatment.
State medical boards play a critical role in protecting people from physicians whose current mental health conditions may risk harming patients, Dr. Haynes said. But they must balance that against the consequences of overly intrusive questioning. Florida’s current application wording may deter physicians from seeking care if they develop conditions such as depression, she said.
“It’s the fear of what’s going to happen if I have to check yes,” Dr. Haynes said in her YouTube video. Physicians will wonder how getting treatment could put their license and their employment at risk, she added.
“It’s really important that [state medical boards] are asking the questions that matter and those questions are questions that talk about impairment, and not just having a history,” Dr. Haynes said.
Members of Florida’s board of medicine last year favorably discussed making such a change during a rules/legislative committee meeting, but have not yet implemented it. The board has postponed further discussion of this topic until its December meeting, said Brad Dalton, a spokesman for the Florida Department of Health.
Dr. Haynes said in an email that she will continue to press for changes in her state’s licensing application.
There’s been a wave of reconsideration of these kinds of questions, spurred by FSMB efforts, which included the board’s offering specific advice to state medical boards about questions on licensing applications as part of a set of recommendations on addressing physician wellness and burnout last year in a report.
Boards have the option of omitting or dropping specific inquiries about mental health. But if they choose to retain this question, FSMB recommends using this phrasing: “Are you currently suffering from any condition for which you are not being appropriately treated that impairs your judgment or that would otherwise adversely affect your ability to practice medicine in a competent, ethical and professional manner? (Yes/No)” the report says.
The staff of the Medical Board of California cited FSMB’s recommendation in a January 2019 report about revising the state’s approach to asking physicians about their mental health. In May 2019, the board voted unanimously to revise its questions on physicians’ mental health, narrowing its inquiry on the licensing application to focus on current impairment.
“There are many doctors who do not seek treatment or find it threatening to seek treatment, because they are concerned that they may lose their license,” Peter Yellowlees, MBBS, MD, chief wellness officer and a professor of psychiatry at University of California, Davis, said in a public comment offered at the meeting. “This is ultimately all about patient safety.”
said Joe Knickrehm, a spokesman for the FSMB. Over the past few years, Kentucky, New Hampshire, New Mexico, North Carolina, North Dakota, Ohio, Vermont and Washington have made changes to their licensing questions, he added.
Still, state boards are not moving quickly enough to remove or revise these questions about mental health, said Katherine J. Gold, MD MSW, associate professor in the department of family medicine at University of Michigan, Ann Arbor. She has studied how these queries can deter physicians from seeking treatment for mental illness. A past treatment for postpartum depression may have no bearing on a physician’s practice, yet members of many state medical boards hesitate to alter their approach, she noted.
Groups including the American Medical Association (AMA) and the American Psychiatric Association also have pushed in recent years for changes in state rules about what a physician has to disclose about mental health, with the FSMB taking a lead in these efforts. In many cases, physicians face questions about their mental health that are beyond the limits of standards set by the Americans With Disabilities Act, according to an article published this year in the FSMB’s Journal of Medical Regulation. As of 2017, a review of questions on initial licensure applications for all 50 states and the District of Columbia showed that 32 licensing boards ask questions beyond the limits of ADA standards, the article said.
Consequences of “reporting stable and easily treatable conditions such as anxiety or depression to a state licensing board can range from a physician simply being required to submit a letter from their primary care provider documenting fitness to practice, to a request to appear before the board, to being required to undergo and pay for an examination by a board-appointed physician,” wrote Catherine M. Welcher, an AMA senior policy analyst, and coauthors of the paper, including Humayun J. Chaudhry, DO, FSMB’s CEO.
Nearly 40% of physicians surveyed about medical licensing questions (2,325 of 5,829) indicated they would be reluctant to seek formal medical care for a mental health condition because of potential repercussions, Liselotte N. Dyrbye, MD, and coauthors reported in 2017 in the Mayo Clinic Proceedings.
“You’re sort of stuck. You recognize you have a problem and you want to seek care so that you can get better, but if you do that, you might have to tell the board,” Dr. Dyrbye said in an interview. “You could be mandated potentially to have an outside psychiatric evaluation, to have your personal medical records reviewed by people other than your personal physician.”
Rising levels of burnout among physicians make it critical to remove obstacles to care, said Arthur S. Hengerer, MD, a former chair of the FSMB who has been a leader in efforts to revise medical licensing questions.

“The only thing that really brings joy to medicine are the relationships you develop with patients, the trust that comes from that, and when you can’t develop relationships because you can’t spend the time, because you are clicking and documenting, it hurts,” said Andrew Lamb, MD, who aided in successful efforts to change North Carolina’s licensing questions.
North Carolina approached this issue in steps. In 2017, it removed a question about mental health from its annual physician license renewal form. In 2018, the North Carolina Medical Board voted to drop questions about mental and physical health from physician license applications.
“It’s hoped that removing this question will encourage clinicians to get the help they need, without fear it will compromise their chances to obtain a professional license,” the board said in an annual report.
Other states are retaining these questions, but allowing physicians to check “no” if they are in an approved program for mental health treatment. The North Dakota Board of Medicine took this approach.
“Because our mission is protection of the public, we didn’t feel that we could eliminate that question,” Bonnie Storbakken, JD, executive secretary of the North Dakota Board of Medicine, said in an interview.
The North Dakota board voted in March 2018 to change its question on mental health after the FSMB spurred a national dialogue on this issue, she said. The North Dakota board opted to allow physicians to check ‘no’ on the question about current mental health and or substance issues if they had voluntarily sought assistance from the North Dakota Professional Health Program or a professional health program in another state. The approach preserves anonymity. The question as previously phrased made some physicians feel they had to report their treatment, she said.
In Alabama, physicians also have the option to check “no” on questions about whether they are undergoing mental health treatment if they are participating in a professional assistance program. Mark Jackson, executive director of the Medical Association of Alabama, said he hopes more states give physicians this option for seeking help not only with mental health issues, but with addiction as well.
“You need to get people in and get them help and not jeopardize their career in the process. Society in general has got that problem going on and physicians are not immune from that,” Mr. Jackson said. “Hopefully other states will see that we need to do something to make sure that the physician population is taken care of.”
Pamela Wible, MD, an advocate for physicians’ mental health, did her own review of state board’s licensing questions. In an assessment posted on her website, Dr. Wible, who practices family medicine, gives 13 states an “A” grade for having no mental health questions or one or two straightforward current questions about impairment that do not mention mental health. “Connecticut, Hawaii, Michigan, and New York are the most physician-friendly of all states with no mental health or impairment questions,” Dr. Wible wrote.
The Texas Medical Board earned a “C” grade in Dr. Wible’s survey. In May 2019, that board’s chairman, Sherif Zaafran, MD, wrote about how his organization had taken a new look at the licensing questions, which were last revised in 2014. While the board has not decided on any immediate changes, it is “maintaining an open dialogue to address future concerns,” Dr. Zaafran wrote in the board’s bulletin.
The Texas Medical Board’s physician licensing form, as posted on its website, includes a broadly phrased question on mental health: “Within the past five (5) years, have you been diagnosed with or treated for any: psychotic disorder, delusional disorder, mood disorder, major depression, personality disorder, or any other mental condition which impaired or does impair your behavior, judgment, or ability to function in school or work?”
Physicians who have had treatment for mental health conditions then must submit another form. They may have to provide details on diagnosis, prognosis, and medications prescribed. Compliance requirements include counseling records, contracts with impairment support groups, and records on file with law enforcement agencies and licensing agencies.
State officials are trying to figure out how best to learn of potential impairments that could pose a risk for patients, without scaring physicians away from getting the care they themselves need said Stephen ‘Brint’ Carlton, JD, executive director of the Texas Medical Board.
Delicia M. Haynes, MD, wants the Florida Board of Medicine to take another look at how its licensing applications query physicians about their mental health history. Her state’s board is one of several nationwide that has, in recent years, mulled whether the phrasing of its questions poses an unintended hurdle for physicians who need help for conditions such as depression.
The Federation of State Medical Boards (FSMB) recommends limiting such queries, if they must be asked, to questions about potential current impairment. But the Florida license application takes a more sweeping approach that may discourage physicians from seeking treatment, Dr. Haynes, founder and CEO of Family First Health Center in Daytona Beach, Fla., said in a video. She describes herself as a direct primary care physician.
In the video, which was posted on YouTube in January, 2019, Dr. Haynes discussed her own experience with having been treated for depression and then needing to report that to the state board. Florida’s license application asks if physicians have been treated within the past 5 years for a mental disorder that has impaired the ability to practice medicine. Those who have had such a condition may need to give the board an explanation providing the names of the physicians, therapists, and counselors they have seen, as well as details and dates for the institutions where they received treatment.
State medical boards play a critical role in protecting people from physicians whose current mental health conditions may risk harming patients, Dr. Haynes said. But they must balance that against the consequences of overly intrusive questioning. Florida’s current application wording may deter physicians from seeking care if they develop conditions such as depression, she said.
“It’s the fear of what’s going to happen if I have to check yes,” Dr. Haynes said in her YouTube video. Physicians will wonder how getting treatment could put their license and their employment at risk, she added.
“It’s really important that [state medical boards] are asking the questions that matter and those questions are questions that talk about impairment, and not just having a history,” Dr. Haynes said.
Members of Florida’s board of medicine last year favorably discussed making such a change during a rules/legislative committee meeting, but have not yet implemented it. The board has postponed further discussion of this topic until its December meeting, said Brad Dalton, a spokesman for the Florida Department of Health.
Dr. Haynes said in an email that she will continue to press for changes in her state’s licensing application.
There’s been a wave of reconsideration of these kinds of questions, spurred by FSMB efforts, which included the board’s offering specific advice to state medical boards about questions on licensing applications as part of a set of recommendations on addressing physician wellness and burnout last year in a report.
Boards have the option of omitting or dropping specific inquiries about mental health. But if they choose to retain this question, FSMB recommends using this phrasing: “Are you currently suffering from any condition for which you are not being appropriately treated that impairs your judgment or that would otherwise adversely affect your ability to practice medicine in a competent, ethical and professional manner? (Yes/No)” the report says.
The staff of the Medical Board of California cited FSMB’s recommendation in a January 2019 report about revising the state’s approach to asking physicians about their mental health. In May 2019, the board voted unanimously to revise its questions on physicians’ mental health, narrowing its inquiry on the licensing application to focus on current impairment.
“There are many doctors who do not seek treatment or find it threatening to seek treatment, because they are concerned that they may lose their license,” Peter Yellowlees, MBBS, MD, chief wellness officer and a professor of psychiatry at University of California, Davis, said in a public comment offered at the meeting. “This is ultimately all about patient safety.”
said Joe Knickrehm, a spokesman for the FSMB. Over the past few years, Kentucky, New Hampshire, New Mexico, North Carolina, North Dakota, Ohio, Vermont and Washington have made changes to their licensing questions, he added.
Still, state boards are not moving quickly enough to remove or revise these questions about mental health, said Katherine J. Gold, MD MSW, associate professor in the department of family medicine at University of Michigan, Ann Arbor. She has studied how these queries can deter physicians from seeking treatment for mental illness. A past treatment for postpartum depression may have no bearing on a physician’s practice, yet members of many state medical boards hesitate to alter their approach, she noted.
Groups including the American Medical Association (AMA) and the American Psychiatric Association also have pushed in recent years for changes in state rules about what a physician has to disclose about mental health, with the FSMB taking a lead in these efforts. In many cases, physicians face questions about their mental health that are beyond the limits of standards set by the Americans With Disabilities Act, according to an article published this year in the FSMB’s Journal of Medical Regulation. As of 2017, a review of questions on initial licensure applications for all 50 states and the District of Columbia showed that 32 licensing boards ask questions beyond the limits of ADA standards, the article said.
Consequences of “reporting stable and easily treatable conditions such as anxiety or depression to a state licensing board can range from a physician simply being required to submit a letter from their primary care provider documenting fitness to practice, to a request to appear before the board, to being required to undergo and pay for an examination by a board-appointed physician,” wrote Catherine M. Welcher, an AMA senior policy analyst, and coauthors of the paper, including Humayun J. Chaudhry, DO, FSMB’s CEO.
Nearly 40% of physicians surveyed about medical licensing questions (2,325 of 5,829) indicated they would be reluctant to seek formal medical care for a mental health condition because of potential repercussions, Liselotte N. Dyrbye, MD, and coauthors reported in 2017 in the Mayo Clinic Proceedings.
“You’re sort of stuck. You recognize you have a problem and you want to seek care so that you can get better, but if you do that, you might have to tell the board,” Dr. Dyrbye said in an interview. “You could be mandated potentially to have an outside psychiatric evaluation, to have your personal medical records reviewed by people other than your personal physician.”
Rising levels of burnout among physicians make it critical to remove obstacles to care, said Arthur S. Hengerer, MD, a former chair of the FSMB who has been a leader in efforts to revise medical licensing questions.
“The only thing that really brings joy to medicine are the relationships you develop with patients, the trust that comes from that, and when you can’t develop relationships because you can’t spend the time, because you are clicking and documenting, it hurts,” said Andrew Lamb, MD, who aided in successful efforts to change North Carolina’s licensing questions.
North Carolina approached this issue in steps. In 2017, it removed a question about mental health from its annual physician license renewal form. In 2018, the North Carolina Medical Board voted to drop questions about mental and physical health from physician license applications.
“It’s hoped that removing this question will encourage clinicians to get the help they need, without fear it will compromise their chances to obtain a professional license,” the board said in an annual report.
Other states are retaining these questions, but allowing physicians to check “no” if they are in an approved program for mental health treatment. The North Dakota Board of Medicine took this approach.
“Because our mission is protection of the public, we didn’t feel that we could eliminate that question,” Bonnie Storbakken, JD, executive secretary of the North Dakota Board of Medicine, said in an interview.
The North Dakota board voted in March 2018 to change its question on mental health after the FSMB spurred a national dialogue on this issue, she said. The North Dakota board opted to allow physicians to check ‘no’ on the question about current mental health and or substance issues if they had voluntarily sought assistance from the North Dakota Professional Health Program or a professional health program in another state. The approach preserves anonymity. The question as previously phrased made some physicians feel they had to report their treatment, she said.
In Alabama, physicians also have the option to check “no” on questions about whether they are undergoing mental health treatment if they are participating in a professional assistance program. Mark Jackson, executive director of the Medical Association of Alabama, said he hopes more states give physicians this option for seeking help not only with mental health issues, but with addiction as well.
“You need to get people in and get them help and not jeopardize their career in the process. Society in general has got that problem going on and physicians are not immune from that,” Mr. Jackson said. “Hopefully other states will see that we need to do something to make sure that the physician population is taken care of.”
Pamela Wible, MD, an advocate for physicians’ mental health, did her own review of state board’s licensing questions. In an assessment posted on her website, Dr. Wible, who practices family medicine, gives 13 states an “A” grade for having no mental health questions or one or two straightforward current questions about impairment that do not mention mental health. “Connecticut, Hawaii, Michigan, and New York are the most physician-friendly of all states with no mental health or impairment questions,” Dr. Wible wrote.
The Texas Medical Board earned a “C” grade in Dr. Wible’s survey. In May 2019, that board’s chairman, Sherif Zaafran, MD, wrote about how his organization had taken a new look at the licensing questions, which were last revised in 2014. While the board has not decided on any immediate changes, it is “maintaining an open dialogue to address future concerns,” Dr. Zaafran wrote in the board’s bulletin.
The Texas Medical Board’s physician licensing form, as posted on its website, includes a broadly phrased question on mental health: “Within the past five (5) years, have you been diagnosed with or treated for any: psychotic disorder, delusional disorder, mood disorder, major depression, personality disorder, or any other mental condition which impaired or does impair your behavior, judgment, or ability to function in school or work?”
Physicians who have had treatment for mental health conditions then must submit another form. They may have to provide details on diagnosis, prognosis, and medications prescribed. Compliance requirements include counseling records, contracts with impairment support groups, and records on file with law enforcement agencies and licensing agencies.
State officials are trying to figure out how best to learn of potential impairments that could pose a risk for patients, without scaring physicians away from getting the care they themselves need said Stephen ‘Brint’ Carlton, JD, executive director of the Texas Medical Board.
Delicia M. Haynes, MD, wants the Florida Board of Medicine to take another look at how its licensing applications query physicians about their mental health history. Her state’s board is one of several nationwide that has, in recent years, mulled whether the phrasing of its questions poses an unintended hurdle for physicians who need help for conditions such as depression.
The Federation of State Medical Boards (FSMB) recommends limiting such queries, if they must be asked, to questions about potential current impairment. But the Florida license application takes a more sweeping approach that may discourage physicians from seeking treatment, Dr. Haynes, founder and CEO of Family First Health Center in Daytona Beach, Fla., said in a video. She describes herself as a direct primary care physician.
In the video, which was posted on YouTube in January, 2019, Dr. Haynes discussed her own experience with having been treated for depression and then needing to report that to the state board. Florida’s license application asks if physicians have been treated within the past 5 years for a mental disorder that has impaired the ability to practice medicine. Those who have had such a condition may need to give the board an explanation providing the names of the physicians, therapists, and counselors they have seen, as well as details and dates for the institutions where they received treatment.
State medical boards play a critical role in protecting people from physicians whose current mental health conditions may risk harming patients, Dr. Haynes said. But they must balance that against the consequences of overly intrusive questioning. Florida’s current application wording may deter physicians from seeking care if they develop conditions such as depression, she said.
“It’s the fear of what’s going to happen if I have to check yes,” Dr. Haynes said in her YouTube video. Physicians will wonder how getting treatment could put their license and their employment at risk, she added.
“It’s really important that [state medical boards] are asking the questions that matter and those questions are questions that talk about impairment, and not just having a history,” Dr. Haynes said.
Members of Florida’s board of medicine last year favorably discussed making such a change during a rules/legislative committee meeting, but have not yet implemented it. The board has postponed further discussion of this topic until its December meeting, said Brad Dalton, a spokesman for the Florida Department of Health.
Dr. Haynes said in an email that she will continue to press for changes in her state’s licensing application.
There’s been a wave of reconsideration of these kinds of questions, spurred by FSMB efforts, which included the board’s offering specific advice to state medical boards about questions on licensing applications as part of a set of recommendations on addressing physician wellness and burnout last year in a report.
Boards have the option of omitting or dropping specific inquiries about mental health. But if they choose to retain this question, FSMB recommends using this phrasing: “Are you currently suffering from any condition for which you are not being appropriately treated that impairs your judgment or that would otherwise adversely affect your ability to practice medicine in a competent, ethical and professional manner? (Yes/No)” the report says.
The staff of the Medical Board of California cited FSMB’s recommendation in a January 2019 report about revising the state’s approach to asking physicians about their mental health. In May 2019, the board voted unanimously to revise its questions on physicians’ mental health, narrowing its inquiry on the licensing application to focus on current impairment.
“There are many doctors who do not seek treatment or find it threatening to seek treatment, because they are concerned that they may lose their license,” Peter Yellowlees, MBBS, MD, chief wellness officer and a professor of psychiatry at University of California, Davis, said in a public comment offered at the meeting. “This is ultimately all about patient safety.”
said Joe Knickrehm, a spokesman for the FSMB. Over the past few years, Kentucky, New Hampshire, New Mexico, North Carolina, North Dakota, Ohio, Vermont and Washington have made changes to their licensing questions, he added.
Still, state boards are not moving quickly enough to remove or revise these questions about mental health, said Katherine J. Gold, MD MSW, associate professor in the department of family medicine at University of Michigan, Ann Arbor. She has studied how these queries can deter physicians from seeking treatment for mental illness. A past treatment for postpartum depression may have no bearing on a physician’s practice, yet members of many state medical boards hesitate to alter their approach, she noted.
Groups including the American Medical Association (AMA) and the American Psychiatric Association also have pushed in recent years for changes in state rules about what a physician has to disclose about mental health, with the FSMB taking a lead in these efforts. In many cases, physicians face questions about their mental health that are beyond the limits of standards set by the Americans With Disabilities Act, according to an article published this year in the FSMB’s Journal of Medical Regulation. As of 2017, a review of questions on initial licensure applications for all 50 states and the District of Columbia showed that 32 licensing boards ask questions beyond the limits of ADA standards, the article said.
Consequences of “reporting stable and easily treatable conditions such as anxiety or depression to a state licensing board can range from a physician simply being required to submit a letter from their primary care provider documenting fitness to practice, to a request to appear before the board, to being required to undergo and pay for an examination by a board-appointed physician,” wrote Catherine M. Welcher, an AMA senior policy analyst, and coauthors of the paper, including Humayun J. Chaudhry, DO, FSMB’s CEO.
Nearly 40% of physicians surveyed about medical licensing questions (2,325 of 5,829) indicated they would be reluctant to seek formal medical care for a mental health condition because of potential repercussions, Liselotte N. Dyrbye, MD, and coauthors reported in 2017 in the Mayo Clinic Proceedings.
“You’re sort of stuck. You recognize you have a problem and you want to seek care so that you can get better, but if you do that, you might have to tell the board,” Dr. Dyrbye said in an interview. “You could be mandated potentially to have an outside psychiatric evaluation, to have your personal medical records reviewed by people other than your personal physician.”
Rising levels of burnout among physicians make it critical to remove obstacles to care, said Arthur S. Hengerer, MD, a former chair of the FSMB who has been a leader in efforts to revise medical licensing questions.
“The only thing that really brings joy to medicine are the relationships you develop with patients, the trust that comes from that, and when you can’t develop relationships because you can’t spend the time, because you are clicking and documenting, it hurts,” said Andrew Lamb, MD, who aided in successful efforts to change North Carolina’s licensing questions.
North Carolina approached this issue in steps. In 2017, it removed a question about mental health from its annual physician license renewal form. In 2018, the North Carolina Medical Board voted to drop questions about mental and physical health from physician license applications.
“It’s hoped that removing this question will encourage clinicians to get the help they need, without fear it will compromise their chances to obtain a professional license,” the board said in an annual report.
Other states are retaining these questions, but allowing physicians to check “no” if they are in an approved program for mental health treatment. The North Dakota Board of Medicine took this approach.
“Because our mission is protection of the public, we didn’t feel that we could eliminate that question,” Bonnie Storbakken, JD, executive secretary of the North Dakota Board of Medicine, said in an interview.
The North Dakota board voted in March 2018 to change its question on mental health after the FSMB spurred a national dialogue on this issue, she said. The North Dakota board opted to allow physicians to check ‘no’ on the question about current mental health and or substance issues if they had voluntarily sought assistance from the North Dakota Professional Health Program or a professional health program in another state. The approach preserves anonymity. The question as previously phrased made some physicians feel they had to report their treatment, she said.
In Alabama, physicians also have the option to check “no” on questions about whether they are undergoing mental health treatment if they are participating in a professional assistance program. Mark Jackson, executive director of the Medical Association of Alabama, said he hopes more states give physicians this option for seeking help not only with mental health issues, but with addiction as well.
“You need to get people in and get them help and not jeopardize their career in the process. Society in general has got that problem going on and physicians are not immune from that,” Mr. Jackson said. “Hopefully other states will see that we need to do something to make sure that the physician population is taken care of.”
Pamela Wible, MD, an advocate for physicians’ mental health, did her own review of state board’s licensing questions. In an assessment posted on her website, Dr. Wible, who practices family medicine, gives 13 states an “A” grade for having no mental health questions or one or two straightforward current questions about impairment that do not mention mental health. “Connecticut, Hawaii, Michigan, and New York are the most physician-friendly of all states with no mental health or impairment questions,” Dr. Wible wrote.
The Texas Medical Board earned a “C” grade in Dr. Wible’s survey. In May 2019, that board’s chairman, Sherif Zaafran, MD, wrote about how his organization had taken a new look at the licensing questions, which were last revised in 2014. While the board has not decided on any immediate changes, it is “maintaining an open dialogue to address future concerns,” Dr. Zaafran wrote in the board’s bulletin.
The Texas Medical Board’s physician licensing form, as posted on its website, includes a broadly phrased question on mental health: “Within the past five (5) years, have you been diagnosed with or treated for any: psychotic disorder, delusional disorder, mood disorder, major depression, personality disorder, or any other mental condition which impaired or does impair your behavior, judgment, or ability to function in school or work?”
Physicians who have had treatment for mental health conditions then must submit another form. They may have to provide details on diagnosis, prognosis, and medications prescribed. Compliance requirements include counseling records, contracts with impairment support groups, and records on file with law enforcement agencies and licensing agencies.
State officials are trying to figure out how best to learn of potential impairments that could pose a risk for patients, without scaring physicians away from getting the care they themselves need said Stephen ‘Brint’ Carlton, JD, executive director of the Texas Medical Board.
Patient-reported outcomes are here to stay
WASHINGTON – The federal official who helps oversee Medicare’s use of quality measures predicted a continued emphasis on patient-reported outcomes in the assessments of physician performance.
Reena Duseja, MD, chief medical officer for quality measurement at the Centers for Medicare & Medicaid Services, said she has seen “more emphasis” in her 2 years with the agency in collecting outcome measures, including ones reported by patients. In doing this, CMS officials are also looking to identify the core elements that willl be part of patient-reported outcomes (PROs).
“We really have to get better at standardization,” Dr. Duseja said during a policy summit sponsored by the National Comprehensive Cancer Network (NCCN). “There is room for improvement there. We’re continuing to think of ways that we can support that.”
She also said the CMS is working, in general, to try to give physicians feedback sooner on how they are faring on measurements.
“The commitment of our agency is trying to think about how we collect data in a way that shortens the cycle of measure development” and speeds the delivery of this data back to providers, Dr. Duseja said.
Her fellow panelists discussed the difficulties in designing PRO measures, including the need to account for special challenges for patients living in or near poverty. Avoiding emergency department visits and hospitalizations, for example, may be a key priority for people who are paid hourly wages, said Kashyap Patel, MD, managing partner of Carolina Blood and Cancer Care in Rock Hill, S.C. These patients will not only face the inconvenience and cost of a hospital stay, but will also lose wages from missed work. He urged policymakers to take these factors into consideration in designing quality measures, and not to forget that “there is a human being behind this.”
Ronald S. Walters, MD, associate head of the Institute for Cancer Care Innovation at the University of Texas MD Anderson Cancer Center, and chair of NCCN’s board of directors, cautioned against continued attempts to devise a “Nirvana” list of outcome measures that can be universally applied. Instead, payers may be better off with a “mix and match” approach. Certain measures may be used across the board, such as pain and quality of life metrics, while other measures could be more tailored.

Dr. Walters also called out a missed opportunity to tie PROs to Medicare payment in the area of chimeric antigen receptor (CAR) T-cell therapy.
In 2018, the CMS indicated it was considering the use of PROs in connection with CAR T-cell payment. The CMS asked its Medicare Evidence Development and Coverage Advisory Committee (MEDCAC) to consider the role of PROs in connection with payment for CAR T-cell therapy. At an August 2018 meeting, MEDCAC panelists generally expressed confidence in PROs in a series of votes about the use of this approach to quality measurement in cancer trials.
But drugmakers and physician groups raised strong objections at the MEDCAC meeting. In its national coverage decision on CAR T, issued in August 2019, the CMS said it had received many comments on PROs “ranging from support of their collection to recommendations for additional assessment tools to request to remove PRO requirements.” The CMS opted at this time to encourage participation in the Center for International Blood and Marrow Transplantation Research (CIBMTR) database “that currently collects health outcomes (and aims to collect patient reported outcomes in the future) on patients who have received CAR T-cell treatments.”
For Dr. Walters, this setback for the use of PROs in CAR T therapy payment is telling, as the treatment is known to produce serious side effects and is administered in well-controlled circumstances.
“If you can’t organize collecting patient-reported outcomes after CAR T cell, that kind of tells you the state of where we are on collecting them on everybody,” Dr. Walters said.
WASHINGTON – The federal official who helps oversee Medicare’s use of quality measures predicted a continued emphasis on patient-reported outcomes in the assessments of physician performance.
Reena Duseja, MD, chief medical officer for quality measurement at the Centers for Medicare & Medicaid Services, said she has seen “more emphasis” in her 2 years with the agency in collecting outcome measures, including ones reported by patients. In doing this, CMS officials are also looking to identify the core elements that willl be part of patient-reported outcomes (PROs).
“We really have to get better at standardization,” Dr. Duseja said during a policy summit sponsored by the National Comprehensive Cancer Network (NCCN). “There is room for improvement there. We’re continuing to think of ways that we can support that.”
She also said the CMS is working, in general, to try to give physicians feedback sooner on how they are faring on measurements.
“The commitment of our agency is trying to think about how we collect data in a way that shortens the cycle of measure development” and speeds the delivery of this data back to providers, Dr. Duseja said.
Her fellow panelists discussed the difficulties in designing PRO measures, including the need to account for special challenges for patients living in or near poverty. Avoiding emergency department visits and hospitalizations, for example, may be a key priority for people who are paid hourly wages, said Kashyap Patel, MD, managing partner of Carolina Blood and Cancer Care in Rock Hill, S.C. These patients will not only face the inconvenience and cost of a hospital stay, but will also lose wages from missed work. He urged policymakers to take these factors into consideration in designing quality measures, and not to forget that “there is a human being behind this.”
Ronald S. Walters, MD, associate head of the Institute for Cancer Care Innovation at the University of Texas MD Anderson Cancer Center, and chair of NCCN’s board of directors, cautioned against continued attempts to devise a “Nirvana” list of outcome measures that can be universally applied. Instead, payers may be better off with a “mix and match” approach. Certain measures may be used across the board, such as pain and quality of life metrics, while other measures could be more tailored.
Dr. Walters also called out a missed opportunity to tie PROs to Medicare payment in the area of chimeric antigen receptor (CAR) T-cell therapy.
In 2018, the CMS indicated it was considering the use of PROs in connection with CAR T-cell payment. The CMS asked its Medicare Evidence Development and Coverage Advisory Committee (MEDCAC) to consider the role of PROs in connection with payment for CAR T-cell therapy. At an August 2018 meeting, MEDCAC panelists generally expressed confidence in PROs in a series of votes about the use of this approach to quality measurement in cancer trials.
But drugmakers and physician groups raised strong objections at the MEDCAC meeting. In its national coverage decision on CAR T, issued in August 2019, the CMS said it had received many comments on PROs “ranging from support of their collection to recommendations for additional assessment tools to request to remove PRO requirements.” The CMS opted at this time to encourage participation in the Center for International Blood and Marrow Transplantation Research (CIBMTR) database “that currently collects health outcomes (and aims to collect patient reported outcomes in the future) on patients who have received CAR T-cell treatments.”
For Dr. Walters, this setback for the use of PROs in CAR T therapy payment is telling, as the treatment is known to produce serious side effects and is administered in well-controlled circumstances.
“If you can’t organize collecting patient-reported outcomes after CAR T cell, that kind of tells you the state of where we are on collecting them on everybody,” Dr. Walters said.
WASHINGTON – The federal official who helps oversee Medicare’s use of quality measures predicted a continued emphasis on patient-reported outcomes in the assessments of physician performance.
Reena Duseja, MD, chief medical officer for quality measurement at the Centers for Medicare & Medicaid Services, said she has seen “more emphasis” in her 2 years with the agency in collecting outcome measures, including ones reported by patients. In doing this, CMS officials are also looking to identify the core elements that willl be part of patient-reported outcomes (PROs).
“We really have to get better at standardization,” Dr. Duseja said during a policy summit sponsored by the National Comprehensive Cancer Network (NCCN). “There is room for improvement there. We’re continuing to think of ways that we can support that.”
She also said the CMS is working, in general, to try to give physicians feedback sooner on how they are faring on measurements.
“The commitment of our agency is trying to think about how we collect data in a way that shortens the cycle of measure development” and speeds the delivery of this data back to providers, Dr. Duseja said.
Her fellow panelists discussed the difficulties in designing PRO measures, including the need to account for special challenges for patients living in or near poverty. Avoiding emergency department visits and hospitalizations, for example, may be a key priority for people who are paid hourly wages, said Kashyap Patel, MD, managing partner of Carolina Blood and Cancer Care in Rock Hill, S.C. These patients will not only face the inconvenience and cost of a hospital stay, but will also lose wages from missed work. He urged policymakers to take these factors into consideration in designing quality measures, and not to forget that “there is a human being behind this.”
Ronald S. Walters, MD, associate head of the Institute for Cancer Care Innovation at the University of Texas MD Anderson Cancer Center, and chair of NCCN’s board of directors, cautioned against continued attempts to devise a “Nirvana” list of outcome measures that can be universally applied. Instead, payers may be better off with a “mix and match” approach. Certain measures may be used across the board, such as pain and quality of life metrics, while other measures could be more tailored.
Dr. Walters also called out a missed opportunity to tie PROs to Medicare payment in the area of chimeric antigen receptor (CAR) T-cell therapy.
In 2018, the CMS indicated it was considering the use of PROs in connection with CAR T-cell payment. The CMS asked its Medicare Evidence Development and Coverage Advisory Committee (MEDCAC) to consider the role of PROs in connection with payment for CAR T-cell therapy. At an August 2018 meeting, MEDCAC panelists generally expressed confidence in PROs in a series of votes about the use of this approach to quality measurement in cancer trials.
But drugmakers and physician groups raised strong objections at the MEDCAC meeting. In its national coverage decision on CAR T, issued in August 2019, the CMS said it had received many comments on PROs “ranging from support of their collection to recommendations for additional assessment tools to request to remove PRO requirements.” The CMS opted at this time to encourage participation in the Center for International Blood and Marrow Transplantation Research (CIBMTR) database “that currently collects health outcomes (and aims to collect patient reported outcomes in the future) on patients who have received CAR T-cell treatments.”
For Dr. Walters, this setback for the use of PROs in CAR T therapy payment is telling, as the treatment is known to produce serious side effects and is administered in well-controlled circumstances.
“If you can’t organize collecting patient-reported outcomes after CAR T cell, that kind of tells you the state of where we are on collecting them on everybody,” Dr. Walters said.
REPORTING FROM NCCN POLICY SUMMIT 2019
CMS weighs extension of Oncology Care Model
WASHINGTON – Federal officials are considering how they might extend a test of payment approaches meant to spur more coordinated cancer care beyond the program’s current 2021 end date.

Alexandra Chong, PhD, the team lead for the Oncology Care Model at the Center for Medicare and Medicaid Innovation (CMMI), said her agency found strong support for sustaining this kind of effort at “practice transformation.” The Oncology Care Model, which kicked off in 2016, involves about 175 practices and 10 insurers.
“As we look forward, we certainly recognize and appreciate the importance of the impact that [the Oncology Care Model] made and the desire for a continuation, either in an iteration of the current model or a different model,” Dr. Chong said during a panel discussion at a policy summit sponsored by the National Comprehensive Cancer Network (NCCN).
Dr. Chong said the Centers for Medicare & Medicaid Services is aware that there is “a lot of interest” in the future of the program. “We have heard from our practices that this model has been an opportunity for them to provide the care that they really want to provide,” Dr. Chong said.
A fellow panelist at the NCCN event, Kerin Adelson, MD, chief quality officer of Yale’s Smilow Cancer Hospital, New Haven, Conn., pressed for continuation of this kind of a payment test. Smilow has used revenue from the Oncology Care Model program to develop dashboards to measure individual clinicians’ patterns of care and share data with them, Dr. Adelson said.
The changes have been “transformative,” including allowing for hiring of additional support staff, Dr. Adelson said.
“I am terrified about what’s going to happen at the end of the program if there is not another model to transition to,” Dr. Adelson said. “We will potentially be looking at layoffs of these people who are so incredible and have done so much for the care we are giving.”
In May 2019, some of Dr. Chong’s CMMI colleagues published a report in the Journal of the National Cancer Institute about the experiences of clinics participating in the Oncology Care Model. For instance, the U.S. Oncology Network used Monthly Enhanced Oncology Services [MEOS] payments through the program for new hires such as navigators, social workers, additional advanced practice providers, “and even data analysts,” according to the report.
Still, there have been “growing pains” with the start-up of the model, Dr. Chong said. Fellow participants on the NCCN panel cited the introduction of costly new drugs as one wrinkle in the early days of the Oncology Care Model. Another is that physicians in practices signed up for the program sometimes are unaware of it.
The American Society of Clinical Oncology also is advocating for a continuation of a model along the lines of the Oncology Care Model.
“It would be good to see it continue. We’re certainly supportive of continuing to work on this model and refine it and see where it goes,” Stephen S. Grubbs, MD, ASCO’s vice president for clinical affairs, said in an interview.

Dr. Grubbs said that CMS’s Oncology Care Model could not be greatly expanded in its current form, as it imposes a steep administrative burden for both practices who participate, as well as for CMMI. The current model also does not serve smaller and rural practices well, he said.
ASCO is refining its own proposal for an alternative payment approach. The group plans to present its Patient-Centered Oncology Payment (PCOP) model, first published in 2015, before an influential federal advisory group, the Physician-Focused Payment Model Technical Advisory Committee (PTAC), at a meeting in 2020.
WASHINGTON – Federal officials are considering how they might extend a test of payment approaches meant to spur more coordinated cancer care beyond the program’s current 2021 end date.
Alexandra Chong, PhD, the team lead for the Oncology Care Model at the Center for Medicare and Medicaid Innovation (CMMI), said her agency found strong support for sustaining this kind of effort at “practice transformation.” The Oncology Care Model, which kicked off in 2016, involves about 175 practices and 10 insurers.
“As we look forward, we certainly recognize and appreciate the importance of the impact that [the Oncology Care Model] made and the desire for a continuation, either in an iteration of the current model or a different model,” Dr. Chong said during a panel discussion at a policy summit sponsored by the National Comprehensive Cancer Network (NCCN).
Dr. Chong said the Centers for Medicare & Medicaid Services is aware that there is “a lot of interest” in the future of the program. “We have heard from our practices that this model has been an opportunity for them to provide the care that they really want to provide,” Dr. Chong said.
A fellow panelist at the NCCN event, Kerin Adelson, MD, chief quality officer of Yale’s Smilow Cancer Hospital, New Haven, Conn., pressed for continuation of this kind of a payment test. Smilow has used revenue from the Oncology Care Model program to develop dashboards to measure individual clinicians’ patterns of care and share data with them, Dr. Adelson said.
The changes have been “transformative,” including allowing for hiring of additional support staff, Dr. Adelson said.
“I am terrified about what’s going to happen at the end of the program if there is not another model to transition to,” Dr. Adelson said. “We will potentially be looking at layoffs of these people who are so incredible and have done so much for the care we are giving.”
In May 2019, some of Dr. Chong’s CMMI colleagues published a report in the Journal of the National Cancer Institute about the experiences of clinics participating in the Oncology Care Model. For instance, the U.S. Oncology Network used Monthly Enhanced Oncology Services [MEOS] payments through the program for new hires such as navigators, social workers, additional advanced practice providers, “and even data analysts,” according to the report.
Still, there have been “growing pains” with the start-up of the model, Dr. Chong said. Fellow participants on the NCCN panel cited the introduction of costly new drugs as one wrinkle in the early days of the Oncology Care Model. Another is that physicians in practices signed up for the program sometimes are unaware of it.
The American Society of Clinical Oncology also is advocating for a continuation of a model along the lines of the Oncology Care Model.
“It would be good to see it continue. We’re certainly supportive of continuing to work on this model and refine it and see where it goes,” Stephen S. Grubbs, MD, ASCO’s vice president for clinical affairs, said in an interview.
Dr. Grubbs said that CMS’s Oncology Care Model could not be greatly expanded in its current form, as it imposes a steep administrative burden for both practices who participate, as well as for CMMI. The current model also does not serve smaller and rural practices well, he said.
ASCO is refining its own proposal for an alternative payment approach. The group plans to present its Patient-Centered Oncology Payment (PCOP) model, first published in 2015, before an influential federal advisory group, the Physician-Focused Payment Model Technical Advisory Committee (PTAC), at a meeting in 2020.
WASHINGTON – Federal officials are considering how they might extend a test of payment approaches meant to spur more coordinated cancer care beyond the program’s current 2021 end date.
Alexandra Chong, PhD, the team lead for the Oncology Care Model at the Center for Medicare and Medicaid Innovation (CMMI), said her agency found strong support for sustaining this kind of effort at “practice transformation.” The Oncology Care Model, which kicked off in 2016, involves about 175 practices and 10 insurers.
“As we look forward, we certainly recognize and appreciate the importance of the impact that [the Oncology Care Model] made and the desire for a continuation, either in an iteration of the current model or a different model,” Dr. Chong said during a panel discussion at a policy summit sponsored by the National Comprehensive Cancer Network (NCCN).
Dr. Chong said the Centers for Medicare & Medicaid Services is aware that there is “a lot of interest” in the future of the program. “We have heard from our practices that this model has been an opportunity for them to provide the care that they really want to provide,” Dr. Chong said.
A fellow panelist at the NCCN event, Kerin Adelson, MD, chief quality officer of Yale’s Smilow Cancer Hospital, New Haven, Conn., pressed for continuation of this kind of a payment test. Smilow has used revenue from the Oncology Care Model program to develop dashboards to measure individual clinicians’ patterns of care and share data with them, Dr. Adelson said.
The changes have been “transformative,” including allowing for hiring of additional support staff, Dr. Adelson said.
“I am terrified about what’s going to happen at the end of the program if there is not another model to transition to,” Dr. Adelson said. “We will potentially be looking at layoffs of these people who are so incredible and have done so much for the care we are giving.”
In May 2019, some of Dr. Chong’s CMMI colleagues published a report in the Journal of the National Cancer Institute about the experiences of clinics participating in the Oncology Care Model. For instance, the U.S. Oncology Network used Monthly Enhanced Oncology Services [MEOS] payments through the program for new hires such as navigators, social workers, additional advanced practice providers, “and even data analysts,” according to the report.
Still, there have been “growing pains” with the start-up of the model, Dr. Chong said. Fellow participants on the NCCN panel cited the introduction of costly new drugs as one wrinkle in the early days of the Oncology Care Model. Another is that physicians in practices signed up for the program sometimes are unaware of it.
The American Society of Clinical Oncology also is advocating for a continuation of a model along the lines of the Oncology Care Model.
“It would be good to see it continue. We’re certainly supportive of continuing to work on this model and refine it and see where it goes,” Stephen S. Grubbs, MD, ASCO’s vice president for clinical affairs, said in an interview.
Dr. Grubbs said that CMS’s Oncology Care Model could not be greatly expanded in its current form, as it imposes a steep administrative burden for both practices who participate, as well as for CMMI. The current model also does not serve smaller and rural practices well, he said.
ASCO is refining its own proposal for an alternative payment approach. The group plans to present its Patient-Centered Oncology Payment (PCOP) model, first published in 2015, before an influential federal advisory group, the Physician-Focused Payment Model Technical Advisory Committee (PTAC), at a meeting in 2020.
REPORTING FROM NCCN POLICY SUMMIT 2019
Lumateperone schizophrenia drug seems to hit snag
FDA cancels lumateperone advisory panel
U.S. regulators canceled a July 31, 2019, advisory committee about lumateperone, an experimental schizophrenia drug that has had some mixed results in testing.
On July 23, the Food and Drug Administration announced the cancellation of the Psychopharmacologic Drugs Advisory Committee meeting it had previously called for to review the new drug application for lumateperone. The agency said the meeting was canceled because of “new information regarding the application.” The FDA said it was continuing to evaluate the application and would, as needed, announce a future meeting on it.
The developer of lumateperone, Intra-Cellular Therapies, issued its own statement on July 23, noting that a meeting had been scheduled with the FDA “shortly” and that an update would be provided after the meeting. The New York–based firm also said it recently had provided additional information to the FDA to meet agency requests. This information was related to “nonclinical studies.”
“The FDA canceled the advisory committee meeting to allow sufficient time to review this new and any forthcoming information as they continue” to review the new drug application for lumateperone, Intra-Cellular said in the July 23 statement. The company also said there may be an extension of the FDA’s Sept. 27, 2019, target action date on the lumateperone application.
Investors viewed this as bad news. Shares of Intra-Cellular on July 23 dropped from an opening price of $11.90 to a closing one of $8.19. On July 29, they closed at $8.12.
Still, it is unclear how the FDA will decide on the lumateperone application and whether the agency will call another advisory committee meeting on it.
Last year, the FDA accepted the application for lumateperone, a once-daily treatment, Intra-Cellular said. The agency had in 2017 given a fast-track designation to lumateperone for the treatment of schizophrenia.
Lumateperone is the lead product for the company.
On the company’s website, Intra-Cellular says three large randomized, double-blind, placebo-controlled trials have been done for lumateperone as a schizophrenia drug. In two of these studies, results for lumateperone at a 60-mg dose showed a “statistically significant separation from placebo on the primary endpoint, the Positive and Negative Syndrome Scale or PANSS total score.”
In a recent routine filing with the Securities and Exchange Commission, Intra-Cellular said it was having an “ongoing dialogue” with the FDA about lumateperone. The company in 2016 had announced that, in a phase 3 study known as ITI-007-302, lumateperone had not separated from placebo on the primary endpoint, change from baseline on the PANSS total score, in the predefined patient population. The active control for ITI-007-302, risperidone, did separate from placebo.
In the recent SEC filing, Intra-Cellular said the FDA already has confirmed that the results of ITI-007-302 did not preclude the submission of a new drug application.
Intra-Cellular also said “lumateperone was statistically significantly better than risperidone on key safety and tolerability parameters, and exhibited a safety profile similar to placebo” in the 302 study. Lumateperone’s failure to best placebo in the 302 test was “in part due to an unusually high placebo response at certain sites.”
FDA cancels lumateperone advisory panel
FDA cancels lumateperone advisory panel
U.S. regulators canceled a July 31, 2019, advisory committee about lumateperone, an experimental schizophrenia drug that has had some mixed results in testing.
On July 23, the Food and Drug Administration announced the cancellation of the Psychopharmacologic Drugs Advisory Committee meeting it had previously called for to review the new drug application for lumateperone. The agency said the meeting was canceled because of “new information regarding the application.” The FDA said it was continuing to evaluate the application and would, as needed, announce a future meeting on it.
The developer of lumateperone, Intra-Cellular Therapies, issued its own statement on July 23, noting that a meeting had been scheduled with the FDA “shortly” and that an update would be provided after the meeting. The New York–based firm also said it recently had provided additional information to the FDA to meet agency requests. This information was related to “nonclinical studies.”
“The FDA canceled the advisory committee meeting to allow sufficient time to review this new and any forthcoming information as they continue” to review the new drug application for lumateperone, Intra-Cellular said in the July 23 statement. The company also said there may be an extension of the FDA’s Sept. 27, 2019, target action date on the lumateperone application.
Investors viewed this as bad news. Shares of Intra-Cellular on July 23 dropped from an opening price of $11.90 to a closing one of $8.19. On July 29, they closed at $8.12.
Still, it is unclear how the FDA will decide on the lumateperone application and whether the agency will call another advisory committee meeting on it.
Last year, the FDA accepted the application for lumateperone, a once-daily treatment, Intra-Cellular said. The agency had in 2017 given a fast-track designation to lumateperone for the treatment of schizophrenia.
Lumateperone is the lead product for the company.
On the company’s website, Intra-Cellular says three large randomized, double-blind, placebo-controlled trials have been done for lumateperone as a schizophrenia drug. In two of these studies, results for lumateperone at a 60-mg dose showed a “statistically significant separation from placebo on the primary endpoint, the Positive and Negative Syndrome Scale or PANSS total score.”
In a recent routine filing with the Securities and Exchange Commission, Intra-Cellular said it was having an “ongoing dialogue” with the FDA about lumateperone. The company in 2016 had announced that, in a phase 3 study known as ITI-007-302, lumateperone had not separated from placebo on the primary endpoint, change from baseline on the PANSS total score, in the predefined patient population. The active control for ITI-007-302, risperidone, did separate from placebo.
In the recent SEC filing, Intra-Cellular said the FDA already has confirmed that the results of ITI-007-302 did not preclude the submission of a new drug application.
Intra-Cellular also said “lumateperone was statistically significantly better than risperidone on key safety and tolerability parameters, and exhibited a safety profile similar to placebo” in the 302 study. Lumateperone’s failure to best placebo in the 302 test was “in part due to an unusually high placebo response at certain sites.”
U.S. regulators canceled a July 31, 2019, advisory committee about lumateperone, an experimental schizophrenia drug that has had some mixed results in testing.
On July 23, the Food and Drug Administration announced the cancellation of the Psychopharmacologic Drugs Advisory Committee meeting it had previously called for to review the new drug application for lumateperone. The agency said the meeting was canceled because of “new information regarding the application.” The FDA said it was continuing to evaluate the application and would, as needed, announce a future meeting on it.
The developer of lumateperone, Intra-Cellular Therapies, issued its own statement on July 23, noting that a meeting had been scheduled with the FDA “shortly” and that an update would be provided after the meeting. The New York–based firm also said it recently had provided additional information to the FDA to meet agency requests. This information was related to “nonclinical studies.”
“The FDA canceled the advisory committee meeting to allow sufficient time to review this new and any forthcoming information as they continue” to review the new drug application for lumateperone, Intra-Cellular said in the July 23 statement. The company also said there may be an extension of the FDA’s Sept. 27, 2019, target action date on the lumateperone application.
Investors viewed this as bad news. Shares of Intra-Cellular on July 23 dropped from an opening price of $11.90 to a closing one of $8.19. On July 29, they closed at $8.12.
Still, it is unclear how the FDA will decide on the lumateperone application and whether the agency will call another advisory committee meeting on it.
Last year, the FDA accepted the application for lumateperone, a once-daily treatment, Intra-Cellular said. The agency had in 2017 given a fast-track designation to lumateperone for the treatment of schizophrenia.
Lumateperone is the lead product for the company.
On the company’s website, Intra-Cellular says three large randomized, double-blind, placebo-controlled trials have been done for lumateperone as a schizophrenia drug. In two of these studies, results for lumateperone at a 60-mg dose showed a “statistically significant separation from placebo on the primary endpoint, the Positive and Negative Syndrome Scale or PANSS total score.”
In a recent routine filing with the Securities and Exchange Commission, Intra-Cellular said it was having an “ongoing dialogue” with the FDA about lumateperone. The company in 2016 had announced that, in a phase 3 study known as ITI-007-302, lumateperone had not separated from placebo on the primary endpoint, change from baseline on the PANSS total score, in the predefined patient population. The active control for ITI-007-302, risperidone, did separate from placebo.
In the recent SEC filing, Intra-Cellular said the FDA already has confirmed that the results of ITI-007-302 did not preclude the submission of a new drug application.
Intra-Cellular also said “lumateperone was statistically significantly better than risperidone on key safety and tolerability parameters, and exhibited a safety profile similar to placebo” in the 302 study. Lumateperone’s failure to best placebo in the 302 test was “in part due to an unusually high placebo response at certain sites.”
Congress’ first pediatrician settles into new job
WASHINGTON – Kim Schrier, MD, the first pediatrician elected to the U.S. House of Representatives, describes her decision to seek a seat in Congress as a natural extension of her work caring for children.

Following the 2016 election, she said she became concerned about what she saw as rising threats to the health of families. The Republican congressional majority made several attempts to undo the Affordable Care Act. In an interview with Pediatric News, Dr. Schrier, a Democrat who represents Washington’s 8th district, said she worried about a rollback of the insurance protections the 2010 law created for people with preexisting conditions. She understood the need for these guarantees of access to care both as a physician and as a patient – Dr. Schrier was diagnosed with type 1 diabetes as a teenager.
Her concerns extended beyond the health care and insurance. Efforts to weaken environmental protections could create new risks for children’s health, she said. Dr. Schrier also worried about attempts to erode Roe v. Wade and to reduce funding for programs such as SNAP supplemental food assistance.
“I felt like every one of those elements was under assault and so who better to speak to that than the pediatrician with her own preexisting condition?” she said.
Dr. Schrier currently is the only female physician serving in Congress. “It’s exciting to be here and it’s exciting to bring that lens that no one else really has,” she said.
Still, the decision to run was not easy.
“I’ve left a practice that I love. I had no intention to run for office ever in my life.”
Dr. Schrier earned an undergraduate degree in astrophysics at the University of California, Berkeley. She worked for about a year at the Environmental Protection Agency before earning her medical degree at the University of California, Davis, and completing her residency at Stanford (Calif.) University. She was working as a pediatrician in the Seattle-based Virginia Mason Health System when she decided to run for Congress.
Her husband, David, and son, Sam, are staying on in the family’s home of Sammamish as she settles into her new work as a legislator. She said she returns home often.
“I go back and forth every week. I just didn’t see it working all that well with having them here,” she said. “I work from 8:00 or 9:00 in the morning until 9:00 at night and would never see them anyway.”
Dr. Schrier has long used an insulin pump and glucose monitors to keep her diabetes in check.
“It is a bit more difficult because my days are less predictable” when working in the Capitol, she said. “So I carry granola bars around in my purse.”
Bipartisan bill
Dr. Schrier said she is taking her time in deciding on the first health care legislation that she will introduce in the House. She’s considering measures that would focus on children’s health, possibly in connection with nutrition or with protection of those in immigrant communities.
“I’m looking at something that is uniquely me and know that a lot of my colleagues are going to be working on things like affordability of medications and I will partner with them,” she said.
Dr. Schrier said that she supports creating a way for people to buy into Medicare coverage, paying for this government insurance on a sliding scale. The American public is getting more comfortable with the idea of expanding access to Medicare, which “is incredibly popular but now exclusively available for seniors and other selected groups,” she said.
In her view, an expanded Medicare would stand among private insurance options, allowing for a gradual change.
“It’s a way that we could start tomorrow to let people buy in, as opposed to having a 5- or 10-year rollout that something as big and daunting as a Medicare-for-all plan would require,” she said.
But Dr. Schrier said she won’t start her legislative career with such a sweeping goal as a Medicare buy-in bill.
“I think it would be jumping the gun to come in right away with my own health plan because I really want to collaborate” with fellow lawmakers, she said.
Winning Republican support for her future legislation also is important to Dr. Schrier. “I want to make sure that I have the support of a lot of my colleagues,” she said. “I would like to see a successful first bill.
“The advantage of coming at this as a pediatrician and taking on a first bill or bills that relate to children is that both sides of the aisle can get behind what is good for families and children,” Dr. Schrier said. “I think that’s hard to really argue with.”
Dr. Schrier cosponsored her first piece of legislation in February, reaching across the aisle to work with Rep. Dan Newhouse (R-Wash.) to introduce H.R. 1048, a bill to authorize the next phase of a water resource management plan for Washington’s Yakima River basin.
Beyond that effort, Dr. Schrier said she’s seeking areas of common ground with Republican members of her state delegation. She had dinner in January with two Republican members of the Washington delegation – Rep. Cathy McMorris Rodgers and Rep. Jaime Herrera Beutler. Rep. McMorris Rodgers’ website describes her as a leader in the disabilities community and the only member of Congress to give birth three times while in office, while Rep. Herrera Beutler has spoken openly about her daughter’s battle with Potter’s syndrome, in which the kidneys fail to develop.
Within her party, Dr. Schrier is allied with the moderate New Democrat Coalition. Also in the coalition are two fellow physicians, Ami Bera, MD, and Raul Ruiz, MD, both from California. The New Democrat Coalition also has several lawmakers who flipped House seats from Republican control to Democratic in the 2018 election, a group that includes Dr. Schrier and Reps. Jennifer Wexton (D-VA) and Mikie Sherrill (D-NJ). It was the victories of these moderates that gave the Democrats control of the House. Yet, they get far less media attention than do House Democratic freshmen who are politically further to the left. These members, including Rep. Alexandria Ocasio-Cortez (D-N.Y.), tend to hail from districts where Republicans stand little chance of winning.
Dr. Schrier said the Democratic party, and Congress as a whole, should focus on issues such as affordable health care, on which the country can unite.
“If you’re looking for collaboration, that just doesn’t make the headlines and yet it is the absolute best thing for the country,” she said, adding that most Americans have more centrist views. “So we should really be governing to the middle and that’s how we can move our country forward.”
WASHINGTON – Kim Schrier, MD, the first pediatrician elected to the U.S. House of Representatives, describes her decision to seek a seat in Congress as a natural extension of her work caring for children.
Following the 2016 election, she said she became concerned about what she saw as rising threats to the health of families. The Republican congressional majority made several attempts to undo the Affordable Care Act. In an interview with Pediatric News, Dr. Schrier, a Democrat who represents Washington’s 8th district, said she worried about a rollback of the insurance protections the 2010 law created for people with preexisting conditions. She understood the need for these guarantees of access to care both as a physician and as a patient – Dr. Schrier was diagnosed with type 1 diabetes as a teenager.
Her concerns extended beyond the health care and insurance. Efforts to weaken environmental protections could create new risks for children’s health, she said. Dr. Schrier also worried about attempts to erode Roe v. Wade and to reduce funding for programs such as SNAP supplemental food assistance.
“I felt like every one of those elements was under assault and so who better to speak to that than the pediatrician with her own preexisting condition?” she said.
Dr. Schrier currently is the only female physician serving in Congress. “It’s exciting to be here and it’s exciting to bring that lens that no one else really has,” she said.
Still, the decision to run was not easy.
“I’ve left a practice that I love. I had no intention to run for office ever in my life.”
Dr. Schrier earned an undergraduate degree in astrophysics at the University of California, Berkeley. She worked for about a year at the Environmental Protection Agency before earning her medical degree at the University of California, Davis, and completing her residency at Stanford (Calif.) University. She was working as a pediatrician in the Seattle-based Virginia Mason Health System when she decided to run for Congress.
Her husband, David, and son, Sam, are staying on in the family’s home of Sammamish as she settles into her new work as a legislator. She said she returns home often.
“I go back and forth every week. I just didn’t see it working all that well with having them here,” she said. “I work from 8:00 or 9:00 in the morning until 9:00 at night and would never see them anyway.”
Dr. Schrier has long used an insulin pump and glucose monitors to keep her diabetes in check.
“It is a bit more difficult because my days are less predictable” when working in the Capitol, she said. “So I carry granola bars around in my purse.”
Bipartisan bill
Dr. Schrier said she is taking her time in deciding on the first health care legislation that she will introduce in the House. She’s considering measures that would focus on children’s health, possibly in connection with nutrition or with protection of those in immigrant communities.
“I’m looking at something that is uniquely me and know that a lot of my colleagues are going to be working on things like affordability of medications and I will partner with them,” she said.
Dr. Schrier said that she supports creating a way for people to buy into Medicare coverage, paying for this government insurance on a sliding scale. The American public is getting more comfortable with the idea of expanding access to Medicare, which “is incredibly popular but now exclusively available for seniors and other selected groups,” she said.
In her view, an expanded Medicare would stand among private insurance options, allowing for a gradual change.
“It’s a way that we could start tomorrow to let people buy in, as opposed to having a 5- or 10-year rollout that something as big and daunting as a Medicare-for-all plan would require,” she said.
But Dr. Schrier said she won’t start her legislative career with such a sweeping goal as a Medicare buy-in bill.
“I think it would be jumping the gun to come in right away with my own health plan because I really want to collaborate” with fellow lawmakers, she said.
Winning Republican support for her future legislation also is important to Dr. Schrier. “I want to make sure that I have the support of a lot of my colleagues,” she said. “I would like to see a successful first bill.
“The advantage of coming at this as a pediatrician and taking on a first bill or bills that relate to children is that both sides of the aisle can get behind what is good for families and children,” Dr. Schrier said. “I think that’s hard to really argue with.”
Dr. Schrier cosponsored her first piece of legislation in February, reaching across the aisle to work with Rep. Dan Newhouse (R-Wash.) to introduce H.R. 1048, a bill to authorize the next phase of a water resource management plan for Washington’s Yakima River basin.
Beyond that effort, Dr. Schrier said she’s seeking areas of common ground with Republican members of her state delegation. She had dinner in January with two Republican members of the Washington delegation – Rep. Cathy McMorris Rodgers and Rep. Jaime Herrera Beutler. Rep. McMorris Rodgers’ website describes her as a leader in the disabilities community and the only member of Congress to give birth three times while in office, while Rep. Herrera Beutler has spoken openly about her daughter’s battle with Potter’s syndrome, in which the kidneys fail to develop.
Within her party, Dr. Schrier is allied with the moderate New Democrat Coalition. Also in the coalition are two fellow physicians, Ami Bera, MD, and Raul Ruiz, MD, both from California. The New Democrat Coalition also has several lawmakers who flipped House seats from Republican control to Democratic in the 2018 election, a group that includes Dr. Schrier and Reps. Jennifer Wexton (D-VA) and Mikie Sherrill (D-NJ). It was the victories of these moderates that gave the Democrats control of the House. Yet, they get far less media attention than do House Democratic freshmen who are politically further to the left. These members, including Rep. Alexandria Ocasio-Cortez (D-N.Y.), tend to hail from districts where Republicans stand little chance of winning.
Dr. Schrier said the Democratic party, and Congress as a whole, should focus on issues such as affordable health care, on which the country can unite.
“If you’re looking for collaboration, that just doesn’t make the headlines and yet it is the absolute best thing for the country,” she said, adding that most Americans have more centrist views. “So we should really be governing to the middle and that’s how we can move our country forward.”
WASHINGTON – Kim Schrier, MD, the first pediatrician elected to the U.S. House of Representatives, describes her decision to seek a seat in Congress as a natural extension of her work caring for children.
Following the 2016 election, she said she became concerned about what she saw as rising threats to the health of families. The Republican congressional majority made several attempts to undo the Affordable Care Act. In an interview with Pediatric News, Dr. Schrier, a Democrat who represents Washington’s 8th district, said she worried about a rollback of the insurance protections the 2010 law created for people with preexisting conditions. She understood the need for these guarantees of access to care both as a physician and as a patient – Dr. Schrier was diagnosed with type 1 diabetes as a teenager.
Her concerns extended beyond the health care and insurance. Efforts to weaken environmental protections could create new risks for children’s health, she said. Dr. Schrier also worried about attempts to erode Roe v. Wade and to reduce funding for programs such as SNAP supplemental food assistance.
“I felt like every one of those elements was under assault and so who better to speak to that than the pediatrician with her own preexisting condition?” she said.
Dr. Schrier currently is the only female physician serving in Congress. “It’s exciting to be here and it’s exciting to bring that lens that no one else really has,” she said.
Still, the decision to run was not easy.
“I’ve left a practice that I love. I had no intention to run for office ever in my life.”
Dr. Schrier earned an undergraduate degree in astrophysics at the University of California, Berkeley. She worked for about a year at the Environmental Protection Agency before earning her medical degree at the University of California, Davis, and completing her residency at Stanford (Calif.) University. She was working as a pediatrician in the Seattle-based Virginia Mason Health System when she decided to run for Congress.
Her husband, David, and son, Sam, are staying on in the family’s home of Sammamish as she settles into her new work as a legislator. She said she returns home often.
“I go back and forth every week. I just didn’t see it working all that well with having them here,” she said. “I work from 8:00 or 9:00 in the morning until 9:00 at night and would never see them anyway.”
Dr. Schrier has long used an insulin pump and glucose monitors to keep her diabetes in check.
“It is a bit more difficult because my days are less predictable” when working in the Capitol, she said. “So I carry granola bars around in my purse.”
Bipartisan bill
Dr. Schrier said she is taking her time in deciding on the first health care legislation that she will introduce in the House. She’s considering measures that would focus on children’s health, possibly in connection with nutrition or with protection of those in immigrant communities.
“I’m looking at something that is uniquely me and know that a lot of my colleagues are going to be working on things like affordability of medications and I will partner with them,” she said.
Dr. Schrier said that she supports creating a way for people to buy into Medicare coverage, paying for this government insurance on a sliding scale. The American public is getting more comfortable with the idea of expanding access to Medicare, which “is incredibly popular but now exclusively available for seniors and other selected groups,” she said.
In her view, an expanded Medicare would stand among private insurance options, allowing for a gradual change.
“It’s a way that we could start tomorrow to let people buy in, as opposed to having a 5- or 10-year rollout that something as big and daunting as a Medicare-for-all plan would require,” she said.
But Dr. Schrier said she won’t start her legislative career with such a sweeping goal as a Medicare buy-in bill.
“I think it would be jumping the gun to come in right away with my own health plan because I really want to collaborate” with fellow lawmakers, she said.
Winning Republican support for her future legislation also is important to Dr. Schrier. “I want to make sure that I have the support of a lot of my colleagues,” she said. “I would like to see a successful first bill.
“The advantage of coming at this as a pediatrician and taking on a first bill or bills that relate to children is that both sides of the aisle can get behind what is good for families and children,” Dr. Schrier said. “I think that’s hard to really argue with.”
Dr. Schrier cosponsored her first piece of legislation in February, reaching across the aisle to work with Rep. Dan Newhouse (R-Wash.) to introduce H.R. 1048, a bill to authorize the next phase of a water resource management plan for Washington’s Yakima River basin.
Beyond that effort, Dr. Schrier said she’s seeking areas of common ground with Republican members of her state delegation. She had dinner in January with two Republican members of the Washington delegation – Rep. Cathy McMorris Rodgers and Rep. Jaime Herrera Beutler. Rep. McMorris Rodgers’ website describes her as a leader in the disabilities community and the only member of Congress to give birth three times while in office, while Rep. Herrera Beutler has spoken openly about her daughter’s battle with Potter’s syndrome, in which the kidneys fail to develop.
Within her party, Dr. Schrier is allied with the moderate New Democrat Coalition. Also in the coalition are two fellow physicians, Ami Bera, MD, and Raul Ruiz, MD, both from California. The New Democrat Coalition also has several lawmakers who flipped House seats from Republican control to Democratic in the 2018 election, a group that includes Dr. Schrier and Reps. Jennifer Wexton (D-VA) and Mikie Sherrill (D-NJ). It was the victories of these moderates that gave the Democrats control of the House. Yet, they get far less media attention than do House Democratic freshmen who are politically further to the left. These members, including Rep. Alexandria Ocasio-Cortez (D-N.Y.), tend to hail from districts where Republicans stand little chance of winning.
Dr. Schrier said the Democratic party, and Congress as a whole, should focus on issues such as affordable health care, on which the country can unite.
“If you’re looking for collaboration, that just doesn’t make the headlines and yet it is the absolute best thing for the country,” she said, adding that most Americans have more centrist views. “So we should really be governing to the middle and that’s how we can move our country forward.”
Brain injury in sickle cell merits more attention
BETHESDA, MD. – The risk of brain damage from sickle cell disease (SCD) merits more attention, even with progress made in recent decades to prevent strokes, according to Lori Jordan, MD, PhD, of Vanderbilt University, Nashville, Tenn.

“Whether we can see it or not, the same injury we’ve been talking about in the kidney and the liver and other places is occurring in the brain” with sickle cell disease, Dr. Jordan said at Sickle Cell in Focus, a conference held by the National Institutes of Health.
The concern about long-term brain injury reflects major shifts in SCD treatment. Improved medical care has transformed SCD from a disease that often resulted in an early death to more of a chronic condition, Dr. Jordan said, citing research that shows a survival rate of roughly 99% to age 18 years (Br J Haematol. 2016 Jun;173[6]:927-37).
One of the major success stories in SCD treatment also has been using primary prevention steps to cut the risk of overt stroke at least 10-fold, Dr. Jordan said. Primary prevention includes annual scans with transcranial Doppler ultrasound to identify children with SCD at high risk of stroke.
“What’s not changing is that there is silent injury that accumulates” and can cause lifelong harm, she said. “We want to protect our patients long term so that they can have a successful adult life, not just a successful childhood.”
Research done by one of Dr. Jordan’s colleagues at Vanderbilt, Michael R. DeBaun, MD, showed that regular blood-transfusion therapy significantly reduced the incidence of the recurrence of brain infarct in children with sickle cell anemia. (N Engl J Med. 2014 Aug 21;371[8]:699-710).
But the lessons from the work of Dr. DeBaun and his colleagues with their Silent Cerebral Infarct Multi-Center Clinical (SIT) Trial have not yet been fully adopted, Dr. Jordan said. That’s partly due to the inconvenience and cost of routinely administered blood transfusions to prevent silent cerebral infarcts, which, when used long term, cause side effects, she said.
Dr. Jordan said there’s growing interest in identifying patients at high risk for stroke and moving them toward stem cell transplant, though studies are ongoing. She urged greater attention to the high lifetime costs of strokes and other cerebrovascular complications, particularly in children and young adults.
While some of the brain infarcts are small and don’t result in focal weakness of the body, these “silent infarcts” do produce cognitive effects that reduce function, school performance, employment, and quality of life, she said.
“The injury to the brain is present, whether we can see it or not,” Dr. Jordan said. “In these precious patients, slow cognitive decline isn’t acceptable, frankly.”
Dr. Jordan reported having received funding from the American Heart Association and the National Institutes of Health for stroke prevention studies in SCD.
BETHESDA, MD. – The risk of brain damage from sickle cell disease (SCD) merits more attention, even with progress made in recent decades to prevent strokes, according to Lori Jordan, MD, PhD, of Vanderbilt University, Nashville, Tenn.
“Whether we can see it or not, the same injury we’ve been talking about in the kidney and the liver and other places is occurring in the brain” with sickle cell disease, Dr. Jordan said at Sickle Cell in Focus, a conference held by the National Institutes of Health.
The concern about long-term brain injury reflects major shifts in SCD treatment. Improved medical care has transformed SCD from a disease that often resulted in an early death to more of a chronic condition, Dr. Jordan said, citing research that shows a survival rate of roughly 99% to age 18 years (Br J Haematol. 2016 Jun;173[6]:927-37).
One of the major success stories in SCD treatment also has been using primary prevention steps to cut the risk of overt stroke at least 10-fold, Dr. Jordan said. Primary prevention includes annual scans with transcranial Doppler ultrasound to identify children with SCD at high risk of stroke.
“What’s not changing is that there is silent injury that accumulates” and can cause lifelong harm, she said. “We want to protect our patients long term so that they can have a successful adult life, not just a successful childhood.”
Research done by one of Dr. Jordan’s colleagues at Vanderbilt, Michael R. DeBaun, MD, showed that regular blood-transfusion therapy significantly reduced the incidence of the recurrence of brain infarct in children with sickle cell anemia. (N Engl J Med. 2014 Aug 21;371[8]:699-710).
But the lessons from the work of Dr. DeBaun and his colleagues with their Silent Cerebral Infarct Multi-Center Clinical (SIT) Trial have not yet been fully adopted, Dr. Jordan said. That’s partly due to the inconvenience and cost of routinely administered blood transfusions to prevent silent cerebral infarcts, which, when used long term, cause side effects, she said.
Dr. Jordan said there’s growing interest in identifying patients at high risk for stroke and moving them toward stem cell transplant, though studies are ongoing. She urged greater attention to the high lifetime costs of strokes and other cerebrovascular complications, particularly in children and young adults.
While some of the brain infarcts are small and don’t result in focal weakness of the body, these “silent infarcts” do produce cognitive effects that reduce function, school performance, employment, and quality of life, she said.
“The injury to the brain is present, whether we can see it or not,” Dr. Jordan said. “In these precious patients, slow cognitive decline isn’t acceptable, frankly.”
Dr. Jordan reported having received funding from the American Heart Association and the National Institutes of Health for stroke prevention studies in SCD.
BETHESDA, MD. – The risk of brain damage from sickle cell disease (SCD) merits more attention, even with progress made in recent decades to prevent strokes, according to Lori Jordan, MD, PhD, of Vanderbilt University, Nashville, Tenn.
“Whether we can see it or not, the same injury we’ve been talking about in the kidney and the liver and other places is occurring in the brain” with sickle cell disease, Dr. Jordan said at Sickle Cell in Focus, a conference held by the National Institutes of Health.
The concern about long-term brain injury reflects major shifts in SCD treatment. Improved medical care has transformed SCD from a disease that often resulted in an early death to more of a chronic condition, Dr. Jordan said, citing research that shows a survival rate of roughly 99% to age 18 years (Br J Haematol. 2016 Jun;173[6]:927-37).
One of the major success stories in SCD treatment also has been using primary prevention steps to cut the risk of overt stroke at least 10-fold, Dr. Jordan said. Primary prevention includes annual scans with transcranial Doppler ultrasound to identify children with SCD at high risk of stroke.
“What’s not changing is that there is silent injury that accumulates” and can cause lifelong harm, she said. “We want to protect our patients long term so that they can have a successful adult life, not just a successful childhood.”
Research done by one of Dr. Jordan’s colleagues at Vanderbilt, Michael R. DeBaun, MD, showed that regular blood-transfusion therapy significantly reduced the incidence of the recurrence of brain infarct in children with sickle cell anemia. (N Engl J Med. 2014 Aug 21;371[8]:699-710).
But the lessons from the work of Dr. DeBaun and his colleagues with their Silent Cerebral Infarct Multi-Center Clinical (SIT) Trial have not yet been fully adopted, Dr. Jordan said. That’s partly due to the inconvenience and cost of routinely administered blood transfusions to prevent silent cerebral infarcts, which, when used long term, cause side effects, she said.
Dr. Jordan said there’s growing interest in identifying patients at high risk for stroke and moving them toward stem cell transplant, though studies are ongoing. She urged greater attention to the high lifetime costs of strokes and other cerebrovascular complications, particularly in children and young adults.
While some of the brain infarcts are small and don’t result in focal weakness of the body, these “silent infarcts” do produce cognitive effects that reduce function, school performance, employment, and quality of life, she said.
“The injury to the brain is present, whether we can see it or not,” Dr. Jordan said. “In these precious patients, slow cognitive decline isn’t acceptable, frankly.”
Dr. Jordan reported having received funding from the American Heart Association and the National Institutes of Health for stroke prevention studies in SCD.
EXPERT ANALYSIS FROM SICKLE CELL IN FOCUS